Note: Descriptions are shown in the official language in which they were submitted.
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
METHODS AND COMPOUNDS FOR TREATING RETINOL-RELATED DISEASES
CROSS REFERENCE TO RELATED APPLICATIONS
This application claims the benefit of U.S. Provisional Application No.
60/975,765 titled
Methods and Compounds for Treating Retinol-Related Diseases, filed on
September 27, 2007, the
disclosure of which is hereby incorporated by reference in its entirety.
FIELD OF THE INVENTION
[0001] The methods and compositions described herein are directed to the
treatment of retinol-related
diseases in a subject by modulating the activity or availability of serum
retinol, retinol-binding protein
(RBP) and/or transthyretin (TTR) in the subject.
BACKGROUND OF THE INVENTION
[0002] Retinoids are essential for maintenance of normal growth, development,
immunity,
reproduction, vision and other physiological processes. Conversely, abnormal
production or
processing of retinoids correlates with the manifestation of disease
processes.
[0003] For example, more than 100 million of the world's children are vitamin-
A deficient, causing
blindness and death among these children. Excess vitamin-A levels in target
organs and tissues, such
as the eye, may also cause a variety of retinal diseases, including macular
degeneration. A large
variety of conditions, generally referred to as vitreoretinal diseases, can
affect the vitreous and retina
that lie on the back part of the eye, including the retinopathies and macular
degenerations and
dystrophies. Macular degeneration is a group of eye diseases that is the
leading cause of blindness for
those aged 55 and older in the United States, affecting more than 10 million
Americans. Some studies
predict a six-fold increase in the number of new cases of macular degeneration
over the next decade,
taking on the characteristics of an epidemic. Age-related macular degeneration
or dystrophy, a
particularly debilitating disease, leads to gradual loss of vision and
eventually severe damage to the
central vision.
[0004] There are two general categories of age-related macular degeneration:
the wet and dry forms.
Dry macular degeneration, which accounts for about 90 percent of all cases, is
also known as atrophic,
nonexudative, or drusenoid macular degeneration. With dry macular
degeneration, drusen typically
accumulates beneath the RPE tissue in the retina. Vision loss can then occur
when drusen interfere
with the function of photoreceptors in the macula. This form of macular
degeneration results in the
gradual loss of vision over many years.
[0005] Wet macular degeneration, which accounts for about 10 percent of cases,
is also known as
choroidal neovascularization, subretinal neovascularization, exudative, or
disciform degeneration. In
wet macular degeneration, abnormal blood vessel growth can form beneath the
macula; these vessels
can leak blood and fluid into the macula and damage photoreceptor cells.
Studies have shown that the
dry form of macular degeneration can lead to the wet form of macular
degeneration. The wet form of
macular degeneration can progress rapidly and cause severe damage to central
vision.
-1-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
SUMMARY OF THE INVENTION
[0006] Presented herein are methods, compounds, and compositions for the
treatment of retinol-
related diseases in a human subject or patient. Such diseases are macular
degenerations, macular
dystrophies and retinal dystrophies, including dry-form macular degenerations,
geographic atrophy,
and/or photoreceptor degeneration. Also presented herein are methods,
compounds, and compositions
for the treatment of hyperretinolemia (excess serum retinol levels) in a human
subject or patient. Also
presented herein are methods, compounds and compositions for lowering levels
of serum retinol, a
serum RBP (retinol binding protein), and/or a serum TTR (transthyretin) in a
human subject or patient.
Also presented herein are methods, compounds, and compositions for the
treatment of vitreoretinal
diseases such that the level of serum retinol in the body of a patient is
lowered. In some embodiments,
the vitreoretinal diseases are macular degenerations, macular dystrophies and
retinal dystrophies. In
some embodiment, the vitreoretinal diseases are dry form macular degeneration,
photoreceptor
degeneration, geographic atrophy, macular dystrophies, diabetic retinopathy,
wet form of macular
degeneration, retinopathy of prematurity, and/or retinitis pigmentosa.
[0007] In one aspect is a pharmaceutical composition comprising a compound of
Formula (I):
X ~
A/\B' E
D
Formula (I);
wherein:
A is 0, NH, or S;
-
B is a bond,-(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
D is isopropyl, isobutyl, sec-butyl, tert-butyl, neopentyl, sec-pentyl,
isopentyl,
cyclopropyl, cyclobutyl, cyclopentyl, methylenecyclopropyl,
methylenecyclobutyl,
methylenecyclopentyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR1 R, NR'-(C=O)-R, -(C1-C7)alkyl-(C=O)-OR, or -(C1-C7)alkyl-(C=O)-NR1R;
G
RisHor
G is -OR1, -(Ci-C6)alkyl, -(Ci-C6)alkyl-OR1, halogen, -COzR', -(Ci-C6)alkyl-
COzR1,
NHR1, -(Ci-C6)alkyl-NHR1, -(C=O)NHR', -(C1-C6)alkyl-(C=0)NHR1, -NHR'(C=0)Rl, -
(Ci-
C6)alkyl-NHR'(C=O)Rl;
R' is H or (Ci-C6)alkyl;
X is a halogen;
-2-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof; and
a pharmaceutically acceptable excipient. In one embodiment, the pharmaceutical
composition is an
oral pharmaeutical composition for systemic administration of the compound of
Formula (I).
[0008] In one embodiment is a pharmaceutical composition comprising a compound
having the
structure of Formula (II):
C1 ~
BE
Formula (II);
wherein:
A is 0, NH, or S;
-
B is a bond, -(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR1R, NR'-(C=O)-R, -(C1-C7)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NR'R;
G
R is H or 15 G is -OR1, -(Ci-C6)alkyl, -(Ci-C6)alkyl-OR1, halogen, -COzR', -
(Ci-C6)alkyl-COzR1,
NHR1, -(Ci-C6)alkyl-NHR1, -(C=O)NHR', -(C1-C6)alkyl-(C=O)NHR1, -NHR'(C=0)Rl, -
(Ci-
C6)alkyl-NHR'(C=O)Rl;
R' is H or (Ci-C6)alkyl;
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof; and
a pharmaceutically acceptable excipient. In one embodiment, the pharmaceutical
composition is an
oral pharmaeutical composition for systemic administration of the compound of
Formula (II).
[0009] In another embodiment is a pharmaceutical composition comprising a
compound having the
structure of Formula (II) wherein A is O. In a further embodiment is a
pharmaceutical composition
comprising a compound having the structure of Formula (II) wherein B is -
(CHz)õ and n is 1-6, or B is
-(C3-Cg)cycloalkyl. In yet a further embodiment is a pharmaceutical
composition comprising a
compound having the structure of Formula (II) wherein E is (C=O)-OR, a
carboxylic acid bioisostere,
-(C=O)-NR'R, -(C1-C7)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NR'R. In one
embodiment is a
pharmaceutical composition comprising a compound having the structure of
Formula (II) wherein A is
0, B is (C3-Cg)cycloalkyl, E is (C=O)-OR, and R is H. In a further embodiment,
B is cyclohexyl, and
R is H. In yet a further embodiment, B is cyclopentyl and R is H. In yet a
further embodiment is a
-3-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
C1
I O
O OH
compound having the structure . In another embodiment is a
C1
I O
O OH
compound having the structure
[0010] In one embodiment is a pharmaceutical composition comprising a compound
having the
G
structure of Formula (II) wherein R is
[0011] In another embodiment is a pharmaceutical composition comprising a
compound having the
structure of Formula (II) wherein E is (C=O)-OR. In a further embodiment is a
pharmaceutical
composition comprising a compound having the structure of Formula (II) wherein
R is H. In yet a
further embodiment is a composition comprising a compound of Formula (I)
selected from the group
consisting of: 5-(2-tert-butyl-4-chlorophenoxy)-N-(4-
hydroxyphenyl)pentanamide, 7-(2-tert-butyl-4-
chlorophenoxy)-N-(4-hydroxyphenyl)heptanamide, 4-(5-(2-tert-butyl-4-
chlorophenoxy)pentanamido)benzoic acid,4-(3-((2-tert-butyl-4-
chlorophenoxy)methyl)cyclopentanamido)benzoic acid, 5-(2-tert-butyl-4-
chlorophenoxy)pentanoic
acid, 4-(2-tert-butyl-4-chlorophenoxy)butanoic acid, 2-(3-((2-tert-butyl-4-
chlorophenoxy)methyl)cyclopentyl)acetic acid, 7-(2-tert-butyl-4-
chlorophenoxy)heptanoic acid, 4-(5-
(2-tert-butyl-4-chlorophenoxy)pentanamido)benzamide, 3-((2-tert-butyl-4-
chlorophenoxy)methyl)cyclohexanecarboxylic acid, 3-((2-tert-butyl-4-
chlorophenoxy)methyl)cyclopentanecarboxylic acid, 3-((2-tert-butyl-4-
chlorophenylamino)methyl)cyclopentanamide, 4-(3-((2-tert-butyl-4-
chlorophenoxy)methyl)cyclopentanecarboxamido)benzoic acid, and 5-(2-tert-butyl-
4-
chlorophenylthio)pentanoic acid
[0012] In one embodiment is a pharmaceutical composition comprising a compound
of Formula (I)
that inhibits retinol-retinol binding protein-transthyretin complex formation
wherein the IC50 of the
inhibition is less than about 5 M. In a further embodiment is a pharmaceutical
composition
comprising a compound of Formula (I) wherein the IC50 of the inhibition is
less than about 1 M. In
another embodiment is a pharmaceutical composition comprising a compound of
Formula (I) that
inhibits cytochrome P450 to less than about 50%. In yet a further embodiment
is a pharmaceutical
composition comprising a compound of Formula (I) that inhibits cytochrome P450
to less than about
10%. In yet another embodiment is a pharmaceutical composition comprising a
compound of Formula
-4-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
(I) wherein the compound of Formula (I) is used for the treatment of a
vitreoretinal disease. In a
further embodiment is a pharmaceutical composition comprising a compound of
Formula (I) wherein
the vitreoretinal disease is selected from the group consisting of: dry form
macular degeneration,
photoreceptor degeneration, geographic atrophy, macular dystrophies, diabetic
retinopathy, wet form
of macular degeneration, retinopathy of prematurity, and retinitis pigmentosa.
[0013] In one aspect is a pharmaceutical composition comprising a compound of
Formula (I):
X
i A/"'B'E
D
Formula (I);
wherein:
A is O, NH, or S;
-
B is a bond, -(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
D is isopropyl, isobutyl, sec-butyl, tert-butyl, neopentyl, sec-pentyl,
isopentyl,
cyclopropyl, cyclobutyl, cyclopentyl, methylenecyclopropyl,
methylenecyclobutyl,
methylenecyclopentyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR'R, NR'-(C=O)-R, -(C1-C7)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NR'R;
R is H, an optionally substituted aryl, or an optionally substituted
heteroaryl;
X is a halogen;
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof; and
a pharmaceutically acceptable excipient or carrier. In one embodiment, the
pharmaceutical
composition is an oral pharmaeutical composition for systemic administration
of the compound of
Formula (I).
[0014] In one embodiment is a pharmaceutical composition comprising a compound
having the
structure of Formula (II):
C1
BE
Formula (II);
wherein:
A is 0, NH, or S;
-
B is a bond, -(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
-5-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR1 R, NR'-(C=O)-R, -(C1-C7)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NR'R;
R is H, an optionally substituted aryl, or an optionally substituted
heteroaryl;
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof; and
a pharmaceutically acceptable excipient or carrier. In one embodiment, the
pharmaceutical
composition is an oral pharmaeutical composition for systemic administration
of the compound of
Formula (II).
[0015] In one embodiment is a pharmaceutical composition comprising a compound
having the
structure of Formula (II) wherein A is 0, B is (C3-Cg)cycloalkyl, E is (C=0)-
OR, and R is H; or an
active metabolite, or a pharmaceutically acceptable prodrug, salt, or solvate
thereof; and a
pharmaceutically acceptable excipient or carrier. In a further embodiment, is
a pharmaceutical
composition comprising a compound having the structure of Formula (II) wherein
A is 0, B is
cyclohexyl, and R is H; or an active metabolite, or a pharmaceutically
acceptable prodrug, salt, or
solvate thereof; and a pharmaceutically acceptable excipient or carrier. In
yet a further embodiment, is
a pharmaceutical composition comprising a compound having the structure of
Formula (II) wherein A
is 0, B is cyclopentyl and R is H; or an active metabolite, or a
pharmaceutically acceptable prodrug,
salt, or solvate thereof; and a pharmaceutically acceptable excipient or
carrier. In yet a further
embodiment is a pharmaceutical composition comprising a compound having the
structure
Cl
O
O OH
; or an active metabolite, or a pharmaceutically acceptable prodrug,
salt, or solvate thereof; and a pharmaceutically acceptable excipient or
carrier. In another embodiment
is a pharmaceutical composition comprising a compound having the structure
Cl
O
O OH
; or an active metabolite, or a pharmaceutically acceptable prodrug,
salt, or solvate thereof; and a pharmaceutically acceptable excipient or
carrier.
[0016] In another embodiment is a pharmaceutical composition comprising a
compound of Formula
(I) wherein the composition is in an amount sufficient to modulate the serum
retinol or ocular tissue
retinol level or activity in a mammal.
[0017] In one aspect is a method of treating dry form macular degeneration,
photoreceptor
degeneration, geographic atrophy, macular dystrophies, diabetic retinopathy,
wet form of macular
degeneration, retinopathy of prematurity, or retinitis pigmentosa in a patient
in need, comprising
administering to the patient a therapeutically effective amount of a compound
of Formula (I):
-6-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
X
A/\B'E
D
Formula (I);
wherein:
A is 0, NH, or S;
-
B is a bond,-(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
D is isopropyl, isobutyl, sec-butyl, tert-butyl, cyclopropyl, cyclobutyl,
cyclopentyl,
methylenecyclopropyl, methylenecyclobutyl, methylenecyclopentyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR1 R, NR'-(C=O)-R, -(C1-C7)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NR'R;
R is H, an optionally substituted aryl, an optionally substituted
heterocycloalkyl, or an
optionally substituted heteroaryl;
X is a halogen;
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof. In
one embodiment, the therapeutically effective amount of a compound of Formula
(I) is
provided in the form of an oral pharmaeutical composition for systemic
administration of the
compound.
[0018] In one embodiment is a method of treating dry form macular
degeneration, photoreceptor
degeneration, geographic atrophy, macular dystrophies, diabetic retinopathy,
wet form of macular
degeneration, retinopathy of prematurity, and retinitis pigmentosa in a
patient in need, comprising
administering to the patient a therapeutically effective amount of a compound
of Formula (II):
C1
I BE
Formula (II);
wherein:
A is O, NH, or S;
-
B is a bond, -(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR1 R, NR'-(C=O)-R, -(C1-C7)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NR'R;
R is H, an optionally substituted aryl, an optionally substituted
heterocycloalkyl, or an
optionally substituted heteroaryl;
-7-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof. In
one embodiment, the therapeutically effective amount of a compound of Formula
(II) is
provided in the form of an oral pharmaeutical composition for systemic
administration of the
compound.
[0019] In one aspect is a method of treating a vitreoretinal disease,
comprising administering to a
mammal a therapeutically effective amount of a compound of Formula (I):
X
A/\B' E
D
Formula (I);
wherein:
A is O, NH, or S;
-
B is a bond,-(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
D is isopropyl, isobutyl, sec-butyl, tert-butyl, cyclopropyl, cyclobutyl,
cyclopentyl,
methylenecyclopropyl, methylenecyclobutyl, methylenecyclopentyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR1R, NR'-(C=O)-R, -(C1-C7)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NR'R;
R is H, an optionally substituted aryl, an optionally substituted
heterocycloalkyl, or an
optionally substituted heteroaryl;
X is a halogen;
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof;
wherein the compound of Formula (I) modulates a retinol binding protein level
or activity in
the mammal. In one embodiment, the therapeutically effective amount of a
compound of
Formula (I) is provided in the form of an oral pharmaeutical composition for
systemic
administration of the compound.
[0020] In one embodiment is a method of treating a vitreoretinal disease
comprising administering to
a mammal a therapeutically effective amount of a compound of Formula (II):
C1
I BE
Formula (II);
wherein:
A is O, NH, or S;
-8-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
-
B is a bond, -(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR1 R, NR'-(C=O)-R, -(C1-C7)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NR'R;
R is H, an optionally substituted aryl, an optionally substituted
heterocycloalkyl, or an
optionally substituted heteroaryl;
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof;
wherein the compound of Formula (I) modulates a retinol binding protein level
or activity in
the mammal. In one embodiment, the therapeutically effective amount of a
compound of Formula (II)
is provided in the form of an oral pharmaeutical composition for systemic
administration of the
compound.
[0021] In another embodiment is a method of treating a vitreoretinal disease
comprising
administering to a mammal a therapeutically effective amount of a compound of
Formula (I) or (II)
wherein the retinol binding protein is RBP4. In a further embodiment is a
method of treating a
vitreoretinal disease comprising administering to a mammal a therapeutically
effective amount of a
compound of Formula (I) or (II) wherein the vitreoretinal disease is dry form
macular degeneration,
photoreceptor degeneration, geographic atrophy, macular dystrophies, diabetic
retinopathy, wet form
of macular degeneration, retinopathy of prematurity, and retinitis pigmentosa.
[0022] In one aspect is a method of lowering serum retinol binding protein or
transthyretin levels or
activity in a mammal, comprising administering a therapeutically effective
amount of a compound of
Formula (I):
X
A/\B' E
D
Formula (I)
wherein:
A is O, NH, or S;
-
B is a bond,-(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
D is isopropyl, isobutyl, sec-butyl, tert-butyl, cyclopropyl, cyclobutyl,
cyclopentyl,
methylenecyclopropyl, methylenecyclobutyl, methylenecyclopentyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR1 R, NR'-(C=O)-R, -(C1-C7)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NR'R;
R is H, an optionally substituted aryl, an optionally substituted
heterocycloalkyl, or an
optionally substituted heteroaryl;
X is a halogen;
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof;
-9-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
wherein the compound of Formula (I) modulates a serum retinol binding protein
or
transthyretin level or activity in the mammal. In one embodiment, the
therapeutically effective amount
of a compound of Formula (I) is provided in the form of an oral pharmaeutical
composition for
systemic administration of the compound.
[0023] In one aspect is a method of lowering serum retinol binding protein or
transthyretin levels or
activity in a mammal comprising administering a therapeutically effective
amount of a compound of
Formula (II):
C1 ~
BE
Formula (II);
wherein:
A is 0, NH, or S;
-
B is a bond, -(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR1R, NR'-(C=O)-R, -(C1-C7)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NR'R;
R is H, an optionally substituted aryl, an optionally substituted
heterocycloalkyl, or an
optionally substituted heteroaryl;
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof;
wherein the compound of Formula (II) modulates a serum retinol binding protein
or
transthyretin level or activity in the mammal. In one embodiment, the
therapeutically effective amount
of a compound of Formula (II) is provided in the form of an oral pharmaeutical
composition for
systemic administration of the compound.
[0024] In yet a further embodiment is a method of lowering a serum retinol
binding protein or
transthyretin level or activity in a mammal comprising administering a
therapeutically effective
amount of a compound of Formula (I) or (II) wherein the serum retinol binding
protein is RBP4. In
one embodiment is a method of lowering serum retinol binding protein or
transthyretin levels or
activity in a mammal comprising administering a therapeutically effective
amount of a compound of
Formula (I) or (II) wherein the compound of Formula (I) or (II) inhibits
transcription of retinol binding
protein or transthyretin in the mammal. In a further embodiment is a method of
lowering serum retinol
binding protein or transthyretin levels or activity in a mammal comprising
administering a
therapeutically effective amount of a compound of Formula (I) or (II) wherein
the compound of
Formula (I) or (II) inhibits translation of retinol binding protein or
transthyretin in the mammal. In yet
another embodiment is a method of lowering serum retinol binding protein or
transthyretin levels or
activity in a mammal comprising administering a therapeutically effective
amount of a compound of
-10-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
Formula (I) or (II) wherein the compound of Formula (I) or (II) inhibits the
binding of retinol to retinol
binding protein. In one embodiment is a method of lowering serum retinol
binding protein or
transthyretin levels or activity in a mammal comprising administering a
therapeutically effective
amount of a compound of Formula (I) or (II) wherein the compound of Formula
(I) or (II) inhibits the
binding of retinol binding protein to transthyretin. In another embodiment is
a method of lowering
serum retinol binding protein or transthyretin levels or activity in a mammal
comprising administering
a therapeutically effective amount of a compound of Formula (I) or (II)
wherein the compound of
Formula (I) or (II) increases retinol binding protein or transthyretin
clearance in the mammal.
[0025] In one aspect is a method for treating a retinol-related disease in a
mammal, comprising
administering to the mammal a therapeutically effective amount of a compound
of Formula (I):
X ~
B' E
D
Formula (I)
wherein:
A is 0, NH, or S;
-
B is a bond, -(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
D is isopropyl, isobutyl, sec-butyl, tert-butyl, cyclopropyl, cyclobutyl,
cyclopentyl,
methylenecyclopropyl, methylenecyclobutyl, methylenecyclopentyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR1 R, NR'-(C=O)-R, -(C1-C7)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NR'R;
R is H, an optionally substituted aryl, an optionally substituted
heterocycloalkyl, or an
optionally substituted heteroaryl;
X is a halogen;
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof. In
one embodiment, the therapeutically effective amount of a compound of Formula
(I) is provided in the
form of an oral pharmaeutical composition for systemic administration of the
compound.
[0026] In one embodiment is a method for treating a retinol-related disease in
a mammal comprising
administering a therapeutically effective amount of a compound of Formula
(II):
C1
I B.1E
Formula (II);
wherein:
A is 0, NH, or S;
-11-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
-
B is a bond, -(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR1 R, NR'-(C=O)-R, -(C1-C7)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NR'R;
R is H, an optionally substituted aryl, an optionally substituted
heterocycloalkyl, or an
optionally substituted heteroaryl;
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof. In
one embodiment, the therapeutically effective amount of a compound of Formula
(II) is provided in
the form of an oral pharmaeutical composition for systemic administration of
the compound.
[0027] In another embodiment is a method for treating a retinol-related
disease in a mammal,
comprising administering to the mammal a therapeutically effective amount of a
compound of
Formula (I) or (II) wherein the retinol-related disease is hyperostosis,
idiopathic intracranial
hypertension, amyloidosis, Alzheimer's disease, and Alstrom-Hallgren syndrome.
[0028] In one aspect is a method for reducing serum retinol levels in a mammal
with dry form age
related macular degeneration comprising administering to the mammal a
therapeutically effective
amount of a compound of Formula (I):
X
A/\B'E
D
Formula (I)
wherein:
A is O, NH, or S;
-
B is a bond, -(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
D is isopropyl, isobutyl, sec-butyl, tert-butyl, cyclopropyl, cyclobutyl,
cyclopentyl,
methylenecyclopropyl, methylenecyclobutyl, methylenecyclopentyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=0)-
NR1 R, NR'-(C=O)-R, -(C1-C7)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NR'R;
R is H, an optionally substituted aryl, an optionally substituted
heterocycloalkyl, or an
optionally substituted heteroaryl;
X is a halogen;
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof;
wherein the compound of Formula (I) does not directly modulate the activity of
an enzyme or
protein in the visual cycle. In one embodiment, the therapeutically effective
amount of a compound of
Formula (I) is provided in the form of an oral pharmaeutical composition for
systemic administration
of the compound.
-12-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
[0029] In one embodiment is a method for reducing serum retinol levels in a
mammal with dry form
age related macular degeneration comprising administering a therapeutically
effective amount of a
compound of Formula (II):
C1
I BE
Formula (II);
wherein:
A is 0, NH, or S;
-
B is a bond, -(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR1 R, NR'-(C=O)-R, -(C1-C7)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NR'R;
R is H, an optionally substituted aryl, an optionally substituted
heterocycloalkyl, or an
optionally substituted heteroaryl;
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof;
wherein the compound of Formula (II) does not directly modulate the activity
of an enzyme or
protein in the visual cycle. In one embodiment, the therapeutically effective
amount of a compound of
Formula (II) is provided in the form of an oral pharmaeutical composition for
systemic administration
of the compound.
[0030] In another embodiment is a method for reducing serum retinol levels in
a mammal with dry
form age related macular degeneration comprising administering to the mammal a
compound of
Formula (I) or (II) wherein the compound of Formula (I) or (II) does not
directly inhibit or bind to an
enzyme or protein in the visual cycle.
[0031] In a further embodiment is a method for reducing a serum retinol level
in a mammal with dry
form age related macular degeneration comprising administering to the mammal a
compound of
Formula (I) or (II) wherein the compound of Formula (I) or (II) does not
affect the rate of rhodopsin
regeneration.
[0032] In yet another embodiment is a method for reducing a serum retinol
level in a mammal with
dry form age related macular degeneration comprising administering to the
mammal a compound of
Formula (I) or (II) wherein the compound of Formula (I) or (II) does not
worsen delayed dark
adaptation.
[0033] In yet a further embodiment is a method for reducing a serum retinol
level in a mammal with
dry form age related macular degeneration comprising administering to the
mammal a compound of
Formula (I) or (II) wherein the compound of Formula (I) or (II) limits the
spread of geographic
atrophy or photoreceptor degeneration.
-13-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
[0034] In one aspect is a method for treating hyperretinolemia comprising
administering to a mammal
a therapeutically effective amount of a compound of Formula (I):
X
A/\B' E
D
Formula (I)
wherein:
A is 0, NH, or S;
-
B is a bond, -(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
D is isopropyl, isobutyl, sec-butyl, tert-butyl, cyclopropyl, cyclobutyl,
cyclopentyl,
methylenecyclopropyl, methylenecyclobutyl, methylenecyclopentyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR1 R, NR'-(C=O)-R, -(C1-C7)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NR'R;
R is H, an optionally substituted aryl, an optionally substituted
heterocycloalkyl, or an
optionally substituted heteroaryl;
X is a halogen;
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof. In
one embodiment, the therapeutically effective amount of a compound of Formula
(I) is provided in the
form of an oral pharmaeutical composition for systemic administration of the
compound.
[0035] In another embodiment is a method for treating hyperretinolemia
comprising administering a
therapeutically effective amount of a compound of Formula (II):
C1 ~
BE
Formula (II);
wherein:
A is 0, NH, or S;
-
B is a bond, -(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR1 R, NR'-(C=O)-R, -(C1-C7)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NR'R;
R is H, an optionally substituted aryl, an optionally substituted
heterocycloalkyl, or an
optionally substituted heteroaryl;
-14-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
or an active metabolite, or a pharmaceutically acceptable prodrug or solvate
thereof. In one
embodiment, the therapeutically effective amount of a compound of Formula (II)
is provided in the
form of an oral pharmaeutical composition for systemic administration of the
compound.
[0036] In a further embodiment is a method for treating hyperretinolemia
comprising administering to
a mammal a compound of Formula (I) or (II) wherein hyperretinolemia is
associated with a
vitreoretinal disease.
[0037] In a yet another embodiment is a method for treating hyperretinolemia
comprising
administering to a mammal a compound of Formula (I) or (II) wherein
hyperretinolemia is associated
with diabetes or Alzheimer's disease.
[0038] In one aspect is a method for treating a retinol-related disease in a
mammal, comprising
administering to the mammal an effective amount of a compound of Formula (I):
X ~
B' E
D
Formula (I)
wherein:
A is O, NH, or S;
-
B is a bond, -(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
D is isopropyl, isobutyl, sec-butyl, tert-butyl, cyclopropyl, cyclobutyl,
cyclopentyl,
methylenecyclopropyl, methylenecyclobutyl, methylenecyclopentyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR1R, NR'-(C=O)-R, -(C1-C7)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NR'R;
R is H, an optionally substituted aryl, an optionally substituted
heterocycloalkyl, or an
optionally substituted heteroaryl;
X is a halogen;
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof;
wherein the compound of Formula (I) inhibits a retinol-retinol binding protein-
transthyretin
complex formation; wherein an IC50 inhibition is less than about 5 M. In one
embodiment, the
effective amount of a compound of Formula (I) is provided in the form of an
oral pharmaeutical
composition for systemic administration of the compound.
[0039] In one embodiment is a method for treating a retinol-related disease in
a mammal comprising
administering to the mammal an effective amount of a compound of Formula (II):
C1 ~
BE
-15-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
Formula (II);
wherein:
A is 0, NH, or S;
-
B is a bond,-(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR1 R, NR'-(C=O)-R, -(C1-C7)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NR'R;
R is H, an optionally substituted aryl, an optionally substituted
heterocycloalkyl, or an
optionally substituted heteroaryl;
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof;
wherein the compound of Formula (II) inhibits the retinol-retinol binding
protein-transthyretin
complex formation; wherein the ICSO inhibition is less than about 5 M. In one
embodiment, the
effective amount of a compound of Formula (II) is provided in the form of an
oral pharmaeutical
composition for systemic administration of the compound.
[0040] In one embodiment is a method for treating a retinol-related disease in
a mammal, comprising
administering to the mammal an effective amount of a compound of Formula (I)
or (II), wherein the
compound of Formula (I) or (II) inhibits the retinol-retinol binding protein-
transthyretin complex
formation; wherein the IC50 inhibition is less than about 1 M.
[0041] In another embodiment is a method for treating a retinol-related
disease in a mammal,
comprising administering to the mammal an effective amount of a compound of
Formula (I) or (II),
wherein the compound of Formula (I) or (II) inhibits the retinol-retinol
binding protein-transthyretin
complex formation and wherein the compound of Formula (I) or (II) further
inhibits cytochrome P450
at less than about 50%.
[0042] In another embodiment is a method for treating a retinol-related
disease in a mammal,
comprising administering to the mammal an effective amount of a compound of
Formula (I) or (II),
wherein the compound of Formula (I) or (II) inhibits the retinol-retinol
binding protein-transthyretin
complex formation and wherein the compound of Formula (I) or (II) further
inhibits cytochrome P450
inhibition at less than about 10%.
[0043] In one aspect is a method for treating Type I or Type II diabetes in a
mammal, comprising
administering to the mammal a therapeutically effective amount of a compound
of Formula (I):
X
A/\B' E
D
Formula (I)
wherein:
A is 0, NH, or S;
-16-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
-
B is a bond,-(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
D is isopropyl, isobutyl, sec-butyl, tert-butyl, cyclopropyl, cyclobutyl,
cyclopentyl,
methylenecyclopropyl, methylenecyclobutyl, methylenecyclopentyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR1 R, NR'-(C=O)-R, -(C1-C7)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NR'R;
R is H, an optionally substituted aryl, an optionally substituted
heterocycloalkyl, or an
optionally substituted heteroaryl;
X is a halogen;
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof;
wherein the compound of Formula (I) modulates retinol binding protein or
transthyretin levels
or activity in the mammal. In one embodiment, the therapeutically effective
amount of a compound of
Formula (I) is provided in the form of an oral pharmaeutical composition for
systemic administration
of the compound.
