Note: Descriptions are shown in the official language in which they were submitted.
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-1-
GLUCAGON ANTAGONISTS
BACKGROUND
Native glucagon is a 29 amino acid peptide that regulates blood glucose levels
through enhanced synthesis and mobilization of glucose in the liver.
Consequently, the
suppression of endogenous glucagon action has been a target for the
development of drugs to
treat conditions characterized by excessive glucose production, such as
diabetes.
Glucagon generally functions as a counter-regulatory hormone, opposing the
actions
of insulin, to maintain the level of blood glucose, particularly in instances
of hypoglycemia.
However, in some patients with Type 1 or Type 2 diabetes, absolute or relative
elevated
glucagon levels have been shown to contribute to the hyperglycemic state. Both
in healthy
control animals,as well as in animal models of Type 1 and Type 2 diabetes,
removal. of
circulating glucagon with selective and specific antibodies has resulted in
reduction of the
glycemic level'(Brand et al., Diabetologia 37, 985 (1994); Diabetes 43, [suppl
1], 172A
(1994); Am. J. Physiol. 269, E469-E477 (1995); Diabetes 44 [suppl 1], 134A
(1995); .
Diabetes 45, 1076 (1996)). These studies suggest that glucagon antagonism
could be useful
in glycemic control of diabetes.
Glucagon exerts its action by binding to and activating its receptor, which is
part of
the glucagon-secretin branch of the 7-transmembrane G-protein coupled receptor
family.
The receptor functions by an activation of the adenylyl cyclase resulting in
increased cAMP
levels. Previous reports have identified peptide-based, (see Unson, C. G. et
al. (1989) J. Biol.
Chem. 264, 789-94, Ahn, J. et al. (2001) J. Peptide Research 58, 151-8 and Ahn
J. et al.
(2001) J. Med. Chem. 44, 1372-9) as well as nucleotide-based glucagon
antagonists (Sloop
K. et al. (2004) J. Clinical Invest. 113, 1571-81). Peptide-based inhibition
acts at the level of
receptor binding while the latter functions by suppressing intracellular mRNA
specific to the
glucagon receptor.
Inhibitors of the glucagon receptor have been described, and are generally
based on
the amino acid sequence of glucagon. Several analogues in which one or more
amino acids
were either deleted or substituted to produce potent antagonists of glucagon
receptor have
been described, for example, [des HisI] [Glue]-glucagon amide (Unson et al.,
(1989) Peptides
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-2-
10, 1171; Post et al., (1993) Proc. Natl. Acad. Sci. USA 90, 1662), des His1,
Phe6 [Glu9]-
glucagon amide (Azizh et al., (1995) Bioorg. & Med. Chem. Lett. 16, 1849) and
Nle9,
Ala11,16-glucagon amide (Unson et al. (1994) J. Biol. Chem. 269(17), 12548).
Other
analogues include substitutions at positions 4 (Ahn J Met al. (2001) J. Pept.
Res. 58(2):151-
8), 1 (Dharanipragada, R. et al. (1993) Int. J. Pept. Res. 42(1): 68-77) and
4, 5, 12, 17 and 18
in the glucagon sequence (Gysin B et al. 1986. Biochemistry. 25(25):8278-84).
As described herein, high potency glucagon antagonists are provided that
represent
modifications of the native glucagon peptide. More particularly, the novel
glucagon
antagonist represent novel chemical modifications of the N-terminus of the
native glucagon
sequence, producing a highly specific antagonist that exhibits no apparent
agonist activity.
These compounds can be used in any setting where the suppression of glucagon
agonism is
desired. In accordance with one embodiment the compounds can be used in the
treatment of
diabetes.
SUMMARY
In accordance with one embodiment, analogs of glucagon are provided that have
pure
glucagon antagonist activities. The glucagon antagonists would be used in any
setting where
the suppression of glucagon agonism is desired. The most immediate and obvious
use would
be in the treatment of diabetes where glucagon antagonism has been
demonstrated in pre-
clinical models of hyperglycemia to yield a lowering of blood glucose. These
glucagon
antagonists can be further modified to improve the biophysical stability
and/or aqueous
solubility of the compounds while maintaining the antagonist activity of the
parent
compound.
In accordance with one embodiment a glucagon antagonist is provided comprising
a
native glucagon peptide that has been modified by the deletion of the first 2 -
5 amino acids
residues from the N-terminus, and a substitution of the aspartic acid at
position 9 of the
native peptide with an amino acid selected from the group consisting of
glutamic acid,
homoglutamic acid, (3-homoglutamic acid, a sulfonic acid derivative of
cysteine, or an
alkylcarboxylate derivative of cysteine having the structure of:
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-3-
H2N COOH
H2C
X5
COOH,
wherein X5 is CI-C4 alkyl, C2-C4 alkenyl, or C2-C4 alkynyl.
In one embodiment, the sulfonic acid derivative.of cysteine is cysteic acid or
homocysteic acid. In one embodiment the natural carboxylic acid of the C-
terminal amino
acid is replaced with a charge-neutral group, such as an amide or ester.
In another embodiment a glucagon antagonist is provided (referred to herein as
the
PLA6 analog) comprising a native glucagon peptide that has been modified by
the deletion of
the first 5 amino acids residues from the N-terminus, and modification of the
remaining N-
terminal amino acid (phenylalanine) to replace the native N-terminal amino
group with a
hydroxyl group (i.e., an N-terminal phenyl-lactic acid (PLA)). The PLA6 analog
increases
the affinity threefold, and the potency of antagonism, relative to an analog
that comprises a
deletion of the first five amino acids but retains the N-terminal native
phenyalanine. In a
further embodiment the aspartic acid residue at position nine of the native
protein is
constituted with an amino acid selected from the group consisting of aspartic
acid, glutamic
acid, homoglutamic acid, (3-homoglutamic acid, a sulfonic acid derivative of
cysteine, or an
alkylcarboxylate derivative of cysteine having the structure of:
H2N COOH
H2C
X5
COOH,
wherein X5 is CI-C4 alkyl, C2-C4 alkenyl, or C2-C4 alkynyl.
In one embodiment the glucagon antagonist comprises a sequence selected from
the
group consisting of SEQ ID NO: 6, SEQ ID NO: 7, SEQ ID NO: 8, SEQ ID NO: 36,
SEQ ID
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-4-
NO: 37 and SEQ ID NO: 38, and in one embodiment the glucagon antagonist
comprises the
sequence of SEQ ID NO: 7 or 8.
In accordance with one embodiment a glucagon antagonist is provided comprising
the
sequence of SEQ ID NO: 7 or SEQ ID NO: 37 wherein a polyethylene glycol chain
is
covalently linked to an amino acid at position 11, 12, 16, 19 or 24, or at the
N- or C-terminal
amino acid of the peptide. In one embodiment a glucagon antagonist is provided
comprising
the sequence of SEQ ID NO: 9, SEQ ID NO: 10, SEQ ID NO: 11 or SEQ ID NO: 12,
wherein a polyethylene glycol chain is covalently linked to an amino. acid at
position 11 of
SEQ ID NO: 9, position 16 of SEQ ID NO: 10, position 19 of SEQ ID NO: 11 or at
both
positions 11 and 19 for SEQ ID NO: 12. In one embodiment a single polyethylene
glycol
chain having a molecular weight selected from the range of about 1,000 to
about 5,000
Daltons is covalently bound to the glucagon antagonist peptide. In another
embodiment a
single polyethylene glycol chain having a molecular weight of at least about
20,000 Daltons
is covalently bound to the glucagon antagonist peptide. Alternatively the
glucagon
antagonist comprises the sequence of SEQ ID NO: 12 and has a polyethylene
glycol chain
covalently bound to both amino acid position 11 and 19, wherein the combined
molecular
weight of the two polyethylene chains is either about 1,000 to about 5,000
Daltons or is
greater than about 20,000 Daltons.
In one embodiment a glucagon antagonist is provided wherein the carboxy
terminal
amino acid of the peptide of SEQ ID NO: 7, SEQ ID NO: 8, SEQ ID NO: 36, SEQ ID
NO:
39 or SEQ ID NO: 40 is covalently bound to a second peptide comprising the
sequence of
SEQ ID NO: 19 (GPSSGAPPPS). These compounds may be further modified by the
covalent linkage of a PEG group to the glucagon analog at a position selected
from 11, 16, 19
for SEQ ID NO: 7, SEQ ID NO: 8, SEQ ID NO: 36, SEQ ID NO: 39 or SEQ ID NO: 40.
In
one embodiment a glucagon antagonist is provided wherein the carboxy terminal
amino acid
of the peptide of SEQ ID NO: 7, SEQ ID NO: 8, SEQ ID NO: 36, SEQ ID NO: 39 or
SEQ
ID NO: 38 is covalently bound to a second peptide comprising the sequence of
SEQ ID NO:
53.
In one embodiment, the glucagon antagonist comprises the structure A-B-C as
described herein, wherein A is selected from the group consisting of:
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-5-
(i) phenyl lactic acid (PLA);
(ii) an oxy derivative of PLA;
(iii) a peptide of 2 to 6 amino acids in which two consecutive amino
acids of the peptide are linked via an ester or ether bond;
B represents amino acids i to 26 of SEQ ID NO: 1, wherein i is 3, 4, 5, 6, or
7,
optionally comprising one or more amino acid modifications, as further
described herein;
and C is selected from the group consisting of:
(x) X;
(xi) X-Y;
(xii) X-Y-Z; and
(xiii) X-Y-Z-R10,
wherein X is Met, Leu, or Nle; Y is Asn or a charged amino acid; Z is Thr,
Gly, Cys, Lys,
ornithine (Orn), homocysteine, acetyl phenylalanine (Ac-Phe), or a charged
amino acid;
wherein R10 is selected from a group consisting of SEQ ID NOs: 19-21 and 53;
and
(xiv) any of (x) to (xiii) in which the C-terminal carboxylate is
replaced with an amide.
In another embodiment the solubility of any of the preceding glucagon
antagonists
which lacks one to five N-terminal amino acids of native glucagon and/or
comprises PLA
can be improved by modifying the peptide by substitutions and/or additions
that introduce a
charged amino acid into the C-terminal portion of the peptide, preferably at a
position C-
terminal to position 22 of the glucagon antagonist. Optionally, one, two or
three charged
amino acids may be introduced within the C-terminal portion, preferably C-
terminal to
position 22 (position 27 of native glucagon). In accordance with one
embodiment the native
amino acid(s) at positions 23 and/or 24 are substituted with a charged amino
acid, and/or in a
further embodiment one to three charged amino acids are also added to the C-
terminus of the
peptide. In exemplary embodiments, one, two or all of the charged amino acids
are
negatively charged. In one embodiment the glucagon antagonist of SEQ ID NO: 7
or SEQ
ID NO: 8 is further modified to include an acidic amino acid substitution at
position 23
and/or 24. In one embodiment the acidic amino acid is an aspartic acid or
glutamic acid
residue, and the glucagon antagonist comprises the sequence of SEQ ID NO: 41.
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-6-
In a further embodiment the stability of the glucagon antagonists which lack
amino
acids 1-5 of native glucagon and/or comprises PLA at physiological pH can be
improved by
substituting the aspartic acid residue at position 10 (position 15 of native
glucagon) with an
amino acid selected from the group consisting of Glu, cysteic acid,
homoglutamic acid or
homocysteic acid. In accordance with one embodiment such a glucagon antagonist
comprises the sequence of SEQ ID NO: 36 or SEQ ID NO: 40.
Any of the glucagon antagonists described herein may be further modified to
comprise one or more a, a-disubstituted amino acids at positions that retain
the desired
activity. In some embodiments, one, two, three, four or more of positions 16,
17, 18, 20, 21,
24 or 29 (according to the amino acid numbering of wild type glucagon) of the
glucagon
antagonist is substituted with an a, a-disubstituted amino acid. For example,
the glucagon
antagonist can comprise a substitution of position 16 (according to the amino
acid numbering
of wild type glucagon) with amino iso-butyric acid (AIB). In some embodiments,
one, two,
three or more of positions 16, 20, 21 or 24 (according to the amino acid
numbering of wild
type glucagon) are substituted with AIB. In a specific aspect, the glucagon
antagonist
comprising one or more a, a-disubstituted amino acids further comprises a C-
terminal
carboxylate.
Any of the glucagon antagonists described herein may be further modified to
comprise an acyl group and/or an alkyl group, which acyl or alkyl group is
attached to an
amino acid of a spacer or the glucagon antagonist via an ester, ether,
thioether, amide, or
alkyl amine linkage, as described herein.
Any of the glucagon antagonists described herein may be further modified by
truncation or deletion of one or two amino acids of the C-terminus of the
glucagon peptide
(i.e., position 29 or positions 28 and 29 of the native glucagon peptide).
Additional modifications, e.g. conservative substitutions, may be made to the
glucagon antagonist that still allow it to retain its glucagon antagonist
activity. Thus, the
invention contemplates that any of the glucagon analogs disclosed herein can
be further
modified to comprise up to 2, 3, 4, 5, 6, 7, 8 or 9 further amino acid
modifications, and still
retain the desired level of activity of a glucagon antagonist at the glucagon
receptor.
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-7-
Dimers of the glucagon antagonists disclosed herein are also encompassed by
the
present disclosure. In one embodiment a glucagon antagonist dimer is provided
comprising
two peptides independently selected form the group consisting of SEQ ID NO: 7,
SEQ ID
NO: 8, SEQ ID NO: 36, SEQ ID NO: 37, SEQ ID NO: 39 and SEQ ID NO: 40, wherein
the
two glucagon antagonists are bound to one another through a linker
independently bound to
position 11 or 19 of the respective peptide chains.
In accordance with one embodiment a pharmaceutical composition is provided
comprising the novel glucagon antagonists disclosed herein. In one embodiment
the
pharmaceutical compositions comprise solutions that are sterilized and
contained within
various packages. The pharmaceutical compositions can be further packaged as
part of a kit
that includes a disposable device for administering the composition to a
patient.
In accordance with one embodiment a method of rapidly treating hyperglycemia
using a pre-formulated aqueous composition is provided. The method comprises
the step of
administering an effective amount of an aqueous solution comprising a novel
modified
glucagon antagonist of the present disclosure. In one embodiment the glucagon
antagonist is
pegylated and the PEG chain has a molecular weight of about 500 to about 5,000
Daltons. In
one embodiment the modified glucagon solution is prepackaged in a device that
is used to
administer the composition to the patient suffering from hyperglycemia.
In accordance with one embodiment an improved method of regulating blood
glucose
levels in insulin dependent patients is provided. The method comprises the
steps of
administering insulin in an amount therapeutically effective for the control
of diabetes and
administering a novel modified glucagon antagonist of the present disclosure,
wherein said
administering steps are conducted within twelve hours of each other. In one
embodiment the
glucagon antagonist and the insulin are co-administered as a single
composition, wherein the
glucagon peptide is pegylated with a PEG chain having a molecular weight
selected from the
range of about 5,000 to about 40,000 Daltons.
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-8-
BRIEF DESCRIPTION OF THE DRAWINGS
Fig. 1 is a bar graph representing the stability of Glucagon Cys21-
maleimidoPEG5K at
37 C incubated for 24, 48, 72, 96, 144 and 166 hours, respectively.
Fig. 2 represents data generated from HPLC analysis of Glucagon Cys21-
maleimidoPEG5K at pH 5 incubated at 37 C for 24, 72 or 144 hours,
respectively.
Fig. 3 represents data generated from a binding assay for the listed N-
terminally
truncated sulfonic acid glucagon antagonist analogs.
Fig. 4 represents data generated measuring glucagon receptor antagonism of the
listed
peptide antagonists, as measure by CAMP production.
Fig. 5A & 5B represents data generated measuring glucagon receptor antagonism
of
the listed glucagon analogs, as measure by cAMP production. More particularly,
Fig. 5A
compares induction of the glucagon receptor by glucagon analogs [E9]G(2-29) =,
[hC9(SO3H)]G(2-29) =, [hC9(SO3H)]G(4-29) V, [hC9(SO3H)]G(5-29) -4 and
[hC9(SO3H)]G(6-29) t, relative to native glucagon U. Fig. 5B provides data
regarding the
inhibitory effect of the same analogs on the induction of the glucagon
receptor by 0.2 nM of
glucagon. Abbreviations: E9 = a substitution of glutamic acid at position 9
relative to native
glucagon; G(X-29) = native glucagon N-terminally truncated by X - 1 amino
acids;
hC9(SO3H) = homocysteic acid at position 9, relative to native glucagon.
Fig. 6 represents the synthetic scheme for the synthesis of Fmoc-
homoCys(SO3Na)-
OH. Fmoc-homoCys(SO3Na)-OH dissolves well in DMF or NMP and can be directly
incorporated using DIC/HOBT or HBTU/HOBT during the automated peptide
synthesis. All
hC9(SO3H)-based glucagon antagonist analogs were synthesized by direct
incorporation of
the sodium form of Fmoc-homoCys(SO3Na)-OH in solid phase peptide synthesis.
Fig. 7 represents data comparing the binding affinity and glucagon receptor
activity
of glucagon antagonists that differ based on modifications to the N-terminal
amino acid
("residue 6" of native glucagon).
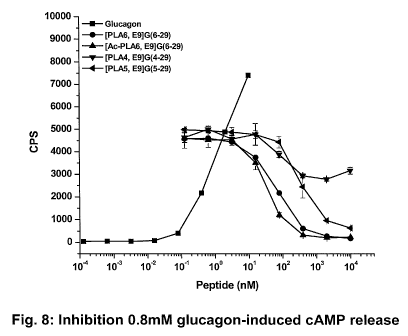
Fig. 8 represents data generated measuring the inhibitory effect of the listed
PLA
glucagon analog antagonists on the induction of the glucagon receptor by 0.8
nM of
glucagon, as measure by cAMP production. The tested PLA analogs include [PLA6,
E9]G(6-29) =, [Ac-PLA6, E9]G(6-29) A, [PLA4, E9]G(4-29) V, [PLA5, E9]G(5-29)
t,
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-9-
compared to the activity of native glucagon ^ alone. Abbreviations: E9 = a
substitution of
glutamic acid at position 9 relative to native glucagon; G(Ac-PLA) =
acetylated PLA.
DETAILED DESCRIPTION
DEFINITIONS
In describing and claiming the invention, the following terminology will be
used in
accordance with the definitions set forth below.
As used herein, the term "pharmaceutically acceptable carrier" includes any of
the
standard pharmaceutical carriers, such as a phosphate buffered saline
solution, water,
emulsions such as an oil/water or water/oil emulsion, and various types of
wetting agents.
The term also encompasses any of the agents approved by a regulatory agency of
the US
Federal government or listed in the US Pharmacopeia for use in animals,
including humans.
As used herein the term "pharmaceutically acceptable salt" refers to salts of
compounds that retain the biological activity of the parent compound, and
which are not
biologically or otherwise undesirable. Many of the compounds disclosed herein
are capable
of forming acid and/or base salts by virtue of the presence of amino and/or
carboxyl groups
or groups similar thereto.
Pharmaceutically acceptable base addition salts can be prepared from inorganic
and
organic bases. Salts derived from inorganic bases, include by way of example
only, sodium,
potassium, lithium, ammonium, calcium and magnesium salts. Salts derived from
organic
bases include, but are not limited to, salts of primary, secondary and
tertiary amines.
Pharmaceutically acceptable acid addition salts may be prepared from inorganic
and
organic acids. Salts derived from inorganic acids include hydrochloric acid,
hydrobromic
acid, sulfuric acid, nitric acid, phosphoric acid, and the like. Salts derived
from organic acids
include acetic acid, propionic acid, glycolic acid, pyruvic acid, oxalic acid,
malic acid,
malonic acid, succinic acid, maleic acid, fumaric acid, tartaric acid, citric
acid, benzoic acid,
cinnamic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, p-
toluene-sulfonic
acid, salicylic acid, and the like.
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-10-
As used herein, the term "treating" includes prophylaxis of the specific
disorder or
condition, or alleviation of the symptoms associated with a specific disorder
or condition
and/or preventing or eliminating said symptoms.
As used herein an "effective" amount or a "therapeutically effective amount"
of a
glucagon antagonist refers to a nontoxic but sufficient amount of the peptide
to provide the
desired effect. For example one desired effect would be the prevention or
treatment of
hyperglycemia. The amount that is "effective" will vary from subject to
subject, depending
on the age and general condition of the individual, mode of administration,
and the like.
Thus, it is not always possible to specify an exact "effective amount."
However, an
appropriate "effective" amount in any individual case may be determined by one
of ordinary
skill in the art using routine experimentation.
The term, "parenteral" means not through the alimentary canal but by some
other
route such as subcutaneous, intramuscular, intraspinal, or intravenous.
A "glucagon peptide" as used herein includes any peptide comprising, either
the
amino acid sequence of SEQ ID NO: 1, or any derivative of the amino acid
sequence of SEQ
ID NO: 1, including amino acid substitutions, or post translational
modifications (e.g.
methylation, acylation, ubiquitination and the like) of the peptide, that
stimulates glucagon or
GLP- 1 receptor activity, as measured by cAMP production using the assay
described in
Example 13.
The term "glucagon antagonist" refers to a compound that counteracts glucagon
activity or prevents glucagon function. For example, a glucagon antagonist
exhibits at least
60% inhibition (e.g., at least 70% inhibition) and preferably, at least 80%
inhibition, of the
maximum response achieved by glucagon at the glucagon receptor. In one
embodiment, the
glucagon antagonist exhibits at least 90% inhibition of the maximum response
achieved by
glucagon at the glucagon receptor. In a specific embodiment, the glucagon
antagonist
exhibits 100% inhibition of the maximum response achieved by glucagon at the
glucagon
receptor. Additionally, a glucagon antagonist at a concentration of about 1 M
exhibits less
than about 20% of the maximum agonist activity achieved by glucagon at the
glucagon
receptor. In one embodiment, the glucagon antagonist exhibits less than about
10% of the
maximum agonist activity achieved by glucagon at the glucagon receptor. In a
specific
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-11-
embodiment, the glucagon antagonist exhibits less than about 5% of the maximum
agonist
activity achieved by glucagon at the glucagon receptor. In yet another
specific embodiment,
the glucagon antagonist exhibits 0% of the maximum agonist activity achieved
by glucagon
at the glucagon receptor.
A "pure glucagon antagonist" is a glucagon antagonist that does not produce
any
detected stimulation of glucagon or GLP-1 receptor activity, as measured by
cAMP
production using a validated in vitro model assay, such as that described in
Example 13. For
example, a pure glucagon antagonist exhibits.less than about 5% (e.g., less
than about 4%,
less than about 3%, less than about 2%, less than about 1%, about 0%) of the
maximum
agonist activity achieved by glucagon at the glucagon receptor and exhibits
less than about
5% (e.g., less than about 4%, less than about 3%, less than about 2%, less
than about 1%, and
about 0%) of the maximum agonist activity achieved by GLP-1 at the GLP-1
receptor.
As used herein a "derivative glucagon antagonist" is a peptide that shares
greater than
60% amino acid sequence identity with the amino acid of SEQ ID NO: 1, but has
been
modified to exhibit glucagon antagonist activity. Such modifications include
amino acid
substitutions or deletions, or post translational modifications (e.g.
methylation, amidation,
acylation, ubiquitination, pegylation and the like) of the peptide In one
example, a derivative
glucagon antagonist comprises the glucagon peptide of SEQ ID NO: 1 that has
been modified
to have the first five amino acid residues deleted from the N-terminus, and
has the amino
group of the remaining N-terminal amino acid (phenylalanine) substituted with
a hydroxyl
group.
As used herein an amino acid "modification" refers to a substitution, addition
or
deletion of an amino acid, and includes substitution with or addition of any
of the 20 amino
acids commonly found in human proteins, as well as atypical or non-naturally
occurring
amino acids. Commercial sources of atypical amino acids include Sigma-Aldrich
(Milwaukee, WI), ChemPep Inc. (Miami, FL), and Genzyme Pharmaceuticals
(Cambridge,
MA). Atypical amino acids may be purchased from commercial suppliers,
synthesized de
novo, or chemically modified or derivatized from naturally occurring amino
acids.
As used herein an amino acid "substitution" refers to the replacement of one
amino acid
residue by a different amino acid residue.
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-12-
As used herein, the term "conservative amino acid substitution" is defined
herein as
exchanges within one of the following five groups:
1. Small aliphatic, nonpolar or slightly polar residues:
Ala, Ser, Thr, Pro, Gly;
II. Polar, negatively charged residues and their amides:
Asp, Asn, Glu, Gln;
III. Polar, positively charged residues:
His, Arg, Lys; Ornithine (Orn)
IV. Large, aliphatic, nonpolar residues:
Met, Leu, Ile, Val, Cys, Norleucine (Nle), homocysteine
V. Large, aromatic residues:
Phe, Tyr, Trp, acetyl phenylalanine
As used herein the general term "polyethylene glycol" or "PEG", refers to
mixtures of
condensation polymers of ethylene oxide and water, in a branched or straight
chain,
represented by the general formula H(OCH2CH2)nOH, wherein n is at least 9.
Absent any
further characterization, the term is intended to include polymers of ethylene
glycol with an
average total molecular weight selected from the range of 500 to 40,000
Daltons.
"Polyethylene glycol" or "PEG" is used in combination with a numeric suffix to
indicate the
approximate average molecular weight thereof. For example, PEG-5,000 refers to
polyethylene glycol having a total molecular weight average of about 5,000.
As used herein the term "pegylated" and like terms refers to a compound that
has
been modified from its native state by linking a polyethylene glycol polymer
to the
compound. A "pegylated glucagon antagonist" is a glucagon antagonist that has
a PEG chain
covalently bound to the glucagon antagonist.
As used herein a general reference to a peptide is intended to encompass
peptides that
have modified amino and carboxy termini. For example, an amino acid chain
comprising an
amide group in place of the terminal carboxylic acid is intended to be
encompassed by an
amino acid sequence designating the standard amino acids.
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-13-
As used herein a "linker" is a bond, molecule or group of molecules that binds
two
separate entities to one another. Linkers may provide for optimal spacing of
the two entities
or may further supply a labile linkage that allows the two entities to be
separated from each
other. Labile linkages include photocleavable groups, acid-labile moieties,
base-labile
moieties and enzyme-cleavable groups.
As used herein a "dimer" is a complex comprising two subunits covalently bound
to
one another via a linker. The term dimer, when used absent any qualifying
language,
encompasses both homodimers and heterodimers. A homodimer comprises two
identical
subunits, whereas a heterodimer comprises two subunits that differ, although
the two
subunits are substantially similar to one another.
As used herein the term "charged amino acid" refers to an amino acid that
comprises
a side chain that is negatively charged (i.e., de-protonated) or positively
charged (i.e.,
protonated) in aqueous solution at physiological pH. For example negatively
charged amino
acids include aspartic acid, glutamic acid, cysteic acid, homocysteic acid,
and homoglutamic
acid, whereas positively charged amino acids include arginine, lysine and
histidine. Charged
amino acids include the charged amino acids among the 20 amino acids commonly
found in
human proteins, as well as atypical or non-naturally occurring amino acids.
As used herein a "sulfonic acid derivative of cysteine" refers to compounds of
the
general structure:
H2N COON
X6
~SO3
wherein X6 is C1-C4 alkyl, C2-C4 alkene or C2-C4 alkynyl.
The term "C1-Cn alkyl" wherein n can be from 1 through 6, as used herein,
represents
a branched or linear alkyl group having from one to the specified number of
carbon atoms.
Typical C1-C6 alkyl groups include, but are not limited to, methyl, ethyl, n-
propyl, iso-
propyl, butyl, iso-butyl, sec-butyl, tert-butyl, pentyl, hexyl and the like.
The terms "C2-Cn alkenyl" wherein n can be from 2 through 6, as used herein,
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-14-
represents an olefinically unsaturated branched or linear group having from 2
to the specified
number of carbon atoms and at least one double bond. Examples of such groups
include, but
are not limited to, 1-propenyl, 2-propenyl (-CH2-CH=CH2), 1,3-butadienyl, (-
CH=CHCH=CH2), 1-butenyl (-CH=CHCH2CH3), hexenyl, pentenyl, and the like.
The term "C2-Cn alkynyl" wherein n can be from 2 to 6, refers to an
unsaturated
branched or linear group having from 2 to n carbon atoms and at least one
triple bond.
Examples of such groups include, but are not limited to, 1-propynyl, 2-
propynyl, 1-butynyl,
2-butynyl, 1-pentynyl, and the like.
As used herein the term "pH stabilized glucagon antagonist" refers to a
glucagon
analog that exhibits superior stability and solubility, relative to native
glucagon, in aqueous
buffers in the broadest pH range used for pharmacological purposes.
As used herein the term "acidic. amino acid" refers to an amino acid that
comprises a
second acidic moiety, including for example, a carboxylic acid or sulfonic
acid group.
As used herein the term "patient" without further designation is intended to
encompass any warm blooded vertebrate domesticated animal (including for
example, but
not limited to livestock, horses, cats, dogs and other pets) and humans.
