Note: Descriptions are shown in the official language in which they were submitted.
CA 02729546 2010-12-24
COMPOUNDS AND COMPOSITIONS AS KINASE INHIBITORS
Technical Field
[0002] The invention relates to protein kinase inhibitors, more particularly
novel
pyrimidine derivatives and pharmaceutical compositions thereof, and their use
as
pharmaceuticals.
Background Art
[0003] Insulin-like growth factor (IGF-1) signaling is highly implicated in
cancer, with
the IGF-1 receptor (IGF-IR) as the predominating factor. IGR-1R is important
for tumor
transformation and survival of malignant cells, but is only partially involved
in normal cell
growth. Targeting of IGF-1R has been suggested to be a promising option for
cancer
therapy. (Larsson et al., Br. J. Cancer 92:2097-2101 (2005)).
[0004] Anaplastic lymphoma kinase (ALK), a member of the insulin receptor
superfamily of receptor tyrosine kinases, has been implicated in oncogenesis
in
hematopoietic and non-hematopoietic tumors. The aberrant expression of full-
length ALK
receptor proteins has been reported in neuroblastomas and glioblastomas; and
ALK fusion
proteins have occurred in anaplastic large cell lymphoma. The study of ALK
fusion
proteins has also raised the possibility of new therapeutic treatments for
patients with
ALK-positive malignancies. (Pulford et al., Cell. Mol. Life Sci. 61:2939-2953
(2004)).
[0005] Because of the emerging disease-related roles of IGF-lR and ALK, there
is a
continuing need for compounds which may be useful for treating and preventing
a disease
which responds to inhibition of IGF-1R and ALK.
Disclosure of the Invention
[0006] The invention relates to novel pyrimidine derivatives and
pharmaceutical
compositions thereof, and their use as pharmaceuticals.
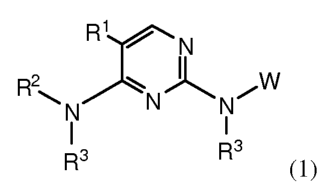
[0007] In one aspect, the invention provides a compound of Formula (1):
1
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
R1 N
R2 rW
~N N N
I
R3 R3
(1)
or a physiologically acceptable salt thereof;
I
X (R4)1-3
o
N
wherein W is \\(R4)m, R5 or W';
W' is pyridyl, isoquinolinyl, quinoliny, naphthalenyl, cinnolin-5-yl or [3-(C1-
6
alkyl)-(2,3,4,5-tetrahydro-1H-benzo[d]azepin-7y1], each of which is optionally
substituted with 1-3 R9; and said pyridyl, isoquinolinyl, quinolinyl and
napthalenyl
NR6
are each substituted on a ring carbon with
X is -C(R)=N-O-R7, C(O)NRR7, C(O)NR-(CR2)õ-NRR7, -(CR2)pNRR wherein
two R groups together with N in NRR form a 5-6 membered ring containing 1-3
heteroatoms selected from N, 0 and S and optionallysubstituted with 1-3 R9, or
a C5-7
carbocycle optionally substituted with oxo, =N-OH or R9; or X is quinolinyl,
(1,2,3,4-
tetrahydroisoquinolin-6-yl) or a 5-6 membered heteroaryl having 1-3
heteroatoms
selected from N, 0 and S, each of which is optionally substituted with 1-3 R9;
R1 is halo, Cl-6 alkyl, or a halo-substituted C1-6 alkyl;
R2 is a 5-6 membered heteroaryl having 1-3 heteroatoms selected from N, 0
and S, and is optionally substituted with C1-6 alkyl, C1-6 haloalkyl or C3-7
cycloalkyl;
each R3 is H;
R4 is halo, hydroxyl, C1-6 alkyl, C1-6 alkoxy, halo-substituted C1-6 alkyl,
halo-
substituted C1-6 alkoxy, cyano or C(O)Oo-1R8;
N R 6
R5isHor
R6 is H, Cl-6 alkyl, C2-6 alkenyl or C2-6 alkynyl, each of which may be
optionally substituted with halo and/or hydroxyl groups; -(CR2)p-OR 7, -(CR2)p-
2
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
CH(OH)C1F2t+1 wherein t is 1-3, (CR2)p-CN; (CR2)p-NR(R7), -(CR2)p C(O)OR7,
(CR2)pNR(CR2)pOR7,
(CR2)pNR-L-C(O)R8, C(O)(CR2)gOR8, -C(O)O-(CR2)p-NRR7, -C(O) -(CR2)p-
OR7, L-Y,
-L-C(O)R7, -L-C(O)-NRR7, -L-C(O)-NR-(CR2)p-NRR7,-L-
C(O)NR(CR2)pOR7,
-L-C(O)-(CR2)g-NR-C(O)-R8, -L-C(O)NR(CR2)pSR7, -L-
C(O)NR(CR2)pS(O)1-2R8,
-L-S(O)2R8, -L-S(0)2-(CR2)g-NRR7, -L-S(O)2NR(CR2)pNR(R7) or
-L-S(O)2NR(CR2)pOR7;
alternatively, R6 is a radical selected from formula (a), (b), (c) or (d):
-2 R11 R12 R15 12 R11 R12 R15
S N- R16 SO2 L N- R16
o-1 `S~ 0-1
-2 R13 R14 1-2 // % R13 R14
(a) O (b) (c) O O (d)
R10 is 0, S, NR17 wherein R17 is H, C1-6 alkyl, SO2R8a or CO2R8a;
R11 R12 R13 R14 R15 and R16 are independently selected from H; C1-6 alkoxy;
C1-6 alkyl, C2-6 alkenyl or C2-6 alkynyl, each of which may be optionally
substituted
with halo, amino or hydroxyl groups; or R11 and R12, R12 and R15 R15 and R16
R13
and R14, or R13 and R15 together with the atoms to which they are attached may
form a
3-7 membered saturated, unsaturated or partially unsaturated ring containing 1-
3
heteroatoms selected from N, 0 and S, and optionally substituted with oxo and
1-3 R9
groups;
L is (CR2)1-4 or a bond;
Y is C3-7 carbocyclic ring, C6-1o aryl, or a 5-10 membered heteroaryl or 4-10
membered heterocyclic ring, each of which is optionally substituted with 1-3
R9
groups;
R7, R8 and Rga are independently C1-6 alkyl, C2-6 alkenyl or C2-6 alkynyl,
each
of which may be optionally substituted with halo, NRR7a, hydroxyl or cyano;
(CR2)gY
or C1-6 alkoxy; or R7 is H;
R9 is R4, C(O)NRR7 or NRR7;
R and R7a are independently H or C1-6 alkyl;
3
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
R and R7 together with N in each NRR7, and R and R7a together with N in
NRR7a may form a 5-6 membered ring containing 1-3 heteroatoms selected from N,
0
and S, and optionally substituted with oxo and 1-3 R4 groups;
m is 2-4;
n and p are independently 1-4; and
q is 0-4.
[0008] In the above Formula (1), R2 may be pyrazolyl or isoxazolyl, each of
which is substituted with C1_6 alkyl or C3_7 cycloalkyl.
[0009] In one embodiment, the invention provides a compound of Formula (2):
- N R1 /
HN N R3 R3
(2)
wherein W is W';
W' is pyridyl optionally substituted with C1_6 alkyl, isoquinolinyl,
quinolinyl,
naphthalenyl, cinnolin-5-yl optionally substituted with C1_6 alkyl or [3-(C1_6
alkyl)-
(2,3,4,5-tetrahydro-1H-benzo[d]azepin-7y1]; and said pyridyl, isoquinolinyl,
N R6
quinolinyl and napthalenyl are each substituted on a ring carbon with
R6 is H, C1_6 alkyl, C2_6 alkenyl or C2_6 alkynyl, each of which may be
optionally substituted with halo, amino, hydroxyl or alkoxy; -(CR2)p-
CH(OH)CrF2t+i
wherein t is 1,
-L-C(O)-NRR7 or -L-S(O)2R8
L is (CR2)1_4;
R and R7 are independently H or C1.6 alkyl;
R8 is C1.6 alkyl; and
R1 and R3 are as defined in Formula (1).
[0010] In another embodiment, the invention provides a compound of Formula
(3):
4
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
Z-N R1 N
R3
N N N
R3 R4
N
R5
(3)
wherein Z is NH or 0;
R4 is halo or C1_6 alkyl;
N R6
R5 is H or R6 is H; and
R1 and R3 are as defined in Formula (1).
[0011] In yet another embodiment, the invention provides a compound of Formula
(4):
R4c
N-N R1 / N I
N R4b
A"K N
R3 R3 R4a 4
O
wherein one of R4a R4b and R4c is H and the others are independently halo, C1-
6 alkyl, C1.6 alkoxy, halo-substituted C1.6 alkyl or halo-substituted C1.6
alkoxy; and
X is as defined in Formula (1).
[0012] In the above Formula (4), X may be -C(R)=N-0-R7, C(O)NRR7, C(O)NR-
(CR2)õ-NRR7 or -(CR2)pNRR wherein two R groups together with N in NRR form
morpholinyl;
R7 is H or C1.6 alkyl optionally substituted with hydroxyl or NRR7a;
each R is H or C1_6 alkyl;
R and R7 together with N in each NRR7 and R and R7a together with N in
NRR7a may form a 5-6 membered ring containing 1-2 heteroatoms selected from N,
0
and S; and n and p are independently 1-4. Alternatively, X may be quinolinyl,
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
(1,2,3,4-tetrahydroisoquinolin-6-yl) or a 5-6 membered heteroaryl selected
from
pyrazolyl, pyridyl, thiophenyl, furanyl, imidazolyl, isoxazolyl, oxazolyl or
thiaxolyl,
each of which is optionally substituted with C1_6 alkyl, hydroxyl, or
C(O)NRR7; R7 is
H or C1_6 alkyl; and R is H or C1_6 alkyl.
[0013] In another embodiment, the invention provides a compound of Formula
(5):
R4c
E
N -N R1~ IIN OP, 'N N
Z~t!' IC
N R4b
R3 R3 R4a 5
wherein one of R4a R4b and R4c is H and the others are independently halo, C1-
6 alkyl, C1_6 alkoxy, halo-substituted C1_6 alkyl or halo-substituted C1_6
alkoxy;
Ring E is a C6 carbocycle optionally substituted with oxo, =N-OH or R9;
R9 is hydroxyl or NRR';
R is H or C1.6 alkyl;
R7 is C1_6 alkyl or (CR2)qY and Y is C3 cycloalkyl;
alternatively, R and R7 together with N in NRR7 forms morpholinyl,
piperidinyl, piperazinyl, (C1.6 alkyl)-piperazinyl, or pyrrolidinyl, each of
which is
optionally substituted with hydroxyl; and
R1 and R3 are as defined in Formual (1).
[0014] In the above Formula (4) and (5), R4b may be H. In other examples, R4a
and R4c are independently halo, C1.6 alkyl, C1.6 alkoxy, halo-substituted C1.6
alkyl or
halo-substituted C1.6 alkoxy.
[0015] In the above Formula (1)-(5), R1 is chloro or a halo-substituted C1_6
alkyl.
In other examples, R3 is H.
[0016] In another aspect, the present invention provides pharmaceutical
compositions comprising a compound having Formula (1), (2), (3), (4) or (5),
and a
physiologically acceptable excipient.
[0017] In yet another aspect, the invention provides methods for inhibiting
IGF-
1R in a cell, comprising contacting the cell with an effective amount of a
compound
having Formula (1), (2), (3), (4) or (5) or a pharmaceutical composition
thereof.
6
CA 02729546 2010-12-24
[0018] The invention also provides methods to treat, ameliorate or prevent a
condition
which responds to inhibition of IGF-IR or anaplastic lymphoma kinase (ALK) in
a
mammal suffering from said condition, comprising administering to the mammal a
therapeutically effective amount of a compound having Formula (1), (2), (3),
(4) or (5) or a
pharmaceutical composition thereof, and optionally in combination with a
second
therapeutic agent. Alternatively, the present invention provides for the use
of a compound
having Formula (1), (2), (3), (4) or (5), and optionally in combination with a
second
therapeutic agent, for treating a condition mediated by IGF-1R or ALK and for
manufacture of a medicament for such treating. The compounds of the invention
may be
administered, for example, to a mammal suffering from an autoimmune disease, a
transplantation disease, an infectious disease or a cell proliferative
disorder. In particular
examples, the compounds of the invention may be used alone or in combination
with a
chemotherapeutic agent to treat a cell proliferative disorder, including but
not limited to,
multiple myeloma, neuroblastoma, synovial, hepatocellular, Ewing's Sarcoma or
a solid
tumor selected from a osteosarcoma, melanoma, and tumor of breast, renal,
prostate,
colorectal, thyroid, ovarian, pancreatic, lung, uterine or gastrointestinal
tumor.
Definitions
[0019] "Alkyl" refers to a moiety and as a structural element of other groups,
for
example halo-substituted-alkyl and alkoxy, and may be straight-chained or
branched. An
optionally substituted alkyl, alkenyl or alkynyl as used herein may be
optionally
halogenated (e.g., CF3), or may have one or more carbons that is substituted
or replaced
with a heteroatom, such as NR, 0 or S (e.g., -OCH2CH2O-, alkylthiols,
thioalkoxy,
alkylamines, etc).
[0020] "Aryl" refers to a monocyclic or fused bicyclic aromatic ring
containing carbon
atoms. "Arylene" means a divalent radical derived from an aryl group. For
example, an
aryl group may be phenyl, indenyl, indanyl, naphthyl, or 1,2,3,4-
tetrahydronaphthalenyl,
which may be optionally substituted in the ortho, meta or para position.
[0021] "Heteroaryl" as used herein is as defined for aryl above, where one or
more of
the ring members is a heteroatom. For example, a heteroaryl substituent for
use in the
compounds of the invention may be a monocyclic or bicyclic 5-10 membered
heteroaryl
containing 1-4 heteroatoms selected from N, 0, and S.
7
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
Examples of heteroaryls include but are not limited to pyridyl, pyrazinyl,
indolyl,
indazolyl, quinoxalinyl, quinolinyl, benzofuranyl, benzopyranyl,
benzothiopyranyl,
benzo[1,3]dioxole, imidazolyl, benzo-imidazolyl, pyrimidinyl, furanyl,
oxazolyl,
isoxazolyl, triazolyl, benzotriazolyl, tetrazolyl, pyrazolyl, thienyl,
pyrrolyl,
isoquinolinyl, purinyl, thiazolyl, tetrazinyl, benzothiazolyl, oxadiazolyl,
benzoxadiazolyl, etc.
[0022] A "carbocyclic ring" as used herein refers to a saturated or partially
unsaturated, monocyclic, fused bicyclic or bridged polycyclic ring containing
carbon
atoms, which may optionally be substituted, for example, with =0. Examples of
carbocyclic rings include but are not limited to cyclopropyl, cyclobutyl,
cyclopentyl,
cyclohexyl, cyclopropylene, cyclohexanone, etc.
[0023] A "heterocyclic ring" as used herein is as defined for a carbocyclic
ring
above, wherein one or more ring carbons is a heteroatom. For example, a
heterocyclic
ring for use in the compounds of the invention may be a 4-7 membered
heterocyclic
ring containing 1-3 heteroatoms selected from N, 0 and S, or a combination
thereof
such as -S(O) or -S(0)2-. Examples of heterocyclic rings include but are not
limited
to azetidinyl, morpholino, pyrrolidinyl, pyrrolidinyl-2-one, piperazinyl,
piperidinyl,
piperidinylone, 1,4-dioxa-8-aza-spiro[4.5]dec-8-yl, 1,2,3,4-
tetrahydroquinolinyl, etc.
Heterocyclic rings as used herein may encompass bicyclic amines and bicyclic
diamines.
[0024] As used herein, an H atom in any substituent groups (e.g., CH2)
encompasses all suitable isotopic variations, e.g., H, 2H and 3H.
[0025] The term "pharmaceutical combination" as used herein refers to a
product
obtained from mixing or combining active ingredients, and includes both fixed
and
non-fixed combinations of the active ingredients. The term "fixed combination"
means that the active ingredients, e.g. a compound of Formula (1) and a co-
agent, are
both administered to a patient simultaneously in the form of a single entity
or dosage.
The term "non-fixed combination" means that the active ingredients, e.g. a
compound
of Formula (1) and a co-agent, are both administered to a patient as separate
entities
either simultaneously, concurrently or sequentially with no specific time
limits,
wherein such administration provides therapeutically effective levels of the
active
ingredients in the body of the patient. The latter also applies to cocktail
therapy, e.g.
the administration of three or more active ingredients.
8
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
[0026] "Mammal" refers to any animal classified as a mammal, including humans,
domestic and farm animals, and zoo, sports, or pet animals, such as dogs,
cats, cattle,
horses, sheep, pigs, goats, rabbits, etc. In particular examples, the mammal
is human.
[0027] The term "administration" or "administering" of the subject compound
means providing a compound of the invention and prodrugs thereof to a subject
in
need of treatment. Administration "in combination with" one or more further
therapeutic agents includes simultaneous (concurrent) and consecutive
administration
in any order, and in any route of administration.
[0028] An "effective amount" of a compound is an amount sufficient to carry
out
a specifically stated purpose. An "effective amount" may be determined
empirically
and in a routine manner, in relation to the stated purpose.
[0029] The term "therapeutically effective amount" refers to an amount of a
compound (e.g., an IGF-1R antagonist) effective to "treat" an IGF-1R-mediated
disorder in a subject or mammal. In the case of cancer, the therapeutically
effective
amount of the drug may reduce the number of cancer cells; reduce the tumor
size;
inhibit (i.e., slow to some extent and preferably stop) cancer cell
infiltration into
peripheral organs; inhibit (i.e., slow to some extent and preferably stop)
tumor
metastasis; inhibit, to some extent, tumor growth; and/or relieve to some
extent one or
more of the symptoms associated with the cancer. See the definition herein of
"treating". To the extent the drug may prevent growth and/or kill existing
cancer cells,
it may be cytostatic and/or cytotoxic.
[0030] The term "cancer" refers to the physiological condition in mammals that
is
typically characterized by unregulated cell growth/proliferation. Examples of
cancer
include, but are not limited to: carcinoma, lymphoma, blastoma, and leukemia.
More
particular examples of cancers include, but are not limited to: chronic
lymphocytic
leukemia (CLL), lung, including non small cell (NSCLC), breast, ovarian,
cervical,
endometrial, prostate, colorectal, intestinal carcinoid, bladder, gastric,
pancreatic,
hepatic (hepatocellular), hepatoblastoma, esophageal, pulmonary
adenocarcinoma,
mesothelioma, synovial sarcoma, osteosarcoma, head and neck squamous cell
carcinoma, juvenile nasopharyngeal angiofibromas, liposarcoma, thyroid,
melanoma,
basal cell carcinoma (BCC), medulloblastoma and desmoid.
[0031] "Treating" or "treatment" or "alleviation" refers to both therapeutic
treatment and prophylactic or preventative measures, wherein the object is to
prevent
9
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
or slow down (lessen) the targeted pathologic disease or condition or
disorder. Those
in need of treatment include those already with the disorder as well as those
prone to
having the disorder or those in whom the disorder is to be prevented
(prophylaxis).
When the IGF-1R-mediated disorder is cancer, a subject or mammal is
successfully
"treated" or shows a reduced tumor burden if, after receiving a therapeutic
amount of
an IGF-1R antagonist according to the methods of the present invention, the
patient
shows observable and/or measurable reduction in or absence of one or more of
the
following: reduction in the number of cancer cells or absence of the cancer
cells;
reduction in the tumor size; inhibition (i.e., slow to some extent and
preferably stop)
of cancer cell infiltration into peripheral organs including the spread of
cancer into
soft tissue and bone; inhibition (i.e., slow to some extent and preferably
stop) of tumor
metastasis; inhibition, to some extent, of tumor growth; and/or relief to some
extent,
one or more of the symptoms associated with the specific cancer; reduced
morbidity
and mortality, and improvement in quality of life issues. To the extent the
IGF-1R
antagonist may prevent growth and/or kill existing cancer cells, it may be
cytostatic
and/or cytotoxic. Reduction of these signs or symptoms may also be felt by the
patient.
[0032] "Carriers" as used herein include pharmaceutically acceptable carriers,
excipients, or stabilizers which are nontoxic to the cell or mammal being
exposed
thereto at the dosages and concentrations employed. Often the physiologically
acceptable carrier is an aqueous pH buffered solution. Examples of
physiologically
acceptable carriers include buffers such as phosphate, citrate, and other
organic acids;
antioxidants including ascorbic acid; low molecular weight (less than about 10
residues) polypeptide; proteins, such as serum albumin, gelatin, or
immunoglobulins;
hydrophilic polymers such as polyvinylpyrrolidone; amino acids such as
glycine,
glutamine, asparagine, arginine or lysine; monosaccharides, disaccharides, and
other
carbohydrates including glucose, mannose, or dextrins; chelating agents such
as
EDTA; sugar alcohols such as mannitol or sorbitol; salt-forming counterions
such as
sodium; and/or nonionic surfactants such as TWEEN , polyethylene glycol (PEG),
and PLURONICS .
