Note: Descriptions are shown in the official language in which they were submitted.
CA 02753381 2011-09-13
WO 99/57134 PCT/US99/09637
PROTEIN PURIFICATION BY ION EXCHANGE CHROMATOGRAPHY
BACKGROUND OF THE INVENTION
Field of the Invention
This invention relates generally to protein purification. In particular. the
invention relates to a method
for purifying a polypeptide (e.g. an antibody) from a composition comprising
the polypeptide and at least one
contaminant using the method of ion exchange chromatography.
Description of Related Art
The large-scale. economic purification of proteins is increasingly an
important problem for the
biotechnology industry. Generally, proteins are produced by cell culture,
using eithermammalian or bacterial cell
lines engineered to produce the protein of interest by insertion of a
recombinant plasmid containing the gene for
that protein. Since the cell lines used are living organisms, they must be fed
with a complex growth medium,
containing sugars. amino acids, and growth factors, usually supplied from
preparations of animal serum.
Separation of the desired protein from the mixture of compounds fed to the
cells and from the by-products of the
cells themselves to a purity sufficient for use as a human therapeutic poses a
formidable challenge.
Procedures for purification of proteins from cell debris initially depend on
the site of expression of the
protein. Some proteins can be caused to be secreted directly from the cell
into the surrounding growth media.
others are made intracellularly. For the latter proteins. the first step of a
purification process involves lysis of the
cell, which can be done by a variety of methods, including mechanical shear,
osmotic shock, or enzymatic
treatment,:. Such disruption releases the entire contents of the cell into the
homogenate, and in addition produces
subcellular fragments that are difficult to remove due to their small size.
These are generally removed by
differential centrifugation or by filtration. The same problem arises,
although on a smaller scale, with directly
secreted proteins due to the natural death of cells and release of
intracellular host cell proteins in the course of
the protein production run.
Once a clarified solution containing the protein of interest has been
obtained. its separation from the
other proteins produced by the cell is usually attempted using a combination
of different chromatography
techniques. These techniques separate mixtures of proteins on the basis of
their charge, degree of hydrophobicity.
or size. Several different chromatography resins are available for each of
these techniques, allowing accurate
tailoring of the purification scheme to the particular protein involved. The
essence of each of these separation
methods is that proteins can be caused either to move at different rates down
a long column, achieving a physical
separation that increases as they pass further down the column. or to adhere
selectively to the separation medium,
being then differentially eluted by different solvents. In some cases, the
desired protein is separated from
impurities when the impurities specifically adhere to the column. and the
protein of interest does not, that is, the
protein of interest is present in the "flow-through".
Ion exchange chromatography is a chromatographic technique that is commonly
used for the purification
of proteins. In ion exchange chromatography, charged patches on the surface of
the solute are attracted by
opposite charges attached to a chromatography matrix, provided the ionic
strength of the surrounding buffer is
low. Elution is generally achieved by increasing the ionic strength (i.e.
conductivity) of the buffer to compete with
the solute for the charged sites of the ion exchange matrix. Changing the pH
and thereby altering the charge of
1
CA 02753381 2011-09-13
WO 99/57134 PCTIUS99/09637
the solute is another way to achieve elution of the solute. The change in
conductivity or pH may be gradual
(gradient elution) or stepwise (step elution). In the past, these changes have
been progressive; i.e., the pH or
conductivity is increased or decreased in a single direction.
SUMMARY OF THE INVENTION
The present invention provides an ion exchange chromatographic method wherein
a polypeptide of
interest is bound to the ion exchange material at an initial conductivity or
pH and then the ion exchange material
is washed with an intermediate buffer at a different conductivity or pH, or
both. At a specific point following this
intermediate wash, and contrary to ion exchange chromatography standard
practice, the ion exchange material
is washed with a wash buffer where the change in conductivity or pH. or both,
from the intermediate buffer to
the wash buffer is in an opposite direction to the change in conductivity or
pH, or both, achieved in the previous
steps. Only after washing with the wash buffer, is the ion exchange material
prepared for the polypeptide
molecule of interest to be eluted by the application of the elution buffer
having a conductivity or pH, or both,
which differ from the conductivity or pH, or both, of the buffers used in
previous steps.
This novel approach to ion exchange chromatography is particularly useful in
situations where a product
molecule must be separated from a very closely related contaminant molecule at
full manufacturing scale, where
both purity and high recovery of polypeptide product are desired.
Accordingly, the invention provides a method for purifying a polypeptide from
a composition
comprising the polypeptide and a contaminant, which method comprises the
followi=g steps performed
sequentially:
(a) binding the polypeptide to an ion exchange material using a loading
buffer, wherein the loading
buffer is at a first conductivity and pH;
(b) washing the ion exchange material with an intermediate buffer at a second
conductivity and/or pH
so as to elute the contaminant from the ion exchange material-,
(c) washing the ion exchange material with a wash buffer which is at a third
conductivity and/or pH,
wherein the change in conductivity and/or pH from the intermediate buffer to
the wash buffer is in an opposite
direction to the change in conductivity and/or pH from the loading buffer to
the intermediate buffer; and
(d) washing the ion exchange material with an elution buffer at a fourth
conductivity and/or pH so as
to elute the polypeptide from the ion exchange material. The first
conductivity and/or pH may be the same as
the third conductivity and/or pH.
Where the ion exchange material comprises a cation exchange resin, the
conductivity and/or pH of the
intermediate buffer is/are preferably greater than the conductivity and/or pH
of the loading buffer; the
conductivity and/or pH of the wash buffer is/are preferably less than the
conductivity and/or pH of the
intermediate buffer: and the conductivity and/or pH of the elution buffer
is/are preferably greater than the
conductivity and/or pH of the intermediate buffer. Preferably, the
conductivity and/or pH of the wash buffer is/are
about the same as the conductivity and/or pH of the loading buffer.
Preferably elution of the contaminant and of the polypeptide is achieved by
modifying the conductivity
of the intermediate buffer and of the elution buffer, respectively, while
keeping the pH of these buffers
approximately the same.
The invention also provides a method for purifying a polypeptide from a
composition comprising the
2
CA 02753381 2011-09-13
WO 99/57134 PCT/US99/09637
polypeptide and a contaminant, which method comprises the following steps
performed sequentially:
(a) binding the polypeptide to a cation exchange material using a loading
buffer, wherein the loading
buffer is at a first conductivity and pH;
(b) washing the cation exchange material with an intermediate buffer at a
second conductivity and/or
pH which is greater than that of the loading buffer so as to elute the
contaminant from the ion exchange material,
(c) washing the cation exchange material with a wash buffer which is at a
third conductivity and/or pH
which is less than that of the intermediate buffer, and
(d) washing the cation exchange material with an elution buffer at a fourth
conductivity and/or pH which
is greater than that of the intermediate buffer so as to elute the polypeptide
from the ion exchange material.
In addition, the invention provides a method for purifying an antibody from a
composition comprising
the antibody and a contaminant, which method comprises loading the composition
onto a cation exchange resin,
wherein the amount of antibody loaded onto the cation exchange resin is from
about 20mg to about 35mg of the
antibody per mL of cation exchange resin and, optionally, further comprising
eluting the antibody from the cation
exchange resin. The method preferably further comprises an intermediate wash
step for eluting one or more
contaminants from the ion exchange resin. This intermediate wash step usually
precedes the step of eluting the
antibody.
The invention further provides a composition comprising a mixture of anti-HER2
antibody and one or
more acidic variants thereof, wherein the amount of the acidic variant(s) in
the composition is less than about 25%
and preferably less than about 20%, e.g. in the range from about 1% to about
18%. Optionally, the composition
further comprises a pharmaceutically acceptable carrier.
Brief Description Of The Drawings
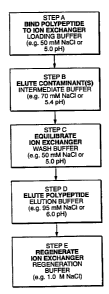
Figure 1 is a flow diagram showing how one could perform cation exchange
chromatography by altering
conductivity (e.g. to the NaCl concentrations of Example I below) or by
altering pH (e.g. to the pH values as
shown in the flow diagram).
Figure 2 is a flow diagram showing how one could perform anion exchange
chromatography by altering
conductivity (e.g. to the NaCl concentrations as depicted in the figure) or by
altering pH (e.g. to the pH values
as shown).
Figure 3 is an absorbance trace from a cation exchange chromatography run of
Example I at full
manufacturing scale. Points at which the column is washed with the different
buffers described herein are marked
with arrows.
Figure 4 depicts recombinant humanized anti-HER2 monoclonal antibody (rhuMAb
HER2) recovered
in each chromatography fraction (calculated as the percentage of the sum total
of all fractions of the relevant
chromatography). Flow through, wash steps, and prepool fractions are all
effluent samples collected from the
onset of load to the initiation of pooling. The pool fraction is the five
column volume effluent sample of elution
starting at the leading shoulder's inflection point. The regeneration fraction
contains effluent captured from the
end of pooling to the end of regeneration.
Figure 5 shows the quality of rhuMAb HER2 in each cation exchange
chromatography pool sample as
evaluated by carboxy sulfon cation exchange high pressure liquid
chromatography(CSx HPIEX). Peaks a, b, and
I are deamidated forms of rhuMAb HER2. Peak 3 is nondeamidated rhuMAb HER2.
Peak 4 is a combination
3
CA 02753381 2011-09-13
WO 99/57134 PCT/US99/09637
of C-terminal Lysine containing and iso-aspartate variants of rhuMAb HER2.
Figure 6 shows the absorbance (280 nm) profiles of the 0.025 M MES / 0.070 M
NaCl, pH 5.6 wash for
each chromatography. The mass of rhuMAb HER2 applied to the cation exchange
resin effects the peak's
absorbance level at the apex as well as the amount of buffer required to reach
the apex. Due to minor peaks which
occur (as best seen in the 30 mg/mL load) in this wash, the apex is defined as
absorbance levels of at least 0.5
absorbance units (AU).
