Note: Descriptions are shown in the official language in which they were submitted.
CA 02748289 2011-06-23
=
Preparation Method of Dihydroindene Amide Compounds, their Pharmaceutical
Compositions Containing Compounds thereof and Use as Protein Kinase Inhibitor
TECHNICAL FIELD OF THE INVENTION
The present invention relates to a novel kind of dihydroindene amide compounds
or
pharmaceutically acceptable salts thereof, their preparation methods,
pharmaceutical
compositions containing the compounds, the methods for their use in the
prevention or
treatment of the diseases associated with the abnormal activities of protein
kinases,
especially for the diseases associated with the abnormal activities of Abl,
Bcr-Abl, c-Kit and
PDGFR, and their use for manufacturing of a medicament for the prevention or
treatment of
the said diseases.
BACKGROUND OF THE INVENTION
Protein kinases are the enzymes which transfer a phosphate group from a
nucleoside
triphosphate to certain serine, threonine or tyrosine residues. Protein
phosphorylation
causes the activation of signal transduction pathways, which play crucial
roles in various
biological processes, including cell growth, metabolism, differentiation and
death. It is known
that abnormal signals caused by abnormal or improper activities of protein
kinases are
related to a number of diseases, including cancer, inflammation, autoimmune
disease,
metabolic disease, infection, central nervous system disease, cardiovascular
disease and so
on. Thus, protein kinases are attractive targets for drug development (Cohen,
Nat Rev. Drug
Discovery 2002, 1, 309).
The abl gene and the bcr gene are normal genes located on chromosome 9 and 22,
respectively. Two fusion genes are created by the reciprocal translocation
between these two
genes: the bcr-abl gene located on chromosome 22q- and the abl-bcr gene
located on
chromosome 9q+. The protein of 210kD (p210Bcr-Abl) is encoded by the bcr-abl
gene on the
Philadelphia chromosome. The Abl part of the Bcr-Abl protein comprising the
Abl tyrosine
kinase is strictly regulated in the prototype c-Abl but continuously activated
in the Bcr-Abl
fusion protein, which results in cell growth disorder. The Bcr-Abl protein can
be found in 95%
of the patients with Chronic Myelogenous Leukemia (CML) and in 10-25% of the
patients
with Acute Lymphoblastic Leukemia (ALL). lmatinib, brand-named as Gleevec, is
a Bcr-Abl
tyrosine kinase inhibitor and has been clinically proven to be an effective
formulation for the
1
CA 02748289 2011-06-23
treatment of CML. (Druker et al. N. Engl. J. Med. 2006, 355, 2408). However,
despite
continuous treatment using lmatinib, some patients with CMLs are recurrent at
the terminal
phase or the blast crisis phase due to drug resistance. The molecular basis of
drug resistance
is that imatinib-resistant mutants arise in the kinase domain of the Bcr-Abl
protein. To date,
more than 22 mutants have been reported and the most common ones are M244V,
G250E,
Q252H, Y253H, E255K, E255V, F311L, T351I, F317L, F359V, V379I, L387M, H396P,
H396R and
etc. (Nardi, et al. Curr. Opin. Hematol. 2004, //, 35).
c-Kit (CD117, stem cell factor receptor), encoded by the c-kit proto-oncogene,
is a kind of
growth factor receptor with tyrosine kinase activity. It can be activated upon
binding to stem
cell factor (SCF). Mutations in c-kit result in continuous activation of the
function of c-Kit
tyrosine kinase, which further causes the activity of tyrosine kinase
independent on ligands,
the autophosphorylation of c-Kit, and the deregulation of cell proliferation.
Overexpression
and mutations of c-Kit are found in most gastrointestinal stromal tumors
(GIST).
Gastrointestinal stromal tumors are a series of mesenchymal tumors which arise
from the
precursors of gastrointestinal tract tissue cells. They mainly occur in the
middle-aged and the
old population. About 70% of the tumors occur in the stomach, 20-30% of the
tumors occur
in the small intestine and less than 10% of the tumors occur in the esophagus,
colon and
rectum. As known to all, gastrointestinal stromal tumors are resistant to
classical
chemotherapy but can be treated effectively by inhibiting c-Kit using
Imatinib, which
suggests that c-Kit plays a vital role in the pathogenesis of these diseases
(Joensuu et al. N.
Engl. J. Med. 2001, 344, 1052). c-Kit is overexpressed and mutates in other
various human
cancers as well, including mast cell tumor, neuroblastoma, germ cell tumor,
melanoma, small
cell lung cancer, breast cancer, oophoroma and acute myeloid leukaemia (see
Edling et al. Int.
J. Biochem. Cell Biol. 2007, 39, 1995; Lennartsson et al. Curr. Cancer Drug
Targets, 2006, 6,
65).
In addition to the role in cancers, SCF/c-Kit also related to autoimmune or
inflammatory
diseases. SCF is expressed by various structural and inflammatory cells in the
breathing
passage. A number of pathways are activated by the combination of SCF and c-
Kit, including
the pathways involving Phosphoinositide-3 (PI3) kinase, phospholipase C (PLC)-
gamma, Src
protein kinase, Janus kinase (JAK) / signal transducers and activators of
transcription (STAT)
and mitogen-activated protein (MAP) kinase. Suppression of the SCF / c-Kit
pathway can
dramatically lower the level of histamine, reduce the penetration of mast
cells and
2
CA 02748289 2011-06-23
eosinophilic granulocytes, and decrease the release of interleukin (IL)-4 and
the
over-reactivity of the breathing passages. SCF/c-Kit is therefore a potential
treatment target,
which can control the number of mast cells and eosinophilic granulocytes, and
can control
the activation of autoimmune or inflammatory diseases, including scytitis,
rheumatoid
arthritis, allergic rhinitis, asthma, ankylosing spondylitis, psoriasis and
Crohn disease (see
Reber et al. Eur J. Pharmacol. 2006, 533, 327; Paniagua et al. Nat. Clin.
Prac. Rheum. 2007, 3,
190).
Platelet-derived growth factor receptors (PDGFR), such as PDGFR-a and PDGFR-
13, are
transmembrane tyrosine kinase receptors, whose ligands are formed by two A
chains
(PDGF-A), or two B chains (PDGF-B), or a heterodimer of one A chain and one B
chain
(PDGF-AB). Platelet-derived growth factor receptors are dimerized upon ligands
binding,
followed ,by activation of its tyrosine kinase and signaling to downstream. In
vivo animal
studies on PDGFs and PDGFRs reveal that PDGFR-a signaling plays a role in the
development
of gastrulation, cranial and cardiac neural crest, gonad, lung, intestine,
skin, central nervous
system and bone. Similarly, the role of PDGFR-13 signaling in angiogenesis and
early
hematopoiesis has been revealed as well. Platelet-derived growth factor
signaling is
associated with a number of diseases. Autocrine activation of growth factor
signaling
pathway relates to some gliomatosis cerebri, myeloproliferative disease,
tumor, multiple
myeloma, and sarcoma including dermatofibrosarcoma protuberans. Paracrine
growth factor
signaling is usually found in epithelial cancer,. It initiates the inhalation
of matrix therein, and
may participate in the epithelial-mesenchymal transition and thus affect
tumor's
development, angiogenesis, invasion and metastasis. Platelet-derived growth
factors drive
organic pathological changes of vascular disease, such as atheromatosis,
arteriostenosis,
pulmonary hypertension, retinal disease, and hepatofibrosis including
pulmonary interstitial
fibrosis, hepatocirrhosis, scleriasis, glomerulosclerosis and myocardial
fibrosis (see Andrae et
al. Gene Dev. 2008, 22, 1276). Therefore, the suppression of PDGFR can prevent
and treat the
above-mentioned diseases. Additionally, the suppression of PDGFR can also
treat a variety of
autoimmune or inflammatory diseases including diabetes, particularly Type-I
diabetes,
rheumatoid arthritis, psoriasis, Crohn disease and etc (Paniagua et al. Nat.
Clin. Prac. Rheum.
2007, 3, 190; Louvet et al. Proc. Natl. Acad. Sci. USA, 2008, 105, 18895).
The invention provides a novel kind of dihydroindene amide derivatives, which
can inhibit
the activity of protein kinases, especially one or more protein kinases
described above. These
3
CA 02748289 2011-06-23
compounds will therefore be useful to prevent or treat the diseases associated
with the
abnormality or disorder in the activity of protein kinases, especially the
diseases associated
with abnormality in the activity of.Abl, Bcr-Abl, c-Kit and PDGFR protein
kinases.
DESCRIPTION OF THE INVENTION
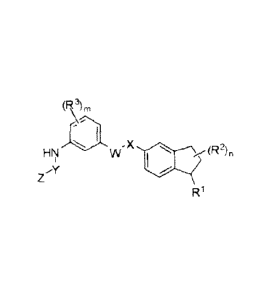
The invention provides the compounds of Formula l:
(R)õ,
x
R'
Formula l
or pharmaceutically acceptable salts or prodrugs thereof, wherein:
R1 is a saturated cyclic amino-group, which can be optionally substituted by
1, 2, 3 or 4 Ria;
Rla is H, halogen, cyano-group, C1_6 alkyl, C16 hydroxyalkyl, C16haloalkyl,
C1cyanoalkyl,
NRbRc, NRbC(0)Rd, NRbS(0)2Rd, C(0)NRbRc, C(0)Rd, C(0)0R8, S(0)2Rd, C2-6
alkenyl, C2-6
alkynyl, aryl, heteroaryl, cycloalkyl, or heterocycloalkyl, wherein the said
C1.6 alkyl, C2-6
alkenyl, C2-6 alkynyl, aryl, heteroaryl, cycloalkyl and heterocycloalkyl can
be optionally
substituted by 1, 2 or 3 groups independently selected from cyano-group,
halogen, OR', SR%
NRb12`, NRb(CO)Rd, NRbS(0)2Rd, C(0)NRbIlc, S(0)2NRbRc, C(0)Rd, C(0)011',
S(0)2Rd, C1-6 haloalkyl,
C1_6 hydroxyalkyl, C1-6 cyanoalkyl, aryl, heteroaryl, cycloalkyl, and
heterocycloalkyl;
Alternatively, two Rla groups taken together with the atoms attached to them
can form a
cycloalkyl or heterocycloalkyl of 3, 4, 5, 6 or 7-membered ring, and can be
optionally
substituted by 1, 2 or 3 groups independently selected from cyano-group,
halogen, OR% SR%
NRb(CO)Rd, NRbS(0)2Rd, C(0)NRIW, S(0)2NRbR`, C(0)Rd, C(0)01e, S(0)2Rd, C1_6
haloalkyl,
hydroxyalkyl, C1_6 cyanoalkyl, C2-6 alkenyl, C2-6 alkynyl, aryl, heteroaryl,
cycloalkyl, and
heterocycloalkyl;
R2 is H, halogen, cyano-group, OR', SRa, NRbRc, C1.6 alkyl, C1-6 hydroxyalkyl,
C1-6 haloalkyl, C1-6
cyanoalkyl, C2-6 alkenyl, C2_6 alkynyl; Alternatively, two R2 groups taken
together with the
atoms attached to them can form a cycloalkyl and heterocycloalkyl of 3, 4, 5,
6 or
4
CA 02748289 2011-06-23
7-membered ring, and can be optionally substituted by 1, 2 or 3 groups
independently
selected from cyano-group, halogen, ORa, SRa, NRbRc, NRb(CO)Rd, NRbS(0)2Rd,
C(0)NRble,
S(0)2NRblic, c(0)Rd, c(0)0Ra, S(0)2Rd, C1.6 haloalkyl, C1_6 hydroxyalkyl,
C1..6 cyanoalkyl, C2-6
alkenyl, and C2_6 alkynyl;
R3 is H, halogen, cyano-group, ORa, SRa, NFeRc, C1_6 alkyl, C16 hydroxyalkyl,
C16haloalkyl, C1_6
cyanoalkyl, C2_6 alkenyl, C2_6 alkynyl, cycloalkyl, or heterocycloalkyl.
Alternatively, two R3
groups taken together with =the atoms attached to them can form a cycloalkyl
and
heterocycloalkyl of 5, 6 or 7-membered ring, and can be optionally substituted
by 1, 2 or 3
groups independently selected from halogen, cyano-group, ORa, SRa, NRbRc, C1-6
alkyl, C1_6
haloalkyl, C1_6 hydroxyalkyl, C1_6 cyanoalkyl, C2_6 alkenyl, and C2-6 alkynyl;
W-X is amide bond;
Y is heteroaryl, which can be optionally substituted by 1, 2 or 3 R5;
Z is heterocycloalkyl or heteroaryl, which can be optionally substituted by 1,
2 or 3 R5;
R4 and R5 are independently selected from halogen, cyano-group, ORa, SR%
NRblic, C1_6 alkyl,
C1_6 hydroxyalkyl, C1_6 haloalkyl, C1_6 cyanoalkyl, C2.6 alkenyl, C2_6
alkynyl, NRb(CO)Rd, C(0)NRbil`,
NRbS(0)2Rd, S(0)2NRb11`, C(0)Rd, C(0)0Ra, S(0)2Rd, cycloalkyl,
heterocycloalkyl, aryl and
heteroaryl. Alternatively, two R4 or two R5 groups taken together with the
atoms attached to
them respectively can form a cycloalkyl or heterocycloalkyl of 5, 6 or 7-
membered ring, and
can be optionally substituted by 1, 2 or 3 groups independently selected from
halogen,
cyano-group, Ofta, SRa, NRbI3`, C1_6 alkyl, C1_6 haloalkyl, C1_6 hydroxyalkyl,
C1.6 cyanoalkyl, C2-6
alkenyl, and C2_6 alkynyl;
Rb, RC and Rd are independently selected from H, C1_6 alkyl, C1_6 haloalkyl,
C1_6 hydroxyalkyl,
= C1_6 cyanoalkyl, C2-6 alkenyl, C2_6 alkynyl, cycloalkyl,
heterocycloalkyl, aryl, and heteroaryl.
Alternatively, the Rb and R` groups taken together with the nitrogen atom
attached to them
can form a heterocycloalkyl of 4, 5, 6 or 7-membered ring, and can be
optionally
= substitued by 1, 2 or 3 groups independently selected from halogen, cyano-
group, C1_6 alkyl,
C1_6 haloalkyl, C1_6 hydroxyalkyl, C1.6 cyanoalkyl, C2_6 alkenyl, C2.6
alkynyl, cycloalkyl,
= heterocycloalkyl, aryl, and heteroaryl;
CA 02748289 2011-06-23
n is an integer from zero to four;
m is an integer from zero to two.
