Note: Descriptions are shown in the official language in which they were submitted.
DEMANDE OU BREVET VOLUMINEUX
LA PRRSENTE PARTIE DE CETTE DEMANDE OU CE BREVET COMPREND
PLUS D'UN TOME.
CECI EST LE TOME 1 DE 2
CONTENANT LES PAGES 1 A 394
NOTE : Pour les tomes additionels, veuillez contacter le Bureau canadien des
brevets
JUMBO APPLICATIONS/PATENTS
THIS SECTION OF THE APPLICATION/PATENT CONTAINS MORE THAN ONE
VOLUME
THIS IS VOLUME 1 OF 2
CONTAINING PAGES 1 TO 394
NOTE: For additional volumes, please contact the Canadian Patent Office
NOM DU FICHIER / FILE NAME:
NOTE POUR LE TOME / VOLUME NOTE:
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
BIARYL PDE4 INHIBITORS FOR TREATING INFLAMMATION
Field of the Invention
[0001] The present invention relates to a chemical genus of biaryl inhibitors
of
phosphodiesterase-4 (PDE4) useful for the treatment and prevention of stroke,
myocardial infarct, cardiovascular inflammatory diseases and central nervous
system
disorders.
Background of the Invention
[0002] PDE4 is the major cAMP-metabolizing enzyme found in inflammatory and
immune cells. PDE4 inhibitors have proven potential as anti-inflammatory
drugs,
especially in inflammatory pulmonary diseases such as asthma, COPD and
rhinitis. They
suppress the release of cytokines and other inflammatory signals and inhibit
the
production of reactive oxygen species. A large number of PDE4 inhibitors have
been
developed for a variety of clinical indications (Torphy and Page. 2000. TIPS
21, 157-
159; Burnouf and Pruniaux. 2002. Curr. Pharm. Design 8, 1255-1296; Lipworth.
2005.
Lancet 365, 167-175). To quote from a recent article in the British Journal of
Pharmacology, "PDE4 inhibitors have been in development as a novel anti-
inflammatory
therapy since the 1980s with asthma and chronic obstructive pulmonary disease
(COPD)
being primary indications. Despite initial optimism, none have yet reached the
market. In
most cases, the development of PDE4 inhibitors of various structural classes,
including
cilomilast, filaminast, lirimilast, piclamilast, tofimilast..... has been
discontinued due to
lack of efficacy. A primary problem is the low therapeutic ratio of these
compounds,
which severely limits the dose that can be given. Indeed, for many of these
compounds it
is likely that the maximum tolerated dose is either sub-therapeutic or at the
very bottom
of the efficacy dose-response curve. Therefore, the challenge is to overcome
this
limitation." [Giembycz, Brit.J.Pharmacol. 155, 288-290 (2008)]. Many of the
PDE4
inhibitors of the prior art have not reached the market because of the adverse
side effect
of emesis (Giembycz 2005. Curr. Opin. Pharm. 5, 238-244). Analysis of all
known
PDE4 inhibitors suggests that they are competitive with cAMP and bind within
the active
site (Houslay et al. 2005. DDT 10, 1503-1519); this may explain their narrow
therapeutic ratio. The compounds of the present invention are non-competitive
inhibitors
1
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
of cAMP while being gene-specific inhibitors (PDE4D), and, based on the target
rationale and in vitro potency, a person of skill in the art would expect the
compounds to
be useful as anti-inflammatory agents for the treatment, amelioration or
prevention of
inflammatory diseases and of complications arising therefrom and useful as CNS
agents
for amelioration of the cognitive decline in Alzheimer's disease, Parkinson's
disease, the
treatment of schizophrenia and depression, and neuroprotective in Huntington's
disease.
Summary of the Invention
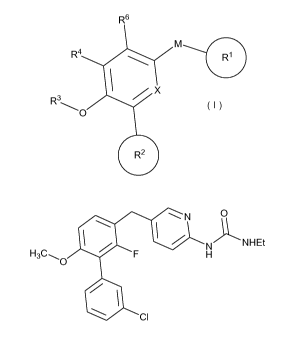
[0003] The present invention relates to compounds exhibiting PDE4 enzyme
inhibition,
having the general formula I
R6
M
R4
X
R3
'___ O
R2
[0004] In these compounds,
R1 is an optionally substituted carbocycle or optionally substituted
heterocycle of three
or fewer rings;
R2 is an optionally substituted carbocycle or optionally substituted
heterocycle of two or
fewer rings;
R3 is chosen from H, -C(=O)NH2, -(Ci-C6)alkyl, halo(Ci-C6)alkyl, -(C1-C6)alkyl-
R30
-(C2-C6)alkyl-R31, and saturated 4- or 5-membered heterocycle optionally
substituted
with methyl;
R30 is chosen from -C(=O)NH2 and 4- or 5-membered heterocycle optionally
substituted
with methyl;
R31 is chosen from (Cl-C4)alkoxy, amino, hydroxy, (Cl-C6)alkylamino and di(Cl-
C6)alkylamino;
R4 is chosen from H and F;
2
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
R6 is chosen from H, (Ci-C6)alkyl and halogen;
X is N, N-*O, or C-R5;
R5 is chosen from H, halogen, OH, (Ci-C6)alkyl, (Ci-C6)alkoxy, CF3, CN, NH2,
CH2OH, CH2NH2 and C=CH; and
M is chosen from direct bond, -C(R20)(R21)-, -0-, -NR22-, -S(O)ri , -C(=O)-,
-C(R20)(R21)C(R20)(R21)-, -C(R20)=C(R21)-, -C(R20)(R21)-O-, -C(R20)(R21)-NR22-
, -
C(R20)(R2)-S(O)ri , -C(R20)(R21)-C(=O)-, -O-C(R20)(R21)-, -NR22_C(R20)(R21)
-S(O)ri C(R20)(R21)-, -C(=O)-C(R20)(R21)- and
A
is a five or six-membered ring optionally substituted with methyl;
n is zero, one or two; and
R20, R21 and R22 are selected independently in each occurrence from H and (Cl-
C4)alkyl.
[0005] The present invention also relates to two subgenera of compounds of
formula I.
The first, in which X is N or N-*O, is represented by the formulae:
R6 R6
M M
R4 q~N \ R~ R4
R3 R3
~O '__ O
0
R2 R2
3
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[0006] The second, in which X is CR5 is represented by the formula:
R6
4
R3~ R5
0
R2
[0007] The present invention also relates to pharmaceutical compositions
comprising a
pharmaceutically acceptable carrier and a therapeutically effective amount of
at least one
compound of the general formula I described above. When the compound is
present as a
salt, the salt should be a pharmaceutically acceptable salt.
[0008] In a third aspect, the invention relates to methods for the treatment
or prophylaxis
of a disease or condition mediated by phosphodiesterase-4. The methods
comprise
administering to a mammal a therapeutically effective amount of a compound
having the
general formula I. The disease or condition may be related to allergic, acute
or chronic
inflammation. The disease may be, for example, atherosclerosis, thrombosis,
stroke,
acute coronary syndrome, stable angina, peripheral vascular disease, critical
leg
ischemia, intermittent claudication, abdominal aortic aneurysm or myocardial
infarction.
[0009] Selective PDE4 inhibitors of the invention are expected to be useful in
improving cognition and thus useful for treating learning disorders, memory
loss and
other cognitive dysfunctions. Selective PDE4 inhibitors of the invention are
also useful
for treating asthma and Chronic Obstructive Pulmonary Disease (COPD).
Compounds
of the invention, which inhibit tumor growth and metastases, also find utility
in the
treatment and prevention of cancer, including esophageal cancer, brain cancer,
pancreatic
cancer, and colon cancer.
[0010] These and other embodiments of the present invention will become
apparent
in conjunction with the description and claims that follow.
4
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Detailed Description of the Invention
[0011] Throughout this specification the substituents are defined when
introduced and
retain their definitions.
[0012] Unless otherwise specified, alkyl is intended to include linear,
branched, or cyclic
hydrocarbon structures and combinations thereof. A combination would be, for
example, cyclopropylmethyl. Lower alkyl refers to alkyl groups of from 1 to 6
carbon
atoms. Examples of lower alkyl groups include methyl, ethyl, propyl,
isopropyl, butyl, s-
and t-butyl and the like. Preferred alkyl groups are those of C20 or below; Ci
to C8 are
more preferred. Cycloalkyl is a subset of alkyl and includes cyclic
hydrocarbon groups
of from 3 to 8 carbon atoms. Examples of cycloalkyl groups include c-propyl, c-
butyl, c-
pentyl, norbornyl and the like.
[0013] C1 to C20 hydrocarbon includes alkyl, cycloalkyl, polycycloalkyl,
alkenyl,
alkynyl, aryl and combinations thereof. Examples include benzyl, phenethyl,
cyclohexylmethyl, camphoryl and naphthylethyl. Hydrocarbon refers to any
substituent
comprised of hydrogen and carbon as the only elemental constituents.
[0014] Unless otherwise specified, the term "carbocycle" is intended to
include ring
systems in which the ring atoms are all carbon but of any oxidation state.
Thus (C3-C10)
carbocycle refers to both non-aromatic and aromatic systems, including such
systems as
cyclopropane, benzene, cyclopentene and cyclohexene; (C8-C12) carbopolycycle
refers to
such systems as norbornane, decalin, indane and naphthalene. Carbocycle, if
not
otherwise limited, refers to monocycles, bicycles and polycycles.
[0015] Alkoxy or alkoxyl refers to groups of from 1 to 8 carbon atoms of a
straight,
branched or cyclic configuration and combinations thereof attached to the
parent
structure through an oxygen. Examples include methoxy, ethoxy, propoxy,
isopropoxy,
cyclopropyloxy, cyclohexyloxy and the like. Lower-alkoxy refers to groups
containing
one to four carbons. For the purpose of this application, alkoxy and lower
alkoxy
include methylenedioxy and ethylenedioxy. Alkoxyalkyl refers to ether groups
of from 3
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
to 8 atoms of a straight, branched, cyclic configuration and combinations
thereof
attached to the parent structure through an alkyl. Examples include
methoxymethyl,
methoxyethyl, ethoxypropyl, and the like. Alkoxyaryl refers to alkoxy
substituents
attached to an aryl, wherein the aryl is attached to the parent structure.
Arylalkoxy refers
to aryl substituents attached to an oxygen, wherein the oxygen is attached to
the parent
structure. Substituted arylalkoxy refers to a substituted aryl substituent
attached to an
oxygen, wherein the oxygen is attached to the parent structure.
[0016] Oxaalkyl refers to alkyl residues in which one or more carbons (and
their
associated hydrogens) have been replaced by oxygen. Examples include
methoxypropoxy; 3,6,9-trioxadecyl; 2,6,7-trioxabicyclo[2.2.2]octane and the
like. The
term oxaalkyl is intended as it is understood in the art [see Naming and
Indexing of
Chemical Substances for Chemical Abstracts, published by the American Chemical
Society, 196, but without the restriction of 127(a)], i.e. it refers to
compounds in which
the oxygen is bonded via a single bond to its adjacent atoms (forming ether
bonds); it
does not refer to doubly bonded oxygen, as would be found in carbonyl groups.
Similarly, thiaalkyl and azaalkyl refer to alkyl residues in which one or more
carbons has
been replaced by sulfur or nitrogen, respectively. Examples include
ethylaminoethyl and
methylthiopropyl.
[0017] Unless otherwise specified, acyl refers to formyl and to groups of 1,
2, 3, 4, 5, 6,
7 and 8 carbon atoms of a straight, branched, cyclic configuration, saturated,
unsaturated
and aromatic and combinations thereof, attached to the parent structure
through a
carbonyl functionality. One or more carbons in the acyl residue may be
replaced by
nitrogen, oxygen or sulfur as long as the point of attachment to the parent
remains at the
carbonyl. Examples include acetyl, benzoyl, propionyl, isobutyryl, t-
butoxycarbonyl,
benzyloxycarbonyl and the like. Lower-acyl refers to groups containing one to
four
carbons. The double bonded oxygen, when referred to as a substituent itself is
called
"oxo".
[0018] Aryl and heteroaryl mean (i) a phenyl group (or benzene) or a
monocyclic 5- or
6-membered heteroaromatic ring containing 1-4 heteroatoms selected from 0, N,
or S;
(ii) a bicyclic 9- or 10-membered aromatic or heteroaromatic ring system
containing 0-4
6
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
heteroatoms selected from 0, N, or S; or (iii) a tricyclic 13- or 14-membered
aromatic or
heteroaromatic ring system containing 0-5 heteroatoms selected from 0, N, or
S. Aryl,
as understood herein, includes residues in which one or more rings are
aromatic, but not
all need be. Thus aromatic 6- to 14-membered carbocyclic rings include, e.g.,
benzene,
naphthalene, indane, tetralin, and fluorene and the 5- to 10-membered aromatic
heterocyclic rings include, e.g., imidazole, pyridine, indole, thiophene,
benzopyranone,
thiazole, furan, benzimidazole, quinoline, isoquinoline, quinoxaline,
pyrimidine,
pyrazine, tetrazole and pyrazole.
[0019] Arylalkyl refers to a substituent in which an aryl residue is attached
to the parent
structure through alkyl. Examples are benzyl, phenethyl and the like.
Heteroarylalkyl
refers to a substituent in which a heteroaryl residue is attached to the
parent structure
through alkyl. In one embodiment, the alkyl group of an arylalkyl or a
heteroarylalkyl is
an alkyl group of from 1 to 6 carbons. Examples include, e.g.,
pyridinylmethyl,
pyrimidinylethyl and the like.
[0020] Heterocycle means a cycloalkyl or aryl carbocycle residue in which from
one to
three carbons is replaced by a heteroatom selected from the group consisting
of N, 0 and
S. The nitrogen and sulfur heteroatoms may optionally be oxidized, and the
nitrogen
heteroatom may optionally be quaternized. Unless otherwise specified, a
heterocycle
may be non-aromatic or aromatic. It is to be noted that heteroaryl is a subset
of
heterocycle in which the heterocycle is aromatic. Examples of heterocyclic
residues that
fall within the scope of the invention include pyrazole, pyrrole, indole,
quinoline,
isoquinoline, tetrahydroisoquinoline, benzofuran, benzodioxan, benzodioxole
(commonly referred to as methylenedioxyphenyl, when occurring as a
substituent),
morpholine, thiazole, pyridine (including 2-oxopyridine), pyridine N-oxide,
pyrimidine,
thiophene (i.e. thiene), furan, oxazole, oxazoline, oxazolidine,
isoxazolidine, isoxazole,
dioxane, azetidine, piperazine, piperidine, pyrrolidine, pyridazine, azepine,
pyrazolidine,
imidazole, imidazoline, imidazolidine, imidazolopyridine, pyrazine,
thiazolidine,
isothiazole, 1,2-thiazine-1,1-dioxide, quinuclidine, isothiazolidine,
benzimidazole,
thiadiazole, benzopyran, benzothiazole, benzotriazole, benzoxazole,
tetrahydrofuran,
tetrahydropyran, benzothiene, thiamorpholine, thiamorpholine sulfoxide,
thiamorpholine
sulfone, oxadiazole, triazole, tetrazole, isatin (dioxoindole), phthalimide
7
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
(dioxoisoindole), pyrrolopyridine, triazolopyridine and the dihydro and
tetrahydro
congeners of the fully unsaturated ring systems among the foregoing.
[0021] An oxygen heterocycle is a heterocycle containing at least one oxygen
in the ring;
it may contain additional oxygens, as well as other heteroatoms. Oxygen
heterocycles
found in the examples of the invention include tetrahydrofuran, benzodioxole,
morpholine, isoxazole and 2,6,7-trioxabicyclo[2.2.2]octane. A sulphur
heterocycle is a
heterocycle containing at least one sulphur in the ring; it may contain
additional
sulphurs, as well as other heteroatoms. A nitrogen heterocycle is a
heterocycle
containing at least one nitrogen in the ring; it may contain additional
nitrogens, as well as
other heteroatoms.
[0022] As used herein, the term "optionally substituted" may be used
interchangeably
with "unsubstituted or substituted". The term "substituted" refers to the
replacement of
one or more hydrogen atoms in a specified group with a specified radical. For
example,
substituted alkyl, aryl, cycloalkyl, heterocyclyl etc. refer to alkyl, aryl,
cycloalkyl, or
heterocyclyl wherein up to three H atoms in each residue are replaced with
halogen,
haloalkyl, alkyl, acyl, alkoxyalkyl, hydroxyalkyl, carbonyl (i.e. oxo),
phenyl, heteroaryl,
benzenesulfonyl, hydroxy, alkoxy, haloalkoxy, oxaalkyl, carboxy,
alkoxycarbonyl [-
C(=O)O-alkyl], alkoxycarbonylamino [ -NHC(=O)O-alkyl],
alkoxycarbonylaminoalkyl [
-alkyl-NHC(=O)O-alkyl], carboxyalkylcarbonylamino [ -NHC(=O)-alkyl-COOH],
carboxamido [-C(=O)NH2], aminocarbonyloxy [-OC(=O)NH2], alkylaminocarbonyl [-
C(=O)NH-alkyl], dialkylaminocarbonyl [-C(=O)N(alkyl)2], aminocarbonylalkyl [-
alkyl-
C(=O)NH2], cyano, acetoxy, nitro, amino, alkylamino, dialkylamino, aminoalkyl,
(alkyl)(aryl)aminoalkyl, alkylaminoalkyl (including cycloalkylaminoalkyl),
dialkylaminoalkyl, dialkylaminoalkoxy, alkyl(hydroxyalkyl)amino,
heterocyclylalkoxy,
mercapto, alkylthio, alkylsulfonyl, alkylsulfonylamino, alkylsulfinyl,
alkylsulfonyl,
arylthio, arylsulfonyl, arylsulfonylamino, arylsulfinyl, arylsulfonyl,
acylaminoalkyl,
acylaminoalkoxy, acylamino, amidino, aryl, benzyl, heterocyclyl,
heterocyclylalkyl,
phenoxy, benzyloxy, heteroaryloxy, heterocyclylamino, hydroxyimino,
alkoxyimino,
oxaalkyl, aminosulfonyl, trityl, amidino, guanidino, ureido, -NHC(=O)NHalkyl,
-NHC(=O)NH-heterocyclyl, -alkyl-NHC(=O)N(alkyl)2,
heterocyclylalkylcarbonylamino,
benzyloxyphenyl, and benzyloxy. Although oxo is included among the
substituents
8
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
referred to in "optionally substituted", it will be appreciated by persons of
skill in the art
that, because oxo is a divalent radical, there are circumstances in which it
will not be
appropriate as a substituent (e.g. on phenyl). Additional substituents that
are considered
within the scope of the term, particularly for R', are the are the residues of
amino acids,
amino acid amides, protected residues of aminoacids and their amides, and N-
methylated
(mono- or di-, as appropriate) amino acids and amino acid amides.
[0023] For the purpose of R', the substituents alkyl, acyl, alkoxyalkyl,
hydroxyloweralkyl, phenyl, heteroaryl, benzenesulfonyl, loweralkoxy,
haloalkoxy,
oxaalkyl, alkoxycarbonyl, alkoxycarbonylamino, carboxamido,
alkylaminocarbonyl,
amino, alkylamino, (alkyl)(aryl)aminoalkyl, alkylaminoalkyl,
heterocyclylalkoxy,
alkylthio, sulfonylamino, alkylsulfinyl, alkylsulfonyl, acylaminoalkyl,
acylaminoalkoxy,
acylamino, amidino, aryl, benzyl, heterocyclyl, heterocyclylalkyl,
heterocyclylalkoxy,
phenoxy, benzyloxy, heteroaryloxy, heterocyclylamino, oxaalkyl, aminosulfonyl,
amidino, guanidino, ureido, benzyloxyphenyl, and benzyloxy may be further
substituted
with one or two substituents from the list of substituents above. Substituents
that are
considered within the scope of the term, particularly for R', are the are the
residues of
amino acids, amino acid amides and protected residues of aminoacids and their
amides,
as well as the following specific residues: -CH3, -CH2CF3, -CF3, -CHO, -COOH, -
CN,
halogen, -OH, , -OEt, -C(=O)NH2, -C(=O)NHEt, -C(=O)NMe2 -COOCH3, -COOEt,
-CH2NHC(=O)NH2, -CH(CH3)NHC(=O)NH2, -CH2NHC(=O)H, -CH2NHC(=O)CH3,
-CH2C(=O)NH2, -CH2COOH, -CH2COOEt, -CH2NHC(=O)OEt, -CH2NHC(=O)O-
C6H5, -CH2NHC(=O)C(=O)NH2, -CHzNHC(=O)NHEt, -C(CH3)20H,
-CH2NHC(=O)N(CH3)2, -CH2NHC(=O)NHCH3, -CH2NH2, -CH(CH3)NH2,
-C(CH3)2NH2, -CH2OH, -CH2CH2OH, -CH2NHSO2CH3, -CH2OC(=O)NHEt, -OCH3,
-OC(=O)NH2, -OCH2CH2N(CH3)2, -OCH2CH2OCH3, -NHC(=O)NH2, -NHC(=O)NHEt,
-NHCH3, -NHEt, -NH(tBoc), -NHCH2COOH ("residue of glycine"), -N(CH3)CH2COOH
("residue of N-methylglycine"), -NHC(=O)NHCH2CH2C1, -NHSO2NH2, -NHEt,
-N(CH3)2, -NH2,, -NH(CH3)C(=O)NH2, -NHSO2CH3, -N(SO2CH3)2, -NHC(=O)OCH3,
-NHC(=O)OtBu, -NHC(=O)CH3, -SO2NH2, -NHC(=O)CH2CH2COOH,
-NHC(=O)NHCH2COOH, -CH2NHCHO, -NHC(=O)NHCH2COOEt,
-NHC(=O)NH(CH2)3COOEt, -NHC(=O)NH(CH2)2COOEt, -N(CH3)CH2CH2OH,
-NHC(=O)OEt, -N(Et)C(=O)OEt, -NHC(=O)NH(CH2)2COOH, -NHC(=O)CH2N(CH3)2,
9
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
-NHC(=O)NH(CH2)3000H, -NHC(=O)CH2NH2, -NHC(=O)CH2CH2NH2,
-NHC(=O)CH2NH(tBoc),
rN'CH3
~ A"- N NJ r N NO
~
`~ o 0 N LZ 0
0
N ~ N N
`22,~~NH2 `22 I-S, N
0 0 O CH3 and 0~~ . The term
"a residue of an amino acid, amino acid amide", etc. refers to an amino acid
etc. minus
the functional groups that are considered part of the bond to the parent
structure. For
example, in the molecule P-143 illustrated below:
O
~N CH30 H TO tBu
P-
143
N02
after one subtracts the hydrogen that connects (BOC)glycinamide to the phenyl
ring, the
structure of A that remains is:
0
",[~ N O
~-HN ~1 tBu
0 This is not sensu stricto a protected amino acid amide,
since it lacks the hydrogen on the C-terminal amide. This and similar
structures that lack
atoms at the points of attachment (e.g. the OH of COOH or the H of NH2) are
referred to
herein as "residues" of their respective parents.
[0024] The terms "haloalkyl" and "haloalkoxy" mean alkyl or alkoxy,
respectively,
substituted with one or more halogen atoms. The terms "alkylcarbonyl" and
"alkoxycarbonyl" mean -C(=O)alkyl or -C(O)alkoxy, respectively.
[0025] The term "halogen" means fluorine, chlorine, bromine or iodine. In one
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
embodiment, halogen may be fluorine or chlorine.
[0026] Substituents R" are generally defined when introduced and retain that
definition
throughout the specification and in all independent claims.
[0027] In the characterization of some of the substituents, it is recited that
certain
substituents may combine to form rings. Unless stated otherwise, it is
intended that such
rings may exhibit various degrees of unsaturation (from fully saturated to
fully
unsaturated), may include heteroatoms and maybe substituted with lower alkyl
or
alkoxy.
[0028] It will be recognized that the compounds of this invention can exist in
radiolabeled form, i.e., the compounds may contain one or more atoms
containing an
atomic mass or mass number different from the atomic mass or mass number
usually
found in nature. Radioisotopes of hydrogen, carbon, phosphorous, fluorine, and
chlorine
include 2H, 3H 13C 14C 15N 355, 18F, and 36C1, respectively. Compounds that
contain
those radioisotopes and/or other radioisotopes of other atoms are within the
scope of this
invention. Tritiated, i.e. 3H, and carbon-14, i.e., 14C, radioisotopes are
particularly
preferred for their ease in preparation and detectability. Compounds that
contain
isotopes 11C 13N 150 and 18F are well suited for positron emission tomography.
Radiolabeled compounds of formula I of this invention and prodrugs thereof can
generally be prepared by methods well known to those skilled in the art.
Conveniently,
such radiolabeled compounds can be prepared by carrying out the procedures
disclosed
in the Examples and Schemes by substituting a readily available radiolabeled
reagent for
a non-radiolabeled reagent.
[0029] As used herein (particularly in the claims), and as would be understood
by the
person of skill in the art, the recitation of "a compound" is intended to
include salts,
solvates, co-crystals and inclusion complexes of that compound as well as any
stereoisomeric form, or a mixture of any such forms of that compound in any
ratio.
Thus, in accordance with some embodiments of the invention, a compound as
described
herein, including in the contexts of pharmaceutical compositions, methods of
treatment,
and compounds per se, is provided as the salt form. Thus, for example, the
recitation "a
11
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
compound of formula P' as depicted above, in which Rt is imidazolyl, would
include
imidazolium salts. In a particular embodiment, the term "compound of formula
I" refers
to the compound or a pharmaceutically acceptable salt thereof.
[0030] The compounds described herein may contain asymmetric centers and may
thus
give rise to enantiomers, diastereomers, and other stereoisomeric forms. Each
chiral
center may be defined, in terms of absolute stereochemistry, as (R)- or (S)-.
The present
invention is meant to include all such possible isomers, in any ratio from
racemic to
optically pure forms. Optically active (R)- and (S)- isomers may be prepared
using
chiral synthons or chiral reagents, or resolved using conventional techniques.
The prefix
"rac" refers to a racemate. When the compounds described herein contain
olefinic
double bonds or other centers of geometric asymmetry, and unless specified
otherwise, it
is intended that the compounds include both E and Z geometric isomers. The
representation of the configuration of any carbon-carbon double bond appearing
herein is
selected for convenience only, and unless explicitly stated, is not intended
to designate a
particular configuration. Thus a carbon-carbon double bond depicted
arbitrarily as E
may be Z, E, or a mixture of the two in any proportion. Likewise, all
tautomeric forms
are also intended to be included.
[0031] The term "solvate" refers to a compound of Formula I in the solid
state, wherein
molecules of a suitable solvent are incorporated in the crystal lattice. A
suitable solvent
for therapeutic administration is physiologically tolerable at the dosage
administered.
Examples of suitable solvents for therapeutic administration are ethanol and
water. When
water is the solvent, the solvate is referred to as a hydrate. In general,
solvates are
formed by dissolving the compound in the appropriate solvent and isolating the
solvate
by cooling or using an antisolvent. The solvate is typically dried or
azeotroped under
ambient conditions. Inclusion complexes are described in Remington: The
Science and
Practice of Pharmacy 19th Ed. (1995) volume 1, page 176-177, which is
incorporated
herein by reference. The most commonly employed inclusion complexes are those
with
cyclodextrins, and all cyclodextrin complexes, natural and synthetic, are
specifically
encompassed within the claims.
[0032] The term "pharmaceutically acceptable salt" refers to salts prepared
from
12
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
pharmaceutically acceptable non-toxic acids or bases including inorganic acids
and bases
and organic acids and bases. When the compounds of the present invention are
basic,
salts may be prepared from pharmaceutically acceptable non-toxic acids
including
inorganic and organic acids. Suitable pharmaceutically acceptable anions for
the
compounds of the present invention include acetate, benzenesulfonate
(besylate),
benzoate, bicarbonate, bisulfate, carbonate, camphorsulfonate, citrate,
ethanesulfonate,
fumarate, gluconate, glutamate, glycolate, bromide, chloride, isethionate,
lactate,
maleate, malate, mandelate, methanesulfonate, mucate, nitrate, pamoate,
pantothenate,
phosphate, succinate, sulfate, tartrate, trifluoroacetate, p-toluenesulfonate,
acetamidobenzoate, adipate, alginate, aminosalicylate,
anhydromethylenecitrate,
ascorbate, aspartate, calcium edetate, camphorate, camsylate, caprate,
caproate,
caprylate, cinnamate, cyclamate, dichloroacetate, edetate (EDTA), edisylate,
embonate,
estolate, esylate, fluoride, formate, gentisate, gluceptate, glucuronate,
glycerophosphate,
glycolate, glycollylarsanilate, hexylresorcinate, hippurate,
hydroxynaphthoate, iodide,
lactobionate, malonate, mesylate, napadisylate, napsylate, nicotinate, oleate,
orotate,
oxalate, oxoglutarate, palmitate, pectinate, pectinate polymer,
phenylethylbarbiturate,
picrate, pidolate, propionate, rhodanide, salicylate, sebacate, stearate,
tannate, theoclate,
tosylate and the like. The desired salt may be obtained by ion exchange of
whatever
counter ion is obtained in the synthesis of the quat. These methods are well
known to
persons of skill. Although pharmaceutically acceptable counter ions will be
preferred for
preparing pharmaceutical formulations, other anions are quite acceptable as
synthetic
intermediates. When the compounds contain an acidic side chain, suitable
pharmaceutically acceptable base addition salts for the compounds of the
present
invention include metallic salts made from aluminum, calcium, lithium,
magnesium,
potassium, sodium and zinc or organic salts made from lysine, N,N'-
dibenzylethylenediamine, chloroprocaine, choline, diethanolamine,
ethylenediamine,
meglumine (N-methylglucamine) and procaine.
[0033] The graphic representations of racemic, ambiscalemic and scalemic or
enantiomerically pure compounds used herein are taken from Maehr J. Chem. Ed.
62,
114-120 (1985): solid and broken wedges are used to denote the absolute
configuration
of a chiral element; wavy lines and single thin lines indicate disavowal of
any
stereochemical implication which the bond it represents could generate; solid
and broken
13
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
bold lines are geometric descriptors indicating the relative configuration
shown but
denoting racemic character; and wedge outlines and dotted or broken lines
denote
enantiomerically pure compounds of indeterminate absolute configuration.
[0034] Terminology related to "protecting", "deprotecting" and "protected"
functionalities occurs throughout this application. Such terminology is well
understood
by persons of skill in the art and is used in the context of processes that
involve
sequential treatment with a series of reagents. In that context, a protecting
group refers
to a group, which is used to mask a functionality during a process step in
which it would
otherwise react, but in which reaction is undesirable. The protecting group
prevents
reaction at that step, but may be subsequently removed to expose the original
functionality. The removal or "deprotection" occurs after the completion of
the reaction
or reactions in which the functionality would interfere. Thus, when a sequence
of
reagents is specified, as it is in the processes of the invention, the person
of ordinary skill
can readily envision those groups that would be suitable as "protecting
groups". Suitable
groups for that purpose are discussed in standard textbooks in the field of
chemistry,
such as Protective Groups in Organic Synthesis by T.W. Greene [John Wiley &
Sons,
New York, 1991 ], which is incorporated herein by reference.
[0035] A comprehensive list of abbreviations utilized by organic chemists
appears in the
first issue of each volume of the Journal of Organic Chemistry. The list,
which is
typically presented in a table entitled "Standard List of Abbreviations", is
incorporated
herein by reference.
[0036] In general, the compounds of the present invention may be prepared by
the
methods illustrated in the general reaction schemes as, for example, described
below, or
by modifications thereof, using readily available starting materials, reagents
and
conventional synthesis procedures. In these reactions, it is also possible to
make use of
variants that are in themselves known, but are not mentioned here. The
starting
materials, are either commercially available, synthesized as described in the
examples or
may be obtained by the methods well known to persons of skill in the art.
[0037] PDE4 inhibitors have been shown to be effective therapeutic agents in
clinical
studies. For example, administration of cilomilast and roflumilast (PDE4
inhibitors) to
14
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
patients suffering from asthma and COPD showed initially excellent results,
although the
effect of cilomilast disappeared on long-term trial [Lipworth, Lancet 365, 167-
175
(2005)]. Genetic studies have clearly demonstrated an association between
PDE4D and
ischemic stroke (Gretarsdottir et al. 2003. Nature Genetics. 35, 1-8). L-
454,560, a
selective PDE4 inhibitor has been shown to improve learning in a rat model in
vivo
[Huang et al. Biochemical Pharmacology 73, 1971-1981 (2007)]. This suggests
that
selective PDE4 inhibitors will be useful in treating learning disorders,
memory loss (e.g.
Alzheimer's disease) and other cognitive dysfunctions. Rolipram, another
selective
PDE4 inhibitor, has been shown to enhance cognition in multiple rodent models
[Blokland et al., Current Pharmaceutical Design 12, 2511-2523 (2006)] as well
as in
primates [Rutten et al., 2008, Psychopharmacology 196, 643-648 (2008)].
Rolipram also
improves the outcome in two separate studies in mice in vivo in models
accepted by
persons of skill in the art as predictive of utility in schizophrenia [Kanes
et al.,
Neuroscience 144, 23 9-246 (2007); Davis and Gould, Behav.Neurosci. 119, 595-
602
(2005)]. Rolipram has also been shown to exhibit a neuroprotective effect in a
rat model
of Huntington's disease [DeMarch et al. Neurobiol.Dis. 25, 266-273 (2007)].
This
suggests that PDE4 modulators will be useful for treating many CNS disorders.
Selective PDE4 inhibitors (e.g. rolipram) are also useful for treating bone
loss [Yao et
al., J.Musculoskelet.Neuronal Interact. 7, 119-130 (2007)].
[0038] Additionally, a PDE4 inhibitor, YM976, was shown to ameliorate the
effects of
experimentally-induced interstitial cystitis in rats, resulting in a decrease
in the frequency
of urination and an increase in the volume of urine at each time of urination
[Kitta et al.,
BJU Int. 102, 1472-1476 (2008)]. Another PDE4 inhibitor, IC485, was shown to
be
equally efficacious as tolteradine tartrate, a marketed drug for treating
overactive
bladder, in a rodent model of obstructive bladder [Kaiho et al. BJU Int. 101,
615-20
(2008)]. These findings suggest that PDE4 inhibitors will be useful in
treating symptoms
of bladder overactivity, inflammation and pain.
[0039] In addition to the foregoing studies demonstrating utility in in vivo
models, a
number of authors have suggested connections between PDE4 inhibition and
putative
utilities as antidepressants [Houslay et al., Drug Discov Today 10, 1503-1519
(2005);
Polesskaya et al., Biol.Psychiatr. 61, 56-64 (2007); anon. Current
Opin.Invetig.Drugs 5,
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
34-39 (2004)] and as anxiolytics [Zhang et al., Neuropsychopharmacology Aug
15, 2007
Epub; Cherry et al., Biochim.Biophys.Acta 1518, 27-35 (2001)]. Rolipram has
been
shown in human clinical trials to ameliorate depression [Hebenstreit et al.,
Pharmacopsychiat. 22, 156-160 (1989)]. Other possible utilities may include
Pick's
disease and epilepsy.
[0040] Furthermore, the compounds, compositions and methods of the present
invention
may be useful in treating cancer. Phosphodiesterase activity has been shown to
be
associated with hematological malignancies [Lerner et al., Biochem.J. 393, 21-
41
(2006); Ogawa et al., Blood 99, 3390-3397 (2002)]. The compounds may also be
administered to overcome cognitive impairment induced by one or more of the
following
agents, alcohol, amphetamine, antipsychotic medication, anti-retroviral
therapy, MDMA
( 3,4-methylenedioxy-N-methylamphetamine, cannabis, cocaine, delta-9
tetrahydrocannabinol, dexamphetamine, haloperidol, heroin and other opiates,
ketamine
and metamphetamine.
[0041] Furthermore, the compounds, compositions and methods of the present
invention,
particularly when radiolabeled as described above or labeled by methods well-
known in
the art with florescent and spin labels, may be employed as imaging agents and
in other
ways for diagnosis and/or treatment. Moreover, immobilization of compounds of
the
invention on solid support could be of utility for affinity purification and
modification of
compounds of the invention with chemically active groups may be used for
protein
labeling.
[0042] For many of the utilities outlined above, it maybe advantageous to
administer
compounds of the general formula I together with cholinesterase inhibitors
(e.g. tacrine,
huperzine, donepezil); NMDA antagonists (e.g. lanicemine, remacemide,
neramexane,
memantine); calpain inhibitors (e.g. CEP-3122); antioxidants (e.g.vitamin E,
coenzyme
Q10) and agents that have shown clinical efficacy but whose mechanism is
unclear (e.g.
dimebon). Compounds of formula I may also be administered together with one or
more
of the following agents to improve cognition: amisulpride, atomoxetine,
bromocryptine,
buspirone, caffeine, chlorpromazine, clonidine, clozapine, diazepam,
flumazenil,
fluoxetine, galantamine, guanfacine, methylphenidate, idazoxan, modafinil,
olanzapine,
16
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
paroxetine, pergolide, phenserine, quetiapine, risperidone, rivastigmine,
SGS742 and
sulpiride.
[0043] The terms "methods of treating or preventing" mean amelioration,
prevention or
relief from the symptoms and/or effects associated with disorders. The term
"preventing"
as used herein refers to administering a medicament beforehand to forestall or
obtund an
acute episode. The person of ordinary skill in the medical art (to which the
present
method claims are directed) recognizes that the term "prevent" is not an
absolute term.
In the medical art it is understood to refer to the prophylactic
administration of a drug to
substantially diminish the likelihood or seriousness of a condition, and this
is the sense
intended in applicants' claims. As used herein, reference to "treatment" of a
patient is
intended to include prophylaxis.
[0044] The term "mammal" is used in its dictionary sense. Humans are included
in the
group of mammals, and humans would be the preferred subjects of the methods.
[0045] The cognitive impairment to be treated may arise from one or more of
the
following disorders, which may not in themselves be necessarily associated
with PDE4
abnormality: acute pain, AD/HD - Attention deficit hyperactivity disorder,
AIDS
dementia complex, alcoholism, amphetamine addiction, amygdalo-hippocampectomy,
anorexia nervosa, anterior parietal damage, antisocial behavior, antisocial
personality
disorder, anxiety, autism, basal ganglia lesions, bipolar disorder, borderline
personality
disorder, camptocormia, capgras syndrome, carcinoid syndrome, carotid
endarterectomy
surgery, chronic drug misuse, chronic fatigue syndrome, chronic occupational
solvent
encephalopathy, chronic pain, brain ischemia, coronary artery bypass surgery,
critical
illness requiring intensive care, dementia Alzheimer-type (DAT), dementia Lewy
Body
type, dementia of frontal type, dementia caused by ischemia, dental pain,
developmental
dyslexia, diabetes, dorsolateral frontal cortical compression, Down's
Syndrome, drug
abuse, dysexecutive syndrome, fibromyalgia, frontal lobe damage, frontal lobe
excision,
frontal variant frontotemporal dementia, gluten ataxia, hallucinosis, head
injury, hearing
loss, heart disease, heart failure, heavy social drinking, hepatic
encephalopathy, heroin
addiction, herpes encephalitis, hippocampal atrophy, HIV/AIDS, Huntington's
disease,
hydrocephalus, hypercortisolemia, hyperostosis frontalis interna,
hypertension,
17
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
idiopathic pain, insomnia, Korsakoff syndrome, late paraphrenia, lead
exposure, left
ventricular systolic dysfunction, orbitofrontal cortex lesion, liver failure,
long term health
effects of diving, Machado-Joseph disease, mad hatter's disease, manic
depression,
melancholia, mercury poisoning, mild cognitive impairment (MCI), mild
cognitive
impairment (MCI) of aging, motor neuron disease, multiple sclerosis, multiple
system
atrophy, narcolepsy, neuronal migration disorders, normal pressure
hydrocephalus,
obsessive compulsive disorder, organophosphate pesticide exposure, panic
disorder,
paraphrenia, Parkinson's disease, periventricular brain insult, personality
disorder,
gasoline sniffing, phenylketonuria, post-concussion syndrome, premature birth
needing
intensive care, premenstrual dysphoric disorder, progressive supranuclear
palsy,
psychopathy, psychosis, questionable dementia, renal cancer, Roifman syndrome,
schizoaffective disorder, schizophrenia, seasonal affective disorder, self
harm, semantic
dementia, specific language impairment, social withdrawal in schizophrenia,
solvent
encephalopathy, spina bifida, Steele-Richardson-Olzsewski syndrome, stiff
person
syndrome, striatocapsular infarct, subarachnoid hemorrhage, substance abuse,
tardive
dyskinesia, temporal lobe excision, temporal lobe lesion, tinnitus, Tourette's
syndrome,
transient cerebral ischemia, traumatic brain injury, trichotillomania,
tuberous sclerosis,
and white matter lesions.
[0046] While it may be possible for compounds of formula Ito be administered
as the
raw chemical, it will often be preferable to present them as part of a
pharmaceutical
composition. In accordance with an embodiment of the present invention there
is
provided a pharmaceutical composition comprising a compound of formula I or a
pharmaceutically acceptable salt thereof, together with one or more
pharmaceutically
carriers thereof and optionally one or more other therapeutic ingredients. The
carrier(s)
must be "acceptable" in the sense of being compatible with the other
ingredients of the
formulation and not deleterious to the recipient thereof Furthermore, when
reference is
made in an independent claim to a compound or a pharmaceutically acceptable
salt
thereof, it will be understood that claims which depend from that independent
claim
which refer to such a compound also include pharmaceutically acceptable salts
of the
compound, even if explicit reference is not made to the salts in the dependent
claim.
18
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[0047] The formulations include those suitable for oral, parenteral (including
subcutaneous, intradermal, intramuscular, intravenous and intraarticular),
rectal and
topical (including dermal, buccal, sublingual and intraocular) administration.
The most
suitable route may depend upon the condition and disorder of the recipient.
The
formulations may conveniently be presented in unit dosage form and maybe
prepared by
any of the methods well known in the art of pharmacy. Such methods include the
step of
bringing into association a compound of formula I or a pharmaceutically
acceptable salt
or solvate thereof ("active ingredient") with the carrier, which constitutes
one or more
accessory ingredients. In general, the formulations are prepared by uniformly
and
intimately bringing into association the active ingredient with liquid
carriers or finely
divided solid carriers or both and then, if necessary, shaping the product
into the desired
formulation.
[0048] Formulations suitable for oral administration may be presented as
discrete units
such as capsules, cachets or tablets each containing a predetermined amount of
the active
ingredient; as a powder or granules; as a solution or a suspension in an
aqueous liquid or
a non-aqueous liquid; or as an oil-in-water liquid emulsion or a water-in-oil
liquid
emulsion. The active ingredient may also be presented as a bolus, electuary or
paste.
[0049] A tablet may be made by compression or molding, optionally with one or
more
accessory ingredients. Compressed tablets may be prepared by compressing in a
suitable
machine the active ingredient in a free-flowing form such as a powder or
granules,
optionally mixed with a binder, lubricant, inert diluent, lubricating, surface
active or
dispersing agent. Molded tablets may be made by molding in a suitable machine
a
mixture of the powdered compound moistened with an inert liquid diluent. The
tablets
may optionally be coated or scored and may be formulated so as to provide
sustained,
delayed or controlled release of the active ingredient therein. The
pharmaceutical
compositions may include a "pharmaceutically acceptable inert carrier", and
this
expression is intended to include one or more inert excipients, which include
starches,
polyols, granulating agents, microcrystalline cellulose, diluents, lubricants,
binders,
disintegrating agents, and the like. If desired, tablet dosages of the
disclosed
compositions may be coated by standard aqueous or nonaqueous techniques,
"Pharmaceutically acceptable carrier" also encompasses controlled release
means.
19
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[0050] Pharmaceutical compositions may also optionally include other
therapeutic
ingredients, anti-caking agents, preservatives, sweetening agents, colorants,
flavors,
desiccants, plasticizers, dyes, and the like. Any such optional ingredient
must be
compatible with the compound of formula Ito insure the stability of the
formulation.
The composition may contain other additives as needed, including for example
lactose,
glucose, fructose, galactose, trehalose, sucrose, maltose, raffinose,
maltitol, melezitose,
stachyose, lactitol, palatinite, starch, xylitol, mannitol, myoinositol, and
the like, and
hydrates thereof, and amino acids, for example alanine, glycine and betaine,
and peptides
and proteins, for example albumen.
[0051] Examples of excipients for use as the pharmaceutically acceptable
carriers and
the pharmaceutically acceptable inert carriers and the aforementioned
additional
ingredients include, but are not limited to binders, fillers, disintegrants,
lubricants, anti-
microbial agents, and coating agents.
[0052] The dose range for adult humans is generally from 0.005 mg to 10 g/day
orally.
Tablets or other forms of presentation provided in discrete units may
conveniently
contain an amount of compound of formula I which is effective at such dosage
or as a
multiple of the same, for instance, units containing 5 mg to 500 mg, usually
around 10
mg to 200 mg. The precise amount of compound administered to a patient will be
the
responsibility of the attendant physician. However, the dose employed will
depend on a
number of factors, including the age and sex of the patient, the precise
disorder being
treated, and its severity.
[0053] A dosage unit (e.g. an oral dosage unit) can include from, for example,
1 to 30
mg,1to40mg,1to100mg,1to300mg,1to500mg,2to500mg,3to100mg,5to
20 mg,5to100mg(e.g.1mg,2mg,3mg,4mg,5mg,6mg,7mg,8mg,9mg,10mg,
11 mg, 12 mg, 13 mg, 14 mg, 15 mg, 16 mg, 17 mg, 18 mg, 19 mg, 20 mg, 25 mg,
30
mg, 35 mg, 40 mg, 45 mg, 50 mg, 55 mg, 60 mg, 65 mg, 70 mg, 75 mg, 80 mg, 85
mg,
90 mg, 95 mg, 100 mg, 150 mg, 200 mg, 250 mg, 300 mg, 350 mg, 400 mg, 450 mg,
500
mg) of a compound described herein.
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[0054] For additional information about pharmaceutical compositions and their
formulation, see, for example, Remington: The Science and Practice of
Pharmacy, 20th
Edition, 2000. The agents can be administered, e.g., by intravenous injection,
intramuscular injection, subcutaneous injection, intraperitoneal injection,
topical,
sublingual, intraarticular (in the joints), intradermal, buccal, ophthalmic
(including
intraocular), intranasaly (including using a cannula), or by other routes. The
agents can
be administered orally, e.g., as a tablet or cachet containing a predetermined
amount of
the active ingredient, gel, pellet, paste, syrup, bolus, electuary, slurry,
capsule, powder,
granules, as a solution or a suspension in an aqueous liquid or a non-aqueous
liquid, as
an oil-in-water liquid emulsion or a water-in-oil liquid emulsion, via a
micellar
formulation (see, e.g. WO 97/11682) via a liposomal formulation (see, e.g., EP
736299,WO 99/59550 and WO 97/13500), via formulations described in WO
03/094886
or in some other form. The agents can also be administered transdermally (i.e.
via
reservoir-type or matrix-type patches, microneedles, thermal poration,
hypodermic
needles, iontophoresis, electroporation, ultrasound or other forms of
sonophoresis, jet
injection, or a combination of any of the preceding methods (Prausnitz et al.
2004,
Nature Reviews Drug Discovery 3:115)). The agents can be administered locally,
for
example, at the site of injury to an injured blood vessel. The agents can be
coated on a
stent. The agents can be administered using high-velocity transdermal particle
injection
techniques using the hydrogel particle formulation described in U.S.
20020061336.
Additional particle formulations are described in WO 00/45792, WO 00/53160,
and WO
02/19989. An example of a transdermal formulation containing plaster and the
absorption promoter dimethylisosorbide can be found in WO 89/04179. WO
96/11705
provides formulations suitable for transdermal administration. The agents can
be
administered in the form a suppository or by other vaginal or rectal means.
The agents
can be administered in a transmembrane formulation as described in WO
90/07923. The
agents can be administered non-invasively via the dehydrated particles
described in U.S.
6,485,706. The agent can be administered in an enteric-coated drug formulation
as
described in WO 02/49621. The agents can be administered intranasaly using the
formulation described in U.S. 5,179,079. Formulations suitable for parenteral
injection
are described in WO 00/62759. The agents can be administered using the casein
formulation described in U.S. 20030206939 and WO 00/06108. The agents can be
administered using the particulate formulations described in U.S. 20020034536.
21
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[0055] The agents, alone or in combination with other suitable components, can
be
administered by pulmonary route utilizing several techniques including but not
limited to
intratracheal instillation (delivery of solution into the lungs by syringe),
intratracheal
delivery of liposomes, insufflation (administration of powder formulation by
syringe or
any other similar device into the lungs) and aerosol inhalation. Aerosols
(e.g., jet or
ultrasonic nebulizers, metered-dose inhalers (MDIs), and dry-Powder inhalers
(DPIs))
can also be used in intranasal applications. Aerosol formulations are stable
dispersions
or suspensions of solid material and liquid droplets in a gaseous medium and
can be
placed into pressurized acceptable propellants, such as hydrofluoroalkanes
(HFAs, i.e.
HFA-134a and HFA-227, or a mixture thereof), dichlorodifluoromethane (or other
chlorofluorocarbon propellants such as a mixture of Propellants 11, 12, and/or
114),
propane, nitrogen, and the like. Pulmonary formulations may include permeation
enhancers such as fatty acids, and saccharides, chelating agents, enzyme
inhibitors (e.g.,
protease inhibitors), adjuvants (e.g., glycocholate, surfactin, span 85, and
nafamostat),
preservatives (e.g., benzalkonium chloride or chlorobutanol), and ethanol
(normally up
to 5% but possibly up to 20%, by weight). Ethanol is commonly included in
aerosol
compositions as it can improve the function of the metering valve and in some
cases also
improve the stability of the dispersion. Pulmonary formulations may also
include
surfactants which include but are not limited to bile salts and those
described in U.S.
6,524,557 and references therein. The surfactants described in U.S. 6,524,557,
e.g., a
C8-C16 fatty acid salt, a bile salt, a phospholipid, or alkyl saccharide are
advantageous in
that some of them also reportedly enhance absorption of the compound in the
formulation. Also suitable in the invention are dry powder formulations
comprising a
therapeutically effective amount of active compound blended with an
appropriate carrier
and adapted for use in connection with a dry-Powder inhaler. Absorption
enhancers
which can be added to dry powder formulations of the present invention include
those
described in U.S. 6,632,456. WO 02/080884 describes new methods for the
surface
modification of powders. Aerosol formulations may include U.S. 5,230,884, U.S.
5,292,499, WO 017/8694, WO 01/78696, U.S. 2003019437, U. S. 20030165436, and
WO 96/40089 (which includes vegetable oil). Sustained release formulations
suitable
for inhalation are described in U.S. 20010036481A1, 20030232019A1, and U.S.
20040018243A1 as well as in WO 01/13891, WO 02/067902, WO 03/072080, and WO
03/079885. Pulmonary formulations containing microparticles are described in
WO
22
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
03/015750, U.S. 20030008013, and WO 00/00176. Pulmonary formulations
containing
stable glassy state powder are described in U.S. 20020141945 and U.S.
6,309,671. Other
aerosol formulations are described in EP 1338272A1 WO 90/09781, U. S.
5,348,730,
U.S. 6,436,367, WO 91/04011, and U.S. 6,294,153 and U.S. 6,290,987 describes a
liposomal based formulation that can be administered via aerosol or other
means.
Powder formulations for inhalation are described in U.S. 20030053960 and WO
01/60341. The agents can be administered intranasally as described in U.S.
20010038824.
[0056] Solutions of medicament in buffered saline and similar vehicles are
commonly
employed to generate an aerosol in a nebulizer. Simple nebulizers operate on
Bernoulli's
principle and employ a stream of air or oxygen to generate the spray
particles. More
complex nebulizers employ ultrasound to create the spray particles. Both types
are well
known in the art and are described in standard textbooks of pharmacy such as
Sprowls'
American Pharmacy and Remington's The Science and Practice of Pharmacy. Other
devices for generating aerosols employ compressed gases, usually
hydrofluorocarbons
and chlorofluorocarbons, which are mixed with the medicament and any necessary
excipients in a pressurized container, these devices are likewise described in
standard
textbooks such as Sprowls and Remington.
[0057] The agent can be incorporated into a liposome to improve half-life. The
agent
can also be conjugated to polyethylene glycol (PEG) chains. Methods for
pegylation and
additional formulations containing PEG-conjugates (i.e. PEG-based hydrogels,
PEG
modified liposomes) can be found in Harris and Chess, Nature Reviews Drug
Discovery
2:214-221 and the references therein. The agent can be administered via a
nanocochleate
or cochleate delivery vehicle (BioDelivery Sciences International). The agents
can be
delivered transmucosally (i.e. across a mucosal surface such as the vagina,
eye or nose)
using formulations such as that described in U.S. 5,204,108. The agents can be
formulated in microcapsules as described in WO 88/01165. The agent can be
administered intra-orally using the formulations described in U.S.
20020055496, WO
00/47203, and U.S. 6,495,120. The agent can be delivered using nanoemulsion
formulations described in WO 01/91728A2.
23
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[0058] In general, compounds of formula I may be prepared by the methods
illustrated in
the general reaction schemes as, for example, described below, or by
modifications
thereof, using readily available starting materials, reagents and conventional
synthesis
procedures. In these reactions, it is also possible to make use of variants
that are in
themselves known, but are not mentioned here.
[0059] The present invention relates to compounds exhibiting PDE4 enzyme
inhibition,
having the general formula I
R6
M
R4
/ X
R3
~O
R2
[0060] In one embodiment, X is CRS, R2 is pyrazolyl or substituted phenyl and
R3 is
other than H. In another embodiment, M is chosen from direct bond, -CH2-, -
CH(OH)-, -
C[(CH3)(OH)]-, -C[(CH3)(NH2)]-, -C(=O)-, -0-, -NH-, -N(CH3)-, -S(O)ri , -CH2NH-
,
CH3-N
-CH2CH2-, -CH=CH-, -CH2S(O)ri , -CH20- and
24
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[0061] There are two subgenera of compounds of formula I. The first, in which
X is N
or N-*O, is represented by the formulae:
R6 R6
M M
R4 \ R~ R4
q~N
R3 R3
~O '-- O
O
R2 R2
[0062] The second, in which X is CR5 is represented by the formula:
R6
R5
O
JR R3
[0063] In one embodiment, R1 is a substituted phenyl. In another embodiment,
R1 is
phenyl and R2 is pyrazolyl or substituted phenyl. In another embodiment, R1 is
substituted phenyl and R2 is pyrazolyl or substituted phenyl.
[0064] In another embodiment, R1 is an unsubstituted heterocycle. In another
embodiment, R1 is a substituted heterocycle. In both heterocycle embodiments,
the
heterocycle may be chosen from pyrazole, pyrrole, indole, quinoline,
isoquinoline,
tetrahydroisoquinoline, benzofuran, benzodioxan, benzodioxole, morpholine,
thiazole,
pyridine, pyridine N-oxide, pyrimidine, thiene, furan, oxazole, oxazoline,
oxazolidine,
isoxazolidine, isoxazole, dioxane, azetidine, piperazine, piperidine,
pyrrolidine,
pyridazine, azepine, pyrazolidine, imidazole, imidazoline, imidazolidine,
purine,
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
imidazolopyridine, pyrazine, thiazolidine, isothiazole, 1,2-thiazine-1,1-
dioxide, 2,6,7-
trioxabicyclo [2.2.2 ]octane, quinuclidine, isothiazolidine, benzimidazole,
thiadiazole,
benzopyran, benzothiazole, benzotriazole, benzoxazole, benzoxadiazole,
tetrahydrofuran,
tetrahydropyran, benzothiene, thiamorpholine, thiamorpholine sulfoxide,
thiamorpholine
sulfone, oxadiazole, triazole, tetrazole, isoindole, pyrrolopyridine,
triazolopyridine and
the dihydro and tetrahydro congeners thereof, whether specifically named or
not. For
example, a dihydro congener of indole would be indoline or dihydroindole; a
tetrahydro
congener of pyridine would be piperidine. In a further embodiment, R1 is an
optionally
substituted heterocycle chosen from pyrazole, benzodioxole, morpholine,
thiazole,
pyridine, pyridine N-oxide, pyrimidine, thiene, oxazolidine, isoxazole,
azetidine,
piperazine, pyrrolidine, imidazole, imidazolidine, imidazolopyridine,
pyrazine, 1,2-
thiazine-1,1-dioxide, benzimidazole, thiadiazole, benzotriazole, benzoxazole,
oxadiazole,
triazole, tetrazole, isoindole, pyrrolopyridine, triazolopyridine and their
dihydro and
tetrahydro congeners.
[0065] In an embodiment, the substituted phenyl or substituted heterocycle is
substituted with a substituent chosen from halogen, haloalkyl, alkyl, acyl,
alkoxyalkyl,
hydroxyalkyl, carbonyl, phenyl, heteroaryl, benzenesulfonyl, hydroxy, alkoxy,
haloalkoxy, oxaalkyl, carboxy, alkoxycarbonyl, alkoxycarbonylalkyl,
alkoxycarbonylamino, carboxyalkyl, alkoxycarbonylaminoalkyl,
carboxyalkylcarbonylamino, carboxamido, aminocarbonyloxy, alkylaminocarbonyl,
dialkylaminocarbonyl, aminocarbonylalkyl, cyano, acetoxy, nitro, amino,
alkylamino,
dialkylamino, aminoalkyl, (alkyl)(aryl)aminoalkyl, alkylaminoalkyl,
dialkylaminoalkyl,
dialkylaminoalkoxy, alkyl(hydroxyalkyl)amino, heterocyclylalkoxy, mercapto,
alkylthio,
alkylsulfonyl, alkylsulfonylamino, alkylsulfinyl, alkylsulfonyl, arylthio,
arylsulfonyl,
arylsulfonylamino, arylsulfinyl, arylsulfonyl, acylaminoalkyl,
acylaminoalkoxy,
acylamino, amidino, aryl, benzyl, heterocyclyl, heterocyclylalkyl, phenoxy,
benzyloxy,
heteroaryloxy, heterocyclylamino, hydroxyimino, alkoxyimino, oxaalkyl,
aminosulfonyl,
trityl, amidino, guanidino, ureido, -NHC(=O)NHalkyl, -NHC(=O)NH-heterocyclyl,
-alkyl-NHC(=O)N(alkyl)2, heterocyclylalkylcarbonylamino, benzyloxyphenyl,
benzyloxy, the residues of amino acids, amino acid amides, protected residues
of
aminoacids, protected residues of amino acid amides, N-methylated amino acids
and N-
methylated amino acid amides. Exemplary amino acids are glycine, alanine and
proline.
26
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[0066] In another embodiment, the substituted phenyl or substituted
heterocycle is
substituted with a substituent chosen from -CH3, -CH2CF3, -CF3, -CHO, -000H, -
CN,
halogen, -OH, , -OEt, -C(=O)NH2, -C(=O)NHEt, -C(=O)NMe2 -COOCH3, -COOEt,
-CH2NHC(=O)NH2, -CH(CH3)NHC(=O)NH2, -CH2NHC(=O)H, -CH2NHC(=O)CH3,
-CH2C(=O)NH2, -CH2COOH, -CH2COOEt, -CH2NHC(=O)OEt, -CH2NHC(=O)O-
C6H5, -CH2NHC(=O)C(=O)NH2, -CHzNHC(=O)NHEt, -C(CH3)20H,
-CH2NHC(=O)N(CH3)2, -CH2NHC(=O)NHCH3, -CH2NH2, -CH(CH3)NH2,
-C(CH3)2NH2, -CH2OH, -CH2CH2OH, -CH2NHSO2CH3, -CH2OC(=O)NHEt, -OCH3,
-OC(=O)NH2, -OCH2CH2N(CH3)2, -OCH2CH2OCH3, -NHC(=O)NH2, -NHC(=O)NHEt,
-NHCH3, -NHEt, -NH(tBoc), -NHCH2COOH, -N(CH3)CH2COOH,
-NHC(=O)NHCH2CH2C1, -NHSO2NH2, -NHEt, -N(CH3)2, -NH2,,
-NH(CH3)C(=O)NH2, -NHSO2CH3, -N(SO2CH3)2, -NHC(=O)OCH3, -NHC(=O)OtBu,
-NHC(=O)CH3, -SO2NH2, -NHC(=O)CH2CH2OOOH, -NHC(=O)NHCH2COOH,
-CH2NHCHO, -NHC(=O)NHCH2COOEt, -NHC(=O)NH(CH2)3COOEt,
-NHC(=O)NH(CH2)2COOEt, -N(CH3)CH2CH2OH, -NHC(=O)OEt, -N(Et)C(=O)OEt,
-NHC(=O)NH(CH2)2COOH, -NHC(=O)CH2N(CH3)2, -NHC(=O)NH(CH2)3COOH,
-NHC(=O)CH2NH2, -NHC(=O)CH2CH2NH2, -NHC(=O)CH2NH(tBoc),
NICH3
H 3 H
0,3 S~~ N N N NO
`4 o 0 0
NH2 VN N~0
,N II J
O O O CH3 andlslO~~N~/
One embodiment of compounds of the first genus are those in which R3 is methyl
or
fluoromethyl; R6 is H; and M is -CH2- or -CH2O-.
[0067] In an embodiment, R2 is chosen from optionally substituted phenyl,
optionally substituted monocyclic unsaturated heterocycle, unsubstituted
bicyclic
unsaturated heterocycle and fluoro-substituted bicyclic unsaturated
heterocycle. In a
further embodiment, R2 is chosen from optionally substituted phenyl, indole,
benzodioxole, benzoxadiazole, benzodioxan, benzimidazole, oxadiazole,
pyrazole,
pyridine and pyridine N-oxide. In a further embodiment, R2 is chosen from meta-
substituted phenyl, indole, benzodioxole, 2,2-difluorobenzodioxole,
benzooxadiazole,
27
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
benzimidazole, 5-(pyridin-4-yl)[1,2,4]oxadiazole, 5-(pyridin-4-
yl)[1,3,4]oxadiazole,
benzodioxan, 4-chloropyrazole, 4-(pyridin-4-yl)pyrazole, 6-chloropyridine, 3-
(trifluoromethyl)pyrazole, and pyridine N-oxide.
[0068] In another embodiment, R2 is substituted phenyl:
R6
M
R1
R3~ R5
0
R7
R13
8
wherein R7 is chosen from hydrogen, halogen, nitro, cyano, halo(Ci-C6)alkyl,
hydroxy,
(C1-C6)alkoxy, (C1-C6)oxaalkyl, carboxy, (C1-C6)alkoxycarbonyl, aminocarbonyl
(-
CONH2), (Ci-C6)alkylaminocarbonyl, acyl, hydroxy(Ci-C6)alkyl, halo(Ci-
C6)alkoxy,
amino(Ci-C6)alkyl, amino, (C1-C6)alkylamino, di[(C1-C6)alkyl]amino, mercapto,
(Cl-
C6)alkylthio, (C1-C6)alkylsulfinyl, (C1-C6)alkylsulfonyl, (C1-
C6)alkylsulfonamido,
acylamino, amidino, phenyl, benzyl, hetercyclyl, phenoxy, benzyloxy, and
heteroaryloxy; and
R8 and R13 are chosen independently from H and F. In a further embodiment, R8
and R13
are H and R7 is chosen from hydrogen, fluoro, chloro, bromo, nitro, cyano,
acetyl,
trifluoromethyl, methoxy, trifluoromethoxy, oxadiazolyl, tetrazolyl,
methylthio,
methanesulfinyl, methanesulfonyl, methansulfonamido, amino, methoxymethyl,
hydroxyethyl, and morpholinyl.
[0069] In another embodiment, R1 is chosen from optionally substituted phenyl,
optionally substituted five membered heteroaryl, optionally substituted six-
membered
heteroaryl, optionally substituted 4-7 membered non-aryl heterocycle, and
optionally
28
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
substituted fused bicycle.
[0070] For example, R1 may be chosen from optionally substituted phenyls;
optionally substituted five membered heteroaryls selected from thiazoles,
thiadiazoles,
pyrazoles, oxadiazole, isoxazoles, triazoles, imidazoles, thiophenes,
tetrazoles and
oxazoles; optionally substituted six membered hereroaryls selected from
pyridines,
pyrimidines, pyridazinones, pyrimidinone, pyridinone, pyrazines and diazines;
optionally
substituted 5-and 6- membered non-aryl heterocyclics selected from
tetrahydrothiophenes, piperazine, oxazolidinones, imidazolidinones,
morpholines,
piperidines, pyrrolidinones, pyrrolidinediones, pyrrolidines, piperidinones,
piperidinediones and trioxa-bicyclo[2.2.2]octanes; and optionally substituted
fused
bicycles selected from benzoxazolones, indoles, isoindolinediones, 2H-
pyrrolopyridinediones, purines, indolinediones, triazolopyridinones,
benzimidazoles,
benzoxadiazoles, quinolines and quinolones; wherein the substituents are
chosen
independently from hydrogen, halogen, halo(Ci-C6)alkyl, hydroxyl, (C1-
C6)alkoxy,
carboxy, (C1-C6)alkoxycarbonyl, aminocarbonyl (-CONH2), (C1-
C6)alkylaminocarbonyl,
cyano, carbonyl (oxo), acyl, hydroxy(Ci-C6)alkyl, halo (C 1 -C6)alkoxy,
amino(Ci-
C6)alkyl, (C1-C6)alkoxy(Ci-C6)alkyl, nitro, amino, (C1-C6)alkylamino, di[(Ci-
C6)alkyl]amino, mercapto, (Ci-C6)alkylthio, sulfoxide, sulfone, sulfonate,
sulfonimide,
acylamino, amidino, phenyl, benzyl, heteroaryl, phenoxy, benzyloxy,
heteroaryloxy,
aminocarbonyl(Ci-C6)alkyl, (Ci-C6)alkoxycarbonyl(Ci-C6)alkyl, carboxy(Ci-
C6)alkyl,
formylamino(Ci-C6)alkyl, carboxy(Ci-C6)alkylamino, -(CH2)pNR 12CO-(CH2)gNR9Ri0
-NHSO2R'1, -OCH2CH2NR9R10 -NHSO2NR9Ri0, -SO2NR9R", -(CH2)p-NHCOR9,
OCONR9R10 and NR12000Ri 1;
R3 is chosen from -CH3, -CH2CH3, -CF3, -CHF2 and -CH2F;
R5 is chosen from H, -F, -OH, -CH3, -OCH3, -CF3, -CN, -NH2 and -C=CH;
R2 is
(a) phenyl and R7 is chosen from H, halogen, nitro, acetyl, hydroxyethyl, -
NH2, -
SCH3, methoxycarbonyl, -SOCH3, -SO2CH3, -OCH3, -OCF3, -CN, -CF3, -CH2OCH3 ; or
(b) benzoxadiazole, benzodioxole, 2,2-difluorobenzodioxole, benzoxadiazole,
benzodioxan, benzimidazole, oxadiazole, pyrazole, pyridine and pyridine N-
oxide;
R9 is chosen from H, (Ci-C6)alkyl, halo(Ci-C6)alkyl, (Ci-C6)alkoxycarbonyl,
carboxy(Ci-C6)alkyl, (Ci-C6)alkoxycarboxy(Ci-C6)alkyl;
29
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
R10 is H, (Ci-C6)alkyl, or taken together, or
R9 and R10 together form a heterocycle optionally substituted with (Ci-
C6)alkyl;
pis0or1,
gis0,1or2,
R" is linear (Cl-C6)alkyl,
R12 is H or (Cl-C6)alkyl; or
two adjacent substituents together form an optionally substituted fused
heterocyclic ring.
When R5 is H, R3 is -CH3 and R1 is substituted or unsubstituted pyrazole,
compounds
that have been tested and found active are those in which R8 is -NO2 or R8
represents two
adjacent substituents that form an optionally substituted, fused heterocycle.
When R5 is
SOH
N
H, R3 is -CH3 R2 is -CF3 and R1 is 0 , the compound does not appear to be
active in initial screening. When R5 is H, R3 is -CH3, R2 is -NO2, and R1 is
0
--N
NHz
C
0 \\ , the compound does not appear to be active in initial screening. And,
0
when R5 is H, R3 is -CH3, R2 is -OCH3 or -COCH3, and R1 is , the compound
does not appear to be active in initial screening.
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[0071] One embodiment of compounds of the second genus has the formula
R6
M R14
a
I R15
R3 0 R5
R27
R28
wherein
Ria is phenyl, five-membered heteroaryl, six-membered heteroaryl, 4-7 membered
non-
aryl heterocycle or fused bicycle;
R14 is chosen from H, -CH2NHC(=O)NH2, -NHC(=O)NH2, -NHC(=O)NHEt, -CH3,
-CH2CF3, -CH2NHC(=O)CH3, -NHCH3, -NHEt, -NH(tBoc), -CHO,
-NHC(=O)NHCH2CH2C1, -NHSO2NH2, -NHEt, -N(CH3)2, -NH2, -000H,
-C(=O)NH2, -CH2C(=O)NH2, -CH2COOH, -CH2COOEt, -CN, -OCH3,
-OC(=O)NH2, -NH(CH3)C(=O)NH2, halogen, -CH2NHC(=O)OEt, -NHSO2CH3,
-N(SO2CH3)2, -NHC(=O)OCH3, -OH, -CH2NHC(=O)N(CH3)2, -CH2NH2,
-CH2OH, -CH2CH2OH, -SO2NH2, -NHC(=O)NHCH2COOH, -CH2NHCHO,
-NHC(=O)NHCH2COOEt, -COOCH3,, -COOEt, -NHC(=O)NH(CH2)3COOEt,
-NHC(=O)NH(CH2)2COOEt, -NH(Et)C(=O)OEt, -NHC(=O)NH(CH2)2COOH,
-CH2NHSO2CH3, -OEt, -NHC(=O)CH2N(CH3)2, -NHC(=O)NH(CH2)3COOH,
-NHC(=O)CH2NH2, -NHC(=O)CH2CH2NH2, -NHC(=O)CH2NH(tBoc),
-OCH2CH2N(CH3)2, -OCH2CH2OCH3, 3'-nitro-6-methoxybiphenyl-3-ylmethyl,
tetrahydroimidazol-2-on-1-yl, 3-methyltetrahydroimidazol-2-one-1-yl, pyrazol-
31
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
N'CH3
IS,
1-yl, O
`,N N 00
O and O ;
R'5 is chosen from H, NO2, OH, NH2, and -NHSO2NH2; or
R'5 together with R14 forms methylene dioxy;
R27 is chosen from hydrogen, halogen, nitro, cyano, halo(Cl-C6)alkyl, hydroxy,
(Cl-
C6)alkoxy, (C1-C6)oxaalkyl, carboxy, (C1-C6)alkoxycarbonyl, aminocarbonyl (-
CONH2), (Cl-C6)alkylaminocarbonyl, acyl, hydroxy(Cl-C6)alkyl, halo(Cl-
C6)alkoxy, amino(Cl-C6)alkyl, amino, (Cl-C6)alkylamino, di[(Cl-
C6)alkyl] amino, mercapto, (Cl-C6)alkylthio, (Cl-C6)alkylsulfinyl, (Cl-
C6)alkylsulfonyl, (Cl-C6)alkylsulfonamido, acylamino, amidino, phenyl, benzyl,
hetercyclyl, phenoxy, benzyloxy, and heteroaryloxy;
R28 is chosen from H and F, or
R27 together with R28 forms a five-membered ring.
[0072] In further embodiments R27 and R28 represent a fused heterocycle at 3-
and 4-
positions so that the residue formed from R27 and R28 together with the phenyl
to which
they are attached is chosen from:
O \ \I N
! - / H1 - and N /
[0073] In other embodiments, R27 is chosen from halogen, nitro, acetyl,
hydroxyethyl,
amino, methylthio, trifluoromethyl, methoxymethyl, methoxycarbonyl,
trifluoromethoxy,
cyano and 1,3,4-thiadiazol-2-yl, or taken together R7 and R8 are
methylenedioxy or
difluoromethylenedioxy. In the foregoing embodiments, R1 may be chosen from a
benzene ring, a triazole, a pyridine or pyridine-N-oxide, a pyrazole, a
tetrahydrothiophene, an imidazole, a pyrimidine, a thiadiazole, and an
imidazopyridine.
32
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[0074] In an embodiment of the invention, R5 is fluoro, H, CN or OH. In other
embodiments, R3 is methyl or fluoromethyl.
[0075] Another embodiment of compounds of the invention have the formula
M Y
R16
R3
'--- 0
R27a
[0076] In these compounds R3 is methyl or fluorinated methyl; Y is CH or N;
R27a is
chosen from halogen, cyano, acetyl, methylthio, nitro and trifluoromethyl; and
R16 is
R20
chosen from -NR 17C(=O)NR18R19 and
N
wherein is a 4-7 membered ring heterocycle attached through its
nitrogen; R17, and R'8 are independently chosen from H, (Cl-C6)alkyl and
halo(Cl-
C6)alkyl; R19 is chosen from H, (Cl-C6)alkyl, halo(Cl-C6)alkyl, -[(C1-
C6)alkyl]000H,
and -[(C1-C6)alkyl]000(C1-C6)alkyl; and R20 is chosen from a carboxylic acid,
a
carboxamide, a carboxylic ester, a primary, secondary or tertiary alcohol and
a primary,
secondary or tertiary amine. Examples of a carboxylic acid, a carboxamide, a
carboxylic
ester, a primary, secondary or tertiary alcohol and a primary, secondary or
tertiary amine
include -000H, -CONH2, -CONHCH3, -CON(CH3)2, -COOCH3, -CH2OH,
33
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
-CH(CH3)OH, -C(CH3)20H, -CH2NH2, -CH(CH3)NH2 and -C(CH3)2NH2. In a further
embodiment, X is CH, CF or N-->O; M is -CH2- or -S-; R27a is chosen from
chloro,
cyano, acetyl and methylthio; and R16 is chosen from -NR17C(=O)NR'8R19
N Y NR19
N~
R2
0 and . In a further embodiment, Y is
CH; M is -CH2-; R27a is chloro; and R16 is -NR 17C(=O)NR18R19. In yet a
further
embodiment, R16 is -NR 17C(=O)NR18R19 and R17, R18 and R19 are all hydrogen.
[0077] Examples of the foregoing substituents on phenyl, five-membered
heteroaryl, six-
membered heteroaryl, 4-7 membered non-aryl heterocycle or fused bicycles (Rh)
may be
described by the formula VI:
R6
M R14
a
R15
R3'___ R5
O
JR
R13 \/\ R8
VI
in which
R14 is chosen from H, -CH2NHC(=O)NH2, -NHC(=O)NH2, -NHC(=O)NHEt, -CH3,
-CH2CF3, -CH2NHC(=O)CH3, -NHCH3, -NHEt, -NH(tBoc), -CHO,
-NHC(=O)NHCH2CH2C1, -NHSO2NH2, -NHEt, -N(CH3)2, -NH2, -000H,
-C(=O)NH2, -CH2C(=O)NH2, -CH2COOH, -CH2COOEt, -CN, -OCH3,
-OC(=O)NH2, -NH(CH3)C(=O)NH2, halogen, -CH2NHC(=O)OEt, -NHSO2CH3,
-N(SO2CH3)2, -NHC(=O)OCH3, -OH, -CH2NHC(=O)N(CH3)2, -CH2NH2,
-CH2OH, -CH2CH2OH, -SO2NH2, -NHC(=O)NHCH2COOH, -CH2NHCHO,
-NHC(=O)NHCH2COOEt, -COOCH3,, -COOEt, -NHC(=O)NH(CH2)3COOEt,
34
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
-NHC(=O)NH(CH2)2COOEt, -NH(Et)C(=O)OEt, -NHC(=O)NH(CH2)2COOH,
-CH2NHSO2CH3, -OEt, -NHC(=O)CH2N(CH3)2, -NHC(=O)NH(CH2)3COOH,
-NHC(=O)CH2NH2, -NHC(=O)CH2CH2NH2, -NHC(=O)CH2NH(tBoc),
-OCH2CH2N(CH3)2, -OCH2CH2OCH3, 3'-nitro-6-methoxybiphenyl-3-ylmethyl,
tetrahydroimidazol-2-on-l-yl, 3 -methyltetrahydroimidazol-2-one-l-yl, pyrazol-
~N'CH3
~ ~~N\ /N J N
1-yl, O O
`,N N O
z
O and O
R15 is chosen from H, NO2, OH, NI-12, and -NHS02NH2; or
R'5 together with R14 forms methylene dioxy.
[0078] In certain embodiments, R3 is chosen from -CH3, -CH2CH3, -CF3, -CHF2
and -
CH2F. In certain embodiments, R5 is chosen from H, -F, -OH, -CH3, -OCH3, -CF3,
-CN,
-NH2 and -C--CH. In certain embodiments, R7, is chosen from H, halogen, nitro,
acetyl,
hydroxyethyl, -NH2, -SCH3, methoxycarbonyl, -SOCH3, -SO2CH3, -OCH3, -OCF3, -
CN,
-CF3, -CH2OCH3 and oxadiazole, and a fused heterocycle at 3- and 4- positions.
[0079] In certain embodiments of the compounds I, R5 may be fluoro, H, CN or
OR In
certain embodiments of the compounds I, R3 may be methyl or fluoromethyl.
(Fluoromethyl is intended to include CHF2, CH2F and CF3.)
[0080] All of the compounds, except for those identified in paragraph [0069],
falling
within the foregoing parent genus I and its subgenera are useful as PDE4
inhibitors, but
not all the compounds are novel. In particular, certain known species fall
within the
genus I, although no utility in inhibiting PDE4 has been suggested for these
species. It
may be found upon examination that compounds that have been excluded from the
claims are patentable to the inventors in this application; it may also be
found that
additional species and genera not presently excluded are not patentable to the
inventors
in this application. In either case, the exclusion of species and genera in
applicants'
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
claims are to be considered artifacts of patent prosecution and not reflective
of the
inventors' concept or description of their invention. The invention, in a
composition
aspect, is all active compounds of formula I except those that are in the
public's
possession. In particular, a search of the literature indicates that certain
compounds in
which X is N, R3 is methyl, M is CH2, R2 is a five-membered ring heterocycle,
and R1 is
a substituted tetralin are known. Similarly certain compounds in which X is N,
R3 is
methyl, M is CH2, R1 is a five-membered ring heterocycle, and R2 is a
substituted tetralin
are known.
[0081] In general, the compounds of the present invention may be prepared by
the
methods illustrated in the general reaction schemes as, for example, described
below, or
by modifications thereof, using readily available starting materials, reagents
and
conventional synthesis procedures.
[0082] Generally compounds of the Formula I, where R2 is a substituted aryl /
heteroaryl
and the two biaryl groups are linked by a C-C bond, may be prepared from
appropriately
functionalized alkoxy-aryl ether derivatives containing desirable
functionalities W,
where W may for example be CH, N, COH, CF, etc (Route A, Scheme Al). The
biaryl
portion can be constructed first, typically via Suzuki or Stille coupling (G1 -
> G2). In
such case either Y = halogen or OS02R (OTf, ONf) and the other reagent would
be R2-
B(OR)2 or R2-SnR3' or vise versa, where R2-halogen is coupled with G1
containing
boronate/boronic acid or trialkyltin as Y. When A is a carbon derived
substituent, e.g.
CH3, CH2OH, CO2R", CN etc. these groups are converted to provide intermediate
G3
where D is either a halogen or OTf, ONf, or OCOOR" (carbonate) such that
substituent
(Rl) is introduced by employing a transition-metal catalyzed coupling
reactions such as
Suzuki, Stille or Negishi reaction. An alternative route to compounds of type
G4
involves essentially reversing the order of incorporation of R1 and R2
fragments. The
route B, as highlighted in Scheme Al, allows formation of G6, where the R2
fragment is
introduced at later stage in the sequence, analogous to chemistries employed
for G1-G2,
for examples Suzuki, Stille coupling.
36
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[0083] Scheme Al
R6 R6
A R6
[A] q CT~M'D
R3.0 .W R3. 0
CYW R3.0 W
Y G7 R2 G2 R2 G3
[Bl
R6 R6
R6
C R1 C M'R1
6 M'D M
R3. W R3.0 R3.0 I . W
0 Y R2
Y G5 G6 G4
[0084] One may attach Rl, which may be aryl, heterocyclic, acyclic, aliphatic,
or any
other desirable variety of functionality, to the central aromatic ring (Ar) by
a wide rage
of tether groups M. The central aromatic ring (Ar) may be a biaryl ring system
with a R2
group already attached, or the R2 group may be attached subsequent to that of
Rl. The
linker group M may be a linear chain of one or more atoms consisting of C, N,
0, or S.
The linker group M may also consist of functionalities including, but not
limited to
amide, sulfonamide, sulfone, or ketone. It is evident to one skilled in the
art that many of
these exemplified functional groups may be attached in more than one way, for
example,
Ar-CH2-O-Rl or Ar-O-CH2-Rl. Considering those compounds with M groups such as
S or 0, the heteroatom may originally be in the Ar or the R1 group. Some
examples of
how the two groups can then be joined are nucleophilic aromatic substitution,
metal-
promoted coupling, and nucleophilic displacement (Scheme A2). Chemical
reactivity of
the Ar and R1 groups should of course be considered when determining which
partner
contains the linker heteroatom, and which shall serve as the reaction partner.
For
example, it is well understood that in aromatic ring systems an electron-
withdrawing
group para- or ortho- to a leaving group (e.g. halogen) allows susceptibility
of the
aromatic halogen for nucleophilic displacement. Thus, Arl group containing
NO2,
CO2R, ketone, CN etc. would allow formation of aryl-M-aryl(heteroaryl)
intermediates.
The linker group M may also be subject to further elaboration. For example,
sulfides
may be oxidized to sulfoxides and sulfones and amines may be subjected to
alkylation or
reductive amination. Well known synthetic transformations can be used to
create tether
37
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
groups M such as ether amide, sulfonamide, and the like. The functional group
location
in the precursor Ar and RI groups can be used to dictate the nature and type
of the
linkage (e.g. alternative ethers), as mentioned above.
[0085] Scheme A2
R6 R6
M. R1
R1IO I W
R3,0 CW1
G7 (Y/R2) Z-Ar(EWG) (Y/R2)
G8
(A=OH, SH e.g.) (e.g. M= 0, S(O)n=0-2)
[0086] The key fragments Ar and R1 could also be joined using non transition-
metal
catalyzed C-C bond forming coupling reactions. When A=H, Fridele-Crafts
acylation or
alkylation can be employed to combine the Ar and RI groups. Given the chemical
reactivity of the aromatic para-methoxy group in the Ar ring, Friedel-Crafts
acylation
would typically involve a suitably elaborated R1-0001 (acid chloride) group to
form
compound G10. In this case J = CO, which can be reduced to the secondary
alcohol
(typically with hydride-based reducing agents). If a R'MgX or other such
organometallic agent is used, the J = CO group can be transformed into the
tertiary
alcohol with simultaneous addition of an R' group. In another variation, the J
= CO
group can be converted into the imine or oxime using standard procedures and
addition
of an R'MgX-type reagent results in the tertiary amine derivative. If desired,
the J = CO
group can be reduced, using a number of well established methods, to the CH2
group
(M). In another variation, when A=H, an aldehyde group can be introduced using
either
Vilsmeier reaction or using Lewis acid (TiC14, BF3.OEt2 etc.) mediated
reaction with
dichloromethylmethyl. The aldehyde functionality can subsequently be
transformed into
a suitable transition-metal catalyzed coupling reaction partner. Alternatively
the aldehye
could be used for Wittig reaction forming olefin or CH2CH2 linkage to
incorporate RI.
In yet another variation, substituent "A" can be various types of carbonyl
(aldehyde or
ketone) or imine groups. In one example, addition of a suitably elaborated
organometallic RI group (e.g. R1-MgX) to aldehyde G9 (A = CHO) would result in
G10
38
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
with J = C(H)OH. Reduction of this alcohol gives rise to Gil with M = CH2.
Similar
types of transformations could be employed by one skilled in the art if G9
contained A =
ketone or imine.
[0087] Scheme A3
A A=H
I~r Friedel-Crafts, Formytion J, R1 M, R1
R3.0 . W R3. I. W R3. . W
A = CHO, COR, C=N-Rb O Reduction, 0
(Y/R 2) Metal-mediated addition (Y/R2) RM addition (Y/R2)
(Grignard)
G9 or Wittig Reaction G10 G11
[0088] Alternatively, the C-C bond forming reaction between the Ar and RI
groups
could be accomplished by displacement of a leaving group on the RI by a
nucleophile
present in the tether region M (Scheme G4) of G12. The activating group could
be either
removed to provide Z=H (Z=CO2R - decarboxylation of or Z=CN, decyanation) or
these
could be further transformed to other functional groups e.g. Z = CH2)H or
CH2NH2.
When M-Z is CH2-halide or CH2-O-sulfonate, RI fragment can be introduced via
formation of ether linkage. This allows attachment of R1 to the central
aromatic ring by
spacers (M) of varying lengths and compositions. (Scheme A4)
[0089] Scheme A4
R6 z R6 z R6
R1 M'R1 T) ~
R3.0 I . W R3.0 I W R3.0 I . W
R2 Z=CO2Ra, CN R2 R2
G12 G13 G14
[0090] The R1 group could also be assembled form an acyclic intermediate to
form a
heterocylic or heteroaromatic ring. Examples of these chemistries include
formation of
5-membered heteroaryls such as oxadiazole, thiadiazole, triazole (G17) form
acyl
hydrazide (G16); thiazole from 2-halo-ketone or dipolar cycloaddition
reactions from
olefin or acetylic group to form 5-membered heterocycles or 5-membered
heteroaryls
(G18) [Scheme A5]. Alternatively the 6-membered heteroaryl or heterocyclic
rings could
39
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
be formed using Diels-Alder or hetero- Diels-Alder chemistries using
appropriately
substituted alkyl aryl ether bearing either a dienophile or a diene
functionalities. The
necessary acyclic precursors could be synthesized by standard methods
according to
previously described intermediates (e.g. aldehyde, alkyl halide) schemes.
[0091] Scheme A5
R6 0 R6
U
N R3. I. W N-N Ra
L=C02H O
R6 ~ :2) L G17 U=O,S,NR
R3.0 I W L= CH=CH2 or C=CH
(Y/R2) R6
G15 \\a~ I \ x', - single or double bond
Nitrile-oxide R1 `0 W
or nitrone
e.g. isoxazole, isoxazoline etc.
R2 G18
[0092] When the R2 group is linked to the Ar group thru a heteroatom (N),
these biaryl
systems could be prepared by organometallic mediated aza-coupling reactions or
other
nucleophilic aromatic substitution-based procedures (Scheme G6). The Ar-(N)R2
biaryl
may be formed from intermediate G6 where R1 group is already in place.
Alternatively,
the (N)R2 ring can be added to the central Ar ring first, RI can be attached
through a
variety of means using approaches described in previous schemes. Examples of
(HN)R2
heteroaryl or heterocyclic rings include, but not limited to, imidazole,
pyrrole, pyrazole,
pyrrolidine, or triazole. The R2 functional group can be fully elaborated
prior to addition
of the (N)R2 to the Ar, or suitably elaborated after formation of the key C-N
bond.
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[0093] Scheme A6
R6 R6
M, R1 I M, R1
R3.0 -W R3.0 ,W
Y G6 0 G19
[0094] The diverse selection of substituents present in R1 and R2 could be
formed by
standard functional group transformations that are well know in the art. Some
of these
include formation of amide, sulfonamide, ureas, imidazolone, oxazolones,
carbamates
from the R2, R3, or Ar ring fragments bearing appropriate amine, carboxylic
acid,
alcohol, or phenol groups. A particularly useful aromatic ring
functionalization
technique, in which either the R2 or RI rings can be employed, is the
nucleophilic
displacement of ortho-halo N-containing aromatic rings (G20, scheme A7).
Examples of
ring substrates useful in this type of transformation include 2-halo-pyridine,
2-halo-
pyrimidine and 2-halo-imidazole. Additionally, other leaving groups besides
halogens
(X) may be used such as sulfonate esters (OTf, ONf). These displacement
reactions can
be carried out using alkali or tertiary amine bases, or could be mediated
through the use
of an organometallic reagent such as palladium or aluminum reagents. Examples
of
nucleophiles (R") useful in this type of transformation include amines
(primary,
secondary, acyclic, or cyclic), alcohols, phenols, NH-containing heterocycle
groups
(imidazole, or pyrrazole) groups capable of performing nucleophilic
displacement.
[0095] Scheme A7
R6 R6
M (e.g. Q=F, Br) M IN- R3.0 CW ~/ Q H-Nuc R3. OI W/ Nuc
Y/R2 Y/R2
G20 G21
[0096] When RI group contains additional functional groups, such as amine,
ester/acid
/alcohols many of which may have be masked or protected during the previous
41
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
chemistries, these could be used for further functional group manipulations. A
wide
variety of modifications of R1 functionalities may be achieved using well
established
synthetic procedures including, but not limited to, alkylation, reductive
amination,
nucleophilic displacement, cyclization, saponification, and
oxidation/reduction.
Additionally, like these functional group manipulations, Arl mono-cyclic maybe
further
transformed to a bi-cyclic ring. Examples of such ring transformations may be
represented by elaboration of pyridine derivatives to imidazo[1,2-a]pyridine
and
imidazo[1,5-a]pyridine. These functional group manipulations and bicyclic ring
elaborations maybe accomplished at any chemically suitable point in the
synthesis prior
to or post incorporation of R2 or other synthetic transformations.
[0097] These above transformations could be carried out from alkylated phenols
containing or lacking fluoro substituents in the central Ar ring. Several of
these
approaches are also applicable to 3 -alkoxy pyridines as the Ar ring starting
materials.
The non-limiting specific examples described in later schemes are meant to
serve as
examples of the broad scope of possible reactions. Similarly, analogs where W
=
CH2OH, COOH, CN, CONH2 etc. (or suitably protected precursors) could be
derived by
following similar chemistries (schemes Al-A7) and these functional groups
could be
derived form an ester or amide derived starting material.
[0098] The following examples of compounds of the invention were prepared.
Table 1.
R6
R3,0 X / R.
Rb
Except Examplees P-093 and P-094 where R6=F, for all other Examplees in Table
1 R6=H.
Example X R3 M Rb Ra
P-001 C-H CH3 CH2 3-NO2 4-F
P-002 C-H CH3 CH2 3-CO2CH3 4-F
P-003 C-H CH3 CH2 3,4-OCH2O 4-F
P-004 C-H CH3 CH2 3,4-NON 4-F
P-013 C-H CH3 CH2 3,4-NON 3,4-NON
42
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example X R3 M Rb Ra
P-021 N CH3 CH2 3-NO2 3,4-OCH2O
P-023 N CH3 CH2 3-NO2 4-F
P-049 C-F CH3 C-0 3-NO2 4-F
P-050 C-F CH3 CH2 3-NO2 4-F
P-051 C-F CH3 CH(OH) 3-NO2 4-F
P-054 N-O CH3 CH2 3-NO2 4-F
P-057 N CHF2 CH2 3-NO2 4-F
P-065 N CH3 CH2 3-CF3 4-F
P-067 C-H CHF2 CH2 3-NO2 4-F
P-079 C-H CF3 CH(OH) 3-NO2 4-F
P-080 C-H CF3 CH2 3-NO2 4-F
P-093 C-H CH3 CH(OH) 3-NO2 4-F
P-094 C-H CH3 CH2 3-NO2 4-F
P-095 C-OCH3 CH3 C-0 3-NO2 4-F
P-096 C-OH CH3 C-0 3-NO2 4-F
P-097 C-OCH3 CH3 CH2 3-NO2 4-F
P-098 C-F CH3 CH2 3-NH2 4-F
P-099 C-OH CH3 CH2 3-NO2 4-F
P-102 C-H CH3 CH2 3-NO2 3-OH
P-103 C-H CH3 CH2 3-NO2 3-CH2OH
P-105 C-H CH3 CH2 3-NO2 2-OH
P-107 C-F CH3 CH2 3-NO2 4-NHCOCH3
P-112 C-H CH3 CH2 3-NO2 4-NHCOOC(CH3)3
P-113 C-H CH3 CH2 3-NO2 4-NH2
P-114 C-H CH3 CH2 3-NO2 4-NHCOCH3
P-116 C-F CHF2 CH2 3-NO2 4-F
P-117 C-H CH3 CH(OH) 3-[2-Oxadiazole] 4-F
P-118 C-H CH3 CH2 3-[2-Oxadiazole] 4-F
P-119 C-H CH3 CH2 3-NO2 2-OH, 4-F
P-121 C-H CH3 CH2 3-NO2 N(S02CH3)2
P-122 C-H CH3 CH2 3-NO2 NHCOCH2CH2COOH
P-123 C-H CH3 CH2 3-NO2 4-NHSO2CH3
P-133 C-H CH3 CH2 3-NO2 2-CH2NH2
P-134 C-H CH3 CH2 3-NO2 2-CH2OH
P-135 C-H CH3 CH2 3-NO2 3-CH2OH, 4-F
P-136 C-F CH3 CH2 3-NO2 4-NH2
P-137 C-OH CH3 CH(OH) 3-NO2 4-CN
P-138 C-OH CH3 CH2 3-NO2 4-CN
P-139 C-F CH3 CH2 3-Br 4-F
P-140 C-H CH3 CH2 3-000H3 4-F
P-141 C-H CH3 CH2 3,4-OCF2O 4-F
P-142 C-H CH3 CH2 3-NO2 4-OH
P-143 C-H CH3 CH2 3-NO2 4-NHCOCH2NHCOOC(CH3)3
P-144 C-H CH3 CH2 3-NO2 4-NHCOCH2N(CH3)2
P-145 C-H CH3 CH2 3-NO2 4-NHCOCH2NH2
P-146 C-H CH3 CH2 3-NO2 4-NHCOCH2CH2N(CH2)5
P-147 C-F CH3 CH2 3-NO2 4-NHCOCH2N(CH3)2
P-148 C-F CH3 CH2 3-NO2 4-CH2NH2
P-151 C-H CH3 CH2 3-SCH3 4-F
P-157 C-OH CH3 CH2 3-NO2 4-N(CH3)2
43
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example X R3 M Rb Ra
P-158 C-H CH3 CH2 3-NO2 2-CH2NHCHO
P-159 C-H CH3 CH2 3-NO2 4-NHCOCH2CH2NH2
P-160 C-H CH3 CH2 3-NO2 3-CH2NHCOOC(CH3)3
P-161 C-H CH3 CH2 3-NO2 3-CH2NH2
P-163 C-CN CH3 CH2 3-NO2 4-F
P-164 C-H CH3 CH2 3-NO2 4-OCH2CH2N(CH3)2
P-165 C-H CH3 CH2 3-NO2 4-OCH2CH2-N(pyrrolidine)
P-166 C-H CH3 CH2 3-NO2 4-OCH2CH2-N(piperidine)
P-167 C-F CH3 CH2 3-NH2 4-NHCOCH3
P-168 C-F CH3 CH2 3-Br 4-NHCOCH3
P-170 C-H CH3 CH2 3-COCH3,4- 4-F
OCH3
P-171 C-H CH3 CH2 3-NO2 4-OCH2CH2-N(morpholine)
P-173 C-OH CH3 CH2 3-NO2 4-(N-pyrazole)
P-175 C-F CH3 CH2 3-NO2 4-CH2NHCOCH2N(CH3)2
P-176 C-NH2 CH3 CH2 3-NO2 4-F
P-180 C-OH CH3 CH2 3-NO2 4-CHO
P-181 C-F CH3 CH2 3-NO2 4-NHCONH2
P-183 C-OH CH3 CH2 3-NO2 4-CH2OH
P-187 C-F CH3 CH2 3-CI 4-F
P-189 C-H CH3 CH2-S 3-NO2 4-F
P-190 C-OH CH3 CH2 3-NO2 4-NHCOCH3
P-191 C-H CH3 CH2-SO2 3-NO2 4-F
P-192 C-H CH3 CH2-SO 3-NO2 4-F
P-193 C-H CH3 0 3-NO2 4-F
P-194 C-F CH3 CH2-O 3-Br 4-NHCOCH3
P-199 C-F CH3 CH2-O 3-Br 4-NH2
P-200 C-F CH3 CH2 3-Br 4-NHSO2CH3
P-202 C-F CH3 CH2-O 3-Br 4-NHSO2CH3
P-216 C-F CH3 CH2 3-CI 4-OH
P-217 C-F CH3 CH2 3-CI 3-OH
P-219 C-F CH3 CH2 3-CI 4-OCH2CH2-N(morpholine)
P-220 C-F CH3 CH2 3-CI 3-OCH2CH2-N(morpholine)
P-221 C-F CH3 CH2 3-CI 4-OCONH2
P-222 C-F CH3 CH2 3-CI 3-OCONH2
P-227 C-F CH3 CH2 3-CI 4-NHSO2CH3
P-228 C-F CH3 CH2 3-CI 4-NHCOCH3
P-230 C-H CH3 0 3-CI 4-F
P-231 C-F CH3 CH2-O 3-CI 3-NHCOCH3
P-232 C-F CH3 CH2-O 3-CI 3-NH2
P-233 C-F CH3 CH2 3-CI 3-NHCOCH3
P-234 C-F CH3 CH2 3-CI 3-NH2
P-237 C-F CH3 CH2 3-NO2 4-CH2NHCONH2
P-238 C-F CH3 CH2 3-CI 4-NHCOCH2N(CH3)2
P-239 C-F CH3 CH2 3-CI 4-NHSO2NH2
P-240 C-H CH3 CH2 3-CI 4-CONH2
P-241 C-H CH3 CH2 3-CI 4-N(CH3)2
P-242 C-F CH3 CH2 3-CI 3,4-OCH2O
P-243 C-F CH3 CH2 3-CI 4-NHCONH2
P-246 C-H CH3 CH2 3-CI 4-NHSO2CH3
44
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example X R3 M Rb Ra
P-247 C-H CH3 CH2 3-CI 4-NHCH2CH3
P-249 C-F CH3 CH2 3-000H3 4-NHSO2CH3
P-250 C-F CH3 CH2 3-CI 4-NHCONHCH2CH3
P-254 C-F CH3 0 3-CI 4-NHSO2CH3
P-255 C-H CH3 CH2 3-CI 3-NH2
P-256 C-F CH3 CH2 3-CI 3-OH, 4-NH2
P-257 C-F CH3 CH2 3-CI 3-OH, 4-NO2
P-260 C-H CH3 NH 3-000H3 4-NH2
P-261 C-F CH3 CH2 3-000H3 4-NH2
P-262 C-F CH3 CH2 3-CI 3-OH, 4-NHCONH2
P-264 C-F CH3 CH2 3-CI 3-OH, 4-NHSO2CH3
P-265 C-F CH3 CH2 3-CI 3-OSO2CH3, 4-NHSO2CH3
P-266 C-H CH3 CH2 3-CI 4-SO2NH2
P-267 C-H CH3 NH 3-000H3 4-NHSO2CH3
P-268 C-H CH3 NH 3-000H3 4-NHCONH2
P-269 C-F CH3 CH2 3-000H3 4-NHCONH2
P-270 C-H CH3 CH2 3-CI 3-NHCONH2
P-271 C-H CH3 CH2 3-CI 3-NHSO2CH3
P-273 C-F CH3 CH2 3-CI 4-NHCOCH2NH2
P-274 C-F CH3 CH2 3-CI 3-F, 4-CN
P-275 C-F CH3 CH2 3-CI 3-F, 4-CH2NH2
P-276 C-F CH3 CH2 3-CI 3-OH, 4-NHSO2NH2
P-280 C-H CH3 NH 3-CI 4-NH2
P-282 C-H CH3 CH2 3-CI 3-CONH2
P-283 C-F CH3 0 3-000H3 4-NH2
P-286 C-F CH3 CH2 3-CI 3-F, 4-CH2NHSO2CH3
P-287 C-F CH3 CH2 3-CI 3-F, 4-CH2NHCONH2
P-288 C-H CH3 N(CH3) 3-CI 4-NH2
P-289 C-F CH3 CH2 3-CI 4-NHCOCH2NHCH3
P-293 C-H CH3 N(CH3) 3-CI 4-NHCONH2
P-294 C-H CH3 NH 3-CI 4-NHCONH2
P-295 C-F CH3 CH2 3-CI 3-OH, 4-CN
P-296 C-F CH3 CH2 3-CI 3-OH, 4-CH2NH2
P-299 C-F CH3 CH2 3-CI 3-OH, 4-CH2NHSO2CH3
P-300 C-F CH3 CH2 3-CI 3-F, 4-CH2NHCOCH3
P-301 C-F CH3 CH2 3-CI 3-OH, 4-CH2NHCONH2
P-302 C-F CH3 CH2 3-CI 3-OH, 4-CH2NHCOCH3
P-303 C-F CH3 CH2 3-CI 4-N(CH3)2
P-304 C-F CH3 CH2 3-000H3 4-SO2NH2
P-305 C-F CH3 CH2 3-000H3 4-CONH2
P-311 C-F CH3 CH2 3-CI 4-NHCH3
P-312 C-F CH3 CH2 3-000H3 4-NHSO2NH2
P-313 C-F CHF2 CH2 3-Br 4-NHCOCH3
P-316 C-F CHF2 CH2 3-NO2 4-NHCONH2
P-317 C-F CHF2 CH2 3-Br 4-NH2
P-319 C-F CH3 CH2 3-000H3 4-COOCH3
P-320 C-F CH3 CH2 3-000H3 4-COOH
P-322 C-F CH2CH3 CH2 3-Br 4-NHCOCH3
P-323 C-F CHF2 CH2 3-Br 4-NHCONH2
P-324 C-F CH3 CH2 3-CI 3-OH, 5-NH2
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example X R3 M Rb Ra
P-326 C-F CH3 CH2 3-000H3 4-N(CH3)2
P-329 C-F CH3 CH2 3-000H3 3-F, 4-CH2NHCOCH3
P-330 C-F CH3 CH2 3-000H3 3-F, 4-CH2NHCONH2
P-331 C-F CH3 CH2 3-000H3 4-NHCH3
P-334 C-F CH2CH3 CH2 3-000H3 4-NH2
P-335 C-F CH2CH3 CH2 3-000H3 4-NHCONH2
P-338 C-F CH3 CH2 3-000H3 4-N(CH3)CONH2
P-340 C-F CH3 CH2 3-CH(OH)CH3 4-NHCONH2
P-347 C-F CH3 CH2 3-Br 4-NHCONH2
P-348 C-F CH3 CH2 3-Br 4-NHCONHCH2COOH
P-349 C-F CH3 CH2 3-Br 4-NHCONHCH2CH2COOH
P-376 N CH3 CH2 3-CI 4-NHCONH2
P-378 C-H CH3 CH2 3-CI 4-NHCONH2
P-380 C-H CH3 CH2 N1-(3-CI 4-F
Pyrrazole)
P-381 C-F CH3 CH2 N-Pyrrolidine 4-NHCOCH3
P-385 C-H CH3 CH2 N-morpholine 4-F
P-390 C-F CH3 CH2 N-morpholine 4-NHCOCH3
P-394 C-H CH3 CH2 3-CN 4-NHCONH2
P-404 C-F CH3 CH2 3-CI 4-(Nl-tetrazole)
P-413 C-F CH3 CH2 3-CN 4-NHCONH2
P-418 C-F CH3 CH-CH 3-CI 4-F
P-419 C-F CH3 CH-CH 3-CI 4-F
P-420 C-H CHF2 CH2 3-CI 4-NHCONH2
P-421 CH H CH2 3-CI 4-NHCONH2
P-434 C-F CH3 C-0 3-CI 4-NHCONH2
P-441 C-F CH3 C(CH3)(OH) 3-CI 4-NHCONH2
P-447 C-H CH3 S 3-CI 4-NH2
P-448 C-H CH3 S 3-CI 4-NHCONH-Et
P-449 C-H CH3 SO 3-CI 4-NHCONH-Et
P-450 C-H CH3 S02 3-CI 4-NHCONH-Et
P-451 C-F H CH2 3-NO2 4-F
P-453 C-F CH3 CH2 3-Br 4-NH2
P-454 C-H CH3 CH2 3-CI 4-NH(2-Thiazolyl)
P-466 C-F CH3 CH2 3-CI 4-NH(2-Thiazolyl)
P-467 C-H CH3 CH2 3-CI 4-NH(N-Methyl 2- imidazolyl)
P-468 C-F H CH2 3-CI 4-NHCOCH3
P-476 C-H CH3 CH2 3-CI 4-NHCH3
P-477 C-H CH3 CH2 3-CI 3-NHCOCH3
P-453 C-F CH3 CH2 3Br 4-NH2
P-494 C-H H CH2 3-N02 4-F
P-496 C-F H CH2 3-Br 4-F
P-497 C-F CH3 CH2 3-Br 4-F
P-498 C-H CH3 CH2 3-N02 4-OH
P-501 C-H CH3 CH2 3-N02 2-CH2NHCOH
P-502 C-F CH3 CH2 3-N02 4-CH2NHCOCH2NMe2
P-505 C-F CH3 CH2-NH 3-CI 4-NHACc
P-508 C-F H CH2 3-N02 4-NHCONH2
P-516 C-H CD3 CH2 3-CI 4-NHCONH2
P-530 C-F CH3 C CH3 OH 3-CI 4-NHAc
P-532 C-F CH3 CD2 3-CI 4-NHCONH2
46
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example X R3 M Rb Ra
P-537 C-F CH3 CD(OH) 3-CI 4-NO2
P-538 C-F CH3 CD(OH) 3-CI 4-NH2
P-539 C-F CH3 CD(OH) 3-CI 4-NHCONH2
P-540 C-H CH3 S 3-CI 4-NHCONH2
P-541 C-H CH2CH3 CH2 3-CI 4-NHCONH2
P-542 H2NCH2C
C-H H2- CH2 3-CI 4-NHCONH2
P-543 2-
(Tetarhydr
ofuranyl)C
C-H H2 CH2 3-CI 4-NHCONH2
P-547 C-H CH3 CD2 3-CI 4-NHCONH2
P-548 (Me)CNCH
C-H 2CH2- CH2 3-CI 4-NHCONH2
P-550 C-H CH3 CH2 3-CI 4-NHCONH2
P-553 3-
C-H thiaten I CH2 3-CI 4-NHCONH2
P-554 3-
C-H azetidin I CH2 3-CI 4-NHCONH2
P-555 3-
C-H (oxetanyl) CH2 3-CI 4-NHCONH2
P-556 3-(N-
methyl
pyrrolidinyl
C-H CH2 3-CI 4-NHCONH2
P-557 H2NCOCH
C-H 2- CH2 3-CI 4-NHCONH2
P-558 C-H CH3 C =0 3-CI
P-560 C(CH3)(OH) -
C-F CH3 enantiomer-A 3-CI 4-NHCOAc
P-561 C(CH3)(OH) -
C-F CH3 enantiomer-B 3-CI 4-NHCOAc
P-562 3-
C-H Oxetan I CH2 3-CI 4-NH2
P-563 ((S)-1-
pyrrolidin-
C-H 2-CH2- CH2 3-CI 4-NHCONH2
P-564 C[(CH2CH2)
N(CH3))CH2
C-H CH3 CH2)] 3-CI 4-F
P-565 3-(N-
emthyl
C-H p rrolidine CH2 3-CI 4-NHCONH2
P-566 C-H CH3 CH2 3-CI NHCONH 2-Tetrah drofuran
P-575 C-F CH3 CH2-O 3-CI 4-NO2
P-576 C-F CH3 CH2-O 3-CI 4-NH2
P-578 C-H CH3 CO 3,4-OCH2CH2O- 4-NH2
P-579 C-H CH3 CO 3-F 4-NH2
P-580 C-H CH3 CO 3,4-F2 4-NH2
P-583 C-H CH3 CD(OH) 3-CI 4-NO2
P-584 C-H CH3 CD(OH) 3-CI 4-NHCONH2
P-589 C-F CH3 CH2-O 3-CI 4-NHCONH2
P-594 C-F CH3 CH-O 3-CI 4-NHAc
P-620 C-F H CH2 3-Br 4-NHAc
47
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[0099] Table 2. Arl=Het(6)-Ra
MYP;
II Ra
R3,0 X Q.V;W
Rb
Example X R3 M Rb P U V W Ra
P-008 C-H CH3 CH2 3-NO2 CH CH CH CH N -
P-009 C-H CH3 CH2 3-NO2 N CH NH CH CO -
P-011 C-H CH3 CH2 3,4-NON CH CH CH CH N -
P-012 C-H CH3 CH2 3,4-NON N NH CH CH CO -
P-015 C-H CH3 0 3-NO2 N CH NH CH CO -
P-016 C-OH CH3 C=0 3-NO2 CH CH CH CH N -
P-017 C-OH CH3 CH2 3-NO2 CH CH CH CH N -
CH2CH
P-018 C-H 20CH3 CH2 3-NO2 N CH N CH CO U)-CH2CH2OCH3
P-019 N CH3 CH2 3-NO2 CH CH CH CH N -
P-020 C-H CONH2 CH2 3-NO2 N CH NH CH CO -
P-106 C-OH CH3 CH2 3-NO2 CH CH N CH CH -
P-109 C-OH CH3 CH2 3-NO2 C CH N CH CH (P)-CI
P-110 C-H CH3 CH2 3-NO2 CH CH N CH C (W)-NHCO2-tBu
P-111 C-H CH3 CH2 3-NO2 CH CH N CH C (W)-NH2
P-124 C-OH CH3 C-0 3-NO2 CH CH N CH C (W)-OCH3
P-125 C-OH CH3 CH2 3-NO2 CH CH N CH C (W)-OCH3
P-126 C-OH CH3 CH2 3-NO2 CH CH N CH C (W)-C1
C (W)-
P-150 C-OH CH3 CH2 3-NO2 CH CH N CH OCH2CH2N(CH3)2
P-152 C-F CH3 CH2 3-NO2 CH CH N CH C (w)-c1
P-177 C-OH CH3 CH2 3-NO2 CH CH N CH C (W)-NHCH2CO2H
P-178 C-OH CH3 CH2 3-NO2 CO CH NH CH CH -
P-182 C-OH CH3 CH2 3-NH2 C CH N CH CH P -Cl
P-185 C-OH CH3 CH2 3-NH2 CO CH NH CH CH -
CH2-
P-224 C-H CH3 0 3-NO2 CH N CH CH CH -
P-225 C-F CH3 CH2 3-CI CH CH N CH C (W)-OCH3
P-226 C-F CH3 CH2 3-CI CH CH N CH C W)-Cl
P-252 C-F CH3 CH2 3-CI CH CH N CH C (W)-NH2
P-258 C-F CH3 CH2 3-CI CH CH N CH C W)-NHSO2CH3
P-259 C-F CH3 CH2 3-CI CH CH N CH C (W)-NHCONH2
P-272 C-F CH3 CH2 3-CI CH CH N CH C W)-NHSO2NH2
P-277 C-F CH3 CH2 3-000H3 CH CH N CH C (W)-NH2
P-278 C-F CH3 CH2 3-CI CH CH N N C (W)-NH2
CH2CH
P-279 C-F 3 CH2 3-CI CH CH N CH C (W)-NHCONH2
48
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example X R3 M Rb P Q U V W Ra
P-281 C-F CH3 CH2 3-000H3 CH CH N N C (W)-NH2
P-284 C-F CH3 CH2 3-000H3 CH CH N N C W)-NHSO2CH3
P-290 C-F CH3 CH2 3-000H3 CH CH N CH C (W)-NHCONH2
P-291 C-F CH3 CH2 3-Cl CH CH N N C (W)-NHCONH2
P-292 C-F CH3 CH2 3-Cl CH CH N N C (W)-NHSO2CH3
P-297 C-F CH3 CH2 3-000H3 CH CH N CH C W)-NHSO2CH3
P-298 C-F CH3 CH2 3-Cl CH CH N N C W-NHSO2NH2
P-307 C-F CH3 CH2 3-000H3 CH CH N CH C (W)-NHSO2NH2
P-308 C-F CH3 CH2 3-Cl CH CH CH N-O C (W)-NHCONH2
P-309 C-F CH3 CH2 3-Cl CH CH N N-O C (W)-NHCONH2
P-314 C-F CH3 CH2 3-000H3 CH CH N N C W)-NHSO2NH2
P-315 C-F CH3 CH2 3-000H3 CH CH N N C (W)-NHCONHEt
P-318 C-F CH3 CH2 3-Cl CH CH CH N C (W)-NHEt
P-321 C-F CH3 CH2 3-000H3 CH CH N CH C W)-CO2CH3
P-325 C-F CH3 CH2 3-000H3 CH CH N N C (W)-N(CH3)2
P-327 C-F CH3 CH2 3-000H3 CH CH N CH C (W)-CO2H
P-328 C-F CH3 CH2 3-000H3 CH CH N CH C (W)-CONH2
P-332 C-F CH3 CH2 3-000H3 CH CH N CH C (W)-N(CH3)2
P-336 C-F CH3 CH2 3-000H3 CH CH N CH C (W)-NHCH3
P-337 C-F CH3 CH2 3-000H3 CH CH N CH C (W)-NHCONHEt
P-339 C-F CH3 CH2 3-000H3 CH CH N CH C (W)-NHEt
P-344 C-F CH3 CH2 3-Cl CH CH N CH C (W)-CH2NH2
P-345 C-F CH3 CH2 3-000H3 CH CH N CH C (W)-NHCOCH3
P-355 C-F CH3 CH2 3-Cl CH CH N CH C (W)-CN
P-356 C-F CH3 CH2 3-Cl CH CH N CH C (W)-NHCONHEt
P-357 C-F CH3 CH2 3-Cl CH CH N CH C W-NHCO2Et
(W)-
NHCONHCH2CO2
P-358 C-F CH3 CH2 3-Cl CH CH N CH C H
P-359 C-F CH3 CH2 3-Cl CH CH N CH C (W)-N(Et)C02Et
(W)-
NHCONHCH2CH2
P-360 C-F CH3 CH2 3-Cl CH CH N CH C CO2Et
(W)-1N-2-
P-361 C-F CH3 CH2 3-000H3 CH CH N CH C imidazolidinone
P-362 C-F CH3 CH2 3-000H3 CH CH N N C (W)-NHCONH2
(W)-
NHCONHCH2CH2
P-365 C-F CH3 CH2 3-Cl CH CH N CH C CO2H
(W)-
NHCONHCH2CH2
P-366 C-F CH3 CH2 3-000H3 CH CH N N C Cl
P-367 C-F CH3 CH2 3-Cl CH CH N CH C W)-CH2NHCO2Et
(W)-
P-368 C-F CH3 CH2 3-Cl CH CH N CH C CH2NHCONH2
(W)-
CH2NHCON(CH3)
P-371 C-F CH3 CH2 3-Cl CH CH N CH C 2
(W)-
P-372 C-F CH3 CH2 3-Cl CH CH N CH C CH2NHCONHEt
(W)-CH2NHCO(4-
P-373 C-F CH3 CH2 3-Cl CH CH N CH C Me-1N-Piperazine)
49
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example X R3 M Rb P Q U V W Ra
(W)-CH2-1N(-2-
P-374 C-F CH3 CH2 3-Cl CH CH N CH C imidazolidinone)
(W)-CH2-1 N((N3-
methyl-(2-
P-375 C-F CH3 CH2 3-Cl CH CH N CH C imidazolidinone)
P-377 C-F CH3 CH2 3-Cl CH CH N CH C (W)-N(morpholine)
P-379 N CH3 CH2 3-Cl CH CH N CH C (W)-NH2
P-382 C-F CH3 CH2 3-Cl CH CH N CH C W -CON CH3 2
(W)-CO-
N(Piperidine-4-
P-383 C-F CH3 CH2 3-Cl CH CH N CH C CO2Et
(W)-CO-
N(Piperidine-4 -
P-384 C-F CH3 CH2 3-Cl CH CH N CH C COOH)
P-386 N CH3 CH2 3-Cl CH CH N CH C W)-NHCONHEt
P-387 C-F CH3 CH2 3-Cl CH CH N CH C (W)-NHCOCH3
(W)-1N-2-
P-388 C-F CH3 CH2 3-Cl CH CH N CH C imidazolidinone
P-392 C-F CH3 CH2 3-Cl CH CH N N C W)-Cl
(W)-N-[3-OH-
P-393 C-F CH3 CH2 3-Cl CH CH N N C Azetidine]
P-395 C-F CH3 CH2 CN CH CH N N C (W)-NH2
P-397 C-F CH3 CH2 3-Cl CH CH N-O CH C (W)-NHCONHEt
P-398 C-F CH3 CH2 CN CH CH N CH C (W)-NHCO2Et
P-399 C-F CH3 CH2 CN CH CH N N C (W)-NHCONHEt
P-400 C-H CH3 CH2 CN CH CH N CH C (W)-NHCONHEt
P-401 C-F CH3 CH2 3-Cl CH CH N CH C (W)-CONH2
P-402 C-F CH3 CH2 3-Cl CH CH N CH C (W)-Br
P-403 C-F CH3 CH2 3-Cl CH CH N CH C (W)-NHCH2CO2H
P-405 C-F CH3 CH2 CN CH CH N CH C (W)-NHCONHEt
(W)-O-[(S)-(N-Me-
P-406 C-F CH3 CH2 3-Cl CH CH N N C 3-pyrrolidine)]
(W)-4-OH-3-
P-407 C-F CH3 CH2 3-Cl CH CH N CH C dine
P-408 C-F CH3 CH2 3-Cl CH CH N CH C (W)-OH
(W)-
N(COCH3)CH2CO
P-409 C-F CH3 CH2 3-Cl CH CH N CH C 2H
P-410 C-F CH3 CH2 3-Cl CH CH N CH C W)-4-Br-3- dine
(W)-
P-411 C-F CH3 CH2 3-Cl CH CH N CH C NHCH2CH2OH
(W)-1N-1,2,4-
P-412 C-F CH3 CH2 3-Cl CH CH N CH C triazole
(W)-CO- 1 N-
pyrrolidine-3-
P-414 C-F CH3 CH2 3-Cl CH CH N CH C CO2H)
P-415 C-F CH3 CH2 CN CH CH N CH (W)-CONH2
P-416 C-F CH3 CH2 3-Cl C CH N N CH (P)-Cl
(W)-N-[3-OH-
P-417 C-F CH3 CH2 CN CH CH N N C Azetidine]
P-422 C-F CH3 CH2 3-Cl CH CH N CH C W)-1N-tetrazole
P-423 C-F CH3 CH2 3-Cl CH CH N CH C W)-1N- i erazine
P-426 C-F CH3 CH2 3-Cl CH CH N N C (W)-1N-(S)-Proline
(W)-1N-(piperidine-
P-427 C-F CH3 CH2 3-Cl CH CH N N C 3-CO2H)
P-428 C-F CH3 CH2 3-Cl CH CH N CH C (W)-1H-5-tetrazolyl
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example X R3 M Rb P Q U V W Ra
(W)-1N-(piperazine-
P-431 C-F CH3 CH2 3-Cl CH CH N CH C 4-CO2Et
(W)-1N-(piperazine-
P-432 C-F CH3 CH2 3-Cl CH CH N CH C 4-CONH2)
P-433 C-F CH3 CH2 3-Cl CH CH C CH N U)-CN
P-437 C-F CH3 CH2 3-Cl CH CH C CH N U)-CH2NH2
(W)-1N-
(pyrrolidine-3-
P-438 C-F CH3 CH2 3-Cl CH CH N N C CO2H)
P-439 C-F CH3 0 CN CH CH N CH C (W)-NH2
P-440 C-F CH3 0 CN CH CH N CH C (W)-NHCONHEt
(W)-1N-
(pyrrolidine-3-
P-442 C-F CH3 CH2 3-Cl CH CH N N C CO2Me
P-446 (W)- N(azetidine-4-
C-F CH3 CH2 3-Cl CH CH CH N C COOH)
P-455 C-F CH3 CH2 3-Cl CH CH N N C (W)-sarcosine
P-456 C-F CH3 CH2 3-Cl CH CH CH N C (W)-F
P-457 (W)-N(azetidine-3-
C-F CH3 CH2 3-Cl CH CH CH N C OH)
P-458 (W)-
N(CH3)CH2CH2O
C-F CH3 CH2 3-Cl CH CH N N C H
P-459 (W)-
C-F CH3 CH2 3-Cl CH CH CH N C CH2NH000O2Et
P-460 (W)-
C-F CH3 CH2 3-Cl CH CH CH N C CH2NHCONH-Me
P-461 (W)-N 1(N3-ethyl-
C-F CH3 CH2 3-Cl CH CH CH N C imidazolidin-2-one)
P-462 (W)-
C-F CH3 CH2 3-Cl CH CH CH N(-O) C CH2NHCONH-Et
P-463 (W)-CH2- N-
C-F CH3 CH2 3-Cl CH CH CH N(-O) C Oxazolidin-2-one
P-464 (W)-N(azetidine-
C-F CH3 CH2 3-Cl CH CH N N C (W)-COOH)
P-465 (W)-CH2- N-
C-F CH3 CH2 3-Cl CH CH CH N C Oxazolidin-2-one
P-469 (W)-N-Oxazolidin-
C-F CH3 CH2 3-Cl CH CH CH N C 2-one
P-470 (W)-N 1(N3-Methyl-
C-F CH3 CH2 3-Cl CH CH CH N C imidazolidin-2-one)
P-471 (W)-
C-F CH3 CH2 3-Cl CH CH CH N C NHCH2CONH2
P-472 (W)-
C-F CH3 CH2 3-Cl CH CH CH N C CONHSO2CH3
P-473 C-F CH3 CH2 3-Cl CH CH CH N C (W)-N(CH3)2
P-478 C-F CH3 CH2 3-Cl N CH CH CH C (W)-NH2
P-488 C-H H CH2 3-NO2 N CH NH CH C(=O) -
P-499 C-H H CH2 3-NO2 CH CH NH CH C (W)-OCH3
P-500 C-H H CH2 3-NO2 CH CH N CH C (W)-Cl
P-507 C-F H CH2 3-Cl CH CH N CH C (W)-NHCONH2
P-513 C-
C-F CH3 CH2 Tetrazole CH CH N CH C (W)-NHCONH-Et
P-514 (W)-
[N(CH2CH2)2N]-
C-F CH3 CH2 3-Cl CH CH N CH C CONHEt
P-515 C-F CH3 CH2 3-Cl CH CH C CH N U)-N-Pi erazin l
P-518 (U)-
[N(CH2CH2)2N]-
C-F CH3 CH2 3-Cl CH CH C CH N CONH2
P-519 (U)-
C-F CH3 CH2 3-Cl CH CH C CH N [N(CH2CH2)2N]-
51
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example X R3 M Rb P Q U V W Ra
CONH-Et
P-520 (W)-CH2NHCOO-
C-F CH3 CH2 3-Cl CH CH N CH C Ph
P-521 (W)-
CH2NHCOCOCNH
C-F CH3 CH2 3-Cl CH CH N CH C 2
P-523 (W)-O-CH2-[(S)-2-
C-F CH3 CH2 3-Cl CH CH N CH C Pyrrolidine]
P-531 CH2- (U)-CH2NHCONH-
C-F CH3 0 3-CN CH CH C CH N Et
P-533 (W)-O-CH2-[(S)-2-
C-F CH3 CH2 3-Cl CH CH N CH C
P-567 C-H CH3 CH2 3-Cl CH CH N CH C W)-CH2OH
P-568 C-H CH3 CH2 3-Cl CH CH N CH C (W)-N-Azetidine
P-569 C-F CH3 CH2 3-Cl CH CH N CH C (W)-N-Azetidine
P-570 (W)-CH2OCO-NH-
C-H CH3 CH2 3-Cl CH CH N CH C Et
P-571 (W)-N-Azetidine-2-
C-F CH3 CH2 3-Cl CH CH N CH C Carboxamide
P-572 C-H CH3 CH2 3-Cl CH CH N CH C W -N-mo holine
P-573 C-F CH3 CH2 3-Cl CH CH N CH C (W)-N-imidazole
P-574 CH2-
C-F CH3 0 3-Cl N CH CH CH C (W)-Br
P-577 (W)-N-[(2,5-
dimethyl)pyrrolidine
C-F CH3 CH2 3-Cl CH CH N CH C ]
P-581 (W)-N-
(pyrrolidine(2-
C-F CH3 CH2 3-Cl CH CH N CH C C(Me2)OH)]
P-582 C-F CH3 CH2 3-Cl CH CH N CH C (W)-N-(pyrrolidine)
P-585 CH2-
C-F CH3 0 3-Cl CH CH N CH C W)-Cl
P-586 CH2-
C-F CH3 0 3-Cl CH CH N CH C (W)-N-Azetidine
P-587 C-F CH3 CH2 3-Cl CH CH N CH C
(W)-C(CH3)20H
P-588 (W)-
C-F CH3 CH2 3-Cl CH CH N CH C OCH2CH2NMe2
P-590 C-H CH3 CH2 3-Cl CH CH N CH C (W)-CN
P-591 C-H CH3 CH2 3-Cl CH CH N CH C (W)-CH2NH2
P-592 (W)-
C-H CH3 CH2 3-Cl CH CH N CH C CH2NHCOOPh
P-593 (W)-
C-H CH3 CH2 3-Cl CH CH N CH C CH2NHCONHCH3
P-595 C-F CH3 CH2 3-Cl CH CH N CH C (W)-C(CH3)2NH2
P-596 C-H CF2H CH2 3-Cl CH CH N CH C (W)-CH2NH2
P-597 (W)-
C-H CH3 CH2 3-Cl CH CH N CH C CH2NHCONH-Et
P-598 (W)-
C-H CF2H CH2 3-Cl CH CH N CH C CH2NHCONH-Me
P-599 C-F CH3 CH2 3-Cl CH CH N CH C (W)-CH2OH
P-600 C-H CH3 CH2 3-Cl CH CH N CH C W -C CH3 2NH2
P-601 (W)-
N[pyrrolidine(3-
C-F CH3 CH2 3-Cl CH CH N CH C h droh 1
P-602 (W)-
N[pyrrolidine(3-
C-H CH3 CH2 3-Cl CH CH N CH C h droh 1
P-603 CH2-
C-H CH3 0 3-Cl CH CH CH C N (V)-Cl
P-604 CH2-
C-F CH3 0 3-Cl CH CH CH C N (V)-Cl
52
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example X R3 M Rb P Q U V W Ra
P-605 C-H CH3 CH2 3-CI CH CH N CH C (W)-C(CH3)20H
P-606 C-F CH3 CH2 3-CI CH CH N CH C (W)-CH(CH3)NH2
P-607 CH2-
C-H CH3 0 3-CI CH CH CH C N (V)-N(pyrrolidine)
P-608 CH2-
C-F CH3 0 3-CI CH CH CH C N V)-N olidine)
P-609 (W)-CH(CH3)-
C-H CH3 CH2 3-CI CH CH N CH C N(azetidine)
P-610 C-H CF2H CH2 3-CI CH CH N CH C
(W)-C(CH3)20H
P-611 C-H CF2H CH2 3-CI CH CH N CH C (W)-NH2
P-612 (W)-N[(S)proline-2-
C-F CH3 CH2 3-CI CH CH N CH C amide
P-613 (W)-
CH(CH3)NHCONH
C-F CH3 CH2 3-CI CH CH N CH C 2
P-614 C-H CF2H CH2 3-CI CH CH N CH C (W)-N-Azetidine
P-615 (W)-N[Azetidine-2-
C-F CH3 CH2 3-CI CH CH N CH C R)-carboxamide)]
P-616 C-H CF2H CH2 3-CI CH CH N CH C (W)-NHCONH2
P-617 (W)-N[Azetidine-2-
C-F CH3 CH2 3-CI CH CH N CH C (S)-carboxamide)]
P-618 (W)-
C-H CF2H CH2 3-CI CH CH N CH C NHCOCH2NMe2
P-619 C-F CH3 CH2 3-CI CH CH N N C (W)-NHCONH-Et
P-621 (W)-
N(CH3)CH2CONH
C-H CF2H CH2 3-CI CH CH N CH C 2
P-622 (W)-
N(CH3)CH2CONH
C-H CH3 CH2 3-CI CH CH N CH C 2
[00100] Table 3.