[0033] A "chemotherapeutic agent" is a chemical compound useful in the
treatment of cancer. Examples of chemotherapeutic agents include alkylating
agents
such as thiotepa and CYTOXAN cyclosphosphamide; alkyl sulfonates such as
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
busulfan, improsulfan and piposulfan; aziridines such as benzodopa,
carboquone,
meturedopa, and uredopa; ethylenimines and methylamelamines including
altretamine, triethylenemelamine, trietylenephosphoramide,
triethiylenethiophosphoramide and trimethylolomelamine; acetogenins
(especially
bullatacin and bullatacinone); delta-9-tetrahydrocannabinol (dronabinol,
MARINOL ); beta-lapachone; lapachol; colchicines; betulinic acid; a
camptothecin
(including the synthetic analogue topotecan (HYCAMTIN ), CPT-11 (irinotecan,
CAMPTOSAR ), acetylcamptothecin, scopolectin, and 9-aminocamptothecin);
bryostatin; callystatin; CC-1065 (including its adozelesin, carzelesin and
bizelesin
synthetic analogues); podophyllotoxin; podophyllinic acid; teniposide;
cryptophycins
(particularly cryptophycin 1 and cryptophycin 8); dolastatin; duocarmycin
(including
the synthetic analogues, KW-2189 and CB1-TM1); eleutherobin; pancratistatin; a
sarcodictyin; spongistatin; nitrogen mustards such as chlorambucil,
chlornaphazine,
cholophosphamide, estramustine, ifosfamide, mechlorethamine, mechlorethamine
oxide hydrochloride, melphalan, novembichin, phenesterine, prednimustine,
trofosfamide, uracil mustard; nitrosureas such as carmustine, chlorozotocin,
fotemustine, lomustine, nimustine, and ranimnustine; antibiotics such as the
enediyne
antibiotics (e.g., calicheamicin, especially calicheamicin gamma 11 and
calicheamicin
omegall (see, e.g., Agnew, Chem Intl. Ed. Engl., 33: 183-186 (1994));
dynemicin,
including dynemicin A; an esperamicin; as well as neocarzinostatin chromophore
and
related chromoprotein enediyne antiobiotic chromophores), aclacinomysins,
actinomycin, authramycin, azaserine, bleomycins, cactinomycin, carabicin,
caminomycin, carzinophilin, chromomycinis, dactinomycin, daunorubicin,
detorubicin, 6-diazo-5-oxo-L-norleucine, ADRIAMYCIN doxorubicin (including
morpholino-doxorubicin, cyanomorpholino-doxorubicin, 2-pyrrolino-doxorubicin
and
deoxydoxorubicin), epirubicin, esorubicin, idarubicin, marcellomycin,
mitomycins
such as mitomycin C, mycophenolic acid, nogalamycin, olivomycins, peplomycin,
potfiromycin, puromycin, quelamycin, rodorubicin, streptonigrin, streptozocin,
tubercidin, ubenimex, zinostatin, zorubicin; anti-metabolites such as
methotrexate and
5-fluorouracil (5-FU); folic acid analogues such as denopterin, methotrexate,
pteropterin, trimetrexate; purine analogs such as fludarabine, 6-
mercaptopurine,
thiamiprine, thioguanine; pyrimidine analogs such as ancitabine, azacitidine,
6-
azauridine, carmofur, cytarabine, dideoxyuridine, doxifluridine, enocitabine,
11
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
floxuridine; androgens such as calusterone, dromostanolone propionate,
epitiostanol,
mepitiostane, testolactone; anti-adrenals such as aminoglutethimide, mitotane,
trilostane; folic acid replenisher such as frolinic acid; aceglatone;
aldophosphamide
glycoside; aminolevulinic acid; eniluracil; amsacrine; bestrabucil;
bisantrene;
edatraxate; defofamine; demecolcine; diaziquone; elfomithine; elliptinium
acetate; an
epothilone; etoglucid; gallium nitrate; hydroxyurea; lentinan; lonidainine;
maytansinoids such as maytansine and ansamitocins; mitoguazone; mitoxantrone;
mopidanmol; nitraerine; pentostatin; phenamet; pirarubicin; losoxantrone; 2-
ethylhydrazide; procarbazine; PSK polysaccharide complex (JHS Natural
Products,
Eugene, Oreg.); razoxane; rhizoxin; sizofuran; spirogermanium; tenuazonic
acid;
triaziquone; 2,2',2"-trichlorotriethylamine; trichothecenes (especially T-2
toxin,
verracurin A, roridin A and anguidine); urethan; vindesine (ELDISINE ,
FILDESIN ); dacarbazine; mannomustine; mitobronitol; mitolactol; pipobroman;
gacytosine; arabinoside ("Ara-C"); thiotepa; taxoids, e.g., TAXOL paclitaxel
(Bristol-Myers Squibb Oncology, Princeton, N.J.), ABRAXANETM Cremophor-free,
albumin-engineered nanoparticle formulation of paclitaxel (American
Pharmaceutical
Partners, Schaumberg, Ill.), and TAXOTERE doxetaxel (Rhone-Poulenc Rorer,
Antony, France); chloranbucil; gemcitabine (GEMZAR ); 6-thioguanine;
mercaptopurine; methotrexate; platinum analogs such as cisplatin and
carboplatin;
vinblastine (VELBAN ); platinum; etoposide (VP-16); ifosfamide; mitoxantrone;
vincristine (ONCOVIN ); oxaliplatin; leucovovin; vinorelbine (NAVELBINE );
novantrone; edatrexate; daunomycin; aminopterin; ibandronate; topoisomerase
inhibitor RFS 2000; difluoromethylornithine (DMFO); retinoids such as retinoic
acid;
capecitabine (XELODA ); pharmaceutically acceptable salts, acids or
derivatives of
any of the above; as well as combinations of two or more of the above such as
CHOP,
an abbreviation for a combined therapy of cyclophosphamide, doxorubicin,
vincristine, and prednisolone, and FOLFOX, an abbreviation for a treatment
regimen
with oxaliplatin (ELOXATINTM) combined with 5-FU and leucovovin.
[0034] Furthermore, a "chemotherapeutic agent" may include anti-hormonal
agents that act to regulate, reduce, block, or inhibit the effects of hormones
that can
promote the growth of cancer, and are often in the form of systemic, or whole-
body
treatment. They may be hormones themselves. Examples include anti-estrogens
and
selective estrogen receptor modulators (SERMs), including, for example,
tamoxifen
12
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
(including NOLVADEX tamoxifen), EVISTA raloxifene, droloxifene, 4-
hydroxytamoxifen, trioxifene, keoxifene, LY117018, onapristone, and FARESTON
toremifene; anti-progesterones; estrogen receptor down-regulators (ERDs);
agents that
function to suppress or shut down the ovaries, for example, leutinizing
hormone-
releasing hormone (LHRH) agonists such as LUPRON and ELIGARD leuprolide
acetate, goserelin acetate, buserelin acetate and tripterelin; other anti-
androgens such
as flutamide, nilutamide and bicalutamide; and aromatase inhibitors that
inhibit the
enzyme aromatase, which regulates estrogen production in the adrenal glands,
such as,
for example, 4(5)-imidazoles, aminoglutethimide, MEGASE megestrol acetate,
AROMASIN exemestane, formestanie, fadrozole, RIVISOR vorozole,
FEMARA letrozole, and ARIMIDEX anastrozole. In addition, such definition of
chemotherapeutic agents includes bisphosphonates such as clodronate (for
example,
BONEFOS or OSTAC ), DIDROCAL etidronate, NE-58095, ZOMETA
zoledronic acid/zoledronate, FOSAMAX alendronate, AREDIA pamidronate,
SKELID tiludronate, or ACTONEL risedronate; as well as troxacitabine (a 1,3-
dioxolane nucleoside cytosine analog); antisense oligonucleotides,
particularly those
that inhibit expression of genes in signaling pathways implicated in abherant
cell
proliferation, such as, for example, PKC-alpha, Raf, H-Ras, and epidermal
growth
factor receptor (EGF-R); vaccines such as THERATOPE vaccine and gene therapy
vaccines, for example, ALLOVECTIN vaccine, LEUVECTIN vaccine, and
VAXID vaccine; LURTOTECAN topoisomerase 1 inhibitor; ABARELIX
rmRH; lapatinib ditosylate (an ErbB-2 and EGFR dual tyrosine kinase small-
molecule
inhibitor also known as GW572016); and pharmaceutically acceptable salts,
acids or
derivatives of any of the above.
Modes of Carrying Out the Invention
[0035] The invention provides novel pyrimidine derivatives and pharmaceutical
compositions thereof, and methods for using such compounds.
[0036] In one aspect, the invention provides a compound of Formula (1):
R1 N
R2 rW
~N N N
I
R3 R3
(1)
13
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
or a physiologically acceptable salt thereof;
JwIv
x I (R4)1-3
~
N
wherein W is \\(R 4)m, R5 or W';
W' is pyridyl, isoquinolinyl, quinoliny, naphthalenyl, cinnolin-5-yl or [3-(C1-
6
alkyl)-(2,3,4,5-tetrahydro-1H-benzo[d]azepin-7y1], each of which is optionally
substituted with 1-3 R9; and said pyridyl, isoquinolinyl, quinolinyl and
napthalenyl
NR6
are each substituted on a ring carbon with
X is -C(R)=N-O-R7, C(O)NRR7, C(O)NR-(CR2)õ-NRR7, -(CR2)pNRR wherein
two R groups together with N in NRR form a 5-6 membered ring containing 1-3
heteroatoms selected from N, 0 and S and optionallysubstituted with 1-3 R9, or
a C5-7
carbocycle optionally substituted with oxo, =N-OH or R9; or X is quinolinyl,
(1,2,3,4-
tetrahydroisoquinolin-6-yl) or a 5-6 membered heteroaryl having 1-3
heteroatoms
selected from N, 0 and S, each of which is optionally substituted with 1-3 R9;
R1 is halo, C1-6 alkyl, or a halo-substituted C1-6 alkyl;
R2 is a 5-6 membered heteroaryl having 1-3 heteroatoms selected from N, 0
and S, and is optionally substituted with C1-6 alkyl, C1-6 haloalkyl or C3-7
cycloalkyl;
each R3 is H;
R4 is halo, hydroxyl, C1-6 alkyl, C1-6 alkoxy, halo-substituted C1-6 alkyl,
halo-
substituted C1-6 alkoxy, cyano or C(O)Oo-1R8;
N R 6
R5 is H or R6 is H, Cl-6 alkyl, C2-6 alkenyl or C2-6 alkynyl, each of which
may be
optionally substituted with halo and/or hydroxyl groups; -(CR2)p-OR 7, -(CR2)p-
CH(OH)CrF2t+1 wherein t is 1-3, (CR2)p-CN; (CR2)p-NR(R7), -(CR2)p-C(O)OR7,
(CR2)pNR(CR2)pOR7,
(CR2)pNR-L-C(O)R8, C(O)(CR2)gOR8, -C(0)0-(CR2)p-NRR7, -C(O) -(CR2)p-
OR7, L-Y,
14
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
-L-C(O)R7, -L-C(O)-NRR7, -L-C(O)-NR-(CR2)p-NRR7,-L-
C(O)NR(CR2)pOR7,
-L-C(O)-(CR2)q-NR-C(O)-R8, -L-C(O)NR(CR2)pSR7, -L-
C(O)NR(CR2)pS(O)1-2R8,
-L-S(O)2R8, -L-S(0)2-(CR2)q-NRR7, -L-S(O)2NR(CR2)pNR(R7) or
-L-S(O)2NR(CR2)pOR7;
alternatively, R6 is a radical selected from formula (a), (b), (c) or (d):
-2 R11 R12 R15 12 R11 R12 R15
S N-R16 SO2 L N-R16
o-1 `S~ 0-1
-2 R13 R14 1-2 // % R13 R14
(a) O (b) (c) O O (d)
R10 is 0, S, NR17 wherein R17 is H, C1-6 alkyl, SO2R8a or CO2R8a;
R11 R12 R13 R14 R15 and R16 are independently selected from H; C1-6 alkoxy;
C1-6 alkyl, C2-6 alkenyl or C2-6 alkynyl, each of which may be optionally
substituted
with halo, amino or hydroxyl groups; or R11 and R12, R12 and R15 R15 and R16
R13
and R14, or R13 and R15 together with the atoms to which they are attached may
form a
3-7 membered saturated, unsaturated or partially unsaturated ring containing 1-
3
heteroatoms selected from N, 0 and S, and optionally substituted with oxo and
1-3 R9
groups;
L is (CR2)1-4 or a bond;
Y is C3-7 carbocyclic ring, C6-1o aryl, or a 5-10 membered heteroaryl or 4-10
membered heterocyclic ring, each of which is optionally substituted with 1-3
R9
groups;
R7, R8 and R8a are independently C1-6 alkyl, C2-6 alkenyl or C2-6 alkynyl,
each
of which may be optionally substituted with halo, NRR7a, hydroxyl or cyano;
(CR2)gY
or C1-6 alkoxy; or R7 is H;
R9 is R4, C(O)NRR7 or NRR7;
R and R7a are independently H or C1-6 alkyl;
R and R7 together with N in each NRR7, and R and R7a together with N in
NRR7a may form a 5-6 membered ring containing 1-3 heteroatoms selected from N,
0
and S, and optionally substituted with oxo and 1-3 R4 groups;
m is 2-4;
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
n and p are independently 1-4; and
q is 0-4.
[0037] In one embodiment, the invention provides a compound of Formula (2):
HN-N XN ,~!I~
N N -~' 1
R3 R3
(2)
wherein W is W';
W' is pyridyl optionally substituted with C1_6 alkyl, isoquinolinyl,
quinolinyl,
naphthalenyl, cinnolin-5-yl optionally substituted with C1_6 alkyl or [3-(C1.6
alkyl)-
(2,3,4,5-tetrahydro-1H-benzo[d]azepin-7y1]; and said pyridyl, isoquinolinyl,
N R6
quinolinyl and napthalenyl are each substituted on a ring carbon with
R6 is H, C1_6 alkyl, C2_6 alkenyl or C2_6 alkynyl, each of which may be
optionally substituted with halo, amino, hydroxyl or alkoxy; -(CR2)p-
CH(OH)C,F2 +1
wherein t is 1,
-L-C(O)-NRR7 or -L-S(O)2R8
L is (CR2)1_4;
R and R7 are independently H or C1_6 alkyl;
R8 is C1_6 alkyl; and
R1 and R3 are as defined in Formula (1).
[0038] In another embodiment, the invention provides a compound of Formula
(3):
Z-N R1
N
R3
N N N
R3 R4
N
R5
(3)
16
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
wherein Z is NH or 0;
R4 is halo or C1_6 alkyl;
N R6
R5 is H or R6 is H; and
R1 and R3 are as defined in Formula (1).
[0039] In yet another embodiment, the invention provides a compound of Formula
(4):
R4c
N-N R1 / N I
N R4b
A"K N
R3 R3 R4a 4
O
wherein one of R4a R4b and R4c is H and the others are independently halo, C1-
6 alkyl, C1.6 alkoxy, halo-substituted C1.6 alkyl or halo-substituted C1.6
alkoxy; and
X is as defined in Formula (1).
[0040] In another embodiment, the invention provides a compound of Formula
(5):
R4c
E
N -N R1~ IIN
Z~t!' IC
N R4b
'N N
R3 R3 R4a 5
wherein one of R4a R4b and R4c is H and the others are independently halo, C1-
6 alkyl, C1_6 alkoxy, halo-substituted C1_6 alkyl or halo-substituted C1_6
alkoxy;
Ring E is a C6 carbocycle optionally substituted with oxo, =N-OH or R9;
R9 is hydroxyl or NRR';
R is H or C1_6 alkyl;
R7 is C1_6 alkyl or (CR2)qY and Y is C3 cycloalkyl;
alternatively, R and R7 together with N in NRR7 forms morpholinyl,
piperidinyl, piperazinyl, (C1.6 alkyl)-piperazinyl, or pyrrolidinyl, each of
which is
optionally substituted with hydroxyl; and
17
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
R1 and R3 are as defined in Formual (1).
[0041] In each of the above formula, any asymmetric carbon atoms may be
present in the (R)-, (S)-or (R,S)-configuration. The compounds may thus be
present
as mixtures of isomers or as pure isomers, for example, as pure enantiomers or
diastereomers. The invention further encompasses possible tautomers of the
inventive
compounds.
[0042] Any formula given herein is also intended to represent unlabeled forms
as
well as isotopically labeled forms of the compounds. Isotopically labeled
compounds
have structures depicted by the formulas given herein, except that one or more
atoms
are replaced by an atom having a selected atomic mass or mass number. Examples
of
isotopes that can be incorporated into compounds of the invention include
isotopes of
hydrogen, carbon, nitrogen, oxygen, phosphorous, fluorine, and chlorine, such
as 2H,
3H 11C 13C 14C 15N 18F 31P 32P 35S 36C1 1251 respectively.
[0043] The invention includes various isotopically labeled compounds as
defined
herein, for example, those into which radioactive isotopes such as 3H, 13C,
and 14C ,
are present. Such isotopically labelled compounds are useful in metabolic
studies
(with, for example, 14C), reaction kinetic studies (with, for example 2H or
3H),
detection or imaging techniques, such as positron emission tomography (PET) or
single-photon emission computed tomography (SPECT) including drug or substrate
tissue distribution assays, or in radioactive treatment of patients. In other
examples, an
18F or labeled compound may be used for PET or SPECT studies. Isotopic
variations
of the compounds have the potential to change a compound's metabolic fate
and/or
create small changes in physical properties such as hydrophobicity, and the
like.
Isotopic variations also have the potential to enhance efficacy and safety,
enhance
bioavailability and half-life, alter protein binding, change biodistribution,
increase the
proportion of active metabolites and/or decrease the formation of reactive or
toxic
metabolites. Isotopically labeled compounds of this invention and prodrugs
thereof
can generally be prepared by carrying out the procedures disclosed in the
schemes or
in the examples and preparations described below by substituting a readily
available
isotopically labeled reagent for a non-isotopically labeled reagent.
[0044] In each of the above formula, each optionally substituted moiety may be
substituted with C1.6 alkyl, C2.6 alkenyl or C3.6 alkynyl, each of which may
be
optionally halogenated or optionally having a carbon that may be replaced or
18
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
substituted with N, S, 0, or a combination thereof (for example, hydroxylCi-
C8alkyl,
Ci-C8alkoxyCi-C8alkyl); halo, amino, amidino, C1_6 alkoxy; hydroxyl,
methylenedioxy, carboxy; C1_8 alkylcarbonyl, C1_8 alkoxycarbonyl, carbamoyl,
C1_8
alkylcarbamoyl, sulfamoyl, cyano, oxo, nitro, or an optionally substituted
carbocyclic
ring, heterocyclic ring, aryl or heteroaryl as previously described.
Pharmacology and Utility
[0045] The compounds of the invention and their pharmaceutically acceptable
salts exhibit valuable pharmacological properties when tested in vitro in cell-
free
kinase assays and in cellular assays, and are therefore useful as
pharmaceuticals.
[0046] In one aspect, the compounds of the invention may inhibit insulin like
growth-factor receptor 1 (IGF-1R), and may be useful in the treatment of IGF-1
R
mediated diseases. Examples of IGF-1R mediated diseases include but are not
limited
to proliferative diseases, such as tumors, for example breast, renal,
prostate,
colorectal, thyroid, ovarian, pancreas, neuronal, lung, uterine and gastro
intestinal
tumors, as well as osteosarcomas and melanomas. The efficacy of the compounds
of
the invention as inhibitors of IGF-1R tyrosine kinase activity may be
demonstrated
using a cellular capture ELISA. In this assay, the activity of the compounds
of the
invention against (IGF-1)-induced autophosphorylation of the IGF-1R is
determined.
[0047] In another aspect, the compounds of the invention may inhibit the
tyrosine
kinase activity of anaplastic lymphoma kinase (ALK) and the fusion protein of
NPM-
ALK. This protein tyrosine kinase results from a gene fusion of nucleophosmin
(NPM) and ALK, rendering the protein tyrosine kinase activity of ALK ligand
independent. NPM-ALK plays a key role in signal transmission in a number of
hematopoetic and other human cells leading to hematological and neoplastic
diseases,
for example in anaplastic large-cell lymphoma (ALCL) and non-Hodgkin's
lymphomas (NHL), specifically in ALK+NHL or Alkomas, in inflammatory
myofibroblastic tumors (IMT) and neuroblastomas. (Duyster et al. 2001 Oncogene
20, 5623-5637). In addition to NPM-ALK, other gene fusions have been
identified in
human hematological and neoplastic diseases; for example, TPM3-ALK (a fusion
of
nonmuscle tropomyosin with ALK).
