Note: Descriptions are shown in the official language in which they were submitted.
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
1
NOUVELLES COMBINAISONS DE COMPOSES HETEROCYCLIQUES AZOTES
ANTIBACTERIENS AVEC D'AUTRES COMPOSES ANTIBACTERIENS ET LEUR
UTILISATION COMME MEDICAMENTS
L'invention concerne la combinaison de composés
hétérocycliques azotés antibactériens avec d'autres composés
antibactériens et leur utilisation comme médicaments.
La demanderesse a découvert que de nouvelles combinaisons
des composés de formule (I) décrits et revendiqués dans la
demande française 07 02663, avec d'autres composés
antibactériens, possèdent des propriétés antibactériennes d'un
niveau tout à fait intéressant, manifestent par là un effet de
synergie aussi remarquable qu'inattendu.
Le caractère unique des combinaisons synergiques de
l'invention réside en particulier dans le fait qu'elles
présentent une excellente activité sur Pseudomonas aeruginosa
et Enterobacteriaceae, qui sont des souches bactériennes
fréquemment rencontrées dans les infections nocosomiales ainsi
que chez les patients souffrant de mucoviscidose.
Cette activité particulièrement intéressante et inattendue
n'est pas présente dans les composés de l'art antérieur et
notamment ceux de la demande WO 02/100860 qui décrit des
composés comportant d'autres groupes R1 que ceux des composés
hétérocycliques azotés répondant à la formule (I) définie ci-
après.
Ces composés de formule (I) se sont montrés actifs sur des
modèles d'infection animale, y compris sur des souches
habituellement résistantes aux antibiotiques communément
utilisés. Ils sont capables de contrecarrer les principaux
mécanismes de résistance des bactéries que sont les (3-
lactamases, les pompes à efflux et les mutations des porines.
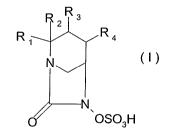
Ces composés répondent à la formule suivante :
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
2
R 2 R3
R R4
N (I)
N
OSO3H
dans laquelle R1 représente un radical (CH2) n-NH2 ou (CH2) n-
NHR, R étant un alkyle (C1-C6) et n étant égal à 1 ou 2;
R2 représente un atome d'hydrogène ;
R3 et R4 forment ensemble un hétérocycle azoté à caractère
aromatique à 5 sommets renfermant 1, 2 ou 3 atomes d'azote
éventuellement substitué par un ou plusieurs groupements R',
R' étant choisi dans le groupe constitué par un atome
d'hydrogène et les radicaux alkyle renfermant de 1 à 6 atomes
de carbone,
sous forme libre, de zwitterions, et sous forme de sels avec
les bases et les acides minéraux ou organiques
pharmaceutiquement acceptables.
La demanderesse a découvert que les composés de formule
générale (I) potentialisent l'activité de composés
antibactériens existants, en particulier sur Pseudomonas
aeruginosa et Enterobacteriaceae.
L'invention a ainsi pour objet la combinaison d'un composé
de formule générale (I) telle que définie ci-dessus, sous
forme libre, de zwitterions, ou sous forme de sels avec les
bases et les acides minéraux ou organiques pharmaceutiquement
acceptables, avec un autre composé antibactérien.
Par autre composé antibactérien, on entend notamment un
beta lactame, un monobactame ou une pénicilline, le cas
échéant combiné à un inhibiteur de beta lactamases, un
aminoglycoside, une glycylcycline, une tétracycline, une
quinolone, un glycopeptide, un lipopeptide, un macrolide, un
ketolide, un lincosamide, une streptogramine, une
oxazolidinone, une polymyxine et d'autres composés connus
doués d'une activité thérapeutique sur Pseudomonas aeruginosa
et Enterobacteriaceae.
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
3
A titre d'exemples d'aminoglycosides qui peuvent être
mentionnés on peut citer amikacine, gentamycine et
tobramycine.
A titre d'exemples de beta lactames on peut citer les
Carbapenèmes tels que Imipénème, Méropénème, Ertapénème et
le composé connu sous l'appellation PZ-601, les
Céphalosporines telles que Céfazoline, Céfépime, Céfotaxime,
Céfoxitine, Ceftaroline, Ceftazidime, Ceftibiprole,
Ceftriaxone, Céfuroxime et Céphalexine, les Monobactames tels
que l'Aztréoname, les Pénicillines et les combinaisons avec
des inhibiteurs de beta-lactamases telles que Amoxicilline,
Amoxicilline/Clavulanate, Ampicilline, Ampicilline/Sulbactame,
Oxacilline, Pipéracilline, Pipéracilline/Tazobactame,
Ticarcilline, Ticarcillin/Clavulanate et Pénicilline.
A titre d'exemples de glycylcycline et tétracycline on peut
citer Doxycycline, Minocycline, Tétracycline et Tigécycline.
A titre d'exemples de Quinolones on peut citer
Ciprofloxacine, Gatifloxacine, Grépafloxacine, Lévofloxacine,
Moxifloxacine et Ofloxacine.
A titre d'exemples de Macrolide et Kétolide on peut citer
Azithromycine, Clarithromycine, Roxythromycine et
Télithromycine.
A titre d'exemples de Polymyxine on peut citer Colistine et
Polymyxine B.
On peut encore citer d'autres composés antibactériens tels
que Fosfomycine et l'association
Triméthoprim/Sulfaméthoxazole.
Dans les composés de fomule générale (I), par radical alkyle
renfermant de 1 à 6 atomes de carbone, on entend notamment le
radical méthyle, éthyle, propyle, isopropyle, ainsi que butyle,
pentyle ou hexyle linéaire ou ramifié.
Par radical al=kényle renfermant de 2 à 6 atomes de
carbone, on entend notamment le radical allyle et les radicaux
butényle, pentényle et hexényle linéaires ou ramifiés.
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
4
Par hétérocycle à caractère aromatique, on entend
notamment ceux choisis dans la liste qui suit, les deux
liaisons symbolisant la jonction avec le cycle azoté (R3R4)
N
N N
1/ 1 1111q
N
N- N N N--\
N
N=N
N
Parmi les sels d'acides des produits de formule (I), on
peut citer entre autres, ceux formés avec les acides minéraux,
tels que les acides chlorhydrique, bromhydrique, iodhydrique,
sulfurique ou phosphorique ou avec les acides organiques comme
l'acide formique, acétique, trifluoroacétique, propionique,
benzoïque, maléique, fumarique, succinique, tartrique, citrique,
oxalique, glyoxylique, aspartique, alcanésulfoniques, tels que
les acides méthane et éthane sulfoniques, arylsulfoniques tels
que les acides benzène et paratoluènesulfoniques.
Parmi les sels de bases des produits de formule (I), on peut
citer, entre autres, ceux formés avec les bases minérales telles
que, par exemple, l'hydroxyde de sodium, de potassium, de
lithium, de calcium, de magnésium ou d'ammonium ou avec les bases
organiques telles que, par exemple, la méthylamine, la
propylamine, la triméthylamine, la diéthylamine, la
triéthylamine, la N,N-diméthyléthanolamine, le tris
(hydroxyméthyl) amino méthane, l'éthanolamine, la pyridine, la
picoline, la dicyclohexylamine, la morpholine, la benzylamine, la
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
procaïne, la lysine, l'arginine, l'histidine, la N-
méthylglucamine, ou encore les sels de phosphonium, tels que les
alkyl-phosphonium, les aryl-phosphoniums, les alkyl-aryl-
phosphonium, les alkényl-aryl-phosphonium ou les sels d'ammoniums
5 quaternaires tels que le sel de tétra-n-butyl-ammonium.
Parmi les combinaisons synergiques telles que définies plus
haut, l'invention a notamment pour objet celles renfermant des
composés de formule (I) dans lesquels R3 et R4 forment ensemble un
hétérocycle pyrazolyle ou triazolyle, éventuellement substitué.
Parmi ces combinaisons, l'invention a notamment pour objet
celles renfermant des composés dans lesquels R1 est choisi
dans le groupe constitué par les groupements (CH2)n-NH2 et
(CH2)n-NHCH3i n étant tel que défini précédemment,
l'hétérocycle formé par R3 et R4 est substitué par un radical
alkyle (C1-C6) .
Parmi ces combinaisons, l'invention a plus
particulièrement pour objet celles renfermant des composés
dans lesquels R1 représente un radical (CH2)n-NH2 ou
(CH2) n-NHCH3i n étant tel que défini précédemment et R3 et R4
forment ensemble un cycle pyrazolyle substitué par un radical
alkyle (C1-C6) .
Parmi ces combinaisons, l'invention a tout
particulièrement pour objet celles renfermant un composé de
formule (I) choisi parmi:
- la trans 8-(aminométhyl)-4,8-dihydro-l-méthyl-5-
(sulfooxy)- 4,7-méthano-7H-pyrazolo[3,4-e] [1,3]diazépin-
6(5H)-one,
- la trans 8-(aminométhyl)-4,8-dihydro-5-(sulfooxy)-
4,7-méthano-7H-pyrazolo[3,4-e] [1,3]diazépin-6(5H)-one,
- la trans 8-(méthylaminométhyl)-4,8-dihydro-5-(sulfooxy)-
4,7-méthano-7H-pyrazolo[3,4-e] [1,3]diazépin-6(5H)-one,
sous forme libre, de zwittterion et de sels avec les bases et
les acides minéraux ou organiques pharmaceutiquement
acceptables.
Parmi les combinaisons telles que définies plus haut,
l'invention a notamment pour objet celles renfermant des composés
antibactériens choisis parmi les beta-lactames ou les
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
6
pénicillines le cas échéant combinés à des inhibiteurs de beta
lactamases, et les polymyxines.
Parmi ces combinaisons, l'invention a notamment pour objet
celles renfermant des composés antibactériens choisis parmi la
Tobramycine, le Méropénème, l'Aztréoname, le Céfépime, le
Ceftazidime, la Pipéracilline le cas échéant combinée au
Tazobactame,la Colistine et la Polymyxine B.
Les composés de formule (I) peuvent être préparés par un
procédé comportant :
a) une étape au cours de laquelle on fait réagir, avec un
agent de carbonylation, le cas échéant en présence
d'une base, un composé de formule (II)
R' 1
HN R3
(II)
R4
H
dans laquelle
R'1 représente un radical CN, COOH protégé, COOR ou
(CH2) nR'5,
R'5 est un radical OH protégé, CN NH2 ou NHR protégé, CO2H
protégé, C02R
n, R, R3 et R4 sont tels que définis ci-dessus, le
substituant aminoalkyle éventuellement présent sur
l'hétérocycle formé par R3 et R4 étant alors le cas échéant
protégé,
ZH représente un groupement -NHOH protégé, en vue
d'obtenir un composé intermédiaire de formule (III)
R' 1
XR3
( III )
R4
X2
dans laquelle
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
7
R' 1, R3 et R4 ont les mêmes significations que ci-dessus et
soit X1 est un atome d'hydrogène ou un groupe protecteur et X2
représente un groupement -Z-C0-X3r X3 représentant le reste de
l'agent de carbonylation, soit X2 est un groupement -ZH et X1
représente un groupement C0-X3, X3 étant défini comme ci-dessus ;
b) une étape au cours de laquelle on cyclise l'intermédiaire
obtenu précédemment, en présence d'une base ;
c) le cas échéant, l'étape a) est précédée et/ou l'étape
b) est suivie de l'une ou de plusieurs des réactions
suivantes, dans un ordre approprié :
- protection des fonctions réactives,
- déprotection des fonctions réactives,
- estérification
- saponification,
- sulfatation,
- réduction d'esters,
- alkylation,
- carbamoylation,
- formation d'un groupe azido,
- réduction d'un azido en amine,
- salification,
- échange d'ions,
- dédoublement ou séparation de diastéréoisomères.
