Note: Descriptions are shown in the official language in which they were submitted.
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
1
Removal of Metals from Complex Ores
Field of the Invention.
This invention relates to a method and device for the removal of metals
from their naturally occurring ores and mineral concentrates preferably using
vitrification processes.
Background Art.
The extraction and subsequent processing of metals from their
naturally occurring ores is well known. Such processes include high
temperature
reduction smelting, chemical reduction, acid leach, gravity separation, hydro
metallurgy, pyrometallurgy, pyro/hydrometallurgy, flotation, hydrolysis, and
electrowinning. .
For example, high temperature and pressure concentrated sulphuric
acid leach processes are currently used to extract nickel and cobalt from
nickel laterite
ores. The high cost of corrosive chemicals, their logistics and safety, can
sometimes
lead to marginal profitability and questionable environmental acceptability.
Such
processes have also required the design and utilisation of equipment that is
relatively
new to the scale and utility demanded by the mining industry. Very high
capital costs
of the required high pressure autoclave support equipment and also long lead
times of
existing commercial extraction systems which are also prone to inefficiencies
and
substantial maintenance costs, make them less commercially preferred.
By way of background information and as outlined in United States
Patent 6,261,527 Arroyo, et al. July 17, 2001, known reserves of nickel and
cobalt in
laterites comprising limonite and saprolite ore, are far greater than the
rapidly
depleting reserves in sulphide ores. However, processing laterite ores is
difficult by
conventional techniques. A number of new hydrometallurgical processes are
being
developed for the extraction of nickel and cobalt from nickeliferous laterite
ores.
Many of these processes require the dissolution of the ore matrix with
sulphuric acid
at high temperature (245 C-270 C.) and pressure (525-785psig), followed by
solid-
liquid separation and neutralization of residual free acid present at ambient
pressure.
This is known as the "Moa Bay Process".
In this process, the nickeliferous ore is first made into a pulp having a
solids content of about 40% before leaching at high temperature and pressure.
During
pressure leaching most metals dissolve and iron and aluminium are rejected by
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
2
hydrolysis to hematite and alunite, respectively. After leaching, the pulp is
cooled and
washed by counter current decantation and the solids are directed to tailing
treatment.
Excess acid is neutralized and the remaining iron and aluminium are
precipitated as
hydroxides with the addition of coral mud. Nickel and cobalt are subsequently
recovered via sulphide precipitation.
Variations of the aforementioned high-pressure acid leach (HPAL) are
exemplified in U.S. Pat. No. 4,044,096.
U.S. Pat. No. 3,804,613 teaches a method of high-pressure acid
leaching of saprolite ore at relatively low acid/ore ratios by preconditioning
the
saprolite with recycled leach liquor from the high-pressure step. This HPAL
process
is suitable for the treatment of high iron ores containing 40wt % iron or
higher.
Lateritic ores with an iron content less than 40 wt % contain a higher
proportion of
magnesium, which consumes higher acid volumes, and are therefore not
economically
suitable for direct high pressure leaching.
U.S. Pat. No. 3,991,159 teaches the use of saprolite ore to neutralize
acid resulting from the high-pressure acid leach of limonite ore. Leaching of
the
saprolite fraction is carried out at high temperature (150 C -250 C) and
pressure for
effective iron and aluminium rejection, but with relatively low nickel
extraction from
the saprolite ore.
In another process, U.S. Pat. No. 4,097,575 teaches saprolite ore
roasting at 500 C-750 C under oxidizing conditions to increase its acid
neutralization
capacity before contact with HPAL liquors.
While prior art HPAL methods obtain a high extraction of nickel and
cobalt, they require the use of expensive equipment and sophisticated
construction
materials to withstand the use of concentrated acid at the high temperatures
needed
(200 C - 300 C), and particularly the high pressure. Furthermore, part of the
rejected
iron and aluminium are in the form of hydroxides, which are difficult to deal
with. For
example, U.S. Pat. No. 4,062,924 describes a method for leaching limonite ores
in
acidic media at temperatures up to 110 C and in the presence of hydrogen
sulphide gas
to precipitate dissolved nickel and cobalt. Most dissolved iron is also
reduced to the
divalent oxidation state in this process, consuming very high amounts of the
reducing
gas in addition to high acid consumption. U.S. Pat. No. 4,065,542 teaches a
similar
method. In this process, ferrous iron produced by the method described above
is used
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
3
to leach metal values from manganiferous sea nodules.
U.S. Pat. No. 4,511,540 illustrates a way to recover nickel and cobalt
from ores with a manganiferous matrix by leaching with sulphuric acid in the
presence of sulphur dioxide gas at temperatures below the boiling point of the
liquid
solution.
U.S. Pat. No. 3,793,432 describes a limonite ore leached with sulphuric
acid at a pH below 1.5, while simultaneously adding alkaline iron-
precipitating
agents. The process is carried out at atmospheric pressures, but requires
leaching
times in excess of 40 hours and usually from 60 to 100 hours for efficient
nickel
extraction and iron precipitation.
U.S. Pat. No. 4,410,498 teaches a method to leach saprolite ore with
sulphuric acid at atmospheric pressure, while adding a reducing agent to
maintain the
redox potential between 400 and 600 in V.
In another process, described in U.S. Pat. No. 5,571,308, nickel and
cobalt are leached from saprolite ore by contact with a mineral acid at room
temperature or in the temperature range of 60 C-SO C.
To anyone skilled in the art it should be obvious from the above
process descriptions that using current nickel laterite acid leach techniques,
metal
recovery efficiencies are still below both commercial and theoretical
expectations.
The complicated chemistry and mechanisms of unlocking metals or their ions
from
ore matrices using acid leach techniques, is at the core of recovery
difficulties.
It will be clearly understood that, if a prior art publication is referred to
herein, this reference does not constitute an admission that the publication
forms part
of the common general knowledge in the art in Australia or in any other
country.
Summary of the Invention.
The present invention is directed to a method for the removal of metals
from complex ores, which may at least partially overcome at least one of the
abovementioned disadvantages or provide the consumer with a useful or
commercial
choice.
With the foregoing in view, the present invention in one form, resides
broadly in a method of removal of a metal from an ore, including the steps of:
1. melting a target metal-containing mixture in a heated process vessel at a
minimum temperature to form a homogeneous melt,
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
4
in the presence of an appropriate flux material to lower the melting
temperature of the
target metal-containing mixture to produce a molten ionic matrix and
2. cooling the matrix.
The matrix will typically be cooled rapidly to preserve the target
properties of the matrix being both hygroscopic and amorphic. This rapid
cooling
process can be achieved by air cooling or fluid quenching using water, saline
solution
or a supersaline ground water as examples.
According to an alternative embodiment, the invention resides in a
method of removal of a target metal from a target metal-containing mixture,
including
the steps of
1. melting a target metal-containing mixture in a heated process vessel at a
minimum temperature to form a homogeneous molten ionic matrix containing
at least the target metal,
in the presence of an appropriate flux material to produce a molten ionic
matrix and
2. cooling the matrix.
It is preferred that silica, normally present as silicate will be present in
the molten ionic matrix in the order of approximately 30% of the total by
mass. If not
present due to the composition of the target metal-containing mixture of
appropriate
flux material, silica may be added through the addition of silica sand for
example.
The invention as described in the above statements particularly and this
specification generally has the distinct advantage of being able to treat the
full
mineralogical range of nickel laterites, which conventional processes such as
the
Caron and HPAL processes cannot.
According to a further alternative embodiment, the invention resides in
an apparatus for the removal of a target metal from a target metal-containing
mixture
by formation of a molten ionic matrix, the apparatus including a heated
process vessel
without pressure to melt a target metal-containing mixture at a minimum
temperature
in the presence of an appropriate flux material including a glass-forming
silica to
lower the melting temperature of the target metal-containing mixture, to form
a
homogeneous molten ionic matrix containing at least the target metal and an
associated cooling assembly adapted to cool the molten ionic matrix.
According to a particularly preferred embodiment of invention, process
raw materials, including said naturally occurring ore bodies containing at
least one
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
target metal or metal compound, are combined with fluxing agents to promote
melting
to form a fluid 'glass' body. Said molten 'glass' is further heated and/or
given
sufficient residence time to substantially assimilate said target metal
compounds and
to degrade or destroy existing mineralogical bonds. Ionic and/or colloidal
forms of the
target metals will then effectively be in solution within said molten glass
body.
Further components within the novel 'glass' formulations will control the role
of any
one or more of metal or other ions introduced from the naturally occurring ore
bodies.
