Note: Descriptions are shown in the official language in which they were submitted.
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
COMBINATION ANTIBODIES FOR THE TREATMENT AND PREVENTION OF
DISEASE CAUSED BY BACILLUS ANTHRACIS AND RELATED BACTERIA AND
THEIR TOXINS
REFERENCE TO RELATED APPLICATIONS
[01] This application claims the benefit of U.S. Provisional Application No.
61/144,507 filed January 14, 2009 the contents of which is incorporated by
reference in its
entirety.
FIELD OF THE INVENTION
[02] The present invention relates to compositions and methods for the
treatment
and prevention of disease caused by Bacillus anthracis (anthrax) or a
bacterium which
produces toxins or toxin components homologous to those produced by B.
anthracis, or
disease caused by the toxins or toxin components themselves, using a
combination of at least
two neutralizing monoclonal antibodies.
BACKGROUND OF THE INVENTION
[03] Bacillus anthracis, the etiologic agent of anthrax, is a gram-positive,
rod
shaped, aerobic and/or facultative anaerobic, spore-forming bacterium that can
cause human
disease via the gastrointestinal, cutaneous, or inhalation routes. The
incubation period
usually varies from 12 hours to 5 days depending upon the dose received. The
onset can be
longer following inhalation exposure and some reports suggest a delayed onset
of several
weeks in low dose exposure or following removal of therapeutic intervention.
With an
anthrax inhalation, the initial clinical signs and symptoms are nonspecific
and may include
malaise, headache, fever, nausea, and vomiting. These are followed by a sudden
onset of
respiratory distress with dyspnea, stridor, cyanosis and chest pain. The onset
of respiratory
distress is followed by shock and death with high mortality.
[04] Anthrax is considered a serious biological terrorist and military threat
due to
the highly lethal effects when exposure is by inhalation (approaching 100
percent lethality)
and the stability of the B. anthracis spore. The virulence of B. anthracis is
based on two
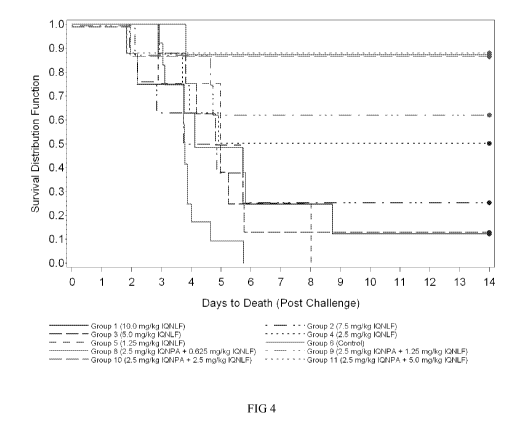
virulence factors: encapsulation (prevention of phagocytosis) and the
production of two
interlinked toxins, lethal toxin and edema toxin. Three exoprotein components,
protective
antigen (PA), lethal factor (LF), and edema factor (EF), interact to form the
two toxins. PA
-1-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
combines with lethal factor to produce lethal toxin and with edema factor to
produce edema
toxin. PA binds to host cells and is cleaved, exposing binding sites for which
lethal factor
and edema factor compete. The current consensus is that the cleaved PA forms a
channel
into the cell, allowing lethal toxin (PA-LF) or edema toxin (PA-EF) to enter.
[05] The PA monomer consists of four functional domains: domain 1 (residues 1-
258), domain 2 (residues 259-487), domain 3 (residues 488-595), and domain 4
(residues
596-735). Domain 1, the amino terminal domain, contains a furin protease
cleavage site.
Cleavage of Domain 1 releases a 20 kilodalton fragment (PA20) which triggers
heptamerization of the remainder of the protein at the cell surface. Domain 2
assists in
heptamerization and, along with domain III, forms a heptameric pore on the
cell surface that
allows binding of LF or EF, enabling endocytosis of the toxin complex into the
cell. Domain
4 contains the host cell receptor binding site.
[06] It is generally believed that lethal toxin is responsible for the
majority of the
tissue damage and systemic shock that occurs as the infection progresses, but
the mechanism
is not clearly understood. Internalization and translocation of the lethal
factor into the cytosol
occurs when the PA protein binds to it cell surface receptor. The highly
specific LF enzyme
has four domains (1-4). Domain III has a hydrophobic core (282-382) and
contains a five-
tandem repeat 101 amino acid sequence. Assembly and cellular internalization
of lethal toxin
results in increased permeability to sodium and potassium ions followed by ATP
hydrolysis
which inhibits macromolecular synthesis and leads to cell death.
[07] As disease progresses, lethal toxin will eventually accumulate to a level
at
which antibiotics are no longer effective, even though the bacteria is
sensitive to the
antibiotic. This means that antibiotics treatment must be started during the
very early stages
of infection in order to potentially be successful. Since a biological attack
is likely to occur
without warning, such early treatment will often be impossible. It is
therefore important to
develop methods that neutralize the effects of lethal toxin. One approach is
vaccination with
toxin components. This approach has the disadvantage of being effective only
for those well
advanced in a vaccination program (vaccination currently takes approximately
12 months to
become effective) and for those with a highly competent immune system. Thus,
vaccination
is ineffective as a post-exposure means of treatment. Other therapeutic
strategies are needed
to neutralize the devastating effects of lethal toxin during the post-exposure
treatment
window.
[08] Bacteria other than B. anthracis may contain B. anthracis virulence
genes. In
other words, other bacteria may contain genes that produce proteins homologous
to those of
-2-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
B. anthracis for encapsulation and the production of toxins, such as PA, LF,
and EF. An
example is the PA protein of Bacillus cereus G9241 and the homologous proteins
of B.
thuringiensis and C. perfringens (see Hoffmaster et al., Proc. Natl. Acad.
Sci. U. S. A. (2004)
101:8449-8454; Hoffmaster et al., J. Clin. Microbiol. (2006) 44:3352-3360; and
Petosa et al.,
Nature (1997) 385:833-838).
[09] The currently recommended post-exposure treatment for anthrax is a
combination of antibiotics (ciprofloxacin or doxycycline), licensed human
vaccine (AVA),
and, in severe cases, intravenously administered preformed human polyclonal
anthrax
immunoglobulin (AIGIV) derived from immunized donors. AIGIV has a number of
advantages. It provides instant protection, is likely to be effective during
mid- to advanced-
stage disease, is equally effective against antibiotic-resistant strains,
results in minimal
adverse reactions, has a prolonged serum half-life, and targets multiple
epitopes, making it
difficult to subvert its efficacy. However, despite these advantages, AIGIV
suffers from
several serious drawbacks that prevent its usefulness on a large scale. First,
AIGIV therapy
requires the maintenance of stocks of antibodies having high toxin
neutralization activity.
These stocks must be obtained from an immunologically diverse population of
donors, and
must be constantly renewed.
[10] The present invention provides an alternative approach which utilizes a
combination of antibodies with neutralizing activity against both protective
antigen and lethal
factor for the prevention and treatment of disease caused by Bacillus
anthracis or a bacterium
which produces toxins or toxin components homologous to those produced by B.
anthracis,
or disease caused by the toxins or toxin components themselves.
SUMMARY OF THE INVENTION
[11] The present invention provides methods and compositions for the treatment
and prevention of disease caused by bacterial infection, particularly
infection by B. anthracis
or a bacterium which produces toxins or toxin components homologous to those
produced by
B. anthracis, or disease caused by the toxins or toxin components themselves.
The methods
and compositions of the invention comprise a combination of at least two
neutralizing
antibodies, preferably monoclonal antibodies, most preferably human monoclonal
antibodies,
each of which binds to a different bacterial antigen. Preferably, the antigens
are selected
from the protective antigen (PA), lethal factor (LF), and edema factor (EF) of
B. anthracis, or
a homolog of any of the foregoing.
-3-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
[12] The methods and compositions of the invention offer enhanced protection
against infection when administered prophylactically and provide an increased
probability of
survival when administered therapeutically. The approach of combining at least
two
antibodies having different antigen specificities provides broader protection
than a single
antibody or single antigen approach. The methods and compositions of the
invention are
more likely than single antibody approaches to be effective against B.
anthracis, including
variations in bacterial strains and escape mutants, as well as against other
bacteria which
produce toxins or toxin components homologous to those produced by B.
anthracis. In
addition, the methods and compositions of the invention advantageously extend
the treatment
window for subjects exposed to B. anthracis, or to bacteria which produce
toxins or toxin
components homologous to those produced by B. anthracis, or to the toxins or
toxin
components themselves, in the absence of bacteria, thereby improving the
probability of
survival. The compositions and methods of the invention also provide
significant cost
reductions and reduced health risks compared to mass vaccination strategies
because the
present invention targets treatment to those who have been exposed or are
likely to be
exposed to B. anthracis toxins, toxin components, or homologs thereof.
[13] The invention provides a method for the treatment of disease caused by B.
anthracis toxins, toxin components, or homologs thereof, in a subject in need
of such
treatment comprising administering to the subject at least two neutralizing
monoclonal
antibodies, or antigen binding fragments thereof, wherein each of the
antibodies has affinity
for a different bacterial antigen selected from the protective antigen (PA),
lethal factor (LF),
and edema factor (EF) of B. anthracis, or a homolog of any of the foregoing.
In a preferred
embodiment, one of the at least two antibodies has affinity for an epitope of
PA within
domain 4 of PA, most preferably within amino acid residues 679-693 of domain
4. In
another preferred embodiment, one of the at least two antibodies has affinity
for an epitope of
LF within domain 1 of LF.
[14] In one embodiment, the disease is caused by a bacteria. In a specific
embodiment, the disease is caused by a bacteria selected from the group
consisting of B.
anthracis, B. cereus, B. thuringiensis, and C. perfringens. In one embodiment,
the disease is
caused by a bacterium, or a combination of different bacteria which produce
one or more
proteins homologous to one or more of the PA, LF, and EF proteins of B.
anthracis. In
another embodiment, the disease results from toxemia caused by one or more
bacterial toxins
comprising one or more of PA, LF and EF, or a homolog of any of the foregoing.
In
accordance with this embodiment, toxemia may occur in the presence or absence
of bacteria.
-4-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
[15] In one embodiment, the antibodies are human monoclonal antibodies. In
another embodiment, the antibodies are humanized monoclonal antibodies.
[16] In one embodiment, the affinity (Ka) of each antibody for its antigen is
from
107 M_1 to 1010 M-1. Preferably, the affinity (Ka) of each antibody for its
antigen is from 109
M-1 to 1010 M-1
[17] In one embodiment, one of the at least two antibodies is an anti-PA
antibody
which neutralizes the protective antigen protein of B. anthracis, or a homolog
thereof. In one
embodiment, the anti-PA antibody competitively inhibits the binding of the
protective antigen
protein of B. anthracis, or a homolog thereof, to the monoclonal antibody
IQNPA. In another
embodiment, the anti-PA antibody competitively inhibits the binding of a
polypeptide
comprising SEQ ID NO:17 or 18 to the monoclonal antibody IQNPA.
[18] In one embodiment, the anti-PA antibody comprises a variable heavy chain
domain (VH) having three complementarity determining regions (CDR), each CDR
comprising the following amino acid sequence: VH CDR1 : KKPGA (SEQ ID NO:5);
VH
CDR2: SNAIQWVRQAPGQRLEW (SEQ ID NO:6); and VH CDR3: YMELSSLR (SEQ
ID NO:7). In another embodiment, the anti-PA antibody comprises a variable
light chain
domain (VL) having three CDRs, each CDR comprising the following amino acid
sequence:
VL CDR1 : LTQSPGTLSLS (SEQ ID NO:8); VL CDR2: SYSSLAW (SEQ ID NO:9); and
VL CDR3: GPDFTLTIS (SEQ ID NO:10). In a particular embodiment, the anti-PA
antibody comprises six CDRs, each comprising the following amino acid
sequence: VH
CDR1: SEQ ID NO:5, VH CDR2: SEQ ID NO:6, VH CDR3: SEQ ID NO:7, VL CDR1:
SEQ ID NO:8, VL CDR2: SEQ ID NO:9, and VL CDR3: SEQ ID NO:10.
[19] In a specific embodiment, the anti-PA antibody is the human monoclonal
antibody IQNPA.
[20] In one embodiment, one of the at least two antibodies is an anti-LF
antibody
which neutralizes the lethal factor protein of B. anthracis, or a homolog
thereof. In one
embodiment, the anti-LF antibody competitively inhibits the binding of the
lethal factor
protein of B. anthracis, or a homolog thereof, to the monoclonal antibody
IQNLF. In another
embodiment, the anti-LF antibody competitively inhibits the binding of a
polypeptide
comprising SEQ ID NO:19 to the monoclonal antibody IQNLF.
[21] In one embodiment, the anti-LF antibody comprises a variable heavy chain
domain (VH) having three complementarity determining regions (CDR), each CDR
comprising the following amino acid sequence: VH CDR1 : VQPGG (SEQ ID NO: 11),
VH
CDR2: SYAMSWVRQAPGKGLEW (SEQ ID NO:12), and VH CDR3: YMQMNSL (SEQ
-5-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
ID NO:13). In another embodiment, the anti-LF antibody comprises a variable
light chain
domain (VL) having three CDRs, each CDR comprising the following amino acid
sequence:
VL CDR1 : TQSPDFQSVSP (SEQ ID NO:14), VL CDR2: SSLHWYQ (SEQ ID NO:15),
and VL CDR3: DFTLTINSL (SEQ ID NO: 16). In a particular embodiment, the
antibody
comprises six CDRs, each comprising the following amino acid sequence: VH CDR1
: SEQ
ID NO: 11, VH CDR2: SEQ ID NO:12, VH CDR3: SEQ ID NO:13, VL CDR1: SEQ ID
NO:14, VL CDR2: SEQ ID NO:15, and VL CDR3: SEQ ID NO:16.
[22] In a specific embodiment, the anti-LF antibody is the human monoclonal
antibody IQNLF.
[23] In one embodiment, one of the at least two neutralizing monoclonal
antibodies
is an anti-PA antibody which neutralizes the protective antigen protein of B.
anthracis, or a
homolog thereof, and the other antibody is an anti-LF antibody which
neutralizes the lethal
factor protein of B. anthracis, or a homolog thereof. In a specific
embodiment, the antibodies
are the human monoclonal antibodies, IQNPA and IQNLF.
[24] In one embodiment, each antibody is administered at a dose of from 1 to
20
mg/kg body weight of the subject. In another embodiment, one antibody is
administered at a
dose of from 1 to 10 mg/kg body weight of the subject. In another embodiment,
one antibody
is administered at a dose of from 2.5 to 15 mg/kg body weight of the subject.
In one
embodiment, the doses of the at least two antibodies are administered
separately. In another
embodiment, the doses of the at least two antibodies are administered at
substantially the
same time. In certain embodiments, each dose is in a separate composition. In
other
embodiments, the doses are contained in the same composition.
[25] In one embodiment, the antibodies are administered to the subject after
the
subject's exposure to B. anthracis toxins, toxin components, or homologs
thereof. In one
embodiment, the antibodies are administered to the subject between 0 and 48
hours after the
subject's exposure to B. anthracis toxins, toxin components, or homologs
thereof. In another
embodiment, the antibodies are administered to the subject 48 hours after the
subject's
exposure to B. anthracis toxins, toxin components, or homologs thereof. Such
exposure may
be in the form of exposure to B. anthracis or to a bacterium that produces
toxins or toxin
components homologous to those produced by B. anthracis. In an alternative
embodiment,
such exposure is in the form of exposure to the B. anthracis toxins or toxin
components
themselves, or homologs thereof, in the absence of bacteria.
-6-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
[26] In one embodiment, the method further comprises administering to the
subject
an antibacterial agent. In a specific embodiment, the antibacterial agent is
levofloxacin,
ciprofloxacin, or doxycycline.
[27] The invention also provides a method for the prevention of disease caused
by
B. anthracis toxins, toxin components, or homologs thereof, in a subject in
need of such
prevention comprising administering to the subject at least two neutralizing
monoclonal
antibodies, or antigen binding fragments thereof, wherein each of the
antibodies has affinity
for a different bacterial antigen selected from the protective antigen (PA),
lethal factor (LF),
and edema factor (EF) of B. anthracis, or a homolog of any of the foregoing,
and wherein the
antibodies are administered prior to the subject's exposure to the B.
anthracis toxins, toxin
components, or homologs thereof.
[28] In one embodiment, the disease caused by a bacteria. In a specific
embodiment, the disease is caused by a bacteria selected from the group
consisting of B.
anthracis, B. cereus, B. thuringiensis, and C. perfringens. In other
embodiments, the disease
is caused by a bacterium, or a combination of different bacteria, which
produce factors
homologous to one or more of the PA, LF, and EF proteins of B. anthracis. In
another
embodiment, the disease results from toxemia caused by one or more bacterial
toxins
comprising one or more of PA, LF and EF, or a homolog of any of the foregoing.
In
accordance with this embodiment, toxemia may occur in the presence or absence
of bacteria.
[29] In one embodiment, the antibodies are human monoclonal antibodies. In
another embodiment, the antibodies are humanized monoclonal antibodies.
[30] In one embodiment, the affinity (Ka) of each antibody for its antigen is
from
107 M_1 to 1010 M-1. Preferably, the affinity (Ka) of each antibody for its
antigen is from 109
M-1 to 1010 M-1
[31] In one embodiment, one of the at least two antibodies is an anti-PA
antibody
which neutralizes the protective antigen protein of B. anthracis, or a homolog
thereof. In
another embodiment, the anti-PA antibody competitively inhibits the binding of
the
protective antigen protein of B. anthracis, or a homolog thereof, to the
monoclonal antibody
IQNPA. In another embodiment, the anti-PA antibody competitively inhibits the
binding of a
polypeptide comprising SEQ ID NO: 17 or 18 to the monoclonal antibody IQNPA.
[32] In one embodiment, the anti-PA antibody comprises a variable heavy chain
domain (VH) having three complementarity determining regions (CDR), each CDR
comprising the following amino acid sequence: VH CDR1 : KKPGA (SEQ ID NO:5);
VH
CDR2: SNAIQWVRQAPGQRLEW (SEQ ID NO:6); and VH CDR3: YMELSSLR (SEQ
-7-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
ID NO:7). In another embodiment, the anti-PA antibody comprises a variable
light chain
domain (VL) having three CDRs, each CDR comprising the following amino acid
sequence:
VL CDR1 : LTQSPGTLSLS (SEQ ID NO:8); VL CDR2: SYSSLAW (SEQ ID NO:9); and
VL CDR3: GPDFTLTIS (SEQ ID NO:10). In a particular embodiment, the anti-PA
antibody comprises six CDRs, each comprising the following amino acid
sequence: VH
CDR1: SEQ ID NO:5, VH CDR2: SEQ ID NO:6, VH CDR3: SEQ ID NO:7, VL CDR1:
SEQ ID NO:8, VL CDR2: SEQ ID NO:9, and VL CDR3: SEQ ID NO:10.
[33] In a specific embodiment, the anti-PA antibody is the human monoclonal
antibody IQNPA.
[34] In one embodiment, one of the at least two antibodies is an anti-LF
antibody
which neutralizes the lethal factor protein of B. anthracis, or a homolog
thereof. In another
embodiment, the anti-LF antibody competitively inhibits the binding of the
lethal factor
protein of B. anthracis, or a homolog thereof, to the monoclonal antibody
IQNLF. In another
embodiment, the anti-LF antibody competitively inhibits the binding of a
polypeptide
comprising SEQ ID NO:19 to the monoclonal antibody IQNLF.