Among the compounds of Formula I and salts or prodrugs thereof, the preferred
compounds
in present invention are of Formula II:
40 N T
OI
HN
H
z
R
Formula II
or pharmaceutically acceptable salts or prodrugs thereof, wherein:
R1 is a saturated cyclic amino-group, which can be selected from piperidinyl,
piperazinyl,
pyrrolidinyl, azetidinyl and morpholinyl, each group can be optionally
substituted by 1, 2, 3,
or 4 Ria;
Rla is H, halogen, cyano-group, C16 alkyl, C16 hydroxyalkyl, C1-6 haloalkyl,
C1.6 cyanoalkyl,
SRa, NRbRc, NRbC(0)Rd, NRbS(0)2Rd, C(0)NRb11`, C(0)Rd, C(0)0Ra, S(0)2Rd, C2-6
alkenyl, C2-6
alkynyl, aryl, heteroaryl, cycloalkyl, or heterocycloalkyl, wherein the said
C1_6 alkyl, C2-6
alkenyl, C2.6 alkynyl, aryl, heteroaryl, cycloalkyl, and heterocycloalkyl can
be optionally
substituted by 1, 2 or 3 groups independently selected from cyano-group,
halogen, 012a, SRa,
NRbRc, NRb(CO)Rd, NRbS(0)2Rd, C(0)NRbR`, S(0)2NRblic, C(0)Rd, C(0)0Ra,
S(0)2Rd, C1-6 haloalkyl,
C1.6 hydroxyalkyl, C1_6 cyanoalkyl, aryl, heteroaryl, cycloalkyl, and
heterocycloalkyl;
Alternatively, two Ria groups taken together with the atoms attached to them
can form a
cycloalkyl or heterocycloalkyl of 3, 4, 5, 6 or 7-membered ring, and can be
optionally
substituted by 1, 2 or 3 groups independently selected from cyano-group,
halogen, Ole, SRa,
N RbRC, N Rb(CO)Rd, N R'S(0)2R', C(0)NRbRc, S(0)2NRbRc, C(0)Rd, C(0)011a,
S(0)2Rd, C1.6 haloalkyl,
hydroxyalkyl, C1.6 cyanoalkyl, C2.6 alkenyl, C2.6 alkynyl, aryl, heteroaryl,
cycloalkyl, and
heterocycloalkyl;
Y is selected from pyridyl, pyrimidyl, pyridazinyl, pyrazinyl, triazinyl,
thiazolyl, isothiazolyl,
6
CA 02748289 2011-06-23
imidazolyl, oxazolyl, isoxazolyl, triazolyl or pyrazolyl, and can be
optionally substituted by 1,
2, or 3 R4;
Z is selected from pyridyl, pyrimidyl, pyridazinyl, pyrazinyl, triazinyl,
thiazolyl, isothiazolyl,
imidazolyl, oxazolyl, isoxazolyl, triazolyl, pyrazolyl, azotic oxazolyl,
pyrindol,
pyrrolo-pyrinnidyl, pyrazolo-pyridyl, pyrazolo-pyrimidyl, quinolyl,
isoquinolyl, quinazolyl,
piperazinyl or morpholinyl, and can be optionally substituted by 1, 2, or 3
R5;
R4 and R5 are independently selected from halogen, cyano-group, ORa, SRa,
NRble, C1_6 alkyl,
C1.6 hydroxyalkyl, C1.6 haloalkyl, C1_6 cyanoalkyl, C2_6 alkenyl, C2_6
alkynyl, NRb(CO)Rd, C(0)NRble,
NRbS(0)2Rd, S(0)2NRbR', C(0)Rd, C(0)0Ra, S(0)2Rd, cycloalkyl,
heterocycloalkyl, aryl, and
heteroaryl. Alternatively, two R4 or two R5 groups taken together with the
atoms attached to
them respectively, can form a cycloalkyl and heterocycloalkyl of 5, 6 or 7-
membered ring,
and can be optionally substituted by 1, 2 or 3 groups independently selected
from halogen,
cyano-group, ORa, SR', NRbRc, C1_6 alkyl, C16 haloalkyl, C1-6 hydroxyalkyl,
C16cyanoaikyl, C2-6
alkenyl, and C2.6 alkynyl;
le, Rb, le and Rd are independently selected from H, C1_6 alkyl, C1.6
haloalkyl, C16hydroxyaikyl,
C1_6 cyanoalkyl, C2_6 alkenyl, C2_6 alkynyl, aryl, heteroaryl, cycloalkyl, and
heterocycloalkyl.
Alternatively, Rb and R` taken together with the nitrogen atom attached to
them respectively,
can form a heterocycloalkyl of 4, 5, 6 or 7-membered ring, and can be
optionally substituted
by 1, 2 or 3 groups independently selected from halogen, cyano-group, C1-6
alkyl, C1-6
haloalkyl, C1_6 hydroxyalkyl, C1_6 cyanoalkyl, C2-6 alkerwl, C2-6 alkynyl,
aryl, heteroaryl, cycloalkyl,
and heterocycloalkyl;
= Among the compounds of Formula l and salt or prodrug thereof, the more
preferred
compounds in present invention are of Formula Ila:
0
HN
z.)I( N 00
,N,R7
Rot )),
Formula Ila
7
CA 02748289 2011-06-23
or pharmaceutically acceptable salts or prodrugs thereof, wherein:
R6 and 137 are independently selected from H, halogen, cyano-group, C1-6
alkyl, C1.-6
hydroxyalkyl, C1_6 haloalkyl, C1..6 cyanoalkyl, C2_6 alkenyl, C2_6 alkynyl.
Alternatively, R6 and 137
taken together with the atom attached to them can form a carbocycle or
heterocycle of 5, 6
or 7-membered ring, and can be optionally substituted by 1, 2 or 3 groups
independently
selected from halogen, cyano-group, OR', Sfe, Nfiblic, C1_6 alkyl, C1_6
hydroxyalkyl, C1.6
haloalkyl, C1-6 cyanoalkyl, C2-6 alkenyl, and C2_6 alkynyl;
R8 is H, C1_6 alkyl, C2_6 hydroxyalkyl, C2-6 haloalkyl, C1-6 haloalkyl,
C(0)NRbRc, C(0)Rd, C(0)0Ra,
S(0)2Rd, C3-6 alkenyl, C3_6 alkynyl, aryl, heteroaryl, cycloalkyl, and
heterocycloalkyl. Wherein
the said C1_6 alkyl, C3.6 alkenyl, C3-6 alkynyl, aryl, heteroaryl, cycloalkyl
and heterocycloalkyl
can be optionally substituted by 1, 2 or 3 groups independently selected from
halogen,
cyano-group, ORa, SRa, and NRbitc;
Y is selected from pyridyl, pyrimidyl, pyridazinyl, pyrazinyl, triazinyl,
thiazolyl, isothiazolyl,
imidazolyl, oxazolyl, isoxazolyl, triazolyl or pyrazolyl, and can be
optionally substituted by 1,
2, or 3 R4;
Z is selected from pyridyl, pyrimidyl, pyridazinyl, pyrazinyl, triazinyl,
thiazolyl, isothiazolyl,
imidazolyl, oxazolyl, isoxazolyl, triazolyl, pyrazolyl, azotic oxazolyl,
pyrindol,
pyrrolo-pyrimidyl, pyrazolo-pyridyl, pyrazolo-pyrimidyl, quinolyl,
isoquinolyl, quinazolyl,
piperazinyl or morpholinyl, and can be optionally substituted by 1, 2, or 3
R5;
R4 and R5 are independently selected from halogen, cyano-group, Ole, SR',
NRbR`, C1_6 alkyl,
C16hydroxyalkyl, C1.6haloalkyl, C16cyanoalkyl, C2.6 alkenyl, C2_6 alkynyl,
NRb(CO)Rd, C(0)NRbRc,
NRbs(0)2-d,
S(0)2NRbric, C(0)Rd, C(0)0R3, S(0)2Rd, cycloalkyl, heterocycloalkyl, aryl, and
heteroaryl. Alternatively, two R4 or two R5 groups taken together with the
atoms attached to
them respectively, can form a cycloalkyl and heterocycloalkyl of 5, 6 or 7-
membered ring,
and can be optionally substitued by 1, 2 or 3 groups independently selected
from halogen,
cyano-group, ORa, SR', NeRc, C1..6 alkyl, C16 haloalkyl, C16hydroxyalkyl, C1_6
cyanoalkyl, C2_6
alkenyl, and C2-6 alkynyl;
le, Rb, 12` and Rd are independently selected from H, C1_6 alkyl, C1.6
haloalkyl, C16hydroxyalkyl,
8
CA 02748289 2011-06-23
C1_6 cyanoaikyl, C2_6 alkenyl, C2_6 alkynyl, aryl, heteroaryl, cycloalkyl, and
heterocycloalkyl.
Alternatively, Rb and II taken together with the nitrogen atom attached to
them respectively,
can form a heterocycloalkyl of 4, 5, 6 or 7-membered ring, and can be
optionally substituted
by 1, 2 or 3 groups independently selected from halogen, cyano-group, C1.6
alkyl, C1-6
haloalkyl, C16hydroxyaikyl, C16cyanoalkyl, C2_6 alkenyl, C2_6 alkynyl, aryl,
heteroaryl,
and heterocycloalkyl;
p is an integer from zero to two.
Among the compounds of Formula I and salts or prodrugs thereof, other more
preferred
compounds in present invention are of Formula Ilb:
N
Hy
H SOO
z
116
R \HD%R9
q
Formula Ilb
or pharmaceutically acceptable salts or prodrugs thereof, wherein:
R9 and Rware independently selected from H, C1-6 alkyl, C2.6 hydroxyalkyl, C2-
6 haloalkyl, C1-6
haloalkyl, C(0)NRIalic, C(0)Rd, C(0)011a, S(0)2Rd, C3-6 alkenyl, C3_6 alkynyl,
aryl, heteroaryl,
cycloalkyl, and heterocycloalkyl. Wherein the said C1_6 alkyl, C3.6 alkenyl,
C3_6 alkynyl, aryl,
heteroaryl, cycloalkyl and heterocycloalkyl can be optionally substituted by
1, 2 or 3 groups
independently selected from halogen, cyano-group, ORa, SW,and NRblIc.
Alternatively, R9 and
R19 taken together with the atom attached to them can form a cycloalkyl or
heterocycloalkyl
of 5, 6 or 7-membered ring, and can be optionally substituted by 1, 2 or 3
groups
independently selected from halogen, cyano-group, ORa, SRa, NRbRc, C1_6 alkyl,
C1-6
hydroxyalkyl, C1-6 haloalkyl, C1_6 cyanoalkyl, C2-6 alkenyl, C2-6 alkynyl,
aryl, heteroaryl, cycloalkyl,
and heterocycloalkyl;
R11 is H, halogen, cyano-group, ORa, 5Ra, NRbRc, C1.6 alkyl, C3_6
hydroxyalkyl, C1-6 halOalkYl, C1-6
cyanoalkyl, C2-6 alkenyl, C2-6 alkynyl;
9
CA 02748289 2011-06-23
Y is selected from pyridyl, pyrimidyl, pyridazinyl, pyrazinyl, triazinyl,
thiazolyl, isothiazolyl,
imidazolyl, oxazolyl, isoxazolyl, triazolyl or pyrazolyl, and can be
optionally substituted by 1,
2, or 3 R4;
Z is selected from pyridyl, pyrimidyl, pyridazinyl, pyrazinyl, triazinyl,
thiazolyl, isothiazolyl,
imidazolyl, oxazolyl, isoxazolyl, triazolyl, pyrazolyl, azotic oxazolyl,
pyrindol,
pyrrolo-pyrimidyl, pyrazolo-pyridyl, pyrazolo-pyrimidyl, quinolyl,
isoquinolyl, quinazolyl,
piperazinyl or morpholinyl, and can be optionally substituted by 1, 2, or 3
R5;
R4 and R5 are independently selected from halogen, cyano-group, ORa, Sle,
NRbR`, C1_6 alkyl,
C1_6 hydroxyalkyl, C1-6 haloalkyl, C1_6 cyanoalkyl, C2.6 alkenyl, C2-6
alkynyl, NRb(CO)Rd, C(0)NRb13`,
K
NRbs(0.2.-.d,
) S(0)2NRbl3`,
C(0)Rd, C(0)0Ra, S(0)2Rd, cycloalkyl, heterocycloalkyl, aryl, and
heteroaryl. Alternatively, two R4 or two R5 groups taken together with the
atoms attached to
them respectively, can form a cycloalkyl and heterocycloalkyl of 5, 6 or 7-
membered ring,
and can be optionally substituted by 1, 2 or 3 groups independently selected
from halogen,
cyano-group, ORa, SR', NRbR`, C1_6 alkyl, C1_6 haloalkyl, C1_6 hydroxyalkyl,
C1-6 cyanoalkyl, C2-6
alkenyl, and C2-6 alkynyl;
Ra, Rb, Rc and Rd are independently selected from H, C1-6 alkyl, C1-6
haloalkyl, C1-6 hydroxyalkyl,
C1_6 cyanoalkyl, C2_6 alkenyl, C2.6 alkynyl, aryl, heteroaryl, cycloalkyl, and
heterocycloalkyl. And
Rb and 11` taken together with the nitrogen atom attached to them
respectively, can form a
heterocycloalkyl of 4, 5, 6 or 7-membered ring, and can be optionally
substituted by 1, 2 or 3
groups independently selected from halogen, cyano-group, C1_6 alkyl, C1_6
haloalkyl,
hydroxyalkyl, C1_6 cyanoalkyl, C2_6 alkenyl, C2_6 alkynyl, aryl, heteroaryl,
cycloalkyl, and
heterocycloalkyl;
q is an integer from zero to three.
The other aspect of the invention provides a method to regulate the activity
of protein
kinases, wherein the said method includes exposing the mentioned protein
kinase to the
above-mentioned compounds or pharmaceutically acceptable salts or prodrugs
thereof.
Preferably, the mentioned protein kinases are selected from Abl, Bcr-Abl, c-
Kit and PDGFR.
Also, the mentioned protein kinases include mutated kinases, which are
seclected from
CA 02748289 2011-06-23
mutated Abl kinase, Bcr-Abl kinase, c-Kit kinase and PDGFR kinase.
Another aspect of the invention provides the use of the above-mentioned
compounds or
pharmaceutically acceptable salts thereof for manufacturing of a medicament
for treating
diseases or disorders associated with the activities of protein kinases or
abnormal cell
proliferation.
Yet another aspect of the invention provides a method to treat patients'
diseases or
disorders associated with the activities of kinases, including the
administration of effective
doses of the above-mentioned compounds or pharmaceutically acceptable salts or
prodrugs
thereof to the patients.
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENTS
Illustrative embodiments will be described in details below. However, these
embodiments
are only for demonstration but not intended to restrict the scope of the
invention.
As used herein, the following definitions shall apply unless otherwise
indicated.
"Halogen" comprises fluorine (F), chlorine (Cl), bromine (Br) and iodine (I).
"Alkyl" refers to straight-chained or branched-chain saturated hydrocarbon
groups. Examples
of alkyl include C1_20 alkyl, preferably C1.6 alkyl, such as methyl (Me),
ethyl (Et), propyl (such as
n-propyl and isopropyl), butyl (such as n-butyl, isobutyl and t-butyl), amyl
(such as n-amyl,
isoamyl and neoamyl), n-hexyl and etc. In each substituted alkyl or alkyl-
substituted group
mentioned below, "alkyl" has the same definition as the above.
"Hydroxyalkyl" refers to alkyl substituted by hydroxyl.
"Haloalkyl" refers to alkyl substituted by one or more halogens, such as CH2F,
CHF2, CF3, C2F5,
CCI3 and etc.
=
"Cyanoalkyl" or "cyano-substituted alkyl" refers to alkyl substituted by cyano-
group.
11
CA 02748289 2011-06-23
"Alkenyl" refers to alkyl having one or more carbon-carbon double bonds, such
as vinyl,
propenyl, 1, 3-butadienyl, cis-butenyl, trans-butenyl and etc.