Figures 7A and 7B show the amino acid sequences of humMAb4D5-8 light chain
(SEQ ID NO: I) and
humMAb4D5-8 heavy chain (SEQ ID NO:2), respectively.
Detailed Description Of The Preferred Embodiments
Definitions:
The "composition" to be purified herein comprises the polypeptide of interest
and one or more
contaminants. The composition maybe "partially purified" (i.e. having been
subjected to one or more purification
steps, such as Protein A Chromatography as in Example I below) or may be
obtained directly from a host cell
or organism producing the polypeptide (e.g. the composition may comprise
harvested cell culture fluid).
As used herein, "polypeptide" refers generally to peptides and proteins having
more than about ten
amino acids. Preferably, the polypeptide is a mammalian protein, examples of
which include renin; a growth
hormone, including human growth hormone and bovine growth hormone; growth
hormone releasing factor,
parathyroid hormone; thyroid stimulating hormone; lipoproteins; alpha- I -
antitrypsin; insulin A-chain; insulin B-
chain; proinsulin; follicle stimulating hormone; calcitonin; luteinizing
hormone; glucagon; clotting factors such
as factor VIIIC, factor IX. tissue factor, and von Willebrands factor; anti-
clotting factors such as Protein C; atrial
natriuretic factor; lung surfactant; a plasminogen activator, such as
urokinase or human urine or tissue-type
plasminogen activator (t-PA); bombesin; thrombin; hemopoietic growth factor;
tumor necrosis factor-alpha and
-beta; enkephalinase. RANTES (regulated on activation normally T-cell
expressed and secreted); human
macrophage inflammatory protein (MIP-I-alpha); a serum albumin such as human
serum albumin; Muellerian-
inhibiting substance. relaxin A-chain; relaxin B-chain; prorelaxin; mouse
gonadotropin-associated peptide: a
microbial protein, such as beta-lactamase; DNase; lgE; a cytotoxic T-
lymphocyte associated antigen (CTLA),
such as CTLA-4; inhibin; activin: vascular endothelial growth factor (VEGF);
receptors for hormones or growth
factors; Protein A or D; rheumatoid factors; a neurotrophic factor such as
bone-derived neurotrophic factor
(BDNF), neurotrophin-3, -4, -5, or -6 (NT-3, NT-4, NT-5, or NT-6), or a nerve
growth factor such as NGF-Q;
platelet-derived growth factor (PDGF); fibroblast growth factor such as aFGF
and bFGF, epidermal growth factor
(EGF); transforming growth factor (TGF) such as TGF-alpha and TGF-beta,
including TGF-R 1, TGF- 32, TGF-
03, TGF-$ 4, or TGF-(35; insulin-like growth factor-I and -II (IGF-I and IGF-
II); des(1-3)-IGF-I (brain IGF-I),
insulin-like growth factor binding proteins (IGFBPs); CD proteins such as CD3,
CD4, CD8, CD19 and CD20;
erythropoietin; osteoinductive factors; immunotoxins; a bone morphogenetic
protein (BMP); an interferon such
as interferon-alpha, -beta, and -gamma; colony stimulating factors (CSFs),
e.g., M-CSF, GM-CSF, and G-CSF;
interleukins (ILs), e.g., IL-I to IL- 10; superoxide dismutase; T-cell
receptors; surface membrane proteins; decay
accelerating factor; viral antigen such as, for example, a portion of the AIDS
envelope; transport proteins; homing
receptors; addressins; regulatory proteins; integrins such as CDI I a, CDI I
b, CDI Ic, CD 18, an ICAM, VLA-4
and VCAM; a tumor associated antigen such as HER2, HER3 or HER4 receptor; and
fragments and/or variants
4
CA 02753381 2011-09-13
WO 99/57134 PCT/US99109637
of any of the above-listed polypeptides. Most preferred is a full length
antibody that binds human HER2.
A "contaminant" is a material that is different from the desired polypeptide
product. The contaminant
may be a variant of the desired polypeptide (e.g. a deamidated variant or an
amino-aspartate variant of the desired
polypeptide) or another polypeptide, nucleic acid, endotoxin etc.
A "variant" or "amino acid sequence variant" of a starting polypeptide is a
polypeptide that comprises
an amino acid sequence different from that of the starting polypeptide.
Generally, a variant will possess at least
80% sequence identity, preferably at least 90% sequence identity, more
preferably at least 95% sequence identity,
and most preferably at least 98% sequence identity with the native
polypeptide. Percentage sequence identity is
determined, for example, by the Fitch et at., Proc. Nutt. Acad. Sci. USA
80:1382-1386 (1983), version of the
algorithm described by Needleman et at., J. Mol. Biol. 48:443-453 (1970),
after aligning the sequences to provide
for maximum homology. Amino acid sequence variants of a polypeptide may be
prepared by introducing
appropriate nucleotide changes into DNA encoding the polypeptide, or by
peptide synthesis. Such variants
include, for example, deletions from, and/or insertions into and/or
substitutions of, residues within the amino acid
sequence of the polypeptide of interest. Any combination of deletion,
insertion, and substitution is made to arrive
at the final construct, provided that the final construct possesses the
desired characteristics. The amino acid
changes also may alter post-translational processes of the polypeptide, such
as changing the number or position
of glycosylation sites. Methods for generating amino acid sequence variants of
polypeptides are described in US
Pat 5,534,615
An "acidic variant" is a variant of a polypeptide of interest which is more
acidic (e.g. as determined by
cation exchange chromatography) than the polypeptide of interest. An example
of an acidic variant is a
deamidated variant.
A "deamidated" variant of a polypeptide molecule is a polypeptide wherein one
or more asparagine
residue(s) of the original polypeptide have been converted to aspartate, i.e.
the neutral amide side chain has been
converted to a residue with an overall acidic character. Deamidated humMAb4D5
antibody from the Example
below has Asn30 in CDRI of either or both of the VL regions thereof converted
to aspartate. The term
"deamidated human DNase" as used herein means human DNase that is deamidated
at the asparagine residue that
occurs at position 74 in the amino acid sequence of native mature human DNase
(US Patent 5,279,823
The term "mixture" as used herein in reference to a composition comprising an
anti-HER2 antibody,
means the presence of both the desired anti-HER2 antibody and one or more
acidic variants thereof. The acidic
variants may comprise predominantly deamidated anti-HER2 antibody, with minor
amounts of other acidic
variant(s). It has been found, for example, that in preparationsof anti-HER2
antibody obtained from recombinant
expression, as much as about 25% of the anti-HER2 antibody is deamidated.
In preferred embodiments of the invention, the polypeptide is a recombinant
polypeptide. A
"recombinant polypeptide" is one which has been produced in a host cell which
has been transformed or
transfected with nucleic acid encoding the polypeptide, or produces the
polypeptide as a result of homologous
recombination. "Transformation" and "transfection" are used interchangeably to
refer to the process of
introducing nucleic acid into a cell. Following transformation or
transfection, the nucleic acid may integrate into
5
CA 02753381 2011-09-13
WO 99/57134 PCT/US99/09637
the host cell genome, or may exist as an extrachromosomalelement, The "host
cell" includes a cell in in vitro cell
culture as well a cell within a host animal. Methods for recombinant
production of polypeptides are described
in US Pat 5,534,615
The term "antibody" is used in the broadest sense and specifically covers
monoclonal antibodies
(including full length monoclonal antibodies), polyclonal antibodies,
multispecific antibodies (e.g., bispecific
antibodies), and antibody fragments so long as they exhibit the desired
biological activity.
The antibody herein is directed against an "antigen" of interest. Preferably,
the antigen is a biologically
important polypeptide and administration of the antibody to a mammal suffering
from a disease or disorder can
result in a therapeutic benefit in that mammal. However, antibodies directed
against nonpolypeptide antigens
(such as tumor-associatedglycolipid antigens; see US Patent 5,091,178) are
also contemplated. Where the antigen
is a polypeptide, it may be a transmembrane molecule (e.g. receptor) or ligand
such as a growth factor. Exemplary
antigens include those polypeptides discussed above. Preferred molecular
targets for antibodies encompassed by
the present invention include CD polypeptides such as CD3, CD4, CD8, CD 19,
CD20 and CD34; members of
the HER receptor family such as the EGF receptor, HER2, HER3 or HER4 receptor;
cell adhesion molecules such
as LFA- 1, Mac1, p150,95, VLA-4, ICAM- 1, VCA M and av/b3 integrin including
either a or b subunits thereof
(e.g. anti-CD] 1 a, anti-CD18 or anti-CD 1 1 b antibodies); growth factors
such as VEGF; IgE; blood group antigens;
flk2/flt3 receptor; obesity (OB) receptor; nip! receptor; CTLA-4; polypeptide
C etc. Soluble antigens or fragments
thereof, optionally conjugated to other molecules, can be used as immunogens
for generating antibodies. For
transmembrane molecules, such as receptors, fragments of these (e.g. the
extracellular domain of a receptor) can
be used as the immunogen. Alternatively, cells expressing the transmembrane
molecule can be used as the
immunogen. Such cells can be derived from a natural source (e.g. cancer cell
lines) or may be cells which have
been transformed by recombinant techniques to express the transmembrane
molecule.
The term "monoclonal antibody" as used herein refers to an antibody obtained
from a population of
substantially homogeneous antibodies, i.e., the individual antibodies
comprising the population are identical
except for possible naturally occurring mutations that may be present in minor
amounts. Monoclonal antibodies
are highly specific. being directed against a single antigenic site.
