Note: Descriptions are shown in the official language in which they were submitted.
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
PROCESS FOR THE PREPARATION OF SUBSTITUTED PHENYLALANINES
1. FIELD OF THE INVENTION
This invention relates to synthetic processes used to make substituted
phenylalanine-
based compounds.
2. BACKGROUND
The neurotransmitter serotonin [5-hydroxytryptamine (5-HT)] is involved in
multiple
central nervous facets of mood control and in regulating sleep, anxiety,
alcoholism, drug
abuse, food intake, and sexual behavior. In peripheral tissues, serotonin is
reportedly
implicated in the regulation of vascular tone, gut motility, primary
hemostasis, and cell-
mediated immune responses. Walther, D.J., et at., Science 299:76 (2003).
The enzyme tryptophan hydroxylase (TPH) catalyzes the rate limiting step of
the
biosynthesis of serotonin. Two isoforms of TPH have been reported: TPH1, which
is
expressed in the periphery, primarily in the gastrointestinal (GI) tract, and;
TPH2, which is
expressed in the brain. Id. The isoform TPH1 is encoded by the tphl gene; TPH2
is encoded
by the tph2 gene. Id.
Mice genetically deficient for the tphl gene ("knockout mice") have been
reported.
In one case, the mice reportedly expressed normal amounts of serotonin in
classical
serotonergic brain regions, but largely lacked serotonin in the periphery. Id.
In another, the
knockout mice exhibited abnormal cardiac activity, which was attributed to a
lack of
peripheral serotonin. Cote, F., et al., PNAS 100(23):13525-13530 (2003).
Because serotonin is involved in so many biochemical processes, drugs that
affect
serotonin levels are often attended by adverse effects. Thus, a need exists
for new methods of
affecting serotonin levels.
1
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
3. SUMMARY OF THE INVENTION
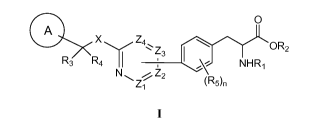
This invention encompasses the preparation of compounds of formula I:
O
A
X Z4\ OR2
Z3
R3 R4 NHR,
N~ iZ2
Z~ (R5)n
I
and pharmaceutically acceptable salts and solvates thereof, wherein the
various substituents
are defined herein. When administered to mammals, preferred compounds of this
formula
inhibit TPH (e.g., TPH 1), and may be useful in the treatment of various
diseases and
disorders.
This invention is also directed to various intermediates that are useful in
the synthesis
of compounds of formula I.
4. DETAILED DESCRIPTION
This invention is based on the discovery of a novel process that can be used
to
efficiently prepare compounds of formula I. When administered to mammals,
preferred
compounds of formula I inhibit peripheral TPH, and may be used in the
treatment of various
diseases and disorders, including disorders of the GI tract. See generally,
U.S. patent
application no. 11/638,677, filed December 12, 2008.
4.1. Definitions
Unless otherwise indicated, the term "alkenyl" means a straight chain,
branched
and/or cyclic hydrocarbon having from 2 to 20 (e.g., 2 to 10 or 2 to 6) carbon
atoms, and
including at least one carbon-carbon double bond. Representative alkenyl
moieties include
vinyl, allyl, 1-butenyl, 2-butenyl, isobutylenyl, 1-pentenyl, 2-pentenyl, 3-
methyl-l-butenyl,
2-methyl-2-butenyl, 2,3-dimethyl-2-butenyl, 1-hexenyl, 2-hexenyl, 3-hexenyl, 1-
heptenyl, 2-
heptenyl, 3-heptenyl, 1-octenyl, 2-octenyl, 3-octenyl, 1-nonenyl, 2-nonenyl, 3-
nonenyl, 1-
decenyl, 2-decenyl and 3-decenyl.
Unless otherwise indicated, the term "alkyl" means a straight chain, branched
and/or
cyclic ("cycloalkyl") hydrocarbon having from 1 to 20 (e.g., 1 to 10 or 1 to
4) carbon atoms.
Alkyl moieties having from 1 to 4 carbons are referred to as "lower alkyl."
Examples of
alkyl groups include, but are not limited to, methyl, ethyl, propyl,
isopropyl, n-butyl, t-butyl,
2
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
isobutyl, pentyl, hexyl, isohexyl, heptyl, 4,4-dimethylpentyl, octyl, 2,2,4-
trimethylpentyl,
nonyl, decyl, undecyl and dodecyl. Cycloalkyl moieties may be monocyclic or
multicyclic,
and examples include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and
adamantyl.
Additional examples of alkyl moieties have linear, branched and/or cyclic
portions (e.g., 1-
ethyl-4-methyl-cyclohexyl). The term "alkyl" includes saturated hydrocarbons
as well as
alkenyl and alkynyl moieties.
Unless otherwise indicated, the term "alkylaryl" or "alkyl-aryl" means an
alkyl
moiety bound to an aryl moiety.
Unless otherwise indicated, the term "alkylheteroaryl" or "alkyl-heteroaryl"
means an
alkyl moiety bound to a heteroaryl moiety.
Unless otherwise indicated, the term "alkylheterocycle" or "alkyl-heterocycle"
means
an alkyl moiety bound to a heterocycle moiety.
Unless otherwise indicated, the term "alkynyl" means a straight chain,
branched or
cyclic hydrocarbon having from 2 to 20 (e.g., 2 to 20 or 2 to 6) carbon atoms,
and including
at least one carbon-carbon triple bond. Representative alkynyl moieties
include acetylenyl,
propynyl, 1-butynyl, 2-butynyl, 1-pentynyl, 2-pentynyl, 3-methyl-l-butynyl, 4-
pentynyl,
1-hexynyl, 2-hexynyl, 5-hexynyl, 1-heptynyl, 2-heptynyl, 6-heptynyl, 1-
octynyl, 2-octynyl,
7-octynyl, 1-nonynyl, 2-nonynyl, 8-nonynyl, 1-decynyl, 2-decynyl and 9-
decynyl.
Unless otherwise indicated, the term "alkoxy" means an -0-alkyl group.
Examples
of alkoxy groups include, but are not limited to, -OCH3, -OCH2CH3, -
O(CH2)2CH3,
-O(CH2)3CH3, -O(CH2)4CH3, and -O(CH2)5CH3.
Unless otherwise indicated, the term "aryl" means an aromatic ring or an
aromatic or
partially aromatic ring system composed of carbon and hydrogen atoms. An aryl
moiety may
comprise multiple rings bound or fused together. Examples of aryl moieties
include, but are
not limited to, anthracenyl, azulenyl, biphenyl, fluorenyl, indan, indenyl,
naphthyl,
phenanthrenyl, phenyl, 1,2,3,4-tetrahydro-naphthalene, and tolyl.
Unless otherwise indicated, the term "arylalkyl" or "aryl-alkyl" means an aryl
moiety
bound to an alkyl moiety.
Unless otherwise indicated, the terms "halogen" and "halo" encompass fluorine,
chlorine, bromine, and iodine.
Unless otherwise indicated, the term "heteroalkyl" refers to an alkyl moiety
in which
at least one of its carbon atoms has been replaced with a heteroatom (e.g., N,
0 or S).
Unless otherwise indicated, the term "heteroaryl" means an aryl moiety wherein
at
least one of its carbon atoms has been replaced with a heteroatom (e.g., N, 0
or S).
3
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
Examples include, but are not limited to, acridinyl, benzimidazolyl,
benzofuranyl,
benzoisothiazolyl, benzoisoxazolyl, benzoquinazolinyl, benzothiazolyl,
benzoxazolyl, furyl,
imidazolyl, indolyl, isothiazolyl, isoxazolyl, oxadiazolyl, oxazolyl,
phthalazinyl, pyrazinyl,
pyrazolyl, pyridazinyl, pyridyl, pyrimidinyl, pyrimidyl, pyrrolyl,
quinazolinyl, quinolinyl,
tetrazolyl, thiazolyl, and triazinyl.
Unless otherwise indicated, the term "heteroarylalkyl" or "heteroaryl-alkyl"
means a
heteroaryl moiety bound to an alkyl moiety.
Unless otherwise indicated, the term "heterocycle" refers to an aromatic,
partially
aromatic or non-aromatic monocyclic or polycyclic ring or ring system
comprised of carbon,
hydrogen and at least one heteroatom (e.g., N, 0 or S). A heterocycle may
comprise multiple
(i.e., two or more) rings fused or bound together. Heterocycles include
heteroaryls.
Examples include, but are not limited to, benzo[1,3]dioxolyl, 2,3-dihydro-
benzo[1,4]dioxinyl,
cinnolinyl, furanyl, hydantoinyl, morpholinyl, oxetanyl, oxiranyl,
piperazinyl, piperidinyl,
pyrrolidinonyl, pyrrolidinyl, tetrahydrofuranyl, tetrahydropyranyl,
tetrahydropyridinyl,
tetrahydropyrimidinyl, tetrahydrothiophenyl, tetrahydrothiopyranyl and
valerolactamyl.
Unless otherwise indicated, the term "heterocyclealkyl" or "heterocycle-alkyl"
refers
to a heterocycle moiety bound to an alkyl moiety.
Unless otherwise indicated, the term "heterocycloalkyl" refers to a non-
aromatic
heterocycle.
Unless otherwise indicated, the term "heterocycloalkylalkyl" or
"heterocycloalkyl-
alkyl" refers to a heterocycloalkyl moiety bound to an alkyl moiety.
Unless otherwise indicated, the term "pharmaceutically acceptable salts"
refers to
salts prepared from pharmaceutically acceptable non-toxic acids or bases
including inorganic
acids and bases and organic acids and bases. Suitable pharmaceutically
acceptable base
addition salts include, but are not limited to, metallic salts made from
aluminum, calcium,
lithium, magnesium, potassium, sodium and zinc or organic salts made from
lysine, N,N'-
dibenzylethylenediamine, chloroprocaine, choline, diethanolamine,
ethylenediamine,
meglumine (N-methylglucamine) and procaine. Suitable non-toxic acids include,
but are not
limited to, inorganic and organic acids such as acetic, alginic, anthranilic,
benzenesulfonic,
benzoic, camphorsulfonic, citric, ethenesulfonic, formic, fumaric, furoic,
galacturonic,
gluconic, glucuronic, glutamic, glycolic, hydrobromic, hydrochloric,
isethionic, lactic,
maleic, malic, mandelic, methanesulfonic, mucic, nitric, pamoic, pantothenic,
phenylacetic,
phosphoric, propionic, salicylic, stearic, succinic, sulfanilic, sulfuric,
tartaric acid, and p-
toluenesulfonic acid. Specific non-toxic acids include hydrochloric,
hydrobromic,
4
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
phosphoric, sulfuric, and methanesulfonic acids. Examples of specific salts
thus include
hydrochloride and mesylate salts. Others are well known in the art. See, e.g.,
Remington' s
Pharmaceutical Sciences (18th ed., Mack Publishing, Easton PA: 1990) and
Remington: The
Science and Practice of Pharmacy (19th ed., Mack Publishing, Easton PA: 1995).
Unless otherwise indicated, the term "protecting group" or "protective group,"
when
used to refer to part of a molecule subjected to a chemical reaction, means a
chemical moiety
that is not reactive under the conditions of that chemical reaction, and which
may be removed
to provide a moiety that is reactive under those conditions. Protecting groups
are well known
in the art. See, e.g., Greene, T.W. and Wuts, P.G.M., Protective Groups in
Organic Synthesis
(3rd ed., John Wiley & Sons: 1999); Larock, R.C., Comprehensive Organic
Transformations
(2"d ed., John Wiley & Sons: 1999).
Unless otherwise indicated, the term "pseudohalogen" refers to a polyatomic
anion
that resembles a halide ion in its acid-base, substitution, and redox
chemistry, generally has
low basicity, and forms a free radical under atom transfer radical
polymerization conditions.
Examples of pseudohalogens include azide ions, cyanide, cyanate, thiocyanate,
thiosulfate,
sulfonates, and sulfonyl halides.
Unless otherwise indicated, the term "stereoisomeric mixture" encompasses
racemic
mixtures as well as stereomerically enriched mixtures (e.g., R/S = 30/70,
35/65, 40/60, 45/55,
55/45, 60/40, 65/35 and 70/30).
Unless otherwise indicated, the term "stereomerically pure" means a
composition that
comprises one stereoisomer of a compound and is substantially free of other
stereoisomers of
that compound. For example, a stereomerically pure composition of a compound
having one
stereocenter will be substantially free of the opposite stereoisomer of the
compound. A
stereomerically pure composition of a compound having two stereocenters will
be
substantially free of other diastereomers of the compound. A typical
stereomerically pure
compound comprises greater than about 80% by weight of one stereoisomer of the
compound
and less than about 20% by weight of other stereoisomers of the compound,
greater than
about 90% by weight of one stereoisomer of the compound and less than about
10% by
weight of the other stereoisomers of the compound, greater than about 95% by
weight of one
stereoisomer of the compound and less than about 5% by weight of the other
stereoisomers of
the compound, greater than about 97% by weight of one stereoisomer of the
compound and
less than about 3% by weight of the other stereoisomers of the compound, or
greater than
about 99% by weight of one stereoisomer of the compound and less than about I%
by weight
of the other stereoisomers of the compound.