[35] In one embodiment, the anti-LF antibody comprises a variable heavy chain
domain (VH) having three complementarity determining regions (CDR), each CDR
comprising the following amino acid sequence: VH CDR1 : VQPGG (SEQ ID NO: 11),
VH
CDR2: SYAMSWVRQAPGKGLEW (SEQ ID NO:12), and VH CDR3: YMQMNSL (SEQ
ID NO:13). In another embodiment, the anti-LF antibody comprises a variable
light chain
domain (VL) having three CDRs, each CDR comprising the following amino acid
sequence:
VL CDR1 : TQSPDFQSVSP (SEQ ID NO:14), VL CDR2: SSLHWYQ (SEQ ID NO:15),
and VL CDR3: DFTLTINSL (SEQ ID NO: 16). In a particular embodiment, the
antibody
comprises six CDRs, each comprising the following amino acid sequence: VH CDR1
: SEQ
ID NO: 11, VH CDR2: SEQ ID NO:12, VH CDR3: SEQ ID NO:13, VL CDR1: SEQ ID
NO:14, VL CDR2: SEQ ID NO:15, and VL CDR3: SEQ ID NO:16.
[36] In a specific embodiment, the anti-LF antibody is the human monoclonal
antibody IQNLF.
[37] In one embodiment, one of the at least two monoclonal antibodies is an
anti-
PA antibody which neutralizes the protective antigen protein of B. anthracis,
or a homolog
thereof, and the other antibody is an anti-LF antibody which neutralizes the
lethal factor
protein of B. anthracis, or a homolog thereof. In a specific embodiment, the
antibodies are
the human monoclonal antibodies, IQNPA and IQNLF.
-8-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
[38] In one embodiment, each antibody is administered at a dose of from 1 to
20
mg/kg body weight of the subject. In another embodiment, one antibody is
administered at a
dose of from 1 to 10 mg/kg body weight of the subject. In another embodiment,
one antibody
is administered at a dose of from 2.5 to 15 mg/kg body weight of the subject.
In one
embodiment, the doses of the at least two antibodies are administered
separately. In another
embodiment, the doses of the at least two antibodies are administered at
substantially the
same time. In certain embodiments, each dose is in a separate composition. In
other
embodiments, the doses are contained in the same composition.
[39] The invention also provides a pharmaceutical composition comprising at
least
two neutralizing monoclonal antibodies, or antigen binding fragments thereof,
wherein each
of the antibodies has affinity for a different bacterial antigen selected from
the protective
antigen (PA), lethal factor (LF), and edema factor (EF) of B. anthracis, or a
homolog of any
of the foregoing, and a pharmaceutically acceptable excipient or carrier.
[40] In one embodiment, the composition comprises an anti-PA antibody which
neutralizes the protective antigen protein of B. anthracis, or a homolog
thereof. In one
embodiment, the anti-PA antibody competitively inhibits the binding of the
protective antigen
protein of B. anthracis, or a homolog thereof, to the monoclonal antibody
IQNPA. In another
embodiment, the anti-PA antibody competitively inhibits the binding of a
polypeptide
comprising SEQ ID NO: 17 or 18 to the monoclonal antibody IQNPA. In one
embodiment,
the anti-PA antibody comprises a variable heavy chain domain (VH) having three
complementarity determining regions (CDR), each CDR comprising the following
amino
acid sequence: VH CDR1: KKPGA (SEQ ID NO:5); VH CDR2:
SNAIQWVRQAPGQRLEW (SEQ ID NO:6); and VH CDR3: YMELSSLR (SEQ ID NO:7).
In another embodiment, the anti-PA antibody comprises a variable light chain
domain (VL)
having three CDRs, each CDR comprising the following amino acid sequence: VL
CDR1 :
LTQSPGTLSLS (SEQ ID NO:8); VL CDR2: SYSSLAW (SEQ ID NO:9); and VL CDR3:
GPDFTLTIS (SEQ ID NO:10). In a particular embodiment, the anti-PA antibody
comprises
six CDRs, each comprising the following amino acid sequence: VH CDR1 : SEQ ID
NO:5,
VH CDR2: SEQ ID NO:6, VH CDR3: SEQ ID NO:7, VL CDR1: SEQ ID NO:8, VL CDR2:
SEQ ID NO:9, and VL CDR3: SEQ ID NO:10. Ina specific embodiment, the anti-PA
antibody is the human monoclonal antibody IQNPA.
[41] In another embodiment, the composition comprises an anti-LF antibody
which
neutralizes the lethal factor protein of B. anthracis, or a homolog thereof.
In one
embodiment, the anti-LF antibody competitively inhibits the binding of the
lethal factor
-9-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
protein of B. anthracis, or a homolog thereof, to the monoclonal antibody
IQNLF. In another
embodiment, the anti-LF antibody competitively inhibits the binding of a
polypeptide
comprising SEQ ID NO:19 to the monoclonal antibody IQNLF. In one embodiment,
the
anti-LF antibody comprises a variable heavy chain domain (VH) having three
complementarity determining regions (CDR), each CDR comprising the following
amino
acid sequence: VH CDR1: VQPGG (SEQ ID NO: 11), VH CDR2:
SYAMSWVRQAPGKGLEW (SEQ ID NO:12), and VH CDR3: YMQMNSL (SEQ ID
NO: 13). In another embodiment, the anti-LF antibody comprises a variable
light chain
domain (VL) having three CDRs, each CDR comprising the following amino acid
sequence:
VL CDR1 : TQSPDFQSVSP (SEQ ID NO:14), VL CDR2: SSLHWYQ (SEQ ID NO:15),
and VL CDR3: DFTLTINSL (SEQ ID NO: 16). In a particular embodiment, the
antibody
comprises six CDRs, each comprising the following amino acid sequence: VH CDR1
: SEQ
ID NO: 11, VH CDR2: SEQ ID NO:12, VH CDR3: SEQ ID NO:13, VL CDR1: SEQ ID
NO:14, VL CDR2: SEQ ID NO:15, and VL CDR3: SEQ ID NO:16.
[42] In a specific embodiment, the composition comprises the monoclonal IQNPA
antibody and the monoclonal IQNLF antibody.
[43] In one embodiment, one of the at least two neutralizing monoclonal
antibodies
in the composition is an anti-PA antibody which neutralizes the protective
antigen protein of
B. anthracis, or a homolog thereof, and the other antibody is an anti-LF
antibody which
neutralizes the lethal factor protein of B. anthracis, or a homolog thereof.
In a specific
embodiment, the antibodies are the human monoclonal antibodies, IQNPA and
IQNLF.
[44] In one embodiment, the composition further comprises at least one
antibacterial agent. Preferably, the at least one antibacterial agent is
selected from
ciprofloxacin, doxycycline, or levofloxacin.
BRIEF DESCRIPTION OF THE FIGURES
[45] Figure 1: Kaplan-Meier curves representing time-to-death and survival
data
for each group of animals in Example 1.4 (Pre-Exposure Efficacy).
[46] Fi ure 2: Pharmacokinetic profiles of IQNPA and IQNLF antibodies as
determined in diluted rabbit serum. Concentrations were back-calculated from
the ELISA
values using a 4 parameter fit method and then expressed as ng/mL in 100%
rabbit serum.
[47] Figure 3: Kaplan-Meier curves representing time-to-death and survival
data
for each group of animals in Example 1.5 (Post-Exposure Efficacy - Experiment
1). IQNPA:
-10-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
red, Groups 1-3; IQNLF: blue, Groups 4-6; IQNPA+IQNLF: green, Groups 8-12;
control:
gray, Group 7.
[48] Fib: Kaplan-Meier curves representing time-to-death and survival data
for each group of animals in Example 1.6 (Post-Exposure Efficacy - Experiment
2).
[49] Fib: Estimated logistic regression curves for each treatment (IQNLF and
IQNPA+IQNLF) in Example 1.6. Points show the proportion of animals that
survived for
each group.
[50] Fib: Estimated logistic regression curves for the IQNLF and combined
treatments in Example 1.6. Points show the proportion of animals that survived
for each
group.
[51] Fib: Estimated logistic regression curves for each treatment (IQNPA,
IQNLF, IQNPA+IQNLF) in Example 1.7 (Post-Exposure Efficacy - Experiment 3).
[52] Fib: Kaplan-Meier curves representing time-to-death and survival data
for each group in Example 1.7.
[53] Fib: Estimated logistic regression curves for each treatment (IQNLF or
IQNPA+IQNLF) in Example 1.8. Points show the proportion of animals that
survived for
each dose group and treatment involving IQNLF.
[54] Fi_u: Estimated logistic regression curves for each treatment (IQNPA or
IQNPA+IQNLF) in Example 1.8. Points show the proportion of animals that
survived for
each dose group and treatment involving IQNPA.
DETAILED DESCRIPTION OF THE INVENTION
[55] The methods and compositions of the invention offer enhanced protection
against bacterial infection or toxemia (which may occur in the presence or
absence of a
bacterial infection) caused by B. anthracis or a bacterium which produces
toxins or toxin
components homologous to those produced by B. anthracis, or disease caused by
the toxins
or toxin components themselves, when administered before exposure and provide
an
increased probability of survival when administered following exposure to the
bacteria,
bacterial toxins, or their component proteins. The invention combines at least
two
neutralizing monoclonal antibodies, each having a different antigen
specificity. Preferably,
each of the at least two antibodies has affinity for a different bacterial
antigen selected from
the protective antigen (PA), lethal factor (LF), and edema factor (EF) of B.
anthracis, or a
homolog of any of the foregoing.
-11-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
[56] In one embodiment, at least one of the antibodies binds to an epitope of
the PA
protein of B. anthracis, or a homolog thereof, that includes one or more amino
acids within
one of the following groups of amino acids (with reference to Genebank
Accession No.
P13423): Group 1 (amino acids 121-150); Group 2 (amino acids 143-158); Group 3
(amino
acids 421-440); Group 4 (amino acids 339-359) and Group 5 (amino acids 678-
697). In a
preferred embodiment, at least one of the antibodies binds to an epitope of PA
that includes
one or more amino acids within at least one of the following groups of amino
acids (with
reference to Genebank Accession No. P13423): Group 6 (Phe-342, Phe-343, Asp-
344);
Group 7 (Trp-375, Met-379, and Leu-381); Group 8 (Phe-581, Phe-583, Ile-591,
Leu-595,
and Ile-603); Group 9 (Pro-213, Leu-216, Phe-231, Leu-232, Pro-234, Ile-236,
Ile-239, Trp-
255, and Phe-265) and Group 10 (Asn-686 and any residue from Lys-708 to Asn-
722). In
another embodiment, at least one of the antibodies binds to an epitope of LF
that includes one
or more amino acids within any of domains 1 to 4 of the LF protein of B.
anthracis, or a
homolog thereof. Epitopes may comprise or consist of one or more linear
polypeptide
fragments of a protein.
[57] As used herein, "neutralizes" or "neutralizing" in the context of
antibodies
against a bacterium, or against a bacterial toxin or its component, means that
the antibody
inhibits the ability of the bacterium or the toxin to cause disease. The
neutralizing activity of
an antibody derives from its ability to bind to a bacterial antigen,
particularly a bacterial
protein necessary for virulence. In the context of toxins and their
components, the antibodies
may neutralize, for example, by preventing or reversing the assembly of toxin
components to
form a functional toxin, or by disabling the toxin or toxin component from
exerting its
biological activity. For example, in the case of the PA toxin, an antibody may
inhibit
cleavage of the PA monomer, or it may inhibit the formation of the PA
heptamer, or the
antibody may block the binding of LF or EF to the PA heptamer. The
neutralizing activity of
an antibody can be measured, for example, as the ability of the antibody to
block entry of the
bacteria into cells, to block replication of the bacteria within cells, to
enhance the uptake
and/or intracellular killing of the bacteria by cells of the immune system,
such as
macrophages, as well as the ability of the antibody to prevent or ameliorate
the clinical
symptoms of disease caused by bacterial infection and/or toxemia in a mammal.
The
neutralizing activity of an antibody against a bacterial toxin can also be
measured more
directly, for example, using a toxin neutralization assay. Such assays are
known in the art
and are described, for example in Albrecht et at., Infect. Immunity, (2007)
75:5425-5433 and
Li et at., J. Immunol. Methods, (2008) 333:89-106.
-12-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
[58] As used herein, the term "homolog" refers to a protein having an amino
acid
sequence which differs from the sequence of the corresponding B. anthracis
protein, PA, LF,
or EF, but in which the differences are such that the protein retains the
function and/or
antigenic character of the corresponding B. anthracis protein. Thus, a homolog
of PA, LF, or
EF may be produced by a bacteria other than B. anthracis. Homology is
typically determined
on the basis of sequence similarity or sequence identity. In certain
embodiments, a
homologous protein is one which shares at least 70%, at least 80%, at least
90%, or at least
95% sequence identity over its entire length to a B. anthracis protein
selected from PA, LF,
and EF. Most preferably, the homolog is at least 98% identical over its entire
length to the
corresponding B. anthracis protein. In other embodiments, the homologous
protein shares
high sequence identity to a B. anthracis protein selected from PA, LF, and EF,
over one or
more regions smaller than its entire length. Preferably, these regions
correspond to one or
more functional domains. Thus, in one embodiment, a PA homolog shares at least
70%, at
least 80%, at least 90%, at least 95%, or at least 98% sequence identity to
the PA protein of
B. anthracis in one or more functional domains selected from the group
consisting of domain
1 (residues 1-258), domain 2 (residues 259-487), domain 3 (residues 488-595),
and domain 4
(residues 596-735), with reference to the amino acid sequence of the PA
protein of B.
anthracis given in GENBANK ACCESSION NO: P 13423. In another embodiment, an LF
homolog shares at least 70%, at least 80%, at least 90%, at least 95%, or at
least 98%
sequence identity to the LF protein of B. anthracis in one or more functional
domains
selected from the group consisting of domain 1, 2, 3, and 4, with reference to
the amino acid
sequence of the LF protein of B. anthracis given in GENBANK ACCESSION NO:
YPO 1 6503.
[59] Also encompassed are derivatives and analogs of the B. anthracis proteins
PA,
LF, and EF, and their homologs. Such derivatives and analogs may be full
length or other
than full length, if the derivative or analog contains a modified amino acid.
Derivatives or
analogs include, e.g., molecules including regions that are substantially
homologous to the
PA, LF, or EF proteins, in various embodiments, by at least about 70%, 80%, or
95%, 98%,
or even 99% identity over an amino acid sequence of identical size or when
compared to an
aligned sequence in which the alignment is done using sequence analysis
software, such as,
for example, the Sequence Analysis Software Package of the Genetics Computer
Group,
University of Wisconsin Biotechnology Center, 1710 University Avenue, Madison,
Wis.
53705, with the default parameters therein.
-13-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
[60] In the case of polypeptide sequences which are less than 100% identical
to a
reference B. anthracis sequence, the non-identical positions are preferably,
but not
necessarily, conservative substitutions of the corresponding residue(s) in the
reference
sequence. Conservative substitutions typically include substitutions within
the following
groups: glycine and alanine; valine, isoleucine, and leucine; aspartic acid
and glutamic acid;
asparagine and glutamine; serine and threonine; lysine and arginine; and
phenylalanine and
tyrosine. Conservative amino acid changes also refer to changes between amino
acids of
broadly similar molecular properties, e.g, substitutions within the aliphatic
group alanine,
valine, leucine and isoleucine. A substitution of glycine for an aliphatic
amino acid is also a
conservative substitution. Other conservative substitutions include those
within the sulfur-
containing group methionine and cysteine. Preferred conservative substitution
groups are
aspartate-glutamate; asparagine-glutamine; valine-leucine-isoleucine; alanine-
valine;
phenylalanine-tyrosine; and lysine-arginine. Preferably, a substitution other
than a
conservative amino acid substitution is made outside of a functional domain of
the reference
protein, e.g., outside of domains 1-4 of either PA or LF.
[61] Where a particular polypeptide is said to have a specific percent
identity to a
reference polypeptide of a defined length, the percent identity is relative to
the reference
peptide. Thus, a peptide that is 50% identical to a reference polypeptide that
is 100 amino
acids long can be a 50 amino acid polypeptide that is completely identical to
a 50 amino acid
long portion of the reference polypeptide. It might also be a 100 amino acid
long polypeptide,
which is 50% identical to the reference polypeptide over its entire length. Of
course, other
polypeptides will meet the same criteria.
[62] The skilled artisan will appreciate that bacterial strains other than B.
anthracis
may contain B. anthracis virulence genes. In other words, other bacterial
strains may contain
genes that produce virulence proteins which are the same or homologous to
those proteins of
B. anthracis which are responsible for virulence. For example, other bacterial
strains may
produce proteins identical or homologous to the PA, LF, or EF proteins
produced by B.
anthracis. Accordingly, the antibodies for use in the methods and compositions
of the
invention include antibodies that neutralize bacteria other than B. anthracis.
The dual
antibody approach of the present invention can thus be used in the prophylaxis
and treatment
of disease caused by such other bacteria, including, but not limited to, B.
thuringiensis, C.
perfringens, and B. cereus, as well as for the prophylaxis and treatment of
disease resulting
from toxemia caused by exposure to bacterial toxins or toxin components that
are identical or
homologous to the PA, LF, and/or EF proteins of B. anthracis.
-14-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
[63] Preferably, the antibodies for use in the methods and compositions of the
invention bind to at least one, and most preferably two, of the B. anthracis
toxin components,
PA, LF, and EF, or a homolog of any of the foregoing. Thus, in a preferred
embodiment, the
methods and compositions of the invention provide a combination of at least
two antibodies,
each antibody having affinity for a different antigen selected from the B.
anthracis toxin
components, PA, LF, and EF, or a homolog of any of the foregoing. Preferably,
at least one
antibody has affinity for PA, or a homolog thereof, and another antibody has
affinity for LF,
or a homolog thereof.
1.1 Antibodies
[64] The antibodies for use in the methods and compositions of the invention
are
monoclonal antibodies. The terms "antibody" and "antibodies" refer to fully
human
antibodies, humanized antibodies, camelised antibodies, chimeric antibodies,
CDR-grafted
antibodies, single-chain Fvs (scFv), disulfide-linked Fvs (sdFv), Fab
fragments, F(ab')
fragments, and antigen-binding fragments of any of the foregoing. In
particular, the
antibodies include immunoglobulin molecules and antigen-binding active
fragments of
immunoglobulin molecules, i.e., molecules that contain an antigen binding
site. Such
fragments may or may not be fused to another immunoglobulin domain including,
but not
limited to, an Fc region or fragment thereof. The skilled person will
appreciate that other
fusion products may be generated, including but not limited to, scFv-Fc
fusions, variable
region (e.g., VL and VH)-Fc fusions, and scFv-scFv-Fc fusions. Immunoglobulin
molecules
can be of any type, including, IgG, IgE, IgM, IgD, IgA and IgY, and of any
class, including
IgGi, IgG2, IgG3, IgG4, IgAi and IgA2), or of any subclass. Preferably, the
monoclonal
antibodies for use in the methods and compositions of the invention are IgG
antibodies.
[65] The antibodies for use in the methods and compositions of the invention
bind
to an antigen selected from PA, LF, or EF, or homologs thereof. Preferably, an
antibody for
use in the methods and compositions of the invention binds with high affinity
to the
protective antigen (PA) or the lethal factor protein (LF) of B. anthracis, B.
cereus, B.
thuringiensis, C. perfringens, or a homolog of any of the foregoing.