Furthermore, in contrast to conventional
(polyclonal) antibody preparations which typically include different
antibodies directed against different
determinants (epitopes), each monoclonal antibody is directed against a single
determinant on the antigen. The
modifier "monoclonal" indicates the character of the antibody as being
obtained from a substantially
homogeneous population of antibodies, and is not to be construed as requiring
production of the antibody by any
particular method. For example, the monoclonal antibodies to be used in
accordance with the present invention
may be made by the hybridoma method first described by Kohler et al., Nature
256:495 (1975), or may be made
by recombinant DNA methods (see, e.g., U.S. Patent No. 4,816,567). In a
further embodiment, "monoclonal
antibodies" can be isolated from antibody phage libraries generated using the
techniques described in McCafferty
et al., Nature, 348:552-554 (1990). Clackson et al., Nature, 352:624-628
(1991) and Marks et a!., J, Mot. Biol.,
222:581-597 (1991) describe the isolation of murine and human antibodies,
respectively, using phage libraries.
Subsequent publications describe the production of high affinity (nM range)
human antibodies by chain shuffling
(Marks et al., Bio/Technology, 10:779-783 (1992)), as well as combinatorial
infection and in vivo recombination
as a strategy for constructing very large phage libraries (Waterhouse et al.,
Nuc. Acids. Res., 21:2265-2266
6
CA 02753381 2011-09-13
WO 99/57134 PCTIUS99/09637
(1993)). Thus, these techniques are viable alternatives to traditional
monoclonal antibody hybridoma techniques
for isolation of monoclonal antibodies. Alternatively, it is now possible to
produce transgenic animals (e.g., mice)
that are capable, upon immunization, of producing a full repertoire of human
antibodies in the absence of
endogenous immunoglobulin production. For example, it has been described that
the homozygous deletion of
the antibody heavy-chain joining region (JH) gene in chimeric and germ-line
mutant mice results in complete
inhibition of endogenous antibody production. Transfer of the human germ-line
immunoglobulin gene array in
such germ-line mutant mice will result in the production of human antibodies
upon antigen challenge. See, e.g.,
Jakobovits et al., Proc. Natl. Acad. Sci. USA, 90:2551 (1993); Jakobovits et
a!., Nature, 362:255-258 (1993);
Bruggermann et al.. Year in bnmuno., 7:33 (1993); and Duchosal et al. Nature
355:258 (1992).
The monoclonal antibodies herein specifically include "chimeric" antibodies
(immunoglobulins) in
which a portion of the heavy and/or light chain is identical with or
homologous to corresponding sequences in
antibodies derived from a particular species or belonging to a particular
antibody class or subclass, while the
remainder of the chain(s) is identical with or homologous to corresponding
sequences in antibodies derived from
another species or belonging to another antibody class or subclass, as well as
fragments of such antibodies, so
long as they exhibit the desired biological activity (U.S. Patent No.
4,816,567; and Morrison et a!.. Proc. Natl.
Acad Sci. USA 81:6851-6855 (1984)).
The term "hypervariable region" when used herein refers to the amino acid
residues of an antibody
which are responsible for antigen-binding. The hypervariable region comprises
amino acid residues from a
"complementaritydetermining region" or "CDR" (i.e. residues 24-34 (L I ), 50-
56 (L2) and 89-97 (L3) in the light
chain variable domain and 31-35 (H1), 50-65 (H2) and 95-102 (H3) in the heavy
chain variable domain; Kabat
et al.. Sequences of Polypeptides of Immunological Interest, 5th Ed. Public
Health Service. National Institutes
of Health, Bethesda. MD. (1991)) and/or those residues from a "hypervariable
loop" (i.e. residues 26-32 (L1),
50-52 (L2) and 91-96 (L3) in the light chain variable domain and 26-32 (H 1),
53-55 (H2) and 96-101(H3) in the
heavy chain variable domain; Chothia and Lesk J. Mol. Biol. 196:901-917
(1987)). "Framework" or "FR" residues
are those variable domain residues other than the hypervariable region
residues as herein defined. The CDR and
FR residues of the rhuMAb HER2 antibody of the example below (humAb4D5-8) are
identified in Carter et al.,
Proc. Nail. Acad Sci. USA, 89:4285 (1992).
"Humanized" forms of non-human (e.g., murine) antibodies are chimeric
antibodies that contain minimal
sequence derived from non-human immunoglobulin. For the most part, humanized
antibodies are human
immunoglobulins(recipient antibody) in which residues from a hypervariable
region of the recipient are replaced
by residues from a hypervariable region of a non-human species (donor
antibody) such as mouse, rat, rabbit or
nonhuman primate having the desired specificity, affinity, and capacity. in
some instances, Fv framework region
(FR) residues of the human immunoglobulin are replaced by corresponding non-
human residues. Furthermore,
humanized antibodies may comprise residues which are not found in the
recipient antibody or in the donor
antibody. These modifications are made to further refine antibody performance.
In general, the humanized
antibody will comprise substantially all of at least one, and typically two,
variable domains, in which all or
substantially all of the hypervariable loops correspond to those of a non-
human immunoglobulin and all or
substantially all of the FR regions are those of a human immunoglobulin
sequence. The humanized antibody
7
CA 02753381 2011-09-13
WO 99/57134 PCT/1JS99/09637
optionally also will comprise at least a portion of an immunogiobulin constant
region (Fc). typically that of a
human immunoglobulin.
The choice of human variable domains, both light and heavy, to be used in
making the humanized
antibodies is very important to reduce antigenicity. According to the so-
called "best-fit" method, the sequence
of the variable domain of a rodent antibody is screened against the entire
library of known human variable-
domain sequences. The human sequence which is closest to that of the rodent is
then accepted as the human
framework (FR) for the humanized antibody (Sims et at, J. Immunol., 151:2296
(1993); Chothia et at, J. Mal.
Biol., 196:901 (1987)).
Another method uses a particular framework derived from the consensus sequence
of all human
antibodies of a particular subgroup of light or heavy chains. The same
framework may be used for several
different humanized antibodies (Carter et a!., Proc. Natl. Acad. Sci. USA,
89:4285 (1992); Presta ei al., J.
lmmnol.. 151:2623 (1993)).
It is further important that antibodies be humanized with retention of high
affinity for the antigen and
other favorable biological properties. To achieve this goal, according to a
preferred method, humanized antibodies
are prepared by a process of analysis of the parental sequences and various
conceptual humanized products using
three-dimensional models of the parental and humanized sequences. Three-
dimensional immunoglobuiin models
are commonly available and are familiar to those skilled in the art. Computer
programs are available which
illustrate and display probable three-dimensionalconformationalstructuresof
selected candidate immunoglobuiin
sequences. Inspection of these displays permits analysis of the likely role of
the residues in the functioning of the
candidate immunoglobulin sequence. i.e., the analysis of residues that
influence the ability of the candidate
immunogiobulin to bind its antigen. In this way, FR residues can be selected
and combined from the recipient
and import sequences so that the desired antibody characteristic, such as
increased affinity for the target
antigen(s), is achieved. In general, the CDR residues are directly and most
substantially involved in influencing
antigen binding.
"Antibody fragments" comprise a portion of a full length antibody, generally
the antigen binding or
variable region thereof. Examples of antibody fragments include Fab, Fab',
F(ab'),, and Fv fragments; diabodies;
linear antibodies; single-chain antibody molecules; and
multispecificantibodies formed from antibody fragments.
Various techniques have been developed for the production of antibody
fragments. Traditionally, these fragments
were derived via proteolytic digestion of intact antibodies (see, e.g.,
Morimoto et al., Journal of Biochemical and
Biophysical Methods 24:107-117 (1992) and Brennan et a!., Science, 229:81
(1985)). However, these fragments
can now be produced directly by recombinant host cells. For example, the
antibody fragments can be isolated
from the antibody phage libraries discussed above. Alternatively, Fab'-SH
fragments can be directly recovered
from E. coil and chemically coupled to form F(ab')2 fragments (Carter et al.,
Bio/Technology 10:163-167 (1992)).
In another embodiment, the F(ab')2 is formed using the leucine zipper GCN4 to
promote assembly of the F(ab')2
molecule. According to another approach, F(ab')2 fragments can be isolated
directly from recombinant host cell
culture. Other techniques for the production of antibody fragments will be
apparent to the skilled practitioner.
in other embodiments, the antibody of choice is a single chain Fv fragment
(scFv). See WO 93/16185.
8
CA 02753381 2011-09-13
WO 99/57134 PCTIUS99/09637
"Single-chain Fv" or "sFv" antibody fragments comprise the VH and VL domains
of antibody. wherein these
domains are present in a single polypeptide chain. Generally, the Fv
polypeptide further comprises a polypeptide
linker between the VH and VL domains which enables the sFv to form the desired
structure for antigen binding.
For a review of sFv see Pluckthun in The Pharmacology of Monoclonal
Antibodies, vol. 113, Rosenburg and
Moore eds. Springer-Verlag, New York, pp. 269-315 (1994).
The term "diabodies" refers to small antibody fragments with two antigen-
bindingsites, which fragments
comprise a heavy chain variable domain (VH) connected to a light chain
variable domain (V1) in the same
polypeptide chain (VH - VL). By using a linker that is too short to allow
pairing between the two domains on the
same chain, the domains are forced to pair with the complementary domains of
another chain and create two
antigen-binding sites. Diabodies are described more fully in, for example, EP
404,097; WO 93/11161; and
Hollinger et al., Proc. Natl. Acad. Sci. USA 90:6444-6448 (1993).
The expression "linear antibodies" when used throughout this application
refers to the antibodies
described in Zapata et al. Polvpeptide Eng. 8(10):1057-1062 (1995). Briefly,
these antibodies comprise a pair
of tandem Fd segments (VH-CHI-VH-CH1) which form a pair of antigen binding
regions. Linear antibodies can
be bispecific or monospecific.