M P. Ra
R3,0 X Q-V
Rb
Ex. Core R3 M Rb P Q U V Ra
No.
P-062 C-H CH3 CH2 3,4-OCH2O C(CH3) C(CH3) N 0 -
C-H CH3 CH(OH 3,4-OCH2O N(CH3) CH N CH
P-073 ) -
P-075 C-H CH3 CH2 3,4-OCH2O N(CH3) CH N CH -
P-087 C-H CH3 CH2 3-NO2 C(CH3) C(CH3) N N )-CH2CO2CH2CH3
P-088 C-H CH3 CH2 3-NO2 C(CH3) C(CH3) N N )-CH2CH2OH
P-089 C-H CH3 CH2 3-NO2 C(CH3) C(CH3) NH N
P-090 C-H CH3 CH2 3-NO2 C(CH3) C(CH3) N N (U)-CH2CF3
P-100 C-H CH3 CH2 3-NO2 C(CH3) C(CH3) N N (U)-CH2COOH
p-101 C-H CH3 CH2 3-NO2 C(CH3) C(CH3) N N -CH2CONH2
53
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Ex. Core R3 M Rb P Q U V Ra
No.
P-115 C-H CH3 CH2 3,4-OCF2O N CH N CH (P)-CH3
P-128 C-F CH3 C-0 3-NO2 S CH CH CH -
C-OH CH3 C-0 3-NO2 0 CH N CH
P-129 -
P-130 C-OH CH3 CH2 3-NO2 0 CH N CH -
P-174 C-OH CH3 CH2 3-NO2 C C N 0 (P)-CH3, (Q)-CH3
P-201 C-H CHF2 CH2 3,4-OCH2O N CH N CH (P)-CH3
P-306 C-H CH3 CH2 3-CI 0 N C N (U)-NH2
P-310 C-F CH3 CH2 3-CI S CH C CH (U)-OCH2CH3
P-333 C-F CH3 CH2 3-CI S N C N (U)-NH2
P-341 C-F CH3 CH2 3-CI N CH C S -NH2
P-342 C-F CH3 CH2 3-CI S N C N (U)-NHCOOCH3
P-343 C-F CH3 CH2 3-CI S N C N (U)-NHCONHCH2CH3
P-346 C-F CH3 CH2 3-CI N CH C S (U)-NHCONHCH2CH3
C-F CH3 CH2 3-CI S N C N (U)-
NHCONHCH2000CH2C
P-350 H3
C-F CH3 CH2 3-CI S N C N (U)-
NHCONHCH2CH2000C
P-351 H2CH3
P-352 C-F CH3 CH2 3-CI S N C N (U)-NHCONH2
P-353 C-F CH3 CH2 3-CI S N C N (U)-NHCONHCH2COOH
C-F CH3 CH2 3-CI S N C N (U)-
P-354 NHCONHCH2CH2COOH
C-F CH3 CH2 3-CI S N C N (U)-
NHCONH(CH2)3COOCH
P-363 2CH3
C-F CH3 CH2 3-CI S N C N (U)-
P-364 NHCONH(CH2)3COOH
P-369 C-F CH3 CH2 3-CI S CH C N (U)-NH2
[00101] Table 4.
M
NRc
H3C.0 I X
Rb
Ex.No. X M Rb N(Rc)
P-005 C-H CH2 3-NO2 1N-1,2,4-Triazole
P-006 C-H CH2 3,4-NON 1N-Imidazole
P-007 C-H CH2 3-NO2 1N-Benzotriazole
P-010 C-H CH2 3-NO2 N(Indole)
P-014 C-H CH2 3,4-NON 1N-Benzimidazole
P-022 C-H CH2 3-CO2CH3 1N-1,2,4-Triazole
P-024 N CH2 3-NO2 1N-1,2,4-Triazole
P-026 N CH2 3-NO2 N Mo holine)
P-029 C-H CH2 3-NO2 N 2-P rrolidinone)
P-031 N CH2 3-NO2 N 2-P rrolidinone)
54
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Ex.No. X M Rb N(Rc)
P-033 N CH2 3-NO2 1N-imidazolidin-2-one
P-034 C-H CH2 3-NO2 1N-2-imidazolidin-2-one
P-035 C-H CH2 3-NO2 N-1,4-Butanesultam
P-036 C-H CH2 3-NO2 N-Succinimide
P-037 C-H CH2 3,4-OCH2O 1N-Imidazole
P-039 C-H CH2 3-NO2 N-Saccharin
P-040 C-H CH2 3-NO2 N-Glutarimide
P-041 C-H CH2 3-NO2 N-Isatin
P-042 C-H CH2 3-NO2 N-Phthalimide
P-044 C-H CH2 3,4-OCH2O 1N-1,2,4-Triazole
P-045 C-H CH2 3,4-OCH2O 1N-2-Pyrrolidinone
P-046 C-H CH2 3-CO2CH3, 4-F 1N-1,2,4-Triazole
P-048 C-H CH2 3-CO2CH3, 2-F 1N-1,2,4-Triazole
P-052 C-H CH2 3,4-OCH2O N-Phthalimide
P-053 C-H CH2 3,4-OCH2O 1N-Succinimide
P-055 C-H CH2 3-NO2 N-3,4-P ridinedicarboximide
P-056 C-H CH2 3-NO2 9N-2-Amino-6-chloropurine
P-058 C-H CH2 3-F 1N-1,2,4-Triazole
P-059 C-H CH2 3-NO2 9N-Guanine
P-060 C-H CH2 3,4-F2 1N-1,2,4-Triazole
P-061 C-H CH2 3,4,5-F3 1N-1,2,4-Triazole
P-063 C-H CH2 3-CF3 1N-1,2,4-Triazole
P-064 N CH2 3-CF3 1N-1,2,4-Triazole
P-066 C-H CH2 3-NO2 4N-1,2,4-Triazole
P-068 N CH2 3-CF3 N-L-4-H drox roline methyl ester
P-069 C-H CH2 3-NO2 N-L-4-Hydroxyproline methyl ester
P-070 C-H CH2 3-NO2 N- S -3-P rrolidinol
P-071 C-H CH2 3-NO2 N- R)-3-P rrolidinol
P-072 C-H CH2 3-NO2 N-D-Prolinol
P-074 C-H CH2 3,4-OCH2O 1N-Pyrazole
P-076 C-H CH2 3-NO2 3N-2-Oxazolidinone
P-077 C-H CH2 3-NO2 1N-Pyrazole
P-078 C-H CH2 3-CF3 N- R)-3-P rrolidinol
P-081 C-H CH2 3-CN 1N-1,2,4-Triazole
P-082 C-H CH2 3-000H3 1N-1,2,4-Triazole
P-083 C-H CH2 3-CF3 3N-2-Oxazolidinone
P-084 C-H CH2 3-OCF3 1N-1,2,4-Triazole
P-085 C-H CH2 3-SCH3 1N-1,2,4-Triazole
P-086 C-H CH2 3-OCH3 1N-1,2,4-Triazole
P-104 C-H CH2 3-NO2 N-2-Pyridone
P-108 C-H CH2 3-CH2OCH3 1N-1,2,4-Triazole
P-131 C-H CH2 3-NO2 N- S)-5- H drox meth 1)- rrolidin-2-one
P-132 C-H CH2 3-NO2 N- R -5- H drox meth 1- rrolidin-2-one
P-153 C-H CH2 3-NO2 1N-5- H drox meth l)-imidazole
P-154 C-H CH2 3-NO2 1N-4- H drox meth l)-imidazole
P-155 C-H CH2 3-NO2 N- (S)-5-(Aminomethyl)-pyrrolidin-2-one
P-156 C-H CH2 3-NO2 N- R)-5- Aminometh 1)- rrolidin-2-one
P-162 C-H CH2 H 1N-2-Pyrrolidinone
P-169 C-H CH2 3-SOCH3 1N-1,2,4-Triazole
P-172 C-H CH2 3-000H3 1N-2-Pyrrolidinone
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Ex.No. X M Rb N(Rc)
P-179 C-H CH2 3-000H3 N-2-Pyridone
P-184 C-H CH2 3-OCH3 1N-2-Pyrrolidinone
P-186 C-H CH2 3-[2-Oxadiazole] 1N-2-Pyrrolidinone
P-188 C-H CH2 3-NO2 1N-2- i eridinone
P-195 C-H CH2 3-NO2 1N-5-Carbonitrile-pyridin-2-one
P-196 C-H CH2 3-NO2 1N-4-Carbonitrile- ridin-2-one
P-197 C-H CH2 3-NO2 2N-6-Methyl pyridazin-3-one
P-198 C-H CH2 3-NO2 1N-5-Amino- ridin-2-one
P-203 C-F CH2NH 3-Cl N- 4- acetamido)-hen l)
P-204 C-H CH2 3-NO2 2N-6-Methoxyl pyridazin-3-one
P-205 C-H CH2 3-NO2 2N-Pyridazinone
P-206 C-H CH2 3-NO2 1N-3-Methoxy- pyridin-2-one
P-207 C-H CH2 3-NO2 1N-4-Carboxamide- pyridin-2-one
P-208 C-H CH2 3-NO2 1N-P idin-2-one-5-urea
P-209 C-H CH2 3-Cl N-Pyrrolidin-2-one
P-210 C-H CH2 3-NO2 1N-P idin-2-one-4-carbox lic acid
P-211 C-H CH2 3-NO2 1N-Pyridin-2-one-5-acetamide
P-212 C-H CH2 3-NO2 1N-P idin-2-one-5-carboxamide
P-215 C-H CH2 3-NO2 1N-4-Amino- ridin-2-one
P-218 C-H CH2 3-Cl N-Pyrrolidin-2-one
P-223 C-H CH2 3,4-F2 N-Pyrrolidin-2-one
P-229 C-OH CH2 3-NO2 N-Pyrrolidin-2-one
P-235 C-H CH2 3-Cl 1N-5-Bromo-2- imidinone
P-236 C-H CH2 3-NO2 1N-6-Amino- ridin-2-one
P-244 C-H CH2CH2 3-NO2 CH2-1N-P rrolidin-2-one
P-245 C-H CH2 SCH3 1N-Pyrrolidin-2-one
P-248 C-H CH2 3-NO2 N- S -4-H drox - rrolid-2-one
P-253 C-H CH2 3-NO2 N-[ S)-P rrolid-2-one-4-carbamate
P-370 C-F CH2 3-Cl 1N- Piperazine-4-carboxamide
P-424 C-F CH2 3-Cl 1N-4- 4-Amino hen l) pyrazole
P-435 C-F CH2 3-Cl 1N-4- 4-Urea hen l) pyrazole
P-436 C-F CH2 3-Cl 1N-4- 4-Eth lurea hen l) pyrazole
P-479 C-F CH2 3-Cl Nl-imidazol-4- l]- idine
P-481 C-H CH2 314(=N-O-N=) 1N-(12,4)Triazole
P-482 C-H CH2 3-NO2 1N-Benztriazole
P-483 C-H CH2 3-NO2 1N-benzimidazole
P-486 C-H CH2 3,4- CHCHN H 1N-(12,4)Triazole
P-492 C-H CH2 3-NO2 N-Glutarmide
P-493 C-H CH2 3-NO2 N- R)- rolinol
P-495 C-H CH2 3-SO2CH3 1N-(12,4)Triazole
P-503 C-H CH2 3-NO2 1N-Pyrrolidin-2-one
P-504 C-OH CH2 3-NO2 1N-P olidin-2-one
P-511 C-F CH2 3-Cl 1N 4 4-nitro hen 1 azole
P-512 C-F CH2 3-Cl 1N[4 4- NHCO2Et) hen 1 razole]
P-517 C-F ch2 3-Cl 1N[3 3- id l)imidazole]
P-524 C-H bond 3-Cl 1N(7-aminobenzimidazole)
P-527 C-F CH2 3-Cl 1N[4 2- id l)imidazole]
P-528 C-H CH2 3-Cl 1N[4 4- id l)imidazole]
P-534 C-F CH2 3-Cl 1N[imidazole-4-carboxamide]
P-535 C-F CH2 3-Cl 1N[imidazole-5-carboxamide]
56
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Ex.No. X M Rb N(Rc)
P-536 C-F CH2 3-CI 1N[4-nitro-imidazole]
P-544 C-F CH2 3-CI 1N[3 2- idin 1) azole]
P-545 C-F CH2 3-CI 1N[3 2- azin 1) azole]
P-546 C-F CH2 3-CI 1N[4 6- imidin 1) razole]
P-549 C-F CH2 3-CI 1N 2 3- idin 1 imidazole
P-551 C-F CH2 3-CI 1N[3-trifluorometh l-5- 2- idin 1)- azole]
P-552 C-F CH2 3-CI 1N[3-trifluoromethyl-5-(3-pyridinyl)-pyrazole]
P-559 C-F CH2 3-CI 1N 2 2-thien limidazole
P-529 C-H bond 3-CI 1N[ 7-NHCONHEt)benzimidazole]
P-623 C-F CH2 3-CI 4N-Piperazin-2-one
[00102] Table 5.
~ M
Ra
R3,o I X
Rb
Ex.No. X R3 M Rb Ra
P-038 N CH3 CH2 3-NO2 O-C clo entane
P-091 C-H CH3 CH2 3-CF3 3-O-2-C to entene-1-one
P-092 C-H CH3 CH2 3-NO2 3-O-2-C to entene-1-one
P-127 C-F CH3 CH2 3-NO2 2-Tetrah drothio hene
P-120 C-H CH3 CH2 3-NO2 1-(4-Methyl-2,6,7-trioxa-bicyclo[2.2.2]octane)
[00103] Table 6.
I M,Ra
R3,O X
Rb
Ex.No. X R3 M Rb Ra
P-025 N CH3 CH2 3-NO2 OCH3
P-030 N CH3 CH2 3-NO2 OCH(CH3)2
P-149 C-OH CH3 CH2 3-NO2 OH
P-027 C-H CH3 CH2 3-NO2 CONHCH3
P-491 N CH3 CH2 3-NO2 OH
57
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00104] Table 7.
Ra
R3.0 X
Rb
All of the Examplees in the table below, R3=CH3
Ex.No. X Rb Ra
N"
O
P-028 C-H 3-NO2 Phenyl
0 H
O,gN
O
P-032 C-H 3-NO2 Phenyl
O
N
O N
P-043 C-H 3-N02 Phenyl
"
Nom( `)
P-047 C-H 3-NO2 Phenyl
~~N
O
P-213 C-H 3-N02 Phenyl NH2
* ~ N 0
O N O
P-214 C-H 3-NO2 Phenyl 00
,s 0
H
14- N
P-251 C-F 3-C1 Phenyl H
j 0>==0
P-263 C-F 3-ClPhenyl N
58
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Ex.No. X Rb Ra
0
/~-NH2
P-285 C-F 3-C1 Phenyl
O
0
\\ \
P-425 C-F 3-C1Phenyl N O-Et
N,~O
P-429 C-F 3-C1Phenyl N N
N,,~~O
P-430 C-F 3-C1Phenyl N O
0
N
P-445 C-F 3-C1 Phenyl
0
OH
N
N
P-480 C-F 3-CI Phenyl
[00105] Table 8.
R3,0 I X
RaH
Rb
Ex.No. X Rb Ra
,N
N
\L
P-389 C-F N 4-F Phenyl
N
I
P-391 C-H C i 4-F Phenyl
N,
/N
P-396 C-F CF3 4-F Phenyl
59
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Ex.No. X Rb Ra
N
P-475 C-H 0 4-F Phenyl
N~
P-380 C-F C1 4-F Phenyl
N
N~
F
P-396 C-H F 4-F Phenyl
NN
P-525 C-H DN 4-F Phenyl
NCO
P-526 C-H N 4-F Phenyl
P-474 C-H 3- ridin l 4-F Phenyl
P-475 C-H 3- idin I N-oxide 4-F Phenyl
[00106] In another aspect the present invention provides a pharmaceutical
composition comprising a pharmaceutically acceptable carrier and a
therapeutically
effective amount of at least one compound as described above.
[00107] Methods of the invention parallel the compositions and formulations.
The
methods comprise administering to a patient in need of treatment a
therapeutically
effective amount of a compound according to the invention. The present
invention also
provides a method for inhibiting phosphodiesterase 4.
[00108] In-vitro assay for PDE4 enzymes. The in-vitro activity of PDE4 enzymes
and
the in-vitro potency of therapeutic agents described in the present invention
were
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
measured using a real-time, enzyme-coupled spectrophotometric assay. By using
three
different coupling enzymes, the product of the PDE4 reaction is coupled to the
oxidation
of the reduced form 0-nicotinamide adenine dinucleotide (NADH), which
dissipation can
be monitored spectrophotmetrically at 340 nM.
Assay description. Buffer A containing 50 mM Tris, pH 8.0, 16 mM MgC12 and 80
mM
KC1 is prepared and stored at room temperature. Buffer B containing 50 mM
Tris, pH
8.0 is prepared and stored at toom temperature. Stock solutions of the
following reagents
are prepared in Buffer B and stored at -20 C: Adenosine-5'-triphosphate (ATP),
cyclic
adenosine-5'-monophosphate (cAMP), phosphoenolpyruvate (PEP) and NADH. An
assay mix is prepared by mixing Buffer A, trichloroethylphosphine (TCEP), ATP,
PEP,
NADH, myokinase (MK), pyruvate kinase (PK), lactate dehydroganese (LDH) and
PDE4 to a final volume of 20 mL, which is enough for a single 96-well assay
plate.
Assay mix (180 L) and test article (10 L) in 1:1 DMSO/H20 mixture is pre-
incubated
at room temperature for 10 min. The enzymatic reaction is initiated by
addition of cAMP
(10 L). Final concentration of all components in the assay (200 L/well) are
as follows:
mM MgC12, 50 mM KC1, 5 mM TCEP, 2.5% DMSO, 0.4 mM NADH, 1 mM PEP,
0.04 mM ATP, 5 units MK, 1 unit PK, 1 unit LDH and appropriate amount of PDE4.
Reaction progress curves are monitored in a plate reader capable of measuring
light
absorbance at 340 nM. A decrease in light absorbance at 340 nm is due to
oxidation of
NADH. Positive controls containing no test article and negative controls
containing no
test article and no cAMP are included on every assay plate. Reaction rates are
determined from the slopes of the linear portions of the progress curves. All
data is
percent normalized with respect to controls and presented as percent
inhibition.
[00109] The results of testing of representative species are shown below in
Tables 9
and 10. The activities are designated A = < 5 M, B = 5-20 M, C = 20-40 M.
[00110] Table 9
CmpdNo hPDE4D CmpdNo hPDE4D CmpdNo hPDE4D
P-001 A P-195 B P-410 A
P-002 A P-196 A P-411 A
P-003 A P-197 B P-412 A
P-004 A P-199 A P-413 A
P-005 A P-200 A P-415 A
61
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
CmpdNo hPDE4D CmpdNo hPDE4D CmpdNo hPDE4D
P-006 A P-201 B P-416 A
P-007 A P-203 A P-417 A
P-008 A P-204 B P-418 B
P-009 A P-205 A P-419 B
P-010 A P-206 A P-420 A
P-011 A P-207 B P-421 A
P-012 B P-208 B P-422 A
P-014 B P-209 B P-423 B
P-015 A P-211 B P-424 A
P-017 A P-212 B P-429 A
P-018 B P-213 B P-431 A
P-019 A P-215 B P-432 A
P-020 B P-216 A P-433 A
P-021 A P-217 A P-434 A
P-022 A P-218 A P-435 A
P-023 A P-219 A P-437 A
P-024 A P-220 A P-439 A
P-025 B P-221 A P-440 A
P-026 B P-222 A P-441 A
P-027 A P-223 B P-445 A
P-028 A P-224 B P-447 A
P-029 A P-225 A P-448 A
P-030 B P-226 A P-449 A
P-033 B P-227 A P-450 A
P-034 A P-228 A P-451 A
P-035 A P-229 A P-453 A
P-036 A P-230 B P-454 A
P-037 A P-231 A P-456 A
P-038 B P-232 A P-457 A
P-040 B P-233 A P-458 A
P-041 B P-234 A P-460 A
P-042 A P-235 B P-461 A
P-043 A P-236 C P-462 A
P-044 A P-237 A P-463 A
P-045 C P-238 A P-465 A
P-046 A P-240 A P-466 A
P-047 B P-241 A P-467 A
P-048 A P-242 A P-468 A
P-049 A P-243 A P-469 A
P-050 A P-244 A P-470 A
P-051 A P-245 B P-471 A
P-052 A P-246 B P-473 A
P-053 B P-247 A P-474 A
P-054 A P-248 A P-475 B
P-055 A P-249 A P-476 A
P-057 A P-250 A P-477 A
62
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
CmpdNo hPDE4D CmpdNo hPDE4D CmpdNo hPDE4D
P-058 A P-251 A P-478 A
P-060 A P-252 A P-479 A
P-061 A P-254 B P-481 A
P-062 A P-255 A P-483 A
P-063 A P-256 A P-486 C
P-064 B P-257 A P-488 A
P-065 A P-258 A P-491 C
P-066 A P-259 A P-492 C
P-067 A P-260 A P-493 C
P-070 A P-261 A P-494 A
P-071 A P-262 A P-495 C
P-072 B P-263 A P-496 A
P-073 A P-264 A P-497 A
P-074 A P-266 A P-499 A
P-075 A P-267 A P-500 A
P-076 A P-268 A P-501 B
P-077 A P-269 A P-502 A
P-078 B P-270 A P-503 B
P-079 A P-271 B P-504 A
P-080 A P-273 A P-505 B
P-081 A P-274 A P-507 A
P-082 A P-275 A P-508 A
P-083 B P-277 A P-513 B
P-084 A P-278 A P-514 A
P-085 A P-279 A P-515 A
P-086 A P-280 A P-516 A
P-087 A P-281 A P-517 A
P-088 A P-282 A P-518 A
P-089 A P-283 A P-519 A
P-090 A P-284 A P-520 A
P-091 A P-285 A P-521 A
P-092 A P-286 A P-523 A
P-093 A P-287 A P-524 A
P-094 A P-288 A P-525 A
P-095 C P-289 A P-526 B
P-097 A P-290 A P-527 A
P-098 A P-291 A P-528 A
P-099 A P-292 A P-530 A
P-101 A P-293 A P-531 A
P-102 A P-294 A P-532 A
P-103 A P-295 A P-533 A
P-104 A P-296 A P-534 A
P-105 A P-297 A P-535 A
P-106 A P-299 A P-536 A
P-107 A P-300 A P-537 A
P-108 A P-301 A P-538 A
63
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
CmpdNo hPDE4D CmpdNo hPDE4D CmpdNo hPDE4D
P-109 A P-302 A P-539 A
P-111 A P-303 A P-540 A
P-112 A P-304 A P-541 A
P-113 A P-305 A P-542 C
P-114 A P-306 B P-543 A
P-115 A P-310 A P-544 A
P-116 A P-311 A P-545 A
P-117 A P-313 A P-546 A
P-118 A P-315 A P-547 A
P-119 A P-316 A P-548 B
P-120 A P-317 A P-549 A
P-121 A P-318 A P-550 A
P-122 A P-322 A P-552 A
P-123 A P-323 A P-553 A
P-125 A P-325 A P-554 B
P-126 A P-326 A P-555 A
P-127 A P-328 A P-556 B
P-128 A P-329 A P-557 A
P-130 A P-330 A P-558 A
P-131 B P-331 A P-559 A
P-132 C P-332 A P-560 B
P-133 B P-333 A P-561 A
P-134 A P-334 A P-562 A
P-135 A P-335 A P-563 A
P-136 A P-336 A P-564 B
P-137 A P-337 A P-565 B
P-138 A P-338 A P-566 A
P-139 A P-339 A P-567 A
P-140 A P-340 A P-568 A
P-141 A P-341 A P-569 A
P-142 A P-342 A P-570 A
P-143 A P-343 A P-571 A
P-144 A P-344 A P-572 A
P-145 A P-345 A P-573 A
P-146 A P-346 A P-574 A
P-147 A P-347 A P-575 B
P-148 A P-352 A P-576 A
P-149 A P-355 A P-577 A
P-150 B P-356 A P-578 A
P-151 A P-357 A P-579 A
P-152 A P-359 A P-580 A
P-153 A P-361 A P-581 A
P-154 A P-362 A P-582 A
P-155 C P-366 A P-583 A
P-156 A P-367 A P-584 B
P-157 A P-368 A P-585 A
64
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
CmpdNo hPDE4D CmpdNo hPDE4D CmpdNo hPDE4D
P-158 B P-369 A P-586 A
P-159 A P-370 A P-587 A
P-160 B P-371 A P-588 A
P-161 B P-372 A P-589 A
P-162 B P-373 A P-590 A
P-163 A P-374 A P-591 A
P-164 A P-375 A P-592 A
P-165 A P-376 A P-593 A
P-166 A P-377 A P-594 A
P-167 A P-378 A P-595 A
P-168 A P-379 A P-596 A
P-169 B P-380 A P-597 A
P-170 A P-381 A P-598 A
P-171 A P-382 A P-599 A
P-172 A P-385 B P-600 A
P-173 A P-386 A P-601 A
P-174 A P-387 A P-602 A
P-175 A P-388 A P-603 A
P-176 A P-389 B P-604 A
P-177 A P-390 B P-605 A
P-178 A P-391 A P-606 A
P-179 B P-392 A P-607 A
P-180 A P-393 A P-608 A
P-181 A P-394 A P-609 A
P-182 B P-395 A P-610 A
P-183 A P-396 A P-611 A
P-184 B P-397 A P-612 A
P-185 B P-398 A P-613 A
P-186 C P-399 A P-614 A
P-187 A P-400 A P-615 A
P-188 A P-401 A P-616 A
P-189 B P-402 A P-617 A
P-190 A P-404 A P-618 A
P-192 B P-405 A P-619 A
P-193 A P-406 A P-620 A
P-194 A P-407 A P-623 A
P-621 A P-622 A
[00111] Table 10.
PDE4B Activity, where A<5 uM, B=5-20 uM, C=21-40 uM.
Cmpd hPDE4B Cmpd hPDE4B Cmpd hPDE4B
No No No
P-001 A P-182 C P-375 A
P-002 A P-183 A P-376 A
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Cmpd hPDE4B Cmpd hPDE4B Cmpd hPDE4B
No No No
P-004 B P-184 B P-378 A
P-006 A P-185 C P-379 A
P-007 B P-186 C P-382 A
P-008 A P-187 A P-386 A
P-009 A P-188 A P-387 A
P-010 A P-190 A P-388 A
P-011 A P-192 B P-392 A
P-012 B P-193 A P-393 A
P-014 B P-195 C P-394 A
P-015 C P-196 B P-395 A
P-017 A P-197 B P-397 A
P-019 A P-199 A P-398 A
P-020 C P-200 A P-399 A
P-021 B P-201 C P-400 A
P-022 B P-204 C P-401 A
P-023 B P-205 A P-402 A
P-024 C P-206 A P-404 A
P-025 C P-207 A P-405 A
P-027 B P-208 B P-406 A
P-028 A P-209 A P-411 A
P-029 A P-211 B P-412 A
P-030 C P-212 B P-413 A
P-031 C P-213 A P-415 A
P-033 B P-215 B P-416 C
P-034 A P-216 A P-417 A
P-035 A P-217 A P-420 A
P-036 B P-218 A P-421 A
P-037 A P-221 A P-422 A
P-038 C P-222 A P-429 A
P-040 B P-223 B P-432 A
P-041 A P-224 B P-433 A
P-042 B P-225 A P-437 A
P-043 B P-226 B P-439 A
P-044 A P-227 A P-440 B
P-045 B P-228 A P-441 B
P-046 B P-229 A P-445 B
P-047 A P-230 C P-447 A
P-049 A P-233 A P-448 A
P-050 A P-234 A P-451 A
P-051 B P-235 B P-453 A
P-053 C P-236 C P-456 A
P-054 A P-237 A P-457 A
P-055 B P-240 B P-458 A
P-057 A P-242 A P-460 A
P-058 B P-243 A P-461 A
P-060 B P-244 A P-462 A
P-061 B P-245 A P-463 A
P-062 A P-246 A P-465 A
P-063 B P-248 A P-467 A
66
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Cmpd hPDE4B Cmpd hPDE4B Cmpd hPDE4B
No No No
P-066 A P-249 A P-468 A
P-067 A P-250 A P-469 A
P-070 C P-251 A P-470 A
P-071 C P-252 A P-471 A
P-072 C P-253 C P-473 A
P-073 C P-254 A P-478 A
P-074 B P-255 A P-479 B
P-075 A P-256 B P-481 B
P-076 A P-258 A P-488 B
P-077 A P-259 A P-494 A
P-079 A P-260 B P-496 A
P-081 B P-261 A P-498 A
P-082 B P-262 A P-499 B
P-083 B P-263 A P-500 B
P-084 B P-264 B P-502 A
P-085 A P-266 A P-503 B
P-086 B P-267 A P-504 B
P-087 A P-268 B P-507 B
P-088 A P-269 A P-508 B
P-089 A P-270 B P-513 C
P-090 A P-274 A P-515 A
P-091 C P-275 B P-516 A
P-092 A P-277 A P-517 B
P-093 B P-278 A P-518 A
P-094 A P-279 A P-519 A
P-099 A P-280 A P-521 A
P-101 A P-281 A P-523 A
P-102 A P-282 B P-524 A
P-103 A P-283 A P-526 B
P-104 A P-284 A P-527 B
P-105 A P-285 B P-530 C
P-106 A P-287 A P-531 A
P-107 A P-290 A P-532 A
P-109 A P-291 A P-533 A
P-111 A P-292 B P-534 A
P-112 A P-295 C P-535 A
P-113 A P-296 C P-536 A
P-114 A P-297 B P-539 C
P-115 A P-299 B P-540 A
P-116 A P-300 A P-541 A
P-117 C P-301 A P-542 C
P-118 A P-302 A P-544 A
P-119 A P-303 A P-545 A
P-120 B P-304 A P-547 A
P-122 B P-305 B P-549 B
P-123 A P-310 A P-550 A
P-125 A P-311 A P-553 A
P-126 A P-313 A P-555 A
P-127 A P-315 A P-559 A
67
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Cmpd hPDE4B Cmpd hPDE4B Cmpd hPDE4B
No No No
P-128 A P-316 A P-560 B
P-130 A P-317 A P-562 A
P-131 B P-318 A P-563 B
P-133 B P-323 A P-564 C
P-134 B P-324 C P-565 C
P-135 A P-325 A P-566 A
P-136 A P-328 A P-567 A
P-138 A P-329 A P-568 A
P-140 A P-330 A P-569 A
P-142 A P-331 A P-570 A
P-143 A P-332 A P-571 A
P-144 A P-333 A P-573 A
P-145 A P-334 A P-575 A
P-146 B P-335 A P-576 A
P-147 A P-336 A P-578 A
P-148 A P-337 A P-579 A
P-149 B P-338 A P-582 A
P-150 B P-339 A P-583 B
P-152 A P-341 A P-585 A
P-153 B P-342 A P-587 A
P-154 A P-343 A P-588 A
P-155 B P-344 A P-590 A
P-156 A P-345 A P-591 A
P-159 A P-346 B P-592 A
P-161 B P-347 A P-593 A
P-162 B P-352 A P-594 A
P-164 B P-355 A P-596 A
P-165 B P-356 A P-597 A
P-166 A P-357 A P-598 A
P-168 A P-359 B P-599 A
P-171 A P-361 A P-600 C
P-172 A P-362 A P-603 A
P-173 A P-366 A P-604 A
P-174 A P-367 A P-605 A
P-175 A P-368 A P-606 A
P-176 A P-369 A P-610 A
P-177 A P-370 B P-611 A
P-178 A P-371 A P-612 A
P-179 B P-372 A P-619 A
P-180 A P-373 B P-620 C
P-181 A P-374 A P-621 A
P-623 B
[00112] The activity of PDE4 inhibitors decribed in the present invention was
also
measured using in an ex-vivo assay measuring leukotriene E4 (LTE4) in human
whole
blood after Sephadex stimulation. The anti-inflammatory activity of
therapeutic agents
of the present invention is demonstrated by the inhibition of eosinophil
activation as
68
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
measured by sephadex bead stimulated LTE4 production in whole human blood. For
each sample, 356 p l of heparinized human whole blood (Vacutainer tube #6480)
is added
to wells of a 96 well plate. Then, 4 l of a series of compound dilutions (in
DMSO) are
added in triplicates, suspension mixed and allowed to incubate at 37 C for 15
min with
gentle shaking. After that, blood samples are stimulated by adding 40 L of
Sephadex G-
15 beads (Sigma-Aldrich, Sweden). The beads are predissolved in PBS (0.16 g/
mL
PBS). After mixing, the suspension is incubated at 37 C for 90 min. Then, 8
L of 15%
EDTA/PBS is added to each sample, mixed and plate centrifuged for 5 min at 115
x g at
21 C and supernatants taken. In each plate, 10 positive controls and 10
negative controls
are used, containing DMSO instead of compound solution. The positive controls
are
stimulated with Sephadex as described for the samples, and in the negative
controls
(unstimulated), Sephadex solution is replaced by PBS. LTE4 levels in the
resulting
plasma samples are determined using a commercial enzyme-linked immunoassay
(Cayman Chemical Company, Ann Arbor, MI) according to the manufacturer's
instructions. Examples P-050, P-075, P-107, P-113, P-136, P-139, P-140, P-156,
P-163,
P-168, P-175, P-181, P-187, P-200, P-221, P-222, P-227, P-237, P-239, P-242, P-
243, P-
250, P-269, P-287, P-312, P-315, P-318, P-325, P-328, P-330, P-332, P-336, P-
337, P-
338, P-339, P-342, P-356, P-378, P-382, P-403, P-405, P-409, P-415, P-420 and
P-439
all showed IC50 < 1 M in this ex-vivo assay, whereas example P-358 had IC50 >
1 M.
Persons of skill in the art accept that positive results in PDE4 models are
predictive of
therapeutic utility as discussed above.
[00113] The following specific non-limiting examples are illustrative of the
synthesis
of compounds of the invention.
69
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00114] General Scheme 1
H2P Y
Y
halogenation Y R1-X Y R2 R1,0 _N R1_ _N
O
halogenation
HO HO N R1 -0 N coupling
X X reaction R2
R2
1-2
H2P-R3 R3 1-3 1-4
R1.0 N 1~5
Y = CH3, CH2OH, CH2Br, CH2OCO2CH3
coupling X = Br, I, Cl
reaction R1 = H, Me, CHF2, CF3
R2 P = halogen, boronate
R2 = Cl, CF3, CH3CO, ON, N02, Br, fused heterocycle
R3 = substituted aryl groups
1-6
[00115] Example 1. Preparation of P-065
I""I~
0 N / F
F
F F
P-065
[00116] Synthesis of 2-bromo-6-methyl-pyridin-3-ol (1-2, X = Br, Y = CH3): To
6-
methyl-pyridin-3-ol (I-1, Y = CH3, 5.0 g, 45.82 mmol) in pyridine (15 mL) was
added
bromine (3.66 g, 22.91 mmol). The reaction was stirred at room temperature
under N2
for 20 h. The crude reaction mixture was poured on to crushed ice-water (300
mL),
stirred for 3 h. The mixture was extracted with ethyl acetate (5 x 100 mL) and
the
combined organic extracts were washed with brine, dried over Na2SO4, filtered,
and
concentrated to afford 6.3 g (73%) of 2-bromo-6-methyl-pyridin-3-ol (1-2, X =
Br, Y =
CH3) as light yellow solid.
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00117] Synthesis of 2-bromo-3-methoxy-6-methyl-pyridine (1-3, X = Br, Y = R1=
CH3):: To the 2-bromo-6-methyl-pyridin-3-ol (1-2) 6.0 g, 31.91 mmol) and K2CO3
(8.82
g, 63.82 mmol) in acetone (100 mL) was added Mel (6.79 g, 479.87 mmol). The
reaction
was stirred at 45 C under N2 for 20 h. The reaction was cooled to room
temperature,
filtered and concentrated. The residue was purified by silica gel column
chromatography
using 1:1 dichloromethane-hexanes as eluent to afford 2.34 g (36%) of 2-bromo-
3-
methoxy-6-methyl-pyridine (1-3, X = Br, Y = R1= CH3) as off-white solid.
[00118] Synthesis of 3-methoxy-6-methyl-2-(3-trifluoromethyl-phenyl)-pyridine
(1-4,
Y = R1= CH3, R2= CF3): To the 2-bromo-3-methoxy-6-methyl-pyridine synthesized
above (1.2 g, 5.94 mmol), 3-trifluoromethylphenylboronic acid (1.69 g, 8.91
mmol),
PPh3 (0.31 g, 1.19 mmol), K2CO3 (2.46 g, 17.82 mmol) and Pd(OAc)2 (0.13 g,
0.59
mmol) was added DME (15 mL), and EtOH-H20 (1:1, 6 mL). Ar gas was bubbled
through the stirred reaction for 5 min. The reaction was stirred at 80 C
under Ar for 20
h. The reaction was cooled to room temperature, concentrated, and H2O and
dichloromethane (40 mL each) were added. The organic layer was separated and
the
aqueous layer was extracted with dichloromethane (2 x 25 mL). The combined
organic
extracts were dried with Na2SO4, filtered, and concentrated. The residue was
purified by
silica gel column chromatography using 1:1 dichloromethane-hexanes then
dichloromethane to afford 1.36 g (86%) of 3-methoxy-6-methyl-2-(3-
trifluoromethyl-
phenyl)-pyridine (1-4, Y = R1= CH3, R2 = CF3) as a light yellow solid.
[00119] Synthesis of 6-bromomethyl-3-methoxy-2-(3-trifluoromethyl-phenyl)-
pyridine (1-5, R1= CH3, R2 = CF3, Y = CH2Br). To the 3-methoxy-6-methyl-2-(3-
trifluoromethyl-phenyl)-pyridine synthesized above (1.3 g, 4.86 mmol) and NBS
(1.04 g,
5.83 mmol) in CC14 (25 mL) was added benzoyl peroxide (0.12 g, 0.49 mmol). The
reaction was stirred at 80 C under N2 for 20 h. The reaction was cooled to
room
temperature and concentrated. The residue was dissolved in mixture of
dichloromethane
and hexanes (1:1, 8 mL) and purified by silica gel column chromatography using
1:1
dichloromethane-hexanes to afford 0.74 g (44%) of 6-bromomethyl-3-methoxy-2-(3-
trifluoromethyl-phenyl)-pyridine as off-white solid.
[00120] Synthesis of 6-(4-fluoro-benzyl)-3-methoxy-2-(3-trifluoromethyl -
phenyl)-
71
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
pyridine (P-065) : To the 6-bromomethyl-3-methoxy-2-(3-trifluoromethyl-phenyl)-
pyridine synthesized above (0.2 g, 0.58 mmol), 4-fluorophenylboronic acid
(0.12 g, 0.87
mmol), PPh3 (0.03 g, 0.12 mmol), K3PO4 (0.37 g, 1.73 mmol) and Pd(OAc)2 (0.013
g,
0.058 mmol) was added DME (4.0 mL), and EtOH-H20 (1:1, 1.0 mL). The reaction
was
stirred at 80 C for 20 h. The reaction was cooled to room temperature,
concentrated. The
residue was purified by silica gel column chromatography using 1:1
dichloromethane-
hexanes then dichloromethane to afford 0.056 g (22%) of 6-(4-fluoro-benzyl)-3-
methoxy-2-(3-trifluoromethyl -phenyl)-pyridine (P-065) as a clear viscous
liquid. 1H
NMR (CDC13, 400 MHz): 8.24 (s, 1 H), 8.14 (d, J= 8.0 Hz, 1 H), 7.6-7.64 (m, 1
H),
7.52-7.65 (m, 1 H), 7.2-7.34 (m, 4 H), 6.96-7.05 (m, 2 H), 4.14 (s, 2 H), 3.85
(s, 3 H);
MS(APCI+): 362.1 (M+1), LC-MS: 97.2%.
[00121] The following compounds were prepared according to general scheme 1,
analogous to the preparation of P-065.
P-005 P-060 P-169 P-248
P-010 P-061 P-184 P-253
P-014 P-063 P-188 P-370
P-019 P-064 P-195 P-376
P-021 P-068 P-196 P-379
P-022 P-069 P-197 P-386
P-023 P-070 P-198 P-422
P-024 P-071 P-204 P-424
P-026 P-072 P-205 P-435
P-028 P-076 P-206 P-436
P-029 P-078 P-207
P-031 P-081 P-208
P-033 P-082 P-209
P-034 P-083 P-210
P-084 P-085 P-211
P-035 P-086 P-212
P-036 P-104 P-213
P-044 P-108 P-214
P-045 P-131 P-215
P-046 P-132 P-218
P-048 P-153 P-229
P-053 P-154 P-233
72
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
P-054 P-155 P-235
P-057 P-156 P-236
P-058 P-162 P-245
[00122] General Scheme 2.
O
/ _ Ho \ alkylation reduction acylation R2
HO Ii / N..O R ` O ~t\O / NHz ~t\O
NH2 O X p N X X
X
1-7 1-8 1-9 1-10 1-11
Y = NH2, CN
CR2 Suzuki R2 X = Br, I, Cl
reduction R1 = H, Me, CHF2, CF3
R,~O Y Coupling R,~O Y R3 = Cl, CH3CO, CN, N02, Br, fused heterocycle
X R2 = substituted aryl groups
R3
1-12
1-13
Example 2. Preparation of P-176.
I\
O / NH2 F
\ I .0
N
I_
O
P-176
[00123] Synthesis of 2-bromo-3-nitro-phenol (1-8, X = Br): (Prepared by a
modification of reported procedure, J. Org. Chem. 1988, 53, pp 1170-1176). To
2-amino-
3-nitro-phenol (24.9 mmol, 1.0 eq.) in 24 mL of water and 12 mL of 1,4-dioxane
at
reflux, was added 13 mL of HBr (48% aq.) over 10 minutes. The resulting
solution was
refluxed for an additional 15 minutes, and cooled 0-5 C. A solution of sodium
nitrite
(24.4 mmol, 0.98 eq.) in 20 mL of water was added over 10 minutes, and stirred
for 15
minutes. The reaction mixture was then heated to 60 C for 15 minutes, and
allowed to
cool naturally to room temperature, and stirred for 16 hours. The reaction
mixture was
then extracted with two portions of diethyl ether, and the combined ethereal
layers
washed with brine, dried over magnesium sulfate, filtered through a layer of
celite and
73
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
concentrated. The residue was diluted with dichloromethane (with -0.1% MeOH),
and
purified via silica gel plug filtration with dichloromethane to yield 2-bromo-
3-nitro-
phenol (1-8, X = Br) as a pale orange-brown solid. Yield: 50%"H NMR (400 MHz;
CDC13): 6.07 (s, 1H), 7.25 (dd, J= 8.4, 1.2 Hz, 1H), 7.37 (t, J= 8.0 Hz, 1H),
7.48 (dd, J
= 8.0, 1.6 Hz, 1 H) ppm.
[00124] Synthesis of 2-bromo-3-nitro-anisole (1-9, X = Br, R, = CH3): : To a
solution
of 2-bromo-3-nitro-phenol synthesized above (11.5 mmol, 1.0 eq.) in DMF at
room
temperature was added cesium carbonate (13.8 mmol, 1.2 eq.), followed by
iodomethane
(33.7 mmol, 2.9 eq.), and the resultant mixture was stirred at room
temperature for 16
hours. The reaction mixture was poured into water, stirred for 2 hours,
filtered, the cake
washed with two portions of water, and the resultant solid dried to afford 2-
bromo-3-
nitro-anisole (1-9, X = Br, R, = CH3) as a pale orange solid. Yield: 97%; 1H
NMR (400
MHz; CDC13): 3.97 (s, 3H), 7.07 (dd, J= 8.4, 1.2 Hz, 1H), 7.32 (dd, J= 8.0,
1.6 Hz, 1H),
7.40 (t, J= 8.0 Hz, 1H) ppm.
[00125] Synthesis of 2-bromo-3-methoxy-aniline (1-10, X = Br, R, = CH3):
(Prepared
by a modification of reported procedure WO Patent: W02006/7700). To a solution
of 2-
bromo-3-nitro-anisole synthesized above (10.3 mmol, 1.0 eq.) in absolute
ethanol and
glacial acetic acid at room temperature was added iron powder (42.1 mmol, 4.1
eq). The
resultant mixture was heated to reflux for 1.5 hours, and then cooled to room
temperature. The reaction mixture was diluted with water, and solid sodium
carbonate
was added until the pH was 6-7. Dichloromethane was added, and the mixture was
filtered through celite. The dried (sodium sulfate) organics were concentrated
to afford
2-bromo-3-methoxy-aniline (1-10, X = Br, R, = CH3) as an oil. Yield: 90%; 1H
NMR
(400 MHz; CDC13): 3.87 (s, 3H), 4.16 (br s, 2H), 6.31 (dd, J= 8.0, 1.2 Hz,
1H), 6.42 (dt,
J = 8.0, 0.8, 0.4 Hz, 1 H), 7.05 (t, J= 8.0 Hz, 1 H) ppm.
[00126] Synthesis of 2 -amino -3 -bromo-4 -methoxy-phenyl) -(4 -fluoro-phenyl)-
methanone (I-11, X = Br, Y = NH2, R, = CH3, R2 = 4-fluorophenyl) : To a
solution of
boron trichloride (1.0 M in heptane; 10.0 mmol; 1.1 eq.) at 0 C in
tetrachloroethane was
added a solution of 2-bromo-3-methoxy-aniline synthesized above (9.09 mmol; 1
eq.) in
tetrachloroethane over 1 minute. The resultant mixture was stirred at the same
74
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
temperature for 10 minutes, and to it was added a solution of 4-
fluorobenzonitrile (18.2
mmol; 2 eq.) in tetrachloroethane and aluminum chloride (10.0 mmol; 1.1 eq.).
The
reaction was heated to 110 C for 5 hours, and allowed to cool to room
temperature and
stir for 16 hours. The reaction mixture was added 10 mL of 3N HC1, and the
resultant
mixture was heated to 90 C for 1 hour, and cooled to room temperature. The pH
was
adjusted with 6N NaOH to 11-12, and extracted with dichloromethane. The
organics
were washed with a brine solution, dried over magnesium sulfate, and filtered.
The
filtrate was concentrated, and the residue purified via silica gel
chromatography using
10% hexanes in dichloromethane as eluent to afford 2-amino-3-bromo-4-methoxy-
phenyl)-(4-fluoro-phenyl)-methanone (I-11, X = Br, Y = NH2, R, = CH3, R2 = 4-
fluorophenyl) in 65% yield.; 1H NMR (400 MHz; CDC13): 3.95 (s, 3H), 6.25 (d,
J= 8.8
Hz, 1H), 6.85 (br s, 2H), 7.11-7.16 (M, 2H), 7.42 (d, J= 8.8 Hz, 1H), 7.60-
7.64 (m, 2H)
ppm.
[00127] Synthesis of 2-bromo-6-(4-fluoro-benzyl)-3-methoxy-aniline (1-12, X =
Br, Y
= NH2, R, = CH3, R2 = 4-fluorophenyl) : To a solution of trifluoroacetic acid
(21.6
mmol; 10 eq.) in dichloromethane at -15 C was added in portions sodium
borohydride
(8.06 mmol; 3.7 eq.) while maintaining internal bath temperature between -15
and -
20 C. (caution, strong gas evolution) The reaction mixture was allowed to warm
to 0-
C, and a solution of the above ketone (2.16 mmol; 1.0 eq) in dichloromethane
was
added over approximately 5 minutes. The resultant mixture was allowed to stir
at room
temperature for 16 hours, and was quenched with 5% aq. NaHCO3, and extracted
with
ethyl acetate. The combined organics were washed with water, brine, and dried
over
magnesium sulfate, and concentrated. (TLC analysis indicated a mixture of
starting
ketone and desired methylene product) The residue was taken into THF, and
treated
with BH3=THF (15 mmol) at room temperature for 1 hour. The excess borane was
quenched with methanol, and the mixture concentrated. Three additional
portions of
methanol were added, and the mixture concentrated. The residue was purified
via flash
chromatography on silica gel using 1:1 dichloromethane-hexanes as eluent to
give 2-
bromo-6-(4-fluoro-benzyl)-3-methoxy-aniline (I-12, X = Br, Y = NH2, R, = CH3,
R2 = 4-
fluorophenyl) in 40% yield.; 1H NMR (400 MHz; CDC13): 3.86 (s, 2H), 3.87 (s,
3H),
4.07 (br s, 2H), 6.33 (d, J= 8.4 Hz, 1H), 6.93 (d, J= 8.4 Hz, 1H), 7.98 (ddd,
J= 8.8, 8.4,
2.0 Hz, 2H), 7.12 (ddd, J= 8.0, 6.4, 1.0 Hz, 2H) ppm.
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00128] Synthesis of 3-(4-fluoro-benzyl)-6-methoxy-3'-nitro-biphenyl-2-ylamine
(P-
176): A mixture of 2-bromo-6-(4-fluoro-benzyl)-3-methoxy-aniline synthesized
above
(0.403 mmol; 1.0 eq.), 4-fluorophenylboronic acid (0.605 mmol; 1.5 eq), and 2
M K2C03
(650 mL, 3.2 eq) in dioxane was degassed with nitrogen for 10 minutes, and
tetrakis(triphenylphosphine)palladium was added, and the resultant mixture
degassed for
an additional 5 minutes. The reaction was stirred at 70 C for 16 hours, and
cooled to
room temperature. The reaction was diluted with water, and extracted with two
portions
of ethyl acetate. The combined organics were washed with three portions of
water,
brine, dried over magnesium sulfate, and filtered through celite. The residue
was
purified via flash chromatography on silica gel using 20% acetone in hexane as
eluent to
give 3-(4-fluoro-benzyl)-6-methoxy-3'-nitro-biphenyl-2-ylamine (P-176) in 46 %
yield.;
1H NMR (400 MHz; CDC13): 3.70 (s, 3H), 3.86 (s, 2H), 6.42 (d, J= 8.4 Hz, 1H),
6.97-
7.02 (m, 2H), 7.04 (d, J= 8.4 Hz, 1H), 7.17 (dd, J= 5.2, 3.2 Hz, 2H), 7.60-
7.67 (m, 2H),
8.18-8.20 (m, 2H) ppm; LC/MS (86.9 %); (ESI+): Found 353.6 (M + 1), Calcd
352.4
m/z.
[00129] General Scheme 3.
R4 OH
R3
R1.O Y
HO OH Suzuki
Coupling R1,0 Y R2 1.20
R1,0 Y + reduction
Br 6~R2 R2 1-16
1-14 1-15 + O R3
O Friedel-Crafts KR3 reduction R1 -o Y
halogenation
R3 OH Acylation R1,o~ Y
R3 CI
1-17 R2
Y = H, F, OH 1-18 L
R1 = H, Me, CHF2, CF3 R2 I-19
R2 = Cl, CH3CO, CN, N02, Br, NH2, fused heterocycle 1-21
R3 = substituted aryl groups
R4 = H, Me
76
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 3. Preparation of P-404.
C F N,N
N
N
CI
P-404
[00130] Synthesis of 3'-chloro-6-fluoro-2-methoxy-biphenyl (1-16, Ri = CH3, R2
= Cl,
Y = F): To 2-bromo-3-fluoroanisole (1.0 g, 4.88 mmol), 3-chlorophenylboronic
acid
(0.91 g, 5.88 mmol), PPh3 (0.64 g, 2.44 mmol), K2CO3 (0.27 g, 1.95 mmol) and
Pd(OAc)2 (0.13 g, 0.58 mmol) was added dioxane (8 mL), and EtOH-H20 (1:1, 4
mL).
Ar gas was bubbled through the stirred reaction for 5 min. The reaction was
heated at
180 C using microwave oven (Biotage Intiator II) for 20 min. The reaction was
cooled
to room temperature, combined with another 0.5 g scale run, concentrated. The
residue
was purified by silica gel column chromatography using 1:1 dichloromethane-
hexanes to
afford 1.33 g (77%) 3'-chloro-6-fluoro-2-methoxy-biphenyl (1-16, Ri = CH3, R2
= Cl, Y
= F) as a viscous liquid.
[00131] Synthesis of N-[4-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-carbonyl)-
phenyl]-acetamide (I-19, Ri = CH3, R2 = Cl, Y = F, R3 = 4-acetylaminophenyl):
To a
stirred suspension of 4-acetylaminobenzoic acid (Aldrich, 1.32 g, 5.58 mmol)
in
anhydrous THE (20 mL) was added SOC12 (1.19 g, 10.04 mmol) and DMF (4 drops).
The reaction mixture was stirred at room temperature for 3 h, concentrated
under vacuum
to afford 4-acetylaminobenzoyl chloride (I-18, R3 = 4-acetylaminophenyl) as a
light
yellow solid.
[00132] To a stirred solution of nitrobenzene (12 mL) was added A1C13 (2.23 g,
16.73
mmol) portion wise over 10 min, then the solution was stirred at room
temperature for 20
min. A solution of 4-acetylaminobenzoyl chloride synthesized above (1.32 g,
5.58 mmol)
in dichloromethane (4 mL) was added one portion to this reaction mixture,
stirred for 72
h. The reaction mixture was poured on to crushed ice-water (250 mL), Extracted
with
dichloromethane (2 x 60 mL). The combined organic layers were washed with
brine (60
77
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
mL), dried (Na2SO4), filtered and then purified by silica gel column
chromatography
using dichloromethane then 3% methanol-dichloromethane to afford 1.97 g (89%)
N-[4-
(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-carbonyl)-phenyl]-acetamide (1-19, Ri
= CH3,
R2 = Cl, Y = F, R3 = 4-acetylaminophenyl) as light brown solid.
[00133] Synthesis of (4-amino-phenyl)-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-
yl)-
methanone hydrochloride (1-19, R3 = 4-aminophenyl). To a stirred suspension of
N-[4-
(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-carbonyl)-phenyl]-acetamide
synthesized
above (1.75 g, 4.44 mmol) in ethanol (40 mL) was added con. HC1(40 mL). The
reaction
was refluxed for 2 h, cooled to room temperature, filtered, washed with water
than
hexanes to afford 0.82 g (48%) of (4-amino-phenyl)-(3'-chloro-2-fluoro-6-
methoxy-
biphenyl-3-yl)-methanone hydrochloride as light yellow solid.
[00134] Synthesis of (3'-chloro-2-fluoro-6-methoxy-biphenyl-3-yl)-(4-tetrazol-
l-yl-
phenyl)-methanone: To a stirred suspension of (4-amino-phenyl)-(3'-chloro-2-
fluoro-6-
methoxy-biphenyl-3-yl)-methanone hydrochloride synthesized above (0.4 g, 1.02
mmol)
and sodium azide (0.2 g, 3.06 mmol) in glacial acetic acid (10 mL) was added
trimethylorthoformate (0.32 g, 3.06 mmol). The reaction was stirred at room
temperature
for 3 h, diluted with cold water (60 mL), than basified with ammonium
hydroxide
solution (28%). Extracted with dichloromethane (2 x 40 mL), the combined
dichloromethane solution was washed with brine (40 ml), dried with Na2SO4,
filtered,
and concentrated to afford 0.38 g (90%) of (3'-chloro-2-fluoro-6-methoxy-
biphenyl-3-
yl)-(4-tetrazol-l-yl-phenyl)-methanone as light yellow solid.
[00135] Synthesis of 1-[4-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
phenyl]-1H-tetrazole (P-404): To (3'-chloro-2-fluoro-6-methoxy-biphenyl-3-yl)-
(4-
tetrazol-l-yl-phenyl)-methanone synthesized above (0.1 g, 0.24 mmol) in TFA
(1.5 mL)
was added triethylsilane (0.28 g, 2.4 mmol). The reaction mixture was stirred
at room
temperature for 20 h. The reaction mixture was cooled to 0 C, diluted with
water (3
mL), basified with ammonium hydroxide solution (28%), filtered, washed with
water
dried to afford 0.08 g (81%) of 1-[4-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-
ylmethyl)-phenyl]-1H-tetrazole (P-404) as off-white solid. 1H NMR (DMSO-d6,
400
MHz): 7.2- 7.4 (m, 6 H), 7.04-7.12 (s, 3 H), 6.7 (d, J= 8.4 Hz, 1 H), 3.95 (s,
2 H), 3.77
78
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
(s, 3 H) ppm; MS(APCI+): 365.1 (M-28), LC-MS: 95.9%.
[00136] The following compounds were prepared by incorporation of various R3
groups analogous to the preparation of P-404.
P-434 P-003 P-187 P-152
P-441 P-004 P-051 P-124
P-099 P-013 P-256 P-125
P-137 P-032 P-257 P-106
P-138 P-095 P-262 P-109
P-157 P-096 P-263 P-126
P-173 P-097 P-264 P-150
P-180 P-049 P-265 P-177
P-183 P-050 P-276 P-178
P-190 P-116 P-285 P-182
P-001 P-098 P-016 P-185
P-002 P-139 P-017 P-163
[00137] General Scheme 4.
O
o c
~I 1 ci H reduction SOH Coupling
R1.0 Y R1, Y R1, 0 0 Y
X Lewis Acid, DCM H P R2
X X
1-22 1-23 1-24
1-25
OH leaving z R3
R1, group Suzuki
O Y formation R1 ,0 Y Coupling R1 Y Y = H F, OH, NH2, CN, OCH3, OEt
X = (Br, I, Cl), boronate
Z = Br, Cl, OCOZCH3
R1 = H, Me, CHF2, CF3
R2 R2 R2 P = halogen, (boronate)
R2 = Cl, CH3CO, CN, N02, Br, fused heterocycle
1-26 1-27 R3 = substituted aryl groups
1-28
79
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00138] Scheme 5.
O
~ SOH OH
~
O F O F -O~ F O F
Br Br Br 4
1-32
CI
1-29 1-30 1-31
O F N=O O g F
O
CI 1-34 CI 1-33
O F N NH
H O F NH2
O F H
P-243 O 7 P-238
Cl
1 Cl I-35 CI
S
O i F NN
H
P-443
CI
Example 4. Preparation of P-443.
[00139] Synthesis of 3-bromo-2-fluoro-4-methoxy-benzaldehyde (1-30). In a 3-
necked 250 mL round-bottomed flask equipped with nitrogen lines and a stir bar
was
placed 2-bromo-l-fluoro-3-methoxy-benzene (1-29, 2.0 g, 9.75 mmol) and
dichloromethane (48 mL). The solution was cooled in an ice water bath for 15
minutes
and then titanium tetrachloride (5.02 mL, 45.8 mmol) and dichloromethyl methyl
ether
(1.32 mL, 14.6 mmol) were added and the reaction mixture was allowed to warm
to
room temperature and react for 2 hours. The reaction mixture was slowly added
to ice
water (250 mL) and extracted with dichloromethane (2 x 100 mL). The organic
portions
were combined, washed with a saturated sodium bicarbonate solution (75 mL),
water (75
mL) and brine (75 mL), dried (MgS04) and concentrated. The crude material was
triturated with hexanes (15 mL) to produce 1.67 g of 3-bromo-2-fluoro-4-
methoxy-
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
benzaldehyde (1-30) as an off-white solid in 74% yield. MS(ESI+): 233.2 (M+)
[00140] Synthesis of (3-bromo-2-fluoro-4-methoxy-phenyl)-methanol (I-31). In a
100
mL round bottomed flask equipped with a stir bar was placed 3-bromo-2-fluoro-4-
methoxy-benzaldehyde (1-30,1.67 g, 7.17 mmol), methanol (12 mL),
dichloromethane
(12 mL) and sodium borohydride. The reaction mixture was allowed to stir at
room
temperature for 17 hours, quenched with water (10 mL) and 1M HC1(5 mL) and
extracted with dichloromethane (2 x 30 mL). The organic portions were
combined,
washed with brine (30 mL), dried (MgSO4) and concentrated. The crude material
was
triturated with hexanes (15 mL) to produce 955 mg (57%) of (3-bromo-2-fluoro-4-
methoxy-phenyl)-methanol (I-31) as a white solid.
[00141] Synthesis of (3'-chloro-2-fluoro-6-methoxy-biphenyl-3-yl)-methanol (1-
32).
Into a 100 mL round bottom flask was added (3-bromo-2-fluoro-4-methoxy-phenyl)-
methanol (1-31, 1.04 g, 4.0 mmol), 3-chlorophenylboronic acid (0.76 g, 4.8
mmol),
Pd(PPh3)4 (0.45 g, 0.41 mmol), Na2CO3 (6 mL, 2M aq), toluene, (32 mL), and
EtOH (11
mL). The reaction was degassed with N2, then stirred at 80 C for 24 hours.
Water was
added and the product was extracted with ethyl acetate. The combined organics
were
concentrated and filtered through a SiO2 plug eluting with 50% ethyl
acetate/hexanes.
The solid was triturated with ether and filtered. The filtrate was
concentrated and
triturated with ether and filtered. The filter cakes were combined and
purified by flash
column chromatography eluting with 20% acetone/hexanes to give (3'-chloro-2-
fluoro-6-
methoxy-biphenyl-3-yl)-methanol (I-32, 0.79 g, 67%) as a white solid.
[00142] Synthesis of 3-bromomethyl-3'-chloro-2-fluoro-6-methoxy-biphenyl (1-
33).
Into a 250 mL round bottom flask was added (3'-Chloro-2-fluoro-6-methoxy-
biphenyl-3 -
yl)-methanol synthesized above (1.21 g, 4.54 mmol), dichloromethane (20 mL),
PPh3
(1.19 g, 4.54 mmol), and the solution was cooled to 0 C. NBS (0.81 g, 4.54
mmol) was
added and the reaction stirred for 2 hours at 0 C. The organics were washed
with H2O
and concentrated. The residue was purified by flash column chromatography
eluting
with 8% ethyl acetate/hexanes to give 3-bromomethyl-3'-chloro-2-fluoro-6-
methoxy-
biphenyl (1-33, 956 mg, 64%) as an off-white solid.
81
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00143] Synthesis of 3'-chloro-2-fluoro-6-methoxy-3-(4-nitro-benzyl)-biphenyl
(1-34).
In a 40 mL vial equipped with a stir bar was placed 3-bromomethyl-3'-chloro-2-
fluoro-6-
methoxy-biphenyl (1-33, 400 mg, 1.21 mmol), 4-nitrophenylboronic acid (420 mg,
1.45
mmol), potassium phosphate (tribasic) (514 mg, 2.42 mmol), dimethoxyethane
(3.5 mL)
and 50% aqueous ethanol (3.5 mL). After degassing with nitrogen for 15
minutes,
tetrakis(triphenylphosphine)palladium(0) (140 mg, 0.121 mmol) was added. The
mixture was heated to 60 C for 18 hours and then the palladium catalyst was
removed
by filtering through Celite. To the filtrate were added water (50 mL) and a
saturated
ammonium chloride solution (50 mL). After extracting with ethyl acetate (3 x
50 mL),
the organic portions were combined, washed with brine (75 mL), dried (Mg504)
and
concentrated. The crude material was purified by silica gel chromatography
utilizing
20% ethyl acetate/hexanes as eluent to produce 342 mg (76%) of 3'-chloro-2-
fluoro-6-
methoxy-3-(4-nitro-benzyl)-biphenyl (1-34) as a yellow solid. MS(APCI-): 370.1
(M-1).
[00144] Synthesis of 4-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
phenylamine (I-35): In an 18 mL vial equipped with a stir bar was placed iron
powder
(179 mg, 3.20 mmol), ethanol (5.0 mL) and water (1.2 mL). The mixture was
heated to
85 C in an oil bath and then the above product 3'-Chloro-2-fluoro-6-methoxy-3-
(4-nitro-
benzyl)-biphenyl (6) (340 mg, 0.914 mmol) was added and the reaction was
continued at
85 C for 2 hours. The reaction mixture was cooled to room temperature and
filtered
through Celite. To the filtrate was added water (50 mL) and extractions were
performed
with ethyl acetate (2 x 60 mL). The organic portions were combined, washed
with brine
(50 mL), dried (Mg504) and concentrated to produce 245 mg of 4-(3'-chloro-2-
fluoro-6-
methoxy-biphenyl-3-ylmethyl)-phenylamine (I-35) as a yellow, viscous oil in
79% yield.
MS(APCI+): 342.0 (M+1).
[00145] Synthesis of [4-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
phenyl]-
thiazol-2-yl-amine.(P-443): In an 8 mL vial equipped with a stir bar was
placed 4-(3'-
chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-phenylamine (1-35) synthesized
above
(100 mg, 0.293 mmol), 2-bromothiazole (52.1 L, 0.585 mmol), 10% aqueous
ethanol
(1.5 mL) and concentrated hydrochloric acid (48.8 L, 0.585 mmol). The mixture
was
heated to 90 C for 18 hours and then cooled to room temperature. After water
(30 mL)
and 5% aqueous potassium carbonate (30 mL) were added, the aqueous portion was
82
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
extracted with ethyl acetate (2 x 35 mL) and the organic portions were
combined,
washed brine (30 mL), dried (magnesium sulfate) and concentrated. The crude
material
was purified by column chromatography utilizing 3% acetone/dichloromethane as
the
eluent to produce 57 mg of [4-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-
ylmethyl)-
phenyl]-thiazol-2-yl-amine.(P-443) as white solid in 45% yield. 'H NMR (400
MHz,
CDC13) 6 3.75 (s, 3 H), 3.94 (s, 2 H), 6.62 (d, J= 4 Hz, 1 H), 6.70 (dd, J= 8,
1 Hz, 1 H),
7.10 (t, J= 9 Hz, 1 H), 7.20-7.22 (m, 2 H), 7.26-7.40 (m, 8 H) ppm; MS(APCI+):
425.0
(M+1), LC-MS: 89%
Example 5. Preparation of P-238.
[00146] Synthesis of N-[4-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
phenyl]-2-dimethylamino-acetamide (P-238). In a 8 ml vial was charged with 4-
(3'-
chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-phenylamine (1-35) (70 mg, 0.2
mmol,
HC1 salt), N,N-dimethylglycine/HC1 salt (31 mg, 0.3 mmol, 1.5 eq.) EDCI (80
mg, 0.4
mmol, 2 eq.), HOBt (41 mg, 0.3 mmol, 1.5 eq.), Et3N (0.2 ml, 1.43 mmol, 5.3
eq.), N, N-
dimethylforamide (2m1). The resulting mixture was stirred at rt overnight. The
mixture
was poured into water and extracted with EtOAc. Evaporation of solvent gave a
residue,
which was purified by chromatography on silica gel using dichloromethane in
methanol
(33:1) as eluent to give the free base product, which was converted into HC1
salt by
treating with 2N HC1 in ether. 65 mg of N-[4-(3'-chloro-2-fluoro-6-methoxy-
biphenyl-3-
ylmethyl)-phenyl]-2-dimethylamino-acetamide (P-238) as HC1 salt in 75% yield.
1H
NMR (CDC13, 400 MHz): 9.04 (br s, 1 H), 7.46 - 7.52 (m, 2 H), 7.37 - 7.43 (m,
1 H),
7.24-7.36 (m, 3 H), 7.14 - 7.23 (m, 2 H), 7.01 - 7.12 (m,1H),6.64-
6.74(m,1H),3.86
- 3.98 (m, 2 H), 3.73 (s, 3 H), 3.05 (s, 2 H), 2.36 (s, 6 H) ppm; LCMS: 96%.
Example 6. Preparation of P-243.
[00147] Synthesis of [4-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
phenyl]-
urea (P-243). A mixture of 4-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-
ylmethyl)-
phenylamine (1-35) (100 mg, 0.26 mmol, 1 eq.), sodium cyanate (74 mg, 0.52
mmol, 2
eq.) in HOAc (1 ml) and water (1 ml) was sonicated at rt for 20 min. then was
shaken at
rt overnight. The mixture was diluted with water. The precipitate was
collected by
83
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
filtration and washed with water. After dried, 71 mg of crude product
containing a less
polar by product (presumably the corresponding acetamide) was obtained.
Trituration of
the crude product with acetone/hexane gave 56 mg of [4-(3'-chloro-2-fluoro-6-
methoxy-
biphenyl-3-ylmethyl)-phenyl]-urea (P-243) as white solid in 56 % yield. 1H NMR
(DMSO-d6, 400 MHz): 8.42 (s, 1 H), 7.40 - 7.47 (m, 2 H), 7.36 (m, 1H), 7.23-
7.32 (m,
4 H), 7.07 (m, 2 H), 6.92 (m, 1 H), 5.76 (s, 2 H), 3.83 (s, 2 H), 3.72 (s, 3
H) ppm.
[00148] Scheme 6.
N CO2H
NHz ~O F ~O F ~~ F ~~ F NJ
cIP-252 CI 1-32 CI ~cI P-446
P-456
N O FO
~O ,F ~NS
H
N O
CI G 11 N
N N H O
-O F N OD O F CN
P-258 ~O F i NHz O F NJAOEt
O
CI 1-37 CI
cI CI 1-40
P-355
P-344 c
OEt
I ., N //O O O~ N
~OJ F fN NHz N \r0y OH N
O gF NH O F
N
P-429 ~N ~ I-41
cI P-367 CI
CI P-480 0
NHz
CI
N N
O F
CI P-445
Example 7. Preparation of P-252
[00149] Synthesis of 5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-
2-ylamine P-252. A solution of 1-145 (1.20 g, 3.70 mmol) and 2-aminopyridine-5-
boronic acid pinacol ester (894 mg, 4.06 mmol) in N,N-dimethylformamide (8 mL)
was
degassed using a nitrogen stream for 10 min. To the solution was added
potassium
carbonate (1.54 g, 11.1 mmol), allylpalladium(II)chloride dimer (203 mg, 0.555
mmol),
and bis(diphenylphosphino)pentane (489 mg, 1.11 mmol) under nitrogen and the
suspension was stirred at 65 C under nitrogen for 15 h. To the reaction was
added ethyl
acetate (50 mL) and water (50 mL) and the biphasic suspension was filtered
through
celite (-15 g). The celite was washed with ethyl acetate (2 x 20 mL), and
water (2 x 20
mL) and the filtrate was separated. The aqueous layer was extracted with ethyl
acetate
84
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
(100 mL) and the organic extracts were combined. The organic solution was
washed
with water (200 mL) and brine (200 mL), dried over sodium sulfate, filtered,
and the
solvent removed under vacuum. The residue was purified by flash silica gel
column
chromatography (10-33% acetone in dichloromethane), triturated in diethyl
ether (5 mL),
filtered, washed with hexanes (5 mL) and diethyl ether (2 mL) to give P-252
(190 mg, 15
% yield) as a beige powder. 1H NMR (400 MHz, CDC13): 7.97 (d, J = 2.4 Hz, 1H),
7.39-7.27 (m, 5H), 7.07 (t, J = 8.6 Hz, 1H), 6.69 (dd, J = 8.8 Hz, 1.2 Hz,
1H), 6.45 (d, J
= 8.8 Hz, 1H), 4.32 (s, 2H), 3.82 (s, 2H), 3.75 (s, 3H) ppm. LCMS = 96.6%
purity.
MS(APCI+) = 343.0 (M+1).
Example 8. Preparation of P-258.
[00150] Synthesis of N-[5-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-2-yl]-methanesulfonamide (P-258). To a solution of 5-(3'-chloro-2-
fluoro-6-
methoxy-biphenyl-3-ylmethyl)-pyridin-2-ylamine (P-252) synthesized above (70
mg,
0.18 mmol) in pyridine (2 ml) was added methanesulfonyl chloride (23 mg, 0.20
mmol)
at 0 C under nitrogen, and stirred at room temperature for 20 h. The reaction
mixture
was diluted with water, neutralized with 6N HC1, extracted with ethyl acetate,
washed
with water and brine, and dried over Na2SO4. After it was concentrated in
vacuo, the
residue was purified by a chromatography on silica gel to yield N-[5-(3'-
chloro-2-fluoro-
6-methoxy-biphenyl-3-ylmethyl)-pyridin-2-yl]-methanesulfonamide (P-258) (25
mg,
32%). 1H NMR (400 MHz, CDC13) 8.15 (s, 1 H), 7.58 (dd, J= 8.8, 2.1 Hz, 1 H),
7.38 (s,
1H),7.34(t,J=6.6Hz,3H),7.24-7.31(m,2H),7.11(t,J= 8.5 Hz,1H),6.73(d,J=
8.5 Hz, 1 H), 3.89 (s, 2 H), 3.77 (s, 3 H), 3.09 (s, 3 H) ppm.
Example 9. Preparation of P-429.
[00151] Synthesis of 6-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
imidazo[1,2-a]pyridine-2-carboxylic acid ethyl ester (I-37). Into a 20 mL vial
with stir
bar was added 5-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-pyridin-2-
ylamine
(P-252, 565 mg, 1.65 mmol), ethyl bromopyruvate (0.52 mL, 4.12 mmol), and 5 mL
of
DME. The reaction was stirred at room temperature for 18 hours, then basified
with
NaHCO3 (aq. sat). The product was extracted with ethyl acetate and
concentrated.
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Purification by flash column chromatography (5% acetone/dichloromethane)
provided a
tan solid which was triturated with ether to obtain 6-(3'-chloro-2-fluoro-6-
methoxy-
biphenyl-3-ylmethyl)-imidazo[1,2-a]pyridine-2-carboxylic acid ethyl ester (1-
37) (198
mg, 27%) as an off-white solid.
[00152] Synthesis of 6-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
imidazo[1,2-a]pyridine-2-carboxylic acid amide (P-429). Into an 8 mL vial was
added 6-
(3'-chloro-2-fluoro-6-methoxy-biphenyl-3 -ylmethyl)-imidazo [ 1,2-a]pyridine-2-
carboxylic acid ethyl ester (1-37) synthesized above (37 mg, 0.084 mmol) and 2
mL of
7N NH3/MeOH. After stirring for 20 hours at 60 C, the reaction was
concentrated.
Trituration with ether provided 6-(3'-chloro-2 -fluoro-6-methoxy-biphenyl-3 -
ylmethyl)-
imidazo[ 1,2-a]pyridine-2-carboxylic acid amide (P-429) (24.5 mg, 71%) as a
white solid.
1H-NMR (400 MHz, DMSO-d6): 8.42 (s, 1H), 8.30 (s, 1H), 7.64 (br s, 1H), 7.53
(d, J=
4.6 Hz, 1H), 7.42 (m, 5H), 7.30 (m, 1H), 7.23 (dd, 1H, J= 9.6, 1.6 Hz), 6.97
(d, 1H), 8.4
Hz), 3.95 (s, 2H), 3.74 (s, 3H) ppm. LC/MS = 83.4%, 410.0 (APCI+).
Example 10. Preparation of P-456.
[00153] Synthesis of 5-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-2-
fluoro-
pyridine (P-456). A flask was charged with 3-Bromomethyl-3'-chloro-2-fluoro-6-
methoxy-biphenyl (1-32, 3.3g, 10 mmol), 2-fluoro-pyridine-5-boronic acid (1.4
g, 10
mmol), toluene (40 mL), 2M aq. Na2CO3 (10 mL, 20 mmol), ethanol (10 mL) and
Pd(PPh3)4 (577mg, 0.5mmol). The reaction mixture was bubbled with nitrogen gas
for 5
minutes. Then the yellow reaction mixture was stirred at 80 C. After
overnight stirring
the reaction mixture was cooled to room temperature and concentrated in-vacuo.
The
residue was diluted in EtOAc (20mL) and washed with water (30 mL). The aqueous
layer was extracted with EtOAc (2 x 20mL). The combined organic layers were
washed
with brine (50 mL), dried over Na2S04 and concentrated in-vacuo. The crude was
purified by silica gel column chromatography, eluted with hexane/EtOAc (9:1)
to
produce 3.23 g (93% yield) of 5-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-
ylmethyl)-2-
fluoro-pyridine (P-456) as a colorless solid. 1H NMR (400 MHz, DMSO-d6) 3.73
(s, 3
H),3.98(s,2H),6.95(d,J=8.6Hz,1H),7.11(dd,J=8.4,2.8Hz,1H),7.25-7.51
(m, 5 H), 7.82 (td, J= 8.2, 2.4 Hz, 1 H), 8.14 (s, 1 H), 8.32 (s, 1 H) ppm.
86
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 11. Preparation of P-446.
[00154] Synthesis of 1-[5-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-2-yl]-azetidine-2-carboxylic acid (P-446). A vial was charged with D,L-
azetidine-2-carboxylic acid (56 mg, 0.56 mmol) and DMF (lmL). Then NaH (60%
dispersion in mineral oil, 33 mg, 0.84 mmol) was added slowly (gas evolution).
After
2min. of stirring at room temperature was added 5-(3'-chloro-2-fluoro-6-
methoxy-
biphenyl-3-ylmethyl)-2-fluoro-pyridine (P-456) . The heterogeneous white
reaction
mixture was stirred and heated at 120 C. After overnight stirring the reaction
mixture
was cooled to rt and was diluted in EtOAc (5mL). The mixture was poured into a
separatory funnel with 0.5M HC1(lmL) and water (5mL). The aqueous layer was
extracted with EtOAc (3 x 5mL). The combined organic layers were washed with
brine
(15 mL), dried over Na2SO4 and concentrated in-vacuo. The crude was purified
by
silica gel column chromatography, eluted with dichloromethane in methanol
(9:1) to
produce 7.7 mg (6% yield) of 1-[5-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-
ylmethyl)-
pyridin-2-yl]-azetidine-2-carboxylic acid (P-446) as a cream solid.
Example 12. Preparation of P-445.
[00155] Synthesis of N-[5-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-2-ylmethyl]-oxalamic acid ethyl ester (1-40). Into a 20 mL vial was
added [5-(3'-
chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-pyridin-2-ylmethyl]-carbamic
acid
ethyl ester (P-367) (152 mg, 0.43 mmol), dichloromethane (4 mL), TEA (0.11 mL,
0.85
mmol). The solution was cooled to 0 C and Ethyl chlorooxoacetate (71 uL, 0.64
mmol)
was added. After 20 minutes at room temperature, the organic solution was
washed with
H2O and brine, and then concentrated. Ethyl acetate was added to the residue,
which
produced a solid and was filtered. The solid was triturated with ether and
dried to give
N-[5-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-pyridin-2-ylmethyl]-
oxalamic
acid ethyl ester (1-40) (69 mg, 35%) as a gray-blue solid.
[00156] Synthesis of 6-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
imidazo[1,5-a]pyridine-3-carboxylic acid ethyl ester (1-41). Into a 4 mL vial
was added
87
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
the above compound (12) (44 mg, 0.096 mmol), dichloromethane (1 mL), pyridine
(31
uL, 0.48 mmol), and POC13 (13 uL, 0.14 mmol). The reaction was stirred for 18
hours at
room temperature and then H2O was added. The product was extracted with
dichloromethane and then concentrated. Purification using FCC eluting eluting
with
20% acetone/hexane provided a yellow semi-solid which was triturated with
hexane to
give 6-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-imidazo[1,5-
a]pyridine-3-
carboxylic acid ethyl ester (I-41) (8.2 mg, 19%) as a tan solid.
[00157] Synthesis of 6-(3'-chloro-2fluoro-6- methoxy-biphenyl-3-ylmethyl)-
imidazo[1,5-a]pyridine-3-carboxylic acid amide (P-445). Into a 4 mL vial was
added I-
41 (7 mg, 0.016 mmol) and 2 mL of 7N NH3/MeOH. The reaction was stirred at 60
C
for 18 hours, and then concentrated to afford 6-(3'-chloro-2-fluoro-6-methoxy-
biphenyl-
3-ylmethyl)-imidazo[1,5-a]pyridine-3-carboxylic acid amide (P-445) (6.6 mg,
99%) as a
tan solid. 1H NMR (400 MHz,DMSO-d6) 9.29 (s, 1 H), 7.82 (br s, 1 H), 7.71 (d,
J=
9.3 Hz, 1 H), 7.52 (s, 1 H), 7.49 - 7.33 (m, 5 H), 7.33 - 7.26 (m, 1 H), 6.95
(s, 2 H), 3.97
(s, 2 H), 3.74 (s, 3 H) ppm. LC/MS = 88.6%, 410.0 (APCI+).
[00158] Scheme 7.
Br \ O I / I / x
~O H NHZ
O.B "a N O NH O / / NH2
Br
Br H z
CI
1-42 1-43 1-44
P-378
Example 13. Preparation of P-378
[00159] Synthesis of [4-(3-bromo-4-methoxy-benzyl)-phenyl]-urea (1-44). To a
40
mL vial equipped with a teflon screw cap and a magnetic stir bar was added 2-
bromo-4-
bromomethyl-1-methoxybenzene (649 mg, 2.32 mmol), [4-(4,4,5,5-tetramethyl-
[1,3,2]dioxaborolan-2-yl)-phenyl]-urea (577 mg, 2.20 mmol) and potassium
phosphate
(933 mg, 4.40 mmol). To the vial was then added dimethoxyethane (15 mL),
ethanol
(3.7 mL) and water (3.7 mL). To this stirring solution was added
tetrakis(triphenylphosphine) palladium (127 mg, 0.11 mmol) and the solution
was
degassed by bubbling N2 gas through the solution for 20 min. The vial was
capped and
88
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
placed in an oil bath with stirring at 65 C for 12.5 h. The cooled reaction
mixture was
concentrated under a stream of N2 gas to a total volume of -5 mL and then
diluted with
ethyl acetate (20 mL) and water (10 mL). Upon shaking a white solid
precipitates. The
solid is filtered and dried to afford 272 mg (34%) of [4-(3-bromo-4-methoxy-
benzyl)-
phenyl]-urea (1-44) as a white solid.
[00160] Synthesis of [4-(3'-chloro-6-methoxy-biphenyl-3-ylmethyl)-phenyl]-urea
(P-
378). To a 20 mL vial equipped with a magnetic stir bar and a screw cap was
added [4-
(3-bromo-4-methoxy-benzyl)-phenyl]-urea (1-44) synthesized above compound (250
mg,
0.746 mmol), dimethoxyethane (5 mL), ethanol (1 mL) and water (1 mL). To this
mixture were added 3-chlorophenylboronic acid (140 mg, 0.895 mmol), potassium
phosphate (316 mg, 1.49 mmol) and tetrakis(triphenylphosphine) palladium (30
mg,
0.0254 mmol). The stirring reaction mixture was degassed by bubbling N2 gas
through
the solution for 10 min. The vial was capped and placed in an oil bath with
stirring at 80
C for 16 h. The cooled reaction mixture was concentrated to dryness, then
diluted with
water (5 mL) and ethyl acetate (15 mL). The aqueous layer was extracted with
ethyl
acetate (2 x 5 mL) and the combined organic extracts were dried (Na2SO4),
filtered and
concentrated under a stream of N2 gas. The residue was purified by flash
chromatography on silica gel (35 g) utilizing 9:1 dichloromethane/acetone as
eluent.
Fractions pure by TLC were combined and concentrated to give 139 mg (5 1%) of
[4-(3'-
chloro-6-methoxy-biphenyl-3-ylmethyl)-phenyl]-urea (P-378) as an off-white
solid. 1H
NMR (400 MHz, DMSO-d6) 6 3.74 (s, 3H), 3.83 (s, 2H), 5.75 (br s, 2H), 7.03 (d,
J= 8.4
Hz, 1H), 7.08 (m, 2H), 7.15 (d, J= 2.4 Hz, 1H), 7.18 (dd, J= 8.4, 2.4 Hz, 1H),
7.28 (m,
2H), 7.36-7.41 (m, 3H), 7.48 (m, 1H), 8.40 (br s, 1H) ppm.
MS(APCI+): 367.0 (M++1) ;LC-MS: 95.9 % purity.
[00161] The following compounds were prepared analogous to the Examples shown
above.
P-008 P-199 P-296 P-339 P-392
P-011 P-202 P-297 P-340 P-394
P-067 P-216 P-298 P-344 P-395
P-102 P-217 P-299 P-345 P-399
P-103 P-219 P-300 P-347 P-400
P-105 P-220 P-301 P-348 P-401
89
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
P-110 P-221 P-302 P-349 P-402
P-111 P-222 P-304 P-355 P-403
P-112 P-225 P-305 P-356 P-407
P-113 P-226 P-308 P-357 P-408
P-114 P-231 P-309 P-358 P-409
P-119 P-232 P-312 P-359 P-410
P-121 P-233 P-313 P-360 P-411
P-122 P-234 P-314 P-361 P-412
P-123 P-240 P-315 P-362 P-413
P-134 P-242 P-316 P-365 P-414
P-135 P-252 P-317 P-366 P415
P-140 P-258 P-318 P-367 P-420
P-141 P-259 P-319 P-368 P-421
P-142 P-261 P-320 P-371 P-423
P-143 P-266 P-321 P-372 P-428
P-144 P-272 P-322 P-373 P-431
P-145 P-274 P-323 P-374 P-432
P-146 P-275 P-324 P-375 P-433
P-151 P-277 P-325 P-377 P-437
P-159 P-278 P-327 P-378 P-269
P-164 P-279 P-328 P-381 P-239
P-165 P-281 P-329 P-382
P-166 P-284 P-330 P-383
P-170 P-286 P-331 P-384
P-171 P-287 P-334 P-385
P-189 P-290 P-335 P-387
P-191 P-291 P-336 P-388
P-192 P-292 P-337 P-390
P-194 P-295 P-338 P-391
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00162] General Scheme 8.
z
z Q
R1.0 Suzuki lR3
X Y Coupling R1'0 Y R3 coupling 0
1-45 R1
PH2
R2 1-48 R2
1-46 1-47 1-49
R2
Y=H,F
Q X = Br, I, Cl, boronate
R3 Z = OH, NH2, N02
R1 0 Y R1 = H, Me, CHF2, CF3
P = halogen, boronate
Q=0,NR',S
R2 = Cl, CH3CO, CN, N02, Br, fused heterocycle
R2 R3 = substituted groups
I-50
Example 14. Preparation of P-268
H
N \ IOI
O 14- N H NH2
O
P-268
[00163] Synthesis of 3-iodo-4-methoxy-phenylamine (1-45, X = I, Y = H, Z =
NH2, R1
= CH3): Ina 3-neck 250 mL round-bottomed flask equipped with a stir bar,
condenser
and N2 lines was placed iron powder (3.50 g, 62.7 mmol), ammonium chloride
(4.88 g,
91.3 mmol), ethanol (72 mL) and water (23 mL). The mixture was heated to 85 C
and
then 2-iodo-1-methoxy-4-nitro-benzene (5.0 g, 17.9 mmol) was added portion
wise over
a period of about 2 minutes. The mixture was allowed to stir at 85 C for 2
hours and
then filtered through celite. The celite was washed with EtOH (100 mL) and the
filtrate
was concentrated. To the concentrated material was added water (100 mL) and
ethyl
acetate (150 mL). The organic portion was removed and the aqueous portion was
re-
extracted with ethyl acetate (150 mL). The organic portions were combined,
washed
with brine (150 mL), dried (MgS04) and concentrated. The crude material was
purified
91
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
by column chromatography utilizing 50% EtOAc/hexanes as the eluent to produce
3.92 g
of 3 -iodo-4-methoxy-phenylamine (1-45, X = I, Y = H, Z = NH2, Ri = CH3) as a
brown
semi-solid in 88% yield. MS(ESI+): 250.1 (M+1)
[00164] Synthesis of 1-(5'-amino-2'-methoxy-biphenyl-3-yl)-ethanone (1-47, Y =
H, Z
= NH2, Ri = CH3, R2 = COCH3): In a 3-neck 100 mL round-bottomed flask equipped
with a condenser, stir bar and N2 lines was placed 3-iodo-4-methoxy-
phenylamine
synthesized above (2.92 g, 11.7 mmol), 3-acetlylphenylboronic acid (2.11 g,
12.9 mmol),
potassium carbonate (4.85 g, 35.1 mmol), triphenylphosphine (921 mg, 3.51
mmol), 1,4-
dioxane (23 mL), 50% aqueous ethanol (23 mL) followed by palladium (II)
acetate (263
mg, 1.17 mmol). The mixture was heated to 90 C for 16 hours and then cooled
to room
temperature. The palladium catalyst was removed via filtration and to the
filtrate was
added 1M HC1(50 mL) and water (50 mL). The aqueous portion was Extracted with
ethyl acetate(2 x 75 mL), the organic portions were combined, washed with
brine (75
mL), dried (Mg504) and concentrated. The crude material was purified by column
chromatography utilizing 50% EtOAc/hexanes as the eluent to produce 1.18 g of
(5'-
amino-2'-methoxy-biphenyl-3 -yl)-ethanone (1-47, Y = H, Z = NH2, Ri = CH3, R2
=
COCH3) as a pale orange oil in 42% yield. MS(APCI+): 242.0 (M+1).
[00165] Synthesis of 1-[2'-methoxy-5'-(4-nitro-phenylamino)-biphenyl-3-yl]-
ethanone
(1-49, Y = H, Q = NH, Ri = CH3, R2 = COCH3, R3 = NO2) : To a 40 mL vial
equipped
with a stir bar was placed 1-iodo-4-nitrobenzene (1.26 g, 5.07 mmol), cesium
carbonate
(2.20 g, 6.76 mmol), ( )-2,2'-bis(diphenylphosphino)-1,1'-binaphthalene (316
mg, 0.507
mmol), and a solution of (5'-amino-2'-methoxy-biphenyl-3-yl)-ethanone (1-47, Y
= H, Z
= NH2, Ri = CH3, R2 = COCH3) synthesized above (816 mg, 3.38 mmol) in toluene
(13.5
mL). The mixture was stirred for 10 minutes and then
tris(dibenzylideneacetone)dipalladium(0) (310 mg, 0.338 mmol) and the mixture
was
heated to 110 C for 16 hours. The reaction was cooled to room temperature and
then
filtered through Celite. The filtrate was treated with water (40 mL), 1M
HC1(40
mL) and then Extracted with ethyl acetate (2 x 75 mL). The organic portions
were
combined, washed with brine (75 mL), dried (Mg504) and concentrated. The crude
material was purified by column chromatography utilizing 35% EtOAc/hexanes as
the
eluent to produce 277 mg of 1-[2'-methoxy-5'-(4-nitro-phenylamino)-biphenyl-3-
yl]-
92
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
ethanone (1-49, Y = H, Q = NH, Ri = CH3, R2 = COCH3, R3 = NO2) as a dark
orange
solid in 23% yield.
[00166] Synthesis of 1-[5'-(4-amino-phenylamino)-2'-methoxy-biphenyl-3-yl]-
ethanone hydrochloride: In an 18 mL vial equipped with a stir bar was placed
iron
powder (148 mg, 2.66 mmol), ammonium chloride (207 mg, 3.87 mmol), absolute
EtOH
(3.1 mL) and water (1.0 mL). The mixture was heated to 85 C and then was
added -[2'-
methoxy-5'-(4-nitro-phenylamino)-biphenyl-3-yl]-ethanone (1-49, Y = H, Q = NH,
Ri =
CH3, R2 = COCH3, R3 = NO2) synthesized above (275 mg, 0.759 mmol) was added
and
the mixture was heated for 2 hours. The reaction was cooled to room
temperature,
filtered through Celite and Extracted with ethyl acetate (2 x 40 mL). The
organic
portions were combined, washed with brine (40 mL), dried (MgSO4) and
concentrated.
The crude material was purified by column chromatography utilizing 75%
EtOAc/hexanes as the eluent to produce 207 mg of the free base as a dark
orange oil in
82% yield. The free base was treated with 4.0 M HC1 in 1,4-dioxane (1.0 mL)
and
stirred for 3 hours at room temperature. The reaction mixture was treated with
diethyl
ether (4 mL) and the solid was collected via suction filtration. After washing
the solid
with diethyl ether (3 x 2 mL), 100 mg of 1-[5'-(4-amino-phenylamino)-2'-
methoxy-
biphenyl-3-yl]-ethanone hydrochloride was isolated as a brown solid in 44%
yield.
MS(APCI-): 366.9 (M-2); LC-MS: 85%
[00167] Synthesis of [4-(3'-acetyl-6-methoxy-biphenyl-3-ylamino)-phenyl]-urea
(P-
268): In an 8 mL vial equipped with a stir bar was placed 1-[5'-(4-amino-
phenylamino)-
2'-methoxy-biphenyl-3-yl]-ethanone synthesized above (free base) (60.0 mg,
0.180
mmol), water (600 L), acetic acid (300 L) and sodium cyanate (46.8 mg, 0.720
mmol).
The mixture was stirred at room temperature for 4 hours and then water (20 mL)
was
added followed by an P-traction with (2 x 30 mL). The organic portions were
combined, washed with brine (30 mL), dried (MgSO4) and concentrated. The crude
material was purified by column utilizing 75% acetone/DCM as the eluent to
produce 32
mg of [4-(3'-acetyl-6-methoxy-biphenyl-3-ylamino)-phenyl]-urea (P-268) as a
light
brown solid in 47% yield. MS(APCI+): 376.1 (M+1); LC-MS: 94%.
93
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00168] Scheme 9.
S
O NCO
II + O, N..O NCO
O Br O 11 O
SH 0-N O 19 21
17 18 20 CI 1-53
1-51 1-52
O
O - S Q
S S
O NH O NH,
O NH CIH
II
O~`NH O NH
P-448 CI
CI P-450 CI
P-447
O
S
~S
~ ~I ~ II I
O O NH
O NH 0 NH
\ CI P-449 CI ,
P-540
Example 15. Preparation of P-447.
[00169] 1 -Methoxy-4-phenmylsulfanyl-(4'-nitrobenzene) (I-51). A solution of 4-
methoxybenzenethiol (500 mg, 3.57 mmol) and 4-iodonitrobenzene (1.07 g, 4.28
mmol) in dimethylformamide (10 mL) was stirred at room temperature. To the
orange
solution was added solid cesium carbonate (3.48 g, 10.7 mmol). The resulting
purple
solution was stirred at room temperature overnight. The solution was diluted
with ethyl
acetate (50 mL) and washed with water (50 mL). The aqueous wash was extracted
with
ethyl acetate (50 mL). The organic washes were combined, washed with saturated
aqueous sodium bicarbonate (100 mL), water (100 mL), and brine (100 mL), dried
over
sodium sulfate, decanted, and the solvent removed under vacuum. The resultant
solid
was purified by flash silica gel column chromatography using 10% ethyl acetate
in
hexanes as eluant to give product I-51 (933 mg, 99% yield).
[00170] 2-Bromo-methoxy-4-(4'-nitro-phenyl-sulfanyl)-benzene (1-52). A 10% v/v
solution of bromine (1 mL) in glacial acetic (9 mL) acid was prepared. A
solution of I-51
(780 mg, 2.99 mmol) in glacial acetic acid (7.8 mL) was stirred at room
temperature. To
94
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
this solution was added the 10% bromine in acetic acid solution (3.53 mL total
solution,
6.87 mmol of bromine). The reaction was allowed to stir at room temperature
for 2 h.
The reaction was combined with a previous pilot run of this reaction (100 mg
0.383
mmol). The previous run had been reacted under the same conditions and TLC
showed
similar results. The combined reactions were diluted with ethyl acetate (100
mL), and
washed with water (100 mL). The organic extract was then washed with saturated
aqueous sodium bicarbonate (2 x 100 mL), water (100 mL), and brine (100 mL),
dried
over sodium sulfate, decanted, and removed under vacuum. The resulting yellow
residue
was purified by flash silica gel column chromatography (10% ethyl acetate in
hexanes as
the eluant) to give 1-52 (550 mg, 48% overall yield) and a second less pure
crop (420
mg).
[00171] 3'-Chloro-2-methxoy-5-(4-nitro-phenyl-sulfonyl)-biphenyl (1-53). A
solution
of the above compound 1-52 (550 mg, 1.62 mmol) and 3-chlorobenzeneboronic acid
(278
mg, 1.78 mmol) in toluene (6 mL) was degassed with a nitrogen stream for 15
min. To
the reaction was added ethanol (800 uL) and 2 M aqueous sodium carbonate
solution
(1.6 mL) and the reaction was degassed under the nitrogen stream. To the
reaction was
added tetrakis(triphenylphosphine)palladium(0) (93.4 mg, 8.08 x 10-2 mmol) and
the
reaction was heated to 100 C under nitrogen for 5 h. The reaction was cooled
to room
temperature and diluted with ethyl acetate (50 mL). The organic suspension was
washed
with water (50 mL). The aqueous wash was extracted into ethyl acetate (50 mL),
the
organic extractions combined, washed with water (2 x 30 mL) and brine (30 mL),
dried
over sodium sulfate, filtered, and the solvent removed under vacuum. The
resulting crude
red oil was purified by flash silica gel column chromatography (10% ethyl
acetate in
hexanes) to give 1-53 (329.9 mg, 55% yield) as a yellow powder.
[00172] 4-(3'-Chloro-6-methoxy-biphenyl-3-ylsulfanyl)-phenylaammonium chloride
(P-447). A suspension of compound 1-53 (315 mg, 0.847 mmol), iron powder (166
mg,
2.97 mmol) and solid ammonium chloride (231 mg, 4.32 mmol) in ethanol (5 mL)
and
water (1.5 mL) was heated to 80 C for 21 h. The solvent was removed under
vacuum
and the dark residue was dissolved in ethyl acetate (30 mL) and water (30 mL).
The
layers were separated, and the aqueous layer was extracted with ethyl acetate
(30 mL).
The organic extracts were combined, washed with water (50 mL) and brine (50
mL),
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
dried over sodium sulfate, filtered, and the solvent removed under vacuum. The
residue
was dried under high vacuum overnight. The orange oil was dissolved in dioxane
(2
mL), stirred, and 4 N hydrogen chloride in dioxane (1 mL) was added. The
reaction was
stirred for 3 h at room temperature, and the solvent was removed under vacuum
to give
product (P-447) (257.8 mg, 81% yield) as a brown powder. 'H NMR (400 MHz,
DMSO-d6) 7.95-7.49 (m, 1H), 7.46-7.37 (m, 3H), 7.34 (dd, J= 8.6, 2.2 Hz, 1H),
7.26-
7.23 (m, 3H), 7.16 (d, J= 8.8 Hz, 1H), 6.96-6.95 (m, 2H), 3.78 (s, 3H) ppm.
LCMS = 97.5 % purity. MS(APCI+) = 342.0 (M+1).
Example 16. Preparation of P-448.
[00173] 1-[4-(3'-Chloro-6-methoxy-biphenyl-3-ylsulfanyl)-phenyl]-3-ethyl urea
(P-
448). A solution of the above compound (P-447) (195 mg, 0.520 mmol) in
pyridine (3
mL) was stirred at room temperature. To the reaction was added ethyl
isocyanate (110
mg, 1.56 mmol). The solution was stirred at room temperature for 22.5 h. The
reaction
was diluted in ethyl acetate (50 mL), and washed with water (50 mL). The
aqueous wash
was extracted into ethyl acetate (50 mL), and the organic extracts combined.
The ethyl
acetate solution was washed with 1 N aqueous hydrochloric acid (50 mL), water
(2 x 50
mL), and brine (50 mL), dried over sodium sulfate, and the solvent removed
under
vacuum. The resultant solid was triturated with diethyl ether (5 mL),
filtered, washed
with diethyl ether (2 x 3 mL), and dried to give product (P-448) (109.2 mg,
51% yield).
'H NMR (400 MHz, DMSO-d6) 7.49-7.48 (m, 1H), 7.39-7.29 (m, 5H), 7.26-7.19 (m,
4H), 6.94 (d, J= 8.8 Hz, 1H), 6.19 (s, 1H), 4.60 (m, 1H), 3.82 (s, 3H), 3.32-
3.25 (m,
2H), 1.15 (t, J = 7.4 Hz, 3H) ppm. LCMS = 97.3% purity. MS (APCI+) = 413.0
(M+1).
Example 17. Preparation of P-449.
[00174] 1-[4-(3'-Chloro-6-methoxy-biphenyl-3-sulfinyl)-phenyl]-3-ethyl urea (P-
449). A solution of 10% v/v 30% aqueous hydrogen peroxide (w/w) (1 mL) in
glacial
acetic acid (9 mL) was made. To a slurry of the above compound (P-448) (41.3
mg,
0.100 mmol) in acetic acid (300 uL) was added the hydrogen peroxide solution
(96.7 uL,
0.100 mmol hydrogen peroxide). The solution was stirred for 1 hour at room
temperature. The solvent was removed under vacuum, and the residue purified by
silica
96
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
gel thin layer chromatography (eluting with 10% acetone in dichloromethane),
triturated
with diethyl ether (5 mL), filtered, and dried to give the product (P-449)
(29.3 mg, 68%
yield) as a brown gum. 1H NMR (400 MHz, DMSO-d6) 7.55-7.44 (m, 5H), 7.36-7.32
(m, 5H), 7.20 (s, 1H), 7.04 (d, J= 8.4 Hz, 1H), 5.27 (m, 1H), 3.84 (s, 3H),
3.29-3.22 (m,
2H), 1.12 (t, J= 7.4 Hz, 1H) ppm. LCMS = 97.6% purity. MS(APCI+)= 429.1 (M+1).
Example 18. Preparation of P-450.
[00175] 1-[4-(3'-Chloro-6-methoxy-biphenyl-3-sulfonyl)-phenyl]-3-ethyl-urea (P-
450). A solution of 1-[4-(3'-Chloro-6-methoxy-biphenyl-3-ylsulfanyl)-phenyl]-3-
ethyl
urea (P-448) (41.3 mg, 0.100 mmol) in glacial acetic acid (300 uL) was stirred
at room
temperature. To the solution was added 290 uL of 10% v/v (30% w/w hydrogen
peroxide
in water) in acetic acid. The resulting solution was stirred for 1.5 h at room
temperature.
The solvent was removed under vacuum, and the residue was purified by silica
gel thin
layer chromatography (eluting with 25% acetone in dichloromethane) and dried
to give
the product (P-450) (20.2 mg, 45% yield) as a yellow grease. 1H NMR (400 MHz,
DMSO-d6) 7.55-7.44 (m, 5H), 7.35-7.31 (m, 4H), 7.28 (s, 1H), 7.04 (d, J= 8.4
Hz, 1H),
5.32 (t, J= 5.6 Hz, 1H), 3.84 (s, 3H), 3.28-3.21 (m, 2H), 1.11 (t, J= 7.4 Hz,
1H) ppm.
LCMS = 97.9% purity. MS (APCI+)= 473.1 (M +28), 443.0 (M+1).
Example 19. Preparation of P-540.
[00176] P-540 Synthesis of [4-(3'-Chloro-6-methoxy-biphenyl-3-ylsulfanyl)-
phenyl]-
urea. A solution of PR195 (200 mg, 0.585 mmol) and sodium cyanate (76 mg, 1.17
mmol) in water (10 mL) and glacial acetic acid (5 mL) was stirred at room
temperature
overnight. Water (20 mL) was added to the gummy suspension, and the reaction
was
extracted into ethyl acetate (2 x 20 mL). The organic extracts were combined,
washed
with 1 N aqueous hydrochloric acid (2 x 20 mL), saturated aqueous sodium
bicarbonate
(2 x 20 mL), water (20 mL), and brine (20 mL), dried over sodium sulfate, and
the
solvent removed under vacuum to give crude PR199 as a yellow gum. The crude
material was purified by preparatory thin layer chromatography (silica)
eluting with
dichloromethane and developed twice to give PR199 (53.1 mg, 22.5% yield) as a
white
powder. 1H NMR (400 MHz, CDC13) 7.49-7.48 (m, 1H), 7.41-7.28 (m, 5H), 7.23 (s,
4H),
97
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
6.95 (d, J= 8.0 Hz, 1H), 6.31 (s, 1H), 4.57 (s, 2H), 3.83 (s, 3H) ppm. LCMS =
95.3%
purity. MS = 385.1 (M+1).