[0044] In one embodiment is a method for treating Type I or Type II diabetes
in a mammal,
comprising administering to the mammal a therapeutically effective amount of a
compound of
Formula (II):
C1 ~
BE
Formula (II);
wherein:
A is 0, NH, or S;
-
B is a bond,-(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR1 R, NR'-(C=O)-R, -(C1-C7)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NR'R;
R is H, an optionally substituted aryl, an optionally substituted
heterocycloalkyl, or an
optionally substituted heteroaryl;
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof;
wherein the compound of Formula (II) modulates retinol binding protein or
transthyretin
levels or activity in the mammal. In one embodiment, the therapeutically
effective amount of a
compound of Formula (II) is provided in the form of an oral pharmaeutical
composition for systemic
administration of the compound.
-17-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
[0045] In one aspect is a method for reducing the serum retinol or ocular
tissue retinol levels in a
mammal, comprising administering to the mammal a therapeutically effective
amount of a compound
of Formula (I):
X ~
B' E
D
Formula (I)
wherein:
A is 0, NH, or S;
-
B is a bond,-(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
D is isopropyl, isobutyl, sec-butyl, tert-butyl, cyclopropyl, cyclobutyl,
cyclopentyl,
methylenecyclopropyl, methylenecyclobutyl, methylenecyclopentyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR'R, NR'-(C=O)-R, -(C1-C7)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NR'R;
R is H, an optionally substituted aryl, an optionally substituted
heterocycloalkyl, or an
optionally substituted heteroaryl;
X is a halogen;
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof. In
one embodiment, the therapeutically effective amount of a compound of Formula
(I) is provided in the
form of an oral pharmaeutical composition for systemic administration of the
compound.
[0046] In one embodiment is a method for reducing the serum retinol or ocular
tissue retinol levels in
a mammal, comprising administering to the mammal a therapeutically effective
amount of a
compound of Formula (II):
C1
I BE
Formula (II);
wherein:
A is 0, NH, or S;
-
B is a bond, -(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR1 R, NR'-(C=O)-R, -(C1-C7)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NR'R;
R is H, an optionally substituted aryl, an optionally substituted
heterocycloalkyl, or an
optionally substituted heteroaryl;
-18-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof. In
one embodiment, the therapeutically effective amount of a compound of Formula
(II) is provided in
the form of an oral pharmaeutical composition for systemic administration of
the compound.
[0047] In another embodiment is a method for reducing the serum retinol or
ocular tissue retinol
levels in a mammal, comprising administering to the mammal an effective amount
of a compound of
Formula (I) or (II) wherein the mammal is a human.
[0048] In a further embodiment is a method for reducing the serum retinol or
ocular tissue retinol
levels in a mammal, comprising administering to the mammal an effective amount
of a compound of
Formula (I) or (II) wherein the serum retinol or ocular tissue retinol level
is reduced by at least 20%.
[0049] In yet a further embodiment is a method for reducing the serum retinol
or ocular tissue retinol
levels in a mammal, comprising administering to the mammal an effective amount
of a compound of
Formula (I) or (II) further comprising administering at least one additional
agent selected from the
group consisting of an inducer of nitric oxide production, an anti-
inflammatory agent, a
physiologically acceptable antioxidant, a physiologically acceptable mineral,
a negatively charged
phospholipid, a carotenoid, a statin, an anti-angiogenic drug, a matrix
metalloproteinase inhibitor,
resveratrol and other trans-stilbene compounds, and 13-cis-retinoic acid.
[0050] In one aspect is a composition comprising a compound of Formula (I):
X ~
A/\B' E
D
Formula (I);
wherein:
A is 0, NH, or S;
-
B is a bond, -(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
with the proviso
that -(C2-C7)heteroalkyl cannot contain a nitrogen atom, -C(=O)-, -5-, -S(=O)-
, -S(=0)2-, -
NR'(C=O)-, -(C=O)NR'-, S(=O)zNR'-, -NR'S(=O)z, -O(C=O)NR'-, -NR'(C=O)O-, -
O(C=0)O-, -NR'(C=O)NR'-, -(C=0)O-, -O(C=O)-;
D is isopropyl, isobutyl, sec-butyl, tert-butyl, cyclopropyl, cyclobutyl,
cyclopentyl,
methylenecyclopropyl, methylenecyclobutyl, methylenecyclopentyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR1R, NR'-(C=O)-R; -(C1-C7)alkyl-(C=O)-OR, -(Ci-C7)alkyl-(C=O)-NRlR, Ci-C4
alkyl, C2-
C4 alkynyl, C3-C6 cycloalkyl, Ci-C4alkyl-(C3-C6cycloalkyl), aryl, heteroaryl,
substituted
heteroaryl, substituted aryl, arylalkyl, -C(O)R2 , hydroxy-(Ci-C6 alkyl),
arloxy, halo, Ci-C6-
haloalkyl, cyano, hydroxy, nitro, -O-C(O)NR2R3, -NR2R3(C=O)OR', -SO2NR2R3,
with the
proviso that heteroaryl cannot contain a nitrogen atom;
-19-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
R 2 and R3 are each independently selected from among H, Cl-C6 alkyl, and C3-
C6
cycloalkyl;
R is H, an optionally substituted aryl, or an optionally substituted
heteroaryl with the
proviso that when B is -S-, R cannot be pyrimidine; and when B is -(C2-
C7)alkyl, R cannot be
imidazole;
R' is H or (Ci-C6)alkyl;
X is a halogen;
x with the proviso that E cannot be D
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof.
[0051] In one embodiment is a composition comprising a compound of Formula
(I):
X
A/\B' E
D
Formula (I);
wherein:
A is 0, NH, or S;
-
B is a bond, -(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
with the proviso
that -(C2-C7)heteroalkyl cannot contain a nitrogen atom, -C(=O)-, -5-, -S(=O)-
, -S(=0)2-, -
NR'(C=O)-, -(C=O)NR'-, S(=O)zNR'-, -NR'S(=O)z, -O(C=O)NR'-, -NR'(C=O)O-, -
O(C=0)O-, -NR'(C=O)NR'-, -(C=0)O-, -O(C=O)-;
D is tert-butyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR1 R, NR'-(C=O)-R; -(C1-C7)alkyl-(C=O)-OR, -(Ci-C7)alkyl-(C=O)-NR1 R, Ci-C4
alkyl, C2-
C4 alkynyl, C3-C6 cycloalkyl, Ci-C4alkyl-(C3-C6cycloalkyl), aryl, substituted
aryl, arylalkyl, -
C(O)R2 , hydroxy-(Ci-C6 alkyl), arloxy, halo, Ci-C6-haloalkyl, cyano, hydroxy,
nitro, -O-
C(O)NR2 R3, -NR2R3(C=0)OR', -SO2NR2 R3;
R 2 and R3 are each independently selected from among H, Cl-C6 alkyl, and C3-
C6
cycloalkyl;
R is H, an optionally substituted aryl, or an optionally substituted
heteroaryl with the
proviso that when B is -S-, R cannot be pyrimidine; and when B is -(C2-
C7)alkyl, R cannot be
imidazole;
R' is H or (Ci-C6)alkyl;
X is Cl;
-20-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
with the proviso that the compound of Formula (I) cannot be a dimer;
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof.
[0052] A pharmaceutical composition comprising the compound of Formula (I):
X
A/\B' E
D
Formula (I);
wherein:
A is 0, NH, or S;
-
B is a bond, -(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
with the proviso
that -(C2-C7)heteroalkyl cannot contain a nitrogen atom, -C(=O)-, -5-, -S(=O)-
, -S(=0)2-, -
NR'(C=O)-, -(C=O)NR'-, S(=O)zNR'-, -NR'S(=O)z, -O(C=O)NR'-, -NR'(C=O)O-, -
O(C=0)O-, -NR'(C=O)NR'-, -(C=0)O-, -O(C=O)-;
D is isopropyl, isobutyl, sec-butyl, tert-butyl, cyclopropyl, cyclobutyl,
cyclopentyl,
methylenecyclopropyl, methylenecyclobutyl, methylenecyclopentyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR'R, NR'-(C=O)-R; -(C1-C7)alkyl-(C=O)-OR, -(Ci-C7)alkyl-(C=O)-NR'R, Ci-C4
alkyl, C2-
C4 alkynyl, C3-C6 cycloalkyl, Ci-C4alkyl-(C3-C6cycloalkyl), aryl, substituted
aryl, arylalkyl, -
C(O)R2 , hydroxy-(Ci-C6 alkyl), arloxy, halo, Ci-C6-haloalkyl, cyano, hydroxy,
nitro, -O-
C(O)NR2 R3, -NR2R3(C=0)OR', -SO2NR2 R3;
R2 and R3 are each independently selected from among H, Ci-C6 alkyl, and C3-C6
cycloalkyl;
R is H, an optionally substituted aryl, or an optionally substituted
heteroaryl with the
proviso that when B is -S-, R cannot be pyrimidine; and when B is -(C2-
C7)alkyl, R cannot be
imidazole;
R' is H or (Ci-C6)alkyl;
X is a halogen;
with the proviso that the compound of Formula (I) cannot be a dimer;
and a pharmaceutically acceptable carrier or excipient. In one embodiment, the
pharmaceutical composition is an oral pharmaeutical composition for systemic
administration of the compound of Formula (I).
[0053] In some embodiments are compositions wherein E is (C=O)-OR, -O-(C=O)-R,
-(C=0)-R, -
OR, a carboxylic acid bioisostere, -(C=O)-NR'R, NR'-(C=O)-R; -(Ci-C-,)alkyl-
(C=O)-OR, or -(Ci-
C-,)alkyl-(C=O)-NR'R. In other embodiments are compositions wherein X is Cl
and D is isopropyl,
tert-butyl or cyclopropyl. In further embodiments are compositions wherein D
is tert-butyl and X is Cl.
In some embodiments are compositions wherein B is -(CHz)õ and n is 1-6, or B
is -(C3-Cg)cycloalkyl.
-21-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
In some embodiments are compositions wherein A is O. In other embodiments are
compositions
wherein A is NH or S.
[0054] In some embodiments are methods for treatment described herein
comprising administering to
a mammal a therapeutically effective amount of the composition of the compound
of Formula (I):
X ~
B' E
D
Formula (I);
wherein:
A is 0, NH, or S;
-
B is a bond, -(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
with the proviso
that -(C2-C7)heteroalkyl cannot contain a nitrogen atom, -C(=O)-, -5-, -S(=O)-
, -S(=0)2-, -
NR'(C=O)-, -(C=O)NR'-, S(=O)zNR'-, -NR'S(=O)z, -O(C=O)NR'-, -NR'(C=O)O-, -
O(C=0)O-, -NR'(C=O)NR'-, -(C=0)O-, -O(C=O)-;
D is tert-butyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR'R, NR'-(C=O)-R; -(C1-C7)alkyl-(C=O)-OR, -(Ci-C7)alkyl-(C=O)-NR'R, Ci-C4
alkyl, C2-
C4 alkynyl, C3-C6 cycloalkyl, Ci-C4alkyl-(C3-C6cycloalkyl), aryl, substituted
aryl, arylalkyl, -
C(O)R2 , hydroxy-(Ci-C6 alkyl), arloxy, halo, Ci-C6-haloalkyl, cyano, hydroxy,
nitro, -O-
C(O)NR2 R3, -NR2R3(C=0)OR', -SO2NR2 R3;
R2 and R3 are each independently selected from among H, Ci-C6 alkyl, and C3-C6
cycloalkyl;
R is H, an optionally substituted aryl, or an optionally substituted
heteroaryl with the
proviso that when B is -S-, R cannot be pyrimidine; and when B is -(C2-
C7)alkyl, R cannot be
imidazole;
R' is H or (Ci-C6)alkyl;
X is Cl;
with the proviso that the compound of Formula (I) cannot be a dimer;
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof.
[0055] In one aspect is a compound of Formula (I):
x
B'E
D
Formula (I);
-22-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
wherein:
A is O;
-
B is a bond, -(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
with the proviso
that -(C2-C7)heteroalkyl cannot contain a nitrogen atom, -C(=O)-, -S-, -S(=O)-
, -S(=O)z-, -
NR'(C=O)-, -(C=O)NR'-, S(=O)zNR'-, -NR'S(=O)z, -O(C=O)NR'-, -NR'(C=O)O-, -
O(C=0)O-, -NR'(C=O)NR'-, -(C=0)O-, -O(C=O)-;
D is isopropyl, isobutyl, sec-butyl, tert-butyl, cyclopropyl, cyclobutyl,
cyclopentyl,
methylenecyclopropyl, methylenecyclobutyl, methylenecyclopentyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR1R, NR'-(C=O)-R; -(C1-C7)alkyl-(C=O)-OR, -(Ci-C7)alkyl-(C=O)-NRlR, Ci-C4
alkyl, C2-
C4 alkynyl, C3-C6 cycloalkyl, Ci-C4alkyl-(C3-C6cycloalkyl), aryl, substituted
aryl, arylalkyl, -
C(O)R2 , hydroxy-(Ci-C6 alkyl), arloxy, halo, Ci-C6-haloalkyl, cyano, hydroxy,
nitro, -O-
C(O)NR2 R3, -NR2(C=O)OR',or -SO2NR2 R3;
R2 and R3 are each independently selected from among H, Ci-C6 alkyl, and C3-C6
cycloalkyl;
R is H, -(C2-C7)alkyl, an optionally substituted aryl, or an optionally
substituted
heteroaryl with the proviso that when B is -S-, R cannot be pyrimidine; and
when B is -(C2-
C7)alkyl, R cannot be imidazole;
R' is H or (Ci-C6)alkyl; and
X is a halogen;
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof.
[0056] In some embodiments is a compound of Formula (I), wherein B is -(CHz)õ
and n is 1-6, or B
is -(C3-Cg)cycloalkyl. In other embodiments, E is (C=O)-OR, a carboxylic acid
bioisostere, -(C=O)-
NR1R, -(Ci-C7)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NRlR. In further
embodiments is a
compound of Formula (I), wherein D is isopropyl, tert-butyl or cyclopropyl. In
some embodiments, X
is Cl and D is tert-butyl.
[0057] In another aspect is a compound of Formula (I):
X
~ A/\B'E
D
Formula (I);
wherein:
A is NH, or S;
-
B is a bond, -(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
-23-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
D is isopropyl, isobutyl, sec-butyl, tert-butyl, cyclopropyl, cyclobutyl,
cyclopentyl,
methylenecyclopropyl, methylenecyclobutyl, methylenecyclopentyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR'R, NR'-(C=O)-R; -(C1-C7)alkyl-(C=O)-OR, -(C1-C7)alkyl-(C=O)-NR1R, Ci-C4
alkyl, C2-
C4 alkynyl, C3-C6 cycloalkyl, Ci-C4alkyl-(C3-C6cycloalkyl), aryl, substituted
aryl, arylalkyl, -
C(O)R2 , hydroxy-(Ci-C6 alkyl), arloxy, halo, Ci-C6-haloalkyl, cyano, hydroxy,
nitro, -O-
C(O)NR2 R3, -NR2(C=O)OR', or -SO2NR 2R3;
R 2 and R3 are each independently selected from among H, Cl-C6 alkyl, and C3-
C6
cycloalkyl;
R is H, -(C2-C7)alkyl, an optionally substituted aryl, or an optionally
substituted
heteroaryl;
R' is H or (Ci-C6)alkyl; and
X is a halogen;
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof.
[0058] In some embodiments is a compound of Formula (I), wherein B is -(CHz)õ
and n is 1-6, or B
is -(C3-Cg)cycloalkyl. In other embodiments, E is (C=O)-OR, a carboxylic acid
bioisostere, -(C=O)-
NR1 R, -(Ci-C-,)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NR1 R. In further
embodiments is a
compound of Formula (I), wherein D is isopropyl, tert-butyl or cyclopropyl. In
some embodiments, X
is Cl and D is tert-butyl.
[0059] Other objects, features and advantages of the methods, compounds, and
compositions
described herein will become apparent from the following detailed description.
It should be
understood, however, that the detailed description and the specific examples,
while indicating specific
embodiments, are given by way of illustration only.
BRIEF DESCRIPTION OF THE FIGURES
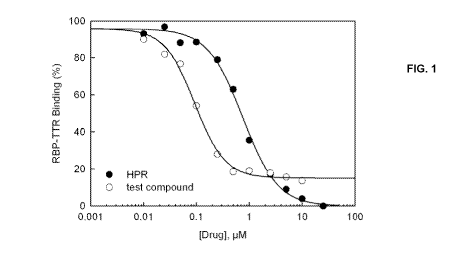
[0060] Figure 1. Dose-response relationship of a test compound compared to
fenretinide (HPR). The
data show that the test compound (with a chemical structure consistent with
Formula I) is
approximately 7-fold more potent than HPR at disrupting RBP4-TTR interaction
(ICSO = 0.1 vs. IC50
=
0.7, respectively).
[0061] Figure 2. HPLC analysis of serum retinol. Mice were treated with either
DMSO or a
compound having the structure of Formula I. Whole blood was collected from
tails veins and serum
was prepared. Serum samples were analyzed by HPLC. Representative
chromatographic tracings are
shown for a mouse receiving DMSO (panel A) and a mouse receiving the test
compound (panel B).
The retinol peak and its absorbance spectra are indicated in the plot.
[0062] Figure 3. Steady state serum concentrations of retinol and the test
compound. Mice were
administered daily doses of a test compound having the structure of Formula
I(20 mg/kg/day, i.p. in
DMSO). Following 28 days of treatment, blood samples were drawn and serum was
prepared for
-24-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
analysis by HPLC. A representative chromatogram which shows the presence of
the test compound
(detected at 280nm, dashed trace) and retinol (detected at 325 nm, solid
trace) is provided.
[0063] Figure 4. Immunoblot detection and densitometric quantitation of RBP4.
Western blot
detection of RBP4 in serum of mice treated with either DMSO (upper panel,
lanes 1- 5) or a test
compound having a structure consistent with Formula I (panel A, lanes 6 - 10).
Pixel density of the
bands shown lanes 1- 5 of panel A were determined and the mean pixel density
was taken as 100%.
The relative level of RBP4 in mice treated with the test compound was also
determined by pixel
densitometry. These data are shown in the histogram.
[0064] Figure 5. Chromatographic separation and identification of toxic
retinal fluorophores. Lipid
soluble components were extracted from the eyecups of an abca4 null mutant
mouse (BL6/129, aged 6
months). The eyecup extract was analyzed by HPLC with on-line absorbance
(solid trace) and
fluorescence (dashed trace) detection. The indicated fluorophores (A2E
precursors and A2E) are not
present in age-matched wild-type mice.
[0065] Figure 6. Genetic modulation of RBP4 in abca4 mutant mice: effects on
serum retinol, RBP4
and toxic retinal fluorophores. A line of mice which expresses heterozygous
mutations in RBP4 and
ABCA4 (RBP4+/-, ABCA4+/-) was generated in order to determine if genetic
reduction of RBP4
would be sufficient to reduce serum levels of retinol, RBP4 and toxic retinal
fluorophores within the
RPE. Serum retinol concentrations in RBP4+/-, ABCA4+/- mice were reduced by
greater than 50%
compared to mice with the normal complement of RBP (RBP4+/+, ABCA4-/-). This
degree of retinol
reduction is comparable to that observed in HPR-treated RBP4+/+, ABCA4-/- mice
(panel A).
Reductions in serum retinol correlated directly with reductions in RBP4, as
determined by immunoblot
analysis. Western blot identification of RBP4 in the various mouse lines is
shown in panel B.
Histological analysis were also performed. Tissue sections from RBP4+/+,
ABCA4+/- mice (panel C),
RBP4+/-, ABCA4+/- (panel D) and RBP4+/+, ABCA4+/+ (panel E) were analyzed by
fluorescence
microscopy. It is clear from this analysis that lipofuscin fluorophores
(indicated by white arrows) are
significantly reduced in mice with lower steady-state levels of RBP4 and
retinol.
[0066] Figure 7. Dose-response relationship of test compounds relative to
fenretinide (HPR). The
data show that the test compounds (test compound 2 having the chemical name 3-
((2-tert-butyl-4-
chlorophenoxy)methyl)cyclohexanecarboxylic acid and test compound 3 having the
chemical
name 3-((2-tert-butyl-4-chlorophenoxy)methyl)cyclopentanecarboxylic acid) are
effective at
disrupting RBP4-TTR interaction (IC50 = 0.25 - 0.70 M).
[0067] Figure 8. Cytochrome P450 inhibition profile for test compounds and
fenretinide (HPR). A
panel of six cytochrome P450 isozymes were screened to explore potential
toxicity associated with the
test compounds (test compound 2 having the chemical name 3-((2-tert-butyl-4-
chlorophenoxy)methyl)cyclohexanecarboxylic acid and test compound 3 having the
chemical
name 3-((2-tert-butyl-4-chlorophenoxy)methyl)cyclopentanecarboxylic acid) and
HPR (present
-25-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
at 2 M in the assays). The data show very little inhibition of the cytochrome
P450 isozymes by the
test compounds.
[0068] Figure 9. Quantitation of ocular retinoids following treatment with
test compound.
ABCA4-/- mice were administered either ATRP (Control) or ATRP + SIR- 1047
daily for 20 days (n =
3 mice/group). On the final day of dosing, a trace amount of [3H]ATROL (0.32
pmol, 8 Ci) in 100 1
corn oil was administered to all animals. Eyes were enucleated 5 hours later.
One eye from each
animal was used for retinoid analysis and the other eye was used for analysis
of A2E and related
fluorophores as described in the methods. The data show a marked reduction in
uptake of [3H]ATROL
in the animals treated with test compound. Analysis of each retinoid species
showed that the precursor
substrate for visual chromophore biosynthesis (ATRE) and the immediate
precursor for A2E
biosynthesis (AT-Ox) are significantly reduced.
[0069] Figure 10. Effect of test compound on reducing total fluorophore levels
in ABCA4-/-
mice. Mice were treated as described in Figure 1(above). At the end of the
study, one eye from each
animal was used to measure total fluorophore levels. Briefly, one whole eye
was homogenized in 1 ml
phosphate buffer saline (50 mM Na2HPO4, 150 mM NaC1, pH 7.8). Following
homogenization, 1 ml
methanol was added and the samples were mixed thoroughly. The mixtures were
incubated at room
temperature for 5 min and extracted twice with 2 ml hexane. The extracts were
concentrated to - 400
l for fluorescence measurements. Corrected fluorescence spectra were obtained
using a Spex
Fluorolog-3 spectrofluorimeter (Jobin Yvon Horiba, Edison, NJ) operated in
ratio mode. The samples
were excited at 488 nm and emissions at 500-700 m were monitored. The data
show a profound
reduction on total fluorophore levels in mice treated with the test compound.
[0070] Figure 11. Effect of test compound on reducing A2E and the A2E
precursor, A2PE-H2 in
ABCA4-/- mice. Mice were treated as described in Figure 1(above). At the end
of the study, one eye
from each animal was used to measure A2E and A2PE-H2. Briefly, one whole eye
was homogenized
in 1 ml phosphate buffer saline (50 mM Na2HPO4, 150 mM NaC1, pH 7.8).
Following
homogenization, 1 ml methanol was added and the samples were mixed thoroughly.
The mixtures
were incubated at room temperature for 5 min and extracted twice with 2 ml
hexane. The solvent was
evaporated under a stream of nitrogen and the sample residues were
reconstituted in 200 l
isopropanol (IPA) for analysis by HPLC. Fluorophores were separated on a
Zorbax RX-Si15- m
column (250 x 4.6-mm) equilibrated with a phospholipid moble phase
(hexane:IPA:ethano1:25mM
phosphate buffer: acetic acid 485:376:100:37.5:0.275 v:v) at a flow rate of 1
mUmin. The data show a
dramatic reduction in both A2E and its precursor in mice treated with test
compound compared to
vehicle-treated mice.
DETAILED DESCRIPTION OF THE INVENTION
[0071] Reference will now be made in detail to embodiments of the methods and
compositions
disclosed herein. Examples of the embodiments are illustrated in the following
Examples section.
-26-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
[0072] Of interest, the compounds of Formula (I) and (II) are used to provide
benefit to patients
suffering from or susceptible to various vitreoretinal diseases, including but
not limited to, macular
degeneration and dystrophy. Compounds of Formula (I) and (II) provide at least
one of the following
benefits to such patients: reduction in the level of serum retinol or RBP,
modulation in the level of
transthyretin, reduction in the formation of drusen, reduction in the
formation of the retinol-retinol-
binding protein complex, and reduction in the formation of the retinol-binding
protein-transthyretin
complex. As used herein, RBP refers to the protein RBP4.
[0073] The use of compounds of Formula (I) and (II) also includes as a
preventative therapy for wet
form age-related macular degeneration. In addition, the compounds of Formula
(I) and (II) provide
further therapeutic effect for wet-form age-related macular degeneration
because such compounds
additionally have anti-angiogenic activity.
The Visual Cycle
[0074] The vertebrate retina contains two types of photoreceptor cells - rods
and cones. Rods are
specialized for vision under low light conditions. Cones are less sensitive,
provide vision at high
temporal and spatial resolutions, and afford color perception. Under daylight
conditions, the rod
response is saturated and vision is mediated entirely by cones. Both cell
types contain a structure
called the outer segment comprising a stack of membranous discs. The reactions
of visual transduction
take place on the surfaces of these discs. The first step in vision is
absorption of a photon by an opsin-
pigment molecule (rhodopsin), which involves 11-cis to all-trans isomerization
of the chromophore.
Before light sensitivity can be regained, the resulting all-trans-retinal must
be converted back 11-cis-
retinal in a multi-enzyme process which takes place in the retinal pigment
epithelium, a monolayer of
cells adjacent to the retina.
Macular or Retinal Degenerations and Dystrophies
[0075] Macular degeneration (also referred to as retinal degeneration) is a
disease of the eye that
involves deterioration of the macula, the central portion of the retina.
Approximately 85% to 90% of
the cases of macular degeneration are the "dry" (atrophic or non-neovascular)
type. In dry macular
degeneration, the deterioration of the retina is associated with the formation
of small yellow deposits,
known as drusen, under the macula; in addition, the accumulation of lipofuscin
in the RPE leads to
photoreceptor degeneration and geographic atrophy. This phenomena leads to a
thinning and
degeneration of the macula. The location and amount of thinning in the retina
caused by the drusen
directly correlates to the amount of central vision loss. Degeneration of the
pigmented layer of the
retina and photoreceptors overlying drusen become atrophic and can cause a
slow loss of central
vision. Ultimately, loss of retinal pigment epithelium and underlying
photoreceptor cells results in
geographic atrophy. Administration of at least one compound having the
structure of Formula (I) or
(II) to a mammal reduces the formation of, or limit the spread of,
photoreceptor degeneration and/or
geographic atrophy in the eye of the mammal. By way of example only,
administration of a compound
-27-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
of Formula (I) or (II) to a mammal, is used to treat photoreceptor
degeneration and/or geographic
atrophy in the eye of the mammal.
[0076] In "wet" macular degeneration new blood vessels form (i.e.,
neovascularization) to improve
the blood supply to retinal tissue, specifically beneath the macula, a portion
of the retina that is
responsible for our sharp central vision. The new vessels are easily damaged
and sometimes rupture,
causing bleeding and injury to the surrounding tissue. Although wet macular
degeneration only occurs
in about 10 percent of all macular degeneration cases, it accounts for
approximately 90% of macular
degeneration-related blindness. Neovascularization can lead to rapid loss of
vision and eventual
scarring of the retinal tissues and bleeding in the eye. This scar tissue and
blood produces a dark,
distorted area in the vision, often rendering the eye legally blind. Wet
macular degeneration usually
starts with distortion in the central field of vision. Straight lines become
wavy. Many people with
macular degeneration also report having blurred vision and blank spots
(scotoma) in their visual field.
Growth promoting proteins called vascular endothelial growth factor, or VEGF,
have been targeted for
triggering this abnormal vessel growth in the eye. This discovery has lead to
aggressive research of
experimental drugs that inhibit or block VEGF. Studies have shown that anti-
VEGF agents blocks and
prevents abnormal blood vessel growth. Such anti-VEGF agents stop or inhibit
VEGF stimulation, so
there is less growth of blood vessels. Such anti-VEGF agents may also be
successful in anti-
angiogenesis or blocking VEGF's ability to induce blood vessel growth beneath
the retina, as well as
blood vessel leakiness. In one embodiment, administration of at least one
compound having the
structure of Formula (I) or (II) to a mammal reduces the formation of, or
limit the spread of, wet-form
age-related macular degeneration in the eye of the mammal. By way of example
only, administration
of a compound of Formula (I) or (II) to a mammal, is used to treat wet-form
age-related macular
degeneration in the eye of the mammal. Similarly, the compounds of Formula (I)
or (II) are used to
treat choroidal neovascularization and the formation of abnormal blood vessels
beneath the macula of
the eye of a mammal. In one embodiment, such therapeutic benefits result from
a number of effects:
lowering of serum retinol and thus ocular retinol levels; anti-angiogenic
activity, and/or the quelling of
geographic atrophy.
[0077] Stargardt Disease is a macular dystrophy that manifests as a recessive
form of macular
degeneration with an onset during childhood. See e.g., Allikmets et al.,
Science, 277:1805-07 (1997);
Lewis et al., Am. J. Hum. Genet., 64:422-34 (1999); Stone et al., Nature
Genetics, 20:328-29 (1998);
Allikmets, Am. J. Hum. Gen., 67:793-799 (2000); Klevering, et al,
Ophthalmology, 111:546-553
(2004). Stargardt Disease is characterized clinically by progressive loss of
central vision and
progressive atrophy of the RPE overlying the macula. Mutations in the human
ABCA4 gene for Rim
Protein (RmP) are responsible for Stargardt Disease. Early in the disease
course, patients show
delayed dark adaptation but otherwise normal rod function. Histologically,
Stargardt Disease is
associated with deposition of lipofuscin pigment granules in RPE cells.
-28-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
[0078] Mutations in ABCA4 have also been implicated in recessive retinitis
pigmentosa, see, e.g.,
Cremers et al., Hum. Mol. Genet., 7:355-62 (1998), recessive cone-rod
dystrophy, see id., and non-
exudative age-related macular degeneration, see e.g., Allikmets et al.,
Science, 277:1805-07 (1997);
Lewis et al., Am. J. Hum. Genet., 64:422-34 (1999), although the prevalence of
ABCA4 mutations in
AMD is still uncertain. See Stone et al., Nature Genetics, 20:328-29 (1998);
Allikmets, Am. J. Hum.
Gen., 67:793-799 (2000); Klevering, et al, Ophthalmology, 111:546-553 (2004).
Similar to Stargardt
Disease, these diseases are associated with delayed rod dark-adaptation. See
Steinmetz et al., Brit. J.
Ophthalm., 77:549-54 (1993). Lipofuscin deposition in RPE cells is also seen
prominently in AMD,
see Kliffen et al., Microsc. Res. Tech., 36:106-22 (1997) and some cases of
retinitis pigmentosa. See
Bergsma et al., Nature, 265:62-67 (1977). In addition, an autosomal dominant
form of Stargardt
Disease is caused by mutations in the ELOV4 gene. See Karan, et al., Proc.
Natl. Acad. Sci. (2005).
