Note: Descriptions are shown in the official language in which they were submitted.
CA 02751392 2016-06-02
1
CRYSTALLINE FORM OF R)-3-(4-(2-(2-METHYLTETRAZOL-5-YL)PYRIDIN- 5-YL)-
3-FLUOROPHENYL)-5-HYDROXYMETHYL OXAZOLIDIN-2-ONE DIHYDROGEN
PHOSPHATE
BACKGROUND OF THE INVENTION
Field of the Invention
[0002] The
present disclosure relates to a crystalline form of (R)-3-(4-(2-(2-
methyltetrazol-5 -yppyridin-5 -y1)-3 -fluoropheny1)-5-hydroxymethyl
oxazolidin-2-one
dihydrogen phosphate, and methods of making and using the crystalline form.
The
crystalline form may be used as a pharmaceutically active compound in
compositions that are
useful in impeding the growth of bacteria or treating patients suffering from
bacterial
infections.
Description of the Related Art
[0003] US
Patent Publication No. 20070155798 recently disclosed a series of
potently anti-bacterial oxazolidinones including
\
N
wherein R = H, PO(OH)2, and PO(ONa)2.
[0004]
Although this patent application discloses methods of making compounds
such as the free acid (wherein R = PO(OH)2) and the disodium salt (wherein R
=P0(0Na)2),
there is no indication that any of the compounds were stably crystallized or
purified. In
addition, these processes include the use of reagents which are highly
corrosive, such as
trichloroacetic acid, or explosive, such as ethyl ether, and therefore are not
suitable for
commercial use. As discussed below in more detail, attempts to crystallize the
disodium salt
by the instant inventors resulted in a highly hygroscopic, unstable
crystalline salt form which
turned amorphous upon drying.
[0005]
There is a need in the art for a stable, non-hygroscopic crystalline form of
the free acid (wherein R = PO(OH)2) or a salt thereof that can be accurately
poured and
weighed for use in pharmaceutical formulations. Also, it would be advantageous
if the
CA 02751392 2017-01-31
2
crystalline form did not form a large number of polymorphs, as the number of
polymorphs
hinders the ability to reproducibly provide the identical polymorph during
manufacturing.
Making a particular crystalline form having these properties is an empirical
process, and one
skilled in the art would be unable to predict among the free acid form of the
pharmaceutical
compound or one of the corresponding salts, which would crystallize, if at
all, under which
crystallization conditions. In addition, one skilled in the art would be
unable to predict which
crystalline form would have the beneficial properties of stability,
pourability, non-
hygroscopicity and reproducibility.
100061 In addition, improved methods of making the free acid are
disclosed in US
Patent Application No. 12/577,089, which is assigned to Trius Therapeutics,
Inc. Difficulties
in filtering crystalline material and processing the crystalline material into
dosage forms, such
as tablets, have arisen because the free acid forms fine particles which delay
processing time.
100071 In addition, it would be advantageous to have a purified compound
that is
suitable for pharmaceutical compositions.
SUMMARY OF THE INVENTION
[00081 Surprisingly, a crystalline (R)-3-(4-(2-(2-methyltetrazol-5-
yppyridin-5-y1)-
3-fluoropheny1)-5-hydroxymethyl oxazolidin-2-one dihydrogen phosphate 1 (R
P0(011)2),
was more stable and non-hygroscopic than the salt forms that were tested. In
addition, unlike
typical crystallizations, where the crystallization conditions, such as the
solvent and
temperature conditions, determine the particular crystalline form, the same
crystalline form of
1 (R = P0(014)2) was produced using many solvent and crystallization
conditions.
Therefore, this crystalline form was stable, was made reproducibly, and
reduced the chances that other polymorphs would form
contaminating impurities during production. However, in all preliminary
testing, the free
acid crystallized as fine particles, making filtering and processing
difficult.
0
NN,N
I \ N
(1)
CA 02751392 2017-01-31
3
[0009] To overcome difficulties in filtering and processing crystalline (R)-
3-(4-
(2-(2-methyltetrazol-5-yl)pyridin-5-y1)-3-fluoropheny1)-5-hydroxymethyl
oxazolidin-2-one
dihydrogen phosphate 1 (R = PO(OH)2), processes described herein result in
reduced filtering time, avoid more toxic solvents, and increased ease of
preparing dosage forms such as tablets. It has been found that implementing
various
processes can control the particle size distribution of the resulting
material, which is useful
for making the crystalline form, and for commercial production and
pharmaceutical use.
Surprisingly, the process for increasing the particle size reduces the amount
of the dimer
impurity, in comparison to the process for making the free acid disclosed in
US Patent
Application No. 12/577,089. Thus, various methods of making and using the
crystalline
form are also provided.
[0010] In addition, by using methods of making the free acid disclosed in
US
Patent Application No. 12/577,089, which is assigned to the same assignee as
in the present
application, and by using the crystallization methods described herein, a
crystalline free acid
having at least 96% purity by weight may be formed that comprises a compound
having the
following formula:
0
NN
N
N CI
(hereinafter "the chloro impurity"), i.e., (R)-5-(chloromethyl)-3-(3-fluoro-4-
(6-(2-methy1-
2H-tetrazol-5-yOpyridin-3-yl)phenypoxazolidin-2-one in an amount less than 1%.
[0011] Similarly, by using methods of making the free acid disclosed in US
Patent Application No. 12/577,089, which is assigned to the same assignee as
in the present
application, and by using the crystallization methods described herein, a
crystalline free acid
having at least 96% purity by weight may be formed that comprises a compound
having the
following formula:
0
zN
CA 02751392 2011-08-02
WO 2010/091131 PCT/US2010/023122
4
(hereinafter "TR-700"), i.e., 5R)-3 -{3 -Flu oro-4-16-(2-methy1-2H-1 ,2,3 ,4-
tetrazol-5 -y1)-
pyridin-3 -yll -phenyl}-5-hydroxymethy1-1,3 -ox az olidin-2-one, in an amount
less than 1%.
[0012] The crystalline free acid may have one or more of the attributes
described
herein.
[0013] In some aspects, a purified crystalline (R)-3-(4-(2-(2-
methyltetrazol-5-y1)-
pyridin-5-y1)-3-fluoropheny1)-5-hydroxymethyl oxazolidin-2-one dihydrogen
phosphate, i.e.,
the free acid, has a purity of at least about 96% by weight. In some
embodiments, the
crystalline free acid has a median volume diameter of at least about 1.0pm.
[0014] In some embodiments, pharmaceutical composition comprises the
free
acid or a salt thereof and at least one pharmaceutically acceptable carrier,
excipient or
diluent.
[0015] In some embodiments, a method of treating a bacterial infection
comprises
administering an effective amount of the crystalline free acid, or a salt
thereof to a subject in
need thereof. Methods may also include comprise treating a bacterial infection
comprising
administering the free acid, pharmaceutical composition thereof or a salt to a
subject in need
thereof.
[0016] In some aspects, processes for making the free acid comprise
drying
crystallized (R)-3-(4-(2-(2-methyltetrazol-5 -yl)pyridin-5 -y1)-3-flu
oropheny1)-5 -hydroxy-
methyl oxazolidin-2-one dihydrogen phosphate, or a pharmaceutical composition
comprising
the salt thereof.
[0017] These and other embodiments are described in greater detail
below.
BRIEF DESCRIPTION OF THE DRAWINGS
[0018] Figure 1 the FT-Raman spectrum of crystalline 1 (R = PO(OH)2).
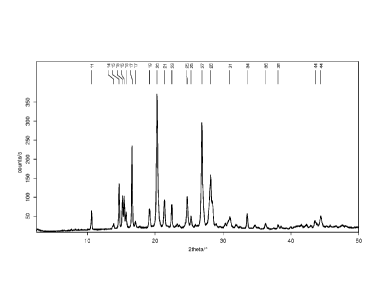
[0019] Figure 2 shows the X-ray powder pattern of crystalline 1 (R =
PO(OH)2).
[0020] Figure 3 shows the differential scanning calorimetry (DSC)
thermogram
of crystalline 1 (R = PO(OH)2).
[0021] Figure 4 shows the 1H NMR spectrum of 1 (R = PO(OH)2).
[0022] Figure 5 depicts the TG-FTIR diagram of crystalline 1 (R =
PO(OH)2).
[0023] Figure 6 is a diagram showing the dynamic vapor sorption (DVS)
behavior of crystalline 1 (R = PO(OH)2).
CA 02751392 2011-08-02
WO 2010/091131 PCT/US2010/023122
[0024] Figure 7 is a manufacturing process schematic for 1 (R =
PO(OH)2) (TR-
701 FA) in a tablet dosage form.
[0025] Figure 8 is a manufacturing process schematic for 1 (R =
PO(OH)2) (TR-
701 FA) Compounding Solution for Lyophilization.
[0026] Figure 9 is a manufacturing process schematic for 1 (R =
PO(OH)2) (TR-
701 FA) for Injection, 200 mg/vial: sterile filtering, filling, and
lyophilization.
[0027] Figure 10 is a representative particle size distribution of
crystalline free
acid without regard to controlling particle size distribution as also
described herein.
[0028] Figure 11 is a representative particle size distribution of
crystalline free
acid made using laboratory processes to control particle size described
herein.
[0029] Figure 12 is a representative particle size distribution of
crystalline free
acid made using scaled up manufacturing processes to control particle size
described herein.
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENT
[0030] (R)-3-(4-(2-(2-methyltetrazol-5 -yl)pyridin-5 -y1)-3 -flu
oropheny1)-5 -
hydroxymethyl oxazolidin-2-one dihydrogen phosphate 1 (R = PO(OH)2), which is
sometimes referred to herein as the "free acid" or "TR-701 FA," and several
salts thereof
were prepared under various crystallization conditions to determine which of
the materials
would form the most stable and least hygroscopic crystalline compound. The
empirical
process of making crystalline forms of the free acid and salts thereof
resulted in the selection
of a crystalline free acid that, in addition to superior stability and non-
hygroscopicity, was
reproducibly made under various crystallization conditions, which was
subsequently purified
and dried.
[0031] Specifically, most of the salts that were evaluated were
difficult to prepare
in a crystalline form or were otherwise unstable, such as in a purified or
dried form. For
example, with respect to the mono-sodium salt, the formation of a stable
hydrate was not
detected. Also, the material contained over 10% by weight of water and
therefore the
material was very hygroscopic, and thus not suitable for the desired use.
[0032] A disodium salt crystalline hydrate was formed, but was unstable
and
contained 19.6% by weight of water. The disodium salt, however, was very
soluble. Drying
the hydrate resulted in amorphous samples. The water content of an amorphous
sample was
about 6.2% by weight.