Comme agent de carbonylation, on peut mettre en ouvre un
réactif tel que le phosgène, le diphosgène, le triphosgène, un
chloroformiate d'aryle tel que le chloroformiate de phényle ou
de p-nitrophényle, un chloroformiate d'aralkyle tel que
chloroformiate de benzyle, un chloroformiate d'alkyle ou
d'alkényle tel que le chloroformiate de méthyle ou d'allyle,
un dicarbonate d'alkyle tel que le dicarbonate de tert-butyle,
le carbonyl-diimidazole et leurs mélanges, le disphosgène
étant préféré.
La réaction a lieu de préférence en présence d'une base ou
d'un mélange de bases qui neutralise l'acide formé. Elle peut
notamment être une amine telle que la triethylamine, la
diisopropyléthylamine, la pyridine, la diméthylaminopyridine.
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
8
Toutefois, on peut également opérer en utilisant le produit de
départ de formule II comme base. On en utilise alors un excès.
Le cas échéant, le produit de formule II est mis en oeuvre
sous la forme d'un sel d'acide, par exemple un chlorhydrate ou
un trifluoroacétate.
Comme base dans l'étape b), on peut également utiliser les
amines, ou encore les hydrures, les alcoolates, les amidures
ou carbonates de métaux alcalins ou alcalino-terreux.
Les amines peuvent être choisies par exemple dans la liste
ci-dessus.
Comme hydrure on peut notamment utiliser l'hydrure de
sodium ou de potassium.
Comme alcoolate de métal alcalin, on utilise de préférence
le t-butylate de potassium.
Comme amidure de métal alcalin on peut notamment utiliser
le bis(triméthylsilyl) amidure de lithium.
Comme carbonate, on peut notamment utiliser le carbonate
ou bicarbonate de sodium ou de potassium.
Le cas échéant, l'intermédiaire de formule III peut être
obtenu sous forme d'un sel d'acide généré lors de la réaction
de carbonylation et notamment un chlorhydrate. Il est ensuite
mis en oeuvre dans la réaction de cyclisation sous cette forme.
De préférence, la cyclisation est effectuée sans isolement
de l'intermédiaire de formule III.
Les réactions mentionnées à l'étape c) sont d'une manière
générale des réactions classiques, bien connues de l'homme du
métier. Des exemples de conditions utilisées sont décrits dans
la demande WO 02/100860 ou encore dans la demande 04/052891.
Les fonctions réactives qu'il convient, le cas échéant, de
protéger sont les fonctions acides carboxyliques, amines,
amides, hydroxy et hydroxylamines.
La protection de la fonction acide est notamment effectuée
sous forme d'esters d'alkyle, d'esters allyliques, de benzyle,
benzhydryle ou p-nitrobenzyle.
La déprotection est effectuée par saponification,
hydrolyse acide, hydrogénolyse, ou encore clivage à l'aide de
complexes solubles du Palladium 0.
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
9
Des exemples de ces protections et déprotections sont
fournis dans la demande WO 02/100860.
La protection des amines, des azotes hétérocycliques et
des amides est notamment effectuée, selon les cas, sous forme
de dérivés benzylés ou tritylés, sous forme de carbamates,
notamment d'allyle, benzyle, phényle ou tertbutyle, ou encore
sous forme de dérivés silylés tels que les dérivés tertbutyle
diméthyl, triméthyl, triphényl ou encore diphényl tertbutyl-
silyle, ou de dérivés phénylsulfonylalkyle ou cyanoalkyle.
La déprotection est effectuée, selon la nature du
groupement protecteur, par le sodium ou le lithium dans
l'ammoniac liquide, par hydrogénolyse ou à l'aide de complexes
solubles du Palladium O, par action d'un acide, ou par action
du fluorure de tétrabutylammonium ou de bases fortes telles
que l hydrure de sodium ou le t.butylate de potassium.
La protection des hydroxylamines est effectuée notamment
sous forme d'éthers de benzyle ou d'allyle.
Le clivage des éthers est effectué par hydrogénolyse ou à
l'aide de complexes solubles du Palladium O.
La protection des alcools et des phénols est effectuée de
manière classique, sous forme d'éthers, d'esters ou de
carbonates. Les éthers peuvent être des éthers d'alkyle ou
d'alkoxyalkyle, de préférence des éthers de méthyle ou de
méthoxyéthoxyméthyle, des éthers d'aryle ou de préférence
d'aralkyle, par exemple de benzyle, ou des éthers silylés, par
exemple les dérivés silylés cités plus haut. Les esters
peuvent être n'importe quel ester clivable connu de l'homme du
métier et de préférence l'acétate, le propionate ou le
benzoate ou p-nitrobenzoate. Les carbonates peuvent être par
exemple des carbonates de méthyle, tertbutyle, allyle, benzyle
ou p-nitrobenzyle.
La déprotection est effectuée par les moyens connus de
l'homme du métier, notamment la saponification, l'hydrogénolyse,
le clivage par des complexes solubles du Palladium O,
l'hydrolyse en milieu acide ou encore, pour les dérivés silylés,
le traitement par le fluorure de tétrabutylammmonium.
Des exemples sont fournis dans la partie expérimentale.
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
La réaction de sulfatation est effectuée par action des
complexes S03-amines tels que S03-pyridine ou S03-
diméthylformamide, en opérant dans la pyridine, le sel formé,
par exemple le sel de pyridine, pouvant ensuite être échangé
5 par exemple par un sel d'une autre amine, d'un ammonium
quaternaire ou d'un métal alcalin. Un exemple est fourni dans
la partie expérimentale.
La réaction d'alkylation est effectuée par action sur les
dérivés hydroxylés, les énolates d'esters ou de cétones, les
10 amines ou les azotes hétérocycliques, selon les cas, d'un
sulfate d'alkyle ou d'un halogènure d'alkyle ou d'alkyle
substitué, notamment par un radical carboxy libre ou
estérifié. Des réactions d'alkylation peuvent également être
réalisées par amination réductrice.
La salification par les acides est le cas échéant réalisée
par addition d'un acide en phase soluble au composé. La
salification par les bases de la fonction sulfooxy peut être
réalisée à partir du sel de pyridinium obtenu lors de l'action
du complexe S03-pyridine et on obtient les autres sels à partir
de ce sel de pyridinium. On peut encore opérer par échange
d'ions sur résine.
La réaction de carbamoylation peut être réalisée par la
mis en oeuvre d'un chloroformiate ou d'un réactif type Boc-ON
puis d'une amine ou, le cas échéant, d'ammoniac.
L'introduction d'un groupe azido peut être effectuée par
exemple par action d'azoture de sodium sur un intermédiaire de
type mésylate ou par des réactions de type Mitsunobu.
La réduction d'un groupe azide peut être effectuée par
action de trialkyl ou triarylphosphine.
La séparation des énantiomères et diastéréoisomères peut
être réalisée selon les techniques connues de l'homme du
métier, notamment la chromatographie.
Outre via les procédés décrits précédemment, des composés
de formule (I) peuvent être obtenus par des méthodes qui
utilisent au départ un composé de formule (II) dans laquelle
R'1, R3, R4 et HZ ont les valeurs qui conduisent directement
(sans transformation) à celles des composés que l'on souhaite
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
11
préparer. Le cas échéant, celles de ces valeurs qui
renfermeraient des fonctions réactives telles que mentionnées
plus haut sont alors protégées, la déprotection intervenant à
l'issue de l'étape de cyclisation b ou à tout autre moment
opportun dans la synthèse. Les protections et déprotections
sont alors réalisées comme décrit ci-dessus.
Le composé de formule (II) peut être obtenu par un procédé
selon lequel on traite un composé de formule (IV):
R'1
ADN R3
( IV )
R4
dans laquelle R' 1r R3 et R4 sont définis comme précédemment, et
A représente un atome d'hydrogène ou un groupement protecteur
de l'azote, par un agent de réduction, pour obtenir un composé
de formule (V) :
R'
A"N R3
(V)
R4
H
dans laquelle A, R'1, R3 et R4 conservent leur signification
précitée, dans lequel, le cas échéant, l'on remplace le
groupement OH par un groupe partant, pour obtenir un composé
de formule (VI)
R'
ADN R3
( VI )
R4
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
12
dans laquelle A, R'1, R3 et R4 conservent leur signification
précitée et R9 représente un groupe partant, que l'on traite
par un composé de formule Z1H2 dans laquelle Z1 représente un
groupement -HN-OH protégé puis, le cas échéant, par un agent
de déprotection de l'atome d'azote approprié.
Le composé de formule (II) peut encore être obtenu par un
procédé selon lequel on traite un composé de formule (IV)
telle que définie précédemment, par l'hydroxylamine protégée
au niveau de l'hydroxy, pour obtenir un composé de formule
(VII)
R'
ADN R3
( VII )
R4
OH protégé
dans laquelle A, R' 1, R' 2, R3, R' 4, n et R'8 sont définis comme
précédemment, que l'on fait réagir avec un agent de réduction
pour obtenir un composé de formule (VIII)
R' 1
ADN R3
(VIII)
R4
ZH
dans laquelle A, R' 1, R3, R4, n" et ZH sont définis comme
précédemment, que l'on traite, le cas échéant, par un agent de
déprotection de l'atome d'azote approprié.
Le groupement protecteur de l'azote est notamment l'un de
ceux qui sont cités plus haut.
L'agent de réduction est notamment un borohydrure alcalin.
Le groupe partant est notamment un sulfonate, par exemple
un mésylate ou un tosylate, obtenu par action de chlorure de
sulfonyle correspondant en présence d'une base, ou un
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
13
halogène, plus particulièrement un chlore, un brome ou un
iode, obtenu par exemple par action du chlorure de thionyle ou
de P (C6H5) 3CBr4 ou PBr3 ou, dans le cas d'un atome d' iode, par
action d'un iodure alcalin sur un sulfonate.
L'agent de déprotection est notamment l'un de ceux
mentionnés plus haut.
L'agent de réduction que l'on fait agir sur le composé de
formule (VII) est notamment un cyano ou un acétoxyborohydrure
de sodium.
Comme indiqué plus haut, les composés de formule générale
(I) potentialisent l'activité de composés antibactériens
existants, en particulier sur Pseudomonas aeruginosa et
Enterobacteriaceae ainsi que sur des modèles d'infection
animale par des souches résistantes aux agents antibactériens
communément utilisés. Une telle activité antibiotique
remarquable et inattendue n'avait pas été observée pour les
composés de l'art antérieur.
Ces propriétés rendent aptes les combinaisons synergiques
selon l'invention à être utilisées comme médicaments, en
particulier dans le traitement des infections sévères à
Pseudomonas et Enterobacteriaceae, notamment des infections
nosocomiales et, d'une manière générale, des infections
majeures chez les sujets à risques. Il peut en particulier
s'agir d'infections des voies respiratoires, par exemple la
pneumonie aiguë ou les infections chroniques des voies
respiratoires inférieures, les infections du sang, par exemple
les septicémies, les infections aiguës ou chroniques des voies
urinaires, celles du système auditif, par exemple l'otite
externe maligne, ou l'otite chronique suppurante, celles de la
peau et des tissus mous, par exemple les dermatites, les
plaies infectées, la folliculite, la pyodermite, les formes
rebelles d'acnée, les infections des yeux, par exemple
l'ulcère de la cornée, celles du système nerveux, notamment
les méningites et les abcès du cerveau, les infections
cardiaques telles que l'endocardite, les infections des os et
des articulations telles que la pyoarthrose sténoarticulaire,
l'ostéomyélite vertébrale, la symphysite pubienne, les
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
14
infections du tube gastro-intestinal, telles que
l'entérocolite nécrosante et les infections péri-rectales.