"Multispecific antibodies" have binding specificities for at least two
different epitopes, where the
epitopes are usually from different antigens. While such molecules normally
will only bind two antigens (i.e.
bispecific antibodies. BsAbs), antibodies with additional specificities such
as trispecific antibodies are
encompassed by this expression when used herein. Examples of BsAbs include
those with one arm directed
against a tumor cell antigen and the other arm directed against a cytotoxic
trigger molecule such as anti-
FcyRI/anti-CD15, anti-p185HER2 /FcyRIII (CD16), anti-CD3/anti-malignant B-cell
(ID10), anti-CD3/anti-
p185HER, anti-CD3/anti-p97, anti-CD3/anti-renal cell carcinoma, anti-CD3/anti-
OVCAR-3, anti-CD3/L-D1
(anti-colon carcinoma), anti-CD3/anti-melanocytestimulating hormone analog,
anti-EGF receptor/anti-CD3.anti-
CD3/anti-CAMA 1. anti-CD3/anti-CD 19, anti-C D3/MoV 18, anti-neural cell
ahesion molecule (NCAM yanti-CD3,
anti-folate binding protein (FBP)/anti-CD3,anti-pan carcinoma associated
antigen (AMOC-3 I )/anti-CD3; BsAbs
with one arm which binds specifically to a tumor antigen and one arm which
binds to a toxin such as anti-
saporin/anti-Id-1, anti-CD22/anti-saporin. anti-CD7/anti-saporin, anti-
CD38/anti-saporin, anti-CEA/anti-ricin A
chain, anti-interferon-a(IFN-a)/anti-hybridoma idiotype, anti-CEA/anti-vinca
alkaloid; BsAbs for converting
enzyme activated prodrugs such as anti-CD30/anti-alkaline phosphatase (which
catalyzes conversion of
mitomycin phosphate prodrug to mitomycin alcohol); BsAbs which can be used as
fibrinolytic agents such as
anti-fibrin/anti-tissue plasminogen activator (tPA), anti-fibrin/anti-
urokinase-type plasminogen activator (uPA);
BsAbs for targeting immune complexes to cell surface receptors such as anti-
low density lipoprotein (LDL)Ianti-
Fc receptor(e.g. FcyRl, or FcyRlII); BsAbs for use in therapy of infectious
diseases such as anti-CD3/anti-herpes
simplex virus (HSV), anti-T-cell receptor:CD3 complex/anti-influenza, anti-
FcyR/anti-HIV; BsAbs for tumor
detection in vitro or in vivo such as anti-CEA/anti-EOTUBE, anti-CEA/anti-
DPTA, anti-p 185 HER2 /anti-hapten;
BsAbs as vaccine adjuvants; and BsAbs as diagnostic tools such as anti-rabbit
IgG/anti-ferritin, anti-horse radish
9
CA 02753381 2011-09-13
WO 99/57134 PCT/US99/09637
peroxidase (HRP)/anti-hormone. anti-somatostatin/anti-substance P. anti-
HRP/anti-FITC, anti-CEA/anti-(i-
galactosidase. Examples of trispecific antibodies include anti-CD3/anti-
CD4/anti-CD37,anti-CD3/anti-CD5/anti-
CD37 and anti-CD3/anti-CD8/anti-CD37. Bispecific antibodies can be prepared as
full length antibodies or
antibody fragments (e.g. F(ab')2 bispecific antibodies).
Methods for making bispecific antibodies are known in the art. Traditional
production of full length
bispecific antibodies is based on the coexpression of two immunoglobulin heavy
chain-light chain pairs, where
the two chains have different specificities (Millstein et a!., Nature, 305:537-
539 (1983)). Because of the random
assortment of immunoglobulin heavy and light chains, these hybridomas
(quadromas) produce a potential mixture
of 10 different antibody molecules, of which only one has the correct
bispecific structure. Purification of the
correct molecule, which is usually done by affinity chromatography steps, is
rather cumbersome, and the product
yields are low. Similar procedures are disclosed in WO 93/08829, and in
Traunecker el al., EMBOJ., 10:3655-
3659(1991).
According to a different approach, antibody variable domains with the desired
binding specificities
(antibody-antigen combining sites) are fused to immunoglobulin constant domain
sequences. The fusion
preferably is with an immunoglobulin heavy chain constant domain, comprising
at least part of the hinge, CH2,
and CH3 regions. It is preferred to have the first heavy-chain constant region
(CH 1) containing the site necessary
for light chain binding, present in at least one of the fusions. DNAs encoding
the immunoglobulin heavy chain
fusions and, if desired. the immunoglobulin light chair., are inserted into
separate expression vectors, and are co-
transfected into a suitable host organism. This provides for great flexibility
in adjusting the mutual proportions
of the three polypeptide fragments in embodiments when unequal ratios of the
three polypeptide chains used in
the construction provide the optimum yields. It is, however, possible to
insert the coding sequences for two or
alI three polypeptide chains in one expression vector when the expression of
at least two polypeptide chains in
equal ratios results in high yields or when the ratios are of no particular
significance.
In a preferred embodiment of this approach. the bispecific antibodies are
composed of a hybrid
immunoglobulin heavy chain with a first binding specificity in one arm, and a
hybrid immunoglobulin heavy
chain-light chain pair (providinga second binding specificity) in the other
arm. It was found that this asymmetric
structure facilitates the separation of the desired bispecific compound from
unwanted immunoglobulin chain
combinations, as the presence of an immunoglobulin light chain in only one
half of the bispecific molecule
provides for a facile way of separation. This approach is disclosed in WO
94104690. For further details of
generating bispecific antibodies see, for example, Suresh et at., Methods in
Enzymology, 121:210 (1986).
According to another approach described in W096/27011, the interface between a
pair of antibody
molecules can be engineered to maximize the percentage ofheterodimerswhich are
recovered from recombinant
cell culture. The preferred interface comprises at least a part of the CH3
domain of an antibody constant domain.
In this method, one or more small amino acid side chains from the interface of
the first antibody molecule are
replaced with larger side chains (e.g. tyrosine or tryptophan). Compensatory
"cavities" of identical or similar size
to the large side chain(s) are created on the interface of the second antibody
molecule by replacing large amino
acid side chains with smaller ones (e.g. alanine or threonine). This provides
a mechanism for increasing the yield
of the heterodimer over other unwanted end-products such as homodimers.
CA 02753381 2011-09-13
WO 99/57134 PCT/US99/09637
Bispecific antibodies include cross-linked or "heteroconjugate" antibodies.
For example, one of the
antibodies in the heteroconjugatecan be coupled to avidin. the other to
biotin. Such antibodies have. for example,
been proposed to target immune system cells to unwanted cells (US Patent No.
4,676,980), and for treatment of
HIV infection (WO 91/00360, WO 92/200373, and EP 03089). Heteroconjugate
antibodies may be made using
any convenient cross-linking methods. Suitable cross-linking agents are well
known in the art, and are disclosed
in US Patent No. 4,676,980, along with a number of cross-linking techniques.
Techniques for generating bispeciftc antibodies from antibody fragments have
also been described in
the literature. For example, bispecific antibodies can be prepared using
chemical linkage. Brennan at at. Science,
229: 81 (1985) describe a procedure wherein intact antibodies are
proteolytically cleaved to generate F(ab')2
fragments. These fragments are reduced in the presence of the dithiol
complexing agent sodium arsenite to
stabilize vicinal dithiols and prevent intermolecular disulfide formation. The
Fab' fragments generated are then
converted to thionitrobenzoate (TNB) derivatives. One of the Fab'-TNB
derivatives is then reconverted to the
Fab'-thiol by reduction with mercaptoethylamine and is mixed with an equimolar
amount of the other Fab'-TNB
derivative to form the bispecific antibody. The bispecific antibodies produced
can be used as agents for the
selective immobilization of enzymes.
Recent progress has facilitated the direct recovery of Fab'-SH fragments from
E. colt, which can be
chemically coupled to form bispecific antibodies. Shalaby et al., J, Erp.
Med., 175: 217-225 (1992) describe the
production of a fully humanized bispeciftc antibody F(ab')2 molecule. Each Fab
' fragment was separately secreted
from E. colt and subjected to directed chemical coupling in vitro to form the
bispecific antibody.
Various techniques for making and isolating bispecific antibody fragments
directly from recombinant
cell culture have also been described. For example, bispecific antibodies have
been produced using leucine
zippers. Kostelny et al., J. Immunol., 148(5):1547-1553(1992). The leucine
zipper peptides from the Fos and Jun
proteins were linked to the Fab' portions of two different antibodies by gene
fusion. The antibody homodimers
were reduced at the hinge region to form monomers and then re-oxidized to form
the antibody heterodimers. This
method can also be utilized for the production of antibody homodimers. The
"diabody" technology described by
Hollinger et al.. Proc. Natl. Acad Sci. USA, 90:6444-6448 (1993) has provided
an alternative mechanism for
making bispecific antibody fragments. The fragments comprise a heavy-chain
variable domain (VH) connected
to a light-chain variable domain (VL) by a linker which is too short to allow
pairing between the two domains
on the same chain. Accordingly, the VH and VL domains of one fragment are
forced to pair with the
complementary VL and VH domains of another fragment, thereby forming two
antigen-binding sites. Another
strategy for making bispecific antibody fragments by the use of single-chain
Fv (sFv) dimers has also been
reported. See Gruber et al., J. Immunol., 152:5368 (1994).
Antibodies with more than two valencies are contemplated. For example,
trispecific antibodies can be
prepared. Tutt et al. J. Immunol. 147: 60 (1991).
The phrase "ion exchange material" refers to a solid phase which is negatively
charged (i.e. a cation
exchange resin) or positively charged (i.e. an anion exchange resin). The
charge may be provided by attaching
11
CA 02753381 2011-09-13
WO 99/57134 PCT/US99/09637
one or more charged ligands to the solid phase, e.g. by covalent linking.
Alternatively, or in addition, the charge
may be an inherent property of the solid phase (e.g. as is the case for
silica, which has an overall negative charge).
By "solid phase" is meant a non-aqueous matrix to which one or more charged
ligands can adhere. The
solid phase may be a purification column, a discontinuous phase of discrete
particles, a membrane, or filter etc.