[66] Affinity is a measure of the strength of binding between an antibody and
an
antigen. Affinity can be expressed in several ways. One way is in terms of the
dissociation
constant (Kd) of the interaction. Kd can be measured by routine methods,
include equilibrium
dialysis or by directly measuring the rates of antigen-antibody dissociation
and association,
the koff and koõrates, respectively (see e.g., Nature, 1993 361:186-87). The
ratio of koff/koõ
-15-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
cancels all parameters not related to affinity, and is equal to the
dissociation constant Kd (see,
generally, Davies et at., Annual Rev Biochem, 1990 59:439-473). Thus, a
smaller Kd means a
higher affinity. Another expression of affinity is Ka, which is the inverse of
Kd, or koõ/koff.
Thus, a higher Ka means a higher affinity. A high affinity antibody for use in
the
compositions and methods of the invention is an antibody that binds to an
antigen of B.
anthracis with a Kd in the picomolar (pM, 10-12 M) or nanomolar (nM, 10-9 M)
range, or with
a Ka of at least 107 M_1 or, preferably, from 109 M-1 to 1010 M-1
[67] In one embodiment, the antibody binds with a Kd of from 1 to 100 pM, from
100 to 250 pM, from 250 to 500 pM, or from 500 to 1000 pM. In another
embodiment, the
antibody binds with a Kd from 1 to 100 nM, from 100 to 250 nM, from 250 to 500
nM, or
from 500 to 1000 nM. Preferably, the antibody binds with a Kd from 1 to 200 pM
or from 1
to 200 nM.
[68] In another embodiment, the antibody binds to the antigen with an affinity
constant (Ka) of at least 107 M-1, preferably with a Ka of from 107 M-1 to 108
M-1, from 108 M-1
to 109 M-1, from 109 M-1 to 1010 M-1, or from 1010 M-1 to 1011 M-1. In a
preferred embodiment,
at least one antibody of the combination binds to its antigen with an affinity
of from 109 M-1
to 1010 M-1
[69] The monoclonal antibodies useful in the methods and compositions of the
invention include chimeric, human, and humanized antibodies, and antigen-
binding
fragments thereof, which exhibit low toxicity when administered to a subject,
preferably a
human subject. Toxicity in the context of antibody therapy in a human subject
includes, for
example, a human anti-murine antibody response (where the antibody is murine)
and a
human anti-chimeric antibody response (where the antibody is chimeric).
Preferably, the
antibodies are monoclonal human or humanized antibodies, or antigen-binding
fragements
thereof.
[70] Antigen-binding fragments of the antibodies include, for example, Fab,
Fab',
F(ab')2 and Fv fragments. These fragments lack the heavy chain constant
fragment (Fc) of an
intact antibody and are sometimes preferred because they tend to clear more
rapidly from the
circulation and have less non-specific binding than an intact antibody. Such
fragments are
produced from intact antibodies using methods well known in the art, for
example by
proteolytic cleavage with enzymes such as papain (to produce Fab fragments) or
pepsin (to
produce F(ab')2 fragments). Preferably, an antigen-binding fragment is a dimer
of heavy
chains (a camelised antibody), a single-chain Fvs (scFv), a disulfide-linked
Fvs (sdFv), a Fab
fragment, or a F(ab') fragment.
-16-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
[71] Preferably, the antibodies for use in the methods and compositions of the
invention are monoclonal antibodies. A monoclonal antibody is derived from a
substantially
homogeneous population of antibodies specific to a particular antigen, which
population
contains substantially similar epitope binding sites. Such antibodies may be
of any
immunoglobulin class including IgG, IgM, IgE, IgA, and any subclass thereof.
Methods for
monoclonal antibody production are well known in the art. Preferably, a
monoclonal
antibody for use in the methods and compositions of the invention is produced
using
hybridoma technology.
[72] A human antibody is one in which all of the sequences arise from human
genes. Human antibodies include antibodies having the amino acid sequence of a
human
immunoglobulin and include antibodies isolated from human immunoglobulin
libraries or
from mice that express antibodies from human genes. For example, the human
heavy and
light chain immunoglobulin gene complexes may be introduced randomly or by
homologous
recombination into mouse embryonic stem cells. Alternatively, the human
variable region,
constant region, and diversity region may be introduced into mouse embryonic
stem cells in
addition to the human heavy and light chain genes. The mouse heavy and light
chain
immunoglobulin genes may be rendered non-functional separately or
simultaneously with the
introduction of human immunoglobulin loci by homologous recombination. In
particular,
homozygous deletion of the JH region prevents endogenous antibody production.
The
modified embryonic stem cells are expanded and microinjected into blastocysts
to produce
chimeric mice. The chimeric mice are then bred to produce homozygous
offspring, which
express human antibodies. The transgenic mice are immunized in the normal
fashion with a
selected antigen. Monoclonal antibodies directed against the antigen can be
obtained from
the immunized, transgenic mice using conventional hybridoma technology. The
human
immunoglobulin transgenes harbored by the transgenic mice rearrange during B
cell
differentiation, and subsequently undergo class switching and somatic
mutation. Thus, using
such a technique, it is possible to produce therapeutically useful IgG, IgA,
IgM and IgE
antibodies. For an overview of this technology for producing human antibodies,
see Lonberg
and Huszar, 1995, Int. Rev. Immunol. 13:65-93. For a detailed discussion of
this technology
for producing human antibodies and human monoclonal antibodies and protocols
for
producing such antibodies, see e.g., International Publication Nos. WO
98/24893, WO
96/34096, and WO 96/33735; and U.S. Patent Nos. 5,413,923, 5,625,126,
5,633,425,
5,569,825, 5,661,016, 5,545,806, 5,814,318, and 5,939,598. In addition,
companies such as
Abgenix, Inc. (Freemont, Calif.) and Genpharm (San Jose, Calif.) can be
engaged to provide
-17-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
human antibodies directed against a selected antigen using technology similar
to that
described above.
[73] Human antibodies can also be derived from phage display of human antibody
fragments. In phage display methods, functional antibody domains are displayed
on the
surface of phage particles, which carry the polynucleotide sequences encoding
them. In
particular, DNA sequences encoding variable heavy and variable light domains
are amplified
from animal cDNA libraries (e.g., human or murine cDNA libraries of lymphoid
tissues).
The DNA encoding the variable heavy and variable light domains are recombined
together
with an scFv linker by PCR and cloned into a phagemid vector. The vector is
electroporated
in E. coli and the E. coli is infected with helper phage. The phage used in
these methods are
typically filamentous phage including fd and M13. Phage expressing an antigen
binding
domain that binds to the antigen epitope of interest can be selected or
identified with antigen,
e.g., using labeled antigen or antigen bound or captured to a solid surface or
bead. Examples
of phage display methods include those disclosed in Brinkman et at., 1995, J.
Immunol.
Methods 182:41-50; Ames et at., 1995, J. Immunol. Methods 184:177;
Kettleborough et at.,
1994, Eur. J. Immunol. 24:952-958; Persic et at., 1997, Gene 187:9; Burton et
at., 1994, Adv.
Immunol. 57:191-280; International Application No. PCT/GB91/01134;
International
Application Publication Nos. WO 90/02809, WO 91/10737, WO 92/01047, WO
92/18619,
WO 93/1 1236, WO 95/15982, WO 95/20401, and W097/13844; and U.S. Patent Nos.
5,698,426, 5,223,409, 5,403,484, 5,580,717, 5,427,908, 5,750,753, 5,821,047,
5,571,698,
5,427,908, 5,516,637, 5,780,225, 5,658,727, 5,733,743 and 5,969,108.
Preferably, after
phage selection, the antibody coding regions from the phage are isolated and
used to generate
whole antibodies, including human antibodies as described in the above
references.
[74] A humanized antibody is an antibody which comprises a framework region
having substantially the same amino acid sequence as human receptor
immunoglobulin and a
complementarity determing region ("CDR") having substantially the same amino
acid
sequence as a non-human donor immunoglobulin. A humanized antibody comprises
substantially all of at least one, and typically two, variable domains (Fab,
Fab', F(ab')2, Fv) in
which all or substantially all of the CDR regions correspond to those of the
non-human donor
immunoglobulin (i.e., the donor antibody) and all or substantially all of the
framework
regions of the human acceptor immunoglobulin. The acceptor may comprise or
consist of a
consensus sequence of human immunoglobulins. Preferably, a humanized antibody
also
comprises at least a portion of an immunoglobulin constant region (Fc),
typically that of a
human immunoglobulin. Ordinarily, the antibody will contain a light chain and
at least the
-18-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
variable domain of a heavy chain. The antibody also may include the CH1,
hinge, CH2,
CH3, and CH4 regions of the heavy chain. The humanized antibody can be
selected from
any class of immunoglobulins, including IgM, IgG, IgD, IgA and IgE, and any
isotype,
including IgGI, IgG2, IgG3 and IgG4. The framework and CDR regions of a
humanized
antibody need not correspond precisely to the donor and acceptor sequences,
e.g., the donor
CDR or the acceptor framework may be mutagenized by substitution, insertion or
deletion of
at least one residue. Such mutations, however, will not be extensive. Usually,
at least 75%
of the humanized antibody residues will correspond to those of the acceptor
framework and
donor CDR sequences, more often 90%, and most preferably greater than 95%. A
humanized
antibody can be produced using variety of techniques known in the art,
including but not
limited to, CDR-grafting (see e.g., European Patent No. EP 239,400;
International
Publication No. WO 91/09967; and U.S. Patent Nos. 5,225,539, 5,530,101, and
5,585,089),
veneering or resurfacing (see e.g., European Patent Nos. EP 592,106 and EP
519,596; Padlan,
1991, Mol. Immunol. 28:489-498; Studnicka et at., 1994, Prot. Eng. 7:805-814;
and Roguska
et at., 1994, Proc. Natl. Acad. Sci. U.S.A. 91:969-973), chain shuffling (see
e.g., U.S. Pat.
No. 5,565,332), and techniques disclosed in, e.g., U.S. Patent. Nos.
6,407,213, 5,766,886,
International Publication No. WO 9317105, Tan et at., 2002, J. Immunol.
169:1119-25,
Caldas et at., 2000, Protein Eng. 13:353-60, Morea et at., 2000, Methods
20:267-79, Baca et
at., 1997, J. Biol. Chem. 272:10678-84, Roguska et at., 1996, Protein Eng.
9:895-904, Couto
et al., 1995, Cancer Res. 55:5973s-5977s, Couto et al., 1995, Cancer Res.
55:1717-22,
Sandhu, 1994, Gene 150:409-10, and Pedersen et at., 1994, J. Mol. Biol.
235:959-73. Often,
framework residues in the framework regions will be substituted with the
corresponding
residue from the donor antibody to alter, preferably improve, antigen binding.
These
framework substitutions are identified by methods well known in the art, e.g
by modeling of
the interactions of the CDR and framework residues to identify framework
residues important
for antigen binding and sequence comparison to identify unusual framework
residues at
particular positions. (See, e.g., Queen et at., U.S. Pat. No. 5,585,089; and
Riechmann et at.,
1988, Nature 332:323, which are incorporated herein by reference in their
entireties).
[75] A chimeric antibody comprises non-human variable region sequences and
human constant region sequences. A chimeric antibody may be monovalent,
divalent or
polyvalent. A monovalent chimeric antibody is a dimer formed by a chimeric
heavy chain
associated through disulfide bridges with a chimeric light chain. A divalent
chimeric
antibody is a tetramer formed by two heavy-light chain dimers associated
through at least one
-19-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
disulfide bridge. A polyvalent chimeric antibody can also be produced, for
example, by
employing a heavy chain constant region that aggregates (e.g., from an IgM
heavy chain).
[76] A "camelised" antibody is one having a functional antigen binding site
comprising only the heavy chain variable domains (VH), rather than the
conventional antigen
binding site which comprises both the heavy and the light chain variable
domains (VL).
Preferably, a camelised antibody comprises one or two VH domains and no VL
domains.
Preferably, a camelised antibody comprises two VH domains. Methods for making
camelised antibodies are known in the art. See, for example, Riechmann et at.,
J. Immunol.
Methods, 1999 231:25-38, and U.S. Patent Application Publication Nos. US
2004137570 and
US 2004142432.
[77] The antibodies for use in the methods and compositions of the invention
may
be produced by recombinant expression using techniques known in the art. In
one
embodiment, the nucleic acid sequences used for recombinant expression are
those described
in U.S. Patent Application Publication No. 20060258842, published November 16,
2006, and
in Albrecht et at., Infection and Immunity 2007 75:5425-5433.
[78] According to the present methods, a combination of at least two
antibodies is
administered to a subject in need of treatment or prevention of disease caused
by B.
anthracis, or a bacterium which produces toxins or toxin components homologous
to those
produced by B. anthracis, or disease caused by the toxins or toxin components
themselves.
The antibodies of the combination may bind to the same or a different
bacterial antigen,
however at least two antibodies of the combination bind to a different
bacterial antigen. In a
preferred embodiment, each of the at least two antibodies binds to a different
antigen selected
from the protective antigen (PA), lethal factor (LF), and edema factor (EF) of
B. anthracis, or
a homolog of any of the foregoing.
[79] The antibodies suitable for use in the methods and compositions of the
invention are preferably human monoclonal antibodies. Human monoclonal
antibodies
suitable for use in the claimed methods include the anti-PA and anti-LF
antiobides described,
for example, in U.S. Patent Application Publication No. 20060258842, published
November
16, 2006, and in Albrecht et at., Infection and Immunity 2007 75:5425-5433.
[80] In one embodiment, at least one antibody is an anti-PA antibody which
binds
to the protective antigen (PA) of B. anthracis, or a homolog thereof, with an
affinity (Ka) of
at least 107 M-1, preferably with a Ka of from 107 M_1 to 108 M-1, from 108 M-
1 to 109 M-1, from
109 M-1 to 1010 M-1, or from 1010 M-1 to 1011 M-1. Preferably, the antibody
binds to PA with a
Ka of from 109 M-1 to 1010 M-1, or from 1010 M-1 to 1011 M-1
-20-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
[81] In one embodiment, at least one antibody is an anti-EF antibody which
binds
to the edema factor protein (EF) of B. anthracis, or a homolog thereof, with
an affinity (Ka)
of at least 107 M-1, preferably with a Ka of from 107 M_1 to 108 M-1, from 108
M_1 to 109 M-1,
from 109 M-1 to 1010 M-1, or from 1010 M-1 to 1011 M-1. Preferably, the
antibody binds to EF
with a Ka of from 109 M-1 to 1010 M-1, or from 1010 M-1 to 1011 M-1
[82] In one embodiment, at least one antibody is an anti-LF antibody which
binds
to the lethal factor protein (LF) of B. anthracis, or a homolog thereof, with
an affinity (Ka) of
at least 107 M-1, preferably with a Ka of from 107 M-1 to 108 M-1, from 108 M-
1 to 109 M-1, from
109 M-1 to 1010 M-1, or from 1010 M-1 to 1011 M-1. Preferably, the antibody
binds to LF with a
Ka of from 109 M-1 to 1010 M-1, or from 1010 M-1 to 1011 M-1
[83] In a specific embodiment, at least two of the antibodies of the
combination are
the antibodies IQNPA and IQNLF described in U.S. Patent Application
Publication No.
20060258842, published November 16, 2006, and in Albrecht et at., Infection
and Immunity
2007 75:5425-5433. The IQNPA antibody binds to the B. anthracis protective
antigen (PA),
specifically to domain IV of the PA protein. The IQNLF antibody binds to B.
anthracis
lethal factor (LF), specifically to domain I of the LF protein. These
antibodies were produced
by collecting blood samples from healthy individuals immunized with the United
Kingdom-
licensed anthrax vaccine following annual booster immunizations. Samples
demonstrating
anthrax lethal toxin-neutralizing activity in cytotoxicity assays were
selected for hybridoma
development using a polyethylene glycol-based variant of the hybridoma
electrofusion
technology described by H. Groen and H. H. Westra (U.S. Patent Application
Serial Nos.
60/710,626 and 11/072,102). Hybridoma fusions were screened for expression of
anti-PA-
and anti-LF-specific antibodies by enzyme-linked immunosorbent assays
(ELISAs).
Hybridoma clones producing anti-PA and anti-LF monoclonal antibody IgG were
expanded
and stabilized, and the antibodies were evaluated for anthrax lethal toxin
neutralization.
Candidate anti-PA and anti-LF antibodies were isotyped using a human Ig
subclass ELISA
kit (Invitrogen, Carlsbad, CA).
[84] The IQNPA and IQNLF antibodies are produced by stable hybridoma cell
lines designated and , respectively. The hybridomas and
, were deposited on , pursuant to the requirements of the Budapest
Treaty on the International Recognition of the Deposit of Microorganisms for
the Purposes of
Patent Procedure with the American Type Culture Collection (ATCC), 10801
University
Boulevard, Manassas, Virginia 20110-2209, under ATCC Designation Nos. and
respectively. During the pendency of the subject application, access to the
deposit
-21-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
shall be afforded to the Commissioner upon request. All restrictions upon
public access to
this deposit shall be removed upon the grant of a patent on this application
and the deposits
shall be replaced if viable samples cannot be made by the depository named
hereinabove.
[85] The gamma heavy chain and kappa light chain sequences of the IQNPA and
IQNLF antibodies are provided below.
[86] >IQNPA H.gamma. amino acid sequence: (SEQ ID NO: 1)
MDWIWRILFLVAAATGAHSQVQLVQSGAEVKKPGASVKVSCKASGYTFTSNAIQW
VRQAPGQRLEWVGWINGGDGNTKYSQKFQGRVTISRDISASTAYMELSSLRSEDTA
VYYCARHRLQRGGFDPWGQGTLVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLV
KDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNH
KPSNTKVDKRVEPKSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCV
VVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNG
KEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYP
SDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHE
ALHNHYTQKSL SLSPGK
[87] >IQNPA L.kappa. amino acid sequence: (SEQ ID NO: 2)
MEAPAQLLFLLLLWLPDTTGEIVLTQSPGTLSLSPGERATLSCRASQSVSYSSLAWYQ
QKPGQAPSLLIYGASSRATGIPDRFSGSGSGPDFTLTISRLEPEDFAVYYCQHYGNSPY
TFGQGTKLEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNA
LQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFN
RGEC
[88] >IQNLF H.gamma. amino acid sequence: (SEQ ID NO: 3)
MELGLCWLFLVAILKGVQCEVQLLESGGGLVQPGGSLRLSCSGSGFMFSSYAMSWV
RQAPGKGLEWV SGISGSGGTTNYADSVKGRFTISRDNSKNTLYMQMNSLRAEDTAV
YYCAKDGVYGRLGGSDYWGQGTLVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCL
VKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVN
HKPSNTKVDKKVEPKSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTC
VVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLN
GKEYKCKV SNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTKNQV SLTCLVKGFY
PSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMH
EGLHNHY- TQK SLSLSPGK
[89] >IQNLF L.kappa. amino acid sequence: (SEQ ID NO: 4)
MLPSQLIGFLLLWVPASRGEIVLTQSPDFQSVSPKEKVTITCRASQSVGSSLHWYQQK
PDQSPKLLIKYASQSFSGVPSRFSGSGSGTDFTLTINSLETEDAATYYCHQSSSLPLTFG
-22-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
GGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQS
GNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSF- NRG
EC
[90] In one embodiment, at least one antibody of the combination is an anti-PA
antibody which neutralizes the protective antigen (PA) and comprises a heavy
chain amino
acid sequences comprising SEQ ID NO: 1. In another embodiment, the anti-PA
antibody
comprises a light chain amino acid sequence comprising SEQ ID NO: 2. In a
particular
embodiment, the anti-PA antibody comprises a heavy chain amino acid sequence
comprising
SEQ ID NO: 1 and a light chain amino acid sequence comprising SEQ ID NO: 2.