[0079] In addition, there are several types of macular degenerations that
affect children, teenagers or
adults that are commonly known as early onset or juvenile macular
degeneration. Many of these types
are hereditary and are looked upon as macular dystrophies instead of
degeneration. Some examples of
macular dystrophies include: Cone-Rod Dystrophy, Corneal Dystrophy, Fuch's
Dystrophy, Sorsby's
Macular Dystrophy, Best Disease, and Juvenile Retinoschisis, as well as
Stargardt Disease.
Modulation of Vitamin A levels
[0080] Vitamin A (all-trans retinol) is a vital cellular nutrient which cannot
be synthesized de novo
and therefore must be obtained from dietary sources. Vitamin A is a generic
term which may designate
any compound possessing the biological activity, including binding activity,
of retinol. One retinol
equivalent (RE) is the specific biologic activity of 1 g of all-trans retinol
(3.33 IU) or 6 g (10 IU) of
beta-carotene. Beta-carotene, retinol and retinal (vitamin A aldehyde) all
possess effective and reliable
vitamin A activity. Each of these compounds are derived from the plant
precursor molecule, carotene
(a member of a family of molecules known as carotenoids). Beta-carotene, which
consists of two
molecules of retinal linked at their aldehyde ends, is also referred to as the
provitamin form of vitamin
A.
[0081] Ingested (3-carotene is cleaved in the lumen of the intestine by (3 -
carotene dioxygenase to
yield retinal. Retinal is reduced to retinol by retinaldehyde reductase, an
NADPH requiring enzyme
within the intestines, and thereafter esterified to palmitic acid.
[0082] Following digestion, retinol in food material is transported to the
liver bound to lipid
aggregates. See Bellovino et al., Mol. Aspects Med., 24:411-20 (2003). Once in
the liver, retinol forms
a complex with retinol binding protein (RBP) and is then secreted into the
blood circulation. Before
the retinol-RBP holoprotein can be delivered to extra-hepatic target tissues,
such as by way of
example, the eye, it must bind with transthyretin (TTR). Zanotti and Berni,
Vitam. Horm., 69:271-95
(2004). It is this secondary complex which allows retinol to remain in the
circulation for prolonged
periods. Association with TTR facilitates RBP release from hepatocytes, and
prevents renal filtration
of the RBP-retinol complex. The retinol-RBP-TTR complex is delivered to target
tissues where retinol
-29-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
is taken up and utilized for various cellular processes. Delivery of retinol
to cells through the
circulation by the RBP-TTR complex is the major pathway through which cells
and tissue acquire
retinol.
[0083] Retinol uptake from its complexed retinol-RBP-TTR form into cells
occurs by binding of RBP
to cellular receptors on target cells. This interaction leads to endocytosis
of the RBP-receptor complex
and subsequent release of retinol from the complex, or binding of retinol to
cellular retinol binding
proteins (CRBP), and subsequent release of apoRBP by the cells into the
plasma. Other pathways
contemplate alternative mechanisms for the entry of retinol into cells,
including uptake of retinol alone
into the cell. See Blomhoff (1994) for review.
[0084] The methods, compounds, and compositions described herein are useful
for the modulation of
vitamin A levels in a mammalian subject. In particular, modulation of vitamin
A levels occurs through
the regulation of retinol binding protein (RBP) and transthyretin (TTR)
availability or activity in a
mammal. The methods, compounds, and compositions described herein provide for
the modulation of
RBP and TTR levels or activity in a mammalian subject, and subsequently
modulation of vitamin A
levels. Increases or decreases in vitamin A levels in a subject have effects
on retinol availability in
target organs and tissues. Therefore, providing a means of modulating retinol
or retinol derivative
availability will correspondingly modulate disease conditions caused by a lack
of or excess in local
retinol or retinol derivative concentrations in the target organs and tissues.
In addition, the therapeutic
methods described herein are used for the treatment of hyperretinolemia, in
which excessive levels of
serum retinol lead to vitreoretinal diseases, or symptoms associated with
vitreoretinal diseases (e.g.,
formation of lipofuscin or drusen).
[0085] For example, A2E, the major fluorophore of lipofuscin, is formed in
macular or retinal
degeneration or dystrophy, including age-related macular degeneration and
Stargardt Disease, due to
excess production of the visual-cycle retinoid, all-trans-retinaldehyde, a
precursor of A2E. Reduction
of vitamin A and all-trans retinaldehyde in the retina, therefore, will be
beneficial in reducing A2E and
lipofuscin build-up, and treatment of age-related macular degeneration.
[0086] Modulators (e.g. compounds of Formula (I) and (II)) that inhibit
delivery of retinol to cells
either through interruption of binding of retinol to apo RBP or holo RBP (RBP
+ retinol) to its
transport protein, TTR, or the increased renal excretion of RBP and TTR,
therefore, are useful in
decreasing serum vitamin A levels, and buildup of retinol and its derivatives
in target tissues such as
the eye.
[0087] Similarly, modulators which affect the availability of the retinol
transport proteins, retinol
binding protein (RBP) and transthyretin (TTR), are useful in decreasing serum
vitamin A levels, and
buildup of retinol (e.g., hyperretinolemia) and its derivatives and physical
manifestations in target
tissues, such as the eye. TTR, for example, has been shown to be a component
of Drusen constituents,
suggesting a direct involvement of TTR in age-related macular degeneration.
Mullins, RF, FASEB J.
14:835-846 (2000); Pfeffer BA, et al., Molecular Vision 10:23-30 (2004).
-30-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
[0088] The same approach to modulation of RBP and/or TTR levels or activity in
a mammal is
expected to find use in the treatment of metabolic disorders, such as type I
or type II diabetes (obese
and/or non-obese), IIH, bone-related disorders, such as hyperostosis, protein
misfolding and
aggregation diseases, such as systemic amyloidoses and Alzheimer's disease,
and Alstrom-Hallgren
syndrome.
[0089] One embodiment of the methods, compounds, and compositions disclosed
herein, therefore,
provides for the modulation of RBP or TTR levels or activity in a mammal by
administering to a
mammal a therapeutically effective amount of at least one of the compounds of
Formula (I) or
Formula (II).
Retinol Binding Protein (RBP) and Transthyretin (TTR)
[0090] Retinol binding protein, or RBP, is a single polypeptide chain, with a
molecular weight of
approximately 21 kD. RBP has been cloned and sequenced, and its amino acid
sequence determined.
Colantuni et al., Nuc. Acids Res., 11:7769-7776 (1983). The three-dimensional
structure of RBP
reveals a specialized hydrophobic pocket designed to bind and protect the fat-
soluble vitamin retinol.
Newcomer et al., EMBO J., 3:1451-1454 (1984). In in vitro experiments,
cultured hepatocytes have
been shown to synthesize and secrete RBP. Blaner, W.S., Endocrine Rev., 10:308-
316 (1989).
Subsequent experiments have demonstrated that many cells contain mRNA for RBP,
suggesting a
widespread distribution of RBP synthesis throughout the body. See Blaner
(1989). Most of the RBP
secreted by the liver contains retinol in a 1:1 molar ratio, and retinol
binding to RBP is required for
normal RBP secretion.
[0091] In cells, RBP tightly binds to retinol in the endoplasmic reticulum,
where it is found in high
concentrations. Binding of retinol to RBP initiates a translocation of retinol-
RBP from endoplasmic
reticulum to the Golgi complex, followed by secretion of retinol-RBP from the
cells. RBP secreted
from hepatocytes also assists in the transfer of retinol from hepatocytes to
stellate cells, where direct
secretion of retinol-RBP into plasma takes place.
[0092] In plasma, approximately 95% of the plasma RBP is associated with
transthyretin (TTR) in a
1:1 mol/mol ratio, wherein essentially all of the plasma vitamin A is bound to
RBP. TTR is a well-
characterized plasma protein consisting of four identical subunits with a
molecular weight of 54,980
Daltons. The full three-dimensional structure, elucidated by X-ray
diffraction, reveals extensive (3-
sheets arranged tetrahedrally. Blake et al., J. Mol. Biol., 121:339-356
(1978). A channel runs through
the center of the tetramer which contains two binding sites for thyroxine.
However, only one thyroxine
molecule appears to be bound normally to TTR due to negative cooperativity.
The complexation of
TTR to RBP-retinol is thought to reduce the glomerular filtration of retinol,
thereby increasing the
half-life of retinol and RBP in plasma by about threefold.
Modulation of RBP or TTR Binding or Clearance in a Subject
[0093] Before retinol bound to RBP is transported in the blood stream for
delivery to the eye, it must
be complexed with TTR. It is this secondary complex which allows retinol to
remain in the circulation
-31-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
for prolonged periods. In the absence of TTR, the retinol-RBP complex is
rapidly excreted in the
urine. Similarly, in the absence of RBP, retinol transport in the blood stream
and uptake by cells is
diminished.
[0094] Another embodiment described herein, therefore, is to modulate
availability of RBP or TTR
for complexing to retinol or retinol-RBP in the blood stream by lowering RBP
or TTR binding
characteristics or clearance rates. As mentioned above, the TTR binding to RBP
holoprotein decreases
the clearance rate of RBP and retinol. Therefore, by modulating either RBP or
TTR availability or
activity, retinol levels will be likewise modulated in a subject in need
thereof.
[0095] For example, antagonists of retinol binding to RBP are used in the
methods, compounds, and
compositions disclosed herein. An antagonist of retinol binding to RBP
includes compounds of
Formula (I) or (II) which compete with the binding of retinol to RBP.
[0096] As mentioned above, one means by which RBP binding to retinol is
modulated is to
competitively bind compounds of Formula (I) or (II). Therefore, one embodiment
of the methods and
compositions disclosed herein provides for lowering RBP levels or activity via
compounds having the
structure of Formula (I):
X
A/\B'E
D
Formula (I);
wherein:
A is 0, NH, or S;
-
B is a bond,-(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
D is isopropyl, isobutyl, sec-butyl, tert-butyl, cyclopropyl, cyclobutyl,
cyclopentyl,
methylenecyclopropyl, methylenecyclobutyl, methylenecyclopentyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR1 R, NR'-(C=O)-R, -(C1-C7)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NR'R;
R is H, an optionally substituted aryl, or an optionally substituted
heteroaryl;
X is a halogen;
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof;
wherein the compound of Formula (I) modulates RBP levels or activity;
[0097] Another embodiment of the methods and compositions disclosed herein
provides for lowering
RBP levels or activity via compounds having the structure of Formula (II):
-32-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
C1
I BE
Formula (II);
wherein:
A is 0, NH, or S;
-
B is a bond, -(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR1 R, NR'-(C=O)-R, -(C1-C7)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NR'R;
G
R is H or 10 G is -OR', -(Ci-C6)alkyl, -(Ci-C6)alkyl-OR1, halogen, -CO2R1, -
(Ci-C6)alkyl-COzR1,
NHR1, -(Ci-C6)alkyl-NHR1, -(C=O)NHR', -(C1-C6)alkyl-(C=O)NHR1, -NHR'(C=0)Rl, -
(Ci-
C6)alkyl-NHR'(C=O)Rl;
R' is H or (Ci-C6)alkyl;
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof;
wherein the compound of Formula (II) modulates RBP levels of activity.
Detection of modulator activity
[0098] In some embodiments, the compounds and compositions disclosed herein
are also be used in
assays for detecting perturbations in RBP or TTR availability through
conventional means. For
example, a subject is treated with any of the compounds or compositions
disclosed herein, and RBP or
TTR levels quantified using conventional assay techniques. See Sundaram, M.,
et al., Biochem. J.
362:265-271 (2002). For example, a typical non-competitive sandwich assay is
an assay disclosed in
U.S. Pat. No. 4,486,530, incorporated herein by reference. In this method, a
sandwich complex, for
example an immune complex, is formed in an assay medium. The complex comprises
the analyte, a
first antibody, or binding member, that binds to the analyte and a second
antibody, or binding member
that binds to the analyte or a complex of the analyte and the first antibody,
or binding member.
Subsequently, the sandwich complex is detected and is related to the presence
and/or amount of
analyte in the sample. The sandwich complex is detected by virtue of the
presence in the complex of a
label wherein either or both the first antibody and the second antibody, or
binding members, contain
labels or substituents capable of combining with labels. For example, the
sample is plasma, blood,
feces, tissue, mucus, tears, saliva, or urine, for example for detecting
modulation of clearance rates for
-33-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
RBP or TTR. For a more detailed discussion of this approach see U.S. Pat. Nos.
Re 29,169 and
4,474,878, the relevant disclosures of which are incorporated herein by
reference.
[0099] In a variation of the above sandwich assay, the sample in a suitable
medium is contacted with
labeled antibody or binding member for the analyte and incubated for a period
of time. Then, the
medium is contacted with a support to which is bound a second antibody, or
binding member, for the
analyte. After an incubation period, the support is separated from the medium
and washed to remove
unbound reagents. The support or the medium is examined for the presence of
the label, which is
related to the presence or amount of analyte. For a more detailed discussion
of this approach see U.S.
Pat. No. 4,098,876, the relevant disclosure of which is incorporated herein by
reference.
[00100] In some embodiments, the modulators disclosed herein are also used in
in vitro assays for
detecting perturbations in RBP or TTR activity. For example, the modulator is
added to a sample
comprising RBP, TTR and retinol to detect complex disruption. A component, for
example, RBP,
TTR, retinol or the modulator, is labeled to determine if disruption of
complex formation occurs. For
example, complex formation and subsequent disruption is detected and/or
measured through
conventional means, such as the sandwich assays disclosed above. Other
detection systems are also
used to detect modulation of RBP or TTR binding, for example, FRET detection
of RBP-TTR-retinol
complex formation. See U.S. Provisional Patent Application No. 60/625,532
"Fluorescence Assay for
Modulators of Retinol Binding," herein incorporated by reference.
[00101] In addition, other potential modulators which include, but are not
limited to, small molecules,
polypeptides, nucleic acids and antibodies, are also screened using the in
vitro detection methods
described above. For example, the methods and compositions described herein is
used to screen small
molecule libraries, nucleic acid libraries, peptide libraries or antibody
libraries in conjunction with the
teachings disclosed herein. Methods for screening libraries, such as
combinatorial libraries and other
libraries disclosed above, are found, for example, in U.S. Pat. Nos.
5,591,646; 5,866,341; and
6,343,257, which are hereby incorporated by reference.
In vivo detection of modulator activity
[00102] In addition to the in vitro methods disclosed above, in some
embodiments, the methods and
compositions disclosed herein are used in conjunction with in vivo detection
and/or quantitation of
modulator activity on TTR or RBP availability. For example, labeled TTR or RBP
is injected into a
subject, wherein a candidate modulator added before, during or after the
injection of the labeled TTR
or RBP. The subject is a mammal, for example a human; however other mammals,
such as primates,
horse, dog, sheep, goat, rabbit, mice or rats are further examples. A
biological sample is then removed
from the subject and the label detected to determine TTR or RBP availability.
A biological sample
includes, but is not limited to, plasma, blood, urine, feces, mucus, tissue,
tears or saliva. Detection of
the labeled reagents disclosed herein takes place using any of the
conventional means, depending upon
the nature of the label. Examples of monitoring devices for chemiluminescence,
radiolabels and other
-34-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
labeling compounds are found in U.S. Pats. No. 4,618,485; 5,981,202, the
relevant disclosures of
which are herein incorporated by reference.
Hyperretinolemia
[00103] Retinol is a fat-soluble, antioxidant vitamin. At proper levels,
retinol is important in vision and
bone growth but can be problematic if they are present in excess.
Hyperretinolemia is the presence of
elevated or abnormal levels of retinol in the blood and is thought to be
related to diseases and
conditions such as Type I and Type II diabetes, hyperostosis, such as, diffuse
idiopathic skeletal
hyperostosis (DISH), and vitreoretinal diseases, such as macular degeneration.
The methods,
compounds, and compositions described herein, which reduce the level of
retinol in the blood are used
to treat such diseases and conditions. Also, the methods, compounds, and
compositions, presented
herein, are effective in reducing retinol availability in target organs and
tissues such as the eye. By
reducing retinol concentrations in the blood the availability of retinol in
organs such as the liver may
modulate the formation of the retinol-retinol-binding protein complex and/or
formation of the retinol-
retinol-binding protein-transthyretin complex. In one embodiment, is a method
of treating a patient
with hyperretinolemia comprising administering a therapeutically effective
amount of a compound of
Formula (I) or (II) wherein the compound of Formula (I) or (II) reduces the
serum levels or activity of
retinol.
Metabolic Disorders
[00104] Metabolic disorders, including Type I and Type II diabetes mellitus
(obese and/or non-obese),
have also been associated with abnormal retinol levels.
Type I Diabetes (Insulin-Dependent Diabetes Mellitus)
[00105] Type I diabetes is a severe form of diabetes. If left untreated, type
I diabetes results in ketosis
of the patient and rapid degeneration. Approximately 10-20% of diabetic
patients are classified as type
I, comprising mainly young individuals. Non-obese adults also comprise type I
diabetic patients,
although at fewer numbers.
[00106] Type I diabetes is a catabolic disorder, where circulating levels of
insulin are virtually absent
and plasma glucagon levels elevated. Type I diabetes is believed to have auto-
immune origins,
possibly resulting from an infectious or toxic environmental insult to the
pancreatic B cells in affected
individuals. In support of the auto-immune theory, autoantibodies to insulin
and islet cells have been
detected in type I diabetes patients, as compared to non-diabetic individuals.
[00107] Lower levels of retinol, with observed decreases in retinol binding
protein (RBP) levels and
increased urinary excretion of RBP, has been correlated with type I diabetes
in juveniles. See Basu,
TK, et al. Am. J. Clin. Nutr. 50:329-331 (1989); Durbey, SW et al., Diabetes
Care 20:84-89 (1997).
The lower levels of retinol and RBP are accompanied by a concomitant decrease
in zinc metabolism, a
factor necessary for the synthesis of RBP in hepatic cells. See Cunningham,
JJ, et al. Metabolism
42:1558-1562 (1994). In contrast, tocopherol, or vitamin E levels, are
unchanged in type I diabetic
patients. See Basu, TK et al (1989).
-35-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
[00108] The lower levels of retinol are observed despite elevated levels of
vitamin A in hepatic storage
cells. See Tuitoek PJ, et al. Br. J. Nutr. 75: 615-622 (1996). Studies
demonstrating the linkage between
vitamin A status and insulin secretion show that only insulin treatment can
relieve the suppressed
levels of vitamin A in type I diabetic subjects. Tuitoek, PJ et al., J. Clin.
Biochem. Nutr. 19:165-169
(1996). In contrast, dietary supplementation of vitamin A does not normalize
metabolic availability of
vitamin A. Id.
[00109] These studies demonstrate the interconnection between vitamin A and
insulin regulation of
glucose transport into muscle and adipocyte cells. Further studies have
strengthened this
interconnection by demonstrating the requirement of vitamin A for normal
insulin secretion. See
Chertow, BS, et al., J. Clin. Invest. 79:163-169 (1987). Retinol was shown to
be necessary for insulin
release from vitamin A-deficient perfused islet cells. Id. In vivo experiments
demonstrated that
vitamin-A deficient rats had impaired glucose-induced acute insulin release,
which only improved
with vitamin A repletion. Id. Vitamin A may exert its effects on insulin
secretion through activation of
transglutaminase activity in islet and insulin-secreting cells, see Driscoll
HK, et al., Pancreas 15:69-77
(1997), and is needed for fetal islet development and prevention of glucose
intolerance in adults, see
Matthews, KA et al., J. Nutr. 134:1958-1963 (2004), further strengthening the
role of vitamin A and
retinol in insulin release and regulation of blood glucose levels in diabetic
patients. Presented herein
are methods, compounds, and compositions, for the treatment of Type I diabetes
using a compound of
Formula (I) and (II) wherein the levels or activity of retinol and/or RBP are
modulated.
Type II Diabetes (Non-Insulin Dependent Diabetes Mellitus)
[00110] Type II diabetes comprises a heterogeneous group of the milder forms
of diabetes. Type II
diabetes usually occurs in adults, but occasionally may have its onset in
childhood.
[00111] Type II diabetics classically exhibit insulin insensitivity in
response to elevated plasma
glucose levels. Up to 85% of type II diabetics are obese, having an
insensitivity to endogenous insulin
that is positively correlated with the presence of an abdominal distribution
of fat. Causes of insulin
insensitivity are linked with post-receptor defect in insulin action. This is
associated with over
distended cellular storage depots (e.g. distended adipocytes and over
nourished liver and muscle cells)
and a reduced ability to clear nutrients from the circulation after meals. The
subsequent
hyperinsulinism can also result in a further down-regulation of cellular
insulin receptors. Furthermore,
glucose transporter proteins (e.g. GLUT4) are also down-regulated upon
continuous activation,
leading to an aggravation of hyperglycemic conditions in patients.
[00112] In contrast to type I diabetes, type II diabetic patients exhibit
elevated levels of RBP
selectively, with normal to increased levels of retinol observed. See Sasaki,
H et al., "Am. J. Med. Sci.
310:177-82 (1995); Basualdo CG, et al. J. Am Coll. Nutr. 16:39-45 (1997);
Abahausain, MA et al.,
Eur. J. Clin. Nutr. 53: 630-635 (1999). Retinoic acid (all trans RA and 13-cis
RA) levels were also
decreased in patients with type II diabetes. Yamakoshi, Y et al., Biol. Pharm.
Bu1125:1268-1271
(2002). Levels of other vitamins, including vitamin E (tocopherol) and
carotenoids were unchanged in
-36-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
both diabetic and control groups, as well as levels of zinc, albumin and TTR,
which influence vitamin
A metabolism. Id.
[00113] This selective increase in RBP levels in type II diabetics, combined
with the selective decrease
of RBP in type I diabetics, supports the role of RBP and vitamin A in insulin
control of blood glucose
levels. The increased RBP levels have been attributed to the increased insulin
levels
(hyperinsulinemia) in diabetic patients. Basualdo et al. (1997). RBP levels
have also been linked to the
severity of hyperglycemia in patients. Id. Retinoids have previously been
shown to increase insulin
sensitivity in humans. See Hartmann, D. et al. Eur. J. Clin. Pharmacol. 42:523-
8 (1992). The inverse
correlation of RBP levels with insulin sensitivity in type I and type II
diabetics indicates a therapeutic
means of controlling insulin sensitivity in mammalian subjects.
Retinol Binding Protein 4 (RBP 4)
[00114] Retinol binding protein 4 (RBP4) is an adipocyte-secreted protein
recognized for its role in the
transport of vitamin A. Studies have shown that elevated levels of serum RBP4
can help indicate an
early stage development of insulin resistance, a major cause of type II
diabetes. See Kahn et al., 354
New Eng. J. Med. 2552-63 (2006). Experiments in mice also suggest that
elevated RBP4 levels cause
insulin resistance. Moreover, serum RBP4 levels correlate with the magnitude
of insulin resistance in
subjects with obesity impaired glucose tolerance, or type II diabetes and in
non-obese, non-diabetic
subjects with a strong family history of type II diabetes. Elevated serum RBP4
has been associated
with components of the metabolic syndrome, including increased body-mass
index, waist-to-hip ratio,
serum triglyceride levels, and systolic blood pressure and decreased high-
density lipoprotein
cholesterol levels.
[00115] Research also suggests that the amount of RBP4 in the blood reflects
the amount of fat
surrounding the abdominal organs indicating that RBP4 might be used as a
biomarker for
cardiovascular risk. As levels of RBP4 increase so do the levels of "inter-
abdominal fat" linked to an
increased risk for heart diseases and type II diabetes, since increased "inter-
abdominal fat" is
associated with cardiovascular risk. Studies also show that the gene
expression of RBP4 increases
more in visceral adipose tissue-the adipose tissue surrounding the internal
organs-than it is in the
subcutaneous adipose tissue. Thus, levels of RPB4 are higher in humans who
have a "visceral pattern"
of obesity compared with people that have a subcutaneous pattern of obesity.
[00116] Presented herein are compounds of Formula (I) and (II) that reduce
excess serum levels of
RBP4. In one embodiment is a compound of Formula (I):
X ~
A/\B' E
D
Formula (I);
wherein:
A is O, NH, or S;
-37-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
-
B is a bond, -(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
D is isopropyl, isobutyl, sec-butyl, tert-butyl, cyclopropyl, cyclobutyl,
cyclopentyl,
methylenecyclopropyl, methylenecyclobutyl, methylenecyclopentyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR1 R, NR'-(C=O)-R, -(Ci-C7)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NR'R;
G
RisHor
G is -OR1, -(Ci-C6)alkyl, -(Ci-C6)alkyl-OR', halogen, -COzRl, -(Ci-C6)alkyl-
COzR',
NHR1, -(Ci-C6)alkyl-NHR', -(C=O)NHR', -(Ci-C6)alkyl-(C=O)NHR', -NHR'(C=0)Rl, -
(Ci-
C6)alkyl-NHR'(C=O)Rl;
R' is H or (Ci-C6)alkyl;
X is a halogen;
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof;
wherein the compound of Formula (I) reduces the serum levels of RBP4.
[00117] In another embodiment is a compound of Formula (II):
C1 ~
BE
Formula (II);
wherein:
A is 0, NH, or S;
-
B is a bond, -(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR1 R, NR'-(C=O)-R, -(Ci-C7)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NR'R;
G
RisHor
G is -OR1, -(Ci-C6)alkyl, -(Ci-C6)alkyl-OR', halogen, -COzRl, -(Ci-C6)alkyl-
COzR',
NHR1, -(Ci-C6)alkyl-NHR', -(C=O)NHR', -(Ci-C6)alkyl-(C=O)NHR', -NHR'(C=0)Rl, -
(Ci-
C6)alkyl-NHR'(C=O)Rl;
R' is H or (Ci-C6)alkyl;
-38-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof;
wherein the compound of Formula (II) reduces the levels of serum RBP4.
[00118] In yet another embodiment is a method for treating diabetes in a
patient comprising
administering to the patient a therapeutically effective amount of a compound
of Formula (II):
X ~
B' E
D
Formula (I);
wherein:
A is 0, NH, or S;
-
B is a bond, -(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
D is isopropyl, isobutyl, sec-butyl, tert-butyl, cyclopropyl, cyclobutyl,
cyclopentyl,
methylenecyclopropyl, methylenecyclobutyl, methylenecyclopentyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR1R, NR'-(C=O)-R, -(C1-C7)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NR'R;
R is H, an optionally substituted aryl, or an optionally substituted
heteroaryl;
X is a halogen;
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof;
wherein administering a therapeutically effective amount of the compound of
Formula (I) lowers the
levels of RBP4. In yet another embodiment is a method for treating ophthalmic
conditions in a
patient, such as by way of example only, macular degeneration, comprising
administering to the
patient a therapeutically effective amount of a compound of Formula (I) or
(II) wherein the level of
RBP4 is reduced. In a further embodiment is a method for lowering RBP4 in
serum comprising
administering a compound of Formula (I) or (II). In yet a further embodiment
is a method for
lowering RBP4 in tissue, such as by way of example only, adipose tissue,
comprising administering
a compound of Formula (I) or (II).
[00119] In one embodiment is a method for treating type I or type II diabetes
in a patient comprising
administering to the patient a therapeutically effective amount of a compound
of Formula (I) or (II)
wherein the therapeutically effective amount of the compound of Formula (I) or
(II) modulates RBP4
in adipose tissue. In a further embodiment is a method for treating type I or
type II diabetes in a patient
comprising administering to the patient a therapeutically effective amount of
a compound of Formula
(I) or (II) wherein levels of RBP4 are reduced such that reduction in RBP4
levels increases insulin
sensitization. In another embodiment is a method for treating type I or type
II diabetes in a patient
comprising administering to the patient a therapeutically effective amount of
a compound of Formula
(II):
-39-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
C1
I BE
Formula (II);
wherein:
A is 0, NH, or S;
-
B is a bond, -(C2-C7)alkyl, -(C2-C7)alkenyl, -(C3-Cg)cycloalkyl, -(C2-
C7)heteroalkyl,
(C3-Cg)heterocycloalkyl, -(C3-Cg)cycloalkenyl, -(C3-Cg)heterocycloalkenyl;
E is (C=O)-OR, -O-(C=O)-R, -(C=O)-R, -OR, a carboxylic acid bioisostere, -
(C=O)-
NR1 R, NR'-(C=O)-R, -(C1-C7)alkyl-(C=O)-OR, or -(Ci-C7)alkyl-(C=O)-NR'R;
R is H, an optionally substituted aryl, or an optionally substituted
heteroaryl;
or an active metabolite, or a pharmaceutically acceptable prodrug, salt, or
solvate thereof;
wherein levels of RBP4 are reduced such that reduction in RBP4 levels
increases insulin
sensitization.
Idiopathic Intracranial Hypertension (IIH)
[00120] IIH, also known as pseudotumor cerebri (PTC), is a condition of high
pressure in the fluid
around the brain without an identifiable causative agent. The condition exists
mostly in women in their
childbearing years. The symptoms often start or worsen during a period of
weight gain. Typical
symptoms include headaches, pulse synchronous tinnitus and visual problems
(papilledema), which
may lead to severe and permanent visual loss in untreated cases.
[00121] Although the etiology of IIH is unknown, excess vitamin A levels are a
candidate because the
symptoms and signs of hypervitaminosis A mimic those of IIH. Studies have
shown that serum retinol
levels are significantly higher in patients with IIH than in control groups,
despite the showing of no
significant differences in vitamin A ingestion or retinyl ester concentration
in both groups. See
Jacobson, DM et al., Neurology, 54:2192-3 (1999). Included herein, are
methods, compounds, and
compositions for the treatment of IIH using compounds of Formula (I) and (II).
Bone-Related Disorders
[00122] Hyperostosis is a condition where an excessive growth of bone occurs.
This condition may
lead to formation of a mass projecting from a normal bone, seen in numerous
musculoskeletal
disorders. Diffuse idiopathic skeletal hyperostosis (DISH) is a form of
hyperostosis, characterized by
flowing calcification and ossification of vertebral bodies. Radiographic
abnormalities in DISH patients
are observed most commonly in the thoracic spine, leading to the presence of a
radiodense shield in
front of the vertebral column. Ossification of the posterior longitudinal
ligament (OPLL) is also
associated with increased frequency in patients with DISH, in addition to
cervical cord compromise as
a result of hyperostosis or ossification of spinal ligaments. Other disorders
accompanying hyperostosis
or DISH patients includes acute fracture and pseudoarthrosis of the spine.
-40-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
[00123] Although the pathogenesis of DISH and OPLL are presently unknown, both
disorders have
been associated with high levels of serum retinol and RBP. See Kodama, T et
al., In vivo 12:339-344
(1998); Kilcoyne, RF, J. Am. Acad. Dermatol. 19:212-216 (1988), suggesting a
possible role for
vitamin A in the pathogenesis of DISH and OPLL. Other studies have shown the
occurrence of
congenital functional RBP deficiency with abnormal levels of retinol and RBP
levels in a hyperostosis
patient. De Bandt, M., et al., J. Rheumatol. 22:1395-8 (1995). Medical
accounts also report the
occurrence of hypervitaminosis A with degenerative joint disease in an elderly
patient. See Romero,
JB et al., Bull Hosp. Jt. Dis. 54:169-174 (1996). Thus, the methods,
compounds, and compositions
described herein are used to treat bone-related disorders, such as, by way of
example only,
hyperostosis, using compounds of Formula (I) or (II) wherein the level of
serum retinol and RBP are
modulated.