As used herein, the term "about" as used herein means greater or lesser than
the value
or range of values stated by 10 percent, but is not intended to designate any
value or range of
values to only this broader definition. Each value or range of values preceded
by the term
"about" is also intended to encompass the embodiment of the stated absolute
value or range
of values.
EMBODIMENTS
Disclosed herein are glucagon antagonists that are highly specific to glucagon
suppression and possess no apparent agonist activity. Such glucagon
antagonists are utilized
in any setting where the suppression of glucagon's agonism is desired. For
example glucagon
antagonists can be used in the treatment of diabetes where glucagon antagonism
has been
demonstrated in pre-clinical models of hyperglycemia to yield a lowering of
blood glucose.
Specific analogs of glucagon have been developed wherein the normally
occurring
aspartic acid at position nine has been substituted with glutamic acid or a
cysteic acid-based
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-15-
derivative. More particularly, deletion of the first amino acid (des-His) and
substitution of
the aspartic acid at position 9 with glutamic acid produces a glucagon
antagonist. Glucagon
derivatives having sulfonic acid substituents substituted at amino acid
position nine of
glucagon perform similarly to the carboxylic acid-based amino acids but with a
few critical
differences in relation to physical properties such as solubility. Homocysteic
acid (hCysSO3)
when substituted for the isosteric glutamic acid at position nine in the
conventional des-His,
Glu9 glucagon antagonist retains a partial antagonist and weak agonist.
Surprisingly, applicants have discovered that the removal of additional N-
terminal
residues, including for example, the removal of the first five amino acids
(yielding des(1-5),
and substitution of position 9 (according to the numbering of SEQ ID NO: 1)
with
hCys(SO3), homoglutamic acid, P-homoglutamic acid, or an alkylcarboxylate
derivative of
cysteine having the structure of:
COOH
H:2N Y
H2C
X5
\COOH,
wherein X5 is C1-C4 alkyl, C2-C4 alkenyl, or C2-C4 alkynyl, provides a
compound that
performs as a hormonal antagonist that is highly specific, potent and without
contaminating
agonist properties. Accordingly, glucagon derivative peptides that exhibit
pure glucagon
antagonist activity are disclosed herein. In accordance with one embodiment
the glucagon
antagonist exhibits activity that reduces glucagon receptor glucagon-induced
cAMP
production by a maximum of at least 50% when the glucagon receptor is
contacted
simultaneously with 0.8nM of glucagon and the glucagon antagonist, as measured
by cAMP
production in an in vitro assay. In one embodiment, the glucagon antagonist
reduces
glucagon receptor glucagon-induced cAMP production by a maximum amount of at
least
80%.
In accordance with one embodiment a derivative glucagon antagonist is provided
that
comprises a glucagon peptide modified, relative to the wild type sequence of
SEQ ID NO: 1,
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-16-
by the deletion of two to five amino acid residues from the N-terminus and
substitution of the
aspartic acid residue at position nine of the native protein with a glutamic
acid,
homoglutamic acid, (3-homoglutamic acid, a sulfonic acid derivative of
cysteine, or an
alkylcarboxylate derivative of cysteine having the structure of:
H2N COOH
H2C
X5
COOH,
wherein X5 is C1-C4 alkyl, C2-C4 alkenyl, or C2-C4 alkynyl.
In one specific embodiment, the glucagon antagonist comprising the deletion of
two
to five amino acid residues from the N-terminus and substitution of the Asp at
position 9 of
the native glucagon, is further modified by up to three amino acid
modifications. For
example, the glucagon antagonist may comprise one, two, or three conservative
amino acid
modifications. Alternatively or additionally, the glucagon antagonist may
comprise one or
more amino acid modifications selected from the group consisting of:
A. substitution of one or two amino acids at positions 10, 20, and
24, (according to the amino acid numbering of SEQ ID NO: 1),
or the N- or C-terminal amino acid of the glucagon antagonist
with an amino acid covalently attached to an acyl group or
alkyl group via an ester, ether, thioether, amide, or alkyl amine
linkage;
B. substitution of one or two amino acids at positions 16, 17, 20,
21, and 24 (according to the amino acid numbering of SEQ ID
NO: 1), or the N- or C-terminal amino acid of the glucagon
antagonist with an amino acid selected from the group
consisting of: Cys, Lys, ornithine, homocysteine, and acetyl-
phenylalanine (Ac-Phe), wherein the amino acid of the group is
covalently bonded to a hydrophilic moiety;
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-17-
C. addition of an amino acid covalently bonded to a hydrophilic
moiety to the N- or C-terminus of the glucagon antagonist;
D. substitutution of Asp at position 15 (according to the
numbering of SEQ ID NO: 1) with cysteic acid, glutamic acid,
homoglutamic acid, and homocysteic acid;
E. substitution of Ser at position 16 (according to the numbering
of SEQ ID NO: 1) with cysteic acid, glutamic acid,
homoglutamic acid, and homocysteic acid;
F. substitution with AIB at one or more of positions 16, 20, 21,
and 24 according to the amino acid numbering of SEQ ID NO:
1;
G. deletion of the amino acid at position 29 or the amino acids at
positions 28 and 29, according to the numbering of SEQ ID
NO: 1;
H. substitution of each or both of the Asn at position 28 and the
Thr at position 29 (according to the amino acid numbering of
SEQ ID NO: 1) with charged amino acids; and/or addition of
one to two charged amino acids at the C-terminus of SEQ ID
NO: 1;
I. substitution of the Met at position 27 (according to the
numbering of SEQ ID NO: 1) with Leu or norleucine;
J. addition of a peptide having the amino acid sequence of any of
SEQ ID NOs: 19-21 and 53 to the C-terminus of SEQ ID NO:
1; wherein Thr at position 29 (according to the numbering of
SEQ ID NO: 1) is Thr or Gly; and
K. replacement of the C-terminal carboxylate with an amide or
ester.
In a specific embodiment, the glucagon antagonist comprises an amino acid
modification of A, B, or C, as described above, or a combination thereof. In
yet another
specific embodiment, the glucagon antagonist further comprises an amino acid
modification
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-18-
of any of D to K as described above, or a combination thereof, in addition to
the amino acid
modification(s) of A, B, and/or C.
In one embodiment the glucagon antagonist comprises a glucagon peptide,
wherein
the first 5 amino acids have been removed from the N-terminus, and the
remaining N-
terminal amino group has been replaced with a hydroxyl group (the "PLA6
analog"),
producing the peptide of SEQ ID NO: 39. Applicants have found that
substitution of phenyl-
lactic acid for phenylalanine in glucagon antagonist analogs that have the
first five amino
acids deleted and substitution of a glutamic acid at position 9 (relative to
native glucagon)
further enhances the potency of those glucagon antagonist analogs.
In one embodiment the glucagon antagonist peptide of SEQ ID NO: 39 is further
modified by substituting the aspartic acid residue at position four (position
9 of the native
.glucagon) with an amino acid of the general structure:
H2N COOH
X6
S03
wherein X6 is C1-C3 alkyl, C2-C3 alkene or C2-C3 alkynyl, and in one
embodiment X is C1-C3
alkyl, and in another embodiment X is C2 alkyl. In one embodiment the glucagon
antagonist
comprises a glucagon peptide, wherein the first 5 amino acids have been
removed from the
N-terminus, and the aspartic acid residue at position four (position 9 of the
native glucagon)
has been substituted with cysteic acid or homocysteic acid. In one embodiment
the glucagon
antagonist comprises a glucagon peptide comprising an amino acid sequence
selected from
the group consisting of SEQ ID NO: 39, SEQ ID NO: 7 and SEQ ID NO: 8. In one
embodiment the glucagon antagonist comprises an amino acid sequence selected
from the
group consisting of SEQ ID NO: 8, wherein the amino acid at position four is
homocysteic
acid.
In another embodiment, the glucagon antagonist of SEQ ID NO: 39 is further
modified by substituting the aspartic acid residue at position four (position
9 of the native
glucagon) with glutamic acid, homoglutamic acid, (3-homoglutamic acid, or an
alkylcarboxylate derivative of cysteine having the structure of:
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-19-
H2N COOH
H2C
X5
COOH,
wherein X5 is C1-C4 alkyl, C2-C4 alkenyl, or C2-C4 alkynyl. In a specific
embodiment, X is C1 or C2 alkyl.
However, applicants have discovered that with the substitution of the N-
terminal
phenylalanine with PLA in a desl-5 glucagon analog (i.e., a glucagon analog
having the first
five amino acids deleted), further substitution of the native aspartic acid
residue at position
four (position 9 of the native glucagon) is not required to produce an analog
that exhibits
pure antagonism. This result is surprising in light of the prior art teachings
that the native
aspartic acid residue at position four must substituted to produce high
affinity and potent
antagonists of glucagon (2-29) analogs. The use of the PLA substitution
improves the
relative potency of the Asp9 analog to a point comparable to that of the G1u9
and
hCys(SO3H)9 analogs. (see Tables 6 and 7 in the Examples).
Substitution of the phenylalanine residue with other phenylalanine analogs,
including
3,4-2F-phenylalnine (3,4-2F-Phe), 2-naphthyalanine (2-Nal), N-acyl-
phenylalanine (Ac-Phe),
alpha-methylhydrocinnamic acid (MCA) and benzylmalonic acid (BMA) did not
perform as
potently as the PLA substitution (see Fig. 7).
Substituting PLA at sites other than at position six (according to the amino
acid
numbering of native glucagon), including at positions 4 and 5 reveals that the
PLA6 analog is
an appreciably more potent antagonist than glucagon analogs having a slightly
extended N-
terminus (see the results presented in Table 8 and Fig. 8). The results
presented in Fig. 8 also
demonstrate that acylation of the PLA hydroxy group does not affect PLA6
analog potency.
Accordingly, the present invention also includes analogs wherein the N-
terminal amino
group is substituted with an acylated and alkylated "O-terminal" peptides.
Furthermore, the PLA6 substitution not only increases the potency of the
antagonist
but also serves a critical role in pegylation. The PLA6 analogs can be
selectively pegylated
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-20-
without restoration of glucagon agonism. In the absence of the PLA
substitution, pegylation
of the analog surprisingly induces glucagon agonism. This glucagon agonism is
not seen in
the pegylated PLA6 analogs. Several sites for pegylation were investigated
including
positions 3, 6 and 19 (positions 8, 11 and 19 of native glucagon) and at the N-
terminal amino
acid residue (see Table 12). In one embodiment the pegylation is at position
19 (position 24
of native glucagon) as that site exhibits the most potent and selective
glucagon antagonism.
In one embodiment, the glucagon antagonist comprises the general structure of
A-B-
C, wherein A is selected from the group consisting of:
(i) phenyl lactic acid (PLA);
(ii) an oxy derivative of PLA;
(iii) a peptide of 2 to 6 amino acids in which two consecutive amino
acids of the peptide are linked via an ester or ether bond;
B represents amino acids i to 26 of SEQ ID NO: 1, wherein i is 3, 4, 5, 6, or
7,
optionally comprising one or more amino acid modifications selected from the
group
consisting of:
(iv) Asp at position 9 (according to the amino acid numbering of
SEQ ID NO: 1) is substituted with a Glu, a sulfonic acid
derivative of Cys, homoglutamic acid, (3-homoglutamic acid, or
an alkylcarboxylate derivative of cysteine having the structure
of:
H2N COON
H2C
X5
COOH,
wherein X5 is C1-C4 alkyl, C2-C4 alkenyl, or C2-C4 alkynyl.
(v) substitution of one or two amino acids at positions 10, 20, and
24, (according to the amino acid numbering of SEQ ID NO: 1)
with an amino acid covalently attached to an acyl or alkyl
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-21-
group via an ester, ether, thioether, amide, or alkyl amine
linkage;
(vi) substitution of one or two amino acids at positions 16, 17, 20,
21, and 24 (according to the amino acid numbering of SEQ ID
NO: 1) with an amino acid selected from the group consisting
of: Cys, Lys, ornithine, homocysteine, and acetyl-
phenylalanine (Ac-Phe), wherein the amino acid of the group is
covalently attached to a hydrophilic moiety;
(vii) Asp at position 15 (according to the numbering of SEQ ID NO:
1) is substituted with cysteic acid, glutamic acid, homoglutamic
acid, and homocysteic acid;
(viii) Ser at position 16 (according to the numbering of SEQ ID NO:
1) is substituted with cysteic acid, glutamic acid, homoglutamic
acid, and homocysteic acid;
(ix) substitution with AIB at one or more of positions 16, 20, 21,
and 24 according to the amino acid numbering of SEQ ID NO:
1;
and C is selected from the group consisting of:
(x) X;
(xi) X-Y;
(xii) X-Y-Z; and
(xiii) X-Y-Z-R10,
wherein X is Met, Leu, or Nle; Y is Asn or a charged amino acid; Z is Thr,
Gly, Cys, Lys,
ornithine (Orn), homocysteine, acetyl phenylalanine (Ac-Phe), or a charged
amino acid;
wherein RIO is selected from a group consisting of SEQ ID NOs: 19-21 and 53;
and
(xiv) any of (x) to (xiii) in which the C-terminal carboxylate is
replaced with an amide.
In a specific aspect, the glucagon antagonist comprises an oxy derivative of
PLA. As
used herein "oxy derivative of PLA" refers to a compound comprising a modified
structure
of PLA in which the hydroxyl group has been replaced with O-R11, wherein R11
is a chemical
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-22-
moiety. In this regard, the oxy derivative of PLA can be, for example, an
ester of PLA or an
ether of PLA.
Methods of making oxy derivatives of PLA are known in the art. For example,
when
the oxy derivative is an ester of PLA, the ester may be formed by upon
reaction of the
hydroxyl of PLA with a carbonyl bearing a nucleophile. The nucleophile can be
any suitable
nucleophile, including, but not limited to an amine or hydroxyl. Accordingly,
the ester of
PLA can comprise the structure of Formula IV:
O
R7
-5 SSJ
Formula IV
wherein R7 is an ester formed upon reaction of the hydroxyl of PLA with a
carbonyl
bearing a nucleophile.
The carbonyl bearing a nucleophile (which reacts with the hydroxyl of PLA to
form
an ester) can be, for example, a carboxylic acid, a carboxylic acid
derivative, or an activated
ester of a carboxylic acid. The carboxylic acid derivative can be, but is not
limited to, an
acyl chloride, an acid anhydride, an amide, an ester, or a nitrile. The
activated ester of a
carboxylic acid can be, for example, N-hydroxysuccinimide (NHS), tosylate
(Tos), a
carbodiimide, or a hexafluorophosphate. In some embodiments, the carbodiimide
is 1,3-
dicyclohexylcarbodiimide (DCC), 1,1'-carbonyldiimidazole (CDI), 1-ethyl-3-(3-
dimethylaminopropyl)carbodiimide hydrochloride (EDC), or 1,3-
diisopropylcarbodiimide
(DICD). In some embodiments, the hexafluorophosphate is selected from a group
consisting
of hexafluorophosphate benzotriazol-1-yl-oxy-tris(dimethylamino)phosphonium
hexafluorophosphate (BOP), benzotriazol-l-yl- oxytripyrrolidinophosphonium
hexafluorophosphate (PyBOP), 2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl
uronium
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-23-
hexafluorophosphate (HATU), and o-benzotriazole-N,N,N',N'-tetramethyl-uronium-
hexafluoro-phosphate (HBTU).
Methods of making ethers from reaction with a hydroxyl group (e.g., the
hydroxyl of
PLA) also are known in the art. For example, the hydroxyl group of PLA may be
reacted
with a halogenated alkyl or tosylated alkyl alcohol to form an ether bond.
Generally, the chemical moiety of R11 is one which does not decrease the
activity of
the glucagon antagonist. In some embodiments, the chemical moiety enhances the
activity,
stability, and/or solubility of the glucagon antagonist.
In a specific embodiment, the chemical moiety bound to PLA via an oxygen-
containing bond (e.g., via an ester or ether bond) is a polymer (e.g., a
polyalkylene glycol), a
carbohydrate, an amino acid, a peptide, or a lipid, e.g., a fatty acid or a
steroid.
In a specific embodiment, the chemical moiety is an amino acid, which,
optionally, is
a part of a peptide, such that Formula IV is a depsipeptide. In this regard,
PLA may be at a
position other than the N-terminal amino acid residue of the glucagon
antagonist, such that
the glucagon antagonist comprises one or more (e.g., 1, 2, 3, 4, 5, 6, or
more) amino acids N-
terminal to the PLA residue. For example, the glucagon antagonist can comprise
PLA at
position n, wherein n is 2, 3, 4, 5, or 6 of the glucagon antagonist.
The amino acids N-terminal to the PLA residue may be synthetic or naturally-
occurring. In a specific embodiment, the amino acids which are N-terminal PLA
are
naturally-occurring amino acids. In one embodiment, the amino acids which are
N-terminal
to PLA are the N-terminal amino acids of native glucagon. For example, the
glucagon
antagonist can comprise at the N-terminus the amino acid sequence of any of
SEQ ID NOs:
54-58, wherein PLA is linked to threonine via an ester bond:
SEQ ID NO: 54 His-Ser-Gln-Gly-Thr-PLA
SEQ ID NO: 55 Ser-Gln-Gly-Thr-PLA
SEQ ID NO: 56 Gln-Gly-Thr-PLA
SEQ ID NO: 57 Gly-Thr-PLA
SEQ ID NO: 58 Thr-PLA
In an alternative embodiment, one or more of the N-terminal amino acids may be
substituted with an amino acid other than the amino acid of native glucagon.
For example,
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-24-
when the glucagon antagonist comprises PLA as the amino acid at position 5 or
6, the amino
acid at position 1 and/or position 2 may be an amino acid which reduces
susceptibility to
cleavage by dipeptidyl peptidase IV. More particularly, in some embodiments,
position 1 of
the glucagon antagonist is an amino acid selected from the group consisting of
D-histidine,
alpha, alpha-dimethyl imidiazole acetic acid (DMIA), N-methyl histidine, alpha-
methyl
histidine, imidazole acetic acid, desaminohistidine, hydroxyl-histidine,
acetyl-histidine and
homo-histidine. More particularly, in some embodiments, position 2 of the
antagonist
peptide is an amino acid selected from the group consisting of D-serine, D-
alanine, valine,
glycine, N-methyl serine, N-methyl alanine, and aminoisobutyric acid (AIB).
Also, for
example, when the glucagon antagonist comprises PLA as the amino acid at
position 4, 5, or
6, the amino acid at position 3 of the glucagon antagonist may be glutamic
acid, as opposed
to the native glutamine residue of native glucagon. In an exemplary embodiment
of the
invention, the glucagon antagonist comprises at the N-terminus the amino acid
sequence of
any of SEQ ID NOs: 59-61.
With respect to the glucagon antagonists comprising a compound of Formula IV,
the
polymer may be any polymer, provided that it can react with the hydroxyl group
of PLA.
The polymer may be one that naturally or normally comprises a carbonyl bearing
a
nucleophile. Alternatively, the polymer may be one which was derivatized to
comprise the
carbonyl bearing the carbonyl. The polymer may be a derivatized polymer of any
of:
polyamides, polycarbonates, polyalkylenes and derivatives thereof including,
polyalkylene
glycols, polyalkylene oxides, polyalkylene terepthalates, polymers of acrylic
and methacrylic
esters, including poly(methyl methacrylate), poly(ethyl methacrylate),
poly(butylmethacrylate), poly(isobutyl methacrylate), poly(hexylmethacrylate),
poly(isodecyl methacrylate), poly(lauryl methacrylate), poly(phenyl
methacrylate),
poly(methyl acrylate), poly(isopropyl acrylate), poly(isobutyl acrylate), and
poly(octadecyl
acrylate), polyvinyl polymers including polyvinyl alcohols, polyvinyl ethers,
polyvinyl
esters, polyvinyl halides, poly(vinyl acetate), and polyvinylpyrrolidone,
polyglycolides,
polysiloxanes, polyurethanes and co-polymers thereof, celluloses including
alkyl cellulose,
hydroxyalkyl celluloses, cellulose ethers, cellulose esters, nitro celluloses,
methyl cellulose,
ethyl cellulose, hydroxypropyl cellulose, hydroxy-propyl methyl cellulose,
hydroxybutyl
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-25-
methyl cellulose, cellulose acetate, cellulose propionate, cellulose acetate
butyrate, cellulose
acetate phthalate, carboxylethyl cellulose, cellulose triacetate, and
cellulose sulphate sodium
salt, polypropylene, polyethylenes including poly(ethylene glycol),
poly(ethylene oxide), and
poly(ethylene terephthalate), and polystyrene.
The polymer can be a biodegradable polymer, including a synthetic
biodegradable
polymer (e.g., polymers of lactic acid and glycolic acid, polyanhydrides,
poly(ortho)esters,
polyurethanes, poly(butic acid), poly(valeric acid), and poly(lactide-
cocaprolactone)), and a
natural biodegradable polymer (e.g., alginate and other polysaccharides
including dextran
and cellulose, collagen, chemical derivatives thereof (substitutions,
additions of chemical
groups, for example, alkyl, alkylene, hydroxylations, oxidations, and other
modifications
routinely made by those skilled in the art), albumin and other hydrophilic
proteins (e.g., zein
and other prolamines and hydrophobic proteins)), as well as.any copolymer or
mixture
thereof. In general, these materials degrade either by enzymatic hydrolysis or
exposure to
water in vivo, by surface or bulk erosion.
The polymer can be a bioadhesive polymer, such as a bioerodible hydrogel
described
by H. S. Sawhney, C. P. Pathak and J. A. Hubbell in Macromolecules, 1993, 26,
581-587, the
teachings of which are incorporated herein, polyhyaluronic acids, casein,
gelatin, glutin,
polyanhydrides, polyacrylic acid, alginate, chitosan, poly(methyl
methacrylates), poly(ethyl ..
methacrylates), poly(butylmethacrylate), poly(isobutyl methacrylate),
poly(hexylmethacrylate), poly(isodecyl methacrylate), poly(lauryl
methacrylate),
poly(phenyl methacrylate), poly(methyl acrylate), poly(isopropyl acrylate),
poly(isobutyl
acrylate), and poly(octadecyl acrylate).
In one embodiment, the polymer is a water-soluble polymer. Suitable water-
soluble
polymers are known in the art and include, for example, polyvinylpyrrolidone,
hydroxypropyl cellulose (HPC; Klucel), hydroxypropyl methylcellulose (HPMC;
Methocel),
nitrocellulose, hydroxypropyl ethylcellulose, hydroxypropyl butylcellulose,
hydroxypropyl
pentylcellulose, methyl cellulose, ethylcellulose (Ethocel), hydroxyethyl
cellulose, various
alkyl celluloses and hydroxyalkyl celluloses, various cellulose ethers,
cellulose acetate,
carboxymethyl cellulose, sodium carboxymethyl cellulose, calcium carboxymethyl
cellulose,
vinyl acetate/crotonic acid copolymers, poly-hydroxyalkyl methacrylate,
hydroxymethyl
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-26-
methacrylate, methacrylic acid copolymers, polymethacrylic acid,
polymethylmethacrylate,
maleic anhydride/methyl vinyl ether copolymers, poly vinyl alcohol, sodium and
calcium
polyacrylic acid, polyacrylic acid, acidic carboxy polymers,
carboxypolymethylene,
carboxyvinyl polymers, polyoxyethylene polyoxypropylene copolymer,
polymethylvinylether co-maleic anhydride, carboxymethylamide, potassium
methacrylate
divinylbenzene co-polymer, polyoxyethyleneglycols, polyethylene oxide, and
derivatives,
salts, and combinations thereof.
In a specific embodiment, the polymer is a polyalkylene glycol, including, for
example, polyethylene glycol (PEG).
The carbohydrate may be any carbohydrate provided that it comprises or is made
to
comprise a carbonyl with an alpha leaving group. The carbohydrate, for
example, may be
one which has been derivatized to comprise a carbonyl with an alpha leaving
group. In this
regard, the carbohydrate may be a derivatized form of a monosaccharide (e.g.,
glucose,
galactose, fructose), a disaccharide (e.g., sucrose, lactose, maltose), an
oligosaccharide (e.g.,
raffinose, stachyose), a polysaccharide (a starch, amylase, amylopectin,
cellulose, chitin,
callose, laminarin, xylan, mannan, fucoidan, galactomannan.
With respect to the glucagon antagonists comprising a compound of Formula IV,
the
lipid may.be any lipid comprising a carbonyl with an alpha leaving group. The
lipid, for
example, may be one which is derivatized to comprise the carbonyl. In this
regard, the lipid,
may be a derivative of a fatty acid (e.g., a C4-C30 fatty acid, eicosanoid,
prostaglandin,
leukotriene, thromboxane, N-acyl ethanolamine), glycerolipid (e.g., mono-, di-
, tri-
substituted glycerols), glycerophospholipid (e.g., phosphatidylcholine,
phosphatidylinositol,
phosphatidylethanolamine, phosphatidylserine), sphingolipid (e.g.,
sphingosine, ceramide),
sterol lipid (e.g., steroid, cholesterol), prenol lipid, saccharolipid, or a
polyketide.
oil, wax, cholesterol, sterol, fat-soluble vitamin, monoglyceride,
diglyceride, triglyceride, a
phospholipid.
In one embodiment, R7 has a molecular weight of about 100 kDa or less, e.g.,
about
90 kDa or less, about 80 kDa or less, about 70 kDa or less, about 60 kDa or
less, about 50
kDa or less, about 40 kDa or less. Accordingly, R7 can have a molecular weight
of about 35
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-27-
kDa or less, about 30 kDa or less, about 25 kDa or less, about 20 kDa or less,
about 15 kDa
or less, about 10 kDa or less, about 5 kDa or less, or about 1 kDa.
In an alternative embodiment, the glucagon antagonist comprises as A, a
peptide of 2
to 6 amino acids in which two consecutive amino acids of the peptide are
linked via an ester
or ether bond. The ester or ether bond may be, e.g., between amino acids 2 and
3, 3 and 4, 4
and 5, or 5 and 6. Optionally the peptide may be further modified by covalent
linkage to
another chemical moiety including linkage to a polymer (e.g. a hydrophilic
polymer),
alkylation, or acylation.
The peptide may comprise any amino acids, synthetic or naturally occurring,
provided that at least two consecutive amino acids of the peptide are linked
via an ester or
ether bond. In a specific embodiment, the peptide comprises amino acids of
native glucagon.
For example, the peptide can comprise j to 6 of native glucagon (SEQ ID NO:
1), wherein j is
1, 2, 3, 4, or 5. Alternatively, the peptide can comprise an amino acid
sequence based on the
N-terminus of SEQ ID NO: 1 with one or more amino acid modifications. The
amino acid at
position 1 and/or position 2 may be an amino acid which reduces susceptibility
to cleavage
by dipeptidyl peptidase IV. For instance, the peptide can comprise at position
1 of the
glucagon antagonist an amino acid selected from the group consisting of D-
histidine, alpha,
alpha-dimethyl imidiazole acetic acid.(DMIA), N-methyl histidine, alpha-methyl
histidine,
imidazole acetic acid, desaminohistidine, hydroxyl-histidine, acetyl-histidine
and homo-
histidine. More particularly, in some embodiments, position 2 of the
antagonist peptide is an
amino acid selected from the group consisting of D-serine, D-alanine, valine,
glycine, N-
methyl serine, N-methyl alanine, and aminoisobutyric acid (AIB). Also, for
example, the
amino acid at position 3 of the glucagon antagonist may be glutamic acid, as
opposed to the
native glutamine residue of native glucagon. Accordingly, the glucagon
antagonist can
comprise an amino acid sequence of:
Xaal-Xaa2-Xaa3-Thr-Gly-Phe (SEQ ID NO: 68);
Xaa2-Xaa3-Thr-Gly-Phe (SEQ ID NO: 69); or
Xaa3-Thr-Gly-Phe (SEQ ID NO: 70);
wherein Xaal is selected from a group consisting of: His, D-histidine, alpha,
alpha-
dimethyl imidiazole acetic acid (DMIA), N-methyl histidine, alpha-methyl
histidine,
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-28-
imidazole acetic acid, desaminohistidine, hydroxyl-histidine, acetyl-histidine
and homo-
histidine; Xaa2 is selected from a group consisting of: Ser, D-serine, D-
alanine, valine,
glycine, N-methyl serine, N-methyl alanine, and aminoisobutyric acid (AIB);
and Xaa3 is Gln
or Glu.
The present invention also encompasses embodiments wherein the C-terminal
amino
acid of the glucagon antagonists have an amide group substituting for the
carboxylic acid
group that is present on the native amino acid.
The glucagon antagonists can be further modified to improve the peptide's
solubility
in aqueous solutions at physiological pH, while retaining the glucagon
antagonist activity.
