Note: Descriptions are shown in the official language in which they were submitted.
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
HYDROPHILIC POROUS SUBSTRATES
Technical Field
The present disclosure relates to hydrophilic, porous substrates, and methods
for
preparing the same.
Background
There is a need in the art for porous polymeric substrates having enhanced
hydrophilicity. Further, there is a need in the art for methods of making
polymeric
substrates having enhanced hydrophilicity from hydrophobic polymers.
Summary of the Invention
The present invention is directed to hydrophilic substrates and methods of
making
hydrophilic substrates. More specifically, the hydrophilic substrates include
a hydrophobic
porous base substrate that has been modified to provide the requisite
hydrophilicity.
Methods of making a hydrophilic substrate are provided. In some embodiments,
the method comprises:
1) providing a porous base substrate having interstitial and
outer surfaces;
2) imbibing the porous base substrate with a first solution to form an
imbibed
porous base substrate, the first solution comprising (a) at least one grafting
monomer
having an acrylate group and a photoinitiator group and (b) one or more
monomers having
at least one acrylate group and at least one additional ethylenically
unsaturated, free-
radically polymerizable group; and optionally (c) one or more additional
monomers
having at least one free-radically polymerizable group and a hydrophilic
group; wherein at
least one of (b) or (c) monomers are hydrophilic.
3) exposing the imbibed porous base substrate to a controlled
amount of
electron beam radiation so as to form a first functionalized substrate
comprising grafted
photoinitiator group attached to the surfaces of the porous base substrate,
and
4) exposing the porous base substrate comprising grafted photoinitiator
groups
to a controlled amount of UV radiation to polymerize or crosslink the
remaining
ethylenically unsaturated, free-radically polymerizable groups.
- 1 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
An article is provided that comprises (a) a first grafted species comprising
the
reaction product of a monomer having an acrylate group and a photoinitiator
group; and
(b) a second species comprising the reaction product of a monomers having at
least one
acrylate group and at least one additional ethylenically unsaturated, free-
radically
polymerizable group and optionally (c) a third species comprising the reaction
product of
monomers having at least one ethylenically unsaturated, free-radically
polymerizable
group and a hydrophilic group, with the surfaces of the porous base substrate
upon
exposure to an electron beam and UV irradiation. At least one of (b) or (c)
monomers is
hydrophilic. Any free ethylenically unsaturated groups that remain ungrafted
to the porous
base substrate may crosslink upon subsequent exposure to UV radiation.
With respect to the method and article, all or a portion of the acrylate
groups of the
photoinitiator monomer (a) will be grafted to the surface of the porous base
substrate upon
e-beam irradiation. The unreacted photoinitiator monomers may be subsequently
incorporated into the growing polymer chain on exposure to UV radiation. The
remaining
(b) and (c) monomers may be directly grafted to the surfaces (for example by
grafting of
an acrylate group), or indirectly grafted by incorporation into the growing
polymer chain
on exposure to UV radiation.
These and other features and advantages of the present invention will become
apparent after a review of the following detailed description of the disclosed
embodiments
and the appended claims.
Brief Description of the Drawings
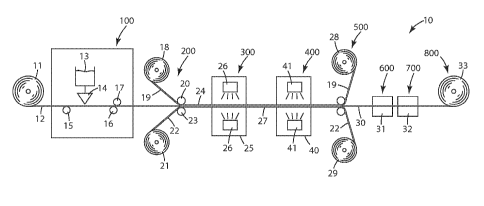
FIG. 1 depicts exemplary method steps for making hydrophilic substrates of the
present invention.
Detailed Description of the Invention
In the article and methods of this invention, hydrophilic porous articles are
provided by a two-step process of e-beam grafting of monomers and subsequent
UV
crosslinking of free, ungrafted ethylenically unsaturated polymerizable
groups.
Compared to the porous base substrate before surface modification, the
functionalized substrate typically has hydrophilicity. The hydrophilic porous
substrate
comprises a number of components including, but not limited to, (1) a porous
base
- 2 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
substrate having interstitial and outer surfaces and (2) the UV initiated
reaction product of
(a) a grafted photoinitiator group extending from the surfaces of the porous
base substrate,
(b) one or more monomers having at least one acrylate group and at least one
additional
free-radically polymerizable group; and optionally (c) one or more additional
monomers
having at least one free-radically polymerizable group and a hydrophilic
group; wherein at
least one of (b) or (c) monomers are hydrophilic.
Suitable porous base substrates include, but are not limited to, porous
membranes,
porous nonwoven webs, and porous fibers. The porous base substrate may be
formed from
any suitable thermoplastic polymeric material. Suitable polymeric materials
include, but
are not limited to, polyolefins, poly(isoprenes), poly(butadienes),
fluorinated polymers,
chlorinated polymers, polyamides, polyimides, polyethers, poly(ether
sulfones),
poly(sulfones), poly(vinyl acetates), copolymers of vinyl acetate,
poly(phosphazenes),
poly(vinyl esters), poly(vinyl ethers), poly(vinyl alcohols), and
poly(carbonates).
Suitable polyolefins include, but are not limited to, poly(ethylene),
poly(propylene), poly(1-butene), copolymers of ethylene and propylene, alpha
olefin
copolymers (such as copolymers of ethylene or propylene with 1-butene, 1-
hexene, 1-
octene, and 1-decene), poly(ethylene-co-l-butene) and poly(ethylene-co-l-
butene-co-l-
hexene).
Suitable fluorinated polymers include, but are not limited to, poly(vinyl
fluoride),
poly(vinylidene fluoride), copolymers of vinylidene fluoride (such as
poly(vinylidene
fluoride-co-hexafluoropropylene), and copolymers of chlorotrifluoroethylene
(such as
poly(ethylene-co-chlorotrifluoroethylene).
Suitable polyamides include, but are not limited to, poly(imino(1-
oxohexamethylene)), poly(iminoadipoyliminohexamethylene),
poly(iminoadipoyliminodecamethylene), and polycaprolactam. Suitable polyimides
include, but are not limited to, poly(pyromellitimide).
Suitable poly(ether sulfones) include, but are not limited to,
poly(diphenylether
sulfone) and poly(diphenylsulfone-co-diphenylene oxide sulfone).
Suitable copolymers of vinyl acetate include, but are not limited to,
poly(ethylene-
co-vinyl acetate) and such copolymers in which at least some of the acetate
groups have
been hydrolyzed to afford various poly(vinyl alcohols).
- 3 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
Preferably, the porous base substrate is formed from a propylene homo- or
copolymers, most preferably propylene homopolymers. Polypropylene polymers are
often
a material of choice for porous articles, such as nonwovens and microporous
films, due to
properties such as non-toxicity, inertness, low cost, and the ease with which
it can be
extruded, molded, and formed into articles. However, polypropylene is
hydrophobic.
While it is desirable to render hydrophobic polymers such as polypropylene
hydrophilic,
polypropylene treated with ionizing radiation is subject to degradation, e.g.,
embrittlement,
discoloration, and thermal sensitivity, during or subsequent to irradiation,
which therefore
limits the ability to render such thermoplastic polymers hydrophilic by e-beam
grafting.
For radiation sensitive substrates, such as polypropylene, the present
invention
overcomes such polymer degradation by using a low dose of electron beam
radiation to
graft photoinitiator groups and optionally grafting other hydrophilic monomers
on a
portion of the surface, then polymerizing or crosslinking any ungrafted,
unreacted
ethylenically unsaturated groups by UV radiation.
In one exemplary embodiment, the porous base substrate comprises a microporous
base substrate having an average pore size that is typically less than about
1.0 microns.
Suitable microporous base substrates include, but are not limited to,
microporous
membranes, microporous nonwoven webs, and microporous fibers. The microporous
base
substrate is often initially hydrophobic and is rendered hydrophilic by the
methods
described herein.
In some embodiments, the porous base substrate is a microporous membrane such
as a thermally-induced phase separation (TIPS) membrane. TIPS membranes are
often
prepared by forming a homogenous solution of a thermoplastic material and a
second
material above the melting point of the thermoplastic material. Upon cooling,
the
thermoplastic material crystallizes and phase separates from the second
material. The
crystallized thermoplastic material is often stretched. The second material is
optionally
removed either before or after stretching. Microporous membrane are further
disclosed in
U.S. Patent Nos. 4,539,256 (Shipman), 4,726,989 (Mrozinski), 4,867,881
(Kinzer),
5,120,594 (Mrozinski), 5,260,360 (Mrozinski et al.), and 5,962,544 (Waller),
all of which
are assigned to 3M Company (St. Paul, MN). Further, the microporous film can
be
prepared from ethylene-vinyl alcohol copolymers as described in U.S. Patent
No.
5,962,544 (Waller).
- 4 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
Some exemplary TIPS membrane comprise poly(vinylidene fluoride) (i.e., PVDF),
polyolefins such as polyethylene homo- or copolymers or polypropylene homo- or
copolymers, vinyl-containing polymers or copolymers such as ethylene-vinyl
alcohol
copolymers and butadiene-containing polymers or copolymers, and acrylate-
containing
polymers or copolymers. For some applications, a TIPS membrane comprising PVDF
is
particularly desirable. TIPS membranes comprising PVDF are further described
in U.S.
Patent Application Publication No. 2005/0058821, which is assigned to 3M
Company (St.
Paul, MN).
In other embodiments, the porous base substrate is a nonwoven web which may
include nonwoven webs manufactured by any of the commonly known processes for
producing nonwoven webs. As used herein, the term "nonwoven web" refers to a
fabric
that has a structure of individual fibers or filaments which are randomly
and/or
unidirectionally interlaid in a mat-like fashion.
For example, the fibrous nonwoven web can be made by carded, air laid,
spunlaced, spunbonding or melt-blowing techniques or combinations thereof
Spunbonded
fibers are typically small diameter fibers that are formed by extruding molten
thermoplastic polymer as filaments from a plurality of fine, usually circular
capillaries of a
spinneret with the diameter of the extruded fibers being rapidly reduced.
Meltblown fibers
are typically formed by extruding the molten thermoplastic material through a
plurality of
fine, usually circular, die capillaries as molten threads or filaments into a
high velocity,
usually heated gas (e.g. air) stream which attenuates the filaments of molten
thermoplastic
material to reduce their diameter. Thereafter, the meltblown fibers are
carried by the high
velocity gas stream and are deposited on a collecting surface to from a web of
randomly
disbursed meltblown fibers. Any of the non-woven webs may be made from a
single type
of fiber or two or more fibers that differ in the type of thermoplastic
polymer and/or
thickness.
Further details on the manufacturing method of non-woven webs of this
invention
may be found in Wente, Superfine Thermoplastic Fibers, 48 INDUS. ENG. CHEM.
1342(1956), or in Wente et al., Manufacture Of Superfine Organic Fibers,
(Naval
Research Laboratories Report No. 4364, 1954).