Protein Misfolding and Aggregation Diseases
[00124] Protein misfolding and aggregation has been linked to several
diseases, generally known as
the amyloidoses, including Alzeheimer's disease, Parkinson's disease and
systemic amyloidosis.
These diseases occur with misfolding of the secondary protein structure, in
which a normally soluble
protein forms insoluble extracellular fibril deposits of (3-sheet-rich
structures referred to as amyloid
fibrils, which causes organ dysfunction. Twenty different fibril proteins,
including transthyretin
(TTR), have been described in human amyloidosis, each with a different
clinical picture.
[00125] Wild-type TTR proteins are involved in the development of senile
systemic amyloidosis, a
sporadic disorder resulting from the deposition of TTR fibrils in cardiac
tissues. Mutant TTR proteins,
in contrast, are associated with familial amyloidotic polyneuropathy and
cardiomyopathy, which
deposits primarily affect the peripheral and autonomic nervous system, and
heart. The mechanisms
responsible for tissue selectivity deposition are currently unknown. In
amyloidosis formation, TTR
associates with fibril formation in its monomer form. Compounds which promote
stabilization of TTR
tetramers, such as the small molecules resveratrol and biarylamine, inhibit
amyloid fibril formation in
vitro. See Reixach, N. et al., PNAS 101:2817-2822 (2004).
[00126] Transthyretin is also implicated in Alzheimer's disease, but in
contrast to the formation of
amyloid fibrils in amyloidosis, TTR inhibits amyloid beta protein formation
both in vitro and in vivo.
See Schwartzman, AL et al., Amyloid. 11:1-9 (2004); Stein, TD and Johnson, JA,
J. Neurosci.
22:7380-7388 (2002). Vitamin A also has been shown to exhibit anti-
amyloidogenic and amyloid-beta
fibril destabilizing effects in vitro. See Ono, K., et al., Exp. Neurol.
189:380-392 (2004).
Cystic Fibrosis
[00127] Cystic fibrosis is a lethal hereditary disease wherein the primary
cause of mortality is due to
excessive lung inflammation associated with recurrent bacterial infections by
Pseudomonas
aeruginosa. Cystic fibrosis is caused by a mutation in the cystic fibrosis
transmembrane conductance
regulator gene (CFTR). The product of this gene is a chloride ion channel
important in creating sweat,
digestive juices, and mucus. The CFTR gene is located at the q31.21ocus of
chromosome 7 and
-41-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
creates a protein which is 1,480 amino acids long. A common mutation, AF508,
is the deletion of three
nucleotides that results in a loss of the amino acid phenylalanine at the 508
position of the protein.
Subsequently, AF508 creates a protein which does not fold normally and is
degraded by the cell. Also,
the protein created by the CFTR gene acts as a chloride channel connecting the
cytoplasm to the
surrounding fluid. Mutation of the CFTR protein traps chloride ions outside
the cell. Exclusion of
chloride ions into the cytoplasm attracts sodium ions and this combination
forms a salt which is lost in
high amounts in the sweat of individuals with cystic fibrosis. Some studies
suggest that the CFTR
protein failure leads to an increase in sodium and chloride uptake which
increases water reabsorption,
thereby causing dehydration and thick mucus.
[00128] Treatment of cystic fibrosis is concentrated on the treatment of lung
damage caused by thick
mucus and infection. Antibiotics such as vancomycin and tobramycin are used
when there has been a
decline in lung function. Nasal steroids such as fluticasone have been
employed to decrease nasal
inflammation. In other cases, sinus surgery is used to alleviate nasal
obstruction and limit further
infections.
[00129] The regulation of ceramide levels appears to be important for
efficient clearance of bacteria
from infected lungs. Presented herein are methods for treating cystic fibrosis
comprising administering
a compound of Formula (I) or Formula (II) wherein the compound aids in the
clearance of bacterial
burden through the mediation of ceramide production. In some embodiments are
methods for treating
bacterial infections related to cystic fibrosis comprising administering
compounds of Formula (I) or
Formula (II) wherein the compound of Formula (I) or Formula (II) assists in
the clearance of bacteria
from infected lungs. In other embodiments, the bacteria is a gram-negative
bacteria. In further
embodiments the gram-negative bacteria is pseudomonas aeruginosa. In another
embodiment is a
method for treating cystic fibrosis comprising administering a compound of
Formula (I) or Formula
(II) wherein the compound of Formula (I) or (II) corrects the ceramide
deficiency in cystic fibrosis
related organs. In yet other embodiments, the cystic fibrosis related organ is
the lungs.
Alstrom-Hallgren Syndrome
[00130] Alstrom-Hallgren syndrome (also known as Alstr6m syndrome) is a rare
autosomal recessive
disorder affecting children at a very early age. Symptoms include blindness or
severe vision
impairment in infancy associated with cone-rod dystrophy, deafness, obesity
onset during the first
year, development of type II diabetes mellitus and severe insulin resistance,
acanthosis nigricans
(development of dark patches of skin) hypergonadotrophic hypogonadism and
thyroid deficiencies.
[00131] Mutations linked to Alstr6m syndrome were localized to a 14.9 cM
region on chromosome 2p.
Collin, GB et al., Hum. Mol. Gen. 6:213-219 (1997). Other than treating
individual symptomatic
manifestations of the disease, there are currently no therapeutic treatments
available for Alstr6m
syndrome patients. In some embodiments are methods, compounds, and
compositions used in the
treatment of Alstrom-Hallgren syndrome using compounds having the structure of
Formula (I) and
(II).
-42-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
Definitions
[00132] An "alkoxy" group refers to an (alkyl)O- group, where alkyl is as
defined herein.
[00133] An "alkyl" group refers to an aliphatic hydrocarbon group. The alkyl
moiety includes a
"saturated alkyl" group, which means that it does not contain any alkene or
alkyne moieties. The alkyl
moiety includes an "unsaturated alkyl" moiety, which means that it contains at
least one alkene or
alkyne moiety. An "alkene" moiety refers to a group consisting of at least two
carbon atoms and at
least one carbon-carbon double bond, and an "alkyne" moiety refers to a group
consisting of at least
two carbon atoms and at least one carbon-carbon triple bond. The alkyl moiety,
whether saturated or
unsaturated, incluces branched, straight chain, or cyclic.
[00134] The "alkyl" moiety includes moieties with 1 to 10 carbon atoms
(whenever it appears herein, a
numerical range such as "1 to 10" refers to each integer in the given range;
e.g., "1 to 10 carbon
atoms" means that the alkyl group consists of 1 carbon atom, 2 carbon atoms, 3
carbon atoms, etc., up
to and including 10 carbon atoms, although the present definition also covers
the occurrence of the
term "alkyl" where no numerical range is designated). The alkyl group could
also be a "lower alkyl"
having 1 to 5 carbon atoms. The alkyl group of the compounds described herein
may be designated as
"Ci-C4 alkyl" or similar designations. By way of example only, "Ci-C4 alkyl"
indicates that there are
one to four carbon atoms in the alkyl chain, i.e., the alkyl chain is selected
from the group consisting
of methyl, ethyl, propyl, iso-propyl, n-butyl, iso-butyl, sec-butyl, and t-
butyl. Typical alkyl groups
include, but are in no way limited to, methyl, ethyl, propyl, isopropyl,
butyl, isobutyl, tertiary butyl,
pentyl, hexyl, ethenyl, propenyl, butenyl, cyclopropyl, cyclobutyl,
cyclopentyl, cyclohexyl, and the
like.
[00135] The term "alkenyl" refers to a type of alkyl group in which the first
two atoms of the alkyl
group form a double bond that is not part of an aromatic group. That is, an
alkenyl group begins with
the atoms -C(R)=C-R, wherein R refers to the remaining portions of the alkenyl
group, which is either
the same or different. Non-limiting examples of an alkenyl group include -
CH=CH, -C(CH3)=CH, -
CH=CCH3 and -C(CH3)=CCH3. The alkenyl moiety includes branched, straight
chain, or cyclic (in
which case, it is also known as a"cycloalkenyP' group).
[00136] An "amide" is a chemical moiety with formula -C(O)NHR or -NHC(O)R,
where R is selected
from the group consisting of alkyl, cycloalkyl, aryl, heteroaryl (bonded
through a ring carbon) and
heteroalicyclic (bonded through a ring carbon). An amide includes an amino
acid or a peptide
molecule attached to a compound of Formula (I), thereby forming a prodrug. Any
amine, hydroxy, or
carboxyl side chain on the compounds described herein can be amidified. The
procedures and specific
groups to make such amides are found in reference sources such as Greene and
Wuts, Protective
Groups in Organic Synthesis, 3rd Ed., John Wiley & Sons, New York, NY, 1999,
which is
incorporated herein by reference.
-43-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
[00137] The term "aromatic" or "aryl" refers to an aromatic group which has at
least one ring having a
conjugated pi electron system and includes both carbocyclic aryl (e.g.,
phenyl) and heterocyclic aryl
(or "heteroaryl" or "heteroaromatic") groups (e.g., pyridine). The term
includes monocyclic or fused-
ring polycyclic (i.e., rings which share adjacent pairs of carbon atoms)
groups. The term "carbocyclic"
refers to a compound which contains one or more covalently closed ring
structures, and that the atoms
forming the backbone of the ring are all carbon atoms. The term thus
distinguishes carbocyclic from
heterocyclic rings in which the ring backbone contains at least one atom which
is different from
carbon.
[00138] The term "cycloalkyl" refers to a monocyclic or polycyclic radical
that contains only carbon
and hydrogen, and includes saturated, partially unsaturated, or fully
unsaturated. Cycloalkyl groups
include groups having from 3 to 10 ring atoms. Illustrative examples of
cycloalkyl groups include the
following moieties:
A , E>, CO
>EO , 0, 0, 0, CC)
C'O ~ ~~
~
and the like.
[00139] The term "halo" or, alternatively, "halogen" means fluoro, chloro,
bromo or iodo. Preferred
halo groups are fluoro, chloro and bromo.
[00140] The terms "heteroalkyl" "heteroalkenyl" and "heteroalkynyl" include
optionally substituted
alkyl, alkenyl and alkynyl radicals and which have one or more skeletal chain
atoms selected from an
atom other than carbon, e.g., oxygen, nitrogen, sulfur, phosphorus or
combinations thereof.
[00141] The terms "heteroaryl" or, alternatively, "heteroaromatic" refers to
an aryl group that includes
one or more ring heteroatoms selected from nitrogen, oxygen and sulfur. An N-
containing
"heteroaromatic" or "heteroaryl" moiety refers to an aromatic group in which
at least one of the
skeletal atoms of the ring is a nitrogen atom. The polycyclic heteroaryl group
includes fused or non-
fused. Illustrative examples of heteroaryl groups include the following
moieties:
NN NN \ N \ S \ N
N N N I ~ I ~ I //
N
-44-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
OO? N,\> > N
9 \
N,N N,N CN N / N) N~N rN)
\ ~ \ ~ ) N N N N
S
a N
N
S and the like.
[00142] The term "moiety" refers to a specific segment or functional group of
a molecule. Chemical
moieties are often recognized chemical entities embedded in or appended to a
molecule.
[00143] The term "bond" or "single bond" refers to a chemical bond between two
atoms, or two
moieties when the atoms joined by the bond are considered to be part of larger
substructure.
[00144] The term "carboxylic acid bioisostere" means a moiety that can replace
a carboxylic acid
group. The bioisostere contains an exchange of an atom or groups of atoms with
another, broadly
similar, atom or groups of atoms and maintains similar biological activity by
mimicking the spatial
arrangement, electronic properties, or some other physicochemical property of
the carboxylic acid
group. Thus, for example, tetrazole, sulfonic acid, and sulfonamide are
carboxylic acid bioisosteres.
[00145] The term "optionally substituted" means that the referenced group
includes substituted with
one or more additional group(s) individually and independently selected from
alkyl, cycloalkyl, aryl,
heteroaryl, heteroalicyclic, hydroxy, alkoxy, aryloxy, mercapto, alkylthio,
arylthio, cyano, halo,
carbonyl, thiocarbonyl, isocyanato, thiocyanato, isothiocyanato, nitro,
perhaloalkyl, perfluoroalkyl,
silyl, and amino, including mono- and di-substituted amino groups, and the
protected derivatives
thereof. Examples of protecting groups are found in references such as Greene
and Wuts, above.
[00146] In some embodiments, the compounds presented herein possess one or
more chiral centers and
each center in the R or S configuration. The compounds presented herein
include all diastereomeric,
enantiomeric, and epimeric forms as well as the appropriate mixtures thereof.
Stereoisomers are
obtained, if desired, for example, by the separation of stereoisomers by
chiral chromatographic
columns.
[00147] The methods and formulations described herein include the use of N-
oxides, crystalline forms
(also known as polymorphs), or pharmaceutically acceptable salts of compounds
having the structure
of Formula (I), as well as active metabolites of these compounds having the
same type of activity. In
some situations, compounds exist as tautomers. All tautomers are included
within the scope of the
compounds presented herein. In addition, the compounds described herein exist
in unsolvated as well
as solvated forms with pharmaceutically acceptable solvents such as water,
ethanol, and the like. The
solvated forms of the compounds presented herein are also considered to be
disclosed herein.
-45-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
Pharmaceutical Compositions
[00148] Another aspect are pharmaceutical compositions comprising a compound
of Formula (I) or
(II) and a pharmaceutically acceptable diluent, excipient, or carrier.
[00149] The term "pharmaceutical composition" refers to a mixture of a
compound of Formula (I) with
other chemical components, such as carriers, stabilizers, diluents, dispersing
agents, suspending
agents, thickening agents, and/or excipients. The pharmaceutical composition
facilitates administration
of the compound to an organism. Techniques of administering a compound
include: intravenous, oral,
aerosol, parenteral, ophthalmic, pulmonary and topical administration.
[00150] The term "carrier" refers to relatively nontoxic chemical compounds or
agents that facilitate
the incorporation of a compound into cells or tissues.
[00151] The term "diluent" refers to chemical compounds that are used to
dilute the compound of
interest prior to delivery. Diluents are also used to stabilize compounds
because they provide a more
stable environment. Salts dissolved in buffered solutions (which also provide
pH control or
maintenance) are utilized as diluents, including, but not limited to a
phosphate buffered saline
solution.
[00152] The term "physiologically acceptable" refers to a material, such as a
carrier or diluent, which
does not abrogate the biological activity or properties of the compound, and
is nontoxic.
[00153] The term "pharmaceutically acceptable salt" refers to a formulation of
a compound that does
not cause significant irritation to an organism to which it is administered
and does not abrogate the
biological activity and properties of the compound. In one example,
pharmaceutically acceptable salts
are obtained by reacting a compound of Formula (I) with acids such as
hydrochloric acid, hydrobromic
acid, sulfuric acid, nitric acid, phosphoric acid, methanesulfonic acid,
ethanesulfonic acid, p-
toluenesulfonic acid, salicylic acid and the like. Pharmaceutically acceptable
salts are also obtained by
reacting a compound of Formula (I) with a base to form a salt such as an
ammonium salt, an alkali
metal salt, such as a sodium or a potassium salt, an alkaline earth metal
salt, such as a calcium or a
magnesium salt, a salt of organic bases such as dicyclohexylamine, N-methyl-D-
glucamine,
tris(hydroxymethyl)methylamine, and salts with amino acids such as arginine,
lysine, and the like.
[00154] A "metabolite" of a compound disclosed herein is a derivative of that
compound that is
formed when the compound is metabolized. The term "active metabolite" refers
to a biologically
active derivative of a compound that is formed when the compound is
metabolized. The term
"metabolized" refers to the sum of the processes (including, but not limited
to, hydrolysis reactions
and reactions catalyzed by enzymes) by which a particular substance is changed
by an organism. Thus,
enzymes may produce specific structural alterations to a compound. For
example, cytochrome P450
catalyzes a variety of oxidative and reductive reactions while uridine
diphosphate
glucuronyltransferases catalyze the transfer of an activated glucuronic-acid
molecule to aromatic
alcohols, aliphatic alcohols, carboxylic acids, amines and free sulphydryl
groups. Further information
-46-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
on metabolism is obtained from The Pharmacological Basis of Therapeutics, 9th
Edition, McGraw-
Hill (1996).
[00155] In some embodiments, metabolites of the compounds disclosed herein are
identified either by
administration of compounds to a host and analysis of tissue samples from the
host, or by incubation
of compounds with hepatic cells in vitro and analysis of the resulting
compounds.
[00156] A "prodrug" refers to an agent that is converted into the parent drug
in vivo. Prodrugs are
often useful because, in some situations, they are easier to administer than
the parent drug. In some
examples, they are, for instance, bioavailable by oral administration whereas
the parent is not. For
example, the prodrug also has improved solubility in pharmaceutical
compositions over the parent
drug. An example, without limitation, of a prodrug is a compound of Formula
(I) which is
administered as an ester (the "prodrug") to facilitate transmittal across a
cell membrane where water
solubility is detrimental to mobility but which then is metabolically
hydrolyzed to the carboxylic acid,
the active entity, once inside the cell where water-solubility is beneficial.
A further example of a
prodrug is a short peptide (polyaminoacid) bonded to an acid group where the
peptide is metabolized
to reveal the active moiety.
[00157] In some embodiements, the compounds described herein are administered
to a human patient
per se, or in pharmaceutical compositions where they are mixed with other
active ingredients, as in
combination therapy, or suitable carrier(s) or excipient(s). Techniques for
formulation and
administration of the compounds of the instant application are found in
"Remington: The Science and
Practice of Pharmacy," 20th ed. (2000).
Routes of Administration
[00158] Suitable routes of administration are, for example, include oral,
rectal, vaginal, transmucosal,
transdermal, pulmonary, or intestinal administration; parenteral delivery,
including intramuscular,
subcutaneous, intravenous, intramedullary injections, as well as intrathecal,
direct intraventricular,
intraperitoneal, or intranasal injections.
[00159] Alternately, one administers the compound in a local rather than
systemic manner, for
example, via injection of the compound directly into an organ, often in a
depot or sustained release
formulation. The liposomes will be targeted to and taken up selectively by the
organ. In addition, for
example, the drug is provided in the form of a rapid release formulation, in
the form of an extended
release formulation, or in the form of an intermediate release formulation.
Composition/Formulation
[00160] For example, pharmaceutical compositions comprising a compound of
Formula (I) or (II) are
manufactured by means of mixing, dissolving, granulating, dragee-making,
levigating, emulsifying,
encapsulating, entrapping or compression processes.
[00161] For example, pharmaceutical compositions are formulated in
conventional manner using one
or more physiologically acceptable carriers comprising excipients and
auxiliaries which facilitate
-47-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
processing of the active compounds into preparations which are used
pharmaceutically. Proper
formulation is dependent upon the route of administration chosen.
[00162] In some embodiments, the compounds of Formula (I) or (II) are
administered in a variety of
ways, including systemically, such as orally or intravenously.
[00163] Useful compositions also include solubilizing agents to aid in the
solubility of a compound of
Formula (I) or (II). The term "solubilizing agent" generally includes agents
that result in formation of
a micellar solution or a true solution of the agent. In some embodiments,
certain acceptable nonionic
surfactants, for example polysorbate 80, are useful as solubilizing agents,
similarly, acceptable
glycols, polyglycols, e.g., polyethylene glyco1400, and glycol ethers.
[00164] Useful compositions also include one or more pH adjusting agents or
buffering agents,
including acids such as acetic, boric, citric, lactic, phosphoric and
hydrochloric acids; bases such as
sodium hydroxide, sodium phosphate, sodium borate, sodium citrate, sodium
acetate, sodium lactate
and tris-hydroxymethylaminomethane; and buffers such as citrate/dextrose,
sodium bicarbonate and
ammonium chloride. Such acids, bases and buffers are included in an amount
required to maintain pH
of the composition in an acceptable range.
[00165] Useful compositions also include one or more acceptable salts in an
amount required to bring
osmolality of the composition into an acceptable range. Such salts include
those having sodium,
potassium or ammonium cations and chloride, citrate, ascorbate, borate,
phosphate, bicarbonate,
sulfate, thiosulfate or bisulfite anions; suitable salts include sodium
chloride, potassium chloride,
sodium thiosulfate, sodium bisulfite and ammonium sulfate.
[00166] Other useful compositions also include one or more acceptable
preservatives to inhibit
microbial activity. Suitable preservatives include mercury-containing
substances such as merfen and
thiomersal; stabilized chlorine dioxide; and quaternary ammonium compounds
such as benzalkonium
chloride, cetyltrimethylammonium bromide and cetylpyridinium chloride.
[00167] In some embodiments, aqueous suspension compositions are packaged in
single-dose non-
reclosable containers. Alternatively, multiple-dose reclosable containers are
used, in which case it is
typical to include a preservative in the composition.
[00168] A pharmaceutical carrier for the hydrophobic compounds of Formula (I)
or (II) is a cosolvent
system comprising benzyl alcohol, a nonpolar surfactant, a water-miscible
organic polymer, and an
aqueous phase. The cosolvent system includes a 10% ethanol, 10% polyethylene
glyco1300, 10%
polyethylene glyco140 castor oil (PEG-40 castor oil) with 70% aqueous
solution. This cosolvent
system dissolves hydrophobic compounds well, and itself produces low toxicity
upon systemic
administration. For example, the proportions of a cosolvent system are varied
considerably without
destroying its solubility and toxicity characteristics. Furthermore, for
example, the identity of the
cosolvent components are varied: for example, other low-toxicity nonpolar
surfactants are used instead
of PEG-40 castor oil, the fraction size of polyethylene glyco1300 is varied;
in some embodiments,
-48-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
other biocompatible polymers replace polyethylene glycol, e.g., polyvinyl
pyrrolidone; and other
sugars or polysaccharides maybe included in the aqueous solution.
[00169] Alternatively, other delivery systems for hydrophobic pharmaceutical
compounds are
employed. Liposomes and emulsions are examples of delivery vehicles or
carriers for hydrophobic
drugs. In some embodiments, certain organic solvents such as N-
methylpyrrolidone also are employed,
although usually at the cost of greater toxicity. Additionally, the compounds
for example, are
delivered using a sustained-release system, such as semipermeable matrices of
solid hydrophobic
polymers containing the therapeutic agent. Sustained-release capsules,
depending on their chemical
nature, release the compounds for a few weeks up to over 100 days. Depending
on the chemical nature
and the biological stability of the therapeutic reagent, additional strategies
for protein stabilization are
employed.
[00170] All of the formulations described herein benefit from antioxidants,
metal chelating agents,
thiol containing compounds and other general stabilizing agents. Examples of
such stabilizing agents,
include, but are not limited to: (a) about 0.5% to about 2% w/v glycerol, (b)
about 0.1% to about 1%
w/v methionine, (c) about 0.1% to about 2% w/v monothioglycerol, (d) about 1
mM to about 10 mM
EDTA, (e) about 0.01% to about 2% w/v ascorbic acid, (f) 0.003% to about 0.02%
w/v polysorbate
80, (g) 0.00 1% to about 0.05% w/v. polysorbate 20, (h) arginine, (i) heparin,
(j) dextran sulfate, (k)
cyclodextrins, (1) pentosan polysulfate and other heparinoids, (m) divalent
cations such as magnesium
and zinc; or (n) combinations thereof.
[00171] In some embodiments, compounds of Formula (I) or (II) are provided as
salts with
pharmaceutically compatible counterions. In some embodiments, pharmaceutically
compatible salts
are formed with many acids, including but not limited to hydrochloric,
sulfuric, acetic, lactic, tartaric,
malic, succinic, etc. Salts tend to be more soluble in aqueous or other
protonic solvents than are the
corresponding free acid or base forms.
Oral Administration
[00172] In some embodiments, the pharmaceutical compositions provided herein
are provided in solid,
semisolid, or liquid dosage forms for oral administration. As used herein,
oral administration also
include buccal, lingual, and sublingual administration. Suitable oral dosage
forms include, but are not
limited to, tablets, capsules, pills, troches, lozenges, pastilles, cachets,
pellets, medicated chewing
gum, granules, bulk powders, effervescent or non-effervescent powders or
granules, solutions,
emulsions, suspensions, solutions, wafers, sprinkles, elixirs, and syrups. In
addition to the active
ingredient(s), in some embodiments, the pharmaceutical compositions contain
one or more
pharmaceutically acceptable carriers or excipients, including, but not limited
to, binders, fillers,
diluents, disintegrants, wetting agents, lubricants, glidants, coloring
agents, dye-migration inhibitors,
sweetening agents, and flavoring agents.
[00173] Binders or granulators impart cohesiveness to a tablet to ensure the
tablet remaining intact
after compression. Suitable binders or granulators include, but are not
limited to, starches, such as corn
-49-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
starch, potato starch, and pre-gelatinized starch (e.g., STARCH 1500);
gelatin; sugars, such as sucrose,
glucose, dextrose, molasses, and lactose; natural and synthetic gums, such as
acacia, alginic acid,
alginates, extract of Irish moss, Panwar gum, ghatti gum, mucilage of isabgol
husks,
carboxymethylcellulose, methylcellulose, polyvinylpyrrolidone (PVP), Veegum,
larch arabogalactan,
powdered tragacanth, and guar gum; celluloses, such as ethyl cellulose,
cellulose acetate,
carboxymethyl cellulose calcium, sodium carboxymethyl cellulose, methyl
cellulose,
hydroxyethylcellulose (HEC), hydroxypropylcellulose (HPC), hydroxypropyl
methyl cellulose
(HPMC); microcrystalline celluloses, such as AVICEL-PH-101, AVICEL-PH-103,
AVICEL RC-581,
AVICEL-PH-105 (FMC Corp., Marcus Hook, PA); and mixtures thereof. Suitable
fillers include, but
are not limited to, talc, calcium carbonate, microcrystalline cellulose,
powdered cellulose, dextrates,
kaolin, mannitol, silicic acid, sorbitol, starch, pre-gelatinized starch, and
mixtures thereof. In some
embodiments, the binder or filler are present from about 50 to about 99% by
weight in the
pharmaceutical compositions provided herein.
[00174] Suitable diluents include, but are not limited to, dicalcium
phosphate, calcium sulfate, lactose,
sorbitol, sucrose, inositol, cellulose, kaolin, mannitol, sodium chloride, dry
starch, and powdered
sugar. Certain diluents, such as mannitol, lactose, sorbitol, sucrose, and
inositol, when present in
sufficient quantity, impart properties to some compressed tablets that permit
disintegration in the
mouth by chewing. In some embodiments, such compressed tablets are used as
chewable tablets.
[00175] Suitable disintegrants include, but are not limited to, agar;
bentonite; celluloses, such as
methylcellulose and carboxymethylcellulose; wood products; natural sponge;
cation-exchange resins;
alginic acid; gums, such as guar gum and Veegum HV; citrus pulp; cross-linked
celluloses, such as
croscarmellose; cross-linked polymers, such as crospovidone; cross-linked
starches; calcium
carbonate; microcrystalline cellulose, such as sodium starch glycolate;
polacrilin potassium; starches,
such as corn starch, potato starch, tapioca starch, and pre-gelatinized
starch; clays; aligns; and
mixtures thereof. The amount of disintegrant in the pharmaceutical
compositions provided herein
varies upon the type of formulation. In some embodiments, the pharmaceutical
compositions provided
herein contain from about 0.5 to about 15% or from about 1 to about 5% by
weight of a disintegrant.
[00176] Suitable lubricants include, but are not limited to, calcium stearate;
magnesium stearate;
mineral oil; light mineral oil; glycerin; sorbitol; mannitol; glycols, such as
glycerol behenate and
polyethylene glycol (PEG); stearic acid; sodium lauryl sulfate; talc;
hydrogenated vegetable oil,
including peanut oil, cottonseed oil, sunflower oil, sesame oil, olive oil,
corn oil, and soybean oil; zinc
stearate; ethyl oleate; ethyl laureate; agar; starch; lycopodium; silica or
silica gels, such as AEROSIL
200 (W.R. Grace Co., Baltimore, MD) and CAB-O-SIL (Cabot Co. of Boston, MA);
and mixtures
thereof. In some embodiments, the pharmaceutical compositions provided herein
contain about 0.1 to
about 5% by weight of a lubricant.
[00177] Suitable glidants include colloidal silicon dioxide, CAB-O-SIL (Cabot
Co. of Boston, MA),
and asbestos-free talc. Coloring agents include any of the approved,
certified, water soluble FD&C
-50-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
dyes, and water insoluble FD&C dyes suspended on alumina hydrate, and color
lakes and mixtures
thereof. A color lake is the combination by adsorption of a water-soluble dye
to a hydrous oxide of a
heavy metal, resulting in an insoluble form of the dye. Flavoring agents
include natural flavors
extracted from plants, such as fruits, and synthetic blends of compounds which
produce a pleasant
taste sensation, such as peppermint and methyl salicylate. Sweetening agents
include sucrose, lactose,
mannitol, syrups, glycerin, and artificial sweeteners, such as saccharin and
aspartame. Suitable
emulsifying agents include gelatin, acacia, tragacanth, bentonite, and
surfactants, such as
polyoxyethylene sorbitan monooleate (TWEEN 20), polyoxyethylene sorbitan
monooleate 80
(TWEEN 80), and triethanolamine oleate. Suspending and dispersing agents
include sodium
carboxymethylcellulose, pectin, tragacanth, Veegum, acacia, sodium
carbomethylcellulose,
hydroxypropyl methylcellulose, and polyvinylpyrolidone. Preservatives include
glycerin, methyl and
propylparaben, benzoic acid, sodium benzoate and alcohol. Wetting agents
include propylene glycol
monostearate, sorbitan monooleate, diethylene glycol monolaurate, and
polyoxyethylene lauryl ether.
Solvents include glycerin, sorbitol, ethyl alcohol, and syrup. Examples of non-
aqueous liquids utilized
in emulsions include mineral oil and cottonseed oil. Organic acids include
citric and tartaric acid.
Sources of carbon dioxide include sodium bicarbonate and sodium carbonate.
[00178] In some embodiments, pharmaceutical preparations which are used orally
include push-fit
capsules made of gelatin, including by way of example only, soft, sealed
capsules made of gelatin and
a plasticizer, such as glycerol or sorbitol; or hard-gel capsules or tablets.
In some embodiments, the
push-fit capsules contain the active ingredients in admixture with filler such
as lactose, binders such as
starches, and/or lubricants such as talc or magnesium stearate and,
optionally, stabilizers. In some
embodiments, soft capsules, the active compounds are dissolved or suspended in
suitable liquids, such
as fatty oils, liquid paraffin, or liquid polyethylene glycols. In addition,
in some embodiments,
stabilizers are added. All formulations for oral administration should be in
dosages suitable for such
administration.
[00179] For buccal or sublingual administration, in some embodiments, the
compositions take the form
of tablets, lozenges, or gels formulated in conventional manner.