CA 02751392 2017-01-31
6
[0033] A crystalline solid material was not isolated for a di-potassium.
[0034] A hemi-calcium salt was prepared as a crystal, however, it was
unsuitably
hygroscopic.
[0035] A hemi-magnesium salt crystalline material was formed and
appeared to
contain various hydrates of a salt, and therefore, the presence of various
polymorphs would
render it less desirable for use in a formulation. In one experiment, a
magnesium salt had a
melting point of 152.8 C, which in this case indicated that this material was
less stable in
comparison to the free acid.
[0036] The free acid formed crystals, which were non-hygroscopic upon
filtering
and drying, which showed an aqueous solubility of 0.1 mg/ml (pH = 3.2 of the
saturated
solution). The crystalline material's melting point was approximately 255-258
C, and
therefore was stable at a relatively high temperature.
[0037] Generally, the crystallization conditions are usually critical
for forming a
particular polymorph; however, surprisingly, the same free acid polymorph was
formed
under all of the various conditions in which the crystalline free acid was
formed.
[0038] In some embodiments, the crystalline material is non-hygroscopic,
so it
does not readily take up and retain water from the atmosphere. In some
embodiments, "non-
hygroscopic" material has a water content of less than about 5%, 4%, 3%, 2%,
1%, 0.9%,
0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%, or 0.1% water by weight.
[0039] The free acid can be used to make both a solid formulation
and an intravenous (IV) formulation. During the evaluation, it was found that
the disodium salt, although unsuitable for solid compositions such as tablets,
was very
soluble and therefore suitable for IV formulations. Thus, in another
embodiment, a sterile
lyophilized powder for injection is made by forming a disodium salt in situ
with sodium
hydroxide and lyophilizing the resulting solution. The disodium salt is highly
soluble and
therefore is advantageous to reconstitute in sterile water to yield a
solution. In some
embodiments, the resulting solution may be added to an intravenous bag. The
bag may
contain an isotonic solution such as 0.9% sodium chloride or 5% dextrose.
[0040] In some embodiments, the salt solution, such as a disodium or
monosodium salt, can be lyophilized by freezing the solution in a lyophilizer
to about -50 to
¨30 C at about 0.1 to 1 degree/minute and holding it for about 200-700 minutes
at which
CA 02751392 2017-01-31
7
point the chamber in the lyophilizer is evacuated to approximately 200-250
millitorr and the
temperature is ramped up to about -30 to about -10 C at about 0.5 to about 3
degrees/minute.
The product is held at -30 to about -10 C for about 1000-2500 minutes and then
the
temperature is ramped up to about 21-35 C at about 0.1 to 1 degrees/minute and
held for
1000-2500 minutes to give the finished product.
[0041] In
embodiments of some preparation methods, the crystalline free acid
(R)-3-(4-(2-(2-methyltetrazol-5-yppyridin-5-y1)-3-fluoropheny1)-5-
hydroxymethyl
oxazolidin-2-one dihydrogen phosphate 1 (R = P0(OH)2) can be prepared by
acidification of
an aqueous solution of the corresponding salt, such as the disodium salt 1 (R
= P0(ONa)2).
[0042] A salt of
the free acid 1 (R = P0(OH)2) can be used to regenerate the
free acid by acidification. In some embodiments, the salt is an alkali metal
or an alkaline
earth metal. In other embodiments, the salt is an alkali metal salt, such as a
disodium salt of 1
(R = P0(01)2).
[0043] It was found
that the choice of acid is not critical. An acid that is
sufficiently acidic to doubly protonate the phosphate disodium salt 1 (R =
PO(ONa)2), or
other salt, to yield the free acid 1 (R = P0(OH)2) can be used. In some
embodiments, the acid
is HC1, HBr, or I-12SO4.
[0044] After
dissolving the salt of the (R)-3-(4-(2-(2-methyl¨itetrazol-5-y1)-
pyridin-5-y1)-3-fluoro-phenyl)-5-hydroxy-methyl oxazolidin-2-one dihydrogen
phosphate,
and after, acidifying the salt solution to form crystals, the crystals may be
filtered from the
supernatant. In some embodiments, wet crystals may be dried, for example by
using a
vacuum or lyophilizing the crystals.
[0045] In some
embodiments, crystalline refers to uniformly crystalline material
of crystalline (R)-3-(4-(2-
(2-methyltetrazol-5-yepyridin-5-y1)-3-fluoropheny1)-5-
hydroxymethyl oxazolidin-2-one dihydrogen phosphate, such as substantially
pure crystals.
[0046] The terms
"approximately, "about," and "substantially" as used herein
represent an amount close to the stated amount that still performs the desired
function or
achieves the desired result. For example, the terms "approximately," "about"
and
"substantially" may refer to an amount that is within less than 10% of, within
less than 5%
of, within less than 1% of, within less than 0.1% of, and within less than
0.01% of the stated
amount.
CA 02751392 2011-08-02
WO 2010/091131 PCT/US2010/023122
8
[0047] For instance, in the pharmaceutical industry, it standard
practice to
provide substantially pure material when formulating pharmaceutical
compositions.
Therefore, in some embodiments, "substantially pure" refers to the amount of
purity required
for formulating pharmaceuticals, which may include, for example, a small
amount of
amorphous material or other material, wherein the material may still achieve
sufficient
pourability, lack of hygroscopicity, and purity suitable for pharmaceutical
use. In some
embodiments, the crystalline free acid that is substantially pure contains at
least about 96%
crystalline free acid by weight, such as at least about 96.1%, 96.2%, 96.3%,
96.4%, 96.5%,
96.6%, 96.7%, 96.8%, 96.9%, 97%, 97.1%, 97.2%, 97.3%, 97.4%, 97.5%, 97.6%,
97.7%,
97.8%, 97.9%, 98%, 98.1%, 98.2%, 98.3%, 98.4%, 98.5%, 98.6%, 98.7%, 98.8%,
98.9%,
99%, 99.1%, 99.2%, 99.3%, 99.4%, 99.5%, 99.6%, 99.7%, 99.8%, 99.9%, or 100%
crystalline free acid by weight. In some embodiments, the di- or mono-sodium
salt in
formulations described herein have at least about 96%, 96.1%, 96.2%, 96.3%,
96.4%, 96.5%,
96.6%, 96.7%, 96.8%, 96.9%, 97%, 97.1%, 97.2%, 97.3%, 97.4%, 97.5%, 97.6%,
97.7%,
97.8%, 97.9%, 98%, 98.1%, 98.2%, 98.3%, 98.4%, 98.5%, 98.6%, 98.7%, 98.8%,
98.9%,
99%, 99.1%, 99.2%, 99.3%, 99.4%, 99.5%, 99.6%, 99.7%, 99.8%, 99.9%, or 100%
crystalline salt by weight. In formulating pharmaceuticals, it is useful to
provide a non-
sticky solid crystalline (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-y1)-3-
fluoropheny1)-5-
hydroxymethyl oxazolidin-2-one dihydrogen phosphate that can be poured and
accurately
weighed for use in, for example, tablets and capsules. Therefore, in some
embodiments, the
crystalline material is in a pourable form such that the particles do not
strongly adhere to
each other or the vessel in which it is contained, such that it is capable of
uniformly and
steadily flowing from a vessel.
[0048] Preparation of the free acid (R)-3-(4-(2-(2-methyltetrazol-5-
yl)pyridin-5-
y1)-3-fluoropheny1)-5-hydroxymethyl oxazolidin-2-one dihydrogen phosphate 1 (R
=
PO(OH)2), and of its disodium salt 1 (R = PO(ONa)2) is described in US Patent
Publ. No.
2007/0155798 and US Patent Application No. 12/577,089, the latter of which is
assigned to
the same assignee as in the present application.
[0049] In embodiments of some preparation methods, the crystalline free
acid
(R)-3-(4-(2-(2-methyltetrazol-5-yepyridin-5-y1)-3-fluoropheny1)-5-
hydroxymethyl
CA 02751392 2017-01-31
9
oxazolidin-2-one dihydrogen phosphate 1 (R = P0(0IO2) can be prepared by
acidification of
an aqueous solution of the corresponding salt, such as the disodium salt 1 (R
= P0(ONa)2).
[0050] A salt of the free acid 1 (R = P0(OH)2) can be used to regenerate
the
free acid by acidification. In some embodiments, the salt is an alkali metal
or an alkaline
earth metal. In other embodiments, the salt is an alkali metal salt, such as a
disodium salt of 1
(R = P0(OH)2).
[0051] In additional embodiments of some preparation methods, the free
acid
itself can be used to prepare the crystalline form by dissolution in a
dissolution solvent, such
as a dipolar aprotic solvent, for example dimethyl sulfoxide (DMSO) or 1-
methy1-2-
pyrrolidone (NMP) followed by addition of a crystallization-inducing solvent
such as
ethanol, acetone, acetonitrile, dioxane, heptanes, isopropyl alcohol,
methanol,
tetrahydrofuran, toluene, water, dichloromethane, methyl isobutyl ketone and
ethyl acetate.
In some embodiments, the dissolution and the crystallization-inducing solvents
can be either
a pure solvent or a mixture of pure solvents, and can be either in the form of
a liquid, a vapor,
or a second layer. In some embodiments of the latter two cases, the
crystallization-inducing
solvent can be employed according to the vapor diffusion method of growing
crystals, or the
solvent-layering method, both of which are well-known to those of skill in the
art.
[0052] In further embodiments of some preparation methods, the free acid
can be
dissolved in at least one dipolar aprotic solvent such as DMSO or NMP at an
elevated
temperature, and crystalline free acid 1 (R = P0(011)2) obtained by cooling of
the resulting
solution, according to methods well-known to those of skill in the art. The
solvent can either
be pure, or itself a mixture of pure solvents
[0053] In formulating pharmaceuticals, it is useful to provide a solid
crystalline
compound that can be easily formed into dosage forms, for example, tablets. In
addition, it is
useful to shorten the length of time necessary to make a compound. To address
these needs,
in some embodiments, a method of making crystalline 1 (R = P0(OH)2) that
results in
increased particle size are disclosed that decrease the filtering time caused
by
fine particles that slow down the filtering step. In further embodiments,
crystalline 1 (R =
P0(OH)2) has a particular particle size distribution, for example, that
directly results from the
method without relying on sieving the material solely to obtain the particle
size distribution.