Introduction of hydrophilic groups at positions corresponding to positions 1,
16, 17, 20, 21,
24 and 29 of the native peptide, or at the C-terminus, can improve the
solubility of the
resulting glucagon antagonist in solutions having a physiological pH, while
retaining the
parent compounds antagonist activity. Therefore, in one embodiment the
presently disclosed
glucagon antagonists are further modified to comprise one or more hydrophilic
groups
covalently linked to the side chains of amino acids corresponding to amino
acid positions 1,
16, 17, 20, 21, 24 and 29 of the native glucagon peptide or of the N- or C-
terminal amino
acid. In a further embodiment the side chains of amino acids corresponding to
amino acid
positions 16 and 24 of the native glucagon peptide are covalently bound to
hydrophilic
groups, and in one embodiment the hydrophilic group is polyethylene glycol
(PEG).
In some embodiments, wherein the glucagon antagonist is PEGylated, the
glucagon
antagonist comprises the shortened glucagon peptides, specifically 6-29 where
the "N-
terminal" amino acid is PLA (phenyl-lactic acid). Such glucagon derivatives
exhibit unique
virtues. They are more potent peptides than those with the native N-terminal
phenylalanine
and they suppress any glucagon agonism that results from pegylation, something
not seen
with the native phenylalanine. Finally, while the current literature
establishes that a
substitution of the native aspartic acid at position 9 is required for
antagonist activity,
applicants have discovered the surprising result that such a substitution is
no longer required
in the PLA6-(6-29) glucagon analogs.
In one embodiment an amino acid of the glucagon antagonist is substituted with
at
least one cysteine residue, wherein the side chain of the cysteine residue is
further modified
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-29-
with a thiol reactive reagent, including for example, maleimido, vinyl
sulfone, 2-pyridylthio,
haloalkyl, and haloacyl. These thiol reactive reagents may contain carboxy,
keto, hydroxyl,
and ether groups as well as other hydrophilic moieties such as polyethylene
glycol units. In
an alternative embodiment, an amino acid of the glucagon antagonist is
substituted with
lysine, and the side chain of the substituting lysine residue is further
modified using amine
reactive reagents such as active esters (succinimido, anhydride, etc) of
carboxylic acids or
aldehydes of hydrophilic moieties such as polyethylene glycol. In accordance
with one
embodiment the lysine residue corresponding to position 12 of the native
peptide is
substituted with arginine and a single lysine substitution is inserted for one
of the amino
acids corresponding to position 1, 16, 17, 20, 21, 24 or 29 of the native
peptide, or a lysine is
added to the N- or C-terminus of the glucagon antagonist.
In another embodiment the methionine residue corresponding to position 27 of
the
native peptide is changed to leucine or norleucine to prevent oxidative
degradation of the
peptide.
In some embodiments, the glucagon antagonists described herein are further
modified
by truncation or deletion of one or two amino acids of the C-terminus of the
glucagon peptide
(i.e., truncation of the amino acid at position 29 or at positions 28 and 29
of native glucagon)
without affecting activity and/or potency at the glucagon receptor. In this
regard, the
glucagon antagonist described herein can, for example, consist essentially of
or consist of
amino acids 1-27, 1-28, 2-27, 2-28, 3-27, 3-28, 4-27, 4-28, 5-27, 5-28, 6-27,
or 6-28 of the
native glucagon peptide (SEQ ID NO: 1) with one or more modifications
resulting in
glucagon antagonistic activity as described herein.
The presently disclosed glucagon antagonists also encompass amino acid
substitutions at positions that are known not to be critical to the function
of the glucagon
peptide. In one embodiment the substitutions are conservative amino acid
substitutions at
one, two or three positions selected from the group consisting of 2, 5, 6, 7,
8, 9, 12, 13, 14,
15, 16, 19, 22, 23 or 24 of SEQ ID NO: 39. In one embodiment the glucagon
antagonist
comprises a derivative peptide of SEQ ID NO: 42 wherein the glucagon peptide
comprises a
further amino acid substitution relative to SEQ ID NO: 42 at one to three
amino acid
positions selected from positions 2, 5, 6, 8, 9, 12, 13 and 14. In one
embodiment the
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-30-
substitutions at positions 2, 5, 6, 8, 9, 12, 13 and 14 of SEQ ID NO: 42 are
conservative
amino acid substitutions. In one embodiment the amino acids corresponding to
positions 16,
17, 20, 21, 24 or 29 of the native peptide, and more particularly at position
21 and/or 24 are
substituted with cysteine or lysine, wherein a PEG chain is covalently
attached to the
substituted cysteine or lysine residue.
In those embodiments wherein the glucagon antagonist comprises a polyethylene
glycol chain, the polyethylene chain may be in the form of a straight chain or
it may be
branched. In accordance with one embodiment the polyethylene glycol chain has
an average
molecular weight selected from the range of about 500 to about 10,000 Daltons.
In one
embodiment the polyethylene glycol chain has an average molecular weight
selected from
the range of about 1,000 to about 5,000 Daltons. In one embodiment the
polyethylene glycol
chain has an average molecular weight selected from the range of about 1,000
to about 5,000
Daltons. In one embodiment the polyethylene glycol chain has an average
molecular weight
selected of about 1,000 to about 2,000 Daltons. In one embodiment the
polyethylene glycol
chain has an average molecular weight of about 1,000 Daltons.
In accordance with one embodiment the modified glucagon antagonist comprises
two
or more polyethylene chains covalently bound to the peptide wherein the total
molecular
weight of the glucagon chains is about 1,000 to about 5,000 Daltons. In one
embodiment the
pegylated glucagon antagonist comprises a peptide selected from the group
consisting of
SEQ ID NO: 12, and SEQ ID NO: 22, wherein said peptide comprise a polyethylene
glycol
chain linked to the amino acid at positions 11 and 19 and the combined
molecular weight of
the two PEG chains is about 1,000 to about 5,000 Daltons.
In accordance with one embodiment a glucagon antagonist is provided comprising
a
modified glucagon peptide selected from the group consisting of:
Rl-Phe-Thr-Ser-Xaa-Tyr-Ser-Xaa-Tyr-Leu-Xaa-Xaa-Arg-Arg-Ala-Gln-Asp-Phe-Val-
Gln-Trp-Leu- Xaa-Asn-Thr-R2 (SEQ ID NO: 9),
Rl-Phe-Thr-Ser-Xaa-Tyr-Ser-Xaa-Tyr-Leu-Asp-Ser-Arg-Arg-Ala-Gln-Xaa-Phe-V al-
Gln-Trp-Leu- Xaa-Asn-Thr-R2 (SEQ ID NO: 10),
R 1-Phe-Thr-Ser-Xaa-Tyr-Ser-Xaa-Tyr-Leu-Asp-Ser-Arg-Arg-Al a-Gln-Asp-Phe-V al-
Xaa-Trp-Leu-Xaa-Asn-Thr-R2 (SEQ ID NO: 11) and
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-31-
R1-Phe- Thr-Ser-Xaa-Tyr-Ser-Xaa-Tyr-Leu-Asp-Ser- Arg-Arg-Ala-Gln-Xaa-Phe-
Val-Xaa-Trp-Leu- Xaa-Asn-Thr-R2 (SEQ ID NO: 12),
wherein Xaa at position 4 = aspartic acid, glutamic acid, cysteic acid or
homocysteic acid,
Xaa at position 7 = Lys or Arg, Xaa at position 10 is aspartic acid, cysteic
acid, glutamic
acid, homoglutamic acid and homocysteic acid; Xaa at position 11 is Ser, Lys,
Cys, Orn,
homocysteine or acetyl phenylalanine, Xaa at position 16 is Asp, Lys, Cys,
Orn,
homocysteine or acetyl phenylalanine a and Xaa at position 19 is Gln, Lys,
Cys, Orn,
homocysteine and acetyl phenylalanine, Xaa at position 22 = Met, Leu or Nle,
R1 is OH or
NH2, and R2 is COOH or CONH2, wherein the peptide is pegylated at position 11
for SEQ ID
NO: 9, at position 16 for SEQ ID NO: 10, position 19 for SEQ ID NO: 11 and at
positions 16
and 19 of SEQ ID NO: 12, with the proviso that when Xaa at position 4 =
aspartic acid then
Rl is OH. In accordance with one embodiment the peptide comprises the sequence
of SEQ
ID NO: 9, SEQ ID NO: 10 or SEQ ID NO: 11, wherein R1 is OH and R2 is CONH2. In
one
embodiment the peptide comprises the sequence of SEQ ID NO: 9, SEQ ID NO: 10
or SEQ
ID NO: 11, wherein R1 is OH, R2 is CONH2 and the amino acid at position 4 is
aspartic acid,
and in a further embodiment such peptides comprise a carboxy terminal
extension
comprising the sequence of SEQ ID NO: 19.
In accordance with one embodiment the peptide comprises a sequence selected
from
the group consisting of SEQ ID NO: 9, SEQ ID NO: 10, SEQ ID NO: 13, SEQ ID NO:
14,
and SEQ ID NO: 16, wherein the peptide is pegylated at position 11 for SEQ ID
NO: 9 and
SEQ ID NO: 13, pegylated at position 16 for SEQ ID NO: 10, and pegylated at
position 19
for SEQ ID NO: 10 and SEQ ID NO: 14. In one embodiment the glucagon agonist
comprises the peptide of SEQ ID NO: 13 or SEQ ID NO: 14. In one embodiment the
C-
terminal amino acid of the glucagon antagonists disclosed herein have an amide
group in
place of the carboxylic acid group that is present on the native amino acid.
In accordance
with one embodiment the glucagon antagonist comprises the sequence of SEQ ID
NO: 18.
In accordance with one embodiment, a glucagon antagonist is provided wherein a
plasma protein has been covalently linked to an amino acid side chain of the
peptide to
improve the solubility, stability and/or pharmacokinetics of the glucagon
peptide. For
example, serum albumin can be covalently bound to the glucagon antagonists
presented
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-32-
herein. In one embodiment the plasma protein is covalently bound to an amino
acid
corresponding to position 16, 17, 20, 21, 24 or 29 of the native glucagon
peptide. More
particularly, in one embodiment the plasmid protein is bound to an amino acid
corresponding
to position 16 or 24 of the native glucagon peptide, wherein the glucagon
antagonist
comprises the sequence of SEQ ID NO: 3, SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO:
6,
SEQ ID NO: 7, SEQ ID NO: 8, SEQ ID NO: 9, SEQ ID NO: 11, SEQ ID NO: 12, SEQ ID
NO: 22, SEQ ID NO: 23, SEQ ID NO: 24, SEQ ID NO: 25, SEQ ID NO: 26, SEQ ID NO:
27, SEQ ID NO: 28, SEQ ID NO: 36 and SEQ ID NO: 39. In one embodiment the
glucagon.
>
antagonist comprises a peptide selected from the group consisting of SEQ ID
NO: 9, SEQ ID
NO: 10, SEQ ID NO: 11 and SEQ ID NO: 12.
In accordance with one embodiment, a glucagon antagonist is provided wherein a
linear. amino acid sequence representing the Fc portion of an immunoglobin
molecule has:
been covalently linked to an amino acid side chain of a glucagon antagonist
disclosed herein
to improve the solubility, stability and/or pharmacokinetics of the glucagon
peptide. For
example, the amino acid sequence representing the Fc portion of an
immunoglobin molecule
can be covalently bound to position 11, 12, 15, 16, 19,.21 or 24 of the
glucagon peptide of
SEQ ID NO: 7, SEQ ID NO: 39, or a glucagon analog thereof. In one embodiment
the Fc
peptide is covalently bound to position 11 or 19 of the glucagon antagonist of
SEQ ID NO: 6,
SEQ ID NO: 7, SEQ ID NO: 8 or SEQ ID NO: 36. The Fc portion is usually
isolated from
IgG, but the Fc peptide fragment from any immunoglobin should function
equivalently. In
one embodiment the glucagon peptide is selected from the group consisting of
SEQ ID NO:
3, SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 7 SEQ ID NO: 8, and SEQ ID NO: 39,
wherein the Fc portion is linked to the corresponding position of 16, 17, 20,
21, 24 or 29 of
the native glucagon peptide. In one embodiment the glucagon analog comprises a
glucagon
peptide selected from the group consisting of SEQ ID NO: 9, SEQ ID NO: 10, SEQ
ID NO:
11 and SEQ ID NO: 12, wherein the Fc peptide is bound to the side chain of the
amino acid
located at position 11, 16 or 19 of SEQ ID NO: 9, SEQ ID NO: 10, SEQ ID NO:
11,
respectively, and at both positions 11 and 19 for SEQ ID NO: 12.
The present disclosure also encompasses other conjugates in which glucagon
peptides of the invention are linked, optionally via covalent bonding and
optionally via a
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-33-
linker, to a conjugate. Linkage can be accomplished by covalent chemical
bonds, physical
forces such electrostatic, hydrogen, ionic, van der Waals, or hydrophobic or
hydrophilic
interactions. A variety of non-covalent coupling systems may be used,
including biotin-
avidin, ligand/receptor, enzyme/substrate, nucleic acid/nucleic acid binding
protein,
lipid/lipid binding protein, cellular adhesion molecule partners; or any
binding partners or
fragments thereof which have affinity for each other.
Exemplary conjugates include but are not limited to a heterologous peptide or
polypeptide (including for example, a plasma protein), a targeting agent, an
immunoglobulin
or portion thereof (e.g. variable region, CDR, or Fc region), a diagnostic
label such as a
radioisotope, fluorophore or enzymatic label, a polymer including water
soluble polymers, or
other therapeutic or diagnostic agents. In one embodiment a conjugate is
provided
comprising a glucagon peptide of the present invention and a plasma protein,
wherein the
plasma protein is selected form the group consisting of albumin, transferin,
fibrinogen and
glubulins. In one embodiment the plasma protein moiety of the conjugate is
albumin or
transferin. In some embodiments, the linker comprises a chain of atoms from 1
to about 60,
or 1 to 30 atoms or longer, 2 to 5 atoms, 2 to 10 atoms, 5 to 10 atoms, or 10
to 20 atoms long.
In some embodiments, the chain atoms are all carbon atoms. In some
embodiments, the
chain atoms in the backbone of the linker are selected from the group
consisting of C, 0, N,
and S. Chain atoms and linkers may be selected according to their expected
solubility
(hydrophilicity) so as to provide a more soluble conjugate. In some
embodiments, the linker
provides a functional group that is subject to cleavage by an enzyme or other
catalyst or
hydrolytic conditions found in the target tissue or organ or cell. In some
embodiments, the
length of the linker is long enough to reduce the potential for steric
hindrance. If the linker is
a covalent bond or a peptidyl bond and the conjugate is a polypeptide, the
entire conjugate
can be a fusion protein. Such peptidyl linkers may be any length. Exemplary
linkers are
from about 1 to 50 amino acids in length, 5 to 50, 3 to 5, 5 to 10, 5 to 15,
or 10 to 30 amino
acids in length. Such fusion proteins may alternatively be produced by
recombinant genetic
engineering methods known to one of ordinary skill in the art.
The Asp-Ser sequence at position 15-16 of native glucagon has been identified
as a
uniquely unstable dipeptide that leads to premature chemical cleavage of the
native hormone
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-34-
in aqueous buffers. For example, when maintained at 0.01N HCl at 37 C for 2
weeks, more
than 50% of the native glucagon may be cleaved into fragments. The two
liberated cleavage
peptides 1-15 and 16-29 are devoid of glucagon-like biological activity and
thus represent a
limitation on the aqueous pre-formulation of glucagon and its related analogs.
The selective
chemical substitution of the Asp at position 15 of the native glucagon peptide
with Glu has
been observed to virtually eliminate chemical cleavage of the 15-16 peptide
bond.
Accordingly, it is expected that the glucagon antagonists of the present
invention can
be similarly modified to decrease their susceptibility to premature chemical
cleavage in
aqueous buffers. In accordance with one embodiment the glucagon antagonists
described
herein can be further modified to enhance their stability in aqueous solutions
by replacing the
native aspartic amino acid, located at position 15 of the native glucagon
peptide, with an
amino. acid selected from the group consisting of cysteic acid; glutamic acid,
homoglutamic:
acid and-homocysteic acid. In accordance with one embodiment the aspartic acid
residue at
position 10 of the glucagon antagonist of SEQ ID NO: 39 can be substituted
with an amino
acid selected from the group consisting of cysteic acid, glutamic acid,
homoglutamic acid
and homocysteic acid, and in one embodiment the native aspartic acid at
position 10 of SEQ
ID NO: 39 is replaced with glutamic acid. In accordance with one embodiment a
glucagon
antagonist having improved stability in aqueous solutions is provided wherein
the antagonist
comprises a sequence selected from the group consisting of SEQ ID NO: 36, SEQ
ID NO: 40
and SEQ ID NO: 42. In a further embodiment the glucagon antagonist is
amidated.
In accordance with one embodiment, increased stability by way of reduced
degradation of the glucagon antagonist described herein may also be achieved
by substitution
of the serine at position 16 (according to the numbering of native glucagon)
with glutamic
acid, cysteic acid, homo-glutamic acid,or homo-cysteic acid. In a specific
embodiment, the
serine at position 16 (according to the native glucagon sequence numbering) is
replaced with
glutamic acid. In a more specific aspect, the glucagon antagonist comprising
such a
modification comprises a C-terminal carboxylate and is not amidated.
In accordance with one embodiment, a glucagon antagonist is provided
comprising a
glucagon peptide selected from the group consisting of SEQ ID NO: 7, SEQ ID
NO: 36, SEQ
ID NO: 39, SEQ ID NO: 40, SEQ ID NO: 41, SEQ ID NO: 42, SEQ ID NO: 43 and SEQ
ID
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-35-
NO: 44, further modified by one or more additional amino acid substitutions at
positions
corresponding to positions 11, 12, 15, 16, 19 and/or 24 of the native glucagon
peptide,
wherein the amino acid substitutions comprise a substitution with an amino
acid having a
side chain suitable for crosslinking with hydrophilic moieties, including for
example, PEG.
The native glucagon peptide can be substituted with a naturally occurring
amino acid or a
synthetic (non-naturally occurring) amino acid. Synthetic or non-naturally
occurring amino
acids refer to amino acids that do not naturally occur in vivo but which,
nevertheless, can be
incorporated into the peptide structures described herein. In one embodiment a
glucagon
antagonist is provided wherein the peptide comprises the sequence of SEQ ID
NO: 7, SEQ
ID NO: 36, SEQ ID NO: 39, SEQ ID NO: 40, SEQ ID NO: 41, SEQ ID NO: 42, SEQ ID
NO: 43 and SEQ ID NO: 44, and further comprises a polyethylene chain bound to
corresponding position 21 or 24 of the native glucagon peptide. In a further
embodiment the
C-terminus of the glucagon peptide is modified to replace the carboxylic acid
group with an
amide group.
Applicants have also discovered that native glucagon can be modified by
introducing
charge at its carboxy terminus to enhance the solubility of the peptide while
retaining the
agonist properties of the peptide. The enhanced solubility allows for the
preparation and
storage of glucagon solutions at near neutral pH. Formulating glucagon
solutions at
relatively neutral pHs (e.g. pH of about 6.0 to about 8.0) improves the long
term stability of
the glucagon peptides.
Again, applicants anticipate that the glucagon antagonists disclosed herein
can be
similarly modified to enhance their solubility in aqueous solutions at
relatively neutral pH
(e.g. pH of about 6.0 to about 8.0) while retaining the antagonist properties
of the parent
protein. Accordingly, one embodiment of the present invention is directed to a
glucagon
antagonist of SEQ ID NO: 39 that has been further modified relative to the
native amino
acids present at positions 6-29 of the wild type glucagon (SEQ ID NO: 1) to
add charge to
the peptide by the substitution of native non-charged amino acids with charged
amino acids,
or the addition of charged amino acids to the carboxy terminus. In accordance
with one
embodiment, one to three of the non-charged native amino acids of the glucagon
antagonist
of SEQ ID NO: 39 are replaced with a charged amino acid. In one embodiment the
charged
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-36-
amino acid is selected from the group consisting of lysine, arginine,
histidine, aspartic acid
and glutamic acid. More particularly, applicants have discovered that
substituting the
normally occurring amino acid at corresponding position 28 and/or 29 relative
to native
glucagon with charged amino acids, and/or the addition of one to two charged
amino acids at
the carboxy terminus of the glucagon peptide, enhances the solubility and
stability of the
glucagon peptides in aqueous solutions at physiologically relevant pHs (i.e.,
a pH of about
6.5 to about 7.5). Accordingly, such modifications of the glucagon antagonist
disclosed
herein are anticipated to have a similar effect on the solubility in aqueous
solutions,
particularly at a pH ranging from about 5.5 to about 8.0, while retaining the
parent peptide's
biological activity
In accordance with one embodiment the glucagon antagonist of SEQ ID NO: 39 is
modified by the substitution of the native amino acid at corresponding
position 28 and/or 29
relative to native glucagon with a negatively charged amino acid (e.g.,
aspartic acid or
glutamic acid) and optionally the addition of a negatively charged amino acid
(e.g., aspartic
acid or glutamic acid) to the carboxy terminus of the peptide. In an
alternative embodiment
the native glucagon peptide of SEQ ID NO: 39 is modified by the substitution
of the native
amino acid at corresponding position 29 relative to native glucagon with a
positively charged
amino acid (e.g., lysine, arginine or histidine) and optionally the addition
of one or two
positively charged amino acid (e.g., lysine, arginine or histidine) on the
carboxy terminus of
the peptide. In accordance with one embodiment a glucagon analog having
improved
solubility and stability is provided wherein the analog comprises the amino
acid sequence of
SEQ ID NO: 41 with the proviso that at least one amino acids at position, 23,
or 24 of SEQ
ID NO: 41 is substituted with an acidic amino acid, and/or an additional
acidic amino acid is
added at the carboxy terminus of SEQ ID NO: 41. In one embodiment the acidic
amino acids
are independently selected from the group consisting of Asp, Glu, cysteic acid
and
homocysteic acid.
In accordance with one embodiment a glucagon antagonist having improved
solubility and stability is provided wherein the antagonist comprises the
amino acid sequence
of SEQ ID NO: 41, SEQ ID NO: 42, SEQ ID NO: 43 or SEQ ID NO: 44, wherein at
least
one of the amino acids at positions 23 or 24 is substituted with a non-native
amino acid
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-37-
residue (i.e. at least one amino acid present at position 23 or 24 of the
analog is an acidic
amino acid different from the amino acid present at the corresponding position
in SEQ ID
NO: 7). In accordance with one embodiment a glucagon agonist is provided
comprising the
sequence of SEQ ID NO: 41 or 42 with the proviso that when the amino acid at
position 23 is
asparagine and the amino acid at position 24 is threonine, the peptide further
comprises one
to two amino acids, independently selected from the group consisting of Lys,
Arg, His, Asp
or Glu, added to the carboxy terminus of the glucagon antagonist.
The present disclosure also encompasses glucagon antagonist fusion peptides
wherein
a second peptide has been fused to the c-terminus of the glucagon antagonist.
More
particularly, the fusion peptide may comprise a glucagon antagonist peptide of
SEQ ID NO:
44 that further comprises an amino acid sequence of SEQ ID NO: 19
(GPSSGAPPPS), SEQ
ID NO: 20. (Lys Arg Asn Arg Asn Asn Ile Ala) or SEQ:ID NO: 21 (Lys Arg Asn
Arg)
linked to.the c-terminal amino acid of the glucagon antagonist. In one
embodiment the
amino acid sequence of SEQ ID NO: 19 (GPSSGAPPPS) is bound to amino acid 24 of
the
glucagon antagonist of SEQ ID NO: 42 through a peptide bond. In another
embodiment the
fusion peptide comprises a glucagon antagonist peptide of SEQ ID NO: 7, SEQ ID
NO: 36,
SEQ ID NO: 39, SEQ ID NO: 40, SEQ ID NO: 41 or SEQ ID NO: 43 that further
comprises
an amino acid sequence of SEQ ID NO: 19 (GPSSGAPPPS) linked to amino acid 24
of the
glucagon antagonist. In another embodiment the fusion peptide comprises a
glucagon
antagonist peptide of SEQ ID NO: 7, SEQ ID NO: 36, SEQ ID NO: 37, SEQ ID NO:
38,
SEQ ID NO: 39, SEQ ID NO: 41 or SEQ ID NO: 43 that further comprises an amino
acid
sequence of SEQ ID NO: 20, SEQ ID NO: 21 or SEQ ID NO: 53 linked to amino acid
24 of
the glucagon antagonist. In one embodiment the glucagon antagonist fusion
peptide
comprises a sequence selected from the group consisting of SEQ ID NO: 46 and
SEQ ID
NO 47. In a further embodiment the C-terminus of the fusion peptide is
modified to replace
the carboxylic acid group with an amide group.
In one embodiment a glucagon antagonist fusion peptide is provided wherein the
glucagon antagonist portion of the fusion peptide is selected from the group
consisting of
SEQ ID NO: 3, SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 6, SEQ ID NO: 7, SEQ ID
NO:
8, SEQ ID NO: 9, SEQ ID NO: 11, SEQ ID NO: 12, SEQ ID NO: 13, SEQ ID NO: 14,
SEQ
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-38-
ID NO: 15, SEQ ID NO: 10, SEQ ID NO: 16, SEQ ID NO: 17, SEQ ID NO: 18 and SEQ
ID
NO: 39 and the sequence of SEQ ID NO: 19 is fused to the carboxy terminus of
the glucagon
antagonist portion, and wherein the PEG chain, when present, is selected from
the range of
500 to 40,000 Daltons. More particularly, in one embodiment the glucagon
antagonist
segment is selected from the group consisting of SEQ ID NO: 13, SEQ ID NO: 14,
SEQ ID
NO: 15, SEQ ID NO: 16, SEQ ID NO: 46 and SEQ ID NO: 47 wherein the PEG chain
is
selected from the range of about 500 to about 5,000 Daltons, and more
particularly, in one
embodiment the PEG chain is about 1,000 Daltons. In a further embodiment the C-
terminus
is modified to replace the carboxylic acid group with an amide group.
The glucagon antagonist may further comprise one to two charged amino acids
added
to the carboxy terminus. In one embodiment, wherein one to two charged amino
acids are
added to the carboxy terminus of SEQ ID NO: 44, the amino acids are negatively
charged
amino acids, including for example glutamic acid and aspartic acid. In one
embodiment, the
glucagon antagonist comprises the sequence of SEQ ID NO: 42 wherein at least
one of
corresponding positions 27 and 28 relative to the native glucagon peptide
comprises an
amino acid selected from the group consisting of aspartic acid and glutamic
acid and wherein
SEQ ID NO: 42 is optionally modified to include an addition one to two
negatively charged
amino acids added to the carboxy terminus. In one embodiment the negatively
charged
amino acids are glutamic acid or aspartic acid.
In another embodiment the solubility of the glucagon antagonist of SEQ ID NO:
42
can be improved by covalently linking a hydrophilic moiety to an amino acid
residue at
position 11, 12, 15, 16, 19 or 24, and in one embodiment the hydrophilic
moiety is linked to
an amino acid at position 11, 16 or 19, and in a further embodiment the
hydrophilic moiety is
linked to amino acid 19. In one embodiment the hydrophilic moiety is a plasma
protein or
the Fc portion of an immunoglobin, and in an alternative embodiment the
hydrophilic moiety
is a hydrophilic hydrocarbon chain. In one embodiment the hydrophilic moiety
is
polyethylene glycol, having a molecular weight selected from the range of
about 1,000 to
about 5,000 Daltons. In another embodiment the hydrophilic moiety is
polyethylene glycol,
having a molecular weight of at least about 20,000 Daltons. In one embodiment
the
polyethylene modified glucagon antagonist comprises the amino acids sequence
of SEQ ID
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-39-
NO: 9, SEQ ID NO: 10, SEQ ID NO: 11, SEQ ID NO: 12, SEQ ID NO: 43, SEQ ID NO:
44
or SEQ ID NO: 45.
In one embodiment, the glucagon antagonist comprises the general structure of
A-B-
C, wherein A is selected from the group consisting of:
(i) phenyl lactic acid (PLA);
(ii) an oxy derivative of PLA;
(iii) a peptide of 2 to 6 amino acids in which two consecutive amino
acids of the peptide are linked via an ester or ether bond;
B represents amino acids i to 26 of SEQ ID NO: 1, wherein i is 3, 4, 5, 6, or
7,
optionally comprising one or more amino acid modifications selected from the
group
consisting of:
(iv) Asp at position 9 (according to the amino acid numbering of
SEQ IDNO: 1) is substituted with a Glu, a sulfonic acid
derivative of Cys, homoglutamic acid, (3-homoglutamic acid, or
an alkylcarboxylate derivative of cysteine having the structure
of:
COOH
H2N Y
H2C
X5
1~11 COOH
wherein X5 is C1-C4 alkyl, C2-C4 alkenyl, or C2-C4 alkynyl.