The functionalized substrate has grafted species attached to the surfaces of
the
porous base substrate which includes (a) at least one photoinitiator group (or
the reaction
- 5 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
product thereof), (b) at least one ethylenically unsaturated group (or the
reaction product
thereof) and (c) optionally other hydrophilic groups, wherein at least one of
(b) or (c) is a
hydrophilic group. The grafting of monomers to the surface of the porous base
substrate
results in a hydrophilic surface imparted to an otherwise hydrophobic base
substrate. The
hydrophilic monomer, whether "(b)" or "(c)", are used in amounts sufficient to
render the
porous substrate wettable as described herein.
The monomers that are grafted to the surface of the porous base substrates
usually
have both (a) an acrylate group for grafting by e-beam and (b) at least one
additional
function group thereon, which includes (a) a photoinitiator group to initiate
the
crosslinking on exposure to UV radiation, (b) an acrylate or a non-acrylate,
free-radically
polymerizable ethylenically unsaturated group for subsequent crosslinking and
optionally
(c) a hydrophilic group.
Acrylate groups are preferred for direct grafting of the monomer to the porous
substrate surface due to the greater reactivity of such acrylates on exposure
to e-beam
irradiation. However, not all such acrylate groups may be directly grafted
(i.e. forming a
covalent bond with the porous surface); some may remain free, and are
subsequently
"indirectly grafted" by incorporation into the polymer chain on exposure to UV
radiation.
Other ethylenically unsaturated groups, such as (meth)acrylamides,
methacrylates, vinyl
and vinyloxy groups, allyl and allyloxy groups, and acetylenic groups are less
reactive
during e-beam, and are less likely to be directly grafted to the porous base
substrate.
Therefore a portion of such non-acrylate groups may be directly grafted, but
largely
remain unreacted, and are indirectly grafted to the substrate by incorporation
into the
polymer chain during UV initiated polymerization.
The photoinitiator monomers may be directly grafted onto interstitial and
outer
surfaces of the porous base substrate to provide the requisite grafted
photoinitiator group
via the acrylate group. The "(b)" monomers, in addition to the acrylate group,
the free-
radically polymerizable groups of monomer (b) are typically other
ethylenically
unsaturated groups such as a (meth)acrylamides, methacrylates, vinyl groups
and
acetylenic groups having reduced reactivity during grafting, and are therefore
free and
unreacted for the subsequent UV initiated polymerization and crosslinking. The
acrylate
group of the "(b)" monomers typically can directly graft (i.e. forming a
covalent bond) to
the surface of the porous base substrate when exposed to an electron beam.
That is,
- 6 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
reaction of acrylate groups of the (b) monomers with the surface of the porous
base
substrate in the presence of the electron beam results in the formation of
ethylenically
unsaturated groups directly grafted to the porous base substrate via the
acrylate group.
A third grafting monomer "(c)" may also be grafted via an acrylate group, and
may
provide hydrophilic groups to the surfaces of the porous base substrate. In
other
embodiments the third monomer may have an ethylenically unsaturated group of
reduced
reactivity during the grafting step, but is subsequently incorporated by free-
radical
polymerization during the UV curing step (indirectly grafted). At least one of
the
monomers (b) and (c) is a hydrophilic monomer.
The grafting photoinitiator monomers include an acrylate group and a
photoinitiator group and may be represented by the generalized formula:
HO
______________ I I I 0 R4_ PI
I where;
R4 is a divalent linking group connecting the acrylate group with the PI
group, and
PI is a photoinitiator represented by the structure:
0
0 ii 2
C¨R
II, wherein R2 is
1
R1
R1
\
R1
R1
E.3
\ / \ / and ¨C¨Ix
1 3
R
wherein Rl is H or a C1 to C4 alkyl group,
each R3 is independently a hydroxyl group, a phenyl group, a C1 to C6 alkyl
group, or a
C1 to C6 alkoxy group. Such photoinitiator monomers are described, for
example, in U.S.
Patent Nos. 5,902,836 (Babu et al.) and 5,506,279 (Babu et al.). Further
details regarding
the linking R4 group may be found with reference to the method of preparing
the
photoinitiator grafting monomer herein, and in the cited references.
- 7 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
In certain preferred embodiments, the photoinitiator monomers may be of the
hydrogen-abstraction type represented by the general formula:
CH2=CHX[¨(M
-E11¨ i)a_(M2)b_(M3)0 ] 0 G PI1
P ix
X' is 0 or NH;
p is 0 or 1;
o is 0 or an integer from 1 to 5;
a, b, and c are independently 0 or 1;
M1 is CH2 or S4R1)2 ;
M2 is C(R1)2 or Si(R1)2;
M3 is ¨0-, -NH-, -C(0)-, -C(0)0-, -C(0)NH-, or -0C(0)NH-;
Each R1 is independently H or a Ci to C4 alkyl group;
G is a covalent bond, -(CH2)d-, or -(CH2)d0- where d is an integer from 1 to
4, preferably
from 1 to 2;
PI' is a radiation-sensitive hydrogen abstracting group having the general
formula:
0
¨,-,A, __________ II p13
, .,
I 12
R
xiv
in which Ar is a substituted arene having 6 to 12 carbon atoms, preferably a
benzenetriyl
group;
R12 is hydrogen, a C1 to C12 alkyl group, a C1 to C12 alkoxy group, or a
phenyl group; and
R13 is a C1 to C6 alkyl group, a cycloalkyl group having 3 to 14 carbon atoms,
or
R14
=
R15
wherein R14 and R15 are independently selected from hydrogen, Cl to C12 alkyl
groups, Cl
to C12 alkoxy groups, and phenyl groups.
Included among those hydrogen abstracting photoinitiator monomers encompassed
by Formula IX are those where PI' is a moiety derived from one of the
following
- 8 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
compounds (or a substituted derivative thereof), the bond to G is preferably
located para to
the bridging carbonyl group: benzophenone, anthraquinone, 5,12-
naphthacenequinone,
aceanthracenequinone, benz(A)anthracene-7,12-dione, 1,4-chrysenequinone, 6,13-
pentacenequinone, 5,7,12,14-pentacenetetrone, 9-fluorenone, anthrone,
xanthone,
thioxanthone, acridone, dibenzosuberone, acetophenone, and chromone. The
synthesis of
the formula XIII monomers is described in U.S. 5,773,485 (Bennett et al).
The weight percentage of the photoinitiator monomers in the imbibing solution
can
be at least about 0.01%, and preferably at least about 0.15%, and no more than
about
2.5%, preferably no more than about 1%, relative to the total weight of other
monomers
(i.e. "(b)" and "(c)" monomers). It will be understood that all or a portion
of the
photoinitiator monomers may be directly grafted to the surfaces of the base
substrate upon
exposure to e-beam irradiation. Those unreacted, ungrafted photoinitiator
monomers will
be incorporated into the growing polymer chain on exposure to UV radiation,
thereby
indirectly grafting the monomers to the porous base substrate.
A variety of photoinitiator grafting monomers can be made by reacting 1) an
acrylate monomer comprising a first reactive functional group with 2) a
compound that
comprises a radiation-sensitive group (photoinitiator group) and second
reactive functional
group, the two functional groups being co-reactive with each other. Preferred
co-reactive
compounds are ethylenically unsaturated aliphatic, cycloaliphatic, and
aromatic
compounds having up to 36 carbon atoms, optionally one or more oxygen and/or
nitrogen
atoms, and at least one reactive functional group. When the first and second
functional
groups react, they form a covalent bond and link the co-reactive compounds.
Examples of useful reactive functional groups include hydroxyl, amino,
oxazolinyl, oxazolonyl, acetyl, acetonyl, carboxyl, isocyanato, epoxy,
aziridinyl, acyl
halide, and cyclic anhydride groups. Where the pendent reactive functional
group is an
isocyanato functional group, the co-reactive functional group preferably
comprises a
amino, carboxyl, or hydroxyl group. Where pendent reactive functional group
comprises a
hydroxyl group, the co-reactive functional group preferably comprises a
carboxyl,
isocyanato, epoxy, anhydride, acyl halide, or oxazolinyl group. Where the
pendent
reactive functional group comprises a carboxyl group, the co-reactive
functional group
preferably comprises a hydroxyl, amino, epoxy, vinyloxy, or oxazolinyl group.
- 9 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
Representative examples of acrylate compounds having a reactive functional
group
include hydroxyalkyl acrylates such as 2-hydroxyethyl acrylate and 2-(2-
hydroxyethoxy)ethyl acrylate; aminoalkyl acrylates such as 3-aminopropyl
acrylate;
oxazolinyl compounds such as 2-etheny1-1,3-oxazolin-5-one and 2-propeny1-4,4-
dimethyl-
1,3-oxazolin-5-one; carboxy-substituted compounds such as acrylic acid and 4-
carboxybenzyl acrylate; isocyanato-substituted compounds such as
isocyanatoethyl
acrylate and 4-isocyanatocyclohexyl acrylate; epoxy-substituted compounds such
as
glycidyl acrylate; aziridinyl-substituted compounds such as N-
acryloylaziridine; and
acryloyl halides.
Representative examples of co-reactive compounds include functional group-
substituted compounds such as 1-(4-hydroxypheny1)-2,2-dimethoxyethanone, 1
4442-
hydroxyethyl)pheny1]-2,2-dimethoxyethanone, (4-isocyanatopheny1)-2,2-dimethoxy-
2-
phenylethanone, 1- {4-[2-(2,3-epoxypropoxy)pheny1]} -2,2-dimethy1-2-
hydroxyethanone,
1 -[4-(2-aminoethoxy)pheny1]-2,2-dimethoxyethanone, and 1 44-
(carbomethoxy)pheny1]-
2,2-dimethoxyethanone.
It will be understood that all or a portion of the acrylate groups of the
photoinitiator
monomer may be directly grafted to the surface of the porous substrate on
exposure of e-
beam irradiation. Those ungrafted, free acrylate groups may be subsequently
indirectly
grafted to the substrate by incorporation into the polymer chain on UV
initiated
polymerization.
The second grafting monomers comprises (a) one or more acrylate groups for
grafting and (b) one or more second, ethylenically unsaturated, free-radically
polymerizable groups for subsequent crosslinking. The second ethylenically
unsaturated
group may be an acrylate or a non-acrylate; i.e. other ethylenically
unsaturated groups
having reduced reactivity relative to the acrylate group during the e-beam
grafting step.
Preferably the second ethylenically unsaturated group is a non-acrylate group
and is left
largely free and unreacted during the grafting step for subsequent UV
crosslinking. Useful
second, non-acrylate ethylenically unsaturated groups include methacrylates,
(meth)acrylamides, vinyl groups, vinyloxy, acetylenic groups, allyl and
allyloxy groups.