[00180] It should be understood that in some embodiments, many carriers and
excipients serve several
functions, even within the same formulation.
[00181] In some embodiments, the pharmaceutical compositions provided herein
are provided as
compressed tablets, tablet triturates, chewable lozenges, rapidly dissolving
tablets, multiple
compressed tablets, or enteric-coating tablets, sugar-coated, or film-coated
tablets. Enteric-coated
tablets are compressed tablets coated with substances that resist the action
of stomach acid but
dissolve or disintegrate in the intestine, thus protecting the active
ingredients from the acidic
environment of the stomach. Enteric-coatings include, but are not limited to,
fatty acids, fats,
phenylsalicylate, waxes, shellac, ammoniated shellac, and cellulose acetate
phthalates. Sugar-coated
tablets are compressed tablets surrounded by a sugar coating, which beneficial
in some embodiments,
-51-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
in covering up objectionable tastes or odors and in protecting the tablets
from oxidation. Film-coated
tablets are compressed tablets that are covered with a thin layer or film of a
water-soluble material.
Film coatings include, but are not limited to, hydroxyethylcellulose, sodium
carboxymethylcellulose,
polyethylene glyco14000, and cellulose acetate phthalate. Film coating imparts
the same general
characteristics as sugar coating. Multiple compressed tablets are compressed
tablets made by more
than one compression cycle, including layered tablets, and press-coated or dry-
coated tablets.
[00182] In some embodiments, the tablet dosage forms are prepared from the
active ingredient in
powdered, crystalline, or granular forms, alone or in combination with one or
more carriers or
excipients described herein, including binders, disintegrants, controlled-
release polymers, lubricants,
diluents, and/or colorants. Flavoring and sweetening agents are especially
useful in the formation of
chewable tablets and lozenges.
[00183] In some embodiments, the pharmaceutical compositions provided herein
are provided as soft
or hard capsules, which are made from gelatin, methylcellulose, starch, or
calcium alginate. The hard
gelatin capsule, also known as the dry-filled capsule (DFC), consists of two
sections, one slipping over
the other, thus completely enclosing the active ingredient. The soft elastic
capsule (SEC) is a soft,
globular shell, such as a gelatin shell, which is plasticized by the addition
of glycerin, sorbitol, or a
similar polyol. In some embodiments, the soft gelatin shells contain a
preservative to prevent the
growth of microorganisms. Suitable preservatives are those as described
herein, including methyl- and
propyl-parabens, and sorbic acid. In some embodiments, the liquid, semisolid,
and solid dosage forms
provided herein are encapsulated in a capsule. Suitable liquid and semisolid
dosage forms include
solutions and suspensions in propylene carbonate, vegetable oils, or
triglycerides. Capsules containing
such solutions are prepared, for example, as described in U.S. Pat. Nos.
4,328,245; 4,409,239; and
4,410,545. In some embodiments, the capsules are coated in order to modify or
sustain dissolution of
the active ingredient.
[00184] In some embodiments, the pharmaceutical compositions provided herein
are provided in liquid
and semisolid dosage forms, including emulsions, solutions, suspensions,
elixirs, and syrups. An
emulsion is a two-phase system, in which one liquid is dispersed in the form
of small globules
throughout another liquid, which includes oil-in-water or water-in-oil.
Emulsions include a
pharmaceutically acceptable non-aqueous liquids or solvent, emulsifying agent,
and preservative.
Suspensions include a pharmaceutically acceptable suspending agent and
preservative. Aqueous
alcoholic solutions include a pharmaceutically acceptable acetal, such as a
di(lower alkyl) acetal of a
lower alkyl aldehyde (the term "lower" means an alkyl having between 1 and 6
carbon atoms), e.g.,
acetaldehyde diethyl acetal; and a water-miscible solvent having one or more
hydroxyl groups, such as
propylene glycol and ethanol. Elixirs are clear, sweetened, and hydroalcoholic
solutions. Syrups are
concentrated aqueous solutions of a sugar, for example, sucrose, and also
contain a preservative. For a
liquid dosage form, for example, a solution in a polyethylene glycol is
diluted with a sufficient
-52-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
quantity of a pharmaceutically acceptable liquid carrier, e.g., water, to be
measured conveniently for
administration.
[00185] Other useful liquid and semisolid dosage forms include, but are not
limited to, those
containing the active ingredient(s) provided herein, and a dialkylated mono-
or poly-alkylene glycol,
including, 1,2-dimethoxymethane, diglyme, triglyme, tetraglyme, polyethylene
glycol-350-dimethyl
ether, polyethylene glycol-550-dimethyl ether, polyethylene glycol-750-
dimethyl ether, wherein 350,
550, and 750 refer to the approximate average molecular weight of the
polyethylene glycol. These
formulations in some embodiments, further comprise one or more antioxidants,
such as butylated
hydroxytoluene (BHT), butylated hydroxyanisole (BHA), propyl gallate, vitamin
E, hydroquinone,
hydroxycoumarins, ethanolamine, lecithin, cephalin, ascorbic acid, malic acid,
sorbitol, phosphoric
acid, bisulfite, sodium metabisulfite, thiodipropionic acid and its esters,
and dithiocarbamates.
[00186] In some embodiments, the pharmaceutical compositions provided herein
for oral
administration are also provided in the forms of liposomes, micelles,
microspheres, or nanosystems. In
some embodiments, micellar dosage forms are prepared as described in U.S. Pat.
No. 6,350,458.
[00187] In some embodiments, the pharmaceutical compositions provided herein
are provided as non-
effervescent or effervescent, granules and powders, to be reconstituted into a
liquid dosage form. In
some embodiments, pharmaceutically acceptable carriers and excipients used in
the non-effervescent
granules or powders include diluents, sweeteners, and wetting agents. In some
embodiments,
pharmaceutically acceptable carriers and excipients used in the effervescent
granules or powders
include organic acids and a source of carbon dioxide.
[00188] In some embodiments, coloring and flavoring agents are used in all of
the above dosage forms.
[00189] In some embodiments, the pharmaceutical compositions provided herein
are formulated as
immediate or modified release dosage forms, including delayed-, sustained,
pulsed-, controlled,
targeted-, and programmed-release forms.
Parenteral Administration
[00190] In some embodiments, the pharmaceutical compositions provided herein
are administered
parenterally by injection, infusion, or implantation, for local or systemic
administration. Parenteral
administration, as used herein, include intravenous, intraarterial,
intraperitoneal, intrathecal,
intraventricular, intraurethral, intrasternal, intracranial, intramuscular,
intrasynovial, and subcutaneous
administration.
[00191] In some embodiments, the pharmaceutical compositions provided herein
are formulated in any
dosage forms that are suitable for parenteral administration, including
solutions, suspensions,
emulsions, micelles, liposomes, microspheres, nanosystems, and solid forms
suitable for solutions or
suspensions in liquid prior to injection. In some embodiments, such dosage
forms are prepared
according to conventional methods (see, e.g., Remington: The Science and
Practice of Pharmacy).
[00192] In some embodiments, the pharmaceutical compositions intended for
parenteral administration
include one or more pharmaceutically acceptable carriers and excipients,
including, but not limited to,
-53-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
aqueous vehicles, water-miscible vehicles, non-aqueous vehicles, antimicrobial
agents or preservatives
against the growth of microorganisms, stabilizers, solubility enhancers,
isotonic agents, buffering
agents, antioxidants, local anesthetics, suspending and dispersing agents,
wetting or emulsifying
agents, complexing agents, sequestering or chelating agents, cryoprotectants,
lyoprotectants,
thickening agents, pH adjusting agents, and inert gases.
[00193] Suitable aqueous vehicles include, but are not limited to, water,
saline, physiological saline or
phosphate buffered saline (PBS), sodium chloride injection, Ringers injection,
isotonic dextrose
injection, sterile water injection, dextrose and lactated Ringers injection.
Non-aqueous vehicles
include, but are not limited to, fixed oils of vegetable origin, castor oil,
corn oil, cottonseed oil, olive
oil, peanut oil, peppermint oil, safflower oil, sesame oil, soybean oil,
hydrogenated vegetable oils,
hydrogenated soybean oil, and medium-chain triglycerides of coconut oil, and
palm seed oil. Water-
miscible vehicles include, but are not limited to, ethanol, 1,3-butanediol,
liquid polyethylene glycol
(e.g., polyethylene glyco1300 and polyethylene glyco1400), propylene glycol,
glycerin, N-methyl-2-
pyrrolidone, dimethylacetamide, and dimethylsulfoxide.
[00194] Suitable antimicrobial agents or preservatives include, but are not
limited to, phenols, cresols,
mercurials, benzyl alcohol, chlorobutanol, methyl and propyl p-
hydroxybenzates, thimerosal,
benzalkonium chloride, benzethonium chloride, methyl- and propyl-parabens, and
sorbic acid.
Suitable isotonic agents include, but are not limited to, sodium chloride,
glycerin, and dextrose.
Suitable buffering agents include, but are not limited to, phosphate and
citrate. Suitable antioxidants
are those as described herein, including bisulfite and sodium metabisulfite.
Suitable local anesthetics
include, but are not limited to, procaine hydrochloride. Suitable suspending
and dispersing agents are
those as described herein, including sodium carboxymethylcelluose,
hydroxypropyl methylcellulose,
and polyvinylpyrrolidone. Suitable emulsifying agents include those described
herein, including
polyoxyethylene sorbitan monolaurate, polyoxyethylene sorbitan monooleate 80,
and triethanolamine
oleate. Suitable sequestering or chelating agents include, but are not limited
to EDTA. Suitable pH
adjusting agents include, but are not limited to, sodium hydroxide,
hydrochloric acid, citric acid, and
lactic acid. Suitable complexing agents include, but are not limited to,
cyclodextrins, including a-
cyclodextrin, (3-cyclodextrin, hydroxypropyl-(3-cyclodextrin, sulfobutylether-
(3-cyclodextrin, and
sulfobutylether 7-(3-cyclodextrin (CAPTISOL , CyDex, Lenexa, KS).
[00195] In some embodiments, the pharmaceutical compositions provided herein
are formulated for
single or multiple dosage administration. The single dosage formulations are
packaged in an ampule, a
vial, or a syringe. The multiple dosage parenteral formulations must contain
an antimicrobial agent at
bacteriostatic or fungistatic concentrations. All parenteral formulations must
be sterile.
[00196] In one embodiment, the pharmaceutical compositions are provided as
ready-to-use sterile
solutions. In another embodiment, the pharmaceutical compositions are provided
as sterile dry soluble
products, including lyophilized powders and hypodermic tablets, to be
reconstituted with a vehicle
prior to use. In yet another embodiment, the pharmaceutical compositions are
provided as ready-to-use
-54-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
sterile suspensions. In yet another embodiment, the pharmaceutical
compositions are provided as
sterile dry insoluble products to be reconstituted with a vehicle prior to
use. In still another
embodiment, the pharmaceutical compositions are provided as ready-to-use
sterile emulsions.
[00197] In some embodiments, the pharmaceutical compositions provided herein
are formulated as
immediate or modified release dosage forms, including delayed-, sustained,
pulsed-, controlled,
targeted-, and programmed-release forms.
[00198] In some embodiments, the pharmaceutical compositions are formulated as
a suspension, solid,
semi-solid, or thixotropic liquid, for administration as an implanted depot.
In one embodiment, the
pharmaceutical compositions provided herein are dispersed in a solid inner
matrix, which is
surrounded by an outer polymeric membrane that is insoluble in body fluids but
allows the active
ingredient in the pharmaceutical compositions diffuse through.
[00199] Suitable inner matrixes include polymethylmethacrylate,
polybutylmethacrylate, plasticized or
unplasticized polyvinylchloride, plasticized nylon, plasticized
polyethyleneterephthalate, natural
rubber, polyisoprene, polyisobutylene, polybutadiene, polyethylene, ethylene-
vinylacetate copolymers,
silicone rubbers, polydimethylsiloxanes, silicone carbonate copolymers,
hydrophilic polymers, such as
hydrogels of esters of acrylic and methacrylic acid, collagen, cross-linked
polyvinylalcohol, and cross-
linked partially hydrolyzed polyvinyl acetate.
[00200] Suitable outer polymeric membranes include polyethylene,
polypropylene, ethylene/propylene
copolymers, ethylene/ethyl acrylate copolymers, ethylene/vinylacetate
copolymers, silicone rubbers,
polydimethyl siloxanes, neoprene rubber, chlorinated polyethylene,
polyvinylchloride, vinylchloride
copolymers with vinyl acetate, vinylidene chloride, ethylene and propylene,
ionomer polyethylene
terephthalate, butyl rubber epichlorohydrin rubbers, ethylene/vinyl alcohol
copolymer, ethylene/vinyl
acetate/vinyl alcohol terpolymer, and ethylene/vinyloxyethanol copolymer.
[00201] In some embodiments, for intravenous injections, compounds of Formula
(I) or (II) are
formulated in aqueous solutions, preferably in physiologically compatible
buffers such as Hank's
solution, Ringer's solution, or physiological saline buffer. For transmucosal
administration, penetrants
appropriate to the barrier to be permeated are used in the formulation. For
other parenteral injections,
appropriate formulations in some embodiments, include aqueous or nonaqueous
solutions, preferably
with physiologically compatible buffers or excipients.
[00202] The compounds in some embodiments, are formulated for parenteral
administration by
injection, e.g., by bolus injection or continuous infusion. Formulations for
injection in some
embodiments, are presented in unit dosage form, e.g., in ampoules or in multi-
dose containers, with an
added preservative. The compositions in some embodiments, take such forms as
suspensions,
solutions or emulsions in oily or aqueous vehicles, and contain formulatory
agents such as suspending,
stabilizing and/or dispersing agents.
[00203] Pharmaceutical formulations for parenteral administration include
aqueous solutions of the
active compounds in water-soluble form. Additionally, suspensions of the
active compounds in some
-55-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
embodiments, are prepared as appropriate oily injection suspensions. Suitable
lipophilic solvents or
vehicles include fatty oils such as sesame oil, or synthetic fatty acid
esters, such as ethyl oleate or
triglycerides, or liposomes. Aqueous injection suspensions, in some
embodiments, contain substances
which increase the viscosity of the suspension, such as sodium carboxymethyl
cellulose, sorbitol, or
dextran. Optionally, the suspension also contains suitable stabilizers or
agents which increase the
solubility of the compounds to allow for the preparation of highly
concentrated solutions.
[00204] Alternatively, the active ingredient is in powder form for
constitution with a suitable vehicle,
e.g., sterile pyrogen-free water, before use.
[00205] The compounds in some embodiments, are formulated in rectal
compositions such as rectal
gels, rectal foam, rectal aerosols, suppositories or retention enemas, e.g.,
containing conventional
suppository bases such as cocoa butter or other glycerides.
[00206] In addition to the formulations described previously, the compounds,
in some embodiments,
are formulated as a depot preparation. Such long acting formulations in some
embodiments, are
administered by implantation (for example subcutaneously or intramuscularly)
or by intramuscular
injection. Thus, for example, the compounds are formulated with suitable
polymeric or hydrophobic
materials (for example as an emulsion in an acceptable oil) or ion exchange
resins, or as sparingly
soluble derivatives, for example, as a sparingly soluble salt.
[00207] Injectable depot forms in some embodiments, are made by forming
microencapsulated
matrices (also known as microencapsule matrices) of the compound of Formula
(I) or (II) in
biodegradable polymers. Depending upon the ratio of drug to polymer and the
nature of the particular
polymer employed, the rate of drug release is controlled. Depot injectable
formulations in some
embodiments, are prepared by entrapping the drug in liposomes or
microemulsions. By way of
example only, posterior juxtascleral depots are used as a mode of
administration for compounds
having the structure of Formula (I) or (II). The sclera is a thin avascular
layer, comprised of highly
ordered collagen network surrounding most of vertebrate eye. Since the sclera
is avascular it can be
utilized as a natural storage depot from which injected material cannot
rapidly removed or cleared
from the eye. In some embodiments, the formulation used for administration of
the compound into the
scleral layer of the eye is any form suitable for application into the sclera
by injection through a
cannula with small diameter suitable for injection into the scleral layer.
Examples for injectable
application forms are solutions, suspensions or colloidal suspensions.
Topical Administration
[00208] The pharmaceutical compositions provided herein in some embodiments,
are administered
topically to the skin, orifices, or mucosa. The topical administration, as
used herein, include
(intra)dermal, conjuctival, intracorneal, intraocular, ophthalmic, auricular,
transdermal, nasal, vaginal,
uretheral, respiratory, and rectal administration.
[00209] The pharmaceutical compositions provided herein in some embodiments,
are formulated in
any dosage forms that are suitable for topical administration for local or
systemic effect, including
-56-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
emulsions, solutions, suspensions, creams, gels, hydrogels, ointments, dusting
powders, dressings,
elixirs, lotions, suspensions, tinctures, pastes, foams, films, aerosols,
irrigations, sprays, suppositories,
bandages, dermal patches. The topical formulation of the pharmaceutical
compositions provided
herein in some embodiments, also comprise liposomes, micelles, microspheres,
nanospheres or
nanoparticles, and mixtures thereof.
[00210] Another useful formulation for administration of compounds having the
structure of Formula
(I) or (II) employs transdermal delivery devices ("patches"). Such transdermal
patches in some
embodiments, are used to provide continuous or discontinuous infusion of the
compounds of the
presented herein in controlled amounts. An example of the construction and use
of transdermal
patches for the delivery of pharmaceutical agents is found in U.S. Pat. No.
5,023,252. Such patches in
some embodiments, are constructed for continuous, pulsatile, or on demand
delivery of
pharmaceutical agents. Still further, in some embodiments, transdermal
delivery of the compounds of
Formula (I) or (II) is accomplished by means of iontophoretic patches and the
like. Transdermal
patches provide controlled delivery of the compounds. In some embodiments, the
rate of absorption is
slowed by using rate-controlling membranes or by trapping the compound within
a polymer matrix or
gel. Conversely, in some embodiments, absorption enhancers are used to
increase absorption. In some
embodiments, formulations suitable for transdermal administration are
presented as discrete patches
and are lipophilic emulsions or buffered, aqueous solutions, dissolved and/or
dispersed in a polymer or
an adhesive.
[00211] Pharmaceutically acceptable carriers and excipients suitable for use
in the topical formulations
provided herein include, but are not limited to, aqueous vehicles, water-
miscible vehicles, non-
aqueous vehicles, antimicrobial agents or preservatives against the growth of
microorganisms,
stabilizers, solubility enhancers, isotonic agents, buffering agents,
antioxidants, local anesthetics,
suspending and dispersing agents, wetting or emulsifying agents, complexing
agents, sequestering or
chelating agents, penetration enhancers, cryopretectants, lyoprotectants,
thickening agents, and inert
gases.
[00212] In some embodiments, he pharmaceutical compositions are administered
topically by
electroporation, iontophoresis, phonophoresis, sonophoresis and microneedle or
needle-free injection,
such as POWDERJECTTM (Chiron Corp., Emeryville, CA), and BIOJECTTM (Bioject
Medical
Technologies Inc., Tualatin, OR).
[00213] In some embodiments, the pharmaceutical compositions provided herein
are provided in the
forms of ointments, creams, and gels. Suitable ointment vehicles include
oleaginous or hydrocarbon
vehicles, including such as lard, benzoinated lard, olive oil, cottonseed oil,
and other oils, white
petrolatum; emulsifiable or absorption vehicles, such as hydrophilic
petrolatum, hydroxystearin
sulfate, and anhydrous lanolin; water-removable vehicles, such as hydrophilic
ointment; water-soluble
ointment vehicles, including polyethylene glycols of varying molecular weight;
emulsion vehicles,
either water-in-oil (W/O) emulsions or oil-in-water (O/W) emulsions, including
cetyl alcohol, glyceryl
-57-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
monostearate, lanolin, and stearic acid (see, Remington: The Science and
Practice of Pharmacy,
supra). These vehicles are emollient but generally require addition of
antioxidants and preservatives.
[00214] In some embodiments, suitable cream base are oil-in-water or water-in-
oil. In some
embodiments, cream vehicles are water-washable, and contain an oil phase, an
emulsifier, and an
aqueous phase. The oil phase is also called the "internaP" phase, which is
generally comprised of
petrolatum and a fatty alcohol such as cetyl or stearyl alcohol. The aqueous
phase usually, although
not necessarily, exceeds the oil phase in volume, and generally contains a
humectant. The emulsifier in
a cream formulation includes a nonionic, anionic, cationic, or amphoteric
surfactant.
[00215] Gels are semisolid, suspension-type systems. Single-phase gels contain
organic
macromolecules distributed substantially uniformly throughout the liquid
carrier. Suitable gelling
agents include crosslinked acrylic acid polymers, such as carbomers,
carboxypolyalkylenes,
Carbopol ; hydrophilic polymers, such as polyethylene oxides, polyoxyethylene-
polyoxypropylene
copolymers, and polyvinylalcohol; cellulosic polymers, such as hydroxypropyl
cellulose, hydroxyethyl
cellulose, hydroxypropyl methylcellulose, hydroxypropyl methylcellulose
phthalate, and
methylcellulose; gums, such as tragacanth and xanthan gum; sodium alginate;
and gelatin. In some
embodiments, in order to prepare a uniform gel, dispersing agents such as
alcohol or glycerin are
added, or the gelling agent dispersed by trituration, mechanical mixing,
and/or stirring.
[00216] In some embodiments, the pharmaceutical compositions provided herein
are administered
rectally, urethrally, vaginally, or perivaginally in the forms of
suppositories, pessaries, bougies,
poultices or cataplasm, pastes, powders, dressings, creams, plasters,
contraceptives, ointments,
solutions, emulsions, suspensions, tampons, gels, foams, sprays, or enemas. In
some embodiments,
these dosage forms are manufactured using conventional processes as described
in Remington: The
Science and Practice of Pharmacy, supra.
[00217] Rectal, urethral, and vaginal suppositories are solid bodies for
insertion into body orifices,
which are solid at ordinary temperatures but melt or soften at body
temperature to release the active
ingredient(s) inside the orifices. Pharmaceutically acceptable carriers
utilized in rectal and vaginal
suppositories include bases or vehicles, such as stiffening agents, which
produce a melting point in the
proximity of body temperature, when formulated with the pharmaceutical
compositions provided
herein; and antioxidants as described herein, including bisulfite and sodium
metabisulfite. Suitable
vehicles include, but are not limited to, cocoa butter (theobroma oil),
glycerin-gelatin, carbowax
(polyoxyethylene glycol), spermaceti, paraffin, white and yellow wax, and
appropriate mixtures of
mono-, di- and triglycerides of fatty acids, hydrogels, such as polyvinyl
alcohol, hydroxyethyl
methacrylate, polyacrylic acid; glycerinated gelatin.. Combinations of the
various vehicles in some
embodiments, are used. Rectal and vaginal suppositories in some embodiments,
are prepared by the
compressed method or molding. The typical weight of a rectal and vaginal
suppository is about 2 to
about 3 g.
-58-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
[00218] In some embodiments, the pharmaceutical compositions provided herein
are administered
ophthalmically in the forms of solutions, suspensions, ointments, emulsions,
gel-forming solutions,
powders for solutions, gels, ocular inserts, and implants.
[00219] In some embodiments, the pharmaceutical compositions provided herein
are administered
intranasally or by inhalation to the respiratory tract. The pharmaceutical
compositions in some
embodiments, are provided in the form of an aerosol or solution for delivery
using a pressurized
container, pump, spray, atomizer, such as an atomizer using
electrohydrodynamics to produce a fine
mist, or nebulizer, alone or in combination with a suitable propellant, such
as 1, 1, 1,2-tetrafluoroethane
or 1,1,1,2,3,3,3-heptafluoropropane. The pharmaceutical compositions in some
embodiments, are
provided as a dry powder for insufflation, alone or in combination with an
inert carrier such as lactose
or phospholipids; and nasal drops. For intranasal use, the powder in some
embodiments, comprises a
bioadhesive agent, including chitosan or cyclodextrin.
[00220] In some embodiments, solutions or suspensions for use in a pressurized
container, pump,
spray, atomizer, or nebulizer are formulated to contain ethanol, aqueous
ethanol, or a suitable
alternative agent for dispersing, solubilizing, or extending release of the
active ingredient provided
herein, a propellant as solvent; and/or an surfactant, such as sorbitan
trioleate, oleic acid, or an
oligolactic acid.
[00221] In some embodiments, the pharmaceutical compositions provided herein
are micronized to a
size suitable for delivery by inhalation, such as about 50 micrometers or
less, or about 10 micrometers
or less. Particles of such sizes in some embodiments, are prepared using a
comminuting method, such
as spiral jet milling, fluid bed jet milling, supercritical fluid processing
to form nanoparticles, high
pressure homogenization, or spray drying.
[00222] In some embodiments, capsules, blisters and cartridges for use in an
inhaler or insufflator are
formulated to contain a powder mix of the pharmaceutical compositions provided
herein; a suitable
powder base, such as lactose or starch; and a performance modifier, such as l-
leucine, mannitol, or
magnesium stearate. The lactose includes an anhydrous form or the form of the
monohydrate. Other
suitable excipients or carriers include dextran, glucose, maltose, sorbitol,
xylitol, fructose, sucrose, and
trehalose. In some embodiments, the pharmaceutical compositions provided
herein for
inhaled/intranasal administration further comprise a suitable flavor, such as
menthol and levomenthol,
or sweeteners, such as saccharin or saccharin sodium.
[00223] In some embodiments, the pharmaceutical compositions provided herein
for topical
administration are formulated to be immediate release or modified release,
including delayed-,
sustained-, pulsed-, controlled-, targeted, and programmed release.
Modified Release
[00224] The pharmaceutical compositions provided herein in some embodiments,
are formulated as a
modified release dosage form. As used herein, the term "modified release"
refers to a dosage form in
which the rate or place of release of the active ingredient(s) is different
from that of an immediate
-59-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
dosage form when administered by the same route. Modified release dosage forms
include delayed-,
extended-, prolonged-, sustained-, pulsatile-, controlled-, accelerated- and
fast-, targeted-,
programmed-release, and gastric retention dosage forms. The pharmaceutical
compositions in
modified release dosage forms are prepared using a variety of modified release
devices and methods,
including, but not limited to, matrix controlled release devices, osmotic
controlled release devices,
multiparticulate controlled release devices, ion-exchange resins, enteric
coatings, multilayered
coatings, microspheres, liposomes, and combinations thereof. In some
embodiments, the release rate
of the active ingredient(s) are modified by varying the particle sizes and
polymorphorism of the active
ingredient(s).
[00225] Examples of modified release include, but are not limited to, those
described in U.S. Pat. Nos.:
3,845,770; 3,916,899; 3,536,809; 3,598,123; 4,008,719; 5,674,533; 5,059,595;
5,591,767; 5,120,548;
5,073,543; 5,639,476; 5,354,556; 5,639,480; 5,733,566; 5,739,108; 5,891,474;
5,922,356; 5,972,891;
5,980,945; 5,993,855; 6,045,830; 6,087,324; 6,113,943; 6,197,350; 6,248,363;
6,264,970; 6,267,981;
6,376,461; 6,419,961; 6,589,548; 6,613,358; and 6,699,500.
1. Matrix Controlled Release Devices
[00226] The pharmaceutical compositions provided herein in a modified release
dosage form in some
embodiments, are fabricated using a matrix controlled release device (see,
e.g., Takada et al in
"Encyclopedia of Controlled Drug Delivery," Vol. 2, Mathiowitz ed., Wiley,
1999).
[00227] In one embodiment, the pharmaceutical compositions provided herein in
a modified release
dosage form is formulated using an erodible matrix device, which is water-
swellable, erodible, or
soluble polymers, including synthetic polymers, and naturally occurring
polymers and derivatives,
such as polysaccharides and proteins.
[00228] Materials useful in forming an erodible matrix include, but are not
limited to, chitin, chitosan,
dextran, and pullulan; gum agar, gum arabic, gum karaya, locust bean gum, gum
tragacanth,
carrageenans, gum ghatti, guar gum, xanthan gum, and scleroglucan; starches,
such as dextrin and
maltodextrin; hydrophilic colloids, such as pectin; phosphatides, such as
lecithin; alginates; propylene
glycol alginate; gelatin; collagen; and cellulosics, such as ethyl cellulose
(EC), methylethyl cellulose
(MEC), carboxymethyl cellulose (CMC), CMEC, hydroxyethyl cellulose (HEC),
hydroxypropyl
cellulose (HPC), cellulose acetate (CA), cellulose propionate (CP), cellulose
butyrate (CB), cellulose
acetate butyrate (CAB), CAP, CAT, hydroxypropyl methyl cellulose (HPMC),
HPMCP, HPMCAS,
hydroxypropyl methyl cellulose acetate trimellitate (HPMCAT), and ethylhydroxy
ethylcellulose
(EHEC); polyvinyl pyrrolidone; polyvinyl alcohol; polyvinyl acetate; glycerol
fatty acid esters;
polyacrylamide; polyacrylic acid; copolymers of ethacrylic acid or methacrylic
acid (EUDRAGIT ,
Rohm America, Inc., Piscataway, NJ); poly(2-hydroxyethyl-methacrylate);
polylactides; copolymers
of L-glutamic acid and ethyl-L-glutamate; degradable lactic acid-glycolic acid
copolymers; poly-D-(-
)-3-hydroxybutyric acid; and other acrylic acid derivatives, such as
homopolymers and copolymers of
-60-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
butylmethacrylate, methylmethacrylate, ethylmethacrylate, ethylacrylate, (2-
dimethylaminoethyl)methacrylate, and (trimethylaminoethyl)methacrylate
chloride.
[00229] In further embodiments, the pharmaceutical compositions are formulated
with a non-erodible
matrix device. The active ingredient(s) is dissolved or dispersed in an inert
matrix and is released
primarily by diffusion through the inert matrix once administered. Materials
suitable for use as a non-
erodible matrix device included, but are not limited to, insoluble plastics,
such as polyethylene,
polypropylene, polyisoprene, polyisobutylene, polybutadiene,
polymethylmethacrylate,
polybutylmethacrylate, chlorinated polyethylene, polyvinylchloride, methyl
acrylate-methyl
methacrylate copolymers, ethylene-vinylacetate copolymers, ethylene/propylene
copolymers,
ethylene/ethyl acrylate copolymers, vinylchloride copolymers with vinyl
acetate, vinylidene chloride,
ethylene and propylene, ionomer polyethylene terephthalate, butyl rubber
epichlorohydrin rubbers,
ethylene/vinyl alcohol copolymer, ethylene/vinyl acetate/vinyl alcohol
terpolymer, and
ethylene/vinyloxyethanol copolymer, polyvinyl chloride, plasticized nylon,
plasticized
polyethyleneterephthalate, natural rubber, silicone rubbers,
polydimethylsiloxanes, silicone carbonate
copolymers, and ; hydrophilic polymers, such as ethyl cellulose, cellulose
acetate, crospovidone, and
cross-linked partially hydrolyzed polyvinyl acetate,; and fatty compounds,
such as carnauba wax,
microcrystalline wax, and triglycerides.