CA 02751392 2011-08-02
WO 2010/091131 PCT/US2010/023122
[0054] To this end, in some embodiments, the resulting larger particle
size of the
crystalline 1 (R = PO(OH)2) may be made by a high temperature precipitation
procedure. In
addition, in embodiments wherein an acid is used to form the free acid from
the salt, it was
found that the increasing the rate at which the reaction mixture was added to
the acid affects
the particle size and makes the particles larger. Thus, in some embodiments,
the reaction
mixture may be contacted to the acid solution as fast as possible, such that
there is essentially
immediate contact with the acid solution. In conventional methods, the
reaction mixture
made contact with the acid solution more slowly, because the acid solution was
added to the
reaction mixture and therefore the reaction mixture may not contact the acid
solution until
some time after addition of the acid solution, causing much smaller particle
size. It was
found that reversing the step, that is, adding the reaction mixture to the
acid solution, will
allow the reaction mixture to effectively immediately contact the acid over
the course of
introducing the reaction mixture to the acidic solution, which results in
larger particle size
material. Thus, in some embodiments, immediate contact is made by adding the
reaction
mixture to the acid solution. The reaction mixture may be pumped into the acid
solution
over time, for example, over a few hours, such as 1-4 hours.
[0055] In some embodiments, an aqueous ethanol- or THF-containing
solution of
TR-701FA may be prepared by adding a sodium bicarbonate solution, for example,
a 2-10%
solution by weight, such as a 5% solution. In some embodiments, the solution
may be added
to an aqueous acidic solution and ethanol or THF to form the free acid. In
some
embodiments, from about 0.5-10, about 1.5-3.0, or about 2.2 equivalents of 1 M
HC1 may be
used. In addition, in some embodiments, about 1-10 volumes, about 2-6 volumes,
or about 4
volumes of ethanol may be used. THF may also be used. In some embodiments, the
solution
including the hydrochloric acid and ethanol may be maintained at about 40-100
C, about 60-
70 C, or about 65 to 70 C. The acid and alcohol may be adjusted. The TR-701FA
crystallized during this addition with a reduced amount of fines in the
product in comparison
to previously disclosed methods.
[0056] In some embodiments, the ethanol or THF prevents the free acid
from
gelling during the process.
[0057] Typical particle size distribution is measured using a laser
diffraction
particle size analyzer, namely a Malvern Mastersizer. D10 (pm) represents the
diameter
CA 02751392 2011-08-02
WO 2010/091131 PCT/US2010/023122
11
below which lies 10% of the total particle volume. D50 (pm) is the median
volume diameter.
D90 (pm) is the diameter below which lies 90% of the total particle volume.
[0058] In some embodiments, when the particle size is not controlled,
10% of the
total particle volume may have a diameter of less than about 0.28pm, the
median volume
diameter may be about 0.79 pm, and 90% of the total particle volume may have a
diameter of
less than about 0.44pm. By controlling (increasing) the particle size using
methods disclosed
herein, the particles are significantly larger overall.
[0059] In some embodiments, when the particle size is controlled using
the
methods described herein to increase particle size, 10% of the total particle
volume may have
an average diameter of at least about 0.5 pm, and/or the median volume
diameter may be at
least about 1.0pm, and/or 90% of the total particle volume may have an average
diameter of
at least about 45pm. In some embodiments, when the particle size is controlled
(to increase
particle size), 10% of the total particle volume may have an average diameter
of about 0.5-
10pm, such as about 1-5pm. For example, when the particle size is controlled
(to increase
particle size), 10% of the total particle volume may have an average diameter
of about 0.5,
0.6, 0.7, 0.8, 0.9, 1.0, 1.1, 1.2, 1.3, 1.4, 1.5, 1.6, 1.7, 1.8, 1.9, 2.0,
2.1, 2.2, 2.3, 2.4, 2.5, 2.6,
2.7, 2.8, 2.9, 3.0, 3.1, 3.2, 3.3, 3.4, 3.5, 3.6, 3.7, 3.8, 3.9, 4.0, 4.1,
4.2, 4.3, 4.4, 4.5, 4.6, 4.7,
4.8, 4.9, 5.0, 5.1, 5.2, 5.3, 5.4, 5.5, 5.6, 5.7, 5.8, 5.9, 6.0, 6.1, 6.2,
6.3, 6.4, 6.5, 6.6, 6.7, 6.8,
6.9, 7.0, 7.1, 7.2, 7.3, 7.4, 7.5, 7.6, 7.7, 7.8, 7.9, 8 .0, 8.1, 8.2, 8.3,
8.4, 8.5, 8.6, 8.7, 8.8, 8.9,
9.0, 9.1, 9.2, 9.3, 9.4, 9.5, 9.6, 9.7, 9.8, 9.9, or 10.0pm.
[0060] In some embodiments, when the particle size is controlled (to
increase
particle size), the median volume diameter may be greater than about 1.0pm,
and have an
average median volume diameter about 1-44pm, about 1-40pm, about 10-35pm,
about 20-
30pm, or about 25-29, such as about 27pm. In some embodiments, when the
particle size is
controlled to increase particle size, the average median volume diameter may
be about 1, 2,
3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23,
24, 25, 26, 27, 28, 29,
30, 31, 32, 33, 34, 35, 36, 38, 39, 40, 41, 42, 43, or 44pm. For example, the
average median
volume diameter may be about 25, 25.1, 25.2, 25.3, 25.4, 25.5, 25.6, 25.7,
25.8, 25.9, 26,
26.1, 26.2, 26.3, 26.4, 26.5, 26.6, 26.7, 26.8, 26.9, 27, 27.1, 27.2, 27.3,
27.4, 27.5, 27.6, 27.7,
27.8, 27.9, 28, 28.1, 28.2, 28.3, 28.4, 28.5, 28.6, 28.7, 28.8, 28 .9, or 29
pm.
CA 02751392 2011-08-02
WO 2010/091131 PCT/US2010/023122
12
[0061] In some embodiments, when the particle size is controlled (to
increase
particle size), 90% of the total particle volume may have an average diameter
of the least
about 45pm such as about 45-100, about 45-80, about 55-75, or about 64-68 such
as about
66. In some embodiments, when the particle size is controlled, 90% of the
total particle
volume may have an average diameter of about 45, 46, 47, 48, 49, 50, 51, 52,
53, 54, 55, 56,
57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75,
76, 77, 78, 79, 80, 81,
82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, or 100
pm. For example,
90% of the total particle volume may have an average diameter of about 64,
64.1, 64.2, 64.3,
64.4, 64.5, 64.6, 64.7, 64.8, 64.9, 65, 65.1, 65.2, 65.3, 65.4, 65.5, 65.6,
65.7, 65.8, 65.9, 66,
66.1, 66.2, 66.3, 66.4, 66.5, 66.6, 66.7, 66.8, 66.9, 67, 67.1, 67.2, 67.3,
67.4, 67.5, 67.6, 67.7,
67.8, 67.9, or 68pm.
[0062] The crystalline free acid 1 (R = PO(OH)2) may be characterized
in having
the FT-Raman for example as shown in Figure 1, and the X-ray powder
diffraction for
example as shown in Figure 2, with the corresponding numerical data for
example as shown
in Table 1 and Table 2 respectively. Figure 3, Figure 4, Figure 5 and Figure 6
show
examples of the differential scanning calorimetry (DSC) thermogram, solution
1H NMR
spectrum, the TG-FTIR diagram, and the dynamic vapor sorption (DVS) behavior
of
crystalline 1 (R = PO(OH)2) respectively.
Table 1 FT-Raman spectroscopic data for crystalline free acid 1 (R = PO(OH)2)
Wavenumber (cm-1) Absolute Intensity
1612.9 2.57
1579.0 0.38
1521.3 0.25
1455.8 0.26
1404.9 0.39
1324.4 0.82
1274.7 0.24
CA 02751392 2011-08-02
WO 2010/091131
PCT/US2010/023122
13
1149.9 0.17
1018.3 0.22
Table 2 X-ray powder pattern diffraction data for crystalline free acid 1 (R =
PO(OH)2)
Angle 2Thetar Intensity / %
10.6 17
13.7 6
13.9 8
14.7 38
15.2 28
15.4 28
15.7 16
16.6 65
17.1 10
19.2 19
20.3 100
21.4 25
22.4 23
23.2 8
23.6 7
24.7 29
25.3 14
25.9 8
26.8 82
CA 02751392 2011-08-02
WO 2010/091131 PCT/US2010/023122
14
28.2 44
28.4 24
29.0 8
30.3 8
30.8 11
31.0 13
31.9 8
33.5 17
34.7 7
[0063] In
some embodiments, the distinguishing peaks for the crystalline free
acid comprise the following peaks: 14.7 , 15.2 , 16.6 , 20.3 , 26.8 , and 28.2
.
[0064] In
other embodiments, the distinguishing peaks for the crystalline free acid
comprise the following peaks: 10.6 , 13.9 , 14.7 , 15.2 , 16.6 , 20.3 , 26.8 ,
and 28.2 .
[0065] In
some embodiments, the crystalline free acid comprises impurities that
are present in less than 1% of the purified crystalline free acid. These
impurities include
0
- /\N N
/ N
\------.....õ..OH
F
i.e., 5R)-3-
{ 3-Fluoro-4-116-(2-methy1-2H-1,2,3,4-tetrazol-5-y1)-pyridin-3-yll -phenyl } -
5-
hydroxymethy1-1,3-oxazolidin-2-one ("TR-700") and/or
0
N--------"N
L / \ / N
F
i.e., (R)-5-
(chloromethyl)-3-(3-fluoro-4-(6-(2-methy1-2H-tetrazol-5-yl)pyridin-3-
yl)phenyeoxazolidin-2-one ("chloro impurity").
CA 02751392 2017-01-31
[0066] Of the conventionally produced material having impurities that
were
identified using HPLC in Example 15, at least 2% by weight of the chloro
impurity was
present. In purified crystalline free acid made using the method of making the
free acid
disclosed in US Patent Application No. 12/577,089, which is assigned to the
same assignee
as in the present application, and the crystallization methods disclosed
herein, the chloro
impurity was present in less than about 1% by weight, such as less than about
0.9%, 0.8%,
0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%, or 0.1% by weight of the of the
crystalline free acid.
In some embodiments the chloro impurity may be reduced to lower than 0.1% by
weight, such as, less than about 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%,
0.03%, 0.02%,
or 0.01% by weight of the crystalline free acid. In some embodiments, the
purified
crystalline free acid is substantially free of the chloro impurity.