Useful second grafting monomers may have the generalized structure:
[CH2=CH-C(0)-0]a-R5-Q-Zb, III
where Z is an acrylate or non-acrylate, ethylenically unsaturated
polymerizable group,
- 10 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
Q is a divalent linking group selected from a covalent bond "-", ¨0-, -NR'- ,
¨0O2- and
¨CONR1-, where R' isH or C1-C4 alkyl; and
R5 is an alkylene group of valence a+b, and optionally containing one or more
catenary
oxygen atoms and/or one or more hydroxyl groups; and a and b are each at least
one.
Preferably the Z group is a non-acrylate of reduced reactivity that is
indirectly grafted into
the polymer chain during UV initiated polymerization.
In certain embodiments, R5 is a poly(alkylene oxide group) to provide the
desired
hydrophilicity, and is of the formula:
Z-Q-(CH(R1)-CH2-0)õ-C(0)-CH=CH2, IV
wherein Z is an acrylate or non-acrylate, polymerizable ethylenically
unsaturated group,
R1 is a H or a C1 to C4 alkyl group, and n is from 2 to 100, preferably 5 to
20, and Q is a
divalent linking group selected from a covalent bond "-", ¨0-, -NR'- , ¨0O2-
and ¨
CONR1-, where R' isH or C1-C4 alkyl. Preferably the Z group is a non-acrylate
of reduced
reactivity that is indirectly grafted into the polymer chain during UV
initiated
polymerization.
In one embodiment, the poly(alkylene oxide) group (depicted as -(CH(R1)-CH2-
Q)õ-) is a poly(ethylene oxide) (co)polymer. In another embodiment, the
pendent
poly(alkylene oxide) group is a poly(ethylene oxide-co-propylene oxide)
copolymer. Such
copolymers may be block copolymers, random copolymers, or gradient copolymers.
Suitable monomers having a first acrylate group for grafting and a second
ethylenically unsaturated group for subsequent UV crosslinking include, but
are not
limited to, polyalkylene glycol acrylate methacrylate including those derived
from
polyethylene glycol and polypropylene glycol acrylated monomers.
In another embodiment, the second monomer is a partially acrylated polyol,
having
at least one acrylate groups and at least one other ethylenically unsaturated
polymerizable
group, which is preferably not a acrylate group and may be selected from
methacrylates,
(meth)acrylamides, vinyl groups, vinyloxy, acetylenic groups, allyl and
allyloxy groups.
Such partially acrylated polyols may have one of more free hydroxyl groups.
Polyols useful in the present invention include aliphatic, cycloaliphatic, or
alkanol-
substituted arene polyols, or mixtures thereof having from about 2 to about 18
carbon
atoms and two to five, preferably two to four hydroxyl groups.
-11-
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
Examples of useful polyols include 1,2-ethanediol, 1,2-propanediol, 1,3-
propanediol, 1,4-butanediol, 1,3-butanediol, 2-methyl-1,3-propanediol, 2,2-
dimethy1-1,3-
propanediol, 2-ethyl-1,6-hexanediol, 1,5-pentanediol, 1,6-hexanediol, 1,8-
octanediol,
neopentyl glycol, glycerol, trimethylolpropane, 1,2,6-hexanetriol,
trimethylolethane,
pentaerythritol, quinitol, mannitol, sorbitol, diethlene glycol, triethylene
glycol,
tetraethylene glycol, 2-ethyl-2-(hydroxymethyl)-1,3-propanediol, 2-ethyl-1,3-
pentanediol,
1,4-cyclohexanedimethanol, 1,4-benzenedimethanol, and polyalkoxylated
bisphenol A
derivatives. Most preferably "(b)" monomers are those monoacrylates of
glycerol having a
free hydroxyl group and a methacrylate group such as 3-(acryloxy)-2-
hydroxypropylmethacrylate).
In some preferred embodiments, the ethylenically unsaturated groups of the
"(b)"
and "(c)" monomers are chosen to be efficiently copolymerizable with each
other. That is,
it is preferred that each of the "(b)" and "(c)" monomers have the same
ethylenically
unsaturated groups.
In one exemplary embodiment, the grafted species result from the reaction of a
polyethylene glycol acrylate monomer of Formulas III or IV with the porous
base
substrate upon exposure to an electron beam. These grafting monomers can be
used to
change a hydrophobic porous base substrate into a hydrophilic functionalized
substrate
due to the presence of the poly(alkylene oxide) group. The resulting
hydrophilic can have
a number of desired properties such as instant wettability following exposure
to 1N NaOH
for 20 hours as described in more detail below.
The optional third monomer ("(c)", hydrophilic monomer) comprises at least one
acrylate or other non-acrylate, ethylenically unsaturated group of reduced
reactivity, and a
hydrophilic group, such as an ionic group, for providing hydrophilicity to the
substrate. If
the optional third monomer contains an acrylate group, it may be directly
grafted to the
surfaces of the porous bases substrate. If it contains a non-acrylate,
ethylenically
unsaturated group it will remain largely unreacted during the grafting step,
and will be
incorporated during the UV polymerization step. It will be understood that all
or a portion
of the acrylate groups may be directly grafted to the porous substrate, and a
portion may
be unreacted, but will be indirectly grafted into the polymer upon UV
initiated irradiation.
Conversely, a portion of other ethylenically unsaturated groups of reduced
reactivity may
- 12 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
be directly grafted, but such groups generally remain largely unreacted during
the grafting
step and are indirectly grafted into the polymer upon UV initiated
irradiation.
The ionic groups may be neutral, have a positive charge, a negative charge, or
a
combination thereof. With some suitable ionic monomers, the ionic group can be
neutral
or charged depending on the pH conditions. This class of monomers is typically
used to
impart a desired hydrophilicity to the porous base substrate in addition to
the second
monomer.
In some preferred embodiments, the third monomer may have an acrylate group,
or
other ethylenically unsaturated groups of reduced reactivity, and a
poly(alkylene oxide)
group; e.g. monoacrylated poly(alkylene oxide compounds, where the terminus is
a
hydroxy group, or an alkyl ether group.
In some embodiments the ionic monomers having a negative charge include
(meth)acrylamidosulfonic acids of Formula II or salts thereof.
0
= I I L Y¨S03H
V
wherein, Y is a straight or branched alkylene (e.g., an alkylenes having 1 to
10 carbon
atoms, 1 to 6 carbon atoms, or 1 to 4 carbon atoms) and L is oxy or ¨NR'-,
where Rl is H
or C1-C4 alkyl-;. Exemplary ionic monomers according to Formula I include, but
are not
limited to, N-acrylamidomethanesulfonic acid, 2-acrylamidoethanesulfonic acid,
2-
acrylamido-2-methyl-1-propanesulfonic acid, and 2-methacrylamido-2-methy1-1-
propanesulfonic acid. Salts of these acidic monomers can also be used. Counter
ions for
the salts can be, for example, ammonium ions, potassium ions, lithium ions, or
sodium
ions. It will be understood with respect to Formula V that the grafting
acrylate group may
be replaced by another ethylenically unsaturated group of reduced reactivity
for
subsequent incorporation (indirect grafting) during UV initiated
polymerization.
Other suitable ionic grafting monomers having a negative charge include
sulfonic
acids such as vinylsulfonic acid and 4-styrenesulfonic acid;
(meth)acrylamidophosphonic
acids such as (meth)acrylamidoalkylphosphonic acids (e.g., 2-
(meth)acrylamidoethylphosphonic acid and 3-(meth)acrylamidopropylphosphonic
acid;
acrylic acid and methacrylic acid; and carboxyalkyl(meth)acrylates such as 2-
carboxyethyl(meth)acrylate, and 3-carboxypropyl(meth)acrylate. Still other
suitable acidic
monomers include (meth)acryloylamino acids, such as those described in U.S.
Patent No.
- 13 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
4,157,418 (Heilmann). Exemplary (meth)acryloylamino acids include, but are not
limited
to, N-acryloylglycine, N-acryloylaspartic acid, N-acryloy1-13-alanine, and 2-
acrylamidoglycolic acid. Salts of any of these acidic monomers can also be
used.
Some exemplary ionic grafting monomers that are capable of providing a
positive
charge are amino (meth)acrylates or amino (meth)acrylamides of Formula II or
quaternary
ammonium salts thereof. The counter ions of the quaternary ammonium salts are
often
halides, sulfates, phosphates, nitrates, and the like.
0
= I I L Y¨N (R5)2
VI
where L is oxy or ¨NR'-, where Rl is H or C1-C4 alkyl-; and Y is an alkylene
(e.g., an
alkylene having 1 to 10 carbon atoms, 1 to 6, or 1 to 4 carbon atoms). Each R5
is
independently hydrogen, alkyl, hydroxyalkyl (i.e., an alkyl substituted with a
hydroxy), or
aminoalkyl (i.e., an alkyl substituted with an amino). Alternatively, the two
R5 groups
taken together with the nitrogen atom to which they are attached can form a
heterocyclic
group that is aromatic, partially unsaturated (i.e., unsaturated but not
aromatic), or
saturated, wherein the heterocyclic group can optionally be fused to a second
ring that is
aromatic (e.g., benzene), partially unsaturated (e.g., cyclohexene), or
saturated (e.g.,
cyclohexane).
It will be understood with respect to Formula VI that the grafting acrylate
group
may be replaced by another ethylenically unsaturated group of reduced
reactivity, such as
methacrylate, (meth)acrylamide, vinyl, vinyloxy, ally, alloxy, and acetylenyl
for
subsequent incorporation (indirect grafting) during UV initiated
polymerization.
In some embodiments of Formula VI, both R5 groups are hydrogen. In other
embodiments, one R5 group is hydrogen and the other is an alkyl having 1 to
10, 1 to 6, or
1 to 4 carbon atoms. In still other embodiments, at least one of R5 groups is
a hydroxy
alkyl or an amino alkyl that have 1 to 10, 1 to 6, or 1 to 4 carbon atoms with
the hydroxy
or amino group being positioned on any of the carbon atoms of the alkyl group.
In yet
other embodiments, the R5 groups combine with the nitrogen atom to which they
are
attached to form a heterocyclic group. The heterocyclic group includes at
least one
nitrogen atom and can contain other heteroatoms such as oxygen or sulfur.
Exemplary
heterocyclic groups include, but are not limited to imidazolyl. The
heterocyclic group can
be fused to an additional ring such as a benzene, cyclohexene, or cyclohexane.
Exemplary
- 14 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
heterocyclic groups fused to an additional ring include, but are not limited
to,
benzoimidazolyl.