5
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
Unless otherwise indicated, the term "substituted," when used to describe a
chemical
structure or moiety, refers to a derivative of that structure or moiety
wherein one or more of
its hydrogen atoms is substituted with a chemical moiety or functional group
such as, but not
limited to, alcohol, aldehylde, alkoxy, alkanoyloxy, alkoxycarbonyl, alkenyl,
alkyl (e.g.,
methyl, ethyl, propyl, t-butyl), alkynyl, alkylcarbonyloxy (-OC(O)alkyl),
amide (-C(O)NH-
alkyl- or -alky1NHC(O)alkyl), amidinyl (-C(NH)NH-alkyl or -C(NR)NH2), amine
(primary,
secondary and tertiary such as alkylamino, arylamino, arylalkylamino), aroyl,
aryl, aryloxy,
azo, carbamoyl (-NHC(O)O-alkyl- or -OC(O)NH-alkyl), carbamyl (e.g., CONH2, as
well as
CONH-alkyl, CONH-aryl, and CONH-arylalkyl), carbonyl, carboxyl, carboxylic
acid,
carboxylic acid anhydride, carboxylic acid chloride, cyan, ester, epoxide,
ether (e.g.,
methoxy, ethoxy), guanidino, halo, haloalkyl (e.g., -CC13, -CF3, -C(CF3)3),
heteroalkyl,
hemiacetal, imine (primary and secondary), isocyanate, isothiocyanate, ketone,
nitrile, nitro,
oxo, phosphodiester, sulfide, sulfonamido (e.g., SO2NH2), sulfone, sulfonyl
(including
alkylsulfonyl, arylsulfonyl and arylalkylsulfonyl), sulfoxide, thiol (e.g.,
sulfhydryl, thioether)
and urea (-NHCONH-alkyl-).
Unless otherwise indicated, the term "include" has the same meaning as
"include, but
are not limited to," and the term "includes" has the same meaning as
"includes, but is not
limited to." Similarly, the term "such as" has the same meaning as the term
"such as, but not
limited to."
Unless otherwise indicated, one or more adjectives immediately preceding a
series of
nouns is to be construed as applying to each of the nouns. For example, the
phrase
"optionally substituted alky, aryl, or heteroaryl" has the same meaning as
"optionally
substituted alky, optionally substituted aryl, or optionally substituted
heteroaryl."
Unless otherwise indicated, a structure or name of a compound or genus of
compounds encompasses all forms of that compound or genus of compounds, and
all
compositions comprising that compound or genus of compounds.
It should be noted that a chemical moiety that forms part of a larger compound
may
be described herein using a name commonly accorded it when it exists as a
single molecule
or a name commonly accorded its radical. For example, the terms "pyridine" and
"pyridyl"
are accorded the same meaning when used to describe a moiety attached to other
chemical
moieties. Thus, the two phrases "XOH, wherein X is pyridyl" and "XOH, wherein
X is
pyridine" are accorded the same meaning, and encompass the compounds pyridin-2-
ol,
pyridin-3-ol and pyridin-4-ol.
6
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
It should also be noted that if the stereochemistry of a structure or a
portion of a
structure is not indicated with, for example, bold or dashed lines, the
structure or the portion
of the structure is to be interpreted as encompassing all stereoisomers of it.
Moreover, any
atom shown in a drawing with unsatisfied valences is assumed to be attached to
enough
hydrogen atoms to satisfy the valences. In addition, chemical bonds depicted
with one solid
line parallel to one dashed line encompass both single and double (e.g.,
aromatic) bonds, if
valences permit.
4.2. Methods of Synthesis
This invention encompasses the preparation of compounds of formula I:
O
A
X Z4\ OR2
Z3
R3 R4 NHR1
N~ iZ2
(R5n
Z1 I
and pharmaceutically acceptable salts and solvates thereof, wherein the
various substituents
are defined herein. The invention is particularly directed to the synthesis of
compounds of
formulae 1(b), 1(c), 1(d) and I(e):
O
R3 R4 OH
O NH2
(R11)m
11 (R12)p N N
R7
1(b)
O
R3 R4 OH
O NH2
(R11)m
(R12)p N N
R7
I(c)
7
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
0
R3 Rq OH
NH2
(R11)m
, N
(R12)p N 'T
R7
I(d)
0
CF3 OH
.,,,,,,10 NH2 11 H3CO N N
"~r
NH2
I(e)
In one aspect of the invention, the synthesis of such compounds is achieved
via a compound
of formula I(a):
O
A
X-,T,- Zq
Z3 P3
R3 R4 N NP1P2
Z2
Z1 (R5)n
I(a)
One embodiment of the invention encompasses a method of preparing a compound
of
formula I(a), which comprises contacting a compound of formula II:
R3 R4
XH
A
II
with a compound of formula III:
0
1 qZ3 P3
Y1 Z
NP1 P2
N Z2
Z1 (R5)n
III
8
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
under conditions sufficient for the formation of the compound of formula 1(a),
wherein: A is
optionally substituted cycloalkyl, aryl, or heterocycle; X is 0, S, or NR6; Y1
is halogen or
pseudohalogen; one of Z1, Z2, Z3, and Z4 is a carbon atom attached to the
adjacent optionally
substituted phenyl moiety, and the others are each independently CR7 or N; P1
is R1 or a
protecting group; P2 is a protecting group; P3 is OR2, SR2, NR9R10, NHNHR9, or
a protecting
group; R1 is hydrogen or optionally substituted alkyl, alkyl-aryl, alkyl-
heterocycle, aryl, or
heterocycle; R2 is hydrogen or optionally substituted alkyl, alkyl-aryl, alkyl-
heterocycle, aryl,
or heterocycle; R3 is hydrogen, cyan, or optionally substituted alkyl or aryl;
R4 is hydrogen,
cyano, or optionally substituted alkyl or aryl; each R5 is independently
hydrogen, cyan,
nitro, halogen, ORg, NR9R10, or optionally substituted alkyl, alkyl-aryl or
alkyl-heterocycle;
R6 is hydrogen or optionally substituted alkyl or aryl; each R7 is
independently hydrogen,
cyan, nitro, halogen, ORg, NR9R10, or optionally substituted alkyl, alkyl-aryl
or alkyl-
heterocycle; each Rs is independently hydrogen or optionally substituted
alkyl, alkyl-aryl or
alkyl-heterocycle; each R9 is independently hydrogen, a protecting group, or
optionally
substituted alkyl, alkyl-aryl or alkyl-heterocycle; each Rio is independently
hydrogen, a
protecting group, or optionally substituted alkyl, alkyl-aryl or alkyl-
heterocycle; and n is 1-4.
In one embodiment, P3 is OR2. In another, R2 is hydrogen. In another, Zi is
CRS. In
another, R7is NR9R10. In another, R9 is hydrogen. In another, Rio is hydrogen.
In another,
Z2 is N. In another, Z3 is a carbon atom attached to the adjacent optionally
substituted phenyl
moiety. In another, Z4 is CRS. In another, R7is hydrogen. In another, n is 1.
In another, R5
is hydrogen. In another, X is O. In another, R3 is hydrogen. In another, R4 is
optionally
substituted alkyl. In another, R4 is -CF3. In another, A is optionally
substituted biphenyl.
In a particular embodiment, the compound of formula II is of formula 11(a):
R3
R4
OH
(R11)m
(R12)p
11(a)
wherein: R11 is independently hydrogen, cyan, nitro, halogen, ORS, NR9R10, or
optionally
substituted alkyl, alkyl-aryl or alkyl-heterocycle; each R12 is independently
hydrogen, cyan,
nitro, halogen, ORg, NR9R10, or optionally substituted alkyl, alkyl-aryl or
alkyl-heterocycle;
m is 1-5; and p is 1-4. In another, the compound of formula 11(a) is of
formula 11(b):
9
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
R3
R4
OH
(R11). 11(b)
In another embodiment, the compound of formula III is of formula 111(a):
0
P3
Y1 / NP1P2
N N
R7
111(a)
In another, the compound of formula 111(a) is of formula 111(b):
0
P3
Y1 / NP1P2
NN
NH2
111(b)
In a particular embodiment, the compound of formula II is prepared by
contacting a
compound of formula IV:
& B(OR)2
IV
with a compound of formula V:
R3 R4
XH
Y2 A2
V
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
under conditions sufficient for the formation of the compound of formula II,
wherein: Al is
optionally substituted cycloalkyl, aryl, or heterocycle; A2 is optionally
substituted cycloalkyl,
aryl, or heterocycle; Y2 is halogen or pseudohalogen; and each R is
independently hydrogen,
optionally substituted alkyl, alkyl-aryl, alkyl-heterocycle, aryl, or
heterocycle, or are taken
together with the oxygen atoms to which they are attached to provide a cyclic
dioxaborolane.
In one embodiment, Al is optionally substituted phenyl. In another, Al is
anisole. In
another, A2 is optionally substituted phenyl. In another, A2 is phenyl. In
another, R3 is
hydrogen. In another, R4 is optionally substituted alkyl. In another, R4 is -
CF3. In another,
Xis0.
In a particular embodiment, the compound of formula IV is of formula IV(a):
B(OR)2
(R11)m
IV(a)
wherein: each R11 is independently hydrogen, cyan, nitro, halogen, ORS,
NRqRjO, or
optionally substituted alkyl, alkyl-aryl or alkyl-heterocycle; and m is 1-5.
In another, the
compound of formula IV(a) is of formula IV(b):
P-B(OR)2
H3CO
IV(b)
In another, the compound of formula V is of formula V(a):
R3
R4
OH
Y2
(R12)p
V(a)
wherein: each R12 is independently hydrogen, cyan, nitro, halogen, ORS,
NRqRjO, or
optionally substituted alkyl, alkyl-aryl or alkyl-heterocycle; and p is 1-4.
In one embodiment,
the compound of formula V(a) is of formula V(b):
11
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
CF3
OH
Y2
V(b)
In a particular embodiment, the compound of formula 111(a) is prepared by
contacting
a compound of formula VI:
0
P3
(R'O)2B NP1 P2
(R5)n
VI
with a compound of formula VII:
YJ Z4\ Z3
Y
Ys
N1-1 ,Z2
Zi
VII
under conditions sufficient for the formation of the compound of formula III,
wherein: Y3 is
halogen or pseudohalogen; and each R' is independently hydrogen or optionally
substituted
alkyl, alkyl-aryl, alkyl-heterocycle, aryl, or heterocycle, or are taken
together with the oxygen
atoms to which they are attached to provide a cyclic dioxaborolane.
In one embodiment, n is 1. In another, R5 is hydrogen. In another, Zi is CRS.
In
another, R7 is NR9Rio. In another, R9 is hydrogen. In another, Rio is
hydrogen. In another,
Z2 is N. In another, Z3 is a carbon atom attached to the adjacent optionally
substituted phenyl
moiety. In another, Z4 is CRS. In another, R7 is hydrogen.
In a particular embodiment, the compound of formula VI is of formula VI(a):
0
P3
,JC
NP1 P2
(R'O)2B
VI(a)
12
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
In another, the compound of formula VI(a) is of formula VI(b):
O
P3
O1B NP,P2
VI(b)
In another, the compound of formula VI(a) is of formula VI(c):
0
~ P3
HO B / NP1P2
HO
VI(c)
In another, the compound of formula VII is of formula VII(a):
Y, s
NN
R7
VII(a)
In another, the compound of formula VII(a) is of formula VII(b):
Y, s
NN
NH2
VII(b)
One embodiment of the invention comprises deprotecting the compound of formula
1(a) to provide a compound of formula I:
O
A
X Z4\ OR2
Z3
R3 R4 NHR,
15 zi (R5)n
I
13
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
wherein: R1 is hydrogen or optionally substituted alkyl, alkyl-aryl, alkyl-
heterocycle, aryl, or
heterocycle. In a particular embodiment, the compound of formula I is of
formula 1(b):
O
R3 R4 OH
0 NH2
(R11)m
(R12)p N N
R7
1(b)
wherein: each R11 is independently hydrogen, cyan, nitro, halogen, ORS,
NRqRjo, or
optionally substituted alkyl, alkyl-aryl or alkyl-heterocycle; each R12 is
independently
hydrogen, cyan, nitro, halogen, ORg, NR9R10, or optionally substituted alkyl,
alkyl-aryl or
alkyl-heterocycle; m is 1-5; and p is 1-4.
