Note: Descriptions are shown in the official language in which they were submitted.
CA 02747845 2011-08-05
1
COMPOSITIONS AND METHODS
FOR DERMALLY TREATING PAIN
This application is a divisional of Canadian patent application Serial No.
2,633,515 filed internationally on December 14, 2006 and entered nationally on
June 13, 2008.
FIELD OF THE INVENTION
The present invention relates generally to formulations for topical delivery
of
various therapeutic agents including antiviral and anti-pain agents, as well
as
placebo formulations, and uses for such formulations. More particularly, the
present invention relates to adhesive formulations having a viscosity suitable
for
application to a skin surface and which form a transdermal drug-delivering
solidified layer on the skin.
BACKGROUND OF THE INVENTION
Pain can be caused by a variety of sources. For example, neuropathic
pain can be caused by diseases such as viral infections and diabetes. For
example, post herpetic neuralgia is caused by herpes viral infection and
typically
causes moderate to severe pain in the Infected skin area to the subject.
Topical
products, such as creams or patches containing appropriate drugs, may be
used to control neuropathic pain; however, patches and traditional semisolid
formulations such as creams and ointments both have significant shortcomings.
Semisolid formulations usually contain solvent(s), such as water and ethanol,
which are volatile and thus evaporate shortly after application. The
evaporation
of such solvents can cause significant decrease or even termination of dermal
drug delivery, which can be undesirable in many cases. Additionally, semisolid
formulations are often "rubbed into" the -skin, which does not necessarily
mean
the drug formulation is actually delivered into the skin- Instead, this phrase
often means that a very thin layer of the drug formulation is applied onto the
surface of the skin. Such thin layers of traditional semisolid formulations
applied
CA 02747845 2011-08-05
2
to the skin may not contain sufficient quantity of the active drug to achieve
sustained delivery over long periods of time, which can be desirable in
treating
neuropathic pain. Additionally, traditional semisolid formulations are often
subject to unintentional removal due to contact with objects such as clothing,
which may compromise the sustained delivery and/or undesirably soil clothing.
The musculoskeletal system is also a common source of pain. However,
as with neuropathic paint, current topical dosage forms for those drugs are
not
typically adequate for this application. For example, semisolid NSAID. and
local
anesthetic formulations, such as creams and gels, usually contain solvent(s),
such as water and ethanol, which are volatile and thus evaporate shortly after
application. The evaporation of such solvents can cause significant decrease
or
even termination of topical drug absorption
A patch containing an appropriate drug can be used to treat neuropathic
or musculoskeletal pain. However, subjects often have to cut the patch to fit
the
shape and size of the skin area to be treated, which is inconvenient. Another
shortcoming of patches is that they are usually neither sufficiently
stretchable
nor flexible for every application location. If the patch is applied on a skin
area
that is significantly stretched during body movements, such as joints and
muscles, separation between the patch and skin may occur, thereby
compromising the delivery of the drug. In addition, a patch on a skin surface
may hinder the expansion of the skin during body movements and cause
discomfort and/or aggravate pain. For these additional reasons, patches are
not ideal dosage forms for skin areas subject to expansion and stretching
during
body movements.
With regard to liquid reservoir patches, even when a drug is compatible
with a particular liquid or semisolid solvent system carried by the thin bag
of the
patch, the solvent system still has to be compatible to the adhesive layer
coated
on the permeable or the semi-permeable membrane otherwise the drug may be
adversely affected by the adhesive layer or the drug/solvent system may reduce
the tackiness of the-adhesive layer. In addition to these dosage form
considerations, reservoir patches are usually more expensive to manufacture
than matrix patches.
CA 02747845 2011-08-05
3
Another shortcoming of dermal (including iransdermal) patches Is that
they are usually not stretchable or flexible, as the backing film (in matrix
patches) and the thin fluid bag (in reservoir patches) are typically made of
polyethylene or polyester, both of which are relatively non-stretchable
materials.
if the patch Is applied on a skin area that is significantly stretched during
body
movements, such as a joint, separation between the patch and skin may occur,
thereby compromising the delivery of the drug. In addition, a patch present on
a
skin surface may hinder the expansion of the skin during body movements and
cause discomfort. For these additional reasons, patches are not ideal dosage
forms for skin areas over muscle and joints that are subject to expansion and
stretch during body movements.
In view of the shortcomings of the current delivery systems, it would be
desirable to provide systems and/or methods that I) can provide more sustained
delivery of pain relieving drugs such as NSAIDs, local anesthetics, or certain
steroids over long periods of time; ii) are not vulnerable to unintentional
removal
by contact with clothing, other objects, or people for the duration of the
application time; iii) can be applied to a.skin area subject to stretch and
expansion without causing discomfort or poor contact to skin; and/or iv) can
be
conveniently removed after application and use.
SUMMARY OF THE INVENTION
It has been recognized that it would be advantageous to treat
neuropathic and/or musculoskeletal pain by providing topical delivery of drugs
from certain classes, e.g., NSAID, local anesthetic, or steroid formulations,
etc,
in the form of adhesive solidifying formulations having a viscosity suitable
for
application to a skin surface as a layer and which form a drug-delivering
solidified adhesive layer on the skin. In one embodiment, a formulation for
treating musculoskeletal or neuropathic pain can comprise a drug suitable for
treating muscuioskeletal or neuropathic pain, a solvent vehicle, and a
solidifying
agent. The solvent vehicle can comprise a volatile solvent system comprising
at
least one volatile solvent, and a non-volatile solvent system comprising at
least
CA 02747845 2011-08-05
4
one non-volatile solvent, wherein the non-volatile solvent system Is capable
of
facilitating transdermal delivery of the drug at a therapeutically effective
rate
over a sustained period of time. The formulation can have a viscosity suitable
for application and adhesion to a skin surface prior to evaporation of the
volatile
solvent system, and further, the formulation applied to the skin surface can
form
a solidified layer after at least partial evaporation of the volatile solvent
system.
The drug can continue to be delivered at the therapeutically effective rate to
treat musculoskeletal pain or inflammation after the volatile solvent system
is at
least-substantially evaporated.
in another embodiment, a method of dermally delivering a drug for
treating musculoskeletal or neuropathic pain can comprise applying a
formulation to a skin surface. The formulation can be a formulation as
described in the previous embodiment.- Additional steps include solidifying
the
formulation to form a solidified layer on the skin surface by at least partial
evaporation of the volatile solvent system; and dermally delivering the drug
from
the solidified layer to the skin surface at therapeutically effective rates
for
treating the pain or inflammation of joints or muscles over a sustained period
of
time.
In another embodiment, a solidified layer for treating pain can comprise a
drug effective for treating musculoskeletal or neuropathic'pain, a non-
volatile
solvent system, and a solidifying agent. The non-volatile solvent system can
include at least one non-volatile solvent, wherein the non-volatile solvent
system
is capable of facilitating the delivery of the drug at therapeutically
effective rates
over a sustained period of time. Additionally, the solidified layer preferably
can
be stretchable by 5% in at least one direction without cracking, breaking,
and/or
separating from a skin surface to which the layer is applied.
Additional features and advantages of the Invention will be apparent from
the following detailed description and figures which illustrate, by way of
example, features of the invention.
The methods described herein of delivering a drug also encompass uses of
said drug for the purposes described herein.
CA 02747845 2011-08-05
4a
According to another aspect, the invention relates to a pharmaceutical
formulation for
topical delivery of an antiviral agent, comprising:
a) a drug comprising an antiviral agent;
b) a solvent vehicle, comprising:
i) a volatile solvent system including at least one volatile solvent, and
ii) a non-volatile solvent system including at least one non-volatile
solvent,
wherein the volatile solvent system comprises one or more solvents consisting
of
water or a solvent more volatile than water; and
c) a solidifying agent.
The above-described pharmaceutical formulation has a viscosity suitable for
application and adhesion to a skin surface as a layer prior to evaporation of
the volatile
solvent system, the layer applied to the skin surface forms a solidified layer
after at least
partial evaporation of the volatile solvent system, and the drug continues to
be dermally
delivered at a therapeutically effective rate after the volatile solvent
system is evaporated,
wherein the solidified layer, upon formation, is a soft, coherent solid that
can be peeled from
a skin surface as a single piece or as only a few large pieces relative to the
application size.
According to another aspect, the invention relates to a solidified layer for
topical
delivery of an antiviral agent, comprising:
a) a drug comprising an antiviral agent,
b) a non-volatile solvent system including at least one non-volatile solvent,
and
c) a solidifying agent.
The above-described solidified layer is capable of adhering to a skin surface
and
delivering the drug across the skin surface at therapeutically effective rates
over a sustained
period of time. The solidified layer is a soft, coherent solid that can be
peeled from a skin
surface as a single piece or as only a few large pieces relative to the
application size.
According to another aspect, the invention relates to a placebo formulation
for dermal
application, comprising:
a) a solvent vehicle, comprising:
i) a volatile solvent system including at least one volatile solvent, and
ii) a non-volatile solvent system including at least one non-volatile
solvent,
CA 02747845 2011-08-05
4b
wherein the volatile solvent system comprises one or more solvents consisting
of
water or a solvent more volatile than water, and
b) a solidifying agent.
The above-described placebo formulation has a viscosity suitable for
application and
adhesion to a skin surface as a layer prior to evaporation of the volatile
solvent system, the
layer applied to the skin surface forms a solidified layer after at least
partial evaporation of
the volatile solvent system, wherein the solidified layer, upon formation, is
a soft, coherent
solid that can be peeled from a skin surface as a single piece or as only a
few large pieces
relative to the application size. The invention further relates to a
solidified layer of a placebo
formulation comprising (a) and (b) as described above, wherein the solidified
layer is
capable of adhering to a skin surface, and is a soft, coherent solid that can
be peeled from a
skin surface as a single piece or as only a few large pieces relative to the
application size.
CA 02747845 2011-08-05
BRIEF DESCRIPTION OF THE DRAWINGS
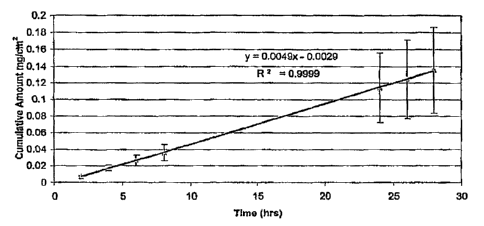
FIG. 1 is a graphical representation of the cumulative amount of
diclofenac delivered transdermally across human cadaver skin over time from a
5 formulation in accordance with embodiments of the present invention where
steady-state delivery is shown over 28 hours; and
FIG. 2 is a graphical representation of the cumulative amount of
ropivacaine delivered transdermally across human cadaver skin over time from
a formulation with similar composition in accordance with embodiments of the
present invention, where steady-state delivery is shown over 30 hours.
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENT(S)
Before particular embodiments of the present invention are disclosed and
described, it is to be understood that this invention is not limited to the
particular
process and materials disclosed herein as such may vary to some degree. It is
also to be understood that the terminology used herein is used for the purpose
of describing particular embodiments only and is not intended to be limiting,
as
the scope- of the present invention will be defined only by the appended
claims
and equivalents thereof.
In describing and claiming the present invention, the following
terminology will be used.
The singular forms "a," "an," and "the" include plural referents unless the
context clearly dictates otherwise. Thus, for example, reference to "a drug"
includes reference to one or more of such compositions.
"Skin" is defined to include human skin (intact, diseased, ulcerous, or
broken), finger and toe nail surfaces, and mucosal surfaces that are usually
at
least partially exposed to air such as lips, genital and anal mucosa, and
nasal
and oral mucosa.
The term "musculoskeletal pain or inflammation" includes pain and/or
inflammation of joints, tendons, ligaments, muscles, bones, synovial fluids,
and/or soft tissues which are part of the musculoskeletal system.
CA 02747845 2011-08-05
6
The term "neuropathic pain" includes pain associated with the nervous system,
including the brain, spinal cord, or the peripheral nervous system.
Neuropathic pain can be
chronic or acute and can occur as a result of trauma, disease, or other
factors.
The term "drug(s)" refers to active agents that can be used with the
formulations of
the present invention and includes active agents that are effective in
treating either neuro-
pathic or musculoskeletal pain. Examples of drugs which can be used to treat
musculo-
skeletal pain include NSAIDs, local anesthetics, steroid drugs, and/or 5-HT2A
receptor
antagonists. An example of a 5-HT2A receptor antagonist includes but is not
limited to
ketanserin. Examples of NSAIDS include but are not limited to ketoprofen,
piroxicam,
diclofenac, indomethacin, and COX inhibitors. Examples of local anesthetics
include but are
not limited to lidocaine, bupivacaine, ropivacaine, and tetracaine. Examples
of steroid drugs
for use in the present invention include but are not limited to dexamethasone,
hydro-
cortisone, prednisone, prednisolone, methylprednisolone, halobetasol
propionate,
betamethasone dipropionate, betamethasone, prodrugs thereof, or combinations
thereof.
Examples of drugs suitable for treating neuropathic pain include, without
limitation, local
anesthetics including lidocaine, bupivacaine, ropivacaine, and tetracaine;
steroids including
dexamethasone; alpha-2 agonists including clonidine; tricyclic anti-
depressants including
amitriptyline, anticonvulsants, N-methyl-D-aspartate (NMDA) antagonists
including dextro-
methorphan, memantine, amantadine, ketamine, methadone, dextropropoxyphene,
and
ketobemidone; steroids; 5-HT2A receptor antagonist including ketanserin; or
combinations
thereof. "Drug(s)" also includes antiviral agents such as acyclovir,
penciclovir, famciclovir,
or valacyclovir.
When referring generally to a "drug," it is understood that there are various
forms of
a given drug, and those various forms are expressly included. In accordance
with this,
various drug forms include polymorphs, salts, hydrates, solvates, and
cocrystals. For some
drugs, one physical form of a drug may possess better physical-chemical
properties making
it more amenable for getting to, into, or through the skin, and this
particular form is defined
as the "physical form favorable for dermal delivery." For example the steady
state flux
CA 02747845 2011-08-05
7
of diclofenac sodium from flux enabling non-volatile solvents is much higher
than the steady state flux of diclofenac acid from the same flux enabling non-
volatile solvents. It is therefore desirable to evaluate the flux of the
physical
forms of a drug from non-volatile solvents to select a desirable physical
form/non-volatile solvent combination.
The term "NSAID" or "non-steroidal anti-inflammatory drug" include all the
non-steroidal anti-inflammatory agents, general COX inhibitors, COX-2
selective.
inhibitors, and COX-3 selective Inhibitors.
The phrases "dermal drug delivery" or "dermal delivery of drug(s)" shall
include both transdermal and topical drug delivery, and Includes the delivery
of
drug(s) to, through, or into the skin. "Transdermal delivery" of drug can be
targeted to skin tissues just under the skin, regional tissues or organs under
the
skin, systemic circulation, and/or the central nervous system.
The term "flux" such as in the context of "dermal flux" or "transdermal
flux," respectively, refers to the quantity of the drug permeated into or
across
skin per unit area per unit time. A typical unit of flux is microgram per
square
centimeter per hour. One way to measure flux is to place the formulation on a
known skin area of'a human volunteer and measure how much drug can
permeate into or across skin within certain time constraints. Various methods
(in viva methods) might be used for the measurements as well. The method
described in Example I or other similar method (in vitro methods) can also be
used to measure flux. Although an in vitro method uses human epidermal
membrane obtained from a cadaver, or freshly separated skin tissue from
hairless mice rather than measure drug flux across the skin using human
volunteers, it Is gQnerally accepted by those skilled in the art that results
from a
properly designed and executed in vitro test can be used to estimate or
predict
the results of an in vivo test with reasonable reliability. Therefore, "Ã lux"
values
set forth herein can mean that measured by either in vivo or in vitro methods.