Examples of materials for forming the solid phase include polysaccharides
(such as agarose and cellulose); and
other mechanically stable matrices such as silica (e.g controlled pore glass),
poly(styrenedivinyl)benzene,
polyacrylamide, ceramic particles and derivatives of any of the above.
A "cation exchange resin" refers to a solid phase which is negatively charged,
and which thus has free
cations for exchange with cations in an aqueous solution passed over or
through the solid phase. A negatively
charged ligand attached to the solid phase to form the cation exchange resin
may, e.g., be a carboxylate or
sulfonate. Commercially available cation exchange resins include carboxy-
methyl-cellulose, BAKERBOND
ABXTM, sulphopropyl(SP) immobilizedon agarose (e.g. SP-SEPHAROSEFAST FLOWT"'
or SP-SEPHAROSE
HIGH PERFORMANCETM, from Pharmacia) and sulphonyl immobilizedon agarose (e.g.
S-SEPHAROSE FAST
FLOWTM from Pharmacia).
The term "anion exchange resin" is used herein to refer to a solid phase which
is positively charged, e .g.
having one or more positively charged ligands, such as quaternary amino
groups, attached thereto. Commercially
available anion exchange resins include DEAE cellulose, QAE SEPHADEXTM and
FAST Q SEPHAROSETM
(Pharmacia).
A "buffer" is a solution that resists changes in pH by the action of its acid-
base conjugate components.
Various buffers which can be employed depending, for example, on the desired
pH of the buffer are described
in Buffers. A Guide for the Preparation and Use of Buffers in Biological
Systems, Gueffroy, D., Ed. Calbiochem
Corporation (1975). In one embodiment, the buffer has a pH in the range from
about 5 to about 7 (e.g. as in
Example I below). Examples of buffers that will control the pH in this range
include MES. MOPS, MOPSO,
phosphate, acetate, citrate, succinate. and ammonium buffers, as well as
combinations of these.
The "loading buffer" is that which is used to load the composition comprising
the polypeptide molecule
of interest and one or more contaminants onto the ion exchange resin. The
loading buffer has a conductivity
and/or pH such that the polypeptide molecule of interest (and generally one or
more contaminants) is/are bound
to the ion exchange resin.
The "intermediate buffer" is used to elute one or more contaminants from the
ion exchange resin, prior
to eluting the polypeptide molecule of interest. The conductivity and/or pH of
the intermediate buffer is/are such
that the contaminant is eluted from the ion exchange resin, but not
significant amounts of the polypeptide of
interest.
The term "wash buffer" when used herein refers to a buffer used to wash or re-
equilibrate the ion
exchange resin, prior to eluting the polypeptide molecule of interest.
Conveniently, the wash buffer and loading
buffer may be the same, but this is not required.
The "elution buffer" is used to elute the polypeptide of interest from the
solid phase. The conductivity
and/or pH of the elution buffer is/are such that the polypeptide of interest
is eluted from the ion exchange resin.
A "regeneration buffer" may be used to regenerate the ion exchange resin such
that it can be re-used. The
regeneration buffer has a conductivity and/or pH as required to remove
substantially all contaminants and the
12
CA 02753381 2011-09-13
WO 99/57134 PCTIUS99109637
polypeptide of interest from the ion exchange resin.
The term "conductivity" refers to the ability of an aqueous solution to
conduct an electric current
between two electrodes. In solution, the current flows by ion transport.
Therefore, with an increasing amount of
ions present in the aqueous solution, the solution will have a higher
conductivity. The unit of measurement for
conductivity is mmhos (mS/cm), and can be measured using a conductivity meter
sold, e.g., by Orion. The
conductivity of a solution may be altered by changing the concentration of
ions therein. For example, the
concentration of a buffering agent and/or concentration of a salt (e.g. NaCl
or KCI) in the solution may be altered
in order to achieve the desired conductivity. Preferably, the salt
concentration of the various buffers is modified
to achieve the desired conductivity as in the Example below,
By "purifying" a polypeptide from a composition comprising the polypeptide and
one or more
contaminants is meant increasing the degree of purity of the polypeptide in
the composition by removing
(completely or partially) at least one contaminant from the composition. A
"purification step" may be part of an
overall purification process resulting in a "homogeneous" composition, which
is used herein to refer to a
composition comprising at least about 70% by weight of the polypeptide of
interest, based on total weight of the
composition, preferably at least about 80% by weight.
Unless indicated otherwise, the term "HER2" when used herein refers to human
HER2 protein and
"HER2" refers to human HER2 gene. The human HER2 gene and HER2 protein are
described in Semba et a!.,
PNAS (USA) 82:6497-6501 (1985) and Yamamoto et al. Nature 319:230-234 (1986)
(Genebank accession
number X03363), for example.
The term "humMAb4D5-8" when used herein refers to a humanized anti-HER2
antibody comprising
the light chain amino acid sequence of SEQ ID NO:1 and the heavy chain amino
acid sequence of SEQ ID NO:2
or amino acid sequence variants thereof which retain the ability to bind HER2
and inhibit growth of tumor cells
which overexpress HER2 (see US Patent 5,677,171 ).
The "pI" or "isoeiectric point" of a polypeptide refer to the pH at which the
polypeptide's positive
charge balances its negative charge. pl can be calculated from the net charge
of the amino acid residues of the
polypeptide or can be determined by isoelectric focussing (e.g. using CSx
chromatography as in the Example
below).
By "binding" a molecule to an ion exchange material is meant exposing the
molecule to the ion
exchange material under appropriate conditions (pH/conductivity) such that the
molecule is reversibly
immobilized in or on the ion exchange material by virtue of ionic interactions
between the molecule and a
charged group or charged groups of the ion exchange material.
By "washing" the ion exchange material is meant passing an appropriate buffer
through or over the ion
exchange material.
To "elute" a molecule (e.g. polypeptide or contaminant) from an ion exchange
material is meant to
remove the molecule therefrom by altering the ionic strength of the buffer
surrounding the ion exchange material
such that the buffer competes with the molecule for the charged sites on the
ion exchange material.
"Treatment" refers to both therapeutic treatment and prophylactic or
preventative measures. Those in
need of treatment include those already with the disorder as well as those in
which the disorder is to be prevented.
A "disorder" is any condition that would benefit from treatment with the
polypeptidepurified as described herein.
13
CA 02753381 2011-09-13
WO 99/57134 PCT/US99/09637
This includes chronic and acute disorders or diseases including those
pathological conditions which predispose
the mammal to the disorder in question.
The word "label" when used herein refers to a detectable compound or
composition which is conjugated
directly or indirectly to the polypeptide. The label may be itself be
detectable (e.g., radioisotope labels or
fluorescent labels) or, in the case of an enzymatic label, may catalyze
chemical alteration of a substrate compound
or composition which is detectable.
The term "cytotoxic agent" as used herein refers to a substance that inhibits
or prevents the function of
cells and/or causes destruction of cells. The term is intended to include
radioactive isotopes (e.g. I 13 t, I12S, Y90
and Ref 86), chemotherapeutic agents, and toxins such as enzymatically active
toxins of bacterial, fungal, plant
or animal origin, or fragments thereof.
A "chemotherapeutic agent" is a chemical compound useful in the treatment of
cancer. Examples of
chemotherapeutic agents include adriamycin, doxorubicin, epirubicin, 5-
fluorouracil, cytosine arabinoside ("Ara-
T11
C"), cyclophosphamide, thiotepa, busulfan, cytoxin, taxoids, e.g. paclitaxel
(TAXOL , Bristol-Myers Squibb
Oncology, Princeton, NJ), and doxetaxel, toxotere, methotrexate, cisplatin,
melphalan, vinblastine, bleomycin,
etoposide, ifosfamide, mitomycin C, mitoxantrone, vincristine, vinorelbine,
carboplatin, teniposide, daunomycin,
carminomycin, aminopterin, dactinomycin, mitomycins, esperamicins (see U.S.
Pat. No. 4,675,187), melphalan
and other related nitrogen mustards. Also included in this definition are
hormonal agents that act to regulate or
inhibit hormone action on tumors, such as tamoxifen and onapristone.
Modes for Carrying Out the Invention
The invention herein provides a method for purifying a polypeptide from a
composition (e.g. an aqueous
solution) comprising the polypeptide and one or more contaminants. The
composition is generally one resulting
from the recombinant production of the polypeptide, but may be that resulting
from production of the polypeptide
by peptide synthesis (or other synthetic means) or the polypeptide may be
purified from a native source of the
polypeptide. Preferably the polypeptide is an antibody, e.g. one which binds
the HER2 antigen.
For recombinant production of the polypeptide, the nucleic acid encoding it is
isolated and inserted into
a replicable vector for further cloning (amplification of the DNA) or for
expression. DNA encoding the
polypeptide is readily isolated and sequenced using conventional procedures
(e.gõ where the polypeptide is an
antibody by using oligonucleotide probes that are capable of binding
specifically to genes encoding the heavy
and light chains of the antibody). Many vectors are available. The vector
components generally include, but are
not limited to, one or more of the following: a signal sequence, an origin of
replication, one or more marker
genes, an enhancer element, a promoter, and a transcription termination
sequence (e.g. as described in US Patent
5,534,615 )=
Suitable host cells for cloning or expressing the DNA in the vectors herein
are the prokaryote, yeast, or
higher eukaryote cells described above. Suitable prokaryotes for this purpose
include eubacteria, such as Gram-
negative or Gram-positive organisms, for example, Enterobacteriaceae such as
Escherichia, e.g., E. coli,
Enterobacter, Erwinia, Klebsiella, Proteus, Salmonella, e.g., Salmonella
typhimurium, Serratia, e.g., Serratia
marcescans, and Shigella, as well as Bacilli such as B. subtilis and B.
licheniformis (e.g., B. licheniformis 41 P
disclosed in DD 266,710 published 12 April 1989), Pseudomonas such as P.
aeruginosa, and Streptomyces. One
14
CA 02753381 2011-09-13
WO 99/57134 PCT/US99/09637
preferred E. coil cloning host is E. coli 294 (ATCC 31,446), although other
strains such as E. coli B, E. coli
X1776 (ATCC 31,537), and E. coil W3110 (ATCC 27,325) are suitable. These
examples are illustrative rather
than limiting.