[00230] In a matrix controlled release system, the desired release kinetics is
controllable, for example,
via the polymer type employed, the polymer viscosity, the particle sizes of
the polymer and/or the
active ingredient(s), the ratio of the active ingredient(s) versus the
polymer, and other excipients or
carriers in the compositions.
[00231] The pharmaceutical compositions provided herein in a modified release
dosage form in some
embodiments, are prepared by methods, including direct compression, dry or wet
granulation followed
by compression, melt-granulation followed by compression.
2. Osmotic Controlled Release Devices
[00232] The pharmaceutical compositions provided herein in a modified release
dosage form in some
embodiments, are fabricated using an osmotic controlled release device,
including one-chamber
system, two-chamber system, asymmetric membrane technology (AMT), and
extruding core system
(ECS). In general, such devices have at least two components: (a) the core
which contains the active
ingredient(s); and (b) a semipermeable membrane with at least one delivery
port, which encapsulates
the core. The semipermeable membrane controls the influx of water to the core
from an aqueous
environment of use so as to cause drug release by extrusion through the
delivery port(s).
[00233] In addition to the active ingredient(s), the core of the osmotic
device optionally includes an
osmotic agent, which creates a driving force for transport of water from the
environment of use into
the core of the device. One class of osmotic agents water-swellable
hydrophilic polymers, which are
also referred to as "osmopolymers" and "hydrogels," including, but not limited
to, hydrophilic vinyl
and acrylic polymers, polysaccharides such as calcium alginate, polyethylene
oxide (PEO),
-61-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
polyethylene glycol (PEG), polypropylene glycol (PPG), poly(2-hydroxyethyl
methacrylate),
poly(acrylic) acid, poly(methacrylic) acid, polyvinylpyrrolidone (PVP),
crosslinked PVP, polyvinyl
alcohol (PVA), PVA/PVP copolymers, PVA/PVP copolymers with hydrophobic
monomers such as
methyl methacrylate and vinyl acetate, hydrophilic polyurethanes containing
large PEO blocks,
sodium croscarmellose, carrageenan, hydroxyethyl cellulose (HEC),
hydroxypropyl cellulose (HPC),
hydroxypropyl methyl cellulose (HPMC), carboxymethyl cellulose (CMC) and
carboxyethyl, cellulose
(CEC), sodium alginate, polycarbophil, gelatin, xanthan gum, and sodium starch
glycolate.
[00234] The other class of osmotic agents are osmogens, which are capable of
imbibing water to affect
an osmotic pressure gradient across the barrier of the surrounding coating.
Suitable osmogens include,
but are not limited to, inorganic salts, such as magnesium sulfate, magnesium
chloride, calcium
chloride, sodium chloride, lithium chloride, potassium sulfate, potassium
phosphates, sodium
carbonate, sodium sulfite, lithium sulfate, potassium chloride, and sodium
sulfate; sugars, such as
dextrose, fructose, glucose, inositol, lactose, maltose, mannitol, raffinose,
sorbitol, sucrose, trehalose,
and xylitol,; organic acids, such as ascorbic acid, benzoic acid, fumaric
acid, citric acid, maleic acid,
sebacic acid, sorbic acid, adipic acid, edetic acid, glutamic acid, p-
tolunesulfonic acid, succinic acid,
and tartaric acid; urea; and mixtures thereof.
[00235] Osmotic agents of different dissolution rates in some embodiments, are
employed to influence
how rapidly the active ingredient(s) is initially delivered from the dosage
form. For example,
amorphous sugars, such as Mannogeme EZ (SPI Pharma, Lewes, DE) are used to
provide faster
delivery during the first couple of hours to promptly produce the desired
therapeutic effect, and
gradually and continually release of the remaining amount to maintain the
desired level of therapeutic
or prophylactic effect over an extended period of time. In this case, the
active ingredient(s) is released
at such a rate to replace the amount of the active ingredient metabolized and
excreted.
[00236] The core in some embodiments, also includes a wide variety of other
excipients and carriers as
described herein to enhance the performance of the dosage form or to promote
stability or processing.
[00237] Materials useful in forming the semipermeable membrane include various
grades of acrylics,
vinyls, ethers, polyamides, polyesters, and cellulosic derivatives that are
water-permeable and water-
insoluble at physiologically relevant pHs, or are susceptible to being
rendered water-insoluble by
chemical alteration, such as crosslinking. Examples of suitable polymers
useful in forming the coating,
include plasticized, unplasticized, and reinforced cellulose acetate (CA),
cellulose diacetate, cellulose
triacetate, CA propionate, cellulose nitrate, cellulose acetate butyrate
(CAB), CA ethyl carbamate,
CAP, CA methyl carbamate, CA succinate, cellulose acetate trimellitate (CAT),
CA
dimethylaminoacetate, CA ethyl carbonate, CA chloroacetate, CA ethyl oxalate,
CA methyl sulfonate,
CA butyl sulfonate, CA p-toluene sulfonate, agar acetate, amylose triacetate,
beta glucan acetate, beta
glucan triacetate, acetaldehyde dimethyl acetate, triacetate of locust bean
gum, hydroxlated ethylene-
vinylacetate, EC, PEG, PPG, PEG/PPG copolymers, PVP, HEC, HPC, CMC, CMEC,
HPMC,
HPMCP, HPMCAS, HPMCAT, poly(acrylic) acids and esters and poly-(methacrylic)
acids and esters
-62-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
and copolymers thereof, starch, dextran, dextrin, chitosan, collagen, gelatin,
polyalkenes, polyethers,
polysulfones, polyethersulfones, polystyrenes, polyvinyl halides, polyvinyl
esters and ethers, natural
waxes, and synthetic waxes.
[00238] Semipermeable membrane in some embodiments, is a hydrophobic
microporous membrane,
wherein the pores are substantially filled with a gas and are not wetted by
the aqueous medium but are
permeable to water vapor, as disclosed in U.S. Pat. No. 5,798,119. Such
hydrophobic but water-vapor
permeable membrane are typically composed of hydrophobic polymers such as
polyalkenes,
polyethylene, polypropylene, polytetrafluoroethylene, polyacrylic acid
derivatives, polyethers,
polysulfones, polyethersulfones, polystyrenes, polyvinyl halides,
polyvinylidene fluoride, polyvinyl
esters and ethers, natural waxes, and synthetic waxes.
[00239] The delivery port(s) on the semipermeable membrane in some
embodiments, is formed post-
coating by mechanical or laser drilling. Delivery port(s) in some embodiments,
is formed in situ by
erosion of a plug of water-soluble material or by rupture of a thinner portion
of the membrane over an
indentation in the core. In addition, delivery ports are formed during coating
process, as in the case of
asymmetric membrane coatings of the type disclosed in U.S. Pat. Nos. 5,612,059
and 5,698,220.
[00240] The total amount of the active ingredient(s) released and the release
rate are substantially
modulated, in some embodiments, via the thickness and porosity of the
semipermeable membrane, the
composition of the core, and the number, size, and position of the delivery
ports.
[00241] The pharmaceutical compositions in an osmotic controlled-release
dosage form in some
embodiments, further comprise additional conventional excipients or carriers
as described herein to
promote performance or processing of the formulation.
[00242] In some embodiments, the osmotic controlled-release dosage forms are
prepared according to
conventional methods and techniques (see, e.g., Remington: The Science and
Practice of Pharmacy;
Santus and Baker, J. Controlled Release 1995, 35, 1-21; Verma et al., Drug
Development and
Industrial Pharmacy 2000, 26, 695-708; Verma et al., J. Controlled Release
2002, 79, 7-27).
[00243] In certain embodiments, the pharmaceutical compositions provided
herein are formulated as
AMT controlled-release dosage form, which comprises an asymmetric osmotic
membrane that coats a
core comprising the active ingredient(s) and other pharmaceutically acceptable
excipients or carriers.
See, U.S. Pat. No. 5,612,059 and WO 2002/17918. In some embodiments, the AMT
controlled-release
dosage forms are prepared using methods such as direct compression, dry
granulation, wet
granulation, and a dip-coating method.
[00244] In certain embodiments, the pharmaceutical compositions provided
herein are formulated as
ESC controlled-release dosage form, which comprises an osmotic membrane that
coats a core
comprising the active ingredient(s), a hydroxylethyl cellulose, and other
pharmaceutically acceptable
excipients or carriers.
-63-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
3. Multiparticulate Controlled Release Devices
[00245] The pharmaceutical compositions provided herein in a modified release
dosage form in some
embodiments, are fabricated a multiparticulate controlled release device,
which comprises a
multiplicity of particles, granules, or pellets, ranging from about 10 m to
about 3 mm, about 50 m to
about 2.5 mm, or from about 100 m to about 1 mm in diameter. Such
multiparticulates in some
embodiments, are made by the processes such as wet-and dry-granulation,
extrusion/spheronization,
roller-compaction, melt-congealing, and by spray-coating seed cores. See, for
example,
Multiparticulate Oral Drug Delivery; Marcel Dekker: 1994; and Pharmaceutical
Pelletization
Technology; Marcel Dekker: 1989.
[00246] For administration by inhalation, the compounds of Formula (I) or (II)
are conveniently
delivered in the form of an aerosol spray presentation from pressurized packs
or a nebuliser, with the
use of a suitable propellant, e.g., dichlorodifluoromethane,
trichlorofluoromethane,
dichlorotetrafluoroethane, carbon dioxide or other suitable gas. In the case
of a pressurized aerosol the
dosage unit in some embodiments, is etermined by providing a valve to deliver
a metered amount.
Capsules and cartridges of, e.g., gelatin for use in an inhaler or insufflator
are formulated containing a
powder mix of the compound and a suitable powder base such as lactose or
starch.
Methods of Treatment, Dosages and Combination Therapies
[00247] The term "mammal" means all mammals including humans. Mammals include,
by way of
example only, humans, non-human primates, cows, dogs, cats, goats, sheep,
pigs, lagomorphs, rats,
mice and rabbits.
[00248] The term "effective amount" as used herein refers to that amount of
the compound being
administered which will relieve to some extent one or more of the symptoms of
the disease, condition
or disorder being treated.
[00249] The term "indirectly modulates" as used herein refers to an agent that
reduces serum retinol
levels in a mammal, resulting in a reduction of retinol levels in the eye of
the mammal. Such a
reduction in ocular retinol levels leads to a modulation of the activity of
visual cycle proteins. This
form of modulation (arises via a decrease in ocular serum retinol levels) is
referred to herein as
indirect modulation.
[00250] In some embodiments, the compositions containing the compound(s)
described herein are
administered for prophylactic and/or therapeutic treatments. The term
"treating" is used to refer to
either prophylactic and/or therapeutic treatments. In therapeutic
applications, the compositions are
administered to a patient already suffering from a disease, condition or
disorder, in an amount
sufficient to cure or at least partially arrest the symptoms of the disease,
disorder or condition.
Amounts effective for this use will depend on the severity and course of the
disease, disorder or
condition, previous therapy, the patient's health status and response to the
drugs, and the judgment of
the treating physician.
-64-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
[00251] In prophylactic applications, compositions containing the compounds
described herein are
administered to a patient susceptible to or otherwise at risk of a particular
disease, disorder or
condition. Such an amount is defined to be a"prophylactically effective amount
or dose." In this use,
the precise amounts also depend on the patient's state of health, weight, and
the like.
[00252] The terms "enhance" or "enhancing" means to increase or prolong either
in potency or
duration a desired effect. Thus, in regard to enhancing the effect of
therapeutic agents, the term
"enhancing" refers to the ability to increase or prolong, either in potency or
duration, the effect of
other therapeutic agents on a system. An "enhancing-effective amount," as used
herein, refers to an
amount adequate to enhance the effect of another therapeutic agent in a
desired system. When used in
a patient, amounts effective for this use will depend on the severity and
course of the disease, disorder
or condition, previous therapy, the patient's health status and response to
the drugs, and the judgment
of the treating physician.
[00253] In the case wherein the patient's condition does not improve, upon the
doctor's discretion the
administration of the compounds is administered chronically, that is, for an
extended period of time,
including throughout the duration of the patient's life in order to ameliorate
or otherwise control or
limit the symptoms of the patient's disease or condition.
[00254] In the case wherein the patient's status does improve, upon the
doctor's discretion the
administration of the compounds is given continuously or temporarily suspended
for a certain length
of time (i.e., a "drug holiday").
[00255] Once improvement of the patient's conditions has occurred, a
maintenance dose is
administered if necessary. Subsequently, in some embodiments, the dosage or
the frequency of
administration, or both, are reduced, as a function of the symptoms, to a
level at which the improved
disease, disorder or condition is retained. In some embodiments, patients
require intermittent treatment
on a long-term basis upon any recurrence of symptoms.
[00256] The amount of a given agent that will correspond to such an amount
will vary depending upon
factors such as the particular compound, disease condition and its severity,
the identity (e.g., weight)
of the subject or host in need of treatment, but is determined according to
the particular circumstances
surrounding the case, including, e.g., the specific agent being administered,
the route of
administration, the condition being treated, and the subject or host being
treated. In general, however,
doses employed for adult human treatment will typically be in the range of
about 0.02 to about 5000
mg per day. In other embodiments, the doses employed for adult human treatment
is in the range of
about 1 to about 1500 mg per day. The desired dose is conveniently presented
in some embodiments,
in a single dose or as divided doses administered simultaneously (or over a
short period of time) or at
appropriate intervals, for example as two, three, four or more sub-doses per
day.
[00257] In certain instances, it is appropriate to administer at least one of
the compounds described
herein (or a pharmaceutically acceptable salt, ester, amide, prodrug, or
solvate) in combination with
another therapeutic agent. By way of example only, if one of the side effects
experienced by a patient
-65-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
upon receiving one of the compounds herein is inflammation, then it is
appropriate to administer an
anti-inflammatory agent in combination with the initial therapeutic agent. Or,
by way of example only,
the therapeutic effectiveness of one of the compounds described herein is
enhanced by administration
of an adjuvant (i.e., by itself the adjuvant has minimal therapeutic benefit,
but in combination with
another therapeutic agent, the overall therapeutic benefit to the patient is
enhanced). Or, by way of
example only, the benefit of experienced by a patient is increased by
administering one of the
compounds described herein with another therapeutic agent (which also includes
a therapeutic
regimen) that also has therapeutic benefit. By way of example only, in a
treatment for macular
degeneration involving administration of one of the compounds described
herein, increased
therapeutic benefit results by also providing the patient with other
therapeutic agents or therapies for
macular degeneration. In any case, regardless of the disease, disorder or
condition being treated, the
overall benefit experienced by the patient is simply additive of the two
therapeutic agents or the
patient experiences a synergistic benefit.
[00258] Also presented herein are combination therapies which include the use
of a compound of
Formula (I) or (II) with modulators of serum retinol levels or activity. In
one embodiment the
combination comprises a compound of Formula (I) or (II) and a compound of
Formula (III):
O
Xi
R
Formula (III)
wherein Xl is selected from the group consisting of NR2, 0, S, CHR2; R' is
(CHR2)X L'-R3, wherein x
is 0, 1, 2, or 3; Ll is a single bond or -C(O)-; R2 is a moiety selected from
the group consisting of H,
(Ci-C4)alkyl, F, (Ci-C4)fluoroalkyl, (Ci-C4)alkoxy, -C(O)OH, -C(O)-NH2, -(Ci-
C4)alkylamine, -C(O)-
(Ci-C4)alkyl, -C(O)-(Ci-C4)fluoroalkyl, -C(O)-(Ci-C4)alkylamine, and -C(O)-(Ci-
C4)alkoxy; and R3 is
H or a moiety, optionally substituted with 1-3 independently selected
substituents, selected from the
group consisting of (C2-C7)alkenyl, (C2-C7)alkynyl, aryl, (C3-C7)cycloalkyl,
(C5-C7)cycloalkenyl, and
a heterocycle; or an active metabolite, or a pharmaceutically acceptable
prodrug or solvate thereof. In
another embodiment, the combination comprises a compound of Formula (I) or
(II) and a compound
of Formula (III) wherein the combination modulates serum retinol levels or
activity. In a further
embodiment is a combination comprising a compound of Formula (I) or (II) and a
compound of
Formula (III) wherein Xl is NR2 and R2 is H or (Ci-C4)alkyl. In yet a further
embodiment is a
combination comprising a compound of Formula (I) or (II) and a compound of
Formula (III) wherein
x is 0. In another embodiment is a combination comprising a compound of
Formula (I) or (II) and a
compound of Formula (III) wherein x is 1 and Ll is -C(O)-. In yet another
embodiment is a
combination comprising a compound of Formula (I) or (II) and a compound of
Formula (III) wherein
R3 is an optionally substituted heteroaryl. In a further embodiment is a
combination comprising a
-66-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
compound of Formula (I) or (II) and a compound of Formula (III) wherein the
compound of Formula
0
\ \ \ \ NH
(III) is oH or an active metabolite, or a pharmaceutically
acceptable prodrug or solvate thereof. In one embodiment is a combination
comprising a compound of
Formula (I) or (II) and a compound of Formula (III) wherein the compound of
Formula (III) is 4-
hydroxyphenylretinamide.
[00259] Specific, non-limiting examples of combination therapies include use
of at least one
compound of Formula (I) or (II) with nitric oxide (NO) inducers, statins,
negatively charged
phospholipids, anti-oxidants, minerals, anti-inflammatory agents, anti-
angiogenic agents, matrix
metalloproteinase inhibitors, and carotenoids. In several instances, suitable
combination agents fall
within multiple categories (by way of example only, lutein is an anti-oxidant
and a carotenoid).
Further, the compounds of Formula (I) or (II) in some embodiments, are
administered with additional
agents that may provide benefit to the patient, including by way of example
only cyclosporin A.
[00260] In addition, the compounds of Formula (I) and (II) in some
embodiments, are used in
combination with procedures that provides additional or synergistic benefit to
the patient, including,
by way of example only, the use of extracorporeal rheopheresis (also known as
membrane differential
filtration), the use of implantable miniature telescopes, laser
photocoagulation of drusen, and
microstimulation therapy.
[00261] The use of anti-oxidants has been shown to benefit patients with
macular degenerations and
dystrophies. See, e.g., Arch. Ophthalmol., 119: 1417-36 (2001); Sparrow, et
al., J. Biol. Chem.,
278:18207-13 (2003). Examples of suitable anti-oxidants that could be used in
combination with at
least one compound having the structure of Formula (I) or (II) include vitamin
C, vitamin E, beta-
carotene and other carotenoids, coenzyme Q, 4-hydroxy-2,2,6,6-
tetramethylpiperidine-N-oxyl (also
known as Tempol), lutein, butylated hydroxytoluene, resveratrol, a trolox
analogue (PNU-83836-E),
and bilberry extract.
[00262] The use of certain minerals has also been shown to benefit patients
with macular
degenerations and dystrophies. See, e.g., Arch. Ophthalmol., 119: 1417-36
(2001). Examples of
suitable minerals that could be used in combination with at least one compound
having the structure of
Formula (I) or (II) include copper-containing minerals, such as cupric oxide
(by way of example only);
zinc-containing minerals, such as zinc oxide (by way of example only); and
selenium-containing
compounds.
[00263] The use of certain negatively-charged phospholipids has also been
shown to benefit patients
with macular degenerations and dystrophies. See, e.g., Shaban & Richter, Biol.
Chem., 383:537-45
-67-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
(2002); Shaban, et al., Exp. Eye Res., 75:99-108 (2002). Examples of suitable
negatively charged
phospholipids that could be used in combination with at least one compound
having the structure of
Formula (I) or (II) include cardiolipin and phosphatidylglycerol. Positively-
charged and/or neutral
phospholipids in some embodiments, also provide benefit for patients with
macular degenerations and
dystrophies when used in combination with compounds having the structure of
Formula (I) or (II).
[00264] The use of certain carotenoids has been correlated with the
maintenance of photoprotection
necessary in photoreceptor cells. Carotenoids are naturally-occurring yellow
to red pigments of the
terpenoid group that can be found in plants, algae, bacteria, and certain
animals, such as birds and
shellfish. Carotenoids are a large class of molecules in which more than 600
naturally occurring
carotenoids have been identified. Carotenoids include hydrocarbons (carotenes)
and their oxygenated,
alcoholic derivatives (xanthophylls). They include actinioerythrol,
astaxanthin, canthaxanthin,
capsanthin, capsorubin, (3-8'-apo-carotenal (apo-carotenal), (3-12'-apo-
carotenal, a-carotene, (3-
carotene, "carotene" (a mixture of a- and (3-carotenes), y-carotenes, (3 -
cyrptoxanthin, lutein, lycopene,
violerythrin, zeaxanthin, and esters of hydroxyl- or carboxyl-containing
members thereof. Many of the
carotenoids occur in nature as cis- and trans-isomeric forms, while synthetic
compounds are
frequently racemic mixtures.
[00265] In humans, the retina selectively accumulates mainly two carotenoids:
zeaxanthin and lutein.
These two carotenoids are thought to aid in protecting the retina because they
are powerful
antioxidants and absorb blue light. Studies with quails establish that groups
raised on carotenoid-
deficient diets had retinas with low concentrations of zeaxanthin and suffered
severe light damage, as
evidenced by a very high number of apoptotic photoreceptor cells, while the
group with high
zeaxanthin concentrations had minimal damage. Examples of suitable carotenoids
for in combination
with at least one compound having the structure of Formula (I) include lutein
and zeaxanthin, as well
as any of the aforementioned carotenoids.
[00266] Suitable nitric oxide inducers include compounds that stimulate
endogenous NO or elevate
levels of endogenous endothelium-derived relaxing factor (EDRF) in vivo or are
substrates for nitric
oxide synthase. Such compounds include, for example, L-arginine, L-
homoarginine, and N-hydroxy-
L-arginine, including their nitrosated and nitrosylated analogs (e.g.,
nitrosated L-arginine, nitrosylated
L-arginine, nitrosated N-hydroxy-L-arginine, nitrosylated N-hydroxy-L-
arginine, nitrosated L-
homoarginine and nitrosylated L-homoarginine), precursors of L-arginine and/or
physiologically
acceptable salts thereof, including, for example, citrulline, ornithine,
glutamine, lysine, polypeptides
comprising at least one of these amino acids, inhibitors of the enzyme
arginase (e.g., N-hydroxy-L-
arginine and 2(S)-amino-6-boronohexanoic acid) and the substrates for nitric
oxide synthase,
cytokines, adenosine, bradykinin, calreticulin, bisacodyl, and
phenolphthalein. EDRF is a vascular
relaxing factor secreted by the endothelium, and has been identified as nitric
oxide or a closely related
derivative thereof (Palmer et al, Nature, 327:524-526 (1987); Ignarro et al,
Proc. Natl. Acad. Sci.
USA, 84:9265-9269 (1987)).
-68-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
[00267] Statins serve as lipid-lowering agents and/or suitable nitric oxide
inducers. In addition, a
relationship has been demonstrated between statin use and delayed onset or
development of macular
degeneration. G. McGwin, et al., British Journal of Ophthalmology, 87:1121-25
(2003). Statins can
thus provide benefit to a patient suffering from an ophthalmic condition (such
as the macular
degenerations and dystrophies, and the retinal dystrophies) when administered
in combination with
compounds of Formula (I) or (II). Suitable statins include, by way of example
only, rosuvastatin,
pitivastatin, simvastatin, pravastatin, cerivastatin, mevastatin, velostatin,
fluvastatin, compactin,
lovastatin, dalvastatin, fluindostatin, atorvastatin, atorvastatin calcium
(which is the hemicalcium salt
of atorvastatin), and dihydrocompactin.
[00268] Suitable anti-inflammatory agents with which the Compounds of Formula
(I) or (II) in some
embodiments, are used include, by way of example only, aspirin and other
salicylates, cromolyn,
nedocromil, theophylline, zileuton, zafirlukast, montelukast, pranlukast,
indomethacin, and
lipoxygenase inhibitors; non-steroidal antiinflammatory drugs (NSAIDs) (such
as ibuprofen and
naproxin); prednisone, dexamethasone, cyclooxygenase inhibitors (i.e., COX-1
and/or COX-2
inhibitors such as NaproxenTM, or CelebrexTM); statins (by way of example
only, rosuvastatin,
pitivastatin, simvastatin, pravastatin, cerivastatin, mevastatin, velostatin,
fluvastatin, compactin,
lovastatin, dalvastatin, fluindostatin, atorvastatin, atorvastatin calcium
(which is the hemicalcium salt
of atorvastatin), and dihydrocompactin); and disassociated steroids.
[00269] Suitable matrix metalloproteinases (MMPs) inhibitors in some
embodiments, are administered
in combination with compounds of Formula (I) or (II) in order to treat
ophthalmic conditions or
symptoms associated with macular or retinal degenerations. MMPs hydrolyze most
components of the
extracellular matrix. These proteinases play a central role in many biological
processes such as normal
tissue remodeling, embryogenesis, wound healing and angiogenesis. However,
excessive expression of
MMP has been observed in many disease states, including macular degeneration.
Many MMPs have
been identified, most of which are multidomain zinc endopeptidases. A number
of metalloproteinase
inhibitors are known (see for example the review of MMP inhibitors by
Whittaker M. et al, Chemical
Reviews 99(9):2735-2776 (1999)). Representative examples of MMP Inhibitors
include Tissue
Inhibitors of Metalloproteinases (TIMPs) (e.g., TIMP-1, TIMP-2, TIMP-3, or
TIMP-4), az-
macroglobulin, tetracyclines (e.g., tetracycline, minocycline, and
doxycycline), hydroxamates (e.g.,
BATIMASTAT, MARIMISTAT and TROCADE), chelators (e.g., EDTA, cysteine,
acetylcysteine, D-
penicillamine, and gold salts), synthetic MMP fragments, succinyl
mercaptopurines,
phosphonamidates, and hydroxaminic acids. Examples of MMP inhibitors that are
used in
combination with compounds of Formula (I) or (II) include, by way of example
only, any of the
aforementioned inhibitors.
[00270] The use of antiangiogenic or anti-VEGF drugs has also been shown to
provide benefit for
patients with macular degenerations and dystrophies. Examples of suitable
antiangiogenic or anti-
VEGF drugs that could be used in combination with at least one compound having
the structure of
-69-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
Formula (I) include Rhufab V2 (LucentisTM), Tryptophanyl-tRNA synthetase
(TrpRS), Eye001 (Anti-
VEGF Pegylated Aptamer), squalamine, RetaaneTM 15mg (anecortave acetate for
depot suspension;
Alcon, Inc.), Combretastatin A4 Prodrug (CA4P), MacugenTM, MifeprexTM
(mifepristone - ru486),
subtenon triamcinolone acetonide, intravitreal crystalline triamcinolone
acetonide, Prinomastat
(AG3340 - synthetic matrix metalloproteinase inhibitor, Pfizer), fluocinolone
acetonide (including
fluocinolone intraocular implant, Bausch & Lomb/Control Delivery Systems),
VEGFR inhibitors
(Sugen), and VEGF-Trap (Regeneron/Aventis). Resveratrol, which can be
extracted from walnuts or
the skins of red grapes, has demonstrated anti-angiogenic activity and in some
embodiments, is used
as the second or additional agent for the combination therapies described
herein. Furthermore, other
trans-stilbene compounds are expected to exhibit similar activity.
[00271] In some embodiments, other pharmaceutical therapies that have been
used to relieve visual
impairment are used in combination with at least one compound of Formula (I)
or (II). Such
treatments include but are not limited to agents such as VisudyneTM with use
of a non-thermal laser,
PKC 412, Endovion (NeuroSearch A/S), neurotrophic factors, including by way of
example Glial
Derived Neurotrophic Factor and Ciliary Neurotrophic Factor, diatazem,
dorzolamide, Phototrop, 9-
cis-retinal, eye medication (including Echo Therapy) including phospholine
iodide or echothiophate or
carbonic anhydrase inhibitors, AE-941 (AEterna Laboratories, Inc.), Sirna-027
(Sirna Therapeutics,
Inc.), pegaptanib (NeXstar Pharmaceuticals/Gilead Sciences), neurotrophins
(including, by way of
example only, NT-4/5, Genentech), Cand5 (Acuity Pharmaceuticals), ranibizumab
(Genentech), INS-
37217 (Inspire Pharmaceuticals), integrin antagonists (including those from
Jerini AG and Abbott
Laboratories), EG-3306 (Ark Therapeutics Ltd.), BDM-E (BioDiem Ltd.),
thalidomide (as used, for
example, by EntreMed, Inc.), cardiotrophin-1 (Genentech), 2-methoxyestradiol
(Allergan/Oculex),
DL-8234 (Toray Industries), NTC-200 (Neurotech), tetrathiomolybdate
(University of Michigan),
LYN-002 (Lynkeus Biotech), microalgal compound (Aquasearch/Albany, Mera
Pharmaceuticals), D-
9120 (Celltech Group plc), ATX-S 10 (Hamamatsu Photonics), TGF-beta 2
(Genzyme/Celtrix),
tyrosine kinase inhibitors (Allergan, SUGEN, Pfizer), NX-278-L (NeXstar
Pharmaceuticals/Gilead
Sciences), Opt-24 (OPTIS France SA), retinal cell ganglion neuroprotectants
(Cogent Neurosciences),
N-nitropyrazole derivatives (Texas A&M University System), KP- 102 (Krenitsky
Pharmaceuticals),
and cyclosporin A. See U.S. Patent Application Publication No. 20040092435.
[00272] In any case, the multiple therapeutic agents (one of which is one of
the compounds described
herein) in some embodiments, are administered in any order or even
simultaneously. If
simultaneously, the multiple therapeutic agents in some embodiments, is
provided in a single, unified
form, or in multiple forms (by way of example only, either as a single pill or
as two separate pills).
One of the therapeutic agents in some embodiments, is given in multiple doses,
or both may be given
as multiple doses. If not simultaneous, the timing between the multiple doses
varies from more than
zero weeks to less than four weeks. In addition, the combination methods,
compositions and
formulations are not to be limited to the use of only two agents; we envision
the use of multiple
-70-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
therapeutic combinations. By way of example only, a compound having the
structure of Formula (I) or
(II) is provided with at least one antioxidant and at least one negatively
charged phospholipid; or a
compound having the structure of Formula (I) or (II) is provided with at least
one antioxidant and at
least one inducer of nitric oxide production; or a compound having the
structure of Formula (I) or (II)
is provided with at least one inducer of nitric oxide productions and at least
one negatively charged
phospholipid; and so forth.