In one embodiment, the compound of formula 1(b) is of formula 1(c), 1(d) or
I(e):
O
R3 R4 OH
0 NH2
(R11)m
11 (R12)p N N
R7
I(c)
O
R3 R4 OH
NH2 11
(R11) m
(R12)p N N
R7
1(d)
O
CF3 OH
.1""110 NH2
H3CO N r N
NH2
I(e)
14
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
Certain embodiments of the invention can be understood with reference to
Scheme 1:
0
Y1 Z4\\ Z
P I ,3
3 + I Y3
(RO)2B NP1P2 NZ1 Z2
(R5)n
VI VII
0
1 4__ \
Z3 Ps
Y1 Z
NP1P2 0
N i Z2
Z1 (R5)n Y1 Z4 OR2
III _ / Z3
~I" ~ NP1P2
N . Z2
Z1 (R5)n
0
R3 R4 Y1 Y Z4\~ P
3
XH + Z3
NP1P2
A N i Z2
Z1 III (RA,
0
A
X Zq\ Z Ps
3
R3 R4 \ NP1P2
N Z2
Z1 (R5)n
I(a) 0
A
X Z4-Z OR2
3
R3 R4 NHR1
N~ iZ2
Z1 I (R5)n
Scheme 1
In this approach, compounds of general formulae VI and VII are coupled under
conditions
suitable for the formation of a compound of formula III (e.g., contact with a
transition metal
catalyst, a base, and a solvent or solvent mixture with water), moieties of
which may be
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
deprotected if appropriate. The compound of formula III is then coupled with a
compound of
formula II under conditions sufficient to provide a compound of formula I(a)
(e.g.,
nucleophilic substitution conditions), which is deprotected (e.g., by
hydrolysis under acidic or
basic conditions) to afford the compound of general formula I.
A more specific adaptation of the approach shown in Scheme 1 is provided
below.
Scheme 2(a) shows the preparation of two intermediate compounds:
C B(OR)2
R3 R4 I Rs R4
OH (R11). OH
Y2
(R12)p (R11). (R12)p
11(a)
Y13 O
O N~N
P3 R Y1 \ NP1P2
CC Ps
(R'O)2B NP1P2 N N (R5),,
(RA,
O R7
Y1
\ \ NP1P2
"-)A OH
C
Y1
N / N (RA,
R7
Scheme 2(a)
Conditions sufficient for the formation of the compound of formula 11(a)
include the use of a
transition metal catalyst, a base, and a solvent or solvent mixture with
water. The
intermediate compounds are coupled as shown below in Scheme 2(b), to provide a
compound
that is deprotected to provide the final product:
16
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
0
OH
R3 R4 CT 11-~ OH
NPlP2
CY I + Y1 Y11 YC(R5),,
N , N (R1 1)m (R12)p
R7
O
R3 R4
C OH
O NP1P2
(R5)n
/ N
R1 1)m (R12)p N `I'
0
R7 R3 R4
OH
O \ ~\ NH2
N /N (RA,
(R5)n (R1)m
R7
Scheme 2(b):
Various reaction conditions may be used in this approach to obtain the desired
product. As those skilled in the art will immediately recognize, preferred
reaction conditions
may depend on the specific compounds involved. In one embodiment of the
invention, Y1 is
Cl. In another, Y2 is Br. In another, Y3 is Cl. In another, R is hydrogen. In
another, R' is
hydrogen. In another, both R' are taken together with the oxygen atoms to
which they are
attached to provide 4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl.
In another embodiment, each protecting group is independently aryl-alkyl,
heteroaryl-
alkyl, or -C(O)R13, wherein R13 is alkyl, aryl-alkyl, aryl, heterocycle,
alkoxy, aryloxy, or
aryl-alkoxy. Examples of protecting groups include benzyl, diphenylmethyl,
trityl, Cbz, Boc,
Fmoc, methoxycarbonyl, ethoxycarbonyl, and pthalimido.
In addition to the various synthetic methods disclosed herein, this invention
encompasses novel compounds that can be used to prepare compounds of formula
I.
Examples include compounds of the formula:
17
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
0
OH
/ NP1P2
CI 11
N`/N
NH2
and salts and solvates thereof, wherein: Pi is R1, -C(O)R13, or optionally
substituted alkyl,
alkyl-aryl, alkyl-heterocycle, aryl, or heterocycle; P2 is -C(O)R13 or
optionally substituted
alkyl, alkyl-aryl, alkyl-heterocycle, aryl, or heterocycle; R1 is hydrogen or
optionally
substituted alkyl, alkyl-aryl, alkyl-heterocycle, aryl, or heterocycle; and
each R13 is
independently alkyl, aryl-alkyl, aryl, heterocycle, alkoxy, aryloxy, or aryl-
alkoxy.
In one embodiment, P1 is hydrogen. In another, P2 is benzyl, diphenylmethyl,
trityl,
Cbz, Boc, Fmoc, methoxycarbonyl, ethoxycarbonyl, or pthalimido. A particular
compound is
of the formula:
0
OH
CI NHBoc
NN
NH2
The invention also encompasses compounds of the formula:
O
CF3 AOH
.1""'10 NP, P2
H3CO N N
I
""Ir
NH2
and salts and solvates thereof, wherein P1 is R1, -C(O)R13, or optionally
substituted alkyl,
alkyl-aryl, alkyl-heterocycle, aryl, or heterocycle; P2 is -C(O)R13 or
optionally substituted
alkyl, alkyl-aryl, alkyl-heterocycle, aryl, or heterocycle; R1 is hydrogen or
optionally
substituted alkyl, alkyl-aryl, alkyl-heterocycle, aryl, or heterocycle; and
each R13 is
independently alkyl, aryl-alkyl, aryl, heterocycle, alkoxy, aryloxy, or aryl-
alkoxy.
18
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
In one embodiment, Pi is hydrogen. In another, P2 is benzyl, diphenylmethyl,
trityl,
Cbz, Boc, Fmoc, methoxycarbonyl, ethoxycarbonyl, or pthalimido. A particular
compound is
of the formula:
O
CF3 AOH
""'O NHBoc
H3CO
/ N N
I() NH2
This invention also encompasses compounds of the formula:
CF3
OH
H3CO /
and salts and solvates thereof.
5. EXAMPLES
The following non-limiting examples describe the synthesis of (S)-2-amino-3-(4-
(2-
amino-6-((R)-2,2,2-trifluoro-l-(3'-methoxybiphenyl-4-yl)ethoxy)pyrimidin-4-
yl)phenyl)-
propanoic acid.
Generally, intermediate compounds 3 and 8 are first prepared, as shown below
in
Schemes 3(a) and (b):
B(OH)2 CF3
H
~ jO1:: 1
CF3 CF3
OMe /
O OH I /
Br Br
OMe
1 2 3
Scheme 3(a)
19
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
O 0
OMe OMe
HO / NHBoc TfO ja~ NHBoc
4 5 O
O
OH
OMe
016 NHBoc
01, / NHBoc
B
7
6 0
OH
CI / NHBoc
11
N\/N
' 8
NH2
Scheme 3(b)
An alternate synthesis of compound 8 is shown in Scheme 3(c):
0 0
OH OH
HO"B NH2 HO,B NHBoc
I I
OH OH
11 12 0
OH
CI NHBoc
NN
8
NH2
Scheme 3(c)
The intermediates are then coupled as shown below in Scheme 3(d):
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
CF3 O
OH OH
CI NHBoc
NN
/ IY
OMe 3 NH2 8
O
CF3
OH
O NHBoc
N / N
NH2
O
OMe CF3 OH
9
NH2 11 / N N
NH2
OMe
Scheme 3(d)
In the following examples, yields of various reactions are reported on a molar
basis.
Unless otherwise indicated, reagents are commercially available and may be
purchased from
5 Sigma-Aldrich Company, Inc. (Milwaukee, WI, USA).
5.1. Preparation of (R)-1-(4-Bromophenyl)-2,2,2-trifluoroethanol (2)
CF3 CF3
O I
1 OH
Br Br 2
This compound was prepared based on a literature procedure (Ohkuma, et at. J.
Am.
Chem. Soc., 1998, 120, 13529-13530). To a 1 L high pressure vessel was charged
4-bromo-
10 trifluoroacetophenone (1, Wilmington PharmaTech, Delaware, 100.0 g, 395
mmol),
potassium tert-butoxide (1 M solution in 2-methyl-2-propanol, 5.0 ml, 10.0
mmol, 0.025 eq),
and catalyst [(trans)-RuC12[(R)-Xyl-P-Phos][(R)-DIAPEN] (Johnson Matthey, New
Jersey,
200 mg, 0.16 mmol, 0.04% mol). The mixture was dissolved in anhydrous 2-
propanol (175
ml) and the entire vessel was purged with argon by 3 vacuum-thaw cycles. The
reaction
21
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
mixture was then purged with hydrogen by 3 vacuum-thaw cycles. The reaction
was carried
out under 60 psi hydrogen atmosphere. After 24 hours of stirring and no more
hydrogen
consumption, the reaction was deemed complete by GC-MS analysis (no more
starting
ketone). The contents of the reaction vessel were transferred to a round
bottom flask with
MeOH rinsing (3 x 20 ml), and concentrated under reduced pressure until no
more solvent
was distilling off. The resulting orange-brown oil was then dissolved in
heptane (1000 ml)
and washed with water (2 x 100 ml), brine (100 ml) and dried over sodium
sulfate. To the
dried organic layer was added Darco activated charcoal (20 g) and Hyflo
Super Cel (20 g)
and the mixture was heated at 70 C for 1 hours. The mixture was filtered hot
to give a light
yellow solution. The filtrate was concentrated under reduced pressure with
heating (- 50 -
60 C) until no more solvent was distilling. The resulting yellow oil was
dissolved in 60 C
warm heptane (350 ml) and allowed to stir while cooling. As the temperature
cooled to rt.,
white solid began to precipitate. After 4 hours of stirring, the solids were
filtered and dried to
give the titled product (63.5 g, 63%, >99% ee) as a white powder. m.p.: 56.7
C. [a] = -30.1
(cl.09, ethanol). GC-MS (CI): MH+ = 255.8. 1H NMR (CDC13) 6 7.58 (m, 2H), 7.42
(d, J=
8.3 Hz, 2H), 5.00 (m, 1H), 2.62 (d, J= 4.3 Hz, 1H). 13CNMR (CDC13): 6 133.2,
132.2,
129.5, 125.7, 124.3 (q, J= 282 Hz), 72.6 (q, J= 32 Hz). 19F NMR (CDC13): 6 -
78.5 (d, J
5.6 Hz).
5.2. (S)-1-(4-Bromophenyl)-2,2,2-tritluoroethanol
Using a procedure similar to the above example, the titled compound was
prepared
using catalyst [(trans)-RuC12[(S)-Xyl-P-Phos][(S)-DIAPEN] (Johnson Matthey,
New Jersey).
5.3. (R)-2,2,2-Tritluoro-l-(p-tolyl)ethanol
CF3 CF3
O OH
Similarly, 2,2,2,-trifluoro-l-(p-tolyl)ethanone was hydrogenated using
catalyst
[(trans)-RuC12[(R)-Xyl-P-Phos][(R)-DIAPEN] to give the titled compound. m.p.:
44.2 C.
1H NMR (CDC13): 6 7.38 (d, J= 6.0 Hz, 2H), 7.25 (d, J= 6.0 Hz, 2H), 5.00 (dq,
J1= 6.6 Hz,
J2 = 3.3 Hz, 1H), 2.49 (d, J = 3.8 Hz, 1H), 2.42 (s, 3H).
22
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
5.4. (S)-2,2,2-Trifluoro-l-(p-tolyl)ethanol
Similarly, the titled compound was prepared using catalyst [(trans)-RuCl2[(S)-
Xyl-P-
Phos] [(S)-DIAPEN] .
5.5. (R)-2,2,2-Trifluoro-l-(3'-methoxybiphenyl-4-yl)ethanol (3)
CF3
CF3 B(OH)2 OH
SOH +
Br
2 We
OMe
To a stirred solution of (R)-1-(4-bromophenyl)-2,2,2-trifluoroethanol (2, 69g,
0.27
mol, > 99% ee), 3-methoxy phenylboronic acid (Matrix, 51 g, 0.34 mol, 97%
purity), and
bis(triphenylphosphine)palladium(II) dichloride (0.95g, 0.5% mol) in ethanol
(560 ml) was
added a solution of potassium carbonate (112 g, 0.81 mol) in water (140 ml)
under nitrogen.