In addition to prokaryotes, eukaryotic microbes such as filamentous fungi or
yeast are suitable cloning
or expression hosts for polypeptide encoding vectors. Saccharomycescerevisiae,
or common baker's yeast, is the
most commonly used among lower eukaryotic host microorganisms. However, a
number of other genera, species,
and strains are commonly available and useful herein, such as
Schizosaccharomycespombe, Kluyveromyces hosts
such as, e.g., K. lactis, K. fragilis (ATCC 12,424), K. bulgaricus (ATCC
16,045), K. wickerami (ATCC 24,178),
K. waltii (ATCC 56,500), K. drosophilarum (ATCC 36,906), K. thermotolerans,
and K. marxianus; yarrowia
(EP 402,226); Pichiapastoris (EP 183,070); Candida; Trichoderma reesia (EP
244,234); Neurospora crassa;
Schwanniomyces such as Schwanniomyces occidentalis; and filamentous fungi such
as, e.g., Neurospora,
Penicillium, Tolvpocladium, and Aspergillus hosts such as A. nidulans and A.
niger.
Suitable host cells for the expression of glycosylated polypeptide are derived
from multicellular
organisms. Examples of invertebrate cells include plant and insect cells.
Numerous baculoviral strains and
variants and corresponding permissive insect host cells from hosts such as
Spodopterafrugiperda (caterpillar),
Aedes aegypti (mosquito), Aedes albopictus (mosquito), Drosophila melanogaster
(fruitfly), and Bombyx mori
have been identified. A variety of viral strains for transfection are publicly
available, e.g., the L-1 variant of
Autographa californica NPV and the Bm-5 strain of Bombyx mori NPV, and such
viruses may be used as the
virus herein according to the present invention, particularly for transfection
of Spodopterafrugiperda cells. Plant
cell cultures of cotton, corn, potato, soybean, petunia, tomato, and tobacco
can also be utilized as hosts.
However, interest has been greatest in vertebrate cells, and propagation of
vertebrate cells in culture
(tissue culture) has become a routine procedure. Examples of useful mammalian
host cell lines are monkey kidney
CVI line transformed by SV40 (COS-7. ATCC CRL 1651); human embryonic kidney
line (293 or 293 cells
subcloned for growth in suspension culture. Graham et al., J. Gen Virol. 36:59
(1977)); baby hamster kidney cells
(BHK, ATCC CCL 10); Chinese hamster ovary cells/-DHFR (CHO, Urlaub et al.,
Proc. Natl. Acad. Sci. USA
77:4216 (1980)); mouse sertoli cells (TM4, Mather, Biol. Reprod 23:243-251
(1980)); monkey kidney cells (CV I
ATCC CCL 70); African green monkey kidney cells (VERO-76, ATCC CRL- 1587);
human cervical carcinoma
cells (HELA, ATCC CCL 2); canine kidney cells (MDCK, ATCC CCL 34); buffalo rat
liver cells (BRL 3A.
ATCC CRL 1442); human lung cells (W138, ATCC CCL 75); human liver cells (Hep
G2, HB 8065); mouse
mammary tumor (MMT 060562, ATCC CCL51); TRI cells (Mather et al., Annals N.Y.
Acad Sci. 383:44-68
(1982)); MRC 5 cells; FS4 cells; and a human hepatoma line (Hep G2).
Host cells are transformed with the above-described expression or cloning
vectors for polypeptide
production and cultured in conventional nutrient media modified as appropriate
for inducing promoters, selecting
transformants, or amplifying the genes encoding the desired sequences.
The host cells used to produce the polypeptide of this invention may be
cultured in a variety of media.
Commercially available media such as Ham's F 10 (Sigma), Minimal Essential
Medium ((MEM), (Sigma), RPMI-
1640 (Sigma), and Dulbecco's Modified Eagle's Medium ((DMEM), Sigma) are
suitable for culturing the host
cells. In addition, any of the media described in Ham et a!., Meth. Enz. 58:44
(1979), Barnes et a!., Anal.
Biochem.102:255 (1980), U.S. Pat. Nos. 4,767.704; 4,657,866; 4,927,762;
4,560,655; or 5,122,469; WO
CA 02753381 2011-09-13
WO 99/57134 PCT/US99/09637
90/03430; WO 87/00195; or U.S. Patent Re. 30,985 may be used as culture media
for the host cells. Any of these
media may be supplementedas necessary with hormones and/or other growth
factors (such as insulin, transferrin,
or epidermal growth factor), salts (such as sodium chloride, calcium,
magnesium. and phosphate), buffers (such
as HEPES), nucleotides (such as adenosine and thymidine), antibiotics (such as
GENTAMYCINT"" drug), trace
elements (defined as inorganic compounds usually present at final
concentrations in the micromolar range), and
glucose or an equivalent energy source. Any other necessary supplements may
also be included at appropriate
concentrations that would be known to those skilled in the art. The culture
conditions, such as temperature, pH,
and the like, are those previously used with the host cell selected for
expression, and will be apparent to the
ordinarily skilled artisan.
When using recombinant techniques, the polypeptidecan be produced
intracellularly, in the periplasmic
space, or directly secreted into the medium. If the polypeptide is produced
intracellularly, as a first step, the
particulate debris, either host cells or lysed cells (e.g. resulting from
homogenization), is removed, for example,
by centrifugation or ultrafiltration. Where the polypeptide is secreted into
the medium, supernatants from such
expression systems are generally first concentrated using a commercially
available protein concentration filter,
for example, an Amicon or Millipore Pellicon ultrafiltration unit.
The polypeptide is then subjected to one or more purification steps, including
the ion exchange
chromatography method as claimed herein. Examples of additional purification
procedures which may be
performed prior to, during, or following the ion exchange chromatography
method include fractionation on a
hydrophobic interaction chromatography (e.g. on phenyl sepharose), ethanol
precipitation, isoelectric focusing,
Reverse Phase HPLC, chromatography on silica, chromatography on HEPARIN
SEPHAROSET", further anion
exchange chromatography and/or further cation exchange chromatography,
chromatofocusing, SDS-PAGE,
ammonium sulfate precipitation, hydroxylapatite chromatography, gel
electrophoresis, dialysis, and affinity
chromatography(e.g. using protein A, protein G, an antibody, a specific
substrate, ligand or antigen as the capture
reagent).
Ion exchange chromatography is performed as claimed herein. A decision is
first made as to whether
an anion or cation exchange resin is to be employed. In general, a cation
exchange resin may be used for
polypeptides with pi's greater than about 7 and an anion exchange resin may be
used for polypeptides with pi's
less than about 7.
The anion or cation exchange resin is prepared according to known methods.
Usually, an equilibration
buffer is passed through the ion exchange resin prior to loading the
composition comprising the polypeptide and
one or more contaminants onto the resin. Conveniently, the equilibration
buffer is the same as the loading buffer,
but this is not required.
The various buffers used for the chromatography depend, for example, on
whether a cation or anion
exchange resin is employed. This is shown more clearly in the flow diagrams of
Figures I and 2.
With particular reference to Figure I, which shows exemplary steps to be
performed where a cation
exchange resin is used, the pH and/or conductivityof each buffer is/are
increased relative to the preceding buffer,
except for the wash buffer where the conductivity and/or pH is/are less than
the conductivity and/or pH of the
preceding intermediate buffer. The aqueous solution comprising the polypeptide
of interest and contaminant(s)
is loaded onto the cation exchange resin using the loading buffer that is at a
pH and/or conductivity such that the
16
CA 02753381 2011-09-13
WO 99/57134 PCT/US99/09637
polypeptide and the contaminant bind to the cation exchange resin. As in the
Example below, the loading buffer
may be at a first low conductivity (e.g. from about 5.2 to about 6.6 mmhos).
An exemplary pH for the loading
buffer may be about 5.0 (see Fig. 1). From about 20mg/mL to about 35mg/mL of
the polypeptide (e.g. of a full
length antibody) may, for example, be loaded on the ion exchange resin.
The cation exchange resin is then washed with an intermediate buffer which is
at a second conductivity
and/or pH so as to essentially elute the contaminant, but not a substantial
amount of the polypeptide of interest.
This may be achieved by increasing the conductivity or pH, or both, of the
intermediate buffer. The change from
loading buffer to intermediate buffer may be step-wise or gradual as desired.
In the Example herein, the
intermediate buffer had a greater conductivity than that of the loading buffer
(i.e. the intermediate buffer's
conductivity was in the range from about 7.3 to about 8.4 mmhos).
Alternatively, as shown in Figure 1, the pH
of the intermediate buffer may exceed that of the loading buffer in this
embodiment of the invention, where a
cation exchange resin is used. For example, the intermediate buffer may have a
pH of about 5.4.
Following washing with the intermediate buffer, the cation exchange resin is
washed or re-equilibrated
with the wash buffer which has a conductivity or pH. or both, which is/are
less than that of the intermediate buffer
(i.e. the conductivity, or pH, or both, is/are changed in an opposite, i.e.
reverse, direction to the preceding step,
unlike ion exchange chromatography steps in the literature). In the Example
below, the wash buffer had about
the same conductivity as the loading buffer (i.e. in the range from about 5.2
to about 6.6 mmhos) and its
conductivity was, therefore, less than that of the intermediate buffer. In
another embodiment, one may reduce the
conductivity of the wash buffer to a conductivity that is less than, or
greater than, that of the loading buffer,
provided the conductivity of the wash buffer is less than that of the
intermediate buffer. In another embodiment,
the pH of the wash buffer may be less than the pH of the intermediate buffer
(e.g. the pH of the wash buffer may
about 5.0). The change in conductivity and/or pH of the wash buffer compared
to the intermediate buffer may
be achieved by step-wise or gradual change of either or both of these
parameters.