Exemplary amino acrylates (i.e., L in Formula VI is oxy) include N,N-
dialkylaminoalkyl acrylates such as, for example, N,N-
dimethylaminoethylacrylate, N,N-
dimethylaminoethylacrylate, N,N-diethylaminoethyl acylate, N,N-
diethylaminoethylacrylate, N,N-dimethylaminopropylacrylate, N,N-
dimethylaminopropylacrylate, N-tert-butylaminopropylmethacrylate, N-tert-
butylaminopropylacrylate and the like.
Exemplary amino (meth)acrylamides, that would be incorporated during the UV
polymerization, (i.e., L in Formula VI is ¨NR'-) include, for example, N-(3-
aminopropyl)methacrylamide, N-(3-aminopropyl)acrylamide, N-[3-
(dimethylamino)propyl]methacrylamide, N-(3-imidazolylpropyl)methacrylamide, N-
(3-
imidazolylpropyl)acrylamide, N-(2-imidazolylethyl)methacrylamide, N-(1,1-
dimethy1-3-
imidazoylpropyl)methacrylamide, N-(1,1-dimethy1-3-imidazoylpropyl)acrylamide,
N-(3-
benzoimidazolylpropyl)acrylamide, and N-(3-
benzoimidazolylpropyl)methacrylamide.
Exemplary quaternary salts of the ionic monomers of Formula VI include, but
are
not limited to, (meth)acrylamidoalkyltrimethylammonium salts (e.g., 3-
methacrylamidopropyltrimethylammonium chloride and 3-
acrylamidopropyltrimethylammonium chloride) and
(meth)acryloxyalkyltrimethylammonium salts (e.g., 2-
acryloxyethyltrimethylammonium
chloride, 2-methacryloxyethyltrimethylammonium chloride, 3-methacryloxy-2-
hydroxypropyltrimethylammonium chloride, 3-acryloxy-2-
hydroxypropyltrimethylammonium chloride, and 2-acryloxyethyltrimethylammonium
methyl sulfate).
Other monomers that can provide positively charged groups to an ion exchange
resin include the dialkylaminoalkylamine adducts of alkenylazlactones (e.g., 2-
(diethylamino)ethylamine, (2-aminoethyl)trimethylammonium chloride, and 3-
(dimethylamino)propylamine adducts of vinyldimethylazlactone) and diallylamine
monomers (e.g., diallylammonium chloride and diallyldimethylammonium
chloride).
A third monomer, that may be incorporated by grafting or by subsequent UV
polymerization are poly(alkylene oxide) monomers having at least one acrylate
or non-
- 15 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
acrylate ethylenically unsaturated group and a non-polymerizable terminus.
Such
monomers are of the general formula:
R1-0-(CH(R1)-CH2-0)õ-C(0)-C(R1)=CH2, VII,
wherein each R1 is independently H or C1-C4 alkyl.
As described in further detail below, functionalized substrates of the present
invention may be prepared using above-described monomers to provide
hydrophilicity to
the surface of a porous base substrate. When two or more of the above-
described
monomers are used to alter the surface properties of a porous base substrate,
the
monomers may be grafted onto the porous base substrate in a single reaction
step (i.e., the
two or more grafting monomers are all present upon exposure to an electron
beam) or in
sequential reaction steps (i.e., a first grafting photoinitiator monomer
"(a)", and present
upon a first exposure to an electron beam and a second grafting monomer "(b)
and/or (c)"
is present upon a second exposure to the electron beam). Similarly, all of
such monomers
(a), (b) and (c) may be present during a first grafting step and directly
grafted, or indirectly
grafted by incorporation during the subsequent UV initiated polymerization.
Alternatively,
all or a potion of such monomers may be imbibed in a first step, or in
subsequent imbibing
steps.
In some embodiments, the grafted species imparts a hydrophilic character to
the
functionalized substrate that contains a porous base substrate that has a
hydrophobic
character prior to surface modification. The hydrophilic character of the
functionalized
substrate results from the reaction of the porous base substrate with the "(b)
and/or (c)"
monomers that contain a hydrophilic group upon exposure to an electron beam
and UV
initiated polymerization.
As previously described, all or a portion of the photoinitiator a) monomers
will be directly grafted to the substrate. Subsequently the additional b)
and/or c)
monomers may be indirectly grafted: the additional monomer may be grafted via
the
residue of the photoinitator and the base substrate. This may be illustrated
with
reference to Formula VIII where a hydrophilic c) monomer of Formula VII is
indirectly grafter via the residue of a photoinitiator:
Ri 0
III
¨PI*¨CH2CH-C¨(OCH2CH(R1)n¨OR1
VIII, wherein
each R1 is independently H or C1-C4 alkyl;
- 16 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
n is from 2 to 100; and
PI* is the residue of a photoinitiator grafted to the substrate surface.
For example a grafting photoinitator monomer such as 2-
propenoylaminoethanoic acid; 2-(4-(2- hydroxy-2 methylpropanoyl)phenoxy)ethyl
ester may be grafted to a substrate surface using ionizing radiation such as e-
beam
energy. In the presence of UV, the photoinitiator undergoes alpha cleavage to
two
radicals. In the presence of the ligand monomer, or other monomers, the
radical may
add to the ethylenically unsaturated group (such as the depicted acryloyl
group) to
indirectly graft the ligand monomer to the substrate surface via the residue
of the
photoinitator as shown in formula I and illustrated in Scheme I below. It will
be
further understood that the radical addition product of the a), b) and/or c)
monomers
may further copolymerize with additional a), b) and/or c) monomers to produce
a
grafted polymer.
- 17 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
Scheme I
0 CH3 0 0 CH3
CH2=CHILNH II 0 CH2CH20¨(0) II OH
CH3 CH3
1 graffing
0
CH3 0 0 CH3
CH2-CH2ILNH II 0 CH2CH20¨(0) II OH
CH3 CH3
1 uv
CH3
0 0 0 CH3
CH2-CH2rNH II 0 CH2CH20
0 I . + . OH
CH3 CH3
1 addition
0 CH3 0 0
Ri 0
I
CH2-CH2rNH II 0 CH2CH20 0 CH2CH11L(OCH2CH(R1)¨OR1
CH3
It will be further understood that the grafting process will yield a radical
species, having a radical on the carbon alpha to the carbonyl of the
hydrophilic
monomer of Formula VII, that may further polymerize with one of more
additional
"b)" monomers, one of more photoinitiator "a)" monomers, one or more "c)"
monomers, resulting in a grafted polymers having these groups pendent from the
polymer chain as simply illustrated below. The formation of grafted polymer
chains
significantly increases the density of the desired ligand groups, and the
efficiency of
binding.
Substrate-(M)_(Mb)x_04%
In the formula, the -(MPI)- represent the residue of the grafted
photoinitiator
monomer (as illustrated in Scheme I), the -(IVIN represents the polymerized b)
monomer (such as in Formulas III or IV), having "x" polymerized monomer units,
where x is at least one and preferably at least two, -(IVIc)y represents the
polymerized
monomer "c)" (such as Formulas V to VII) , having y polymerized monomer units,
- 18-
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
where y may be zero and is preferably at least 1. The polymer may be random or
block, and the "b)" monomer, having two polymerizable groups may provide a
crosslink between polymer chains. The polymer may be directly grafted via the
residue of the photoinitiator, as shown in Scheme I, or may be directly
grafted via the
"b)" monomers or the "c)" monomers, as described herein. The polymer may
further
comprise polymerized photoinitiator monomer units from unreacted, ungrafted
photoinitiator monomers.
The above-described hydrophilic substrates may be prepared using a combination
of process steps. The method comprises:
1) providing a porous base substrate having interstitial and outer
surfaces;
2) imbibing the porous base substrate with a solution to form an imbibed
porous base substrate, the first solution comprising (a) at least one grafting
monomer
having an acrylate group and a photoinitiator group and optionally (b) one or
more
monomers having at least one acrylate group and at least one additional
ethylenically
unsaturated, free-radically polymerizable group; and optionally (c) one or
more additional
monomers having at least one ethylenically unsaturated, free-radically
polymerizable
group and a hydrophilic group; wherein at least one of (b) or (c) monomers are
hydrophilic. The imbibing step may comprise a single solution or multiple
solutions.
3) exposing the imbibed porous base substrate to a controlled amount of
electron beam radiation so as to form a first functionalized substrate
comprising grafted
photoinitiator group attached to the surfaces of the porous base substrate,
and
4) exposing the porous base substrate comprising grafted photoinitiator
groups
to a controlled amount of UV radiation to crosslink the remaining free-
radically
polymerizable groups.
With respect to the above-described method, the substrate may be imbibed with
a
first solution comprising the photoinitiator "a)" monomers and the "b)"
monomers and
optionally the "c)" monomers, subsequently exposed to the e-beam radiation and
then
crosslinked by UV radiation. Alternatively the method may comprise first
imbibing the
porous base substrate with the photoinitator "a)" monomer, exposing the
imbibed substrate
to e-beam radiation to graft the photoinitiator "a)" monomer to the surface of
the substrate,
subsequently imbibing the grafted porous base substrate with a second imbibing
solution
comprising the "b)" and optionally the "c)" monomers, then exposing the
imbibed
- 19 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
substrate to UV radiation to indirectly graft the "b)" and the "c)" monomers
to the
substrate.
Some of the porous base substrates used in this embodiment can be porous,
microporous, nonwoven, or a combination thereof
One exemplary method for making functionalized substrates is depicted in FIG.
1.
As shown in FIG. 1, exemplary method 10 comprises the following steps: an
imbibing
step 100, a sandwiching step 200, an irradiation step 300, a UV initiated
polymerization
step 400, a peeling step 500, a wash/rinse step 600, a drying step 700, and a
take-up step
800. Each of these exemplary steps is described in further detail below.
Methods of making functionalized substrates of the present invention may
comprise one or more of the following steps.
Imbibing Step
As shown in FIG. 1, a roll 11 comprising a porous base substrate 12 may be
unwound so that porous base substrate 12 enters into imbibing step 100. In
imbibing step
100, porous base substrate 12 is brought into contact or into proximity with
applicator 14
that is connected to a reservoir of solution 13 containing one or more
grafting monomers.
Rollers 15 and 16 guide porous base substrate 12 past applicator 14 so that
porous base
substrate 12 is exposed to solution 13 for a desired amount of time.
Typically, the
exposure time of the porous base substrate 12 to solution 13 is up to about
1.0 minutes,
more typically, less than about 15 seconds. Porous base substrate 12 usually
proceeds
through imbibing step 100 and to irradiation step 300 in less than 1 minute.
In some
imbibing steps, the porous base substrate 12 is saturated with the solution
13.
As discussed above, solution 13 may comprise one or more grafting suitable for
grafting onto interstitial and outer surfaces of porous base substrate 12. Any
of the
exemplary grafting monomers described above can be included in solution 13. In
addition
to grafting monomers, solution 13 can contain other materials such as, for
example, one or
more other non-grafting monomers for UV curing, and solvents. The
concentration of each
grafting monomer in solution 13 may vary depending on a number of factors
including,
but not limited to, the grafting monomer or monomers in solution 13, the
extent of grafting
desired, the reactivity of the grafting monomer(s), and the solvent used.
