Note: Descriptions are shown in the official language in which they were submitted.
CA 02749797 2011-07-14
1
D E S C R I P T I O N
Title of the Invention
COMPOUND HAVING TUMOR-RESIDENT PROPERTY
Technical Field
The present invention relates to novel compound
having a tumor-resident property.
Background Art
In the medical field, MRI, PET, X-raying, CT, and
the like are used as diagnostic imaging techniques.
Methods for detection and diagnosis using these
techniques have been developed and used. In the
methods, such substances that give rise to a difference
in X-ray absorption in comparison with the surrounding
tissues, substances that have an influence on nuclear
magnetic resonance, or the like are used as a contrast
agent. Such a contrast agent provides contrast for X-
ray or MRI imaging by distribution in tissues and
generates an image of the tissue shape.
In recent years, due to MRI, PET, X-raying, CT,
and the like, not only a technique for obtaining
information of the tissue pattern based on the
aforementioned contrast but also a technique for
detecting a tumor itself has been developed. Contrast
agents used in such techniques are, for example, those
prepared by labeling molecules like glucose, which is
CA 02749797 2011-07-14
2
easily taken into carcinoma tissues whose metabolism is
enhanced compared to normal cells, by a radioactive
substance, fluorescent substance, magnetic substance,
or the like and those prepared by labeling, in the same
manner, molecules such as sugar chains specifically
existing on a carcinoma cell surface and proteins
aggregating by targeting at antigens. These contrast
agents are utilized for detection and/or imaging of
carcinoma tissues by a detector such as PET, MRI, or CT
scanner.
On the other hand, as a delivery method of a drug
to carcinoma tissues, technique of a drug delivery
system delivering by encapsulating drugs such as an
anticancer agent in minute particles or associating
therewith has been developed. Such a drug delivery
system is, for example, a system in which molecules
having an affinity with tumor tissues and molecules
preventing from being eliminated as a foreign substance
are incorporated into the particle surface.
Summary of the Invention
[PROBLEMS TO BE SOLVED BY THE INVENTION]
An object of the present invention is to provide a
novel compound which specifically resides in a tumor.
Another object of the present invention is to
provide a method for allowing the compound to reside in
a tumor.
Another object of the present invention is to
CA 02749797 2011-07-14
3
provide a method for detecting tumor with use of the
compound.
Another object of the present invention is to
provide a method for diagnosing tumor with use of the
compound.
Another object of the present invention is to
provide a method for treating tumor with use of the
compound.
[MEANS FOR SOLVING THE PROBLEMS]
The aforementioned problem can be solved by the
present invention described below.
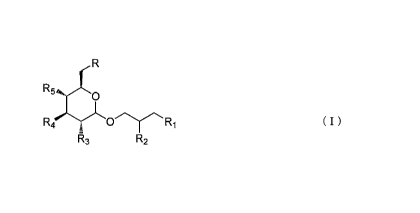
(1) A compound represented by chemical
formula (I) :
R
R5O
,
R4 O'yRi ( I )
R3 R2
wherein R is an anionic group binding to hydrogen,
R1 is OH, OCOH, OCO(CH2)hCH3, or an acting group,
h being an integer of 0 or more,
R2 is H, OH, OCOH, OCO(CH2)iCH3, or an acting
group, i being an integer of 0 or more,
R3 is OH, S03H, or an acting group,
R4 is OH, S03H, or an acting group, and
R5 is OH, S03H, or an acting group,
at least one of R1, R2, R3, R4, and R5 containing
an acting group,
CA 02749797 2011-07-14
4
or pharmaceutically acceptable salts thereof.
(2) A compound represented by chemical
formula (II):
R
e14,... O
Xk
R4 O"~--r R, (II)
R3 R2 i
wherein R is an anionic group binding to hydrogen,
R1 is OH, OCOH, OCO(CH2)hCH3, or an acting group,
h being an integer of 0 or more,
R2 is H, OH, OCOH, OCO(CH2)iCH3, or an acting
group, i being an integer of 0 or more,
R3 is OH, S03H, or an acting group,
R4 is OH, S03H, or an acting group,
R5 is OH, S03H, or an acting group,
j is an integer of 1 or more,
X is an acting group or a cationic group, and
k is an integer of 1 or more,
at least one of R1, R2, R3, R4, R5, and X
containing an acting group,
or pharmaceutically acceptable salts thereof.
(3) A method for allowing the compounds
represented by formula (I) or (II) to reside in a
tumor, wherein the compound or salts described in (1)
or (2) are administered to a subject.
(4) A method for detecting tumor, comprising:
administering the compound or salts described in
CA 02749797 2011-07-14
(1) or (2) to a subject, wherein the acting group is a
labeled substance;
detecting the labeled substance when the compound
or salts exist in a tumor in a higher concentration
5 than in other tissues except a tumor; and
detecting tumor in the subject based on results of
the detecting.
(5) A method for diagnosing tumor, comprising:
administering the compound or salts described in
(1) or (2) to a subject, wherein the acting group is a
labeled substance;
detecting the labeled substance when the compound
or salts exist in a tumor in a higher concentration
than in other tissues except a tumor; and
diagnosing presence or absence of tumor in the
subject based on results of the detecting.
(6) A method for treating tumor, comprising
administering the compound or salts described in (1) or
(2) to a subject, wherein the acting group is an
antitumor substance.
[EFFECTS OF THE INVENTION]
According to an aspect of the present invention, a
novel compound which specifically resides in a tumor is
provided.
According to an aspect of the present invention, a
method for allowing the compound to reside in a tumor
is provided.
CA 02749797 2011-07-14
6
Further, according to an aspect of the present
invention a method for detecting tumor with use of the
compound is provided.
According to another aspect of the present
invention, a method for diagnosing tumor with use of
the compound is provided.
Furthermore, according to another aspect of the
present invention, a method for treating tumor with use
of the compound is provided,
Brief Description of the Drawings
FIG. 1 is a graph showing retention of biotinated
aSQMG C18:0 in a tumor.
FIG. 2 is a graph showing retention of biotinated
aSQAP C18:0 in a tumor.
FIG. 3 is a concentration-time transition curve in
blood plasma and organs of nude mice to which aSQMG
C18:0 was administered intraperitoneally.
FIG. 4 is a concentration-time transition curve in
blood plasma and organs of nude mice to which aSQAP
C18:0 was administered intraperitoneally.
FIG. 5 is a concentration-time transition curve in
blood plasma and organs of nude mice to which aSQAP
C18:0 was administered intravenously.
The Embodiments for Carrying Out the Invention
As a result of diligent study by the present
inventors, a novel compound which specifically
distributes in a tumor was found.
CA 02749797 2011-07-14
7
According to one embodiment of the present
invention, the novel compound is a compound represented
by the following formula (I):
R
R5 O
R4 O'--r R, ( I )
R3 R2
wherein R is an anionic group binding to hydrogen,
Rl is OH, OCOH, OCO(CH2)hCH3, or an acting group,
h being an integer of 0 or more, preferably from 0 to
30, more preferably from 0 to 26, further preferably
from 1 to 22,
R2 is H, OH, OCOH, OCO(CH2)iCH3, or an acting
group, i being an integer of 0 or more, preferably from
0 to 30, more preferably from 0 to 26, further
preferably from 1 to 22,
R3 is OH, S03H, or an acting group,
R4 is OH, S03H, or an acting group, and
R5 is OH, S03H, or an acting group,
at least one of R1, R2, R3, R4, and R5 containing
an acting group, preferably any one of R1, R2, R3, R4,
and R5 containing an acting group,
or pharmaceutically acceptable salts thereof.
According to one embodiment, the compound of the
present invention may be the compound of formula (I),
wherein R is S03H,
R3 is OH,
CA 02749797 2011-07-14
8
R4 is OH,
R5 is OH,
R1 is OH, OCOH, OCO(CH2)hCH3 (an acting group),
0- (an acting group), OCO- (an acting group), or
OCO(CH2)hCH2- (an acting group), h being an integer of
0 or more, preferably from 0 to 30, more preferably
from 0 to 26, further preferably from 1 to 22, and
R2 is H, OH, OCOH, OCO(CH2)iCH3, an acting group,
0- (an acting group), OCO- (an acting group), or
OCO(CH2)iCH2- (an acting group), i being an integer of
0 or more, preferably from 0 to 30, more preferably
from 0 to 26, further preferably from 1 to 22,
at least one of R1 and R2 containing an acting
group,
or pharmaceutically acceptable salts thereof.
According to a further embodiment, the compound of
the present invention is the compound represented by
formula (I),
wherein R is S03H,
R1 is OH, OCOH, OCO(CH2)hCH3 or an acting group, h
being an integer of 0 or more, preferably from 0 to 30,
more preferably from 0 to 26, further preferably from 1
to 22,
R2 is H, OH, OCOH, OCO(CH2)iCH3, or an acting
group, i being an integer of 0 or more, preferably from
0 to 30, more preferably from 0 to 26, further
preferably from 1 to 22,
CA 02749797 2011-07-14
9
R3 = R4 = R5 is OH, or at least one of R3, R4, and
R5 is an active agent and the others are OH,
at least one of R1, R2, R3, R4, and R5 containing
an acting group,
or pharmaceutically acceptable salts thereof.
In the compound of the formula (I), when Rl and/or
R2 is OCO(CH2)hCH3 or OCO(CH2)iCH3, they may be a
linear or branched, saturated or unsaturated fatty
acid.
Further, according to a further embodiment, the
compound of the present invention may be
sulfopyranosyl(acyl)glycerol,
sufopyranosyl(acyl)propanediol, or sulfoquinovosylacyl
propanediol, wherin at least one of the side residues
binds to an acting group, or pharmaceutically
acceptable salts thereof.
In the compound of the aforementioned formula (I),
when a sugar backbone constituting pyranoside,
pyranose, exists, the pyranose may be a-D-glucose,
R-D-glucose, a-D-galactose, R-D-galactose, a-D-mannose,
R-D-mannose, and the like.
These sugar backbones of the pyranoside may be in
either configuration of a boat or a chair form, or a
mixed form. However, a chair form is more preferable
from the viewpoint of stability.
In the compound of formula (I), when there is an
asymmetrical carbon, the absolute configuration may be
CA 02749797 2011-07-14
S or R. For example, when the compound comprises
sulfopyranosyl(acyl)glycerol and an acting group, the
carbon at position 2 of the glycerol portion is an
asymmetrical carbon. Also in this case the absolute
5 configuration may be S or R, or a mixed form.
When the compound of formula (I) comprises
sulfoquinovosylacyl propanediol and an acting group,
the quinovose ring contained therein may be in a boat
or chair form, or a mixed form. However, in general a
10 chair form is preferable due to the stability.
Further, the steric configuration of the propanediol
with respect to the quinovose ring may be a-anomer or
R-anomer, or the mixed configuration.
In the compound of formula (I), examples of the
anionic group binding to hydrogen include, but not
limited to, SO3H, OSO3H, P03H2, CO2H, and the like,
preferably SO3H, OSO3H, P03H2, more preferably SO3H.
The term "acting group" used herein refers to a
group exhibiting a desired action of delivering to a
tumor. The compound of formula (I) may contains, but
not limited to, an active substance such as a labeled
substance and an antitumor substance as an acting
group. The active substance is maintained by a
covalent bond in any position in the compound
represented by formula (I). The acting group may be
contained in one or more in one molecule of the
compound of formula (I). When two or more acting
CA 02749797 2011-07-14
11
groups exist in one molecule of the compound of
formula (I), these acting groups may be identical or
different from each other or one another.
The acting group may be one of a coupled pair
which is capable of specifically coupling to the other.
Examples of such a coupled pair include, but not
limited to, biotin and avidin, an antigen and a
specific antibody against the antigen, and an antigen
and a specific receptor with respect to the antigen.
In the case of such a compound, the compound of the
present invention comprising one of the coupled pair as
an acting group is administered to a subject and then
the other of the pair, with which a drug or labeled
substance, is associated is administered to the subject
after desired time interval, thereby specifically
delivering the drug or labeled substance to tumor.
The acting group may or may not include a linker
for linking the active substance and a glycolipid
portion.
The term "labeled substance" used herein is a
marker which is used in technique for diagnosing tumor
utilizing imaging technique which is generally used in
noninvasive diagnosis. The labeled substance may be
any known vital-stainable labeled substance. For
example, such a labeled substance includes, but not
limited to, a radioactive substance, paramagnetic
metal, radiopaque metal, and pigment.
CA 02749797 2011-07-14
12
Examples of the radioactive substance include, but
not limited to, 11C, 13N, 150, 18F, 58Co, 59Fe, 62Cu,
64Cu, 67Ga, 68Ga, 75Br, 76Br, 77Br, 82Br, 89Sr, 90Y,
1111, 113Sn 117mSn, 153Sm, 165Dy, 166Ho, 169Er, 186Re,
201T1, 212Bi, and 213Bi.
Examples of the paramagnetic metal include, but
not limited to, Cr (III), Mn (III), Fe (II), Fe (III),
Pr (III), Nd (III), Sm (III), Yb (III), Gd (III), Tb
(III), Dy (III), Ho (III), and Er (III).
Examples of the radiopaque metal include, but are
not limited to, I, Bi, W, Ta, Hf, La, Ln, Ba, Mo, Nb,
Zr, Sr, and the like.
Examples of the pigment may include, but not
limited to, a known pigment for vital staining, for
example, a pigment such as fluorescent pigment. For
example, the fluorescent pigment includes those having
the following skeleton as a fluorescent chromophore
group: naphthalene, anthracene, quinoline, acridine,
coumarin, salicylic acid, anthranilic acid, NBD
(7-nitrobenz-2-oxa-1,3-diazole), stilbene, resorufin,
BODIPY (4,4-difluoro-4-bora-3a,4a-diaza-s-indecene),
carbocyanine, and thiacarbocyanine,
tetramethylindocarbocyanine. Examples of the
fluorescent pigment include the substances disclosed in
R. P. Haugland, Molecular Probes Handbook, 6th edition.
The term `antitumor substance' used herein may be
a substance which attacks tumor by cytotoxicity.
CA 02749797 2011-07-14
13
Therefore, the antitumor substance may be any known
substance which is used for treating a tumor. The
antitumor substance may be, for example, but not
limited to, an alkylating agent, antimetabolite,
anticancer antibiotic, metallic complex, plant
alkanoids, topoisomerase inhibitor, microtubular
polymerization inhibitor, microtubular depolymerization
inhibitor, molecular target drug, and hormonal agent.
Examples of the antitumor substance include, but not
limited to, Aceglatone, Aclarubicin hydrochloride,
Actinomycin D (referred to also as Dactinomycin),
Amrubicin hydrochloride, Anastrozole, Arsenic trioxide,
Asparaginase (referred to also as L-Asparaginase),
Bicalutamide, Bleomycin hydrochloride, Bortezomib,
Buslufan, Capecitabine, Carboplatin, Carboquone,
Carmofur, Celmoleukin, Cisplatin, Cladribine,
Cyclophosphamide, Cytarabine, Dacarbazine, Daunorubicin
hydrochloride (referred to also as Daunomycin),
Docetaxcel, Doxifluridine, Doxorubicin hydrochloride
(referred to also as Adriamycin), Enocitabine,
Epirubicin hydrochloride, Erlotinib hydrochloride,
Estramustine phosphate sodium hydrate, Etoposide,
Exemestane, Fadrozole hydrochloride hydrate,
Fludarabine phosphate, Fluorouracil, Flutamide,
Gefitinib, Gemcitabine hydrochloride, Goserelin
acetate, Hydroxycarbamide (refered to also as
Hydroxyurea), Idarubicin hydrochloride, Ifosfamide,
CA 02749797 2011-07-14
14
Imatinib mesylate, Irinotecan hydrochloride, Letrozole,
Leuprorelin acetate, Medroxyprogestrone acetate,
Meiphalan, Mepitiostane, Mercaptopurine hydrate,
Methotrexate, Mitomycin C, Mitotane, Mitoxantrone
hydrochloride, Nedaplatin, Nelarabine,
Neocarzinostatine, Nimustine hydrochloride, Octreotide
acetate, Oxaliplatin, Paclitaxel, Pemetrexed disodium,
Pentostatin, Peplomycin sulfate, Pirarubicin, Porfimer
sodium, Procarbazine hydrochloride, Ranimustine,
Sizofiran, Sobuzoxane, Sorafenib tosylate, Talaporfin
sodium, Tamibarotene, Tamoxifen citrate, Teceleukin,
Tegafur, Temozolomide, Thiotepa, Topotecan
hydrochloride (referred to also as Nogitecan
hydrochloride), Toremifene citrate, Tretinoin,
Ubenimex, Vinblastine sulfate, Vincristine sulfate,
Vindesine sulfate, Vinorelbine ditartrate, and
Zinostatin stimalamer.
The active substance may be any known substance
which is used for treating tumor. For example, it may
be a thermal neutron capturing substance, and examples
thereof include compounds comprising an element such as
B and Gd.
The compound according to the present invention
may be pharmaceutically acceptable salts of the
compound represented by formula (I). Such salts may be
those obtained by binding an ionic group generated by
ionization of at least one of the side residues
CA 02749797 2011-07-14
represented by R, R1, R2, R3, R4, and/ or R5 of the
compound of formula (I) and an ion which is capable of
binding to the ionic group through an ionic bond.
Further, the ionized compound of formula (I) may bind
5 to the ionized substance with the ratio of one molecule
with respect to one molecule, that is, 1 : 1, or n : m.
Here, m and n are an integer of 1 or more. For
example, the salt may consist of plural molecules of
the ionized compounds of formula (I) and one molecule
10 of an ionic substance. The salt may consist of one
molecule of the ionized compound of formula (I) and
plural molecules of the ionic substances.
When the ionized compound of formula (I) has
negative charge, the ionic substance forming a pair
15 therewith may be a cation having positive charge. The
cation may be a metal ion including monovalent metal
ions such as Na+ and K+ and a divalent metal ion such
as Ca2+. For example, the cation may be, but not
limited to, lithium, sodium, potassium, calcium,
magnesium, manganese, iron, zinc, copper, strontium,
lead, silver, barium, aluminum, chromium, cobalt,
nickel, ammonium, monoalkylammonium, dialkylammonium,
trialkylammoniumu, tetraalkylammonium,
monohydroxyalkylammonium dihydroxyalkylammonium,
trihydroxyalkylammonium, and tetrahydroxyalkylammonium,
and the like.
Further, the compound according to the present
CA 02749797 2011-07-14
16
invention may be a complex. Such a complex is a
compound represented by the following formula (II):
R
Xk
[::OR1] N.. O
II
R3 R2
wherein R, R1, R2, R3, R4, and R5 are as defined
in formula (I),
j is an integer of one or more, and
X is an acting group or a cationic group, and
k is an integer of one or more,
preferably at least one of R, R1, R2, R3, R4, R5,
and X being an acting group,
or pharmaceutically acceptable salts thereof.
Here, the cationic group may be a cation having
positive charge. The cationic group may be a metal ion
including monovalent metal ions such as Na+ and K+ and
a divalent metal ion such as Ca2+. For example, the
cationic group may be, but not limited to, lithium,
sodium, potassium, calcium, magnesium, manganese, iron,
zinc, copper, strontium, lead, silver, barium,
aluminum, chromium, cobalt, nickel, ammonium,
monoalkylammonium, dialkylammonium, trialkylammoniumu,
tetraalkylammonium, monohydroxyalkylammonium,
dihydroxyalkylammonium, trihydroxyalkylammonium, and
tetrahydroxyalkylammonium.
When X of formula (II) contains an acting group,
CA 02749797 2011-07-14
17
the acting group may be a similar active substance to
those defined in formula (I), and in this case the
active substance and glycolipid may bind to each other
through a coordinate bond.
The compounds of formulae (I) and (II) and
pharmaceutically acceptable salts thereof can be
synthesized by utilizing a known reaction pathway.
Nonexclusive examples of the compound according to
the present invention are described below as compounds
of formulae (1)-(13).
0
SO3Na
0
N'" ~-
H NO OH (1)
HO 0" OCOC17H35
0 0
N/ SO3Na
0 (2)
H HO OH
HOO`\OCOC17H35
0
N~' \ SONa
H OCOC H
17 35
O-"OCOC17H35
(3)
0 0
5O3Na
0
H HO 0000 H
17 35
W*
I HO0-, OCOC17H35
(4)
CA 02749797 2011-07-14
18
SO3Na
0 0
H0 0
H 0`OGOC17H35 (5)
I ~ 1
1 ~
SO3Na
0 0
HT10 0
HO 0``OCOC17H35 (6)
SO3Na
0
H NO OCOC17H
Hoo"~035\ J I (~ )
0
S03Na
O I I
H HO OCOC H35
HO 0,-. o
O I (8)
SO3Na
0
HOHO 18F
OH
0 OCOC17H35 (9)
SO3Na
0
HO HO OH
185 0 OCOG17H35 (10)
CA 02749797 2011-07-14
19
S03Na
HO 0
HO OCOC H
OH 17 35
O 18F
(11)
H
HN jNa
0 H ' H 0 H N 0
HOO\~/OCOC17H35 (12)
H
HN O 0
SO3Na
0 H H 0 Him ~\p0 0
OH
HOO'- iOCOC,,H35 (13)
The compound according to the present invention
exhibits a resident property for a long period in tumor
tissues in comparison with other tissues. Namely,
other tissues and tumor tissues greatly differ from
each other in the distribution concentration.
Therefore, administration of the compound according to
the present invention, that is, the compound of
formula (I), the pharmaceutically acceptable salts
thereof, or the compound of formula (II) to the subject
allows the compound of formula (I) or formula (II) or
the salts reside therein. Thus, according to the
present invention, a method for allowing the compound
of formula (I), formula (II), or pharmaceutically
acceptable salts thereof reside in a tumor is also
provided.