The term "flux-enabling" with respect to the non-volatile solvent system
(or solidified layer including the same) refers to a non-volatile solvent
system
(including one or more non-volatile solvents) selected or formulated
specifically
to be able to provide therapeutically effective flux for a particular drug(s).
For
CA 02747845 2011-08-05
8
topically or regionally delivered drugs, a flux enabling non-volatile solvent
system is defined as a non-volatile solvent system which, alone without the
help
of any other ingredients, is capable of delivering therapeutic sufficient
levels of
the drug across, onto or Into the subject's skin when the non-volatile solvent-
system is saturated with the drug. For systemically targeted drugs, a flux
enabling non-volatile solvent system Is a non-volatile solvent system that can
provide therapetucially sufficient daily doses over 24 hours when'the non-
volatile solvent system is saturated with the drug and is in full contact with
the
subject's skin with no more than 500 cm2 contact area. Preferably, the contact
area for the non-volatile solvent system Is no more than 100 cm2. Testing
using
this saturated drug-in-solvent state can be used to measure the maximum flux-
generating ability of a non-volatile solvent system. To determine flux, the
drug
solvent mixture needs to be kept on the skin for a clinically sufficient
amount.of
time. In reality, it may be difficult to keep a liquid solvent on the skin of
a human
volunteer for an extended period of time. Therefore, an alternative method to
determine whether a solvent system is "flux-enabling" Is to measure the in
vitro
drug permeation across the hairless mouse skin or human cadaver skin using
the apparatus and method described in Example 1. This and similar methods
are commonly used by those skilled in the art to evaluate permeability and
feasibility of formulations. Alternatively, whether a non-volatile solvent
system is
flux-enabling can be tested on the skin of a live human subject with means to
maintain the non-volatile solvent system with saturated drug on the skin, and
such means may not be practical for a product. For example, the non-volatile
solvent system with saturated drug can be soaked into an absorbent fabric
material which is then applied on the skin and covered with a protective
membrane. Such a system Is not practical as a pharmaceutical product,. but is
appropriate for testing whether a non-volatile solvent system has the
Intrinsic
ability to provide sufficient drug flux, or whether it is flux-enabling.
It is also noted that once the formulation forms a solidified layer, the
solidified layer can also be "flux enabling" for the drug while some of the
non-
volatile solvents remain in the solidified layer, even after the volatile
solvents
(including water) have been substantially evaporated.
CA 02747845 2011-08-05
9
For lidocalne base, a.non-volatile solvent system would be "flux enabling"
if it is capable of generating a flux of at least about 20 mcg/cm2/hour in a
setup
same or similar to that described in Example 1. For tetracaine and ropivacaine
.
bases, a non-volatile solvent system would be "flux enabling" if it Is capable
of
generating a flux of at least about 5 mcg/cm2/hour in a setup the same or
similar
to that described in Example 1. For ketoprofen and diclofenac, a non-volatile
solvent system would be "flux enabling" if it is capable of generating a flux
of at
least about 5 mcg/cm2/hour in the same or similar setup to that described In
Example 1.
For example, the importance of selecting an appropriate non-volatile
solvent Is demonstrated In Table 1. The flux of ropivacaine (a local
anesthetic
agent effective in treating neuropathic pain) from saturated glycerol,
isostearic
acid (ISA) alone and ISA+trolamine, and ISA+trolamine peel are presented in
Table 1. Flux values were generated in an in vitro experiment described below
in Example 1. The estimated therapeutically beneficial ropicavaine flux Is 5-
10
mcg/cm2/h.
Table 1
Non-volatile solvent In vitro flux me /cm /h
ISA 112
ISA + 20% Trolamine 43* 7
ISA+Trolamine peel 32:t 2
Glycerol 1.2 0.7
Estimated therapeutically beneficial flux = 5-10
me /cm2/h
In vitro flux values represent the mean and st. dev. of three determinations.
In vitro flux results of ropivacaine from I=SA, and ISA+trolamine are examples
of
a suitable non-volatile solvent and glycerol Is an example of an unsuitable
non-
volatile solvent. When Incorporated into a peel formulation, the suitable non-
volatile solvent dictates the flux-generating power of the formulation. It
should
be noted that a "non-volatile solvent system suitable for the selected drug"
can
be a single chemical substance or a mixture of two or more chemical
substances. As can be seen above, the non-volatile solvent system of
CA 02747845 2011-08-05
ISA+trolamine can generate more flux than the non-volatile solvent system of
pure ISA, though both are probably suitable for certain applications.
The phrase "effective amount," "therapeutically effective amount,"
"therapeutically effective, rate(s)," or the like, as it relates to a drug,
refers to
5 sufficient amounts or delivery rates of a drug which achieves any
appreciable
level of therapeutic results in treating a condition for which the drug is
being
delivered. It is understood- that "appreciable level of therapeutic results"
may or
may not meet any government agencies' efficacy standards for approving the
commercialization of a product. It is understood that various biological
factors
10 may affect the ability of a substance to perform its intended task.
Therefore, an
"effective amount," "therapeutically effective amount," or "therapeutically
effective rate(s)" may be dependent in some Instances on such biological
factors to some degree. However, for each drug, there is usually a consensus
among those skilled in the art on the range of doses or fluxes that are
sufficient
in most subjects. Further, while the achievement of therapeutic effects may be
measured by a physician or other qualified medical personnel using evaluations
known In the art, It Is recognized that Individual variation and response to
treatments may make the achievement of therapeutic effects a subjective
decision. The determination of a therapeutically effective amount or delivery
rate Is well within the ordinary skill In the art of pharmaceutical sciences
and
medicine.
"Therapeutically effective flux" is defined as the permeation flux of the
selected 'drug that delivers sufficient, amount of drug into or across the
skin to be
clinically beneficial in that some of the patient population can obtain some
degree of benefit from the drug flux. It does not necessarily mean that most
of
the patient population can obtain some degree of benefit or the benefit is
high
enough to be deemed "effective" by relevant government agencies or the
medical profession. More specifically, for drugs that target skin or regional
tissues or organs close to the skin surface (such as joints, certain muscles,
or
tissues/organs that are at least partially within 5 cm of the skin surface),
"therapeutically effective flux" refers to the drug flux that can deliver a
sufficient
amount of the drug Into the target tissues within a clinically reasonable
amount
CA 02747845 2011-12-20
11
of time. For drugs that target the systemic circulation, "therapeutically
effective
flux" refers to drug flux that, via clinically reasonable skin contact area,
can
deliver sufficient amounts of the selected drug to generate clinically
beneficial
plasma or blood drug concentrations within a clinically reasonable time.
Clinically reasonable skin contact area is'defined as a size of skin
application
area that most subjects would accept. Typically, a skin contact area of 400
cm2
or less Is considered reasonable. Therefore, in order to deliver 4000 mcg of a
drug to the systemic circulation via a 400 cm2 skin contact area over 10
hours,
the flux needs to be at least 4000_mcg1400cm2/10 hour, which equals 1
mcg/cm2/hr. By this definition, different drugs have different
"therapeutically
effective flux." Additionally, therapeutically effective flux may be different
in
different subjects and or at different times for even the same subject.
However,
for each drug, there is usually a consensus among the skilled in the art on
the
range of doses-or fluxes that are sufficient in most subjects at most times.
The term "plasticizing", "plasticizing" in relation to non-volatile solvent
(or a non-volatile solvent system) and the solidifying agent is defined as a
non-
volatile solvent (or a non-volatile solvent system) that acts as a plasticizer
for
the solidifying agent. A "plasticizer" is an agent which Is capable of
providing
the flexibility and/or elasticity of the solidified formulation layer after
the volatile
solvent system has at least substantially evaporated. Plasticizers also have
the
capability to reduce the brittleness of solidified formulation by making it
more
flexible and/or elastic. For example, propylene glycol is a plasticizing non-
volatile solvent for a solidifying formulation with ketoprofen as the drug and
polyvinyl alcohol as the selected solidifying agent. However, propylene gtycoi
In
a solidifying formulation of ketoprofen with Gantrez S-97TM or Avalure UR
405TH as
solidifying agents does not provide the same plasticizing effect. The
combination of propylene glycol and Gantrez S-97 or Avalure UR 405 is less
compatible and results in less desirable formulation fol= topical
applications.
Therefore, whether a given non-volatile solvent is "plasticizing" depends on
which solidifying agent(s) is selected.
It should be noted that "flux-enabling non-volatile solvent," "flux-enabling,
plasticizing non-volatile solvent," or "high flux-enabling non-volatile
solvent" can
CA 02747845 2011-08-05
12
be a single chemical substance or a mixture of two or more chemical
substances. For example, the steady state flux value for clobetasol propionate
in Table C is a 9:1 for propylene glycol:isostearic acid mixture that
generated
much higher clobetasol flux than propylene glycol or ISA alone (see Table B).
Therefore, the 9:1 propylene glycol:isostearlc acid mixture is a "high flux-
enabling non-volatile solvent" but propylene glycol or isostearic acid alone
Is
not.
The term "adhesion" or "adhesive" when referring to a solidified layer
herein refers to sufficient adhesion between the solidified layer and the skin
so
that the layer does not fall off the skin during Intended use on most
subjects.
Thus, "adhesive" or the like when used to describe the solidified layer means
the solidified layer is adhesive to the body surface to which the Initial
formulation
layer was originally applied (before the evaporation of the volatile
solvent(s)). In
one embodiment, it does not mean the solidified layer is adhesive on the
opposing side. In addition, it should be noted that whether a solidified layer
can
adhere to a skin surface for the desired extended period of time partially
depends on the condition of the body surface. For example, -excessively
sweating or oily skin, or oily substances on the skin surface may make the
solidified layer less adhesive to the skin. Therefore, the adhesive solidified
layer
of the current invention may not be able to maintain perfect contact with the
body surface and deliver the drug over a sustained period of time for every
subject under any conditions on the body surface. A standard is that it
maintains
good contact with most of the body surface, e.g. 70% of the total area, over
the
specified period of time for most subjects under normal conditions of the body
surface and external environment.
The terms "flexible," "elastic," "elasticity," or the like, as used herein
refer
to sufficient elasticity of the solidified layer so that it Is not broken if
it is
stretched in at least one direction by up to about 5%, and often to about 10%
or
even greater. For example, a solidified layer that exhibits acceptably
elasticity
and adhesion to skin can be attached to human skin over a flexible skin
location, e.g., elbow, finger, wrist, neck, lower back, lips, knee, etc., and
will
remain substantially intact on the skin upon stretching of the skin. It should
be
CA 02747845 2011-08-05
. ;1
13
noted that the solidified layers of the present invention do not necessarily
have
to have any elasticity in some embodiments.
The term "peelable," when used to describe the solidified layer, means
the solidified layer can be lifted from the skin surface in one large piece or
several large pieces, as opposed to many small pieces or crumbs.
The term "sustained" relates to therapeutically effective rates of dermal
drug delivery for a continuous period of time of at least 30 minutes, and in
some
embodiments, periods of time of at least about 2 hours, 4 hours, 8 hours, 12
hours, 24 hours, or longer.
The use of the term "substantially" when referring to the evaporation of
the volatile solvents means that a majority of the volatile solvents which
were
included in the initial formulation have evaporated. Similarly, when a
solidified
layer Is said to be "substantially devoid" of volatile solvents, including
water, the
solidified layer has less than 10 wt%, and preferably less than 5 wt%, of the
volatile solvents in the solidified layer as a whole.
:`Volatile solvent system" can be a single solvent or a mixture of solvents
that are volatile, including water and solvents that-are more volatile than
water.
Non-limiting examples of volatile solvents that can be used in the present
invention include denatured alcohol, methanol, ethanol,
isopropyl alcohol, water, propanol, C4-C6 hydrocarbons, butane, isobutene,
pentane, hexane, acetone, ethyl acetate, fluro-chioro-
hydrocarbons, methyl ethyl ketone, methyl ether,
hydrofluorocarbons, ethyl ether, 1,1,1,2 tetrafluorethane
1,1,1,2,3,3,3-heptafluoropropane, 1,1,1,3,3,3 hexafluoropropane, or
combinations thereof.
"Non-volatile solvent system" can be a single solvent or mixture of
solvents that are less volatile than water. It can also contain substances
that
are solid or liquid at room temperatures, such as pH or ion-pairing agents.
After
evaporation of the volatile solvent system, most of the non-volatile solvent
system should remain in the solidified layer for an amount of time sufficient
to
dermally delivery a given drug to, into, or through the skin of a subject at a
sufficient flux for a period of time to provide a therapeutic effect. In some
CA 02747845 2011-08-05
14
embodiments, in order to obtain desired permeability for an active drug and/or
compatibility with solidifying agents or other ingredients of the formulation,
a
mixture of two or more non-volatile solvents can be used to form the non-
volatile
solvent system. In one embodiment, the combination of two or more non-
volatile solvents to form a solvent system provides a higher transdermal flux
for
a drug than the flux provided for the drug by each of the non-volatile
solvents
Individually. The non-volatile solvent system may also serve as a plasticizer
of
the solidified layer, so that the solldified layer is elastic and flexible.
The term "solvent vehicle" describes compositions that Include both a
volatile solvent system and non-volatile solvent system. The volatile solvent
system is chosen so as to evaporate from the adhesive peelable formulation
quickly to form a solidified layer, and the non-volatile solvent system Is
formulated or chosen to substantially remain as part of the solidified layer
after
volatile solvent system evaporation so as to provide continued delivery of the
drug. Typically, the drug can be partially or completely dissolved in the
solvent
vehicle or formulation as a whole. Likewise, the drug can also be partially or
completely solubilizable in the non-volatile solvent system once the volatile
solvent system is evaporated. Formulations in which the drug is only partially
dissolved In the non-volatile solvent system after the evaporation of the
volatile
solvent system have the potential to maintain longer duration of sustained
delivery, as the undissolved drug can dissolve Into the non-volatile solvent
system as the dissolved drug is being depleted from the solidified layer
during
drug delivery.
"Adhesive solidifying formulation" or "solidifying formulation" refers to a
composition that has a viscosity suitable for application to a skin surface
prior to
evaporation of its volatile solvent(s), and which can become a solidified
layer
after evaporation of at least a portion of the volatile solvent(s). The
solidified
layer, once formed, can be very durable. In one embodiment, once solidified on
a skin surface, the formulation can form a peel. The peel can be a soft,
coherent solid that can be removed by peeling large pieces from the skin
relative to the size of the applied formulation, and often, can be peeled from
the
skin as a single piece. The application viscosity is typically more viscous
than a
CA 02747845 2011-08-05
wafer-like liquid, but less viscous than a soft solid. Examples of preferred
viscosities include materials that have consistencies similar to pastes, gels,
ointments, and the like, e.g., viscous liquids that flow but are not subject
to
spilling. Thus, when a composition is said to have a viscosity "suitable for
5 application" to a skin surface, this means the composition has a viscosity
that is
high enough so that the composition does not substantially run off the skin
after
being applied to skin, but also has a low enough viscosity so that it can be
easily spread onto the skin. A viscosity range that meets this definition can
be
from about 100 cP to about 3,000,000 cP (centipoises), and more preferably
10 from about 1,000 cP to about 1,000,000 cP.