[0048] The inhibition of ALK tyrosine kinase activity may be demonstrated
using
known methods, for example using the recombinant kinase domain of the ALK in
analogy to the VEGF-R kinase assay described in J. Wood et al. Cancer Res. 60,
19
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
2178-2189 (2000). In general, in vitro enzyme assays using GST-ALK protein
tyrosine kinase are performed in 96-well plates as a filter binding assay in
20 mM Tris
HC1, pH = 7.5, 3 mM MgC12, 10 mM MnC12, 1 mM DTT, 0.1 O/assay (=30 l) [y-
33P]-ATP, 2 M ATP, 3 g/mL poly (Glu, Tyr 4:1) Poly-EY (Sigma P-0275), 1 %
DMSO, 25 ng ALK enzyme. Assays are incubated for 10 min at ambient
temperature.
Reactions are terminated by adding 50 l of 125 mM EDTA, and the reaction
mixture
is transferred onto a MAIP Multiscreen plate (Millipore, Bedford, MA, USA),
previously wet with methanol, and rehydrated for 5 min with H20. Following
washing (0.5 % H3PO4), plates are counted in a liquid scintillation counter.
IC50
values are calculated by linear regression analysis of the percentage
inhibition.
[0049] The compounds of the invention may potently inhibit the growth of human
NPM-ALK overexpressing murine BaF3 cells (DSMZ Deutsche Sammiung von
Mikroorganismen and Zelikulturen GmbH, Germany). The expression of NPM-ALK
may be achieved by transfecting the BaF3 cell line with an expression vector
pClneoTm (Promega Corp., Madison WI, USA) coding for NPM-ALK and subsequent
selection of G418 resistant cells. Non-transfected BaF3 cells depend on IL-3
for cell
survival. In contrast, NPM-ALK expressing BaF3 cells (named BaF3-NPM-ALK
hereinafter) can proliferate in the absence of IL-3 because they obtain
proliferative
signal through NPM-ALK kinase. Putative inhibitors of the NPM-ALK kinase
therefore abolish the growth signal and may result in antiproliferative
activity. The
antiproliferative activity of putative inhibitors of the NPM-ALK kinase can
however
be overcome by addition of IL-3, which provides growth signals through an NPM-
ALK independent mechanism. An analogous cell system using FLT3 kinase has also
been described (see, E Weisberg et al. Cancer Cell; 1, 433-443 (2002)).
[0050] The inhibitory activity of the compounds of the invention may be
determined as follows. In general, BaF3-NPM-ALK cells (15,000/microtitre plate
well) are transferred to 96-well microtitre plates. Test compounds dissolved
in
dimethyl sulfoxide (DMSO) are added in a series of concentrations (dilution
series) in
such a manner that the final concentration of DMSO is not greater than 1 %
(v/v).
After the addition, the plates are incubated for two days during which the
control
cultures without test compound are able to undergo two cell-division cycles.
The
growth of the BaF3-NPM-ALK cells is measured by means of YOPROTM staining [T
Idziorek et al. J. Immunol. Methods; 185: 249-258 (1995)]: 25 l of lysis
buffer
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
comprising 20 mM sodium citrate, pH 4.0, 26.8 mM sodium chloride, 0.4 % NP40,
20
mM EDTA and 20 mM is added to each well. Cell lysis is completed within 60 min
at room temperature and total amount of YOPROTm bound to DNA is determined by
measurement using the Cytofluor II 96-well reader (PerSeptive Biosystems) with
the
following settings: Excitation (nm) 485/20 and Emission (nm) 530/25.
[0051] The compounds of the invention may also be useful in the treatment
and/or
prevention of acute or chronic inflammatory diseases or disorders or
autoimmune
diseases e.g. rheumatoid arthritis, osteoarthritis, systemic lupus
erythematosus,
Hashimoto's thyroiditis, multiple sclerosis, myasthenia gravis, diabetes (type
I and II)
and the disorders associated therewith, respiratory diseases such as asthma or
inflammatory liver injury, inflammatory glomerular injury, cutaneous
manifestations
of immunologically-mediated disorders or illnesses, inflammatory and
hyperproliferative skin diseases (such as psoriasis, atopic dermatitis,
allergic contact
dermatitis, irritant contact dermatitis and further eczematous dermatitis,
seborrhoeic
dermatitis), s inflammatory eye diseases, e.g. Sjoegren's syndrome,
keratoconjunctivitis or uveitis, inflammatory bowel disease, Crohn's disease
or
ulcerative colitis.
[0052] In accordance with the foregoing, the present invention provides:
(1) a compound of the invention for use as a pharmaceutical;
(2) a compound of the invention for use as an IGF-1R inhibitor, for example
for use in any of the particular indications hereinbefore set forth;
(3) a pharmaceutical composition, e.g. for use in any of the indications
herein
before set forth, comprising a compound of the invention as active ingredient
together
with one or more pharmaceutically acceptable diluents or carriers;
(4) a method for the treatment of any particular indication set forth
hereinbefore in a subject in need thereof which comprises administering an
effective
amount of a compound of the invention or a pharmaceutical composition
comprising
same;
(5) the use of a compound of the invention for the manufacture of a
medicament for the treatment or prevention of a disease or condition in which
IGF-1R
activation plays a role or is implicated;
(6) the method as defined above under (4) comprising co-administration, e.g.
concomitantly or in sequence, of a therapeutically effective amount of a
compound of
21
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
the invention and one or more further drug substances, said further drug
substance
being useful in any of the particular indications set forth hereinbefore;
(7) a combination comprising a therapeutically effective amount of a
compound of the invention and one or more further drug substances, said
further drug
substance being useful in any of the particular indications set forth
hereinbefore;
(8) use of a compound of the invention for the manufacture of a medicament
for the treatment or prevention of a disease which responds to inhibition of
the
anaplastic lymphoma kinase;
(9) the use according to (8), wherein the disease to be treated is selected
from
anaplastic large cell lymphoma, non-Hodgkin's lymphomas, inflammatory
myofibroblastic tumors, neuroblastomas and neoplastic diseases;
(10) the use according to (8) or (9), wherein the compound is of Formula (1),
(2), (3), (4) or (5), or any one of the examples, or a pharmaceutically
acceptable salt
thereof;
(11) a method for the treatment of a disease which responds to inhibition of
the anaplastic lymphoma kinase, especially a disease selected from anaplastic
large-
cell lymphoma, non Hodgkin's lymphomas, inflammatory myofibroblastic tumors,
neuroblastomas and neoplastic diseases, comprising administering an effective
amount of a compound of the invention or a pharmaceutically acceptable salt
thereof.
Administration and Pharmaceutical Compositions
[0053] In general, compounds of the invention will be administered in
therapeutically effective amounts via any of the usual and acceptable modes
known in
the art, either singly or in combination with one or more therapeutic agents.
A
therapeutically effective amount may vary widely depending on the severity of
the
disease, the age and relative health of the subject, the potency of the
compound used
and other factors known to those of ordinary skill in the art. For example,
for the
treatment of neoplastic diseases and immune system disorders, the required
dosage
will also vary depending on the mode of administration, the particular
condition to be
treated and the effect desired.
[0054] In general, satisfactory results are indicated to be obtained
systemically at
daily dosages of from about 0.01 to about 100 mg/kg per body weight, or
particularly,
from about 0.03 to 2.5 mg/kg per body weight. An indicated daily dosage in the
larger mammal, e.g. humans, may be in the range from about 0.5 mg to about
2000
22
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
mg, or more particularly, from about 0.5 mg to about 100 mg, conveniently
administered, for example, in divided doses up to four times a day or in
retard form.
Suitable unit dosage forms for oral administration comprise from ca. 1 to 50
mg active
ingredient.
[0055] Compounds of the invention may be administered as pharmaceutical
compositions by any conventional route; for example, enterally, e.g., orally,
e.g., in
the form of tablets or capsules; parenterally, e.g., in the form of injectable
solutions or
suspensions; or topically, e.g., in the form of lotions, gels, ointments or
creams, or in a
nasal or suppository form.
[0056] Pharmaceutical compositions comprising a compound of the present
invention in free form or in a pharmaceutically acceptable salt form in
association
with at least one pharmaceutically acceptable carrier or diluent may be
manufactured
in a conventional manner by mixing, granulating, coating, dissolving or
lyophilizing
processes. For example, pharmaceutical compositions comprising a compound of
the
invention in association with at least one pharmaceutical acceptable carrier
or diluent
may be manufactured in conventional manner by mixing with a pharmaceutically
acceptable carrier or diluent. Unit dosage forms for oral administration
contain, for
example, from about 0.1 mg to about 500 mg of active substance.
[0057] In one embodiment, the pharmaceutical compositions are solutions of the
active ingredient, including suspensions or dispersions, such as isotonic
aqueous
solutions. In the case of lyophilized compositions comprising the active
ingredient
alone or together with a carrier such as mannitol, dispersions or suspensions
can be
made up before use. The pharmaceutical compositions may be sterilized and/or
contain adjuvants, such as preserving, stabilizing, wetting or emulsifying
agents,
solution promoters, salts for regulating the osmotic pressure and/or buffers.
Suitable
preservatives include but are not limited to antioxidants such as ascorbic
acid, or
microbicides, such as sorbic acid or benzoic acid. The solutions or
suspensions may
further comprise viscosity-increasing agents, including but not limited to,
sodium
carboxymethylcellulose, carboxymethylcellulose, dextran, polyvinylpyrrolidone,
gelatins, or solubilizers, e.g. Tween 80 (polyoxyethylene(20)sorbitan mono-
oleate).
[0058] Suspensions in oil may comprise as the oil component the vegetable,
synthetic, or semi-synthetic oils customary for injection purposes. Examples
include
liquid fatty acid esters that contain as the acid component a long-chained
fatty acid
23
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
having from 8 to 22 carbon atoms, or in some embodiments, from 12 to 22 carbon
atoms. Suitable liquid fatty acid esters include but are not limited to lauric
acid,
tridecylic acid, myristic acid, pentadecylic acid, palmitic acid, margaric
acid, stearic
acid, arachidic acid, behenic acid or corresponding unsaturated acids, for
example
oleic acid, elaidic acid, erucic acid, brassidic acid and linoleic acid, and
if desired,
may contain antioxidants, for example vitamin E, 3-carotene or 3,5-di-tert-
butyl-
hydroxytoluene. The alcohol component of these fatty acid esters may have six
carbon atoms and may be monovalent or polyvalent, for example a mono-, di- or
trivalent, alcohol. Suitable alcohol components include but are not limited to
methanol, ethanol, propanol, butanol or pentanol or isomers thereof; glycol
and
glycerol.
[0059] Other suitable fatty acid esters include but are not limited ethyl-
oleate,
isopropyl myristate, isopropyl palmitate, LABRAFIL M 2375, (polyoxyethylene
glycerol), LABRAFIL M 1944 CS (unsaturated polyglycolized glycerides prepared
by alcoholysis of apricot kernel oil and comprising glycerides and
polyethylene glycol
ester), LABRASOLTm (saturated polyglycolized glycerides prepared by
alcoholysis of
TCM and comprising glycerides and polyethylene glycol ester; all available
from
GaKefosse, France), and/or MIGLYOL 812 (triglyceride of saturated fatty acids
of
chain length C8 to C12 from Hills AG, Germany), and vegetable oils such as
cottonseed oil, almond oil, olive oil, castor oil, sesame oil, soybean oil, or
groundnut
oil.
[0060] Pharmaceutical compositions for oral administration may be obtained,
for
example, by combining the active ingredient with one or more solid carriers,
and if
desired, granulating a resulting mixture, and processing the mixture or
granules by the
inclusion of additional excipients, to form tablets or tablet cores.
[0061] Suitable carriers include but are not limited to fillers, such as
sugars, for
example lactose, saccharose, mannitol or sorbitol, cellulose preparations,
and/or
calcium phosphates, for example tricalcium phosphate or calcium hydrogen
phosphate, and also binders, such as starches, for example corn, wheat, rice
or potato
starch, methylcellulose, hydroxypropyl methylcellulose, sodium
carboxymethylcellulose, and/or polyvinylpyrrolidone, and/or, if desired,
disintegrators, such as the above-mentioned starches, carboxymethyl starch,
crosslinked polyvinylpyrrolidone, alginic acid or a salt thereof, such as
sodium
24
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
alginate. Additional excipients include flow conditioners and lubricants, for
example
silicic acid, talc, stearic acid or salts thereof, such as magnesium or
calcium stearate,
and/or polyethylene glycol, or derivatives thereof.
[0062] Tablet cores may be provided with suitable, optionally enteric,
coatings
through the use of, inter alia, concentrated sugar solutions which may
comprise gum
arable, talc, polyvinylpyrrolidone, polyethylene glycol and/or titanium
dioxide, or
coating solutions in suitable organic solvents or solvent mixtures, or, for
the
preparation of enteric coatings, solutions of suitable cellulose preparations,
such as
acetylcellulose phthalate or hydroxypropylmethylcellulose phthalate. Dyes or
pigments may be added to the tablets or tablet coatings, for example for
identification
purposes or to indicate different doses of active ingredient.
[0063] Pharmaceutical compositions for oral administration may also include
hard
capsules comprising gelatin or soft-sealed capsules comprising gelatin and a
plasticizer, such as glycerol or sorbitol. The hard capsules may contain the
active
ingredient in the form of granules, for example in admixture with fillers,
such as corn
starch, binders, and/or glidants, such as talc or magnesium stearate, and
optionally
stabilizers. In soft capsules, the active ingredient may be dissolved or
suspended in
suitable liquid excipients, such as fatty oils, paraffin oil or liquid
polyethylene glycols
or fatty acid esters of ethylene or propylene glycol, to which stabilizers and
detergents, for example of the polyoxyethylene sorbitan fatty acid ester type,
may also
be added.
[0064] Pharmaceutical compositions suitable for rectal administration are, for
example, suppositories comprising a combination of the active ingredient and a
suppository base. Suitable suppository bases are, for example, natural or
synthetic
triglycerides, paraffin hydrocarbons, polyethylene glycols or higher alkanols.
[0065] Pharmaceutical compositions suitable for parenteral administration may
comprise aqueous solutions of an active ingredient in water-soluble form, for
example
of a water-soluble salt, or aqueous injection suspensions that contain
viscosity-
increasing substances, for example sodium carboxymethylcellulose, sorbitol
and/or
dextran, and, if desired, stabilizers. The active ingredient, optionally
together with
excipients, can also be in the form of a lyophilizate and can be made into a
solution
before parenteral administration by the addition of suitable solvents.
Solutions such
as are used, for example, for parenteral administration can also be employed
as
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
infusion solutions. The manufacture of injectable preparations is usually
carried out
under sterile conditions, as is the filling, for example, into ampoules or
vials, and the
sealing of the containers.
[0066] The compounds of the invention may be administered as the sole active
ingredient, or together with other drugs useful against neoplastic diseases or
useful in
immunomodulating regimens. For example, the compounds of the invention may be
used in accordance with the invention in combination with pharmaceutical
compositions effective in various diseases as described above, e.g. with
cyclophosphamide, 5-fluorouracil, fludarabine, gemcitabine, cisplatinum,
carboplatin,
vincristine, vinblastine, etoposide, irinotecan, paclitaxel, docetaxel,
rituxan,
doxorubicine, gefitinib, or imatinib; or also with cyclosporins, rapamycins,
ascomycins or their immunosuppressive analogs, e.g. cyclosporin A, cyclosporin
G,
FK-506, sirolimus or everolimus, corticosteroids, e.g. prednisone,
cyclophosphamide,
azathioprene, methotrexate, gold salts, sulfasalazine, antimalarials,
brequinar,
leflunomide, mizoribine, mycophenolic acid, mycophenolate, mofetil, 15-
deoxyspergualine, immuno-suppressive monoclonal antibodies, e.g. monoclonal
antibodies to leukocyte receptors, e.g. MHC, CD2, CD3, CD4, CD7, CD25, CD28, I
CD40, CD45, CD58, CD80, CD86, CD152, CD137, CD154, ICOS, LFA-1, VLA-4 or
their ligands, or other immunomodulatory compounds, e.g. CTLA41g.
[0067] The invention also provides for a pharmaceutical combinations, e.g. a
kit,
comprising a) a first agent which is a compound of the invention as disclosed
herein,
in free form or in pharmaceutically acceptable salt form, and b) at least one
co-agent.
The kit can comprise instructions for its administration.
Processes for Making Compounds of the Invention
[0068] General procedures for preparing compounds of the invention are
described in the Examples, infra. In the reactions described, reactive
functional
groups, for example hydroxy, amino, imino, thio or carboxy groups, where these
are
desired in the final product, may be protected to avoid their unwanted
participation in
the reactions. Conventional protecting groups may be used in accordance with
standard practice (see e.g., T.W. Greene and P. G. M. Wuts in "Protective
Groups in
Organic Chemistry", John Wiley and Sons, 1991).
[0069] The compounds of the invention, including their salts, are also
obtainable
in the form of hydrates, or their crystals may include for example the solvent
used for
26
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
crystallization (present as solvates). Salts can usually be converted to
compounds in
free form, e.g., by treating with suitable basic agents, for example with
alkali metal
carbonates, alkali metal hydrogen carbonates, or alkali metal hydroxides, such
as
potassium carbonate or sodium hydroxide. A compound of the invention in a base
addition salt form may be converted to the corresponding free acid by treating
with a
suitable acid (e.g., hydrochloric acid, etc.). In view of the close
relationship between
the novel compounds in free form and those in the form of their salts,
including those
salts that may be used as intermediates, for example in the purification or
identification of the novel compounds, any reference to the free compounds is
to be
understood as referring also to the corresponding salts, as appropriate.
[0070] Salts of the inventive compounds with a salt-forming group may be
prepared in a manner known per se. Acid addition salts of compounds of Formula
(1),
(2A), (2B), (3A) and (3B), may thus be obtained by treatment with an acid or
with a
suitable anion exchange reagent. Pharmaceutically acceptable salts of the
compounds
of the invention may be formed, for example, as acid addition salts, with
organic or
inorganic acids, from compounds of Formula (1), (2A), (2B), (3A) and (3B),
with a
basic nitrogen atom.
[0071] Suitable inorganic acids include, but are not limited to, halogen
acids, such
as hydrochloric acid, sulfuric acid, or phosphoric acid. Suitable organic
acids include,
but are not limited to, carboxylic, phosphoric, sulfonic or sulfamic acids,
for example
acetic acid, propionic acid, octanoic acid, decanoic acid, dodecanoic acid,
glycolic
acid, lactic acid, fumaric acid, succinic acid, adipic acid, pimelic acid,
suberic acid,
azelaic acid,-malic acid, tartaric acid, citric acid, amino acids, such as
glutamic acid or
aspartic acid, maleic acid, hydroxymaleic acid, methylmaleic acid,
cyclohexanecarboxylic acid, adamantanecarboxylic acid, benzoic acid, salicylic
acid,
4 aminosalicylic acid, phthalic acid, phenylacetic acid, mandelic acid,
cinnamic acid,
methane-or ethane-sulfonic acid, 2-hydroxyethanesulfonic acid, ethane-1,2-
disulfonic
acid, benzenesulfonic acid, 2-naphthalenesulfonic acid, 1,5-naphthalene-
disuifonic
acid, 2-, 3-or 4 methylbenzenesulfonic acid, methylsulfuric acid,
ethylsulfuric acid,
dodecylsulfuric acid, N cyclohexylsulfamic acid, N-methyl-, N-ethyl-or N-
propyl-
sulfamic acid, or other organic protonic acids, such as ascorbic acid. For
isolation or
purification purposes, it is also possible to use pharmaceutically
unacceptable salts,
for example picrates or perchlorates. For therapeutic use, only
pharmaceutically
27
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
acceptable salts or free compounds are employed (where applicable in the form
of
pharmaceutical preparations).
[0072] Compounds of the invention in unoxidized form may be prepared from N-
oxides of compounds of the invention by treating with a reducing agent (e.g.,
sulfur,
sulfur dioxide, triphenyl phosphine, lithium borohydride, sodium borohydride,
phosphorus trichloride, tribromide, or the like) in a suitable inert organic
solvent (e.g.
acetonitrile, ethanol, aqueous dioxane, or the like) at 0 to 80 C.