After the wash step of the preceding paragraph, the cation exchange resin is
prepared for elution of the
desired polypeptide molecule therefrom. This is achieved using an elution
buffer that has a pH and/or
conductivity such that the desired polypeptide no longer binds to the cation
exchange resin and therefore is eluted
therefrom. The pH and/or conductivity of the elution buffer generally
exceed(s) the pH and/or conductivity of
the loading buffer, the intermediate buffer and the wash buffer used in the
previous steps. In the Example below,
the conductivity of the elution buffer was in the range from about 10.0 to
about 11.0 mmhos. Alternatively, or
in addition, the pH of the elution buffer may be increased relative to the
wash buffer and to the intermediate
buffer (for example, the pH of the elution buffer may about 6.0). The change
in conductivity and/or pH may be
step-wise or gradual, as desired. Hence, the desired polypeptide is retrieved
from the cation exchange resin at this
stage in the method.
in an alternative embodiment, the ion exchange material comprises an anion
exchange resin. This
embodiment of the invention is depicted in Figure 2 herein. As illustrated in
this figure, the changes in
conductivity are generally as described above with respect to a cation
exchange resin. However, the direction
of change in pH is different for an anion exchange resin. For example, if
elution of the contaminant(s) and
polypeptide are to be achieved by altering pH, the loading buffer has a first
pH and the pH is decreased in the
intermediate buffer so as to elute the contaminant or contaminants. In the
third step, the column is washed/re-
17
CA 02753381 2011-09-13
WO 99/57134 PCT/US99/09637
equilibrated with the wash buffer and the change in conductivity or pH, or
both, is in the opposite direction to
that of the previous step. Hence, the pH may be increased in the wash buffer,
compared to the intermediate buffer.
Following this step, the polypeptide of interest is eluted from the anion
exchange resin using an elution buffer
at a fourth conductivity and/or pH. If pH is altered, it will normally be less
than the pH of the loading buffer, the
intermediate buffer and the wash buffer. The change in pH and/or conductivity
in progressive buffers can, as
explained above, be step-wise or gradual.
In the preferred embodiment of the invention, a single parameter (i.e. either
conductivity or pH) is
changed to achieve elution of both the polypeptide and contaminant, while the
other parameter (i.e. pH or
conductivity, respectively) remains about constant. For example, while the
conductivity of the various buffers
(loading buffer, intermediate buffer, wash buffer and/or elution buffer) may
differ, the pH's thereof may be
essentially the same.
In an optional embodiment of the invention, the ion exchange resin is
regenerated with a regeneration
buffer after elution of the polypeptide, such that the column can be re-used.
Generally, the conductivity and/or
pH of the regeneration buffer is/are such that substantially all contaminants
and the polypeptide of interest are
eluted from the ion exchange resin. Generally, the regeneration buffer has a
very high conductivity for eluting
contaminants and polypeptide from the ion exchange resin.
The method herein is particularly useful for resolving a polypeptide molecule
of interest from at least
one contaminant, where the contaminant and polypeptide molecule of interest
differ only slightly in ionic charge.
For example, the pl's of the polypeptide and contaminant may be only "slightly
different", for example they may
differ by only about 0.05 to about 0.2 pi units. In the Example below, this
method could be used to resolve an
anti-HER2 antibody having a p1 of 8.87, from a singly-deamidated variant
thereof having a p1 of 8.79.
Alternativeiy,the method may be used to resolve a deamidated DNase. for
example, from nondeamidated DNase.
In another embodiment, the method may be used to resolve a polypeptide from a
glycosylation variant thereof,
e.g. for resolving a variant of a polypeptide having a different distribution
of sialic acid compared to the
nonvariant polypeptide.
The polypeptide preparation obtained according to the ion exchange
chromatography method herein
may be subjected to additional purification steps, if necessary. Exemplary
further purification steps have been
discussed above.
Optionally, the polypeptide is conjugated to one or more heterologous
molecules as desired. The
heterologous molecule may, for example, be one which increases the serum half-
life of the polypeptide (e.g.
polyethylene glycol, PEG), or it may be a label (e.g. an enzyme, fluorescent
label and/or radionuclide) or a
cytotoxic molecule (e.g. a toxin, chemotherapeutic drug, or radioactive
isotope etc).
A therapeutic formulation comprising the polypeptide, optionally conjugated
with a heterologous
molecule, may be prepared by mixing the polypeptide having the desired degree
of purity with optional
pharmaceutically acceptable carriers, excipients or stabilizers (Remington's
PharmaceuticaiSciences 16th edition,
Osol, A. Ed. (1980)), in the form of lyophilized formulationsor aqueous
solutions. "Pharmaceuticallyacceptable"
carriers, excipients, or stabilizers are nontoxic to recipients at the dosages
and concentrations employed, and
include buffers such as phosphate, citrate, and other organic acids;
antioxidants including ascorbic acid and
methionine; preservatives (such as octadecyldimethylbenzyl ammonium chloride;
hexamethonium chloride;
18
CA 02753381 2011-09-13
WO 99/57134 PCT/US99/09637
benzalkonium chloride, benzethonium chloride; phenol, butyl or benzyl alcohol;
alkyl parabens such as methyl
or propyl paraben; catechol; resorcinol; cyclohexanol; 3-pentanol; and m-
cresol); low molecularweight (less than
about 10 residues) polypeptide; proteins, such as serum albumin, gelatin, or
immunoglobulins; hydrophilic
polymers such as polyvinylpyrrolidone; amino acids such as glycine, glutamine,
asparagine, histidine, arginine,
or lysine; monosaccharides, disaccharides, and other carbohydrates including
glucose, mannose, or dextrins;
chelating agents such as EDTA; sugars such as sucrose, mannitol, trehalose or
sorbitol; salt-forming counter-ions
such as sodium; metal complexes (e.g., Zn-protein complexes); and/or non-ionic
surfactants such as TWEENrm,
PLURONICSTM or polyethylene glycol (PEG). The humMAb4D5-8 antibody of
particular interest herein may
be prepared as a lyophilized formulation, e.g. as described in WO 97/04801
The formulation herein may also contain more than one active compound as
necessary for the particular
indication being treated, preferably those with complementary activities that
do not adversely affect each other.
Such molecules are suitably present in combination in amounts that are
effective for the purpose intended. For
example, for an anti-HER2 antibody a chemotherapeutic agent, such as a taxoid
or tamoxifen, may be added to
the formulation.
The active ingredients may also be entrapped in microcapsule prepared, for
example, by coacervation
techniques or by interfacial polymerization, for example,
hydroxymethylcellulose or gelatin-microcapsule and
poly-(methylmethacylate)microcapsule,respectively, in colloidal drug
deliverysystems (for example, liposomes,
albumin microspheres, microemulsions,nano-particlesand nanocapsules)or in
macroemulsions.Such techniques
are disclosed in Remington's Pharmaceutical Sciences 16th edition, Osol, A.
Ed. (1980).
The formulation to be used for in vivo administration must be sterile. This is
readily accomplished by
filtration through sterile filtration membranes.
Sustained-release preparations may be prepared, Suitable examples of sustained-
release preparations
include semipermeable matrices of solid hydrophobic polymers containing the
polypeptide variant, which
matrices are in the form of shaped articles, e.g., films, or microcapsule.
Examples of sustained-release matrices
include polyesters, hydrogels (for example, poly(2-hydroxyethyl-methacrylate),
or poly(vinyla)cohol)),
polylactides (U.S. Pat. No. 3,773,919), copolymers of L-glutamic acid and y
ethyl-L-glutamate, non-degradable
ethylene-vinyl acetate, degradable lactic acid-glycolic acid copolymers such
as the LUPRON DEPOTTM
(injectable microspheres composed of lactic acid-glycolic acid copolymer and
leuprolide acetate), and poly-D-(-)-
3-hydroxybutyric acid.
The polypeptide purified as disclosed herein or the composition comprising the
polypeptide and a
pharmaceutically acceptable carrier is then used for various diagnostic,
therapeutic or other uses known for such
polypeptides and compositions. For example, the polypeptide may be used to
treat a disorder in a mammal by
administering a therapeutically effective amount of the polypeptide to the
mammal.
The following examples are offered by way of illustration and not by way of
limitation. The disclosures
of all citations in the specification are expressly incorporated herein by
reference.
EXAMPLE I
Full length human IgG rhuMAb HER2 (humAb4D5-8 in Carter et a!, Proc. Nat!.
Acad. Sci. 89: 4285-
4289 (1992) comprising the light chain amino acid sequence of SEQ ID NO:1 and
heavy chain amino acid
19
CA 02753381 2011-09-13
WO 99/57134 PCT/US"/09637
sequence of SEQ ID NO:2) was produced recombinantly in CHO cells. Following
protein production and
secretion to the cell culture medium, the CHO cells were separated from the
cell culture medium by tangential
flow filtration (PROSTACK '). Protein A chromatography was then performed by
applying the Harvested Cell
Culture Fluid (HCCF) from the CHO cells directly to an equilibrated PROSEP
ATIA column (Bioprocessing, Ltd).
Following Protein A chromatography, cation exchange chromatography was
performed using a
sulphopropyl (SP)-SEPHAROSE FAST FLOWT" (SPSFF) column (Pharmacia) to further
separate the desired
anti-HER2 antibody molecule. The chromatography operation was performed in
bind and elute mode.