Such further components encourage specific metal ions to be available either
as
network extenders or network modifiers.
Typically the cooling process "locks" the form of the ionic matrix in
which the target metal has been liberated from less desirable materials. By
controlling
such matrix positioning as part of the fluxing/fusing/residence process,
specific target
metal ions may be subsequently retrieved from the 'glass' or solidified body
by
downstream processes, including by solvent and/or electrolytic treatments.
Other
downstream processes may include ion exchange process steps.
The objective is to produce a homogeneous melt with the silica of the
ore and the added flux, typically NaOH or Na2CO3, combining to produce a
soluble
water glass according to the equation:
2NaOH + Si02 c Na 2SiO3 + H2O (or as shown in some literature, Na2O.SiO2.)
This water glass is a most aggressive agent which completely dissolves
the target-metal containing material in the furnace, converting the target
metal when
molten, to an amorphous soluble admixture or glass. The quenched product
remains
amorphous but importantly is hygroscopic. This material preferably quickly
breaks
down in a water quench to become a formless sludge
Typically the flux will be added to a process vessel and then heated to
form a molten flux pool with the target metal-containing mixture gradually
added to
the molten flux pool.
Silica, boron, arsenic and phosphorous exemplify the primary
characteristics of glass network formers. For example, with a silica-based
network
each oxygen atom is shared between two silicon atoms, and each silicon atom
located
between four oxygen atoms allowing the formation of complex three-dimensional
networks. In this form the resultant glass (100% fused silica) is amorphous
and
structurally very strong.
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
6
Network modifiers such as metal ions may be introduced, as oxides,
into a silica-based network. These metal ions typically occupy open spaces
bounded
by the silicon-oxygen three-dimensional network, as it does so the metal ion
tends to
remove/displace at least one of the silicon-oxygen bonds of the network.
Furthermore
the bond between metallic ion and the network oxygen atom is non-directional
resulting in a weaker and less viscous structure.
The metal ions can be introduced into a glass batch material as salts,
for example: sodium carbonate, calcium carbonate, calcium phosphate, magnesium
carbonate, zinc carbonate, nickel oxide, potassium carbonate, alumina, and the
like.
These components may be sourced from naturally occurring compounds, for
example;
magnesium and calcium introduced as dolomite, potassium and aluminium as
feldspar, and fluorine introduced along with sodium and aluminium as cryolite,
and
sodium as trona and nephelene syenite.
It should be made clear that according to the present invention, the
aforesaid metal compounds, sourced from naturally occurring materials, ores
and/
concentrates, are fluxed and assimilated as ions into, what are basically, a
range of
temporary at least partially vitrified structures.
The composition of the ore will typically be dependant upon the target
metal and/or the feed stock target metal-containing mixture and the
composition of
the flux material will be similarly dependent.
Particularly preferred fluxes are sodium carbonate and/or caustic soda.
The composition of the flux material may be or include, but not be
limited to, naturally occurring compounds whose compositions range as follows
as an
example for nickel laterites:
Nickel 0.4-3.0%
Cobalt 0.04-1.6%
Aluminium 0.5-6.00%
Magnesium 0.4-48.0%
Silica 8.0-60.0%
Calcium 0.01-1.5%
Chromium 0.2-3.0%
Iron 8.0-55.0%
Manganese 0.05-10.0%
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
7
Zinc 0.05-0.1%
Copper 0.05-0.1%
A further preferred composition for nickel laterite may be:
Nickel 0.6-4.0%
Aluminium 0.5-18%
Magnesium 0.4-38%
Silica 1.3-55%
Calcium 0.01-50%
Chromium 0.2%-3.0%
Iron 8.0-85%
Manganese 0.12-1.5%
Typically, the metals will be present in the composition in an oxide or
silicate form, for example aluminium present as aluminium oxide/silicate,
magnesium
present as magnesium oxide/silicate, calcium present as calcium
oxide/silicate,
chromium present as chromium oxide/silicate, iron present as ferric or ferrous
oxide/silicate and maganese present as manganese oxide/silicate. This is
particularly
the case when the ore is a laterite.
Table 1.
Component b/Zn Lead Zinc Cu Ore Cu n 7n
Ore zone. cone. Cone. Ore cone.
b% 7.12 72.05 1.50 8.71 0.015 0.023 3.1
n% 8.80 .77 9.80 10.9 0.025 0.034 50.2
Cu% 0.142 1.21 0.140 0.081 7.3 3.75 0.153
g g/t 73.70 771.00 7.00
Au g/t 0.15 1.15 0.16
S% 7.50 16.90 31.50 18.1 30.2 8.88 31.6
s% 0.091 0.155 0.105 0.052 0.144 0.157 0.024
Sb% 0.0213 0.17 0.018 0.051 0.007 0.012 0.033
Cd% 0.0308 0.018 0.17
3i% 0.0017 0.0157 0.0005
e% 7.49 .90 11.50 15.4 6.7 9.05 8.37
n% 1.13 0.36 1.19
i% 0.0022 0.0065 0.003
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
8
Co% 0.0074 0.0095 0.022 0.01 0.094 0.094 0.008
g% 0.0004 0.0005 0.0016
0.451 0.010 0.010
1% .056 0.060 .105
Si02% 17.32 1.31 1.82 17.6 11.00 62.7 .33
1203% 5.87 0.27 0.37 .9 0.41 .72 0.49
g0% 0.67 0.03 0.05 3.62 1.62 1.06 0.39
CaO% 1.33 0.25 0.33 7.11 0.135 .31 0.47
20% 0.94 0.03 0.04
a20% 0.11 0.01 0.01
i02% 0.195 0.001 0.002
2O3% 0.349 0.20 0.020
Further novel temporary 'glass' flux compositions can incorporate
naturally occurring ores and ore concentrates. By way of example, Table 1
illustrates
the mechanical and chemical concentration steps from example specific ore
bodies.
Metal content in ores/concentrates can occur within any extensive range to the
above.
A further embodiment of the invention is the treatment of sulphide ores
where the grain size of individual particles is so fine that particle
liberation cannot be
successfully achieved with conventional grinding practices without generating
excessive amounts of non-target material which forms slime which inhibit
selective
flotation separation processes. Target metal losses to tails become high in
such
circumstances.
The present invention allows for a coarser grind to produce a rougher
concentrate which, in that state, has limited market potential, however making
an
ideal feed for present process. Metal losses to tails in this process are
significantly
reduced.
By way of example, a large silver-lead-zinc deposit with a very fine
grain size has grade, recovery and losses as shown below:
Ore Grade Recovery Losses to Tails % Loss
Silver 41g 29g 12g 30%
Lead 4.1% 2.66% 1.44% 35%
Zinc 9.2% 6.9% 2.3% 25%
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
9
While the invention process is able to assimilate the majority of the ore
components, said invention process is equally able to assimilate the
components of
ore concentrates.
The process of the present invention can be used to treat ores known as
refractory ores. An example of such an ore an/or concentrate occurs with gold
and/or
silver ores where the noble metals are locked within crystal lattice
structures or
sulphides such as pyrite and arsenopyrite. When these noble metals are
contained in
such a manner, their extraction by conventional cyanide processes is limited.
The
process of the present invention allows the liberation of both gold and silver
into the
melt matrix in their ionic form, so presenting such metals in a form which is
readily
recoverable using downstream processes such as cyanide separation.
The invention process is equally able to assimilate metals and
compounds from mine tailings and other waste/effluent type sources.
It should be understood that the invention process is most appropriate
in the extraction of metals from mined ore, however it may be noted that in
many
existing metal processing steps where it is difficult to separate individual
metals from
alloys or mixtures, said invention process will provide an effective
separation means.
In existing processes like smelting and similar metal extraction
techniques, said invention process may provide an intermediate step or indeed
alternative extraction method. This is particularly true when considering high
melting
point metals such as platinum, palladium, rhodium, niobium, tantalum, tungsten
and
the like.
Ores such as tantalite, niobium, columbite, tin and mixtures of these
metals in an ore may be introduced into said invention process to form a
molten ionic
mixture with a base composition in a range as follows:
Manganese oxide5-12%
Iron oxide 5-15%
Niobium oxide 50-80%
Tantalum oxide 65-90%
Tin oxide 50-80%
Another molten ionic composition for use with the invention process
includes the following or similar base components:
Copper 1.0%-4.0%
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
Uranium 0.1%-1.5%
Gold 0.2g/t-10.Og/t
Silver 3 Og/t-200.0g/t
Platinum 0.1 g/t-7g/t
Palladium 0.1 g/t-7g/t
Yet another mixed sulphide concentrate with the following base
composition has been treated by the process of the present invention.