[0073] Prodrug derivatives of the compounds of the invention may be prepared
by
methods known to those of ordinary skill in the art (e.g., for further details
see
Saulnier et al., (1994), Bioorganic and Medicinal Chemistry Letters, Vol. 4,
p. 1985).
For example, appropriate prodrugs may be prepared by reacting a non-
derivatized
compound of the invention with a suitable carbamylating agent (e.g., 1,1-
acyloxyalkylcarbanochloridate, para-nitrophenyl carbonate, or the like).
[0074] Protected derivatives of the compounds of the invention may be made by
means known to those of ordinary skill in the art. A detailed description of
techniques
applicable to the creation of protecting groups and their removal may be found
in T.
W. Greene, "Protecting Groups in Organic Chemistry", 3rd edition, John Wiley
and
Sons, Inc., 1999.
[0075] Compounds of the invention may be prepared as their individual
stereoisomers by reacting a racemic mixture of the compound with an optically
active
resolving agent to form a pair of diastereoisomeric compounds, separating the
diastereomers and recovering the optically pure enantiomers. Resolution of
enantiomers may be carried out using covalent diastereomeric derivatives of
the
compounds of the invention, or by using dissociable complexes (e.g.,
crystalline
diastereomeric salts). Diastereomers have distinct physical properties (e.g.,
melting
points, boiling points, solubilities, reactivity, etc.) and may be readily
separated by
taking advantage of these dissimilarities. The diastereomers may be separated
by
fractionated crystallization, chromatography, or by separation/resolution
techniques
based upon differences in solubility. The optically pure enantiomer is then
recovered,
along with the resolving agent, by any practical means that would not result
in
racemization. A more detailed description of the techniques applicable to the
resolution of stereoisomers of compounds from their racemic mixture may be
found in
28
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
Jean Jacques, Andre Collet, Samuel H. Wilen, "Enantiomers, Racemates and
Resolutions", John Wiley And Sons, Inc., 1981.
[0076] In summary, the compounds of the invention may be made by a process as
described in the Examples; and
(a) optionally converting a compound of the invention into a pharmaceutically
acceptable salt;
(b) optionally converting a salt form of a compound of the invention to a non-
salt form;
(c) optionally converting an unoxidized form of a compound of the invention
into a pharmaceutically acceptable N-oxide;
(d) optionally converting an N-oxide form of a compound of the invention to
its unoxidized form;
(e) optionally resolving an individual isomer of a compound of the invention
from a mixture of isomers;
(f) optionally converting a non-derivatized compound of the invention into a
pharmaceutically acceptable prodrug derivative; and
(g) optionally converting a prodrug derivative of a compound of the invention
to its non-derivatized form.
[0077] Insofar as the production of the starting materials is not particularly
described, the compounds are known or can be prepared analogously to methods
known in the art or as disclosed in the Examples hereinafter. One of skill in
the art
will appreciate that the above transformations are only representative of
methods for
preparation of the compounds of the present invention, and that other well
known
methods can similarly be used. The present invention is further exemplified,
but not
limited, by the following and Examples that illustrate the preparation of the
compounds of the invention.
Intermediate 1
2,5-dichloro-N-(5-methyl- IH-pyrazol-3- l)p rimidin-4-amine
HN-N CI
N N CI
H
29
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
[0078] A mixture of 5-methyl-1H-pyrazol-3-amine (3.00 g, 30.9 mmol), 2,4,5-
trichloropyrimidine (5.67 g, 30.9 mmol, 1 equiv.) and Na2CO3 (3.60 g, 34.0
mmol, 1.1
equiv.) in EtOH (100 mL) was heated at 40 C for 24 h. The solvent was removed
in
vacuo. The resulting residue was partitioned between EtOAc (350 mL) and water
(100
mL). The EtOAc layer was washed with water (3x), saturated aqueous NaCl (lx)
and
dried over Na2SO4. The resulting EtOAc solution was concentrated in vacuo,
providing the product 2,5-dichloro-N-(5-methyl-iH-pyrazol-3-yl)pyrimidin-4-
amine;
ESMS m/z 244.0 (M + H+).
Intermediate 2
2-chloro-N-(5-meth pyrazol-3-yl)-5-(trifluoromethyl)pyrimidin-4-amine
F
F
F N
HN N CI
N
HN
[0079] A mixture of 2,4-dichloro-5-(trifluoromethyl)pyrimidine (1.06 g, 4.86
mmol), 5-methyl-1H-pyrazol-3-amine (472.2 mg, 4.86 mmol) and sodium carbonate
(2.06 g, 19.4 mmol) in 100 mL of EtOH was stirred at room temperature
overnight.
The reaction mixture was concentrated in vacuo. The crude solid was
partitioned
between EtOAc and water. The combined organic extracts were dried (Na2SO4),
concentrated in vacuo, and purified by silica chromatography (EtOAc/ hexanes:
1/1)
to afford 2-chloro-N-(5-methyl-iH-pyrazol-3-yl)-5-(trifluoromethyl)pyrimidin-4-
amine; ESMS m/z 278.0 (M + H+).
Intermediate 3
2,5-dimethyl-4-(3-methylisoxazol-5-yl)aniline
\
O
H2N
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
Step 1: 1-(2,5-dimethyl-4-nitrophenyl)ethanone
[0080] To a mixture of 1-Bromo-2,5-dimethyl-4-nitrobenzene (1 g, 4.34 mmol)
and tributyl(1-ethoxyvinyl)tin (1.88 g, 5.2 mmol) in DMF (20 mL), was added
tetrakis(triphenylphospine) palladium (0) (250 mg, 5 % mmol). The reaction
tube was
sealed, the mixture was purged with N2 for 3 min and then heated at 90 C under
N2 for
overnight. The reaction was cooled to room temperature and poured into aqueous
HCl
(1N, 100 mL). The mixture was stirred for 1 hour, and then extracted with
ethyl
acetate (3 x 100 mL). The organic extracts were combined, washed with brine
and
concentrated. The crude product was purified with silica gel chromatography
(20 %
ethyl acetate in hexanes) to afford 1-(2,5-dimethyl-4-nitrophenyl)ethanone as
a yellow
solid.
Step 2: 1-(2,5-dimethyl-4-nitrophenyl)-3-(dimethylamino)but-2-en-1-one
[0081] A mixture of 1-(2,5-dimethyl-4-nitrophenyl)ethanone (Step 1, 300 mg,
1.55 mmol) and 1,1-dimethoxy-N,N-dimethylethanamine (1 mL) was heated in a
microwave at 130 C for 10 min. The crude product was purified with silica gel
chromatography (60% ethyl acetate in hexanes) to afford 1-(2,5-dimethyl-4-
nitrophenyl)-3-(dimethylamino)but-2-en-1-one as a yellow solid.
Steps 3 and 4: 2,5-dimethyl-4-(3-methylisoxazol-5-yl)aniline
[0082] A mixture of 1-(2,5-dimethyl-4-nitrophenyl)-3-(dimethylamino)but-2-en-
1-one (Step 2, 100 mg, 0.38 mmol) and hydroxylamine monohydrochloride (132 mg,
1.9 mmol) in ethanol (3 mL) was heated in a microwave at 100 C for 15 min.
The
obtained 5-(2,5-dimethyl-4-nitrophenyl)-3-methylisoxazole was dissolved in
methanol
(10 mL). To this solution was added Pd/C (10%). The reaction mixture was
degassed
and purged with H2 for several times and stirred under H2 (1 atm.) overnight.
The
mixture was filtered and concentrated to afford 2,5-dimethyl-4-(3-
methylisoxazol-5-
yl)aniline. ESMS m/z 203 (M+ H+).
Intermediate 4
2,5 -dimethyl-4- (oxazol-5 -yl) aniline
31
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
N
0
H2N
[0083] Step 1: To a mixture of 2,5-dimethyl-4-nitrobenzaldehyde (75 mg, 0.42
mmol) and toluenesulfonylmethyl isocyanide (TOSMIC) (98 mg, 0.5 mmol) in
methanol (2 mL), was added sodium methoxide (68 mg, 1.26 mmol). The mixture
was
sealed and heated at 90 C for 15 hr. The reaction mixture was concentrated
and
partitioned between water and ethyl acetate. The organic layer was separated,
dried
over sodium sulfate and concentrated. The 5-(2,5-Dimethyl-4-
nitrophenyl)oxazole
obtained was used in the next step without purification.
[0084] Step 2: The 5-(2,5-dimethyl-4-nitrophenyl)oxazole obtained in the last
step
was dissolved in methanol (10 mL). To the solution was added Pd/C (10%). The
reaction mixture was degassed and purged with H2 for several times and then
stirred
under 1 atm. hydrogen gas overnight. The mixture was filtered and concentrated
to
afford 2,5-dimethyl-4-(oxazol-5-yl)aniline as a white solid. ESMS m/z 189 (M+
H+).
Intermediate 5
2,5-Dimethyl-4-(3-methyl- I H-pyrazol-5-yl)aniline
,N
H
H2N
[0085] A mixture of 1-(2,5-dimethyl-4-nitrophenyl)-3-(dimethylamino)but-2-en-
1-one (100mg, 0.38 mmol) and hydrazine (60 uL, 1.9 mmol) in ethanol (3 mL) was
heated in a microwave at 100 C for 15 min. The obtained 5-(2,5-dimethyl-4-
nitrophenyl)-3-methylpyrazol was dissolved in methanol (10 mL). To the
solution was
added Pd/C (10%). The reaction mixture was degassed and purged with H2 for
several
times and stirred under 1 atm. hydrogen gas overnight. The mixture was
filtered and
concentrated to afford 2,5-Dimethyl-4-(3-methyl-1H-pyrazol-5-yl)aniline. ESMS
m/z
202 (M+ H+).
Intermediate 6
2,5-Dimethyl-4-(2-methylthiazol-4-yl)aniline
32
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
I S
N
H2N
[0086] Step 1: To a mixture of 1-(2,5-dimethyl-4-nitrophenyl)ethanone (300 mg,
1.55 mmol) in HBr (48%) (5 mL)/methanol (2.4 mL) was added bromine (250 mg,
1.55 mmol). The mixture was stirred at room temperature for 4 hrs. The mixture
was
diluted with water and extracted with ethyl acetate (2 x 20 mL). The organic
layer was
washed with brine and concentrated. The residue was purified with silica gel
column
chromatography (10 % ethyl acetate in hexanes) to afford 2-bromo-l-(2,5-
dimethyl-4-
nitrophenyl)ethanone as a white solid.
[0087] Step 2: A mixture of 2-bromo-l-(2,5-dimethyl-4-nitrophenyl)ethanone (70
mg, 0.26 mmol) and ethanethioamide (30 mg, 0.4 mmol) in ethanol (2 mL) was
heated in a microwave at 150 C for 20 min. The obtained 4-(2,5-dimethyl-4-
nitrophenyl)-2-methylthiazole was dissolved in methanol (10 mL). To the
solution
was added Pd/C (10%). The reaction mixture was degassed and purged with H2 for
several times and stirred under 1 atm. hydrogen gas overnight. The mixture was
filtered and concentrated to afford 2,5 -dimethyl-4- (2-methylthiazol-4-yl)
aniline.
ESMS m/z 219 (M+ H+).
Intermediate 7
2,5-Dimethyl-4-(1-methyl-lH-imidazol-2-yl)aniline
N
N
H2N
[0088] To a solution of 2-(2,5-dimethyl-4-nitrophenyl)-1H-imidazole (50 mg,
0.23 mmol) in DMF (2 mL) was added NaH (11 mg, 0.46 mmol) at 0 C. After
stirring for 15 min, iodomethane (65 mg, 046 mmol) was added dropwise. The
mixture was stirred at 0 C for 1 hr, and quenched by adding saturated
ammonium
chloride aqueous solution. The mixture was extracted with ethyl acetate (3 x
15 mL).
The organic layer was washed with brine and dried over sodium sulfate. After
concentration, the residue was dissolved in methanol (10 mL). To the solution
was
33
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
added Pd/C (10%). The reaction mixture was degassed and purged with H2 for
several
times and stirred under 1 atm. hydrogen gas overnight. The mixture was
filtered and
concentrated to afford 2,5-dimethyl-4-(1-methyl-lH-imidazol-2-yl)aniline. ESMS
m/z 202 (M+ H+).
Intermediate 8
2,5-dimethyl-4-(pyridin-3-yl) aniline
N
H2N
[0089] A suspension of 4-bromo-2,5-dimethylaniline (4.00 g, 20 mmol), pyridin-
3-ylboronic acid (2.70 g, 11 mmol), Pd2(dba)3 (0.55 g, 0.6 mmol), 2-
dicyclohexylphosphino-2',6'-dimethoxybiphenyl (0.98 g, 1.2 mmol) and Na2CO3
(10.6 g, 100 mmol) in n-BuOH (50 mL) was degassed by a stream of argon gas for
15
min. The reaction flask was sealed and placed in a pre-heated oil bath (115
C). After
stirring overnight, the reaction was cooled and filtered. The filter cake was
washed
with DCM and the filtrate was concentrated in vacuo. The resulting residue was
dissolved in EtOAc (150 mL). EtOAC is sequentially washed with water (20 mL),
brine (20 mL), dried over Na2SO4 and evaporated. The crude product was
purified by
silica chromatography (0-50% EtOAC in hexanes gradient) to give 2,5-dimethyl-4-
(pyridin-3-yl)aniline as a yellow solid; ESMS m/z 199.1 (M+H+).
Intermediate 9
8-(2,5-dimethyl-4-nitrophenyl)-1,4-dixoaspiro [4.5]dec-7-ene
N 02
LO
[0090] A mixture of 4,4,4,4-tetramethyl-2-(1,4-dioxaspiro[4.5]dec-7-en-8-yl)-
1,3,2-dixoaborolane (200 mg, 0.86 mmol), 1-bromo-2,5-dimethyl-4-nitrobenzene
(228
34
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
mg, 0.86 mmol), tetrakis(triphenylphosphine)palladium(0) (98 mg, 0.09 mmol),
and
cesium fluoride (392 mg, 2.58 mmol) in a mixture of 1,2-dimethoxyethane (2 mL)
and
methanol (1 mL) was degassed for 5 min, and then heated at 130 C in a
microwave
reactor for 15 min. The reaction was concentrated in vacuo, and purified by
silica
chromatography (EtOAC/hexanes: 3/7) to afford 8-(2,5-dimethyl-4-nitrophenyl)-
1,4-
dixoaspiro[4.5]dec-7-ene; ESMS m/z 290.2 (M + H+).
Intermediate 10
2,5 -dimethyl-4- (1,4-dioxaspiro [4.51 decan- 8 -yl) aniline
NH2
O(--/p
[0091] A mixture of 8-(2,5-dimethyl-4-nitrophenyl)-1,4-dioxaspiro[4.5]decane
(130.2 mg, 0.45 mmol) and 10% Pd-C (13.0 mg) in MeOH (20 mL) was degassed and
then reacted under 1 atm. H2. Upon reaction completion, the Pd-C was removed
by
filtration and the filtrate was concentrated in vacuo to afford 2,5-dimethyl-4-
(1,4-
dioxaspiro[4.5] decan-8-yl)aniline; ESMS m/z 262.2 (M + H+).
Intermediate 11
2-fluoro-5 -methyl-4- (1,4-dioxaspiro [4.51 dec-7 -en- 8 -yl) aniline
NH2
F
O
[0092] A mixture of 4,4,4,4-tetramethyl-2-(1,4-dioxaspiro[4.5]dec-7-en-8-yl)-
1,3,2-dioxaborolane (532.0 mg, 2.0 mmol), 4-bromo-2-fluoro-5-methylaniline
(406.0
mg, 2.0 mmol), tetrakis(triphenylphosphine)palladium(0) (231.1 mg, 0.2 mmol),
cesium fluoride (912.0 mg, 6.0 mmol), 1,2-dimethoxyethane (4 mL) and methanol
(2
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
mL) was degassed for 5 min and then heated at 130 C in a microwave reactor for
15
min. The reaction was concentrated in vacuo, and purified by silica
chromatography
(EtOAC / Hexanes: 3/7) to afford 2-fluoro-5-methyl-4-(1,4-dioxaspiro[4.5]dec-7-
en-
8-yl)aniline; ESMS m/z 264.1 (M + H+).
Intermediate 12
2-fluoro- 5 -dimethyl-4- (1,4-dioxaspiro [4.51 decan- 8 -yl) aniline
NH2
F
O O
Li
[0093] A mixture of 2-fluoro-5-methyl-4-(1,4-dioxaspiro[4.5]dec-7-en-8-
yl)aniline (130 mg, 0.50 mmol) and 10% Pd-C (13.0 mg) in MeOH (20 mL) was
degassed and then reacted under 1 atm. H2. Upon reaction completion, the Pd-C
was
removed by filtration and the filtrate was concentrated in vacuo to afford 2-
fluoro-5-
dimethyl-4-(1,4-dioxaspiro[4.5]decan-8-yl)aniline; ESMS m/z 266.2 (M + H+).
Intermediate 13
4-(5-fluoro-2-methyl-4-(4-(5-methyl- IH-pyrazol-3-ylamino)-5-
(trifluorometh l)p rimidin-2-ylamino)phenyl)cyclohexanone
F :1:0 j
- I
HN
[0094] A mixture of 2-fluoro-5 -methyl-4- (1,4-dioxaspiro [4.5 ] decan- 8 -yl)
aniline
(318.6 mg, 1.2 mmol), 2-chloro-N-(5-methyl-1H-pyrazol-3-yl)-5-
(trifluoromethyl)pyrimidin-4-amine (333.0 mg, 1.2 mmol) and HC1(4 N in water,
0.3 mL, 1.2 mmol) in i-PrOH (4.0 mL) was heated at 125 C in an oil bath
overnight.
The reaction mixture was concentrated in vacuo. The resulting crude mixture
was
36
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
dissolved in THE (4 mL), MeOH (2 mL) and HCl (4N in water, 0.3 mL, 1.2 mmol),
and stirred at room temperature for an additional 2 h. The reaction mixture
was
concentrated in vacuo, and purified by silica chromatography (0-100% EtOAC in
hexanes gradient) to afford 4-(5-fluoro-2-methyl-4-(4-(5-methyl-1H-pyrazol-3-
ylamino)-5-(trifluoromethyl)pyrimidin-2-ylamino)phenyl)cyclohexanone; ESMS m/z
463.2 (M + H+).
Intermediate 14
N-(2,5-dichloropyrimidin-4-yl)-5-methylisoxazol-3-amine
a I \ N
HN N CI
N
O
[0095] The mixture 5-methylisoxazol-3-amine (98 mg, 1.0 mmol), 2,4,5-
trichloropyrimidine (344 L, 3.0 mmol), and sodium carbonate (106 mg, 1.0
mmol) in
3 mL of EtOH was heated at 60 C over night. The reaction mixture was
concentrated
and then partitioned between EtOAc and brine. The collected organic extracts
were
dried (Na2SO4), concentrated in vacuo, and purified with silica gel
chromatography
(MeOH/DCM: 1/9) to afford N-(2,5-dichloropyrimidin-4-yl)-5-methylisoxazol-3-
amine; ESMS m/z 245.0 (M + H+).
Intermediate 15
2,5-dichloro-N- (5-cyclopropyl-1 H-pyrazol-3-yl)pyrimidin-4-amine
a xC'
N~~
HN
[0096] A mixture of 5-cyclopropyl-1H-pyrazol-3-amine (246 mg, 2.00 mmol),
2,4,5-trichloropyrimidine (367 mg, 2.00 mmol, 1 equiv.) and Na2CO3 (233 mg,
2.20
mmol, 1.1 equiv.) in EtOH (10 mL) was heated at 40 C for 16 h. The crude
reaction
mixture was diluted with EtOAc and sequentially washed with: water (3x) and
37
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
saturated aqueous NaCl (lx). The resulting EtOAc layer was dried over Na2SO4
and
then concentrated in vacuo, providing 2,5-dichloro-N-(5-cyclopropyl-lH-pyrazol-
3-
yl)pyrimidin-4-amine; ESMS m/z 270.0 (M + H+).
Intermediate 16
tert-Butyl 4- (6- amino- 5 -fluoro- l-oxoisoindolin-2-l)piperidine- -
carboxylate
O
\/-
~O
N
0
N
H2N
F
Step 1: 4-(2-chloro-4-fluoro-5-nitrobenzamido)piperidine-l-
carboxylate
[0097] Under N2, to the solution of 2-chloro-4-fluoro-5-nitro-benzoic acid
(1.0 g,
4.56 mmol in 40 mL of DCM at 0 C are added thionyl chloride (1.0 mL, 13.68
mmol) and 0.1 mL of DMF sequentially. The reaction mixture was warmed
gradually
to room temperature and stirred overnight. The reaction mixture was
concentrated in
vacuo to afford 2-chloro-4-fluoro-5-nitrobenzoyl chloride. The crude was
dissolved
in 40 mL of DCM, and 1-boc-4-aminopiperidine (910 mg, 4.56 mmol) and
triethylamine (1.29 mL, 9.12 mmol) were added to the solution sequentially.