The resulting mixture was heated at 75 C for 1 hour and deemed complete by GC-
MS or
TLC. After reaction mixture was cooled to 40 C, it was filtered through a pad
of Celite,
washed with methanol (3x100 ml). The filtrate was diluted with 100 ml of water
and
concentrated. The resulting syrup was dissolved in 700 ml of ethyl acetate and
washed with 1
N sodium hydroxide (2x100 ml), water (2x100 ml) and brine (1x100 ml). The
organic layer
was heated with activated carbon (14 g) and Hyflo Super Cel (14 g) at 60 C for
1 hours. This
mixture was filtered hot and washed with ethyl acetate (100 ml) and then
concentrated to a
syrup. This syrup was immediately dissolved in 1% ethyl acetate/heptane (700
ml) and stirred
for 4 hours. The resulting slurry was filtered and dried to give the titled
compound as a white
crystalline solid (3, 68 g, 89% yield, >99% ee)
Alternative crystallization method: The crude product syrup/solid (10 g) was
dissolved in MTBE (10 ml) and diluted with heptane (200 ml). The solution was
concentrated to about 70 ml under reduced pressure. This mixture was stirred
at room
temperature overnight and the resulting slurry was filtered and dried to give
the title
compound (3, 8.8 g) as a white crystalline solid. m.p.:107.6 C. [a] = -31.85
(c 1.067,
ethanol). LC-MS (ESI): MH+ = 283.1. 1H NMR (CDC13): 6 7.66 (m, 2H), 7.56 (d,
J= 8.2
Hz, 2H), 7.42 (t, J = 7.8 Hz, 2H), 7.20 (m, 1 H), 7.14 (m, 1 H), 6.95 (m, 1
H), 5.82 (q, J = 6.6
Hz, 1H), 3.85 (s, 3H), 2.63 (br s, 1H). 13C NMR (CDC13): 6 160.3, 142.6,
142.2, 133.5, 130.3,
128.3, 127.8, 124.8 (q, J= 282 Hz), 120.1, 113.4, 113.3, 73.0 (q, J= 32 Hz ),
55.7. 19F NMR
23
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
(CDC13): 6 -78.3 (d, J= 6.4 Hz). Residual palladium: 11 ppm. Anal. Calcd for
C15H13F302:
C, 63.83; hours, 4.64. Found: C, 63.78; hours, 4.60.
5.6. (R)-2,2,2-Trifluoro-l-(3'-methoxybiphenyl-4-yl)ethanol (3)
A 22-L, round-bottom flask equipped with a mechanical stirrer, a thermocouple
attached to a temperature controller, and a condenser with a nitrogen line was
charged with
compound 2 (1.00 kg, 1 wt, 3.92 mol) and ethanol (4.5 L, 4.5 vol). The mixture
was sparged
with nitrogen for 10 minutes and (Ph3P)2PdC12 (12.6 g, 0.0126 wt, Strem) was
added.
Following additional sparging with nitrogen, a solution of K2C03 (1.63 kg, 3
equiv) in water
(2 vol) was added. The mixture was heated to 75 C under nitrogen and then
approximately
20% of a solution of 3-methoxy phenylboronic acid (715 g, 4.70 mol, 1.2 equiv,
Usun) in
ethanol (4.5 vol) was added via a peristaltic pump. After 20 minutes, an in-
process control
(IPC) sample was taken and showed that the boronic acid had been consumed.
This process
was repeated until all of the boronic acid was added. After stirring for a
further 20 minutes,
HPLC analysis showed that the reaction was complete. The heat was switched off
and at
69 C, water (3.6 vol) was added. The reaction mixture was then filtered at 50
C through a
pad of celite (Celpure P300, 0.15 wt., Sigma) and the filter cake was washed
with methanol
(2 x 2.5 vol). The filtrate was concentrated under reduced pressure at 40-45 C
to 5 vol. The
slurry was then transferred to a reparatory funnel and MTBE (10 vol) was
added. The
mixture was then washed with a 50% solution of sodium hydroxide (0.6 vol).
After stirring,
the layers were separated and the aqueous phase was extracted with MTBE (1.5
vol). The
organic extracts were combined and washed with water (1 vol) followed by 20%
aqueous
sodium chloride (1 vol) to provide 11.9 volumes of organic product solution.
The solution
was transferred to a reactor, treated with a slurry of Darco G-60 (0.3 wt) in
MTBE (1 vol) and
heated to 50 C. After 90 minutes, the mixture was filtered through a pad of
Celpure P300
(0.15 wt) and washed with MTBE (2 x 3 vol).
The filtrate (14.8 vol) was transferred to a reactor and distilled under
vacuum at 45 C
to remove MTBE. The filtrate was reduced to 6.7 volumes over 2.5 hours and
then heptane
(3.15 vol) was added. The solution was further distilled at 50 C to 6.7 vol
over 1 hours and
then additional heptane (3.15 vol) was added. The solution was concentrated to
6.7 vol at
55 C over 1.5 hours and then heptane was added (3.15 vol). Precipitation was
observed
immediately and the distillation was continued under vacuum at 60 C. After 2.5
hours, the
distillation was stopped (7 vol remaining), the heat was switched off and the
batch was
cooled overnight to ambient temperature. The batch was filtered at 24 C and
washed with
24
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
heptane (1.5 vol). The solids were dried at room temperature under vacuum over
the
weekend to provide 799.7 g of 3 as a white solid [72% yield, >99% (AUC)].
5.7. (R)-2,2,2-Trifluoro-l-(3'-fluorobiphenyl-4-yl)ethanol
CF3
CF3 B(OH)2 OH
\ OH + / I \
Br
2 F
F
Similar to the above procedure, the title compound was prepared from (R)-1-(4-
bromophenyl)-2,2,2-trifluoroethanol (2) and 3-fluorophenylboronic acid. 1H NMR
(CDC13):
6 7.62(d, J = 6.0 Hz, 2H), 7.56 (d, J = 6.3 Hz, 2H), 7.42 (m, 2H), 7.28 (m,
1H), 7.06 (m, 1H),
5.82 (q, J = 5.1 Hz, 1H).
5.8. (S)-Methyl2-(tent-butoxycarbonylamino)-3-(4-
(trifluoromethylsulfonyloxy)phenyl)propanoate (5)
0 0
OMe OMe
HO NHBoc TfO NHBoc
4 5
This compound was prepared based on a literature procedure (Shieh, et at. J.
Org.
Chem., 1992, 57, 379-381). To a solution of Boc-Tyr-OMe (4, Bachem,
California, 100 g,
0.34 mol) and N-methylmorpholine (51 g, 1.5 eq) in dichloromethane (1000 ml)
was added
triflic anhydride (100 g, 1.05 eq) over 2 hours at -5 to -15 C. The resulting
red solution was
stirred at -10 C for 10 minutes. HPLC analysis showed complete disappearance
of starting
material. The reaction was quenched with 10% citric acid (500 ml). The organic
layer was
washed with 10% citric acid (500 ml) followed by water (500 ml). The resulting
light pink
solution was concentrated under reduced pressure to 200 ml. This was diluted
with
acetonitrile (600 ml) and further concentrated to a 200 g solution. This
solution was used in
the next step without further purification. Estimated yield is 98% by
stripping a sample to
dryness to give a low melting pale yellow solid. LC-MS (ESI): MH+ = 428.0,
MNH4+ =
445Ø 1H NMR (CDC13) 6 7.16 (m, 4H), 4.95 (d, J= 7.1 Hz, 1H), 4.53 (m, 1H),
3.64 (s, 3H),
3.10 (dd, Ji = 5.7 Hz, J2 = 13.8 Hz, 1H), 2.97 (dd, Ji = 6.3 Hz, J2 = 13.6 Hz,
1H), 1.34 (s, 9H).
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
13C NMR (CDC13) 6 172.3, 155.4, 149.0, 137.4, 131.5, 121.7, 119.1 (q, J= 321
Hz), 80.54,
54.62, 52.7, 38.3, 28.6. 19F NMR (CDC13) 6 -73.4.
5.9. (S)-2-(Tert-butoxycarbonylamino)-3-(4-(4,4,5,5-tetramethyl-1,3,2-
dioxaborolan-2-yl)phenyl)propanoic acid (7)
0 0
0
OMe OH
OMe B / NHBoc ,B ~NH B oNHBo
TfO c ,
0 6 0 7
5
This ester compound 6 was prepared based on a literature procedure (Firooznia,
et at.,
Tetrahedron Lett., 1999, 40, 213-216). Bis(pinacolato)diboron (90 g, 1.1 eq),
potassium
acetate (63 g, 2 eq), tricyclohexylphosphine (2.3 g, 2.5% mol), and palladium
acetate (0.72 g,
1 mol%) were mixed in acetonitrile (950 ml) and the resulting mixture stirred
at room
temperature for 5 minutes. The above triflate (5) solution (190 g, 0.32 mol)
was added and
the resulting mixture was heated at 80 C for 1 hours and cooled. HPLC showed
complete
consumption of the starting material. The reaction mixture was quenched with
aqueous
potassium bicarbonate solution (57 g in 475 ml water) and resulting mixture
was stirred at
room temperature for 30 minutes. The mixture was filtered through a pad of 20
cellulose to
remove palladium black. A sample of the organic layer was concentrated and
purified by
column chromatography (gradient: 1:10 to 1:4 ethyl acetate/hexanes) to give
the ester
compound 6 as a clear oil. LC-MS (ESI): MH+ = 406.2, MNH4+ = 423.2, MzH+ =
811.5,
M2NH4+ = 428.5. 1H NMR (CDC13) 6 7.76 (d, J= 8.l Hz, 2H), 7.15 (d, J= 7.6 Hz,
2H),
4.96 (d, J= 7.3 Hz, 1H), 4.60 (m, 1H), 3.72 (s, 3H), 3.13 (m, 2H), 1.44 (s,
9H), 1.36 (s, 12H).
The above organic layer of 6 was stirred with aqueous lithium hydroxide
solution (23
g in 500 ml water) at room temperature for 30 minutes. The pH of the resulting
slurry was
adjusted to about 10 with 6 N hydrochloric acid and filtered. The cake was
washed with
water (200 ml). Acetonitrile was removed from the filtrate under reduced
pressure to give an
aqueous slurry (950 ml, additional water was added during distillation). The
slurry was
filtered through a pad of 20 cellulose and washed with water (200 ml). The
filtrate was
washed with MTBE (500 ml) and rediluted with 700 ml MTBE. The mixture was
acidified
to pH about 4.5 with 6 N hydrochloric acid. The organic layer was washed with
water (500
ml) and concentrated under reduced pressure to the titled product (7) as a
brown oil (206 g,
95% yield based on estimated purity by NMR). The crude product was used
directly in the
following step. LC-MS (ESI): MH+ = 392.2, MNH4+ = 409.2, MzH+ = 783.4, M2NH4+
_
26
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
800.4. 'H NMR (CDC13) 6 7.95 (br s, 1H), 7.76 (d, J= 7.8 Hz, 2H), 7.21 (d, J=
7.6 Hz, 2H),
5.03 (d, J= 7.8 Hz, 1H), 4.62 (m, 1H), 3.18 (m, 2H), 1.43 (s, 9H), 1.35 (s,
12H). 13C NMR
(CDC13) 6 175.8, 155.7, 139.7, 135.4, 129.2, 84.2, 80.5, 54.5, 38.3, 28.7,
25.2.
Compound 7 can optionally isolated by crystallization. Thus, the above MTBE
solution of 7 can be dried with anhydrous Na2SO4 and concentrated to about 1.0
vol under
vacuum. Heptane (2.5 vol) was added and concentrated to about 1.5 vol under
vacuum.
Heptane (4.2 vol) was added slowly at 3642 C followed by cooling slowly to 5-
10 C. The
resulting slurry is filtered, washed by heptane, and dried under vacuum at 20-
30 C to give the
product 7 in about 76% yield.
5.10. Alternative Crystallization of (S)-2-(Tert-butoxycarbonylamino)-3-(4-
(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)propanoic acid (7)
A 1 L jacketed three-necked round bottom flask with mechanical stirrer, rubber
septum with temperature probe, and gas bubbler was charged with 100ml of an
ethanol
solution containing 50.88 g 7. The solution was set stirring under nitrogen,
diluted with 35m1
ethanol, then with 50m12-propanol, and was heated to -60 C. Then 250m1 water
were added
to reach the cloudy point and the turbid solution was held at -60 C for 75
minutes followed
by cooling to -10 C over -1.5hrs. After 45 minutes, the mixture was biphasic
and was
diluted with an additional 30m12-propanol. The mixture was stirred under
nitrogen at 10 C
overnight and the resulting white fine suspension was filtered. The collected
solids were
washed with 100ml 9:1 water:2-propanol and were dried in vacuo at -50-60 C to
give 39.88g
7 as a chalky white powder (78% recovery). The solid was in the filtrate was
filtered and
dried to afford 4.51 g of a pale yellow granular solid. HPLC suggested this
material was
mostly the boronic acid 12.