Concentrate 1 Concentrate 2
Au g/t 60 6.5
Ag ppm 783 148
Cu% 10.7 1.2
Zn% 2.2 1.2
Fe% 13.5 20.7
As% 3.4 0.4
S% 24.1 25.1
Si02% 30.0 40.0
With sufficient applied temperature and the presence of fluxing agents
most metals and indeed gases (including water vapour, chlorine, fluorine,
hydrogen
and the like) can be incorporated within a molten ionic matrix rather than
expelled
within the exhaust gases.
The molten ionic compositions of the present invention are designed to
selectively place network formers and network modifiers found in naturally
occurring
ores into relatively unstable positions within the molten ionic matrix. This
is
preferably achieved by a combination of temperature control and chemical
reaction.
Chemical reactions are initiated by the inclusion of phosphorous,
chlorine, fluorine, sulphur, calcium, sodium, vanadium, lithium, potassium,
boron,
barium, zinc, and arsenic, either individually or in combination.
Although it is easier to use and calculate the effects of such
components from their oxides, the major molten ionic materials used according
to the
present invention have natural bulk sources, for example:
Phosphorous Apatite
Boron & sodium & water Kemite
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
11
Boron & calcium & water Colemanite
Soda Ash Trona
Arsenic Arsenopyrite
with fluorides and chlorides providing both free and dissolved gases, and
sulphates
both sulphur and gas.
One important feature of the present invention is the treatment of
sulphide ores including those ores where arsenic is present. The process of
the
present invention is an immersion process. New feed additions or ores are
immediately immersed into an existing melt. This process considerably limits
the
emissions of sulphur and arsenic in furnace exhaust gases. Further, the
invention
utililises the ability of arsenic, like silica and borax for example to act as
a glass
former. Hence the arsenic contributes the formation of the glass in the melt.
Such a
glass is rendered hygroscopic by including an extra 5 to 8% of sodium
carbonate.
A further preferred flux forming matrix used according to the invention
has a composition as follows for nickel laterites:
Nickel 0.4-3.0%
Cobalt 0.04-1.6 %
Aluminium 0.5-6.0%
Magnesium 0.4-48.0%
Silica 8.0-60.0%
Calcium 0.01-0.5%
Chromium 0.2-3.0%
Iron 8.0-55.0%
Manganese 0.05-10.0%
Zinc 0.05-0.10%
Copper 0.05-0.1%
Phosphorous 0.0-30.0%
Boron 0.0-40.0%
Sodium 15.0-70.0%
Chlorine 0.05-12.0%
Water 0.02-10.0%
Yet another embodiment of said flux forming composition includes but
is not limited to the following components:
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
12
Phosphorous 10.0-30.0%
Boron 10.0-40.0%
Sodium 15.0-30.0%
Chlorine 0.05-12.0%
Lithium 0.5-15.0%
Water 0.02-3.5%
To this basic formula range can be added between 10% and 60% of
most naturally occurring metal ores.
The process of the present invention is also suitable for treatment of
high grade silver ore in which the sliver is contained within rich silica, the
latter up to
95% silica.
The inclusion of other aforementioned ionic matrix forming materials
will consequently enable selective extraction of target metals. For example
the
inclusion of arsenic and/or lithium compounds will assist ion mobility and
general
network sharing capacity.
The product ionic melt formed according to the method of the present
invention may be soluble in a range of solvents including water, acids, and
alkali
solutions such as ammonia and ammonia carbonate. Other solvents may be used
including but not limited to fused salts, and gases including but not limited
to, steam,
chlorine, sulphur dioxide, and carbon monoxide.
The melt product of the present invention will typically be hygroscopic
due in large part to the composition of the flux material chosen. One of the
by-
products of the process of the present invention is silica gel which may be
used or on-
sold. The silica gel is typically formed when the carbon dioxide of the
exhaust gases
is passed through a solution containing sodium silicate.
According to preferred embodiments of the present invention, one or
more "flux" material is included in order to achieve more or less complete
melting of
any "glass" which may form in the furnace and/or the target metal containing
material
at a lower temperature that would otherwise be needed in the absence of the
flux
material. For example, for common glasses such as soda-lime-silica, sodium and
calcium are the fluxing agents for the silica component which itself would
otherwise
require a melting temperature of 1,700 C. The fluxing effect of calcium and
sodium
reduces that temperature by over 800 C.
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
13
Typically, the flux/target metal containing material ratio is between
approximately 1:0.5 at the lower end and 1:3 at the upper end with an optimum
ratio
for nickel laterite in particular being approximately 1:1.
It is further preferred that when nickel laterites are to be treated, up to
approximately 10% sulphur may be added to the melt as elemental sulphur or in
a
sulphide such as iron pyrite.
FLUXES
As mentioned briefly above, the purpose of the flux is preferably to
reduce the melting point of the target material. The melting point of
laterites can for
example, be reduced from approximately 1800 C to under 950 C.
The flux can also act as high temperature solvents. Again, with laterites
in a melt, they dissolve silica and silicates containing the nickel and
cobalt, also the
oxides and silicates of Fe, Al, Mg, Mn, and Ca.
Other examples of particular fluxes which may be used include soda
ash, soft cake, potassium hydrate, ammonium nitrate, Borax or similar
compounds.
Typically, flux compounds are provided as low melting point temperature molten
solid-phase compounds.
Using Na20 based fluxes as Na2CO3 or NaOH, the presence of silica or
silicates preferably provides the glass former.
It is preferred that the glass product created is soluble in water. This is
typically provided by the glass former materials, Na2O and SiO2 combination
forming
Na2SiO3 (water glass), which is the aggressive destroyer of laterites and
itself is
soluble in water.
The preferred fluxes for treatment of nickel laterite are NaOH and
Na2CO3. This choice is based on:
= NaOH can be manufactured on site
= NaOH changes to Na2CO3 in the presence of CO2
= Both supply the Na2O required to produce the water soluble water
glass, fundamental to the disintegration of almost all rocks, including
laterites.
NaOH has:
= The ability to be made onsite by electrolysis of common salt, NaCl, as found
as rich, hypersaline brine ground water beneath the WA laterites, which may
also provide a source of NaCl.
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
14
= Ability to be regenerated on site , with the presence of CO2 converting the
NaOH to Na2CO3 according to the equation :
2NaOH + CO2 >>> Na2CO3 + H2O
The chemical reaction for the conversion of a laterite to a diffused salt
commences with, for example:
Na2CO3 >>> Na2O + C02, and
2NaOH >>> Na2O + H2O
The Na2CO3 and NaOH fluxes generate Na2SiO3 (water glass),
according to the equations:
Na2CO3 + Si02 >>> Na2SiO3 + C02, and
2NaOH + Si02 >>> Na2SiO3 + H2O
Though Na2CO3 (or NaOH) alone do not dissolve Fe203 or MgO,
however in combination with Si02 (to form Na2SiO3) these iron and magnesium
compounds are readily dissolved in the melt.
The two main iron oxides (FeO and Fe203) are soluble in Na2SiO3.
Most FeO will be oxidized in the furnace to Fe2O3.
The reaction of the breaking of the strong silica and silicate bonds, Si -
0 - Si is:
Si-0-Si+Na2O>>>2Si-0 +2Na+.
It should be noted that the Si - 0 - Si reaction shows the breaking of
only one Si- Si bond when every Si has a total of four bonds. However, in the
present
invention, because of the excess of soda, more bonds will typically be broken.
The Na2Si03, itself soluble in water, attacks and renders soluble
everything, including all the key components of laterites. These include both
the
oxides and silicates of Fe, Al, Mg, Mn and Ca. Aluminium silicate is of course
the
major component of clay minerals, which in turn contains nickel in some of the
laterite deposits. Many laterite deposits contain both nickel and cobalt
predominantly
in clays (aluminum silicates) as well as in the iron oxides of limonite and
goethite.
One of the major and higher grade sources of nickel is saprolite and
serpentine/garnerite ores. These ores are not processed by the HPAL system
because
they are rich in magnesium and magnesium consumes excess amounts of the
sulphuric acid added to the autoclave. These ores can be processed by the
method of
the present invention.
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
Na2CO3 and NaOH in combination with Si02 not only create fast
dissolution of the rock material in any raw ore used but also increases
solubility of the
fused salt in water. The Na2O is typically highly mobile in the melt.
The melt if homogeneous, is a sound indication both the Ni and Co
have been released and are in their ionic form.