After
stirring at room temperature 1 h, the reaction mixture was washed with 20 mL
of
water. The organic extract was dried (Na2SO4), followed by concentration in
vacuo to
afford tert-Butyl4-(2-chloro-4-fluoro-5-nitrobenzamido)piperidine-1-
carboxylate;
ESMS m/z 402.1 (M + H+).
Step 2: 4-(4-fluoro-5-nitro-2-vinylbenzamido)piperidine-l-
l
carboxylate.
[0098] Under nitrogen, the mixture of tert-butyl 4-(2-chloro-4-fluoro-5-
nitrobenzamido)piperidine-l-carboxylate (Step 1, 1.52 g, 3.88 mmol),
vinylboronic
acid dibutyl ester (0.86 mL, 3.88 mmol),
dichlorobis(triphenylphosphine)palladium
38
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
(II) (136 mg, 0.2 mmol) and sodium carbonate (2.9 g, 27.2 mmol) in THE (40 mL)
and water (10 mL) was heated at 90 C overnight. The reaction mixture was
cooled to
room temperature, partitioned between EtOAc and brine. The organic extracts
were
dried (Na2SO4), concentrated, and purified by silica gel chromatography
(EtOAc/Hexanes: 1/4) to afford tert-Butyl4-(4-fluoro-5-nitro-2-
vinylbenzamido)piperidine-l-carboxylate; ESMS m/z 394.2 (M + H+).
Step 3: 4-(5-fluoro-6-nitro-l-oxoisoindolin-2-yl)piperidine-l-
carboxylate
[0099] tert-Butyl4-(4-fluoro-5-nitro-2-vinylbenzamido)piperidine-l-carboxylate
(Step 2, 1.1 g, 2.77 mmol) in 50 mL of DCM is chilled to -78 C. Ozone was
passed
through the solution until the starting material was consumed, and then
nitrogen was
passed through the solution for 5 min. The reaction mixture was warmed to room
temperature. Triphenylphosphine-resin (2.77 g) in 10 mL of DCM was added, and
was stirred for another 1.5 h. The resin was filtered off, and the solution
was
concentrated in vacuo. The resulting crude was dissolved in DCM (15 mL), and
to
this solution were added TFA (15 mL) and triethylsilane (1.0 mL, 5.9 mmol)
sequentially. The reaction was stirred at room temperature 2 h. After
concentration,
the reaction crude was poured into 10 mL of water, neutralized to pH 8 with
sat. aq.
NaHCO3, followed by addition of (Boc)20 (603 mg, 2.77 mmol) in 10 mL of DCM.
The reaction was stirred at room temperature for 1.5 h, then extracted with
DCM. The
organic extracts were dried (Na2SO4), concentrated in vacuo, and purified by
silica gel
chromatography (EtOAc/Hexanes: 1/4) to give tert-Butyl4-(5-fluoro-6-nitro-l-
oxoisoindolin-2-yl)piperidine-l-carboxylate; ESMS m/z 380.2 (M + H+).
Step 4: tert-Butyl4-(6-amino- 5-fluoro-l-oxoisoindolin-2-yl)piperidine-l-
carbox,
[0100] The mixture of tert-butyl 4-(5-fluoro-6-nitro-l-oxoisoindolin-2-
yl)piperidine-1-carboxylate (Step 3, 240 mg, 0.72 mmol), Pd (10% wt on active
carbon, 24 mg) and MeOH (20 mL) was evacuated to remove air, and then the
reaction was stirred under a hydrogen balloon until the starting material is
consumed.
Pd/C was filtered off and the solution was concentrated in vacuo to afford
tert-Butyl
4-(6-amino-5-fluoro-l-oxoisoindolin-2-yl)piperidine-l-carboxylate; ESMS m/z
350.2
(M + H+).
39
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
Intermediate 17
tert-butyl-4-(4-aminonaphthalen-1-yl)piperidine-l-carboxylate
BOC
i
N
/ I \
NH2
Step 1: tert-butyl-4-(4-aminonaphthalen-1-yl)-5,6-dihydropyridine-1(2H)-
carboxylate
[0101] A mixture of 4-bromonaphthalen-l-amine (1.0 g, 4.5 mmol), tent-butyl-4-
(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-5,6-dihydropyridine-1(2H)-
carboxylate
(1.7 g, 5.4 mmol), Pd(PPh3)4 (26.01 mg, 0.02 mmol) and Na2CO3 (3.34 g, 31.5
mmol)
in 10 mL of DMF and 5 mL of water was degassed and purged with nitrogen. The
reaction was heated at 100 C for 5 h, and cooled to room temperature. The
reaction
mixture was partitioned between EtOAc and water. The combined organic extracts
were dried (Na2SO4), concentrated in vacuo and purified by silica gel
chromatography
(MeOH/DCM: 5/95) to afford tent-butyl-4-(4-aminonaphthalen-1-yl)-5,6-
dihydropyridine-1(2H)-carboxylate.
Step 2: tert-butyl-4-(4-aminonaphthalen-1-yl)piperidine-l-carboxylate
[0102] To a solution of tent-butyl-4-(4-aminonaphthalen-1-yl)-5,6-
dihydropyridine-1(2H)-carboxylate in 50 mL of MeOH was added 10 wt% Pd-C (100
mg). The reaction was degassed to remove air and stirred under 1 atm. H2 until
the
starting material is consumed. The Pd-C was removed by filtration and the
resulting
solution was concentrated in vacuo to afford tent-butyl-4-(4-aminonaphthalen-l-
yl)piperidine-l-carboxylate.
Intermediate 18
3-ethyl-2,3 ,4,5-tetrahydro-lH-benzo [dl azepin-7-amine
H2N ):)ON-\
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
[0103] 3-ethyl-2,3,4,5-tetrahydro-lH-benzo[d]azepin-7-amine was synthesized by
the method described in the literature reference: Pecherer, B. et al. J.
Heterocyclic
Chem 1971, 8(5), 779-783, in conjunction with standard synthetic methodology.
Example 1
1-(4-(5-chloro-4-(5-meth pyrazol-3-ylamino)pyrimidin-2-ylamino)-2,5-
dimethylphenyl)ethanone O-2-h, d~yethyloxime (28)
HO~O.N I I CI N-NH
N N N
H H
[0104] Step 1: To a solution of N2-(4-bromo-2,5-dimethylphenyl)-5-chloro-N4-(5-
methyl-1H-pyrazol-3-yl)pyrimidine-2,4-diamine (280 mg, 0.69 mmol) in THE (3
mL)
was added p-TSA (119 mg, 0.69 mmol) and 3,4-dihydro-2H-pyran (348 mg, 2.86
mmol). The mixture was stirred at room temperature for 14 h and then poured
into
saturated aqueous NaHCO3 solution (10 mL). The resulting mixture was extracted
with EtOAc (3 x 10 mL) and the combined organic layers were concentrated. The
resulting residue was purified by flash column chromatography (silica gel, 0-
50%EtOAc in hexanes gradient) to provide N2-(4-bromo-2,5-dimethylphenyl)-5-
chloro-N4-(5-methyl- l-(tetrahydro-2H-pyran-2-yl)-1H-pyrazol-3-yl)pyrimidine-
2,4-
diamine as a white solid; ESMS m/z 491.1 (M + H+).
[0105] Step 2: A mixture of N2-(4-bromo-2,5-dimethylphenyl)-5-chloro-N4-(5-
methyl-l-(tetrahydro-2H-pyran-2-yl)-1H-pyrazol-3-yl)pyrimidine-2,4-diamine
(166
mg, 0.34 mmol), tributyl(1-ethoxyvinyl)stannane (146 mg, 0.41 mmol) and
Pd(PPh3)4
(39 mg, 0.034 mmol) in toluene (2 mL) was degassed and heated at 100 C under
N2
for 14 h. After cooling down to room temperature, the mixture was
concentrated. The
resulting residue was redissolved in acetonitrile (2 mL) and treated with IN
HCl (2
mL) for 14 h. After extraction with EtOAc (3 x 15 mL), the combined organic
layers
were treated with saturated aqueous KF (10 mL). The resulted organic layer was
collected and treated with brine and dried over MgS04. After removing the
drying
agent by filtration, the filtrate was concentrated and purified by flash
column
chromatography (silica gel, 0-100% EtOAc in hexanes gradient) to provide 1-(4-
(5-
chloro-4-(5-methyl-1H-pyrazol-3-ylamino)pyrimidin-2-ylamino)-2,5-
dimethylphenyl)ethanone as a white solid; ESMS m/z 371.1 (M + H+).
41
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
[0106] Step 3: To a solution of 1-(4-(5-chloro-4-(5-methyl-1H-pyrazol-3-
ylamino)pyrimidin-2-ylamino)-2,5-dimethylphenyl)ethanone (60 mg, 0.16 mmol) in
MeOH (1 mL) was added AcOH (15 mg, 0.25 mmol), followed by the addition of 2-
(aminooxy)ethanol (20 mg, 0.26 mmol). The mixture was heated to 60 C for 14 h
and cooled down to room temperature. The mixture was then purified directly by
preparative RP-HPLC to provide 1-(4-(5-chloro-4-(5-methyl-1H-pyrazol-3-
ylamino)pyrimidin-2-ylamino)-2,5-dimethylphenyl)ethanone O-2-hydroxyethyl
oxime; ESMS m/z 430.2 (M + H+).
Example 2
4-(5-chloro-445-meth pyrazol-3-ylamino)pyrimidin-2-ylamino)-N-(2-
(dimethylamino)ethyl)-2,5-dimethylbenzamide (31)
~N\ 0
CI N-NH
H taN 11
/
N N
H H
[0107] To a solution of 4-(5-chloro-4-(5-methyl-l-(tetrahydro-2H-pyran-2-yl)-
1H-pyrazol-3-ylamino)pyrimidin-2-ylamino)-2,5-dimethylbenzoic acid (35.0 mg,
0.077 mmol) in DMF (0.5 mL) was added HATU (29.1 mg, 0.077 mmol), DIEA
(26.7 L, 0.153 mmol) and N1,N1-dimethylethane-l,2-diamine (25.2 L, 0.230
mmol). The reaction mixture was stirred at room temperature overnight. IN HCI
aqueous solution (0.5 mL) was then added and the reaction mixture was heated
at 100
C for 1.5 hours to remove the tetrahydro-2H-pyran-2-yl protecting group. The
mixture was purified by preparative RP-HPLC to provide 4-(5-chloro-4-(5-methyl-
1H-pyrazol-3-ylamino)pyrimidin-2-ylamino)-N-(2-(dimethylamino)ethyl)-2,5-
dimethylbenzamide; ESMS m/z 443.2 (M + H+).
Example 3
4-(4-(5-chloro-4-(5-meth pyrazol-3-ylamino)pyrimidin-2-ylamino)-2,5-
dimethylphenyl)cyclohexanone oxime (33)
42
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
HN,
N NH N,OH
CI
~N \
i
N N I
H
[0108] To a solution of 4-(4-(5-chloro-4-(5-methyl-1H-pyrazol-3-
ylamino)pyrimidin-2-ylamino)-2,5-dimethylphenyl)cyclohexanone (15 mg, 0.033
mmol) in MeOH (0.5 mL) was added NaOAc (6 mg, 0.073 mmol) and NH2OH HCl
(5 mg, 0.073 mmol). The resulting mixture was stirred at 70 C for 2 h. and
then
cooled down to room temperature. The mixture was purified directly by
preparative
RP-HPLC to provide 4-(4-(5-chloro-4-(5-methyl-1H-pyrazol-3-ylamino)pyrimidin-2-
ylamino)-2,5-dimethylphenyl)-cyclohexanone oxime; ESMS m/z 440.2 (M + H+).
Examples 4 and 5
5-chloro-N2-(2,5-dimethyl-4-(4-trans-morpholinocyclohexyl)phenyl)-N4-(5-
methyl-IH-pyrazol-3-yl)pyrimidine-2,4-diamine (37)
H
CIN,N
\ N
NH N")
H
HN-N O
5-chloro-N2-(2,5-dimethyl-4-(4-cis-morpholinocyclohexyl)phenyl)-N4-(5-
methyl-IH-pyrazol-3-yl)pyrimidine-2,4-diamine (38)
H
N~N
C I \ N \ I ~.,,
NH
I
HN-N O
[0109] Step 1: A mixture of 1-bromo-2,5-dimethyl-4-nitrobenzene (100 mg, 0.43
mmol), 4,4,5,5-tetramethyl-2-(1,4-dioxaspiro[4.5]dec-7-en-8-yl)-1,3,2-
dioxaborolane
(112 mg, 0.43 mmol), Pd(PPh3)4 (49 mg, 0.043 mmol) and CsF (196 mg, 1.29 mmol)
43
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
in a mixture of dimethyl ethylene glycol and water (2:1, 1.5 mL) was degassed
and
heated under N2 at 130 C in microwave reactor for 15 min. After cooling down
to
room temperature, the reaction mixture was treated with saturated NH4C1
aqueous
solution (5 mL) and extracted with EtOAc (3 x 4 mL). The combined organic
layers
were concentrated and purified by flash column chromatography (silica gel, 0%-
30%
EtOAc in hexanes gradient) to provide 8-(2,5-dimethyl-4-nitrophenyl)-1,4-
dioxaspiro[4.5]dec-7-ene as a white solid; ESMS m/z 290.1 (M + H+).
[0110] Step 2: A mixture of 8-(2,5-Dimethyl-4-nitrophenyl)-1,4-
dioxaspiro[4.5]dec-7-ene (105 mg, 0.36 mmol) and Pd/C (10 mg) in EtOH was
degassed and stirred under 1 atm. H2 at room temperature for 14 h. Pd/C was
removed
by filtration and filtrate was concentrated to provide 2,5-dimethyl-4-(1,4-
dioxaspiro[4.5]decan-8-yl)aniline, which was used in the next step without
further
purification; ESMS m/z 262.2 (M + H+).
[0111] Step 3: A mixture of 2,5-dimethyl-4-(1,4-dioxaspiro[4.5]decan-8-
yl)aniline (86 mg, 0.33 mmol) and 2,5-dichloro-N-(5-methyl-iH-pyrazol-3-
yl)pyrimidin-4-amine (104 mg, 0.43 mmol) in 'PrOH (3 mL) was treated with HCl
(82
L, 4N in dioxane, 0.33mmol) and heated to 125 C in a sealed tube for 14 h.
After
cooling down to room temperature, the mixture was concentrated and was treated
with acetone (2 mL) and HCl (330 L, 4N in dioxane) at room temperature for 5
h.
The mixture was then treated with saturated NaHCO3 aqueous solution (10 mL)
and
extracted with EtOAc (3 x 10 mL). The organic layers were combined,
concentrated
and purified by flash column chromatography (silica gel, 0%-100% EtOAc in
hexanes
gradient) to provide 4-(4-(5-chloro-4-(5-methyl-1H-pyrazol-3-ylamino)pyrimidin-
2-
ylamino)-2,5-dimethylphenyl)cyclohexanone as white solid; ESMS m/z 425.2 (M +
H+).
[0112] Step 4: To a solution of 4-(4-(5-chloro-4-(5-methyl-1H-pyrazol-3-
ylamino)pyrimidin-2-ylamino)-2,5-dimethylphenyl)cyclohexanone (30 mg, 0.071
mmol) in 1,2-dichloroethane (1 mL) was added morpholine (9 mg, 0.11 mmol)
followed by AcOH (6.5 mg, 0.11 mmol) and 4A molecular sieves. The mixture was
stirred at room temperature for 1 h before the addition of sodium
triacetoxyborohydride (22.5 mg, 0.11 mmol). The mixture was stirred at room
temperature for 14 h at which point additional morpholine (5 mg, 0.058 mmol)
and
sodium triacetoxyborohydride (10 mg, 0.047 mmol) was added and stirred at room
44
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
temperture for an additional 5 h. The reaction mixture was then treated with
saturated
NH4C1 aqueous solution (3 mL) and extracted with EtOAc (3 x 4 mL). The
combined
organic layers were concentrated and purified by preparative silica thin layer
chromatography (7% MeOH/CH2C12 with 0.2% NH3) to provide 5-chloro-N2-(2,5-
dimethyl-4-(4-cis-morpholinocyclo-hexyl)phenyl)-N4-(5-methyl-1H-pyrazol-3-
yl)pyrimidine-2,4-diamine as the less polar isomer; iH NMR (400 MHz, DMSO-d6)
6
12.01 (br, 1H), 8.49 (br, 2H), 7.98 (s, 1H), 7.10 (s, 1H), 7.00 (s, 1H), 6.13
(br, 1H),
3.64 (m, 4H), 2.75 (m, 1H), 2.41 (m, 4H), 2.24 (s, 3H), 2.00-2.20 (m, 9H),
1.70-1.85
(m, 2H), 1.35-1.55 (m, 4H); ESMS m/z 496.3 (M + H+) and 5-chloro-N2-(2,5-
dimethyl-4-(4-trans-morpholinocyclohexyl)phenyl)-N4-(5-methyl-1H-pyrazol-3-
yl)pyrimidine-2,4-diamine as the more polar isomer; iH NMR (400 MHz, DMSO-d6)
6 12.01 (br, 1H), 8.47 (br, 2H), 7.98 (s, 1H), 7.14 (s, 1H), 7.02 (s, 1H),
6.13 (br, 1H),
3.57 (br, 4H), 2.60 (m, 1H), 2.50 (br, 4H), 2.32 (m, 1H), 2.22(s, 3H), 2.13
(s, 3H),
2.10 (s, 3H), 1.93 (b, 2H), 1.79 (b, 2H), 1.30-1.55 (m, 4H); ESMS m/z 496.3 (M
+
H+).
Example 6
5-Chloro-N2-(2,5-dimethyl-4-(morpholinomethl)phenyl)-N4-(5-meth, ll
pyrazol-3-yl)pyrimidine-2,4-diamine (39)
HN-N CIrN N
N N N
H H
[0113] Step 1: A mixture of 2,5-dichloro-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-
4-amine (732 mg, 3 mmol), 4-bromo-2,5-dimethylaniline (554 mg, 2 mmol) and
conc.
aqueous HCl (1.5 mL) in iso-propanol (15 mL) was heated in a microwave for 40
min
at 130 V. LCMS showed that the reaction was not complete, and additional2,5-
dichloro-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (732 mg, 3 mmol) was
added to the reaction. The reaction was again heated in a microwave for an
additional
60 min at 130 V. The crude reaction mixture was then diluted with EtOAc (100
mL),
sequentially washed with saturated aqueous NaHCO3 (2x20 mL) and brine (10 mL),
dried over Na2CO3 and concentrated in vacuo. The crude product was purified by
silica chromatography (0-10% MeOH in EtOAc gradient with 1% NH3 additive) to
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
give N2-(4-bromo-2,5-dimethylphenyl)-5-chloro-N4-(5-methyl-1H-pyrazol-3-
yl)pyrimidine-2,4-diamine as an off-white solid; ESMS m/z 407.2 (M+H+).
[0114] Step 2: A mixture of N2-(4-bromo-2,5-dimethylphenyl)-5-chloro-N4-(5-
methyl-1H-pyrazol-3-yl)pyrimidine-2,4-diamine (41 mg, 0.1 mmol), potassium 1-
trifluoroboratomethylmorpholine (43 mg, 0.2 mmol), Pd(OAc)2 (3 mg, 0.013
mmol),
Xantphos (12 mg, 0.025 mmol) and Cs2CO3 (98 mg, 0.3 mmol) in THE (1 mL)/H20
(0.1 mL) was degassed by a stream of argon gas. The vial was sealed and heated
in a
microwave for 30 min at 160 T. Additional potassium 1-trifluoroborato-
methylmorpholine (69 mg), Xantphos (18 mg) and Cs2CO3 (98 mg) were added. The
reaction was again heated in the microwave for an additional 30 min at 160 V.
The
crude product is purified by RP-HPLC to give 5-Chloro-N2-(2,5-dimethyl-4-
(morpholinomethyl)phenyl)-N4-(5-methyl-iH-pyrazol-3-yl)pyrimidine-2,4-diamine
as a white powder; ESMS m/z 428.2 (M+H+).
Example 7
4-(2,5-dimethyl-4-(4-(5-meth pyrazol-3-ylamino)-5-(trifluoromethyl)pyrimidin-
2-ylamino)phenyl)cyclohexanone (40)
F O
F
F
HN N N
H
N
HN
[0115] A mixture of 2,5-dimethyl-4-(1,4-dioxaspiro[4.5]decan-8-
yl)aniline(111.3
mg, 0.43 mmol), 2-chloro-N-(5-methyl-1H-pyrazol-3-yl)-5-
(trifluoromethyl)pyrimidin-4-amine (119.6 mg, 0.43 mmol), HCl (4 N in water,
0.11
mL, 0.43 mmol) in i-PrOH (4.0 mL) was heated at 125 C in an oil bath over
night.