(v) substitution of one or two amino acids at positions 10, 20, and
24, (according to the amino acid numbering of SEQ ID NO: 1)
with an amino acid covalently attached to an acyl or alkyl
group via an ester, ether, thioether, amide, or alkyl amine
linkage;
(vi) substitution of one or two amino acids at positions 16, 17, 20,
21, and 24 (according to the amino acid numbering of SEQ ID
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-40-
NO: 1) with an amino acid selected from the group consisting
of: Cys, Lys, ornithine, homocysteine, and acetyl-
phenylalanine (Ac-Phe), wherein the amino acid of the group is
covalently attached to a hydrophilic moiety;
(vii) Asp at position 15 (according to the numbering of SEQ ID NO:
1) is substituted with cysteic acid, glutamic acid, homoglutamic
acid, and homocysteic acid;
(viii) Ser at position 16 (according to the numbering of SEQ ID NO:
1) is substituted with cysteic acid, glutamic acid, homoglutamic
acid, and homocysteic acid;
(ix) substitution with AIB at one or more of positions 16, 20, 21,
and 24 according to the amino acid numbering of SEQ ID NO:
l;
and C is selected from the group consisting of:
(x) X;
(xi) X-Y;
(xii) X-Y-Z; and
(xiii) X-Y-Z-R10,
wherein X is Met, Leu, or Nle; Y is Asn or a charged amino acid; Z is Thr,
Gly, Cys, Lys,
ornithine (Orn), homocysteine, acetyl phenylalanine (Ac-Phe), or a charged
amino acid;
wherein RIO is selected from a group consisting of SEQ ID NOs: 19-21 and 53;
and
(xiv) any of (x) to (xiii) in which the C-terminal carboxylate is
replaced with an amide.
In a specific aspect, the glucagon antagonist comprises an oxy derivative of
PLA,
e.g., an ester of PLA or an ether of PLA. In one embodiment, the ester of PLA
comprises the
structure of Formula IV:
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-41-
O
R7
Formula IV
wherein R7 is an ester formed upon reaction of the hydroxyl of PLA with a
carbonyl
bearing a nucleophile.
In a specific embodiment, the oxy derivative of PLA comprises a chemical
moiety
selected from the group consisting of a polymer (e.g., a polyalkylene glycol),
a carbohydrate,
an amino acid, a peptide, and a lipid, e.g., a fatty acid or a steroid,
wherein the chemical
moiety is linked to PLA via an oxygen-containing bond (e.g., an ester or ether
bond).
In a specific embodiment, the chemical moiety is an amino acid, which,
optionally, is
a part of a peptide, such that Formula IV is a depsipeptide. In this regard,
PLA may be at a
position other than the N-terminal amino acid residue of the glucagon
antagonist, such that
the glucagon antagonist comprises one or more (e.g., 1, 2, 3, 4, 5, 6, or
more) amino acids N-
terminal to the PLA residue. For example, the glucagon antagonist can comprise
PLA at
position n, wherein n is 2, 3, 4, 5, or 6 of the glucagon antagonist.
The amino acids N-terminal to the PLA residue may be synthetic or naturally-
occurring. In a specific embodiment, the amino acids which are N-terminal to
PLA are
naturally-occurring amino acids. In one embodiment, the amino acids which are
N-terminal
to PLA are the N-terminal amino acids of native glucagon. For example, the
glucagon
antagonist can comprise at the N-terminus the amino acid sequence of any of
SEQ ID NOs:
54-58, wherein PLA is linked to threonine via an ester bond:
SEQ ID NO: 54 His-Ser-Gln-Gly-Thr-PLA
SEQ ID NO: 55 Ser-Gln-Gly-Thr-PLA
SEQ ID NO: 56 Gln-Gly-Thr-PLA
SEQ ID NO: 57 Gly-Thr-PLA
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-42-
SEQ ID NO: 58 Thr-PLA
In an alternative embodiment, one or more of the N-terminal amino acids may be
substituted with an amino acid other than the amino acid of native glucagon.
For example,
when the glucagon antagonist comprises PLA as the amino acid at position 5 or
6, the amino
acid at position 1 and/or position 2 may be an amino acid which reduces
susceptibility to
cleavage by dipeptidyl peptidase IV. More particularly, in some embodiments,
position 1 of
the glucagon antagonist is an amino acid selected from the group consisting of
D-histidine,
alpha, alpha-dimethyl imidiazole acetic acid (DMIA), N-methyl histidine, alpha-
methyl
histidine, imidazole acetic acid, desaminohistidine, hydroxyl-histidine,
acetyl-histidine and
homo-histidine. More particularly, in some embodiments, position 2 of the
antagonist
peptide is an amino acid selected from the group consisting of D-serine, D-
alanine, valine,
glycine, N-methyl serine, N-methyl alanine, and aminoisobutyric acid (AIB).
Also, for
example, when the glucagon antagonist comprises PLA as the amino acid at
position 4, 5, or
6, the amino acid at position 3 of the glucagon antagonist may be glutamic
acid, as opposed
to the native glutamine residue of native glucagon. In an exemplary embodiment
of the
invention, the glucagon antagonist comprises at the N-terminus the amino acid
sequence of
any of SEQ ID NOs: 59-61.
With respect to the glucagon antagonists comprising a compound of Formula IV,
the
polymer may be any polymer, provided that it can react with the hydroxyl of
PLA. The
polymer may be one that naturally or normally comprises a carbonyl with
nucleophile, for
example. Alternatively, the polymer may be one which was derivatized to
comprise the
carbonyl bearing a nucleophile. The polymer may be a derivatized polymer of
any of:
polyamides, polycarbonates, polyalkylenes and derivatives thereof including,
polyalkylene
glycols, polyalkylene oxides, polyalkylene terepthalates, polymers of acrylic
and methacrylic
esters, including poly(methyl methacrylate), poly(ethyl methacrylate),
poly(butylmethacrylate), poly(isobutyl methacrylate), poly(hexylmethacrylate),
poly(isodecyl methacrylate), poly(lauryl methacrylate), poly(phenyl
methacrylate),
poly(methyl acrylate), poly(isopropyl acrylate), poly(isobutyl acrylate), and
poly(octadecyl
acrylate), polyvinyl polymers including polyvinyl alcohols, polyvinyl ethers,
polyvinyl
esters, polyvinyl halides, poly(vinyl acetate), and polyvinylpyrrolidone,
polyglycolides,
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-43-
polysiloxanes, polyurethanes and co-polymers thereof, celluloses including
alkyl cellulose,
hydroxyalkyl celluloses, cellulose ethers, cellulose esters, nitro celluloses,
methyl cellulose,
ethyl cellulose, hydroxypropyl cellulose, hydroxy-propyl methyl cellulose,
hydroxybutyl
methyl cellulose, cellulose acetate, cellulose propionate, cellulose acetate
butyrate, cellulose
acetate phthalate, carboxylethyl cellulose, cellulose triacetate, and
cellulose sulphate sodium
salt, polypropylene, polyethylenes including poly(ethylene glycol),
poly(ethylene oxide), and
polyethylene terephthalate), and polystyrene.
The polymer can be a biodegradable polymer, including asynthetic biodegradable
polymer (e.g., polymers of lactic acid and glycolic acid, polyanhydrides,
poly(ortho)esters,
polyurethanes, poly(butic acid), poly(valeric acid), and poly(lactide-
cocaprolactone)), and a
natural biodegradable polymer (e.g., alginate and other polysaccharides
including dextran
and cellulose, collagen, chemical derivatives thereof (substitutions,
additions of chemical
groups, for example, alkyl, alkylene, hydroxylations, oxidations, and other
modifications
routinely made by those skilled in the art), albumin and other hydrophilic
proteins (e.g., zein
and other prolamines and hydrophobic proteins)), as well as any copolymer or
mixture
thereof. In general, these materials degrade either by enzymatic hydrolysis or
exposure to
water in vivo, by surface or bulk erosion.
The polymer can be a bioadhesive polymer, such as a bioerodible hydrogel
described
by H. S. Sawhney, C. P. Pathak and J. A. Hubbell in Macromolecules, 1993, 26,
581-587, the
teachings of which are incorporated herein, polyhyaluronic acids, casein,
gelatin, glutin,
polyanhydrides, polyacrylic acid, alginate, chitosan, poly(methyl
methacrylates), poly(ethyl
methacrylates), poly(butylmethacrylate), poly(isobutyl methacrylate),
poly(hexylmethacrylate), poly(isodecyl methacrylate), poly(lauryl
methacrylate),
poly(phenyl methacrylate), poly(methyl acrylate), poly(isopropyl acrylate),
poly(isobutyl
acrylate), and poly(octadecyl acrylate).
In one embodiment, the polymer is a water-soluble polymer. Suitable water-
soluble
polymers are known in the art and include, for example, polyvinylpyrrolidone,
hydroxypropyl cellulose (HPC; Kiucel), hydroxypropyl methylcellulose (HPMC;
Methocel),
nitrocellulose, hydroxypropyl ethylcellulose, hydroxypropyl butylcellulose,
hydroxypropyl
pentylcellulose, methyl cellulose, ethylcellulose (Ethocel), hydroxyethyl
cellulose, various
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-44-
alkyl celluloses and hydroxyalkyl celluloses, various cellulose ethers,
cellulose acetate,
carboxymethyl cellulose, sodium carboxymethyl cellulose, calcium carboxymethyl
cellulose,
vinyl acetate/crotonic acid copolymers, poly-hydroxyalkyl methacrylate,
hydroxymethyl
methacrylate, methacrylic acid copolymers, polymethacrylic acid,
polymethylmethacrylate,
maleic anhydride/methyl vinyl ether copolymers, poly vinyl alcohol, sodium and
calcium
polyacrylic acid, polyacrylic acid, acidic carboxy polymers,
carboxypolymethylene,
carboxyvinyl polymers, polyoxyethylene polyoxypropylene copolymer,
polymethylvinylether co-maleic anhydride, carboxymethylamide, potassium
methacrylate
divinylbenzene co-polymer, polyoxyethyleneglycols, polyethylene oxide, and
derivatives,
salts, and combinations thereof.
In a specific embodiment, the polymer is a polyalkylene glycol, including, for
example, polyethylene glycol (PEG).
The carbohydrate may be any carbohydrate provided that it comprises or is made
to
comprise a carbonyl with an alpha leaving group. The carbohydrate, for
example, may be
one which has been derivatized to comprise a carbonyl with an alpha leaving
group. In this
regard, the carbohydrate may be a derivatized form of a monosaccharide (e.g.,
glucose,
galactose, fructose), a disaccharide (e.g., sucrose, lactose, maltose), an
oligosaccharide (e.g.,.
raffinose, stachyose), a polysaccharide (a starch, amylase, amylopectin,
cellulose, chitin,
callose, laminarin, xylan, mannan, fucoidan, galactomannan.
With respect to the glucagon antagonists comprising a compound of Formula IV,
the
lipid may be any lipid comprising a carbonyl with an alpha leaving group. The
lipid, for
example, may be one which is derivatized to comprise the carbonyl. In this
regard, the lipid,
may be a derivative of a fatty acid (e.g., a C4-C30 fatty acid, eicosanoid,
prostaglandin,
leukotriene, thromboxane, N-acyl ethanolamine), glycerolipid (e.g., mono-, di-
, tri-
substituted glycerols), glycerophospholipid (e.g., phosphatidylcholine,
phosphatidylinositol,
phosphatidylethanolamine, phosphatidylserine), sphingolipid (e.g.,
sphingosine, ceramide),
sterol lipid (e.g., steroid, cholesterol), prenol lipid, saccharolipid, or a
polyketide.
oil, wax, cholesterol, sterol, fat-soluble vitamin, monoglyceride,
diglyceride, triglyceride, a
phospholipid.
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-45-
In one embodiment, R7 has a molecular weight of about 40 kDa or less.
Accordingly,
R7 has a molecular weight of about 35 kDa or less, about 30 kDa or less, about
25 kDa or
less, about 20 kDa or less, about 15 kDa or less, about 10 kDa or less, about
5 kDa or less, or
about 1 kDa.
In an alternative embodiment, the glucagon antagonist comprises as A a peptide
of 2
to 6 amino acids in which two consecutive amino acids of the peptide are
linked via an ester
or ether bond. The peptide may comprise any amino acids, synthetic or
naturally occurring,
provided that at least two consecutive amino acids of the peptide are linked
via an ester or
ether bond. In a specific embodiment, the peptide of A comprises amino acids
of native
glucagon. For example, the peptide can comprise j to 6 of native glucagon (SEQ
ID NO: 1),
wherein j is 1, 2, 3, 4, or 5. Alternatively, the peptide can comprise an
amino acid sequence
based on the N-terminus of SEQ ID NO: 1 with one or more amino acid
modifications. For
instance, the peptide can comprise.at position 1 of the glucagon antagonist an
amino acid
selected from the group consisting of D-histidine, alpha, alpha-dimethyl
imidiazole acetic
acid (DMIA), N-methyl histidine, alpha-methyl histidine, imidazole acetic
acid,
desaminohistidine, hydroxyl-histidine, acetyl-histidine and homo-histidine.
More
particularly, in some embodiments, position 2 of the antagonist peptide is an
amino acid
selected from the group consisting of D-serine, D-alanine, valine, glycine, N-
methyl serine,
N-methyl alanine, and aminoisobutyric acid (AIB). Also, for example, the amino
acid at
position 3 of the glucagon antagonist may be glutamic acid, as opposed to the
native
glutamine residue of native glucagon. Accordingly, the glucagon antagonist can
comprise an
amino acid sequence of:
Xaal-Xaa2-Xaa3-Thr-Gly-Phe (SEQ ID NO: 68);
Xaa2-Xaa3-Thr-Gly-Phe (SEQ ID NO: 69); or
Xaa3-Thr-Gly-Phe (SEQ ID NO: 70);
wherein Xaal is selected from a group consisting of: His, D-histidine, alpha,
alpha-
dimethyl imidiazole acetic acid (DMIA), N-methyl histidine, alpha-methyl
histidine,
imidazole acetic acid, desaminohistidine, hydroxyl-histidine, acetyl-histidine
and homo-
histidine; Xaa2 is selected from a group consisting of. Ser, D-serine, D-
alanine, valine,
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-46-
glycine, N-methyl serine, N-methyl alanine, and aminoisobutyric acid (AIB);
and Xaa3 is Gln
or Glu.
With regard to the glucagon antagonist comprising the general structure A-B-C,
B
represents amino acids of native glucagon, e.g., i to 26 of SEQ ID NO: 1,
wherein i is 3, 4, 5,
6, or 7, optionally comprising one or more amino acid modifications. In a
specific
embodiment, B represents amino acids 7 to 26 of SEQ ID NO: 1, optionally
further modified.
In one embodiment, B is modified by up to three amino acid modifications. For
example, B, which represents native amino acid sequence of SEQ ID NO: 1 is
modified by
one or more conservative amino acid modifications.
In another embodiment, B comprises one or more amino acid modifications
selected
from the group consisting of (iv) to (ix), as described herein. In a specific
embodiment, B
comprises one or both of the amino acid modifications (v) and (vi): In a
further specific
embodiment, B comprises one or a combination of amino acid modifications
selected from
the group consisting of (iv), (vii), (viii), and (ix), in addition to (v) and
(vi).
In another specific embodiment, the glucagon antagonist comprises one or more
charged amino acids at the C-terminus. For example, Y and/or Z can be a
charged amino
acid, e.g., Lys, Arg, His, Asp, and Glu. In yet another embodiment, the
glucagon antagonist
comprises one to two charged amino acids (e.g., Lys, Arg, His, Asp, and Glu) C-
terminal to
Z. In a specific aspect, Z followed by one to two charged amino acids does not
comprise
RIO.
The glucagon antagonist in one embodiment comprises a hydrophilic moiety
covalently bound to an amino acid residue of the glucagon antagonist, as
described herein.
For example, the glucagon antagonist can comprise a hydrophilic moiety
covalently attached
to an amino acid at position 1, 16, 20, 21, or 24 according to the numbering
of SEQ ID NO:
1. In another embodiment, the hydrophilic moiety is attached to the C-terminal
amino acid
of the glucagon antagonist, which in some cases, is 1 or 11 amino acids C-
terminal to Z. In
yet another embodiment, the hydrophilic moiety is attached to PLA, when A is
PLA, PLA-
Phe, or PLA-Thr-Phe, wherein PLA is modified to comprise the hydrophilic
moiety. In
another embodiment, an amino acid comprising a hydrophilic moiety is added to
the N- or C-
terminus of the glucagon antagonist.
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-47-
In another embodiment, the glucagon antagonist comprises an acyl group or
alkyl
group as described herein. For example, the acylation or alkylation can occur
off the side
chain of the amino acid at position 10, 20, or 24, according to the numbering
of SEQ ID NO:
1. In an alternative embodiment, the the acylation or alkylation occurs off
the side chain of
the C-terminal amino acid of the glucagon antagonist, which in some cases, is
1 or 11 amino
acids C-terminal to Z. In yet another embodiment, when A is PLA, PLA-Phe, or
PLA-Thr-
Phe, the PLA is modified to comprise an acyl or alkyl group.
In certain embodiments of the invention, the glucagon antagonist comprises the
amino acid sequence of any of SEQ ID NOs: 62, 64-67, and 71.
The disclosed glucagon antagonists are believed to be suitable for any use
that has
previously been described for other glucagon antagonists. Accordingly, the
modified
glucagon peptides described herein can be used to. treat hyperglycemia, or
treat other
metabolic diseases that result from high blood levels of glucagon or high
blood glucose
levels. In accordance with one embodiment the patient to be treated using the
glucagon
antagonists disclosed herein is a domesticated animal, and in another
embodiment the patient
to be treated is a human. Studies suggest that lack of glucagon suppression in
diabetic
patients contributes to postprandial hyperglycemia in partvia accelerated
glycogenolysis.
Analysis of blood glucose during an. Oral Glucose Tolerance Test (OGTT), and
in the
presence or absence of somatostatin-induced glucagon suppression, has shown a
significant
increase in glucose in subjects with higher glucagon levels. Accordingly, the
glucagon
antagonist of the present invention can be used to treat hyperglycemia, and
are expected to be
useful for treating a variety of types of diabetes including diabetes mellitus
type I, diabetes
mellitus type II, or gestational diabetes, either insulin-dependent or non-
insulin-dependent,
and reducing complications of diabetes including nephropathy, retinopathy and
vascular
disease.
Exendin-4, is a peptide made up of 39 amino acids. It is a powerful stimulator
of a
receptor known as GLP-l. This peptide has also been reported to suppress
appetite and
induce weight loss. Applicants have found that the terminal sequence of
Exendin-4 when
added at the carboxy terminus of glucagon improves the solubility and
stability of glucagon
without compromising the bioactivity of glucagon. In accordance with one
embodiment the
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-48-
glucagon antagonist disclosed herein are administered to patients as a method
of reducing
appetite or promoting loss of body weight. In accordance with one embodiment
the patient is
a domesticated animal, and in another embodiment the patient to be treated is
a human. In
one embodiment the terminal ten amino acids of Exendin-4 (i.e. the sequence of
SEQ ID NO:
19 (GPSSGAPPPS)) are linked to the carboxy terminus of a glucagon antagonists
disclosed
herein. These fusion proteins are anticipated to have pharmacological activity
for
suppressing appetite and inducing weight loss/weight maintenance. In
accordance with one
embodiment the glucagon antagonists disclosed herein can be further modified
to include the
amino acid sequence of SEQ ID NO: 19 (GPSSGAPPPS) linked to amino acid 24 of
the
glucagon antagonist of SEQ ID NO: 42 and administered to individuals to induce
weight loss
or assist in weight maintenance. More particularly, the glucagon peptide
comprises a
sequence selected from the group consisting of SEQ .ID NO:. 2, SEQ ID NO: 3,
SEQ ID NO:
4 SEQ ID NO: 5, SEQ ID NO: 6, SEQ ID NO: 7; SEQ IDNO: 8, SEQ ID, NO: 36, SEQ
ID
NO: 39, SEQ ID NO: 40 SEQ ID NO: 41, SEQ ID NO: 42, SEQ ID NO: 43 and SEQ ID
NO:
44 and further comprising the amino acid sequence of SEQ ID NO: 19
(GPSSGAPPPS)
linked to amino acid 24 of the glucagon antagonist is used to suppress
appetite and inducing
weight loss/weight maintenance. In one embodiment the administered glucagon
antagonist
comprises the sequence of SEQ ID NO: 46 or SEQ ID NO: 47.
Such methods for reducing appetite or promoting loss of body weight are
expected to
be useful in reducing body weight, preventing weight gain, or treating obesity
of various
causes, including drug-induced obesity, and reducing complications associated
with obesity
including vascular disease (coronary artery disease, stroke, peripheral
vascular disease,
ischemia reperfusion, etc.), hypertension, onset of diabetes type II,
hyperlipidemia and
musculoskeletal diseases.
The glucagon peptides of the invention may be administered alone or in
combination
with other anti-diabetic or anti-obesity agents. Anti-diabetic agents known in
the art or under
investigation include insulin, sulfonylureas, such as tolbutamide (Orinase),
acetohexamide
(Dymelor), tolazamide (Tolinase), chlorpropamide (Diabinese), glipizide
(Glucotrol),
glyburide (Diabeta, Micronase, Glynase), glimepiride (Amaryl), or gliclazide
(Diamicron);
meglitinides, such as repaglinide (Prandin) or nateglinide (Starlix);
biguanides such as
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-49-
metformin (Glucophage) or phenformin; thiazolidinediones such as rosiglitazone
(Avandia),
pioglitazone (Actos), or troglitazone (Rezulin), or other PPARy inhibitors;
alpha glucosidase
inhibitors that inhibit carbohydrate digestion, such as miglitol (Glyset),
acarbose
(Precose/Glucobay); exenatide (Byetta) or pramlintide; Dipeptidyl peptidase-4
(DPP-4)
inhibitors such as vildagliptin or sitagliptin; SGLT (sodium-dependent glucose
transporter 1)
inhibitors; or FBPase (fructose 1,6-bisphosphatase) inhibitors.
Anti-obesity agents known in the art or under investigation include appetite
suppressants, including phenethylamine type stimulants, phentermine
(optionally with
fenfluramine or dexfenfluramine), diethylpropion (Tenuate ), phendimetrazine
(Prelu-2 ,
Bontril ), benzphetamine (Didrex ), sibutramine (Meridia , Reductil );
rimonabant
(Acomplia ), other cannabinoid receptor antagonists; oxyntomodulin; fluoxetine
hydrochloride (Prozac); Qnexa (topiramate and phentermine), Excalia (bupropion
and
zonisamide) or Contrave (bupropion and naltrexone); or lipase inhibitors,
similar to xenical
(Orlistat) or Cetilistat (also known as ATL-962), or GT 389-255.
The glucagon antagonists of the present invention can also be administered to
patients
suffering from catabolic wasting. It is estimated that over half of cancer
patients experience
catabolic wasting which is characterized by unintended and progressive weight
loss,
weakness, and low body fat and muscle. The syndrome is equally common in AIDS
patients
and can also be.present in bacterial and parasitic diseases, rheumatoid
arthritis, and chronic
diseases of the bowel, liver, lungs, and heart. It is usually associated with
anorexia and can
manifest as a condition in aging or as a result of physical trauma. Catabolic
wasting is a
symptom that diminishes the quality of life, worsens the underlying condition,
and is a major
cause of death. Applicants anticipate that the glucagon antagonists disclosed
herein can be
administered to patients to treat catabolic wasting.
Pharmaceutical compositions comprising the glucagon antagonists disclosed
herein
can be formulated and administered to patients to using standard
pharmaeuctically acceptable
carriers and routes of administration known to those skilled in the art.
Accordingly the
present disclosure also encompasses pharmaceutical compositions comprising one
or more of
the glucagon antagonists disclosed herein in combination with a
pharmaceutically acceptable
carrier. The pharmaceutical compositions may comprise the glucagon antagonists
as the sole
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-50-
pharmaceutically active component, or the glucagon antagonists can be combined
with one
or more additional active agents. In accordance with one embodiment a
composition is
provided comprising a glucagon antagonist of the present invention and a
compound that
activates the GLP-1 receptor (such as GLP-1, a GLP-1 analog, an exendin-4
analog, or
derivatives thereof). In accordance with one embodiment a composition is
provided
comprising a glucagon antagonist of the present invention and insulin or an
insulin analog.
Alternatively, a composition provided for inducing weight loss or preventing
weight gain can
be provided that comprises the sequence of SEQ ID NO: 42 further comprising
the amino
acid sequence of SEQ ID NO: 19 (GPSSGAPPPS) linked to amino acid 24 of SEQ ID
NO:
42, and an anti-obesity peptide. Suitable anti-obesity peptides include those
disclosed in US
patents 5,691,309, 6,436,435 or US Patent application 20050176643, and
including, but
limited to GLP-1, GIP (Gastric Inhibitory Polypeptide), MP1 , PYY, MC-4,
Leptin.
In accordance with one embodiment a pharmaceutical composition is provided
comprising any of the novel glucagon peptides disclosed herein, preferably
sterile and
preferably at a purity level of at least 90%, 91%, 92%, 93%, 94%, 95%, 96%,
97%, 98% or
99%, and a pharmaceutically acceptable diluent, carrier or excipient. Such
compositions
may contain a glucagon peptide at a concentration of at least 0.5 mg/ml, 1
mg/ml, 2 mg/ml, 3
mg/ml ,.4 mg/ml, 5 mg/ml, 6 mg/ml, 7 mg/ml, 8 mg/ml, 9 mg/ml, 10 mg/ml, 11
mg/ml, 12
mg/ml, 13 mg/ml, 14 mg/ml, 15 mg/ml, 16 mg/ml, 17 mg/ml, 18 mg/ml, 19 mg/ml,
20
mg/ml, 21 mg/ml, 22 mg/ml, 23 mg/ml, 24 mg/ml, 25 mg/ml or higher. In one
embodiment
the pharmaceutical compositions comprise aqueous solutions that are sterilized
and
optionally stored within various containers. The compounds of the present
invention can be
used in accordance with one embodiment to prepare pre-formulated solutions
ready for
injection. In other embodiments the pharmaceutical compositions comprise a
lyophilized
powder. The pharmaceutical compositions can be further packaged as part of a
kit that
includes a disposable device for administering the composition to a patient.
The containers or
kits may be labeled for storage at ambient room temperature or at refrigerated
temperature.
All therapeutic methods, pharmaceutical compositions, kits and other similar
embodiments described herein contemplate that the use of the term glucagon
antagonist
includes all pharmaceutically acceptable salts thereof.
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-51-
Pegylating glucagon antagonists can improve the aqueous solubility of the
antagonists. However, increasing the length of the PEG chain, or attaching
multiple PEG
chains to the peptide, such that the total molecular weight of the linked PEG
is greater than
5,000 Daltons, begins to delay the time action of the modified glucagon
antagonist. In
accordance with one embodiment, a glucagon antagonist is provided wherein the
peptide
comprises one or more polyethylene glycol chains, wherein the total molecular
weight of the
linked PEG is greater than 5,000 Daltons, and in one embodiment is greater
than 10,000
Daltons. Such modified glucagon antagonists have a delayed time of activity
but without
loss of bioactivity. Accordingly, such compounds can be administered
prophylactically to
extend the effect of the administered glucagon antagonist.
In one embodiment the pegylated glucagon antagonist comprises a peptide
selected
from, the group consisting of SEQ ID NO: 8, SEQ ID NO: 9, SEQ IDNO: 10, SEQ
IDNO:
11, SEQ ID NO: 12, SEQ ID NO: 43, SEQ ID NO: 44 and SEQ ID NO: 45, wherein the
side
chain of an amino acid residue at position 11, 16 or 19 of the peptide is
covalently bound to
one or more polyethylene glycol chains, wherein the total molecular weight of
the PEG
chain(s) is greater than about 10,000 Daltons. In one embodiment the molecular
weight of
the PEG chain(s) is greater than 10,000 and less than or equal to 40,000
Daltons. In one
embodiment, the pegylated glucagon antagonist comprises the peptide of SEQ ID
NO: 9 or
SEQ ID NO: 43, wherein an amino acid residue at position 11 of the peptide is
covalently
linked to a polyethylene glycol chain having a molecular weight selected from
the range of
about 10,000 to about 40,000 Daltons. In one embodiment, the pegylated
glucagon
antagonist comprises the peptide of SEQ ID NO: 10 or SEQ ID NO: 44, wherein an
amino
acid residue at position 16 of the peptide is covalently linked to a
polyethylene glycol chain
having a molecular weight selected from the range of about 10,000 to about
40,000 Daltons.
In one embodiment, the pegylated glucagon antagonist comprises the peptide of
SEQ ID NO:
11 or SEQ ID NO: 45, wherein an amino acid residue at position 19 of the
peptide is
covalently linked to a polyethylene glycol chain having a molecular weight
selected from the
range of about 10,000 to about 40,000 Daltons. In another embodiment the
pegylated
glucagon antagonist comprises the peptide of SEQ ID NO: 13, SEQ ID NO. 14, SEQ
ID NO:
15 or SEQ ID NO: 16 wherein the covalently linked PEG chain has a molecular
weight of at
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-52-
least about 10,000 Daltons, and in one embodiment the molecular weight of the
PEG is
selected from the range of about 20,000 to about 40,000 Daltons. In another
embodiment the
pegylated glucagon agonist comprises the peptide of SEQ ID NO: 12, SEQ ID NO:
17 or
SEQ ID NO: 22, wherein a PEG chain is covalently linked to the amino acid
residue at
position 11 and at position 19, wherein the combined molecular weight of the
two PEG
chains is at least about 10,000 Daltons.