[00177] The following compounds were prepared analogous to the Examples shown
above.
P-260 P-294 P-193 P-283
P-267 P-288 P-230 P-254
P-280 P-293 P-240
[00178] General Scheme 10.
Br Suzuki transition-metal
Coupling R2 catalyzed coupling R1 R2
R1, R1- -O R2
O OH O N
Br Br
HO-B R1 = H, CH3 NV
R2=F
1-54 1-56 R3 = H, CF3 R3 R4
R2 R4 = CI, 4-pyridyl 1-57
1-55
Example 20. Preparation of P-380
Me,O I I F
N
N\
CI
P-380
[00179] 2-Bromo-4-(4-fluoro-benzyl)-1-methoxy-benzene: In a 250 mL round-
bottomed flask equipped with a stir bar was placed 3-bromo-4-methoxybenzyl
bromide
(5.0 g, 17.9 mmol), 4-fluorophenylboronic acid (2.76 g, 19.7 mmol), potassium
phosphate (tribasic) (7.60 g, 35.8 mmol), dimethoxyethane (30 mL) and 50%
aqueous
ethanol (30 mL). The mixture was degassed with nitrogen for 30 minutes and
then
added tetrakis(triphenylphosphine)palladium(0) (5.17 g, 4.48 mmol). The
mixture was
98
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
heated to 60 C for 4 hours and then the palladium catalyst was filtered off.
To the
filtrate were added water (100 mL) and a saturated ammonium chloride solution
(150
mL). After extracting with ethyl acetate (3 x 100 mL), the organic portions
were
combined, washed with brine (150 mL), dried (MgSO4) and concentrated. The
crude
material was purified by column utilizing 5% EtOAc/hexanes as the eluent to
produce
2.12 g of 2-bromo-4-(4-fluoro-benzyl)-1-methoxy-benzene as a colorless oil in
40%
yield.
[00180] 4-Chloro-l-[5-(4-fluoro-benzyl)-2-methoxy-phenyl]-1H-pyrazole (P-380):
In
a 2-5 mL microwave vial equipped with a stir bar was placed the above product
(250 mg,
0.847 mmol), 4-chloro-lH-pyrazole (172 mg, 1.69 mmol), potassium carbonate
(234 mg,
1.69 mmol), CuI (48.4 mg, 0.254 mmol) and N-methyl-2-pyrrolidone (2.8 mL). The
mixture was heated to 190 C in a Biotage Initiator microwave reactor for 1
hour. The
mixture was quenched with water (30 mL) and a saturated ammonium chloride
solution
(30 mL) followed by an P-traction with ethyl acetate (2 x 30 mL). The organic
portions
of the product (P-380) as a viscous, yellow oil in 35% yield. 'H NMR (400 MHz,
CDC13)
6 3.87 (s, 3 H), 3.93 (s, 2 H), 6.94-6.98 (m, 3 H), 7.08 (dd, J= 8, 2 Hz, 1
H), 7.12-7.16
(m, 2 H), 7.55 (d, J= 2 Hz, 1 H), 7.60 (s, 1 H), 8.04 (s, 1 H) ppm.
MS(APCI+): 317.0 (M+1); LC-MS: 97%
Example 21. Preparation of P-389.
O I I F
,N
N
N
P-389
[00181] Synthesis of 4-{1-[5-(4-Fluoro-benzyl)-2-methoxy-phenyl]-1H-pyrazol-4-
yl}-pyridine (P-3 89). In a 2-5 mL microwave vial equipped with a stir bar was
placed I-
185 (250 mg, 0.847 mmol), 4-(1H-pyrazol-4-yl)-pyridine (245 mg, 1.69 mmol),
99
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
potassium carbonate (234 mg, 1.69 mmol) and N-methyl-2-pyrrolidone (2.8 mL).
The
mixture was degassed with nitrogen for 10 minutes and then CuI (48.4 mg, 0.254
mmol)
was added. The mixture was heated to 190 C in a Biotage Initiator microwave
reactor
for 1 hour. The mixture was quenched with water (50 mL) and a saturated
ammonium
chloride solution (50 mL) followed by an extraction with ethyl acetate (2 x
100 mL).
The organic portions were combined, dried (MgSO4) and concentrated. The
residue was
purified via column chromatography utilizing 15% acetone/dichloromethane as
the
eluent. After a failed recrystallization attempt with dichlormethane/hexanes,
41 mg of P-
389 as a viscous, yellow oil in 13% yield. 'H NMR (400 MHz, CDC13) 6 3.91 (s,
3 H),
3.96 (s, 2 H), 6.95-7.02 (m, 3 H), 7.10-7.18 (m, 3 H), 7.42-7.45 (m, 2 H),
7.62 (d, J= 2
Hz, 1 H), 8.04 (s, 1 H), 8.41 (s, 1 H), 8.57-8.59 (m, 2 H) ppm. MS(APCI+):
360.1
(M+1); LC-MS: 90%.
Example 22. Preparation of P-396.
O I I F
,N
N\
F
F F
P-396
[00182] Synthesis of 1-[5-(4-Fluoro-benzyl)-2-methoxy-phenyl]-3-
trifluoromethyl-
1H-pyrazole (P-396). In a 2-5 mL microwave vial equipped with a stir bar was
placed I-
185 (250 mg, 0.847 mmol), 3-trifluoromethyl-1H-pyrazole (230 mg, 1.69 mmol),
potassium carbonate (234 mg, 1.69 mmol) and N-methyl-2-pyrrolidone (2.8 mL).
The
mixture was degassed with nitrogen for 10 minutes and then CuI (48.4 mg, 0.254
mmol)
was added. The mixture was heated to 190 C in a Biotage Initiator microwave
reactor
for 1 hour. The mixture was quenched with water (30 mL) and a saturated
ammonium
chloride solution (30 mL) followed by an extraction with ethyl acetate (2 x 60
mL). The
organic portions were combined, dried (MgS04) and concentrated. The residue
was
purified via column chromatography utilizing 20% ethyl acetate/hexanes as the
eluent to
produce 20 mg of P-396 as a viscous, yellow oil in 7% yield. 'H NMR (400 MHz,
100
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
CDC13) 6 3.86 (s, 3 H), 3.94 (s, 2 H), 6.65 (d, J= 2 Hz, 1 H), 6.95-6.99 (m, 3
H), 7.10-
7.16 (m, 3 H), 7.57 (d, J= 2 Hz, 1 H), 8.03 (m, 1 H) ppm. MS(APCI+): 351.0
(M+1);
LC-MS: 92%.
[00183] General Scheme 11.
O OH
O
PH, Suzuki H R3
H R3
Coupling R1, R1 O R1.
R1_ + i 0 0
O organometallic
X R2 reagent deoxygenation
1-58 R2 R2 R2
1-59
X, P = halogen, boronate 1-60 1-61
R1 = H, Me, CHF2, CF3 1-62
R2 = CI, CH3CO, CN, N02, Br, fused heterocycle
R3 = substituted groups
Example 23. Preparation of P-117.
OH
Me.O V0~ F
N-N
P-117
[00184] 2-(3-Iodo-phenyl)-[1,3,4]oxadiazole: A mixture containing 3-iodo
benzoylhydrazide (4.5 g, 17.17 mmol) and 85.6 ml of triethylorthoformate (30
equivalents) was refluxed for 16 hours. The reaction mixture was concentrated
to a solid
residue and triturated with 15 ml mixture of diethyl ether/ heptane= 1: 1.
After filtration
and drying, 3.9 g white solid was obtained. The mother liquor was concentrated
and
recrystallized from a mixture of methanol/ water= 1:3 to give another 200 mg
of the
product, a total of 4.1 g.
[00185] 6-Methoxy-3'-[1,3,4]oxadiazol-2-yl-biphenyl3-carbaldehyde. A mixture
of 5-
formyl-2-methoxyphenyl boronic acid (414 mg, 2.29 mmol), the above product, 2-
(3-
iodo-phenyl)-[1,3,4]oxadiazole, (626 mg, 2.29 mmol), aqueous 2N K2C03 (3.4 mL,
3
101
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
equivalents), Pd(PPh3)2C12 (50 mg, 0.068 mmol) in DME (15 mL) was stirred at
room
80 C for 7 hours. Reaction mixture was cooled to r.t., then it was diluted
with ethyl
acetate (45 mL) and washed with water, brine and dried over Na2SO4. After
removal of
solvent, 700 mg of crude was obtained. Purification by column chromatography
gave
350 mg of the product (Yield: 55%).
[00186] (4-Fluoro-phenyl)-(6-methoxy-3'-[ 1,3,4]oxadiazol-2-yl-biphenyl-3-yl)-
methanol (P-117). To a solution of the above aldehyde, 6-Methoxy-3'-
[1,3,4]oxadiazol-
2-yl-biphenyl3-carbaldehyde ( 86 mg, 0.307 mmol) in anhydrous THE (1 mL) was
added
dropwise a solution of 4-fluorophenyl magnesium bromide in THE (0.46 ml, 1M)
at -78
C. After the addition was complete, the resulting mixture was allowed to warm
to room
temperature and stirred for 40 minutes. Then saturated NH4C1 aq. was added.
The
mixture was extracted with EtOAc (2 x 10 ml). The combined organic layers were
washed with water and dried over Na2SO4. Removal of solvent gave a residue,
which
was purified by chromatography on silica gel to give 112 mg of the product (P-
117). 1H
NMR (400 MHz, CDC13) 6 ppm 3.82 (s, 3H), 5.85 (s, 1H), 6.97(d, J= 8.4 Hz, 1H),
6.98- 7.05 (m, 2H), 7.31- 7.39 (m, 4H), 7.54 (t, 1H), 7.67- 7.7 (m, 1H), 8.01-
8.04 (m,
1H), 8.23 (s, 1H), 8.46 (s, 1H) ppm; LCMS (ESI+): 377 (M+1).
[00187] The following compounds were prepared analogous to the example shown
above.
Example 24. Preparation of P-118.
[00188] 2-[5'-(4-Fluoro-benzyl)-2'-methoxy-biphenyl-3-yl]-[1,3,4]oxadiazole (P-
118).
1H NMR (400 MHz, CDC13) 6 3.80 (s, 3H), 3.95 (s, 2H), 6.93(d, J= 8.4 Hz, 1H),
6.97-
6.99 (m, 2H), 7.13- 7.17 (m, 4H), 7.54 (t, 1H), 7.68- 7.7 (m, 1H), 8.01- 8.03
(m, 1H),
8.23 (s, 1H), 8.46 (s, 1H) ppm; MS(APCI+): 361 (M+1), LCMS: 96.7%.
Example 25. Preparation of P-093.
[00189] (4-Fluoro-6-methoxy-3'-nitro-biphenyl-3-yl)-(4-fluoro-phenyl)-methanol
(P-
093). 1H NMR (400 MHz, CDC13): 6 8.35 (t, J= 2.0 Hz, 1 H), 8.15-8.19 (m, 1 H),
7.75
102
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
- 7.79 (m, 1 H), 7.55 (t, J= 8.0 Hz, 1 H), 7.44 (d, J= 8.4 Hz, 1 H), 7.38-7.41
(m, 2 H),
7.01 - 7.06 (m, 2 H), 6.72 (d, J= 12 Hz, 1 H), 6.13 (br s, 1 H), 3.82 (s, 3 H)
ppm.
Calc.373.36; APCT(M-2): 371.0; >85%.
Example 26. Preparation of P-094.
[00190] 4-Fluoro-5-(4-fluoro-benzyl)-2-methoxy-3'-nitro-biphenyl (P-094). 'H
NMR
(400 MHz, CDC13): 6 = 8.33 (br s, 1 H), 8.15-8.17 (m, 1 H), 7.75 (d, J= 8.4
Hz, 1 H),
7.53(t,J=8.0Hz,1H),7.12-7.18(m,2H),.7.10(d,J=8.4Hz,1H),.6.97(t,J=8.4
Hz, 2 H), 6.74 (d, J= 11.6 Hz, 1 H), 3.95 (s, 2 H), 3.81 (s, 3 H) ppm.
Calc.357.36;
APCT(M-1): 356.1, -(M-2):355.2,93%.
Example 27. Preparation of P-079.
[00191] (4-Fluoro-phenyl)-(3'-nitro-6-trifl uoromethoxy-biphenyl-3-yl)-
methanol (P-
079). 'H NMR (400 MHz, CDC13) 6 2.29 (1H, d, J = 3.2 Hz), 5.91 (1H, d, J= 3.2
Hz),
7.06 (2H, m), 7.37 (3H, m), 7.44 (1H, dd, J= 8.4, 2.4 Hz), 7.49 (1H, d, J= 2
Hz), 7.61
(1H, dd, J= 8 and 8 Hz), 7.78 (1H, m), 8.25 (1H, m), 8.32 (1H, m) ppm.
Example 28. Preparation of P-080.
[00192] 5-(4-Fluoro-benzyl)-3'-nitro-2-trifluoromethoxy-biphenyl (P-080).
'H NMR (400 MHz, CDC13) 6 4.02 (2H, s), 7.01 (2H, m), 7.16 (2H, m), 7.24 (2H,
m),
7.31 (1H, m), 7.60 (1H, dd, J= 8, 8 Hz), 7.77 (1H, m), 8.24 (1H, m), 8.30 (1H,
m)
ppm.
103
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00193] General Scheme 12.
N
Suzuki
iN N nucleophilic
R1, R1 Coupling R1,0 displacement
,
O Halogenation 0
X
1-63 R2
1-64 1-65
N
N_N,R3
N NH decyanation R10' 0
R1,0 0 X = halogen, boronate
R1 = H, Me, CH2CH2OCH3, CONH2
R2 = Cl, CH3CO, CN, N02, Br, fused heterocycle
R2 R3 = substituted groups
R2 1-67
1-66
Example 29. Preparation of P-009.
N, NH
I 0 O
N02
P-009
[00194] (3-Bromo-4-methoxy-phenyl)-acetonitrile; To a mixture of compound 4-
methoxyphenyl acetonitrile (5.88 g, 40 mmol), KBr (9.52 g, 80 mmol),
tetrabutylammonium chloride (332 mg, 1.2 mmol) in dichloroethane (80 mL), was
added
21% w/w nitric acid (48 g, 160 mmol). The reaction mixture was stirred at rt
for 20 h,
diluted with dichloromethane (80 mL) and washed with sat. NaHCO3 aq. (2 x 50
mL),
water (2 x 50 mL), brine and dried over Na2SO4. After removal of solvent under
vacuum, the residue was triturated with Et20 (10 ml)/hexanes (40 ml) to give
6.32 g of
product; Yield: 70 %.
[00195] (6-Methoxy-3'-nitro-biphenyl-3-yl)-acetonitrile; A reaction mixture of
the
above product (2.26 g, 10 mmol), 3-nitrophenyl boronic acid (1.67 g, 10 mmol),
104
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
triphenylphosphine (262 mg, 1 mmol), K2CO3 (4.14 g, 30 mmol), Pd(OAc)2 (112
mg, 0.5
mmol) in 1,2-dimethoxyethane (80 mL), ethanol (10 mL) and water (10 mL) was
stirred
at 80 C for 20 hrs under Ar. After removal of solvent under vacuum, reaction
mixture
was diluted with ethyl acetate (80 mL) and washed with water (2 x 40 mL),
brine and
dried over Na2SO4. After removal of solvent, the residue was triturated with
Et20 (20
mL) to give 2.18 g of product; Yield: 81 %.
[00196] (6-Chloro-pyridazin-3-yl)-(6-methoxy-3'-nitro-biphenyl-3-yl)-
acetonitrile;
To a mixture of the above product (1072 mg, 4 mmol) 3,6-dicloropyridazine
(1311 mg,
8.8 mmol) in DMF (15 mL), was added portion wise NaH (400 mg, 60% in oil, 10
mmol) at 0 C under Ar. The reaction mixture was stirred at 0 C for 1 hr and
allowed to
slowly warm to rt and then stirred at rt for 16 hrs. Reaction mixture was
cooled to 0 C
and added sat. NH4C1 aq. (50 mL)/ water (150 mL) and stirred at rt for 10 min.
The
resulting solid was filtered, dissolved in ethyl acetate (80 mL) and then
washed with
water (2 x 40 mL), brine, and dried over Na2SO4. After removal of solvent, the
residue
was purified by silica gel column chromatography with ethyl acetate / hexane
as eluent
to give 1.4 g of product; Yield: 92 %.
[00197] 6-(6-Methoxy-3'-nitro-biphenyl-3 -ylmethyl)-2H-pyridazin-3 -one (P-
009). A
reaction mixture of the above product (600 mg, 1.58 mmol) in acetic acid (10
mL),
concentrated HC1(20 mL) and water (10 mL) was refluxed for 18 hrs. After
cooling to
r.t., water (200 mL) was added to reaction mixture and stirred at rt. for 20
min. The
resulted solid was filtered and dried at r.t. over night. The solid was
dissolved in
dichloromethane (50 ml) and filtered off solid. After removal of
dichloromethane, the
solid was washed with diethyl ether (20 mL) to give 310 mg of product (P-009);
Yield:
58%; 'H NMR (400 MHz, CDC13) 6 3.83 (3H, s), 3.92 (2H, s), 6.88 (1H, d, J= 9.6
Hz),
6.98 (1H, d, J= 8.4 Hz), 7.13 (1H, d, J= 9.6 Hz), 7.18 (1H, m), 7.22 (1H, dd,
J= 8.4.
2 Hz), 7.56 (1H, dd, J= 8, 8 Hz), 7.82 (1H, m), 8.18 (1H, m) ppm; MS(ESI+):
338.6
(M+1); LC-MS: 96 %.
[00198] The following compounds were prepared analogous to the example shown
above.
105
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 30. Preparation of P-012.
[00199] 6-(3 -Benzo[ 1,2,5]oxadiazol-5-yl-4-methoxy-benzyl)-2H-pyridazin-3 -
one (P-
012). 'H NMR (400 MHz, CDC13) 6, 3.85 (3H, s), 3.92 (2H, s), 6.89 (1H, d, J=
9.6
Hz), 6.99 (1H, d, J= 8.4 Hz), 7.14 (1H, d, J= 9.6 Hz), 7.23 (1H, d, J= 2 Hz),
7.27 (1H,
m), 7.60 (1H, m), 7.81 (1H, m), 7.85 (1H, m), 10.42 (1H, br s).
Example 30. Preparation of P-018.
[00200] 6-[6-(2-Methoxy-ethoxy)-3'-nitro-biphenyl-3-ylmethyl]-2-(2-methoxy-
ethyl)-
2H-pyridazin-3-one (P-018). 1H NMR (400 MHz, CDC13) 6 3.34 (3H, s), 3.36 (3H,
s),
3.68 (2H, m), 3.80 (2H, t, J = 5.5 Hz), 3.92 (2H, s), 4.14 (2H, m), 4.36 (2H,
t, J= 5.5
Hz), 6.84(1H,d,J=9.5Hz), 6.98(1H,d,J=8Hz), 7.05(1H,d,J=9.5Hz), 7.19 -
7.22 (2H, m), 7.55 (1H, dd, J = 8 and 8 Hz), 7.87 (1H, m), 8.17 (1H, m), 8.49
(1H, m).
Example 31. Preparation of P-020.
[00201] Carbamic acid 3'-nitro-5-(6-oxo-1,6-dihydro-pyridazin-3-ylmethyl)-
biphenyl-
2-yl ester (P-020). 1H NMR (400 MHz, DMSO-d6) 6, 3.92 (2H, s), 6.82 (1H, dd, J-
9.6 and 2.4 Hz), 6.82 (1H, br s), 7.15 (1H, br s), 7.18 (1H, J= 8.4 Hz), 7.32
(1H, dd, J
= 8 and 2 Hz), 7.41 (1H, d, J= 10 Hz), 7.44 (1H, d, J= 2Hz), 7.75 (1H, dd, J=
8 and 8
Hz), 7.88 (1H, m).
[00202] General Scheme 13
R3
Znucleophilic
R1.0 Y displacement R1.0 Y Y= H, F, OH, NH2, CN, OCH3, OEt
Z = Br, CI, OCOZCH3
R1 = H, Me, CHF2, CF3
R2 = Cl, CH3CO, CN, N02, Br, fused heterocycle
R2 R2 R3 = substituted aryl groups
1-68 1-69
Example 32. Preparation of P-224.
106
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
~I
O ~N
Br N A92COs
i
O
O + HO Benzene
\I I Nto
N I -
O- 0
1-70 P-224
[00203] Synthesis of 2-(6-Methoxy-3'-nitro-biphenyl-3-ylmethoxy)-pyridine (P-
224).
Into a 20 mL vial with stir bar was added 1-70 (230 mg, 0.71 mg), 2-
hydroxypyridine (91
mg, 0.59 mmol), Ag2CO3 (236 mg, 0.89 mmol), and 4 mL of benzene. The reaction
was
stirred for 18 hours at 80 C protected from light. An additional 204 mg of 2-
hydroxypyridine and 515 mg of Ag2CO3 was added and the reaction stirred for 3
more
days at 80 C. The reaction was filtered and the filtrate was concentrated.
The residue
was purified by flash column chromatography eluting with 20% ethyl
acetate/hexanes to
give 63 mg (32%) of P-224 as a light yellow oil. 1H NMR (400 MHz, CDC13) 8.42
(s, 1
H),8.22-8.13(m,2H),7.87(d,J=7.8Hz,1H),7.63-7.53 (m, 2 H), 7.52 - 7.44 (m, 2
H), 7.02 (d, J= 8.3 Hz, 1 H), 6.89 (dd, J= 5.4, 6.2 Hz, 1 H), 6.80 (d, J= 8.3
Hz, 1 H),
5.37 (s, 2 H), 3.85 (s, 3 H) ppm. LC/MS = 90.0%, 337.1 (APCI+).
Example 33. Preparation of P-479.
N
N
O fF -N
P-479
[00204] 3-[l-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-1H-imidazol-4-
yl]-
pyridine (P-479). A flask was charged with 3-(1H-Imidazol-4-yl)-pyridine
(165mg,
0.75mmol) and THE (5mL). Then NaH (60% dispersion in mineral oil, 60 mg, 1.5
mmol) was added slowly (gas evolution). After 2min. of stirring at room
temperature 3-
Bromomethyl-3'-chloro-2-fluoro-6-methoxy-biphenyl (165mg, 0.5 mmol) was added.
The reaction mixture was stirred at rt. After overnight stirring the reaction
mixture was
107
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
quenched with water (20 mL). The mixture was extracted with EtOAc (3 x 10 mL).
The
combined organic layers were washed with brine (50 mL), dried over Na2SO4 and
concentrated in-vacuo. The crude was purified by silica gel column
chromatography,
eluted with dichloromethane/MeOH (200:5) to produce 77.1 mg (39% yield) of the
product (P-479) as a pale yellow solid. 'H NMR (400 MHz, DMSO-d6) 6 3.75 (s, 3
H),
5.26 (s, 2 H), 7.02 (d, J=,8.59 Hz,1H),7.28-7.50 (m, 6 H), 7.78 - 7.88 (m, 2
H), 8.04 -
8. 11 (m, 1 H), 8.38 (dd, J= 4.7, 1.2 Hz, 1 H), 8.96 (d, J= 1.5 Hz, 1 H) ppm.
[00205] Scheme 14.
Br OH O 0
p
O 0 ~O _ \O O r'or O. + O O O
UN to N J i t P-120
O - N N
0 1-72 0
O
1-70 1-71 1-73
Example 34. Preparation of P-120.
[00206] (6-Methoxy-3'-nitro-biphenyl-3-yl)-acetic acid (1-71) Into a 100 mL
round
bottom flask with stir bar was added 5-bromomethyl-2-methoxy-3'-nitro-biphenyl
(1-70)
(0.60 g, 1.86 mmol), NaCN (0.18 g, 3.72 mmol), and dry DMF (20 mL). The
reaction
was stirred at room temperature for 20 hours after which 20 mL of water and 20
mL of
dichloromethane were added. The layers were separated and the aqueous was
extracted
with 20 mL of dichloromethane. The combined organic were washed with water (3
x 20
mL) and concentrated to afford the corresponding nitrile which was used as is
in the next
reaction. Into a 100 mL round bottom flask with a stir bar was added the crude
nitrile,
mL of ethanol, and 5 mL of 2 N KOH. The suspension dissolved upon heating and
the solution was stirred at reflux for 18 hours. The reaction was cooled to
room
temperature and the ethanol was removed under reduced pressure. The remaining
aqueous portion was washed with 2 x 10 mL of dichloromethane then acidified to
pH = 1
using 6 N HC1. The precipitate that formed was filtered and washed with water
(2 x 10
mL). After drying in a 40 C vacuum oven for 4 hours, 0.32 g (60%) of 1-71 was
obtained as a yellow solid.
108
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00207] (6-Methoxy-3'-nitro-biphenyl-3-yl)-acetic acid 3-methyl-oxetan-3-
ylmethyl
ester (1-73). Into a 20 mL vial with stir bar were added the above acid (1-71)
(0.12 g,
1.15 mmol), 2 mL dichloromethane, 3-Hydroxymethyl-3-methyl-oxetane (0.30 g,
1.04
mmol), DMAP (38 mg, 0.31 mmol), DCC (0.24 g, 1.15 mmol), and 2 mL of
dichloromethane. After the reaction was stirred at room temperature for 30
minutes, 10
mL of hexanes were added and the reaction was stirred an additional 10
minutes. The
suspension was filtered through celite and the celite pad was washed with 5:1
hexane:dichloromethane (3 x 10 mL). The filtrate was concentrated and the
residue
purified by flash column chromatography using 30% ethyl acetate/hexanes to
give 0.33
g (85%) of 1-73 as a colorless semi-solid.
[00208] 1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-4-methyl-2,6,7-trioxa-
bicyclo[2.2.2] octane (P-120); Into a 100 mL round bottom flask with stir bar
was added
1-73 (0.32 g, 0.86 mmol) and 10 mL of dry dichloromethane. The solution was
cooled to
-15 C and BF3-OEt2 (270 uL, 2.2 mmol) was added. The reaction was allowed to
slowly attain room temperature over 4 hours then cooled again to 0 C.
Triethylamine
(0.48 mL, 3.45 mmol) was added and the reaction was concentrated. The product
was
purified by flash column chromatography using 2% ethyl acetate in 50% ethyl
acetate/hexanes to afford 9.8 mg (3%) of the product (P-120) as a colorless
oil. 1H NMR
(400 MHz, CDC13) 2.99 (s, 3 H), 3.81 (s, 3 H), 3.90 (s, 6 H), 6.93 (d, J= 8.3
Hz, 1 H),
7.27-7.35(m,2H),7.54(t,J=8.0Hz,1H),7.87(d,J= 7.8 Hz,2H),8.15(dd,J=8.3,
1.3 Hz, 2 H), 8.43 (t, J= 1.8 Hz, 1 H) ppm. MS (APCI+) 372.1.
109
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00209] General Scheme 15.
OH N N
Br
cyclization
R1, / O 1. halogenation 1, O R1, S R
O Y O Y O Y
2. bromomethylation
R2 1-74 R2 1-75 R2 I-76
Y = H, F, OH, NH2, ON, OCH3, OEt R = H, CONHCH2CH3
Z = Br, Cl, 0002CH3
R1 = H, Me, CHF2, CF3
R2 = Cl, CH3CO, ON, N02, Br, fused heterocycle
Example 35. Preparation of P-341
N
S>- NH2
O F
CI
P-341
[00210] 1-Bromo-3-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-yl)-propan-2-one;
Into
a 20 mL vial with stir bar was added (3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-
yl)-
acetic acid (1-74, R1 = Me, Y = F) (306 mg, 1.04 mmol), dichloroethane (4 mL),
thionyl
chloride (151 uL, 2.08 mmol) and the solution was stirred at 80 C for 1 hour.
The
solution was cooled to 0 C, 4 mL of dichloromethane were added, and a
solution of
(trimethylsilyl)diazomethane (1.56 mL, 3.11 mmol, 2.0 M in ether) was added
and the
reaction stirred at room temperature for 16 hours. The reaction was cooled to
0 C and
0.5 mL HBr (48% in H20) was added. After 30 minutes at 0 C, Na2CO3 was added
until bubbling ceased and the reaction was dried by the addition of Na2SO4.
The
suspension was filtered and concentrated to yield product (390 mg, 99%) as a
brown oil,
which was used as is.
[00211] 4-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-thiazol-2-ylamine
(P-
341). Into a 20 mL vial were added the above compound (390 mg, 1.05 mmol),
ethanol
(4 mL), and thiourea (160 mg, 2.10 mmol). The reaction was stirred for 2 hours
at 75 C,
then cooled to room temperature. Water (10 mL)and 10 mL of brine were added
and the
110
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
product was extracted with ethyl acetate. The organics were dried with Na2SO4,
filtered,
and concentrated. The residue was purified by flash column chromatography
eluting
with 3% methanol/dichloromethane to afford crude product as a yellow solid.
The solid
was triturated with 1:5 ether:hexane to give pure product (135 mg, 37%) as a
tan solid.
The solid was dissolved in ether:THF and a solution of 2.0 M HC1/ether were
added.
The suspension was concentrated to give the title compound (P-341). 'H NMR
(400
MHz,DMSO-d6) 6 8.89 (br s, 1 H), 7.52 - 7.42 (m, 2 H), 7.38 (s, 1 H), 7.37 -
7.27 (m, 2
H), 6.98 (d, J= 8.6 Hz, 1 H), 6.42 (s, 1 H), 4.00 - 3.55 (br s, 2H), 3.87 (s,
2 H), 3.75 (s, 3
H). MS: 349.0 (APCI+).
Example 36. Preparation of P-346.
I>- N N
O F S 0
CI
P-346
[00212] 1-[4-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-thiazol-2-yl]-
3-
ethyl-urea (P-346). Into an 8 mL vial was added P-341 (40 mg, 0.11 mmol),
pyridine
(2 mL), and ethylisocyanate (24 mg, 0.34 mmol). The solution was stirred at 50
C for 3
days, then an additional 40 mg ethyl isocyanate was added and the reaction was
stirred at
80 C for 2 hours. After cooling to room temperature, water was added and the
product
was extracted with ethyl acetate. The organics were concentrated and the
residue was
purified by flash column chromatography eluting with 15 - 20% acetone/hexane
to give
the product (P-346) (31 mg, 64%) as an off-white solid. 1H NMR (400 MHz,CDC13)
8
10.42 (br s, 1H), 7.45 - 7.42 (m, 2H), 7.37 (s, 1H), 7.30 - 7.25 (m, 2H), 6.93
(d, J= 8.4
Hz, 1H), 6.57 (s, 2H), 3.86 (s, 2H), 3.73 (s, 3H), 3.14 - 3.11 (m, 2H), 1.04
(t, J= 7.2 Hz,
3H). LC/MS = 95.4%, 420.0 (APCI+).
111
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00213] Scheme 16.
Br H p S N S
O I F S II N~ O F Nr h-O TF >,-NH,
+ HSn- , N O O O N CIH
CI CI P-369
1-33 1-77
1-78
Example 37. Preparation of P-369.
[00214] [5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-thiazol-2-yl]-
carbamic acid tert-butyl ester (1-78). Into an 8 mL vial was added 1-33 (70
mg, 0.21
mmol), (5-tributylstannanyl-thiazol-2-yl)-carbamic acid tert-butyl ester (1-
77, 104 mg,
0.21 mmol), 4A molecular sieves (100 mg), and THE (2 mL). The mixture was
degassed
for 10 minutes with N2 and then Pd(PPh3)4 (25 mg, 0.021 mmol) was added. After
stirring for 18 hours at 80 C, the suspension was filtered and the filter
cake was washed
with EtOAc. The filtrate was washed with water and then concentrated.
Purification by
flash column chromatography (15% acetone/hexanes) provided 1-78 (11 mg, 12%)
as a
white semi-solid.
[00215] 5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-thiazol-2-ylamine
(P-
369). Into a 4 mL vial was added 1-78 (10 mg, 0.022 mmol) and 1 mL of 4N
HC1/dioxane. After stirring for 8.5 hours the reaction was concentrated to
obtain P-369
(6.8 mg, 77 %) 1H NMR (400 MHz, DMSO-d6) 9.69 (br s, 2 H), 7.52 - 7.45 (m, 2
H),
7.43 - 7.36 (m, 2 H), 7.32 (d, J= 4.6 Hz, 1 H), 7.29 (d, J= 6.3 Hz, 1 H), 7.08
- 7.01 (m, 2
H), 5.29 (s, 2 H), 3.77 (s, 3 H) ppm. LC/MS = 94.4%, 349.0 (APCI+).
112
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00216] General Scheme 17.
OH
T'o~N~ R 1. 0 N.
0 Y cyclization R1. N R
O R2 1-79 R2
1-80
Y = H, F, OH, NH2, ON, OCH3, OEt
Z = Br, Cl, 0002CH3
R1 = H, Me, CHF2, CF3
R2 = Cl, CH3CO, ON, N02, Br, fused heterocycle
R = urea derivatives
Example 38. Preparation of P-333.
OH S
~ I I ~--NH
O I / F O O / F NLN
~ OI \
c1 p-333
1-79
[00217] 5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-[1,3,4]thiadiazol-
2-
ylamine (P-333). A mixture of (3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-yl)-
acetic
acid (1-79) (0.998 mmol, 1.0 eq.) and thiosemicarbazide (2.99 mmol; 3 eq.) in
excess
phosphorous oxychloride was heated to 120 C for 45 minutes, and allowed to
cool to
room temperature. The resultant mixture was added to water, and extracted with
2
portions of ethyl acetate. The organics were washed with brine, and dried over
magnesium sulfate. The residue was purified via flash chromatography on silica
gel
using 5% (1N NH3 in MeOH) in dichloromethane as eluent to afford the desired
product
(P-333) in 39% yield.
[00218] 'HNMR (DMSO-d6, 400 MHz): 3.75 (s, 3H), 4.17 (s, 2H), 6.98 (d, J= 8.0
Hz, 1H), 7.29 (d, J= 6.4 Hz, 1H), 7.37-7.41 (m, 2H), 7.43-7.49 (m, 2H), 7.58-
7.76 (br s,
2H)
113
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 39. Preparation of P-342.
S
S'N
/- NH2 TF
O
/ F NLN a o~ O N 0
O
Benzene, reflux CI
P-333 P-342
[00219] Synthesis of Methyl [5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-
ylmethyl)-
[1,3,4]thiadiazol-2-yl]-carbamate (P-342). To P-333 (0.286 mmol; 1.0 eq.) was
added a
solution of methyl chloroformate (0.388 mmol; 1.4 eq.) in benzene. The mixture
was
heated to reflux for 10 h, and allowed to cool to room temperature. The
resultant
mixture was diluted with ethyl ether, filtered, and washed with ethyl ether.
The solids
were dried at 30-35 C under vacuum for 4 h to afford the title compound, P-
342, in 41%
yield.
[00220] 'H NMR (400 MHz, DMSO-d6): 3.73 (s, 3H), 3.75 (s, 3H), 4.32 (s, 2H),
4.74
(br s, 1H), 6.99 (d, J= 8.8 Hz, 1H), 7.29 (d, J= 6.4 Hz, 1H), 7.38 (s, 1H)
7.42-7.47 (m,
3H) ppm. LC/MS(94.0%) APCI+ found: 408.0; calc'd: 407.9 m/z
Example 40. Preparation of P-343.
S
~-NH _
, 2 O-- TF >_N H
O N N
fF
O ON Pyridine P-333 P-343
[00221] Synthesis of 1-[5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
[1,3,4]thiadiazol-2-yl]-3-ethyl-urea (P-343). To a solution of P-333 (0.214
mmol; 1.0
eq.) in pyridine was added ethyl isocyanate (0.643 mmol; 3 eq.), and the
resultant
114
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
solution stirred at room temperature for 4 days. The reaction was diluted with
water, and
stirred for 1 h. The solids were filtered, washed with water, and dried under
high
vacuum to afford the title compound, P-343, in 47% yield. 1H NMR (400 MHz,
DMSO-
d6): 1.04 (t, J= 7.2 Hz, 3H), 3.12 (q, J= 5.6 Hz, 2H), 3.75 (s, 3H), 4.26 (s,
2H), 6.53 (br
s, 1H), 6.99 (d, J= 8.8 Hz, 1H), 7.29 (dd, J= 6.4, 1.2 Hz, 1H), 7.38 (s, 1H),
7.40-7.48
(M, 3H), 10.71 (s, 1H) ppm. LC/MS = 94.8% purity APCI+ found: 421.0; calc'd:
420.9
m/z
Example 41. Preparation of P-352.
S S H
I _ // NHZ N, - / N
O / F N N + O/ /Si" THE O I / F N. NH2
\ CI P-333 \ CI
P-352
[00222] Synthesis of [5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
[1,3,4]thiadiazol-2-yl]-urea (P-352). To a solution of P-333 (0.177 mmol; 1
eq) in THE
at room temperature was added trimethylsilyl isocyanate (0.886 mmol; 5 eq),
and the
resultant solution was stirred overnight. The reaction was then heated to 60 C
for 5 h,
additional trimethylsilyl isocyanate (0.460 mmol; 2.6 eq) was added, and the
reaction
allowed to proceed overnight at 60 C. The reaction was cooled, and poured
into excess
aqueous 5% sodium bicarbonate. The resultant suspension was stirred at room
temperature for 1 h, filtered, and washed with water. The solid was dried in
vacuo to
obtain the title compound, P-352, as a white solid in 46% yield. 1H NMR (400
MHz,
DMSO-d6): 3.75 (s, 3H), 4.26 (s, 2H), 6.30 (br s, 1H), 6.99 (d, J= 8.4 Hz,
1H), 7.29 (d, J
= 6.4 Hz, 1H), 7.38 (s, 1H), 7.40-7.48 (m, 4H), 10.72 (s, 2H) ppm. LC/MS =
91.1%
purity. APCI+ found: 393.0; calc'd: 392.8 m/z.
115
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00223] General Scheme 18.
OH
acylation 1 -O bromination \ 0 ~
II II hydrolysis
R1,0_ .Y R1.0- Y O R1,0 0
1-84 X
1-85 1-86
B(OH)2
6 165 0 R3
OH PO R3 R1,
R1. X R3 R1, O Y
O X Y nucleophilic X coupli Coupling 0 Y
R2
1-87 displacement
Y = H F 1-88
X = Br, I, Cl 1-89
R1 = H, Me, CHF2, CF3
R2 = Cl, CH3CO, CN, N02, Br, fused heterocycle
R3 = substituted aryl groups
Example 42. Preparation of P-015.
ONNNIH
0 O
NO
2
P-015
[00224] Acetic acid 4-methoxy-phenyl ester (1-85, RI = Me, Y = H); A mixture
of 4-
methoxyphenol (6.2 g, 50 mmol), K2CO3 (10 g, 72 mmol) and acetic anhydride
(6.12 g,
60 mmol) in acetone (150 ml) was stirred at RT. over night. The solid was
filtered off
and washed with acetone (50 ml). After removal of acetone, 8.6 g of product
was
obtained. Yield: 100 %.
[00225] Acetic acid 3-bromo-4-methoxy-phenyl ester (1-86, RI = Me, Y = Br); To
a
mixture of the above product (4.15g, 25 mmol) in acetonitrile (100 ml), was
added NBS
(5.34g, 30 mmol) at rt. The reaction mixture was stirred at rt. over night and
more NBS
(5.34 g, 30 mmol) was added and the reaction mixture was stirred at 75 C over
night.
After removal of solvent, the crude product directly went to next step.
116
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00226] 3-Bromo-4-methoxy-phenol (1-87, RI = Me, Y = Br):; A mixture of crude
the above product (25 mmol) in MeOH (100 mL) and 2N NaOH aq. (30 mL) was
stirred
at r.t. for 40 min. The pH of reaction mixture was adjusted to acidic by
adding 2N HC1
aq. and extracted with dichloromethane (4 x 60 mL). The dichloromethane layer
was
dried over Na2SO4. After removal of solvent, the residue was purified by
silica gel
column chromatography with dichloromethane as eluent to give 2.9 g of product
Yield:
57% (for two steps)
[00227] 3-(3-Bromo-4-methoxy-phenoxy)-6-chloro-pyridazine; To a mixture of the
above product (1420 mg, 7 mmol) 3,6-dicloropyridazine (1252 mg, 8.4 mmol) in
DMSO
(15 ml), was added K2CO3 (1159 mg, 8.4 mmol) at rt. The reaction mixture was
stirred at
110 oC for 3 hr under Ar. After cooling to rt, water (50 mL) was added to
reaction
mixture and extracted with ethyl acetate (3x 50 mL). The combined ethyl
acetate layer
was washed with water (3 x 50 mL), brine and dried over Na2SO4. After removal
of
solvent, the solid was washed with diether (20 mL) to give 1650 mg of the
product
Yield: 75%
[00228] 6-(3-Bromo-4-methoxy-phenoxy)-2H-pyridazin-3-one; A reaction mixture
of
the above product (455 mg, 1.4 mmol) in acetic acid (10 mL), was stirred at
110 C for 7
hrs. After removal of solvent, the solid was washed with ethyl acetate (10 mL)
to give
340 mg of product. Yield: 82 %; MS(ESI+): 297.3 (M+1); LC-MS: 94 %.
[00229] 6-(6-Methoxy-3'-nitro-biphenyl-3 -yloxy)-2H-pyridazin-3 -one (P-015);
A
reaction mixture of the above product (85 mg, 0.29 mmol), 3-nitrophenyl-
boronic acid
(72 mg, 0.43 mmol), triphenylphosphine (16 mg, 0.06 mmol), K2CO3 (124 mg, 0.9
mmol), Pd(OAc)2 (7 mg, 0.03 mmol) in 1,2-dimethoxyethane (6 ml), ethanol (0.5
ml)
and water (0.5 ml) was stirred at 80 C for 20 hrs under Ar. Reaction mixture
was diluted
with water (40 ml) and extracted with ethyl acetate (2 x 40 mL). The combined
organic
phase was washed with water (2 x 30 mL), brine and dried over Na2SO4. After
removal
of solvent, the residue was purified by silica gel column chromatography with
ethyl
acetate/hexane as eluent to give 39 mg g of the product (P-015) Yield: 40 %.
1H NMR
(400MHz,CDC13) 8 = 3.85 (3H, s), 7.03 (1H, d, J= 10 Hz), 7.04 (1H, d, J= 8.8
Hz),
7.16 - 7.20 (2H, m), 7.22 (1H, d, J= 10 Hz), 7.57 (1H, dd, J= 8, 8 Hz), 7.86
(1H, m),
117
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
8.19 (1H, m), 8.41 (1H, m), 9.62 (1H, br s). MS(ESI+): 340.6 (M+1) ; LC-MS: 92
%.
[00230] General Scheme 19.
OH
OH HO,B,OH
OqF + Tetrakis(triphenylphosphine)palladium(O) F
O ~ i I
Br Tolune, Ethanol
I-88a N Sodium Carbonate(aq)
A
I-89a N
~ O
Br Copper(I)chloride N
I-89a 2,2,6,6-Tetra methyl-heptane-3,5-dione O F O N O
N Cesium Carbonate 0
N-Methylpyrolidine
O 0 A N
I-89b
1.) Iron powder
Ammonium Chloride N O N O
O
Ethanol, Water
0 O F NH3 Ethyl Isocyanate O F - N N
H H
2.) Hydrocloric Acid Cl Pyridine
Diethyl Ether ~
N
P-439
P-440
Example 43. Preparation of P-439 and P-440
[00231] Synthesis of 2'-Fluoro-3'-hydroxy-6'-methoxy-biphenyl-3-carbonitrile
(1-89,
R1 = Me, R2 = 3-CN, R3 = H). A solution of I-88a (2.00 g, 8.22 mmol) and 3-
cyanophenylboronic acid (1.45 g, 9.87 mmol) in toluene (30 mL) was purged with
a
nitrogen stream for 15 min To the solution was added ethanol (5 mL) and 2 M
aqueous
sodium carbonate (8..2 mL), a suspension formed and
palladium(0)tetrakis(triphenylphosphine) (475 mg, 0.411 mmol) was added. The
reaction
was heated to 108 C and stirred at this temperature overnight. The reaction
was diluted
with ethyl acetate (200 mL) and water (200 mL). The layers were separated and
the
aqueous layer extracted with ethyl acetate (200 mL). The organic extracts were
combined, washed with water (400 mL) and brine (300 mL), dried over sodium
sulfate,
118
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
filtered, and the solvent removed under vacuum to give crude product. The
crude
material was purified by flash silica gel column chromatography (0-5% methanol
in
dichloromethane) to give I-89a (1.53 g, 77% yield) as a beige powder.
1H NMR (400 MHz CDC13) d: 7.74-7.73 (m, 1H), 7.67-7.64 (m, 2H), 7.53 (t, J=
7.8 Hz,
1H), 7.01 (t, J= 9.4 Hz, 1H), 6.691 (dd, J= 9.2, 2.0 Hz, 1H), 4.91 (br m, 1H),
3.77 (s,
3H) ppm. LCMS = 100 % purity. MS(APCI-) = 224.0 (M-19).
[00232] Synthesis of 2'-Fluoro-6'-methoxy-3'-(6-nitro-pyridin-3-yloxy)-
biphenyl-3-
carbonitrile (I-89b). A solution of I-89a (500 mg, 2.06 mmol) and 5-bromo-2-
nitropyridine (379 mg, 1.87 mmol) in n-methylpyrrolidine (10 mL) was purged
with a
nitrogen stream. To the solution was added cesium carbonate (1.22 g, 3.74
mmol),
copper(I)chloride (92.4 mg, 0.934 mmol), and 2,2,6,6-tetramethyl-3,5-
heptanedione
(43.1 mg, 0.234 mmol) under nitrogen. The reaction was heated to 60 C
overnight. The
reaction was diluted with ethyl acetate (75 mL) and water (75 mL), and the
layers
separated. The aqueous wash was extracted with ethyl acetate (75 mL), and the
organic
extracts combined. The organic extracts were washed with 1 N aqueous sodium
hydroxide (100 mL), water (100 mL), and brine (100 mL), dried over sodium
sulfate,
decanted, and the solvent removed under reduced pressure to give crude
product. The
material was purified by flash silica gel column chromatography (0-50% ethyl
acetate in
hexanes) to give I-89b (541.8 mg, 79% yield) as a yellow powder. 1H NMR (400
MHz,
CDC13) 8.34 (d, J= 2.8 Hz, 1H), 8.26 (d, J= 8.8 Hz, 1H), 7.74 (m, 1H), 7.69-
7.64 (m,
2H), 7.55 (t, J= 7.8 Hz, 1H), 7.42 (dd, J = 8.8, 2.4 Hz, 1H), 7.26 (t, J= 9.0
Hz, 1H), 6.86
(dd, J= 9.2, 1.6 Hz, 1H), 3.85 (s, 3H) ppm. LCMS = 74.2 % purity. MS (APCI+) =
(M-
29).
[00233] Synthesis of 5-(3'-Cyano-2-fluoro-6-methoxy-biphenyl-3-yloxy)-pyridin-
2-
yl-ammonium chloride (P-439).. A suspension of I-89b (RI = Me, R2 = 3-CN, R3 =
2-
N02-3-pyridyl) (250 mg, 0.684 mmol) and tin(II),chloride (567 mg, 2.51 mmol)
in
isopropyl alcohol (2.5 mL) and concentrated hydrochloric acid (1.25 mL) was
stirred at
reflux for 3 h. To the reaction was added ethyl acetate (50 mL) and aqueous
saturated
sodium bicarbonate (50 mL). The layers were separated and the aqueous layer
extracted
with ethyl acetate (50 mL). The combined organic extracts were washed with
water (50
mL) and brine (50 mL), dried over sodium sulfate, filtered, and the solvent
removed
119
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
under vacuum. The residue was purified by flash silica gel column
chromatography
(eluting with 50% ethyl acetate in toluene) to give the free base as an orange
gum. The
gum was dissolved in dioxane (1 mL) and 4 N hydrochloric acid in dioxane was
added (2
mL). The suspension was stirred for 2 h at room temperature and subsequently
the
solvent was removed under reduced pressure. The residue was triturated with
diethyl
ether (5 mL), filtered, and washed with diethyl ether (2 x 1 mL) to give P-439
(55.1 mg,
22% yield) as a pale yellow solid.
1H NMR (400 MHz d6-DMSO) d: 7.89-7.84 (m, 4H), 7.75 (d, J= 7.6 Hz, 1H), 7.68
(t,
J= 8.2 Hz, 1H), 7.32 (t, J= 9.4 Hz, 1H), 7.02-6.99 (m, 2H), 3.77 (s, 3H). LCMS
= 96.2
% purity. MS(APCI+) = 336.1 (M+1).
[00234] Synthesis of 1-[5-(3'-Cyano-2-fluoro-6-methoxy-biphenyl-3-yloxy)-
pyridin-
2-yl]-3-ethyl-urea (P-440).. A solution of P-439 (85.0 mg, 0.230 mmol) and
ethyl
isocyanate (49.0 mg, 0.690 mmol) in pyridine (1.5 mL) was stirred at room
temperature
over night. The reaction was neutralized with water (50 mL) and extracted with
ethyl
acetate (2 x 50 mL). The combined extracts were washed with water (50 mL) and
brine
(50 mL), dried over sodium sulfate, filtered, and the solvent removed under
vacuum. The
residue was purified by preparatory silica gel thin layer chromatography (5%
acetone in
dichloromethane), triturated with diethyl ether (3 mL), filtered, and washed
with diethyl
ether (2 mL) to give P-440 (32.0 mg, 34% yield) as a white powder.
1H NMR (400 MHz, DMSO-d6) 9.10 (s, 1H), 7.99 (t, J= 1.8 Hz, 1H), 7.90 (s, 1H),
7.87
(dt, J= 7.5 Hz, 1.50 Hz, 1H), 7.76 (d, J= 8.0 Hz, 1H), 7.69-7.65 (m, 2H), 7.46
(d, J=
2.00 Hz, 1H), 7.25 (t, J= 9.2 Hz, 1H), 6.99 (dd, J= 9.2 Hz, 1.60 Hz, 1H), 3.76
(s, 2H),
3.31 (s, 3H), 3.15 (2H), 1.07 (t, J= 7.4 Hz, 3H).
LCMS = 97.9 % purity. MS(APCI+) = 407.1 (M+1).
[00235] General Scheme 20.
N-R2
form lation O Suzuki O Reductive H
Y Amination R1.
R1,O Y R1,O ( oouling R1,0 Y O Y
Y
Br Br
R1 R1
R2 = aryl, sulfonyl group
1-90 1-91 1-92 1-93
120
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 44. Preparation of P-203.
H
N
H
HO F
~I
\ CI
P-203
[00236] 3 -Bromo-2-fluoro-4-methoxy-benzaldehyde and N-{4-[(3'-Chloro-2-fluoro-
6-
methoxy-biphenyl-3-ylmethyl)-amino]-phenyl}-acetamide To a hot (80 C)
solution of
hexamethylenetetramine (13.7 g, 97.5 mmol) in TFA (50 ml) was added a solution
of 3-
bromo-2-fluoro-4-methoxy-benzene (10 g, 48.8 mmol) in TFA (40 ml) over 1 hour.
The
resultant solution was continued to stir at 80 C for 1 h. After it was cooled
to room
temperature, the reaction mixture was poured to water, and stirred for 30 min.
The
products were collected on a filter and dried in vacuo to yield desired
aldehyde (I-91)
and a dimeric amine impurity (total weight: 1 Ig), as an inseperable mixture .
1H-NMR
indicated the molar ratio of these two products is about 1:1.
[00237] 3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-carbaldehyde (1-92), A mixture
of
the above products (1.0 g, 4.3 mmol), 3-chlorophenylboronic acid (1.34 g, 8.6
mmol),
K2C03 (1.36 g, 12.9 mmol) and (Ph3P)4Pd (1.49 g, 1.29 mmol) in dioxane/H20
(5:1, 40
ml) was heated to 85 C for 20 h under nitrogen. After it was cooled to room
temperature, the mixture was diluted with water, extracted with ethyl acetate,
washed
with water and brine, and dried over Na2SO4. After it was concentrated in
vacuo, the
residue was purified by a column chromatography on silica gel to yield the
title
compound (1-92) (0.4 g).
[00238] 3-Hydroxymethyl-6-methoxy-3'-nitro-biphenyl-2-ol To a solution of
above
product (1-92) (0.4 g, 1.5 mmol) in methanol (8 ml) was added 4'-
aminoacetanilide (0.34
g, 2.3 mmol) and p-toluenesulfonic acid mono-hydrate (0.013 g, 0.08 mmol). The
resulting mixture was stirred at room temperature for 20 h. After the solvent
was
121
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
removed, the residue was dissolved in acetic acid (4 ml), and sodium
cyanoboronhydride
(0.28 g, 4.5 mmol) at 0 C. The resultant stirred at room temperature for 2 h.
The
mixture was poured into ice-water, extracted with ethyl acetate, washed with
water and
brine, and dried over Na2SO4. After it was concentrated in vacuo, the residue
was
purified by a column chromatography on silica gel to yield the product (1-93,
RI = Me,
Y = F) (0.4 g, 67%).
[00239] N-{4-[(3'-Chloro-2-fluoro-6-hydroxy-biphenyl-3-ylmethyl)-amino]-
phenyl}-
acetamide (P-203). To a solution of the above product (0.17 g, 0.43 mmol) in
methylene
chloride (20 mL) was added BBr3 (0.32 g, 1.28 mmol) at -78 C. After it was
stirred at -
78 C, the resultant was allowed to warm to room temperature, and continued to
stir for 3
h. The reaction was quenched with ice-water and basified with Na2CO3. The
resulting
mixture was extracted with ethyl acetate, washed with brine, and dried over
Na2SO4.
The solvent was removed in vacuo to yield the product P-203 (150 mg, 95%). 1H
NMR
(400 MHz, DMSO-d6) 10.06 (br s, 1 H), 7.46 - 7.61 (m, 4 H), 7.40 - 7.47 (m, 2
H), 7.30
(s, 1 H), 7.25 (d, J= 6.3 Hz, 1 H), 7.07 - 7.16 (m, 2 H), 7.01 (d, J= 8.7 Hz,
1 H), 4.40 (s,
2 H), 2.03 (s, 3 H) ppm.
[00240] The following compound was prepared by procedure similar to the one
described above.
Example 45. Preparation of P-25 1.
[00241] N- {4- [(3 '-Chloro-2-fluoro-6-methoxy-biphenyl-3 -ylmethyl)-
sulfamoyl]-
phenyl}-acetamide (P-251) 1H NMR (400 MHz, CDC13) 7.71 (d, J= 8.7 Hz, 2 H),
7.58
(d,J=8.7Hz,2H),7.45(brs,1H),7.26-7.37 (m,2H),7.17-7.25(m,2H),7.12-
7.19(m,1H),6.68(d,J,=,8.6Hz,1H)4.83(t,J=6.2Hz,1H) 4.19 (d, J,=,6.3 Hz, 2 H)
3.75 (s, 3 H) 2.21 (s, 3 H) ppm.
122
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00242] General Scheme 21.
Suzuki Br
bromination alkylation \ Coupling R10 N _ R1, O
g,-N
HO yN R1,0 I yN R2 R2
1-94 1-95 1-96 1-97
\ 0.R3
R1 = H, Me, CHF2, CF3
alkylation RI, 0 I N R2 = CI, CH3CO, CN, N02, Br, fused heterocycle
30 R3 = alkyl group
R2
1-98
Example 46. Preparation of P-025.
0 N
6'N CO
0
P-025
[00243] 2-iodo-3-methoxy-6-methyl-pyridine (1-95): To 2-iodo-6-methyl-pyridin-
3-ol
(1-94) (1.0 g, 4.25 mmol) and K2C03 (1.18 g, 8.51 mmol) in acetone (20 mL) was
added
Mel (0.91 g, 6.38 mmol). The reaction was stirred at 45 C under N2 for 20 h.
The
reaction was cooled to room temperature and concentrated. The residue was
purified by
silica gel column chromatography using dichloromethane to afford 1.04 g (98%)
of the
product (1-95, RI = Me) as light yellow solid.
[00244] 3-methoxy-6-methyl-2-(3-nitro-phenyl)-pyridine (1-96): To the above
product (1-95, RI = Me) (0.5 g, 2.0 mmol), 3-nitrophenylboronic acid (2) (0.5
g, 3.06
mmol), PPh3 (0.11 g, 0.4 mmol), K2CO3 (0.83 g, 6.0 mmol) and Pd(OAc)2 (0.045
g, 0.2
mmol) was added DME (16 mL), and EtOH-H20 (1:1, 4 mL). Ar gas was bubbled
through the stirred reaction for 5 min. The reaction was stirred at 60 C
under Ar for 18
h. The reaction was cooled to room temperature, concentrated, and H2O and
123
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
dichloromethane (40 mL each) were added. The organic layer was separated and
the
aqueous layer was extracted with dichloromethane (2 x 25 mL). The combined
organic
extracts were dried with Na2SO4, filtered, and concentrated. The residue was
purified by
silica gel column chromatography using 1:1 dichloromethane-hexanes then
dichloromethane to afford 0.22 g (44%) of the product (1-96, RI = Me) as a
light yellow
solid.
[00245] 6 -bromomethyl-3 -methoxy-2 -(3 -nitro -phenyl)-pyridine (1-97): To
the above
product (1-96, R1 = Me) (0.21 g, 0.86 mmol) and NBS (0.17 g, 0.95 mmol) in
CC14 (10
mL) was added benzoylperoxide (0.02 g, 0.08 mmol). The reaction was stirred at
60 C
under N2 for 18 h. The reaction was cooled to room temperature and
concentrated. The
residue was dissolved in mixture of dichloromethane and hexanes (1:1, 8 mL)
and
purified by silica gel column chromatography using 1:1 dichloromethane/hexanes
then
dichloromethane to afford 0.15 g (55%) of the product as a light brown solid.
[00246] 3-methoxy-6-methoxymethyl-2-(3-nitro-phenyl)-pyridine (P-025): To the
above product (0.08 g, 0.25 mmol), methanol (0.11 g, 2.5 mmol), and CsCO3
(0.24 g,
0.74 mmol) was added DMF (2 mL). The vial was capped and stirred at room
temperature for 20 h. The reaction was diluted with 1:1 ethyl acetate-H20 (60
mL). The
organic layer was separated and the aqueous was extracted with ethyl acetate
(2 x 20
mL). The combined organic extracts were dried with Na2SO4, filtered, and
concentrated
to afford 0.066 g (97%) of the product (P-025) as off white solid. 'H NMR
(CDC13,
400MHz): 8.88 (dd, J= 2.0, 1.6 Hz, 1 H), 8.3-8.35 (m, 1 H), 8.2-8.24 (m, 1 H),
7.59 (t, J
= 8.0 Hz, 1 H), 7.44 (d, J = 8.8 Hz, 1 H), 7.3 7 (d, J = 8.4 Hz, 1 H), 3.61
(s, 2 H), 3.92
(s, 3 H), 3.50 (s, 3 H); MS(APCI+): 275.1 (M+1), LC-MS: 96.1%.
[00247] Scheme 22.
O
Br O- %O O
O O 30 O
B(OH)2 (~~F
F F F F
F F
FF F P-091 F
1-99 1-100 1-101
124
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 47. Preparation of P-09 1.
[00248] 2-methoxy-5-methyl-3'-trifluoromethyl-biphenyl (I-100) A reaction
mixture
of 2-methoxy-5-methylphenyl boronic acid (1-99) (5 g, 30.3 mmol), 3-
iodobenzenetrifluoride (8.24 g, 30.3 mmol), K2CO3 (8.3 g, 60.6 mmol),
palladium
acetate (350 mg, 1.5 mmol) in methanol (200 ml) and water (40 ml) was stirred
at room
temperature over night. Reaction mixture was concentrated to a 1/3, and then
it was
diluted with ethyl acetate (300 ml) and washed with 0.6 N sodium hydrogen
sulfate
solution (300 ml), water (2x 150 ml), brine and dried over Na2SO4. After
removal of
solvent, 7.75 g of product (1-100) was obtained as oil. Yield: 100 %.
[00249] 5-Bromomethyl-2-methoxy-3'-trifluoromethyl-biphenyl (1-101); To a 250
mL
round bottom flask equipped with stirring bar was added I-100 (5 g, 18.79
mmol) and
CC14 (125 mL). To this solution lg of NBS and 100 mg of AIBN were added. The
flask
was connected to a condenser and the mixture was reflux under the sun lamp for
one
hour. To the reaction mixture lg of NBS and 100 mg AIBN were
[00250] added and this stirred at reflux, under N2 for 2 more hours. After 2
hours
another portion of NBS (1.3 g) and 108 mg of AIBN were added and mixture was
refluxed for 2 more hours. The reaction mixture was cooled to RT, concentrated
to half
and filtered off. The solid was washed with 100 mL of CC14. After removal of
solvent
6.76 g of crude product (I-101) was obtained.
[00251] 3-(6-Methoxy-3'-trifluoromethyl-biphenyl-3-ylmethoxy)-cyclopent-2-
enone
(P-091). To a solution of 1,3- cyclopentanedione (110 mg, 1.11 mmol) in 1.5 ml
anhydrous DMF was added at 0 C NaH (60% dispersion in mineral oil, 45 mg, 1.11
mmol). The suspension was stirred for 30 minutes, then a solution of 1-101
(350 mg, 0.76
mmol) in 1.5 ml DMF was added. The reaction mixture was stirred at RT,
overnight. The
reaction mixture was quenched with saturated NH4C1 solution and extracted with
methylene chloride. Combined organic layers were washed with brine, dried over
sodium sulfate, filtered, concentrated to give 300 mg crude. Purification was
done using
preparative silica gel plate (1500 um) to afford 125 mg of product P-091. 1H
NMR
(CDC13, 400MHz): 2.45 (t, 2H), 2.66 (t, 2H), 3.05(d, 1H), 3.84 (s, 3H), 5(s,
2H), 5.43 (s,
125
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
1H), 7.02 (d, J = 8.4 Hz, 1H), 7.33 (d, J= 2Hz, 1H), 7.38 (dd, J= 8.4, 2.4Hz,
1H), 7.53 (t,
1H), 7.6 (d, J= 8.4Hz, 1H), 7.7 (d, J= 8.4Hz, 1H), 7.77 (s, 1H). LCMS (APCI+):
363
(M+1), 90%.
Example 48. Preparation of P-092.
Br 0 O O
0 + K2C03 0
DMF NA 0 N.0
0 p
1-70
P-092
[00252] Synthesis of 3-(6-Methoxy-3'-nitro-biphenyl-3-ylmethoxy)-cyclopent-2-
enone (P-092). Into a 50 mL round bottom flask with stir bar added 1-70 (284
mg, 0.83
mmol), 1,3-cyclopentadione (217 mg, 2.21 mmol), K2CO3 (305 mg, 2.21 mmol), and
5
mL DMF. The reaction was stirred at room temperature for 18 hours. 5 mL of
dichloromethane and 5 mL water were added. The layers were separated and the
aqueous extracted with 10 mL dichloromethane. The combined organics were
washed
with water (4 x 10 mL), dried over sodium sulfate, and concentrated. The
residue was
purified by flash column chromatography using 0-2% methanol/dichloromethane.
Obtained 112 mg (37%) P-092 as a white solid. 'H NMR (400 MHz, CDC13) 2.41 -
2.54 (m, 2 H) 2.60 - 2.71 (m, 2 H) 3.86 (s, 3 H) 5.01 (s, 2 H) 5.44 (s, 1 H)
7.04 (d, J=8.5
Hz, 1 H) 7.37 (d, J=2.2 Hz, 1 H) 7.41 (dd, J=8.5, 2.2 Hz, 1 H) 7.58 (t, J=8.0
Hz, 1 H)
7.86 (d, J=7.8 Hz, 1 H) 8.20 (dd, J=8.3, 1.3 Hz, 1 H) 8.41 (t, J=1.7 Hz, 1 H).
LC/MS =
92.0%, 340.1 (APCI+)
[00253] Scheme 23
0
32 S S
j ~ \\ / - &
O F S CI O F Et3SiH/TFA 0 F
P-128
N N
D 33 N O
31 0 0 0
1-102 1-103
126
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 49. Preparation of P-128.
[00254] (2-fluoro-6-methoxy-3'-nitro-biphenyl-3-yl)-thiophen-2-yl-methanone (1-
103)
To a 25 mL vial which contained a solution of thiophene-2-carbonyl chloride
(90 mg, 0.6
mmol) in nitrobenzene (0.5 mL) was added A1C13 (75 mg, 0.75 mmol) at -10 T.
After
stirring at 0 C for 2 h, 6-Fluoro-2-methoxy-3'-nitro-biphenyl (31) (125 mg,
0.5 mmol) in
nitrobenzene (0.5 mL) was added at rt. The reaction mixture was allowed to
stir at rt for
24 hours. The reaction mixture was cooled to -10 C and quenched with ice-
water (10
mL, extracted with ethyl acetate (10 mL), washed with water (2x10 mL), NaHCO3
(sat.,
mL), brine (30 mL) and dried over Na2SO4. After removal of solvent, the crude
was
purified by crystallization from ether-hexane to give 100 mg of 1-103 in 60%
yield.
LCMS: Calc.357.4; APCI-(M):356.9, 342(M-16-1) 97.4%
[00255] 2-(2-fluoro-6-methoxy-3'-nitro-biphenyl-3-ylmethyl)-tetrahydro-
thiophene
(P-128). To a 25 mL vial which contained compound 1-103 (90 mg, 0.25 mmol) in
triethylsilane (1 mL) was added TFA (1 mL) at -10 T. The reaction mixture was
allowed to warm to rt and stir at rt for 72 h. The reaction mixture was poured
onto 30
mL ice-water, extracted with ethyl acetate (3 x 30 mL), washed with NaHCO3
(sat. 30
mL), water (20 mL), brine (30 mL) and dried over Na2SO4. After removal of
solvent,
the residue was purified by silica gel column chromatography with ethyl
acetate /hexane
as eluent to give 30 mg of product (P-128) in 30% yield. 1H NMR (CDC13,400
MHz):
8.33 (br s, 1 H), 8.22 - 8.26 (m, 1 H), 7.59-7.76 (m, 5 H), 7.15 (t, J= 4.0
Hz, 1 H), 6.91
(d, J= 9.2 Hz, 1 H), 3.89 (s, 3 H) LCMS: Calc.347.4; APCI-(M) 347.0: 99%.
Example 50. Preparation of P-481
Pd(dba)2
PPh3 N-N\\
N,N HO,B.OH Cs2CO3 N
0
Br DMF
N 0 \N
N-O
N-O
1-104 1-105 P-481
127
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00256] Synthesis of 5-(2-Methoxy-5-[1,2,4]triazol-1-ylmethyl-phenyl)-
benzo[ 1,2,5]oxadiazole (P-481).. A suspension of 1-(3-Bromo-4-methoxy-benzyl)-
1H-
[1,2,4]triazole (1-104, 402 mg, 1.50 mmol), benzo[1,2,5]oxadiazole-5-boronic
acid (I-
105, 246 mg, 1.50 mmol), palladium(0) bis(dibenzylideneacetone) (43 mg, 0.075
mmol),
and triphenyl phosphine (39.3 mg, 0.15 mmol) in dimethylformamide (15 mL) and
1 M
aqueous cesium carbonate (4.5 mL, 4.5 mmol) was heated to 85 C with stirring
overnight. The solvent was removed under vacuum and the residue suspended in
ethyl
acetate (15 mL). The organic suspension was washed with water (3 x 15 mL) and
brine,
dried over sodium sulfate, decolorized over activated carbon, filtered, and
the solvent
removed under vacuum to give crude material. The residue was purified by
recyrstalization from dichloromethane (2 mL) and hexanes (10 mL) to give P-481
(180
mg, 39% yield). 1H NMR (400 MHz, CDC13) 6 ppm 3.88 (s, 4 H) 5.51 (s, 2 H) 7.06
(d, J
=7.8Hz,1H)7.41-7.54(m,3H)7.59(d,J=9.3Hz,1H)7.71-8.03(m,3H)8.25(s,
1 H) 9.34 (br. s., 1 H), LCMS = 95.7 % purity.
Example 51. Preparation of P-482
Pd(dba)2
-
HO, OH PPh3 N
N NaCO g' 2 O N
O N;N
Br 6 1.0 Dimethylformamide
N 4 / N "0
O
1-106 P-482
[00257] Synthesis of 1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-1H-
benzotriazole
(P-482).. A suspension of 1-(3-Bromo-4-methoxy-benzyl)-1H-benzotriazole (1-
106)
(477 mg, 1.50 mmol), 3-nitrophenylboronic acid (250 mg, 1.50 mmol),
palladium(0)
bis(dibenzylideneacetone) (43 mg, 0.075 mmol), and triphenyl phosphine (39 mg,
0.15
mmol) in dimethylformamide (10 mL) and 1 M aqueous sodium carbonate (4.5 mL,
4.5
mmol) was heated to 85 C with stirring overnight. The solvent was removed
under
vacuum and the residue suspended in ethyl acetate (20 mL). The organic
suspension was
washed with water (3 x 20 mL) and brine, dried over sodium sulfate,
decolorized over
128
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
activated carbon, and the solvent removed under vacuum to give crude material.
The
residue was purified dissolving in ethyl acetate (5 mL) and adding hexanes (25
mL) until
a solid formed. This was repeated 3 times to give P-482 (210 mg, 39% yield).
1H NMR
(400 MHz, CDC13) 'H NMR (400 MHz, CDC13): 3.82 (s, 3H), 5.86 (s, 2H), 6.98 (d,
J=
8.2 Hz, 1H),7.30-7.37(m,2H),7.37-7.52(m,3H),7.52- 7.59 (m,1H),7.75(d,J=
7.8 Hz, 1H), 8.12 (d, J= 8.3 Hz, 1H), 8.18 (dd, J= 8.2, 1.2 Hz, 1H), 8.35 (d,
J= 1.6 Hz,
1H) ppm. LCMS = 93.9% purity.
[00258] Scheme 24.
Potassium Bromide a.)Sodium Hydride
Br Nitric Acid r Br N b.) Potassium Carbonate
0 Dichloroethane '0 +
Tetrabutyl ammonium chloride Br X Dimethylformamide
1-42
HO,B-OH N /
~ ~ p Paladium(II)Bis(triphenylphospine)dichloride 0 ~X
+
O
y Dioxane
Br Z CsCO3 (aq) i
A Y
X= C 1-107 1-108 z
X=N
X=C,Y,Z=H,N02
X = N, Y,Z = H, N02
X = N, Y, Z = Dioxazole
P-010, P-483, P-014
Example 52. Preparation of P-010, P-483, P-014
[00259] Synthesis of 2-Bromo-4-bromomethyl-1-methoxy-benzene (1-42). To
solution
of potassium bromide (29.6 g, 248 mmol) in nitric acid (21% by volume, 149 g,
497
mmol), was added dichloroethane (188 mL) and tetrabutylammonium chloride (1.04
g,
3.73 mmol) followed by 4-(bromomethyl)anisol (25.0 g, 124 mmol) in
dichloroethane
(62 mL), and the reaction was stirred at room temperature for 5 h. The organic
layer was
separated, washed with water (2 x 100 mL, 2 x 150 mL), 2% aqueous potassium
carbonate (150 mL), dried over sodium sulfate, and the solvent removed under
vacuum.
The residue was purified by being run through a flash silica gel plug (10%
ethyl acetate
in hexanes) to give the title compound (1-42) (9.22 g, 26% yield). 'H NMR (400
MHz,
CDC13) 6: 7.59 (d, 1 H), 7.30 (dd, J= 8.2, 2.2 Hz, 1 H), 6.85 (d, J= 8.4 Hz, 1
H), 4.44 (s,
2 H), 4.44 (s, 3 H).
129
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00260] Synthesis of 1-(3-Bromo-4-methoxy-benzyl)-1H-indole (I-107, X= CH). A
solution of indole (250 mg, 2.14 mmol) in DMF (15 mL) was cooled in an ice
bath. To
this solution was added sodium hydride (64.3 mg, 2.68 mmol), followed by 2-
Bromo-4-
bromomethyl-1-methoxy-benzene (500 mg, 1.79 mmol). The reaction was warmed to
room temperature and stirred overnight. To the reaction was added saturated
aqueous
ammonium chloride (75 mL) and the layers separated. The organic extract was
washed
with saturated aqueous ammonium chloride (2 x 75 mL), water (3 x 75 mL) and
brine
(50 mL). The extract was dried over sodium sulfate, and the solvent removed
under
reduced pressure. To residue was purified by flash silica gel column
chromatography
eluting with 1:3 hexanes: dichloromethane to give the title compound (1-107, X
=
CH)(490 mg, 87% yield) which was taken into further reactions.
1H NMR (400 MHz CDC13) d: 7.659-7.635 (m, 1H), 7.391 (dd, J = 1.60 Hz, 0.80
Hz,
1H), 7.215-7.092 (m, 4H), 6.996-6.969 (m, 1H), 6.794 (d, J = 8.40 Hz, 1H),
6.549 (dd, J
= 3.00 Hz, 1.00 Hz, 1H), 5.234 (s, 2H), 3.850 (s, 3H).
[00261] Synthesis of 1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-1H-indole (P-
010).
A suspension of 1-(3-Bromo-4-methoxy-benzyl)-1H-indole (150 mg, 0.474 mmol),
palladium bis(triphenylphosphine)dichloride (13.3 mg, 0.0 190 mmol), and 3-
nitrophenylboronic acid (94.9 mg, 0.569 mmol) in dioxane (10 mL) and 1 M
aqueous
sodium carbonate (1.1 mL) was stirred at 85 C for 22 h. To the reaction was
added ethyl
acetate (30 mL). The organic suspension was washed with water (4 x 30 mL),
brine (2 x
30 mL), dried over sodium sulfate, and the solvent removed under vacuum. The
residue
was purified by flash silica gel column chromatography (10% ethyl acetate in
hexanes as
the elutant) to yield 1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-1H-indole P-0
10 (23.6
mg, 17% yield).
[00262] 1H NMR (400 MHz, CDC13) d: 8.36 (t, J = 1.60, 1H), 8.16 (dt, J = 6.40,
0.90,
1H),7.76 (dd, J = 6.40, 0.80, 1H),7.65 (d, J= 6.40, 1H),7.53 (t, J= 6.20,
1H),7.326 (d, J
= 6.40, 1H), 7.26 - 7.10 (m, 5H), 6.92 (d, J = 7.20, 1H), 6.55 (d, J = 2.40,
1H), 5.32 (s,
2H), 3.797 (s, 3H). LCMS = 87.7% purity. MS(APCI+) = 359.1 (M+1).
[00263] Synthesis of 1-(3-Bromo-4-methoxy-benzyl)-1H-benzoimidazole (I-107, X
=
130
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
N). A suspension of IH-benzoimidazole (317 mg, 2.68 mmol) and potassium
carbonate
(495 mg, 3.58 mmol) in DMF (10 mL) was stirred at 45 C for 1 h. To the heated
suspension was added 2-Bromo-4-bromomethyl-1-methoxy-benzene (500 mg, 1.79
mmol) and the reaction was stirred at 45 C for 4 h, cooled to room
temperature and
stirred at room temperature overnight. About half of the solvent was removed
under
vacuum, and ethyl acetate (30 mL) was added. The organic solution was washed
with
saturated aqueous ammonium chloride (3 x 30 mL), water (2 x 15 mL), and brine
(15
mL). The organic extract was dried over anhydrous sodium sulfate and the
solvent
removed under vacuum to give crude product as an orange oil. The product was
purified
by flash silica gel column chromatography eluting with 5% methanol in
dichloromethane
to give the title compound (282.5 mg; 48% yield) and a second crop of 50% pure
2-
Bromo-4-bromomethyl-1-methoxy-benzene (207.1 mg).
[00264] 1H NMR (400 MHz CDC13) 7.93 (s, 1H0 7.84-7.82 (m, 1H), 7.45 (d, J =
2.40 Hz, 1H), 7.30-7.26 (m, 3H) 7.07 (dd, J = 8.60 Hz, 2.20 Hz, 1H), 6.84 (d,
J = 8.40
Hz, 1H), 5.28 (s, 2H), 3.87 (s, 3H).
[00265] Synthesis of 1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-1H-
benzoimidazole
(P-483).. A suspension of 1-(3-Bromo-4-methoxy-benzyl)-1H-benzoimidazole (I-
107, X
= N) (250 mg, 0.788 mmol), 3-nitrophenylboronic acid (132 mg, 0.788 mg),
triphenylphosphine (20.6 mg, 0.0788 mmol), solid potassium carbonate (326 mg,
2.37
mmol) in 1,2-dimethoxyethane (6 mL), water (1 mL), and ethanol (1 mL) was
flushed
with argon gas and the palladium acetate (8.9 mg, 0.0394 mmol) was added. The
reaction was heated to 80 C over night. Additional palladium acetate (8.9 mg,
0.039
mmol) and triphenylphosphine (20.6 mg, 0.0788 mmol) was added and the mixture
was
stirred an additional night at 80 C. The solvent was removed under vacuum and
taken up
in ethyl acetate (50 mL). The organic solution was washed with saturated
aqueous
ammonium chloride (2 x 75 mL), water (3 x 75 mL) water, and the combined
aqueous
layers were extracted with ethyl acetate (50 mL). The combined organic
extracts were
washed with brine (75 mL), decolorized using activated carbon, dried over
sodium
sulfate, and the solvent removed under vacuum. The residue was taken up in
dichloromethane (5 mL), hexanes (20 mL) were added and the yellow powder was
filtered to give crude P-483. This was purified by flash silica gel column
131
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
chromatography eluting with 10-20% acetone in dichloromethane, and the excess
boronic acid removed by taking up the residue in dichloromethane (3 mL), and
washing
with 1 N aqueous sodium hydroxide solution. This wash was extracted into
dichloromethane (5 mL), the combined extracts dried over sodium sulfate, and
the
solvent removed under vacuum to give P-483 (23.9 mg, 8.4% yield) as an orange
tacky
powder.
[00266] 1H NMR (400 MHz, DMSO-d6) d: 8.44 (s, 1H), 8.27 (t, J = 2.00 Hz, 1H),
8.21-8.178 (m, 1H), 7.924-7.898 (m, 1H), 7.711 (t, J = 8.00 Hz, 1H), 7.646-
7.597 (m,
2H), 7.523 (d, J = 2.40 Hz, 1H), 7.398 (dd, J = 8.40 Hz, 2.00 Hz, 1H), 7.219-
7.127 (m,
3H), 5.485 (s, 2H), 3.764 (s, 3H). LCMS = 93.1% purity. MS(ESI+) = 360.9
(M+1).
[00267] Synthesis 5-(5-Benzoimidazol-1-ylmethyl-2-methoxy-phenyl)-
benzo[ 1,2,5]oxadiazole (P-014). A suspension of 1-(3-Bromo-4-methoxy-benzyl)-
1H-
benzoimidazole (I-107, X = N) (250 mg, 0.778 mmol), 1 N aqueous cesium
carbonate
(2.4 mL), and triphenylphosphine (20.7 mg, 0.0788 mmol) in DMF (5 mL) was
stirred.
To the suspension was added benzo[1,3]dioxol-5-yl-boronic acid (155 mg, 0.946
mmol),
the reaction purged with nitrogen, and bis(dibenzylideneacetone)palladium(0)
(22.7 mg,
0.0394 mmol) was added under nitrogen. The reaction was stirred at 100 C
overnight.
After cooling to room temperature, ethyl acetate (50 mL) was added. The
organic
suspension was washed with saturated aqueous ammonium chloride (2 x 50 mL), 1
N
aqueous sodium hydroxide (3 x 50 mL), water (50 mL), and brine (50 mL),
decolorized
over activated carbon, dried over sodium sulfate, and removed under vacuum to
give
crude product. The product was purified by flash silica gel column
chromatography
eluting with 1% methanol in dichloromethane to give P-014 (102.1 mg, 36%
yield) as an
off white powder. 1H NMR (400 MHz, d6-DMSO) d: 8.446 (s, 1H), 8.054-8.020 (m,
2H), 7.715 (dd, J = 9.60 Hz, 1.60 Hz, 1H), 7.651-7.649 (m, 2H), 7.605 (d, J =
2.40 Hz,
1H), 7.440 (dd, J = 8.60 Hz, 2.20 Hz, 1H), 7.244-7.147 (m, 3H), 5.485 (s, 2H),
3.787 (s,
3H).
LCMS = 99.5 % purity. MS(ESI+) = 357.8 (M+1).
132
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 53. Preparation of P-005
CI H Cesium Carbonate N~N>
+ N, - ~O L- N
Br N DMF
Br
1-109 1-110
HO, OH Palladium(II)acetate - . N'N
B' Triphenyl Phosphine L
Potassium Carbonate --O N O N
1-110 + HCI
+.O 1,2-dimethoxyethane
N Methanol / water .O 6,
0 N N
0
0 0
P-005 P-005-HCI
[00268] Synthesis of 2-Bromo-4-chloromethyl-1-methoxy-benzene (I-109). 1-109
was synthesized from 4-chloromethylanisole (25.0 g, 159.6 mmol) using the same
conditions as 1-42, and was purified by dissolution in diethyl ether (50 mL)
and hexanes
(50 mL) and crystallization by removing the diethyl ether under vacuum to give
I-109
(19.1 g, 51% yield) as a yellow powder.
[00269] 1H NMR (400 MHz CDC13) d: 7.584 (d, J = 2.40 Hz, 1H), 7.296 (dd, J =
8.40 Hz, 2.00 Hz, 1H), 6.871 (d, J = 8.40 Hz, 1H), 4.520 (s, 2H), 3.901 (s,
3H).
[00270] Synthesis of 1-(3-Bromo-4-methoxy-benzyl)-1H-[l,2,4]triazole (I-110).
A
suspension of 1,2,4-triazole (5.72 g, 82.8 mmol), 3-bromo-4-
methoxybenzylchloride (I-
109) (13.0 g, 55.2 mmol), and solid cesium carbonate (27.0 g, 82.8 mmol) in
DMF (225
mL) was stirred at room temperature overnight. The reaction was diluted with
ethyl
acetate (400 mL) Dilute with 400 ml EA, and washed with water (300 mL). The
aqueous
wash was extracted with ethyl acetate (2 x 150 mL), and all the ethyl acetate
extracts
combined, washed with saturated aqueous ammonium chloride (3 x 300 mL), water
(2 x
300 mL), and brine (300 mL), dried over sodium sulfate and the solvent removed
under
vacuum to give I-110 as a yellow oil (11.39 g; 77% yield) which solidified to
a greasy
yellow solid.
133
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00271] 'H NMR (400 MHz CDC13) d: 8.060 (s, 1H), 7.969 (s, 1H), 7.492 (d, J =
2.00
Hz, 1H), 7.212 (dd, J = 8.60 Hz, 2.20 Hz, 1H), 6.890 (d, J = 8.40 Hz, 1H).
[00272] Synthesis of 1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-1H-
1,2,4]triazole
(P-005). A solution of 1-110 (12.8 g, 47.6 mmol) and 3-nitrophenylboronic acid
(9.53 g,
57.1 mmol) in DMF (250 mL) was purged with nitrogen for 10 min at room
temperautre
and 1 N aqueous sodium carbonate (143 mL), triphenylphosphine (2.49 g, 9.51
mmol),
and bis(dibenzylideneacetone)palladium(0) (2.73 g, 4.75 mmol under nitrogen.
The
reaction was heated to 80 C and stirred overnight. The reaction did not go to
completion, so it was heated to 100 C with vigorous stirring for 2 h. The
diluted with
ethyl acetate (1 L) and washed with water (3 x 1 L) and brine (500 mL), dried
over
sodium sulfate and the solvent removed under vacuum to give a brown oil. The
oil was
purified by silica gel column chromatography eluting with 0-5% acetone in
dichloromethane, and the residue dissolved in ethyl acetate (300 mL), washed
with water
(2 x 300 mL), and brine (300 mL) to give a 30% mixture of I-110 and P-005.
This
mixture was added to a solution of 3-nitrophenylboronic acid (8.00 g, 47.9
mmol) and
triphenylphosphine (2.24 g, 8.58 mmol) in 1,2-dimethoxyethane (150 mL). Argon
was
bubbled through for 10 min, and methanol (15 mL), water (15 mL), solid
potassium
carbonate (17.8 g, 129 mmol), and palladium acetate (960 mg, 4.29 mmol) were
added
under argon gas, and the argon stream was continued for 10 min. The reaction
stirred at
80 C under nitrogen overnight. The solvent was removed under vacuum, and the
residue
dissolved in ethyl acetate (300 mL) and washed with water (300 mL). The water
was
extracted with ethyl acetate (2 x 300 mL), and the extracts combined. The
organic
extracts were washed with water (2 x 500 mL), saturated aqueous sodium
bicarbonate (2
x 500 mL), and brine (500 mL), dried over sodium sulfate, and removed under
vacuum
to give crude product as a brown oil. The product was purified by flash silica
gel column
chromatography eluting with 0-25% acetone in dichloromethane to give slightly
impure
P-005. The material was triturated with dichloromethane (30 mL) in hexanes
(300 mL),
then in dichloromethane (15 mL) and hexanes (10 mL) and washed with hexanes
(20
mL) to give pure P-005 (5.26 g, 36% yield) as a white powder. 1H NMR (400
MHz,CDC13): 3.84 (s, 3H), 5.34 (s, 2 H), 7.02 (d, J= 8.5 Hz, 1 H), 7.28 (d, 1
H), 7.32
(dd, J= 8.5, 2.2 Hz, 1 H), 7.57 (t, J= 8.0 Hz, 1 H), 7.80 (d, J= 7.8 Hz, 1 H),
7.97 (s, 1
H), 8.09 (s, 1 H), 8.19 (dd, J= 8.3, 1.3 Hz, 1 H), 8.38 (t, J= 1.7 Hz, 1 H)
ppm. LCMS =
134
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
100.0 % purity. MS(APCI+) = 311.1 (M+1).
HPLC (220 nm); 99.95%. [Mobile Phase A and Mobile Phase B = Water and
Acetonitrile, Symmetry C18, (250 x 4.6 mm, 5 um), Flow=1.0 mL/min, Inj. Wash =
ACN, Inj. Vol.= 10 uL. Retention time = 22.16 min]
[00273] Elemental Analysis (Calc): C 61.93, H 4.55, N 18.05 (Found), C 61.92,
H
4.62, N 17.88.
[00274] Synthesis of 1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-1H-
1,2,4]triazole
hydrochloride salt (P-005). A solution of I-111 (200 mg, 0.645 mmol) in
tetrahydrofuran
(6 mL) was stirred at room temperature. To this was added 2 M hydrogen
chloride in
diethyl ether at room temperature. The reaction turned cloudy after 10 sec.
The reaction
was allowed to stir at room temperature for 45 min, and the reaction filtered
and washed
with diethyl ether to give P-005 (189.0 mg, 85% yield) as a white solid.
[00275] 1H NMR.(400 MHz d6-DMSO) d: 8.736 (s, 1H), 8.282 (t, J = 2.00 Hz, 1H),
8.219-8.192 (m, 1H), 8.026 (s, 1H), 7.941-7.922 (m, 1H), 7.724 (t, J = 8.00
Hz, 1H),
7.434 (d, J = 2.40 Hz, 1H), 7.389 (dd, J = 8.40 Hz, 2.40 Hz, 1H), 7.175 (d, J
= 8.80 Hz,
1H), 5.417 (s, 2H), 3.797 (s, 3H).
LCMS = 100.0 % purity. MS(APCI+) = 311.1 (M+1).
Elemental Analysis (Calc): C, 55.42 H 4.36, N 16.16, Cl 10.22 (Found): C 55.23
, H
4.39, N 16.00, Cl 11.06
Example 54. Preparation of P-486
O N
N
H
P-486
[00276] Synthesis of 5-(2-Methoxy-5-[1,2,4]triazol-1-ylmethyl-phenyl)-1H-
indole (P-
486). P-486 was synthesized from IH-indole-3-boronic acid (240 mg, 1.49 mmol)
and I-
135
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
110 (200 mg, 0.746 mmol) using conditions similar to those that were used for
I-111.
The reaction was worked up by diluting the reaction with ethyl acetate (50
mL), washing
with 1 N aqueous sodium hydroxide (3x 30 mL), water (2 x 30 mL), and brine (30
mL).
The organic portion was concentrated and the residue was purified by flash
silica gel
column chromatography eluting with 2% methanol in dichloromethane, followed by
flash silica gel column chromatography eluting wit 0-20% ethyl acetate in
hexanes, and
then separation on a silica gel preparatory plate eluting with 50% ethyl
acetate in
hexanes to give P-486 (51.6 mg, 23% yield).
[00277] 1H NMR (400 MHz, CDC13) 8 ppm 3.82 (s, 3 H) 5.33 (s, 2 H) 6.58 (br.
s., 1
H)6.98(d,J=8.33Hz,1H)7.18-7.25(m,2H)7.29-7.35 (m, 2 H) 7.38 - 7.44 (m,1H)
7.73 (s, 1 H) 7.96 (s, 1 H) 8.06 (s, 1 H) 8.17 (br. s., 1 H)
LCMS = 100.0 % purity. MS(APCI+) = 305.1 (M+1).
Example 55. Preparation of P-488
N_NH N_NH N'NH N_NO
O O BBr3 HO K2C03 0 o +
iIIii}0
MEK I N,~ I ~
O O p O
P-009 P-488 P-487 P-018
[00278] Synthesis of 6-(6-Hydroxy-3'-nitro-biphenyl-3-yl methyl)-2H-pyridazin-
3-
one (P-488). To a mixture of compound P-009 (260 mg, 0.77 mmol) in
dichloroethane
(15 ml), boron tribromide (1 M in dichloromethane, 2.3 ml, 2.3 mmol) was added
at -70
C. The reaction mixture was allowed to slowly warm up to rt and then stirred
at rt for 2
hrs. Water (100 ml) and sat. NaHCO3 aq. (20 ml) were added to the reaction
mixture and
it was stirred at rt for 1 hr. The resulting solid was filtered and washed
with water (50
ml) to give 236 mg (95%) of P-488.
[00279] 'H-NMR (400 MHz, DMSO-d6) 3.83 (2H, s), 6.79 (1H, dd, J = 9.6 and 2
Hz), 6.94 (1H, d, J = 8.4 Hz), 7.10 (1H, dd, J = 8.4 and 2 Hz), 7.29 (1H, d, J
= 2.4 Hz),
7.33(1H,d,J=9.6Hz), 7.70(1H,dd,J=8and8Hz), 8.00(1H,m), 8.16(1H,m),
8.40 (1H, m), 9.83 (1H, s), 12.80 (1H, br). MS(ESI-): 322.5 (M-1) LC-MS: 97 %.
136
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 56. Preparation of P-487 and P-018
[00280] Synthesis of 6-[6-(2-Methoxy-ethoxy)-3'-nitro-biphenyl-3-ylmethyl]-2H-
pyridazin-3-one P-018 and 6-[6-(2-Methoxy-ethoxy)-3'-nitro-biphenyl-3-
ylmethyl]-2-(2-
methoxy-ethyl)-2H-pyridazin-3-one P-487. A reaction mixture of compound P-488
(97
mg, 0.3 mmol), Bromoethyl methyl ether (63 mg, 0.45 mg) and K2CO3 (124 mg, 0.9
mmol) in 2-butanone (15 ml) was stirred at 80 C under argon for 18 hrs. After
cooling
to rt, water (10 ml) was added to the reaction mixture and the pH was adjusted
to acidic
by addition of 2 N HC1 aq. and then extracted with ethyl acetate (40 ml). The
organic
layer was washed with water (20 ml), brine, and dried over Na2SO4. After
removal of
solvent, the residue was purified by silica gel column chromatography with
ethyl acetate.
Hexane as eluent to give 30 mg of product P-487 (3a) and 20 mg of product P-
018 (3b).
[00281] P-487: 'H-NMR (400 MHz, DMSO-d6) 3.24 (3H, s), 3.61 (2H, m), 3.89
(2H, s), 4.15 (2H, m), 6.81 (1H, d, J = 9.6 Hz), 7.13 (1H, d, J = 8.8 Hz),
7.26 (1H, dd,
J = 8.8 and 2 Hz), 7.35 (2H, m), 7.71 (1H, dd, J= 8 and 8 Hz), 8.00 (1H, m),
8.18 (1H,
m), 8.43 (1H, m) ppm. MS(ESI-): 380.6 (M-1) LC-MS: 98 %.
[00282] P-018: 'H-NMR (400 MHz, CDC13) 3.34 (3H, s), 3.36 (3H, s), 3.68 (2H,
m), 3.80 (2H, t, J = 5.5 Hz), 3.92 (2H, s), 4.14 (2H, m), 4.36 (2H, t, J = 5.5
Hz), 6.84
(1H,d,J=9.5Hz), 6.98(1H,d,J=8Hz), 7.05(1H,d,J=9.5Hz), 7.19-7.22(2H,
m), 7.55 (1H, dd, J = 8 and 8 Hz), 7.87 (1H, m), 8.17 (1H, m), 8.49 (1H, m)
ppm.
MS(APCI+): 440.1 (M+1)
137
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00283] Scheme 25.
o i 0
Br
3
\ \
\ Pd(OAc)z -'0 / 0- 6'-'C
ciH O / OH I N
/ N~ O + ~0 \ 011 KzCOI, PPh3 A
DME .0 DCM I \
O HO~ 'OH EtOH-H20 N / O
O
O
1-81 1-112
NaBH4 O 7H~'
TFA \
/ N
0
P-017
Example 57. Preparation of P-017
[00284] Synthesis of 2,6-dimethoxy-3'-nitro-biphenyl (I-81). To 1-bromo-3-
nitrobenzene (2.02 g, 10.0 mmol), 2,6-dimethoxyphenylboronic acid (2.70 g,
15.0
mmol), triphenylphosphine (0.52 g, 2.0 mmol), K2C03 (4.14 g, 30.0 mmol) and
palladium(II) acetate (0.009 g, 0.04 mmol) was added DME (80 mL) and EtOH/H20
(1: 1, 20 mL). Argon gas was bubbled through the stirred reaction for 5 min.
The
reaction was stirred at 80 C under argon for 20 h. The reaction was cooled to
room
temperature, concentrated, and H2O (60 mL) and dichloromethane (80 mL) were
added.
The layers were separated and the aqueous was extracted with dichloromethane
(2 x 40
mL). The combined organic extracts were dried with Na2S04, filtered, and
concentrated.
The residue was purified by silica gel column chromatography using 10% ethyl
acetate/hexanes as eluent to afford 1.69 g (65%) of I-81 as a white solid.
[00285] Synthesis of (2-hydroxy-6-methoxy-3'-nitro-biphenyl-3-yl)-pyridin-4-yl-
methanone (I-112). I-81 (0.26 g, 1.0 mmol), 4-nicotinyl chloride (0.18 g, 1.0
mmol), and
dichloromethane (2 mL) were stirred for 5 min at room temperature. A1C13 (0.33
g, 2.47
mmol) was added in portions over 30 min under argon, then argon gas was
bubbled
through the reaction mixture for an additional 2 min. The vial was capped and
stirred at
138
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
room temperature for 30 min, then at 50 C for 4 h. The reaction was cooled to
room
temperature and poured onto cooled concentrated HC1(3 mL). The resulting
aqueous
mixture was extracted with dichloromethane (2 x 30 mL), the aqueous layer was
separated, made basic through the addition of 50% aqueous NaOH, and extracted
with
ethyl acetate (2 x 30 mL). The combined organics were dried with Na2SO4,
filtered, and
concentrated. The residue was purified by silica gel column chromatography
using 2%
McOH/dichloromethane as eluent to afford 0.02 g (6%) of I-112 as an off-white
solid.
1H NMR (CDC13, 400 MHz): 12.53 (s, 1H), 8.84 (dd, J= 4.4, 1.6 Hz, 2 H), 8.3-
8.32 (m,
1 H), 8.21-8.25 (m, 1 H), 7.74-7.78 (m, 1 H), 7.48-7.63 (m, 4 H), 6.61 (d, J=
8.8 Hz, 1
H), 3.87 (s, 3 H); MS(ESI-): 349.3 (M-1), LC-MS: 94.3%.
[00286] Synthesis of 6-methoxy-3'-nitro-3-pyridin-4-ylmethyl-biphenyl-2-ol (P-
017).
To a cooled (0 oC) and stirred solution of TFA (2.5 ml) under N2 was added
NaBH4 (0.12
g, 3.09 mmol) in portions over 20 min. The reaction mixture was warmed to 15
C, and a
solution of I-112 (0.055 g, 0.15 mmol) in dichloromethane (2.5 mL), was added
over 30
min. The reaction was stirred at room temperature for 20 h, poured onto ice-
water (10
mL), made basic (pH 8-10) through the addition of 50% aqueous NaOH, and
extracted
with ethyl acetate (3 x 40 mL). The combined organic extracts were washed with
brine
(60 mL), dried with Na2SO4, filtered, and concentrated. The residue was
purified by
silica gel column chromatography using 2% MeOH/dichloromethane as eluent to
afford
0.039 g (74%) of P-017 as a foamy solid. 1H NMR (CDC13, 400MHz): 8.41 (d, J=
4.4
Hz, 1 H), 8.2-8.26 (m, 2 H), 7.6-7.74 (m, 2 H), 7.1-7.18 (m, 3 H), 6.57 (d, J=
8.0 Hz, 1
H), 5.3 9s, 1 H), 3.96 (s, 2 H), 3.73 (s, 3 H); MS(ESI-): 335.1 (M-1), LC-MS:
92.5%.
139
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 58. Preparation of P-019.
[00287] Scheme 26.
HO..OH
B
Br HO. OH
:: NBS/CCI 2
HO one 1O N Pd(OAc), / P c ,4 PPh
K2CO3, PPh3 .O KsPOa B
O N_ DME
EtOH-H 20 01- U EtOH-H20
1-95
1-113 1-114
I\
O IN iN
N~.O
O
P-019
[00288] Synthesis of 2-iodo-3-methoxy-6-methyl-pyridine (1-95). To 2-iodo-6-
methyl-pyridin-3-ol (1.0 g, 4.25 mmol) and K2C03 (1.18 g, 8.51 mmol) in
acetone (20
mL) was added Mel (0.91 g, 6.38 mmol). The reaction was stirred at 45 C under
N2 for
20 h. The reaction was cooled to room temperature and concentrated. The
residue was
purified by silica gel column chromatography using dichloromethane to afford
1.04 g
(98%) of 1-95 as light yellow solid.
[00289] Synthesis of 3-methoxy-6-methyl-2-(3-nitro-phenyl)-pyridine (I-113).
To I-
95 (0.5 g, 2.0 mmol), 3-nitrophenylboronic acid (0.5 g, 3.06 mmol),
triphenylphosphine
(0.11 g, 0.4 mmol), K2C03 (0.83 g, 6.0 mmol) and palladium(II) acetate (0.045
g, 0.2
mmol) was added DME (16 mL), and EtOH-H20 (1:1, 4 mL). Argon gas was bubbled
through the stirred reaction for 5 min. The reaction was stirred at 60 C
under argon for
18 h. The reaction was cooled to room temperature, concentrated, and H2O and
dichloromethane (40 mL each) were added. The organic layer was separated and
the
aqueous layer was extracted with dichloromethane (2 x 25 mL). The combined
organic
extracts were dried with Na2SO4, filtered, and concentrated. The residue was
purified by
silica gel column chromatography using 1:1 dichloromethane-hexanes then
dichloromethane to afford 0.22 g (44%) of I-113 as a light yellow solid.
140
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00290] Synthesis of 6-bromomethyl-3 -methoxy-2-(3 -nitro-phenyl)-pyridine (I-
114).
To 1-113 (0.21 g, 0.86 mmol) and NBS (0.17 g, 0.95 mmol) in CC14 (10 mL) was
added
benzoylperoxide (0.02 g, 0.08 mmol). The reaction was stirred at 60 C under
N2 for 18
h. The reaction was cooled to room temperature and concentrated. The residue
was
dissolved in a mixture of dichloromethane and hexanes (1:1, 8 mL) and purified
by silica
gel column chromatography using 1:1 dichloromethane:hexanes then
dichloromethane to
afford 0.15 g (55%) of I-114 as a light brown solid.
[00291] Synthesis of 3-methoxy-2-(3-nitro-phenyl)-6-pyridin-4-ylmethyl-
pyridine (P-
019). To 1-114 (0.1 g, 0.31 mmol), 4-pyridylboronic acid (0.057 g, 0.46 mmol),
Triphenylphosphine (0.008 g, 0.031 mmol), K3PO4 (0.13 g, 0.62 mmol) and
palladium(II)acetate (0.004 g, 0.015 mmol) was added DME (4 mL), and EtOH-H20
(1:1, 1 mL). Argon gas was bubbled through the stirred reaction for 5 min. The
reaction
was stirred at 80 C under argon for 18 h. The reaction was cooled to room
temperature,
concentrated, and diluted with H2O and dichloromethane (40 mL each). The
organic
layer was separated and the aqueous was extracted with dichloromethane (2 x 25
mL).
The combined organic extracts were dried with Na2SO4, filtered, and
concentrated. The
residue was purified by preparative TLC using 3% MeOH in dichloromethane to
afford
0.051 g (51%) of P-019 as a light brown viscous liquid. 1H NMR (CDC13,
400MHz):
8.89 (dd, J= 2.0, 1.6 Hz, 1 H), 8.52-8.55 (m, 1 H), 8.31-8.34 (m, 1 H), 8.2-
8.24 (m, 1
H), 7.64-7.7 (m, 1 H), 7.58 (t, J= 8.0 Hz, 1 H), 7.44-7.49 (m, 1 H), 7.2-7.3
(m, 2 H),
7.12 (d, J= 8.4 Hz, 1 H), 4.16 (s, 2 H), 3.9 (s, 3 H); MS(APCI+): 322.1 (M+1),
LC-MS:
95.6%.
Example 59. Preparation of P-020.
NNH OIII N~NH
HO O CI-S(O)2-NCO H2N O O
N +O CH2CI2 N +O
O O
P-488 P-020
[00292] Synthesis of Carbamic acid 3'-nitro-5-(6-oxo-1,6-dihydro-pyridazin-3-
141
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
ylmethyl)-biphenyl-2-yl ester (P-020). A reaction mixture of compound P-488
(97 mg,
0.3 mmol) and chlorosulfonyl isocyanate (64 mg, 0.45 mg) in dichloromethane
(15 ml)
was stirred at rt under argon for 3 days. The reaction mixture was diluted
with water (40
ml) and washed with dichloromethane (40 mL) and ethyl acetate (40 mL). Water
was
removal under vacuum, the residue was stirred with acetone (40 mL), and the
resulting
solid was filtered and washed with water (30 mL) to give 20 mg of product P-
020. The
acetone mother liquid was concentrated to 5 ml and the resulting solid was
filtered to
give another 40 mg of product P-020.
[00293] Yield: 55 %'H-NMR (400 MHz, DMSO-d6) 3.92 (2H, s), 6.82 (1H, dd, J=
9.6 and 2.4 Hz), 6.82 (1H, br), 7.15 (1H, br), 7.18 (1H, J= 8.4 Hz), 7.32 (1H,
dd, J= 8
and 2 Hz), 7.41 (1H, d, J= 10 Hz), 7.44 (1H, d, J= 2Hz), 7.75 (1H, dd, J= 8
and 8
Hz), 7.88 (1H, m). MS(ESI+): 367.5 (M+1) LC-MS: 92%.
Example 60. Preparation of P-02 1.
OH a
HO B,ao
B r 4 \)
I \ \ /
O N Pd(OAc)Z O IN O
K3PO,, PPh3
/ DME
NCO EtOH-HZO I O
i- N'
O 0
1-114 P-021
[00294] Synthesis of 6-benzo[1,3]dioxol-5-ylmethyl-3-methoxy-2 -(3-nitro-
phenyl)-
pyridine (P-021). To I-114 (0.1 g, 0.31 mmol), 3,4-methylenedioxyphenylboronic
acid
(0.077 g, 0.46 mmol), Triphenylphosphine (0.008 g, 0.031 mmol), K3PO4 (0.13 g,
0.62
mmol) and palladium(II)acetate (0.004 g, 0.015 mmol) was added DME (4 mL), and
EtOH-H20 (1:1, 1 mL). Argon gas was bubbled through the stirred reaction for 5
min.
The reaction was stirred at 80 C under argon for 18 h. The reaction was
cooled to room
temperature, concentrated, and diluted with H2O and dichloromethane (40 mL
each).
The organic layer was separated and the aqueous was extracted with
dichloromethane (2
x 25 mL). The combined organic extracts were dried with Na2S04, filtered, and
concentrated. The residue was purified by silica gel column chromatography
using 1:1
142
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
dichloromethane-hexanes then dichloromethane to afford 0.08 g (71%) of P-021
as a
light brown viscous liquid. 1H NMR (CDC13, 400 MHz): 8.89 (dd, J= 2.0, 1.6 Hz,
1 H),
8.34-8.37 (m, 1 H), 8.2-8.24 (m, 1 H), 7.59 (t, J= 8.0 Hz, 1 H), 7.22-7.28 (m,
2 H), 7.09
(d, J= 8.4 Hz, 1 H), 6.78-6.8 (m, 2 H), 5.93 (s, 2 H), 4.09 (s, 2 H), 3.88 (s,
3 H) ppm.
MS(APCI+): 365.1 (M+1), LC-MS: 94.3%.
Example 61. Preparation of P-491.
OH
Br b
\ OH
0 N Cs003 ~0 1 iN
DMF
/1 /
\ N`.O \ I NCO
O
O
1-114 P-491
[00295] Synthesis of [5 -methoxy-6-(3 -nitro-phenyl)-pyridin-2 -yl] -methanol
(P-491).
To I-114 (0.06 g, 0.19 mmol), cyclopentanol (0.032 g, 0.37 mmol), and CsCO3
(0.18 g,
0.56 mmol) was added DMF (2.5 mL). The vial was capped and stirred at room
temperature for 20 h. The reaction was diluted with 1:1 ethyl acetate-H20 (60
mL). The
organic layer was separated and the aqueous was extracted with ethyl acetate
(2 x 20
mL). The combined organic extracts were dried with Na2SO4, filtered, and
concentrated.
The residue was purified by preparative TLC using 2% MeOH in dichloromethane
to
afford 0.015 g (3 1%) of P-491 as a viscous liquid. 1H NMR (CDC13, 400 MHz):
8.88
(dd, J= 2.0, 1.6 Hz, 1 H), 8.32-8.36 (m, 1 H), 8.22-8.26 (m, 1 H), 7.62 (t, J=
8.0 Hz, 1
H), 7.39 (d, J= 8.4 Hz, 1 H), 7.29 (d, J= 8.4 Hz, 1 H), 4.78 (d, J= 4.8 Hz, 2
H), 3.94
(s, 3 H), 3.37 (t, J= 9.2 Hz, 1 H); MS(APCI+): 261.1 (M+1), LC-MS: 100%.
Example 62. Preparation of P-023.
OH
HO.2y+~ 1 1 \
\ Br ~F
I .N
0 N Pd(OAc)Z O F
K3PO4, PPh3
/ 1 DME \ I 0
\ N~'O DOH-HZO N
0- O
1-114 P-023
143
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00296] Synthesis of 6-(4-fluoro-b enzyl)-3 -methoxy-2 -(3-nitro-phenyl)-
pyridine (P-
023). To 1-114 (0.05 g, 0.15 mmol), 4-fluorophenylboronic acid (1) (0.032 g,
0.23
mmol), triphenylphosphine (0.004 g, 0.015 mmol), K3PO4 (0.066 g, 0.31 mmol),
and
palladium(II)acetate (0.002 g, 0.008 mmol) was added DME (1.8 mL), and EtOH-
H20
(1:1, 0.6 mL). The reaction was stirred at 160 C for 5 min using Biotage-60
Microwave
Synthsizer. The reaction was cooled to room temperature and concentrated. The
residue
was purified by silica gel column chromatography using 1:1 dichloromethane-
hexanes
then dichloromethane to afford 0.024 g (46%) of P-023 as a light brown viscous
liquid.
1H NMR (CDC13, 400 MHz): 8.89 (dd, J= 2.0, 1.6 Hz, 1 H), 8.32-8.36 (m, 1 H),
8.2-
8.24 (m, 1 H), 7.59 (t, J= 8.4 Hz, 1 H), 7.22-7.3 (m, 3 H), 7.07 (d, J= 8.4
Hz, 1 H),
6.96-7.04 (m, 2 H), 4.14 (s, 2 H), 3.88 (s, 3 H); MS(APCI+): 339.1 (M+1), LC-
MS:
100%.
Example 63. Preparation of P-024.
N,,
j Br L
0 N CS2CO3 0 iN N
DMF
/ I
\ N`"0 NCO
0 0
1-114 P-024
[00297] Synthesis of 3-methoxy-2-(3-nitro-phenyl)-6-[1,2,4]triazol-1-ylmethyl-
pyridine (P-024). To 1-114 (0.055 g, 0.17 mmol), 1H-[1,2,4]triazole (0.018 g,
0.26
mmol), and CsCO3 (0.17 g, 0.51 mmol) was added DMF (2 mL). The vial was capped
and stirred at room temperature for 20 h. The reaction was diluted with 1:1
ethyl acetate-
H20 (60 mL). The organic layer was separated and the aqueous was extracted
with ethyl
acetate (2 x 20 mL). The combined organic extracts were dried with Na2SO4,
filtered,
and concentrated. The residue was purified by preparative TLC using 3% MeOH in
dichloromethane to afford 0.043 g (8 1%) of P-024 as an off white solid. 1H
NMR
(CDC13, 400 MHz): 8.87 (dd, J= 2.0, 1.6 Hz, 1 H), 8.22-8.3 (m, 3 H), 7.99 (s,
1 H), 7.6
(t, J= 8.0 Hz, 1 H), 7.34 (d, J= 8.4 Hz, 1 H), 7.25 (d, J= 8.4 Hz, 1 H), 5.5
(s, 2 H),
144
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
3.92 (s, 3 H); MS(APCI+): 312.1 (M+1), LC-MS: 100%.
Example 64. Preparation of P-026.
CN
J
Br 0 N
0 N ~~
Cs2CO3 O
DMF
~p
N N
O 0
1-114 P-026
[00298] Synthesis of 4-[5-methoxy-6-(3-nitro-phenyl)-pyridin-2-ylmethyl]-
morpholine (P-026). To I-114 (0.06 g, 0.19 mmol), morpholine (0.032 g, 0.37
mmol),
and Cs2CO3 (0.18 g, 0.56 mmol) was added DMF (2.5 mL). The vial was capped and
stirred at room temperature for 20 h. The reaction was diluted with 1:1 ethyl
acetate-H20
(60 mL). The organic layer was separated and the aqueous was extracted with
ethyl
acetate (2 x 20 mL). The combined organic extracts were dried with Na2SO4,
filtered,
and concentrated. The residue was purified by silica gel column chromatography
using
5% methanol in dichloromethane to afford 0.051 g (80%) of P-026 as a viscous
liquid.
1H NMR (CDC13, 400 MHz): 8.86 (dd, J= 2.4, 1.6 Hz, 1 H), 8.3-8.34 (m, 1 H),
8.2-8.24
(m, 1 H), 7.59 (t, J= 8.4 Hz, 1 H), 7.45 (d, J= 8.4 Hz, 1 H), 7.34 (d, J= 8.4
Hz, 1 H),
3.91 (s, 3 H), 3.75 (t, J= 4.4 Hz, 4 H), 3.71 (s, 2 H), 2.56 (t, J= 4.4 Hz, 4
H);
MS(APCI+): 330.9 (M+1), LC-MS: 97%.
Example 65. Preparation of P-030.
Br 0
N
0 NaH 0 iN
DMF
N~.O I N0
0
0
1-114 P-030
145
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00299] Synthesis of 6-isopropoxymethyl-3 -methoxy-2 -(3-nitro-phenyl)-
pyridine (P-
030). To a cooled (0 C) and stirred suspension of NaH (0.016 g, 0.39 mmol) in
DMF
(2.0 mL) was added a solution of isopropanol (0.033 g, 0.56 mmol) in DMF (0.5
ml).
The reaction mixture was slowly warmed to room temperature and stirred for 2
h. After
cooling to (0 C), a solution of I-114 (0.06 g, 0.19 mmol) in DMF (1.0 mL) was
added
over 5 min. The reaction mixture was slowly warmed to room temperature and
stirred for
20 h. The reaction was poured on to crushed ice-water and extracted with ethyl
acetate
(2 x 40 mL). The combined organic extracts were dried with Na2SO41 filtered,
and
concentrated. The residue was purified by silica gel column chromatography
using
dichloromethane to afford 0.027 g (47%) of P-030 as a viscous liquid. 1H NMR
(CDC13,
400 MHz): 8.86 (dd, J= 2.0, 1.6 Hz, 1 H), 8.33 (dt, J= 8.0, 1.2 Hz, 1 H), 8.2-
8.25 (m, 1
H), 7.59 (t, J= 8.0 Hz, 1 H), 7.49 (d, J= 8.4 Hz, 1 H), 7.36 (d, J= 8.8 Hz, 1
H), 4.66
(s, 2 H), 3.91 (s, 3 H), 3.72-3.81 (m, 1 H), 1.27 (d, J= 6.0 Hz, 6 H);
MS(APCI+): 303.1
(M+1), LC-MS: 100%.
Example 66. Preparation of P-03 1.
0 0
I N~
Br [~JN
0 g'_IN 0N ~/
CS2C03 ND
N" DMF 0
O- O
1-114 P-031
[00300] Synthesis of 1-[5-methoxy-6-(3-nitro-phenyl)-pyridin-2-ylmethyl]-
pyrrolidin-
2-one (P-03 1). To I-114 (0.08 g, 0.25 mmol), pyrrolidone (0.042 g, 0.5 mmol),
and
Cs2CO3 (0.24 g, 0.74 mmol) was added DMF (3 mL). The vial was capped and
stirred at
room temperature for 20 h. The reaction was diluted with 1:1 ethyl acetate-H20
(60 mL).
The organic layer was separated and the aqueous was extracted with ethyl
acetate (2 x 20
mL). The combined organic extracts were dried with Na2S04, filtered, and
concentrated.
The residue was purified by prep TLC using 5% methanol in dichloromethane to
afford
0.054 g (78%) of P-031 as a viscous liquid. 1H NMR (CDC13, 400 MHz): 8.88-8.9
(m, 1
146
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
H), 8.36 (dt, J= 8.0, 1.2 Hz, 1 H), 8.2-8.25 (m, 1 H), 7.6 (t, J= 8.0 Hz, 1
H), 7.26-7.34
(m,2H),4.61(s,2H),3.92(s,3H),3.49(t,J= 7.2 Hz, 2 H), 2.47 (t, J= 8.02 Hz, 2
H), 2.0-2.2 (m, 2 H); MS(APCI+): 328.8 (M+1), LC-MS: 94.0%.
Example 67. Preparation of P-033.
0 0
Br
O JrI N
H~ \ VNH
KZC03 '-0 N
&N DMF
* I t.O
O N I-
O
1-114 P-033
[00301] Synthesis of 1-[5-methoxy-6-(3-nitro-phenyl)-pyridin-2-ylmethyl]-
imidazolidin-2-one (P-033). To I-114 (0.08 g, 0.25 mmol), imidazol-2-one
(0.053 g,
0.62 mmol), and K2CO3 (0.086 g, 0.62 mmol) was added DMF (3 mL). The vial was
capped and heated at 80 C for 20 h. The reaction was diluted with 1:1 ethyl
acetate-H20
(60 mL). The organic layer was separated and the aqueous was extracted with
ethyl
acetate (2 x 20 mL). The combined organic extracts were dried with Na2SO4,
filtered,
and concentrated. The residue was purified by prep TLC using 5% methanol in
dichloromethane to afford 0.015 g (18%) of P-033 as an off white foamy solid.
