Note: Descriptions are shown in the official language in which they were submitted.
CA 02719597 2010-10-29
-1-
METHODS AND APPARATUS FOR NANOPARTICLE-ASSISTED NUCLEIC
ACID HYBRIDIZATION AND MICROARRAY ANALYSIS
Technical Field
[0001] The present invention relates to methods and apparatus for nucleic acid
hybridization and microarray analysis.
Background
[0002] The ability to detect specific nucleic acid sequences is important for
clinical diagnosis, biomedical research, food and drug testing, environmental
monitoring and industrial applications. For example, detection of variations
in
nucleic acid sequences (e.g., single nucleotide polymorphisms (SNPs) in the
human genome) is important for studying the genetic basis of inherited
diseases
and for facilitating individualized medicine.
[0003] Nucleic acid hybridization assays have been used for detection of
specific nucleic acid sequences. The principle of nucleic acid hybridization
has
also allowed the development of microarray technology. Microarray technology
provides parallel nucleic acid hybridizations for a large number of
immobilized
oligonucleotides or DNA on a relatively small surface.
[0004] However, a central challenge to conventional nucleic acid
hybridization assays and microarray technology is achieving a high degree of
discrimination between a target sequence that is perfectly matched to a probe
and
a sequence having a single base-pair mismatch to the probe. In conventional
microarray assays, single-base-pair discrimination is typically achieved by
adjusting the hybridization conditions (e.g., increasing incubation
temperature, or
lowering ionic strength, or adding additives such as formamide). In order to
reduce or minimize non-specific binding between mismatched target and probe
sequences, conventional microarray analysis is typically performed using an
incubation temperature in the range of 40 to 65 C. This usually requires the
use
of temperature control equipment, for example, an incubation oven. Further,
conventional microarray analysis is typically performed at elevated
temperatures
CA 02719597 2010-10-29
-2-
and the incubation requires an extended period of time, for example, from 8 to
20
hours. Additionally, because microarray analysis involves a large set of
different
probes having different sequences on a single glass slide, it is difficult to
find an
optimum incubation temperature that would provide good binding specificity for
all the target/probe pairs.
[0005] Nanoparticles often exhibit properties that differ significantly from
those observed in bulk materials. Some of the properties of nanoparticles are
their
(1) small size (typically 1-100 nm), (2) large surface-to-volume ratio, and
(3)
unusual target binding properties.
[0006] One of the unusual properties about nanoparticles, such as gold
nanoparticles, is that single-stranded nucleic acid molecules can non-
covalently
bind or adsorb to nanoparticles, whereas double-stranded nucleic acid
molecules
generally do not: Huixiang Li and Lewis Rothberg, 2004, Proceedings of the
National Academy of Science, Vol. 101, No. 39, 14036-14039, which is hereby
incorporated by reference. Silver nanoparticles have also been shown to behave
like gold nanoparticles in terms of non-covalent binding with single-stranded
nucleic acid molecules: Chen et al., Analyst, (2010), 135, 1066-1069, which is
hereby incorporated by reference. It should be noted that this type of binding
between single-stranded nucleic acid molecules and nanoparticles is non-
covalent
in nature and does not require the formation of covalent bonds.
[0007] The different propensities of single-stranded and double-stranded
nucleic acid molecules to non-covalently adsorb to nanoparticles arise because
single-stranded nucleic acid molecules can uncoil sufficiently to expose their
bases, whereas double-stranded nucleic acid molecules have a stable double-
helix
that presents the negatively charged phosphate backbone. Gold or silver
nanoparticles in solution are typically stabilized by adsorbed negatively
charged
ions (e.g., citrate) whose repulsion prevents the nanoparticles from
aggregating.
Repulsion between the charged phosphate backbone of double-stranded nucleic
acid molecules and the adsorbed citrate ions dominates the electrostatic
interaction
between the nanoparticles and double-stranded nucleic acid molecules so that
double-stranded nucleic acid molecules will not adsorb to the nanoparticles.
In
CA 02719597 2010-10-29
-3-
contrast, because the single-stranded nucleic acid molecules are sufficiently
flexible to partially uncoil the bases, they can be exposed to the
nanoparticles.
Under these conditions, the negative charge on the phosphate backbone is
sufficiently distant so that attractive electrostatic interactions, van der
Waals
forces and hydrophobic interactions between the bases and the gold
nanoparticles
are sufficient to cause single-stranded nucleic acid molecules to stick to the
nanoparticles. The same mechanism is not operative with double-stranded
nucleic
acid molecules because the duplex structure does not permit the uncoiling
needed
to expose the bases: Li and Rothberg, 2004, supra.
[00081 Rothberg et al. have developed a method for detection of DNA
sequences based on the interaction of single-stranded DNA molecules and gold
nanoparticles: Li and Rothberg, supra; Rothberg et al., US Patent Application
Publications US 2005/0059042 and US 2006/0166249. The Rothberg method
requires a target single-stranded DNA molecule to be first exposed to a probe
single-stranded DNA molecule in solution, and then exposed to gold
nanoparticles. If the target molecule hybridizes to the probe molecule in
solution,
the target molecule will not subsequently adsorb to the gold nanoparticles. If
the
target molecule does not hybridize to the probe molecule in solution, the
target
molecule will then adsorb to the gold nanoparticles. The differences may be
detected by adding salt to the solution, which results in color changes as
gold
nanoparticles that are not stabilized by single-stranded DNA molecules
aggregate
under high salt conditions.
[00091 In the Rothberg method, the hybridization of target and probe DNA
molecules occurs in the absence of gold nanoparticles. In other words, gold
nanoparticles are not added before the hybridization reaction, but are added
after
hybridization to adsorb unhybridized single-stranded DNA molecules.
Furthermore, in order to distinguish single-base-pair mismatches, the Rothberg
method requires a dehybridization step which appears to require elevated
temperatures (e.g., above the melting temperature of mismatched duplexes, but
below the melting temperature of perfectly matched duplexes). Furthermore, the
Rothberg method does not use immobilized DNA molecules.
CA 02719597 2010-10-29
-4-
[0010J Based on the foregoing, it would be desirable to provide nucleic acid
hybridization and microarray methods and apparatus that provide a high degree
of
discrimination between a perfectly matched target sequence and a sequence with
a
single base-pair mismatch. In particular, it would be desirable to provide
methods
and apparatus which utilize the unique property of nanoparticles, while
avoiding
the need for elevated temperatures. For example, it would be desirable to
provide
such methods and apparatus wherein the incubation step can be performed at
ambient temperature without the use of any specialized heating equipment.
Brief Description of Drawings
[0011] In drawings which show non-limiting embodiments of the invention:
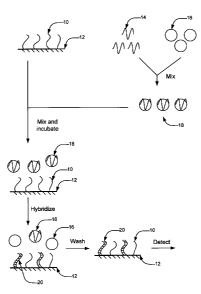
[0012] FIG. 1 illustrates a nucleic acid hybridization method according to an
example embodiment of the invention.
[0013] FIG. 1A illustrates a nucleic acid hybridization method according to an
example embodiment of the invention, wherein the target nucleic acid molecules
are labeled with a detectable label.
[0014] FIGS. 2A-C illustrate probe nucleic acid molecules immobilized on
substrate surfaces.
[0015] FIG. 3 illustrates a nucleic-acid-hybridization-based method to
distinguish between two different nucleic acid molecules according to an
example
embodiment of the invention.
[0016] FIG. 4 illustrates an alternative nucleic-acid-hybridization-based
method to distinguish between two different nucleic acid molecules according
to
an example embodiment of the invention.
[0017] FIG. 5A illustrates a method for microarray analysis according to an
example embodiment of the invention.
[0018] FIG. 5B illustrates a method for microarray analysis according to an
CA 02719597 2010-10-29
-5-
example embodiment of the invention, wherein the microarray comprises line
arrays.
100191 FIG. 6 illustrates a method for microarray analysis according to an
example embodiment of the invention.
[00201 FIG. 7 is a schematic view showing the assembly steps for fabricating
a microarray device by combining a test chip and first and second channel
plates.
[00211 FIG. 8A is a schematic view of a first channel plate having a plurality
of first microfluidic channels configured in a radial pattern.
100221 FIG. 8B is a schematic view of second channel plate having a plurality
of second microfluidic channels configured in a spiral pattern.
[00231 FIG. 8C is schematic view showing a first dimensional, centrifugal
force (F) used to distribute liquids in the radially configured first
microfluidic
channels of FIG. 8A.
100241 FIG. 8D is a schematic view showing a second dimensional centrifugal
force used to distribute liquids in the spiral second microfluidic channels of
FIG.
8B.
[00251 FIG. 8E is a schematic view showing the first channel plate sealingly
connected to a test chip;
[0026] FIG. 8F is a schematic view, showing the second channel plate
sealingly connected to the test chip after removal of the first channel plate.
10027] FIG. 8G is a schematic view of a test chip showing positive test
results
at select microarray test positions after removal of the second channel plate.
[00281 FIG. 9A is a schematic view of a blank test chip.
CA 02719597 2010-10-29
-6-
[0029] FIG. 9(B1) is a schematic view of a first channel plate having a
plurality of first microfluidic channels configured in a right spiral pattern.
[0030] FIG. 9(B2) is a schematic view of a second channel plate having a
plurality of second microfluidic channels configured in a left spiral pattern.
[00311 FIG. 9C is a schematic view showing the intersecting reagent
distribution patterns on the test chip.
100321 FIG. 9D is a schematic view showing positive test results at microarray
test positions located at the intersections between the reagent distribution
patterns
of FIG. 9C.
100331 FIG. I OA is a plan view of a first channel plate having a plurality of
closely spaced first microfluidic channels configured in a right spiral
pattern. The
inset shows selected channels in fluid communication with fluid inlet
reservoirs.
100341 FIG. I OB is a plan view of a second channel plate having a plurality
of
closely spaced second microfluidic channels conFig.d in a left spiral pattern.
The
inset shows selected channels in fluid communication with fluid inlet
reservoirs.