The reaction mixture was concentrated in vacuo. The crude product was
dissolved in
THE (2 mL), MeOH (1 mL) and HCl (4N in water, 0.11 mL, 0.43 mmol), and stirred
at room temperature for 2 h. The reaction mixture was concentrated in vacuo,
followed by purification by silica chromatography (0-100% EtOAc in hexanes
gradient) to afford 4-(2,5-dimethyl-4-(4-(5-methyl-1H-pyrazol-3-ylamino)-5-
(trifluoromethyl)pyrimidin-2-ylamino)phenyl)cyclohexanone; ESMS m/z 459.2 (M +
H+).
46
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
Example 8
trans-4-(2,5-dimethyl-4-(4-(5-methyl-IH-pyrazol-3-ylamino)-5-(trifluoromethyl)
pyrimidin-2-ylamino)phenyl)cyclohexanol (41)
F ,%0H
F
F ~N
HN N N
H
N
HN
[0116] To a solution of 4-(2,5-dimethyl-4-(4-(5-methyl-iH-pyrazol-3-ylamino)-5-
(trifluoromethyl)pyrimidin-2-ylamino)phenyl)cyclohexanone (100 mg, 0.22 mmol)
in
MeOH (15 mL) was added NaBH4 (33.2 mg, 0.87 mmol). The reaction was stirred 30
for min., concentrated and purified by silica chromatography (MeOH / DCM:
8:92) to
afford trans-4-(2,5-dimethyl-4-(4-(5-methyl-1H-pyrazol-3-ylamino)-5-
(trifluoromethyl)pyrimidin-2-ylamino)phenyl)cyclohexanol; Rf. 0.35 (silica;
MeOH/DCM 8:92); ESMS m/z 461.2 (M + H+).
Example 9
cis-4-(2,5-dimethyl-4-(4-(5-methyl- pyrazol-3-ylamino)-5-
(trifluoromethyl)pyrimidin-2-ylamino)phenyl)cyclohexanol (42)
F ..O H
F
F N '~~~
HN N N
H
N
HN
[0117] To a solution of 4-(2,5-dimethyl-4-(4-(5-methyl-iH-pyrazol-3-ylamino)-5-
(trifluoromethyl)pyrimidin-2-ylamino)phenyl)cyclohexanone (100 mg, 0.22 mmol)
in
MeOH (15 mL) was added NaBH4 (33.2 mg, 0.87 mmol). The reaction was stirred
for 30 mins., concentrated and purified by silica chromatography (MeOH / DCM:
8:92) to afford cis-4-(2,5-dimethyl-1H-pyrazol-3-ylamino)-5-
(trifluoromethyl)pyrimidin-2-ylamino)phenyl)cyclohexanol; Rf. 0.45 (silica;
MeOH/DCM 8:92); ESMS m/z 461.2 (M + H+).
47
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
Example 10
2-(2-fluoro-5-methyl-4-(cis-4-(piperidin-1-~yclohexl)phenyl)-N4
N -(5-meth, ll
pyrazol-3-yl)-5-(trifluoromethyl)pyrimidine-2,4-diamine (62)
N
F
F
HN N N
H F
N
HN
[0118] A mixture of 4-(5-fluoro-2-methyl-4-(4-(5-methyl-iH-pyrazol-3-ylamino)-
5-(trifluoromethyl)pyrimidin-2-ylamino)phenyl)cyclohexanone (30 mg, 0.065
mmol),
piperidine (30 L, 0.174 mmol) and acetic acid (75 L, 0.13 mmol) was stirred
at
room temperature for 1 h. Sodium triacetoxyborohydride (27.4 mg, 0.13 mmol)
was
added to the reaction and the reaction mixture was stirred overnight. The
mixture was
concentrated and purified by silica chromatography (MeOH/DCM 8:92) to afford
N2-
(2-fluoro-5-methyl-4-(cis-4-(piperidin-1-yl)cyclohexyl)phenyl)-N4-(5-methyl-1
H-
pyrazol-3-yl)-5-(trifluoromethyl)pyrimidine-2,4-diamine; RI. 0.41 (Silica;
MeOH/DCM 8:92); ESMS m/z 532.3 (M + H+).
Example 11
2-(2-fluoro-5-methyl-4-(trans-4-(piperidin-1-~yclohex~phenyl)-N4
N -(5-meth
1H-pyrazol-3-yl)-5-(trifluoromethyl)pyrimidine-2,4-diamine (63)
,.N
F
F N
1
HN N N
H F
N
HN
[0119] A mixture of 4-(5-fluoro-2-methyl-4-(4-(5-methyl-iH-pyrazol-3-ylamino)-
5-(trifluoromethyl)pyrimidin-2-ylamino)phenyl)cyclohexanone (30 mg, 0.065
mmol),
piperidine (30 L, 0.174 mmol) and acetic acid (75 L, 0.13 mmol) was stirred
at
room temperature for 1 h. Sodium triacetoxyborohydride (27.4 mg, 0.13 mmol)
was
48
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
added to the reaction and the reaction mixture was stirred overnight. The
mixture was
concentrated and purified by silica chromatography (MeOH/DCM 8:92) to afford
N2-
(2-fluoro-5-methyl-4-(trans-4-(piperidin-1-yl)cyclohexyl)phenyl)-N4-(5-methyl-
1 H-
pyrazol-3-yl)-5-(trifluoromethyl)pyrimidine-2,4-diamine; Rf. 0.32 (Silica;
MeOH/DCM 8:92); ESMS m/z 532.3 (M + H+).
Example 12
2-(2,5-dimethyl-4-(cis-4-morpholinocyclohex~phenyl)-N4
N -(5-methyl- IH-pyrazol-
3-yl)-5-(trifluoromethyl)pyrimidine-2,4-diamine (66)
F N
F
HN N N
H
N/
HN
[0120] A mixture of 4-(2,5-dimethyl-4-(4-(5-methyl-iH-pyrazol-3-ylamino)-5-
(trifluoromethyl)pyrimidin-2-ylamino)phenyl)cyclohexanone (40 mg, 0.087 mmol),
morpholine (15 L, 0.174 mmol) and acetic acid (75 L, 0.13 mmol) was stirred
at
room temperature for 1 h. Sodium cyanoborohydride (8.2 mg, 0.13 mmol) was
added
to the reaction and the reaction mixture was stirred overnight. The mixture
was
concentrated and purified by silica chromatography (MeOH/DCM 8:92) to afford
N2-
(2,5-dimethyl-4-(cis-4-morpholinocyclohexyl)phenyl)-N4-(5-methyl-1H-pyrazol-3-
yl)-5-(trifluoromethyl)pyrimidine-2,4-diamine; Rf. 0.50 (Silica; MeOH/DCM
8:92);
ESMS m/z 530.3 (M + H+).
Example 13
2-(2,5-dimethyl-4-(trans-4-morpholinocyclohexl)phenyl)-N4
N -(5-meth
pyrazol-3-yl)-5-(trifluorometh l)p rimidine-2,4-diamine (67)
49
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
F N
F SIN
HN N N
H
N
HN
[0121] A mixture of 4-(2,5-dimethyl-4-(4-(5-methyl-iH-pyrazol-3-ylamino)-5-
(trifluoromethyl)pyrimidin-2-ylamino)phenyl)cyclohexanone (40 mg, 0.087 mmol),
morpholine (15 L, 0.174 mmol) and acetic acid (75 L, 0.13 mmol) was stirred
at
room temperature for 1 h. Sodium cyanoborohydride (8.2 mg, 0.13 mmol) was
added
to the reaction and the reaction mixture was stirred overnight. The mixture
was
concentrated and purified by silica chromatography (MeOH/DCM 8:92) to afford
N2-
(2,5-dimethyl-4-(trans-4-morpholinocyclohexyl)phenyl)-N4-(5-methyl-1H-pyrazol-
3-
yl)-5-(trifluoromethyl)pyrimidine-2,4-diamine; RI. 0.38 (Silica; MeOH/DCM
8:92);
ESMS m/z 530.3 (M + H+).
[0122] The following compounds in Table 1 are obtained by repeating the
procedures described in examples above and using appropriate starting
materials.
Table 1
STRUCTURE NMR or ESMS IGF1R
Ba/F3
IC50
(uM)
1 N/ ESMS m/z 413.1 (M + 0.067
A H+).
HN-NCI
N N N
H H
5-chloro-N2-(2-fluoro-5-methyl-4-(1-methyl-1 H-
pyrazol-4-yl)phenyl)-N4-(5 -methyl-1 H-pyrazol-3 -
yl)pyrimidine-2,4-diamine
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
STRUCTURE NMR or ESMS IGF1R
Ba/F3
IC50
(um)
2 N ESMS m/z 410.1 (M + 0.103
~ H+).
HN- Cl N N N
H H F
5-chloro-N2-(2-fluoro-5-methyl-4-(pyridin-4-
yl)phenyl)-N4-(5-methyl-1 H-pyrazol-3-
yl)pyrimidine-2,4-diamine
3 N ESMS m/z 410.1 (M + 0.067
H+).
HN- CI
N N N
H H
-chloro-N2-(2-fluoro-5 -methyl-4-(pyridin-3 -
yl)phenyl)-N4-(5-methyl-1 H-pyrazol-3-
yl)pyrimidine-2,4-diamine
4 1H NMR (400 MHz, 0.089
N i DMSO-d6) 6 9.22 (br,
H 2H), 8.85 (m, 1H), 8.63
HN -NCI N / (m, 1H), 8.16 (s, 1H),
\ 8.09 (m, 1H), 8.07 (m,
H H 1H), 7.85 (br, 1H), 7.27
F (m, 1H), 6.19 (s, 1H),
5-(4-(5-chloro-4-(5-methyl-lH-pyrazol-3- 2.84 (d, 3H), 2.20 (s,
ylamino)pyrimidin-2-ylamino)-5-fluoro-2- 3H), 2.15 (s, 3H); ESMS
methylphenyl)-N-methylpicolinamide m/z 467.1 (M + H+).
5 N ESMS m/z 424.1 (M + 0.137
H+).
HN- CI /
N N N
H H F
5-chloro-N2-(2-fluoro-5-methyl-4-(6-
methylpyridin-3-yl)phenyl)-N4-(5-methyl-lH-
pyrazol-3 -yl)pyrimidine-2, 4-diamine
51
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
STRUCTURE NMR or ESMS IGF1R
Ba/F3
IC50
(um)
6 N ESMS m/z 410.1 (M + 0.294
H+).
/
HN- CI N
N N N
H H F
5-chloro-N2-(2-fluoro-5-methyl-4-(pyridin-2-
yl)phenyl)-N4-(5-methyl-1 H-pyrazol-3-
yl)pyrimidine-2,4-diamine
7 N \ ESMS m/z 460.1 (M + 0.579
H+).
HN- CI /
N N N
H H F
5-chloro-N2-(2-fluoro-5-methyl-4-(quinolin-3-
yl)phenyl)-N4-(5-methyl-1 H-pyrazol-3-
yl)pyrimidine-2,4-diamine
8 N OH ESMS m/z 426.1 (M + 0.055
HN- CI H+).
N N N
H H F
5-(4-(5-chloro-4-(5-methyl-lH-pyrazol-3-
ylamino)pyrimidin-2-ylamino) -5 -fluoro-2-
methylphenyl)pyridin-2-ol
9 ESMS m/z 424.1 (M + 0.094
H+).
HN- Cl N N N
H H F
5-chloro-N2-(2-fluoro-5-methyl-4-(2-
methylpyridin-4-yl)phenyl)-N4-(5-methyl-1 H-
pyrazol-3 -yl)pyrimidine-2, 4-diamine
52
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
STRUCTURE NMR or ESMS IGF1R
Ba/F3
IC50
(um)
S ESMS m/z 415.1 (M + 0.833
HN-NCI H+).
N N N
H H F
5-chloro-N2-(2-fluoro-5-methyl-4-(thiophen-3-
yl)phenyl)-N4-(5-methyl -1 H-pyrazol-3-
yl)pyrimidine-2,4-diamine
11 0 ESMS m/z 399.1 (M + 0.683
HN-NCI N H+).
N N N
H H F
5-chloro-N2-(2-fluoro-4-(furan-3-yl)-5-
methylphenyl)-N4-(5-methyl-lH-pyrazol-3-
yl)pyrimidine-2,4-diamine
12 N 1H NMR (400 MHz, 0.025
I 'N DMSO-d6) 6 9.67(br,
HN-NCI 2H), 9.30 (br, 1H), 8.12
(s, 1H), 7.96 (s, 1H),
H N N 7.68 (s, 1H), 7.31 (s,
1H), 7.28 (s, 1H), 6.97
5-chloro-N2-(2,5-dimethyl-4-(1-methyl-1H- (s, 1H), 3.89 (s, 3H),
pyrazol-4-yl)phenyl)-N4-(5-methyl-lH-pyrazol-3- 2.33 (s, 3H), 2.18 (s,
yl)pyrimidine-2,4-diamine 3H), 2. 07 (s, 3H);
ESMS m/z 409.1 (M +
H+)
13 N ESMS m/z 406.1 (M + 0.029
H+).
HN- CI
N
N N N
H H
5-chloro-N2-(2-fluoro-4-(furan-3-yl)-5-
methylphenyl)-N4-(5-methyl-lH-pyrazol-3-
yl)pyrimidine-2,4-diamine
53
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
STRUCTURE NMR or ESMS IGF1R
Ba/F3
IC50
(um)
14 0 ESMS m/z 463.2 (M + 0.015
~N NH H+).
HN-N CI \
N N N
H H
5-(4-(5-chloro-4-(5-methyl-lH-pyrazol-3-
ylamino)pyrimidin-2-ylamino)-2,5-
dimethylphenyl)-N-methylpicolinamide
15 ESMS m/z 420.2 (M + 0.039
~ H+).
HN- Cl
\
N N N
H H
5-chloro-N2-(2,5-dimethyl-4-(2-methylpyridin-4-
yl)phenyl)-N4-(5-methyl-1 H-pyrazol-3-
yl)pyrimidine-2,4-diamine
16 N ESMS m/z 420.2 (M + 0.030
H+).
HN-NCI / INII
N N N
H H
5-chloro-N2-(2,5-dimethyl-4-(6-methylpyridin-3-
yl)phenyl)-N4-(5-methyl-1 H-pyrazol-3-
yl)pyrimidine-2,4-diamine
17 HN 1H NMR (400 MHz, 0.068
HN-N Cl N N DMSO-d6) 6 9.01 (br,
I~ 2h), 8.83 (s, lh), 8.12 (s,
H N H 1H), 7.83 (s, 2H(, 7.78
(s, 1H), 7.45 (s, 1H),
N2-(4-(1H-imidazol-2-yl)-2,5-dimethylphenyl)-5- 6.20 (s, 1H), 2.30 (s,
chloro-N4-(5-methyl-lH-pyrazol-3-yl)pyrimidine- 3H), 2.29 (s, 3H), 2.15
2,4-diamine (s, 3H); ESMS m/z
395(M + H+).
54
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
STRUCTURE NMR or ESMS IGF1R
Ba/F3
IC50
(um)
18 o-N 1H NMR (400 MHz, 0.276
HN-N / DMSO-d6) 6 9.3 (br,
N I 2H), 9.0 (br, 1H), 8.13
H N H (s, 1H), 7.58 (s, 1H),
7.55 (s, 1H), 6.64 (s,
5-chloro-N2-(2,5-dimethyl-4-(3-methylisoxazol-5- 1H), 6.07 (s, 1H), 2.39
yl)phenyl)-N4-(5-methyl-1H-pyrazol-3- (s, 3H), 2.30 (s, 3H),
yl)pyrimidine-2,4-diamine 2.25 (s, 3H), 2.09 (s,
3H); ESMS m/z 410.1
(M + H+).
19 HN ESMS m/z 399.1 (M + 0.288
HN- CI N H+).
N N N
H H F
5-chloro-N2-(2-fluoro-4-(1 H-imidazol-2-yl)-5-
methylphenyl)-N4-(5-methyl-1H-pyrazol-3-
yl)pyrimidine-2,4-diamine
20 OWN 1H NMR (400 MHz, 0.109
HN-NCI DMSO-d6) cS 9.40 (br,
2H), 9.14 (br 1H), 8.47
H N H (s, 1H) 8.14 (s, 1H),
7.56 (s, 1H), 7.48 (s,
5-chloro-N2-(2,5-dimethyl-4-(oxazol-5-yl)phenyl)- 1H), 7.45 (s, 1H), 6.03
N4-(5-methyl-1H-pyrazol-3-yl)pyrimidine-2,4- (s, 1H), 2.39 (s, 3H),
diamine 2.24 (s, 3H), 2.06 (s,
3H); ESMS m/z 396.1
(M + H+).
21 O--\\ ESMS m/z 412.1 (M + 0.169
HN-NCI / IIN / N H+).
N N N
H H OMe
5-chloro-N2-(2-methoxy-5-methyl-4-(oxazol-5-
yl)phenyl)-N4-(5-methyl-1 H-pyrazol-3-
yl)pyrimidine-2,4-diamine
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
STRUCTURE NMR or ESMS IGF1R
Ba/F3
IC50
(um)
22 HN-N ESMS m/z 409.2 (M + 0.166
H N-NCI / INII H+).
N N N
H H
5-chloro-N2-(2,5-dimethyl-4-(3-methyl-lH-
pyrazol-5-yl)phenyl)-N4-(5-methyl-lH-pyrazol-3-
yl)pyrimidine-2,4-diamine
23 -N 1H NMR (400 MHz,
HN-NCIN DMSO-d6) 6 9.3 (br,
1 \ I 2H), 9.0 (br, 1H), 8.13
H N H (s, 1H), 7.58 (s, 1H),
7.55 (s, 1H), 6.64 (s,
5-chloro-N2-(2,5-dimethyl-4-(3-methylisoxazol-5- 1H), 6.07 (s, 1H), 2.39
yl)phenyl)-N4-(5-methyl-lH-pyrazol-3- (s, 3H), 2.30 (s, 3H),
yl)pyrimidine-2,4-diamine 2.25 (s, 3H), 2.09 (s,
3H); ESMS m/z 410.1
(M + H+).
24 s 1H NMR (400 MHz, 0.106
HN-NCI N i I N~ DMSO-d6) 9.56 (br,
1~ v 2H), 9.25 (br, 1H), 8.14
H N N (s, 2H), 7.56 (s, 1H),
7.53 (s, 1H), 7.37 (s,
5-chloro-N2-(2,5-dimethyl-4-(2-methylthiazol-4- 1H), 6.07 (s, 1H), 2.71
yl)phenyl)-N4-(5-methyl-lH-pyrazol-3- (s, 3H), 2.37 (s, 3H),
yl)pyrimidine-2,4-diamine 2.21 (s, 3H), 2.09 (s,
3H); ESMS m/z 426.1
(M + H+).
25 N ESMS m/z 409.2 (M + 0.071
H+).
H\_N CI N
N N N
H H
5-chloro-N2-(2,5-dimethyl-4-(1-methyl-lH-
imidazol-2-yl)phenyl)-N4-(5-methyl-1 H-pyrazol-
3-yl)pyrimidine-2,4-diamine
56
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
STRUCTURE NMR or ESMS IGF1R
Ba/F3
IC50
(um)
26 HN 1H NMR (400 MHz,
HN-N 01N / N DMSO-d6) 9.01 (br,
k I
2h), 8.83 (s, lh), 8.12 (s,
H N N 1H), 7.83 (s, 2H(, 7.78
(s, 1H), 7.45 (s, 1H),
N2-(4-(1H-imidazol-2-yl)-2,5-dimethylphenyl)-5- 6.20 (s, 1H), 2.30 (s,
chloro-N4-(5-methyl-iH-pyrazol-3-yl)pyrimidine- 3H), 2.29 (s, 3H), 2.15
2,4-diamine (s, 3H); ESMS m/z
395.1 (M + H+).