In some embodiments of the present invention it may be desirable to add
an additional agent or substance to the formulation so as to provide enhanced
or increased adhesive characteristics- The additional adhesive agent or
substance can be an additional non-volatile solvent or an additional
solidifying
15 agent. Non-limiting examples of substances which might be used as
additional
adhesion enhancing agents include copolymers of methylvinyl ether and malefic
anhydride (Gantrez polymers), polyethylene glycol and polyvinyl pyrrolidone,
gelatin, low molecular weight polyisobutylene rubber, copolymer of acrylsan
alkyl/octylacrylamido (Dermacryl 79), and various aliphatic resins and
aromatic
resins.
The terms "washable," "washing" or "removed by washing" when used
with respect to the adhesive formulations of the present invention refers to
the
ability of the adhesive formulation to be removed by the application of
a.washing
solvent using a normal or medium amount of washing force. The required force
to remove the formulations by washing should not cause significant skin
irritation or abrasion. Generally, gentle washing force accompanied by the
application of an appropriate washing solvent is sufficient to remove the
adhesive formulations disclosed herein. The solvents which can be used for
removing by washing the formulations of the present invention are numerous,
but preferably are chosen from commonly acceptable solvents including the
volatile solvents listed herein. Preferred washing solvents do not
significantly
irritate human skin and are generally available to the average subject.
CA 02747845 2011-08-05
16
Examples of washing solvents include but are not limited to water, ethanol,
methanol, isopropyl alcohol, acetone, ethyl acetate, propanol, or combinations
thereof. In aspect of the invention the washing solvents can be selected from
the group consisting of water, ethanol, Isopropyl alcohol, or combinations
thereof. Surfactants can also be used in some embodiments.
An acceptable length of time as it relates to "drying time" refers to the
time it takes for the formulation to form a non-messy solidified surface after
application on skin under standard skin and ambient conditions, and with
standard testing procedure. It. is noted that the word "drying time" in this
application does not mean the'time it takes to completely evaporate off the
volatile solvent(s). Instead, it means the time it takes to form the non-messy
solidified surface as described above.
"Standard skin" is defined as dry, healthy human skin with a surface
temperature of between about 30 C to about 36 C. Standard ambient
conditions are defined by the temperature. range of from 20 C to 25 C and a
relative humidity range of from 20% to 80%. The term "standard skin" in no way
limits the types of skin or skin conditions on which the formulations of the
present invention can be used. The formulations of the present invention can
be used to treat all types of "skin," including undamaged (standard skin),
diseased skin, or damaged skin. Although skin conditions having different
characteristics can be treated using the formulations of the present
Invention,
the use of the term "standard skin" is used merely as a standard to test the
compositions of the varying embodiments of the present Invention. As a
practical.matter, formulations that perform well (e.g., solidify, provide
therapeutically effective flux, etc.) on standard skin can also perform well
diseased or damaged skin.
The "standard testing procedure" or "standard testing condition" is as
follows: To standard skin at standard ambient conditions is applied an
approximately 0.1 mm layer of the adhesive solidifying formulation and the
drying time is measured. The drying time is defined as the time It takes for
the
formulation to form a non-messy surface such that the formulation does not
lose
CA 02747845 2011-08-05
17
mass by adhesion to a piece of 100% cotton cloth pressed onto the formulation
surface with a pressure of between about 5 and about 10 g/cm2 for 5 seconds.
"Solidified layer" describes the solidified or dried layer of an adhesive
solidifying formulation after at least a portion of the volatile solvent
system has
evaporated. The solidified layer remains adhered to the skin, and Is
preferably
capable of maintaining good contact with the subject's skin for substantially
the
entire duration of application under standard skin and ambient conditions. The
solidified layer also preferably exhibits sufficient tensile strength so that
it can be
peeled off the skin at the end of the application in one piece or several
large
pieces (s opposed to a layer with weak tensile strength that breaks Into many
small pieces or crumbles when removed from the skin).
As used herein, a plurality of drugs, compounds, and/or solvents may be
presented in a common list for convenience. However, these lists should be
construed as though each member of the list is individually identified as a
separate and unique member. Thus, no individual member of such list should
be construed as a de facto equivalent of any other member of the same list
solely based on their presentation In a common group without indications to
the
contrary.
Concentrations, amounts, and other numerical data may be expressed or
presented herein in a range format. It is to be understood that such a range
format is used merely for convenience and brevity and thus should be
Interpreted flexibly to include not only the numerical values explicitly
recited as
the limits of the range, but also to Include all the Individual numerical
values or
sub-ranges encompassed within that range as if each numerical value and sub-
range. is explicitly recited. As an illustration, a numerical range of "about
0.01 to
2.0 mm" should be interpreted to Include not only the explicitly recited
values of
about 0.01 mm to about 2.0 mm, but also include individual values and sub-
ranges within the indicated range. Thus, included in this numerical range are
individual values such as 0.5, 0.7, and 1.5, and sub-ranges such as from 0.5
to
1.7, 0.7 to 1.5, and from 1.0 to 1.5, etc. This same principle applies to
ranges
reciting only one numerical value. Furthermore, such an interpretation should
CA 02747845 2011-08-05
18
apply regardless of the breadth of the range or the characteristics being
described.
With these definitions in mind, In one embodiment, a formulation for
treating musculoskeletal pain or Inflammation can comprise a drug suitable for
treating musculoskeletal pain or Inflammation, a solvent vehicle, and a
solidifying agent. The solvent vehicle can comprise a volatile solvent system
including at least one volatile solvent, and a non-volatile solvent system
including at least one non-volatile solvent, wherein the non-volatile solvent
system is capable of facilitating transdermal delivery of the drug at a
therapeutically effective rate over a sustained period of time. The
formulation
can have a viscosity suitable for application and adhesion to a skin surface
as a
layer prior to evaporation of the volatile solvent system, and further, the
formulation applied to the skin surface can form a solidified layer after at
least
partial evaporation of the volatile solvent system. The drug can continue to
be
delivered at the therapeutically effective rate to treat musculoskeletal pain
or
Inflammation after the volatile solvent system is at least substantially
evaporated.
in another embodiment, a method of dermally delivering a drug for
treating pain or inflammation of joints or muscles can comprise applying an
adhesive solidifying formulation to a skin surface adjacent to the tissue
suffering
from the pain or inflammation (for example, the skin surface of a knee
suffering
from arthritis or the skin of lower back which is suffering from lower back
pain) .
The adhesive solidifying formulation can comprise a drug suitable for treating
musculoskeletal pain or inflammation, a solvent vehicle, and a solidifying
agent.
The solvent vehicle can comprise a volatile solvent system including at least
one volatile solvent, and a non-volatile solvent system including at least one
non-volatile solvent, wherein the non-volatile solvent system is capable of
facilitating dermal delivery of the drug at a therapeutically effective rate
over a
sustained period of time. The formulation can have a viscosity suitable for
application and adhesion to the skin surface prior to evaporation of the
volatile
solvent system. Additional steps include solidifying the formulation to form a
solidified layer on the skin surface by at least partial evaporation of the
volatile
CA 02747845 2011-08-05
19
solvent system; and dermally delivering the drug from the solidified layer to
the
skin surface at therapeutically effective rates for treating the pain or
inflammation of joints or muscles over a sustained period of time.
In another embodiment, a solidified layer for treating musculoskeletal
pain or Inflammation can comprise a drug effective for treating
musculoskeletal
pain or inflammation, a non-volatile solvent system, and a solidifying agent.
The
non-volatile solvent system can include at least one non-volatile solvent,
wherein the non-volatile solvent system is capable of facilitating the
delivery of
the drug at therapeutically effective rates over a sustained period of time.
Additionally, the solidified layer can be stretchable by 5% In at least one
direction without cracking, breaking, and/or separating from a skin surface to
which the layer is applied.
In another embodiment, a formulation for treating musculoskeletal pain or
inflammation can comprise ropivacalne, a solvent vehicle, and a solidifying
agent. The solvent vehicle can Include a volatile solvent system including at
least one volatile solvent, and a non-volatile solvent system including at
least
one solvent selected from the group consisting of triacetin, span 20,
isostearic
acid, or combinations thereof. The ropivacalne can either be in base or salt
form. The formulation has a viscosity suitable for application to a skin
surface
prior to evaporation of the volatile, solvent system, and can be applied to
the
skin surface to form a solidified, coherent, flexible, and continuous layer
after at
least partial evaporation of the volatile solvent system. Further, the
ropivacaine
can continue to be delivered at a transdermal flux of at least 5 mcg/cm2/hour
after the volatile solvent system is at least substantially all evaporated. In
another embodiment, the transdermal flux can be at least 10 mcglcm2/hour after
the volatile solvent system Is at least substantially all evaporated from the
solidified layer.
In another embodiment, a formulation for treating musculoskeletal pain or
inflammation can comprise lidocaine, a solvent vehicle, and a solidifying
agent.
The solvent vehicle can Include a volatile solvent system Including at least
one
volatile solvent, and a non-volatile solvent system including at least one
solvent
selected from the group consisting of propylene glycol and dipropylene glycol.
CA 02747845 2011-08-05
The lidocalne can be in either base or salt form. The formulation can have a
viscosity suitable for application to a skin surface prior to evaporation of
the
volatile solvent system, and can be applied to the skin surface to form a
solidified, coherent, flexible and continuous layer after at least partial
6 evaporation of the volatile solvent system. The lidocalne can continue to be
delivered at a transdermal flux of at least 20 mcg/cm2/hour after the volatile
solvent system Is at least substantially all evaporated fro the solidified
layer.
In another embodiment, a formulation for treating musculoskeletal pain or
inflammation can comprise ketoprofen, a solvent vehicle, and a solidifying
10 agent. The solidifying agent can comprise a volatile solvent system
including at
least one volatile solvent, and a non-volatile solvent system including at
least
one solvent selected from the group consisting of propylene glycol and
glycerol,
Isostearic acid, and triacetin. The ketoprofen can be in either base or salt
form.
The formulation can have a viscosity suitable for application to a skin
surface
15 prior to evaporation of the volatile solvent system, and can be applied to
the
skin surface to form a solidified, coherent, flexible and continuous layer
after at
least partial evaporation of the volatile solvent system. The ketoprofen can
continue to be delivered at a transdermal flux of at least 10 mcg/cm2/hour
after
the volatile solvent system is at least substantially all evaporated fro the
20 solidified layer.
In still another embodiment, a formulation for treating musculoskeletai
pain or inflammation can comprise tetracaine, a solvent vehicle, and a
solidifying agent. The solvent vehicle can comprise a volatile solvent system
including at least one volatile solvent, and a non-volatile solvent system
including at least one solvent selected from the group consisting of propylene
glycol and isostearic acid. The tetracaine can be in either base or salt form.
The formulation can have a viscosity suitable for application to a skin
surface
prior to evaporation of the volatile solvent system, and can be applied to the
skin surface to form a solidified, coherent, flexible and continuous layer
after at
least partial evaporation of the volatile solvent system. The tetracaine can
continue to be delivered.at a transdermal flux of at least 5 mcg/cm2/hour
after
CA 02747845 2011-08-05
21
the volatile solvent system is at least substantially all evaporated fro the
solidified layer.
. In yet another embodiment, a formulation for treating musculoskeletal
pain or Inflammation can comprise lidocaine and tetracaine, a solvent vehicle,
and a solidifying agent. The solvent vehicle can comprise volatile solvent
system Including at least one volatile solvent, and a non-volatile solvent
system
Including at least one solvent selected from the group consisting of propylene
glycol and dipropylene glycol, and Isostearic acid. The tetracaine and
lidocaine
can be in either base or salt form. ,The formulation can have a viscosity
suitable
for application to a skin surface prior to evaporation of the volatile solvent
system, and can be applied to the skin surface to form a solidified, coherent,
flexible and continuous layer after at least partial evaporation of the
volatile
solvent system. The tetracaine and lidocaine can continue to be delivered at a
transdermal flux of at least 5 mcg/cm2/hour, respectively, after the volatile
solvent system Is at least substantially all evaporated from the solidified
layer.
In another embodiment, a formulation for treating musculoskeletal pain or
inflammation, can comprise a drug include at least one member from the group
consisting of lidocaine, tetracaine, ropivacaine,'ketoprofen, diclofenac, or
combinations thereof; a solvent vehicle; and a solidifying agent. The solvent
vehicle can comprise a volatile solvent system including a volatile solvent
whose
boiling point is below 20 C, and a non-volatile solvent system comprising at
least one non-volatile solvent. The formulation can have a viscosity suitable
for
application to a skin surface prior to evaporation of the volatile solvent
system,
and can be applied to the skin surface to a solidified, coherent, flexible and
continuous layer after at least partial evaporation of the volatile solvent
system.
The drug can continue to be delivered at a therapeutically effective rate
after the
volatile solvent system is at least substantially all evaporated.
In another embodiment, a formulation for treating neuropathic pain can
comprise a drug suitable for treating neuropathlc pain, a solvent vehicle, and
a
solidifying agent. The solvent vehicle can include a volatile solvent system
comprising at least one volatile solvent, and a non-volatile solvent system
comprising at least one non-volatile solvent. The non-volatile solvent system
CA 02747845 2011-08-05
22
facilitates dermal delivery of the drug at a therapeutically effective rate
over a
sustained period of time. The formulation can have a viscosity suitable for
application and adhesion to a skin surface as a layer prior to evaporation'of
the
volatile solvent system. The formulation applied to the skin surface can form
a
solidified layer after at least partial evaporation of the volatile solvent
system.
Further; the drug can continue to be delivered at the therapeutically
effective
rate after the volatile solvent system is at least substantially evaporated.
In another embodiment, a method for treating neuropathic pain can
comprise the step of applying a layer of an adhesive formulation to a skin
surface of a subject. The formulation can comprise a drug suitable for
treating
neuropathic pain, a solvent vehicle, and a solidifying agent. The solvent
vehicle
can comprise a volatile solvent system including at least one volatile
solvent,
and a non-volatile solvent system including at least one non-volatile solvent.
The non-volatile solvent system facilitates dermal delivery of the drug at a
therapeutically effective rate over a sustained period of time. The
formulation
can have a viscosity suitable for application and adhesion to a skin surface
prior
to evaporation of the volatile solvent system. Additional steps include
solidifying
the formulation to form a solidified layer on the skin surface by at least
partial
evaporation of the volatile solvent system; and dermally delivering the drug
from
the solidified layer to the subject. at therapeutically effective rates over a
sustained period of time to reduce the neuropathic pain.
In another embodiment, a solidified layer for treating neuropathic pain
can comprise a drug suitable for treating neuropathic pain, a non-volatile
solvent
system suitable for the drug, and a solidifying agent. The solidified layer
can
have sufficient elasticity, flexibility, and adhesion to the skin so that it
is not
separated from the skin even If the skin surface is stretched or bent during_a
subject's normal daily activities. For example, the solidified layer can be
stretchable by 5% in one direction without cracking, breaking, and/or
separating
from a skin surface to which the layer is applied.
In another embodiment, a formulation for treating neuropathic pain can
comprise ropivacaine, a solvent vehicle, and a solidifying agent. The solvent
vehicle can comprise a volatile solvent system including at least one volatile
CA 02747845 2011-08-05
23
solvent, and a non-volatile solvent system can include at least one solvent
selected from the group consisting of an amine base, triacetin, span 20,
isostearic acid, or a mixture thereof. The solidifying agent can include butyl
and
methyl methacrylate copolymers. The formulation can have a viscosity suitable
for application to a skin surface as a layer prior to evaporation of the
volatile
solvent system. Further, formulation layer applied to the skin surface can
form a
solidified, coherent, flexible, and continuous layer after at least partial
evaporation of the volatile solvent system. The ropivapaine can also continue
to .
be delivered at a therapeutically effective rate after the volatile solvent
system Is
at least substantially all evaporated. .