[0067] Of the conventionally produced material having impurities that
were
identified using HPLC in Example 15, at least about 1% by weight of the TR-700
impurity
was present. In purified crystalline free acid made using the method of making
the free acid
disclosed in US Patent Application No. 12/577,089, which is assigned to the
same assignee
as in the present application, and the crystallization methods disclosed
herein, the TR-700
impurity was present in less than about 1% by weight. In some embodiments, the
crystalline
free acid contains less than about 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%,
0.2%, or 0.1%
by weight of the TR-700 impurity. In some embodiments, the crystalline free
acid is
substantially free of the TR-700 impurity.
[0068] In addition, purified crystalline free acid made using the method
of
making the free acid disclosed in US Patent Application No. 12/577,089, which
is assigned
to the same assignee as in the present application, and the crystallization
methods disclosed
herein, may also be distinguished from the conventionally produced crystalline
free acid by
the presence of the following compounds. For example, the following impurities
were not
found in a sample of conventionally produced crystalline free acid as shown in
Example 15:
HN0
HO/
(hereinafter "des-methyl TR-701"), i.e., dihydrogen ((5R)-3- { 3 -fluoro-4-[6-
(2H-1,2,3,4-
tetraz o1-5-y1)-3-pyridinyllphenyl )-2-oxo-1,3-oxazolan-5-yl)methyl phosphate;
CA 02751392 2011-08-02
WO 2010/091131 PCT/US2010/023122
16
0
0% OH
,.-N ¨ )\----0
N
P
/ \
0
/ 11 \-----c.,, OH
V N N
F
0
N,..-N ¨ )\----0 OH
/ ii N .-- ON.õ,,,
---- ,--- H
c.)0
F
(hereinafter "overalkylated-phosphorylated impurity"), i.e., 514(3-(3-fluoro-4-
(6-(2-
methyl-2H-tetrazol-5-yepyridin-3-yepheny1)-2-oxooxazolidin-5-yl)methoxy)-3-
hydroxypropan-2-y1 dihydrogen phosphate and,
34(3-(3-fluoro-4-(6-(2-methy1-2H-tetrazol-5-yl)pyridin-3-yepheny1)-2-
oxooxazolidin-5-
yl)methoxy)-2-hydroxypropyl dihydrogen phosphate;
0
N.!-N1 )\----0
IV, / \ / . N OH
F
% õ...0
0"--P\
OH
\N
=
/ \ F
-N
S \ N
N\
(hereinafter "one of the 0A-700 mixed di ester") i.e., 3-11(5R)-3-{3-fluoro-4-
16-(2-
methy1-2H-tetrazol-5-yepyridin-3-yflphenyl }-2-oxo-1,3-oxazolidin-5-Amethoxy} -
2-
hydroxypropyl 1(5R)-3-{ 3-fluoro-4-16-(2-methy1-2H-tetrazol-5-yepyridin-3-
yflphenyl } -2-
oxo-1,3-oxazolidin-5-yflmethyl hydrogen phosphate; and/or
CA 02751392 2016-06-02
17
z ----N
--N
N
0
HO
L.C.0
HON /0
/150
0
N% \
/ N
(hereinafter "another of the 0A-700 mixed di ester") i.e., 2- { [(5R)-3-13-
fluoro-4-[6-
(2-methy1-2H-tetrazol-5-yl)pyridin-3-yl]pheny11-2-oxo-1,3-oxazolidin-5-
ylimethoxyl -1-
hydroxyethyl [(5R)-3- { 3-fluoro-446-(2-methy1-2H-tetrazol-5-yl)pyridin-3 -
yl]phenyl } -2-oxo-
1 ,3-oxazolidin-5-yllmethyl hydrogen phosphate.
[0069] Those skilled in the art will appreciate that various isotopically-
substituted
variants (through, e.g., substitution of deuterium for hydrogen, 13C for
carbon, 15N for
nitrogen, or 32P for phosphorus) can also be readily produced. All such
variants are
contemplated within the scope of this disclosure.
[0070] In various embodiments, the purified crystallized free acid
disclosed
herein can be used alone, in combination with other compounds disclosed
herein, or in
combination with one or more other agents active in the therapeutic areas
described herein.
[0071] In another aspect, the present disclosure relates to a
pharmaceutical
composition comprising one or more physiologically acceptable surface active
agents,
additional carriers, diluents, excipients, smoothing agents, suspension
agents, film forming
substances, and coating assistants, or a combination thereof; and a
composition disclosed
herein. Acceptable additional carriers or diluents for therapeutic use are
well known in the
pharmaceutical art, and are described, for example, in Remington's
Pharmaceutical Sciences,
18th Ed., Mack Publishing Co., Easton, PA (1990). Preservatives, stabilizers,
dyes,
sweeteners, fragrances, flavoring agents, and the like may be provided in the
pharmaceutical
composition. For example, sodium benzoate,
CA 02751392 2011-08-02
WO 2010/091131 PCT/US2010/023122
18
ascorbic acid and esters of p-hydroxybenzoic acid may be added as
preservatives. In
addition, antioxidants and suspending agents may be used. In various
embodiments,
alcohols, esters, sulfated aliphatic alcohols, and the like may be used as
surface active agents;
sucrose, glucose, lactose, starch, microcrystalline cellulose, crystallized
cellulose, mannitol,
light anhydrous silicate, magnesium aluminate, magnesium metasilicate
aluminate, synthetic
aluminum silicate, calcium carbonate, sodium acid carbonate, calcium hydrogen
phosphate,
calcium carboxymethyl cellulose, and the like may be used as excipients;
magnesium
stearate, talc, hardened oil and the like may be used as smoothing agents;
coconut oil, olive
oil, sesame oil, peanut oil, soya may be used as suspension agents or
lubricants; cellulose
acetate phthalate as a derivative of a carbohydrate such as cellulose or
sugar, or
methylacetate-methacrylate copolymer as a derivative of polyvinyl may be used
as
suspension agents; and plasticizers such as ester phthalates and the like may
be used as
suspension agents.
[0072] The term "pharmaceutical composition" refers to a mixture of a
compound
disclosed herein with other chemical components, such as diluents or
additional carriers. The
pharmaceutical composition facilitates administration of the compound to an
organism.
Multiple techniques of administering a pharmaceutical composition exist in the
art including,
but not limited to, oral, injection, aerosol, parenteral, and topical
administration.
Pharmaceutical compositions can also be obtained by reacting the free acid
with inorganic or
organic bases such as sodium hydroxide or magnesium hydroxide. In some
embodiments,
pharmaceutically acceptable salts of the compounds disclosed herein (e.g., as
made in situ
during the manufacture of an intravenous formulation) are provided. In some
embodiments,
sodium hydroxide is used to prepare a lyophilized powder the formulation that
comprises a
salt of the free acid, which is produced in situ.
[0073] The term "carrier" refers to a chemical compound that
facilitates the
incorporation of a compound into cells or tissues.
[0074] The term "diluent" refers to chemical compounds diluted in water
that will
dissolve the composition of interest as well as stabilize the biologically
active form of the
compound. Salts dissolved in buffered solutions are utilized as diluents in
the art. One
commonly used buffered solution is phosphate buffered saline because it mimics
the salt
conditions of human blood. Since buffer salts can control the pH of a solution
at low
CA 02751392 2011-08-02
WO 2010/091131 PCT/US2010/023122
19
concentrations, a buffered diluent rarely modifies the biological activity of
a compound. As
used herein, an "excipient" refers to an inert substance that is added to a
composition to
provide, without limitation, bulk, consistency, stability, binding ability,
lubrication,
disintegrating ability, etc., to the composition. A "diluent" is a type of
excipient.
[0075] The term "physiologically acceptable" refers to a carrier or
diluent that
does not abrogate the biological activity and properties of the compound.
[0076] The pharmaceutical compounds described herein can be
administered to a
human patient per se, or in pharmaceutical compositions where they are mixed
with other
active ingredients, as in combination therapy, or suitable carriers or
excipient(s). Techniques
for formulation and administration of the compounds of the instant application
may be found
in "Remington's Pharmaceutical Sciences," Mack Publishing Co., Easton, PA,
18th edition,
1990.
[0077] Suitable routes of administration may, for example, include
oral, rectal,
transmucosal, topical, or intestinal administration; parenteral delivery,
including
intramuscular, subcutaneous, intravenous, intramedullary injections, as well
as intrathecal,
direct intraventricular, intraperitoneal, intranasal, or intraocular
injections. The compound
can also be administered in sustained or controlled release dosage forms,
including depot
injections, osmotic pumps, pills, transdermal (including electrotransport)
patches, and the
like, for prolonged and/or timed, pulsed administration at a predetermined
rate.
[0078] The pharmaceutical compositions of the present invention may be
manufactured in a manner that is itself known, e.g., by means of conventional
mixing,
dissolving, granulating, dragee-making, levigating, emulsifying,
encapsulating, entrapping or
tabletting processes.
[0079] Pharmaceutical compositions may be formulated in any
conventional
manner using one or more physiologically acceptable carriers comprising
excipients and
auxiliaries which facilitate processing of the active compounds into
preparations which can be
used pharmaceutically. Proper formulation is dependent upon the route of
administration
chosen. Any of the well-known techniques, diluents, carriers, and excipients
may be used as
suitable and as understood in the art; e.g., in Remington's Pharmaceutical
Sciences, above.
[0080] Injectables can be prepared in conventional forms, either as
liquid
solutions or suspensions, solid forms suitable for solution or suspension in
liquid prior to
CA 02751392 2011-08-02
WO 2010/091131 PCT/US2010/023122
injection, or as emulsions. Suitable excipients are, for example, water,
saline, dextrose,
mannitol, lactose, lecithin, albumin, sodium glutamate, cysteine
hydrochloride, and the like.
In addition, if desired, the injectable pharmaceutical compositions may
contain minor
amounts of nontoxic auxiliary substances, such as wetting agents, pH buffering
agents, and
the like. Physiologically compatible buffers include, but are not limited to,
Hanks's solution,
Ringer's solution, or physiological saline buffer. If
desired, absorption enhancing
preparations may be utilized.
[0081] For
transmucosal administration, penetrants appropriate to the barrier to be
permeated may be used in the formulation.
[0082]
Pharmaceutical formulations for parenteral administration, e.g., by bolus
injection or continuous infusion, include aqueous solutions of the active
compounds in water-
soluble form. Additionally, suspensions of the active compounds may be
prepared as
appropriate oily injection suspensions. Aqueous injection suspensions may
contain
substances which increase the viscosity of the suspension, such as sodium
carboxymethyl
cellulose, sorbitol, or dextran. Optionally, the suspension may also contain
suitable
stabilizers or agents that increase the solubility of the compounds to allow
for the preparation
of highly concentrated solutions. Formulations for injection may be presented
in unit dosage
form, e.g., in ampoules or in multi-dose containers, with an added
preservative. The
compositions may take such forms as suspensions, solutions or emulsions in
oily or aqueous
vehicles, and may contain formulatory agents such as suspending, stabilizing
and/or
dispersing agents. Alternatively, the active ingredient may be in powder form
for
constitution with a suitable vehicle, e.g., sterile pyrogen-free water, before
use.