If the nickel is not contained within the silica, say within an oxide, then
an acidic oxide such as Si02, (orB2O3, or P205) may be required. However, the
water
glass melt will typically dissolve the nickel and cobalt contained in non-
silicate forms
and converts these elements in the melt to ions, which make them far more
accessible
for downstream processing.
It is important to obtain the correct balance of target-metal containing
mixture and flux to ensure the combination provides a homogeneous and low
viscosity melt and produces a pour which is hygroscopic.
Fluxes are one of the most important elements to the method of the
present invention, including considerations such as:
= The optimum flux for the target metal containing material.
= The optimum target metal containing material/flux ratio
= The flux recovery/regeneration/recycle processes.
When using Na2CO3 or NaOH as the flux, a large part of the vitrified
structure or melt does not dissolve in water, just disintegrates as Si02,
Fe203, MnO,
CaO and A1203, and/or hydroxides or carbonates of these elements, the latter
to form
a formless sludge. In a very basic environment, very small amounts of the Si02
and
A1203 may dissolve.
The high temperature solvents for the ore should also be soluble in
water, as are Na2CO3, NaOH and Na2Si03.
The introduction of carbon monoxide, or sulphur into the furnace
would reduce the nickel and cobalt to metals. However, CO is dangerous and
should
not be considered.
Cr203 does not dissolve easily in Na2SiO3. This can be improved by
increasing the Na2CO3, or Si02.
The substitution of NaOH for Na2CO3 can be viewed when examining
the molar weights. For example: Na2CO3 has a molar wt of 106g/mol and NaOH of
40g/mol. Therefore the molar equivalent of NaOH to Na2CO3 = 40 X 2 = 80 to
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
16
106g/mol. This means less NaOH is required than Na2CO3 in the ratio of 80 to
106 or
1 tol.33.
NaCl has little value in dissolving silica. Also, it will not dissolve
Fe203.
Borax as a flux material dissolves most components and reduces
viscosity and increases the solubility of the glass. It also promotes a
homogeneous
pour. Even though borax appears to be cost prohibitive when compared with
Na2CO3
or NaOH, it is possible to partially recover borax by evaporation.
For the higher magnesium ores such as serpentine/garnerite (MgO 25 -
38%), a little more silica and Na2CO3 or NaOH will be required to dissolve the
magnesium.
The use of natural trona compared to the artificial Na2CO3 should have
little influence in changing the glass chemistry and therefore on the process
of the
present invention. Trona, if readily available must be considered.
Dolomite does not make a good glass and importantly, it cannot
dissolve silicates.
Preferred Flux Regime for Nickel Laterite
A laterite, LH2, sourced from Western Australia is treated as follows:
LH2
Laterite 130g
Na2CO3 130g or 1. to 1
The Na2CO3 is added initially as IOOg to form the initial melt pool. To this
melt, 3
separate lots of LH2 are added, together with l Og Na2CO3 with each LH2
addition.
A 50%mol/50%mol ratio of CaC12/NaCl may be more effective for a
nickel laterite target metal containing material and may be effective at
reducing the
furnace operating temperature to 650 C-850 C. Other components that may be
used
in this flux include CaC12 or Na2CO3 to replace the CaC12.
FURNACE
The furnace of the present invention needs only to melt, dissolve then
discharge. Therefore the comparative energy costs and resident times are much
lower
for the furnace of the invention. This equates to furnace production being
much higher
and unit costs much lower for the present invention when compared to an
equivalent
sized glass furnace.
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
17
Furnace operating temperatures are expected to range from 980-
1100 C. This temperature will be dependent on the combinations of ore types,
flux
addition and particle size of the feed.
The furnace will preferably have similar refractory liners, waste-heat
regeneration, exhaust, feed and heat supply systems and automated controls as
for a
production glass furnace. A heat regeneration/capture system may be provided.
The furnace will typically include one or more electrodes. The
electrodes may be AC to boost the melt and stirring process or DC which may
assist
in metal recovery and/or the destruction of mineralogical bonds in the melt.
The
electrodes may be static or movable to provide either static or moving heat
zones in
the melt.
The electrodes may require additional protection from the aggressive
environment in which they are used, said protection typically in the form of a
layer of
porous but toughened ceramic material.
At least some of the electrodes may be provided with AC power in
order to supply bursts of heat to the melt if required.
The presence of C02 in the exhaust gases of the furnace or the heating
process for the furnace may be important as this gas can then be used as a
part of the
flux regeneration process.
QUENCHING
In a preferred embodiment, once metal fluxing and ion assimilation has
taken place, said specific metal ions may be extracted from the glass body by
quenching the molten `glass' in neutral pH aqueous solutions or in aqueous
solutions
containing acids or alkalis. The resultant liquor is preferably an
electrolyte;
consequently direct electrowinning techniques may be applied during quenching
or
soon thereafter.
One particularly preferred quench fluid is brine or salt water.
As an alternative to quenching the melt, the melt exiting the furnace
may be "sheet formed" to produce a thin layer of the melt. This can be
accomplished
in a number of ways including pouring the melt onto a tray and
allowing/forcing the
melt layer to cool rapidly with air cooling and/or water sprays. Typically,
any water
sprays will be directed so as not to wet the pour. The water sprays will
therefore
preferably be from beneath the melt or indirect.
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
18
Such unquenched material to be passed through a crusher, such as a
rolls crusher. A solvent such as water is added and the mix vigorously
agitated for up
to fifteen minutes to convert the crushed particles to a formless sludge.
Post quench treatment may comprise sequential pH adjustment with or
without inclusive electrowinning mechanisms.
A preferred quenching option utilises a fused salt bath containing
molten sodium chloride and/or potassium or sodium nitrate; said bath may or
may not
incorporate electrolysing electrodes.
Another preferred quench procedure incorporates a molten metal bath
containing appropriate metals either independently or in combination with
other
metals or salts; said bath may or may not incorporate stirring means,
preferably in the
form of moveable paddles.
Once the molten material enters the quench bath, immediate chemical
changes occur. A large part of the glass will not dissolve in water but just
precipitate
as Si02, Fe203, MnO, CaO, MgO and A1202 or as carbonates or hydroxides of
these
metals. It is important that all the glass should be hygroscopic and
disintegrate.
Because of the strong basic condition of the melt, many of the components of
the melt
are precipitated to their oxides, hydroxides or carbonates. The changes
include nickel
and cobalt. This is because nickel and cobalt ions can not exist in water and
immediately precipitates to Ni(OH)2 and Co(OH)2 respectively. Basic oxides,
carbonates or even silicates might form. The key is that nickel and cobalt
compounds
are finely dispersed in the sludge. A likely equation is as follows:
Ni2+ + 20H - >>> Ni(OH)2.
Therefore, the majority of the nickel and cobalt will be found in the
sludge and not the liquid. Similarly, the Fe, Mn, Ca, Mg and Al all
precipitate to
oxides or hydroxides. This means the quenched material will divide into a
sludge and
solution. The soluble compounds include unused flux and Na2SiO3 will be in the
solution. The sludge should be of a formless nature.
Importantly, the nickel and cobalt in the sludge would be in a fine and
more reactive form than when in the laterite and therefore far more accessible
to
downstream processing, including by solvents. An example of a reaction is:
Ni2+ + 20H >>> Ni(OH)2.
The Ni2+ is released by the Na2SiO3, and the OH" is derived from the
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
19
basic Na2SiO3 solution.
The volume of the quench solution should be in the order of 1 to 1 to
the product mixture exiting the furnace and may require replacement or
replenishment.
On some occasions, the quench water may be laced with salt. There are
no obvious signs of this salt having any influence on the solubility of the
pour.
However, the salt will typically influence the recovery/regeneration of the
Na2CO3.
A water quench dissolves only the soluble compounds. The Na
compounds dissolved may later be recovered by evaporation. If NaCl is also
present,
this should be separated first, possibly by fractionation crystallization or
by chemical
separation.
With the laterites, after an apparent successful run, the quenched
product is coloured a dark olive green. However, with exposure to the
atmosphere, or
water, the colour, and appearance, quickly changes to become a dark sticky
toffee.
This is a feature of the hygroscopic nature of the flux-laden poured material.
This
hygroscopic form is a preferred requirement.
A preferred pour, which should flow like medium to runny honey, will
typically at best, make a slight sizzle when hitting the water. Again, the
latter is a
desirable feature as it indicates the stream of the poured material into the
quench is
thin, and quenching therefore is likely to be immediate and fully effective.
The solids of the quenched product, when mixed with water, may be
allowed to sit for only a relatively short period before pressure or vacuum
filtration.