[91] In one embodiment, at least one antibody of the combination is an anti-LF
antibody which neutralizes the lethal factor protein (LF) and comprises a
heavy chain amino
acid sequence comprising SEQ ID NO: 3. In another embodiment, the anti-LF
antibody
comprises a light chain amino acid sequence comprising SEQ ID NO: 4. In a
particular
embodiment, In another embodiment, the anti-LF antibody comprises a heavy
chain amino
acid sequence comprising SEQ ID NO: 3 and a light chain amino acid sequence
comprising
SEQ ID NO: 4.
[92] In one embodiment, the anti-PA antibody comprises a variable heavy chain
domain (VH) having three complementarity determining regions (CDR), each CDR
comprising the following amino acid sequence: VH CDR1 : KKPGA (SEQ ID NO:5);
VH
CDR2: SNAIQWVRQAPGQRLEW (SEQ ID NO:6); and VH CDR3: YMELSSLR (SEQ
ID NO:7). In another embodiment, the anti-PA antibody comprises a variable
light chain
domain (VL) having three CDRs, each CDR comprising the following amino acid
sequence:
VL CDR1 : LTQSPGTLSLS (SEQ ID NO:8); VL CDR2: SYSSLAW (SEQ ID NO:9); and
VL CDR3: GPDFTLTIS (SEQ ID NO:10). In a particular embodiment, the anti-PA
antibody comprises all six of the preceding CDRs.
[93] In one embodiment, the anti-LF antibody comprises a VH domain having
three CDRs, each CDR comprising the following amino acid sequence: VH CDR1:
VQPGG
(SEQ ID NO: 11); VH CDR2: SYAMSWVRQAPGKGLEW (SEQ ID NO: 12); and VH
CDR3: YMQMNSL (SEQ ID NO:13). In another embodiment, the anti-LF antibody
comprises a VL domain having three CDRs, each CDR comprising the following
amino acid
sequence: VL CDR1: TQSPDFQSVSP (SEQ ID NO:14); VL CDR2: SSLHWYQ (SEQ ID
NO:15); and VL CDR3: DFTLTINSL (SEQ ID NO:16). In a particular embodiment, the
anti-LF antibody comprises all six of the preceding CDRs.
-23-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
[94] In one embodiment, the anti-PA antibody binds to a protective antigen
(PA)
polypeptide comprising or consisting of the following amino acid sequence:
NNIAVGADES
VVKEAHREVI NSSTEGLLLN IDKDIRKILS GYIVEIEDTE
GLKEVINDRYDMLNISSLRQ DGKTFIDFKK YNDKLPLYIS NPNYKVNVYA
VTKENTIINP SENGDTSTNG IKKILIFSKK GYEIG (SEQ ID NO:17). In another
embodiment, the anti-PA antibody binds to a protective antigen (PA)
polypeptide comprising
or consisting of the following amino acid sequence: TNIYTVLDKI KLNAKMNILI
RDKRFHYDRN NIAVGADESV VKEAHREVIN SSTEGLLLNI DKDIRKILSG
YIVEIEDTEG LKEVINDRYD MLNISSLRQD GKTFIDFKKY NDKLPLYISN
PNYKVNVYAV TKENTIINPS ENGDTSTNGI KKILIFSKKG YEIG (SEQ ID NO:18).
[95] In one embodiment, the anti-PA antibody competitively inhibits the
binding of
the monoclonal antibody IQNPA to the protective antigen protein of B.
anthracis, or a
homolog thereof. In another embodiment, the anti-PA antibody competitively
inhibits the
binding of a polypeptide comprising SEQ ID NO: 17 or 18 to the monoclonal
antibody
IQNPA.
[96] In one embodiment, the anti-LF antibody binds to a lethal factor (LF)
polypeptide comprising or consisting of the following amino acid sequence:
ERNKTQEEHLK EIMKHIVKIE VKGEEAVKKE AAEKLLEKVP SDVLEMYKAI
GGKIYIVDGD ITKHISLEAL SEDKKKIKDI YGKDALLHEH YVYAKEGYEP
VLVIQSSEDY VENTEKALNV YYEIGKILSR DILSKINQPY QKFLDVLNTI
KNASDSDGQD LLFTNQLKEH PTDFSVEFLE QNSNEVQEVF AKAFAYYIEP
QHRDVLQLYA PEAFNYMDKF NEQEINLSLE ELKDQ (SEQ ID NO: 19).
[97] In one embodiment, the anti-LF antibody competitively inhibits the
binding of
the monoclonal antibody IQNLF to the lethal factor protein of B. anthracis, or
a homolog
thereof. In another embodiment, the anti-LF antibody competitively inhibits
binding of the
monoclonal antibody IQNLF to a polypeptide comprising SEQ ID NO: 19.
[98] Methods for determining antibody specificity and affinity by competitive
inhibition are known in the art, for example, such methods can be found in
Harlow, et at.,
Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold
Spring
Harbor, N.Y., 1988; Colligan et at., eds., Current Protocols in Immunology,
Greene
Publishing Assoc. and Wiley Interscience, N.Y., (1992, 1993); and Muller,
Meth. Enzymol.
92:589 601 (1983).
[99] Preferably, the antibodies for use in the methods and compositions of the
invention are isolated or purified. An "isolated" or "purified" antibody is
substantially free
-24-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
of cellular material or other contaminating proteins from the cell or tissue
source from which
the antibody is derived, or substantially free of chemical precursors or other
chemicals when
chemically synthesized. The language "substantially free of cellular material"
includes
preparations in which the antibody is separated from cellular components of
the cells from
which it is isolated or recombinantly produced. Thus, antibody that is
substantially free of
cellular material includes preparations having less than about 30%, or about
20%, or about
10%, or about 5%, or about 1% (by dry weight) of heterologous protein (also
referred to
herein as a "contaminating protein"). When the antibody is recombinantly
produced, it is also
preferably substantially free of culture medium, e.g., culture medium
represents less than
about 20%, or about 10%, or about 5%, or about I% of the volume of the protein
preparation.
When the antibody is produced by chemical synthesis, it is preferably
substantially free of
chemical precursors or other chemicals, e.g., it is separated from chemical
precursors or other
chemicals that are involved in the synthesis of the protein. Accordingly such
preparations of
antibody have less than about 30%, or about 20%, or about 10%, or about 5%, or
about 1%
(by dry weight) of chemical precursors or compounds other than the antibody of
interest.
1.1.1 Compositions
[100] The present invention also provides compositions comprising a
combination
of at least two of the antibodies described above in Section 1.1. Preferably,
a composition
comprising the antibodies is suitable for administration to a human subject.
In one
embodiment, the composition is a pharmaceutical composition comprising at
least two
antibodies, an anti-PA antibody and an anti-LF antibody, and one or more
pharmaceutically
acceptable carriers or excipients. In one embodiment, the composition is
formulated as a
liquid. In another embodiment, the composition is lyophilized.
[101] The term excipient broadly refers to a biologically inactive substance
used in
combination with the active agents, i.e., the antibodies, of the composition.
An excipient can
be used, for example, as a solubilizing agent, a stabilizing agent, a
surfactant, a demulcent, a
viscosity agent, a diluent, an inert carrier, a preservative, a binder, a
disintegrant, a coating
agent, a flavoring agent, or a coloring agent. Preferably, at least one
excipient is chosen to
provide one or more beneficial physical properties to the composition, such as
increased
stability and/or solubility of the active agent(s). A "pharmaceutically
acceptable" excipient is
one that has been approved by a state or federal regulatory agency for use in
animals, and
preferably for use in humans, or is listed in the U.S. Pharmacopia, the
European Pharmacopia
-25-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
or another generally recognized pharmacopia for use in animals, and preferably
for use in
humans.
[102] Examples of carriers that may be used in the compositions of the present
invention include water, mixtures of water and water-miscible solvents, such
as C l - to C7-
alkanols, vegetable oils or mineral oils comprising from 0.5 to 5% non-toxic
water-soluble
polymers, natural products, such as gelatin, alginates, pectins, tragacanth,
karaya gum,
xanthan gum, carrageenin, agar and acacia, starch derivatives, such as starch
acetate and
hydroxypropyl starch, and also other synthetic products, such as polyvinyl
alcohol,
polyvinylpyrrolidone, polyvinyl methyl ether, polyethylene oxide, preferably
cross-linked
polyacrylic acid, such as neutral Carbopol, or mixtures of those polymers. The
concentration
of the carrier is, typically, from 1 to 100000 times the concentration of the
active ingredient.
[103] Further examples of excipients include certain inert proteins such as
albumins;
hydrophilic polymers such as polyvinylpyrrolidone; amino acids such as
aspartic acid (which
may alternatively be referred to as aspartate), glutamic acid (which may
alternatively be
referred to as glutamate), lysine, arginine, glycine, and histidine; fatty
acids and
phospholipids such as alkyl sulfonates and caprylate; surfactants such as
sodium dodecyl
sulphate and polysorbate; nonionic surfactants such as such as TWEEN ,
PLURONICS , or
a polyethylene glycol (PEG) designatied 200, 300, 400, or 600; a Carbowax
designated 1000,
1500, 4000, 6000, and 10000; carbohydrates such as glucose, sucrose, mannose,
maltose,
trehalose, and dextrins, including cyclodextrins; polyols such as mannitol and
sorbitol;
chelating agents such as EDTA; and salt-forming counter-ions such as sodium.
[104] In one embodiment, the pharmaceutical composition further comprises one
or
more additional therapeutic agents. In a preferred embodiment, the one or more
additional
therapeutic agents is selected from an antibiotic, preferably ciprofloxacin or
doxycycline.
1.2 Methods of Use
[105] The present invention provides methods for the prevention and treatment
of
disease caused by B. anthracis or a bacterium which produces toxins or toxin
components
homologous to those produced by B. anthracis, or disease caused by the toxins
or toxin
components themselves, in a subject in need thereof by administering at least
two
neutralizing monoclonal antibodies to the subject, each having a different
antigen specificity.
Each of the at least two antibodies has affinity for a different bacterial
antigen selected from
the protective antigen (PA), lethal factor (LF), and edema factor (EF) of B.
anthracis, or a
homolog of any of the foregoing. Preferably, at least one antibody is an anti-
protective
-26-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
antigen (PA) antibody. The combination of antibodies is administered to a
subject either
prophylactically or therapeutically. A subject in need of prophylactic
treatment is one who
has been exposed to B. anthracis or a bacterium which produces toxins or toxin
components
homologous to those produced by B. anthracis, or to the toxins or toxin
components
themselves, but has not developed any clinical signs of infection (post-
exposure prophylaxis),
or one who is likely to be exposed in the near future (pre-exposure
prophylaxis). In one
embodiment, prophylactic treatment is administered to a subject who is
asymptomatic
following exposure. In another embodiment, prophylactic treatment is
administered to a
subject prior to exposure. In the context of these embodiments, prophylactic
treatment results
in an inhibition or delay in the onset or progression of at least one clinical
symptom
associated with the bacterial infection. A subject in need of therapeutic
treatment is one who
already presents with one or more clinical symptoms of bacterial infection. In
one
embodiment, therapeutic treatment is administered to a subject following
exposure who
presents with one or more clinical signs or symptoms of the bacterial
infection or toxemia,
which may occur in the absence of bacteria.
[106] The invention also provides methods for increasing the survival odds for
a
subject who has been exposed to B. anthracis or a bacterium which produces
toxins or toxin
components homologous to the virulence factors produced by B. anthracis or to
the toxins or
toxin components themselves, in the absence of bacteria, by administering a
combination of
at least two antibodies to the subject, preferably an anti-PA antibody and an
anti-LF antibody.
In one embodiment, the antibodies are administered as part of a therapeutic
regimen that
includes antibiotics, preferably ciprofloxacin and/or doxycycline. Combination
therapy with
antibiotics is discussed in more detail below in Section 1.2.2.
1.2.1 Administration and Dosages
[107] The antibodies of the present invention can be administered to a subject
either
separately or together. Preferably, the antibodies are administered at the
same time or at
substantially the same time. The subject may be any mammal, including, for
example, a
mouse, a rat, a rabbit, a dog, a pig, a non-human primate, or a human.
Preferably, the subject
is human. In certain embodiments, one or more of the antibodies is
administered in
combination with one or more additional therapeutic agents, preferably one or
more
antibiotics, as described below in Section 1.2.2. The dosage administered will
vary
depending upon known factors such as the pharmacodynamic characteristics of
the particular
-27-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
antibodies, the mode and route of administration, and the age, health, and
weight of the
subject.
[108] Dosage preferably reflects the total amount of antibody administered to
the
subject. Exemplary doses include 1 to 20 mg of antibody per kg (mg/kg) of body
weight or
about 1 to 10 mg/kg of body weight. In a specific embodiment, the total amount
of antibody
administered is about 2.5 mg/kg, about 5 mg/kg, about 10 mg/kg, about 12
mg/kg, about 14
mg/kg, about 16 mg/kg, about 18 mg/kg, or about 20 mg/kg of the subject's body
weight.
[109] The amount of each antibody administered will usually be different, and
depends on the efficacy of the particular combination of antibodies. The
effective amount of
each antibody is determined using routine methods for determining optimal dose
and
efficacy. Typically, the amount of each antibody administered will be in the
range of 1 to 20
mg/kg, preferably 1 to 10 mg/kg, and most preferably 1 to 5 mg/kg body weight.
In one
embodiment, the antibody combination comprises the IQNPA and IQNLF antibodies,
wherein the IQNPA antibody is administered at a dosage of from 1 to 5 mg/kg
body weight
and the IQNLF antibody is administered at a dosage of from 1 to 10 mg/kg. In a
particular
embodiment, the IQNPA antibody is administered at a dosage of 2.5 mg/kg.
Preferably, the
total amount of antibody administered is from 1 to 10 mg/kg.
[110] The antibodies for use in the methods of the invention are preferably
formulated for intravenous, intramuscular, or subcutaneous administration. In
certain
embodiments, the antibodies are formulated for administration by injection
through another
route, such as intradermal or transdermal. In one embodiment, the antibodies
are formulated
for intravenous administration. However, the antibodies may be formulated for
any suitable
route of administration.
[111] An effective amount of the antibodies is the amount sufficient to reduce
the
severity of the disease caused by B. anthracis or a bacterium which produces
toxins or toxin
components homologous to those produced by B. anthracis, or disease caused by
the toxins
or toxin components themselves, the amount sufficient to prevent the incidence
or
advancement of the disease, or the amount sufficient to enhance or improve the
therapeutic
effect(s) of another therapy or therapeutic agent. Preferably, the effective
amount is the
amount sufficient to prevent mortality or to expand the treatment window for a
subject who
has been exposed to B. anthracis or a bacterium which produces toxins or toxin
components
homologous to those produced by B. anthracis or to the toxins or toxin
components
themselves, in the absence of bacteria.
-28-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
[112] The antibodies of the present invention can be administered either as
individual therapeutic agents or in combination with other therapeutic agents.
The dosage
administered will vary depending upon known factors such as the
pharmacodynamic
characteristics of the particular agent, and its mode and route of
administration; age, health,
and weight of the recipient; nature and extent of symptoms, kind of concurrent
treatment,
frequency of treatment, and the effect desired.
[113] Examples of dosing regimens that can be used in the methods of the
invention
include, but are not limited to, once daily, three times weekly
(intermittent), weekly, or every
14 days. In certain embodiments, dosing regimens include, but are not limited
to, monthly
dosing or dosing every 6-8 weeks. In a preferred embodiment of pre-exposure
treatment, the
regimen includes dosing once every 2 to 4 weeks. In a preferred embodiment of
post-
exposure treatment, a single dose is administered as soon as possible
following exposure. In
one embodiment of post-exposure treatment, the regimen further includes
another dose about
2 weeks following exposure. In another embodiment, the regimen includes dosing
once a
week for 4 to 8 weeks following exposure.
1.2.2 Combination Therapy
[114] In certain embodiments, the antibodies are administered as part of a
therapeutic regimen which includes antibacterial agents. Antibacterial agents,
including
antibiotics, that can be used in combination with the antibodies of the
invention include,
without limitation, aminoglycoside antibiotics, glycopeptides, amphenicol
antibiotics,
ansamycin antibiotics, cephalosporins, cephamycins oxazolidinones,
penicillins, quinolones,
streptogamins, tetracyclins, and analogs thereof. Preferably, the antibody
combinations of
the invention are administered as part of a therapeutic regimen that includes
ciprofloxacin
and doxycycline.
[115] In one embodiment, the regimen includes administration of an
antibacterial
agent at a dosage of from 10-50 mg/kg/day, preferably 20, 25, 30, or 35
mg/kg/day, for 30-90
days, preferably for 60-90 days.
[116] In one embodiment, the antibacterial agent is selected from the group
consisting of ampicillin, amoxicillin, ciprofloxacin, gentamycin, kanamycin,
neomycin,
penicillin G, streptomycin, sulfanilamide, and vancomycin.
[117] In one embodiment, the antibacterial agent is selected from the group
consisting of azithromycin, cefonicid, cefotetan, cephalothin, cephamycin,
chlortetracycline,
-29-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
clarithromycin, clindamycin, cycloserine, dalfopristin, doxycycline,
erythromycin, linezolid,
mupirocin, oxytetracycline, quinupristin, rifampin, spectinomycin, and
trimethoprim
[118] Additional, non-limiting examples of antibacterial agents for use in
combination with the antibodies of the invention include the following:
aminoglycoside
antibiotics (e.g., apramycin, arbekacin, bambermycins, butirosin, dibekacin,
neomycin,
neomycin, undecylenate, netilmicin, paromomycin, ribostamycin, sisomicin, and
spectinomycin), amphenicol antibiotics (e.g., azidamfenicol, chloramphenicol,
florfenicol,
and thiamphenicol), ansamycin antibiotics (e.g., rifamide and rifampin),
carbacephems (e.g.,
loracarbef), carbapenems (e.g., biapenem and imipenem), cephalosporins (e.g.,
cefaclor,
cefadroxil, cefamandole, cefatrizine, cefazedone, cefozopran, cefpimizole,
cefpiramide, and
cefpirome), cephamycins (e.g., cefbuperazone, cefinetazole, and cefminox),
folic acid
analogs (e.g., trimethoprim), glycopeptides (e.g., vancomycin), lincosamides
(e.g.,
clindamycin, and lincomycin), macrolides (e.g., azithromycin, carbomycin,
clarithomycin,
dirithromycin, erythromycin, and erythromycin acistrate), monobactams (e.g.,
aztreonam,
carumonam, and tigemonam), nitrofurans (e.g., furaltadone, and furazolium
chloride),
oxacephems (e.g., flomoxef, and moxalactam), oxazolidinones (e.g., linezolid),
penicillins
(e.g., amdinocillin, amdinocillin pivoxil, amoxicillin, bacampicillin,
benzylpenicillinic acid,
benzylpenicillin sodium, epicillin, fenbenicillin, floxacillin, penamccillin,
penethamate
hydriodide, penicillin o benethamine, penicillin 0, penicillin V, penicillin V
benzathine,
penicillin V hydrabamine, penimepicycline, and phencihicillin potassium),
quinolones and
analogs thereof (e.g., cinoxacin, ciprofloxacin, clinafloxacin, flumequine,
grepagloxacin,
levofloxacin, and moxifloxacin), streptogramins (e.g., quinupristin and
dalfopristin),
sulfonamides (e.g., acetyl sulfamethoxypyrazine, benzylsulfamide,
noprylsulfamide,
phthalylsulfacetamide, sulfachrysoidine, and sulfacytine), sulfones (e.g.,
diathymosulfone,
glucosulfone sodium, and solasulfone), and tetracyclines (e.g., apicycline,
chlortetracycline,
clomocycline, and demeclocycline). Additional examples include cycloserine,
mupirocin,
tuberin amphomycin, bacitracin, capreomycin, colistin, enduracidin,
enviomycin, and 2,4
diaminopyrimidines (e.g., brodimoprim).