As is described above, the extinction time of the
CA 02749797 2011-07-14
compound according to the present invention in the
subject differs between a tumor tissue and other
tissues, and the compound resides for a longer period
in a tumor in comparison with other tissues. Namely,
5 the extinction time of the compound in a tumor is
longer than that in other tissues except a tumor.
Further, the compound contains an acting group in
the structure. Thus, the compound enables detection,
diagnosis, and treatment of tumor by taking advantage
10 of the effect exhibited by the tumor-resident property
and the acting group of the compound. In other words,
the compound according to the present invention can be
utilized as diagnostic agent, therapeutic agent, and
contrast agent for tumor
15 When tumor is detected, a labeled substance may be
selected as the acting group. When the compound or the
salts exist in a higher concentration in a tumor than
in other tissues except a tumor after administration of
the compound or the pharmaceutically acceptable salts
20 containing a labeled substance to a subject, it is
possible to detect the labeled substance and, based on
the detection results, presence or absence of tumor in
the subject and/or portion and tissues having tumor.
Similarly, when a labeled substance is selected as
the acting group, it is possible, to diagnose presence
or absence of tumor in the subject and/or to diagnose
which tissue has tumor, based on detection results by
CA 02749797 2011-07-14
21
detecting the labeled substance at the time when the
compound or pharmaceutical acceptable salts thereof
exist in a higher concentration in tumor than in other
tissues except a tumor, after administering the
compound or the salts containing the labeled substance.
The detection of the labeled substance may be
carried out by any known method in accordance with the
type of the labeled substance contained as an acting
group.
For example, when the labeled substance is a
radioactive substance, the labeled substance may be
detected by a diagnostic imaging method such as PET and
SPECT and/or a detection method using a gamma probe.
In this case the compound according to the present
invention may be recognized as a radioactive diagnostic
agent.
When the labeled substance is a paramagnetic
material, the labeled substance may be detected by a
known method utilizing magnetic nuclear resonance such
as MRI. In this case, the compound according to the
present invention may be recognized as a nuclear
magnetic resonance diagnostic agent.
When the labeled substance is a radiopaque
substance, the labeled substance may be detected by
either known CT or X-ray photography. In this case,
the compound according to the present invention may be
recognized as an X-ray contrast agent.
CA 02749797 2011-07-14
22
When the labeled substance is a fluorescent
pigment, the labeled substance may be detected by a
known fluorescent detection method. In this case, the
compound according to the present invention may be
recognized as a fluorescent diagnostic agent.
Further, the detection may be carried out by a
noninvasive means, but not limited to the above-
mentioned detection means. Here, noninvasive means may
be those which can be carried out without surgical
procedure to the subject. From the viewpoint of QOL of
the subject, it is desirable to detect a labeled
substance by such a noninvasive means.
Use of an antitumor substance as the active agent
enables treatment of tumor. Such a method may comprise
administrating the compound of formula (I) or (II) or
pharmaceutically acceptable salts thereof to a subject.
The compound according to the present invention which
is used for such a method can be recognized as an
anticancer agent or an antitumor agent.
Further, also when the substance contained as the
acting group is a thermal neutron capturing substance,
treatment of tumor can be carried out. Such a method
may comprise administering the compound of formula (I)
.or (II) or pharmaceutically acceptable salts thereof
and irradiating neutrons at the time when the compounds
or salts exist in a higher concentration in a tumor
than tissues except a tumor. The neutron irradiation
CA 02749797 2011-07-14
23
can be carried out by any known means. The compound
according to the present invention which is used for
such a method may be recognized as a cancer treatment
agent or a tumor treatment agent.
The conditions of the aforementioned radiation
irradiation and administration of the compound
according to the present invention, as is well-known in
the field of radiation therapy, can be selected
appropriately by healthcare professionals and other
professionals, depending on the type of radiation
source, radiation method, radiation part, and radiation
time; the type of the compound, administration route,
and administration timing; type and severity degree of
diseases to be treated; age, body weight, health
condition, and clinical record of the subject to be
radiated; and the like.
The term `subject' used herein means humans;
animals other than humans, for example, mammals such
as, cats, dogs, horses, cattle, sheep, mice, rats, and
rabbits; reptiles; amphibians; fish; and birds.
As is described above, the compound according to
the present invention can be used as an active
ingredient such as a diagnostic agent, therapeutic
agent, and contrast agent. The compound can be
administered, for example, orally and parenterally.
Further, the compound can be combined with an
appropriate excipient, diluent, or the like which is
CA 02749797 2011-07-14
24
pharmaceutically acceptable in accordance with the
administration route thereof, thereby making a
pharmaceutical preparation.
Examples of dosage forms suitable for oral
administration include, but not limited to, solid,
semi-solid, liquid, and gas forms, and specific
examples of thereof include tablets, capsules, powders,
granules, solutions, suspending agents, syrups, and
elixirs.
For manufacture and formulation of the compound
into tablets, capsules, powders, granules, solutions,
suspending agents, and the like, the compound may be
mixed to a binder, tablet disintegrant, lubricant
agent, and the like, and further, according to need,
mixed to diluents, buffers, wetting agents,
preservation agents, flavoring agents, and the like
with use of a known method. For example, the binder
includes crystalline cellulose, cellulose derivatives,
cornstarch, and gelatin. The tablet disintegrant
includes, for example, cornstarch, potato starch, and
carboxymethylcellulose sodium. The lubricant agent
includes, for example, talc, and magnesium stearate.
Conventionally-used additives such as lactose and
mannitol may be further used.
Further, the compound in the form of liquid or
minute powder may be filled in a non-pressurized
container such as an aerosol container and nebulizer
CA 02749797 2011-07-14
together with gaseous or liquid air spray or, according
to need, together with a known auxiliary agent such as
a moistening agent, to administer it in the form of an
aerosol agent or inhalant. As the air spray,
5 pressurized gas such as dichlorofluoromethane, propane,
and nitrogen may be used.
When a pharmaceutical preparation containing the
compound as an active ingredient is parenterally
administered, it may be given by, for example, rectal
10 administration, or injection.
When it is given by rectal administration, it may
be administered as a suppository. With respect to the
suppository, the pharmaceutically active substance can
be mixed to an excipient such as cacao butter, carbon
15 wax, and polyethylene glycol which are melted at a body
temperature but are solidified at room temperature and
are formed by a known method, thereby making a
preparation.
Administration by injection may be conducted
20 through, for example, hypodermic, intradermal,
intravenous, or intramuscular injection. With respect
to these preparations for injection, the compound of
the present invention is dissolved, suspended, or
emulsified in an aqueous or non-aqueous solvent such as
25 plant oil, synthetic resin acid glyceride, higher fatty
acid esters, and propylene glycol, thereby making a
preparation, if desired, together with a conventionally
CA 02749797 2011-07-14
26
used additive such as a solubilizing agent,
osmoregulatory agent, emulsifying agent, stabilizing
agent, and preservative.
In order to make the compound according to the
present invention into a form such as solution,
suspension, syrup, and elixir, a pharmaceutically
acceptable solvent such as injectable sterilized water
and normal saline solution may be used.
The compound according to the present invention
may be also combined with a pharmaceutically acceptable
compound having other activity, thereby making a
pharmaceutical preparation.
The pharmaceutical preparation according to the
present invention may be appropriately established and
adjusted according to the dosage form, administration
route, tumor type and location to be detected, and
degree and stage of diseases to be treated. For
example, the effective dose of the compound according
to the present invention may be, but not limited to,
from 0.01 to 1000 mg/kg body weight per day via oral
administration, from 0.01 to 500 mg/kg body weight per
day via injection, and from 0.01 to 500 mg/kg body
weight per day via rectal administration.
The compound according to the present invention
can be used for treating tumor. Examples of tumor
include, but not limited to: neurogenic tumor such as
cerebral tumorquamous cell carcinoma and adenocarcinoma
CA 02749797 2011-07-14
27
such as head and neck cancer, skin cancer, esophagus
cancer, thyroid cancer, stomach cancer, lung cancer,
gellbladder cancer, biliary tract cancer, pancreas
cancer, liver cancer, prostate cancer, uterus cancer,
ovarian cancer, breast cancer, kidney cancer, bladder
cancer, and colon cancer; and melanoma, osteoma, soft
tissue tumor, and lymphoma, leukemia, and myeloma. The
term "treatment" used herein refers to the reduction,
destruction, and/or inhibition of enhancement of the
above-described tumor.
The pharmaceutical preparation according to the
present invention may contain, as an active ingredient,
an effective dose of at least one or more selected from
the group consisting of the compounds represented by
formula (I) and formula (II) and pharmaceutically
acceptable salts thereof. The pharmaceutical
preparation may contain plural kinds of different
compounds among the compounds according to the present
invention. In addition, the compound according to the
present invention may be combined with other
radiosensitizer, and antitumor agent, or other
substances having pharmacological activity and/or
pharmaceutical activity without affecting its activity.
The sulfoquionovosylacyl propanediol, an example
of glycolipid portion contained in the compound
according to the present invention, may be hereinafter
referred to as "SQAP." In a term "aSQAP Cp:q", "a"
CA 02749797 2011-07-14
28
represents an a anomer, and "Cp:q" represents that the
number of carbon atoms contained in the R1 group of
SQAP is "p", and the number of double bonds is "q",
wherein "p" is an integer of 1 or more, and "q" is an
integer of 0 or more. Accordingly, for example, "aSQAP
C18:0" represents an a anomer of sulfoquinovosylacyl
propanediol wherein the number of carbon atoms
contained in R1 of the compound is 18, and the number
of double bonds is 0.
Further, the sulfoquionovosylacylglycerol, an
example of glycolipid portion contained in the compound
according to the present invention, may be hereinafter
referred to as "SQMG." In a term "aSQMG Cris", "a"
represents an a anomer, and "Cr:s" represents that the
number of carbon atoms contained in the R1 group of
SQMG is "r", and the number of double bonds is "s",
wherein "r" is an integer of 1 or more, and "s" is an
integer of 0 or more. Accordingly, for example, "aSQMG
C18:0" represents an a anomer of
sulfoquinovosylacylglycerol wherein the number of
carbon atoms contained in R1 of the compound is 18, and
the number of double bonds is 0.
EXAMPLES
Example 1. Synthesis examples
Synthesis Example 1
The steps for preparing the sulfopyranosyl
glycerol derivative according to the present invention
CA 02749797 2011-07-14
29
is described in the following scheme 1 taking, as an
example, a-sulfoquinovosylacylgrlycerol biotin
derivative, which may be referred to as biotinated
aSQMG hereinafter.
CA 02749797 2011-07-14
_ 1T.4
I"'1 CL
f \/
N 0
fo
0 0
f3=
0
ZT
z o
< t U+ t,
W
I?
O Q
f293 C= 0
qC,
0 0 0 p
F
Cl)
i
CA 02749797 2011-07-14
31
0
T 0 o -~
1 T
w z o
0
0 ~.
O tta 0
cn 0 N
in
0
0
0 00
zx
0
0
0
0
i
0-z
0
0
of
r' f r, o Y z
O 0 z=
m o 0
0 d
Q 0 0
Fc
Z= Z3: o
0
C)
0
CA 02749797 2011-07-14
32
Each of the steps is described below in detail.
Route Al: 1-0-allyl-4,6-0-benzylidene-a-D-
glucopyranoside (1-2)
The compound (1-1) (100 g, 555 mmol) was suspended
in allyl alcohol (500 mL), to which
trifluoromethanesulfonic acid (1.00 mL) was added at
0 C, and the reaction liquid was vigorously stirred at
80 C for 48 hours. After the sufficient progress of
the reaction was confirmed, triethylamine (3 mL) was
added to stop the reaction, and the reaction liquid was
concentrated under reduced pressure. Subsequently, the
residue was suspended in anhydrous acetonitrile
(500 mL), to which benzaldehyde dimethyl acetal (127 g,
1.5 equivalents) and p-toluenesulfonic acid monohydrate
(5.28 g, 0.05 equivalent) were added. The reaction
liquid was stirred at 40 C for 4 hours, to which
triethylamine (10 mL) was added to stop the reaction,
and the reaction liquid was concentrated under reduced
pressure. The residue was poured to hexane (2000 mL)
and water (500 mL), and the mixed liquid was vigorously
stirred. The generated precipitate was collected by
filtration, and rinsed with water and hexane. The
precipitate was crystallized from heated ethanol twice
to obtain the title compound (1-2) in the form of
colorless needle crystals {34.5 g (112 mmol), 20.2%
yield}.
LRMS 331 m/Z (M+Na)+.
CA 02749797 2011-07-14
33
Route B1; 1-0-allyl-2,3-di-0-benzyl-4,6-0-
benzylidene-a-D-glucopyranoside (1-3)
To a solution of the compound (1-2) (30.0 g,
97.3 mmol) in anhydrous N,N-dimethylformamide (DMF,
300 mL), added were benzyl bromide (41.6 g, 2.5
equivalents) and sodium hydroxide (11.7 g, 3.0
equivalents), and the reaction liquid was vigorously
stirred at room temperature for 24 hours. After the
sufficient progress of the reaction was confirmed, the
reaction liquid was poured to chilled water (900 mL),
and extracted with ethyl acetate (3 x 300 mL). The
organic layers were combined and washed with saturated
saline (2 x 100 mL), dried with sodium sulfate,
filtered, and concentrated under reduced pressure. The
obtained residue was crystallized from heated ethanol
twice to obtain the title compound (1-3) in the form of
colorless needle crystals (33.5 g). The filtrate was
concentrated, purified with silica gel chromatography
(hexane-ethyl acetate, 15:1 - 10:1 - 8:1), crystallized
from heated ethanol to obtain the compound (1-3)
(6.63 g) {40.1 g (82.1 mmol) in total, 84.4% yield}
LRMS 511 m/Z (M+Na)+.
Route Cl; 1-0-allyl-2,3-di-0-benzyl-a-D-
glucopyranoside (1-4)
The compound (1-3) (15.6 g, 32.0 mmol) was
dissolved in acetic acid (90 mL). Subsequently,
distilled water (50 mL) was further added to the
CA 02749797 2011-07-14
34
solution, and the mixture was stirred for 1 hour under
heating and reflux. After the sufficient progress of
the reaction was confirmed, the reaction liquid cooled
to at room temperature, and the solvent was removed by
evaporation. Distilled water (15 mL) was then added to
the solution, and further concentrated under reduced
pressure four times. The concentrate was purified with
silica gel flash chromatography (hexane-ethyl acetate,
4:1 - 2:1 - 1:1 - 1:2) to obtain the title compound
(1-4) {12.1 g (30.2 mmol), 94.5% yield}.
LRMS 423 m/Z (M+Na)+.
Route D1 D;l-O-allyl-2,3,4-di-O-benzyl-6-0-tosyl-
a-D-glucopyranoside (1-5)
To a solution of the compound (1-4) (12.1 g,
30.2 mmol) in anhydrous pyridine (120 mL), added were
p-toluenesulfonyl chloride (7.5 g, 39.3 mmol) and 4-
dimethylaminopyridine (369 mg, 3.0 mmol), and the
reaction liquid was stirred at 0 C for 16 hours. After
the sufficient progress of the reaction was confirmed
the reaction liquid was then poured slowly into iced
water (100 mL), and the aqueous layers were extracted
with ethyl acetate (3 x 200 mL). The organic layers
were combined, washed with 1N HC1 solution until pH
becomes 4, washed with a saturated sodium hydrogen
carbonate solution (2 x 100 mL) and saturated saline
(2 x 100 mL), dried with sodium sulfate, filtered, and
concentrated under reduced pressure. The concentrate
CA 02749797 2011-07-14
was purified with silica gel flash chromatography
(hexane-ethyl acetate, 6:1 4:1 2:1) to obtain the
title compound (1-5).
{15.3 g (27.6 mmol), 91.4% yield}.
5 LRMS 577 m/Z (M+Na)+.
Route El; 1-O-allyl-2,3-di-O-benzyl-4-0-
(carbobenzoxy-R-alanyl)-6-0-tosyl-a-D-glucopyranoside
(1-6)
The compound (1-5) (15.3 g, 27.6 mmol), N-
10 carbobenzoxy-(3-alanine(12.32g, 55.2mmol), 1-ethyl-3-(3-
dimethylaminopropyl)carbodiimide hydrochloride salt
(EDCI=HCI) (15.8 mg, 82.8 mmol), and 4-
dimethylaminopyridine (6.7 g, 55.2 mmol) were dissolved
in the mixed solution of anhydrous dichloromethane
15 (200 mL) and anhydrous pyridine (50 mL) and reacted at
room temperature for 18 hours. After the sufficient
progress of the reaction was confirmed, water (10 mL)
was poured into the reaction liquid to stop the
reaction, and the solution was concentrated under
20 reduced pressure. The obtained residue was purified
with silica gel chromatography (hexane-ethyl acetate,
4:1 3:1 2:1 3:2) to obtain the title compound
(1-6) [17.1 g (22.5 mmol), 81.5% yield].
LRMS 782 m/z (M+Na)+.
25 Route Fl; 1-0-allyl-2,3-di-0-benzyl-4-0-
(carbobenzoxy-3-alanyl)-6-0-thioacetyl-a-D-
glucopyranoside (1-7)
CA 02749797 2011-07-14
36
To a solution of the compound (1-6) (17.1 g,
22.5 mmol) in anhydrous N,N-dimethylforamide (300 mL),
added was potassium thioacetate (5.1 g, 45.0 mmol), and
the mixture was stirred at 90 C for 3 hours. After the
sufficient progress of the reaction was confirmed, the
reaction liquid was poured to chilled water (400 mL),
and extracted with ethyl acetate (3 x 150 mL). The
organic layers were combined and washed with
saturated saline (2 x 100 mL), dried with sodium
sulfate, filtered, and concentrated under reduced
pressure. The obtained residue was purified with
silica gel chromatography (hexane-ethyl acetate,
4:1 - 3:1 - 2:1 -, 3:2) to obtain the title compound
(1-7) {14.2 g (20.3 mmol), 90.0% yield).
LRMS 686 m/Z (M+Na)+.
Route G1; 3-0-[2,3-di-O-benzyl-4-0-(carbobenzoxy-
R-alanyl)-6-thioacetyl-a-D-quinovopyranosyl]-glycerol
(1-8)
The compound (1-7) (14.2 g, 20.4 mmol) was
dissolved in t-butylalcohol : distilled water = 4 : 1
solution (200 mL). To the solution, added were 0.04 M
osmium tetraoxide-t-butyl alcohol (3 mL) and
trimethylamine N-oxide (3.4 g, 30.5 mmol), stirred with
a stirrer at room temperature for 24 hours. After the
sufficient progress of the reaction was confirmed, 3 g
of actived charcoal was added and stirred for
minutes to adsorb the catalyst, and the catalyst was
CA 02749797 2011-07-14
37
removed by suction filtration through celite in
Kiriyama funnel, followed by three times of washing of
the remained reaction product on the celite with ethyl
acetate and collecting thereof. Distilled water
(200 mL) was added to the collected filtrate and
extracted with ethyl acetate (3 x 150 mL). The organic
layers were combined and washed with saturated saline
(2 x 100 mL), dried with sodium sulfate, filtered,
and concentrated under reduced pressure. The
obtained residue was purified with silica
gel chromatography (hexane-ethyl acetate,
2:1 - 1:1 - 2:3 - 1:2 - 1:4 - 1:8) to obtain the title
compound (1-8) {13.2 g (18.9 mmol), 93.0% yield}.
LRMS 720 m/Z (M+Na)+.
Route H1; 3-0-[2,3-di-0-benzyl-4-0-(carbobenzoxy-
R-alanyl)-6-thioacetyl-a-D-quinovopyranosyl]-1-0-
stearoyl-glycerol (1-9)
To a mixed solution of anhydrous dichloromethane
(200 mL) and anhydrous pyridine (50 mL) of the compound
(1-8) (13.1 g, 18.8 mmol), added were stearoylchloride
(8.5 g, 28.2 mmol), and the mixture was stirred at room
temperature for 2 hours. After the sufficient progress
of the reaction was confirmed, methanol (5 mL) was
added to stop the reaction, and concentrated under
reduced pressure. The residue was suspended in a minor
amount of ethyl acetate, poured to water (200 mL), and
extracted with ethyl acetate (3 x 100 mL). The organic
CA 02749797 2011-07-14
38
layers were combined, washed with saturated saline
(2 x 100 mL), dried with sodium sulfate, filtered, and
concentrated under reduced pressure. The obtained
residue was purified with silica gel chromatography
(hexane-ethyl acetate, 6:1 -+ 4:1 - 2:1 - 3:2 - 1:1) to
obtain the title compound (1-9) in the form of a
colorless oily substance {7.1 g (7.4 mmol), 39.4%
yield}.
LRMS 987 m/Z (M+Na)+.
Route I1; 3-O-[2,3-di-O-benzyl-4-0-(carbobenzoxy-
R-alanyl)-6-sulfo-a-D-quinovopyranosyl]-1-0-stearoyl-
glycerol (1-10)
To a solution of the compound (1-9) (7.1 g,
7.4 mmol) in acetic acid (160 g, 4 mol), added were
Oxone (18.1 g, 29.5 mmol) and potassium acetate
(4.0 g), and the mixture was vigorously stirred at room
temperature for 48 hours. After the sufficient
progress of the reaction was confirmed, the reaction
liquid was poured to a chilled 7.5 M sodium hydroxide
solution (500 mL), and extracted with ethyl acetate
(4 x 100 mL). The organic layers were combined, washed
with saturated sodium hydrogen carbonate solution
(2 x 100 mL) and saturated saline solution
(2 x 100 mL), dried with sodium sulfate, filtered,
and concentrated under reduced pressure. The
obtained residue was purified with silica
gel chromatography (chloroform-methanol,
CA 02749797 2011-07-14
39
100:1 - 50:1 - 20:1 - 15:1 -+ 12:1) to obtain the title
compound (1-10) in the form of a colorless waxy
substance {5.2 g (5.24 mmol), 71.2% yield}.
LRMS 968 m/Z (M-Na)-.