27 F N N ESMS m/z 412.8 (M + 0.549
\ Y H+)'
N / CI
N/ ~ HN
N t"NH
-chloro-N2-(5 -fluoro-2-methyl-4-(1-methyl-1 H-
pyrazol-4-yl)phenyl)-N4-(5 -methyl-1 H-pyrazol-3 -
yl)pyrimidine-2,4-diamine
28 ESMS m/z 430.2 (M + 0.141
HO---,- ~N NNJN H+)
H H
1-(4-(5-chloro-4-(5-methyl-1H-pyrazol-3-
ylamino)pyrimidin-2-ylamino)-2,5-
dimethylphenyl)ethanone O-2-hydroxyethyl
oxime
29 ESMS m/z 386.1 (M + 0.051
HON CI N-NH H+)
N N N
H H
1-(4-(5-chloro-4-(5-methyl-1H-pyrazol-3-
ylamino)pyrimidin-2-ylamino)-2,5-
dimethylphenyl)ethanone oxime
57
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
STRUCTURE NMR or ESMS IGF1R
Ba/F3
IC50
(um)
30 0 ESMS m/z 400.2 (M + 0.539
\ 1 . , C' ~ ~1 1 4,./~
N N N
H H
4-(5-chloro-4-(5-methyl-iH-pyrazol-3-
ylamino)pyrimidin-2-ylamino)-N,N,2,5-
tetramethylbenzamide
31 I ESMS m/z 443.2 (M + 0.659
/N1 0 H+)
CI _NH
H \ I NJ1NJ, N N
H H
4-(5-chloro-4-(5-methyl-iH-pyrazol-3-
ylamino)pyrimidin-2-ylamino)-N-(2-
(dimethylamino)ethyl)-2,5-dimethylbenzamide
32 HN ESMS m/z 442.2 (M + 0.451
H+)
N
H
Air N
Ny N
HN
r o
NJ
O
(4-(5-chloro-4-(5-methyl-iH-pyrazol-3-
ylamino)pyrimidin-2-ylamino)-2,5-
dimethylphenyl) (morpholino)methanone
33 ESMS m/z 440.2 (M + 0.015
H+)
HN
N NH aN'OH
CI
I \
NN
N
H
4-(4-(5-chloro-4-(5-methyl-iH-pyrazol-3-
ylamino)pyrimidin-2-ylamino)-2,5-
dimethylphenyl)cyclohexanone oxime
58
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
STRUCTURE NMR or ESMS IGF1R
Ba/F3
IC50
(um)
34 ESMS m/z 400.2 (M + 0.269 '101 cl N N N
H H
1-(4-(5-chloro-4-(5-methyl-1H-pyrazol-3-
ylamino)pyrimidin-2-ylamino)-2,5-
dimethylphenyl)ethanone O-methyl oxime
35 H H ESMS m/z 457.2 (M + 0.121
/ NYNYN I I H+)
HN-N ClD IIN N-0i N,,
1-(4-(5-chloro-4-(5-methyl-1H-pyrazol-3-
ylamino)pyrimidin-2-ylamino)-2,5-
dimethylphenyl)ethanone 0-2-
(dimethylamino)ethyl oxime
36 ESMS m/z 499.2 (M + 0.187
l N -NH H+)
(N/\/O,N I N N N
H H
1-(4-(5-chloro-4-(5-methyl-1H-pyrazol-3-
ylamino)pyrimidin-2-ylamino)-2,5-
dimethylphenyl)ethanone 0-2-
morpholinoethyl oxime
37 ESMS m/z 496.3 (M + 0.011
HN, H+)
N NH
cI
N N
H
Trans -5 -chloro-N2- (2,5 -dimethyl-4- (4-
morpholinocyclohexyl)phenyl)-N4-(5-methyl-
1 H-pyrazol-3-yl)pyrimidine-2,4-diamine
59
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
STRUCTURE NMR or ESMS IGF1R
Ba/F3
IC50
(um)
38 ESMS m/z 496.3 (M + 0.009
H+)
HN,
N NH
C I IN
N N
H
Cis -5 -chloro-N2- (2,5 -dimethyl-4- (4-
morpholinocyclohexyl)phenyl)-N4-(5-methyl-
1 H-pyrazol-3-yl)pyrimidine-2,4-diamine
39 N NII Cl N-NH ESMS m/z 428.2 (M + 0.151
OJ ')C:CN H)
H N H
5-chloro-N2-(2,5-dimethyl-4-
(morpholinomethyl)phenyl)-N4-(5-methyl-1H-
pyrazol-3-yl)pyrimidine-2,4-diamine
40 ESMS m/z 459.2 (M + 0.049
,NH H+)
FHN N
F
N
F \ I \
NN
H
4-(2,5-dimethyl-4-(4-(5-methyl-1H-pyrazol-3-
ylamino)-5 -(trifluoromethyl)pyrimidin-2-
ylamino)phenyl)cyclohexanone
41 ESMS m/z 461.2 (M + 0.025
H+)
F F HN 4NH ,,OH
F N I \
N N
H
trans -4-(2,5-dimethyl-4-(4-(5-methyl-1H-
pyrazol-3-ylamino)-5-
(trifluoromethyl)pyrimidin-2-
ylamino)phenyl)cyclohexanol
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
STRUCTURE NMR or ESMS IGF1R
Ba/F3
IC50
(um)
42 ESMS m/z 461.2 (M + 0.053
H+)
F F HN NH ,,OH
N
N
F I \ I \~`
N N
H
Cis-4-(2,5-dimethyl-4-(4-(5-methyl-lH-
pyrazol-3-ylamino)-5-
(trifluoromethyl)pyrimidin-2-
ylamino)phenyl)cyclohexanol
43 ESMS m/z 494.3 (M + 0.008
H+)
HN, N
N NH
CI N
N N
H
Cis-5-chloro-N2-(2,5-dimethyl-4-(4-
(piperidin-1-yl)cyclohexyl)phenyl)-N4-(5-
methyl-1H-pyrazol-3-yl)pyrimidine-2,4-
diamine
44 ESMS m/z 494.3 (M + 0.007
H+)
HN, %N
N NH
CI
N I
N N
H
Trans -5-chloro-N2-(2,5-dimethyl-4-(4-
(piperidin-1-yl)cyclohexyl)phenyl)-N4-(5-
methyl-1H-pyrazol-3-yl)pyrimidine-2,4-
diamine
61
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
STRUCTURE NMR or ESMS IGF1R
Ba/F3
IC50
(um)
45 ESMS m/z 427.2 (M + 0.014
H+)
HO NH
HN N
''/ NY
N~N
C'
H
trans-4-(4-(5-chloro-4-(5-methyl-lH-pyrazol-
3-ylamino)pyrimidin-2-ylamino)-2,5-
dimethylphenyl)cyclohexanol
46 ESMS m/z 466.2 (M + 0.016
H H+)
HN,
N NH
CI
N
N N
H
Trans- 5-chloro-N2-(4-(4-
(cyclopropylamino)cyclohexyl)-2,5-
dimethylphenyl)-N4-(5-methyl-1H-pyrazol-3-
yl)pyrimidine-2,4-diamine
47 ESMS m/z 466.2 (M + 0.017
H+)
HN, H
N NH
N
N N
H
Cis-5-chloro-N2-(4-(4-
(cyclopropylamino)cyclohexyl)-2,5-
dimethylphenyl)-N4-(5-methyl-1H-pyrazol-3-
yl)pyrimidine-2,4-diamine
62
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
STRUCTURE NMR or ESMS IGF1R
Ba/F3
IC50
(um)
48 ESMS m/z 498.3 (M + 0.021
H+)
HN, N
N NH
CI I N
N N
H F
Cis-5-chloro-N2-(2-fluoro-5-methyl-4-(4-
(piperidin-1-yl)cyclohexyl)phenyl)-N4-(5-
methyl-1H-pyrazol-3-yl)pyrimidine-2,4-
diamine
49 ESMS m/z 498.3 (M + 0.040
rD H+)
HN, N
N ~NH
CI
N N
H
Trans -5-chloro-N2-(2-fluoro-5-methyl-4-(4-
(piperidin-1-yl)cyclohexyl)phenyl)-N4-(5-
methyl-1H-pyrazol-3-yl)pyrimidine-2,4-
diamine
50 ESMS m/z 500.2 (M + 0.031
_ ro H+)
HN, N
N NH
CI
N N
H F
Trans -5 -chloro-N2- (2-fluoro-5 -methyl-4- (4-
morpholinocyclohexyl)phenyl)-N4-(5-methyl-
1 H-pyrazol-3-yl)pyrimidine-2,4-diamine
63
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
STRUCTURE NMR or ESMS IGF1R
Ba/F3
IC50
(um)
51 ESMS m/z 500.2 (M + 0.400
ro H+)
HN N NH
CI I ~N I ~~,.=
N N
H F
Cis -5 -chloro-N2- (2-fluoro- 5 -methyl-4- (4-
morpholinocyclohexyl)phenyl)-N4-(5-methyl-
1 H-pyrazol-3-yl)pyrimidine-2,4-diamine
52 ESMS m/z 513.3 (M + 0.024 N H N\ T/N , ~ N H+)
HNN CI" N F
H
H
Cis -5 -chloro-N2- (2-fluoro- 5 -methyl-4- (4- (4-
methylpiperazin- l -yl)cyclohexyl)phenyl)-N4-
(5-methyl-iH-pyrazol-3-yl)pyrimidine-2,4-
diamine
53 ESMS m/z 513.3 (M + 0.025
~ ' N N\N llz~ ~ N H+)
T N
H N I N
CIS\i F H
H
Trans- 5 -chloro-N2- (2-fluoro- 5 -methyl-4- (4- (4-
methylpiperazin- l -yl)cyclohexyl)phenyl)-N4-
(5-methyl-iH-pyrazol-3-yl)pyrimidine-2,4-
diamine H 54 N H NON OH
~ ESMS m/z 500.2 (M + 0.016
/ ., H+)
HN-N CI" F
H
H
(S)-1-((Cis)-4-(4-(5-chloro-4-(5-methyl-1H-
pyrazol-3-ylamino)pyrimidin-2-ylamino)-5-
fluoro-2-methylphenyl)cyclohexyl)pyrrolidin-
3-ol
64
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
STRUCTURE NMR or ESMS IGF1R
Ba/F3
IC50
(um)
55 H H ,oH
ESMS m/z 500.2 (M + 0.032
/ ' N N 'r I H~, H+)
HN'N I N /
Cl F OOH
(S)-1-((Trans)-4-(4-(5-chloro-4-(5-methyl-1H-
pyrazol-3-ylamino)pyrimidin-2-ylamino)-5-
fluoro-2-methylphenyl)cyclohexyl)pyrrolidin-
3-ol
56 H H ESMS m/z 486.3 (M + 0.025
H+)
/ ~ N ~NN N-/
HNN N
Cl F H
5-chloro-N2-(4-((cis)-4-
(diethylamino)cyclohexyl)-2-fluoro-5-
methylphenyl)-N4-(5-methyl-1H-pyrazol-3-
yl)pyrimidine-2,4-diamine
57 H N H ESMS m/z 486.3 (M + 0.031
Y l H)
HN-N
Cl N F H
H
5-chloro-N2-(4-((trans)-4-
(diethylamino)cyclohexyl)-2-fluoro-5-
methylphenyl)-N4-(5-methyl-1H-pyrazol-3-
yl)pyrimidine-2,4-diamine
58 H H NH ESMS m/z 499.2 (M + 0.017
N NN r I J H+)
N
HNN Cl N F
H
H
5-chloro-N2-(2-fluoro-5-methyl-4-((cis)-4-
(piperazin-1-yl)cyclohexyl)phenyl)-N4-(5-
methyl-1H-pyrazol-3-yl)pyrimidine-2,4-
diamine
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
STRUCTURE NMR or ESMS IGF1R
Ba/F3
IC50
(um)
59 H H NH ESMS m/z 499.2 (M + 0.039
N N N H+)
N
HN-N C~" N F
H
H
5-chloro-N2-(2-fluoro-5-methyl-4-((trans)-4-
(piperazin-1-yl)cyclohexyl)phenyl)-N4-(5-
methyl-1 H-pyrazol-3-yl)pyrimidine-2,4-
diamine
60 F F ESMS m/z 528.3 (M + 0.004
F HN -<N-NH H+)
N
N=
HN 01,,=0= INO
N2-(2,5-dimethyl-4-((cis)-4-(piperidin-1-
yl)cyclohexyl)phenyl)-N4-(5-methyl-1 H-
pyrazol-3-yl)-5- (trifluoromethyl)pyrimidine-
2,4-diamine
61 F F ESMS m/z 528.3 (M + 0.003
F HN N-NH H+)
N --~
HN ",N,
N2-(2,5-dimethyl-4-((trans)-4-(piperidin-1-
yl)cyclohexyl)phenyl)-N4-(5-methyl-1 H-
pyrazol-3-yl)-5- (trifluoromethyl)pyrimidine-
2,4-diamine
62 F F ESMS m/z 532.3 (M + 0.018
F HN N-NH H+)
N
N=~
HN õO, ENO
F
N2-(2-fluoro-5-methyl-4-((cis)-4-(piperidin-1-
yl)cyclohexyl)phenyl)-N4-(5-methyl-1 H-
pyrazol-3-yl)-5- (trifluoromethyl)pyrimidine-
2,4-diamine
66
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
STRUCTURE NMR or ESMS IGF1R
Ba/F3
IC50
(um)
63 F F ESMS m/z 532.3 (M + 0.016
/\ N~
F HN \ NH H+)
N
N=<
HN ..IN. )
F ~/
N2-(2-fluoro-5-methyl-4-((trans)-4-(piperidin-
1-yl)cyclohexyl)phenyl)-N4-(5-methyl-1H-
pyrazol-3-yl)-5- (trifluoromethyl)pyrimidine-
2,4-diamine
64 lH F F ESMS m/z 534.3 (M + 0.05
H.,, F N H+)
rN F
H N NH
O
N
HN
N2-(2-fluoro-5-methyl-4-((cis)-4-
morpholinocyclohexyl)phenyl)-N4- (5-methyl-
1H-pyrazol-3-yl)-5-
(trifluoromethyl)pyrimidine-2,4-diamine
65 H F F ESMS m/z 534.3 (M + 0.032
H.,,~~ F H+)
N jj F
~ H N NH
O
A
N2-(2-fluoro-5-methyl-4-((trans)-4-
morpholinocyclohexyl)phenyl)-N4- (5-methyl-
1H-pyrazol-3-yl)-5-
(trifluoromethyl)pyrimidine-2,4-diamine
67
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
STRUCTURE NMR or ESMS IGF1R
Ba/F3
IC50
(um)
66 F F ESMS m/z 530.3 (M + 0.014
F HN NH H+)
/ N~
HN - O NO
N2- (2,5 -dimethyl-4- ((cis) -4-
morpholinocyclohexyl)phenyl)-N4-(5-methyl-
1H-pyrazol-3-yl)-5-
(trifluoromethyl)pyrimidine-2,4-diamine
67 F F ESMS m/z 530.3 (M + 0.009
F H N NH H+)
/ N~
N
N HN 0 ."N O
C/
N2-(2,5-dimethyl-4-((trans)-4-
morpholinocyclohexyl)phenyl)-N4- (5-methyl-
1H-pyrazol-3-yl)-5-
(trifluoromethyl)pyrimidine-2,4-diamine
68 lH F F ESMS m/z 550.3 (M + 0.12
N
H F - H+)
HO N F
N N NH
H
HHO.' H N
HN
(3S,4S)-1-((cis)-4-(5-fluoro-2-methyl-4-(4-(5-
methyl-1H-pyrazol-3-ylamino)-5-
(trifluoromethyl)pyrimidin-2-
ylamino)phenyl)cyclohexyl)pyrrolidine-3,4-
diol
68
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
STRUCTURE NMR or ESMS IGF1R
Ba/F3
IC50
(um)
69 H F F ESMS m/z 550.3 (M + 0.302
H~~ / F N )F H+)
HO \ N N NH
HH N
H
O H N
HN
(3S,4S)-1-((trans)-4-(5-fluoro-2-methyl-4-(4-
(5-methyl-1H-pyrazol-3-ylamino)-5-
(trifluoromethyl)pyrimidin-2-
ylamino)phenyl)cyclohexyl)pyrrolidine-3,4-
diol
H F F ESMS m/z 550.3 (M + 0.248
0,
F
H 0l N ~aN N NH
H
HH O ;H N
HN /
(3R,4R)-1-((trans)-4-(5-fluoro-2-methyl-4-(4-
(5-methyl-iH-pyrazol-3-ylamino)-5-
(trifluoromethyl)pyrimidin-2-
ylamino)phenyl)cyclohexyl)pyrrolidine-3,4-
diol
71 vH F F ESMS m/z 550.3 (M + 0.109
F
HOB9 ,
N N NH
H
HO H N4 /
HN
(3R,4R)-1-((cis)-4-(5-fluoro-2-methyl-4-(4-
(5-methyl-iH-pyrazol-3-ylamino)-5-
(trifluoromethyl)pyrimidin-2-
ylamino)phenyl)cyclohexyl)pyrrolidine-3,4-
diol
69
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
STRUCTURE NMR or ESMS IGF1R
Ba/F3
IC50
(um)
72 H N H ESMS m/z 431.2 (M + 0.099
~ i Y- Y, N::,,:::: H)
HN-N CI N F =0IOH
~=
Trans-4-(4-(5-chloro-4-(5-methyl-1H-pyrazol-
3-ylamino)pyrimidin-2-ylamino)-5-fluoro-2-
methylphenyl)cyclohexanol
73 H N H ESMS m/z 431.2 (M + 0.032
i,Y H)
HNN CI N F / ,,,.O-OH
H
Cis-4-(4-(5-chloro-4-(5-methyl-iH-pyrazol-3-
ylamino)pyrimidin-2-ylamino)-5-fluoro-2-
methylphenyl)cyclohexanol
74 ESMS m/z 460.2 (M + 0.087
H+)
HN,
N NH NH
CI
N N
H
5-chloro-N2-(2,5-dimethyl-4-(1,2,3,4-
tetrahydroisoquinolin-6-yl)phenyl)-N4-(5-
methyl-1H-pyrazol-3-yl)pyrimidine-2,4-
diamine
75 ESMS m/z 474.2 (M + 0.054
H+)
HN,
N NH I NN HN
CI
N N
H
5-chloro-N2-(2,5-dimethyl-4-(2-methyl-
1,2,3,4-tetrahydroisoquinolin-6-yl)phenyl)-N4-
(5-methyl-iH-pyrazol-3-yl)pyrimidine-2,4-
diamine
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
Example 14
6-(5-chloro-445-meth pyrazol-3-ylamino)pyrimidin-2-ylamino)-5-fluoro-2-
(piperidin-4-yl)isoindolin- l-one (79)
NH
O
N
HN-NCI
N N N
H H F
[0123] To the solution of tert-butyl 4- (6- amino- 5 -fluoro -I -oxoisoindolin-
2-
yl)piperidine-l-carboxylate (Intermediate 20, 18 mg, 0.05 mmol) and 2,5-
dichloro-4-
(5-methyl-iH-pyrazol-3-yl)pyrimidine (12 mg, 0.05 mmol) in 1 mL of i-PrOH was
added 5 drops of cone. aqueous HC1. The reaction mixture was heated at 150 C
in a
microwave reactor for 20 min, followed by concentration and purification with
preparative RP-HPLC to afford 6-(5-chloro-4-(5-methyl-iH-pyrazol-3-
ylamino)pyrimidin-2-ylamino)-5-fluoro-2-(piperidin-4-yl)isoindolin-l-one; ESMS
m/z 457.2 (M + H+).
Example 15
6-(5-chloro-4-(5-methylisoxazol-3-ylamino)pyrimidin-2-ylamino)-5-fluoro-2-
(piperidin-4-yl)isoindolin- l-one (80)
NH
O
N
O-NCI / II IN
N N N
H H F
[0124] To the solution of tert-butyl 4-(6-amino-5-fluoro-l-oxoisoindolin-2-
yl)piperidine-1-carboxylate (35 mg, 0.1 mmol) and 5-chloro-4-(5-methylisoxazol-
3-
yl)pyrimidin-2-amine (25 mg, 0.1 mmol) in 1 mL of i-PrOH was added 5 drops of
cone. aqueous HC1. The reaction mixture was heated at 150 C in microwave
reactor
for 20 min, followed by concentration and purification with preparative RP-
HPLC to
71
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
afford 6-(5-chloro-4-(5-methylisoxazol-3-ylamino)pyrimidin-2-ylamino)-5-fluoro-
2-
(piperidin-4-yl)isoindolin-l-one; ESMS m/z 458.1 (M + H+).
Example 16
2-(4-(6-(5-Chloro-4-(5-methyl- IH-pyrazol-3-ylamino)pyrimidin-2-ylamino)-4-
methylpyridin-3-l)piperidin-1-yl)acetamide (84)
H2NyN
0 /
N N NI/H
N N N
H H
[0125] Step 1: To a mixture of 5-bromo-4-methylpyridin-2-amine (200 mg, 1.07
mmol), tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-5,6-
dihydropyridine- 1(2H)-carboxylate (370 mg, 1.2 mmol) and sodium carbonate
(400
mg, 1.28 mmol) in DMF/H20 (8/2 mL) was added tetrakis(triphenylphosphine)
palladium (0) (62 mg, 5% mmol). The reaction tube is sealed, the mixture was
purged
with N2 for 3 min and then heated at 100 C under N2 for overnight. The
reaction was
cooled to room temperature and poured into saturated aqueous ammonia chloride
solution. The crude reaction mixture was extracted with ethyl acetate (3 x 15
mL).