1.3 Kits
[119] The present invention provides a pharmaceutical pack or kit comprising
one or
more containers filled with an antibody composition of the invention. In one
embodiment,
the composition is an aqueous formulation. In one embodiment, the composition
is
lyophilized. In preferred embodiments, the liquid or lyophilized composition
is sterile. In
-30-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
one embodiment, the kit comprises a liquid or lyophilized composition of the
invention, in
one or more containers, and one or more other prophylactic or therapeutic
agents useful for
the treatment of a bacterial infection or toxemia. The one or more other
prophylactic or
therapeutic agents may be in the same container as the antibody composition,
or in one or
more other containers. Preferably, the one or more other prophylactic or
therapeutic agents
comprises an antibiotic, preferably ciprofloxacin and/or doxycycline.
[120] In certain embodiments, the kit further comprises instructions for use
in the
treatment of anthrax (e.g., using the antibody compositions of the invention
alone or in
combination with another prophylactic or therapeutic agent), as well as side
effects and
dosage information for one or more routes of administration. Optionally
associated with such
container(s) is a notice in the form prescribed by a governmental agency
regulating the
manufacture, use or sale of pharmaceuticals or biological products, which
notice reflects
approval by the agency of manufacture, use or sale for human administration.
While the
instructional materials typically comprise written or printed materials they
are not limited to
such. Any medium capable of storing such instructions and communicating them
to an end
user is contemplated by this invention. Such media include, but are not
limited to, electronic
storage media (e.g., magnetic discs, tapes, cartridges, chips), optical media
(e.g. CD ROM),
and the like. Such media may include addresses to internet sites that provide
such
instructional materials.
[121] In another embodiment, this invention provides kits for the packaging
and/or
storage and/or use of the antibody composition described herein, as well as
kits for the
practice of the methods described herein. The kits can be designed to
facilitate one or more
aspects of shipping, use, and storage.
[122] All publications and patents mentioned herein are hereby incorporated by
reference in their entirety as if each individual publication or patent was
specifically and
individually indicated to be incorporated by reference.
EXAMPLE S
1.4 Pre-Exposure Efficacy
[123] The objective of this study was to examine the ability of two
antibodies,
IQNPA and IQNLF, when administered prior to exposure, to protect against death
due to
inhalational anthrax in New Zealand White rabbits. The average aerosol
challenge dose for
this study was 132 21 LD50s with a range of 99 - 199 LD50s. Body weight,
body
-31-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
temperature, clinical observations, and bacteremia were all examined during
the course of
this study. The data, discussed in more detail below, demonstrated that both
antibodies were
able to prolong survival following infection with B. anthracis. Treatment did
not affect the
weight gain or loss seen in animals following infection, nor was there any
significant change
in body temperature during the course of infection (data not shown).
1.4.1 Results
1.4.1.1 Survival
[124] Pre-treatment with either IQNLF or IQNPA prolonged survival to 5.8 and
8.12
days, respectively, on average. In the control group, the average time to
death was 4.41 days.
In addition, both antibodies increased the survival rate following exposure.
While no animals
survived in the untreated control group, 100% of animals survived for the 14
day study period
in the groups receiving either 5 mg/kg or 2.5 mg/kg IQNPA. 75% survival was
seen in the
groups receiving either 10 mg/kg or 1.25 mg/kg IQNPA as well as in the group
receiving 10
mg/ml IQNLF.
[125] Figure 1 shows a Kaplan-Meier curve representing time-to-death and
survival
data for each group. The survival data are tabulated below in Table 1.
Table 1: Percentage of animals surviving for 14 days
Dose (mg/kg) IONPA % Survival (n=4) IONLF % Survival (n=4)
75 75
5 100 50
2.5 100 25
1.25 75 50
0.625 25 50
[126] The survival rate of the IQNPA treatment groups receiving 5.0 mg/kg and
2.5
mg/kg was significantly higher than controls by the Fisher's exact test. Due
to the small
group size (4 animals per group), a statistically significant effect was not
observed when the
more stringent Bonferroni-Holm adjustment was used.
1.4.1.2 Presence of Bacteria in Blood
[127] The proportion of animals that were bacteremic at any time point post-
challenge is given in Table 2 below, along with the 95 percent confidence
interval. Twenty-
three out of twenty five animals that survived to study day 14 were not
bacteremic. Fifteen
out of seventeen animals that died or were euthanized prior to study day 14
were bacteremic
(by culture). Only five of the animals were bacteremic on study day 2.
-32-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
[128] 25 of the 44 animals (56%) were never positive for bacteria in the blood
and
23 of these 25 animals survived through the end of the study. The plated
impinger samples
were positive for bacterial growth, confirming that the animals were exposed
to B. anthracis.
The lack of detectable bacteria in the blood may indicate that the circulating
levels of
antibody inhibited bacterial growth enough to push the amount of bacteria
below the level of
detection.
[129] Although no statistically significant difference (by Fisher's exact
test) was
apparent between the control and treatment groups, this result is likely due
to the relatively
small number of animals (4) per group. The was a statistically significant
relationship
between death and bacteremia (approximately 88 percent of animals that were
bacteremic at
any time point died).
Table 2: Proportion of animals bacteremic
One-sided Fisher's Exact P-
Treatment # Proportion value, Comparison
Group Bacteremic/ Bacteremic (95% to Grou 6
Test Dose Total Confidence Bonferroni-
Interval) Unadjusted Holm
Material mg/kg Adjusted
1 IQNPA 10.0 3/4 0.75 (0.01, 0.81) 0.7857 1.0000
2 IQNPA 5.0 0/4 0.00 (0.40, 1.00) 0.0714 0.7143
3 IQNPA 2.5 0/4 0.00 (0.40, 1.00) 0.0714 0.7143
4 IQNPA 1.25 0/4 0.00 (0.40, 1.00) 0.0714 0.7143
IQNPA 0.625 3/4 0.75 (0.01, 0.81) 0.7857 1.0000
6 Control 1.0 ml/kg 3/3' 1.00 (0.29, 1.00)
7 IQNLF 10.0 1/4 0.25 (0.19, 0.99) 0.2429 1.0000
8 IQNLF 5.0 1/4 0.25 (0.19, 0.99) 0.2429 1.0000
9 IQNLF 2.5 2/3' 0.67 (0.01, 0.91) 0.5000 1.0000
IQNLF 1.25 2/4 0.50 (0.07, 0.93) 0.5000 1.0000
11 IQNLF 0.625 2/4 0.50 (0.07, 0.93) 0.5000 1.0000
Bacteremia sample could not be processed for one animal because sample was
lost due to broken tube.
Therefore, there were only three animals in this group where presence of
bacteremia could be determined at all
time points.
f Bacteremia sample could not be drawn for animal K94338. Therefore, there
were only three animals in
this group where presence of bacteremia could be determined at all time
points.
1.4.1.3 Pharmacokinetic Profiles
[130] Sera drawn from the animals were frozen and shipped to IQ Corp. for
pharmacokinetic analysis. At IQ Corporation, the sera were defrosted and
subjected to a
quantitative ELISA for IQNPA and IQNLF. Sera from the control rabbits scored
below the
-33-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
lower level of quantification of the ELISA. The pharmacokinetic profiles of
the groups
injected with (10, 5, 2.5 and 0.625 mg/kg) IQNPA or IQNLF are plotted in the
Figure 2. The
profiles show a normal decrease of antibody concentrations in time. The mean
half-life of the
IQNPA antibody was 61.4 hrs. +/- 11 hrs; the mean half-life of the IQNLF
antibody was 39.5
hrs +/- 5.2 hrs.
1.4.1.4 Clinical Observations
[131] Clinical observations were taken from day 0 through day 14 or time of
death.
All animals were in good health prior to challenge. Lethargy, stool
abnormalities (soft stool,
diarrhea, and no stool) and lack of eating were the most common clinical
observations noted
post-challenge. Sixty five percent (13/20) of rabbits receiving IQNPA
demonstrated
abnormal clinical observations including diarrhea, not eating and lethargy.
Seventy-five
percent (15/20) of rabbits receiving IQNLF presented with some form of
clinical
manifestation including diarrhea, not eating, soft stool, and lethargy. Three
quarters of the
control animals presented with some form of clinical manifestation including
not eating and
lethargy.
1.4.2 Methods
[132] Study Design: All rabbit studies were performed at Battelle Biomedical
Research Center, located at State Route 142, West Jefferson, OH 43162. Forty-
four (22 male
and 22 female) specified pathogen free New Zealand white rabbits (purchased
from Covance
Laboratories) weighing between 2.0 - 4.0 kg at the time of randomization were
placed in the
study. Four additional rabbits were housed as extras until the end of the
study. All animals
were free of malformations and clinical symptoms of disease prior to placement
on study.
Prior to treatment, rabbits were assigned to one of eleven groups (four
rabbits per group), one
of two aerosol challenge days (two rabbits per group per day), and a challenge
order per day.
Randomization occurred based on animal weights.
[133] Pre-Exposure Dosing: Approximately twenty-four hours prior to challenge,
rabbits were intravenously administered one of two antibodies at doses of
10.0, 5.0, 2.5, 1.25,
or 0.625 mg/kg IQNPA or 10.0, 5.0, 2.5, 1.25, or 0.625 IQNLF (see Table 1,
supra). Groups
1-5 received the IQNPA antibody at doses ranging from 10.0 mg/kg to 0.625
mg/kg. Animals
in group 6 received buffer only as a control. Groups 7-11 received the IQNLF
antibody at
doses ranging from 10.0 mg/kg to 0.625 mg/kg. All four rabbits in each group
received the
-34-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
indicated doses. Administration of the appropriate dose of either IQNPA or
IQNLF was
verified by documentation kept during the process of administering.
[134] Aerosol Challenge: This study required two aerosol challenge days with
22
rabbits challenged per day. The overall average dose for the two study days
was 132 LD50s
with an average challenge dose of 133 23 LD50s for the first day and 131
19 LD50s for
the second day. The mass-median aerodynamic diameter for challenge material
aerosols on
day one was 1.17 m and the mass-median aerodynamic diameter for challenge
material
aerosols on day two was 1.18 gm. Rabbits were transported into the BL-3
facility 6 days
prior to challenge to allow time for acclimation. On Study Day 0, rabbits were
placed into a
plethysmography chamber and passed into a Class III cabinet system, and
aerosol challenged
with a targeted dose of 100 LD5Os B. anthracis (Ames strain) spores
aerosolized by a
Collision nebulizer. Aerosol concentrations of B. anthracis were quantified by
determination
of cfu. Effluent streams were collected directly from an animal exposure port
by an in-line
impinger (Model 7541, Ace Glass Incorporated). Serial dilutions of impinger
samples were
plated and enumerated for challenge dose assessment.
[135] Blood Collection: Blood was drawn from the medial auricular artery or
the
marginal ear vein. Oil of wintergreen (topical) or acepromazine (1-5 mg/kg)
subcutaneously)
was used to facilitate blood sampling via the ear. Amounts of blood collected
were within
the guidelines established by the Battelle IACUC, derived in part from the
Canadian Guide to
the Care and Use of Experimental Animals.
[136] Bacteremia (Culture): Blood collected in EDTA tubes on study days 0, 2,
14
and/or time of death were cultured, by streaking -40 gl of whole blood over
blood agar
plates, to determine the presence or absence of B. anthracis.
[137] Clinical Observations: Animals were monitored twice daily for abnormal
clinical signs (such as respiratory distress, inappetence, inactivity,
seizures and moribundity)
until Study Day 14. On study day 13, AM observations were not recorded for
four of the
rabbits. Any rabbits that were moribund, as assessed by a highly trained life
sciences
technician, Battelle veterinarian, or Study Director, were euthanized.
[138] Sera Collection and Shipment: Approximately 2.0 ml of whole blood was
collected into SST tubes on the days 0, 1, 2, 7, 14 and time of death. This
blood was
processed and the serum collected. Serum was then filtered, and checked for
sterility for
shipment to IQ Corporation for serological analysis. When possible, a terminal
sample was
taken from any animal found dead or found to be moribund prior to euthanasia.
-35-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
1.5 Post-Exposure Efficacy - Experiment 1
[139] The objective of this study was to examine the ability of two
antibodies,
IQNPA and IQNLF, when administered after exposure to B. anthracis, either
alone or in
combination, to protect against death due to inhalational anthrax in rabbits.
In rabbits, a body
temperature increase of about 2 degrees Farenheit is observed at the start of
the symptomatic
period, which occurs about 25-29 hours post-exposure. Thus, in the following
three
experiments, treatment during the period of time from 0 to 24 hours following
exposure is
considered prophylactic treatment. Treatment after 32 hours is considered
therapeutic
treatment.
[140] The average aerosol challenge dose for this study was 132 30 LD50s,
with
an average challenge dose of 144 28 LD50s for the first day of challenges
and an average
dose of 119 31 LD50s for the second day of challenges. Body weight, body
temperature,
clinical observations and bacteremia were all examined during the course of
this study. The
data, discussed in more detail below, demonstrate that both the IQNPA and
IQNLF
antibodies increase survival when administered 24 hours following exposure to
inhalational
anthrax. The data further demonstrate that the combination of both antibodies
resulted in an
increased probability of survival compared to either antibody administered
alone.
1.5.1 Results
1.5.1.1 Survival
[141] In general, animals treated with either the IQNPA or IQNLF antibodies,
or
their combination, survived longer than untreated controls. The average time
to death for the
control animals was 3.87 days. The average time to death in the IQNLF
treatment group was
4.72 days; whereas the average time to death in the IQNPA treatment group was
5.52 days.
The average time to death for animals receiving a combination of antibodies
was 4.53, 3.24,
and 6.06 days for treatment groups having doses of IQNPA + IQNLF of 1.25 +
3.75, 0.625 +
1.88, and 0.3125 mg/kg + 0.94 mg/kg.
[142] The highest dose of IQNPA, 5.0 mg/kg, resulted in 100% survival. The
lower
IQNPA doses, 2.5 mg/kg and 1.25 mg/kg, resulted in 50% and 33% survival,
respectively.
The two highest doses of IQNLF (15 mg/kg and 7.5 mg/kg) resulted in 66%
survival and the
lowest dose (3.75 mg/kg) resulted in 33% survival. None of the animals in the
control group
survived. Table 3 shows the survival data and results of the Fisher's Exact
Test for each
treatment group.
-36-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
[143] The data further show that when administered in combination, the two
antibodies are capable of working in a coordinated manner to eradicate
infection.
Importantly, 50% survival was observed in the Group 11 even at doses as low as
0.625 mg/kg
IQNPA and 1.88 mg/kg IQNLF. The data also show that survival is dose-
dependent. Figure
3 shows the Kaplan-Meier curves for each group.
-37-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
Table 3: Survival Data
One-sided Fisher's Exact I
Treatment Survival Rate value, Comparison to the
Group # Survived/ (95% Confidence Control Grou (Group 7)
Dose Total Interval) Bonferroni
Test Material (mg/kg) Unadjusted Holm
Adjusted
1 IQNPA 5.0 6/6 1.00 (0.54, 1.00) 0.0011* 0.0119*
2 IQNPA 2.5 3/6 0.50 (0.12, 0.88) 0.0909 0.6364
3 IQNPA 1.25 2/6 0.33 (0.04, 0.78) 0.2273 1.0000
4 IQNLF 15 1/6 0.17 (0.00, 0.64) 0.5000 1.0000
IQNLF 7.5 1/6 0.17 (0.00, 0.64) 0.5000 1.0000
6 IQNLF 3.75 2/6 0.33 (0.04, 0.78) 0.2273 1.0000
7 Control PBS Alone 0/6 0.00 (0.00, 0.46)
8 IQNPA + IQNLF 5.0+15 6/6 1.00 (0.54, 1.00) 0.0011* 0.0119*
9 IQNPA + IQNLF 2.5+7.5 6/6 1.00 (0.54, 1.00) 0.0011* 0.0119*
IQNPA + IQNLF 1.25+3.75 4/6 0.67 (0.22, 0.96) 0.0303* 0.2424
11 IQNPA + IQNLF 0.625+1.88 3/6 0.50 (0.12, 0.88) 0.0909 0.6364
12 IQNPA + IQNLF 0.3125+0.94 2/6 0.33 (0.04, 0.78) 0.2273 1.0000
Table 4: Percentage Survival
.................. ...........
............................y..,~.................................
...........................................
ÃÃGruà E#lentÃÃÃLF ÃÃÃrÃriui
:;:;
...............................................................................
.......................................................
...............................................................................
..........................................................
...............................................................................
...........................................................
...............................................................................
..........................................................
...............................................................................
...........................................................
1 IQNPA 5.00 100 [144] Statistical analysis
2 IQNPA 2.50 50
showed a significant difference for
3 IQNPA 1.25 33
4 IQNLF 15.00 16 Groups 1, 8, and 9 when compared
5 IQNLF 7.50 16 to the control group using a
6 IQNLF 3.75 33 Bonferroni Holm adjustment to
7 Control 1 mL/kg 0 control the overall level of
8 IQNPA + IQNLF 5.0 + 15.0 100
9 IQNPA + IQNLF 2.5 + 7.5 100 significance at 0.05. When total
10 IQNPA + IQNLF 1.25 + 3.75 66 antibody dose was modeled against
11 IQNPA + IQNLF 0.625 + 1.88 50 the probability of survival, the
12 IQNPA + IQNLF 0.3125 + 0.94 33
protection provided by the IQNPA
antibody alone was not significantly different from that of the combined
antibodies.
However, the protection provided by the IQNLF antibody alone was significantly
less than
that provided by either the IQNPA alone or the combined antibodies. When the
dose of
IQNPA or IQNLF antibody alone was modeled separately against the probability
of survival,
the combined antibodies provided significantly greater protection than either
of the single
antibodies. The odds of survival for animals treated with the combined
antibodies were about
-38-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
9.6 times higher than for the IQNPA antibody alone and about 25.6 times higher
than for the
IQNLF antibody alone.
1.5.1.2 Presence of Bacteria in Blood
[145] Table 5 shows the proportion of animals that were bacteremic at any time
point during the 14 day study period with a 95 percent binomial confidence
interval. About
half (51 % or 37/72) of the challenged animals were bacteremic on day 1. All
but three of the
animals that died or were euthanized prior to study day 14 were bacteremic.
The majority of
the surviving animals were not bacteremic on day 14 as determined by blood
culture. Of
these surviving animals, 33% (12/36) were bacteremic at some time point.