"Alkynyl" refers to alkyl having one or more carbon-carbon triple bonds, such
as acetylenyl,
propinyl and etc.
"Cycloalkyl" refers to non-aromatic carbon ring, including cycloalkyl,
cycloalkenyl and
cycloalkynyl. Cycloalkyl can have monocyclic or polycyclic ring system (such
as having 2, 3 or
4 fused rings), including spirocycles. Cycloalkyl can have 3-20 carbon atoms,
as well as 0, 1, 2
or 3 double bonds and/or 0, 1, or 2 triple bonds. Cycloalkyl can also comprise
a ring of one or
more fused aromatic rings (i.e. with a shared bond), for example, pentane,
pentene, hexane
and the like substituted by benzene derivatives. Cycloalkyl having one or more
fused
aromatic rings can be attached to other groups via the aromatic ring moiety or
the
non-aromatic ring moiety. Examples of cycloalkyl include cyclopropyl,
cyclobutyl, cyclopentyl,
cyclohexyl, cycloheptyl, cyclopentenyl, cyclohexenyl, cyclohexadienyl,
cycloheptatrienyl,
adamantyl and etc.
"Heterocycloalkyl" refers to a non-aromatic ring, wherein one or more atoms in
the ring are
heteroatoms such as N, 0 or S. Heterocycloalkyl can comprise a monocyclic or
polycyclic ring
system (such as having 2, 3 or 4 fused rings), including spirocycles.
Preferred examples of
heterocycloalkyl include, but are not limited to aziridine, .azetidine,
tetrahydrofuran,
tetrahydrothiophene, pyrrolidine, oxazolidine, thiazolidine, imidazolidine,
isoxazolidine,
isothiazolidine, pyrazolidine, morpholine, thiomorpholine, piperazine,
piperidine and etc.
Heterocycloalkyl can also comprise a heterocyclic ring of one or more fused
aromatic rings
(i.e. with a shared bond), for example, 2, 3-dihydrobenzofuran, 1,3-
benzodioxolane,
benzo-1,4-dioxan, methylphthalimide, and naphthalimide. Heterocycloalkyl
having one or
more fused aromatic rings can be attached to other groups via the aromatic
ring moiety or
the non-aromatic ring moiety.
"Aromatic ring" refers to monocyclic or polycyclic (such as having 2, 3 or 4
fused rings)
aromatic carbohydrate, such as benzene, naphthalene, anthracene, phenanthrene
and etc.
=
"Hetero-aromatic ring" refers to aromatic heterocycles at least comprising one
ring-membered heteroatom, such as S, 0 or N. Hetero-aromatic ring may comprise
a
12
CA 02748289 2011-06-23
monocyclic or polycyclic ring system (such as containing 2, 3 or 4 fused
rings). Any
ring-membered nitrogen atom in the hetero-aromatic ring may be oxidized to
form nitrogen
oxide. Preferred hetero-aromatic rings include, but are not limited to
pyridine, pyrimidine,
pyrazine, pyridazine, triazine, furan, thiofuran, imidazole, triazole,
tetrazole, thiazole,
isothiazole, 1,2,4-thiadiazole, pyrrole, pyrromonazole, oxazole, isoxazole,
oxadiazole,
benzofuran, benzothiophene, benzothiazole, indole, indazole, quinoline,
isoquinoline, purine,
carbazole, benzimidazole, pyrindol, pyrrolo-
pyrimidine, pyrazolo-pyridine,
pyrazolo-pyrimidine and etc.
"Optionally" means that the event or situation described subsequently may or
may not
happen. The mentioned description includes examples of the event or situation
described
herein when it happens or when it does not happen.
"Effective therapeutic dose" refers to administering effective amount of the
compounds of
the formula to mammal in need for sufficient treatment. Effective therapeutic
dose are
subjected to change, depended on the specific activity of medicament, and the
age,
physiologic condition, other diseases and nutritional status of the patient.
In addition,
determination of effective therapeutic dose to be used will be affected by the
other possible
medical therapy which the patient receives in the meantime.
"Treatment" means any therapy to treat diseases in mammals, including:
(i) Preventing disease, i.e. resulting in no development of clinical symptoms
of disease;
(ii) Repressing disease, i.e. keeping clinical symptoms from developing; and
/or
(iii) Alleviating disease, i.e. resulting in elimination of clinical symptoms.
In many cases, the compound of the present invention may form acidic and/or
basic salt due
to the existence of amino and/or carboxyl group or the like.
"Compound" described herein refers to all stereo isomers, geometric isomers,
dynamic
isomers and isotopes.
The compound of the present invention may be asymmetric, for example, having
one or
more stereo isomers. Unless otherwise indicated, all stereo isomers are
included, such as
enantiomers and diastereomers. The compound comprising asymmetrically
substituted
13
CA 02748289 2011-06-23
carbon atom can be isolated in optically active-pure or racemic forms. The
optically active
form can be separated from the racemic mixture, or synthesized by utilizing
chiral materials
or chiral reagents.
The compound of the present invention also includes dynamic isomers. The
dynamic isomer
form is derived from a swap between a single bond and the adjacent double
bonds,
accompanied by the migration of a proton.
The compound of the present invention also includes the final compound or the
intermediate thereof which comprises isotope atoms. Isotope atoms have the
same atomic
number but different mass number. For example, the isotopes of hydrogen
include
deuterium and tritium.
The compound of the present invention also includes pharmaceutically
acceptable salts
which mean the basic groups in parent compounds are converted into the salt
form.
Pharmaceutically acceptable salts include, but are not limited to the
inorganic or organic
acid salts of basic group such as amidocyanogen. The pharmaceutically
acceptable salt
herein can be synthesized from its parent compound, i.e. the basic group in
the parent
compound reacts with 1-4 equivalent of acid in solvent systems. The suitable
salts were
enumerated in Remington's Pharmaceutical Sciences, 17th ed, Mark Publishing
Company,
Easton, Pa, 1985, p. 1418 and Journal of Pharmaceutical Science, 66, 2 (1977).
The pharmaceutically acceptable acid addition salts can be prepared from
inorganic or
organic acids. The acid addition salts can be derived from the inorganic acids
including
hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric
acid and etc. The
acid addition salts can be derived from the organic acids including acetic
acid, propionic acid,
glycolic acid, pyruvic acid, oxalic acid, malic acid, malonic acid, succinic
acid, maleic acid,
fumaric acid, tartaric acid, citric acid, benzoic acid, cinnamic acid,
mandelic acid,
methanesulfonic acid, ethyl sulfonic acid, toluene-p-sulfonic acid, salicylic
acid, and etc.
As used herein, "pharmaceutically acceptable carriers" include any and all
solvents,
dispersive media, coats, anti-bacterial or anti-fungal agents, isotonic agents
or absorption
retardants and so on. Such media and agents used in pharmaceutically active
substances are
well known in the art. The uses of them in therapeutic compositions are
predictable, unless
14
o
CA 02748289 2011-06-23
=
any common media or agents are incompatible with the active substances.
Additional active
ingredients can be incorporated into the compositions as well.
The compositions herein are preferably prepared in unit dosage form. The term
"unit dosage
form" refers to physically discrete unit of a single dose which is suitable to
be administrated
to human or other mammal subjects. As to achieve required effective treatment,
based on
collocation, each unit contains predetermined quantity of active substances,
as well as
relevant suitable pharmaceutical excipients (such as tablet, capsule,
ampoule). The
compounds of Formula I are effective in a broad dose range and are usually
administrated in
effective amount. Preferably, with regard to oral administration, each dose
unit contains 10
mg to 2 g of the compounds of Formula I, more preferably 10mg to 700 mg; while
with
regard to parenteral administration, each dose unit contains preferably 10 mg
to 700 mg of
the compounds of Formula I, more preferably 50 mg to 200 mg. However, it
should be
appreciated that the actual administration amount of the compounds of Formula
I is
determined by physicians based on the relevant conditions, including the
disease to be
treated, the administration route to be selected, the actual compound and its
relative
activity to be given, as well as the age, body weight, response and the
severity of symptoms
- of the patient, and etc.
To prepare solid composition such as tablets, the main active components are
mixed with
the pharmaceutical excipients (or carriers) to form solid pre-prepared
composition, wherein
the homogeneous mixture of the compounds of the present invention is
contained. When
these pre-prepared compositions are called as homogeneous mixture, it means
that the
active components are evenly dispersed in the whole composition, which allows
the
composition to be easily divided into unit dosage form with the same efficacy,
such as tablet,
pill or capsule.
The tablet or pill of this invention may be coated or compounded in other
patterns so as to
provide a dosage form which has an advantage to prolong the efficacy, or to
protect the
tablet or pill against the acidic environment in the stomach. For example, a
tablet or pill may
comprise internal dose and external dose components, wherein the latter exists
in the form
of coating on the top of the former. These two kinds of components can be
separated by an
enteric layer, which is to prevent the breakup in the stomach and to allow the
internal
component to enter the duodenum wholly or to be released slowly. A variety of
materials
CA 02748289 2011-06-23
can be used as the enteric layer or coating and the said materials include
polymeric acids as
well as the mixtures of the polymeric acids and the following materials, such
as shellac,
hexadecanol and cellulose acetate.
The compositions used in inhalation or insufflation include pharmaceutically
acceptable
aqueous or organic solvents, or the solutions and suspensions of the mixture,
as well as
pulvis. Liquid or solid compositions can comprise the suitable pharmaceutical
excipients as
mentioned above. Preferably, these compositions are administrated via oral
route or nasal
respiration so as to get partial or systemic effects. The compositions in the
preferred
pharmaceutically acceptable solvents can be atomized by using inert gases: the
atomized
solution may be sucked directly into an atomization device, or alternatively
the atomization
device may be connected to a face tent or intermittent positive pressure
breathing machine.
The compositions of solutions, suspensions, or pulvis can be administrated by
a device in a
suitable route, preferably oral or nasal route, of delivering dosage forms.
In this invention, the compounds and the pharmaceutically acceptable salts
thereof also
include the forms of solvates or hydrates. Generally, the forms of solvates or
hydrates are
equal to the forms of non-solvates or non-hydrates, and covered in the scope
of the
invention. Some compounds in the present invention can probably exist in the
form of
polycrystal or amorphism. In short, all of the physical forms possess the
equal uses and are
covered in the scope of the invention.
This invention also includes the prodrugs of the compounds. Prodrug is a
pharmacological
substance, derived from the parent drug, and will be metabolized into the
parent drug once
entering the body. The prodrug can be prepared by substituting one or, more
functional
groups in the parent drug, which will be released once the substituted groups
are degraded
in vivo. The preparation and the usage of prodrugs can be found in T. Higuchi
and V. Stella,
"Pro-drugs as Novel Delivery Systems", Vol. 14 of the A.C.S. Symposium Series,
and
Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American
Pharmaceutial
Association and Pergmon Press, 1987.
This invention also provides pharmaceutical compositions comprising the
compounds of =
Formula l or pharmaceutically acceptable salts or prodrugs thereof, and at
least one kind of
pharmaceutically acceptable carrier. The pharmaceutical compositions of the
present
16
CA 02748289 2011-06-23
invention can be administrated by the route of oral, injection, spray
inhalational,
extraepithelial, rectal, nasal, vaginal, celiac, reservoir-embedded, or
transdermal patch and
etc.
On the other hand, this invention also provides the method to regulate protein
kinase
activity by utilizing the compound of Formula l. The term "regulate kinase
activity" herein
means that the activity of protein kinases is reduced to some extent once the
kinases
exposing to dihydroindene amide compounds of the present invention, compared
to the
activity without exposing to the compounds. This invention therefore provides
a method to
regulate protein kinase activity by exposing protein kinases to dihydroindene
amide
compounds.
Specifically, the protein kinases described in the invention are protein
tyrosine kinases,
including Abl, Bcr-Abl, c-Kit and PDGFR.
In addition, the protein kinases in this invention also include mutated
kinases, such as
mutated Abl and Bcr-Abl kinases, mutated c-Kit kinases and mutated PDGFR
kinases. The
mutated Abl and Bcr-Abl kinases include, for example, one or more of the
following mutants:
M244V, G250E, Q252H, Y253F, Y253H, E255K, E255V, F311L, T351I, F317L, M351T,
F359V,
V379I, L387M, H396P, H396R and etc.
On the other hand, this invention provides the method to treat diseases or
disorders in
which protein kinase activity can be regulated. The diseases and disorders
associated with
protein kinase activity include cancer, inflammation, autoimmune disease,
metabolic disease,
infection, central nervous system disease, cardiovascular disease and etc.
One aspect of this invention is that the compounds herein can be used to treat
the diseases
or disorders associated with abnormal cell proliferation, such as cancer
including leukemia,
myeloproliferative disease, hematonosis, gastrointestinal stromal tumor, colon
cancer, breast
cancer, stomach cancer, oophoroma, cervical cancer, lung cancer, kidney
cancer, prostate
cancer, bladder cancer, pancreas cancer, neuroblastoma, mast cell tumor,
encephaloma,
germ cell tumor, melanoma, malignant tumor, and sarcoma, such as
dermatofibrosarcoma
protuberans and etc.
17
i
CA 02748289 2011-06-23
=
One aspect of this invention is that the compounds herein can be used to treat
the diseases
associated with autoimmune diseases or inflammatory diseases, including
diabetes, scytitis,
rheumatoid arthritis, allergic rhinitis, asthma, ankylosing spondylitis,
psoriasis, Crohn disease
and etc.
One aspect of this invention is that the compounds herein can be used to treat
vascular
diseases such as atheromatosis, hemadostenosis, pulmonary hypertension and
retinal
disease, as well as fibrosis diseases such as pulmonary interstitial fibrosis,
hepatofibrosis,
hepatocirrhosis, scleriasis, glomerulosclerosis, myocardial fibrosis and etc.
Another aspect of this invention relates to the methods of preparing the
compounds of
Formula I. The compounds in this invention can be prepared by the following
methods and
procedures.
sysyrk,,,r--\ CuCN NC
NaBI-14
.1 = >
I q
FO Me0H k=s,""==
õcc
1.1 ON
sociz cyclic amino-group NC`r"-µr"\ Na0H/1.120
CH202 K-CO
= 3, CN 3 ;2. SO C12110 (30 H
1-3 1.4
.õ-
k400
1-3
Scheme 1
The intermediate formula 1-5 can be prepared as shown in Scheme 1.
5-Bromo-2,3-dihydroinden-1-one and CuCN may be refluxed in DMF to obtain the
cyano
intermediate 1-1. Intermediate 1-1 may be reduced to the alcoholic
intermediate 1-2 by
treating with a reductant such as sodium borohydride in a solvent system such
as methanol.
Intermediate 1-2 can react with thionyl chloride to get a chlorine group which
will be
replaced with a cyclic amino-group in the presence of triethylamine or
potassium carbonate
to obtain intermediate 1-4. The cyano-group in intermediate 1-4 may be
hydrolyzed to obtain
a carboxylic acid, which will subsequently be treated with methanol and
thionyl chloride to
obtain compound 1-5. The two enantiomers of compound 1-4 or compound 1-5 can
be
separated by chiral high performance liquid chromatography or by
crystallization using
camphorsulfonic acid.