Typically, the
concentration of each monomer in solution 13 ranges from about 1 wt% to about
100 wt%,
- 20 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
desirably, from about 5 wt% to about 30 wt%, and more desirably from about 10
wt% to
about 20 wt% based on a total weight of solution 13.
Once porous base substrate 12 has been imbibed in solution 13 for a desired
period
of time, the porous base substrate 12 is directed toward sandwiching step 200
via guide
roller 17. Guide roller 17 may be used to meter excess solution 13 from the
imbibed
porous base substrate 12 if so desired. Alternately, rollers (not shown) could
be used to
squeeze air bubbles and excess solution 13 from the imbibed porous base
substrate 12.
Typically, porous base substrate 12 enters sandwiching step 200 in a
substantially
saturated condition (i.e., porous base substrate 12 contains a maximum amount
of solution
13 or close to a maximum amount) wherein substantially all of the interstitial
and outer
surfaces of porous base substrate 12 are coated with solution 13.
It should be noted that imbibing step 100 is only one possible method of
introducing solution 13 into porous base substrate 12. Other suitable methods
include, but
are not limited to, a spray coating method, flood coating method, knife
coating, etc.
Sandwiching Step
In sandwiching step 200, imbibed porous base substrate 12 is sandwiched (i.e.,
positioned) between a removable carrier layer 22 and a removable cover layer
19 to form
multilayer sandwich structure 24. As shown in exemplary method 10, removable
cover
layer 19 may be unwound from roll 18 and brought into contact with an outer
surface of
imbibed porous base substrate 12 via roller 20, while removable carrier layer
22 may be
unwound from roll 21 and brought into contact with an opposite outer surface
of imbibed
porous base substrate 12 via roller 23. Rollers 20 and 23 form a gap that may
be used to
regulate the amount of imbibing solution 13 imparted to the porous substrate.
Removable cover layer 19 and removable carrier layer 22 may comprise any inert
sheet material that is capable of providing temporary protection to
functionalized substrate
(i.e., grafted porous base substrate 12) from direct exposure to oxygen upon
exiting
chamber 25. Suitable inert sheet materials for forming removable cover layer
19 and
removable carrier layer 22 include, but are not limited to, polyethylene
terephthalate film
material, other aromatic polymer film materials, and any other non-reactive
polymer film
30 material. In some embodiments, removable carrier layer 22 may be
selected from
materials that are transparent to UV radiation. Once assembled, multilayer
sandwich
structure 24 proceeds to irradiation step 300.
-21 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
In irradiation step 300, multilayer sandwich structure 24 is exposed to a
sufficient
quantity of radiation so as to graft one or more monomers within solution 13
onto
interstitial and outer surfaces of porous base substrate 12 so as to form
multilayer
sandwich structure 27 comprising functionalized substrate 30 sandwiched
between
removable carrier layer 22 and removable cover layer 19. As shown in exemplary
method
10, multilayer sandwich structure 24 proceeds through chamber 25, which
contains at least
one device 26 capable of providing a sufficient dose of radiation. A single
device 26 is
capable of providing a sufficient dose of radiation, although two or more
devices 26 may
be used especially for relatively thick porous base substrates 12. Typically,
chamber 25
comprises an inert atmosphere such as nitrogen, carbon dioxide, helium, argon,
etc. with a
minimal amount of oxygen, which is known to inhibit free-radical
polymerization. In
embodiments wherein porous base substrate 12 is irradiated without removable
cover layer
19, the amount of oxygen within chamber 25 can be more of a concern. When
removable
carrier layer 22 and removable cover layer 19 cover the porous base substrate
12, exposure
to oxygen within chamber 25 is minimal.
The irradiation step 300 provides the further advantage of converting any
dissolved
oxygen to peroxy compounds, which would interfere with the subsequent UV
initiated
polymerization. Therefore the e-beam irradiation step 300 facilitates the
subsequent UV
initiation 400 by the removal of oxygen.
Although other sources of irradiation may be used, desirably device 26
comprises
an electron beam source. Electron beams (e-beams) are generally produced by
applying
high voltage to tungsten wire filaments retained between a repeller plate and
an extractor
grid within a vacuum chamber maintained at about 10-6 Torr. The filaments are
heated at
high current to produce electrons. The electrons are guided and accelerated by
the repeller
plate and extractor grid towards a thin window of metal foil. The accelerated
electrons,
traveling at speeds in excess of 107 meters/second (m/sec) and possessing
about 100 to
300 kilo-electron volts (keV), pass out of the vacuum chamber through the foil
window
and penetrate whatever material is positioned immediately beyond the foil
window.
The quantity of electrons generated is directly related to the extractor grid
voltage.
As extractor grid voltage is increased, the quantities of electrons drawn from
the tungsten
wire filaments increase. E-beam processing can be extremely precise when under
- 22 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
computer control, such that an exact dose and dose rate of electrons can be
directed
against multilayer sandwich structure 24.
Electron beam generators are commercially available from a variety of sources,
including the ESI "ELECTROCURE" EB SYSTEM from Energy Sciences, Inc.
(Wilmington, MA), and the BROADBEAM EB PROCESSOR from PCT Engineered
Systems, LLC (Davenport, IA). For any given piece of equipment and irradiation
sample
location, the dosage delivered can be measured in accordance with ASTM E-1275
entitled
"Practice for Use of a Radiochromic Film Dosimetry System." By altering
extractor grid
voltage, beam diameter and/or distance to the source, various dose rates can
be obtained.
The temperature within chamber 25 is desirably maintained at an ambient
temperature by conventional means. Without intending to be limited to any
particular
mechanism, it is believed that the exposure of the imbibed porous base
substrate to an
electron beam results in free radical initiation on the substrate which can
then react with
monomers having a double bond such as monomers having an ethylenically
unsaturated
group.
The total dose received by multilayer sandwich structure 24 primarily affects
the
extent to which the grafting monomer is grafted onto the porous base
substrate. In general,
it is desirable and typical to convert at least 10 wt%, desirably 20 wt%, even
more
desirably greater than 50 wt% of the grafting monomers added during the
imbibing step to
grafted species. Further, it is desirable and typical to graft as much as
about 5 wt%,
desirably as much as about 10 wt%, more desirably as much as about 20 wt% (or
as much
as about 100 wt%) of one or more grafting monomers added during the imbibing
step onto
porous base substrate 12, based on a total weight of porous base substrate 12.
Dose is
dependent upon a number of processing parameters, including voltage, speed and
beam
current. Dose can be conveniently regulated by controlling line speed (i.e.,
the speed with
which multilayer sandwich structure 24 passes under device 26), and the
current supplied
to the extractor grid. A target dose (e.g., < 10 kGy) can be conveniently
calculated by
multiplying an experimentally measured coefficient (a machine constant) by the
beam
current and dividing by the web speed to determine the exposure. The machine
constant
varies as a function of beam voltage.
While the controlled amount of electron beam radiation exposure is dependent
upon the residence time, as a general matter, the monomers imbibed on the
porous
- 23 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
base substrate 12 that is part of multilayer sandwich structure 24 will
generally be
significantly grafted upon receiving a controlled amount of dosage ranging
from a
minimum dosage of about 1 kilograys (kGy) to a maximum dosage of less than
about
50 kGy, depending on the particular polymer. For propylene polymers the amount
typically ranges from a minimum dosage of about 1 kilograys (kGy) to a maximum
dosage of less than about 10 kGy. Typically, the total controlled amount of
dosage
ranges from less than about 9 kGy to about 7 kGy for propylene polymers to
avoid
degradation. Less radiation sensitive polymers such as nylons or PVDF may be
subjected to higher dosages, typically 10 to 70 kGy.
While low dose rates and longer residence times are preferred for radiation
grafting, practical operation may necessitate speeds that force higher dose
rates and shorter
residence. Exclusion of oxygen in a multilayer sandwich allows free radical
chemistry to
continue after E-beam exposure for duration sufficient to improve the grafting
yield.
UV curing step
In UV irradiation step 400, multilayer sandwich structure 24 is exposed to a
sufficient quantity of ultraviolet radiation so as to initiate free radical
polymerization from
the grafted photoinitiator monomer and any free, unreacted acrylate groups
and/or
ethylenically unsaturated groups. The polymerization of the unreacted
ethylenically
unsaturated groups onto the grafted interstitial and outer surfaces of porous
base substrate
12 forms multilayer sandwich structure 27 comprising functionalized substrate
30
sandwiched between removable carrier layer 22 and removable cover layer 19. As
shown
in exemplary method 10, multilayer sandwich structure 24 proceeds through
chamber 40,
which contains at least one device 41 capable of providing a sufficient dose
of UV
radiation. A single device 41 is capable of providing a sufficient dose of
radiation,
although two or more devices 41 may be used especially for relatively thick
porous base
substrates 12 or to double the lamp output. Upon UV irradiation, essentially
all remaining
acrylate and non-acrylate groups are incorporated into a polymer coating on
the surfaces
of the base substrate 12, rendering it hydrophilic
Typically, chamber 40 comprises an inert atmosphere such as nitrogen, carbon
dioxide, helium, argon, etc. with a minimal amount of oxygen, which is known
to inhibit
free-radical polymerization. In embodiments wherein porous base substrate 12
is irradiated
without removable cover layer 19, the amount of oxygen within chamber 25 can
be more
- 24 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
of a concern. When removable carrier layer 22 and removable cover layer 19
cover the
porous base substrate 12, exposure to oxygen within chamber 25 is minimal.
UV light sources can be of two types: 1) relatively low light intensity
sources such
as blacklights which provide generally 10 mW/cm2 or less (as measured in
accordance
with procedures approved by the United States National Institute of Standards
and
Technology as, for example, with a UVIMAPTm UM 365 L-S radiometer manufactured
by
Electronic Instrumentation & Technology, Inc., in Sterling, VA) over a
wavelength range
of 280 to 400 nanometers and 2) relatively high light intensity sources such
as medium
pressure mercury lamps which provide intensities generally greater than 10
mW/cm2,
preferably between 15 and 450 mW/cm2. Where actinic radiation is used to fully
or
partially crosslink the oligomer composition, high intensities and short
exposure times are
preferred. For example, an intensity of 600 mW/cm2 and an exposure time of
about 1
second may be used successfully. Intensities can range from about 0.1 to about
150
mW/cm2, preferably from about 0.5 to about 100 mW/cm2, and more preferably
from
about 0.5 to about 50 mW/cm2.