In another embodiment, a formulation for treating neuropathic pain
associated with viral infections can comprise a drug, a solvent vehicle, and a
solidifying agent. The drug can include at least one member selected from the
group consisting of acyclovir, valacyclovir, and pencyclovir. The solvent
vehicle
can include a volatile solvent system Including at least one volatile solvent,
and
a non-volatile solvent system comprising at. least one solvent selected from
the
group of oleic acid, isostearic acid, and olive oil. The solidifying agent can
be
selected from the group consisting of ethyl acrylate-methyl methacrylate-
trimethylammonioethyl methacrylate chloride copolymers, butyl and methyl
methacryllae copolymers, and ethyl cellulose. The formulation can have a
viscosity suitable for application to a skin surface as a layer prior to
evaporation
of the volatile solvent system. Further, formulation applied to the skin
surface
can form a solidified, coherent, flexible, and continuous layer after at least
partial evaporation of the volatile solvent system. The drug can also continue
to
be delivered at a therapeutically effective rate after the volatile solvent
system is
at least substantially all evaporated.
In another embodiment, a formulation for treating neuropathic pain can
comprise a local anesthetic selected from the group consisting of lidocaine,
tetracaine, and a combination thereof; a solvent vehicle; and a solidifying
agent.
The solvent vehicle can comprise a volatile solvent system including at least
one volatile solvent, and a non-volatile solvent system including at least one
solvent selected from the group consisting of propylene glycol and'dipropylene
CA 02747845 2011-08-05
24
glycol. The local anesthetic can be in either base or salt form, and the
formulation can have a viscosity suitable for application to a skin surface
prior to
evaporation of the volatile solvent system. The formulation applied to the
skin
surface can form a solidified, coherent, flexible and continuous layer after
at
least partial evaporation of the volatile solvent system, and the local
anesthetic
can continue to be delivered at a therapeutically effective rate after the
volatile
solvent system Is at least substantially all evaporated.
In another embodiment, a formulation for treating neuropathic pain can
comprise a drug selected from the group consisting of amitriptyline, ketamine,
and combinations thereof; a solvent vehicle; and a solidifying agent. The
solvent vehicle can comprise a volatile solvent system including at least one
volatile solvent, and a non-volatile solvent system comprising at least one
non-
volatile solvent. The formulation can have a viscosity suitable for
application to
a skin surface prior to evaporation of the volatile solvent system. The
formulation applied to the skin surface can form a solidified, coherent,
flexible
and continuous layer after at least partial evaporation of the volatile
solvent
system. Further, the drug can continue to be delivered at a therapeutically
effective rate after the volatile solvent system is at least substantially all
evaporated.
In another embodiment, a formulation for treating neuropathic pain can
comprise a drug selected from the group consisting of lidocaine, tetracaine,
ropivacaine, amitriptyline, ketamine, and combinations thereof; a solvent
vehicle; and a solidifying agent. The solvent vehicle can include a volatile
solvent system comprising a volatile solvent whose boiling point is below 20
C,
and a non-volatile solvent system comprising at least one non-volatile
solvent.
The formulation can have a viscosity suitable for application to a skin
surface
prior to evaporation of the volatile solvent system, and when applied to the
skin
surface, can form a solidified, coherent, flexible and continuous layer after
at
least partial evaporation of the volatile solvent system. The drug can
continue
to be delivered at a therapeutically effective rate after the volatile solvent
system
is at least substantially all evaporated.
CA 02747845 2011-08-05
Thus, the present invention is related to novel formulations, methods, and
solidified
layers that are typically in the initial form of semi-solids (including
creams, gels, pastes,
ointments, and other viscous liquids), which can be easily applied onto the
skin as a layer,
and can quickly (from 15 seconds to 4 minutes under standard skin and ambient
conditions)
5 to moderately quickly (from 4 to 15 minutes under standard skin and ambient
conditions)
change into a solidified layer, e.g., a coherent and soft solid layer for drug
delivery for
reducing musculoskeletal pain. The solidified layer thus formed is capable of
delivering drug
into or across the skin at therapeutically effective rates, over a sustained
period of time,
e.g., hours to tens of hours, so that most of the drug delivery occurs after
the solidified layer
10 is formed. Additionally, the solidified layer typically adheres to the
skin, but has a solidified,
minimally-adhering, outer surface which is formed relatively soon after
application and
which does not substantially transfer to or otherwise soil clothing or other
objects that a
subject is wearing or that the solidified layer may inadvertently contact. The
solidified layer
can also be formulated such that it is highly flexible and stretchable, and
thus capable of
15 maintaining good contact with the skin surface, even if the skin is
stretched during body
movement, such as at a knee, finger, elbow, wrist, finger, hip, neck, back,
joints, or other
areas where skin is typically stretched.
In selecting or formulating the various components that can be used, e.g.,
drug,
solvent vehicle of volatile solvent system and non-volatile solvent system,
solidifying
20 agent(s), etc., certain variables can be considered. For example, the
volatile solvent system
can be selected from pharmaceutically or cosmetically acceptable solvents
known in the art,
including water and solvents at least as volatile as water. In one embodiment
of the present
invention, the volatile solvent system can include ethanol, isopropyl alcohol,
water, dimethyl
ether, diethyl ether, butane, propane, isobutene, 1,1, difluoroethane, 1,1,1,2
tetrafluoro-
25 ethane, 1,1,1,2,3,3,3-heptafluoropropane, 1,1,1,3,3,3 hexafluoropropane,
ethyl acetate,
acetone, or combinations thereof. In another embodiment of the present
invention, the
volatile solvent system can include denatured alcohol, methanol, propanol,
isobutene,
pentane, hexane, cytopentasiloxane, cyclomethicone, methyl ethyl ketone,
butanol, butyl
alcohol, acetyl monoglycerides, alkyl dioxolanes, coriander oil,
cyclomethicone, dietthylene
glycol monoethyl ether, milk, olive alcohol, vinyl acetate, hexylene glycerol,
methacrylic
acid, or combinations thereof. The volatile solvent system can
CA 02747845 2011-08-05
26
include a mixture or combination of any of the volatile solvents set forth in
the
embodiments above.
These volatile solvents should be chosen to be compatible with the -rest
of the formulation. It is desirable to use an appropriate weight percentage of
the
volatile solvent(s) In the formulation. Too much of the volatile solvent
system
prolongs the drying time. Too little of the volatile solvent system can make
it
difficult to spread the formulation on the skin. For most formulations, the
weight
percentage of the volatile solvent(s) can be from about 10 wt% to about 85
wt%,
and more preferably from about 20 wt% to about 50 wt%.
The non-volatile solvent system can also be chosen or formulated to be
compatible with the solidifying agent, the drug, the volatile solvent, and any
other Ingredients that may be present. For example, the solidifying -agent can
be chosen so that it Is dispersible or soluble In the non-volatile solvent
system.
Most non-volatile solvent systems and solvent vehicles as a whole will be
formulated appropriately after experimentation. For instance, certain drugs
have good solubility in poly ethylene glycol (PEG) having a molecular weight
of
400 (PEG 400, non-volatile solvent) but poor'solubility in glycerol (non-
volatile
solvent) and water (volatile solvent). However, PEG 400 cannot effectively
dissolve poly vinyl alcohol (PVA), and thus, is not very compatible alone with
PVA, a solidifying agent. In order to dissolve sufficient amount of an active
drug
and use PVA as a solidifying agent at the same time, a non-solvent system
including PEG 400 and glycerol (compatible with PVA) in an appropriate ratio
can-be formulated, achieving a compatibility compromise. As a further example
of compatibility, non-volatile solvent/solidifying agent incompatibility is
observed
when Span 20 is formulated into a formulation containing PVA. With this
combination, Span 20 can separate out of the formulation and form an oily
layer
on the surface of the solidified layer. Thus, appropriate solidifying
agent/non-
volatile solvent selections are desirable in developing a viable formulation
and
compatible combinations.
.30 Non-volatile solvent(s) that can be used alone or in combination to form
non-volatile solvent systems can be selected from a variety of
pharmaceutically
acceptable liquids. In one embodiment of the present invention, the non-
volatile
CA 02747845 2011-08-05
27
solvent system can include glycerol, propylene glycol, isostearic acid, oleic
acid,
propylene glycol; trolamine, tromethamine, triacetin, sorbitan monolaurate,
sorbitan monoolate, sorbitan monopalmitate, or combinations thereof.
In another embodiment the non-volatile solvent system can include benzoic
acid, dibutyl sebecate, diglycerides, dipropylene glycol, eugenol,
fatty acids such as coconut oil, fish oil, palm oil, grape seed oil, isopropyl
myristate, mineral oil, oleyl alcohol, vitamin E, triglycerides, sorbitan
fatty acid
surfactants, triethyl citrate, or combinations thereof. In a further
embodiment the
non-volatile solvent system can include 1,2,6-hexanetriol, alkyltriols,
alkyldiols,
tocopherol, p-propenylanisole, anise
oil, apricot oil, dimethyl Isosorbide, alkyl glucoside, benzyl alcohol, bees
wax,
benzyl benzoate, butylene glycol, caprylic/capric triglyceride, caramel,
cassia oil,
castor oil, cinnamaldehyde, cinnamon oil, clove oil, coconut oil, cocoa
butter,
cocoglycerides, coriander oil, corn oil, corn syrup, cottonseed oil,
cresol, diacetin, diacetylated monoglycerides, diethanolamine,
diglycerides, ethylene glycol, eucalyptus oil,
fat, fatty alcohols, flavors, liquid sugars ginger extract,- glycerin, high
fructose
corn syrup, hydrogenated castor oil, IP palmitate, lemon oil, lime oil,
limonene,
monoacetin, monoglycerides, nutmeg oil, octyldodecanol,
orange oil, palm oil, peanut oil, PEG vegetable oil, peppermint oil,
petrolatum,
phenol, pine needle oil, polypropylene glycol, sesame oil, spearmint oil,
soybean oil, vegetable oil, vegetable shortening, wax, 2-(2-
(octadecyloxy)ethoxy)ethanol, benzyl benzoate, butylated hydroxyanisole,
candelilla wax, carnauba wax, ceteareth-20, cetyl alcohol, polyglyceryl,
dipolyhydroxy stearate, PEG-7 hydrogenated castor oil, diethyl phthalate,
diethyl
sebacate, dimethicone, dimethyl phthalate, PEG Fatty acid esters such as PEG-
stearate, PEG - oleate, PEG- laurate, PEG fatty acid diesters such as PEG-
dioleate, PEG- distearate, PEG-castor oil, glyceryl behenate, PEG glycerol
fatty
acid esters such as PEG glyceryl laurate, PEG glyceryl stearate, PEG glyceryl
oleate, lanolin, lauric diethanolamide, lauryl lactate, lauryl
sulphate, medronic acid, multisterol extract, myristyl alcohol,
neutral off, PEG-octyl phenyl ether, PEG -alkyl ethers such as PEG-cetyl
ether,
CA 02747845 2011-08-05
28
PEG-stearyl ether, PEG- sorbitan fatty acid esters such as PEG-sorbitan
diisosterate, PEG-
sorbitan monostearate, propylene glycol fatty acid esters such as propylene
glycol stearate,
propylene glycol, caprylate/caprate, sodium pyrrolidone carboxylate, sorbitol,
squalene,
stear-o-wet, triglycerides, alkyl aryl polyether alcohols, polyoxyethylene
derivatives of
sorbitan-ethers, saturated polyglycolyzed C8-C10 glycerides, N-methyl
pyrrolidone, honey,
polyoxyethylated glycerides, dimethyl sulfoxide, azone and related compounds,
dimethyl-
formamide, N-methyl formamide, fatty acid esters, fatty alcohol ethers, alkyl-
amides (N,N-
dimethylalkylamides), N-methyl pyrrolidone related compounds, ethyl oleate,
polyglycerized
fatty acids, glycerol monooleate, glyceryl monomyristate, glycerol esters of
fatty acids, silk
amino acids, PPG-3 benzyl ether myristate, Di-PPG2 myreth 10-adipate,
honeyquat,
sodium pyroglutamic acid, abyssinica oil, dimethicone, macadamia nut oil,
limnanthes alba
seed oil, cetearyl alcohol, PEG-50 shea butter, shea butter, aloe vera juice,
phenyl
trimethicone, hydrolyzed wheat protein, iso-amyl acetate, chlorobutanol,
turpentine,
cytopentasiloxane, cyclomethicone, or combinations thereof. In yet a further
embodiment
the non-volatile solvent system can include a combination or mixture of non-
volatile solvents
set forth in the any of the above discussed embodiments.
In addition to these and other considerations, the non-volatile solvent system
can
also serve as plasticizer in the adhesive formulation so that when the
solidified layer,is
formed, the layer is flexible, stretchable, and/or otherwise skin friendly.
Plasticizers also
have the capability to reduce the brittleness of solidified formulation by
making it more
flexible and/or elastic. For example, propylene glycol is a plasticizing non-
volatile solvent for
a solidified layer with polyvinyl alcohol as the selected solidifying agent
and ketoprofen as
the drug. However, propylene glycol in a solidifying formulation with Gantrez
S-97 or
Avelure UR 405 as solidifying agents does not provide the same plasticizing
effect. There-
fore, whether a given non-volatile solvent is "plasticizing" depends on which
solidifying
agent(s) is selected.
Certain volatile and/or nonvolatile solvent(s) that are irritating to the skin
may be
desirable to use to achieve the desired solubility and/or permeability of the
drug. It is also
desirable to add compounds that are both capable of
CA 02747845 2011-08-05
29
preventing or reducing skin irritation and are compatible with the
formulation.
For example, in a formulation where the volatile solvent is capable of
irritating
the skin, it would be helpful to use a non-volatile solvent that Is capable of
reducing skin irritation. Examples of solvents that are known to'be capable of
preventing or reducing skin irritation include, but are not limited to,
glycerin,
honey, and propylene glycol.
The'formulations of the present invention may also contain two or more
non-volatile solvents that independently are not adequate non-volatile
solvents
for a. drug but when formulated together become an adequate non-volatile
solvent. One possible reason for these initially non adequate non-volatile
solvents to become adequate non-volatile solvents when formulated together
may be due to the optimization of the Ionization state of the drug to a
physical
form which has higher flux or the non-volatile solvents act in some other
synergistic manner. One further benefit of the mixing of the non-volatile
solvents is that it may optimize the pH of the formulation or the skin tissues
under the formulation layer to minimize irritation. Examples of suitable
combinations of non-volatile solvents that result in an adequate non-volatile
solvent system Include but are not limited to isostearic acid /trolamine,
Isostearic
acid /dlisopropyl amine, oleic acid/trolamine, and propylene glycol
/isostearic
acid.