The glucagon antagonists disclosed herein can be combined with other active
agents,
including for example, insulin, to treat diseases or conditions that are
characterized by
excessive glucagon activity. In one embodiment, glucagon antagonists that have
been
modified to be covalently bound to a PEG chain having a molecular weight of
greater than
10,000 Daltons can be administered in conjunction with insulin to help to
maintain stable
blood glucose levels in diabetics. The glucagon antagonists of the present
disclosure can be
co-administered with insulin as a single composition, simultaneously
administered as
separate solutions, or alternatively, the insulin and the glucagon antagonist
can be
administered at different times relative to one another. In one embodiment the
composition
comprising. insulin and the composition comprising the glucagon antagonist are
administered
within 12 hours of one another. The exact ratio of the glucagon antagonist
relative to the
administered insulin will be dependent in part on determining the glucagon
levels of the
patient, and can be determined through routine experimentation.
The present disclosure also encompasses multimers of the modified glucagon
antagonists disclosed herein. Two or more of the modified glucagon peptides
can be linked
together using standard linking agents and procedures known to those skilled
in the art. For
example, dimers can be formed between two modified glucagon antagonists
through the use
of bifunctional thiol crosslinkers and bi-functional amine crosslinkers,
particularly for
glucagon antagonists that have been substituted (at positions 11, 16 or 19,
for example) with
cysteine, lysine ornithine, homocysteine or acetyl phenylalanine residues
(e.g. SEQ ID NO:
9, SEQ ID NO: 10, SEQ ID NO: 11 and SEQ ID NO: 12). The dimer can be a
homodimer or
alternatively can be a heterodimer. In one embodiment the dimer is formed
between two
glucagon antagonists independently selected from the group consisting of SEQ
ID NO: 8,
SEQ IDNO: 9, SEQ IDNO: 10, SEQ ID NO: 11, SEQ IDNO: 12, SEQ IDNO: 45, SEQ ID
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-53-
NO: 46, or SEQ ID NO: 47, wherein the two peptides are linked to one another
via a linker
attached to position 11 of each peptide, 16 of each peptide, or position 19 of
each peptide or
any combination thereof. In one embodiment the linkage is a disulfide linkage
between a
Cys 11 to Cys 11 or a Cys 19 to Cys 19 or a Cys 11 to Cys 19 residue of the
respective glucagon
antagonist peptides.
Similarly, a dimer can be formed between two glucagon antagonist peptides
independently selected form the group consisting of SEQ ID NO: 3, SEQ ID NO:
4, SEQ ID
NO: 5, SEQ ID NO: 6, SEQ ID NO: 7, SEQ ID NO: 8, SEQ ID NO: 9, SEQ ID NO: 10,
SEQ
ID NO: 11, SEQ ID NO: 12, SEQ ID NO: 36, SEQ ID NO: 37, SEQ ID NO: 38, SEQ ID
NO: 39 and SEQ ID NO: 42 wherein the linkage is formed between amino acid
positions
independently selected from positions 16, 21 and 24 with respect to the native
glucagon
peptide.
In accordance with one embodiment a glucagon antagonist dimer is provided
comprising two glucagon antagonists, each comprising the sequence of SEQ ID
NO: 46,
wherein the two antagonists are linked to one another by a disulfide bond
through amino acid
position 25. In another embodiment a glucagon antagonist dimer is provided
comprising two
glucagon antagonists, each comprising the sequence of SEQ ID NO: 47, wherein
the two
antagonists are linked to one another by a disulfide bond through amino acid
position 35. In
one embodiment the dimer is formed from glucagon antagonists of SEQ ID NO: 46
and SEQ
ID NO: 47 wherein the amino acid at position 10 is glutamic acid.
In one embodiment the dimer comprises a homodimer of a glucagon antagonist
fusion
peptide selected from the group consisting of SEQ ID NO: 7, SEQ ID NO: 8, SEQ
ID NO:
36, SEQ ID NO: 37, SEQID NO: 40, SEQ ID NO: 39, SEQ ID NO: 40, SEQ ID NO: 41,
SEQ ID NO: 42 and pharmaceutically acceptable salts of said glucagon
antagonists. In
accordance with one embodiment a dimer is provided comprising a first glucagon
antagonist
bound to a second glucagon antagonist via a linker, wherein the first and
second glucagon
peptides of the dimer are independently selected from the group consisting of
SEQ ID NO: 7,
SEQ ID NO: 8, SEQ ID NO: 36, SEQ ID NO: 37, SEQ ID NO: 39, SEQ ID NO: 40, SEQ
ID NO: 41, , and SEQ ID NO: 42, and pharmaceutically acceptable salts of said
glucagon
polypeptides. In another embodiment the first and second glucagon peptides of
the dimer are
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-54-
independently selected from the group consisting of SEQ ID NO: 7, SEQ ID NO:
8, SEQ ID
NO: 36 and SEQ ID NO: 39.
In another embodiment the dimer comprises a homodimer of a glucagon antagonist
selected from the group consisting of SEQ ID NO: 23, SEQ ID NO: 24, SEQ ID NO:
25,
SEQ ID NO: 26, SEQ ID NO: 27, SEQ ID NO: 28, SEQ ID NO: 29, SEQ ID NO: 30, SEQ
ID NO: 31. In another embodiment, a glucagon antagonist dimer is provided
wherein the
first and second glucagon peptides of the dimer comprise an amino acid
sequence
independently selected from the group consisting of SEQ ID NO: 23, SEQ ID NO:
24, SEQ
ID NO: 25, SEQ ID NO: 26, SEQ ID NO: 27 and SEQ ID NO: 28. In another
embodiment
the dimer comprises a homodimer of a glucagon antagonist selected from the
group
consisting of SEQ ID NO: 9, SEQ ID NO: 11 and SEQ ID NO: 12, wherein the
glucagon
peptide further comprises a polyethylene glycol chain covalently bound to
position 11 or 19
of the glucagon peptide.
The modified glucagon peptides of the present invention can be provided in
accordance with one embodiment as part of a kit. In one embodiment a kit for
administering
a glucagon agonist to a patient in need thereof is provided wherein the kit
comprises a
modified glucagon antagonist selected from the group consisting of
1) a glucagon antagonist comprising an amino acid sequence selected from the
group
consisting of SEQ ID NO: 7, SEQ ID NO: 8, SEQ ID NO: 36, SEQ ID NO: 37, SEQ ID
NO:
38, SEQ ID NO: 39, SEQ ID NO: 40, SEQ ID NO: 41, SEQ ID NO: 42, SEQ ID NO: 43
and
SEQ ID NO: 44.
2) a pegylated glucagon antagonist, wherein the PEG chain is covalently bound
to
position 11,12, 15, 16, 19, 24 or 35 of a glucagon antagonist comprising an
amino acid
sequence selected from the group consisting of SEQ ID NO: 9, SEQ ID NO: 10,
SEQ ID
NO: 11, SEQ ID NO: 12, SEQ ID NO: 13, SEQ ID NO: 14, SEQ ID NO: 15, SEQ ID NO:
16, SEQ ID NO: 46 and SEQ ID NO: 47, wherein the PEG chain has a molecular
weight of
about 500 to about 40,000 Daltons;
3) a fusion peptide comprising a glucagon antagonist selected from the group
consisting of SEQ ID NO: 7, SEQ ID NO: 8, SEQ ID NO: 36, SEQ ID NO: 39, SEQ ID
NO:
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-55-
40, SEQ ID NO: 41, SEQ ID NO: 42, SEQ ID NO: 43 and SEQ ID NO: 44, and the
peptide
of SEQ ID NO: 19 fused to the terminal amino acid of said glucagon antagonist;
and
4) a pegylated glucagon antagonist, further comprising an amino acid sequence
of
SEQ ID NO: 19 (GPSSGAPPPS) linked to the carboxy terminus of the glucagon
antagonist,
wherein the covalently bound PEG chain has a molecular weight of about 500 to
about
40,000 Daltons.
In one embodiment the kit is provided with a device for administering the
glucagon
antagonist composition to a patient. The kit may further include a variety of
containers, e.g.,
vials, tubes, bottles, and the like. Preferably, the kits will also include
instructions for use.
In accordance with one embodiment the device of the kit is an aerosol
dispensing device,
wherein the composition is prepackaged within the aerosol device. In another
embodiment
the kit comprises a syringe and a needle, and in. one embodiment the glucagon
antagonist
composition is prepackaged within the syringe.
Incorporation of Alpha, Alpha Di-Substituted Amino Acids
In accordance with one embodiment, a glucagon antagonist is provided
comprising
(either by amino acid substitution or insertion) one or more a, a-
disubstituted amino acids at
the C-terminal portion of the glucagon peptide (around amino acids 12-29
according to the
amino acid numbering of wild type glucagon). In a specific embodiment, the a,
a-
disubstituted amino acid is one of amino iso-butyric acid (AIB), an amino acid
disubstituted
with the same or a different group selected from methyl, ethyl, propyl, and n-
butyl, or with a
cyclooctane or cycloheptane (e.g., 1-aminocyclooctane-1-carboxylic acid). In
some
embodiments, one, two, three, four or more of positions 16, 17, 18, 20, 21, 24
or 29
(according to the amino acid numbering of wild type glucagon) is substituted
with an a, a-
disubstituted amino acid. In a specific embodiment, one, two, three or all of
positions 16, 20,
21, and 24 (according to the amino acid numbering of wild type glucagon) are
substituted
with AIB. In a specific aspect, wherein the glucagon antagonist comprises one
or more a, a-
disubstituted amino acids, e.g., AIB, the glucagon antagonist comprises a C-
terminal
carboxylate and is not amidated at the C-terminus.
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-56-
Linkage of hydrophilic moieties
In one embodiment, the solubility of the glucagon antagonists disclosed herein
are
enhanced by the covalent linkage of a hydrophilic moiety to the peptide.
Hydrophilic
moieties can be attached to the glucagon antagonists under any suitable
conditions used to
react a protein with an activated polymer molecule. Any means known in the art
can be
used, including via acylation, reductive alkylation, Michael addition, thiol
alkylation or other
chemoselective conjugation/ligation methods through a reactive group on the
PEG moiety
(e.g., an aldehyde, amino, ester, thiol, a-haloacetyl, maleimido or hydrazino
group) to a
reactive group on the target compound (e.g., an aldehyde, amino, ester, thiol,
a-haloacetyl,
.10 maleimido or hydrazino group). Activating groups which can be used to link
the water
soluble polymer to one or more proteins include without limitation sulfone,
maleimide,
sulfkzydryl; thiol; triflate, tresylate, azidirine, oxirane and 5=.pyridyl. If
attached to the
antagonist peptide by reductive alkylation, the polymer selected should have a
single reactive
aldehyde so that the degree of polymerization is controlled. See, for example,
Kinstler et al.,
Adv. Drug. Delivery Rev. 54: 477-485 (2002); Roberts et al., Adv. Drug
Delivery Rev. 54:
459-476 (2002); and Zalipsky et al., Adv. Drug Delivery Rev. 16: 157-182
(1995).
Suitable hydrophilic moieties include polyethylene glycol (PEG), polypropylene
glycol, polyoxyethylated polyols (e.g., POG), polyoxyethylated sorbitol,
polyoxyethylated
glucose, polyoxyethylated glycerol (POG), polyoxyalkylenes, polyethylene
glycol
propionaldehyde, copolymers of ethylene glycol/propylene glycol, monomethoxy-
polyethylene glycol, mono-(C1-C10) alkoxy- or aryloxy-polyethylene glycol,
carboxymethylcellulose, polyacetals, polyvinyl alcohol (PVA), polyvinyl
pyrrolidone, poly-
1, 3-dioxolane, poly-1,3,6-trioxane, ethylene/malefic anhydride copolymer,
poly (.beta.-amino
acids) (either homopolymers or random copolymers), poly(n-vinyl
pyrrolidone)polyethylene
glycol, propropylene glycol homopolymers (PPG) and other polyakylene oxides,
polypropylene oxide/ethylene oxide copolymers, colonic acids or other
polysaccharide
polymers, Ficoll or dextran and mixtures thereof.
The hydrophilic moiety, e.g., polyethylene glycol chain in accordance with
some
embodiments has a molecular weight selected from the range of about 500 to
about 40,000
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-57-
Daltons. In one embodiment the hydrophilic moiety, e.g. PEG, has a molecular
weight
selected from the range of about 500 to about 5,000 Daltons, or about 1,000 to
about 5,000
Daltons. In another embodiment the hydrophilic moiety, e.g., PEG, has a
molecular weight
of about 10,000 to about 20,000 Daltons. In yet other exemplary embodiments
the
hydrophilic moiety, e.g., PEG, has a molecular weight of about 20,000 to about
40,000
Daltons.
In one embodiment dextrans are used as the hydrophilic moiety. Dextrans are
polysaccharide polymers of glucose subunits, predominantly linked by al-6
linkages.
Dextran is available in many molecular weight ranges, e.g., about 1 kD to
about 100 kD, or
from about 5, 10, 15 or 20 kD to about 20, 30, 40, 50, 60, 70, 80 or 90 kD.
Linear or branched polymers are contemplated. Resulting preparations of
conjugates
may be essentially monodisperse or polydisperse, and may have about 0.5, 0.7,
1, 1.2, 1.5 or
2 polymer moieties per antagonist peptide.
In one embodiment the hydrophilic moiety is a polyethylene glycol (PEG) chain,
optionally linked to the antagonist at one or more of positions 1, 1.6, 17,
21, 24, 29 (according
to the amino acid numbering of wild type glucagon), a position within a C-
terminal
extension, e.g., 30, or at the N- or C-terminal amino acid. In some
embodiments, the native
amino acid at that position is substituted with an amino acid having a side
chain suitable for
crosslinking with hydrophilic moieties, to facilitate linkage of the
hydrophilic moiety to the
antagonist. In exemplary embodiments, the native amino acid at that position
is substituted
with Lys, Cys, Orn, homocysteine, or acetyl-phenylalanine residue. In other
embodiments,
an amino acid modified to comprise a hydrophilic group is added to the peptide
at the N- or
C-terminus.
Conjugates and fusions
The present disclosure also encompasses other conjugates in which glucagon
antagonists of the invention are linked, optionally via covalent bonding and
optionally via a
linker, to a conjugate moiety. Linkage can be accomplished by covalent
chemical bonds,
physical forces such electrostatic, hydrogen, ionic, van der Waals, or
hydrophobic or
hydrophilic interactions. A variety of non-covalent coupling systems may be
used, including
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-58-
biotin-avidin, ligand/receptor, enzyme/substrate, nucleic acid/nucleic acid
binding protein,
lipid/lipid binding protein, cellular adhesion molecule partners; or any
binding partners or
fragments thereof which have affinity for each other.
The antagonist can be linked to conjugate moieties via direct covalent linkage
by
reacting targeted amino acid residues of the antagonist with an organic
derivatizing agent that
is capable of reacting with selected side chains or the N- or C-terminal
residues of these
targeted amino acids. Reactive groups on the antagonist or conjugate include,
e.g., an
aldehyde, amino, ester, thiol, a-haloacetyl, maleimido or hydrazino group.
Derivatizing
agents include, for example, maleimidobenzoyl sulfosuccinimide ester
(conjugation through
cysteine residues), N-hydroxysuccinimide (through lysine residues),
glutaraldehyde, succinic
anhydride or other agents known in the art. Alternatively, the conjugate
moieties can be
linked to the antagonist indirectly through intermediate carriers, such as
polysaccharide or
polypeptide carriers. Examples of polysaccharide carriers include
aminodextran. Examples
of suitable polypeptide carriers include polylysine, polyglutamic acid,
polyaspartic acid, co-
polymers thereof, and mixed polymers of these amino acids and others, e.g.,
serines, to
confer desirable solubility properties on the resultant loaded carrier.
Cysteinyl residues most commonly are reacted with a-haloacetates (and
corresponding amines), such as chloroacetic acid or chloroacetamide, to give
carboxymethyl
or carboxyamidomethyl derivatives. Cysteinyl residues also are derivatized by
reaction with
bromotrifluoroacetone, alpha-bromo-(3-(5-imidozoyl)propionic acid,
chloroacetyl phosphate,
N-alkylmaleimides, 3-nitro-2-pyridyl disulfide, methyl 2-pyridyl disulfide, p-
chloromercuribenzoate, 2-chloromercuri-4-nitrophenol, or chloro-7-nitrobenzo-2-
oxa-1,3-
diazole.
Histidyl residues are derivatized by reaction with diethylpyrocarbonate at pH
5.5-7.0
because this agent is relatively specific for the histidyl side chain. Para-
bromophenacyl
bromide also is useful; the reaction is preferably performed in 0.1 M sodium
cacodylate at
pH 6Ø
Lysinyl and amino-terminal residues are reacted with succinic or other
carboxylic
acid anhydrides. Derivatization with these agents has the effect of reversing
the charge of the
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-59-
lysinyl residues. Other suitable reagents for derivatizing alpha-amino-
containing residues
include imidoesters such as methyl picolinimidate, pyridoxal phosphate,
pyridoxal,
chloroborohydride, trinitrobenzenesulfonic acid, O-methylisourea, 2,4-
pentanedione, and
transaminase-catalyzed reaction with glyoxylate.
Arginyl residues are modified by reaction with one or several conventional
reagents,
among them phenylglyoxal, 2,3-butanedione, 1,2-cyclohexanedione, and
ninhydrin.
Derivatization of arginine residues requires that the reaction be performed in
alkaline
conditions because of the high pKa of the guanidine functional group.
Furthermore, these
reagents may react with the groups of lysine as well as the arginine epsilon-
amino group.
The specific modification of tyrosyl residues may be made, with particular
interest in
introducing spectral labels into tyrosyl residues by reaction with aromatic
diazonium
compounds or tetranitromethane. Most commonly, N-acetylimidizole and
tetranitromethane
are used to form O-acetyl tyrosyl species and 3-nitro derivatives,
respectively.
Carboxyl side groups (aspartyl or glutamyl) are selectively modified by
reaction with
carbodiimides (R-N=C=N-R'), where R and R' are different alkyl groups, such as
1-
cyclohexyl-3-(2-morpholinyl-4-ethyl) carbodiimide or 1-ethyl-3-(4-azonia-4,4-
dimethylpentyl) carbodiimide. Furthermore, aspartyl and glutamyl residues are
converted to
asparaginyl and glutaminyl residues by reaction with ammonium ions.
Other modifications include hydroxylation of proline and lysine,
phosphorylation of
hydroxyl groups of seryl or threonyl residues, methylation of the alpha-amino
groups of
lysine, arginine, and histidine side chains (T. E. Creighton, Proteins:
Structure and Molecular
Properties, W.H. Freeman & Co., San Francisco, pp. 79-86 (1983)), deamidation
of
asparagines or glutamine, acetylation of the N-terminal amine, and/or
amidation or
esterification of the C-terminal carboxylic acid group.
Another type of covalent modification involves chemically or enzymatically
coupling
glycosides to the antagonist. Sugar(s) may be attached to (a) arginine and
histidine, (b) free
carboxyl groups, (c) free sulfhydryl groups such as those of cysteine, (d)
free hydroxyl
groups such as those of serine, threonine, or hydroxyproline, (e) aromatic
residues such as
those of tyrosine, or tryptophan, or (f) the amide group of glutamine. These
methods are
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-60-
described in W087/05330 published 11 Sep. 1987, and in Aplin and Wriston, CRC
Crit.
Rev. Biochem., pp. 259-306 (1981).
Exemplary conjugate moieties that can be linked to any of the glucagon
antagonists
described herein include but are not limited to a heterologous peptide or
polypeptide
(including for example, a plasma protein), a targeting agent, an
immunoglobulin or portion
thereof (e.g. variable region, CDR, or Fc region), a diagnostic label such as
a radioisotope,
fluorophore or enzymatic label, a polymer including water soluble polymers, or
other
therapeutic or diagnostic agents. In one embodiment a conjugate is provided
comprising a
glucagon antagonist of the present invention and a plasma protein, wherein the
plasma
protein is selected form the group consisting of albumin, transferin,
fibrinogen and globulins.
In some embodiments, the linker comprises a chain of atoms from 1 to about 60,
or 1
to 30 atoms or longer, 2 to 5 atoms, 2 to 10 atoms, 5 to 10 atoms, or 10 to 20
atoms long. In
some embodiments, the chain atoms are all carbon atoms. In some embodiments,
the chain
atoms in the backbone of the linker are selected from the group consisting of
C, 0, N, and S.
Chain atoms and linkers may be selected according to their expected solubility
(hydrophilicity) so as to provide a more soluble conjugate. In some
embodiments, the linker
provides a functional group that is subject to cleavage by an enzyme or other
catalyst or
hydrolytic conditions found in the target tissue or organ or cell. In some
embodiments, the
length of the linker is long enough to reduce the potential for steric
hindrance. If the linker is
a covalent bond or a peptidyl bond and the conjugate is a polypeptide, the
entire conjugate
can be a fusion protein. Such peptidyl linkers may be any length. Exemplary
linkers are
from about 1 to 50 amino acids in length, 5 to 50, 3 to 5, 5 to 10, 5 to 15,
or 10 to 30 amino
acids in length. Such fusion proteins may alternatively be produced by
recombinant genetic
engineering methods known to one of ordinary skill in the art.
As noted above, in some embodiments, the glucagon antagonists are conjugated,
e.g.,
fused to an immunoglobulin or portion thereof (e.g. variable region, CDR, or
Fc region).
Known types of immunoglobulins (Ig) include IgG, IgA, IgE, IgD or IgM. The Fc
region is
a C-terminal region of an Ig heavy chain, which is responsible for binding to
Fc receptors
that carry out activities such as recycling (which results in prolonged half-
life), antibody
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-61-
dependent cell-mediated cytotoxicity (ADCC), and complement dependent
cytotoxicity
(CDC).
For example, according to some definitions the human IgG heavy chain Fc region
stretches from Cys226 to the C-terminus of the heavy chain. The "hinge region"
generally
extends from Glu216 to Pro230 of human IgGI (hinge regions of other IgG
isotypes may be
aligned with the IgGI sequence by aligning the cysteines involved in cysteine
bonding). The
Fc region of an IgG includes two constant domains, CH2 and CH3. The CH2 domain
of a
human IgG Fc region usually extends from amino acids 231 to amino acid 341.
The CH3
domain of a human IgG Fc region usually extends from amino acids 342 to 447.
References
made to amino acid numbering of immunoglobulins or immunoglobulin fragments,
or
regions, are all based on Kabat et al. 1991, Sequences of Proteins of
Immunological Interest,
U.S. Department of Public Health, Bethesda, Md. In a related embodiments, the
Fc region
may comprise, one or more native or modified constant regions from an
immunoglobulin
heavy chain, other than CH1, for example, the CH2 and CH3 regions of IgG and
IgA, or the
CH3 and CH4 regions of IgE.
Suitable conjugate moieties include portions of immunoglobulin sequence that
include the FcRn binding site. FcRn, a salvage receptor, is responsible for
recycling
immunoglobulins and returning them to circulation in blood. The region of the
Fc portion of
IgG that binds to the FcRn receptor has been described based on X-ray
crystallography
(Burmeister et al. 1994, Nature 372:379). The major contact area of the Fc
with the FcRn is
near the junction of the CH2 and CH3 domains. Fc-FcRn contacts are all within
a single Ig
heavy chain. The major contact sites include amino acid residues 248, 250-257,
272, 285,
288, 290-291, 308-311, and 314 of the CH2 domain and amino acid residues 385-
387, 428,
and 433-436 of the CH3 domain.
Some conjugate moieties may or may not include FcyR binding site(s). FcyR are
responsible for ADCC and CDC. Examples of positions within the Fc region that
make a
direct contact with FcyR are amino acids 234-239 (lower hinge region), amino
acids 265-269
(B/C loop), amino acids 297-299 (C'/E loop), and amino acids 327-332 (F/G)
loop
(Sondermann et al., Nature 406: 267-273, 2000). The lower hinge region of IgE
has also
been implicated in the FcRI binding (Henry, et al., Biochemistry 36, 15568-
15578, 1997).
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-62-
Residues involved in IgA receptor binding are described in Lewis et al., (J
Immunol.
175:6694-701, 2005). Amino acid residues involved in IgE receptor binding are
described in
Sayers et al. (J Biol Chem. 279(34):35320-5, 2004).
Amino acid modifications may be made to the Fc region of an immunoglobulin.
Such variant Fc regions comprise at least one amino acid modification in the
CH3 domain of
the Fc region (residues 342-447) and/or at least one amino acid modification
in the CH2
domain of the Fc region (residues 231-341). Mutations believed to impart an
increased
affinity for FcRn include T256A, T307A, E380A, and N434A (Shields et al. 2001,
J. Biol.
Chem. 276:6591). Other mutations may reduce binding of the Fc region to FcyRI,
FcyRIIA,
FcyRIIB, and/or FcyRIIIA without significantly reducing affinity for FcRn. For
example,
substitution of the Asn at position 297 of the Fc region with Ala or another
amino acid
removes a highly conserved N-glycosylation site and may result in reduced
immunogenicity
with concomitant prolonged half-life. of the Fc region, as well as reduced
binding to FcyRs
(Routledge et al. 1995, Transplantation 60:847; Friend et al. 1999,
Transplantation 68:1632;
Shields et al. 1995, J. Biol. Chem. 276:659 1). Amino acid modifications at
positions 233-
236 of IgG1 have been made that reduce binding to FcyRs (Ward and Ghetie 1995,
Therapeutic Immunology 2:77 and Armour et al. 1999, Eur. J. Immunol. 29:2613).
Some
exemplary amino acid substitutions are described in US Patents 7,355,008 and
7,381,408,
each incorporated by reference herein in its entirety.
The present disclosure also encompasses glucagon fusion peptides or proteins
wherein a second peptide or polypeptide has been fused to a terminus, e.g.,
the carboxy
terminus of the glucagon antagonist. In some embodiments the second peptide
added to the
carboxy terminus of the glucagon antagonist is GPSSGAPPPS, KRNRNNIA or KRNR
linked to amino acid 29 of the glucagon antagonist (according to the amino
acid numbering
of wild type glucagon). In other embodiments, the second peptide is
XGPSSGAPPPS,
wherein X is selected from one of the 20 common amino acids, e.g., glutamic
acid, aspartic
acid or glycine. In one embodiment X represents an amino acid, for example
Cys, that
further comprises a hydrophilic moiety covalently linked to the side chain of
that amino acid.
Such C-terminal extensions improve solubility and also can improve glucagon or
GLP-1
activity. In some embodiments wherein the glucagon antagonist further
comprises a carboxy
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-63-
terminal extension, the carboxy terminal amino acid of the extension ends in
an amide group
or an ester group rather than a carboxylic acid.
In some embodiments, e.g., in glucagon antagonists which comprise the C-
terminal
extension, the threonine at position 29 (according to the amino acid numbering
of wild type
glucagon) is replaced with a glycine. For example, a glucagon antagonist
having a glycine
substitution for threonine at position 29 and comprising the C-terminal
extension of
GPSSGAPPPS is four times as potent at the GLP- 1 receptor as native glucagon
modified to
comprise the same C-terminal extension. This T29G substitution can be used in
conjunction
with other modifications disclosed herein to enhance the affinity of the
glucagon antagonists
for the GLP- 1 receptor.
In some embodiments an amino acid is added to the C-terminus, and the
additional
amino-acid is selected from the group,consisting of glutamic acid, aspartic
acid and glycine.
The present disclosure also encompasses multimers of the modified glucagon
antagonists disclosed herein. Two or more of the modified glucagon antagonists
can be
linked together using standard linking agents and procedures known to those
skilled in the
art. For example, dimers can be formed between two modified glucagon
antagonists through
the use of bifunctional thiol crosslinkers and bi-functional amine
crosslinkers, particularly for
the glucagon antagonists that have been substituted with cysteine, lysine
ornithine,
homocysteine or acetyl phenylalanine residues.
Acylation and alkylation
In accordance with some embodiments, the glucagon antagonists disclosed herein
are
modified to comprise an acyl group or alkyl group. Acylation or alkylation can
increase the
half-life of the glucagon antagonists in circulation. Acylation or alkylation
can
advantageously delay the onset of action and/or extend the duration of action
at the glucagon
and/or GLP- 1 receptors and/or improve resistance to proteases such as DPP-IV
and/or
improve solubility. In some embodiments, the potency of the acylated glucagon
antagonists
is comparable to the unacylated versions of the glucagon antagonists. Glucagon
antagonists
may be acylated or alkylated at the same amino acid position where a
hydrophilic moiety is
linked, or at a different amino acid position.
