Note: Descriptions are shown in the official language in which they were submitted.
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
TITLE: Vector Encoding Therapeutic Polypeptide and Safety Elements to
Clear Transduced Cells
Government Interest
These studies were supported in part by a grant from the National
Institutes of Health (HL70569). The United States government may have rights
in this disclosure.
FIELD OF THE APPLICATION
The disclosure relates to compositions comprising a vector encoding
safety elements to clear transduced cells.
BACKGROUND OF THE APPLICATION
Gene therapy has been used successfully to treat a number of inherited
disorders. 1,2 Although many viral and non-viral gene delivery alternatives
exist,
retroviral vectors offer the advantages of stable integration into host
genomes,
the ability to infect a wide variety of cell types, and relatively high levels
of
transgene expression.3 Concerns regarding the safety of integrating vectors
have been prompted, however, by the development of leukemia in three X-
linked severe combined immunodeficiency patients in a recent clinical trial
using an oncoretroviral vector.4 A variety of explanations for this outcome
have
been proposed, but the exact mechanism of leukemogenesis has remained
unresolved, as no other clinical trials have reported this type of adverse
event.5,6
Despite this outcome, retroviral gene therapy continues because
of the conceptual effectiveness of the treatment and the fact that gene
therapy is
the only potential cure available for many disorders such as X-linked severe
combined immunodeficiency.
Integrating viral vectors are still a good choice for gene therapy because
they offer fairly efficient transduction and consistent long-term gene
expression.
Much research has been directed towards improving vector design to increase
1
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
safety and reliability. Therefore, the development of improved vectors and
viable
alternative safety strategies is exceedingly important and timely.
One example of a disease targeted for gene therapy is Fabry disease, a
lysosomal storage disorder resulting from a deficiency of a-galactosidase A (a-
gal A) activity. Fabry disease is a good candidate for gene therapy because
there is reduced neurological involvement in contrast to many other lysosomal
storage disorders, and supra-physiological levels of a-galA are well-
tolerated.8
Gene therapy for Fabry disease by introducing aa-galactosidase A (a-gal
A) activity, has the potential to provide a cure for the disorder with a
single
treatment. Despite modifications to existing vectors, concerns have arisen
regarding the risk of genotoxicity associated with the use of retroviruses.
There
remains a need for suitable gene therapy vectors for Fabry disease and other
enzyme deficiency diseases.
SUMMARY OF THE INVENTION
Incorporating an effective suicide gene into a therapeutic vector ensures
that any malignant clones arising from deleterious insertion of the vector are
specifically killed. Likewise, such a control schema is useful as an inserted
safety
component for a variety of transplants, including stem cell transplants
reducing
teratomas, for example, should these outgrowth events develop. A suicide gene
schema us also useful to control post-transplant complications such as Graft v
Host disease. The invention provides vectors with improved safety elements to
effectively clear transduced cells to further decrease risk to the patient.
The disclosure provides a novel strategy for improving the safety of
therapeutic integration vectors. This novel strategy has great utility, as a
variety
of cell surface proteins are readily incorporated into various retroviral
vectors in
combination with any therapeutic transgene. Using this system adds another
safety mechanism to current and future retroviral gene transfer systems and
transplant schemas of a variety of manifestations.
2
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
The disclosure provides a composition comprising:
a stably integrating delivery vector;
an activator polynucleotide encoding a polypeptide that converts a
prodrug to a drug; and/or
a docking polynucleotide encoding a docking polypeptide that
selectively binds a toxic binding agent.
In one embodiment, the activator polynucleotide is deoxycytidine kinase.
In one embodiment the activator polynucleotide is thymidylate kinase. In
another
embodiment the activator polynucleotide is a modified thymidylate kinase. In
another embodiment the activator polynucleotide is thymidine kinase (tk). In a
further embodiment the tk is herpes simplex virus-tk (HSV-tk). In another
embodiment, the tk is Equine Herpes Virus Type 4 (EHV4-tk). Mutations,
variants, and derivatives thereof that maintain kinase activity are also
included.
The application further provides a suicide gene therapy safety system
comprising a stably integrating delivery vector comprising an activator
polynucleotide encoding a polypeptide that converts a prodrug to a drug and/or
a
docking polynucleotide encoding a docking polypeptide that selectively binds a
toxic binding agent. In one embodiment the docking polypeptide is a cell
surface
protein. The system further comprises, a prodrug that is converted to a drug
by
the activator polynucleotide and a toxic binding polypeptide that binds the
docking polypeptide. In one embodiment, the docking polynucleotide is a
polynucleotide that encodes a cell surface polypeptide or cell surface marker.
In
one embodiment, the docking polynucleotide is CD25. In another embodiment,
the docking polynucleotide is truncated CD19. In other embodiments, the
docking polynucleotide is selected from the group consisting of CD19,
truncated
CD19, EGFP, CD25, LNGFR, truncated LNGFR, CD24, truncated CD34, EpoR,
HSA and CD20.
In one embodiment the toxic binding agent is an antibody. In another
embodiment, the toxic binding agent is an antibody conjugated to a toxin. In
3
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
certain embodiments the toxin comprises saporin, other cytotoxic polypeptides,
cytotoxic chemicals, radionuclides, etc. In a further embodiment, the antibody
is
an anti-CD25 antibody. In another embodiment, the antibody is an anti-CD19
antibody. In other embodiments, the antibody binds CD19, truncated CD19,
EGFP, CD25, LNGFR, truncated LNGFR, CD24, truncated CD34, EpoR, HSA or
CD20.
In one embodiment, the delivery vector is an integrating vector. In another
embodiment, the delivery vector is a retroviral vector such as an
oncoretroviral or
lentiviral vector. In other embodiments, the delivery vector is a foamy virus.
In
yet other embodiments, the delivery vector is a transposon such as Sleeping
Beauty (Discovery Genomics, Inc.; US Patent No. 6,489,458).
The disclosure also provides a method of expressing an activator
polynucleotide and a docking polynucleotide in a mammalian cell comprising
contacting the mammalian cell with a composition of the disclosure. In other
embodiments, the disclosure provides a method of additionally expressing a
therapeutic polypeptide.
In one embodiment the mammalian cell is selected from the group
comprising a stem cell, a hematopoietic cell, a T cell and a human cell.
Any stem cell or ES cell or iPS cell that is transplanted benefits from
having this safety system to decrease the risk of aberrant cell growth when
cells
are placed out of their normal context. A therapeutic gene is optionally
provided.
The safety system described herein is also useful in BMT and DLI. One can use
direct tumor injection to administer the safety system herein described;
addition
of the prodrug will then kill transduced and neighboring cells.
The system is also useful to remove any transplanted cell in any
transplantation setting that has lost effectiveness or actually becomes
deleterious
to the host by any mechanism.
In another embodiment, the application discloses compositions and
systems further comprising a therapeutic polynucleotide. In one embodiment the
therapeutic polynucleotide is a-galactosidase A(aGal A).
Another aspect of the disclosure provides a kit comprising the composition
4
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
or system previously described, optionally additionally comprising
instructions for
use according to a method described herein.
Other features and advantages of the present disclosure will become
apparent from the following detailed description. It should be understood,
however, that the detailed description and the specific examples while
indicating
preferred embodiments of the disclosure are given by way of illustration only,
since various changes and modifications within the scope of the disclosure
will
become apparent to those skilled in the art from this detailed description.
BRIEF DESCRIPTION OF THE DRAWINGS
Preferred embodiments of the disclosure will be described in relation to
the drawings in which:
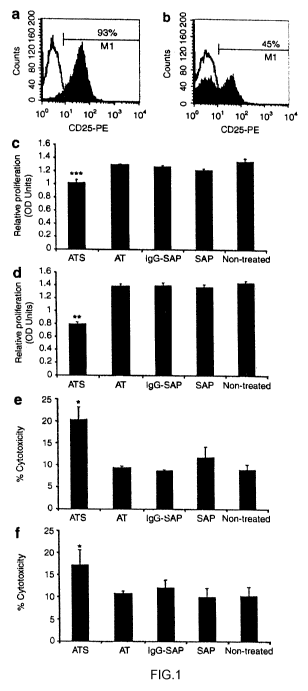
Figure 1 In vitro clearance of C1498 cells expressing a broad
concentration range of human CD25 (huCD25) molecules by anti-Tac saporin
(ATS). C1498 cells were infected with LV/a-gal A/huCD25 and then sorted by
magnetic activated cell sorting to isolate a pool of cells that express
huCD25.
Shown are two cell populations that are (a) 90% and (b) 45% positive for
huCD25 expression as measured by flow cytometry analysis. Cells were
treated with 5 nM of each reagent. (c, d) Cell proliferation was assessed by
MIT
[3-(4,5-dimethyl-2-thiazolyl)-2,5- diphenyl-2H-tetrazolium bromide] assays 72
hours later. (e, f) Cytoxicity was assessed by measurement of lactate
dehydrogenase (LDH) release 48 hours later. AT, anti-Tac; SAP, saporin; IgG-
SAP, IgG-saporin (isotypec control immunotoxin). Error bars represent SD. *P
< 0.05, **P < 0.01, ***P < 0.001 forATS compared with all other groups.
Figure 2 In vitro clearance of a C1498/CD25 clone by anti-Tac-
saporin (ATS). (a) Representative flow cytometry analysis of a derived single
cell clone of C1498/huCD25 cells. Cells were transduced with LV/a-gal A/CD25
and single-cell clones were isolated by flow cytometry on the basis of human
CD25 (huCD25) expression. (b) Proliferation of C1498/ huCD25 and non-
transduced (NT) cells after incubation with ATS or control reagents for 72
hours, as measured by MIT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
tetrazolium bromide] assay. (c) Cell death, measured by a lactate
dehydrogenase (LDH) release assay. Error bars represent SD. ***P < 0.001 for
ATS compared with all other groups.
Figure 3 The in vivo effect of different antibody doses on plasma
human CD25 (huCD25) levels. Fabry mice were transplanted with 1 X 106
C1498/huCD25 cells and treated with 5 pg anti-Tac-saporin (ATS), 20 pg ATS,
or 20 pg saporin (SAP) 2 days after cell transplantation. Plasma was collected
from the peripheral blood 18 days after cell transplantation and analyzed for
levels of soluble huCD25. n = 3 per group.
Figure 4 Anti-Tac-saporin (ATS) and anti-Tac (AT) treatment in a
human CD25 (huCD25)-expressing myeloid leukemia model. Fabry mice
were transplanted with C1498/huCD25 cells and treated with immunotoxins on
days 2, 4, and 6. On day 18, plasma was analyzed for (a) soluble huCD25
levels by enzyme-linked immunosorbent assay and (b) a-galactosidase A (a-
gal A) activity. Error bars represent SEM. n = 6 in all groups, except for the
untreated group (n = 8) and the wildtype (WT) group (n = 4). (c) Kaplan-Meier
survival curve for treated and control mice.
Figure 5 Bone marrow transplantation model. Bone marrow
mononuclear cells (BMMNCs) were harvested from Fabry mice and transduced
using supernatant from E86/pMFG/a-gal AIRES/huCD25 clone 21 (n = 24) or
E86/pUMFG/enYFP (n = 6). Forty-eight hours after transduction, BMMNCs were
analyzed for expression of (a) human CD25 (huCD25) or (b) enhanced yellow
fluorescent protein (enYFP). Transduced cells were transplanted into lethally
irradiated recipient Fabry mice. (c) Eight weeks after transplant, plasma of
recipient mice was analyzed for a-galactosidase A (a-gal A) activity. Error
bars
represent SEM.
Figure 6 Clearance of retrovirally transduced bone marrow-derived
cells by anti-Tac-saporin (ATS) and anti-Tac (AT). Nine weeks after bone
marrow transplantation with cells transduced with either E86/ pMFG/a-gal
AIRES/huCD25 clone2l or E86/pUMFG/enYFP, mice were treated with either
ATS, AT, or immunogloblin (Ig)G Ab conjugated to SAP (IgG-SAP). Peripheral
6
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
blood was collected 1 week later and analyzed for (a) levels of soluble human
CD25 (huCD25) in the plasma and (b) expression of huCD25 on mononuclear
cells. Values are expressed as percentage reduction compared with pre-
treatment values (measured at week 8). (c) Expression of enhanced yellow
fluorescent protein (enYFP) on peripheral blood mononuclear cells (PBMNCs)
over the course of the experiment. Error bars represent SEM. n = 5 in all
groups
except ATS (n = 4) and green fluorescent protein (n = 6).
Figure 7 Systemic effect of anti-Tac-saporin (ATS) treatment on a-
galactosidase A (a-gal A) activity. Twelve weeks after bone marrow
transplantation and three weeks after the first treatment with immunotoxin,
mice were killed and a-gal activity was measured in various tissues: (a)
peripheral blood mononuclear cells, (b) liver, (c) spleen. Error bars
represent
SEM. n = 5 in all groups except ATS (n = 4) and green fluorescent protein (n =
6).
DETAILED DESCRIPTION OF THE INVENTION
The disclosure relates to the use of a cell surface antigen such as
huCD25 in a gene expression cassette as a safety mechanism for retroviral
vectors.
The inventors have demonstrated that a targeted antibody and/or
targeted immunotoxin reduces tumor burden and selectively clear transduced
hematopoietic cells that express a target antigen, thus acting as a built-in
safety mechanism for gene therapy vectors. The inventors show that anti-CD25
antibody and/or an anti-CD25 conjugated immunotoxin, specifically targets and
eliminate transduced leukemia cells expressing CD25.
In one embodiment, the disclosure provides a combination of a novel
prodrug/enzyme and a docking polypeptide/toxic binding agent for suicide gene
therapy, for example for use in transplant schemas. The disclosure also
provides
the combination of a therapeutic gene, a novel prodrug/enzyme and a docking
polypeptide/toxic binding agent for gene therapy. In one embodiment,
catalytically improved variants of human tmpk and a CD25 docking
7
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
polynucleotide are delivered into target cells by novel lentiviruses (LVs)
providing
the ability to selectively clear these cells in vitro and in vivo by
administering the
prodrug AZT and/or a CD25 toxic binding agent. Catalytically improved variants
of human tmpk, methods of delivering said modified variants are disclosed in
US
11/559,757, US 12/052,565 filed March 20, 2008 and US provisional application
61/038,398 filed March20, 2008 each of which are herein incorporated by
reference in its entirety and in Sato et al. Engineered Human tmpk/AZT As a
Novel Enzyme/Prodrug Axis for Suicide Gene Therapy. Mol Ther. 2007
doi:10.1038/mt.sj.6300122. Other genes, such as dck, HSV-tk, EHV4-tk and
derivatives, are useful with other prodrugs. In addition, a cell surface
protein
(marker), such as truncated CD19, CD19, CD20, HSA, truncated LNGFR, CD34,
CD24 or CD25 - is delivered into target cells which allows for detecting
and/or
isolating transduced cells and can further provide the ability to selectively
clear
these cells in vitro and in vivo by administering a toxic binding agent such
as an
antibody alone or comprised in an immunotoxin (antibody conjugated to a toxin)
directed against the docking polypeptide such as a cell surface protein.
In an alternate embodiment the activator polynucleotide and docking
polynucleotide are fused so as to produce a fusion polypeptide upon
expression.
In one embodiment, truncated CD19 is fused to a modified mammalian tmpk. As
the docking polynucleotide is fused to the activator polynucleotide such as
mammalian modified tmpk, permissive cells transfected or transduced with such
a construct will express tmpk and the docking polynucleotide. This is useful
for a
number of applications including ensuring that all cells isolated using the
docking
polynucleotide express both the tmpk safety component and the docking
polypeptide safety component. A docking polynucleotide fused to tmpk is
alternatively referred to as tmpk/ docking polynucleotide fusions.
These suicide genes are efficiently transferred into mammalian T cells and
cell lines. In other embodiments, these suicide genes are efficiently
transferred
into ES cells IPS cells, mesenchymal stem cells, bone marrow stroma cells,
endothelial progenitor cells, hematopoietic stem cells, any other stem cell
for
transplantation, etc.
8
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
The disclosure provides the first gene therapy methods and vectors using
both suicide genes and docking polypeptides such as encoded by cell surface
genes recognized by immunotoxins to provide more effective clearance of
transduced cells. In addition, this system is useful to endow stem cells (both
embryonic and of later ontogeny) for in clinical transplantation, for example,
with
a reliable safety system.
Safety Systems and Vector Constructs
The disclosure provides safety systems comprising combinations of a
novel prodrug/enzyme and a docking polypeptide/toxic binding agent for suicide
gene therapy. Such safety systems are useful, for example in clearing cells in
transplant schemas, for example in the event of a transplant adverse event.
The
disclosure also provides the combination of a therapeutic gene, a novel
prodrug/enzyme and a docking polypeptide/toxic binding agent for gene therapy.
Certain embodiments of the disclosure optionally comprise a vector construct
including i) an activator gene and/or ii) DNA encoding a cell surface protein
recognized by a toxic binding agent.
i) The Activator Gene - Conversion of Prodrug to Drug to Kill
Transduced Cells
The term "activator gene" or "activator polynucleotide" also referred to as
a 'cell fate control gene' as used herein refers to a safety element
comprising a
polynucleotide encoding a polypeptide that catalytically converts or aids in
the
conversion of a prodrug to a drug, such that administration of the prodrug is
cytotoxic to cells expressing the activator gene. The term "suicide gene" is
used
interchangeably with "activator gene" herein.
The activator genes of the disclosure such as modified mammalian tmpk
work by increasing phosphorylation of prodrugs such as AZT. For example, the
prodrug AZT is converted through a series of phosphorylation steps into AZT-
triphosphate (AZT-TP)12. This is the active metabolite that inhibits
replication of
the human immunodeficiency virus (HIV)13-15, and to a lesser extent, DNA
replication in eukaryotic cells16. Safety profiles for this compound are well
known
9
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
and concentrations of AZT in the bloodstream of AIDS patients being treated
with
this agent can reach high levels. The rate-limiting step in the conversion of
AZT
to the toxic AZT-TP form is the intermediate step of phosphorylation of AZT-
monophosphate (AZT-MP) to AZT-diphosphate (AZT-DP) catalyzed by the
cellular thymidylate kinase (tmpk), which has a low enzymatic efficiency for
AZT-
MP17. Accumulation of AZT-metabolites in the cells of AZT-treated AIDS
patients
reportedly induces toxic mitochondrial myopathy18-22. To harness this dual
toxicity
of AZT-TP, the disclosure uses any suitable suicide gene encoding a
polypeptide
that converts prodrug to drug.
Tmpk
An example of a useful safety element comprises a nucleic acid encoding
mammalian or human tmpk. In order to improve the processing of AZT-MP to
AZT-DP, thereby increasing intracellular AZT-TP concentrations, minimally
modified tmpk mutants with approximately 200-fold enhanced activity for AZT-MP
have been engineered (Brundiers R, Lavie A, Veit T, Reinstein J, Schlichting
I,
Ostermann N, et al. Modifying human thymidylate kinase to potentiate
azidothymidine activation. J Biol Chem. 1999; 274: 35289-35292; Ostermann N,
Lavie A, Padiyar S, Brundiers R, Veit T, Reinstein J, et al. Potentiating AZT
activation: structures of wild-type and mutant human thymidylate kinase
suggest
reasons for the mutants' improved kinetics with the HIV prodrug metabolite
AZTM P. J Mol Biol. 2000; 304: 43-53).
Thymidylate kinase is a kinase that catalyzes the addition of a phosphoryl
group to thymidylate as well as thymidine analogs such as AZT. Several wild-
type human sequences have been reported. SEQ ID NOS: 1, 3, 5 and 7 are
reported nucleotide sequences of human thymidylate kinase (SEQ ID NO: 7 does
not have a stop codon). The different sequences represent natural polymorphic
variations present in the population and it will be recognized in the art that
future
identified molecules with polymorphic variations will also be considered to be
wildtype tmpk. SEQ ID NO: 9 is the reported mouse thymidylate kinase
sequence. The mouse sequence shares 82% nucleotide identity, 81% amino
acid identity and several residues that have been identified as limiting the
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
nucleoside analog activity of the human tmpk enzyme and which result in
increased enzymatic activity when modified, are conserved in the murine
sequence. The corresponding amino acid sequences are reported in SEQ ID
NOS: 2, 4, 6, 8, and 10. SEQ ID NO: 2 provides the amino acid sequence for the
wild-type tmpk polynucleotide described in SEQ ID NO: 1; SEQ ID NO: 4
provides the amino acid sequence for the wild-type tmpk polynucleotide
reported
in SEQ ID NO: 3, SEQ ID NO: 6 provides the amino acid sequence for the wild-
type tmpk polynucleotide described in SEQ ID NO: 5; SEQ ID NO: 8 provides the
putative sequence of the wild-type tmpk polynucleotide reported in SEQ ID NO:
7; and SEQ ID NO: 10 provides the amino acid sequence of the wild-type murine
tmpk polynucleotide described in SEQ ID NO: 9. Modified tmpk molecules and
mutant tmpk refer to mammalian tmpk molecules that have been modified
compared to wild-type. Among the mutant tmpks, some of these showed a
superior enzymatic activity to convert deoxy-thymidine-monophosphate (dTMP)
to dTMP-diphosphate (dTDP) or AZT-MP to AZT-DP. Increased kinase activity
relative to wild-type refers to modified tmpk molecules that exhibit improved
enzymatic kinetics compared to tmpk wild-type. The improved activity comprises
increases in binding and or enzymatic turnover to convert the monophosphate-
form of the substrate of tmpk to the diphosphate form.
Mutations which show superior enzymatic activity included the F105Y
mutant (SEQ ID NO: 11, SEQ ID NO: 21), R16GLL mutant (SEQ ID NO: 12, SEQ
ID NO: 22) and the R200A mutant (SEQ ID NOS: 15 and 16).
One aspect of the invention provides delivery vectors comprising modified
tmpk enzymes with increased nucleoside analog kinase activity relative to wild-
type. In one aspect, the modification that increases tmpk nucleoside analog
kinase activity comprises one or more deletions. The deletions are optionally
internal or optionally result in a truncated variant. In an alternate
embodiment the
modification that increases tmpk nucleoside analog kinase activity comprises
one
or more point mutations. In another embodiment an exogenous sequence
replaces an endogenous sequence. For example, in one embodiment all or part
of the large lid domain of human tmpk (SEQ ID NO:20) is replaced with all or
part
11
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
of the large lid domain of a different species. In one embodiment the
different
species is a bacteria species. In one embodiment, all or part of the large lid
domain of human tmpk (SEQ ID NO:20) is replaced with all or part of the large
lid
domain of E. coli tmpk (SEQ ID NO:17). In another embodiment, residues 145-
148 of SEQ ID NO: 1 (AFGH) are replaced with all or part of the small lid
region
of E. coil residues 151-156 in SEQ ID NO: 17 (RARGEL). In another embodiment
the modified tmpk is selected from the group including the F105Y mutant (SEQ
ID NO: 11, SEQ ID NO: 21), R16GLL mutant (SEQ ID NO: 12, SEQ ID NO: 22),
a tmpk molecule modified by the substitution of all or part of a bacterial
large lid
domain such as the E. coli large lid domain in SEQ ID NO: 17, a tmpk molecule
modified by the substitution of all or part of a bacterial small lid domain
such as
the E. coli small lid domain at 151-156 of SEQ ID NO: 17, and the R200A mutant
(SEQ ID NOS: 15 and 16).
In another embodiment, the exogenous sequence is optionally
synthesized or obtained from a non-mammalian thymidylate kinase such as a
bacterial thymidylate kinase. As used herein a modified mammalian tmpk
molecule includes a modified tmpk molecule that comprises non-mammalian
sequences such as all or part of either a large lid domain or a small lid
domain
sequence from bacteria such as E. coll. A variant may comprise one or more of
the aforementioned modifications. Examples of modifications are described
above.
A person skilled in the art will recognize that conservative amino acid
substitutions as well as additions/deletions or a number of divergent amino
acid
sequences can be used are readily made to the disclosed sequences and are
within the scope of the present disclosure.
A "conservative amino acid substitution" as used herein, is one in which
one amino acid residue is replaced with another amino acid residue without
abolishing the protein's desired properties. Conservative amino acid
substitutions
are known in the art. For example, conservative substitutions include
substituting
an amino acid in one of the following groups for another amino acid in the
same
12
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
group: alanine (A), serine (S), and threonine (T); aspartic acid (D) and
glutamic
acid (E); asparagine (N) and glutamine (Q); arginine (R) and lysine (L);
isoleucine
(I), leucine (L), methionine (M), valine (V); and phenylalanine (F), tyrosine
(Y),
and tryptophan (W).
Also included are tmpk sequences with sequence identity with the tmpk
sequences provided below. In one embodiment, the tmpk has 60-70%, 70-80%,
90-95%, 95-99% or 99-99.9% sequence identity with a tmpk described herein.
The term "sequence identity" as used herein refers to the percentage of
sequence identity between two polypeptide sequences or two nucleic acid
sequences. To determine the percent identity of two amino acid sequences or of
two nucleic acid sequences, the sequences are aligned for optimal comparison
purposes (e.g., gaps can be introduced in the sequence of a first amino acid
or
nucleic acid sequence for optimal alignment with a second amino acid or
nucleic
acid sequence). The amino acid residues or nucleotides at corresponding amino
acid positions or nucleotide positions are then compared. When a position in
the
first sequence is occupied by the same amino acid residue or nucleotide as the
corresponding position in the second sequence, then the molecules are
identical
at that position. The percent identity between the two sequences is a function
of
the number of identical positions shared by the sequences (i.e., %
identity=number of identical overlapping positions/total number of
positions×100%). In one embodiment, the two sequences are the same
length. The determination of percent identity between two sequences can also
be accomplished using a mathematical algorithm. A preferred, non-limiting
example of a mathematical algorithm utilized for the comparison of two
sequences is the algorithm of Karlin and Altschul, 1990, Proc. Natl. Acad.
Sci.
U.S.A. 87:2264-2268, modified as in Karlin and Altschul, 1993, Proc. Natl.
Acad.
Sci. U.S.A. 90:5873-5877. Such an algorithm is incorporated into the NBLAST
and XBLAST programs of Altschul et al., 1990, J. Mol. Biol. 215:403. BLAST
nucleotide searches can be performed with the NBLAST nucleotide program
parameters set, e.g., for score=100, wordlength=12 to obtain nucleotide
sequences homologous to a nucleic acid molecules of the present application.
13
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
BLAST protein searches can be performed with the XBLAST program
parameters set, e.g., to score-50, wordlength=3 to obtain amino acid sequences
homologous to a protein molecule of the present invention. To obtain gapped
alignments for comparison purposes, Gapped BLAST can be utilized as
described in Altschul et al., 1997, Nucleic Acids Res. 25:3389-3402.
Alternatively, PSI-BLAST can be used to perform an iterated search which
detects distant relationships between molecules (Id.). When utilizing BLAST,
Gapped BLAST, and PSI-Blast programs, the default parameters of the
respective programs (e.g., of XBLAST and NBLAST) can be used (see, e.g., the
NCBI website). The percent identity between two sequences can be determined
using techniques similar to those described above, with or without allowing
gaps.
In calculating percent identity, typically only exact matches are counted.