18
CA 02748289 2011-06-23
NaBH4Br
,-"=\
)1? s`r--.\ S0012
L.N1e011 CH2C12
oH CI
2-1 2.2
C0/R01-1/NEIAMS0/
cyclic amino-group pci(0A02/cippp
......................................... RO
)
K2co,/cH3cN or BULYTHF/CO2
or :NE=i3lEt0H R' W
2-3 2-4
Scheme 2
Alternatively, the 131-subsituted 2,3-dihydroindene carboxylic acid (or its
ester) can be
prepared by the method depicted in Scheme 2. 5-Bromo-2,3-dihydroinden-1-one
may be
reduced to the alcoholic intermediate 2-1 by treating with a reductant such as
sodium
borohydride in a solvent system such as methanol. After the hydroxyl group of
intermediate 2-1 is converted into chlorine by thionyl chloride, the chloride
2-2 can be
replaced with a cyclic amino-group by using triethylamine or potassium
carbonate as a base
to obtain intermediate 2-3. Intermediate 2-3 can react with CO utilizing
palladium, such as
palladium diacetate / 1,3-bis-(phenylphosphine) propane (dppp) or bis-
(triphenylphosphine)
palladium dichloride (11)[(PP113)2PdC12] as catalysts to obtain the
intermediate of formula 2-4
as a mixture of two enantiomers. When R of compound 2-4 is H, compound 2-4 can
be
obtained by treating compound 2-3 with butyllithium followed by quenching the
reaction
with carbon dioxide. The compound 2-3 and the two enantiomers of compound 2-4
can be
separated by chiral high performance liquid chromatography or using chiral
acids, such as by
crystallization using camphorsulfonic acid.
NaOHooNaOH
Me OH
R4 =
3-13-2
aniline derivatives/
`.ttialkylaluminium coupling agents/
aniline derivatives/Et3N
)1,
aniline ..
.derivatives HN N
= pyndine
z,V "
R R1
3-3 3-4
Scheme 3
Final compounds of formula 3-4 can be prepared as shown in Scheme 3. The
carboxylic ester
of intermediate 3-1 can be hydrolyzed by an alkali such as sodium hydroxide
into carboxylic
acid 3-2, which can be then condensed with an aniline derivative to obtain
final compounds
19
CA 02748289 2011-06-23
=
of formula 3-4 using coupling agents such as
benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorophosphate (BOP)
or
0-(7-azabenzotriazol-1-y1)-N,N,N',N'- tetra methyluronium hexafluorophosphate
(HATU).
Addtionally, carboxylic acid 3-2 may be treated with thionyl chloride to form
an acid chloride
3-3, which can then react with an aniline derivative to obtain compounds of
formula 3-4.
Final compounds of formula 3-4 may also be obtained via the reaction between
the ester 3-1
and an aniline derivative utilizing trialkylaluminium such as
trinnethylaluminium or
triethylaluminium as the coupling agent.
EXAMPLE 1
Preparation of 1-(4-methylpiperazin-1-yI)-N-(4-methyl-3-[(4-pyridin-3-
ylpyrimidin-2-yl)amino]
phenyl)-2,3-dihydro-1H-inden-5-carboxamide.
T
HN,
VI 40.
N
I I
'.1\1
Step A: 1-0xo-2,3-dihydro-1H-inden-5-carbonitrile
NC 411111,1"
0
5-Bromo-2,3-dihydro-1H-inden-1-one (21.1 g, 100 mmol ) and cupric cyanide
(17.9 g, 200
mmol) were. mixed in 200 ml of dimethylformamide and stirred overnight at 140
C. After the
solution was cooled down to room temperature, 500 ml of ethyl acetate was
added and the
precipitate was removed by filteration using kieselguhr. The solid was rinsed
with ethyl
acetate for several times. The pooled filtrates were washed with 1 N
hydrochloric acid twice
and then with brine for 3 times, dried over anhydrous magnesium sulfate,
filtered, and
concentrated. The crude product was purified on silica gel, eluting with ethyl
acetate/
hexanes (1:2), to obtain 7.9 g of the desired compound (50% yield).
MS(M+1)=158.05.
Step B: 1-Hydroxy-2,3-dihydro-1H-inden-5-carbonitrile
CA 02748289 2011-06-23
NC se
OH
1-0xo-2,3-dihydro-1H-inden-5-carbonitrile (7.85 g, 50 mmol) was dissolved in
50 mL of
methanol . To it was added sodium borohydride (2.3 g, 60 mmol) gradually
within about 30
minutes. The solution was concentrated after being stirred for 2 hours. The
residue was
dissolved in ethyl acetate and the solution obtained was washed with sodium
bicarbonate
twice and then with brine twice, dried over magnesium sulfate, filtered, and
concentrated to
obtain 8 g of the desired compound (100% yield). MS(M+1)=160.07.
Step C: 1-(4-Methylpiperazin-1-yI)- 2,3-di hydro-1H-indene-5-carbonitrile
= NC
N
1-Hydroxy-2,3-dihydro-1H-indene-5-carbonitrile (4.77 g, 30 mmol) was dissolved
in 10 mL of
methylene dichloride. While cooled 6n ice, thionyl chloride (6.6 ml, 90 mmol)
was added
dropwise within about 15 minutes. The solution was concentrated after being
stirred for 3
hours. The residue was dissolved in ethyl acetate and the solution obtained
was washsed
with cooled brine for 3 times, dried over anhydrous magnesium sulfate, and
concentrated to
obtain 1-chloro-2,3-dihydro-1H-indene-5-carbonitrile.
1-Chloro-2,3-dihydro-1H-indene-5-carbonitrile obtained was dissolved in 80 mL
of
acetonitrile and then 1-methyl piperazine (6 g, 60 mmol) as well as potassium
carbonate
(4.14 g, 30 mmol) are added. After the solution was stirred overnight at 60
C, the
acetonitrile was removed by concentration under reduced pressure. Ethyl
acetate was then
added. The solution obtained was washed with brine for 3 times, dried over
magnesium
sulfate, concentrated, and purified on silica gel using 5% methanol /
methylene dichloride as
the eluent to obtain 4.3 g of the desired compound (60% yield).
MS(M+1)=242.16.
Step D: Methyl 1-(4-methylpiperazine-1-yI)-2,3-dihydro-1H-indene-5-carboxylate
21
CA 02748289 2011-06-23
0.
1-(4-Methylpiperazin-1-yI)-2,3-dihydro-1H-indene-5-carbonitrile (2.41 g, 10
mmol) was
dissolved in 10 mL of a 2 N solution of sodium hydroxide. The solution was
stirred overnight
at 100 C and then concentrated. Followed by vacuum desiccation, the solid was
suspended
in 30 mL methanol. Thionyl chloride (3.3 mL) was added dropwise with stirring
within 1 hour.
The mixture was refluxed overnight and then concentrated. Water was firstly
added, and
then potassium carbonate was added to make the solution turned into basic. The
solution
was extracted with ethyl acetate for 3 times. The pooled extracts were washed
with brine,
dried over magnesium sulfate, and then concentrated. It was further purified
by silica gel
column using 5% methanol / methylene dichloride as the eluent to obtain 2.1 g
(77% yield)
of the subtitle compound. MS(M+1)=275.17.
Step E: Preparation of 1-(4-
methylpiperazine-1-y1)-N-(4-methy1-3-[(4-pyridin-3-y1
pyrimidin-2-ypa min 0] ph enyI)-2,3-di hydro-1H-indene-5-carboxamideamide
40 T
HN
iNdi se
N
N
Methyl 1-(4-methylpiperazin-1-yI)-2,3-dihydro-1H-indene-5-carboxylate (1.37 g,
5 mmol) and
4-methyl-N(3)-(4-pyridin-3-ylpyrimidin-2-yl)pheny1-1,3-diamine (Szakacs et al.
J. Med. Chem.
2005, 48:249) (1.66 g, 6 mmol) was suspended in 30 mL of toluene. A solution
of 2 M
trimethylalurninium in toluene (5 mL, 10 mmol) was added and the mixture was
stirred
overnight at 50 C. The reaction was incomplete. Another batch of 2 M
trimethylaluminium
in toluene (3 mL, 6 mmol) was then added. The mixture was cooled on ice after
being stirred
overnight at 60 C. Saturated potassium sodium tartrate aqueous solution (50
mL) was added
with stirring. The solution was extracted with methylene dichloride (3 x 100
mL). The pooled
extracts were washed with sodium bicarbonate (100 mL) and then with brine (2 x
100 mL),
dried over magnesium sulfate, and then concentrated. It was further purified
by silica gel
22
CA 02748289 2011-06-23
column using 50% ethyl acetate/ methylene dichloride/ 5-10% triethylamine as
the eluent to
obtain 1.5 g (58% yield) of the title compound. MS(M+1)=520.27. 1FINMR (DMSO-
d6, PPm):
8 10.10 (s, 1H); 9.20 (s, 1H); 8.95, (s, 1H); 8.62 (d, .1= 4.8 Hz, 1H); 8.42
(d, 1= 4.8 Hz, 1H); 8.40
(d, J= 9.0 Hz, 1H); 8.00 (s, 1H); 7.75 (s, 1H); 7.72 (d, J=9.0 Hz, 1H); 7.45
(dd, J=8.2 Hz, 4.8 Hz,
1H); 7.40 (d, 1=8.0 Hz, 1H); 7.38 (d, 1=4.8 Hz, 1H); 7.28 (d, J=9.0 Hz, 1H);
7.15 (d, J=9.0 Hz, 1H);
4.26 (t, J=9.0 Hz, 1H); 2.2-2.9 (m, 1H); 2.15 (s, 3H); 2.08 (s, 3H); 2.0 (m,
2H).
EXAMPLE 2
Preparation of tert-butyl 4-{54({4-methyl-3-[(4-pyridin-3-ylpyrimidin-2-
yl)amino]phenyll
amino)carbonyI)-2,3-di hydro-1-H-inden-1-yllpiperazin-1-carboxylate
40 7
HN
N
OS
/1--\
Boc
Step A: 5-Bromo-2,3-dihydro-1H-inden-1-ol
Br sie
OH
5-Bromo-2,3-dihydro-1H-inden-1-one (210 g, 1000 mmol) was suspended in 1 L
methanol,
and sodium borohydride (41.6 g, 1100 mmol) was added gradually within about 1
hour with
stirring. The solvent was removed at 50 C under reduced pressure after being
stirred for
another 1 hour. Ethyl acetate (1 L) was added followed by saturated sodium
bicarbonate
solution (500 mL). After being stirred for some time, the solution was
transferred to a
separatory funnel and the aqueous phase was removed. The organic phase was
washed with
saturated sodium bicarbonate solution twice and with brine twice, dried
(magnesium sulfate)
and finally concentrated to obtain 198 g (93%) of the subtitle compound.
Step B: 5-Bromo-1-chloro-2,3-dihydro-1H-indene
23
CA 02748289 2011-06-23
Br se
CI
5-Bromo-2,3-dihydro-1H-inden-1-ol (198 g, 934 mmol) was dissolved in 500 mL of
methylene
dichloride. While cooled on ice, thionyl chloride (275 mL, 3770 mmol) was
added to the
methylene chloride solution dropwise within about 2 hours. The solution was
concentrated
at 30 C under reduced pressure condition after being stirred for 2 hours at
room
temperature. The residue was dissolved in ethyl acetate (1 L) and the solution
obtained was
washed with ice-cold water (3 x 500 mL) and with brine (2 x 300 mL), dried
over magnesium
sulfate, and concentrated to obtain 5-bomo-1-chloro-2,3-dihydro-1H-indene.
Step C: tert-Butyl 4-(5-Bromo-2,3-dihydro-1H-inden-1-y1) piperazin-l-
carboxylate
Br
Akbak
Boc
5-Bromo-1-chloro-2,3-dihydro-1H-indene (10 g, 43 mmol) was dissolved in 80 mL
of
acetonitrile, and sodium carbonate (4.8 g, 45 mmol) was added followed by tert-
butyl
piperazin-1-carboxylate (9.7 g, 52 mmol). The mixture was stirred at 60 C
overnight. The
insoluble substance was removed by filtration and the filtrate was
concentrated. The residue
was separated by silica gel column using ethyl acetate / hexanes (1:2 to 1:1)
as the eluent to
obtain 12 g (72% yield) of the subtitle compound. MS(M+1)=381.11, 383.11.
Step D: tert-Butyl 4-[5-( Ethoxycarbony1)-2,3-di hydro-1H-inden-1-y1)
piperazin-l-carboxylate
0
Boc
tert-Butyl 4-(5-bromo-2,3-dihydro-1H-inden-1-y1) piperazin-1-carboxylate (11
g, 28.87 mmol)
was dissolved in ethanol (50 mL), and dimethyl sulfoxide (5 mL) and
triethylamine (5 mL)
were added. The system was vacuumized and charged with N2. Palladium acetate
(2 g) and
24
CA 02748289 2011-06-23
1,3-bis(diphenylphosphino)propane (3 g) were added. The system was vacuumized
and
charged with N2. The system was vacuumized once again and stirred at 100 C
for 24 hours
with inserting CO balloons. After being cooled down to the room temperature,
the mixture
was filtered by kieselguhr which was then rinsed thoroughly with ethanol. The
filtrate was
concentrated. The residue was dissolved in ethyl acetate (500 mL) and the
solution obtained
was washed with brine (3 x 200 mL), dried over magnesium sulfate, concentrated
and finally
separated by silica gel column using ethyl acetate / hexanes (1:2 to 1:1) as
the eluent to
obtain 8.5 g (79% yield) of the subtitle compound. MS(M+1)=375.22.
Step E: 1-[4-(BOC) piperazin-1-y1]-2,3-dihydro-1H-indene-5-carboxylic acid
HO Se
\-H
Boc
tert-Butyl 4-[5-(ethoxycarbonyI)-2,3-dihydro-1H-inden-1-yl)piperazin-1-
carboxylate (8 g,
21.36 mmol) was dissolved in 20 mL of methanol, and 30 mL of sodium hydroxide
(1 N) was
added. The solution was stirred overnight at room temperature and at 50 C for
another 2
hours, and then concentrated. The residue was dissolved in water (50 mL) and
the obtained
solution was acidified to pH 5 with 1 N HCI and then extracted with ethyl
acetate (3 x 100
mL). The extract liquor was pooled together, dried over magnesium sulfate, and
concentrated to obtain the subtitle compound. MS(M+1)=347.19.