[0083] For
oral administration, the composition can be formulated readily by
combining the compositions of interest with pharmaceutically acceptable
carriers well known
in the art. Such carriers, which may be used in addition to the cationic
polymeric carrier,
enable the compositions of the invention to be formulated as tablets, pills,
dragees, capsules,
liquids, gels, syrups, slurries, suspensions and the like, for oral ingestion
by a patient to be
treated. Pharmaceutical preparations for oral use can be obtained by combining
the active
compounds with solid excipient, optionally grinding a resulting mixture, and
processing the
mixture of granules, after adding suitable auxiliaries, if desired, to obtain
tablets or dragee
cores. Suitable excipients are, in particular, fillers such as sugars,
including lactose, sucrose,
CA 02751392 2011-08-02
WO 2010/091131 PCT/US2010/023122
21
mannitol, or sorbitol; cellulose preparations such as, for example, maize
starch, wheat starch,
rice starch, potato starch, gelatin, gum tragacanth, methyl cellulose,
hydroxypropylmethyl-
cellulose, sodium carboxymethylcellulose, and/or polyvinylpyrrolidone (PVP),
e.g.,
Povidone. If desired, disintegrating agents may be added, such as the cross-
linked
polyvinylpyrrolidone (e.g. Crospovidone), agar, or alginic acid or a salt
thereof such as
sodium alginate. Dragee cores are provided with suitable coatings. For this
purpose,
concentrated sugar solutions may be used, which may optionally contain gum
arabic, talc,
polyvinyl pyrrolidone, carbopol gel, polyethylene glycol, and/or titanium
dioxide, lacquer
solutions, and suitable organic solvents or solvent mixtures. Dyestuffs or
pigments may be
added to the tablets or dragee coatings for identification or to characterize
different
combinations of active compound doses. For this purpose, concentrated sugar
solutions may
be used, which may optionally contain gum arabic, talc, polyvinyl pyrrolidone,
carbopol gel,
polyethylene glycol, and/or titanium dioxide, lacquer solutions, and suitable
organic solvents
or solvent mixtures. Dyestuffs or pigments may be added to the tablets or
dragee coatings
for identification or to characterize different combinations of active
compound doses.
[0084] Pharmaceutical preparations which can be used orally include
push-fit
capsules made of gelatin, as well as soft, sealed capsules made of gelatin and
a plasticizer,
such as glycerol or sorbitol. The push-fit capsules can contain the active
ingredients in
admixture with filler such as lactose, binders such as starches, and/or
lubricants such as talc
or magnesium stearate and, optionally, stabilizers. In soft capsules, the
active compounds
may be dissolved or suspended in suitable liquids, such as fatty oils, liquid
paraffin, or liquid
polyethylene glycols. In addition, stabilizers may be added. All formulations
for oral
administration should be in dosages suitable for such administration.
[0085] For buccal administration, the compositions may take the form of
tablets
or lozenges formulated in conventional manner.
[0086] For administration by inhalation, the composition can be
conveniently
delivered in the form of an aerosol spray presentation from pressurized packs
or a nebulizer,
with the use of a suitable propellant, e.g., dichlorodifluoromethane,
trichlorofluoromethane,
dichlorotetrafluoroethane, carbon dioxide or other suitable gas. In the case
of a pressurized
aerosol the dosage unit may be determined by providing a valve to deliver a
metered amount.
Capsules and cartridges of, e.g., gelatin for use in an inhaler or insufflator
may be formulated
CA 02751392 2016-06-02
22
containing a powder mix of the compound and a suitable powder base such as
lactose or
starch.
100871 Further disclosed herein are various pharmaceutical
compositions well
known in the pharmaceutical art for uses that include intraocular, intranasal,
and
intraauricular delivery. Suitable penetrants for these uses are generally
known in the art.
Such suitable pharmaceutical formulations are most often and preferably
formulated to be
sterile, isotonic and buffered for stability and comfort. Pharmaceutical
compositions for
intranasal delivery may also include drops and .sprays often prepared to
simulate in many
respects nasal secretions to ensure maintenance of normal ciliary action. As
disclosed in
Remington's Pharmaceutical Sciences, 18th Ed., Mack Publishing Co., Easton, PA
(1990),
and well-known to those skilled in the art, suitable formulations are most
often and preferably
isotonic, slightly buffered to maintain a pH of 5.5 to 6.5, and most often and
preferably
include antimicrobial preservatives and appropriate drug stabilizers.
Pharmaceutical
formulations for intraauricular delivery include suspensions and ointments for
topical
application in the ear. Common solvents for such aural formulations include
glycerin and
water.
100881 The compositions may also be formulated in rectal compositions
such as
suppositories or retention enemas, e.g., containing conventional suppository
bases such as
cocoa butter or other glycerides.
[0089] In addition to the formulations described previously, the
compositions may
also be formulated as a depot preparation. Such long acting formulations may
be
administered by implantation (for example subcutaneously or intramuscularly)
or by
intramuscular injection. Thus, for example, the compounds may be formulated
with suitable
polymeric or hydrophobic materials (for example as an emulsion in an
acceptable oil) or ion
exchange resins, or as sparingly soluble derivatives, for example, as a
sparingly soluble salt.
[0090] For hydrophobic compounds, a suitable pharmaceutical carrier
may be a
cosolvent system comprising benzyl alcohol, a nonpolar surfactant, a water-
miscible organic
polymer, and an aqueous phase. A common cosolvent system used is the VPD co-
solvent
system, which is a solution of 3% w/v benzyl alcohol, 8% w/v of the nonpolar
surfactant
Polysorbate 8OTM, and 65% w/v polyethylene glycol 300, made up to volume in
absolute
ethanol. Naturally, the proportions of a co-solvent system may be varied
considerably
CA 02751392 2017-01-31
23
without destroying its solubility and toxicity characteristics. Furthermore,
the identity of the
co-solvent components may be varied: for example, other low-toxicity nonpolar
surfactants
may be used instead of POLYSORBATE 8OTM; the fraction size of polyethylene
glycol may
be varied; other biocompatible polymers may replace polyethylene glycol, e.g.,
polyvinyl
pyrrolidone; and other sugars or polysaccharides may substitute for dextrose.
[0091] Methods for treating bacterial infections may include
administering a
therapeutically effective amount of the therapeutic compounds as described
herein. Treating
a bacterial infection may also include prophylactically administering the
therapeutic
compounds to prevent infection or the spread of an infection in a subject at
imminent risk of
infection, such as a subject receiving or about to undergo surgery, an
immunocompromised
subject, or subject otherwise at risk of an infection if the compound was not
administered.
The compounds show inhibitory activity against a broad spectrum of bacteria,
against
methicillin resistant Staphylococcus aureus (MRSA) and vancomycin resistant
Enterococci
(VRE) and have relative antibiotic activity with a relatively low
concentration
thereof or in vivo. Further, the compounds of the present invention may exert
antibacterial activity versus various human and animal pathogens, including
Gram-positive
bacteria such as Staphylococi, Enterococci and Streptococi, anaerobic
microorganisms such
as Bacteroides and Clostridia, and acid-resistant microorganisms such as
Mycobacterium
tuberculosis and Mycobacterium avium. In an embodiment, the bacterial
infection that may
be treated or ameliorated is MRSA.
[0092] The compositions or pharmaceutical compositions described herein
may
be administered to the subject by any suitable means. Non-limiting examples of
methods of
administration include, among others, (a) administration though oral pathways,
which
administration includes administration in capsule, tablet, granule, spray,
syrup, or other such
forms; (b) administration through non-oral pathways such as rectal, vaginal,
intraurethral,
intraocular, intranasal, or intraauricular, which administration includes
administration as an
aqueous suspension, an oily preparation or the like or as a drip, spray,
suppository, salve,
ointment or the like; (c) administration via injection, subcutaneously,
intraperitoneally,
intravenously, intramuscularly, intradermally, intraorbitally,
intracapsularly, intraspinally,
intrasternally, or the like, including infusion pump delivery; as well as (d)
administration
CA 02751392 2011-08-02
WO 2010/091131 PCT/US2010/023122
24
topically; as deemed appropriate by those of skill in the art for bringing the
active compound
into contact with living tissue.
[0093] Pharmaceutical compositions suitable for administration include
compositions where the active ingredients are contained in an amount effective
to achieve its
intended purpose. The therapeutically effective amount of the compounds
disclosed herein
required as a dose will depend on the route of administration, the type of
animal, including
human, being treated, and the physical characteristics of the specific animal
under
consideration. The dose can be tailored to achieve a desired effect, but will
depend on such
factors as weight, diet, concurrent medication and other factors which those
skilled in the
medical arts will recognize. More specifically, a therapeutically effective
amount means an
amount of compound effective to prevent, alleviate or ameliorate symptoms of
disease or
prolong the survival of the subject being treated. Determination of a
therapeutically effective
amount is well within the capability of those skilled in the art, especially
in light of the
detailed disclosure provided herein.
[0094] As will be readily apparent to one skilled in the art, the
useful in vivo
dosage to be administered and the particular mode of administration will vary
depending
upon the age, weight and mammalian species treated, the particular compounds
employed,
and the specific use for which these compounds are employed. The determination
of
effective dosage levels, that is the dosage levels necessary to achieve the
desired result, can
be accomplished by one skilled in the art using routine pharmacological
methods. Typically,
human clinical applications of products are commenced at lower dosage levels,
with dosage
level being increased until the desired effect is achieved. Alternatively,
acceptable in vitro
studies can be used to establish useful doses and routes of administration of
the compositions
identified by the present methods using established pharmacological methods.
[0095] In non-human animal studies, applications of potential products
are
commenced at higher dosage levels, with dosage being decreased until the
desired effect is
no longer achieved adverse side effects disappear. The dosage may range
broadly,
depending upon the desired effects and the therapeutic indication. Typically,
dosages may be
about 10 microgram/kg to about 100 mg/kg body weight, preferably about 100
microgram/kg
to about 10 mg/kg body weight. Alternatively dosages may be based and
calculated upon the
surface area of the patient, as understood by those of skill in the art.
CA 02751392 2016-06-02
[0096] The exact formulation, route of administration and dosage for
the
pharmaceutical compositions of the present invention can be chosen by the
individual
physician in view of the patient's condition. (See e.g., Fingl et al. 1975, in
"The
Pharmacological Basis of Therapeutics", with particular reference to Ch. 1, p.