The selection of the solidifying agent can also be carried out in
consideration of the other components present in the adhesive formulation. An
appropriate solidifying agent is compatible with the formulation such that the
formulation is In liquid or semi-liquid state (e.g. cream, paste, gel,
ointment)
before any evaporation of the volatile solvent(s) and becomes a soft, coherent
adhesive solidified layer after the evaporation of at least some of the
volatile
solvent(s). The solidifying agent can be selected or formulated to be
compatible
with the drug and the solvent vehicle (including the volatile solvent(s) and
the
non-volatile solvent system), as well as provide desired physical properties
to
the solidified layer once it is formed. Depending on the drug, solvent
vehicle,
and/or other components that may be present, the solidifying agent can be
selected from a varisty'of agents. In one embodiment, the solidifying agent
can
CA 02747845 2011-12-20
include polyvinyl alcohol with a MW range of 20,000-70,000 (AmrescoTM), esters
of
polyvinylmethylether/maleic anhydride copolymer (ISP Gantrez ES-425TH and
Gantrez
ES-225 TM) with a MW range of 80,000-160,000, neutral copolymer of butyl
methacrylate
and methyl methacrylate (Degussa Plastoid BTM) with a MW range of 120,000-
180,000,
5 dimethylaminoethyl methacrylate-butyl methacrylatemethyl methacrylate
copolymer
(Degussa Eudragit E100TM) with a MW range of 100,000-200,000, ethyl acrylate-
methyl
methacrylate-trimethylammonioethyl methacrylate chloride copolymer with a MW
greater
than 5,000 or similar MW to Eudragit RLPOTM (DegussaTM), ZeinTM (prolamine)
with a
MW greater than 5,000 such as Zein with a MW around 35,000 (Freeman
industries),
10 pregelatinized starch having a MW similar to Instant Pure-Cote B793TM
(Grain Process-
ing Corporation), ethyl cellulose MW greater than 5,000 or MW similar to
AqualonTM EC
N7, N10, N14, N22, N50, or N100 (Hercules), fish gelatin having a MW 20,000-
250,000
(Norland Products), gelatin, other animal sources with MW greater than 5,000,
acryiates/octylacrylamide copolymer MW greater than 5,000 or MW similar to
National
15 StarchTM, and Chemical Dermacry lTM 79.
In another embodiment, the solidifying agent can include ethyl cellulose,
hydroxy
ethyl cellulose, hydroxy methyl cellulose, hydroxy oropyl cellulose,
hydroxypropyl methyl
cellulose, carboxymethyl cellulose, methyl cellulose, polyether amides, corn
starch, pre-
gelatinized corn starch, polyether amides, shellac, polyvinyl pyrrolidone,
polyisobutylene
20 rubber, polyvinyl acetate phthalate, or combinations thereof. In a further
embodiment the
solidifying agent can include ammonia methacrylate, carrageenan, cellulose
acetate
phthalate aqueous such as CAPNFTM from Eastman, carboxy polymethylene,
cellulose
acetate (microcrystalline), cellulose polymers, divinyl benzene styrene,
ethylene vinyl
acetate, silicone, guar gum, guar rosin, gluten, casein, calcium caseinate,
ammonium
25 caseinate, sodium caseinate, potassium caseinate, methyl acrylate,
microcrystalline wax,
polyvinyl acetate, PVP ethyl cellulose, acrylate, PEG/PVP, xantham gum,
trirnethyl
siloxysilicate, maleic acid/anhydride copolymers, polacrilin, poloxamer,
polyethylene
oxide, poly glactic acid/poly-I-lactic acid, turpene resin, locust bean gum,
acrylic
copolymers, polyurethane dispersions, dextrin, polyvinyl alcohol-polyethylene
glycol
30 copolymers,
CA 02747845 2011-12-20
31
methyacrylic acid-ethyl acrylate copolymers such as BASF's KollicoatTM
polymers,
methacrylic acid and methacrylate based polymers such as poly(methacrylic
acid), or combinations thereof. In another embodiment, the solidifying agent
can include a combination of solidifying agents set forth in the any of the
above
discussed embodiments. Other polymers may also be suitable as the solidifying
agent, depending on the solvent vehicle components, the drug, and the specific
functional requirements of the given formulation. Other.polymers may also be
suitable as the solidifying agent, depending on the solvent vehicle
components,
the drug, and the specific functional requirements of the given formulation.
In some embodiments of the present invention, it may be desirable to add
an additional agent or substance to the formulation so as to provide enhanced
or increased adhesive characteristics. The additional adhesive agent or
substance can be an additional non-volatile solvent or an additional
solidifying
agent. Non-limiting examples of substances which might be used as additional
adhesion enhancing agents include copolymers of,methylvinyl ether and maleic
anhydride (Gantrez polymers), polyethylene glycol and polyvinyl pyrrolldone,
gelatin, low molecular weight polyisobutylene rubber, Copolymer of Acryisan
alkyl/Octylacry lam ido (Dermacryl 79TM), and various aliphatic resins and
aromatic
resins.
The non-volatile solvent system and the solidifying agent are preferably
compatible with each other.. Compatibility can be defined as i) the
solidifying
agent does not substantially negatively influence the function of the non-
volatile
solvent system; ii) the solidifying agent can-hold the non-volatile solvent
system,
in the. solidified layer so that substantially no non-volatile solvent oozes
out of
the layer, and iii) the solidified layer formed with the selected non-volatile
solvent system and the solidifying agent has-acceptable flexibility, rigidity,
tensile strength, elasticity, and adhesiveness. The weight ratio of the non-
volatile solvent system to the solidifying agent can be from about 0.1:1 to
about
10:1, or from about 0.5:1 to about 2:1.
The thickness of the formulation layer applied on the skin should also be
appropriate for a given formulation and desired drug delivery considerations.
if
the layer is too thin, the amount of the drug may not be sufficient to support
CA 02747845 2011-08-05
32
sustained delivery over the desired length of time. If the layer is too thick,
it may
take too long to form a non-messy outer surface of the soli difled.layer. If
the
drug is very potent and the solidified layer has very high tensile strength, a
layer
as thin as 0.01 mm may be sufficient. If the drug has rather low potency and
the
solidified layer has low tensile strength, a layer as thick as 2-3 mm may be
needed. Thus, for most drugs and formulations, the appropriate thickness can
be from about 0.01 mm to about=3 mm, but more typically, from about 0.05 mm
to about 1 mm.
In some embodiments, the flexibility and stretchability of a solidified layer,
or optionally solidified peelable layer, can be desirable. Skin areas over
joints
and certain muscle groups are often significantly stretched during body
movements. Such movement prevents non-stretchable patches from
maintaining good skin contact. Lotions, ointments, creams, gels, pastes, or
the
like also may not be suitable for use for the reasons cited above. = As such,
in
transdermal delivery of NSAIbs and other drugs for treating musculoskeletal
pain in joints and/or muscles, the solidifying formulations of the present
invention can offer unique advantages and benefits.
A further feature of the solid-forming formulations is related to the drying
time. If a formulation dries too quickly, the user may not have sufficient
time to
spread the formulation into a thin layer on the skin surface before the
formulation is solidified, leading to poor skin contact. If the formulation
dries too
slowly, the subject may have to wait a long time before resuming normal
activities (e.g. putting clothing on) that may remove un-solidified
formulation.
Thus, it is desirable for the drying time to be longer than about 15 seconds
but
shorter than about 15 minutes, and preferably from about 0.5 minutes to about
4
minutes.
Another feature of the formulations of the current invention is related to
solidifying formulations comprising a drug for musculoskeletal pain or
Inflammation of joint or muscles, a non-volatile solvent system comprising at
least one non-volatile solvent, a solidifying agent, and a volatile solvent
system
comprising a volatile solvent whose boiling point Is below 20 C (such a
solvent
is referred to as gaseous volatile solvent). The formulation can be stored In
a
CA 02747845 2011-08-05
33
pressurized container and be sprayed on the skin surface with the help of the
gaseous volatile solvent. Some hydrofluorocarbons commonly used as gaseous
volatile solvents in pharmaceutical or cosmetic Industries can work In this
design. More specifically, the gaseous volatile solvents may include, but not
limited to dimethyl ether, butane, 1,1, Difluoroethane, 1,1,1,2
tetrafluorethane,
1, 1, 1,2,3,3,3-heptafluoropropane, 1,1,1,3,3,3 hexafluoropropane, or a
mixture
thereof. The formulation may also be expelled out of the container and applied
on the skin via a manual pump. Formulations including a gaseous volatile
solvent are expected to "dry" much faster. Spraying the formulation onto the
skin
suffering from musculoskeletal pain or inflammation of joints or muscles can
avoid touching the skin with an applicator which can cause discomfort to
hypersensitive skin and provide an easier means of application of the
formulation to a body surface which is inconvenient to reach with an
applicator.
The formulations of the current invention may further comprise a pH
modifying agent for adjusting the pH of the formulation to a point or-a range
most suitable for the delivery of the drug. This feature can be important for
a
drug that is ionizable.
The adhesion to skin and elasticity of the material is such that the
solidified layer may not easily separate from the skin. For example, in one
embodiment, the solidified layer can be stretched in at least one direction by
up
to about 5% or even 10% or more without cracking, breaking, or separating form
a skin surface to which the solidified layer is applied.
These and other advantage can be summarized by the following non-
limiting application embodiments. The solidified formulation layer of the
present
invention can be prepared in an -initial form that is easy to apply as a
semisolid
dosage form. Additionally, the dosage form can be applied to be relatively
thick
and can contain much more active drug than a typical layer of traditional
cream,
gel, lotion, ointment, paste,=etc., and further, is not as subject to
unintentional
removal. After the evaporation of the volatile solvent(s) and the formation of
the solidified layer, the drug in the solidified layer can be delivered at
desired
delivery rates over sustained periods of time. Further, as the solidified
layer
remains adhesive and can be peelable, easy removal of the solidified layer can
CA 02747845 2011-08-05
34
occur, usually without the aid of a solvent or surfactant. In some
embodiments,
the adhesion to skin and elasticity of the material is such that the
solidified layer
will not separate from the skin upon skin stretching at highly stretchable
skin
areas, such as over joints and muscles. For example, In one embodiment, the
solidified layer can be stretched by 5% or even 10%=or greater in one
direction
without cracking, breaking, and/or separating form a skin surface to which the
solidified layer Is applied. Specific examples of applications that can
benefit
from the systems, formulations, and methods of the present invention are as
follows. In one embodiment, a solidified layer including ketoprofen,
diclofanec,
or another NSAID, or Ildocaine, ropivacaine, or another local anesthetic, can
be
formulated for treating acute injuries of joints such as joints of the angle,
knee,
wrist, back, hip, and fingers. In another embodiment, a solidified layer with
the
same active drugs can be used to treat chronic disorders, such as arthritis
(including osteoarthr(tls and rheumatoid arthritis) Induced pain of the finger
and/or toe joints.
Still another embodiment involves a formulation containing a drug
selected from the NSAID class, such as ketoprofen, piroxicam, diolofenac, and
indomethacin, which is applied topically to treat symptoms of back pain,
muscle
tension, or myofascial pain or a combination thereof. The NSAID is gradually
released from the formulation to provide pain relief over a sustained period
of
time. The formulation can become a coherent, soft solid after about 5 minutes
and remains adhered to the body surface for the length of its application. It
Is
easily removed any time after drying without leaving residual-formulation on
the
skin surface.
. In another embodiment, solidifying formulations for the delivery of drugs
that treat the causes or symptoms of diseases involving joints and muscles can
also benefit from the systems, formulations, and methods of the present
invention. Such diseases that may be applicable include, but not limited to,
osteoarthritis (OA), rheumatoid arthritis (RA), joint and skeletal pain of
various
other causes, myofascial pain, muscular pain, and sports injuries. Drugs or
drug classes that can be used for such applications include, but are not
limited
to, non-steroidal anti-inflammatory drugs (NSAIDs) such as ketoprofen,
CA 02747845 2011-08-05
piroxicam, diclofenac, and indomethacin; COX inhibitors such as non-selective
COX inhibitors, COX-2 selective NSAIDs and agents, COX-3 selective NSAIDs
and agents; local anesthetics such as lidocaine, bupivacaine, ropivacaine, and
tetracalne; 5HT-2A receptor antagonists such as ketanserin; and steroids such
5 as dexamethasone, hydrocortisone, prednisone, prednisolone,
methylprednisolone, halobetasol propionate, betamethasone dipropionate,
betamethasone, prodrugs thereof, or combinations thereof.
The solidifying formulations and the methods of the current invention are
expected, to be particularly useful for treating inflammation and/or pain of
small
10 joints such as the joints of toes, wrists, ankles, elbow, and especially
fingers, as
well as chronic musculoskeletal pain that is not necessarily associated. with
inflammation. Because the pathway from the skin surface to the joints are
shorter for smaller joints, therapeutically beneficial amounts of the drugs
are
more likely reach smaller joints before being taken away by the blood
15 circulation. In addition, as the fingers are often used, bent, and
contacted by
many objects during normal activities, it is difficult to keep a conventional
dosage form or formulation, such as a patch or cream, on the fingers.
Furthermore, some physical therapy devices, such as ThermaCareTM heating
pads, are too big for finger joints. Therefore, there are many unmet needs for
20 treating the pain or inflammation of finger joints. By applying a drug
formulation
to the skin overlying affected joints or muscles, the drug can penetrate the
skin
and directly enter the target tissues (before being taken away by the blood
circulation) and establish therapeutic local tissue concentrations without
causing
significantly high systemic drug concentrations that are associated with
adverse
25 side effects. Under such a scenario, it would be easier to deliver the
drugs into
the tissues of the smaller joints, including the joints of the wrist, elbow,
ankle,
toe, and particularly the finger, than to that of larger joints such as knees
and
hips, due to the short pathway between the skin surface and the small joints.
Therefore, one method of the present invention uses the solidifying
formulations
30 containing NSAID(s), local anesthetic(s), and/or steroid(s) for treating
inflammation or pain of small joints, and particularly of finger joints. This
being
stated, treatment of larger joints or areas of the body can also be treated,
such
CA 02747845 2011-08-05
36
as the back, neck, shoulder, or hip, is also efficacious. Another embodiment
entails a solidifying formulation containing a -drug from the class of alpha-2
antagonists which is applied topically to treat neuropathic pain. The alpha-2
agonist is gradually released from the formulation to provide pain relief over
a
sustained period of time. The formulation can become a coherent, soft solid
and remains adhered to the body surface for the length of its application. It
is
easily removed after drying without leaving residual formulation on the skin
surface.
Another embodiment involves a formulation containing capsaicin which is
applied topically to treat neuropathic pain. The capsaicin is gradually
released
from the formulation for treating this pain over a sustained period of time.
The
formulation can become a coherent, soft solid and remain adhered to the body
surface for the length of its application. It is easily removed any time after
drying without leaving residual formulation on the skin surface.
A further embodiment involves a solidified formulation containing at least
one alpha-2 agonist drug, at least one tricyclic antidepressant agent, and/or
at
least one local anesthetic drug which Is applied topically to treat
neuropathic
pain. The drugs are gradually released from the formulation to provide pain
relief over a sustained period of time. The formulation can become a coherent,
soft solid and remain adhered to the body surface for the length of its
application. It is easily removed any time after drying without leaving
residual
formulation on the skin surface.
In another embodiment, the delivery of drugs for treating neuropathic
pain can also benefit from the methods, systems, and formulations of the
present invention. A patch containing a local anesthetic agent is used for
treating neuropathic pain, such as pain caused by post-herpetic neuralgia. Due
to the limitations of the patch as discussed above, a solidified layer
prepared in
accordance with the present invention provides some unique benefits, as well
as provide a potentially less expensive alternative to the use of such a
patch.
Possible drugs delivered for such applications include, but are not limited
to,
local anesthetics such as lidocaine, prilocalne, tetracaine, bupivicalne,
CA 02747845 2011-08-05
37
etidocalne; and other drugs including ketamine, amitriptyline, capsaicin,
tricyclic
antidepressants, alpha-2 agonists such as clonidine, or combinations thereof.
In one embodiment, the drug can be an antiviral agent and the solidified
layer is capable of generating a flux of the antiviral agent of at least 2
mcg/cm2/h. In another embodiment, the drug can be a local anesthetic and the
solidified layer is capable of generating a flux of the local anesthetic of at
least 5
mcg/cm2/h. In another embodiment, the drug can be an alpha-2 agonist and the
solidified layer is capable of generating a flux of the alpha-2 agonist of at
leastl
mcg/cm2/h. In another embodiment, the drug is capsalcin and the solidified
layer is capable of generating a flux of capsaicin of at least 5 mcg/cm2/h. In
yet
a further embodiment, the drug is ketamine and the solidified layer is capable
of
generating a flux of ketamine of at least 1 mcg/cm2/h.