5.11. (S)-3-(4-(2-Amino-6-chloropyrimidin-4-yl)phenyl)-2-(tert-
butoxycarbonylamino)propanoic acid (8)
0 0
OH OH
B / NHBoc CI / NHBoc
O N iN
7 8
NH2
The above crude compound 7 (0.32 mol) was dissolved in ethanol (800 ml) and
resulting solution was concentrated under reduced pressure to about 700 ml and
diluted with
ethanol (1300 ml). To this solution was added 2-amino-4,6-dichloropyrimidine
(74 g, 1.4
27
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
eq), bis(triphenylphosphine)palladium(II) dichloride (2.3 g, 1 mol%), and
aqueous potassium
bicarbonate solution (97 g, 3 eq, 380 ml water). This mixture was heated at 75-
80 C for 2
hours, at which time HPLC analysis showed complete consumption of the starting
material.
Ethanol was removed from the filtrate under reduced pressure to give an
aqueous slurry (600
ml, additional water was added during distillation). The slurry was filtered
and washed with
200 ml water. The cake was dried at 50 C under vacuum to give recovered 2-
amino-4,6-
dichloropyrimidine as a tan solid (30 g, 41% of original charge). 1H NMR (DMSO-
d6) 6
7.58 (br s, 2H), 6.84 (s, lH). 13C NMR (DMSO-d6) 6 162.8, 160.9, 107.5. The
filtrate was
washed with ethyl acetate (400 ml) and diluted with 3:1 THF/MTBE (600 ml). The
mixture
was acidified to pH about 3.5. The organic layer was washed with brine (300
ml) and
concentrated to give the crude product 8 as a red oil (180 g). This oil was
redissolved in THE
(300 ml), polish-filtered, and washed with THE (100 ml). The filtrate was
diluted with
isopropanol (400 ml) and the mixture was distilled atmospherically to about
300 ml. More
isopropanol (400 ml) was added and distillation continued until the volume
reached about
500 ml. The mixture was then cooled over 1 hours to 45 C and held for 2 hours
before it was
cooled to room temperature over 1 hours. After 1 hours hold, the slurry was
filtered, washed
with isopropanol (150 ml), and dried at 50 C under vacuum to give the product
8 as a light
pink solid (46.2 g, 37% yield from Boc-Tyr-OMe, 4). Purity: 93.4% by HPLC.
Chiral
purity: >99% ee. Chiral analysis was performed on the corresponding methyl
ester
derivative, which was prepared using trimethylsilyldiazomethane. An analytical
pure sample
was obtained by column chromatography (gradient 1:20 to 1:10
methanol/dichloromethane).
LC-MS (ESI) MH+ = 393.1, MH++ acetonitrile = 434.1, M2H+ = 785.3. 1H NMR (DMSO-
d6) 6 12.60 (s, 1 H), 8.02 (d, J = 8.3 Hz, 2H), 7.3 8 (d, J = 8.1 Hz, 2H),
7.23 (s, 1 H), 7.13 (br s,
2H), 3.09 (dd, Jl = 4.4 Hz, J2 = 13.5 Hz, I H), 2.91 (dd, Ji = 10.5 Hz, J2 =
13.8 Hz, I H), 1.32
(s, 9H). 13C NMR (DMSO-d6) 6 173.4, 165.8, 163.5, 161.0, 155.4, 141.4, 134.0,
129.4,
126.8, 104.4, 78.0, 54.8, 36.2, 28.1. Anal. Calcd for C18H21C1N404: C, 55.03;
hours, 5.39; N,
14.26. Found: C, 54.76; hours, 5.65; N, 14.09.
HPLC analysis of the above mother liquor against an standard solution of
compound
8 showed additional 38 g product 8 (30% yield from Boc-Tyr-OMe, 4). Product 8
can be
partially recovered by further concentration of the mother liquor to give a
total yield of 60%
from Boc-Tyr-OMe, 4.
28
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
5.12. (S)-3-(4-(2-Amino-6-chloropyrimidin-4-yl)phenyl)-2-(tert-
butoxycarbonylamino)propanoic acid (8)
A 22-L, round-bottom flask equipped with a mechanical stirrer, a thermocouple
attached to a temperature controller, and a condenser with a nitrogen line was
charged with
compound 7 (850 g, 1 wt, 2.17 mol), 2-amino-4,6-dichloropyrimidine (712.3 g, 2
equiv,
Usun), and ethanol (13.6 L, 16 vol). The slurry was sparged with nitrogen for
10 minutes;
then (Ph3)2PdC12 (18.3 g, 0.021 wt, Strem) was added and nitrogen sparging was
continued
for 10 minutes. A solution of potassium bicarbonate (783 g, 3.6 equiv) in
water (3.2 L, 3.7
vol) was then charged to the reactor whereupon gas evolution was observed. The
mixture
was heated to 75 C for a total of 11.5 hours and then cooled to 45 C
overnight. HPLC
analysis after 9.5 hours at 75 C indicated that there were about 3.0% of 7
remaining (by
conversion). The reaction was cooled to 45 C and stirred overnight whereupon
HPLC
analysis indicated that there was <1.0% of 7 remaining.
The batch was then distilled under reduced pressure at 45 C over a period of
15 hours
to afford 4-5 L of a yellow slurry. The batch was then allowed to cool
overnight. Water was
added (3 vol) and after heating to 45 C, distillation was continued for 1
hours until no more
distillate was collected. The vacuum was released and water (3 vol) was added
to the batch.
After allowing to settle, the batch was filtered through a slurry of cellulose
powder (20
micron, 0.2 wt.) in water (1 vol). Water (2 vol) was added to the remaining
solids/slurry in
the reactor and this was filtered through a sintered glass funnel. This
filtrate was then further
filtered through the cellulose pad to afforded 11.2 L of product solution
(13.2 vol).
The solution was then transferred to a reparatory funnel containing EtOAc (3.3
vol).
After stirring and separating, the aqueous phase was transferred to a 22-L
reactor and then a
solution of PBu3 (212 ml, 0.25 vol, 97%) in EtOAc (3.5 vol) was charged to the
reactor. The
solution was heated at 50 C for 2.5 hours. Additional EtOAc (3.3 vol) was
added to the
reactor and the contents were charged to a reparatory funnel and the two
phases separated.
The aqueous phase (41 C) was charged back to the reparatory funnel and washed
with
additional EtOAc (3.3 vol). The two phases were separated and then the aqueous
phase was
charged to a 22-L reactor and heated to 45 C. Heptane (5 vol) was added to the
reactor, the
contents of the reactor were transferred to a reparatory funnel and the two
phases were
separated. The aqueous phase (11.2 L, 13.2 vol) was charged to the 22-L
reactor, diluted to
14 vol with water and then a slurry of Darco G-60 (0.2 wt) in water (1 vol)
was charged to
the reactor. The mixture was heated to 60 C and stirred at 60 C for 2 hours.
The heat was
29
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
switched off and the batch was stirred over the weekend. The batch was
filtered through a
pad of Celpure P300 (0.2 wt, Sigma) and washed with water (2 x 1.2 vol).
A 22-L, round-bottom flask equipped with a mechanical stirrer, a thermocouple
attached to a temperature controller, and pH probe attached to a pH meter was
charged with
citric acid (127.5 g, 0.15 wt) and water (2 vol). The solution was heated to
40 C and the pH
of the solution was adjusted to 4.0 with a 2 M solution of sodium hydroxide. A
solution of
citric acid (40 wt%, 2 L) was charged to an addition funnel and was attached
to the reactor.
The basic solution of 8 was then transferred via peristaltic pump through an
in-line filter to
the citric acid solution and the pH was maintained at pH 4.0 with the 40%
citric acid solution.
Once the addition was complete, the batch was heated to 60 C and stirred for 2
hours. The
batch was then cooled overnight and the solids were filtered at 29 C. The cake
was washed
with water (2 x 2.5 vol) and then dried at 45-50 C for 24 hours to provide 720
g of 8 (84%
yield) with a purity of 85.9% (AUC).
5.13. Alternate Preparation of (S)-3-(4-(2-Amino-6-chloropyrimidin-4-
yl)phenyl)-2-(tert-butoxycarbonylamino)propanoic acid (8)
To a 5-L three-necked flask with a thermometer controller, a mechanical
stirrer, and a
condenser protected under N2 was charged EtOH (570g), THE (1330g), boronic
acid 7 (100g,
255.6mmol, 1.0 eq.), 2-amino-4,6-dichloropyrimidine 126g (768.3mmol, 3.0 eq.),
PPh3
(0.87g, 3.32mmol, 1.3mol%) and Pd(OAc)2 (0.373g, 1.66mmol, 0.65mo1%). The
flask was
then degassed by three vacuum/nitrogen fill cycles and stirred at about 10-20
C for about 10-
20 minutes. To the stirring solution was added an aqueous solution of KHCO3
(92g, 918.9
mmol, 3.6 eq in 579 g of water) over about 30-50 minutes to control the
evolution of CO2
gas). The flask was then degassed again by three vacuum/nitrogen fill cycles.
The resulting
solution was then refluxed at about 6872 C for 2123 hours and the reaction was
determined to be complete base on HPLC analysis. The reaction mixture was
concentrated to
-750 mL under reduced pressure at 3540 C and then flushed with water (300 mL x
2). The
concentrated solution was diluted with water (600 g) and stirred for 1530
minutes at
2025 C. After filtration, the filter cake was washed with water (200 g x 2).
The filtrate was
extracted with EtOAc (500 g x 2). The combined EtOAc layer was washed with
water (300
g). THE (1000 g) and toluene (730 g) were then added to the combined aqueous
layer and
the mixture was acidified to pH 2.53.5 with 6 N HCl (-100 g) at 2025 C. The
layers were
separated and NaC1(500 g) was added to the aqueous layer then extracted with
EtOAc (400
g). The combined organic layer was treated with active carbon (50 g) at 6568 C
for 8-10
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
hours twice. The resulting mixture was concentrated under vacuum to -300 mL at
3040 C
and then flushed with toluene (500 g). The resulting mixture was cooled to 0-5
C and stirred
for 6080 minutes at 0-5 C, and then filtered. The wet cake was washed with
toluene (43 g)
and dried in vacuum oven at 4045 C for 12 hours to afford 82.3g of the
monochloride
toluene solvate 8 as an off-white solid in 66% yield corrected for 80w% purity
(96.3A%).
Alternatively, the non-solvated product can be isolated from acetonitrile (50g
scale of
boronic acid 7). For example, the combined organic extraction was concentrated
under
vacuum to -150 mL (3.OX) at 40 C, followed by addition of 500 g (l OX) of
CH3CN, and
then concentrated to -250 mL (5.OX). The resulting slurry was stirred for 2
hours at 60 C,
and then filtered. The wet cake was washed with 50 g (1.OX) of CH3CN twice and
dried in
vacuum oven at 40 C to afford 37.3 g of the desired product as white powder in
72% yield
after correcting for w% purity (97.2 A%, 96.9 wt%, Pd: 22 ppm).
5.14. Optional Purification of (S)-3-(4-(2-Amino-6-chloropyrimidin-4-
yl)phenyl)-2-(tent-butoxycarbonylamino)propanoic acid (8)
O
O OH
HO _ / I I OH CI I I / NHBoc
NHBoc NHBoc N N
N N HNT N~NH2
NH2 I N
A CI B
The crude 8 as prepared from examples 5.11 or 5.12 is impure and usually
contains
about 6% of the diacid impurity (A) and about 4% amination product (B). This
material can
be used directly in the next step or it can optional purified by the following
methods.
Method 1. To a 3-necked 250 ml RB flask was added crude 8 (10.0 g, 25.4 mmol,
90% pure, with 6% A and 4% B), i-PrOH/toluene (1:1, 80 ml / 80 ml, 8x / 8x)
and tent -
butylamine (13.4 ml, 5.0 equiv). The resulting mixture was stirred and heated
at 78 C for 1
hour and then slowly cooled to 0 C, and stirred for another hour. The solids
were collected
by filtration and the cake was washed with 20 ml of i-PrOH /toluene (1:3). The
cake was
dried under vacuum to constant weight to provide the desired tert-butylamine
salt of 8 as a
pale yellow solid (8.8 g, 74% yield, 94% pure, 3% A, 3% B).
To a 3-necked 250 ml RB flask was added the tert-butylamine salt of 8 (20.0 g,
42.9
mmol) and followed by H2O / THE / toluene (400 ml / 200 ml / 160 ml, 20x / l
Ox / 8x). The
31
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
resulting mixture was heated to 60 C and slowly added 6M HC1 until pH of the
mixture
reached 4Ø The mixture was cooled to room temperature and the organic layer
was
separated. The organic layer was washed with H2O (100 ml, 5x) and concentrated
by rotary
evaporating to around 160 ml of overall volume. The solids were collected by
filtration and
the cake was washed with 20 ml of toluene. The cake was dried under vacuum to
constant
weight to provide 8 as a pale yellow solid (15.0 g, 89% yield, 94% pure, 3% A,
3% B).