[0035] FIG. IOC is a plan view showing the intersecting reagent distribution
patterns applied to a test chip. The inset shows selected test positions
formed by
the intersections of the first and second reagent distribution patterns.
[0036] FIG. 11 illustrates a microfluidic microarray method using straight
microchannels: (a) creation of a DNA probe line array on aldehyde-modified
glass
slide via straight microchamnels; (b) hybridization of DNA samples in straight
channels orthogonal to the straight probe lines printed on the glass slide.
[00371 FIG. 12 illustrates an example PDMS microfluidic microarray
assembly: (a) a first channel plate used for probe immobilization; (b) a
second
channel plate used for sample hybridization; (c) a cross-sectional view of the
16
microchannels along line AB in (b).
CA 02719597 2010-10-29
= -7-
[00381 FIG. 13 illustrates non-covalent association and dissociation between
DNA molecules and gold nanoparticles (GNPs). (a) GNP-DNA complexes from
the incubation of target DNA with GNP; (b) perfectly matched target DNAs
desorbed from GNPs and hybridized to the surface immobilized probes. (c)
mismatched DNAs remained bound to GNPs and were washed away.
100391 FIG. 14 shows (a) images of hybridized patches of perfectly matched
(PM) and single-base-pair-mismatched (MM) target oligonucleotides in
triplicate.
Here, the oligonucleotides (oligos) were preincubated with GNPs (5 nm) at
different ratios. (b) Discrimination ratios between PM and MM duplexes. The
discrimination ratios were calculated by dividing the signal of PM DNAs with
that
of MM DNAs (the higher ratio, the better discrimination). (c) The fluorescent
hybridization signals from the images in (a), and the results at
oligo/GNPs=1:1 are
expanded and shown in the right inset.
[0040] FIG. 15 shows (a) images of hybridized patches of PM and MM (264-
bp) PCR products or amplicons in triplicate. Here, the amplicons were
preincubated with GNPs (5 nm) at different ratios. (b) Discrimination ratios
between PM and MM amplicons. The discrimination ratios were calculated by
dividing the signal of PM DNAs with that of MM DNAs.
Detailed Description
[00411 Throughout the following description, specific details are set forth in
order to provide a more thorough understanding of the invention. However, the
invention may be practiced without these particulars. In other instances, well
known elements have not been shown or described in detail to avoid
unnecessarily
obscuring the invention. Accordingly, the specification and drawings are to be
regarded in an illustrative, rather than a restrictive, sense.
Nanoparticle-Assisted Nucleic Acid Hybridization
100421 One aspect of the invention provides a method for nanoparticle-
assisted nucleic acid hybridization analysis. The method has many
applications.
CA 02719597 2010-10-29
-8-
For example, the method can be used to assist the detection of the presence or
absence of a specific target nucleic acid molecule in a sample or test
solution.
[0043] An example embodiment is schematically illustrated in FIG. 1. In a
first step, probe nucleic acid molecules 10 are immobilized on a surface 12.
In a
second step, target nucleic acid molecules 14 are mixed with nanoparticles 16
to
form complexes 18 comprising nanoparticles 16 non-covalently associated with
target nucleic acid molecules 14. As described further below, nanoparticles 16
may comprise gold nanoparticles, for example. Nanoparticles 16 should be in
suspended, non-aggregated form, or deaggregated under suitable conditions. In
a
third step, complexes 18 comprising nanoparticles 16 non-covalently associated
with target nucleic acid molecules 14 are mixed with the probe nucleic acid
molecules 10 immobilized on surface 12 and incubated for a period of time. As
described further below, the incubation may be performed at ambient
temperature
or room temperature, for example. In a fourth step, a determination is made as
to
whether at least some of target nucleic acid molecules 14 have hybridized with
probe nucleic acid molecules 10 to form a hybridization duplex 20 comprising a
strand from target nucleic acid molecules 14 and a strand from probe nucleic
acid
molecules 10 and the level of hybridization. Each of these step are described
in
further detail below.
[0044] In the first step of FIG. 1, probe nucleic acid molecules 10 are
immobilized on surface 12. Surface 12 may be a solid surface, or a semi-solid
surface. For example, surface 12 may be a gel, polyacrylamide, agar, agarose,
or
gelatin. Surface 12 may be made of solid or curable materials, for example,
glass,
silicon, plastic, polymer, cellulose, etc. The solid surface may be, for
example, a
solid surface inside a test tube or a microfluidic channel, or on a glass
slide, a test
chip, a microarray chip, a microtiter plate, a nylon membrane, a film, or a
bead.
Surface 12 may be substantially flat (see FIG. 2A), or curved, for example,
the
surface of a spherical bead (see FIG. 2B) or a well of a microtiter plate (see
FIG.
2C). Surface 12 may also be coated or conjugated with one or more compounds,
for example, surface 12 may be aldehyde-functionalized. If surface 12 is a
solid
surface on a bead, the bead may be magnetic or magnetically attractable.
CA 02719597 2010-10-29
-9-
100451 There are a number of ways to immobilize or tether probe nucleic acid
molecules 10 on surface 12. One approach is in situ synthesis, wherein probe
nucleic acid molecules 10 are synthesized directly base by base on surface 12.
Another approach is to spot or print probe nucleic acid molecules 10 on
surface 12
using contact or non-contact printing methods. Other methods of immobilizing
probe nucleic acid molecules 10 are known to persons skilled in the art. For
example, immobilization can be achieved by chemical, mechanical, or
biochemical methods such as covalent binding, adsorption, polymer
encapsulation
and so forth. As described further below, one example method of chemical
immobilization is Schiff-base linkage formed between an aminated DNA or
oligonucleotides probe and an aldehyde-functionalized glass surface.
[00461 Probe nucleic acid molecules 10 are typically single-stranded, or
comprise at least a single-stranded region. In some embodiments, probe nucleic
acid molecules 10 may also comprise a double-stranded region, or a triple-
stranded region. Probe nucleic acid molecules 10 may be formed from
oligonucleotides, DNA, RNA, or peptide nucleic acid (PNA). They may include
both natural or artificial or synthetic nucleic acids. They may include
genomic
DNA or even a chromosome preparation (e.g., a chromosome preparation suitable
for fluorescent in situ hybridization (FISH)). They may be synthesized or
generated or amplified using standard procedures known to those skilled in the
art
or ordered from commercial vendors. Standard molecular biology methods for
probe preparation can be found in Sambrook and Russel, Molecular Cloning: A
Laboratory Manual, Cold Spring Harbor Laboratory Press, 2001, 3r`' edition,
which is hereby incorporated by reference.
10047] In some embodiments, probe nucleic acid molecules 10 may comprise
DNA fragments having a length in the 1Kb to 50 Kb range. In some
embodiments, probe nucleic acid molecules 10 are between 10 and 1000
nucleotides in length. In some embodiments, probe nucleic acid molecules 10
are
between 10 and 100 nucleotides in length. In some embodiments, probe nucleic
acid molecules 10 are between 10 and 50 nucleotides in length. For
oligonucleotides probes that are synthesized in situ, the maximum length is
typically 80 to 100 nucleotides. For probes that are not synthesized in situ,
they
CA 02719597 2010-10-29
-10-
can be more than 1000 nucleotides in length.
[0048] In the second step of FIG. 1, a sample solution containing complexes
18 comprising nanoparticles 16 non-covalently associated with target nucleic
acid
molecules 14 is provided. To prepare the sample solution, a first solution
comprising target nucleic acid molecules 14 is mixed with a second solution
comprising suspended non-aggregated nanoparticles 16. Target nucleic acid
molecules 14 may be oligonucleotides, or DNA, or RNA, or PNA. Target nucleic
acid molecules 14 may be single-stranded or double-stranded, or combinations
thereof. As described in the background section, single-stranded nucleic acid
molecules would bind non-covalently to nanoparticles, and double-stranded
nucleic acid molecules would not bind to nanoparticles.
(0049] In some embodiments, a heat denaturing step is performed after target
nucleic acid molecules 14 are mixed with nanoparticles 16. This is typically
done
if target nucleic acid molecules 14 are double-stranded, or comprise secondary
structures (e.g., hairpins). For example, target nucleic acid molecules 14 can
be
denatured at an elevated temperature sufficient to separate them into single-
stranded molecules or to remove the secondary structures so that target
nucleic
acid molecules 14 can non-covalently associate with or adsorb to the
nanoparticles. For example, target nucleic acid molecules 14 may be denatured
at
a temperature between 85 to 100 C, for a period of time between 10 seconds to
5
minutes. For example, target nucleic acid molecules 14 may be denatured at 95
C
for 3 minutes. The incubation at the elevated temperature may promote the
binding of single-stranded nucleic acid molecules 14 to nanoparticles 16.
After
formation of complex 18, the reaction solution may be snap chilled in an ice-
water
bath (e.g., at 4 C) to prevent renaturation of single-stranded molecules or
reformation of secondary structures.
[0050] The principle behind the non-covalent association between
nanoparticles and single-stranded nucleic acid is briefly described in the
background section above. Without wishing to be bound by any particular
theory,
it is likely that uncoiled single-stranded nucleic acid molecules bind or
adsorb to
nanoparticles through hydrophobic forces and/or van der Waals forces between
CA 02719597 2010-10-29
-11-
the bases of the uncoiled single-stranded nucleic acid molecules and the
nanoparticles. In contrast, double-stranded nucleic acid molecules do not bind
to
nanoparticles because the bases of the double-stranded nucleic acid molecules
are
not exposed and therefore not available for the aforementioned hydrophobic or
van der Waals interactions. The non-covalent binding of single-stranded
nucleic
acid molecules to nanoparticles can be relatively tight and stable. However,
this
binding is reversible, and single-stranded nucleic acid molecules may
dissociate
from the nanoparticles under suitable conditions. As described further below,
single-stranded nucleic acid molecules non-covalently bound to nanoparticles
may
dissociate from the nanoparticles and hybridize with complementary single-
stranded nucleic acid molecules to form duplexes.