[00273] In addition, the compounds of Formula (I) or (II) in some embodiments,
are used in
combination with procedures that provide additional or synergistic benefit to
the patient. Procedures to
relieve visual impairment include but are not limited to `limited retinal
translocation', photodynamic
therapy (including, by way of example only, receptor-targeted PDT, Bristol-
Myers Squibb, Co.;
porfimer sodium for injection with PDT; verteporfin, QLT Inc.; rostaporfin
with PDT, Miravent
Medical Technologies; talaporfin sodium with PDT, Nippon Petroleum; motexafin
lutetium,
Pharmacyclics,, Inc.), antisense oligonucleotides (including, by way of
example, products tested by
Novagali Pharma SA and ISIS- 13650, Isis Pharmaceuticals), laser
photocoagulation, drusen lasering,
macular hole surgery, macular translocation surgery, implantable miniature
telescopes, Phi-Motion
Angiography (also known as Micro-Laser Therapy and Feeder Vessel Treatment),
Proton Beam
Therapy, microstimulation therapy, Retinal Detachment and Vitreous Surgery,
Scleral Buckle,
Submacular Surgery, Transpupillary Thermotherapy, Photosystem I therapy, use
of RNA interference
(RNAi), extracorporeal rheopheresis (also known as membrane differential
filtration and
Rheotherapy), microchip implantation, stem cell therapy, gene replacement
therapy, ribozyme gene
therapy (including gene therapy for hypoxia response element, Oxford
Biomedica; Lentipak, Genetix;
PDEF gene therapy, GenVec), photoreceptor/retinal cells transplantation
(including transplantable
retinal epithelial cells, Diacrin, Inc.; retinal cell transplant, Cell
Genesys, Inc.), and acupuncture.
[00274] Further combinations that are used to benefit an individual include
using genetic testing to
determine whether that individual is a carrier of a mutant gene that is
correlated with certain
ophthalmic conditions. By way of example only, defects in the human ABCA4 gene
are thought to be
associated with five distinct retinal phenotypes including Stargardt disease,
cone-rod dystrophy, age-
related macular degeneration and retinitis pigmentosa. See e.g., Allikmets et
al., Science, 277:1805-07
(1997); Lewis et al., Am. J. Hum. Genet., 64:422-34 (1999); Stone et al.,
Nature Genetics, 20:328-29
(1998); Allikmets, Am. J. Hum. Gen., 67:793-799 (2000); Klevering, et al,
Ophthalmology, 111:546-
553 (2004). In addition, an autosomal dominant form of Stargardt Disease is
caused by mutations in
the ELOV4 gene. See Karan, et al., Proc. Natl. Acad. Sci. (2005). Patients
possessing any of these
mutations are expected to find therapeutic and/or prophylactic benefit in the
methods described herein.
[00275] In addition, in some embodiments, compounds of Formula (I) or (II) or
other agents that result
in the reduction of serum retinol levels are administered with (meaning
before, during or after) agents
that treat or alleviate side effects arising from serum retinol reduction.
Such side effects include dry
skin and dry eye. Accordingly, agents that alleviate or treat either dry skin
or dry eye are administered
-71-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
in some embodiments, with compounds of Formula (I) or (II) or other agents
that reduce serum retinol
levels.
EXAMPLES
[00276] The following examples provide illustrative methods for the synthesis
of compounds of
Formula (I) or (II). These examples are provided for illustrative purposes
only and not to limit the
scope of the claims provided herein.
Analytical LC/MS methods:
[00277] Method A:
Instrument: Waters UPLC/MS with UV detector (220 nM) and MS detector (ESI).
HPLC column: Waters Acquity BEH C18 1.7 m 2.1 mm x 50 mm.
HPLC Gradient: 0.6 mL/min, from 95:5 20 mM ammonium formate buffer (brought to
pH 7.4
with ammonium hydroxide): acetonitrile to 20:80 ammonium formate buffer:
acetonitrile in 1.5 min,
maintaining for 1.3 min.
[00278] Method B:
Instrument: Waters LC/MS with DAD detector (220 and 254 nM) and MS detector
(ESI).
HPLC column: Merck LiChroCART 30-4 Purospher STAR RP-18, endcapped, 3 m 4.6 mm
x
50 mm.
HPLC Gradient: 1.5 mL/min, from 95:5 20 mM ammonium formate buffer (brought to
pH 7.4
with ammonium hydroxide): acetonitrile to 5:95 ammonium formate
buffer:acetonitrile in 2.5 min,
maintaining for 1.8 min.
[00279] Method C:
Instrument: Waters Alliance with UV detector (220 nM) and MS detector (ESI).
HPLC column: Waters XTerra MS C18, 5 m, 4.6mm x 50 mm.
HPLC Gradient: 2 mL/min, from 95:5 water + 5% formic acid:acetonitrile + 5%
formic acid
for 0.5 min. then to 5:95 aqueous:organic in 5 min, maintaining for 0.5 min.
Example 1: Synthesis of 5-(2-tert-Butyl-4-chlorophenoxy)pentanoic acid
ci ci I~ o ci I~ o
OH O~-~Oi O OH
1a 1-1
[00280] Methyl 5-bromovalerate (1.52 mL, 2.09 g, 10.7 mmol,), 2-tert-butyl-4-
chloro-phenol (2.4 g,
13 mmol) and potassium carbonate (1.4 g, 10.1 mmol) were suspended in dry DMF
(10 mL) and
stirred for 2 h at 120 C. To the resulting mixture water was added (80 mL)
and extracted with ethyl
acetate (3 x 20 mL). The organic layers were washed two times with 10% sodium
hydroxide (2 x 20
mL), water (1 x 20 mL), dried over sodium sulfate and evaporated in vacuo. To
afford 3.46 g (quant)
crude la that was used in the next step without further purification. 'H NMR
(400 MHz, CDC13) b
-72-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
ppm 7.21 (d, J= 2.6 Hz, 1 H), 7.10 (dd, J= 8.6, 2.6 Hz, 1 H), 6.75 (d, J= 8.6
Hz, 1 H), 3.97 (t, J= 5.8
Hz, 2H), 2.46 (t, J= 6.9 Hz, 2H), 1.84-1.94 (m, 4H), 1.36 (s, 9H).
[00281] A mixture of la (3.46 g), 10% sodium hydroxide (10 mL) in dioxane (50
mL) was stirred
overnight at room temperature. The dioxane was removed in vacuo, the remaining
mixture was diluted
with water and extracted with ethyl acetate (2 x 20 mL). The pH of the aqueous
layer was adjusted to 4
with concentrated hydrochloric acid, and extracted with chloroform ( 1 x 20
mL). The combined
organic layers were dried over sodium sulfate and evaporated in vacuo. The
product was triturated
with hexane to yield 1.87 g (61% calculated from methyl 5-bromovalerate) white
crystalline 1-1. mp.
95.4-96.4 C, 'H NMR (400 MHz, CDC13) b ppm 7.21 (d, J= 2.6 Hz, 1H), 7.10 (dd,
J= 8.6, 2.6 Hz,
1H), 6.75 (d, J= 8.6 Hz, 1H), 3.97 (t, J= 5.8 Hz, 2H), 2.46 (t, J= 6.9 Hz,
2H), 1.84-1.94 (m, 4H),
1.36 (s, 9H).
[00282] The following compounds were made by the above procedure using 2-tert-
butyl-4-
chlorophenol and the appropriate methanesulfonyl ester.
No. NMR
ci 'H NMR (400 MHz, CDC13) S ppm 7.23 (d, J= 2.6
~ Hz, 1 H), 7.12 (dd, J= 2.6, 8.7 Hz, 1 H), 6.76 (d,
1-2 oH 8.7 Hz, 1H), 3.81 (d, J= 5.9 Hz, 1H), 2.39-2.50
(m, 1H), 2.23 (m, 1H), 2.10 (m, 1H), 1.89-1.98 (m,
3H), 1.41-1.45 (m, 1H), 1.38 (s, 9H), 1.26-1.34 (m,
1H), 1.08-1.18 (m, 1H).
ci
10 1 H NMR (400 MHz, CDC13) S ppm 7.23 (d, J= 2.6
OH Hz, 1 H), 7.12 (dd, J= 2.6, 8.7 Hz, 1 H), 6.76 (d, J
cr, 1-3 = 8.7 Hz, 1H), 3.85-3.92 (m, 2H), 2.88-2.98 (m,
1H), 2.47-2.68 (m, 1H), 2.19-2.35 (m, 1H), 1.92-
2.14 (m, 3H), 1.50-1.84 (m, 2H), 1.38 (s, 9H)
[00283] The following compounds were made by the above procedure using 2-tert-
butyl-4-
chlorophenol, the appropriate bromoester and cesium carbonate as the base in
the first step and lithium
hydroxide in the second step.
No. NMR
ci 1 H NMR (300 MHz, CDC13) S ppm
7.21 (d, J= 3 Hz, 1 H), 7.10 (dd, J= 3,
ox 9 Hz, 1H), 6.75 (d, 9 Hz, 1H), 4.01 (t,
1-4 J= 6 Hz, 2H), 2.63 (t, J= 6 Hz, 2H),
0 2.18 (quint, J= 6 Hz, 2H), 1.32 (s,
9H).
-73-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
q 1 H NMR (300 MHz, CDC13) S ppm
0 7.21 (d, J= 3 Hz, 1 H), 7.10 (dd, J= 3,
9 Hz, 1H), 6.75 (d, 9 Hz, 1H), 3.94 (t,
15 OH J= 6 Hz, 2H), 2.37 (t, J= 6 Hz, 2H),
1.86 (quint, J= 6 Hz, 2H), 1.68
(quint, J= 6 Hz, 2H), 1.4-1.6 (m, 4H),
1.32 s,9H.
Example 2: Synthesis of 5-(2-tert-Butyl-4-chlorophenoxy)-N-(4-
hydroxyphenyl)pentanamide
CI O CI O / OH
~ ~ I
~~OH Ol N
O
1-1 2-1
[00284] A mixture of 1-1 (0.3 g, 1.05 mmol) and carbonyl diimidazole (0.18 g,
1.11 mmol) was stirred
in dichloroethane (2.5 mL) for 30 minutes, then 4-aminophenol (0.136 g, 1.25
mmol) and N,N-
diisopropylethylamine (220 L, 1.25 mmol) was added. The resulting solution
was stirred for
additional 4 h. The solvent was evaporated, the remaining mixture was diluted
with 20 mL ether and
washed with 1M hydrochloric acid (1 x 20mL), the organic layer was dried over
sodium sulfate and
the solvent evaporated. The product was triturated with diisopropyl ether to
give 2-1 (6 mg, 25%).
Example 3: Synthesis of Methyl 4-(5-(2-tert-butyl-4-
chlorophenoxy)pentanamido)benzoate
0
ci I~ O ci I~ O / I Oi
O~OH O~ " N \
1-1 3-1
[00285] A mixture of 1-1 (0.25 g, 0.88 mmol) and oxalyl chloride (0.12 g, 0.95
mmol) in
dichloroethane (5 mL) was stirred for 48 h at room temperature. The solvent
was removed in vacuo
and the residue was dissolved in dichloroethane. To this solution methyl 4-
aminobenzoate (0.146 g,
0.96 mmol) and N,N-diisopropylethylamine (350 L, 2.0 mmol) were added and
stirred overnight at
room temperature. The mixture was washed with 20 mL 10% hydrochloric acid, the
organic layer was
dried over sodium sulfate and the solvent was removed in vacuo to give 0.25 g
(69%) 3-1. LCMS:
method A, Rt: 1.85 min, M+H=418.
[00286] The following compounds were made by the above procedure using either
oxalyl chloride or
thionyl chloride.
-74-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
No. MW MH+ Rt LCMS
Method
ci I ~ O / OH
3-2 H 418 418 1.85 A
ci a coonb
0- KO,
3-3 H 390 390 1.64 A
G
O
Goavle
H
3-4 424 424 3.63 B
a a coaw
3-5 424 424 284 B
a
o a1 3-6 444 444 2.12 A
G GJGvIe
G
H
3-7 416 416 1.84 A
-75-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
CI I\ O / I OH
3-8 H 450 450 1.98 A
a O / I COaVIe
\
3-9 H 416 416 1.85 A
a
0 a1
\
H
3-10 458 458 2.14 A
Example 4: Synthesis of Methyl3-(3-((2-tert-butyl-4-
chlorophenoxy)methyl)cyclopentane-
carboxamido)cyclohexanecarboxylate
CI o - CI o JO----~ N COOMe
1-2 4-1
[00287] A solution of 1-2 (0.109 g, 0.354 mmol), N-hydroxybenzotriazole (0.053
g, 0.35 mmol) and
EDC HC1(0.067 g, 0.35 mmol) in dry DMF was stirred for 40 minutes at room
temperature, then
methyl 3-aminocyclohexanecarboxylate hydrochloride (0.075 g, 0.385 mmol) and
triethylamine (54
L, 0.385 mmol) was added and stirring was continued for 4 h. The reaction
mixture was diluted with
5% sodium bicarbonate (20 mL) and extracted with dichloromethane (3 x 20 mL).
The combined
organic layers were washed with 10% hydrochloric acid (1 x 20 mL), brine (2 x
20 mL) and dried over
sodium sulfate. The solvent was removed in vacuo, the oily residue was
purified by flash
chromatography on Kiese1ge160H using chloroform as an eluent to yield 0.1 g
(63%) 4-1 as a pale
yellow oil. LCMS: method A, Rt: 1.98 min, M+H=450).
[00288] The following compounds were made by the above procedure using the
appropriate amine.
-76-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
[00289]
No. MW MH+ Rt LCMS
Method
a O / I oH
4-2 H 402 402 5 C
a coavle
a
H
4-3 464 464 1.99 A
ci O
cooNb
4-4 H 464 464 2.02 A
Example 5: Synthesis of 4-(5-(2-tert-Butyl-4-chlorophenoxy)pentanamido)benzoic
acid
O O
CI O / I 0 CI O OH
O N \ O N
3-1 5-1
[00290] A mixture of 3-1 (0.25 g, 0.59 mmol) and sodium hydroxide (1M, 3 mL)
in dioxane (4 mL)
was stirred overnight at room temperature. The pH was adjusted to 4 with 1 M
hydrochloric acid and
the solution was extracted with 20 mL dichloromethane. The organic layer was
dried over sodium
sulfate, and the solvent was removed in vacuo to yield 5-1 (0.2 g, 83%). LCMS:
method B, Rt: 2.08
min, M+H20=421.
-77-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
[00291] The following compounds were made by the above procedure from the
appropriate ester.
No. MW MH+ Rt LCMS
Method
a cocH
0 wj:::),
H
5-2 410 410 1.94 B
a
cocH
0
H
5-3 410 410 4.41 A
a o / I cocH
H
5-4 444 444 1.67 A
a cocH
O
H
5-5 450 450 1.61 A
[00292] The following compounds were made by the above procedure from the
appropriate ester,
heating at 50 C during the hydrolysis step.
No. MW MH+ Rt LCMS
Method
coCH
5-6 436 436 1.62 A
-78-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
0
CI 0 OH
5-7 ~~ 430 430 1.62 A
H
a
CoCH
H
5-8 436 436 1.6 A
a
a
CocH
H
5-9 450 450 1.63 A
Example 6: Synthesis of N-(4-(2-tert-Butyl-4-chlorophenoxy)butyl)-4-hydroxy-3-
(morpholinomethyl)-benzamide
CI
H H
HO,~~N, Boc OMs--~~N, Boc ON, Boc
6a 6b
CI OH
CI
O~~NH2
O N
0
Jl
``
6c 6-1
[00293] To a solution of Boc-4-aminobutanol (5.07 g, 26.8 mmol) and
triethylamine (7.5 mL, 53.6
mol) in dichloroethane (60 mL) was added methanesulfonyl chloride (2.3 mL,
29.5 mmol) at 0 C.
After the addition was completed, the mixture was stirred for additional 10
minutes. The reaction
mixture was washed with 30 mL cold saturated sodium bicarbonate and the
organic layer was dried
over magnesium sulfate. The solvent was evaporated to give 9.9 g crude 6a as
yellow oil. This crude
product was used in the next step.
[00294] To a mixture of 2-tert-butyl-4-chlorophenol (4.0 g, 21.7 mmol) and
potassium carbonate (6.0
g, 43.4 mol) in dry DMF (40 mL) was added a solution of 24a (9.9 g, mmol) in
dry DMF (40 mL)
dropwise at 60 C. The resulting mixture was stirred overnight at 60 C. The
mixture was poured into
-79-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
water, extracted with 20 mL ethyl acetate, the organic layer was dried over
sodium sulfate, and the
solvent was removed in vacuo. The crude product (8.3 g) was purified by column
chromatography
using a gradient elution with hexane/dichloromethane (1:1) then
dichloromethane to give 2.41 g(31%)
6b.
[00295] A mixture of 6b (2.41 g, 6.8 mmol) and HC1 in dioxane (15 mL, 6.5M) in
ethyl acetate was
stirred overnight. The mixture was evaporated and the residue was
recrystallized from a mixture of
diethyl ether and ethyl acetate to yield 1.41 g (81%) 6c.
[00296] A mixture of 4-(2-tert-butyl-4-chloro-phenoxy)-butylamine
hydrochloride (0.102 g, 0.35
mmol), 4-hydroxy-3-morpholin-4-ylmethyl-benzoic acid (0.091 g, 0.385 mmol),
triethylamine (54 L,
0.385 mmol), N-hydroxybenzotriazole (0.059 g, 0.385 mmol), EDC HC1(0.074 g,
0.385 mmol) in dry
DMF was stirred for 3 h at room temperature. 20 mL of a 5% sodium bicarbonate
solution was added,
and the reaction was extracted with dichloromethane (3 x 20 mL). The combined
organic layers were
washed with 10% hydrochloric acid (1 x 20 mL), brine (2 x 20 mL) and dried
over sodium sulfate.
The solvent was removed in vacuo, the residue was triturated with a mixture of
diethyl ether and ethyl
acetate. The crude product was purified by flash chromatography (Kiese1ge160H)
using
chloroform:methanol (10:3) as an eluent. Yield: 0.070 g (42%) 6-1. LCMS:
method A, Rt: 1.80 min,
M+H=475).
[00297] The following compounds were made by the above procedure, using the
appropriate
acid.
No. MW MH+ Rt LCMS
Method
a I \ / I cH
H
6-2 O 489 489 1.88 A
G / I OH
H
/ N \ F
6-3 0 394 394 1.72 A
a / I OH
H
N \ F 0--~ 6-4 0 408 408 1.84 A
-80-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
C1 OH
H
N F
I,,[::::(
6-5 o F F 444 444 1.81 A
CI OH
H
F
6-6 o F F 458 458 1.88 A
Example 7: Synthesis of 4-(4-(2-tert-Butyl-4-
chlorophenoxy)butylcarbamoyl)benzoic acid
~
0
Ci C~
ON H2 N \ ~ O
O
6c 7a
O
CI / OH
ON \ ~
O
7-1
[00298] A mixture of 6c (0.117 g, 0.40 mmol), terephthalic acid mono-tert-
butyl ester (0.098 g, 0.44
mmol), triethylamine (62 L, 0.44 mmol), N-hydroxybenzotriazole (0.067 g, 0.44
mmol) and EDC
HC1(0.084 g, 0.44 mmol) in dry DMF (5 mL) was stirred for 1 h at room
temperature. 5% Sodium
bicarbonate solution was added (20 mL), and the mixture was extracted with
dichloromethane (3 x 20
mL). The combined organic layers were washed with 10% hydrochloric acid (1 x
20 mL), brine (2 x
20 mL) and dried over sodium sulfate. The solvent was removed in vacuo to give
0.246 g crude 7a.
[00299] The ester 7a was dissolved in 5 mL dioxane, 4 mL of 6.5 M hydrogen
chloride in dioxane was
added and the mixture was stirred at room temperature for 24 h. The reaction
mixture was poured into
water and extracted with dichloromethane (2 x 20mL). The combined organic
layers were washed with
water (1 x 20 mL), dried over sodium sulfate, and the solvent was evaporated.
The product 7-1 was
crystallized from ether/hexane, filtered and washed several times with
ether/hexane to give 0.127
(59%) white crystalline 25-1. LCMS: method A, Rt: 1.36 min, M+H=404.
[00300] The following compound was made by the above procedure.
-81-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
[00301]
No. MW MH+ Rt LCMS
Method
a aOa-i
H
7-2 0 418 418 1.39 A
Example 8
[00302] The following compounds are made by the procedures described herein.
No. MW
CI 0
8-1 O OH 310
CI
0
8-2 O OH 294
CI
0
8-3 O OH 268
CI
O
8-4 O O H 270
CI
0
8-5 O ~ OH 294
CI
8-6 ~ O OH 324
O
CI
8-7 O 1NH2 307
O
-82-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
NH2
CI
0 O
8-8 402
N
H
CI
O
8-9 H NH2 308
CI
H O
8-10 N OH 295
CI
8-11 N OH 309
H
CI
O
8-12 N OH 323
H
CI
O
8-13 N OH 283
H
CI O H
I O I
8-14 N ~ N 356
H H
CI / I COOH
8-15 N N \ 416
H
O
CI
OH
8-16 g O 300
CI
O
8-17 S OH 326
-83-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
CI
8-18 S OH 326
CI
8-19 S O OH 342
O
CI
O
8-20 S ~ O H 310
O
CI
8-21 0 \ I OH 405
S N
H
CI
H COOH
/
8-22 S N \ ~ 433
CI O
8-23 O _1~e OH 338
[00303] The following examples provide illustrative methods for testing the
effectiveness and safety of
the compounds of Formula (I) or (II). These examples are provided for
illustrative purposes only and
not to limit the scope of the claims provided herein.
Mice and Rat Studies
[00304] The optimal dose of compounds of Formula (I) or (II) to block
formation of A2E in abca4-/-
mice is determined using a standard dose escalation study. In order to
determine the range of doses
which will be employed, a high throughput, in vitro assay which detects
modulators that inhibit RBP-
TTR interaction has been developed. Drug concentrations which cause a 50%
inhibition of RBP-TTR
interaction (the ICSO values) are calculated from the data. In these
experiments, fenretinide is employed
as a positive control as it is known to be a potent inhibitor of RBP-TTR
interaction. Representative
data from a typical dose-response experiment is shown in Figure 1. An
illustrative in vivo approach,
utilizing a compound having the structure of Formula (I) or (II) is presented
below.
[00305] The effects of compounds of Formula (I) or (II) on all-trans-retinal
in retinas from light-
adapted mice is determined at doses that bracket the human therapeutic dose.
The method includes
treating mice with a single morning intraperitoneal dose. An increased
frequency of injections in some
-84-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
embodiments, is required to maintain reduced levels of all-trans-retinal in
the retina throughout the
day.
[00306] ABCA4 Knockout Mice. ABCA4 encodes rim protein (RmP), an ATP-binding
cassette (ABC)
transporter in the outer-segment discs of rod and cone photoreceptors. The
transported substrate for
RmP is unknown. Mice generated with a knockout mutation in the abca4 gene, see
Weng et al., Cell,
98:13-23 (1999), are useful for the study of RmP function as well as for an in
vivo screening of the
effectiveness for candidate substances. These animals manifest the complex
ocular phenotype: (i) slow
photoreceptor degeneration, (ii) delayed recovery of rod sensitivity following
light exposure, (iii)
elevated atRAL and reduced atROL in photoreceptor outer-segments following a
photobleach, (iv)
constitutively elevated phosphatidylethanolamine (PE) in outer-segments, and
(v) accumulation of
lipofuscin in RPE cells. See Weng et al., Cell, 98:13-23 (1999).
[00307] Rates of photoreceptor degeneration are monitored in treated and
untreated wild-type and
abca4-1- mice by two techniques. One is the study of mice at different times
by ERG analysis and is
adopted from a clinical diagnostic procedure. See Weng et al., Cell, 98:13-23
(1999). An electrode is
placed on the corneal surface of an anesthetized mouse and the electrical
response to a light flash is
recorded from the retina. Amplitude of the a,-wave, which results from light-
induced hyperpolarization
of photoreceptors, is a sensitive indicator of photoreceptor degeneration. See
Kedzierski et al., Invest.
Ophthalmol. Vis. Sci., 38:498-509 (1997). ERGs are done on live animals. In
some embodiments, the
same mouse is analyzed repeatedly during a time-course study. The definitive
technique for
quantitating photoreceptor degeneration is histological analysis of retinal
sections. The number of
photoreceptors remaining in the retina at each time point will be determined
by counting the rows of
photoreceptor nuclei in the outer nuclear layer.
[00308] Tissue Extraction. Eye samples are thawed on ice in 1 ml of PBS, pH
7.2 and homogenized by
hand using a Duall glass-glass homogenizer. The sample is further homogenized
following the
addition of 1 ml chloroform/methanol (2:1, v/v). The sample is transferred to
a boroscilicate tube and
lipids are extracted into 4 mls of chloroform. The organic extract is washed
with 3 mls water and the
samples are then centrifuged at 3,000 x g, 10 min. The chloroform phase is
decanted and the aqueous
phase is re-extracted with another 4 mls of chloroform. Following
centrifugation, the chloroform
phases are combined and the samples were taken to dryness under nitrogen gas.
Samples residues are
resuspended in 200 12-propanol and analyzed by HPLC as described below.
[00309] HPLC Analysis. Chromatographic separations are achieved on an Agilent
Zorbax Rx-Sil
Column (5 m, 4.6 X 250 mm) using an Agilent 1100 series liquid chromatograph
equipped with
fluorescence and diode array detectors. The mobile phase is delivered at a
desired rate of a determined
volume per min. Sample peak identification is made by comparison to retention
time and absorbance
spectra of authentic standards. Data is reported as peak fluorescence (L.U.)
obtained from the
fluorescence detector.
-85-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
[00310] Administration of a compound of Formula (I) or (II) to an experimental
group of mice and
administration of DMSO alone to a control group of mice is performed and
assayed for accumulation
of A2E. The experimental group is given about 2.5 to about 20 mg/ kg of a
compound of Formula (I)
or (II) per day in about 10 to about 25 l of DMSO. Higher dosages are tested
if no effect is seen with
the highest dose of about 50 mg/kg. The control group is given 10 to 25 l
injections of DMSO alone.
Mice are given either experimental or control substances by intraperitoneal
(i.p.) injection
administered during chronic dosing regimes.
[00311] To assay for the accumulation of A2E in abca4-/- mice RPE, 2.5 to 20
mg/kg of a compound
of Formula (I) or (II) is provided by i.p. injection per day to 2-month old
abca4-/- mice. After a
predetermined period, both experimental and control mice are killed and the
levels of A2E in the RPE
are measured by HPLC.
Example 9: Effect of Test Compounds on Rod Cell Death or Rod Functional
Impairment
[00312] Administration of a compound of Formula (I) or (II) to an experimental
group of mice and
administration of DMSO alone to a control group of mice is performed and
assayed for the effects of
the test compound on rod cell death or rod functional impairment. The
experimental group is given
about 2.5 to about 20 mg/kg of a compound of Formula (I) or (II) per day in
about 10 to about 25 l of
DMSO. Higher dosages are tested if no effect is seen with the highest dose of
about 50 mg/kg. The
control group is given about 10 to about 25 l injections of DMSO alone. Mice
are administered either
experimental or control substances by i.p. injection for various experimental
time periods.
Alternatively, mice are implanted with a pump which delivers either
experimental or control
substances at a rate of about 0.25 Uhr for various experimental time periods.
[00313] Mice that are treated to about 2.5 to about 20 mg/kg of a compound of
Formula (I) or (II) per
day for approximately 8 weeks are assayed for the effects of a compound of
Formula (I) or (II) on rod
cell death or rod functional impairment by monitoring ERG recordings and
performing retinal
histology.
Example 10: Testing for Protection from Light Damage
[00314] The following study is adapted from Sieving, P.A., et al, Proc. Natl.
Acad. Sci., 98:1835-40
(2001). For chronic light-exposure studies, Sprague-Dawley male 7-wk-old
albino rats are housed in
12:12 h light/dark cycle of 51ux fluorescent white light. Injections of about
20 to about 50 mg/kg a
compound of Formula (I) or (II) by i.p. injection in 0.18 ml DMSO are given
three times daily to
chronic rats for 8 wk. Controls receive 0.18 ml DMSO by i.p. injection. Rats
are killed 2 d after final
injections. Higher dosages are tested if no effect is seen with the highest
dose of about 50 mg/kg.
[00315] For acute light-exposure studies, rats are dark-adapted overnight and
given a single i.p.
injection of the test compound about 20 to about 50 mg/kg in 0.18 ml DMSO
under dim red light and
kept in darkness for 1 h before being exposed to the bleaching light before
ERG measurements. Rats
exposed to 2,0001ux white fluorescent light for 48 h. ERGs are recorded 7 d
later, and histology is
performed immediately.
-86-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
[00316] Rats are euthanized and eyes are removed. Column cell counts of outer
nuclear layer thickness
and rod outer segment (ROS) length are measured every 200 m across both
hemispheres, and the
numbers are averaged to obtain a measure of cellular changes across the entire
retina. ERGs are
recorded from chronic rats at 4 and 8 wks of treatment. In acute rodents, rod
recovery from bleaching
light is tracked by dark-adapted ERGs by using stimuli that elicit no cone
contribution. Cone recovery
is tracked with photopic ERGs. Prior to ERGs, animals are prepared in dim red
light and
anaesthetized. Pupils are dilated and ERGs are recorded from both eyes
simultaneously by using gold-
wire corneal loops.
Example 11: Combination Therapy Involving Fenretinide
[00317] Mice and/or rats are tested in the manner described in Examples 8 and
9, but with an
additional two arms. In one of the additional arms, groups of mice and/or rats
are treated with
increasing doses of Fenretinide, from about 5 mg/kg per day to about 50 mg/kg
per day. In the second
additional arm, groups of mice and/or rats are treated with a combination of
about 20 mg/kg per day of
the test compound and increasing doses of Fenretinide, from about 5 mg/kg per
day to about 50 mg/kg
per day. The benefits of the combination therapy are assayed as described in
Examples 8 and 9.
Example 12: Efficacy of Test Compounds on the Accumulation of Lipofuscin
(and/or A2E) in
abca4 null Mutant Mice: Phase I - Dose Response and Effect on Serum Retinol.
[00318] The effect of a compound of Formula (I) or (II) on reducing serum
retinol in animals and
human subjects led us to explore the possibility that reductions in lipofuscin
and the toxic bis-retinoid
conjugate, A2E, is realized. The rationale for this approach is based upon two
independent lines of
scientific evidence: 1) reduction in ocular vitamin A concentration via
inhibition of a known visual
cycle enzyme (11-cis retinol dehydrogenase) results in profound reductions in
lipofuscin and A2E; 2)
animals maintained on a vitamin A deficient diet demonstrate dramatic
reductions in lipofuscin
accumulation. Thus, the objective for this example is to examine the effect of
a compound of Formula
(I) or (II) in an animal model which demonstrates massive accumulation of
lipofuscin and A2E in
ocular tissue, the abca4 null mutant mouse.
[00319] Initial studies begin by examining the effect of a compound of Formula
(I) or (II) on serum
retinol. Animals are divided into groups and given either DMSO, about 20 mg/kg
of a compound of
Formula (I) or (II) for at least 14 days. At the end of the study period,
blood is collected from the
animals, sera are prepared and an acetonitrile extract of the serum is
analyzed by reverse phase
LC/MS. UV-visible spectral and mass/charge analyses are performed to confirm
the identity of the
eluted peaks. Representative chromatographic data which show serum retinol
levels in mice treated
with either DMSO or a compound having the structure of Formula I are provided
in Figure 9. The
steady state serum concentrations of the test compound (structure consistent
with Formula I) are also
determined by HPLC (see Figure 3). Additionally, levels of RBP4 in serum are
quantified by
immunoblot (see Figure 4).