1H NMR
(CDC13, 400 MHz): 8.88-8.95 (m, 1 H), 8.36 (dt, J= 7.6, 1.6 Hz, 1 H), 8.2-8.24
(m, 1
H), 7.6 (t, J= 8.4 Hz, 1 H), 7.35 (s, 2 H), 4.55 (s, 1 H), 4.53 (s, 2 H), 3.92
(s, 3 H), 3.44-
3.58 (m, 4 H); MS(APCI+): 329.0 (M+1), LC-MS: 90.7%.
147
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 68. Preparation of P-038.
Br OH ON
O N O IN
NaH
DMF
6'N NCO
O O
I- I-
1-114
P-038
[00302] Synthesis of 6-cyc lopentylo xymethyl-3 -methoxy-2 -(3-nitro-phenyl)-
pyridine
(P-038). To a cooled (0 C) and stirred suspension of NaH (0.016 g, 0.39 mmol)
in DMF
(2.0 mL) was added a solution of cyclopentanol (0.048 g, 0.56 mmol) in DMF
(0.5 ml).
The reaction mixture was slowly warmed to room temperature and stirred for 2
h. After
cooling to 0 C, a solution of I-114 (0.06 g, 0.19 mmol) in DMF (1.0 mL) was
added
over 5 min. The reaction mixture was slowly warmed to room temperature and
stirred for
20 h. The reaction was poured on to crushed ice-water and extracted with ethyl
acetate
(2 x 40 mL). The combined organic extracts were dried with Na2SO4, filtered,
and
concentrated. The residue was purified by silica gel column chromatography
using
dichloromethane to afford 0.047 g (77%) of P-038 as a viscous liquid. 1H NMR
(CDC13,
400 MHz): 8.86 (dd, J= 2.4, 1.6 Hz, 1 H), 8.3-8.34 (m, 1 H), 8.2-8.24 (m, 1
H), 7.59 (t,
J= 8.0 Hz, 1 H), 7.47 (d, J= 8.4 Hz, 1 H), 7.36 (d, J= 8.4 Hz, 1 H), 4.63 (s,
2 H),
4.07-4.12 (m, 1 H), 3.91 (s, 3 H), 1.7-1.84 (m, 8 H); MS(APCI+): 329.7 (M+1),
LC-MS:
96.1%.
148
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 69. Preparation of P-064.
[00303] Scheme 27.
2 \ \ Br
BrZ _ I \ CH31/KZC03 I \ F- ~O I iN NBS/CCI4 O iN
HO iN Pyridine HO iN Acetone O iN Pd(OAc), /
1 Br Br KZCOT PPh3
DME F \ I F
EtOH-H20 F F F F
1-115 1-116
1-117 1-118
N L N
Nr) ~)
N O IN N
CS2CO3
\ I F
DMF FF
P-064
[00304] Synthesis of 2-bromo-6-methyl-pyridin-3-ol (1-115). To 6-methyl-
pyridin-3-
ol (5.0 g, 45.82 mmol) in pyridine (15 mL) was added bromine (3.66 g, 22.91
mmol).
The reaction was stirred at room temperature under N2 for 20 h. The crude
reaction
mixture was poured on to crushed ice-water (300 mL) and stirred for 3 h. The
reaction
was extracted with ethyl acetate (5 x 100 mL), washed with brine, dried with
Na2S04,
filtered, and concentrated to afford 6.3 g (73 8%) of I-115 as light yellow
solid.
[00305] Synthesis of 2-bromo-3-methoxy-6-methyl-pyridine (I-116). To crude I-
115
(6.0 g, 31.91 mmol) and K2CO3 (8.82 g, 63.82 mmol) in acetone (100 mL) was
added
methyl iodide (6.79 g, 479.87 mmol). The reaction was stirred at 45 C under
N2 for 20
h. The reaction was cooled to room temperature, filtered and concentrated. The
residue
was purified by silica gel column chromatography using 1:1 dichloromethane-
hexanes to
afford 2.34 g (36%) of I-116 as an off-white solid.
[00306] Synthesis of 3-methoxy-6-methyl-2-(3-trifluoromethyl-phenyl)-pyridine
(I-
117). To 1-116 (1.2 g, 5.94 mmol), 3-trifluoromethylphenylboronic acid (1.69
g, 8.91
mmol), Triphenylphosphine (0.31 g, 1.19 mmol), K2CO3 (2.46 g, 17.82 mmol) and
palladium(II)acetate (0.13 g, 0.59 mmol) was added DME (15 mL) and EtOH-H20
(1:1,
6 mL). Argon gas was bubbled through the stirred reaction for 5 min. The
reaction was
stirred at 80 C under argon for 20 h. The reaction was cooled to room
temperature,
149
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
concentrated, and H2O and dichloromethane (40 mL each) were added. The organic
layer was separated and the aqueous layer was extracted with dichloromethane
(2 x 25
mL). The combined organic extracts were dried with Na2SO4, filtered, and
concentrated.
The residue was purified by silica gel column chromatography using 1:1
dichloromethane-hexanes then dichloromethane to afford 1.36 g (86%) of I-117
as a light
yellow solid.
[00307] Synthesis of 6-bromomethyl-3-methoxy-2-(3-trifluoromethyl-phenyl)-
pyridine (1-118). To 1-117 (1.3 g, 4.86 mmol) and NBS (1.04 g, 5.83 mmol) in
CC14 (25
mL) was added benzoyl peroxide (0.12 g, 0.49 mmol). The reaction was stirred
at 80 C
under N2 for 20 h. The reaction was cooled to room temperature and
concentrated. The
residue was dissolved in a mixture of dichloromethane and hexanes (1:1, 8 mL)
and
purified by silica gel column chromatography using 1:1 dichloromethane-hexanes
to
afford 0.74 g (44%) of 1-118 as an off-white solid.
[00308] Synthesis of 3-methoxy-6-[1,2,4]triazol-1-ylmethyl-2-(3-
trifluoromethyl-
phenyl)-pyridine(P-064). To I-118 (0.2 g, 0.58 mmol), 1H-[1,2,4]triazole
(0.048 g, 0.26
mmol), and Cs2CO3 (0.56 g, 1.73 mmol) was added DMF (4 mL). The vial was
capped
and stirred at room temperature for 20 h. The reaction was diluted with
crushed ice-H2O
(60 mL), stirred for 5 h, filtered, and dried to afford 0.14 g (73%) of P-064
as an off
white solid. 1H NMR (CDC13, 400MHz): 8.28 (s, 1 H), 8.21 9s, 1 H), 8.09 (d, J=
7.6
Hz, 1 H), 7.99 (s, 1 H), 7.65 (d, J= 8.0 Hz, 1 H), 7.52-7.59 (m, 1 H), 7.31
(d, J= 8.4
Hz, 1 H), 7.22 (d, J= 8.4 Hz, 1 H), 5.49 (s, 2 H), 3.89 (s, 3 H) ppm.
MS(APCI+): 335.1
(M+1), LC-MS: 91.4%.
150
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00309] Scheme 28.
O
Cl O N
+ HN Sodium Hydride
Br p DMF Br
1-109 1-119
O
HO, OH Palladium(II)acetate
B Triphenylphosphine O O
1-119 +
Potassium Carbonate
p 1,2-dimethoxyethane
O Ethanol, Water O
A O-J
P-492
Example 70. Preparation of P-492
[00310] Synthesis of 1-(3-Bromo-4-methoxy-benzyl)-piperidine-2,6-dione (1-
119). A
suspension of sodium hydride (56.0 mg, 2.34 mmol) in anhydrous DMF (12 mL) was
stirred under nitrogen for 5 min. To the suspension was added piperidine-2,6-
dione (264
mg, 2.34 mmol) and the reaction was stirred under nitrogen for 5 additional
minutes.
After gas evolution ceased, 1-109 (500 mg, 2.12 mmol) was added and the
reaction
stirred for 24 It under nitrogen at ambient temperature. The reaction was
diluted with
saturated aqueous ammonium chloride (100 mL) and the suspension was extracted
with
ethyl acetate (100 mL). The purple organic extract was washed with water (3 x
50 mL)
water, saturated aqueous ammonium chloride (3 x 50 mL), brine (50 mL) dried
over
anhydrous sodium sulfate, and the solvent removed under vacuum to afford 476.1
mg of
I-119 as a purple powder in 72% yield. 'H NMR (400 MHz, CDC13) 7.59 (d, J=
2.40
Hz, 1H), 7.33 (dd, J= 8.4 Hz, 2.0 Hz, 1H), 6.80 (d, J= 8.40 Hz, 1H), 4.85 (s,
2H), 3.86
(s, 3H), 2.67 (t, J= 6.6 Hz, 4H), 1.95-1.92 (m, 2H).
[00311] Synthesis of 1-(3-Benzo[1,3]dioxol-5-yl-4-methoxy-benzyl)-piperidine-
2,6-
dione (P-492). A solution of I-119 (200 mg, 0641 mmol) and benzo[1,3]dioxol-5-
yl-
boronic acid (117 mg, 0.705 mmol) in 1,4-dioxane were degassed with a nitrogen
stream
151
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
for 10 min. Subsequently, triphenylphosphine (33.5 mg, 0.128 mmol), solid
potassium
carbonate (265 mg, 1.92 mmol) and a mixture of ethanol and water (1:1, 1 mL)
was
added, and the reaction was stirred under nitrogen for 10 min. To the reaction
was added
palladium(II)acetate, and the reaction heated to 80 C with stirring overnight.
The
solvent was removed under vacuum and the residue was suspended in saturated
aqueous
ammonium chloride (50 mL), and the aqueous slurry was extracted with ethyl
acetate (2
x 50 mL). The combined extracts were washed with water (3 x 50 mL) and brine
(30
mL), dried over sodium sulfate, decolorized using activated charcoal, and the
solvent
removed under vacuum. The residue was purified by chromatography on flash
silica gel
eluting with 0-10% acetone in dichloromethane followed by trituration with
diethyl ether
(10 mL) to afford 50.9 mg of P-492 as an off white solid in 22% yield). 'H NMR
(CDC13 400 MHz). 7.29 (m, 2H), 7.03 (d, J = 1.6 Hz, 1H), 6.95 (dd, J = 8.1 Hz,
J = 1.7
Hz, 1H), 6.86 (t, J = 8.2 Hz, 2H), 5.98 (s, 2H), 4.92 (s, 2H), 3.78 (s, 3H),
2.66 (t, J = 6.6
Hz, 4H), 1.93 (m, 2H). LCMS = 100.0 % purity. MS (APCI-) 350.0 (M-H).
Example 71. Preparation of P-070.
Br No
O + HN3 Cs2CO3 O
'OH
OH DMF
N N
O O-
1-114 P-070
[00312] Synthesis of (S)-1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-pyrrolidin-
3-ol
(P-070). Into a 20 mL vial with stir bar was added 1-114 (0.30 g, 0.93 mmol),
(S)-
pyrrolidin-3-ol (54 mg, 0.62 mmol), Cs2CO3 (0.20 g, 0.62 mmol), and DMF (2
mL). The
reaction was stirred for 4 days at room temperature and then 10 mL water was
added.
The product was extracted with ethyl acetate (3 x 10 mL) and the organics were
combined and conecentrated. The residue was purified by flash column
chromatography
using 0-5% methanol/dichloromethane to aford 27.7 mg (14%) of P-070 as a
yellow oil.
1H NMR (400 MHz, CDC13) 8 1.75 (m, 1 H) 2.14 - 2.27 (m, 1 H) 2.29 - 2.41 (m, 1
H)
2.56 (dd, J=10.1, 5.1 Hz, 1 H) 2.68 (d, J=9.8 Hz, 1 H) 2.88 (td, J=8.6, 5.3
Hz, 1 H) 3.63
(s,2H)3.83(s,3H)4.29-4.43(m,1H)6.96(d,J=8.3Hz,1H)7.28-7.38(m,2H)
152
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
7.56 (t, J=8.0 Hz, 1 H) 7.86 (d, J=7.7 Hz, 1 H) 8.17 (dd, J=8.2, 1.9 Hz, 1 H)
8.42 (s, 1 H)
ppm. LGMS = 98.5%, 329.1 (APCI+)
Example 72. Preparation of P-071.
Br
N
0 + HNN CsZCO3 10
OH 30
OH DMF
N N
0 O-
1-114 P-071
[00313] Synthesis of (R)-1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-pyrrolidin-
3-ol
(P-071). The same procedure was used as described for P-070 except using (R)-
Pyrrolidin-3-ol. P-071 was obtained 44.2 mg (22%) as a yellow oil. 'H NMR (400
MHz, CDC13) 1.75 (m, 1 H) 2.13 - 2.28 (m, 1 H) 2.29 - 2.41 (m, 1 H) 2.56 (dd,
J=10.0,
5.0 Hz, 1 H) 2.69 (d, J=9.8 Hz, 1 H) 2.88 (td, J=8.6, 5.2 Hz, 1 H) 3.63 (s, 2
H) 3.83 (s, 3
H)4.29-4.41(m,1H)6.96(d,J=8.3Hz,1H)7.28-7.38 (m,2H)7.56(t,J=8.0Hz,1
H) 7.86 (d, J=7.8 Hz, 1 H) 8.17 (dd, J=8.2, 1.3 Hz, 1 H) 8.42 (t, J=1.7 Hz, 1
H) ppm.
LGMS = 99.0%, 329.1 (APCI+)
Example 73. Preparation of P-493.
OH
Br OH
N
0 + HN Cs2CO3 0
/ I DMF
N NtO
1-114 P-493
[00314] Synthesis of [(R)-1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-
pyrrolidin-2-
yl]-methanol (P-493). The same procedure was used as described for P-070
except
using (R)-1-Pyrrolidin-2-yl-methanol to obtain P-493 (110 mg, 52%). 'H NMR
(400
MHz, CDC13) 1.63 - 1.77 (m, 2 H) 1.78 - 1.89 (m,1H)1.89-2.03 (m,1H)2.28-2.37
153
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
(m, 1 H) 2.74 (m, 1 H) 3.01 (ddd, J=9.1, 6.0, 3.4 Hz, 1 H) 3.37 (d, J=13.0 Hz,
1 H) 3.44
(dd, J=10.7, 2.0 Hz, 1 H) 3.66 (dd, J=10.7, 3.5 Hz, 1 H) 3.83 (s, 3 H) 3.95
(d, J=13.0
Hz, 1 H) 6.97 (d, J=8.3 Hz, 1 H) 7.25 (d, J=2.0 Hz, 1 H) 7.32 (dd, J=8.3, 2.0
Hz, 1 H)
7.56(t,J=8.0Hz,1H)7.85(d,J=7.8Hz,1H)8.18(dd,J=8.3,1.3 Hz,1H)8.41(t,
J=1.8 Hz, 1 H) ppm. LGMS = 98.9%, 343.1 (APCI+)
Example 74. Preparation of P-072.
SOH
Br SOH
N
Cs2CO3 O
O HN
DMF
N N
O O-
1-114 P-072
[00315] Synthesis of [(S)-1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-
pyrrolidin-2-
yl]-methanol (P-072). The same procedure was used as described for P-070
except
using (R)-1-Pyrrolidin-2-yl-methanol to obtain P-072 (68 mg, 32%). 'H NMR (400
MHz, CDC13) 1.63 - 1.78 (m, 2 H) 1.84 (td, J=13.2, 5.6 Hz, 1 H) 1.89 - 2.02
(m, 1 H)
2.33 (q,J=9.0Hz,1H)2.69-2.82(m,1H)2.95-3.07 (m,1H)3.38(d,J=12.9Hz,1
H)3.44(d,J=10.6Hz,1H)3.66(dd,J=10.7,3.4Hz,1H)3.83(s,3H)3.96(d,J=13.0
Hz,1H)6.97(d,J=8.3Hz,1H)7.24-7.26(m,1H)7.29- 7.36 (m,1H)7.56(t,J=8.0
Hz, 1 H) 7.85 (d, J=7.8 Hz, 1 H) 8.18 (dd, J=8.2, 1.3 Hz, 1 H) 8.41 (t, J=1.7
Hz, 1 H)
ppm. LGMS = 98.8%, 343.1 (APCI+)
Example 75. Preparation of P-076.
II
Br 0
O N0
NaH
O k
H v
LJ THE O
N
N 1J
0
O
1-114 P-076
154
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00316] Synthesis of 3 -(6-Methoxy-3'-nitro-b iphenyl-3 -ylmethyl)-oxazolidin-
2 -one
(P-076). Into a dry 100 mL round bottom flask with stir bar was added
oxazolidinone
(0.15 g, 1.74 mmol) and dry THE (8 mL). The solution was cooled to 0 C and
NaH (83
mg, 2.09 mmol) was added. The suspension was stirred for 10 minutes at 0 C
and 20
minutes at room temperature, and then cooled to 0 C. I-114 (0.56 g, 1.74
mmol) in 2
mL dry THE was added to the above mixture and the reaction was stirred for 16
hours at
room temperature. 10 mL of aqueous saturated NH4C1 was added and the THE was
removed under reduced pressure. An additional 10 mL of water was added and the
product was extracted with ethyl acetate (3 x 10 mL). The combined organics
were dried
over sodium sulfate, concentrated, and purified by flash column chromatography
using
35%-75% ethyl acetate/hexanes to obtain 94 mg (16%) of P-076 as an oil. 'H NMR
(400
MHz, CDC13) 3.47 (t, J=8.1 Hz, 2 H) 3.84 (s, 3 H) 4.32 (t, J=7.8 Hz, 2 H) 4.44
(s, 2 H)
7.00 (d, J=8.4 Hz, 1 H) 7.26 (s, 1 H) 7.33 (dd, J=8.5, 2.2 Hz, 1 H) 7.57 (t,
J=8.0 Hz, 1 H)
7.83 (d, J=7.7 Hz, 1 H) 8.19 (dd, J=8.2, 1.3 Hz, 1 H) 8.40 (t, J=1.8 Hz, 1 H)
ppm.
LC/MS = 98.0 %1328.1 (APCI-)
Example 76. Preparation of P-001.
[00317] Scheme 29.
"Br
` ~ Pd(OAc)Z O 9~N
Brz O B(OH)z NOz K2C03 CCIa D
MeOH/H20 1O N
O_ 4 O1-69
1-70
B(OH)2
/ F O F BBr3 HO F CCIF2000Na Fx0 i i F
Pd(OAc)21 PPh3 DCM NaOH
K3P04 / NCO NOD DMF NCO
DME/EtOH/H20 O O O
P-001 P-494 P-067
[00318] Synthesis of 2-Methoxy-5-methyl-3'-nitro-biphenyl (1-69): A reaction
mixture of 2-methoxy-5-methylphenyl boronic acid (1.65 g, 10 mmol), 3-nitro-
iodobenzen (2.49 g, 10 mmol), K2CO3 (2.76 g, 20 mmol), palladium(II)acetate
(112 mg,
0.5 mmol) in methanol (75 ml) and water (15 ml) was stirred at rt overnight.
The
reaction mixture was diluted with ethyl acetate (300 ml) washed with diluted
Na2S207
155
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
aq., water, brine, and dried over Na2SO4. After removal of solvent, 2.5 g
(100%) of 1-69
was obtained.
[00319] Synthesis of 5-Bromomethyl-2-methoxy-3'-nitro-biphenyl (1-70): To a
mixture of compound 1-69 (2.43 g, 10 mmol) in carbon tetrachloride (150 mL),
was
added bromine (1.76 g, 11 mmol) at rt. The reaction mixture was stirred at 80
C under a
sun lamp for 1 hr. After removal of solvent, the residue was washed with
diethylether
(15 mL)/Hexane (15 mL) to give 2.1 g (65%) of compound 1-70.
[00320] Synthesis of 5-(4-Fluoro-benzyl)-2-methoxy-3'-nitro-biphenyl (P-001):
A
reaction mixture of 1-70 (300 mg, 0.93 mmol), 4-fluorophenyl-boronic acid (196
mg, 1.4
mmol), triphenylphosphine (52 mg, 0.2 mmol), K3PO4 (394 mg, 1.86 mmol),
palladium(II)acetate (22 mg, 0.1 mmol) in DME (5 ml), ethanol (0.5 ml) and
water (0.5
ml) was stirred at 80 C overnight under argon. The reaction mixture was
diluted with
diethylether (40 ml), washed with water, brine, and dried over Na2SO4. After
removal of
solvent, the residue was purified by silica gel column chromatography with
ethyl acetate
/Hexane as eluent to give 200 mg (60%) of P-001 'H NMR (400 MHz, DMSO-d6):
3.77
(s, 3H), 3.94 (s, 2H), 7.05 - 7.14 (m, 3H), 7.22 - 7.34 (m, 4H), 7.65 - 7.78
(m, 1H), 7.92
(d, J= 7.8 Hz, 1 H), 8.18 (dd, J= 8.2, 1.34 Hz, 1 H), 8.28 (t, J= 1.8 Hz, 1 H)
ppm.
LCMS = 97.4% purity TSI(+) = 308.6 (M-29).
Example 77. Preparation of P-494.
[00321] Synthesis of 5-(4-Fluoro-benzyl)-3'-nitro-biphenyl-2-ol (P-494): To a
mixture of compound P-001 (200 mg, 0.59 mmol) in dichloromethane (15 ml) was
added
BBr3 (1M in dichloromethane, 1.78 ml, 1.78 mmol) at -78 C under N2. The
reaction
mixture was stirred at -78 C to rt overnight. The reaction mixture was
diluted with
water and extracted with dichloromethane (2 x 20 ml). The dichloromethane
layer was
washed with water (2 x 40 ml), brine, and dried over Na2SO4. After removal of
solvent,
the residue was purified by silica gel column chromatography with ethyl
acetate /Hexane
as eluent to give 180 mg (94%) of P-494. 'H-NMR (400 MHz, CDC13) 3.94 (2H, s),
4.83 (1H, s), 6.87 (1H, d, J = 8 Hz), 6.99 (2H, m), 7.07 - 7.17 (4H, m), 7.60
(1H, dd, J
= 8 and 8 Hz), 7.85 (1H, m), 8.21 (1H, m), 8.40 (1H, m). MS(APCI-): 322.1 (M-
1) LC-
156
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
MS: 97 %.
Example 78. Preparation of P-067.
[00322] Synthesis of (7) 2-Difluoromethoxy-5-(4-fluoro-benzyl)-3'-nitro-
biphenyl (P-
067): To a mixture of compound P-494 (100 mg, 0.3 mmol) and NaOH (40 mg, 1
mmol) in DMF (6 ml), was added sodium chlorodifluoroacetate (228 mg, 1.5
mmol).
The reaction mixture was stirred at 50 C overnight. After removal of solvent,
the
residue was diluted with water (40 mL) and extracted with dichloromethane (2 x
30 mL).
The combined organic extracts were washed with water (4 x 30 ml), brine, and
dried
over Na2SO4. After removal of solvent, the residue was purified by silica gel
column
chromatography with dichloromethane /Hexane as eluent to give 45 mg (40%) of P-
067.
'H-NMR (400 MHz, CDC13) 4.00 (2H, s), 6.37 (1H, t, J= 7,3 Hz), 7.00 (2H, m),
7.15
(2H, m), 7.22 (1H, m), 7.59 (1H, dd, J= 8 and 8 Hz), 7.80 (1H, m), 8.22 (1H,
m), 8.32
(1H, m) ppm. MS(APCI-): 373.1 (M-1) LC-MS: 98 %.
[00323] Example 79. Preparation of P-022.
O N
O"
0
P-022
[00324] Synthesis of 2'-Methoxy-5'-[ 1,2,4]triazol-1-ylmethyl-biphenyl-3 -
carboxylic
acid methyl ester (P-022). A nitrogen stream was bubbled through a solution of
I-110
(810 mg, 3.02 mmol) and 3-methoxycarbonylphenylboronic acid (816 mg, 4.53
mmol) in
1,4-dioxane (50 mL) for 45 min. To this solution was added
bis(triphenylphosphine)palladium(II) dichloride (106 mg, 0.151 mmol) and 1 M
aqueous
sodium carbonate (9 mL) under nitrogen. The reaction was heated to 85 C
overnight.
The reaction was cooled to room temperature and diluted with ethyl acetate
(150 mL).
The organic solution was washed with water (5 x 100 mL), brine (100 mL), dried
over
157
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
sodium sulfate, and the solvent removed under vacuum. The residue was purified
by
flash silica gel column chromatography eluting with 0-2% methanol in
dichloromethane
followed by flash silica gel column chromatography with 50% ethyl acetate in
hexanes,
and the residue wash dissolved in ethyl acetate (10 mL). The organic solution
was
washed with water (2 x 10 mL) and the wash extracted with ethyl acetate (2 x
10 mL).
The combined ethyl acetate extracts were washed with water (3 x 10 mL), dried
over
sodium sulfate, and the solvent removed under vacuum. The product was
recrystalized
by dissolving in hot diethyl ether (2 mL), adding hexanes (3 mL), and removing
the
diethyl ether under a nitrogen stream. The product was filtered and washed
with hexanes
(3 x 3 mL) to give P-022 (233.6 mg, 23.9 % yield) as a white solid.
[00325] 'H NMR (400 MHz, CDC13) d: 8.15 (t, J= 1.60 Hz, 1H), 8.066 (s, 1H),
8.019-7.999 (m, 1H), 7.97 (s, 1H), 7.69-7.67 (m, 1H), 7.47 (t, J = 7.60 Hz,
1H), 7.29-
7.26 (m, 2H), 6.989 (d, J = 9.20, 1H), 5.33 (s, 2H), 3.93 (s, 3H), 3.82 (s,
3H) ppm.
[00326] LCMS = 99.0% purity. MS(APCI+) = 324.1 (M+1).
Example 80. Preparation of P-044.
N,N.
0 LN
O
O-J
P-044
[00327] Synthesis of 1-(3-Benzo[1,3]dioxol-5-yl-4-methoxy-benzyl)-1H-
[1,2,4]triazole (P-044). A nitrogen stream was bubbled through a solution of I-
110
(500 mg, 1.87 mmol) and benzo[1,3]dioxol-5-yl-boronic acid (345 mg, 2.08 mmol)
in
DMF (40 mL) for 10 min. To the solution was added
bis(dibenzylideneacetone)palladium(0) (120 mg, 0.208 mmol), triphenylphosphine
(109
mg, 0.416 mmol), and 1 M aqueous sodium carbonate (6.25 mL) under nitrogen.
The
reaction was heated to 80 C under a nitrogen atmosphere and stirred for 16h.
Approximately three quarters of the solvent was removed under vacuum and the
158
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
remaining suspension was diluted with ethyl acetate (50 mL). The organic
solution was
washed with water (3 x 50 mL), saturated aqueous sodium bicarbonate (50 mL),
brine
(50 mL), dried over sodium sulfate, and the solvent removed under reduced
pressure.
The product was twice purified by flash silica gel column chromatography
eluting with
0-1% methanol in dichloromethane, followed by 0-25% acetone in dichloromethane
to
give P-044 (296.1 mg, 51 % yield) as a yellow oil. 1H NMR (400 MHz, CDC13)
8.05 (s,
1H), 7.96 (s, 1H), 7.24-7.21 (m, 2H), 7.01 (d, J = 2.0 Hz, 1H), 6.98-6.89
(2H), 6.85 (d, J
= 8.0 Hz, 1H), 5.99 (s, 2H), 5.31 (s, 2H), 3.82 (s, 3H) ppm. LCMS = 100.0%
purity.
MS(APCI+) = 310.1 (M+1). HPLC (220 nm); 97.7%. [Water and acetonitrile with
0.05% trifluroacetic acid, Column: Symmetry C18 (250 x 4.6 mm, 5 um),
Gradient, Flow
= 1.0 ml/min, Wash = CAN, Inj vol. = 10ul, Retention time = 21.2 min]
[00328] Example 81. Preparation of P-058.
O N
F
P-058
[00329] Synthesis of 1-(3'-Fluoro-6-methoxy-biphenyl-3-ylmethyl)-1H-
[1,2,4]triazole
(P-058). A nitrogen stream was bubbled through a solution of 1-110 (300 mg,
1.12
mmol) and 3-fluorophenylboronic acid (172 mg, 1.23 mmol) in 1,2-
dimethoxyethane (5
mL) for 10 min. To this solution was added triphenylphosphine (58.8 mg, 0.112
mmol),
solid potassium carbonate (463 mg, 3.36 mmol), ethanol (1 mL), water (1 mL),
and
palladium(II)acetate (25.1 mg, 0.112 mmol) under nitrogen. The reaction was
stirred at
100 C for 18 h. The solvent was removed under vacuum and the residue taken up
in
ethyl acetate (50 mL). The organic solution was washed with saturated aqueous
ammonium chloride (50 mL), the residual palladium was filtered, and the
organic extract
was washed with water (2 x 50 mL), brine (50 mL), dried over sodium sulfate,
and
removed under vacuum. The crude product was purified by flash silica gel
column
chromatography eluting with 5% methanol in dichloromethane, followed by a
second
159
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
flash silica gel column eluting with 5% acetone in dichloromethane. The solid
thus
obtained was recrystallized in diethyl ether (5 mL) and hexanes (10 mL),
filtered, and
washed with hexanes (5 mL) to give P-058 (65 mg; 21% yield) as a white powder.
1H
NMR (400 MHz CDC13): 8 8.07 (s, 1H), 7.97 (s, 1H), 7.36 (dt, J= 8.0 Hz, J= 6.0
Hz,
1H), 7.25 (m, 2H), 7.03 (m, 1H), 6.98 (d, J= 8.4 Hz, 1H), 5.32 (s, 2H), 3.83
(s, 3H)
ppm. LCMS = 100.0 % purity. MS:(APCI+) = 284.1 (M+1).
Example 82. Preparation of P-081.
O N
P-081
[00330] Synthesis of 2'-Methoxy-5'-[ 1,2,4]triazol-1-ylmethyl-biphenyl-3 -
carbonitrile
(P-081). A nitrogen stream was bubbled through a solution of 1-110 (200 mg,
0.746
mmol) and 3-cyanophenylboronic acid (121 mg, 0.821 mmol) in 1,2-
dimethoxyethane (5
mL) for 15 min. To the solution was added ethanol (0.5 mL) and water (0.5 mL)
and
degassing was continued for 5 min. To the solution was added solid potassium
carbonate
(309 mg, 2.24 mmol), triphenylphosphine (39.1 mg, 0.149 mmol), and
palladium(II)acetate (16.7 mg, 0.0746 mmol) simultaneously under nitrogen. The
reaction was heated to 80 C and stirred with heating overnight. The reaction
was cooled
to room temperature and the solvent removed under vacuum. The residue was
diluted
with ethyl acetate (30 mL), washed with water (2 x 30 mL), the spent catalyst
was
filtered off, and the combined aqueous layers extracted with ethyl acetate (30
mL). The
combined organic extracts were washed with saturated aqueous ammonium chloride
(50
mL), water (2 x 50 mL), brine (50 mL), dried over sodium sulfate, and the
solvent
removed under vacuum. The residue was purified by flash silica gel column
chromatography eluting with 3-15% acetone in dichloromethane to give a white
solid,
which was triturated with diethyl ether (10 mL), filtered and washed with
diethyl ether (2
x 5 mL) to give P-081 (112.8 mg, 52% yield) as a white solid.
[00331] 1 H NMR (400 MHz CDC13) 8.08 (s, 1H), 7.97 (s, 1H), 7.81 (t, J = 1.8
Hz,
160
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
1H), 7.69 (dt, J = 8.0 Hz, 1.5, 1H), 7.61 (dt, J = 8.0 Hz, 1.3, 1H), 7.50 (t,
J = 7.8 Hz, 1H),
7.31 (dd, J = 8.6, 2.2 Hz, 1 H), 7.22 (d, J = 2.0 Hz, 1 H), 7.00 (d, J = 8.4
Hz, 1 H), 5.3 3 (s,
2H), 3.83 (s, 3H) ppm. MS(ESI+)= 291.4 (M+1).
[00332] Example 83. Preparation of P-082.
O N
O
P-082
[00333] Synthesis of 1-(2'-Methoxy-5'-[1,2,4]triazol-1-ylmethyl-biphenyl-3-yl)-
ethanone (P-082). A nitrogen stream was bubbled through a solution of I-110
(200 mg,
0.746 mmol) and 3-acetylphenlyboronic acid (135 mg, 0.821 mmol) in 1,2-
dimethoxyethane (5 mL) for 20 min. To the solution was added ethanol (0.5 mL)
and
water (0.5 mL) and degassing was continued for 5 min. To the solution was
added solid
potassium carbonate (309 mg, 2.24 mmol), triphenylphosphine (39.1 mg, 0.149
mmol)
and palladium(II)acetate (16.7 mg, 0.0746 mmol) simultaneously under nitrogen.
The
reaction was heated to 80 C and stirred with heating overnight. The reaction
was cooled
to room temperature and the solvent removed under vacuum. The residue was
diluted
with ethyl acetate (50 mL). The organic solution was washed with water (3 x 50
mL),
saturated aqueous ammonium chloride (50 mL), brine (50 mL), dried over sodium
sulfate, decolorized in activated carbon, and the solvent removed under
vacuum. The
residue was purified by flash silica gel column chromatography eluting with 3-
15%
acetone in dichloromethane followed by silica gel preparatory plate eluting
with 10%
acetone in dichloromethane to give P-082 (109.1 mg, 48% yield) as a tacky gum.
1H
NMR (400 MHz, CDC13) 8.08 - 8.07 (m, 2H), 7.97 (s, 1H), 7.94-7.92 (m, 1 H),
7.70-
7.68 (m, 1H), 7.50 (t, J = 7.8 Hz, 1H), 7.30-7.26 (m, 2H), 67.00 (d, J = 8.0
Hz, 1H), 5.33
(s, 2H), 3.83 (s, 3H), 2.63 (s, 3H) ppm. LCMS = 99.5% purity. MS(APCI+)= 308.1
(M+1).
161
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 84. Preparation of P-084.
O N
OoF
P-084
[00334] Synthesis of 1-(6-Methoxy-3'-trifluoromethoxy-biphenyl-3-ylmethyl)-1H-
[1,2,4]triazole (P-084). P-084 was synthesized from 1-110 (200 mg, 0.746 mmol)
and
3-trifluoromethoxyphenylboronic acid (169 mg, 0.821 mmol) by the same reaction
conditions that were used for P-08 1. After removal of the solvent under
vacuum, the
residue was diluted with ethyl acetate (30 mL) and washed with water (30 mL).
The
aqueous wash was extracted with ethyl acetate (30 mL), and the combined
organic
extracts were washed with water (2 x 50 mL), saturated aqueous ammonium
chloride (50
mL), brine (50 mL), dried over sodium sulfate, decolorized over activated
charcoal, and
the solvent removed under vacuum. The residue was purified by flash silica gel
column
chromatography eluting with 5% acetone in dichloromethane to give P-084 as a
yellow
oil (187.3 mg, 54% yield). 1H NMR (400 MHz, DMSO-d6) 8.45 (s, 1H), 7.87 (s,
1H),
7.57-7.53 (m, 2H), 7.48-7.48 (brm, 1H), 7.43-7.39 (m, 2H), 7.31-7.29 (m, 1H),
7.14 (d, J
= 8.4 Hz, 1H), 5.45 (s, 2H), 3.84 (s, 3H) ppm.LCMS = 100.0% purity. MS(APCI+)=
350.1 (M+1).
Example 85. Preparation P-085.
O N
S
P-085
[00335] Synthesis of 1-(6-Methoxy-3'-methylsulfanyl-biphenyl-3-ylmethyl)-1H-
162
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[1,2,4]triazole (P-085). P-085 was synthesized from 1-110 (200 mg, 0.746 mmol)
and
3-methylsulfanylphenylboronic acid (138 mg, 0.821 mmol) using the same
conditions as
P-081. The reaction was worked up by diluting with ethyl acetate (30 mL) and
washing
with water (30 mL). The aqueous wash was extracted with ethyl acetate (30 mL),
and the
organic extractions combined, washed with water (2 x 50 mL), saturated aqueous
ammonium chloride (50 mL), brine (50 mL), decolorized with activated carbon,
dried
over sodium sulfate, and the solvent removed under vacuum. The residue was
purified by
flash silica gel column chromatography eluting with 5% acetone in
dichloromethane to
give P-085 (117.1 mg, 50% yield) as a yellow gum. 1H NMR (400 MHz, CDC13) 8.06
(s, 1H), 7.97 (s, 1H), 7.37 (t, J= 1.4 Hz, 1H), 7.32 (t, J= 7.6 Hz, 1H), 7.26-
7.23 (m, 4H),
6.97 (d, J= 8.4 Hz, 1H), 5.32 (s, 2H), 3.81 (s, 3H), 2.50 (s, 3H) ppm.
[00336] LCMS = 100.0% purity. MS(APCI+)= 312.1 (M+1).
Example 86. Preparation of P-086.
O N
O
P-086
[00337] Synthesis of 1-(6,3'-Dimethoxy-biphenyl-3-ylmethyl)-1H-[l,2,4]triazole
(P-
086). P-086 was synthesized from 1-110 (200 mg, 0.746 mmol) and 3-
methoxyphenylbronic acid (125 mg, 0.821 mmol) using the same conditions as P-
081.
The reaction was cooled to room temperature and the solvent removed under
vacuum.
The residue was suspended in ethyl acetate (30 mL), washed with water (30 mL),
the
aqueous wash extracted with ethyl acetate (30 mL), and the organic extracts
combined.
The organic extracts were washed with water (3 x 30 mL), saturated aqueous
ammonium
chloride (30 mL), brine (30 mL), decolorized with activated carbon, dried over
sodium
sulfate, and the solvent removed under vacuum. The product was purified by
flash silica
gel column chromatography eluting with 5% acetone in dichloromethane to obtain
P-086
(148.9 mg, 68% yield) as a yellow oil. 1H NMR (400 MHz, acetone-d6) 8.45 (s,
1H),
163
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
7.86 (s, 1H), 7.36-7.28 (m, 3H), 7.10-7.03 (m, 3H), 6.91-6.88 (m, 1H), 5.43
(s, 2H), 3.82
(s, 6H) ppm. LCMS= 100% purity MS(APCI+) = 296.1 (M+1).
Example 87. Preparation of P-102.
~O
Br II
0
0
Pd(OAc)2
K3PO4 PPh3
0 DME
EtOH, H2O
0 0
1-70 P-102
[00338] Synthesis of 3-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-phenol (P-
102).
Into a 20 mL vial with stir bar was added 1-70 (0.30 g, 0.93 mmol), 3-
hydroxyphenylboronic acid (0.19 g, 1.40 mmol), triphenylphosphine (49 mg, 0.19
mmol), K3PO4 (0.40 g, 1.86 mmol), DME (5 mL), water (0.5 mL), and ethanol (0.5
mL).
N2 gas was bubbled through the stirred reaction for 10 minutes. Palladium(II)
acetate (21
mg, 0.09 mmol) was added and N2 was bubbled through for an additional 5
minutes.
The reaction was stirred at 80 C under N2 for 18 hours. The reaction was
cooled to
room temperature and 20 mL of water and 20 mL of ethyl acetate were added. The
layers were separated and the aqueous was extracted with ethyl acetate (3 x 15
mL). The
organics were combined, dried with sodium sulfate, and concentrated. The
residue was
purified by flash column chromatography using 15% ethyl acetate/Hexanes to
obtain 177
mg (57%) of P-102 as a light-yellow oil. 'H NMR (400 MHz, CDC13) 3.81 (s, 3 H)
3.93
(s, 2 H) 4.72 (s, 1 H) 6.62 - 6.72 (m, 2 H) 6.79 (d, J=7.7 Hz, 1 H) 6.94 (d,
J=8.3 Hz, 1
H) 7.11 - 7.23 (m, 3 H) 7.54 (t, J=8.0 Hz, 1 H) 7.82 (d, J=7.8 Hz, 1 H) 8.16
(dd, J=8.2,
1.34 Hz, 1 H) 8.39 (t, J=1.8 Hz, 1 H) ppm. MS: (APCI-) 335.1
164
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 88. Preparation of P-103.
Br HO,B,OH
Pd(OAc)2
K3PO4 PPh3
~ OH
N OH EtOH, H2O I N
0 '0
1-70 1-120 P-103
[00339] Synthesis of [3 -(6 -Methoxy-3'-nitro-b iphenyl-3 -ylmethyl)-phenyl] -
methanol
(P-103). The same procedure that was used for P-102 was used, except using 3-
hydroxybenzylboronic acid. The title compound P-103 was obtained (209 mg, 64%)
as a
yellow oil. 1H NMR (400 MHz, CDC13) 1.62 (t, J=5.8 Hz, 1 H) 3.81 (s, 3 H) 3.99
(s, 2
H)4.67(d,J=5.6Hz,2H)6.94(d,J=8.3Hz,1H)7.10-7.24 (m, 5 H) 7.27 - 7.34 (m,
1 H) 7.54 (t, J=8.0 Hz, 1 H) 7.83 (d, J=7.7 Hz, 1 H) 8.16 (dd, J=8.3, 1.0 Hz,
1 H) 8.38
(s, 1 H) ppm. LC/MS = 96.8%, (APCI-) 349.1
Example 89. Preparation of P-104.
0
Br gNa
+ HO N N
O 0
1-70 P-104
[00340] Synthesis of 1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-1H-pyridin-2-
one
(P-104). Into a 20 mL vial with stir bar was added 2-hydroxypyridine (74 mg,
0.78
mmol), K2CO3 (0.24 g, 1.71 mmol), 1-70 (0.30 g, 0.94 mmol), and 3 mL of DME.
The
mixture was stirred at 80 C for 18 hours, and then cooled to room
temperature, filtered
to remove the solids, and concentrated. The residue was purified by flash
column
chromatography using 30% - 75% ethyl acetate/hexanes to obtain 124 mg (40%) of
P-
104 as a tan-colored solid. 'H NMR (500 MHz ,CDC13) 8.38 (t, J= 1.9 Hz, 1 H),
8.17
165
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
(dd,J=1.3,8.2Hz,1H),7.81(d,J=7.8Hz,1H), 7.56 (t, J= 7.9 Hz,1H),7.40-7.27
(m,5H),6.99(d,J=8.6Hz,1H),6.60(d,J=8.9Hz,1H),6.17(td,J=1.2,6.7Hz,1
H), 5.14 (s, 2 H), 3.82 (s, 3 H) ppm. 13C NMR (125 MHz,CDC13) 8 162.8, 156.4,
148.3,
13 9.8, 13 9.6, 13 7.3, 13 5.8, 13 0.9, 13 0.1, 12 9.3, 12 9.1, 12 8.7, 124.7,
122.2, 121.6, 111.9,
106.5, 55.9, 51.7 ppm. LC/MS = 96.7%, 337.1 (APCI+).
Example 90. Preparation of P-105.
OH
Br HO,B.OH
O
K3PO4 PPh3
A DME
0 EtOH, H2O I N
O
1-70 P-105
[00341] Synthesis of 2-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-phenol (P-
105),
Into a 20 mL vial with stir bar was added 1-70 (0.30 g, 0.93 mmol), 2-
hydroxyphenylboronic acid (GS39) (0.19 g, 1.40 mmol), triphenylphosphine (49
mg,
0.19 mmol), K3PO4 (0.40 g, 1.86 mmol), DME (5 mL), water (0.5 mL), and ethanol
(0.5
mL). N2 gas was bubbled through the stirred reaction for 10 minutes.
palladium(II)
acetate (21 mg, 0.09 mmol) was added and N2 was bubbled through for an
additional 5
minutes. The vial was capped and the reaction was stirred at 80 C for 18
hours. The
reaction was cooled to room temperature and 5 mL of water and 5 mL of ethyl
acetate
were added. The layers were separated and the aqueous was extracted with ethyl
acetate
(3 x 10 mL). The organics were combined, dried with sodium sulfate, and
concentrated.
The residue was purified by flash column chromatography using 15% ethyl
acetate/hexanes to afford 120 mg (38%) of P-105 as a yellow oil. 1H NMR (400
MHz,
CDC13) 3.80 (s, 3 H) 3.99 (s, 2 H) 4.74 (s, 1 H) 6.78 (d, J=7.8 Hz, 1 H) 6.90
(t, J=7.5
Hz,1H)6.94(d,J=8.2Hz,1H)7.09-7.17(m,2 H) 7.19 - 7.25 (m, 2 H) 7.53 (t, J=8.0
Hz, 1 H) 7.82 (d, J=7.8 Hz, 1 H) 8.15 (dd, J=8.2, 1.2 Hz, 1 H) 8.38 (t, J=1.8
Hz, 1 H)
ppm. MS: 334.1 (APCI-)
166
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 91. Preparation of P-119.
OH
Br HO,B.OH
Pd(OAc)2
0 + OH 0 I I F
K,PO4 PPh3
DME \
N DOH, H2O A
I
0
1-70 P-119
[00342] Synthesis of 5-Fluoro-2-(6-methoxy-3'-nitro-biphenyl-3-ylmethyl)-
phenol (P-
119). Into a 20 mL vial with stir bar was added 1-70 (0.30 g, 0.93 mmol), 4-
fluoro-2-
hydroxyphenylboronic acid (0.22 g, 1.40 mmol), triphenylphosphine (49 mg, 0.19
mmol), K3PO4 (0.40 g, 1.86 mmol), DME (5 mL), water (0.5 mL), and ethanol (0.5
mL).
N2 gas was bubbled through the stirred reaction for 10 minutes. palladium(II)
acetate (21
mg, 0.09 mmol) was added and N2 was bubbled through for an additional 5
minutes.
The vial was capped and the reaction was stirred at 80 C for 18 hours. The
reaction was
cooled to room temperature and 5 mL of water and 5 mL of ethyl acetate were
added.
The layers were separated and the aqueous was extracted with ethyl acetate (3
x 10 mL).
The organics were combined, dried with sodium sulfate, and concentrated. The
residue
was purified by flash column chromatography using 15% ethyl acetate/hexanes
followed
by preparative TLC using 1:1 dichloromethane/Hexanes to obtain 24.3 mg (7%) of
P-119
as a light-yellow oil. 'H NMR (400 MHz, CDC13) 3.81 (s, 3 H) 3.94 (s, 2 H)
5.07 (br s,
1 H) 6.55 (dd, J=9.8, 2.6 Hz, 1 H) 6.61 (td, J=8.4, 2.55 Hz, 1 H) 6.94 (d,
J=8.3 Hz, 1
H)7.07(dd,J=8.3,6.7Hz,1H)7.17-7.24(m,2H)7.54 (t,J=8.0Hz,1H)7.81(d,J
=7.7 Hz, 1 H) 8.16 (dd, J=8.3, 1.3 Hz, 1 H) 8.38 (t, J=1.8 Hz, 1 H) ppm. LC/MS
=
97.2%, 352.1 (APCI-)
167
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 92. Preparation of P-134
O 0,
I Br HO,B.OH
O
K;PO4 PPh3
I / A DME
0 DOH, H2O N
O
1-70 1-121
[00343] Synthesis of 2-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-benzoic acid
methyl
ester (I-121). Into a 20 mL vial with stir bar was added 1-70 (0.30 g, 0.93
mmol), 2-
methoxycarbonylphenylboronic acid (0.18 g, 1.02 mmol), triphenylphosphine (49
mg,
0.19 mmol), K3PO4 (0.40 g, 1.86 mmol), DME (5 mL), water (0.5 mL), and ethanol
(0.5
mL). N2 gas was bubbled through the stirred reaction for 10 minutes.
Palladium(II)
acetate (21 mg, 0.09 mmol) was added and N2 was bubbled through for an
additional 5
minutes. The vial was capped and the reaction was stirred at 80 C for 18
hours. The
reaction was cooled to room temperature and 5 mL of water and 5 mL of ethyl
acetate
were added. The layers were separated and the aqueous was extracted with ethyl
acetate
(3 x 10 mL). The organics were combined, dried with sodium sulfate, and
concentrated.
The residue was purified by flash column chromatography using 10%
acetone/hexanes to
afford 241.0 mg (69%) of 1-121 as a colorless oil.
0 0, off
DIBAL-H
o THE '-0
N I i N-D
0
1-121 P-134
[00344] [2-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-phenyl]-methanol (P-134).
Into
a 20 mL vial with stir bar was added 1-121 (126.5 mg, 0.34 mmol) and 4 mL dry
THF.
The solution was cooled to 0 C and DIBAL-H (0.84 mL, 0.84 mmol, 1.0 M in
hexane)
was added. The reaction was stirred at 0 C for 30 minutes. Aqueous 1 N HC1(1
mL)
168
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
was added followed by 5 mL of water. The aqueous solution was extracted with
ethyl
acetate (3 x 10 mL). The organic were combined, dried over sodium sulfate, and
concentrated. The product was purified by flash column chromatography using
12%
acetone/hexane to afford 71 mg (66%) of P-134 as a colorless oil. 1H NMR (400
MHz,
CDC13) 1.44 (t, J= 5.9 Hz, 1 H), 3.80 (s, 3 H), 4.10 (s, 2 H), 4.70 (d, J= 5.8
Hz, 2 H),
6.92 (d, J= 8.9 Hz, 1H),7.10-7.16 (m, 2 H), 7.16 - 7.22 (m, 1H),7.27-
7.30(m,2H),
7.39 - 7.45 (m, 1 H), 7.53 (t, J= 8.0 Hz, 1 H), 7.80 (d, J= 7.7 Hz, 1 H), 8.15
(dd, J= 8.2,
1.3 Hz, 1 H), 8.38 (t, J= 1.7 Hz, 1 H). LC/MS = 99.9%, 349.1 (APCI-).
Example 93. Preparation of P-108
N-N\
O I/ LN
O`11
P-108
[00345] Synthesis of 1-(6-Methoxy-3'-methoxymethyl-biphenyl-3-ylmethyl)-1H-
[1,2,4]triazole (P-108). P-108 was synthesized from 1-110 (200 mg, 0.746 mmol)
and 3-
methoxymethylphenylboronic acid (136 mg, 0.821 mmol) using the same conditions
as
P-08 1. The reaction was cooled to room temperature and the solvent removed
under
vacuum. The residue was suspended in ethyl acetate (30 mL), washed with water
(30
mL), the aqueous wash extracted with ethyl acetate (30 mL), and the organic
extracts
combined. The organic extracts were washed with water (3 x 30 mL), saturated
aqueous
ammonium chloride (2 x 30 mL), brine (30 mL), decolorized with activated
carbon,
dried over sodium sulfate, and the solvent removed under vacuum. The product
was
purified by flash silica gel column chromatography eluting with 0-25% acetone
in
dichloromethane, and was run on a silica gel preparatory plate eluting with 1%
acetone
in dichloromethane for three developments to obtain P-108 (49.0 mg, 19% yield)
as a
yellow oil.
[00346] 1H NMR (CDC13) d: 8.444 (s, 1H), 7.849 (s, 1H), 7.44-7.354 (m, 4H),
7.295-
169
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
7.280 (m, 2H), 7.104-7.086 (m 1H), 5.435 (s, 2H), 4.472 (s, 2H), 3.805 (s,
3H), 3.350 (s,
3H).
[00347] LCMS =100.0% purity. MS(APCI+) =310.1 (M+1).
Example 94. Preparation of P-495
O N
O=O
P-495
[00348] Synthesis of 1-(3'-Methanesulfonyl-6-methoxy-biphenyl-3-ylmethyl)-1H-
[1,2,4]triazole (P-495). P-495 was synthesized from 1-110 (200 mg, 0.746 mmol)
and 3-
methanesulfonylphenylboronic acid (136 mg, 0.821 mmol) by a similar proceedure
to P-
081 . Upon completion, the residue was suspended in ethyl acetate (20 mL),
washed
with (20 mL), and the aqueous wash was extracted with ethyl acetate (2 x 30
mL). The
organic extracts were combined and washed with water (3 x 50 mL), saturated
aqueous
ammonium chloride (2 x 50 mL), and brine (50 mL), dried over sodium sulfate,
and the
solvent removed under vacuum. The impure product was purified by flash silica
gel
column chromatography eluting with 5-15% acetone in dichloromethane, and by
separation on a silica gel preparatory plate eluting with 5% acetone in
dichloromethane
to give P-495 (145.0 mg, 57% yield). 1H NMR (400 MHz, CDC13) d: 8.09-8.08 (m,
2H),
7.97 (s, 1H), 7.97-7.90(m, 1H), 7.79-7.77 (m, 1H), 7.60 (t, J = 6.4 Hz, 1H),
7.32-7.26 (m,
2H), 7.01 (d, J = 6.8 Hz, 1H), 5.34 (s, 2H), 3.83 (s, 3H), 3.09 (s, 3H).
[00349] LCMS =100.0% purity. MS(APCI+) =344.0 (M+1).
170
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 95. Preparation of P-163.
[00350] Scheme 30.
HO, B.OH
O O
NaNO2/Cu(I)CN TES/TFA I \ I \ "
/ I / I / F O F Pd(OAc),
O HZ F HCI O \N N KZCO3, PPh3
Br N Br
Br
Dioxane
1-122 EtOH-H20
1-123 1-124
O F
N~
O
P-163
[00351] Synthesis of 2-bromo-6-(4-fluoro-benzoyl)-3-methoxy-benzonitrile (I-
123).
To a cooled (0 C) solution of (2-amino-3-bromo-4-methoxy-phenyl)-(4-fluoro-
phenyl)-
methanone (I-122, 0.32 g, 1.0 mmol) in con. HC1(1.5 mL) was added a solution
of
NaNO2 (0.065 g, 0.95 mmol). The reaction mixture was stirred for 10 min at 0
C, then
added to a suspension of Cu(I)CN (0.031 g, 1.2 mmol) in water (0.5 mL) and
toluene (1
mL) over 5 min at 0 T. The reaction mixture was slowly warmed to room
temperature,
stirred at room temperature for 2 h, then at 50 C for 30 min. The reaction
was extracted
with dichloromethane (3 x 5 mL), and the combined organic extracts were washed
with
brine, dried with Na2SO4, filtered, and concentrated under vacuum. The residue
was
purified by silica gel column chromatography using 1:1 dichloromethane-hexanes
then
dichloromethane to afford I-123 (0.201 g, 61% yield) as white solid.
[00352] Synthesis of 2-bromo-6-(4-fluoro-benzyl)-3-methoxy-benzonitrile (I-
124): To
a solution of I-123 (0.15 g, 0.45 mmol) in trifluoroacetic acid (1.0 mL) was
added
triethylsilane (0.55 g, 4.5 mmol). The reaction mixture was stirred at room
temperature
for 20 h, then concentrated under vacuum to afford I-124 (0.14 g, 94% yield)
as white
solid.
[00353] Synthesis of 3-(4-fluoro-benzyl)-6-methoxy-3'-nitro-biphenyl-2-
carbonitrile
171
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
(P-163): To 1-124 (0.14 g, 0.42 mmol), 3-nitrophenylboronic acid (0.07 g, 0.44
mmol),
PPh3 (0.05 g, 0.21 mmol), K2C03 (0.02 g, 0.16 mmol) and Pd(OAc)2 (0.01 g, 0.06
mmol)
was added dioxane (10 mL) and EtOH-H20 (1:1, 5 mL). The reaction was degassed
with an Argon stream for 5 min. The reaction was then stirred at 85 C under
Ar for 18 h.
The reaction was cooled to room temperature, concentrated under vacuum, and
H2O (40
mL) and dichloromethane (40 mL) were added. The organic layer was separated
and the
aqueous layer was extracted with dichloromethane (2 x 25 mL). The combined
organic
extracts were dried with Na2SO4, filtered, and concentrated under vacuum. The
residue
was purified by silica gel column chromatography using dichloromethane to
afford P-
163 (0.07 g, 46% yield) as an off-white solid. 1H NMR (CDC13, 400MHz): 8.12-
8.26 (m,
2 H), 7.54-7.62 (m, 2 H), 7.1-7.2 (m, 3 H), 6.9-7.04 (m, 2 H), 6.86 (d, J= 8.4
Hz, 1 H),
4.07 (s, 2 H), 3.71 (s, 3 H); MS(APCI+): 383.1 (M+1).
Example 96. Preparation of P-169
N -N
N' N~~
0 LN Hyrodgen Peroxide (30% in water) '-0 N
Glacial Acetic Acid Nz~
I / S~ Si
11
0
P-085 P-169
[00354] Synthesis of 1-(3'-Methanesulfinyl-6-methoxy-biphenyl-3-ylmethyl)-1H-
[1,2,4]triazole (P-169). A 30% hydrogen peroxide by weight in water solution
(1 mL)
was diluted with glacial acetic acid (9 mL). A solution of P-085 (38.1 mg,
0.111 mmol)
in glacial acetic acid (300 uL) was stirred at room temperature, and the
hydrogen
peroxide solution was added (107 uL total solution, 3.78 mg hydrogen peroxide,
0.111
mmol) dropwise. The reaction was stirred for 1 h at room temperature.
Following
completion, solid sodium carbonate was added (-100 mg) to the mixture. The
reaction
was diluted with water (500 uL), and extracted with ethyl acetate (3 x 1 mL).
The
organic extracts were combined, dried over sodium sulfate, and the solvent
removed
under vacuum. The residue was dried under high vacuum overnight to give P-169
(25.6
mg, 70% yield).
[00355] 1H NMR (400 MHz, CDC13) 8.08 (s, 1H), 7.97 (s, 1H), 7.79-7.78 (m, 1H),
172
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
7.64-7.54 (m, 3H), 7.31-7.28 (m, 2H), 7.00 (d, J = 8.40 Hz, 1H), 5.33 (s, 2H),
3.83 (s,
3H), 2.76 (s, 3H) ppm. LCMS = 96.4% purity. MS(APCI+) = 328.1 (M+1).
Example 97. Preparation of P-530
0 0
Fe, NH4CI
O gF N.O O ~ ~ F NHz
0 EtOH, H2O
CI CI
1-126 1-127
[00356] Synthesis of (4-Amino-phenyl)-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-
yl)-methanone (I-127). A suspension of I-126 (108 mg, 0.28 mmol), Fe powder(55
mg,
0.98 mmol), NH4C1(75 mg, 1.40 mmol), in ethanol (3 mL), and water (1 mL) was
stirred
for 2 hours at 80 C. Upon completion the reaction was filtered through
Celite, washed
with ethyl acetate, and concentrated under vacuum. The residue was taken up in
ethyl
acetate and washed with saturated aqueous NaHCO3 . The combined extracts were
concentrated under vacuum to afford compound 1-127 (90 mg, 90% yield) as a
yellow
gum.
0
O
O
Ac
20 O F N
O F NH2 H
pyridine Nz~
CI
CI
1-127 1-128
[00357] Synthesis of N-[4-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-carbonyl)-
phenyl]-acetamide (I-128).A solution of 1-127 (88 mg, 0.25 mmol) in pyridine
(2mL),
and acetic anhydride (35 uL, 0.37 mmol) was stirred at room temperature for 2
hours.
The reaction was diluted with ethyl acetate, and was washed with IN aqueous
HC1. The
combined extracts were dried over Na2SO4 and concentrated to a solid which was
triturated with diethyl ether to give I-128 (21 mg, 21% yield) as a white
solid.
173
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
0
OH
O
O
O F N
O F H I
Nz~ H
CI CI
1-128 P-530
[00358] Synthesis of N-{4-[1-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-yl)-1-
hydroxy-ethyl]-phenyl}-acetamide (P-530). A mixture of 1-128 (18.4 mg, 0.046
mmol)
and methylmagnesium bromide (46 uL, 0.14 mmol, 3.0M in ether) in THE (1 mL)
was
stirred at room temperature for 10 min, The reaction was quenched by the
addition of
saturated aqueous NH4C1.. The reaction was extracted with ethyl acetate, and
the
combined extracts washed with brine, dried over Na2SO4, and concentrated under
vacuum. The residue was filtered through Si02 to remove any remaining
inorganic
impurities to give P-530 (4.6 mg, 24% yield) as a white solid. 'H NMR (400
MHz,
,DMSO-d6) 9.85 (s, 1 H), 7.70 (t, J= 9.1 Hz, 1 H), 7.50 - 7.34 (m, 4 H), 7.29 -
7.21 (m,
3 H), 7.21 - 7.12 (m, 1 H), 6.97 (d, J= 8.9 Hz, 1 H), 3.74 (s, 3 H), 2.00 (s,
3 H), 1.82 (s,
3 H)
[00359] Purification of P-560 and P-561
OH OH HO
O F H O F H + O F H
CI
CI CI
P-530 P-560 P-561
[00360] The enantiomers of P-530 were separated by semi-preparative chiral
HPLC.
The sample for injections was dissolved in warm EtOH. The column used was a
Chiralpack AD (250 X 20 mm, 10 um). The mobile phase was 15% EtOH, 85% hexane
(with 0.2% DEA), isocratic at 9.9 mL/min. The injection volume was 385 uL and
the
174
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
fractions were collected by manually changing fractions. The run time was 60
minutes
and the UV detection was set at 254 nm. The retention time of the first
enantiomer
collected was 33.2 min which is P-560, and the second enantiomer was 41.5 min
which
is P-561.
[00361] The chiral purity of each fraction was then determined by analytical
scale
chiral HPLC using a Chiralpack AD column (250 x 4.6 mm, 5um). The mobile phase
was 15% EtOH, 85% hexane (with 0.2% DEA), isocratic at 1.0 mL/min. The
injection
volume was 10 uL, the run time was 30 min, and the UV detection was set at 254
nm. A
sample of the original racemic mixture was injected and the retention times
were 12.3
and 14.9 minutes, respectively. The retention time of P-560 was 12.3 minutes
and the
enantiomeric excess was determined to be 100.0%. The retention time of P-561
was
15.0 minutes and the enantiomeric excess was determined to be 99.2%.
[00362] The absolute stereochemistry of P-560 and P-561 is not known. The
stereochemistry of the structures was drawn arbitrarily.
[00363] N- {4-[(R)-1-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-yl)-1-hydroxy-
ethyl]-
phenyl}-acetamide (P-560).
[00364] 'H NMR (400 MHz ,DMSO-d6) 9.85 (s, 1 H), 7.70 (t, J= 9.1 Hz, 1 H),
7.44
(d,J=8.7Hz,2H),7.41-7.37(m,2H),7.29-7.22(m,3H),7.19-7.14(m,1H),6.97
(d, J= 8.9 Hz, 1 H), 5.67 (br. s., 1 H), 3.74 (s, 3 H), 2.00 (s, 3 H), 1.82
(s, 3 H)
[00365] N-{4-[(S)-1-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-yl)-1-hydroxy-
ethyl]-
phenyl}-acetamide (P-561).
[00366] 'H NMR (400 MHz ,DMSO-d6) 9.85 (s, 1 H), 7.70 (t, J= 9.1 Hz, 1 H),
7.44
(d,J=8.6Hz,2H),7.41-7.36(m,2H),7.29-7.21(m,3H),7.20-7.15(m,1H),6.97
(d, J= 8.9 Hz, 1 H), 5.67 (br s, 1 H), 3.74 (s, 3 H), 2.00 (s, 3 H), 1.82 (s,
3 H)
175
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 98. Preparation of P-547
[00367] Scheme 31.
HO- OH
O
(~Ci
0-9
I CI
I-16a
O D
II D
~-O ~O
O N O N
O O
CI 1-129 Cl 1-130
D D
kD D
O NH2 O NNH2
H
CI 1-131 CI P-547
[00368] Synthesis of 3'-Chloro-2-methoxy-biphenyl (1-16). A suspension of 2-
iodoanisol (3.00 g, 12.8 mmol), 3-chlorophenylboronic acid (2.41 g, 15.4
mmol), and
potassium carbonate (3.54 g, 25.6 mmol) in water (10 mL) and methanol (50 mL)
was
purged with a nitrogen stream for 20 min. To the suspension was added
palladium(II)
acetate (57.6 mg, 0.2564 mmol) and the solution was stirred at room
temperature
overnight (18 h). To the reaction was added ethyl acetate (100 mL) and water
(100 mL).
The layers were separated, the aqueous layer extracted with ethyl acetate (100
mL), and
the organic extracts were combined. The organic solution was washed with water
(2 x
150 mL) and brine (150 mL), decolorized with activated charcoal, dried over
sodium
sulfate, and the solvent removed under reduced pressure. The resulting residue
was
purified by flash silica gel column chromatography eluting with 10-30% ethyl
acetate in
hexanes to give I-16 ( 2.50 g, 89% yield) as colorless oil. 1H NMR (400 MHz
CDC13)
7.53 (t, J = 1.8 Hz, 1H), 7.40 (dt, J = 7.2 Hz, 1.6 Hz, 1H), 7.38-7.28 (m,
4H), 7.05-6.97
(m, 2H), 3.82 (s, 3H).
176
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00369] Synthesis of (3'-Chloro-6-methoxy-biphenyl-3-yl)-(4-nitro-phenyl)-
methanone (1-129). A solution of I-16a (1.50 g, 6.86 mmol) in nitrobenzene (6
mL) was
cooled to 0 C in an ice water bath. Aluminum trichloride (1.10 g, 8.23 mmol)
was added
portionwise, and the solution stirred at 0 C for 1 h. To the solution was
added, 4-nitro-
benzoylchloride (1.53 g, 8.23 mmol) and the reaction was stirred for 20 h
allowing
warming to room temperature. The solution was poured into 100 mL of an ice-
water
mixture and stirred for 2 h. The yellow oil was extracted into ethyl acetate
(2 x 100 mL),
and the organic extracts combined. The extracts were washed with saturated
aqueous
sodium bicarbonate (100 mL), water (2 x 150 mL), and brine (150 mL), dried
over
sodium sulfate, and the solvent removed under vacuum to give crude 1-129 The
resultant
oil was purified by flash silica gel column chromatography (10-33% ethyl
acetate in
hexanes) to give 1-129 (1.66 g, 66% yield).
[00370] 1H NMR (400 MHz CDC13) d: 8.35-8.34 (m, 2H), 7.92-7.91 (m, 2H), 7.84-
7.81 (m, 2H), 7.51-7.51 (m, 1H), 7.37-7.34 (m, 3H), 7.08 (d, J = 6.8 Hz, 1H),
3.94 (s,
3H).
MS(APCI+) = 304.1 (M - 63.0)
[00371] Synthesis of 3'-Chloro-2-methoxy-5-[bisdeutero-(4-nitro-phenyl)-
methyl]-
biphenyl (I-130). A solution of 1-129 (500 mg, 1.36 mmol) in dichloromethane
(10 mL)
was purged with nitrogen. To the solution was added duetro-trifluoroacetic
acid (3.91 g,
34.0 mmol) and the resultant orange solution was cooled to 0 C in a ice water
bath. To
the solution was slowly added sodium borodeuteride (569 mg, 13.6 mmol) portion
wise
over 45 min. The reaction was stirred overnight allowing the mixture to warm
to room
temperature. The reaction was basified to pH 9 with saturated aqueous sodium
bicarbonate, and extracted into ethyl acetate (75 mL). The extract was washed
with water
(2 x 50 mL) and brine (50 mL), dried over sodium sulfate, and the solvent
removed
under vacuum. The crude material was purified by flash silica gel column
chromatography (10% ethyl acetate in hexanes) to give I-130 (500 mg,
quantitative
yield.)
[00372] 1H NMR (400 MHz CDC13) 8.16-8.14 (m, 2H), 7.489-7.48 (m, 1H), 7.37-
7..26 (m, 5H), 7.13 (dd, J = 8.4 Hz, 2.4 Hz, 1H), 7.10 (d, J = 2.4 Hz, 1H),
6.93 (d, J =
177
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
8.4 Hz, 1H), 3.80 (s, 3H).
[00373] Synthesis of 4-[Bisdeutero-(3'-chloro-6-methoxy-biphenyl-3-yl)-methyl]-
phenylamine (I-131). A suspension of 1-130 (440 mg, 1.24 mmol), iron powder
(241 mg,
4.33 mmol), and ammonium chloride (337 mg, 6.31 mmol) in ethanol (5 mL) and
water
(1.6 mL) was stirred at room temperature for 1 h and at 100 C for 30 min. The
ethanol
was removed under reduced pressure, the reaction diluted with water (50 mL),
and
extracted with ethyl acetate (2 x 50 mL). The combined extracts were washed
with water
(2 x 50 mL) and brine (50 mL), dried over sodium sulfate, and the solvent
removed
under vacuum to give I-131 (279 mg, 70% yield) as an orange oil.
[00374] 1H NMR (400 MHz CDC13) d: 7.43 (m, 1H), 7.31-7.19 (m, 3H), 7.07-7.04
(m, 2H), 6.93-6.910 (m, 2H), 6.82 (d, J = 8.4 Hz, 1H), 6.57-6.55 (m, 2H), 3.72
(s, 2H),
3.50 (s, 2H).
[00375] Synthesis of {4-[Bisduetero-(3'-chloro-6-methoxy-biphenyl-3-yl)-
methyl]-
phenyl}-urea (P-547). A suspension of 1-131 (270 mg, 0.829 mmol) and sodium
cyanate
(107.7 mg, 1.66 mmol) in water (15 mL) and glacial acetic acid (7.5 mL) was
stirred at
room temperature over night. To the solution was added aqueous saturated
sodium
bicarbonate (10 mL), and the reaction was extracted with ethyl acetate (50 mL,
25 mL).
The extracts were combined, washed with water (50 mL) and brine (50 mL), dried
over
sodium sulfate, and the solvent removed under vacuum to give P-547 as an
orange gum.
The material was purified by flash silica gel column chromatography (eluting
with 10-
25% ethyl acetate in dichloromethane) and recrystallized by in water (10 mL)
and
isopropanol (8 mL) to give P-547 (111 mg, 36% yield) as a white powder.
[00376] 1H NMR (400 MHz, d6-DMSO): 8.40 (s, 1H), 7.48-7.36 (m, 4H), 7.29-7.27
(m, 2H), 7.20-7.10 (m, 2H0, 7.09-7.08 (m, 2H), 7.03 (d, J = 8.8 Hz, 1H), 5.75
(s, 2H),
3.74 (s, 3H).
LCMS = 96.9% purity. MS(APCI+) = 369.1 (M+1), 326.1 (M-42.0).
HPLC (254 nm); 96.6%. [Mobile Phase A and Mobile Phase B = Water and
Acetonitrile, Symmetry C18, (250 x 4.6 mm, 5 um), Flow=1.0 mL/min, Inj. Wash =
ACN, Inj. Vol.= 10 uL. Retention time = 28.37 min]
178
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00377] Example 99. Preparation of P-537, P-538, and P-539.
[00378] Scheme 32
O HO D
O F+ I 0 AICI3, 0 F N NaBD4 0 F N
O
O CI nitrobenzene O
THF/D20
CI CI P-537
6~cl
1-132 1-133
Fe/NH4CI/ H2/Pd/C/EA
O
HO\ D
O F NH2 i
O F NH2
CI I-134
CI
o
N
py/THF P-538
O HO D
0
O NaBD4
O F N U NH2 THF/D20 O F H~NH2
CI
CI
P-434
P-539
[00379] Synthesis of (3'-chloro-2-fluoro-6-methoxy-biphenyl-3-yl)-(4-nitro-
phenyl)-
methanone (1-133). To a solution of 4-nitro-benzoyl chloride (1900 mg, 10.2
mmol) in
nitrobenzene (6 mL) was added aluminum trichloride (1360 mg, 10.2 mmol) at 0
T.
The mixture was allowed to warm to room temperature and stirred at room
temperature
for 1 h, cooled to 0 C, and 3'-chloro-6-fluoro-2-methoxy-biphenyl (2.01 g,
8.5 mmol) in
nitrobenzene (1 mL) was added at 0 T. The reaction mixture was allowed to warm
to
room temperature and stirred 24 h. The reaction mixture was cooled to -10 C
and
quenched with ice-water (50 mL), extracted with ethyl acetate (2 x 25 mL),
washed with
water (2 x 10 mL), saturated aqueuos sodium bicarbonate (10 mL), brine (30 mL)
, and
dried over Na2SO4.. After filtration and removal of solvent, the crude was
purified by
crystallization from ether-hexane to give 1-133 (3.00 g, 91% yield)..
179
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00380] Synthesis of deutero-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-yl)-(4-
nitro-
phenyl)-methanol (P-537). To a mixture of 1-133 (500 mg, 1.25 mmol) in THE-D2O
(1:1, 20 mL) was added NaBD4 (190 mg, 3.1 mmol) at 0 T. The reaction mixture
was
allowed to warm to room temperautre and stirred for 24 h. The mixture was
poured into
ice-water (100 mL), neutralized with aqueous saturated NH4C1(5 mL), extracted
with
ethyl acetate (3 x 30 mL), washed with water (20 mL), brine (30 mL) and dried
over
Na2SO4. After filtration and removal of solvent, the residue was purified by
silica gel
column chromatography with ethyl acetate-hexane as eluent to give P-537 (350
mg, 70%
yield).
[00381] 1H NMR (CDC13 ,400 MHz) 8.21 (d, J = 8.8 Hz, 2 H), 7.62 (d, J = 8.8
Hz, 2
H), 7.23-7.37 (m, 5 H), 6.78 (dd, J = 8.8, 1.2 Hz, 1 H), 3.77 (s, 3 H), 2.39
(s, 1 H).
Cale. 388.8; APCI+(M-OH): 371.1, 100%.
[00382] Synthesis of deutero-(4-amino-phenyl)-(3'-chloro-2-fluoro-6-methoxy-
biphenyl-3-yl)-methanol (P-538). To a mixture of P-537 (300 mg, 0.7 mmol) in
ethyl
acetate (10 mL) was added Pd/C (10%, 450 mg, 4 mmol) at room temperature. The
reaction vessle was sealed and the mixture was shacken under a Hydrogen
atmosphere
(30 psi) for 60 min. The solid was filtered, and the filtrate was concentrated
to give crude
product. The crude was purified by silica gel column chromatography with ethyl
acetate-
hexane as eluent and then was further purified by a preparation TLC to yield P-
53 8 (45
mg, 16 % yield).
[00383] 1H NMR (CDC13,400 MHz) 7.46 (t, J = 8.4 Hz, 1 H), 7.23-7.37 (m, 4 H),
7.18 (d, J = 8.4 Hz, 2 H), 6.77 (dd, J = 8.8, 1.2
Hz,1H),6.55(d,J=8.4Hz,2H),3.77
(s, 3 H), 2.10 (s, 1 H). Calc.358.8; APCI+(M-OH): 341.1, 91%.
[00384] Synthesis of (4-amino-phenyl)-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-
yl)-
methanone (I-134). To a solution of I-133 (500 mg, 1.2 mmol) in EtOH-H20 (1:1,
15
mL) was added solid NH4C1(200 mg, 4 mmol) and iron powder (150 mg, 3 mmol),
and
the reaction was stirred at room temperature for 72 h. The mixture was poured
into water
(50 mL), extracted with ethyl acetate (3 x 30 mL), washed with water (20 mL)
and brine
(30 mL), and dried over Na2SO4, and filtrered. The solvent was rermoved under
vacuum
180
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
to yield 1-134 (380 mg, 82% yield).
[00385] Synthesis of [4-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-carbonyl)-
phenyl]-
urea (P-434). To a solution of 1-134 (220 mg, 0.7 mmol) in pyridine (1 mL) and
THE (4
mL) was added trimethylsilylisocyanate (1 mL, excess) and the reaction stirred
at room
temperature for 36 h. The mixture was poured into 25 mL ice-water solution. To
the
suspension was added saturated aqueous sodium bicarbonate (5 mL), and the
resultant
mixture was stirred at room temperature for 2 h. The mixture was extracted
with ethyl
acetate (3 x 30 mL), washed with water (20 mL), and brine (30 mL), dried over
Na2S04,
and filtred. The solvent was removed under vacuum, and the crude product was
purified
by silica gel column chromatography with ethyl acetate-hexane as eluent to
yield P-434
(50 mg, 20 % yield).
[00386] Synthesis of {4-[(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-yl)-hydroxy-
deutero-methyl]-phenyl} -urea (P-539). To a mixture of P-434 (50 mg, 0.13
mmol) in
THE (5 mL) and D20 (2 mL) was added NaBD4 (50 mg, 0.25 mmol) at 0 T. The
reaction mixture was allowed to warm to room temperature and stirred for 24 h.
The
reaction mixture was poured into 20 mL ice-water, neutralized with NH4C1(sat.
2 mL),
extracted with ethyl acetate (3x10 mL), and washed with water (10 mL) and
brine (10
mL) . The organic layer was dried over Na2S04, filtered, and the solvent
removed under
vacuum.. After removal of solvent, the residue was purified by a preparation
TLC to
give P-539 (12 mg, 26% yield). 1H NMR (DMSO-d6,400 MHz) d: 8.45 (s, 1 H), 7.49
(t,J=8.8Hz,1H),7.41-7.44(m,2H),7.24-7.34(m,4H),7.17(d,J=8.8Hz,1H),
6.97 (d, J = 9.2 Hz, 1 H) 5.79 (s, 1 H), 3.73 (s, 3 H) ppm. Calc.401.85;
APCI+(M-OH):
384.1, 98.6%.
Example 100. Preparation of P-541
O
KZC03
HO N ~ NHZ O H~NH2
H Acetone, EtI
CI
CI
P-421 P-541
181
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00387] [4-(3'-Chloro-6-ethoxy-biphenyl-3-ylmethyl)-phenyl]-urea (P-541). A
suspension of
P-421 (66 mg, 0.19 mmol), K2C03 (39 mg, 0.28 mmol), and EtI (23 uL, 0.28 mmol)
in acetone
(2 mL) was stirred for 16 hours at 50 C. An additional 50 uL of EtI and 80 mg
of K2C03 were
added amd the reaction was stirred for 4 It at 50 C. To the reaction was
added 80 mg of Cs2CO3
and 50 uL of EtI, the reaction stirred 2 It at 60 C, and the reaction was
concentrated. The crude
product was purificated by flash column chromatography eluting with 20% - 30%
Acetone/DCM
afforded the title compound P-541 (37 mg, 51 %) as a white solid. 'H NMR (400
MHz,DMSO-
d6) 6 = 8.40 (s, 1 H), 7.53 (s, 1 H), 7.45 - 7.39 (m, 2 H), 7.39 - 7.34 (m, 1
H), 7.28 (d, J= 8.3 Hz,
2 H), 7.19 - 7.13 (m, 2 H), 7.09 (d, J= 8.5 Hz, 2 H), 7.01 (d, J= 8.1 Hz, 1
H), 5.75 (s, 2 H), 4.01
(q, J= 7.0 Hz, 2 H), 3.82 (s, 2 H), 1.31 - 1.18 (m, 3 H). LC/MS = 96.5%, 381.1
(APCI+).
Example 101. Preparation of P-542
I I x o
Cs2c03 _ H N I I
HO N NHZ 2 O H~NHZ
H Acetone
CI
CI
P-421 P-542
[00388] {4-[6-(2-Amino-ethoxy)-3'-chloro-biphenyl-3-ylmethyl]-phenyl}-urea (P-
542). A suspension of P-421 (100 mg, 0.28 mmol), 2-bromoethylamine
hydrobromide
(204 mg, 8.8 mmol), Cs2CO3 (600 mg, 18.4 mmol), and sodium iodide (20 mg) in
acetone (10 mL) was stirred at reflux for 20 h. The reaction mixture was then
portioned
between dichloromethane and water, and the organic layer was extracted with IN
NaOH
followed by 1 N HCl. The acidic extract was adjusted to pH 10 by addition of
NaOH
followed by extraction with dichloromethane. The solvent was removed in vacuo
and
the residue was chromatographed with dichloromethane:methanol (9:1) to yield P-
542 (9
mg, 8.1% yield) as a solid.
[00389] 1H NMR (CDC13) 7.50 - 7.52 (m, 1H), 7.28 - 7.32 (m, 2H), 7.17 - 7.23
(m,
3H),7.11-7.13(m,2H),7.09-7.11(m,1H),6.89-6.92(m,2H), 3.96 (t, J=5Hz,2H),
3.93 (s, 2H), 2.99 (t, J = 5 Hz, 2H), APCI (M+1; 396.1) LCMS 95%.
182
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 102. Preparation of P-543
/ I I \ KzC03 O 0
HO \ Q H N A, NH2 O / H~NHz
DMF
CI
CI
P-421 P-543
[00390] {4-[3'-Chloro-6-(tetrahydro-furan- 2-ylmethoxy)-biphenyl-3-ylmethyl]
phenyl}-urea (P-543). A mixture of phenol P-421(100 mg, 0.28 mmol),
tetrahydrofurfuryl bromide (50 mg, 0.28 mmol), and K2CO3 (78 mg, 0.56 mmol) in
DMF
(2 mL) was heated at 80 C for 24 h. To this mixture, K2CO3 (78 mg), NaI (10
mg), and
tetrahydrofurfuryl bromide (50 mg) were added and the reaction was run at 100
C for 6
h. The mixture was cooled to room temperature and patitioned between
dichloromethane
and water, the dichloromethane layer was washed with brine, dried over Na2SO4,
filtered, and concentrated. The residue was purified by preparative plate with
dichloromethane:methanol (95:5) to yield P-543 (55 mg, 45% yield).
[00391] 'H NMR (400 MHz, DMSO-d6) 8.39 (s, 1H), 7.58 (s, 1H), 7.37 - 7.43 (m,
3H), 7.28 (d, J= 7.2Hz, 1H), 7.14 - 7.17 (m, 2H), 7.08 (d, J= 7.2 Hz, 2H),
7.02 (d, J=
7.2 Hz, 1H), 4.07 (m, 1H), 3.94 (t, J= 5 Hz, 2H), 3.82 (s, 2H), 3.6- 3.69 (m,
2H), 1.88-
1.93 (m,1H),1.73-1.77(m,2H),1.62-1.69(m,1H).
APCI (M+1; 437) LCMS 93.5%;
Example 103. Preparation of P-548 and P-557
183
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00392] Scheme 33.
p IR p
HO I I NANH z Cesium Carbonate p I I / N)~ NH
H + Br^R H z
Acne
A
\ CI R = CH2N(CH3)2 CI
R = CONH2
P-548 R = CH2N(CH3)2
P-421 P-557 R = CONH2
iN
O I I NNH
H z
CI
P-548
[00393] Synthesis of {4-[3'-Chloro-6-(2-dimethylamino-ethoxy)-biphenyl-3-
ylmethyl]-phenyl}-urea (P-548). A suspension of P-421 (100 mg, 0.283 mmol),
dimethylaminoethylbromide hydrobromide (198 mg, 0.850 mmol), and cesium
carbonate
(600 mg, 1.84 mmol) in acetone (10 mL) was stirred at reflux for 17 h. The
suspension
was diluted with dichloromethane (50 mL), washed with 0.5 N aqueous
hydrochloric
acid (100 mL), and the solvent removed under vacuum. The crude orange oil was
purified by preparatory thin layer chromatography (silica gel) eluting with
10% acetone
in dichloromethane followed by eluting with 10% methanol in dichloromethane to
give
P-548 (13.4 mg, 11% yield) as a colorless gum.
[00394] 1H NMR (400 MHz CDC13) d: 7.54 (m, 1H), 7.39-7.36 (m, 1H), 7.31-7.08
(m, 8H), 6.90-6.89 (m, 1H), 6.23 (s, 1H), 4.57 (s, 2H), 4.05 (t, J = 5.8 Hz,
2H), 3.92 (s,
2H), 2.68 (t, J = 5.60 Hz, 2H), 2.27 (s, 6H).
LCMS = 97.3% purity. MS(APCI+) = 424.2 (M+1)
184
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
IOI
H2N\ r O NNH2
O
CI
P-557
[00395] Synthesis of 2-[3'-Chloro-5-(4-ureido-benzyl)-biphenyl-2-yloxy]-
acetamide
(P-557). A suspension of P-421 (150 mg, 0.425 mmol), 2-bromoacetamide (175 mg,
1.28
mmol), and cesium carbonate (900 mg, 2.76 mmol) in acetone (15 mL) was stirred
at
reflux overnight. The solvent was removed under a nitrogen stream, and the dry
residue
was suspended in ethyl acetate (50 mL). The organic solution was washed with
water (50
mL) and brine (15 mL), dried over sodium sulfate, decanted, and the solvent
removed
under vacuum to give crude product. The solid was triturated in a mixture of
dichloromethane (5 mL), methanol (5 mL), and acetone (2 mL) to give P-557
(58.9 mg,
34% yield) as a white powder.
[00396] 1H NMR (400 MHz, DMSO-d6) 8.40 (s, 1H), 7.65 (t, J= 2.0 Hz, 1H), 7.52-
7.50 (m, 1H), 7.41 (t, J= 7.2 Hz, 1H), 7.43-7.37 (m, 2H), 7.28 (d, J= 8.4 Hz,
1H), 7.20-
7.15 (m, 3H), 7.09 (d, J= 8.4 Hz, 1H), 6.90 (d, J= 8.0 Hz, 1H), 5.75 (s, 2H),
4.42 (s,
2H), 3.83 (s, 2H). LCMS = 93.63% purity. MS(APCI+) = 410.1 (M+1).
Example 104. Preparation of P-554
\ I I/ x :::O3 bocN3 / I I O H N O
HO N NH2 O iNH2 3O 1NH2
H H
CI CI CI
P-421 1-136 P-554
3-[3'-Chloro-5-(4-ureido-benzyl)-biphenyl-2-yloxy]-azetidine-l-carboxylic acid
tert-
butyl ester (1-136). A mixture of phenol P-421 (150 mg, 0.42 mmol), 3-
methanesulfonyloxy-azetidine-l-carboxylic acid t-butyl ester (200 mg, 0.79
mmol), and
Cs2CO3 (277 mg, 0.85 mmol) in DMF (2 mL) was heated at 100 C for 18 h. The
185
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
mixture was cooled to room temperature and partitioned between dichloromethane
and
water. The water layer was extracted twice with dichloromethane. The combined
organic
layers were washed with brine, dried over Na2SO4, filtered, and concentrated.
The
residue was purified by preparative plate with dichloromethane:methanol (95:5)
to yield
1-136 (200 mg, quantitative yield) as an oil.
[00397] {4-[6-(Azetidin-3-yloxy)-3'-chloro-biphenyl-3-ylmethyl]-phenyl}-urea
(P-
554). To a solution of 1-136 (200 mg, 0.393 mmol) dichloromethane (2 mL), at 0
C was
added trifluoroacetic acid (0.5 mL). The reaction mixture was stirred at room
temperature overnight. The reaction mixture was concentrated under vacuum,
diluted
with water, saturated aqueous Na2HCO3 (0.5 mL) was added and the mixture was
extracted with dichloromethane (3 x 5 mL). The combined organic layers were
dried
over Na2SO4, filtered, and concentrated under vacuum The residue was purified
by
preparative plate thin layer chromatography eluting with
dichloromethane:methanol
(95:5) to yield P-554 (20 mg, 12 % yield) as a solid.
[00398] 'H NMR (400 MHz, CDC13) 7.51 (t, 1H), 7.38 - 7.41 (m, 1H), 7.27- 7.33
(m,
2H), 7.12 - 7.22 (m, 5H), 7.06 (dd, J= 8.4, 2.4 Hz, 1H), 6.58 (d, J= 8.4 Hz,
1H), 6.22
(br s, 1H), 4.97 (m, 1H), 4.56 (br s, 1H), 3.91 (s, 2H), 3.85- 3.89 (t, 2H),
3.73- 3.75 (m,
2H), 3.48- 3.52 (m, 1H) ppm.
APCI (M+1; 408) LCMS 97%;
Example 105. Preparation of P-553
O
O N II NH2
Sa
H
CI
P-553
[00399] {4-[3'-Chloro-6-(thietan-3-yloxy)-biphenyl-3-ylmethyl]-phenyl}-urea P-
553). Compound P-553 was synthesized by a route analogous to that reported for
P-
554 (Example 104).
186
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00400] 1H NMR (CDC13) 6:7.48 (t, 1H), 7.34 - 7.38 (m, 1H), 7.27- 7.33 (m,
2H),
7.12 - 7.22 (m, 5H), 7.06 (dd, J= 8.4, 2.4 Hz, 1H), 6.73 (d, J= 8.4 Hz, 1H),
6.28 (bs, 1H),
5.19 (m, 1H), 4.59 (bs, 1H), 3.92 (s, 2H), 3.46- 3.53 (t, 2H), 3.23- 3.31 (m,
2H).
APCI (M+1; 425)
Example 106. Preparation of P-555
O
O O NNH2
\~\ H
CI
P-555
[00401] {4-[3'-Chloro-6-(oxetan-3-yloxy)-biphenyl-3-ylmethyl]-phenyl}-urea (P-
555). Compound P-555 was synthesized by a route analogous to that reported for
P-
554.. 1H NMR (DMSO-d6) S 8.39 (s, 1H), 7.57 (t, 1H), 7.38 - 7.48 (m, 3H), 7.28
(d, J
= 8.4Hz, 2H), 7.22 (d, J= 2.4 Hz, 1H), 7.09 - 7.14 (m, 1H), 7.08 (d, J= 8.4
Hz, 2H),
6.62 (d, J = 8.4 Hz, 1H), 5.75 (s, 2H), 5.25 (m, 1H), 4.87 (t, J = 5 Hz, 2H),
4.46 (m,
2H), 3.82 (s, 2H).
APCI (M+1; 409) LCMS 97%.
Example 107. Preparation of P-556
CIO O
NANH2
H
CI
P-556
[00402] 4-[3'-Chloro-6-((S)-1-methyl-pyrrolidin-3-yloxy)-biphenyl-3-ylmethyl]-
phenyl}-urea (P-556). Compound P-556 was synthesized by a route analogous to
that reported for P-554 (Example 104).
1H NMR (400 MHz, CDC13) 7.51 (t, 1H), 7.37 - 7.39 (m, 1H), 7.27- 7.33 (m, 2H),
187
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
7.12-7.22(m,SH),7.06(dd,J=8.4,2.4Hz,1H),6.8 (d, J = 8.4 Hz,1H),6.29(brs,
1H), 4.76 (m, 1H), 4.59 (br s, 1H), 3.91 (s, 2H), 3.02 (m, 1H), 2.6 (t, 2H),
2.35 (s, 3H),
2.17- 2.24 (m, 2H). APCI (M+1; 436)
Example 108. Preparation of P-562
O
a
O I I NH2
CI
P-562
[00403] 4-[3'-Chloro-6-(oxetan-3-yloxy)-biphenyl-3-ylmethyl]-phenylamine (P-
562). Compound P-562 was synthesized by a route analogous to that reported for
P-554 (Example 104).
[00404] 1H NMR (400 MHz, DMSO-d6) 7.56 (t, 1H), 7.57 (t, 1H), 7.37 - 7.48 (m,
3H), 7.18 (d, J= 2 Hz, 1H), 7.01 (dd, J= 8.4, 2 Hz, 1H), 6.88 (d, J= 8.4 Hz,
2H), 6.61
(d, J= 8.4 Hz, 1H), 6.47 (d, J= 8.4 Hz, 1H), 5.75 (s, 2H), 5.25 (m, 1H), 4.93
(br s, 2H),
4.87 (t, J= 5 Hz, 2H), 4.46 (m, 2H), 3.72(s, 2H) ppm. APCI (M+1; 366) LCMS
97%;
Example 109. Preparation of P-563
NH
O
O I I NANH2
H
CI
P-563
[00405] {4-[3'-Chloro-6-((S)-1-pyrrolidin-2-ylmethoxy)-biphenyl-3-ylmethyl]-
phenyl}-urea (P-563). Compound P-562 was synthesized by a route analogous to
188
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
that reported for P-554 (Example 104).
[00406] 1H NMR (400 MHz, DMSO-d6) 9.25 (br s, 1H), 8.77 (br s, 1H), 8.49 (s,
1H),
7.57 (s, 1H), 7.37 - 7.46 (m, 3H), 7.28 (d, J= 8.4 Hz, 1H), 7.19 (overlap,
2H), 7.08 (d,
overlap, J= 8.4 Hz, 3H), 4.09- 4.22 (m, 2H), 3.84 (overlap, 1H), 3.13 (m, 1H),
2.97 (m,
1H), 2.05 (m, 1H), 1.71- 1.83 (m, 3H) ppm.
Example 110. Preparation of P-565
N 0
0 I I NA, NH
H z
CI
P-565
[00407] {4-[3'-Chloro-6-((R)-1-methyl-pyrrolidin-3-yloxy)-biphenyl-3-ylmethyl]-
phenyl}-urea (P-565). Compound P-562 was synthesized by a route analogous to
that reported for P-554 (Example 104).
[00408] 1H NMR (400 MHz, CDC13) 7.51 (t, 1H), 7.35 - 7.38 (m, 1H), 7.23- 7.31
(m,
4H), 7.13 (d, J= 8.4 Hz), 7.11 (s, 1H), 7.06 (dd, J= 8.4, 2.4 Hz, 1H), 6.95
(br s, 1H),
6.78 (d, J= 8.4 Hz, 1H), 4.8 (br s, 2H), 4.75(m, 1H), 3.89 (s, 2H), 3.06 (t,
2H), 2.62 (m,
2H), 2.36 (s, 3H), 2.17- 2.24 (m, 1H), 1.93- 1.98 (m, 1H) ppm. APCI (M+1; 436)
LCMS
98%.
Example 111. Preparation of P-550
189
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00409] Scheme 34.
HO.B3OH
kH I ~ F 0
F I Br Br2/AcOH I Br H2N O F q-~ONJL0 CI3 A
'O i ' O NH 'O H NHz 2 Br (Ph3P)4Pd/KZCO3 Br H (Ph3P)4Pd/KZCO3
Toluene/ Ethanol Toluene/ Ethanol CI
MR 131 MR 132
MR 133
1-137 1-138
P-550
[00410] Synthesis of 1-bromo-5-bromomethyl-3-fluoro-2-methoxy-benzene (1-137).
To a heated (80 C) and stirred solution of 4-bromomethyl-2-fluoro-1-methoxy-
benzene
(1.0 g, 4.57 mmol) in acetic acid (4 mL) was added a solution of bromine (1.09
g, 6.85
mmol) in acetic acid (2 mL) over 30 min. The reaction mixture was stirred at
80 C for
18 h, cooled to room temperature, and poured on to crushed ice-water (50 mL).
Ammonium hydroxide solution (28%) was added to pH 8, extracted with
dichloromethane (2 x 40 mL), washed with water, dried over Na2S04, filtered,
and
concentrated under vacuum. The residue was purified by silica gel column
chromatography using 5% ethyl acetate in hexanes to afford I-137 (0.43 g,
31%yield) as
a viscous liquid.
[00411] Synthesis of 4-(3 -bromo-5 -fluoro-4-methoxy-benzyl)-phenyl] urea (I-
138):
To 1-137 (0.2 g, 0.67 mmol), [4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-
phenyl]-
urea (0.19 g, 0.74 mmol), and Pd(Ph3P)4 (0.04 g, 0.04 mmol), was added toluene
(8 mL),
EtOH (2 mL) and 2 M aqueous Na2CO3 solution (0.7 mL, 1.4 mmol). The suspension
was degassed by bubbling argon gas for 15 min. The reaction was stirred at 60
C under
an argon atmosphere for 24 h. The reaction was cooled to room temperature, and
H2O
(20 mL) and ethyl acetate (30 mL) were added. The layers were separated and
the
aqueous wash extracted with ethyl acetate (2 x 20 mL). The combined organic
extracts
were dried over Na2SO4, filtered, and concentratedunder vacuum. The residue
was
triturated with dichloromethane to afford I-138 (0.18 g, 77% yield) as yellow
solid.
190
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00412] Synthesis of [4-(3'-chloro-5-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
phenyl]-
urea (P-550): To a suspension of I-138 (0.11 g, 0.31 mmol), 3-
chlorophenylboronic acid
(0.05 g, 0.34 mmol) and Pd(Ph3P)4 (0.02 g, 0.02 mmol) was added toluene (6
mL), EtOH
(1.5 mL) and 2 M aqueous NaCO3 solution (0.31 mL, 0.62 mmol). The reaction was
degassed with an argon stream for 15 min. The reaction was stirred at 80 C
under an
argon atmosphere for 18 h. The reaction was cooled to room temperature,and H2O
(20
mL) and ethyl acetate (20 mL) were added. The layers were separated and the
aqueous
wash was extracted with ethyl acetate (2 x 10 mL). The combined organic
extracts were
dried with Na2S04, filtered, and concentrated under vacuum. The residue was
purified by
preparative thin layer chromatography using 8% methanol in dichloromethane to
afford
P-550 (0.064 g, 53% yield) as an off-white solid. 1H NMR (DMSO-d6, 400 MHz):
8.42
(s, 1 H), 7.41-7.54 (m, 4 H), 7.30 (d, J= 8.4 Hz, 2 H), 7.1-7.17 (m, 3 H),
7.08 (s, 1 H),
5.77 (s, 2 H), 3.85 (s, 2 H), 3.64 (s, 3 H); MS(APCI+): 485.1 (M+1), LC-MS:
99.1%;
HPLC 96.8% pure.
Example 112. Preparation of P-558
O
Fe/NH4CI
O N~.O O NH2
O
CI CI
1-129 P-558
[00413] Synthesis of (4-amino-phenyl)-(3'-chloro-6-methoxy-biphenyl-3-yl)-
methanone (P-558). To a solution of 1-129 (500 mg, 1.3 mmol) in EtOH-H20 (1:1,
15
mL) was added NH4C1(200 mg, 4 mmol) and iron powder (150 mg, 3 mmol). The
reaction mixture was stirred at room temperature for 72 h. The mixture was
poured into
water (50 mL), extracted with ethyl acetate (3 x 30 mL), washed with water (20
mL) and
brine (30 mL), dried over Na2SO4, and filtered. The solvent was rermoved under
vacuum to yield P-558 (420 mg, 85% yield). 1H NMR (DMSO-d6 ,400MHz) 7.70 (dd,
J
=8.4,2.4Hz,1H),7.40-7.57(m,7H),7.25(d,J=8.8Hz,1H),6.61(d,J=8.8Hz,2
191
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
H), 6.09 (s, 2 H), 3.88 (s, 3 H) ppm. Calc.337.8; APCI+(M+1): 338, 98%.
Example 113. P-566
O NN 0
H H
CI
P-566
[00414] 1-[4-(3'-Chloro-6-methoxy-biphenyl-3-ylmethyl)-phenyl]-3-(tetrahydro-
furan-2-yl)-urea (P-566). 1H NMR (DMSO-d6,400 MHz): 8.27 (s, 1 H), 7.47 - 7.50
(m,
1H),7.34-7.45 (m, 3 H), 7.27 - 7.32 (m, 2 H), 7.09 - 7.21 (m, 4 H), 7.03 (d, J
= 8.4 Hz,
1 H), 6.71 (d, J = 9.3 Hz, 1 H), 5.52 - 5.48 (m, 1 H), 3.84 (s, 2 H), 3.73 (s,
3 H), 3.70 -
3.79(m,1H),3.62-3.69(m,1H),1.99- 2.11 (m,1H),1.79-1.91(m,2H),1.57-1.69
(m, 1 H) ppm.
Example 114. Preparation of P-523 and P-533
[00415] Scheme 35.
-N 'N O~\rO
N-boc-D-prolinol
O F F Sodium Hydride O F N
Dimethylformamide
A
Cl Cl
1-139
P456
H
~N H O N
N
O F 0 IN Ethyl Isocyanate O
Trifluoroacetic acid
Dichloromethane, Water Pyridine O F
Cl
(ACI
P-523
P-533
[00416] Synthesis of (R)-2-[5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-
ylmethyl)-
pyridin-2-yloxymethyl]-pyrrolidine-l-carboxylic acid tert-butyl ester (I-139).
A mixture
192
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
of N-boc-D-prolinol (225 mg, 1.12 mmol) and sodium hydride (60% weight
dispersion,
66 mg, 1.68 mmol) in dimethylformamide (2 mL) was stirred until gas evolution
ceased.
After 2 min of stirring P-456 (194 mg, 0.56 mmol) was added, and the reaction
heated at
120 C overnight. The reaction was cooled to room temperature, diluted ethyl
acetate (10
mL), washed with water (10 mL), and the aqueous wash extracted with ethyl
acetate (25
mL). The combined extracts were dried over sodium sulfate, filtered, and the
solvent
removed under vacuum. The residue was purified by silica gel column
chromatography
(9:1 hexanes/ethyl acetate) to give I-139 (60.4 mg, 25% yield) as a thick
colorless oil.
[00417] Synthesis of 5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-2-
((R)-1-
pyrrolidin-2-ylmethoxy)-pyridine (P-523). A biphasic solution of I-139 (60 mg,
0.11
mmol) and trifluoroacetic acid (851 mg, 0.75 mmol) in dichloromethane (2 mL)
and
water (0.5 mL) was heated to 80 C for 4 h. The mixture was concentrated under
vacuum, and the residue purified by silica gel column chromatography (9:1
dichloromethane/methanol) to give P-523 (32.4 mg, 69% yield). LCMS = 100%
purity.
[00418] Synthesis of (R)-2-[5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-
ylmethyl)-
pyridin-2-yloxymethyl]-pyrrolidine-1-carboxylic acid ethylamide (P-533). A
solution of
P-523 (235 mg, 0.55 mmol) and ethyl isocyanate (0.2 mL, 2.8 mmol) in pyridine
(1 mL)
was stirred at room temperature overnight. The reaction was diluted with water
(5 mL),
extracted with ethyl acetate (2 x 3 mL), and the extracts combined. The
organic solution
was dried over sodium sulfate, filtered, and the solvent removed under vacuum.
The
residue was purified by silica gel column chromatography (19:1
dichloromethane/methanol) to give P-533 (92.1 mg, 34% yield). A portion of P-
533
(19.0 mg) was futher purified by a sodium bicarbonate wash, and silica gel
preparatory
thin layer chromatography (9:1 dichloromethane/methanol) to give (14.1 mg). 1H
NMR
(400 MHz, DMSO-d6) 1.03 (t, J= 7.2 Hz, 3 H), 1.80 - 1.95 (m, 4 H), 3.07 (m, 2
H), 3.18
(m, 1H), 3.30 (m, 1H), 3.72 (s, 3H), 3.87 (s, 2H), 3.97 (m, 1H), 4.05 (m, 1H),
4.28 (m,
1H), 6.22 (t, J= 5.2 Hz, 1H), 6.76 (d, J= 8.4 Hz, 1 H), 6.93 (d, J= 8.8 Hz, 1
H), 7.26 -
7.32(m,2H),7.36(s,1H),7.39-7.50(m,2H), 7.55(dd,J=8.4,2.2Hz,1H),8.03(d,
J= 2.01 Hz, 1 H) ppm. MS(APCI+): 498.2 (M+1), LC-MS: 92.2%.
193
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 115. Preparation of P-568
N
O F O~ N
DBU
~CI CI
1-140 P-568
[00419] Synthesis of 2-azetidin-l-yl-5-(3'-chloro-6-methoxy-biphenyl-3-
ylmethyl)-
pyridine (P-568): To a mixture of 1-140 (0.15 g, 0.46 mmol) and azetidine
(0.09 g, 0.92
mmol) was added 1,8-diazabicyclo[5.4.0]undec-7-ene (0.35 g, 2.29 mmol). The
reaction
mixture was stirred and at 85 C for 15 min. The reaction was then cooled to
room
temperature, diluted with dichloromethane (6 mL), washed with 0.5 N aqueous
HC1(2 x
4 mL), dried over Na2SO4, filtered, and concentrated. The residue was purified
by silica
gel column chromatography using 3% methanol in dichloromethane followed by
preparative thin layer chromatography using 5% methanol in dichloromethane to
afford
P-568 (0.05 g, 30% yield) as a viscous liquid. 1H NMR (DMSO-d6, 400 MHz): 7.99
(d,
J= 1.6 Hz, 1 H), 7.34-7.49 (m, 5 H), 7.14-7.2 (m, 2 H), 7.04 (d, J= 8.8 Hz, 1
H), 6.28
(dd, J= 8.4, 0.8 Hz, 1 H), 3.86 (t, J= 7.2 Hz, 4 H), 3.77 (s, 2 H), 3.74 (s, 3
H), 2.22-
2.32 (m, 2 H); MS(APCI+): 365.1 (M+1), LC-MS: 100%.
Example 116. Preparation of P-571
1, 1 N OH
O % F' N IBCF/NMM O F I `NN NH
2
CI NH4OH
CI
MR 128
MR 140
1-141
P-571
[00420] Synthesis of 1-[5-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-2-yl]-azetidine-2-carboxylic acid amide (P-571): To a cooled (0 C)
solution of I-
141 (0.23g, 0.54 mmol) in THE (10 mL) was added N-methylmorpholine (0.05 g,
0.54
mmol). The reaction mixture was stirred for 5 min, and isobutylchloroformate
(0.07 g,
194
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
0.54 mmol) was added. The reaction was stirred at 0 C for 45 min, and
ammonium
hydroxide (28%, 4.0 mL) was added. The reaction was warmed to room
temperature,
stirred for 1.5 h, and diluted with water (5 mL). The organic layer was
separated, the
aqueous layer extracted with ethyl acetate (2 x 20 mL). The combined organic
layers
were dried over Na2SO4, filtered, and concentrated. The residue was purified
by silica gel
column chromatography using 5% methanol in dichloromethane, and preparative
thin
layer chromatography using 5% methanol in dichloromethane to afford P-571
(0.03g,
13% yield) as off-white solid. 1H NMR (DMSO-d6, 400 MHz): 7.99 (d, J= 2.0 Hz,
1
H), 7.51 (br s, 1 H), 7.4-7.48 (m, 3 H), 7.36 (s, 1 H), 7.24-7.3 (m, 2 H),
7.14 (br s, 1 H),
6.92 (d, J= 8.8 Hz, 1 H), 6.34 (d, J= 8.4 Hz, 1 H), 4.39 (dd, J= 9.2, 6.4 Hz,
1 H), 3.7-
3.9 (m, 2 H), 3.8 (s, 2 H), 3.72 (s, 3 H), 2.26-2.46 (m, 2 H); MS(APCI+):
426.1 (M+1),
LC-MS: 94.3%.
Example 117. Preparation of P-572
H
N
/ Cod N
0 ~I
O F 0
DBU CI
CI
1-140 P-572
[00421] Synthesis of 4-[5-(3'-chloro-6-methoxy-biphenyl-3-ylmethyl)-pyridin-2-
yl]-
morpholine (P-572). To 1-140 (0.15 g, 0.46 mmol) and morpholine (0.08 g, 0.92
mmol)
was added 1,8-diazabicyclo[5.4.0]undec-7-ene(0.35 g, 2.29 mmol). The reaction
mixture
was stirred at 160 C for 6 h. The reaction was cooled to room temperature,
diluted with
dichloromethane (6 mL), washed with 0.5 N aqueous HC1(2 x 4 mL), dried over
Na2SO4, filtered, and concentrated under vacuum. The residue was purified by
silica gel
column chromatography using 3% methanol in dichloromethane to afford P-572
(0.042
g, 23% yield) as a viscous liquid. 1H NMR (DMSO-d6, 400 MHz): 8.07 (d, J= 2.8
Hz,
1 H), 7.38-7.45 (m, 5 H), 7.16-7.24 (m, 2 H), 7.04 (d, J= 8.4 Hz, 1 H), 6.76
(d, J= 8.8
Hz, 1 H), 3.8 (s, 2 H), 3.74 (s, 3 H), 3.67 (t, J= 4.8 Hz, 4 H), 3.36 (t, J=
4.8 Hz, 4H);
MS(APCI+): 395.1 (M+1), LC-MS: 95.1%.
195
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 118. Preparation of P-581
I.N OH
'N
OFN O MeMgBr
OgF NL
C I
CI
MR 138 MR 142
1-142 P-581
[00422] Synthesis of 2-{(S)-1-[5-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-
ylmethyl)-pyridin-2-yl]-pyrrolidin-2-yl}-propan-2-ol (P-581). To a cooled (0
C) and
solution of I-142 (0.05 g, 0.11 mmol) in THE (8.0 mL) was added
methylmagnesium
bromide (3M sol, 0.26 mL, 0.88 mmol). The reaction mixture was slowly warmed
to
room temperature, and stirred for 3 h. The reaction was then cooled to 0 C,
and
saturated aqueous ammonium chloride solution (5 mL), was added. The aqueous
suspensions was extracted with diethyl ether (2 x 30 mL), washed with brine
(10 mL),
dried over Na2SO4, filtered, and concentrated under vacuum to afford P-581
(0.046 g,
88% yield) as an off-white solid. 1H NMR (DMSO-d6, 400 MHz): 7.90 (d, J= 2.4
Hz, 1
H), 7.75 (br s, 1 H), 7.36-7.48 (m, 4 H), 7.26-7.32 (m, 2 H), 6.92 (d, J= 8.8
Hz, 1 H),
6.61 (d, J= 8.8 Hz, 1 H), 6.33 (br s, 1 H), 4.0-4.08 (m, 1 H), 3.79 (s, 2 H),
3.72 (s, 3 H),
3.3-3.45 (m, 2 H), 1.7-2.0 (m, 4 H), 1.11 (s, 3 H), 0.98 (s, 3 H); MS(APCI+):
455.1
(M+1), LC-MS: 97.5%.
Example 119. Preparation of P-601
H
N
N I I N
F F 0 F NO-OH
DBU
CI CI
P-456 P-601
[00423] Synthesis of 1-[5-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
196
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
pyridin-2-yl]-pyrrolidin-3-ol (P-601). To 5-(3'-chloro-2-fluoro-6-methoxy-
biphenyl-3-
ylmethyl)-2-fluoro-pyridine (P-456, 0.3 g, 0.87 mmol) and 3-pyrrolidinol (2)
(0.15 g,
1.74 mmol) was added 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (0.66 g, 4.34
mmol).
The reaction mixture was stirred and heated at 100 C for 2.5 h, cooled to
room
temperature, diluted with dichloromethane (8 mL), washed with 0.5 N HC1(2 x 4
mL),
dried with Na2SO4, filtered, and concentrated. The residue was purified by
silica gel
column chromatography using 5% methanol in dichloromethane to afford 0.27 g
(75%)
of P-601 as white solid. 1H NMR (DMSO-d6, 400 MHz): 7.95 (s, 1 H), 7.2-7.46
(m, 6
H), 6.91 (d, J= 8.4 Hz, 1 H), 6.35 (d, J= 8.4 Hz, 1 H), 4.9 (d, J= 3.6 Hz, 1
H), 4.32-
4.4 (m, 1 H), 3.76 (s, 2 H), 3.71(s, 3 H), 3.12-3.45 (m, 4 H), 1.8-2.05 (m, 2
H) ppm;
MS(APCI+): 413.1 (M+1), LC-MS: 91.6%.
Example 120. Preparation of P-602
H
N
N q N
O F OH 0 N OH
DBU
CI CI
1-140 P-602
[00424] Synthesis of 1-[5-(3'-chloro-6-methoxy-biphenyl-3-ylmethyl)-pyridin-2-
yl]-
pyrrolidin-3-ol (P-602): To 5-(3'-chloro-6-methoxy-biphenyl-3-ylmethyl)-2-
fluoro-
pyridine (1-140, 0.22 g, 0.67 mmol) and 3-pyrrolidinol (2) (0.12 g, 1.34 mmol)
was
added 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (0.51 g, 3.36 mmol). The
reaction
mixture was stirred and heated at 100 C for 1.5 h, cooled to room
temperature, diluted
with dichloromethane (8 mL), washed with 0.5 N HC1(2 x 4 mL), dried with
Na2SO4,
filtered, and concentrated. The residue was purified by silica gel column
chromatography
using 5% methanol in dichloromethane to afford 0.13 g (49%) of P-602 as white
solid.
1H NMR (DMSO-d6, 400 MHz): 7.9 (d, J= 2.4 Hz, 1 H), 7.3-7.49 (m, 5 H), 7.12-
7.2
(m, 2 H), 7.03 (d, J= 8.4 Hz, 1 H), 6.34 (d, J= 8.8 Hz, 1 H), 4.89 (d, J= 3.6
Hz, 1 H),
4.35 (br s, 1 H), 3.76 (s, 2 H), 3.73 (s, 3 H), 3.2-3.45 (m, 4 H), 1.8-2.05
(m, 2 H) ppm;
MS(APCI+): 395.1 (M+1), LC-MS: 99.1%.
197
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 121. Preparation of P-612
0
N OH N O N 0 F N D IBCF/DIPEA ~'O F N
NH3/MeOH
CI CI
1-143 P-612
[00425] Synthesis of (S)-1-[5-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-
ylmethyl)-
pyridin-2-yl]-pyrrolidine-2-carboxylic acid amide (P-612): To a cooled (0-5
C) and
stirred solution of (S)-1-[5-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-
ylmethyl)-pyridin-
2-yl]-pyrrolidine-2-carboxylic acid (1-143, 0.15 g, 0.34 mmol) in THE (2 mL)
was added
diisopropylethylamine (0.05 g, 0.37 mmol). The reaction mixture was stirred
for 5 min,
isobutylchloroformate (0.05 g, 0.37 mmol) was added, stirred at 0-5 C for 45
min.
Ammonium hydroxide (28%, 4.0 mL) was added, the reaction warmed to ambient
temperature, stirred for 1.5 h. The reaction mixture was diluted with water (5
mL). The
organic layer was separated, the aqueous layer was extracted with ethyl
acetate (2 x 20
mL). The combined organic layers were dried with Na2S04, filtered, and
concentrated.
The residue was purified by silica gel column chromatography using 5% methanol
in
dichloromethane followed by preparative thin layer chromatography using 5%
methanol
in dichloromethane to afford 0.074g (49%) of P-612 as an off-white solid. 1H
NMR
(DMSO-d6, 400 MHz): 8.27 (d, J= 2.4 Hz, 1 H), 7.34-7.46 (m, 4 H), 8.2 (d, J=
8.8 Hz,
1 H), 7.65 (dd, J= 8.8, 2.4 Hz, 1 H), 7.4-7.46 (m, 2 H), 7.37 (br s, 1 H),
7.26-7.38 (m, 2
H), 6.94 (d, J= 8.0 Hz, 1 H), 3.92-3.98 (m 3 H), 3.93 (s, 2 H), 3.73 (s, 3 H),
2.55 (t, J=
8.4 Hz, 2 H), 1.98-2.06 (m, 2 H) ppm; MS(APCI+): 441.1 (M+1), LC-MS: 97.0%;
HPLC 97.1% pure.
198
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 122. Preparation of P-615
IN N
-OH NH
2
0 F N~ IBCF/DIPEA F N
NH3/MeOH
CI CI
1-143 P-615
[00426] Synthesis of (S)-1-[5-(3'-chloro-2-fluoro-6methoxy-biphenyl-3-
ylmethyl)-
pyridin-2-yl]-azetidine-2-carboxylic acid amide (P-615). To a cooled (0-5 C)
and stirred
solution of (S)-1-[5-(3'-chloro-2-fluoro-6methoxy-biphenyl-3-ylmethyl)-pyridin-
2-yl]-
azetidine-2-carboxylic acid (I-143, 0.22 g, 0.54 mmol) in THE (2.5 mL) was
added N,N-
diisopropylethylamine (0.07 g, 0.57 mmol). The reaction mixture was stirred
for 5 min,
isobutylchloroformate (0.08 g, 0.57 mmol) was added, stirred at 0-5 C for 45
min.