[00511 It should be emphasized that the binding between the nanoparticles and
the single-stranded nucleic acid molecules in the present invention does not
require covalent links between the nanoparticles and the single-stranded
nucleic
acid molecules. Therefore, it is not necessary for nanoparticles 16 to be
covalently functionalized. Nor does it require target nucleic acid molecules
14 to
be thiol-modified in order to be bound to nanoparticles 16.
100521 Nanoparticles 16 may be sized between I and 100 nanometers. They
may be spherical or rod-shaped or of other shapes. Nanoparticles 16 may be
coated with negatively charged ions. As mentioned earlier, the negatively
charged
ions may help prevent aggregation of nanoparticles 16. Nanoparticles 16 may be
formed of a metal, a semiconductor, or an uncharged substrate, such as glass,
or
combinations thereof. Nanoparticles 16 may be sized between 1 and 50 nm, or
between 20 and 30 nm, or between 10 to 20 nm, or between I to 10 nm, or
between 3.0 to 5.5 nm. Nanoparticles 16 may have a mean particle size of 5.0
nm.
Nanoparticles may have a coefficient of variance of particle size that is less
than
15% of the mean particle size.
[00531 Metal nanoparticles 16 may be formed of a conductive metal or metal
alloy that allows a nanoparticle 16 to be capable of non-covalently
associating
with a single-stranded nucleic acid molecule. Prior to use in the present
invention,
it should be appreciated that the colloidal suspension should maintain the
metal
CA 02719597 2010-10-29
-12-
nanoparticles in a stable environment in which they are substantially free of
aggregation. The metal nanoparticles should not significantly associate with
double-stranded nucleic acid molecules. Example metal nanoparticles include,
without limitation, gold nanoparticles, silver nanoparticles, platinum
nanoparticles, mixed metal nanoparticles (e.g., gold shell surrounding a
silver
core), and combinations thereof. The metal nanoparticles may be magnetic or
magnetically attractable, for example, formed of an inner core such as cobalt
and
an outer layer such as gold.
100541 Preparation of colloidal metal nanoparticle suspensions can be carried
out according to known procedures, e.g., Grabar et al., Anal. Chem. 67:735-743
(1995), which is hereby incorporated by reference in its entirety. Metal
nanoparticles may be stabilized in the solution by negatively charged anions,
such
as citrate, acetate, carbonate, phosphate, oxalate, sulfate, or nitrate.
100551 In some embodiments, nanoparticles 16 comprise gold nanoparticles.
Preparation of gold nanoparticles can be carried out according to known
procedures, e.g., J. Turkevich, P. C. Stevenson, J. Hillier, Discuss. Faraday.
Soc.
1951, 11, 55-75; J. Kimling, M. Maier, B. Okenve, V. Kotaidis, H. Ballot, A.
Plech, J. Phys. Chem. B 2006, 110, 15700-15707; G. Frens, Colloid & Polymer
Science 1972, 250, 736-741; G. Frens, Nature (London), Phys. Sci. 1973, 241,
20-
22; J. W. Slot and H.J. Geuze, Eur. J. Cell Biol. 38, 87 (1985); M.C. Daniels
and
D. Astruc, Chem. Rev. (Washington DC) 104, 293 (2004), each of which is
hereby incorporated by reference in its entirety. Briefly, gold nanoparticles
are
typically produced in a liquid by reduction of chloroauric acid (HAuC14).
After
dissolving HAuC14, the solution is rapidly stirred while a reducing agent is
added.
This causes Au(III) ions to be reduced to neutral gold atoms. As more and more
of
these gold atoms form, the solution becomes supersaturated, and gold gradually
starts to precipitate in the form of sub-nanometer particles. The rest of the
gold
atoms that form stick to the existing particles, and, if the solution is
stirred
vigorously enough, the particles will be fairly uniform in size. The anions in
gold
nanoparticle preparation also prevent the gold nanoparticles from aggregating.
These anions may include citrate, acetate, carbonate, phosphate, oxalate,
sulfate,
or nitrate.
CA 02719597 2010-10-29
-13-
[0056] Alternatively, nanoparticles 16 can be purchased from commercial
sources. For example, gold nanoparticles can be purchased from Sigma Life
Sciences. The gold nanoparticles from Sigma Life Sciences may be in aqueous
solution and may have a particle size in the range of 3.0 to 5.5 nm and a mean
particle size of 5.0 nm.
[0057] Nanoparticles 16 may also be formed of an uncharged substrate (e.g.,
glass). The substrate may be charged using anions or polyanions. The anions or
polyanions can be coupled to the substrate (e.g., glass) using standard glass
binding chemistry. Example anions include, without limitation, citrate,
acetate,
carbonate, dihydrogen phosphate, oxalate, sulfate, and nitrate. Example
polyanions include, without limitation, poly(2-acrylamido-2-methyl-l-
propanesulfonic acid), poly(acrylic acid), poly(anetholesulfonic acid),
poly(anilinesulfonic acid), poly(sodium 4-styrenesulfonate), poly(4-
styrenesulfonic acid), and poly(vinylsulfonic acid). Other anions and
polyanions
may also be employed.
[0058] Although some of the examples in the present disclosure describe
experiments performed using gold nanoparticles, it will be appreciated by
those
skilled in the art that other nanoparticles having similar properties may also
be
used. For example, silver nanoparticles have been shown to behave like gold
nanoparticles in terms of non-covalent binding with single-stranded nucleic
acid
molecules: Chen et al., Analyst, (2010), 135, 1066-1069, which is hereby
incorporated by reference.
[0059) Target nucleic acid molecules 14 may be oligonucleotides,
deoxyribonucleic acid (DNA), ribonucleic acid (RNA) or an artificial or
synthetic
nucleic acid. They may be synthesized or generated or amplified using standard
procedures known to those skilled in the art or ordered from commercial
vendors.
Target nucleic acid molecules 14 may be isolated directly from samples (e.g.,
cells, tissues, cell extracts, tissue culture media, bodily fluids,
environmental
samples, other biological samples etc.), or they may first be amplified by
polymerase chain reaction (PCR) or reverse-transcription PCR (RT-PCR). Target
CA 02719597 2010-10-29
-14-
nucleic acid molecules 14 may comprise synthetic, natural, or structurally
modified nucleoside bases. Target nucleic acid molecule 14 can also be from
any
source organism (e.g., human or another animal, virus, bacteria, insect,
plant,
etc.).
[00601 Target nucleic acid molecules 14 may be of any length that is suitable
for non-covalent association with nanoparticles. In some embodiments, target
nucleic acid molecules 14 are between 10 to 1000 nucleotides or base-pairs in
length. In some embodiments, target nucleic acid molecules 14 may exceed 1000
nucleotides or base-pair in length. Target nucleic acid molecules 14 may
comprise single-stranded molecules, or double stranded molecules, or
combinations thereof. In some embodiments, target nucleic acid molecules 14
may comprise a single-stranded region and a double-stranded region. Target
nucleic acid molecules 14 may be purified or isolated molecules, or may be
present in a solution or sample that comprises other molecules or
contaminants.
Target nucleic acid molecules 14 may comprise nucleic acid molecules having
different sequences (e.g., a mixture of genomic DNA molecules, a mixture of
different PCR products, a mixture of eDNA molecules, or a mixture comprising
two related DNA sequences differing by a single base-pair). Target nucleic
acid
molecules 14 may be from a single sample source or may be from two or more
sample sources (e.g., pooled eDNA molecules from two types of cells, one being
stern cell, the other being differentiated cell, or genomic DNA from two human
individuals).
100611 Target nucleic acid molecules 14 can either be unlabeled or they can be
conjugated or otherwise coupled to a detectable label 22. FIG. IA shows a
method that is the same as the method illustrated in FIG. 1 except that target
nucleic acid molecules 14 are labeled with detectable label 22. Suitable
detectable
labels 22 include, without limitation, fluorescent labels, redox
(electrochemical)
labels, and radioactive labels.
[0062] Coupling of a fluorescent label to nucleic acid molecules can be
achieved using known nucleic acid-binding chemistry or by physical means, such
as through ionic, covalent or other forces well known in the art (see, e.g.,
CA 02719597 2010-10-29
-15-
Dattagupta et al., Analytical Biochemistry 177:85-89 (1989); Saiki et al.,
Proc.
Natl. Acad. Sci. USA 86:6230-6234 (1989); Gravitt et al., J. Clin. Micro.
36:3020-
3027 (1998), each of which is hereby incorporated by reference in its
entirety).
Either a terminal base or another base near the terminal base can be bound to
the
fluorescent label. For example, a terminal nucleotide base of the target
nucleic
acid molecules can be modified to contain a reactive group, such as (without
limitation) carboxyl, amino, hydroxyl, thiol, or the like.
[0063] The fluorescent label can be any fluorophore that can be conjugated to
a nucleic acid and preferably has a photoluminescent property that can be
detected
and easily identified with appropriate detection equipment. Exemplary
fluorescent
labels include, without limitation, fluorescent dyes, semiconductor quantum
dots,
lanthanide atom-containing complexes, and fluorescent proteins. Example
fluorescent dyes include, without limitation, Calcein, FITC, AlexaTM,
Rhodamine
110, 5-FAM, Oregon Green i M 500, Oregon Green I M 488, RiboGreen ' M,
Rhodamine Green-"m, Rhodamine 123, Magnesium Green 1 M, Calcium Green F M,
Cy3 CM, AlexaTM 546, TRITC, Magnesium OrangeTM, Phycoerythrin R&B,
Rhodamine Phalloidin, Calcium Orange'M, Pyronin Y, Rhodamine B, TAMRA,
Rhodamine Red.TM., ROX, Nile Red, YO-PRO ! M-3, R-phycocyanin, C-
Phycocyanin, Cy5 ! M, Thiadicarbocyanine, and Cy5.5 1 M. Other dyes now known
or hereafter developed may similarly be used.