To reduce contact with C02, the washing and filtration processes will
preferably be
conducted in enclosed vessels. The solids before washing and filtration are
preferably
crushed to reduce particle size to <3mm. When wash water is added, agitation
preferably takes place to form both a sludge and solution. Ideally, the
process will be
continuous so that filtering takes place in a matter of hours from the time of
the pour
as this will typically decrease the amount of silica gel present in the mixed
and
quenched product.
Preferably, the furnace temperature and residence time and pour
conditions are such that the powder product is produced.
Forced drying of the sludge may make the nickel and cobalt
compounds less reactive and therefore should be avoided.
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
When attempting to wash the poured product, difficulties may be
encountered due to the formation of insoluble silica gel (Si02). This gel
binds all of
the contents together so that physical separation by filtration is not
possible. When
washing the poured product, the wash solution will typically be maintained at
a pH of
approximately 12 or above, preferably through the addition of NaOH or similar
basic
solution. To overcome an ageing process where colloidal silica will form, even
at this
elevated pH, the wash solution will ideally be filtered within approximately
45minutes of the commencement of washing. Filtration may be assisted either
through pressure of vacuum. The NaOH will typically be recovered at a later
stage in
the flux regeneration process.
The formation of the silica gel is from the dissolution of the Na2SiO3 in
water as:
Na2SiO3 + H2O >>> 2Na+ + Si032_.
The silica gel becomes part of the sludge. This continues to react
further to Si02 but still is in a very finely dispersed reactive form.
The presence of silica gel and sodium silicate are both strongly
indicative of the break-down of the silica/silicate compounds within the
laterite, and
through this, the release of the nickel and cobalt.
As silica gel is very reactive, it may bind some nickel very loosely, if at
all.
Quenching in a reaction vessel, with NH4OH as the quench bath may
be preferred. This should lead to immediate nickel and cobalt extraction,
assisted by
the elevated temperatures within the vessel. The reaction vessel should assist
in the
recovery of the ammonia for reuse.
Chemically, the silica gel should not influence the extraction of the
nickel from the sludge, whilst physically, it typically does.
Warm brine is very aggressive against ordinary steel and aluminum. If
metals are considered as transfer pipes or vessels, some types of stainless
steel, would
be required to replace the above metals. Plastics would be another
alternative.
The NaCl of the brine will typically reduce the stability of silica gel.
The presence of silica gel should not influence the extraction of the
nickel from the sludge. If NaOH is used as the quench this should keep the
silica gel
in solution.
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
21
Nickel and cobalt may appear in trace amounts in the quench bath. This
possibility needs to be monitored to ensure any limits are not exceeded.
However, this
is a positive sign as it demonstrates these elements are in the sludge in a
reactive
form, with even water dissolving them slightly.
If nickel and cobalt are in the flux, this does not constitute a potential
loss as the dried flux is returned as furnace feed.
The nickel and cobalt may form another compound in the quench,
combining with say Fe, Al, Mg, or Si02
It has been noted that when NH4OH has been added to a sludge
containing silica gel, the separation of liquids from solids was not a
problem.
However, when replacing the NH4OH with H2SO4, the total beaker contents became
a
jelly.
The dried, washed sludge will have a higher nickel content than the
dried, unwashed pour. This is simply because the Na2SiO3 and Na2CO3, both
soluble,
would have been removed from the sludge.
As the separation of the silica gel from the sludge may prove difficult,
a physical means of separation such as a centrifuge or a porous membrane, or a
chemical process, such as a flocculent may be used.
The target metal to flux ratio largely depends on the ability for the pour
to disintegrate afterwards in the water quench to form sludge.
The separation of the sludge from the silica gel may be desirable to
capture the target metals and also for the recovery of the silica gel as a by-
product.
Small quantities of Si02 and A1203 as found in the sludge may also
dissolve if the solution is sufficiently basic.
If cost effective acetic acid (CH3COOH) is available as a quench,
further options would be provided.
Where a laterite is deficient in Si02, Na2SiO3 from the quench may be
added to the front of the furnace.
Time between pour and the downstream processing of the quenched
pour may be a significant factor. This is because time may allow changes in
the
chemical composition of the quenched product which in turn may be deleterious
to
downstream processing.
The change of colour of the quenched solution from say a canary
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
22
yellow to dark brown indicates dissolution of the iron, and if this is the
case also the
dissolution of nickel and cobalt is likely.
In order to produce finer particles rather than lumps within the cooled
pour, it is important to have low viscosity and a thin laminar flow of the
pour.
Acetic acid dissolves most metal oxides, including those of nickel and
cobalt in laterites. Si02 is the exception. Acetic acid reacts with Na2CO3
driving out
CO2. When reacting with Na2SIO3, acetic acid produces silica gel. Using acetic
acid
may not be desirable in preference to a solvent that releases the target metal
only,
such as provided by NH4OH
Acetic acid should dissolve even more of the quenched laterite than
H2SO4
Ultrasound may greatly facilitate the disintegration process of glass in
water. Energy costs could be a deterrent.
Phase separation, should it occur after the pour enters the quench
would not be a problem providing all the pour disintegrates into sludge.
The presence of some nickel and cobalt in the quenched water could
indicate one of two things as, a) there are fine nickel and cobalt particles
that have not
been filtered out, or b) nickel and cobalt are so reactive that even water can
dissolve a
little of them.
When quenching the melt it is best to have low viscosity and higher
temperature of the pour immediately before quenching or air cooling.
The Na2O in the melt and the NaOH in the quench solution both have
the same effect. They tend to convert Si02 to water soluble Na2SiO3 according
to the
equations:
Si02 + Na2O >>> Na2SiO3
Si02 + 2NaOH >>> Na2SiO3 + H2O
The difficulty may be that the above reactions are reversible. It is
possible the Si02 has a stronger affinity towards another compound such as
Ni(OH)2.
If the Si02 prefers to bond Ni instead of Na, we need to add sufficient Na is
added to
set the Si02 free.
The addition of Na2CO3 or NaOH to the quench bath could assist in
releasing the Ni and Co from the sludge. The addition of extra Na2CO3 to the
melt
may also assist.
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
23
NaOH if used in the quench bath should keep the silica gel in solution.
Unfortunately, the one experiment conducted using the NaOH as the quench
solution
resulted in quite violent explosions as the pour hit the quench.
The sludge should not contain much Na. If it does it means either the
washing and/or filtering are incomplete or the soluble glass is far from
soluble.
The colour of the sludge is no definite indication that something is
released. There should be very little release in water except soda and water
glass.
Cobalt hydroxide is orange to pink but when in combination with silica it is
blue. If
nickel is the dominant element in a nickel/cobalt mix, the blue colour will
predominate. Nickel hydroxide is green and this colour is maintained in
combination
with silica. However, with NH4OH, nickel becomes ultra marine blue.
From the quench bath, it is also possible to recover:
Na2CO3 (surplus) Solution
Na2SiO3 Solution
NaOH (if this is the flux) Solution
Si02 (silica gel) Solid
Quenching in atmospheric conditions is a preferred option. However,
quenching at elevated or decreased pressures may be used. Oxidizing or
reducing
agents may also be used.
One preferred aspect of the invention method and device incorporates a
moving-heat-zone means in both the primary vitrification furnace and high
temperature quench bath. Said moving-heat-zone utilises conductive ceramic,
e.g.,
silicon carbide, or graphite electrodes. A preferred moving-heat-zone
electrode may
utilise AC or DC currents. Where it is appropriate to use AC currents as a
heating
source, electrode materials may consist of carbon, molybdenum, conductive
ceramic,
tungsten, platinum, niobium, tantalum and the like can be used or alloyed as a
combination or with other suitable materials.
A direct current will normally be used to electrowin the melt but either
an alternative or direct current may be used to break or disrupt bonds in the
melt.
However, a moving-heat-zone electrode assembly manufactured from a
silicon carbide material is a preferred invention device. Said assembly may
comprise
solid structures or a mesh whereby as the assembly is traversed and/or rotated
through
the molten media, the majority of particles will pass through. Larger
particles will of
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
24
course be held in contact with the electrode mesh structure enabling high-
localized
heating to encourage assimilation of said large particles into the molten bulk
media.
The heat zone(s) may alternatively be provided as static heat zones.
METAL RECOVERY
The preferred rapid quench of a homogeneous and low viscosity melt
into a quench bath preserves the ionic conditions of the target metal formed
in the
furnace. Rapid air cooling is also an effective means of producing an amorphic
pour.