Ammonia in methanol (7M sol, 1.0 mL, 7.0 mmol) was added, the reaction warmed
to
room temperature, and stirred for 0.5 h. The reaction mixture was diluted with
water (5
mL). The organic layer was separated, the aqueous layer was extracted with
ethyl acetate
(2 x 20 mL). The combined organic layers were dried with Na2SO4, filtered, and
concentrated. The residue was purified by silica gel column chromatography
using 5%
methanol in dichloromethane, followed by preparative thin layer chromatography
using
5% methanol in dichloromethane to afford 0.06g (27%) of P-615 as an off-white
solid. .
1H NMR (DMSO-d6, 400MHz): 7.99 (d, J= 2.8 Hz, 1 H), 7.51 (br s, 1 H), 7.34-
7.46
(m, 4 H), 7.24-7.3 (m, 2 H), 7.14 (br s, 1 H), 6.92 (d, J= 8.4 Hz, 1 H), 6.35
(d, J= 8.4
Hz, 1 H), 4.39 (dd, J= 9.6, 6.8 Hz, 1 H), 3.7-3.9 (m, 2 H), 3.8 (s, 2 H), 3.72
(s, 3 H),
2.26-2.48 (m, 2 H) ppm; MS(APCI+): 426.6 (M+1), LC-MS: 98.0%.
Example 123. Preparation of P-617
,N O ~N 0
0 I F I N OH IBCF/DIPEA 0 I F I N J-NH2
I NH4OH I
CI CI
MR 129
MR 150
1-144 P-617
199
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00427] Synthesis of (R)-1-[5-(3'-chloro-2-fluoro-6methoxy-biphenyl-3-
ylmethyl)-
pyridin-2-yl]-azetidine-2-carboxylic acid amide (P-617). To a cooled 0-5 C
and stirred
solution of (R)-1-[5-(3'-chloro-2-fluoro-6methoxy-biphenyl-3-ylmethyl)-pyridin-
2-yl]-
azetidine-2-carboxylic acid (I-144, 0.14 g, 0.33 mmol) in THE (2.5 mL) was
added N,N-
diisopropylethylamine (0.09 g, 0.69 mmol). The reaction mixture was stirred
for 5 min,
isobutylchloroformate (0.05 g, 0.36 mmol) was added, and the reaction stirred
at 0-5 C
for 30 min. Ammonium hydroxide solution (28%, 1.0 mL, 8.0 mmol) was added, the
reaction warmed to room temperature, and stirred for 0.5 h. The reaction
mixture was
diluted with water (5 mL). The organic layer was separated, the aqueous layer
was
extracted with ethyl acetate (2 x 20 mL). The combined organic layers were
dried with
Na2SO4, filtered, and concentrated. The residue was purified by silica gel
column
chromatography using 5% methanol in dichloromethane, followed by preparative
thin
layer chromatography using 5% methanol in dichloromethane to afford 0.07g
(47%) of
P-617 as an off-white solid. 1H NMR (DMSO-d6, 400 MHz): 7.99 (d, J= 2.8 Hz, 1
H),
7.51 (br s, 1 H), 7.38-7.46 (m, 3 H), 7.4 (br s, 1 H), 7.24-7.3 (m, 2 H), 7.14
(br s, 1 H),
6.92 (d, J= 8.4 Hz, 1 H), 6.34 (d, J= 8.0 Hz, 1 H), 4.39 (dd, J= 9.6, 6.8 Hz,
1 H), 3.7-
3.9 (m, 2 H), 3.84 (s, 2 H), 3.72 (s, 3 H), 2.26-2.48 (m, 2 H) ppm; MS(APCI+):
426.9
(M+1), LC-MS: 100%.
Example 124. Preparation of P-615-HC1
HCI
-NHZ \ I / I N -NHZ
O F N. 2M HCI-Ether O F N
Ether
CI
CI
P-615 P-615-HCI
[00428] Synthesis of (S)-1-[5-(3'-chloro-2-fluoro-6methoxy-biphenyl-3-
ylmethyl)-
pyridin-2-yl]-azetidine-2-carboxylic acid amide hydrochloride (P-615-HC1). To
a
cooled (0-5 C) and stirred solution of (S)-1-[5-(3'-chloro-2-fluoro-6methoxy-
biphenyl-
3-ylmethyl)-pyridin-2-yl]-azetidine-2-carboxylic acid amide (P-615, 0.04 g,
0.1 mmol)
in ether (2 mL) was added 2M HC1 in ether (0.5 ml, 1.0 mmol), and the
reaction, stirred
200
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
for 1 h. The ether layer was removed, again triturated with ether (2.0 mL),
concentrated
to afford 0.045 g (98%) of P-615-HC1 as a white solid. 1H NMR (DMSO-d6, 400
MHz):
7.92 (d, J= 2.0 Hz, 1 H), 7.78 (d, J= 7.6 Hz,, 1 H), 7.69 (br s, 1 H), 7.26-
7.48 (m, 6 H),
6.95 (d, J= 8.8 Hz, 1 H), 6.72 (d, J= 9.2 Hz, 1 H), 4.8-4.9 (m, 1 H), 4.0-4.18
(m, 2 H),
3.89 (s, 2 H), 3.73 (s, 3 H), 2.64-2.74 (m 1 H), 2.26-2.34 (m, 1 H) ppm;
MS(APCI+):
426.16 (M+1), LC-MS: 98.8%; HPLC 98.3% pure.
Example 125. Preparation of P-617-HC1
CIH
N N O
\ 2 NHZ I I NHZ
O F N 2M HCI-Ether 1O F N
Ether
CI CI
P-617 P-617-H C I
[00429] Synthesis of (R)-1-[5-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-
ylmethyl)-
pyridin-2-yl]-azetidine-2-carboxylic acid amide hydrochloride (P-617-HC1): To
a cooled
(0-5 C) and stirred solution of (R)-1-[5-(3'-chloro-2-fluoro-6methoxy-
biphenyl-3-
ylmethyl)-pyridin-2-yl]-azetidine-2-carboxylic acid (P-617, 0.057 g, 0.13
mmol) in ether
(2 mL) was added 2M HC1 in ether (0.65 ml, 1.3 mmol) and the reaction stirred
for 1 h.
The ether layer was removed, again triturated with ether (2.0 mL),
concentrated to
afford 0.059 g (98%) of P-617-HC1 as a white solid. 1H NMR (DMSO-d6, 400 MHz):
7.92 (d, J= 2.0 Hz, 1 H), 7.78 (d, J= 7.6 Hz,, 1 H), 7.69 (br s, 1 H), 7.26-
7.48 (m, 6 H),
6.95 (d, J= 8.8 Hz, 1 H), 6.72 (d, J= 7.62 Hz, 1 H), 4.8-4.9 (m, 1 H), 4.0-
4.16 (m, 2
H), 3.89 (s, 2 H), 3.73 (s, 3 H), 2.64-2.74 (m 1 H), 2.26-2.36 (m, 1 H) ppm;
MS(APCI+): 426.16 (M+1), LC-MS: 95.8%; HPLC 97.3% pure.
201
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00430] Scheme 36.
O
OIII, H I I N
He B i OMe 0 F 0 F F DBU
[Pd(n3-C3H5)CI]2 /
DPPPent ZZLI
CI
CI KZC03, DMF
1-145 P-456
~N -~
0 F R R= " P-577
NEI P-569 `Nc7 P-582
CI
Example 126. Preparation of P-569
[00431] Synthesis of 5-(3'Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-2-
fluoro-
pyridine(P-456). In a 250 mL round bottomed flask equipped with a condenser,
nitrogen
lines and a stir bar was placed carbonic acid 3'-chloro-2-fluoro-6-methoxy-
biphenyl-3-
ylmethyl methylcarbonate (I-145, 3.61 g, 11.1 mmol), 2-fluoropyridineboronic
acid
(1.72 g, 12.2 mmol), potassium carbonate (4.60 g, 33.3 mmol), 1,5-
bis(diphenylphosphino)pentane (1.47 g, 3.33 mmol) and DMF (56 mL). The
reaction
mixture was degassed for 15 minutes by bubbling nitrogen and then
allylpalladium(II)
chloride dimer (609 mg, 1.67 mmol) was added. The reaction mixture was heated
to 85
C for 18 hours. To the reaction mixture was added water (80 mL), and saturated
ammonium chloride (150 mL). The aqueous portion was extracted with ethyl
acetate (3
x 125 mL). The organic portions were combined, washed with brine (150 mL),
dried
(MgS04) and concentrated. The crude material was purified by column
chromatography
utilizing 10% hexanes/dichloromethane as the eluent to produce 2.45 g of P-456
as a pale
yellow solid in 64% yield.
[00432] Synthesis of 2-Azetidin-1-yl-5-(3'-chloro-2-fluoro-6-methoxy-biphenyl-
3-
ylmethyl)-pyridine (P-569). In an 8 mL vial equipped with a stir bar was
placed 5-(3'-
chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-2-fluoro-pyridine (P-456,150
mg,
0.434 mmol), azetidine (88.0 uL, 1.30 mmol) and 1,8-diazabicyclo[5.4.0]undec-7-
202
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
ene (306 uL, 2.17 mmol). The mixture was heated to 160 C for 1 hour and then
cooled
to room temperature. The reaction mixture was treated with water (4 mL) and 1M
HC1
(6 mL). The aqueous portion was extracted with dichloromethane (2 x 30 mL),
the
organic portions were combined, washed with brine (15 mL), dried (MgSO4) and
concentrated to produce 156 mg of P-569 as a white solid in 94% yield.
[00433] 'H NMR (400 MHz, DMSO-d6) 6 2.37-2.44 (m, 2 H), 3.73 (s, 3 H), 3.87
(s, 2
H),4.19(t,J=8Hz,4H),6.76(bd,J=9Hz,1H),6.95(d,J=9Hz,1H),7.28(bd,J=
7 Hz, 1 H), 7.33 (d, J= 8 Hz, 1 H), 7.36 (bs, 1 H), 7.42-7.48 (m, 2 H), 7.76
(bd, J= 9 Hz,
1 H), 7.86 (s, 1 H) ppm. MS(APCI+): 383.1 (M+1) LC-MS: 96%
Example 127. Preparation of P-577
[00434] Synthesis of 5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-2-
((2S,5S)-2,5-dimethyl-pyrrolidin-1-yl)-pyridine (P-577): In an 8 mL vial
equipped with
a stir bar was placed 5-(3'Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-2-
fluoro-
pyridine (P-456, 100 mg, 0.289 mmol), (2S,5S)-2,5-dimethyl-pyrrolidine (176
mg, 1.30
mmol) and 1,8-diazabicyclo[5.4.0]undec-7-ene (367 uL, 2.60 mmol). The mixture
was
heated to 160 C for 3 hours and then cooled to room temperature. The reaction
mixture
was treated with water (4 mL) and 1M HC1(6 mL). The aqueous portion was
extracted
with dichloromethane (2 x 30 mL), the organic portions were combined, washed
with
brine (15 mL), dried (Mg504) and concentrated. The crude material was purified
by
silica gel column chromatography utilizing 5% acetone/dichloromethane as the
eluent to
produce 10 mg of P-577 as a pale viscous oil. 1H NMR (400 MHz, DMSO-d6) 6 1.05
(d,
J=6Hz,6H),1.56-1.61(m,4H),3.72(s,3H),3.75(s,2H),4.07-4.12 (m,2H),6.38
(d, J= 9 Hz, 1 H), 6.92 (d, J= 8 Hz, 1 H), 7.27-7.32 (m, 3 H), 7.37 (s, 1 H),
7.41-7.47
(m, 2 H), 7.95 (d, J= 2 Hz, 1 H) ppm. MS(APCI+): 425.1 (M+1) LC-MS: 92%
Example 128. Preparation of P-582
[00435] Synthesis of 5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-2-
pyrrolidin-1-yl-pyridine (P-582). In an 8 mL vial equipped with a stir bar was
placed 5-
(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-2-fluoro-pyridine (P-
456,100 mg,
203
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
0.289 mmol), pyrrolidine (60.3 uL, 0.723 mmol) and 1,8-
diazabicyclo[5.4.0]undec-7-
ene (204 uL, 1.45 mmol). The mixture was heated to 160 C for 30 minutes and
then
cooled to room temperature. The reaction mixture was diluted with
dichloromethane (25
mL) and washed with 0.5M HC1(3 x 10 mL). The combined aqueous portions were
extracted with dichloromethane (15 mL), the organic portions were combined,
washed
with brine (15 mL), dried (MgSO4) and concentrated. The crude material was
purified
by silica gel column chromatography utilizing 10% acetone/dichloromethane as
the
eluent to produce 79 mg of P-582 as a pale orange semisolid in 69% yield. 1H
NMR
(400 MHz, DMSO-d6) 6 1.89-1.93 (m, 4 H), 3.31-3.34 (m, 4 H), 3.71 (s, 3 H),
3.77 (s, 2
H), 6.37 (d, J= 9 Hz, 1 H), 6.91 (d, J= 9 Hz, 1 H), 7.23-7.28 (m, 2 H), 7.32
(dd, J= 9, 2
Hz, 1 H), 7.36 (s, 1 H), 7.41-7.47 (m, 2 H), 7.95 (d, J= 2 Hz, 1 H) ppm.
MS(APCI+):
397.1 (M+1) LC-MS: 98%
[00436] Scheme 37.
0
0
~I OOMe B N o N 1. MeMgBr ~I N 0F p O F O- THE 10 F OH
0
[Pd(n3-C3H5)CI]2 2. HCI, Et20
DPPPent CI CI K2CO3, DMF
CI
1-146 P-587
1-145
IiN
LAH,THF 0' F OH
CI
P-599
Example 129. Preparation of P-587
[00437] Synthesis of 5-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridine-
2-carboxylic acid methyl ester (1-146). In a 250 mL round bottomed flask
equipped with
a condenser, nitrogen lines and a stir bar was placed carbonic acid 3'-chloro-
2-fluoro-6-
methoxy-biphenyl-3-ylmethyl methylcarbonate (1-145, 3.60 g, 11.1 mmol), 5-
(4,4,5,5-
Tetramethyl-[1,3,2]dioxaborolan-2-yl)-pyridine-2-carboxylic acid methyl ester
(3.21 g,
12.2 mmol), potassium carbonate (4.60 g, 33.3 mmol), 1,5-
bis(diphenylphosphino)
204
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
pentane (1.47 g, 3.33 mmol) and DMF (56 mL). The reaction mixture was degassed
for
15 minutes by bubbling nitrogen and then allylpalladium(II) chloride dimer
(609 mg,
1.67 mmol) was added. The reaction mixture was heated to 85 C for 8 hours.
After the
addition of water (50 mL) and dichlormethane (100 mL), the mixture was
filtered
through Celite. The layers were separated and the aqueous portion was
extracted with
dichlormethane (75 mL). The organic portions were combined, washed with brine
(150
mL), dried (MgSO4) and concentrated. The crude material was purified by column
chromatography utilizing a gradient elution of 1%, 5%, 10%
acetone/dichloromethane
as the eluent to produce 3.13 g of 1-146 as an orange-yellow viscous oil with
solids
forming in 73% yield.
[00438] Synthesis of 2-[5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-2-yl]-propan-2-ol hydrochloride (P-587). In a 100 mL round bottomed
flask was
placed 5-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-pyridine-2-
carboxylic acid
methyl ester (1-146, 2.0 g, 5.18 mmol) and tetrahydrofuran (26 ml). The
solution was
cooled to 0 C and then a solution of methylmagnesium bromide (3M in diethyl
ether)
(8.6 mL, 25.9 mmol) was added over a 10 minute period. The mixture was stirred
at 0
C for 5 minutes and then at room temperature for 3 hours. The reaction mixture
was
concentrated by a stream of nitrogen, cooled to 0 C and then quenched slowly
with
saturated ammonium chloride (20 mL). The aqueous portion was extracted with
ethyl
acetate (3 x 40 mL), the organic portions were combined, washed with brine (25
mL),
(MgSO4) and concentrated. The crude material was purified by column
chromatography
utilizing 7% acetone/DCM as the eluent to produce a colorless viscous oil.
This product
was combined with material isolated from a 1.04 g run (MCLS1546-106) of the
exact
same reaction conditions and purification (442 mg as a white semi-solid
isolated from
the 1.04 gram run). The combined lots were purified by silica gel column
chromatography utilizing a gradient elution of 50%, then 60% ethyl
acetate/hexanes as
the eluent to produce 1.33 g of P-587 as a colorless, viscous oil in 44%
yield. In an 18
mL vial equipped with a stir bar was placed P-587 (325 mg, 0.842 mmol),
diethyl ether
(3.5 mL) followed by 2M HC1 in diethyl ether (1.5 mL, 3.00 mmol). The mixture
was
stirred for 45 minutes, concentrated by stream of nitrogen and then dried in a
high
vacuum oven set at 40 C for 18 hours to produce 355 mg of P-587 HC1 salt as
an off-
white solid. 1H NMR (400 MHz, DMSO-d6) 6 1.53 (s, 6 H), 3.74 (s, 3 H), 4.13
(s, 2 H),
205
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
6.98 (d, J= 8 Hz, 1 H), 7.29 (bd, J= 7 Hz, 1 H), 7.38 (s, 1 H), 7.40-7.48 (m,
3 H), 7.95
(bd, J= 8 Hz, 1 H), 8.18 (bs, 1 H), 8.56 (s, 1 H) ppm.MS(APCI+): 386.1 (M+1).
HPLC
purity: 98.8%.
Example 130. Preparation of P-599
[00439] Synthesis of [5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-
2-yl]-methanol (P-599). In an 8 mL vial equipped with a stir bar was placed 5-
(3'-chloro-
2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-pyridine-2-carboxylic acid methyl
ester (I-
146, 100 mg, 0.259 mmol) and tetrahydrofuran (1.3 mL). The solution was cooled
to 0
C and then lithium aluminum hydride (39.3 mg, 1.04 mmol) was added resulting
in
strong gas evolution. Then mixture was stirred at 0 C for 1 hour and then
concentrated
with a stream of nitrogen. The solid was diluted with dichloromethane (2 mL)
and then
slowly quenched with water (7 mL). The aqueous portion was extracted with DCM
(3 x
6 mL), the organic portions were combined, filtered through Celite, dried
(MgS04) and
concentrated. The crude material was purified by silica gel column
chromatography
utilizing 30% acetone/dichloromethane to produce 22 mg of P-599 as a pale
orange
viscous oil in 24% yield.
[00440] 'H NMR (400 MHz, DMSO-d6) 6 3.72 (s, 3 H), 3.95 (s, 2 H), 4.51 (s, 2
H),
5.33 (bs, 1 H), 6.94 (d, J= 9 Hz, 1 H), 7.27-7.46 (m, 6 H), 7.61 (bd, J= 8 Hz,
1 H), 8.38
(s, 1 H) ppm. MS(APCI+): 358.1 (M+1) LC-MS: 96%.
Example 131. Preparation of P-588
PF N
0 F NaH, THE 0 F O-__iN
C1 a
P-456 P-588
[00441] Synthesis of {2-[5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-2-yloxy]-ethyl}-dimethyl-amine (P-588). In an 8 mL vial equipped with
a stir
bar was sodium hydride (34.7 mg, 0.867 mmol), THE (1.1 mL) and 2-
diemethylamino-
206
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
ethanol (87.2 uL, 0.867 mmol. The mixture was stirred for 5 minutes and then a
solution
of 5-(3'Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-2-fluoro-pyridine (P-
456, 100
mg, 0.289 mmol) and THE (1.1 mL) was added. After stirring at 60 C for 2
hours, the
reaction was combined with a previous run (30 mg scale) and the quenched with
water
(20 mL). After extractions with ethyl acetate (2 x 30 mL), ), the organic
portions were
combined, dried (MgSO4), concentrated and dried in a high vacuum oven at 60 C
for 18
hours to produce 136 mg of P-588 as a cloudy, pale yellow viscous oil in 87%
yield.
[00442] 'H NMR (400 MHz, DMSO-d6) 6 2.18 (s, 6 H), 2.57 (t, J= 6 Hz, 2 H),
3.72
(s, 3 H), 3.87 (s, 2 H), 4.28 (t, J= 6 Hz, 2 H), 6.73 (d,
J=8Hz,1H),6.93(d,J=8Hz,1
H), 7.27-7.29 (m, 1 H), 7.32 (d, J= 9 Hz, 1 H), 7.37 (s, 1 H), 7.41-7.47 (m, 2
H), 7.53
(dd, J= 8, 2 Hz, 1 H), 8.03 (br s, 1 H). MS(APCI+): 415.1 (M+1)LC-MS: >99%.
Example 132. Preparation of P-595
N fF N
0
F
N McLi 0 NH2
\ I CeCl3, THE CI P-355 P-595
[00443] Synthesis of 1-[5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-2-yl]-1-methyl-ethylamine (P-595). In an 18 mL vial equipped with a
stir bar
was placed cerium (III) chloride (315 mg, 1.28 mmol) which was placed under
and
vacuum and heated with heat gun to remove residual moisture. Then, THE (2 mL)
was
added and the mixture was cooled to 0 C and stirred for 1 hour and then at
room
temperature for 25 minutes. After cooling to -78 C for 15 minutes, methyl
lithium
(1.6M in diethyl ether) (800 uL, 1.28 mmol) was added and the mixture was
stirred at -
78 C for 15 additional minutes. After this time period, a solution of 5-(3'-
Chloro2-
fluoro-6-difluoromethoxy-biphenyl-3-ylmethyl)-pyridine-2-carbonitrile (P-355,
150 mg,
0.425 mmol) and THE (800 uL) was added and the mixture was stirred at -78 C
for 1
hour and then reacted at room temperature for 2 hours. Additional methyl
lithium(1.6M
in diethyl ether) (530 uL, 0.850 mmol) was introduced after cooling to 0 C
and the
207
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
mixture was allowed to react at room temperature for 1 hour. The reaction was
then
cooled to 0 C and quenched with isopropanol (2 mL). After filtration through
Celite,
the filtrate was concentrated and then combined with material from a previous
run (30
mg scale). The combined material was purified by silica gel column
chromatography
utilizing gradient elution of 10% 1M NH3 in MeOH/ dichloromethane and then 20%
to
produce 17 mg of P-595 as a dark orange oil in 8% yield. 1H NMR (400 MHz, DMSO-
d6) 6 1.35 (s, 6 H), 3.72 (s, 3 H), 3.93 (s, 2 H), 6.95 (d, J= 8 Hz, 1 H),
7.27-7.47 (m, 6
H), 7.55 (s, 1 H), 8.40 (s, 1 H) ppm.
MS(APCI+): 385.1 (M+1).
[00444] Scheme 3 8.
F Br F -N IF N
F1~ 0 Pd(PPh3)4 10%Pd/C l NH2
aq. Na2CO3 F O N F O
toluene MeOH, conc HCI
Cl Cl Cl P-596
material from Gary
1-147 1-148
CIH
ci o F ~N F N H H
O /1 H NN
F O ~~ N, O_ McNHZ Y
F~O
TEA, DCM O THF, H2O 0
('t Cl Cl
1-149 P-598
Example 133. Preparation of P-596
[00445] Synthesis of 5-(3'-Chloro-6-difluoromethoxy-biphenyl-3-ylmethyl)-
pyridine-
2-carbonitrile(I-148). Ina 40 mL vial equipped with a stir bar was placed 1-
147 (1.0 g,
2.88 mmol), 5-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-pyridine-2-
carbonitrile
(729 mg, 3.17 mmol), tetrakis(triphenylphosphine)palladium (166 mg, 0.144
mmol),
ethanol (4.4 mL) and toluene (17.6 mL). After stirring for 5 minutes, a 2M
aqueous
solution of sodium carbonate (2.88 mL) was added and the reaction mixture was
degassed by bubbling nitrogen through the mixture. After heating at 80 C for
2.5 hours,
the reaction was filtered through Celite and the filtrate was diluted with
water (40 mL)
and extracted with ethyl acetate (2 x 60 mL). The organic portions were
combined, dried
(MgS04), concentrated and purified by silica gel column chromatography
utilizing 40%
ethyl acetate/hexanes as the eluent to produce 859 mg of I-148 as a yellow
viscous oil in
80% yield.
208
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00446] Synthesis of C-[5-(3'-Chloro-6-difluoromethoxy-biphenyl-3-ylmethyl)-
pyridin-2-yl]-methylamine (P-596). In a 40 mL vial equipped with a stir bar
was placed
5-(3'-Chloro-6-difluoromethoxy-biphenyl-3-ylmethyl)-pyridine-2-carbonitrile (1-
148,
860 mg, 2.32 mmol), methanol (20 mL) concentrated HC1(967 uL, 11.6 mmol) and
10%
Pd/C. The reaction is allowed to stir at ambient temperature for 6 hours under
a
hydrogen atmosphere. After filtering the mixture through Celite, the filtrate
is
concentrated, diluted with 0.5M HC1(20 mL) and water (20 mL) followed by
extractions
with ethyl acetate (2 x 50 mL). The organic portions were combined, dried
(MgSO4),
concentrated and purified by silica gel column chromatography utilizing 10% 1
M NH3 in
MeOH/dichloromethane as the eluent to produce 121 mg of P-596 as a pale yellow
solid
in 14% yield.
[00447] 'H NMR (400 MHz, DMSO-d6) 6 2.12 (bs, 2 H), 3.74 (s, 2 H), 3.99 (s, 2
H),
7.11 (t, J= 74 Hz, 1 H), 7.23 (d, J= 8 Hz, 1 H), 7.32-7.35 (m, 2 H), 7.40-7.51
(m, 5 H),
7.64 (dd, J= 10, 2 Hz, 1 H), 8.45 (s, 1 H) ppm. MS(APCI+): 375.1 (M+1), LC-MS:
99%.
Example 134. Preparation of P-598
[00448] Synthesis of [5-(3'-Chloro-6-difluoromethoxy-biphenyl-3-ylmethyl)-
pyridin-
2-ylmethyl]-carbamic acid phenyl ester (1-149). In an 8 mL vial equipped with
a stir bar
was placed C-[5-(3'-chloro-6-difluoromethoxy-biphenyl-3-ylmethyl)-pyridin-2-
yl]-
methylamine (P-596, 115 mg, 0.307 mmol), dichloromethane (1.5 mL) and
triethylamine
(85.6 uL, 0.614 mmol). The solution was cooled to 0 C and then phenyl
chloroformate
(57.8 uL, 0.461 mmol) was added and the mixture was stirred at 0 C for 15
minutes.
The reaction was quenched with water (3 mL) and then the organic portion was
removed.
The aqueous portion was extracted with dichloromethane (2 mL), the organic
portions
were combined, washed with brine (3 mL), dried (MgSO4), concentrated and
purified by
silica gel column chromatography utilizing 70% ethyl acetate/hexanes as the
eluent to
produce 78 mg of I-149 as a yellow viscous oil in 51% yield.
[00449] 'H NMR (400 MHz, DMSO-d6) 6 4.01 (s, 2 H), 4.32 (d, J= 6 Hz, 2 H),
6.92-
209
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
7.39 (m, 9 H), 7.41-7.48 (m, 4 H), 7.51 (s, 1 H), 7.71 (dd, J= 8, 2 Hz, 1 H),
8.292 (t, J=
6 Hz, 1 H), 8.51 (d, J= 2 Hz, 1 H). MS(APCI+): 495.1 (M+1). LC-MS: >99%.
[00450] Synthesis of 1-[5-(3'-Chloro-6-difluoromethoxy-biphenyl-3-ylmethyl)-
pyridin-2-ylmethyl]-3-methyl-urea hydrochloride (P-598). In an 8 mL vial
equipped
with a stir bar was placed 5-(3'-chloro-6-difluoromethoxy-biphenyl-3-ylmethyl)-
pyridin-
2-ylmethyl]-carbamic acid phenyl ester (I-149, 74 mg, 0.150 mmol),
tetrahydrofuran
(300 uL) and methylamine (40 wt.% in water)(116 uL, 1.50 mmol). The solution
was
stirred at room temperature for 2 hours and then concentrated by a stream of
nitrogen.
The solid was triturated with diethyl ether (2 mL), collected and washed with
diethyl
ether (2 mL) to produce 48 mg of P-598 as a white solid. To P-598 (41 mg,
0.0949) was
added diethyl ether (2 mL) and 2M HC1 in diethyl ether (700 uL). The mixture
was
stirred at ambient temperature for 2 hours, the solids collected, washed with
diethyl ether
(1 mL) and dried in a high vacuum oven to produce 35 mg of P-598 HC1 salt as a
white
solid in 50% yield.
[00451] 'H NMR (400 MHz, DMSO-d6) 6 2.55 (s, 3 H), 4.18 (s, 2 H), 4.46 (s, 2
H),
6.29 (bs, 1 H), 6.78 (bs, 1 H), 7.13 (t, J= 74 Hz, 1 H), 7.27 (d, J= 8 Hz, 1
H), 7.42-7.49
(m, 5 H), 7.52 (s, 1 H), 7.76 (d, J= 8 Hz, 1 H), 8.39 (d, J= 8 Hz, 1 H), 8.79
(s, 1 H).
MS(APCI+): 432.1 (M+1-HC1). LC-MS: >99%.
[00452] Scheme 39.
N CIH
o gF 1. McMgBr 0 F NHZ 2M HCI in Et20 0 F - NHZ
N THF CIH
2. LAH P20
CI CI
P-355 P-606 P-606-diHCI
1. NaOCN CIH
AcOH, H2O H
NYNHZ
O F
2. HCI, Et2O 0
CI
P-613-HCI
210
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 135. Preparation of P-606
[00453] Synthesis of 1-[5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-2-yl]-ethylamine (P-606). In a 40 mL vial equipped with a stir bar was
placed
methylmagnesium bromide (3M in diethyl ether)(1.32 mL, 3.96 mmol), nitrogen
in/out
lines attached and the solution was cooled to 0-5 C. After the addition of a
solution of
5-(3'-Chloro2-fluoro-6-difluoromethoxy-biphenyl-3-ylmethyl)-pyridine-2-
carbonitrile
(P-355, 700 mg, 1.98 mmol) in tetrahydrofuran (10 mL), the reaction was warmed
to
ambient temperature and stirred for 40 minutes. Additional methylmagnesium
bromide
(3M in diethyl ether)(660 uL, 1.98 mmol) was introduced and the reaction was
heated to
60 C for 3 hours. The reaction was cooled to 0-5 C and a slurry of lithium
aluminum
hydride (150 mg, 3.96 mmol) in tetrahydrofuran (1 mL) was added. The reaction
was
heated to 60 C for 1 hour and then partially concentrated, cooled to 0-5 C
and
quenched with water (5 mL) and 1M NaOH (3 mL). After the addition of ethyl
acetate
(10 mL), the reaction was filtered through Celite and the filtrate layers were
separated.
The aqueous portion was extracted with ethyl acetate (30 mL), the organic
portions were
combined, dried (Mg504), concentrated and purified by a 50 gram silica gel
SNAP
cartridge (Biotage SP4 Flash Chromatography instrument) utilizing gradient
elution of 5-
40% MeOH/dichloromethane to produce 124 mg of P-606 as a yellow viscous oil in
17%
yield. 'H NMR (400 MHz, DMSO-d6) 61.26 (d, J= 7 Hz, 3 H), 3.72 (s, 3 H), 3.94
(s, 2
H), 3.99 (q, J= 6 Hz, 1 H), 6.94 (d, J= 8 Hz, 1 H), 7.27-7.29 (m, 1 H), 7.32-
7.38 (m, 3
H), 7.41-7.47 (m, 2 H), 7.57 (dd, J= 8, 2 Hz, 1 H), 8.39 (s, 1 H). MS(APCI+):
372.3
(M+2). LC-MS: 95%
Example 136. Preparation of P-606-diHCl
[00454] Synthesis of 1-[5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-2-yl]-ethylamine dihydrochloride (P-606-diHCl): In an 8 mL vial
equipped with
a stir bar was placed 1-[5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-
2-yl]-ethylamine (P-606, 115 mg, 0.311 mmol), diethyl ether (3 mL), 1,4-
dioxane (300
uL) and 2M HC1 in diethyl ether (600 uL). The mixture was stirred at ambient
temperature for 15 minutes, the solid was collected, washed with diethyl ether
(3 mL)
and dried to produce 98 mg of P-606-diHCl as a light tan solid in 71% yield.
211
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00455] 'H NMR (400 MHz, DMSO-d6) 6 1.47 (d, J= 7 Hz, 3 H), 3.73 (s, 3 H),
4.01
(s, 2 H), 4.46-4.51 (m, 1 H), 6.96 (d, J= 8 Hz, 1 H), 7.27 (d, J= 6 Hz, 1 H),
7.36 (s, 1
H), 7.38-7.48 (m, 4 H), 7.72 (dd, J= 8, 2 Hz, 1 H), 8.33 (bs, 3 H), 8.55 (d,
J= 2 Hz, 1
H).
MS(APCI+): 371.5 (M+1-2HC1)
LC-MS: 92%
Example 137. Preparation of P-613
[00456] Synthesis of {1-[5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-2-yl]-ethyl}-urea hydrochloride (P-613): In an 8 mL vial equipped with
a stir
bar was placed 1-[5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-pyridin-
2-yl]-
ethylamine dihydrochloride (P-606, 55 mg, 0.124 mmol), water (800 uL), glacial
acetic
acid (400 uL) followed by sodium cyanate (32.2 mg, 0.496 mmol). The mixture
was
stirred at room temperature for 18 hours and then slowly quenched with a
saturated
solution of sodium bicarbonate (3 mL) and extracted with dichloromethane (3 x
4 mL).
The organic portions were combined, dried (Mg504), concentrated and purified
by a 10
gram silica gel SNAP cartridge (Biotage SP4 Flash Chromatography instrument)
utilizing gradient elution of 2-20% MeOH/dichloromethane to produce 13 mg of P-
613
(free base) as an off-white solid in 25% yield. To P-613 (free base) (13 mg,
0.0314
mmol) was added diethyl ether (1 mL) and 2M HC1 in diethyl ether (300 UL). The
mixture was allowed to stir at room temperature for 10 minutes, concentrated
and dried
to produce 14 mg of P-613 HC1 salt as an off-white solid in quantitative
yield. 1H NMR
(400 MHz, DMSO-d6) 6 1.36 (d, J= 7 Hz, 3 H), 3.73 (s, 3 H), 4.06 (s, 2 H),
4.82 (m, 1
H), 5.53 (br s, 2 H), 6.69 (br s, 1 H), 6.97 (d, J= 9 Hz, 1 H), 7.29 (br d, J=
6 Hz, 1 H),
7.38-7.47 (m, 4 H), 7.60 (br d, J= 8 Hz, 1 H), 8.02 (br d, J= 8 Hz, 1 H), 8.58
(s, 1 H)
ppm.
[00457] Scheme 40:
212
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
O
F ~a F -O~OMe B " F N
gOH
O
F O pyridine F O O
[Pd(n3-C3H,)CI]2
THE DPPPent
CI
CI \ CI K2CO3, DMF
1-150 1-151 1-152
CIH
1. McMgBr OH
N
THE F 0
2. HCI, E20
CI
P-610-HCI
Example 138. Preparation of P-610
[00458] Synthesis of carbonic acid 3'-chloro-6-difluoromethoxy-biphenyl-3-
ylmethyl
ester methyl ester (1-151). Compound 1-151 was synthesized following the same
procedure as that for the generation of 1-223.
[00459] Synthesis of 5-(3'-Chloro-6-difluoromethoxy-biphenyl-3-ylmethyl)-
pyridine-
2-carboxylic acid methyl ester (1-152). In a 40 mL vial equipped with a stir
bar was
placed carbonic acid 3'-chloro-6-difluoromethoxy-biphenyl-3-ylmethyl ester
methyl ester
(1-151, 700 mg, 2.04 mmol), 5-(4,4,5,5-tetramethyl-[1,3,2] dioxaborolan-2-yl)-
pyridine-
2-carboxylicacid methyl ester (590 mg, 2.24 mmol), potassium carbonate (846
mg, 6.12
mmol), 1,5-bis(diphenylphosphino)pentane (270 mg, 0.612 mmol) and DMF (10 mL).
The reaction mixture was degassed for 15 minutes by bubbling nitrogen and then
allylpalladium(II) chloride dimer (112 mg, 0.306 mmol) was added. The reaction
mixture was heated to 85 C for 4 hours. To the reaction mixture was added
water (40
mL) and ethyl acetate (40 mL) and the mixture was filtered through Celite. The
layers of
the filtrate were separated and the aqueous portion was extracted with ethyl
acetate (40
mL). The organic portions were combined, washed with brine (40 mL), dried
(MgS04)
and concentrated. The crude material was purified by a 50 gram silica gel SNAP
cartridge (Biotage SP4 Flash Chromatography instrument) utilizing gradient
elution of
12-100% ethyl acetate/hexanes to produce 458 mg of 1-152 as a viscous yellow
solid in
56% yield.
[00460] 'H NMR (400 MHz, DMSO-d6) 6 3.86 (s, 3 H), 4.12 (s, 2 H), 7.13 (t, J=
74
213
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Hz, 1 H), 7.25 (d, J= 8 Hz, 1 H), 7.37 (dd, J= 8, 2 Hz, 1 H), 7.57 (dt, J= 7,
2 Hz, 1 H),
7.44-7.52 (m, 4 H), 7.88 (dd, J= 8, 2 Hz, 1 H), 7.99 (d, J= 8 Hz, 1 H), 8.71
(s, 1 H)
ppm.
MS(APCI+): 404.5 (M+1). LC/MS: 98%.
[00461] Synthesis of 2-[5-(3'-Chloro-6-difluoromethoxy-biphenyl-3-ylmethyl)-
pyridin-2-yl]-propan-2-ol hydrochloride (P-610). In a 40 mL vial equipped with
a stir
bar was placed 5-(3'-Chloro-6-difluoromethoxy-biphenyl-3-ylmethyl)-pyridine-2-
carboxylic acid methyl ester (I-152, 385 mg, 0.953 mmol) and tetrahydrofuran
(8 mL).
The solution was cooled to 0 C and then methylmagnesium bromide (3M in
diethyl
ether) (3.2 mL, 9.53 mmol) was added and the reaction was warmed to ambient
temperature and allowed to stir for 2 hours. The reaction was concentrated,
placed in an
ice bath and slowly quenched with a saturated solution of ammonium chloride (5
mL).
After adding dichloromethane (15 mL), the mixture was filtered through Celite,
the
layers of the filtrate were separated and the aqueous portion was extracted
with
dichloromethane (15 mL). The organic portions were combined, washed with brine
(40
mL), dried (MgSO4) and concentrated. The crude material was purified by a 10
gram
silica gel SNAP cartridge (Biotage SP4 Flash Chromatography instrument)
utilizing
gradient elution of 2-20% acetone/dichloromethane to produce 183 mg P-610 as a
colorless oil in 48% yield. To P-610 (175 mg, 0.433 mmol) was added diethyl
ether (1
mL) and 2M HC1 in diethyl ether (700 uL). The mixture was allowed to stir at
room
temperature for 20 minutes, concentrated and dried to produce 139 mg of P-61 O-
HCI as
an off-white solid in 73% yield. 1H NMR (400 MHz, DMSO-d6) 6 1.53 (s, 6 H),
3.82
(bs, 1 H), 4.17 (s, 2 H), 7.14 (t, J= 74 Hz, 1 H), 7.27 (d, J= 9 Hz, 1 H),
7.43-7.54 (m, 6
H), 7.97 (bs, 1 H), 8.32 (bs, 1 H), 8.67 (s, 1 H) ppm. MS(ESI+): 405.6 (M+1-
HC1.
LC/MS: 92%.
214
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00462] Scheme 41.
B
1. I jN
0 NH2
h [Pd(n3-C3H5)C1]2 nN CIH CIH
O We 1. NaOCN F N O
DPPPent
F O K2CO3, DMF F O NH2 AcOH, H2O F0 NxIIII
NH2
H
2. HCI, Et 20 2. HCI, Et20
CI CI CI
I-151 P-611-HCI P-616-HCI
Example 139. Preparation of P-611
[00463] Synthesis of 5-(3'-Chloro-6-difluoromethoxy-biphenyl-3-ylmethyl)-
pyridin-2-
ylamine hydrochloride (P-61 1). In a 40 mL vial equipped with a stir bar was
placed
compound I-151 (1.0 g, 2.92 mmol), 5-(4,4,5,5-tetramethyl-[ 1,3,2]dioxaborolan-
2-yl)-
pyridin-2-ylamine (707 mg, 3.21 mmol), potassium carbonate (1.21 g, 8.76
mmol), 1,5-
bis(diphenylphosphino)pentane (386 mg, 0.876 mmol) and DMF (15 mL). The
reaction
mixture was degassed for 15 minutes by bubbling nitrogen and then
allylpalladium(II)
chloride dimer (160 mg, 0.438 mmol) was added. The reaction mixture was heated
to 85
C for 4 hours. To the reaction mixture was added water (40 mL) and ethyl
acetate (40
mL) and the mixture was filtered through Celite. The layers of the filtrate
were separated
and the aqueous portion was extracted with ethyl acetate (40 mL). The organic
portions
were combined, washed with brine (40 mL), dried (MgS04) and concentrated. The
crude material was purified by a 50 gram silica gel SNAP cartridge (Biotage
SP4 Flash
Chromatography instrument) utilizing gradient elution of 1-10%
methanol/dichloromethane to produce 841 mg of P-611 as a viscous dark yellow
oil in
80% yield. To P-611 (840 mg, 2.33 mmol) was added 1,4-dioxane (8 mL) and 4M
HC1
in 1,4-dioxane (2 mL). The mixture was allowed to stir at room temperature for
20
minutes, concentrated and dried to produce 643 mg of P-611-HC1 as a yellow-
orange
solid in 70% yield. 'H NMR (400 MHz, DMSO-d6) 63.39 (bs, 2 H), 3.89 (s, 2 H),
6.932(s,1H),7.14(t,J=74Hz,1H),7.26(d, J=8Hz,1H),7.36(dd,J=9,2Hz,1
H), 7.42-7.52 (m, 4 H), 7.85 (dd, J= 9, 2 Hz, 1 H), 7.91 (br s, 1 H), 7.94 (br
s, 1 H),
13.71 (br s, 1 H)ppm. LC/MS: 94%.
215
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 140. Preparation of P-616
[00464] Synthesis of [5-(3'-Chloro-6-difluoromethoxy-biphenyl-3-ylmethyl)-
pyridin-
2-yl]-urea (P-616). In an 18 mL vial equipped with a stir bar was placed P-611
(80
mg, 0.222 mmol), glacial acetic acid (1.5 mL), water (750 uL) and sodium
cyanate (57.7
mg, 0.888 mmol). The mixture was heated to 80 C for 3 hours, cooled to room
temperature, diluted with dichloromethane (4 mL) and slowly quenched with a
saturated
solution of sodium bicarbonate (20 mL) to pH 8. The layers were separated and
the
aqueous portion was extracted with dichloromethane (2 x 15 mL). The organic
portions
were combined, washed with brine (15 mL), dried (MgS04) and concentrated. The
crude material was purified by silica gel column chromatography utilizing 5 %
methanol/dichloromethane as the eluent to produce a brown oil. To this
material was
added diethyl ether (1 mL) and 2M HC1 in diethyl ether (500 uL). After
stirring at room
temperature for 30 minutes, the mixture was concentrated and dried to produce
39 mg of
P-616-HC1 as a yellow solid in 40% yield 1H NMR (400 MHz, DMSO-d6) 6 3.89 (s,
2
H), 6.93 (d, J= 9.0 Hz, 1 H), 7.13 (t, J= 74 Hz, 1 H), 7.35 (dd, J= 8, 2 Hz, 1
H), 7.41-
7.52 (m, 4 H), 7.84 (d, J= 2 Hz, 1 H), 7.86-7.90 (m, 2 H), 13.55 (s, 1 H) ppm.
MS(ESI+): 404.5 (M+1-HC1).
[00465] Scheme 42.
0 Q, B
O N
IF 0 We I F F N
J~ / I L I M
F O [Pd(n3-C3H5)CI]2 F O F DBU
DPPPent
K2CO31 DMF CI
CI
1-151 1-153
F F IN
O I / I N NH2
0
CI
P-621
216
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 141. Preparation of P-621
[00466] Synthesis of 5-(3'-Chloro-6-difluoromethoxy-biphenyl-3-ylmethyl)-2-
fluoro-
pyridine (I-153). Compound 1-153 was prepared in a manner analogous to P-611
(Example 138) in 70% yield. MS(ESI+): 364.4 (M+1). LC/MS: 96%.
[00467] Synthesis of 2-{[5-(3'-Chloro-6-difluoromethoxy-biphenyl-3-ylmethyl)-
pyridin-2-yl]-methyl-amino}-acetamide (P-621). In an 8 mL vial equipped with a
stir
bar was placed 5-(3'-Chloro-6-difluoromethoxy-biphenyl-3-ylmethyl)-2-fluoro-
pyridine
(1-153, 210 mg, 0.577 mmol), 2-methylamino-acetamide hydrochloride (216 mg,
1.73
mmol) and 1,8-diazabicyclo[5.4.0]undec-7-ene (488 uL, 3.46 mmol). The mixture
was
heated to 160 C for 3 hours and then diluted with dichloromethane (5 mL). The
organic
portion was washed with 0.5M HC1(3 mL) and the aqueous washes were combined
and
extracted with dichloromethane (2 x 3 mL). The organic portions were combined,
washed with brine (4 mL), dried (MgS04) and concentrated. The crude material
was
purified by Teledyne CombiFlash system utilizing a 12 g RediSepRf silica gel
cartridge
and a gradient elution of 0-30% methanol/dichloromethane, followed by
preparative
TLC (20 x 20 cm, 1000 microns) using 5% methanol/ dichloromethane as the
eluent to
produce 8 mg of P-621?? as a dark brown semi-solid in 3% yield. 'H NMR (400
MHz,
DMSO-d6) 6 2.98 (s, 3 H), 3.83 (s, 2 H), 4.03 (s, 2 H), 6.53 (d, J= 9 Hz, 1
H), 7.10 (t, J
=74Hz,1H),7.21(d,J=8Hz,1H),7.30(dd,J=8,2Hz,1H),7.36(d,J=2Hz,1
H), 7.40-7.50 (m, 5 H), 8.01 (d, J= 2 Hz, 1 H) ppm. MS(APCI+): 432.1 (M+1).
LC/MS:
86%.
Example 142. Preparation of P-618
O
CH F " N O
IF N HO SO2 CI
IN, DCM F N F O N H H CH
2. DIPEA, THE
CI 3. HCI, Et20 CI
P-611 P-618
[00468] Synthesis of N-[5-(3'-Chloro-6-difluoromethoxy-biphenyl-3-ylmethyl)-
217
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
pyridin-2-yl]-2-dimethylamino-acetamide hydrochloride (P-618). In an 18 mL
vial
equipped with a stir bar was placed dimethylglycine (58.5 mg, 0.567 mmol),
dichloromethane (2.8 mL) and thionyl chloride (82.5 uL, 1.13 mmol). The
solution was
stirred at room temperature for 4 hours and then concentrated. To the reaction
mixture
was added a solution of P-611 (150 mg, 0.378 mmol), tetrahydrofuran (2.8 mL)
and
diisopropylethylamine (263 uL, 1.51 mmol). The mixture was stirred at ambient
temperature for 2 hours and then heated to 45 C for 60 hours. The reaction
was
concentrated, diluted with ethyl acetate (3 mL) and washed with water (5 mL).
The
layers were separated and the aqueous portion was extracted with ethyl acetate
(3 x 3
mL). The organic portions were combined, washed with brine (5 mL), dried
(MgSO4)
and concentrated. The crude material was purified by silica gel column
chromatography
utilizing 5% methanol/ dichloromethane (with 1% AcOH) as the eluent, followed
by
preparative TLC (20x20 cm, 1500 microns) using 10% methanol/dichloromethane as
the
eluent to produce 27 mg of P-618 as an orange semi-solid in 16% yield. To P-
618 (45
mg, 0.0561 mmol) was added diethyl ether (1 mL) and 2M HC1 in diethyl ether
(500 uL).
The mixture was allowed to stir at room temperature for 20 minutes,
concentrated and
dried to produce 8 mg of P-618-HC1 as a pale orange solid in 30% yield. 'H NMR
(400
MHz, DMSO-d6) 6 2.86 (d, J= 4 Hz, 6 H), 4.00 (s, 2 H), 4.16 (m, 2 H), 7.17 (t,
J= 74
Hz, 1 H), 7.25 (d, J= 8 Hz, 1 H), 7.34 (dd, J= 8, 2 Hz, 1 H), 7.41-7.51 (m, 5
H), 7.77
(dd, J= 8, 2 Hz, 1 H), 7.97-7.99 (m, 1 H), 8.34 (d, J= 2 Hz, 1 H), 9.82 (bs, 1
H), 11.14
(s, 1 H) ppm. MS(APCI+): 446.1 (M+1).
LC/MS: 96%
Example 143. Preparation of P-622
N N
O F F DBU O F N^ j/NH2
'0
/ I CH NH /
CI HN~ 2 \ CI
O
P-456 P-622
[00469] Synthesis of 2-{[5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
218
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
pyridin-2-yl]-methyl-amino} -acetamide (P-622). In an 8 mL vial equipped with
a stir
bar was placed P-456 (210 mg, 0.607 mmol), 2-methylamino-acetamide
hydrochloride
(227 mg, 1.82 mmol) and 1,8-diazabicyclo[5.4.0]undec-7-ene (513 uL, 3.64
mmol).
The mixture was heated to 160 C for 2.5 hours and then diluted with
dichloromethane
(5 mL). The organic portion was washed with 0.5M HC1(3 x 3 mL) and the aqueous
washes were combined and extracted with dichloromethane (3 x 3 mL). The
organic
portions were combined, washed with brine (4 mL), dried (MgSO4) and
concentrated.
The crude material was purified by Teledyne CombiFlash system utilizing a
RediSepRf
12 g silica gel cartridge and a gradient elution of 0-30%
isopropanol/dichloromethane to
produce material that still contained impurities. The impure material was
purified by
preparative TLC (20 x 20 cm, 1000 microns) using 10% isopropanol/
dichloromethane as
the eluent to produce 89 mg of P-622 as a yellow solid in 35% yield. 'H NMR
(400
MHz, DMSO-d6) 6 2.99 (s, 3 H), 3.71 (s, 3 H), 3.78 (s, 2 H), 4.03 (s, 2 H),
6.53 (d, J= 9
Hz, 1 H), 6.90-6.92 (m, 2 H), 7.23-7.25 (m, 1 H), 7.28 (d, J= 8 Hz, 2 H), 7.34-
7.37 (m, 2
H), 7.40-7.47 (m, 2 H), 7.95 (d, J= 2 Hz, 1 H) ppm. MS(APCI+): 414.1 (M+1).
Example 144. Preparation of P-573
~'r~N H + 0
N DBU O F NON
O K F F CN OH
160 C
CI
CI
P-456 P-573
[00470] Synthesis of 5-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-2-
imidazol-1-yl-pyridine(P-573). To a 20 mL vial which contained P-456 (200 mg,
0.55
mmol) and 1H-imidazole-2-carboxylic acid (202 mg, 1.8 mmol) was added DBU (0.5
mL, excess), at rt. The vial was sealed and the mixture was allowed to heat to
160 C
and stir at 160 C for 2.5 h. The mixture was cooled to rt and then poured
onto 20 mL
ice-water solution, acidified with 2N HC1 to pH=1-2, extracted with ethyl
acetate (3 x 15
mL), washed with water (3 x 15 mL), brine (20 mL) and dried over Na2SO4. After
removal of solvent, the residue was separated by a chromatography on silica
gel with
dichloromathane/iPA as eluent to yield P-573 (60 mg) in 26 % yield.
219
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00471] 1H NMR (DMSO-d6, 400 MHz): 8.48 (s, 1 H), 8.40 (d, J= 2.4 Hz, 1 H),
7.91 (br s, 1 H), 7.85 (dd, J= 8.2, 2.4 Hz, 1 H), 7.74 (d, J= 9.2 Hz, 1 H),
7.27-7.47 (m, 5
H), 7.11 (s, 1 H), 6.86 (d, J= 7.6 Hz, 1 H), 4.01 (s, 2 H), 3.73 (s, 3 H) ppm.
LC/MS: 393.84 Calc.393.8; APCI+(M+1): 394.1, 99%
[00472] Scheme 43.
Br
O N 0 1) Hz/Pd/C
B N Na2C03/tetrakis N EtOH/5% 12NHCI
0 toluene/EtOH/H2
CI 60 C 2) 2N HCI in ether
6CI
1-154 P-590
J N CIH CIAO \ N H
NH 0
0 z O
6CI DCM/TEA
6_CI
P-592
P-591
1) O N 1) MeNH2/THF
~ Py
2) 2N HCI in dioxane
2) 2N HCI in dioxane
CIH
IN CH H N H H
0 NN, 0 NYN
I0I 0
\ CI CI
P-597 P-593
Example 145. Preparation of P-590
[00473] Synthesis of 5-(3'-chloro-6-methoxy-biphenyl-3-ylmethyl)-pyridine-2-
carbonitrile (P-590). To a 100 mL flask which contained the mixture of 1-154
(1324 mg,
4 mmol) and 5-(4,4,5,5-tetramethyl-[1,3,2] dioxaborolan-2-yl)-pyridine-2-
carbonitrile
(1.1 g, 4.4 mmol) in toluene/EtOH/H20 (4/1/1, 25 mL) was added potassium
phosphate
(1..7 g, 8 mmol) and tetrakis(triphenylphosphine)palladium (0) (400 mg, 0.3
mmol)
under nitrogen. The reaction mixture was stirred at 60 C for 4 h. The
reaction mixture
was cooled to ambient temperature, poured onto ice-water solution (100 mL),
and
extracted with ethyl acetate (3 x 30 mL). The combined organic layers was
washed with
water (30 mL), brine (25 mL), dried over Na2SO4, and concentrated in vacuo.
The
220
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
residue was separated by a chromatography on silica gel with ethyl acetate-
hexane as
eluent to afford P-590 (380 mg, 27%). 1H NMR (CDC13, 400 MHz): 8.61 (s, 1 H),
7.61
(d, J = 1.2 Hz, 2 H), 7.46 - 7.48 (m, 1H),7.29-7.37(m,3H),7.11(dd,J=8.4,2.4Hz,
1
H), 7.08 (d, J= 2.4 Hz, 1 H), 6.94 (d, J= 8.4 Hz, 1 H), 4.03 (s, 2 H), 3.81
(s, 3 H) ppm.
Example 146. Preparation of P-591
[00474] Synthesis of C-[5-(3'-hloro-6-methoxy-biphenyl-3-ylmethyl)-pyridin-2-
yl]-
methylamine hydrochloride (P-59 1). To a 50 mL flask which contained P-590
(220 mg,
0.66 mmol) in EtOH (14 mL) was added 12 N HC1(1 mL) and then Pd/C (10%, 200
mg)
at rt. The system was allowed to stir at ambient temperature under a hydrogen
atmosphere (15 psi) for 3 h. The solids were removed by filtration and the
filtrate was
concentrated. The residue was purified via chromatography on silica gel with
ethyl
acetate as eluent to yield P-591 (160 mg) in 70% yield. P-591 (25 mg) was
treated with
2N HC1 in diethyl ether (2 mL) to afford P-591 HC1 salt (16 mg) in 60 % yield.
1H NMR
(DMSO-d6,400 MHz): 8.54 (s, 1 H), 8.26 (br. s, 2 H), 7.76 (d, J= 8.4 Hz, 1 H),
7.39-
7.47 (m, 4 H), 7.22-7.27 (m,2H),7.06(d,J=8.4Hz,1H),4.14(brs,2H),3.99(s,2
H), 3.74 (s, 3 H) ppm.
Calc.338.84; APCI+(M+1): 339.1, 100%.
Example 147. Preparation of P-592
[00475] Synthesis of [5-(3'-Chloro-6-methoxy-biphenyl-3-ylmethyl)-pyridin-2-
ylmethyl]-carbamic acid phenyl ester (P-592). To a 20 mL vial which contained
P-591
(140 mg, 0.4 mmol) and triethylamine (85 mg, 0.8 mmol) in dichloromethane (3
mL)
was added phenyl chloroformate (95 mg, 0.6 mmol) at 0-5 T. The reaction
mixture was
allowed to warm to ambient temperature and stir for 1 h. The mixture was
poured onto
30 mL ice-water solution, extracted with dichloromethane (3 x 15 mL). The
combined
organic layers were washed with water (20 mL), brine (15 mL), dried over
Na2SO4, and
concentrated in vacuo. The residue was purified via chromatography on silica
gel with
ethyl acetate-hexane as eluent to yield 150 mg (79 %) of P-592. 1H NMR (CDC13
,400
MHz): 8.46 (s, 1 H), 7.48-7.52 (m, 2 H), 7.11-7.37 (m, 10 H), 6.92 (d, J= 8.4
Hz, 1 H),
6.13 (br.s, 1 H), 4.55 (d, J= 4.8 Hz, 2 H), 3.97 (s, 2 H), 3.80 (s, 3 H) ppm.
Calc.458.9; APCI+(M+1): 459.1, 100%.
221
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 148. Preparation of P-597
[00476] Synthesis of 1-[5-(3'-chloro-6-methoxy-biphenyl-3-ylmethyl)-pyridin-2-
ylmethyl]-3-ethyl-urea hydrochloride (P-597). To a 20 mL vial which contained
P-591
(66 mg, 0.2 mmol) in pyridine (2 mL) was added isocyanato-ethane (0.2 mL,
excess) at
ambient temperature, and the resultant mixture stirred for 48 h. The mixture
was poured
onto 20 mL ice-water solution and the solid was filtered, washed with water (2
x 20 mL)
and air dried to afford 55 mg of P-597. The free base was treated with 4N HC1
in
dioxane at ambient temperature to afford 55 mf of P-597 HC1 salt in 63 %
yield. 1H
NMR (DMSO-d6,400 MHz). 8.72 (s, 1 H), 8.29 (d, J= 7.6 Hz, 1 H), 7.68 (d, J=
8.8
Hz, 1 H), 7.50 (br s, 1 H), 7.37 - 7.41 (m, 3 H), 7.29-7.31 (m, 2 H), 7.08 (d,
J= 8.8 Hz,
1 H), 6.67 (s, 1 H), 6.36 (s, 1 H), 4.44 (s, 2 H), 4.09 (s, 2 H), 3.75 (s, 3
H), 3.00 (d, J=
7.2 Hz, 2 H), 0.98 (t, J= 7.2 Hz, 3 H) ppm.
Example 149. Preparation of P-593
[00477] Synthesis of 1-[5-(3'-chloro-6-methoxy-biphenyl-3-ylmethyl)-pyridin-2-
ylmethyl]-3-methyl-urea hydrochloride (P-593):. To a 20 mL vial which
contained P-592
(92 mg, 0.2 mmol) in THE (3 mL) was added excess methylamine (40% in H2O, 1
mL),
and the resultant mixture allowed to stir at ambient temperature for 4 h. The
solvent was
removed and diethyl ether (10 mL) was added. The solids were filtered, washed
with
diethyl ether (2 x 5 mL) and air dried to afford 50 mg of P-593. The free base
was
treated with 4 N HC1 in dioxane (0.5 mL) to afford 45 mg of P-593 HC1 salt in
52 %
yield. 1H NMR (DMSO-d6,400 MHz): 8.77 (s, 1 H), 8.41 (d, J=8.4 Hz, 1 H), 7.78
(d,
J=8.4 Hz, 1 H), 7.51 (s, 1 H), 7.31-7.44 (m, 5 H), 7.08 (d, J=8.4 Hz, 1 H),
6.90 (s, 1 H),
4.49 (s, 1 H), 4.12 (s, 3 H), 3.75 (s, 3 H), 2.54 (s, 2 H) ppm. Calc.395.88;
APCI+(M+1):
396.1, 100%.
Example 150. Preparation of P-600
N ~N
0 CeCl3/MeLi 0 NH2
THE
CI CI
P-590 P-600
222
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00478] Synthesis of 1-[5-(3'-chloro-6-methoxy-biphenyl-3-ylmethyl)-pyridin-2-
yl]-
methylamine hydrochloride(P-600). A 50 mL flask which contained CeC13 (370 mg,
1.
mmol) was dried with a heat gun for 0.5 h. After cooling to ambient temeratre,
THE (5
mL ) was added and the mixture was allowed to stir for 2 h, and cooled to -78
T. Methyl
lithium (1 M in ether, 1 mL) was added at -78 C and stirred at -78 C for 1 h
and then
P-590 (170 mg, 0.5 mmol) in THE (0.5 mL) was added. The mixture was allowed to
stir
at -78 C for 0.5 h, allowed to warm to ambient temperature, and stirred for
16 h. The
reaction was quenched with the addition of isopropanol (1 mL). The solids were
removed by filtration, and the filtrate concentrated. The residue was purified
via
chromatography on silica gel with ethyl acetate-EtOH as eluent to yield 30 mg
of P-600
which was treated with 2N HC1 in diethyl ether (2 mL) to afford 15 mg of P-600
HC1 salt
in 8% yield.
[00479] 'H NMR (DMSO-d6,400 MHz): 8.59 (d, J= 2.0 Hz, 1 H), 8.39 (br s, 2 H),
7.80 (dd, J= 8.0, 2.0 Hz, 1 H), 7.54 (d, J= 8.4 Hz, 1 H), 7.48 (br s, 1 H),
7.36 - 7.45 (m,
3H),7.25-7.28(m,2H),7.06(d,J=9.2Hz,1H), 3.99 (s, 2 H), 3.74 (s, 3 H), 1.57
ppm (s, 6 H) ppm. LGMS: Calc.409.9; APCI+(M+1): 410.1, 100%
223
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00480] Scheme 44.
O
O We N
O N O KzCo3DPPPent O O
+ B [Pd(y3CsHs)Cllz
O O' O
CI CI
1-155
1-156
'N N
McMgBr O OH O O
THE
CI CI 1-157
P-605
HCI in ether N_ NaCNBH3
i HCIH MeOH
N CIH N
O OH O N]
6CI I CI
P-609
P-605-HCI
Example 151. Preparation of P-605
[00481] Synthesis of 5-(3'-chloro-6-methoxy-biphenyl-3-ylmethyl)-pyridine-2-
carboxylic methyl ester (1-156). To a 250 mL flask which contained the mixture
of 1-155
(2.0 g, 6.6 mmol) and 5-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-
pyridine-2-
carboxylic acid methyl ester (1.6 g, 6 mmol) in DME (30 mL) was added K2CO3
(2.5 g,
18 mmol), [Pd(y3C3H5C1)]2 (300 mg, 0.8 mmol) and DPPPent (800 mg, 1.9 mmol) at
rt
under nitrogen. The reaction mixture was heated to 85 C and stirred at 85 C
for 16 h.
The reaction mixture was cooled to rt, poured onto ice-water (200 mL), The
semi-solid
which formed was separated from the aq. layer to provide the crude which was
purified
by a chromatography on silica gel with dichloromethane-acetone as eluent to
yield I-156
(1200 mg, 50%)- 1H NMR (CDC13, 400 MHz): 8.64 (s, 1 H), 8.05 (d, J= 8.0 Hz, 1
H),
7.62 (d, J= 8.0 Hz,1H),7.47(s,1H),7.29-7.36 (m,2H),7.11(d,J=8.0Hz,1H),
224
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
7.09 (d, J= 2.0 Hz, 1 H), 6.93 (d, J= 8.4 Hz, 1 H), 4.04 (s, 2 H), 3.99 (s, 3
H), 3.80 (s, 3
H) ppm. LGMS: Calc.367.84; APCI+(M+1): 368.1, 99%.
[00482] Synthesis of 2-[5-(3'-chloro-6-methoxy-biphenyl-3-ylmethyl)-pyridin-2-
yl]-
propan-2-ol HC1 salt (P-605) and 1-[5-(3'-chloro-6-methoxy-biphenyl-3-
ylmethyl)-
pyridin-2-yl]-ethanone (I-157). To a 50 mL dried flask containing I-156 (350
mg, 1
mmol) in THE (5 mL) and cooled to 0 C was added methylmagnesium bromide (3M
in ether, 3 mL, 9 mmol). The reaction mixture was allowed to warm to ambient
temperature and stirred for 16 h, and then poured onto 50 mL ice-water. The
mixture
was neutralized with NH4C1(sat. 10 mL), extracted with ethyl acetate (3 x 20
mL), and
the organics washed with water (20 mL), brine (20 mL) and dried over Na2SO4.
After
removal of solvent, the residue was purified by silica gel column
chromatography with
dichloromethane-acetone as eluent to give 100 mg of the P-605 in 28% yield and
60 mg
of 1-157 in 15% yield. 50 mg of P-605 was treated with 2N HC1 in diethyl ether
(1 mL)
to afford 55 mg of P-605 HC1 salt in 99% yield. 'H NMR (DMSO-d6, 400 MHz):
8.60
(br s, 1 H), 7.85-8.24 (m, 2 H), 7.30 - 7.52 (m, 6 H), 7.08 (d, J=6.8 Hz, 1
H), 4.08 (s, 2
H), 3.75 (s, 3 H), 1.51 ppm (s, 6 H) ppm. LGMS: Calc.367.88; APCI+(M+1):
368.1,
96%.
Example 152. Preparation of P-609
[00483] Synthesis of 2-(1-azetidin-1-yl-ethyl)-5-(3'-chloro-6-methoxy-biphenyl-
3-
ylmethyl)-pyridine (P-609). To a 25 mL vial which contained 1-157 (52mg, 0.15
mmol)
and azetidine HC1 salt (30 mg, 0.3 mmol) in MeOH (2 mL) was added sodium
cyanoborohydride (16 mg, 22 mmol) at 0 T. The reaction mixture was allowed to
warm
to ambient temperature and stir for 72 h. The mixture was poured onto 5 mL 0.5
N
aqueous sodium hydroxide solution and extracted with ethyl acetate (3x10 mL).
The
combined organics were washed with water (10 mL), brine (10 mL) and dried over
Na2SO4. After removal of the solvent, the residue was purified by a
chromatography on
silica gel with dichloromethane-acetone as eluent to afford 30 mg of P-609 in
54% yield.
[00484] 'H NMR (CDC13, 400 MHz): 8.41 (d, J= 2.0 Hz, 1 H), 7.27-7.48 (m, 6 H),
7.10-7.15 (m, 2 H), 6.91 (d, J= 8.0 Hz, 1 H), 3.92 (s, 2 H), 3.79 (s, 3 H),
3.41-3.46 (m, 1
H), 3.10-3.25 (m, 4 H), 2.00-2.07 (m, 2 H), 1.21 (d, J= 6.4 Hz, 3 H) ppm.
225
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 153. Preparation of P-567
Br O toluene, EtOH
0 I 0"B H2O, K3PO4 0 I / I N O
N 0 Pd(PPh3)4 0
CI Cl
1-154 1-158
[00485] Synthesis of 5-(3'-Chloro-6-methoxy-biphenyl-3-ylmethyl)-pyridine-2-
carboxylic acid methyl ester (1-158). Into a 250 mL round-bottomed flask was
added I-
154 (4.89 g, 15.69 mmol), 5-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolan-2-yl)-
pyridine-2-
carboxylic acid methyl ester (4.52 g, 17.26 mmol), toluene (120 mL), EtOH (20
mL),
water (20 mL), and K3PO4 (6.66 g, 31.38 mmol). The suspension was degassed
with N2
for 15 minutes and then Pd(PPh3)4 (1.81 g, 1.57 mmol) was added and the
reaction was
stirred at 80 C for 1 hour. The layers were separated and the aqueous was
extracted
with 50 mL EtOAc. The organics were combined and washed with 50 mL of brine,
dried over Na2SO4, filtered and concentrated. The residue was purified by
flash column
chromatography eluting with 10% acetone/hexanes to afford 890 mg of 1-158 as a
light-
yellow oil inl5% yield..
0 N- 0, LiAIH4, THE I / I OH
0 O N~
I
Cl Cl
1-158 P-567
[00486] [5-(3'-Chloro-6-methoxy-biphenyl-3-ylmethyl)-pyridin-2-yl]-methanol (P-
567). Into a 100 mL round-bottomed flask was added 1-158 (0.71 g, 1.93 mmol),
THE
(20 mL), and the solution was cooled to 0 C. LiAIH4 (0.29 g, 7.72 mmol) was
added
and the reaction was stirred at 0 C for 1 hour. 20 mL of water was slowly
added and the
product was extracted with EtOAc (3 x 20 mL). The organics were combined and
filtered through Celite and then concentrated. The residue was purified by
flash column
chromatography eluting with 20% acetone/hexanes to provide 352 mg of P-567 as
a
226
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
light-yellow oil in 54% yield.. 'H NMR (400 MHz,DMSO-d6) 8.42 (d, J= 1.6 Hz, 1
H), 7.64 (dd, J= 2.0, 7.9 Hz, 1 H), 7.49 (s, 1 H), 7.45 - 7.34 (m, 4 H), 7.25 -
7.20 (m, 2
H), 7.05 (d, J= 8.6 Hz, 1 H), 5.31 (t, J= 5.8 Hz, 1 H), 4.50 (d, J= 5.9 Hz, 2
H), 3.93 (s,
2 H), 3.74 (s, 3 H) ppm. LC/MS = 92.1%, 340.1 (APCI+).
Example 154. Preparation of P-570
H
O N OH EtNCO, toluene "o N OYN
CIo
H
Cl P-567 P-570
[00487] Ethyl-carbamic acid 5-(3'-chloro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-2-
ylmethyl ester hydrochloride (P-570). Into an 18 mL vial was added P-567 (98
mg, 0.29
mmol), toluene (2 mL), and ethyl isocyanate (57 uL, 0.72 mmol). The reaction
was
stirred at 60 C for 6 hours and then concentrated. The solid was triturated
with ether to
obtain 79 mg of P-570 as a white solid. Of this material, 68 mg was dissolved
in 4N
HC1/Dioxane and then concentrated. This solid was triturated with ether,
filtered, and
washed with ether to afford 50 mg of P-570 HC1 salt as a tan solid in 45%
yield. 1H
NMR (400 MHz,DMSO-d6) 8.48 (d, J= 1.5 Hz, 1 H), 7.71 - 7.64 (m, 1 H), 7.49 (s,
1
H), 7.45 - 7.34 (m, 3 H), 7.32 - 7.19 (m, 3 H), 7.05 (d, J= 9.1
Hz,1H),5.01(s,2H),
3.94 (s, 2 H), 3.74 (s, 3 H), 3.07 - 2.95 (m, 2 H), 1.01 (t, J= 7.2 Hz, 3 H)
ppm. LC/MS =
100.0%, 411.0 (APCI+).
Example 155. Preparation of P-534
0 0
~ Br
l i HNN N N N
O F + O NaH, DMF O O + O F
F I i LN
Cl
1-33 1-159 1-160
[00488] 1-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-1H-imidazole-4-
carboxylic acid methyl ester (1-159) and 3-(3'-Chloro-2-fluoro-6-methoxy-
biphenyl-3-
227
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
ylmethyl)-3H-imidazole-4-carboxylic acid methyl ester (1-160). Into an 18 mL
vial was
added methyl 4-imidazole carboxylate (113 mg, 0.90 mmol), DMF (3 mL), and NaH
(43
mg, 1.08 mmol). After 20 minutes at room temperature 1-33 (295 mg, 0.90 mmol)
was
added. The reaction was stirred for 2 hours at room temperature and then water
was
added. The product was extracted with EtOAc and the organics were
concentrated. The
residue was purified by flash column chromatography eluting with 6% - 10%
acetone /
dichloromethane to separate the regioisomers. The 4-substituted ester 1-159
(67 mg,
20%) and the 2-substituted ester 1-160 (79 mg, 23%) were obtained as colorless
oils.
O o
O N
N NH3, McOH LN NHz
O F O F
CI CI
1-159 P-534
[00489] 1-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-1H-imidazole-4-
carboxylic acid amide (P-534). Into an 8 mL vial was added I-159 (30 mg, 0.08
mmol)
and 2 mL of 7N NH3/MeOH. The reaction was stirred at 60 C for 6 days after
which it
was concentrated. Ether was added to form a solid which was filtered and
washed with
ether to afford 8 mg of P-534 as a tan solid in 28% yield. 1H NMR (400 MHz,
DMSO-
d6) 7.77 (s, 1 H), 7.63 (s, 1 H), 7.48 - 7.36 (m, 4 H), 7.29 (d, J= 6.4 Hz, 1
H), 7.26 (br s,
1 H), 7.05 (br s, 1 H), 7.02 (d, J= 8.7 Hz, 1 H), 5.25 (s, 2 H), 3.75 (s, 3 H)
ppm.
Example 156. Preparation of P-535
O O, 0 NH
2
N
O F ~N NH3/MeOH \ I / LN
O F
CI CI
1-160 P-535
[00490] 3-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-3H-imidazole-4-
carboxylic acid amide (P-535). Into an 8 mL vial was added 1-160 (36 mg, 0.096
mmol)
and 2 mL of 7N NH3/MeOH. The reaction was stirred at 60 C for 6 days after
which it
228
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
was concentrated. The solid was triturated with ethe, filtered and washed with
ether to
afford 7 mg of P-535 as a white solid in 20% yield. 'H NMR (400 MHz, DMSO-d6)
7.83 (s, 1 H), 7.75 (br s, 1 H), 7.63 (s, 1 H), 7.51 - 7.41 (m, 2 H), 7.38 (s,
1 H), 7.29 (d, J
= 6.6 Hz, 1 H), 7.19 (br s, 1 H), 7.10 - 7.00 (m, 1 H), 6.94 (d, J= 8.7 Hz, 1
H), 5.59 (s, 2
H), 3.72 (s, 3 H) ppm. LC/MS = 92.2%, 359.1 (APCI-).
Example 157. Preparation of P-536
Br
HNIlk, N N N O F NaH, DMF O I "1 \ `=~
W0 F ~.O
O
CI CI
1-33 P-536
[00491] 1-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-4-nitro-1H-
imidazole
(P-536). Into a 100 mL round bottom flask was added 1-33 (1.0 g, 3.03 mmol),
DMF
(25 mL), and the solution was cooled to 0 C. Ssodium hydride (145 mg, 3.64
mmol) was
added followed by 4-nitro-1H-imidazole (377 mg, 3.34 mmol). The reaction was
stirred
at 0 C for 2 hours and then 10 mL of water was added. The product was
extracted with
3 x15 mL EtOAc and the combined organics were concentrated. The product was
purified by flash column chromatography eluting with 20% acetone/hexanes to
afford
625 mg of P-536 as an amber oil in 57% yield. 'H NMR (400 MHz, DMSO-d6) 8.41
(d,
J=0.9Hz,1H),7.94(s,1H),7.56-7.37(m,4H),7.30(d,J=6.4Hz,1H),7.03(d,J
= 8.6 Hz, 1 H), 5.34 (s, 2 H), 3.76 (s, 3 H) ppm
Example 158. Preparation of P-531
O
O B Pd/C, MeOH
N O'B
N HCI, HZ NH
N z
1-161
[00492] C-[5-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolan-2-yl)-pyridin-2-yl]-
229
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
methylamine (I-161), Into a 100 ml round bottom flask was added 5-(4,4,5,5-
Tetramethyl-[1,3,2]dioxaborolan-2-yl)-pyridine-2-carbonitrile (1.0 g, 0.43
mmol), 20 mL
of MeOH, cone. HC1(1.8 mL, 2.17 mmol), and 10% Pd/C (0.2 g). The reaction was
stirred under a hydrogen atmosphere for 18 hours. The reaction was filtered
through
Celite, washed with MeOH, and then concentrated to a yellow solid. The
material was
used as is.
-~'
O B
EtNCO
NH O B
z H
N pyridine N~NH
N
O
1-161 1-162
[00493] 1 -Ethyl-3-[5-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-pyridin-2-
ylmethyl]-urea (1-162). Into a 50 mL round bottom flask was added C-[5-
(4,4,5,5-
Tetramethyl-[1,3,2]dioxaborolan-2-yl)-pyridin-2-yl]-methylamine (I-161,1 g,
crude),
pyridine (10 mL), and ethyl isocyanate (0.5 mL, 6.5 mmol). The reaction was
stirred at
room temperature for 30 minutes and then NaHCO3 (sat) was added. The product
was
extracted with 3 x 10 mL EtOAc and the combined organics were dried over
Na2SO4,
and concentrated. The residue was purified by flash column chromatography
eluting
with 50% acetone/dichloromethane - 100% MeOH to afford 289 mg of I-162 as a
brown
oil in 22 % yield.
O
Br
H ~
O F OB H Pd(PPh3)4, K,P04 I O
/ N NH
DME, DOH, H2O O F
~I O i
CN
1-163 1-162 ~\N
P-531
[00494] 1-[4-(3'-Cyano-2-fluoro-6-methoxy-biphenyl-3-ylmethoxy)-pyridin-2-
ylmethyl]-3-ethyl-urea (P-53 1). Into an 8 mL vial was added I-163 (125 mg,
0.39
mmol), 1-ethyl-3-[5-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-pyridin-2-
ylmethyl]-
urea (1-162,119 mg, 0.39 mmol), K3P04 (248 mg, 1.17 mmol), DME (2.5 mL), EtOH
(0.5 mL), and water (0.5 mL). The suspension was degassed with N2 and then
Pd(PPh3)4
230
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
was added. The reaction was stirred for 1 hour at 80 C. Water (2 mL) was
added and
the product was extracted with EtOAc (3 x 2 mL). The organic extracts were
combined
and concentrated. The residue was purified by flash column chromatography
eluting
with 25% - 50% acetone/dichloromethane to give a tan-gray solid which was then
triturated with ether. The resulting solid was recrystallized from EtOH to
provide 7 mg
of P-531 as a white solid in 4% yield. 1H NMR (400 MHz, DMSO-d6) 8.26 (d, J=
2.8
Hz, 1 H), 7.89 - 7.82 (m, 2 H), 7.75 - 7.53 (m, 3 H), 7.47 (dd, J= 3.0, 8.6
Hz, 1 H), 7.22
(d, J= 8.6 Hz,1H),7.04(d,J=8.6Hz,1H),6.33 (t, J= 5.8 Hz,1H),6.01(t,J=5.4
Hz, 1 H), 5.16 (s, 2 H), 4.22 (d, J= 5.9 Hz, 2 H), 3.78 (s, 3 H), 3.09 - 2.95
(m, 2 H), 0.99
(t, J= 7.1 Hz, 3 H) ppm. LC/MS = 90.7%, 435.2 (APCI+).
Example 159. Preparation of P-244
; OH OH
BH3 THE
Br THE Br
1-164
[00495] 2-(3-Bromo-4-methoxy-phenyl)-ethanol (1-164). Into a 100 mL round
bottom flask was added (3-bromo-4-methoxy-phenyl)-acetic acid (0.58 g, 2.37
mmol),
THE (10 mL), and the mixture was cooled to 0 C. BH3-THF (10.6 mL, 10.6 mmol,
1.OM in THF) was added and the reaction was stirred at room temperature for 20
hours.
20 mL of MeOH was added and the solvent was removed on a rotary evaporator.
This
was repeated an additional 5 times. The residue was purified by flash column
chromatography eluting with 10- 20% acetone/hexanes to afford 457 mg of I-164
as a
light yellow oil in 83 % yield.
OH CBr4 Br
O PPh3 O
Br DCM Br
1-164 1-165
[00496] 2-Bromo-4-(2-bromo-ethyl)-1-methoxy-benzene (1-165). Into a 100 mL
round bottom flask was added 2-(3-bromo-4-methoxy-phenyl)-ethanol (I-164, 418
mg,
231
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
1.81 mmol), dichloromethane (15 mL), carbon tetrabromide (1.50 g, 4.52 mmol),
and
triphenylphosphine (1.19 g, 4.52 mmol). After stirring for 1 hour at room
temperature
the reaction was concentrated. The residue was purified by flash column
chromatography eluting with hexanes to afford 1.91 g of 1-165 as a colorless
oil, which
was used without further purification.
\ Br N K2C03 \ N
/ O
O / + HO DME
'-0
Br Br
1-165 1-166
[00497] 1- [2 -(3 -Bromo-4-methoxy-phenyl)-ethyl] -1H-pyridin-2-one (1-166).
Into a
100 mL round bottom flask was added 2-bromo-4-(2-bromo-ethyl)-1-methoxy-
benzene
(I-165, 1.82 g, crude), 2-hydroxypyridine (258 mg, 2.72 mmol), K2CO3 (625 mg,
4.53
mmol), and DME (20 mL). After stirring for 18 hours at 80 C the suspension
was
filtered and the filtrate was concentrated. The residue was purified by flash
column
chromatography eluting with 25 - 50% acetone/hexanes to afford 221 mg of 1-166
as a
brown oil in 40 % yield (2 steps).
N I HO.B.OH Pd(OAc)2 N \
0 6'N to KZCO3 PPh3
DME to
Br ON
0
DOH, H2O 1-166 0
P-244
[00498] 1-[2-(6-Methoxy-3'-nitro-biphenyl-3-yl)-ethyl]-1H-pyridin-2-one (P-
244).
Into a 40 mL vial were added 1-[2-(3-bromo-4-methoxy-phenyl)-ethyl]-1H-pyridin-
2-
one (1-166, 210 mg, 0.68 mmol), 3-nitrophenylboronic acid (125 mg, 0.75 mmol),
PPh3
(36 mg, 0.014 mmol), potassium carbonate (283 mg, 2.04 mmol), dimethoxyethane
(10
mL), ethanol (1 mL), and water (1 mL). The suspension was degassed with N2 for
5
minutes and then palladium(II) acetate (15 mg, 0.068 mmol). After degassing
for an
additional 2 minutes the reaction was stirred at 80 C for 18 hours. To the
reaction was
added 5 mL of water and 10 mL of ethyl acetate. The layers were separated and
the
aqueous was extracted with ethyl acetate (3 x10 mL). The organics were
combined and
232
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
concentrated. The residue was purified by flash column chromatography eluting
with 10
- 20% acetone/dichloromethane. The yellow oil which was obtained (136 mg) was
dissolved in 1 mL of ether and allowed to stand at room temperature for 3
days. The tan
solid which formed was filtered, washed with ether, and dried to afford 74 mg
of P-244
in 31% yield. 1H NMR (400 MHz, CDC13) 8 8.30 - 8.29 (m, 1H), 8.18 - 8.15 (m,
1H),
7.81-7.79(m,1H),7.54(t,J=8.0Hz),7.37-7.33(m,1H), 7.19 (dd, J = 8.2, 2.2 Hz,
1H), 7.05 (d, J= 2.4 Hz, 1H), 6.95 - 6.93 (m, 2H), 6.61 (d, J= 9.2 Hz, 1H),
6.06 (td, J =
6.6, 1.2 Hz, 1H), 4.15 (t, J= 7.0 Hz, 2H), 3.82 (s, 3H), 3.07 (t, J= 7.0 Hz,
2H) ppm.
LC/MS = 99.4%, 351.6 (ESI+).
Example 160. Preparation of P-003
\ I \
0 F
O
O-/
P-003
[00499] 5-[5-(4-Fluoro-benzyl)-2-methoxy-phenyl]-benzo[1,3]dioxole (P-003). P-
003 was prepared by according to the method described for P-001. 1H NMR (400
MHz,
CDC13) 3.80 (s, 3 H) 3.93 (s, 2 H) 5.98 (s, 2 H) 6.83 - 6.92 (m, 2 H) 6.92 -
7.05 (m, 4 H)
7.05 - 7.12 (m, 2 H) 7.16 (dd, J= 8.4, 5.6 Hz, 2 H) ppm. LCMS = 94.4% purity.
TSI(+)
= 365.4 (M+29).
Example 161. Preparation of P-004
O I I F
'N
N-O
P-004
[00500] 5-[5-(4-Fluoro-benzyl)-2-methoxy-phenyl]-benzo[1,2,5]oxadiazole (P-
004).
233
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
P-004 was prepared by according to the method described for P-001. 'H NMR (400
MHz, CDC13) 3.84 (s, 3 H), 3.97 (s, 2 H), 6.91 - 7.05 (m, 3 H), 7.13 - 7.24
(m, 4 H),
7.62 (dd, J= 9.3, 1.3 Hz, 1 H) 7.75 - 7.87 (m, 2 H) ppm. LCMS = 96.2% purity.
TSI (+)
= 365.4 (M+3 1).
Example 162. Preparation of P-006
Pd(dba)2
N HO, OH PPh3 LD\,
B' Cs2CO3 0 N
-N +
Br DMF
~ \N 0 \N
N-0
1-167 N-0
1-105 P-006
[00501] Synthesis of 5-(5-Imidazol-1-ylmethyl-2-methoxy-phenyl)-
benzo[ 1,2,5]oxadiazole (P-006). A suspension of 1-(3-bromo-4-methoxy-benzyl)-
1H-
imidazole (1-167, 267 mg, 1.00 mmol), benzo[1,2,5]oxadiazole-5-boronic acid (I-
105,
164 mg, 1.00 mmol), palladium(0)bis(dibenzylideneacetone) (28.7 mg, 0.050
mmol),
and triphenylphosphine (26.2 mg, 0.10 mmol) in dimethylformamide (20 mL) and 1
M
aqueous cesium carbonate (3.0 mL, 3.0 mmol) was heated to 85 C with stirring
overnight. The solvent was removed under vacuum and the residue suspended in
ethyl
acetate (25 mL). The organic suspension was washed with water (3 x 20 mL) and
brine,
dried over sodium sulfate, decolorized over activated carbon, filtered, and
concentrated
under. The residue was purified by slica gel preparatory thin layer
chromatography (ethyl
acetate: dichloromethane 3:1) to give 10.4 mg of P-006 in 9.1% yield. 'H NMR
(400
MHz, CDC13) 3.86 (s, 3 H) 5.13 (s, 2 H) 6.93 (s, 1 H) 7.01 (d, J= 8.32 Hz, 1H)
7.10 (s,
1H)7.17-7.25(m,2H)7.52-7.62(m,2H)7.78-7.85 (m,2H)ppm. LCMS = 100%
purity. APCI(+) = 307.1 (M+1).
234
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00502] Scheme 45.
Pd(dba)2
HO,B,OH PPh3
NaCO
O F + 2 O F
Br N dimethylformamide
4 N
1-168
P-474
McReO3
H202
O F
Dichloromethane
NO
P-475
Example 163. Preparation of P-474
[00503] Synthesis of 3-[5-(4-Fluoro-benzyl)-2-methoxy-phenyl]-pyridine (P-
474). A
suspension of 1-168 (158 mg, 0.53 mmol), 3-pyridineboronic acid (61.5 mg, 0.50
mmol),
palladium(0)bis(dibenzylideneacetone) (14.4 mg, 0.025 mmol), and
triphenylphosphine
(13.1 mg, 0.050 mmol) in dimethylformamide (5 mL) and 1 M aqueous sodium
carbonate (1.5 mL, 1.5 mmol) was heated to 85 C with stirring overnight. The
solvent
was removed under vacuum and the residue suspended in ethyl acetate (15 mL).
The
organic suspension was washed with water (3 x 15 mL) and brine, dried over
sodium
sulfate and the solvent removed under vacuum to give crude material. The
residue was
purified by silica gel preparatory thin layer chromatography to afford 69.1 mg
(47%) of
P-474
LCMS = 94.6% purity.
Example 164. Preparation of P-475
[00504] Synthesis of 3-[5-(4-Fluoro-benzyl)-2-methoxy-phenyl]-pyridine 1-oxide
(P-
475). A vial was charged with P-474 (60 mg, 0.20 mmol), methyl ruthenium oxide
(2.5
235
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
mg, 0.010 mmol), 30% aqueous hydrogen peroxide (0.5 mL), and dichloromethane
(1.0
mL). The reaction was allowed to stir at room temperature for 3 days. The
biphasic
mixture was treated with catalytic amount of manganese dioxide (1.7 mg, 0.02
mmol)
and carefully stirred until the oxygen evolution ceased (1 h). The phases were
separated,
the aqueous layer extracted into dichloromethane (2 x 1 mL), the organic
layers
combined, dried over sodium sulfate, and the solvent removed under vacuum to
afford
20.9 mg of P-475 in 34% yield. 1H NMR (400 MHz, CDC13) 3.82 (s, 3 H) 3.94 (s,
2
H) 6.89 - 7.24 (m, 8 H) 7.41 (d, J= 8.1 Hz, 1 H) 8.16 (d, J= 6.3 Hz, 1 H) 8.44
(s, 1 H)
ppm. LCMS = 92.0 % purity.
Example 165. Preparation of P-007
Pd(dba)2
HO, OH PPh3 N
N NaCO Ni
;
O NN B 2 O
Br O Dimethylformamide
N 0 N"0
1-169 O
P-007
[00505] Synthesis of 1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-1H-
benzotriazole
(P-007). A suspension of 1-169 (477 mg, 1.50 mmol), 3-nitrophenylboronic acid
(250
mg, 1.50 mmol), palladium(0) bis(dibenzylideneacetone) (43 mg, 0.075 mmol),
and
triphenyl phosphine (39 mg, 0.15 mmol) in dimethylformamide (10 mL) and 1 M
aqueous sodium carbonate (4.5 mL, 4.5 mmol) was heated to 85 C with stirring
overnight. The solvent was removed under vacuum and the residue suspended in
ethyl
acetate (20 mL). The organic suspension was washed with water (3 x 20 mL) and
brine,
dried over sodium sulfate, decolorized over activated carbon, filtered and the
solvent
removed under vacuum to give a residue. The residue was purified dissolving in
ethyl
acetate (5 mL) and adding hexanes (25 mL) until a solid formed. This was
repeated 3
times to afford 210 mg of P-007 in 39% yield. 'H NMR (400 MHz, CDC13) 3.83 (s,
3
H),5.87(s,2H),6.99(d,J=8.2Hz,1H),7.31-7.52(m,4H),7.56(t,J=7.9Hz,1H),
7.76 (d, J= 7.8 Hz, 1 H), 8.13 (d, J= 8.3 Hz, 1 H), 8.19 (dd, J= 8.2, 1.2 Hz,
1 H), 8.36
(d, J= 1.6 Hz, 1 H) ppm. LCMS = 93.9% purity. APCI(+) = 361.10 (M+1).
236
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 166. Preparation of P-037
Pd(dba)2
PPh3 N
\ I j ~ HO,B,OH Na2CO3 O ~N
N +
DMF
Br
O 0
1-167 0--" O
O-1
P-037
[00506] Synthesis of 1-(3-Benzo[1,3]dioxol-5-yl-4-methoxy-benzyl)-1H-imidazole
(P-037). To a solution of 1-167 (484 mg, 1.80 mmol) and benzo[1,3]dioxole-5-
boronic
acid (332 mg, 2.00 mmol) were added palladium(0) bis(dibenzylideneacetone)
(57.5 mg,
0.100 mmol), and triphenylphosphine (52.4 mg, 0.200 mmol) in dimethylformamide
(40
mL) and 1 M aqueous sodium carbonate (6.0 mL, 6.0 mmol) was heated to 80 C
with
stirring overnight. The solvent was removed under vacuum and the residue
suspended in
ethyl acetate (30 mL). The organic suspension was washed with water (3 x 30
mL) and
brine, dried over sodium sulfate, decolorized over activated carbon, filtered,
and the
solvent removed under vacuum to give a residue. The residue was purified by
reverse
phase (water: acetonitrile 3:1 to 1:1) followed by extraction with
dichloromethane and
removal of solvent under reduced pressure to afford 69.6 mg of P-037 as a
clear viscous
oil in 13% yield. 1H NMR (400 MHz, CDC13) 3.81 (s, 3 H), 5.08 (s, 2 H), 5.99
(s, 2
H),6.82-6.96(m,4H),7.00(d,J=1.5Hz,1H),7.09(d, J= 10.2 Hz,3H),7.55(s,1
H) ppm. LCMS = 100% purity. APCI(+) = 309.10 (M+1).
Example 167. Preparation of P-040
0
Br
N
\I ~ ~
O O NaH
O O
N
+ H Dimethylformamide
O
N NI;O
1-70
P-040
237
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00507] Synthesis of 1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-piperidine-2,6-
dione (P-040). To a solution of 1-70 (161 mg, 0.500 mmol) and glutarimide (113
mg,
1.00 mmol) in dimethyl formamide (1.5 mL) was added sodium hydride (60% weight
dispersion, 40 mg, 1.00 mmol) at -78 C. After hydrogen gas evolution ceased
the
reaction was stirred at 120 C overnight. The reaction mixture was filtered,
and the
filtrate concentrated under reduced pressure. The residue was diluted with
ethyl acetate
(15 mL), washed with water, brine, dried over sodium sulfate, filtered, and
the solvent
removed under vacuum. The product was purified by trituration with hexanes (50
mL)
and dichloromethane (2 mL) followed by silica gel column chromatography (50%
ethyl
acetate in hexanes) to afford 79.8 mg of P-040 in 45% yield. 1H NMR (400 MHz,
CDC13) 1.94 (quintet, J= 6.4 Hz, 2H), 2.67 (t, J = 6.4 Hz, 4H), 3.81 (s, 3H),
4.94 (s,
2H), 6.92 (d, J= 8.4 Hz, 1H), 7.39 (d, J= 2.4 Hz, 1H), 7.44 (dd, J= 8.4, 2.0
Hz, 1H),
7.55 (t, J= 8.0 Hz, 1H), 7.81 (ddd, J= 8.0, 1.6, 1.2 Hz, 1H), 8.16 (ddd, J=
8.4, 2.4, 1.2
Hz, 1H), 8.39 (t, J= 2.0 Hz, 1H) ppm. LCMS = 92% purity.
Example 168. Preparation of P-041
Br
^N ~
O 0
p NaH 0
+ HN Dimethylformamide 0
O_ N~.O
1-70 0
P-041
[00508] Synthesis of 1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-1H-indole-2,3-
dione (P-041). To a solution of 1-70 (98 mg, 0.300 mmol) and isatin (88 mg,
0.60 mmol)
in dimethyl formamide (1.0 mL) was added sodium hydride (60% weight
dispersion, 24
mg, 0.6 mmol) at -78 C. After hydrogen gas evolution ceased the reaction was
stirred at
room temperature overnight. The reaction mixture was filtered, and the
filtrate
concentrated under reduced pressure. The residue was diluted with ethyl
acetate (15 mL),
washed with water, brine, dried over sodium sulfate, filtered, and the solvent
removed
under vacuum. The crude product was purified by silica gel column
chromatography (4:1
ethyl acetate : hexanes) to afford 45.5 mg of P-041 in 39.1% yield). 1H NMR
(400 MHz,
CDC13) 3.82 (s, 3 H), 4.93 (s, 2 H), 6.85 (d, J= 7.9 Hz, 1 H), 6.98 (d, J= 8.6
Hz, 1 H),
7.07 - 7.14 (m,1H),7.29-7.40(m,2H),7.49-7.65 (m,4H),7.79(d,J=7.8Hz,1H),
238
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
8.18 (s, 1 H), 8.37 (t, J= 1.74 Hz, 1 H) ppm. Turb. Spray (+) = 389.60 (M+1)
Example 169. Preparation of P-042
0
Br
O H O
NaH O
+ O \ Dimethylformamide
\ I IJ
N / \ I N.~O
O
1-70
P-042
[00509] Synthesis of 2-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-isoindole-1,3-
dione
(P-042). To a solution of 1-70 (161 mg, 0.500 mmol) and phthalimide (147 mg,
1.00
mmol) in dimethyl formamide (1.5 mL) was added sodium hydride (60% weight
dispersion, 40 mg, 1.0 mmol) at -78 C. After hydrogen gas evolution ceased the
reaction
was stirred at 80 C overnight. The reaction mixture was filtered, and the
filtrate
concentrated under reduced pressure. The residue was diluted with ethyl
acetate (15 mL),
washed with water, brine, dried over sodium sulfate, filtered, and the solvent
removed
under vacuum. The crude product was purified by trituration several times in
hexanes
(100 mL) and ethyl acetate (5 mL) to afford 43.8 mg of P-042 as a grey-white
solid in
26% yield. 'H NMR (400 MHz, CDC13) 3.80 (s, 3 H), 4.84 (s, 2 H), 6.95 (d,
J=,8.5
Hz, 1 H), 7.43 (d, J= 2.2 Hz, 1 H), 7.49 (dd, J= 8.5, 2.2 Hz, 1 H), 7.55 (t,
J= 8.0 Hz, 1
H),7.65-7.76(m,2H),7.77-7.87(m,3H),8.17(dd,J=8.2,1.3Hz,1H),8.39(t,J=
1.8 Hz, 1 H) ppm. LCMS = 97.1 % purity. APCI (+) = 359.1 (M-29).
Example 170. Preparation of P-043
0
Br
O
O NaH -O O N
+ O \ Dimethylformamide
'N -.0 N Np
1-70 O
P-043
239
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00510] Synthesis of 2-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-pyrrolo[3,4-
c]pyridine-1,3-dione (P-043). To a solution of 1-70 (161 mg, 0.500 mmol) and
pyridinecarboximide (148 mg, 1.00 mmol) in dimethyl formamide (1.5 mL) was
added
sodium hydride (60% weight dispersion, 40 mg, 1.0 mmol) at -78 C. After
hydrogen gas
evolution ceased the reaction was stirred at 80 C overnight. The reaction was
diluted
with ethyl acetate (15 mL), washed with water, brine, decolorized with
activated
charcoal, dried over sodium sulfate, filtered, and the solvent removed under
vacuum. The
crude product was purified by trituration several times in hexanes (100 mL)
and ethyl
acetate (5 mL) to afford 20.9 mg of P-043 as a grey-white solid in 11 % yield.
'H NMR
(400 MHz, CDC13) 3.81 (s, 3 H), 4.86 (s, 2 H), 6.96 (d, J = 8.5 Hz, 1 H), 7.43
(d, J= 2.2
Hz, 1 H), 7.45 - 7.52 (m, 1 H), 7.56 (t, J= 8.0 Hz, 1 H), 7.71 - 7.83 (m, 3
H), 8.18 (d, J=
8.3 Hz, 1 H), 8.38 (s, 1 H), 9.06 (d, J= 4.70 Hz, 1 H), 9.08 - 9.17 (m, 2 H),
9.20 (s, 1 H)
ppm. LCMS = 95.2% purity. APCI(-)= 389.1(M).
Example 171. Preparation of P-047
O
Br
~I \ N
0 H O N
NaH 0 =~~
+ N x N Dimethylformamide
N .0 N p
O
1-70 0
P-047
[00511] Synthesis of 2-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-2H-
[ 1,2,4]triazolo[4,3-a]pyridin-3-one (P-047. To a solution of 1-70 (161 mg,
0.500 mmol)
and triazolepyridinone (135 mg, 1.00 mmol) in dimethyl formamide (1.5 mL) was
added
sodium hydride (60% weight dispersion, 40 mg, 1.0 mmol) at -78 C. After
hydrogen gas
evolution ceased the reaction was stirred at 75 C for 4 h. The reaction was
diluted water
(15 mL). The resultant precipitate was isolated, dissolved in ethyl acetate
(10 mL),
decolorized with activated carbon, dried over sodium sulfate, filtered, and
the solvent
removed under vacuum to afford 43.2 mg of P-047 as a cream colored solid in
23%
yield). 'H NMR (400 MHz, CDC13) 3.82 (s, 3 H), 5.16 (s, 2 H), 6.48 (ddd, J=
7.1,
4.0,3.2Hz,1H),6.99(d,J=8.5Hz,1H),7.04-7.13(m,2 H), 7.41 (d, J= 2.2Hz,1
H), 7.47(dd,J=8.5,2.2Hz,1H),7.55(t,J=8.0Hz,1H), 7.73 - 7.86 (m, 2 H), 8.17
240
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
(dd, J= 8.2, 1.2 Hz, 1 H), 8.39 (t, J= 1.8 Hz, 1 H) ppm. LCMS =98.0% purity.
APCI(+) = 377.1 (M+1).
Example 172. Preparation of P-052
0
CI
O H O K 2CO3 O I/ N
O
+ O Dimethylformamide
O_/ O
O-J
1-168
P-052
[00512] Synthesis of 2-(3-Benzo[1,3]dioxol-5-yl-4-methoxy-benzyl)-isoindole-
1,3-
dione (P-052). A suspension of phthalimide (147 mg, 1.00 mmol), 1-168 (138 mg,
0.500
mmol), and solid potassium carbonate (138 mg, 1.00 mmol) was stirred over 72
h. Water
was added, and a precipitate was formed. The suspension was stirred for 10
min, and the
solid collected and dissolved in ethyl acetate (20 mL). The organic solution
was dried
over sodium sulfate, filtered, and the solvent removed under vacuum to afford
173 mg of
P-052 in 93% yield. 'H NMR (400 MHz, CDC13) 3.78 (s, 3 H), 4.82 (s, 2 H), 5.91
-
6.00(m,2H),6.80-6.98(m,3H),7.02(d,J=1.3Hz,1H), 7.35 - 7.43 (m, 2 H), 7.70
(dd, J= 5.4, 3.0 Hz, 2 H), 7.80 - 7.88 (m, 2 H) ppm. LCMS = 95.6% purity.
Example 173. Preparation of P-055
0
Br
\
O N O K 2CO3 O I / N
+ O N Dimethylformamide
N
\ I N
O
1-70 O
P-055
[00513] Synthesis of 6-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-pyrrolo[3,4-
b]pyridine-5,7-dione (P-055). A suspension of 1-70 (322 mg, 1.00 mmol),
quinolinimide (148 mg, 1.00 mmol), and solid potassium carbonate (276 mg, 2.00
mmol)
241
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
in dimethyl formamide (20 mL) was stirred at room temperature overnight. The
reaction
was diluted water, stirred for 10 min, and the resultant precipitate was
isolated. The solid
was dissolved in ethyl acetate (20 mL), dried over sodium sulfate, filtered,
and the
solvent removed under vacuum to afford 145 mg of P-055 as a pale yellow solid
in 37%
yield. 1H NMR (400 MHz, CDC13) 3.81 (s, 3 H), 4.91 (s, 2 H), 6.96 (d, J= 8.5
Hz, 1
H),7.45(d,J=2.2Hz,1H),7.48-7.66 (m,3H),7.80(d,J=7.8Hz,1H),8.10-8.22
(m,2H),8.39(t,J=1.8Hz,1H),8.96(dd,J=4.9,1.4Hz,1H)ppm.LCMS=95%
purity. TSI (+) = 390.40 (M+1).
Example 174. Preparation of P-062
Palladium(II)acetate
Potassium Carbonate O
0-9 + Methanol, Water
HO' OH ~NIO - NCO
O
O
1-169
[00514] Synthesis of 2-Methoxy-3'-nitro-biphenyl (1-169). A suspension of 2-
methoxyphenylboronic acid (911 mg, 6.00 mmol), 3-nitro-iodobenzne (1.24 g,
5.00
mmol), palladium(II) acetate (22 mg, 0.10 mmol), and solid potassium carbonate
(1.38 g,
10. mmol), in methanol (25 mL), and water (5 mL) was stirred at room
temperature
overnight. The reaction was diluted with ethyl acetate (50 mL), washed with
water (2 x
50 mL) and brine, dried over sodium sulfate, filtered, and the solvent removed
under
vacuum to give crude product. The product was purified by silica gel column
chromatography (hexanes/ethyl acetate 9:1) to afford 940 mg of I-169 as a
white solid in
82% yield. 'H NMR (400 MHz, CDC13) 3.84 (s, 3 H), 6.96 - 7.14 (m, 2 H), 7.30 -
7.44
(m,2H),7.56(t,J=8.0Hz,1H),7.86(d,J=7.7Hz,1H),8.17(dd,J=8.2,1.2Hz,1
H), 8.42 (s, 1 H) ppm.
242
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
CI Pd(dba)2
PPh3 ri
O NNa2CO3 O g OH Dimethylformamide
O HO Water
O-J 4 O
1-168 P-062
[00515] Synthesis of 4-(3-Benzo[1,3]dioxol-5-yl-4-methoxy-benzyl)-3,5-dimethyl-
isoxazole (P-062). To a solution of 1-168 (138 mg, 0.500 mmol) and 3,5-
dimethyl-
isoxazole-4-boronic acid (70 mg, 0.500 mmol) in dimethyl formamide (5 mL) were
added palladium(O) bis(dibenzylideneacetone (14 mg, 0.025 mmol),
triphenylphosphine
(13 mg, 0.0500 mmol) and 1 M aqueous sodium carbonate (1.5 mL, 1.5 mmol). The
resultant suspension was stirred at 85 C for 72 h. The reaction was
concentrated under
reduced pressure, diluted with ethyl acetate (10 mL), washed with water (3 x
10 mL) and
brine, dried over sodium sulfate, filtered, and the solvent removed under
reduced
pressure. The crude residue was purified by silica gel preparatory thin layer
chromatography eluting with 1:1 hexanes and dichloromethane to afford 16.2 mg
(10%)
of P-062. 'H NMR (400 MHz, CDC13) 2.11 (s, 3 H), 2.31 (s, 3 H), 3.64 (s, 2 H),
3.79
(s, 3 H), 5.98 (s, 2 H), 6.80 - 6.94 (m, 3 H), 6.94 - 7.05 (m, 3 H) ppm. LCMS
= 100%
purity. APCI(+)= 338.10 (M+1).
Example 175. Preparation of P-066
O
N NHZ
O O
Hydrazine Hydrate
O Ethanol
N_' O N'0
O p-
P-052 1-170
[00516] Synthesis of C-(6-Methoxy-3'-nitro-biphenyl-3-yl)-methylamine (1-170):
A
suspension of P-052 (700 mg, 1.80 mmol) and hydrazine hydrate (0.35 mL. 7.2
mmol) in
ethanol (60 mL) was stirred at reflux for 6 h. The reaction was cooled to room
temperature, filtered, and the solvent was evaporated. The residue was
dissolved in
243
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
ethanol (15 mL), precipitated with water (50 mL), and filtered to afford 97.9
mg as a
white solid in 21% yield.
NH2 NON
O N,N-dimethylformamide dimethyl acetal O LN
Acetonitrile, Acetic Acid, Hydrazine
N O N O
O O
1-170 P-066
[00517] Synthesis of 4-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-4H-
[1,2,4]triazole
(P-066). A solution of formic hydrazine (25 mg, 0.41 mmol) and N,N-
dimethylformamide dimethyl acetal (0.050 mL, 0.41 mmol) in acetonitrile (0.5
mL) was
stirred at 50 C for 30 min. To the solution was added 1-170 (98 mg, 0.38 mmol)
and
acetic acid (0.5 mL). The reaction was then stirred at 160 C for 6h. The
reaction was
cooled to room temperature and the concentrated under vacuum. The reaction was
diluted with water (30 mL) and extracted with dichloromethane (2 x 30 mL), the
combined extracts were washed with brine, dried over sodium sulfate, filtered,
and the
solvent removed under vacuum. The crude material was purified by silica gel
preparatory
thin layer chromatography (9:1 dichloromethane:methanol) to afford 19.4 mg of
P-066 in
17% yield. 1H NMR (400 MHz, CDC13): 3.86 (s, 3H), 5.19 (s, 2H), 7.03 (d, J=
8.5 Hz,
1H), 7.17 - 7.25 (m, 2H), 7.58 (t, J= 8.0 Hz, 1H), 7.79 (d, J= 7.8 Hz, 1H),
8.16 - 8.24
(m, 3H), 8.37 (t, J= 1.9 Hz, 1H) ppm. LCMS = 94.0% purity. APCI(+) =31 1.1
(M+1).
[00518] Scheme 46.
OH
rNN NN 1.) n-butyl litium N
Tetrahydrofuran O Triethylsilane O
o Trifluoroacetic acid
2.) H O
o O_/ O-J
i I P-073 P-075
o
o-/
1-170
244
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 176. Preparation of P-073
[00519] Synthesis of (3-Benzo[1,3]dioxol-5-yl-4-methoxy-phenyl)-(2-methyl-2H-
pyrazol-3-yl)-methanol (P-073). A solution of 1-methyl pyrazole (123 mg, 1.50
mmol) in
tetrahydrofuran (10 mL) was cooled to 0 C in an ice bath, and n-butyl lithium
(2.5 M in
hexanes, 0.80 mL, 2.0 mmol) was added. The yellow solution was stirred at 0 C
for 30
min followed by the addition of 1-170 (256 mg, 1.00 mmol), and the resultant
pale green
solution stirred for 2 h. The reaction was diluted with water (50 mL), and
extracted with
ethyl acetate (2 x 30 mL). The combined extracts were washed with brine, dried
over
sodium sulfate, filtered, and concentrated under reduced pressure. The crude
material
was purified by silica gel column chromatography (1:1 hexanes:dichloromethane)
to
afford 259.1 mg of P-073 as a pale yellow solid in 77% yield. 'H NMR (400 MHz,
CDC13): 3.81 (s, 3H), 3.83 (s, 3H), 5.90 (s, 1H), 5.98 (s, 2H), 6.12 (d, J=
1.6 Hz, 1H),
6.82-6.89(m,1H),6.91-6.98(m,2H),7.03(d,J=1.5Hz,1H),7.29 (s, 2 H), 7.40 (d,
1 H) ppm. LCMS = 98.9%; APCI(+) =339.1 (M +1).
Example 177. Preparation of P-075
[00520] Synthesis of 5-(3-Benzo[1,3]dioxol-5-yl-4-methoxy-benzyl)-1-methyl-lH-
pyrazole (P-075). To a solution of P-073 (169 mg, 0.500 mmol) in
trifluoroacetic acid
(2.0 mL), was added triethylsilane (0.50 mL, 3.0 mmol). The reaction was
stirred at
room temperature overnight. The reaction was diluted with water (10 mL) and
extracted
with dichloromethane (2 x 10 mL). The combined organic extracts were washed
with
brine, dried over sodium sulfate, filtered, and the solvent removed under
vacuum. The
crude product was purified by silica gel column chromatography (20% methanol
in
dichloromethane) to afford 39.1 mg (24%) of P-075. 'H NMR (400 MHz, CDC13)
7.40
(d, J= 1.7 Hz, 1H), 7.07 (d, J= 2.3 Hz, 1H), 7.02 - 7.06 (m, J= 2.4 Hz, 1H),
7.02(d, J=
1.7 Hz, 1H), 6.91 - 6.94 (m, 1H), 6.90 (s, 1H), 6.86 (t, J= 8.5 Hz, 2H), 5.98
(s, 2H), 3.95
(s, 2H), 3.80 (s, 3H),3.74 (s, 3H) ppm.
245
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 178. Preparation of P-074
CI
I \ Nom/
0 N N\ K2CO3
O
Dimethylformamide
O
O
1-168 P-074
[00521] Synthesis of 1-(3-Benzo[1,3]dioxol-5-yl-4-methoxy-benzyl)-1H-pyrazole
(P-
074). A suspension of pyrazole (136 mg, 2.00 mmol), 1-168 (108 mg, 1.00 mmol),
and
solid potassium carbonate (276 mg, 2.00 mmol) was stirred at room temperature
overnight. Water (30 mL) was added, and the suspension extracted with ethyl
acetate (2
x 30 mL). The combined organic layers were washed with brine, dried over
sodium
sulfate, filtered, and the solvent removed under vacuum. The product was
purified by
silica gel column chromatography (10% ethyl acetate in hexanes) to afford 44.4
mg of P-
074 as a pale yellow oil in 14% yield. 'H NMR (400 MHz, CDC13) 7.54 (d, J= 1.8
Hz,
1H), 7.38 (d, J= 2.3 Hz, 1H), 7.17 (dd, J= 4.4, 2.2 Hz, 2H), 7.01 (d, J= 1.7
Hz, 1H),
6.91 - 6.94 (m, 2H), 6.82 - 6.87 (m, 1H), 6.26 (t, J= 2.1 Hz, 1H), 5.98 (s,
2H), 5.28 (s,
2H), 3.80 (s,3H)
Example 179. Preparation of P-077
Br
"N
N~~
O H K2CO3
N-N O
Dimethylformamide
N_ \ NCO
O 6
1-70
P-077
[00522] Synthesis of 1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-1H-pyrazole (P-
077). A suspension of 1-70 (322 mg, 1.00 mmol), pyrazole (136 mg, 2.00 mmol),
and
solid potassium carbonate (276 mg, 2.00 mmol) in dimethyl formamide (10 mL)
was
stirred at room temperature overnight. The reaction was diluted water (30 mL),
and
246
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
extracted with ethyl acetate (2 x 30 mL).The combined extracts were washed
with brine,
dried over sodium sulfate, filtered, and the solvent removed under vacuum. The
residue
was purified by silica gel column chromatography (15% ethyl acetate in
hexanes) to give
P-077 (148 mg, 48% yield) as a white solid. 'H NMR (400 MHz, DMSO-d6): 8.27
(s,
1H), 8.17 - 8.22 (m, 1H), 7.89 - 7.94 (m, 1H), 7.83 (d, J= 2.2 Hz, 1H), 7.72
(t, J= 8.0
Hz, 1H), 7.44 (d, J= 1.7 Hz, 1H), 7.35 (d, J= 2.1 Hz, 1H), 7.28 - 7.33 (m,
1H), 7.15 (d,
J= 8.5 Hz, 1H), 6.19 - 6.30 (m, 1H), 5.32 (s, 2H), 3.79 (s, 3H) ppm.
LCMS = 99.7% purity. APCI (+) = 310.1 (M+1)
Example 180. Preparation of P-087
I "~ O
O O N
+ CI N0 O O
NH s + O 1,2-Dimethoxyethane
N A
O I N
1-171 6-
P-087
[00523] Synthesis of [4-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-3,5-dimethyl-
pyrazol-1-yl]-acetic acid ethyl ester (P-087). To a solution of 1-171 (102 mg,
0.300
mmol) in 1,2-dimethoxyethane (5 mL), were added ethyl hydrazinoacetate
hydrochloride
(93 mg, 0.60 mmol) and 4 Angstrom molecular sieves (200 mg), and the reaction
was
stirred at reflux for 3 h. The hot suspension was filtered and the solvent
removed under
vacuum. The residue was dissolved in dichloromethane (10 mL) and washed with
water
(30 mL). The aqueous wash was extracted with dichloromethane (2 x 30 mL), and
the
extracts combined. The organic solution was washed with brine, dried over
sodium
sulfate, filtered, and the solvent removed under vacuum. The residue was
purified by
silica gel column chromatography (2:1 hexanes:ethyl acetate) to give P-087
(97.1 mg,
76% yield). 1H NMR (400 MHz, CDC13): 8.38 (t, J= 1.9 Hz, 1H), 8.15 (dd, J=
8.2, 2.3
Hz, 1H), 7.79 - 7.82 (m, 1H), 7.53 - 7.56 (m, 1H), 7.07 - 7.10 (m, 2H), 6.90
(d, J= 9.1
Hz, 1H), 4.79 (s, 2H), 4.21 (q, J= 7.1 Hz, 2H), 3.80 (s, 3H), 3.74 (s, 2H),
2.13 (s, 3H),
2.13 (s, 3H), 1.26 (t, J= 7.1 Hz, 3H) ppm. LCMS: 98.7% purity. APCI(+) = 310.1
(M-
29)
247
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 181. Preparation of P-088
1 o
O O I i N
O
+ H
_O H 2 N'N0 1,2-Dimethoxyethane
N 4
O I N
1-171 O
P-088
[00524] Synthesis of 2-[4-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-3,5-
dimethyl-
pyrazol-1-yl]-ethanol (P-088). To a solution of 1-171 (102 mg, 0.300 mmol) in
1,2-
dimethoxyethane (5 mL), were added 2-hydroxyethyl hydrazine (0.037 mL, 0.60
mmol)
and 4 Angstrom molecular sieves (200 mg), and the reaction was stirred at
reflux for 3 h.