La présente invention a donc également pour objet, à titre
de médicaments et notamment de médicaments antibiotiques, les
combinaisons synergiques telles que définies ci-dessus.
Parmi ces combinaisons, l'invention a notamment pour
objet, à titre de médicaments, celles renfermant des composés
de formule (I) dans lesquels R3 et R4 forment ensemble un
hétérocycle pyrazolyle ou triazolyle, éventuellement
substitué, et parmi ceux-ci, ceux dans lesquels R1 est choisi
dans le groupe constitué par les groupements (CH2)n-NH2 et
(CH2)n-NHCH3r n étant tel que défini précédemment,
l'hétérocycle formé par R3 et R4 est substitué par un radical
alkyle (C1-C6) .
Parmi ces combinaisons, l'invention a plus
particulièrement pour objet, à titre de médicaments, celles
renfermant des composés dans lesquels R1 représente un radical
(CH2)n-NH2 ou (CH2)n-NHCH3, n étant tel que défini précédemment
et R3 et R4 forment ensemble un cycle pyrazolyle substitué par
un radical alkyle (C1-C6).
Parmi ces combinaisons, l'invention a tout
particulièrement pour objet, à titre de médicament, celles
renfermant l'un au moins des composés dont les noms suivent:
- la trans 8-(aminométhyl)-4,8-dihydro-1-méthyl-5-
(sulfooxy)- 4,7-méthano-7H-pyrazolo[3,4-e] [1,3]diazépin-
6(5H)-one,
- la trans 8-(aminométhyl)-4,8-dihydro-5-(sulfooxy)-
4,7-méthano-7H-pyrazolo[3,4-e] [1,3]diazépin-6(5H)-one,
ou
- la trans 8-(méthylaminométhyl)-4,8-dihydro-5-(sulfooxy)-
4,7-méthano-7H-pyrazolo[3,4-e] [1,3]diazépin-6(5H)-one,
sous forme libre, de zwittterion et de sels avec les bases et
les acides minéraux ou organiques pharmaceutiquement
acceptables.
Parmi ces combinaisons, l'invention a notamment pour
objet, à titre de médicaments, celles renfermant des composés
antibactériens choisis parmi les aminoglycosides, les beta-
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
lactames, les pénicillines le cas échéant combinées à des
inhibiteurs de beta lactamases, et les polymyxines.
Parmi ces combinaisons, l'invention a notamment pour objet,
à titre ' de médicaments, celles renfermant des composés
5 antibactériens choisis parmi la Tobramycine, le Méropénème, le
Céfépime, le Ceftazidime, l'Aztréoname, la Lévofloxacine, la
Pipéracilline le cas échéant combinée au Tazobactame,la
Colistine et la Polymyxine B.
L'invention a aussi pour objet les compositions
10 pharmaceutiques renfermant comme principes actifs, une
combinaison synergique telle que définie ci-dessus.
Ces compositions peuvent être administrées par voie
buccale, rectale, parentérale, notamment intramusculaire, ou
par voie locale en application topique sur la peau et les
15 muqueuses.
Les compositions selon l'invention peuvent être solides ou
liquides et se présenter sous les formes pharmaceutiques
couramment utilisées en médecine humaine comme par exemple,
les comprimés simples ou dragéifiés, les gélules, les
granulés, les suppositoires, les préparations injectables, les
pommades, les crèmes, les gels; elles sont préparées selon les
méthodes usuelles. Le ou les principes actifs peuvent y être
incorporés à des excipients habituellement employés dans ces
compositions pharmaceutiques, tels que le talc, la gomme
arabique, le lactose, l'amidon, le stéarate de magnésium, le
beurre de cacao, les véhicules aqueux ou non, les corps gras
d'origine animale ou végétale, les dérivés paraffiniques, les
glycols, les divers agents mouillants, dispersants ou
émulsifiants, les conservateurs.
Ces compositions peuvent notamment se présenter sous forme
d'un lyophilisat destiné à être dissout extemporanément dans
un véhicule approprié, par exemple, de l'eau stérile
apyrogène.
Les compositions selon l'invention comprennent donc au
moins deux principes actifs, et ceux-ci peuvent être
administrés simultanément, séparément ou d'une manière
échelonnée dans le temps. Elles peuvent par exemple se
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
16
présenter sous forme d'un kit , permettant l'administration
d'un composé de formule générale (I) et celle d'un autre
composé antibactérien séparément.
La dose en composé de formule (I) administrée est variable
selon le niveau et la nature de l'affection traitée, le sujet
en cause, la voie d'administration et l'autre produit
antibactérien considéré. Elle peut être, par exemple, comprise
entre 0,25 g et 10 g par jour, par voie orale chez l'homme,
avec le produit décrit à l'exemple 1 ou encore comprise entre
0,25 g et 10 g par jour par voie intramusculaire ou intra-
veineuse.
La dose en l'autre composé antibactérien est aussi
variable selon l'affection traitée, le sujet en cause, la voie
d'administration et le produit considéré, mais suit,
généralement les doses habituelles prescrites par les
pratitiens, par exemple comme décrit dans la publication de
référence Vidal. Cette dose peut aller jusqu'à 10 g par jour,
voire davantage. Néanmoins, par suite de la potentialisation
apportée par les composés de formule générale (I) aux autres
composés antibacteriéns, les doses de ceux-ci au sein de la
combinaision peuvent être réduites par rapport aux doses
standards.
Les combinaisons selon l'invention peuvent également être
utilisées comme désinfectants des instruments chirurgicaux.
Les exemples suivants illustrent la préparation de
composés de formule (I). Les autres composés antibactériens
sont quant à eux connus et commerciaux.
EXEMPLES
Exemple 1 : sel de sodium et de trifluoroacétate de la trans
8-(aminométhyl)-4,8-dihydro-1-méthyl-5-(sulfooxy)-4,7-méthano-
7H-pyrazolo[3,4-e] [1,3] diazépin-6(5H)-one
Stade A :
4,7-dihydro-1-méthyl-4-((phénylméthoxy)amino)-1H-
pyrazolo[3,4-c]pyridine-6(5H),7-dicarboxylate de 6-(1,1-
diméthyléthyle) et de 7-méthyle (B)
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
17
Le dérivé A (4,7-dihydro-4-hydroxy-l-methyl-lH-
pyrazolo[3,4-c]pyridine-6 (5H),7-dicarboxylate de 6-(1,1-
diméthyléthyle) et de 7-méthyle, décrit dans la demande
WO 02100860 (10 g, 32.12 mmol) est mis en suspension dans le
dichlorométhane (100 ml) à température ambiante sous azote et
sous agitation. La suspension se dissout après l'ajout de
triethylamine (14.30 ml, 10.28 mmol, 3.2eq). Au milieu
réactionnel refroidi à -78 C est additionné goutte à goutte une
solution de chlorure de méthane sulfonyle (11.4 ml, 96.36 mmol,
3eq) dans le dichlorométhane (12 ml, 1 volume). Après 30 min de
contact, l'alcool A est complètement transformé en mésylate.
Une solution de 0-benzyl-hydroxylamine dans le
dichlorométhane est fraîchement préparée à partir du
chlorhydrate de O-benzylhydroxylamine (25.4 g, 160.6 mmol,
5eq). Le chlorhydrate de 0-benzylhydroxylamine est dissout dans
un mélange de dichlorométhane (100 ml) et d'eau (50 ml). Une
solution de soude 2N (85 ml, 176.66 mmol) est ajoutée à 0 C.
Après 10 min de contact et décantation, la phase organique est
séchée sur sulfate de magnésium durant 45 min, puis concentrée
à demi volume. L'addition de cette solution au mésylate préparé
ci-dessus se fait à -78 C goutte à goutte sur 1 heure. Le
mélange réactionnel est agité en laissant la température
remonter progressivement à l'ambiante. On traite par addition
d'eau (200 ml) et dilue le milieu avec du dichlorométhane
(100 ml), agite, décante puis réextrait la phase aqueuse au
dichlorométhane. La phase organique est lavée avec une solution
de NaCl saturée (200 ml), séchée, puis concentrée à sec. On
récupère une poudre amorphe blanche , qui après chromatographie
délivre le dérivé B attendu (8.25 g, 66%).
MS (ES (+)) : m/z [M+] = 417.2
1H NMR (400MHz, CDC13): un diastéréoisomère (2 rotamères) 8 (ppm)
= 1.43 (s, 9H, tBu), 3.15 (dd, 1H, N-CH2-CH-N), 3.68 / 3.70 (s,
3H, CH3), 3.84(s, 3H, CH3), 3.98 (m, 2H, N-CH2-CH-N), 4.6-4.8
(massif, 3H, NH-O-CH2-Ph et N-CH2-CH-N), 5.40 / 5.8 (s, 1H, CH-
C02Me), 7.22-7.31(massif, 5H, Ph), 7.40(s, 1H, H pyrazole)
Stade B
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
18
Trans 1-methyl-6-oxo-5-(phénylméthoxy)-4,5,6,8-tétrahydro-4,7-
méthano-1H-pyrazolo[3,4-e] [1,3]diazépine-8 (7H) carboxylate
de méthyle (C)
Une solution 4N de HCl/ dioxane (400 ml,l5eq) est coulée
sur une solution de B (21 g, 50.42 mmol) dissout dans le
dioxane (50 ml) à température ambiante. Le mélange réactionnel
est agité pendant 30 min, puis le dioxane est évaporé. Le
résidu est repris sous agitation dans un mélange d'eau (100
ml) et d'acétate d'éthyle (500 ml). Une solution d'ammoniaque
concentrée à 20% (42 ml) est ajoutée à 0 C. L'agitation est
poursuivie pendant 30 min. Après décantation la phase aqueuse
est ré-extraite avec de l'acetate d'éthyle (2*300 ml), la
dernière extraction étant réalisée après saturation de la
phase aqueuse avec du NaCl. La phase organique est séchée puis
concentrée. On obtient l'intermédiaire pipéridine déprotégée
sous forme d'une huile jaune (m=15.7 g, 98%) qui est repris
dans l'acétonitrile (400 ml). A ce mélange refroidi à 0 C,
sont ajoutés la triéthylamine (21 ml, 151.2 mmol, 3eq), puis
le diphosgène (3.04 ml, 25.2 mmol, 0.5eq) coulé goutte à
goutte sur 30 min. Après une nuit de contact à température
ambiante, le milieu est concentré puis repris avec de
l'acétate d'éthyle (500 ml) et traité avec une solution
d'acide tartrique 10% (200 ml). Le mélange est agité, décanté.
La phase organique est lavée par une solution d'acide
tartrique 10% (2*200 ml), par une solution de NaCl saturée,
puis séchée et concentrée sous pression réduite. Le produit
blanc obtenu (m=15.3 g, 89%) est repris dans le
dichlorométhane (150 ml). Du 1-8-Diazabicyclo[5.4.0]undéc-7-
ène (7.53 ml,50.04 mmol) est ajouté goutte à goutte. Le
mélange est agité pendant 2h, traité avec de l'eau (200 ml),
agité, décanté. La phase organique est lavée avec de l'eau
(2*200 ml), puis avec une solution de NaCl saturée (1*200 ml),
et séchée sur MgSO4r puis concentrée à sec.