In a water quench bath the nickel and cobalt ions are immediately
precipitated as hydroxides or carbonates.
As well as the nickel and cobalt precipitating out, so too do other
elements and compounds in the laterite, namely the oxides of: Al, Mg, Fe, Mn,
Ca,
and Si02, all combining to form a formless sludge.
In this sludge the nickel and cobalt are in a finely divided form and are
more readily accessible for downstream processing.
The form of the target metal in the sludge can be any one or more of:
hydroxide, oxide, carbonate, silicate, or perhaps occurring as a chemical
compound
with another element, such as Fe. The form of the target metal is an important
step in
the process of deciding the path of recovery.
Once this form has been determined, the downstream steps in isolating
and recovering the target metal can then be taken.
There are a number of options available. These include the use of
ammonia or ammonia carbonates, H2SO4 or other specialized solvents. Ammonia,
as
NH4OH, has advantages as it can be recycled.
The possibility of using ion exchanges to capture the nickel and cobalt
once in ionic form needs to be considered with Na+ as a possible exchange ion.
The
clinoptilolite from natural zeolite may be used.
There may be electrowinning of the melt in the furnace as well as
possibly in the quench bath. Electrowinning the melt will preferably take
place by
providing electrodes in the main production furnace which may be truncated
towards
the discharge end of the furnace to supply extra electrode space, or by
discharging the
formed melt into at least one secondary furnace specifically equipped and
designed
with electrodes for electrowinning the product melt. Any secondary furnaces
may be
provided in parallel to provide the flexibility of having an operating
secondary
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
furnace and an inoperative secondary furnace to allow for the replacement of
the
electrodes whilst maintaining output.
The preferred method for electrowinning in the melt is to switch the
electrodes provided in the furnace to provide additional heat in the melt, to
DC
current. As well as target metal recovery, this will typically break any of
the
remaining strong mineralogical bonds of the ore. This will materially assist
with the
capture of unrecovered target metals in conventional downstream processes.
In an alternative embodiment, of the target metal may be recovered
using a forced precipitation mechanism. For example, addition of iron/steel to
the
melt will typically cause or force precipitation of the nickel and cobalt
contained in a
nickel laterite melt, into a relatively pure although molten metal form. Using
iron/steel will typically precipitate the nickel and cobalt from a nickel
laterite melt in
preference to any other metals contained in the melt.
Normally, the molten metal nickel and cobalt precipitating out will
settle to the base of the melt pool on the basis of density where these target
metals
may be drawn off separately from other waste or less desired metal components
of the
melt.
Without wishing to be limited by theory, iron, nickel and cobalt have
similar atomic weights. Therefore, the quantity of iron/steel added to the
melt in order
to force precipitation of the nickel and cobalt will normally be at a rate
substantially
equivalent to the combined percentage content of nickel and cobalt in the ore.
The
approximation of the percentage content of nickel and cobalt in the ore can be
obtained by sample in the feedstock. In principle, the same addition protocol
can be
used for other target metals based on the addition of a precipitate forming
material of
a substantially similar molecular weight.
For example, if the nickel and cobalt content in the ore are
approximately 1.6 and 0.07 weight percent respectively, then the amount of
iron/steel
to be added is approximately equivalent to 1.67 weight percent. A redundancy
margin
in order to ensure as much as possible that the precipitate forming material
contact the
target metal may be built in of approximately 5% of the precipitate forming
material,
above the approximate weight percent of the target metals.
It is further preferred that the precipitate forming material is added in
particulate form in order to increase the surface area per unit volume of the
precipitate
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
26
forming material. Increased mobility of the precipitate forming material
through the
melt may be achieved through agitation and/or stirring of the melt after the
addition of
precipitate forming material thereto.
FLUX REGENERATION
With the very high levels of flux which will likely be required for the
successful melting of target metal containing ores, it is important to the
present
invention, that the fluxes can be generated and/or regenerated on site.
As an example, there are two preferred flux options for nickel laterite
and sulphide ores. These are Na2CO3 and NaOH, both suppliers of the required
Na2O
to the melt and through its combining with the essential silica, forms the
soluble
Na2SiO3.
Sodium Carbonate (Na2CO3)
There are two well known artificial manufacturing processes, named
after the inventors. These are the Solvay and Hue processes. Both processes
use salt to
produce Na2CO3.
The basic process is:
NaCl + NH3 + CO2 + H2O >>> NaHCO3 + NH4C1
The NaHCO3 precipitated from this reaction is dried to produce:
2NaHCO3 >>> Na2CO3 + C02-
The NH4C1 is sold as a fertilizer.
Note: Additional to NaCl necessary for the manufacture of both Na2CO3 and
NaOH,
Na2CO3 also requires CO2 and NH3. The burning of limestone is necessary to
produce
CO2. The manufacture of NH3 requires a complex plant to separately supply each
of
the nitrogen and hydrogen. An extensive industrial chemical process facility
is
therefore necessary to produce Na2CO3. An advantage is that the NH3 is
recovered.
On-Site regeneration
The key ingredients for the on-site manufacture of Na2CO3 are CO2 and
sodium, the latter produced from Na2SiO3, a by-product of the process of the
present
invention when used to treat nickel laterite and/or sulphide
ores/concentrates.
One source of the CO2 is from the exhaust gases from the production
furnace. Where necessary, additional CO2 may be required from other on-site
industrial exhaust systems.
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
27
The energy sources of the production furnace can be varied. These will
typically include fuel oil, powdered coal, natural gas and a diesel/sump oil
mix.
With methane as an example:
CH4 + 202 >>> CO2 + 2H20
The CO2 used in regeneration does not have to be pure. Traces of 02,
N2, NO and SO2 do not have to be removed. This simply means the exhaust gas
can
be captured and applied. There will also be large quantities of nitrogen,
resulting from
the use of air in combustion.
Bubbling CO2 through the quench solution and sludge therefore
preferably accomplishes two things, a) precipitates the silica gel and b)
regenerates
the Na2SiO3 to Na2CO3.
Basic Steps
1 Collect the exhaust gases, and specifically the CO2 component.
2 The sludge wash-water will contain Na2SiO3, plus any remaining Na2CO3, both
soluble in water.
3 Pass the CO2 through this solution, to produce Na2CO3 according to the
equation:
Na2SiO3 + CO2 >>> Na2CO3 + Si02
The insoluble SiO2 precipitates as silica gel. This should be collected and
stored.
4 Evaporate off the water to recover the Na2CO3.
In the presence of any CO2, including atmospheric furnace combustion, the
NaOH is converted to Na2CO3 as :
2NaOH + CO2 >>> Na2CO3 + H2O.
Accordingly in practical terms the NaOH cannot be recycled. This
factor has no impact on the nature of the flux as it is the Na2O that is
required.
Sodium Hydroxide (NaOH)
Manufactured by the electrolysis of brine (NaCI). This is known as the
"chloralkali"
process.
The electrolysis formula are:
NaCl >>> Na+ + Cl- (dissolution in water)
Na+ + e" (reaction on the cathode)
2C1 + 2e >>> Cl (reaction on the anode)
2Na + 2H2O >>> 2NaOH + H2
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
28
In the presence of any C02, including atmospheric C02, the NaOH is
converted to Na2CO3 as :
2NaOH + CO2 >>> Na2CO3 + H2O
Accordingly NaOH cannot be recycled as noted above.
To recover the Na2CO3/NaOH, first precipitate the silica gel. (This
precipitation is a common industrial procedure). After precipitation of the
silica gel,
the solution and gel are separated by such processes as a centrifuge or
decanatation.
The Na2CO3 remains in solution, the later recovered by evaporation.
Production of NaOH on site normally uses brine or saltwater. One by-
product of this process is chlorine gas which may find an integrated use in
the process
of the present invention through conversion to HCl to leach Ni and Co from the
quench sludge, when treating laterites.
A most preferred process of the present invention normally comprises
the following steps:
1. Formulation and preparation of an appropriate temporary' glass'
composition.
2. Introduce composition into a glass melting type furnace.
3. Apply fluxing and fusing temperature (950 C-1150 C) using gas, fuel oil,
electricity, or other available energy source via appropriate burners and/or
electrodes.
4. Once the bulk 'glass' is molten, apply current to said moving-heat-zone
assembly and traverse it through the molten section of the furnace contents so
that extra localised heat is applied (approx. 1,250 C). This could be
performed
at least twice to achieve optimum feed material assimilation.
5. Once assimilation is complete the furnace exit gate is opened to allow a
controlled flow of said molten temporary' glass' into the quenching tank, or
rapid air cool.