The hot suspension was filtered and the solvent removed under vacuum. The
residue was
dissolved in dichloromethane (10 mL) and washed with water (30 mL). The
aqueous
wash was extracted with dichloromethane (2 x 30 mL), and the extracts
combined. The
organic solution was washed with brine, dried over sodium sulfate, filtered,
and the
solvent removed under vacuum. The crude material was purified by silica gel
column
chromatography (2:1:0.3 hexanes:ethyl acetate: methanol) to give P-088 (61.0
mg, 53%
yield) as a yellow solid. 'H NMR (400 MHz, CDC13): 8.37 (t, J= 2.0 Hz, 1H),
8.16 (dt,
J= 8.2, 1.1 Hz, 1H), 7.81 (dt, J= 7.7, 0.8 Hz, 1H), 7.54 - 7.57 (m, 1H), 7.06 -
7.10 (m,
2H), 6.91 (d, J= 9.1 Hz, 1H), 4.05 - 4.09 (m, 2H), 3.95 - 4.02 (m, 2H), 3.80
(s, 3H), 3.73
(s, 2H), 2.14 (s, 3H), 2.16 (s, 3H) ppm. LCMS: 98.2% purity. APCI(+) = 382.1
(M+1)
Example 182. Preparation of P-089
NH
V"'_N4
O O O + 1,2-Dimethoxyethane NOD N'NH2 4 1-171 P-089
248
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00525] Synthesis of 4-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-3,5-dimethyl-
lH-
pyrazole (P-089). To a solution of 1-171 (102 mg, 0.300 mmol) in 1,2-
dimethoxyethane
(5 mL), were added hydrazine (0.029 mL, 0.60 mmol) and 4 Angstrom molecular
sieves
(200 mg), and the reaction was stirred at reflux for 3 h. The hot suspension
was filtered
and the solvent removed under vacuum. The residue was dissolved in
dichloromethane
(10 mL) and washed with water (30 mL). The aqueous wash was extracted with
dichloromethane (2 x 30 mL), and the extracts combined. The organic solution
was
washed with brine, dried over sodium sulfate, filtered, and the solvent
removed under
vacuum. The crude material was purified by silica gel column chromatography
(1:1
hexanes:ethyl acetate) to give P-089 (67.8 mg, 67% yield) 1H NMR (400 MHz,
CDC13):
8.38 (s, 1H), 8.13 - 8.20 (m, 1H), 7.75 - 7.83 (m, 1H), 7.50 - 7.58 (m, 1H),
7.05 -
7.13,(m, 2H), 6.91 (d, J= 8.1 Hz, 1H), 3.80 (s, 3H), 3.74 (s, 2H), 2.18 (s,
6H) ppm.
LCMS = 97.2% purity. APCI (+) = 338.1 (M+1).
Example 183. Preparation of P-090
o
N
O O I / 'N F
+ H FF O F F
H N Nv F 1,2-Dimethoxyethane
N z
N10
1-171 6-
[00526] P-090
Synthesis of 4-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-3,5-dimethyl-l-
(2,2,2-trifluoro-ethyl)-1H-pyrazole (P-090). To a solution of 1-171 (102 mg,
0.300
mmol) in 1,2-dimethoxyethane (5 mL), were added 2,2,2-trifluoroethyl hydrazine
(0.098
mL, 0.60 mmol) and 4 Angstrom molecular sieves (200 mg), and the reaction was
stirred
at reflux for 3 h. The hot suspension was filtered and the solvent removed
under vacuum.
The residue was dissolved in dichloromethane (10 mL) and washed with water (30
mL).
The aqueous wash was extracted with dichloromethane (2 x 30 mL), and the
extracts
combined. The organic solution was washed with brine, dried over sodium
sulfate,
filtered, and the solvent removed under vacuum. The crude material was
purified by
silica gel column chromatography (5:1 hexanes:ethyl acetate) to give P-090
(64.5 mg,
249
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
51% yield) as a yellow-red oil. 1H NMR (400 MHz, CDC13): 8.38 (t, J= 2.0 Hz,
1H),
8.16 (ddd, J= 8.2, 2.3, 1.1 Hz, 1H), 7.78 - 7.81 (m, 1H), 7.52 -7.55 (m, 1H),
7.04 - 7.07
(m, 2H), 6.91 (d, J= 9.1 Hz, 1H), 4.58 (q, J= 8.4 Hz, 2H), 3.78 - 3.81 (m,
3H), 3.73 (s,
2H), 2.20 (s, 3H), 2.14 (s, 3H) ppm. LCMS = 97.6% purity. APCI (+) = 420.1
(M+1).
Example 184. Preparation of P-101
N
N
O V_~~N N
O O Ammonia o ~-NH2
O
Ethanol
N"O O 6-
P-087 P-101
[00527] Synthesis of 2-[4-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-3,5-
dimethyl-
pyrazol-1-yl]-acetamide (P-101). To a solution of P-087 (212 mg, 0.500 mmol)
in
methanol (5 mL) was added ammonia (7 M in methanol, 0.5 mL, 3.5 mmol) and the
solution was stirred at room temperature overnight. The resulting suspension
was
concentrated under vacuum, and dissolved in ethyl acetate (10 mL). The
reaction was
washed with water (10 mL) and brine (10 mL), dried over sodium sulfate,
filtered, and
the solvent removed under vacuum. The resulting solid was triturated in
dichloromethane
(lmL) in hexanes (10 mL) to give P-101 (85.8 mg, 44% yield) as a white solid.
1H NMR
(400 MHz, CDC13) 8.31 - 8.37 (m, 1H), 8.14 - 8.20 (m, 1H), 7.77 - 7.84 (m,
1H), 7.51 -
7.59 (m, 1H), 7.01- 7.10 (m, 2H), 6.91 (d, J= 8.1 Hz, 1H), 6.05 (br s, 1H),
5.39 (br s,
1H), 4.68 (s, 2H), 3.80 (s, 3H), 3.73 (s, 2H), 2.17 (s, 3H), 2.16 (s, 3H) ppm.
LCMS
=98.2% purity. APCI (+) = 395.1 (M+1).
OH I
1.) n-butyl litium ~ N N I /N
N" Tetrahydrofuran / Triethylsilane 0
L /N O
0 Trifluoroacetic acid
2.) H 0
0 O
o ~/
1-172 O\ F F F
1-173 F P-115
0
O F
F
250
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 185. Preparation of P-115
[00528] Synthesis of 3-(2,2-Difluoro-benzo[1,3]dioxol-5-yl)-4-methoxy-
benzaldehyde (1-172). To a solution of 5-formyl-2-methoxy phenyl boronic acid
(1.0 g,
5.5 mmol) in water (6 mL) and methanol (30 mL), was added 5-bromo-2,2-difluoro-
1,3-
benzodioxole (0.97 mL, 7.2 mmol), solid potassium carbonate (1.5 g, 11 mmol),
and
palladium(II) acetate (25 mg, 0.11 mmol). The reaction was stirred at room
temperature
for 16 h. The black mixture was diluted with water (3 0 mL) and extracted with
ethyl
acetate (2 x 30 mL). The combined extracts were decolorized with activated
charcoal,
dried over sodium sulfate, filtered, and concentrated to 20 mL under vacuum.
The
solution was purfied by silca gel column chromatography eluting with
hexanes/ethyl
acetate (11:1) to give I-172 (620 mg, 39% yield) as a white solid.
[00529] Synthesis of [3-(2,2-Difluoro-benzo[1,3]dioxol-5-yl)-4-methoxy-phenyl]-
(2-
methyl-2H-pyrazol-3-yl)-methanol (I-173). A solution of 1-methyl pyrazole (123
mg,
1.5 mmol) under a positive nitrogen atmosphere was cooled to 0 C in an ice
water bath.
To the stirring solution was added n-butyl lithium (2.5 M in hexanes, 0.80 mL,
2.0
mmol) The reaction mixture was stirred at 0 C for 30 min, and 1-172 (292 mg,
1.0
mmol) was added in one portion. The reaction was stirred an additional 2 h.
The reaction
was diluted with water (50 mL), and extracted with ethyl acetate (2 x 30 mL).
The
combined extracts were washed with brine, dried over sodium sulfate, filtered,
and the
solvent removed under reduced pressure to give I-173 (220 mg, 59% yield) as a
yellow
solid.
[00530] Synthesis of 5-[3-(2,2-Difluoro-benzo[1,3]dioxol-5-yl)-4-methoxy-
benzyl]-1-
methyl-1H-pyrazole (P-115). To a solution of 1-173 (187 mg, 0.500 mmol) in
trifluoroacetic acid (2.0 mL), was added triethyl silane (0.50 mL, 3.0 mmol).
The
reaction was stirred at room temperature overnight. The reaction was diluted
with water
(10 mL) and extracted with dichloromethane (2 x 10 mL). The combined extracts
were
washed with brine, dried over sodium sulfate, filtered, and the solvent
removed under
vacuum. The residue was purified by silica gel column chromatography (5:2
hexanes/ethyl acetate) and silica gel preparatory thin layer chromatography
(20:1
dichloromethane/methanol) to give P-115 (29.9 mg, 17% yield). 'H NMR (400 MHz,
251
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
CDC13): 3.74 (s, 3 H), 3.81 (s, 3 H), 3.96 (s, 2 H), 6.02 (d, J=1.6 Hz, 1H),
6.91 (d, J=8.3
Hz, 1 H), 7.00 - 7.17 (m, 4 H), 7.24 (d, J=1.5 Hz, 1 H), 7.40 (d, J=1.6 Hz, 1
H) ppm.
LCMS = 100 % purity. APCI (+) = 359.1 (M+1).
Example 186. Preparation of P-201
N HO- OH Pd[P(Ph3)]CIZ F N
F N + Na2C03(aq) FO
FO Xylene, Water
Br O 4
1-174
O-/
P-201
[00531] Synthesis of 5-(3-Benzo[1,3]dioxol-5-yl-4-difluoromethoxy-benzyl)-1-
methyl-lH-pyrazole (P-20 1). A suspension of benzo[ 1,3 ]dioxole-5-boronic
acid (108
mg, 0.65 mmol), 1-174 (158 mg, 0.500 mmol), palladium(II)
triphenylphosphindichloride
(35 mg, 0.050 mmol), and 1M aqueous sodium carbontate (1.0 mL, 1.00 mmol) in
xylene (3 mL) was stirred at 150 C for 24 h. The reaction was diluted with
ethyl acetate
(10 mL), washed with water (3 x 10 mL) and brine, dried over sodium sulfate,
filtered,
and the solvent removed under vacumm. The residue was purified by silica gel
column
chromatography (4:1 hexanes/ethyl acetate), and then by silica gel preparatory
thin layer
chromatography (5:1 dichloromethane/ acetone) to give P-201 (13.4 mg, 7%
yield).
[00532] 1H NMR (400 MHz, CDC13): 3.64 (s, 3 H), 4.05 (s, 2 H), 5.82 (s, 1 H),
5.95
(s, 2 H), 6.63 -6.94 (m, 7 H), 7.59 (s, 1 H) ppm. LCMS = 99% purity. APCI (-)
= 321.1
(M-3 7).
Example 187. Preparation of P-306
-fOH H 0
0 Hydrazine hydrate N NH2 N } NH2
0 0 Cyanogenbromide 0 N
O
A 1,2-dimethoxyethane
CI CI
CI
1-175 1-176 P-306
[00533] Synthesis of 5-(3'-Chloro-6-methoxy-biphenyl-3-ylmethyl)-
[1,3,4]oxadiazol-
252
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
2-ylamine (P-306). A solution of 1-176 (277 mg, 1.00 mmol) in hydrazine
hydrate (0.5
mL) was stirred at 100 C overnight. The reaction was concentrated under vacuum
to
obtain a gummy white solid. The crude material was dissolved in 1,2-
dimethoxyethane
(0.5 mL) and cyanogen bromide (212 mg, 2.00 mmol) was added. The reaction was
stirred at room temperature for 3 h. The reaction was diluted with
dichloromethane (5
mL) and washed with 1 M aqueous sodium hydroxide (5 mL). The organic layer was
separated, dried over sodium sulfate, filtered and concentrated under vacuum.
The
residue was purified by silica gel preparatory thin layer chromatography
(hexanes/ethyl
acetate) to give P-306 (64.2 mg, 30% yield). 'H NMR (400 MHz, DMSO-d6): 3.77
(s, 3
H), 4.01 (s, 2 H), 6.87 (s, 2 H), 7.10 (d, J=8.5 Hz, 1 H), 7.21 -7.29 (m, 2
H), 7.36 - 7.47
(m, 3 H), 7.50 (s, 1 H) ppm. LCMS = 97.1% purity. APCI(+) = 316 (M).
Example 188. Preparation of P-393
HO
HCI
T-F N NH ~N
~OCI K2C03 O F I NN
a OH
I dimethylformamide
CI
1-177 P-393
[00534] Synthesis 1-[5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyrimidin-2-yl]-azetidin-3-ol (P-393 ). A suspension of 1-177 (72 mg, 0.20
mmol),
azetidin-3-ol hydrochloride (44 mg, 0.40 mmol) and potassium carbonate (55 mg,
0.40
mmol) in dimethyl formamide (1.5 mL) was stirred at 80 C overnight. The
mixture was
cooled to room temperature, diluted with water (5 mL), and extracted with
diethyl ether
(3 x 5 mL). The combined extracts were concentrated, and the crude material
purified by
silica gel column chromatography (2:1 hexanes/ethyl acetate) to give P-393
(73.3 mg,
92% yield) as a cream white solid. 1H NMR (400 MHz, CDC13): 3.75 (s, 3 H),
3.77 (s, 2
H), 3.98 (dd, J= 10.3, 4.3 Hz, 2 H), 4.34 - 4.43 (m, 2 H),
4.77(brs,1H),6.70(d,J=8.3
Hz, 1 H), 7.08 (t, J= 8.6 Hz, 1 H), 7.26 - 7.40 (m, 4 H), 8.21 (s, 2 H) ppm.
LCMS = 100% purity. APCI (+) = 400 (M).
253
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 189. Preparation of P-397
N 0 0 N+.O O
) H O.O H O F H ) H
O F H
diethyl ether
CI
CI
P-356 P-397
[00535] Synthesis of 1-[5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-1-
oxy-pyridin-2-yl]-3-ethyl-urea (P-397). A solution of P-356 (82.7 mg, 0.200
mmol) in
diethyl ether (5 mL) was stirred at room temperature with peracetic acid (32%
wt., 0.06
mL, 0.300 mmol) for 3 h. The mixture was concentrated and the residue purified
by
silica gel column chromatography (19:1 dichloromethane/methanol) to give P-397
(27.6
mg, 32% yield).
[00536] 'H NMR (400 MHz, CDC13): 1.12 (t, 3 H) 3.17 - 3.35 (m, 2 H), 3.72 -
3.80
(m, 3 H), 3.87 (s, 2 H), 6.64 - 6.78 (m, 1 H), 6.86 (br s, 1 H), 7.02 - 7.17
(m, 1 H), 7.22
(s, 1 H), 7.29 - 7.43 (m, 3 H), 7.93 (s, 1 H), 8.36 (d, J= 8.9 Hz, 1 H), 9.73
(s, 1 H) ppm.
LCMS = 100% purity. APCI (-) = 428.1 (M-2).
Example 190. Preparation of P-398
N O N iNH2 O O
H
pyridine,
tetrahydrofuran
1-178 P-398
[00537] Synthesis of [5-(3'-Cyano-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-
2-yl]-carbamic acid ethyl ester (P-398). A solution of 1-178 (166 mg, 0.500
mmol) in
tetrahydrofuran was cooled to 0 T. To the reaction was added pyridine (0.08
mL, 1
mmol), and the reaction stirred for 10 min. To the solution was added ethyl
chloroformate (0.09 mmol, 1 mmol) slowly, and the yellow suspension was
stirred at
254
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
room temperature overnight. The reaction was allowed to sit after the addition
of water
(10 mL), and the solid that settled to the bottom was collected by filtration.
The solid
was suspended in dimethyl sulfoxide (5 mL) and water (5 mL), and filtered to
give P-398
(14.6 mg, 7.2% yield) as a white solid. 'H NMR (400 MHz, CDC13): 1.31 (t, J=
7.1 Hz,
3 H), 3.76 (s, 3 H), 3.91 (s, 2 H), 4.23 (q, J= 7.1 Hz, 2 H), 6.73 (d, J= 8.6
Hz,1H),7.12
(t,J=8.5Hz,1H),7.32-7.41(m,1H),7.44-7.56(m,2H),7.63(d,J=7.8Hz,2H),
7.70 (s, 1 H), 7.88 (d, J= 8.5 Hz, 1 H), 8.12 (s, 1 H) ppm. LCMS = 94.0 %
purity.
APCI(+) = 406.1 (M+1).
Example 191. Preparation of P-405-HC1
N O \N OII N O
O F NHZ N O F NJ H 4 N HCI in dioxane O F NN
pyridine, diethyl ether H H
tetrahydrofuran
~ ~ HCI
N P-405 N
1-178 P-405-HCI
[00538] Synthesis of 1-[5-(3'-Cyano-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-2-yl]-3-ethyl-urea hydrochloride salt (P-405-HC1). A solution of 1-178
(166 mg,
0.500 mmol) and ethyl isocyanate (0.2 mL, 2.5 mmol) in pyridine (0.5 mL) was
stirred at
room temperature overnight. To the solution was added water (5 mL), and the
reaction
stirred for 1 h. The solid that formed was filtered, triturated with
tetrahydrofuran and
water to give a residue. This residue was purified by silica gel column
chromatography
(100:1 dichloromethane/methanol) to give impure P-405 (131 mg, 65% yield). The
whole of the material was suspended in diethyl ether (1.5 mL) and 4 N hydrogen
chloride in dioxane (0.75 mL, 3.0 mmol) was added. Additional hydrogen
chloride
solution was added (2.25 mL, 9 mmol). The reaction mixture was allowed to stir
at room
temperature overnight. The precipitate was filtered and dried to give P-405-
HC1(52.5
mg, 24% yield).
[00539] 'H NMR (400 MHz, DMSO-d6): 1.09 (t, J= 7.2 Hz, 3 H), 3.12 - 3.25 (m, 2
H), 3.74 (s, 3 H), 3.94 (s, 2 H), 6.97 (d, J= 8.7 Hz, 1 H), 7.25 - 7.47 (m, 2
H), 7.53 - 7.92
(m, 5 H), 8.10 (s, 1 H) ppm. LCMS = 100% purity. APCI (+) = 405.1 (M+1).
255
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 192. Preparation of P-406
H 0õ,.,0
~N N
O F NCI Sodium Hydride I / I
Tetrahydrofuran O F N O
\ CI I CI
1-177 P-406
[00540] Synthesis of 5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-2-
((R)-1-
methyl-pyrrolidin-3-yloxy)-pyrimidine (P-406). To a solution of I-177 (65 mg,
0.18
mmol) and (R)-(-)-methyl-3-pyrrolidinol (0.039 mL, 0.36 mmol) in
tetrahydrofuran (1
mL) was added NaH (60% weight dispersion, 29 mg, 0.72 mmol) slowly. Once gas
evolution ceased, the reaction was stirred at 80 C for 4 h. The reaction was
cooled to
room temperature and diluted with water (10 mL). The mixture was extracted
with ethyl
acetate (3 x 2 mL), the pH of the aqueous layer adjusted to pH 7 by addition
of 1 M
aqueous hydrochloric acid, and the aqueous mixture again extracted with ethyl
acetate (2
x 2 mL). The combined extracts were dried over sodium sulfate, filtered and
the solvent
removed under vacuum. The residue was purified by silica gel column
chromatography
(9:1 dichloromethane / methanol) to give P-406 (46.8 mg, 61% yield) as a thick
pale
yellow oil.
[00541] 'H NMR (400 MHz, CDC13): 2.03 - 2.48 (m, 2 H), 2.54 (s, 3 H), 2.72 -
2.93
(m, 2 H), 3.00 (br s, 1 H), 3.47 (dd, J= 11.4, 5.9 Hz, 1 H), 3.77 (s, 3 H),
3.87 (s, 2 H),
5.45 (t, J= 6.4 Hz, 1 H), 6.73 (d, J= 8.5 Hz, 1 H), 7.12 (t, J= 8.5 Hz, 1 H),
7.29 - 7.41
(m, 3 H), 8.37 (s, 2 H) ppm. LCMS = 100% purity. MS (ESI+) 428.1 (M+H)..
256
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 193. Preparation of P-417
HO
HCI
N
\ I j I N NH TFN-1j"N
O F N~ CI K2CO3 O dimethylformamide OH
N
1-177 P-417
[00542] Synthesis of 2'-Fluoro-3'-[2-(3-hydroxy-azetidin-1-yl)-pyrimidin-5-
ylmethyl]-
6'-methoxy-biphenyl-3-carbonitrile (P-417). To a stirred solution of I-177 (61
mg, 0.17
mmol) and azetidin-3-ol hydrochloride (40 mg, 0.34 mmol) in dimethylformamide
(0.5
mL) was added solid potassium carbonate (48 mg, 0.34 mmol), and the pale
yellow
solution was subsequently stirred at 80 C overnight. The reaction was cooled
to room
temperature, diluted with water (15 mL), stirred for 30 min at room
temperature, and the
resulting precipitate collected by filtration. The crude was purified by
trituration in
dichloromethane/diethyl ether/ hexanes to give P-417 (26 mg, 39% yield) as a
white-
cream solid. 1H NMR (400 MHz, DMSO-d6): 3.68 - 3.74 (m, 5 H), 3.77 (s, 2 H),
4.08 -
4.26 (m, 2 H), 4.52 (br s, 1 H), 5.63 (d, J= 6.4 Hz, 1 H), 6.95 (d, J= 8.7 Hz,
1 H), 7.32
(t, J= 8.7 Hz, 1 H), 7.54 - 7.74 (m, 2 H), 7.74 - 7.91 (m, 2 H), 8.24 (s, 2 H)
ppm.
LCMS =100% purity. APCI (+) = 391.1 (M+1).
Example 194. Preparation of P-513
IN 0 N 0
NaN3
O F N N Ammonium Chloride O F N)~ N
H H H H
I
\
Dimethylformamide H
N I N
N-N'
P-405 P-513
[00543] Synthesis of 1-Ethyl-3-{5-[2-fluoro-6-methoxy-3'-(1H-tetrazol-5-yl)-
biphenyl-3-ylmethyl]-pyridin-2-yl}-urea (P-513). A mixture of P-405 (405 mg,
1.00
mmol), sodium azide (325 mg, 5.00 mmol), and ammonium chloride (374 mg, 7.00
mmol) in dimethylformamide (10 mL) was stirred at 80 C overnight. The
reaction was
cooled to room temperature, diluted with ethyl acetate (30 mL), and washed
with water
257
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
(50 mL). The aqueous wash was extracted with ethyl acetate (2 x 30 mL), the
extracts
combined, dried over sodium sulfate, and the solvent removed under vacuum. The
residue was purified by silica gel column chromatography (19: Ito 4:1
dichloromethane/methanol) to give P-513 (11.5 mg, 2.5% yield) as an off-white
solid.
'H NMR (400 MHz, DMSO-d6): 1.05 - 1.10 (m, 3 H), 3.11 - 3.21 (m, 2 H), 3.72
(s, 3 H),
3.88 (s, 2 H), 6.93 (d, J= 8.6 Hz, 1 H), 7.10 - 7.37 (m, 3 H), 7.42 (s, 1 H),
7.54 (s, 1 H),
7.90 (br s, 2 H), 8.07 (s, 2 H), 9.09 (s, 1 H) ppm. LCMS =95.9% yield.APCI (+)
_
448.1
Example 195. Preparation of P-456
Br
IN
O / F HO,B,OH Palladium(0)tetrakis(triphenyl phosphine) \
Sodium Carbonate O F F
+ Toluene, Ethanol, Water
\ I CI I ~N 0
F Cl
1-33
P-456
[00544] Synthesis of 5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-2-
fluoro-
pyridine (P-456). A suspension of 1-33 (824 mg, 2.50 mmol), 2-fluoropyridin-5-
boronic
acid (352 mg, 2.50 mmol), 2 M aqueous sodium carbonate (2.5 mL, 5.00 mmol),
and
palladium(0)tetrakis(triphenylphosphine) (144 mg, 0.125 mmol) in toluene (10
mL) and
ethanol (2.5 mL) was stirred at 80 C overnight under a high pressure nitrogen
atmosphere. The reaction was cooled to room temperature, diluted with water
(10 mL),
and extracted with ethyl acetate (2 x 20 mL). The combined extracts were dried
over
sodium sulfate, filtered, and concentrated under vacuum. The residue was
purified by
silica gel column chromatography (4:1 hexanes/ethyl acetate) to give P-456
(695 mg,
78% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6): 3.73 (s, 3 H), 3.98
(s, 2
H), 6.95 (d, J = 8.6 Hz, 1 H), 7.11 (dd, J = 8.4, 2.7 Hz, 1 H), 7.25 - 7.51
(m, 5 H), 7.82
(td, J = 8.2, 2.4 Hz, 1 H), 8.14 (s, 1 H), 8.32 (s 1H) ppm. LCMS = 97.4%
purity. APCI
(+) = 346 (M)
Example 196. Preparation of P-457
258
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
\ N Azetidin-3-ol N
O - F F Sodium Hydride O F N
Dimethylformamide ~OH
I o
CI CI
P-456 P-457
[00545] Synthesis of 1-[5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-2-yl]-azetidin-3-ol (P-457). A mixture of azetidin-3-ol (56 mg, 0.56
mmol) and
sodium hydride (60% weight dispersion, 33 mg, 0.84 mmol) in dimethylformamide
(1
mL) was stirred under gas evolution ceased. After 2 min of stirring P-456 (97
mg, 0.28
mmol) was added, and the reaction heated at 140 C overnight. The reaction was
cooled
to room temperature, diluted with water (5 mL), and extracted with ethyl
acetate (2 x 5
mL). The combined extracts were dried over sodium sulfate, filtered, and the
solvent
removed under vacuum. The residue was purified by silica gel column
chromatography
(1:2 hexanes/ethyl acetate followed by 9:1 dichloromethane/methanol) and
trituration in
ethyl acetate and hexanes to give P-457 (22.2 mg, 19.9% yield) as a cream
colored solid.
[00546] 'H NMR (400 MHz, DMSO-d6): 3.61 (dd, 2 H), 3.68 - 3.74 (m, 3 H), 3.78
(s,
2 H), 4.09 (t, J = 7.5 Hz, 2H), 4.47 - 4.60 (m, 1 H), 5.59 (d, J = 6.6 Hz, 1
H), 6.33 (d, J
=8.5Hz,1H),6.92(d,J=8.6Hz,1H),7.20-7.32(m,2H),7.32-7.39(m,2H),7.39
7.50 (m, 2 H), 7.96 (d, J = 1.6 Hz, 1 H) ppm. LCMS = 98.5% purity. APCI (+) =
399
(M).
259
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00547] Scheme 47.
~N
N HO,OH Palladium(0)tetrakis(triphenylphosphine) N
+ B' Potassium Phosphate
O O
'WO
Br O~=O 1,2-Dimethoxyethane, Water, Ethanol O
(~Cl A
1-179 Cl
P-522
F~N
Tin(II)Chloride Dihydrate _
O O
Ethanol, Tetrahydrofuran NH2 NH
O~
NH
'Cl Cl
P-524 P-529
Example 197. Preparation of P-522
[00548] Synthesis of 1-(3'-Chloro-6-methoxy-biphenyl-3-yl)-6-nitro-1H-
benzoimidazole (P-522). A suspension of 1-179 (328 mg, 1.00 mmol), potassium
phosphate (424 mg, 2.00 mmol), and 3-chlorophenylboronic acid (313 mg, 1.30
mmol)
in ethanol (1 mL), water (1 mL), and 1,2-dimethoxyethane (2 mL) was degassed
with
nitrogen stream for 15 min. To the mixture was added
palladium(0)tetrakis(triphenylphosphine), and the reaction stirred at 80 C
overnight. The
reaction was cooled to room temperature, basified with 1 M aqueous sodium
hydroxide
(2 mL), diluted with water (15 mL), and extracted with ethyl acetate (2 x 15
mL). The
combined organic layers were dried over sodium sulfate, filtered, and
concentrated under
vacuum. The crude was purified by silica gel column chromatography (4:1 to 1:1
hexanes/ethyl acetate) to give P-522 (310 mg, 82% yield) as a pale yellow
solid. 1H
NMR (400 MHz, DMSO-d6) 3.90 (s, 3 H), 7.36 - 7.51 (m, 3 H), 7.60 (d, J = 7.4
Hz, 1
H), 7.64 - 7.77 (m, 3 H), 7.81 (d, J = 9.1 Hz, 1 H), 8.23 (dd, J= 9.1, 2.2 Hz,
1 H), 8.67
(d, J = 2.0 Hz, 1 H), 8.88 (s, 1 H) ppm.
LCMS = 96.1% purity. APCI (+) = 380 (M).
260
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 198. Preparation of P-524
[00549] Synthesis of 3-(3'-Chloro-6-methoxy-biphenyl-3-yl)-3H-benzoimidazol-5-
ylamine (P-524). A solution of P-522 (310 mg, 0.811 mmol) in ethanol (577 mg)
and
tetrahydrofuran (5 mL) was stirred at room temperature. To the solution was
added
tin(II) chloride hydrate (900 mg, 4.00 mmol) and the reaction stirred at 80 C
for 3 h.
The reaction was then cooled to room temperature, and concentrated in vacuo.
The
residue was diluted with chloroform (10 mL), washed with 1 N aqueous sodium
hydroxide (4 mL), water, and brine, dried over sodium sulfate, filtered, and
the solvent
removed under vacuum. The residue was purified by silica gel column
chromatography
(9:1 dichloromethane/methanol) to give P-524 (55.3 mg, 19% yield) as off-white
solid.
[00550] 'H NMR (400 MHz, DMSO-d6): 3.86 (s, 3 H), 4.87 (s, 2 H), 6.66 (dd, J =
8.6,1.9Hz,1H),6.86(d,J=1.9Hz,1H),7.25-7.35(m,2H),7.38-7.65 (m, 4 H),
7.66 (s, 1 H), 8.30 (s, 1 H) ppm. LCMS = 93.9% purity. APCI (+) = 380 (M).
Example 199. Preparation of P-529
[00551] Synthesis of 1-[3-(3'-Chloro-6-methoxy-biphenyl-3-yl)-3H-benzoimidazol-
5-
yl]-3-ethyl-urea (P-529). A solution of P-524 (35 mg, 0.10 mmol) and ethyl
isocyanate
(0.04 mL, 0.5 mmol) in pyridine (0.2 mL) was stirred at room temperature
overnight.
The reaction was diluted with water (2 mL), extracted with ethyl acetate (2 x
2mL), and
the extracts combined. The extracts were dried over sodium sulfate, filtered,
and the
solvent removed under vacuum. The residue was purified by trituration in ethyl
acetate
and diethyl ether to give P-529 (19.0 mg, 45% yield) as an off-white solid. 1H
NMR
(400 MHz, DMSO-d6): 1.07 (t, J = 7.2 Hz, 3 H), 3.02 - 3.20 (m, 2 H), 3.88 (s,
3 H), 5.96
- 6.09 (m, 1 H), 7.23 (dd, J= 8.7, 1.9 Hz, 1 H), 7.35 (d, J= 8.9 Hz, 1 H),
7.40 - 7.54 (m,
3 H), 7.54 - 7.62 (m, 2 H), 7.62 - 7.73 (m, 2 H), 7.89 (d, J = 1.7 Hz, 1 H),
8.42 (s, 1 H),
8.46 (s, 1 H) ppm.
LCMS = 94.17% purity. APCI (+) = 421.1 (M).
261
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 200. Preparation of P-473
N
N I \ I ~
Sodium Hydride
O F F O F N
+
HN Dimethylformamide
O CI
CI
O
P-456 P-473
[00552] Synthesis of [5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-
2-yl]-dimethyl-amine (P-473). To a solution of pyrrolidine-3-carboxylic acid
methyl
ester (185 mg, 1.12 mmol), was added sodium hydride (60% weight dispersion, 66
mg,
1.68 mmol). After 2 min of stirring, P456 (194 mg, 0.56 mmol) was added, and
the
reaction stirred for 10 min in a microwave reactor at 240 C and 15 bar. The
reaction was
diluted with ethyl acetate (5 mL), washed with 0.1 N aqueous hydrochloric acid
(5 mL),
and the aqueous layer was extracted with ethyl acetate (3 x 5 mL). The
combined
extracts were washed with brine, dried over sodium sulfate, filtered, and the
solvent
removed under vacuum. The crude material was purified by silica gel column
chromatography (9:1 ethyl acetate/ hexanes) to give P-473 (80.7 mg, 39%
yield). 1H
NMR (400 MHz, DMSO-d6): 2.96 (s, 6 H), 3.71 (s, 3 H), 3.77 (s, 2 H), 6.57 (d,
J = 8.6
Hz,1H),6.91(d,J=8.6Hz,1H),7.18-7.50 (m,6H),7.98(d,J=2.0Hz,1H)ppm.
LCMS = 96.9% purity. APCI (+) = 371.1 (M).
Example 201. Preparation of P-029
O
Br
H Sodium Hydride N
O O
LJ DMF
N~:O I N O
1-70 P-029 0
[00553] Synthesis of 1-(6-Methoxy-3'-nitro-b iphenyl-3 -ylmethyl)-pyrrolidin-2
-one (P-
029). A suspension of sodium hydride (15.6 mg, 0.652 mmol) in DMF (10 mL) was
allowed to stir under nitrogen for 15 min. To the suspension was added 2-
pyrrolidone
262
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
(55.5 mg, 0.652 mmol) and the reaction allowed to stir at room temperature for
15 min
under nitrogen. To the reaction was added 1-70 (200 mg, 0.621 mmol) and the
reaction
stirred under nitrogen at ambient temperature for 16 h. The reaction was
diluted with
saturated aqueous ammonium chloride (50 mL), extracted with ethyl acetate (50
mL),
and the layers separated. The organic extract was washed with saturated
aqueous
ammonium chloride (2 x 50 mL), water (3 x 50 mL), brine (50 mL), dried over
sodium
sulfate, and the solvent removed under vacuum. The product was purified by
separation
on a silica gel preparatory plate eluting with 10% methanol in dichloromethane
to give
P-029 (108 mg, 53% yield) as a yellow gum. 'H NMR (400 MHz, CDC13): 8.40 (t,
J=
2.2 Hz, 1H), 8.20 - 8.17 (m, 1H), 7.82 (dt, J= 7.6 Hz, 1.2 Hz, 1H), 7.56 (t,
J= 8.0 Hz,
1H), 7.28 (dd, J= 8.4 Hz, 2.4 Hz, 1H), 7.21 (d, J= 2.4 Hz, 1H), 6.97 (d, J=
8.8 Hz,
1H), 4.45 (s, 2H), 3.83 (s, 3H), 3.30 (t, J= 7.0 Hz, 2H), 2.44 (t, J= 8.2 Hz,
2H), 2.03-
1.99 (m, 2 H) ppm.
LCMS = 93.9 % purity. MS(ESI+) = 327.7 (M+1).
Example 202. Preparation of P-034
0
Br NA
O I / + H Potassium Carbonate \O / L__JNH
C N
DMF
A
N O N. 1-70 O
P-034
[00554] Synthesis of 1-(6-Methoxy-3'-nitro-biphenyl-3 -ylmethyl)-imidazolidin-
2 -one
(P-034). A suspension of potassium carbonate (160 mg, 1.16 mmol), 2-
imidazolidone
(104 mg, 1.21 mmol), and 1-70 (150 mg, 0.466 mmol) in DMF (10 mL) was heated
to 80
C overnight. The reaction was cooled to room temperature, ethyl acetate (30
mL) and
saturated aqueous ammonium chloride (30 mL) were added, and the layers
separated.
The extract was washed with saturated aqueous ammonium chloride (2 x 30 mL),
water
(2 x 30 mL), brine (30 mL), dried over sodium sulfate, and the solvent removed
under
vacuum. The crude product was purified by silica gel preparatory TLC to give P-
034
(30.2 mg, 20 % yield) as a white powder. 1H NMR (400 MHz, CDC13): 8.41 (t, J=
2.0
Hz, 1H), 8.19-8.16 (m, 1H), 7.84 (dt, J= 7.9 Hz, 1.5 Hz, H), 7.56 (t, J= 8.0
Hz, 1H),
263
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
7.32 (dd, J= 8.2 Hz, 2.2 Hz, 1H), 7.25 (d, J= 2.4 Hz, 1H), 6.98 (d, J = 8.4
Hz, 1H), 4.37
(s, 2H), 4.32 (brs, 1H), 3.84 (s, 3H), 3.41-3.33 (m, 4H) ppm.
LCMS = 86.3% purity. MS(ESI+) = 328.3 (M+1).
Example 203. Preparation of P-035
0
III
Br S
S Sodium Hydride N
O + HNo O
DMF
N.O I N O 1-70 O
P-035
[00555] Synthesis of 2-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-
[1,2]thiazinane
1,1-dioxide (P-035). A suspension of sodium hydride (11.7 mg, 0.489 mmol) in
DMF (8
mL) was stirred under nitrogen for 5 min. To the suspension was added 1,4-
butanesultam
(66.1 mg, 0.489 mnmol) under nitrogen, and the reaction was stirred at room
temperature
for 30 min. To the reaction was added 1-70 (150 mg, 0.466 mmol), and the
reaction
stirred overnight at room temperature under nitrogen. The reaction was diluted
with ethyl
acetate (40 mL), washed with saturated aqueous ammonium chloride (2 x 50 mL),
water
(2 x 50 mL), brine (40 mL), dried over sodium sulfate, and the solvent removed
under
vacuum. The residue was purified by silica gel preparatory TLC eluting with
dichloromethane to give P-035 (99.4 mg; 57 % yield) as an off white powder. 1H
NMR
(400 MHz, CDC13): 8.41 (t, J= 1.8 Hz, 1H), 8.20-8.17 (m, 1H), 7.85-7.83 (m,
1H), 7.57
(t, J= 8.0 Hz, 1H), 7.37 (dd, J = 8.4 Hz, 2.4 Hz, 1H), 7.30 (d, J= 2.0 Hz,
1H), 7.00 (d, J
= 8.4 Hz, 1H), 4.30 (s, 2H), 3.84 (s, 3H), 3.27-3.24 (m, 2H), 2.11-3.08 (m,
2H), 2.24-
2.21 (m, 2H), 1.66-1.570 (m, 2H) ppm. LCMS = 98.9 % purity. MS(APCI-) = 376.1
(M).
264
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 204. Preparation of P-036
O
Br
O O H O Sodium Hydride N
+ O O
DMF
NCO Ni.O
O 1-
1-70 P-036 O
[00556] Synthesis of 1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-pyrrolidine-
2,5-
dione (P-036). To a solution of pyrrolidine-2,5-dione (67.2 mg, 0.679 mmol) in
DMF (10
mL) was added sodium hydride (16.3 mg, 0.679 mmol) and the reaction was
stirred
under nitrogen for 15 min until gas evolution ceased. To the reaction was
added 1-70
(100 mg, 0.310 mmol) and the reaction was stirred under nitrogen at room
temperature
for 16 h. The reaction was diluted with ethyl acetate, the organic layer
washed with water
(2 x 50 mL) and saturated aqueous ammonium chloride (2 x 50 mL), and the
aqueous
washes were combined. The aqueous washes were extracted with ethyl acetate (50
mL)
and the organic extracts combined. The combined extracts were washed with
brine (50
mL), dried over sodium sulfate, filtered and the solvent removed under vacuum.
The
resulting yellow oil was purified by silica preparatory TLC eluting with
dichloromethane
to give P-036 (68.6 mg; 65% yield) as a yellow oil. 'H NMR (400 MHz, CDC13):
8.39
(t, J= 2.0 Hz, 1H), 8.18 (ddd, J = 8.2, 2.2, 2.0 Hz, 1H), 7.80 (dt, J = 8.0,
1.3 Hz, 1H),
7.56 (t, J = 8.0 Hz, 1H) 7.45 (dd, J = 7.2, 2.4 Hz, 1H), 7.39 (d, J = 2.4 Hz,
1H), 6.95 (d,
J = 8.4 Hz, 1H), 4.66 (s, 2H), 3.82 (s, 3H), 2.71 (s, 4H) ppm. MS (ESI+)=
341.4 (M+1).
LCMS = 97.5% purity.
Example 205. Preparation of P-045
HO.B.OH
i O O
O O O N~
Br N Palladium(II)Bis
0 + HN\/ Sodium Hydride i J (Triphenylphosphine)dichloride 0
DMF 0
Br Br Sodium Carbonate
1-42 1-181 Dioxane / Water
0 O-J
P-045
265
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00557] Synthesis of 1-(3 -Bromo-4-methoxy-benzyl)-pyrrolidin-2 -one (1-181).
A
suspension of sodium hydride (28.8 mg, 1.20 mmol) in anhydrous DMF (15 mL) was
stirred at room temperature under nitrogen for 5 min. To the reaction was
added 2-
pyrollidone (102 mg, 1.20 mmol) under nitrogen and the reaction stirred for 10
min.
Subsequently 1-42 (320 mg, 1.14 mmol) was added and the reaction was stirred
at room
temperature overnight. The reaction was diluted with ethyl acetate (50 mL).
The organic
material was washed with saturated aqueous ammonium chloride (2 x 30 mL),
water (2 x
30 mL), brine (2 x 30 mL), dried over sodium sulfate and the solvent removed
under
vacuum to give 1-42 (243 mg) which was taken on without purification.
[00558] Synthesis of 1-(3 -Benzo [ 1, 3 ] dioxol-5 -yl-4-methoxy-benzyl)-
pyrrolidin-2 -one
(P-045). A solution of 1-42 (230 mg, 0.809 mmol) and benzo[1,3]dioxol-5-yl-
boronic
acid (201 mg, 1.21 mmol) in 1,4-dioxane (15 mL) was degassed with a nitrogen
stream
for 30 min, and subsequently bis(triphenylphosphine)palladium(II) dichloride
(28.4 mg,
0.0405 mmol) was added under nitrogen. Degassing was continued for 5 min,
followed
by the addition of 1 M aqueous sodium carbonate (2.5 mL). The reaction was
heated to
80 C with stirring overnight under nitrogen. The reaction did not go to
completion, so
additional benzo[1,3]dioxol-5-yl-boronic acid (201 mg, 1.21 mmol) and
bis(triphenylphosphine)palladium(II) dichloride (28.4 mg, 0.0405 mmol) were
added
and the reaction stirred for 5 h at 80 C. The reaction was diluted with ethyl
acetate (40
mL), washed with brine (2 x 40 mL), water (4 x 40 mL), and brine (40 mL). The
solution
was dried over sodium sulfate, filtered and the solvent removed under vacuum
to give a
residue. This material was purified by multiple development silica gel
preparatory plate
thin layer chromatography eluting with 10% methanol in dichloromethane,
followed by
10% acetone in dichloromethane to give P-045 (21.3 mg, 8.1% yield) as a yellow
syrup.
[00559] 'H NMR (400 MHz, CDC13): 7.19-7.17 (m, 2H), 7.04 (d, J= 1.6 Hz, 1H),
6.96-6.85 (m, 3H), 5.99 (s, 2H), 4.42 (s, 2H), 3.81 (s, 3H), 2.43 (t, J= 8.0
Hz, 1H), 2.00-
1.97 (m, 2H) ppm. MS(ESI+) = 326.7 (M+1).
266
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00560] Scheme 48.
O
O
Cl + HN Sodium Hydride
DMF
Br p Br
1-109 1-182
O
HO,B,OH Palladium(II)acetate N
Triphenylphosphine p I i
O
1-182 +
Potassium Carbonate
O 1,2-dimethoxyethane
p-/ Ethanol, Water 0
o p_l
P-053
Example 206. Preparation of P-053
[00561] Synthesis of 1-(3-Bromo-4-methoxy-benzyl)-pyrrolidine-2,5-dione (1-
182): A
suspension of sodium hydride (56.2 mg,2.34 mmol) in anhydrous DMF (20 mL) was
stirred under nitrogen for 5 min. To the suspension was added pyrrolidine-2,5-
dione (231
mg, 2.34 mmol), and and the resulting slurry was stirred for 5 min. After
stirring, I-109
(500 mg, 2.13 mmol) was added under nitrogen, and the reaction was allowed to
stir at
ambient temperature under nitrogen for 17.5 h, and diluted with ethyl acetate
(50 mL).
The organic solution was washed with water (4 x 50 mL), brine (2 x 50 mL),
dried over
anhydrous sodium sulfate, and the solvent removed under vacuum to afford 532.8
mg, I-
182 as a yellow powder in 84% yield. 1H NMR (400 MHz CDC13) d: 7.59 (d, J= 2.4
Hz, 1H), 7.34 (dd, J= 8.4 Hz, 2.0 Hz, 1H), 6.82 (d, J= 8.4 Hz, 1H), 4.569 (s,
2H), 3.870
(s, 3H), 2.7 10 (s, 4H).
[00562] Synthesis of 1-(3-Benzo[1,3]dioxol-5-yl-4-methoxy-benzyl)-pyrrolidine-
2,5-
dione (P-053). A solution of 1-182 (300 mg, 1.01 mmol) and benzo[1,3]dioxol-5-
yl-
boronic acid (183 mg, 1.10 mmol) in 1,4-dioxane (5 mL) were degassed with a
nitrogen
stream for 10 min. Subsequently triphenylphosphine (52.7 mg, 0.201 mmol),
solid
267
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
potassium carbonate (417 mg, 3.02 mmol) and an ethanol in water mixture (1:1,
1 mL)
were added under nitrogen. The reaction was stirred under nitrogen for 5 min,
palladium(II) acetate (22.6 mg, 0.101 mmol) was added, and the reaction was
heated
under nitrogen to 80 C for 19 h. The solvent was removed under vacuum and the
mixture was diluted with saturated aqueous ammonium chloride (50 mL) and ethyl
acetate (50 mL), the layers separated, and the aqueous layer extracted with
ethyl acetate
(50 mL). The combined organic extracts were washed with water (3 x 30 mL),
brine (30
mL), dried over sodium sulfate, and the solvent removed under. The residue was
chromatographed on flash silica gel eluting with 10% acetone in
dichloromethane to
afford 153.7 mg of P-053 as a yellow powder in 45% yield. 1H NMR (CDC13, 400
MHz): 8 7.35 (m, 2H), 7.02 (d, J = 1.6 Hz, 1H), 6.94 (ddd, J = 8.0 Hz, J = 1.6
Hz, J =
0.8 Hz, 1H), 6.87 (m, 2H), 4.63 (s, 2H), 3.79 (s, 3H), 2.70 (s, 4H). LCMS =
91.2%.
MS(ESI+)= 340.3 (M+1). .
[00563] Scheme 49.
HOB,OH Palladium(II)acetate
Cl ~Triphenylphosphine Potassium Bromide
0 + Potassium Phosphate Nitric Acid(aq)
Toluene 0 F 1,2-dichloroethane
F A 1-184 Tetrabutylammonium Chloride
B is(pinacolate )d iboran
[1,1'-bis(diphenylphosphino)ferrocene]dichloropalladium(ll) 0 F
0 -(~~ F
Potassium Acetate B
O O
Br DMF
4
1-185
1-186
Example 207. Preparation of 1-186
[00564] Synthesis of 4-Fluoro-4'-methoxydiphenylmethane (I-184). A suspension
of
4-fluorophenylboronic acid (6.70 g, 47.9 mmol), ground solid potassium
phosphate (13.6
g, 63.9 mmol), triphenylphosphine (168 mg, 0.639 mmol), and
palladium(II)acetate (72
mg, 0.319 mmol) was stirred under nitrogen in toluene (100 mL). Nitrogen was
streamed
through with stirring for 15 min, and subsequently 4-(chloromethyl)anisol
(5.00 g, 31.9
mmol) was added under nitrogen, and the reaction was heated to 80 C
overnight.
Additional palladium(II) acetate (72 mg, 0.319 mmol) was added and the
reaction stirred
268
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
at 80 C for an additional 6 h. The reaction was not complete so a third
portion of
palladium(II)acetate (144 mg, 0.639 mmol) and more triphenylphosphine (336 mg,
1.28
mmol) was added and the reaction stirred at 80 C overnight. The reaction was
diluted
with ethyl acetate (300 mL), washed with 1 N aqueous sodium hydroxide (2 x 300
mL),
water (3 x 300 mL), and brine (2 x 300 mL). The organic extract was dried over
sodium
sulfate and decolorized over activated carbon, filtered and the solvent
removed under.
The residue was purified by flash silica gel column chromatography (10% ethyl
acetate
in hexanes Rf = 0.31) to afford 2.50 g of 1-184 as a clear oil in 36% yield,
which was
taken on without further purification. 1H NMR (400 MHz, CDC13) 7.13-7.06 (m,
4H),
6.976-6.93 (m, 2H), 6.84-6.82 (m, 2H), 3.89 (s, 2H), 3.78 (s, 3H) ppm.
[00565] Synthesis of 2-Bromo-4-(4-fluoro-benzyl)-1-methoxy-benzene (I-185). A
biphasic solution potassium bromide (1.65 g, 13.9 mmol) in 21% (w/v) aqueous
nitric
acid (8.32 g, 27.7 mmol) and 4-fluoro-4-methoxydiphenylmethane (I-184, 1.50 g,
6.94
mmol) and tetrabutylammonium chloride (57.7 mg, 0.208 mmol) in 1,2-
dichloroethane
(16 mL) was stirred at room temperature overnight, and the red suspension
diluted with
dichloromethane (30 mL). The aqueous layer was removed and the organic layer
washed
with aqueous potassium carbonate (2% w/v, 3 x 50 mL), water (2 x 50 mL), and
brine
(50 mL). The reaction was dried over magnesium sulfate, filtered, and the
solvent
removed under vacuum. The crude product was purified by flash silica gel
column
chromatography (10% ethyl acetate in hexanes Rf = 0.39) to 1.24 g of I-185 as
a yellow
oil in 60% yield. 'H NMR (400 MHz, CDC13) d: 7.34 (d, J= 1.6 Hz, 1H), 7.12-
7.10 (m,
2H), 7.05 (dd, J= 6.6 Hz, 1.80 Hz, 1H), 6.99-6.96 (m, 2H), 6.82 (d, J= 6.80
Hz, 1H),
3.867 (s, 5H) ppm.
[00566] Synthesis of 2-[5-(4-Fluoro-benzyl)-2-methoxy-phenyl]-4,4,5,5-
tetramethyl-
[1,3,2]dioxaborolane (I-186). A stream of nitrogen was blown through a
solution of 2-
bromo-4-(4-fluoro-benzyl)-1-methoxy-benzene (I-185, 500 mg, 1.69 mmol) in DMF
(5
mL) for 15 min. Subsequently solid potassium acetate (499 mg, 5.08 mmol), [
1,1'-
bis(diphenylphosphino)ferrocene]dichloropalladium(II) (138 mg, 0.169 mmol),
and
bis(pinocolate)diboron (516 mg, 2.03 mmol) were added under nitrogen. The
reaction
was heated to 85 C 16 h. The reaction was cooled to room temperature, diluted
with
ethyl acetate (50 mL) and water (50 mL). The biphasic suspension was filtered
twice to
269
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
removed spent catalyst and separated. The organic extract was washed with
water (3 x 50
mL) and brine (50 mL), decolorized over activated carbon, dried over sodium
sulfate,
filtered, and the solvent removed under vacuum. The residue was purified by
flash silica
gel column chromatography (10% acetone in hexanes, Rf = 0.10) to afford 419.8
mg of
1-186 as a colorless oil in 72% yield. 'H NMR (400 MHz, CDC13) d: 7.50 (d, J=
2.4
Hz, 1H), 7.14-7.09 (m, 3H), 6.97-6.92 (m, 2H), 6.78 (d, J= 8.4 Hz, 1H), 3.88
(s, 2H),
3.80 (s, 3H), 1.35 (s, 12H) ppm. MS(ESI+) = 341.3 (M-1), 500.7 (M+159).
Example 208. Preparation of 1-187
Br HO,B,OH
1.)t-Butyllithium
2.) Triisopropylborate
0 Diethyl ether 0
0- -78 C 0-
F F F F
PR129
1-187
[00567] Synthesis of 2,2-Difluoro-benzo[1,3]dioxol-5-yl-boranic acid (1-187):
A
solution of tert-butyl lithium (1.15 mL, 1.95 mmol) in anhydrous diethyl ether
(5 mL)
was cooled to -78 C under nitrogen. 5-bromo-2,2-difluoro-benzo[1,3]dioxole
(180 mg,
0.760 mmol) in diethyl ether (0.80 mL) was added, and the reaction was stirred
for 1 h at
-78 C, followed by the addition of triisopropylborate (0.37 mL, 1.60 mmol)
under
nitrogen. The reaction allowed to stir for 1.5 h while allowing to warm to
ambient
temperature. The reaction was poured into 4 N aqueous sodium hydroxide (5 mL),
stirred, and adjusted to pH -1 by the dropwise addition of concentrated
aqueous
hydrochloric acid, and the product extracted with ethyl acetate (10 mL). The
organic
extract was dried over anhydrous sodium sulfate, filtered, and the solvent
removed under
vacuum to afford 140.2 mg of I-187 as a brown solid, which was taken on
without
further purification.
[00568] 1H NMR (400 MHz, DMSO-d6) 7.77-7.74 (m, 1H), 7.67-7.65 (m, 1H), 7.51
(s, 1H).
270
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00569] Scheme 50
O O Br NII N
HN TBSCI, DCM HNJ~~>/ O NaH TBAF
imidazole THE THE
OH
~
OH ores O-Si N o N10 I N'O P-131/P-132
0 0- o-
1-18811-189 1-70
I-190/1-191 I TsCI, DCM
DMAP, TEA
O O O
I 11 II
N PPh3 II ND NaN3 II
E E
O O O
NH2 THF, H2 DMF I
N3 OTs
N N"O N 0
O 0 0
P-155/P-156 1-194/1-195 1-192/1-193
Example 209. Preparation of P-131 and P-132
0 0
HN TBSCI, DCM HN
imidazole
OH OTBS
1-188
[00570] Synthesis of (S)-5 -(tert-Butyl-dimethyl-silanyloxymethyl)-pyrrolidin-
2 -one
(1-188). A solution of (S)-5-hydroxymethyl-pyrrolidin-2-one (1.0 g, 8.69
mmol), DMF
(10 mL), t-butyldimethylsilyl chloride (1.57g, 10.42 mmol), and imidazole
(0.89 g, 13.03
mmol) was stirred at ambient temperature for 4 hours after which time 4 mL of
water
were added. The layers were separated and the aqueous was extracted with
dichloromethane (10 mL). The combined organics were washed with water (15 mL)
and
brine (15 mL), dried with sodium sulfate, filtered and concentrated to afford
1.91 g of I-
188 as a colorless oil in 96% yield.
Ox xo
HN1 ) TBSCI, DCM _ HN1
))____// imidazole )__/
OH OTBS
1-189
[00571] Synthesis of (R)-5-(tert-Butyl-dimethyl-silanyloxymethyl)-pyrrolidin-2-
one
(1-189).
1-189 was prepared in an analogous manner as that for 1-188, starting from (R)-
5-
271
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Hydroxymethyl-pyrrolidin-2-one. 1.91 g of 1-189 was obtained as a colorless
oil in 96%.
0
O Br
'1O I / NaH
+ HN
O
THE
OTBS I N
o` ~
O N
O
1-188 1-70 1-190
[00572] Synthesis of (S)-5-(tert-Butyl-dimethyl-silanyloxymethyl)-1-(6-methoxy-
3'-
nitro-biphenyl-3-ylmethyl)-pyrrolidin-2-one (I-190). Into a 20 mL vial with
stir bar was
added 1-188 (0.28 g, 1.24 mmol), dry THE (4 mL), and the solution cooled to 0
C.
sodium hydride (65 mg, 1.61 mmol) was added and the suspension stirred for 30
minutes
at ambient temperature. After cooling to 0 C, 1-70 (0.40 g, 1.24 mmol) was
added the
reaction stirred at ambient temperature for 18 hours after which 5 mL water
was added.
The product was extracted with ethyl acetate (4 x 15 mL). The organics were
combined
and dried with sodium sulfate, filtered, and concentrated. Flash column
chromatography
purification using 30% ethyl acetate/Hexanes afforded 476 mg of 1-190 as a
colorless oil
in 83% yield.
O
IOxI Br \ N6
HN O NaH
THE
O,
OTBS ~ I N`o 1:0
N
O -
O
1-189 1-70 1-191
[00573] (R)-5-(tert-Butyl-dimethyl-silanyloxymethyl)-1-(6-methoxy-3'-nitro-
biphenyl-3-ylmethyl)-pyrrolidin-2-one (1-191). 1-191 was prepared in an
analagous
manner as that for 1-190, starting from 1-189. 462 mg of 1-191 was obtained as
a
colorless oil in 80% yield.
272
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
O O
N N
~O I
TBAF
_Si THE OH
N Z Nto
0 6-
1-190 P-131
[00574] Synthesis of (S)-5-Hydroxymethyl-l-(6-methoxy-3'-nitro-biphenyl-3-
ylmethyl)-pyrrolidin-2-one (P-131). Into a 50 mL round bottom flask equipped
with a
stir bar was added I-190 (0.45 g, 0.96 mmol), dry THF (5 mL), and the solution
was
cooled to 0 C. TBAF (1.4 mL, 1.43 mmol, 1.0 M in THF) was added and the
reaction
was stirred at 0 C for 30 minutes. 2 mL of saturated aqueous NH4C1 and 2 mL
of water
were added and the product was extracted with ethyl acetate (3 x 10 mL). The
combined
organics were washed with brine, dried over sodium sulfate, filtered, and
concentrated.
The residue was purified by flash column chromatography using 10-25%
acetone/dichloromethane to afford 307 mg (90%) of P-131 as a colorless semi-
solid. 1H
NMR (400 MHz, CDC13) 8.39 (s, 1 H), 8.18 (dd, J= 1.3, 8.3 Hz, 1 H), 7.81 (d,
J= 7.8
Hz, 1 H), 7.56 (t, J= 7.9 Hz, 1 H), 7.32 (dd, J= 1.9, 8.4 Hz, 1 H), 7.25 (d,
J= 2.0 Hz, 1
H),6.97(d,J=8.3Hz,1H),4.81(d,J=15.0Hz,1H),4.29 (d, J= 14.9 Hz,1H),3.83
(s,3H),3.78(d,J=2.5Hz,1H),3.68-3.50 (m,2H),2.65-2.48(m,1H),2.46-2.31
(m, 1 H), 2.16 - 2.03 (m, 1 H), 2.02 - 1.91 (m, 1 H). LC/MS = 100.0%, 357.1
(APCI+).
~O 1~O
O Nom/ TBAF O I N1./
_Si 6 THF OH
O
S N N to
C
0 6-
1-191 P-132
[00575] Synthesis of (R)-5-Hydroxymethyl-l-(6-methoxy-3'-nitro-biphenyl-3-
ylmethyl)-pyrrolidin-2-one (P-132). P-132 was prepared in a similar manner as
that for
P-131, except beginning with I-191. 310 mg (91%) of P-132 was obtained as a
colorless
oil. 'H NMR (400 MHz ,CDC13) 8.39 (t, J= 1.7 Hz, 1 H), 8.18 (dd, J= 1.3, 8.3
Hz, 1
H), 7.82 (d, J= 7.8 Hz, 1 H), 7.56 (t, J= 8.0 Hz, 1 H), 7.32 (dd, J= 2.1, 8.5
Hz, 1 H),
7.25 (d, J= 2.1 Hz,1H),6.97(d,J=8.5Hz,1H),4.80 (d, J= 14.9 Hz,1H),4.30(d,J
=15.0Hz,1H),3.83(s,3H),3.79(dd,J=2.8,11.4Hz,1 H), 3.68- 3.53 (m, 2 H), 2.64
273
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
2.50 (m, 1 H), 2.46 - 2.32 (m, 1 H), 2.15 - 2.03 (m, 1 H), 2.02 - 1.90 (m, 1
H). LC/MS
= 100.0%, 357.1 (APCI+).
Example 210. Preparation of P-155 and P-156
O O
N TsCI, DCM N
7,~H DMAP, TEA OTs
N NtD
O O
P-131 1-192
[00576] Synthesis of Toluene-4-sulfonic acid (S)-1-(6-methoxy-3'-nitro-
biphenyl-3-
ylmethyl)-5-oxo-pyrrolidin-2-ylmethyl ester (1-192). Into a 20 mL vial with
stir bar and
dichloromethane (5 mL) at 0 C was added P-131 (0.25 g, 0.70 mmol), TsC1(0.16
g,
0.84 mmol), triethylamine (0.15 mL, 1.05 mmol), and DMAP (9 mg, 0.07 mmol).
The
reaction was stirred at room temperature for 18 hours and then concentrated.
The residue
was purified by flash column chromatography using 10% acetone/dichloromethane
to
afford 340 mg (95%) of 1-192 as a colorless oil.
O O
N
N TsCI, DCM
DMAP, TEA
OH OTs
\ I N I N'O
0 0
P-132 1-193
[00577] Synthesis of Toluene-4-sulfonic acid (R)-1-(6-methoxy-3'-nitro-
biphenyl-3-
ylmethyl)-5-oxo-pyrrolidin-2-ylmethyl ester (1-193). 1-193 was prepared in a
similar
manner as that for 1-192, except beginning with P-132. 290 mg (92%) of 1-193
was
obtained as a colorless oil.
O O
N NaN3 I N
OTs DMF r-"), N3
N N'O
O O
1-192 1-194
274
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00578] Synthesis of (S)-5-Azidomethyl-l-(6-methoxy-3'-nitro-biphenyl-3-
ylmethyl)-
pyrrolidin-2-one (I-194). Into a 20 mL vial with stir bar was added 1-192
(0.33 g, 0.65
mmol), DMF (4 mL), and NaN3 (63 mg, 0.97 mmol). The reaction was stirred for 1
hour
at 60 C, and then cooled to room temperature. 5 mL of water was added and the
product
was extracted with dichloromethane (2 x 5 mL). The combined organics were
washed
with water (4 x 5 mL), dried over sodium sulfate, filtered, and concentrated
to obtain
0.24 g (97%) of 1-194 as a colorless oil.
O O
N NaN3 I N6
OTs DMF N3
N "I I Nto
O O
1-193 1-195
[00579] Synthesis of (R)-S-Azidomethyl-l-(6-methoxy-3'-nitro-biphenyl-3-
ylmethyl)-
pyrrolidin-2-one (1-195). 1-195 was prepared in a similar manner as that for 1-
194 except
beginning with 1-193. 1-195 (0.20 g, 96%) was obtained as a colorless oil.
O o
N
PPh3 \ I / N
O O
N3 THF, H2O NH2
Z~'No MINA
O O
1-194 P-155
[00580] Synthesis of (S)-5-Aminomethyl-l-(6-methoxy-3'-nitro-biphenyl-3-
ylmethyl)-pyrrolidin-2-one (P-155). Into a 50 mL round bottom flask with stir
bar was
added 1-194 (0.24 g, 0.63 mmol), THF (4 mL), and PPh3 (0.50 g, 1.89 mmol). The
reaction was stirred at room temperature for 18 hours after which 1 mL of
water was
added. After stirring at room temperature for 2 more days, the THF was removed
under
reduced pressure. 1 N HC1 was added to until the aqueous material was pH =1
and the
solution was washed with dichloromethane (3 x 3 mL). The aqueous phase was
basified
with 1 N NaOH to pH = 11 and the product was extracted with dichloromethane (3
x 3
mL) and concentrated. The residue was purified by flash column chromatography
using
5% methanol/dichloromethane and afforded 164 mg (73%) of P-155 as a light-
yellow
oil. 1H NMR (400 MHz,CDC13) 8.39 (s, 1 H), 8.18 (dd, J= 1.3, 8.2 Hz, 1 H),
7.81 (d, J
275
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
= 7.8 Hz, 1 H), 7.56 (t, J= 7.9 Hz, 1 H), 7.30 (dd, J= 1.9, 8.4 Hz, 1 H), 7.23
(d, J= 2.0
Hz,1H),6.96(d,J=8.5Hz,1H),4.85(d,J=14.9Hz,1 H), 4.15 (d, J= 14.9 Hz,1H),
3.83 (s, 3 H), 3.61 - 3.50 (m, 1 H), 2.93 - 2.84 (m, J= 4.8 Hz, 1 H), 2.80
(br. s., 1 H),
2.61 - 2.49 (m, 1 H), 2.47 - 2.35 (m, 1 H), 2.19 - 2.03 (m, 1 H), 1.95 - 1.83
(m, 1 H).
LGMS = 100.0%, 356.1 (APCI+).
o
~
N
I / N
PPh3
I /
,10
O
N3 THF, H2O NHZ
NA NA
O O
1-195 P-156
[00581] Synthesis of (R)-5-Aminomethyl-l-(6-methoxy-3'-nitro-biphenyl-3-
ylmethyl)-pyrrolidin-2-one (P-156). P-156 was prepared in a similar manner as
that for
P-155, except beginning with 1-195. P-156 (80 mg, 43%) was obtained as a light-
yellow
oil. 1H NMR (400 MHz ,CDC13) 8.39 (s, 1 H), 8.18 (dd, J= 1.1, 8.3 Hz, 1 H),
7.81 (d, J
=7.8Hz,1H),7.56(t,J=8.0Hz,1H),7.30(dd,J=1.8,8.4Hz,1H),7.23(d,J=1.9
Hz,1H),6.96(d,J=8.5Hz,1H),4.85(d,J=14.9Hz,1 H), 4.15 (d, J= 14.9 Hz,1H),
3.83 (s, 3 H), 3.61 - 3.49 (m, 1 H), 2.93 - 2.84 (m, 1 H), 2.83 - 2.74 (m, 1
H), 2.61 - 2.49
(m, 1 H), 2.46 - 2.35 (m, 1 H), 2.15 - 2.03 (m, 1 H), 1.95 - 1.83 (m, 1 H).
LGMS =
100.0%, 356.1 (APCI+).
Example 211. Preparation of P-135
0
Br HO,B'OH 0
Pd(OAC)2
K3PO4 PPh3
C02Me DME
0 F EtOH, HZO N A
O
1-70 1-196
[00582] Synthesis of 2-Fluoro-5-(6-methoxy-3'-nitro-biphenyl-3-ylmethyl)-
benzoic
acid methyl ester (1-196). Into a 20 mL vial with stir bar was added 1-70
(0.30 g, 0.93
mmol), 4-Fluoro-3-methoxycarbonylphenylboronic acid (203 mg, 1.02 mmol),
triphenylphosphine (49 mg, 0.19 mmol), K3P04 (0.40 g, 1.86 mmol), DME (5 mL),
water
(0.5 mL), and ethanol (0.5 mL). N2 gas was bubbled through the stirred
reaction for 10
276
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
minutes. Pd(OAc)2 (21 mg, 0.09 mmol) was added and N2 was bubbled through for
an
additional 5 minutes. The vial was capped and the reaction was stirred at 80
C for 18
hours. The reaction was cooled to room temperature and 5 mL of water and 5 mL
of
ethyl acetate were added. The layers were separated and the aqueous was
extracted with
ethyl acetate (3 x 10 mL). The organics were combined, dried with sodium
sulfate, and
concentrated. The residue was purified by flash column chromatography using
10%
acetone/Hexanes to obtain 207 mg (56%) of 1-196 as a colorless oil.
COzMe OH
O
F THE O F
7N"O Dib al
NUJ
J
O O
1-196 P-135
[00583] Synthesis of Fluoro-5-(6-methoxy-3'-nitro-biphenyl-3-ylmethyl)-phenyl]-
methanol (P-135). Into a 20 mL vial with stir bar was added 1-196 (112.3 mg,
0.28
mmol) and 4 mL of dry THF. The solution was cooled to 0 C and DIBAL-H (0.71
mL,
0.71 mmol, 1.0 M sol. in hexane) was added. The reaction was stirred at 0 C
for 1 hour.
Aqueous 1 N HC1(1 mL) was added followed by water (5 mL). The aqueous solution
was extracted with ethyl acetate (3 x 10 mL). The organics were combined,
dried over
sodium sulfate, and concentrated. The product was purified by flash column
chromatography using 10% acetone/hexanes to obtain 53 mg (5 1%) of P-135 as
colorless
oil 1H NMR (400 MHz, CDC13) 1.72 (t, J= 6.2 Hz, 1 H) 3.81 (s, 3 H) 3.95 (s, 2
H) 4.73
(d, J= 6.2 Hz, 2 H) 6.91 - 7.02 (m, 2 H) 7.06 - 7.21 (m,3H)7.23-
7.28(m,1H)7.54(t,
J=8.0Hz,1H)7.82(d,J=7.7Hz,1H)8.16(dd,J=8.3,1.3Hz,1H)8.38(s,1H)
ppm. LC/MS = 100.0%, 367.1 (APCI-).
Example 212. Preparation of P-153 and P-154
Br HNN NN \ N_ ,N
O O
+ K2CO3 O + O-
DMF
NA N.0 O NA
6- O
1-70 1-197 1-198
277
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00584] Synthesis of 1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-1H-imidazole-4-
carboxylic acid methyl ester (1-197) and 1-(6-Methoxy-3'-nitro-biphenyl-3-
ylmethyl)-
1H-imidazole-5-carboxylic acid methyl ester (1-198). Into a 20 mL vial with
stir bar was
added 1-70 (0.30 g, 0.92 mmol), 1H-imidazole-4-carboxylic acid methyl ester
(0.12 g,
0.93 mmol), K2CO3 (0.15 g, 1.12 mmol), and DMF (3 mL). The reaction was
stirred at
room temperature for 20 hours. To the reaction was added 10 mL of
dichloromethane
and 10 mL of water and the layers were separated. The aqueous was extracted
with 10
mL of dichloromethane and the combined organics were washed with water (3 x 10
mL).
The dichloromethane portion was concentrated and purified first by flash
column
chromatography using 0-5% methanol/dichloromethane followed by preparative TLC
using 10% acetone/dichloromethane. 72.4 mg (21%) of 1-197 and 91.1 mg (27%) of
I-
198 were obtained as light-yellow oils.
N~
Dbal
O gIPCN
I N to
O O
1-198 P-153
[00585] Synthesis of [3-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-3H-imidazol-4-
yl]-
methanol (P-153). Into a 20 mL vial with stir bar was added 1-198 (85.5 mg,
0.23 mmol)
and 5 mL of dry THF. The solution was cooled to 0 C and DIBAL-H (0.70 mL,
0.70
mmol, 1.0 M sol. in hexane) was added. The reaction was stirred at room
temperature
for 16 hours and then cooled to 0 C. Aqueous 1 N HC1(1 mL) was added followed
by 3
mL of 1 N NaOH and 5 mL of ethyl acetate. The layers were separated and the
aqueous
was extracted with ethyl acetate (3 x 10 mL). The organic were combined, dried
over
sodium sulfate, and concentrated. The residue was purified by flash column
chromatography using 50% acetone/dichloromethane, followed by flash column
chromatography using 5% methanol/dichloromethane to obtain 52.2 mg (66%) of P-
153
as a tan solid. 1H NMR (400 MHz, DMSO-d6) 3.79 (s, 3 H) 4.38 (d, J= 5.2 Hz, 2
H)
5.13 (t, J= 5.3 Hz, 1 H) 5.21 (s, 2 H) 6.80 (br. s., 1 H) 7.16 (d, J= 8.5 Hz,
1 H) 7.27 (dd,
J=.5,1.9Hz,1H)7.34(d,J=1.9Hz,1H)7.65-7.77 (m,2H)7.92(d,J=7.8Hz,1
H) 8.20 (dd, J= 8.1, 1.7 Hz, 1 H) 8.28 (s, 1 H) ppm. LC/MS = 99.9%, 340.1
(APCI+)
278
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
N
THE MeO HO
fZtl.to N Dibal fto
O O
1-197 P-154
[00586] Synthesis of [1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-1H-imidazol-4-
yl]-
methanol (P-154). P-154 was prepared in a similar manner as that for P-153
except
beginning with 1-197. P-154 (45.0 mg, 71%) was obtained as a tan solid. 1H NMR
(400
MHz, DMSO-d6) 3.79 (s, 3 H) 4.28 (d, J=5.5 Hz, 2 H) 4.75 (t, J=5.6 Hz, 1 H)
5.12 (s, 2
H) 7.03 (s, 1 H) 7.16 (d, J=8. 5 Hz, 1 H) 7.3 8 (dd, J=8.5, 2.1 Hz, 1 H) 7.44
(d, J=2.2 Hz,
1 H) 7.68 (d, J=0.8 Hz, 1 H) 7.72 (t, J=8.0 Hz, 1 H) 7.94 (d, J=7.9 Hz, 1 H)
8.20 (dd,
J=8.2, 1.5 Hz, 1 H) 8.29 (t, J=1.7 Hz, 1 H). LC/MS = 99.9%, 340.1 (APCI+).
[00587] General Scheme 51.
HO, OH
B' Palladium(II)acetate
Triphenylphosphine 0 F
O I / I F ~
Potassium carbonate
Br Ar2 Water, ethanol,
1,2-Dimethoxyethane
1-185 Ar2
Example 213. Preparation of P-140
O F
I I
O
P-140
[00588] Synthesis of 1-[5'-(4-Fluoro-benzyl)-2'-methoxy-biphenyl-3-yl]-
ethanone (P-
140). A nitrogen stream was bubbled through a solution of 1-185 (150 mg, 0.508
mmol)
and 3-acetylphenylboronic acid (91.7 mg, 0.559 mmol) in 1,2-dimethoxyethane (4
mL)
for 15 min, followed by the addition of ethanol:water (1:1, 1 mL). The
nitrogen stream
was continued for 10 min, and solid potassium carbonate (210 mg, 1.52 mmol),
triphenylphosphine (26.8 mg, 0.102 mmol), and palladium(II) acetate (11.4 mg,
0.0508
279
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
mmol) were added. The reaction was heated to 80 C and heated with stirring
overnight.
The reaction was diluted with ethyl acetate (50 mL) and saturated aqueous
ammonium
chloride (50 mL), filtered twice, and separated. The organic extract was
washed with
water (3 x 50 mL), brine (50 mL), dried over anhydrous sodium sulfate,
filtered, and the
solvent removed under vacuum. The reaction was purified by flash silica gel
column
chromatography (5% ethyl acetate in hexanes, Rf = 0.10 in 10% ethyl acetate in
hexanes)
to give P-140 (88.0 mg, 52% yield) as a colorless oil. 1H NMR (400 MHz, DMSO-
d6)
d: 7.98 (t, J= 1.6 Hz, 1H), 7.91 (dt, J= 7.9 Hz, 1.4 Hz, 1H), 7.70 (dt, J= 8.0
Hz, 1.5 Hz,
1H), 7.56 (t, J= 7.6 Hz, 1H), 7.30-7.30 (m, 2H), 7.23-7.21 (m, 2H), 7.12-7.05
(m, 3H),
3.93 (s, 2H), 3.74 (s, 3H), 2.60 (s, 3H) ppm.LCMS = 98.5% purity. MS(APCI+)=
335.1
(M+1).
Example 214. Preparation of P-141
O F
I I
O
O~
F F
P-141
[00589] Synthesis of 2,2-Difluoro-5-[5-(4-fluoro-benzyl)-2-methoxy-phenyl]-
benzo[1,3]dioxole (P-141). A nitrogen stream was bubbled through a solution of
1-185
(114 mg, 0.385 mmol) and 1-187 (140 mg, 0.693 mmol) in 1,2-dimethoxy ethane (3
mL)
for 20 min, followed by addition of ethanol and water solution (1:1, 0.75 mL)
and the
nitrogen stream was continued for 5 min. To the reaction was added solid
potassium
carbonate (160 mg, 1.16 mmol), triphenyl phospine (20.2 mg, 0.0770 mmol), and
palladium(II) acetate (8.6 mg, 0.0385 mmol) under nitrogen, the reaction was
heated to
80 C, and stirred at this temperature overnight. The solvent was removed under
vacuum,
the residue dissolved in ethyl acetate (20 mL) and water (20 mL), the biphasic
solution
was filtered, and the two layers separated. The aqueous wash was extracted
into ethyl
acetate (20 mL), and the combined extracts were washed with water (3 x 50 mL),
saturated aqueous ammonium chloride (50 mL), dried over sodium sulfate,
decolorized
280
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
with activated carbon, filtered, and the solvent removed under vacuum. The
residue was
purified by flash silica gel column chromatography (5% acetone in
dichloromethane, Rf=
0.40 in 10% acetone in dichloromethane) followed by preparatory silica TLC
(2.5%
acetone in dichloromethane) to give P-141 (96.8 mg, 68% yield). 'H NMR (400
MHz,
DMSO-d6) 7.48 (d, J= 1.6 Hz, 1H), 7.42 (d, J= 8.4 Hz, 1H), 7.30-7.179 (m, 5H),
7.11-7.03 (m, 3H), 3.90 (s, 2H), 3.74 (s, 3H) ppm. MS(ESI+)=371.8 (M-1), 353.0
(M-
19).
Example 215. Preparation of P-151
O I I F
S
P-151
[00590] Synthesis of 5-(4-Fluoro-benzyl)-2-methoxy-3'-methylsulfanyl-biphenyl
(P-
151). P-151 was synthesized from 1-185 (160 mg, 0.542 mmol) and 3-
methylsulfanylphenylboronic acid (100 mg, 0.596 mmol) using the same condition
as
described for P-141. To the reaction vial was added ethyl acetate (50 mL) and
water (50
mL), the biphasic solution filtered, and the layers separated. The organic
extract was
washed with water (3 x 50 mL), aqueous saturated ammonium chloride (50 mL),
brine
(50 mL), dried over sodium sulfate, filtered, and the solvent removed under
vacuum. The
product was purified by flash silica gel column chromatography (5% acetone in
hexanes)
followed by preparatory silica gel TLC (2.5% acetone in hexanes) to give P-151
(47.6
mg, 26% yield). 1H NMR (400 MHz, DMSO-d6) 7.35-7.27 (m, 4H), 7.22-7.16 (m,
4H),
7.11-7.07 (m, 2H), 7.03 (d, J= 8.4 Hz, 1H), 3.909 (s, 2H), 3.73 (s, 2H). LCMS
= 99.4%
purity. MS(APCI+) = 338.1 (M).
281
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 216. Preparation of P-170
Br Palladium(II)acetate
O F Triphenylphosphine 0 F
O~B,O 0 Potassium Carbonate
Ethanol, Water, O
1,2-Dimethoxyethane
A
1-186
P-170
[00591] Synthesis of 1-[5'-(4-Fluoro-benzyl)-4,2'-dimethoxy-biphenyl-3-yl]-
ethanone
(P-170). A nitrogen steam was bubbled through a solution of 1-186 (150 mg,
0.438
mmol) and 1-(5-bromo-2-methoxy-phenyl)-ethanone (110 mg, 0.482 mmol) in 1,2-
dimethoxy ethane (4 mL) for 10 min, followed by the addition of an ethanol and
water
solution (1:1, 1 mL). Nitrogen bubbling was continued for 5 min, palladium(II)
acetate
(9.8 mg, 0.044 mmol), triphenylphosphine (23.0 mg, 0.0877 mmol), and solid
potassium
carbonate (181 mg, 1.32 mmol) were added under nitrogen, and the reaction was
heated
at 80 C under nitrogen for 16 h. The solvent was removed under vacuum and
ethyl
acetate (50 mL) was added. The organic solution was washed with water (3 x 50
mL)
and brine (50 mL). The organic extract was removed under vacuum. The residue
was
purified by two silica gel preparatory TLC plates (10% ethyl acetate in hexane
with 2
developments followed by 5% acetone in hexanes with 6 developments) to give P-
170
(43.0 mg, 27% yield) as an orange oil. 1H NMR (400 MHz, CDC13): 7.63-7.60 (m,
2H),
7.29-7.25 (m, 2H), 7.21-7.07 (m, 5H), 7.02 (d, J= 8.4 Hz, 1H), 3.92 (s, 3H),
3.91 (s,
2H), 3.32 (s, 3H), 2.55 (s, 3H). LCMS = 100.0% purity. MS(APCI+)= 365.1 (M+1).
[00592] General Scheme 52.
0
~ CI BrZ Br NaH N Pd(OAc)Z N
O AcOH O THE O K,PO4 PPh, 30 \ O
Br
O Br DME
1-42 HN 1-181 EtOH, H2O Q-1 R
Example 217. Preparation of P-162
[00593] Synthesis of 2-Bromo-4-bromomethyl-1-methoxy-benzene (1-42). Into a 1
L
round bottom flask was added 4-methoxybenzyl chloride (19.7 g, 125.8 mmol),
glacial
282
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
acetic acid (400 mL), and Br2 (9.1 mL, 177.5 mmol, dropwise over 5 minutes).
The
brown solution was stirred at room temperature for 20 hours and then poured
into a 10%
(w/v) aqueous NaHSO3 solution (2000 mL). After stirring at room temperature
for 30
minutes, the white solid was filtered through a coarse glass-frit and the
solids were
washed with water (4 x 500 mL). The solid was dried in a vacuum dessicator for
16
hours. 1-42 (24.5 g, 74%) was obtained as a white solid.
O
IOxI I / Br N~(
V~
HN + 0 NaH "j?
LJ Br THE Br
1-42 1-181
[00594] Synthesis of 1-(3 -Bromo-4-methoxy-benzyl)-pyrrolidin-2 -one (1-181).
Into a
dry 100 mL round bottom flask was added pyrrolidin-2-one (0.20 g, 2.34 mmol),
dry
DMF (20 mL), and the solution was cooled to 0 C. NaH (110 mg, 2.76 mmol) was
added and the suspension was stirred at room temperature for 30 minutes. 1-42
(0.50 g,
2.12 mmol) was added and the reaction was stirred at 80 C for 30 minutes and
then
cooled to room temperature. After extraction with dichloromethane (2 x 20 mL),
the
combined organics were washed with water (4 x 20 mL), dried over sodium
sulfate,
filtered, and concentrated. The residue was purifed by flash column
chromatography
using 10% acetone/dichloromethane to obtain 482 mg (80%) of 1-181 as a light-
yellow
oil.
O O
6 HO. OH
N B Pd(OAc)Z N
Br I K3P04 PPh3
DME
1-181 DOH, H2O
P-162
[00595] Synthesis of 1-(6-Methoxy-biphenyl-3 -ylmethyl)-pyrrolidin-2 -one (P-
162).
Into a 20 mL vial with stir bar was added 1-181 (95.5 mg, 0.34 mmol),
phenylboronic
acid (45 mg, 0.37 mmol), triphenylphosphine (18 mg, 0.07 mmol), K2CO3 (139 mg,
1.01
mmol), DME (5 mL), water (0.5 mL), and ethanol (0.5 mL). N2 gas was bubbled
through the stirred reaction for 10 minutes. Palladium(II) acetate (8 mg, 0.03
mmol) was
283
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
added and N2 was bubbled through for an additional 5 minutes. The vial was
capped and
the reaction was stirred at 80 C for 18 hours. The reaction was cooled to
room
temperature and 5 mL of water and 5 mL of ethyl acetate were added. The layers
were
separated and the aqueous was extracted with ethyl acetate (3 x 10 mL). The
organics
were combined, dried with sodium sulfate, concentrated, and the residue was
purified by
prep TLC using 10% acetone/dichloromethane to obtain 38.9 mg (41%) of P-162 as
a
light-yellow oil. 'H NMR (400 MHz, CDC13) 1.98 (quin, J= 7.6 Hz, 2 H) 2.43 (t,
J-
8.1 Hz, 2 H) 3.29 (t, J= 7.1 Hz, 2 H) 3.80 (s, 3 H) 4.43 (s, 2 H) 6.94 (d, J=
8.2 Hz, I H)
7.17-7.24(m,2H)7.29-7.36(m,1H)7.40(t,J= 7.5 Hz, 2 H) 7.51 (d, J= 7.3 Hz, 2
H) ppm. LC/MS = 100.0%, 282.1 (APCI+).
Example 218. Preparation of P-184
0
O HO. OH
B Pd(OAc)Z N
N6 + 1 `11 30 0
O O / K2CO1 PPh3
Br DME
EtOH, H2O
1-181 P-184
[00596] Synthesis of 1-(6,3'-Dimethoxy-b iphenyl-3 -ylmethyl)-pyrrolidin-2 -
one (P-
184). Into a 20 mL vial with stir bar was added 1-181 (0.21 g, 0.73 mmol), 3-
methoxyphenylboronic acid (0.11 g, 0.73 mmol), triphenylphosphine (39 mg, 0.15
mmol), K2CO3 (0.31 g, 2.22 mmol), DME (5 mL), water (0.5 mL), and ethanol (0.5
mL).
N2 gas was bubbled through the stirred reaction for 10 minutes. Palladium(II)
acetate (17
mg, 0.07 mmol) was added and N2 was bubbled through the reaction for an
additional 5
minutes. The reaction was stirred at 80 C for 18 hours under N2. The reaction
was
cooled to room temperature and 5 mL of water and 5 mL of ethyl acetate were
added.
The layers were separated and the aqueous was extracted with ethyl acetate (3
x 10 mL).
The organics were combined, dried with sodium sulfate, and concentrated. The
residue
was purified by flash column chromatography using 10% acetone/dichloromethane
to
obtain 40.2 mg (17%) of P-184 as a brown oil. 'H NMR (400 MHz, CDC13) 1.98
(quin,
J= 7.6 Hz, 2 H) 2.43 (t, J= 8.1 Hz, 2 H) 3.28 (t, J= 7.1 Hz, 2 H) 3.80 (s, 3
H) 3.84 (s, 3
H) 4.43 (s, 2 H) 6.88 (dd, J= 8.3, 2.0 Hz, 1 H) 6.93 (d, J= 8.2 Hz, 1 H) 7.04 -
7.11 (m, 2
284
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
H)7.17-7.23(m,2H)7.32(t,J=7.9Hz,1H)ppm. LGMS=100.0%,312.1
(APCI+).
Example 219. Preparation of P-503
O O
1 NL I \ N6
O BBr3 HO
DCM
N Nt 0
O O
P-029 P-503
[00597] Synthesis of 1-(6-Hydroxy-3'-nitro-biphenyl-3-ylmethyl)-pyrrolidin-2-
one (P-
503). Into a 50 mL round bottom flask with stir bar was added P-029 (324.5 mg,
0.99
mmol) and dichloromethane (10 mL). The solution was cooled to 0 C and BBr3
(3.0
mL, 2.98 mmol, 1.0 M in dichloromethane) was added. The reaction was stirred
at room
temperature for 18 hours and then water was added. The layers were separated
and the
aqueous was extracted with dichloromethane (4 x 15 mL). The organics were
combined,
dried with sodium sulfate, and concentrated. The solid was triturated with
ether and
284.4 mg (92%) of P-503 was obtained as a tan-colored solid. 'H NMR (400 MHz,
DMSO-d6) 1.90 (quin, J= 7.5 Hz, 2 H) 2.26 (t, J= 8.1 Hz, 2 H) 3.24 (t, J= 7.0
Hz, 2 H)
4.32(s,2H)6.97(d,J=8.3Hz,1H)7.11(dd,J=8.3,2.0Hz,1H)7.25(d,J=2.0Hz,
1 H) 7.71 (t, J= 8.0 Hz, 1 H) 8.00 (d, J= 7.8 Hz, 1 H) 8.17 (dd, J= 8.2, 1.6
Hz, 1 H)
8.40 (t, J= 1.7 Hz, 1 H) 9.91 (br. s., 1 H) ppm. LGMS = 98.4%, 313.1 (APCI+).
Example 220. Preparation of P-188
O
Br N
O + HND NaH O
O 1~/I TH
N I N
O O
1-70 P-188
Synthesis of 1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-piperidin-2-one (P-
188). Into
a 50 mL round bottom flask with stir bar was added 2-Piperidone (88 mg, 0.89
mmol),
dry THE (5 mL), NaH (42 mg, 1.05 mmol), and the suspension was stirred for 10
minutes at room temperature. 1-70 (261 mg, 0.81 mmol) was added and the
reaction was
285
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
stirred for 18 hours at room temperature. To the reaction was added 10 mL of
H2O and
the THE was removed by rotary evaporation. The product was extracted with
ethyl
acetate (3 x 10 mL) and the combined organics were washed with 20 mL of brine,
dried
over Na2SO4, and concentrated. The residue was purified by flash column
chromatography using 5 - 10% acetone/dichloromethane to obtain 201 mg (73%) of
P-
188 as a light-yellow semi-solid. 1H NMR (500 MHz,CDC13) 8.40 - 8.39 (m, 1H),
8.18
- 8.15 (m, 1H), 7.84 - 7.82 (m, 1H), 7.56 (t, J= 8.0 Hz), 7.31 (dd, J= 8.5,
2.0 Hz, 1H),
7.24 - 7.23 (m, 1H), 6.97 (d, J= 8.0 Hz, 1H), 4.59 (s, 2H), 3.83 (s, 3H), 3.26
- 3.24 (m,
2H), 2.47 - 2.44 (m, 2H), 1.80 - 1.79 (m, 4H) ppm. 13C-NMR (100 MHz, CDC13) 8
170.0, 155.9, 148.2, 140.1, 135.8, 130.7, 130.3, 129.9, 129.0, 128.3, 124.7,
122.0, 111.7,
55.9, 49.6, 47.4, 32.6, 23.4, 21.5 ppm. LC/MS = 100.0%, 341.1 (APCI+).
Example 221. Preparation of P-192
F
O Br HS S
TEA
Br F DCM Br
1-42 1-199
[00598] Synthesis of 2-Bromo-4-(4-fluoro-phenylsulfanylmethyl)-1-methoxy-
benzene
(1-199). Into a 20 mL vial with stir bar was added 1-42 (0.50 g, 1.90 mmol), 4-
fluorothiophenol (0.27 g, 2.09 mmol), dichloromethane (5 mL), and TEA (0.43
mL, 3.14
mmol). The solution was stirred at room temperature for 16 hours and then
concentrated. The residue was purified by flash column chromatography using
15%
acetone/hexane to obtain I-199 which was used without further purification in
the next
step.
F OH
F
B S
~ a HO' / Pd(OAc)Z, KZC03 , I i
S + O
O + PPh3, DME, DOH, HZO
Br 01:0 N
1-199 1-200 b-
[00599] Synthesis of 5-(4-Fluoro-phenylsulfanylmethyl)-2-methoxy-3'-nitro-
biphenyl
286
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
(1-200). Into a 20 mL vial with stir bar was added I-199 (0.40 g, 1.22 mmol),
3-
nitrophenylboronic acid (0.20 g, 1.22 mmol), K2C03 (0.51 g, 3.67 mmol),
triphenylphosphine (64 mg, 0.24 mmol), DME (5 mL), EtOH (0.5 mL), and H2O (0.5
mL). The mixture was degassed with N2 for 10 minutes and then palladium(II)
acetate
(27 mg, 0.12 mmol) was added. The reaction was stirred at 80 C for 18 hours
and then
filtered through Celite and concentrated. The residue was purified by flash
column
chromatography using 10% acetone/dichloromethane, then by flash column
chromatography using 12% acetone/hexane, followed by preparative layer
chromatography using 15% ethyl acetate/hexane to give 102 mg (23%) of 1-200 as
a
light-yellow oil. 1H NMR (400 MHz,CDC13) 8.33 (s, 1 H), 8.17 (dd, J= 1.3, 8.3
Hz, 1
H),7.78(d,J=7.8Hz,1H),7.55(t,J=8.0Hz,1H),7.35- 7.27 (m, 2 H), 7.23 (dd, J=
2.1,8.5Hz,1H),7.15(d,J=2.1Hz,1H),6.98(t,J=8.7Hz,2H),6.91(d,J=8.5Hz,
1 H), 4.04 (s, 2 H), 3.82 (s, 3 H) ppm. LC/MS = 369.0 (APCI-).
i I F ~F
\ S mCPBA, DCM \ I % O S.O
O
N to
N
b- p-
1-200 1-201
[00600] Synthesis of 5-(4-Fluoro-benzenesulfonylmethyl)-2-methoxy-3'-nitro-
biphenyl (I-201). Into a 20 mL vial with stir bar was added 1-200 (38.4 mg,
0.10 mmol),
dichloromethane (2 mL), and mCPBA (45 mg, 0.26 mmol). The reaction was stirred
at
room temperature for 30 minutes and then 5 mL of saturated NaHCO3 (aq.) was
added.
The product was extracted with dichloromethane (2 x 5 mL) and then
concentrated.
Purification by preparative layer chromatography using 35% acetone/hexane gave
18.3
mg (44%) of 1-201 as a colorless semi-solid. 1H NMR (400 MHz,CDC13) 8.22 (d,
J=
1.6Hz,1H),8.21-8.14(m,1H),7.76-7.67 (m,3H),7.55(t,J=8.1Hz,1H),7.23-
7.15 (m, 3 H), 7.01 (d, J= 2.3 Hz, 1 H), 6.94 (d, J= 8.5 Hz, 1 H), 4.30 (s, 2
H), 3.84 (s, 3
H) ppm. LC/MS = 100.0%, 401.0 (APCI-)
287
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
i I F ~F
S\
H202, AcOH S \
p O
N N to
b- p-
1-200 P-192
[00601] Synthesis of 5-(4-Fluoro-benzenesulfinylmethyl)-2-methoxy-3'-nitro-
biphenyl (P-192). Into a 20 mL vial with stir bar was added 1-200 (35.5 mg,
0.096
mmol), glacial AcOH (0.5 mL), and H202 (1.1 mL, 0.096 mmol, 0.088M solution in
AcOH)). The reaction was stirred at room temperature for 1 hour and then 10 mL
of
saturated NaHCO3 (aq.) and 1 mL of IN NaOH were added until pH = 10. The
product
was extracted with dichloromethane (3 x 5 mL) and then concentrated.
Purification by
preparative layer chromatography using 35% acetone/hexane gave 21.0 mg (57%)
of P-
192 as a colorless semi-solid. 1H NMR (400 MHz,CDC13) 8.24 (t, J= 1.8 Hz, 1
H),
8.17(dd,J=1.3,8.3Hz,1H),7.71(d,J=7.6Hz,1H),7.54(t,J=8.0Hz,1H),7.45-
7.36(m,2H),7.17(t,J=8.5Hz,2H),7.07(dd,J=2.1,8.5 Hz,1H),6.93(d,J=8.5
Hz, 1 H), 6.85 (d, J= 2.3 Hz, 1 H), 4.03 (d, J= 2.7 Hz, 2 H), 3.83 (s, 3 H)
ppm. LC/MS
= 100.0%, 386.0 (APCI+).
Example 222. Preparation of P-195
O
\ % Br + N N K2CO3 \p ,
O Br HO DME Br CN
1-42 1-202
[00602] Synthesis of 1-(3-Bromo-4-methoxy-benzyl)-6-oxo-1,6-dihydro-pyridine-3-
carbonitrile (1-202). Into a 100 mL round-bottomed flask with a stir bar was
added 1-42
(2.59 g, 9.84 mmol), 6-hydroxy-nicotinonitrile (985 mg, 8.20 mmol), K2C03
(2.49 g,
18.0 mmol), and DME (30 mL). The reaction was stirred at 80 C for 20 hours
and then
cooled to room temperature and filtered. The filtrate was concentrated and the
resulting
light-pink solid was triturated with 40 mL of ether to obtain 529 mg (20%) of
1-202 as a
white solid.
288
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
0
0 HO. OH
N
Pd(OAc)Z
0 1-
~O I I bo KZCOI PPh3
Br N DME N
EtOH, H
20 N
0
1-202
P-195
[00603] Synthesis of 1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-6-oxo-1,6-
dihydro-
pyridine-3-carbonitrile (P-195). Into a 50 mL round bottom flask with a stir
bar was
added 1-202 (510 mg, 1.60 mmol), 3-nitrophenylboronic acid (0.40 g, 2.40
mmol),
triphenylphosphine (84 mg, 0.32 mmol), K2CO3 (0.66 g, 4.79 mmol), DME (10 mL),
H2O (1 mL), EtOH (1 mL), and the suspension was degassed with N2 for 5
minutes. To
this was added palladium(II) acetate (36 mg, 0.16 mmol) and the reaction was
stirred at
80 C for 18 hours. The reaction was cooled to rt and 50 mL of H2O was added
and the
product was extracted with ethyl acetate (3 x 20 mL). The combined organics
were
washed with brine, dried over Na2SO4, filtered, and concentrated. The
resulting solid
was triturated with ether (2 x 25 mL) to afford 391 mg (68%) of P-195 as a tan
solid. 1H
NMR (400 MHz ,DMSO-d6) 8.84 (d, J= 2.1 Hz, 1 H), 8.30 (s, 1 H), 8.21 (d, J=
8.2 Hz,
1 H), 7.94 (d, J= 7.6 Hz, 1 H), 7.73 (t, J= 8.0 Hz, 1 H), 7.67 (dd, J= 2.4,
9.5 Hz, 1 H),
7.52(d,J=1.9Hz,1H),7.47(dd,J=1.9,8.5 Hz,1H),7.16(d,J=8.6Hz,1H),6.51
(d, J= 9.5 Hz, 1 H), 5.09 (s, 2 H), 3.80 (s, 3 H) ppm. LGMS = 93.4%, 362.6
(ESI+).
Example 223. Preparation of P-196
O
Br + N KzCOI N
O HO N DME O N
tD Z Nto
N
p- O
1-70 P-196
[00604] Synthesis of 1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-2-oxo-1,2-
dihydro-
pyridine-4-carbonitrile (P-196). Into a 100 mL round bottom flask with a stir
bar was
added 1-70 (2.76 g, 8.55 mmol), 2-hydroxy-isonicotinonitrile (934 mg, 7.78
mmol),
K2C03 (2.36 g, 17.11 mmol), and DME (30 mL). The suspension was stirred for 18
hours at 80 C and then at rt for 2 days. The reaction was filtered, the
filtrate was
289
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
concentrated, and the resulting solid was triturated with ether to give 2.12 g
(75%) of P-
196 as a yellow solid. 'H NMR (400 MHz,DMSO-d6) 8.29 (s, 1 H), 8.20 (dd, J=
1.3,
8.3 Hz, 1 H), 8.12 (d, J= 7.1 Hz, 1 H), 7.93 (d, J= 7.8 Hz, 1 H), 7.73 (t, J=
8.0 Hz, 1
H), 7.49 (d, J= 2.0 Hz, 1 H), 7.43 (dd, J= 1.9, 8.5 Hz, 1 H), 7.16 (d, J= 8.6
Hz, 1 H),
7.04 (d, J= 1.3 Hz, 1 H), 6.56 (dd, J= 1.7, 7.0 Hz, 1 H), 5.12 (s, 2 H), 3.79
(s, 3 H) ppm.
LGMS = 96.1%, 361.0 (APCI-).
Example 224. Preparation of P-207
O O
HCI N
O I - '1O l i \ I O
N NH2
to ;0
N
N
O 6-
P-196 P-207
[00605] Synthesis of 1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-2-oxo-1,2-
dihydro-
pyridine-4-carboxylic acid amide (P-207). Into a 20 mL vial with a stir bar
was added P-
196 (132 mg, 0.37 mmol) and concentrated HC1(2 mL). The reaction was stirred
at rt
for 18 hours, basified with 4N aqueous NaOH, and the product was extracted
with ethyl
acetate (3 x 5 mL). The combined organics were concentrated to half their
original
volume and 5 mL of hexane was added. The resulting solids were filtered,
washed with
hexane, and triturated with ether to afford 29.8 mg (22%) of P-207 as a tan
solid. 1H
NMR (400 MHz,DMSO-d6) 8.28 (s, 1 H), 8.20 (dd, J= 1.4, 8.3 Hz, 1 H), 8.05 (br.
s., 1
H), 7.93 (t, J= 7.4 Hz, 2 H), 7.72 (t, J= 8.0 Hz, 1 H), 7.61 (br s, 1 H), 7.48
(d, J= 2.0
Hz, 1 H), 7.42 (dd, J= 2.1, 8.5 Hz, 1 H), 7.15 (d, J= 8.5 Hz, 1 H), 6.84 (d,
J= 1.5 Hz, 1
H), 6.54 (dd, J= 1.8, 7.0 Hz, 1 H), 5.10 (s, 2 H), 3.79 (s, 3 H) ppm. MS:
380.1 (APCI+).
Example 225. Preparation of P-208
O
Br + N+ K CO
3 N
\
N \ O - /
O DME O
Br HO Br O'N'O
1-42 1-135
290
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00606] Synthesis of 1-(3-Bromo-4-methoxy-benzyl)-5-nitro-1H-pyridin-2-one (I-
135). Into a 100 mL round-bottomed flask with a stir bar was added 1-42 (3.0
g, 11.41
mmol), 5-nitro-pyridin-2-ol (1.60 g, 11.41 mmol), K2CO3 (3.47 g, 25.10 mmol),
and 30
mL of DME. The reaction was stirred at 80 C for 18 hours and then cooled to
rt and
filtered. The filtrate was concentrated and the resulting solid was triturated
with 50 mL
of ether to give 3.01 g (78%) of I-135 as a yellow solid.
O o
N I HZ Pd/C I ~ N
McOH
Br O-N.O Br NH2
1-135 1-203
[00607] Synthesis of 5-Amino-1 -(3-bromo-4-methoxy-benzyl)-1H-pyridin-2-one (I-
203). Into a 250 mL round bottom flask with a stir bar was added 1-135 (1.93
g, 5.69
mmol), 75 mL of MeOH, and 400 mg of 10% Pd/C. The suspension was stirred under
a
H2 atmosphere for 1 hour, and then filtered through Celite and concentrated.
To the
residue was added 30 mL of IN HC1 and the reaction was washed with
dichloromethane
(3 x 20 mL). The aqueous portion was basified with IN NaOH until pH = 10, then
extracted with dichloromethane (3 x 15 mL). The organics were dried with
Na2SO4 and
concentrated to give 825 mg of 1-203 as a light-green solid which used without
further
purification.
0
0 HO. OH
I \ N
Pd(OAc)2
N +
~O I I K2C0 P O bo- NH
Br NHZ O DME 2
EtOH, H
20 N
0
1-203
1-204
[00608] Synthesis of 5-Amino-l-(6-methoxy-3'-nitro-biphenyl-3-ylmethyl)-1H-
pyridin-2-one (1-204). Into a 100 mL round bottom flask with a stir bar was
added 1-203
(802 mg, 2.59 mmol), 3-nitrophenylboronic acid (433 mg, 2.59 mmol),
triphenylphosphine (136 mg, 0.52 mmol), K2C03 (1.07 g, 7.77 mmol), DME (15
mL),
H2O (1.5 mL), EtOH (1.5 mL), and the suspension was degassed with N2 for 10
minutes.
291
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
To this was added palladium(II) acetate (58 mg, 0.26 mmol) and the reaction
was stirred
at 80 C for 18 hours. After cooling to rt 5 mL of aqueous IN NaOH was added
and the
product was extracted with ethyl acetate (3 x 15 mL). The combined organics
were
filtered through Celite and concentrated. The residue was purified by flash
column
chromatography using 5% MeOH/dichloromethane to give 185 mg (21%) of 1-204 as
a
brown solid. 1H NMR (400 MHz,DMSO-d6) 8.28 (s, 1 H), 8.20 (dd, J= 1.3, 8.2 Hz,
1
H),7.92(d,J=7.8Hz,1H),7.72(t,J=8.0Hz,1H),7.40(d,J=1.9Hz,1H),7.35
(dd, J= 1.9, 8.5 Hz, 1 H), 7.15 (d, J= 8.6 Hz, 1 H), 7.05 (dd, J= 2.9, 9.5 Hz,
1 H), 6.91
(d, J= 2.8 Hz, 1 H), 6.29 (d, J= 9.5 Hz, 1 H), 4.99 (s, 2 H), 4.29 (br. s., 2
H), 3.79 (s, 3
H) ppm. LC/MS = 100.0%, 352.1 (APCI+).
O H
N NaOCN N NYNHZ
O l i I O O O
NH z AcOH, H2O
i i
N N
O O
1-204 P-208
[00609] Synthesis of [1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-6-oxo-1,6-
dihydro-
pyridin-3-yl]-urea (P-208). Into a 20 mL vial with a stir bar was added 1-204
(36.5 mg,
0.10 mmol), NaOCN (13.5 mg, 0.21 mmol), H2O (2 mL), and 1 mL glacial acetic
acid.
The reaction was stirred as 40 C for 2 hours, then cooled to r.t. and 10 mL
of H2O was
added. The suspension was filtered to obtain 19.8 mg (48%) of P-208 as a gray
solid.
1H NMR (400 MHz,DMSO-d6) 8.29 (s, 1 H), 8.20 (d, J= 8.2 Hz, 1 H), 8.03 (s, 1
H),
7.92 (d, J= 7.6 Hz, 1 H), 7.88 (d, J= 2.3 Hz, 1 H), 7.72 (t, J= 7.9 Hz, 1 H),
7.44 (s, 1
H), 7.40 - 7.35 (m, 1 H), 7.31 (dd, J= 2.5, 9.5 Hz, 1 H), 7.15 (d, J= 8.5 Hz,
1 H), 6.37
(d, J= 9.7 Hz, 1 H), 5.79 (s, 2 H), 5.07 (s, 2 H), 3.79 (s, 3 H) ppm. LC/MS =
90.5%,
395.1 (APCI+).
292
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 226. Preparation of P-212
O
N CN N NH2
O O HCI O O
OND N
6- 6-
P-195 P-212
[00610] Synthesis of 1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-6-oxo-1,6-
dihydro-
pyridine-3-carboxylic acid amide (P-212). Into an 8 mL vial with a stir bar
was added P-
195 (44 mg, 0.12 mmol) and concentrated HC1(2 mL). The reaction was stirred at
r.t.
for 24 hours. To the reaction was added 5 mL of ethyl acetate and the solution
was
basified with aqueous 4N NaOH. The ethyl acetate layer was removed and
concentrated.
The resulting solid was triturated with ether to obtain 15.6 mg (34%) of P-212
as a tan
solid. 1H NMR (400 MHz,DMSO-d6) 8.52 (d, J= 2.4 Hz, 1 H), 8.32 - 8.26 (m, 1
H),
8.20(dd,J=1.3,8.3Hz,1H),7.93(d,J=7.8Hz,1H),7.85(dd,J=2.5,9.5Hz,1H),
7.76 (br. s., 1 H), 7.73 (t, J= 8.1 Hz, 1 H), 7.49 (d, J= 2.0 Hz, 1 H), 7.44
(dd, J= 2.0,
8.6Hz,1H),7.28(br.s.,1H),7.16(d,J=8.6Hz,1H),6.42(d,J=9.5Hz,1H),5.14
(s, 2 H), 3.79 (s, 3 H) ppm. LGMS = 96.4%, 380.1 (APCI+).
Example 227. Preparation of P-213
O o
N I BH3 THE N
N NH2
~ I Nto "I Nto
o- o-
P-196 P-213
[00611] Synthesis of 4-Aminomethyl-l-(6-methoxy-3'-nitro-biphenyl-3-ylmethyl)-
1H-pyridin-2-one (P-213). Into a 20 mL vial with a stir bar with P-196 (144
mg, 0.40
mmol) and 3 mL of THE was added BH3-THF (2.0 mL, 2.0 mmol, 1.OM solution in
THF). The solution was stirred at 60 C for 4 hours, then cooled to 0 C and a
solution of
2.0 M HC1 in ether was added slowly, followed by 5 mL of methanol. The
solution was
stirred at room temperature for 16 hours and then concentrated. To the residue
was
added aqueous IN HC1 and it was washed with dichloromethane, then basified
with
293
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
aqueous 4N NaOH and extracted with dichloromethane and concentrated. The
residue
was purified by flash column chromatography eluting with 10 - 17%
methanol/dichloromethane and afforded 7.2 mg (5%) of P-213 as a tan solid. 1H
NMR
(400 MHz,DMSO-d6) 8.27 (s, 1 H), 8.23 - 8.17 (m, 1 H), 7.92 (d, J= 7.6 Hz, 1
H), 7.79
-7.67(m,2H),7.45(d,J=1.9Hz,1H),7.39(dd,J=1.8,8.4 Hz,1H),7.14(d,J=8.5
Hz, 1 H), 6.35 (s, 1 H), 6.19 (dd, J= 1.5, 7.0 Hz, 1 H), 5.05 (s, 2 H), 3.78
(s, 2 H), 3.51
(s, 2 H) ppm. LC/MS = 98.4 %, 366.1 (APCI+).
Example 228. Preparation of P-215
N \ NN"~ O
O O OH DPPA O I O' v N 0 H
TEA
Nto t-BuOH
Zt, to 1-289 1-206
[00612] Synthesis of [1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-2-oxo-1,2-
dihydro-
pyridin-4-yl]-carbamic acid tert-butyl ester (1-206). Into a 25 mL round
bottom flask
was placed 1-289 (97.5 mg, 0.26 mmol), diphenylphosphoryl azide (71 mg, 0.26
mmol),
triethylamine (26 mg, 0.26 mmol), and t-BuOH (5 mL). The solution was refluxed
for
16 hours, then cooled to room temperature. The reaction was diluted with ethyl
acetate,
washed with saturated aqueous NaHCO3, and brine. After concentration of the
organics,
the residue was purified by flash column chromatography eluting with 40%
acetone/hexane to afford 61 mg (53%) of 1-206 as a colorless oil. 1H NMR (400
MHz,
DMSO-d6) 9.60 (s, 1 H), 8.27 (s, 1 H), 8.20 (dd, J= 1.6, 8.2 Hz, 1 H), 7.91
(d, J= 7.8
Hz, 1 H), 7.78 - 7.65 (m, 2 H), 7.40 (d, J= 2.0 Hz, 1 H), 7.36 (dd, J= 2.0,
8.5 Hz, 1 H),
7.14 (d, J= 8.5 Hz, 1 H), 6.53 (d, J= 2.1 Hz, 1 H), 6.34 (dd, J= 2.3, 7.5 Hz,
1 H), 4.99
(s, 2 H), 3.78 (s, 3 H), 1.45 (s, 9 H) ppm. LC/MS = 93.0%, 452.1 (APCI+).
N O N
O O i Hx0 TFA O I O NH2
+ DCM
o
N Np
O O
1-206 P-215
294
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00613] Synthesis fo 4-Amino-l-(6-methoxy-3'-nitro-biphenyl-3-ylmethyl)-1H-
pyridin-2-one (P-215 ). Into a 20 mL vial with a stir bar was added 1-206 (50
mg, 0.11
mmol), dichloromethane (2 mL), and 1 mL of TFA. The reaction was stirred at
room
temperature for 4 hours and then concentrated. The residue was diluted with
aqueous IN
HC1 and washed with dichloromethane. The aqueous portion was basified with
aqueous
4N NaOH, extracted with dichloromethane, and concentrated to afford 22 mg
(56%) of
P-215 as a tan solid. 'H NMR (400 MHz, DMSO-d6) 8.27 (s, 1 H), 8.23 - 8.16 (m,
1
H), 7.91 (d, J= 7.6 Hz, 1 H), 7.72 (t, J= 8.0 Hz, 1 H), 7.41 (d, J= 7.4 Hz, 1
H), 7.38 -
7.30 (m, 2 H), 7.13 (d, J= 8.5 Hz, 1 H), 5.99 (s, 2 H), 5.66 (dd, J= 2.3, 7.4
Hz, 1 H),
5.24 (d, J= 2.3 Hz, 1 H), 4.90 (s, 2 H), 3.78 (s, 3 H) ppm. LGMS = 97.8%,
352.1
(APCI+).
Example 229. Preparation of P-209
O
Br + N KZC03 N 11 HO DME
"O
Br Br
1-45 1-205
[00614] Synthesis of 1-(3-Bromo-4-methoxy-benzyl)-1H-pyridin-2-one (1-205).
Into
a 100 mL round bottom flask with a stir bar was added 1-45 (2.56 g, 9.73
mmol), 2-
hydroxypyridine (926 mg, 9.73 mmol), K2CO3 (2.96 g, 21.41 mmol), and 30 mL of
DME. The suspension was stirred at 80 C for 18 hours and then r.t. for 2 days
after
which it was filtered and concentrated. The solid was triturated with ether to
afford 2.20
g (77%) of 1-205 as a white solid.
0
0 HO. OH
Pd(OAc)Z N
\ I CI KZC03 PPh3
Br DME
EtOH, H2O CI
1-205
P-209
[00615] Synthesis of 1-(3'-Chloro-6-methoxy-biphenyl-3-ylmethyl)-1H-pyridin-2-
one
(P-209). Into a 40 mL vial with a stir bar was added 1-205 (250 mg, 0.81
mmol), 3-
chlorophenylboronic acid (126 mg, 0.81 mmol), triphenylphosphine (42 mg, 0.16
mmol),
295
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
K2C03 (335 mg, 2.43 mmol), DME (10 mL), H2O (1 mL), EtOH (1 mL), and the
suspension was degassed with N2 for 10 minutes. To this was added
palladium(II)
acetate (18 mg, 0.08 mmol) and the reaction was stirred at 80 C for 18 hours.
After
cooling to r.t., 10 mL of H2O was added and the product was extracted with
ethyl acetate
(3 x 15 mL). The combined organics were concentrated. The residue was purified
by
flash column chromatography using 20% - 35% acetone/hexanes to give 241 mg (9
1%)
of P-209 as a light-yellow oil. 1H NMR (400 MHz, DMSO-d6) 7.84 (dd, J= 1.3,
6.6
Hz,1H),7.66-7.52(m,1H),7.51-7.31(m,6H), 7.10 (d, J= 9.1 Hz,1H),6.39(d,J
= 9.1 Hz, 1 H), 6.22 (t, J= 6.5 Hz, 1 H), 5.06 (s, 2 H), 3.76 (s, 3 H) ppm.
LC/MS =
97.6%, 326.2 (APCI+).
[00616] Scheme 53.
Br 4-Fluorophenol O
Br Cupric Iodide
Palladium(II)acetate Dimethylglycine HCl
O + Potassium Carbonate 0 Cesium Carbonate O F
6'R Methanol 1,4-Dioxane
HO g OH R= Cl Water A
R=N02 R R
1-207 R = NO2 P-193 R = NO2
1-208R=Cl P-230R=Cl
Example 230. Preparation of P-193
[00617] Synthesis of 5-Bromo-2-methoxy-3'-nitro-biphenyl (1-207). A suspension
of
5-bromo-2-methoxyphenylboronic acid (500 mg, 2.17 mmol), 3-iodonitrobenzene
(647
mg, 2.60 mmol), and potassium carbonate (599 mg, 4.33 mmol) in methanol (10
mL)
and water (2 mL) was degassed for 30 min under a nitrogen stream followed by
the
addition of palladium(II) acetate (9.72 mg, 0.0433 mmol). The reaction was
stirred at
room temperature for 4 h. The methanol was removed under reduced pressure,
ethyl
acetate (50 mL) and water (50 mL) were added, and the biphasic suspension was
filtered.
The layers were separated and the organic was washed with water (50 mL). The
aqueous
washes were combined and extracted with ethyl acetate (2 x 50 mL), and the
organic
extracts combined. The organic solution was washed with water (3 x 50 mL),
saturated
aqueous sodium bicarbonate (50 mL), brine, dried over sodium sulfate,
filtered, and the
solvent removed under vacuum to give a crude brown gum. The product was
purified by
flash silica gel column chromatography (5% ethyl acetate in hexanes), and
crystallized
296
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
by dissolving in diethyl ether (20 mL) and hexanes (50 mL), removing one half
of the
solvent under vacuum, filtering and washing with hexanes (2 x 10 mL) to give 1-
207
(417.8 mg, 63% yield). 'H NMR (400 MHz, CDC13) 8.38 (t, J= 1.8 Hz, 1H), 8.21-
8.19
(m, 1H), 7.81 (dt, J= 7.5 Hz, 1.3 Hz, 1H), 7.58 (t, J= 8.0 Hz, 1H), 7.50-7.45
(m, 2H),
6.90 (d, J= 8.8 Hz, 1H), 3.82 (s, 3H) ppm.