-87-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
[00320] Administration of an agent or agents that lower the levels of serum
retinol in a patient without
modulating at least one enzyme in the visual cycle is expected to provide a
treatment for macular
and/or retinal dystrophies and degenerations or the symptoms associated
thereof. Assays, such as those
described herein, are used in some embodiments, to select further agents
possessing this action,
including agents selected from compounds having the structure of Formula (I)
or (II).
Example 13: Efficacy of Test Compounds on the Accumulation of Lipofuscin
(and/or A2E) in
abca4 null Mutant Mice: Phase II - Chronic Treatment of abca4 Null Mutant
Mice.
[00321] Studies are performed to evaluate the effects of a compound of Formula
(I) or (II) on
reduction of A2E and A2E precursors in abca4 null mutant mice. A compound of
Formula (I) or (II) is
administered in DMSO (about 20 mg/kg, ip) to abca4 null mutant mice (BL6/129,
aged 2 months)
daily for a period of at least 28 days. Control age/strain matched mice
receive only the DMSO vehicle.
Mice are sampled at 0, 14, and 28 days (n = 3 per group), the eyes are
enucleated and chloroform-
soluble constituents (lipids, retinoids and lipid-retinoid conjugates) are
extracted. Mice are sacrificed
by cervical dislocation, the eyes are enucleated and individually snap frozen
in cryo vials. The sample
extracts are then analyzed by HPLC with on-line fluorescence detection. A
representative
chromatographic tracing obtained from an eyecup extract of an abca4 null
mutant mouse in shown in
Figure 5. A similar study is undertaken to ascertain effects of treatment with
a compound of Formula
(I) or (II) on the electroretinographic and morphological phenotypes.
Example 14: Combination Therapy Involving Test Compound and a Statin
[00322] Mice and/or rats are tested in the manner described in Examples 8 and
9, but with an
additional two arms. In one of the additional arms, groups of mice and/or rats
are treated with a
suitable statin such as: Lipitor (Atorvastatin), Mevacor (Lovastatin),
Pravachol (Pravastatin
sodium), ZocorTM (Simvastatin), Leschol (fluvastatin sodium) and the like with
optimal dosage based
on weight. In the second additional arm, groups of mice and/or rats are
treated with a combination of
about 20 mg/kg per day of a compound of Formula (I) or (II) and increasing
doses of the statin used in
the previous step. Suggested human dosages of such statins are for example:
Lipitor (Atorvastatin)
about 10 to about 80 mg/day, Mevacor (Lovastatin) about 10 to about 80 mg/day,
Pravachol
(Pravastatin sodium) about 10 to about 40 mg/day, ZocorTM (Simvastatin) about
5 to about 80 mg/day,
Leschol (fluvastatin sodium) about 20 to about 80 mg/day. Dosage of statins
for mice and/or rat
subjects should be calculated based on weight. The benefits of the combination
therapy are assayed as
described in Examples 8 and 9.
Example 15: Combination Therapy Involving Test Compound, Vitamins and Minerals
[00323] Mice and/or rats are tested in the manner described in Example 13, but
with selected vitamins
and minerals. Administration of a compound of Formula (I) or (II) in
combination with vitamins and
minerals is either orally or parenterally administered at amounts effective to
inhibit the development or
reoccurrence of macular degeneration. Test dosages are initially in the range
of about 20 mg/kg per
day of a compound of Formula (I) or (II) with about 100 to about 1000 mg
vitamin C, about 100 to
-88-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
about 600 mg vitamin E, about 10,000 to about 40,000 IU vitamin A, about 50 to
about 200 mg zinc
and about 1 to about 5 mg copper for 15 to 20 weeks. The benefits of the
combination therapy are
assayed as described in Examples 8 and 9.
Example 16: Analysis of Serum Retinol as a Function of the Concentration of
Test Compound
[00324] ABCA4 null mutant mice are given the indicated dose of a compound of
Formula (I) or (II) in
DMSO (i.p.) daily for 28 days (n = 4 mice per dosage group). At the end of the
study period, blood
samples are taken and serum is prepared. Following acetonitrile precipitation
of serum proteins, the
concentrations of retinol and of a compound of Formula (I) or (II) are
determined from the soluble
phase by LC/MS. Identification of the eluted compounds is confirmed by UV-vis
absorption
spectroscopy and co-elution of sample peaks with authentic standards (see
Figure 2).
Example 17: Identification of Compounds that bind to TTR and/or Inhibit Gene
Expression of
TTR
[00325] Purified TTR polypeptides comprising a glutathione-S-transferase
protein and adsorbed onto
glutathione-derivatized wells of 96-well microtiter plates are contacted with
test compounds from a
small molecule library at pH 7.0 in a physiological buffer solution. Purified
TTR polypeptides have
been described in the art. See U.S. Patent App. No. 20020160394, herein
incorporated by reference.
The test compounds in some embodiments, comprise a fluorescent tag. The
samples are incubated for
5 minutes to one hour. Control samples are incubated in the absence of a test
compound.
[00326] The buffer solution containing the test compounds is washed from the
wells. Binding of a test
compound to a TTR polypeptide is detected by fluorescence measurements of the
contents of the
wells. A test compound that increases the fluorescence in a well by at least
15% relative to
fluorescence of a well in which a test compound is not incubated is identified
as a compound which
binds to a TTR polypeptide.
[00327] The identified test compound in some embodiments, are administered to
a culture of human
cells transfected with a TTR expression construct and incubated at 37 C for
10 to 45 minutes. A
culture of the same type of cells that have not been transfected is incubated
for the same time without
the test compound to provide a negative control.
[00328] RNA is then isolated from the two cultures as described in Chirgwin et
al., Biochem. 18,
5294-99, 1979). Northern blots are prepared using 20 to 30 g total RNA and
hybridized with a 32P-
labeled TTR-specific probe. Probes for detecting TTR mRNA transcripts have
been described
previously. A test compound that decreases the TTR-specific signal relative to
the signal obtained in
the absence of the test compound is identified as an inhibitor of TTR gene
expression.
Example 18: Identification of Compounds that bind to RBP and/or Inhibit Gene
Expression of
RBP
[00329] Purified apo RBP are contacted with test compounds from a small
molecule library at pH 7.0
in a physiological buffer solution. Purified apo RBP have been described in
the art. See U.S. Patent
App. No. 20030119715, herein incorporated by reference. The test compounds in
some embodiments,
-89-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
comprise a fluorescent tag. The samples are incubated for 5 minutes to one
hour. Control samples are
incubated in the absence of a test compound. In some embodiments, competition
assays in the
presence of holo RBP (RBP complexed with retinol) are also performed.
[00330] The buffer solution containing the test compounds is washed from the
wells. Binding of a test
compound to apo RBP is detected by fluorescence measurements of the contents
of the wells. A test
compound that increases the fluorescence in a well by at least 15% relative to
fluorescence of a well in
which a test compound is not incubated is identified as a compound which binds
to apo RBP.
[00331] The identified test compound are administered to a culture of human
cells transfected with an
RBP expression construct and incubated at 37 C for 10 to 45 minutes. A
culture of the same type of
cells that have not been transfected is incubated for the same time without
the test compound to
provide a negative control.
[00332] RNA is then isolated from the two cultures as described in Chirgwin et
al., Biochem. 18,
5294-99, 1979). Northern blots are prepared using about 20 to about 30 g
total RNA and hybridized
with a 32P-labeled RBP-specific probe. A test compound that decreases the RBP-
specific signal
relative to the signal obtained in the absence of the test compound is
identified as an inhibitor of RBP
gene expression.
Example 19: Further Analysis of the Effect of Test Compound on Serum Retinol,
Eyecup
Retinoids, and A2E Levels
[00333] Compound of Formula (I) or (II) Treatments. A compound of Formula (I)
or (II) is
administered daily (about 1.5 to about 15 g/ l in 25 l DMSO, i.p.) to ABCA4-
/- mice for 28 days.
Mice 1- 2 months of age at study onset and are either pigmented (129/SV) or
albino (BALB/c)
strains. Mice are raised under 12-hr cyclic light/dark (30 - 501ux) during the
treatment period and are
anesthetized by i.p. injection of ketamine (about 200 mg/kg) plus xylazine
(about 10 mg/kg) before
death by cervical dislocation.
[00334] Analysis of Serum Retinol. Whole blood is collected from tail veins of
test compound-treated
mice 18 hrs. following the final test compound dose (i.e., at day 28). Serum
is obtained from whole
blood following centrifugation at 1,500 x g, 10 min. Serum proteins are
precipitated with the addition
of an equivolume of ice-cold acetonitrile and centrifugation (10,000 x g, 10
min). An aliquot is
removed from the soluble phase and analyzed by HPLC using an Agilent 1100
series capillary liquid
chromatograph equipped with a diode-array detector. Chromatography is
performed as described
above.
[00335] Extraction and Analysis of Retinoids and A2E. Steady-state levels of
retinoids and A2E in
eyecups of ABCA4-/- mice are determined following daily administration (28
days) of a compound of
Formula (I) or (II). Mice are sacrificed, the eyes enucleated, and the
posterior portion of each eye is
used for extraction of retinoids or A2E. Methodologies used for extraction of
retinoids and A2E from
eye tissue and HPLC analysis techniques have been described. See, e.g., Mata
NL, Weng J, Travis
GH. Biosynthesis of a major lipofuscin fluorophore in mice and humans with
ABCR-mediated retinal
-90-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
and macular degeneration. Proc Natl Acad Sci USA. 2000;97:7154-7159; Weng J,
et al.; Cell.
1999;98:13-23; Mata NL, et al.; Invest. Ophthalmol. Visual Sci. 2001;42:1685-
1690. All samples are
analyzed by HPLC using absorbance and fluorescence detection. In these
analyses, a column
thermostat is employed to maintain the solvent and column temperature at 40 C.
Identity of the
indicated compounds is confirmed by on-line spectral analysis and by co-
elution with authentic
standards.
[00336] Correlation between Serum Retinol, Ocular Retinoids, and A2E. Data
collected
demonstrates a direct correlation between reduction in serum retinol and a
reduction in the level of
retinoids and the level of A2E in the eyecups of mammals. Notably serum
retinol reduction tracks, in a
dose-dependent manner, both ocular retinoid levels and ocular A2E levels. For
example, a compound
of Formula (I) or (II) lower serum retinol levels in mammals, showing that a
reduction of serum retinol
effects the level of materials (e.g., A2E) associated with retinopathy and
macular
degenerations/dystrophies. Accordingly, agents, such as those described
herein, that cause serum
retinol reductions also are used, in some embodiments, to reduce A2E and
retinoid levels in the eye,
and further, be used to treat lipofuscin-based retinal diseases, e.g.,
retinopathies and macular
degenerations/dystrophies, in the mammal.
Example 20: Validation of RBP as a Therapeutic Target for Arresting
Accumulation of A2E
[00337] Exploration of a non-pharmacological means of reducing lipofuscin
fluorophores in order to
validate our therapeutic approach based upon reduction of RBP levels in a
patient will be performed.
In this study, RBP protein levels will be reduced through genetic
manipulation. Two new lines of mice
expressing heterozygous mutations in retinol binding protein (RBP4) are
generated. The first line
carries a heterozygous mutation only at the RBP locus (RBP+/-); the second
line carries heterozygous
mutations at both ABCA4 and RBP4 loci (RBP4+/-, ABCA4+/-). The RBP+/- mice
will be wild type at
the ABCA4 locus and, therefore, will not accumulate excessive amounts of A2E
fluorophores.
However, ABCA4+/- mice will accumulate A2E fluorophores at levels which are
approximately 50%
of that observed in ABCA4-/- (null homozygous) mice. At issue is whether the
reduced expression of
RBP in the RBP4+/-, ABCA4+/- mice will have an effect on the accumulation of
toxic retinal
fluorophores (e.g., A2E). Figure 6 (panel A) illustrates a dramatic reduction
(> 50%) in serum retinol
in RBP4+/-, ABCA4+/- mice compared to RBP4 +/+, ABCA4-/- mice. The degree of
retinol reduction
in the RBP4+/-, ABCA4+/- mice is comparable to that observed in RBP4+/+, ABCA4-
/- mice which
have been administered HPR for a 28-day period. Immunoblot analysis of RBP4
levels in the sera of
these mice is consistent with the retinol data (Figure 6, panel B).
[00338] The levels of A2E and precursor fluorophores (A2PE and A2PE-H2) in
mice with genetically
reduced expression of RBP4 (RBP4+/-, ABCA4+/-) are monitored monthly over a
three month period
and compared to the fluorophore levels in RBP4+/+, ABCA4+/- mice. Overall, the
RBP4+/-, ABCA4+/-
mice demonstrate a- 70% reduction in total fluorophore level relative to the
levels present in
ABCA4+/- mice (panels D and C, respectively). In fact, the measured
fluorophore levels in the
-91-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
RBP4+/-, ABCA4+/- mice approach that observed in wild-type mice (compare
panels D and E). These
data validate RBP as a therapeutic target for reducing fluorophore levels in
the eye. Further, these data
will demonstrate that agents or methods that inhibit the transcription or
translation of RBP in a patient
also (a) reduce serum retinol levels in that patient, and (b) provide a
therapeutic benefit in the retinol-
related diseases described herein. Further, agents or methods that enhance the
clearance of RBP in a
patient will also produce such effects and benefits.
Example 21: Quantitation of ocular retinoids
[00339] ABCA4-/- mice were administered either ATRP (Control) or ATRP + test
compound daily
for 20 days (n = 3 mice/group). On the final day of dosing, a trace amount of
[3H]ATROL (0.32 pmol,
8 Ci) in 100 1 corn oil was administered to all animals. Eyes were enucleated
5 hours later. One eye
from each animal was used for retinoid analysis and the other eye was used for
analysis of A2E and
related fluorophores as described in the methods. The data show a marked
reduction in uptake of
[3H]ATROL in the animals treated with test compound [Figure 9]. Analysis of
each retinoid species
showed that the precursor substrate for visual chromophore biosynthesis (ATRE)
and the immediate
precursor for A2E biosynthesis (AT-Ox) are significantly reduced.
Example 22: Effect of test compound in reducing total fluorophore levels
[00340] Effect of test compound on reducing total fluorophore levels in ABCA4-
/- mice. Mice
were treated as described in Example 13 above. At the end of the study, one
eye from each animal was
used to measure total fluorophore levels. Briefly, one whole eye was
homogenized in 1 ml phosphate
buffer saline (50 mM NazHPO4, 150 mM NaC1, pH 7.8). Following homogenization,
1 ml methanol
was added and the samples were mixed thoroughly. The mixtures were incubated
at room temperature
for 5 min and extracted twice with 2 ml hexane. The extracts were concentrated
to - 400 l for
fluorescence measurements. Corrected fluorescence spectra were obtained using
a Spex Fluorolog-3
spectrofluorimeter (Jobin Yvon Horiba, Edison, NJ) operated in ratio mode. The
samples were excited
at 488 nm and emissions at 500-700 m were monitored. The data show a profound
reduction on total
fluorophore levels in mice treated with the test compound [Figure 10].
Example 23: Effect of test compound on reducing A2E and A2E precursor
[00341] Mice were treated as described in Example 13 above. At the end of the
study, one eye
from each animal was used to measure A2E and A2PE-H2. Briefly, one whole eye
was homogenized
in 1 ml phosphate buffer saline (50 mM Na2HPO4, 150 mM NaC1, pH 7.8).
Following
homogenization, 1 ml methanol was added and the samples were mixed thoroughly.
The mixtures
were incubated at room temperature for 5 min and extracted twice with 2 ml
hexane. The solvent was
evaporated under a stream of nitrogen and the sample residues were
reconstituted in 200 l
isopropanol (IPA) for analysis by HPLC. Fluorophores were separated on a
Zorbax RX-Si15- m
column (250 x 4.6-mm) equilibrated with a phospholipid moble phase
(hexane:IPA:ethano1:25mM
phosphate buffer: acetic acid 485:376:100:37.5:0.275 v:v) at a flow rate of 1
mUmin. The data show a
-92-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
dramatic reduction in both A2E and its precursor in mice treated with test
compound compared to
vehicle-treated mice [Figure 11 ].
Human Studies
[00342] Detection of Macular or Retinal Degeneration. Identification of
abnormal blood vessels in the
eye is done, for example with an angiogram. This identification helps
determine which patients are
candidates for the use of a candidate substance or other treatment method to
hinder or prevent further
vision loss. Angiograms are useful for follow-up of treatment as well as for
future evaluation of any
new vessel growth.
[00343] A fluorescein angiogram (fluorescein angiography, fluorescein
angioscopy) is a technique for
the visualization of choroidal and retinal circulation at the back of the eye.
Fluorescein dye is injected
intravenously followed by multiframe photography (angiography),
ophthalmoscopic evaluation
(angioscopy), or by a Heidelberg retina angiograph (a confocal scanning laser
system). Additionally,
the retina is examined by OCT, a non-invasive way to obtain high-resolution
cross-sectional images of
the retina. Fluorescein angiograms are used in the evaluation of a wide range
of retinal and choroidal
diseases through the analysis of leakage or possible damage to the blood
vessels that feed the retina. It
has also been used to evaluate abnormalities of the optic nerve and iris by
Berkow et al., Am. J.
Ophthalmol. 97:143-7 (1984).
[00344] Similarly, angiograms using indocyanine green are used for the
visualization circulation at the
back of the eye. Wherein fluorescein is more efficient for studying retinal
circulation, indocyanine is
better for observing the deeper choroidal blood vessel layer. The use of
indocyanine angiography is
helpful when neovascularization is not observed with fluorescein dye alone.
[00345] Appropriate human doses for compounds having the structure of Formula
(I) will be
determined using a standard dose escalation study.
Example 24: Clinical Trials for Testing the Efficacy of Test Compounds to
Treat Macular
Degeneration
[00346] For pre-testing, all human patients will undergo a routine
ophthalmologic examination
including fluorescein angiography, measurement of visual acuity,
electrophysiologic parameters and
biochemical and rheologic parameters. Inclusion criteria are as follows:
visual acuity between 20/160
and 20/32 in at least one eye and signs of AMD such as drusen, areolar
atrophy, pigment clumping,
pigment epithelium detachment, or subretinal neovascularization. Patients that
are pregnant or actively
breast-feeding children are excluded from the study. Other exclusion criteria
include previous
vitrectomy or other AMD surgical intervention, severe scarring or severe
concurrent ocular disease
(uncontrolled glaucoma).
[00347] A group of patients diagnosed with macular degeneration, or who have
progressive formations
of A2E, lipofuscin, or drusen in their eyes are divided into a control group
and an experimental group.
A compound of Formula (I) or (II) is administered to the experimental group on
a daily basis. A
-93-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
placebo is administered to the control group in the same regime as the test
compound is administered
to the experimental group.
[00348] A compound of Formula (I) or (II) or placebo is administered to a
patient either orally or
parenterally at amounts effective to inhibit the development or reoccurrence
of macular degeneration.
Effective dosage amounts are in the range of from about 1 to about 4000 mg/m2
up to three times a
day.
[00349] One method for measuring progression of macular degeneration in both
control and
experimental groups is the best corrected visual acuity as measured by Early
Treatment Diabetic
Retinopathy Study (ETDRS) charts (Lighthouse, Long Island, NY) using line
assessment and the
forced choice method (Ferris et al. Am J Ophthalmol, 94:97-98 (1982)). Visual
acuity is recorded in
1ogMAR. The change of one line on the ETDRS chart is equivalent to 0.1 1ogMAR.
Further typical
methods for measuring progression of macular degeneration in both control and
experimental groups
include use of visual field examinations, including but not limited to a
Humphrey visual field
examination, microperimetry (using, e.g., Micro Perimeter MP-1 from NIDEK) and
measuring/monitoring of autofluorescence of certain compounds in the eye of
the patient.
[00350] Additional methods for measuring progression of macular degeneration
in both control and
experimental groups include taking fundus photographs, observing changes in
autofluorescence over
time using a Heidelberg retina angiograph (or alternatively, techniques
described in M. Hammer, et al.
Ophthalmologe 2004 Apr. 7 [Epub ahead of patent]), and taking fluorescein
angiograms at baseline,
three, six, nine and twelve months at follow-up visits. Documentation of
morphologic changes include
changes in (a) drusen size, character, and distribution; (b) development and
progression of choroidal
neovascularization; (c) other interval fundus changes or abnormalities; (d)
reading speed and/or
reading acuity; (e) scotoma size; or (f) the size and number of the geographic
atrophy lesions. In
addition, Amsler Grid Test and color testing are optionally administered.
[00351] To assess statistically visual improvement during drug administration,
examiners use the
ETDRS (LogMAR) chart and a standardized refraction and visual acuity protocol.
Evaluation of the
mean ETDRS (LogMAR) best corrected visual acuity (BCVA) from baseline through
the available
post-treatment interval visits aids in determining statistical visual
improvement.
[00352] To assess the ANOVA (analysis of variance between groups) between the
control and
experimental group, the mean changes in ETDRS (LogMAR) visual acuity from
baseline through the
available post-treatment interval visits are compared using two-group ANOVA
with repeated
measures analysis with unstructured covariance using SAS/STAT Software (SAS
Institutes Inc, Cary,
North Carolina).
[00353] The serum retinol levels are assessed as follows: after acetonitrile
precipitation of serum
proteins, the concentrations of retinol are determined from the soluble phase
by LC/MS. Alternatively,
the serum retinol levels are assessed as described in Driskell et al., J
Chromatogr, 1982, 231, 439-444
or Futterman et al., Invest. Ophthalmol Vis Sci, 1975, 14, 125.
-94-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
[00354] Toxicity evaluation after the commencement of the study includes check
ups every three
months during the subsequent year, every four months the year after and
subsequently every six
months. Plasma levels of the test compound and its metabolite, serum retinol
and/or RBP are also
assessed during these visits. The toxicity evaluation includes patients using
a compound of Formula (I)
or (II) as well as the patients in the control group.
Example 25: Activity of non-retinoid modulators on RBP-TTR and CYP450
Table 1:
Compound of Formula (I) Apparent CYP
IC50 Inhibition
5-(2-tert-butyl-4-chlorophenoxy)-N-(4- 1 M -50%
hydroxyphenyl)pentanamide,
7-(2-tert-butyl-4-chlorophenoxy)-N-(4- 1 M -40%
hydroxyphenyl)heptanamide
2-(4-(5-(2-tert-butyl-4-chlorophenoxy)pentanamido)phenyl)acetic 1 M <10%
acid
3-((2-tert-butyl-4-chlorophenoxy)methyl)-N-(4- 5 M <10%
hydroxyphenyl)cyclopentanecarboxamide
4-(5-(2-tert-butyl-4-chlorophenoxy)pentanamido)benzoic acid 5 M <10%
4-(3-((2-tert-butyl-4- 5 M <10%
chloro henox meth 1 c clo entanamido benzoic acid
5-(2-tert-butyl-4-chlorophenoxy)pentanoic acid 0.1 M <10%
4-(2-tert-butyl-4-chlorophenoxy)butanoic acid 1 M -15%
2-(3-((2-tert-butyl-4-chlorophenoxy)methyl)cyclopentyl)acetic NT NT
acid
7-(2-tert-butyl-4-chlorophenoxy)heptanoic acid 0.5 M -15%
4-(5-(2-tert-butyl-4-chlorophenoxy)pentanamido)benzamide NT NT
3-((2-tert-butyl-4-chlorophenoxy)methyl)cyclohexanecarboxylic 0.16 M <10%
acid
3-((2-tert-butyl-4-chlorophenoxy)methyl)cyclopentanecarboxylic 0.4 M <10%
acid
3-((2-tert-butyl-4-chlorophenylamino)methyl)cyclopentanamide NT NT
5-(2-tert-butyl-4-chlorophenylthio)pentanoic acid NT NT
NT: Not Tested
[00355] Table 1 shows IC50 values and cytochrome P450 inhibition profiles of
representative
compounds having the structure of Formula (I) and (II).
-95-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
Example 26: Testing for the Efficacy of Compounds of Formula (1) or (II) to
Reduce A2E
Production
[00356] The same protocol design, including pre-testing, administration,
dosing and toxicity
evaluation protocols, that are described in Example 24 are also used to test
for the efficacy of
compounds of Formula (I) or (II) in reducing or otherwise limiting the
production of A2E in the eye of
a patient.
[00357] Methods for measuring or monitoring formation of A2E include the
measuring/monitoring of
autofluorescence of certain compounds in the eye of the patient, the use of
visual acuity and visual
field examinations (including, by way of example, microperimetry), reading
speed and/or reading
acuity examinations, measurements on the size and number of scotoma and/or
geographic atrophic
lesions, as described in Example 23. The statistical analyses described in
Example 23 is employed.
Example 27: Testing for the Efficacy of Compounds of Formula (I) or (11) to
Reduce Lipofuscin
Production
[00358] The same protocol design, including pre-testing, administration,
dosing and toxicity
evaluation protocols, that are described in Example 24 are also used to test
for the efficacy of
compounds of Formula (I) or (II) in reducing or otherwise limiting the
production of lipofuscin in the
eye of a patient. The statistical analyses described in Example 24 are
employed.
[00359] Tests that are used as surrogate markers for the efficacy of a
particular treatment include the
use of visual acuity and visual field examinations (including, by way of
example, microperimetry),
reading speed and/or reading acuity examinations, measurements on the size and
number of scotoma
and/or geographic atrophic lesions, and the measuring/monitoring of
autofluorescence of certain
compounds in the eye of the patient, as described in Example 24.
Example 28: Testing for the Efficacy of Compounds of Formula (I) or (11) to
Reduce Drusen
Production
[00360] The same protocol design, including pre-testing, administration,
dosing and toxicity
evaluation protocols, that are described in Example 24 are also used to test
for the efficacy of
compounds of Formula (I) or (II) in reducing or otherwise limiting the
production or formation of
drusen in the eye of a patient. The statistical analyses described in Example
24 are employed.
[00361] Methods for measuring progressive formations of drusen in both control
and experimental
groups include taking fundus photographs and fluorescein angiograms at
baseline, three, six, nine and
twelve months at follow-up visits. Documentation of morphologic changes
include changes in (a)
drusen size, character, and distribution (b) development and progression of
choroidal
neovascularization and (c) other interval fundus changes or abnormalities.
Other tests that are used as
surrogate markers for the efficacy of a particular treatment include the use
of visual acuity and visual
field examinations (including, by way of example, microperimetry), reading
speed and/or reading
acuity examinations, measurements on the size and number of scotoma and/or
geographic atrophic
-96-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
lesions, and the measuring/monitoring of autofluorescence of certain compounds
in the eye of the
patient, as described in Example 23.
Example 29: Genetic Testing for Macular Dystrophies
[00362] Defects in the human ABCA4 gene are thought to be associated with five
distinct retinal
phenotypes including Stargardt Disease, cone-rod dystrophy, age-related
macular degeneration (both
dry form and wet form) and retinitis pigmentosa. See e.g., Allikmets et al.,
Science, 277:1805-07
(1997); Lewis et al., Am. J. Hum. Genet., 64:422-34 (1999); Stone et al.,
Nature Genetics, 20:328-29
(1998); Allikmets, Am. J. Hum. Gen., 67:793-799 (2000); Klevering, et al,
Ophthalmology, 111:546-
553 (2004). In addition, an autosomal dominant form of Stargardt Disease is
caused by mutations in
the ELOV4 gene. See Karan, et al., Proc. Natl. Acad. Sci. (2005). Patients are
diagnosed as having
Stargardt Disease by any of the following examples of assays:
- A direct-sequencing mutation detection strategy which involves sequencing
all exons
and flanking intron regions of ABCA4 or ELOV4 for sequence mutation(s);
- Genomic Southern analysis;
- Microarray assays that include all known ABCA4 or ELOV4 variants; and
- Analysis by liquid chromatography tandem mass spectrometry coupled with
immunocytochemical analysis using antibodies and Western analysis.
Fundus photographs, fluorescein angiograms, and scanning laser ophthalmoscope
imaging
along with the history of the patient and his or her family are methods to
anticipate and/or confirm
diagnosis.
Example 30: Formulations
Example 30a: Oral Composition
[00363] To prepare a pharmaceutical composition for oral delivery, 100 mg of a
compound of any of
Formula (I), or Formula (II) is mixed with 750 mg of starch. The mixture is
incorporated into an oral
dosage unit for, such as a hard gelatin capsule, which is suitable for oral
administration.
Example 30b: Parenteral Composition
[00364] To prepare a parenteral pharmaceutical composition suitable for
administration by injection,
100 mg of a water-soluble salt of a compound of any of Formula (I), or Formula
(II) is dissolved in
DMSO and then mixed with 10 mL of 0.9% sterile saline. The mixture is
incorporated into a dosage
unit form suitable for administration by injection.
Example 30c: Sublingual (Hard Lozenge) Composition
[00365] To prepare a pharmaceutical composition for buccal delivery, such as a
hard lozenge, mix 100
mg of a compound of any of Formula (I), or Formula (II) is mixed with 420 mg
of powdered sugar,
1.6 mL of light corn syrup, 2.4 mL distilled water, and 0.42 mL mint extract.
The mixture is gently
blended and poured into a mold to form a lozenge suitable for buccal
administration.
Example 30d: Inhalation Composition
-97-
CA 02699773 2010-03-16
WO 2009/042444 PCT/US2008/076499
[00366] To prepare a pharmaceutical composition for inhalation delivery, 20 mg
of a compound of any
of Formula (I), or Formula (II) is mixed with 50 mg of anhydrous citric acid
and 100 mL of 0.9%
sodium chloride solution. The mixture is incorporated into an inhalation
delivery unit, such as a
nebulizer, which is suitable for inhalation administration.
Example 30e: Rectal Gel Composition
[00367] To prepare a pharmaceutical composition for rectal delivery, 100 mg of
a compound of any of
Formula (I), or Formula (II) is mixed with 2.5 g of methylcelluose (1500 mPa),
100 mg of
methylparapen, 5 g of glycerin and 100 mL of purified water. The resulting gel
mixture is then
incorporated into rectal delivery units, such as syringes, which are suitable
for rectal administration.
Example 30f: Topical Gel Composition
[00368] To prepare a pharmaceutical topical gel composition, 100 mg of a
compound of any of
Formula (I), or Formula (II) is mixed with 1.75 g of hydroxypropyl celluose,
10 mL of propylene
glycol, 10 mL of isopropyl myristate and 100 mL of purified alcohol USP. The
resulting gel mixture is
then incorporated into containers, such as tubes, which are suitable for
topicl administration.
Example 30g: Ophthalmic Solution Composition
[00369] To prepare a pharmaceutical opthalmic solution composition, 100 mg of
a compound of any of
Formula (I), or Formula (II) is mixed with 0.9 g of NaC1 in 100 mL of purified
water and filtered using
a 0.2 micron filter. The resulting isotonic solution is then incorporated into
ophthalmic delivery units,
such as eye drop containers, which are suitable for ophthalmic administration.
[00370] The examples and embodiments described herein are for illustrative
purposes only and various
modifications or changes suggested are to be included within the spirit and
purview of this application
and scope of the appended claims.
-98-