On récupère le dérivé C attendu (m=14.72 g, 85%), sous
forme d'un solide blanc
MS (ES (+)) : m/z [M+]=343
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
19
1H NMR (400MHz, CDC13): 6 (ppm) = 3.25 (d, 1H, N-CH2-CH-N),
3.45 (d, 1H, N-CH2-CH-N), 3.80 (s, 3H, CH3), 3.88 (s, 3H,
CH3), 3.9 (s,1H, N-CH2-CH-N), 4.7 (d, 1H, N-0-CH2-Ph), 5.02
(d, 1H, N-0-CH2-Ph), 5.22 (s; 1H, CH-C02Me), 7.39-7.43(massif,
6H, H pyrazole + Ph)
Stade C
4,8-dihydro-8-(hydroxyméthyl)-1-méthyl-5-(phénylméthoxy)-4,7-
méthano-7H-pyrazolo[3,4-e] [1,3] diazépin-6(5H)-one (D)
Une solution de C (5g, 14.60 mmol) dans un mélange de
tétrahydrofurane (150 ml) / méthanol (50 ml) anhydres, sous
azote et sous agitation, est refroidie à -10 C. Du borohydrure
de lithium (668 mg, 30.67 mmol, 1.2eq) est ajouté au milieu
réactionnel. Après 2 h d'agitation à -10 C, 1.2eq
supplémentaires de LiBH4 sont ajoutés. La réaction est traitée
à froid 2 h plus tard par une solution à 10% de NaH2PO4. Le
tétrahydrofurane et le méthanol sont évaporés sous pression
réduite(200 mbar, 40 C). Le mélange résiduel est repris avec
de l'acétate d'éthyle (200 ml), agité et décanté. La phase
aqueuse est réextraite avec de l'acétate d'éthyle (100 ml). La
phase organique est séchée sur sulfate de magnésium puis
concentrée à sec. La poudre jaune clair obtenue ,(6.6 g) est
chromatographiée sur silice (éluant-acétate d'éthyle) pour
donner le dérivé D (3.2 g,10.18 mmol, 64%).
MS (ES (+) ) : m/z [M+] =315
'H NMR (400 MHz, DMSO-d6) : 6 (ppm) = 3.16 (dd, 1H, N-CH2-CH-N),
3.48 (d, 1H, N-CH2-CH-N), 3.71 (s, 3H, CH3), 3.81-3.91
(massif, 2H, CH2OH), 4.44 (m, 1H, N-CH2-CH-N), 4.48 (m, 1H,
CHCH2OH), 4.88 (m, 2H, N-O-CH2-Ph), 5.20 (m, 1H, OH), 7.35-
7.40 (massif, 6H, H pyrazole + Ph).
Stade D :
Trans 4,8-dihydro-1-méthyl-8-[(méthylsulfonyl)oxyméthyl)]- 5-
(phénylméthoxy)-4,7-méthano-7H-pyrazolo[3,4-e] [1,3] diazépin-
6 (5H) -one (E)
Le dérivé D (2.76 g, 8.78 mmol) est mis en solution dans
du dichioromethane (100 ml) à température ambiante sous azote
et sous agitation. Après refroidissement à 0 C, on ajoute de
la triethylamine (1.83 ml, 13.17 mmol, 1.5eq), puis goutte à
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
goutte une solution de chlorure de mesyle (1.61 g, 14.05 mmol)
dans le dichioromethane (100 ml) . Le bain de glace est retiré
en fin d'addition. Au bout d'une heure de contact à
température ambiante, la réaction est traitée sous agitation
5 par une solution à 10% de NaH2PO4 (80 ml). Après agitation et
décantation, la phase aqueuse est réextraite au
dichlorométhane (50 ml) . La phase organique est séchée, puis
concentrée sous pression réduite pour donner le dérivé attendu
(3.44 g, rendement quantitatif).
10 MS (ES (+) ) : m/z [Mf]=393
1H NMR (400 MHz, DMSO-d6) : 8 (ppm) = 3.23 (dd, 1H, N-CH2-CH-N) ,
3.26 (s, 3H, CH3), 3.45 (d, 1H, N-CH2-CH-N), 3.76(s, 3H, CH3),
4.52 (m, 1H, N-CH2-CH-N), 4.58 (dd, 1H, CH-CH2-OMs), 4.66 (dd,
1H, CH-CH2-OMs), 4.88 (m, 3H, CHCH2OMs et N-O-CH2-Ph), 7.35-
15 7.45 (massif, 6H, H pyrazole + Ph)
Stade E :
trans 8-(azidométhyl)-4,8-dihydro-1-méthyl-5-(phénylméthoxy)-
4,7-méthano-7H-pyrazolo[3,4-e] [1,3], diazépin-6(5H)-one (F)
De l'azidure de sodium est ajouté en une seule fois
20 (1.71 g, 26.3 mmol) à une solution de E (3.44 g, 8.78 mmol)
dans le dimethylformamide (70 ml) à température ambiante sous
azote et sous agitation. Le milieu réactionnel est chauffé à
65 C toute une nuit, puis traité par une solution aqueuse à 10%
de NaH2PO4 (50 ml). Après agitation et décantation, la phase
aqueuse est réextraite au dichloromethane (2*50 ml). La phase
organique est séchée, puis concentrée sous pression réduite
pour donner 3.96 g de dérivé F attendu (3 g, 8.78 mmol).
MS (ES (+) ) : m/z [M+] =340
iH NMR (400 MHz, DMSO-d6) : 8 (ppm) =3.20 (dd, 1H, N-CH2-CH-N),
3.48 (d, 1H, N-CH2-CH-N), 3.66 (dd, 1H, CH-CH2-N3), 3.72 (s,
3H, CH3), 3.92 (dd, 1H, CH-CH2-N3), 4.50(d, 1H, N-CH2-CH-N),
4.76 (dd, 1H, CHCH2ON3), 4.89 (m, 2H, N-O-CH2-Ph), 7.35-7.45
(massif, 6H, H pyrazole + Ph)
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
21
Stade F
trans [[4,5,6,8-tétrahydro-1-méthyl-6-oxo-5-(phénylméthoxy)-4,7-
méthano-7H-pyrazolo[3,4-e] [1,3] diazépin-8-yl]méthyl]-carbamate
de'1,1-diméthyléthyle (G)
Une solution molaire de trimethylphosphine (3.4 ml,
3.4 mmol) est ajoutée goutte à goutte à une solution de F
(1.15 g, 3.39 mmol) dans un mélange toluène (5 ml) et
tetrahydrofurane (5 ml) à température ambiante sous azote et
sous agitation. Apres 3 H de contact, une solution de BOC-ON
(0.92 g, 3.6 mmol) dans le tetrahydrofurane (10 ml) est ajoutée
goutte à goutte au milieu réactionnel refroidi à 0 C.
L'agitation est poursuivie pendant 3 h à température ambiante.
Le milieu réactionnel est traité par une solution aqueuse à 10%
de NaHCO3 (50 ml). Après agitation et décantation, la phase
aqueuse est réextraite à l'acétate d'éthyle (50 ml). La phase
organique est séchée, puis concentrée sous pression réduite
pour donner une huile (2.2 g). Le produit brut est
chromatographié sur colonne de silice (éluant cyclohexane/
acétate d'éthyle 5/5). On obtient le produit attendu (0.62 g,
1.49 mmol, 70%).
MS (ES (+) ) : m/z [M+] =414
'H NMR (400MHz, CDC13) : 6 (ppm) = 1.39 (s, 9H, tBu) , 3.05 (dd,
1H, N-CH2-CH-N), 3.19 (dd, 1H, CH-CH2-NHBOC), 3.27 (dd, 1H, N-
CH2-CH-N), 3.72 (s, 3H, CH3), 3.78(m, 1H, CH-CH2-NHBOC), 3.88
(d, 1H, N-CH2-CH-N), 4.48 (dd , 1H, CHCH2NHBOC), 4.79 (d, 1H,
N-0-CH2-Ph), 4.92 (d, 1H, N-0-CH2-Ph), 5.18 (m, 1H, H mobile),
7.35 (s, 1H, H pyrazole), 7.37-7.48 (massif, 5H, Ph)
Stade G :
Sel de pyridinium du trans [[4,5,6,8-tétrahydro-1-méthyl-6-oxo-
5-(sulfooxy)-4,7-méthano-7H-pyrazolo[3,4-e] [1,3] diazépin-8-
yl]méthyl]-carbamate de 1,1-diméthyléthyle (H).
Du palladium 10% sur charbon (140 mg) est ajouté à une
solution de G (0.6 g, 1.45 mmol) dans le méthanol (10 ml). Le
milieu réactionnel est hydrogéné pendant 3h. Le méthanol est
ensuite évaporé sous pression réduite pour donner
l'intermédiaire débenzylé.
MS (ES (+) ) : m/z [M+] =324
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
22
L'intermédiaire débenzylé est repris dans la pyridine
(3 ml) en présence du complexe pyridine / sulfure de trioxyde
(462 mg, 2.9 mmol). La réaction est maintenue sous agitation à
température ambiante pendant une nuit. Lee milieu est ensuite
concentré sous pression réduite. Le brut réactionnel est
chromatographié sur colonne de silice (éluant dichloromethane
100% puis gradient avec du méthanol de 5% à 20%) pour donner
le dérivé H (0.49 g, 1.25 mmol, 84%).
MS (ES (+) ) : m/z [M-]=402
'H NMR (400 MHz, DMSO-d6) (ppm) =1.41 (s, 9H, tBu), 3.30-
3.80 (massif, 4H, 2 CH2), 3.72(s, 3H, CH3), 4.42 (dd, 1H,
CHCH2ONHBOC), 4.64 (d, 1H, N-CH2-CH-N), 7.21 (m, 1H, H
mobile), 7.35 (s, 1H, H pyrazole), 8.02 (dd, 2H, pyridine),
8.54 (m, 1H, pyridine), 8.91 (m, 2H, pyridine)
Stade H :
Sel de sodium de la trans [[4,5,6,8-tétrahydro-1-méthyl-6-oxo-
5-(sulfooxy)-4,7-méthano-7H-pyrazolo[3,4-e] [1,3] diazépin-8-
yl]méthyl]-carbamate de 1,1-diméthyléthyle (I)
Une suspension de 60 g de résine DOWEX 50WX8 dans une
solution de soude 2N (300 ml) est agitée pendant une heure,
puis versée sur une colonne à chromatographie. On élue à l'eau
déminéralisée jusqu'à pH neutre, puis conditionne la colonne
avec un mélange eau/THF 90/10. Le dérivé H (0.49 g, 1.01 mmol)
est dissout dans un minimum d'eau, déposé sur la colonne, puis
élué avec un mélange eau/THF 90/10. Les fractions contenant le
substrat sont réunies et congelées. La solution congelée est
lyophilisée pour conduire au produit I attendu (0.44 g,
1.03 mmol, 100%).
MS (ES (-) ) : m/z [M-]=402
'H NMR (400 MHz, DMSO-d6) : 5 (ppm) =1.39 (s, 9H, tBu), 3.30-3.72
(m, 7H, 2 CH2, CH3), 4.42 (m, 1H, CHCH2ONHBOC), 4.64 (s, 1H, N-
CH2-CH-N),7.16 (m, 1H, H mobile), 7.35 (s, 1H, H pyrazole).
Stade I :
Sel de sodium et de trifluoroacétate de la trans 8-
(aminométhyl)-4,8-dihydro-l-méthyl-5-(sulfooxy)-4,7-méthano-
7H-pyrazolo [3, 4-e] [1,3] diazépin-6 (5H) -one (J)
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
23
Une solution d'acide trifluoroacétique (10 ml) dans le
dichloromethane (10 ml) est coulée goutte à goutte sur une
solution de I (0.15 g, 0.35 mmol) dans le dichloromethane
(5 ml) sous azote et refroidie à 0 C. La réaction est
maintenue sous agitation pendant 1h à température ambiante. Le
mélange est évaporé à sec et repris dans un minimum d'eau. La
solution est congelée puis lyophilisée pour donner le dérivé J
attendu (193 mg, 0.35 mmol, 100%).