[146] For Group 1, which received 5.0 mg/kg IQNPA, 3/6 animals were bacteremic
on study days 1 and 2, but negative at the end of study (day 14). The other
three animals in
this group were not bacteremic at any time point. These data indicate that, at
least in these
animals, the IQNPA antibody was able to suppress bacterial proliferation. The
fact that three
of the animals in this group were not found to be bacteremic does not mean
that they were not
exposed to B. anthracis. Exposure was confirmed by the plate counts for the
impinger
samples taken during the exposure process. It is possible that IQNPA has
completely cleared
the infection when given 24 after exposure.
[147] For Groups 8 and 9, all surviving animals were bacteremia negative at
the end
of the study. For Group 8, 83% (5/6) of the animals were not bacteremic at any
time point.
For Group 9, 66% (4/6) of the animals were not bacteremic at any time point.
As discussed
above, these data indicate that the antibodies were able to suppress bacterial
proliferation.
Many of the animals receiving lower doses of the antibodies were not only
bacteremic on
study day 1, but also at the time of euthanasia (or when found dead) prior to
day 14.
-39-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
Table 5: Incidence of Bacteremia
One-sided Fisher's Exact
Treatment Proportion P-value, Comparison to
# Bacteremic the Control Group
Group Bacteremic/ o Grou p 7
(95 /o Confidence
Test Dose Total Interval) Bonferroni-
Materia Unadjusted Holm
I (mg/kg) Adjusted
1 IQNPA 5.0 3/6 0.50 (0.12, 0.88) 0.0909 0.8182
2 IQNPA 2.5 3/6 0.50 (0.12, 0.88) 0.0909 0.8182
3 IQNPA 1.25 5/6 0.83 (0.36, 1.00) 0.5000 1.0000
4 IQNLF 15 5/6 0.83 (0.36, 1.00) 0.5000 1.0000
IQNLF 7.5 5/6 0.83 (0.36, 1.00) 0.5000 1.0000
6 IQNLF 3.75 5/6 0.83 (0.36, 1.00) 0.5000 1.0000
7 Control PBS Alone 6/6 1.00 (0.54, 1.00)
8 IQNPA + IQNLF 5.0+15 1/6 0.17 (0.00, 0.64) 0.0076* 0.0833
9 IQNPA + 2.5+7.5 2/6 0.33 (0.04, 0.78) 0.0303* 0.3030
IQNLF
IQNPA + 1.25+3.75 4/6 0.67 (0.22, 0.96) 0.2273 1.0000
IQNLF
11 IQNPA + 0.625+1.88 3/6 0.50 (0.12, 0.88) 0.0909 0.8182
IQNLF
12 IQNPA + 0.3125+0.94 5/6 0.83 (0.36, 1.00) 0.5000 1.0000
IQNLF
[148] Since the level of bacteria in the blood was often undetectable, a
statistically
significant correlation between bacteremia and treatment could not be
determined. However,
there was sufficient evidence (p value<O. 000 1) to conclude that whether an
animal was
bacteremic at any time point was related to whether the animal died.
Approximately 72
percent of animals that were bacteremic at any time point died and 94 percent
of animals that
died were bacteremic at some point.
1.5.1.3 Clinical Observations
[149] Clinical observations were done from day 0 through day 14 or at time of
death. Lethargy, stool abnormalities (soft stool, diarrhea, and no stool), and
lack of eating
were the most common clinical observations noted during the post-challenge
observation
period. One hundred percent of the control animals displayed clinical symptoms
including
-40-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
not eating and lethargy from day 2 post-challenge until death. In Group 1 (5
mg/kg IQNPA),
66% (4/6) of animals displayed clinical symptoms for 6/14 days of the post-
challenge period.
In Group 2 (2.5 mg/kg IQNPA), 83% (5/6) of animals displayed clinical symptoms
for 8/14
days. In Group 3 (1.25 mg/kg IQNPA), 100% (6/6) of animals displayed clinical
symptoms
on 9/14 days. In Group 8 (5.0 IQNPA + 15.0 IQNLF), 50% (3/6) of animals
displayed
clinical symptoms on 5/14 days. In Group 9 (2.5 IQNPA + 7.5 IQNLF), 66% (4/6)
of
animals displayed clinical symptoms on 5/14 days. In Group 10 (1.25 IQNPA +
3.75
IQNLF), 66% (4/6) of animals displayed clinical symptoms on 9/14 days. In
Group 11
(0.625 IQNPA + 1.88 IQNLF), 83% (5/6) of animals displayed clinical signs on
9/14 days.
In Group 12 (0.3125 IQNPA + 0.94 IQNLF), 83% (5/6) of animals displayed
clinical signs
on 9/14 days. These data indicate that all animals have gone through a
symptomatic period.
1.5.2 Methods
[150] Test System: Seventy-two (36 male and 36 female) specific pathogen free
New Zealand white rabbits (purchased from Covance Laboratories) weighing
between 2.0 to
4.0 kg at the time of randomization that were in good health were placed on
study. Six
additional rabbits were housed as extras until the completion of the study.
Prior to challenge,
rabbits were assigned to one of twelve groups (six rabbits per group) based on
animal
weights, one of three aerosol challenge days (two rabbits per group per day)
and a challenge
order per day. Randomization was based on animal weights. The study was
continued for 14
days.
[151] Aerosol Challenge: This study required two aerosol challenge days with
36
rabbits challenged per day. Rabbits were transported into the BL-3 facility 6
days prior to
challenge to allow time for acclimation. On Study Day 0, rabbits were placed
into a
plethysmography chamber and passed into a Class III cabinet system, and
aerosol challenged
with a targeted dose of 100 LD5Os B. anthracis (Ames strain) spores
aerosolized by a
Collision nebulizer. Aerosol concentrations of B. anthracis were quantified by
determination
of cfu. effluent streams collected directly from an animal exposure port by an
in-line
impinger (Model 7541, Ace Glass Incorporated). Serial dilutions of impinger
samples were
plated and enumerated. The overall average dose for the two study days was 132
30 LD50s
with an average challenge dose of 144 28 LD50s for the first day of
challenges and an
average dose of 119 31 LD5Os for the second day of challenges. The mass-
median
aerodynamic diameter for challenge material aerosols on day one was 1.14 m
and the mass-
-41-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
median aerodynamic diameter for challenge material aerosols on day two was
1.13 gm (as
determined with an Aerodynamic Particle Sizer (APS model 3321, TSI Inc, St.
Paul, MN).
[152] Post-Exposure Dosing: Approximately twenty-four hours post-challenge
with
B. anthracis, the rabbits were administered one of two antibodies at varying
doses, or in
combination (see Table 3, supra). Groups 1-3 received IQNPA at doses ranging
from 5
mg/kg to 1.25 mg/kg. Groups 4-6 received IQNLF at doses ranging from 15.0
mg/kg to 3.75
mg/kg. Group 7 received buffer only as a control. Groups 8-12 received
decreasing doses of
the combination treatment (IQNPA + IQNLF). All six rabbits in each group
received the
indicated doses as indicated by signed paperwork filled out during the dosing
process. All
treatments were via a single bolus dose.
[153] Blood Collection: Blood samples were collected on days -1, 1 (prior to
treatment), 2 and 14, or at time of death. Blood was drawn from the marginal
ear vein
according. Oil of wintergreen (topical) or acepromazine (1-5 mg/kg
subcutaneously) was
utilized to facilitate blood sampling via the ear. Amounts of blood collected
fell within the
guidelines established by the Battelle IACUC, derived in part from the
Canadian Guide to the
Care and Use of Experimental Animals.
[154] Bacteremia (Culture): Blood collected in EDTA tubes on study days -1, 1,
2,
14 and/or time of death were cultured by streaking -40 gl of whole blood over
blood agar
plates, to determine the presence or absence of B. anthracis bacteremia.
[155] Sera Collection and Shipment: Approximately 2.0 ml of whole blood was
collected into SST tubes. This blood was processed and the serum collected.
Serum was then
filtered, and checked for sterility for shipment to IQ Corporation for
serological analysis.
When possible, a terminal sample was taken from any animal found dead or found
to be
moribund prior to euthanasia.
[156] Clinical Observations: Animals were monitored twice daily by laboratory
animal personnel near the beginning and end of each workday for abnormal
clinical signs
(such as respiratory distress, inappetence, inactivity, seizures and
moribundity) until Study
Day 14. Any rabbits that were moribund, as assessed by a highly trained life
sciences
technician, Battelle veterinarian, or Study Director, were euthanized.
1.6 Post-Exposure Efficacy - Experiment 2
[157] The main objective of this study was to further examine the efficacy of
the
combined treatment of IQNLF and IQNPA against inhalational anthrax infection.
The target
infectious dose for this study was 100 LD5Os; the average aerosol challenge
dose for the
-42-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
study was 91 27 with a range of 47 - 149 LD50s. The log-rank test applied to
the time-to-
death data showed that the pooled control group was significantly less
protected than groups
treated with combined antibodies when time to death was considered in addition
to the
overall survival rates. Overall, IQNLF alone did not provide as high a level
of protection as
the combined treatment.
1.6.1 Results
1.6.1.1 Survival
[158] Fifty-seven percent (46/80, regardless of treatment) of the challenged
animals
succumbed to the infection, with an average time to death of approximately
4.08 days. For
the control group, 100% (12/12) of animals succumbed to infection with an
average time to
death of 3.8 days. For Groups 1-5 (IQNLF alone at 10.00, 7.50, 5.00, 2.50, and
1.25 mg/kg,
respectively), 87.5% (7/8), 75% (6/8), 87.5% (7/8), 50% (4/8), and 100% (4/4)
of the animals
succumbed to disease with an average time to death of 4.6, 3.5, 4.5, 3.8, and
5.39 days
respectively. For Groups 8-11 (IQNPA + IQNLF in combination at 2.5 + 0.625,
2.5 + 1.25,
2.5 + 2.5, and 2.5 + 5.0 mg/kg respectively), 25% (2/8), 38% (3/8), 12.5%
(1/8), and 0 %
(0/8) of the animals succumbed to disease. Figure 4 is a Kaplan-Meier curve
representing
time-to-death and survival data for each group.
[159] Table 6 summarizes the survival data for each group. Confidence
intervals for
the survival rates and the results of Fisher's exact test comparisons of
survival rates to the
control group are also provided. According to the unadjusted p values from
Fisher's exact
test, treatment group 4 (IQNLF, 2.5 mg/kg) as well as groups 8 through 11 (all
groups with
combined antibody doses of IQNPA and IQNLF) had significantly higher survival
rates than
the pooled control group (group 6). When a Bonferroni Holm adjustment was used
to control
the overall level of significance at 0.05, the same groups (4 and 8 through
11) had a
significantly higher survival rate than the control group.
-43-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
Table 6: Survival Rates
One-sided Fisher's Exact
Treatment P-value, Comparison to
oup 6
No. Survival Rate
Group Dose Survived (95% Confidenc
Test / Total e Interval) Unadjusted Material ( 9)k nadjusted Adjusted
1 IQNLF 10.0 1/8 0.13 (0.00, 0.53) 0.2000 0.6000
2 IQNLF 7.5 2/8 0.25 (0.03, 0.65) 0.0737 0.2947
3 IQNLF 5.0 1/8 0.13 (0.00, 0.53) 0.2000 0.6000
4 IQNLF 2.5 4/8 0.50 (0.16, 0.84) 0.0072* 0.0361*
IQNLF 1.25 0/4 0.00 (0.00, 0.60) 0.5000 0.6000
6 Control PBS 1 0/12 0.00 (0.00, 0.26)
mL/kg EM PA + 8 IQNNLF 0 625* 7/8 0.88 (0.47, 1.00) <0.0001* 0.0005*
9 IQNNLF 1.25* 5/8 0.63 (0.24, 0.91) 0.0018* 0.0108*
1 IQ
IQNNPA + LF .5 7/8 0.88 (0.47, 1.00) <0.0001* 0.0005*
I 2.5*
11 IQNNLF 5 0* 7/8 0.88 (0.47, 1.00) <0.0001* 0.0005*
Table 7: Percent Survival
...............................................................................
...............................................................................
..................................
...............................................................................
..............................................................................
1 IQNLF 24 10.00 12.5
2 IQNLF 24 7.50 25
3 IQNLF 24 5.00 12.5
4 IQNLF 24 2.50 50
5 IQNLF 24 1.25 0
6 Control 24 1.0 mL/kg PBS 0
8 IQNPA + IQNLF 24 2.5 + 0.625 87.5
9 IQNPA + IQNLF 24 2.5 + 1.25 62.5
10 IQNPA + IQNLF 24 2.5 + 2.5 87.5
11 IQNPA + IQNLF 24 2.5 + 5.0 87.5
[160] Table 8 shows the estimates and p values for the effects included in the
final
logistic regression model that models survival with effects for the base 10
log transformed
combined antibody dose and treatment (IQNLF or IQNPA + IQNLF). The interaction
-44-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
between dose and treatment was not significant (p value=0.5023) and so was not
included in
the final model. The effect for the log transformed dose was not statistically
significant (p
value=0.8460). Thus, there was not a statistically significant relationship
between treatment
dose and probability of survival. However, the overall effect for treatment
was statistically
significant at the 0.05 level (p value<0.0001) indicating that survival rates
differed among the
two treatments (IQNLF and combined IQNPA+IQNLF). Table 9 shows the odds ratios
for
the model.
Table 8: Effects included in Logistic Regression
Model Fitted to Combined Antibody Dose and
Treatment (IQNLF or IQNLF + IQNLP)
Effect P-value
Intercept 0.7604 Table 9:
Treatment <0.0001* Summary of Odds
Logio(Dose) 0.8460 Ratios for Logistic
Regression Model
Fitted to
Combined Antibody Dose and Treatment
Treatment Group Comparison Odds Ratio P-Value
(IQNLF+IQNPA) vs. IQNLF 15.15 <0.0001*
Log lo(Dose) 0.79 0.8460
[161] The data show that the odds of survival for animals treated with both
antibodies (IQNPA+IQNLF) are about 15 times higher than for animals treated
with the
IQNLF antibody alone. Figure 5 plots the estimated logistic regression curves
for each
treatment along with points showing the proportion of animals that survived
for each dose
group and treatment.
[162] Table 10 shows the estimates and p values for the effects included in
the
logistic regression model fitted to the base 10 log transformed IQNLF dose and
an indicator
for treatment (IQNLF or IQNPA+IQNLF). The interaction between IQNLF dose and
treatment was not significant (p value=0.5480) and so was not included in the
final model.
The effect for the log transformed IQNLF dose was not statistically
significant (p
value=0.9788). The treatment effect was statistically significant at the 0.05
level (p
value=0.0002) indicating that survival rates differed among the two treatments
(IQNLF and
combined IQNPA+IQNLF). Table 11 presents the odds ratios from the model. The
odds of
survival for animals treated with both antibodies (IQNPA+IQNLF) are about 15
times higher
-45-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
for animals treated with IQNLF alone. Figure 6 plots the estimated logistic
regression curves
for the IQNLF and combined treatments along with points showing the proportion
of animals
that survived for each group.
Table 10: Effects included in Logistic
Regression Model Fitted to IQNLF Dose and
Treatment
Effect P-value
Intercept 0.8243
Treatment 0.0002*
Loglo(IQNLF Dose) 0.9788
Table 11: Summary of Odds Ratios for Logistic Regression Model Fitted
to IQNLF Dose and Treatment
Treatment Group Comparison Odds P-value
Ratio
IQNPA+IQNLF vs. IQNLF 15.00 0.0002*
Loglo(IQNLF Dose) 0.98 0.9788
[163] Overall, the results of this study suggest that there is no significant
dose-
response relationship. However, there is a significant treatment effect with
the combined
antibody (IQNPA+IQNLF) treatment resulting in a higher probability of
survival. The group
that received no treatment (group 6) was significantly less protected than all
treatment groups
except groups treated with 10mg/kg IQNLF (group 1) , 7.5mg/kg IQNLF (group 2),
and
2.5mg/kg IQNLF (group 4) when time to death was considered in addition to the
overall
survival rates. Groups 8, 10, and 11 which were treated with both antibodies
and where most
animals survived also showed significantly greater protection than groups 1,
2, 3, and 5
which were treated only with IQNLF and where at most two animals survived.
[164] IQNLF as stand alone treatment is not very protective. The IQNPA+IQNLF
combination, however, is very successful in protecting from death due to
anthrax challenge
24 hrs before treatment. The combination treatment provided a 15 times higher
chance of
survival in this post-exposure aerosol challenge model. Although the increased
protection by
the IQNPA+IQNLF combination was significant, there was no dose dependency (all
but one
combination group demonstrated 87.5% survival). The results of this study
suggest that the
IQNPA+IQNLF combination represents an improvement over the IQNLF treatment
alone.
-46-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
1.6.1.2 Presence of Bacteria in Blood
[165] Table 12 illustrates the proportion of animals that were bacteremic at
any time
point post-challenge along with the 95 percent confidence interval. All
animals surviving to
study day 14 were negative for bacteria on study day 14. However, only 32%
(11/34) of
these surviving animals were positive at any time point. All except two
animals that died or
were euthanized prior to study day 14, were positive. While only 2.7% (1/36)
of the animals
were bacteremia positive on study day 7, 32.8% (25/76) of the animals were
bacteremia
positive on study day 2 and fifty-three percent (42/80) of the animals were
bacteremia
positive just prior to treatment.
[166] The proportion of animals that were bacteremic at any time point in
groups 4,
8, 9 and 11 was significantly lower than the pooled control group by Fisher's
exact test.
When the more stringent Bonferroni Holm adjustment was used to control the
overall level of
significance at 0.05, only group 8 was significantly different from controls
(Table 12). There
was a statistically significant correlation between testing positive for
bacteria in the blood at
any time point and death. Approximately 81 percent of animals that were
bacteremic at any
time point died and 100 percent of animals that died were bacteremic at some
point. Table 13
is a frequency table that summarizes the relationship between an animal being
bacteremic at
any time point and death that includes day of death measurements. According to
Fisher's two
sided exact test of independence, the hypothesis that death and whether an
animal was
bacteremic at any time point was independent was rejected (p value<0.0001).
-47-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
Table 12: Proportion of Animals that were Bacteremic at Any Time Point and 95
Percent
Binomial Confidence Interval
One-sided Fisher's Exact
Treatment No Proportion P-value, Comparison to
Group Bacteremic Bacteremic Grou 6
(95% Confidence Bonferroni.