1). Typically,
the dose range of the composition administered to the patient can be from
about 0.5 to about
1000 mg/kg of the patient's body weight. The dosage may be a single one or a
series of two
or more given in the course of one or more days, as is needed by the patient.
In instances
where human dosages for compounds have been established for at least some
condition, the
present invention will use those same dosages, or dosages that are about 0.1%
to about 500%,
more preferably about 25% to about 250% of the established human dosage. Where
no
human dosage is established, as will be the case for newly-discovered
pharmaceutical
compositions, a suitable human dosage can be inferred from ED50 or ID50
values, or other
appropriate values derived from in vitro or in vivo studies, as qualified by
toxicity studies and
efficacy studies in animals.
[0097] It should be noted that the attending physician would know how
to and
when to terminate, interrupt, or adjust administration due to toxicity or
organ dysfunctions.
Conversely, the attending physician would also know to adjust treatment to
higher levels if
the clinical response were not adequate (precluding toxicity). The magnitude
of an
administrated dose in the management of the disorder of interest will vary
with the severity of
the condition to be treated and to the route of administration. The severity
of the condition
may, for example, be evaluated, in part, by standard prognostic evaluation
methods. Further,
the dose and perhaps dose frequency will also vary according to the age, body
weight, and
response of the individual patient. A program comparable to that discussed
above may be
used in veterinary medicine.
[0098] Although the exact dosage will be determined on a drug-by-drug
basis, in
most cases, some generalizations regarding the dosage can be made. The daily
dosage
regimen for an adult human patient may be, for example, an oral dose of about
0.1 mg to
2000 mg of each active ingredient, preferably about 1 mg to about 500 mg, e.g.
5 to 200 mg.
In other embodiments, an intravenous, subcutaneous, or intramuscular dose of
each active
ingredient of about 0.01 mg to about 100 mg, preferably about 0.1 mg to about
60 mg, e.g.
CA 02751392 2011-08-02
WO 2010/091131 PCT/US2010/023122
26
about 1 to about 40 mg is used. In cases of administration of a
pharmaceutically acceptable
salt, dosages may be calculated as the free base. In some embodiments, the
composition is
administered 1 to 4 times per day. Alternatively the compositions of the
invention may be
administered by continuous intravenous infusion, preferably at a dose of each
active
ingredient up to about 1000 mg per day. As will be understood by those of
skill in the art, in
certain situations it may be necessary to administer the compounds disclosed
herein in
amounts that exceed, or even far exceed, the above-stated, preferred dosage
range in order to
effectively and aggressively treat particularly aggressive diseases or
infections. In some
embodiments, the compounds will be administered for a period of continuous
therapy, for
example for a week or more, or for months or years.
[0099] Dosage amount and interval may be adjusted individually to
provide
plasma levels of the active moiety which are sufficient to maintain the
modulating effects, or
minimal effective concentration (MEC). The MEC will vary for each compound but
can be
estimated from in vitro data. Dosages necessary to achieve the MEC will depend
on
individual characteristics and route of administration. However, HPLC assays
or bioassays
can be used to determine plasma concentrations.
[0100] Dosage intervals can also be determined using MEC value.
Compositions
should be administered using a regimen which maintains plasma levels above the
MEC for
10-90% of the time, preferably between 30-90% and most preferably between 50-
90%.
[0101] In cases of local administration or selective uptake, the
effective local
concentration of the drug may not be related to plasma concentration.
[0102] The amount of composition administered may be dependent on the
subject
being treated, on the subject's weight, the severity of the infection, the
manner of
administration and the judgment of the prescribing physician.
[0103] Compositions disclosed herein can be evaluated for efficacy and
toxicity
using known methods. For example, the toxicology of the compound may be
established by
determining in vitro toxicity towards a cell line, such as a mammalian, and
preferably human,
cell line. The results of such studies are often predictive of toxicity in
animals, such as
mammals, or more specifically, humans. Alternatively, the toxicity of
particular compounds
in an animal model, such as mice, rats, rabbits, or monkeys, may be determined
using known
methods. The efficacy of a particular compound may be established using
several
CA 02751392 2016-06-02
27
recognized methods, such as in vitro methods, animal models, or human clinical
trials.
Recognized in vitro models exist for nearly every class of condition.
Similarly, acceptable
animal models may be used to establish efficacy of chemicals to treat such
conditions. When
selecting a model to determine efficacy, the skilled artisan can be guided by
the state of the
art to choose an appropriate model, dose, and route of administration, and
regime. Of course,
human clinical trials can also be used to determine the efficacy of a compound
in humans.
[0104] The compositions may, if desired, be presented in a pack or
dispenser
device which may contain one or more unit dosage forms containing the active
ingredient.
The pack may for example comprise metal or plastic foil, such as a blister
pack. The pack or
dispenser device may be accompanied by instructions for administration. The
pack or
dispenser may also be accompanied with a notice associated with the container
in form
prescribed by a governmental agency regulating the manufacture, use, or sale
of
pharmaceuticals, which notice is reflective of approval by the agency of the
form of the drug
for human or veterinary administration. Such notice, for example, may be the
labeling
approved by the U.S. Food and Drug Administration for prescription drugs, or
the approved
product insert. Compositions comprising a compound of the invention formulated
in a
compatible pharmaceutical carrier may also be prepared, placed in an
appropriate container,
and labeled for treatment of an indicated condition.
A. Examples
1. Instrumentation
[0105] Raman microscopy was performed on a Renishaw System 1000, with
stabilized diode laser 385 nm excitation and a NIR enhanced Peltier-cooled
charge coupled
device camera as detector. Measurements were carried out with 50x or a long
working
distance 20x objective over a frequency range of 2000-100 cm-I.
[0106] FT-Raman spectra were obtained on a Bruker RFS100 spectrometer
with
Nd:YAG 1064 nrn excitation, 100 mW laser power, and a Ge detector. Sixty-four
scans were
recorded over the range 25-3500 cm-I, at 2 cm' resolution.
[0107] Bruker D8; Bragg-Brentano, reflection geometry; Copper K(alpha)
radiation, 40 kV/ 40 mA; variable divergence slit; LynxEyeTM detector with 3
window; step
size, 0.02-'2; step time, 37 s. The samples were rotated (0.5 rps) during the
measurement.
CA 02751392 2011-08-02
WO 2010/091131 PCT/US2010/023122
28
[0108] Sample preparation: The samples were generally prepared without
any
special treatment other than the application of slight pressure to get a flat
surface. Silicon
single crystal sample holder types: a) standard holder for polymorph
screening, 0.1 mm deep,
less than 20 mg sample required; b) 0.5 mm deep, 12 mm cavity diameter, ca. 40
mg
required; c) 1.0 mm deep, 12 mm cavity diameter, ca. 80 mg required. Normally
samples
were measured uncovered. Kapton foil or PMMA "dome" covers are always
indicated on the
diffractogram with the sample identification.
2. Preparation of Crystalline Free Acid 1 (R = PO(OH)/)
Example 1
[0109] A solution of 1 (R = PO(ONa)2) was prepared in H20 and 1 M HC1
added
to give a fine suspension, which, after addition of tetrahydrofuran (THF), was
stirred and
filtered. The resulting crystalline solid 1 (R = PO(OH)2) was dried in vacuum,
and
characterized by FT-Raman (FTR) (Figure 1), X-ray powder diffraction (XRPD,
Malvern
Mastersizer) (Figure 2), thermogravimetry-Fourier transform infrared
spectroscopy (TG-
FTIR), and differential scanning calorimetry (DSC). DSC measurement showed a
melting
point at 256.9 C followed by a decomposition of the sample (Fig. 3).
Example 2
[0110] To 1 (R = PO(ONa)2) (2 g) dissolved in 10 mL H20 was slowly
added
HCI (6 mL; 1 M) to yield a fine suspension of a light yellow solid. After
addition of a further
mL H20 and 20 mL THF the suspension was filtered and dried in vacuum.
Example 3
[0111] To 1 (R = PO(ONa)2) (2 g) dissolved in 10 mL H20 was slowly
added
HC1 (8 mL; 1 M) to give a fine suspension of a light yellow solid, to which a
further 25 mL
H20 were added. The solid was filtered, washed with 10 mL 0.1 M HCI and 100 mL
water
and dried in vacuum.
Example 4
[0112] To 1 (R = PO(ONa)2) (5 g) dissolved in 30 mL water was added 15
mL
HCI (1 M) and 30 mL of THF to produce a light yellow suspension, which was was
stirred
30 mm at room temperature and filtered. The resulting solid was suspended in
150 mL water
and stirred 60 min at room temperature. Then 50 mL THF were added and the
suspension
CA 02751392 2011-08-02
WO 2010/091131 PCT/US2010/023122
29
was stirred 18 h. The suspension was filtered and the solid was washed with 10
mL HC1 (0.1
M) and 100 mL water and dried in vacuum (15 h).
Example 5
[0113] To 1 (R = PO(ONa)2) (2 g) dissolved in 15 mL water was slowly
added
HC1 (6 mL; 1 M) to give a light yellow suspension. After addition of 20 mL THF
and 60 mL
water the suspension was stirred 18 hours, filtered, and the solid was stirred
again in 6 mL
HCI (1 M) for 15 min. Afterwards the suspension was filtered and the solid was
dried in
vacuum.
Example 6
[0114] To 1 (R = PO(ONa)2) (3 g) dissolved in 35 mL water was added HCI
(9
mL; 1 M) to yield a light yellow suspension. After addition of 20 mL THF the
suspension
was stirred 30 min at room temperature and then filtered. The resulting solid
was washed
with 20 mL HC1 (0.1 M) and water and dried in vacuum.
Example 7
[0115] Solid dihydrogen phosphate is added to a volume of DMSO or N-
methylpyrrolidinone at about 50 C until no more salt dissolves. The solution
containing
suspended salt is then heated further just until the remaining solid
dissolves, and the solution
filtered while hot and allowed to cool undisturbed, when it deposits crystals
of the
dihydrogen phosphate.
Example 8
[0116] A solution of the dihydrogen phosphate is prepared in DMSO or N-
methylpyrrolidinone and filtered. To the filtered solution is added ethanol
with stirring until
the solution becomes cloudy. Stirring is then discontinued, and a layer of
ethanol carefully
placed on top of the cloudy solution, which is allowed to sit undisturbed,
when it deposits
crystals of the dihydrogen phosphate.