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-64-
In some embodiments, the invention provides a glucagon antagonist modified to
comprise an acyl group or alkyl group covalently linked to the amino acid at
position 10 of
the glucagon antagonist (according to the amino acid numbering of wild type
glucagon). The
glucagon antagonist may further comprise a spacer between the amino acid at
position 10 of
the glucagon antagonist and the acyl group or alkyl group. In some
embodiments, the acyl
group is a fatty acid or bile acid, or salt thereof, e.g. a C4 to C30 fatty
acid, a C8 to C24 fatty
acid, cholic acid, a C4 to C30 alkyl, a C8 to C24 alkyl, or an alkyl
comprising a steroid
moiety of a bile acid. The spacer is any moiety with suitable reactive groups
for attaching
acyl or alkyl groups. In exemplary embodiments, the spacer comprises an amino
acid, a
dipeptide, or a tripeptide, or a hydrophilic bifunctional spacer. In some
embodiments, the
spacer is selected from the group consisting of: Trp, Glu, Asp, Cys and a
spacer comprising
NH2(CH2CH2O)n(CH2)mCOOH, wherein m is any integer from ..1.to 6 and n is any
integer
from 2 to 12. Such acylated or alkylated glucagon antagonists may .also
further comprise a
hydrophilic moiety, optionally a polyethylene glycol. Any of the foregoing
glucagon
antagonists may comprise two acyl groups or two alkyl groups, or a combination
thereof.
Acylation can be carried out at any positions within the glucagon antagonist,
including any of positions 1-29, a position within a C-terminal extension, or
the C-terminal
amino acid, provided that glucagon antagonist activity (and optionally GLP- 1
activity) is
retained. Nonlimiting examples include positions 5, 10, 11, 12, 13, 14, 16,
17, 18, 19, 20, 21,
24, 27, 28, or 29 (according to the amino acid numbering of wild type
glucagon). The acyl
group can be covalently linked directly to an amino acid of the glucagon
antagonist, or
indirectly to an amino acid of the glucagon antagonist via a spacer, wherein
the spacer is
positioned between the amino acid of the glucagon antagonist and the acyl
group. Glucagon
antagonists may be acylated at the same amino acid position where a
hydrophilic moiety is
linked, or at a different amino acid position. Nonlimiting examples include
acylation at
position 10 (according to the amino acid numbering of wild type glucagon) and
pegylation at
one or more positions in the C-terminal portion of the glucagon antagonist,
e.g., position 24,
28 or 29 (according to the amino acid numbering of wild type glucagon), within
a C-terminal
extension, or at the C-terminus (e.g., through adding a C-terminal Cys).
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-65-
In a specific aspect of the invention, the glucagon antagonist is modified to
comprise
an acyl group by direct acylation of an amine, hydroxyl, or thiol of a side
chain of an amino
acid of the glucagon antagonist. In some embodiments, the glucagon antagonist
is directly
acylated through the side chain amine, hydroxyl, or thiol of an amino acid. In
some
embodiments, acylation is at position 10, 20, 24, or 29 (according to the
amino acid
numbering of wild type glucagon). In this regard, the acylated glucagon
antagonist can
comprise the amino acid sequence of SEQ ID NO : 2, or a modified amino acid
sequence
thereof comprising one or more of the amino acid modifications described
herein, with at
least one of the amino acids at positions 10, 20, 24, and 29 (according to the
amino acid
numbering of wild type glucagon) modified to any amino acid comprising a side
chain
amine, hydroxyl, or thiol. In some specific embodiments of the invention, the
direct
acylationofthe glucagon antagonist occurs through the side chain amine,
hydroxyl, or thiol.
of the:amino acid at position 10 (according to the amino acid numbering of
wild type
glucagon).
In some embodiments, the amino acid comprising a side chain amine is an amino
acid
of Formula I:
H
H2N C COOH
(CH
Al
NH2
wherein n = 1 to 4
[Formula I]
In some exemplary embodiments, the amino acid of Formula I, is the amino acid
wherein n is
4 (Lys) or n is 3 (Orn).
In other embodiments, the amino acid comprising a side chain hydroxyl is an
amino
acid of Formula II:
H
H2N C COOH
I
(CH2)n
I
OH
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-66-
wherein n = 1 to 4
[Formula II]
In some exemplary embodiments, the amino acid of Formula II is the amino acid
wherein n
is 1 (Ser).
In yet other embodiments, the amino acid comprising a side chain thiol is an
amino
acid of Formula III:
H
H2N i COOH
(CH2)n
SH
wherein n = 1 to 4
[Formula III]
In some exemplary embodiments, the amino acid of Formula II is the amino acid
wherein n
is 1 (Cys).
In one embodiment of the invention, the acylated glucagon antagonist comprises
a
spacer between the antagonist and the acyl group. In some embodiments, the
glucagon
antagonist is covalently bound to the spacer, which is covalently bound to the
acyl group. In
some exemplary embodiments, the glucagon antagonist is modified to comprise an
acyl
group by acylation of an amine, hydroxyl, or thiol of a spacer, which spacer
is attached to a
side chain of an amino acid at position 10, 20, 24, or 29 (according to the
amino acid
numbering of wild type glucagon), or at the C-terminal amino acid of the
glucagon
antagonist. The amino acid to which the spacer is attached can be any amino
acid
comprising a moiety which permits linkage to the spacer. For example, an amino
acid
comprising a side chain NI12, -OH, or -COOH (e.g., Lys, Orn, Ser, Asp, or Glu)
is suitable.
In this respect, the acylated glucagon antagonist can comprise the amino acid
sequence of
SEQ ID NO: 1, or a modified amino acid sequence thereof comprising one or more
of the
amino acid modifications described herein, with at least one of the amino
acids at positions
10, 20, 24, and 29 (according to the amino acid numbering of wild type
glucagon) modified
to any amino acid comprising a side chain amine, hydroxyl, or carboxylate.
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-67-
In some embodiments, the spacer is an amino acid comprising a side chain
amine,
hydroxyl, or thiol, or a dipeptide or tripeptide comprising an amino acid
comprising a side
chain amine, hydroxyl, or thiol.
When acylation occurs through an amine group of a spacer the acylation can
occur
through the alpha amine of the amino acid or a side chain amine. In the
instance in which the
alpha amine is acylated, the spacer amino acid can be any amino acid. For
example, the
spacer amino acid can be a hydrophobic amino acid, e.g., Gly, Ala, Val, Leu,
Ile, Trp, Met,
Phe, Tyr. Alternatively, the spacer amino acid can be an acidic residue, e.g.,
Asp and Glu.
In the instance in which the side chain amine of the spacer amino acid is
acylated, the spacer
amino acid is an amino acid comprising a side chain amine, e.g., an amino acid
of Formula I
(e.g., Lys or Orn). In this instance, it is possible for both the alpha amine
and the side chain
amine of the spacer amino acid to be acylated, such. that the glucagon
antagonist is
diacylated. Embodiments of the invention include such diacylated molecules.
When acylation occurs through a hydroxyl group of a spacer, the amino acid or
one of
the amino acids of the dipeptide or tripeptide can be an amino acid of Formula
II. In a
specific exemplary embodiment, the amino acid is Ser.
When acylation occurs through a thiol group of a spacer, the amino acid or one
of the
amino acids of the dipeptide or tripeptide can be an amino acid of Formula
III. In a specific
exemplary embodiment, the amino acid is Cys.
In one embodiment, the spacer comprises a hydrophilic bifunctional spacer. In
a
specific embodiment, the spacer comprises an amino poly(alkyloxy)carboxylate.
In this
regard, the spacer can comprise, for example, NH2(CH2CH2O)õ (CH2)m000H,
wherein m is
any integer from 1 to 6 and n is any integer from 2 to 12, such as, e.g., 8-
amino-3,6-
dioxaoctanoic acid, which is commercially available from Peptides
International, Inc.
(Louisville, KY).
Suitable methods of peptide acylation via amines, hydroxyls, and thiols are
known in
the art. See, for example, Example 19 (for methods of acylating through an
amine), Miller,
Biochem Biophys Res Commun 218: 377-382 (1996); Shimohigashi and Stammer, Int
J Pept
Protein Res 19: 54-62 (1982); and Previero et al., Biochim Biophys Acta 263: 7-
13 (1972)
(for methods of acylating through a hydroxyl); and San and Silvius, J Pept Res
66: 169-180
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-68-
(2005) (for methods of acylating through a thiol); Bioconjugate Chem.
"Chemical
Modifications of Proteins: History and Applications" pages 1, 2-12 (1990);
Hashimoto et al.,
Pharmacuetical Res. "Synthesis of Palmitoyl Derivatives of Insulin and their
Biological
Activity" Vol. 6, No: 2 pp.171-176 (1989)..
The acyl group of the acylated glucagon antagonist can be of any size, e.g.,
any
length carbon chain, and can be linear or branched. In some specific
embodiments of the
invention, the acyl group is a C4 to C30 fatty acid. For example, the acyl
group can be any
of a C4 fatty acid, C6 fatty acid, C8 fatty acid, C10 fatty acid, C12 fatty
acid, C14 fatty acid,
C16 fatty acid, C18 fatty acid, C20 fatty acid, C22 fatty acid, C24 fatty
acid, C26 fatty acid,
C28 fatty acid, or a C30 fatty acid. In some embodiments, the acyl group is a
C8 to C20 fatty
acid, e.g., a C14 fatty acid or a C16 fatty acid.
In an alternative embodiment, the acyl group. is a bile acid. The bile acid
can be any
suitable bile acid, including, but not limited to, cholic acid,
chenodeoxycholic acid,
deoxycholic acid, lithocholic acid, taurocholic acid, glycocholic acid, and
cholesterol acid.
The acylated glucagon antagonists described herein can be further modified to
comprise a hydrophilic moiety. In some specific embodiments the hydrophilic
moiety can
comprise a polyethylene glycol (PEG) chain.- The incorporation of a
hydrophilic moiety can
be accomplished through any suitable. means, such as any of the methods
described herein.
In this regard, the acylated glucagon antagonist can. comprise SEQ ID NO: 1,
including any
of the modifications described herein, in which (a) at least one of the amino
acids at position
10, 20, 24, and 29 (according to the amino acid numbering of wild type
glucagon) comprise
an acyl group and (b) at least one of the amino acids at position 16, 17, 21,
24, or 29
(according to the amino acid numbering of wild type glucagon), a position
within a C-
terminal extension, or the C-terminal amino acid are modified to a Cys, Lys,
Orn, homo-Cys,
or Ac-Phe, and the side chain of the amino acid is covalently bonded to a
hydrophilic moiety
(e.g., PEG). In some embodiments, the acyl group is attached to position 10
(according to
the amino acid numbering of wild type glucagon), optionally via a spacer
comprising Cys,
Lys, Orn, homo-Cys, or Ac-Phe, and the hydrophilic moiety is incorporated at a
Cys residue
at position 24 (according to the amino acid numbering of wild type glucagon).
Alternatively, the acylated glucagon antagonist can comprise a spacer, wherein
the
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-69-
spacer is both acylated and modified to comprise the hydrophilic moiety.
Nonlimiting
examples of suitable spacers include a spacer comprising one or more amino
acids selected
from the group consisting of Cys, Lys, Om, homo-Cys, and Ac-Phe.
In accordance with one embodiment, the glucagon antagonist is modified to
comprise
an alkyl group which is attached to the glucagon antagonist via an ester,
ether, thioether,
amide, or alkyl amine linkage for purposes of prolonging half-life in
circulation and/or
delaying the onset of and/or extending the duration of action and/or improving
resistance to
proteases such as DPP-IV.
Alkylation can be carried out at any positions within the glucagon antagonist,
including any of positions 1-29, a position within a C-terminal extension, or
the C-terminal
amino acid, provided that the glucagon antagonist activity (and optionally GLP-
1 activity) is
retained.! Nonlimiting examples include positions 5, 10, 11 12,13, 14, 16, 17,
18, 19, 20, 21,
24, 27, 28, or 29 (according to the amino acid numbering of wild type
glucagon). The alkyl
group can be covalently linked directly to an amino acid of the glucagon
antagonist, or
indirectly to an amino acid of the glucagon antagonist via a spacer, wherein
the spacer is
positioned between the amino acid of the glucagon antagonist and the alkyl
group. Glucagon
antagonists may be alkylated at the same amino acid position where a
hydrophilic moiety is
linked, or at a different amino acid position. Nonlimiting examples include
alkylation at
position 10 (according to the amino acid numbering of wild type glucagon) and
pegylation at
one or more positions in the C-terminal portion of the glucagon antagonist,
e.g., position 24,
28 or 29 (according to the amino acid numbering of wild type glucagon), within
a C-terminal
extension, or at the C-terminus (e.g., through adding a C-terminal Cys).
In a specific aspect of the invention, the glucagon antagonist is modified to
comprise
an alkyl group by direct alkylation of an amine, hydroxyl, or thiol of a side
chain of an amino
acid of the glucagon antagonist. In some embodiments, the glucagon antagonist
is directly
alkylated through the side chain amine, hydroxyl, or thiol of an amino acid.
In some
embodiments, alkylation is at position 10, 20, 24, or 29 (according to the
amino acid
numbering of wild type glucagon). In this regard, the alkylated glucagon
antagonist can
comprise the amino acid sequence of SEQ ID NO : 2, or a modified amino acid
sequence
thereof comprising one or more of the amino acid modifications described
herein, with at
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-70-
least one of the amino acids at positions 10, 20, 24, and 29 (according to the
amino acid
numbering of wild type glucagon) modified to any amino acid comprising a side
chain
amine, hydroxyl, or thiol. In some specific embodiments of the invention, the
direct
alkylation of the glucagon antagonist occurs through the side chain amine,
hydroxyl, or thiol
of the amino acid at position 10 (according to the amino acid numbering of
wild type
glucagon).
In some embodiments, the amino acid comprising a side chain amine is an amino
acid
of Formula I. In some exemplary embodiments, the amino acid of Formula I, is
the amino
acid wherein n is 4 (Lys) or n is 3 (Orn).
In other embodiments, the amino acid comprising a side chain hydroxyl is an
amino
acid of Formula II. In some exemplary embodiments, the amino acid of Formula
II is the
amino acid wherein n is 1 (Ser).
In yet other embodiments, the amino, acid comprising a side chain thiol is an
amino
acid of Formula III. In some exemplary embodiments, the amino acid of Formula
II is the
amino acid wherein n is 1 (Cys).
In one embodiment of the invention, the alkylated glucagon antagonist
comprises a
spacer between the antagonist and the alkyl group. In some embodiments, the
glucagon
antagonist is covalently bound to the spacer, which is. covalently bound to
the alkyl group. In
some exemplary embodiments, the glucagon antagonist is modified to comprise an
alkyl
group by alkylation of an amine, hydroxyl, or thiol of a spacer, which spacer
is attached to a
side chain of an amino acid at position 10, 20, 24, or 29 of the glucagon
antagonist
(according to the amino acid numbering of wild type glucagon). The amino acid
to which
the spacer is attached can be any amino acid comprising a moiety which permits
linkage to
the spacer. For example, an amino acid comprising a side chain NH2, -OH, or -
COOH (e.g.,
Lys, Om, Ser, Asp, or Glu) is suitable. In this respect, the alkylated
glucagon antagonist can
comprise the amino acid sequence of SEQ ID NO: 1, or a modified amino acid
sequence
thereof comprising one or more of the amino acid modifications described
herein, with at
least one of the amino acids at positions 10, 20, 24, and 29 (according to the
amino acid
numbering of wild type glucagon) modified to any amino acid comprising a side
chain
amine, hydroxyl, or carboxylate.
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-71-
In some embodiments, the spacer is an amino acid comprising a side chain
amine,
hydroxyl, or thiol or a dipeptide or tripeptide comprising an amino acid
comprising a side
chain amine, hydroxyl, or thiol.
When alkylation occurs through an amine group of a spacer the alkylation can
occur
through the alpha amine of the amino acid or a side chain amine. In the
instance in which the
alpha amine is alkylated, the spacer amino acid can be any amino acid. For
example, the
spacer amino acid can be a hydrophobic amino acid, e.g., Gly, Ala, Val, Leu,
Ile, Trp, Met,
Phe, Tyr. Alternatively, the spacer amino acid can be an acidic residue, e.g.,
Asp and Glu.
In the instance in which the side chain amine of the spacer amino acid is
alkylated, the spacer
amino acid is an amino acid comprising a side chain amine, e.g., an amino acid
of Formula I
(e.g., Lys or Orn). In this instance, it is possible for both the alpha amine
and the side chain
amine of the spacer amino acid to be alkylated, such that the glucagon
antagonist is
dialkylated. Embodiments of the invention include such dialkylated molecules.
When alkylation occurs through a hydroxyl group of a spacer, the amino acid or
one
of the amino acids of the dipeptide or tripeptide can be an amino acid of
Formula II. In a
specific exemplary embodiment, the amino acid is Ser.
When acylation occurs through a.thiol group of spacer, the amino acid or one
of the
amino acids of the dipeptide or tripeptide can be an amino acid of Formula
III.. In a specific
exemplary embodiment, the amino acid is Cys.
In one embodiment, the spacer comprises a hydrophilic bifunctional spacer. In
a
specific embodiment, the spacer comprises an amino poly(alkyloxy)carboxylate.
In this
regard, the spacer can comprise, for example, NH2(CH2CH2O)n(CH2)mCOOH, wherein
m is
any integer from 1 to 6 and n is any integer from 2 to 12, such as, e.g., 8-
amino-3,6-
dioxaoctanoic acid, which is commercially available from Peptides
International, Inc.
(Louisville, KY).
Suitable methods of peptide alkylation via amines, hydroxyls, and thiols are
known in
the art. For example, a Williamson ether synthesis can be used to form an
ether linkage
between the glucagon antagonist and the alkyl group. Also, a nucleophilic
substitution
reaction of the peptide with an alkyl halide can result in any of an ester,
ether, thioether,
amide, or alkyl amine linkage.
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-72-
The alkyl group of the alkylated glucagon antagonist can be of any size, e.g.,
any
length carbon chain, and can be linear or branched. In some embodiments of the
invention,
the alkyl group is a Cl to C30 alkyl. For example, the alkyl group can be any
of a C4 alkyl,
C6 alkyl, C8 alkyl, C10 alkyl, C12 alkyl, C14 alkyl, C16 alkyl, C18 alkyl, C20
alkyl, C22
alkyl, C24 alkyl, C26 alkyl, C28 alkyl, or a C30 alkyl. In some embodiments,
the alkyl
group is a C8 to C20 alkyl, e.g., a C14 alkyl or a C16 alkyl.
In some specific embodiments, the alkyl group comprises a steroid moiety of a
bile
acid, e.g., cholic acid, chenodeoxycholic acid, deoxycholic acid, lithocholic
acid, taurocholic
acid, glycocholic acid, and cholesterol acid.
The alkylated glucagon antagonists described herein can be further modified to
comprise a hydrophilic moiety. In some specific embodiments the hydrophilic
moiety can
comprise a polyethylene glycol (PEG) chain. The. incorporation of a
hydrophilic moiety can
be accomplished through any suitable means, such::as any of the methods
described herein.
In this regard, the alkylated glucagon antagonist can comprise SEQ ID NO: 1,
or a modified
amino acid sequence thereof comprising one or more of the amino acid
modifications
described herein, in which (a) at least one of the amino acids at position 10,
20, 24, and 29
(according to the amino acid numbering of wild type glucagon) comprise an
alkyl group ' and
(b) at least one of the amino acids at position 16, 17, 21, 24, and 29
(according to the amino
acid numbering of wild type glucagon), a position within a C-terminal
extension or the C-
terminal amino acid are modified to a Cys, Lys, Orn, homo-Cys, or Ac-Phe, and
the side
chain of the amino acid is covalently bonded to a hydrophilic moiety (e.g.,
PEG). In some
embodiments, the alkyl group is attached to position 10 (according to the
amino acid
numbering of wild type glucagon), optionally via a spacer comprising Cys, Lys,
Orn, homo-
Cys, or Ac-Phe, and the hydrophilic moiety is incorporated at a Cys residue at
position 24
(according to the amino acid numbering of wild type glucagon).
Alternatively, the alkylated glucagon antagonist can comprise a spacer,
wherein the
spacer is both alkylated and modified to comprise the hydrophilic moiety.
Nonlimiting
examples of suitable spacers include a spacer comprising one or more amino
acids selected
from the group consisting of Cys, Lys, Orn, homo-Cys, and Ac-Phe.
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-73-
EXAMPLES
The compounds of this invention may be prepared by standard synthetic methods,
recombinant DNA techniques, or any other methods of preparing peptides and
fusion
proteins. Although certain non-natural amino acids cannot be expressed by
standard
recombinant DNA techniques, techniques for their preparation are known in the
art.
Compounds of this invention that encompass non-peptide portions may be
synthesized by
standard organic chemistry reactions, in addition to standard peptide
chemistry reactions
when applicable.
General Synthesis Protocol:
Glucagon analogs were synthesized using HBTU-activated "Fast Boc" single
coupling starting from 0.2mmole of Boc Thr(OBzl)Pam: resin on a modified
Applied
Biosystem 430 A peptide synthesizer. Boc amino acids andHBTU were obtained
from
Midwest Biotech (Fishers, IN). Side chain protecting groups, used were:
Arg(Tos),
Asn(Xan), Asp(OcHex), Cys(pMeBzl), His(Bom), Lys(2C1-Z), Ser(OBzl), Thr(OBzl),
Tyr(2Br-Z), and Trp(CHO). The side-chain protecting group on the N-terminal
His was Boc.
Each completed peptidyl resin was treated with a solution of 20% piperdine in
dimethylformamide to remove the formyl group from the tryptophan. Liquid
hydrogen
fluoride cleavages were performed in the presence of p-cresol and dimethyl
sulfide. The
cleavage was run for 1 hour in an ice bath using an HF apparatus (Penninsula
Labs). After
evaporation of the HF, the residue was suspended in diethyl ether and the
solid materials
were filtered. Each peptide was extracted into 30-70m1 aqueous acetic acid and
a diluted
aliquot was analyzed by HPLC [Beckman System Gold, 0.46x5cm Zorbax C8,
lml/min,
45C, 214nm, A buffer =0.1%TFA, B=0.1%TFA/90%acetonitrile, gradient of 10% to
80%B
over 10min].
Purification was done on a FPLC over a 2.2 x 25 cm Kromasil C 18 column while
monitoring the UV at 214nm and collecting 5 minute fractions. The homogeneous
fractions
were combined and lyophilized to give a product purity of >95%. The correct
molecular
mass and purity were confirmed using MALDI-mass spectral analysis.
General Pegylation Protocol: (Cys-maleimido)
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-74-
Typically, the glucagon Cys analog is dissolved in phosphate buffered saline
(5-
10mg/ml) and 0.O1M ethylenediamine tetraacetic acid is added (10-15% of total
volume).
Excess (2-fold) maleimido methoxyPEG reagent (Nektar) is added and the
reaction stirred at
room temp while monitoring reaction progress by HPLC. After 8-24hrs, the
reaction
mixture, is acidified and loaded onto a preparative reverse phase column for
purification
using 0.1 %TFA/acetonitrile gradient. The appropriate fractions were combined
and
lyophilized to give the desired pegylated derivatives.
EXAMPLE 1
Synthesis of Glucagon Cys17(1-29) and Similar MonoCys Analogs
0.2mmole Boc Thr(OBzI) Pam resin (SynChem Inc) in a 60ml reaction vessel and
the
following sequence was. entered and run on a modified Applied Biosystems 430A
Peptide
Synthesizer using FastBoc HBTU-activated single couplings.
HSQGTFTSDYSKYLDSCRAQDFVQWLMNT=(SEQ ID NO: 32)
The following side chain protecting groups were used: Arg(Tos), Asp(OcHex),
Asn(Xan),
Cys(pMeBzl), Glu(OcHex), His(Boc), Lys(2Cl-Z), Ser(Bzl), Thr(Bzl), Trp(CHO),
and
Tyr(Br-Z). The completed peptidyl resin was treated with 20%
piperidine/dimethylformamide to remove the Trp formyl protection then
transferred to an HF
reaction vessel and dried in vacuo. 1.Oml p-cresol and 0.5 ml dimehyl sulfide
were added
along with a magnetic stir bar. The vessel was attached to the HF apparatus
(Pennisula
Labs), cooled in a dry ice/methanol bath, evacuated, and aprox. 10ml liquid
hydrogen
fluoride was condensed in. The reaction was stirred in an ice bath for lhr
then the HF was
removed in vacuo. The residue was suspended in ethyl ether; the solids were
filtered,
washed with ether, and the peptide extracted into 50 ml aqueous acetic acid.
An analytical
HPLC was run [0.46 x 5 cm Zorbax C8, 1 ml/min, 45C, 214nm, A buffer of
0.1%TFA, B
buffer of 0. 1 %TFA/90%ACN, gradient=10%B to 80%B over 10min.] with a small
sample of
the cleavage extract. The remaining extract was loaded onto a 2.2 x 25cm
Kromasil C18
preparative reverse phase column and an acetonitrile gradient was run using a
Pharmacia
FPLC system. 5min fractions were collected while monitoring the UV at 214nm
(2.OA).
A=0. l %TFA, B=0.1%TFA/50%acetonitrile. Gradient = 30%B to 100%B over 450min.
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-75-
The fractions containing the purest product (48-52) were combined frozen, and
lyophilized to give 30.1mg. An HPLC analysis of the product demonstrated a
purity of
>90% and MALDI mass spectral analysis demonstrated the desired mass of 3429.7.
Glucagon Cys21, Glucagon Cys24, and Glucagon Cys29 were similarly prepared.
EXAMPLE 2
Synthesis of Glucagon-Cex and Other C-Terminal Extended Analogs.
285mg (0.2mmole) methoxybenzhydrylamine resin (Midwest Biotech) was placed in
a 60m1 reaction vessel and the following sequence was entered and run on a
modified
Applied Biosystems 430A peptide synthesizer using FastBoc HBTU-activated
single
couplings.
HSQGTFTSDYSKYLDSRRAQDFVQWLMNTGPSSGAPPPS (SEQ ID NO: 33)
The following side chain protecting groups were used: Arg(Tos), Asp(OcHex),
Asn(Xan),
Cys(pMeBzl), Glu(OcHex), His(Boc), Lys(2C1-Z), Ser(Bzl), Thr(Bzl), Trp(CHO),
and
Tyr(Br-Z). The completed peptidyl resin was treated with 20%
piperidine/dimethylformamide to remove the Trp formyl protection then
transferred to HF
reaction vessel and dried in vacuo. 1.0ml p-cresol and 0.5 ml dimehyl sulfide
were added
along with a magnetic stir bar. The vessel was attached to the HF apparatus
(Pennisula
Labs), cooled in a dry ice/methanol bath, evacuated, and aprox. lOml liquid
hydrogen
fluoride was condensed in. The reaction was stirred in an ice bath for lhr
then the HF was
removed in vacuo. The residue was suspended in ethyl ether; the solids were
filtered,
washed with ether, and the peptide extracted into 50 ml aqueous acetic acid.
An analytical
HPLC was run [0.46 x 5 cm Zorbax C8, 1 ml/min, 45C, 214nm, A buffer of
0.1%TFA, B
buffer of 0. 1 %TFA/90%ACN, gradient=10%B to 80%B over 10min.] on an aliquot
of the
cleavage extract. The extract was loaded onto a 2.2 x 25cm Kromasil C18
preparative
reverse phase column and an acetonitrile gradient was run for elution using a
Pharmacia
FPLC system. 5min fractions were collected while monitoring the UV at 214nm
(2.OA).
A=0.1 %TFA, B=0.1 %TFA/50%acetonitrile. Gradient = 30%B to 100%B over 450min.
Fractions 58-65 were combined, frozen and lyophilized to give 198.1mg.
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-76-
HPLC analysis of the product showed a purity of greater than 95%. MALDI mass
spectral analysis showed the presence of the desired theoretical mass of
4316.7 with the
product as a C-terminal amide. Oxyntomodulin and oxyntomodulin-KRNR were
similarly
prepared as the C-terminal carboxylic acids starting with the appropriately
loaded PAM-
resin.
EXAMPLE 3
Glucagon Cys17 Mal-PEG-5K
15.1mg of Glucagon Cys17(1-29) and 27.3mg methoxy poly(ethyleneglycol)
maleimide avg. M.W.5000 (mPEG-Mal-5000,Nektar Therapeutics) were dissolved in
3.5ml
phosphate buffered saline (PBS) and 0.5m1 0.01M ethylenediamine tetraacetic
acid (EDTA)
was added. The reaction was stirred at room temperature and the progress of
the reaction
was.monitored by HPLC analysis [0.46 x 5 cm Zorbax C8, lml/min,45C, 214nm
(0.5A), .. 0
A=0.1%TFA, B=0.1%TFA/90%ACN, gradient=10%B to 80%B over 10min.].
After 5 hours, the reaction mixture was loaded onto 2.2 x 25 cm Kromasil C18
preparastive
reverse phase column. An acetonitrile gradient was run on a Pharmacia FPLC
while
monitoring the UV wavelength at 214nm and collecting 5 min fractions.