6. In some instances said quench tank may contain a concentrated brine
solution
and/or sodium hydroxide, or a mild acid solution for example HCI. It may be
necessary depending on metal extraction selectivity to use a fused salt such
as
potassium nitrate as a quenching medium. On other occasions it may be useful
to quench with a molten metal for example tin or lead.
7. Generally the result of said quenching cause the precipitation of the
majority
of melt components out of the melt. The quench solution may additionally be
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
29
an electrolyte in which electrowinning techniques can immediately or
subsequently be applied. Electrolyte thus generated can be conveniently
pumped to extra electrowinning tanks, or for storage and additional physical
and chemical treatments.
8. Further chemical and heat treatments can be applied to the electrolyte to
assist
the sequential removal of mineral or metallic constituents.
9. Many of the residual constituents of the original "glass" composition can
either be directly recycled into the process and/or utilized as non-toxic
aggregate materials for agricultural and building applications including
pozzolanic substances including silica.
The process of the present invention is designed to allow balanced
output-input-replenish schedules; fusing chamber and quench tank capacities
are
therefore increased accordingly.
It is preferred that the apparatus and method of the present invention
are used to treat platinum group elements (PGE), refractory gold, base metal
concentrates and high silicon silver amongst other metals and ores. The
invention
provides the capability of treating the full and extensive range of nickel
laterite ore
types, PGE's and refractory gold, a situation not possible with any one
existing
treatment and recovery process.
The present process also gives the ability to separate many rare earth
metals both from each other and from the accompanying ore matrix. Rare earths
that
may be separated using the present invention include lanthanum, cerium,
praseodymium. Neodymium, promethium, samarium, europium, gadolinium, terbium,
dysprosium, holmium, erbium, thulium, ytterbium, lutetium, yttrium, and
scandium.
The process may also be used on environmentally dangerous metals such as
cadmium
and mercury. Further, the separation of magnesium from the mineral magnesite
has
been proven.
Typically, the process for the separation of rare earth metals will
include mixing of the rare earth-containing ore with a flux such as borax,
sodium
carbonate or caustic soda with approximately 35% silica to form a "glass" at a
furnace
temperature of between approximately 1050 C to 1350 C.
There are three separate likely metal recovery methods with the present
invention. These include:
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
1 from the cathode;
2 from the solution, and
3 from the sludge (there are current methods of heap-leaching nickel
laterite ores. Because of the tight mineralogical bonds these heap-
leaching methods so far, are proving to be inefficient and bordering on
being uneconomical, even at high metal prices. However, with the
furnace treatment of the present invention alone, and specifically if
electrowinning in the melt is included, the target metals become far
simpler to capture and recover.
Addition of iron to the melt preferably precipitates nickel and cobalt.
Where used in this specification, the term "glass" is not used to convey
a crystalline structure but rather an ionic melt structure which when cooled
is
hygroscopic and typically amorphous. It may be partially vitrified when cooled
but
typically when an appropriate flux material is used, will typically retain a
non-solid
and non-crystalline structure.
Brief Description of the Drawings.
Various embodiments of the invention will be described with reference
to the following drawings, in which:
Figure 1 is a sectional side view of a process apparatus according to a
preferred embodiment of the present invention.
Figure 2 is a sectional plan view of the process apparatus illustrated in
Figure 1.
Figure 3 is a sectional side view of a process apparatus according to a
second preferred embodiment of the present invention.
Figure 4 is a sectional plan view of the process apparatus illustrated in
Figure 3.
Figure 5 is a sectional side view of a process apparatus according to a
third preferred embodiment of the present invention.
Figure 6 is a sectional plan view of the process apparatus illustrated in
Figure 5.
Figure 7 is the front page of a result sheet from a trial of a preferred
embodiment of the method of the present invention.
Figure 8 is the reverse of the result sheet illustrating Figure 7.
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
31
Figure 9 is the front page of a result sheet from a trial of a preferred
embodiment of the method of the present invention.
Figure 10 is the reverse of the result sheet illustrating Figure 9.
Figure 11 is the front page of a result sheet from a trial of a preferred
embodiment of the method of the present invention.
Figure 12 is the reverse of the result sheet illustrating Figure 11.
Figure 13 is the front page of a result sheet from a trial of a preferred
embodiment of the method of the present invention.
Figure 14 is the reverse of the result sheet illustrating Figure 13.
Figure 15 is the front page of a result sheet from a trial of a preferred
embodiment of the method of the present invention.
Figure 16 is the reverse of the result sheet illustrating Figure 15.
Detailed Description of the Preferred Embodiment.
According to a particularly preferred embodiment, a method and
device for the removal of metals from their naturally occurring ores and
mineral
concentrates using vitrification processes are provided.
The only ores considered in this discussion of the operation of a
preferred embodiment, are nickel/cobalt laterites even though the process can
similarly be used to treat sulphide ores/concentrates including refractory
gold and
refractory gold with arsenopyrite present.
Testing has been performed to date with three laterites, two from India
and one from Western Australia. Initial focus has been on laterites from
Western
Australia.
Two separate samples, LH and LH2 respectively have been used. A
typical component analysis of LH2 is as follows:
Ni CoO Cr203 A1203 CaO MgO Fe203 Si02 LOI %
1.73 0.074 0.10 0.93 0.04 3.15 14.31 50.69 19.2
The composition of the glass forming substrate used in the illustrative
embodiment for the nickel laterite processed is as follows:
Nickel 0.4-3.0%
Cobalt 0.04-1.6 %
Aluminium 0.5-6.00%
Magnesium 0.4-48.0%
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
32
Silica 8.0-60.0%
Calcium 0.01-0.5%
Chromium 0.2-3.0%
Iron 8.0-55.0%
Manganese 0.05-10.0%
Zinc 0.05-0.1%
Copper 0.05-0.1%
The process to which the nickel laterite is subjected is as follows:
1. Preparation of the appropriate temporary' glass' composition as above.
2. Introduce the composition into a glass melting type furnace.
3. Apply fluxing and fusing temperature (950 C-1150 C) using gas, fuel oil,
electricity, or other available energy source via appropriate burners and/or
electrodes.
4. Once the bulk material is molten, apply current to said moving-heat-zone
assembly and traverse it through the molten section of the furnace contents so
that extra localised heat is applied (approx. 1,250 C). This could be
performed
at least twice to achieve optimum feed material assimilation.
5. Once assimilation is complete the furnace exit gate is opened to allow a
controlled flow of said molten temporary' glass' into the quenching tank or
rapid air cooling.
6. In this illustrative embodiment, the quench tank contains a brine solution.
7. The result of said quenching cause the precipitation of the majority of
melt
components out of the melt.
Three alternative forms of a production apparatus used in the above
process, are illustrated in Figures 1 to 6.
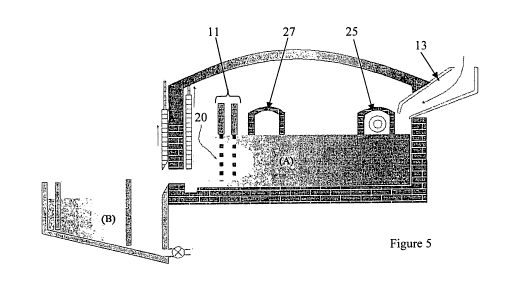
Figure 1 shows both a cross section and plan view of a first preferred
embodiment of a `glass' fusing chamber or furnace A and quenching tank B. In
Figures 1 to 6, the following components of the system are present:
molten `glass'.
11 electrode assembly creating a moving-heat-zone or high temperature
zone
13 raw material entry point
14 inner glass exit plug
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
33
15 outer glass exit plug
16 electrowinning assembly
17 electrowinning assembly
18 quenching fluid/electrolyte (g,h,i, options only)
19 metal tank material (with corrosive materials this is an option only)
20 high temperature zone
21 quenching fluid/electrolyte outlet valve
Figures 1 and 2 show a cross section view of the furnace or melting
chamber and quenching tank with ionic melt exit plugs 14 and 15 in their open
positions allowing the ionic melt 10 to enter quenching tank B. Moving-heat-
zone
electrode assembly 11 is shown in its exit position after at least two
traverses of the
fusing chamber A.
The embodiment of the invention illustrated in Figures 1 and 2 also has
a pair of burners 25 for heat addition to the furnace A and an exhaust 26 for
heat
capture.
Also illustrated in embodiment of Figures 1 and 2 is a subsurface gas
inlet system 23 which although illustrated closer to the discharge end of the
furnace
A, will normally be located towards the inlet end. The subsurface gas inlet
system 23
is provided to promote mixing in the furnace A. It may use air and/or steam
and will
allow for pulsed or intermittent injections as well as steady flow injection.