[0064] The molar ratio of single-stranded target nucleic acid molecules 14 to
nanoparticles 16 can be varied within relatively wide limits. In some
embodiments, the molar ratio of single-stranded target nucleic acid molecules
14
to nanoparticles 16 may be in the range of 4 to 1. For example, the molar
ratio
may be 4:1, 3:1, 2:1, or 1:1. As described further below, a lower molar ratio
may
result in a higher degree of discrimination between perfect match (PM) and
single-
base-pair mismatch (MM) sequences in a hybridization reaction.
[0065] In some embodiments, complexes 18 comprising nanoparticles 16 non-
covalently associated with single-stranded target nucleic acid molecules 14
are
used immediately after formation for hybridization with probe nucleic acid
molecules 10 immobilized on surface 12, as described later. In some
CA 02719597 2010-10-29
-16-
embodiments, complexes 18 may be stored at a low temperature (e.g., 4 C) for
future use. For example, complexes 18 comprising gold nanoparticles 16 non-
covalently associated with single-stranded target nucleic acid molecules 14
remain
stable at 4 C for at least 48 hours. In some embodiments, sample solutions
containing complexes 18 may be diluted to a desired concentration before
hybridization.
[00661 In the third step of FIG. 1, the sample solution containing complexes
18 comprising nanoparticles 16 non-covalently associated with single-stranded
target nucleic acid molecules 14 are mixed with probe nucleic acid molecules
10
immobilized on surface 12 and incubated for a period of time. Because the non-
covalent binding between a nanoparticle and a single-stranded nucleic acid
molecule is relatively stable, a single-stranded target nucleic acid molecule
would
typically only dissociate from the nanoparticle if the target nucleic acid
molecule
has a sequence that is complementary to the sequence of a probe nucleic acid
molecule and the probe nucleic acid molecule is available to hybridize with
the
target nucleic acid molecule. If the target nucleic acid molecule has a
sequence
that is perfectly complementary (i.e., a perfect match) to the sequence of the
immobilized probe nucleic acid molecule, the target nucleic acid molecule
would
tend to dissociate from the nanoparticles and hybridize with the immobilized
probe nucleic acid molecule. If the target nucleic acid molecule has a
sequence
that is non-complementary and unrelated to the sequence of the immobilized
probe nucleic acid molecule, the target nucleic acid molecules would tend to
remain associated with the nanoparticles and not hybridize with the
immobilized
probe nucleic acid molecule. If the target nucleic acid molecule has a
sequence
that is related to the sequence of the immobilized probe nucleic acid molecule
but
has one or a few base-pair mismatches to the sequence of the immobilized probe
nucleic acid molecule, the target nucleic acid molecules would not tend to
dissociate from the nanoparticles and hybridize with the immobilized probe
nucleic acid molecule.
[00671 In other words, nanoparticles 16 serve as a carrier or competitor in
the
hybridization process, and probe nucleic acid molecules 10 and nanoparticles
16
compete for binding with target nucleic acid molecules 14. Since binding of
CA 02719597 2010-10-29
-17-
either probe nucleic acid molecules 10 or nanoparticles 16 with target nucleic
acid
molecules 14 are reversible processes, the amount of target nucleic acid
molecules
14 that would dissociate from nanoparticles 16 and hybridize with probed
nucleic
acid molecules 10 would largely depend on the thermodynamic stability of
hybridization duplexes 20. Typically, hybridization duplexes composed of two
perfectly matched strands are thermodynamically more stable (i.e., having
lower
Gibbs free energy) than hybridization duplexes composed of two mismatched
strands. The nanoparticles therefore could be used to discriminate between
nucleic acid molecules having a sequence complementary to the sequence of
probe nucleic acid molecules and nucleic acid molecules having a sequence not
complementary to the sequence of the probe nucleic acid molecules.
100681 In some embodiments, the hybridization step is performed at elevated
temperatures (e.g., above ambient temperature or room temperature). For
example, the hybridization step may be performed at a temperature between 30
to
70 C, and in an incubation oven. However, this is not necessary. In some
embodiments, the hybridization step is performed at ambient temperature or
room
temperature without the use of any specialized equipment for temperature
control.
For example, the hybridization step may be performed at a temperature between
20 to 30 C, for example, between 22 and 26 C, or between 22 and 24 C, or
between 24 and 26 C.
[00691 As described further later, the method provides a high degree of
discrimination between nucleic acid molecules having a sequence complementary
(i.e., perfect match) to the sequence of probe nucleic acid molecules and
nucleic
acid molecules having a sequence that has a single-base-pair mismatch to the
sequence of the probe nucleic acid molecules without the use of elevated
temperatures or a heating equipment.
[00701 Buffer conditions for hybridization are well known to those skilled in
the art and can be varied within relatively wide limits. The term
hybridization
stringency refers to the degree to which hybridization conditions disfavor the
formation of hybrids containing mismatched nucleotides, thereby promoting the
formation of perfectly matched hybrids; with higher stringency correlated with
a
CA 02719597 2010-10-29
-18-
lower tolerance for mismatched hybrids. Factors that affect the stringency of
hybridization include, but are not limited to, pH, ionic strength,
concentration of
organic solvents such as formarnide and dimethylsulfoxide, and concentration
of
sodium dodecyl sulfate (SDS). As is well known to those of skilled in the art,
hybridization stringency is increased by higher temperatures, lower ionic
strengths, and lower solvent concentrations. See, for example, M. A. Innis et
al.
(eds.) PCR Protocols, Academic Press, San Diego, 1990; B. D. Hames et al.
(eds.)
Nucleic Acid Hybridisation: A Practical Approach, IRL Press, Oxford, 1985,
which are hereby incorporated by reference in their entirety.
100711 In some embodiments, the hybridization buffer comprises 1.0 X saline-
sodium citrate (SSC) and 0.2% sodium dodecyl sulfate (SDS). It will be
appreciated by those skilled in the art that buffers comprising very high salt
(e.g.,
Na) concentrations should typically be avoided. For example, buffers
comprising
Na+ concentrations greater than 0.3 M should typically be avoided. This is to
prevent the premature aggregation of the nanoparticles, although nanoparticle
aggregation is typically inhibited by the non-covalently associated single-
stranded
nucleic acid molecules. In other words, single-stranded nucleic acid molecules
stabilize the nanoparticles in the suspended, non-aggregated form. It will be
appreciated by those skilled in the art that many other hybridization buffers
can
also be used.
[00721 After incubation, surface 12 with immobilized probe nucleic acid
molecules 10 and hybridization duplexes 20, if any, is washed with a wash
solution. The wash buffer may be the same or different from the hybridization
buffer. The wash step washes away target nucleic acid molecules 14 that remain
associated with nanoparticles 16 as well as nanoparticles 16. Target nucleic
acid
molecules 14 that have hybridized with probe nucleic acid molecules 10 as
hybridization duplexes 20 would remain on surface 12.
100731 In the fourth step of FIG. 1, a determination is made as to whether at
least some of target nucleic acid molecules 14 have hybridized with probe
nucleic
acid molecules 12 to form hybridization duplexes 20 and the level of
hybridization. In some embodiments, surface 12 is dried before a detection
CA 02719597 2010-10-29
19-
method is applied. In some embodiments, a detection method may be applied
without drying surface 12.
100741 This determination or detection may be qualitative or quantitative. A
large number of methods are available to detect or quantify hybridization
duplexes
20 on surface 12. For example, if target nucleic acid molecules 14 were
fluorescently labeled prior to mixing and incubation (FIG. IA), surface 12 can
be
scanned for fluorescence emissions. For example, a confocal laser fluorescent
scanner may be used. If both the target and the probe are fluorescently
labeled, a
detection method called fluorescence resonance energy transfer (FRET) may be
used. If the probe is a chromosome preparation, detection methods suitable for
fluorescent in situ hybridization (FISH) may be used. Alternatively, if the
target
nucleic acid molecules comprise redox labels, or radioactive labels, other
methods
may be used to detect the level of hybridization. These methods are well known
to those skilled in the art.
Distinguishing Between Two Different Nucleic Acid Molecules
[00751 The above described nanoparticle assisted hybridization method can be
used to distinguish between two different nucleic acid molecules. In some
embodiments, the method can be used to distinguish between two related nucleic
acid sequences. In some embodiments, the method can be used to distinguish
between two related nucleic acid sequences that differ by two or more
nucleotides.
In some embodiments, the method can be used to distinguish between two related
nucleic acid sequences that differ by a single nucleotide. It is often
advantageous
to distinguish between two or more nucleic acid sequences which are related
but
which differ by a single nucleotide. For example, single nucleotide
polymorphisms (SNPs) in human and other mammalian genomes are
characterized by a sequence difference of a single nucleotide. Also, many
mutations of clinical significance differ by only a single nucleotide.
[00761 One example embodiment is described here, which is a method for
distinguishing between two different nucleic acid samples. As illustrated in
FIG.
3, the method involves performing two nanoparticle-assisted hybridization
assays
CA 02719597 2010-10-29
-20-
in parallel with two different nucleic acid samples 14A, 14B. Since the method
is
based on the already described nanoparticle-assisted hybridization method,
only
key steps are described below and the reader can refer to the previous section
for
further details.
[00771 First, probe nucleic acid molecules 10 are provided and immobilized
on a first surface 12A and a second surface 12B. Typically, an equal amount of
the probe nucleic acid molecules 10 are immobilized on first surface 12A and
second surface 12B.
[00781 Second, first and second nucleic acid samples 14A and 14B are mixed
separately with nanoparticles 16 under identical or comparable conditions to
form
nucleic acid-nanoparticle complexes 18A and 18B. As described earlier, only
single-stranded nucleic acid molecules bind or adsorb to nanoparticles 16.
Therefore, if the first and second nucleic acid samples 14A and 14B are double-
stranded, they need to be heat denatured into single-stranded molecules. These
two parallel reactions should be performed under identical or comparable
conditions. In some embodiments, the amounts and concentrations of the first
and
second nucleic acid samples 14A and 14B are comparable. In some embodiments,
one of the nucleic acid samples 14A and 14B may be a positive control, i.e.,
having a sequence that is known to be complementary to the sequence of the
probe nucleic acid molecules 10. In some embodiments, one of the nucleic acid
samples 14A and 14B may be a negative control, i.e., having a sequence that is
known to be not complementary or unrelated to the sequence of the probe
nucleic
acid molecules 10.