The SPSFF column was prepared for load by sequential washes with regeneration
buffer (0.025 M MES
/ 1.0 M NaCl, pH 5.6) followed by equilibration buffer (0.025 M MES / 50 mM
NaCl, pH 5.6). The column was
then loaded with Protein A pool adjusted to a pH of 5.60 0.05 and a
conductivity of 5.8 0.2 mmhos. Prior to
elution, the column was washed in three steps: (1) loading buffer (0.025 M MES
/ 50 mM NaCl, pH 5.6) for a
minimum of I column volume; (2) intermediate buffer (0.025 M MES / 70 mM NaCI,
pH 5.6) until an apex of
a 280 nm peak was reached; and (3) wash buffer (0.025 M MES / 50 mM NaCl, pH
5.6) for a minimum of 1.2
column volumes. rhuMAb HER2 was then eluted from the column with elution
buffer (0.025 M MES / 95 mM
NaCl, pH 5.6). The elution 280 nm profile has a shoulder on the leading edge
(Figure 3). At the inflection point
of this shoulder, pooling starts and continues for an additional 5 column
volumes. The column was then
regenerated with regeneration buffer (0.025 M MES / 1.0 M NaCl, pH 5.6).
Materials and Methods
Column and Load Preparation: A reduced-scale SPSFF column was packed. The
dimensions were:
27.0 mL volume, 1.0 cm diameter and 34.5 cm bed height. The pH of an aliquot
of Protein A pool was titered
to 5.6 with 1.5 M Tris base. The conductivity of the pool was reduced by the
addition of an equal volume of
sterile water for injection (SWFI).
Chromatography: The chromatography runs for this study were performed with
Pharmacia's
UNICORNTM FPLC system. The equilibration, load, and initial wash steps were
performed at a linear flow rate
of 200 cm/h. All chromatography steps were performed at a linear flow rate of
100 cm/h. The sequence of
chromatography steps are defined in Table 1. A total of six chromatography
runs were performed with load
densities of 15, 20, 25, 30, 35, and 40 mg of rhuMAb HER2 per mL of SPSFF
resin.
CA 02753381 2011-09-13
WO 99/57134 PCTIUS99/09637
Table I - Chromatography Steps'
Chromatography Step Buffer Approximate Endpoint
2
Equilibration: Part 1 0.025 M MES 11.0 M NaCl, pH 5.6 2 CV
Equilibration: Part 2 0.025 M MES / 0.05 M NaCl. pH 5.6 pH: 5.6:t 0.1
Cond.: 5.8 t 0.2 mmhos
Load Adjusted Protein A Pool As Required
Wash 1 0.025 M MES / 0.05 M NaCI, pH 5.6 1.5 CV
Wash 2 0.025 M MES 10.07 M NaCl, pH 5.6 Apex of Peak
Wash 3 0.025 M MES / 0.05 M NaCl. pH 5.6 2 CV
Elution: Prepool 0.025 M MES 10.095 M NaCl, pH 5.6 To Leading Shoulder's
Inflection Point (-1.2 CV)
Elution: Pool 0.025 M MES 10.095 M NaCl, pH 5.6 5 CV
Regeneration 0.025 M MES / 1.0 M NaCl, pH 5.6 2 CV
1. The equilibration of the resin was performed in manual mode; the remaining
steps were executed
from a Pharmacia Unicorn Program.
2. CV = column volume(s).
Total Protein: The protein concentration of each chromatography fraction (flow
through, wash steps,
elution prepool, elution pool, and regeneration) was determined by
spectrophometric scans of each sample. The
results were used to calculate product recovery yields. The extinction
coefficient for rhuMAb HER2 is 1.45.
Calculations used to derive the results (Figure 4) are:
Protein Concentration (mg/mL) = 280 nm x Dilution Factor
1.45
Protein Mass (mg) in Each Fraction = Protein Concentration (mg/mL) x Fraction
Volume (mL)
Fraction Mass (mg)
Yield (%) = x 100
Total Mass (mg)
Determination of rhuMAb HER2 Antibody Variants (CSx HPIEX): The rhuMAb HER2
SPSFF
chromatography column resolves antibody variants. Fractions from each of the
study chromatographies were
tested for the relative amount of variant antibody by CSx HPIEX
chromatography. A BAKERBOND WIDE-
PORET"" CSx HPIEX column (4.6 x 250mm) was run at I mUmin at 55 C. The mobile
phase was formed from
a tertiary gradient (Table 2).
21
CA 02753381 2011-09-13
WO 99/57134 PCT/US99/09637
Table 2 - Gradient Scheme
rime (min) % A %a /. C
0 - Initial Conditions 49 1 50
10.0 40 10 50
50.0 33 17 50
50.2 49 1 50
70.0 49 1 50
The column is run at I mUmin at 55 C.
The A buffer was 0.025 M MES, pH 5.9; the B buffer was I M Ammonium Acetate,
pH 7.0; and the
C solution was sterile water for injection. The column was equilibrated with
the gradient's initial conditions (49
% A. 1% B; and 50% C) and 200 tI of sample, diluted with SWFI and containing <
300 g protein, was injected.
Each resulting chromatogram was integrated to determine the percent area of
each peak for each fraction (Table
3 and Figure 5).
Table 3 - CSx HPIEX analysis of rhuMAb HER2
CSx Peak rhuMAb HER2 Variant
a & b Light Chain: Asn ---*Asp deamidation
- and -
Other unidentifiable variation by tryptic map
1 Light Chain: Asn -, Asp deamidation
3 Fully Processed Antibody
102
4 Heavy Chain: Asp -+ Iso-Asp
- and / or -
450
Heavy Chain: An Additional Lys
102
Others Heavy Chain: Asp -- Succinimide
- and/or -
Multiple permutations found in Peaks 1 and 4
10 Chromatograms Compared.- The absorbance data (AU 280 nm) from each
chromatography file was
exported from Unicorn in ASCII format. The data from the 0.025 M MES / 0.07 M
NaCl. pH 5.6 wash was
translated into Excel format and copied into KALEIDAGRAPHTM. Using
KALEIDAGRAPHTM, the wash profiles
were overlaid (Figure 6) and compared to each other.
RESULTS AND DISCUSSION-
15 Deamidated and other acidic variants of rhuMAb HER2 were produced when the
antibody was made
22
CA 02753381 2011-09-13
WO 99/57134 PCT/US99/09637
by recombinant DNA technology (see e.g., CSx peaks a, b and I in Figure 5).
The deamidated and other acidic
variants constituted about 25% (calculated as area under the integrated curve
or profile obtained by CSx
chromatography) of the composition obtained from the initial Protein A
chromatography step. It was discovered
that the ion exchange method described herein could be used to substantially
reduce the amount of deamidated
and other acidic variants in the anti-HER2 composition, i.e. to about 13% or
less (i.e. the amount of acidic
variants in the preparation subjected to cation exchange chromatography as
described herein was decreased by
about 50% or more).
An absorbance trace from a cation exchange column run performed as described
above is shown in
Figure 3. This method resolved a deamidated variant of anti-HER2 antibody that
differed only slightly from
nondeamidated anti-HER2 antibody. The increase in conductivity from the
initial conditions to the intermediate
wash began to elute the deamidated anti-HER2 antibody. However, continued
washing at this conductivity was
found to elute nondeamidated anti-HER2 antibody, resulting in a loss of
product. Proceeding directly from the
intermediate buffer to the elution buffer was observed to result in either an
unacceptably low removal of
deamidated anti-HER2 antibody from the product if pooling began early or
unacceptably low yields of anti-HER2
antibody product if pooling was delayed until the deamidated anti-HER2
antibody was reduced. It was discovered
that by going back to lower conductivity as used initially, the elution of
deamidated anti-HER2 antibody
continued, without significant anti-HER2 antibody product elution.
The effect of rhuMAb HER2 load on (a) buffer requirements, (b) product
recovery in the pool and (c)
product quality in the pool was evaluated.
At load densities of 15 mg/mL up to 35 mg/mL. the product yield in the elution
pool is approximately
75%. For the load density of 40 mg/mL, the product yield in the pool dropped
to 65% (Figure 4). This reduced
recovery in the pool is largely attributed to an increased antibody in the two
wash steps (at 70 mM NaCl and SO
mM NaCl, respectively).
The quality of rhuMAb HER2 in all the elution pools is equivalent as
determined by CSx HPIEX
analysis (Figure 5). Compared to the load material: there is an enrichment of
the nondeamidated antibody (Peak
3), no change in the amount [so-Asp 102 or Lys 450 antibody (Peak 4), and a
reduction of the amount of Asp30
deamidated antibody (Peaks a. b, 1 and others).
The quality of rhuMAb HER2 in these cation pools is improved through the
intermediate wash step. As
the mass of rhuMAb HER2 bound to the resin increases, the intermediate buffer
volume consumption needed to
reach the apex of the 280 nm peak decreases. The buffer volume required for a
40 mg/mL load density is
approximately 2.5 column volumes. The buffer volume required for a 15 mg/mL
load density is approximately
15 column volumes. The exact increase of buffer requirement is not linearwith
the 5 mg/mL incremental changes
between these two extremes. The greatest increase is seen between the load
densities of 20 mg/mL and 15 mg/mL.
Here the requirement doubles from 7.5 column volumes to the previously
mentioned 15 column volumes of
buffer. If the apex of the 70 mM NaCl wash peak is reached, however, the
product quality is equivalent for any
of load densities examined.
This study determined how much rhuMAb HER2 can be loaded onto the SPSFF resin.
Between the
ranges of 15 to 40 mg of antibody per mL of resin, there is no difference in
the quality of rhuMAb HER2
23
CA 02753381 2011-09-13
WO 99/57134 PCTIUS99/09637
recovered in the elution pool. The quantity of rhuMAb HER2 recovered, however,
is reduced by approximately
10% when the resin is loaded with greater than 35 mg/mL. For consistent yields
it is recommended that 35 mg/mL
be set as the maximum load for manufacture of rhuMAb HER2. Furthermore, due to
the substantial increase in
the 70 mM NaCl wash volume requirement between the 20 and 15 mg/mL, it is
recommended that 20 mg/mL
be set as the minimal load for manufacture of rhuMAb HER2.
24