Step F: tert-Butyl 4-{54({4-Methyl-3-[(4-pyridin-3-ylpyrimidin-2-
ypamino]phenyl}amino)
carbonyl]-2,3-di hydro-1H-i nden-1-yll pi perazin-1-carboxylate
I
HN
N
N
V..11)
Boc
1[4-(BOC)piperazin-1-y1]-2,3-dihydro-1H-indene-5-carboxylic acid (7.4 g, 21.36
mmol) and
4-methyl-N(3)-[(4-pyridin-3-ylpyrimidin-2-yl)pheny1-1,3-diamine (6.1 g, 22
mmol) were
CA 02748289 2011-06-23
dissolved in 20 mL of N,N-dimethylformamide. Both triethylamine (8.9 mL, 64
mmol) and
0-(7-azabenzotriazol-1-y1)-N,N,N',N'-tetramethyluronium hexafluorophosphate
(9.5 g, 25
mmol) were added. The solution was stirred overnight at room temperature and
then brine
(100 mL) was added followed by ethyl acetate (200 mL). The aqueous phase was
removed
and the ethyl acetate layer was washed with brine (3 x 100 mL). Then the
solution was dried
over magnesium sulfate, concentrated and finally separated by silica gel
column using
methanol/methylene chloride (1:2 to 1:1) as the eluent to obtain 9.5 g (73%
yield) of the title
compound. MS(M+1)=606. 31. 1HNMR (DMSO-c1-6, ppm): 6 10.15 (s, 1H); 9.25 (s,
1H); 8.99 (s,
1H); 8.67 (d, J= 4.8 Hz, 1H); 8.50 (d, J= 5.2 Hz, 1H); 8.46 (d, 1= 8.4 Hz,
1H); 8.05 (s, 1H); 7.78 (s,
1H); 7.76 (d, J=8.0 Hz, 1H); 7.50 (dd, 1=8.0 Hz, 4.8 Hz, 1H); 7.46 (d, 1=8.4
Hz, 1H); 7.41 (d,
J=5.2 Hz, 1H); 7.38 (d, J=7.6 Hz, 1H); 7.18 (d, J=8.8 Hz, 1H); 4.35 (t,1=7.2
Hz, 1H); 3.30 (m, 3H);
3.05 (m, 1H); 2.08 (s, 2H); 2.42 (m, 2H); 2.30 (m, 2H); 2.20 (s, 3H); 2.04 (m,
2H); 1.36 (s, 9H).
EXAMPLE 3
Preparation of N-(4-
methy1-3-[(4-pyridin-3-ylpyrimidin-2-ypamino]pheny1)-1-
piperazin-1-y1-2,3-dihydro-1H-indene-5-carboxamide
HN
H sot
N". N
I
tert-Butyl 4-15-[({4-
methyl-3-[(4-pyridin-3-ylpyrimidin-2-yl)amino]phenyl}amino)
carbonyl]-2,3-dihydro-1H-inden-1-yllpiperazin-1-carboxylate (2 g, 3.3 nnmol)
was dissolved in
= 4 N HC1 in dioxane (10 mL). The solution was concentrated to obtain the
solid product after
being stirred at room temperature for 3 hours. The product (100 mg) was
purified by high
performance liquid chromatography at pH = 10 to obtain the subtitle compound.
MS(M+1)=506.26. 1HNMR (DMSO-d6, ppm): 6 10.08 (s, 1H); 9.20 (s, 1H); 8.93 (s,
1H); 8.62
(d, J= 4.8 Hz, 1H); 8.44 (d, J= 5.2 Hz, 1H); 8.40 (d, 1= 8.0 Hz, 1H); 8.00 (s,
1H); 7.72 (s, 1H); 7.70
(d, J=8.0 Hz, 1H); 7.45 (dd, J=8.2 Hz, 4.8 Hz, 1H); 7.41 (d, J=8.2 Hz, 1H);
7.36 (d, J=5.2 Hz, 1H);
7.31 (d,1=8.0 Hz, 1H); 7.12 (d, 1=8.8 Hz, 1H); 4.22 (t, J=6.8 Hz, 1H); 2.80
(m, 2H); 2.60 (m, 4H);
2.35 (m, 21-1); 2.22 (m, 2H); 2.15 (s, 3H); 2.00 (m, 2H).
EXAMPLE 4
26
CA 02748289 2011-06-23
Preparation of 1-(4-ethylpiperazin-1-y1)-N-(4-methy1-3-[(4-pyridin-3-
ylpyrimidin-2-yl)amino]
phenyl)-2,3- dihydro-1H-indene-5-carboxamide
O
el =
H.11 FN1
N N
0))
N-(4-Methy1-3-[(4-pyridin-3-ylpyrimidin-2-yl)amino]pheny1)-1-piperazin-1-y1-
2,3-
dihydro-1H-indene-5-carbxamide tetrahydrochloride (100 mg, 0.15 mmol) was
dissolved in
DMF (2 mL) and triethylamine (101 mg, 1 mmol) was added followed by
acetaldehyde (26 mg,
0.6 mmol). After the solution was stirred for 20 minutes, sodium
triacetoxyborohydride (128
mg, 0.6 mmol) was added. The solution obtained was stirred overnight at room
temperature
and then purified by high performance liquid chromatography at pH=10 to obtain
50 mg
(63% yield) of the title compound. MS(M+1)=523.29. 11-1NMR (DMSO-d6, ppm): 8
10.14 (s,
1H); 9.25 (s, 1H); 8.98 (s, 1H); 8.67 (d, 1= 4.8 Hz, 1H); 8.49 (d, J= 5.2 Hz,
1H); 8.46 (d, J= 8.6 Hz,
1H); 8.05 (s, 1H); 7.77 (s, 1H); 7.75 (d, J=8.8 Hz, 1H); 7.50 (dd, J=8.0 Hz,
4.8 Hz, 1H); 7.46 (d,
J=8.2 Hz, 1H); 7.41 (d, J=5.2 Hz, 1H); 7.35 (d, J=7.6 Hz, 1H); 7.17 (d, 1=8.8
Hz, 1H); 4.31 (t,-
1=6.8 Hz, 1H); 2.2-3.0 (m, 12H); 2.20 (s, 3H); 2. 03 (m, 2H); 0.95 (t, 1=7.0
Hz, 3H).
EXAMPLE 5
Preparation of 1-(4-isopropylpiperazin-1-y1)-N-(4-methy1-3-[(4-pyridin-3-
ylpyrimidin-2-y1)
amino]phenyI)-2,3-dihydro-1H-indene-5-carboxamide
= 111
NHO
N
/11-\
r\J
2--
N-(4-Methy1-3-[(4-pyridin-3-ylpyrimidin-2-yl)amino]pheny1)-1-piperazin-1-y1-
2,3-
dihydro-1H-indene-5-carboxamide tetra hydrochloride (100 mg, 0.15 mmol) was
dissolved in
DMF (2 mL), and triethylamine (101 mg, 1 mmol) was added followed by acetone
(35 mg, 0.6
mmol). After the solution was stirred for 20 minutes, sodium
triacetoxyborohydride (128 mg,
0.6 mmol) was added. The solution obtained was stirred overnight at room
temperature and
27
CA 02748289 2011-06-23
then purified by high performance liquid chromatography at pH=10 to obtain 58
mg (71%
yield) of the title compound. MS(M+1)=548.31. 11-4N1MR (DM50-d6, ppm): 8 10.14
(s, 1H);
9.26 (s, 1H); 8.98 (s, 1H); 8.67 (d, 1= 4.8 Hz, 1H); 8.49 (d, 1= 4.8 Hz, 1H);
8.46 (d, J= 8.4 Hz);
8.05 (s, 1H); 7.77 (s, 1H); 7.74 (d, J=8.0 Hz, 1H); 7.51 (dd, J=8.0 & 4.8 Hz,
4.8 Hz, 1H); 7.46 (d,
1=8.2 Hz, 1H); 7.41 (d, J=5.2 Hz, 1H); 7.35 (d, J=7.6 Hz, 1H); 7.17 (d, J=8.4
Hz, 1H); 4.30 (t,
J=7.0 Hz, 1H); 2.91 (m, 12H); 2.81 (s, 3H); 2.3-2.6 (m, 9H); 2.02 (m, 2H);
0.92 (t, J=6.4 Hz,6H).
EXAMPLE 6
Preparation of 1-(4-(2-hydroxyethylpiperazin-1-y1)-N-(4-methy1-3-[(4-
pyridin-3-y1
pyrimidin-2-yl)amino]phenyI)-2,3-dihydro-1H-indene-5-carboxamide
t17, [_11
N
I
(j--)
NL\
OH
N-(4-Methyl-3-[(4-pyridin-3-ylpyrimidin-2-yl)amino]pheny1)-1-piperazin-1-y1-
2,3-
dihydro-1H-indene-5-carboxamide tetrahydrochloride (100 mg, 0.15 mmol) was
dissolved in
DMF (2 mL), and triethylamine (101 mg, 1 mmol) was added followed by {[tert-
butyl
(dimethyl)silyl]oxolacetaldehyde (100 mg, 0.6 mmol). After the solution was
stirred for 20
minutes, sodium triacetoxyborohydride (128 mg, 0.6 mmol) was added. The
solution
obtained was stirred overnight at room temperature and then purified by high
performance
liquid chromatography. The dried product was dissolved in 2 mL of methylene
chloride / 2 ml
trifluoroacetic acid. The solution was concentrated after being stirred
overnight and purified
by high performance liquid chromatography at pH=10 to obtain 38 mg (46% yield)
of the title
compound. MS(M+1)=550.29. iFINMR (DMSO-d6, ppm): 6 10.16 (s, 1H); 9.23 (s,
1H); 8.97 (s,
1H); 8.65 (d, J= 4.4 Hz, 1H); 8.48 (d, J= 6.0 Hz, 1H); 8.46 (d, J= 4.8 Hz);
8.02 (s, 1H); 7.82 (m,
2H); 7.51 (m, 1H); 7.40 (m, 2H); 7.14 (d, 1=8.4 Hz, 1H); 2.6-3.7 (m, 17H);
2.16 (s, 3H).
EXAMPLE 7
Preparation of 1-44-acetylpiperazin-1-y1)-N-(4-methyl-3-[(4-pyridin-3-
ylpyrimidin-2-y1)amino]
phenyI)-2,3- dihydro-1H-indene-5-carboxamide
28
CA 02748289 2011-06-23
40 0
HN 11
N N
1
N-(4-Methyl-3-[(4-pyridin-3-ylpyrimidin-2-yl)amino]phenyl)-1-piperazin-1-y1-
2,3-
dihydro-1H-indene-5-carboxamide tetra hydrochloride (100 mg, 0.15 mmol) was
dissolved in
DMF (2 mL), and triethylamine (101 mg, 1 mmol) was added followed by acetyl
chloride (16
mg, 0.2 mmol) while being cooled in an ice bath. After being stirred for 20
minutes, the
solution obtained was purified by high performance liquid chromatography at
pH=10 to
obtain 45 mg (55% yield) of the title compound. MS(+1)=548.27. 11-INMR (DMSO-
d6, PPm):
8 10.15 (s, 1H); 9.26 (s, 1H); 8.99 (s, 1H); 8.66 (d, J= 4.8 Hz, 1H); 8.49 (d,
J= 5.2 Hz, 1H); 8.45
(d, J= 8.4 Hz); 8.05 (s, 1H); 7.79 (s, 1H); 7.76 (d, J=8.0 Hz, 1H); 7.50
(dd,J=8.0 & 4.8 Hz, 1H);
4.37 (t, J=7.0 Hz, 1H); 3.42 (m, 1H); 3.40 (m, 3H); 2.91 (m, 1H); 2.83 (m,
1H); 2.2-2.5 (m, 4H);
2.20 (s, 3H); 2.06 (m, 2H); 1.95 (s, 3H).
EXAMPLE 8
Preparation of N43-(4,5'-bipyrimidine-2-ylamino)-4-methylpheny1]-1-(4-
methylpiperazin-1
-yI)- 2,3- dihydro-1H-indene-5-carboxamide
N N
IffC)
Step A: 5-Acetylpyrimidine
o
5-Bromopyrimidine (3.18 g, 20 mmol) was dissolved in 50 mL of tetrahydrofuran.
While
cooled to -78 C, 15 mL of 1.6 M n-butyllithium in hexane solution was added
dropwise
with stirring. After the solution was stirred for 30 minutes, a solution of N-
methoxyl-N-
methylacetamide (2.58 g, 25 mmol) in tetrahydrofuran solution (10 mL) was
added slowly.
29
CA 02748289 2011-06-23
The mixture was stirred at -78 C for 1 hour and then allowed to be warmed
slowly. When
the temperature of the mixture was at 0 C , aqueous ammonium chloride
solution was
added. The solution obtained was extracted with ethyl acetate for 3 times. The
pooled
extracts were washed with brine, dried over magnesium sulfate, concentrated
under reduced
pressure, and purified by silica gel column chromatography using 5% methanol /
methylene
chloride as eluent to obtain 1 g of the title compound (45% yield).
MS(M+1)=123.05.
Step 8: (2E)-3-(Dimethylamino)-1-pyrimidin-5-ylprop-2-en-1-one
rj
5-Acetylpyrimidine (1 g, 8.2 mmol) and N,N-dimethylformamide dimethyl acetal
(1.3 g, 11
mmol) were dissolved in 20 mL of isopropanol. The solution was stirred at 100
C for 24
hours, cooled to room temperature, and concentrated under reduced pressure.
Ethyl ether
was then added to the residue. After being cooled in an ice bath for a couple
of hours, the
solid was collected by filtration, rinsed with cold ethyl ether, dried in
vacuum to obtain 1 g
(59% yield) of the title compound. MS(M+1)=178Ø
Step C: N-(2-Methyl-5-nitropheny1)-4,5'-bipyrimidine-2-amine
N N NO2
(2E)-3-(Dimethylamino)-1-pyrimidin-5-ylprop-2-en-1-one (1 g, 5.6 mmol) and
N-(2-methyl-5-nitrophenyl)guanidine nitrate (1.44 g, 5.6 mmol) (Z. Szakacs et
al., J. Med.
Chem. 2005, 48, 249) were suspended in 20 mL of isopropanol. Sodium hydroxide
(0.28 g, 7
mmol) was then added. The mixture solution was stirred overnight and cooled to
room
temperature. The solid was collected by filtration and rinsed with isopropanol
and diethyl
ether. The filtrate was concentrated under reduced pressure and the residue
was dissolved
in 15 mL of isopropanol. The solution obtained was refluxed overnight and
cooled to room
temperature. The solid was collected by filtration and rinsed with isopropanol
and diethyl
ether. The pooled solid was rinsed with water and diethyl ether, and dried in
vacuum to
CA 02748289 2011-06-23
obtain 1.2 g (70% yield) of the title compound. MS(M+1)=309.10.
Step D: N(3)-4,5'-Bipyrimidin-2-y1-4-methylbenzene-1,3-diamine
N
N N NH2
Stannous chloride dihydrate (3.6 g, 16 mmol) was dissolved in 10 mL of
concentrated
hydrochloric acid. The solution was added to N-(2-methy1-5-nitropheny1)-4,5'-
bipyrimidin-
2-amine with violent stirring. The mixture was poured into ice-cold water
after being stirred
for 2 hours. It was then neutralized to pH > 8 with sodium carbonate and
extracted with
ethyl acetate for 4 times. The pooled extracts were washed with brine, dried
over
magnesium sulfate and finally concentrated under reduced pressure to obtain
0.7 g of the.
title compound. MS(M+1)=279.13.
Step E: N43-(4,5'-Bipyrimidin-2-ylamino)-4-methylpheny1]-1-(4-methylpiperazin-
1-y1)-2,3-
dihydro-1H-indene-5-carboxamide
0
doe
N N
cr_N-)N
Methyl 1-(4-methylpiperazin-1-yI)-2,3-dihydro-1H-indene-5-carboxylate (823 g,
3 mmol) and
N(3)-4,5'-bipyrimidin-2-y1-4-methyl benzene-1,3-diamine (973 g, 3.5 mmol) were
suspended
in 15 mL of toluene, and then a 2 M trimethylaluminium solution (3 mL, 6 mmol)
was added.
The mixture was stirred overnight at 50 C and another 2 M trimethylaluminium
solution (2
mL, 4 mmol) was added. The solution was stirred overnight at 60 C and then
cooled in an
ice bath. Saturated aqueous solution of potassium sodium tartrate was added
with stirring.