Example 9
[0117] A solution of the dihydrogen phosphate is prepared in DMSO or N-
methylpyrrolidinone and filtered. The filtered solution is then exposed to
vapor of ethanol,
for example by placing an open container of the solution and an open container
of ethanol
together in a sealed vessel such that the two containers share a common
headspace inside the
CA 02751392 2011-08-02
WO 2010/091131 PCT/US2010/023122
vessel. On standing the container with the solution deposits crystals of the
dihydrogen
phosphate.
Example 10
[0118] A solution of a salt of the dihydrogen phosphate, such as the
mono- or
disodium phosphate, is prepared. Such a solution can be prepared by such
methods as simply
dissolving a sample of the solid disodium phosphate in water, or by adding the
dihydrogen
phosphate to an aqueous solution of a base sufficiently strong to
substantially deprotonate the
dihydrogen phosphate. Identification of an appropriate base is a routine
matter for the
practicing chemist. Typically the resulting solution of the salt of the
dihydrogen phosphate is
then filtered, and to the filtrate is added an acid to reprotonate the salt
and induce
crystallization of the dihydrogen phosphate. In a typical example, the
dihydrogen phosphate
is added to an aqueous solution containing NaOH or Na2CO3 to yield a solution
of the
disodium phosphate, to which after filtration is added aqueous or gaseous HC1
to regenerate
the dihydrogen phosphate, which deposits as crystals.
[0119] For pharmaceutical purposes it is advantageous to use
pharmaceutically-
acceptable acids and bases in this process, such as those compiled in Handbook
of
Pharmaceutical Salts: Properties, Selection and Use. (P. Heinrich Stahl and
Camille G.
Wermuth, eds.) International Union of Pure and Applied Chemistry, Wiley-VCH
2002 and
L.D. Bighley, S.M. Berge, D.C. Monkhouse, in "Encyclopedia of Pharmaceutical
Technology'. Eds. J. Swarbrick and J.C. Boylan, Vol. 13, Marcel Dekker, Inc.,
New York,
Basel, Hong Kong 1995, pp. 453-499 discusses such salts in detail.
[0120] As those skilled in the art will appreciate, elements of the
methods above
can be combined. For example, a solution of the dihydrogen phosphate in DMSO
or N-
methylpyrrolidinone can be prepared at one temperature, a second solvent such
as ethanol
added, and the resulting solution allowed to cool. Similarly, mixtures of
solvents can be used
instead of pure solvents, as is well-known to those skilled in crystallizing
compounds.
Furthermore, other solvents and mixtures thereof can also be used.
[0121] Elemental analysis for C17H16FN606P (measured/calculated) C 43.9
(44.8); H 3.6 (3.7); N 18.1 (18.4); 0 21.2 (22.1); F 4.2 (4.2); P 6.7 (6.8).
Example 11
CA 02751392 2011-08-02
WO 2010/091131 PCT/US2010/023122
31
[0122] The particle size was measured using a Malvern Mastersizer. The
sampling instructions that were consistent with the instrument manufacturer's
instructions
were followed. The sample was prepared by suspending in 1-2 mL of deionized
water and
sonicating for 3 minutes.
[0123] An exemplary particle size distribution of crystalline material such
as
those described in Examples 1-10 above is set forth in Figure 10 and Table 3
below:
Table 3 Typical Particle Size Distribution (uncontrolled process)
Lot 02090054 D10 (urn) D50 (urn) D90 (urn)
Average 0.28 0.79 44
Example 12 Particle-Size Adjustment Experimental
[0124] A 22-L reactor was charged with 1 M HC1 (1.95 L, 2.2 equivalents)
and
ethanol (1.6 L, 4 volumes), and the solution was heated to 70 C. A separate
12-L reactor
equipped with a gas bubbler to monitor gas evolution was charged with TR-701FA
110.4 kg,
AMRI lot # DUG-AH-166(2)1, water (2.8 L, 7 vol), and ethanol (0.4 L, 1 vol).
The slurry
was stirred at ambient temperature and 5 wt % aqueous NaHCO3 was added via
peristaltic
pump over 30 minutes. No foaming was observed, however the gas evolution was
vigorous
as observed through the gas bubbler. Upon completion of the addition, the
clear yellow
solution was pH 6.6. The aqueous TR-701 solution was added via peristaltic
pump to the
ethanol/HC1 solution over 90 minutes. Upon completion of the addition, the pH
of the
reaction mixture was 1.9 and the reaction mixture was cooled to 30 C. A
sample of the
slurry was withdrawn for analysis by optical microscopy. The slurry was
filtered through a
polypropylene filter cloth and the reactor and filter cake were rinsed with
water (5 volumes)
and acetone (5 volumes). The total filtration time including the washes was 12
minutes. The
solids were dried under high vacuum at 50 C to afford 391.7 g of
reprecipitated TR-701FA
(98% yield). Analysis by 1H NMR was consistent with the assigned structure.
HPLC
analysis (Method A): 98.8% (AUC) tR = 5.2 mm. The level of residual ethanol by
1H NMR
analysis was 0.03%, the water content was 0.15% by Karl Fischer titration, and
the sodium
content was 5 ppm.
[0125] The particle size was measured using a Malvern Mastersizer laser
scattering microscopy. The sampling instructions that were consistent with the
instrument
CA 02751392 2016-06-02
32
manufacturer's instructions were followed. The sample was prepared by by
suspending in 1-2
mL of deionized water and sonicating for 3 minutes. The laser diffraction data
is set forth in
Figure 11 and in Table 4 below.
Table 4
Lot JAS-I-45 D10 (um) D50 (urn) D90 (um)
Average 0.45 14.13 38.42
[0126] In another experiment, the typical particle size distribution
using a
controlled method, such as provided in this example, is set forth Figure 12
and in Table 5
below:
Table 5: Typical Particle Size Distribution (using particle size control
process)
Lot 0209118 D10 (pm) D50 (pm) D90 (pm)
Average 3.3 27 66
Range for Application 1-5 1-40 45-80
[0127] The immediate release formulation and the intravenous
formulation
described in Examples 13-14 below were made using the crystalline free acid
wherein the
particle size was controlled.
Example 13 Immediate Release Formulation
[0128] The qualitative and quantitative formulation of immediate
release (R)-3-
(4-(2-(2-methyltetrazol-5-yl)pyridin-5-y1)-3-fluoropheny1)-5-hydroxymethyl
oxazolidin-2-one
dihydrogen phosphate 1 (R = P0(011)2) tablets ("Torezolid Phosphate Tablets"),
200 mg, is
presented in Table 6. All components used in the manufacturing are listed with
the quality
standard, function, and weight percent of each individual component. The
listing is inclusive
of all materials used during the manufacture of the drug product whether or
not they are
present in the finished product.
Table 6 Composition of Torezolid Phosphate Tablets, 200 mg
Ingredient Quality Function 200 mg Tablet
Standard Weight
(mg/unit) (w/w)
Torezolid Phosphate' In-house Active Ingredient 200 50.0
Microcrystalline Cellulose NF Diluent 78.0 19.5
(AvicelTM PH-101)
Mannito12 NF Diluent 78.0 19.5
(Mannogene EZ Spray
Dried)
CA 02751392 2011-08-02
WO 2010/091131 PCT/US2010/023122
33
Povidone NF Binder 16.0 4.0
(Plasdone K-29/32)
Crospovidone NF. Disintegrant 24.0 6.0
(Kollidon CL)
Purified Water2 USP Granulating
Medium
Magnesium Stearate NF Lubricant 4.0 1.0
(HyQual(D) Vegetable
Source
Total Core Tablet Weight' 400.0 100.0
Opadry II Yellow Colored Film 14.0 3.4
Coat
Purified Water3 USP Film Coating
Medium
Total Weight 414.0 103.4
Abbreviations: NF = National Formulary; USP = United States Pharmacopeia
1The actual amount of torezolid phosphate is adjusted based on potency of the
drug substance lot
used.
2The actual amount of mannitol is adjusted based on amount of the drug
substance used.
'Removed during processing.
Example 14 Powder and Formulation for Injection
[0129] (R)-3-(4-(2-(2-methyltetrazol-5-yepyridin-5-y1)-3-fluoropheny1)-
5-
hydroxymethyl oxazolidin-2-one dihydrogen phosphate 1 (R = PO(OH)2) ")
("Torezolid
Phosphate for Injection" or "TR-701 FA for Injection"), 200 mg/vial, was
prepared in a
formulation as a sterile lyophilized powder for injection. TR-701 FA for
Injection is
formulated in situ as the disodium salt using sodium hydroxide to take
advantage of its
superior aqueous solubility (> 130 mg/mL).
[0130] TR-701 FA for Injection, 200 mg/vial, is to be reconstituted
with 4 mL of
Sterile Water for Injection (WFI), USP to yield a 50 mg/mL solution. The
appropriate
clinical dose volume is to be withdrawn from the vial and added to an
intravenous (IV)
non-di(2-ethylhexyl)phthalate (DEHP) bag containing either 0.9% Sodium
Chloride
Injection, USP (saline) or 5% Dextrose Injection, USP (dextrose). The
resulting IV solution
is to be infused using a non-DEHP solution set with a 0.22 um in-line filter.
[0131] The unit composition of TR-701 FA Compounding Solution for
Lyophilization is presented in Table 7 and the unit composition of TR-701 FA
for Injection,
200 mg/vial is presented in Table 8.
CA 02751392 2011-08-02
WO 2010/091131 PCT/US2010/023122
34
Table 7. Unit Composition of TR-701 FA Compounding Solution for
Lyophilization
Component Function Theoretical Quantity
TR-701 FA Drug Substance 100 mg/mL
Mannitol, Powder, USP Bulking Agent 50 mg/mL
Sodium Hydroxide, USP In-situ salt formation, qs for pH adjustment to
7.75
pH adjustment
Hydrochloric Acid, NF pH adjustment qs for pH adjustment to
7.75
Water for Injection, USP/EP Manufacturing solvent qs to 1.0 mL
Table 8. Unit Composition of TR-701 FA for Injection, 200 mg/vial
Component Function Theoretical Quantity
TR-701 FA Drug Substance 210 mg a
Mannitol, Powder, USP Bulking Agent 105 mg
Sodium Hydroxide, USP In-situ salt formation, qs for pH adjustment
to 7.75
pH adjustment
Hydrochloric Acid, NF pH adjustment qs for pH adjustment to
7.75
Water for Injection, USP/EP b Manufacturing solvent qs to 2.1 mL
a A volume equivalent to 210 mg of TR-701 FA is filled into each vial to so
that reconstitution of the vial with
4.0 n-IL of Water for Injection (a final volume of 4.2 mL is obtained due to
volume displacement of the
dissolved solids) results in a 50 mg/mL solution of TR-701 FA that will allow
withdrawal of the label contents.