Route J1;
3-0-[4-0-(3-alanyl)-6-sulfo-a-D-quinovopyranosyl]-
1-0-stearoyl-glycerol (1-11)
To a solution of the compound (1-10) (490 mg,
494 pmol) in methanol (20 mL) and acetic acid (0.6 mL),
added was 10% palladium-activated carbon (300 mg), and
the mixture was stirred at room temperature for
48 hours in a hydrogen gas atmosphere. After the
sufficient progress of the reaction was confirmed,
palladium-activated carbon was filtrated off, and the
filtrate was concentrated under reduced pressure. The
obtained residue was purified with silica gel
chromatography (chloroform-methanol-distilled water,
100:25:3 - 65:25:4 65:35:5 - 65:45:6) to obtain the
title compound (1-11) {221 mg (327 pmol), 66.1% yield}.
LRMS 654 m/Z (M-Na)-.
Route K1; 3-O-[4-0-(biotinyl-3-alanyl)-6-sulfo-a-
D-quinovopyranosyl]-1-0-stearoyl-glycerol (1-12)
The compound (1-11) (273.2 mg, 403 pmol) was
dissolved in a mixture solution of anhydrous N,N-
dimethylformamide (20 mL) and triethylamine (1 mL), and
biotion-p-nitrophenylester (162 mg, 443 pmol) was added
and reacted at room temperature for 24 hours while
CA 02749797 2011-07-14
stirring with a stirrer. After the sufficient progress
of the reaction was confirmed, toluene and methanol
were added and the solvent was distilled away and
concentrated under reduced pressure while azeotropy.
5 The obtained residue was purified with silica gel
chromatography (chloroform-methanol-distilled water,
40:10:1.2 - 70:30:2.6 - 30:10:1.5 -4 65:25:4) to obtain
the title compound (1-12) {320.8 mg (355 pmol), 88.0%
yield}.
10 LRMS 880 m/Z (M-Na)-.
Synthesis Example 2
A process for producing the sulfopyranosylacyl
propanediol derivative according to an embodiment of
the present invention is illustrated in the following
15 scheme 2, giving an a-sulfoquinovosylacyl propanediol
biotin derivative, which may be referred to as
biotinated aSQAP hereinafter, as an example thereof:
CA 02749797 2011-07-14
41
04
co LL
N
tV
0 0 0
a o
0
4 0 0
m
o00
oo :c 0o0
m ~- =
CL
Q U w
CV
c%j
0
0 o
0
N o 0 0
co
o o
am
o o )0Ta
0c
zor ~-c
CA 02749797 2011-07-14
42
N
z
z .^~
N
o
o o U
co N
N N
z z
p 0 cn >=O
ftcj 4 n z z
m
o m QOI
zm zz
N
o
~ zz
z
(~ N
0 0 >z=0 o
N N
o Q a o Q Q
4 Fa m
~a o ~
dcm 4 cm E I
m zm
CA 02749797 2011-07-14
43
The following will describe a synthesis example
through individual steps in detail:
<Synthesis Example>
Route A2; 1-0-allyl-4,6-0-benzylidene-a-D-
glucopyranoside (2-2):
Trifluoromethanesulfonic acid (1.00 mL) was added
to a liquid suspension of D-glucose (2-1) (100 g,
555 mmol) in allyl alcohol (500 mL) at 0 C, and the
reaction liquid was vigorously stirred at 80 C for
48 hours. After the sufficient progress of the
reaction was confirmed, thereto was added triethylamine
(3 mL) to terminate the reaction. The resultant was
concentrated under reduced pressure. Next, the residue
was suspended in anhydrous acetonitrile (500 mL), and
thereto were added benzaldehydedimethylacetal (127 g,
1.5 equivalent) and p-toluenesulfonic acid monohydrate
(5.28 g, 0.05 equivalent). The reaction liquid was
stirred at 40 C for 4 hours, and then thereto was added
triethylamine (10 mL) to terminate the reaction. The
resultant was concentrated under reduced pressure. The
residue was poured into hexane (200 mL) and water
(500 mL), and then the mixed solution was vigorously
stirred. The resultant precipitate was filtrated, and
rinsed with water and hexane. The precipitate was
crystallized two times from heated ethanol to yield the
title compound (2-2) as colorless needle crystals
{34.5 g (112 mmol), 20.2% yield). [a]23D +97.5 (cl.00
CA 02749797 2011-07-14
44
CH3OH), LRMS m/z 331 [M + Na] +, mp 139-141 C
1H NMR (400MHz, CD3OD); 6 7.51-7.47 (m, 2H, ArH)77.37-7.32 (m, 3H, ArH), 5.99
(dddd, 1H, J=17.2, 10.5,
6.08, 5 . 32Hz, H2), 5.56 (s, 1H, PhCH), 5.36 (dq, 1H,
J=17.3, 1.68Hz, H3a), 5.20 (ddt, 1H, J=10.4, 1.80,
1.28Hz, H3b), 4.88 (d, 1H, J=3.86Hz, Hl'), 4.25-4.18
(m, 2H, Hla & H6'a), 4.07 (ddt, 1H, J=13.0, 6.10,
1.36Hz, H1b), 3.85 (t, 1H, J=9.38Hz, H3'), 3.81-3.71
(m, 2H, H5' & H6'b), 3.52 (dd, 1H, J=9.38, 3.86Hz,
H2'), 3.45 (t, 1H, J=9.24Hz, H4')
13C NMR (100MHz, CD3OD); 6 139.1 (Ar-ipso), 135.4
(C2), 129.9 (Ar), 129.0 (Ar), 127.5 (Ar), 117.8 (C3),
103.0 (PhCH), 100.0 (Cl'), 82.9 (C4'), 74.0 (C2'), 72.0
(C3'), 69.9 (C6'), 69.7 (Cl), 64.1 (C5).
Route B2; 1-O-allyl-2,3-di-O-benzyl-4,6-0-
benzylidene-a-D-glucopyranoside (2-3):
To a solution of the compound (2-2) (30.0 g,
97.3 mmol) in anhydrous N,N-dimethylformamide (DMF,
300 mL) were added benzylbromide (41.6 g, 2.5
equivalent) and sodium hydroxide (11.7 g, 3.0
equivalent), and the reaction solution was vigorously
stirred at room temperature for 24 hours. After the
sufficient progress of the reaction was confirmed, the
reaction solution was poured into cold water (900 mL),
and the resultant was extracted with ethyl acetate
(3 x 300 mL). The organic layers were combined with
each other, and the combination was washed with
CA 02749797 2011-07-14
saturated saline (2 x 100 mL), dried over sodium
sulfate, filtrated and then concentrated under reduced
pressure. The resultant residue was crystallized
two times from heated ethanol to yield the title
5 compound (2-3) as a colorless needle crystal (33.5 g).
The filtrate was concentrated, purified by silica gel
chromatography (hexane/ethyl acetate, 15:1-410:1--*8:1),
and then crystallized from heated ethanol to yield the
same compound (2-3) (6.63 g) {total amount: 40.1 g
10 (82.1 mmol), 84.4% yield}. [a]26D -1.46 (cl.03 CHC13),
LRMS m/z 511 [M + Na]+, mp 86-87 C
1H NMR (400MHz, CDC13); 5 7.50-7.47 (m, 2H, ArH),
7.40-7.24 (m, 13H, ArH), 5.94 (dddd, 1H, J=17.0, 10.4,
6.70, 5.24Hz, H2), 5.56 (s, 1H, PhCH), 5.33 (dq, 1H,
15 J=17.2, 1.56Hz, H3a), 5.24 (ddt, 1H, J=10.3, 1.56,
1.12Hz, H3b), 4.92 (d, 1H, J=11.2Hz, ArCH2), 4.84 (d,
1H, J=11.2Hz, ArCH2), 4.83 (d, 1H, J=12.lHz, ArCH2)44.80 (d, 1H, J=3.76Hz,
Hl'), 4.68 (d, 1H, J=12. 1Hz,
ArCH2), 4.26 (dd, 1H, J=10.2, 4.84Hz, H6'a), 4.18 (ddt,
20 1H, J=12.9, 5.18, 1.40Hz, H1a), 4.79 (t, 1H, J=9.3OHz,
H3'), 4.03 (ddt, 1H, J=12.9, 6.68, 1.20Hz, Hlb), 3.89
(dt, 1H, J=9.96, 4.80Hz, H5'), 3.70 (t, 1H, J=10.3Hz,
H6' b) , 3.61 (t, 1H, J=9.44Hz, H4), 3.57 (dd, 1H,
J=8.72, 3.80Hz, H2')
25 13C NMR (100MHz, CDC13); 5 138.7 (Ar-ipso), 138.1
(Ar-ipso), 137.3 (Ar-ipso), 133.5 (C2), 128.9-127.5 (m,
Ar), 126.0 (Ar), 118.4 (C3), 101.2 (PhCH), 96.7 (Cl'),
CA 02749797 2011-07-14
46
82.1 (C3'), 79.1 (C2'), 78.6 (C4'), 75.3 (ArCH2), 73.6
(ArCH2), 69.0 (C6'), 68.4 (Cl), 62.5 (C5').
Route C2; 1-0-ally-2,3,4-tri-0-benzyl-a-D-
glucopyranoside (2-4):
The compound (2-3) (20.0 g, 40.9 mmol) was added
to a mixed solution of anhydrous dichloromethane
(100 mL) and anhydrous diethyl ether (100 mL) wherein
lithium aluminum hydride (2.02 g, 1.3 equivalent) was
suspended. Next, to the reaction solution was added
200 mL of a solution of aluminum chloride (7.09 g, 1.03
equivalent) in anhydrous diethyl ether, and then the
solution was stirred for 4 hours while heated and
refluxed. After the sufficient progress of the
reaction was confirmed, water (10 mL) was slowly added
thereto, dropwise. After one night, the precipitate
was filtrated, and rinsed with diethyl ether. The
filtrate was washed with water (2 x 100 mL), and then
the water layers were combined with each other. The
combination was extracted with diethyl ether
(2 x 100 mL). The organic layers were combined with
each other, and the combination was washed with
saturated saline (2 x 200 mL), dried over sodium
sulfate, filtrated, and concentrated under reduced
pressure. The resultant residue was purified by silica
gel chromatography (hexane/ethyl acetate,
5:1-X4:1--*3:1,2:1) to yield the title compound (2-4) as a
colorless oily substance {18.1 g (36.9 mmol), 90.2%
CA 02749797 2011-07-14
47
yield}. [a]22D +45.0 (cl.21 CHC13), LRMS m/z 513
[M + Na]+
1H NMR (400MHz, CDC13); 5 7.37-7.26 (m, 15H, ArH),
5.92 (dddd, 1H, J=17.1, 10.4, 6.66, 5.24Hz, H2), 5.31
(dq, 1H, J=17.2, 1.52Hz, H3a), 5.22 (ddt, 1H, J=10.3,
1.46, 1.10Hz, H3b), 5.00 (d, 1H, J=10.9Hz, ArCH2), 4.89
(d, 1H, J=11.OHz, ArCH2), 4.84 (d, 1H, J=10.9Hz,
ArCH2), 4.77 (d, 1H, J=12.OHz, ArCH2), 4.77 (d, 1H,
J=3.60Hz, Hl'), 4.65 (d, 1H, J=12.lHz, ArCH2), 4.64 (d,
1H, J=11.OHz, ArCH2), 4.14 (ddt, 1H, J=12.9, 5.22,
1.34Hz, Hla), 4.04 (t, 1H, J=9.36Hz, H3'), 3.99 (ddt,
1H, J=12.9, 6.64, 1.08Hz, H1b), 3.79-3.66 (m, 3H,
H5' & H6'a & H6'b), 3.54 (t, 1H, J=9.28Hz, H4'), 3.51
(dd, 1H, J=9.60, 3.64Hz, H2'), 1.69 (t, 1H, J=12.OHz,
61-OH)
13C NMR (100MHz, CDC13); 5 138.7 (Ar-ipso), 138.1
(Ar-ipso), 138.1 (Ar-ipso), 133.6 (C2), 128.4-127.6 (m,
Ar), 118.3 (C3), 95.6 (Cl'), 81.9 (C3'), 79.9 (C2'),
77.3 (C4'), 75.7 (ArCH2), 75.0 (ArCH2), 73.2 (ArCH2)220 70.8 (C5), 68.2 (Cl),
61.7 (C6).
Route D2; 1-O-ally-2,3,4-tri-O-benzyl-6-O-tosyl-a-
D-glucopyranoside (2-5):
To a solution of the compound (2-4) (25.1 g,
51.2 mmol) in anhydrous pyridine (250 mL) were added p-
toluenesulfonyl chloride (14.6 g, 1.5 equivalent) and
4-dimethylaminopyridine (626 mg, 0.1 equivalent), and
the reaction solution was stirred at room temperature
CA 02749797 2011-07-14
48
for 16 hours. After the sufficient progress of the
reaction was confirmed, water was added thereto so as
to terminate the reaction. The reaction solution was
concentrated under reduced pressure. A small amount of
ethyl acetate was used to suspend the residue, and the
suspended residue was poured into 0.5 M hydrochloric
acid (200 mL). The resultant was extracted with ethyl
acetate (3 x 200 mL). The organic layers were combined
with each other, and the combination was washed with an
aqueous saturated sodium hydrogen carbonate solution
(2 x 100 mL) and saturated saline (2 x 100 mL), dried
over sodium sulfate, filtrated and then concentrated
under reduced pressure. The resultant residue was
crystallized two times from heated ethanol to yield the
title compound (2-5) as a colorless needle crystal
(25.0 g). The filtrate was concentrated and purified
by silica gel chromatography (hexane/ethyl acetate,
5:1-X4:1->3:1) to yield the same compound (2-5) (4.00 g)
{total amount: 29.0 g (45.0 mmol), 87.9% yield}.
[a]25D +32.1 (cl.02 CHC13), LRMS m/z 667 [M+Na]+, mp
86-87 C
1H NMR (400MHz, CDC13); 6 7.76 (ddd, 2H, J=8.32,
1.96, 1.76Hz, ArH), 7.35-7.26 (m, 15H, ArH), 7.17-7.12
(m, 2H, ArH), 5.88 (dddd, 1H, J=17.2, 10.3, 6.62,
5.24Hz, H2), 5.28 (dq, 1H, J=17.2, 1.56Hz, H3a), 5.20
(ddt, 1H, J=10.3, 1.60, 1.12Hz, H3b), 4.99 (d, 1H,
J=10.9Hz, ArCH2), 4.82 (d, 1H, J=10.6Hz, ArCH2), 4.78
CA 02749797 2011-07-14
49
(d, 1H, J=10.8Hz, ArCH2), 4.74 (d, 1H, J=12.1Hz,
ArCH2), 4.72 (d, 1H, J=3.58Hz, H1'), 4.62 (d, 1H,
J=12.lHz, ArCH2), 4.42 (d, 1H, J=10.6Hz, ArCH2), 4.22
(dd, 1H, J=10.5, 4.20Hz, H6'a), 4.16 (dd, 1H, J=10.5,
2.12Hz, H6'b), 4.07 (ddt, 1H, J=12.9, 5.24, 1.40Hz,
Hla), 3.98 (t, 1H, J=9.24Hz, H3'), 3.93 (ddt, 1H,
J=12.9, 6.64, 1.16Hz, Hlb), 3.81 (ddd, 1H, J=10.1,
4.12, 2.04Hz, H5'), 3.48 (dd, 1H, J=9.62, 3.58Hz, H2'),
3.45 (dd, 1H, J=10.0, 8.90Hz, H4'), 2.39 (s, 3H, Ts-Me)
13C NMR (100MHz, CDC13); 6 144.8 (Ar-ipso), 138.5
(Ar-ipso), 137.9 (Ar-ipso), 137.7 (Ar-ipso), 133.4
(C2), 132.8 (Ar-ipso), 129.8 Ar(), 128.4-127.6 (m, Ar),
118.4 (C3), 95.4 (Cl'), 81.8 (C3'), 79.6 (C2'), 76.9
(C4'), 75.7 (ArCH2), 75.0 (ArCH2), 73.2 (ArCH2), 68.6
(C5'), 68.5 (C6'), 68.3 (Cl), 21.6 (Ts-Me).
Route E2; 1-O-(2,3,4-tri-O-benzyl-6-O-tosyl-a-D-
glucopyranosyl)propane-1,3-diol (2-6):
To a solution of the compound (2-5) (29.0 g,
45.0 mmol) in anhydrous tetrahydrofuran (THF, 150 mL)
was added a 0.5 M solution (180 mL, 90.0 mmol) of 9-
borabicyclo[3,3,1]nonane (9-BBN) in tetrahydrofuran at
0 C under an atmosphere of argon. After 1 hour, the
temperature of the reaction solution was returned to
room temperature, and subsequently the solution was
stirred for 10 hours. The reaction solution was again
cooled to 0 C. Thereto were successively added water
(20 mL), a 3 M solution (70 mL) of sodium hydroxide,
CA 02749797 2011-07-14
and a 35% solution (70 mL) of hydrogen peroxide in
water. After 1 hour, the temperature was returned to
room temperature. The solution was then stirred for
12 hours. After the sufficient progress of the
5 reaction was confirmed, this solution was extracted
with ethyl acetate (3 x 100 mL). The organic layers
were combined with each other, and the combination was
washed with saturated saline (2 x 100 mL), dried over
sodium sulfate, filtrated, and then concentrated under
10 reduced pressure. The resultant residue was purified
by silica gel chromatography (hexane/ethyl acetate,
3:2-+1:1-->2:3) to yield the title compound (2-6) as a
colorless oily substance {28.2 g (42.5 mmol), 94.4%
yield}. [a]24D +26.6 (cl.02 CHC13), LRMS m/z 685
15 IN + Na]+
1H NMR (400MHz, CDC13); 5 7.76-7.74 (m, 2H, ArH),
7.35-7.26 (m, 15H, ArH), 7.16-7.12 (m, 2H, ArH), 4.94
(d, 1H, J=10.9Hz, ArCH2), 4.82 (d, 1H, J=10.7Hz,
ArCH2), 4.77 (d, 1H, J=10.9Hz, ArCH2), 4.75 (d, 1H,
20 J=12.OHz, ArCH2), 4.61 (d, 1H, J=12.OHz, ArCH2), 4.61
(d, 1H, J=3.64Hz, H1'), 4.43 (d, 1H, J=10.7Hz, ArCH2),
4.20-4.13 (m, 2H, H6'a & H6'b), 3.92 (t, 1H, J=9.24Hz,
H3'), 3.84-3.74 (m, 4H, Hla & H3a & H3b & H5'),
3.48-3.40 (m, 3H, H1b & H2' & H4'), 2.52 (t, 1H,
25 J=4.74Hz, 3-OH), 2.39 (s, 3H, Ts-Me), 1.88-1.75 (m, 2H,
H2a & H2b)
13C NMR (100MHz, CDC13); 5 144.8 (Ar-ipso), 138.4
CA 02749797 2011-07-14
51
(Ar-ipso), 137.9 (Ar-ipso), 137.6 (Ar-ipso), 132.7
(Ar-ipso), 129.8 (Ar), 128.5-127.6 (m, Ar), 97.1 (Cl'),
81.8 (C3'), 79.5 (C2'), 76.8 (C4'), 75.6 (ArCH2), 75.0
(ArCH2), 73.4 (ArCH2), 68.7 (C5'), 68.6 (C6'), 67.5
(Cl), 61.5 (C3), 31.5 (C2), 21.6 (Ts-Me).
Route F2; 1-0-(2,3,4-tri-0-benzyl-6-thioacetyl-a-
D-quinovopyranosyl)-propane-1,3-diol (2-7):
Potassium thioacetate (7.28 g, 1.5 equivalent) was
added to a solution of the compound (2-6) (28.2 g,
42.5 mmol) in anhydrous DMF (300 ml), and the solution
was stirred at 90 C for 3 hours. After the sufficient
progress of the reaction was confirmed, the reaction
solution was poured into cold water (900 mL), and the
resultant was extracted with ethyl acetate
(3 x 300 mL). The organic layers were combined with
each other, and the combination was washed with
saturated saline (2 x 200 mL), dried over sodium
sulfate, filtrated and concentrated under reduced
pressure. The resultant residue was purified by silica
gel chromatography (hexane/ethyl acetate,
2:1,3:2--,1:1---~2:3) to yield the title compound (2-7) as a
light brown oily substance {21.9 g (38.6 mmol), 90.8%
yield}. [a]23D +33.0 (cl.02 CHC13), LRMS m/z 584
[M + Na]+
lH NMR (400MHz, CDC13); 5 7.37-7.24 (m, 15H, ArH),
4.95 (d, 1H, J=10.8Hz, ArCH2), 4.89 (d, 1H, J=10.6Hz,
ArCH2), 4.80 (d, 1H, J=10.8Hz, ArCH2), 4.77 (d, 1H,
CA 02749797 2011-07-14
52
J=12.1Hz, ArCH2), 4.63 (d, 1H, J=12.0Hz, ArCH2), 4.63
(d, 1H, J=3.52Hz, Hl'), 4.61 (d, 1H, J=10.7Hz, ArCH2),
3.94 (t, 1H, J=9.22Hz, H3'), 3.88 (ddd, 1H, J=9.86,
6.10, 4.88Hz, Hla), 3.83-3.73 (m, 3H, H3a & H3b & H5'),
3.50 (dd, 1H, J=9.60, 3.64Hz, H2'), 3.45 (ddd, 1H,
J=9.92, 5.24, 2.28Hz, Hib), 3.41 (dd, 1H, J=13.6,
3.00Hz, H6'a), 3.30 (dd, 1H, J=9.54, 9.06Hz, H4'), 3.02
(dd, 1H, J=13.7, 7.64Hz, H6'b), 2.67 (br, 1H, 3-OH),
2.32 (s, 3H, SAc-Me), 1.92-1.78 (m, 2H, H2a & H2b)
13C NMR (100MHz, CDC13); 5 195.0 (SAC-C=0), 138.5
(Ar-ipso), 137.9 (Ar-ipso), 137.8 (Ar-ipso),
128.5-127.6 (m, Ar), 96.9 (Cl'), 81.8 (C3'), 80.4
(C4'), 79.8 (C2'), 75.7 (ArCH2), 75.2 (ArCH2), 73.4
(ArCH2), 69.5 (C5'), 67.2 (Cl), 61.5 (C3), 31.5 (C2),
30.8 (C6'), 30.5 (SAc-Me).