Method 2. To a 3-necked 250 ml RB flask was added crude 8 (20.0 g, 42.9 mmol,
90% pure, with 6% A and 4% B) and followed by THE / toluene (200 ml / 160 ml,
l Ox / 8x).
The resulting mixture was heated to 60 C for 1 hour and cooled to room
temperature. THE
was removed by rotary evaporating to around 160 ml of overall volume. The
solids were
collected by filtration and the cake was washed with 20 ml of toluene. The
cake was dried
under vacuum to constant weight to provide 8 as a pale yellow solid (11.8 g,
70% yield,
92.8% pure, 6.0% A, 1.3% B).
To a 3-necked 250 ml RB flask was added the above 8 (10.0 g, 25.4 mmol) and
tent -
butylamine (13.4 ml, 5 equiv) followed by i-PrOH / toluene (1:1, 80 ml / 80
ml, 8x / 8x). The
resulting mixture was heated to clear (78 C) for 1 hour, slowly cooled to 0
C, and stirred at
0 C for another 1 hour. The solids were collected by filtration and the cake
was washed with
ml of i-PrOH / toluene (1:3). The cake was dried under vacuum to constant
weight to
provide the tert-butylamine salt of 8 as a pale yellow solid (9.7 g, 82%
yield, 96% pure, 3.3%
20 A, 0.6% B).
To a 3-necked 250 ml RB flask was added the tert-butylamine salt of 8 (20.0 g,
42.9
mmol) and followed by H2O / THE / toluene (400 ml / 200 ml / 160 ml, 20x / l
Ox / 8x). The
resulting mixture was heated to 60 C and slowly added 6M HC1 until pH of the
mixture
reached 4Ø The mixture was cooled to room temperature and the aqueous layer
was
separated. The organic layer was washed with H2O (100 ml, 5x) and concentrated
by rotary
evaporating to around 160m1 of overall volume. The solids were collected by
filtration and
the cake was washed with 20 ml of toluene. The cake was dried under vacuum to
constant
weight to provide 8 as a pale yellow solid (15 g, 88% yield, 96% pure, 3.3% A,
0.5% B).
Method 3. To a 3-necked 3 L RB flask was added the aqueous solution of the
potassium salt containing -50 g 8 (90 %, 6 % A, 4 % B, all normalized AUC) and
followed
by THE / toluene (500 ml / 400 ml, l Ox / 8x). The resulting mixture was
heated to 60 C and
slowly added 6M HC1 until pH of the mixture reached 4Ø The mixture was
cooled to room
temperature and the aqueous layer was separated. The organic layer was washed
with H2O
32
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
(250 ml, 5x) and concentrated by rotary evaporating to around 400m1 of overall
volume to
afford a slurry of 8 in -8x toluene.
To a 3-necked 3 L RB flask was added the slurry (in 8x toluene, 400 ml) and
tert-
butylamine (67 ml, 5.0 equiv) followed by i-PrOH (400 ml, 8x). The resulting
mixture was
heated at 78 C for 1 hour, cooled to 0 C, and stirred at 0 C for another 1
hour. The solids
were collected by filtration and the cake was washed with 100 ml of i-PrOH /
toluene (1:3).
The cake was dried under vacuum to constant weight to provide the tert-
butylamine salt of 8
as a pale yellow solid (42.4 g, 72% yield, 95% pure, 3.2% A, 1.9% B).
To a 3-necked 250 ml RB flask was added the tert-butylamine salt of 8 (42.4 g,
91.0
mmol) and followed by H2O / THE / toluene (1000 ml / 500 ml / 400 ml, 20x / l
Ox / 8x). The
resulting mixture was heated to 60 C and slowly added 6M HC1 until pH reached
4Ø The
mixture was cooled to room temperature. The organic layer was separated and
washed with
H2O (250 ml, 5x). The organic solution was concentrated by rotary evaporating
to -400 ml
of overall volume. The solids were collected by filtration and the cake was
washed with 100
ml of toluene. The cake was dried under vacuum to constant weight to provide 8
as a pale
yellow solid (35.4g, 89.5% yield, 96% pure, 2.9% A, 1.6% B).
Method 4. To a test tube was added 8 (198.6 mg, 0.5 mmol) and cinchonidine
(167.1
mg) followed by acetonitrile (7.5 ml). The resulting mixture was heated to
clear and cooled
to room temperature, and stirred for another 2 hours. The solids were
collected by filtration
and the cake was washed with 1 ml of MTBE. The cake was dried under vacuum to
constant
weight to provide the final product (208 mg, 68% yield, 92% pure, 4.4% A, 1.4%
B).
5.15. Preparation of (S)-3-(4-(2-Amino-6-chloropyrimidin-4-yl)phenyl)-2-(tert-
butoxycarbonylamino)propanoic acid (8) using potassium carbonate as
base
To a 500 ml 3-neck round-bottom flask equipped with a mechanical stirrer, a
thermocontroller was charged 2-amino-4,6-dichloropyrimidine (12.57 g, 1.5
equiv), boronate
compound 7 (20.00 g, 51.1 mmol), potassium carbonate (21.19g, 3.0 equiv) and
ethanol/water (200 ml, 5:1 by volume). The mixture was stirred and the
catalyst
bis(triphenylphosphine)palladium(II) dichloride (359 mg, 1 mol%) was added.
The mixture
was heated to 80 C and stirred for 2 hours. The reaction was cooled to room
temperature and
diluted with water (100 ml). The mixture was then concentrated under reduced
pressure to
remove most of ethanol and 1 N NaOH (60 ml) was added. The mixture was
extracted twice
with ethyl acetate (2x200 ml) and the aqueous layer was acidified to pH -3
using 1 N HC1.
33
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
The mixture was extracted with ethyl acetate twice (200 ml and 100 ml,
respectively) and the
combined organic layers were concentrated and the residue was purified by
column
chromatography (gradient 1:20 to 1:10 methanol/dichloromethane) to afford
compound 8 as a
pale yellow solid (15.92 g, 79%).
5.16. Preparation of (S)-3-(4-(2-Amino-6-chloropyrimidin-4-yl)phenyl)-2-(tert-
butoxycarbonylamino)propanoic acid (8) using the lithium salt of (S)-2-
(tent-butoxycarbonylamino)-3-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-
2-yl)phenyl)propanoic acid (7)
0 0
OOH
NHBoc NHBoc
0 N N
8
NH2
During preparation of compound 7, the isolation of the free acid can be
optionally
omitted. Thus, an aqueous solution of the lithium salt of compound 7 in 100 ml
water,
prepared from 5.0 g of Boc-Tyr-OMe (4, 17 mmol), was mixed 2-amino-4,6-
dichloropyrimidine (3.3 g, 1.2 eq), potassium bicarbonate (5.0 g, 3 eq),
bis(triphenylphosphine)palladium(II) dichloride (60 mg, 0.5 mol%), and 100 ml
ethanol. The
resulting mixture was heated at 70 C for 5 hours. Additional 2-amino-4,6-
dichloropyrimidine (1.1 g, 0.4 eq) was added and heating was continued at 70 C
for 2 hours
more. HPLC analysis showed about 94% conversion. Upon cooling and filtration,
the
filtrate was analyzed by HPLC against a standard solution of compound 8. The
assay
indicated 3.9 g compound 8 was contained in the solution (59% yield from
compound 4).
5.17. Alternative procedure for preparation of (S)-3-(4-(2-Amino-6-
chloropyrimidin-4-yl)phenyl)-2-(tent-butoxycarbonylamino)propanoic
acid (8) using potassium carbonate as base
O O O
OH OH OH
HOB NH2 HOB I / NHBoc CI NHBoc
OH OH N N
11 12 8
NH2
The boronic acid compound 11 (Ryscor Science, Inc., North Carolina, 1.0 g, 4.8
mmol) and potassium carbonate (1.32 g, 2 eq) were mixed in aqueous ethanol (15
ml ethanol
and 8 ml water). Di-tert-butyldicarbonate (1.25 g, 1.2 eq) was added in one
portion. After 30
34
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
minutes agitation at room temperature, HPLC analysis showed complete
consumption of the
starting compound 11. The 2-amino-4,6-dichloropyrimidine (1.18 g, 1.5 eq) and
the catalyst
bis(triphenylphosphine)palladium(II) dichloride (34 mg, 1 mol%) were added and
the
resulting mixture was heated at 65-70 C for 3 hours. HPLC analysis showed
complete
consumption of compound 12. After concentration and filtration, HPLC analysis
of the
resulting aqueous solution against a standard solution of compound 8 showed
1.26 g
compound 8 (67% yield).
5.18. Alternative procedure for preparation of (S)-3-(4-(2-Amino-6-
chloropyrimidin-4-yl)phenyl)-2-(tent-butoxycarbonylamino)propanoic
acid (8) using potassium carbonate/potassium bicarbonate as base
O O O
OH OH OH
HOB 01!!: NH2 HOB NHBoc CI I / NHBoc
I
OH OH N N
11 12 8
NH2
The boronic acid compound 11 (10 g, 48 mmol) and potassium bicarbonate (14.4
g, 3
eq) were mixed in aqueous ethanol (250 ml ethanol and 50 ml water). Di-tert-
butyldicarbonate (12.5 g, 1.2 eq) was added in one portion. HPLC analysis
indicated that the
reaction was not complete after overnight stirring at room temperature
Potassium carbonate
(6.6 g, 1.0 eq) and additional di-tert-butyldicarbonate (3.1 g, 0.3 eq) were
added. After 2.5
hours agitation at room temperature, HPLC analysis showed complete consumption
of the
starting compound 11. The 2-amino-4,6-dichloropyrimidine (11.8 g, 1.5 eq) and
the catalyst
bis(triphenylphosphine)-palladium(II) dichloride (0.34 g, 1 mol%) were added
and the
resulting mixture was heated at 75-80 C for 2 hours. HPLC analysis showed
complete
consumption of compound 12. The mixture was concentrated under reduced
pressure and
filtered. The filtrate was washed with ethyl acetate (200 ml) and diluted with
3:1
THF/MTBE (120 ml). This mixture was acidified to pH about 2.4 by 6 N
hydrochloric acid.
The organic layer was washed with brine and concentrated under reduced
pressure. The
residue was precipitated in isopropanol, filtered, and dried at 50 C under
vacuum to give
compound 8 as an off-white solid (9.0 g, 48% yield). Purity: 92.9% by HPLC
analysis.
Concentration of the mother liquor yielded and additional 2.2 g off-white
powder (12%
yield). Purity: 93.6% by HPLC analysis.
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
5.19. (S)-3-(4-(2-Amino-6-((R)-2,2,2-trifluoro-l-(3'-methoxybiphenyl-4-
yl)ethoxy)pyrimidin-4-yl)phenyl)-2-(tent-butoxycarbonylamino)propanoic
acid
CF3 O O
CF3 OH
OH I OH
+ CI / NHBoc O NHBoc
N N N`/N
r
OMe 3 NH2 8 NHz 9
OMe
To a 250 ml 3-neck round-bottom flask equipped with a mechanical stirrer, a
thermocontroller was charged monochloride 8 (20.39 g, 51.9 mmol), alcohol 3
(17.58 g, 1.2
equiv), cesium carbonate (84.55, 5.0 equiv) and dioxane (205 ml). The mixture
was heated to
100 C and stirred for 17 hours. The reaction was cooled to room temperature
and diluted
with water (80 ml). Two phases were split and the organic layer was collected
and diluted
with ethyl acetate (200 ml), washed with a mixture of brine (50 ml) and 1 N
HC1(50 ml).
The organic layer was concentrated and the residue was purified by column
chromatography
(gradient: 1:30 to 1:20 methanol/dichloromethane and 0.5% acetic acid) to
afford compound
9 as a yellow solid. This solid was recrystallized from EtOH and heptane to
give 21.78 g pale
yellow solid. Further crystallization of the mother liquor gave 2.00 g pale
yellow solid
(overall 23.78 g, 72% yield). Chiral analysis of the corresponding methyl
ester derivative,
prepared using trimethylsilyldiazomethane, showed no detectable amount of the
diastereomers. LC-MS (ESI): MH+ = 639.2. 1H NMR (DMSO-d6) 6 12.60 (br s, 1H),
8.00 (d,
J= 8.0 Hz, 2H), 7.77 (d, J= 8.0 Hz, 2H), 7.67 (d, J= 8.0 Hz, 2H), 7.37 (m,
3H), 7.21 (m,
2H), 7.13 (d, J= 8.0 Hz, 1H), 6.96 (m, 1H), 6.84 (m, 2H), 6.75 (s, 2H, 4.15
(m, 1H), 3.82 (s,
3H), 3.10 (dd, J= 13.6, 4.4 Hz, 1H), 2.89 (dd, J= 13.6, 10.4 Hz, 1H), 1.32 (s,
9H). 13C NMR
(DMSO-d6) 6 173.4, 168.4, 166.1, 162.9, 159.7, 155.4, 141.5, 140.8, 134.8,
130.7, 130.0,
129.3, 128.4, 127.2, 126.6, 124.1 (q, J= 281 Hz), 119.1, 113.4, 112.3, 91.3,
78.0, 71.3 (q, J=
Hz), 55.1, 54.9, 36.2, 28.1. 19F NMR (DMSO-d6): 6 -74.6 (d, J= 7.2 Hz). Anal.