100791 Third, the two complexes 18A and 18B from the previous step are
separately mixed and incubated with the immobilized nucleic acid probe 10 in a
suitable hybridization buffer. If one of the nucleic acid samples 14A and 14B
has
a sequence that is complementary to the sequence of the probe nucleic acid
molecules 10, it would dissociate from nanoparticles 16 and hybridize with
probe
nucleic acid molecules 10. As mentioned before, this step may be performed at
room temperature (e.g., 22-26 C), because of the higher level of
discrimination
provided by the nanoparticles against hybridization between mismatched nucleic
CA 02719597 2010-10-29
-21 -
acid molecules. After the incubation is completed, surfaces 12A, 12B with the
immobilized probe nucleic acid molecules 10 and hybridization duplexes 20, if
any, are washed with a wash buffer. These two parallel reactions should be
performed under identical or comparable conditions.
[0080] Fourth, determinations are made as to how much of the first nucleic
acid sample 14A and how much of the second nucleic acid 14B sample is
hybridized with probe 10. The results can be used to infer if one of the
nucleic
acid samples 14A, 14B is complementary or non-complementary to probe 10.
[0081] In other words, two parallel and separate nanoparticle-assisted
hybridization reactions are carried out, one using a first nucleic acid sample
14A
and a probe 10, the other using a second nucleic acid sample 14B and the same
probe 10, under identical or comparable conditions.
10082] The inventors have determined, as described further later, that use of
nanoparticles increases the discrimination ratio between a perfectly matched
(PM)
sequence and a single base-pair mismatch (MM) sequence. In the present
disclosure, the term "discrimination ratio" refers to the ratio between the
degree of
hybridization of the PM sequence with the probe and the degree of
hybridization
of the MM sequence with the probe. When both the PM and MM sequences are
fluorescently labeled, the discrimination ratio may be calculated based on the
ratio
between the measured fluorescence for the PM sequence and the measured
fluorescence for the MM sequence upon hybridization with the probe.
[0083] Because use of nanoparticles increases the discrimination ratio, it is
possible to perform the incubation step at a temperature that is lower than
conventional hybridization or DNA microarray protocols and achieve the same
degree of discrimination. In some embodiments, the hybridization step is
performed at ambient temperature or room temperature without the use of any
specialized equipment for temperature control. For example, the hybridization
step may be performed at a temperature between 20 to 30 degrees Celsius, for
example, between 22 and 26 degrees Celsius, or between 22 and 24 degrees
Celsius, or between 24 and 26 degrees Celsius. Under these ambient or room
CA 02719597 2010-10-29
-22-
temperature incubation conditions, the discrimination ratio may be greater
than
5:1, for example, greater than 10:1, or 15:1, or 20:1, or 25:1.
[0084] Another example embodiment is described below, which is also a
method for distinguishing two different nucleic acid samples and is also a
variation of the earlier described nanoparticles-assisted hybridization
method. As
illustrated in FIG. 4, the method involves incubating two different nucleic
acid
samples in a single hybridization solution.
[0085] First, probe nucleic acid molecules 10 are provided and are
immobilized on a surface 12 as described above.
[0086] Second, the first and second nucleic samples 14A, 14B are separately
labeled with two respective detectable labels 22A, 22B (e.g., two different
fluorescent labels). For example, first nucleic acid sample 14A may be labeled
with Cy3 and second nucleic acid sample may be labeled with Cy5. Then the
first
and second nucleic samples 14A, 14B are mixed in a single sample solution. The
amounts and concentrations of the first and second nucleic acid samples 14A,
14B
should be comparable. In some embodiments, there is an equal molar
concentration of the first and second nucleic acid samples 14A, 14B in the
sample
solution. In some embodiments, one of the nucleic acid samples 14A and 14B
may be a positive control, i.e., having a sequence that is known to be
complementary to the sequence of the probe nucleic acid molecules 10. In some
embodiments, one of the nucleic acid samples 14A and 14B may be a negative
control, i.e., having a sequence that is known to be not complementary or
unrelated to the sequence of the probe nucleic acid molecules 10.
[0087] The sample solution is then incubated with the immobilized probe
nucleic acid molecules 10 in a single reaction. As mentioned before, the
incubation can be performed at room temperature.
[0088] After incubation and washing, a determination is made as to how much
of the first sample nucleic acid 14A and how much of the second sample nucleic
acid 14B is hybridized with the probe 10. For example, this can be determined
by
CA 02719597 2010-10-29
-23-
measuring the intensity of the two fluorescent labels. The results can be used
to
infer if one of the nucleic acid samples 14A, 14B is complementary or non-
complementary to probe 10.
[00891 When two nucleic acid samples 14A, 14B are mixed in a single
solution and each is conjugated to a different fluorescent label 22A, 22B, it
is
preferable that the fluorescent labels 22A, 22B can be distinguished from one
another using appropriate detection equipment. That is, the fluorescent
emissions
of one fluorescent label should not overlap or interfere with the fluorescent
emissions of another fluorescent label. For example, one fluorescent label may
be
Cy3 and the other fluorescent label may by C5. Other example pairs of
fluorescent labels that are suitable for this purpose are well-known to those
skilled
in the art.
Use of Nanoparticle-Assisted Hybridization with Microarrays
[0090] The nanoparticle-assisted hybridization method can also be applied to
microarray technology. A DNA microarray is a multiplex technology commonly
used in molecular biology. It consists of an arrayed series of tens, hundreds,
thousands, or even tens of thousands of microscopic spots of picomoles (10- 12
moles) of oligonucleotides or DNA probes, each having a specific nucleotide
sequence. These can be a short section of a gene or other DNA element that are
used to hybridize a sample (e.g., cDNA or genomic DNA or RNA) under high-
stringency conditions. Probe-target hybridization is usually detected and
quantified by detection of fluorophore-, silver-, or chemilurninescence-
labeled
targets to determine relative abundance of nucleic acid sequences in the
sample.
Since an array can contain tens, hundreds, thousands, or even tens of
thousands of
probes, a microarray experiment can accomplish many genetic tests in parallel.
Therefore arrays have dramatically accelerated many types of investigation.
[0091] In standard microarrays, the probes are typically attached via surface
engineering to a solid surface by a covalent bond to a chemical matrix (via
epoxy-
silane, amino-silane, lysine, polyacrylamide or others). The solid surface can
be
glass or a silicon chip, for example, in microarray chips produced by
Affymetrix.
CA 02719597 2010-10-29
-24-
Other microarray platforms, such as Illumina, may use microscopic beads,
instead
of the large solid support.
[00921 DNA microarrays can be used to measure changes in gene expression
levels, to detect single nucleotide polymorphisms (SNPs), or to genotype or
sequence mutant genomes. Procedures for using DNA microarrays are well-
known to those skilled in the art: e.g., David Bowtell and Joseph Sambrook,
DNA
Microarrays: A Molecular Cloning Manual, Cold Spring Harbor Laboratory Press;
1st edition (2002), which is hereby incorporated by reference.
[00931 An example embodiment according to the present invention is
described below. First, a surface 12 is provided. Surface 12 may be any
surface
suitable for microarrays. Surface 12 may comprise a glass slide, a glass chip,
a
compact disc, a plate, a membrane, a film, or beads, etc.
[00941 Second, a plurality of nucleic acid probes 10 is immobilized on surface
12 at spaced-apart positions. These positions may be discrete spots or lines
or
some other patterns. FIG. 5A shows an example microarray 24 having a dot array
pattern. FIG. 5B shows an example microarray 24 having a line pattern. As
mentioned before, there are a number of ways to immobilize or tether probe
nucleic acid molecules 10 on surface 12. One approach is in situ synthesis,
wherein the probe nucleic acid molecules 10 are synthesized directly base by
base
on surface 12. Another approach is to spot or print the probe nucleic acid
molecules 10 on surface 12 using contact or non-contact printing methods.
Another approach is chemical immobilization which is based on Schiff-base
linkage formed between aminated probe and aldehyde-functionalized surface.
This approach is described in detail later.
100951 Third, a sample solution is prepared and incubated with the probes.
The sample solution contains complexes 18 comprising sample nucleic acid
molecules 14 non-covalently associated with nanoparticles 16. Sample nucleic
acid molecules 14 may be obtained from a single sample source, or from two
different sample sources (for example, a healthy cell and a diseased cell). If
the
sample nucleic acid molecules 14 are obtained from two different sources, they
CA 02719597 2010-10-29
-25-
may be labeled with two different detectable labels 22 (e.g., two different
fluorophores) and then mixed together. Sample nucleic acid molecules 14 are
then mixed with nanoparticles 16 to form a sample solution. Sample nucleic
acid
molecules 14 may be heat denatured to separate into single-stranded molecules
before non-covalently binding or adsorbing to nanoparticles 16.
[0096] Fourth, the sample solution is incubated with microarray 24.
Because nanoparticles 16 can increase the discrimination ratio between
perfectly
matched and mismatched sequences, the requirement for thermal stringency is
reduced or eliminated. In some embodiments, the hybridization step is
performed
at ambient temperature or room temperature. For example, the hybridization
step
may be performed at a temperature between 20 to 30 degrees Celsius, for
example, between 22 and 26 degrees Celsius, or between 22 and 24 degrees
Celsius, or between 24 and 26 degrees Celsius. Advantageously, the use of an
incubation oven that provides elevated temperatures is not required. After
incubation is completed, microarray 24 is washed to remove the nanoparticles
16
and un-hybridized nucleic acid molecules 14.
[0097] Fifth, after incubation and washing, a determination is made as to the
level of hybridization with each of the probes 10. Suitable microarray
scanners
can be used and computer software can be used to analyze the microarray data.
For example, the microarray data can be analyzed to infer gene expression
levels,
or the presence or absence of specific DNA sequences (e.g, SNP) in a sample.