Route G2; 3-0-(2,3,4-tri-O-benzyl-6-sulfo-a-D-
quinovopyranosyl)-propane-1,3-diol sodium salt (2-8):
To a solution of the compound (2-7) (500 mg,
0.88 mmol) in acetic acid (5 mL) were added Oxone
(1.63 g, 2.65 mmol) and potassium acetate (25 mg). The
solution was vigorously stirred at room temperature for
2 days. After the sufficient progress of the reaction
was confirmed, the reaction solution was poured into a
7.5 M cold solution (15 mL) of sodium hydroxide, and
the resultant was extracted with chloroform
(4 x 30 mL). The organic layers were combined with
each other, and the combination was washed with
CA 02749797 2011-07-14
53
saturated aqueous sodium hydrogen carbonate (2 x 20 mL)
and saturated saline (2 x 20 mL), dried over sodium
sulfate, filtrated and concentrated under reduced
pressure. The resultant residue was further dissolved
in methanol (20 mL), and thereto was added an
appropriate amount of sodium methoxide (NaOMe). The
reactive components were then caused to react with each
other at room temperature for 3 hours. While the
reaction solution was cooled with ice, cold water was
added to the solution to terminate the reaction. The
solution was then concentrated under reduced pressure.
The concentrated substance was purified by silica gel
chromatography (chloroform/methanol, 100:0--,3:1) to
yield the title compound (2-8) as a colorless waxy
substance {498 mg (0.87 mmol), 98.7% yield}. LRMS m/z
571 [M - Na] -.
Route H2; 3-0-(2,3,4-tri-0-benzyl-6-sulfo-a-D-
quinovopyranosyl)-1-0-(carbobenzoxy-3-alanyl)-propane-
1,3-diol sodium salt (2-9):
Into an anhydrous DMF (15 mL) mixed solution were
dissolved the compound (2-8) (900 mg, 1.51 mmol), N-
carbobenzoxy-[i-alanine (440 mg, 1.97 mmol), 1-ethyl-3-
(3-dimethylaminopropyl)carbodiimide hydrochloride
(EDCI=HC1) (871 mg, 4.55 mmol), and 4-
dimethylaminopyridine (55 mg, 0.15 mmol), and then this
solution was left to stand overnight so that reaction
between the reactive components would proceed. After
CA 02749797 2011-07-14
54
the sufficient progress of the reaction was confirmed,
water (1 mL) was poured into the reaction solution to
terminate the reaction. Thereafter, the solution was
concentrated under reduced pressure. The resultant
residue was purified by silica gel chromatography
(chloroform/methanol, 100:012:1) to yield the title
compound (2-9) {517 mg (0.65 mmol), 42.8% yield}. LRMS
m/z 776 [M - Na]-.
Route 12; 3-0-(6-sulfo-a-D-quinovopyranosyl)-1-0-
(R-alanyl)-propane-l,3-diol sodium salt (1-10):
To a solution of the compound (2-9) (517 mg,
0.65 mmol) in methanol (3.0 mL), dichloromethane
(3.0 mL) and acetic acid (0.2 mL) was added 20%
palladium hydroxide-activated carbon (50 mg), and then
this solution was left to stand overnight at room
temperature in an atmosphere of hydrogen so that
reaction between the reactive components would proceed.
Since the reaction did not advance sufficiently,
thereto were further added methanol (3.0 mL) and 10%
palladium-activated carbon (50 mg). The solution was
then stirred at room temperature under an atmosphere of
hydrogen for 2 days. After the sufficient progress of
the reaction was confirmed, the palladium-activated
carbon was filtrated off with celite, and the filtrate
was concentrated under reduced pressure. The resultant
residue was purified by silica gel chromatography
(chloroform/methanol, 4:1-42:1-1:1-2:3) to yield the
CA 02749797 2011-07-14
title compound (2-10) {250.8 mg (0.63 mmol), 98.2%
yield}. LRMS m/z 372 [M - Na]-.
Route J2; 3-0-(6-sulfo-a-D-quinovopyranosyl)-1-0-
(biotinyl-R-alanyl)-propane-l,3-diol calcium salt
5 (1-11):
The compound (2-10) (226 mg, 0.57 mmol) was
dissolved in a mixed solution of anhydrous DMF (5 mL)
and triethylamine (2 mL), and then thereto was added a
solution wherein a biotin-p-nitrophenylester(313 mg,
10 0.86 mg) was dissolved in anhydrous DMF (2 mL). The
solution was stirred, at room temperature with a
stirrer so that the reactive components would react
with each other, and left to stand overnight. After
the sufficient progress of the reaction was confirmed,
15 toluene and methanol were added to the solution. Under
reduced pressure, the solvents were then removed by
evaporation to concentrate the solution, while
azeotropy. The resultant residue was purified by
silica gel chromatography (chloroform/methanol,
20 10:11:4) to yield a sodium salt thereof. Into the
column was filled an ion exchange resin (Amberlite
(registered trade name) IR-120, 15.7 mL) changed to a
calcium form with a solution of calcium chloride in
water. A solution of the sodium salt was repeatedly
25 passed through the column to change the sodium salt
into a calcium salt. The title compound (2-11) {210 mg
(340 pmol), 59.5% yield} was produced as a result of
CA 02749797 2011-07-14
56
this method.
LRMS m/z 598 [M - H]-, m/z 638 [M - H + Ca]+.
Synthesis Example 3
A process for producing the
sulfopyranosylacylglycerol derivative according to an
embodiment of the present invention is illustrated in
the following scheme 3, giving an a-
sulfoquinovosylmonoacylglycerol monoiodide derivative
as an example thereof:
CA 02749797 2011-07-14
57
M 0 L
ry
0
-
CY)
on
0 0
o z
O o
o
= c~
0 c:
0=<
o
a
< C w
M
t`7
C o o 0
O a
C
4 m
CY? 0
E
N
U
(I)
CA 02749797 2011-07-14
58
a
z.
O M 0 U
0
r-ti = p `7
f 0
co a'
Q
O
...,O
O O
0 o 0
V) 0
m 8 o
Fc"
0a a a ax
0
xx zx
a =~ zx
a 0 0
a
U
M a cfa
C3
o
U
~o o U a T a
m O M O
O O
fn 0
O ciqC
z2 4x
CA 02749797 2011-07-14
59
The following will describe a synthesis example
through individual steps in detail:
<Synthesis Example>
Route A3; 1-0-allyl-4,6-0-benzylidene-a-D-
glucopyranoside (3-2):
Trifluoromethanesulfonic acid (1.00 mL) was added
to a liquid suspension of the compound (3-1) (100 g,
555 mmol) in allyl alcohol (500 mL) at 0 C, and the
reaction liquid was vigorously stirred at 80 C for
48 hours. After the sufficient progress of the
reaction was confirmed, thereto was added triethylamine
(3 mL) to terminate the reaction. The resultant was
concentrated under reduced pressure. Next, the residue
was suspended in anhydrous acetonitrile (500 mL), and
thereto were added benzaldehydedimethylacetal (127 g,
1.5 equivalent) and p-toluenesulfonic acid monohydrate
(5.28 g, 0.05 equivalent). The reaction liquid was
stirred at 40 C for 4 hours, and then thereto was added
triethylamine (10 mL) to terminate the reaction. The
resultant was concentrated under reduced pressure. The
residue was poured into hexane (2000 mL) and water
(500 mL), and then the mixed liquid was vigorously
stirred. The resultant precipitate was filtrated, and
rinsed with water and hexane. The precipitate was
crystallized two times from heated ethanol to yield the
title compound (3-2) as a colorless needle crystal
{34.5 g (112 mmol), 20.2% yield}.
CA 02749797 2011-07-14
LRMS m/z 331 [M+Na]+.
Route B3; 1-0-allyl-2,3-di-0-benzyl-4,6-0-
benzylidene-a-D-glucopyranoside (3-3):
To a solution of the compound (3-2) (30.0 g,
5 97.3 mmol) in anhydrous N,N-dimethylformamide (DMF,
300 mL) were added benzylbromide (41.6 g, 2.5
equivalent) and sodium hydroxide (11.7 g, 3.0
equivalent), and the reaction solution was vigorously
stirred at room temperature for 24 hours. After the
10 sufficient progress of the reaction was confirmed, the
reaction solution was poured into cold water (900 mL),
and the resultant was extracted with ethyl acetate
(3 x 300 mL). The. organic layers were combined with
each other, and the combination was washed with
15 saturated saline (2 x 100 mL), dried over sodium
sulfate, filtrated and then concentrated under reduced
pressure. The resultant residue was crystallized
two times from heated ethanol to yield the title
compound (3-3) as a colorless needle crystal (33.5 g).
20 The filtrate was concentrated, purified by silica gel
chromatography (hexane/ethyl acetate, 15:1-10:1-8:1),
and then crystallized from heated ethanol to yield the
same compound (3-3) (6.63 g) {total amount: 40.1 g
(82.1 mmol), 84.4% yield}.
25 LRMS m/z 511 [M+Na]+.
Route C3; 1-0-ally-2,3-di-0-benzyl-a-D-
glucopyranoside (3-4):
CA 02749797 2011-07-14
61
The compound (3-3) (15.6 g, 32.0 mmol) was
dissolved in acetic acid (90 mL), and further distilled
water (50 mL) was added thereto. The solution was
stirred for 1 hour while heated and refluxed. After
the sufficient progress of the reaction was confirmed,
the solution was cooled to room temperature, and then
the solvent was removed by evaporation. Distilled
water (15 mL) was added thereto, and the solution was
again concentrated under reduced pressure; this
operation was repeated 4 times. Thereafter, the
resultant was purified by silica gel flash
chromatography (hexane/ethyl acetate, 4:1-2:1-1:1-1:2)
to yield the title compound (3-4) {12.1 g (30.2 mmol),
94.5% yield}.
LRMS m/z 423 [M+Na]+.
Route D3; 1-0-ally-2,3,4-di-0-benzyl-6-0-tosyl-a-
D-glucopyranoside (3-5):
To a solution of the compound (3-4) (12.1 g,
30.2 mmol) in anhydrous pyridine (120 mL) were added p-
toluenesulfonyl chloride (7.5 g, 39.3 mmol) and 4-
dimethylaminopyridine (369 mg, 3.0 mmol), and the
reaction solution was stirred at 0 C for 16 hours.
After the sufficient progress of the reaction was
confirmed, the reaction solution was slowly poured into
ice water (100 mL). The water layer was subjected to
extraction with ethyl acetate (3 x 200 mL). The
organic layers were combined with each other. The
CA 02749797 2011-07-14
62
combination was washed with an aqueous 1 N HC1 solution
until the pH became 4, washed with saturated aqueous
sodium hydrogen carbonate (2 x 100 mL) and saturated
saline (2 x 100 mL), dried over sodium sulfate,
filtrated and then concentrated under reduced pressure.
The concentrated product was purified by silica gel
flash chromatography (hexane/ethyl acetate,
6:l-4:l---3:l) to yield the title compound (3-5) (15.3 g
(27.6 mmol), 91.4% yield}.
LRMS m/z 577 [M+Na]+.
Route E3; 1-0-allyl-2,3-di-0-benzyl-4-0-
(carbobenzoxy-R-alanyl)-6-0-tosyl-a-D-glucopyranoside
(3-6) :
Into a mixed solution of anhydrous dichloromethane
(200 mL) and anhydrous pyridine (50 mL) were dissolved
the compound (3-5) (15.3 g, 27.6 mmol), N-carbobenzoxy-
R-alanine (12.32 g, 55.2 mmol), 1-ethyl-3-(3-
dimethylaminopropyl)carbodiimide hydrochloride
(EDCI=HC1) (15.8 mg, 82.8 mmol) and 4-
dimethylaminopyridine (6.7 g, 55.2 mmol), and then the
reactive components were caused to react with each
other at room temperature for 18 hours. After the
sufficient progress of the reaction was confirmed,
water (10 mL) was poured into the reaction solution to
terminate the reaction. Thereafter, the solution was
concentrated under reduced pressure. The resultant
residue was purified by silica gel chromatography
CA 02749797 2011-07-14
63
(hexane/ethyl acetate, 4:1-3:1,2:1-,3:2) to yield the
title compound (3-6) {17.1 g (22.5 mmol), 81.5% yield}.
LRMS m/z 782 [M+Na]+.
Route F3; 1-0-allyl-2,3-di-O-benzyl-4-0-
(carbobenzoxy-R-alanyl)-6-thioacetyl-a-D-
glucopyranoside (3-7):
Potassium thioacetate (5.1 g, 45.0 mmol) was added
to a solution of the compound (3-6) (17.1 g, 22.5 mmol)
in anhydrous N,N-dimethylformamide (300 mL), and the
solution was stirred at 90 C for 3 hours. After the
sufficient progress of the reaction was confirmed, the
reaction solution was poured into cold water (400 mL),
and the resultant was extracted with ethyl acetate
(3 x 150 mL). The organic layers were combined with
each other, and the combination was washed with
saturated saline (2 x 100 mL), dried over sodium
sulfate, filtrated and concentrated under reduced
pressure. The resultant residue was purified by silica
gel chromatography (hexane/ethyl acetate,
4:1-*3:1-2:13:2) to yield the title compound (3-7)
{14.2 g (20.3 mmol), 90.0% yield}.
LRMS m/z 686 [M+Na]+.
Route G3; 3-O-[2,3-di-O-benzyl-4-O-(carbobenzoxy-
R-alanyl)-6-thioacetyl-a-D-quinovopyranosyl-glycerol
(3-8):
The. compound (3-7) (14.2 g, 20.4 mmol) was
dissolved in a solution (200 mL) of t-butyl alcohol and
CA 02749797 2011-07-14
64
distilled water (4:1), and then thereto were added a
0.04 M osmium tetraoxide solution in t-butyl alcohol
(3 mL) and trimethylamine N-oxide (3.4 g, 30.5 mmol).
The solution was stirred with a stirrer at room
temperature for 24 hours. After the sufficient
progress of the reaction was confirmed, 3 g of
activated carbon was added thereto and then the
solution was stirred for 30 minutes to cause the
catalyst to be adsorbed on the carbon. The solution
was subjected to suction filtration through a Kiriyama
funnel containing celite, so as to remove the catalyst.
The reaction product remaining on the celite was washed
three times with ethyl acetate so as to be collected.
Distilled water (200 mL) was added to the collected
filtrate, and the resultant was extracted with ethyl
acetate (3 x 150 mL). The organic layers were combined
with each other, and the combination was washed with
saturated saline (2 x 100 mL), dried over sodium
sulfate, filtrated and concentrated under reduced
pressure. The resultant residue was purified by silica
gel chromatography (hexane/ethyl acetate,
2:1-+1:12:3-1:21:4-1:8) to yield the title compound
(3-8) (13.2 g (18.9 mmol), 93.0% yield).
LRMS m/z 720 [M+Na]+.
Route H3; 3-0-[2,3-di-O-benzyl-4-0-(carbobenzoxy-
R-alanyl)-6-thioacetyl-a-D-quinovopyranosyl]-1-0-
stearoyl-glycerol (3-9):
CA 02749797 2011-07-14
Stearoyl chloride (8.5 g, 28.2 mmol) was added to
a mixed solution of the compound (3-8) (13.1 g,
18.8 mmol) in anhydrous dichloromethane (200 mL) and
anhydrous pyridine (50 mL), and then the solution was
5 stirred at room temperature for 2 hours. After the
sufficient progress of the reaction was confirmed,
methanol (5 mL) was added thereto so as to terminate
the reaction. The solution was concentrated under
reduced pressure. A small amount of ethyl acetate was
10 used to suspend the residue, and the suspended residue
was poured into water (200 mL). The resultant was
extracted with ethyl acetate (3 x 100 mL). The organic
layers were combined with each other, and the
combination was washed with saturated saline
15 (2 x 100 mL), dried over sodium sulfate, filtrated and
then concentrated under reduced pressure. The
resultant residue was purified by silica gel
chromatography (hexane/ethyl acetate,
6:1-4:1,2:1~3:2-1:1) to yield the title compound (3-9)
20 as a colorless oily substance {7.1 g (7.4 mmol), 39.4%
yield}.
LRMS m/z 987 [M+Na]+.
Route 13; 3-O-[2,3-di-O-benzyl-4-0-(carbobenzoxy-
R-alanyl)-6-sulfo-a-D-quinovopyranosyl]-1-0-stearoyl-
25 glycerol (3-10):
Oxone (18.1 g, 29.5 mmol) and potassium acetate
(4.0 g) were added to a solution of the compound (3-9)
CA 02749797 2011-07-14
66
( 7 . 1 g, 7 . 4 mmol) in acetic acid (160 g, 4 mol), and
then the solution was vigorously stirred at room
temperature for 48 hours. After the sufficient
progress of the reaction was confirmed, the reaction
solution was poured into a cold 7.5 M sodium hydroxide
solution (500 mL) and the resultant was extracted with
ethyl acetate (4 x 100 mL). The organic layers were
combined with each other, and the combination was
washed with saturated aqueous sodium hydrogen carbonate
(2 x 100 mL) and saturated saline (2 x 100 mL), dried
over sodium sulfate, filtrated and then concentrated
under reduced pressure. The resultant residue was
purified by silica gel chromatography
(chloroform/methanol, 100:1-X50:1--X20:1-X15:1-->12:1) to
yield the title compound (3-10) as a colorless waxy
substance {5.2 g (5.24 mmol), 71.2% yield}.
LRMS m/z 968 [M-Na]-.
Route J3; 3-0-[4-0-(13-alanyl)-6-sulfo-a-D-
quinovopyranosyl]-1-0-stearoyl-glycerol (3-11):
To a solution of the compound (3-10) (490 mg,
494 pmol) in methanol (20 mL) and acetic acid (0.6 mL)
was added 10% palladium-activated carbon (300 mg), and
then the solution was stirred at room temperature under
an atmosphere of hydrogen gas for 48 hours. After the
sufficient progress of the reaction was confirmed, the
palladium-activated carbon was filtrated off. The
filtrate was concentrated under reduced pressure. The
CA 02749797 2011-07-14
67
resultant residue was purified by silica gel
chromatography (chloroform/methanol/distilled water,
100:25:3-65:25:4-+65:35:5-65:45:6) to yield the title
compound (3-11) {221 mg (327 pmol), 66.1% yield}.
LRMS m/z 654 [M-Na]-.
Route K3; 3-0-[4-0-(4-iodobenzoyl-R-alanyl)-6-
sulfo-a-D-quinovopyranosyl]-1-0-stearoyl-glycerol (3-
12):
The compound (3-11) (221 mg, 327 pmol) was
dissolved in a mixed solution of anhydrous
dichloromethane (15 mL), pyridine (5 mL) and
triethylamine (2 mL), and thereto was added (4-
iodobenzoyl)-p-nitrophenyl ester (241 mg, 653 pmol).
While the solution was stirred with a stirrer, the
reactive components were caused to react with each
other at room temperature for 24 hours. After the
sufficient progress of the reaction was confirmed,
toluene and methanol were added to the solution. Under
reduced pressure, the solvents were then removed by
evaporation to concentrate the solution, while
azeotropy. The resultant residue was purified by
silica gel chromatography
(chloroform/methanol/distilled water,
30:5:0.5-30:6:0.7-30:8:09-30:10:1.1) to yield the title
compound (3-12) {262 mg (238 pmol), 72.8% yield}.
LRMS m/z 884 [M - Na]-.
CA 02749797 2011-07-14
68
Synthesis Example 4
A process for producing the
sulfopyranosylacylglycerol derivative according to an
embodiment of the present invention is illustrated in
the following scheme 4, giving an a-
sulfoquinovosylmonoacylglycerol triiodide derivative as
an example thereof:
CA 02749797 2011-07-14
69
LLt
Itt
O O
)
O O
N to
p
Om
O
C7 O
O
Z2
O
O D Ofn
a.
1 Cll$ / LO
Q
a o
o W 0
T Q= a
2 0 0 cz
m
~ am
CL
Q)
U
t!)
CA 02749797 2011-07-14
u~
rf
Z
yr
h
o x
'ct O
0 r^~. 0
00 d' o dam' p " ` o
O 0
O 0 O
m m O
0
o I.0=.I
zx zz
d~ O~ zs
O O 0
0
0
rr 0 Itr z:
0 0 0
^ 0
rl T
d O O
cn o
O o 0 o
C) ; a m ~' FS
o Oz
o
Zr Zr
o o
0
z
N
CA 02749797 2011-07-14
71
The following will describe a synthesis example
through the individual steps in detail:
<Synthesis Example>
Route A4; 1-0-allyl-4,6-0-benzylidene-a-D-
glucopyranoside (4-2):
Trifluoromethanesulfonic acid (1.00 mL) was added
to a liquid suspension of the compound (4-1) (100 g,
555 mmol) in allyl alcohol (500 mL) at 0 C, and the
reaction liquid was vigorously stirred at 80 C for
48 hours. After the sufficient progress of the
reaction was confirmed, thereto was added triethylamine
(3 mL) to terminate the reaction. The resultant was
concentrated under reduced pressure. Next, the residue
was suspended in anhydrous acetonitrile (500 mL), and
thereto were added benzaldehydedimethylacetal (127 g,
1.5 equivalent) and p-toluenesulfonic acid monohydrate
(5.28 g, 0.05 equivalent). The resultant liquid was
stirred at 40 C for 4 hours, and then thereto was added
triethylamine (10 mL) to terminate the reaction. The
resultant was concentrated under reduced pressure. The
residue was poured into hexane (2000 mL) and water
(500 mL), and then the mixed liquid was vigorously
stirred. The resultant precipitate was filtrated, and
rinsed with water and hexane. The precipitate was
crystallized two times from heated ethanol to yield the
title compound (4-2) as a colorless needle crystal
{34.5 g (112 mmol), 20.2% yield}.