Solidifying formulations of the current Invention that comprise two or
more active drugs may provide additional benefits. For example, a formulation
for treating neuropathic pain in accordance with the current invention may
include lidocalne and tetracalne. The iidocaine and tetracain can be present
in
either the salt form or in the base form. Preferably, the non-volatile solvent
system includes at least one of propylene glycol and dipropylene glycol, and
isostearic acid. Similar formulations may comprise other combinations of
drugs,
such as amitriptyline and ketamine, amitriptyline and a "local anesthetic,
etc.
As a further note, it is a unique feature of the solidified layers of the
present invention that they can keep a substantial amount of the non-volatile
solvent system, which is optimized for delivering the drug, on the body
surface.
This feature can provide unique advantages over existing products. For
example, in some semi-solid formulations, upon application to a skin surface
the
volatile solvents quickly evaporate and the formulation layer solidifies into
a hard
lacquer-like layer. The drug molecules are immobilized in the hard lacquer
layer
and are substantially unavailable for delivery into the skin surface. As a
result, it
is believed that the delivery of the drug is not sustained over a long period
of
time. In contrast to this type of formulation, the solidified layers formed
using
the formulations of the present invention keep the drug molecules quite mobile
CA 02747845 2011-08-05
38
in the non-volatile solvent system which is in contact with the skin surface,
thus
ensuring sustained delivery.
EXAMPLES
The following examples illustrate the embodiments of the invention that
are presently best known. However, it is to be understood that the following
are
only exemplary or illustrative of the application of the principles of the
present
invention. Numerous modifications and alternative compositions, methods, and
systems may be devised by those skilled in the art without departing from the
spirit and scope of the present invention. The appended claims are intended to
cover such modifications and arrangements- Thus, while the present invention
has been described above with particularity, the following examples provide
further detail in connection with what are presently deemed to be the most
practical and'preferred embodiments of the invention.
Example 1
Hairless mouse skin (HMS) or human epidermal membrane (HEM) is
used as the model membranes as noted for the in vitro flux studies described
in
herein. Hairless mouse skin (HMS) is used as the model membrane for the in
vitro flux studies described in herein. Freshly separated epidermis removed
from the abdomen of a hairless mouse is mounted carefully between the donor
and receiver chambers of a Franz diffusion cell. The receiver chamber is
filled
with pH 7.4 phosphate buffered saline (PBS). The experiment is initiated by
placing test formulations on the stratum corneum (SC) of the skin sample.
Franz cells are placed in a heating block maintained at 37 C and the HMS
temperature is maintained at 35 C. At predetermined time intervals, 800 L
aliquots are withdrawn and replaced with fresh PBS solution. Skin flux
( g/cm2/h) is determined from the steady-state slope of a plot of the
cumulative
. amount of permeation versus time. It is to be noted that human'cadaver skin
can be used as the model membrane for the in vitro flux studies as well. The
CA 02747845 2011-08-05
39
mounting of the skin and the sampling techniques used as the same as
described above for the HMS studies.
Example 2
Formulations of ropivacaine (base) in various non-volatile solvent
systems are evaluated. Excess ropivacaine is present. The permeation of
ropivacaine from the test formulations through HMS is presented in Table 2
below.
Table 2
Non-volatile solvent system Skin Flux*
nic lcmZ/h
Glycerol 1.2 0.7
Tween 20 2.4 0.1
Mineral Oil 8.9 0.6
ISA lsostearic Acid) 11 :t2
Span 20 26 8
* Skin flux measurements represent the mean and standard
deviation of three determinations. Flux measurements reported
were determined from the linear region of the cumulative amount
versus time plots. The linear region was observed to be between
4-8 hours. If experimental conditions allowed, the steady-state
delivery would likely continue well beyond 8 hours.
Steady state flux of ropivacaine base from the above non-volatile solvents are
obtained by placing 200 mcL on the stratum corneum side (donor) of hairless
mouse skin. The in vitro studies are carried out as described in Example 1.
20. From Table 2, the non-volatile solvents glycerol, and Tween 20 had low
steady
state flux values and would not be considered "flux-enabling". However,
mineral
oil and isostearic acid are flux-enabling solvents and are good candidates for
evaluation with solidifying agents and volatile solvents to design an
acceptable
peel formulation. Surprisingly Span 20 has much higher steady state flux
values
and would also qualify as a high flux-enabling solvent.
Example 3
Formulations of diclofenac sodium in various non-volatile solvent systems
are evaluated. Excess diclofenac sodium is present. The permeation of
CA 02747845 2011-08-05
diclodenac sodium from the test formulations through HMS is presented in
Table 3 below.
Table 3
Non-volatile solvent system Skin Flux*
me !cm !h
Glycerol . 1.7 t 0.3
isopropyl Myristate 13* 3
Ethyl Oleate 14:t 4
Propylene Glycol 30 t 30
San 20 98 t 20
* Skin flux measurements represent the mean and standard
5 deviation of three determinations. Flux measurements reported
were determined from the linear region of the cumulative amount
versus time plots. The linear region was observed to be between
4-8 hours. If experimental conditions allowed, the steady-state
delivery would likely continue well beyond 8 hours.
Steady state flux of diclofenac sodium from the above non-volatile solvents
are
obtained by placing 200 mcL on the stratum comeum side (donor) of hairless
mouse skin. The in vitro studies are carried out as described In Example 1.
From Table 3, the non-volatile solvent glycerol has a steady state flux value
comparable to the estimated therapeutic steady state flux value of 1 mcg/cm2/h
and may be considered a flux-enabling solvent. -However, the steady state flux
values of isopropyl myristate, ethyl oleate, propylene glycol, and Span 20 are
at
least 10 times the flux value reported for glycerol and are considered flux
enabling.
Example 4
Formulations of diclofenac acid in various non-volatile solvent systems
are evaluated. Excess diclofenac acid is present. The permeation of diclofenac
from the test formulations through HMS is presented in Table 4 below.
Table 4
Non-volatile solvent system Skin Flux*
me /cmz/h
Glycerol 0
Isopropyl Myristate 8:t 3
Eth l Oleate 7 t 3
Propylene Glycol 5 :t2
CA 02747845 2011-08-05
41
Span 20 3t1
* Skin flux measurements represent the mean and standard
deviation of three determinations. Flux measurements reported
were determined from the linear region of the cumulative amount
versus time plots. The linear region was observed to be between
4-8 hours. If experimental conditions allowed, the steady-state
delivery would likely continue well beyond 8 hours.
Steady state flux of diclofenac acid from the above non-volatile solvents are
obtained by placing 200 mcL on the stratum corneum side (donor) of hairless
mouse skin. The in vitro studies are carried out as described In Example 1.
From Table 4, the non-volatile solvent glycerol has no reported steady state
flux
value and is not considered a flux enabling non-volatile solvent viable non-
volatile solvent candidate. However, the steady state flux values of isopropyl
myristate, ethyl oleate, propylene glycol, and Span 20 are no-more than 10
times the flux value reported for currently available marketed products, and
as
such, could be considered flux-enabling solvents. It should be noted that the
steady state flux values for diclofenac acid from each of the above non-
volatile
solvents are much lower than the steady state flux values obtained with
diclofenac sodium. Therefore, if therapeutically effective flux values need to
be
increased, utilizing a flux-enabling non-volatile solvent and the salt form of
diclofenac would likely yield higher steady state flux values than using the
acid
form of diclofenac.
Examples 5-7
Prototype peel formulations are prepared as follows. Several peel
formulations are prepared in accordance with embodiments of the present
invention in accordance with Table 5, as follows:
Table 5 .
Example 5 6 7
% by weight
Volatile Solvents
Ethanol 21 24 18.5
Water 32 28
Solidifying agents
Eudra it RL-PO 40
Eudragit E-100 18.5
CA 02747845 2011-08-05
42
Polyvinyl Alcohol 21 18.5
Non-volatile solvents
Glycerol 12
Propylene Glycol 21 4
Isostearic Acid 13
San 20 11
Trolamine -4
Drug
Ketoprofen
Roplvacaine 3
Diciofenac Na 5.5
Peel formulations of Examples 5-7 are prepared in the following manner:
= The solidifying agents are dissolved in the volatile solvent (e.g., dissolve
polyvinyl alcohol in water, Eudragit polymers in ethanol),
5 = The non-volatile solvent is mixed with the solidifying agent/volatile
solvent
mixture.
= The resulting solution is vigorously mixed well for several minutes.
= The drug is then added and the peel formulation is mixed again for
several minutes.
In all the examples noted above, the flux-enabling non-volatile
solvent/solidifying agent/volatile solvent combination is compatible as
evidenced
by a homogeneous, single phase system that exhibited appropriate drying time,
and provided a stretchable peel and steady state flux for the drug (see
Example
8 below).
Example 8
The formulations of the examples are tested in a hairless mouse skin
(HMS) or human epidermal membrane (HEM) in vitro model described in
Example 1. Table 6 shows data obtained using the experimental process
outlined above.
Table 6 - Stead -state flux (J)
X
Formulation 1crn21h
Example 5 35.-t 20***
Example 6 32 t 2***
Example-7 5 at 2****
CA 02747845 2011-08-05
43
Skin flux measurements represent the mean and standard
deviation of three determinations.
***Flux measurements across HMS reported were determined
from the linear region of the cumulative amount versus time plots. The
linear region was observed to be between 4-8 hours. If experimental
conditions allowed, the steady-state delivery would likely continue well
beyond 8 hours.
****Flux measurements across HEM reported were determined
from the linear region of the cumulative amount versus time plots. The
linear region was observed to be between 6-28 hours. If the experiment
was continued It Is anticipated the steady state would continue.
In all cases in Table 6, the flux enabling non-volatile solvents in the
formulation
resulted in therapeutically effective flux for each of the formulations
studied.
FIGS. 1 and 2 provide a graphical representation of the cumulative
amount of diciofenac and ropivacaine, respectively, delivered transdermally
across human cadaver skin. The formulations tested were similar to those
described in Examples 6 and 7. In these particularly embodiments, steady-state
delivery is shown over 28 hours, and over 30 hours, repsectively.
Example 9
A placebo formulation with the following composition: -10.4% polyvinyl
alcohol, 10.4% polyethylene glycol 400, 10.4% polyvinyl pyrrolidone K-90,
10.4% glycerol, 27.1 % water, and 31.3% ethanol-was applied onto a human
skin surface at an elbow joint and a finger joint, resulting in a thin,
transparent,
flexible, and stretchable film. After a few minutes of evaporation of the
volatile
solvents (ethanol and water), a solidified layer that was peelable was formed.
The stretchable peel had good adhesion to the skin and did not separate from
the skin on joints when bent, and could easily be peeled away from the
skin. Addition of an active drug into this placebo formulation is not expected
to
significantly change the physical properties of the initial formulation or the
solidified layer, as the concentration of the active drug as a percentage of
the
total weight of the formulation is typically small.
CA 02747845 2011-08-05
44
Examples 10-12
Three formulations are applied on the stratum comeum side of freshly
separated hairless mouse skin. The in vitro flux is determined for each
formulation as outlined in Example 1. =The formulation compositions are noted
in Table 7 below.
Table 7
Example 10 11 12
b weight
PVA 15 15 15
Water 23 23 23
Ethylcellulose ECN-1 00 11 11 11
Ethanol 33 '33 33
Span 20 11
Polyethylene GI col 400 11
Tween 40 11
Tromethamine 4 4 4
Ropivacaine HCI 3 3 3
Avg. Flux* me cm /h 15 t.1 4.7 * 0.3 3.4 7t 0.7
* Flux values represent the mean and standard deviation of three
determinations. Flux measurements reported were determined from the
linear region of the cumulative amount versus time plots. The linear
region was observed to be between 4-9 hours. If the experiment was
continued it is anticipated the steady state would continue.
Since all three formulations have the exact same compositions of
solidifying agent, volatile solvents, and flux-enabling non-volatile solvent.
The
only difference is which flux-enabling non-volatile solvent is used it is
reasonable to conclude that for ropivacaine HCI that Example 10 is flux
enabling.
Examples 13-14
A peel-forming formulation for dermal delivery of ropivacaine is prepared
which includes a specified amount of ropivacaine in an excipient mixture to
form
an adhesive formulation in accordance with embodiments of the present
invention. The peel formulations contained the following components:
CA 02747845 2011-08-05
Table 8 Ropivacalne eelable formulations
Ingredients* Examples
13 14
Eudragit RL-100 39.6% 39.6%
Ethanol 23.7% 23.6%
ISA lsostearic Acid) 13.5% 13.5%
PG (Propylene Glycol) 7.9% 4.0%
Trolamine 4.0% 4.0%
Glycerol 7.9% 11.9%
Ro ivacaine 3.4% 3.4%
Ingredients are noted as weight percent..
These formulations are applied to HMS skin as described in Example 1, and the
5 ropivacaine flux is measured. A summary of the results from in vitro flux
studies
carried out with the formulations in Examples 13 and 14 Is listed In Table 9.
Table 9 - Steady-state flux of ropivacaine through hairless mouse skin
from various adhesive eelable formulations at 35 C
Formulation Average flux
me !cm 1h
Example 13 36;t 5
Example 14 32:t 2
10 " The flux values represent the mean and SD of three
determinations
Regarding the formulation described In Examples 13 and 14, ethanol'is used as
the volatile solvent, and the ISA, glycerol, and PG mixture is used as the non-
15 volatile solvent system. Through experimentation, it is determined that ISA
and
propylene glycol used together to provide the appropriate flux for the drug,
while
being compatible with the Eudragit RL-1 00 solidifying agent. Further, In this
embodiment, ISA, PG and glycerol serve as a plasticizer in the peelable
formulation after the ethanol. (volatile solvent) has evaporated. The steady
state
20 flux of ropivacalne from formulation Examples 13 and 14 demonstrate the
importance of the non-volatile solvent in dictating the flux-generating power
of
the entire formulation.
CA 02747845 2011-08-05
46
Example 15
The effect of solubility on permeation, compatibility between the non-
volatile solvent system and the solidifying agent is shown in this Example.
Ropivacaine base solubility in isostearic acid (ISA) is experimentally
determined
to be slightly above 1:4, meaning 1 gram ropivacalne base can completely
dissolve in 4 gram isostearic acid. In one experiment, two solutions are made:
Solution A includes 1 part ropivacaine base and 4 parts Isostearic acid.