The quenching tank B has a liquid outlet 21 and a sludge removal
outlet 22. The sludge removal outlet 22 is located in a lower portion of the
tank B
below a grading grill 24 which will typically act to break down any larger
sludge
lumps that may form upon entry of the molten mixture 10 into the quenching
tank B.
Figures 3 and 4 illustrate an alternative embodiment of the production
furnace and quenching tank. In these Figures, the preferred moving electrode
assembly 11 to create a higher temperature area within the melt is illustrated
as is the
moving heat zone 20 which is created. In most other respects, the embodiment
illustrated in Figures 3 and 4 is similar to that illustrated in Figures 1 and
2.
Figure 5 and 6 show views of an ionic melt chamber and quenching
tank, illustrating exhaust heat recuperation/regeneration chamber 27 and
burner
assembly 25. It should be noted that multiple burner/exhaust assemblies can be
employed for larger capacity chambers and/or to increase heat transfer
efficiencies.
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
34
All proof of concept experiments to date (over 300 separate runs) have
been conducted in a small crucible furnace. This furnace is fire-brick lined,
insulated,
all encased in a steel liner. This furnace is designed to take a ceramic
crucible that can
comfortably take a 300g charge. The furnace has a central, hinged lid, with a
central
port. This port serves three main purposes. The first is to observe the
charge, the
second provides access to a stirring rod while the third is to provide the one
and only
exhaust.
The furnace is fired by propane gas, using conventional household
cylinders. The gas enters through a lower, tangential port. The gas is
assisted by a
small electrically powered centrifugal blower which allows temperature to
reach up to
1150 C. This blower is normally activated after the furnace temperature
reaches
300 C. A removable thermocouple is located opposite to the gas port. This
thermocouple in turn is wired to a wall-mounted temperature display panel.
It is in this test furnace that the results illustrated in Figures 7 to 16
have been obtained to prove the invention.
The first step is to add the flux only then melt so generating an initial
molten flux pool.
Coarser particles (2-8m) have proved more reactive, as observed by
increased effervescence (efc), when compared to the additions of finer
particles. Thus
ore feed of a production furnace will typically not have to be ground. The
avoidance
of grinding becomes a major cost benefit of the present invention. Crushing to
10 mm
should be adequate. The highly aggressive nature of the flux suggests the feed
size
could even be greater than 10mm.
The procedure is to add the laterite and additional flux in a number of
smaller amounts, stirring after each addition.
Fresh additions tend to fuse into `lumps'. When breaking these sintered
lumps, some of the fines of the core become airborne. In a production furnace,
this
may constitute losses. However, on a mass scale, with a thin feed stream and
air jet
agitation, this loss will not occur.
The crucible stirring rod, after each stirring, quickly gains a thick wad
of crucible contents on the tip. This wad, immediately after each stirring, is
broken off
and returned to the crucible and allowed to re-melt.
Frequently, immediately prior to the pour, there is effervescence and/or
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
bubbling within the crucible contents. This suggests the crucible contents
have not yet
reached their final temperature and digestion is not complete. Providing the
laterite
particles have been digested, the pour is homogeneous and fluid, and the final
temperature has been reached, the pour should proceed. This suggests that
increased
residence time has little benefit.
The aim is to have the shortest furnace residence time as possible, then
expel. This allows for increasing furnace production rates and efficiencies.
The commercial/production aspects of the balance between increased
furnace temperatures and residence time will need to be carefully assessed on
a case
by case basis. Current findings are showing the time span between the first
ore
addition and pour to be under 40 minutes. The time span between the last ore
addition
and pour is 5-10 minutes.
The pour should be homogeneous. This illustrates the ore to flux ratios
are in balance. In earlier times, the pour of the ore contents was preceded by
an initial
run of fluxes, which cooled to a canary-yellow mass. This non-homogeneous pour
indicated either a surplus of flux in the mix, or the wrong fluxes that were
not
contributing to the break-down of the ore particles, e.g., excessive amounts
of sodium
chloride. Analysis of this flux showed the presence of only trace quantities
of nickel.
Just prior to the pour, it is desirable to ensure the temperature is
elevated to plus 1000 C. This will assist in reducing the viscosity of the
pour
approximating a medium to runny honey-like consistency.
Production Furnace
Calculations have been made of the size of a possible production
furnace according to the present invention. Based on a production rate of 3
dry tonnes
of laterite per minute, or 4,300 dry tonnes per day and a total residency time
of 35
minutes, the furnace size calculates at an internal area of 7 x 20 meters.
Furnace design and construction engineers have provided a pre-
feasibility estimate for an equipped, installed and commissioned 7 x 18 meter
internal
dimensions production furnace with an estimate of natural gas consumption for
the
furnace at 40,000 cubic meters per day.
Therefore, for a furnace size expanded from 7x 18 to 7x 20 meters
50,000 cubic meters per day should be allowed.
The increased throughput of the furnace of the present invention
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
36
compared to a conventional glass furnace suggests increased wear. Therefore,
provision needs to be made both in initial construction and major maintenance
programmes to accommodate this aspect. The operating life of a production
furnace
before re-lining is required, is expected to be in excess of four years.
The feed into the furnace will be along the lines of a conventional large
scale glass furnace. The ore plus any fluxes will be pre-mixed.
In a production furnace, the feed will be pre-heated with waste heat.
The ore feed moisture content of Western Australia laterites can range from 25
-
30%. Some of this moisture will be lost in the pre-heating. However, the
addition of
moist ore directly into the furnace is not expected to create problems. In
fact, the
generation of steam may also assist with the stirring.
A practical method to assist mixing of the contents of the furnace is to
have floor-mounted air jets. These jets would typically be fed with a constant
low
pressure, with periodic pulses of higher pressure. In practice these
subsurface jets will
be located towards the feed end, rather than the discharge end.
The use of sub- surface low pressure air, possibly with steam included,
will assist in mixing the furnace contents. Pulsing of the introduced
air/steam would
assist even further.
The production furnace design contains electrodes protruding into the
melt. Glass conducts electricity in the molten state, but not as a solid.
These
electrodes may be either AC to boost the melt and stirring process, or DC. DC
electrodes have recently been trialed in the crucible experiments. Duration in
the melt
has been as little as 8 minutes. The most recent trial of LH2 material was 10
minutes
at 2 volts and 3 amps. These electrodes were both carbon, but the cathode
should be
of another element, such as molybdenum. The purpose of the DC electrodes is
primarily to assist in the breaking of any of the silica bonds of the
compounds within
the melt.
It is also preferred that the electrodes be oriented horizontally rather
than vertically.
Electrowinning (EW) in the melt may be used as a means of the direct
winning of both the nickel and cobalt onto the cathode provided that the
electrowinning of iron can be minimised.
In order to protect the electrodes in a high production furnace, the
CA 02731859 2011-01-24
WO 2010/009512 PCT/AU2009/000947
37
electrodes may be encased within a loose sleeve of a high quality, porous
ceramic.
Electrodes used so far have been carbon. As carbon electrodes burn in
the air component of the furnace atmosphere other electrodes such as
molybdenum
will be required in production furnaces.
The use of a rotary furnace has been considered. The only possible use
of a rotary furnace would be to pre-heat the feed to the main furnace.
However, it
would appear that through conventional pre-heat processes, a rotary furnace
would
not be required.
It may be required that furnace gases be cleaned of solids by
electrostatic precipitators. Also HCI, SO2 and NO gases may need to be
recovered for
environmental reasons.
In the present specification and claims (if any), the word "comprising"
and its derivatives including "comprises" and "comprise" include each of the
stated
integers but does not exclude the inclusion of one or more further integers.
Reference throughout this specification to "one embodiment" or "an
embodiment" means that a particular feature, structure, or characteristic
described in
connection with the embodiment is included in at least one embodiment of the
present
invention. Thus, the appearance of the phrases "in one embodiment" or "in an
embodiment" in various places throughout this specification are not
necessarily all
referring to the same embodiment. Furthermore, the particular features,
structures, or
characteristics may be combined in any suitable manner in one or more
combinations.
In compliance with the statute, the invention has been described in
language more or less specific to structural or methodical features. It is to
be
understood that the invention is not limited to specific features shown or
described
since the means herein described comprises preferred forms of putting the
invention
into effect. The invention is, therefore, claimed in any of its forms or
modifications
within the proper scope of the appended claims (if any) appropriately
interpreted by
those skilled in the art.