CA 02749797 2011-07-14
72
LRMS m/z 331 [M+Na]+.
Route B4; 1-0-allyl-2,3-di-0-benzyl-4,6-0-
benzylidene-a-D-glucopyranoside (4-3):
To a solution of the compound (4-2) (30.0 g,
97.3 mmol) in anhydrous N,N-dimethylformamide (DMF,
300 mL) were added benzylbromide (41.6 g, 2.5
equivalent) and sodium hydroxide (11.7 g, 3.0
equivalent), and the reaction solution was vigorously
stirred at room temperature for 24 hours. After the
sufficient progress of the reaction was confirmed, the
reaction solution was poured into cold water (900 mL),
and the resultant was extracted with ethyl acetate
(3 x 300 mL). The organic layers were combined with
each other, and the combination was washed with
saturated saline (2 x 100 mL), dried over sodium
sulfate, filtrated and then concentrated under reduced
pressure. The resultant residue was crystallized
two times from heated ethanol to yield the title
compound (4-3) as a colorless needle crystal (33.5 g).
The filtrate was concentrated, purified by silica gel
chromatography (hexane/ethyl acetate, 15:1-10:1-8:1),
and then crystallized from heated ethanol to yield the
same title compound (4-3) (6.63 g) {total amount:
40.1 g (82.1 mmol), 84.4% yield}.
LRMS m/z 511 [M+Na]+.
Route C4; 1-0-ally-2,3-di-0-benzyl-a-D-
glucopyranoside (4-4):
CA 02749797 2011-07-14
73
The compound (4-3) (15.6 g, 32.0 mmol) was added
to acetic acid (90 mL), and further distilled water
(50 mL) was added thereto. The solution was stirred
for 1 hour while heated and refluxed. After the
sufficient progress of the reaction was confirmed, the
solution was cooled to room temperature, and then the
solvent was removed by evaporation. Distilled water
(15 mL) was added thereto, and the solution was again
concentrated under reduced pressure; this operation was
repeated 4 times. Thereafter, the resultant was
purified by silica gel flash chromatography
(hexane/ethyl acetate, 4:1-2:1-1:11:2) to yield the
title compound (4-4) {12.1 g (30.2 mmol), 94.5% yield}.
LRMS m/z 423 [M+Na]+.
Route D4; 1-0-ally-2,3,4-di-0-benzyl-6-0-tosyl-a-
D-glucopyranoside (4-5):
To a solution of the compound (4-4) (12.1 g,
30.2 mmol) in anhydrous pyridine (120 mL) were added p-
toluenesulfonyl chloride (7.5 g, 39.3 mmol) and 4-
dimethylaminopyridine (369 mg, 3.0 mmol), and the
reaction solution was stirred at 0 C for 16 hours.
After the sufficient progress of the reaction was
confirmed, the reaction solution was slowly poured into
ice water (100 mL). The water layer was subjected to
extraction with ethyl acetate (3 x 200 mL). The
organic layers were combined with each other. The
combination was washed with an aqueous 1 N HC1 solution
CA 02749797 2011-07-14
74
until the pH become 4, washed with saturated aqueous
sodium hydrogen carbonate (2 x 100 mL) and saturated
saline (2 x 100 mL), dried over sodium sulfate,
filtrated and then concentrated under reduced pressure.
The concentrated product was purified by silica gel
flash chromatography (hexane/ethyl acetate,
6:1-X4:1->2:1) to yield the title compound (4-5) {15.3 g
(27.6 mmol), 91.4% yield).
LRMS m/z 577 [M+Na]+.
Route E4; 1-0-allyl-2,3-di-0-benzyl-4-0-
(carbobenzoxy-(3-alanyl)-6-0-tosyl-a-D-glucopyranoside
(4-6) :
Into a mixed solution of anhydrous dichloromethane
(200 mL) and anhydrous pyridine (50 mL) were dissolved
the compound (4-5) (15.3 g, 27.6 mmol), N-carbobenzoxy-
3-alanine (12.32 g, 55.2 mmol), 1-ethyl-3-(3-
dimethylaminopropyl)carbodiimide hydrochloride
(EDCI=HCl) (15.8 mg, 82.8 mmol) and 4-
dimethylaminopyridine (6.7 g, 55.2 mmol), and then the
reactive components were caused to react with each
other at room temperature for 18 hours. After the
sufficient progress of the reaction was confirmed,
water (10 mL) was poured into the reaction solution to
terminate the reaction. Thereafter, the solution was
concentrated under reduced pressure. The resultant
residue was purified by silica gel chromatography
(hexane/ethyl acetate, 4 : 1-*3 : 1-*2 : 1,3 : 2) to yield the
CA 02749797 2011-07-14
title compound (4-6) {17.1 g (22.5 mmol), 81.5% yield}.
LRMS m/z 782 [M+Na]+.
Route F4; 1-0-allyl-2,3-di-O-benzyl-4-O-
(carbobenzoxy-R-alanyl)-6-thioacetyl-a-D-
5 glucopyranoside (4-7):
Potassium thioacetate (5.1 g, 45.0 mmol) was added
to a solution of the compound (4-6) (17.1 g, 22.5 mmol)
in anhydrous N,N-dimethylformamide (300 mL), and the
solution was stirred at 90 C for 3 hours. After the
10 sufficient progress of the reaction was confirmed, the
reaction solution was poured into cold water (400 mL),
and the resultant was extracted with ethyl acetate
(3 x 150 mL). The organic layers were combined with
each other, and the combination was washed with
15 saturated saline (2 x 100 mL), dried over sodium
sulfate, filtrated and concentrated under reduced
pressure. The resultant residue was purified by silica
gel chromatography (hexane/ethyl acetate,
4:1-*3:1-2:1_3:2) to yield the title compound (4-7)
20 {14.2 g (20.3 mmol), 90.0% yield}.
LRMS m/z 686 [M+Na]+.
Route G4; 3-0-[2,3-di-O-benzyl-4-0-(carbobenzoxy-
R-alanyl)-6-thioacetyl-a-D-quinovopyranosyl-glycerol
(4-8):
25 The compound (4-7) (14.2 g, 20.4 mmol) was
dissolved in a solution (200 mL) of t-butyl alcohol and
distilled water (4:1), and then thereto were added a
CA 02749797 2011-07-14
76
0.04 M osmium tetraoxide solution in t-butyl alcohol
(3 mL) and trimethylamine N-oxide (3.4 g, 30.5 mmol).
The solution was stirred with a stirrer at room
temperature for 24 hours. After the sufficient
progress of the reaction was confirmed, 3 g of
activated carbon was added thereto and then the
solution was stirred for 30 minutes to cause the
catalyst to be adsorbed on the carbon. The solution
was subjected to suction filtration through a Kiriyama
funnel containing celite, so as to remove the catalyst.
The reaction product remaining on the celite was washed
three times with ethyl acetate so as to be collected.
Distilled water (200 mL) was added to the collected
filtrate, and the resultant was extracted with ethyl
acetate (3 x 150 mL). The organic layers were combined
with each other, and the combination was washed with
saturated saline (2 x 100 mL), dried over sodium
sulfate, filtrated and concentrated under reduced
pressure. The resultant residue was purified by silica
gel chromatography (hexane/ethyl acetate,
2:11:1,2:3,1:2,1:4,1:8) to yield the title compound
(4-8) {13.2 g (18.9 mmol), 93.0% yield}.
LRMS m/z 720 [M+Na]+.
Route H4; 3-O-[2,3-di-O-benzyl-4-0-(carbobenzoxy-
P-alanyl)-6-thioacetyl-a-D-quinovopyranosyl]-1-0-
stearoyl-glycerol (4-9):
Stearoyl chloride (8.5 g, 28.2 mmol) was added to
CA 02749797 2011-07-14
77
a mixed solution of the compound (4-8) (13.1 g,
18.8 mmol) in anhydrous dichloromethane (200 mL) and
anhydrous pyridine (50 mL), and then the solution was
stirred at room temperature for 2 hours. After the
sufficient progress of the reaction was confirmed,
methanol (5 mL) was added thereto so as to terminate
the reaction. The solution was concentrated under
reduced pressure. A small amount of ethyl acetate was
used to suspend the residue, and the suspended residue
was poured into water (200 mL). The resultant was
extracted with ethyl acetate (3 x 100 mL). The organic
layers were combined with each other, and the
combination was washed with saturated saline
(2 x 100 mL), dried over sodium sulfate, filtrated and
then concentrated under reduced pressure. The
resultant residue was purified by silica gel
chromatography (hexane/ethyl acetate,
6:1-4:12:1-3:2-1:1) to yield the title compound (4-9)
as a colorless oily substance {7.1 g (7.4 mmol), 39.4%
yield}.
LRMS m/z 987 [M+Na]+.
Route 14; 3-O-[2,3-di-O-benzyl-4-0-(carbobenzoxy-
R-alanyl)-6-sulfo-a-D-quinovopyranosyl]-1-0-stearoyl-
glycerol (4-10):
Oxone (18.1 g, 29.5 mmol) and potassium acetate
(4.0 g) were added to a solution of the compound (4-9)
(7.1 g, 7.4 mmol) in acetic acid (160 g, 4 mol), and
CA 02749797 2011-07-14
78
then the solution was vigorously stirred at room
temperature for 48 hours. After the sufficient
progress of the reaction was confirmed, the reaction
solution was poured into a cold 7.5 M sodium hydroxide
solution (500 mL) and the resultant was extracted with
ethyl acetate (4 x 100 mL). The organic layers were
combined with each other, and the combination was
washed with saturated aqueous sodium hydrogen carbonate
(2 x 100 mL) and saturated saline (2 x 100 mL), dried
over sodium sulfate, filtrated and then concentrated
under reduced pressure. The resultant residue was
purified by silica gel chromatography
(chloroform/methanol, 100:1-+50:1-20:1-15:1-+12:1) to
yield the title compound (4-10) as a colorless waxy
substance {5.2 g (5.24 mmol), 71.2% yield}.
LRMS m/z 968 [M-Na]-.
Route J4; 3-0-[4-0-(8-alanyl)-6-sulfo-a-D-
quinovopyranosyl]-1-0-stearoyl-glycerol (4-11):
To a solution of the compound (4-10) (191 mg,
193 pmol) in methanol (10 mL), dichloromethane (10 mL)
and acetic acid (0.2 mL) was added 10% palladium-
activated carbon (160 mg), and then the solution was
stirred at room temperature under an atmosphere of
hydrogen gas for 48 hours. After the sufficient
progress of the reaction was confirmed, the palladium-
activated carbon was filtrated off. The filtrate was
concentrated under reduced pressure. The resultant
CA 02749797 2011-07-14
79
residue was purified by silica gel chromatography
(chloroform/methanol/distilled water,
100:25:3-65:25:4,65:35:5,65:45:6) to yield the title
compound (4-11).
LRMS m/z 654 [M-Na]-.
Route K4; 3-0-[4-0-(2,3,5-triiodobenzoyl-R-
alanyl)-6-sulfo-a-D-quinovopyranosyl]-1-0-stearoyl-
glycerol (4-12):
Into a mixed solution of anhydrous dichloromethane
(10 mL) and anhydrous pyridine (5 mL) were dissolved
the compound (4-11), 2,3,5-triiodobenzoic acid (192 mg,
385 pmol), 1-ethyl-3-(3-
dimethylaminopropyl)carbodiimide hydrochloride
(EDCI=HC1) (73.8 mg, 385 pmol) and 4-
dimethylaminopyridine (2 g, 19 pmol), and then the
reactive components were caused to react with each
other at room temperature for 18 hours. After the
sufficient progress of the reaction was confirmed,
water (10 mL) was poured into the reaction solution to
terminate the reaction. Thereafter, the solution was
concentrated under reduced pressure. The resultant
residue was purified by silica gel chromatography
(chloroform/methanol/distilled water,
50:10:140:10:1.2-30:10:1.5,20:10:2) to yield the title
compound (4-12) {81 mg (70 pmol), 36.3% yield, which
was the yield through the two routes J4 and K4}.
LRMS m/z 1136 [M - Na]-.
CA 02749797 2011-07-14
Synthesis Example 5
A process for producing the
sulfopyranosylacylglycerol derivative according to an
embodiment of the present invention is illustrated in
5 the following scheme 5, giving an
a-sulfoquinovosyldiacylglycerol monoiodide derivative
as an example thereof:
CA 02749797 2011-07-14
81
GZ tit.
cn
I
u)
1-11
1
O O
N O
O
to %
u L.+
Oro
0 O
o O
O
O
fO O O=<
y
0
Q.
LO U")
`~ U W
IC) LO LO
v Q ~ ~
Q O O
0 r- a
fl3C qC
'ca co
0 0
ax:
n = ca 000
cv
IL
U
Ul
CA 02749797 2011-07-14
82
S
0 s
0
0 0 (j
O vs Q
lid {3 n U
Lot
r. x O U N tJ
W) O. i O O
LO ltd
O C) O
O O O O O
0
om m O
0 o
O
z:r z2
o OT`C zz
0 0 0
17
V
Y/ X
Ul) r. ,=~ o l O
rf = Q
_ v
U o. 0 u
00. V O
< LO
m
O
O O ~]
o U)
o
0
0
ox
Z3:
z~
~ O
O 0
CA 02749797 2011-07-14
83
The following will describe a synthesis example
through the individual steps in detail:
<Synthesis Example>
Route A5; 1-0-allyl-4,6-0-benzylidene-a-D-
glucopyranoside (5-2):
Trifluoromethanesulfonic acid (1.00 mL) was added
to a liquid suspension of the compound (5-1) (100 g,
555 mmol) in allyl alcohol (500 mL) at 0 C, and the
reaction liquid was vigorously stirred at 80 C for
48 hours. After the sufficient progress of the
reaction was confirmed, thereto was added triethylamine
(3 mL) to terminate the reaction. The resultant was
concentrated under reduced pressure. Next, the residue
was suspended in anhydrous acetonitrile (500 mL), and
thereto were added benzaldehydedimethylacetal (127 g,
1.5 equivalent) and p-toluenesulfonic acid monohydrate
(5.28 g, 0.05 equivalent). The resultant liquid was
stirred at 40 C for 4 hours, and then thereto was added
triethylamine (10 mL) to terminate the reaction. The
resultant was concentrated under reduced pressure. The
residue was poured into hexane (2000 mL) and water
(500 mL), and then the mixed liquid was vigorously
stirred. The resultant precipitate was filtrated, and
rinsed with water and hexane. The precipitate was
crystallized two times from heated ethanol to yield the
title compound (5-2) as a colorless needle crystal
{34.5 g (112 mmol), 20.2% yield}.
CA 02749797 2011-07-14
84
LRMS m/z 331 [M+Na]+.
Route B5; 1-0-allyl-2,3-di-0-benzyl-4,6-0-
benzylidene-a-D-glucopyranoside (5-3):
To a solution of the compound (5-2) (30.0 g,
97.3 mmol) in anhydrous N,N-dimethylformamide (DMF,
300 mL) were added benzylbromide (41.6 g, 2.5
equivalent) and sodium hydroxide (11.7 g, 3.0
equivalent), and the reaction solution was vigorously
stirred at room temperature for 24 hours. After the
sufficient progress of the reaction was confirmed, the
reaction solution was poured into cold water (900 mL),
and the resultant was extracted with ethyl acetate
(3 x 300 mL). The organic layers were combined with
each other, and the combination was washed with
saturated saline (2 x 100 mL), dried over sodium
sulfate, filtrated and then concentrated under reduced
pressure. The resultant residue was crystallized
two times from heated ethanol to yield the title
compound (5-3) as a colorless needle crystal (33.5 g).
The filtrate was concentrated, purified by silica gel
chromatography (hexane/ethyl acetate, 15:1-10:18:1),
and then crystallized from heated ethanol to yield the
same compound (5-3) (6.63 g) {total amount: 40.1 g
(82.1 mmol), 84.4% yield}.
LRMS m/z 511 [M+Na]+.
Route C5; 1-0-ally-2,3-di-0-benzyl-a-D-
glucopyranoside (5-4):
CA 02749797 2011-07-14
The compound (5-3) (15.6 g, 32.0 mmol) was
dissolved in acetic acid (90 mL), and further distilled
water (50 mL) was added thereto. The solution was
stirred for 1 hour while heated and refluxed. After
5 the sufficient progress of the reaction was confirmed,
the solution was cooled to room temperature, and then
the solvent was removed by evaporation. Distilled
water (15 mL) was added thereto, and the solution was
again concentrated under reduced pressure; this
10 operation was repeated 4 times. Thereafter, the
resultant was purified by silica gel flash
chromatography (hexane/ethyl acetate, 4:1-2:1-1:1-1:2)
to yield the title compound (5-4) {12.1 g (30.2 mmol),
94.5% yield}.
15 LRMS m/z 423 [M+Na]+.
Route D5; 1-0-ally-2,3,4-di-0-benzyl-6-0-tosyl-a-
D-glucopyranoside (5-5):
To a solution of the compound (5-4) (12.1 g,
30.2 mmol) in anhydrous pyridine (120 mL) were added p-
20 toluenesulfonyl chloride (7.5 g, 39.3 mmol) and 4-
dimethylaminopyridine (369 mg, 3.0 mmol), and the
reaction solution was stirred at 0 C for 16 hours.
After the sufficient progress of the reaction was
confirmed, the reaction solution was slowly poured into
25 ice water (100 mL). The water layer was subjected to
extraction with ethyl acetate (3 x 200 mL). The
organic layers were combined with each other. The
CA 02749797 2011-07-14
86
combination was washed with an aqueous 1 N HC1 solution
until the pH became 4, washed with saturated aqueous
sodium hydrogen carbonate (2 x 100 mL) and saturated
saline (2 x 100 mL), dried over sodium sulfate,
filtrated and then concentrated under reduced pressure.
The concentrated product was purified by silica gel
flash chromatography (hexane/ethyl acetate,
6:1-*4:1,2:1) to yield the title compound (5-5) {15.3 g
(27.6 mmol), 91.4% yield}.
LRMS m/z 577 [M+Na]+.
Route E5; 1-0-allyl-2,3-di-0-benzyl-4-0-
(carbobenzoxy-(3-alanyl)-6-0-tosyl-a-D-glucopyranoside
(5-6) :
Into a mixed solution of anhydrous dichloromethane
(200 mL) and anhydrous pyridine (50 mL) were dissolved
the compound (5-5) (15.3 g, 27.6 mmol), N-carbobenzoxy-
(3-alanine (12.32 g, 55.2 mmol), 1-ethyl-3-(3-
dimethylaminopropyl)carbodiimide hydrochloride
(EDCI=HC1) (15.8 mg, 82.8 mmol) and 4-
dimethylaminopyridine (6.7 g, 55.2 mmol), and then the
reactive components were caused to react with each
other at room temperature for 18 hours. After the
sufficient progress of the reaction was confirmed,
water (10 mL) was poured into the reaction solution to
terminate the reaction. Thereafter, the solution was
concentrated under reduced pressure. The resultant
residue was purified by silica gel chromatography
CA 02749797 2011-07-14
87
(hexane/ethyl acetate, 4:1-X3:1-X2:1--+3:2) to yield the
title compound (5-6) {17.1 g (22.5 mmol), 81.5% yield}.
LRMS m/z 782 [M+Na]+.
Route F5; 1-0-allyl-2,3-di-O-benzyl-4-0-
(carbobenzoxy-[3-alanyl)-6-thioacetyl-a-D-
glucopyranoside (5-7):
Potassium thioacetate (5.1 g, 45.0 mmol) was added
to a solution of the compound (5-6) (17.1 g, 22.5 mmol)
in anhydrous N,N-dimethylformamide (300 mL), and the
solution was stirred at 90 C for 3 hours. After the
sufficient progress of the reaction was confirmed, the
reaction solution was poured into cold water (400 mL),
and the resultant was extracted with ethyl acetate
(3 x 150 mL). The organic layers were combined with
each other, and the combination was washed with
saturated saline (2 x 100 mL), dried over sodium
sulfate, filtrated and concentrated under reduced
pressure. The resultant residue was purified by silica
gel chromatography (hexane/ethyl acetate,
4:1--,3:1,2:1,3:2) to yield the title compound (5-7)
{14.2 g (20.3 mmol), 90.0% yield}.
LRMS m/z 686 [M+Na]+.
Route G5; 3-O-[2,3-di-O-benzyl-4-0-(carbobenzoxy-
P-alanyl)-6-thioacetyl-a-D-quinovopyranosyl-glycerol
(5-8):
The compound (5-7) (14.2 g, 20.4 mmol) was
dissolved in a solution (200 mL) of t-butyl alcohol and
CA 02749797 2011-07-14
88
distilled water (4:1), and then thereto were added a
0.04 M osmium tetraoxide solution in t-butyl alcohol
(3 mL) and trimethylamine N-oxide (3.4 g, 30.5 mmol).
The solution was stirred with a stirrer at room
temperature for 24 hours. After the sufficient
progress of the reaction was confirmed, 3 g of
activated carbon was added thereto and then the
solution was stirred for 30 minutes to cause the
catalyst to be adsorbed on the carbon. The solution
was subjected to suction filtration through a Kiriyama
funnel containing celite, so as to remove the catalyst.
The reaction product remaining on the celite was washed
three times with ethyl acetate so as to be collected.