The solution obtained was extracted with methylene chloride (3 x 100 mL). The
pooled
extracts were washed with sodium bicarbonate (100 mL) and with brine (2 x 100
ml), dried
o
over magnesium sulfate, concentrated, and purified by silica gel column
chromatography
using 50% ethyl acetate / methylene chloride / 5-10% triethylamine as eluent
to obtain 702
mg of the title compound (45% yield). MS(M+1)=527.27. 11-1NMR (DMSO-d6, ppm):
5 10.10
31
CA 02748289 2011-06-23
(s, 1H); 9.46 (s, 2H); 9.28 (s, 1H); 9.08 (s, 1H); 8.50 (d, J= 5.7 Hz, 1H);
8.04 (s, 1H); 7.74 (s,
1H);7.70 (d, J= 9.0 Hz, 1H); 7.46 (d, 1= 5.7 Hz, 1H); 7.42 (d, 1=9.0 Hz, 1H);
7.32 (d, J=9.0 Hz,
1H); 7.15 (d, J=9.0 Hz, 1H); 4.25 (t, J=5.7 Hz, 1H); 2.2-2.9 (m, 10H); 2.15
(s, 3H); 2.07 (s, 3H);
2.0 (m, 2H).
EXAMPLE 9
Preparation of 1-[(35)-3-
(dimethylamino)pyrrolidin-1-y11-N-{4-methy1-34(4-pyridin-3-y1
pyrimidin-2-yl)aminolpheny11-2,3-dihydro-1H-indene-5-carboxamide
o
õP.
N
;.
(r =-=,=
Step A: (3S)-1-(5-Bromo-2,3-dihydro-1H-inden-1-y1)-N,N-dimethylpyrrolidin-3-
amine
Br
5-Bromo-1-chloro-2,3-di hydro-1H-indene (2.03 g, 8.76 mmol)
and
(3S)-N,N-2,5-dimethylpyrrolidin-3-amine (1 g, 8.76 mmol) were dissolved in 30
mIL of
acetonitrile, and potassium carbonate (1.81 g, 13.14 mmol) was then added. The
mixture
was stirred overnight at 60 C and then concentrated. The residue was
dissolved in ethyl
acetate. The solution was washed with brine for 3 times, dried over magnesium
sulfate, and
then concentrated. It was further purified by silica gel column chromatography
using ethyl
acetate / methylene chloride / triethylamine / methanol (10:10:1:1) as eluent
to obtain 1.3 g
of the title compound (48% yield). MS(M+1)=309.0, 311Ø
Step B: Methyl 1-[(35)-3-(N,N-Dimethylamino)pyrrolidin-1-y1]-2,3-dihydro-1H-
indene
-5-carboxylate
rif
32
CA 02748289 2011-06-23
=
(3S)-1-(5-Bromo-2,3-dihydro-1H-inden-l-yI)-N,N-2,5-dimethylpYrrolidin-3-amine
(1.3 g, 4.2
mmol) was dissolved in 30 mL of methanol, 5 mL of dimethylsulfoxide and 7 mL
of
triethylamine. The reaction flask was vacuumized and then charged with N2.
Palladium
acetate (0.24 g, 1 mmol) plus 1,3-bis(diphenylphosphino)propane (0.5 g, 1.5
mmol) were
then added. The mixed solution was stirred at 80 C for 2 days in the presence
of CO. After
being cooled to room temperature, it was filtered and concentrated. The
residue was
dissolved in ethyl acetate. The solution obtained was washed with brine for 3
times, dried
over magnesium sulfate, and then concentrated. It was further purified by
silica gel column
chromatography using ethyl acetate / methylene chloride / triethylamine
(10:10:1) as eluent
to obtain 0.7 g of the title compound (58% yield). MS(M+1)=289.1.
Step C: 1-[(35)-3-(Dimethylamino)pyrrolidin-1-yll-N-(4-methyl-3-[(4-pyridin-3-
ylpyrimidin-2-
yl) amino] phenyl}-2,3-dihydroindene-5-carboxamide
HA) nl>
sts1
11
Methyl 1-[(3S)-3-(N,N-dimethylamino)pyrrolidin-1-y11-2,3-dihydroindene-5-
carboxylate (0.2 g,
0.69 mmol) and 4-methyl-N(3)-(4-pyridin-3-ylpyrimidin-2-yl)pheny1-1,3-diamine
(0.22 g, 0.8
o
mmol) were dissolved in 5 mL toluene. A solution of 2 M trimethylaluminium in
toluene (1.3
ml, 2.6 mmol) was then added. The mixture was stirred at 60 C for 2 days and
then cooled in
an iCe bath. Potassium sodium tartrate in aqueous solution (15 mL) followed by
methylene
chloride (50 mL) was then added. The organic phase was separated and the
aqueous phase
was extracted with methylene chloride twice. The pooled organic phase was
washed with
brine, dried over magnesium sulfate, and then concentrated. It was further
purified by high
performance liquid chromatography to obtain 0.22 g (60% yield) of the title
compound.
MS(M+1)=534.29. 11-INMR (CD30D, ppm): 8 9.19 (s, 1H); 8.54 (d, J= 5.2 Hz, 1H);
8.50 (d, 1=
8.4 Hz, 1H); 8.36 (d, J= 5.2 Hz, 1H); 8.10 (s, 1H); 7.72 (s, 1H); 7.67 (d, J=
8.4 Hz, 1H); 7.44 (d, 1=
5.2 Hz, 1H); 7.41 (d, J= 8.4 Hz, 1H); 7.32 (d, J= 8.4 Hz, 1H); 7.28 (d, J= 5.2
Hz, 1H); 7.15 (d, 1=
8.4 Hz, 1H); 4.18 (m, 2H); 3.01 (m, 1H); 2.90 (m, 1H); 2.80 (m, 2H); 2.72 (m,
2H); 2.60 (m, 1H);
2.37 (m, 1H); 2.22 (s, 3H); 2.16 (m, 1H); 2.14 (s, 6H); 1.95 (m, 1H); 1.62 (m,
1H).
33
CA 02748289 2011-06-23
EXAMPLE 10
Preparation of 1-[(3R)-3-(dimiethylamino)pyrrolidin-1-yli-N-{4-methy1-3-
[(4-pyridin-3-y1
pyrimidin-2-yl)amino]phenyI}-2,3-dihydro-1H-indene-5-carboxamide
411 T
HN
011.
N
I
The title compound was prepared according to the method described in EXAMPLE
9. MS(M+1)=
524.29. 1HNMR(CD30D, ppm): 6 9.19 (s, 1H); 8.54 (d, J=5.2 Hz, 1H); 8.50 (d,
1=8.8 Hz, 1H); 8.36 (d,
J=5.2 Hz, 1H); 8.10 (s, 1H); 7.72 (s, 1H); 7.6.7 (d, J=7.2 Hz, 1H); 7.44 (d,
J=7.2 Hz, 1H); 7.41 (d,
1=8.8 Hz, 1H); 7.32 (d, 1=7.2 Hz, 1H); 7.28 (d, J=5.2 Hz, 1H); 7.15 (d, J=7.2
Hz, 1H); 4.18 (m, 1H);
3.02 (m, 1H); 2.95 (m, 1H); 2.85 (m, 2H); 2.75 (m, 2H); 2.65 (m, 1H), 2.39 (m,
1H); 2.24 (s, 3H);
2.20 (m, 1H); 2.15 (s, 3H); 1.98 (m, 1H); 1.65 (m, 1H).
EXAMPLE 11
Preparation of (15)-1-(4-methylpiperazin-1-y1)-N-(4-methyl-3-[(4-pyridin-
3-ylpyrimidin-2-y1)
aminolphehyl)-2,3-dihydro-1H-indene-5-carboxamide
N
'
1
Step A: 1-((1S)-5-Bromo-2,3-dihydro-1H-indene-1-yI)-4-methylpiperazine
I
5-Bromo-1-chloro-2,3-dihydro-1H-indene (220 g, 950 mmol) was dissolved in
acetonitrile (1 L),
and 1-methylpiperazine (150 g, 1500 mmol) was added followed by potassium
carbonate (131 g,
950 mmol). The mixture was stirred overnight at 60 C. The solid was removed
by filtration and
the filtrate was concentrated. The residue was dissolved in ethyl acetate (1
L) and the solution
obtained was washed with sodium hydroxide twice (2 x 300 mL) and with brine
for 3 times (3 x
34
CA 02748289 2011-06-23
300 mL), dried over magnesium sulfate, concentrated and purified by silica gel
column
chromatography using 5% methanol / methylene chloride as eluent to obtain 202
g of the
product (72% yield). MS(M+1)=295.07, 297.07.
The product obtained (202 g, 684.6 mmol) was dissolved in 2000 mL of methanol
and then
(1S)-(+)-10-camphorsulfonic acid (318 g, 1369 mmol) was added followed by 4000
mL of
isopropanol. The solution was refluxed with heating for 10 minutes, and then
stirred overnight at
room temperature. The solid was collected by filtration. When there was no
more liquid dripping
off, the solid was rinsed with isopropanol and then dissolved in 600 mL of
methanol. After
isopropanol (1500 ml) was added, the solution was heated under reflux for 15
minutes and
then stirred overnight at room temperature. The solid was collected by
filtration. When there
was no more liquid dripping dif, the solid was washed with isopropanol and
then dissolved in 1
N sodium hydroxide (600 mL). The solution was stirred for 30 minutes and then
extracted with
ethyl acetate for 3 times (3 x 300 mL). The pooled extracts were washed with 1
N sodium
hydroxide (300 mL) and with brine (2 x 300 mL), dried over magnesium sulfate,
and then
concentrated to obtain 50 g of the title compound. Its chiral purity was
99.7%, measured by
chiral high performance liquid chromatography. X-ray monocrystal structure
analysis of the title
compound indicated that the chiral center at the 1 position of 2,3-dihydro-1H-
indene is of S
configuration. MS(M+1)=295.07, 297.07.
Step B: Ethyl (1S)-1-(4-Methylpiperazin-1-y1)-2,3- dihydro-1H-indene -5-
carboxylate
)1
"Y, N\
=
1-((1S)-5-Bromo-2,3-dihydro-1H-inden-1-yI)-4-methylpiperazine (29.6 g, 100
mmol) was dissolved
in 300 mL of ethanol, 30 mL of DMSO and 42 mL of triethylamine. The system was
vacuumized
and charged with N2. After palladium acetate (2.4 g, 10 mmol) and
1,3-bis(diphenylphosphino)propane (3.3 g, 10 mmol) were added, the system was
vacuumized and charged with N2. After being vacuumized once again, the mixture
was stirred
at 90 C for 2 days under CO. After being cooled to the room temperature, the
solution was
filtered by kieselguhr and then concentrated. The residue was dissolved in
ethyl acetate (500
ml) and the solution obtained was washed with brine (3 x 200 mL), dried over
magnesium
sulfate, concentrated, and finally separated by silica gel column
chromatography using 50%
o
CA 02748289 2011-06-23
=
ethyl acetate / 45% methylene chloride / 5% triethylamine as the eluent to
obtain 17.3 g of the
title compound (60% yield). MS(M+1)=289.18.
Step C: (1S)-1-(4-
Methylpiperazin-1-yI)-N-(4-methyl-3-[(4-Pyridin-3-ylpyrimidin-2-yl)amino]
phenyl)-2,3-dihydro-1H-indene-5-carboxamide
;Lis,* O.
n))
Ethyl (15)-1-(4-methylpiperazin-1-yI)-2,3-dihydro-1H-indene-5-carboxylate (7.2
g, 25 mmol)
and 4-methyl-N(3)-(4-pyridin-3-ylpyrimidin-2-yl)pheny1-1,3-diamine (8.3 g, 30
mmol) were
dissolved in 150 mL of toluene, and a solution of 2 M trimethylaluminium in
toluene (20 m L,
40 mmol) was then added. The solution obtained was stirred overnight at 50 C,
and then 20
ml of 2 M rimethylaluminium in toluene was added. After being stirred at 60 C
for another
24 hours, the solution was cooled in ice bath and potassium sodium tartrate in
aqueous
solution (200 ml) followed by methylene chloride (300 mL) was then added. The
organic
phase was separated and the aqueous phase was extracted with methylene
chloride twice.
The pooled extract was washed with brine twice, dried over magnesium sulfate,
and then
concentrated. It was further purified by silica gel column chromatography
using 50% ethyl
acetate / methylene chloride / 5-10% triethylamine as eluent to obtain 7.5 g
(58% yield) of
the title compound. MS(M+1)=520.27. 11-INMR (DMSO-d6, ppm): 5 10.10 (s, 1H);
9.20 (s, 1H);
8.95 (s, 1H); 8.66 (d, 1= 6.0 Hz, 1H); 8.48 (d, .1= 6.0 10 Hz, 1H); 8.43 (d,
1= 8.4 Hz, 1H); 8.02 (s,
1H); 7.77 (s, 1H); 7.74 (d, J= 1H); 7.48 (dd,
1H); 7.42 (dd, 1H);7.40 (d, 1= 6.0 Hz, 1H);
7.32 (d, J=8.4 Hz, 1H); 7.18 (d, J=8.4 Hz, 1H); 4.26 (t, J=8.4 Hz, 1H); 2.2-
3.0 (m, 10H):, 2.20 (s,
3H); 2.12 (s, 3H); 2.02 (m, 2H).
EXAMPLE 12
Preparation of (1R)-1-(4-methylpiperazin-1-y1)-N-(4-methy1-3-[(4-pyridin-3-
ylpyrimidin-2-y1)
amino]phenyI)-2,3-dihydro-1H-indene-5-carboxamide
36
CA 02748289 2011-06-23
H N
N ..rs1
1
Step A: 1-((1R)-5-Bromo-2,3-dihydro-1H-inden-1-yI)-4-methylpiperazine
Br io
In the Step A of EXAMPLE 11, the methanol / isopropanol filtrate containing
1-(5-bromo-2,3-dihydro-1H-inden-1-yI)-4-methylpiperazine and (1S)-(+)-10-
camphorsulfonic
acid was concentrated under reduced pressure. The residue was dissolved in 1 L
of sodium
hydroxide (1 N). After being stirred for 30 minutes, the solution was
extracted with ethyl
acetate (3 x 300 mL). The pooled extract was washed with 1 N sodium hydroxite
(300 mL)
and with brine (3 x 300 mL), dried over magnesium sulfate and then
concentrated to obtain
140 g (474 mmol) of 1-(5-bromo-2,3-dihydro-1H-inden-1-y1)-4-methylpiperazine,
wherein the
dominating isomer is R-enantiomer. The residue was dissolved in 1.4 L of
methanol, and
(1R)-(-)-10-camphorsulfonic acid (220 g, 948 mmol) was added followed by 2.8 L
of
isopropanol. The solution obtained was heated under reflux for 15 minutes and
then stirred
overnight at room temperature. The solid was collected by filtration. When
there was no more
liquid dripping off, the solid was rinsed with isopropanol and then dissolved
in 600 mL of
methanol. After isopropanol (1500 mL) was added, the solution was heated under
reflux for 15
minutes and then stirred overnight at room temperature. The solid was
collected by filtration.
Without liquid dripping off, the solid was rinsed with isopropanol and then
dissolved in 800
mL of sodium hydroxide (1 N). The mixture was stirred for 30 minutes and then
extracted
with ethyl acetate for 3 times (3 x 300 mL). The pooled extract was washed
with 1 N sodium
hydroxide (500 mL) and with brine (2 x 400 ml), dried over magnesium sulfate,
and then
concentrated to obtain 60 g of the title compound. Its chiral purity is 99.8%,
measured by
chiral high performance liquid chromatography. MS(M+1)=295.07, 297.07.
Step B: Ethyl (1R)-1-(4-methylpiperazin-1-y1)-2,3-dihydro-1H-indene-5-
carboxylate =
37
CA 02748289 2011-06-23
Starting from 1-((1R)-5-bromo-23-dihydro-1H-inden-1-yI)-4-methylpiperazine,
the title
compound was obtained according to the procedure described in Step B of
EXAMPLE 11.
MS(M+1)=289.18.
Step C: (1R)-1-(4-Methylpiperazin-1-yI)-N-(4-methyl-3-[(4-pyridin-3-
ylpyrimidin-2-yl)amino]
phenyl)-2,3-dihydro-1H-indene-5-carboxamide
I,
H
. (
1=1
The title compound was obtained by condensation of ethyl (1R)-1-(4-
methylpiperazin-1-yI)-
2,3-dihydro-1H-indene-5-carboxylate and 4-methyl-N(3)-(4-pyridin-3-
ylpyrimidin-2-y1)
phenyl-1,3-diamine, according to the procedure described in Step C of EXAMPLE
11.
MS(M+1)=520.27. iHNMR (DMSO-d6, ppm): 8 10.15 (s, 1H); 9.22 (s, 1H); 8.98 (s,
1H); 8.64
(d, J= 6.0 Hz, 1H); 8.46 (d, J= 6.0 Hz, 1H); 8.42 (d, J= 8.4 Hz, 1H); 8.02 (s,
1H); 7.75 (s, 1H); 7.72
(d, J= 9.0 Hz, 1H); 7.50 (dd, 1H); 7.45 (dd, 1H);7.40 (d, J= 5.4 Hz, 1H); 7.35
(d, J=8.4 Hz, 1H);
7.18 (d, 1=9.0 Hz, 1H); 4.26 (t, J=6.0 Hz, 1H); 2.2-3.0 (m, 10H); 2.20 (s,
3H); 2.12 (s, 3H); 2.3 (m,
2H).
EXAMPLE 13
Preparation of (15)-N-[3-(4,5'-bipyrimidin-2-ylamino)-4-methylpheny1]-1-(4-
methylpiperazin
-1-yI)-2,3-di hydro-1H-indene-5-ca rboxamide
[,.41 0111
1
38
CA 02748289 2011-06-23
The title compound was obtained by condensation of ethyl (1S)-1-(4-
methylpiperazin-1-yI)-
2,3-dihydro-1H-indene-5-carboxylate and
N-(3),4,5'-bipyrimidin-2-y1-4-methylpheny1-1,3-diamine according to the
procedure
described in Step C of EXAMPLE 11. MS(M+1)=521.27. 11-11\1MR (DMSO-d6, ppm): 6
10.10 (s,
1H); 9.40 (s, 2H); 9.28 (s, 1H); 9.08 (s, 1H); 8.50 (d, J= 4.8 Hz, 1H); 8.04
(s, 1H); 7.74 (s, 1H);
7.70 (d, J= 9.0 Hz, 1H); 7.46 (d, J= 4.8 Hz, 1H); 7.42 (d, J= 7.8 Hz, 1H);
7.32 (d, J=7.8 Hz, 1H);
7.15 (d, J=9.0 Hz, 1H); 4.25 (t, 1=7.8 Hz, 1H); 2.2-2.9 (m, 10H); 2.15 (s,
3H); 2.07 (s, 3H); 2.0 (m,
2H).
EXAMPLE 14
Preparation of (1R)-1\143-(4,5'-bipyrimidin-2-ylamino)-4-methylpheny11-1-(4-
methylpiperazin
-1-yI)-2,3-dihydro-1H-indene-5-carboxamide
H ilµ
N=.:% 'N
The title compound was obtained by condensation
of ethyl
(1R)-1-(4-methylpiperazin-1-yI)-2,3-dihydroindene-5-carboxylate and N-(3)-4,5'-
bipyrimidin-2
-y1-4-methylpheny1-1,3-diamine, according to the procedure described in Step C
of EXAMPLE
11. MS(M+1)=521.27. iHNMR (Dm50-d6, ppm): 6 10.10 (s, 1H); 9.40 (s, 2H); 9.28
(s, 1H);
9.08 (s, 1H); 8.50 (d, J= 5.7 Hz, 1H); 8.04 (s, 1H); 7.74 (s, 1H); 7.70 (d, J=
8.4 Hz, 1H); 7.46 (d,
1= 5.7 Hz, 1H); 7.42 (d, .1= 8.4 Hz, 1H); 7.32 (d, J=8.40 Hz, 111); 7.15 (d,
1=8.4 Hz, 1H); 4.25 (t,
J=7.5 Hz, 1H); 2.2-2.9 (m, 10H); 2.15 (s, 3H); 2.07 (s, 3H); 2.0 (m, 2H).
EXAMPLE 15
Preparation of (1S)-1-(4-methylpiperazin-l-y1)-N-(4-methy1-3-[(4-pyridin-4-
ylpyrimidin-2-y1)
amino]phenyI)-2,3-dihydro-1H-indene-5-carboxamide
HN
.1,
N
I
39
CA 02748289 2011-06-23
Step A: N-(2-Methyl-5-nitropheny1)-4-pyridin-4-ylpyrimidine-2-amine
40/
N NO2
The title compound was prepared by condensation reaction between
(2E)-3-(dimethylamino)-1-pyridin-4-ylprop-2-en-1-one and
N-(2-methyl-5-nitrophenyl)guanidine nitrate, according to the procedure
described in Step C of
EXAMPLE 8. MS(M+1)=308.11.
Step 8: 4-Methyl-N(3)-(4-pyridin-4-ylpyrimidin-2-yl)benzne-1,3-diamine
N
N NH2
The title compound was prepared by reduction of N-(2-methyl-5-nitropheny1)-4-
pyridin-4-y1
pyrimidin-2-amine, according to the procedure described in Step D of EXAMPLE
8.
MS(M+1)=278.13.
Step C: (15)-1-(4-
Methylpiperazin-1-y1)-N-(4-methy1-3-[(4-pyridin-4-ylpyrimidin-2-yl)amino]
phenyl)-2,3-dihydro-1H-indene-5-carboxamide
40 N
HN
4100.
N H N
(N--\
The title compound was prepared by condensation' reaction between ethyl
(1.5)-1-(4-methylpiperzin-1-y1)-2,3-dihydro-1H-indene-5-carboxylate and
4-methyl-N(3)-(4-pyridin-4-ylpyrimicR1-2-yl)benzne-1,3-diamine, according to
the procedure
described in Step C of EXAMPLE 11. MS(M+1)=520.27. 11-1NMR(DMSO-d6, ppm): 6
10.14 (s, 1H);
9.04(s, 1H); 8.07 (d, 1=4.4 Hz, 2H); 8.55 (d, J=4.8Hz, 1H); 8.06 (s, 1H); 8.04
(d, 1=4.4Hz, 2H); 7.78 (s,
1H); 7.75 (d, J=8.8Hz, 1H); 7.45 (d, 1=7.6 Hz, 1H); 7,44 (d, 1=4.8 Hz, 11-4);
7.35 (d, 1=7.6 Hz, 1H);
CA 02748289 2011-06-23
=
7.18 (d, J8.8 Hz, 1H); 4.31 (t, J=7.2 Hz, 1H); 2.0-3.0 (m, 10H); 2.19 (s, 3H);
2.12 (s, 3H); 2.04 (m,
2H).
EXAMPLE 16
Preparation of (1S)-1-(4-methylpiperazin-1-y1)-N-(4-methyl-3-[(4-pyridin-
3-ylpyrimidin-2-y1)
aminolpheny1)-2,3-dihydro-1H-indene-5-carboxamide sulfate
a0
HN 41Pr N 0111
H
N
1.5H2SO4
Step A: (1S)-1-(4-Methylpiperazin-1-yI)-2,3-dihydroindene-5-carboxylic acid
o
HO
'&.(\ipH r-v\p
--
N
Under the protection of N2, 1-((15)-5-bromo-2,3:dihydro-1H-inden-1-y1)-4-
nnethylpiperazine (660
g, 2.235 mol) and THF (3.3 L) were added to a 10 L three-neck flask, and the
solution was stirred
until dissolved. The system temperature was cooled to -78 C in liquid
nitrogen-acetone bath.
n-butyllithium (n-BuLi) (2.5 M in hexane solution) (1072 mL, 2.682 mol, 1.2-
fold) was added
dropwise into the solution at -78 C ¨ -82 C. After being stirred for 10
minutes, when the LC-MS
assay shown that the reaction of tl(Z raw materials was completed, dry ice
(170 g, 3.86 mol,
1.73-fold) was added carefully. The solution was then stirred for 10 minutes
at -60 C ¨ -75 C.
After the reaction was completed, the cold bath was removed and aqueous
solution of 2 N HCI
was then added to adjust the pH value till pH=2. Most of the water was removed
using rotary
evaporator. The system was further dried overnight at 50 C ¨ 60 C in a
vacuum drying oven to
obtain the title compound (1289 g, the actual product of 583 g providing 100%
yield). This crude
product was used in the next step of reaction directly.
Step B: (1S)-1-(4-Methylpiperazin-1-yI)-2,3-dihydro-indene-5-carbonyl chloride
hydrochloride
CI-Arr's 2HCI
L.;
41
CA 02748289 2011-06-23
S0Cl2 (2.5 L) was added to a 5 L three-neck flask. (1S)-1-(4-Methylpiperazin-1-
yI)-2,3-
dihydro-1H-indene-5-carboxylic acid (1289 g, the actual maximum content of 583
g, equivalent to
2.235 mol) was then added in batches within 1 hour. The solution was heated
under reflux
overnight and then cooled to room temperature. Most of S0Cl2 was removed using
rotary
evaporator. After ethyl acetate (1.5 L) was added, the solution was cooled to
0 C, filtered with
suction to obtain the white solid which was then dried in vacuum to get the
title compound
(about 1325 g, 100% yield).
Step C: (1S)-1-(4-Methylpiperazin-1-yI)-N-(4-methyl-3-[(4-pyridin-3-
ylpyrimidin-2-yl)amino]
phenyl)-2,3-dihydro-1H-indene-5-carboxamide
40 0
HN
ri
N N
C_N)
N-(5-Amino-2-methylphenyI)-4-(3-pyridy1)-2-aminopyrimidine (681 g, 2.46 mol,
1.1-fold) was
dissolved in pyridine (3 L). (1S)-1-(4-Methylpiperazin-1-yI)-2,3-dihydro-1H-
indene-5-carbonyl
chloride hydrochloride (1325 g, the actual maximum content of 626 g,
equivalent to 2.235 mol,
1-fold) was added slowly within 30 minutes with stirring. The solution turned
very hot since the
reaction was severe but there was no need to have it cool down. After the
solution was stirred
overnight at room temperature, the reaction solution was added to 2 N aqueous
solution of
sodium hydroxide (2 L) with stirring, immediately followed by addition of
methylene dichloride (2
L). After being stirred for a while, the solution was transferred to a 5 L
separatory funnel and then
methylene chloride layer was separated. The water phase was extracted with
methylene chloride
(2x 500 mL). The extracts were pooled, dried over anhydrous magnesium sulfate,
and then
concentrated. The residue was dissolved in methylene dichloride and then
purified by silica gel
column chromatography using methylene chloride / 5% methanol / 1%
triethylamine as
eluent. The fractions containing the desired product were pooled and
concentrated under
reduced pressure. The residue was'bissolved in 1 L of warm ethyl acetate and
crystals were
precipitated after stirring. The solid was collected by filtration and dried
in vacuum at 50 C to
obtain the title product (617 g, 53% total yield of the three steps).
MS(M+1)=520.27.
Step D: (1S)-1-(4-Methylpiperazin-1-y1)-N-(4-methyl-3-[(4-pyridin-3-
ylpyrimidin-2-ypamino]
42
CA 02748289 2011-06-23
phenyl)-2,3-dihydro-1H-indene-5-carboxa mide su lfate
gin 0
HN
H so
N
N2
15H2SO4
(15)-1-(4-Methylpiperazin-1-y1)-N-(4-methyl-3-[(4-pyridin-3-ylpyrimidin-2-
ypamino]pheny1)-2,3-
Dihydro-1H-indene-5-carboxamide (248 g, 0.477 mol) was dissolved in ethanol
(4.76 L). After
being stirred for 20 minutes, the solution was filtered with suction. The
filtrate was added to a 20
L three-neck flask. Sulfuric acid solution (46.74 g, 0.477 mol, 1-fold)
diluted with ethanol (532 ml)
was added slowly through dropping funnel with thorough stirring to form a
yellowish suspension.
Ethanol (9.5 L) was then supplemented. The mixture was heated under reflux for
2 hours until it
became a milky-white suspension. The suspension was cooled to room temperature
statically,
filtered with suction, and then dried to obtain the title product (183 g,
57.5%). The filtrate was
adjusted to basic (pH=11) with aqueous solution of NaOH and then extracted
with methylene
dichloride (4 x 200 mL). The extract was dried and concentrated to reclaim the
(1S)-1-(4-methyl
piperazin-1-y1)-N-(4-methyl-3-[(4-pyridin-3-ylpyrimidin-2-yl)a mino]pheny1)-
2,3-dihydro-1H-inden
e-5-carboxamide (98 g, 39.5%). Melting point of the title product: 187 ¨ 189
C. Elemental
analysis of C311136N70751.5, calculating value: C 55.84; H 5.44; N 14.71;
testing value: C 55.72; H
5.70; N 14.40.
The regulation of protein kinase activity and inhibition of cell proliferation
by the compounds
of this invention may be tested by the procedures demonstrated below.
EXAMPLE A: Assay of enzyme activity of Abl, c-Kit and PDGFR kinases
The activity of the compounds of this invention on Abl, c-Kit and PDGFR
kinases was
tested by Mobility Shift Assay (MSA). ATP concentration is at Km of each
kinase, i.e. Abl Km
ATP= 12 M, c-Kit Km ATP=87 jtM, PDGFR Km ATP= 38 M.
Materials: Abl (purchased from Ca rna, Lot No. 06C13S-2988C); c-Kit (purchased
from BPS, Cat.
No. 40250, Lot No. 1003); PDGFR (purchased from BPS, Cat. No. 40263, Lot No.
1001); DMSO
(purchased from Sigma, Cat. No. D2650, Lot No. 474382); 96-well culture plate
(purchased
from Corning, Cat. No. 3365, Lot No. 22008026); 384-well culture plate
(purchased from
43
CA 02748289 2011-06-23
Corning, Cat. No. 3573, Lot No. 12608008); Staurosporine (purchased from
Sigma, Cat. No.
54400-1MG, Lot No. 046K4080).
Methods:
1. Preparation of kinase buffer and stop buffer;
(1) Kinase buffer: 62.5 mM HEPES, pH 7.5; 0.001875% Brij-35; 12.5 mM MgC12;
2.5 mM
DTT;
(2) Stop buffer: 100 mM HEPES, pH. 7.5; 0.015% Brij-35; 0.2% coating agent #3;
50 mM
E DTA;
2. The compound was dissolved in DMSO and then serial dilutions were prepared;
3. Preparation of kinase solution: Kinase solution was obtained by dissolving
kinases in the
kinase buffer mentioned above. With regard to c-Kit kinase, the pre-activation
treatments
should be carried out as follows..700 nM c-Kit, 2 mM ATP, 4 mM DTT and 10 mM
MgC12
were dissolved in kinase buffer. After being incubated at 28 C for 15
minutes, the solution
was added to the kinase buffer;
4. Preparation of polypeptide solution: Polypeptide FAM and ATP were dissolved
in kinase
= buffer;
5. The kinase solution was transferred to a culture plate and incubated_for 10
minutes at
room temperature. The final concentrations of Abl, c-Kit and PDGFR were 0.45
nM, 12
nM, 8 nM, respectively;
6. The polypeptide solution was transferred to the culture plate. The final
concentrations of
ATP in the Abl, c-Kit and PDGFR circumstances were 12 1AM, 87 and 38 ,IM,
respectively. The final concentrations of MgC12 in all circumstances were all
10 mM;
7. The mixture in each well of the plate was incubated at 28 C, for 1 hour
for Abl, 40 minutes
for c-Kit and 5 hours for PDGFR. The stop buffer was then added to terminate
the
reaction;
8. Data were collected in Caliper and then input into the software XLfit to
calculate IC50
va lu es.
The required concentrations (IC50, nM) of each compound of this invention to
result in 50%
inhibition rate were listed in Table 1. Meanwhile, IC50 values of lmatinib
leading to
inhibition of these three kinases in the same experimental condition were also
listed in Table
1 for convenient comparison. Use staurosporine as positive control in this
assay.
44
CA 02748289 2011-06-23
Table 1
IC 50 (nM)
Compound
Abl c-Kit . PDGFR
EXAMPLE 1 5.8 22 17
EXAMPLE 2 2044 1186 233
EXAMPLE 3 6.2 16 12
EXAMPLE 4 6.3 26 15
EXAMPLE 5 5.4 23 18
EXAMPLE 6 12 32 20
EXAMPLE 7 411 529 41
EXAMPLE 8 5.3 32 22
EXAMPLE 9 121 51 15
EXAMPLE 10 70 32 21
EXAMPLE 11 2.2 8.5 9.6
EXAMPLE 12 256 2251 53
EXAMPLE 13 2.4 15 13
EXAMPLE 14 266 2260 65
EXAMPLE 15 23 526 24 -
Imatinib 207 703 39
Staurosporine 162 2.0 0.50
As shown in Table 1, the compounds of the invention exhibited very high
inhibitory activity
against Abl, c-Kit and PDGFR: IC50 values ranged from 2.2 nM to 2044 nM in
inhibiting Abl;
IC50 values ranged from 8.5 nM to 2260 nM in inhibiting c-Kit; IC50 values
ranged from 9.6
nM to 233 nM in inhibiting PDGFR. Except for EXAMPLEs 2, 7, 12 and 14, the
compounds of
the invention had higher activity than Imatinib in inhibiting these three
kinases.
EXAMPLE B: Test of kinase activity of Abl and c-Kit mutants
Inhibitory activity of the compounds of this invention on Abl, c-Kit and PDGFR
mutant
kinases was tested by the phosphor isotope-labelled ATP (33P-ATP) assay.
1. Substrate solution was prepared using newly-prepared reaction buffer. The
buffer includes:
20 mM HEPES, pH 7,5, 10 mM MgC12, 1 mM EGTA, 0.02% Brij 35, 0.02 mg/ml BSA,
0.1 mM
CA 02748289 2011-06-23
Na3VO4, 2 mM DTT, 1% DMSO. As for c-Kit and c-Kit (V654A), additionally, 2 mM
MnCl2
was added into the buffer;
2. Required co-enzymes were added to the above substrate solution;
3. Kinases were added and mixed gently;
4. The tested compound was dissolved in DMSO, and then added to the above
kinase
solution by using Acoustic technique (Echo550, nanoliter range) and incubated
for 20
minutes;
5. 33P-ATP was added to the above reaction mixture to initiate the reaction;
6. The mixture was incubated for 2 hours at room temperature;
7. The activity of kinases was tested by filtration-combination;
8. The data were processed in Excel and the control data were subtracted. A
curve was drawn
by the GraphPad Prism software to get the IC50 value.
IC50 values of the compound of EXAMPLE 11 which can inhibit Abl and 7 mutants
thereof, as
well as c-Kit and 5 mutants thereof were listed in Table 2. Meanwhile, IC50
values of
Nilotinib leading to inhibition of these mutants in the same experimental
condition were
also listed in Table 2 for convenient comparison. Use staurosporine as
positive control in
these assays.
Table 2
ATP Concentration EXAMPLE 11 Nilotinib Staurosporine
Kinase
(11M) IC50 (nM) IC50 (nM) IC50 (nM)
Abl 10 0.21 1.19 14.5
Abl (T3151) 10 14090 >20000 3.38
Abl (E225K) 10 3.72 = 36.3 27.0
Abl (G250E) 10 2.94 24.8 5.68
Abl (H396P) 10 0.38 2.99 9.02
Abl (M351T) 10 0.29 1.89 9.33
Abl (Q252H) 10 0.42 4.66 4.47
Abl (Y253F) 10 0.71 5.13 15.4
c-Kit 30 132 302 8,41
c-Kit
30 83.6 574 <1.0
(D816H)
o
c-Kit " 30 1738 >20000 <1.0
46
CA 02748289 2011-06-23
(D816V)
c-Kit (T6701) 30 2057 >20000 2.67
c-Kit
30 1.77 16.5 <1:0
(V560G)
c-Kit (V654A) 30 969 13940 1.16
As shown in Table 2, the compound of EXAMPLE 11 possessed higher inhibitory
activity in
inhibiting the mutants of Abl and c-Kit than Nilotinib. Nilotinib (brand-named
as Tasigna) had
a good effect on treating those leukemia patients who gain resistance to
Imatinib
(brand-named as Gleevec). As the compound of EXAMPLE 11 had higher effect on
inhibition
of the Imatinib mutants in the test than Nilotinib, the compounds of the
invention will have
more effective results in treating the leukemia patients resistant to
Imatinib. The c-Kit
mutants exist widely in gastrointeQinal stromal tumor, mast cell disease and
acute myeloid
leukaemia. As shown in Table 2, the compound of EXAMPLE 11 had a good effect
in inhibiting
all the c-Kit mutants. Thus, the compound of the invention can be applied to
treat
gastrointestinal stromal tumor, mast cell disease, acute myeloid leukaemia and
etc.
EXAMPLE C: K562 cell assay
The inhibitory activity of the compounds of the invention on the growth of
chronic myeloid
leukaemia cell K562 was tested using a CellTiter-Glo assay.
Materials: K562 cell strain (purchased from ATCC, Cat. No. CCL-243, Lot No.
50644810);
IMDM (purchased from Invitrogen, Cat. No. 12440-053); fetal bovine serum
(purchased from
Invitrogen, Eat. No. 10099141, Lot No. 613866); DMSO (purchased from Sigma,
Cat. No.
D2650, Lot No. 077k2357); 96-well culture plate (purchase from Corning, Cat.
No. 3903); 15
ml centrifuge tube (purchased from Greiner, Cat. No. 0703115, Lot No. 2012-
01); cell viability
assay kit (CellTiter-Glo) (purchased from Promega, Cat. No. G7571, Lot No.
256984);
Staurosporine (purchased from Sigma, Cat. No. 54400-1MG, Lot No. 046K4080).
Methods:
=
1. plating cell
(1) Preparation of complete medium: Complete medium was composed of 90% IMDM
and
10% fetal bovine serum which were mixed thoroughly;
(2) The cell strain in good growth status was selected;
47
CA 02748289 2011-06-23
(3) The cell suspension was transferred to centrifuge tubes using piette and
then centrifuged
at 800-1000RPM for 3-5 minutes;
(4) The supernatant in the tube was removed using pipette;
(5) A proper volume of the medium was added to the tube and the cells were
resuspended
by pipetting up and down gently;
(6) The cells were counted using blood counting chamber;
(7) The cell suspension was adjusted to the cell concentration of 4x 104 cells
/ml;
(8) The cell suspension was added to a 96-well bottom-transparent culture
plate, 100 I per
well, i.e. 4000 cells per well. The plate was incubated overnight with CO2 in
an incubator.
2. Preparation and addition of the compound
c,4
(1) The compounds were dissolved in DMSO and then diluted with DMSO to 10
different
Concentrations;
(2) 0.54 of the compound solution was transferred to the culture plate;
(3) The culture plate was incubated at 37 C in the incubator for 72 hours.
3. Test and analysis
(1) The cellular morphology was observed in inverted microscope;
(2) 1004 of the cell viability assay reagent was added to each well;
(3) The plate was shaked for 2 minutes on a shaker, allowing cell lysis;
(4) The plate was kept at room temperature for 10 minutes to stabilize
luminescence signals;
(5) A white membrane was attached to the plate bottom and the plate was
detected using
Flexstation 3 (luminescence, integration time 500 ms);
(6) The results were recorded and then analyzed.
The required concentrations (IC50, nM) of the compounds of EXAMPLE 3, 11, 12,
13 and 15
in this invention to result in 50% inhibition rate were showed in Table 3.
Meanwhile, IC50
value of Imatinib inhibiting K562 cell growth in the same experimental
condition was also
included in Table 3 for convenient comparison. Use staurosporine as positive
control in this
assay.
= Table 3
EXAMPLE 3 11 12 13 15 lmatinib Staurosporine
1050 (nM) 12 3.2 208 2.2 35 206 139
48
CA 02748289 2011-06-23
As shown in Table 3, the compounds of EXAMPLE 3, 11, 12, 13, and 15 exhibted
very high
inhibitory activity on the growth of chronic myeloid leukaemia cell K562.
Except for EXAMPLE
12, the required concentrations (IC50, nM) of the compounds of EXAMPLE 3, 11,
13 and 15
to lead to 50% inhibition rate in inhibiting the growth of K562 cells were
much lower than
that of Imatinib (130.05). The compound of EXAMPLE 12 was the optical
enantiomer of
EXAMPLE 11. Although its inhibitory activity on the growth of K562 was 65-fold
lower than
that of EXAMPLE 11, it was as potent as Imatinib. This suggests that the
compounds of this
invention can be used to treat chronic myeloid leukaemia effectively.
EXAMPLE D: Assay of K562, KU812, MEG-01, Kasumi-1 and Sup-B15 cell strains
This invention also tested the inhibitory activity of the compounds of this
invention on the
growth of chronic myeloid leukaemia cell K562, KU812, and MEG-01, acute
myeloid
leukaemia cell Kasumi-1 and acute lymphatic leukemia cell Sup-B15.
- Materials: SpectraMAX Plus Micr9plate Spectrophotometer Mode 3011 (purchased
from
Molecular Devices Corp, California, USA); Water-Jacketed CO2 Incubator
(purchased from
Therma, USA); inverted microscope, Chongguang XDS-1B (Chongqing Optical &
Electrical
Instrument Co.,Ltd., Chongqing, China); CellTiter 96 Aqueous MTS reagent
powder
(purchased from Promega, Cat. No. G1112); Phenazine methosulfate (PMS)
(purchased from
Sigma, Product No. P9625); RPMI1640 (purchased from GIBCO, USA, Cat. No. 31800-
022);
IMDM (purchased from GIBCO, USA, Cat. No. 12200-036); fetal bovine serum (FBS)
= (purchased from GIBCO, USA, Cat. No. FCS100).
Methods:
1. Preparation of assay solution
(1) Preparation of PMS solution: PMS was dissolved in DPBS to give a
concentration of 0.92
g/ml. The solution was then filtered into a sterile and lightproof container;
(2) Preparation of MTS solution: a) 21 ml DPBS was added to a lightproof
container; b) 42
mg MTS powder was weighted and then added to DPBS; c) the above were mixed on
a
electromagnetic stirrer until the powder was dissolved; d) the pH value was
measured. The
preferred value should be between 6.0 and 6.5. If the pH was higher than 6.5,
it should be
adjust to 6.5 with 1 N HCI; e) the solution was filtered into a sterile and
lightproof container;
49
CA 02748289 2011-06-23
(3) Preparation of MTS/PMS mixture: a) 2 ml of the MTS solution was
transferred to a tube;
b) 100 IA of the PMS solution was added to the tube; c) the tube was vortexed
gently to mix
the solution thoroughly.
2. Plating cell
(1) The cells were counted using blood counting chamber after the cells grew
to certain
number;
(2) The cell concentration was adjusted to 2.78 x 104 cell/ml with RPMI1640
medium
=
containing 10% FBS (K562, KU812, MEG-01 or Kasumi-1 cells) or IMDM medium
containing 0.05 mM 2-mercaptoethanol and 20% FBS (Sup-815 cell);
(3) 180 [IL of the cell suspension was added to each well of 96-well culture
plate with the
final cell density of 5x 103per well.
3. Preparation and addition of the compounds
(1) The tested compounds were dissolved in DMSO and then diluted to 10
different
concentration;
(2) 20 pLof each concentration was transferred to each well containing the
cell suspension
already (3 wells for each concentration);
(3) The plate was incubated for 72 hours at 37 C, 5% CO2 and 95% humidity.
4. Test and analysis
(1) 40 1.11_ of the MTS/PMS solution was pipetted into each well containing
200 piL medium to
give the final volume per well of 240 1.;
(2) The plate was incubated for 1-4 hours at 37 C, 5% CO2 and 95% humidity;
(3) The absorptions were recorded at wavelength of 490 nm using Spectra Max
Plus;
(4) IC50 values were calculated using the 5th versions Gra phPad Prism
software.
The required concentrations (1050, nM) of the compounds of EXAMPLE 16 to lead
to 50%
inhibition rate in inhibiting K562, KU812, MEG-01, Kasumi-1 and Sup-B15 cell
strains were
showed in Table 4. Meanwhile, the inhibition activity of Imatinib and
Nilotinib in the same
experimental condition was also included in this table for convenient
comparison. Use
=
staurosporine as positive control in this assay.
Table 4
CA 02748289 2015-01-06
IC50 (nM)
Cell strain
EXAMPLE 16 Imatinib Nilotinib Staurosporine
K562 0.25 121 6.26 71.5
KU812 0.024 51.4 2.10 9.57
MEG-01 0.085 19.3 1.65 8.97
Kasumi-1 11.2 297 22.3 1.04
Sup-B15 39.6 382 135 6.84
As shown in Table 4, the compound of EXAMPLE 16 exhibited very high inhibitory
activity on
the growth of the chronic myeloid leukaemia cell strains K562, KU812 and MEG-
01, the acute
myeloid leukaemia cell strain Kasumi-1 and the acute lymphatic leukemia cell
strain Sup-B15.
Its IC50 values ranged from 0.024 nM to 39.6 nM. Additionally, the activity of
the compound
of EXAMPLE 16 in inhibiting the growth of these cell strains was higher than
that of either
Imatinib or Nilotinib. These results suggest that the compounds of this
invention can be used
to treat chronic myeloid leukaemia, acute myeloid leukaemia and acute
lymphatic leukemia
effectively.
While this invention has been described together with the illustrated
embodiment, the
scope of the claims should not be limited by the preferred embodiments set
forth in the
Examples, but should be given the broadest interpretation consistent with the
description as
a whole.
51