[00981 FIG. 6 illustrates an example embodiment, where the sample nucleic
acid molecules are obtained from two separate sources. In FIG. 6, Sample A may
be cancer cells, and Sample B may be normal cells. RNA is isolated from Sample
A and Sample B and reverse transcription is performed to generate cDNA. The
eDNA from Sample A is labeled with a first detectable label (e.g., Cy3), and
the
eDNA from Sample B is labeled with a second detectable label (e.g., Cy5). The
two eDNA samples are then combined and mixed with nanoparticles, heat
denatured, and then incubated with a microarray. As will be apparent to those
skilled in the art, other forms of DNA or RNA or oligonucleotides samples may
be used. For example, the sample DNA may comprise genomic DNA,
CA 02719597 2010-10-29
-26-
mitochondrial DNA or chloroplast DNA.
[0099] One of the advantages of the present invention in microarray analysis
is that the use of nanoparticles non-covalently associated with sample nucleic
acid
molecules could obviate the need to maintain high temperatures throughout the
hybridization process, resulting in reduced cost and analysis time. In
addition, the
nanoparticle-assisted approach may normalize the reaction conditions across
different probe/target pairs, allowing for high-throughput analysis of
multiple
DNA samples simultaneously. In contrast, in conventional microarray analysis,
different probe/target pairs may require different optimal elevated
hybridization
temperature and a single optimal hybridization is not available across
different
probe/target pairs.
Use of Nanoparlicle-Assisted Hybridization with MMA
[001001 One aspect of the invention provides nanoparticle-assisted
hybridization methods in association with a microfluidic microarray assembly
(MMA) or a microchannel plate assembly. MMA and microchannel plate
assemblies are described in WO 2006/060922 and L. Wang and P. C. I-I..Li, J.
Agric. Food. Chem. 55, 10509 (2007), which are hereby incorporated by
reference
in their entirety. It should be noted that both MMA and microchannel plate
assembly can be considered to be a subset of microarrays, and that
microchannel
plate assembly can be considered to be a subset of MMA.
1001011 The general concept of an embodiment of MMA is shown in FIGS. 7
and 8. In this embodiment a microfluidic microarray assembly (MMA) 30 is
illustrated which is produced by the combination of a test chip ("common
chip")
32 and a first channel plate 34 and/or a second channel plate 36. As described
in
detail below, channel plates 34, 36 may be each separately connected to test
chip
32 in consecutive order, to deliver reagents to test chip 32 (such as probes
or test
samples) in predetermined patterns defined by microfluidic channel patterns.
That
is, in one example, first channel plate 34 is first sealingly connected to
test chip 32
to deliver a plurality of probes thereto. First channel plate 34 is then
removed
from test chip 32 and second channel plate 36 is sealingly connected to test
chip
CA 02719597 2010-10-29
-27-
32 to deliver a plurality of samples thereto. MMA thus enables the efficient
formation of high density multi-probe, multi-sample microarrays by employing
microfluidics.
1001021 As shown best in FIGS. 7 and 8, first channel plate 34 has a plurality
of first microfluidic channels (or "microchannels") 38 arranged in a first
predetermined reagent pattern 38A, such as a radial pattern comprising a
plurality
of linear, radially extending segments. In the example of FIGS. 7 and 8, first
channel plate 34 has 24 separate radially extending microfluidic channels 38.
Similarly, second channel plate 36 has a plurality of second microfluidic
channels
40 arranged in a second predetermined reagent pattern 40A, such as a spiral
pattern. In the example of FIGS. 7 and 8, second channel plate 36 shown in
FIGS.
7 and 8 has 4 separate spiral microfluidic channels 40.
[001031 Reservoirs are located at each end of microfluidic channels 38, 40 in
fluid communication therewith. More particularly, each first microfluidic
channel
38 has an inlet reservoir 42 at one end thereof and an outlet reservoir 44 at
the
other end thereof and each second microfluidic channel 40 has an inlet
reservoir
46 at one end thereof and an outlet reservoir 48 at the other end thereof
(FIGS. 8A
and 8B). In the case of high density microarrays, the inlet and/pr outlet
reservoirs
may be staggered in rows to fit within the available space on MMA 30 (FIG.
10).
[001041 First and second predetermined reagent patterns 38A, 40A, and hence
the geometric configurations of first and second microfluidic channels 38, 40,
preferably differ. For example, first predetermined reagent pattern 38A may be
a
radial pattern and second predetermined reagent pattern 40A may be a spiral
pattern, or vice versa. This results in an intersecting pattern of reagent
deposition
on test chip 32 when each of the channel plates 34, 36 is consecutively sealed
to
test chip 32 and reagents are flowed through microfluidic channels 38, 40.
1001051 For example, in FIGS. 7 and 8E, when first channel plate 34 is sealed
with test chip 32, one or more first reagents can be loaded into inlet
reservoirs 42
and flowed through first microfluidic channels 38 to outlet reservoirs 44.
This
results in the distribution of the first reagent in a radial pattern 38A on
test chip
CA 02719597 2010-10-29
-28-
32. As described below, the first reagent is then immobilized on test chip 32
and
first channel plate 34 is removed. Second channel plate 36 is then sealed to
test
chip 32 (FIGS. 7 and 8F). One or more second reagents are loaded into inlet
reservoirs 46 and flowed through second microfluidic channels 40 to outlet
reservoirs 48. This results in the distribution of the second reagent in a
spiral
pattern 40A on test chip 32. The intersection points between first and second
predetermined patterns 38A, 40A (in this case the radial pattern and the
spiral
pattern) defines a plurality of microarray test positions 50 on test chip 32.
If the
first reagent reacts with the second reagent at select test positions 52, a
positive
test result is obtained (FIG. 8G). For example, as discussed further below, a
positive test result could indicate reaction (e.g. hybridization) between the
first
reagent and the second reagent, formation of a reaction product, modification
of a
biochemical or cellular parameter or the like.
[001061 The number of microarray test positions 50 which are created from the
intersection points of first and second predetermined reagent patterns 38A,
40A on
test chip 32 depends upon the number and configuration of microfluidic
channels
38, 40 on first and second channel plates 34, 36, respectively. For example,
in this
embodiment, each line of the first reagent pattern produced by first
microfluidic
channels 38 intersects only once with each line of the second reagent pattern
produced by second microfluidic channels 40. Thus, if first channel plate 34
has x
microfluidic channels 38 and second channel plate 36 has y microfluidic
channels
40, the resulting microarray has x*y number of intersection points or test
positions
50. In FIGS. 7 and 8, first channel plate 34 has x=24 radial microfluidic
channels
38 and second channel plate 36 has y=4 spiral microfluidic channels 40. The
resulting microarray has 24*4=96 test positions 50 on test chip 32.
Preferably,
there is only one intersection point between each line of the first reagent
pattern
produced by first microfluidic channels 38 and each line of the second reagent
pattern produced by second microfluidic channels 40. However, it is possible
to
design first and second channel plates 34, 36 with first and second
predetermined
reagent patterns 38A, 40A having more than one intersection, point between
each
set of lines. Further, the first and second reagent distribution patterns
formed on
test chip 32 may in some cases comprise a plurality of discrete reagent spots
rather
than a continuous line or lines of reagent.
CA 02719597 2010-10-29
-29-
1001071 FIG. 10 shows diagrams of first and second channel plates 34, 36
having 384 high density microfluidic channels. FIG. IOA shows a first channel
plate 34 having 384 microfluidic channels 38 arranged in a right spiral
pattern.
The inset shows inlet reservoir 42 for loading a first reagent thereinto. FIG.
I OB
shows second channel plate 36 having 384 microfluidic channels 40 arranged in
a
left spiral pattern. The inset shows inlet reservoir 46 for loading a second
reagent
thereinto. FIG. I OC shows the intersection points of the two spiral patterns
of first
and second channel plates 34, 36. The intersection points define a dense
microarray of 147456 (384*384) test positions 50.
[00108] In alternative embodiments, the MMA 30 may be formed from the
assembly of one or more additional channel plates. Such additional channel
plates
may comprise microfluidic channels arranged in a similar pattern to either
first or
second channel plates 34, 36, or the microfluidic channels may be arranged in
other patterns, and may be used to deliver additional reagents, reagent
primers or
other reagent modifiers, detectors or other materials to test positions 50 on
test
chip 32.
[001091 Various means may be used to induce and regulate the flow of
reagent(s) deposited on chip 32 for the purpose of microarray formation and
testing. In use, after first channel plate 34 is sealed with test chip 32, one
or more
first reagents are loaded into inlet reservoirs 42 of first microfluidic
channels 38.
To initiate the flow of and to distribute the first reagents in first
microfluidic
channels 38, a force is applied to MMA 30 (FIG. 8E). Various types of forces
may
be applied to MMA 30 to induce fluid flow, such as centrifugal force applied
by
spinning MMA 30. The first reagents are then immobilized or fixed on test chip
32. Immobilization of the first reagent may be achieved by various techniques
which are known to persons, skilled in the art. For example, immobilization
can
be achieved by chemical, mechanical, or biochemical methods such as covalent
binding, adsorption, cellular adhesion, protein-protein interactions, polymer
encapsulation and so forth. As described further below, one example of
chemical
immobilization is Schiff-base linkage formed between amine and aldehyde groups
on test chip 32. The first reagent may comprise probe nucleic acid molecules.
CA 02719597 2010-10-29
-30-
X00110] After the first reagent is distributed and immobilized on test chip 32
as
described above, first channel plate 34 is then removed. In the next step,
second
channel plate 36 is sealed with test chip 32. One or more second reagents are
loaded into inlet reservoirs 46 of second microfluidic channels 40. A force is
applied to MMA 30 (FIG. 8F) to cause the second reagents to flow and become
distributed through second microfluidic channels 40. If necessary, a priming
reagent or other reagent for modifying or labeling the second reagents may
also be
applied through second microfluidic channels 40. At test positions 50, the
first
reagents are exposed to the second reagents. If the first and second reagents
are
capable of reacting with one another, this results in a positive test reaction
at
select test positions 52.
[00111] In a further step, the positive test reactions between the first and
second reagents are detected using methods which are well known in the art.
For
example, fluorescence labeling, biotin labeling, reflectance measurements, and
so
forth can be used. In addition, novel detection methods such as surface
plasmon
resonance may also be used.
[001121 Once reagents arc loaded into one or more inlet reservoirs 42, 46,
various means may be used to induce fluid flow through microfluidic channels
38,
40, including the application of centrifugal, electrokinetic or hydrodynamic
forces.
The application of centrifugal force, sometimes referred to as "centrifugal
pumping", provides particular advantages. Centrifugal force may be simply
applied by spinning MMA 30 in a disc spinner and avoids the need for
complicated fluid handling interfaces. As shown in FIG. 8, distribution of
reagents
by application of centrifugal force is possible for microfluidic channels 38,
40
arranged in either a radial pattern or a spiral pattern. More particularly,
when first
channel plate 34 having first microfluidic channels 38 arranged in a radial
pattern
38A is sealed against test chip 32, direct centrifugal force (F) is used to
distribute
the first reagent through microfluidic channels 38 by loading MMA 30 in a
spinning device and spinning MMA 30 (FIGS. 8C and 8E). When second channel
plate 36 having second microfluidic channels 40 arranged in a spiral pattern
40A
is sealed against test chip 32 and the resulting MMA 30 is spun in a spinning
CA 02719597 2010-10-29
-31 -
device, a component of centrifugal force is used to distribute the second
reagent
through second microfluidic channels 40 (FIGS. 8D and 8F).
[00113] When centrifugal force is used, reagents are loaded into inlet
reservoirs
42, 46 at locations near the centre of channel plates 34, 36 respectively. To
ensure
that all the liquids in inlet reservoirs 42, 46 are distributed into first and
second
microfluidic channels 38, 40 without spillage, and are retained in outlet
reservoirs
44, 48 while spinning the chip, inlet and outlet reservoirs 42, 46, 44, 48 may
be
disposed at an oblique angle (for example, <90 degrees relative to the central
axis
of the channel plate). In different embodiments, the reservoirs can carry
between
0.1 microlitres and 100 microlitres of reagent depending on the size of
channel
plates 34, 36 and microfluidic channels 38, 40 formed therein. In one
embodiment, the microfluidic channels 38, 40 may be on the order of
approximately 60 .tm wide and approximately 20 m deep, although many
variations are possible. When MMA 30 is spun, the fluid in the inlet
reservoirs 42,
46 is driven into first or second microfluidic channels 38, 40. The fluid then
moves outwardly along first or second microfluidic channels 38, 40 until it
reaches corresponding outlet reservoirs 44, 48 near the periphery of MMA 30,
thereby distributing the reagents along the length of microfluidic channels
38, 40.
[00114] The flow speeds of the reagents in first or second microfluidic
channels 38, 40 can be controlled by adjusting the rotation speed of MMA 30.
For
example, the flow speeds can be between 200 rpm and 10,000 rpm. Thus, the
residence time or the reaction time of reagents can be controlled, i.e. the
time can
be adjusted to be long enough to allow for reactions, but short enough to save
analysis time.
[00115] As discussed above, first and/or second microfluidic channels 38, 40
may be arranged in a spiral shape in one embodiment of the invention. It will
be
appreciated by persons skilled in the art that any type of spiral shape may be
used.
However, to achieve uniform and quantitative hybridization (or other types of
reactions), it is desirable to ensure an approximately constant flow velocity
of
liquid reagents in the spiral microfluidic channels 38, 40. If the sample
volume of
the reagents is many times larger than the channel volume, this constant
velocity
CA 02719597 2010-10-29
-32-
design for spiral microfluidic channels may not be necessary because there is
continuous liquid flow in the microfluidic channels. However, when a small
volume of reagent is used (e.g. 1 L), an approximately constant flow velocity
of
liquid reagents is desirable.
[00116] In an example embodiment, MMA 30 is used for testing nucleic acid
hybridations, such as DNAs, RNAs, cDNAs or other nucleic acids. For example,
the first reagent may comprise DNA probes while the second reagent may
comprise samples for testing. In the first step, first channel plate 34 having
first
microfluidic channels 38 arranged in first predetermined pattern 38A, such as
a
radial pattern, is sealed with test chip 32, such as an aldehyde glass slide.
Next,
solutions of aminated DNA probes are loaded into inlet reservoirs 32 and
distributed through first microfluidic channels 38 using centrifugal force as
described above. The DNA probes become immobilized onto test chip 32 due to
Schiff-base linkage formed between amine and aldehyde groups. The DNA probes
will form an array on test chip 32 in the same pattern as first predetermined
pattern 38A. First channel plate 34 is then removed from test chip 32 and the
procedure for reduction of Schiff-base linkages and excess aldehyde moieties
is
performed. Other methods for immobilizing or fixing the probes to the test
chip 32
can also be used.
[00117] In the second step, second channel plate 36 having second microfluidic
channels 40 arranged in second predetermined pattern 40A, such as a spiral
pattern, is sealed against test chip 32, and samples are introduced into inlet
reservoirs 46 and distributed through second microfluidic channels 40 using
centrifugal force. As the samples flow through second microfluidic channels 40
of
second channel plate 36, the probes are exposed to the samples at test
positions
50. Any samples which are complementary to any of the probes become
hybridized at select test positions 50, thus indicating a positive test
result. In the
final step, detection of hybridization of samples on test chip 32, with or
without
removing second channel plate 36, is then conducted.
[00118] In some embodiments, the sample or second reagent comprises
complexes 18 comprising nanoparticles 16 that are non-covalently associated
with
CA 02719597 2010-10-29
- 33 -
single-stranded nucleic acid molecules 14. Use of nanoparticles (e.g., gold
nanoparticles) in hybridization assays have been described in earlier sections
of
this disclosure. In some embodiments, the use of nanoparticle 16 increases the
degree of discrimination between a perfectly matched target sequence and a
mismatched sequence in hybridization assays. In some embodiments, the
incubation of complexes 18 with probe nucleic acid molecules 10 is performed
at
ambient temperature or room temperature. For example, the hybridization step
may be performed at a temperature between 20 to 30 degrees Celsius, for
example, between 22 and 26 degrees Celsius, or between 22 and 24 degrees
Celsius, or between 24 and 26 degrees Celsius. Advantageously, the use of an
incubation oven that provides elevated temperatures is not required.
[00119] To detect hybridized samples on test chip 32, samples could be
labeled, and only hybridized samples will remain bound to test chip 32 and be
detected. For instance, the sample can be fluorescently labeled in which only
the
hybridized regions are fluorescent, or the sample can be biotin-labeled in
which
strept(avidin)-tagged microbeads, after binding, can be detected by
reflectance
measurement. Alternatively, a detection probe which interacts with hybridized
samples only, but not to probes, could be used to detect hybridization. Other
methods of detecting hybridized samples are known to persons skilled in the
art.
[00120] FIG. 11 illustrates another example of MMA comprising parallel
straight microchannels. The non-centrifugal MMA method consists of two steps
of an assembly process (see FIG. 11). In the first step, channel plate I is
assembled with the glass chip via reversible bonding. Aminated DNA probes are
introduced into the microchannels and are immobilized on the glass chip. A
line
microarray of probes is thus created. After plate 1 is peeled off, channel
plate 2 is
then assembled with the same glass chip. The linear microchannels in plate 2
are
orthogonal to the linear microchannels in plate 1. The sample solution that
flows
through the microchannels in plate 2 will intersect the line microarray, and
hybridization assays are then performed at the intersections. Typically, a
very
small sample volume (<1 L) is need for the hybridization assay using this
MMA.
The MMA also prevents evaporation and cross- contamination of sample
solutions. In some embodiments, the MMA may be PDMS channel plates.
CA 02719597 2010-10-29
-34-
Examples
[00121] The following examples are provided to illustrate embodiments of the
present invention but they are by no means intended to limit its scope.
[00122] Surface Modification of Glass Chips. The glass substrates were
chemically modified to produce aldehyde-functionalized surfaces using an
established procedure (see H. Wang et al., Nucleic Acid Research, 2002, 30, 1-
9,
which is hereby incorporated by reference). Briefly, plain glass slides were
cleaned with a 10% NaOH solution for 10 min at - 100 C. After being rinsed
with
distilled water, the slides were treated with a piranha solution (70:30 v/v
sulfuric
acid to 30% hydrogen peroxide) for 1 h at -80 C. The slides were then rinsed
with water and dried under a stream of nitrogen. The cleaned slides were
treated
with a mixture of ethanol/H20/APTES (95:3:2 by volume) for 2 h under stirring,
rinsed with 95% ethanol and deionized H2O, dried under nitrogen, and baked at
120 C for 1 h. The aminated glass slides were then immersed in 5%
glutaraldehyde in a lOx phosphate-buffered saline (PBS) solution overnight and
washed with acetone and deionized H2O. After being dried in a nitrogen gas
stream, the aldehyde-modified glass slides were stored in a dark place at 4 C
before probe printing.
[00123] Fabrication of the PDMS-glass microchip. PDMS channel plates
consisting of 16 parallel microchannels were fabricated, as follows. A 2 in. x
2 in.
PDMS channel plate was fabricated using a photolithographic method. The
channel pattern was designed using Visual Basic (Microsoft) and was printed on
a
transparency to create the photomask at a resolution of 3368 dpi. Molding
masters
were fabricated in a modular clean room (577 series, Clean Air Products,
Minneapolis, MN). First, a 4 in. silicon wafer was spin-coated with a layer of
SU-
8 photoresist by a spin coater (WS-400, Laurell Technologies Corp., North
Wales,
PA). Then the channel patterns were created on the SU-8 coated wafer with the
photomask using a UV exposure system (model LS-150-3, Bachur & Associates,
San Jose, CA). The SU-8-coated wafer was developed to produce the molding
CA 02719597 2010-10-29
-35-
master. PDMS prepolymer was cast against the molding master and cured at 50 C
for 12 h to yield an elastomeric channel plate. The width of the straight
channels
was 300 m, and the channel height was 20 m. The length of the straight
section
of each channel was 30 mm. Solution reservoirs (1 mm in diameter) at both ends
of channels were punched on the PDMS channel plate using a flat-end syringe
needle.
[001241 Probe line array creation. The glass slides were chemically modified
to produce amine or aldehyde-functionalized surfaces. As shown in FIG. 11, the
PDMS channel plate was sealed against a glass slide to assemble the microchip.
Then, 0.8 L of probe DNA prepared in the spotting solution (1.0 M NaCl + 0.15
M NaHCO3) was added into the inlet reservoirs using a micropipet. The probe
solution was filled through the channels by applying vacuum pumping at the
outlets. With incubation at room temperature for 30 min, covalent Schiff
linkage
was formed between the amine ends of the probe oligonucelotides and the
aldehyde groups on the glass surface. After the microchannels had been washed
with I L of washing solution (0.15% Triton-X 100, 1.0 M NaCl, and 0.15 M
NaHCO3), the PDMS channel plate was then peeled off and the glass slide was
chemically reduced with a NaBH4 solution (100 mg of NaBH4 dissolved in 30
mL of 1 x PBS and 10 mL of 95% EtOH) for 15 min to reduce the Schiff linkage
to the more stable C-N single bond. The glass chip was then rinsed with
deionized
water for 2 min and dried by nitrogen gas and was ready for hybridization. All
procedures were conducted at room temperature.
[001251 DNA samples. Oligonucleotides were synthesized and modified by
Sigma-Genosys Oakville, ON, Canada or International DNA Technologies,
Coralville, IA. 21-mer DNA probes were modified with an amine group at the 5'-
end. Target oligonucleotides are 50-mer with Cy5 dye at the 5' -ends. The
central
21 bases are complementary (perfect match) or one base-pair mismatch to the
sequences of the probe molecules.
[001261 The 21-mer probe sequence is CGCCAGAGAATACCAAAACTC.
The sequence of the perfectly matched 50-mer oligonucleotide is CGACATTAA
TAAAAAGAGTTTTGGTATTCTCTGGCGAGCATACAAGGCCC. The
CA 02719597 2010-10-29
-36-
sequence of the single-base-pair mismatched 50-mer oligonucleotide is
CGACATTAATAAAAAGAGTTTTGGTTTTCTCTGGCGAGCATACAAGGC
Cc.
[00127] Two 264 bp PCR products or amplicons were amplified from genomic
DNA samples, and were labeled with Cy5 dyes. The central sequences of the
sense strand of perfect matched PCR amplicons are complementary to the
sequences of probe molecules, while the mismatched amplicons have one base-
pair difference from that of the perfect matched ones.
[00128] The sequence of the perfectly matched PCR product is:
1 TTACAGAGTT CATGCCCGAA AGGGTAGACC TCCCACCCTT GTGTATTATT ACTTTGTTGC
61 TTTGGCGAGC TGCCTTCGGG CCTTGTATGC TCGCCAGAGA ATACCP.P.AAC TCTTTTTATT
121 PATGTCGTCT GAGTACTATA TAATAGTTAA AACTTTCAAC AACGGATCTC TTGGTTCTGG
181 CATCGATGAA GPACGCAGCG kA_T1TGCGATA AGTAATGTGA ATTGCAGAAT TCAGTGAATC
241 ATCGAATCTT TGAACGCACA
[00129] The sequence of the single-base-pair mismatched PCR product is:
1 TTACAGAGTT CATGCCCGAA AGGGTAGACC TCCCACCCTT GTGTATTATT ACTTTGTTGC
61 TTTGGCGAGC TGCCTTCGGG CCTTGTATGC TCGCCAGAGA AAACCAAAAC TCTTTTTATT
121 AATGTCGTCT GAGTACTATA TAATAGTTAA AACTTTCAAC P.ACGGATCTC TTGGTTCTGG
181 CATCGATGAA GAACGCAGCG AAATGCGATA AGTAATGTGA ATTGCAGAAT TCAGTGAATC
241 ATCGAATCTT TGAACGCACA
[00130] Preparation of DNA-GNP (Gold Nanoparticle) conjugates. Sample
DNA molecules were bound to GNPs to form DNA-GNP conjugates before
hybridizations. GNP solutions (5 nm in average diameter, Sigma life science)
were added into the DNA samples (oligonucelotides or PCR products) in water.
In the case of PCR products, the mixtures were incubated at 95 C to denature
and
uncoil the DNA chains and so ss-DNA molecules were produced for binding to
GNP noncovalently. The DNA-GNP conjugates were snap cooled in an ice-water
bath before the hybridization experiments. All the conjugate solutions were
diluted to the desired concentration before hybridization.
CA 02719597 2010-10-29
-37-
[00131] Sample hybridization and result read-out by fluorescent scanning.
For DNA target hybridization, the glass chip with probe line arrays was sealed
against the second PDMS channel plate. The straight channels were orthogonal
to
the printed probe lines on the slide. The DNA samples (oligonucelotides or PCR
products) were prepared in the hybridization buffer (IX SSC+0.2% SDS). 1.0 I
DNA targets were added to the inlet reservoirs. Sample solutions in different
reservoirs were then pumped into the channels by vacuum suction simultaneously
applied at the 16 channel outlets. Hybridizations were achieved at the
intersections
between complementary DNA targets in solution and probe lines, showing the
hybridization patches of 300 x 300 m2. The microchannels were rinsed
immediately with 2 l hybridization buffer following hybridization.
[00132] Following the hybridization and washing procedures, the glass slide
was scanned on a confocal laser fluorescent scanner (Typhoon 9410, Molecular
Dynamics, Amersham Biosystems) at 25 m resolution. The excitation and
emission wavelengths are 633 and 670 nm, respectively. The photomultiplier
tube
voltage was set to 600 V. The scanned image was analyzed by IMAGEQUANT
5.2 software. The average fluorescent signals were measured in relative
fluorescent unit.
[00133] Single base-pair discrimination. The principle of nanoparticle-
assisted DNA discrimination was illustrated in FIG. 13. This is based on non-
covalent binding between GNPs and targets, in competition with that between
targets and immobilized probes. This non-covalent binding is thought to act
between GNPs and nitrogen bases on the DNA chains, and can be resulted from
both hydrophobic interaction and electrostatic interaction. Here, target DNA
labeled with fluorescent molecules were first incubated with GNPs solutions at
high temperature in solution. The solution of GNP-DNA conjugates were then
applied to DNA probes preprinted microfluidically on the glass slide through
the
microfluidic method.
[00134] Because the base-pair interaction between matched DNA chains is
strong, sample DNA molecules could desorb from GNPs and were hybridized
CA 02719597 2010-10-29
-38-
with the immobilized probes. The read-out of the hybridization signals were
achieved through the fluorescent labels on the sample DNA molecules. On the
contrary, mismatched DNA shows much less binding energy with the probes and
thus still bind with GNPs. The conjugates would be washed away in the
microfluidic flow and discrimination was made.
[00135] The nanoparticle-assisted discrimination method was shown with two
50-mer oligonucleotides with one-base difference in center. The samples
hybridized with the same probe molecules to produce two types of duplexes,
namely, the PM duplex and the MM duplex. As shown in the images in FIG.
14(a), without pre-incubation with GNPs, the hybridization signals from
mismatched targets are very close to those obtained from complementary
targets,
indicating a high degree of non-specific binding between the MM sequence and
the probe. The discrimination ratio, which is the hybridization signal ratio
of PM
duplexes over MM duplexes, is around 1.4. With the use of GNP conjugates
instead of free DNA molecules, the discrimination ratio was raised up to 6.8.
A
clear discrimination between two oligonucleotides was observed from the images
in FIG. 14(a).
[00136] The effect of the molar ratio between GNPs and target oligonucleotides
on hybridization signals and discrimination ratios was also investigated. The
molar concentration of GNPs can be calculated from the total gold
concentration
as well as the size of the GNPs. For 5 run diameter GNPs in our study, the
particle
molar concentration is around 86 nM. Conjugates of different GNP/DNA ratios
were thus prepared in this manner. FIG. 14(c) compares different hybridization
intensities from conjugates of different oligo/GNP ratios (2:1 and 1:1,
respectively). It was found that the more GNPs were incubated with DNA
samples, the weaker were the hybridization signals. This observation could be
explained by the relatively strong binding between GNPs and DNA as well as the
slow kinetics of desorption. Despite the reduction in the fluorescent
intensities,
GNPs do enhance the discrimination of single base-pair mismatch, and the
hybridization signals are still adequate as shown in both the images and the
inset
graph in FIG. 14(c).
CA 02719597 2010-10-29
-39-
[00137] The nanoparticle-assisted microfluidic method was applied to the
room-temperature discrimination of two related Botrytis subspecies, B. cinerea
and B. squamosa. The two PCR amplicons differ in only one base pair in the
middle of the 264 bp long sequence. The amplicons were first incubated at 95
C
with GNPs. This incubation serves for two purposes: one is to denature double-
strand amplicons as the usual procedures and another is to promote the
subsequent
binding of ss-DNA to GNPs. The later snap chilling procedures (at 4 C)
prevented the renaturation of ss-DNA molecules. Although both of the two
complementary strands were bound to GNPs and coexisted in the same solutions,
they can be still used as samples for later microarray hybridization because
the
renaturation between long ss-DNA of high complexity is much slower than that
between long ss-DNA with short oligonucleotide probes. The discrimination
ratio
without the use of GNPs is 3.6 at room temperature while it goes up to 27.7
with
the assistance of nanoparticles at room temperature. (FIG. 15). Without the
use
of GNP and using temperature stringency at 50 C, the discrimination ratio is
6.7.
This indicates that the nanoparticle-assisted method has not only improved
discrimination, but also alleviated the need of high temperature and related
heating devices in the microfluidic chip applications.
[00138] As will be apparent to those skilled in the art in the light of the
foregoing disclosure, many alterations and modifications are possible in the
practice of this invention without departing from the spirit or scope thereof.