Distilled water (200 mL) was added to the collected
filtrate, and the resultant was extracted with ethyl
acetate (3 x 150 mL). The organic layers were combined
with each other, and the combination was washed with
saturated saline (2 x 100 mL), dried over sodium
sulfate, filtrated and concentrated under reduced
pressure. The resultant residue was purified by silica
gel chromatography (hexane/ethyl acetate,
2:1-1:1-2:3-1:2-1:4-1:8) to yield the title compound
(5-8) {13.2 g (18.9 mmol), 93.0% yield}.
LRMS m/z 720 [M+Na]+.
Route H5; 3-0-[2,3-di-O-benzyl-4-0-(carbobenzoxy-
a-alanyl)-6-thioacetyl-a-D-quinovopyranosyl]-1,2-di-0-
stearoyl-glycerol (5-9):
CA 02749797 2011-07-14
89
Stearoyl chloride (8.5 g, 28.2 mmol) was added to
a mixed solution of the compound (5-8) (13.1 g,
18.8 mmol) in anhydrous dichloromethane (200 mL) and
anhydrous pyridine (50 mL), and then the solution was
stirred at room temperature for 2 hours. After the
sufficient progress of the reaction was confirmed,
methanol (5 mL) was added thereto so as to terminate
the reaction. The solution was concentrated under
reduced pressure. A small amount of ethyl acetate was
used to suspend the residue, and the suspended residue
was poured into water (200 mL). The resultant was
extracted with ethyl acetate (3 x 100 mL). The organic
layers were combined with each other, and the
combination was washed with saturated saline
(2 x 100 mL), dried over sodium sulfate, filtrated and
then concentrated under reduced pressure. The
resultant residue was purified by silica gel
chromatography (hexane/ethyl acetate,
6:1-4:1-2:1-3:21:1) to yield the title compound (5-9)
as a colorless oily substance {12.2 g (9.9 mmol), 52.8%
yield}.
LRMS m/z 1253 [M+Na]+.
Route 15; 3-O-[2,3-di-O-benzyl-4-0-(carbobenzoxy-
R-alanyl)-6-sulfo-a-D-quinovopyranosyl]-1,2-di-0-
stearoyl-glycerol (5-10):
Oxone (24.4 g, 39.7 mmol) and potassium acetate
(3.8 g) were added to a solution of the compound (5-9)
CA 02749797 2011-07-14
(12.2 g, 9.9 mmol) in acetic acid (150 g, 9.9 mol), and
then the solution was vigorously stirred at 45 C for
48 hours. After the sufficient progress of the
reaction was confirmed, the reaction solution was
5 poured into a cold 7.5 M sodium hydroxide solution
(500 mL) and the resultant was extracted with ethyl
acetate (4 x 100 mL). The organic layers were combined
with each other, and the combination was washed with
saturated aqueous sodium hydrogen carbonate
10 (2 x 100 mL) and saturated saline (2 x 100 mL), dried
over sodium sulfate, filtrated and then concentrated
under reduced pressure. The resultant residue was
purified by silica gel chromatography
(chloroform/methanol, 100:1-50:1-20:1-15:1-12:1) to
15 yield the title compound (5-10) as a colorless waxy
substance {8.6 g (6.97 mmol), 70.3% yield}.
LRMS m/z 1235 [M-Na]-.
Route J5; 3-0-[4-0-(3-alanyl)-6-sulfo-a-D-
quinovopyranosyl]-1,2-di-0-stearoyl-glycerol sodium
20 salt (5-11):
To a solution of the compound (5-10) (480.8 mg,
382 pmol) in methanol (20 mL), chloroform (10 mL) and
acetic acid (0.5 mL) was added 10% palladium-activated
carbon (300 mg), and then the solution was stirred at
25 room temperature under an atmosphere of hydrogen gas
for 48 hours. After the sufficient progress of the
reaction was confirmed, the palladium-activated carbon
CA 02749797 2011-07-14
91
was filtrated off. The filtrate was concentrated under
reduced pressure. The resultant residue was purified
by silica gel chromatography
(chloroform/methanol/distilled water,
100:25:3-465:25:4---65:35:5-65:45:6) to yield the title
compound (5-11).
LRMS m/z 921 [M-Na]-.
Route K5; 3-0-[4-0-(4-iodobenzoyl-fi-alanyl)-6-
sulfo-a-D-quinovopyranosyl]-1,2-di-O-stearoyl-glycero l
sodium salt (5-12):
The compound (5-11) was dissolved in a mixed
solution of anhydrous dichloromethane (15 mL), pyridine
(5 mL) and triethylamine (2 mL), and thereto was added
(4-iodobenzoyl) p-nitrophenyl ester (635 mg,
1.72 mmol). While the solution was stirred with a
stirrer, the reactive components were caused to react
with each other at room temperature for 24 hours.
After the sufficient progress of the reaction was
confirmed, toluene and methanol were added to the
solution. Under reduced pressure, the solvents were
then removed by evaporation to concentrate the
solution, while azeotropy. The resultant residue was
purified by silica gel chromatography
(chloroform/methanol/distilled water,
80:10:0.8-70:10:1.0-65:10:1.0-60:10:1.0) to yield the
title compound (5-12) {279 mg (238 pmol), 55.2% yield,
which was the yield through the two reactions J5
CA 02749797 2011-07-14
92
and K5}.
LRMS m/z 1151 [M - Na]-.
Synthesis Example 6
A process for producing the
sulfopyranosylacylglycerol derivative according to an
embodiment of the present invention is illustrated in
the following scheme 6, giving an a-
sulfoquinovosylacylglycerol monoiodide derivative as an
example thereof:
CA 02749797 2011-07-14
93
co to
co a
N d' t?
to
0 r~ 0 0
,
0 0
m
0 0
oOO 0Z 0co
w }"
Q
Q U ww
C'') Lt)
= Cwt) to
0
0
a
o o
C 0 0
0 0
~ c a:
0
W
0
z
u
CA 02749797 2011-07-14
94
tot
M
o =
U ,- U
co 0
ca 0
O z 0
cQi~ O O O
0 0
r-I
C
~m om
z
r T N.
o
{.3
-~ O co 0 0 v O 0
O _ f 0
0 Lc~~
O W 0
z
O 'mi
0
W Co COP ~0
CA 02749797 2011-07-14
The following will describe a synthesis example
through the individual steps in detail:
<Synthesis Example>
Route A6; 1-0-allyl-4,6-0-benzylidene-a-D-
5 glucopyranoside (6-2):
Trifluoromethanesulfonic acid (1.00 mL) was added
to a liquid suspension of the compound (6-1) (100 g,
555 mmol) in allyl alcohol (500 mL) at 0 C, and the
reaction liquid was vigorously stirred at 80 C for
10 48 hours. After the sufficient progress of the
reaction was confirmed, thereto was added triethylamine
(3 mL) to terminate the reaction. The resultant was
concentrated under reduced pressure. Next, the residue
was suspended in anhydrous acetonitrile (500 mL), and
15 thereto were added benzaldehydedimethylacetal (127 g,
1.5 equivalent) and p-toluenesulfonic acid monohydrate
(5.28 g, 0.05 equivalent). The resultant liquid was
stirred at 40 C for 4 hours, and then thereto was added
triethylamine (10 mL) to terminate the reaction. The
20 resultant was concentrated under reduced pressure. The
residue was poured into hexane (2000 mL) and water
(500 mL), and then the mixed liquid was vigorously
stirred. The resultant precipitate was filtrated, and
rinsed with water and hexane. The precipitate was
25 crystallized two times from heated ethanol to yield the
title compound (6-2) as a colorless needle crystal
{34.5 g (112 mmol), 20.2% yield}.
CA 02749797 2011-07-14
96
LRMS m/z 331 [M+Na]+.
Route B6; 1-0-(2-propenyl)-a-D-glucose (6-3):
The compound (6-2) (10.7 g, 34.7 mmol) was
dissolved in a solution (260 mL) of acetic acid and
water (8:5), and the reactive components were caused to
react with each other at 100 C for 1 hour. The
solution was concentrated under reduced pressure, and
purified by silica gel flash chromatography
(dichloromethane/methanol, 6:1) to yield the title
compound (6-3) (6.3 g (28.6 mmol), 82.4% yield).
Route C6; 1-0-(2-propenyl)-6-0-(4-tolylsulfonyl)-
a-D-glucose (6-4):
The compound (6-3) (6.3 g, 28.6 mmol) was
dissolved in dry pyridine (200 mL), and thereto were
added p-dimethylaminopyridine (DMAP) (195 mg), and p-
toluenesulfonyl chloride (7.0 g). While the solution
was stirred, the reactive components were caused to
react with each other at room temperature for 16 hours.
Thereafter, thereto was added cold distilled water
(20 mL) to terminate the reaction. The resultant was
extracted with ethyl acetate (3 x 200 mL), and the
organic layers were combined with each other. The
combination was neutralized to a pH of 4 with 1.0 M
hydrochloric acid and 0.1 M hydrochloric acid, washed
with saturated saline (2 x 200 mL), dried over
anhydrous sodium sulfate, filtrated, concentrated under
reduced pressure, and then purified by silica gel flash
CA 02749797 2011-07-14
97
chromatography (dichloromethane/methanol, 20:1) to
yield the title compound (6-4) (8.6 mg (23.0 mmol),
83.8% yield).
Route D6; 2,3,4-tri-O-(t-butyldimethylsilyl)-1-0-
(2-propenyl)-6-0-(4-tolylsulfonyl)-a-D-glucose (6-5):
The compound (6-4) (11.2 g, 29.9 mmol) was
dissolved in dry dichloromethane (25 mL), and then
thereto were added t-butyldimethylsilyltrifluoromethane
sulfonate (23.8 g) and 2,6-lutidine (14.4 g). While
the solution was stirred under the flow of nitrogen
gas, the reaction components were caused to react with
each other for 16 hours. Thereafter, dichloromethane
(150 mL) was added thereto so as to terminate the
reaction. The solution was washed with saturated
saline (2 x 100 mL), dried over anhydrous sodium
sulfate, filtrated, concentrated under reduced
pressure, and then purified by silica gel flash
chromatography (hexane/ethyl acetate, 30:1) to yield
the title compound (6-5) (19.6 g (27.4 mmol), 91.6%
yield).
Route E6; 2,3,4-tri-O-(t-butyldimethylsilyl)-1-0-
(2-propenyl)-6-deoxy-6-acetylthio-a-D-glucose (6-6):
The compound (6-5) (7.9 g, 11.0 mmol) was
dissolved in dry ethanol (20 mL), and thereto were
added potassium thioacetate (1.8 g). While the
solution was stirred under reflux conditions, the
reactive components were caused to react with each for
CA 02749797 2011-07-14
98
3 hours. Thereafter, thereto was added cold distilled
water (100 mL) to terminate the reaction. The
resultant was extracted with ethyl acetate
(3 x 200 mL), and the organic layers were combined with
each other. The combination was washed with saturated
saline (2 x 200 mL), dried over anhydrous sodium
sulfate, filtrated, concentrated under reduced
pressure, and then purified by silica gel flash
chromatography (hexane/ethyl acetate, 50:1) to yield
the title compound (6-6) (5.6 g (9.02 mmol), 82.0%
yield).
Route F6; 3-O-[2,3,4-tri-O-(t-butyldimethylsilyl)-
6-deoxy-6-acetylthio-a-D-glucopyranosyl]-glycerol
(6-7):
The compound (6-6) (5.6 g, 9.02 mmol) was
dissolved in a solution of t-butyl alcohol and water
(4:1), and then thereto were added trimethylamine N-
oxide dehydrate (1.5 g) and a 0.04 M osmium tetraoxide
solution in t-butyl alcohol (15 mL). While the
solution was stirred, the reactive components were
caused to react with each other at room temperature for
22 hours. Thereafter, activated carbon (15 g) was
added thereto. While stirred, the solution was allowed
to stand at room temperature for 1.5 hours to cause
osmium tetraoxide to be adsorbed thereon. The solution
was then subjected to suction filtration. Next, cold
distilled water (200 mL) was added thereto so as to
CA 02749797 2011-07-14
99
terminate the reaction. The resultant was extracted
with ethyl acetate (3 x 200 mL). The organic layers
were combined with each other, and the combination was
washed with saturated saline (2 x 300 mL), dried over
anhydrous sodium sulfate, filtrated, concentrated under
reduced pressure, and then purified by silica gel
chromatography (hexane/ethyl acetate, 3:11:2) to yield
the title compound (6-7) {5.2 g (7.94 mmol), 88.0%
yield}.
Route G6; 3-0-[2,3,4-0-tert-butyldimethylsilyl-6-
thioacetyl-a-D-quinovopyranosyl]-1-0-stearoyl-glycerol
(6-8):
Stearoyl chloride (692 mg, 2.3 mmol) was added to
a mixed solution of the compound (6-7) (1.4 g,
6.1 mmol) in anhydrous dichloromethane (20 mL) and
anhydrous pyridine (5 mL), and then the solution was
stirred at room temperature for 2 hours. After the
sufficient progress of the reaction was confirmed,
methanol (1 mL) was added thereto so as to terminate
the reaction. The solution was concentrated under
reduced pressure. A small amount of ethyl acetate was
used to suspend the residue, and the suspended residue
was poured into water (20 mL). The resultant was
extracted with ethyl acetate (3 x 20 mL). The organic
layers were combined with each other, and the
combination was washed with saturated saline
(2 x 20 mL), dried over sodium sulfate, filtrated and
CA 02749797 2011-07-14
100
then concentrated under reduced pressure. The
resultant residue was purified by silica gel
chromatography (hexane/ethyl acetate, 10:1-48:1-6:1-4:1)
to yield the title compound (6-8) as a colorless oily
substance {l.6 g (1.7 mmol), 83.6% yield}.
LRMS m/z 944 [M+Na]+.
Route H6; 3-0-[2,3,4-0-tert-butyldimethylsilyl-6-
thioacetyl-a-Dquinovopyranosyl]-1-0-stearoyl-2-0-(4-
iodobenzoyl)-glycerol (6-9):
4-Benzoyl iodochloride (555 mg, 2.1 mmol) was
added to a mixed solution of the compound (6-8) (1.6 g,
1.7 mmol) in anhydrous dichloromethane (20 mL) and
anhydrous pyridine (5 mL), and then the solution was
stirred at room temperature for 2 hours. After the
sufficient progress of the reaction was confirmed,
methanol (1 mL) was added thereto so as to terminate
the reaction. The solution was concentrated under
reduced pressure. A small amount of ethyl acetate was
used to suspend the residue, and the suspended residue
was poured into water (20 mL). The resultant was
extracted with ethyl acetate (3 x 20 mL). The organic
layers were combined with each other, and the
combination was washed with saturated saline
(2 x 20 mL), dried over sodium sulfate, filtrated and
then concentrated under reduced pressure. The
resultant residue was purified by silica gel
chromatography (hexane/ethyl acetate,
CA 02749797 2011-07-14
101
12:1-10:1-8:1-*6:1-+5:1-4:1) to yield the title compound
(6-9) as a colorless oily substance {l.7 g (1.5 mmol),
85.0% yield}.
LRMS m/z 1174 [M+Na]+.
Route 16; 3-0-[2,3,4-0-tert-butyldimethylsilyl-6-
sulfo-a-D-quinovopyranosyl]-1-0-stearoyl-2-0-(4-
iodobenzoyl)-glycerol (6-10):
Oxone (2.7 g, 4.4 mmol) and potassium acetate
(1.25 g) were added to a solution of the compound (6-9)
(1.7 g, 1.5 mmol) in acetic acid (50 mL), and then the
solution was vigorously stirred at room temperature for
48 hours. After the sufficient progress of the
reaction was confirmed, ethyl acetate (200 mL) was
poured into the reaction solution. The solution was
washed with saturated saline (2 x 50 mL), dried over
sodium sulfate, and filtrated. Thereafter, ethyl
acetate was removed therefrom under reduced pressure.
The resultant residue containing acetic acid was
obtained as the title compound (6-10), and was to be
used as it was for the next reaction.
Route J6; 3-0-[6-sulfo-a-D-quinovopyranosyl]-1-0-
stearoyl-2-0-(4-iodobenzoyl)-glycerol (6-11):
Tetrahydrofuran (1.5 mL) and water (1.5 mL) were
added to the mixed solution containing the compound
(6-10) and acetic acid, and further trifluoroacetic
acid (0.3 mL) was added thereto. The solution was then
stirred at room temperature for 48 hours. After the
CA 02749797 2011-07-14
102
sufficient progress of the reaction was confirmed, the
solution was concentrated under reduced pressure. The
resultant residue was purified by silica gel
chromatography (chloroform/methanol/distilled water,
100:10:0.5-80:10:0.5,60:10:1-,40:10:160:25:2) to yield
the title compound (6-11) {752 mg (899 pmol) through
the two reactions in the routes 16 and J6, 60.9%
yield}.
LRMS m/z 813 [M-Na]-.
Synthesis Example 7
A process for producing the
sulfopyranosylacylglycerol derivative according to an
embodiment of the present invention is illustrated in
the following scheme 7, giving an a-
sulfoquinovosylacylglycerol triiodide derivative as an
example thereof:
CA 02749797 2011-07-14
103
m t~i.
c%j
0
0
Q qfC 0 4 q
C,
00 0 r 02 C z tH
M
n
Q U w
'-' M tt}
n N ' I
z
0
0
0
0 0 o_
~ O q..'Ca
v ox m
U H
s F-
CA 02749797 2011-07-14
104
M M
/~ rr t~T
U n
co O
0 - O O
p O
O O" O
O O
in co
0 0
m o
2
O
tv~ O O O O O
O p O
< 0 O O O 0 0 O
U) 0
W 2
r, C
OV) O
U)CO OM
CA 02749797 2011-07-14
105
The following will describe a synthesis example
through the individual steps in detail:
<Synthesis Example>
The compound (7-8) was yielded by the same process
as in the routes A6 to G6 of Synthesis Example 6.
Route H7; 3-0-[2,3,4-0-tert-butyldimethylsilyl-6-
thioacetyl-a-Dquinovopyranosyl]-1-O-stearoyl-2-O-
(2,3,5-triiodobenzoyl)-glycerol (7-9):
Into anhydrous dichloromethane (10 mL) were
dissolved the compound (7-8) (467 mg, 507 pmol), 2,3,5-
triiodobenzoic acid (303.8 mg, 608 pmol), di-2-pyridyl
carbonate (DPC) (219 mg, 1013 pmol), and 4-
dimethylaminopyridine (6.2 mg, 51 pmol). The reactive
components were caused to react with each other at room
temperature for 48 hours. After the sufficient
progress of the reaction was confirmed, water (30 mL)
was poured into the reaction solution to terminate the
reaction. The resultant was then extracted with
dichloromethane (3 x 20 mL). The organic layers were
combined with each other, and the combination was
washed with saturated saline (2 x 20 mL), dried over
sodium sulfate, filtrated and then concentrated under
reduced pressure. The resultant residue was purified
by silica gel chromatography (hexane/ethyl acetate,
12:l-10:l-8:1-6:1-5:l-4:l) to yield the title compound
(7-9) as a colorless oily substance {245 mg (174 pmol),
34.4% yield}.
CA 02749797 2011-07-14
106
LRMS m/z 1425 [M + Na]-.
Route 17; 3-0-[2,3,4-0-tert-butyldimethylsilyl-6-
sulfo-a-D-quinovopyranosyl]-1-0-stearoyl-2-O-(2,3,5-
triiodobenzoyl)-glycerol (7-10):
Oxone (322 mg, 523 pmol) and potassium acetate
(250 mg) were added to a solution of the compound (7-9)
(245 mg, 174 pmol) in acetic acid (10 mL), and then the
solution was vigorously stirred at room temperature for
24 hours. After the sufficient progress of the
reaction was confirmed, ethyl acetate (50 mL) was
poured into the reaction solution. The solution was
washed with saturated saline (2 x 20 mL), and the
organic layer was dried over sodium sulfate, filtrated,
and then concentrated under reduced pressure. The
resultant residue containing acetic acid was to be
used, as it was, as the title compound (7-10) for the
next reaction.
Route J7; 3-0-[6-sulfo-a-D-quinovopyranosyl]-1-0-
stearoyl-2-0-(2,3,5-triiodobenzoyl)-glycerol (7-11):
Tetrahydrofuran (5 mL), acetic acid (10 mL) and
water (5 mL) were added to the mixed solution
containing the compound (7-10) yielded through the
route 17 and acetic acid, and further trifluoroacetic
acid (1 mL) was added thereto. The solution was then
stirred at room temperature for 48 hours. After the
sufficient progress of the reaction was confirmed, the
solution was concentrated under reduced pressure. The
CA 02749797 2011-07-14
107
resultant residue was purified by silica gel
chromatography (chloroform/methanol/distilled water,
100:10:0.5-80:10:0.5-+60:10:1-40:10:1-60:25:2) to yield
the title compound (7-11) {102 mg (94 pmol) through the
two reactions in the routes I7 and J7, 53.7% yield}.
LRMS m/z 1065 [M-Na]-.