Solution B includes 1 part ropivacaine base, 4 parts isostearic acid, and 1
part
trolamine. (all parts are in weight). All ropivacaine in Solution A is
dissolved, but
only a portion of ropivacaine in solution B is dissolved. The transdermal flux
across hairless mouse skin generated by the solutions is measured by a typical
Franz Cell system, with the following results:
Table 10 - Flux across hairless mouse skin, in vitro, in iglhr/cm2
Cell I Cell2 Cell3 Average
Solution A 13.1 9.9 9.1 10.7
Solution B 43.2 35.0 50.0 42.7
As can be seen, the flux generated by Solution B is about 4 times that of
Solution A. These results demonstrate that the addition of the ion paring
agent
trolamine significantly increases the transdermal flux. However, the attempt
to
Incorporate this system Into a poly vinyl alcohol (PVA) based peel formulation
failed because the PVA In the formulation acted as a strong pH buffer that
inhibited the effect of trolamine. Addition of more trolamine, In attempt to
over-
power the pH buffer capacity of PVA, caused the loss of the desired
solidifying
property of PVA (in other words, a non-volatile solvent system containing ISA
and too much trolamine Is not compatible with PVA). When PVA is replaced by
another solidifying agent, Eudragit RL 100 (Rohm & Haas), the effect of
trolamine is not inhibited and formulations capable of generating fluxes
around
pg/hr/cm2 were obtained. A by product of the addition of trolamine, ISA, and
Eudragit RL 100 is that a precipitate forms from the ionic interaction of the
three
components. The latter Example produced a better formulation in terms of flux
30 and wear properties, but the precipitation still demonstrates the need for
CA 02747845 2011-08-05
47
improvement. In an effort to eliminate the ionic interaction between non-
volatile
solvent and solidifying agent the trolamine, ISA mixture was added to
Pla'stoid B
polymer in isopropanol. However, in this instance the trolamine was found to
be
incompatible with the Plastoid B polymer and the base was changed to
trilsopropanolamine. This combination eliminated the precipitate formed when
the Eudragit RL 100 polymer was used and produced a clear formulation that
was capable of generated flux values around 30 pg/hr/cm2 . This demonstrates
the importance of compatibility between the non-volatile solvent system and
the
solidifying agent.
Example 16
A solidifying formulation for dermal delivery of ropivacaine is prepared
from the following ingredients:
Table 11 - Ropivacalne solidifying formulation components
Ingredients* Example
1 t3 .
Ropivacaine HCI 0.096
Eudragit RL- 100 1.0
Ethanol 0.7
Isostearic Acid 0.34
Glycerol 0.3
Pro lene=GI cot 0.1
Trolamine Ø15
*Ingredients are noted as parts by weight.
The ingredients listed above are combined according to the following
procedure.
The Eudragit RL-1 00 and ethanol are combined In a glass jar and heated to
about 60 C until the Eudragit RL-100 is completely dissolved. Once the
Eudragit solution cooled to room temperature, the appropriate amount of
ropivacaine HCl is added and mixed thoroughly for 1 minute. To this solution,
isostearic acid (ISA) is added and the mixture is stirred vigorously for 2-3
minutes. One hour later, the solution is vigorously mixed again for 2-3
minutes.
To this solution, glycerol, propylene' glycol, and trolarnine are added in
sequential order. After addition of each ingredient the solution is stirred
for 1
minute.
CA 02747845 2011-08-05
48
Example 17
The formulation prepared in accordance with Example 16 is applied to
HMS as described in Example 1, and the ropivacaine flux was measured. A
summary of the results is listed in Table 12, as follows:
Table 12 - Steady-state flux of ropivacaine through hairless mouse skin
from various adhesive peelable formulations at 35 C
Formulation Average flux
me /cm2/h*
Example 16 43* 4
* The flux values represent the mean and SD of three
determinations
The ropivacalne peel formulation prepared in accordance with Example 16
possessed acceptable application properties, e.g., ease of removal of peel
from
the sample tube, ease of spreading on intended skin application site, etc.,
and
forms a solidified film in 2-3 minutes after being applied to normal human
skin
surface as a thin layer with a thickness of about 0.1 mm. The solidified layer
becomes more easily peelable in 2 hours, and the peel remains affixed to the
skin surface without any unintended removal of the peel for at least 12 hours.
At the end of intended use, the peel is easily removed in one continuous
piece.
Example 18
A solidifying formulation for dermal delivery of lidocaine (base) is
prepared which includes a saturated amount of lidocaine in an excipient
mixture
to form an adhesive formulation in accordance with embodiments of the present
invention. The peel formulation is prepared from the ingredients as shown in
Table 13.
Table 13 - Lldocaine solidifying -formulation components.
Ingredients* Example
18
PVA 11.7
Eud ra it E-100** 11.7
PVP-K90 5.8
CA 02747845 2011-08-05
49
Glycerol 8.8
PEG-400 8.8
Water 23.8
Ethanol 23.8
Lidocaine 5.6
*Ingredients are noted as weight percent.
** from Rohm & Haas.
Table 14 Steady-state flux of lidocalne through hairless mouse. skin
from various adhesive solidi . in formulations at 35 C
Formulation Average flux
me Icm Ih
Example 18 47:t 3
The adhesive formulation of lidocalne formulation in the present Example
18 has similar physical properties to the examples noted above. The
transdermai flux across hairless mouse skin is acceptable and steady-state
delivery is maintained over 8 hours.
Examples 19-22
Solidifying formulations for dermal delivery of ropivacaine are prepared
which includes an excipient mixture to form an adhesive solidifying
formulation
in accordance with embodiments of the present Invention. The peel
formulations are prepared from the ingredients as shown In Table 15;
Table 15 - Ro lvacaine' HCI solidifying formulation components
Ingredients* Exam le
'19 20 21 22 .
Ropivacaine HCI 0.31 0.31 0.31 0.31
Iso ro anol 2 2 2.2 2
Water 0.125 0.125 0.125 0.125
Isostearic Acid 0.36 0.66 0.41 0
Trilso ro anolamine 0.31 0.34 0.34 0.34
Triacetin 0.17 0.19 0, 0.19
Span 20 0.34 0 0.37 0.66
Plastoid B** 1 1 1 1
Ingredients are noted as parts by weight.
*"` from Degussa.
CA 02747845 2011-08-05
The ingredients listed above are combined according to the following
procedure.
The ropivacaine HCl, water, and triisopropanolamine are combined in a glass
jar
and mixed until the drug is dissolved. Then the isostearic acid, triacetin,
Span
20, and isopropanol. are added to the formulation and mixed well. The polymer
5 Plastoid B Is added last and heated to about 60 C until the Plastoid B Is
completely dissolved. Once the polymer solution cooled to room temperature,
the formulation is stirred vigorously for 2-3 minutes.
The formulations in Table 15 are applied to HMS according to Example 1,
and the: flux of ropivacaine was measured. The results are summarized in Table
'10 16:
Table 16 - Steady-state flux of ropivacaine HCI through hairless mouse skin
from various adhesive solidifying formulations at 35 C
Formulation Average flux
me !cm !h
Example 19 56* 2
Example 20 39 6
Example 21 31 6
Example 22 37 9
The flux of Examples 19-22 show the importance of the triacetin, isostearic
acid,
15 Span 20 combination in the formulation. In Examples 20-22 formulations were
made without Span 20, triacetin, and isostearic acid respectively. The in
vitro
flux of ropivacaine was Impacted. The synergistic combination of the non
volatile solvents is an important In obtaining the maximum In vitro flux of
ropivacalne.
Exam le 23
This solidifying formulation has the following ingredients in the indicated
weight parts: .
Table 17
Ethyl Dermacryl Isostearic
PVA Water Cellulose 79 = Ethanol Acid Glycerol Ropivacaine
ECN-7 (National (iSA)
A ualon Starch
1 1.5 0.25 0.35 0.85 0.8 0.35 0.3
CA 02747845 2011-08-05
51
In this formulation, polyvinyl alcohol (USP grade MW 31,000-50,000, from
Amresco) Is .a solidifying agent, ethyl cellulose and Dermacryl 79 are
auxiliary
solidifying agents. Isostearic acid and glycerol form the non-volatile solvent
system while ethanol and water form the volatile solvent system. Ropivacalne
Is
the drug.
Procedures of making the formulation:
1. Ropivacaine is mixed with ISA.
2. Ethyl cellulose and Dermacryl 79 are dissolved in ethanol.
3. PVA is dissolved in water at temperature of about 60-70 C.
4. All of the above mixtures are combined together in one container and
glycerol is added and the whole mixture is mixed well.
The resulting formulation is a viscous fluid. When a layer of about 0.1 mm
thick
is applied on skin, a non-tacky surface is formed in less than 2 minutes.
Examples 24-27
A stretchable adhesive formulation for transdermal delivery of ketoprofen
(which is suitable for delivery via skin for treating inflammation or pain of
joints
and muscles) is prepared which includes saturated amount of ketoprofen in an
excipient mixture (more ketoprofen than that can be dissolved in the excipient
mixture) to form an adhesive formulation, some of which is prepared in
accordance with embodiments of the present invention. The excipient mixture,
which is a viscous and transparent fluid, is prepared using the ingredients as
shown in Table 18.
CA 02747845 2011-08-05
52
Table =18 - Ketoprofen solidifying formulation components
Ingredients* Examples
24 25 26 27
PVA (Polyvinyl Alcohol) 10.4 21.4 21.1 21.2
PEG-400 (Polyethylene Glycol) 10.4 10.8 2.9 18.6
PVP-K90 Pol in I P rrolidone 10.4 0.0 0.0 0.0
Glycerol 10.4 10.8 19.0 2.9
Water 27.1 57.0 57.0 67.3
Ethanol 31.3 0 .0 0
Ketoprofen saturated saturated saturated saturated
` Ingredients are noted as % by weight.
Each of the compositions of Examples 24-27 were studied for flux of
ketoprofen,
as shown in Table 19, as follows:
Table 19 - Steady-state flux of Ketoprofen through hairless mouse skin
from various adhesive formulations at 35 C
Formulation Average flux
me lcm !h
Example 24 8:t 3
Example 25 21 j:6
Example 26 3 1
Exam le 27 1 = t 0.4
* Skin flux measurements represent the mean and standard
deviation of three determinations. Flux measurements reported were
determined from the linear region of the cumulative amount versus time
plots. The linear region was observed to be between 4-8 hours. If
experimental conditions allowed the steady state flux would extend
beyond the 8 hours measured.
Regarding formulation described in Example 24, ethanol and water formed the
volatile solvent system, while a 1:1 mixture of glycerol and PEG 400 formed
the
non-volatile solvent system. Through experimentation, It is determined that
PEG 400 is a slightly better solvent than glycerol for ketoprofen, while
glycerol is
much more compatible with PVA than PEG 400. Thus, the non-volatile solvent
system of glycerol and PEG 400 are used together to provide a non-volatile
solvent system for the drug, while being reasonably compatible with PVA. In
additional detail with respect to the formulation in Example 24, PVA and PVP
act as the solidifying agents. Further, in this embodiment, glycerol and PEG
400 also serve as plasticizers in the adhesive formulation formed after the
CA 02747845 2011-08-05
53
evaporation of the volatile solvents. Without the presence of glycerol and PEG
400, a film formed by PVA and PVP alone would be rigid and non-stretchable.
Regarding the formulation of Example 25, the adhesive peelabie
formation formed has similar physical properties as that of Example 24, though
the transdermal flux across hairless mouse skin is higher. This suggests that
the solidifying agent,. 1:1 PVA:PVP-K-90 in Example 24 and pure PVA in
Example 25, have an Impact on permeation.
The formulation in Example 26 delivers less ketoprofen than the
formulations of Examples 24 or 25 The formulation of Example 27 delivers
much less ketoprofen than the formulations in Examples 24 and 25. One
possible reason for the reduced flux is believed to be the reduced permeation
driving force caused by the high concentration of PEG 400 in the non-volatile
solvent system, which resulted in too high of solubility for ketoprofen.
The only significant difference among the formulations in Examples 25,
26, and 27, respectively, Is with respect to the non-volatile solvent system,
or
more specifically, the PEG 400:glyceroi weight ratio. These results reflect
the
impact of the non-volatile solvent system on skin flux.
Example 28
A stretchable adhesive formulation for transdermal delivery of ketoprolen
(which is suitable for delivery via skin for treating inflammation or paiff'of
joints
and muscles) is prepared which includes ketoprofen in an excipient mixture to
form an adhesive formulation, some of which is prepared in accordance with
embodiments of the present invention. The peel formulation is prepared from
the Ingredients as shown in Table 20.
Table 20 - Ketoprofen solidifying formulation com onents
Ingredients'" Example Example
29 30
PVA 22.1 18.9
Water 30.9 37.9
Fumed Silica 3.0
Glycerol 11.1 9.5
Propylene glycol 17.7 15.2
Gantrez ES-425 4.4 3.8
CA 02747845 2011-08-05
54
Ethanol 8.8 7.6
Ketoprofen 5.0 4.2
*Ingredients are noted as weight percent.
Table 21 - Steady-state flux of Ketoprofen through hairless mouse skin
from an adhesive solidi in formulations at 35 C
Formulation Average flux
me !cm 1h
Example 29 25 6
Example 30 27 t 2
* Skin flux measurements represent the mean and standard
deviation of three determinations. Flux measurements reported were
determined from the linear region of the cumulative amount versus time
plots. The linear region was observed to be between 4-8 hours. If
experimental conditions allowed the steady state flux would extend
beyond the 8 hours measured.
Example 29-31
A stretchable adhesive formulation for transdermal delivery of ketoprofen
(which is suitable for delivery via skin on joints and muscles) is prepared
which
includes saturated amount of ketoprofen in an excipient mixture (more
ketoprofen than that can be dissolved in the excipient mixture) to form an
adhesive formulation, some of which are prepared in accordance with
embodiments of the present Invention. The excipient mixture, which is a
viscous and transparent fluid, Is prepared using the ingredients as shown in
Table 22.
Table 22
Ingredients" Examples
29 30 31
Eu ra it RL-PO 28.06 27.7 27.5
Ethanol 40.07 39.5 39.5
Glycerol 27.40 13.9
Polyethylene Glycol 400 PEG 13.9 28.
Keto rofen 4.5 5 5
Peel formulations of Examples 29-31 are prepared in the following manner:
The solidifying agents are dissolved in the volatile solvent (i.e., dissolve
Eudragit polymers in ethanol).
CA 02747845 2011-08-05
= The flux adequate non-volatile solvent (glycerol, PEG) is mixed together
with the solidifying agent/volatile solvent mixture.
= The resulting solution is vigorously mixed for several minutes.
= Drug is then added and the formulation is mixed again for several
5 minutes.
Example 32
The formulations prepared in accordance with Example 29-31 are applied
to HMS as described in Example 1, and the ketoprofen flux is measured. A
10 summary of the results is listed in Table 23, as follows:
Table 23 - Stead -state flux of keto rofen through hairless mouse skin
Formulation Average flux
me lcm2/h*
.
Example 29 15:1:7
Example 30 10 t 3
Example 31 4 1
* Skin flux measurements represent the mean and standard
deviation of three determinations. Flux measurements reported were
15 determined from the linear region of the cumulative amount versus time
plots. The .linear region was observed to be between 4-8 hours. If
experimental conditions allowed the steady state flux would extend
beyond the 8 hours measured.
20 The ketoprofen adhesive solidifying formulations prepared in accordance
with
Examples 29-30 possessed acceptable solidified film properties (e.g., formed a
solidified layer In 2-3 minutes). With Example 31, the ketoprofen formulation
does not form a solidified layer 30 minutes after application. This
demonstrates
that order to obtain desired flux and wear properties in a peel formulation, a
25 delicate balance between solidifying agents, non-volatile solvents, and
volatile
solvents is evaluated and considered in developing a formulation.
Example 33
An adhesive solidifying formulation for transdermal delivery of ketoprofen,
30 which can form elastic solidified layers and is suitable for delivery via
skin on
joints and muscles, Is prepared which includes saturated amount of ketoprofen
CA 02747845 2011-08-05
56
in an excipient mixture (more ketoprofen than that can be dissolved in the
excipient mixture) to form an adhesive formulation, some of which are prepared
in accordance with embodiments of the present invention. The exciplent
mixture, which is a viscous and transparent fluid, Is prepared using the
ingredients as shown in Table 24.