Test Material ( gskg) / Total Interval) Unadjusted Holm
Adjusted
1 IQNLF 10.0 7/8 0.88 (0.47, 1.00) 0.4000 1.0000
2 IQNLF 7.5 6/8 0.75 (0.35, 0.97) 0.1474 0.7368
3 IQNLF 5.0 7/8 0.88 (0.47, 1.00) 0.4000 1.0000
4 IQNLF 2.5 5/8 0.63 (0.24, 0.91) 0.0491* 0.2947
IQNLF 1.25 4/4 1.00 (0.40, 1.00) 1.0000 1.0000
6 Control PBS 1 mL/kg 12/12 1.00 (0.74, 1.00)
8 IQNPA + IQNLF 2.5 + 0.625* 2/8 0.25 (0.03, 0.65) 0.0007* 0.0065*
9 IQNPA + IQNLF 2.5 + 1.25* 4/8 0.50 (0.16, 0.84) 0.0144* 0.1156
IQNPA + IQNLF 2.5 + 2.5* 6/8 0.75 (0.35, 0.97) 0.1474 0.7368
11 IQNPA + IQNLF 2.5 + 5.0* 4/8 0.50 (0.16, 0.84) 0.0144* 0.1156
Table 13: Frequency Table of Animals Bacteremic at Any
Time Point Versus Survival Status (Alive or Dead)
Survival Status
Survived Died
Bacteremia
Positive 11 46
Negative 23 0
1.6.1.3 Clinical Observations
[167] Clinical observations were documented from day 0 through day 14 or time
of
death. Lethargy, stool abnormalities (soft stool, diarrhea, and no stool), and
lack of eating
were the most common clinical observations noted during the post-challenge
observation
period. All of the control animals displayed clinical symptoms including not
eating, lethargy
or no stool from day 2 post-challenge until death. All animals receiving
10.00, 7.50, 5.00,
2.50, or 1.25 mg/kg IQNLF displayed clinical symptoms such as not eating,
lethargy,
lacrimation, soft stool, labored breathing, and/or no stool as early as Study
Day 2. While
animals receiving the combination treatment displayed clinical symptoms such
as not eating,
lacrimation, soft stool, and lethargy, the symptoms themselves as well as the
length of time
the symptoms last, was shortened considerably. By study day 8, 14 out of the
16 remaining
animals in these groups were completely normal.
-48-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
1.6.2 Methods
[168] Test S,, sue: Eighty (40 male and 40 female) specific pathogen free New
Zealand white rabbits (purchased from Covance Laboratories), weighing between
2.72 to
3.96 kg at the time of randomization and which were in good health were placed
on study.
Eighty rabbits were ordered, therefore, there would be no replacements in the
event that a
rabbit was to be removed from the study.
[169] Aerosol Challenge: This study required two aerosol challenge days with
40
rabbits challenged per day. The first 40 rabbits were challenged and treated
and followed for
14 days and then the second 40 rabbits arrived, were challenged, treated, and
followed for 14
days. Thus, there were two separate randomizations performed. Prior to
challenge day A,
rabbits were assigned to one of six groups based on animal study day -8
weights, and a
challenge order per day. The day of aerosol challenge was considered Day 0.
The first group
of 40 rabbits was randomized according to Table 14A and the second group of 40
rabbits was
randomized according to Table 14B. All rabbits to be challenged on the second
of the two
challenge days were randomized by Study Day -7 weights. Rabbits were
transported into the
BL3 facility immediately upon arrival for quarantine. On Study Day 0, rabbits
were placed
into a plethysmography chamber and passed into a Class III cabinet system, and
challenged
with a targeted aerosol dose of 100 LD5Os B. anthracis (Ames strain) spores.
The
concentrations of B. anthracis inhaled by the rabbits was determined from the
number of B.
anthracis spores collected directly from an animal exposure port by an in-line
impinger
(Model 7541, Ace Glass Incorporated). Serial dilutions of impinger samples
were plated and
enumerated. The inhaled dose was calculated using the number of CFU/liter of
air multiplied
by the respiratory volume of the rabbits. The overall average dose for the two
aerosol
challenge days was 91 27 LD50s with an average challenge dose of 115 34
LD50s for the
first day of challenges and an average dose of 66 19 LD50s for the second
day of
challenges. The mass-median aerodynamic diameter for challenge material
aerosols on day
one was 1.18 m and the mass-median aerodynamic diameter for challenge
material aerosols
on day two was 1.15 m (as determined with an Aerodynamic Particle Sizer (APS
model
3321, TSI Inc, St. Paul, MN)).
-49-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
Table 14A: Study Desi n Part I
...............................................................................
...............................................................................
.........................................................
>::>:::>:>::# f :::::::::>:' t11 Ã.....:............... !3' ill
::::::::::::::::::::::::::
:::::..::: .:::IY:: 9.::::::
...................... .....................................................
.::::::::::::::::::.:::::::::::::::::::::::::::::::::::::::::::::::::::.:::::::
:::::::::::::::::.............:.....
1 IQNLF 8 I.V. 10 +24 -7 1` 2, 7 14 or TOD
2 IQNLF 8 I.V. 7.5 +24 -7 1` 2, 7, 14 or TOD
3 IQNLF 8 I.V. 5 +24 -7, 1`, 2, 7, 14 or TOD
4 IQNLF 8 I.V. 2.5 +24 -7, 1` 2, 7, 14 or TOD
IQNLF 4 I.V. 1.25 +24 -7, 1` 2, 7, 14 or TOD
6 PBS Only 4 I.V. 1 mL/kg +24 -7, 1`, 2, 7, 14 or TOD
a Single bolus dose
b Treatment time to be 24 hours post-exposure f 15 minutes
Blood to be collected immediately prior to treatment
Table 14B: Study Design Part II
...............................................................................
...............................................................................
...................................................................
...............................................................................
............................................................ .
:::::>::::::: 66 :::::::>::>::::>::::>:::: >::::>::::>:::: ::: rent m : :>::>:
> :>::>::>:>::>::>::>::::>::>::>::>::>::>:::>::::>::>::>:>::>::>::
iii cu >
'''''>>'':::::>::::::>::::>::>::>:::>::::>:::::>::::>:::::T:a:>::>:::>::>::>:::
:>: ilatn:::>av::>::>::>::>::>::>:
::::>::::>::::>::::>::::>::::>::::::>::::>::::>::::>::::>::::>::::>::::>::::>::
::::::>::re o...:......:............:>117. :: C >:>:>::.;:::
:.;::>:>::::>::>::>::>::>:;..:..:.;:.;::;
:
..................................................
::::::::>::::............d.......................
7:;:;:;; PBS Only:*;;:;:;: ;:;:;::4;:;:;:. ;::I.V IN. .1 mL/kg +24 -7 1c, 2,
7, 14 or TOD
8 IQNPA + IQNLF 8 .V. 2.5 + 0.625d +24 -7, 1` 2, 7, 14 or TOD
9 IQNPA + IQNLF 8 .V. 2.5 + 1.25d +24 -7, 1`, 2, 7, 14 or TOD
IQNPA + IQNLF 8 .V. 2.5 + 2.5d +24 -7 1` 2, 7, 14 or TOD
11 IQNPA + IQNLF 8 .V. 2.5 + 5.0d +24 -7, 1` 2, 7, 14 or TOD
12 PBS Only 4 I.V. 1 mL/k +24 -7, 1`, 2, 7, 14 or TOD
a Single bolus dose
b Treatment time to be 24 hours post-exposure f 15 minutes
Blood to be collected immediately prior to treatment
a IQNPA dose concentration remained the same at 2.5 mg/kg while the IQNLF dose
varied accordingly
See Deviation (DR-5813)
[170] Post-Exposure Dosing: Approximately twenty-four hours post-challenge
with
B. anthracis, the rabbits were administered antibodies according to Tables 14A
and B. Doses
were given via a single bolus intravenous injection.
[171] Blood Collection: Blood samples were collected according to Tables 14A
and
B. Blood was drawn from the marginal ear vein according to SOP BBRC.VII-020.
Oil of
wintergreen (topical) or acepromazine (1-5 mg/kg subcutaneously) was utilized
to facilitate
blood sampling via the ear. Amounts of blood collected fell within the
guidelines established
by the Battelle IACUC, derived in part from the Canadian Guide to the Care and
Use of
Experimental Animals.
[172] Bacteremia (Culture): Blood collected in EDTA tubes on days -7, just
prior to
treatment, 2, 14 and/or at time of death was cultured by streaking -40 gl of
whole blood over
blood ager plates, to determine the presence or absence of B. anthracis
bacteremia.
[173] Sera Collection and Shipment: Approximately 2.0 ml of whole blood was
collected into SST tubes on study days -7, 14 or at time of death. This blood
was processed
and the serum collected. Serum was then filtered and checked for sterility for
shipment to IQ
-50-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
Corporation for serological analysis. When possible, a terminal sample was
taken from any
animal found dead or found to be moribund prior to euthanasia.
[174] Clinical Observations: Animals were monitored twice daily by laboratory
animal personnel near the beginning and end of each workday for abnormal
clinical signs
(such as respiratory distress, inappetence, inactivity, seizures and
moribundity) until Study
Day 14. Any rabbits that were moribund, as assessed by a highly trained life
sciences
technician, Battelle veterinarian, or Study Director, were euthanized.
1.7 Post-Exposure Efficacy - Experiment 3
[175] The objective of this study was to determine whether treatment with the
IQNPA and IQNLF antibodies, alone or in combination, could extend the window
for
treatment following inhalational infection with B. anthracis. The IQNPA and
IQNLF
antibodies were given alone or in combination at 24, 32, 40, and 48 hours post-
challenge.
The target infectious dose for this study was 100 LD5Os; the overall average
dose for the
three study days was 100 25 LD50s with an average challenge dose of 91 28
LD50s for
the first day of challenges and an average dose of 102 19 LD50s for the
second day of
challenges and an average dose of 106 29 LD50s for the third day of
challenge.
[176] While survival was the key objective of this study, several other
parameters
including temperature, body weight, clinical observations, and bacteremia were
also
examined.
1.7.1 Results
1.7.1.1 Survival
[177] The results from the logistic regression model fitted to the survival
data shows
that there was a significant treatment effect. Groups treated with the
combination of IQNPA
and IQNLF antibodies had significantly greater odds of survival than either
antibody alone
(Figure 7). Fisher's exact test showed that three of the groups treated with
both IQNPA and
IQNLF antibodies (groups 8, 9, and 11) had significantly greater survival
rates than the
control group. When a Bonferroni Holm adjustment was used to control the
overall level of
significance at 0.05, only the group treated with IQNPA+IQNLF at treatment
time 24 hrs
(group 8) was significantly different from the control group (group 7). Figure
8 shows the
Kaplan-Meier survival curves for each group.
-51-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
Table 15: Dosing Schedule
Group nr IQNPA IQNLF IQNPA + IQNLF
2.5 m/k @ 7.5 mmg/kg @ 2.5 + 7.5 mmg/kg @
1 +24 hrs - -
2 +32 hrs - -
3 +40 hrs - -
4 - +24 hrs -
- +32 hrs -
6 - +40 hrs -
7 PBS control 1 mL/k @ +24 hrs
8 - - +24 hrs
9 - - +32 hrs
- - +40 hrs
11 - - +48 hrs
[178] The log rank test applied to the time to death data showed a
statistically
significant difference over the control group for Group 5 (IQNLF at 32 hrs),
Group 8
(IQNPA+IQNLF at 24 hrs), and Group 9 (IQNPA+IQNLF at 32 hrs) when time to
death was
considered in addition to the overall survival rates and a Bonferroni Holm
adjustment was
used to control the overall level of significance at 0.05.
Tablel6: Survival Rate and Results of Fisher's Exact Test Comparison for Each
Treatment Group
One-sided Fisher's Exact
Treatment No. Survival Rate P-value, Comparison to
roup Survived (95% Confiden oup 7
Dose Treatment / Total ce Interval) Bonferroni-
Test Material (mg/kg) Time Unadjusted Holm Adjuste
(hours)
1 IQNPA 2.5 24 3/6 0.50 (0.12, 0.88) 0.0909 0.5455
2 IQNPA 2.5 32 2/6 0.33 (0.04, 0.78) 0.2273 0.6818
3 IQNPA 2.5 40 3/6 0.50 (0.12, 0.88) 0.0909 0.5455
4 IQNLF 7.5 24 4/6 0.67 (0.22, 0.96) 0.0303* 0.2727
5 IQNLF 7.5 32 1/6 0.17 (0.00, 0.64) 0.5000 1.0000
6 IQNLF 7.5 40 1/6 0.17 (0.00, 0.64) 0.5000 1.0000
7 Control PBS Alone 24 0/6 0.00 0.00 0.46
................ .................
8 IQNPA + IQNLF 2.5+7.5 24 6/6 1.00 (0.54, 1.00) 0.0011* 0.0108*
9 IQNPA + IQNLF 2.5+7.5 32 4/6 0.67 (0.22, 0.96) 0.0303* 0.2727
10 IQNPA + IQNLF 2.5+7.5 40 3/6 0.50 (0.12, 0.88) 0.0909 0.5455
11 IQNPA + IQNLF 2.5+7.5 48 4/6 0.67 (0.22, 0.96 0.0303* 0.2727
-52-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
Table 17: Percent Survival
...............................................................................
...............................................................................
..............................
...............................................................................
...............................................................................
..................................
.......................................................................
n:::::>:>::>::>:::::>::>::>::>:::>::::>::::>::::>::::>::::>::::>::::>::::::::>:
:::>::::>::::>::::>::::>::::>::::>::::>::::>::::>::::>::::>::::>:
...............................................................................
................
...............................................................................
...............................................................................
..................................
...............................................................................
...............................................................................
..................................
...............................................................................
...............................................................................
.................................
................. :..
....... ..................................................
.........................................
...............................................................................
...............
...............................................................................
..........................................................................
1 IQNPA 24 2.5 50
2 IQNPA 32 2.50 33
3 IQNPA 40 2.5 50
4 IQNLF 24 7.5 66
IQNLF 32 7.50 16
6 IQNLF 40 7.5 16
7 Control 24 1 mL/kg 0
8 IQNPA + IQNLF 24 2.5 + 7.5 100
9 I NPA+I NLF 32 2.5+7.5 66
I NPA+I NLF 40 2.5+7.5 50
11 IQNPA + IQNLF 48 2.5 + 7.5 66
Table 18: Unadjusted p-values from the pairwise log-rank test comparing time-
to-death
and overall survival between all groups
Group 1 2 3 4 5 6 7 8 9 10
2 0.5213
3 0.7426 0.3468
4 0.5126 0.1967 0.6475
703 0.7054 0.3271 0.1171
5 06
6 0.5759 0.8336 0.2578 0.1171 0.6451
7 0.1093 0.3685 0.0117* 0.0111* 0.0046* 0.0271*
8 0.0554 0.0183* 0.0554 0.1385 0.0043* 0.0043* 0.0005*
9 0.3912 0.1265 0.5901 0.9193 0.0817 0.0576 0.0016* 0.1385
10 0.9467 0.4297 0.8065 0.5126 0.6703 0.5097 0.0486* 0.0554 0.3912
11 0.5901 0.2955 0.7206 0.9193 0.2387 0.2387 0.0498* 0.1385 0.8395 0.6606
1.7.1.2 Presence of Bacteria in Serum
[179] 58% (38/66) of the rabbits in this study were bacteremic at some point
(either
during the study, or at the time of death) while the remaining 42% (28/66) was
never blood
culture positive at any time during the study. All rabbits were exposed to a
100 LD50 aerosol
dose (based on the plate counts for the impinger samples taken during the
exposure process).
Thus, it is unlikely that they were not infected. The negative blood culture
results are most
likely due to a concentration of bacteria at the time of collection that was
below the limits of
detection (CFU/ml). Unless the IQNPA and/or IQNLF antibodies can completely
eliminate
both bacteria and toxin, one would expect to see a bacteremia positive result
at some point
during the study; typically early after infection. However, these results may
indicate that the
antibodies were able to suppress bacterial growth in the blood below the level
of detection.
-53-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
The inability to detect bacteria in the blood precluded a determination of
whether the
antibodies, alone or the combination, were able to clear the bacteria.
[180] Table 19 summarizes the bacteremia results for all animals of this
study.
Twenty-nine percent (21/72) of the challenged animals were bacteremic on day
1. All
animals that survived to study day 14 were negative. 16% (5/31) of these
surviving animals
had positive blood cultures at any time during the study. In contrast, all
except three of the
animals which died or were euthanized prior to study day 14 were bacteremic.
Three of the
six animals (50%) that received 2.5 mg/kg IQNPA 24 hours post-challenge were
blood
culture negative on study day 14 while only 2/6 and 3/6 of the animals
receiving 2.5 mg/kg
IQNPA 32 and 40 hours post-exposure were blood culture negative at the end of
the study.
Sixty-six percent (4/6) of the rabbits receiving 7.5 mg/kg IQNLF 24 hours post-
challenge
were blood culture negative at the end of the study (day 14). Only one rabbit
from each of the
other two IQNLF treatment groups survived and both were blood culture
negative. One
hundred percent (6/6) of the rabbits receiving the combination treatment 24
hours post-
challenge were blood culture negative at the end of the study. Sixty-six
percent (4/6), 50%
(3/6), and 66% (4/6) of the rabbits receiving the combination treatment 32,
40, and 48 hours
post-challenge were blood culture negative at the end of the study.
Table 19: Proportion of Animals that were Bacteremic at Any Time Point and 95%
Binomial Confidence Interval
One-sided Fisher's Exac
Treatment No Proportion P-value, Comparison to
Group Bacteremic Bacteremic Grou 7
Dose Treatme / Total (95% Confiden Bonferron
Test Material (mg/kg) nt Time ce Interval) Unadjusted Holm
(hours) Adjusted
1 IQNPA 2.5 24 3/6 0.50 (0.12, 0.88) 0.2727 1.0000
2 IQNPA 2.5 32 4/6 0.67 (0.22, 0.96) 0.5000 1.0000
3 IQNPA 2.5 40 3/6 0.50 (0.12, 0.88) 0.2727 1.0000
4 IQNLF 7.5 24 3/6 0.50 (0.12, 0.88) 0.2727 1.0000
IQNLF 7.5 32 5/6 0.83 (0.36, 1.00) 0.7727 1.0000
6 IQNLF 7.5 40 5/6 0.83 (0.36, 1.00) 0.7727 1.0000
7 Control PBS Alone 24 5/6 0.83 0.36 1.00
8 IQNPA + IQNLF 2.5+7.5 24 3/6 0.50 (0.12, 0.88) 0.2727 1.0000
9 IQNPA + IQNLF 2.5+7.5 32 2/6 0.33 (0.04, 0.78) 0.1212 1.0000
IQNPA + IQNLF 2.5+7.5 40 3/6 0.50 (0.12, 0.88) 0.2727 1.0000
11 IQNPA + IQNLF 2.5+7.5 48 2/6 0.33 (0.04, 0.78 0.1212 1.0000
1.7.1.3 Clinical Observations
[181] Clinical observations were recorded from day 0 through day 14 or time of
death. Lethargy, stool abnormalities (soft stool, diarrhea, and no stool), and
lack of eating
-54-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
were the most common clinical observations noted during the post-challenge
observation
period. One hundred percent of the control animals displayed clinical symptoms
including
not eating, lethargy, no stool, and lacrimation from day 3 post-challenge
until death. For
Groups 1 (2.5 mg/kg IQNPA at 24 hrs) and 2 (2.5 mg/kg IQNPA at 32 hrs ), 66%
(4/6) of
animals displayed clinical symptoms on 3/14 days during the post-challenge
observation
period. For Group 3 (2.5 mg/kg IQNPA at 40 hrs), 100% (6/6) of animals
displayed clinical
symptoms on 6/14 days. For Group 8 (IQNPA + IQNLF at 24 hrs), 50% (3/6) of
animals
displayed clinical symptoms on 9/14 days. For Group 9 (IQNPA + IQNLF at 32
hrs), 66%
(4/6) of animals displayed clinical symptoms on 6/14 days. For Group 10 (IQNPA
+ IQNLF
at 40 hrs), 83% (5/6) of animals displayed clinical symptoms on 12/14 days.
For Group 11
(IQNPA + IQNLF at 48 hrs), 66% (4/6) of animals displayed clinical signs on
10/14 days.