Synthesis Example 8
A process for producing the
sulfopyranosylacylglycerol derivative according to an
embodiment of the present invention is illustrated in
the following scheme 8, giving a R-
sulfoquinovosylmonoacylglycerol monoiodide derivative
as an example thereof:
CA 02749797 2011-07-14
108
00
co
uco-f Cn t~ 2:
I
co
v
/ 0
! - o
0
co 0
0 0 0 00
co
0
S _ m
0 0 0 44~)
z
00 z
aka `i 0=~
Q o
r
a
U w coot
Un
co
U ~--~
0 0 0
0 0 0 a O
T m m
00
=0,= 0
cca
0=
U
CA 02749797 2011-07-14
109
x
0
ti
U
O 0
0
0
f 0_
CC) Q
O
vi 0 J
x r
0 0 0 0
Om .. ~. co
0
co r N ..i
Q
v 0 00
0 Q2
U 0
Zx 0
0=<
0 O o
ZS
0 0 0
d rI
~` v3 0 Q
Q Q
m
CO t
0 co
ca m
0 0
x
6
0
,-, X
ai 0 0
o
m o
co o
Q
M
00
Q
co O
Zx
0
x
O coot ox
O
Z
2
CA 02749797 2011-07-14
110
The following will describe a synthesis example
through the individual steps in detail:
<Synthesis Example>
Route A8; 2,3,4,6-tetra-0-acetyl-1-0-allyl-R-D-
glucopyranoside (8-2):
A solution (500 mL) of the compound (8-1) (50.0 g,
128 mmol), allyl alcohol (22.3 g, 384 mmol), and zinc
chloride (17.4 g, 128 mmol) in toluene was vigorously
stirred at 80 C under an atmosphere of nitrogen for
48 hours. After the sufficient progress of the
reaction was confirmed, the temperature of the solution
was returned to room temperature. The solution was
then washed with saturated aqueous sodium hydrogen
carbonate (2 x 100 mL) and saturated saline
(2 x 100 mL), dried over sodium sulphate, filtrated and
then concentrated under reduced pressure. The
concentrated product was crystallized two times from
heated ethanol to yield the title compound (8-2) as a
colorless needle crystal {24.8 g (64 mmol), 50.0%
yield}.
LRMS m/z 411 [M+Na]+.
Route B8; 1-0-allyl-3-D-glucopyranoside (8-3):
The compound (8-2) (21.0 g, 54 mmol) was dissolved
in methanol (200 mL). While the solution was stirred
at room temperature under an atmosphere of nitrogen,
thereto was added a 28% solution (1 mL) of sodium
methylate in methanol. The solution was then stirred
CA 02749797 2011-07-14
111
at room temperature for 2 hours. After the sufficient
progress of the reaction was confirmed, the reaction
was terminated. The reaction solution was
concentrated, as it was, under reduced pressure, and a
crude product of the resultant title compound (8-3) was
to be used for the next reaction.
LRMS m/z 243 [M+Na]+.
Route C8; 1-0-allyl-4,6-0-benzylidene-R-D-
glucopyranoside (8-4):
The compound (8-3) (11.9 g, 54 mmol) was suspended
in anhydrous acetonitrile (60 mL), and thereto were
added benzaldehyde dimethylacetal (16.4 g, 2.0
equivalent) and p-toluenesulfonic acid monohydrate
(1.0 g, 0.05 equivalent). The reaction liquid was
stirred at 40 C for 4 hours, and then triethylamine
(0.5 mL) was added thereto so as to terminate the
reaction. The resultant was concentrated under reduced
pressure. The concentrated product was dissolved in a
small amount of ethyl acetate, and then cold water
(200 mL) was added thereto. The resultant was
extracted with ethyl acetate (3 x 100 mL). The organic
layers were combined with each other, and the
combination was washed with saturated saline
(2 x 50 mL), dried over sodium sulfate, filtrated and
then concentrated under reduced pressure. The
resultant residue was crystallized two times from
heated ethanol to yield the title compound (8-4) as a
CA 02749797 2011-07-14
112
colorless needle crystal. The filtrate was further
concentrated, purified by silica gel chromatography
(chloroform/methanol, 100:1-50:1,25:1-20:1), and then
crystallized from heated ethanol to yield the same
compound (8-4) {11.6 g (37.6 mmol), 69.6% yield}.
LRMS m/z 311 [M+Na]+.
Route D8; 1-0-allyl-2,3-di-0-benzyl-4,6-0-
benzylidene-3-D-glucopyranoside (8-5):
To a solution of the compound (8-4) (11.6 g,
37.6 mmol) in anhydrous N,N-dimethylformamide (DMF,
100 mL) were added benzylchloride (4.76 g, 4
equivalent) and sodium hydroxide powder (4.5 g, 3.0
equivalent), and the reaction solution was vigorously
stirred at room temperature for 24 hours. After the
sufficient progress of the reaction was confirmed, the
reaction solution was poured into cold water (100 mL),
and the resultant was extracted with ethyl acetate
(3 x 50 mL). The organic layers were combined with
each other, and the combination was washed with
saturated saline (2 x 30 mL), dried over sodium
sulfate, filtrated and then concentrated under reduced
pressure. The resultant residue was crystallized
two times from heated ethanol to yield the title
compound (8-5) as a colorless needle crystal. The
filtrate was concentrated, purified by silica gel
chromatography (hexane/ethyl acetate,
12:1.10:1-8:1-6:1-4:1), and then crystallized from
CA 02749797 2011-07-14
113
heated ethanol to yield the same compound (8-5).
The resultant compound was collected into a
flask, and was to be used as it was for the next
reaction.
LRMS m/z 511 [M+Na]+.
Route E8; 1-0-ally-2,3-di-O-benzyl-R-D-
glucopyranoside (8-6):
The compound (8-5) yielded through the route D8
was dissolved in acetic acid (72 mL), and further
distilled water (40 mL) was added thereto. The
solution was stirred for 1 hour while heated and
refluxed. After the sufficient progress of the
reaction was confirmed, the solution was cooled to room
temperature, and then the solvent was removed by
evaporation. Distilled water (15 mL) was added
thereto, and the solution was again concentrated under
reduced pressure; this operation was repeated 4 times.
Thereafter, the resultant residue was dissolved in
methanol (150 mL). While the solution was stirred at
room temperature under an atmosphere of nitrogen,
thereto was added a 28% solution (0.7 mL) of sodium
methylate in methanol. The solution was then stirred
at room temperature for 2 hours. After the sufficient
progress of the reaction was confirmed, the reaction
was terminated. The reaction solution was
concentrated, as it was, under reduced pressure, and
purified by silica gel flash chromatography
CA 02749797 2011-07-14
114
(hexane/ethyl acetate, 4:1-3:1->2:1,3:2-X1:1->2:3,1:2) to
yield the title compound (8-6) {9.3 g (23.2 mmol),
61.7% yield, which was the yield through the two
reactions in the routes D8 and E8}.
LRMS m/z 423 [M+Na]+.
Route F8; 1-0-ally-2,3,4-di-0-benzyl-6-0-tosyl-3-
D-glucopyranoside (8-7):
To a solution of the compound (8-6) (9.3 g,
23.2 mmol) in anhydrous pyridine (120 mL) were added p-
toluenesulfonyl chloride (5.8 g, 30.2 mmol) and 4-
dimethylaminopyridine (283 mg, 2.3 mmol), and the
reaction solution was stirred at 0 C for 16 hours.
After the sufficient progress of the reaction was
confirmed, the reaction solution was slowly poured into
ice water (100 mL). The water layer was subjected to
extraction with ethyl acetate (3 x 200 mL). The
organic layers were combined with each other. The
combination was washed with an aqueous 1 N HC1 solution
until the pH became 4, washed with saturated aqueous
sodium hydrogen carbonate (2 x 100 mL) and saturated
saline (2 x 100 mL), dried over sodium sulfate,
filtrated and then concentrated under reduced pressure.
The concentrated product was purified by silica gel
flash chromatography (hexane/ethyl acetate,
6:1,4:1,2:1) to yield the title compound (8-7) {12.3 g
(22.2 mmol), 95.7% yield}.
LRMS m/z 577 [M+Na]+.
CA 02749797 2011-07-14
115
Route G8; 1-0-allyl-2,3-di-O-benzyl-4-0-
(carbobenzoxy-R-alanyl)-6-O-tosyl-R-D-glucopyranoside
(8-8):
Into a mixed solution of anhydrous dichloromethane
(200 mL) and anhydrous pyridine (50 mL) were dissolved
the compound (8-7) (12.3 g, 22.2 mmol), N-carbobenzoxy-
R-alanine (12.1 g, 44.4 mmol),
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
hydrochloride (EDCI=HCl) (17.0 mg, 88.8 mmol) and 4-
dimethylaminopyridine (5.4 g, 44.4 mmol), and then the
reactive components were caused to react with each
other at room temperature for 18 hours. After the
sufficient progress of the reaction was confirmed,
water (10 mL) was poured into the reaction solution to
terminate the reaction. Thereafter, the solution was
concentrated under reduced pressure. The resultant
residue was purified by silica gel chromatography
(hexane/ethyl acetate, 4:1-3:1-2:1-3:2) to yield the
title compound (8-8) {15.7 g (20.7 mmol), 93.0% yield}.
LRMS m/z 782 [M+Na]+.
Route H8; 1-0-allyl-2,3-di-O-benzyl-4-0-
(carbobenzoxy-R-alanyl)-6-thioacetyl-R-D-
glucopyranoside (8-9):
Potassium thioacetate (4.7 g, 41.3 mmol) was added
to a solution of the compound (8-8) (15.7 g, 20.7 mmol)
in anhydrous N,N-dimethylformamide (200 mL), and the
solution was stirred at 90 C for 3 hours. After the
CA 02749797 2011-07-14
116
sufficient progress of the reaction was confirmed, the
reaction solution was poured into cold water (400 mL),
and the resultant was extracted with ethyl acetate
(3 x 150 mL). The organic layers were combined with
each other, and the combination was washed with
saturated saline (2 x 100 mL), dried over sodium
sulfate, filtrated and concentrated under reduced
pressure. The resultant residue was purified by silica
gel chromatography (hexane/ethyl acetate,
4:1->3:1-2:1-43:2) to yield the title compound (8-9)
{13.6 g (20.5 mmol), 99.2% yield}.
LRMS m/z 686 [M+Na]+.
Route 18; 3-O-[2,3-di-O-benzyl-4-0-(carbobenzoxy-
(3-alanyl)-6-thioacetyl-(3-D-quinovopyranosyl-glycerol
(8-10) :
The compound (8-9) (13.6 g, 20.5 mmol) was
dissolved in a solution (200 mL) of t-butyl alcohol and
distilled water (4:1), and then thereto were added a
0.04 M osmium tetraoxide solution in t-butyl alcohol
(5 mL) and trimethylamine N-oxide (3.4 g, 30.7 mmol).
The solution was stirred with a stirrer at room
temperature for 24 hours. After the sufficient
progress of the reaction was confirmed, 3 g of
activated carbon was added thereto and then the
solution was stirred for 30 minutes to cause the
catalyst to be adsorbed on the carbon. The solution
was subjected to suction filtration through a Kiriyama
CA 02749797 2011-07-14
117
funnel wherein celite was laid, so as to remove the
catalyst. The reaction product remaining on the celite
was washed three times with ethyl acetate so as to be
collected. Distilled water (200 mL) was added to the
collected filtrate, and the resultant was extracted
with ethyl acetate (3 x 150 mL). The organic layers
were combined with each other, and the combination was
washed with saturated saline (2 x 100 mL), dried over
sodium sulfate, filtrated and concentrated under
reduced pressure. The resultant residue was purified
by silica gel chromatography (toluene/ethyl acetate,
2:1-3:2-4l:1~2:3,1:2-1:4) to yield the title compound
(8-10) {11.7 g (16.8 mmol), 81.8% yield}.
LRMS m/z 720 [M+Na]+.
Route J8; 3-O-[2,3-di-O-benzyl-4-0-(carbobenzoxy-
R-alanyl)-6-thioacetyl-3-D-quinovopyranosyl]-1-0-
stearoyl-glycerol (8-11):
Stearoyl chloride (5.4 g, 26.8 mmol) was added to
a mixed solution of the compound (8-10) (11.7 g,
16.8 mmol) in anhydrous dichloromethane (200 mL) and
anhydrous pyridine (50 mL), and then the solution was
stirred at 0 C for 2 hours. After the sufficient
progress of the reaction was confirmed, methanol (5 mL)
was added thereto so as to terminate the reaction. The
solution was concentrated under reduced pressure. A
small amount of ethyl acetate was used to suspend the
residue, and the suspended residue was poured into
CA 02749797 2011-07-14
118
water (200 mL). The resultant was extracted with ethyl
acetate (3 x 100 mL). The organic layers were combined
with each other, and the combination was washed with
saturated saline (2 x 100 mL), dried over sodium
sulfate, filtrated and then concentrated under reduced
pressure. The resultant residue was purified by silica
gel chromatography (hexane/ethyl acetate,
6:1-4:1-+2:1-+3:2-1:1) to yield the title compound (8-11)
as a colorless oily substance {10.7 g (11.1 mmol),
66.2% yield}.
LRMS m/z 987 [M+Na]+.
Route K8; 3-O-[2,3-di-O-benzyl-4-0-(carbobenzoxy-
R-alanyl)-6-sulfo-R-D-quinovopyranosyl]-1-0-stearoyl-
glycerol (8-12):
Oxone (27.3 g, 44.4 mmol) and potassium acetate
(3.7 g) were added to a solution of the compound (8-11)
(10.7 g, 11.1 mmol) in acetic acid (147 g, 2.5 mol),
and then the solution was vigorously stirred at room
temperature for 48 hours. After the sufficient
progress of the reaction was confirmed, the reaction
solution was poured into a cold 7.5 M sodium hydroxide
solution (500 mL) and the resultant was extracted with
ethyl acetate (4 x 100 mL). The organic layers were
combined with each other, and the combination was
washed with saturated aqueous sodium hydrogen carbonate
(2 x 100 mL) and saturated saline (2 x 100 mL), dried
over sodium sulfate, filtrated and then concentrated
CA 02749797 2011-07-14
119
under reduced pressure. The resultant residue was
purified by silica gel chromatography
(chloroform/methanol,
100:1,50:1-,20:1~15:1~12.5:1,10:1,8:1) to yield the
title compound (8-12) as a colorless waxy substance.
LRMS m/z 968 [M-Na]-.
Route L8; 3-0-[4-0-(3-alanyl)-6-sulfo-R-D-
quinovopyranosyl]-1-0-stearoyl-glycerol (8-13):
To a solution of the compound (8-12) (756 mg,
762 pmol) in methanol (10 mL), dichloromethane (10 mL)
and acetic acid (0.5 mL) was added 10% palladium
hydroxide-activated carbon (835 mg), and then the
solution was stirred at room temperature under an
atmosphere of hydrogen gas for 48 hours. After the
sufficient progress of the reaction was confirmed, the
palladium-activated carbon was filtrated off. The
filtrate was concentrated under reduced pressure. The
resultant residue (crude product of the compound (8-
13)) was to be used as it was for the next reaction.
LRMS m/z 654 [M-Na]-.
Route M8; 3-0-[4-0-(4-iodobenzoyl-3-alanyl)-6-
sulfo-3-D-quinovopyranosyl]-1-0-stearoyl-glycerol
(8-14):
The crude product of the compound (8-13) yielded
through the route L8 was dissolved in a mixed solution
of anhydrous dichloromethane (15 mL), pyridine (5 mL),
and triethylamine (2 mL). Thereto was added
CA 02749797 2011-07-14
120
(4-iodobenzoyl)-p-nitrophenyl ester (563 mg, 1.5 mmol).
While the solution was stirred with a stirrer, the
reactive components were caused to react with each
other at room temperature for 24 hours. After the
sufficient progress of the reaction was confirmed,
toluene and methanol were added thereto. Under reduced
pressure, the solvents were then removed by evaporation
to concentrate the solution, while azeotropy. The
resultant residue was purified by silica gel
chromatography (chloroform/methanol/distilled water,
70:10:1-60:10:1.150:10:1.2-40:10:2-30:10:1.5-65:25:4)
to yield the title compound (8-14) {442.5 mg
(487.4 pmol), 64.0% yield}.
LRMS m/z 884 [M - Na]-.
Synthesis Example 9
A process for producing the
sulfopyranosylacylglycerol derivative according to an
embodiment of the present invention is illustrated in
the following scheme 9, giving a R-
sulfoquinovosyldiacylglycerol monoiodide derivative as
an example thereof:
CA 02749797 2011-07-14
121
rn rn rn C)
01 LL
S
00
rn
m / c / m / o
O a
C
0 0 0 0
am
a 0 0 0 0
s m
U o0 0O
< cg a) o
d
= O
LL
d U W ~
LO a)
rn rn
a O 0 a
0 a o 0 0
rn
m mm
E a
a.
U
CA 02749797 2011-07-14
122
0
i
0 bo
= o
v o
Q U
i o
rn O
O Y J O 1T
ao ~
OC o a
a
o ax
zz a x a o
a 0
0 0
ca
0 zx
o O O.
a (50
0 a
O W
am
/
a O
^ 1~
a> o
ci
O= O= 0 U7
0 0 0
0
0
am
zx z
o oxx
O
`= z
N
x
CA 02749797 2011-07-14
123
The following will describe a synthesis example
through the individual steps in detail:
<Synthesis Example>
Route A9; 2,3,4,6-tetra-0-acetyl-1-0-allyl-R-D-
glucopyranoside (9-2):
A solution (500 mL) of the compound (9-1) (50.0 g,
128 mmol), allyl alcohol (22.3 g, 384 mmol), and zinc
chloride (17.4 g, 128 mmol) in toluene was vigorously
stirred at 80 C under an atmosphere of nitrogen for
48 hours. After the sufficient progress of the
reaction was confirmed, the temperature of the solution
was returned to room temperature. The solution was
then washed with saturated aqueous sodium hydrogen
carbonate (2 x 100 mL) and saturated saline
(2 x 100 mL), dried over sodium sulphate, filtrated and
then concentrated under reduced pressure. The
concentrated product was crystallized two times from
heated ethanol to yield the title compound (9-2) as a
colorless needle crystal {24.8 g (64 mmol), 50.0%
yield}.
LRMS m/z 411 [M+Na]+.
Route B9; 1-0-allyl-[3-D-glucopyranoside (9-3):
The compound (9-2) (21.0 g, 54 mmol) was dissolved
in methanol (200 mL). While the solution was stirred
at room temperature under an atmosphere of nitrogen,
thereto was added a 28% solution (1 mL) of sodium
methylate in methanol. The solution was then stirred
CA 02749797 2011-07-14
124
at room temperature for 2 hours. After the sufficient
progress of the reaction was confirmed, the reaction
was terminated. The reaction solution was
concentrated, as it was, under reduced pressure, and a
crude product of the resultant title compound (9-3) was
to be used for the next reaction.
LRMS m/z 243 [M+Na]+.
Route C9; 1-0-allyl-4,6-0-benzylidene-R-D-
glucopyranoside (9-4):
The compound (9-3) (11.9 g, 54 mmol) was suspended
in anhydrous acetonitrile (60 mL), and thereto were
added benzaldehyde dimethylacetal (16.4 g, 2.0
equivalent) and p-toluenesulfonic acid monohydrate
(1.0 g, 0.05 equivalent). The reaction liquid was
stirred at 40 C for 4 hours, and then triethylamine
(0.5 mL) was added thereto so as to terminate the
reaction. The resultant was concentrated under reduced
pressure. The concentrated product was dissolved in a
small amount of ethyl acetate, and then cold water was
extracted with ethyl acetate (3 x 100 mL). The organic
layers were combined with each other, and the
combination was washed with saturated saline
(2 x 50 mL), dried over sodium sulfate, filtrated and
then concentrated under reduced pressure. The
resultant residue was crystallized two times from
heated ethanol to yield the title compound (9-4) as a
colorless needle crystal (33.5 g). The filtrate was
CA 02749797 2011-07-14
125
further concentrated, purified by silica gel
chromatography (hexane/ethyl acetate, 15:1-410:1-8:1),
and then crystallized from heated ethanol to yield the
same compound (9-3)(6.63 g) {total amount: 40.1 g
(82.1 mmol), 84.4% yield}.
LRMS m/z 511 [M+Na]+.
Route D9; 1-0-allyl-2,3-di-0-benzyl-4,6-0-
benzylidene-R-D-glucopyranoside (9-5):
To a solution of the compound (9-4) (11.6 g,
37.6 mmol) in anhydrous N,N-dimethylformamide (DMF,
100 mL) were added benzylchloride (4.76 g, 4
equivalent) and sodium hydroxide powder (4.5 g, 3.0
equivalent.), and the reaction solution was vigorously
stirred at room temperature for 24 hours. After the
sufficient progress of the reaction was confirmed, the
reaction solution was poured into cold water (100 mL),
and the resultant was extracted with ethyl acetate
(3 x 50 mL). The organic layers were combined with
each other, and the combination was washed with
saturated saline (2 x 30 mL), dried over sodium
sulfate, filtrated and then concentrated under reduced
pressure. The resultant residue was crystallized
two times from heated ethanol to yield the title
compound (9-5) as a colorless needle crystal. The
filtrate was concentrated, purified by silica gel
chromatography (hexane/ethyl acetate,
12:1.10:1,8:1-6:1-4:1), and then crystallized from
CA 02749797 2011-07-14
126
heated ethanol to yield the same compound (9-5). The
resultant compound was collected into a flask, and was
to be used as it was for the next reaction.
LRMS m/z 511 [M+Na]+.
Route E9; 1-0-ally-2,3-di-0-benzyl-R-D-.
glucopyranoside (9-6):
The compound (9-5) yielded through the route D9
was dissolved in acetic acid (72 mL), and further
distilled water (40 mL) was added thereto. The
solution was stirred for 1 hour while heated and
refluxed. After the sufficient progress of the
reaction was confirmed, the solution was cooled to room
temperature, and then the solvent was removed by
evaporation. Distilled water (15 mL) was added
thereto, and the solution was again concentrated under
reduced pressure; this operation was repeated 4 times.
Thereafter, the resultant residue was dissolved in
methanol (150 mL). While the solution was stirred at
room temperature under an atmosphere of nitrogen,
thereto was added a 28% solution (0.7 mL) of sodium
methylate in methanol. The solution was then stirred
at room temperature for 2 hours. After the sufficient
progress of the reaction was confirmed, the reaction
was terminated. The reaction solution was
concentrated, as it was, under reduced pressure, and
purified by silica gel flash chromatography
(hexane/ethyl acetate, 4:1-3:12:1-3:2-+1:1_2:3_*1:2) to
CA 02749797 2011-07-14
127
yield the title compound (9-6) {9.3 g (23.2 mmol),
61.7% yield, which was the yield through the two
reactions in the routes D9 and E9}.
LRMS m/z 423 [M+Na]+.