Peeling Step
Upon exiting chamber 25, multilayer sandwich structure 27 proceeds toward
peeling step 500. In peeling step 500, multilayer sandwich structure 27 is
disassembled by
separating removable carrier layer 22 and removable cover layer 19 from
functionalized
substrate 30. As shown in exemplary method 10, removable cover layer 19 is
separated
from an outer surface of functionalized substrate 30 and taken-up as roll 28,
while
removable carrier layer 22 is separated from an opposite outer surface of
functionalized
substrate 30 and taken-up as roll 29.
In one desired embodiment, after exposure to an electron beam, UV curing, and
exiting chamber 40, removable carrier layer 22 and removable cover layer 19
are allowed
to remain on functionalized substrate 30 for a period of time prior to peeling
step 400 so as
to provide prolonged protection of functionalized substrate 30 from exposure
to oxygen.
Desirably, removable carrier layer 22 and removable cover layer 19 remain on
functionalized substrate 30 for at least 15 seconds, more desirably, for about
30 to about
60 seconds after exiting chamber 25. However, there is no upper time limit
that will
reduce grafting quality and multilayer sandwich structure 27 can remain intact
for an
extended time period as would be the case if batch processing rolls of
multilayer sandwich
- 25 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
structure 27 are prepared. Once multilayer sandwich structure 27 is
disassembled,
functionalized substrate 30 can proceed to an optional washing/rinsing step
600.
In optional washing/rinsing step 600, functionalized substrate 30 is washed or
rinsed one or more times in rinse chamber 31 to remove any unreacted monomers,
solvent
or other reaction by-products from functionalized substrate 30. Typically,
functionalized
substrate 30 is washed or rinsed up to three times using a water rinse, an
alcohol rinse, a
combination of water and alcohol rinses, and/or a solvent rinse (e.g.,
acetone, MEK, etc).
When an alcohol rinse is used, the rinse may include one or more alcohols
including, but
not limited to, isopropanol, methanol, ethanol, or any other alcohol that is
practical to use
and an effective solvent for any residual monomer. In each rinse step,
functionalized
substrate 30 may pass through a rinse bath or a rinse spray.
In optional drying step 700, functionalized substrate 30 is dried to remove
any
rinse solution from functionalized substrate 30. Typically, functionalized
substrate 30 is
dried in oven 32 having a relatively low oven temperature for a desired period
of time
(referred to herein as "oven dwell time"). Oven temperatures typically range
from about
60 C to about 120 C, while oven dwell times typically range from about 120 to
about 600
seconds. Any conventional oven may be used in optional drying step 700 of the
present
invention. Suitable ovens include, but are not limited to, a convection oven.
It should also be noted that in other embodiments drying step 700 can proceed
before washing/rinsing step 600 eliminating volatile components before
extraction of non-
grafted residue.
Following optional drying step 700, dried hydrophilic substrate 30 can be
taken up
in roll form as roll 33 in step 800. Hydrophilic substrate 30 may be stored
for future use in
roll form, used immediately as is, or further processed to further alter the
surface
properties of hydrophilic substrate 30.
In one exemplary embodiment, hydrophilic substrate 30 is further processed to
alter the surface properties of hydrophilic substrate 30. In this embodiment,
functionalized
substrate 30 is processed through a grafting process such as exemplary method
10 for a
second time (or even more times) in order to (i) graft additional grafting
monomers onto
interstitial and outer surfaces of functionalized substrate 30, (ii) graft
additional monomers
onto grafted species extending from interstitial and outer surfaces of
functionalized
substrate 30, or (iii) both (i) and (ii).
- 26 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
For example, in one exemplary embodiment, functionalized substrate 30 is
prepared by imbibing a porous base substrate with a first solution comprising
one or more
grafting monomers in a solvent, wherein the one or more grafting monomers
comprise at
least one grafting monomer having an acrylate group and a photoinitiator group
thereon;
and then exposing the porous base substrate imbibed with the first solution to
a controlled
amount of electron beam radiation so as to graft the photoinitiator monomers
to interstitial
and outer surfaces of the porous base substrate.
The resulting first functionalized substrate is rinsed to remove any unreacted
grafting monomer, and may then subsequently imbibed with a second solution
comprising
one or more grafting monomers in a solvent, wherein the one or more grafting
monomers
comprise at least one grafting monomer having and acrylate group for grafting
and at least
one additional ethylenically unsaturated group for subsequent UV crosslinking;
and then
exposing the first functionalized substrate imbibed with the second solution
to a controlled
amount of electron beam radiation to form a second functionalized substrate
having both
photoinitiator groups and ethylenically unsaturated polymerizable groups.
The further modified functionalized substrate 30 can then proceed through an
optional washing/rinsing step, such as exemplary washing/rinsing step 500 in
exemplary
method 10, and an optional drying step, such as exemplary drying step 600 in
exemplary
method 10. Subsequent to the two-step grafting process, the imbibed substrate
can be
further processed by the UV irradiation step.
In optional heating step (not shown), hydrophilic substrate 30 is heated.
Typically,
during the optional heating step, hydrophilic substrate 30 is subjected to an
oven having an
oven temperature of up to about 120 C depending on a number of factors
including, but
not limited to, the reactants, the porous base substrate, the functional
groups present on the
grafted species, and the dwell time within oven 36. Typically, the oven
temperature used
in optional heating step is 30 C of greater (desirably, 40 C or greater, 50 C
or greater, or
60 C or greater). The oven temperature typically ranges from about 60 C to
about 120 C.
Typically, oven dwell time in optional heating step ranges from about 60
seconds to about
1 hour.
Any conventional oven may be used in the optional heating step of the present
invention, such as optional heating step. Suitable ovens include, but are not
limited to, the
above-described ovens used in optional drying step 600 of exemplary method 10.
- 27 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
Desirably, the oven used in optional heating step 800 of exemplary method 50
comprises a
circulating air oven.
The hydrophilic substrate 33 may be stored for future use in roll form, used
immediately as is, or further processed in one or more additional process
steps (not
shown). Suitable additional process steps may include, but are not limited to,
a reaction
step or a coating step wherein a coating composition is applied to further
hydrophilic
substrate 35, a lamination step wherein one or more additional layers are
temporarily or
permanently joined to further hydrophilic substrate 33, an assembling step
wherein further
hydrophilic substrate 33 is combined with one or more additional components to
form a
finished product (e.g., a filter assembly), a packaging step wherein further
hydrophilic
substrate 33 or a finished product comprising further hydrophilic substrate 33
is packaged
within a desired packaging material (e.g., a polyethylene film or bag), or any
combination
thereof
The methods of making functionalized substrates of the present invention may
also
be described by one or more process parameters including, but not limited to,
the process
parameters provided below.
1. Batch Versus Continuous Process
It should be noted that the methods of making functionalized substrates of the
present invention may be performed using a continuous process, such as
exemplary
method 10 shown in FIG. 1, or alternatively, using a batch process wherein one
or more of
the above-described process steps are performed separate from one another.
Desirably, the
methods of making functionalized substrates are performed using a continuous
process,
such as exemplary method 10 shown in FIG. 1.
2. Line Tension
When using a continuous process, such as exemplary method 10, one or more
drive
rolls (not shown) may be used to move porous base substrate 12 or
functionalized
substrate 30 through the continuous process. The one or more drive rolls
provide sufficient
tension on porous base substrate 12 and functionalized substrate 30 to move
porous base
substrate 12 and functionalized substrate 30 through a given apparatus. Care
should be
taken when determining the amount of tension to apply in order to prevent
shrinkage
and/or tearing of porous base substrate 12 or functionalized substrate 30
during
processing. If a stronger carrier web (e.g., removable carrier layer 22) is
used to convey
- 28 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
porous base substrate 12 or functionalized substrate 30, then the tension load
is easier to
adjust without transmitting the tension load through the substrate itself.
In the exemplary continuous grafting process of the present invention, the one
or
more drive rolls typically operate in a range of 5 to 40 lbs (22 to 178
Newtons) of tension on a (12 inch) 30 cm wide web of porous base substrate 12
or
functionalized substrate 30 in order to move porous base substrate 12 or
functionalized
substrate 30 through a given apparatus, resulting in a tension of 0.7 to 5.9
Newtons per
lineal centimeter of porous base substrate 12 or functionalized substrate 30.
In one desired
embodiment, the one or more drive rolls operate in a range of 1.4 to 3.0
Newtons per
lineal centimeter of porous base substrate 12 or functionalized substrate 30.
3. Line Speed
In the exemplary continuous grafting process of the present invention, the one
or
more drive rolls also provide a desired line speed through a given apparatus.
Desirably,
porous base substrate 12 and functionalized substrate 30 move through a given
apparatus
at a line speed of at least about 1.52 meters/minute (mpm) (5 fpm). In one
desired
embodiment, porous base substrate 12 and functionalized substrate 30 move
through a
given apparatus at a line speed ranging from about 3.05 mpm (10 fpm) to about
30.5 mpm
(100 fpm).
The disclosed methods of making functionalized substrate may be used to
prepare
a variety of hydrophilic substrates. The hydrophilic substrates have a
polymerized coating
derived from grafting followed by UV initiated polymerization from the grafted
photoinitiator (a), the monomer having an ethylenically unsaturated group (b)
, and (c)
optional other hydrophilic monomers that may be grafted or non-grafted.
In any of the above-described methods of making a functionalized substrate,
any of
the above-mentioned porous base substrates, grafting monomers, and reactants
may be
used to form a given functionalized substrate. The porous base substrate is
often in the
form of a porous membrane such as a microporous membrane, a nonwoven web, or
porous
fibers. In some embodiment, the porous base substrate comprises a hydrophobic
microporous membrane formed by a thermally-induced phase separation (TIPS)
method.
In one embodiment, the methods provide a porous article having a hydrophilic
polymer coating on the surface thereof, the polymer coating comprising the UV
polymerization reaction product of a grafted photoinitiator group and one or
more
- 29 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
ethylenically unsaturated polymerizable monomers, which may be ungrafted
acrylate
groups or other non-acrylate ethylenically unsaturated polymerizable groups.
In another
embodiment, the methods provide a porous article having a hydrophilic polymer
coating
on the surface thereof, the polymer coating comprising the UV polymerization
reaction
product of a grafted photoinitiator group, a grafted monomer having and one or
more non-
acrylate ethylenically unsaturated polymerizable groups, and one or more
ethylenically
unsaturated polymerizable monomers.
The method of making a hydrophilic substrate alters the original hydrophobic
nature of the porous base substrate, as the grafted and UV polymerized species
include a
hydrophilic group. In one embodiment, the grafting monomer having a first
grafting
acrylate group and a second non-grafting ethylenically unsaturated group may
comprise a
hydrophilic groups, as illustrated in Formula IV (supra).
For example, poly(alkylene oxide) compounds of Formula IV can be used to
impart a hydrophilic character to a hydrophobic porous base substrate. These
grafting
monomers have an acrylate group, a non-acrylate ethylenically unsaturated
group and a
hydrophilic polyalkylene glycol (i.e., polyalkylene oxide) group.