A=0.1%TFA,
B=0.1%aTFA/50% acetonitrile, gradient= 30%B to 100%B over 450 min. The
fractions
corresponding to the product were combined, frozen and lyophilized to give
25.9 mg.
This product was analyzed on HPLC [0.46 x 5 cm Zorbax C8, 1 ml/min, 45C, 214nm
(0.5A), A=0.1%TFA, B=0.1%TFA/90%ACN, gradient=10%B to 80%B over 10min.] which
showed a purity of aprox. 90%. MALDI (matrix assisted laser desorption
ionization) mass
spectral analysis showed a broad mass range (typical of PEG derivatives) of
8700 to 9500.
This shows an addition to the mass of the starting glucagon peptide (3429) of
approximately
5,000 a.m.u.
EXAMPLE 4
Glucagon Cys21 Mal-PEG-5K
21.6mg of Glucagon Cys21(1-29) and 24mg mPEG-MAL-5000 (Nektar Therapeutics)
were dissolved in 3.5ml phosphate buffered saline (PBS) and 0.5ml 0.01M
ethylene diamine
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-77-
tetraacetic acid (EDTA) was added. The reaction was stirred at room temp.
After 2hrs,
another 12.7 mg of mPEG-MAL-5000 was added. After 8hrs, the reaction mixture
was
loaded onto a 2.2 x 25cm Vydac C18 preparative reverse phase column and an
acetonitrile
gradient was run on a Pharmacia FPLC at 4 ml/min while collecting 5min
fractions.
A=0.1 %TFA, B=0.1 %TFA/50%ACN. Gradient= 20% to 80%B over 450min.
The fractions corresponding to the appearance of product were combined frozen
and
lyophilized to give 34 mg. Analysis of the product by analytical HPLC [0.46 x
5 cm Zorbax
C8, 1 ml/min, 45C, 214nm (0.5A), A=0.1%TFA, B=0. i %TFA/90%ACN, gradient=10%B
to
80%B over 10min.] showed a homogeneous product that was different than
starting glucagon
peptide. MALDI (matrix assisted laser desorption ionization) mass spectral
analysis showed
a broad mass range (typical of PEG derivatives) of 8700 to 9700. This shows an
addition to
the mass of the starting glucagon peptide (3470) of approximately 5,000 a.m.u.
EXAMPLE 5
Glucagon Cys24 Mal-PEG-5K
20.1mg Glucagon C24(1-29) and 39.5mg mPEG-Mal-5000 (Nektar Therapeutics)
were dissolved in 3.5m1 PBS with stirring and 0.5 ml 0.01M EDTA was added. The
reaction
was stirred at room temp for 7 hrs, then another 40 mg of mPEG-Mal-5000 was
added. After
approximately 15 hr, the reaction mixture was loaded onto=a 2.2 x 25 cm Vydac
C18
preparative reverse phase column and an acetontrile gradient was run using a
Pharmacia
FPLC. 5 min. fractions were collected while monitoring the UV at 214nm (2.OA).
A buffer =
0.1 %TFA, B buffer = 0.1 %TFA/50%ACN, gradient = 30%B to 100%B over 450min.
The
fractions corresponding to product were combined, frozen and lyophilized to
give 45.8mg.
MALDI mass spectral analysis showed a typical PEG broad signal with a maximum
at
9175.2 which is approximately 5,000 a.m.u. more than Glucagon C24 (3457.8).
EXAMPLE 6
Glucagon Cys24 Mal-PEG-20K
25.7mg of Glucagon Cys24(1-29) and 40.7mg mPEG-Mal-20K (Nektar Therapeutics)
were dissolved in 3.5m1 PBS with stirring at room temp. and 0.5 ml 0.O1M EDTA
was
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-78-
added. After 6hrs, the ratio of starting material to product was aprox. 60:40
as determined by
HPLC. Another 25.1mg of mPEG-Mal-20K was added and the reaction allowed to
stir
another 16hrs. The product ratio had not significantly improved, so the
reaction mixture was
loaded onto a 2.2 x 25 cm Kromasil C 18 preparative reverse phase column and
purified on a
Pharmacia FPLC using a gradient of 30%B to 100%B over 450min. A buffer =0.1
%TFA, B
buffer = 0.1 %TFA/50%ACN, flow = 4m1/min, and 5 min fractions were collected
while
monitoring the UV at 214nm (2.OA). The fractions containing homogeneous
product were
combined, frozen and lyophilized to give 25.7 mg. Purity as determined by
analytical HPLC
was -90%. A MALDI mass spectral analysis showed a broad peak from 23,000 to
27,000
which is approximately 20,000 a.m.u. more than starting Glucagon C24 (3457.8).
EXAMPLE 7
Glucagon Cys29 Mal-PEG-5K
20.0mg of Glucagon Cys29(1-29) and 24.7 mg mPEG-Mal-5000 (Nektar
Therapeutics) were dissolved in 3.5 ml PBS with stirring at room temperature
and 0.5 ml
0.01M EDTA was added. After 4 hr, another 15.6 mg of mPEG-Mal-5000 was added
to
drive the reaction to completion. After 8 hrs, the reaction mixture was loaded
onto a 2.2 x 25
cm Vydac C18 preparative reverse phase column and an acetonitrile gradient was
run on a
Pharmacia FPLC system. 5 min fractions were collected while monitoring the UV
at 214nm
(2.OA). A=0. 1 %TFA, B=0.1 %TFA/50%ACN. Fractions 75-97 were combined frozen
and
lyophilized to give 40.0 mg of product that is different than recovered
starting material on
HPLC (fractions 58-63). Analysis of the product by analytical HPLC [0.46 x 5
cm Zorbax
C8, 1 ml/min, 45C, 214nm (0.5A), A=0.1%TFA, B=0.1%TFA/90%ACN, gradient=10%B to
80%B over 10min.] showed a purity greater than 95%. MALDI mass spectral
analysis
showed the presence of a PEG component with a mass range of 8,000 to 10,000
(maximum
at 9025.3) which is 5,540 a.m.u. greater than starting material (3484.8).
EXAMPLE 8
Glucagon Cys24 (2-butyrolactone)
To 24.7mg of Glucagon Cys24(1-29) was added 4ml 0.05M ammonium
bicarbonate/50%acetonitrile and 5.5 ul of a solution of 2-bromo-4-
hydroxybutyric acid-y-
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-79-
lactone (100ul in 900u1 acetonitrile). After 3hrs of stirring at room
temperature, another 105
ul of lactone solution was added to the reaction mixture which was stirred
another 15hrs.
The reaction mixture was diluted to 10ml with 10% aqueous acetic acid and was
loaded onto
a 2.2 x 25 cm Kromasil C18 preparative reverse phase column. An acetonitrile
gradient
(20%B to 80%B over 450min) was run on a Pharmacia FPLC while collecting 5min
fractions
and monitoring the UV at 214nm (2.OA). Flow =4m1/min, A=0.1 %TFA,
B=0.1 %TFA/50%ACN. Fractions 74-77 were combined frozen and lyophilized to
give
7.5mg. HPLC analysis showed a purity of 95% and MALDI mass spect analysis
showed a
mass of 3540.7 or 84 mass units more than starting material. This result
consistent with the
addition of a single butyrolactone moiety.
0
H S Q G T F T S D Y S K Y L D S R R A 0 D F V-NW L M N T-coax
I0
Molecular Weight =3541.91 SEQ ID NO: 34
Exact Mass =3538
Molecular Formula =C 155H226N42050S2
EXAMPLE 9
Glucagon Cys24(S-carboxymethyl)
15- 18.1mg of Glucagon Cys24(1-29) was dissolved in 9.4m1'0.1M sodium
phosphate
buffer (pH=9.2) and 0.6ml bromoacetic acid solution (1.3mg/ml in acetonitrile)
was added.
The reaction was stirred at room temperature and the reaction progress was
followed by
analytical HPLC. After lhr another O.lml bromoacetic acid solution was added.
The
reaction was stirred another 60min. then acidified with aqueous acetic acid
and was loaded
onto a 2.2 x 25cm Kromasil C18 preparative reverse phase column for
purification. An
acetonitrile gradient was run on a Pharmacia FPLC (flow = 4m1/min) while
collecting 5min
fractions and monitoring the UV at 214nm (2.OA). A=0.1%TFA, B=0.1%TFA/50%ACN.
Fractions 26-29 were combined frozen and lyophilized to give
several mg of product. Analytical HPLC showed a purity of 90% and MALDI mass
spectral
analysis confirmed a mass of 3515 for the desired product.
EXAMPLE 10
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-80-
Glucagon Cys24 maleimido,PEG-3.4K-dimer
16mg Glucagon Cys24 and 1.02mg Mal-PEG-Mal-3400, poly(ethyleneglycol)-bis-
maleimide avg. M.W. 3400, (Nektar Therpeutics) were dissolved in 3.5 phosphate
buffered
saline and 0.5m1 0.01M EDTA and the reaction was stirred at room temperature.
After 16hrs,
another 16mg of Glucagon Cys24 was added and the stirring continued. After
approximately
40hrs, the reaction mixture was loaded onto a Pharmcia PepRPC 16/10 column and
an
acetonitrile gradient was run on a Pharmacia FPLC while collecting 2min
fractions and
monitoring the UV at 214nm (2.OA). Flow=2ml/min, A=0.1 %TFA, B=0.1
%TFA/50%ACN.
Fractions 69-74 were combined frozen and lyophilized to give 10.4mg.
Analytical HPLC
showed a purity of 90% and MALDI mass spectral analysis shows a component in
the 9500-
11,000 range which is consistent with the desired dimer.
GlucagonCys24(1-29)
\S GlucagonCys24(1-29)
3457.80 O O S
3457.80
3572.00 N
10487.60
O PEG34oo
O
EXAMPLE 11
Glucagon Solubility Assays:
A solution (lmg/ml or 3mg/ml) of glucagon (or an analog) is prepared in 0.01N
HCI.
100ul of stock solution is diluted to lml with 0.01N HC1 and the UV absorbance
(276nm) is
determined. The pH of the remaining stock solution is adjusted to pH7 using
200-250u10.1M Na2HPO4 (pH9.2). The solution is allowed to stand overnight at 4
C then
centrifuged. 100ul of supernatant is then diluted to lml with 0.01N HCI, and
the UV
absorbance is determined (in duplicate).
The initial absorbance reading is compensated for the increase in volume and
the
following calculation is used to establish percent solubility:
Final Absorbance X 100 = percent soluble
Initial Absorbance
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-81-
Results are shown in Table 1 wherein Glucagon-Cex represents wild type
glucagon (SEQ ID
NO: 1) plus a carboxy terminal addition of SEQ ID NO: 19 and Glucagon-Cex R12
represents
SEQ ID NO: 48.
Table 1 Solubility date for glucagon analogs
Analog Percent Soluble
Glucagon 16
Glucagon-Cex, R12 104
Glucagon-Cex 87
Oxyntomodulin 104
Glucagon, Cys17PEG5K 94
Glucagon, Cys21PEG5K 105
Glucagon, Cys24PEG5K 133
EXAMPLE 12
Glucagon Receptor Binding Assay
The affinity of peptides to the glucagon receptor was measured in a
competition
binding assay utilizing scintillation proximity assay technology. Serial 3-
fold dilutions of the
peptides made in scintillation proximity assay buffer (0.05 M Tris-HC1, pH
7.5, 0.15 M
NaCl, 0.1% w/v bovine serum albumin) were mixed in 96 well white/clear bottom
plate
(Corning Inc., Acton, MA) with 0.05 nM (3-[1251]-iodotyrosyl) TyrlO glucagon
(Amersham
Biosciences, Piscataway, NJ), 1-6 micrograms per well, plasma membrane
fragments
prepared from cells over-expressing human glucagon receptor, and 1 mg/well
polyethyleneimine-treated wheat germ agglutinin type A scintillation proximity
assay beads
(Amersham Biosciences, Piscataway, NJ). Upon 5 min shaking at 800 rpm on a
rotary
shaker, the plate was incubated 12h at room temperature and then read on
MicroBetal450
liquid scintillation counter (Perkin-Elmer, Wellesley, MA). Non-specifically
bound (NSB)
radioactivity was measured in the wells with 4 times greater concentration of
"cold" native
ligand than the highest concentration in test samples and total bound
radioactivity was
detected in the wells with no competitor. Percent specific binding was
calculated as
following: % Specific Binding = ((Bound-NSB)/(Total bound- NSB)) X 100. IC50
values
were determined by using Origin software (OriginLab, Northampton, MA).
EXAMPLE 13
Functional Assay- cAMP Synthesis
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-82-
The ability of glucagon analogs to induce cAMP was measured in a firefly
luciferase-
based reporter assay. HEK293 cells co-transfected with either glucagon- or GLP-
1 receptor
and luciferase gene linked to cAMP responsive element were serum deprived by
culturing
16h in DMEM (Invitrogen, Carlsbad, CA) supplemented with 0.25% Bovine Growth
Serum
(HyClone, Logan, UT) and then incubated with serial dilutions of either
glucagon, GLP-1 or
novel glucagon analogs for 5 h at 37 C, 5% CO2 in 96 well poly-D-Lysine-coated
"Biocoat"
plates (BD Biosciences, San Jose, CA). At the end of the incubation 100
microliters of
LucLite luminescence substrate reagent (Perkin-Elmer, Wellesley, MA) were
added to each
well. The plate was shaken briefly, incubated 10 min in the dark and light
output was
measured on MicroBeta-1450 liquid scintillation counter (Perkin-Elmer,
Wellesley, MA).
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-83-
Effective 50% concentrations were calculated by using Origin software
(OriginLab,
Northampton, MA. Results are shown in Fig 3 and in Tables 2 and 3.
Table 2 cAMP Induction by Glucagon Analogs with C-Terminus Extension
cAMP Induction
Peptide Glucagon Receptor GLP-1 Receptor
EC50, nM N* EC50, nM N*
Glucagon 0.22 0.09 14 3.85 1.64 10
GLP-1 2214.00 182.43 2 0.04 0.01 14
Glucagon Cex 0.25 0.15 6 2.75 2.03 7
Oxyntomodulin 3.25 1.65 5 2.53 1.74 5
Oxyntomodulin KRNR 2.77 1.74 4 3.21 0.49 2
Glucagon R12 0.41 0.17 6 0.48 0.11 5
Glucagon R12 Cex 0.35 0.23 10 1.25 0.63 10
Glucagon R12 K20 0.84 0.40 5 0.82 0.49 5
Glucagon R12 K24 1.00 0.39 4 1.25 0.97 5
Glucagon R12 K29 0.81 0.49 5 0.41 0.24 6
Glucagon Amide 0.26 0.15 3 1.90 0.35 2
Oxyntomodulin C24 2.54 0.63 2 5.27 0.26 2
Oxyntomodulin C24 PEG 20K 0.97 0.04 1 1.29 0.11 .1
* number of experiments
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-84-
Table 3 cAMP Induction by Pegylated Glucagon Analogs
cAMP Induction
Peptide Glucagon Receptor GLP-1 Receptor
EC50, nM W EC50, nM N*
Glucagon 0.33 0.23 18 12.71 3.74 2
Glucagon C17 PEG 5K 0.82 0.15 4 55.86 1.13 2
Glucagon C21 PEG 5K 0.37 0.16 6 11.52 3.68 2
Glucagon C24 PEG 5K 0.22 0.10 12 13.65 2.95 4
Glucagon C29 PEG 5K 0.96 0.07 2 12.71 3.74 2
Glucagon C24 PEG 20K 0.08 0.05 3 Not determined
Glucagon C24 Dimer 0.10 0.05 3 Not determined
GLP-1 > 1000 0.05 0.02 4
* - number of experiments
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-85-
EXAMPLE 14
Stability Assay for glucagon Cys-maleimido PEG analogs
Each glucagon analog was dissolved in water or PBS and an initial HPLC
analysis
was conducted. After adjusting the pH (4, 5, 6, 7), the samples were incubated
over a
specified time period at 37 C and re-analyzed by HPLC to determine the
integrity of the
peptide. The concentration of the specific peptide of interest was determined
and the percent
remaining intact was calculated relative to the initial analysis. Results for
Glucagon Cys21-
maleimidoPEG5K are shown in Figs. 1 and 2.
EXAMPLE 15
Glucagon Antagonists
The glucagon antagonists were synthesized using the following general
strategies:
General Peptide Synthesis Protocol with Boc-chemistry strategy:
Glucagon analogs were synthesized using HBTU-activated "Fast Boc" single
coupling starting from 0.2mmole of MBHA resin or first amino acid attached Pam
resin on a
modified Applied Biosystem 430A peptide synthesizer. Boc amino acids and HBTU
were
obtained from Midwest Biotech (Fishers, IN). Side chain protecting groups used
were:
Arg(Tos), Asn(Xan), Asp(OcHex), Cys(pMeBzl), His(Bom), Lys(2Cl-Z), Ser(OBzl),
Thr(OBzl), Tyr(2Br-Z), and Trp(CHO). The N-terminal 3-phenyllactic acid (PLA)
(Aldrich,
Milwaukee, WI) was coupled manually by BEPBT (3-(Diethoxy-phosphoryloxy)-3H-
benzo[d] [ 1,2,3] triazin-4-one, Synchem Inc., Aurora, OH) after the automated
solid phase
synthesis.
After peptide solid phase synthesis, each completed peptidyl resin was treated
with
20% piperdine/DMF to remove the formyl group from the tryptophan. Liquid
hydrogen
fluoride cleavages were performed in the presence of p-cresol and dimethyl
sulfide. The
cleavage was run for 1 hour in an ice bath using an HF apparatus (Penninsula
Labs). After
evaporation of the HF, the residue was suspended in diethyl ether and the
solid materials
were filtered. Each peptide was extracted into 30-70ml aqueous acetic acid and
diluted with
water and lyophilized. Crude peptide was analyzed by analytical HPLC and
Peptide molecule
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-86-
weight was checked by ESI or MALDI-TOF mass spectrometry. Peptide was purified
by the
general HPLC purification procedure.
General Peptide Synthesis Protocol with Fmoc-chemistry Strategy:
Peptides were synthesized on an ABI 433A automated peptide synthesizer using
standard Fmoc chemistry with Rink MBHA amide resin or first amino acid
attached Wang
resin (Novabiochem, San Diego, CA) using DIC/HOBT as coupling reagent. 3-
phenyllactic
acid (PLA) was coupled manually by BEPBT after the automated peptide
synthesis. The side
chain protecting groups of N -Fmoc [N-(9-fluorenyl)methoxycarbonyl] amino
acids were as
follows: Arg, Pmc; Asp, OtBu; Cys, Trt; Gln, Trt; His, Trt; Lys, Boc; Ser,
tBu, Tyr, tBu; and
Trp, Boc (Pmc = 2,2,5,7,8-pentamethylchroman-6-sulfonyl, OtBu = tert-butyl
ester, Trt =
trityl, Boc = tert-butyloxycarbonyl, and tBu = tert-butyl ester). Fmoc-
Cys(SO3Na)-OH and
Fmoc-homoCys(SO3Na)-OH were used for the synthesis of the cysteic acid and
homocysteic
acid containing peptides. Peptides were cleaved from the resin with cleavage
cocktail
containing 85%TFA, 5% phenol, 5% water and 5% thioanisole (2.5% EDT was added
when
peptide contains Cysteine). Crude peptides were precipitated in ether,
centrifuged, and
lyophilized. Peptide was analyzed by analytical HPLC and checked by ESI or
MALDI-TOF
mass spectrometry. Peptide was purified by the general HPLC purification
procedure.
General analytical HPLC procedure:
Analytical HPLC was performed on a Beckman System Gold HPLC system with a
ZORBAX SB-C8 column ( 0.46 x 5cm, 5 m, Agilent) with a gradient elution at a
flow rate
of 1.0 mL/min and monitored at 214nm. The gradients were set up as 10%B to
80%B over
10min and then 10%B for 5min. Buffer A =0.1 %TFA and
B=0.1%TFA/90%acetonitrile.
General Preparative HPLC purification procedure:
The peptides were typically purified on a Waters 600E connected 486 monitor
systems with semi-prepare HPLC column (ZORBAX SB-C8, 21.2x250mm, 7 m, Agilent)
monitored at 214nm or 230nM. Buffer A =0.1%TFA / 10%acetonitrile and
B=0.1 %TFA/90%acetonitrile. The gradients used for the purification were 0-
30%B over 40
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-87-
min, then 30-50%B over 30min at a flow rate of 12m1/min if not specifically
noted. Fractions
were analyzed by analytical HPLC and checked by mass spectrometry. The
fractions over
90% pure were collected, lyophilized and stored. The fractions with purity
between 60-90%
were combined, lyophilized and purified again.
Pegylated glucagon analog deriviatives were prepared in accordance with the
following general Cys-maleimido procedure. Typically, the glucagon Cys analog
is
dissolved in phosphate buffered saline (5-10mg/ml) and 0.01M ethylenediamine
tetraacetic
acid is added (10-15% of total volume). Excess (1.5-2-fold) maleimido
methoxyPEG
reagent (Nektar, Huntsville, AL) is added and the reaction stirred at room
temp while
monitoring reaction progress by HPLC. After 2-24hrs, the reaction mixture, is
acidified and
loaded onto a preparative reverse phase column for purification using 0.1
%TFA/acetonitrile
gradients. The appropriate fractions were combined and lyophilized to give the
desired
pegylated derivatives. For peptides that exhibit low solubility in PBS, the
peptides were
dissolved in 25% acetonitrile water or 4-6M urea buffer with 0.1M Tris (adjust
pH 8.0-8.5)
and reacted with PEG reagents.
Specific examples of compounds synthesized by the methods described above are
provided as follows:
Preparation of Fmoc-homoCys(SO3Na)-OH
L-Homocysteic acid 0.92g (5mmole) (Sigma, St. Louis, MO) and 0.5g (12.5mmole)
NaOH were dissolved in 50ml water cooled in ice bath. A solution of 9-
Fluorenylmethyl
succinimidyl carbonate (Fmoc-OSu) (1.86g, 5.5mmole) in 50m1 dioxane was added
in one
portion. The mixture was stirred at room temperature for over 4h. The mixture
was
evaporated under reduced pressure and 100m1 water was added. The aqueous
solution was
washed with ether and then passed through an ion exchange column (Amberlite IR-
120B,
H+form; GFS Chemicals, Columbus, OH)). The aqueous eluate was lyophilized to
yield a
viscous amorphous Fmoc-homoCys(SO3H)-OH (1.6g, 3.95mmole, yield 79.2%). The
above
free acid was then added 50ml water containing 0. 16g (4mmole) NaOH in ice
bath and
lyophilized to get quantitative Fmoc-homoCys(SO3Na)-OH which can be used
directly in
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-88-
SPPS without further purification. The amorphous lyophilized Fmoc-
homoCys(SO3Na)-OH
is recrystallined in ethanol/acetate ester(2: 1) to yield a crystalline solid
product with m.p. of
215-218 C and ESI-MS 404.2[(M-H)+, acid form]
Synthesis of [PLA6, E9]glucagon(6-29) amide
A peptide sequence TSEYSKYLDSRRAQDFVQWLMNT (SEQ ID NO: 51;
[E3]glucagon(7-29)) was first solid phase synthesized on ABI 433A automated
peptide
synthesizer using 0.lmmole Fmoc/HOBT/DCC chemistry program with 0.lmmole Rink
MBHA amide resin using DIC/HOBT as coupling reagent. The following Fmoc amino
acid
were used: Ala, Arg(Pmc), Asp(OtBu), Asn(Trt), Glu(OtBu), Gln(Trt), Leu,
Lys(Boc), Met,
PLA, Ser(tBu), Thr(tBu), Trp(Boc), Tyr(tBu), and Val. After the automated
synthesis, the
peptidyl resin was coupled manually with 3-phenyllactic acid (83mg, 0.5mmole)
and DEPBT
(150mg; 0.5mmole) in 4m15%DIEA/DMF for about 2h to obtain the peptidyl resin
with the
following sequence: HO-PLA-Thr-Ser-Glu-Tyr-Ser-Lys-Tyr-Leu-Asp-Ser-Arg-Arg-Ala-
Gln-Asp-Phe-Val-Gln-Trp-Leu-Met-Asn-Thr-NH2 (SEQ ID NO: 50).
Peptidyl resin was treated with 8.5ml TFA with addition of 0.5g phenol, 0.5m1
water
and 0.5m1 thioanisole at room temperature for about 2hs. The peptide dissolved
in TFA was
filtered and 40ml ether was added to precipitate the peptide. The crude
peptide were
centrifuged, dissolved in aqueous acetic acid and lyophilized. Crude peptide
yield was
200-250mg and after purification the yield was 25-40mg (10-15% yield totally)
peptide
with 95% purity. The peptide was analyzed in general analytical HPLC showing
retention
time as 7.66min and ESI-MS analysis demonstrated the desired mass of 2986.0
corresponding with the peptide molecular weight 2986.3.
Similar procedure were used to synthesize the peptide [PLA6, D9]glucagon(6-29)
amide with analytical HPLC 7.25min and ESI-MS 2973.5 corresponding to the
calculated
MW 2973.3; [PLA6, D9, D28]glucagon(6-29) amide with analytical HPLC 7.46min
and
ESI-MS 2973.0 corresponding the calculated MW 2973.3; [PLA6, C8, E9]glucagon(6-
29)
amide with analytical HPLC 7.20min and ESI-MS 3002.0 corresponding the
calculated MW
3002.3; [PLA6, E9, C16]glucagon(6-29) amide with analytical HPLC 7.38min and
ESI-MS
3002.0 corresponding the calculated MW 3002.3; [PLA6, E9, C24]glucagon(6-29)
amide
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-89-
with analytical HPLC 7.33min and ESI-MS 2961.0 corresponding the calculated MW
2961.3; [PLA6, D9, C24]glucagon(6-29) amide with analytical HPLC 7.43min and
ESI-MS
2947.0 corresponding the calculated MW 2947.3; [PLA6, E9, C40]glucagon(6-40)
amide
with analytical HPLC 7.28min and MALDI-MS 3925.5 corresponding the calculated
MW
3924.3.
Synthesis of [hCys(SO3H)9]glucagon (6-29) amide
A peptide sequence YSKYLDSRRAQDFVQWLMNT (SEQ ID NO: 51; glucagon(10-
29)) was first solid phase synthesized on ABI 433A automated peptide
synthesizer using
0.lmmole Fmoc/HOBT/DCC chemistry program with 0..lnnnole Rink MBHA amide resin
using DIC/HOBT as coupling reagent. After the automated synthesis, the
peptidyl resin was
coupled: manually with Fmoc-homoCys(SO3Na)-OH (130mg; 0.3mmole),.HOBT (45.2mg,
0.33mole) and DIC (52.Oul, 0.33mole) in 4m1 DMF for about 2h. After the
ninhydrin test,
half potion of the peptidyl resin (0.05mmole) was further assembled
automatically with the
remain 3 amino acid Ser, Thr and Phe to obtain the peptidyl resin with the
following
sequence: H2N-Phe-Thr-Ser-homoCys(SO3H)-Tyr-Ser-Lys-Tyr-Leu-Asp-Ser-Arg-Arg-
Ala-Gln-Asp-Phe-Val-Gln-Trp-Leu-Met-Asn-Thr-NH2 (SEQ ID NO: 52).
The following Fmoc amino acid were used: Ala, Arg(Pmc), Asp(OtBu), Asn(Trt),
Gln(Trt), homoCys(SO3Na), Leu, Lys(Boc), Met, Phe, Ser(tBu), Thr(tBu),
Trp(Boc),
Tyr(tBu), and Val.
Peptidyl resin was treated with 8.5m1 TFA with addition of 0.5g phenol, 0.5m1
water
and 0.5m1 thioanisole at room temperature for about 2hs. The peptide dissolved
in TFA was
filtered and 40m1 ether was added to precipitate the peptide. The crude
peptides were
centrifuged, dissolved in aqueous acetic acid and lyophilized. Crude peptide
yield was
100--130mg, and after purification 15-20mg (10-15% yield totally) peptide was
obtained
with 95% purity. The peptide was analyzed in general analytical HPLC shown
retention time
as 6.73min and ESI-MS analysis demonstrated the desired mass of 3021.0
corresponding
with the peptide molecular weight 3021.3.
Similar procedure to synthesize the [hCys(SO3H)9]glucagon (5-29) amide with
analytical HPLC retention time 6.82min and ESI-MS 3122.5 corresponding the
calculated
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-90-
MW 3122.4; [hCys(S03H)9]glucagon (4-29) amide with analytical HPLC retention
time
6.83min and ESI-MS 3178.5 corresponding the calculated MW 3179.3;
[hCys(S03H)9]glucagon (2-29) amide with analytical HPLC retention time 6.79min
and
ESI-MS 3394.5 corresponding the calculated MW 3394.7; [PLA6,
hCys(S03H)9]glucagon
(6-29) amide with analytical HPLC retention time 7.17min and ESI-MS 3022.0
corresponding the calculated MW 3022.3.