Route F9; 1-0-ally-2,3,4-di-0-benzyl-6-0-tosyl-R-
D-glucopyranoside (9-7):
To a solution of the compound (9-6) (9.3 g,
23.2 mmol) in anhydrous pyridine (120 mL) were added p-
toluenesulfonyl chloride (5.8 g, 30.2 mmol) and 4-
dimethylaminopyridine (283 mg, 2.3 mmol), and the
reaction solution was stirred at 0 C for 16 hours.
After the sufficient progress of the reaction was
confirmed, the reaction solution was slowly poured into
ice water (100 mL). The water layer was subjected to
extraction with ethyl acetate (3 x 200 mL). The
organic layers were combined with each other. The
combination was washed with an aqueous 1 N HC1 solution
until the pH became 4, washed with saturated aqueous
sodium hydrogen carbonate (2 x 100 mL) and saturated
saline (2 x 100 mL), dried over sodium sulfate,
filtrated and then concentrated under reduced pressure.
The concentrated product was purified by silica gel
flash chromatography (hexane/ethyl acetate,
6 : 1-,4 : 1-,2 : 1) to yield the title compound (9-7) (12.3 g
(22.2 mmol), 95.7% yield}.
LRMS m/z 577 [M+Na]+.
Route G9;
CA 02749797 2011-07-14
128
1-O-allyl-2,3-di-O-benzyl-4-0-(carbobenzoxy-R-alanyl)-
6-0-tosyl-R-D-glucopyranoside (9-8):
Into a mixed solution of anhydrous dichloromethane
(200 mL) and anhydrous pyridine (50 mL) were dissolved
the compound (9-8) (12.3 g, 22.2 mmol), N-carbobenzoxy-
R-alanine (12.1 g, 44.4 mmol), 1-ethyl-3-(3-
dimethylaminopropyl)carbodiimide hydrochloride
(EDCI=HC1) (17.0 mg, 88.8 mmol) and 4-
dimethylaminopyridine (5.4 g, 44.4 mmol), and then the
reactive components were caused to react with each
other at room temperature for 18 hours. After the
sufficient progress of the reaction was confirmed,
water (10 mL) was poured into the reaction solution to
terminate the reaction. Thereafter, the solution was
concentrated under reduced pressure. The resultant
residue was purified by silica gel chromatography
(hexane/ethyl acetate, 4:1-3:1-*2:1,3:2) to yield the
title compound (9-8) {15.7 g (20.7 mmol), 93.0% yield}.
LRMS m/z 782 [M+Na]+.
Route H9; 1-0-allyl-2,3-di-0-benzyl-4-0-
(carbobenzoxy-R-alanyl)-6-thioacetyl-R-D-
glucopyranoside (9-9):
Potassium thioacetate (4.7 g, 41.3 mmol) was added
to a solution of the compound (9-8) (15.7 g, 20.7 mmol)
in anhydrous N,N-dimethylformamide (200 mL), and the
solution was stirred at 90 C for 3 hours. After the
sufficient progress of the reaction was confirmed, the
CA 02749797 2011-07-14
129
reaction solution was poured into cold water (400 mL),
and the resultant was extracted with ethyl acetate
(3 x 150 mL). The organic layers were combined with
each other, and the combination was washed with
saturated saline (2 x 100 mL), dried over sodium
sulfate, filtrated and concentrated under reduced
pressure. The resultant residue was purified by silica
gel chromatography (hexane/ethyl acetate,
4:1-3:12:1-3:2) to yield the title compound (9-9)
{13.6 g (20.5 mmol), 99.2% yield}.
LRMS m/z 686 [M+Na]+.
Route 19; 3-O-[2,3-di-O-benzyl-4-0-(carbobenzoxy-
R-alanyl)-6-thioacetyl-R-D-quinovopyranosyl-glycerol
(9-10):
The compound (9-9) (13.6 g, 20.5 mmol) was
dissolved in a solution (200 mL) of t-butyl alcohol and
distilled water (4:1), and then thereto were added a
0.04 M osmium tetraoxide solution in t-butyl alcohol
(5 mL) and trimethylamine N-oxide (3.4 g, 30.7 mmol).
The solution was stirred with a stirrer at room
temperature for 24 hours. After the sufficient
progress of the reaction was confirmed, 3 g of
activated carbon was added thereto and then the
solution was stirred for 30 minutes to cause the
catalyst to be adsorbed on the carbon. The solution
was subjected to suction filtration through a Kiriyama
funnel containing celite, so as to remove the catalyst.
CA 02749797 2011-07-14
130
The reaction product remaining on the celite was washed
three times with ethyl acetate so as to be collected.
Distilled water (200 mL) was added to the collected
filtrate, and the resultant was extracted with ethyl
acetate (3 x 150 mL). The organic layers were combined
with each other, and the combination was washed with
saturated saline (2 x 100 mL), dried over sodium
sulfate, filtrated and concentrated under reduced
pressure. The resultant residue was purified by silica
gel chromatography (toluene/ethyl acetate,
2:13:2-1:1_2:3-1:2-1:4) to yield the title compound
(9-10) {11.7 g (16.8 mmol), 81.8% yield}.
LRMS m/z 720 [M+Na]+.
Route J9; 3-O-[2,3-di-O-benzyl-4-0-(carbobenzoxy-
R-alanyl)-6-thioacetyl-R-D-quinovopyranosyl]-1,2-di-0-
stearoyl-glycerol (9-11):
Stearoyl chloride (5.4 g, 26.8 mmol) was added to
a mixed solution of the compound (9-10) (11.7 g,
16.8 mmol) in anhydrous dichloromethane (200 mL) and
anhydrous pyridine (50 mL), and then the solution was
stirred at 0 C for 2 hours. After the sufficient
progress of the reaction was confirmed, methanol (5 mL)
was added thereto so as to terminate the reaction. The
solution was concentrated under reduced pressure. A
small amount of ethyl acetate was used to suspend the
residue, and the suspended residue was poured into
water (200 mL). The resultant was extracted with ethyl
CA 02749797 2011-07-14
131
acetate (3 x 100 mL). The organic layers were combined
with each other, and the combination was washed with
saturated saline (2 x 100 mL), dried over sodium
sulfate, filtrated and then concentrated under reduced
pressure. The resultant residue was purified by silica
gel chromatography (hexane/ethyl acetate,
6:1-4:1-2:1,3:2-,1:1) to yield the title compound (9-11)
as a colorless oily substance {3.3 g (2.7 mmol), 16.0%
yield}.
LRMS m/z 1253 [M+Na]+.
Route K9; 3-O-[2,3-di-O-benzyl-4-0-(carbobenzoxy-
R-alanyl)-6-sulfo-R-D-quinovopyranosyl]-1,2-di-0-
stearoyl-glycerol (9-12):
Oxone (6.6 g, 10.7 mmol) and potassium acetate
(2.5 g) were added to a solution of the compound (9-11)
(3.3 g, 2.7 mmol) in acetic acid (100 g, 2.5 mol), and
then the solution was vigorously stirred at room
temperature for 48 hours. After the sufficient
progress of the reaction was confirmed, the reaction
solution was poured into a cold 7.5 M sodium hydroxide
solution (100 mL) and the resultant was extracted with
ethyl acetate (4 x 50 mL). The organic layers were
combined with each other, and the combination was
washed with saturated aqueous sodium hydrogen carbonate
(2 x 50 mL) and saturated saline (2 x 50 mL), dried
over sodium sulfate, filtrated and then concentrated
under reduced pressure. The resultant residue was
CA 02749797 2011-07-14
132
purified by silica gel chromatography
(chloroform/methanol,
100: 1--X50: 1,20: 1,15: 1,12 . 5 : 1-110: l- 8 : 1) to yield the
title compound (9-12) as a colorless waxy substance.
LRMS m/z 1235 [M-Na]-.
Route L9; 3-0-[4-0-(8-alanyl)-6-sulfo-R-D-
quinovopyranosyl]-1,2-di-0-stearoyl-glycerol (9-13):
To a solution of the compound (9-12) (345 mg,
274 pmol) in methanol (10 mL), dichloromethane (15 mL)
and acetic acid (0.6 mL) was added 10% palladium
hydroxide-activated carbon (300 mg), and then the
solution was stirred at room temperature under an
atmosphere of hydrogen gas for 48 hours. After the
sufficient progress of the reaction was confirmed, the
palladium-activated carbon was filtrated off. The
filtrate was concentrated under reduced pressure. The
resultant residue (crude product of the compound
(9-13)) was to be used, as it was, as a substance
containing the title compound (9-13) for the next
reaction.
LRMS m/z 921 [M-Na]-.
Route M9; 3-0-[4-0-(4-iodobenzoyl-p-alanyl)-6-
sulfo-(3-D-quinovopyranosyl]-1,2-di-0-stearoyl-glycerol
(9-14) :
The crude product of the compound (9-13) yielded
through the route L9 was dissolved in a mixed solution
of anhydrous dichloromethane (15 mL), pyridine (5 mL),
CA 02749797 2011-07-14
133
and triethylamine (2 mL). Thereto was added (4-
iodobenzoyl)-p-nitrophenyl ester (202 mg, 548 pmol).
While the solution was stirred with a stirrer, the
reactive components were caused to react with each
other at room temperature for 24 hours. After the
sufficient progress of the reaction was confirmed,
toluene and methanol were added thereto. Under reduced
pressure, the solvents were then removed by evaporation
to concentrate the solution, while azeotropy. The
resultant residue was purified by silica gel
chromatography (chloroform/methanol/distilled water,
70:10:1-60:10:1-60:14:1.6-60:18:2,60:22:2.4,65:25:3) to
yield the title compound (9-14) {77.6 mg (82.1 pmol),
30.0% yield}.
LRMS m/z 1151 [M - Na]-.
Example 2. Biological distribution
1) Biotinated aSQMG
1 x106 of human esophageal squamous cell
carcinoma cells TE-8 were transplanted into the right
femoral region of 11 male KSN nude mice (Japan SLC,
Inc.), and the mice were bred to form a tumor mass of
about 300-900 cm3 volume.
Thereafter, biotinated aSQMG C18:0 (formula XI)
was dissolved in physiological saline solution, and
0.1 mL of the solution per administration was
intravenously administered to each group comprising
three mice so that the amount of the biotinated aSQMG
CA 02749797 2011-07-14
134
C18:0 could be 40 mg/kg, 20 mg/kg, and 2 mg/kg. To two
reference tumor bearing mice, physiological saline was
administered. Each mouse was slaughtered 9 hours after
administration, and tumor was extracted and immediately
frozen. Frozen segments having a thickness of 5pm were
prepared from the frozen tumor, and each segment was
stained by avidin which was labeled with fluorescent
pigment TRITC (Tetramethylrhodamine-5-(and-6)-
isothiocyanate). Each stained segment was observed by
fluorescent microscope and subjected to image
processing with Adobe Photoshop after digitization.
In order to evaluate a tumor region which emits
fluorescence due to TRITC, binarization imaging was
performed with use of Photoshop. Thereafter, a portion
(fraction) containing tumor tissues was selected from
the visual field, and the number of pixels of a TRITC
positive region (black color region) and the entire
region were counted and the ratio of the positive
region was calculated according to the formula:
The proportion (%) of biotinated aSQAP detected region
= [the area of the black color region (region
where the biotinated aSQMG was detected)]/[the area of
the whole of the selected fraction] x 100
= [the number of pixels in the TRITC positive
region] / [the number of pixels in the whole
fraction] x 100 (%) formula (A)
In the same manner, a different tumor tissue
CA 02749797 2011-07-14
135
portion was selected, and with respect to 5 positions
the ratio of a TRITC positive region was calculated and
averaged out.
The results are shown in FIG. 1, indicating that
tumor tissues were strongly stained according to the
dose of biotinated (YSQMG C18:0 and the tumor-resident
amount of biotinated aSQMG C18:0 is high.
2) Biotinated aSQAP
Human giant cell lung carcinoma cells Lu65 were
transplanted into six-week-old male KSN nude mice in a
number of 1 x 106 for each of the mice, and the mice
were bred for 14 days to form a tumor mass of about
100-200 mm3 volume in each of the mice.
Thereafter, three mice were assigned to each of
the following four groups (1) to (4):
(1) a control group wherein physiological saline
was administered,
(2) a high dose group wherein biotinated aSQAP was
administered in a dose of 50 mg/kg,
(3) a middle dose group wherein biotinated aSQAP
was administered in a dose of 5 mg/kg, and
(4) a low dose group wherein biotinated aSQAP was
administered in a dose of 1 mg/kg; the used biotinated
aSQAP was the compound represented by the
formula (2-11).
The biotinated aSQAP dissolved in physiological
saline, or only physiological saline for the control
CA 02749797 2011-07-14
136
group was administered to each of the nude mice from
its tail vein so as to give a predetermined amount out
of the above-mentioned amounts. After one hour from
the administration, a tumor was extirpated. The tumor
piece was immersed into an OCT compound, and then
frozen with liquid nitrogen.
The frozen tumor of each of the test animals was
sliced into a thickness of 10 pm, and the slice was
fixed onto a microplate. The region wherein biotin
was distributed was dyed with an Alexa 488 labeled
avidin.
The slice was observed by a fluorescence
microscope, and the slice was made into an image. Data
on the image were input to a computer, and then
positive signals of the image were quantified according
to image analysis software.
Specifically, binary image processing based on
Photoshop was performed to estimate a tumor region
emitting fluorescence through the labeling material
Alexa 488 in the slice. Thereafter, a portion
(fraction) containing tumor tissues was selected
from the visual field, and the number of pixels of
the Alexa 488 positive region (black region) and the
entire region were counted and the ratio of the
positive region was calculated according to the
formula:
CA 02749797 2011-07-14
137
The proportion (%) of biotinated aSQAP detected region
= [the area of the black color region (region
where the biotinated aSQAP was detected)]/[the area of
the whole of the selected fraction] x 100
= [the number of pixels in the Alexa 488 positive
region]/[the number of pixels in the whole
fraction] x 100 (%) formula (B)
In the same way, different tumor tissue fractions
were selected from 5 sites thereof, and about each of
the sites the proportion of the Alexa 488 positive
region was calculated. The resultant proportions were
then averaged.
Further, in the case of using a different labeling
material, substantially the same value can be
calculated in accordance with the following expression:
The proportion (%) of the region where a substance to
be analyzed was detected
= [the area of the region where the signal of the
labeling material was generated (region where the
substance to be analyzed was detected)]/[the area of
the whole of the selected fraction] x 100
= (the number of pixels in the labeling material
positive region)/(the number of pixels in the whole of
the selected fraction) x 100 (o) formula (C)
The results are shown in FIG. 2. In the high dose
group, wherein the biotinated aSQAP was administered in
a dose of 50 mg/kg, the tumor tissues were intensely
CA 02749797 2011-07-14
138
dyed. It was demonstrated that the amount of
biotinated aSQAP in tumor was large.
Example 3. Pharmacokinetics 1
(1) Animal tests
To eight-week-old male KSN nude mice (Japan SLC,
Inc.), 2 x 106 of human colon carcinoma cells (SW480)
were transplanted subcutaneously, two weeks later (body
weight approximately 25 g, tumor volume approximately
200 mm3) aSQMG C18:0 or aSQAP C18:0 was dissolved in
physiological saline solution and administered
intraperitoneally at a dose of 10 mg/kg (0.1 mL).
After the administration, the mice were slaughtered
chronologically with carbon dioxide; 30 minute, 1 hour,
2 hours, 4 hours, 12 hours, and 24 hours after the
administration. Tissues of blood, liver, kidney, lung
and spleen, and tumor tissue were then collected. At
each time when tissues were collected, five animals
were used.
(2) Pretreatment of analytical samples
Blood plasma
Whole blood was collected from postcaval vein with
use of an injection needle wetted with heparin sodium
injection solution (10,000 unit/ 10 mL) to obtain blood
plasma by 20 minutes of centrifugation at 6,000 g. To
90 ijl of blood plasma, 5 pl of an internal standard
compound (aSQMG C16:0), 5 pl of distilled water, and
4 pl of phosphoric acid were added, and denatured
CA 02749797 2011-07-14
139
protein by adding 96 p1 of acetonitrile. To 0.2 mL of
the solution, 600 pi of 10 mM ammonium acetate solution
was added, and stirred sufficiently to obtain
supernatant by centrifugation at 16,000 g for
15 minutes. The supernatant was subjected to solid-
phase extraction operation as described below. The
solid-phase extraction was performed by washing with
1000 pl of 10 mM ammonium acetate aqueous solution and
200 pl of 10 mM ammonium acetate aqueous solution
containing 10% acetonitrile, followed by elution of a
test compound with 200 pl of 10 mM ammonium acetate
aqueous solution containing 70% acetonitrile with use
of Empore (trademark) disc cartridge (3M Company,
product number 4115SD). The compound was used as a
LC/MS analytical sample.
Organ samples
Each organ was extirpated after collection of
blood and immediately frozen on dry ice. The frozen
sample was subjected to frost shattering, and the
weight was measured. Subsequently, 1/2-fold (weight
ratio) internal standard compound (aSQMG C16:0) and
distilled water were added, and 8-fold (weight ratio)
extraction solution (2.5% NP-40, 2% phosphoric acid,
2 mM Eserine aqueous solution) was further added to the
sample, followed by homogenization. The homogenized
solution was subjected to protein denaturation with
acetonitrile and solid phase extraction in the same
CA 02749797 2011-07-14
140
manner as the above-mentioned treatment of blood plasma
to obtain an analytical sample.
(3) Quantitative analysis assay
pl of the analytical sample was separated with
5 a reverse phase HPLC system equipped with Capcell Pak
MG column (particle diameter; 3 pm, column size;
2.1 x 50 mm, Shiseido Co., Ltd.). HPLC was performed
with 68% acetonitrile and a single solvent of 10 mM
ammonium acetate aqueous solution at a flow rate of
10 0.2 mL per minute. Mass spectrum was measured in a
negative mode, and the peak area ratio of the internal
standard compound was calculated by integration of the
peak area from chromatogram corresponding to each ion
by measuring a precursor ion; m/z = 583.1, fragment
ion; m/z = 224.7 and m/z = 298.8 for aSQMG C18:0, a
precursor ion; m/z = 567.1, fragment ion; m/z = 224.7
and m/z = 283.1 for aSQAP C18:0, and a precursor ion;
m/z = 555.1, fragment ion; m/z = 224.7 and m/z = 298.8
for a internal standard compound aSQMG C16:0. As a
method of quantitative analysis, an internal standard
analysis was adopted. More specifically, a standard
curve was drawn based on the peak area ratio of the
internal standard compound by analyzing the sample to
which the test compound was added, to determine the
quantity of the test compound.
FIG. 3 shows a concentration-time transition curve
in blood plasma and organs of nude mice to which aSQMG
CA 02749797 2011-07-14
141
C18:0 was intraperitoneally administered in a ratio of
mg/kg.
As is shown by FIG. 3, in tissues including blood
plasma, liver, kidney, spleen and lung, the highest
5 concentration of aSQMG C18:0 was indicated immediately
after the administration of aSQMG C18:0, thereafter the
concentration of aSQMG C18:0 fell rapidly. In liver,
lung, kidney, and blood plasma, four hours after the
administration and in spleen 12 hours thereafter, the
10 concentration of aSQMG C18:0 fell below the detection
limit. On the other hand, in tumor tissues, the
concentration of aSQMG C18:0 in tissue was low
immediately after administration of aSQMG C18:0 in
comparison with other tissues, but it was detectable
even 24 hours after administration. Therefore, it was
proved that aSQMG C18:0 resides for a long period in
tumor tissues.
FIG. 4 shows a concentration-time transition curve
in blood plasma and organs of nude mice to which aSQAP
C18:0 was intraperitoneally administered in a ratio of
10 mg/kg.
From the results of FIG. 4, in tissues including
blood plasma, liver, kidney, spleen and lung, the
highest concentration of aSQAP C18:0 was indicated
immediately after the administration of aSQAP C18:0,
thereafter the concentration of aSQAP C18:0 fell
rapidly. In spleen, lung, kidney, and blood plasma,
CA 02749797 2011-07-14
142
four hours after the administration and in liver
12 hours thereafter, the concentration of aSQAP C18:0
fell below the detection limit. On the other hand, in
tumor tissues, the concentration of aSQAP C18:0 in
tissue was low immediately after administration of
aSQAP C18:0 in comparison with other tissues, but it
was detectable even 24 hours after administration.
Therefore, it was proved that aSQAP C18:0 resides for a
long period in tumor tissues.
Example 4. Pharmacokinetics 2
The pharmacokinetics of aSQAP C18:0 was tested by
administering 1 mg/kg of aSQAP C18:0 to animals in the
same manner as Example 3 except that human sophageal
squamous cell carcinoma cells TE-8 were used as tumor
of transplanted tumor model and that intravenous
administration was adopted as substitute for
interperitoneal administration.
The results are shown in FIG. 5. As is shown by
FIG. 5, in tissues including blood plasma, liver,
spleen and lung, the highest concentration of aSQAP
C18:0 in tissue was indicated immediately after
administration of aSQAP C18:0, and thereafter aSQAP
C18:0 rapidly fell. In blood plasma, liver, and lung
two hours after the administration, in spleen 24 hours
thereafter, the concentration of aSQAP C18:0 fell below
the detection limit. On the other hand, in comparison
with other organs the concentration in tumor tissue was
CA 02749797 2011-07-14
143
low immediately after the administration of aSQAP
C18:0, but no decrease tendency was observed even after
two hours and the concentration of aSQAP C18:0 was
detectable even after 24 hours. Therefore, it was
proved that aSQAP C18:0 resides for a long period in
tumor tissues.
Industrial Applicability
The novel compound of the present invention
specifically resides in a tumor and is a substance
containing an acting group in the structure.
Therefore, according to the type of the acting group it
can be advantageously used for drug deliver to tumor,
detection and diagnosis of tumor, treatment of tumor,
and the like.