Calcd.
for C33H33F3N406: C, 62.06; hours, 5.21; N, 8.77. Found: C, 62.25; hours,
5.10; N, 8.69.
25 5.20. Preparation of (S)-3-(4-(2-Amino-6-((R)-2,2,2-trifluoro-l-(3'-
methoxybiphenyl-4-yl)ethoxy)pyrimidin-4-yl)phenyl)-2-(tert-
butoxycarbonylamino)propanoic acid under various conditions
Using similar procedure, various reaction conditions were examined. The
results are
listed in the table below.
36
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
entry equiv of 3 base (x, equiv) additive time (h) % conversion
(isolated yield a
1 1.2 Cs2CO3 (5.0) - 17 97 (72)
2 1.2 NaH (5.0) - 1 -b
3 1.2 NaOt-Bu (3.0) - 1
4 1.2 NaOt-Am (3.0) - 1
1.2 DBU (5.0) - 24 0
6 1.2 tetramethyl- - 24 0
guanidine (5.0)
7 1.2 K2C03 (5.0) - 24 0
8 1.2 Cs2CO3 (4.0) - 20 98d
9 1.2 Cs2CO3 (4.0) 10 mol% 17 98d
n-Bu4NHSO4
1.2 Cs2CO3 (3.0) 10 mol% 18 98d
n-Bu4NHSO4
11 1.0 Cs2CO3 (3.0) 10 mol% 18 88d
n-Bu4NHSO4
a All the reactions were run in l Ox dioxane except otherwise noted; b The
starting
material 1 decomposed; 'A complex mixture of starting material, deBoc of
starting material,
product, deBoc of product was observed. d The reaction was run in 5x dioxane.
5 5.21. Alternate Preparation of (S)-3-(4-(2-Amino-6-((R)-2,2,2-trifluoro-l-
(3'-
methoxybiphenyl-4-yl)ethoxy)pyrimidin-4-yl)phenyl)-2-(tert-
butoxycarbonylamino)propanoic acid
CI \ CI
MeO + NYN -
\ OH
NHZ
CF3
0
OH
O,B I / NHBoc
O
Me0 \ / I O 7 Me0 \ / I / I OH
\ O` ^ /CI \ O I \ \ NHBoc
CF3 ~NY,N CF3 NYN
NHZ INHZ
13 9
37
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
A suspension of (R)-2,2,2-trifluoro-l-(3'-methoxybiphenyl-4-yl)ethanol (30 g,
0.106
mol), dichloropyrimidine (34.8 g, 0.212 mol) and cesium carbonate (34.6 g,
0.106 mol) in
1,4-dioxane (300 ml, l OX) was heated to 100 C with good stirring. After
stirred for 4 hours
at 100 C, cesium carbonate (17.3 g, 0.053 mol) was added and further stirred
for 14 hours at
100 C. Cooled to 50 C, water (90 mL, 3X) was added and stirred for 30 minutes
at room
temperature. The organic layer was concentrated to a 5X solution and solid was
removed by
polish filtration. After diluted with toluene (300 mL, l OX) and concentrated
to a 5X solution
and heptane (150 mL, 5X) was added. After stirred for 2 hours at room
temperature,
removed solid by filtration. 1,4-Dioxane was added and concentrated to prepare
a solution of
monochloride 13 in 1,4-dioxane.
To a 15 X solution of 13 (ideal: 0.106 mol) in 1,4-dioxane was added boronic
ester 7
(62.25 g, 0.159 mol), potassium bicarbonate (37.2 g, 0.372 mol) and water (90
mL, 3X) at
room temperature. After degassing (three vacuum/nitrogen refill cycles),
PdC12(PPh3)2 (372
mg, 0.529 mmol) and triphenylphosphine (72 mg, 0.275 mmol) were added. The
reaction
mixture was then stirred for 8 hours at 90 C, cooled to room temperature and
then acidified
with 2 N HCl to pH 3-4. After stirred for 30 minutes at room temperature, the
organic layer
was treated with activated carbon at 50 C for 2 hours. After filtered though a
tight packed
celite, the solution was then concentrated to a 3X solution at reduced
pressure (50 mbar,
40 C). CH3CN (20X) was added and concentrated to -10X at reduced pressure (100
mbar,
40 C) to gave a suspension. It was filtered and the filter cake was washed
with CH3CN
(l OX). The solid was then dried under vacuum at 40 C to obtain 20.1 g of
desired Boc acid 9
as a white solid in 90% overall yield corrected for 98 wt% purity.
5.22. (S)-2-Amino-3-(4-(2-amino-6-((R)-2,2,2-trifluoro-l-(3'-methoxybiphenyl-
4-yl)ethoxy)pyrimidin-4-yl)phenyl)propanoic acid (10)
O O
CF3 OH CF3 OH
O NH2
NHBoc I H2
PIC, NYN PI N
NH2 NH2
OMe 9 OMe 10
To a 500 ml round-bottom flask was added compound 9 (20.00 g, 31.32 mmol) and
THE (100 ml). The solid was dissolved upon stirring and 6 N hydrochloric acid
(100 ml) was
added slowly. The mixture was then stirred at room temperature for 14 hours.
The reaction
was diluted with water (100 ml) and most of THE was removed under reduce
pressure. The
38
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
resulting aqueous solution was then transferred to a 500 ml three-necked round-
bottom flask
equipped with a mechanical stirrer, a pH meter, a thermocontroller and an
addition funnel.
At 60 C, a solution of 50% aqueous sodium hydroxide was added slowly until pH
= 4, then a
solution of 1 N aqueous sodium hydroxide was added until pH reached 6.5. The
mixture was
stirred at 60 C for additional 30 minutes and the solid was collected by
filtration and oven-
dried under vacuum to give compound 10 (16.30 g, 96% yield) as a pale yellow
solid. LC-
MS (ESI): MH+ = 539.1. 1H NMR (DMSO-d6) 6 8.01 (d, J= 8.0 Hz, 2H), 7.76 (d, J=
8.0 Hz,
2H), 7.67 (d, J= 8.0 Hz, 2H), 7.38 (m, 3H), 7.23 (m, 2H), 6.96 (d, J= 8.0 Hz,
1H), 6.81 (m,
3H), 3.81 (s, 3H), 3.59 (br m, 1H), 3.00 (br m, 1H). 13C NMR (DMSO-d6) 169.9,
168.4,
166.1, 162.9, 159.7, 141.5, 140.8, 140.8, 140.0, 134.9, 130.7, 130.0, 129.7,
128.4, 127.2,
126.8, 124.1 (q, J= 281 Hz), 119.1, 113.4, 112.3, 91.2, 71.4 (q, J= 30 Hz),
55.1, 55.0, 36.5.
19F NMR (DMSO-d6): 6 -74.6 (d, J= 6.8 Hz).
5.23. One-pot preparation of (S)-2-Amino-3-(4-(2-amino-6-((R)-2,2,2-trifluoro-
1-(3'-methoxybiphenyl-4-yl)ethoxy)pyrimidin-4-yl)phenyl)propanoic acid
CF3 0
"'OH OH
\ + CII\ NHBoc
N,,/N
OMe 3 NH2 8
O O
CF3 OH CF3 OH
NHBoc O \ I / NH2
NYN NYN
NH2 NH2
OMe OMe
9 10
To a 3-neck 250 ml round-bottom flask equipped with a mechanical stirrer, a
thermocontroller, was charged compound 3 (8.62 g, 1.2 equiv), 8 (10.00 g,
25.46 mmol),
tetrabutylammonium bisulfate (0.86 g, 10 mol%), and cesium carbonate (29.04 g,
3.5 equiv).
Dioxane (50 ml) was added and the resulting mixture was heated at 100 C for 18
hours.
HPLC analysis showed 99% conversion of the starting material 8. The mixture
was cooled
down to 60 C and water (60 ml) was added. The top organic layer was diluted
with THE (80
ml), washed with brine (50 ml), transferred to a 500 ml round-bottom flask,
and 80 ml of 6 N
hydrochloric acid was added. The mixture was stirred at room temperature for
16 hours. LC-
MS analysis of the reaction mixture showed complete consumption of the
intermediate
39
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
compound 9. The reaction mixture was transferred to a 500 ml separatory
funnel. The
round-bottom flask was washed with water (2 x 40 ml) and the washes were also
transferred
to the funnel. The mixture was washed with ethyl acetate (2 x 100 ml) and the
aqueous layer
was collected and concentrated at 40 C (bath temperature) under 80 mbar vacuum
to remove
any remaining organic solvents. The resulting aqueous solution was then
transferred to a 500
ml three-necked round-bottom flask equipped with a mechanical stirrer, a pH
meter, a
thermocontroller and an addition funnel. At 60 C, a solution of 50% aqueous
sodium
hydroxide solution was added slowly until pH = 4, then a solution of IN
aqueous sodium
hydroxide was added until pH reached 6.5. The mixture was stirred at 60 C for
additional 30
minutes and the yellow solids were collected by filtration. HPLC analysis of
this solid
showed a purity of about 95%. The solids were dried under vacuum at 50 C
overnight to give
the crude product compound 10 as a yellow solid (9.48 g, 69% overall yield).
The above solids (9.48 g) were transferred to a 500 ml round-bottom flask and
water
(95 ml) was added. The mixture was heated at 80 C (bath temperature) and THE
(40 ml) was
added dissolve the solids. Most of THE was then removed under vacuum at 80 C.
The
precipitate was added acetonitrile (80 ml) and was stirred at 80 C for 2
hours, cooled down to
room temperature and then stirred at 0 C for 30 minutes. The solid was
collected by
filtration, washed with water (2 x 50 ml) to give compound 10 as a pale yellow
solid (8.53 g,
90% recovery, 62% overall yield). HPLC analysis showed a purity greater than
99%.
5.24. One-pot preparation of (S)-2-Amino-3-(4-(2-amino-6-((R)-2,2,2-trifluoro-
1-(3'-methoxybiphenyl-4-yl)ethoxy)pyrimidin-4-yl)phenyl)propanoic acid
A 22-L, round-bottom flask equipped with a mechanical stirrer, a thermocouple
attached to a temperature controller, and a condenser with a nitrogen line was
charged with
1,4-dioxane (4 vol) followed by the addition of Cs2CO3 (2.03 kg, 3.5 equiv),
compound 3
(603 g, 1.2 equiv) and tetrabutylammonium bisulfate (102.8 g, 0.147 wt). The
slurry was
slowly heated to 70 C and then a slurry of compound 8 (700.0 g, 1.782 mol, 1
wt) in 1,4-
dioxane (1.5 vol) was added in three portions over 10 minutes. The beaker
containing 8 was
rinsed with 1,4-dioxane (0.5 vol) and added to the reactor. The reaction
became thick briefly
after stirring for 15-30 minutes but the entire batch was stirrable. The
controller was heated
at 78 C overnight followed by heating at 98 C for 8 hours then 85 C overnight.
HPLC
analysis indicated that there were 2.1 % of 8 remaining. The reaction was
quenched at 78 C
with water (6 vol) and then cooled further. At 42 C, the batch was transferred
to a separatory
funnel and the two phases separated. The organic phase was then diluted with
THE (8 vol)
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
and washed with brine (5 vol). The phases were separated and the organic phase
was washed
with brine (5 vol). The phases were separated and the organic phase (9.5 L)
was transferred
to a 22-L reactor. A solution of 6 N HC1(11.4 vol) was added and the batch was
heated at
40-45 C for 2 hours. HPLC analysis indicated that the reaction was complete
and Darco G-
60 (0.33 wt.) and water (2 vol) were added. The batch was stirred at 40 C over
the weekend
and then heated to 60 C. The reaction mixture was filtered at 60 C through
PTFE cloth and
the reactor was rinsed with water (6 vol). The rinse was heated to 60 C and
washed through
the Darco pad. The filtrate was then passed through a 0.3- m in-line filter
and washed with
IPAc twice (10 vol, 8.8 vol). The aqueous phase was then concentrated under
reduced
pressure at 45 C using a 20-L, rotary evaporator until the mixture turned
cloudy (2-3 h). The
volume of distillate collected was approximately 3.3 L. The batch was then
transferred back
to a 22-L reactor and held at 40 C overnight.