Synthesis of [PLA6, E9, C24(1.2K)]glucagon (6-29) amide
20mg (0.00675mmole) [PLA6, E9, C24]glucagon (6-29) amide and 12.5mg
(O.Olmmole) m-dPEGTM24 -MAL(MW 1239, Quanta biodesign Ltd. Powell, OH) were
dissolved in 9ml 25% acetonitrile water and lml 1M Tris base buffer (adjust pH
to 8.0-8.5).
The reaction was stirred at room temperature and the progress of the reaction
was monitored
by analytical HPLC. After no initial product was detected on HPLC (about after
2hrs), the
reaction mixture was directly purified by preparative HPLC.
After lyophilized, about 10 -12 mg [PLA6, E9, C24(1.2K)]glucagon (6-29) amide
was obtained which analytical HPLC analysis shown retention time as 7.48min
and ESI-MS
4218.5 corresponding the calculated [M +H20] 4218Ø
A similar procedure was used to synthesize the [C5(1.2K), E9]glucagon (5-29)
amide
which analytical HPLC analysis shown retention time as 7.25min and ESI-MS
4327.5
corresponding the calculated MW 4327.8; [C8(1.2K), E9]glucagon (6-29) amide
which
analytical HPLC analysis shown retention time as 7.25min and ESI-MS 4260.0
corresponding the calculated [M +H20] 4259.0
Synthesis of [PLA6, E9, C24(20K)]glucagon (6-29) amide
15mg (0.005mmole) [PLA6, E9, C24]glucagon (6-29) amide and 140mg
(0.006mmole) 20K mPEG-MAL(MW -22k, Nektar, Huntsville, AL) were dissolved in
9ml
25% acetonitrile water and lml 1M Tris base buffer (adjust pH to 8.0-8.5). The
reaction was
stirred at room temperature and the progress of the reaction was monitored by
analytical
HPLC. After no initial product was detected on HPLC (about after 6hrs), the
reaction
mixture was directly purified by preparative HPLC. The fractions were checked
by
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-91-
analytical HPLC at 214nm and also measured by UV at 280nm. The fractions with
90%
HPLC purity and also with high absorption (A280nm=1Ø2.0) in UV measurement
were
combined and lyophilized. About 60 -80 mg [PLA6, E9, C24(20K)]glucagon (6-29)
amide
can be obtained which analytical HPLC analysis shown retention time as 8.5-8.6
min and
MALDI-MS shown broad mass spectrometry at 24K-26K.
A similar procedure was used to synthesize [PLA6, C8(20K), E9]glucagon (6-29)
amide, [PLA6, E9, C16(20K)]glucagon (6-29) amide, [PLA6, E9, C40(20K)]glucagon
(6-40)
amide, [PLA6, D9, C16(20K)]glucagon (6-29) amide and [PLA6, D9,
C24(20K)]glucagon
(6-29) amide.
Synthesis of [PLA6, E9, C24(40K)]glucagon (6-29) amide
15mg ( 0.005mmole) [PLA6, E9, C24]glucagon (6-29) amide and 240mg
(0.006mmole) 40K mPEG-MAL(MW -40k, Chirotech Technology Ltd., Cambs CB4 OWG,
German) were dissolved in 18ml 25% acetonitrile water and 2m1 1M Tris base
buffer (adjust
pH to 8.0-8.5). The reaction was stirred at room temperature and the progress
of the reaction
was monitored by analytical HPLC. After no initial product was detected on
HPLC (about
after 6hrs), the reaction mixture was directly purified by preparative HPLC.
The fractions
were checked by analytical HPLC at 214nm and also measured by UV at 280nm. The
fractions with 90% HPLC purity and also with high absorption (A280nm=1.0--2.0)
in UV
measurement were combined and lyophilized. About 100 -120 mg [PLA6, E9,
C24(40K)]glucagon (6-29) amide can be obtained which analytical HPLC analysis
shown
retention time as 8.60-8.8 min.
A similar procedure was used to synthesize [PLA6, C8(40K), E9]glucagon (6-29)
amide, [PLA6, E9, C16(40K)]glucagon (6-29) amide and [PLA6, E9,
C40(40K)]glucagon
(6-40) amide, [PLA6, D9, C16(40K)]glucagon (6-29) amide and [PLA6, D9,
C24(40K)]glucagon (6-29) amide.
Synthesis of Dimer[PLA6, E9, C24]glucagon (6-29) amide
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-92-
20mg (0.00675mmole) [PLA6, E9, C24]glucagon (6-29) amide was dissolved in 6m1
PBS buffer, lml 1M Tris base (adjust pH 8.0-8.5) and 3 ml DMSO. The reaction
mixture
was stirred in an open air container and monitored by analytical HPLC every
2hr. After the
initial product (HPLC RT 7.4min) was gone and the dimer product (HPLC RT
7.9min) was
the dominate product (after 12hr), the mixture was diluted with 0.1%TFA10%
acetonitrile
water and directly purified by preparative HPLC. After lyophilized about 6-8mg
Dimer[PLA6, E9, C24]glucagon (6-29) amide was obtained with ESI-MS 5920.0
corresponding the calculated MW 5920.6.
A similar procedure was used to synthesize the Dimer[C9]glucagon(6-29) amide
with
ESI-MS 5916.0 corresponding the calculated MW 5916.6 and Dimer[C5,
E9]glucagon(5-29)
amide with ESI-MS 6174.0 corresponding the calculated MW 6174.8.
EXAMPLE 16
Antagonist Activities of the Glucagon Analogs
The receptor binding, cAMP induction and cAMP inhibition of glucagon and
various
glucagon derivative inhibitors were compared. The assays for measuring
receptor binding
and CAMP induction and cAMP inhibition were conducted using the assay system
essentially
as disclosed in Examples 12 and 13, respectively.
Specific glucagon analogs have been prepared that exhibit glucagon antagonist
activity. Such compounds differ from native glucagon in that they do not
possess the native
N-terminal residues and have a glutamic acid substitution at position 9
relative to native
glucagon. Table 4 provides glucagon receptor affinity and antagonist activity
for several
specific glucagon analog antagonists.
Table 4:
Glutamic acid modified N-truncated glucagon analogs and their glucagon
antagonism
activities
Peptide Receptor cAMP
Binding Inhibition
IC50(nM) IC50(nM)
Glucagon 1-2.5 N/A
[G1u ]Glucagon(aa2-29)-NH2 14 partial antagonist
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-93-
[G1u ]Glucagon(aa4-29)-NH2 136 128
[G1u ]Glucagon(aa5-29)-NH2 37 74
[G1u ]Glucagon(aa6-29)-NH2 36 97
Glu is glutamic acid at position 9 according to the numbering of native
glucagon.
As the data in Table 5 indicates, a set of hCys9-based antagonists do not
perform as
potently or selectively as the previously reported Glu9-based antagonists.
Compounds 513
and 6B demonstrate some level of antagonism but only at concentrations that
are threefold
higher than their effective dose as an agonist. However, when N-terminal amino
acids (in
addition to the amino acid at position 1 of native glucagon) are removed, the
potency of the
hCys9-based glucagon antagonists is enhanced (See Table 8).
Table 5
Receptor Binding and cAMP Inhibition by Glucagon Antagonist Analogs
Cmpd. Peptide Receptor cAMP cAMP
# Binding Induction Inhibition
IC50(n-M) EC50(nM) IC50(nM)
Glucagon 1.75-0.31 0.21 0.11 N/A
[desHis, Glu ]Glucagon-NH2 36.90 0.32 65 37 1862 1234
[desHis , GO, Phe 21 , Leu ]Glucagon-NH2 12.59 0.41 81 23 N/A*
5 [desHis , desPhe 6 ] Glucagon-NH2 129.55 44. 1178 105 N/A*
9
6 [desHis , Leu , G1u ] Glucagon-NH2 36.88 0.03 318 112 102 52
25 , Leu ] 13.90 0.37 430 45 N/A*
4B [desHis , hCys (SO3-), Phe
Glucagon-NH2
, 53.32 9.97 3212 368 9217 3176
5B [desHis , desPhe , hCys (SO3-), Phe
Leu27] Glucagon-NH2
6B [desHis , Leu , hCys (SO3-), Phe , Leu ] 1614 1132 4456 1469 21 Glucagon-
NH2
*not an antagonist
amino acid positions according to the numbering of native glucagon indicated
by
superscripted numbers
Glucagon receptor binding affinity of glucagon and glucagon peptides modified
by
truncation of the first amino acid and by substitution at position 9
(according to the amino
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-94-
acid numbering of native glucagon) was analyzed as essentially described in
Example 12,
The results are shown in Table 6.
TABLE 6
peptide no. peptide residue 9 IC50 (nM)
rCOON
Glucagon 1.50 (1.0 -- 2.5)*
H2N COOH
^
Asp
1 [desHis', Glu9]glucagon-NH2 XT COON 14.08 0.34
HzN COON
Glu
2 [hGlu9]Glucagon(aa2-29)-NH2 `OOH 8.10 0.40
COOH
HzN
hGlu
3 [(CSA-1]9]Glucagon(aa2-29)- (SCOOH 12.66 0.13
NH2 H2N COOH
CSA-1
4 [(CSA-2)9]Glucagon(aa2-29)- S COOH 13.28 0.78
NH2 H,N (COOH
CSA-2
[ IN(3-hGlu9]Glucagon(aa2-29) CC OOH 37.10 0.34
"TrlT2 HiN COOH
R-hGlu
6 [(NSG-1)9]Glucagon(aa2-29)- (COON 983 82
NH2 H~NCOOH
NSG-1
[(NSG-2)9]Glucagon(aa2-29)- ~COOH 2348 382
7
NH2 HN ~,000H
NSG-2
*EC50 (nM)
5 hGlu = homoglutamic acid;
amino acid positions according to the numbering of native glucagon indicated
by
superscripted numbers
Several of the modified glucagon based peptides tested, including the peptides
modified at position 9 with Glu, hGlu, CSA-1, CSA-2, and (3-hGlu, exhibited
potent
glucagon antagonist activity.
Glucagon peptides comprising a modified amino acid at positin 9 and having
different
extents of N-terminal truncation were analyzed for glucagon antagonist
activity. The results
of the peptides tested are shown in Table 7.
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-95-
TABLE 7
peptide no. peptide residue 9 IC50 (nM) cAMP
pA26 (I/A)50`
8 [Glu9]Glucagon(aa4-29)- Glu 136.0 17.84 7.05 1.01 1375
NHS
9 [Leu4, Glu9]Glucagon(aa4- Glu 36.38 8.69 NA`' NA
29)-NH2
[Glu9]Glucagon(aa5-29)- Glu 37.38 3.41 6.94 0.34 390
NH2
11 [Glu9]Glucagon(aa6-29)- Glu 36.35 5.23 7.16 0.27 486
NH2
12 [hGlu9]Glucagon(aa6-29)- hGlu 162.9 70.8 6.27 0.11 2361
NH2
13 [(CSA-1)9]Glucagon(aa6- CSA-1 107.3 5.37 6.68 1.05 506
29)-NH2
14 [(CSA-2)9]Glucagon(aa6- CSA-2 146.4 36.9 6.64 0.29 580
29)-NH2
Glucagon(aa6-29)-NH2 Asp 1894 383 6.94 0.63 1730
16 [Lys9]Glucagon(aa6-29)- Lys 5779 1382 6.58 0.60 1990
NH2
17 [Glu9]Glucagon(aa7-29)- Glu >10000 NDe ND
NH2
amino acid positions according to the numbering of native glucagon indicated
by
superscripted numbers
a Data are average STD for at least three independent experiments.
5 b pA2, the negative logarithm of the concentration of the antagonist that
reduce the response
to lunit of the agonist to the response obtained from 0.5 unit of agonist.
Data are average
STD for at least two duplicate experiments.
(UA)50, the inhibition index, the ratio of inhibitor IC50 to the added
constant glucagon (0.1-
0.2nM). Data are average of at least three independent experiments and
normalized by the
10 EC50
d NA, not full antagonist. e ND, not detected.
Fig. 3 presents data measuring the binding affinity of glucagon antagonists,
where the
N-terminus was further shortened with removal of one, three or five amino
acids. More
15 particularly, the binding affinity of hCys9-based glucagon antagonists was
investigated by
measured based on their ability to compete with 1125 labeled glucagon in
binding to the
glucagon receptor. The results demonstrate that the removal of the first
residue reduces
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-96-
affinity but further removal changes the affinity only slightly still yielding
a ligand of
nanomolar affinity. Fig. 4 presents data demonstrating the ability of select
antagonists to
suppress glucagon action in a cAMP bioassay. Surprisingly, the
5-29 hCys9-based glucagon antagonist was found to be more potent and effective
than the
literature standards of G1u9 2-29, or the Leu4, Glu9 4-29. Furthermore, the 5-
29 or 6-29
hCys9 analogs have exhibited pure potent antagonism, without any measurable
agonist
activity.
Table 8 provides glucagon receptor affinity and antagonist activity for
several
homocysteic acid modified truncated glucagon fragment analogs. The desHisl-
based
hCys(S03H)9-based antagonist performs as potently as the previously reported
Glu9- based
antagonist [desHis', Glu9]glucagon peptides. The further shortened hCys(SO3H)9-
based
glucagon antagonists with removal of three, four or five amino acids were
studied. The
receptor binding results demonstrate that the removal of the first residue
reduces affinity of
the compound for the glucagon receptor, but further removal changes the
affinity only
slightly, and still yields a ligand of nanomolar affinity.
Table 8: Homocysteic acid modified truncated glucagon fragment analogs and
their
glucagon antagonism activities
Peptide IC50(nM) cAMP
pA2 IC50(nM)
Glucagon 1.0-2.5 (EC50)
[desHis', Glu9]glucagon-NH2 14.08 0.34 NA 1089
(partial antagonist)
[hCys9(SO3H)]Glucagon(aa2- 13.16 1.0 NA 146.6
29)NH2 (partial antagonist)
[hCys9(SO3H)]Glucagon(aa4- 41.55 4.79 7.22 1.09 68.4
29)-NH2
[hCys9(SO3H)]Glucagon(aa5- 33.85 9.38 6.77 0.33 98.3
29)-NH2
[hCys9(SO3H)]Glucagon(aa6-29)- 59.11 18.10 7.16 0.51 133.4
NH2
amino acid positions according to the numbering of native glucagon indicated
by
superscripted numbers
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-97-
As shown in the data presented in Figs. 5A and 513, the 4-29, 5-29 and 6-29
hCys(SO3H)9-based peptides are surprisingly all full antagonists of glucagon
action, while
the 2-29 peptides were far less effective in fully suppressing glucagon
activity. Fig. 5A
demonstrates that the inability of the 2-29 peptides to fully suppress
glucagon action is a
function of the residual glucagon agonism that each of these two peptides
maintains.
Specific analogs of glucagon have also been developed where the normally
occurring
phenylalanine at position six has been substituted with phenyl-lactic acid
(PLA), on a 6-29
shortened glucagon amide backbone. PLA is isoelectronic with phenylalanine
(Phe) but has
no titratable hydrogen. The data presented in Tables 9 & 10 demonstrate that
with the PLA6
substitution, the native Asp9 analog exhibits pure antagonism but the potency
is reduced
relative to that of the Glu9 and hCys(S03H)9. analogs. The literature has
previously, indicated
that the native Asp9 residue has to be changed to Glu9 or h.Cys(S03H)9 for
high affinity and
potent antagonism of glucagon (2-29) analogs. Accordingly, it is surprising
that substitution
of Phe with PLA on a 6-29 shortened glucagon amide backbone improves the
relative
antagonist potency of the analog to a point comparable to that of the Glu9 and
hCys(S03H)9
analogs. More specifically, the PLA6 analog increases the affinity of the
analog for the
glucagon receptor threefold as well as the potency of antagonism relative to
the native Phe6
analog.
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-98-
Table 9: Residue 9 substituted glucagon (6-29) analogs and their glucagon
antagonism
activities
IC50(nM) cAMP
Peptide Residue 9 receptor pA2 IC50(nM)
binding
Glucagon Asp 1.0-2.5 0.050.15
(EC50)
[E9]Glucagon(aa6-29)-NH2 Glu 36.35 5.23 7.16 0.27 97.2
[hCys(S03)9]Glucagon(aa6- hCys(S03) 59.11 18.10 7.16 0.51 133.4
29)-NH2
[hE9]Glucagon(aa6-29)-NH2 hGlu 162.9 70.8 6.27 0.11 472.2
[C9(SCH2OOOH)]Glucagon(aa CSA-1 107.3 5.37 6.68 1.05 101.2
6-29)-NH?
[C9(SCH2CH2OOOH)]Glucagon CSA-2 146.4 36.9 6.64 0.29 = 116
(aa6-29)-NH2
Glucagon(aa6-29)-NH2 Asp 1670 -- 6.94 0.63 346
[K9]Glucagon(aa6-29)-NH2 Lys 3236 -- 6.58 0.60 398
amino acid positions according to the numbering of native glucagon indicated
by
superscripted numbers
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-99-
Table 10: Residue 9 substituted [PLA6]glucagon (6-29) analogs and their
glucagon
antagonism activities
Peptide IC50(nM) IC50(nM) Solubility
(Receptor (cAMP, inhibit (%, pH 6-8)
binding) Glucagon)
0.1 nM or 0.2nM
Glucagon 1.96 0.61 0.09 (EC50)
[PLA6, D9]Glucagon(aa6- 13.85 3.22 6.90 11
29)-NH2
[PLA6, D9]Glucagon(aa6- 15.51 3.86 13.20 96
29)-COOH
[PLA6, E9]Glucagon(aa6- 12.33 2.24 2.39 42.40 11
29)-NH2
[PLA6, 14.20 0.45 40.20
hCys(S03)9] Glucagon(aa6-
29)-NH2
[PLA6, D9, D28] 9.0 1.24 1.32 100
Glucagon(aa6-29)-NH2
[PLA6, E9]Glucagon (aa6- 40.28 11.29 24.75 16
29+CEX)-NH2
amino acid positions according to the numbering of native glucagon indicated
by
superscripted numbers
The effect of the PLA substitution at different positions of glucagon analog,
including
at positions 4 and 5 was also investigated. The data presented in Table 11 and
in Fig. 8
demonstrate that the PLA6 analog is an appreciably more potent antagonist than
the slightly
lengthened peptides. The results presented in Fig 8 also demonstrate that
acylation of the
hydroxyl group does not affect the PLA6 analog potency.
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-100-
Table 11: Analogs with PLA substitution at position 4, 5 and 6 and their
glucagon
antagonism activities
Peptide IC50(nM) IC50(nM)
(Receptor binding) (CAMP, inhibit 0.8mM Glucagon)
Glucagon 1.0-2.5 1.44 (EC50+)
[PLA6, E9]Glucagon(aa6-29)- 12.34 0.13 64.8 3.4
NH2
[Ac-PLA6, E9]Glucagon(aa6- ND 38.1 9.2
29)-NH2
[PLA5' E9]Glucagon(aa5-29)- ND 328 25
NH2
[PLA4, E9]Glucagon(aa4-29)- ND 84.4 19.5
NHZ (partial agonist)
ND:.not detected.
amino acid positions according to the numbering of native glucagon indicated
by
superscripted numbers
The data presented in Table 12 demonstrates that the PLA6 substitution not
only
increases the peptide potency but also serves a critical role in pegylation.
The PLA6 analogs
can be selectively pegylated without restoration of glucagon agonism. The
native Phe6
analogs surprisingly demonstrate a restoration of agonism when pegylated.
However this
restoration of agonism is not observed in the P1a6 peptide analogs. Several
specific
pegylation sites were examined, including amino acid positions 8, 11 and 24
(relative to the
native glucagon peptide). Pegylation at position 24 of the Pla6 analog
exhibits the most
potent and selective glucagon antagonism.
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-101-
Table 12: PEGylated N-terminal truncated glucagon analogs and their glucagon
antagonism activities
Peptide IC50(nM) IC50(nM)
(Receptor binding) (cAMP, inhibit 0.2mM
Glucagon)
[C8(2OkDaPEG), E9]Glucagon(aa6-29)- >1000 no antagonism
NH2
[PLA6, C8(2OkDaPEG), 303 14 236
E9]Gluca on(aa6-29)-NH2
[E9, C 1 1(20kDaPEG)] Glucagon(aa6-29)- >1000 no antagonism
NH2
[PLA6, E9, 776 161 664
C1 1(20kDaPEG)]Glucagon(aa6-29)-NH2
[E9, C24 (20kDaPEG)]Glucagon(aa6-29)- >1000 no antagonism
NH2
[PLA6, E9, 90 7 126
C24(2OkDaPEG)]Glucagon(aa6-29)-NH2
[MCA6, E9, 208 + 57 no antagonism
C24(2OkDaPEG)]Glucagon(aa6-29)-NH2
[C5(1.2kDaPEG), E9]Glucagon(aa5-29) - 1081 268 2281
NH2
[C5(5kDaPEG), E9]Glucagon(aa5-29) - 1608
NH2 634 174
[C5(2OkDaPEG), E9]Glucagon(aa5-29) - 331 74 976
NH2
[d-CysS(2OkDaPEG), E9]Glucagon(aa5- >10000 14764
29) -NH2
[K5(CH2CH2S-2OkDaPEG), >10000 no antagonism
E9]Glucagon(aa5-29) -NH2
3.4kDaPEG-dimer[C5, E9]Glucagon(aa5- 435 256 1343
29) -NH2
[PLA6, C8(1.2kDaPEG), 220 36 no antagonism
E9]Glucagon(aa6-29) -NH2
[PLA6, C8(5kDaPEG), E9]Glucagon(aa6- 948 297 216
29) -NH2
[PLA6, C8(2OkDaPEG), 303 14 92
E9]Glucagon(aa6-29) -NH2
[PLA6, E9 ,C24(1. 4-7 0.4 18
2kDaPEG)]Glucagon(aa6-29) -NH2
[PLA6, E9, 90 7 126
C24(2OkDaPEG)]Glucagon(aa6-29) -NH2
[MCA6, E9, 208 57 no antagonism
C24(2OkDaPEG)]Glucagon(aa6-29) -NH2
[Phe6, E9, >10000 no antagonism
C24(2OkDaPEG)]Gluca on(aa6-29) -NH2
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-102-
EXAMPLE 17
The glucagon antagonists described herein are acylated as follows:
Acylated and/or PEGylated peptides are prepared as follows. Peptides are
synthesized on a solid support resin using either a CS Bio 4886 Peptide
Synthesizer or
Applied Biosystems 430A Peptide Synthesizer. In situ neutralization chemistry
is used as
described by Schnolzer et al., Int. J. Peptide Protein Res. 40: 180-193
(1992). For acylated
peptides, the target amino acid residue to be acylated (e.g., position ten) is
substituted with an
N a -FMOC lysine residue. Treatment of the completed N-terminally BOC
protected peptide
with 20% piperidine in DMF for 30 minutes removes FMOC/formyl groups. Coupling
to the
free E-amino Lys residue is achieved by coupling a ten-fold molar excess of
either an FMOC-
protected spacer amino acid (ex. FMOC-(N-BOC)-Tryptophan-OH) or acyl chain
(ex. C17-
COOH) and PyBOP or DEPBT coupling reagent in DMF/DIEA. Subsequent removal of
the
spacer amino acid's FMOC group is followed by repetition of coupling with an
acyl chain.
Final treatment with 100% TFA results in removal of any side chain protecting
groups and
the N-terminal BOC group. Peptide resins are neutralized with 5% DIEA/DMF,
dried, and
then cleaved from the support using HF/p-cresol, 95:5, at 0 C for one hour.
Following ether
extraction, a 5% HOAc solution is used to solvate the crude peptide. A sample
of the
solution is then verified to contain the correct molecular weight peptide by
ESI-MS. Correct
peptides are purified by RP-HPLC using a linear gradient of 10% CH3CN/0.1% TFA
to
0.1% TFA in 100% CH3CN. A Vydac C18 22 mm x 250 mm protein column is used for
the
purification. Acylated peptide analogs generally complete elution by a buffer
ratio of 20:80.
Portions are pooled together and checked for purity on an analytical RP-HPLC.
Pure
fractions are lyophilized yielding white, solid peptides.
For peptide pegylation, 40 kDa methoxy poly(ethylene glycol) maleimido-
propionamide (Chirotech Technology Ltd.) is reacted with a molar equivalent of
peptide in
7M Urea, 50mM Tris-HC1 buffer using the minimal amount of solvent needed to
dissolve
both peptide and PEG into a clear solution (generally less than 2 mL for a
reaction using 2-3
mg peptide). Vigorous stirring at room temperature is commenced for 4-6 hours
and the
reaction is analyzed by analytical RP-HPLC. PEGylated products appear distinct
from the
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-103-
starting material with decreased retention times. Purification is performed on
a Vydac C4
column with conditions similar to those used for the initial peptide
purification. Elution
occurrs around buffer ratios of 50:50. Fractions of pure PEGylated peptide are
found and
lyophilized.
EXAMPLE 18
The synthesis of glucagon-based depsipeptides [ThrS-O-PLA6, E9]glucagon (2-29)
amide and [Thr5-O-PLA6, E9]glucagon (1-29) amide were carried out as follows:
A peptide sequence HO-PLA-TSEYSKYLDSRRAQDFVQWLMNT [PLA6,
E9]glucagon(6-29) (SEQ ID NO: 71) was synthesized by solid-phase Boc-chemistry
using an
ABI 430A automated peptide synthesizer with 0.2mmole MBHA amide resin and
DEPBT as
coupling reagent. The following Boc amino acids were used: Ala, Arg(Tos),
Asp(OcHx),
Asn(Xan), Glu(OcHx), Gln(Xan), Leu, Lys(2-Cl-Z), Met, PLA, Ser(OBz1),
Thr(OBzl),
Trp(HOC), Tyr(2.6-di-Cl-Bzl), and Val. To this peptide was formed a depsi-
petide (ester
bond) on the resin through manual coupling with a pre-activated symmetrical
anhydride
solution composed of Boc-Thr(OBzl)-OH (2mmole) / DIC (lmmole) / DMAP
(0.2mmole) in
DCM for about 16h. The remaining amino acids were coupled by standard Boc-
chemistry to
obtain the depsipeptidyl resin of the following sequence: Ser-Gln-Gly-Thr-O-
PLA-Thr-Ser-
Glu-Tyr-Ser-Ly s-Tyr-Leu-Asp-Ser-Arg-Arg-Ala-Gln-Asp-Phe-Val-Gln-Trp-Leu-Met-
Asn-
Thr-NH2 (SEQ ID NO: 64).
The peptidyl resin was treated with liquid hydrogen fluoride to cleave the
crude
peptide from the solid support and remove all protecting groups. The
depsipeptide was
purified by preparative HPLC, and analyzed by MS and analytical HPLC. The
purified
peptide demonstrated a single peak in analytical chromatography and the ESI-MS
analysis
yielded the desired mass of 3359.0 which corresponds with the calculated
molecular weight
of 3359.6 daltons.
A similar procedure was used to synthesize the depsipeptide [ThrS-O-PLA6,
E9]glucagon (1-29) amide (SEQ ID NO: 65) with a single addition coupling of an
N-terminal
histidine residue. The purified peptide demonstrated a single peak in
analytical
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-104-
chromatography and the ESI-MS analysis yielded the desired mass of 3495.9
which
corresponds with the calculated molecular weight of 3496.8 daltons.
EXAMPLE 19
The following peptides were synthesized as generally described above and
subsequently tested for the ability to stimulate the GLP-1 receptor by
assaying cAMP release
from cells expressing the human GLP-1 receptor and for the ability to
stimulate the glucagon
receptor by assaying cAMP release from cells expressing the human glucagon
receptor and
stimulated with 0.5 nM glucagon, as generally described in Example 13. The
results of the
assays are shown in Table 13.
CA 02707861 2010-04-09
WO 2009/058662 PCT/US2008/080973
-105-
TABLE 13
eptide Glucagon GLP-1 agonism
antagonism
C50(nM, C50(nM)
inhibit 0.5nM
G)
Glucagon .005 0.008
GLP-1 .005 0.002
[PLA6, E9]Glucagon(6-29) 23.75 4.16 No agonism
LA TSEYSKYLDSRRAQDFVQWLMNT-NH2
(SEQ ID NO: 71)
[PLA6, D9, D28]Glucagon(6-29) 9.03 1.54 746.0 225.7
LA TSDYSKYLDSRRAQDFVQWLMDT-NH2
(SEQ ID NO: 62)
[E9]Glucagon (2-29) 340.0 149.0 2719.8 2136.4
SQGTFTSEYSKYLDSRRAQDFVQ WLMNT-NH2
(SEQ ID NO: 63)
[Thr5-O-PLA6, E9]Glucagon(2-29) 6.49 2.17 1305.6 241.5
S QGT(O *)FTSEYS KYLDSRRAQDFVQ WLMNT-
2 (SEQ ID NO: 64)
[Thr5-O-PLA6, E9]Glucagon(1-29) 14.19 7.89 121.0 35.5
SQGT(O *)FTSEYSKYLDSRRAQDFVQWLMNT-
2 (SEQ ID NO: 65)
(0*) represents a depsipeptide bond.