Water for Injection is essentially removed during lyophilization.
[0132] The typical manufacturing batch formula for TR-701 FA for
Injection,
200 mg/vial is presented in Table 9.
CA 02751392 2011-08-02
WO 2010/091131 PCT/US2010/023122
Table 9. Typical Batch Formula for TR-701 FA for Injection,
200 mg/vial
Material Theoretical Quantity
TR-701 FA 400 g
Mannitol, Powder, USP 200 g
Sodium Hydroxide, NF qs for pH adjustment to pH 7.75
Hydrochloric Acid, NF qs for pH adjustment to pH 7.75
Water for Injection, USP/EP qs 4276 g
Total 4000 mL
(-1900 vials)
a The actual quantity of TR-701 FA drug substance to be weighed is adjusted
based on potency.
[0133] The manufacturing process is summarized below and schematics of the
process for preparing a compounding solution and for sterile filtering,
filling, and
lyophilization are presented Figures 8 and 9.
Compounding Solution
[0134] The compounding solution is prepared in the following sequence:
[0135] Add approximately 50% of the total amount of Water for Injection to
a
tared compounding vessel.
[0136] Add TR-701 FA and slowly neutralize with a solution of sodium
hydroxide while mixing.
[0137] Add and dissolve mannitol with mixing.
[0138] Measure the pH of the resulting solution. If the solution is outside
the
target range of pH 7.70 to 7.80, adjust the pH using either 1N sodium
hydroxide or 1N
hydrochloric acid.
[0139] Add Water for Injection to final volume and mix.
Sterile Filtering/Filling/Lyophilization
[0140] Filter the bulk solution through 2 integrity-tested 0.22 wict
filters in series
and collect the solution in a sterile receiving vessel.
[0141] Add target fill weight of solution into 20 mL vials under aseptic
conditions.
[0142] Partially insert lyophilization stoppers into the vials.
CA 02751392 2011-08-02
WO 2010/091131 PCT/US2010/023122
36
[0143] Lyophilize the vials according to an appropriate cycle.
[0144] At the end of the lyophilization cycle, backfill the chamber
with nitrogen
and stopper vials under partial vacuum.
[0145] Seal vials with flip off caps.
Example 15
[0146] A sample of crystalline free acid which was made according to
a method
of making the free acid disclosed in US Patent Application No. 12/577,089,
which is
assigned to the same assignee as in the present application, and by using the
crystallization
methods described herein, was crystallized according to methods described
herein was
characterized using HPLC and contains various levels of impurities such as
those described
in Table 10 below:
Table 10
Identified Individual Impurities HPLC (TM.1911)
NMT 0.5% Rx600013
NMT 0.5%
Rx600024
NMT 0.5%
Rx600014
NMT 0.2%
Rx600023
NMT 0.5%
Rx600025
NMT 0.5%
Rx600020
NMT 2.0%
Rx600001
NMT 1.5% Rx600022
[0147] In addition, a substantially pure sample of crystalline free
acid which was
made according to processes that were not disclosed in US Patent Publication
No.
20070155798 and was crystallized according to methods described herein
(hereinafter
"ours"), was compared to a sample of material made by Dong-A Pharm. Co.
(hereinafter "the
Dong-A material"), which was given to Trius Therapeutics Inc. in approximately
2007. The
potency of the Dong-A material was approximately 84% by weight of the sample
in
comparison to a substantially pure reference sample; however, the purity of
the crystalline
free acid was 94.1% by weight of the material identified by HPLC as indicated
below.
Therefore, approximately 10% of the impurities in the Dong-A material was not
identified by
HPLC. The purity profile comparison is set forth in Table 11 below:
Table 11
Area Percent
Impurity
Name RRT Dong A ours ID
600011 0.54 0.12 ND DA-1dimer diphos
CA 02751392 2011-08-02
WO 2010/091131
PCT/US2010/023122
37
600013** 0.56 ND 0.08 Des-Me
UNK 0.65 0.07 ND
UNK 0.77 0.34 ND
UNK 0.86-0.88 0.07 0.03
600024 0.91 0.22 0.12 Pyrophosphate
UNK 0.94 0.07 ND
UNK 0.95 0.05 ND
600012 1 94.1 97.1 API
600023 1.08 0.14 ND N-1 Phosphorylated
UNK 1.1 0.06 ND
UNK 1.14 0.05 ND
600025** 1.15 ND 0.27 Over-Alk'd pair
UNK 1.2 0.07 ND
UNK 1.21 0.05 ND
UNK 1.31 0.05 ND
UNK 1.39 0.26 0.04
UNK 1.47 0.35 ND
600020 1.5-1.51 0.2 0.08 Dimer
UNK 1.56 -0.05 ND
1.67-
600001* 1.12 0.63
1.688 TR-700
600022 1.72-1.73 0.28 1.2 Bis
0A-700 mixed di
600042** 1.79 0.12
ND ester
0A-700 mixed di
600043** 1.8 0.15
ND ester
600026* 2.27-2.28 2.17 0.06 Chloro
UNK 2.34 0.05 ND
UNK 2.4 0.06 ND
* equals ours Dong A
** equals impurity present in ours but not Dong A
CA 02751392 2011-08-02
WO 2010/091131 PCT/US2010/023122
38
Organic Impurities in TR-701 FA Drug Substance
Impurity
Structure and Chemical Name
'Name'
Rx600013 0
'Des-methyl TR-
701' NN
HN,
N N \---"ss...õ..ON pip
HO/ NOH
dihydrogen ((5R)-3-{3-fluoro-4-16-(2H-1,2,3,4-tetrazol-5-
y1)-3-pyridinyllphenyl }-2-oxo-1,3-oxazolan-5-yl)methyl
phosphate
Rx600024 0
'Pyrophosphate'
----N ¨
/ \ /
H N
N
HO I
HO
trihydrogen ((5R)-3-13-fluoro-4-16-(1-methy1-1H-1,2,3,4-
tetraazol-5-y1)-3-pyridinyllphenyll -2-oxo-1,3-oxazolan-5-
yl)methyl pyrophosphate
Rx600014
N-1\1 ¨ = OH
'Ring opened' 0
N N N
/Põ
HO X
dihydrogen 3-f 3-fluoro-4- [6-(2-methyl-2H- 1,2,3 ,4-tetraazol-5-
y1)-3-pyridinyl] aniline I -2-hydroxypropyl phosphate
Impurity
Structure and Chemical Name
'Name'
CA 02751392 2011-08-02
WO 2010/091131
PCT/US2010/023122
39
Rx600023 0
`Me-isomer'
)\---0
N''-14 _
N N \ .
II \ / N
, N \----N.-----0 0
\
\ F /PNOH
HO
dihydrogen ((5R)-3-13-fluoro-4-16-(1-methy1-1H-1,2,3,4-
tetraazol-5-y1)-3-pyridinyllphenyll-2-oxo-1,3-oxazolan-5-
y1)methyl phosphate
Rx600025 0
'Oyeralkylated NN-
P
z \
phosphorylated / \ ¨/ . 0 O
NN____H\ -----1..)
.0OH
impurity'
F N
0
N-r-----N - 41 -OH
1
N/ \ / N \ ON__O_______cõ, H 0:----p-
...0H
c)
F
(R)-14(3-(3-fluoro-4-(6-(2-methy1-2H-tetrazol-5-
yepyridin-3-yl)pheny1)-2-oxooxazolidin-5-yl)methoxy)-3-
hydroxypropan-2-y1 dihydrogen phosphate;
(R)-34(3-(3-fluoro-4-(6-(2-methy1-2H-tetrazol-5-
yepyridin-3-yl)pheny1)-2-oxooxazolidin-5-yl)methoxy)-2-
hydroxypropyl dihydrogen phosphate
Impurity
Structure and Chemical Name
'Name'
CA 02751392 2011-08-02
WO 2010/091131
PCT/US2010/023122
Rx600020 0
`Dimer impurity' NN¨
N N cv0
HO'?
r\c)
F \\
0
----N
\
dihydrogen bis-0-0' - R5R)-3- 3-fluoro-4-116-(2-methy1-
2H-1,2,3,4-tetrazol-5-y1)-3-pyridinyllphenyl } -2-ox o-1 ,3-
oxazolidin-5-yll methyl pyrophosphate
Rx600026 0
"Chloro"
/
µrsi /
(R)-5-(chloromethyl)-3-(3-fluoro-4-(6-(2-methy1-2H-
tetrazol-5-yl)pyridin-3-yephenyeoxazolidin-2-one
Rx600001 0
TR-700
I /N
OH
5R)-3- { 3-Fluoro-446-(2-methy1-2H- 1,2,3 ,4-tetrazol-5-y1)-
pyridin-3 -y1{ -phenyl I -5-hydroxymethyl- 1,3-ox azolidin-2-one
Impurity
Structure and Chemical Name
'Name'
CA 02751392 2011-08-02
WO 2010/091131
PCT/US2010/023122
41
Rx600022 0
'Ws phosphate'
N%--N
/
N N
/N
HO (1
N
N
\N _N
/
hydrogen bis-0-0' -[(5R)-3- f 3-fluoro-446-(2-methy1-2H-
1,2,3,4-
tetrazol-5-y1)-3-pyridinyl]pheny11-2-oxo-1,3-oxazolidin-5-yltmethyl
phosphate
Rx600042NN 0
¨
r!, \ / W
OH
N
OH
F
-N
1,7 \N
3-f t(5R)-3- f 3-fluoro-446-(2-methy1-2H-tetrazol-5-yepyridin-3-
yl]phenyl -2-oxo-1,3-oxazolidin-5-yl]methoxy -2-hydroxypropyl
R5R)-3- f 3-fluoro-446-(2-methy1-2H-tetrazol-5-yepyridin-3-
yl]phenyl -2-oxo-1,3-oxazolidin-5-yl]methyl hydrogen phosphate
CA 02751392 2011-08-02
WO 2010/091131
PCT/US2010/023122
42
Impurity
Structure and Chemical Name
'Name'
Rx600043
NN
--N
N
0
HO
HO /0
7%0
0
*N,y0
0
/ N
N-N
2-f R5R)-3- f 3-fluoro-446-(2-methy1-2H-tetrazol-5 -yepyridin-3-
yl]phenyl 1-2-oxo-1,3-oxazolidin-5-yltmethoxy -1-hydroxyethyl
R5R)-3- f 3-fluoro-4- [6-(2-methy1-2H-tetrazol-5-yepyridin-3-
yl]phenyll-2-oxo-1,3-oxazolidin-5-yltmethyl hydrogen phosphate