1.7.2 Methods
[182] Test System: Seventy-two (36 male and 36 female) New Zealand white
rabbits (purchased from Covance Laboratories), specific pathogen free (SPF),
that weighed
between 2.0 to 4.0 kg at the time of randomization and were in good health
were placed on
study. Seventy-eight rabbits were ordered. As all animals were free of
malformations and
illness, the replacement animals were not required, and were transferred to a
training protocol
and used for training purposes.
[183] Aerosol Challenge: This study required three aerosol challenge days with
22
rabbits challenged per day. Rabbits were transported into the BL-3 facility 3
days prior to
challenge to allow time for acclimation. On Study Day 0, rabbits were placed
into a
plethysmography chamber and passed into a Class III cabinet system, and
challenged with a
targeted aerosol dose of 100 LD50s B. anthracis (Ames strain) spores. The
concentrations of
B. anthracis inhaled by the rabbits is determine from the number of B.
anthracis collected
directly from an animal exposure port by an in-line impinger (Model 7541, Ace
Glass
Incorporated). Serial dilutions of impinger samples were plated and enumerated
as per SOP
BBRC.X-054. The inhaled dose was calculated using the number of CFU/liter of
air
multiplied by the respiratory volume of the rabbits. The overall average dose
for the three
study days was 100 25 LD50s with an average challenge dose of 91 28 LD50s
for the
first day of challenges and an average dose of 102 19 LD50s for the second
day of
challenges and an average dose of 106 29 for the third challenge day. The
mass-median
aerodynamic diameter for challenge material aerosols on day one was 1.16 m
and the mass-
median aerodynamic diameter for challenge material aerosols on day two was
1.15 m and
-55-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
the mass-median aerodynamic diameter for challenge material aerosols on day
three was 1.12
m (as determined with an Aerodynamic Particle Sizer (APS model 3321, TSI Inc,
St. Paul,
MN)).
[184] Post-Exposure Dosing: Approximately twenty-four hours post-challenge
with
B. anthracis, the rabbits were administered either IQNPA (2.5 mg/kg), IQNLF
(7.5 mg/kg),
or a combination of the two (see Table 15, supra). Doses were given via a
single bolus
intravenous injection. Briefly, groups one through three received 2.5 mg/kg of
IQNPA at the
indicated times post-challenge, group six received buffer only as a control,
and groups eight
through eleven received 7.5 mg/kg IQNLF at 24, 32, 40, and 48 hours post-
challenge. All
rabbits were treated according to Table 15. The study director visually
verified doses prior to
being administered to ensure the required dose levels were given. Rabbits were
administered
2.5 mg/kg IQNPA at 24, 32, and 40 hours post-challenge; 7.5 mg/kg IQNLF was
administered at 24, 32, and 40 post-challenge; 5.0 + 7.5 mg/kg IQNPA + IQNLF
was
administered 24, 32, 40, and 48 hours after being challenged with B.
anthracis. All
treatments were via a single bolus dose.
[185] Temperature Monitoring: Body temperatures were monitored twice daily via
an implantable programmable temperature transponder chip (IPTT-300, BMDS,
Seaford,
DE). Temperature chips were implanted on or before study day -9 depending on
which day
the animals were to be challenged. Rabbits were sedated with acepromazine (1-5
mg/kg)
prior to implantation of the transponder chips and each rabbit had two chips
injected
subcutaneously (one at shoulder blade level and one at rump level). Recording
of twice daily
baseline body temperature from both transponder chips began on or before study
day -11 and
continued until the morning of each groups day of challenge. Clinical
temperature readings
began in the afternoon of the day of challenge and were taken twice daily for
the duration of
the study. Post-challenge body temperatures were monitored from a single
transponder chip
(rump). All temperature readings were taken prior to treatment with
acepromazine.
[186] Animal Weights: Animals were weighed once daily for the duration of the
study beginning 10 days prior to the first challenge day. Weights were used to
determine the
amount of acepromazine to be administered prior to temperature transponder
implantation.
Study day 0 weights were used to determine the required amount of test/control
article to be
administered.
[187] Blood Collection: Blood samples were collected on time points -1, 0, 2,
and
14 or time of death. Blood was drawn from the marginal ear vein. Oil of
wintergreen
(topical) or acepromazine (1-5 mg/kg subcutaneously) was utilized to
facilitate blood
-56-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
sampling via the ear. Amounts of blood collected fell within the guidelines
established by the
Battelle IACUC, derived in part from the Canadian Guide to the Care and Use of
Experimental Animals.
[188] Bacteremia (Culture): Blood collected in EDTA tubes on days -1, just
prior to
treatment, 2, 14 and/or time of death were cultured, by streaking -40 gl of
while blood over
blood ager plates, to determine the presence or absence of B. anthracis
bacteremia.
[189] Sera Collection and Shipment: Approximately 2.0 ml of whole blood was
collected into SST tubes (see Blood collection). This blood was processed and
the serum
collected. Serum was then filtered, and checked for sterility for shipment to
IQ Corporation
for serological analysis. When possible, a terminal sample was taken from any
animal found
dead or found to be moribund prior to euthanasia.
[190] Clinical Observations: Animals were monitored twice daily by laboratory
animal personnel near the beginning and end of each workday for abnormal
clinical signs
(such as respiratory distress, inappetence, inactivity, seizures and
moribundity) until Study
Day 14. Any rabbits that were moribund, as assessed by a highly trained life
sciences
technician, Battelle veterinarian, or Study Director, were euthanized.
1.8 Overall Survial Odds
[191] An additional statistical analysis was performed to compare the data
generated
across the three different post-exposure studies, Post-Exposure Efficacy
Experiments 1-3.
The data used in this analysis included all groups treated with IQNPA, IQNLF,
or combined
IQNPA+IQNLF where the treatment was administered 24 hours post-challenge. The
groups
included in the analysis from each experiment are set forth in Tables 20-22
below.
-57-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
Table 20: Groups Included from Post-Exposure Efficacy
Ex eriment 1
Group* Treatment
1 IQNPA (5.0 mg/kg)
2 IQNPA (2.5 mg/ kg)
3 IQNPA (1.25 mg/ kg)
4 IQNLF (15 mg/ kg)
IQNLF (7.5 mg/ kg)
6 IQNLF (3.75 mg/ kg)
7 Controls (PBS Alone)
8 IQNPA (5.0 mg/kg) + IQNLF (15 mg/ kg)
9 IQNPA (2.5 mg/kg) + IQNLF (7.5 mg/ kg)
IQNPA (1.25 mg/kg) + IQNLF (3.75 mg/ kg)
11 IQNPA (0.625 mg/kg) + IQNLF (1.88 mg/ kg)
12 IQNPA (0.3125 mg/kg) + IQNLF (0.94 mg/ kg)
Table 21: Groups Included from Post-Exposure Efficacy Experiment 2
Group* Antibody Dose Number of Animals
1 IQNLF 10.0 mg/kg IQNLF 8
2 IQNLF 7.5 mg/kg IQNLF 8
3 IQNLF 5.0 mg/kg IQNLF 8
4 IQNLF 2.5 mg/kg IQNLF 8
5 IQNLF 1.25 mg/kg IQNLF 4
6 PBS (Control) 1 mL/k 4
8 IQNPA + IQNLF 2.5 mg/kg IQNPA + 0.625 mg/kg I NLF 8
9 IQNPA + IQNLF 2.5 mg/kg IQNPA + 1.25 mg/kg IQNLF 8
10 IQNPA + IQNLF 2.5 mg/kg IQNPA + 2.5 mg/kg IQNLF 8
11 IQNPA + IQNLF 2.5 mg/kg IQNPA + 5.0 mg/kg IQNLF 8
12 PBS (Control) 1 mL/kg 8
Table 22: Groups Included from Post-Exposure Efficacy Experiment 3
(highlighted in
re
Group* Antibody Dose (mg/kg) Treatment Time (Hours post-exposure)**
...............................................................................
...............................................................................
.............................................................
...............................................................................
...............................................................................
...........................................................
...............................................................................
...............................................................................
.............................................................
XXXXXXXXXXXXX >> >>> I:NE;- >>> <>>>> >>>>>>>>>>>>>2 >>>>>>>>>>>>
I?
2 IQNPA 2.5 32
3 IQ N PA 2.5 40
...............................................................................
...............................................................................
...........................................................
...............................................................................
...............................................................................
.............................................................
...............................................................................
...............................................................................
......................................................
5 IQNLF 7.5 32
6 IQNLF 7.5 40
7 Vehicle Control 1 mL/kg 24
...............................................................................
...............................................................................
...........................................................
...............................................................................
...............................................................................
.............................................................
...............................................................................
...............................................................................
...........................................................
8 >>>:.NPA?..:: Ikil > 7<5>>>>>>>>>>>>>>>?>>>>>>>> >>>>>
9 IQNPA + IQNLF 2.5 + 7.5 32
10 IQNPA + IQNLF 2.5 + 7.5 40
11 IQNPA + IQNLF 2.5 + 7.5 48
-58-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
[192] Two logistic regression models were fitted to the survival data for all
groups
with 24 hour post-challenge vaccinations across the three studies (no
controls). The first
modeled survival against the base- 10 log-transformed IQNLF dose, an indicator
for treatment
where treatment is defined as either the IQNLF antibody or the combined IQNPA
and IQNLF
antibodies. The second modeled survival against the base- l O log-transformed
IQNPA dose,
an indicator for treatment where treatment is defined as either the IQNPA
antibody or the
combined IQNPA and IQNLF antibodies.
1.8.1 Results
[193] The results indicate that there were significant differences between
treatment
groups and controls in Experiments 1 and 2 and that there was a dose-response
in Experiment
1, but not in experiment 2. Thus, the result show that there was a significant
dose effect in
the study where both IQNPA and IQNLF dosages were variable in the combination
(Experiment 1), but not in the study where the IQNPA dose was fixed and the
IQNLF dose
was varied (Experiment 2). Importantly, the results also indicate that the
odds of survival for
animals treated with both antibodies (IQNPA+IQNLF) are significantly higher
than for
animals treated with either antibody alone. When compared to the IQNPA
antibody, the odds
of survival are 5 times higher and when compared with the IQNLF antibody, the
odds of
survival are 15 times higher. Figures 9 and 10 show the estimated logistic
regression curves
for each treatment.
-59-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
Table 23: Effects included in Logistic
Regression Model Fitted to IQNLF
Dose
Effect P-value
Intercept 0.0008*
Treatment <0.0001*
Logio(IQNLF Dose) 0.0562
* Effect significant at the 0.05 level of significance
Table 24: Summary of Odds Ratios from Logistic Regression Model
Fitted to IQNLF Dose
Treatment Group Odds Ratio P-value
Comparison
(IQNPA+IQNLF) vs IQNLF 15.327* <0.0001
Logio(IQNLF Dose) 3.268 0.0562
*Odds ratio significantly different from 1 at the 0.05 level of significance
Table 25: Effects included in Logistic
Regression Model Fitted to IQNPA
Dose
Effect P-value
Intercept 0.0751
Treatment 0.0068*
Loglo(IQNPA Dose) 0.0001*
* Effect significant at the 0.05 level of significance
Table 26: Summary of Odds Ratios from Logistic Regression Model
Fitted to IQNPA Dose
Treatment Group Odds Ratio P-value
Comparison
(IQNPA+IQNLF) vs IQNPA 4.918* 0.0068
Logio(IQNPA Dose) 30.635* <0.0001
*Odds ratio significantly different from 1 at the 0.05 level of significance
[194] Thus, the additional statistical analysis indicates that the combined
IQNPA+IQNLF antibody treatment represents a statistically significant
improvement over
both treatment with either antibody alone.
1.9 Optimization of Dose-Response
[195] The following describes a series of experiments to determine the optimal
dosages for combination therapy. Intially, dose-response experiments will be
performed to
determine the smallest amount of anti-PA humAb IQNPA that gives the highest
degree of
protection against disease progression. Another set of dose-response
experiments will then
-60-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
be performed using this optimal IQNPA dose along with varying doses of anti-LF
(IQNLF)
antibody to determine the optimal dose of IQNLF in combination with IQNPA.
Finally, a
third set of experiments will be performed to determine the optimal doses for
the combination
of IQNPA and IQNLF and Levoflaxin and to determine whether there are any
negative side
effects when the antibodies are combined with Levoflaxin. It is expected that
no adverse
effects will be manifest.
[196] All experiments will be performed in 2-3 kilogram New Zealand White
rabbits
(F&M). The rabbits will be challenged with 100 times the LD50 equivalent of
Bacillus
anthracis Ames spores (100xLD50) by inhalation or intranasal instillation.
Antibodies will
be administered via a single injection of either (i) IQNPA alone, (ii) IQNLF
alone, or (iii)
either a single injection containing both IQNPA+IQNLF or two separate
injections of IQNPA
and IQNLF alone given at about the same time. Injections will be either
subcutaneous,
intramuscular, or intravenous. In addition, as a control, Levofloxacin (7
mg/kg/d for 6
consecutive days) will be administered at the same time as exposure to anthrax
to
demonstrate that the model can be modulated with agents other than monoclonal
antibodies.
1.9.1 Example 1: Optimization of IQNPA and combination dosages
[197] Time of antibody treatment is the moment where the rabbits start to
become
symptomatic. The symptomatic phase (as measure by the presence of PA in blood
and the
presence of bacteremia) generally starts between 25 and 29 hrs after exposure.
The results of
these studies should demonstrate the added benefit of IQNLF with respect to
survival.
Experiment 1: Dose finding for IQNPA (n=8)
Group IQNPA i.v. IQNLF i.v. Time of
(mg/kg) (mg/kg) treatment
1 saline Symptomatic
2 Levofloxacin 7 mg/kg/d i.m. dO
3 2.5 0 Symptomatic
4 5 0 Symptomatic
7.5 0 Symptomatic
6 10 0 Symptomatic
7 12.5 0 Symptomatic
8 15 0 Symptomatic
Experiment 2: Use of optimal IQNPA dose (A
mg/kg) with different IQNLF dosages
Group IQNPA i.v. IQNLF i.v. Time of
(mg/kg) (mg/kg) treatment
1 saline Symptomatic
-61-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
2 Levofloxacin 7 m /k /d i.m. dO
3 A 0 Symptomatic
4 A 0.5 Symptomatic
A 1.0 Symptomatic
6 A 2.0 Symptomatic
7 A 3.0 Symptomatic
8 A 4.0 Symptomatic
9 A 5.0 Symptomatic
A 7.5 Symptomatic
11 A 10.0 Symptomatic
Experiment 3: Optimal doses of IQNPA+IQNLF (A
mg/kg + B mg/kg) with Levofloxacin
Group IQNPA i.v. IQNLF i.v. Time of Levo i.m. Start of Levo
(mg/kg) (mg/kg) treatment 7 mg/kg/d
1 saline Symptomatic - -
2 saline Symptomatic for 6 days dO
3 A B Symptomatic - -
4 A B Symptomatic for 6 days dO
1.9.2 Example 2: Combination with Levofloxacin
[198] In this example, the start of the Levofloxacin treatment is at the time
the
rabbits become symptomatic. Time of antibody treatment is the moment where the
rabbits
start to become symptomatic. The symptomatic phase (as measure by the presence
of PA in
blood and the presence of bacteremia) generally starts between 25 and 29 hrs
after exposure.
The results should indicate that no significant negative impact of the
antibiotic on the effect
to the antibodies.
[199]
Experiment 1: Dose finding IQNPA +
Levofloxacin (n=8)
Group IQNPA i.v. IQNLF i.v. Time of
m /k (mg/kg) treatment
1 saline Symptomatic
2 Levofloxacin 7 m /k /d i.m. Symptomatic
3 2.5 0 Symptomatic
4 5 0 Symptomatic
5 7.5 0 Symptomatic
6 10 0 Symptomatic
7 12.5 0 Symptomatic
8 15 0 Symptomatic
Experiment 2: Use optimal IQNPA dose (A
mg/kg) and combine with different IQNLF
dosages
Group IQNPA i.v. IQNLF i.v. Time of
m /k (mg/kg) treatment
1 saline Symptomatic
2 Levofloxacin 7 m /k /d i.m. Symptomatic
-62-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
3 A 0 Symptomatic
4 A 0.5 Symptomatic
A 1.0 Symptomatic
6 A 2.0 Symptomatic
7 A 3.0 Symptomatic
8 A 4.0 Symptomatic
9 A 5.0 Symptomatic
A 7.5 Symptomatic
11 A 10.0 Symptomatic
Experiment 3: Use optimal IQNPA+IQNLF
combination (A mg/kg + B mg/kg) and combine
with Levofloxacin
Group IQNPA i.v. IQNLF i.v. Time of Levo i.m. Start of Levo
/kg ) ~ (mg/kg) treatment 7m/k/d
1 saline Symptomatic
-
2 saline Symptomatic for 6 days Symptomatic
3 A B Symptomatic
-
4 A B Symptomatic for 6 days Symptomatic]
1.9.3 Example 3: Combination Antibodies and Levofloxacin well into
symptomatic phase (48 hrs)
[200] Time of antibody treatment is the moment 48 hrs after exposure. The
symptomatic phase (as measure by the presence of PA in blood and the presence
of
bacteremia) generally starts between 25 and 29 hrs after exposure, and 48 hrs
is well into this
symptomatic phase.
Experiment 1: Dose finding IQNPA +
Levofloxacin(n=8)
Group IQNPA i.v. IQNLF i.v. Time of
m /k (mg/kg) treatment (hrs)
1 saline +48
2 Levofloxacin 7 m /k /d i.m. +48
3 2.5 0 +48
4 5 0 +48
5 7.5 0 +48
6 10 0 +48
7 12.5 0 +48
8 15 0 +48
Experiment 2: Use optimal IQNPA dose (A
mg/kg) and combine with different IQNLF
dosages
Group IQNPA i.v. IQNLF i.v. Time of
m /k (mg/kg) treatment (hrs)
1 saline +48
2 Levofloxacin 7 m /k /d i.m. +48
3 A 0 +48
4 A 0.5 +48
5 A 1.0 +48
-63-
CA 02749572 2011-07-12
WO 2010/082134 PCT/IB2010/000146
6 A 2.0 +48
7 A 3.0 +48
8 A 4.0 +48
9 A 5.0 +48
A 7.5 +48
11 A 10.0 +48
Experiment 3: Use optimal IQNPA+IQNLF
combination (A mg/kg + B mg/kg) and combine
with Levofloxacin
Group IQNPA i.v. IQNLF i.v. Time of Levo i.m. Start of Levo
m /kg) ~ (mg/kg) treatment 7 m /k /d
1 saline +48 - -
2 saline +48 for 6 days +48
3 A B +48 - -
4 A B +48 for 6 days +48
EQUIVALENTS
[201] Those skilled in the art will recognize or be able to ascertain using no
more
than routine experimentation, many equivalents to the specific embodiments of
the invention
described herein. Such equivalents are intended to be encompassed by the
following claims.
[202] All references cited herein are incorporated herein by reference in
their
entirety and for all purposes to the same extent as if each individual
publication or patent or
patent application was specifically and individually indicated to be
incorporated by reference
in its entirety for all purposes.
[203] The present invention is not to be limited in scope by the specific
embodiments described herein. Indeed, various modifications of the invention
in addition to
those described herein will become apparent to those skilled in the art from
the foregoing
description and accompanying figures. Such modifications are intended to fall
within the
scope of the appended claims.
-64-