The organic extracts were combined, washed with brine and concentrated. The
crude
product was purified with silica gel column chromatography (80% ethyl acetate
in
hexanes) to afford tert-butyl 4-(6-amino-4-methylpyridin-3-yl)-5,6-
dihydropyridine-
1(2H)-carboxylate as a yellow oil. The obtained oil was dissolved in methanol
(20
mL). To the solution was added Pd/C (10% w/w). The reaction mixture was
degassed
and purged with H2 for several times and then stirred under 1 atm. H2
overnight. The
mixture was filtered and concentrated to afford tert-butyl 4-(6-amino-4-
methylpyridin-3-yl)piperidine-l-carboxylate as a yellow solid; ESMS m/z 236 (M
-
56+ H+).
[0126] Step 2: To a mixture of 2,5-dichloro-N-(5-methyl-l-(tetrahydro-2H-pyran-
2-yl)-1H-pyrazol-3-yl)pyrimidin-4-amine (150 mg, 0.45 mmol), tert-butyl 4-(6-
amino-4-methylpyridin-3-yl)piperidine-l-carboxylate (120 mg, 0.41 mmol),
Xantphos
(24 mg, 0.04 mmol) and cesium carbonate (270 mg, 0.82 mmol) in THE (4 mL) was
added palladium acetate (5 mg, 0.02 mmol). The mixture was purged with
nitrogen
and the tube was sealed. The mixture was heated in an oil bath at 100 C for 5
h. The
mixture was filtered and concentrated. The residue was purified with silica
72
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
chromatography (70% ethyl acetate in hexanes) to afford tert-butyl 4-(6-(5-
chloro-4-
(5-methyl- l-(tetrahydro-2H-pyran-2-yl)-1H-pyrazol-3-ylamino)pyrimidin-2-
ylamino)-4-methylpyridin-3-yl)piperidine-1-carboxylate as a yellow solid; ESMS
m/z
583 (M+ H+).
[0127] Step 3: To a solution of tert-butyl 4-(6-(5-chloro-4-(5-methyl-l-
(tetrahydro-2H-pyran-2-yl)-1H-pyrazol-3-ylamino)pyrimidin-2-ylamino)-4-
methylpyridin-3-yl)piperidine-l-carboxylate in DCM (1 mL), was added TFA (1
mL).
The mixture was stirred for 1 h and concentrated to afford 5-chloro-N4-(5-
methyl-lH-
pyrazol-3-yl)-N2-(4-methyl-5-(piperidin-4-yl)pyridin-2-yl)pyrimidine-2,4-
diamine as
brownish oil. The product was used directly for subsequent reactions without
further
purification.
[0128] Step 4: To a mixture of 5-chloro-N4-(5-methyl-iH-pyrazol-3-yl)-N2-(4-
methyl-5-(piperidin-4-yl)pyridin-2-yl)pyrimidine-2,4-diamine (50 mg, 0.12
mmol)
and triethylamine (50 uL, 0.36 mmol) in DMF (1.5 mL), was added 2-bromo-
acetamide (25 mg, 0.18 mmol). The mixture was stirred at room temperature for
2
hours. The reaction was filtered and the filtrate was purified by RP-HPLC to
afford 2-
(4-(6-(5-Chloro-4-(5-methyl-iH-pyrazol-3-ylamino)pyrimidin-2-ylamino)-4-
methylpyridin-3-yl)piperidin-1-yl)acetamide as a white solid; iH NMR (400 MHz,
MeOD-d4) 8 8.30 (s, 1H), 8.21 (s, 1H), 7.28 (s, 1H), 6.19 (s, 1H), 4.0 (s,
2H), 3.80-
3.73 (m, 2H), 3.33-3.21 (m, 3H), 2.61 (s, 3H), 2.37 (s, 3H), 2.37-2.32 (m,
2H), 2.20 -
2.16 (m, 2H); ESMS m/z 456.2 (M + H+).
Example 17
4_(5-methyl- pyrazol-3-yl)-N2-(4-(piperidin-4 lnaphthalene-l-
yl)pyrimidine-2,4-diamine (85)
NH
HN-N NH N
CI-NH
N
[0129] A mixture of 2,5-dichloro-N-(5-methyl-l-(tetrahydro-2H-pyran-2-yl)-1H-
pyrazol-3-yl)pyrimidin-4-amine (202.7 mg, 0.62 mmol), tent-butyl-4-(4-
aminonaphthalen-1-yl)piperidine-l-carboxylate (202.0 mg, 0.62 mmol), Xantophos
73
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
(35.9 mg, 0.062 mmol), padallium acetate (II) (7.0 mg, 0.031 mmol), and cesium
carbonate (40.3 mg, 0.12 mmol) in 5.0 mL of THE was heated at 150 C in a
microwave reactor for 25 min. The reaction mixture was filtered and the
filtrate was
concentrated in vacuo. The crude product was dissolved in 5 mL of DCM and 4 mL
of TFA. This reaction mixture was stirred at room temperature for 2 h followed
by
concentration in vacuo. The crude product is purified by RP-HPLC to afford 5-
chloro-
N4-(5-methyl-1H-pyrazol-3-yl)-N2-(4-(piperidin-4-yl)naphthalene-1-
yl)pyrimidine-
2,4-diamine; ESMS m/z 434.2 (M + H+)
Example 18
2-(4-(4-(5-chloro-4-(5-meth pyrazol-3-ylamino)pyrimidin-2-
ylamino)naphthalene-1-yl)piperidin-1-yl)acetamide (86)
4
N NH2
HN-N NH N
CI \/\-NH
N
[0130] A mixture of 5-chloro-N4-(5-methyl-1H-pyrazol-3-yl)-N2-(4-(piperidin-4-
yl)naphthalene-1-yl)pyrimidine-2,4-diamine (30.8 mg, 0.07 mmol), 2-
bromoacetamide (19.6 mg, 0.14 mmol) and triethylamine (30.0 L, 0.21 mmol) in
2
mL of DMF was heated at 130 C in a microwave reactor for 20 min. The crude
product was purified by RP-HPLC to afford 2-(4-(4-(5-chloro-4-(5-methyl-1H-
pyrazol-3-ylamino)pyrimidin-2-ylamino)naphthalene-1-yl)piperidin-1-
yl)acetamide;
ESMS m/z 491.2 (M + H+).
Example 19
3-(4-(4-(5-chloro-4-(5-meth pyrazol-3-ylamino)pyrimidin-2-
ylamino)naphthalen-1-yl)piperidin-1-yl)-1,1,1-trifluoropropan-2-ol (88)
74
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
OH
F
N F
F
HN-~ZNH
N CI \NH
N
[0131] A mixture of 5-chloro-N4-(5-methyl-1H-pyrazol-3-yl)-N2-(4-(piperidin-4-
yl)naphthalene-1-yl)pyrimidine-2,4-diamine (30.8 mg, 0.07 mmol) and 2-
(trifluoromethyl)oxirane (39.2 mg, 0.35 mmol) in 1 mL of DMF was stirred at
room
temperature overnight. The crude mixture was purified by RP-HPLC to give 3-(4-
(4-
(5-chloro-4-(5-methyl-1 H-pyrazol-3-ylamino)pyrimidin-2-ylamino)naphthalen- l-
yl)piperidin-l-yl)-1,1,1-trifluoropropan-2-ol; ESMS m/z 546.2 (M + H+).
[0132] The following compounds in Table 2 are obtained by repeating the
procedures described in examples above and using appropriate starting
materials.
Table 2
STRUCTURE NMR or ESMS IGF1R
Ba/F3
IC50
(uM)
76 ESMS m/z 456.2 1.79
(M + H+).
HNC NH2
N NH N"-r
CI N 0
N N N
H
2-(4-(6-(5-chloro-4-(5-methyl-1H-pyrazol-3-
ylamino)pyrimidin-2-ylamino) -2-methylpyridin-3 -
yl)piperidin-l-yl)acetamide
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
STRUCTURE NMR or ESMS IGF1R
Ba/F3
IC50
(um)
77 N ESMS m/z 492.2 3.3
N N N (M + H+).
HN'N CI NY O
N
NH2
2-(4-(8-(5-chloro-4-(5-methyl-1H-pyrazol-3-
ylamino)pyrimidin-2-ylamino)isoquinolin-5 -yl)piperidin-
1-yl)acetamide
78 \ o ESMS m/z 541.2 0.412
-Is
o (M+H+).
N
HN
- N.-NH
N HN /~ CI
N
5-chloro-N4-(5-methyl-1H-pyrazol-3-yl)-N2-(5-(1-(2-
(methylsulfonyl)ethyl)piperidin-4-yl)quinolin-8 -
yl)pyrimidine-2,4-diamine
79 ESMS m/z 457.2 6.4
N (M + H+).
0
N
HI ~ fl I /
N N N
H H
6-(5-chloro-4-(5-methyl-1 H-pyrazol-3-
ylamino)pyrimidin-2-ylamino)-5-fluoro-2-(piperidin-4-
yl)isoindolin-l-one
76
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
STRUCTURE NMR or ESMS IGF1R
Ba/F3
IC50
(um)
80 N H ESMS m/z 458.1 2.65
(M + H+).
O-NCI IIIN
N N N
H H F
0 N
6-(5-chloro-4-(5-methylisoxazol-3-ylamino)pyrimidin-2-
ylamino) -5 -fluoro-2-(piperidin-4-yl)isoindolin-l-one
81 HN~N CI ESMS m/z 400.1 2.30
- N N- NH O F HN NH
HN
6-(5-chloro-4-(5-cyclopropyl-1H-pyrazol-3-
ylamino)pyrimidin-2-ylamino)-5-fluoroisoindolin-l-one
82 H H 0 ESMS m/z 374.1 2.63
' Nx\N'\N / I NH (M + H+).
HNN CI v TN F \
6-(5-chloro-4-(5-methyl-1 H-pyrazol-3-
ylamino)pyrimidin-2-ylamino)-5-fluoroisoindolin-l-one
83 ESMS m/z 367.1 1.45
H H N (M+H+).
N NyN N
CI
H N-N N
5-chloro-N4-(5-methyl-iH-pyrazol-3-yl)-N2-(3-
methylcinnolin-5 -yl)pyrimidine-2,4-diamine
84 H H ESMS m/z 456.2 0.525
) YI
N N
N -N
H CI
N NH2
2-(4-(6-(5-chloro-4-(5-methyl-iH-pyrazol-3-
ylamino)pyrimidin-2-ylamino)-4-methylpyridin-3 -
yl)piperidin- l -yl)acetamide
77
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
STRUCTURE NMR or ESMS IGF1R
Ba/F3
IC50
(um)
85 NH ESMS m/z 434.2 0.493
(M + H+)
HN-N NH N
CI /NH
N
5-chloro-N4-(5-methyl-iH-pyrazol-3-yl)-N2-(4-
(piperidin-4-yl)naphthalen-l-yl)pyrimidine-2,4-
diamine
86 H N 0 ESMS m/z 491.2 0.107
2 (M + H+)
HN1
N NH N
CI
N H
2-(4-(4-(5-chloro-4-(5-methyl-iH-pyrazol-3-
ylamino)pyrimidin-2-ylamino)naphthalen- l -
yl)piperidin- l -yl)acetamide
87 - ESMS m/z 505.2 0.082
(M + H+)
\ / N)
N ~ NH
HN~~ N O
\N Cl N
H
2-(4-(4-(5-chloro-4-(5-methyl-iH-pyrazol-3-
ylamino)pyrimidin-2-ylamino)naphthalen- l -
yl)piperidin- l -yl)-N-methylacetamide
78
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
STRUCTURE NMR or ESMS IGFIR
Ba/F3
IC50
(uM)
88 F F ESMS m/z 546.2 0.346
OH (M+H+)
HN, F
N NH N
CI
~N I \
N H
3-(4-(4-(5-chloro-4-(5-methyl-iH-pyrazol-3-
ylamino)pyrimidin-2-ylamino)naphthalen- l -
yl)piperidin- l-yl)-1,1,1-trifluoropropan-2-ol
89 H H ESMS m/z 398.2 0.088
/ ~ N iN Y N T"N\ (M + H+)
HN-N N
CI
5-chloro-N2-(3-ethyl-2,3,4,5-tetrahydro-lH-
benzo [d] azepin-7-yl)-N4-(5-methyl-iH-pyrazol-3-
yl)pyrimidine-2,4-diamine
Assays
[0133] The IC50 of a drug may be determined constructing a dose-response curve
and examining the effect of different concentrations of antagonist on
reversing agonist
activity. IC50 values may be calculated for a given antagonist by determining
the
concentration needed to inhibit half of the maximum biological response of the
agonist. To calculate IC50 values, a series of dose-response data (e.g., drug
concentrations xl, x2, ...,xn and growth inhibition yl, y2, ...,yn, the values
of y are in
the range of 0-1) is generated. IC50 values may be determined by a computer-
aided
system using the formula:
y = D+((A-D)/( 1+10(x-log(IC50)B)
where A is the ratio of growth inhibition between lowest drug concentration
and control; B is the slope of sigmoidal curvel; and D is the ratio of growth
inhibition
between highest drug concentration and control.
[0134] The IC50 value is given as that concentration of the test compound that
79
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
results in growth inhibition that is 50 % lower than that obtained using the
control
without inhibitor. The compounds of the invention in free form or in
pharmaceutically acceptable salt form may exhibit valuable pharmacological
properties, for example, as indicated by the in vitro tests described in this
application.
In general, compounds of the invention have IC50 values from 1 nM to 10 M. In
some examples, compounds of the invention have IC50 values from 0.01 M to 5
M.
In other examples, compounds of the invention have IC50 values from 0.01 M to
1
M, or more particularly from 1 nM to 1 M. In yet other examples, compounds of
the invention have IC50 values of less than 1 nM or more than 10 M. The
compounds of the invention may exhibit a percentage inhibition of greater than
50%,
or in other embodiments, may exhibit a percentage inhibition greater than
about 70%,
against IGF-1R at 10 M.
Ba/F3 cell line panel and reagents
[0135] Ba/F3 is a murine IL-3-dependent pro-B lymphoma cell line. Parental
Ba/F3 cells are used to generate a panel of sublines whose proliferation and
survival is
rendered IL-3-independent by stable transduction with individual tyrosine
kinases
activated by fusion with the amino-terminal portion of TEL (amino acid 1-375)
or
BCR. In order to generate Ba/F3 cell lines transformed by Tel-Tyrosine Kinase
(TK)
fusions, parental Ba/F3 cells are infected with a retrovirus harboring each
TEL-fusion
kinase and subjected to puromycin selection and IL-3 withdrawal to obtain IL-3-
independent, transformed Ba/F3 cells.
[0136] Each transformed Ba/F3 cells are cultured in RPMI-1640 media (Gibco
Cat #11875093, Carlsbad, CA) supplemented with 10% FBS (Hyclone Cat
#SV30014.03, Logan, UT), 4.5 g/L glucose (Sigma #G5400, St.Louis, MO), 1.5 g/L
sodium bicarbonate (Biowhittaker #17-613E, Walkersville, MD) and Pen/Strep
(Gibco #10378-016, Carlsbad, CA). Cells are splitted twice weekly.
Ba/F3 cell viability inhibition assay
[0137] The potency of test compounds against various Tel-TK transformed Ba/F3
lines is determined as follows. Exponentially growing BaF3 Tel-TK cells are
diluted
in fresh medium to 75,000 cells/mL and seeded into 384-well plates (3750
cells/well)
at 50 pL/well using a pFill liquid dispenser (BioTek, Winooski, VT, USA).
Duplicate
plates are run for each cell line. Test and control compounds are serially
diluted with
CA 02729546 2010-12-24
WO 2009/158431 PCT/US2009/048509
DMSO and arrayed in a polypropylene 384-well plate. 50 nL of compound is
transferred into the assay plates using a pin-transfer device, and the plates
are
incubated at 37 C (5% CO2) for 48 hours. 25 pL Britelite (Perkin Elmer) is
added
and luminescence is quantified using Analyst GT (Molecular Devices). Custom
curve-fitting software is used to produce a logistic fit of percent cell
viability as a
function of the logarithm of inhibitor concentration. The IC50 is interpolated
as the
concentration of compound needed to reduce cell viability to 50% of a DMSO
control. Parental Ba/F3 cells that are maintained and cultured in presence of
IL-3 (1
ng/ml in final) are diluted in fresh medium containing IL-3 (1 ng/ml in final)
to
75,000 cells/mL following the same procedure as described above.
Enzymatic HTRF assay
[0138] IGF-1R and INSR (insulin receptor) are purchased from Upstate.
Following reagents are prepared in-house; 10 x kinase buffer (KB) (200 mM Tris
(pH
7.0), 100 mM MgC12, 30 mM MnC12, 50 nM NaVO4), 10 mM ATP, 100 mg/ml BSA,
0.5 M EDTA, 4 M KF. Proxiplate-384 from Perkin-Elmer is used for set up assay.
All the HTRF reagents including substrate (Biotin-poly-GT (61GTOBLB), Mab PT66-
K, (61T66KLB), Streptavidin-XLe"t (611SAXLB)) are purchased from CIS-US, Inc.
[0139] The substrate/ATP mix is prepared by adding ATP (final concentration, 3
M) and biotinylated poly-GT (final concentration, 10 ng/ l) into lx KB, and
dispensed into Proxiplate-384 at 5 l/well using Fill (Bio-TEK). Serially
diluted
compounds (in DMSO) are transferred into plate using 50 nL pinhead. 5 L of
prepared Enzyme mix (enzyme (final concentration, 5 ng/ l), mixed with BSA and
DTT in Ix KB) is added to initiate kinase reaction using Fill (Bio-TEK).
Assay plate
is incubated at room temperature for 2 hours. Detection mix is prepared by
adding
both Mab PT66-K and Streptavidin-XLe"r into 0.5 x KB solution containing KF
(final
concentration, 125 mM), EDTA (final concentration, 50 mM) and BSA (final
concentration, 100 g/ml) in. At the end of reaction, 10 L of detection mix
is added
and incubated for 30 minutes at room temperature before measurement. HTRF
signal
is detected using Analyst-GT (molecular Devices).
Cancer cell proliferation inhibition assay
[0140] For luciferizing cancer cell line, each cell line is transduced by
ampholytic
81
CA 02729546 2010-12-24
retrovirus carrying both luciferase gene and puromycin-resistant gene whose
expression is
driven by LTR. Briefly, the retroviral vector pMSCV-Puro-Luc is transfected
into Phoenix
cell line using Fugene6 (Roche) according to manufacturer's instruction. Two
days after
transfection, supernatant containing virus is harvested and filtered with 0.2
m filter.
Harvested virus is used immediately or stored at -80'C. For infection,
cultured cancer cells
are harvested and plated (5x105 cells/well in 1 ml medium) on 6-well tissue
culture plate.
For each well, 3m1 virus supernatant is added together with 400 l FBS, 40 l
1 M HEPES
(pH8.0) and 4 l of polybrene (10 g/ml, Specialty media). The plate is
centrifuged down
for 90 minutes at 2500 rpm for spin-infection and is transferred into an
incubator for
overnight infection. Next day, infected cell line is transferred into T-75
flask containing
fresh medium and incubated for one day. Two days after infection, puromycin is
added at
the final concentration of 1 g/ml to begin selection. Within 1-2 weeks,
puromycin-
resistant cell line is established after at least two subsequent splits and is
preserved as
luciferized stock.
[0141] Each cell line is harvested while in log phase growth by trypsinization
and
diluted in respective media to appropriate density prior to plating. Cells are
dispensed
using Fill (BioTeK) at 50 1/well into white walled clear bottom plates
(Greiner - custom
for GNF). Cells are then placed in 37 C incubator supplying 5% CO2 overnight.
Compounds are transferred using 50nL/well Pintool technology via Platemate
(Matrix).
Assay plates are then placed back into the incubator for 3 days. On the third
day
following compound transfer, BRITELITE (Perkin Elmer, diluted according to
manufacturer's suggestion) is added to assay plates and read on Analyst GT
(Molecular
Devices) or Envision (Perkin Elmer). Raw data is generated in RLU.
[0142] It is understood that the examples and embodiments described herein are
for
illustrative purposes only and that various modifications or changes in light
thereof will be
suggested to persons skilled in the art and are to be included within the
spirit and purview
of this application and scope of the appended claims.
82