Table 24
FORMULATIONS
Ingredients* A B C
PVA Ceivol 502 MW 10 000 24.4
PVA (Amresco MW 31,000-
50.,000 . 24.4
PVA (Celvol 623 MW 125,000) 41.7
Water 33.4 33.4 58.3
Ethanol 8.9 8.9
PG 17.8 . 17.8
Glycerol 11.1 11.1
Gantrez ES 425 4.4 4.4
Ingredients are noted in weight percent.
Formulations A and B are prepared in.the following manner:
= PVA (solidifying agent) is dissolved in water.
= The flux adequate non-volatile solvent (glycerol, PG) is mixed together
with the solidifying agent/volatile solvent mixture.
= Then ethanol, and Gantrez ES 425 is added to the mixture.
= The resulting solution Is vigorously mixed for several minutes.
Preparation of the PVA in water solution in Formulation C was not feasible for
this molecular weight of PVA at the percentages noted. Formulation C
demonstrates that the correct polymer molecular weight for PVA is important to
obtain the desired formulation properties.
Formulations A and B are placed on the skin of human volunteers. After
a period of several hours, long enough for the volatile solvent to evaporate,
the
peels were removed by the volunteers and the peelability properties were
evaluated. In all instances the volunteers reported that formulation example A
could not be removed in one or two pieces, but was removed in numerous small
pieces. Formulation example B. removed in one or two pieces. The lack of
cohesion nature of formulation A is attributed to the lower molecular weight
PVA
CA 02747845 2011-08-05
57
sample (Celvol). Low molecular weight PVA does not possess the same
cohesive strength as higher molecular weight PVA material (Amresco) due to
the reduced size of the polymer chain leading to a reduction in the degree of
cross linking and physical interactions between individual PVA polymer chains.
The reduced PVA chain interactions lead to a weaker solidified layer that is
unable to withstand the mechanical forces it is subjected to upon removal,
Example 34-35
A stretchable adhesive formulation for transdermal delivery of ketoprofen
(which is suitable for delivery via skin on Joints and muscles) was evaluated
.
which includes a placebo excipient mixture which will form an adhesive
formulation, some of which are'p'repared In accordance with embodiments of the
present invention. The excipient mixture, which Is a viscous and transparent
fluid, is prepared using the Ingredients as shown in Table 25,
Table 25
Ingredients* E=xamples
34 35
PVA Amresco MW 31,000-50,000) 20.41 21.28
Water 30.61 27.66
Ethanol 20.41 21.28
PG. 20.41 21.28
Glycerol 6.12 6.38
Gantrez S97 2.04 . = 2.13
Ingredients are noted in weight percent.
Peel formulations in Examples 1 and 2 are prepared in the following manner:
= PVA (solidifying agent) is dissolved in water.
= The flux adequate non-volatile solvent (glycerol, PG).is mixed together
with the solidifying agent/volatile solvent mixture.
= Then ethanol, and Gantrez S97 is added to the mixture.
= The resulting solution is vigorously mixed for several minutes.
Formulations above were applied on the forearms of study volunteers
and the drying time was assessed by placing a piece of cotton to the
application
site and then applying a 5 gram weight on the cotton. The cotton and weight
was removed after 5 seconds. This procedure was started approximately 3 - 4
CA 02747845 2011-08-05
58
minutes after application and at 10 to 60 second intervals thereafter until
the
cotton was removed without lifting the peel from the skin or leaving residue
behind. The time when this observation is made is defined as the drying time
for the peel formulation. The results of the study are summarized in Table 26
below.
Table 26
Example Drying Time min
34 7.0
35 6.5
The amount of water In the formulation did not significantly Influence the
time for the formulation to dry. However, it was noted during the study that
the
formulation was difficult to expel from the sample tube. After approximately 4
weeks after the formulation in examples I and 2 were made the sample tubes
were retrieved and were evaluated for ease of dispensing the formulation. It
was noted that the formulation was Impossible to expel from the tube.
Interpolymer complexation between Gantrez S-97 and PVA through electrostatic
interactions, hydrophobic interactions, hydrogen bonding, or Van der Waals
interactions is hypothesized to be the reason(s) for the observed thickening.
Moreover, the extent of this interaction may be dependent on the
stoichiometric
ratio of the two polymers. It is believed that the water content of the,
formulations is too low for obtaining acceptable long term physical stability,
although the formulation shorter term viscosity was' acceptable. This
demonstrates the value of having sufficient amount of the volatile solvent
system in the formulation in some embodiments.
Example 36-39
A stretchable adhesive formulation for transdermal delivery of ketoprofen
(which is suitable for delivery via skin on joints and muscles) was evaluated
which includes an exciplent mixture which will form an adhesive formulation,
some of which are prepared in accordance with embodiments of the present
invention. The excipient mixture, which is a viscous and transparent fluid,
is.
prepared using the ingredients as shown in Table 27.
Table 27
CA 02747845 2011-08-05
59
Ingredients* Examples
36 37 38 39
PVA (Amresco MW 22.1 24.4 22.1 21.1
31,000-50,000)
Water 26.6 29.2 30.9 33.8
Ethanol 12.6 4.2 8.4 8.2
Butanol 0.4 0.5 0.4 0.4
PG 19.9 21.9 17.7 16.9
Glycerol 8.8 9.7 11 10.6
Gantrez ES 425 4.6 5.1 4.4 4.0
Keto =rofen 5.0 5.0 5.1 5.0
* Ingredients are noted in weight percent.
Peel formulations In Examples 1-4 are prepared in the following manner:
= PVA (solidifying agent) Is dissolved In water.
= The flux adequate non-volatile solvent (glycerol, PG) is mixed together
with the solidifying agent/volatile solvent mixture.
= Then ethanol; and Gantrez ES 425 Is added to the mixture.
= The resulting solution Is vigorously mixed for several minutes.
= After mixing, ketoprofen Is added and the final mixture is vigorously
mixed again for several minutes.
Formulations noted above were placed In laminate packaging tubes and
stored at 25 C/60% RH and 40 C/ 75% RH conditions until pulled for testing.
Physical testing was performed on each formulation. Table 28 summarizes the
data generated on each formulation.
Table 28
Example Viscosity*
Storage Cond. cps
T=0 2 weeks 4 weeks 8 weeks 12 weeks 16 weeks
36 96000 670000 >2500000 Not
C/60% RH measured
36 96000 500000 587500 2320000
40 C/75% RH
37 168500 204500 251000 >2500000
25 C/60% RH
37
40 C/75% RH 168500 215000 217500 >2500000
as 23000 -- 25000 36250 76250 57500
25 0160% RH
so 23000 -- 31000 40000 243500 164500
40 C/T5% RH
CA 02747845 2011-08-05
39 11250 13750
25 C/60% RH
39 11250 17500
40 C/75% RH
* Viscosity measured using a RVDV 1 + viscometer at 0.5 rpm.
Examples 36 and 37 had the lowest water content of the four
formulations and within 4 weeks of storage attained high viscosity values. The
5 only difference between Examples 36 and 37 is the amount of ethanol In the
formulations. It was hypothesized that reducing the level of ethanol may
reduce
the physical thickening of the formulation due to an Incompatibility between
the
PVA and ethanol. The viscosity data show that the higher ethanol formulation
(Example 36) had lower initial viscosity, but over the 4 weeks storage the
10 viscosity of both Example 36 and 37 attained viscosity values that were too
high
for a viable formulation. Another hypothesis for the formulation thickening is
that PVA is not compatible in high concentrations when dissolved In water.
-Additional formulations with higher water content were prepared to determine
if
an optimal water amount would keep the formulation from thickening up over
15 time. Example 38 viscosity after 16 weeks has not reached the viscosity
values
of the initial viscosity values of Examples 36 and 37.
Placebo versions of the formulations above were applied on study
volunteers and the drying time was assessed by placing a piece of cotton to
the
application site and then applying a 5 gram weight on the cotton. The cotton
20 and weight was removed after 5 seconds. This procedure was started
approximately 3 - 4 minutes after application and at 10 to 60 second Intervals
thereafter until the cotton was removed without lifting the peel or leaving
residue
behind. The results of the study are summarized in Table 29 below.
25 Table 29
Example Drying Time min *
36 4 min 49 sec
37 5 min 41 sec
38 4 min 27 sec
39 5 min 1 sec
* average dry time value from 12 study subjects.
CA 02747845 2011-08-05
61
The presence of ethanol as a second volatile solvent appears to
significantly reduce the time to dry. In data not shown a local anesthetic
formulation containing only water as the volatile solvent and a ratio of water
to
PVA of 2:1 has a drying time of >15 minutes. Optimizing the ratio and the
presence of an additional volatile solvent In formulations containing water
significantly reduce the drying time. It is hypothesized that the additional
volatile
solvent, in this case ethanol, will hydrogen bond with the water and water
will
escape with the ethanol when evaporating off the skin thereby forming a
solidified layer. This example demonstrates the value of using the right
mixture
and quantities of volatile solvents in the volatile solvent system in certain
embodiments.
Exanigles 40-42
Solidifying formulations for dermal delivery of ropivacaine HCl are
prepared which include excipient mixtures in accordance with embodiments of
the present invention. The formulations are prepared from the ingredients-as
shown in Table 30.
Table 30 - Ro ivacaine HCl solidifying formulation comp nents.
Ingredients" Example
40 41 42
Ropivacalne HCI 6.9 6.5 6.6
Iso ro anol 50.7 45.8 45.9
Water 5.5 5.2 5.2
Isostearic Acid 6.3 6.6 6.6
Triethylamine 3.0
Diiso ro anolamine 3.9
Ce l alcohol 3.3 3.9
Triacetin 2.9 2.6 2.6
Span 20 5.8 5.2 5.2
Plastoid B** 21.9 20.9 21.0
.*Ingredients are noted as weight percent.
from Degussa.
The ingredients listed above are combined according to the following
procedure.
The ropivacaine HCI, water, and the amine base (triethylamine or
diisopropanolamine) are combined in a glass jar and mixed until the drug is
CA 02747845 2011-08-05
62
dissolved. Then the isostearic acid, triacetin, Span 20, and cetyl alcohol
(Examples 41 and 42), or Isopropanol (Example 40) are added to the
formulation and mixed well. The polymer Plastoid B is added last and heated to
about 60 C until the Plastoid B Is completely dissolved. Once the polymer
solution cooled to room temperature, the formulation is stirred vigorously for
2-3
minutes.
The formulations in Table 30 are applied to HMS according to Example 1,
and the flux of ropivacaine was measured. The results are summarized in Table
31:
Table 31 - Steady-state flux of ropivacalne HCl through hairless mouse skin
from various adhesive solidifying formulations at 35 C
x'
Formulation Average flux*
me /cm 2/h*
40 96 14
41 61 t2
42 70*7
Example 43
Solvent formulations of ketoprofen in various non-volatile solvent systems
are evaluated. Excess ketoprofen Is present.
.The permeation of ketoprofen from the test formulations through HMS is
presented in Table 32 below.
Table 32
Non-volatile solvent system Skin FI'ux*
me lcm2lh
Glycerol 2+_1
Polyethylene Glycol 400 5 2
Span 20 1513
Propylene GI col 90 50
Oleic Acid 180 20
* Skin flux measurements represent the mean and standard
deviation of three determinations. Flux measurements reported
were determined from the linear region of the cumulative amount
versus time plots. The linear region was observed to be between
4-8 hours. If experimental conditions allowed, the steady-state
delivery would likely continue well beyond 8 hours.
CA 02747845 2011-08-05
63
Steady state flux of ketoprofen from the above non-volatile solvents are
obtained by placing 200 mcL on the stratum corneum side (donor) of hairless
mouse skin. The in vitro studies are carried out as described in Example 1.
From Table 32, the non-volatile solvents glycerol and polyethylene glycol 400
had low steady state flux values and would not be considered "flux-enabling."
Span 20 maybe considered flux-enabling, and propylene glycol or oleic acid
provided the highest flux and are considered flux-enabling non-volatile
solvent
systems. Assessment of flux-enabling solvents is based on the estimated
therapeutically effective flux of 16mcg/cm2/h for ketoprofen. Steady state
flux
values of a'drug from the non-volatile solvent that are below the
therapeutically
effective flux are not considered flux-enabling while steady state flux values
of a
drug from a non-volatile solvent above the therapeutically effective flux
value is
considered flux-enabling.
Examples 44-47
Solidifying formulations for dermal delivery of amitriptyline and a
combination of amitripyline and ketamine are prepared which include excipient
mixtures to form an adhesive solidifying-formulation In accordance with
embodiments of the present invention. The formulations are prepared from the
, Ingredients as shown in Table 33.
Table 33 - Amitriptyline and amitriptyline/ketamine
solidifying formulation components
ingredients* Exam le
44 45 46 47
Iso ro anol 50.3 48.6 50.8 49.8
Water = 2.7 2.6 2.7 2.7
Isostearic Acid 6.2 6.1 . 6.3 6.2
Trilso ro anoiamine 7.5 = 7.3 7.5 7.4
Triacetin 2.9 2.8 2.9 2.8
Span 20 5.7 5.5 5.8 5.6
Plastold B** 21.7 21.1 22 21.5
Amitri line 2 4
Ketamine 1 2 2 4
*Ingredients are noted as weight percent.
= ** from DeGussa.
CA 02747845 2011-08-05
64
The ingredients listed above are combined according to the following
procedure.
The drug(s), water, and triisopropanofamine are combined In a glass jar and
mixed until the drug is dissolved. Then the isostearic acid, triacetin, Span
20,
6 and isopropanol are added to the formulation and mixed well. The polymer
Plastold B Is added last and heated to, about 60 C until. the Plastold B Is
completely dissolved. Once the polymer solution cooled to room temperature,
the formulation is stirred vigorously for 2-3 minutes.
The formulations in Table 11 are applied to HMS according to Example 1,
and the flux of amitriptyline and%or ketamine was measured. The results are
summarized in Table 34:
Table 34 - Steady-state flux of amitriptyllne and amitriptyline/ketamine
through
hairless mouse skin from various adhesive solidi iri formulations at 35 C
Average Average
amitriptyline
= Formulation ketamine flux
me flcm2lh* mcg/cm/h""
Example-44 3 1 15:1.4
Example 45 7.6 0.2 38 6
Example 46 3* 1
Example 47 8.2*0.7
The adhesive formulation of amitriptyline and amitriptyline/ketamine
formulations in the present example have similar physical properties to the
formulations in examples noted above.
Example 48
A formulation similar to the formulation of Example 18 composition (with
no lidocaine) is applied onto a human skin surface at an elbow joint and a
finger
joint, resulting In a thin, transparent, flexible, and stretchable film. After
a few
minutes of evaporation of the volatile solvents (ethanol and water), a
solidified
layer is formed. The stretchable film has good adhesion to the skin and does
not separate from the skin on joints when bent, and can easily be peeled away
from the skin.
CA 02747845 2011-12-20
Example 49
A number of non-volatile solvents are tested for their flux-enabling ability
for dermal delivery of tetracaine. Each of the solutions in the table below
contain
saturated amounts of tetracalne base. The transdermal flux of tetracalne
5 generated by the saturated solutions are measured by a setup similar to that
in
Example 1. The results are as follows:
Table 35
Non-volatile solvent Flux (laglcm2lh)
Glycerin 3.8 t 2.9
Isostearic acid 60.5 t 15.3
Propylene Glycol 83.9:t 11
Triacetin 5.7:1- 0.7
The importance of selecting a non-volatile solvent system to achieving
10 therapeutically effective dermal drug flux is clearly shown here.
While the invention has been described with reference to certain
preferred embodiments, those skilled in'the art will appreciate that various
modifications, changes, omissions, and substitutions can be made thereto. It
is
15 therefore intended that the invention be limited only by the scope of the
appended
claims.