Alternatively grafting
monomers of Formula II may be used which do not contain the hydrophilic
polyalkylene
glycol (i.e. poly(alkylene oxide)) group. In these instances, hydrophilicity
is imparted
using a third monomer, which may contain a grafting acrylate group or a non-
acrylate
ethylenically unsaturated group, and a hydrophilic group, such as a quaternary
ammonium
group.
The present invention enables the formation of functionalized substrates
having
many of the advantages of a hydrophobic porous bases substrate (e.g., a
hydrophobic
microporous membrane), but with permanent hydrophilicity on the surfaces of
the
functionalized substrate. The present invention reduces or eliminates many of
the known
problems associated with porous bases substrates formed from hydrophilic
polymers
including, but not limited to, hygroexpansive issues; brittleness without
humidification
problems; mechanical strength weakness; and poor solvent, caustic and/or
acidic
resistance. The present invention also enables the formation of functionalized
substrates
having various degrees of hydrophilicity depending on the materials and steps
used to
form a given functionalized substrate.
- 30 -
CA 02702291 2014-09-03
60557-8125
The hydrophilic porous membranes are particularly suited as filter media, such
as
the filter media found in water filtration devices. As the polymer is grafted,
either directly
or indirectly, to render it hydrophilic, the filter media is durable. In many
water filtration
media, such as filter cartridges, the filter media is cleaned or sanitized by
contact or
flushing with aqueous NaOH. The hydrophilic porous substrate described herein,
can be
contacted or flushed with NaOH and retain the hydrophilic properties as
evidenced by the
surface energy and wettability.
The present invention is described above and further illustrated below by way
of
examples, which are not to be construed in any way as imposing limitations
upon the
scope of the invention. On the contrary, it is to be clearly understood that
resort may be
had to various other embodiments, modifications, and equivalents thereof
which, after
reading the description herein, may suggest themselves to those skilled in the
art without
departing from the scope of the present invention.
Examples
Materials
"VAZPIA" refers to 2-propenoylaininoethanoic acid, 2-(4-(2- hydroxy-2
methylpropanoyl)phenoxy)ethyl ester prepared according to Example 1 of U. S.
Patent
No. 5,506,279 (Babu et al.).
"PEG 400" Polyethyleneglycol, molecular weight 400, Aldrich Chemical Co.
"LUCIRIN TPO" is s 2,4,6-trimethylbenzoy diphenyl phosphine oxide, available
from BASF, Charlotte, N.C.
Electron beam irradiation was carried out using a Model CB-300 electron beam
system, obtained from Energy Sciences, Inc., Wilmington, MA. The film samples
were
placed between two sheets of poly(ethylene terephthalate) film for the
irradiation.
The following procedure was adhered to unless otherwise specified. Samples of
film were
, placed between two larger area size pieces of 4-mil thick PET and taped
together at one
end. This sandwich was then opened and the sample film was wetted with monomer
solution and the sandwich reclosed. Trapped air bubbles were removed and
excess liquid
was squeezed out by gently applying a rubber roller over the surface of the
sandwich. The
sandwich was taped to a moving web of PET and conveyed through the electron
beam
processor at a speed of 20 fpm and at a voltage of 300 keV with sufficient
beam current
-31-
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
applied to the cathode to deliver the targeted dose. The beam was calibrated
using thin
film dosimeters, calibrated and traceable to a national standards laboratory
(RISO,
Denmark). In some cases, to lower the overall dose rate and increase residence
time while
under the beam, the dose was fractionated by multiple passes through the beam
to simulate
a longer exposure time more characteristic of electron beams with cathodes
extended in
the web direction (i.e. BroadBeam, etc).
Water Flux Test
Water flux was determined by placing a disk of the test film having a diameter
of
approximately 47 millimeters (1.85 inches) in a Model 4238 Pall Gelman
magnetic filter
holder (available from Pall Corp., East Hills, NY). The filter holder was then
placed on a
filter flask that was attached to a vacuum pump. A vacuum gauge was used to
monitor the
vacuum. Approximately 150 milliliters of water was placed in the filter holder
and then
vacuum was applied. After approximately 50 milliliters of water passed through
the film
(the vacuum gauge at this time indicated approximately 0.83 millimeters of
mercury
(approximately 21 inches of mercury), timing was commenced using a stopwatch.
When
all of the remaining water had passed through the film, timing was stopped.
The water flux
was the time, measured in seconds, that elapsed for 100 milliliters of water
to pass through
the membrane under a vacuum of 0.83 millimeters of mercury.
Average Pore Diameter
The principle for determining average pore diameter is by allowing a wetting
liquid to spontaneously fill the pores in the sample membrane and then using a
non-
reacting gas to displace the liquid from the pores of the membrane, where the
gas pressure
and flow rates are accurately measured. An Automated Capillary Flow Porometer,
model
number APP-1200-AEX with supplied software, Capwin version 6.71.54 from Porous
Materials Inc. (PMI) of Ithaca New York was used to obtain these values.
Fluorinert FC-
43 (available from 3M) was used as the wetting fluid and compressed nitrogen
gas was
used for displacement with a maximum pressure setting of 100 psi (689 kPa).
Penetrating Drop Method:
The surface energy of the samples was measured using. Dyne Test SolutionsTM
available from Jemmco LLC., Mequon WI 53092 (general test method disclosed in
- 32 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
Journal of Membrane Science, 33(1987) 315-328 Wetting Criteria For The
Applicability
of Membrane Distillation). A series of the test solutions of increasing
surface tension are
applied to the samples to until the solution beads up on the sample surface.
The surface
tension is then recorded.
The hydrophilic substrates of the present invention can exhibit various
degrees of
wettability upon exposure to various solutions or solvents. Wettability can
often be
correlated to the hydrophilic character of the hydrophilic substrate. As used
herein, the
term "instant wet" or "instant wettability" refers to the penetration of
droplets of water
into a given substrate as soon as the water contacts the substrate surface,
typically within
less than 1 second. For example, a surface wetting energy of about 72 dynes or
larger
usually results in instant wetting. As used herein, the term "no instant wet"
refers to
penetration of droplets of water into a given substrate but not as soon as the
water contacts
the substrate surface. As used herein, the term "no wetting" refers to the
lack of
penetration of droplets of water into a given substrate. For example, a
surface wetting
energy of about 60 dynes or less usually results in no wetting.
The hydrophilic substrates also exhibit resistance to multiple exposures to
heat.
Example 1
A sample of a thermally induced phase separation (TIPS) microporous
polypropylene film was prepared using the method described in U.S. 4,726,989
(Mrozinski). The TIPS film had the following properties: about 4.5 mils thick,
Gurley (air
flow) about 6 sec/50cc air with an pore size range of about 0.44 0.8 microns,
has a surface
wetting energy of about 35 dynes (using JEMMCO LLC solutions for the
penetrating drop
method) and has a water flux of 25 sec (47 mm holder, 23 in Hg vacuum, IPA
prewet).
The sample was imbibed with a solution of 10% PEG 400 diacrylate with 0.5%
VAZPIA
(added to solids) in methanol. The samples were conveyed through the beam on a
web
carrier and were sandwiched 'wet' between layers of 4 mil PET in order to
delay the
diffusion of oxygen back into the membranes when they exited the beam chamber.
The sandwiched sample was irradiated by E-beam on an ESI CB-300 electron
beam with a dose of 10 kGy set at a voltage of 300 keV. The samples (still
sandwiched)
were then UV irradiated with a Spectroline model SP-100P 365 nm light for 20
minutes.
- 33 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
Following UV irradiation, the grafted, crosslinked TIPS sample was soaked in a
tray of water and exchanged three times with clean water to wash the sample.
The sample
was dried with an air gun with low heat and then heated to 60 C for 1/2 hour
in an oven.
The resulting porous film sample was instantly wettable with water. As used
herein, the
term "instant wet" or "instant wettability" refers to the penetration of
droplets of water
into a given substrate as soon as the water contacts the substrate surface,
typically within
less than 1 second.
Comparative Example 2
This comparative example was prepared as in Example 1 except no VAZPIA was
imbibed and the sample was not subsequently UV irradiated. The sample after
drying and
heating was not spontaneously wettable, indicating the TIPS sample was
insufficiently
grafted to render the film hydrophilic. Using the penetrating drop method for
determining
surface wetting energy, the surface wetting energy was now found to be about
56 dynes.
The increase in surface wetting energy (relative to the starting TIPS sample)
indicates
some grafting to the membrane was initiated.
Example 3
This example was prepared as in Example 1 except the E-beam was 5 kGy. The
sample, after drying and heating, was spontaneously wettable, indicating
sufficient
polymerization of the PEG 400 diacrylate (from UV cure and E-beam grafting) to
render
the film hydrophilic. Using water for the penetrating drop method for
determining surface
wetting energy (desired hydrophilicity) the surface wetting energy was found
to be at or
above 72 dynes.
Comparative Example 4
This comparative example was prepared as in Example 3 except no VAZPIA was
imbibed and the sample was not subsequently UV irradiated. The sample after
drying and
heating was not spontaneously wettable or with vacuum (pressure) assistance,
indicating
insufficient grafting to render the film hydrophilic. Using the penetrating
drop method for
determining surface wetting energy, the surface wetting energy was found to be
about 42
- 34 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
dynes. The increase in surface wetting energy (relative to the starting TIPS
sample)
indicates some grafting to the membrane was initiated.
Comparative Example 5
This comparative example was prepared as in Example 3 except no VAZPIA was
imbibed, the sample was not subsequently UV irradiated and the sample was
irradiated at
20 kGy instead of 11 kGy. The sample after drying and heating was
spontaneously
wettable, indicating sufficient grafting with the extra E-beam radiation to
render the film
hydrophilic. However, physical properties of the PP membrane are compromised ¨
the
strength was poor and would crumble after exposure to heat, indicating polymer
chain
degradation at the indicated e-beam dose.
Example 6
This example was prepared as in Example 1 except only 5% PEG400 diacrylate
was used in the imbibing solution. The sample, after drying and heating, was
not
spontaneously wettable with water, but was wettable with vacuum (pressure)
assistance,
indicating insufficient grafting occurred to render the film instantly
wettable.
Comparative Example 7
This comparative example was prepared as in Example 6 except no VAZPIA was
imbibed and the sample was not subsequently UV irradiated. The sample after
drying and
heating was not spontaneously wettable or with vacuum (pressure) assistance,
indicating
insufficient grafting occurred to render the film hydrophilic.
Example 8
This example was prepared as in Example 1 except the sample was E-beam
processed at about 0.75 Mrads and the imbibing solution contained 10% PEG 400
dimethacrylate, 2% 3-(Acryloxy)-2-hydroxypropylmethacrylate, and VAZPIA (at
.42 % to
monomer weight). The sample after washing was dried by heating in a frame at
60 C for
1/2 hour in an oven and was found to be spontaneously wettable. This indicates
sufficient
E-beam grafting and subsequent polymerization (from UV cure) to render the
film
hydrophilic.
- 35 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
Using water for the penetrating drop method for determining surface wetting
energy (desired hydrophilicity), the surface wetting energy was found to be at
or above 72
dynes. The bubble point pore size was slightly reduced to about .38 microns
and had a
water flux of about 30 seconds when coated and irradiated with the tight pore
size up
(bubble point of .36 microns and water flux of 43 seconds tight pore side
down, (no IPA
pre-wetting needed)). Two separate pieces of this sample were soaked in both
1N HC1 and
1N NaOH at room temperature for 75 hours without a change in the film
properties.
Comparative Example 9
This example was prepared as in Example 8 except no VAZPIA or 3-(Acryloxy)-
2-hydroxypropylmethacrylate was added to the imbibing solution and the sample
was not
subsequently UV irradiated. The sample was dried at 55 C for an hour on an
Emerson
Speed Dryer (Thwing Albert) Model 130. After drying, the porous film was not
spontaneously wettable. The surface wetting energy was found to be 39 dynes.
Despite the
amount of hydrophilicizing monomer (PEG 400 dimethacrylate) used, grafting was
insufficient to render the film hydrophilic because of the reduced reactivity
of the
methacrylate groups in the e-beam grafting step.
Comparative Example 10
This example was prepared as in Example 8 except the sample was irradiated at
20
kGy instead of 7.5 kGy, no VAZPIA or 3-(Acryloxy)-2-hydroxypropylmethacrylate
was
added to the coating solution, the sample was not subsequently UV irradiated
and was
dried on the Speed Dryer as in Example 9. After drying, the sample was not
spontaneously
wettable, indicating insufficient grafting to render the film hydrophilic,
despite with the
extra E-beam radiation, as the methacrylate groups are less reactive than the
acrylate
groups in the grating step (compare with Example 5). However, with the extra
radiation,
the surface wetting energy was found to be slightly better than Example 9,
measured at 45
dynes.
Example 11
This example was prepared as in Example 8 (E-beam dose of 7.5 kGy) except the
VAZPIA was added 1.0 % to monomer weight. After washing, the sample was dried
by
- 36 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
heating in a frame at 60 C for 1/2 hour in an oven and was found to be
spontaneously
wettable, indicating that after E-beam grafting there was sufficient
polymerization (from
UV cure) to render the film hydrophilic.
Using water for the penetrating drop method for determining surface wetting
energy (desired hydrophilicity), the surface wetting energy was found to be at
or above 72
dynes. The bubble point pore size was not reduced and remained at about .44
microns and
had a water flux of about 25 seconds (coated and irradiated with the tight
pore size down).
It is surmised that, during the UV cure, the larger amount of grafted VAZPIA
generated more free radical initiation sites on the membrane substrate. This
effectively
limited the grafted chain length as the supply of monomer in solution was
depleted,
thereby reducing or eliminating pore plugging from the coating while still
remaining very
hydrophilic.
Example 12
This example was prepared as in Example 11 except the VAZPIA was added at .25
% to monomer weight. The sample, after washing, was dried by heating it in a
frame at 60
C for 1/2 hour in an oven. The sample was not found to be spontaneously
wettable,
indicating that after E-beam grafting there was not a sufficiently high enough
concentration of initiation sites for continued polymerization (from UV cure)
to render the
film hydrophilic. Using JEMMCO LLC solutions for the penetrating drop method,
the
surface wetting energy was found to be about 64 dynes.
Example 13
This example was prepared as in Example 1 except the sample was not E-beam
radiated. The imbibing solution contained 10% PEG400 dimethacrylate and 2% 3-
(Acryloxy)-2-hydroxypropylmethacrylate, with 1.0% VAZPIA (to monomer weight)
in
methanol. After 20 minutes of UV cure, the sample, after washing and drying,
was not
spontaneously wettable, indicating polymerization from the UV cure alone was
insufficient to render the film hydrophilic.
- 37 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
Example 14
This example was prepared as in Example 1 except the sample was not E-beam
radiated. The imbibing solution contained 11.5% PEG400 dimethacrylate and 4% 3-
(Acryloxy)-2-hydroxypropylmethacrylate, with 2.0% VAZPIA (to monomer weight)
in
methanol. After 4 minutes of UV cure with Quantum Technologies (Quant 48) UVA
lamps, the sample, after washing and drying, was not spontaneously wettable,
indicating
polymerization from the higher intensity UV cure was insufficient to render
the film
hydrophilic.
Comparative Example 15
This example was prepared as in Example 1 except the sample was not E-beam
radiated. The imbibing solution contained 11.5% PEG400 dimethacrylate and 4% 3-
(Acryloxy)-2-hydroxypropylmethacrylate, with 1.0% Lucerin TPO to monomer
weight (a
non-grafting photoinitiator)) in methanol. After 4 minutes of UV cure with
Quantum
Technologies (Quant 48) UVA lamps, the sample, after washing and drying, was
not
spontaneously wettable. This indicates polymerization from the higher
intensity UV cure
in this system and more efficient photo initiator was not enough to make the
film
hydrophilic.
Example 16
This example was prepared as in Example 1 except the imbibing solution
contained 10% PEG400 dimethacrylate, no 3-(Acryloxy)-2-
hydroxypropylmethacrylate,
with 1.0% VAZPIA (to monomer weight) in methanol. After E-beam and UV
processing,
washing and drying, the sample was not spontaneously wettable, indicating
insufficient E-
beam grafting or polymerization (from the UV cure) occured to render the film
hydrophilic.
Example 17
This example was prepared as in Example 1 except the imbibing solution
contained 11.5% PEG400 dimethacrylate, 4% PEG400 diacrylate with 2.0% VAZPIA
(to
monomer weight) in methanol. After E-beam and UV processing, washing and
drying, the
sample was spontaneously wettable, indicating the E-beam grafting sufficiently
modified
- 38 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
the surface for polymerization from the UV cure to render the film
hydrophilic. The
significance of having a faster grafting acrylate or diacrylate in the coating
formula is
demonstrated.
Comparative Example 18
This example was prepared as in Example 1 except the imbibing solution
contained 11.5% PEG400 dimethacrylate, and 1.4% Lucerin TPO (to monomer
weight) in
methanol. After E-beam and UV processing, washing and drying the sample was
not
spontaneously wettable, indicating the low dose E-beam grafting did not modify
the
surface enough for subsequent polymerization from the UV cure to make the film
hydrophilic, in the absence of a grafting photoinitiator.
Example 19
This example was prepared as in Example 1 except the imbibing solution
contained 12% acrylic acid, 4% 3-(acryloxy)-2-hydroxypropylmethacrylate with
2%
VAZPIA (to monomer weight) in methanol. After E-beam and UV processing,
washing
and drying, the sample was spontaneously wettable, indicating the E-beam
grafting
modified the surface enough for polymerization from the UV cure to render the
film
hydrophilic.
Example 20
This example was prepared as in Example 1, except the imbibing solution
contained 12% acrylic acid and 4% 3-(acryloxy)-2-hydroxypropylmethacrylate,
with 2.0%
VAZPIA (to monomer weight) in methanol. After E-beam and 4 minutes of UV cure
with
Quantum Technologies (Quant 48) UVA lamps, washing and drying, the sample was
spontaneously wettable.
Example 21
This example was prepared as in Example 1 except the the imbibing solution
contained 12% N-vinyl pyrrolidone, 4% 3-(acryloxy)-2-hydroxypropylmethacrylate
with
2% VAZPIA (added to solids) in methanol. After E-beam and UV processing,
washing
and drying, the sample was wettable, but not as complete as other samples.
- 39 -
CA 02702291 2010-04-09
WO 2009/048933
PCT/US2008/079176
Comparative Example 22
This example was prepared as in Example 1 except the sample was not E-beam
radiated, and the imbibing solution contained 12% N-vinyl pyrrolidone and 4% 3-
(Acryloxy)-2-hydroxypropylmethacrylate, with 2.0% VAZPIA (to monomer weight)
in
methanol. After 4 minutes of UV cure with Quantum Technologies (Quant 48) UVA
lamps, washing and drying, the sample was not spontaneously wettable.
Example 23
A sample of TIPS porous polypropylene (PP) film was made using methods
disclosed in U.S. 4,726,989 (Mrozinski), where the oil diluent was extracted
before
stretching. The porous membrane has a surface wetting energy of about 35 dynes
measured using JEMMCO LLC solutions for the Penetrating Drop Method and has a
water flux time of 46 sec (100 ml, 47 mm Gelman Magnetic Filter Funnel (4238),
21
inches Hg vacuum, IPA prewet).
The porous polypropylene TIPS sample was imbibed with a solution of 9% PEG
400 dimethacrylate, 2% 3-(acryloxy)-2-hydroxypropylmethacrylate with 1.0%
VAZPIA
photoinitiator (to monomer weight) in methanol. The PP membrane sample was
sandwiched 'wet' between layers of 4 mil PET film with any excess solution
squeezed out
with a hand held rubber roller. The assembly was conveyed through the beam on
a carrier
web. (The PET covers delay the diffusion of oxygen back into the membranes
when they
exit the beam chamber.) The sandwiched assembly was irradiated by E-beam on an
ESI
CB-300 electron beam with a dose of 7.5 kGy set at a voltage of 300 keV. The
samples
(still sandwiched) were then UV irradiated with a Spectroline model SP-100P
365 nm
light for 10 minutes on each side.
Following UV irradiation, the grafted, crosslinked PP sample was removed from
the PET covers, soaked in a tray of water and that was exchanged three times
with clean
water to wash the sample. The sample was mounted on a frame and dried by
heating to 60
C for 1/2 hour in an oven. The resulting hydrophilic porous film sample was
instantly
wettable with a drop of water. The starting films average pore size was
measured at .51
microns compared with the finished product's average pore size at .56 microns
indicating
- 40 -
CA 02702291 2010-04-09
WO 2009/048933 PCT/US2008/079176
no pore plugging occurred from the grafting process. (The very slight pore
size expansion
is well within experimental error and sampling film variations.)
The framed PP sample was steam autoclaved for three half hour cycles at 121 C
and found to be still instantly water wettable after exposure. A 47 mm disk
was cutout and
the flux time was essentially unchanged measuring 48 seconds. The grafted
hydrophilic PP
disk was then placed into a 40 ml vial and filled with .625 N NaOH and heated
to 60 C
for 300 minutes. The sample was removed from the vial and thoroughly washed
with
water and dried. The sample was still instantly wettable with a drop of water.
-41 -