Phosphorylation of the prodrug leads to its activation and increases its
effectiveness in killing vector transduced cells (also called "suicide gene
therapy"). The disclosure is useful in the event of a transplant related
adverse
event. A transplant related adverse event typically comprises graft versus
host
disease where following T-cell (or other cell) transplant to a recipient the
transplanted cells attack the host. A transplant adverse event also comprises
any
situation where it would be beneficial to eliminate the transplanted cells,
including
where transplanted cells contain integrations that can cause malignant
transformation or any other disease. The transplanted cells express mutant
tmpk
so that upon detection of graft versus host disease, a prodrug such as AZT is
optionally administered to the patient to kill the transplanted cells.
Other Activator Molecules
Other genes are useful with other prodrugs. Nucleic acid encoding dck is
one example of a useful gene. The dck polypeptide catalyzes the
phosphorylation of a range of pyrimidine and purine deoxynucleotides to the
corresponding nucleotide to modify prodrug compounds so that they exhibit an
antineoplastic effect, such as ara-C, aza-CdK, dFdC, cladribine, zalcitabine
and
fludarabine (see eg. US patent no. 6,423,692). In addition, herpes simplex
virus
14
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
type 1 thymidine kinase (HSV-tk),Equine Herpes Virus 4 thymidine kinase
(EHV4-tk) and their derivatives are also useful as suicide genes. The tk gene,
converts the antiviral prodrug ganciclovir (GCV) to its toxic drug form. It
converts
GCV to GCV-MP; this is converted by guanylate kinase to GCV-DP. This is
converted by other cellular kinases to GCV-TP, which intercalates into DNA
upon
upon replication causing termination and eventual cell death. As mentioned
above DCK converts a variety of drugs to a mono-phosphorylated form.
ii) The Docking Polypeptide - Use of a Toxic binding agent to Kill
Transduced Cells
The term "docking polynucleotide" or "docking gene" alternatively referred
to as "targeting polynucleotide" or "targeting gene" as used herein refers to
a
polynucleotide that encodes a polypeptide (herein referred to as a docking
polypeptide) that functions as a cell marker and is accessible to binding by a
toxic binding agent such as an antibody or an immunotoxin. The docking
polynucleotide can comprise a polypeptide that protects cells from a different
drug - such as neomycin phosphotransferase and G418. In certain
embodiments, the docking polynucleotide encodes a cell surface polypeptide.
The polynucleotide optionally provides for a mode of isolating cells
expressing
said docking molecule. The docking molecule is optionally used to select
transduced or transfected cells or to determine the efficiency of cell
transduction
or transfection.
A good docking gene component optionally encodes a polypeptide that is
recognized by an antibody and is useful for enrichment, sorting, tracking, and
also killing such as a cell surface molecule. Such docking gene components
optionally have the additional ability to track cells and ensure that
expression of
the therapeutic safety gene is maintained. A variety of cell surface markers
are
useful in this context: human CD24, murine HSA, human CD25 (huCD25), a
truncated form of LNGFR, and truncated CD34 .
As the docking polypeptide is substantially overexpressed in transduced
cells, said transduced cells are targeted due to mass action effects, i.e.
more
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
conjugated toxin will accumulate on the cells that express more of the cell
surface marker.
CD19 (SEQ ID NOS: 27-28) is a 95-kDa glycoprotein of the
immunoglobulin superfamily. It forms a complex with CD21, CD81, and Leu-13,
and collectively functions to modulate the activation threshold of the B cell
receptor. As expression of CD19 and CD21 is restricted to B cell lineages from
immature progenitors to blasts, it is suitable for use in murine and human T
cells.
To further decrease any signaling capacity from the CD19 molecule, the
cytoplasmic tail has been deleted for the present adaptation. In one
embodiment
truncated CD19 comprises all or a portion of SEQ ID NO: 29. In another
embodiment truncated CD19 comprises all or a portion of SEQ ID NO: 30. In
another embodiment truncated CD19 comprises all or a portion of SEQ ID NO:
31.
Molecules that are useful as cell markers or detection agents comprise
CD19, truncated CD19, CD25 and EGFP, HSA, CD20, GFP, ETC. EGFP is
variably referred to as enGFP or GFP herein. One skilled in the art will
recognize
that other fluorescent molecules are similarly used. These molecules are
optionally fused to tmpk to provide a tmpk/docking fusion molecule.
As mentioned, the docking polynucleotide encodes a molecule that can be
used to isolate transduced or transfected cells. The docking polynucleotide
useful
in vectors optionally comprises modified tmpk or control molecules. Control
molecules include molecules that do not function as suicide gene therapy
molecules which are typically employed to assess the effect of tmpk mutants in
similarly related cells.
In certain embodiments of the disclosure, the docking polynucleotide
encodes a cell surface protein (marker), such as truncated CD19, CD19, CD20
or CD25 is delivered into target cells which provides the ability to
selectively clear
these cells in vitro and in vivo by administering a toxic binding agent such
as an
antibody or fragment thereof or an immunotoxin directed against the cell
surface
protein. The toxic binding agent binds and kills transfected or transduced
cells
expressing the docking polynucleotide.
16
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
The phrase "cell surface protein" or "cell surface polypeptide" as used
herein refers to a polypeptide that is expressed, in whole or in part on the
surface
of a cell. This optionally includes polypeptide fragments that are presented
on
cells as well as polypeptides or fragments thereof that are naturally found on
the
surface of a cell. In the context of a cell modified to express a vector
construct
comprising a docking polypeptide, wherein the docking polypeptide is a cell
surface polypeptide, the cell surface marker need not be native to the cell it
is
being expressed on.
The term "kills" with respect to transfected or transduced cells refers to
inducing cell death through any of a variety of mechanisms including
apoptosis,
necrosis and autophagy. For example an agent that is cytotoxic kills the
cells.
The term "toxic binding agent" as used herein refers to an agent that binds
a docking polypeptide expressed on or in a cell transfected or transduced with
a
composition, system or vector construct described herein, and which is
cytotoxic
to and/or kills said cell.
The inventors have shown that cell clearance is attainable using the AT
antibody and/or the ATS immunotoxin.
The term "antibody" as used herein is intended to include monoclonal
antibodies, polyclonal antibodies, and chimeric antibodies. The antibody may
be
from recombinant sources and/or produced in transgenic animals. The term
"antibody fragment" as used herein is intended to include without limitations
Fab,
Fab', F(ab')2, scFv, dsFv, ds-scFv, dimers, minibodies, diabodies, and
multimers
thereof, multispecific antibody fragments and Domain Antibodies. Antibodies
can
be fragmented using conventional techniques. For example, F(ab')2 fragments
can be generated by treating the antibody with pepsin. The resulting F(ab')2
fragment can be treated to reduce disulfide bridges to produce Fab' fragments.
Papain digestion can lead to the formation of Fab fragments. Fab, Fab' and
F(ab')2, scFv, dsFv, ds-scFv, dimers, minibodies, diabodies, bispecific
antibody
fragments and other fragments can also be synthesized by recombinant
techniques. The term also includes antibodies or antibody fragments that bind
to
the docking polypeptides disclosed herein. A number of clinical antibodies are
17
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
known in the art that can be used with the methods of the application. For
example Herceptin which recognizes a cell surface molecule called HER2/neu
can be employed. Other drugs exist here that recognize CD25 and CD19: Br J
Haematol. 2006 Jul;134(2):157-70. Epub 2006 Jun 12 The anti-CD20 antibody
rituximab augments the immunospecific therapeutic effectiveness of an anti-
CD19 immunotoxin directed against human B-cell lymphoma. Flavell DJ, Warnes
SL, Bryson CJ, Field SA, Noss AL, Packham G, Flavell SU; Clin Cancer Res.
2005 May 1;11(9):3567-73 Anti-CD19-targeted liposomal doxorubicin improves
the therapeutic efficacy in murine B-cell lymphoma and ameliorates the
toxicity of
liposomes with varying drug release rates, Allen TM, Mumbengegwi DR,
Charrois GJ.; Neurodegener Dis. 2008;5(1):23-6, Humanized anti-CD25 antibody
treatment with daclizumab in multiple sclerosis. Martin R.
Monoclonal antibodies against a variety of receptors and molecules are
currently being introduced in clinical medicine. One of these targets is the
interleukin-2 receptor alpha-chain CD25. The humanized monoclonal anti-CD25
antibody daclizumab (Zenapax) has been approved several years ago for the
prevention of allotransplant rejection and adult T cell leukemia. Following
promising observations in uveitis, daclizumab has been tested in a number of
small clinical trials in multiple sclerosis based on the rationale that
blocking CD25
would prevent the expansion of autoreactive T lymphocytes. The data from this
preliminary clinical exploration as well as findings about the mechanism of
action
of anti-CD25 treatment are summarized in this study. Copyright (c) 2008 S.
Karger AG, Basel.
The term "immunotoxin" as used herein means an antibody or fragment
thereof that is cytotoxic and/or an antibody or fragment there of that is
fused to a
toxic agent. Immunotoxins are described in this application and known in the
art,
for example, in US patent application publication no. 20070059275.
Many immunotoxins are approved for use in humans. In one
embodiment the immunotoxin is a murine anti-Tac (AT) monoclonal antibodyl9
fused to saporin (SAP),20 a toxin that irreversibly damages ribosomes by
cleaving adenine molecules from ribosomal RNA.21 The inventors have
18
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
demonstrated both in vitro and in vivo that AT and the AT-SAP (ATS) complex
specifically targets and kill retrovirally transduced cells that express
huCD25.
Importantly, the inventors have achieved enzymatic correction of a mouse
model of Fabry disease using a bicistronic vector of the disclosure and
demonstrate removal of transduced cells using both ATS and AT. As noted
above, the disclosure provides in one embodiment the combination of a
therapeutic gene, a novel prodrug/enzyme and a docking gene/toxic binding
agent for transducing cells and/or clearing transduced cells, for example in
gene
therapy. Catalytically improved activator polynucleotide, such as dck or
variants
of human tmpk, were delivered into target cells by novel lentiviruses (LVs),
and
the ability to selectively clear these cells in vitro and in vivo in response
to
increasing AZT concentrations was thoroughly evaluated. The inventors transfer
these suicide genes and cell surface protein into cells, such as mammalian T
cells and cell lines, preferably human T-cells and cell lines.
Accordingly, the disclosure relates to methods of using a suicide gene
therapy system comprising an activator polynucleotide and/or docking
polynucleotide inserted in transplant cells for treatment of diseases such as
Fabry disease, Farber disease, cancer and controlling transplant-associated
graft
versus host disease. Where the disease being treated results from a gene or
enzyme deficiency, the system optionally comprises a therapeutic gene for gene
therapy. For example, the system used for the treatment of Fabry disease
further
comprises a therapeutic gene, such as alpha-galactosidase A. The system used
for the treatment of Farber disease further comprises a therapeutic gene, acid
ceramidase. A lentivirus is optionally used to deliver an activator
polynucleotide
and docking polynucleotide. Other methods of delivery are also useful for
example onco-retroviral vectors that engineer expression of huCD25 (see Qin et
al. and Medin PNAS 2004).
The compositions and systems disclosed are useful in the event of a
transplant related adverse event. A transplant related adverse event
optionally
comprises a graft versus host disease where following T-cell (or other cell)
transplant to a recipient the transplanted cells attack the host. A transplant
19
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
adverse event also comprises any situation where it would be beneficial to
eliminate the transplanted cells, including where transplanted cells comprise
integrations that can cause disease. In the case of a transplant adverse
event,
the transplanted cells modified to comprise an activator polynucleotide, are
treated with a prodrug that is converted to a cytotoxic drug by the enzyme
encoded by the activator polynucleotide thereby resulting in cell death. In
another embodiment, transplanted cells modified to comprise targeted
polynucleotide are treated with an amino toxin that binds a polypeptide
encoded
by the docking polynucleotide thereby resulting in cell death. In a further
embodiment, transplanted cells which are modified to comprise both an
activator
polynucleotide and a docking polynucleotide are treated with both a prodrug
and
an immunotoxin, such that a dual suicide safety system is utilized. The
methods,
compositions and systems of the disclosure are also useful to terminate
transplanted cells once their primary desired functions are depleted: such as
for
facilitating transplantation of allogeneic organs, facilitating engraftment of
hematopoietic stem cells, or directly attacking solid tumors or leukemias.
In one embodiment, a prodrug such as AZT is administered to the patient
to kill the transplanted cells. In another embodiment, a toxic binding agent,
such
as an antibody or an antibody conjugated to a toxin, is administered to the
subject and bind to the polypeptide, such as a cell surface polypeptide (eg.
CD19, CD20, CD25), produced by a docking polynucleotide. The toxic binding
agent is optionally administered before, concurrently with, or after
administration
of the prodrug.
For cancer treatment, the above method is useful to treat leukemia where
donor transplant cells are used to kill leukemic cells. The transplanted cells
expressing activator polynucleotide and docking polynucleotide are likely to
also
attack the host, so the disclosure allows the transplanted cells to be killed
after
detection of the onset of graft versus host disease.
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
In a variation of the disclosure, vectors comprising activator polynucleotide
and docking polynucleotide are inserted directly into the solid tumor.
Expression
of activator polynucleotide to produce activator polypeptide sensitizes the
immediate cells, and also surrounding cells if a 'bystander effect' is present
as it
is with some enzyme/prodrug combinations such as HSV-tk/GCV, etc., to the
prodrug and expression of docking polynucleotide sensitize the cells to toxic
binding agent.
Additionally, the activator polynucleotides and docking polynucleotide are
useful as a general 'safety component' in gene therapy. For example in
patients
with Severe Combined Immunodeficiency Disease (SCID), gene therapy has
been used successfully to introduce deficient genes however at least one
clinical
trial was halted due to safety concerns arising from inappropriate DNA
integrations. The prior art also includes much discussion about the dangers of
gene therapy due to vector integrations that can cause cancer. The safety
component overcomes this problem by allowing the transplanted cells to be
destroyed upon administration of a prodrug or a toxic binding agent.
(iii) Activator/Docking Fusions
In an alternate embodiment, the activator polynucleotide is fused to the
docking polynucleotide to produce a fusion polypeptide upon expression. The
inventors have made an activator/docking fusion by fusing truncated CD19 with
a
modified mammalian tmpk polynucleotide as described in US provisional
61/038,398 filed March 20, 2008 herein incorporated by reference. As the
fusion
is expressed in all cells, all cells express the docking polypeptide and the
suicide
gene product, the fusion construct provides a dual safety mechanism whereby
each of the modified cells is killed by either a prodrug alone, a toxic
binding agent
alone or a combination thereof. This provides flexibility and ensures that all
cells
modified to express the fusion are killed if required.
The activator/docking fusion is optionally constructed comprising a tmpk
activator. The tmpk component is optionally a N-terminal (or 5') or C-terminal
(or
21
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
3') in continuous or discontinuous relationship to the docking component. For
example, in a continuous relationship the fusion polypeptide can comprise a
tmpk
component fused to a docking polypeptide (e.g. NH2-tmpk-GFP-COOH) or
alternatively can comprise a docking polypeptide component fused to a tmpk
molecule (e.g NH2-GFP-tmpk-COOH). Similarly, a fusion polynucleotide can
comprise a tmpk component fused to a docking polynucleotide (e.g. 5'-tmpkGFP)
or alternatively can comprise a docking component fused to a tmpk molecule
(e.g
5'-GFP-tmpk-3')).
The docking molecule optionally permits isolation of tmpk expressing or
therapeutic gene expressing cells. A person skilled in the art would recognize
that many molecules are useful for fusing to tmpk to permit isolation of
modified
tmpk or control expressing cells. Choice of molecule will depend on the cell
type
to be transfected or transduced. Generally, the docking molecule is not
expressed on the cell type to be transfected or transduced in appreciable
levels
permitting targeting and/or isolation of cells expressing the docking
polynucleotide. In one embodiment the docking polynucleotide encodes a CD19
(SEQ ID NOS: 27-28). In a preferred embodiment, the docking polynucleotide
encodes a truncated CD19 (SEQ ID NOS: 29-31). In an alternate embodiment,
the detection docking polynucleotide encodes CD25. In another embodiment, the
docking polynucleotide encodes a fluorescent protein such as EGFP. In another
embodiment, the molecules encoded by the docking polynucleotide comprise
CD20, CD25, low affinity nerve growth factor receptor (LNGFR), truncated CD34,
or erythropoietin receptor (EpoR).
In addition, the tmpk and docking components are optionally
discontinuous. For example a linker sequence is optionally present between the
tmpk and docking components.
The term "linker sequence" as used in reference to a tmpk/docking fusion
refers to residues that link the tmpk and docking components. In a
polypeptide,
the residues are generally amino acids. In a polynucleotide, the residues are
generally nucleotides. The term "linker sequence" as used in reference to an
activator/docking fusion polypeptide accordingly generally refers to a
sequence
22
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
of amino acids that links the activator and docking components. The term
"linker
sequence" as used in reference to a tmpk/docking fusion polynucleotide
accordingly generally refers to a sequence of nucleotides that link the tmpk
and
docking components. The linker when referring to a polypeptide sequence
optionally comprises 3, 4, 5, 6, 6-10, 10-15 or 15-25 amino acids or longer
and
when referring to a polynucleotide sequence comprises 3-6, 6-12, 18, 12-24, or
24-72 nucleic acid residues or longer. A linker sequence is useful for several
reasons. A linker sequence can be used to facilitate cloning. Further a linker
sequence can provide a gap between the components that facilitates proper
folding and/or activity (e.g. antigenic activity for the docking component
and/or
catalytic activity for the tmpk component). A person skilled in the art will
recognize that a number of linker sequences can be used and a number of linker
sequences are known in the art. The linker sequence can comprise any
sequence of amino acids or nucleotides that is suitable. For example, suitable
refers to the amino acid composition of the linker. For example, uncharged
amino
acids are preferable. Amino acids such as proline which could limit the
flexibility
of the linker are generally not preferred. In one embodiment the components
are
comprised in a discontinuous relationship, the fusion polypeptide optionally
comprises a tmpk component fused to a linker fused to a docking polypeptide
(e.g. NH2-tmpk-linker-GFP) or alternatively comprises a docking polypeptide
component fused to a linker fused to a tmpk molecule (e.g NH2-truncated CD19-
linker-tmpk-COOH). Similarly, a fusion polynucleotide can comprise a tmpk
component fused to a linker fused to a docking polynucleotide (e.g. 5'-tmpk-
linker-GFP') or alternatively can comprise a docking polynucleotide component
fused to a linker fused to a tmpk molecule (e.g 5'-truncated CD19-linker-tmpk-
3';
such as SEQ ID NO: 28, 29, 31 or 37 fused to a linker sequence described
herein fused to SEQ ID NO:36)). The tmpk and docking components are fused in
frame such that both components are expressed together as one continuous
polypeptide sequence in each cell.
Delivery Vectors
23
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
It will be appreciated by one skilled in the art that a variety of delivery
vectors and expression vehicles are usefully employed to introduce a modified
DNA molecule into a cell. Vectors that are useful comprise lentiviruses,
oncoretroviruses, expression plasmids, adenovirus, and adeno-associated
virus. Other delivery vectors that are useful comprise herpes simplex viruses,
transposons, vaccinia viruses, human papilloma virus, Simian immunodeficiency
viruses, HTLV, human foamy virus and variants thereof. Further vectors that
are
useful comprise spumaviruses, mammalian type B retroviruses, mammalian type
C retroviruses, avian type C retroviruses, mammalian type D retroviruses,
HTLV/BLV type retroviruses, and lentiviruses.
Vectors such as those listed above have been employed to introduce DNA
molecules into cells for use in gene therapy. Examples of vectors used to
express DNA in cells include: Kanazawa T, Mizukami H, Okada T, Hanazono Y,
Kume A, Nishino H, Takeuchi K, Kitamura K, Ichimura K, Ozawa K. Suicide gene
therapy using AAV-HSVtk/ganciclovir in combination with irradiation results in
regression of human head and neck cancer xenografts in nude mice. Gene Ther.
2003 Jan;10(1):51-8. Fukui T, Hayashi Y, Kagami H, Yamamoto N, Fukuhara H,
Tohnai I, Ueda M, Mizuno M, Yoshida J Suicide gene therapy for human oral
squamous cell carcinoma cell lines with adeno-associated virus vector. Oral
Oncol. 2001 Apr;37(3):211-5.
Lentiviral vectors
The safety facet of suicide gene therapy relies on efficient delivery and
stable, consistent expression of both the therapeutic and the safety component
genes. LVs transduce a wide range of dividing and non-dividing cell types with
high efficiency, conferring stable, long-term expression of the transgene25
27.
The use of lentivirus-based gene transfer techniques relies on the in vitro
production of recombinant lentiviral particles carrying a highly deleted viral
genome in which the transgene of interest is accommodated. In particular, the
recombinant lentivirus are recovered through the in trans coexpression in a
24
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
permissive cell line of (1) the packaging constructs, i.e., a vector
expressing the
Gag-Pol precursors together with Rev (alternatively expressed in trans); (2) a
vector expressing an envelope receptor, generally of an heterologous nature;
and (3) the transfer vector, consisting in the viral cDNA deprived of all open
reading frames, but maintaining the sequences required for replication,
incapsidation, and expression, in which the sequences to be expressed are
inserted.
In one embodiment the Lentigen lentiviral vector described in Lu, X. et al.
Journal of gene medicine (2004) 6:963-973 is used to express the DNA
molecules.
In one embodiment the disclosure comprises a lentiviral vector expressing
a dck or modified tmpk molecule. In one embodiment the lentiviral vector
comprises a 5'-Long terminal repeat (LTR), HIV signal sequence, HIV Psi signal
5'-splice site (SD), delta-GAG element, Rev Responsive Element (RRE), 3'-
splice
site (SA), Elongation factor (EF) 1-alpha promoter and 3'-Self inactivating
LTR
(SIN-LTR). It will be readily apparent to one skilled in the art that
optionally one
or more of these regions is substituted with another region performing a
similar
function.
Gene therapy requires the transgene product to be expressed at
sufficiently high levels. Enhancer elements can be used to increase expression
of
modified DNA molecules or increase the lentiviral integration efficiency.
Locus
control regions or scaffold attachment regions can also be added to vectors to
mitigate position-mediated expression effects or the likelihood that
dysfunctional
expression of surrounding genes will occur. In one embodiment the lentiviral
vector further comprises a nef sequence. In a preferred embodiment the
lentiviral
further comprises a cPPT sequence which enhances vector integration. The
cPPT acts as a second origin of the (+)-strand DNA synthesis and introduces a
partial strand overlap in the middle of its native HIV genome. The
introduction of
the cPPT sequence in the transfer vector backbone strongly increased the
nuclear transport and the total amount of genome integrated into the DNA of
target cells. In an alternate preferred embodiment, the lentiviral vector
further
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
comprises a Woodchuck Posttranscriptional Regulatory Element (WPRE). The
WPRE acts at the transcriptional level, by promoting nuclear export of
transcripts
and/or by increasing the efficiency of polyadenylation of the nascent
transcript,
thus increasing the total amount of mRNA in the cells. The addition of the
WPRE
to lentiviral vector results in a substantial improvement in the level of
transgene
expression from several different promoters, both in vitro and in vivo. In a
further
preferred embodiment, the lentiviral vector comprises both a cPPT sequence and
WPRE sequence. The vector also comprises in an alternate embodiment an
internal ribosome entry site (IRES) sequence that permits the expression of
multiple polypeptides from a single promoter. In another embodiment the
lentiviral vector comprises a detection cassette. In another embodiment, the
detection cassette comprises a CD19 molecule or fragment thereof. In another
preferred embodiment the plasmid comprises a docking polynucleotide
incorporated into pHR'-cppt-EF-IRES-W-SIN, pH R'-cppt-EF-tmpk(R16GLL)-
IRES-hCD19-W-SIN or pHR'-cppt-EF-tmpk(F105Y)-IRES-hCD19-W-SIN.
Additionally it will be readily apparent to one skilled in the art that
optionally one
or more of these elements can be added or substituted with other regions
performing similar functions.
In addition to IRES sequences, other elements which permit expression of
multiple polypeptides are useful. In one embodiment the vector comprises
multiple promoters that permit expression more than one polypeptide. In
another
embodiment the vector comprises a protein cleavage site that allows expression
of more than one polypeptide. Examples of protein cleavage sites that allow
expression of more than one polypeptide comprise those listed in the following
articles which are incorporated by reference: Retroviral vector-mediated
expression of HoxB4 in hematopoietic cells using a novel coexpression
strategy.
Klump H, Schiedlmeier B, Vogt B, Ryan M, Ostertag W, Baum C. Gene Ther.
200;8(10):811-7; A picornaviral 2A-like sequence-based tricistronic vector
allowing for high-level therapeutic gene expression coupled to a dual-reporter
system Mark J. Osborn, Angela Panoskaltsis-Mortari, Ron T. McElmurry, Scott K.
Bell, Dario A.A. Vignali, Martin D. Ryan, Andrew C. Wilber, R. Scott Mclvor,
26
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
Jakub Tolar and Bruce R. Blazar. Molecular Therapy 2005; 12 (3) , 569-574;
Development of 2A peptide-based strategies in the design of multicistronic
vectors. Szymczak AL, Vignali DA. Expert Opin Biol Ther. 2005; 5(5):627-38;
Correction of multi-gene deficiency in vivo using a single 'self-cleaving' 2A
peptide-based retroviral vector. Szymczak AL, Workman CJ, Wang Y, Vignali
KM, Dilioglou S, Vanin EF, Vignali DA. Nat Biotechnol. 2004;22(5):589-94. It
will
be readily apparent to one skilled in the art that other elements that permit
expression of multiple polypeptides are useful and readily utilized in the
vectors
of the disclosure.
Viral Regulatory Elements
In addition to the viral regulatory elements described above, additional
viral regulatory elements are readiliy included in the vector constructs of
the
application. Viral regulatory elements are components of vehicles used to
introduce nucleic acid molecules into a host cell. The viral regulatory
elements
are optionally retroviral regulatory elements. For example, the viral
regulatory
elements may be the LTR and gag sequences from HIV1, HSC1, or MSCV. The
retroviral regulatory elements may be from lentiviruses or they may be
heterologous sequences identified from genomic regions.
One skilled in the art would also appreciate that as other viral regulatory
elements are identified, these may be used with the nucleic acid molecules of
the
disclosure.
Detection or Selection Cassette
As noted above, the docking polynucleotide produces a docking
polypeptide, such as a cell surface polypeptide (eg. CD19, CD20 or CD25), that
is recognized by a toxic binding agent. An example of a toxic binding agent is
an
antibody such as Herceptin or an antibody conjugated to a toxin. The docking
polypeptide when a cell surface polypeptide is optionally used as a detection
and/or selection cassette is also referred to as a detection or selection
marker. In
27
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
other embodiments the vector construct comprises a detection or selection
marker that is distinct from the docking polypeptide.
In suicide gene therapy, it is typically desirable that the majority of
transduced cells express the suicide gene or genes. This need can be met by co-
introducing a detection marker polynucleotide, which in some cases can be the
same gene as the docking polynucleotide. In other cases, the detection marker
polynucleotide is different than the docking polypeptide recognized by the
toxic
binding agent. Transduced cells are readily identified and enriched based on
expression of this detection marker polynucleotide. A good detection marker
should be inert in itself, devoid of signaling capacity and non-immunogenic. A
variety of detection markers can be used in this context: human CD24, murine
HSA, human CD25 (huCD25) and a truncated form of LNGFR.
A novel truncated form of CD19 (CD19A) is optionally adopted as a
detection marker (SEQ ID NOS: 29-31). CD19 (SEQ ID NOS: 27-28) is a 95-kDa
glycoprotein of the immunoglobulin superfamily. It forms a complex with CD21,
CD81, and Leu-1 3, and collectively functions to modulate the activation
threshold
of the B cell receptor. As expression of CD19 and CD21 is restricted to B cell
lineages from immature progenitors to blasts, it is suitable for use in murine
and
human T cells. To further decrease any signaling capacity from the CD19
molecule, the cytoplasmic tail has been deleted for the present adaptation. In
one
embodiment truncated CD19 comprises all or a portion of SEQ ID NO: 29. In
another embodiment truncated CD19 comprises all or a portion of SEQ ID NO:
30. In another embodiment truncated CD19 comprises all or a portion of SEQ ID
NO: 31.
"Detection cassette" or "detection marker" is used to refer to a
polynucleotide that directs expression of a molecule that acts as a selection
marker and that optionally provides for a mode of isolating cells expressing
said
selection marker. The molecule is optionally used to select transduced or
transfected cells or to determine the efficiency of cell transduction or
transfection.
Molecules that are useful as selection markers or detection agents comprise
CD19, truncated CD19, CD25 and EGFP. EGFP is variably referred to as
28
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
enGFP herein. One skilled in the art will recognize that other fluorescent
molecules are similarly used.
As mentioned, in certain embodiments, the detection cassette is also a
docking gene. In certain embodiments, the detection cassette and docking gene
are different.
As mentioned, the detection cassette encodes a selection molecule that is
typically used to isolate transduced or transfected cells. The detection
cassette is
useful in vectors comprising therapeutic gene and/or an activator
polynucleotide
or control molecules. Control molecules include molecules that do not function
as suicide gene therapy molecules that are typically employed to assess the
effect of mutants in similarly related cells. A person skilled in the art
would
recognize that many molecules are useful to permit isolation of cells. Choice
of
molecule will depend on the cell type to be transfected or transduced. The
detection cassette molecule is not expressed on the cell type to be
transfected or
transduced in appreciable levels permitting isolation of cells expressing the
detection cassette. In one embodiment the detection cassette encodes a CD19
(SEQ ID NOS: 27-28) selection marker. In a preferred embodiment, the detection
cassette encodes a truncated CD19 (SEQ ID NOS: 29-31) selection marker. In
an alternate embodiment, the detection cassette encodes CD25. In another
embodiment, the detection cassette encodes a fluorescent protein such as
EGFP. In another embodiment, the molecules encoded by the detection cassette
comprise CD20, CD25, low affinity nerve growth factor receptor (LNGFR),
truncated CD34, or erythropoietin receptor (EpoR). Additionally, the detection
cassette optionally comprises a drug resistance gene permitting isolation of
transduced or transfected cells by drug selection.
Polynucleotides of Interest /Therapeutic Nucleic Acid Molecules
Cells transfected or transduced in vitro with the vector constructs
described herein are useful for ex vivo gene therapy or as a research tool or
for
29
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
protein production. Nucleic acid molecules described herein are also useful
for
gene therapy by transfecting or transducing cells in vivo to express a
therapeutic
polynucleotide/protein in addition to activator polynucleotide and docking
polynucleotide. The therapeutic polynucleotide is alternatively referred to
herein
as the therapeutic gene, therapeutic cassette and/or therapeutic expression
cassette. For example, if one were to upregulate the expression of a gene, one
could insert the sense polynucleotide into a vector construct described
herein. If
one were to downregulate the expression of the gene, one could insert the
antisense or an siRNA polynucleotide sequence into the therapeutic expression
cassette. Techniques for inserting sense and antisense sequences (or
fragments of these sequences) would be apparent to those skilled in the art.
The
therapeutic nucleic acid molecule or nucleic acid molecule fragment is
optionally
either isolated from a native source (in sense or antisense orientations) or
synthesized. It is also optionally a mutated native or synthetic sequence or a
combination of these.
Examples of therapeutic coding nucleic acid molecules to be expressed
include adenosine deaminase (ADA), yc interleukin receptor subunit, a-
galactosidase A (a-galA), acid ceramidase, galactocerebrosidase, and
transmembrane conductance regulator (CFTR) molecules.
Other molecules may also be introduced. For example T cells may be
genetically modified to express other relevant molecules for therapy such as T
cell receptors. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC,
Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, Zheng Z, Nahvi A,
de Vries CR, Rogers-Freezer LJ, Mavroukakis SA, Rosenberg SA. Cancer
regression in patients after transfer of genetically engineered lymphocytes.
Science. 2006 Oct 6;314(5796):126-9. Epub 2006 Aug 31.PMID: 16946036
[PubMed - indexed for MEDLINE]
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
Pharmaceutical Compositions
Another aspect relates to pharmaceutical compositions comprising the
vector constructs described herein for use in a system comprising a
corresponding
prodrug and/or toxic binding agent. The pharmaceutical compositions of this
disclosure are used to treat patients having diseases, disorders or abnormal
physical states could include an acceptable carrier, auxiliary or excipient.
The pharmaceutical compositions are optionally administered by ex vivo and
in vivo methods such as electroporation, DNA microinjection, liposome DNA
delivery, and virus vectors that have RNA or DNA genomes including retrovirus
vectors, lentivirus vectors, Adenovirus vectors and Adeno-associated virus
(AAV)
vectors, Semliki Forest Virus, Vaccinia virus, Herpes Simplex Virus,
Vesticular
Stomatitis Virus, etc. Derivatives or hybrids of these vectors are also
useful.
Dosages to be administered depend on patient needs, on the desired effect
and on the chosen route of administration. The expression cassettes are
optionally
introduced into the cells or their precursors using ex vivo or in vivo
delivery vehicles
such as liposomes or DNA or RNA virus vectors. They are also optionally
introduced into these cells using physical techniques such as microinjection
or
chemical methods such as coprecipitation.
The pharmaceutical compositions are typically prepared by known methods
for the preparation of pharmaceutically acceptable compositions which are
administered to patients, and such that an effective quantity of the nucleic
acid
molecule is combined in a mixture with a pharmaceutically acceptable vehicle.
Suitable vehicles are described, for example in Remington's Pharmaceutical
Sciences (Remington's Pharmaceutical Sciences, Mack Publishing Company,
Easton, Pa., USA).
On this basis, the pharmaceutical compositions could include an active
compound or substance, such as a nucleic acid molecule, in association with
one
or more pharmaceutically acceptable vehicles or diluents, and contained in
buffered
31
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
solutions with a suitable pH and iso-osmotic with the physiological fluids.
The
methods of combining the expression cassettes with the vehicles or combining
them with diluents is well known to those skilled in the art. The composition
could
include a targeting agent for the transport of the active compound to
specified sites
within cells.
The application also provides compositions comprising the prodrug and/or
toxic binding agent for use with the safety gene therapy system described
herein. In
the event of an adverse transplant or gene therapy related event, or any
situation
where it is desirable to clear cells modified to express the activator and
docking
genes of the disclosure, a subject, preferably a human patient is administered
a
composition comprising a suitable prodrug and/or toxic binding agent. The
prodrug
can be administered contemporaneously with a composition comprising a toxic
binding agent. In other embodiments, the prodrug and toxic binding agent are
administered separately.
The term "treating" or "treatment" as used herein means administering to a
subject a therapeutically effective amount of the compound of the present
application and may consist of a single administration, or alternatively
comprise a
series of applications. For example, the compound of the present application
may
be administered at least once a week. However, in another embodiment, the
compound may be administered to the subject from about one time per week to
about once daily for a given treatment. The length of the treatment period
depends on a variety of factors, such as the severity of the disease, the age
of
the patient, the concentration and the activity of the compounds of the
present
application, or a combination thereof. In one embodiment, the treatment is
chronic treatment and the length of treatment is 1-2 weeks, 2-4 weeks or more
than 4 weeks. The treatment regimen can include repeated treatment schedules.
It will also be appreciated that the effective amount or dosage of the
compound
used for the treatment or prophylaxis may increase or decrease over the course
of a particular treatment or prophylaxis regime. Changes in dosage may result
32
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
and become apparent by standard diagnostic assays known in the art. In some
instances, chronic administration may be required.
As used herein, and as well understood in the art, "treatment" or
"treating" is also an approach for obtaining beneficial or desired results,
including
clinical results. Beneficial or desired clinical results can include, but are
not
limited to, alleviation or amelioration of one or more symptoms or conditions,
diminishment of extent of disease, stabilized (i.e. not worsening) state of
disease,
preventing spread of disease, delay or slowing of disease progression,
amelioration or palliation of the disease state, and remission (whether
partial or
total), whether detectable or undetectable. "Treatment" can also mean
prolonging
survival as compared to expected survival if not receiving treatment. Further
any
of the treatment methods or uses described herein can be formulated alone or
for
contemporaneous administration with other agents or therapies.
As used herein, the phrase "effective amount" or "therapeutically effective
amount" or a "sufficient amount" of a compound or composition of the present
application is a quantity sufficient to, when administered to the subject,
including
a mammal, for example a human, effect beneficial or desired results, including
clinical results, and, as such, an "effective amount" or synonym thereto
depends
upon the context in which it is being applied. For example, in the context of
treating GVHD, it is an amount of the compound sufficient to achieve a
treatment
response as compared to the response obtained without administration of the
compound. The amount of a given compound of the present application that will
correspond to such an amount will vary depending upon various factors, such as
the given drug or compound, the pharmaceutical formulation, the route of
administration, the type of disease or disorder, the identity of the subject
(e.g.
age, sex, weight) or host being treated, and the like, but can nevertheless be
routinely determined by one skilled in the art. Also, as used herein, a
"therapeutically effective amount" of a compound of the present disclosure is
an
amount which results in a beneficial or desired result in a subject as
compared to
a control. As defined herein, a therapeutically effective amount of a compound
of
the present disclosure may be readily determined by one of ordinary skill by
33
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
routine methods known in the art. Dosage regime may be adjusted to provide the
optimum therapeutic response.
The term "subject" as used herein includes all members of the animal
kingdom including mammals, suitably humans including patients.
Compositions comprising the prodrug and/or toxic binding agent can be
administered by various routes. For example, oral formulations of AZT are well
known in the art. Accordingly the prodrug can be administered orally. The
toxic
binding agent can be administered in one embodiment intraperitoneally (i.p.),
intravenously (i.v.) or intratumorally,
In other embodiments, the composition comprises cells modified with the
vector constructs described herein. Such modified cells can be administered
intravenously using methods known in the art i.p., i.v., intratumorally,
stereotactic
injections to a variety of sites, direct injections, intramuscularly, etc.
Host Cells
The disclosure also relates to a host cell (isolated cell in vitro, a cell in
vivo, or a cell treated ex vivo and returned to an in vivo site) containing a
nucleic
acid molecule of the disclosure. Cells transfected with a nucleic acid
molecule
such as a DNA molecule, or transduced with the nucleic acid molecule such as a
DNA or RNA virus vector, are optionally used, for example, in bone marrow or
cord blood cell transplants according to techniques known in the art. Examples
of the use of transduced bone marrow or cord blood cells in transplants are
for ex
vivo gene therapy of Adenosine deaminase (ADA) deficiency. Other cells which
are optionally transfected or transduced either ex vivo or in vivo include
purified
stem cells (of embryonic or later ontogeny), as described above.
Methods of Isolation
In one aspect of the present disclosure, methods for expressing a vector
construct of the disclosure in cells for transplant are provided. After
transduction
or transfection with vectors comprising elements such as the activator
polynucleotide and docking/selection polynucleotide, cells expressing these
34
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
molecules are optionally isolated by a variety of means known in the art. In
certain embodiments, the cells are isolated by cell sorting or flow cytometry
using
an antibody to the detection cassette encoded selection marker. Additionally
cell
sorting is useful to isolate modified cells where the detection cassette is a
fluorescent protein such as EGFP. Cells expressing polynucleotides of the
disclosure are, in an alternate embodiment, isolated using magnetic sorting or
other immuno-selection schemas. Additionally, cells may be isolated by drug
selection. In one embodiment, a vector comprising a drug resistance gene and a
polynucleotides of the disclosure is introduced into cells. Examples of drug
resistance genes include, but are not limited to, neomycin resistance gene,
blasticidin resistance gene (Bsr), hygromycin resistance gene (Hph), puromycin
resistance gene (Pac), Zeocin resistance gene (Sh ble), FHT, bleomycin
resistance gene and ampicillin resistance gene. After transduction or
transfection, modified cells including the drug resistance gene are selected
by
adding the drug that is inactivated by the drug resistance gene. Cells
expressing
the drug resistance gene survive while non-transfected or non-transduced cells
are killed. A person skilled in the art would be familiar with the methods and
reagents required to isolate cells expressing the desired polynucleotides.
Cell Types for Transplant
Compositions and vector constructs of the disclosure are usefully
introduced into any cell type ex vivo where it is desirable to provide a
mechanism
for killing the modified cells. Cell types that are useful in one embodiment
of the
present disclosure include, but are not limited to, stem cells (both embryonic
and
of later ontogeny), cord blood cells, and immune cells such as T cells,
adherent
and non-adherent bone marrow cells and peripheral blood mononuclear cells
including dendritic cells. T-cells are optionally CD4 positive, CD8 positive,
CD4/CD8 double positive, or CD4/CD8 double negative. These latter cells are
useful for inducing tolerance. In addition, T cells are optionally mature T
cells. In
one embodiment T cells are transduced with a vector of the disclosure,
isolated
and transplanted in a host. In another embodiment the T cells are mature T
cells.
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
In an alternate embodiment stem cells are transduced, isolated and
transplanted
in a host.
Cell lines are optionally transduced. For example human T cell leukemia
Jurkat T cells, human erythro-leukemic K562 cells, human prostate cell lines
DU145 and PC3 cells are optionally transduced or transfected with
polynucleotides of the disclosure. Primary human tumor cells can also be
isolated and transduced by this method. With addition of other modulator
polypeptides such as IL-12, these cells can be made to become potent
initiators
of anti-leukemia responses. Endowment of such cells with a suicide mechanism
will allow their selective removal after anti-tumor immune response
initiation.
Such selective killing and engulfment of dying cells by antigen-presenting
cells
can serve to augment the specific anti-tumor response.
Methods of Treatment
The present disclosure provides modified compositions and vector
constructs for treatment of diseases such as Fabry and Farber diseases. The
compositions and vectors are also useful for the reduction of cell
proliferation, for
example for treatment of cancer. The present disclosure also provides methods
of using compositions and vectors of the disclosure for expressing therapeutic
polynucleotides for the reduction of cell proliferation, for example for
treatment of
cancer.
Vector constructs are introduced into cells that are used for transplant or
introduced directly in vivo in mammals, preferably a human. The vector
constructs are typically introduced into cells ex vivo using methods known in
the
art. Methods for introducing vector constructs comprise transduction,
transfection, infection, electroporation. These methods optionally employ
liposomes or liposome like compounds.
In one embodiment, compositions and vectors of the disclosure are used
to treat cancer by adoptive therapy. Adoptive therapy or adoptive
(immuno)therapy refers to the passive transfer of immunologically competent
tumor-reactive cells into the tumor-bearing host to, directly or indirectly,
mediate
36
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
tumor regression. The feasibility of adoptive (immuno)therapy of cancer is
based
on two fundamental observations. The first of these observations is that tumor
cells express unique antigens that can elicit an immune response within the
syngeneic (genetically identical or similar especially with respect to
antigens or
immunological reactions) host. The other is that the immune rejection of
established tumors can be mediated by the adoptive transfer of appropriately
sensitized lymphoid cells. Clinical applications include transfer of
peripheral
blood stem cells following non-myeloablative chemotherapy with or without
radiation in patients with lymphomas, leukemias, and solid tumors.
In one aspect of the present disclosure, donor T cells or stem cells (either
embryonic or of later ontogeny) are transduced with vector constructs of the
disclosure. Cells expressing these vector contructs are isolated and
adoptively
transferred to a host in need of treatment. In one embodiment the bone marrow
of the recipient is T-cell depleted. Methods of adoptive T-cell transfer are
known
in the art (J Translational Medicine, 2005 3(17): doi;0.1186/1479-5876-3-17,
Adoptive T cell therapy: Addressing challenges in cancer immunotherapy.
Cassian Yee). This method is used to treat solid tumors and does not require
targeting the vector construct-transduced expressing T-cells to the tumor
since
the modified T-cells will recognize the different MHC class molecules present
in
the recipient host resulting in cytotoxic killing of tumor cells.
Another aspect of the disclosure provides for the treatment of solid tumors
by injecting activator polynucleotides and docking polynucleotides of the
disclosure and/or vector constructs or compositions comprising the same,
directly
into the tumor. Methods of introducing polynucleotides of the disclosure
directly
in vivo in a mammal, preferably a human, comprise direct viral delivery,
microinjection, in vivo electroporation, and liposome mediated methods.
Activator genes have been introduced by injection directly into the site of a
tumor to examine results of the technique as a cancer therapeutic treatment
(Chevez-Barrios P, Chintagumpala M, Mieler W, Paysse E, Boniuk M, Kozinetz
C, Hurwitz MY, Hurwitz RL. Response of retinoblastoma with vitreous tumor
37
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
seeding to adenovirus-mediated delivery of thymidine kinase followed by
ganciclovir. J Clin Oncol. 2005 Nov 1;23(31):7927-35. Sterman DH, Treat J,
Litzky LA, Amin KM, Coonrod L, Molnar-Kimber K, Recio A, Knox L, Wilson JM,
Albelda SM, Kaiser LR. Adenovirus-mediated herpes simplex virus thymidine
kinase/ganciclovir gene therapy in patients with localized malignancy: results
of a
phase I clinical trial in malignant mesothelioma. Hum Gene Ther. 1998 May
1;9(7):1083-92). The activator polynucleotides and docking polynucleotides of
the present disclosure are optionally introduced directly into the site of a
tumor to
reduce proliferation of tumor cells, for example, to treat cancer.
In one embodiment, cells are transfected or transduced ex vivo with
vectors. In an optional embodiment, the vector comprises a lentiviral vector.
Tissue specific expression
In an alternate embodiment of the disclosure, the modified expressing
cells express activator polynucleotides and docking polynucleotides under the
control of a tissue or cell specific promoter providing expression in a tissue
specific manner. Expression of modified activator polynucleotides and docking
polynucleotides is optionally targeted to tumor cells using promoters that are
active in tumor cells.
Accordingly, in one aspect of the disclosure, delivery vectors comprising
activator polynucleotides and docking polynucleotides molecules are provided
that result in tissue or cell specific expression. Tissue and cell specific
expression
is typically accomplished using promoters operably linked with the activator
polynucleotides and docking polynucleotides, which limit expression to cells
or
tissues. One skilled in the art will recognize that a variety of promoter
sequences
that direct tissue or cell specific expression are useful to direct tissue or
cell
specific expression. For example, one skilled in the art will readily
recognize that
liver specific expression is accomplished using a liver specific promoter.
Expression is readily limited to a variety of cell and tissue types. Examples
include, but are not limited to, liver, heart, pancreas and T cells. Examples
of liver
38
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
specific promoters include, but are not limited to, the transthyretin
promoter,
albumin promoter, alpha feto protein promoter. Examples of other cell specific
promoters include, but are not limited to, islet cell specific promoters such
as the
insulin promoter, and T cell specific promoters such as CD4-promoter. In
another
embodiment, expression is inducible. The hypoxia-inducible promoter is
optionally used to direct expression of a cytoprotective gene such as but not
limited to erythropoietin. Introduction of a cytoprotective gene under the
control of
a inducible promoter such as the hypoxia inducible promoter is useful, to
prevent
the severe tissue damage by hypoxia. Other promoters are also useful. For
example, tet regulator inducible systems can be employed including tet on and
tet off versions. Other analogous activating systems are known in the artlf
the
transduced cells cause some problems, the transduced cells are optionally
cleared (killed) by suicide effect by administering prodrug and/or toxic
binding
agent to the transduced cells.
Tumor cell specific expression is accomplished using a tumor specific
promoter. Tumor specific promoters comprise the progression elevated gene-3
(PEG-3) promoter. This promoter functions selectively in divergence cancer
cells
with limited activity in normal cells, for tumor cell-specific expression.
Other tumor
specific promoters are also known in the art. The transduced tumor cells are
specifically killed by the prodrug and toxic binding agent.
Graft versus Leukemia
In addition, the disclosure provides, in one aspect, a method of treating
leukemia. Donor T cells or stem cells are transduced with vectors comprising
activator polynucleotides and docking polynucleotides, cells expressing said
activator polynucleotides and docking polynucleotides are isolated and
transplanted to a host in need of treatment. The transplanted cells induce a
graft
versus leukemia effect. If the transplanted cells induce graft versus host
disease,
the transplanted cells can be killed by administering a prodrug or toxic
binding
agent.
39
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
Graft versus leukemia refers to using donor transplant cells to kill host
leukemic cells. Introduced cells will often also attack the cancer cells that
still
may be present after transplant. This was first documented in acute leukemia,
and this phenomenon has been called "graft-versus- leukemia" effect. Similar
effects have been observed in malignant lymphoma, myeloma, and even some
solid tumors. For certain diseases, such as chronic myelogenous leukemia
(CML), the graft-versus-leukemia (GvL) effect may well be the most important
reason that allogeneic transplants are successful in curing the disease.
Graft Versus Host Disease (GVHD)
The infusion of donor lymphocytes in allogenic bone marrow transplant
(BMT) recipients provides potent antitumor activity to treat recurrent
malignancies. One complication, however, is severe Graft Versus Host Disease
(GVHD).
Graft versus host disease is a common complication of allogeneic bone
marrow transplantation (BMT). After bone marrow transplantation, T cells
present
in the graft, either as contaminants or intentionally introduced into the
host, attack
the tissues of the transplant recipient. Graft-versus-host disease can occur
even
when HLA-identical siblings are the donors. HLA-identical siblings or HLA-
identical unrelated donors (called a minor mismatch as opposed to differences
in
the HLA antigens, which constitute a major mismatch) often still have
genetically
different proteins that can be presented on the MHC.
Graft versus host disease is a serious complication of transplant and can
lead to death in patients that develop severe graft versus host disease (the
clinical manifestations of graft versus host disease are reviewed in Socie G.
Chronic graft-versus-host disease: clinical features and grading systems. Int
J
Hematol. 2004 Apr;79(3):216-20). Viral thymidine kinase has been introduced
into transplant cells and used in combination with drugs such as ganciclovir
to
determine the results in individuals who develop graft versus host disease.
(Bonini C, Ferrari G, Verzeletti S, Servida P, Zappone E, Ruggieri L, Ponzoni
M,
Rossini S, Mavilio F, Traversari C, Bordignon C HSV-TK gene transfer into
donor
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
lymphocytes for control of allogeneic graft-versus-leukemia. Science. 1997 Jun
13;276(5319):1719-24; Bondanza A, Valtolina V, Magnani Z, Ponzoni M,
Fleischhauer K, Bonyhadi M, Traversari C, Sanvito F, Toma S, Radrizzani M, La
Seta-Catamancio S, Ciceri F, Bordignon C, Bonini CSuicide gene therapy of
graft-versus-host disease induced by central memory human T lymphocytes.
Blood. 2005.)
While donor T-cells are undesirable as effector cells of graft-versus-host-
disease, they are valuable for engraftment by preventing the recipient's
residual
immune system from rejecting the bone marrow graft (host-versus-graft).
Additionally, as bone marrow transplantation is frequently used to cure
malignant
disorders (most prominently the leukemias), donor T-cells have proven to have
a
valuable graft-versus-tumor (GVT, graft versus leukemia described above)
effect.
A great deal of current research on allogeneic bone marrow transplantation
involves attempts to separate the undesirable graft-vs-host-disease aspects of
T-
cell physiology from the desirable graft-versus-tumor effect.
The present disclosure provides, in one embodiment, methods of treating
transplant patients that develop graft versus host disease by administering
compounds described herein (e.g. activator polynucleotides and docking
polynucleotides used in combination with drugs and/or toxic binding agents) to
a
mammal in need thereof. For example, transplant cells modified to express
polypeptides encoded by activator polynucleotides and docking polynucleotides
are treated with prodrug and/or a toxic binding agent to clear the modified
cells.
In another embodiment, the disclosure provides a method of promoting graft
versus tumor effect by administering compounds of the disclosure to a mammal
in need thereof.
Vector constructs containing the nucleic acid molecules of the disclosure are
typically administered to mammals, preferably humans, in gene therapy using
techniques described below. The polypeptides produced from the nucleic acid
molecules are also optionally administered to mammals, preferably humans. The
disclosure relates to a method of medical treatment of a mammal in need
thereof,
41
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
preferably a human, by administering to the mammal a vector of the disclosure
or a
cell containing a vector of the disclosure. A recipient, preferably human, who
develops an adverse event, such as graft versus host disease, is typically
administered a drug, such as AZT, that is a substrate for the polypeptide
produced
by the activator polynucleotides of the disclosure. The subject is also
optionally
administered a toxic binding agent, such as an antibody conjugated t a toxin.
Diseases, such as blood diseases or neural diseases (neurodegenerative), that
are
readily treated are described in this application and known in the art (eg.
diseases,
such as thalassemia or sickle cell anemia that are treated by administering a
globin
gene as described in Canadian patent application no. 2,246,005). Blood
diseases
treatable by stem cell transplant include leukemias, myelodysplastic
syndromes,
stem cell disorders, myeloproliferative disorders, lymphoproliferative
disorders
phagocyte disorders, inherited metabolic disorders, histiocytic disorders,
inherited erythrocyte abnormalities, inherited immune system disorders,
inherited
platelet abnormalities, plasma cell disorders, malignancies (See also, Medical
Professional's Guide to Unrelated Donor Stem Cell Transplants, 4th Edition).
Stem
cell nerve diseases either inherited or acquired to be treated by neural stem
cell
transplantation include diseases resulting in neural cell damage or loss, eg.
paralysis, Parkinson's disease, Alzheimer's disease, ALS, multiple sclerosis,
traumatic injury). The vector constructs of the disclosure are useful for
providing a
stem cell marker and to express genes that cause stem cells to differentiate
(e.g.
growth factor).
The inventors have achieved long-term enzymatic correction and
corresponding lipid reduction in a mouse model of Fabry disease by bone
marrow transplantation (BMT) of transduced cells9-11 and by direct delivery of
lentivirus into neonates.12
The inventors have developed and utilized retroviral vectors that engineer
expression of both a-gal A and human CD25 (huCD25) in a bicistronic
format.13 CD25, also known as the T-cell activation antigen (Tac) and the
interleukin (IL)-2 receptor alpha chain-a,14 is incapable of mediating IL-2
internalization or signaling by itself; however, in tandem with the A-chain of
the
42
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
receptor and the ycchain, it forms the "high-affinity" receptor for IL-2.15
Though
it can be induced upon activation, expression of CD25 is absent on resting T
cells, B cells, monocytes, and CD34+-enriched cells.16,17 Thus, its limited
expression pattern and lack of ability to mediate signaling make it a good
choice as a cell surface marking protein in bicistronic vectors. In previous
studies, huCD25 expression was used functionally to assess viral titers, for
the
enrichment of transgene-positive cells before BMT, and for tracking transduced
cells after BMT.13 As it is cleaved from the IL-2 receptor complex on the cell
surface and can be detected as soluble CD25 (sCD25) in the plasma,18
sCD25 was also used as a surrogate marker to evaluate the level of transgene
expression in an experimental setting.12
The inventors now extend the use of huCD25 expression from bicistronic
retroviral vector constructs into the development and application of a built-
in
safety mechanism within the gene therapy context. Unwanted proliferative
abnormality occurs following retroviral gene transfer, huCD25 can act as a
target antigen to eliminate transduced cells selectively using either
clinically
approved anti-CD25 antibodies or newer, highly potent antibody toxin
conjugates (immunotoxins).
Using a murine leukemia model, the inventors demonstrated that
antibody treatment reduced tumor burden 32-fold and increased survival
compared with untreated mice. Furthermore, after a bone marrow transplant of
therapeutically transduced cells into Fabry mice, antibody treatment reduced
the number of retrovirally transduced huCD25-expressing cells in the
peripheral blood. A systemic loss of transduced cells with functional
consequences was also evident in the liver and spleen.
Gene Therapy
The disclosure includes compositions and methods for providing a coding
nucleic
acid molecule or therapeutic gene to a subject such that expression of the
molecule in
the cells provides the biological activity of the polypeptide encoded by the
coding
43
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
nucleic acid molecule to those cells. A coding nucleic acid as used herein
means a
nucleic acid that comprises nucleotides which specify the amino acid sequence,
or a
portion thereof, of the corresponding protein. A coding sequence may comprise
a start
codon and/or a termination sequence.
The disclosure includes methods and compositions for providing a coding
nucleic
acid molecule to the cells of an individual such that expression of the coding
nucleic
acid molecule in the cells provides the biological activity or phenotype of
the polypeptide
encoded by the coding nucleic acid molecule. The method also relates to a
method for
providing an individual having a disease, disorder or abnormal physical state
with a
biologically active polypeptide by administering a nucleic acid molecule of
the present
disclosure. The method may be performed ex vivo or in vivo. Gene therapy
methods
and compositions are demonstrated, for example, in U.S. Patent Nos. 5,869,040,
5,639,642, 5,928,214, 5,911,983, 5,830,880,5,910,488, 5,854,019, 5,672,344,
5,645,829, 5,741,486, 5,656,465, 5,547,932, 5,529,774, 5,436,146, 5,399,346
and
5,670,488, 5,240,846. The amount of polypeptide will vary with the subject's
needs.
The optimal dosage of vector may be readily determined using empirical
techniques, for
example by escalating doses (see US 5,910,488 for an example of escalating
doses).
Various approaches to gene therapy may be used. The isclosure includes
a process for providing a human with a therapeutic polypeptide including:
introducing human cells into a human, said human cells having been treated in
vitro or ex vivo to insert therein a vector of the disclosure, the human cells
expressing in vivo in said human a therapeutically effective amount of said
therapeutic polypeptide.
The method also relates to a method for producing a stock of recombinant virus
by producing virus suitable for gene therapy comprising modified DNA encoding
a gene
of interest. This method preferably involves transfecting cells permissive for
virus
replication (the virus containing therapeutic gene) and collecting the virus
produced.
Cotransfection (DNA and marker on separate molecules) may be employed (see
eg US 5,928,914 and US 5,817,492). As well, a detection cassette or marker
(such as
44
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
Green Fluorescent Protein marker or a derivative) may be used within the
vector itself
(preferably a viral vector).
Polypeptide Production and Research Tools
A cell line (either an immortalized cell culture or a stem cell culture)
transfected or transduced with a polynucleotide of the disclosure (or
variants) is
useful as a research tool to measure levels of expression of the coding
nucleic
acid molecule and the activity of the polypeptide encoded by the coding
nucleic
acid molecule.
The disclosure includes a method for producing a recombinant host cell
capable of expressing a nucleic acid molecule of the disclosure comprising
introducing into the host cell a vector of the disclosure.
The disclosure also includes a method for expressing a polypeptide in a
host cell of the disclosure including culturing the host cell under conditions
suitable for coding nucleic acid molecule expression. The method typically
provides the phenotype of the polypeptide to the cell.
In these methods, the host cell is optionally a stem cell or a T cell.
Another aspect of the disclosure is an isolated polypeptide produced from
a nucleic acid molecule or vector of the disclosure according to a method of
the
disclosure.
Uses
The application further provides various uses of the safety system and vector
constructs described herein.
Uses of Activator/Docking Polynucleotide
Also provided are a number of uses of suicide gene systems comprising
activator
and docking polynucleotides and activator/docking genes. All the
aforementioned
activator polynucleotides, tmpk variants, delivery vectors, docking
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
polynucleotides, therapeutic genes, methods and composition embodiments are
contemplated for the various uses herein described.
One embodiment provides use of a suicide gene system comprising a
vector construct comprising a stably integrating delivery vector; an activator
polynucleotide such as a modified mammalian thymidylate kinase (tmpk)
polynucleotide; and a docking polynucleotide wherein the docking
polynucleotide
encodes a docking polypeptide, such as truncated CD19 for expressing an
activator polynucleotide such as a modified mammalian tmpk polynucleotide in a
mammalian cell or subject. In one embodiment the truncated CD19 is fused to
the tmpk polynucleotide. In certain embodiments, the vector construct further
comprises a therapeutic gene and the suicide gene system is for expressing the
therapeutic gene. The suicide cell system further comprises use of a prodrug
such as AZT that is converted to a drug by a polypeptide encoded by an
activator
polynucleotide and/or a toxic binding polypeptide that binds the docking
polypeptide.
Another embodiment provides a suicide gene system comprising a vector
construct comprising a stably integrating delivery vector; an activator
polynucleotide such as modified mammalian thymidylate kinase (tmpk)
polynucleotide; and a docking polynucleotide wherein the docking
polynucleotide
encodes a docking polypeptide, for expressing a modified mammalian tmpk
polynucleotide and a docking polynucleotide in a mammalian cell or subject. In
one embodiment, the docking polynucleotide is fused to the tmpk
polynucleotide.
In certain embodiments, the vector construct further comprises a therapeutic
gene and the suicide gene system is for expressing the therapeutic gene. The
suicide gene system further comprises use of a prodrug such as AZT that is
converted to a drug by a polypeptide encoded by an activator polynucleotide or
a
toxic binding polypeptide that binds the docking polypeptide.
A further embodiment provides use of a suicide gene system comprising a
vector construct comprising a stably integrating delivery vector; an activator
46
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
polynucleotide such as modified mammalian thymidylate kinase (tmpk)
polynucleotide; and a docking polynucleotide wherein the docking
polynucleotide
encodes a docking polypeptide, in the manufacture of a medicament for
expressing a modified mammalian tmpk polynucleotide in a mammalian cell or
subject.In one embodiment, the docking polynucleotide is fused to the tmpk
polynucleotide.
Another embodiment provides use of a composition comprising a stably
integrating delivery vector; an activator polynucleotide such as modified
mammalian thymidylate kinase (tmpk) polynucleotide; and a docking
polynucleotide wherein the docking polynucleotide encodes a docking
polypeptide, for expressing a modified mammalian tmpk polynucleotide in a
mammalian cell or subject. In one embodiment, the docking polynucleotide is
fused to the tmpk polynucleotide.
A further embodiment provides a composition comprising a stably
integrating delivery vector; an activator polynucleotide such as a modified
mammalian thymidylate kinase (tmpk) polynucleotide; and a docking
polynucleotide wherein the docking polynucleotide encodes a docking
polypeptide, for expressing a modified mammalian tmpk polynucleotide in a
mammalian cell or subject. In one embodiment, the docking polynucleotide is
fused to the tmpk polynucleotide
Yet a further embodiment provides use of a composition comprising a
stably integrating delivery vector; an activator polynucleotide such as a
modified
mammalian thymidylate kinase (tmpk) polynucleotide; and a docking
polynucleotide wherein the docking polynucleotide encodes a docking
polypeptide in the manufacture of a medicament for expressing a modified
mammalian tmpk polynucleotide in a mammalian cell or subject. In one
embodiment, the docking polynucleotide is fused to the tmpk polynucleotide.
Another embodiment, provides use of a vector construct or composition
comprising a stably integrating delivery vector; an activator polynucleotide
such
as modified mammalian thymidylate kinase (tmpk) polynucleotide; and a docking
polynucleotide wherein the docking polynucleotide encodes a docking
47
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
polypeptide, for gene therapy. In one embodiment, the docking polynucleotide
is
fused to the tmpk polynucleotide.
A further embodiment provides a vector construct or composition
comprising a stably integrating delivery vector; an activator polynucleotide
such
as a modified mammalian thymidylate kinase (tmpk) polynucleotide; and a
docking polynucleotide wherein the docking polynucleotide encodes a docking
polypeptide, for gene therapy. In one embodiment, the docking polynucleotide
is
fused to the tmpk polynucleotide.
Yet a further embodiment provides a vector construct or composition
comprising a stably integrating delivery vector; an activator polynucleotide
such
as a modified mammalian thymidylate kinase (tmpk) polynucleotide; and a
docking polynucleotide wherein the docking polynucleotide encodes a docking
polypeptide, and wherein the docking polynucleotide is fused to the tmpk
polynucleotide for the manufacture of a medicament for gene therapy.
Also provided is use of a vector construct or composition disclosed herein
for treating a disease selected from the group consisting of cancer, GVHD or
diseases resulting from a deficiency of a gene product.
Another embodiment provides a vector construct or composition disclosed
herein for treating a disease selected from the group consisting of cancer,
GVHD
or diseases resulting from a deficiency of a gene product.
A further embodiment provides a vector construct or composition
disclosed herein for the manufacture of a medicament for treating a disease
selected from the group consisting of cancer, GVHD or diseases resulting from
a
deficiency of a gene product.
Another aspect provides use of an effective amount of a prodrug for killing
a cell expressing a modified mammalian tmpk polynucleotide wherein the
expression of the modified mammalian tmpk results from contact with a vector
construct or composition comprising a stably integrating delivery vector; an
48
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
activator polynucleotide such as a modified mammalian thymidylate kinase
(tmpk) polynucleotide; and a docking polynucleotide wherein the docking
polynucleotide encodes a docking polypeptide In one embodiment, the docking
polynucleotide is fused to the tmpk polynucleotide.
One embodiment provides an effective amount of a prodrug for killing a
cell expressing a modified mammalian tmpk polynucleotide wherein the
expression of the modified mammalian tmpk results from contact with a vector
construct or composition comprising a stably integrating delivery vector; an
activator polynucleotide such as a modified mammalian thymidylate kinase
(tmpk) polynucleotide; and a docking polynucleotide wherein the docking
polynucleotide encodes a docking polypeptide. In one embodiment, the docking
polynucleotide is fused to the tmpk polynucleotide.
Yet another embodiment provides use of an effective amount of a prodrug
for the manufacture of a medicament for killing a cell expressing a modified
mammalian tmpk polynucleotide wherein the expression of the modified
mammalian tmpk results from contact with a vector construct or composition
comprising a stably integrating delivery vector; an activator polynucleotide
such
as a modified mammalian thymidylate kinase (tmpk) polynucleotide; and a
docking polynucleotide wherein the docking polynucleotide encodes a docking
polypeptide. In one embodiment the docking polynucleotide is fused to the tmpk
polynucleotide.
The following non-limiting examples are illustrative of the present
disclosure:
Examples
Example 1
Materials and Methods
Cells lines.
The cell lines C1498 (C57BL/6 derived), 293T, 3T3, and HeLa cells (American
Type Culture Collection, Manassas, VA) were maintained in Dulbecco's
modified Eagle's medium supplemented with 10% fetal calf serum (Cansera,
49
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
Rexdale, Ontario), 2 mM 1-glutamine, 1 mM sodium pyruvate, 100 U/ml
penicillin, and 100 mg/ml streptomycin (all from Sigma, Oakville, Ontario) at
37
C in a humidified incubator with 5% C02.
Vector constructs and viral vector production.
The lentiviral vector pHR'cppt-EF-a-gal A-IRES-huCD25-W-SIN (LV/a-gal
A/huCD25) was constructed.12 Virus was produced by co-transfection of the
lentiviral vector with accessory plasmids pMD.G and pCMVVR8.91 into 293T
cells using FuGENE 6 transfection reagent (Roche, Mississauga, Toronto) and
titered on HeLa cells as previously described.48
The ecotropic oncoretroviral packaging cell line E86/pMFG/a-gal A/
IRES/huCD25 clone 21 (RV/a-gal A/huCD25) was constructed to produce virus
engineered to express both a-gal A and huCD25 as previously described.13 As
a control, E86/pUMFG/enYFP (RV/enYFP) was used, which has the same
vector backbone and expresses enYFP.49 Cells (4 x 106) were seeded in 15-
cm dishes, and medium containing virus was harvested after 72 hours. Viral
titer was determined by infection of 3T3 cells.
Infected cells were then analyzed 72 hours later by flow cytometry to
detect either huCD25 or enYFP. huCD25 expression was detected using a
phycoerythrin- conjugated antibody against CD25 (a-CD25-phycoerythrin; BD
Bioscience Canada, Mississauga, Toronto) and enYFP expression was
measured directly. Flow cytometry was performed using the FACSCalibur and
analyzed using the CELLQuest software (BD Bioscience Canada).
Establishment of huCD25-expressing murine leukemia cell line.
C1498 cells were infected with LV/a-gal A/huCD25 at a multiplicity of
infection of 10 productively infectious particles per cell. Cells were re-
suspended in filtered viral supernatant supplemented with 8 pg/ml protamine
sulfate and overlaid onto plates coated with fibronectin (Roche). Infected
C1498
cells were sorted by magnetic activated cell sorting into pools and by flow
cytometry on the basis of expression of huCD25 into single-cell clones
(C1498/huCD25).
In vitro clearance of retrovirally transduced cells.
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
Transduced C1498 cell pools, C1498/huCD25 or C1498 NT, were plated in
triplicate at a density of 1 x 104 cells/well in a 96-well plate in a volume
of 100
pl. C1498/huCD25 cells were incubated with increasing concentrations (0.1-
nM) of either of the following reagents: AT antibody, ATS, control IgG-SAP,
or SAP (Advanced Targeting Systems, San Diego, CA). C1498 NT cells were
treated with ATS at the same concentrations. Cells were incubated at 37 C
and growth inhibition and cell death were then assessed. All treatments were
tested in at least two independent experiments.
To assess growth inhibition, 10 pl of 5 mg/ml MTT labeling reagent
(Sigma) was added to each well 72 hours after seeding cells. Plates were
incubated for 4 hours at 37 C in a humidified incubator with 5% C02. Then
100 pl of solubilizing solution (10% sodium dodecyl sulfate, 0.01 M HCI) was
added and plates were incubated at 37 C overnight.
Cell death was assessed 48 hours after seeding by the measurement
of lactate dehydrogenase release using the CytoTox 96 Non-Radioactive
Cytotoxicity Assay Kit (Promega, Madison, WI) as per the manufacturer's
instructions.
Establishment of in vivo leukemia model.
C1498/huCD25 cells were used to generate a leukemia model in Fabry
mice.50 Mice were lethally irradiated (11 Gy), and 4 hours later 1 x 106
C1498/huCD25 cells were injected into the tail vein along with 1 x 106 fresh
BMMNCs isolated by flushing the femurs and tibias of syngeneic donor Fabry
mice. Control mice were injected with 1 x 106 C1498 NT cells. All recipient
mice were treated with 5 pg ATS or equimolar (24.4 pmol) amounts of either AT
or IgG-SAP on days 2, 4, and 6 after cell transplantation, by injection into
the
intraperitoneal cavity in a volume of 200 pl. Mice were monitored daily for
evidence of disease or distress in compliance with standards set by the Animal
Care Committee of the University Health Network.
In vivo clearance of gene-corrected cells in a BMT model.
Donor Fabry mice were treated with 150 mg/kg 5-fluorouracil (Sigma). Three
days later, BM was isolated by flushing the femurs and tibias of treated donor
51
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
Fabry mice. Mononuclear cells were isolated by centrifugation on Nycoprep and
stimulated for 12 hours in Dulbecco's modified Eagle's medium
supplemented with 10% fetal calf serum, 2 mM 1-glutamine, 1 mM sodium
pyruvate, 100 U/ml penicillin, 100 mg/ml streptomycin, 50 ng/ml stem cell
factor, 20 ng/ml FIt3 ligand, and 20 ng/ml IL-6. All cytokines were obtained
from
R&D Systems (Minneapolis, MN). Cells were transduced twice (at 12-hour
intervals) using supernatant from RV/a-gal A/huCD25 or RV/enYFP producer
cell linesl3 at multiplicities of infection of approximately 3 and 1
infectious
particles per cell, respectively. Infections were performed on plates coated
with
fibronectin (Roche) and viral supernatant was supplemented with the same
cytokine cocktail as above plus 8 pg/ml protamine sulfate (Sigma).
Recipient Fabry mice were lethally irradiated (11 Gy), and 4 hours later
infected cells were injected via the tail vein. Cell doses were 0.4 x 106 and
0.3
x 106 cells/mouse for the groups transplanted with cells infected with RV/a-
gal
A/huCD25 and RV/enYFP, respectively. From 4 weeks after transplantation, PB
cells were monitored every 4 weeks to detect engraftment. Eight weeks after
transplantation, mice were treated intraperitoneally with three doses of 5 pg
ATS or equimolar amounts of either AT or IgG-SAP. Doses were administered
every 2 days. At 10 weeks after transplantation, PB was analyzed for response
to the immunotoxins.
A fourth dose of immunotoxin was administered, as before,11 weeks
after transplantation and the animals were killed 12 weeks after
transplantation.
Soluble human CD25 ELISA. Plasma was isolated from PB of mice by
centrifugation at 16,000 for 20 minutes. The level of sCD25 was measured by a
direct ELISA using the BD OptEIA Human IL-2 sRa ELISA Set (BD Bioscience
Canada) as per the manufacturer's instructions. Each sample was measured
in triplicate. a-gal A activity assay. a-gal A activity was measured by a
microtiter
plate-based fluorometric assay using 5 mM 4-methylumbelliferyl a-
dgalactopyranoside (Research Products International, Mount Prospect, IL) as
the substrate for a-gal A, and 0.1 M N-acetyl-d-galactosamine (Sigma) as an
52
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
inhibitor of a-N-acetylgalactosaminidase, as previously described.9 Plasma
was added directly to the plate in triplicate repeats for each analysis.
For measurement of organ enzyme activity, frozen tissue samples were
homogenized and lysates prepared as previously described.12 Plates were
read on a fluorescence microtiter plate reader (Dynex, Chantilly, VA) against
nine independent dilutions of a 4-methylumbelliferone standard (Sigma). The
protein concentrations of tissue samples were determined using the BCA
Protein Assay Kit (Pierce, Rockford, IL).
Statistical analysis.
Data presented represent means of triplicate determinations for each
sample and are representative of results obtained from independent
experiments that produced similar relative results.
Differences between groups for enzyme assays and ELISAs were
assessed using Student's t-test. The Kaplan-Meier product-limit method was
used to assess the survival of mice and the log-rank statistic was used to
test
differences between groups (Excel, Microsoft Corporation). Values of P < 0.05
were considered to be statistically significant.
Results
In vitro effect of targeting huCD25 with a specific immunotoxin. The inventors
first determined the specificity and efficacy of the huCD25-targeted
immunotoxin ATS. A murine myeloid leukemia cell line, C1498, was infected
with a lentiviral vector pHR'cPPTEF- a-gal A-IRES-huCD25-W-SIN (LV/a-gal
A/huCD25) that is engineered to express both human a-gal A and huCD25.12
Infected pools were enriched for expression of huCD25 by magnetic activated
cell sorting. Two populations of cells were tested that have a broad spectrum
of
huCD25 expression with a 5 nM concentration of each reagent: ATS, AT, control
immunogloblin (Ig)G Ab conjugated to SAP (IgG-SAP), or SAP only. These
populations, shown in Figure la and b, were 90 and 45% positive for huCD25
expression, respectively. MTT [3-(4,5-dimethyl-2-thiazolyl)-2, 5-diphenyl-2H-
tetrazolium bromide] assays confirmed that both populations of cells treated
53
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
with ATS showed reduced proliferation (Figure 1c and d) and increased cell
death as measured by lactate dehydrogenase release (Figure le and f)
compared with cells treated with other reagents. Non-transduced cells did not
show any inhibition of proliferation or increased cytotoxicity when treated
with
ATS.
Next, the inventors tested the ability of ATS to clear a clonal population of
transduced cells. A single-cell clone expressing huCD25 (C1498/huCD25)
was isolated from the infected pool of cells by flow cytometry-based sorting
(Figure 2a). Both C1498/huCD25 and C1498 non-transduced (C1498 NT) cells
were incubated with increasing concentrations of each reagent. The effects on
cellular proliferation and cell killing were then measured. As shown in Figure
2b, inhibition of cellular proliferation was significantly higher (P < 0.001)
when
cells were treated with ATS than when cells were treated with control
reagents.
This effect was specific to cells expressing huCD25, as C1498 NT cells treated
with ATS did not show the same level of impaired growth. Similar results were
obtained from a lactate dehydrogenase assay. At low doses (<1 nM), cell
killing
was higher in cells treated with ATS than in cells treated with control
reagents
(P < 0.001) (Figure 2c).
Clearance of huCD25-expressing cells in vivo Leukemia model.
As a first step toward determining whether treatment with a CD25 antibody or
immunotoxin could clear huCD25-expressing leukemic cells in our mouse
model of Fabry disease, the dose of C1498 leukemia cells to use in this strain
was optimized. Increasing doses (1 X 103 to 1 X 106) of C1498 NT cells were
injected into Fabry mice and the effects were monitored.
Although leukemic cells were not present in the peripheral blood,
systemic subcutaneous invasion were present, splenomegaly, and
lymphoadenopathy, which mimics some leukemic phenotypes.
For cell doses of 1 X 103 and 1 X 104 cells/mouse, it was found that 100
and 70% of mice, respectively, survived the challenge. For higher cell doses
of
1 X 105 and 1 X 106 cells/mouse, 100% of the mice succumbed to the leukemia
within 60 days and 30 days, respectively. To obtain a more clinically relevant
54
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
leukemia model, a cell dose of 1 X 106 cells/mouse for future studies was
chosen as at this higher cell dose the phenotype of the transplanted mice
progressed to the disease state more quickly and aggressively.
As no previous in vivo studies have been carried out with murine ATS
and most studies using other AT derivatives use receptor-saturating doses of
antibody,22 it was next necessary to determine an effective dose of
immunotoxin. The ability of two different doses of ATS to eliminate huCD25-
expressing cells in Fabry mice challenged with C1498/huCD25 leukemia was
tested. Mice were lethally irradiated and injected with 1 X 106 C1498/huCD25
cells and supportive syngeneic BM cells. At days 2, 4, and 6 after leukemic
transplant, animals were injected with either 5 pg ATS or 20 pg ATS, injected
with SAP only, or left untreated (n = 3 per group). Eleven days after
challenge,
blood was sampled and plasma analyzed for levels of sCD25 by enzyme-
linked immunosorbent assay (ELISA). Evaluation of sCD25 levels is a common
method used in the clinical setting to monitor tumor burden and treatment
response in patients with CD25-expressing lymphoma and leukemia.23 This
method also allows sensitive detection of the presence of CD25-positive cells
for such studies as it can reflect contributions from abstruse populations. As
shown in Figure 3, treatment with ATS significantly reduced sCD25 (P < 0.05)
levels compared with animals treated with the control reagent, SAP, and with
those left untreated. As treatment with the lower dose of
pg of ATS had a similar effect to treatment with the 20 pg dose (Figure 3),
the
lower dose of ATS was chose for use in future experiments because this was
more cost-effective and may lower the risk of secondary or non-specific
toxicities.
To test the efficacy of our CD25-targeting approach further, a larger
experiment using 5 pg ATS was then performed. Mice were lethally irradiated
and injected with 1 X 106 C1498/huCD25 or C1498 NT cells along with
supportive syngeneic BM cells via the tail vein. Mice transplanted with
C1498/huCD25 cells were then treated with equimolar amounts of ATS, AT, or
IgG-SAP. Mice transplanted with C1498 NT cells were treated with 5 pg ATS as
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
a control. All animals were bled on days 7, 11, and 18 after transplantation,
and
levels of sCD25 in the plasma were measured by ELISA. As shown in Figure
4a, in mice challenged with C1498/ huCD25 cells, at 18 days after
transplantation average plasma sCD25 levels were significantly lower in
animals treated with ATS (474 pg/ml) and AT (848 pg/ml) than in mice treated
with IgG-SAP (4,762 pg/ml; P < 0.01) or not treated (15,450 pg/ml; P < 0.05).
This indicates a lower tumor burden in mice treated with both CD25-targeted
reagents, ATS and AT.
The inherent a-gal A deficiency of Fabry mice and the fact that the
transplanted tumor cells were engineered to express a- gal A meant that
differences in a-gal A activity itself could be used as another surrogate
marker
of tumor burden. Therefore, plasma a-gal A activity was measured and was
found to be lowest in mice treated with ATS (16 nmol/hour/ml) and AT (21 nmol/
hour/ml) (Figure 4b). These levels were significantly lower than those in mice
that received IgG-SAP (47 nmol/hour/ml; P < 0.001 and P < 0.01, versus ATS
and AT, respectively) or that were left untreated (77 nmol/hour/ml; P < 0.05).
Therefore, both ATS and AT are able to de-bulk tumor burden in this huCD25-
expressing leukemia model.
To determine the ability of anti-CD25 antibodies to affect survival,
animals were monitored daily; a Kaplan-Meier representation of survival is
shown in Figure 4c. In mice treated with ATS, the median survival duration was
29 days. This was significantly higher (P < 0.01) than that seen in mice that
were not treated (median survival = 23 days). Increased survival was also
seen in mice treated with AT (median survival of 30 days, P < 0.05 versus
untreated mice). Therefore, even in the context of a very high leukemic
burden,
treatment with CD25-targeted antibodies increased survival compared with
control treatments. Note that these results are representative of two
independent experiments.
BMT model
The clearance strategy in the context of a therapeutic BMT model was
next tested. BMT is a common gene therapy approach,24 and incorporation of a
56
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
cell surface protein that can be targeted can improve the safety of the
system.
Murine bonemarrow mononuclear cells (BMMNCs) were isolated and infected
twice with one of two ecotropic oncoretroviral vectors, either E86/pMFG/a-gal
AIRES/huCD25 clone 21 (RV/a-gal A/huCD25) orE86/pUMFG/enYFP
(RV/enYFP).13 Flow cytometry analysis of these transduced BMMNCs showed
that cells infected with RV/a-gal A/huCD25 were approximately 30% positive for
expression of huCD25 (Figure 5a) and cells infected with RV/enYFP were
approximately 20% positive for enhanced yellow fluorescent protein(enYFP)
expression (Figure 5b). Cells were then injected into lethally irradiated
Fabry
mice, which were monitored monthly for engraftment.
At 8 weeks after transplantation, plasma from recipient Fabry mice was
analyzed for a-gal A activity and for levels of sCD25. Average plasma a-gal A
activity in mice transplanted with BMMNCs infected with RV/a-gal A/huCD25
was 65 nmol/ hour/ml, approximately sixfold higher than in both control Fabry
mice and mice transplanted with RV/enYFP-infected BMMNCs (Figure 5c). This
indicates that a therapeutic correction twofold higher than in normal C57BL/6
mice was acheived (Figure 5c). At this time, the average level of sCD25 in the
plasma of Fabry mice transplanted with BMMNCs infected with RV/a-gal
A/huCD25 was 1212 370 pg/ml. In contrast, sCD25 was undetectable in mice
transplanted with RV/enYFP-infected cells, in wild-type C57BU6 mice, and in
untreated Fabry mice.
Mice were then treated with either ATS, AT, or IgG-SAP, as in our leukemia
model (see above). Seven days after the third dose of immunotoxin, plasma
was sampled to determine the effect of treatment. Comparisons were made
with pre-treatment values collected for each mouse at 8 weeks after
transplantation.
As shown in Figure 6a, treatment with ATS resulted in lower plasma sCD25
levels than in mice that were treated with IgG-SAP or mice that were not
treated
(P < 0.05). In addition, analysis of huCD25 expression on peripheral blood
(PB)
mononuclear cells by flow cytometry showed that mice treated with ATS had
significantly reduced numbers of huCD25-expressing PB mononuclear
57
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
cells than mice treated with IgG-SAP (P < 0.01) or untreated mice (P < 0.05)
(Figure 6b). Similar effects were observed in mice treated with AT, further
supporting the conceptual ability of targeted anti-CD25 antibodies to
eliminate
retrovirally transduced donor hematopoietic cells in vivo. Expression of enYFP
was monitored before and after treatment with ATS and it was found that levels
remained stable over the course of the experiment (Figure 6c), demonstrating
the specificity of the immunotoxin for cells expressing huCD25.
To examine the effect of a later administration of antibody or immunotoxin,
one
final dose was administered and then mice were killed. Enzyme activity was
measured in various tissues to determine the systemic effect of each reagent.
PB mononuclear cells from mice that were treated with ATS showed
significantly lower (P < 0.05) a-gal A activity than mice treated with IgG-SAP
(Figure 7a). Similarly, a-gal A activity in the livers of mice treated with
ATS was
significantly lower (P < 0.05) than enzyme activity in the livers of mice
treated
with AT or IgG-SAP or untreated mice (Figure 7b). Likewise, in the spleens of
mice treated with ATS, there was significantly lower (P < 0.01) a-gal A
activity
than in IgG-SAP-treated or untreated mice (Figure 7c).
Discussion
Gene therapy is the most promising curative treatment for monogenetic
diseases such as lysosomal storage disorders.25 Although considerable
advances toward the development of retrovirus-based gene therapy strategies
for Fabry disease have been made, concerns remain regarding the safety of
integrating vectors.
To address this issue, a cell surface marker such as huCD25 acts as
an effective built-in safety mechanism in the event of insertional
genotoxicity by
facilitating the clearance of transduced cells with a specifically targeted
immunotoxin. huCD25 in combination with a-galA in studies evaluating the
efficacy of retroviral gene therapy for Fabry disease has been previously
described.12,13 No untoward effects of exogenously expressing this protein
were observed nor have altered therapeutic effects of this surface antigen on
a-
gal A-mediated correction in vivo been seen.
58
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
Monoclonal antibodies have been successfully used in the clinic for
many years to treat hematological malignancies, with minimal toxicity.26,27
For
instance, rituximab, an anti-CD20 antibody, has been used to treat a variety
of
lymphoid malignancies.26,28-30 In addition, a strategy for clearing transduced
hematopoietic cells in vivo using an anti-CD20 Ab was proposed for the
treatment of graft-versus-host disease.31 The premise is that T cells can be
transduced with a viral vector carrying the complementary DNA for CD20 before
BMT, and if graft-versus-host disease occurs, then anti-CD20 antibodies can
be used to eliminate the donor T cells. These studies have shown promising
results in vitro; however, no studies have been carried out to demonstrate
efficacy in vivo.32,33
Aberrant levels of CD25 expression characterize numerous disorders
such as adult T-cell leukemia/lymphoma, Hodgkin's lymphoma, hairy cell
leukemias, and true histiocytic lymphomas.34 Treatment of these diseases
using antibodies against CD25, as well as newer recombinant immunotoxins,
has resulted in complete and partial remissions in patients.34,35 Currently,
anti-CD25 antibodies are widely used for the prevention of renal graft
rejection
and in some cases for prophylactic treatment against graftversus-
host disease.36,37 Furthermore, studies have shown that when anti-CD25
antibodies are used to deplete CD4+CD25+ regulatory T cells, anti-tumor
immunity is enhanced.38-40 These findings provided the rationale for using
anti-CD25 toxinconjugated antibodies to target huCD25.
The inventors have shown both in vitro in cell culture and in vivo in a
Fabry mouse model, that a CD25-targeted treatment can specifically and
effectively kill leukemia cells that express both a therapeutic transgene, a-
gal A,
and huCD25 following infection with a retroviral vector. In a model using
huCD25-expressing C1498 leukemia cells, measurement of sCD25 levels and
a- gal A activity following ATS treatment showed a 32- and 5-fold reduction
over
untreated mice, respectively. Similar results were obtained when mice were
treated with AT. In addition, treatment with either ATS or AT extended
survival by
approximately 26% over mice that were not treated. It was not unexpected that
59
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
despite this increase in survival time, these mice still succumbed to the
leukemia, as a very high tumor dose was chosen for administration. As has
been observed in clinical trials for the treatment of naturally occurring CD25-
expressing leukemias, better outcomes are achievable with multi-modal
therapy.41-43 These results demonstrate proof of principle that the clearance
strategy can de-bulk tumor burden and extend survival.
The clearance strategy is useful as a safety mechanism against retroviral-
induced genotoxicity in hematopoietic stem/progenitor cells. The system was
next examined in a BMT setting in a mouse model of Fabry disease using an
oncoretroviral vector gene delivery vehicle.13 An oncoretroviral vector was
chosen here. Oncoretroviral vectors have a greater propensity for integrating
near transcriptional start sites, proto-oncogenes, and cell cycle regulatory
genes than do lentiviral vectors,44-46 perhaps making them more likely to
cause dysregulation in gene expression leading to leukemias,4 for example.
After our standard gene transfer and BMT protocol in Fabry mice, supra-
physiological levels of a-gal A activity in the plasma of transplanted mice
was
achieved. Anti-CD25-targeted treatment of transplanted mice decreased levels
of a-gal A activity in PB mononuclear cells and decreased expression of
huCD25 on these cells, indicating clearance of the transduced cell population
itself. As expected, a corresponding decrease in the level of sCD25 in the PB
was seen. This corresponds well with data showing a positive correlation
between levels of sCD25 and a-gal A activity in the PB of mice treated with
LV/a-
gal A/huCD25.12
ATS treatment was more effective at clearing transduced cells from the
organs than AT. ATS treatment resulted in a systemic decrease in organ a-gal
A activity, indicating that there was widespread elimination of transduced
cells. The study further provides evidence that ATS is not merely clearing
circulating sCD25 directly but is targeting and killing the CD25-expressing
cells.
This is the first report of an antibody-mediated clearance strategy being
applied to gene therapy in the context of a therapeutic BMT. The complementary
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
DNA for CD25 was recently cloned from the rhesus macaque, which will
facilitate this endeavor.47 A variety of cell surface proteins are readily
incorporated into various retroviral vectors in combination with any
therapeutic
transgene. Using this system will add another safety mechanism to retroviral
gene transfer systems.
Example 2
CD19 Immunotoxin Experiments
The efficacy of a CD19 monoclonal antibody conjugated to an immunotoxin to
specifically clear cells transduced with
pCCL.SIN.cPPT.EF.CD19ATmpkF105YR200A.WPRE will be tested in vitro and
in vivo.
In vitro
Jurkat cells will be transduced with
pCCL.SIN.cPPT.EF.CD19ATmpkF105YR200A.WPRE. The transduced pool of
cells will be enriched for cells expressing CD19 using fluorescence-activated
cell
sorting (FACS). The CD19 enriched population and a non-transduced control
group will be cultured in the presence of various concentrations of CD19
immunotoxin for 2-4 days. Cell proliferation will then be assessed using the
Cell
Titer 96 Aqueous One Solution Cell Proliferation Assay Kit (Promega) and cell
death will be measured using the CytoTox 96 Cytotoxicity Assay Kit (Promega).
It is expected that transduced cells cultured with CD19 immunotoxin will show
a
significant decrease in proliferation and a significant increase in
cytotoxicity
compared to control groups. To model a potential clinical adverse advent in
which a patient might develop a malignancy such as leukemia, these
experiments will also be performed using K562 ereythroid leukemia cells
transduced with pCCL.SIN.cPPT.EF.CD190TmpkF105YR200A.WPRE.
61
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
In vivo
Murine bone marrow cells will be isolated and transduced ex vivo with
pCCL.SIN.cPPT.EF.CD19ATmpkF105YR200A.WPRE. Transduced cells and a
non-transduced control group will then be injected into irradiated non-obese
diabetic/severe combined immunodeficiency (SCID) mice. Approximately 8
weeks post transplant, the levels of CD19 in the peripheral blood will be
measured using flow cytometry. Transplanted mice will then be given an
intraperitoneal injeaction of CD19 immunotoxin or placebo as a control every 2
days for a total of three injections. Clearance of CD19-positive cells in the
peripheral blood will be measured using flow cytometry. It is expected that
mice
transplanted with pCCL.SIN.cPPT.EF.CD19ATmpkF105YR200A.WPRE
transduced cells and treated with CD19 immunotoxin will show significantly
decreased levels of CD19 expressing cells in the peripheral blood compared to
control groups. Again, to model a potential clinical adverse event such as
leukemia, a second experiment in which K562 ereythroid leukemia cells are used
will be done. K562 cells will be transduced with
pCCL.SIN.cPPT.EF.CD19ATmpkF105YR200A.WPRE and enriched for CD19
expression using FACS. The enriched K562 population and a non-transduced
control group will be injected into the flank of NOD/SCID mice. After
transplantation, mice will be given IP injections of CD19 immunotoxin or a
placebo as a control every 2 days for 3 a total of 3 injections. The growth of
the
tumors will be monitored. It is expected that mice transplanted with K562
cells
transduced with pCCL.SIN.cPPT.EF.CD19ATmpkF105YR200A.WPRE and
treated with CD19 immunotoxin will show significantly decreased rates of tumor
growth compared to control groups.
Example 3
CD19 Immunotoxin Experiments
The efficacy of a monoclonal antibody against CD19 conjugated to an
immunotoxin (CD19-IT) to specifically clear cells transduced with
62
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
pCCL.SIN.cPPT.EF.CD19ATmpkF105YR200A.WPRE. will be tested in vitro and
in vivo.
In vitro
Jurkat cells will be transduced with
pCCL.SIN.cPPT.EF.CD19ATmpkF105YR200A.WPRE. The transduced pool of
cells will be enriched for cells expressing CD19 using fluorescence-activated
cell
sorting (FACS). The CD19 enriched population and a non-transduced control
group will be cultured in the presence of various concentrations of CD19-IT
for 2-
4 days. Cell proliferation will then be assessed using the Cell Titer 96
Aqueous
One Solution Cell Proliferation Assay Kit (Promega) and cell death will be
measured using the CytoTox 96 Cytotoxicity Assay Kit (Promega). It is expected
that transduced cells cultured with CD19-IT will show a significant decrease
in
proliferation and a significant increase in cytotoxicity compared to control
groups.
CD19-IT will also be tested in combination with AZT to determine if transduced
cells that are not sensitive to AZT can subsequently be eliminated by
administration of CD19-IT. The CD19 enriched Jurkat population and a non-
transduced control group will be cultured in 1001iM AZT for 4 days with media
and AZT being replaced each day. Cell proliferation and cell death will be
measured as described above. Cells will then be cultured in the presence of
CD19-IT for 2-4 days and proliferation and cell death will be measured again.
It
is expected that approximately 80% of transduced cells will be killed by
treatment
with AZT, and that subsequent culture with CD19-IT will cause a further
significant decrease in viable cell number compared to control groups. To
model
a potential clinical adverse advent in which a patient might develop a
malignancy
such as leukemia, these experiments will also be performed using K562
erythroid
leukemia cells transduced with
pCCL.SIN.cPPT.EF.CD19ATmpkF105YR200A.WPRE to show cell killing.
In vivo
Murine bone marrow cells will be isolated and transduced ex vivo with
pCCL.SIN.cPPT.EF.CD19ATmpkF 105YR200A.WPRE. Transduced cells and a non-
transduced control group will then be injected into irradiated non-obese
63
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
diabetic/severe combined immunodeficiency (NOD/SCID) mice. Approximately 8
weeks post transplant, the levels of CD19 expressing cells in the peripheral
blood
will be measured using flow cytometry. Transplanted mice will then be given an
intraperitoneal (IP) injection of CD19-IT or placebo as a control every 2 days
for a
total of three injections. Clearance of CD19-positive cells in the peripheral
blood
will be measured using flow cytometry. It is expected that mice transplanted
with
pCCL.SIN.cPPT.EF.CD19ATmpkF105YR200A.WPRE transduced cells and treated
with CD19-IT will show significantly decreased levels of CD19 expressing cells
in
the peripheral blood compared to control groups. Another group to be included
in this experiment will examine if treatment with AZT in combination with CD19-
IT
can further reduce levels of CD19 expressing cells in the peripheral blood. In
this
group, mice will receive IP injections of AZT everyday for 14 days. At this
point,
CD19 levels in the peripheral blood will be measured using flow cytometry.
Mice
will then be administered IP injections of CD19-IT every 2 days for a total of
3
injections. CD19 levels in the peripheral blood will then be measured again.
It is
expected that this combination therapy will reduce levels of CD19 expressing
cells in the peripheral blood comapared to groups treated with either AZT or
CD19-IT alone, and to control groups.
Again, to model a potential clinical adverse event such as leukemia, a
second experiment in which K562 erythroid leukemia cells are used will be
performed. K562 cells will be transduced with
pCCL.SIN.cPPT.EF.CD19ATmpkF105YR200A.WPRE and enriched for CD19
expression using FACS. The enriched K562 population and a non-transduced
control group will be injected into the flank of NOD/SCID mice. After
transplantation, mice will be given IP injections of CD19-IT or a placebo as a
control every 2 days for 3 a total of 3 injections. The growth of the tumors
will be
monitored. It is expected that mice transplanted with K562 cells transduced
with
pCCL.SIN.cPPT.EF.CD19ATmpkF105YR200A.WPRE and treated with CD19-IT will
show significantly decreased rates of tumor growth compared to control groups.
Treatment with a combination of AZT and CD19-IT is also tested in a method
similar to that described above and provides cell killing.
64
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
While the present invention has been described with reference to what are
presently considered to be the preferred examples, it is to be understood that
the
invention is not limited to the disclosed examples. To the contrary, the
invention
is intended to cover various modifications and equivalent arrangements
included
within the spirit and scope of the appended claims.
All publications, patents and patent applications are herein incorporated
by reference in their entirety to the same extent as if each individual
publication,
patent or patent application was specifically and individually indicated to be
incorporated by reference in its entirety.
Table of Sequences
SEQUENCES
SEQ ID NO:1
<210> 1
<211> 639
<212> DNA
<213> Homo sapiens
<400> 1
atggcggccc ggcgcggggc tctcatagtg ctggagggcg tggaccgcgc cgggaagagc 60
acgcagagcc gcaagctggt ggaagcgctg tgcgccgcgg gccaccgcgc cgaactgctc 120
cggttcccgg aaagatcaac tgaaatcggc aaacttctga gttcctactt gcaaaagaaa 180
agtgacgtgg aggatcactc ggtgcacctg cttttttctg caaatcgctg ggaacaagtg 240
ccgttaatta aggaaaagtt gagccagggc gtgaccctcg tcgtggacag atacgcattt 300
tctggtgtgg ccttcaccgg tgccaaggag aatttttccc tagattggtg taaacagcca 360
gacgtgggcc ttcccaaacc cgacctggtc ctgttcctcc agttacagct ggcggatgct 420
gccaagcggg gagcgtttgg ccatgagcgc tatgagaacg gggctttcca ggagcgggcg 480
ctccggtgtt tccaccagct catgaaagac acgactttga actggaagat ggtggatgct 540
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
tccaaaagca tcgaagctgt ccatgaggac atccgcgtgc tctctgagga cgccatccgc 600
actgccacag agaagccgct gggggagcta tggaagtga 639
SEQ ID NO:2
<210> 2
<211> 212
<212> PRT
<213> Homo sapiens
<400> 2
Met Ala Ala Arg Arg Gly Ala Leu lie Val Leu Glu Gly Val Asp Arg
1 5 10 15
Ala Gly Lys Ser Thr Gin Ser Arg Lys Leu Val Glu Ala Leu Cys Ala
20 25 30
Ala Gly His Arg Ala Glu Leu Leu Arg Phe Pro Glu Arg Ser Thr Glu
35 40 45
Ile Gly Lys Leu Leu Ser Ser Tyr Leu Gin Lys Lys Ser Asp Val Glu
50 55 60
Asp His Ser Val His Leu Leu Phe Ser Ala Asn Arg Trp Glu Gin Val
65 70 75 80
Pro Leu Ile Lys Glu Lys Leu Ser Gin Gly Val Thr Leu Val Val Asp
85 90 95
Arg Tyr Ala Phe Ser Gly Val Ala Phe Thr Gly Ala Lys Glu Asn Phe
100 105 110
Ser Leu Asp Trp Cys Lys Gin Pro Asp Val Gly Leu Pro Lys Pro Asp
115 120 125
Leu Val Leu Phe Leu Gin Leu Gin Leu Ala Asp Ala Ala Lys Arg Gly
130 135 140
Ala Phe Gly His Glu Arg Tyr Glu Asn Gly Ala Phe Gin Glu Arg Ala
145 150 155 160
Leu Arg Cys Phe His Gin Leu Met Lys Asp Thr Thr Leu Asn Trp Lys
165 170 175
Met Val Asp Ala Ser Lys Ser Ile Glu Ala Val His Glu Asp lie Arg
66
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
180 185 190
Val Leu Ser Glu Asp Ala Ile Arg Thr Ala Thr Glu Lys Pro Leu Gly
195 200 205
Glu Leu Trp Lys
210
SEQ ID NO:3
<210> 3
<211> 639
<212> DNA
<213> Homo sapiens
<400> 3
atggcggccc ggcgcggggc tctcatagtg ctggagggcg tggaccgcgc cgggaagagc 60
acgcagagcc gcaagctggt ggaagcgctg tgcgccgcgg gccaccgcgc cgaactgctc 120
cggttcccgg aaagatcaac tgaaatcggc aaacttctga gttcctactt gcaaaagaaa 180
agtgacgtgg aggatcactc ggtgcacctg cttttttctg caaatcgctg ggaacaagtg 240
ccgttaatta aggaaaagtt gagccagggc gtgaccctcg tcgtggacag atacgcattt 300
tctggtgtgg ccttcaccgg tgccaaggag aatttttccc tagattggtg taaacagcca 360
gacgtgggcc ttcccaaacc cgacctggtc ctgttcctcc agttacagct ggcggatgct 420
gccaagcggg gagcgtttgg ccatgagcgc tatgagaacg gggctttcca ggagcgggcg 480
ctccggtgtt tccaccagct catgaaagac acgactttga actggaagat ggtggatgct 540
tccaaaagca tcgaagctgt ccatgaggac atccgcgtgc tctctgagga cgccatccgc 600
actgccacag agaagccgct gggggagcta tggaagtga 639
SEQ ID NO:4
<210> 4
<211> 212
<212> PRT
<213> Homo sapiens
<400> 4
Met Ala Ala Arg Arg Gly Ala Leu Ile Val Leu Glu Gly Val Asp Arg
67
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
1 5 10 15
Ala Gly Lys Ser Thr Gin Ser Arg Lys Leu Val Glu Ala Leu Cys Ala
20 25 30
Ala Gly His Arg Ala Glu Leu Leu Arg Phe Pro Glu Arg Ser Thr Glu
35 40 45
Ile Gly Lys Leu Leu Ser Ser Tyr Leu Gin Lys Lys Ser Asp Val Glu
50 55 60
Asp His Ser Val His Leu Leu Phe Ser Ala Asn Arg Trp Glu Gin Val
65 70 75 80
Pro Leu Ile Lys Glu Lys Leu Ser Gin Gly Val Thr Leu Val Val Asp
85 90 95
Arg Tyr Ala Phe Ser Gly Val Ala Phe Thr Gly Ala Lys Glu Asn Phe
100 105 110
Ser Leu Asp Trp Cys Lys Gin Pro Asp Val Gly Leu Pro Lys Pro Asp
115 120 125
Leu Val Leu Phe Leu Gin Leu Gin Leu Ala Asp Ala Ala Lys Arg Gly
130 135 140
Ala Phe Gly His Glu Arg Tyr Glu Asn Gly Ala Phe Gin Glu Arg Ala
145 150 155 160
Leu Arg Cys Phe His Gin Leu Met Lys Asp Thr Thr Leu Asn Trp Lys
165 170 175
Met Val Asp Ala Ser Lys Ser Ile Glu Ala Val His Glu Asp Ile Arg
180 185 190
Val Leu Ser Glu Asp Ala Ile Arg Thr Ala Thr Glu Lys Pro Leu Gly
195 200 205
Glu Leu Trp Lys
210
SEQ ID NO:5
<210> 5
<211> 636
<212> DNA
<213> Homo sapiens
68
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
<400> 5
atggcggccc ggcgcggggc tctcatagtg ctggagggcg tggaccgcgc cgggaagagc 60
acgcagagcc gcaagctggt ggaagcgctg tcgcgcgggc caccgcccga actgctccgg 120
ttcccggaaa gatcaactga aatcggcaaa cttctgagtt cctacttgca aaagaaaagt 180
gacgtggagg atcactcggt gcacctgctt ttttctgcaa atcgctggga acaagtgccg 240
ttaattaagg aaaagttgag ccagggcgtg accctcgtcg tggacagata cgcattttct 300
ggtgtggcct tcaccggtgc caaggagaat ttttccctag actggtgtaa acagccagac 360
gtgggccttc ccaaacccga cctggtcctg ttcctccagt tacagctggc ggatgctgcc 420
aagcggggag cgtttggcca tgagcgctat gagaacgggg ctttccagga gcgggcgctc 480
cggtgtttcc accagctcat gaaagacacg actttgaact ggaagatggt ggatgcttcc 540
aaaagactcg aagctgtcca tgaggaactc cgcgtgctct ctgaggacgc catccgcact 600
gccacagaga agccgctggg ggagctatgg aagtga 636
SEQ ID NO:6
<210> 6
<211> 211
<212> PRT
<213> Homo sapiens
<400> 6
Met Ala Ala Arg Arg Gly Ala Leu Ile Val Leu Glu Gly Val Asp Arg
1 5 10 15
Ala Gly Lys Ser Thr Gin Ser Arg Lys Leu Val Glu Ala Leu Ser Arg
20 25 30
Gly Pro Pro Pro Glu Leu Leu Arg Phe Pro Glu Arg Ser Thr Glu Ile
35 40 45
Gly Lys Leu Leu Ser Ser Tyr Leu GIn Lys Lys Ser Asp Val Glu Asp
50 55 60
His Ser Val His Leu Leu Phe Ser Ala Asn Arg Trp Glu GIn Val Pro
65 70 75 80
69
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
Leu lie Lys Glu Lys Leu Ser Gin Gly Val Thr Leu Val Val Asp Arg
85 90 95
Tyr Ala Phe Ser Gly Val Ala Phe Thr Gly Ala Lys Glu Asn Phe Ser
100 105 110
Leu Asp Trp Cys Lys Gin Pro Asp Val Gly Leu Pro Lys Pro Asp Leu
115 120 125
Val Leu Phe Leu Gin Leu Gin Leu Ala Asp Ala Ala Lys Arg Gly Ala
130 135 140
Phe Gly His Glu Arg Tyr Glu Asn Gly Ala Phe Gin Glu Arg Ala Leu
145 150 155 160
Arg Cys Phe His Gin Leu Met Lys Asp Thr Thr Leu Asn Trp Lys Met
165 170 175
Val Asp Ala Ser Lys Arg Leu Glu Ala Val His Glu Glu Leu Arg Val
180 185 190
Leu Ser Glu Asp Ala Ile Arg Thr Ala Thr Glu Lys Pro Leu Gly Glu
195 200 205
Leu Trp Lys
210
SEQ ID NO:7
<210> 7
<211> 639
<212> DNA
<213> Homo sapiens
<400> 7
atggcggccc ggcgcggggc tctcatagtg ctggagggcg tggaccgcgc cgggaagagc 60
acgcagagcc gcaagctggt ggaagcgctg tgcgccgcgg gccaccgcgc cgaactgctc 120
cggttcccgg aaagatcaac tgaaatcggc aaacttctga gttcctactt gcaaaagaaa 180
agtgacgtgg aggatcactc ggtgcacctg cttttttctg caaatcgctg ggaacaagtg 240
ccgttaatta aggaaaagtt gagccagggc gtgaccctcg tcgtggacag atacgcattt 300
tctggtgtgg ccttcaccgg tgccaaggag aatttttccc tagattggtg taaacagcca 360
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
gacgtgggcc ttcccaaacc cgacctggtc ctgttcctcc agttacagct ggcggatgct 420
gccaagcggg gagcgtttgg ccatgagcgc tatgagaacg gggctttcca ggagcgggcg 480
ctccggtgtt tccaccagct catgaaagac acgactttga actggaagat ggtggatgct 540
tccaaaagca tcgaagctgt ccatgaggac atccgcgtgc tctctgagga cgccatccgc 600
actgccacag agaagccgct gggggagcta tggaaggac 639
SEQ ID NO:8
<210> 8
<211> 213
<212> PRT
<213> Homo sapiens
<400> 8
Met Ala Ala Arg Arg Gly Ala Leu Ile Val Leu Glu Gly Val Asp Arg
1 5 10 15
Ala Gly Lys Ser Thr Gin Ser Arg Lys Leu Val Glu Ala Leu Cys Ala
20 25 30
Ala Gly His Arg Ala Glu Leu Leu Arg Phe Pro Glu Arg Ser Thr Glu
35 40 45
Ile Gly Lys Leu Leu Ser Ser Tyr Leu Gin Lys Lys Ser Asp Val Glu
50 55 60
Asp His Ser Val His Leu Leu Phe Ser Ala Asn Arg Trp Glu Gin Val
65 70 75 80
Pro Leu Ile Lys Glu Lys Leu Ser Gin Gly Val Thr Leu Val Val Asp
85 90 95
Arg Tyr Ala Phe Ser Gly Val Ala Phe Thr Gly Ala Lys Glu Asn Phe
100 105 110
Ser Leu Asp Trp Cys Lys Gin Pro Asp Val Gly Leu Pro Lys Pro Asp
115 120 125
Leu Val Leu Phe Leu Gin Leu Gin Leu Ala Asp Ala Ala Lys Arg Gly
130 135 140
71
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
Ala Phe Gly His Glu Arg Tyr Glu Asn Gly Ala Phe GIn Glu Arg Ala
145 150 155 160
Leu Arg Cys Phe His GIn Leu Met Lys Asp Thr Thr Leu Asn Trp Lys
165 170 175
Met Val Asp Ala Ser Lys Ser lie Glu Ala Val His Glu Asp Ile Arg
180 185 190
Val Leu Ser Glu Asp Ala lie Arg Thr Ala Thr Glu Lys Pro Leu Gly
195 200 205
Glu Leu Trp Lys Asp
210
SEQ ID NO:9
<210> 9
<211> 639
<212> DNA
<213> Mus muscuius
<400> 9
atggcgtcgc gtcggggagc gctcatcgtg ctggagggtg tggaccgtgc tggcaagacc 60
acgcagggcc tcaagctggt gaccgcgctg tgcgcctcgg gccacagagc ggagctgctg 120
cgtttccccg aaagatcaac ggaaatcggc aagcttctga attcctactt ggaaaagaaa 180
acggaactag aggatcactc cgtgcacctg ctcttctctg caaaccgctg ggaacaagta 240
ccattaatta aggcgaagtt gaaccagggt gtgacccttg ttttggacag atacgccttt 300
tctggggttg ccttcactgg tgccaaagag aatttttccc tggattggtg taaacaaccg 360
gacgtgggcc ttcccaaacc tgacctgatc ctgttccttc agttacaatt gctggacgct 420
gctgcacggg gagagtttgg ccttgagcga tatgagaccg ggactttcca aaagcaggtt 480
ctgttgtgtt tccagcagct catggaagag aaaaacctca actggaaggt ggttgatgct 540
tccaaaagca ttgaggaagt ccataaagaa atccgtgcac actctgagga cgccatccga 600
aacgctgcac agaggccact gggggagcta tggaaataa 639
SEQ ID NO:10
<210> 10
72
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
<211> 212
<212> PRT
<213> Mus musculus
<400> 10
Met Ala Ser Arg Arg Gly Ala Leu Ile Val Leu Glu Gly Val Asp Arg
1 5 10 15
Ala Gly Lys Thr Thr Gin Gly Leu Lys Leu Val Thr Ala Leu Cys Ala
20 25 30
Ser Gly His Arg Ala Glu Leu Leu Arg Phe Pro Glu Arg Ser Thr Glu
35 40 45
Ile Gly Lys Leu Leu Asn Ser Tyr Leu Glu Lys Lys Thr Glu Leu Glu
50 55 60
Asp His Ser Val His Leu Leu Phe Ser Ala Asn Arg Trp Glu Gin Val
65 70 75 80
Pro Leu Ile Lys Ala Lys Leu Asn Gin Gly Val Thr Leu Val Leu Asp
85 90 95
Arg Tyr Ala Phe Ser Gly Val Ala Phe Thr Gly Ala Lys Glu Asn Phe
100 105 110
Ser Leu Asp Trp Cys Lys Gin Pro Asp Val Gly Leu Pro Lys Pro Asp
115 120 125
Leu Ile Leu Phe Leu Gin Leu Gin Leu Leu Asp Ala Ala Ala Arg Gly
130 135 140
Glu Phe Gly Leu Glu Arg Tyr Glu Thr Gly Thr Phe Gin Lys Gin Val
145 150 155 160
Leu Leu Cys Phe Gin Gin Leu Met Glu Glu Lys Asn Leu Asn Trp Lys
165 170 175
Val Val Asp Ala Ser Lys Ser Ile Glu Glu Val His Lys Glu Ile Arg
180 185 190
Ala His Ser Glu Asp Ala Ile Arg Asn Ala Ala Gin Arg Pro Leu Gly
195 200 205
Glu Leu Trp Lys
73
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
210
SEQ ID NO:11
<210> 11
<211> 212
<212> PRT
<213> Homo sapiens
<400> 11
Met Ala Ala Arg Arg Gly Ala Leu lie Val Leu Glu Gly Val Asp Arg
1 5 10 15
Ala Gly Lys Ser Thr Gin Ser Arg Lys Leu Val Glu Ala Leu Cys Ala
20 25 30
Ala Gly His Arg Ala Glu Leu Leu Arg Phe Pro Glu Arg Ser Thr Glu
35 40 45
Ile Gly Lys Leu Leu Ser Ser Tyr Leu Gin Lys Lys Ser Asp Val Glu
50 55 60
Asp His Ser Val His Leu Leu Phe Ser Ala Asn Arg Trp Glu Gin Val
65 70 75 80
Pro Leu lie Lys Glu Lys Leu Ser Gin Gly Val Thr Leu Val Val Asp
85 90 95
Arg Tyr Ala Phe Ser Gly Val Ala Tyr Thr Gly Ala Lys Glu Asn Phe
100 105 110
Ser Leu Asp Trp Cys Lys Gin Pro Asp Val Gly Leu Pro Lys Pro Asp
115 120 125
Leu Val Leu Phe Leu Gin Leu Gin Leu Ala Asp Ala Ala Lys Arg Gly
130 135 140
Ala Phe Gly His Glu Arg Tyr Glu Asn Gly Ala Phe Gin Glu Arg Ala
145 150 155 160
Leu Arg Cys Phe His Gin Leu Met Lys Asp Thr Thr Leu Asn Trp Lys
165 170 175
Met Val Asp Ala Ser Lys Ser Ile Glu Ala Val His Glu Asp lie Arg
180 185 190
74
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
Val Leu Ser Glu Asp Ala Ile Arg Thr Ala Thr Glu Lys Pro Leu Gly
195 200 205
Glu Leu Trp Lys
210
SEQ ID NO:12
<210> 12
<211> 214
<212> PRT
<213> Homo sapiens
<400> 12
Met Ala Ala Arg Arg Gly Ala Leu Ile Val Leu Glu Gly Val Asp Gly
1 5 10 15
Ala Gly Lys Ser Thr Gin Ser Arg Lys Leu Val Glu Ala Leu Cys Ala
20 25 30
Ala Gly His Arg Ala Glu Leu Leu Arg Phe Pro Glu Arg Ser Thr Glu
35 40 45
Ile Gly Lys Leu Leu Ser Ser Tyr Leu Gin Lys Lys Ser Asp Val Glu
50 55 60
Asp His Ser Val His Leu Leu Phe Ser Ala Asn Arg Trp Glu Gin Val
65 70 75 80
Pro Leu Ile Lys Glu Lys Leu Ser Gin Gly Val Thr Leu Val Val Asp
85 90 95
Arg Tyr Ala Phe Ser Gly Val Ala Phe Thr Gly Ala Lys Glu Asn Phe
100 105 110
Ser Leu Asp Trp Cys Lys Gin Pro Asp Val Gly Leu Pro Lys Pro Asp
115 120 125
Leu Val Leu Phe Leu Gin Leu Thr Pro Glu Val Gly Leu Lys Arg Ala
130 135 140
Arg Ala Arg Gly Gin Leu Asp Arg Tyr Glu Asn Gly Ala Phe Gin Glu
145 150 155 160
Arg Ala Leu Arg Cys Phe His Gin Leu Met Lys Asp Thr Thr Leu Asn
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
165 170 175
Trp Lys Met Val Asp Ala Ser Lys Ser Ile Glu Ala Val His Glu Asp
180 185 190
Ile Arg Val Leu Ser Glu Asp Ala Ile Ala Thr Ala Thr Glu Lys Pro
195 200 205
Leu Gly Glu Leu Trp Lys
210
SEQ ID NO:13
<210> 13
<211> 6811
<212> DNA
<213> Vector
<400> 13
tggaagggct aattcactcc caacgaagac aagatatcct tgatctgtgg atctaccaca 60
cacaaggcta cttccctgat tggcagaact acacaccagg accagggatc agatatccac 120
tgacctttgg atggtgctac aagctagtac cagttgagcc agataaggta gaagaggcca 180
acaaaggaga gaacaccagc ttgttacacc ctgtgagcct gcatggaatg gatgacccgg 240
agagagaagt gttagagtgg aggtttgaca gccgcctagc atttcatcac gtggcccgag 300
agctgcatcc ggagtacttc aagaactgct gatatcgagc ttgctacaag ggactttccg 360
ctggggactt tccagggagg cgtggcctgg gcgggactgg ggagtggcga gccctcagat 420
gctgcatata agcagctgct ttttgcctgt actgggtctc tctggttaga ccagatctga 480
gcctgggagc tctctggcta actagggaac ccactgctta agcctcaata aagcttgcct 540
tgagtgcttc aagtagtgtg tgcccgtctg ttgtgtgact ctggtaacta gagatccctc 600
agaccctttt agtcagtgtg gaaaatctct agcagtggcg cccgaacagg gacttgaaag 660
cgaaagggaa accagaggag ctctctcgac gcaggactcg gcttgctgaa gcgcgcacgg 720
caagaggcga ggggcggcga ctggtgagta cgccaaaaat tttgactagc ggaggctaga 780
aggagagaga tgggtgcgag agcgtcagta ttaagcgggg gagaattaga tcgcgatggg 840
76
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
aaaaaattcg gttaaggcca gggggaaaga aaaaatataa attaaaacat atagtatggg 900
caagcaggga gctagaacga ttcgcagtta atcctggcct gttagaaaga tcagaaggct 960
gtagacaaat actgggacag ctacaaccat cccttcagac aggatcagaa gaacttagat 1020
cattatataa tacagtagca accctctatt gtgtgcatca aaggatagag ataaaagaca 1080
ccaaggaagc tttagacaag atagaggaag agcaaaacaa aagtaagacc accgcacagc 1140
aagcggccgc tgatcttcag acctggagga ggagatatga gggacaattg gagaagtgaa 1200
ttatataaat ataaagtagt aaaaattaa ccattaggag tagcacccac caaggcaaag 1260
agaagagtgg tgcagagaga aaaaagagca gtgggaatag gagctttgtt ccttgggttc 1320
ttgggagcag caggaagcac tatgggcgca gcgtcaatga cgctgacggt acaggccaga 1380
caattattgt ctggtatagt gcagcagcag aacaatttgc tgagggctat tgaggcgcaa 1440
cagcatctgt tgcaactcac agtctggggc atcaagcagc tccaggcaag aatcctggct 1500
gtggaaagat acctaaagga tcaacagctc ctggggattt ggggttgctc tggaaaactc 1560
atttgcacca ctgctgtgcc ttggaatgct agttggagta ataaatctct ggaacagatt 1620
tggaatcaca cgacctggat ggagtgggac agagaaatta acaattacac aagcttaata 1680
cactccttaa ttgaagaatc gcaaaaccag caagaaaaga atgaacaaga attattggaa 1740
gagataaat gggcaagttt gtggaattgg tttaacataa caaattggct gtggtatata 1800
aaattattca taatgatagt aggaggcttg gtaggtttaa gaatagtttt tgctgtactt 1860
tctatagtga atagagttag gcagggatat tcaccattat cgtttcagac ccacctccca 1920
accccgaggg gacccgacag gcccgaagga atagaagaag aaggtggaga gagagacaga
1980
gacagatcca ttcgattagt gaacggatct cgacggtatc gcttttaaaa gaaaaggggg 2040
gattgggggg tacagtgcag gggaaagaat agtagacata atagcaacag acatacaaac 2100
taaagaatta caaaaacaaa ttacaaaaat tcaaaatttt atcgataagc tttgcaaaga 2160
tggataaagt tttaaacaga gaggaatctt tgcagctaat ggaccttcta ggtcttgaaa 2220
77
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
ggagtgggaa ttggctccgg tgcccgtcag tgggcagagc gcacatcgcc cacagtcccc 2280
gagaagttgg ggggaggggt cggcaattga accggtgcct agagaaggtg gcgcggggta 2340
aactgggaaa gtgatgtcgt gtactggctc cgcctttttc ccgagggtgg gggagaaccg 2400
tatataagtg cagtagtcgc cgtgaacgtt ctttttcgca acgggtttgc cgccagaaca 2460
caggtaagtg ccgtgtgtgg ttcccgcggg cctggcctct ttacgggtta tggcccttgc 2520
gtgccttgaa ttacttccac gcccctggct gcagtacgtg attcttgatc ccgagcttcg 2580
ggttggaagt gggtgggaga gttcgaggcc ttgcgcttaa ggagcccctt cgcctcgtgc 2640
ttgagttgag gcctggcctg ggcgctgggg ccgccgcgtg cgaatctggt ggcaccttcg 2700
cgcctgtctc gctgctttcg ataagtctct agccatttaa aatttttgat gacctgctgc 2760
gacgcttttt ttctggcaag atagtcttgt aaatgcgggc caagatctgc acactggtat 2820
ttcggttttt ggggccgcgg gcggcgacgg ggcccgtgcg tcccagcgca catgttcggc 2880
gaggcggggc ctgcgagcgc ggccaccgag aatcggacgg gggtagtctc aagctggccg 2940
gcctgctctg gtgcctggcc tcgcgccgcc gtgtatcgcc ccgccctggg cggcaaggct 3000
ggcccggtcg gcaccagttg cgtgagcgga aagatggccg cttcccggcc ctgctgcagg 3060
gagctcaaaa tggaggacgc ggcgctcggg agagcgggcg ggtgagtcac ccacacaaag 3120
gaaaagggcc tttccgtcct cagccgtcgc ttcatgtgac tccacggagt accgggcgcc 3180
gtccaggcac ctcgattagt tctcgagctt ttggagtacg tcgtctttag gttgggggga 3240
ggggttttat gcgatggagt ttccccacac tgagtgggtg gagactgaag ttaggccagc 3300
ttggcacttg atgtaattct ccttggaatt tgcccttttt gagtttggat cttggttcat 3360
tctcaagcct cagacagtgg ttcaaagttt ttttcttcca tttcaggtgt cgtgagagga 3420
attctgcagt cgagcggagc gcgcgtaata cgactcacta tagggcgcca tgggtaccgg 3480
gccccccctc gatcgaacaa caacaacaat aacacatggt tccgcgtggc tctcatatgg 3540
cggcccggcg cggggctctc atagtgctgg agggcgtgga cggcgccggg aagagcacgc 3600
78
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
agagccgcaa gctggtggaa gcgctgtgcg ccgcgggcca ccgcgccgaa ctgctccggt 3660
tcccggaaag atcaactgaa atcggcaaac ttctgagttc ctacttgcaa aagaaaagtg 3720
acgtggagga tcactcggtg cacctgcttt tttctgcaaa tcgctgggaa caagtgccgt 3780
taattaagga aaagttgagc cagggcgtga ccctcgtcgt ggacagatac gcattttctg 3840
gtgtggcctt caccggtgcc aaggagaatt tttccctaga ctggtgtaaa cagccagacg 3900
tgggccttcc caaacccgac ctggtcctgt tcctgcagtt aactccggaa gttggcttaa 3960
aacgcgcacg tgctcgcggc gagcttgacc gctatgagaa cggggctttc caggagcggg 4020
cgctccggtg tttccaccag ctcatgaaag acacgacttt gaactggaag atggtggatg 4080
cttccaaaag catcgaagct gtccatgagg acatccgcgt gctctctgag gacgccatcg 4140
ccactgccac agagaagccg ctgggggagc tatggaagtg aggatcagtc gacggtatcg 4200
attccccctc tccctccccc ccccctaacg ttactggccg aagccgcttg gaataaggcc 4260
ggtgtgcgtt tgtctatatg ttattttcca ccatattgcc gtcttttggc aatgtgaggg 4320
cccggaaacc tggccctgtc ttcttgacga gcattcctag gggtctttcc cctctcgcca 4380
aaggaatgca aggtctgttg aatgtcgtga aggaagcagt tcctctggaa gcttcttgaa 4440
gacaaacaac gtctgtagcg accctttgca ggcagcggaa ccccccacct ggcgacaggt 4500
gcctctgcgg ccaaaagcca cgtgtataag atacacctgc aaaggcggca caaccccagt 4560
gccacgttgt gagttggata gttgtggaaa gagtcaaatg gctctcctca agcgtattca 4620
acaaggggct gaaggatgcc cagaaggtac cccattgtat gggatctgat ctggggcctc 4680
ggtgcacatg ctttacgtgt gtttagtcga ggttaaaaaa cgtctaggcc ccccgaacca 4740
cggggacgtg gttttccttt gaaaaacacg atgatatcga attcctgcag cccgggggat 4800
ccgccccctc tgaccaccat gccacctcct cgcctcctct tcttcctcct cttcctcacc 4860
cccatggaag tcaggcccga ggaacctcta gtggtgaagg tggaagaggg agataacgct 4920
gtgctgcagt gcctcaaggg gacctcagat ggccccactc agcagctgac ctggtctcgg 4980
79
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
gagtccccgc ttaaaccctt cttaaaactc agcctggggc tgccaggcct gggaatccac 5040
atgaggcccc tggcatcctg gcttttcatc ttcaacgtct ctcaacagat ggggggcttc 5100
tacctgtgcc agccggggcc cccctctgag aaggcctggc agcctggctg gacagtcaat 5160
gtggagggca gcggggagct gttccggtgg aatgtttcgg acctaggtgg cctgggctgt 5220
ggcctgaaga acaggtcctc agagggcccc agctcccctt ccgggaagct catgagcccc 5280
aagctgtatg tgtgggccaa agaccgccct gagatctggg agggagagcc tccgtgtgtc 5340
ccaccgaggg acagcctgaa ccagagcctc agccaggacc tcaccatggc ccctggctcc 5400
acactctggc tgtcctgtgg ggtaccccct gactctgtgt ccaggggccc cctctcctgg 5460
acccatgtgc accccaaggg gcctaagtca ttgctgagcc tagagctgaa ggacgatcgc 5520
ccggccagag atatgtgggt aatggagacg ggtctgttgt tgccccgggc cacagctcaa 5580
gacgctggaa agtattattg tcaccgtggc aacctgacca tgtcattcca cctggagatc 5640
actgctcggc cagtactatg gcactggctg ctgaggactg gtggctggaa ggtctcagct 5700
gtgactttgg cttatctgat cttctgcctg tgttcccttg tgggcattct tcatctttaa 5760
ggcgcgcccc gggatccaag cttcaattgt ggtcactcga caatcaacct ctggattaca 5820
aaatttgtga aagattgact ggtattctta actatgttgc tccttttacg ctatgtggat 5880
acgctgcttt aatgcctttg tatcatgcta ttgcttcccg tatggctttc attttctcct 5940
ccttgtataa atcctggttg ctgtctcttt atgaggagtt gtggcccgtt gtcaggcaac 6000
gtggcgtggt gtgcactgtg tttgctgacg caacccccac tggttggggc attgccacca 6060
cctgtcagct cctcccggg actttcgctt tccccctccc tattgccacg gcggaactca 6120
tcgccgcctg ccttgcccgc tgctggacag gggctcggct gttgggcact gacaattccg 6180
tggtgttgtc ggggaagctg acgtcctttc catggctgct cgcctgtgtt gccacctgga 6240
ttctgcgcgg gacgtccttc tgctacgtcc cttcggccct caatccagcg gaccttcctt 6300
cccgcggcct gctgccggct ctgcggcctc ttccgcgtct tcgccttcgc cctcagacga 6360
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
gtcggatctc cctttgggcc gcctccccgc ctgtctcgag acctagaaaa acatggagca 6420
atcacaagta gcaatacagc agctaccaat gctgattgtg cctggctaga agcacaagag 6480
gaggaggagg tgggttttcc agtcacacct caggtacctt taagaccaat gacttacaag 6540
gcagatctta gccacttttt aaaagaaaag gggggactgg aagggctaat tcactcccaa 6600
cgaagacaag atctgctttt tgcttgtact gggtctctct ggttagacca gatctgagcc 6660
tgggagctct ctggctaact agggaaccca ctgcttaagc ctcaataaag cttgccttga 6720
gtgcttcaag tagtgtgtgc ccgtctgttg tgtgactctg gtaactagag atccctcaga 6780
cccttttagt cagtgtggaa aatctctagc a 6811
SEQ ID NO:14
<210> 14
<211> 6805
<212> DNA
<213> Vector
<400> 14
tggaagggct aattcactcc caacgaagac aagatatcct tgatctgtgg atctaccaca 60
cacaaggcta cttccctgat tggcagaact acacaccagg accagggatc agatatccac 120
tgacctttgg atggtgctac aagctagtac cagttgagcc agataaggta gaagaggcca 180
acaaaggaga gaacaccagc ttgttacacc ctgtgagcct gcatggaatg gatgacccgg 240
agagagaagt gttagagtgg aggtttgaca gccgcctagc atttcatcac gtggcccgag 300
agctgcatcc ggagtacttc aagaactgct gatatcgagc ttgctacaag ggactttccg 360
ctggggactt tccagggagg cgtggcctgg gcgggactgg ggagtggcga gccctcagat 420
gctgcatata agcagctgct ttttgcctgt actgggtctc tctggttaga ccagatctga 480
gcctgggagc tctctggcta actagggaac ccactgctta agcctcaata aagcttgcct 540
tgagtgcttc aagtagtgtg tgcccgtctg ttgtgtgact ctggtaacta gagatccctc 600
agaccctttt agtcagtgtg gaaaatctct agcagtggcg cccgaacagg gacttgaaag 660
81
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
cgaaagggaa accagaggag ctctctcgac gcaggactcg gcttgctgaa gcgcgcacgg 720
caagaggcga ggggcggcga ctggtgagta cgccaaaaat tttgactagc ggaggctaga 780
aggagagaga tgggtgcgag agcgtcagta ttaagcgggg gagaattaga tcgcgatggg 840
aaaaaattcg gttaaggcca gggggaaaga aaaaatataa attaaaacat atagtatggg 900
caagcaggga gctagaacga ttcgcagtta atcctggcct gcagaaaca tcagaaggct 960
gtagacaaat actgggacag ctacaaccat cccttcagac aggatcagaa gaacttagat 1020
cattatataa tacagtagca accctctatt gtgtgcatca aaggatagag ataaaagaca 1080
ccaaggaagc tttagacaag atagaggaag agcaaaacaa aagtaagacc accgcacagc 1140
aagcggccgc tgatcttcag acctggagga ggagatatga gggacaattg gagaagtgaa 1200
ttatataaat ataaagtagt aaaaattgaa ccattaggag tagcacccac caaggcaaag 1260
agaagagtgg tgcagagaga aaaaagagca gtgggaatag gagctttgtt ccttgggttc 1320
ttgggagcag caggaagcac tatgggcgca gcgtcaatga cgctgacggt acaggccaga 1380
caattattgt ctggtatagt gcagcagcag aacaatttgc tgagggctat tgaggcgcaa 1440
cagcatctgt tgcaactcac agtctggggc atcaagcagc tccaggcaag aatcctggct 1500
gtggaaagat acctaaagga tcaacagctc ctggggattt ggggttgctc tggaaaactc 1560
atttgcacca ctgctgtgcc ttggaatgct agttggagta ataaatctct ggaacagatt 1620
tggaatcaca cgacctggat ggagtgggac agagaaatta acaattacac aagcttaata 1680
cactccttaa ttgaagaatc gcaaaaccag caagaaaaga atgaacaaga attattggaa 1740
tagataaat gggcaagttt gtggaattgg tttaacataa caaattggct gtggtatata 1800
aaattattca taatgatagt aggaggcttg gtaggtttaa gaatagtttt tgctgtactt 1860
tctatagtga atagagttag gcagggatat tcaccattat cgtttcagac ccacctccca 1920
accccgaggg gacccgacag gcccgaagga atagaagaag aaggtggaga gagagacaga
1980
gacagatcca ttcgattagt gaacggatct cgacggtatc gcttttaaaa gaaaaggggg 2040
82
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
gattgggggg tacagtgcag gggaaagaat agtagacata atagcaacag acatacaaac 2100
taaagaatta caaaaacaaa ttacaaaaat tcaaaatttt atcgataagc tttgcaaaga 2160
tggataaagt tttaaacaga gaggaatctt tgcagctaat ggaccttcta ggtcttgaaa 2220
ggagtgggaa ttggctccgg tgcccgtcag tgggcagagc gcacatcgcc cacagtcccc 2280
gagaagttgg ggggaggggt cggcaattga accggtgcct agagaaggtg gcgcggggta 2340
aactgggaaa gtgatgtcgt gtactggctc cgcctttttc ccgagggtgg gggagaaccg 2400
tatataagtg cagtagtcgc cgtgaacgtt ctttttcgca acgggtttgc cgccagaaca 2460
caggtaagtg ccgtgtgtgg ttcccgcggg cctggcctct ttacgggtta tggcccttgc 2520
gtgccttgaa ttacttccac gcccctggct gcagtacgtg attcttgatc ccgagcttcg 2580
ggttggaagt gggtgggaga gttcgaggcc ttgcgcttaa ggagcccctt cgcctcgtgc 2640
ttgagttgag gcctggcctg ggcgctgggg ccgccgcgtg cgaatctggt ggcaccttcg 2700
cgcctgtctc gctgctttcg ataagtctct agccatttaa aatttttgat gacctgctgc 2760
gacgcttttt ttctggcaag atagtcttgt aaatgcgggc caagatctgc acactggtat 2820
ttcggttttt ggggccgcgg gcggcgacgg ggcccgtgcg tcccagcgca catgttcggc 2880
gaggcggggc ctgcgagcgc ggccaccgag aatcggacgg gggtagtctc aagctggccg 2940
gcctgctctg gtgcctggcc tcgcgccgcc gtgtatcgcc ccgccctggg cggcaaggct 3000
ggcccggtcg gcaccagttg cgtgagcgga aagatggccg cttcccggcc ctgctgcagg 3060
gagctcaaaa tggaggacgc ggcgctcggg agagcgggcg ggtgagtcac ccacacaaag 3120
gaaaagggcc tttccgtcct cagccgtcgc ttcatgtgac tccacggagt accgggcgcc 3180
gtccaggcac ctcgattagt tctcgagctt ttggagtacg tcgtctttag gttgggggga 3240
ggggttttat gcgatggagt ttccccacac tgagtgggtg gagactgaag ttaggccagc 3300
ttggcacttg atgtaattct ccttggaatt tgcccttttt gagtttggat cttggttcat 3360
tctcaagcct cagacagtgg ttcaaagttt ttttcttcca tttcaggtgt cgtgagagga 3420
83
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
attctgcagt cgagcggagc gcgcgtaata cgactcacta tagggcgcca tgggtaccgg 3480
gccccccctc gatcgaacaa caacaacaat aacacatggt tccgcgtggc tctcatatgg 3540
cggcccggcg cggggctctc atagtgctgg agggcgtgga ccgcgccggg aagagcacgc 3600
agagccgcaa gctggtggaa gcgctgtgcg ccgcgggcca ccgcgccgaa ctgctccggt 3660
tcccggaaag atcaactgaa atcggcaaac ttctgagttc ctacttgcaa aagaaaagtg 3720
acgtggagga tcactcggtg cacctgcttt tttctgcaaa tcgctgggaa caagtgccgt 3780
taattaagga aaagttgagc cagggcgtga ccctcgtcgt ggacagatac gcattttctg 3840
gtgtggccta cacaggtgcc aaggagaatt tttccctaga ctggtgtaaa cagccagacg 3900
tgggccttcc caaacccgac ctggtcctgt tcctccagtt acagctggcg gatgctgcca 3960
agcggggagc gtttggccat gagcgctatg agaacggggc tttccaggag cgggcgctcc 4020
ggtgtttcca ccagctcatg aaagacacga ctttgaactg gaagatggtg gatgcttcca 4080
aaagcatcga agctgtccat gaggacatcc gcgtgctctc tgaggacgcc atcgccactg 4140
ccacagagaa gccgctgggg gagctatgga agtgaggatc agtcgacggt atcgattccc 4200
cctctccctc ccccccccct aacgttactg gccgaagccg cttggaataa ggccggtgtg 4260
cgtttgtcta tatgttattt tccaccatat tgccgtcttt tggcaatgtg agggcccgga 4320
aacctggccc tgtcttcttg acgagcattc ctaggggtct ttcccctctc gccaaaggaa 4380
tgcaaggtct gttgaatgtc gtgaaggaag cagttcctct ggaagcttct tgaagacaaa 4440
caacgtctgt agcgaccctt tgcaggcagc ggaacccccc acctggcgac aggtgcctct 4500
gcggccaaaa gccacgtgta taagatacac ctgcaaaggc ggcacaaccc cagtgccacg 4560
ttgtgagttg gatagttgtg gaaagagtca aatggctctc ctcaagcgta ttcaacaagg 4620
ggctgaagga tgcccagaag gtaccccatt gtatgggatc tgatctgggg cctcggtgca 4680
catgctttac gtgtgtttag tcgaggttaa aaaacgtcta ggccccccga accacgggga 4740
cgtggttttc ctttgaaaaa cacgatgata tcgaattcct gcagcccggg ggatccgccc 4800
84
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
cctctgacca ccatgccacc tcctcgcctc ctcttcttcc tcctcttcct cacccccatg 4860
gaagtcaggc ccgaggaacc tctagtggtg aaggtggaag agggagataa cgctgtgctg 4920
cagtgcctca aggggacctc agatggcccc actcagcagc tgacctggtc tcgggagtcc 4980
ccgcttaaac ccttcttaaa actcagcctg gggctgccag gcctgggaat ccacatgagg 5040
cccctggcat cctggctttt catcttcaac gtctctcaac agatgggggg cttctacctg 5100
tgccagccgg ggcccccctc tgagaaggcc tggcagcctg gctggacagt caatgtggag 5160
ggcagcgggg agctgttccg gtggaatgtt tcggacctag gtggcctggg ctgtggcctg 5220
aagaacaggt cctcagaggg ccccagctcc ccttccggga agctcatgag ccccaagctg 5280
tatgtgtggg ccaaagaccg ccctgagatc tgggagggag agcctccgtg tgtcccaccg 5340
agggacagcc tgaaccagag cctcagccag gacctcacca tggcccctgg ctccacactc 5400
tggctgtcct gtggggtacc ccctgactct gtgtccaggg gccccctctc ctggacccat 5460
gtgcacccca aggggcctaa gtcattgctg agcctagagc tgaaggacga tcgcccggcc 5520
agagatatgt gggtaatgga gacgggtctg ttgttgcccc gggccacagc tcaagacgct 5580
ggaaagtatt attgtcaccg tggcaacctg accatgtcat tccacctgga gatcactgct 5640
cggccagtac tatggcactg gctgctgagg actggtggct ggaaggtctc agctgtgact 5700
ttggcttatc tgatcttctg cctgtgttcc cttgtgggca ttcttcatct ttaaggcgcg 5760
ccccgggatc caagcttcaa ttgtggtcac tcgacaatca acctctggat tacaaaattt 5820
gtgaaagatt gactggtatt cttaactatg ttgctccttt tacgctatgt ggatacgctg 5880
ctttaatgcc tttgtatcat gctattgctt cccgtatggc tttcattttc tcctccttgt 5940
ataaatcctg gttgctgtct ctttatgagg agttgtggcc cgttgtcagg caacgtggcg 6000
tggtgtgcac tgtgtttgct gacgcaaccc ccactggttg gggcattgcc accacctgtc 6060
agctcctttc cgggactttc gctttccccc tccctattgc cacggcggaa ctcatcgccg 6120
cctgccttgc ccgctgctgg acaggggctc ggctgttggg cactgacaat tccgtggtgt 6180
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
tgtcggggaa gctgacgtcc tttccatggc tgctcgcctg tgttgccacc tggattctgc 6240
gcgggacgtc cttctgctac gtcccttcgg ccctcaatcc agcggacctt ccttcccgcg 6300
gcctgctgcc ggctctgcgg cctcttccgc gtcttcgcct tcgccctcag acgagtcgga 6360
tctccctttg ggccgcctcc ccgcctgtct cgagacctag aaaaacatgg agcaatcaca 6420
agtagcaata cagcagctac caatgctgat tgtgcctggc tagaagcaca agaggaggag 6480
gaggtgggtt ttccagtcac acctcaggta cctttaagac caatgactta caaggcagat 6540
cttagccact ttttaaaaga aaagggggga ctggaagggc taattcactc ccaacgaaga 6600
caagatctgc tttttgcttg tactgggtct ctctggttag accagatctg agcctgggag 6660
ctctctggct aactagggaa cccactgctt aagcctcaat aaagcttgcc ttgagtgctt 6720
caagtagtgt gtgcccgtct gttgtgtgac tctggtaact agagatccct cagacccttt 6780
tagtcagtgt ggaaaatctc tagca 6805
SEQ ID NO:15
<210> 15
<211> 638
<212> DNA
<213> Homo sapiens
<400> 15
tggcggcccg gcgcggggct ctcatagtgc tggagggcgt ggaccgcgcc gggaagagca 60
cgcagagccg caagctggtg gaagcgctgt gcgccgcggg ccaccgcgcc gaactgctcc 120
ggttcccgga aagatcaact gaaatcggca aacttctgag ttcctacttg caaaagaaaa 180
gtgacgtgga ggatcactcg gtgcacctgc ttttttctgc aaatcgctgg gaacaagtgc 240
cgttaattaa ggaaaagttg agccagggcg tgaccctcgt cgtggacaga tacgcatttt 300
ctggtgtggc cttcacaggt gccaaggaga atttttccct agactggtgt aaacagccag 360
acgtgggcct tcccaaaccc gacctggtcc tgttcctcca gttacagctg gcggatgctg 420
ccaagcgggg agcgtttggc catgagcgct atgagaacgg ggctttccag gagcgggcgc 480
86
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
tccggtgttt ccaccagctc atgaaagaca cgactttgaa ctggaagatg gtggatgctt 540
ccaaaagcat cgaagctgtc catgaggaca tccgcgtgct ctctgaggac gccatccgca 600
ctgccacaga gaagccgctg ggggagctat ggaagtga 638
SEQ ID NO:16
<210> 16
<211> 211
<212> PRT
<213> Homo sapiens
<400> 16
Met Ala Ala Arg Arg Gly Ala Leu Ile Val Leu Glu Gly Val Asp Arg
1 5 10 15
Ala Gly Lys Ser Thr Gin Ser Arg Lys Leu Val Glu Ala Leu Cys Ala
20 25 30
Ala Gly His Arg Ala Glu Leu Leu Arg Phe Pro Glu Arg Ser Thr Glu
35 40 45
Ile Gly Lys Leu Leu Ser Ser Tyr Gin Lys Lys Ser Asp Val Glu Asp
50 55 60
His Ser Val His Leu Leu Phe Ser Ala Asn Arg Trp Glu Gin Val Pro
65 70 75 80
Leu Ile Lys Glu Lys Leu Ser Gin Gly Val Thr Leu Val Val Asp Arg
85 90 95
Tyr Ala Phe Ser Gly Val Ala Phe Thr Gly Ala Lys Glu Asn Phe Ser
100 105 110
Leu Asp Trp Cys Lys Gin Pro Asp Val Gly Leu Pro Lys Pro Asp Leu
115 120 125
Val Leu Phe Leu Gin Leu Gin Leu Ala Asp Ala Ala Lys Arg Gly Ala
130 135 140
Phe Gly His Glu Arg Tyr Glu Asn Gly Ala Phe Gin Glu Arg Ala Leu
145 150 155 160
Arg Cys Phe His Gin Leu Met Lys Asp Thr Thr Leu Asn Trp Lys Met
87
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
165 170 175
Val Asp Ala Ser Lys Ser Ile Glu Ala Val His Glu Asp Ile Arg Val
180 185 190
Leu Ser Glu Asp Ala Ile Ala Thr Ala Thr Glu Lys Pro Leu Gly Glu
195 200 205
Leu Trp Lys
210
SEQ ID NO:17
Thr Pro Glu Val Gly Leu Lys Arg Ala Arg Ala Arg Gly Glu Leu
1 5 10 15
SEQ ID NO:18
ttttaaaaga aaagggggga ttggggggta cagtgcaggg gaaagaatag tagacataat 60
agcaacagac atacaaacta aagaattaca aaaacaaatt acaaaaattc aaaatttt 118
SEQ ID NO:19
<213> Woodchuck Hepatitus Virus
aatcaacctc tggattacaa aatttgtgaa agattgactg gtattcttaa ctatgttgct 60
ccttttacgc tatgtggata cgctgcttta atgcctttgt atcatgctat tgcttcccgt 120
atggctttca ttttctcctc cttgtataaa tcctggttgc tgtctcttta tgaggagttg 180
tggcccgttg tcaggcaacg tggcgtggtg tgcactgtgt ttgctgacgc aacccccact 240
ggttggggca ttgccaccac ctgtcagctc ctttccggga ctttcgcttt ccccctccct 300
attgccacgg cggaactcat cgccgcctgc cttgcccgct gctggacagg ggctcggctg 360
ttgggcactg acaattccgt ggtgttgtcg gggaagctga cgtcctttcc atggctgctc 420
gcctgtgttg ccacctggat tctgcgcggg acgtccttct gctacgtccc ttcggccctc 480
aatccagcgg accttccttc ccgcggcctg ctgccggctc tgcggcctct tccgcgtctt 540
cgccttcgcc ctcagacgag tcggatctcc ctttgggccg cctccccgcc tg 592
SEQ ID NO:20
88
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
Gln Leu Ala Asp Ala Ala Lys Arg Gly Ala Phe Gly His
1 5 10
SEQ ID NO:21
atggcggccc ggcgcggggc tctcatagtg ctggagggcg tggaccgcgc cgggaagagc 60
acgcagagcc gcaagctggt ggaagcgctg tgcgccgcgg gccaccgcgc cgaactgctc 120
cggttcccgg aaagatcaac tgaaatcggc aaacttctga gttcctactt gcaaaagaaa 180
agtgacgtgg aggatcactc ggtgcacctg cttttttctg caaatcgctg ggaacaagtg 240
ccgttaatta aggaaaagtt gagccagggc gtgaccctcg tcgtggacag atacgcattt 300
tctggtgtgg cctacacagg tgccaaggag aatttttccc tagactggtg taaacagcca 360
gacgtgggcc ttcccaaacc cgacctggtc ctgttcctcc agttacagct ggcggatgct 420
gccaagcggg gagcgtttgg ccatgagcgc tatgagaacg gggctttcca ggagcgggcg 480
ctccggtgtt tccaccagct catgaaagac acgactttga actggaagat ggtggatgct 540
tccaaaagca tcgaagctgt ccatgaggac atccgcgtgc tctctgagga cgccatcgcc 600
actgccacag agaagccgct gggggagcta tggaagtga 639
SEQ ID NO:22
atggcggccc ggcgcggggc tctcatagtg ctggagggcg tggacggcgc cgggaagagc 60
acgcagagcc gcaagctggt ggaagcgctg tgcgccgcgg gccaccgcgc cgaactgctc 120
cggttcccgg aaagatcaac tgaaatcggc aaacttctga gttcctactt gcaaaagaaa 180
agtgacgtgg aggatcactc ggtgcacctg cttttttctg caaatcgctg ggaacaagtg 240
ccgttaatta aggaaaagtt gagccagggc gtgaccctcg tcgtggacag atacgcattt 300
tctggtgtgg ccttcaccgg tgccaaggag aatttttccc tagactggtg taaacagcca 360
gacgtgggcc ttcccaaacc cgacctggtc ctgttcctgc agttaactcc ggaagttggc 420
ttaaaacgcg cacgtgctcg cggcgagctt gaccgctatg agaacggggc tttccaggag 480
cgggcgctcc ggtgtttcca ccagctcatg aaagacacga ctttgaactg gaagatggtg 540
89
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
gatgcttcca aaagcatcga agctgtccat gaggacatcc gcgtgctctc tgaggacgcc 600
atcgccactg ccacagagaa gccgctgggg gagctatgga agtga 645
SEQ ID NO:23
<210> 23
<211> 28
<212> DNA
<213> Homo sapiens
<400> 23
atgccacctc ctcgcctcct cttcttcc 28
SEQ ID NO:24
<210> 24
<211> 22
<212> DNA
<213> Homo sapiens
<400> 24
tcacctggtg ctccaggtgc cc 22
SEQ ID NO:25
<210> 25
<211> 23
<212> DNA
<213> Homo sapiens
<400> 25
ccgccaccgc ggtggagctc cag 23
SEQ ID NO:26
<210> 26
<211> 26
<212> DNA
<213> Homo sapiens
<400> 26
ttaaagatga agaatgccca caaggg 26
SEQ ID NO:27
<210> 27
<211> 1966
<212> DNA
<213> Homo sapiens
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
<400> 27
aggcccctgc ctgccccagc atcccctgcg cgaagctggg tgccccggag agtctgacca 60
ccatgccacc tcctcgcctc ctcttcttcc tcctcttcct cacccccatg gaagtcaggc 120
ccgaggaacc tctagtggtg aaggtggaag agggagataa cgctgtgctg cagtgcctca 180
aggggacctc agatggcccc actcagcagc tgacctggtc tcgggagtcc ccgcttaaac 240
ccttcttaaa actcagcctg gggctgccag gcctgggaat ccacatgagg cccctggcca 300
tctggctttt catcttcaac gtctctcaac agatgggggg cttctacctg tgccagccgg 360
ggcccccctc tgagaaggcc tggcagcctg gctggacagt caatgtggag ggcagcgggg 420
agctgttccg gtggaatgtt tcggacctag gtggcctggg ctgtggcctg aagaacaggt 480
cctcagaggg ccccagctcc ccttccggga agctcatgag ccccaagctg tatgtgtggg 540
ccaaagaccg ccctgagatc tgggagggag agcctccgtg tctcccaccg agggacagcc 600
tgaaccagag cctcagccag gacctcacca tggcccctgg ctccacactc tggctgtcct 660
gtggggtacc ccctgactct gtgtccaggg gccccctctc ctggacccat gtgcacccca 720
aggggcctaa gtcattgctg agcctagagc tgaaggacga tcgcccggcc agagatatgt 780
gggtaatgga gacgggtctg ttgttgcccc gggccacagc tcaagacgct ggaaagtatt 840
attgtcaccg tggcaacctg accatgtcat tccacctgga gatcactgct cggccagtac 900
tatggcactg gctgctgagg actggtggct ggaaggtctc agctgtgact ttggcttatc 960
tgatcttctg cctgtgttcc cttgtgggca ttcttcatct tcaaagagcc ctggtcctga 1020
ggaggaaaag aaagcgaatg actgacccca ccaggagatt cttcaaagtg acgcctcccc 1080
caggaagcgg gccccagaac cagtacggga acgtgctgtc tctccccaca cccacctcag 1140
gcctcggacg cgcccagcgt tgggccgcag gcctgggggg cactgccccg tcttatggaa 1200
acccgagcag cgacgtccag gcggatggag ccttggggtc ccggagcccg ccgggagtgg 1260
gcccagaaga agaggaaggg gagggctatg aggaacctga cagtgaggag gactccgagt 1320
91
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
tctatgagaa cgactccaac cttgggcagg accagctctc ccaggatggc agcggctacg 1380
agaaccctga ggatgagccc ctgggtcctg aggatgaaga ctccttctcc aacgctgagt 1440
cttatgagaa cgaggatgaa gagctgaccc agccggtcgc caggacaatg gacttcctga 1500
gccctcatgg gtcagcctgg gaccccagcc gggaagcaac ctccctgggg tcccagtcct 1560
atgaggatat gagaggaatc ctgtatgcag ccccccagct ccgctccatt cggggccagc 1620
ctggacccaa tcatgaggaa gatgcagact cttatgagaa catggataat cccgatgggc 1680
cagacccagc ctggggagga gggggccgca tgggcacctg gagcaccagg tgatcctcag 1740
gtggccagcc tggatctcct caagtcccca agattcacac ctgactctga aatctgaaga 1800
cctcgagcag atgatgccaa cctctggagc aatgttgctt aggatgtgtg catgtgtgta 1860
agtgtgtgtg tgtgtgtgtg tgtgtataca tgccagtgac acttccagtc ccctttgtat 1920
tccttaaata aactcaatga gctcttccaa aaaaaaaaaa aaaaaa 1966
SEQ ID NO:28
<210> 28
<211> 556
<212> PRT
<213> Homo sapiens
<400> 28
Met Pro Pro Pro Arg Leu Leu Phe Phe Leu Leu Phe Leu Thr Pro Met
1 5 10 15
Glu Val Arg Pro Glu Glu Pro Leu Val Val Lys Val Glu Glu Gly Asp
20 25 30
Asn Ala Val Leu Gin Cys Leu Lys Gly Thr Ser Asp Gly Pro Thr Gin
35 40 45
Gin Leu Thr Trp Ser Arg Glu Ser Pro Leu Lys Pro Phe Leu Lys Leu
50 55 60
Ser Leu Gly Leu Pro Gly Leu Gly Ile His Met Arg Pro Leu Ala Ile
65 70 75 80
Trp Leu Phe Ile Phe Asn Val Ser Gin Gin Met Gly Gly Phe Tyr Leu
92
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
85 90 95
Cys Gin Pro Gly Pro Pro Ser Glu Lys Ala Trp Gin Pro Gly Trp Thr
100 105 110
Val Asn Val Glu Gly Ser Gly Glu Leu Phe Arg Trp Asn Val Ser Asp
115 120 125
Leu Gly Gly Leu Gly Cys Gly Leu Lys Asn Arg Ser Ser Glu Gly Pro
130 135 140
Ser Ser Pro Ser Gly Lys Leu Met Ser Pro Lys Leu Tyr Val Trp Ala
145 150 155 160
Lys Asp Arg Pro Glu Ile Trp Glu Gly Glu Pro Pro Cys Leu Pro Pro
165 170 175
Arg Asp Ser Leu Asn Gin Ser Leu Ser Gin Asp Leu Thr Met Ala Pro
180 185 190
Gly Ser Thr Leu Trp Leu Ser Cys Gly Val Pro Pro Asp Ser Val Ser
195 200 205
Arg Gly Pro Leu Ser Trp Thr His Val His Pro Lys Gly Pro Lys Ser
210 215 220
Leu Leu Ser Leu Glu Leu Lys Asp Asp Arg Pro Ala Arg Asp Met Trp
225 230 235 240
Val Met Glu Thr Gly Leu Leu Leu Pro Arg Ala Thr Ala Gin Asp Ala
245 250 255
Gly Lys Tyr Tyr Cys His Arg Gly Asn Leu Thr Met Ser Phe His Leu
260 265 270
Glu Ile Thr Ala Arg Pro Val Leu Trp His Trp Leu Leu Arg Thr Gly
275 280 285
Gly Trp Lys Val Ser Ala Val Thr Leu Ala Tyr Leu Ile Phe Cys Leu
290 295 300
Cys Ser Leu Val Gly Ile Leu His Leu Gin Arg Ala Leu Val Leu Arg
305 310 315 320
93
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
Arg Lys Arg Lys Arg Met Thr Asp Pro Thr Arg Arg Phe Phe Lys Val
325 330 335
Thr Pro Pro Pro Gly Ser Gly Pro Gin Asn Gin Tyr Gly Asn Val Leu
340 345 350
Ser Leu Pro Thr Pro Thr Ser Gly Leu Gly Arg Ala Gin Arg Trp Ala
355 360 365
Ala Gly Leu Gly Gly Thr Ala Pro Ser Tyr Gly Asn Pro Ser Ser Asp
370 375 380
Val Gin Ala Asp Gly Ala Leu Gly Ser Arg Ser Pro Pro Gly Val Gly
385 390 395 400
Pro Glu Glu Glu Glu Gly Glu Gly Tyr Glu Glu Pro Asp Ser Glu Glu
405 410 415
Asp Ser Glu Phe Tyr Glu Asn Asp Ser Asn Leu Gly Gin Asp Gin Leu
420 425 430
Ser Gin Asp Gly Ser Gly Tyr Glu Asn Pro Glu Asp Glu Pro Leu Gly
435 440 445
Pro Glu Asp Glu Asp Ser Phe Ser Asn Ala Glu Ser Tyr Glu Asn Glu
450 455 460
Asp Glu Glu Leu Thr Gin Pro Val Ala Arg Thr Met Asp Phe Leu Ser
465 470 475 480
Pro His Gly Ser Ala Trp Asp Pro Ser Arg Glu Ala Thr Ser Leu Gly
485 490 495
Ser Gin Ser Tyr Glu Asp Met Arg Gly Ile Leu Tyr Ala Ala Pro Gin
500 505 510
Leu Arg Ser lie Arg Gly Gin Pro Gly Pro Asn His Glu Glu Asp Ala
515 520 525
Asp Ser Tyr Glu Asn Met Asp Asn Pro Asp Gly Pro Asp Pro Ala Trp
530 535 540
Gly Gly Gly Gly Arg Met Gly Thr Trp Ser Thr Arg
545 550 555
94
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
SEQ ID NO:29
<210> 29
<211> 313
<212> PRT
<213> Homo sapiens
<400> 29
Met Pro Pro Pro Arg Leu Leu Phe Phe Leu Leu Phe Leu Thr Pro Met
1 5 10 15
Glu Val Arg Pro Glu Glu Pro Leu Val Val Lys Val Glu Glu Gly Asp
20 25 30
Asn Ala Val Leu GIn Cys Leu Lys Gly Thr Ser Asp Gly Pro Thr GIn
35 40 45
GIn Leu Thr Trp Ser Arg Glu Ser Pro Leu Lys Pro Phe Leu Lys Leu
50 55 60
Ser Leu Gly Leu Pro Gly Leu Gly Ile His Met Arg Pro Leu Ala Ile
65 70 75 80
Trp Leu Phe Ile Phe Asn Val Ser GIn GIn Met Gly Gly Phe Tyr Leu
85 90 95
Cys Gln Pro Gly Pro Pro Ser Glu Lys Ala Trp GIn Pro Gly Trp Thr
100 105 110
Val Asn Val Glu Gly Ser Gly Glu Leu Phe Arg Trp Asn Val Ser Asp
115 120 125
Leu Gly Gly Leu Gly Cys Gly Leu Lys Asn Arg Ser Ser Glu Gly Pro
130 135 140
Ser Ser Pro Ser Gly Lys Leu Met Ser Pro Lys Leu Tyr Val Trp Ala
145 150 155 160
Lys Asp Arg Pro Glu Ile Trp Glu Gly Glu Pro Pro Cys Leu Pro Pro
165 170 175
Arg Asp Ser Leu Asn GIn Ser Leu Ser Gln Asp Leu Thr Met Ala Pro
180 185 190
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
Gly Ser Thr Leu Trp Leu Ser Cys Gly Val Pro Pro Asp Ser Val Ser
195 200 205
Arg Gly Pro Leu Ser Trp Thr His Val His Pro Lys Gly Pro Lys Ser
210 215 220
Leu Leu Ser Leu Glu Leu Lys Asp Asp Arg Pro Ala Arg Asp Met Trp
225 230 235 240
Val Met Glu Thr Gly Leu Leu Leu Pro Arg Ala Thr Ala Gin Asp Ala
245 250 255
Gly Lys Tyr Tyr Cys His Arg Gly Asn Leu Thr Met Ser Phe His Leu
260 265 270
Glu Ile Thr Ala Arg Pro Val Leu Trp His Trp Leu Leu Arg Thr Gly
275 280 285
Gly Trp Lys Val Ser Ala Val Thr Leu Ala Tyr Leu Ile Phe Cys Leu
290 295 300
Cys Ser Leu Val Gly Ile Leu His Leu
305 310
SEQ ID NO:30
<210> 30
<211> 939
<212> DNA
<213> Homo sapiens
<220>
<221> exon
<222> (1)..(939)
<400> 30
atg cca cct cct cgc ctc ctc ttc ftc ctc ctc ftc ctc acc ccc atg 48
Met Pro Pro Pro Arg Leu Leu Phe Phe Leu Leu Phe Leu Thr Pro Met
1 5 10 15
gaa gtc agg ccc gag gaa cct cta gtg gtg aag gtg gaa gag gga gat 96
Glu Val Arg Pro Glu Glu Pro Leu Val Val Lys Val Glu Glu Gly Asp
20 25 30
aac get gtg ctg cag tgc ctc aag ggg acc tca gat ggc ccc act cag 144
Asn Ala Val Leu Gin Cys Leu Lys GIy Thr Ser Asp Gly Pro Thr Gin
35 40 45
96
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
cag ctg acc tgg tct cgg gag tcc ccg ctt aaa ccc ftc tta aaa ctc 192
GIn Leu Thr Trp Ser Arg Glu Ser Pro Leu Lys Pro Phe Leu Lys Leu
50 55 60
agc ctg ggg ctg cca ggc ctg gga atc cac atg agg ccc ctg gcc atc 240
Ser Leu Gly Leu Pro Gly Leu Gly Ile His Met Arg Pro Leu Ala Ile
65 70 75 80
tgg ctt ttc atc ttc aac gtc tct caa cag atg ggg ggc ftc tac ctg 288
Trp Leu Phe Ile Phe Asn Val Ser GIn GIn Met Gly Gly Phe Tyr Leu
85 90 95
tgc cag ccg ggg ccc ccc tct gag aag gcc tgg cag cct ggc tgg aca 336
Cys GIn Pro Gly Pro Pro Ser Glu Lys Ala Trp GIn Pro Gly Trp Thr
100 105 110
gtc aat gtg gag ggc agc ggg gag ctg ftc cgg tgg aat gtt tcg gac 384
Val Asn Val Glu Gly Ser Gly Glu Leu Phe Arg Trp Asn Val Ser Asp
115 120 125
cta ggt ggc ctg ggc tgt ggc ctg aag aac agg tcc tca gag ggc ccc 432
Leu Gly Gly Leu Gly Cys Gly Leu Lys Asn Arg Ser Ser Glu Gly Pro
130 135 140
agc tcc cct tcc ggg aag ctc atg agc ccc aag ctg tat gtg tgg gcc 480
Ser Ser Pro Ser Gly Lys Leu Met Ser Pro Lys Leu Tyr Val Trp Ala
145 150 155 160
aaa gac cgc cct gag atc tgg gag gga gag cct ccg tgt ctc cca ccg 528
Lys Asp Arg Pro Glu Ile Trp Glu Gly Glu Pro Pro Cys Leu Pro Pro
165 170 175
agg gac agc ctg aac cag agc ctc agc cag gac ctc acc atg gcc cct 576
Arg Asp Ser Leu Asn GIn Ser Leu Ser GIn Asp Leu Thr Met Ala Pro
180 185 190
ggc tcc aca ctc tgg ctg tcc tgt ggg gta ccc cct gac tct gtg tcc 624
Gly Ser Thr Leu Trp Leu Ser Cys Gly Val Pro Pro Asp Ser Val Ser
195 200 205
agg ggc ccc ctc tcc tgg acc cat gtg cac ccc aag ggg cct aag tca 672
Arg Gly Pro Leu Ser Trp Thr His Val His Pro Lys Gly Pro Lys Ser
210 215 220
ttg ctg agc cta gag ctg aag gac gat cgc ccg gcc aga gat atg tgg 720
97
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
Leu Leu Ser Leu Glu Leu Lys Asp Asp Arg Pro Ala Arg Asp Met Trp
225 230 235 240
gta atg gag acg ggt ctg ttg ttg ccc cgg gcc aca get caa gac get 768
Val Met Glu Thr Gly Leu Leu Leu Pro Arg Ala Thr Ala Gin Asp Ala
245 250 255
gga aag tat tat tgt cac cgt ggc aac ctg acc atg tca ftc cac ctg 816
Gly Lys Tyr Tyr Cys His Arg Gly Asn Leu Thr Met Ser Phe His Leu
260 265 270
gag atc act get cgg cca gta cta tgg cac tgg ctg ctg agg act ggt 864
Glu lie Thr Ala Arg Pro Val Leu Trp His Trp Leu Leu Arg Thr Gly
275 280 285
ggc tgg aag gtc tca get gtg act ttg get tat ctg atc ftc tgc ctg 912
Gly Trp Lys Val Ser Ala Val Thr Leu Ala Tyr Leu Ile Phe Cys Leu
290 295 300
tgt tcc ctt gtg ggc aft ctt cat ctt 939
Cys Ser Leu Val Gly Ile Leu His Leu
305 310
SEQ ID NO:31
<210> 31
<211> 313
<212> PRT
<213> Homo sapiens
<400> 31
Met Pro Pro Pro Arg Leu Leu Phe Phe Leu Leu Phe Leu Thr Pro Met
1 5 10 15
Glu Val Arg Pro Glu Glu Pro Leu Val Val Lys Val Glu Glu Gly Asp
20 25 30
Asn Ala Val Leu Gin Cys Leu Lys Gly Thr Ser Asp Gly Pro Thr Gin
35 40 45
Gin Leu Thr Trp Ser Arg Glu Ser Pro Leu Lys Pro Phe Leu Lys Leu
50 55 60
Ser Leu Gly Leu Pro Gly Leu Gly Ile His Met Arg Pro Leu Ala Ser
65 70 75 80
98
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
Trp Leu Phe Ile Phe Asn Val Ser Gin Gin Met Gly Gly Phe Tyr Leu
85 90 95
Cys Gin Pro Gly Pro Pro Ser Glu Lys Ala Trp Gin Pro Gly Trp Thr
100 105 110
Val Asn Val Glu Gly Ser Gly Glu Leu Phe Arg Trp Asn Val Ser Asp
115 120 125
Leu Gly Gly Leu Gly Cys Gly Leu Lys Asn Arg Ser Ser Glu Gly Pro
130 135 140
Ser Ser Pro Ser Gly Lys Leu Met Ser Pro Lys Leu Tyr Val Trp Ala
145 150 155 160
Lys Asp Arg Pro Glu Ile Trp Glu Gly Glu Pro Pro Cys Val Pro Pro
165 170 175
Arg Asp Ser Leu Asn Gin Ser Leu Ser Gin Asp Leu Thr Met Ala Pro
180 185 190
Gly Ser Thr Leu Trp Leu Ser Cys Gly Val Pro Pro Asp Ser Val Ser
195 200 205
Arg Gly Pro Leu Ser Trp Thr His Val His Pro Lys Gly Pro Lys Ser
210 215 220
Leu Leu Ser Leu Glu Leu Lys Asp Asp Arg Pro Ala Arg Asp Met Trp
225 230 235 240
Val Met Glu Thr Gly Leu Leu Leu Pro Arg Ala Thr Ala Gin Asp Ala
245 250 255
Gly Lys Tyr Tyr Cys His Arg Gly Asn Leu Thr Met Ser Phe His Leu
260 265 270
Glu Ile Thr Ala Arg Pro Val Leu Trp His Trp Leu Leu Arg Thr Gly
275 280 285
Gly Trp Lys Val Ser Ala Val Thr Leu Ala Tyr Leu Ile Phe Cys Leu
290 295 300
Cys Ser Leu Val Gly Ile Leu His Leu
305 310
SEQ ID NO: 34
99
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
ATGGCGGCCCGGCGCGGGGCTCTCATAGTGCTGGAGGGCGTGGACCGCG
CCGGGAAGAGCACGCAGAGCCGCAAGCTGGTGGAAGCGCTGTGCGCCGC
GGGCCACCGCGCCGAACTGCTCCGGTTCCCGGAAAGATCAACTGAAATCG
GCAAACTTCTGAGTTCCTACTTGCAAAAGAAAAGTGACGTGGAGGATCACTC
GGTGCACCTGCTTTTTTCTGCAAATCGCTGGGAACAAGTGCCGTTAATTAAG
GAAAAGTTGAGCCAGGGCGTGACCCTCGTCGTGGACAGATACGCATTTTCT
GGTGTGGCCTACACAGGTGCCAAGGAGAATTTTTCCCTAGACTGGTGTAAA
CAGCCAGACGTGGGCCTTCCCAAACCCGACCTGGTCCTGTTCCTCCAGTTA
CAGCTGGCGGATGCTGCCAAGCGGGGAGCGTTTGGCCATGAGCGCTATGA
GAACGGGGCTTTCCAGGAGCGGGCGCTCCGGTGTTTCCACCAGCTCATGA
AAGACACGACTTTGAACTGGAAGATGGTGGATGCTTCCAAAAGCATCGAAG
CTGTCCATGAGGACATCCGCGTGCTCTCTGAGGACGCCATCGCCACTGCCA
CAGAGAAGCCGCTGGGGGAGCTATGGAAGTGA
SEQ ID NO: 35
ATGCCACCTCCTCGCCTCCTCTTCTTCCTCCTCTTCCTCACCCCCATGGAAG
TCAGGCCCGAGGAACCTCTAGTGGTGAAGGTGGAAGAGGGAGATAACGCT
GTGCTGCAGTGCCTCAAGGGGACCTCAGATGGCCCCACTCAGCAGCTGAC
CTGGTCTCGGGAGTCCCCGCTTAAACCCTTCTTAAAACTCAGCCTGGGGCT
GCCAGGCCTGGGAATCCACATGAGGCCCCTGGCATCCTGGCTTTTCATCTT
CAACGTCTCTCAACAGATGGGGGGCTTCTACCTGTGCCAGCCGGGGCCCC
CCTCTGAGAAGGCCTGGCAGCCTGGCTGGACAGTCAATGTGGAGGGCAGC
GGGGAGCTGTTCCGGTGGAATGTTTCGGACCTAGGTGGCCTGGGCTGTGG
CCTGAAGAACAGGTCCTCAGAGGGCCCCAGCTCCCCTTCCGGGAAGCTCA
TGAGCCCCAAGCTGTATGTGTGGGCCAAAGACCGCCCTGAGATCTGGGAG
GGAGAGCCTCCGTGTGTCCCACCGAGGGACAGCCTGAACCAGAGCCTCAG
CCAGGACCTCACCATGGCCCCTGGCTCCACACTCTGGCTGTCCTGTGGGG
TACCCCCTGACTCTGTGTCCAGGGGCCCCCTCTCCTGGACCCATGTGCACC
CCAAGGGGCCTAAGTCATTGCTGAGCCTAGAGCTGAAGGACGATCGCCCG
GCCAGAGATATGTGGGTAATGGAGACGGGTCTGTTGTTGCCCCGGGCCAC
AGCTCAAGACGCTGGAAAGTATTATTGTCACCGTGGCAACCTGACCATGTC
ATTCCACCTGGAGATCACTGCTCGGCCAGTACTATGGCACTGGCTGCTGAG
GACTGGTGGCTGGAAGGTCTCAGCTGTGACTTTGGCTTATCTGATCTTCTG
CCTGTGTTCCCTTGTGGGCATTCTTCATCTT
SEQ ID NO:36
MAARRGALIVLEGVDRAGKSTQSRKLVEALCAAGH RAE
LLRFPERSTEIGKLLSSYLQKKSDVEDHSVHLLFSANR
W E Q V P L I KEKLSQGVTLVVDRYAFSGVAYTGAKENFSL
D W C K Q P D V G L P K P D L V L F L Q L Q L A D A A K R G A F G H E R Y
E N G A F Q E R A L R C F H Q L M K D T T L N W K M V D A S K S I EAVH
E D I R V L S E D A I A T A T E K P L G E L W K
SEQ ID NO:37
MPPPRLLFFLLFLTPMEVRPEEPLVVKVEEGDNAVLQC
LKGTSDGPTQQLTWSRESPLKPFLKLSLGLPGLGIHMR
100
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
PLASWLFIFNVSQQMGGFYLCQPGPPSEKAWQPGWTV
NVEGSGELFRWNVSDLGGLGCGLKNRSSEGPSSPSGK
LMSPKLYVWAKDRPEIWEGEPPCVPPRDSLNQSLSQD
LTMAPGSTLWLSCGVPPDSVSRGPLSWTHVHPKGPKS
LLSLELKDDRPARDMWVMETGLLLPRATAQDAGKYYC
HRGNLTMSFHLEITARPVLWHWLLRTGGWKVSAVTLA
YLIFCLCSLVGILHL
SEQ ID NO: 38
MPPPRLLFFLLFLTPMEVRPEEPLVVKVEEGDNAVLQC
LKGTSDGPTQQLTWSRESPLKPFLKLSLGLPGLGIHMR
PLASWLFIFNVSQQMGGFYLCQPGPPSEKAWQPGWTV
NVEGSGELFRWNVSDLGGLGCGLKNRSSEGPSSPSGK
LMSPKLYVWAKDRPEIWEGEPPCVPPRDSLNQSLSQD
LTMAPGSTLWLSCGVPPDSVSRGPLSWTHVHPKGPKS
LLSLELKDDRPARDMWVMETGLLLPRATAQDAGKYYC
H R G N L T M S F H L E I T A R P V L W H W L L R T G G W K V S A V T L A
YLIFCLCSLVGILHLAGGAAGMAARRGALIVLEGVDRAG
KSTQSRKLVEALCAAGHRAELLRFPERSTEIGKLLSSYL
QKKSDVEDHSVHLLFSANRWEQVPLIKEKLSQGVTLVV
D R Y A F S G V A Y T G A K E N F S L D W C K Q P D V G L P K P D L V L F L
QLQLADAAKRGAFGHERYENGAFQERALRCFHQLMKD
TTLNWKMVDASKSIEAVHEDIRVLSEDAIATATEKPLGE
LWK
SEQ ID NO: 39
ATGCCACCTCCTCGCCTCCTCTTCTTCCTCCTCTTCCTCACCCCCATGGAAG
TCAGGCCCGAGGAACCTCTAGTGGTGAAGGTGGAAGAGGGAGATAACGCT
GTGCTGCAGTGCCTCAAGGGGACCTCAGATGGCCCCACTCAGCAGCTGAC
CTGGTCTCGGGAGTCCCCGCTTAAACCCTTCTTAAAACTCAGCCTGGGGCT
GCCAGGCCTGGGAATCCACATGAGGCCC CTGGCATCCTGGCTTTTCATCTT
CAACGTCTCTCAACAGATGGGGGGCTTCTACCTGTGCCAGCCGGGGCCCC
CCTCTGAGAAGGCCTGGCAGCCTGGCTGGACAGTCAATGTGGAGGGCAGC
GGGGAGCTGTTCCGGTGGAATGTTTCGGACCTAGGTGGCCTGGGCTGTGG
CCTGAAGAACAGGTCCTCAGAGGGCCCCAGCTCCCCTTCCGGGAAGCTCA
TGAGCCCCAAGCTGTATGTGTGGGCCAAAGACCGCCCTGAGATCTGGGAG
GGAGAGCCTCCGTGTGTCCCACCGAGGGACAGCCTGAACCAGAGCCTCAG
CCAGGACCTCACCATGGCCCCTGGCTCCACACTCTGGCTGTCCTGTGGGG
TACCCCCTGACTCTGTGTCCAGGGGCCCCCTCTCCTGGACCCATGTGCACC
CCAAGGGGCCTAAGTCATTGCTGAGCCTAGAGCTGAAGGACGATCGCCCG
GCCAGAGATATGTGGGTAATGGAGACGGGTCTGTTGTTGCCCCGGGCCAC
AGCTCAAGACGCTGGAAAGTATTATTGTCACCGTGGCAACCTGACCATGTC
ATTCCACCTGGAGATCACTGCTCGGCCAGTACTATGGCACTGGCTGCTGAG
GACTGGTGGCTGGAAGGTCTCAGCTGTGACTTTGGCTTATCTGATCTTCTG
CCTGTGTTCCCTTGTGGGCATTCTTCATCTTGCCGGCGGGGCTGCAGGGAT
GGCGGCCCGGCGCGGGGCTCTCATAGTGCTGGAGGGCGTGGACCGCGCC
101
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
GGGAAGAGCACGCAGAGCCGCAAGCTGGTGGAAGCGCTGTGCGCCGCGG
GCCACCGCGCCGAACTGCTCCGGTTCCCGGAAAGATCAACTGAAATCGGC
AAACTTCTGAGTTCCTACTTGCAAAAGAAAAGTGACGTGGAGGATCACTCG
GTGCACCTGCTTTTTTCTGCAAATCGCTGGGAACAAGTGCCGTTAATTAAGG
AAAAGTTGAGCCAGGGCGTGACCCTCGTCGTGGACAGATACGCATTTTCTG
GTGTGGCCTACACAGGTGCCAAGGAGAATTTTTCCCTAGACTGGTGTAAAC
AGCCAGACGTGGGCCTTCCCAAACCCGACCTGGTCCTGTTCCTCCAGTTAC
AGCTGGCGGATGCTGCCAAGCGGGGAGCGTTTGGCCATGAGCGCTATGAG
AACGGGGCTTTCCAGGAGCGGGCGCTCCGGTGTTTCCACCAGCTCATGAA
AGACACGACTTTGAACTGGAAGATGGTGGATGCTTCCAAAAGCATCGAAGC
TGTCCATGAGGACATCCGCGTGCTCTCTGAGGACGCCATCGCCACTGCCAC
AGAGAAGCCGCTGGGGGAGCTATGGAAGTGA
References
1. Aiuti, A, Slavin, S, Aker, M, Ficara, F, Deola, S, Mortellaro, A et al.
(2002). Correction of ADA-SCID by stem cell gene therapy combined with
nonmyeloablative conditioning. Science 296: 2410-2413.
2. Gaspar, HB, Parsley, KL, Howe, S, King, D, Gilmour, KC, Sinclair, J et
al. (2004). Gene therapy of X-linked severe combined immunodeficiency by use
of a pseudotyped gammaretroviral vector. Lancet 364: 2181-2187.
3. Robbins, PD and Ghivizzani, SC (1998). Viral vectors for gene therapy.
Pharmacol Ther 80: 35-47.
4. Hacein-Bey-Abina, S, Von Kalle, C, Schmidt, M, McCormack, MP,
Wulffraat, N, Leboulch, P et al. (2003). LMO2-associated clonal T cell
proliferation in two patients after gene therapy for SCID-X1. Science 302: 415-
419.
5. McCormack, MP and Rabbitts, TH (2004). Activation of the T-cell
oncogene LMO2 after gene therapy for X-linked severe combined
immunodeficiency. N Eng/ J Med 350: 913-922.
6. Baum, C, von Kalle, C, Staal, FJ, Li, Z, Fehse, B, Schmidt, M et al.
(2004). Chance or necessity? Insertional mutagenesis in gene therapy and its
consequences. Mol Ther9: 5-13.
7. Brady, RO, Gal, AE, Bradley, RM, Martensson, E, Warshaw, AL and
Laster, L (1967). Enzymatic defect in Fabry's disease. Ceramidetrihexosidase
102
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
deficiency. N Eng! J Med 276: 1163-1167.
8. Kase, R, Shimmoto, M, Itoh, K, Utsumi, K, Kotani, M, Taya, C et al.
(1998). Immunohistochemical characterization of transgenic mice highly
expressing human lysosomal alpha-galactosidase. Biochim Biophys Acta
1406: 260-266.
9. Medin, JA, Tudor, M, Simovitch, R, Quirk, JM, Jacobson, S, Murray, GJ
et al. (1996). Correction in trans for Fabry disease: expression, secretion
and
uptake of alpha-galactosidase A in patient-derived cells driven by a high-
titer
recombinant retroviral vector. Proc Nat! Acad Sci USA 93: 7917-7922.
10. Takenaka, T, Murray, GJ, Qin, G, Quirk, JM, Ohshima, T, Qasba, P et
al. (2000). Long-term enzyme correction and lipid reduction in multiple organs
of primary and secondary transplanted Fabry mice receiving transduced bone
marrow cells. Proc Nat! Acad Sci USA 97: 7515-7520.
11. Yoshimitsu, M, Higuchi, K, Ramsubir, S, Nonaka, T, Rasaiah, VI,
Siatskas, C et al. (2007). Efficient correction of Fabry mice and patient
cells
mediated by lentiviral transduction of hematopoietic stem/progenitor cells.
Gene Ther 14: 256-265.
12. Yoshimitsu, M, Sato, T, Tao, K, Walia, JS, Rasaiah, VI, Sleep, GT et
a!. (2004). Bioluminescent imaging of a marking transgene and correction of
Fabry mice by neonatal injection of recombinant lentiviral vectors. Proc Nat!
Acad Sci USA 101: 16909-16914.
13. Qin, G, Takenaka, T, Telsch, K, Kelley, L, Howard, T, Levade, T et a!.
(2001). Preselective gene therapy for Fabry disease. Proc Natl Acad Sci USA
98: 3428-3433.
14. Taniguchi, T and Minami, Y (1993). The IL-2/IL-2 receptor system: a
current overview. Cell 73: 5-8.
15. Gaffen, SL (2001). Signaling domains of the interleukin 2 receptor.
Cytokine 14: 63-77.
16. Hanisch, UK and Quirion, R (1995). Interleukin-2 as a
neuroregulatory cytokine. Brain Res Brain Res Rev 21: 246-284.
17. David, D, Bani, L, Moreau, JL, Demaison, C, Sun, K, Salvucci, 0 et al.
103
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
(1998). Further analysis of interleukin-2 receptor subunit expression on the
different human peripheral blood mononuclear cell subsets. Blood 91: 165-
172.
18. Rubin, LA, Kurman, CC, Fritz, ME, Biddison, WE, Boutin, B, Yarchoan,
R et al. (1985). Soluble interleukin 2 receptors are released from activated
human lymphoid cells in vitro. J Immunol 135: 3172-3177.
19. Uchiyama, T, Broder, S and Waldmann, TA (1981). A monoclonal
antibody (anti-Tac) reactive with activated and functionally mature human T
cells. I. Production of anti-Tac monoclonal antibody and distribution of Tac
(+)
cells. J Immunol 126:1393-1397.
20. Lappi, DA, Esch, FS, Barbieri, L, Stirpe, F and Soria, M (1985).
Characterization of a Saponaria officinalis seed ribosome-inactivating
protein:
immunoreactivity and sequence homologies. Biochem Biophys Res Commun
129: 934-942.
21. Barbieri, L, Valbonesi, P, Bonora, E, Gorini, P, Bolognesi, A and
Stirpe, F (1997). Polynucleotide:adenosine glycosidase activity of ribosome-
inactivating proteins: effect on DNA, RNA and poly(A). Nucleic Acids Res 25:
518-522.
22. Zhang, Z, Zhang, M, Garmestani, K, Talanov, VS, Plascjak, PS, Beck,
B et al. (2006). Effective treatment of a murine model of adult T-cell
leukemia
using 211At-7G7/B6 and its combination with unmodified anti-Tac (daclizumab)
directed toward CD25. Blood 108: 1007-1012.
23. Perez-Encinas, M, Villamayor, M, Campos, A, Gonzalez, S and Bello,
JL (1998). Tumor burden and serum level of soluble CD25, CD8, CD23, CD54
and CD44 in non-Hodgkin's lymphoma. Haematologica 83: 752-754.
24. Boelens, JJ (2006). Trends in haematopoietic cell transplantation for
inborn errors of metabolism. J Inherit Metab Dis 29: 413-420.
25. Futerman, AH and van Meer, G (2004). The cell biology of lysosomal
storage disorders. Nat Rev Mol Cell Biol 5: 554-565.
26. Dillman, RO (2001). Monoclonal antibody therapy for lymphoma: an
update. Cancer Pract 9: 71-80.
104
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
27. Gokbuget, N and Hoelzer, D (2004). Treatment with monoclonal
antibodies in acute lymphoblastic leukemia: current knowledge and future
prospects. Ann Hematol 83: 201-205.
28. McLaughlin, P, Grillo-Lopez, AJ, Link, BK, Levy, R, Czuczman, MS,
Williams, ME et al. (1998). Rituximab chimeric anti-CD20 monoclonal antibody
therapy for relapsed indolent lymphoma: half of patients respond to a four-
dose
treatment program. J Clin Onco/16: 2825-2833.
29. Grillo-Lopez, AJ, Hedrick, E, Rashford, M and Benyunes, M (2002).
Rituximab: ongoing and future clinical development. Semin Oncol 29: 105-112.
30. Cvetkovic, RS and Perry, CM (2006). Rituximab: a review of its use in
non-Hodgkin's lymphoma and chronic lymphocytic leukaemia. Drugs 66: 791-
820.
31. Introna, M, Barbui, AM, Bambacioni, F, Casati, C, Gaipa, G, Borleri, G
et al. (2000). Genetic modification of human T cells with CD20: a strategy to
purify and lyse transduced cells with anti-CD20 antibodies. Hum Gene Ther 11:
611-620.
32. van Meerten, T, Claessen, MJ, Hagenbeek, A and Ebeling, SB (2006).
The
CD20/alphaCD20 'suicide' system: novel vectors with improved safety and
expression profiles and efficient elimination of CD20-transgenic T cells.
Gene Ther 13: 789-797.
33. Serafini, M, Manganini, M, Borleri, G, Bonamino, M, Imberti, L, Biondi,
A et al. (2004). Characterization of CD20-transduced T lymphocytes as an
alternative suicide gene therapy approach for the treatment of graft-versus-
host
disease. Hum Gene Ther 15: 63-76.
34. Kreitman, RJ, Wilson, WH, White, JD, Stetler-Stevenson, M, Jaffe, ES,
Giardina, S et al. (2000). Phase I trial of recombinant immunotoxin anti-
Tac(Fv)-
PE38 (LMB-2) in patients with hematologic malignancies. J Clin Onco! 18:
1622-1636.
35. Kreitman, RJ, Wilson, WH, Robbins, D, Margulies, I, Stetler-
Stevenson, M, Waldmann, TA et al. (1999). Responses in refractory hairy cell
105
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
leukemia to a recombinant immunotoxin. Blood 94: 3340-3348.
36. Lin, M, Ming, A and Zhao, M (2006). Two-dose basiliximab compared
with two-dose daclizumab in renal transplantation: a clinical study. Clin
Transplant 20: 325-329.
37. Chen, HR, Ji, SQ, Wang, HX, Yan, HM, Zhu, L, Liu, J et al. (2003).
Humanized anti-CD25 monoclonal antibody for prophylaxis of graft-vs-host
disease (GvHD) in haploidentical bone marrow transplantation without ex vivo
T-cell depletion. Exp Hematol31: 1019-1025.
38. Fecci, PE, Sweeney, AE, Grossi, PM, Nair, SK, Learn, CA, Mitchell, DA
et al. (2006). Systemic anti-CD25 monoclonal antibody administration safely
enhances immunity in murine glioma without eliminating regulatory T cells.
Clin Cancer Res 12: 4294-4305.
39. Knutson, KL, Dang, Y, Lu, H, Lukas, J, Almand, B, Gad, E et al.
(2006). IL-2 immunotoxin therapy modulates tumor-associated regulatory T
cells and leads to lasting immune-mediated rejection of breast cancers in neu-
transgenic mice. J Immunol 177: 84-91.
40. Onizuka, S, Tawara, I, Shimizu, J, Sakaguchi, S, Fujita, T and
Nakayama, E (1999). Tumor rejection by in vivo administration of anti-CD25
(interleukin-2 receptor alpha) monoclonal antibody. Cancer Res 59: 3128-
3133.
41. Zhang, M, Zhang, Z, Goldman, CK, Janik, J and Waldmann, TA
(2005). Combination therapy for adult T-cell leukemia-xenografted mice:
flavopiridol and anti-CD25 monoclonal antibody. Blood 105: 1231-1236.
42. Jaeger, G, Neumeister, P, Brezinschek, R, Hofler, G, Quehenberger,
F, Linkesch, W et al. (2002). Rituximab (anti-CD20 monoclonal antibody) as
consolidation of first-line CHOP chemotherapy in patients with follicular
lymphoma: a phase II study. EurJHaematol69: 21-26.
43. Claviez, A, Eckert, C, Seeger, K, Schrauder, A, Schrappe, M, Henze, G
et al. (2006). Rituximab plus chemotherapy in children with relapsed or
refractory CD20-positive B-cell precursor acute lymphoblastic leukemia.
Haematologica 91: 272-273.
106
CA 02719711 2010-09-27
WO 2008/116316 PCT/CA2008/000579
44. Montini, E, Cesana, D, Schmidt, M, Sanvito, F, Ponzoni, M,
Bartholomae, C et al. (2006). Hematopoietic stem cell gene transfer in a tumor-
prone mouse model uncovers low genotoxicity of lentiviral vector integration.
Nat Biotechnol 24: 687-696.
45. De Palma, M, Montini, E, Santoni de Sio, FR, Benedicenti, F, Gentile,
A, Medico, E et al. (2005). Promoter trapping reveals significant differences
in
integration site selection between MLV and HIV vectors in primary
hematopoietic cells. Blood 105: 2307-2315.
46. Wu, X, Li, Y, Crise, B and Burgess, SM (2003). Transcription start
regions in the human genome are favored targets for MLV integration. Science
300: 1749-1751.
47. Silvertown, JD, Walia, JS and Medin, JA (2005). Cloning, sequencing
and characterization of lentiviral-mediated expression of rhesus macaque
(Macaca mulatta) interleukin-2 receptor alpha cDNA. Dev Comp Immunol 29:
989-1002.
48. Naldini, L, Blomer, U, Gallay, P, Ory, D, Mulligan, R, Gage, FH et al.
(1996). In vivo gene delivery and stable transduction of nondividing cells by
a
lentiviral vector. Science 272: 263-267.
49. Medin, JA, Brandt, JE, Rozler, E, Nelson, M, Bartholomew, A, Li, C et
a!. (1999). Ex vivo expansion and genetic marking of primitive human and
baboon hematopoietic cells. Ann NYAcad Sci 872: 233-240; discussion 240-
242.
50. Ohshima, T, Murray, GJ, Swaim, WD, Longenecker, G, Quirk, JM,
Cardarelli, CO et a!. (1997). alpha-Galactosidase A deficient mice: a model of
Fabry disease. Proc Natl Acad Sci USA 94: 2540-2544.
107