MS (ES (-) ) : m/z [M-1=301
'H NMR (400 MHz, DMSO-d6) 6 (ppm) =3.32 (dd, 1H, N-CH2-CH-N),
3.33-3.37 (m, 2H, 2CH), 3.43 (d, 1H, N-CH2-CH-N), 3.74 (s, 3H,
CH3), 4.73 (m, 2H, CH-CH2-NH3+), 7.41(s., 1H, H pyrazole), 8.10
(m, 3H, NH3)
Exemple 2 : Le sel de sodium et de trifluoroacétate de la
trans 8-(amino-méthyl)-4,8-dihydro-5-(sulfooxy)-4,7-méthano-
7H-pyrazolo [3, 4-e] [1,3] diazépin-6 (5H) -one
Stade A :
Trans 4,8-dihydro-8-(hydroxyméthyl)-5-(phénylméthoxy)-4,7-
méthano-7H-pyrazolo [3, 4-e] [1, 3] diazépin-6 (5H) -one
L'ester trans-4,5,6,8-tétrahydro-6-oxo-5-(phénylméthoxy)-
4,7-méthano-7H-pyrazolo[3,4-e][1,3]diazépine-8-carboxylate de
méthyle décrit dans le brevet WO 2004/052891 (Exemple 1, stade
K) (5 g, 15.2 mmol) est mis en solution dans un mélange
méthanol/tétrahydrofurane anhydres 1/1 (100 mL), sous azote.
Du NaBH4 (2.3 g, 60.9 mmol) est alors ajouté par portions.
Après une nuit d'agitation à température ambiante, le mélange
réactionnel est traité avec une solution aqueuse 10% de NaH2PO4
(100 mL). Après évaporation à sec, le mélange réactionnel est
repris dans l'eau. Le précipité formé est agité une nuit dans
la glace, puis filtré et séché au moins 24h sous vide en
présence de P205, pour donner le composé attendu (3.30 g,
11.0 mmol, 72%) sous forme de poudre blanche.
MS (ES(+)) : m/z [M+H]+ = 301
1H RMN (400MHz, DMSO-d6) 6(ppm) = 3.18-3.50 (ABX, 2H, N-CH2-
CH-N), 3.65-3.76 (ABX, 2H, N-CH-CH2-OH), 4.34 (t, 1H, N-CH-CH2-
OH), 4.46 (d, 1H, N-CH2-CH-N), 4.88 (s, 2H, CH2-Ph), 7.29-7.43
(m, 5H, Ph), 7.66 (s, 1H, H pyrazole), 12.72 (broad, 1H, OH).
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
24
Stade B
trans [[4,5,6,8-tétrahydro-6-oxo-5-(phénylméthoxy)-4,7-
méthano-7H-pyrazolo[3,4-e] [1,3] diazépin-8-yl]méthyl]-
carbamate de 1,1-diméthyle
L'alcool obtenu au stade A de l'exemple 2 (1.73 g,
5.76 mmol) est mis en solution dans la pyridine anhydre (35 mL)
sous azote, à 0 C. Puis du chlorure de méthanesulfonyle
(1.78 mL, 23 mmol) est ajouté goutte à goutte. Après 2h30
d'agitation à température ambiante, le mélange réactionnel est
traité avec une solution aqueuse saturée de chlorure d'ammonium
(100 mL), puis extrait à l'acétate d'éthyle. Les phases
organiques combinées sont ensuite lavées 5 fois avec une
solution aqueuse saturée de chlorure d'ammonium, séchées sur
sulfate de sodium, filtrées puis concentrées sous vide pour
donner le dérivé dimésylé attendu sous forme d'huile jaune.
L'intermédiaire dimésylé est mis en solution dans du
diméthylformamide anhydre (45 mL), sous azote, en présence
d'azoture de sodium (1.12 g, 17.3 mmol). Le mélange
réactionnel est chauffé à 70 C pendant 24 heures. Si
nécessaire 1 éq. d'azoture est ajouté pour que la conversion
soit complète. Lorsque la réaction est complète, le mélange
est traité avec une solution aqueuse 10% de NaH2PO4 (100 mL)
puis extrait au dichlorométhane. Les phases organiques
combinées sont séchées sur sulfate de sodium, filtrées puis
concentrées sous vide pour donner l'azoture attendu sous forme
d'huile jaune.
L'intermédiaire est mis en réaction, sous azote, dans
l'éthanol absolu (17.5 mL) . Puis sont ajoutés successivement du
di-tert-butyl dicarbonate (1.38 g, 6.34 mmol), du triéthylsilane
(1.38 mL, 8.64 mmol) et de l'hydroxyde de palladium sur charbon
10% Degussa (52 mg). Après une nuit à température ambiante, le
mélange réactionnel est filtré puis concentré pour donner une
huile jaune brute. Ce brut est purifié par chromatographie sur
colonne de silice (éluant gradient CH2C12/MeOH 100/0 à 95/5 par
1%) pour conduire au composé attendu (1.36 g, 3.40 mmol, 34%)
sous forme de solide blanc.
MS (ES (+)) : m/z [M+H]+ = 401
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
1H RMN (400MHz, MeOH-d4) : ô (ppm) = 1.51 (s, 9H, C(0H3)3), 3.21-
3.59 (m, 4H, N-CH2-CH-N et N-CH-CH2-NHBoc), 4.36 (m, 1H, N-CH-
CH2-OH), 4.46 (m, 1H, N-CH2-CH-N), 4.99 (AB, 2H, CH2-Ph), 7.41-
7. 52 (m, 5H, Ph), 7.63 (s, 1H, H pyrazole).
5 Stade C :
trans [[4,5,6,8-tétrahydro-1-tert-butoxycarbamate-6-oxo-5-
(phényl-méthoxy)-4,7-méthano-7H-pyrazolo[3,4-e] [1,3]
diazépin-8-yl]méthyl]-carbamate de 1,1-diméthyle
Le composé obtenu au stade B de l'exemple 2 (104 mg,
10 0.26 mmol) est mis en solution dans du dichlorométhane anhydre
(2.5 mL) puis du di-tert-butyl dicarbonate (114 mg, 0.52 mmol)
et de la diméthylaminopyridine (32 mg, 0.26 mmol) sont ajoutés
au mélange. Après 1 nuit d'agitation à température ambiante, le
mélange réactionnel est traité avec de l'eau. Les phases sont
15 séparées puis la phase organique est lavée avec une solution
aqueuse saturée de chlorure de sodium, séchée sur sulfate de
sodium, filtrée puis concentrée sous vide. Le brut ainsi obtenu
est purifié par chromatographie sur silice (éluant : CH2C12/AcOEt
90/10) pour donner le produit attendu (76 mg, 0.15 mmol, 59%).
20 MS (ES(+)) : m/z [M+H]+ = 500
Stade D :
sel de pyridinium du trans [[1-tert-butoxycarbamate-4,5,6,8-
tétrahydro-6-oxo-5-(sulfooxy)-4,7-méthano-7H-pyrazolo[3,4-e]
25 [1,3] diazépin-8-yl]méthyl]-carbamate de 1,1-diméthyle
Le composé obtenu au stade C de l'exemple 2 (76 mg,
0.15 mmol) est mis en solution, sous azote, dans un mélange
diméthylformamide/CH2C12 1/3 anhydre (0.87 mL). Du palladium
10% sur charbon à 50% en eau (49 mg) est ajouté. Après trois
purges vide/azote, le mélange réactionnel est placé sous
atmosphère d'hydrogène jusqu'à disparition du produit de
départ en HPLC. Le mélange est alors concentré sous vide puis
co-évaporé trois fois avec du dichlorométhane anhydre, enfin
séché sous cloche à vide en présence de P205 pendant 2h.
Le dérivé débenzylé est repris dans de la pyridine anhydre
(0.43 mL), sous azote, en présence du complexe
pyridine/sulfure de trioxyde (48 mg, 0.30 mmol). Le mélange
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
26
réactionnel est agité à température ambiante jusqu'à
conversion complète en HPLC, puis évaporé à sec après
traitement par ajout d'eau. Le brut ainsi obtenu est purifié
par chromatographie sur silice (éluant CH2C12/MeOH 90/10) pour
donner le composé attendu (47 mg, 0.083 mmol, 55%).
MS (ES(-)) : m/z [M-2*BOC-H]- = 388
1H RMN (400MHz, MeOH-d4) : 8 (ppm) = 1.52 (s, 18H , 2x C (CH3) 3) ,
3.50 (m, 4H, N-CH2-CH-N et CH2-NHBoc), 4.62 (m, 1H, CH-CH2-
NHBoc), 4.85 (d, 1H, N-CH2-CH-N), 7.72 (s, 1H, H pyrazole).
Stade E :
sel de sodium et de trifluoroacétate de la trans [[8-(amino-
méthyl)-4,8-dihydro-5-(sulfooxy)-4,7-méthano-7H-pyrazolo[3,4-
e] [1,3] diazépin-6(5H)-one
Une suspension de 6g de résine DOWEX 50WX8 dans une
solution de soude 2N (30 mL) est agitée à température ambiante
pendant 1h, puis versée sur une colonne à chromatographier.
Après rinçage avec H20 jusqu'à pH neutre, la colonne est
conditionnée avec un mélange THF/H20 10/90. Le dérivé obtenu au
stade D de l'exemple 2 (47 mg, 0.08 mmol) est dissout dans un
minimum de méthanol puis déposé sur la colonne. Après élution
avec un mélange THF/H20 10/90, les fractions contenant le
produit attendu sont rassemblées, congelées, puis lyophilisées
pour conduire au sel de sodium attendu.
Le sel de sodium est repris dans le dichlorométhane
anhydre (1.04 mL) sous azote puis refroidit à 0 C. Une
solution d'acide trifluoroacétique/dichlorométhane anhydre 1/1
(2.04 mL) est ajouté goutte à goutte. Le mélange réactionnel
est ensuite agité à température ambiante pendant 45 min. Après
évaporation à sec puis co-évaporation avec du dichlorométhane
anhydre, le composé est repris dans l'eau (-2 mL) puis congelé
et lyophilisé pour donner le sel attendu (16 mg,0.030 mmol,
36%) sous forme de poudre jaune pâle.
MS (ES(-)) : m/z [M-H]- = 288
1H RMN (400MHz, MeOH-d4) : 8(ppm) = 3.37-3.69 (m, 4H, N-CH2-CH-N
et CH-CH2-NH2), 4.81 (dd, 1H, CH-CH2-NH2), 4.98 (d, 1H, N-CH2-
CH-N), 7.79 (s, 1H, H pyrazole).
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
27
Exemple 3 : Sel de sodium et de trifluoroacétate de la trans
[[8-(méthylaminométhyl)-4,8-dihydro-1-méthyl-5-(sulfooxy)-4,7-
méthano-7H-pyrazolo[3,4-e] [1,3] diazépin-6(5H)-one
Stade A :
Iodure du trans [[[4,5,6,8-tétrahydro-1-méthyl-6-oxo-5-
(phénylméthoxy)-4,7-méthano-7H-pyrazolo[3,4-e] [1,3] diazépin-
8-yl]méthyl]-méthylamino]triméthylphosphonium
Une solution molaire de triméthylphosphine (1.5 mL,
1.5 mmol) est ajoutée goutte à goutte à une solution du dérivé
obtenu au stade E de l'exemple 1 (0.5 g, 1.25 mmol) en
solution dans le tétrahydrofurane (15 mL) à température
ambiante sous azote et sous agitation. Après 2h d'agitation,
l'iodure de méthane (0.21 g, 3.75 mmol) est ajouté au milieu
réactionnel. Il se forme rapidement un précipité jaune clair.
Après une nuit d'agitation à température ambiante, le milieu
réactionnel est concentré sous pression réduite. Le produit
brut est trituré dans dichlorométhane. Le précipité est filtré
pour donner le produit (0.42 g, 1.04 mmol, 84%) attendu sous
forme de sel d'iode de couleur jaunâtre.
MS (ES (+)) : m/z [M+H]+ = 402
1H NMR (400MHz, CDC13) sous forme de 2 conformères (ppm) _
2.04 (s, 3H, CH3P), 2.32 (s, 3H, CH3P), 2.35 (s, 3H, CH3P),
3.03 (s, 3H, P-NCH3 (A) -CH2) , 3.05 (s, 3H, P-NCH3 (B) -CH2) , 3.37
(m, 1H, N-CH2-CH-N ou CH-CH2-N (CH3) P) , 3.44(m, 1H, N-CH2-CH-N ou
CH-CH2-N (CH3) P) , 3.69 (m, 1H, N-CH2-CH-N ou CH-CH2-N (CH3) P) ,
3.82 (s, 3H, CH3) , 3.88 (m, 1H, N-CH2-CH-N ou CH-CH2-N (CH3) P) ,
4. 0 5 (d, 1H, N-CH2-CH-N) , 4. 5 9 (d, 1H, CH-CH2-N (CH3) P) , 4. 8 8
(d, 1H, N-0-CH2-Ph), 5.00 (d, 1H, N-0-CH2-Ph), 7.35 (s, 1H, H
pyrazole), 7.37-7.45 (massif, 5H, Ph)
Stade B :
trans 8-(méthylaminométhyl)-4,8-dihydro-1-méthyl-5-
(phénylméthoxy)-4,7-méthano-7H-pyrazolo[3,4-e] [1,3] diazépin-
6(5H)-one
A une solution aqueuse de carbonate de sodium (2.5N, 9 mL)
est ajouté le dérivé obtenu au stade A de l'exemple 3 (0.42 g,
1.04 mmol). Le milieu réactionnel est agité à 55 C pendant
3h30. Après refroidissement à température ambiante, le milieu
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
28
réactionnel est saturé de chlorure de sodium en présence
d'acétate d'éthyle (25 mL). La phase aqueuse est extraite avec
de l'acétate d'éthyle (3x25 mL). La phase organique est séchée
sur sulfate de magnésium puis concentrée sous pression réduite
pour délivrer une huile jaune (0.26 g). Le brut réactionnel
est purifié par chromatographie sur colonne de silice (éluant
dichlorométhane 100% puis gradient avec du méthanol de 2% à
10%) pour donner le dérivé attendu (0.084 g, 0.256 mmol, 26%).
MS (ES (+) ) : m/z [M+H] 328
'H NMR (400MHz, CDC13) : (ppm) = 2.97-3.00 (dd, 1H, N-CH2-CH-
N), 3.00 (CH-CH2-NCH3), 3.15 (dd, 1H, CH-CH2-NCH3), 3.9 (dd, 1H,
N-CH2-CH-N), 3.75 (s, 3H, CH3), 3.98 (d, 1H, CH-CH2-N (CH3) Boc) ,
4.72 (dd, 1H, N-CH2-CH-N), 4.90 (d, 1H, N-O-CH2-Ph), 5.03 (d,
1H, N-O-CH2-Ph), 7.30 (s, 1H, H pyrazole), 7.34-7.44 (massif,
5H, Ph)
Stade C
trans [[4,5,6,8-tétrahydro-1-méthyl-6-oxo-5-(phénylméthoxy)-
4,7-méthano-7H-pyrazolo[3,4-e] [1,3] diazépin-8-yl]méthyl]-
méthyl-carbamate de 1,1-diméthyléthyle
Le dérivé obtenu au stade B de l'exemple 3 (80 mg, 0.244
mmol) est mis en solution dans le dichlorométhane (1 mL) puis
à température ambiante est additionné successivement de la
triéthyle amine (60 pL, 0.488 mmol) et du di-tert-
butyldicarbonate (106 mg, 0.488 mmol). Après 4h d'agitation à
température ambiante, une solution saturée de chlorure de
sodium (5 mL) est additionnée au milieu réactionnel. La phase
aqueuse est extraite par le dichlorométhane (3x20 mL). La
phase organique est séchée sur sulfate de magnésium puis
concentrée sous pression réduite pour donner une poudre
blanche amorphe (157 mg). Le brut réactionnel est
chromatographié sur colonne de silice (éluant dichlorométhane
100% puis gradient avec de l'acétate d'éthyle de 20% à 30%)
pour donner le dérivé attendu (0.068 g, 0.159 mmol, 60%).
MS (ES (+)) : m/z [M+H]+=428
'H NMR (400MHz, CDC13) : 6 (ppm) = 1.59 (s, 9H, C (CH3) 3) , 3.05
(s, 3H, CH3NBoc-CH2), 3.10 (m, 3H, N-CH2-CH-N, CH-CH2-NBoc),
3.75 (m, 1H, N-CH2-CH-N), 3.85 (s, 3H, CH3), 3.99 (s, 1H, N-
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
29
CH2-CH-N), 4.75 (m, 1H, CH-CH2-N (CH3) Boc) , 4.90 (d, 1H, N-O-CH2-
Ph), 5.02 (d, 1H, N-O-CH2-Ph), 7.37 (s, 1H, H pyrazole), 7.40-
7.46 (massif, 5H, Ph)
Stade D :
Sel de pyridinium du trans [[4,5,6,8-tétrahydro-1-méthyl-6-
oxo-5-(sulfooxy)-4,7-méthano-7H-pyrazolo[3,4-e] [1,3]
diazépin-8-yl]méthyl]-méthyl-carbamate de 1,1-diméthyléthyle
En procédant comme indiqué au stade G de l'exemple 1, le
composé obtenu au stade C de l'exemple 3 (0.068 g, 0.159 mmol)
dans le méthanol (5 mL), en présence de palladium 10% sur
charbon (25 mg) conduisent au produit débenzylé.
MS (ES (+)) : m/z [M+H]+ = 337
L'intermédiaire débenzylé, la pyridine (1 mL), le complexe
pyridine/sulfure de trioxyde (50 mg, 0.318 mmol) conduisent au
sel attendu (0.045 g, 0.090 mmol, 100%).
MS (ES (-) ) : m/z [M-H] - = 416
1H NMR (400 MHz, MeOH-d4) sous forme de 2 conformères : 6 (ppm)
= 1.53 (s, 9H, C(CH3)3, 3.09 (s, 3H, CH3(A)NHBoc), 3.10 (s, 3H,
CH3 (B) NHBoc) , 3.37 (m, 1H, BocN (CH3) -CH2-CH ou N-CH2-CH-N), 3.58
(m, 1H, BocN (CH3) -CH2-CH ou N-CH2-CH-N), 3.75 (s, 3H, CH3), 3.84
(m, 1H, BocN (CH3) -CH2-CH ou N-CH2-CH-N), 3.90(m, 1H, BocN (CH3) -
CH2-CH ou N-CH2-CH-N), 4.90 (m, 2H, N-CH-CH2-N, N-CH2-CH-N +
signal H20), 7.54 (s, 1H, H pyrazole), 8.16 (dd, 2H, pyridine),
8.70 (dd, 2H, pyridine), 8.94 (d, 1H, pyridine)
Stade E :
Sel de sodium de la trans [[4,5,6,8-tétrahydro-1-méthyl-6-oxo-
5-(sulfooxy)-4,7-méthano-7H-pyrazolo[3,4-e] [1,3] diazépin-8-
yl]méthyl]-méthyl-carbamate de 1,1-diméthyléthyle
En procédant comme indiqué au stade H de l'exemple 1, le
sel obtenu au stade D de l'exemple 3 (0.045 g, 0.090 mmol), la
résine DOWEX 50WX8 (30 g) et la soude 2N (150 mL) conduisent
au sel de sodium attendu (0.039 g, 0.090 mmol, 100%).
MS (ES (-)) : m/z [M-H]- = 416
1H NMR (400 MHz, MeOH-d4) sous forme de 2 conformères : 6 (ppm)
=1.56 (s, 9H, C(CH3)3), 3.09(s, 3H, CH3(A)NHBoc), 3.10 (s, 3H,
CH3 (B) NHBoc) , 3.37 (m, 1H, BocN (CH3) -CH2-CH ou N-CH2-CH-N) ,
3.64(m, 1H, BocN (CH3) -CH2-CH ou N-CH2-CH-N), 3.75 (s, 3H, CH3),
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
3.84 (m, 1H, BocN (CH3) -CH2-CH ou N-CHz-CH-N) , 3.93 (m, 1H,
BocN (CH3) -CH2-CH ou N-CH2-CH-N) , 4. 90 (m, 2H, N-CH-CH2-N, N-CH2-
CH-N + signal H20), 7.55 (s, 1H, H pyrazole).
Stade F :
5 Sel de sodium et de trifluoroacétate de la trans 8-
(méthylaminométhyl)-4,8-dihydro-1-méthyl-5-(sulfooxy)-4,7-
méthano- 7H-pyrazolo [ 3, 4-e ] [1,3] diazépin-6 (5H) -one
En procédant comme indiqué au stade I de l'exemple 1, le
sel de sodium obtenu au stade E de l'exemple 3 (0.039 g,
10 0.088 mmol), le dichlorométhane (5 mL) et un mélange d'acide
trifluoroacétique/dichlorométhane anhydre 1/1 (4 mL)
conduisent au produit attendu (39 mg, 0.08 mmol, 100%).
MS (ES (-) ) : m/z [M-H] = 315
1H NMR (400 MHz, DMSO-d6) : 8 (ppm) = 2.76 (s, 3H, CH3NH+2-CH2) ,
15 3.30-3.50 (m, 4H, N-CH2-CH-N, NH+2-CH2-CH) , 3.75 (s, 3H, CH3),
4.74 (m, 1H, N-CH2-CH-N), 4.82 (d, 1H, CH-CH2-NH+2CH3), 7.43 (s,
1H, H pyrazole) , 8.67 (m, 2H, NH3)
Exemple 4: Compositions pharmaceutiques
On a préparé une composition pour injection renfermant:
20 - Composé de l'exemple 1: 300 mg
- Tobramycine : 500 mg
Excipient aqueux stérile: q.s.p. 5 cm3
On a préparé une composition pour injection renfermant:
- Composé de l'exemple 1: 200 mg
25 Ceftazidime : 500mg
Excipient aqueux stérile: q.s.p. 5 cm3
Détermination de l'activité bactéricide
But : On mesure l'activité bactéricide in vitro de
l'antibiotique en mettant en évidence la plus petite
concentration qui permet la survie de 0,001% de bactéries au
bout d'un temps unique donné et dans le temps.
Produits
-On pèse les produits à tester et les solubilise puis la
solution mère obtenue est diluée dans du milieu en fonction
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
31
des concentrations à tester, sachant que chaque dilution sera
introduite sous 0.5m1 dans un volume total de 20m1 soit une
dilution finale au 1/40.
Méthode
On détermine au préalable les Concentrations Minimales
Inhibitrices (CMIs) des produits à tester (produits seuls et
combinaisons).
= Pour chaque concentration de produit à tester ainsi que le
témoin souche, on prépare un erlen contenant 18.5m1 de milieu
Muller-Hinton (Ca2++).
= A partir d'une culture de nuit en bouillon ou d'une
suspension bactérienne de DO (densité optique) = 1, on fait
une dilution au 1/100.
= On met en culture agitée pendant 2h à 37 C.
= On mesure la DO, si DO > 0,5, on dilue au 1/10.
= On ensemence chaque erlen avec 1 ml de la culture agitée ou
de sa dilution, l'inoculum initial devant être lx 106 cfu/ml.
= On ajoute les différentes solutions d'antibiotiques sous un
volume de 0,5ml et 0,5m1 de milieu dans l'erlen témoin.
= Sur un volume de 0.lml, on numère l'erlen témoin = TO.
= On incube sous agitation à 37 C.
= A chaque temps de prélèvement (2, 4, 6, 24, 48 heures), on
prélève un volume de 0.lml de chaque erlen et réalise une
numération.
= On incube toutes les boites de numération (24h - 48h) à
37 C.
Paramètres mesurés
= On compte les colonies.
= On trace les courbes CFU/ml en fonction du temps.
= Effet bactéricide = diminution de 3 log par rapport à
1'inoculum initial.
Bibliographie
PETERSON L.R., SHANHOLTZER C.J.
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
32
Tests for bactericidal effects of antimicrobial agents:
technical performance and clinical relevance.
Clin. Microb. Rev., 1992, 5, 420-432
COURVALIN P., DRUGEON H., FLANDROIS J.P., GOLDSTEIN F.
Bactéricidie. Aspects théoriques et thérapeutiques.
Ed. Maloine, Paris, 1991.
L'activité bactéricide a été évaluée sur une souche sensible
de Pseudomonas aeruginosa (391HT2)
Les CMI sont déterminées sur microplaque:
- Ceftazidime/CAZ: 2 pg/mL
- Ciprofloxacine/CIPRO: 1 pg/mL
- Tobramycine/TOBRA: 1 pg/mL
- Produit de l'exemple 1 (NXL105): 0.25 pg/mL
Pour les essais bactéricides, les CMI sont déterminées dans un
volume de 10 mL - Conditions bactéricides (croissance
bactérienne exponentielle):
- CAZ: 8 pg/mL
- CIPRO: 2 p.g/mL
- TOBRA: 1 pg/mL
- Produit de l'exemple 1: 0.25 pg/mL
Les activités bactéricides qui figurent sur les planches 1 à 3
en annexe sont évaluées après 48 H, soit pour le produit de
l'exemple 1 seul, soit pour une combinaison. Elles montrent
une absence totale de recroissance bactérienne après 48 H pour
les combinaisons.
Mise en évidence de l'activité synergistique - Détermination
des CMI:
Activité in vitro, méthode des dilutions en milieu liquide
On prépare une série de microplaques à 96 puits dans lesquels
on répartit la même quantité de milieu nutritif stérile, on
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
33
distribue dans chaque puit des quantités croissantes du
composé à étudier, à savoir le composé antibactérien seul et
la combinaison selon l'invention avec le composé de formule
(I) de l'exemple 1, dans les proportions respectives 2 :1 et
4 :1, puis chaque plaque est ensemencée avec une souche
bactérienne : Pseudomonas aeruginosa et enterobacteriaceae.
Après incubation de 24 heures à l'étuve à 37 C, l'inhibition
de la croissance est appréciée par transillumination, ce qui
permet de déterminer les concentrations minimales inhibitrices
(C.M.I.) exprimées en g/ml.
Dans l'ensemble des essais ci-dessous (CMI et FIC):
- Ceftazidime = CAZ
- Méropénème = MRP
- Aztréoname = AZT
- Lévofloxacine = LVX
- Composé de l'exemple 1 = Composé A
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
di Co dl N N d' d~ d~ rl H d+ N N "ZP 1-1 H = r-1 rl H H H H
c~ CD
=rl 0
ri
O
R7 U
=r1 ri Ln Ln Ln Ln N Ln u) Ln Ln Ln Ln Ln
N + N N = H N H N d+ ~H N H N
O O O O O O O O O O O O
44
U
N N N N N N N N N N N N N N lO N N N N N N N
M M M M M M M M M M M M N M '-I
W ^ A r-I ^ ~ ^ ^ ^ ^ A A A A M M r-I M M M M M M
r= N Ln N
N 1-1 N M dl r-1 ri N N ri N 1-1 = N r-1 N N N H W
dL A O O
U ri Ln N Ln Ln Ln Ln Ln Ln Ln Ln Ln Ln
0 ,}, == Cl N = H r1 M N N W N c-1 H r-1 N = N = N . . 00
0 O A O O O O Q O O O O O
41
N
r1 N N N N N N N N N N N N N N N N N N lO N Ln
M M W M M M M M M M M M Co W M
A A r1 M A A A A A A A M A A M M M M M -i A O
O O O O O O O O O O O O O O Q O O O O O O O O O O
rl O O O O O O O O O O O O Q O O Ln Q O O O O O O L9
O O O O O O O Ln o O Q Ln O Ln u) N Ln u) Ln Ln Ln Ln O O
O r-) O O O O O O O O O W O
di d~ c-1 N N d1 N N O W r q,
0
u Q Q Q Q Q Q 0 Q Q Q Q Q Q Q Q Q Q Q Q Q Q Q Q Q O
W -}= ri O Q O O O Q O O Q O o O Ln O O O Ln Q O Q Ln O O O Lfl
Qr O O Ln O O O O O Ln O O Q N Ln Ln Ln N Ln Ln Ln N Ln Ln O O
N
i4 dL H O H rl r-I r-i r-1 O N r-I O Q O O Q O O O O O O W Q
W
O Q CD O O O Q O CD O O O O O O O O O O
N N 0 N N N QO QO O N QO O O O O O O O O O O O O 10
W M M M M M O M O O Q O O O O O O O O O O
A A A A A N Lp d- A 00 .-i W dt N N dt N N N N à' N O
rI M rl H M
O O O O O O O O O O O O Q CD Q O O O O O Q Q O Q
O O O O 00 N O O O O O O O O O O O O O O O O O OQ O
O O O Q M O O O O O O Ln O O O O O O O O O O O
0 (A r-I N N N N d1 W r I O rl N ri N N N N N ;H M d1
Ln m O o-1 di O IO M Ln lO Ln Ln l0 0,
M M dt di d' N M W H N O W W d1 O t- N Ln M N H t- N W rn
ri p4 pi pi pi W o, M lO c-i r-1 LO Ln d1 Ln Ln d1 M M M M
P7 PU W pq fY1 M pq m on PQ W 0.l en m m r- 1-1 I .-1 I N N N 0 C7
,-1 H H ,1 H a H a a H-1 ,xi H H rxA ,Hi H _i -1 H ,H1 _l H chi rx-1
rn o- o, orn CI) o- o- rn ol~ o m o- o om m o% o, oi a% o~ rn a% orn m c
M M M M M M M M M M M M M M M M M M M M M M M M M
ni ni ni ni ni ni ni Ld ni ni ns ni ni ni ni Ld ni rd Ld ni ni nS ni rd rd
Ln L) m a) rn m m m m m U) m tn m 0 Co m m m Ln U) U) cn rA Ln
0 0 0 0 O O 0 0 0 0 0 O 0 0 0 0 0 0 0 0 0 0 0 O O
r1 =rl =rl =rl -rl =rl -mi -ri -ri =r1 -ri =H -rl ri =r1 =r1 H -ri -ri -ri -ri
-rl -r1 -r1 -ri -ri
b bL b1 b) tA bi bi bi bL bi bL ci CI b) bi bL bi bi b c t bi bL bi
S-1 ?d ?-1 l-i ~1 l-c l-1 l-1 i1 l-1 l-1 ~1 i-1 }-1 i-1 l-c
cf) W W W d) W W W W W W W W W W W W W W W W W W W W W
ns rd ni rd rd ni ni nf W nf nf nf rd rd rd ni ni ni cd cd nf rd rd rd ni
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
Ln
O
Ln
N
r-I Co N rl rH
N N N N
N M M N M
A A A
Ln
N N N
O
Ln
IV CO N N rl
O
N N N (n
M M (n (n
A A A O
O O O O O
O Oo O W O
O O O O
~ ~ O O N
Lf)
O O O O O
O O O W O
O O O O O
~r O N
O O O
O O N O N
O O Co O M
N N A A
M M OJ
O O Lf) O
N CD CD N O
M CD CD r-1 O
A N N
Co M O N
n CO rl 61
N (n r- N
cli
C7 U C9 O C7
x x x ra x
c--I 1-1 rl N r-I
rn W < rn
( ) M M W M
ro Co ro ro
(n Co m (n (n
Z)l 0) 0)
1~ w a, a, w
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
m m M M M M M
Ln p p LO lD O l0 O tO O 10 O O
rl N N N d' N tb C7 df O O 0 O 0 0 O O
`r 1 II O O il O il O il O I I I I
O V V V V V V V
O
m
Ln M M M M M
U
Q Q
N ' N in O CD O O O O O Q O O
ô + (V ri O r-I 00 N r-I d' O pp O = O= O O O d O O O
o O II 11 O O II O II O II O II II
V V V V V V V
M Ln Ln Ln Ln U, M M
N N N O N N Ln n N N O
N M r M M m M 0 O 1-1 N O H 'q r-1 O O
A A A V O O O O O O O O = V il Il
V
Ln Ln Ln Ln Ul
t=i Ln CD N N N ô N N
= O
Q O O O O O O O
0
li U M M m M M M P,
0 N O Ll ô O O O O N O O
+ =-I N dt dl O O O O O N H O O
1.) O O 11 O O 11 II II II O II II
N V V V V V V V
MI
N N N N N N l0 N LO l0 N N N l0 LO N
M m M m co M N CO M M
A A n n A rI A rI rt M A r1 rI M
M
Ln Ln LI1 U, l0 O N Ln
== N (N ,-I H di N CO N N N .--t O -I H H ra H
O O O O O 11 0
V
0
U !n in l0 Ln O LD Q LO
+ N N O N O Ln Ln U1 O N
== ri r1 ri N c-i d' N ri O '-i H
JJ N O O O il O O 11 O 0 =
114 l0 N V V Z;
N N N N N N N N N N N N N N N N N N N N
M M M M M M M M M M M M M M M M M M m
A A A A A A A A A A A A A A A A A M A A
Q N N N N N N N N N N N N N N N N N
0 0 m M M M M M M M M M M M M M M M M M (Nl
U A A A A A A A n A A A A A A A A
I I H
Il H H + â
Ln +
+ H + +
R$ r i I U7 ri r 1 r f N L1 W N N
Co W W H W W N W x+ + W W
j H H U H en E-1 H H Co H E-1
.â =~ + + + + + + + + + + r-I r-I c-i + +
1 1
Co
N N N N M M M M ri ~ d' .-I dl c-i M r-1 N .moi c-I Ln c-I
1 I I I I I I I I I 1 1 I I 1 I I I I I I 1
U U U U U U U U ~++ U PC >C >C DC
x x N x x x x C n > U > U rC f A r-I Z O O O U u J rn 1 114, ~ PLI
d' H
x
.Ij 4j r- c--I d x
N M M
W l0 l0 4.D O N U) dl d+ W d) N N
cd N l0 Pc r r r U LD =r1 =11 r-1 U1 dl CO ri L: -ri {~ dt
O Ln Ln Ln Co U 0 JJ 4J O N Co Ln I-a il, O 0 O r Ol
r1 ri U Ln A a a FC H CD Id 13, Id r M r 0 1-1 r-i w Ln dl
r~ N ~+ r ,7 U V U ,~ ry' r W W[ a m M r-i E-I x H U H U x H dl
v (U v(U a) v Q) v Q) Q) Q) Q) a~ v v Q) Q) v
R1 RS Itf RS RS 43 Id Id Id Id Id IIj II) cts f ro Id I ro
=r1 =rI =ri =r1 ' f =ri =ri 'rf = I =ri =ri =ri =H =r{ =r-I =ri =H =r( =r1
C,
O Id O O O O O O O O O O O O O O O O O O
U H O O N N U U N N 4I N v QI U O N Q) N N 4) QI
U
=I) H U t ~ C Q, t Z C a q 01 >01 ti a 01 01 a p a C ]
P41 C
a
11, Lt4~14
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
M M M M M M M M M M M M M M M M
O O O O UD UD O O O O UD N U) O W UD O O O O O O O
O (n . p p
M
O O O p 0 p O O O O. '
O 0 0
Il il II II O Q Q il 11 Il II o o O ol O o 0 0 0
V V V V V V V V V V V V V V V V
M M M M ('") M M M M M M M M M M M M M
O O O O U0 Q N l0 O O O O UD Ufl N O U0 O O O O O O O O
O . N p . . . . p O H O - O O O O O O
O O O O O 01 O = O O O O = C. O O O O il il il il Il CD Il Il il Il Il Il Il
Il
Il il il V V V 1 V V V V V V V V V V V V V V
V
M M M M M
U7 0 W U) U) l0 Ln O Ln O W lD Ln O W Ln o lW O O n l0
cli Ln Ln
CM O O O N O d, ( O O O N C' O O N O
p O O
CD 0 0 0 0 0 101 0 0 o II o o o o II II O C
V v v v v
M M U)
Ln Ln Li,
Ln lO N t0 Ln U) Ln N U) Ln O tn lD lfl Cfl O Ln ~[)
N O O H N H H H - m H - N O N O O H
O O O O O O O O O O O O O O O IOVI Q
V
M M M
U7 U7 UO U) ln Ln Ln Ln O M O Ln n
O O O UD UD
U) N N N N U) Ln U7 O O
H H O N N H H H O H O N O O H
CD 0 0 o II Q Il CD II II o II O o
o O O O V O V V V V
W W N N N N UO UD N N W ID N W N W
m H H M H M M Co H H M ^ H H n H M H m m H CD
(q U) Ln
lD Lfl L() L() U) tn U) O Ln Lm
Ln LO Ç\1 Ln Lr)
O U O N N
N N N UO ' N N a' Co O H H H
O O O O O O O O O O H O
O O Il O O O O
Ln LO
U) Ln O Ln M O U) W UD O O
O UO O
N N Ln N U) N N O O
O H N N H CV m N O H N N O O
p O II o 0 o O II ' o o O II 0 0 0 II 11
V O V V O V V v
E-
M
N N N N N N N N N N N N N N N N N N N N lfl N N N N
M M Co M M M M M M M M M M M M M M M M M Co M M M
n n n n n n n A A A A A A n A A n n n n n n
N N N N N N N N N N N N N N N N N N N N N A N
M A M M M M M M M M M M M M M M H M M M M M M H M
n n n n n n n
A A A A A A n
~c EH ~c
W W W U U U + +
0 a a
+ N + + i-1 N H N
H
+ + + 1 I 1 I Co
(Q m N O M N <D N 1
N N N .--I I H H WO H H W Ni W G,
1 1 i 1 >' 1 I I I I H E-i 1 U H i W
H 1 >A
+ O
W W W W U W W W W W + +
H H H H E+ H E-i E-a H W U R+
L1) W O)
-+- + + + + + + + + + H 0) H + H H O N
Ln H N H WH N U) tn N U) Ln N H H H H (Ci 2 M <t'
1 1 1 1 1 1 I 1 1 I I I I U U 1 1 1 1 1 I .C I I I U U
> > .7 .> M > / / > N / > >+ zz.. LL !2 >C ~C CO ?C m X Q. >C S.. Q R
:El Ca Co Ux Co Co O H O O > EU~ H U U U H Co
ü co co in EW-i CCa
xW
Co .ri
(1)
t-I M =,=i C) W
H r- Ln ro H M Co W 0) M CI H C) 0 O
' O N 10 Ln m v' Co M U0 W0 '0' 2 2 J) H Z
cr c l0 ~r ter' N H N N N N H H H H H U H
o ro a) a) v 4)) 0 0 a) Q) a)
ro ro ro ro ro ro ro ro m m m
C C C C C C C C C C C 2i ro N Q) rl 1
0 0 0 0 0 0 0 0 0 0 0 0 U ro ro o
e~ >~ s s o o O 0 1 C[
J J J J J J J J J J 4- JJ n) m 'ri 'f-1 "i -'-I '^I -=-I =r1 .,-1 J J
N N O O N O N N N N Q) ~, ~, O 0 ^1 ^I ^I - ry I ~I H N a)
C) C C C C C C C C C C X x '-I H 0 O O O O 0 O O 1-1 1.4
R R R R R R R R R R R 0 0 0 0 u 0 u 0 o U o 0 4-, l-1
x W W W W W W W W w W C> C)
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
M M M Ln
O O O N
O O O
V V V
m m
O O N
O
O O
o II II O
V V
Ln Ln
c'q O O rH-1
O O O
m
v
M
Ln O
O
m
O N
00 N o m
il A
V
M
CD N
N O Co
~
CO V
N N Qo N
m m H m
n A A
N N N
n M
H a
H +
+ a
U
q
Q)) QQ) N
14 çu
U U U W
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
39
Mise en évidence de l'activité synergistique -
Détermination des Concentrations Inhibitrices
Fractionnées.
(FIC = Fractional Inhibitory Concentrations)
Technique de l'échiquier( checkerboard ) pour la
determination de la synergie des antibiotiques:
Objectif:Le but de l'étude est de déterminer la
concentration d'un composé A, nécessaire pour réduire la
CMI d'un composé B d'un demi, d'un quart, d'un huitième,
d'un seizième et d'un trente deuxième contre des souches
d' espèces enterobacteriaceae et non-enterobacteriaceae
résistantes au composé B.
L'objectif ci-dessus est atteint par la technique dite de
l'échiquier ( checkerboard ). Cette technique est
utilisée pour évaluer les combinaisons antimicrobiennes.
Cette technique consiste à titrer le composé A, un
inhibiteur, dans une série de dilutions (2 en 2) disposées
horizontalement sur une microplaque, tout en titrant en
même temps le composé B dans une série de dilutions
disposées verticalement. La plaque est ensuite inoculée
avec la souche bactérienne et on laisse la bactérie
croitre pendant une nuit. Chaque puit sur l'échiquier de
la microplaque contient une combinaison differente de
concentrations de l'inhibiteur et du composé
antibactérien, ce qui permet une totale détermination
d'une quelconque synergie entre les deux composants.
Lecture des plaques:
La croissance est évaluée pour chaque puit. Les points
terminaux (CMIs) pour lesquels il y a absence de
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
croissance sont déterminés pour chaque rangée et les
concentrations des composés A et B pour chaque puit où il
y a absence de croissance sont ensuite utilisées pour
déterminer les niveaux de synergie.
5
La synergie est représentée par des "indices FIC", FIC
étant la Concentration Inhibitrice Fractionnée
( Fractional Inhibitory Concentration ) de la
combinaison.
Calculs de la "Fractional Inhibitory Concentration" (FIC)
Indice de combinaisons de deux agents antimicrobiens:
(A)/(CMIA) + (B)/(CMIB) = FICA + FICB = indice FIC
(A) est la concentration du composé A dans un puit
correspondant à la plus basse concentration de ce composé
inhibant la croissance dans la rangée.lorsque le puit
contient également le composé B (CMIA) est la plus basse
concentration du composé A seul qui inhibe la croissance.
FICA est la "Fractional Inhibitory Concentration" du
composé A.
(B), (CMIB),and FICB sont définis de la même façon que ci-
dessus, pour le composé B.
Si la valeur de l'indice FIC est <=0,5 on considère qu'on
est en présence d'une synergie.
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
tn Cfl CO Co N M O
M
CD O O CD O O CD O O O Q O O c
O O O O O O O O O O c 0 0 czî
U O MO O O O 0 0 O O O O m Co ce)
O O
X O O c O O O O O O O O O O O
N CXO CO C)) f~ CO CO (0 CO Co CO r- (O O CO Co CNO (OO
O O c a O O O O M O O C 0 c 0
O O OO O O O O O O O O O O O CD CD O
LL tn Ln CO Ln LO Co Ln Ln LO 10 >- 10 CO LO U) ~ Ln O) N- Ln
N N N N N N N N N N M N Ln Il N N N N N N
r r r r ~- r r r ~- M r_ _ r r r
d O O c 0 c O O O O O O 6 6 O c O O
LX n ü) tt) LO Ln LO LO Ln f LO U) tn LO M c-- Ln O to t10 O Ln
N N N N N N N N N N N M N LO N N LO N
O O 6 O O M O O O O c c c d O O O C 0 ô 0
CD CD
Q¾ a Q¾ Q Q a a¾ Q a Q a Q Q a a a
0 aa aaQ aaQ aaaaaQ aaQ a0-a
E E E E E E E E E E E E E E E E E E E E E
c U U V U U 0 U U U (j U U U U U U U U (j U U U
E + + + NN+ + + 11+ + + + + + + + + + + + + + +
(j Q ~ a N N Q N Q N Q N Q O' N
~ U~ Q U~ Q U~ Q U~ Q U~ Q U~ Q U~ Q
0
y-
H H U U H Cq
0 + + O n U
(V N Q Q N d
c I- H
o U U U
.0
d
cc
N
LO (0
Co 0) m
M W
o W Q) = O N
m
Q Co
0
ai
F CO N N
U) CD (b Q) C)
z U U LO
Q)
ci OU Q U O U Q
CA 02740035 2011-04-07
WO 2010/041112 PCT/IB2009/006992
x tn ,n Ln tn
O CV CV O O
CD C7 O O O
x tn 00 00 M C') M
O O O N N O O O N
x O CD O O O O O
.Cu
x x U') ce)
Co CCDD CO C) CD O f- Cf) M M
-0 O O O O O
Ç d 6 O O O c 0 O O O O
lL x O Co tn tn to (fl Co tn tn C0 to tn tn
tn et u) r= `n r- r= Ci) tn r= r= r= r= r=
r r CY) Ce) N r- Co Co CO M C) C`')
O O O O O O O O 6 CD O O CO O
_ tn Y- LO
t0 10 CD OO cli 0 tn OO C0 m tn U-) C'07 tn LC) tn CO
C V c "i C V Ch 6 d ' N C ; 0 0 0 0
CD 0 0 c> O O O
Ç Q Q Q Q Q Q Q Q Q Q Q Q Q Q Q Q Q d
O n- t2 t2 O L1 Q 0 C2 C1 O- C2 O. Q Q. d O.
=4 E E E E E E E E E E E E E E E E E E
O O 0 0 0 0 0 0 0 0 0 0 O 0 0 0 0 0 O
U U U U U U U U U U U U U U U U U U
E + + + + + + + + + + + + 1N+ + + + + +
N U Q(> 4 t> Q Q> Q> a13- > Q13- >
U J U J U J U ry J U J U J
4)
U
(o
â n
Q Q Q + + Q
N (0 (a ttf cl. tZ (U
E E
C
Q) tü
(a
U
m m o
O
CF) m
i 2'
M d q)
E
Q
O j Q) Q) Q Q) cn
~r
Q) â) (b VI
(j) `
U) (13 co (u r ro Cu r
U
M à
LL à à à à à ; ~