[00618] Synthesis of 5-(4-Fluoro-phenoxy)-2-methoxy-3'-nitrobiphenyl (P-193).
To
a suspension of 1-207 (200 mg, 0.649 mmol), 4-fluorophenol (109 mg, 0.974
mmol),
cesium carbonate (423 mg, 1.30 mmol), and N,N-dimethylglycine (20.1 mg, 0.195
mmol) in 1,4-dioxane (2 mL) was added copper(I) iodide (12.4 mg, 0.0649 mmol)
under
argon, and the reaction was heated to 105 C. The reaction was left to stir
overnight
under argon at 105 C . The blue solution was diluted with ethyl acetate (50
mL), washed
with water (2 x 50 mL), brine (50 mL), dried over sodium sulfate, filtered,
and the
solvent removed under vacuum. The residue was purified by flash silica gel
column
chromatography (10% ethyl acetate in hexanes), and was crystallized from
diethyl ether
(5 mL) to give P-193 (61.4 mg, 28% yield) as a white powder. 1 H NMR (400 MHz,
CDC13) 8.40 (t, J= 2.0 Hz, 1H), 8.19-8.16 (m, 1H), 7.82 (dt, J= 7.6 Hz, 1.2
Hz, 1H),
7.56 (t, J= 8.0 Hz, 1H), 7.04-6.95 (m, 7H), 3.83 (s, 3H) ppm. LCMS =98.3%
purity.
MS(APCI-)=339.2 (M).
Example 231. Preparation of P-230
[00619] Synthesis of 5-Bromo-3'-chloro-2-methoxy-biphenyl (1-208). 1-208 was
synthesized from 5-bromo-2-methoxyphenylboronic acid (2.00 g, 8.66 mmol) and 3-
chloroiodobenzene (2.48 g, 10.4 mmol) using the same conditions as for 1-207.
The
reaction time was extended to 16 h. The methanol was removed under vacuum, and
the
residue dissolved in ethyl acetate (100 mL), washed with water (100 mL), and
the
aqueous wash extracted with ethyl acetate (100 mL). The combined organic
extracts
were washed with saturated aqueous sodium bicarbonate (2 x 200 mL), water (2 x
200
mL) and brine (150 mL), dried over sodium sulfate, filtered and the solvent
removed
under vacuum to give 1-208 (2.56 g, 99% yield). 1H NMR (400 MHz, CDC13) 7.49-
7.48 (m, 1H), 7.44-7.40 (m, 2H), 7.36-7.31 (m, 3H), 6.85 (d, J= 8.4 Hz, 1H),
3.80 (s,
3H) ppm.
297
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00620] Synthesis of 3'-Chloro-2-fluoro-3-(4-fluoro-phenoxy)-6-methoxy-
biphenyl
(P-230). To a suspension of 1-208 (200 mg, 0.672 mmol), 4-fluorophenol (113
mg, 1.01
mmol), cesium carbonate (438 mg, 1.34 mmol), and N,N-dimethylglycine
hydrochloride
salt (28.2 mg, 0.202 mmol) in 1,4-dioxane (2 mL) was added copper(I) iodide
(12.8 mg,
0.0672 mmol) under argon. The reaction was heated to 105 C under argon, and
stirred at
105 C overnight. The reaction was diluted with ethyl acetate (20 mL), washed
with
water (20 mL), and the aqueous wash extracted into ethyl acetate (20 mL). The
combined
organic extracts were washed with 1 N aqueous sodium hydroxide (20 mL), 1 N
aqueous
hydrochloric acid (20 mL), water (2 x 20 mL), brine (20 mL), dried over sodium
sulfate,
filtered, and the solvent removed under vacuum. The product was purified by
preparatory
TLC eluting with 5% acetone in hexanes with 3 developments to give P-230
(140.6 mg,
64% yield) as a yellow gummy solid. 1 H NMR (400 MHz, CDC13) 7.51-7.51 (m,
1H),
7.38-7.30 (m, 3H), 7.023-6.93 (m, 7H), 3.81 (s, 3H) ppm. MS(ESI+) = 389.6
(M+61),
328.2 (M), 279.5 (M-49).
Example 232. Preparation of P-236
0 O"
Br O\
O I N
KZCOI
N DME O O
HO
N I N
O
1-70 1-290
[00621] Synthesis of 1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-6-oxo-1,6-
dihydro-
pyridine-2-carboxylic acid methyl ester (1-290). Into a 20 mL vial with stir
bar was
added 1-70 (268 mg, 0.83 mmol), 6-hydroxy-pyridine-2-carboxylic acid methyl
ester (98
mg, 0.64 mmol), K2CO3 (195 mg, 1.41 mmol), and DME (4 mL). The reaction was
stirred at 80 C for 4 hours and then cooled to room temperature and filtered.
The filtrate
was concentrated and purified by flash column chromatography using 15 - 50%
acetone/hexanes to afford 98 mg (39%) of 1-290 as a tan solid.
298
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
0 O, 0 OH
N N
NaOH (aq),,
O O O O
MeOH
Z~' to
N NtD
0 6-
1-290 1-209
[00622] Synthesis of 1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-6-oxo-1,6-
dihydro-
pyridine-2-carboxylic acid (1-209). Into a 20 mL vial with a stir bar was
added 1-290
(92 mg, 0.23 mmol), IN aqueous NaOH (2 mL), and methanol (2 mL). The reaction
was
stirred at 60 C for 18 hours and then cooled to room temperature and washed
with
dichloromethane. The aqueous portion was acidified with 6N aqueous HC1 and
extracted
with ethyl acetate. The organics were dried over Na2SO4 and concentrated to
give 1-209
(61 mg, 69%) as a tan solid. 1H NMR (400 MHz, DMSO-d6) 8.25 (s, 1 H), 8.19
(dd, J
= 1.4, 8.3 Hz, 1 H), 7.88 (d, J= 7.8 Hz, 1 H), 7.72 (t, J= 7.9 Hz, 1 H), 7.48
(dd, J= 6.8,
9.1 Hz, 1 H), 7.24 (d, J= 1.7 Hz, 1 H), 7.21 - 7.15 (m, 1 H), 7.14 - 7.08 (m,
1 H), 6.71
(d, J= 6.6 Hz, 1 H), 6.63 (d, J= 9.0 Hz, 1 H), 5.50 (s, 2 H), 3.77 (s, 3 H)
ppm. LC/MS =
99.9%, 381.0 (APCI+)
O
0 OH
HNxO
N N
O O DPPA O O
TEA
N,~ t-BuOH
O p-
1-209 1-210
[00623] Synthesis of [1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-6-oxo-1,6-
dihydro-
pyridin-2-yl]-carbamic acid tert-butyl ester (I-210). Into a 20 mL vial was
placed I-209
(48 mg, 0.13 mmol), diphenylphosphoryl azide (35 mg, 0.13 mmol), triethylamine
(13
mg, 0.13 mmol), and t-BuOH (4 mL). The solution was stirred at 80 C for 24
hours and
then cooled to room temperature. The solvent was evaporated, the residue
diluted with
saturated aqueous NaHCO3 and extracted with ethyl acetate. The organics were
concentrated and purified by flash column chromatography eluting with 15%
acetone/dichloromethane to give I-210 (43 mg, 75%) as a white solid. 1H NMR
(400
299
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
MHz, DMSO-d6) 9.42 (br s, 1 H), 8.27 (s, 1 H), 8.20 (d, J= 8.2 Hz, 1 H), 7.90
(d, J=
7.6 Hz, 1 H), 7.72 (t, J= 7.9 Hz, 1 H), 7.40 (dd, J= 7.4, 9.0 Hz, 1 H), 7.27 -
7.18 (m, 2
H), 7.12 (d, J= 8.3 Hz, 1 H), 6.31 (d, J= 9.0 Hz, 1 H), 6.14 (d, J= 7.0 Hz, 1
H), 5.28 (br
s, 2 H), 3.76 (s, 3 H), 1.27 (s, 9 H) ppm. LC/MS = 94.2%, 452.1 (APCI+).
O
HNAO'J':~ NH2
N N
TFA
0 O 0 O
CH2CI2 \ I tD
N
N
O- O
1-210 P-236
[00624] Synthesis of 6-Amino-l-(6-methoxy-3'-nitro-biphenyl-3-ylmethyl)-1H-
pyridin-2-one (P-236). Into a 20 mL vial with a stir bar was added I-210 (22
mg, 0.049
mmol), dichloromethane (2 mL), and TFA (2 mL). After 6 hours at room
temperature
the solution was concentrated. The residue was purified by flash column
chromatography using 2 - 5% methanol/dichloromethane to give 12.5 mg (74%) of
P-
236 as a brown solid. 'H NMR (400 MHz, DMSO-d6) 8.27 (s, 1 H), 8.20 (d, J= 8.2
Hz, 1 H), 7.91 (d, J= 7.6 Hz, 1 H), 7.76 - 7.69 (m, 1 H), 7.39 (d, J= 1.7 Hz,
1 H), 7.25
(dd, J= 1.7, 8.5 Hz, 1 H), 7.15 - 7.08 (m, 2 H), 6.44 (s, 1 H), 5.76 (s, 2 H),
5.53 (d, J=
8.7 Hz, 1 H), 5.40 (d, J= 7.6 Hz, 1 H), 5.19 (br s, 1 H), 3.77 (s, 3 H) ppm.
LC/MS =
100.0%, 352.1 (APCI+).
[00625] Scheme 54.
O
O + Pd(OAc)z ~O NBS/CCI4 O PI Br 3 \O I / N
B, CI K2CO3 NaH/DMF
NaH///DMF O'Cl
HO OH MeOH-Water I
~ CI ~ I CI 1-211 1-154 P-218
Example 233. Preparation of P-218
[00626] Synthesis of 3'-chloro-2-methoxy-5-methyl-biphenyl (1-211). To 2-
methoxy-
5-methylboronic acid (4.0 g, 24.1 mmol), 1-chloro-3-iodo-benzene (6.32 g,
26.51 mmol),
K2C03 (8.33 g, 60.25 mmol), and palladium(II) acetate (0.27 g, 1.2 mmol) was
added
300
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
methanol (150 mL), and H2O (30 mL). Argon gas was bubbled through the stirred
reaction for 5 min. The reaction was stirred at room temperature under argon
for 20 h.
The reaction mixture was concentrated, and H2O and dichloromethane (60 mL
each)
were added. The organic layer was separated and the aqueous layer was
extracted with
dichloromethane (2 x 60 mL). The combined organic extracts were dried with
Na2SO4,
filtered, and concentrated. The residue was purified by silica gel column
chromatography using 1:1 dichloromethane-hexanes then dichloromethane to
afford 5.6
g (98%) of 1-211 as a viscous liquid.
[00627] Synthesis of 5-bromomethyl-3'-chloro-2-methoxy-biphenyl (1-154). To I-
211 (2.0 g, 8.59 mmol) and NBS (1.68 g, 9.45 mmol) in CC14 (30 mL) was added
benzoylperoxide (0.1 g, 0.43 mmol). The reaction was stirred at 85 C under N2
for 5 h.
The reaction was cooled to room temperature and concentrated. The residue was
dissolved in mixture of 5% ethyl acetate in hexanes (20 mL) and purified by
silica gel
column chromatography using 5% ethyl acetate in hexanes to afford 2.81 g (98%)
of I-
154 as light yellow viscous liquid.
[00628] Synthesis of 1-(3'-chloro-6-methoxy-b iphenyl-3 -ylmethyl)-pyrrolidin-
2 -one
(P-218). To a solution of 2-pyrrolidone (0.2 g, 2.41 mmol) in DMF (1 mL) was
added to
a cooled (0 C) slurry of NaH (0.1 g, 2.41 mmol) in DMF (3 mL) under Ar. The
reaction
mixture was slowly warmed to room temperature, stirred for 45 min, then cooled
to 0 C.
To this I-154 (0.5 g, 1.6 mmol) in DMF (2 mL) was added over 5 min. The
reaction
mixture was slowly warmed to room temperature, stirred for 20 h, poured on to
crushed-
ice water, and extracted with ethyl acetate (2 x 60 mL). The combined organic
extracts
were dried with Na2SO4, filtered, and concentrated. The residue was dissolved
in
dichloromethane (20 mL) and purified by silica gel column chromatography using
5%
ethyl acetate in hexanes to afford 0.22 g (43%) of P-218 as a viscous liquid.
1H NMR
(CDC13, 400 MHz): 7.49-7.51 (m, 1 H), 7.27-7.4 (m, 4 H), 7.23 (dd, J= 8.4, 2.4
Hz, 1
H), 7.16 (d, J= 2.4 Hz, 1 H), 6.93 (d, J= 8.4 Hz, 2 H), 4.43 (s, 2 H), 3.81
(s, 3 H), 3.28
(t, J= 7.2 Hz, 2 H), 2.44 (t, J= 8.0 Hz, 2 H), 1.95-2.05 (m, 2 H) ppm;
MS(APCI+):
316.1 (M+1), LC-MS: 100%.
301
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 234. Preparation of P-211
H/
NHz N N II
N
0 O Ac2O O O O
DCM,TEA
Z~' to
N- 0-
O
1-204 P-211
[00629] Synthesis of N-[1-(6-Methoxy-3'-nitro-biphenyl-3-ylmethyl)-6-oxo-1,6-
dihydro-pyridin-3-yl]-acetamide (P-211). Into a 20 mL vial with a stir bar was
added I-
204 (113 mg, 0.32 mmol), dichloromethane (2 mL), Ac20 (46 uL, 0.48 mmol), and
TEA
(90 uL, 0.64 mmol). The reaction was stirred at room temperature for 4 hours,
IN
aqueous HC1 was added, the product was extracted with dichloromethane, and the
organics were concentrated. The residue was purified by flash column
chromatography
using 25% - 35% acetone/dichloromethane to afford 12.5 mg (10%) of P-211 as a
tan
solid. 1H NMR (400 MHz, DMSO-d6) 9.66 (s, 1 H), 8.28 (s, 1 H), 8.23 - 8.12 (m,
2 H),
7.92(d,J=7.8Hz,1H),7.72(t,J=8.0Hz,1H),7.43(d,J=1.9Hz,1H),7.41-7.32
(m, 2 H), 7.16 (d, J= 8.5 Hz, 1 H), 6.42 (d, J= 9.5 Hz, 1 H), 5.09 (s, 2 H),
3.79 (s, 3 H),
1.95 (s, 3 H) ppm. LC/MS = 95.5%.
Example 235. Preparation of P-223
O
0 HO,B,OH NN
N Pd(OAC)z 0
0 6+ K2CO3, PPh3
Br F Dioxane
F F
EtOH-H20 F
1-181
P-223
[00630] Synthesis of 1-(3',4'-difluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyrrolidin-2-
one (P-223). To 1-181 (0.15 g, 0.53 mmol), 3,4-difluorophenylboronic acid
(0.13 g, 0.79
mmol), triphenylphosphine (0.07 g, 0.26 mmol), K2C03 (0.03 g, 0.21 mmol) and
palladium(II) acetate (0.014 g, 0.06 mmol) were added dioxane (6 mL) and EtOH-
H20
302
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
(1:1, 3 mL). Argon gas was bubbled through the stirred reaction for 5 min. The
reaction
was stirred at 85 C under argon for 20 h. The reaction was cooled to room
temperature
and concentrated. The residue was purified by silica gel column chromatography
using
30-50% ethyl acetate in hexanes to afford 0.15 g (89%) of P-223 as a light
brown viscous
liquid. 1H NMR (CDC13, 400 MHz): 7.33-7.38 (m 1 H), 7.14-7.24 (m, 4 H), 6.93
(d, J=
8.4 Hz, 1 H), 4.43 (s, 2 H), 3.81 (s, 3 H), 3.29 (t, J= 7.2 Hz, 2 H), 2.44 (t,
J= 8.4 Hz, 2
H), 1.95-2.05 (m, 2 H) ppm; MS(APCI+): 318.1 (M+1), LC-MS: 100%.
Example 236. Preparation of P-240
Br OH Palladium(II)acetate
Triphenylphosphine
O HO Potassium Carbonate O
B
+ O 1,2-Dimethoxyethane NH
>- 2
NH 2 Ethanol, Water
Cl 4 Cl
1-154 P-240
[00631] Synthesis of 4-(3'-Chloro-6-methoxy-biphenyl-3-ylmethyl)-benzamide (P-
240). A suspension of 1-154 (300 mg, 0.963 mmol), 4-aminocarboxyphenylboronic
acid
(238 mg, 1.44 mmol), triphenylphosphine (50.4 mg, 0.193 mmol), and water (1
mL) was
degassed with a nitrogen stream for 30 minutes. To the reaction was added
palladium(II)
acetate (22 mg, 0.0963 mmol) under nitrogen and the reaction was heated to 90
C under
nitrogen with stirring overnight. The solvent was removed under vacuum and the
product
crystallized in hexanes (50 mL). The product was purified by flash silica gel
column
chromatography (5% methanol in dichloromethane) to give P-240 (112 mg, 33%
yield)
as a grey solid. 1 H NMR (400 MHz, CDC13) 7.75-7.73 (m, 2H), 7.49 (t, J= 1.6
Hz,
1H), 7.38-7.27 (m, 5H), 7.14-7.10 (m, 2H), 6.91 (d, J= 8.0 Hz, 1H), 6.0 (br s,
1H), 5.75
(br s, 1H), 4.01 (s, 2H), 3.79 (s, 3H) ppm. LCMS =92.3 % purity. MS(APCI+)=
352.1
(M+1).
303
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 237. Preparation of P-245
O
O HO, OH N6
v
\ I / N ~f Pd(OAc)Z~ fz~
v\
KZCO3, PPh3
O Br \ I SDioxane
EtOH-H20 1-181
P-245
[00632] Synthesis of 1-(6-methoxy-3'-methylsulfanyl-biphenyl-3-ylmethyl)-
pyrrolidin-2-one (P-245). To I-181 (0.15 g, 0.53 mmol), 3-methylsulfanyl-
benzeneboronic acid (0.11 g, 0.639 mmol), triphenylphosphine (0.07 g, 0.26
mmol),
K2C03 (0.03 g, 0.21 mmol) and palladium(II) acetate (0.014 g, 0.06 mmol) were
added
dioxane (6 mL) and EtOH-H20 (1:1, 3 mL). Argon gas was bubbled through the
stirred
reaction for 5 min. The reaction was stirred at 85 C under argon for 20 h.
The reaction
was cooled to room temperature and concentrated. The residue was purified by
silica gel
column chromatography using 15% acetone in dichloromethane then reverse phase
(Cis)
prep TLC using 40% acetonitrile in water then by silica gel column
chromatography
using 5% methanol in dichloromethane to afford 0.013 g (8%) of P-245 as a
viscous
liquid. 1H NMR (CDC13, 400 MHz): 7.39-7.45 (m 1 H), 7.2-7.34 (m, 4 H), 7.17
(d, J=
6.4 Hz,1H),6.93(d,J= 8.4Hz,1H),4.43(s,2H),3.8(s,3H),3.28(t,J= 7.2 Hz, 2
H), 2.43 (t, J= 8.0 Hz, 2 H), 1.95-2.05 (m, 2 H) ppm; MS(APCI+): 328.1 (M+1),
LC-
MS: 93.2%.
Example 238. Preparation of P-248
0
HN TBSCI, DMF 0
HN 3" ~5
OH imidazole
OTBS
1-212
[00633] Synthesis of (S)-4-(tert-Butyl-dimethyl-silanyloxy)-pyrrolidin-2-one
(1-212).
Into a 100 mL round-bottomed flask with a stir bar was added (S)-4-hydroxy-
pyrrolidin-
2-one (3.04 g, 30.07 mmol), TBSC1(4.99 g, 33.07 mmol), imidazole (3.07 g,
45.10
mmol), and DMF (30 mL). After stirring at room temperature for 18 hours, the
reaction
was added to 100 mL of water and stirred for 30 minutes. The solids were
filtered and
washed with water. After drying the solids in a vacuum dessicator for 3 days,
5.97 g
304
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
(92%) of 1-212 was obtained.
O
O N6
O + HN ) NaH O
THE OTBS
OTBS
N
1-212 0-
[00634] 1-70 1-213
[00635] Synthesis of ((S)-4-(tert-Butyl-dimethyl-silanyloxy)-1-(6-methoxy-3'-
nitro-
biphenyl-3-ylmethyl)-pyrrolidin-2-one (I-213). Into a 250 mL round-bottomed
flask
with a stir bar was added 1-212 (2.57 g, 11.95 mmol), THE (150 mL), and the
solution
was cooled to 0 C. NaH (0.56 g, 14.12 mmol) was added and the suspension was
stirred
at room temperature for 30 minutes. To this, 1-70 (3.50 g, 10.86 mmol) was
added and
the reaction was stirred for 20 hours at room temperature after which time 50
mL of
water was added and the product was extracted with ethyl acetate. The organics
were
concentrated to yield 8.9 g of 1-213, which was used as is in the next
reaction.
O o
N6 N6
0 % TBAF
OTBS - O
OH
THE i
N, to
_ N
6-
P-248
[00636] Synthesis of (S)-4-Hydroxy-l-(6-methoxy-3'-nitro-biphenyl-3-ylmethyl)-
pyrrolidin-2-one (P-248). Compound I-213 (8.9 g, 19.5 mmol) from the step
described
above was added to a 100 mL round-bottomed flask, followed by 10 mL of THE and
TBAF (29 mL, 29 mmol, 1.OM in THF). The solution was stirred at room
temperature
for 30 minutes after which time NH4C1 and brine were added. The product was
extracted
with ethyl acetate, dried over Na2S04, filtered and concentrated. Flash column
chromatography purification eluting with 25 - 50% acetone/dichloromethane
followed
by flash column chromatography eluting with 5% methanol/dichloromethane gave
143
mg (4%, 2 steps) of P-248. 'H NMR (400 MHz,CDC13) 8.40 (d, J= 1.6 Hz, 1 H),
8.21
-8.14(m,1H),7.84(d,J=7.8Hz,1H),7.56(t,J= 8.0 Hz,1H),7.31-7.20 (m, 2 H),
305
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
6.98 (d, J= 8.5 Hz, 1 H), 4.63 - 4.48 (m, 2 H), 4.46 - 4.34 (m, 1 H), 3.83 (s,
3 H), 3.56
(dd, J= 5.6, 10.8 Hz, 1 H), 3.24 (dd, J= 1.6, 10.9 Hz, 1 H), 2.76 (dd, J= 6.5,
17.4 Hz, 1
H), 2.45 (dd, J= 2.1, 17.3 Hz, 1 H), 1.83 (d, J= 4.0 Hz, 1 H) ppm. LC/MS =
97.4%,
343.1 (APCI+).
Example 239. Preparation of P-253
O o
N6 chlorosulfonyl- N6
isocyanate O
O O O
OH DCM NH 2
N p- 6-
P-248 P-253
[00637] Synthesis of Carbamic acid (S)-1-(6-methoxy-3'-nitro-biphenyl-3-
ylmethyl)-
5-oxo-pyrrolidin-3-yl ester (P-253). Into a 20 mL vial with a stir bar was
added
chlorosulfonyl isocyanate (42 mg, 0.30 mmol), 1 mL dichloromethane, then P-248
(51.3
mg, 0.15 mmol) in 2 mL of dichloromethane. The reaction was stirred at room
temperature for 18 hours after which 2 mL of water was added and the mixture
was
stirred at room temperature for 30 minutes. The layers were separated and the
aqueous
was extracted with dichloromethane. The combined organics were concentrated
and then
purified by preparative layer TLC using 50% acetone/ dichloromethane to obtain
P-253
(12.4 mg, 21%) as an off-white semi-solid. 'H NMR (400 MHz, DMSO-d6) 8.30 (s,
1
H), 8.20 (dd, J= 2.1, 8.6 Hz, 1 H), 7.94 (d, J= 7.8 Hz, 1 H), 7.72 (t, J= 8.0
Hz, 1 H),
7.33 - 7.26 (m, 2 H), 7.16 (d, J= 9.0 Hz, 1 H), 6.59 (br. s., 2 H), 5.04 (t,
J= 5.8 Hz, 1 H),
4.49 - 4.30 (m, 2 H), 3.80 (s, 3 H), 3.46 - 3.38
(m,1H),3.21(d,J=11.4Hz,1H),2.78
(dd, J= 6.9, 17.5 Hz, 1 H), 2.23 (d, J= 18.1 Hz, 1 H) ppm. LC/MS = 100.0%,
386.1
(APCI+).
306
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 240. Preparation of P-506
0
7 tN
OH
:ILI
No
O
P-506
[00638] Synthesis of (R)-4-Hydroxy-l-(6-methoxy-3'-nitro-biphenyl-3-ylmethyl)-
pyrrolidin-2-one (P-506). P-506 was prepared in a similar manner as that
described
above for P-248 except starting with (R)-4-Hydroxy-pyrrolidin-2-one. 1H NMR
(400
MHz, CDC13) 8.40 (t, J= 1.9 Hz, 1 H), 8.17 (dd, J= 1.7, 8.2 Hz, 1 H), 7.84 (d,
J= 7.8
Hz, 1 H), 7.55 (t, J= 8.0 Hz, 1 H), 7.30 - 7.27 (m, 1 H), 7.22 (d, J= 2.1 Hz,
1 H), 6.97
(d,J=8.5Hz,1H),4.61-4.48(m,2H),4.41(d,J=14.8Hz,1H),3.85-3.81(m,3
H), 3.56 (dd, J= 5.6, 10.9 Hz, 1 H), 3.24 (dd, J= 1.8, 10.8 Hz, 1 H), 2.75
(dd, J= 6.5,
17.3 Hz, 1 H), 2.44 (dd, J= 2.1, 17.4 Hz, 1 H), 2.12 (br s, 1 H) ppm. LC/MS =
95.6%,
343.1 (APCI+).
Example 241. Preparation of P-266
Br OH Palladium(II)acetate
Triphenylphosphine
O
T
O HOB a Potassium Carbonate 0 S'0
+ ;O 1,2-dimethoxyethane NH2
0 NH ethanol, water
Cl z 4 Cl
1-154 P-266
[00639] Synthesis of 4-(3'-Chloro-6-methoxy-biphenyl-3-ylmethyl)-
benzenesulfonamide (P-266). P-266 was synthesized from 1-154 (300 mg, 0.963
mmol)
and 4-aminosulfonylphenylboronic acid pinocolate ester (409 mg, 1.44 mmol)
using the
same conditions as for the synthesis of P-240. The solvent was removed under
vacuum
and the residue suspended in ethyl acetate (20 mL) and water (20 mL). The
layers were
separated and the organic solution was washed with water (40 mL), saturated
aqueous
sodium bicarbonate (40 mL), water (2 x 40 mL), and brine (40 mL), dried over
sodium
sulfate, filtered, and the solvent removed under vacuum. The residue was
purified by
flash silica gel column chromatography (2.5% acetone in dichloromethane) to
give a
material which was then purified by preparatory silica gel TLC (eluting with
2.5 %
307
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
acetone in dichloromethane with 3 developments) to give P-266 (52.2 mg, 14%
yield) as
a clear gum. 1H NMR (400 MHz, CDC13) 7.86-7.83 (m, 2H), 7.49-7.48 (m, 1H),
7.38-
7.29 (m, 5H), 7.14-7.09 (m, 2H), 6.92 (d, J= 8.4 Hz, H), 4.71 (s, 2H), 4.03
(s, 2H), 3.80
(s, 3H) ppm. LCMS = 100.0% purity. MS(APCI-)= 386.0 (M-1), 217.0 (M-171).
Example 242. Preparation of 1-216
O
Hexamethylenetetramine AH Meta-chloroperbenzoic Acid
O F
Br Trifluoroacetic Acid O ~ F Dichloromethane
A Br A
1-30
O H Potassium Hydroxide
O F 0 Ethanol O OH
F
Br Br
1-215 1-216
[00640] Synthesis of 3-Bromo-2-fluoro-4-methoxy-benzaldehyde (I-30). A
solution
of 2-bromo-3-fluoroanisol (5.00 g, 24.4 mmol) in trifluoroacetic acid (25 mL)
was
heated to 80 C and then a solution of hexamethylenetetramine (6.83 g, 48.8
mmol) in
trifluoroacetic acid (25 mL) was added dropwise over 1.5 h. Upon completion of
the
addition, the reaction was stirred at 80 C for 1 h under nitrogen. The excess
trifluoroacetic acid was removed under vacuum, and the pH was adjusted to 7.5-
8.0 by
addition of saturated aqueous potassium carbonate (-100 mL). The white solid
that
formed was filtered to give 1-30 which was used without additional
purification.
[00641] Synthesis of Formic acid 3-bromo-2-fluoro-4-methoxy-phenyl ester (1-
215).
To a solution of 1-30 (4.84 g) in dichloromethane (60 mL) was added meta-
chloroperbenzoic acid (16.09 g) and the reaction was stirred at room
temperature
overnight. The reaction was diluted with dichloromethane (200 mL), washed with
saturated aqueous sodium thiosulfate (300 mL), and extracted into
dichloromethane (2
x100 mL). The combined organic extracts were washed with saturated aqueous
sodium
bicarbonate (3 x 400 mL), water (2 x 400 mL), and brine (400 mL), dried over
sodium
sulfate, decanted, and the solvent removed under vacuum to give I-215 (12:5,
4.05 g)
which was used as is without additional purification. 1H NMR (400 MHz, CDC13)
d:
8.27 (t, J= 0.8 Hz, 1H), 7.10-7.06 (m, 1H), 6.70 (dd, J= 8.2 Hz, 1.0 Hz, 1H),
3.92 (s,
3H) ppm.
308
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00642] Synthesis of 3-Bromo-2-fluoro-4-methoxy-phenol (I-216). To a solution
of I-
215 (4.05 g) in ethyl alcohol (75 mL) was added solid potassium hydroxide
pellets (1.71
g) and the brown solution was stirred at room temperature overnight. The
reaction was
acidified to pH 2 using concentrated hydrochloric acid and the ethanol was
removed
under vacuum. The material was diluted with water (300 mL) and extracted with
dichloromethane (300 mL, 2 x 100 mL). The combined extracts were washed with
brine
(500 mL), dried over sodium sulfate, decanted, and the solvent removed under
vacuum.
The residue was then dissolved in diethyl ether (500 mL) and extracted with 1
M
aqueous sodium hydroxide (300 mL, 2 x 250 mL). The combined aqueous layers
were
acidified to pH 2 with concentrated hydrochloric acid and extracted into
diethyl ether (2
x 300 mL). The combined diethyl ether extracts were washed with water (500 mL)
and
brine (300 mL), dried over sodium sulfate, decanted, and the solvent removed
under
vacuum to give an orange solid. The solid was purified by flash silica gel
column
chromatography eluting with dichloromethane to give I-216 (1.45 g) as a yellow
powder.
1H NMR (400 MHz, CDC13) 6.96-6.91 (m, 1H), 6.62 (dd, J= 9.2 Hz, 2.0 Hz, 1H),
4.78
(d, J= 3.6 Hz, 1H), 3.86 (s, 3H) ppm.
309
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00643] Scheme 55.
OH HO,B,OH OH
+ Tetra kis(triphenylphosphine)palladium(0) -~O F
O F
Br Tolune, Ethanol
R Sodium Carbonate(aq)
1-216 A R
R=CI
R=000H3 1-217R=Cl
1-218 R = COCH3
Copper(I)chloride 0
1-217 or 1.218 + 2,2,6,6-Tetramethyl-heptane-3,5-dione 0 F N+0
Cesium Carbonate 0
~V* N-Methylpyrolidine
0 '0 A R
1-219 R=Cl
1-220 R = COCH3
1.) Iron powder
Ammonium Chloride O O
ethanol, Water 0 F NH Methylsulfonylchloride 0 F ON;S\
3 H
2.) Hydrocloric Acid Cl Chloroform
Diethyl Ether R Cl
P-254
1-221 R=Cl
P-283 R = COCH3
Example 243. Preparation of P-254
[00644] Synthesis of 3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ol (1-217). A
solution
of 1-216 (390 mg, 1.76 mmol) in toluene (6 mL) was degassed with a nitrogen
stream for
min. To this solution was added ethanol (1 mL), 3-chlorophenylboronic acid
(331 mg,
2.12 mmol), 2 M aqueous sodium carbonate (1.76 mL, 3.53 mmol), and the
nitrogen
stream was continued for 15 min. To this suspension was added
tetrakis(triphenylphosphine) palladium(0) (102 mg, 0.0882 mmol) under nitrogen
and the
reaction was heated to 90 C overnight. Additional
tetrakis(triphenylphosphine)palladium(0) (102 mg, 0.0882 mmol) was added under
nitrogen and the reaction reacted at 90 C for 24 h, and a third portion of
tetrakis(triphenylphosphine)palladium(0) (102 mg, 0.0882 mmol) was added and
the
reaction heated to 90 C for 24 hours. Approximately one half of the solvent
was
removed under vacuum and the residual material was diluted with ethyl acetate
(100 mL)
and saturated aqueous ammonium chloride (100 mL). The layers were filtered,
separated,
310
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
and the organic solution was washed with saturated aqueous sodium bicarbonate
(2 x 100
mL), water (100 mL), and brine (50 mL). The solvent was dried over sodium
sulfate,
filtered, and the solvent removed under reduced pressure. The product was
purified by
flash silica gel column chromatography (eluting with 2.5% acetone in
dichloromethane)
to give I-217 (398 mg, 89 % yield). 1H NMR (400 MHz, CDC13) 7.41 (s, 1H), 7.39-
7.28 (m, 3H), 6.97 (t, J= 9.4 Hz, 1H), 6.67 (dd, J= 9.2 Hz, 2.0 Hz, 1H), 4.79
(s, 1H),
3.72 (s, 3H) ppm.
[00645] Synthesis of 3'-Chloro-2-fluoro-6-methoxy-3-(4-nitro-phenoxy)-biphenyl
(I-
219). A suspension of 1-217 (200 mg, 0.792 mmol), 4-iodonitrobenzene (179 mg,
0.720
mmol), cesium carbonate (469 mg, 1.44 mmol), and 2,2,6,6-tetramethylheptane-
3,5-
dione (37 uL, 0.180 mmol) in N-methylpyrrolidone (4 mL) was stirred under
nitrogen.
To this reaction was added copper(I) chloride (35.6 mg, 0.360 mmol) and the
reaction
was heated to 100 C under nitrogen overnight. The reaction was diluted with
ethyl
acetate (20 mL) and washed with water (20 mL). The aqueous wash was extracted
with
ethyl acetate (20 mL) and the organic extracts were combined. The organic
extracts were
washed with water (2 x 20 mL), 1 N aqueous sodium hydroxide (2 x 20 mL), 1 N
aqueous hydrochloric acid (2 x 20 mL), brine (20 mL), dried over sodium
sulfate,
filtered, and the solvent removed under vacuum to give crude I-219 (181.0 mg,
61%
yield) which was used as is in the next reaction.
[00646] Synthesis of 4-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-yloxy)-phenyl-
amine hydrochloride salt (I-221). To a suspension of 1-219 (180 mg, 0.645
mmol) and
iron powder (126 mg, 2.26 mmol) in ethanol (2.6 mL) and water (0.8 mL) was
added
solid ammonium chloride (193 mg, 3.60 mmol). The reaction was purged with
nitrogen,
and heated to 85 C with stirring for 3 h. The solvent was removed under
vacuum and
the material was diluted in ethyl acetate (50 mL) and water (50 mL). The
layers were
separated and the aqueous layer was extracted with ethyl acetate (50 mL). The
combined
organic extracts were washed with water (2 x 50 mL) and brine (50 mL), dried
over
sodium sulfate, filtered, and the solvent removed under reduced pressure. The
product
was purified by flash silica gel column chromatography (5% acetone in
dichloromethane) to give the free base of 1-221 (136 mg). The free base was
dissolved in
diethyl ether (2.5 mL) and 2 N hydrogen chloride in diethyl ether (0.8 mL) was
added
311
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
drop wise. The salt began to precipitate out after 15 min and the suspension
was allowed
to stir for an additional 1.5 h. The solid was filtered and washed with
diethyl ether (3
mL) and hexanes (4 x 5 mL) to give I-221 (104 mg, 47% yield) as a beige
powder. 1H
NMR (400 MHz, DMSO-d6) 9.6 (br s, 2H), 7.51-7.45 (m, 3H), 7.36-7.25 (m, 4H),
7.08-
7.01 (m, 3H), 3.77 (s, 3H) ppm. MS(ESI+)=345.5 (M+1).
[00647] Synthesis of N-[4-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-yloxy)-
phenyl]-
methanesulfonamide(P-254). A suspension of 1-221(50.0 mg, 0.138 mmol) and
pyridine (21.8 mg, 0.276 mmol) in chloroform (1 mL) was stirred for 10 min. To
the
suspension was added methanesulfonylchloride (15.8 mg, 0.138 mmol) and the
reaction
was stirred at room temperature overnight. The solvent was removed under
vacuum and
the residue was purified by preparatory silica gel TLC (5 % acetone in
dichloromethane,
2 developments, 0.5% acetone in dichloromethane, 5 developments, and 5%
acetone in
dichloromethane) to give P-254 (26.1 mg, 47% yield) as a orange gum. 1 H NMR
(400
MHz, CDC13) 7.42-7.42 (m, 1H), 7.35-7.29 (m, 3H), 7.25-7.18 (m, 2H), 7.18 (t,
J= 2.8
Hz, 1H), 7.00 - 6.95 (m, 2H), 6.75 (dd, J= 7.4 Hz, 0.2 Hz, 1H), 6.20 (s, 1H),
3.80 (s,
3H), 2.98 (s 3H) ppm. MS(ESI+)= 421.6 (M), 343.0 (M-79.0).
Example 244. Preparation of P-283
[00648] Synthesis of 1-(2'-Fluoro-3'-hydroxy-6'-methoxy-biphenyl-3-yl)-
ethanone (I-
218). A solution of 1-216 (500 mg, 2.26 mmol) and 3-acetylphenylboronic acid
(446 mg,
2.72 mmol) in toluene (8 mL) was degassed with a nitrogen stream. To this
solution was
added ethanol (1.5 mL) and 2 M aqueous sodium carbonate (2.25 mL, 4.52 mmol),
followed by the addition of tetrakis(triphenylphosphine)palladium(0) (130 mg,
0.113
mmol). The reaction was heated to 100 C for 24 h. An additional portion of
tetrakis(triphenylphosphine)palladium(0) (130 mg, 0.113 mmol) was added under
nitrogen and the reaction was heated to 100 C overnight. An additional
portion of 3-
aceteylphenyl boronic acid (223 mg, 1.36 mmol) was added and the reaction
heated to
100 C an additional 24 hours. The reaction was diluted with ethyl acetate (50
mL) and
water (50 mL), filtered, and the layers separated. The aqueous layer was
extracted with
ethyl acetate (50 mL) and the organic extracts combined. The organic solution
was
washed with water (2 x 100 mL), saturated aqueous sodium bicarbonate (100 mL),
brine
312
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
(50 mL), dried over sodium sulfate, filtered and the solvent removed under
vacuum. The
product was purified by flash silica gel column chromatography (0-5% acetone
in
dichloromethane) to give 1-218 (280 mg, 48% yield) as a brown solid.
[00649] 1H NMR (400 MHz, CDC13) 8.01 (m, 1H), 7.97 (dt, J= 7.9 Hz, 1.5 Hz,
1H),
7.62 (dd, J= 7.6 Hz, 1.2 Hz, 1H), 7.54 (t, J= 7.0 Hz, 1H), 6.99 (t, J= 9.2 Hz,
1H), 6.69
(dd, J= 9.2 Hz, 2.0 Hz, 1H), 4.81 (d, J= 4.4 Hz, 1H), 3.73 (s, 3H), 2.63 (s,
3H) ppm.
[00650] Synthesis of 1-[2'-Fluoro-6'-methoxy-3'-(4-nitro-phenoxy)-biphenyl-3-
yl]-
ethanone (1-220). A solution of 1-218 (275 mg, 1.06 mmol) and 4-
iodonitrobenzene
(264 mg, 1.06 mmol) in N-methylpyrrolidone (5.5 mL) was degassed with a
nitrogen
stream for 1 min. To the solution was added cesium carbonate (689 mg, 2.11
mmol),
copper(I) chloride (52.3 mg, 0.528 mmol), 2,2,6,6-tetramethylheptane-3,5-dione
(48.6
mg, 0.264 mmol), and the reaction was heated to 90 C for 18 h. The reaction
was cooled
to room temperature and the solvent was removed under vacuum. The residue was
taken
up in ethyl acetate (20 mL), washed with water (20 mL), the aqueous wash
extracted
with ethyl acetate (20 mL), and the organic extracts combined. The organic
extracts were
washed with water (2 x 40 mL), 1 N aqueous sodium hydroxide (40 mL), 1 N
aqueous
hydrochloric acid (2 x 40 mL), and brine (50 mL). The solvent was dried over
sodium
sulfate, filtered, and removed under vacuum to give a brown solid. The residue
was
purified by flash silica gel column chromatography (1-5% acetone in
dichloromethane)
followed by flash silica gel column chromatography (30% ethyl acetate in
hexanes) to
give 1-220 (78.4 mg, 19% yield). 1H NMR (400 MHz, CDC13) 8.23-8.20 (m, 2H),
8.02
(m, 1H), 7.98-7.96 (m,1H), 7.62-7.60 (m, 1H), 7.55 (t, J= 7.8 Hz, 1H), 7.20
(t, J= 9.0
Hz, 1H), 7.05-7.02 (m, 2H), 6.84 (dd, J= 9.2 Hz, 1.6 Hz, 1H), 3.83 (s, 3H),
2.63 (s, 3H)
ppm.
[00651] Synthesis of 4-(3'-Acetyl-2-fluoro-6-methoxy-biphenyl-3-yloxy)-phenyl-
amine hydrochloride salt (P-283). A suspension of 1-220 (75.0 mg, 0.197 mmol),
ammonium chloride (53.5 mg, 1.00 mmol), and iron powder (38.5 mg, 0.690 mmol)
in
ethanol (0.8 mL) and water (0.25 mL) was purged with nitrogen and stirred
under
nitrogen for 4 h at 85 C. The solvent was removed under vacuum and the
residual
material was suspended in ethyl acetate (50 mL). The organic suspension was
washed
313
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
with water (50 mL), the aqueous layer was extracted with ethyl acetate (50
mL), and the
organic solutions combined. The combined extracts were washed with water (2 x
50 mL)
and brine (50 mL), dried over sodium sulfate, filtered, and the solvent
removed under
vacuum. The residue was dissolved in diethyl ether (2 mL), and 2 N hydrogen
chloride in
diethyl ether (400 uL) was added. The reaction was stirred for 1 h and
filtered to give P-
283 (57.0 mg, 75% yield) as a white powder.
[00652] 1H NMR (400 MHz, DMSO-d6) d: 7.98 (dt, J= 7.3 Hz, 1.5 Hz, 1H), 7.93
(s,
1H), 7.66-7.59 (m, 2H), 7.34-7.29 (m, 3H), 7.09-7.03 (m, 3H), 3.77 (s, 3H),
2.60 (s, 3H)
ppm. MS(ESI+) = 352.9 (M+1)
[00653] Scheme 56.
0
1,2-Dichloromethylmethylether
Titanium(IV)Chloride H Sodium Borohydride OH _(~~ - - O ICI F Dichloromethane
O F Tetrahydrofuran O F
Br Br Water Br
1-30 1-31
0
HO, OH Palladium(II)Acetate OH LL
B' Triphenylphosphine 0 0
Potassium Carbonate 0 F Chloromethylformate O F
1-31 +
Dioxane, Water, Ethanol TetraPyridine
hydrofuran
O O
1-222 1-223
Example 245. Preparation of 1-223
[00654] Synthesis of 3-Bromo-2-fluoro-4-methoxy-benzaldehyde using
titanium(IV)chloride (1-30). A solution of 2-bromo-3-fluoroanisol (5.00 g,
24.3 mmol)
in dichloromethane (120 mL) was cooled to 0 C in a salt-ice bath and purged
with
nitrogen. The reaction was allowed to stir 15 min under nitrogen. To the
reaction was
added titanium(IV) chloride (23.1 g, 122 mmol), followed by a,a-dichloromethyl-
methyl ether (4.21 g, 36.6 mmol) at 0 C under nitrogen. The reaction was
allowed to
warm to room temperature and stirred for 22 h. The red solution was poured
into ice
water (600 mL), and extracted into dichloromethane (3 x 200 mL). The organic
extracts
were combined, washed with saturated aqueous sodium bicarbonate (2 x 400 mL),
water
(2 x 400 mL), and brine (400 mL), dried over sodium sulfate, filtered, and the
solvent
314
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
was removed under vacuum. The product was dried in a vacuum oven at 80 C
overnight
to give 1-30 (5.75 g, quantitative yield). 1H NMR (400 MHz, CDC13) 10.22 (s,
1H),
7.86 (dd, J= 8.8 Hz, 7.6 Hz, 1H), 6.82 (d, J= 8.4 Hz, 1H), 4.01 (s, 3H) ppm.
[00655] Synthesis of (3-Bromo-2-fluoro-4-methoxy-phenyl)-methanol (I-31). A
suspension of 1-30 (5.20 g, 22.3 mmol) in tetrahydrofuran (40 mL) and water
(40 mL)
was cooled to 0 C and sodium borohydride (2.53 g, 66.9 mmol) was added
portionwise.
The reaction was stirred for 3.5 h allowing the reaction to warm to room
temperature.
The tetrahydrofuran was removed under vacuum, water was added (100 mL) and the
resultant solid was extracted into ethyl acetate (2 x 200 mL). The combined
extracts were
washed with water (2 x 200 mL), brine (200 mL), dried over sodium sulfate,
filtered, and
the solvent removed under vacuum to give 1-31 (4.82 g, 92% yield) as a white
powder.
1H NMR (400 MHz, CDC13) 7.32 (t, J= 8.4 Hz, 1H), 6.71 (dd, J= 8.4 Hz, 1.6 Hz,
1H),
4.72 (d, J= 6.0 Hz, 2H), 3.92 (s, 3H), 1.70 (t, J= 6.0 Hz, 1H) ppm.
[00656] Synthesis of 1-(2'-Fluoro-3'-hydroxymethyl-6'-methoxy-biphenyl-3-yl)-
ethanone (1-222). A solution of 1-31 (4.00 g, 17.0 mmol), 3-
acetylphenylboronic acid
(3.07 g, 18.7 mmol) in ethanol (17.5 mL), water (17.5 mL) and 1,4-dioxane (35
mL) was
degassed with a nitrogen stream for 30 min. To the solution was added
potassium
carbonate (7.06 g, 51.1 mmol), triphenylphosphine (1.34 g, 5.11 mmol), and
palladium(II) acetate (382 mg, 1.70 mmol), and the reaction was stirred under
nitrogen
for 10 min. The reaction was heated to 85 C for 4 h under nitrogen, and
additional
palladium(II) acetate (191 mg, 0.851 mmol) and triphenylphosphine (700 mg,
2.55
mmol) were added. Heating with stirring was continued for 4 h, the reaction
was cooled
to room temperature, and ethyl acetate (300 mL) was added. The reaction was
washed
with water (300 mL), sodium chloride was added (-1 g) and the aqueous wash was
extracted with ethyl acetate (300 mL). The organic extracts were combined,
washed with
water (500 mL) and brine (500 mL), dried over sodium sulfate, decolorized with
activated charcoal, filtered, and the solvent was removed under reduced
pressure. The
crude product was purified by flash silica gel column chromatography (50%
ethyl acetate
in hexanes) to give pure 1-222 (700 mg, 15% yield). 1H NMR (400 MHz, CDC13)
8.00-
8.00 (m, 1H), 7.97 (dt, J= 7.9 Hz, 1.6 Hz, 1H), 7.61 (dd, J= 6.2 Hz, 1.4 Hz,
1H), 7.53 (t,
J= 7.6 Hz, 1H), 7.38 (t, J= 8.6 Hz, 1H), 6.80 (dd, J= 8.8 Hz, 1.2 Hz, 1H),
4.74 (d, J=
315
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
6.0 Hz, 2H), 3.79 (s, 3H), 2.63 (s, 3H), 1.74 (t, J= 6.0 Hz, 1H) ppm.H)
[00657] Synthesis of Carbonic acid 3'-acetyl-2-fluoro-6-methoxy-biphenyl-3-
ylmethyl
ester methyl ester (1-223). A solution of 1-222 (700 mg, 2.55 mmol) and
pyridine (429
uL, 6.64 mmol) in tetrahydrofuran (10 mL) was cooled to 0 C in an ice water
bath. To
the solution was added methylchloroformate (542 uL, 5.61 mmol) under nitrogen.
The
white suspension that formed was stirred at room temperature overnight. The pH
was
adjusted to 1 by the addition of concentrated aqueous hydrochloric acid, and
the solution
was extracted with dichloromethane (2 x 30 mL). The combined organic extracts
were
washed with brine (30 mL), dried over magnesium sulfate, filtered, and the
solvent was
removed under vacuum to give 1-223 (661.3 mg, 78% yield) as an orange syrup.
1H
NMR. (400 MHz, CDC13) 7.98-7.95 (m, 2H), 7.61-7.58 (m, 1H), 7.53 (t, J= 7.6
Hz,
1H), 7.41 (t, J= 8.4 Hz, 1H), 6.79 (dd, J= 8.6 Hz, 1.0 Hz, 1H), 5.22 (s, 2H),
3.81 (s,
3H), 3.79 (s, 3H) ppm.
Example 246. Preparation of P-304
O F S:O
NH2
O
P-304
[00658] Synthesis of 4-(3'-Acetyl-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
benzenesulfinic acid amide (P-304). A suspension of 1-223 (300 mg, 0.928
mmol), 4-
amino sulfonylbenzylboronic pinocolate ester (289 mg, 1.02 mmol), and
potassium
carbonate (385 mg, 2.78 mmol) in dimethylformamide (2 mL) was purged with
nitrogen
and allylpalladium chloride dimer (50.9 mg, 0.139 mmol) and
bis(diphenylphosphino)
pentane (123 mg, 0.278 mmol) were added. The reaction was heated to 65 C
overnight.
To this reaction was added ethyl acetate (25 mL) and water (25 mL) and the
layers
separated. The aqueous layer was extracted with ethyl acetate (2 x 25 mL), and
the
organic extracts were combined and washed with brine (50 mL). The organic
solution
316
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
was dried over magnesium sulfate, filtered, and the solvent removed under
vacuum. The
product was purified by flash silica gel column chromatography (5% acetone in
dichloromethane) followed by trituration with diethyl ether (25 mL) to give P-
304 (117
mg, 30% yield) as a faint yellow powder. 'H NMR (400 MHz, CDC13) 7.99-7.98 (m,
1H), 7.95 (dt, J= 7.6 Hz, 1.4 Hz, 1H), 7.86-7.84 (m, 2H), 7.60-7.58 (m, 2H),
7.52 (t, J=
7.6 Hz, 1H), 7.38 (d, J= 8.4 Hz, 2H), 7.13 (t, J = 8.6 Hz, 1H), 6.75 (d, J =
8.8 Hz, 1H),
4.74 (s, 2H), 4.04 (s, 2H), 3.77 (s, 3H), 2.62 (s, 3H) ppm. LCMS = 100.0%
purity.
MS(APCI+) = 414.0 (M+1).
Example 247. Preparation of P-305
O F O
NH2
O
P-305
[00659] Synthesis of 4-(3'-Acetyl-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
benzamide (P-305). A suspension of 1-223 (300 mg, 0.928 mmol), 4-
aminocarbonylbenzylboronic acid (190 mg, 1.02 mmol), and potassium carbonate
(385
mg, 2.78 mmol) in dimethylformamide (2 mL) was purged with nitrogen and
allylpalladium chloride dimer (50.9 mg, 0.139 mmol) and
bis(diphenylphosphino)pentane (123 mg, 0.278 mmol) were added. The reaction
was
heated to 65 C overnight. To this reaction was added ethyl acetate (25 mL)
and water
(25 mL) and the layers separated. The aqueous layer was extracted with ethyl
acetate (2 x
25 mL), and the organic extracts combined and washed with brine (50 mL). The
organic
solution was dried over magnesium sulfate, filtered, and the solvent removed
under
vacuum to give crude product. The product was purified by flash silica gel
column
chromatography (5-10 % acetone in dichloromethane) followed by trituration
with
diethyl ether (25 mL) to give P-305 (160 mg, 46 % yield) as a white powder. 1H
NMR
(400 MHz, CDC13) 7.99 (m, 1H), 7.95 (dt, J= 7.7 Hz, 1.3 Hz, 1H), 7.75-7.73 (m,
2H),
7.61-7.58 (m, 1H), 7.52 (t, J= 7.6 Hz, 1H), 7.32 (d, J= 8.4 Hz, 2H), 7.11 (t,
J= 8.6 Hz,
1H), 6.73 (d, J= 8.8 Hz, 1H), 5.97 (br s, 1 H), 5.54 (br s, 1H), 4.03 (s, 2H),
3.76 (s, 3H),
317
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
2.62 (s, 3H) ppm. LCMS = 96.5% purity. MS(APCI+) = 378.1 (M+1).
[00660] Example 248. preparation of P-276
0 F 0`11
O
O
P-276
[00661] Synthesis of 4-(3'-Acetyl-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
benzoic
acid methyl ester (P-276). A suspension of 1-223 (300 mg, 0.928 mmol), 4-
methoxycarbonyl-benzylboronic acid (184 mg, 1.02 mmol), and potassium
carbonate
(385 mg, 2.78 mmol) in dimethylformamide (2 mL) was purged with nitrogen and
allylpalladium chloride dimer (50.9 mg, 0.139 mmol) and
bis(diphenylphosphino)pentane (123 mg, 0.278 mmol) were added. The reaction
was
heated to 65 C overnight. To this reaction was added ethyl acetate (5 mL) and
water (5
mL), the layers were filtered through celite,the celite washed with ethyl
acetate (15 mL)
and water (15 mL), and the layers separated. The aqueous layer was extracted
with ethyl
acetate (2 x 50 mL), and the organic extracts combined and washed with brine
(100 mL).
The organic solution was dried over magnesium sulfate, filtered, and the
solvent
removed under vacuum to give crude product. The product was purified by flash
silica
gel column chromatography (25% ethyl acetate in hexanes), followed by a
preparatory
silica gel TLC plate (eluting with 25% ethyl acetate in hexnaes), and
trituration with
diethyl ether (5 mL) to give P-276 (72.4 mg, 20 % yield) as a white powder. 1H
NMR
(400 MHz, CDC13) 7.99-7.94 (m, 4H), 7.59-7.58 (m, 1H), 7.52 (t, J= 7.8 Hz,
1H), 7.30
(d, J= 8.80\ Hz, 2H), 7.11 (t, J= 8.6 Hz, 1H), 6.74-6.72 (m, 1H), 4.03 (s,
2H), 3.90 (s,
3H), 3.76 (s, 3H), 2.62 (s, 3H) ppm. LCMS = 96.6% purity. MS(APCI+)=
394.1(M+2).
318
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 249. Preparation of 1-226
",-N
O I F I O`11
O
O
1-226
[00662] Synthesis of 5-(3'-Acetyl-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridine-
2-carboxylic acid methyl ester (1-226). A suspension of 1-223 (500 mg, 1.50
mmol), 2-
methylcarboxypyridine-5-boronic acid pinocol ester (435 mg, 1.65 mmol), and
potassium carbonate (624 mg, 4.51 mmol) in dimethylformamide (3.5 mL) was
degassed
under a nitrogen stream for 15 min. To this solution was added
bis(diphenylphosphino)pentate (199 mg, 0.451 mmol) and allylpalladium chloride
dimer
(82.7 mg, 0.226 mmol). The reaction was heated to 65 C for 50 h. The
reaction was
diluted with ethyl acetate (50 mL) and filtered through celite. To the
filtrate was added
water (50 mL), and the layers were separated. The aqueous wash was extracted
with
ethyl acetate (2 x 50 mL), and all three organic extracts were combined and
washed with
brine (100 mL). The organic solution was dried over magnesium sulfate,
filtered, and the
solvent removed under vacuum. The residue was purified by flash silica gel
column
chromatography (5% acetone in dichloromethane) followed by trituration with
diethyl
ether (15 mL), filtered, and washed with diethyl ether (5 mL) to give 1-226
(190.6 mg,
32% yield) as a white powder. 1H NMR (400 MHz, CDC13) 8.66 (d, J = 2.0 Hz,
1H),
8.06 (d, J= 8.0 Hz, 1H), 7.97-7.94 (m, 2H), 7.67 (dd, J= 8.2 Hz, 2.2 Hz, 1H),
7.57 (dd,
J= 7.6 Hz, 1.6 Hz, H), 7.52 (t, J= 7.6 Hz, 1H), 7.13 (t, J= 8.6 Hz, 1H), 6.75
(d, J= 8.8
Hz, 1H), 4.05 (s, 2H), 4.00 (s, 3H), 3.77 (s, 3H), 2.62 (s, 3H) ppm. LCMS =
100.0%
purity. MS(APCI+)= 394.1 (M+1).
319
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 250. Preparation of 1-224
0
O F N'J~ O
O
1-224
[00663] Synthesis of [4-(3'-Acetyl-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
phenyl]-
methyl-carbamic acid tert-butyl ester (1-224). To a suspension of 1-223 (500
mg, 1.50
mmol) and 4-(tert-butoxycarbonyl-N-methylamino)phenyl boronic acid (415 mg,
1.65
mmol), and potassium carbonate (624 mg, 4.51 mmol) in dimethylformamide (3.5
mL)
was added bis(diphenylphosphino)pentate (199 mg, 0.451 mmol) and
allylpalladium
chloride dimer (82.7 mg, 0.226 mmol) under nitrogen. The reaction was heated
to 85 C
for 29 h. The reaction was diluted with ethyl acetate (30 mL) and water (30
mL) and
filtered through a Celite plug. The Celite was washed with ethyl acetate (2 x
20 mL) and
water (20 mL), and the layers separated. The aqueous wash was extracted with
ethyl
acetate (2 x 50 mL), and all three organic extracts were combined and washed
with water
(2 x 100 mL) and brine (100 mL). The organic solution was dried over sodium
sulfate,
filtered, and the solvent removed under vacuum. The residue was purified by
flash silica
gel column chromatography (dichloromethane) to give 1-224 (624.3 mg, 90%
yield) as a
yellow oil which was used as is without further purification. iH NMR (400 MHz,
CDC13) 8.00 (d, J= 1.0 Hz, 1H), 7.95 (dt, J= 7.7 Hz, 1.3 Hz, 1H), 7.60 (dd, J=
7.6
Hz, 1.2 Hz, 1H), 7.52 (t, J= 7.6 Hz, 1H), 7.19-7.09 (m, 5H), 6.72 (d, J= 8.4
Hz, 1H),
3.95 (s, 2H), 3.75 (s, 3H), 2.62 (s, 3H), 1.45 (s, 9H) ppm. LCMS = 94.1%
purity.
MS(APCI+) = 364.1 (M-100).
Example 251. Preparation of P-328
1.)Thionyl Chloride
N Toluene
N
O F O NaOH O i F i OH 2)Aqueous Ammonium Hydroxide i NH
Tetrahydrofuran O' F
0 Tetrahydrofuran, i 0 0
Methanol, Water
U-Y
0 0 0
1-226 1-228 P-328
320
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00664] Synthesis of 5-(3'-Acetyl-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridine-
2-carboxylic acid (1-228). A solution of 1-226 (150 mg, 0.381 mmol) in
tetrahydrofuran
(1 mL), methanol (1 mL), water (1 mL) and 1 N aqueous sodium hydroxide (0.763
mL)
was strred at room temperature for 18 h. Approximately one half of the solvent
was
removed under vacuum. The remaining solution was adjusted to pH 3 by addition
of
glacial acetic acid. The suspension was extracted with dichloromethane (10
mL), water
(5 mL) was added to the wash, and the aqueous wash was extracted with
additional
dichloromethane (2 x 10 mL). All three organic extracts were combined, dried
over
magnesium sulfate, filtered, and the solvent was removed under vacuum, and the
residue
was dried under high vacuum for 24 h. The resultant beige syrup crystallized
in diethyl
ether (5 mL), stirred for 30 min, filtered, and washed with hexanes (2 x 2 mL)
to give I-
226 (90.6 mg, 63% yield) as a white powder. 1H NMR (400 MHz, DMSO-d6) 8.62 (d,
J
= 2.0 Hz, 1H), 7.99-7.94 (m, 2H), 7.87 (s, 1H), 7.78 (dd, J= 7.8 Hz, 2.2 Hz,
1H), 7.58
(d, J= 5.2 Hz, 2H), 7.39 (t, J= 8.8 Hz, 1H), 6.98 (d, J= 8.4 Hz, 1H), 4.08 (s,
2H), 3.73
(s, 3H), 2.588 (s, 3H) ppm. LCMS = 100.0 % purity (APCI+). MS(APCI+)=
424.1(M+45), 380.0 (M+1).
[00665] Synthesis of 5-(3'-Acetyl-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridine-
2-carboxylic acid amide (P-328). A solution of 1-228 (50.0 mg, 0.184 mmol) in
toluene
(2 mL) was stirred under nitrogen. To this reaction was added thionyl chloride
(65.7 mg,
0.552 mmol) and the reaction was heated to 100 C for 2 h over which time it
turned
from colorless to a deep red. The solvent and excess thionyl chloride was
removed under
reduced pressure and the residue was dissolved in tetrahydrofuran (2 mL). To
the
solution was then added aqueous ammonium hydroxide (40 uL). The reaction was
stirred
at room temperature for 1 h. The reaction was diluted with ethyl acetate (30
mL) and
washed with saturated aqueous sodium bicarbonate (30 mL). The aqueous wash was
extracted with ethyl acetate (30 mL) and the organic extracts were combined.
The
combined extracts were washed with saturated aqueous sodium bicarbonate (25
mL),
dried over sodium sulfate, filtered, and the solvent removed under vacuum. The
residue
was purified by preparatory silica gel TLC (eluting with 12.5 % acetone in
dichlormethane, 3 developments) to give P-328 (12.6 mg, 18% yield) as a white
powder.
1H NMR.(400 MHz,CDC13) 8.47 (d, J= 2.0 Hz, 1H), 8.13 (d, J= 8.0 Hz, 1H), 7.98-
7.94 (m, 2H), 7.78 (s, 1H), 7.69 (dd, J= 8.0 Hz, 2.0 Hz, 1H), 7.96- 7.58 (m,
1H), 7.52 (t,
321
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
J= 7.6 Hz, 1H), 7.14 (t, J= 8.6 Hz, 1H), 6.76 (d, J= 8.4 Hz, 1H), 5.48 (s,
1H), 4.05 (s,
3H), 3.77 (s, 3H), 2.62 (s, 3H) ppm. LCMS = 95.9% purity. MS(APCI+) = 379.1
(M+1).
Example 252. Preparation of P-324
NH2 NHz
O, NBS Br O,
CH3CN
O o O'N`O
1-229
[00666] Synthesis of 2-Bromo-6-methoxy-4-nitro-phenylamine (1-229). Into a 2 L
round bottom flask with a stir bar was added 2-methoxy-4-nitro-phenylamine
(50.0 g,
297.4 mmol), CH3CN (1 L), and NBS (53.5 g, 297.4 mmol). The reaction was
stirred at
room temperature for 2 hours while protected from light. The reaction was
concentrated
and then 500 mL water was added. The product was extracted with ethyl acetate
and
concentrated. The solid which precipitated from the aqueous washes was
combined with
the solid which resulted from the organic concentration to give 1-229 (59.6 g)
which was
used without further purification.
NH2
Br ~ O, H20 Br\^/O,
EtOHV/
O' O NaNOZ O'N'O
1-229 1-230
[00667] Synthesis of 1-Bromo-3-methoxy-5-nitro-benzene (1-230). Into a 2 L
round-
bottomed flask equipped with a mechanical stirrer was added 1-229 (59.8, 249.2
mmol),
ethanol (167 mL), water (83 mL), and the mixture was cooled to 0 C. H2SO4
(750 mL)
was added followed by NaNO2 (25.8 g, 373.8 mmol) in 75 mL water. The reaction
was
stirred at 0 C for 15 minutes, room temperature for 30 minutes, and 60 C for
15
minutes, after which it was cooled to room temperature and filtered. The
solids were
washed with water, dried in a 40 C vacuum oven for 1 hour, then a vacuum
dessicator
for 5 days. After drying for an additional 6 hours in a 60 C vacuum oven,
47.3 g (84%)
of 1-230 was obtained as a rust-colored solid.
322
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
O
Br OH B I OH
bis(pinacolato) O'
diboron
OA'O PdCl2dppf-CH2CI2 O O
KOAc dioxane
1-231 1-232
[00668] Synthesis of 3 -Nitro -5 -(4,4,5,5 -tetramethyl- [ 1, 3,2]dioxaborolan-
2-yl)-phenol
(1-232). Into a 250 mL round bottom flask was placed 1-231 (2.03 g, 9.31
mmol),
bis(piniacolato)diboron (2.60 g, 10.24 mmol), KOAc (2.74 g, 27.93 mmol), and
50 mL
dioxane. After degassing with N2 for 10 minutes, PdCl2dppf-CH2C12 (0.38 g,
0.47
mmol) was added and the reaction was stirred at 90 C for 20 hours. Most of
the solvent
was removed by rotary evaporation and brine was added. The product was
extracted
with ethyl acetate and the organics were filtered through Celite and
concentrated. The
residue was purified by flash column chromatography eluting with 20% ethyl
acetate/hexanes to afford 1.44 g (58%) of 1-232.
Br OH
0B' O Pd(PPh3)4 I I \
O F 0 F
31
+ NaHCO3
AN
\ I O
CI HO N DME O
DOH, H2O CI
1-33 1-232 1-233
[00669] Synthesis of 3 -(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3 -ylmethyl)-5 -
nitro-
phenol (1-233). Into a 40 mL vial with a stir bar was added 1-33 (597 mg, 1.81
mmol), I-
232 (480 mg, 1.81 mmol), NaHCO3 (456 mg, 5.43 mmol), DME (10 mL), ethanol (1
mL), and water (1 mL). After degassing for 10 minutes,
tetrakis(triphenylphosphine)palladium(0) (208 mg, 0.18 mmol) was added and the
reaction was stirred at 65 C for 18 hours. The reaction was cooled to room
temperature
and diluted with 10 mL water. The product was extracted with ethyl acetate,
dried over
Na2SO4, and concentrated. Flash column chromatography purification of the
residue
with 15 - 20% ethyl acetate/hexanes afforded 175 mg (25%) of 1-233 as a yellow
oil.
323
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
OH
\ I / Fe, NH4CI \ I / OH
O ,F O ,F
O;O DOH, H2O NH2
CI CI
1-233 P-324
[00670] Synthesis of 3-Amino -5-(3'-chloro-2-fluoro-6-methoxy-biphenyl-3-
ylmethyl)-phenol (P-324). Into a 40 mL vial was added 1-233 (175 mg, 0.39
mmol), Fe
powder (89 mg, 1.35 mmol), NH4C1(120 mg, 1.93 mmol), water (1 mL), and ethanol
(3
mL). The suspension was stirred at 80 C for 2 hours, then filtered through
Celite. The
filtrate was diluted with ethyl acetate and washed with water and brine. After
concentrating the organics, the residue was purified by flash column
chromatography
eluting with 20 - 50% ethyl acetate/hexanes to give 104 mg (65%) of P-324 as a
colorless oil. 1H NMR (400MHz, CDC13) 7.40 (s, 1 H), 7.38 - 7.27 (m, 3 H),
7.11 (t, J
=8.6Hz,1H),6.74-6.61(m,2H),6.12(d,J=17.4Hz,2H),6.04(t,J=1.9Hz,1H),
3.80 (s, 2 H), 3.75 (s, 3 H), 3.63 (br s, 2 H) ppm. LC/MS = 97.7%, 358.2
(APCI+).
Example 253. Preparation of P-331 and P-338
1.)Trifluoroacetic Acid ICIII
Dichloromethane i '
C ,, 0 2.)Hydrochloric Acid C ~P MHz O~ F N NHz
Diethyl Ether Y CI_ Potassium Cyanate
Acetic Acid
Water
0 0 0
1-224 P-331 P-338
[00671] Synthesis of [4-(3'-Acetyl-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
phenyl]-
methyl-amine hydrochloride(P-33 1). To a solution of 1-224 (485 mg, 1.05 mmol)
in
dichloromethane (2 mL) was added trifluroacetic acid (1 mL) dropwise and the
reaction
was allowed to stir at room temperature for 4 h. The solvent was removed under
vacuum,
and the residue was dissolved in ethyl acetate (20 mL) and water (10 mL). The
pH of the
aqueous layer was adjusted to 8 using solid sodium bicarbonate. The layers
were
separated and the organic solution was washed with brine. The organic extract
was dried
over sodium sulfate and the solvent was removed under vacuum. The residue was
dried
under high vacuum overnight to give the free base. A portion of the free base
(44.5 mg)
was dissolved in diethyl ether (2 mL). To the solution was added 2 N hydrogen
chloride
in diethyl ether (184 uL, 0.367 mmol) dropwise, and the reaction was stirred
at room
324
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
temperature for 3 h. The resultant solid was filtered and dried under vacuum
in an
abderhaulden apparatus under acetone at reflux to give P-331 (28.8 mg). 1H NMR
(400
MHz, DMSO-d6) 7.94 (m, 1H), 7.86 (s, 1H), 7.58 (d, J= 5.2 Hz, 1H), 7.26 (t, J=
8.8
Hz, 1H), 7.09 (d, J= 8.4 Hz, 2H), 6.93 (d, J= 9.2 Hz, 1H), 6.77 (br m, 2H),
3.84 (s,
2H), 3.71 (s, 3H), 2.72 (s, 3H), 2.59 (s, 3H) ppm.
LCMS = 94.4 % purity. MS(APCI+) = 364.1 (M+1).
[00672] Synthesis of 1-[4-(3'-Acetyl-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
phenyl]-1-methyl-urea (P-338). A suspension of P-331 free base (180 mg, 0.495
mmol)
in glacial acetic acid (1 mL) and water (1 mL) was stirred until a solution
formed. To the
solution was added a solution of potassium cyanate (120 mg, 1.49 mmol) in
water (250
uL), and the solution became cloudy. The reaction was stirred at room
temperature for
2.5 h. The reaction was diluted with diethyl ether (10 mL) and water (5 mL)
and the
layers separated. The organic extract was washed with saturated aqueous sodium
bicarbonate (10 mL), dried over sodium sulfate, filtered, and the solvent
removed under
vacuum. The residue was dissolved in diethyl ether (30 mL), washed with
saturated
aqueous sodium bicarbonate (30 mL) and brine (30 mL), dried over sodium
sulfate,
filtered, and the solvent was removed under vacuum. The residue was purified
by
preparatory silica TLC (10 % acetone in dichloromethane, 2 developments), and
dried
overnight in a vacuum oven at 40 C to give P-338 (96.1 mg, 48% yield) as a
white
powder. 1H NMR (400 MHz CDC13) d: 8.00 (d, J = 1.2 Hz, 1H), 7.95 (dt, J = 7.7
Hz,
1.4 Hz, 1H). 7.61 (dd, J = 7.6 Hz, 1.6 Hz, 1H), 7.52 (t, J = 7.6 Hz, 1H), 7.28
(d, J = 8.4
Hz, 2H), 7.22-7.14 (m, 3H), 6.75 (d, J = 8.4 Hz, 1H), 4.34 (brs, 2H), 3.98 (s,
2H), 3.76
(s, 3H), 3.24 (s, 3H), 2.62 (s, 3H). LCMS = 96.1 % purity. MS(APCI+)= 407.1
(M+1).
325
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00673] Scheme 57.
0II
O 0 N
0 Allylpalldiumchloride dimer
O F B Bis(diphenylphosphino)pentane O F NHZ
+ 0- N
potassium carbonate
NHZ dimethylformate
4
O
1-223 P-277
N O N 0
Chloroethylisocyanate Sodium Carbonate U
0 F NNH ~O F N
L ~N H
P-277 Chloroform H Acetonitrile
Cl
O O
1-237 P-361
Example 254. Preparation of P-277
[00674] Synthesis of 1-[3'-(6-Amino-pyridin-3-ylmethyl)-2'-fluoro-6'-methoxy-
biphenyl-3-yl]-ethanone (P-277). A solution of 1-223 (1.00 g, 3.01 mmol) and 2-
aminopyridine-5-boronic acid pinacol ester (728 mg, 3.31 mmol) in N,N-
dimethylformamide (8 mL) was degassed using a nitrogen stream for 10 min. To
the
solution was added potassium carbonate (1.25 g, 9.03 mmol), allylpalladium(II)
chloride
dimer (165 mg, 0.451 mmol), and bis(diphenylphosphino)pentane (398 mg, 0.903
mmol)
under nitrogen and the suspension was stirred at 65 C under nitrogen for 15 h.
To the
reaction was added ethyl acetate (50 mL) and water (50 mL) and the biphasic
suspension
was filtered through celite (-15 g). The celite was washed with ethyl acetate
(2 x 20 mL),
and water (2 x 20 mL) and the filtrate was separated. The aqueous layer was
extracted
with ethyl acetate (100 mL) and the organic extracts were combined. The
organic
solution was washed with water (200 mL) and brine (200 mL), dried over sodium
sulfate, filtered, and the solvent removed under vacuum. The residue was
purified by
flash silica gel column chromatography (impurities eluted with 50% ethyl
acetate in
hexanes, product eluted with 12.5 % acetone in dichloromethane) to give P-277
(653 mg,
62% yield) as an orange oil. 1H NMR (400 MHz, CDC13) 7.99 (m, 1H), 7.95 (dt, J
=
7.73 Hz, 1.6 Hz, 1H), 7.61-7.59 (m, 1H), 7.52 (t, J = 7.6 Hz, 1H), 7.30 (dd, J
= 8.2 Hz,
326
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
2.2 Hz, 1H), 7.09 (t, J = 8.6 Hz, 1H), 6.71 (dd, J = 8.4 Hz, 0.8 Hz, 1H), 6.46
(d, J = 8.8
Hz, 1H), 4.33 (s, 2H), 3.83 (s, 2H), 3.75 (s, 3H), 2.62 (s, 3H). LCMS = 100.0%
purity.
MS(APCI+) = 351.1 (M+1).
Example 255. Preparation of P-361
[00675] Synthesis of 1-[5-(3'-Acetyl-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-2-yl]-3-(2-chloro-ethyl)-urea (1-237). A solution of P-277 (300 mg,
0.856 mmol)
in chloroform (7.4 mL) was purged with nitrogen for 5 min at room temperature.
To this
solution was added 2-chloroethylisocyanate (90.3 mg, 0.856 mmol), and the
reaction was
stirred at reflux for 24 h. Additional 2-chloroethylisocyanate (220 uL, 2.57
mmol) was
added and the reaction was heated at reflux for an additional 16 h. The
solvent was
removed under vacuum and the resultant red syrup was purified by flash silica
gel
column chromatography (0-25% acetone in dichoromethane) followed by
preparatory
thin layer chromatography to give I-237 (41.7 mg, 11% yield) as a yellow oil.
[00676] Synthesis of 1-[5-(3'-Acetyl-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-2-yl]-imidazolidin-2-one (P-361). A suspension of 1-237 (40.0 mg,
0.0877
mmol) and sodium carbonate (27.9 mg, 0.263 mmol) in acetonitrile (1 mL) was
stirred at
reflux overnight. The reaction was cooled to room temperature and filtered.
The solvent
was removed under vacuum and the resultant yellow oil was crystallized in
diethyl ether
(2 mL), filtered, washed with hexanes (2 x 1 mL) and dried in a vacuum oven
overnight
at 40 C to give P-361 (17.1 mg, 46% yield) as an off white powder. 1H NMR (400
MHz
,CDC13) 8.10 (d, J= 1.6 Hz, 1 H), 7.99 (s, 1 H), 7.95 (d, J= 7.8 Hz, 1 H),
7.63 - 7.48
(m,3H),7.46-7.39(m,1H),7.14-7.05(m,1H),6.97(d, J= 8.3 Hz,1H),6.72(d,J=
8.5 Hz, 1 H), 4.42 (t, J= 7.9 Hz, 2 H), 3.89 (s, 2 H), 3.82 (t, J= 7.9 Hz, 2
H), 3.75 (s, 3
H), 2.62 (s, 3 H) ppm. LCMS = 91.4% purity. MS(APCI+) = 420.1 (M+1).
327
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 256. Preparation of P-355
Br t3 PdClZdppf-CHZCIZ t3
+ OB O _ I O
N O KOAc DMSO N N
1-238
[00677] Synthesis of 5-(4,4,5,5-Tetramethyl-[1,3,2] dioxaborolan-2-yl)-
pyridine-2-
carbonitrile (I-238). Into a 250 mL round-bottomed flask was added 3.0 g of 5-
bromo-
pyridine-2-carbonitrile (3.0 g, 16.39 mmol), bis(piniacolato)diboron (4.58 g,
18.03
mmol), KOAc (5.47 g, 55.74 mmol), and DMSO (100 mL). After degassing for 20
minutes, PdClzdppf-CH2C12 (1.39 g, 1.64 mmol) was added and the solution was
stirred
for 24 hours at 80 C, and then at room temperature for 3 days. 50 mL water
was added
and the product was extracted with ethyl acetate. The combined organics were
washed
with brine, dried over Na2S04 and concentrated. The dark-colored residue was
purified
by flash column chromatography eluting with 20% acetone/hexanes to give a red
solid.
The solid was triturated with hexane to give 1.72 g (46%) of 1-238 as a light-
pink solid.
Br 0 i
+ B_0 ::::;
~ N
CI 1-3
3 1-238 P-355
[00678] Synthesis of [5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-
2-ylmethyl]-carbamic acid tert-butyl ester (P-355). Into a 20 mL vial was
added I-33
(401 mg, 1.22 mmol), I-238 (336 mg, 1.46 mmol), K2CO3 (504 mg, 3.65 mmol), DME
(5 mL), water (0.5 mL), ethanol (0.5 mL), and the suspension was degasses for
15
minutes. Tetrakis(triphenylphosphine)palladium(0) (141 mg, 0.12 mmol) was
added and
the reaction stirred at 80 C for 16 hours. The reaction was diluted with
water and
extracted with ethyl acetate. The organics were concentrated and purified by
flash
column chromatography eluting with 15 - 20% ethyl acetate/hexanes to afford P-
355 (64
mg, 15%) as a light-yellow oil. 1H NMR (400 MHz,CDC13) 8 8.62 (br s, 1 H),
7.68 -
7.56 (m, 2 H), 7.40 - 7.30 (m, 3 H), 7.26 - 7.21
(m,1H),7.13(t,J=8.4Hz,1H),6.75
328
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
(d, J= 8.3 Hz, 1 H), 4.03 (br s, 2 H), 3.78 (s, 3 H) ppm. LC/MS = 98.5%, 353.0
(APCI+).
Example 257. Preparation of P-344
Pd/C, HCI, McOH
O F N N "O F N NH2
H2
CI CI
P-355 P-344
[00679] Synthesis of C-[5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-2-yl]-methylamine (P-344). Into a 100 mL round bottom flask was added
P-355
(0.55 g, 1.56 mmol), methanol (20 mL), concentrated HC1(0.65 mL, 7.79 mmol),
and
10% Pd/C (100 mg). The suspension was stirred under a H2 balloon for 18 hours,
then
filtered through Celite. The filtrate was concentrated. To the solid was added
IN
aqueous NaOH and the product was extracted with dichloromethane. The
dichloromethane was concentrated and purified by flash column chromatography
eluting
with 5-10% methanol/dichloromethane to give P-344 (89 mg, 16%) as a colorless
oil.
'H NMR (400MHz, DMSO-d6) 8.55 (d, J= 1.2 Hz, 1 H), 8.32 (br s, 3 H), 7.71 (dd,
J=
1.9,7.9Hz,1H),7.50-7.37(m,3H),7.36(d,J=5.4Hz,2H),7.27(d,J=6.6Hz,1
H),6.96(d,J=8.6Hz,1H),4.75(brs,2H),4.14(q,J=5.8Hz,2H),4.01(s,2H),
3.73 (s, 3 H) ppm. LC/MS = 95.1%, 357.1 (APCI+).
Example 258. Preparation of P-367
I \ I \ EtOCOCI
NH 2 H
O F N - "O F I N- NYO
CH2CI2, TEA O
CI
CI
P-344 P-367
[00680] Synthesis of [5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-
2-ylmethyl]-carbamic acid ethyl ester (P-367). Into an 8 mL vial was added P-
344 (43
329
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
mg, 0.12 mmol), dichloromethane (2 mL), triethylamine (33 uL, 0.24 mmol). The
solution was cooled to 0 C and ethyl chloroformate (20 mg, 0.18 mmol) was
added.
After 15 minutes at room temperature the solution was concentrated. The
residue was
purified by flash column chromatography eluting with 40 - 60% ethyl acetate to
give 23
mg (54%) of P-367 as a white solid. 1H NMR (400MHz, DMSO-d6) 8.39 (d, J= 1.5
Hz, 1 H), 7.63 (t, J= 6.0 Hz, 1 H), 7.59 (dd, J= 1.7, 8.1 Hz, 1 H), 7.50 -
7.40 (m, 2 H),
7.37 (s, 1 H), 7.33 (t, J= 8.7 Hz, 1 H), 7.28 (d, J= 6.6 Hz, 1 H), 7.20 (d, J=
7.9 Hz, 1
H),6.94(d,J=8.6Hz,1H),4.22(d,J=6.2Hz,2H),4.03- 3.96 (m, 2 H), 3.94 (s, 2
H), 3.72 (s, 3 H), 1.16 (t, J= 7.1 Hz, 3 H) ppm. LGMS = 100.0%, 429.1 (APCI+).
Example 259. Preparation of P-368
\ I TMSNCO, DCM
O / F N NHZ3 O fF NN
YNH2
O
\ I CI P-344 P-368
[00681] Synthesis of [5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-
2-ylmethyl]-urea (P-368). Into an 8 mL vial was added P-344 (43 mg, 0.12
mmol),
dichloromethane (2 mL), and trimethylsilyl isocyanate (49 uL, 0.36 mmol).
After 1 hour
at 35 C, the solution was concentrated. The resulting solid was triturated
with ether to
afford P-368 (25 mg, 52%) as a white solid. 1H NMR (400 MHz, DMSO-d6) 8.44 (s,
1
H),7.68(d,J=7.8Hz,1H),7.48-7.40(m,2H),7.37(s,1 H), 7.34 (t, J= 8.9 Hz,1H),
7.28 (d, J= 7.6 Hz, 2 H), 6.94 (d, J= 8.6 Hz, 1 H), 6.51 (br s, 1 H), 5.64 (br
s, 2 H), 4.25
(d, J= 5.0 Hz, 2 H), 3.97 (s, 2 H), 3.72 (s, 3 H). LGMS = 96.4%, 400.1
(APCI+).
[00682] Example 260. Preparation of P-371
O
O i F N NHZ + DCM, TEA I H
CI N O F N NYN,
HCI o
'I
CI
CI
P-344 P-371
330
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00683] Synthesis of 3-[5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-2-ylmethyl]-1,1-dimethyl-urea hydrochloride (P-371). Into an 8 mL vial
was
added P-344 (23 mg, 0.064 mmol), dichloromethane (1.5 mL), triethylamine (18
uL,
0.13mmol) and the solution was cooled to 0 C. Dimethylcarbamyl chloride (9
uL,
0.097) was added and the reaction stirred for 18 hours at room temperature.
The
dichloromethane solution was washed with water and brine, and then
concentrated. The
residue was purified by flash column chromatography eluting with 5%
methanol/dichloromethane. The colorless oil obtained was triturated with ether
to obtain
a white solid. The resulting solid was dissolved in 2 mL of 4.0 M HC1/dioxane
and
stirred for 2 hours at room temperature. The oil was treated with ether to
form a solid,
which was filtered to obtain P-371 (6.1 mg, 20%). 1H NMR (400 MHz, DMSO-d6)
8.60 (s, 1 H), 8.08 (d, J= 7.4 Hz, 1 H), 7.62 (d, J= 8.1 Hz, 1 H), 7.50 - 7.35
(m, 4 H),
7.28 (d, J= 6.4 Hz, 1 H), 7.11 (br s, 1 H), 6.97 (d, J= 8.6 Hz, 1 H), 4.41 (br
s, 2 H), 4.08
(s, 2 H), 3.73 (s, 3 H), 2.82 (s, 6 H) ppm. LC/MS = 100.0%, 428.1 (APCI+).
Example 261. Preparation of P-372
I NH CH2CI2
O F N 2 I\ I N
N Y NH
~O F
ethylisocyanate O
ci I HCI
ci
P-344 P-372
[00684] Synthesis of 1-[5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-2-ylmethyl]-3-ethyl-urea (P-372). Into an 8 mL vial was added P-344
(23 mg,
0.064 mmol), dichloromethane (1.5 mL), and the solution was cooled to room
temperature. Ethyl isocyanate (8 uL, 0.097 mmol) was added and the reaction
was
stirred for 18 hours at room temperature and then concentrated. To the
resulting solid
was added 4.0 M HC1/dioxane and the solution was stirred for 18 hours at room
temperature and then concentrated. The oil which was obtained was treated with
ether to
form a solid, which was filtered to afford P-372 (15.1 mg, 50%) as a white
solid. 1H
NMR (400 MHz, DMSO-d6) 8.63 (s, 1 H), 8.14 (d, J= 8.3 Hz, 1 H), 7.63 (d, J=
8.2
Hz, 1 H), 7.51 - 7.33 (m, 4 H), 7.28 (d, J= 6.3 Hz, 1 H), 6.98 (d, J= 8.6 Hz,
1 H), 6.62
(br s, 1 H), 6.31 (br s, 1 H), 4.42 (s, 2 H), 4.10 (s, 2 H), 3.74 (s, 3 H),
3.01 (q, J= 7.1 Hz,
331
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
2 H), 0.99 (t, J= 7.1 Hz, 3 H) ppm. MS: 428.1 (APCI+).
Example 262. Preparation of P-373
O
CI N~ N'~
O / F N NH2 + N TEA H J
HCI O F N_ NYN
\ CH2CI2 O
CI
CI
P-344 P-373
[00685] Synthesis of 4-Methyl-piperazine-l-carboxylic acid [5-(3'-chloro-2-
fluoro-6-
methoxy-biphenyl-3-ylmethyl)-pyridin-2-ylmethyl]-amide (P-373). Into an 8 mL
vial
was added P-344 (23 mg, 0.064 mmol), dichloromethane (1.5 mL), triethylamine
(18 uL,
0.13 mmol). The solution was cooled to 0 C and 4-methyl-piperazine-l-carbonyl
chloride (10 mg, 0.097 mmol) was added. After 18 hours at room temperature the
reaction was washed with water, followed by brine. The organics were
concentrated and
purified by flash column chromatography eluting with 5 - 10%
methanol/dichloromethane to afford 28 mg of semi-solid. The semi-solid was
dissolved
in 2 mL of 4.OM HC1/dioxane and stirred at room temperature for 18 hours, and
then
concentrated. Ether was added and a solid formed, which was filtered. To the
solid was
added 5N aqueous NaOH and the product was extracted with dichloromethane and
then
concentrated. Purification of the residue by flash column chromatography
eluting with
10% methanol/dichloromethane afforded P-373 (9.6 mg, 31%) as a colorless oil.
1H
NMR (400 MHz, DMSO-d6) 8.38 (d, J= 1.5 Hz, 1 H), 7.57 (dd, J= 2.0, 8.1 Hz, 1
H),
7.48-7.39(m,2H),7.37(s,1H),7.32(t,J=8.7 Hz,1H),7.28(d,J=6.4Hz,1H),
7.17(d,J=8.1Hz,1H),7.09(t,J=5.8Hz,1H),6.94(d,J=8.5 Hz,1H),4.26(d,J=
5.6 Hz, 2 H), 3.93 (s, 2 H), 3.72 (s, 3 H), 3.17 (d, J= 5.2 Hz, 4 H), 2.29 -
2.20 (m, 4 H),
2.16 (s, 3 H) ppm. LC/MS = 100.0%, 483.1 (APCI+).
332
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 263. Preparation of P-374
NH CH2CI2 H rCI
O fF N z O I F I N. NYNH
chloroethylisocyanate o
CI
P-344 1-239
[00686] Synthesis of 1-(2-Chloro-ethyl)-3-[5-(3'-chloro-2-fluoro-6-methoxy-
biphenyl-
3-ylmethyl)-pyridin-2-ylmethyl]-urea (I-239). Into an 8 mL vial was added P-
344 (63
mg, 0.177 mmol), dichloromethane (2 mL), and the reaction was cooled to 0 C.
Chloroethyl isocyanate (19 mg, 0.177 mmol) was added and the reaction was
stirred at
room temperature for 1 hour and then concentrated to yield I-239 which was
used as is.
\ I% I N H NCI NaH, THE OI/ F N N
O F 0 O
O NH
I
CI GS144 \ CI GS145
1-239 P-374
[00687] Synthesis of 1-[5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-2-ylmethyl]-imidazolidin-2-one (P-374). Into an 8 mL vial was added 1-
239 (82
mg, 0.177 mmol), THE (2 mL), and the suspension was cooled to 0 C. Sodium
hydride
(8 mg, 0.212 mmol) was added and the reaction was stirred at room temperature
for 3
days. An additional 4 mg of sodium hydride was added and the reaction was
stirred at
50 C for 1 hour. Water was added and the product was extracted with ethyl
acetate.
The organics were concentrated and purified by flash column chromatography
eluting
with 25 - 50% acetone/dichloromethane to give P-374 (35 mg, 47%, 2 steps) as a
white
solid. 1H NMR (400 MHz, DMSO-d6) 8.42 (d, J= 1.5 Hz, 1 H), 7.60 (dd, J= 1.9,
7.9
Hz,1H),7.49-7.39(m,2H),7.37(s,1H),7.34(t, J= 8.7 Hz,1H),7.28(d,J=6.7
Hz, 1 H), 7.19 (d, J= 7.9 Hz, 1 H), 6.94 (d, J= 8.6 Hz, 1 H), 6.41 (s, 1 H),
4.28 (s, 2 H),
3.95 (s, 2 H), 3.72 (s, 3 H), 3.31 - 3.19 (m, 4 H) ppm. LC/MS = 100.0%, 426.1
(APCI+).
333
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 264. Preparation of P-375
\ I I N N NH NaH, Mel, THE \ I / N-
O F O O F N_ N
11
O
CI I CI
P-374 P-375
[00688] Synthesis of 1-[5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-2-ylmethyl]-3-methyl-imidazolidin-2-one (P-375). Into an 8 mL vial was
added
P-374 (21 mg, 0.05 mmol), THE (1 mL) and the solution was cooled to 0 C.
Sodium
hydride (3 mg, 0.076 mmol) was added and after 15 minutes at room temperature,
methyl iodide (6 uL, 0.101 mmoL) was added. The reaction was stirred for 30
minutes
at room temperature and then 1 mL of water was added. The THE was evaporated
and
the product was extracted with ethyl acetate and concentrated. The residue was
passed
through a silica gel plug eluting with methanol, which afforded P-375 (18.1
mg, 82%) as
a colorless oil. 1H NMR (400 MHz, DMSO-d6) 8.42 (d, J= 1.6 Hz, 1 H), 7.60 (dd,
J=
2.0,7.9Hz,1H),7.50-7.39(m,2H),7.37(s,1H),7.34(t,J=8.7Hz,1H),7.28(d,J
= 6.4 Hz, 1 H), 7.19 (d, J= 8.1 Hz, 1 H), 6.94 (d, J= 8.5 Hz, 1 H), 4.31 (s, 2
H), 3.95 (s,
2 H), 3.72 (s, 3 H), 3.24 (s, 4 H), 2.67 (s, 3 H) ppm. LC/MS = 97.8%, 440.1
(APCI+).
Example 265. Preparation of P-520
I I NH 2 TEA H
YO~
O / F N z \O fF I NN
\ I CH2CI2 O CI
P-344 P-520
[00689] Synthesis of [5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-
2-ylmethyl]-carbamic acid phenyl ester (P-520). Into an 18 mL vial was added P-
344
(128 mg, 0.36 mmol), TEA (0.10 mL, 0.72 mmol), and dichloromethane (3 mL). The
solution was cooled to 0 C and phenylchloroformate (68 uL, 0.54 mmol) was
added.
After stirring at room temperature for 15 minutes the reaction was
concentrated.
Purification by flash column chromatography (25% - 75% EtOAc/hexanes) afforded
P-
334
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
520 (102 mg, 59%) as an off-white solid. (400MHz, DMSO-d6) 8.44 (s, 1 H), 8.30
(t, J
= 5.9 Hz, 1 H), 7.63 (dd, J= 1.5, 7.9 Hz, 1 H), 7.49 - 7.26 (m, 8 H), 7.24 -
7.17 (m, 1 H),
7.12 (d, J= 7.9 Hz, 2 H), 6.95 (d, J= 8.6 Hz, 1H),4.33(d,J=6.0Hz,2H),3.96(s,2
H), 3.73 (s, 3 H) ppm. LC/MS = 100.0%, 477.1 (APCI+).
Example 266. Preparation of P-460
H
O fF NN O O o I, F I NNON,
CIH o
1 . McNHZ (aq), I
DMSO CI
2. HCI
P-520 P-460
[00690] 1-[5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-pyridin-2-
ylmethyl]-3-methyl-urea hydrochloride (P-460). Into an 8 mL vial was added P-
520 (84
mg, 0.18 mmol), DMSO (2 mL), and 40% aq. MeNH2 (0.14 mL). After stirring for
30
minutes at room temperature, 5 mL of water was added. The product was
extracted with
EtOAc (4 x 2 mL) and the organics were concentrated. The white solid was
triturated
with ether to afford the free base compound. To the free base was added 4N
HC1/dioxane (2 mL) and after stirring for 5 minutes at room temperature the
solution was
concentrated. The residue was triturated with ether to provide P-460 (42 mg,
53%) as a
tan solid. 1H NMR (400 MHz, DMSO-d6) 8.64 (s, 1 H), 8.16 (d, J= 7.9 Hz, 1 H),
7.66
(d,J=8.2Hz,1H),7.51-7.34(m,4H),7.28(d,J=6.4Hz,1H),6.98(d,J=8.6Hz,1
H), 6.80 (br s, 1 H), 6.30 (br s, 1 H), 4.44 (s, 2 H), 4.10 (s, 2 H), 3.74 (s,
3 H), 2.55 (s, 3
H) ppm. LC/MS = 97.5%, 414.0 (APCI+).
Example 267. Preparation of P-461
O FIN ~NH_NaH, Eti, THE \ I I N N-/
O 30 O F N
CIo
H
cI a
P-374 P-461
335
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00691] 1-[5 -(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3 -ylmethyl)-pyridin-2-
ylmethyl]-3 -ethyl-imidazolidin-2 -one hydrochloride (P-461). Into an 18 mL
vial was
added P-374 (65 mg, 0.16 mmol), THE (3 mL), and the solution was cooled to 0
C. NaH
(9 mg, 0.23 mmol) was added and the suspension was stirred at room temperature
for 15
minutes. EtI (25 uL, 0.31 mmol) was added and the reaction was stirred for 1
hour at
room temperature. Water was added and the product was extracted with EtOAc (3
x 3
mL). The organics were washed with water, brine, and concentrated. To the
residue was
added 2 mL of 4N HC1/dioxane. After stirring to dissolve, the solution was
concentrated. The resulting residue was triturated with ether, filtered,
washed with ether,
and dried to give P-461 (51 mg, 65%) as a tan solid. 1H NMR (400 MHz, DMSO-d6)
8.63 (br s, 2 H), 8.07 (br s, 1 H), 7.60 (d, J= 7.9 Hz, 1 H), 7.50 - 7.33 (m,
4 H), 7.29 (d,
J= 5.8 Hz, 1 H), 6.97 (d, J= 8.3 Hz, 1 H), 4.51 (s, 2 H), 4.09 (br s, 2 H),
3.74 (s, 3 H),
3.57 (s, 2 H), 3.31 (s, 2 H), 3.19 - 3. 10 (m, 2 H), 1. 10 - 0.97 (m, 3 H)
ppm. LC/MS=
84.2%, 454.2 (APCI+).
Example 268. Preparation of P-462
H CH2CI2 H
O F N NYNH O F N: NYNH
O mCPBA O O
CI CI
P-372 P-462
[00692] 1-[5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-1-oxy-pyridin-
2-
ylmethyl]-3-ethyl-urea (P-462). Into an 18 mL vial was added P-372 (66 mg,
0.15
mmol), dichloromethane (4 mL), and the solution was cooled to 0 C. mCPBA (69
mg,
0.31 mmol) was added and the reaction was stirred at room temperature for 1
hour after
which 5 mL of saturated aqueous NaHCO3 was added. The layers were separated
and
the organic layer was washed sequentially with 5 mL each of saturated aqueous
NaHCO3, H20, and brine. The residue was then washed with IN NaOH (2 x 5 mL),
water (5 mL), and brine (5 mL). The product was dried over Na2SO4, filtered
and
concentrated to obtain P-462 (14.8 mg, 22%) as a white solid. 1H NMR (400 MHz,
DMSO-d6) 8.19 (s,1H),7.53-7.34 (m,4H),7.29(d,J=6.6Hz,1H),7.25-7.16 (m,
2H),6.96(d,J=8.6Hz,1H),6.38(t,J=6.0Hz,1 H), 6.17 (t, J= 5.4 Hz,1H),4.22
336
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
(d, J= 6.2 Hz, 2 H), 3.91 (s, 2 H), 3.73 (s, 3 H), 3.09 - 2.91 (m, 2 H), 0.97
(t, J= 7.2 Hz,
3 H) ppm. LGMS = 100.0%, 444.1 (APCI+).
Example 269. Preparation of P-463
N NH CHzCIz \ NH
O i F N O F N+ N
O mCPBA 0- O
CI CI
P-374 P-463
[00693] 1-[5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-1-oxy-pyridin-
2-
ylmethyl]-imidazolidin-2-one (P-463). Into an 18 mL vial was added P-374 (62
mg,
0.15 mmol), dichloromethane (4 mL), and the solution was cooled to 0 C. mCPBA
(82
mg, 0.36 mmol) was added and the reaction was stirred at room temperature for
1 hour
after which 5 mL of aqueous IN NaOH was added. The layers were separated and
the
aqueous layer was extracted with dichloromethane (2 x 3 mL) . The organics
were
combined and washed with water (5 mL) and brine (5 mL) and then concentrated.
The
residue was taken up in 5 mL of EtOAc and it was washed with 5 mL of brine,
dried
over Na2SO4, and concentrated to a solid. The residue was triturated with
ether to afford
P-463 (23.5 mg, 35%) as a white solid. 'H NMR (400 MHz, DMSO-d6) 8.23 (s, 1
H),
7.49 - 7.34 (m, 4 H), 7.32 - 7.16 (m, 3 H), 6.96 (d, J= 8.6 Hz, 1 H), 6.56 (s,
1 H), 4.31 (s,
2 H), 3.93 (s, 2 H), 3.73 (s, 3 H), 3.45 - 3.36 (m, 2 H), 3.32 - 3.25 (m, 2 H)
ppm. LGMS
= 93.4%, 442.0 (APCI+).
Example 270. Preparation of P-465
NH O CHzCIz, TEA H
0 F N z "0 F N N~O~'CI lj~ + Cl 0
CI 0
CI CI
P-344 1-240
[00694] [5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-pyridin-2-
ylmethyl]-
carbamic acid 2-chloro-ethyl ester (1-240). Into an 18 mL vial was added P-344
(100
mg, 0.28 mmol), dichloromethane (4 mL), TEA (78 uL, 0.56 mmol), and after the
337
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
solution was cooled to 0 C 2-Chloroethyl chloroformate (43 uL, 0.42 mmol) was
added.
The reaction was stirred at room temperature for 18 hours and then 5 mL of
water was
added. The layers were separated and the aqueous was extracted with 5 mL more
dichloromethane. The organics were combined, washed with 5 mL of water and 5
mL of
brine, dried over Na2SO4, and then concentrated. The residue which was
obtained was
used as is in the next reaction.
H (CI NaH, THE fF O
F N_ NYO O N N
O CH
CI 1-240 P-465
[00695] 3-[5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-pyridin-2-
ylmethyl]-oxazolidin-2-one hydrochloride (P-465). Into an 18 mL vial was added
1-240
(0.28 mmol), THE (3 mL), and NaH (28 mg, 0.70 mmol). The reaction was stirred
at
room temperature for 18 hours, 50 C for 4 hours, and then room temperature
for 3 days.
To the reaction was added 5 mL of water and the product was extracted with
EtOAc.
The organics were washed with brine and then dried over Na2SO4. The residue
was
purified by flash column chromatography eluting with 20% - 50%
acetone/hexanes. The
free base which was obtained was dissolved inl mL of 4N HC1/dioxane and then
concentrated. Compound P-465 was obtained as a tan solid (24.1 mg, 19% for 2
steps).
1H NMR (400 MHz, DMSO-d6) 8.52 (s, 1 H), 7.77 (br. s., 1 H), 7.49 - 7.32 (m, 6
H),
7.27 (br s, 1 H), 6.96 (d, J= 8.6 Hz, 1 H), 4.47 (s, 2 H), 4.35 - 4.24 (m, 2
H), 4.01 (s, 2
H), 3.73 (s, 3 H), 3.57 - 3.50 (m, 2 H) ppm. LC/MS = 100.0%, 427.1 (APCI+).
Example 271. Preparation of P-521
l I NH O CH2CI2, TEA I\ I H o
O F N + C I O O F N NOS
O
O
CI I CI
P-344 1-241
[00696] N-[5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-pyridin-2-
338
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
ylmethyl]-oxalamic acid ethyl ester (I-241). Into an 18 mL vial was added P-
344 (152
mg, 0.43 mmol), TEA (0.11 mL, 0.85 mmol), and dichloromethane (4 mL). The
solution
was cooled to 0 C and ethyl chlorooxoacetate (71 uL, 0.64 mmoL) was added.
After 20
minutes at room temperature the reaction was washed with brine and the
organics were
concentrated. The semi-solid was triturated with 1:1 ether:EtOAc, filtered,
and washed
with EtOAc to provide I-241 (69 mg, 35%) as a gray-blue solid. 'H NMR (400
MHz,
DMSO-d6) 9.38 (t, J= 6.0 Hz, 1 H), 8.41 (s, 1 H), 7.59 (dd, J= 4.0, 8.0 Hz, 2
H), 7.48
- 7.2 5 (m, 8 H), 7.22 (d, J = 8. 1 Hz, 2 H), 6.94 (d, J = 8.6 Hz, 2 H), 4.3 9
(d, J = 6. 0 Hz, 2
H), 4.32 - 4.18 (m, 2 H), 3.94 (s, 2 H), 3.72 (s, 3 H), 1.27 (t, J= 7.1 Hz, 3
H) ppm.
LC/MS = 97.9%, 457.0 (APCI+).
O
N O
O F N N O~ NH3MeOH N
O F N YKNH2
O / O
CI I CI
1-241 P-521
[00697] N-[5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-pyridin-2-
ylmethyl]-oxalamide (P-521). Into an 8 mL vial was added 1-241 (14.8 mg, 0.032
mmol)
and 2 mL of 7N NH3/MeOH. After stirring for 1 hour at room temperature, the
solution
was concentrated to afford P-521 (11.9 mg, 87%) as a tan solid. 'H NMR (400
MHz,
DMSO-d6) 9.15 (t, 1 H), 8.41 (d, J= 1.5 Hz, 1 H), 8.08 (br s, 1 H), 7.82 (br
s, 1 H), 7.59
(dd,J=1.9,8.1Hz,1H),7.48-7.25(m,5H),7.19(d,J=8.1Hz,1H),6.94(d,J=8.6
Hz, 1 H), 4.39 (d, J= 6.2 Hz, 2 H), 3.94 (s, 2 H), 3.72 (s, 3 H) ppm. LC/MS =
98.9%,
428.0 (APCI+).
Example 272. Preparation of 1-145.
O
OH OO
O F Chloromethylformate
O F
Pyridine
Tetrahydrofuran
CI CI
1-32 1-145
[00698] Synthesis of Carbonic acid 3'-chloro-2-fluoro-6-methoxy-biphenyl-3-
339
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
ylmethyl ester methyl ester (1-145). A solution of (3'-Chloro-2-fluoro-6-
methoxy-
biphenyl-3-yl)-methanol (1-32, 3.00g, 11.3 mmol) in pyridine (2.31 g, 29.3
mmol) and
tetrahydrofuran (40 mL) was cooled to 0 C in an ice water bath. The reaction
vessel was
purged with nitrogen and methyl chloroformate (2.34 g, 24.8 mmol) was added.
The ice
bath was removed and the reaction was stirred at room temperature overnight.
The white
suspension was adjusted to pH 2 by addition of 1 N aqueous hydrochloric acid (-
25 mL)
and the yellow biphasic solution was diluted with dichloromethane (200 mL) and
water
(150 mL). The layers were separated, and the aqueous layer was extracted with
dichloromethane (2 x 100 mL). The dichloromethane extracts were combined,
washed
with water (2 x 200 mL) and brine (200 mL), dried over sodium sulfate,
filtered, and the
solvent removed under vacuum to give I-145 (3.84 g, quantitative yield).
[00699] )H NMR (400 MHz, CDC13) 7.41-7.32 (m, 4H), 7.29-7.26 (m, 1H), 6.76
(dd, J= 8.40 Hz, 0.80 Hz, 1H), 5.204 (s, 2H), 3.800 (s, 3H), 3.795 (s, 3H)
ppm.
[00700] LCMS = 98.2% purity. MS(APCI+) = 249.0 (M-78), MS(APCI-) = 249.0 (M-
78).
Example 273. Preparation of P-376
HO 9 OH o_B
Br c 2 IOII
~N 1 -0 -N NBS/CCIa 0 KN H 0 N NxNH,
0 Pd(OAc), (Ph3P)4Pd/KIP04
Br K2CO31 PPh3 DME
CI CI EtOH-H20 CI
1-243 1-244 P-376
[00701] Synthesis of 2-(3-chloro-phenyl)-3-methoxy-6-methyl-pyridine (1-243):
To 2-
bromo-3-methoxy-6-methylpyridine (0.2 g, 1.0 mmol), 3-chlorophenylboronic acid
(1)
(0.19 g, 1.2 mmol), PPh3 (0.13 g, 0.5 mmol), K2CO3 (0.06 g, 0.4 mmol) and
Pd(OAc)2
(0.03 g, 0.12 mmol) was added dioxane (3 mL), and EtOH-H20 (1:1, 1.5 mL). Ar
gas
was bubbled through the stirred reaction for 5 min. The reaction was stirred
at 180 C for
15 m using microwave oven (Biotage Intiator II). The reaction was cooled to
room
temperature, concentrated, and H2O and dichloromethane (40 mL each) were
added. The
340
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
organic layer was separated and the aqueous layer was extracted with
dichloromethane (2
x 25 mL). The combined organic extracts were dried with Na2SO4, filtered, and
concentrated. The residue was purified by silica gel column chromatography
using 1:1
dichloromethane-hexanes then dichloromethane to afford 0.19 g (81%) of 1-243
as a
viscous liquid.
[00702] Synthesis of 6-bromomethyl-2-(3-chloro-phenyl)-3-methoxy-pyridine (I-
244). To 1-243 (1.02 g, 4.36 mmol) and NBS (0.78 g, 4.36 mmol) in CC14 (20 mL)
was
added benzoylperoxide (0.03 g, 0.12 mmol). The reaction was stirred at 80 C
under N2
for 20 h. The reaction was cooled to room temperature and concentrated. The
residue
was dissolved in mixture of dichloromethane and hexanes (1:1, 8 mL ) and
purified by
silica gel column chromatography using 1:1 dichloromethane-hexanes to afford
0.83 g
(61%) of 1-244 as a viscous liquid.
[00703] Synthesis of {4-[6-(3-chloro-phenyl)-5-methoxy-pyridin-2-ylmethyl] -
phenyl}-urea (P-376). To 1-244 (0.31 g, 1.0 mmol), [4-(4,4,5,5-Tetramethyl-
[ 1,3,2] dioxaborolan-2-yl)-phenyl]-urea (0.39 g, 1.5 mmol), (PPh3)4Pd (0. 12
g, 0.1
mmol) and K3PO4 (0.42 g, 2.0 mmol) was added DME (8 mL), and EtOH-H20 (1:1, 4
mL). Ar gas was bubbled through the stirred reaction for 5 min. The reaction
was stirred
at 160 C for 20 m using microwave oven (Biotage Intiator II). The reaction
was cooled
to room temperature, concentrated and H2O and dichloromethane (50 mL each)
were
added. The organic layer was separated and the aqueous layer was extracted
with
dichloromethane (2 x 25 mL). The combined organic extracts were dried with
Na2SO4,
filtered, and concentrated. The residue was purified by silica gel column
chromatography using 3-5% methanol in dichloromethane than triturated with 1:1
ethyl
acetate in hexanes to afford 0.023 g (6%) of P-376 as off-white solid. 1H NMR
(DMSO-
d6, 400 MHz): 8.42 (s, 1 H), 7.84-7.93 (m, 2 H), 7.4-7.54 (m, 3 H), 7.3 (d, J=
8.4 Hz, 2
H), 7.22 (d, J= 8.4 Hz, 1 H), 7.14 (d, J= 8.0 Hz, 2 H), 5.77 (s, 2 H), 3.99
(s, 2 H), 3.83
(s, 3 H) ppm; MS(APCI+): 368.0 (M+1), LC-MS: 92.5%.
341
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 274. Preparation of P-379
o
o_B
Br 0 "' N
O g '-IN NH O iN I NH
z
/ (Ph3P)4Pd/K2P04
DME
CI EtOH-H20 CI
1-244 P-379
[00704] Synthesis of 5-[6-(3-chloro-phenyl)-5-methoxy-pyridin-2-ylmethyl]-
pyridin-
2-ylamine (P-379) To 1-244 (0.1 g, 0.32 mmol), 5-(4,4,5,5-tetramethyl
[1,3,2]dioxaborolan-2-yl)-pyridin-2-ylamine (0.08 g, 0.38 mmol),
(PPh3)4Pd(0.04 g,
0.03 mmol) and K3PO4 (0.14 g, 0.64 mmol) was added DME (3 mL), and EtOH-H20
(1:1, 1.54 mL). Ar gas was bubbled through the stirred reaction for 5 min. The
reaction
was stirred at 150 C for 20 m using microwave oven (Biotage Intiator II). The
reaction
was cooled to room temperature, concentrated. The residue was purified by prep
TLC
using 70% ethyl acetate in hexanes to afford 0.06 g (55%) of P-379 as a
viscous liquid.
1H NMR (CDC13, 400 MHz): 8.03 (d, J= 1.6 Hz, 1 H), 7.93-4 -7.96 (m, 1 H), 7.82-
7.86 (m, 1 H), 7.34-7.58 (m, 3 H), 7.2 (d, J= 8.4 Hz, 1 H), 7.02 (d, J= 8.8
Hz, 1 H),
6.47 (dd, J= 8.4, 0.8 Hz, 1 H), 4.35 (s, 2 H), 4.01 (s, 2 H), 3.84 (s, 3 H)
ppm;
MS(APCI+): 326.1 (M+1), LC-MS: 100%.
Example 275. Preparation of P-386
N 1) Ethylisocyanate I I N
N Pyridine 0 IN N Ilt, N'-"',
O NHz H H
/ CIH
2) 2 M HCI-Ether \ I
CI CI
P-379 P-386
[00705] Synthesis of 1-{5-[6-(3-chloro-phenyl)-5-methoxy-pyridin-2-ylmethyl]-
pyridin-2-yl}-3-ethyl-urea hydrochloride (P-386). To P-379 (0.05 g, 0.15 mmol)
in
pyridine (1.5 mL) was added ethylisocyanate (0.033 g, 0.46 mmol). The reaction
was
stirred at room temperature for 20 h. Water and ethyl acetate (20 mL each)
were added.
The organic layer was separated and the aqueous layer was extracted with ethyl
acetate
342
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
(2 x 10 mL). The combined organic extracts were washed with water (2 x 30 mL),
brine
(20 mL), dried with Na2SO4, filtered, and concentrated. The residue was
dissolved in
ether (2 mL), then 2M HC1 in ether (0.5 ml) was added, stirred for 1 h. The
ether layer
was decanted, triturated with ether (2 x 2 mL), dried to afford 0.045 g (68%)
of P-386 as
light yellow solid. 'H NMR (DMSO-d6, 400 MHz): 10.03 (br s, 1 H), 7.84-7.9 (m,
3 H),
7.76 (br s, 1 H), 7.58 (d, J= 8.8 Hz, 1 H), 7.44 -7.5 (m, 2 H), 7.34 (d, J=
8.4 Hz, 1 H),
7.30 (d, J= 8.8 Hz, 1 H), 4.08 (s, 2 H), 3.85 (s, 3 H), 3.15-3.23 (m, 2 H),
1.08 (t, J-
7.2 Hz, 3 H) ppm; MS(APCI+): 397.1 (M+1), LC-MS: 99%, HPLC 97.9% pure.
Example 276. Preparation of P-099
I \ O
O O AICl3
acyl chloride
O OH F
nitrobenzene
NO2 1-81 NO2
1-246
[00706] Synthesis of (4-Fluoro-phenyl)-(2-hydroxy-6-methoxy-3'-nitro-biphenyl-
3-
yl)-methanone (1-246). In an 8 mL vial equipped with a stir bar was placed
nitrobenzene (1.0 mL) and A1C13 (92.7 mg, 0.695 mmol). After stirring for 5
minutes, 4-
fluorobenzoyl chloride (83.2 L, 0.695 mmol) was added and the mixture was
allowed to
stir for 1 hour at room temperature. Then, 1-81 (150 mg, 0.579 mmol) was added
and the
reaction mixture was stirred at room temperature for 19 hours. The reaction
mixture as
quenched with water (25 mL) and extracted with EtOAc (2 x 30 mL). The
extractions
were combined, washed with brine (30 mL), dried (MgS04) and concentrated to a
yellow
solid. The crude material was triturated with Et20 (5 mL) and the solid was
collected by
suction filtration. After the solid was washed with Et20, 64.5 mg of 1-246 was
isolated
as a light yellow solid in 30% yield. MS(APCI-): 366.0 (M-1)
O
\ ~ \ O OH F
O OH F
NO 2
NO2
1-246 P-099
343
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00707] Synthesis of 3-(4-Fluoro-benzyl)-6-methoxy-3'-nitro-biphenyl-2-ol (P-
099)
In an 8 mL vial equipped with a stir bar was placed 1-246 (60 mg, 0.163 mmol)
and
triethylsilane (350 L). The mixture was cooled in an ice-water bath and then
TFA (350
L) was added. The reaction mixture was warmed to room temperature and reacted
for
17 hours. After this time period, additional triethylsilane (1.1 mL) and TFA
(1.1 mL)
was introduced and the reaction mixture was heated to 60 C in an oil bath for
24 hours.
The reaction mixture was concentrated by a stream of N2, quenched with water
(20 mL)
and extracted with dichloromethane (2 x 30 mL). The organic portions were
combined,
washed with brine (30 mL), dried (MgS04) and concentrated. The residue was
purified
by Si02 column chromatography utilizing 15% EtOAc/hexanes as the eluent to
produce
18.9 mg of P-099 as a viscous, tan oil in 33% yield. 1H NMR (400 MHz, CDC13) 6
3.72
(s, 3 H), 3.94 (s, 2 H), 4.73 (s, 1 H), 6.55 (d, J= 8 Hz, 1 H), 6.96-7.00 (m,
2 H), 7.01 (d,
J= 8 Hz, 1 H), 7.19-7.22 (m, 2 H), 7.63 (t, J= 7 Hz, 1 H), 7.69 (dt, J= 8, 2
Hz, 1 H),
8.22-8.26 (m, 2 H) ppm. MS(APCI-): 352.1 (M-1); LC-MS: 98%.