The batch was heated to 60 C whereupon the batch turned from cloudy to clear.
To a
separate 22-L reactor was charged water (1.6 vol) and 85% phosphoric acid
(0.24 vol) and
the pH was adjusted to 6.5 using a 50% NaOH solution (approximately 0.3 vol).
The acidic
product solution was then transferred via peristaltic pump to the reactor
containing the pH 6.5
buffered solution and the pH was maintained within 6 and 7 through the
addition of 50%
NaOH (approximately 3.5 vol). The temperature of the reactor was maintained
between 55
and 65 C (2-h addition time). Once the addition was complete, the slurry was
heated at 60-
65 C for 90 minutes, filtered, and washed with water (2 x 6.7 vol). The wet
cake was dried
in a vacuum oven at 55 C for 39 hours to afford 635 g of crude 10 as a yellow
solid (66%
yield). The purity of the product was 93.2% (AUC).
5.25. Purification of (S)-2-Amino-3-(4-(2-amino-6-((R)-2,2,2-trifluoro-l-(3'-
methoxybiphenyl-4-yl)ethoxy)pyrimidin-4-yl)phenyl)propanoic acid
A 22-L, round-bottom flask equipped with a mechanical stirrer, a thermocouple
attached to a temperature controller, and a condenser with a nitrogen line was
charged with
crude 10 (630 g) followed by the addition of THE (5 vol). The slurry was
heated to 65 C.
After 30 minutes, a solution of 5-6 N HCl in IPA (0.47 L, 0.746 vol) was added
and the
solids slowly dissolved. The orange solution was heated at 65 C for 30 minutes
IPA (10 vol)
was slowly added maintaining the temperature between 60-70 C. Once the
addition was
complete, the mixture was stirred for 20 minutes and then IPAc (10 vol) was
slowly added
maintaining the temperature between 60-70 C. Once the addition was complete,
the thick
slurry was stirred at 65 C for 1 hours and then cooled to 27 C over 4.5 hours.
The solids
41
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
were filtered and washed with IPA (2 x 3 vol). The product was dried in a
vacuum oven at
55 C for 15 hours to afford 630 g of 10 diHCl salt (88% yield) with a purity
of 95.0% (AUC).
A 12-L, round-bottom flask equipped with a mechanical stirrer, a thermocouple
attached to a temperature controller, and a pH probe attached to a pH meter
was charged with
10 diHCl salt (620 g, 1 wt) followed by an aqueous solution of 1 M NaOH (10
vol). The
mixture was heated to 40 C, stirred until all the solids dissolved (2 h), and
then transferred to
a 10-L carboy. The 12-L, round-bottom flask was washed with water and then 85%
phosphoric acid (124 ml, 0.2 vol) and water (1.3 vol) were charged to the
reactor. The pH
was adjusted to 6.5 using 50% NaOH (0.24 vol) and then heated to 65 C. The
product
solution in the carboy was transferred via peristaltic pump to the pH buffered
solution and the
pH was maintained between 6 and 7 through the addition of an aqueous solution
of 6 M HCl
(0.67 L). Once the addition was complete, the slurry was heated at 65 C for 3
hours and the
solids were filtered. The cake was washed with water (3 x 5 vol) and then
dried in a vacuum
oven at 55 C for 41 hours to afford 473 g of 10 as a light yellow solid (87%
yield) with a
purity of 97.7% (AUC).
5.26. Alternate Preparation of (S)-3-(4-(2-Amino-6-((R)-2,2,2-trifluoro-l-(3'-
methoxybiphenyl-4-yl)ethoxy)pyrimidin-4-yl)phenyl)-2-(tert-
butoxycarbonylamino)propanoic acid
O ~I O
OH MeO \ OH
MeO OH + CI \ I NHBoc \ I O \ NHBoc
I
N~"~ N CF3 N`/ N
CF3 ~"
NH2 NH2
8 9
To a 2-L 3-neck round bottom flask was charged (R)-2,2,2-trifluoro-1-(3'-
methoxybiphenyl-4-yl)ethanol (70.8 g, 251 mmol) and cesium carbonate (milled,
238 g, 730
mmol) followed by 1,4-dioxane (500 mL, 5 X) at 20-25 C. The mixture was heated
to
100 C and a slurry of monochloride 8 (82 w%, 100 g, 209 mmol) in 1,4-dioxane
(250 mL,
2.5 X) was added over 30 minutes at 90-100 C. The reaction mixture was then
stirred at
100 C for 20 hours. The mixture was cooled to 90 C, water (750 mL, 7.5 X) was
added and
the mixture was allowed to cool to room temperature. A solution of di-tent-
butyl dicarbonate
(6.84 g, 31 mmol) in 1,4-dioxane (10 mL) was added and the mixture was aged
for 2 hours.
Toluene (450 mL, 4.5 X) was added and the mixture was stirred, settled. The
aqueous layer
was split off and the organic layer was acidified with 2 N HC1(150 mL, 1.5 X)
to pH = 3-4
at 20-25 C. The organic layer was washed with water (100 mL) and concentrated
to
42
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
200-300 mL (50 mbar, 40 C). It was then flushed with CH3CN (1500 mL, 15 X)
(150 mbar,
45 C) to give a suspension (1000-1200 mL). The mixture was aged at 20-25 C,
filtered,
washed with CH3CN (500 mL, 5 X), dried in a vacuum oven at 40 C with slow
nitrogen
sweep to give the desired Boc-acid (114.6 g, 95.2 A%, 92.5 w%, KF: 0.24%, Pd:
<1 ppm,
79% yield (after purity correction) as an off-white solid.
Scale-up Procedure
To a 20 L reactor equipped with a mechanical stirrer, a temperature controller
and a
nitrogen inlet was charged (R)-2,2,2-trifluoro-1-(3'-methoxybiphenyl-4-
yl)ethanol (343 g,
1.22 mol) and cesium carbonate (milled, 1.15 kg, 3.54 mol) followed by 1,4-
dioxane (2.5 L,
5 X) at 20-25 C. The mixture was heated to 100 C and a suspension of 8 (79.4
w%, 500 g
as-is, 397 g pure, 1.04 mol) in 1,4-dioxane (1.25 L, 2.5 X) was added over 30
minutes at
90-100 C. The reaction mixture was then stirred at 100 C for 22 hours. The
mixture was
cooled to 90 C, water (3.75 L, 7.5 X) was added, the mixture was allowed to
cool to 30 C,
and a solution of di-tent-butyl dicarbonate (33.1 g, 0.15 mol) in 1,4-dioxane
(50 mL, 0.1 X)
was added. After 15 hours agitation at room temperature, toluene (2.25 L, 4.5
X) was added,
stirred for 30 minutes, settled, and the aqueous layer was split off. The
organic layer was
then acidified with 2 N HO (0.78 L, ca. 1.5 X) at 20-25 C to 3.1. The organic
layer was
washed with water (0.50 L, 1 X) and concentrated to a 2-3 X solution (50 mbar,
40 C). It
was then flushed with CH3CN (8 L, 16 X) (100 mbar, 50 C) to give a suspension
(10-12 X).
After stirred at 20-25 C for a few hours, the mixture was filtered and the
filter cake was
washed with CH3CN (3.0 L, 6 X). The solid was then dried in a vacuum oven at
45 C with
slow nitrogen sweep to afford the desired Boc-acid (566 g, 95.6 A%, 94.9 w%,
KF: 0.19%,
83% yield corrected for purity) as a slightly pinkish solid.
5.27. Alternate Preparation of (S)-2-amino-3-(4-(2-amino-6-((R)-2,2,2-
trifluoro-
1-(3'-methoxybiphenyl-4-yl)ethoxy)pyrimidin-4-yl)phenyl)propanoate
De-Boc with HC1
6 N HC1(118 mL, 708 mmol) was added to a slurry of the Boc-acid (150.5 g, -92
w%, 138.5 g active, 217 mmol) in THE (450 mL). The mixture was heated to 55 C
over 1
hour and aged at this temperature for 5 hours. It was cooled to 40 C,
neutralized with 10 N
NaOH (68.5 mL) to pH 6.7. The aqueous layer was split off (132 mL). To the
organic layer
(615 mL) was added water (325 mL) and the mixture was seeded with LX1031-THF
solvate
(1.36 g). The mixture was aged at 30 C overnight and more water (827 mL) was
added over
2 hours. The mixture was aged at 30 C for 2 hours, slowly cooled to 20 C and
aged
43
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
overnight. It was filtered and then washed with 2.5/1 water/THF (525 mL). Loss
in the
mother liquor and wash was 4.5%. The filter cake was pressed with spatula
periodically until
no more liquid came out to give 244 g of wet cake. The cake was loosened and
then dried
under flowing dry nitrogen overnight to give 122.96 g of the title compound as
an off-white
solid (92% yield, KF = 0.5%). HPLC indicated 95.8 A% and 87.2 w% purity. 1H
NMR
showed that it was a partial THF solvate (38 mol%, -5 w% THF).
De-Boc with H2SO4
H2SO4 (5.0 M, 68.5 mL, 2.0 eq) was added to a solution of the Boc-acid (107.3
g, 94
A%, 92.5 w%, 155 mmol corrected for purity) in THF (321 mL). The mixture was
heated to
60 C over 15 minutes and then aged at this temperature for 6 hours. It was
neutralized with
2.0 N NaOH until pH reached 6.6 (336.5 mL NaOH).
The aqueous layer (300 mL) contained no product and was discarded. Water (165
mL) was added to the organic layer and then seeded with (S)-2-amino-3-(4-(2-
amino-6-((R)-
2,2,2-trifluoro-l-(3'-methoxybiphenyl-4-yl)ethoxy)pyrimidin-4-
yl)phenyl)propanoate (0.9 g
THF solvate and 1.5 g non-solvate). It slowly became a thick slurry. The
mixture was aged
at 40 C for 2 hours and more water (377 mL) was added slowly over 3 hours at
40 C. It was
aged at 40 C for 3 hours then slowly cooled to 20 C over 2 hours and stirred
for 40 hours.
The slurry was filtered (25 minutes) and the filter cake was washed with 2.3/1
water/THF
(430 mL) (30 minutes). Yield loss to mother liquor was 2.5%. The filter cake
was then
pressed with spatula repeatedly until the cake could be loosened readily to
give 211 g wet
solid. It was air dried for 2 days to give 83.7 g of crude (S)-2-amino-3-(4-(2-
amino-6-((R)-
2,2,2-trifluoro-l-(3'-methoxybiphenyl-4-yl)ethoxy)pyrimidin-4-
yl)phenyl)propanoate as an
off-white solid (purity: 98.8 A%, 94.2 w%) in 91.6% yield after subtracting
seeds and
corrected for purity. Loss on drying = 60%. KF = 1.4%.
5.28. Preparation of (S)-2-amino-3-(4-(2-amino-6-((R)-2,2,2-trifluoro-l-(3'-
methoxybiphenyl-4-yl)ethoxy)pyrimidin-4-yl)phenyl)propanoate Tosylate
Dihydrate
(S)-2-amino-3-(4-(2-amino-6-((R)-2,2,2-trifluoro- l -(3'-methoxybiphenyl-4-
yl)ethoxv)pyrimidin-4-yl)phenyl)propanoate (120.0 g, 88 w%, 105.6 g active,
196 mmol)
was added to a solution of TsOH=H2O (39.8 g, 209 mmol) in a mixture of THF
(240 mL) and
water (48 mL). The mixture was heated to 50 C to give a homogeneous solution.
Approximately 120 mL of a mixture of ACN/water (1200/60 mL) was added and the
mixture
was seeded with (S)-2-amino-3-(4-(2-amino-6-((R)-2,2,2-trifluoro-1-(3'-
methoxybiphenyl-4-
44
CA 02741563 2011-04-21
WO 2010/047712'CT PCT/US2008/081054
yl)ethoxy)pyrimidin-4-yl)phenyl)propanoate tosylate dihydrate (0.63 g). After
aging for 1
hour at 40 C, a nice slurry was obtained. The remaining ACN/water mixture was
added
slowly over 3 hours at 40 C and the slurry was aged at 40 C for 2 hours then
slowly cooled
to 20 C and aged overnight. The solid was collected by filtration and the
filter cake was
washed with 5/1 ACN/THF with - 5 vol% water (500 mL). Air drying at room
temperature
overnight gave 138.5 g of the product as a white solid (99.5 A%, 93.4% yield
corrected for
purity). Loss in the mother liquor and wash was 6.5%. KF of solid was 4.4%.
All of the publications (e.g., patents and patent applications) disclosed
above are
incorporated herein by reference in their entireties.