Example 277. Preparation of P-137
I \ O
o : 0 AICI3 gOH
acy
l chloride
O N
nitrobenzene
NO2
1-81 NOZ
1-247
[00708] Synthesis of 4-(2-Hydroxy-6-methoxy-3'-nitro-biphenyl-3-carbonyl)-
benzonitrile (1-247). In an 8 mL vial equipped with a stir bar was placed
nitrobenzene
(2.0 mL) and A1C13 (515 mg, 3.86 mmol). After stirring for 5 minutes, 4-
cyanobenzoyl
chloride (83.2 L, 0.695 mmol) was added and the mixture was allowed to stir
for 30
minutes at room temperature. Then, 1-81 (200 mg, 0.771 mmol) was added and the
reaction mixture was stirred at 60 C for 17 hours. The reaction was quenched
with 1M
HC1(4 mL), water was added (20 mL) and then extracted with dichloromethane (2
x 30
mL). The organic portions were combined, washed with brine (30 mL), dried
(MgS04)
and concentrated. The material began to solidify which was increased by the
addition of
hexanes (4 mL). The solid was collected by suction filtration, washed with
hexanes (3 x
344
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
1 mL) to produce 147 mg of 1-247 as a light yellow solid in 51% yield. MS(APCI-
):
373.1 (M-1);
LC-MS: 91%.
0 OH
0 OH 0 OH N / ~\N
N02 N02
1-247 P-137
[00709] Synthesis of 4-[Hydroxy-(2-hydroxy-6-methoxy-3'-nitro-biphenyl-3-yl)-
methyl]-benzonitrile (P-137). In an 8 mL vial equipped with a stir bar was
placed 1-247
(20 mg, 0.0534 mmol), absolute EtOH (300 L), anhydrous THE (400 L) followed
by
NaBH4 (40.5 mg, 1.07 mmol). The reaction mixture was stirred at room
temperature for
17 hours. The reaction was quenched with water (20 mL) and extracted with
dichloromethane (2 x 30 mL). The organic portions were combined, washed with
brine
(30 mL), dried (MgSO4) and concentrated to produce 12.0 mg of P-137 as an off-
white
solid in 63% yield. 'H NMR (400 MHz, CDC13) 6 3.01 (d, J= 3 Hz, 1 H), 3.74 (s,
3 H),
6.08 (m, 1 H), 6.55 (d, J= 8 Hz, 1 H), 6.99 (d, J= 9 Hz, 1 H), 7.13 (s, 1 H),
7.54 (d, J=
8 Hz, 2 H), 7.59 (t, J= 8 Hz, 1 H), 7.67 (d, J= 8 Hz, 2 H), 7.69-7.72 (m, 1
H), 8.19-8.22
(m, 1 H), 8.26-8.27 (m, 1 H) ppm. MS(APCI-): 375.1 (M-1).
Example 278. Preparation of P-138
0
0 OH \~N 0 OH
N
N02 N02
1-247 P-138
[00710] Synthesis of 4-(2-Hydroxy-6-methoxy-3'-nitro-biphenyl-3-ylmethyl)-
345
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
benzonitrile(P-138). In an 8 mL vial equipped with a stir bar was placed 1-247
(60 mg,
0.160 mmol) and triethylsilane (600 L). The reaction mixture was cooled in an
ice-
water bath and then TFA (600 L) was added. The mixture was heated to 70 C
for 23
hours. The reaction mixture was concentrated by a stream of N2 followed by the
addition of water (20 mL) and extraction with dichloromethane (2 x 30 mL). The
organic portions were combined, washed with brine (30 mL), dried (MgSO4) and
concentrated. The residue was purified by Si02 column chromatography utilizing
25%
EtOAc/hexanes as the eluent to produce 21.9 mg of P-138 as a pale yellow solid
in 38%
yield). 1H NMR (400 MHz, CDC13) 6 3.72 (s, 3 H), 4.02 (s, 2 H), 4.77 (s, 1 H),
6.56 (d,
J=8Hz,1H),7.11(d,J=8Hz,1H),7.35(d,J=9 Hz, 2 H), 7.57 (d, J= 8 Hz, 2 H),
7.64-7.70 (m, 2 H), 8.24-8.27 (m, 2 H) ppm. MS(APCI-): 359.1 (M-1), LC-MS:
>99%.
Example 279. Preparation of P-157
1 0
0 0 AICI3
acyl chloride
O OH N
nitrobenzene
N02
N02
1-81
1-248
[00711] Synthesis of (4-Dimethylamino-phenyl)-(2-hydroxy-6-methoxy-3'-nitro-
biphenyl-3-yl)-methanone (I-248). In an 8 mL vial equipped with a stir bar was
placed
nitrobenzene (2.5 mL) and A1C13 (773 mg, 5.80 mmol). After stirring for 5
minutes, 4-
dimethylamino-benzoyl chloride (533 mg, 2.90 mmol) was added and the mixture
was
allowed to stir for 30 minutes at room temperature. Then, 1-81 (300 mg, 1.16
mmol) was
added and the reaction mixture was stirred at 60 C for 23 hours. The reaction
was
quenched with 1M HC1(4 mL), water was added (20 mL) and then extracted with
dichloromethane (2 x 30 mL). The organic portions were combined, washed with
brine
(30 mL), dried (MgS04) and concentrated. The residue was purified by Si02
column
chromatography utilizing 10% EtOAc/hexanes as the eluent to produce 167 mg of
1-248
as a yellow solid in 37% yield.
346
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
O
i i 0 OH I i N
0 OH N
N02
N02
1-248 P-157
[00712] Synthesis of 3-(4-Dimethylamino-benzyl)-6-methoxy-3'-nitro-biphenyl-2-
ol
(P-157). In an 8 mL vial equipped with a stir bar was placed 1-248 (165 mg,
0.420
mmol) and triethylsilane (1.6 mL). The reaction mixture was cooled in an ice-
water bath
and then TFA (1.6 mL) was added. The mixture was heated to 65 C for 17 hours.
The
reaction mixture was concentrated by a stream of N2 followed by the addition
of water
(25 mL) and extraction with dichloromethane (2 x 30 mL). The organic portions
were
combined, washed with brine (30 mL), dried (MgS04) and concentrated. The
residue
was purified by Si02 column chromatography utilizing 50% EtOAc/hexanes as the
eluent to produce 52.4 mg of P-157 as a orange-red viscous oil in 33% yield.
1H NMR
(400 MHz, CDC13) 6 2.91 (s, 6 H), 3.73 (s, 3 H), 3.90 (s, 2 H), 4.86 (s, 1 H),
6.55 (d, J=
8Hz,1H),6.69(2,J=9Hz,2H),7.11-7.15(m,3H),7.57 (t, J=8Hz,1H),7.70(d,J
= 8 Hz, 1 H), 8.17-8.20 (m, 1 H), 8.25 (s, 1 H) ppm. MS(APCI+): 379.1 (M+1);
LC-MS:
>99%.
Example 280. Preparation of P-173
0
I
0 0 AIC13 I \
acyl chloride ~ N. "N
0 OH
nitrobenzene
NO 2
N02
1-81 1-249
[00713] Synthesis of (2-Hydroxy-6-methoxy-3'-nitro-biphenyl-3-yl)-(4-pyrazol-1-
yl-
phenyl)-methanone (1-249). In an 8 mL vial equipped with a stir bar was placed
nitrobenzene (2.4 mL) and A1C13 (515 mg, 3.86 mmol). After stirring for 5
minutes, 4-
pyrazol-1-yl-benzoyl chloride (318 mg, 1.54 mmol) was added and the mixture
was
347
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
allowed to stir for 45 minutes at room temperature. Then, 1-81 (250 mg, 0.964
mmol)
was added and the reaction mixture was stirred at 60 C for 17 hours. The
reaction was
quenched with 1M HC1(10 mL), water was added (15 mL) and then extracted with
dichloromethane (2 x 30 mL). The organic portions were combined, washed with
brine
(30 mL), dried (MgSO4) and concentrated. The residue was purified by Si02
column
chromatography utilizing 50% EtOAc/hexanes as the eluent to produce 257 mg of
1-249
as a yellow solid in 64% yield.
O
0 OH N N\ N,0 OH N. "N,
\I \I
N02 N02
1-249 P-173
[00714] Synthesis of 6-Methoxy-3'-nitro-3-(4-pyrazol-1-yl-benzyl)-biphenyl-2-
ol (P-
173). In an 8 mL vial equipped with a stir bar was placed 1-249 (150 mg, 0.361
mmol)
and triethylsilane (1.3 mL, 8.14). The reaction mixture was cooled in an ice-
water bath
and then TFA (1.3 mL, 17.5 mmol) was added. The mixture was heated to 70 C
for 16
hours. The reaction mixture was concentrated by a stream of N2 followed by the
addition of water (20 mL) and extraction with dichloromethane (2 x 30 mL). The
organic portions were combined, washed with brine (30 mL), dried (MgSO4) and
concentrated. The residue was purified by Si02 column chromatography utilizing
20%
EtOAc/hexanes as the eluent to produce 105 mg of P-173 as a yellow viscous oil
in 33%
yield. 1H NMR (400 MHz, CDC13) 6 3.73 (s, 3 H), 4.01 (s, 2 H), 4.79 (s, 1 H),
6.44-6.45
(m, 1 H), 6.56 (d, J= 8 Hz, 1 H), 7.13 (d, J= 8 Hz, 1 H), 7.33 (d, J= 8 Hz, 2
H), 7.60-
7.65 (m, 3 H), 7.69-7.71 (m, 2 H), 7.88 (d, J= 3 Hz, 1 H), 8.22-8.23 (m, 1 H),
8.24-8.26
(m, 1 H) ppm.
MS(APCI+): 402.1 (M+1); LC-MS: >99%.
348
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 281. Preparation of P-174
0
-N
0 - 0 AICI3 \
acyl chloride
N, OH
nitrobenzene
NO2
\
1-81 NO2
1-250
[00715] Synthesis of [4-(3,5-Dimethyl-isoxazol-4-yl)-phenyl]-(2-hydroxy-6-
methoxy-
3'-nitro-biphenyl-3-yl)-methanone (1-250) In an 8 mL vial equipped with a stir
bar was
placed nitrobenzene (2.4 mL) and A1C13 (515 mg, 3.86 mmol). After stirring for
5
minutes, 3,5-dimethyl-isoxazole-4-carbonyl chloride (246 mg, 1.54 mmol) was
added
and the mixture was allowed to stir for 45 minutes at room temperature. Then,
1-81 (250
mg, 0.964 mmol) was added and the reaction mixture was stirred at 60 C for 17
hours.
The reaction was quenched with 1M HCl (10 mL), water was added (15 mL) and
then
extracted with dichloromethane (2 x 30 mL). The organic portions were
combined,
washed with brine (30 mL), dried (MgS04) and concentrated. The residue was
purified
by Si02 column chromatography utilizing 50% EtOAc/hexanes as the eluent to
produce
194 mg of 1-250 as a light brown solid in 55% yield.
0
-N -N
\ 0 I \ O
O g OH O e OH
N02 N02
1-250 P-174
[00716] Synthesis of 3-[4-(3,5-Dimethyl-isoxazol-4-yl)-benzyl]-6-methoxy-3'-
nitro-
biphenyl-2-ol (P-174). In an 8 mL vial equipped with a stir bar was placed TFA
(114
L, 1.22 mmol) and anhydrous dichloromethane (450 L). The solution was cooled
to
about - 40 to -50 C in an acetone-dry ice bath. Then NaBH4 (46.2 mg, 1.22
mmol) was
added portion wise over 5 minutes. The reaction mixture was warmed to 0 C in
an ice-
water bath and then a solution of 1-250 (45 mg, 0.122 mmol) in anhydrous
dichloromethane (450 L) was added drop wise over 5 minutes. The reaction
mixture
349
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
was warmed to room temperature and reacted for 18 hours. The reaction mixture
was
slowly quenched with water (20 mL) and extracted with dichloromethane (2 x 30
mL).
The organic portions were combined, washed with brine (30 mL), dried (MgSO4)
and
concentrated. The residue was purified by Si02 column chromatography utilizing
30%
EtOAc/hexanes as the eluent to produce 16.9 mg of P-174 as a pale yellow
viscous oil in
39% yield.
[00717] 'H NMR (400 MHz, CDC13) 6 2.16 (s, 3 H), 2.32 (s, 3 H), 3.62 (s, 2 H),
3.71
(s, 3 H), 4.84 (s, 1 H), 6.51 (d, J= 7 Hz, 1 H), 6.95 (d, J= 7 Hz, 1 H), 7.66-
7.70 (m, 2
H), 8.24-8.28 (m, 2 H) ppm. MS(APCI+): 355.1 (M+1); LC-MS: >99%.
Example 282. Preparation of P-180
0 OH H
N 0 OH
N02 N02
P-138 P-180
[00718] Synthesis of 4-(2-Hydroxy-6-methoxy-3'-nitro-biphenyl-3-ylmethyl)-
benzaldehyde(P-180). In an 8 mL vial equipped with a stir bar was placed P-138
(105
mg, 0.291 mmol) and anhydrous THE (950 L). The solution was cooled in an ice-
water
bath for 10 minutes and then DIBAL-H (1.OM in hexanes, 1.46 mL, 1.46 mmol) was
added. The reaction mixture was warmed to room temperature and reacted for 17
hours.
The reaction was cooled in an ice-water bath, quenched slowly with 1 M HC1(4
mL)
followed by the addition of water (20 mL) and extraction with dichloromethane
(2 x 30
mL). The organic portions were combined, washed with brine (30 mL), dried
(MgS04)
and concentrated. The residue was purified by Si02 column chromatography
utilizing
25% EtOAc/hexanes as the eluent to produce 32.4 mg of P-180 as an off-white
solid in
31% yield. 1H NMR (400 MHz, CDC13) 6 3.72 (s, 3 H), 4.05 (s, 2 H), 4.76 (s, 1
H), 6.57
(d, J= 8 Hz, 1 H), 7.13 (d, J= 8 Hz, 1 H), 7.42 (d, J= 8 Hz, 2 H), 7.63-7.70
(m, 2 H),
7.81 (d, J= 8 Hz, 2 H), 8.24-8.26 (m, 2 H), 9.97 (s, 1 H) ppm. MS(APCI-):
362.1 (M-1);
LC-MS: 99%.
350
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 283. Preparation of P-183
0 I OH H 0 OH
0 OH
\ N02 N02
P-180 P-183
[00719] Synthesis of 3-(4-Hydroxymethyl-benzyl)-6-methoxy-3'-nitro-biphenyl-2-
ol(P-183). In an 8 mL vial equipped with a stir bar was placed P-180 (23 mg,
0.0630
mmol) and anhydrous MeOH (250 L). The mixture was cooled in an ice-water bath
and then NaBH4 (11.9 mg, 0.315 mmol) was added upon which the reaction mixture
became a solution. The reaction was stirred for 22 hours at room temperature,
quenched
with 1M aqueous HC1(4 mL) and water (20 mL) and extracted with dichloromethane
(2
x 30 mL). The organic portions were combined, washed with brine (30 mL), dried
(MgSO4) and concentrated. The residue was purified by Si02 column
chromatography
utilizing 50% EtOAc/hexanes as the eluent to produce 15.1 mg of P-183 as an
orange
viscous oil in 65% yield. 'H NMR (400 MHz, CDC13) 6 3.72 (s, 3 H), 3.98 (s, 2
H), 4.67
(s, 2 H), 4.74 (s, 1 H), 6.55 (d, J= 9 Hz, 1 H), 7.13 (d, J= 9 Hz, 1 H), 7.24-
7.32 (m, 4
H), 7.62 (t, J= 8 Hz, 1 H), 7.68-7.71 (m, 1 H), 8.21-8.25 (m, 2 H) ppm.
MS(APCI+):
348.1 (M-17); LC-MS: >99%.
Example 284. Preparation of P-190
O
O I i Oi AICI3
acyl chloride O \ H I ~'_N
H
nitrobenzene
NO2 I-81 NO2
1-251
[00720] Synthesis of N-[4-(2-Hydroxy-6-methoxy-3'-nitro-biphenyl-3-carbonyl)-
phenyl]-acetamide (I-251). In an 18 mL vial equipped with a stir bar was
placed
nitrobenzene (4.6 mL) and A1C13 (1.23 g, 9.24 mmol). After stirring for 5
minutes, 4-
acetylamino-benzoyl chloride (913 mg, 4.62 mmol) was added and the mixture was
allowed to stir for 30 minutes at room temperature. Then, 1-81 (600 mg, 2.31
mmol) was
351
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
added and the reaction mixture was stirred at 70 C for 21 hours. The reaction
was
quenched with 1M aqueous HC1(10 mL), water was added (100 mL) and then
extracted
with dichloromethane (2 x 50 mL). The organic portions were combined, washed
with
brine (200 mL), dried (MgSO4) and concentrated. The residue was triturated
with Et20
(35 mL), collected by suction filtration and washed with Et20 (5 x 2 mL) to
produce 333
mg of 1-251 as a pale orange solid in 35% yield. MS(APCI+): 407.0 (M+1)
O
O
~I I )~' \O OH N
O OH N
H
H 31.
\ I \ NOz
NO z
1-251 P-190
[00721] Synthesis of N-[4-(2-Hydroxy-6-methoxy-3'-nitro-biphenyl-3-ylmethyl)-
phenyl]-acetamide (P-190). In an 8 mL vial equipped with a stir bar was placed
1-251
(200 mg, 0.492 mmol) and triethylsilane (1.80 mL, 11.3). The reaction mixture
was
cooled in an ice-water bath and then TFA (1.80 mL, 24.6 mmol) was added. The
mixture was heated to 70 C for 18 hours. The reaction mixture was
concentrated by a
stream of N2 followed by the addition of water (30 mL) and extraction with
dichloromethane (2 x 30 mL). The organic portions were combined, washed with
brine
(30 mL), dried (MgS04) and concentrated. The crude material was purified by
Si02
column chromatography utilizing 5% 1M NH3 in MeOH/ dichloromethane as the
eluent
to produce 36.6 mg of P-190 as a light yellow solid in 19% yield. 'H NMR (400
MHz,
CDC13) 6 2.16 (s, 3 H), 3.72 (s, 3 H), 3.94 (s, 2 H), 4.74 (s, 1 H), 6.55 (d,
J= 9 Hz, 1 H),
7.09(bs,1H),7.7.11(d,J=8Hz,1H),7.20(d,J=8Hz,2H),7.42(d,J=8Hz,2H),
7.62 (t, J= 8 Hz, 1 H), 7.68-7.70 (m, 1 H), 8.21-8.25 (m, 2 H) ppm. MS(APCI+):
393.1
(M+1); LC-MS: 93%.
352
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00722] Scheme 58.
O NHz NHz
I
N. _ Pd(OAc)2
O NHgCI, Fe 0 JB-20 PPh3 0 i
I JB-22 -O Pdz(dba)3, BINAP
0 EtOH, H2O 1-252 K2CO3 N
JB-19 HO,B.OH EDtOH/HzO JB-23 0 Cs2CO31 toluene
JB-21 O
1-253
H 0
N H H
~ N a N ~
O i N 1. Fe, NHgCI
O EtOH, HZO 0 NHz O R
J B-24
2.41M HCI in H-Cl O R = NHSOZCH3
dioxane
O O
O
1-254 P-260 P-267
Example 285. Preparation of P-260
[00723] Synthesis of 3-Iodo-4-methoxy-phenylamine (I-252). In a 3-neck 250 mL
round-bottomed flask equipped with a stir bar, condenser and N2 lines was
placed iron
powder (3.50 g, 62.7 mmol), ammonium chloride (4.88 g, 91.3 mmol), ethanol (72
mL)
and water (23 mL). The mixture was heated to 85 C and then 2-iodo-l-methoxy-4-
nitro-benzene (5.0 g, 17.9 mmol) was added portion wise over a period of about
2
minutes. The mixture was allowed to stir at 85 C for 2 hours and then
filtered through
Celite. The Celite was washed with EtOH (100 mL) and the filtrate was
concentrated.
To the concentrated material was added water (100 mL) and ethyl acetate (150
mL). The
organic portion was removed and the aqueous portion was re-extracted with
ethyl acetate
(150 mL). The organic portions were combined, washed with brine (150 mL),
dried
(MgS04) and concentrated. The residue was purified by column chromatography
utilizing 50% EtOAc/hexanes as the eluent to produce 3.92 g of 1-252 as a
brown semi-
solid in 88% yield. MS(ESI+): 250.1 (M+1).
[00724] Synthesis of 1-(5'-Amino-2'-methoxy-biphenyl-3-yl)-ethanone (1-253).
In a
3-neck 100 mL round-bottomed flask equipped with a condenser, stir bar and N2
lines
was placed 1-252 (2.92 g, 11.7 mmol), 3-acetlylphenylboronic acid (2.11 g,
12.9 mmol),
353
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
potassium carbonate (4.85 g, 35.1 mmol), triphenylphosphine (921 mg, 3.51
mmol), 1,4-
dioxane (23 mL), 50% aqueous ethanol (23 mL) followed by palladium (II)
acetate (263
mg, 1.17 mmol). The mixture was heated to 90 C for 16 hours and then cooled
to room
temperature. The palladium catalyst was removed via filtration and to the
filtrate was
added 1M aqueous HC1(50 mL) and water (50 mL). The aqueous portion was
extracted
with ethyl acetate (2 x 75 mL), the organic portions were combined, washed
with brine
(75 mL), dried (MgSO4) and concentrated. The residue was purified by column
chromatography utilizing 50% EtOAc/hexanes as the eluent to produce 1.18 g of
1-253
as a pale orange oil in 42% yield. MS(APCI+): 242.0 (M+1).
[00725] Synthesis of 1-[2'-Methoxy-5'-(4-nitro-phenylamino)-biphenyl-3-yl]-
ethanone
(1-254). To a 40 mL vial equipped with a stir bar was placed 1-iodo-4-
nitrobenzene
(1.26 g, 5.07 mmol), cesium carbonate (2.20 g, 6.76 mmol), ( )-2,2'-
bis(diphenylphosphino)-1,1'-binaphthalene (316 mg, 0.507 mmol), and a solution
of I-
253 (816 mg, 3.38 mmol) in toluene (13.5 mL). The mixture was stirred for 10
minutes
and then tris(dibenzylideneacetone)dipalladium(0) (310 mg, 0.338 mmol) and the
mixture was heated to 110 C for 16 hours. The reaction was cooled to room
temperature and then filtered through Celite. The filtrate was treated with
water (40
mL), 1M HC1(40 mL) and then extracted with ethyl acetate (2 x 75 mL). The
organic
portions were combined, washed with brine (75 mL), dried (Mg504) and
concentrated.
The residue was purified by column chromatography utilizing 35% EtOAc/hexanes
as
the eluent to produce 277 mg of 1-254 as a dark orange solid in 23% yield.
[00726] Synthesis of 1-[5'-(4-Amino-phenylamino)-2'-methoxy-biphenyl-3-yl]-
ethanone hydrochloride(P-260). In an 18 mL vial equipped with a stir bar was
placed
iron powder (148 mg, 2.66 mmol), ammonium chloride (207 mg, 3.87 mmol),
absolute
EtOH (3.1 mL) and water (1.0 mL). The mixture was heated to 85 C and then 1-
254
(275 mg, 0.759 mmol) was added and the mixture was heated for 2 hours. The
reaction
was cooled to room temperature, filtered through Celite and extracted with
ethyl acetate
(2 x 40 mL). The organic portions were combined, washed with brine (40 mL),
dried
(Mg504) and concentrated. The residue was purified by column chromatography
utilizing 75% EtOAc/hexanes as the eluent to produce 207 mg of the free base
as a dark
orange oil in 82% yield. The free base was treated with 4.0 M HC1 in 1,4-
dioxane (1.0
354
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
mL) and stirred for 3 hours at room temperature. The reaction mixture was
treated with
diethyl ether (4 mL) and the solid was collected via suction filtration. After
washing the
solid with diethyl ether (3 x 2 mL), 20 mg of P-260 was isolated as a brown
solid in 44%
yield.
[00727] 'H NMR (400 MHz, DMSO-d6) 6 2.61 (s, 3 H), 3.75 (s, 3 H), 4.63 (br s,
1
H), 7.02-7.20 (m, 7H), 7.57 (t, J = 8.0 Hz, 1H), 7.75 (d, J = 8 Hz, 1H), 7.92
(d, J = 8 Hz,
1H), 8.02 (t, J = 2 Hz, 1 H), 9.42 (br s, 3H) ppm. MS(APCI-): 366.9 (M-2)
Example 286. Preparation of P-267
[00728] Synthesis of N-[4-(3'-Acetyl-6-methoxy- biphenyl-3-ylamino)-phenyl]-
methanesulfonamide (P-267). In an 8 mL vial equipped with a stir bar was
placed P-260
(free base) (60.0 mg, 0.180 mmol), anhydrous dichloromethane (600 L),
pyridine (14.6
L, 0.180 mmol), methanesulfonyl chloride (13.9 L, 0.180 mmol). The reaction
mixture was stirred at room temperature for 17 hours and then quenched with 1M
aqueous HC1 to pH 1-2. After adding water (20 mL), an extraction was performed
with
dichloromethane (2 x 30 mL), the organic portions were combined, washed with
brine
(30 mL), dried (MgSO4) and concentrated. The residue was purified by column
utilizing
10% acetone/dichloromethane as the eluent to produce 25 mg of P-267 as an off-
white
solid in 34% yield after drying in a high vacuum oven for 2 hours at 40 C.
[00729] 'H NMR (400 MHz, CDC13) 6 1.2.63 (s, 3 H), 2.96 (s, 3 H), 3.81 (s, 3
H),
6.08 (br s, 1 H), 6.91-6.98 (m, 3 H), 7.11-7.14 (m, 4 H), 7.51 (t, J= 8 Hz, 1
H), 7.72 (dt,
J= 8, 1 Hz, 1 H), 7.92 (d, J= 8 Hz, 1 H), 8.10 (t, J= 2 Hz, 1 H) ppm.
MS(APCI+):
411.1 (M+1)
LC-MS: 97%.
355
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 287. Preparation of P-261
O
9H
O OMe HO.B 0
a NHBoc N,IIII
O F O F Nx0
H
[Pd(n3-C3H5)CI]2
DPPPent
K2CO3, DMF
1-223 0 1-255
[00730] Synthesis of [4-(3'-Acetyl-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
phenyl]-
carbamic acid tert-butyl ester (1-255). In an 8 mL vial equipped with a stir
bar was
placed 1-223 (295 mg, 0.888 mmol), 4-[(tert-butoxycarbonyl)amino]-
phenylboronic acid
(232 mg, 0.977 mmol), potassium carbonate (270 mg, 1.95 mmol), 1,5-
bis(diphenylphosphino)pentane (39.1 mg, 0.0888 mmol), allylpalladium(II)
chloride
dimer (16.2 mg, 0.0444 mmol) and dimethylformamide (1.5 mL). The reaction
mixture
was heated to 80 C for 17 hours. In order to consume residual 1-223,
additional
allylpalladium(II) chloride dimer (32.5 mg, 0.0888 mmol) and 1,5-
bis(diphenylphosphino)pentane (78.2 mg, 0.178 mmol) were added and the
reaction
mixture was allowed to stir at 80 C for 17 hours. The reaction mixture was
filtered
through Celite and to the filtrate was added water (40 mL) and a saturated
ammonium
chloride solution (40 mL). After an extraction with ethyl acetate (2 x 50 mL),
the
organic portions were combined, washed with brine (50 mL), dried (MgS04) and
concentrated. The residue was purified by column chromatography utilizing 30%
EtOAc/hexanes as the eluent to produce 365 mg of I-255 as a pale yellow solid
in 91%
yield.
0
1. TFA
O F N 0 DCM NH
H O F 2
H-CI
2. 2M HCI in
Et20 11
1-255 P-261 0
356
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00731] Synthesis of 1-[3'-(4-Amino-benzyl)-2'-fluoro-6'-methoxy-biphenyl-3-
yl]-
ethanone; hydrochloride (P-261). In an 8 mL vial equipped with a stir bar was
placed I-
255 (265 mg, 0.590 mmol), dichloromethane (2.0 ml) and trifluoroacetic acid
(438 L,
5.90 mmol). The reaction mixture was stirred at room temperature for 4 hours
and then
quenched to pH 7 with a saturated sodium bicarbonate solution. After the
addition of
water (30 mL) and extraction with dichloromethane (2 x 30 mL), the organic
portions
were combined, washed with brine (30 mL), dried (MgS04) and concentrated. The
crude material was treated with diethyl ether (3 mL) and 2.0 M HC1 in diethyl
ether (1
mL) and allowed to stir at room temperature for 2 hours. The solid was
collected an
washed with diethyl ether (3 x 2 mL) to produce 139 mg of P-261 as a pale
orange
powder in 61% yield. 'H NMR (400 MHz, DMSO-d6) 62.59 (s, 3 H), 3.72 (s, 3 H),
3.96(s,2H),6.96(d,J=8Hz,1H),7.20-7.34 (m, 5 H), 7.57 (d, J= 5 Hz, 2 H), 7.85
(bs, 1 H), 7.94-7.97 (m, 1 H) ppm. MS(APCI+): 350.1 (M+1- HC1).
Example 288. Preparation of P-269
O F NH2 0 F H~NH2
H-CI
O O
P-261 P-269
[00732] Synthesis of [4-(3'-Acetyl-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
phenyl]-
urea (P-269). In an 8 mL vial equipped with a stir bar was placed P-261 (31
mg, 0.0803
mmol), water (400 L), acetic acid (200 L) and sodium cyanate (20.9 mg, 0.321
mmol).
The mixture was stirred at room temperature for 4 hours and then water (20 mL)
was
added followed by an extraction with dichloromethane (2 x 30 mL). The organic
portions were combined, washed with a saturated sodium bicarbonate solution
(30 mL)
and brine (30 mL), dried (MgS04) and concentrated. The residue was purified by
column chromatography utilizing 75% acetone/dichloromethane as the eluent to
produce
18 mg of P-269 as an off-white solid in 56% yield. "H NMR (400 MHz, CDC13) 6
2.62
(s, 3 H), 3.75 (s, 3 H), 3.95 (s, 2 H), 4.59 (br s, 2 H), 6.27 (br s, 1 H),
6.72 (d, J= 8 Hz, 1
357
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
H),7.11(t,J=9Hz,1H),7.21(s,4H),7.52(t,J=8Hz,1H),7.60(dd,J=9,1Hz,1
H), 7.95 (d, J= 8 Hz, 1 H), 7.99 (br s, 1 H) ppm. MS(APCI+): 393.1 (M+1); LC-
MS:
95%.
[00733] Scheme 59.
0 0
N O Pd(OAc)z N O _NH,
O J B-28 PPh3 O JB-30 NHgCI, Fe
EtOH, H20 JB-31 N+O Pd2(dba)3, BINAP
1 K2CO3 0
HO_BOH Di xane
EEtOH/H20 - Cl JB-32 O Cs2CO3, toluene
J B-29 C I
Cl 1-256 1-257
H H
N
,0 1. Fe, NHgCI N N 0 -Ct 0 0_ EtOH, H2O O NH2 NaOCN O H~NH2
JB-33 P-294
2. 2M HCI P-280 H-Cl AcOH, H2O Cl ~in Etz0 Cl
Cl
1-258
Example 289. Preparation of P-280
[00734] Synthesis of 3'-Chloro-2-methoxy-5-nitro-biphenyl (1-256). In a 3-neck
500
mL round bottomed flask equipped with a stir bar, condenser and nitrogen lines
was
placed was placed 2-iodo-l-methoxy-4-nitrobenzene (7.0 g, 25.1 mmol), 3-
chlorophenylboronic acid (4.32 g, 27.6 mmol), potassium carbonate(10.4 g, 75.3
mmol),
triphenylphosphine (1.98 g, 7.53 mmol), 1,4-dioxane (50 mL), 50% aqueous EtOH
followed by palladium acetate (564 mg, 2.51 mmol). The mixture was heated to
90 C in
an oil bath for 16 hours and then quenched with 1M aqueous HC1(80 mL). After
the
reaction mixture was filtered through Celite, water (100 mL) and 1M aqueous
HC1(100
mL) were added followed by extractions with ethyl acetate (3 x 150 mL). The
organic
portions were combined, washed with brine (250 mL), dried (MgS04) and
concentrated.
The residue was purified by column utilizing 20% EtOAc/Hexanes as the eluent
to
produce 4.26 g of 1-256 as an orange solid in 64% yield. MS(ESI+): 263.3 (M+).
358
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00735] Synthesis of 3'-Chloro-6-methoxy-biphenyl-3-ylamine (1-257). In a 3-
neck
250 mL round bottomed flask equipped with a stir bar, condenser and nitrogen
lines was
placed iron powder (3.17 g, 56.7 mmol), ethanol (66 mL) and water (21 mL). The
mixture was heated to 85 C in an oil bath and then 1-256 (4.26 g, 16.2 mmol)
was added
and the reaction was continued at 85 C for 2 hours. The reaction mixture was
cooled to
room temperature and filtered through Celite. To the filtrate was added water
(150 mL)
and extractions were performed with ethyl acetate (3 x 100 mL). The organic
portions
were combined, washed with brine (100 mL), dried (Mg504) and concentrated. The
crude material was purified by column utilizing 65% EtOAc/Hexanes as the
eluent to
produce 2.87 g of 1-257 as an orange viscous oil in 76% yield. MS(APCI+):
234.0
(M+1)
[00736] Synthesis of (3'-Chloro-6-methoxy-biphenyl-3-yl)-(4-nitro-phenyl)-
amine (I-
258). In a 3-neck 100 mL round-bottomed flask equipped with a stir bar,
condenser and
nitrogen lines was placed 1-257 (1.2 g, 5.13 mmol), 4-iodo-nitrobenzene (1.92
g, 7.70
mmol), ( )-2,2'-Bis(diphenylphosphino)-1,1'-binaphthalene (480 mg, 0.770
mmol),
cesium carbonate (3.34 g, 10.3 mmol), toluene (21 mL) followed by
tris(dibenzylideneacetone)dipalladium(0) (470 mg, 0.513 mmol). The mixture was
heated to 100 C in an oil bath for 17 hours and then cooled to room
temperature and
quenched with 1M aqueous HC1(50 mL). The reaction mixture was filtered through
Celite and to the filtrate was added water (50 mL) followed by extractions
with ethyl
acetate (2 x 75 mL). The organic portions were combined, washed with brine
(100 mL),
dried (Mg504) and concentrated. The residue was purified by column utilizing
25%
EtOAc/Hexanes as the eluent to produce 1.36 g of 1-258 as a red-orange solid
in 75%
yield.MS(APCI+): 355.1 (M+1)
[00737] Synthesis of N-(3'-Chloro-6-methoxy-biphenyl-3-yl)-benzene-1,4-
diamine;
hydrochloride(P-280). In a 40 mL vial equipped with a stir bar was placed iron
powder
(330 mg, 5.92 mmol), ethanol (6.9 mL) and water (2.2 mL). The mixture was
heated to
85 C in an oil bath and then I-258 (600 mg, 1.69 mmol) was added and the
reaction was
continued at 85 C for 2 hours. The reaction mixture was cooled to room
temperature
and filtered through Celite. To the filtrate was added water (50 mL) and
extractions were
performed with ethyl acetate (2 x 50 mL). The organic portions were combined,
washed
359
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
with brine (50 mL), dried (MgSO4) and concentrated. The residue was purified
by
column utilizing 75% EtOAc/Hexanes as the eluent to produce 480 mg of the free
base
of P-280. To the free base was added Et2O (4 mL) and 2.0 M HC1 in Et20 (2 mL)
and
the mixture was stirred at room temperature for 1 hour. The solid was
collected, washed
with Et20 (10 mL) and dichloromethane (8 mL) and then dried in a high vacuum
oven
set at 35 C for 2 hours to produce 355 mg of P-280 as a pale blue solid in
58% yield.
[00738] 'H NMR (400 MHz, DMSO-d6) 6 3.75 (s, 3 H), 7.02-7.21 (m, 7 H), 7.38-
7.45
(m, 3 H), 7.51-7.53 (m, 1 H), 9.97 (br s, 3 H) ppm. MS(ESI+): 326.4 [(M+1)-
HC1].
LC/MS: 92%.
Example 290. Preparation of P-294
[00739] Synthesis of [4-(3'-Chloro-6-methoxy-biphenyl-3-ylamino)-phenyl]-
urea(P-
294). In an 8 mL vial equipped with a stir bar was placed P-280 (150 mg, 0.415
mmol),
water (2.4 mL), acetic acid (1.2 mL) and sodium cyanate (108 mg, 1.66 mmol).
The
mixture was stirred at room temperature for 72 hours and then water (20 mL)
was added
followed by an extraction with dichloromethane (2 x 30 mL). The organic
portions were
combined, washed with brine (30 mL), dried (MgSO4) and concentrated. The
residue
was purified by column utilizing 40% acetone/dichloromethane as the eluent to
produce
46 mg of P-294 as a light purple solid in 30% yield. 'HNMR (400 MHz, DMSO-d6)
6
3.71 (s, 3 H), 5.67 (br s, 2 H), 6.90-6.92 (m, 3 H), 7.01 (s, 2 H), 7.23 (d,
J= 9 Hz, 2 H),
7.37-7.49 (m, 4 H), 7.68 (s, 1 H), 8.23 (br s, 1 H) ppm. MS(APCI+): 368.1
(M+1); LC-
MS: 95%.
Example 291. Preparation of P-281
1 0
ONH
[Pd(n3-C3H5)CI]2 N
ro OMe DPPPent
O. F K2CO3, DMF O F N NH2
H-Cl
2. 4M HCI in
i J 1,4-dioxane
0 Y
0
1-223 P-281
360
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00740] Synthesis of 1-[3'-(2-Amino-pyrimidin-5-ylmethyl)-2'-fluoro-6'-methoxy-
biphenyl-3-yl]-ethanone hydrochloride(P-281). In an 18 mL vial equipped with a
stir
bar was placed 1-223 (704 mg, 2.12 mmol), 5-(4,4,5,5-tetramethyl-
[1,3,2]dioxaborolan-
2-yl)-pyrimidin-2-ylamine (515 mg, 2.33 mmol), potassium carbonate (879 mg,
6.36
mmol), 1,5-bis(diphenylphosphino)pentane (280 mg, 0.636 mmol),
allylpalladium(II)
chloride dimer (116 mg, 0.318 mmol) and dimethylformamide (4.2 mL). The
reaction
mixture was heated to 70 C for 65 hours. The reaction mixture was filtered
through
Celite and to the filtrate were added water (40 mL) and a saturated ammonium
chloride
solution (40 mL). After an extraction with ethyl acetate (2 x 50 mL), the
organic
portions were combined, washed with brine (50 mL), dried (MgSO4) and
concentrated.
The residue was purified by column chromatography utilizing 20%
acetone/dichloromethane (gradient elution increased to 30%, then 40%
acetone/dichloromethane) as the eluent to produce 341 mg of P-281 an off-white
solid in
46% yield. Then P-281 (20 mg, 0.0569 mmol) was treated with 1,4-dioxane (1 mL)
and
the mixture was heated to form a solution. To this solution was added 4.OM HC1
in 1,4-
dioxane (1 mL) and the mixture was stirred at room temperature for 3 hours.
The solvent
was removed via nitrogen stream and the resulting solid was triturated with
diethyl ether
(1 mL), collected via suction filtration and washed with diethyl ether (3 x 1
mL) to
produce 12 mg of P-281 HC1 salt as a pale yellow solid in 55% yield. 1H NMR
(400
MHz, DMSO-d6) 6 2.59 (s, 3 H), 3.73 (s, 3 H), 3.83 (s, 2 H), 6.97 (d, J= 8 Hz,
1 H),
7.35 (t, J= 9 Hz, 1 H), 7.59 (d, J= 6 Hz, 2 H), 7.88 (bs, 1 H), 7.95-7.97 (m,
1 H), 8.36
(bs, 2 H). MS(APCI +): 352.1 [(M+1)-HC1]; LC-MS: 98%.
Example 292. Preparation of P-284
O
N O~. .
O TFN N H
O
P-284
[00741] Synthesis of N-[5-(3'-Acetyl-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyrimidin-2-yl]-methanesulfonamide(P-284). In an 8 mL vial equipped with a
stir bar
361
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
was placed P-281 (free base) (70.0 mg, 0.199 mmol), pyridine (800 L) and
methane
sulfonyl chloride (15.4 L, 0.199 mmol). The reaction mixture was stirred at
room
temperature for 2 hours and then heated to 50 C for 2 hours and then quenched
with
aqueous 1M HC1 to pH 1-2. After adding water (20 mL), an extraction was
performed
with ethyl acetate (2 x 30 mL), the organic portions were combined, washed
with brine
(30 mL), dried (MgSO4) and concentrated. The residue was purified by column
utilizing
20% acetone/dichloromethane as the eluent to produce 29 mg of P-284 as a light
yellow
solid in 34% yield after drying in a high vacuum oven for 2 hours at 35 C. 'H
NMR
(400 MHz, CDC13) 6 2.62 (s, 3 H), 3.44 (s, 3 H), 3.77 (s, 3 H), 3.91 (s, 2 H),
6.76 (d, J=
8 Hz, 1 H), 7.158 (t, J= 8 Hz, 1 H), 7.53 (t, J= 8 Hz, 1 H), 7.59 (dd, J= 8, 1
Hz, 1 H),
7.95 (t, J= 1 Hz, 1 H), 7.97 (m, 1 H), 8.51 (s, 2 H), 9.85 (br s, 1H) ppm.
MS(APCI+):
430.0 (M+1); LC-MS: 92%. HPLC: 97%.
Example 293. Preparation of P-315
O F N ), A,
H H
O
P-315
[00742] Synthesis of 1-[5-(3'-Acetyl-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyrimidin-2-yl]-3-ethyl-urea (P-315), In an 8 mL vial equipped with a stir bar
was
placed P-281 (free base) (80 mg, 0.228 mmol), pyridine (1.0 mL) and ethyl
isocyanate
(36.1 L, 0.456 mmol). The reaction mixture was stirred at room temperature
for 18
hours. TLC analysis indicated that the reaction mixture consisted of mostly
starting
materials. To the reaction mixture was added ethyl isocyanate (180 L, 2.28
mmol) and
the mixture was heated to 55 C for 65 hours. The reaction mixture was
quenched with
water (3 mL) and the resulting solid was collected, washed with water (3 x 2
mL), ethyl
acetate (3 x 1 mL) and dried in a high vacuum oven set at 40 C for 4 hours to
produce
59 mg of P-315 as a white solid in 61% yield. 1H NMR (400 MHz, DMSO-d6) 6 1.09
(t,
362
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
J=7Hz,3H),2.59(s,3H),3.18-3.26(m,2H),3.73(s,3H),3.90(s,2H),6.97(d,J=
9 Hz, 1 H), 7.36 (t, J= 9 Hz, 1 H), 7.57-7.59 (m, 2 H), 7.88 (s, 1 H), 7.94-
7.97 (m, 1 H),
8.46 (s, 2 H), 8.91 (t, J= 4 Hz, 1 H), 9.61 (s, 1 H) ppm. MS(APCI+): 423.1
(M+1); LC-
MS: 94%, HPLC: 93%.
Example 294. Preparation of P-325
N
O TFN-5 N
O
P-325
[00743] Synthesis of 1-[3'-(2-Dimethylamino-pyrimidin-5-ylmethyl)-2'-fluoro-6'-
methoxy-biphenyl-3-yl]-ethanone (P-325). In an 8 mL vial equipped with a stir
bar was
placed 1-223 (200 mg, 0.602 mmol), dimethyl-[5-(4,4,5,5-tetramethyl-
[1,3,2]dioxaborolan-2-yl)-pyrimidin-2-yl]-amine (165 mg, 0.662 mmol),
potassium
carbonate (250 mg, 1.81 mmol), 1,5-bis(diphenylphosphino)pentane (79.6 mg,
0.181
mmol), allylpalladium(II) chloride dimer (33.0 mg, 0.0903 mmol) and
dimethylformamide (1.2 mL). The reaction mixture was heated to 65 C for 18
hours.
The reaction mixture was filtered through Celite and to the filtrate were
added water (40
mL) and a saturated ammonium chloride solution (40 mL). After an extraction
with
ethyl acetate (2 x 50 mL), the organic portions were combined, washed with
brine (50
mL), dried (MgS04) and concentrated. The residue was purified by column
chromatography utilizing 50% ethyl acetate/hexanes as the eluent to produce 90
mg of P-
325 as a yellow solid in 39% yield. 'H NMR (400 MHz, DMSO-d6) 6 2.59 (s, 3 H),
3.07 (s, 6 H), 3.72 (s, 3 H), 3.77 (s, 2 H), 6.94 (d, J= 9 Hz, 1 H), 7.30 (t,
J= 9 Hz, 1 H),
7.58 (d, J= 5 Hz, 2 H), 7.87 (s, 1 H), 7.94-7.96 (m, 1 H), 8.24 (s, 2 H) ppm.
MS(APCI+): 380.1 (M+1); LC-MS: >99%.
363
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 295. Preparation of P-362
O F N~NNH z
H
O
P-362
[00744] Synthesis of [5-(3'-Acetyl-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyrimidin-2-yl]-urea (P-362). In an 8 mL vial equipped with a stir bar was
placed P-281
(free base) (30 mg, 0.0854 mmol), 1,4-dioxane (800 L) pyridine (34.5 L,
0.427 mmol)
and trimethylsilyl isocyanate (57.8 L, 0.427 mmol). The reaction mixture was
heated to
90 C for 18 hours and then water (15 mL) and 1M HC1(15 mL) were added
followed
by an extraction with dichloromethane (2 x 30 mL). The organic portions were
combined and dried (MgS04) and concentrated. To the residue was added hexanes
until
cloudy and the resulting solid was collected, washed with hexanes (3 x 1 mL)
and dried
in a high vacuum oven set at 40 C for 4 hours to produce 8 mg of P-362 as a
pale yellow
solid in 23% yield. 'H NMR (400 MHz, DMSO-d6) 6 2.59 (s, 3 H), 3.73 (s, 3 H),
3.80
(s, 2 H), 6.96 (d, J= 9 Hz, 1 H), 7.34 (t, J= 9 Hz, 1 H), 7.58-7.59 (m, 2H),
7.87 (s, 1 H),
7.94-7.97 (m, 1 H), 8.29 (s, 2 H) ppm. LC-MS: >99%.
Example 296. Preparation of P-366
O TFNNN~~CI
H H
O
P-366
[00745] Synthesis of 1-[5-(3'-Acetyl-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyrimidin-2-yl]-3-(2-chloro-ethyl)-urea (P-366). In an 8 mL vial equipped with
a stir bar
was placed P-281 (free base) (70 mg, 0.199 mmol), chloroform (1.5 mL) and
364
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
chloroethylisocyanate (17.0 L, 0.199 mmol). The reaction mixture was heated
to 65 C
for 18 hours. TLC analysis indicated a small amount of starting material was
present.
To the reaction mixture was added chloroethylisocyanate (50.9 0 L, 0.597
mmol) and
the mixture was stirred at 75 C for 22 hours. The reaction was quenched with
water (30
mL) and extracted with dichloromethane (2 x 30 mL). The organic portions were
combined, washed with brine (30 mL), dried (MgSO4) and concentrated. The
residue
was purified by column chromatography utilizing 15% acetone/dichlormethane and
then
recrystallized by dissolving the solid with dichloromethane (1 mL) and adding
hexanes
until cloudy. The solid was collected, washed with hexanes (3 x 1 mL) to
produce 9 mg
of P-366 as a white solid in 10% yield. 1H NMR (400 MHz, CDC13) 6 2.62 (s, 3
H),
3.69-3.72 (m, 4 H), 3.77 (s, 3 H), 3.89 (s, 2 H), 6.76 (d,
J=8Hz,1H),7.15(t,J=9Hz,
1 H), 7.45 (s, 1 H), 7.51-7.59 (m, 2 H), 7.94-7.98 (m, 2 H), 8.38 (s, 2 H),
9.36 (br s, 1 H)
ppm. MS(APCI+): 457.1 (M+1).
Example 297. Preparation of P-288
N N
1 +'0 1. Fe, NHaCI N
0 / N+O Mel 0 N DOH, H2O
0 O NH
NaH, DMF 2. 2M HCl in D20 H-Cl
CI CI CI
1-258 1-259 P-288
[00746] Synthesis of (3'-Chloro-6-methoxy-biphenyl-3-yl)-methyl-(4-nitro-
phenyl)-
amine (1-259), In a 40 mL vial equipped with a stir bar was placed I-258 (600
mg, 1.69
mmol), anhydrous DMF (6.8 mL), NaH (60%)(94.6 mg, 2.37 mmol) and methyl iodide
(579 L, 9.30 mmol). The reaction mixture was stirred at 50 C for 17 hours
and then
quenched with water (30 mL) and a saturated ammonium chloride solution (30
mL).
After extracting with EtOAc (2 x 50 mL), the extracts were combined, washed
with brine
(50 mL), dried (MgSO4) and concentrated. The residue was purified by column
utilizing
20% EtOAc/hexanes as the eluent to produce 490 mg of 1-259 as a yellow-orange
solid
in 79% yield.
365
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00747] Synthesis of N-(3'-Chloro-6-methoxy-biphenyl-3-yl)-N-methyl-benzene-
1,4-
diamine; hydrochloride (P-288). In a 40 mL vial equipped with a stir bar was
placed iron
powder (260 mg, 4.66 mmol), ethanol (5.3 mL) and water (1.7 mL). The mixture
was
heated to 85 C in an oil bath and then 1-259 (490 mg, 1.33 mmol) was added
and the
reaction was continued at 85 C for 2 hours. The reaction mixture was cooled
to room
temperature and filtered through Celite. To the filtrate was added water (50
mL) and
extractions were performed with ethyl acetate (2 x 50 mL). The organic
portions were
combined, washed with brine (50 mL), dried (MgSO4) and concentrated. The
residue
was purified by column utilizing 50% EtOAc/hexanes as the eluent to produce
376 mg of
the free base of P-288 as an orange, viscous oil. To the free base was added
Et20 (5 mL)
and 2.0 M HC1 in Et20 (3 mL) and the mixture was stirred at room temperature
for 2
hours. The solid was collected, washed with Et20 (6 mL) and dried in a high
vacuum
oven set at 40 C for 3 hours to produce 225 mg of P-288 as a light brown
solid in 54%
yield. 1H NMR (400 MHz, DMSO-d6) 6 3.25 (s, 3 H), 3.79 (s, 3 H), 6.85 (d, J= 9
Hz, 2
H), 7.12 (d, J= 2 Hz, 1 H), 7.15-7.21 (m, 4 H), 7.38-7.44 (m, 3 H), 7.52-7.54
(m, 1 H),
9.84 (br s, 3 H) ppm.
MS(APCI+): 339.1 (M+1- HC1); LC-MS: 99%.
Example 298. Preparation of P-293
0 / HA, NH2
010I
P-293
[00748] Synthesis of {4-[(3'-Chloro-6-methoxy-biphenyl-3-yl)-methyl-amino] -
phenyl}-urea (P-293). In an 8 mL vial equipped with a stir bar was placed P-
288 (100
mg, 0.266 mmol), water (1.6 mL), acetic acid (800 L) and sodium cyanate (69.2
mg,
1.06 mmol). The mixture was stirred at room temperature for 72 hours and then
water
(30 mL) was added followed by an extraction with dichloromethane (2 x 30 mL).
The
organic portions were combined, washed with brine (30 mL), dried (MgSO4) and
366
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
concentrated. The residue was triturated with diethyl ether (2 mL) to produce
49 mg of
P-293 as an off-white solid in 48% yield. 'H NMR (400 MHz, DMSO-d6) 6 3.19 (s,
3
H), 3.73 (s, 3 H), 5.72 (br s, 2 H), 6.87-6.89 (m, 3 H), 6.94 (dd, J= 9, 3 Hz,
1 H), 7.04
(d, J= 8 Hz, 1 H), 7.28 (d, J= 9 Hz, 2 H), 7.36-7.43 (m, 3 H), 7.48 (br s, 1
H), 8.34 (br s,
1 H) ppm. MS(APCI+): 382.1 (M+1); LC-MS: 98%.
[00749] Scheme 60.
0II
0 e OH
HO B
0 F NO, ~0 F N.0 BBr3
0---- [Pd(n3-C3H5)CI]2 0 DCM
DPPPent
0 K2CO3, DMF
1-223 O I-261
r- a~--
HO F N10 +0 1. Fe, NHaCI
Eti, K2CO3 0 F N EtOH, H20
O O
DMF 2. 2M HCI in E120
E120
1-263
0 1-262 0
IOII
~O F NHZ NaOCN 0 F N NH2
H1 -CI AcOH, H2O
O O
P-334 P-335
Example 299. Preparation of P-334
[00750] Synthesis of 1-[2'-Fluoro-6'-methoxy-3'-(4-nitro-benzyl)-biphenyl-3-
yl]-
ethanone (I-261) In an 18 mL vial equipped with a stir bar was placed 1-223
(600 mg,
1.81 mmol), 4-nitrophenylboronic acid (332 mg, 1.99 mmol), potassium carbonate
(750
mg, 5.43 mmol), 1,5-bis(diphenylphosphino)pentane (239 mg, 0.543 mmol),
allylpalladium(II) chloride dimer (99.3 mg, 0.272 mmol) and dimethylformamide
(3.6
mL). The reaction mixture was heated to 80 C for 18 hours. The reaction
mixture was
filtered through Celite and to the filtrate were added water (40 mL) and a
saturated
ammonium chloride solution (40 mL). After an extraction with ethyl acetate (2
x 50
mL), the organic portions were combined, washed with brine (50 mL), dried
(MgS04)
367
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
and concentrated. The residue was purified by column chromatography utilizing
30%
ethyl acetate/hexanes as the eluent to produce 410 mg of 1-261 as a red-
orange, wet solid
in 60% yield.
MS(ESI-): 378.5 (M-1).
[00751] Synthesis of 1-[2'-Fluoro-6'-hydroxy-3'-(4-nitro-benzyl)-biphenyl-3-
yl]-
ethanone (1-262). In an 18 mL vial equipped with a stir bar was placed 1-261
(351 mg,
0.925 mmol) and dichloromethane (3.0 mL). The solution was cooled to -78 C in
a dry-
ice/acetone bath over 15 minutes and then boron tribromide (1.OM in
dichloromethane)
(2.78 mL, 2.78 mmol) was added and the vial was allowed to gradually warm to
room
temperature over a period of 2 hours. The reaction was poured into ice-water
(40 mL)
and then extracted with dichloromethane (2 x 30 mL). The organic portions were
combined, washed with brine (50 mL), dried (Mg504) and concentrated to produce
274
mg of 1-262 as a brown solid in 81% yield.
[00752] Synthesis of 1-[6'-Ethoxy-2'-fluoro-3'-(4-nitro-benzyl)-biphenyl-3-yl]-
ethanone (1-263). In an 18 mL vial equipped with a stir bar was added 1-262
(200 mg,
0.547 mmol), dimethylformamide (3.6 mL) and iodoethane (131 L, 1.64 mmol).
The
mixture was stirred for 5 minutes and then potassium carbonate (227 mg, 1.64
mmol)
was added and the reaction mixture was stirred at room temperature for 18
hours. The
reaction mixture was quenched with water (30 mL) and acidified to pH 1 with 1M
HC1.
An extraction was performed with dichloromethane (2 x 30 mL) and the organic
portions
were combined, washed with brine (30 mL), dried (Mg504) and concentrated to
produce
185 mg of 1-263 as a red-orange oil in 86% yield.
[00753] Synthesis of 1-[3'-(4-Amino-benzyl)-6'-ethoxy-2'-fluoro-biphenyl-3-yl]-
ethanone; hydrochloride (P-334). In an 18 mL vial equipped with a stir bar was
placed
iron powder (129 mg, 2.31 mmol), ethanol (5.6 mL) and water (850 L). The
mixture
was heated to 85 C in an oil bath and then 1-263 (260 mg, 0.661 mmol) was
added and
the reaction was continued at 85 C for 3 hours. The reaction mixture was
cooled to
room temperature and filtered through Celite. To the filtrate was added water
(50 mL)
and extractions were performed with ethyl acetate (2 x 50 mL). The organic
portions
were combined, washed with brine (50 mL), dried (Mg504) and concentrated. The
368
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
residue was purified by column utilizing 40% EtOAc/hexanes as the eluent. To
this
purified material (free base) was added Et20 (3 mL) and 2.0 M HC1 in Et20 (1.5
mL)
and the mixture was stirred at room temperature for 1 hour. The solid was
collected and
dried in a high vacuum oven set at 40 C for 3 hours to produce 46 mg of P-334
as an
orange solid in 17% yield. 'H NMR (400 MHz, DMSO-d6) 6 1.18 (t, J= 7 Hz, 3 H),
2.59 (s, 3 H), 3.47 (br s, 2 H), 3.96 (s, 2 H), 4.03 (q,
J=7Hz,2H),6.94(d,J=9Hz,1
H), 7.20-7.32 (m, 5 H), 7.55-7.62 (m, 2 H), 7.91 (s, 1 H), 7.94 (dt, J= 7, 2
Hz, 1 H) ppm.
[00754] MS(APCI+): 364.1 (M+1- HC1); LC-MS: 98%.
Example 300. Preparation of P-335
[00755] Synthesis of [4-(3'-Acetyl-6-ethoxy-2-fluoro-biphenyl-3-ylmethyl)-
phenyl]-
urea (P-335). In an 8 mL vial equipped with a stir bar was placed P-334 (38
mg, 0.0950
mmol), water (500 L), acetic acid (250 L) and sodium cyanate (24.7 mg, 0.380
mmol).
The mixture was stirred at room temperature for 18 hours and then water (3 mL)
was
added and then the pH was adjusted to 6 with a saturated sodium bicarbonate
solution.
The resulting solid was collected and dried in a high vacuum oven set at 40 C
for 4
hours to produce 20 mg of P-335 as a pale orange solid in 53% yield. 'H NMR
(400
MHz, CDC13) 6 1.27 (t, J= 7 Hz, 3 H), 2.62 (s, 3 H), 3.94 (s, 2 H), 3.99 (q,
J= 7 Hz, 2
H), 4.65 (s, 2 H), 6.40 (s, 1 H), 6.70 (d, J= 8 Hz, 1 H), 7.078 (t, J= 8 Hz, 1
H), 7.21 (s, 4
H), 7.50 (t, J= 8 Hz, 1 H), 7.62 (dd, J= 8, 1 Hz, 1 H), 7.94 (dt, J= 8, 2 Hz,
1 H), 8.03
(br s, 1 H) ppm.
MS(APCI+): 407.1 (M+1); LC-MS: 94%.
Example 301. Preparation of P-340
O F HNHZ NaBH41 MeOH O F Q 0
HNHZ
JB-46 HCI
O OH
P-269 P-340
369
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00756] Synthesis of {4-[2-Fluoro-3'-(1-hydroxy-ethyl)-6-methoxy-biphenyl-3-
ylmethyl]-phenyl}-urea (P-340). In an 8 mL vial equipped with a stir bar was
placed P-
269 (21 mg, 0.0535 mmol), methanol (200 L) and sodium borohydride (6.1 mg,
0.161
mmol). The mixture was stirred at room temperature for 1 hour and then
quenched with
water (2 mL) and 1M aqueous HC1(4 mL). After stirring for 20 minutes, a
saturated
sodium bicarbonate solution was used to adjust the mixture to pH 7-8. After
extracting
with dichloromethane (2 x 30 mL), the organic portions were combined, dried
(MgS04)
and concentrated. The residue was purified via column chromatography utilizing
40%
acetone/dichloromethane as the eluent produce 10 mg of P-340 as a white solid
in 49%
yield. 1H NMR (400 MHz, DMSO-d6) 6 1.33 (d, J= 6 Hz, 3 H), 3.69 (s, 3 H), 3.83
(s, 2
H), 4.70-4.78 (m, 1 H), 5.15 (d, J= 4 Hz, 1 H), 5.76 (s, 2H), 6.89 (d, J= 9
Hz, 1 H), 7.06
(d, J= 8 Hz, 2 H), 7.13 (d, J= 7 Hz, 1 H), 7.21 (t, J= 9 Hz, 1 H), 7.25-7.36
(m, 4 H),
8.42 (s, 1 H) ppm. MS(APCI+): 377.1 [(M+1)-18]; LC-MS: >99%.
Example 302. Preparation of P-3 81
0 O
N,O F N
O F N H
H
No
Br
P-168 P-381
[00757] Synthesis ofN-[4-(2-Fluoro-6-methoxy-3'-pyrrolidin-l-yl-biphenyl-3-
ylmethyl)-phenyl]-acetamide (P-381). In an 8 mL vial equipped with a stir bar
was
placed P-168 (100 mg, 0.233 mmol), 1,4-dioxane (800 L) and pyrrolidine (117
L, 1.40
mmol). The mixture was degassed with nitrogen for 20 minutes and then
dichlorobis(chloro tert-butyl phosphine) palladium (50.2 mg, 0.0932 mmol) was
added.
The mixture was heated to 85 C for 20 hours and then filtered through Celite.
The
filtrate was treated with water (20 mL) and a saturated ammonium chloride
solution (20
mL). After an extraction with ethyl acetate (2 x 30 mL), the organic portions
were
combined, washed with brine (30 mL), dried (MgS04) and concentrated. The
residue
was purified by column chromatography utilizing 20% acetone/dichloromethane as
the
eluent and to produce 31 mg of P-381 as a pale orange solid in 32% yield. 1H
NMR (400
MHz, DMSO-d6) 6 1.38 (d, J= 14 Hz, 4 H), 2.01 (s, 3 H), 3.24 (m, 4 H), 3.65
(s, 3 H),
370
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
3.84 (s, 2 H), 6.76 (d, 1 H), 6.84 (d, J= 8 Hz, 1 H), 6.93 (t, J= 8 Hz, 1 H),
7.11 (d, J= 8
Hz, 2 H), 7.17 (t, J= 8 Hz, 1 H), 7.23 (br s, 1 H), 7.30-7.32 (m, 1 H), 7.48
(d, J= 8 Hz, 2
H), 9.83 (s, 1 H) ppm. LC-MS: 92%.
Example 303. Preparation of P-390
O
O
O F N
\O F H
H
Br 00
P-168 P-390
[00758] Synthesis of N-[4-(2-Fluoro-6-methoxy-3'-morpholin-4-yl-biphenyl-3-
ylmethyl)-phenyl]-acetamide (P-390). In an 8 mL vial equipped with a stir bar
was
placed P-168 (80 mg, 0.187 mmol), sodium tert-butoxide (27.0 mg, 0.281 mmol),
toluene (700 L), morpholine (19.6 L, 0.224 mmol) and 2,8,9-triisobutyl-
2,5,8,9-
tetraaza-l-phosphabicyclo[3.3.3]undecane (13.3 L, 0.0374 mmol). The mixture
was
degassed with nitrogen for 15 minutes and then tris(dibenzylideneacetone)-
dipalladium(0) (8.56 mg, 0.00935 mmol) was added. The mixture was heated to 90
C
for 18 hours and then filtered through Celite. The filtrate was treated with
water (20 mL)
and a saturated ammonium chloride solution (20 mL). After an extraction with
ethyl
acetate (2 x 30 mL), the organic portions were combined, washed with brine (30
mL),
dried (MgS04) and concentrated. The residue was purified by column
chromatography
utilizing 10% acetone/dichloromethane as the eluent and to produce 21 mg of P-
3 90 as
an off white solid in 26% yield. 1H NMR (400 MHz, DMSO-d6) 6 2.01 (s, 3 H),
3.09 (t,
J=5Hz,4H),3.69(s,3H),3.72(t,J=5Hz,4H),3.85 (s,2H),6.72(d,J=7Hz,1
H), 6.83 (br s, 1 H), 6.87 (d, J= 9 Hz, 1 H), 6.92 (dd, J= 8, 2 Hz, 1 H), 7.13
(d, J= 8 Hz,
2 H), 7.18-7.27 (m, 2 H), 7.47 (d, J= 8 Hz, 2 H), 7.69-7.80 (m, 1 H) ppm.
MS(APCI+):
435.1 (M+1); LC-MS: 95%.
371
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 304. Preparation of P-385
O I I F
P-385
[00759] Synthesis of 4-[5'-(4-Fluoro-benzyl)-2'-methoxy-biphenyl-3-yl]-
morpholine
(P-385). In an 8 mL vial equipped with a stir bar was placed 1-185 (200 mg,
0.678
mmol), 4-[3-(4,4,5,5-tetramethyl-[1,3,2] dioxaborolan-2-yl)-phenyl]-morpholine
(216
mg, 0.746 mmol), potassium carbonate (281 mg, 2.03 mmol), triphenylphosphine
(53.3
mg, 0.203 mmol), 1,4-dioxane (1.1 mL), 50% aqueous ethanol (1.1 mL) followed
by
palladium (II) acetate (15.2 mg, 0.0678 mmol). The mixture was heated to 90 C
for 18
hours and then cooled to room temperature. The palladium catalyst was removed
via
filtration through Celite. To the filtrate were added water (30 mL) and a
saturated
ammonium chloride solution (30 mL). After the aqueous portion was extracted
with
ethyl acetate (2 x 30 mL), the organic portions were combined, washed with
brine (30
mL), dried (MgS04) and concentrated. The residue was purified by column
chromatography utilizing 20% EtOAc/hexanes as the eluent and dried in a high
vacuum
oven set at 40 C for 9 hours to produce 137 mg of P-385 as a yellow viscous
oil in 54%
yield. 1H NMR (400 MHz, CDC13) 6 3.18 (t, J= 5 Hz, 4 H), 3.78 (s, 3 H), 3.87
(t, J= 5
Hz, 4 H), 3.92 (s, 2 H), 6.87-7.01 (m, 5 H), 7.04 (t, J= 2 Hz, 1 H), 7.09 (dd,
J= 8, 3 Hz,
1 H), 7.12-7.17 (m, 3 H), 7.30 (t, J= 8 Hz, 1 H) ppm. MS(APCI+): 378.1 (M+1);
LC-
MS: >99%
372
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 305. Preparation of P-3 91
O F
CI
P-391
[00760] Synthesis of 2-Chloro-6-[5-(4-fluoro-benzyl)-2-methoxy-phenyl]-
pyridine (P-
391). In an 8 mL vial equipped with a stir bar was placed 1-185 (250 mg, 0.847
mmol),
2-chloro-6-(4,4,5,5-tetramethyl-[1,3,2] dioxaborolan-2-yl)-pyridine (243 mg,
1.02 mmol),
sodium tert-butoxide (122 mg, 1.27 mmol) and 1,4-dioxane. The mixture was
degassed
with nitrogen for 20 minutes and then dichlorobis(chloro tert-butyl phosphine)
palladium
(122 mg, 0.127 mmol) was added. The mixture was heated to 90 C for 18 hours
and
then filtered through Celite. The filtrate was treated with water (20 mL) and
a saturated
ammonium chloride solution (20 mL). After an extraction with ethyl acetate (2
x 30
mL), the organic portions were combined, washed with brine (30 mL), dried
(MgSO4)
and concentrated. The residue was purified by column chromatography utilizing
10%
ethyl acetate/hexanes as the eluent. The material was then recrystallized with
diethyl
ether and hexanes to produce 29 mg of P-391 as a white solid in 10% yield.
[00761] 'H NMR (400 MHz, CDC13) 6 3.84 (s, 3 H), 3.96 (s, 2 H), 6.91 (d, J= 8
Hz, 1
H), 6.94-6.98 (m, 2 H), 7.12-7.17 (m, 2 H), 7.23 (dd, J= 8, 1 Hz, 1 H), 7.64
(t, J= 8 Hz,
1 H), 7.69 (d, J= 2 Hz, 1 H), 7.79 (dd, J= 8, 1 Hz, 1 H) ppm. MS(APCI+): 328.0
(M+1);
LC-MS: 97%.
373
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00762] Scheme 61.
O
~-r'OH 11 O OMe B
O E pyridine E NNH 0 F N NH2
ado' [Pd(n3-C3H5)Ci12 P-395
DPPPent
NI THE K2CO3, DMF
N N
1-265
1-266
N O
N 0 F NNJ N
H
pyridine P-399
N
Example 306. Preparation of P-395
[00763] Synthesis of Carbonic acid 3'-cyano-2-fluoro-6-methoxy-biphenyl-3-
ylmethyl
ester methyl ester (1-266). In an 18 mL vial equipped with a stir bar was
placed 1-265
(465 mg, 1.81 mmol), anhydrous tetrahydrofuran (6.0 mL) and pyridine (381 L,
4.71
mmol). The resulting clear solution was cooled in an ice water bath for 10
minutes and
then methyl chloroformate (308 L, 3.98 mmol) was added and reaction mixture
was
slowly warmed to room temperature and reacted for 17 hours. The reaction was
acidified to pH 1 with 1M HC1, water (30 mL) was added followed by an
extraction with
dichlormethane (2 x 60 mL). The organic portions were combined, washed with
brine
(40 mL), dried (MgS04) and concentrated. After drying in a high vacuum oven
for 2
hours at 45 C, 378 mg of 1-266 was isolated as a light yellow in 66% yield.
[00764] Synthesis of 3'-(2-Amino-pyrimidin-5-ylmethyl)-2'-fluoro-6'-methoxy-
biphenyl-3-carbonitrile (P-395). In an 8 mL vial equipped with a stir bar was
placed I-
266 (120 mg, 0.381 mmol), 5-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-
pyrimidin-
2-ylamine (92.7 mg, 0.419 mmol), potassium carbonate (158 mg, 1.14 mmol), 1,5-
bis(diphenylphosphino)pentane (50.3 mg, 0.114 mmol), allylpalladium(II)
chloride
374
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
dimer (20.9 mg, 0.0572 mmol) and dimethylformamide (1.3 mL). The reaction
mixture
was heated to 85 C for 18 hours. The reaction mixture was filtered through
Celite and
to the filtrate were added water (30 mL) and a saturated ammonium chloride
solution (30
mL). After an extraction with ethyl acetate (2 x 30 mL), the organic portions
were
combined, washed with brine (40 mL), dried (MgSO4) and concentrated. The
residue
was purified by column chromatography utilizing 10% acetone/dichloromethane
and
then 30% acetone/dichloromethane as the eluent to produce 62 mg of P-395 as a
pale
yellow solid in 49% yield. 'H NMR (400 MHz, DMSO-d6) 6 3.73 (s, 5 H), 6.47 (s,
2 H),
6.96 (d, J= 8 Hz, 1 H), 7.33 (t, J= 9 Hz, 1 H), 7.61-7.69 (m, 2 H), 7.82-7.84
(m, 2 H),
8.12 (s, 2 H) ppm. MS(APCI+): 335.1 (M+1); LC-MS: 94%.
Example 307. Preparation of P-399
[00765] Synthesis of 1-[5-(3'-Cyano-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyrimidin-2-yl]-3-ethyl-urea (P-399). In an 8 mL vial equipped with a stir bar
was
placed P-3 95 (60 mg, 0.180 mmol), pyridine (600 L) and ethyl isocyanate
(71.2 L,
0.900 mmol). The reaction mixture was stirred at room temperature for 18
hours. TLC
analysis indicated that the reaction mixture consisted of about 50% starting
materials.
To the reaction mixture was added ethyl isocyanate (71.2 L, 0.900 mmol) and
the
mixture was heated to 55 C for 5 hours. The reaction mixture was quenched
with water
(4 mL) and the resulting solid was collected, washed with water (3 x 2 mL),
ethyl acetate
(3 x 2 mL) and dried in a high vacuum oven set at 40 C for 18 hours to
produce 36 mg
of P-399 as a white solid in 49% yield. 1H NMR (400 MHz, DMSO-d6) 6 1.09 (t,
J= 7
Hz, 3 H), 3.19-3.25 (m, 2 H), 3.74 (s, 3 H), 3.89 (s, 2 H), 6.98 (d, J= 9 Hz,
1 H), 7.39 (t,
J= 9 Hz, 1 H), 7.61-7.70 (m, 2 H), 7.83-7.85 (m, 2 H), 8.46 (s, 2 H), 8.91 (t,
J= 6 Hz, 1
H), 9.611 (s, 1 H) ppm. MS(APCI+): 406.1 (M+1); LC-MS: 92%.
375
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00766] Scheme 62
Pd(PPh3)4
Br K3PO4 N-o NN O
O , IIII
DME O NHZ ~0- NN--~
Br JB-61 50% aq. EtOH Br JB-75 pyridine Br H H
JB-76
1-267
material from Gary B jN 1-268
NHz
1-42
Pd(PPh3)4 N O
K3PO4 O N, .N
11
H H
DME P-400
50% aq. EtOH
OH
Hog N
N
Example 308. Preparation of P-400
[00767] Synthesis of 5-(3-Bromo-4-methoxy-benzyl)-pyridin-2-ylamine (1-267) In
a
40 mL vial equipped with a stir bar was placed 1-42 (1.0 g, 3.57 mmol), 5-
(4,4,5,5-
tetramethyl-[1,3,2]dioxaborolan-2-yl)-pyridin-2-ylamine (865 mg, 3.93 mmol),
potassium phosphate (tribasic) (1.52 g, 7.14 mmol), dimethoxyethane (6.0 mL)
and 50%
aqueous ethanol (6.0 mL). The mixture was degassed with nitrogen for 20
minutes and
then add tetrakis(triphenylphosphine)palladium(0) (619 mg, 0.536 mmol). The
mixture
was heated to 60 C for 4 hours and then the palladium catalyst was filtered
off. To the
filtrate were added water (50 mL) and a saturated ammonium chloride solution
(50 mL).
After extracting with ethyl acetate (2 x 60 mL), the organic portions were
combined,
washed with brine (50 mL), dried (MgS04) and concentrated. The residue was
purified
by column utilizing 10% acetone/dichloromethane, 30% acetone/dichloromethane,
40%
acetone/dichloromethane as the eluent to produce 308 mg of 1-267 as a yellow
viscous
oil in 29% yield. MS(APCI+): 295.0 (M+1); LC-MS: >99%.
[00768] Synthesis of 1-[5-(3-Bromo-4-methoxy-benzyl)-pyridin-2-yl]-3-ethyl-
urea (I-
268). In an 8 mL vial equipped with a stir bar was placed 1-267 (300 mg, 1.02
mmol),
pyridine (3.5 mL) and ethyl isocyanate (646 L, 8.16 mmol). The reaction
mixture was
stirred at room temperature for 18 hours. The reaction mixture was quenched
with water
(30 mL) and 1M aqueous HC1(30 mL). After extracting with dichloromethane (2 x
60
mL), the organic portions were washed with 1M aqueous HC1(40 mL), brine (40
mL),
376
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
dried (MgSO4) and concentrated. The crude sticky material was recrystallized
with
diethyl ether (2 mL), ethyl acetate(1 mL) and hexanes (700 L). The resulting
solid was
collected by suction filtration and washed with diethyl ether (2 x lmL) to
produce 49 mg
of 1-268 as an off-white solid in 13% yield. MS(APCI+): 366.0 (M+1); LC-MS:
98%.
[00769] Synthesis of 1-[5-(3'-Cyano-6-methoxy-biphenyl-3-ylmethyl)-pyridin-2-
yl]-
3-ethyl-urea (P-400). In an 8 mL vial equipped with a stir bar was placed 1-
268 (47 mg,
0.129 mmol), 3-cyanophenylboronic acid (22.7 mg, 0.155 mmol), potassium
phosphate
(tribasic) (54.8 mg, 0.258 mmol), dimethoxyethane (250 L) and 50% aqueous
ethanol
(250 L). The mixture was degassed with nitrogen for 15 minutes and then add
tetrakis(triphenylphosphine)palladium(0) (14.9 mg, 0.0129 mmol). The mixture
was
heated to 85 C for 18 hours and then the palladium catalyst was filtered off.
To the
filtrate were added water (20 mL) and a saturated ammonium chloride solution
(20 mL).
After extracting with ethyl acetate (2 x 30 mL), the organic portions were
combined,
washed with brine (30 mL), dried (MgSO4) and concentrated. The residue was
purified
by column utilizing 15% acetone/dichloromethane as the eluent to produce 27 mg
of P-
400 as a white solid in 55% yield. 'H NMR (400 MHz, DMSO-d6) 61.07 (t, J= 7
Hz, 3
H), 3.13-3.20 (m, 2 H), 3.75 (s, 3 H), 3.84 (s, 2 H), 7.07 (d, J= 8 Hz, 1 H),
7.23 (s, 1 H),
7.25 (s, 1 H), 7.55-7.62 (m, 3 H), 7.77-7.82 (m, 1 H), 7.90 (m, 1 H), 8.10 (br
s, 1 H), 9.06
(s, 1 H) ppm. MS(APCI+): 387.1 (M+1); LC-MS: 98%.
Example 309. Preparation of P-433
Bu
Sn2(Bu3)2 Bu ..
N Bu N
N (PPh3)2PdCl2 I N
dioxane
1-214
[00770] Synthesis of 4-Tributylstannanyl-pyridine-2-carbonitrile (1-214). In a
40 mL
vial equipped with a stir bar was placed 4-iodo-pyridine-2-carbonitrile (1.26
g, 5.48
mmol), 1,4-dioxane (18 mL), and bis(tributyl)tin (3.32 mL, 6.58 mmol). After
the
reaction mixture was degassed with nitrogen for 15 minutes,
bis(triphenylphosphine)-
palladium(II) dichloride (192 mg, 0.274 mmol) was added. The reaction mixture
was
377
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
heated to 90 C for 18 hours and then quenched with 2.2 M aqueous potassium
fluoride
(100 ml) in order to consume excess tin reagent. After stirring at room
temperature for 2
hours, the mixture was filtered through Celite. To the filtrated was added
water (50 mL),
followed by extractions with ethyl acetate (5 x 100 mL). The organic portions
were
combined, washed with 2.2 M aqueous potassium fluoride (150 mL), water (150
mL)
and brine (150 mL). After drying the organic portion with magnesium sulfate,
the
material was concentrated and purified by column chromatography utilizing 5%
ethyl
acetate/hexanes as the eluent to produce 1.24 g of 1-214 as a colorless oil in
58% yield.
MS(APCI+): 394.0 (M+1)
Br
BU I Bu
0 F Bu'Sn N (PPh3)2PdCl2 I I N
0 F N
Y + I ~N
dioxane
CI
1-214
CI
1-33
P-433
[00771] Synthesis of 4-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridine-
2-carbonitrile (P-433). In an 18 mL vial equipped with a stir bar was placed 1-
33 (162
mg, 0.492 mmol), 1,4-dioxane (2.3 mL), and I-214 (232 mg, 0.590 mmol). After
the
reaction mixture was degassed with nitrogen for 20 minutes,
bis(triphenylphosphine)-
palladium(II) dichloride (17.3 mg, 0.0246 mmol) was added. The reaction
mixture was
heated to 80 C for 18 hours. To the reaction mixture was added water (40 mL)
followed
by extractions with ethyl acetate (2 x 60 mL). The organic portions were
combined,
washed with brine (40 mL), dried (magnesium sulfate) and concentrated. The
residue
was purified by column chromatography utilizing 30% ethyl acetate/hexanes as
the
eluent to produce 102 mg of P-433 as an orange oil in 59% yield. 'H NMR (400
MHz,
DMSO-d6) 6 3.74 (s, 3 H), 4.06 (s, 2 H), 6.98 (d, J= 9 Hz, 1 H), 7.28-7.30 (m,
1 H),
7.38-7.47 (m, 4 H), 7.56 (d, J= 5 Hz, 1 H), 7.93 (s, 1 H), 8.65 (d, J= 5 Hz, 1
H) ppm.
MS(APCI+): 354.0 (M+1); LC-MS: 97%.
378
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 310. Preparation of P-437
NH2
N, 0 7 1 5111 CI
P-437
[00772] Synthesis of C-[4-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-2-yl]-methylamine (P-437). In an 8 mL vial equipped with a stir bar
was placed
P-433 (95 mg, 0.269 mmol), methanol (2.8 mL), concentrated HC1(113 L, 1.35
mmol),
10% palladium on activated carbon (19.0 mg), followed by attachment of a
balloon of
hydrogen. The mixture was allowed to stir at room temperature for 2 hours and
then
filtered through Celite. To the filtrate was added water (20 mL), the pH was
adjusted to
10-11 with 1M NaOH, and extractions were done with ethyl acetate (2 x 60 mL).
The
organic portions were combined, washed with brine (50 mL), dried (magnesium
sulfate)
and concentrated. The residue was purified by column chromatography utilizing
10%
methanol/dichloromethane to produce 14 mg of P-437 as a black, semi-solid in
14%
yield. 1H NMR (400 MHz, DMSO-d6) 6 3.17 (s, 2 H), 3.74 (s, 3 H), 3.78 (s, 2
H), 3.95
(s, 2 H),6.96 (d, J= 8 Hz, 1 H), 7.06-7.07 (m, 1 H), 7.28-7.47 (m, 6 H), 8.37
(d, J= 5
Hz, 1 H) ppm. MS(APCI+): 358.0 (M+1); LC-MS: 96%.
[00773] Scheme 63
~Br Pd(PPh3)4 Pd(PPh3)4_
O N O
0 K3PO4 N O K3PO4 0-
Br DME Br 0- DME
aq. EtOH aq. EtOH
1-42 1-269 CI
1-270
Br
Fe, NH4CI N~
O NH2 0 N N
EtOH, H2O H
conc. HCI
aq. EtOH
CI CI
1-271 P-454
379
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 311. Preparation of P-454
[00774] Synthesis of 2-Bromo-l-methoxy-4-(4-nitro-benzyl)-benzene (1-269). In
a
100 mL round bottomed flask equipped with a stir bar was placed 1-42 (2.0 g,
7.14
mmol), 4-nitrophenylboronic acid (2.27 g, 7.85 mmol), potassium phosphate
(tribasic)
(3.03 g, 14.3 mmol), dimethoxyethane (12 mL) and 50% aqueous ethanol (12 mL).
The
mixture was degassed with nitrogen for 20 minutes and then added
tetrakis(triphenylphosphine)palladium(0) (825 mg, 0.714 mmol). The mixture was
heated to 60 C for 4 hours and then the palladium catalyst was filtered off.
To the
filtrate were added water (50 mL) and a saturated ammonium chloride solution
(100
mL). After extracting with ethyl acetate (3 x 100 mL), the organic portions
were
combined, washed with brine (150 mL), dried (Mg504) and concentrated. The
residue
was purified by column utilizing 15% ethyl acetate/hexanes as the eluent to
produce 1.06
g of 1-269 as a pale yellow solid in 46% yield. MS(APCI-): 321.2 (M-1).
[00775] Synthesis of 3'-Chloro-2-methoxy-5-(4-nitro-benzyl)-biphenyl (1-270).
In a
40 mL vial equipped with a stir bar was placed 1-269 (960 mg, 2.98 mmol), 3-
chlorophenylboronic acid (559 mg, 3.58 mmol), ), potassium phosphate
(tribasic) (1.27
g, 5.96 mmol), dimethoxyethane (5 mL) and 50% aqueous ethanol (5 mL) and
tetrakis(triphenylphosphine)palladium(0) (344 mg, 0.298 mmol). The mixture was
heated to 67 C for 65 hours and then the palladium catalyst was filtered off.
To the
filtrate were added water (50 mL) and a saturated ammonium chloride solution
(70 mL).
After extracting with ethyl acetate (3 x 75 mL), the organic portions were
combined,
washed with brine (80 mL), dried (Mg504) and concentrated. The residue was
purified
by column utilizing 10% ethyl acetate/hexanes as the eluent to produce 1.05 g
of 1-270 as
a yellow viscous oil in quantitative yield. MS(APCI-): 351.8 (M-2).
[00776] Synthesis of 4-(3'-Chloro-6-methoxy-biphenyl-3-ylmethyl)-phenylamine
(I-
27 1). In a 40 mL vial equipped with a stir bar was placed iron powder (586
mg, 10.5
mmol), ethanol (12 mL) and water (3.8 mL). The mixture was heated to 85 C in
an oil
bath and then 1-270 (1.05 g, 3.00 mmol) was added and the reaction was
continued at 85
C for 2 hours. The reaction mixture was cooled to room temperature and
filtered
through Celite. To the filtrate was added water (60 mL) and extractions were
performed
380
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
with ethyl acetate (2 x 60 mL). The organic portions were combined, washed
with brine
(60 mL), dried (MgSO4) and concentrated to produce 862 mg of 1-271 as a pale
orange
viscous oil in 89% yield. MS(APCI+): 324.1 (M+1); LC-MS: 97%.
[00777] Synthesis of [4-(3'-Chloro-6-methoxy-biphenyl-3-ylmethyl)-phenyl]-
thiazol-
2-yl-amine (P-454). In an 8 mL vial equipped with a stir bar was placed I-271
(70 mg,
0.216 mmol), 2-bromothiazole (38.5 L, 0.432 mmol), 10% aqueous ethanol (1.1
mL)
and concentrated hydrochloric acid (36.0 L, 0.432 mmol). The mixture was
heated to
95 C for 18 hours and then combined with the reaction mixture from a 50 mg
scale
reaction of the exact same type. After water (30 mL) and 5% aqueous potassium
carbonate (30 mL) were added, the aqueous portion was extracted with ethyl
acetate (2 x
30 mL) and the organic portions were combined, washed with water (40 mL),
brine (40
mL), dried (magnesium sulfate) and concentrated. The crude material was
purified by
column chromatography utilizing 3% acetone/dichloromethane as the eluent to
produce
70 mg of P-454 as viscous, light yellow oil in 47% yield. 'H NMR (400 MHz,
CDC13) 6
3.79 (s, 3 H), 3.94 (s, 2 H), 6.61 (d, J= 4 Hz, 1 H), 6.91 (d, J= 8 Hz, 1 H),
7.12-7.19 (m,
H), 7.27-7..33 (m, 4 H), 7.38 (dt, J= 7, 2 Hz, 1 H), 7.50 (t, J= 2 Hz, 1 H).
MS(APCI+): 407.0 (M+1); LC-MS: 99%.
Example 312. Preparation of P-458
N H N
N
O 7FN CI OH O F N N OH
i-PrOH, DIEA
CI CI
1-177 P-458
[00778] Synthesis of 2-{[5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyrimidin-2-yl]-methyl-amino}-ethanol (P-458). In an 8 mL vial equipped with a
stir
bar was placed 1-177 (40 mg, 0.110 mmol), 2-(methylamino)-ethanol (13.2 L,
0.165
mmol), isopropanol (600 L) and diisopropylethylamine (57.5 L, 0.330 mmol).
The
mixture was heated to 80 C for 18 hours, combined with an identical 20 mg
scale
reaction and then treated with water (30 mL), 1M HC1(20 mL) and extracted with
dichloromethane (2 x 50 mL). The organic portions were combined, washed with
brine
381
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
(50 mL), dried (magnesium sulfate) and concentrated. The residue was purified
by silica
gel column chromatography utilizing 10% acetone/dichloromethane as the eluent
to
produce 35 mg of P-458 as a colorless, viscous oil in 52% yield. 'H NMR (400
MHz,
DMSO-d6) 6 3.10 (s, 3 H), 3.52-3.53 (m, 4 H), 3.72 (s, 3 H), 3.75 (s, 2 H),
4.64 (t, J= 5
Hz, 1 H), 6.93 (d, J= 8 Hz, 1 H), 7.27-7.32 (m, 2 H), 7.38 (s, 1 H), 7.43-7.47
(m, 2 H),
8.22 (s, 2 H) ppm.
MS(APCI+): 402.0 (M+1); LC-MS: 99%.
Example 313. Preparation of P-467
Br-,\ N/ N
NH2 N1J--N
H
p-TsOH
toluene
Cl
~OcI
1-271 P-467
[00779] Synthesis of [4-(3'-Chloro-6-methoxy-biphenyl-3-ylmethyl)-phenyl]-(1-
methyl-lH-imidazol-2-yl)-amine (P-467). In an 8 mL vial equipped with a stir
bar was
placed 1-271 (150 mg, 0.463 mmol), 2-bromo-l-methyl-lH-imidazole (67.8 L,
0.695
mmol), p-toluenesulfonic acid (106 mg, 0.556 mmol) and toluene (2.0 mL). The
mixture
was heated to 115 C for 18 hours and then treated with water (30 mL) followed
by
adjustment to pH 10 with 5% aqueous potassium carbonate. After extracting with
ethyl
acetate (2 x 35 mL), the organic portions were combined, washed with brine (50
mL),
dried (MgS04) and concentrated. The residue was purified by silica gel column
chromatography utilizing 5% then 50% acetone/dichloromethane as the eluent to
produce 89 mg of P-467 as a pale orange solid in 47% yield. 1H NMR (400 MHz,
DMSO-d6) 6 3.45 (s, 3 H), 3.74 (s, 3 H), 3.82 (s, 2 H), 6.61 (d, J= 2 Hz, 1
H), 6.81 (d, J
= 2 Hz, 1 H), 7.02-7.07 (m, 3 H), 7.15 (d, J= 2 Hz, 1 H), 7.19 (dd, J= 8, 2
Hz, 1 H),
7.24 (d, J= 8, 2 H), 7.35-7.44 (m, 3 H), 7.48 (bs, 1 H), 8.09 (s, 1 H) ppm;
MS(APCI+):
404.1 (M+1).
LC-MS: >99%.
382
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00780] Scheme 64:
O r-N x O /-0 II
Br B N N x0
0 F NIJ
Br (PPh3)4Pd O F (PPh3)4Pd
JB-94 K3PO4 Br JB-95 K3PO4
1-31 DME, aq. EtOH 1-273 DME, aq. EtOH
0
NO NH
N + NJ
0 FFF FA 0 F N
JB-96 DCM
CI T
CI
1-274
P-515
Example 314. Preparation of P-515
[00781] Synthesis of 4-[4-(3-Bromo-2-fluoro-4-methoxy-benzyl)-pyridin-2-yl]-
piperazine-l-carboxylic acid tert-butyl ester (1-273). In a 40 mL vial
equipped with a
stir bar was placed 1-31 (642 mg, 2.15 mmol), 4-[4-(4,4,5,5-Tetramethyl-
[1,3,2]dioxaborolan-2-yl)-pyridin-2-yl]-piperazine-l-carboxylic acid tert-
butyl ester (922
mg, 2.37 mmol), potassium phosphate (tribasic) (913 mg, 4.30 mmol),
dimethoxyethane
(3.6 mL) and 50% aqueous ethanol (3.6 mL). After degassing with nitrogen for
15
minutes, tetrakis(triphenylphosphine)palladium(0) (248 mg, 0.215 mmol) was
added.
The mixture was heated to 60 C for 5 hours and then the palladium catalyst
was
removed by filtering through Celite. To the filtrate were added water (50 mL)
and a
saturated ammonium chloride solution (50 mL). After extracting with ethyl
acetate (3 x
75 mL), the organic portions were combined, washed with brine (75 mL), dried
(MgS04)
and concentrated. The residue was purified by column utilizing 35% ethyl
acetate/hexanes as the eluent to produce 495 mg of 1-273 as a yellow, viscous
oil in 48%
yield.
MS(APCI+): 480.0 (M+1);
383
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00782] Synthesis of 4-[4-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-2-yl]-piperazine-l-carboxylic acid tert-butyl ester (1-274). In a 18
mL vial
equipped with a stir bar was placed 1-273 (490 mg, 1.02 mmol), 3-chlorophenyl
boronic
acid (191 mg, 1.22 mmol), potassium phosphate (tribasic) (433 mg, 2.04 mmol),
dimethoxyethane (2.0 mL) and 50% aqueous ethanol (2.0 mL). After degassing
with
nitrogen for 15 minutes, tetrakis(triphenylphosphine)palladium(0) (118 mg,
0.102 mmol)
was added. The mixture was heated to 90 C for 22 hours and then the palladium
catalyst was removed by filtering through Celite. To the filtrate was added
water (150
mL) followed by extractions with ethyl acetate (3 x 75 mL). The organic
portions were
combined, washed with brine (75 mL), dried (MgSO4) and concentrated. The
residue
was purified by column utilizing 25% then 35% ethyl acetate/hexanes as the
eluent to
produce 360 mg of 1-274 as a pale yellow solid in 69% yield. MS(APCI+): 512.1
(M+)
[00783] Synthesis of 1-[4-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-2-yl]-piperazine (P-515) In a 40 mL vial equipped with a stir bar was
placed I-
274 (360 mg, 0.703 mmol) and dichlormethane (2.3 mL). The solution was cooled
in an
ice-water bath for 10 minutes and then trifluoroacetic acid (522 L, 7.03
mmol) was
added. The solution was stirred at room temperature for 3 hours and then
concentrated
by a stream of nitrogen. To the oil was added water (50 mL), a saturated
sodium
bicarbonate solution (till pH 8) followed by extractions with dichloromethane
(2 x
75mL). The organic portions were combined, washed with brine (50 mL), dried
(MgSO4) and concentrated. The residue was purified by column utilizing 10%,
15% and
then 20% methanol/dichloromethane as the eluent to produce 120 mg of P-515 as
an off-
white solid in 41% yield. 'H NMR (400 MHz, DMSO-d6) 6 2.80 (t, J= 5 Hz, 4 H),
3.39
(t, J= 5 Hz, 4 H), 3.73 (s, 3 H), 3.84 (s, 2 H), 6.46 (d, J= 5 Hz, 1 H), 6.69
(s, 1 H), 6.94
(d, J= 8 Hz, 1 H), 7.28 (bd, J= 7 Hz, 1 H), 7.32 (t, J= 9 Hz, 1 H), 7.36 (s, 1
H), 7.41-
7.47 (m, 2 H), 7.98 (d, J= 5 Hz, 1 H) ppm. MS(APCI+): 412.1 (M+1); LC-MS:
>99%.
384
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 315. Preparation of P-518
O
rN'J~ NH2
NN
O
TF
P-518
[00784] Synthesis of 4-[4-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-2-yl]-piperazine-l-carboxylic acid amide (P-518). In an 8 mL vial
equipped
with a stir bar was placed P-515 (50 mg, 0.121 mmol), water (800 L), acetic
acid (400
L) and sodium cyanate (31.5 mg, 0.484 mmol). The mixture was stirred at room
temperature for 18 hours and then water (30 mL) was added. After adjusting to
pH 8
with a saturated sodium bicarbonate solution, the aqueous portion was
extracted with
dichloromethane (2 x 30 mL). The organic portions were combined, washed with
brine
(30 mL), dried (MgS04) and concentrated to produce an off-white solid. The
residue
was triturated with diethyl ether (1 mL), collected and dried in a high vacuum
oven for
18 hours to produce 17 mg of P-518 as an off-white solid in 30% yield. 'H NMR
(400
MHz, DMSO-d6) 6 3.36-3.44 (m, 8 H), 3.73 (s, 3 H), 3.85 (s, 2 H), 6.03 (s, 2
H), 6.48 (d,
J= 5 Hz, 1 H), 6.76 (s, 1 H), 6.94 (d, J= 9 Hz, 1 H), 7.28 (d, J= 7 Hz, 1 H),
7.33 (t, J=
9 Hz, 1 H), 7.37 (s, 1 H), 7.41-7.47 (m, 2 H), 8.00 (d, J= 5 Hz, 1 H) ppm.
MS(APCI+):
455.1 (M+1).
Example 316. Preparation of P-519
O
rNAN
NH
J
N
O \
CI
P-519
385
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00785] Synthesis of 4-[4-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-2-yl]-piperazine-l-carboxylic acid ethylamide (P-519). In an 8 mL vial
equipped
with a stir bar was placed P-515 (60 mg, 0.146 mmol), pyridine (500 L) and
ethyl
isocyanate (92.5 L, 1.17 mmol). The mixture was stirred at room temperature
for 18
hours and then treated with water (20 mL) and a saturated sodium bicarbonate
solution
(20 mL). After extractions with dichloromethane (3 x 30 mL), the organic
portions were
combined, washed with 1M HC1(2 x 30 mL), brine (40 mL), dried (Mg504) and
concentrated to produce an off-white solid. The crude material was triturated
with
diethyl ether (1 mL), collected and dried in a high vacuum oven for 18 hours
to produce
22 mg of P-519 in 31% yield. 'H NMR (400 MHz, DMSO-d6) 6 1.02 (t, J= 4 Hz, 3
H),
3.05-3.18 (m, 2 H), 3.42 (br s, 4 H), 3.53 (br s, 4 H), 3.74 (s, 3 H), 3.93
(s, 2 H), 6.57 (m,
2H),6.96(d,J=8Hz,2H),7.28(d,J=7Hz,1H),7.35-7.48(m,3H),7.96(d,J=S
Hz, 1 H) ppm.
MS(APCI+): 483.1 (M+1); LC-MS: 98%.
Example 317. Preparation of P-527
N
N, 0 F N- N
CI
P-527
[00786] Synthesis of 2-[l-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
1H-
pyrazol-4-yl]-pyridine (P-527). In an 8 mL vial equipped with a stir bar was
placed 2-
(1H-pyrazol-4-yl)-pyridine (31.4 mg, 0.216 mmol), DMF (600 L) and sodium
hydride
(60%)(11.9 mg, 0.296 mmol). The mixture was stirred at room temperature for 25
minutes and then a solution of 1-33 (75 mg, 0.228 mmol) and DMF (600 L) was
added
and the reaction mixture was stirred at room temperature for 18 hours. The
reaction was
quenched with water (40 mL) and extracted with dichloromethane (2 x 35 mL).
The
organic portions were combined, washed with brine (40 mL), dried (Mg504) and
concentrated. The residue was purified by silica gel column chromatography
utilizing
30% acetone/dichloromethane to produce 58 mg (65%) of P-527 as a colorless
viscous
oil.
386
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
[00787] 'H NMR (400 MHz, DMSO-d6) 6 3.75 (s, 3 H), 5.39 (s, 2 H), 7.00 (d, J=
9
Hz, 1 H), 7.15-7.18 (m, 1 H), 7.29-7.31 (m, 1 H), 7.36 (d, J= 9 Hz, 1 H), 7.39
(s, 1 H),
7.42-7.49 (m, 2 H), 7.65 (dt, J= 8, 1 Hz, 1 H), 7.74 (td, J= 8, 2 Hz, 1 H),
8.03 (s, 1 H),
8.36 (s, 1 H), 8.48-8.50 (m, 1 H) ppm. MS(APCI+): 394.1 (M+1); LC-MS: 92%.
Example 318. Preparation of P-528
~ N \ \ N
N-
CI
P-528
[00788] Synthesis of 4-[l-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
1H-
pyrazol-4-yl]-pyridine (P-528). In an 8 mL vial equipped with a stir bar was
placed 4-
(1H-pyrazol-4-yl)-pyridine (31.4 mg, 0.216 mmol), DMF (600 L) and sodium
hydride
(60%)(11.9 mg, 0.296 mmol). The mixture was stirred at room temperature for 25
minutes and then a solution of 1-33 (75 mg, 0.228 mmol) and DMF (600 L) was
added
and the reaction mixture was stirred at room temperature for 18 hours. The
reaction was
quenched with water (40 mL) and extracted with dichloromethane (2 x 35 mL).
The
organic portions were combined, washed with brine (40 mL), dried (MgSO4) and
concentrated. The residue was purified by silica gel column chromatography
utilizing
40% acetone/dichloromethane to produce 41 mg of P-528 as a white solid in 46%
yield.
[00789] 'H NMR (400 MHz, DMSO-d6) 6 3.75 (s, 3 H), 5.38 (s, 2 H), 7.00 (d, J=
8
Hz, 1 H), 7.30 (br d, J= 7 Hz, 1 H), 7.36 (d, J= 9 Hz, 1 H), 7.39 (s, 1 H),
7.42-7.48 (m,
2 H), 7.57-7.58 (m, 2 H), 8.09 (s, 1 H), 8.49 (m, 3 H) ppm.
MS(APCI+): 394.1 (M+1); LC-MS: 90%.
387
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 319. Preparation of P-544
N
N, 0 F N
N
CI
P-544
[00790] Synthesis of 2-[1-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
1H-
pyrazol-4-yl]-pyridine hydrochloride salt (P-544). In an 8 mL vial equipped
with a stir
bar was placed 2-(1H-pyrazol-3-yl)-pyridine (41.8 mg, 0.288 mmol), DMF (800
uL) and
sodium hydride (15.8 mg, 0.394 mmol). The reaction mixture was stirred at room
temperature for 25 minutes and then a solution of 1-33 (100 mg, 0.303 mmol) in
DMF
(800 uL) was added and the reaction mixture was stirred at room temperature
for 18
hours. The reaction was quenched with water (20 mL) and extracted with ethyl
acetate
(2 x 30 mL). The organic portions were combined, washed with brine (30 mL),
dried
(MgSO4) and concentrated. The crude material was purified by silica gel column
chromatography utilizing 15% acetone/dicholormethane as the eluent to produce
79 mg
as a colorless, viscous oil. The hydrochloride salt was formed by treated the
purified
product with 1,4-dioxane (500 uL) and 4.OM HC1 in 1,4-dioxane (500 uL). After
stirring
for 1 hour at room temperature the reaction mixture was concentrated and dried
in a high
vacuum oven set at 35 C for 18 hours to provide 80 mg of P-544 as a white
solid in 62%
yield. 1H NMR (400 MHz, DMSO-d6) 6 3.75 (s, 3 H), 5.49 (s, 2 H), 7.01 (d, J= 8
Hz, 1
H), 7.09 (s, 1 H), 7.30 (br d, J= 7 Hz, 1 H), 7.36-7.40 (m, 2 H), 7.43-7.49
(m, 2 H), 7.59
(br s, 1 H), 8.01 (d, J= 2 Hz, 1 H), 8.16 (bs, 2 H), 8.65 (d, J= 5 Hz, 1 H)
ppm.
MS(APCI+): 394.1 (M+1-HC1); LC-MS: >99%.
388
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
Example 320. Preparation of P-545
_N
O F N
CI
P-545
[00791] Synthesis of 2-[l-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
1H-
pyrazol-4-yl]-pyrazine (P-545). Same as procedure for P-544. Target compound
crashed out of DMF with addition of water (6 mL), collect solid, wash with
water (3 x 2
mL), dry in vacuum oven to afford 46 mg (35 %) of P-545 as an off-white solid.
1H
NMR (400 MHz, DMSO-d6) 6 3.75 (s, 3 H), 5.42 (s, 2 H), 7.01 (d, J= 8 Hz, 1 H),
7.30
(br d, J= 7 Hz, 1 H), 7.39 (t, J= 8 Hz, 2 H), 7.43-7.48 (m, 2 H), 8.14 (s, 1
H), 8.41 (d, J
= 2 Hz, 1 H), 8.50 (s, 1 H), 8.54-8.55 (m, 1 H), 8.99 (d, J= 2 Hz, 1 H) ppm.
MS(APCI+): 395.1 (M+1); LC-MS: 99%.
Example 321. Preparation of P-546
NI O F N
CI
P-546
[00792] Synthesis of 4-[l-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
1H-
pyrazol-4-yl]-pyrimidine hydrochloride salt (P-546). Same as procedure for P-
544.
Purification was performed using 30% acetone/dichloromethane as the eluent.
HC1 salt
formation provided 73 mg of P-546 HC1 salt as a white solid in 56% yield. 1H
NMR
(400 MHz, DMSO-d6) 6 3.75 (s, 3 H), 5.43 (s, 2 H), 7.01 (d, J= 8 Hz, 1 H),
7.30 (br d, J
= 7 Hz, 1 H), 7.39 (br s, 1 H), 7.40-7.49 (m, 3 H), 7.79 (dd, J= 6, 1 Hz, 1
H), 8.21 (s, 1
H), 8.61 (s, 1 H), 8.71 (br d, J= 5 Hz, 1 H), 9.07 (s, 1 H) ppm. MS(APCI+):
395.1
389
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
(M+1-HC1);
LC-MS: >99%.
Example 322. Preparation of P-549
N
i N x N
O F
CI
P-549
[00793] Synthesis of 3-[1-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
1H-
imidazol-2-yl]-pyridine hydrochloride salt (P-549). Same as procedure for P-
544.
Purification was performed using 5% methanol/dichloromethane as the eluent.
HC1 salt
formation provided 14 mg of P-549 as a pale yellow solid in 11% yield. 'H NMR
(400
MHz, DMSO-d6) 6 3.74 (s, 3 H), 5.47 (s, 2 H), 6.96 (d, J= 9 Hz, 1 H), 7.21-
7.23 (m, 1
H), 7.28-7.31 (m, 2 H), 7.44-7.46 (m, 2 H), 7.67-7.70 (m, 1 H), 7.88 (s, 2 H),
8.23 (dt, J
= 8, 2 Hz, 1 H), 8.85 (dd, J= 5, 2 Hz, 1 H), 8.94 (d, J= 2 Hz, 1 H) ppm.
MS(APCI+): 394.1 (M+1-HC1); LC-MS: >99%.
Example 323. Preparation of P-559
~7s
N Y
~N
O" F
i
\ Cl
P-559
[00794] Synthesis of 1-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-2-
thiophen-2-yl-1H-imidazole hydrochloride salt (P-559). Same as procedure for P-
544.
Purification performed using 5% methanol/dichloromethane as the eluent. HC1
salt
formation provided 61 mg of P-559 in 46% yield. 1H NMR (400 MHz, DMSO-d6) 6
3.75(s,3H),5.52(s,2H),6.84(d,J=9Hz,1H),7.18(t, J=8Hz,1H),7.27-7.28 (m,
390
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
1 H), 7.31-7.34 (m, 1 H), 7.37 (bs, 1 H), 7.44-7.49 (m, 2 H), 7.70-7.75 (m, 3
H), 8.00 (br
d, J= 4 Hz, 1 H) ppm. MS(APCI+): 399.1 (M+1-HC1); LC-MS: >99%.
Example 324. Preparation of P-551
F F
F
N
0 I / F N-
N
CI
P-551
[00795] Synthesis of 2-[l-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-5-
trifluoromethyl-lH-pyrazol-3-yl]-pyridine hydrochloride salt (P-551). Same as
procedure for P-544. Purification performed using 3% methanol/dichloromethane
as the
eluent. HC1 salt formation provided 49 mg of P-551 as a white solid in 32%
yield.
[00796] 1H NMR (400 MHz, DMSO-d6) 6 3.75 (s, 3 H), 5.58 (s, 2 H), 7.01 (d, J=
8
Hz, 1 H), 7.26 (d, J= 9 Hz, 1 H), 7.28-7.30 (m, 1 H), 7.38 (s, 1 H), 7.44-7.48
(m, 3 H),
7.50 (s, 1 H), 7.94-8.06 (m, 2 H), 8.64-8.66 (m, 1 H) ppm. MS(APCI+): 462.1
(M+1-
HC1);
LC-MS: >99%.
Example 325. Preparation of P-552
~N
CIH
N \
N
o F F F
CI
P-552
[00797] Synthesis of 3-[2-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-5-
trifluoromethyl-2H-pyrazol-3-yl]-pyridine hydrochloride salt (P-552). Same as
procedure for P-544. Purification performed using 20% acetone/dichloromethane
as the
391
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
eluent. HC1 salt formation provided 53 mg of P-552 as an off-white solid in
35% yield.
[00798] 'H NMR (400 MHz, DMSO-d6) 6 3.75 (s, 3 H), 5.58 (s, 2 H), 7.01 (d, J=
9
Hz, 1 H), 7.27-7.31 (m, 2 H), 7.37 (s, 1 H), 7.43-7.49 (m, 2 H), 7.72 (t, J= 8
Hz, 2 H),
8.48 (br d, J= 8 Hz, 1 H), 8.70 (dd, J= 5, 1 Hz, 1 H), 9.17 (d, J= 2 Hz, 1 H)
ppm.
MS(APCI+): 462.1 (M+1-HC1); LC-MS: 98%.
Example 326. Preparation of P-388
N N O rF O
O P NHz O P II NNH O ~NH
\ CI CI CI I
P-252 1-272 P-388
[00799] Synthesis of 1-(2-Chloro-ethyl)-3-[5-(3'-chloro-2-fluoro-6-methoxy-
biphenyl-
3-ylmethyl)-pyridin-2-yl]-urea (1-272).. To a solution of P-252 (180 mg, 0.525
mol) in
chloroform (4 mL) was added 2-chloroethylisocyanate (66.4 mg, 0.630 mmmol)
under
nitrogen. The solution was stirred at room temperature for 48 h. Additional 2-
chloroethylisocyanate (33.2 mg, 315 mmol) was added, and the reaction was
stirred at 80
C overnight. The solvent was removed under vacuum and the crude residue was
purified
by flash silica gel column chromatography (0-5% acetone in dichloromethane) to
give I-
272 (86.9 mg, 37% yield). 'H NMR (400 MHz, CDC13): 9.75 (s, 1H), 8.10 (m, 1H),
7.47-7.38(m,1H),7.38-7.26(m,4H),7.10(t,J=8.4Hz,1H),6.90(s,1H),6.72(d,J=
9.2 Hz, 1H), 6.58 (d, J = 8.4 Hz, 1H), 3.89 (s, 2H), 3.76 (s, 3H), 3.72-3.67
(m, 4H)
ppm.
LCMS = 55.1% purity. MS(APCI+) = 448.0 (M).
[00800] Synthesis of 1-[5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridin-2-yl]-imidazolidin-2-one (P-388). A suspension of 1-272 (85.0 mg,
0.190 mmol)
and sodium carbonate (60.3 mg, 0.569 mmol) in acetonitrile (2 mL) was stirred
at reflux
21 h. The reaction was cooled to room temperature and the sodium carbonate
removed
by filtration. The solid was washed with ethyl acetate (3 x 5 mL) and the
filtrate removed
under vacuum. The residue was purified by silica gel preparatory thin layer
392
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
chromatography eluting with 15% acetone in dichloromethane and using two
developments, triturated in diethyl ether (2 mL), and filtered to give P-388
(11.8 mg,
15% yield) as a red pink powder. 1H NMR (400 MHz, CDC13) d: 8.09 (s, 1H), 7.43-
7.28
(m, 5H), 7.08 (t, J = 8.6 Hz, 1H), 6.97 (d, J = 8.4 Hz, 1H), 6.69 (d, J = 8.8
Hz, 1H),
4.42 (t, J = 8.0 Hz, 2H), 3.88 (s, 2H), 3.82 (t, J = 7.8 Hz, 2H), 3.75 (s, 3H)
ppm. LCMS
= 95.9% purity. MS(APCI+) = 412.1 (M+1).
Example 327. Preparation of 1-275
O O~
fF
O
1-27
[00801] Synthesis of 5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridine-
2-carboxylic acid methyl ester (1-275). 1-275 was synthesized from I-145 (516
mg, 1.59
mmol) and 2-methylcarboxypyridine-5-boronic acid pinocol ester (460 mg, 1.75
mmol)
using the a similar proceedure to P-252. The crude material was purified by
flash silica
gel column chromatography (0-5% acetone in dichloromethane) to give 1-275 (381
mg,
62% yield) as an orange syrup. 1H NMR (400 MHz, CDC13): 8.65 (d, J = 2.4 Hz,
1H),
8.06 (d, J = 8.0 Hz, 1H), 7.66 (dd, J = 8.0 Hz, 2.0 Hz, 1H), 7.37-7.33 (m,
3H), 7.26-
7.24 (m, 1H), 7.11 (t, J = 8.6 Hz, 1H), 6.73 (dd, J = 8.6 Hz, 1.0 Hz, 1H),
4.04 (s, 2H),
4.00 (s, 3H), 3.77 (s, 3H) ppm. LCMS = 96.8% purity. MS(APCI+)= 386.0 (M+1).
Example 328. Preparation of 1-276
fF ~N
I / OH
O
O
1-276
[00802] Synthesis of 5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridine-
2-carboxylic acid (1-276). To a solution of 1-275 (317 mg, 0.822 mmol) in
393
CA 02722582 2010-10-26
WO 2009/067600 PCT/US2008/084193
tetrahydrofuran (2.2 mL), methanol (2.2 mL), and water (2.2 mL) was added 1 N
aqueous sodium hydroxide (1.64 mL, 1.64 mmol). The resultant solution was
stirred at
room temperature for 23 h. The pH was adjusted with glacial acetic acid to pH
4.
Approximately three-fourths of the solvent was removed under vacuum, and the
remaining suspension was diluted with water (10 mL). The suspension was
extracted
with dichloromethane (3 x 10 mL), and the combined extracts were dried over
magnesium sulfate, filtered, and the solvent removed under vacuum to give 1-
276 (250.8
mg, 82% yield) as a beige solid.
[00803] 'H NMR (400 MHz, CDC13): 8.50 (m, 1H), 8.13 (d, J = 7.6 Hz, 1H), 7.76-
7.71 (m, 1H), 7.37-7.32 (m, 3H), 7.26-7.24 (m, 1H), 7.13 (t, J = 8.6 Hz, 1H),
6.74 (d, J
8.8 Hz, 1H), 4.05 (s, 2H), 3.77 (s, 3H) ppm.MS(APCI+) = 327.2 (M-44), MS(APCI-
)=
327.9 (M-44).
Example 329. Preparation of P-382
IN NN
O I / F OH+ HNC - O I / F N
/ O / O
\ I CI \
CI
1-276 P-382
[00804] Synthesis of 5-(3'-Chloro-2-fluoro-6-methoxy-biphenyl-3-ylmethyl)-
pyridine-
2-carboxylic acid dimethylamide (P-382). A solution of 1-276 (50.0 mg, 0.134
mmol),
dimethyl amine (33% by weight in ethanol, 26.6 uL, 0.148 mmol), EDCI (28.4 mg,
0.148 mmol), and HOBt (20.0 mg, 0.148 mmol) was stirred at room temperature
for 36
h. The solvent was removed under vacuum and the residue dissolved in ethyl
acetate (5
mL). The organic solution was washed with water (5 mL), and the aqueous wash
extracted with ethyl acteate (5 mL). The organic extracts were combined,
washed with
aqueous saturated sodium bicarbonate (10 mL), water (5 mL) and brine (5 mL),
dried
over sodium sulfate, filtered, and the solvent removed under vacuum. The crude
yellow
oil was purified by silica gel preparatory thin layer chromatography
(dichloromethane)
and dried under high vacuum at room temperature for 4 h to give P-382 (28.6
mg, 54%
yield) as a clear syrup.' H NMR (400 MHz, CDC13): 8.48 (s, 1H), 7.63-7.58 (m,
2H),
7.39-7.28 (m, 4H), 7.12 (t, J= 8.4 Hz, 1H), 6.73 (d, J= 8.4 Hz, 1H), 4.00 (s,
2H), 3.770
394
DEMANDE OU BREVET VOLUMINEUX
LA PRRSENTE PARTIE DE CETTE DEMANDE OU CE BREVET COMPREND
PLUS D'UN TOME.
CECI EST LE TOME 1 DE 2
CONTENANT LES PAGES 1 A 394
NOTE : Pour les tomes additionels, veuillez contacter le Bureau canadien des
brevets
JUMBO APPLICATIONS/PATENTS
THIS SECTION OF THE APPLICATION/PATENT CONTAINS MORE THAN ONE
VOLUME
THIS IS VOLUME 1 OF 2
CONTAINING PAGES 1 TO 394
NOTE: For additional volumes, please contact the Canadian Patent Office
NOM DU FICHIER / FILE NAME:
NOTE POUR LE TOME / VOLUME NOTE: