Note: Descriptions are shown in the official language in which they were submitted.
CA 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
TITLE OF THE APPLICATION
INHIBITORS OF HEPATITIS C VIRUS REPLICATION
FIELD OF THE INVENTION
The present disclosure relates to antiviral compounds that are useful as
inhibitors
of hepatitis C virus (HCV) replication. The compounds are expected to act on
HCV NS5A (non-
structural 5A) protein. Compositions comprising such compounds, the use of
such compounds
for treating HCV infection and/or reducing the likelihood or severity of
symptoms of HCV
infection, methods for inhibiting the function of the NS5A non-structural
protein, and methods
for inhibiting HCV viral replication and/or viral production are also
provided.
BACKGROUND OF THE INVENTION
Hepatitis C virus (HCV) infection is a major health problem that leads to
chronic
liver disease, such as cirrhosis and hepatocellular carcinoma, in a
substantial number of infected
individuals. Current treatments for HCV infection include immunotherapy with
recombinant
interferon-a alone or in combination with the nucleoside-analog ribavirin.
Several virally-encoded enzymes are putative targets for therapeutic
intervention,
including a metalloprotease (NS2-3), a serine protease (NS3, amino acid
residues 1-180), a
helicase (NS3, full length), an NS3 protease cofactor (NS4A), a membrane
protein (NS4B), a
zinc metalloprotein (NS5A) and an RNA-dependent RNA polymerase (NS5B).
One identified target for therapeutic intervention is HCV NS5A non-structural
protein, which is described, for example, in Seng-Lai Tan & Michael G. Katze,
How Hepatitis C
Virus Counteracts the Interferon Response: The Jury Is Still Out on NS5A, 284
VIROLOGY 1-12
(2001); and in Kyu-Jin Park et al., Hepatitis C Virus NS5A Protein Modulates c-
Jun N-terminal
Kinase through Interaction with Tumor Necrosis Factor Receptor-associated
Factor 2, 278(33)
J. Mo. CHEM. 30711 (2003). A non-structural protein, NS5A is an essential
component for viral
replication and assembly. Mutations in NS5A at or near known sites of
phosphorylation can
affect the ability for high-level replication in cell-culture systems,
suggesting an important role
for NS5A phosphorylation in viral replication efficiency. Inhibitors of the
phosphorylation of
NS5A can lead to reduced viral RNA replication.
NS5A is a zinc metalloprotein organized into three discreet domains. NS5A
localizes to the membrane-associated site of RNA synthesis via an N-terminal
amphipathic a-
-1 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
helix anchor. The crystal structure of domain I demonstrates that NS5A can
exist as a dimer,
with a large putative RNA binding groove located at the interface of the
monomers. Timothy L.
Tellinghuisen et all, Structure of the zinc-binding domain of an essential
component of the
hepatitis C viral replicase, 435(7040) NATURE 374 (2005). Robert A. Love et
at., Crystal
Structure of a Novel Dimeric Form of NS5A Domain I Protein From Hepatitis C
Virus, 89(3) J.
VIROLOGY 4395-403 (2009). The interaction of NS5A with RNA is thought to be
critical for the
function of this protein in RNA replication. No structural information has yet
been obtained for
domains II or III. Recent genetic mapping has shown that although some
residues in domain II
are essential for RNA replication, many portions of domain II and all of
domain III are
dispensable. Timothy L. Tellinghuisen et all, Identification of Residues
Required for RNA
Replication in Domains II and III of the Hepatitis C Virus NS5A Protein, J.
VIROLOGY 1073
(2008). Mutations constructed within domain III result in virus that can
maintain RNA
replication but that produces lower titers of infectious virus in cell
culture, demonstrating a
second distinct role for NS5A after RNA replication has occurred. Timothy L.
Tellinghuisen et
al., Regulation of Hepatitis C Virion Production via Phosphorylation of the
NS5A Protein, 4(3)
PLOS PATHOGENS e1000032 (2008); Nicole Appel et al., Mutational Analysis of
Hepatitis C
Virus Nonstructural Protein 5A: Potential Role of Differential Phosphorylation
in RNA
Replication and Identification of a Genetically Flexible Domain, 79(5) J.
VIROLOGY 3187
(2005). NS5A, unlike the other non-structural proteins, can be trans-
complemented, consistent
with functions outside of the viral replicase. The interaction of NS5A with
numerous host-
signaling pathways has been described (Michael J. Gale Jr. et all, Evidence
That Hepatitis C
Virus Resistance to Interferon Is Mediated through Repression of the PKR
Protein Kinase by the
Nonstructural 5A Protein, 230 VIROLOGY 217 (1997); Andrew Macdonald & Mark
Harris,
Hepatitis C virus NS5A: tales of a promiscuous protein, 85 J. GEN. VIROLOGY
2485 (2004).),
suggesting this protein may modify the host cell environment to a state
favorable for the virus,
events that may require a form of NS5A dissociated from the replication
complex.
There is a clear and long-felt need to develop effective therapeutics for
treatment
of HCV infection_ Specifically, there is a need to develop compounds that are
useful for treating
HCV-infected patients and compounds that selectively inhibit HCV viral
replication.
- 2 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
SUMMARY OF THE INVENTION
The present disclosure relates to novel compounds of formula (I) and/or
pharmaceutically acceptable salts, hydrates, solvates, prodrugs or isomers
thereof. These
compounds are useful, either as compounds or their pharmaceutically acceptable
salts (when
appropriate), in the inhibition of HCV (hepatitis C virus) NS5A (non-
structural 5A) protein, the
prevention or treatment of one or more of the symptoms of HCV infection, the
inhibition of
HCV viral replication and/or HCV viral production, and/or as pharmaceutical
composition
ingredients. As pharmaceutical composition ingredients, these compounds, which
includes
reference to hydrates and solvates of such compounds, and their salts may be
the primary active
therapeutic agent, and, when appropriate, may be combined with other
therapeutic agents
including but not limited to other HCV antivirals, anti-infectives,
immunomodulators, antibiotics
or vaccines.
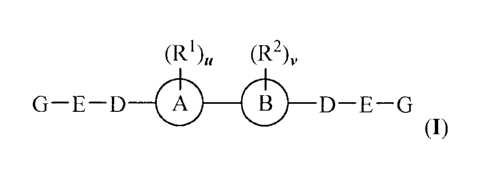
More particularly, the present disclosure relates to a compound of formula
(I):
(R1)õ (R2)v
G¨E¨D 0 0 D¨E¨G
0)
and/or a pharmaceutically acceptable salt thereof, wherein:
is chosen from the group consisting of 9-membered bicyclic aryl ring
systems that contain from 0 to 4 heteroatoms independently chosen from the
group consisting of
N, 0 and S, and that are substituted on C or N atoms by u substituents
each RI is independently chosen from the group consisting of hydrogen,
halogen, -0R3a, -CN, -(CH2)0_6C(0)R3, -CO2R3a, -C(0)N(R3a)2, -SR, -S(0)R3a, -
S(02)R3a,
-(CH2)0_6N(R3a)2, -N(R3a)S 02R3 a, -NR3 a)CO2R3 a, -N(R3a)C(0)R3, -N(R3a)COR3
a,
-N(R3a)C(0)N(R3 a), C1_6alkyl, C3_8carbocyc1e containing from 0 to 3
heteroatoms chosen from N,
0 and S, and phenyl, and the
C3_8carbocycle and phenyl are substituted by from 0 to 3 substitutents
independently
chosen from the group consisting of hydrogen, halogen, -0R3a, -CN, -CO2R3a, -
C(0)N(R3a)2,
-N(R3a)2, -N(R3a)CO2R3a, -SR3a, -S(0)R3a, -S(02)R3a, -N(R3a)S02R3a, -
N(R3a)CO2R3a,
-N(R3a)C(0)N(R3a), C1,6alkyl, -0-C1_6alkyl, -S-C1_6alkyl, and C3_8cycloalkyl,
u is from 0 to 4,
- 3 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
each R3 is independently chosen from the group consisting of hydrogen,
C1.6alky1, -0H, -0-C1_6alkyl and Cmcycloalkyl, and
each R3' is independently chosen from the group consisting of hydrogen,
C1_6alkyl and C3.8cycloalkyl;
(111) is a group chosen from the group consisting of
(a) and
(b) aryl ring systems B' chosen from the group consisting of:
(i) 5- to 7-membered monocyclic ring systems and
(ii) 8- to 10-membered bicyclic ring systems,
and the aryl ring systems B' containing from 0 to 4 heteroatoms
independently chosen from the group consisting of N, 0 and S, and substituted
on C or N atoms
by v substituents R2,
each R2 is independently chosen from the group consisting of
hydrogen, halogen, -Ole', -CN, -CO2R4a, -C(0)R4a, -C(0)N(R4a)2, -N(R4a)2, -
N(R4a)COR4,
-N(R4a)CO2R4a, -
N(R4a)C(0)N(R4a), -N(R4a)S02R4a, -SR4a, -S(0)R4a, -S(02)R4a, Cmalkyl
substituted by from 0 to 4 R4 and C3.8cyc1oalkyl substituted by from 0 to 4
R4,
v is from 0 to 4,
each R4 is independently chosen from the group consisting of
hydrogen, -OH, C1_6alkyl and C3_8cycloalkyl;
each R4a is independently chosen from the group consisting of
hydrogen, C1_6alkyl and C3..gcyc1oalky1;
RI and R2 may be taken together with 0
and to form a 5- to
9-membered carbo cyclic ring containing 1 or 2 heteroatoms independently
chosen from the
group consisting of N, 0 and S;
each D is a group independently chosen from the group consisting of:
(a) a single bond,
(b) -C(0)N(R5)-,
(c) -N(R5)C(0)-, and
(d) a 5- or 6-membered aryl ring system D' containing from 0 to 4
heteroatoms independently chosen from the group consisting of N, 0 and S, and
substituted on C
or N atoms by from 0 to 2 substituents R5,
- 4 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
each R5 is independently chosen from the group consisting of
hydrogen, halogen, -0R6, -CN, -0O2R6, -C(0)N(R6)2, -N(R6)2, -N(R6)COR6, -SR6, -
S(0)R6,
-S(02)R6, -N(R6)S02R6, -NCO2R6, -NC(0)N(R6)2, Ci_6alkyl substituted by from 0
to 3 R6 and
C3_8cycloalkyl substituted by from 0 to 3 R6, and
each R6 is independently chosen from the group consisting of
hydrogen, C1_6a1kyl and C3_8cycloalkyl;
each E is a group independently chosen from the group consisting of:
(a) a single bond,
(b) -(C(R7)2)0_2NR7C(0)00_1-, and
(c) a pyrrolidinyl derivative chosen from the group consisting of:
3\1 R8'
R8c
Raa Rao
Rs.
Raa (R9)o-4 (R9)0-4 j
Rab R8b and
is a bivalent group chosen from -C(0)-, -0O2- and
-C(0)N(R7)-,
J is a fused ring system chosen from the group consisting of 3- to
7-membered carbocycles and 5- or 6-membered aryl rings containing from 0 to 4
heteroatoms
independently chosen from the group consisting of N, 0 and S, and substituted
on C or N atoms
by substituents R9,
each R8' is independently chosen from the group consisting of
hydrogen, halogen, -OH, -0C1_6alkyl and C16alkyl, or two R8a may be taken
together to form
oxo,
each R8b is independently chosen from the group consisting of
hydrogen, halogen, -OH, -0C1_6alkyl and C16alkyl, or two R8b may be taken
together to form
oxo,
each R8' is independently chosen from the group consisting of
hydrogen and C1_6alkyl,
or any two groups selected from R8a, R8b and R8' may be taken
together to form a spiro-bicyclic or bridged bicyclic ring;
each R9 is independently chosen from the group consisting of
hydrogen, halogen, C1.6alkyl, -0-C1_6alkyl, -S-C1_6a1ky1, -NH-C1_6alky1 and -
NHC(0)-Ci_6alkyl,
- 5 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
each R7 is independently chosen from the group consisting of hydrogen,
C1_6a1kyl and phenyl, and the Ci_6alkyl and phenyl are substituted by from 0
to 3 substitutents
independently chosen from the group consisting of hydrogen, halogen,
C1_6alkyl, -0-C1_6alkyl
and -S-Ci_6alkyl; and
each G is independently chosen from the group consisting of:
(a) hydrogen,
(b) -OR1 a,
(c) -CN,
(d) -CO2R10a,
(e) -C(0)N(R1 )2,
(f) -SR1 a,
(g) -S(0)R1 a,
(h) -S(02)RI",
(1) -N(R10)2,
(j) -N(R1))S02R ma,
(k) -NCO2RI
(1) -NC(0)N(R18)2,
(m) C1_6alkyl having 0 to 4 substituents R11,
each R11 is independently chosen from the group consisting of:
(i) -OH,
(ii) -N(R10)2,
(iii) =NR1 ,
(iv) -0-C1_6alkyl,
(v) -C(0)R10
,
(vi) -S-C1_6a1ky1,
(vii) -S02-Ci.6alkyl,
(viii) 3- to 8-membered carbocycles containing from 0 to
3 heteroatoms independently chosen from the group consisting of N, 0 and S,
and having from 0
to 3 substitutents R12 on N or C atoms, and each R12 is independently selected
from the group
consisting of hydrogen, halogen, C1_6a1ky1 having from 0 to 3 substituents
chosen from R1 ,
-0-C1_6a1ky1, -S-C1_6alkyl, -0R10', -CN, -C(0)R10, -CO2R1 a, -C(0)N(R1 )2, -
Sea, -S(0)Rma,
- 6 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
-S(02)ea, -N(R10)S02RICia, -NCO2ea, -NC(0)N(R1 )2 and-N(R1 )2, or two R12 are
taken
together to form oxo, and
(ix) 5- or 6-membered aryl containing from
0 to 3
heteroatoms independently chosen from the group consisting of N, 0 and S, and
having from 0
to 3 substitutents R13 on N or C atoms, and each R13 is independently selected
from the group
consisting of hydrogen, halogen, Ci_6alkyl, -0-C1_6a1ky1 and 3- to 8-membered
carbocycles
containing from 0 to 3 heteroatoms independently chosen from the group
consisting of N, 0 and
S,
(n) 3- to 8-membered carbocycles containing from 0 to 3 heteroatoms
independently chosen from the group consisting of N, 0 and S, and having from
0 to 3
substitutents R1 on N or C atoms; and
(o) aryl ring systems G chosen from the group consisting of:
(i) 5- to 7-membered monocyclic ring systems and
(ii) 8- to 10-membered bicyclic ring systems,
and the aryl ring systems G' containing from 0 to 4 heteroatoms
independently chosen from the group consisting of N, 0 and S, and substituted
on C or N atoms
by 0 to 3 substitutents R1 ;
each R1 is independently chosen from the group consisting of
(i) hydrogen,
(ii) -CN,
(iii) Ci_6alkyl,
(iv) -0-Co_6alkyl,
(v) -S-00.6a1ky1,
(vi) C1_6a1ky1-0-R14,
(vii) -C(0)R14,
(viii) -0O2R14,
(ix) -SO2R14,
(x) -N(R14)2,
(xi) -N(R14)S02R14,
(xii) -NCO2R14,
(xiii) -NC(0)N(R14)2, and
- 7 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
(xiv) 3- to 8-membered carbocycles containing from 0 to 3
heteroatoms independently chosen from the group consisting of N, 0 and S.
or two RI may be taken together to form oxo;
each RI ' is independently chosen from the group consisting of
(i) hydrogen,
(ii) -CN,
(iii) C1_6alkyl,
(iv) Cioalkyl-O-R14,
(v) -C(0)R14,
(vi) -CO2R14,
(vii) -SO2R14,
(x) -N(RI4)2,
(xi) -N(R14)S02R14,
(X11) -NCO2R14,
(xiii) -NC(0)N(R14)2, and
(xiv) 3- to 8-membered carbocycles containing from 0 to 3
heteroatoms independently chosen from the group consisting of N, 0 and S,
and two RI or Rma groups can be taken together with the N to which they
are attached to form a ring, which may be substituted by from 0 to 3
substituents RI4, and
each RI4 is independently chosen from the group consisting of hydrogen,
C1_6alkyl, C3_8cycloalkyl, -(CH2)0,3C3_8cycloalkyl and phenyl.
The present invention also includes pharmaceutical compositions containing a
compound of the present invention and methods of preparing such pharmaceutical
compositions.
The present invention further includes methods of treating or reducing the
likelihood or severity
of HCV infection, methods for inhibiting the function of the NS5A protein, and
methods for
inhibiting HCV viral replication and/or viral production.
Other embodiments, aspects and features of the present invention are either
further described in or will be apparent from the ensuing description,
examples and appended
claims.
- 8 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
DETAILED DESCRIPTION OF THE INVENTION
The present invention includes compounds of formula (I) above, and
pharmaceutically acceptable salts thereof. The compounds of formula (I) are
HCV NS5A
inhibitors.
A first embodiment of the invention relates to compounds having structural
formula (I):
(R)u (R2)v
G¨E¨D 0 D¨E¨G
and/or a pharmaceutically acceptable salt thereof, wherein:
(--) is chosen from the group consisting of 9-membered bicyclic aryl ring
systems that contain from 0 to 4 heteroatoms independently chosen from the
group consisting of
N, 0 and S, and that are substituted on C or N atoms by u substituents RI,
each RI is independently chosen from the group consisting of hydrogen,
halogen, -0R3a, -CN, -C(0)R3, -CO2R3a, -C(0)N(R3a)2, -SR3a, -S(0)R3a, -
S(02)R3a,
-(CH2)0.6N(R3')2, -N(R3a)S02R3a, -N(R3a)CO2R31, -N(R3a)C(0)R3, -N(R3a)COR3a,
-N(R3a)C(0)N(R3a), C3_6a1ky1, Cmcarbocycle containing from 0 to 3 heteroatoms
chosen from N,
0 and S, and phenyl, and the Ci_6alkyl, C34carbocycle and phenyl are
substituted by from 0 to 3
substitutents independently chosen from the group consisting of hydrogen,
halogen, -0R3a, -CN,
-CO2R3a, -C(0)N(R3a)2, -N(R3a)2, -N(R3')CO2R3a, -SR3a, -S(0)R3a, -S(02)R3', -
N(R3a)S02R3a,
-N(R3a)CO2R3a, -N(R3a)C(0)N(R3a), Ci_6alkyl, -0-Ci_6alkyl and -S-Ci_olkyl,
u is from 0 to 4,
each R3 is independently chosen from the group consisting of hydrogen,
Ci_6a1kyl, -OH, -0-C1_6alkyl and C3 _8cycloalkyl, and
each R3' is independently chosen from the group consisting of hydrogen,
Ci_6alkyl and C3_8cycloalkyl;
0 is a group chosen from the group consisting of
(a) and
(b) aryl ring systems B6 chosenfrom the group consisting of:
(i) 5- to 7-membered monocyclic ring systems and
(ii) 8- to 10-membered bicyclic ring systems,
- 9 -
CA 02756172 2013-05-24
and the aryl ring systems B' containing from 0 to 4 heteroatorns
independently chosen from the group consisting of N, 0 and S. and substituted
on C or N atoms
by v substituents R2,
each R2 is independently chosen from the group consisting of
hydrogen, halogen, -OR", -CN, -CO2R48, -C(0)N(R4a)2, -N(R4a)2,
-N(R4a)COR4, -N(R4a)CO2R43, -N(R4a)C(0)N(R4a), _N(R4a)so2R4a,_SR4a,_s(0)R4a,
-S(02)R4a, C1_6alkyl substituted by from 0 to 4 R4 and C3.8cycloalkyl
substituted by from 0 to 4
R4,
v is from 0 to 4,
each R4 is independently chosen from the group consisting of
hydrogen, -OH, Ci_6alkyl and C3.8cycloalkyl;
each R4a is independently chosen from the group consisting of
hydrogen, Ci.6alkyl and C3_8cycloalkyl;
RI and R2 may be taken together with 0
and to form a 5-
to
9-membered carbocyclic ring containing 1 or 2 heteroatoms independently chosen
from the
group consisting of N, 0 and S and wherein said 5- to 9-membered carbocyclic
ring is optionally
substituted with C1_6alkyl, C3_8cyeloalkyi or phenyl;
each D is a group independently chosen from the group consisting of:
(a) a single bond,
(b) -C(0)N(R5)-,
(c) -N(R5)C(0)-, and
(d) a 5- Or 6-membered aryl ring system D' containing
from 0 to 4
heteroatoms independently chosen from the group consisting of N, 0 and S, and
substituted on C
or N atoms by from 0 to 2 substituents R5,
each R5 is independently chosen from the group consisting of
hydrogen, halogen, -0R6, -CN, -0O2R6, -C(0)N(R6)2, -N(R6)2, -N(R6)COR6, -SR6, -
S(0)R6,
-S(02)R6, -N(R6)S02R6, -NCO2R6, -NC(0)N(R6)2, C1_6alkyl substituted by from 0
to 3 R6 and
C3_8cycloalkyl substituted by from 0 to 3 R6, and
each R6 is independently chosen from the group consisting of
hydrogen, Ci_6alkyl and C3,8cycloalkyl;
each E is a group independently chosen from the group consisting of:
(a) a single bond,
-10-
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
(b) -(C(R7)2)0_2NR7C(0)004-, and
(c) a pyrrolidinyl derivative chosen from the group consisting of:
D ,I¨G D ,I¨G
D ,I¨G
N
R-c
Rac
R8a R e8 -õ
Rsb
R8A b Rsc cl.") 9
..(R )04 (10)0-4 j Rs
and
I is a bivalent group chosen from -C(0)-, -0O2- and
-C(0)N(R7)-,
J is a fused ring system chosen from the group consisting of 3- to
7-membered carbocycles and 5- or 6-membered aryl rings containing from 0 to 4
heteroatoms
independently chosen from the group consisting of N, 0 and S, and substituted
on C or N atoms
by substituents R9,
each Rga is independently chosen from the group consisting of
hydrogen, halogen, -OH, -0C1_6a1ky1 and Ci_6alkyl, or two Rga may be taken
together to form
oxo,
each Rgb is independently chosen from the group consisting of
hydrogen, halogen, -OH, -0C1_6alkyl and C1_6alkyl, or two R8b may be taken
together to form
oxo,
each Rgc is independently chosen from the group consisting of
hydrogen and C1_6alky1,
or any two groups selected from Rga, Rgb and Rsc may be taken
together to form a spiro-bicyclic or bridged bicyclic ring;
each R9 is independently chosen from the group consisting of
hydrogen, halogen, C1_6a1ky1, -NH-Ci_6alkyl and -NHC(0)-
C1_6alkyl,
each R7 is independently chosen from the group consisting of hydrogen,
C1_6a1ky1 and phenyl, and the Ci_6alkyl and phenyl are substituted by from 0
to 3 substitutents
independently chosen from the group consisting of hydrogen, halogen,
C1_6alkyl, -0-C1_6alkyl
and -S-Ci_6a1kyl; and
each G is independently chosen from the group consisting of:
(a) hydrogen,
(b) -OR1 a,
(c) -CN,
(d) -CO2R1c4,
- 11 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
(e) -C(0)N(R1 )2,
(f) -SR1 a,
(g) -S(0)R1 a,
(h) -S(02)R1 a,
(i) -N(RI0)2,
-N(R1 )S02R1 a,
(k) -NCO2R1 a,
(1) -NC(0)N(R1 )2,
(m) C1,6alkyl having 0 to 4 substituents R11,
each R11 is independently chosen from the group consisting of:
(i) -011,
(ii) -N(R1 )2,
(iii) =NR1 ,
(iv) -0-C1_6alkyl,
(v) -C(0)R1 ,
(vi) -S-C1_6alkyl,
(vii) -S02-C1_6alkyl,
(viii) 3- to 8-membered carbocycles containing from 0 to
3 heteroatoms independently chosen from the group consisting of N, 0 and S.
and having from 0
to 3 substitutents R12 on N or C atoms, and each R12 is independently selected
from the group
consisting of hydrogen, halogen, C1_6alkyl having from 0 to 3 substituents
chosen from R1 ,
-0-C1.6a1kyl, -S-C6alkyl, -0R1 a, -CN, -C(0)R1 , -CO2Rwa, -C(0)N(R1 )2, -SR1
a, -S(0)Rma,
-S(02)Rwa, -N(R1 )S02R113a, -NCO2R1 a, -NC(0)N(R1 )2 and-N(R1 )2., or two R12
are taken
together to form oxo, and
(ix) 5- or 6-membered aryl containing from 0 to 3
heteroatoms independently chosen from the group consisting of N, 0 and S, and
having from 0
to 3 substitutents R13 on N or C atoms, and each R13 is independently selected
from the group
consisting of hydrogen, halogen, C1_6a1ky1, -0-C1.6a1ky1 and 3- to 8-membered
carbocycles
containing from 0 to 3 heteroatoms independently chosen from the group
consisting of N, 0 and
S,
-12-
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
(n) 3- to 8-membered carbocycles containing from 0 to 3 heteroatoms
independently chosen from the group consisting of N, 0 and S, and having from
0 to 3
substitutents Rl on N or C atoms; and
(o) aryl ring systems G' chosen from the group consisting of:
(i) 5- to 7-membered monocyclic ring systems and
(ii) 8- to 10-membered bicyclic ring systems,
and the aryl ring systems G' containing from 0 to 4 heteroatoms
independently chosen from the group consisting of N, 0 and S, and substituted
on C or N atoms
by 0 to 3 substitutents R1');
each RI is independently chosen from the group consisting of
(i) hydrogen,
(ii) -CN,
(iii) Ci_6alkyl,
(iv) -0-Co_oalkyl,
(v)
(vi) C1_6alky1-0-R14,
(vii) -C(0)R14,
-CO2R14,
(ix) -SO2R14,
(x) -N(R14)2,
(xi) -N(R14)S021Z14,
(xii) -NCO2R14,
(xiii) -NC(0)N(R14)2, and
(xiv) 3- to 8-membered earbocycles containing from 0 to 3
heteroatoms independently chosen from the group consisting of N, 0 and S,
or two RI may be taken together to form oxo;
each ea is independently chosen from the group consisting of
(i) hydrogen,
(ii) -CN,
(iii) Ci_oalkyl,
(iv) Ci..6alkyl-0-R14,
(v) -C(0)R14,
- 13 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
(vi) -0O2R14,
(vii) -S02R14,
(x) -N(R14)2,
(xi) -N(RI4)S02R14,
(xii) -NCO2R14,
(xiii) -NC(0)N(R14)2, and
(xiv) 3- to 8-membered carbocycles containing from 0 to 3
heteroatoms independently chosen from the group consisting of N, 0 and S,
and two R1 or R1'1" groups can be taken together with the N to which they
are attached to form a ring, which may be substituted by from 0 to 3
substituents R14, and
each R14 is independently chosen from the group consisting of hydrogen,
C/_6alkyl, C34cycloalkyl, -(CH2)0_3C3_8cycloalkyl and phenyl. In this
embodiment, all other
groups are as provided in the general formula above.
In a second embodiment of the invention, is chosen from the group
(12.1)o-47 I
consisting of X Y , where each X is
independently chosen from the group
X\ CR1 NR N NR1
CR1
7 ) e
consisting of CR1 and N, Y is chosen from the group consisting of NR', CR1,
N 0 ,
NO
0 N
and s , each RI is independently chosen from the group consisting of hydrogen,
halogen, -0R3a, -CN, -C(0)R3, -0O2R3a, -C(0)N(R3a)2, -SR3a, -S(0)R3a, -
S(02)R3a,
-(CH2)0-6N(R32)2, -N(R3a)S02R3a, -N(R3a)CO2R3a, -N(R3a)C(0)R3, -N(R3a)C0R3a,
-N(R3a)C(0)N(R3a), C1_6alkyl, C3_8carbocycle containing from 0 to 3
heteroatoms chosen from N,
0 and S, and phenyl, and the Ci.6alkyl, C3,8carbocycle and phenyl are
substituted by from 0 to 3
substitutents independently chosen from the group consisting of hydrogen,
halogen, -0R3a, -CN,
-CO2R3a, -C(0)N(R3a)2, -N(R3a)2, -N(R3a)CO2R3a, -SR3a, -S(0)lea, -S(02)R3a, -
N(R3a)S02R3a,
-N(R3a)CO2R3, -N(R3a)C(0)N(R3a), Ci_6alkyl, -0-Ci_6alkyl and -S-Ci_6alkyl,
each R3 is
independently chosen from the group consisting of hydrogen, Ci_6alkyl, -OH, -0-
Ci_6a1kyl and
Cmcycloalkyl, and each R3 is independently chosen from the group consisting of
hydrogen,
-14-
:A 02756172 2011 09 21
WO 2010/111483 PCT/US2010/028653
Ci_6alkyl and C3,8cycloalkyl. In all aspects of this embodiment, all other
groups are as provided
in the general formula above or in the first embodiment above.
In a first aspect of the second embodiment of the invention, is chosen from
RI i RI RI
/ * \ 1 /10
N feN
" / * \ I 1---
- i
N \
the group consisting of H RI / 0 .----0 /
RI i 1 pr
t
N- /........,,N,,
f / SI IN? 1 /....,....õ-N
I \ 1 * >-i
µ
1\1----() H 7 RI / H / RI /
/ / / 11
/ 0
0 No>._
\ 1 0 N NH
S N , and 4111 i
_______________________ 1 , where 0 is substituted
by from 0 to 3 additional Rl, which are as provided above.
In a second aspect of the second embodiment, 0 is chosen from the group
RI / /
RI
/
/ N
N N
N \ N
consisting of H / RI H / RI H
/
H
N
--"---.1.,----- N >---- 1
1
RI and 0 N , where 0 is
substituted by from 0 to 3 additional RI,
/
al1
which are as provided above. In preferred instances of this aspect, 0 is RI
,
where 0 is substituted by from 0 to 3 additional RI, which are as provided
above.
In a third aspect of the second embodiment, 0 is chosen from the group
RI RI
/ , W
N / N
0 \icN ___
1 .,.(.....,.,...rµ)_ 1 ,s' õ..--- 1V \ 0 )
consisting of 0 ,
0\
/ 0 1-1
and N , where 0 is
substituted by from 0 to 3 additional RI, which are as
- 15 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
RI
/ \
provided above. In preferred instances of this aspect, 0 is 0
, where 0 is
substituted by from 0 to 3 additional RI, which are as provided above.
In further aspects of the second embodiment, each Rl is chosen from the group
consisting of hydrogen, halogen, -CN and Ci_6alkyl. In particular, each RI is
chosen from the
group consisting of hydrogen, fluorine and -CN.
In a third embodiment of the invention, C1 is chosen from the group consisting
of
phenyl, pyridinyl, pyrazinyl, pyrimidyl, 1,2,4-triazinyl, pyridazinyl, thiazyl
and 9-
membered bicyclic ring systems that contain from 1 to 3 heteroatoms
independently chosen from
the group consisting of N, 0 and S, v is from 0 to 4, each R2 is independently
chosen from the
group consisting of hydrogen, halogen, -0R4', -CN, -CO2R4a, -C(0)N(R4a)2, -
N(102,
-N(R4a)CO2R4a, -SR4a, -S(0)R4a, -S(02)R4a, -N(R4a)S02R4a, -N(R4a)CO2R4a, -
N(R4a)C(0)N(R4a),
Ci_6alkyl substituted by from 0 to 4 R4 and C34cycloalkyl substituted by from
0 to 4 R4, each R4
is independently chosen from the group consisting of hydrogen, -OH, Ci_6alkyl
and
Cmcycloalkyl, and each R4a is independently chosen from the group consisting
of hydrogen,
Ci..6alkyl and C3_8cycloalkyl. In particular aspects of this embodiment, 01 is
phenyl, v is from
0 to 2, and each le is independently chosen from the group consisting of
fluorine, chlorine, -OH,
-CH3, -OCH3 and -CN. In all aspects of this embodiment, all other groups are
as provided in the
general formula above and/or in the first or second embodiments.
In a fourth embodiment of the invention,
and , taken together
with one
substituent RI and one substituent R2, are represented by a group chosen from
the group
consisting of:
RI
(RI)02
(RI)o 3 L - N (RI)0 \
-3
(10)0- I \
2 ' (R2/0.5 (RI)o-3 \ (R!)
and
L(R0-3 , where W is
q-(Rt.3 --a - 2)
chosen from the group consisting of -(CH2)t-3-, -(CH2)0-2NH(CH2)o-2-,
-(CH2)0..2N(C1_6alky1)(CH2)o-2-, -(CH2)0-20(CH2)0_2- and -(CH2)0_2C(0)(C1-12)0-
2-, where W is
-16-
CA 02756172 2013-05-24
substituted by from 0 to 4 R.', where each led is independently selected from
C1.6alkyl and
C34cycloalkyl; and V is chosen from the group consisting of-C(0)- and -CH2-,
and where V is
-CH2-, V is substituted by from 0 to 2 le, where each R" is independently
selected from the
group consisting of Ci_oalkyl, phenyl and C3.8cycloalkyl. In a first aspect of
this embodiment,
and , taken together with one substituent RI and one substituent
R2, are represented
by a group
R.'
-\
1
(10)0=
(R
113-37/
chosen from the group consisting of \!--w , and
\
o ________________ " . In particular instances of this embodiment, and of
the first aspect of
this embodiment, W is chosen from the group consisting of -CH2-, -NH-, -
N(C1..6alkyl)-, -C(0)-,
-CH2NH-, -CH2N(C1.6alkyl)-, -CH2CH2-, -C(0)CH2-, -CH2C(0)-, -CH20-, -CH2CH2CH2-
,
-C(0)CH2CH2-, -CH2C(0)CH2-, -CH2OCH2-, -CH2CH2C(0)-, -CH2CH20-, -CH2CH2NH-,
-CH2CH2N(C/_6alkyl)-, -CH2NHCH2-, -CH2N(Ci_6alkyl)CH2-, -NHCH2CH2-, and
-N(C1_6a1ky1)CH2CH2-. In all aspects of this embodiment, all other groups are
as provided in the
general formula above.
In a fifth embodiment of the invention, each D is independently chosen from
the
1,1 9-13
1,(\NA
group consisting of a single bond, -C(0)N(R5)-, -NR5C(0)-, \--N \--N
1,1
kor\ kNo,
\--N N-N and N -N , where R5 is independently
chosen from
the group consisting of hydrogen, halogen -0R6, -CN, -0O2R6, -C(0)N(R6)2, -
N(R6)2,
-N(R6)COR6, -SR6, -S(0)R6, -S(02)R6, -N(R6)S02R6, -NCO2R6, -NC(0)N(R6)2,
Ci_6alkyl
substituted by from 0 to 3 substituents R6 and Cmcycloalkyl substituted by
from 0 to 3
substituents R6, and each R6 is independently chosen from the group consisting
of hydrogen,
C1alkyl and C3_8cycloalkyl. In particular aspects of this embodiment, each D
is independently
1.1
chosen from the group consisting of N and N In this embodiment,
all other
-17-
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
groups are as provided in the general formula above and/or in the first
through fourth
embodiments.
In a sixth embodiment of the invention, each E is independently chosen from
the
group consisting of a single bond, -CH2NHC(0)-, -CH2N(CH3)C(0)-, -
C(CH3)HNHC(0)-,
-C(CH3)HN(CH3)C(0)-, -C(CH3)2NHC(0)-, -C(CH3)2N(CH3)C(0)-, -CH2NHC(0)0-,
-CH2N(CH3)C(0)0-, -C(CH3)}1NHC(0)0-, -C(CH3)1IN(CH3)C(0)0-, -C(CH3)2NHC(0)0-,
o,
0 D
D
R8
-C(CH3)2N(CH3)C(0)0-, and R" R" where one of lea and R8b
is
-OH or fluorine. In a first aspect of this embodiment, each E is independently
chosen from the
o,
0 D
0
D Dd¨Se
118'
group consisting of a single bond,
and R" 0 where one of R8a and
R8b is -OH or fluorine. In all aspects of this embodiment, all other groups
are as provided in the
general formula above and/or in the first through fifth embodiments.
In some embodiments, adjacent D and E groups each may be selected to be a
single bond. In such embodiments, D and E are combined to be one single bond,
and all other
groups are as provided in the general formula above and/or in the first,
second, third and fourth
embodiments. That is, where D is a single bond and the adjacent E is a single
bond, or
is connected directly to G by one single bond.
In a seventh embodiment of the invention, each G is independently chosen from
the group consisting of:
(a) Ci_6alkyl having 0 to 4 substituents
(b) 3- to 8-membered carbocycles containing from 0 to 3 heteroatoms
independently chosen from the group consisting of N, 0 and S. and having from
0 to 3
substitutents RI on N or C atoms; and
(c) aryl ring systems G' chosen from the group consisting
of:
(i) 5- to 7-membered monocyclic ring systems and
(ii) 8- to 10-membered bicyclic ring systems,
- 18-
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
and the aryl ring systems G containing from 0 to 4 heteroatoms
independently chosen from the group consisting of N, 0 and S, and substituted
on C or N atoms
by 0 to 3 substitutents R1 . In all aspects of the seventh embodiment, G is
chosen such that
stable compounds result. In all aspects of this seventh embodiment, all other
groups are as
provided in the general foimula above and/or in the first through sixth
embodiments.
In an eighth embodiment, each G is independently chosen from the group
consisting of:
(a) hydrogen,
(b) -CN,
(c) Ci_5alkyl having Ito 3 substituents R11,
each R11 is independently chosen from the group consisting of -OH, -NI-12,
-NCH3H, -N(CH3)2, -N(CH2CH3)2, =NH, =NCH3, -C(0)H, -C(0)0H, -C(0)CH3, -
C(0)0CH3,
-NHC(0)H, -NHC(0)0H, -NHC(0)CH3, -NHC(0)0CH3, cyclopropyl, cyclobutyl,
cyclopentyl,
cyclohexyl, pyranyl, pyrrolidinyl, piperidinyl, oxacyclopentyl, and
oxacyclohexyl, phenyl,
pyridinyl, pyrimidinyl and pyrrolyl, where
the cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, pyranyl,
pyrrolidinyl, piperidinyl, oxacyclopentyl and oxacyclohexyl are substituted by
from 0 to 2
substitutents R12 on N or C atoms, and each R12 is independently selected from
the group
consisting of hydrogen, halogen, carboxy, Ci_6alkyl, -0-Ci_6alkyl and -S-
Ci_oalkyl; and
the phenyl, pyridinyl, pyrirnidinyl and pyn-olyl are substituted by from 0
to 3 substitutents R13 on N or C atoms, and each R13 is independently selected
from the group
consisting of hydrogen, halogen, Ci_6alkyl and 3- to 8-membered cycloalkyl
containing from 0 to
3 heteroatoms independently chosen from the group consisting of N, 0 and S,
(d) cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, pyranyl,
pyrrolidinyl,
piperidinyl, oxacyclopentyl and oxacyclohexyl having from 0 to 3 substitutents
R1 on N or C
atoms, the R1 independently selected from the group consisting of hydrogen,
halogen, carboxy,
C1_6alkyl, -0-Ci_6alkyl, -S-C1..6alkyl, phenyl and benzyl, and
(e) aryl ring systems G chosen from the group consisting of: phenyl,
pyridinyl and 9-membered bicyclic ring systems containing from 0 to 2
heteroatoms
independently chosen from the group consisting of N and 0.
In a first aspect of the eighth embodiment, G is independently chosen from the
group consisting of Ci_4alkyl having 1 to 2 substituents R11, wherein each R11
is independently
-19-
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
chosen from the group consisting of -0H, -NH2, -NCH3H, -N(CH3)2, -N(CH2CH3)2, -
C(0)0CH3,
cyclopropyl, cyclobutyl, cyclopentyl, eyelohexyl, pyranyl, pyrrolidinyl,
piperidinyl,
oxacyclopentyl, oxacyclohexyl, phenyl, pyridinyl, pyrimidinyl and pyrrolyl. In
all aspects of the
eighth embodiment, G is chosen such that stable compounds result. In all
aspects of this eighth
embodiment, all other groups are as provided in the general formula above
and/or in the first
through sixth embodiments.
In a ninth embodiment of the invention,
is chosen from the group consisting
I
of X ,where
each X is independently chosen from the group consisting of CR1 and N,
CRI NR' N
NR1 CR1 N 0
) ) )
Y is chosen from the group consisting of NR', CR , NR', N 0 , 0 N
and s ,
each RI is independently chosen from the group consisting of hydrogen,
halogen,
-0R3a, -CN, -C(0)R3, -CO2R3a, -C(0)N(R3a)2, -SR3a, -S(0)R3, -S(O2)R3', -(CH2)0-
6N(R31,
-N(R3a)S02R3 a, -N(R3 a)CO2R3a, -N(R3a)C(0)R3 -N(R3a)C OR3a, -N(R3a)C(0)N(R3
a), C _6alkyl,
C3_8carbocycle containing from 0 to 3 beteroatoms chosen from N, 0 and S. and
phenyl, and the
Ci_6alkyl, C34carbocycle and phenyl are substituted by from 0 to 3
substitutents independently
chosen from the group consisting of hydrogen, halogen, -0R3a, -CN, -CO2R3a, -
C(0)N(R3a)2,
-N(R3a)2, -N(R3a)CO2R3a, -SR3a, -S(0)R3a, -S(02)R3', -N(R3a)S02R3a, -
N(R3a)CO2R3,
-N(R3')C(0)N(R3 a), C j_6alky1, -0-C1 alkyl and -S-C1_6alkyl,
each R3 is independently chosen from the group consisting of hydrogen,
Ci_6alkyl,
-0H, -0-Ci_6alkyl and C3,8cycloalkyl, and
each R3' is independently chosen from the group consisting of hydrogen,
Ci.6a1kyl
and C3.8cycloalkyl;
is chosen from the group consisting of
phenyl, pyridinyl, pyrazinyl,
pyrimidyl, 1,2,4-triazinyl, pyridazinyl, thiazyl and 9-membered bicyclic ring
systems that
contain from 1 to 3 heteroatoms independently chosen from the group consisting
of N, 0 and S,
v is from 0 to 4,
-20 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
each R2 is independently chosen from the group consisting of hydrogen,
halogen,
_0R4, -CN, -CO2R4a, -C(0)N(R4a)2, -N(R4a)2, -N(R4a)CO2R4a, -SR4a, -S(0)R4a, -
S(02)R4a,
-N(R4a)S02R4a, -N(R4a)CO2R4a, -N(R4a)C(0)N(R4a), Ci_6a1kyl substituted by from
0 to 4 R4 and
C3.8cycloalkyl substituted by from 0 to 4 R4,
each R4 is independently chosen from the group consisting of hydrogen, -OH,
C1_6alkyl and C3_8cycloalkyl, and
each Rla is independently chosen from the group consisting of hydrogen,
Ci_6alkyl
and C3.8cycloalkyl;
wherein each D is independently chosen from the group consisting of a single
CH3
,c\or$4,
kor\
bond, -C(0)N(R5)-, -NR5C(0)-, \--N N-N
and NN , where
R5 is independently chosen from the group consisting of hydrogen,
halogen -0R6, -CN, -0O2R6, -C(0)N(R6)2, -N(R6)2, -N(R6)COR6, -SR6, -S(0)R6, -
S(02)R6,
-N(R6)S02R6, -NCO2R6, -NC(0)N(R6)2, C1_6a1ky1 substituted by from 0 to 3
substituents R6 and
C3_gcycloa1kyl substituted by from 0 to 3 substituents R6, and
each R6 is independently chosen from the group consisting of hydrogen,
C1,6alkyl and C3_8cycloalkyl;
wherein each E is independently chosen from the group consisting of a single
bond, -CH2NHC(0)-, -CH2N(CH3)C(0)-, -C(CH3)HNHC(0)-, -C(C113)}1N(CH3)C(0)-,
-C(CH3)2NHC(0)-, -C(CH3)2N(CH3)C(0)-, -CH2NHC(0)0-, -CH2N(CH3)C(0)0-,
-C(CH3)HNHC(0)0-, -C(CH3)FIN(CH3)C(0)0-, -C(CH3)2NHC(0)0-, -C(CH3)2N(CH3)C(0)0-
,
0µ\ D
C)
D D
G
and R" R" where one of R8a and R8b is -OH or fluorine;
wherein each G is independently chosen from the group consisting of
(a) hydrogen,
(b) -CN,
(c) Ci_salkyl having 1 to 3 substituents RH,
-21-
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
each R11 is independently chosen from the group consisting of -OH, -NH2,
-NCH3H, -N(CH3)2, -N(CH2C1-13)2, =NH, =NCH3, -C(0)H, -C(0)0H, -C(0)C1-13, -
C(0)0CH3,
-NHC(0)H, -NHC(0)0H, -NHC(0)CH3, -NHC(0)0CH3, cyclopropyl, cyclobutyl,
cyclopentyl,
cyclohexyl, pyranyl, pyrrolidinyl, piperidinyl, oxacyclopentyl, and
oxacyclohexyl, phenyl,
pyridinyl, pyrimidinyl and pyrrolyl, where
the cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, pyranyl,
pyrrolidinyl, piperidinyl, oxacyclopentyl and oxacyclohexyl are substituted by
from 0 to 2
substitutents R12 on N or C atoms, and each R12 is independently selected from
the group
consisting of hydrogen, halogen, carboxy, C1_6a1ky1, -0-C1_6alkyl and -S-
Ci_6a1kyl; and
the phenyl, pyridinyl, pyrimidinyl and pyrrolyl are substituted by from 0
to 3 substitutents R13 on N or C atoms, and each R13 is independently selected
from the group
consisting of hydrogen, halogen, C1_6alkyl and 3- to 8-membered cycloalkyl
containing from 0 to
3 heteroatoms independently chosen from the group consisting of N, 0 and S,
(d) cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, pyranyl,
pyrrolidinyl,
piperidinyl, oxacyclopentyl and oxacyclohexyl having from 0 to 3 substitutents
Rl on N or C
atoms, the R1 independently selected from the group consisting of hydrogen,
halogen, carboxy,
C1_6alkyl, -0-Ci.6alkyl, -S-Ci_olkyl, phenyl and benzyl, and
(e) aryl ring systems G' chosen from the group consisting of: phenyl,
pyridinyl and 9-membered bicyclic ring systems containing from 0 to 2
heteroatoms
independently chosen from the group consisting of N and 0. In all aspects of
this embodiment,
all other groups are as provided in the general formula above.
In a tenth embodiment of the invention,
is chosen from the group consisting
R1 RI
of
1\1/ 1\1 Oo)1, *s)land
0\
/
N , where is substituted by from 0 to 3 additional R1; is
phenyl; v is from
0 to 2; each R2 is independently chosen from the group consisting of fluorine,
chlorine,
-OH, -CH3, -OCH3 and -CN; each D is independently chosen from the group
consisting of
N and N ; each E is independently chosen from the group
consisting of a
- 22 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
0,
Do
D
R33-
single bond, and R" R" where one of R8a and R8b is -01-1
or fluorine;
and each G is independently chosen from the group consisting of Cmalkyl having
1 to 2
substituents R11, wherein each R11 is independently chosen from the group
consisting of -OH,
-NH2, -NCH3H, -N(CH3)2, -N(CH2CH3)2, -C(0)0CH3, cyclopropyl, cyclobutyl,
cyclopentyl,
cyclohexyl, pyranyl, pyrrolidinyl, piperidinyl, oxacyclopentyl, oxacyclohexyl,
phenyl, pyridinyl,
pyrimidinyl and pyrrolyl. In all aspects of this embodiment, all other groups
are as provided in
the general formula above or in the eighth embodiment.
In an eleventh embodiment of the invention, the compound having structural
formula (I) is a compound having structural formula (Ia):
G- (R2),
N N
y
N E-G ja)
\
or a pharmaceutically acceptable salt thereof, wherein
Y is substituted by u substituents
RI, and Y is selected from the group consisting of 0 and NR'. In all aspects
of this embodiment,
all other groups are as provided in the general formula above or in any one of
the first through
tenth embodiments.
In a twelfth embodiment of the invention, the compound having structural
folmula (Ia) is a compound having structural formula (1b):
0\ro (Rs,0
N
N
(Ila)
\
or a pharmaceutically acceptable salt thereof, wherein
Y is substituted by u substituents
RI, and Y is selected from the group consisting of 0 and NW. In particular
aspects of this
\
embodiment, Y is substituted by u substituents RI, Y is 0, and both
instances of G are
- 23 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
vvvsnru 0
. In all aspects of this embodiment, all other groups are as provided in the
general
formula above or in the eleventh embodiment.
In a thirteenth embodiment of the invention, the compound having structural
formula (la) is a compound having structural formula (Ib):
(R0)04
N (R2)p
N N
y NNN
(th)
or a pharmaceutically acceptable salt thereof, wherein said Y is
substituted by u
1401 \
substituents Rl, and said Y and said 0, taken together with one
substituent RI and
one substituent R2, are represented by a group chosen from the group
consisting of
RI
(R1)o.2¨
(R1)0 N (R
and (R2L-3. In
001 \
particular aspects of this embodiment, Y and said 0, taken together with
one
substituent RI and one substituent R2, are represented by
RI
¨
(R)o{JiT ________ 11¨
(R2L-3
v¨w wherein V is -CH2-, W is -(CH2)0,20(CH2)o-2-, RI is
fluorine, and both
¨ 0
'1%13LCY
instances of G are H In all aspects of this embodiment, all other
groups are as
provided in the general formula above Or in the eleventh embodiment.
In a fourteenth embodiment of the invention, 0 and , taken together with
one substituent R1 and one substituent R2, are represented by a group chosen
from the group
consisting of:
- 24 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
RI
(R))mr (RI \
I
nR2)o-3 (R1)o aLL \ )o_3-71N
RI
MI) I I \
V-W
N (R I ) 3 and " 0 \ __ (R21-3
where
W is chosen from the group consisting of -(CH2)1-3-, -(CH2)0_2N11(CH2)o-2-,
-(CH2)0_2N(C1.6alkyl)(CH2)0_2-, -(CH2)0_20(CH2)0_2- and -(CH2)0_2C(0)(CH2)0-2-
, where W is
substituted by from 0 to 4 Rw, where each R.`" is independently selected from
C1_6alkyl and
C3_8cycloalkyl; and
V is chosen from the group consisting of -C(0)- and -CH2-, and where V is
-CH2-, V is substituted by from 0 to 2 R", where each Rv is independently
selected from the
group consisting of Ci_6alkyl and C3_8cycloalkyl;
each RI is independently chosen from the group consisting of hydrogen,
halogen,
-0R3, -CN, -C(0)R3, -0O2R3, -C(0)N(R3a)2, -SR3, -S(0)R3, -S(02)R3, -N(R3a)2,
-(012)061\T(R3a)2, -N(R3a)S02R3, -N(R3a)CO2R3, -N(R3a)COR3, -N(R3a)C(0)N(R3a),
C1_6a1ky1,
C3_8carbocycle containing from 0 to 3 heteroatoms chosen from N, 0 and S, and
phenyl, and the
C1_6alkyl, Cmcarbocycle and phenyl are substituted by from 0 to 3
substitutents independently
chosen from the group consisting of hydrogen, halogen, -0R3a, -CN, -CO2R3a, -
C(0)N(R3a)2,
-N(R3a)2, -N(R3a)CO2R3a, -SR3a, -S(0)R3a, -S (02)R3 -N(R3a)S02R3a, -
N(R3a)CO2R3a,
-N(R3a)C(0)N(R3a), Ci_6alkyl, -0-Ci_6alkyl and --S-C1_6alky1,
each R3 is independently chosen from the group consisting of hydrogen,
C1_6a1ky1, -OH, -0-C1_6a1ky1 and C3_8cycloalkyl, and
each Rh is independently chosen from the group consisting of hydrogen,
C1_6alkyl and C3.8cycloa1kyl;
each R2 is independently chosen from the group consisting of hydrogen,
halogen,
-CN, -CO2R4a, -C(0)N(R4a)2, -N(R4a)2, -N(R4a)CO2R4a,K
-s-4a,
S(0)R4a, -S(02)R4a,
-N(R4a)SO2R4a, -N(R4a)CO2R4a, -
N(R4a)C(0)N(R4a), C1_6a1ky1 substituted by from 0 to 4 R4 and
C3_8cycloalkyl substituted by from 0 to 4 R4,
each R4 is independently chosen from the group consisting of hydrogen,
-014, Calkyl and C3_8cycloalkyl, and
-25-
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
each R4a is independently chosen from the group consisting of hydrogen,
C1_6a1ky1 and C3,8cycloalkyl;
wherein each D is independently chosen from the group consisting of a single
yH3
/A
fy,õ kor\
bond, -C(0)N(R5)-, -NR5C(0)-, \--N \-N
N-N
11
and NN ,where
R5 is independently chosen from the group consisting of hydrogen,
halogen -0R6, -CN, -0O2R6, -C(0)N(R6)2, -N(R6)2, -N(R6)COR6, -SR6, -S(0)R6, -
S(02)R6,
-N(R6)S02R6, -NCO2R6, -NC(0)N(R6)2, C1_6alkyl substituted by from 0 to 3
substituents R6 and
C3_8cycloalkyl substituted by from 0 to 3 substituents R6, and
each R6 is independently chosen from the group consisting of hydrogen,
C1_6alkyl and C3.8cycloalkyl;
wherein each E is independently chosen from the group consisting of a single
bond, -CH2NHC(0)-, -CH2N(CH3)C(0)-, -C(CH3)HNHC(0)-, -C(CH3)HN(CH3)C(0)-,
-C(CH3)2NHC(0)-, -C(CH3)2N(C113)C(0)-, -CH2NHC(0)0-, -CH2N(CH3)C(0)0-,
-C(CH3)HNHC(0)0-, -C(CH3)HN(CH3)C(0)0-, -C(CH3)2NHC(0)0-, -C(CH3)2N(C113)C(0)0-
,
D
D G D R8a71
b lea
and R" R" where one of R8a and R8b is -OH or fluorine;
wherein each G is independently chosen from the group consisting of
(a) hydrogen,
(b) -CN,
(c) Ci_salkyl having Ito 3 substituents R",
each R11 is independently chosen from the group consisting of -OH, -NH2,
-NCH3H, -N(CH3)2, -N(CH2CH3)2,
-NCH3, -C(0)H, -C(0)0H, -C(0)CH3, -C(0)0CH3,
-NHC(0)H, -NHC(0)0H, -NHC(0)CH3, -NHC(0)0CH3, cyclopropyl, cyclobutyl,
cyclopentyl,
cyclohexyl, pyranyl, pyrrolidinyl, piperidinyl, oxacyclopentyl, and
oxacyclohexyl, phenyl,
pyridinyl, pyrimidinyl and pyrrolyl, where
the cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, pyranyl,
pyrrolidinyl, piperidinyl, oxacyclopentyl and oxacyclobexyl are substituted by
from 0 to 2
-26-
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
substitutents R12 on N or C atoms, and each R12 is independently selected from
the group
consisting of hydrogen, halogen, carboxy, C1_6alkyl, -0-C 1_6alkyl and ¨S-
C1_6alkyl; and
the phenyl, pyridinyl, pyrimidinyl and pyrroly1 are substituted by from 0
to 3 substitutents R.13 on N or C atoms, and each R13 is independently
selected from the group
consisting of hydrogen, halogen, Ci.6alkyl and 3- to 8-membered cycloalkyl
containing from 0 to
3 heteroatoms independently chosen from the group consisting of N, 0 and S,
(d) cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, pyranyl,
pyrrolidinyl,
piperidinyl, oxacyclopentyl and oxacyclohexyl having from 0 to 3 substitutents
R1 on N or C
atoms, the R.1 independently selected from the group consisting of hydrogen,
halogen, carboxy,
Ci_6alkyl, -0-Ci.6alkyl, -S-C1,6alkyl, phenyl and benzyl, and
(e) aryl ring systems G' chosen from the group consisting of: phenyl,
pyridinyl and 9-membered bicyclic ring systems containing from 0 to 2
heteroatoms
independently chosen from the group consisting of N and O. In all aspects of
this embodiment,
all other groups are as provided in the general formula above.
In another embodiment of the invention, the compound of the invention is
selected from the exemplary species depicted in Examples 1 through 215 shown
below, or
pharmaceutically acceptable salts thereof_
In another embodiment of the invention, for the compounds of formula (I),
variables 0 , , D, E, G, R1, R2, it, v, R3, R3a, R4, R4a, R5, R6, R7, /, j,
R8a, R8b, R8c, R9, Ri0.,
RI Cia, R11, R12, R13, R14, W, le', V, and RV, are selected independently from
each other.
Other embodiments of the present invention include the following:
(a) A pharmaceutical composition comprising an effective amount of a
compound of formula (I) and a pharmaceutically acceptable carrier.
(b) The pharmaceutical composition of (a), further comprising a second
therapeutic agent selected from the group consisting of HCV antiviral agents,
immunomodulators, and anti-infective agents.
(c) The pharmaceutical composition of (b), wherein the HCV antiviral agent
is an antiviral selected from the group consisting of HCV protease inhibitors
and HCV NS5B
polymerase inhibitors.
(d) A pharmaceutical combination that is (i) a compound of formula (I) and
(ii) a second therapeutic agent selected from the group consisting of HCV
antiviral agents,
- 27 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
immunomodulators, and anti-infective agents; wherein the compound of formula
(I) and the
second therapeutic agent are each employed in an amount that renders the
combination effective
for inhibiting HCV NS5A activity, or for treating HCV infection and/or
reducing the likelihood
or severity of symptoms of HCV infection, or for inhibiting HCV viral
replication and/or HCV
viral production in a cell-based system.
(e) The combination of (d), wherein the HCV antiviral agent is an antiviral
selected from the group consisting of HCV protease inhibitors and HCV NS5B
polymerase
inhibitors.
(f) A method of inhibiting HCV NS5A activity in a subject in need thereof,
which comprises administering to the subject an effective amount of a compound
of formula (I).
(g) A method of treating HCV infection and/or reducing the likelihood or
severity of symptoms of HCV infection in a subject in need thereof, which
comprises
administering to the subject an effective amount of a compound of formula (I).
(h) The method of (g), wherein the compound of formula (I) is administered
in combination with an effective amount of at least one second therapeutic
agent selected from
the group consisting of HCV antiviral agents, immunomodulators, and anti-
infective agents.
(i) The method of (h), wherein the HCV antiviral agent is an antiviral
selected from the group consisting of HCV protease inhibitors and HCV NS5B
polymerase
inhibitors.
(j) A method of inhibiting HCV viral replication and/or HCV viral
production
in a cell-based system, which comprises administering to the subject an
effective amount of a
compound of formula (I).
(k) The method of (j), wherein the compound of formula (I)
is administered in
combination with an effective amount of at least one second therapeutic agent
selected from the
group consisting of HCV antiviral agents, immunomodulators, and anti-infective
agents.
(1) The method of (k), wherein the HCV antiviral agent is an
antiviral
selected from the group consisting of HCV protease inhibitors and HCV NS5B
polymerase
inhibitors.
(m) A method of inhibiting HCV NS5A activity in a subject in
need thereof,
which comprises administering to the subject the pharmaceutical composition of
(a), (b), or (c) or
the combination of (d) or (e).
- 28 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
(11) A method of treating HCV infection and/or reducing the
likelihood or
severity of symptoms of HCV infection in a subject in need thereof, which
comprises
administering to the subject the pharmaceutical composition of (a), (b), or
(c) or the combination
of (d) or (e).
(0) A method of inhibiting HCV viral replication and/or HCV viral
production
in a cell-based system, which comprises administering to the subject the
pharmaceutical
composition of (a), (b), or (c) or the combination of (d) or (e).
In the embodiments of the compounds and salts provided above, it is to be
understood that each embodiment may be combined with one or more other
embodiments, to the
extent that such a combination provides a stable compound or salt and is
consistent with the
description of the embodiments. It is further to be understood that the
embodiments of
compositions and methods provided as (a) through (o) above are understood to
include all
embodiments of the compounds and/or salts, including such embodiments as
result from
combinations of embodiments.
The present invention also includes a compound of the present invention for
use
(i) in, (ii) as a medicament for, or (iii) in the preparation of a medicament
for: (a) inhibiting HCV
NS5A activity, or (b) treating HCV infection and/or reducing the likelihood or
severity of
symptoms of HCV infection, or (c) inhibiting HCV viral replication and/or HCV
viral production
in a cell-based system, or (d) use in medicine. In these uses, the compounds
of the present
invention can optionally be employed in combination with one or more second
therapeutic
agents selected from HCV antiviral agents, anti-infective agents, and
immunomodulators.
Additional embodiments of the invention include the pharmaceutical
compositions, combinations and methods set forth in (a)-(o) above and the uses
set forth in the
preceding paragraph, wherein the compound of the present invention employed
therein is a
compound of one of the embodiments, aspects, classes, sub-classes, or features
of the
compounds described above. In all of these embodiments, the compound may
optionally be used
in the form of a pharmaceutically acceptable salt, or may be present in the
form of a solvate or
hydrate as appropriate.
As used herein, all ranges are inclusive, and all sub-ranges are included
within
such ranges, although not necessarily explicitly set forth. In addition, the
term "or," as used
herein, denotes alternatives that may, where appropriate, be combined; that
is, the term "or"
includes each listed alternative separately as well as their combination.
- 29 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
As used herein, the term "alkyl" refers to any linear or branched chain alkyl
group
having a number of carbon atoms in the specified range. Thus, for example,
"C1,6 alkyl" (or
"C1-C6 alkyl") refers to all of the hexyl alkyl and pentyl alkyl isomers as
well as n-, iso-, sec- and
tert-butyl, n- and isopropyl, ethyl and methyl. As another example, "C1,4
alkyl" refers to n-, iso-,
sec- and tert-butyl, n- and isopropyl, ethyl and methyl. Where indicated, "Co"
refers to
hydrogen; thus, for example, "C0.6 alkyl" (or "C0-C6 alkyl") refers to all of
the hexyl alkyl and
pentyl alkyl isomers as well as n-, iso-, sec- and tert-butyl, n- and
isopropyl, ethyl, methyl and
hydrogen. Alkyl groups may be substituted as indicated.
The term "halogenated" refers to a group or molecule in which a hydrogen atom
has been replaced by a halogen. Similarly, the term "haloalkyl" refers to a
halogenated alkyl
group. The term "halogen" (or "halo") refers to atoms of fluorine, chlorine,
bromine and iodine
(alternatively referred to as fluoro, chloro, bromo, and iodo), preferably
fluorine.
The term "alkoxy" refers to an "alkyl-O-" group, where alkyl is as defined
above.
Alkoxy groups may be substituted as indicated.
The term "cycloalkyl" refers to any cyclic ring of an alkane or alkene having
a
number of carbon atoms in the specified range. Thus, for example, "C3.8
cycloalkyl" (or "C3-C8
cycloalkyl") refers to cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl,
cycloheptyl, and
cyclooctyl. The term "cycloalkoxy" refers to a "cycloalkyl-O-" group, whre
cycloalkyl is as
defined above. Cycloalkyl groups may be substituted as indicated.
The term "aryl" (or "aryl ring system") refers to aromatic mono- and poly-
carbocyclic or heterocyclic ring systems wherein the individual carbocyclic
rings in the polyring
systems are fused or attached to each other via a single bond. As used herein,
the teim aryl
includes aromatic mono- and poly-carbocyclic ring systems that include from 0
to 4 heteroatoms
(non-carbon atoms) that are independently chosen from N, 0 and S. Suitable
aryl groups include
phenyl, naphthyl, biphenylenyl, pyridinyl, pyrimidinyl and pyrrolyl, as well
as those discussed
below. Aryl groups may be substituted as indicated. Aryl ring systems may
include, where
appropriate, an indication of the variable to which a particular ring atom is
attached. Unless
otherwise indicated, substituents to the aryl ring systems can be attached to
any ring atom,
provided that such attachment results in founation of a stable ring system.
The term "carbocycle" (and variations thereof such as "carbocyclic") as used
herein, unless otherwise indicated, refers to (i) a C5 to C7 monocyclic,
saturated or unsaturated
ring, or (ii) a Cg to C10 bicyclic saturated or unsaturated ring system. Each
ring in (ii) is either
-30-
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
independent of, or fused to, the other ring, and each ring is saturated or
unsaturated. Carbocycle
groups may be substituted as indicated. When the carbocycles contain one or
more heteroatoms
independently chosen from N, 0 and S, the carbocycles may also be referred to
as
"heterocycles," as defined below. The carbocycle may be attached to the rest
of the molecule at
any carbon or nitrogen atom that results in a stable compound. The fused
bicyclic carbocycles
are a subset of the carbocycles; i.e., the term "fused bicyclic carbocycle"
generally refers to a C8
to C10 bicyclic ring system in which each ring is saturated or unsaturated and
two adjacent
carbon atoms are shared by each of the rings in the ring system. A fused
bicyclic carbocycle in
which both rings are saturated is a saturated bicyclic ring system. Saturated
carbocyclic rings are
also referred to as cycloalkyl rings, e.g., cyclopropyl, cyclobutyl, etc. A
fused bicyclic
carbocycle in which one or both rings are unsaturated is an unsaturated
bicyclic ring system.
Ca.rbocycle ring systems may include, where appropriate, an indication of the
variable to which a
particular ring atom is attached. Unless otherwise indicated, substituents to
the ring systems can
be attached to any ring atom, provided that such attachment results in
formation of a stable ring
system.
Unless indicated otherwise, the term "heterocycle" (and variations thereof
such as
"heterocyclic" or "heterocycly1") broadly refers to (i) a stable 5- to 7-
membered, saturated or
unsaturated monocyclic ring, or (ii) a stable 8- to 10-membered bicyclic ring
system, wherein
each ring in (ii) is independent of, or fused to, the other ring or rings and
each ring is saturated or
unsaturated, and the monocyclic ring or bicyclic ring system contains one or
more heteroatoms
(e.g., from 1 to 6 heteroatoms, or from 1 to 4 heteroatoms) independently
selected from N, 0 and
S and a balance of carbon atoms (the monocyclic ring typically contains at
least one carbon atom
and the bicyclic ring systems typically contain at least two carbon atoms);
and wherein any one
or more of the nitrogen and sulfur heteroatoms is optionally oxidized, and any
one or more of the
nitrogen heteroatoms is optionally quatemized. Unless otherwise specified, the
heterocyclic ring
may be attached at any heteroatom or carbon atom, provided that attachment
results in the
creation of a stable structure. Heterocycle groups may be substituted as
indicated, and unless
otherwise specified, the substituents may be attached to any atom in the ring,
whether a
heteroatom or a carbon atom, provided that a stable chemical structure
results. Representative
examples include pyranyl, piperidinyl, piperazinyl, azepanyl, pyrrolidinyl,
pyrazolidinyl,
imidazolidinyl, oxazolidinyl, isoxazolidinyl, morpholinyl, thiomorpholinyl,
thiazolidinyl,
isothiazolidinyl, and tetrahydrofuryl (or tetrahydrofuranyl). Unless expressly
stated to the
-31 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
contrary, the term "heteroaryl ring system" refers to aryl ring systems, as
defined above, that
include from 1 to 4 heteroatoms (non-carbon atoms) that are independently
chosen from N, 0
and S. In the case of substituted heteraromatic rings containing at least one
nitrogen atom (e.g.,
pyridine), such substitutions can be those resulting in N-oxide formation.
Representative
examples of heteroaromatic rings include pyridyl, pyrrolyl, pyrazinyl,
pyrimidinyl, pyridazinyl,
thienyl (or thiophenyl), thiazolyl, furanyl, imidazolyl, pyrazolyl, triazolyl,
tetrazolyl, oxazolyl,
isooxazolyl, oxadiazolyl, thiazolyl, isothiazolyl, and thiadiazolyl.
Representative examples of
bicyclic heterocycles include benzotriazolyl, indolyl, isoindolyl, indazolyl,
indolinyl,
isoindolinyl, quinoxalinyl, quinazolinyl, cinnolinyl, chromanyl, isochromanyl,
tetrahydroquinolinyl, quinolinyl, tetrahydroisoquinolinyl, isoquinolinyl, 2,3-
dihydrobenzofuranyl, 2,3-dihydrobenzo-1,4-dioxinyl and benzo-1,3-dioxolyl.
Unless otherwise specifically noted as only "substituted", alkyl, cycloalkyl,
and
aryl groups are not substituted. If substituted, preferred substituents are
selected from the group
that includes, but is not limited to, halo, C1-C20 alkyl, -CF3, -N112, -N(C1-
C6 alky1)2, -NO2, oxo,
-CN,-N3, -OH, -0(C1-C6 alkyl), C3-C10 cycloalkyl, C2-C6 alkenyl, C2-C6
alkynyl, (Co-C6 alkyl)
S(0)0..2-, aryl-S(0)0_2-, (Co-C6 alkyl)S(0)0_2(Co-C6 alkyl)-, (Co-C6
alkyl)C(0)NH-, H2N-C(NH)-,
-0(C1-C6 alky1)CF3, (Co-C6 alkyl)C(0)-, (Co-C6 alky1)0C(0)-, (Co-C6alky1)0(Ci-
C6
(Co-C6 alkyl)C(0)1_2(Co-C6 alkyl)-, (C1-C6 alky1)0C(0)NH-, aryl, aralkyl,
heteroaryl,
heterocycloalkyl, halo-aryl, halo-aralkyl, halo-heterocycle and halo-
heterocycloalkyl.
Unless expressly stated to the contrary, all ranges cited herein are
inclusive. For
example, a heteroaryl ring described as containing from "0 to 3 heteroatoms"
means the ring can
contain 0, 1, 2, or 3 heteroatoms. It is also to be understood that any range
cited herein includes
within its scope all of the sub-ranges within that range. The oxidized forms
of the heteroatoms N
and S are also included within the scope of the present invention.
When any variable (for example, RI or R3) occurs more than one time in any
constituent or in formula (I) or in any other formula depicting and describing
compounds of the
invention, its definition on each occurrence is independent of its definition
at every other
occurrence. Also, combinations of substituents and/or variables are
petwissible only if such
combinations result in stable compounds.
Unless expressly stated to the contrary, substitution by a named substituent
is
permitted on any atom provided such substitution is chemically allowed and
results in a stable
compound. A "stable" compound is a compound that can be prepared and isolated
and that has a
- 32 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
structure and properties that remain or can be caused to remain essentially
unchanged for a
period of time sufficient to allow use of the compound for the purposes
described herein (e.g.,
therapeutic or prophylactic administration to a subject).
As used herein, the term "compound" is intended to encompass chemical agents
described by generic formula (I) in all forms, including hydrates and solvates
of such chemical
agents.
In the compounds of generic formula (I), the atoms may exhibit their natural
isotopic abundances, or one or more of the atoms may be artificially enriched
in a particular
isotope having the same atomic number, but an atomic mass or mass number
different from the
atomic mass or mass number predominantly found in nature. The present
invention is meant to
include all suitable isotopic variations of the compounds of generic formula
(I). For example,
different isotopic forms of hydrogen (H) include protium (1H) and deuterium
(2H or D). Protium
is the predominant hydrogen isotope found in nature. Enriching for deuterium
may afford
certain therapeutic advantages, such as increasing in vivo half-life or
reducing dosage
requirements, or may provide a compound useful as a standard for
characterization of biological
samples. Isotopically-enriched compounds within generic formula (I) can be
prepared without
undue experimentation by conventional techniques well known to those skilled
in the art or by
processes analogous to those described in the Schemes and Examples herein
using appropriate
isotopically-enriched reagents and/or intermediates.
As a result of the selection of substituents and substituent patterns, certain
of the
compounds of the present invention can have asymmetric centers and can occur
as mixtures of
stereoisomers, or as individual diastereomers, or enantiomers. All isomeric
forms of these
compounds, whether isolated or in mixtures, are within the scope of the
present invention.
As would be recognized by one of ordinary skill in the art, certain of the
compounds of the present invention can exist as tautomers. For the purposes of
the present
invention a reference to a compound of formula (I) is a reference to the
compound per se, or to
any one of its tautomers per se, or to mixtures of two or more tautomers.
The compounds of the present invention may be administered in the form of
pharmaceutically acceptable salts. The term "pharmaceutically acceptable salt"
refers to a salt
that possesses the effectiveness of the parent compound and that is not
biologically or otherwise
undesirable (e.g., is neither toxic nor otherwise deleterious to the recipient
thereof). Suitable
salts include acid addition salts that may, for example, be formed by mixing a
solution of the
- 33 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
compound of the present invention with a solution of a pharmaceutically
acceptable acid such as
hydrochloric acid, sulfuric acid, acetic acid, trifluoroacetic acid, or
benzoic acid. Many of the
compounds of the invention carry an acidic moiety, in which case suitable
pharmaceutically
acceptable salts thereof can include alkali metal salts (e.g., sodium or
potassium salts), alkaline
earth metal salts (e.g., calcium or magnesium salts), and salts formed with
suitable organic
ligands such as quaternary ammonium salts. Also, in the case of an acid (-
COOH) or alcohol
group being present, pharmaceutically acceptable esters can be employed to
modify the
solubility or hydrolysis characteristics of the compound.
The term "administration" and variants thereof (e.g., "administering" a
compound) in reference to a compound of the invention mean providing the
compound or a
prodrug of the compound to the individual in need of treatment. When a
compound of the
invention or a prodrug thereof is provided in combination with one or more
other active agents
(e.g., antiviral agents useful for treating HCV infection), "administration"
and its variants are
each understood to include concurrent and sequential provision of the compound
or salt (or
hydrate) and other agents.
As used herein, the term "composition" is intended to encompass a product
comprising the specified ingredients, as well as any product that results,
directly or indirectly,
from combining the specified ingredients.
By "pharmaceutically acceptable" is meant that the ingredients of the
pharmaceutical composition must be compatible with each other and not
deleterious to the
recipient thereof.
The tetins "subject" (alternatively referred to herein as "patient") as used
herein,
refer to an animal, preferably a mammal, most preferably a human, who has been
the object of
treatment, observation or experiment.
The term "effective amount" as used herein means that amount of active
compound or pharmaceutical agent that elicits the biological or medicinal
response in a tissue,
system, animal or human that is being sought by a researcher, veterinarian,
medical doctor or
other clinician. In one embodiment, the effective amount is a "therapeutically
effective amount"
for the alleviation of one or more symptoms of the disease or condition being
treated. In another
embodiment, the effective amount is a "prophylactically effective amount" for
reduction of the
severity or likelihood of one or more symptoms of the disease or condition. In
another
embodiment, the effective amount is a "therapeutically effective amount" for
inhibition of HCV
- 34 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
viral replication and/or HCV viral production. The tenu also includes herein
the amount of
active compound sufficient to inhibit HCV NS5A and thereby elicit the response
being sought
(i.e., an "inhibition effective amount"). When the active compound (i.e.,
active ingredient) is
administered as the salt, references to the amount of active ingredient are to
the free acid or free
base form of the compound.
It is understood that claimed compounds cause inhibition in replicon assay
testing.
Thus, compounds described herein are useful for inhibiting HCV replication,
specifically the
NS5A protein. Compounds described herein have different uses, including the
prevention or
treatment of one or more of the symptoms of HCV infection, the inhibition of
HCV viral
replication and/or HCV viral production, and/or as pharmaceutical composition
ingredients.
The compounds of this invention are useful in the preparation and execution of
screening assays for antiviral compounds. For example, the compounds of this
invention are
useful for identifying resistant HCV replicon cell lines harboring mutations
within NS5A, which
are excellent screening tools for more powerful antiviral compounds.
Furthermore, the
compounds of this invention are useful in establishing or determining the
binding site of other
antivirals to the HCV replicase.
For the purposes of inhibiting HCV NS5A protein, treating HCV infection and/or
reducing the likelihood or severity of symptoms of HCV infection and
inhibiting HCV viral
replication and/or HCV viral production, the compounds of the present
invention, optionally in
the form of a salt or a hydrate, can be administered by any means that
produces contact of the
active agent with the agent's site of action. They can be administered by one
or more
conventional means available for use in conjunction with pharmaceuticals,
either as individual
therapeutic agents or in a combination of therapeutic agents. They can be
administered alone,
but typically are administered with a pharmaceutical carrier selected on the
basis of the chosen
route of administration and standard pharmaceutical practice. The compounds of
the invention
can, for example, be administered by one Or more of the following: orally,
parenterally
(including subcutaneous injections, intravenous, intramuscular, intrasternal
injection or infusion
techniques), by inhalation (such as in a spray form), or rectally, in the form
of a unit dosage of a
pharmaceutical composition containing an effective amount of the compound and
conventional
non-toxic pharmaceutically-acceptable carriers, adjuvants and vehicles. Liquid
preparations
suitable for oral administration (e.g., suspensions, syrups, elixirs and the
like) can be prepared
according to techniques known in the art and can employ any of the usual media
such as water,
-35-
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
glycols, oils, alcohols and the like. Solid preparations suitable for oral
administration (e.g.,
powders, pills, capsules and tablets) can be prepared according to techniques
known in the art
and can employ such solid excipients as starches, sugars, kaolin, lubricants,
binders,
disintegrating agents and the like. Parenteral compositions can be prepared
according to
techniques known in the art and typically employ sterile water as a carrier
and optionally other
ingredients, such as solubility aids. Injectable solutions can be prepared
according to methods
known in the art wherein the carrier comprises a saline solution, a glucose
solution or a solution
containing a mixture of saline and glucose. Further description of methods
suitable for use in
preparing pharmaceutical compositions of the present invention and of
ingredients suitable for
use in said compositions is provided in Remington's Pharmaceutical Sciences,
18th edition (ed.
A. R. Gennaro, Mack Publishing Co., 1990).
The compounds of this invention can be administered orally in a dosage range
of
0.001 to 1000 mg/kg of mammal (e.g., human) body weight per day in a single
dose or in divided
doses. One dosage range is 0.01 to 500 mg/kg body weight per day orally in a
single dose or in
divided doses. Another dosage range is 0.1 to 100 mg/kg body weight per day
orally in single or
divided doses. For oral administration, the compositions can be provided in
the form of tablets
or capsules containing 1.0 to 500 mg of the active ingredient, particularly 1,
5, 10, 15, 20, 25, 50,
75, 100, 150, 200, 250, 300, 400, and 500 mg of the active ingredient for the
symptomatic
adjustment of the dosage to the patient to be treated. The specific dose level
and frequency of
dosage for any particular patient may be varied and will depend upon a variety
of factors
including the activity of the specific compound employed, the metabolic
stability and length of
action of that compound, the age, body weight, general health, sex, diet, mode
and time of
administration, rate of excretion, drug combination, the severity of the
particular condition, and
the host undergoing therapy.
As noted above, the present invention also relates to a method of inhibiting
HCV
replicon activity, inhibiting HCV viral replication and/or HCV viral
production, treating HCV
infection and/or reducing the likelihood or severity of symptoms of HCV
infection with a
compound of the present invention in combination with one or more therapeutic
agents and a
pharmaceutical composition comprising a compound of the present invention and
one or more
therapeutic agents selected from the group consisting of a HCV antiviral
agent, an
immunomodulator, and an anti-infective agent. Such therapeutic agents active
against HCV
include, but are not limited to, ribavirin, levovirin, viramidine, thymosin
alpha-1, R7025 (an
-36-
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
enhanced interferon (Roche)), interferon3 interferon-a, pegylated interferon-a
(peginterferon-a), a combination of interferon-a and ribavirin, a combination
of peginterferon-a
and ribavirin, a combination of interferon-a and levovirin, and a combination
of peginterferon-a
and levovirin. The combination of peginterferon-a and ribaviron represents the
current Standard
of Care for HCV treatment. The combination of one or more compounds of the
present
invention with the Standard of Care for HCV treatment, pegylated-interferon
and ribaviron is
specifically contemplated as being encompassed by the present invention.
Interferon-a includes,
but is not limited to, recombinant interferon-a2a (such as ROFERON
interferon), pegylated
interferon-a2a (PEGAsYs), interferon-a2b (such as INTRON-A interferon),
pegylated interferon-
a2b (PEGINTRoN), a recombinant consensus interferon (such as interferon
alphacon-1), albuferon
(interferon-a bound to human serum albumin (Human Genome Sciences)), and a
purified
interferon-a product. Amgen's recombinant consensus interferon has the brand
name INFERGEN.
Levovirin is the L-enantiomer of ribavirin which has shown immunomodulatory
activity similar
to ribavirin. Viramidine represents an analog of ribavirin disclosed in
International Patent
Application Publication WO 01/60379. In accordance with the method of the
present invention,
the individual components of the combination can be administered separately at
different times
during the course of therapy or concurrently in divided or single combination
forms.
Ribavirin, levovirin, and viramidine may exert their anti-HCV effects by
modulating intracellular pools of guanine nucleotides via inhibition of the
intracellular enzyme
inosine monophosphate dehydrogenase (IMPDH). IMPDH is the rate-limiting enzyme
on the
biosynthetic route in de nova guanine nucleotide biosynthesis. Ribavirin is
readily
phosphorylated intracellularly and the monophosphate derivative is an
inhibitor of IMPDH.
Thus, inhibition of IMPDH represents another useful target for the discovery
of inhibitors of
HCV replication. Therefore, the compounds of the present invention may also be
administered
in combination with an inhibitor of IMPDH, such as those disclosed in
International Patent
Application Publications WO 97/41211, WO 01/00622 and WO 00/25780; or
mycophenolate
mofetil. See Anthony C. Allison and Elsie M. Eugui, immunosuppressive and
Other Anti-
Rheurnetic Activities of Mychophenolate Mofetil, 44 (SUPPL.) AGENTS ACTION 165
(1993).
For the treatment of HCV infection, the compounds of the present invention may
also be administered in combination with the antiviral agent polymerase
inhibitor R7128
(Roche).
-37 -
CA 02756172 2014-03-03
The compounds of the present invention may also be combined for the treatment
of HCV infection with antiviral 2'-C-branched ribonucleosides disclosed in
Rogers E. Harry-
O'Kuru et al., A Short, Flexible Route to 2'-C-Branched Ribonucleosides, 62 J.
ORG. CHEM.
1754-59 (1997); Michael S. Wolfe and Rogers E. Harry-O'Kuru, A Consise
Synthesis of 2'-C-
Methylribonucleosides, 36 TET. LETT. 7611-14 (1995); U.S. Patent No.
3,480,613; and
International Patent Application Publications WO 01/90121, WO 01/92282, WO
02/32920,
WO 04/002999, WO 04/003000 and WO 04/002422. Such 2'-C-branched
ribonucleosides
include, but are not limited to, 2'-C-methyl-cytidine, 2'-C-methyl-uridine, 2'-
C-methyl-adenosine,
2'-C-methyl-guanosine, and 9-(2-C-methyl-P-D-ribofuranosyl)-2,6-diaminopurine,
and the
corresponding amino acid ester of the ribose C-2', C-3', and C-5' hydroxyls
and the corresponding
optionally substituted cyclic 1,3-propanediol esters of the 5'-phosphate
derivatives.
For the treatment of HCV infection, the compounds of the present invention may
also be administered in combination with an agent that is an inhibitor of HCV
NS3 serine
protease. HCV NS3 serine protease is an essential viral enzyme and has been
described to be an
excellent target for inhibition of HCV replication. Exemplary substrate and
non-substrate based
inhibitors of HCV NS3 protease inhibitors are disclosed in International
Patent Application
Publications WO 98/22496, WO 98/46630, WO 99/07733, WO 99/07734, WO 99/38888,
WO 99/50230, WO 99/64442, WO 00/09543, WO 00/59929, WO 02/48116, WO 02/48172,
WO 2008/057208 and WO 2008/057209, in British Patent No. GB 2 337 262, and in
U.S. Patent
Nos. 6,323,180, 7,470,664, and 7,012,066 and in Ashok Arasappan et al.,
Discovery of
Narlaprevir (SCH 900518): A Potent, Second Generation HCV NS3 Serine Protease
Inhibitor,
ACS MED. CHEM. LETT. DOI: 10.1021/m19000276 (February 15, 2010),
The compounds of the present invention may also be combined for the treatment
of HCV infection with nucleosides having anti-HCV properties, such as those
disclosed in
International Patent Application Publications WO 02/51425, WO 01/79246, WO
02/32920,
WO 02/48165 and WO 2005/003147 (including R1656, (2'R)-2'-deoxy-2'-fluoro-2'-C-
methylcytidine, shown as compounds 3-6 on page 77); WO 01/68663; WO 99/43691;
WO 02/18404 and WO 2006/021341, and U.S. Patent Application Publication
US 2005/0038240, including 4'-azido nucleosides such as R1626, 4'-
azidocytidine; U.S. Patent
Application Publications US 2002/0019363, US 2003/0236216, US 2004/0006007,
US 2004/0063658 and US 2004/0110717; U.S. Patent Nos. 7,105,499, 7,125,855,
7,202,224; and
- 38 -
CA 02756172 2014-03-03
International Patent Application Publications WO 02/100415, WO 03/026589, WO
03/026675,
WO 03/093290, WO 04/011478, 'WO 04/013300 and WO 04/028481.
For the treatment of HCV infection, the compounds of the present invention may
also be administered in combination with an agent that is an inhibitor of HCV
NS5B polymerase.
Such HCV NS5B polymerase inhibitors that may be used as combination therapy
include, but
are not limited to, those disclosed in International Patent Application
Publications
WO 02/057287, WO 02/057425, WO 03/068244, WO 2004/000858, WO 04/003138 and
WO 2004/007512; U.S. Patent Nos. 6,777,392, 7,105,499, 7,125,855, 7,202,224
and U.S. Patent
Application Publications US 2004/0067901 and US 2004/0110717. Other such HCV
polymerase inhibitors include, but are not limited to, valopicitabine (NM-283;
Idenix) and 2'-F-2'-
beta-methylcytidine (see also WO 2005/003147).
In one embodiment, nucleoside HCV NS513 polymerase inhibitors that are used in
combination with the present HCV inhibitors are selected from the following
compounds: 4-
amino-7-(2-C-methyl-p-D-arabinofuranosy1)-7H-pyrrolo[2,3-djpyrimidine; 4-amino-
7-(2-C-
methyl-P-D-ribofuranosyl)-7H-pyrrolo[2,3-cipyrimidine; 4-methylamino-7-(2-C-
methy1-13-D-
ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidine; 4-dimethylamino-7-(2-C-methyl-P-D-
ribofuranosyl)-7H-pyrrolo[2,3-4pyrimidine; 4-cyclopropylamino-7-(2-C-methyl-P-
D-
riboftiranosyl)-7H-pyrrolo[2,3-4pyrimidine; 4-amino-7-(2-C-viny1-13-D-
ribofuranosyl)-7H-
pyrrolo[2,3-4pyrimidine; 4-amino-7-(2-C-hydroxymethyl-P-D-ribofuranosyl)-7H-
pyrrolo[2,3-
dipyrimidine; 4-amino-7-(2-C-fluoromethy1-13-D-ribofuranosyl)-7H-pyrrolo[2,3-
cipyrimidine; 4-
amino-5-methy1-7-(2-C-methyl-P-D-ribofuranosyl)-7H-pyrrolo[2,3-clpyrimidine; 4-
arnino-7-(2-
C-methyl-P-D-ribofuranosyl)-7H-pyrrolo[2,3-4pyrimidine-5-carboxylic acid; 4-
amino-5-bromo-
7-(2-C-methyl-P-D-ribofuranosyl)-7H-pyrrolo[2,3-cipyrimidine; 4-amino-5-chloro-
7-(2-C-
methyl-P-D-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidine; 4-amino-5-fluoro-7-(2-C-
methy1-13-D-
ribofuranosyl)-7H-pyrrolo[2,3-dipyrimidine; 2,4-diarnino-7-(2-C-methyl-P-D-
ribofuranosyl)-
7H-pyrrolo[2,3-d}pyrimidine; 2-amino-7-(2-C-methyl-P-D-ribofuranosyl)-7H-
pyrrolo[2,3-
cilpyrimidine; 2-amino-4-cyclopropylamino-7-(2-C-methy1-13-D-ribofuranosyl)-7H-
pyrrolo[2,3-
d}pyrimidine; 2-amino-7-(2-C-methyl-P-D-ribofiiranosyl)-7H-pyrrolo[2,3-
d]pyrimidin-4(311)-
one; 4-arnino-7-(2-C-ethyl-13-D-ribofuranosyl)-7H-pyrrolo[2,3-4pyrimidine; 4-
amino-7-(2-C,2-
0-dimethy1-13-D-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidine; 7-(2-C-methy1-13-D-
- 39 -
CA 02756172 2014-03-03
ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidin-4(3H)-one; 2-amino-5-methyl-7-(2-C,
2-0-dimethyl-
P-D-ribofuranosyl)-7H-pyrrolo[2,3-dipyrimidin-4(3.H)-one; 4-amino-7-(3-deoxy-2-
C-methyl-P-
D-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidine; 4-amino-7-(3-deoxy-2-C-methyl-P-
D-
arabinofuranosyl)-7H-pyrrolo[2,3-d]pyrimidine; 4-amino-2-fluoro-7-(2-C-methy1-
13-D-
ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidine; 4-amino-7-(3-C-methy1-13-D-
ribofuranosyl)-7H-
pyrrolo[2,3-d]pyrimidine; 4-amino-7-(3-C-methyl-P-D-xylofuranosyl)-7H-pyrrolof
2,3-
dIpyrimidine; 4-amino-7-(2,4-di-C-methyl-13-D-ribofuranosyl)-7H-pyrrolo[2,3-
dbyrimidine; 4-
amino-7-(3-deoxy-3-fluoro-2-C-methyl-(3-D-ribofuranosyl)-7H-pyrrolo[2,3-
d]pyrimidine; and
the con-esponding 5'-triphosphates; or a pharmaceutically acceptable salt
thereof.
The compounds of the present invention may also be combined for the treatment
of HCV infection with non-nucleoside inhibitors of HCV polymerase such as
those disclosed in
U.S. Patent Applciation Publications US 2006/0100262 and US 2009-0048239;
International
Patent Application Publications WO 01/77091, WO 01/47883, WO 02/04425, WO
02/06246,
WO 02/20497, WO 2005/016927 (in particular JTK003), WO 2004/041201, WO
2006/066079,
WO 2006/066080, WO 2008/075103, WO 2009/010783 and WO 2009/010785.
In one embodiment, non-nucleoside HCV NS5B polymerase inhibitors that are
used in combination with the present HCV NS5A inhibitors are selected from the
following
compounds: 14-cyclohexy1-642-(dimethylamino)ethy1]-7-oxo-5,6,7,8-
tetrahydroindolo[2,1-
a][2,5]benzodiazocine-11-carboxylic acid; 14-cyclohexy1-6-(2-morpholin-4-
ylethyl)-5,6,7,8-
tetrahydroindolo[2,1 -a] [2,5]benzodiazocine-11-carboxylic acid; 14-cyclohexy1-
6-[2-
(dimethylamino)ethy1]-3-methoxy-5,6,7,8-tetrahydroindolo[2,1 -a]
[2,5]benzodiazocine-11-
carboxylic acid; 14-cyclohexy1-3-methoxy-6-methy1-5,6,7,8-tetrahydroindolo[2,1
a] [2,5Thenzodiazocine-11-carboxylic acid; methyl ( {[(14-cyclohexy1-3-methoxy-
6-methyl-
5,6,7,8-tetrahydroindolo[2,1 -a] [ 2,5]benzodiazocin-11-yl)carbonyl]am ino)
sulfonyl)acetate;
({ [(14-cycl ohexy1-3-methoxy-6-methy1-5,6,7,8-tetrahydroindolo[2,1 -a] [2,5]
benzodiazocin-11-
yl)carbonyllamino}sulfonyl)acetic acid; 14-cyclohexyl-N-
[(dimethylamino)sulfony1]-3-
methoxy-6-methy1-5,6,7,8-tetrahydroindolo[2,1-a] [2,5]benzodiazocine-11-
carboxamide; 3-
chloro-14-cyclohexy1-642-(dimethylamino)ethyi]-7-oxo-5,6,7,8-
tetrahydroindolo[2,1-
a][2,5Thenzodiazocine 11-carboxylic acid; Y-(11-carboxy-14-cyclohexyl-7,8-
dihydro-61/-
indolo[1,2-e][1,5Thenzoxazocin-7-y1)-N,N-dinlethylethane-1,2-diaminium
bis(trifluoroacetate);
14-cyclohexy1-7,8-dihydro-6H-indolo[1,2-e][1,5] benzoxazocine-1 I -carboxylic
acid; 14-
- 40 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
cyclohexy1-6-methyl-7-oxo-5,6,7,8-tetrahydroindolo [2,1-a][2,5]benzodiazocine-
11-carboxylic
acid; 14-cyclohexy1-3-methoxy-6-rnethy1-7-oxo-5,6,7,8-tetrahydroindolo[2,1 -
a] [2,5]benzodiazocine-11-carboxylic acid; 14-cyclohexy1-642-
(dimethylamino)ethy1]-3-
methoxy-7-oxo-5,6,7,8-tetrahydroindolo[2,1 - a] [2,5]benzodiazocine-11-
carboxylic acid; 14-
cyclohexy1-6[3.(dimethylamino)propy1]-7-oxo-5,6,7,8-tetrahydroindolo [2,1 -
a] [2,5] benzodiazocine-11 -carboxylic acid; 14-cyclohexy1-7-oxo-6-(2-
piperidin-l-ylethyl)-
5,6,7,8-tetrahydroindolo[2,1 -a] [2,5]benzodiazocine-11-carboxylic acid; 14-
cyclahexy1-6-(2-
morpholin-4-ylethyl)-7-oxo-5,6,7,8-tetrahydroindolo [2,1 - a] [2,5]
benzodiazocine- I 1-carboxylic
acid; 14-cyclohexy1-642-(diethylamino)ethy1]-7-oxo-5,6,7,8-
tetrahydroindolo[2,1 -a]
[2,5]benzodiazocine-11-carboxylic acid; 14-cyclohexy1-6-(1-methylpiperidin-4-
y1)-7-oxo-
5,6,7,8-tetrahydroindolo[2,1 - a] [2,5]benzodiazocine-11-carboxylic acid; 14-
cyclohexyl-N-
[(dimethylamino)sulfony1]-7-oxo-6-(2-piperidin-1-ylethyl)-5,6,7,8-
tetrahydroindolo[2,1 - a]
[2,5] benzodiazocine-11 -carboxamide ; 14-cyclohexy1-6- [2-
(dimethylarnino)ethylj-N-
[(dimethyl anaino)sulfonyl] -7-oxo-5 ,6,7,8-tetrahydroindolo [2,1 - a] [2,5]
benzodiazocine-11
carboxamide; 14-cyclopenty1-642-(dimethylamino)ethyl]-7-oxo-5,6,7,8-
tetrahydroindolo[2,1 -a]
[2,5]benzodiazocine-11-carboxylic acid; 14-cyclohexy1-5,6,7,8-
tetrahydroindolo[2,1 - a]
[2,5]benzodiazocine-11-carboxylic acid; 6-ally1-14-cyclohexy1-3-methoxy-
5,6,7,8-
tetrahydroindolo[2,1 - a] [2,5]benzodiazocine-11-carboxylic acid; 14-
cyclopenty1-642-
(dimethylarnino)ethyl]-5,6,7,8-tetrahydroindolo[2,1 - a] [2,5Thenzodiazocine-
11-carboxylic acid;
14-cyclohexy1-642-(dimethylamino)ethyl)-5,6,7,8-tetrahydroindolo[2,1 -
a][2,5]benzodiazocine-
11-carboxylic acid; 13-cyclohexy1-5-methy1-4,5,6,7-
tetrahydrofuro[3',2':6,7][1,4]diazocino[1,8-
a] indole-10-carboxylic acid; 15-cyclohexy1-642-(dimethylamino)ethy11-7-oxo-
6,7,8,9-
tetrahydro-5H-indolo[2, 1 - a] [2,6]benzodiazonine-12-carboxylic acid; 15-
cyclohexy1-8-oxo-
6,7,8,9-tetrahydro-5H-indolo[2,1-a][2,5]benzodiazonine-12-carboxylic acid; 13-
cyclohexy1-6-
oxo-6,7-dihydro-5H-indolo[1,2-4[1,4]benzodiazepine-10-carboxylic acid; and
pharmaceutically
acceptable salts thereof.
The HCV replicons and NS5A inhibitory activity of the present compounds may
be tested using assays known in the art. HCV inhibitors, such as those
described in the
Examples herein have activities in genotype lb, 2a and la replicon assays of
from about I pM to
about 1 uM. The assay is performed by incubating a replicon harboring cell-
line in the presence
of inhibitor for a set period of time and measuring the effect of the
inhibitor on HCV replicon
replication either directly by quantifying replicon RNA level, or indirectly
by measuring
- 41 -
:A 02756172 2011 09 21
WO 2010/111483 PCT/US2010/028653
enzymatic activity of a co-encoded reporter enzyme such as luciferase or 0-
lactamase. By
performing a series of such measurements at different inhibitor
concentrations, the effective
inhibitory concentration of the inhibitor (EC50 or EC90) is determined. See
Jan M. Vrolijk et al.,
A rep/icons-based bioassay for the measurement of interferons in patients with
chronic hepatitis
C, 110 J. VIROLOGICAL METHODS 201 (2003). Such assays may also be run in an
automated
format for high through-put screening. See Paul Zuck et al., A cell-based fl-
lactamase reporter
gene assay for the identification of inhibitors of hepatitis C virus
replication, 334 ANALYTICAL
BIOCHEMISTRY 344 (2004).
The present invention also includes processes for making compounds of
formula (1). The compounds of the present invention can be readily prepared
according to the
following reaction schemes and examples, or modifications thereof, using
readily available
starting materials, reagents and conventional synthesis procedures. In these
reactions, it is also
possible to make use of variants which are themselves known to those of
ordinary skill in this art,
but are not mentioned in greater detail. Furthermore, other methods for
preparing compounds of
the invention will be readily apparent to the person of ordinary skill in the
art in light of the
following reaction schemes and examples. Unless otherwise indicated, all
variables are as
defined above. The following reaction schemes and examples serve only to
illustrate the
invention and its practice.
General Schemes
Scheme A-1
o,No2 A.20, N1-12
H2 Pd-C1 f HCI,NaNO2 Cy N.NI-12
019.5
H2N N N
2. SnCl2, HCI
1 2 3 4
=I( PPA _ HCI-12N N
A
HTU
I * NH2
N
RCOOH
5 6 7
0
0,
RAU
1-12N N
______________________________________________ MIN IR
NH
N Or 0
RCOOH HATU
8
The synthesis of analogs containing the 4-azaindole core can be accomplished
starting from a suitably protected 2-amino-5-nitropyridine 2, which can then
be reduced by
catalytic hydrogenation in order to convert the resulting free amino group to
its hydrazine by the
action of NaNO2 and SnC12. The resulting pyridylhydrazine can be condensed
with a ketone
-42 -
:A 02756172 2011-09-21
WO 2010/111483 PCT/US2010/028653
then subjected to Fisher indole cyclization conditions to afford the indole 6.
Acidic deprotection
of the acetyl groups can be accomplished by using a strong acid to liberate
the diamine, which
can be selectively coupled on the more reactive aniline nitrogen using
standard coupling agents,
such as HATU. The aminopyridine group can then be acylated with a reagent,
such as acetyl
chloride or a carboxylic acid, in the presence of an amide bond-forming
reagent.
Scheme A-2
----y, "2 NBS
r NH2
II, -,:-1'' DMSO,H20, OC I. ...-CI
N Br N Br
NH,
1 2 Pd(PPh3)2C12, CUI ¨TFAA,Py
,---
I ____________________________________________ * \ / = = NH
DMF E3N
N >,¨ 1,4-
clioxane
H2N
E13N 0 io Br 01
41i ---* 1
AN 5
H
4
3
0õ 0,
Br N.,, i 3N HC1 Br rµ1,.,----- le, NH, HAM
lik Br N
H H H
6
2-Bromo-3-aminopyridines can be coupled to a terminally substituted alkyne
using standard Sonagashira coupling procedures to give intermediates 5, which
can undergo
TFAA-mediated cyclization to provide the 4-azaindole compounds 6. Protecting
groups can be
removed with a strong acid, such as aqueous HC1, and the resulting amine can
be acylated using
an appropriately substituted carboxylic acid and an amide bond forming
reagent, such as HATU.
Scheme B
0
Et$N õ,....,õNi=IPW
I
0 . 0 N-" BuLi ,
_______________________________ x
.
1-12N---NN--- Phil-Irµ N + PivCI N
1 2 H 3
PivHN
/ \ aq HBr H2N õ, ...
*HATU, DI EA
PIvHN * ¨ Ileat
ki , $, 11 NH, ____ p..-
0 NHIptv 11 RCOOH
4 5
R N 0õ H CI 0
).'¨R R N >--R
YH = Nvi NCS . Y i ` ip NH
0 N / N 0 N.'- N
H H
6
The synthesis of scaffolds B containing a 6-azaindole core can be accomplished
by the metallation of the 4-methylpyridine analog 2 with a strong base, such
as BuLi, and
quenching the resulting anion with the acylating agent, such as 3.
Intermediate 4 can be globally
deprotected by the action of a strong acid, such as HBr, to give the diamino
azaindole structure 5.
Both amino groups can be acylated using an appropriately substituted
carboxylic acid and an
-43 -
:A 02756172 2011-09-21
WO 2010/111483 PCT/US2010/028653
amide bond forming reagent, such as HATU. Compounds 6 can be further
finictionalized at the
C-3 indole position with electrophiles, such as NCS.
Scheme C
02N
02N
KI, 1002, H2$04, F120. PdC12(PRI13)2, Et3N, NH KOeu
7
N NH, 1000
NH,
2 NH, 3
0
07N I NH He' \ HATU 02N 112R -C
Nr W
RCOOH N N
4 5
H2N ( \>-R HAT UN NCS
õõ NH RCOON 8
N , I , NH N
----- 8 I W'
N N N
Fl
7 a
Iodo aminopyridines 2 can be coupled to a terminally substituted alkyne using
standard Sonagashira coupling procedures to give intermediates 5, which can
undergo a base-
mediated cyclization using a reagent, such as KOtBu, to provide the 7-
azaindole compounds 4.
Protecting groups can be removed with a strong acid, such as aqueous HC1, and
the resulting
amine can be acylated using an appropriately substituted carboxylic acid and
an amide bond
forming reagent, such as HATU. Compounds 6 can then be reduced using hydrogen
and a
catalyst then coupled a second time with a carboxylic acid and HATU to provide
8. Treatment
of 8 with an electrophilic agent, such as NCS, provides the desired compounds.
Scheme D
0
so Hlar. Dlu19H NiaNH2 Et20
02
02N 07N
1 3
2 4
al,OHMCS' 4
1 OZO0E1 HBr, HOM
1 0 2 CHA 1P
7 I E13N, THF
6 0 R O
5 6 7
1rN04-4:4_0_1,402 Snap R
HART'Cu0.01)111pEA,
0 0
9
C")Y.Pi F-r-N\ NH
tr-Lo 0 f
Compounds in the D series can be synthesized by reacting dicarbonyl
intermediate 2 with a 2-aminopyrirnidine derivative in the presence of a Lewis
acid, such as
-44 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
boron trifluoride etherate. The resulting heterocycle can be alkylated with a
bromoketone analog
of an amino acid, such as proline, in the presence of a tertiary amine base.
The nitro group can
be reduced, and the resulting aniline can be acylated using an appropriately
substituted
carboxylic acid and an amide bond forming reagent, such as HATU, to give the
final products.
Scheme E-1
NH2 0
-ThrOH
ON
02N 4111" NH 02N 10 N\ 4. NH, Boc 0 so N = NH ,,---,
ONlj
112N ip ,= H
OH130c/ N HOR DIPEA
'PA H EDCI, DMF, FT 3
1 2
HO
02N
deprotect )---R ChN 0 N,),_,,G,_ .7---
\> reduce
0
0 IIIN it NP-D 111(--11 __ -.
N
H 11 HATU, DIPEA H
HCI 0---- -R
4 5
OH
N2N ilth N ,-) :rmr
erre 0 CIY1 Ak'' deprotect
4,=P
H
0 I N ElIC: ITINTART N
H
6 / 7 0-AR
14
clirm
H 01101 N\ * NH
_ cY1
0= N C?--C MOON N H n
H
O\ HATU, D1PEA R.--"Lo 0 VP r;' . NT -N
8 0 c))
NO R H,.R
Scaffold E-1 can be prepared by the condensing a benzoic acid derivative, such
as
1, with a phenylenediamine counterpart 2 in the presence of a dehydrating
agent, such as
polyphosphoric acid. The resulting aniline can be acylated using an
appropriately substituted
carboxylic acid, such as N-Boc-L-proline, and an amide bond-forming reagent,
such as HATU,
and can then be subjected to acidic conditions to remove the Boc group.
Compounds 4 can be
coupled again with an appropriately substituted carboxylic acid and an amide
bond-forming
reagent, such as HATU. The nitro group in 5 can be reduced under catalytic
hydrogenating
conditions, and the resulting aniline can be further coupled with various
amines to give the target
compounds.
-45-
:A 02756172 2011 09 21
WO 2010/111483 PCT/US2010/028653
Scheme E-2
No2 No2 HATU N
(L H NHBoc I.,NH2
AcOH
L
OH '*LNIH2 NH,
1 2 3
NO2 NO2
=40:")-0¨NH2 ROOOH N\=
R PCl/C,
HATU, DiPEA
=
Fi 0
4 5
NH2 9 0
Ar¨tcr Ar+NH N ,
N)___0_R ____________ 8
b¨NH N
Fi 8 TEA
6
Scaffold E-2 can be prepared by the reacted with a benzoic acid derivative
with a
phenylenedia.mine analog and an amide bond-forming reagent, such as HATU, to
give amides 3,
which can be cyclodehydrated by heating with a reagent, such as HOAc. The
resulting aniline
can be acylated using an appropriately substituted carboxylic acid and an
amide bond-Twining
reagent, such as HATU, to give intermediates 5. The nitro group can be reduced
under catalytic
hydrogenating conditions, and the resulting aniline can be sulfonylated with
an appropriately
substituted sulfonyl chloride and a tertiary amine base to give the targets.
Scheme F
0.0H
mThr1-1
o H2N NH2 NH2 H2N 40, L.
H2N_ ip
OH PPA 0 HATU
1 2
(N.11rN
4P- oN)--04y----r)y 0=N, *
0 Boc 0
2HC1
4
3
RCOOH
40 =
H
R 0
HATU
0 R
Scaffold F can be prepared by the condensing a benzoic acid derivative, such
as 1,
with an amino phenol counterpart 2 in the presence of a dehydrating agent,
such as
polyphosphoric acid. The resulting aniline can be acylated using an
appropriately substituted
carboxylic acid, such as N-Boc-L-proline, and an amide bond-forming reagent,
such as HATU,
and can then be subjected to acidic conditions to remove the Boe group.
Compounds 4 can be
coupled again with an appropriately substituted carboxylic acid and an amide
bond -forming
reagent, such as HATU.
- 46 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Scheme G-1
HO Br
0211-0--/ so NO2
DBU, 1,4-dioxarie, C" 411111'lh NO2
11141111P NIP
C'2 DPEA, 1,4-o9-n9-
0 H 0
1 2 3
Fe, I-ICI 1." 1111, RDOOH OyN ash N
NH2
M, 0 DIPEA, HATO R 10) 0
4 5
Compounds having the benzofuran structure G can be prepared by reacting an
appropriately substituted salicylaldehyde with a benzyl halide, such as 4-
nitrobenzyl bromide, in
the presence of a tertiary amine base to give ethers 2. The benzylic ethers
can be treated with a
base, such as DBU, and heated to elevated temperatures to effect cyclization
to the benzofurans
3. The nitro groups in 3 can be reduced under catalytic hydrogenating
conditions, and the
resulting anilines can be coupled with various carboxylic acids to give the
target compounds G-
1.
Scheme G-2
o O CHO
Br 10 cH Br Br
NO2
base
1111 H 0 gr o
No2
1 2 3
LiHMDS H211
SO \ NO2 RC0011 RTN Amh.
0 couple 0
4 5
R N
R'CO011 fl
reduce Y 40 it)
0 1\1117 couple Y \ NH
0 0
For differentially substituted compounds (R, R') having the benzofuran
structure
G, the synthesis can be modified by reacting an appropriately substituted
bromo salicylaldehyde
with a benzyl halide, such as 4-nitrobenzyl bromide, in the presence of a
tertiary amine base to
give ethers 2. The benzylic ethers can be treated with a base, such as DBU,
and heated to
elevated temperatures to effect cyclization to the benzofurans 3. The aryl
bromide can be
converted to the aryl amine by reaction with LHMDS and a palladium catalyst to
provide 4,
which can be coupled to an appropriately substituted carboxylic acid to give
5. The nitro group
in 5 can be reduced under catalytic hydrogenating conditions, and the
resulting aniline can be
coupled with a second carboxylic acid analog to give the target compounds G-2.
- 47 -
:A 02756172 2011 09 21
WO 2010/111483 PCT/US2010/028653
Scheme H
0 NO2 Rr ii I + 1)acetone, refIr N \
NO2 __________________________________________________________ SnCl2 )
N NH, Br 2)methanot, Har ''NN
0
= NH2 R'COOH 1 HATU
RI.,...ri \
N N ,-.---,IN
4
Appropriately substituted aminopyrimidines can be cyclodehydrated after
acylation with an appropriately substituted ketone, such as 45-nitro-2-
bromobenzophenone, by
heating in a solvent, such as Me0H, and an acid source, such as HBr. The
resulting heterocyclic
nitro compound can be converted to the aromatic amine by reduction with a
reagent, such as
SnC12. The final compounds H can be obtained by reacting 4 with an
appropriately substituted
carboxylic acid and an amide bond-forming reagent such as HATU.
Scheme I
+ Br a No, thanoe 0,N .,.,
1)actone,fit12; re 0,1 \ 11 NO, __ reduce
\ 2)mel, I i Br ----= --N
NH,
0
1 2 3
0
H
H2Nõ, __,=_, r:j -6 to NH, RC001-1 / HATU RY \ 4.
----1"-
,---14 OL N(14
4
Compounds in scheme 1 can be prepared by reacting the appropriately
substituted
aminopyridine with 4'-nitro-2-bromobenzophenone by heating in a solvent, such
as acetone, then
effecting a cyclodehydration reaction using methanol and an acid source, such
as HBr. The
resulting heterocyclic nitro compound 3 can be converted to the aromatic amine
by reduction
with a reagent such as SnC12. The final compounds can be obtained by reacting
4 with an
appropriately substituted carboxylic acid and an amide bond-forming reagent,
such as HATU.
Scheme J
02N At POBra 02N Br PdOpPf)C12 02N
\ \ NHBoc
0 * lo \ 7 [
H N floe H Soc
H
1 2 (}10}2a \ 0 3
NHBoc
02Nill Ai NH,
Ai
HCI-Me0H , \ / Pd/C N2N NH? io
. ,
4111, N N tilir H2
H H N N IIII"
H H
4 5
H
RGOOH
0
N N
H H
-48-
/102756172201109-21
WO 2010/111483
PCT/US2010/028653
Compounds in scheme .1 can be prepared by coupling indole boronic acids with
an
appropriately substituted 2-bromoindole, such as 2, under standard Suzuki
conditions. The
protecting groups can be removed with HC1, and the nitro group in 4 can be
reduced under
catalytic hydrogenation conditions. The penultimate diamine can be coupled
with an
appropriately substituted carboxylic acid and an amide bond-forming reagent,
such as BOP,
reagent to give compounds with the targeted central scaffold.
Scheme K-1
02N
Br dim
N,NH2 HN 411) Br H+ Br ip NO2 LIHMOS
¨N
2 3
H2N
H2N
reduce -
NO2 __ , \ ,H7"00õo0710
NH
couple R N
4 5
The synthesis of compounds with the indole core scaffold K can be prepared
using standard Fisher indole synthesis protocol starting for an aryl hydrazine
and a ketone such
as 2. Conversion of the aryl bromide to the aryl amine 4 could be effected by
the Pd-catalyzed
reaction with LHMDS. The nitro group could be reduced and the diamine can be
coupled with
an appropriately substituted carboxylic acid and an amide bond-forming
reagent, such as HATU,
to give compounds with the targeted scaffold.
Scheme K-2
No, 40
reduce N112 protect BocHH io Bub B(ONte)a
N /1
1 2 3 Boc
BocNH
\
B(01-1)2 couple B"NH \ 41_4402 deprotect
WHX
Br-4, ., ¨NO2 N Z¨Y
Boo
4
Z¨Y
H2NW=X RCOOH R yN so
reduce
=)_.
(X¨NO2
N \/, NO2
6 7
W=X
acylote
"ICC)
6 N z¨Y
8
In an alternative procedure indoles K can be prepared starting from a suitably
protected and substituted aminoindole 3. Lithiation and quenching with a
boronate ester affords
key intermediate 4, which can be coupled to an appropriately substituted aryl
or heteroaryl halide
20 to provide targets 5. The Boc groups can be removed with acid, and the
resulting aniline can be
- 49 -
1102756172201109-21
WO 2010/111483
PCT/US2010/028653
coupled with an appropriately substituted carboxylic acid and an amide bond-
forming reagent,
such as HATU. The nitro group in 7 can be reduced and coupled in a second
amide coupling
reaction to give the desired compounds.
Scheme L
402. Pd, HAD, Ac20
AcHN n COON ____ AcHN COOH
0
1 2
= fill N'NH2
AcHN .110 )n AcHN A cHN rift 410ip
NH NHAc HCI
104 FlAc MOH
4
3
H2NNN2 HATU =ip NH
NH R000l-1 NI
5 5 6
Tetracyclic indole scaffold L can be prepared as outline in the scheme above.
Cyclization of a carboxylic acid derivative 2 with PPA can provide the ketones
3, which can
participate in a Fischer indole reaction with an appropriately substituted
phenylhydrazine to give
4. The acetamide groups can be deprotected under acidic conditions and the
resulting aryl
10 amines can be coupled with an appropriately substituted carboxylic acid
and an amide bond-
forming reagent, such as HATU, to give compounds with the targeted scaffold.
Scheme M-1
NH7
HATU
0 y NH,OAc
y--ON
HC HO bz
Br lir
1 2 Br NH Cba 1400
3
Pd(cippt)C1 Boo'N
\ = TFA
N
Bac
Cba N`-' H
u 0-1)¨B(OH7
5 Cbz"õ
4 Boo
RCOOH._ RyN =
HATU 0
Cbi
N
H H
6 7
deprotect II sp ft
couple H 11
0
Scaffold M-1 compounds can be prepared by coupling proline 2 to amino ketone
15 1 using standard amide bond-forming procedures to provide 3, which can
be cyclized upon
heating with ammonium acetate at elevated temperatures. Intermediate 4 can be
coupled to
-50-
:A 02756172 2011 09 21
WO 2010/111483 PCT/US2010/028653
indole boronic acids, such as using standard Suzuki-type conditions. The Boc
groups can be
removed with acid, and the resulting aniline can be coupled with an
appropriately substituted
carboxylic acid and an amide bond-forming reagent, such as HATU. The
pyrrolidine protecting
group can be removed under hydrogenating conditions, and the resulting amine
can coupled in a
second amide coupling reaction to give the desired compounds.
Scheme M-2
cbz
Et1AgSr N NH2NH2 N
Cbi Br Cbi * Br
0-
1 2 3
P01(11) &protect
N
(Boc)2Nci,R(011)2 CI4 \
lµ4130c)2
4
H2N ist N,N
\ phz RCOOH 0 N
N _________________ r y
L.
70090e R \ 0- pi:a
5
01, NH a \= \,,H7
1 deprotect oyN 116 N- Os
R
2 R'COOH N
couple
7
Scaffold M-2 compounds can be prepared by reacting praline 1 with an anion of
4-ethynylbenzene to give intermediate ketone 2, which can be cyclized with
hydrazine.
Intermediate 3 can be coupled to indole boronic acids, such as using standard
Suzuki-type
conditions. The Bac groups can be removed with acid and the resulting aniline
can be coupled
with an appropriately substituted carboxylic acid, and an amide bond-forming
reagent such as
HATU. The pyrrolidine protecting group can be removed under hydrogenating
conditions, and
the resulting amine can coupled in a second amide coupling reaction to give
the desired
compounds.
Scheme M-3
cl,4.74H2 cs)4142
N 0 N
'CID7 bbz \Cbz
1 2 3
Scheme 1,4.2 Ryil,\ 0
/
JR
- 51 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Thiazole analogs of scaffold M can be prepared from the cyclocondensation
reaction of Z-pro line thioamide 2 with an alpha-bromoacetophenone. Products 3
can be
processed to the final compounds using methodology similar to that described
in scheme M-2.
Scheme M-4
\
Br 41111". p4H2 NaHCO3 %
THF --Br CL2
Cbz Br
1 2 3
Scheme M-2 0 101\
R 0
N
Imidazole analogs of scaffold M can be prepared from the cyclocondensation
reaction of Z-proline bromornethyl ketone 1 with an aromatic amidine
derivative. Products 3 can
be processed to the final compounds using methodology similar to that
described in scheme M-2.
Scheme M-5
0-s
NBS
N N N +
ot\lio NH, H20 Br *
0 110
NO2
Pd(dppfp, d CLiN
deprole,
\ Mum
o
THF/H,o
0 IMP NO,
5 6
nAN H1.4
coupte RGOOH
N io N
o 0
7
HN
e
,ouP1 y N N \ Ark NI-R
L
deproied NH N dab \ 4. NH
R COOH WI 0 W iP 0 IR'
9
Isomeric imidazoles can be prepared starting from a protected amino acid
aldehyde, such as 1, and glyoxal in the presence of ammonia. Halogenation of
the resulting
imidazole 2 with NBS can be followed by a Pd-catalyzed cross coupling reaction
with a
functionalized indole boronate ester, such as 4. Deprotection, reduction and
coupling with an
appropriately substituted carboxylic acid and an amide bond-forming reagent,
such as HATU,
can provide intermediate compounds 8. A second deprotection/amide-coupling
procedure can
provide the targeted M-5 scaffold.
- 52 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Scheme M-6
0 0 0 0
\ hydrazine, Hr4H, cot,pie
N
H 2 * 3
LOA
t
cyclodehydrate protec B(01-Pr)7
CbZ " Cld I
N.,
4 5
B. P=PHt)212 NR
io
R N 40 ______________________________________
ed w reflux, 24 h
N-N Cbi /
N-N
7 8
reduce NTR to MIR
deproleot
N. N Roc N
9 10
couple
R'COOH H
0
Oxadiazole compounds can be prepared starting from indole hydrazide 2 and
coupling to an amino acid, such as Z-proline. Cyclodehydration of intermediate
3 can be
effected with a reagent, such as TPP/iodine, to give the desired oxadiazole,
which can be
protected on the indole nitrogen with Boc anhydride. Introduction of the
boronic acid functional
group activates compound 6 for coupling with a substituted aryl halide 7 to
give intermediate 8.
Removal of the cbz and Boc groups afford the penultimate structure 10, which
can be coupled
with an appropriately substituted carboxylic acid and an amide bond-forming
reagent to give the
targets M-6.
Scheme M-7
0
HN 101,
c-µ74H
I 40-14
CbZ
Chz Cbz 0
1 2 3
Scheme M-1 , AL
r
8
Liir" N
Oxadiazole analogs of scaffold M can be prepared by cyclocondensation
reactions
of diacylhydrazines 2. Coupling to heterocyclic boronic acids using
methodology similar to that
described in scheme M4 can provide the targeted compounds.
- 53 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Scheme M-8
("
0.,., Oxalyl Chloride 0c N NOS
-.-CN)---("%---Br Na2S03
i DMSO/NE13 B04 0 wis H20 13,4 HN---
11 [I,,' HN---1( )" 001HN-jrc
Boc Br Br
1 2 3 4 5
Br 0
40 0...,....- NBS 0 O, , Br a Br
l'H Os2003/DMF,. I \
_________________________ =-= * Br
0 0 reflux '-'-- 0
Br Br lir OH
6 7 8 9
'5' / Pd(II¶ Na2C00
U 10 a 0-.B 0 , . E,,\0_ ______________ .
THF/H20
Pd(S) /KOAc I dioxane o 0
11
N
N 1.1 I / 111 deprotect N
it / liJy
,
couple
Boo ....-- 0
11--).¶ ' C.--% \ .e
RCOOH
,,N---/ NH H 11 HN__)
12 Bee
13
N \ * / (\.,,,
(....õ) \
N lip = N N_.)
N
R
Double imidazole containing benzofuran compounds can be prepared starting
from a protected amino acid aldehyde, such as 2, and glyoxal in the presence
of ammonia.
Halogenation of the resulting imidazole 3 with NBS can ultimately provide
intermediate 5,
which can be coupled to a functionalized boronate ester, such as 11, to
provide 12. Deprotection
and coupling with an appropriately substituted carboxylic acid and an amide
bond-forming
reagent, such as HATU, can provide the targeted M-8 scaffold.
Scheme M-9
\ pH NO)
1,DA la, Br pdoi)
_________________________________________________________________ * \ (tt
Br-f-al , Bo_pr0)3 Br io ,, , ... Y Br Br ____ *
/ 0 0 OH 0
i 1111"
X 4 X
1 2 3 6
Y---z Z;CY--)
I N deptotect
Pc1(11 ________________________________ * n-4 ___ =\ . Boc
x , .
Na2CO3 N X
0
X Boc X
6 7
RCOOH
H
couple n _____________________
x -KJ iti p 0
8 R X
An alternative synthesis of benzofurans can be realized starting from
benzofuran
1, which can be converted to boronate ester 2, which can then coupled to an
appropriately
-54-
1102756172201109-21
WO 2010/111483 PCT/US2010/028653
substituted aryl halide to afford 5. Intermediate 5 can subsequently be
converted to a
functionalized boronate ester and converted to the final products in a manner
similar to that
described in Scheme M-8.
Scheme M40
45_8,\o--_c_
(5--(:)--
HO Br . Br NH2
MAI OH PPA Br N
III \ it
0 Br 0 .
Pd(II)
1 3
Y
)49
0-43 110 + crj-- POI) L-N.Boo X 411
0\ 41P 13\0-''¨ '
NBec Na,CO2 i
Y--- Y-z
ir
coupE,T Crj\ X so N
Y
N¨/
ROOH Ri'l0 0
R
Benzoxazoles 3 can be prepared starting from a suitable substituted benzoic
acid
and an aminophenol, such as 2, in the presence of polyphosphoric acid. Such
products can be
converted to the corresponding boronate esters using standard procedures.
Intermediates 4 can
subsequently be coupled to a heterocyclic halide in the presence of a Pd(II)
catalyst to provide
compounds 5. Deprotection and coupling with an appropriately substituted
carboxylic acid and
an amide bond-foiming reagent, such as HATU, can provide the targeted M-10
scaffold.
Scheme N4
H HHN4
0 gli N mi
,NH2
8 411 NMP
crowave ______________________________________________ r ....,i.N 0
' PI
H
1 2 3
1
HC H2N NH2 RCOOH HN----iR
I H
____________________________________________ RiN 00---<1 ilS' HATU 0
H W N
H
4 5
The compounds in scheme N-1 can be prepared by heating hydrazines 1. with
ketones 2 in a microwave reactor in a polar aprotic solvent, such as NMP. The
indole
acetamides 3 can be deprotected with strong acid, such as HC1. The resulting
aryl amines can be
coupled with an appropriately substituted carboxylic acid, and an amide bond-
forming reagent,
such as HATU, to give compounds of the targeted scaffold.
- 55 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Scheme N-2
air NHAc
40 i c.,,pd,,,,,3,2.2 , µP InE3r3 _
,,,im
02N NH2 * NHAc
02N
H
I 02N NH2
2 3
___________________ 1 ip
NH, RCOON ' 02N )-"H R 142 t
N 02N N coupie
H H
4 5
C\=-=R ,
0 , iti toi R MOH
R'IN 16I N\ 11 NC31)1¨R
H2N H 6 couple H H
Iodo anilines 1 can be coupled to a terminally substituted alkyne using
standard
Sonagashira coupling procedures to give intermediates 2, which can undergo
cyclization using a
reagent, such as indium bromide, to provide the indole compounds 3. Protecting
groups can be
removed with a strong acid, such as aqueous HC1, and the resulting amine can
be acylated using
an appropriately substituted carboxylic acid and an amide bond-forming
reagent, such as HATU.
Compounds 5 can then be reduced using hydrogen and a catalyst, then coupled a
second time
with a carboxylic acid and HATU to provide the desired compounds.
Scheme N-3
PdC12(PPh3)2
07N si Nii, ,
Cul, E12111_._ 07N--Q ¨
. PdaFeC13
I
* WtO NH, HN4
i H 2 0
0
HN--
N ( HN-4,
10 \ = 0
reducer 0 N\ 1, 3N HO t
02N H2N m
H , 4
H,
H2N 1,1 RFIcAoTolin 1 iii ,,,,,, ip
al. IP *- R N
c ' H H HN--<
H 5
In a slight variation of scheme N-2, iodo anilines 1 can be coupled to a
terminally
substituted alkyne using standard Sonagashira coupling procedures to give
inteimediates 2,
which can undergo cyclization using a reagent such as palladium
chloride/ferric chloride to
provide the indole compounds 3. Compounds 3 can then be reduced using H2, and
the protecting
group can be removed with a strong acid, such as aqueous HCI, and the
resulting amines can be
acylated using an appropriately substituted carboxylic acid and an amide bond-
forming reagent,
such as HATU.
- 56 -
:A 02756172 2011 09 21
WO 2010/111483 PCT/US2010/028653
Scheme 0
Br 40 NH2r> H
NF,2 HO r)
,oe
z-_,õ1 ....
ebz illp 4 c.
1 2 3
B BocHN
r...,cc. <,-D
BocHN Pd Ni N
ri cbiN 0Q1-13(OH)2 I* \ .
N NH '61,z
BocBoo
4 5
K---r-M n
doprOec H2N t 1*\ N, r)
Ala- '7-.---''N couple i
, Bac 0 01
40N ,)
N\ NH cbz
N ir NH L,2.
H li 7
6
F---) H
deprotect Cly id nkrilsi deprolect cijNyr4 I. rifi;
, NN
Boo c, ______________________
14 H
8 9
H
RCOOH Cly N
N'NFI
). R:40 0 0 \ it , R
Scaffold 0 can be prepared by reacting a protected praline compound (such as
Cbz) with a phenylenediamine analog and an amide bond-forming reagent, such as
HATU, to
give amides 3, which can be cyclodehydrated by heating with a reagent, such as
HOAc. The
resulting benzimidazole can be coupled to an indole boronic acid derivative
using standard
Suzuki conditions to provide 5. Removal of the Boc groups with acid provides
7, which can be
acylated using an appropriately substituted carboxylic acid, such as Boc-L-
proline, and an amide
bond-forming reagent, such as HATU, to give intermediates 7. The Cbz group can
be reduced
under catalytic hydrogenating conditions, and the Boc group can be deprotected
with acid to
provide penultimate compounds 9. Amide bond formation between 9 and carboxylic
acids
afford the targeted compounds.
Scheme P-1
H X
H
fir NyAri_40....
-0-
s CITI'y -_ NH
NlioN ---
RCXO Y
0 C 0
R R
Heterocycles can be halogenated at C-3 by the action of electrophilic agents,
such
as N-halosuccinimides, to provide targets P-1.
Scheme P-2
\
Y \ ip NH N SELECIFLuoR
y 1 \ R it __
NH NI¨
R--k, ---k, 0 13,c N
OA
0 c N
H R 0 H R
- 57-
:A 02756172 2011-09-21
WO 2010/111483
PCT/US2010/028653
Heterocycles can be fluorinated at C-3 by the action of electrophilic
fluorinating
agents, such as SELECTFLUOR, to provide targets P-1.
Scheme P-3
H Br 0 ON
o
R 1,1
(k)¨R
R i. ,I,A,.. \H
NH GuCN/DMF , Ry yA, \ it
0
NH
--" 0 8, --- y
8'0 Y C
C-3 halogenated compounds can be converted to the corresponding cyano analogs
by cyanating agents, such as CuCN.
Scheme P4
0 IT'
CNII1N
IR--- 0 a \ 11 ZnC12/MeMgBr. 0
0 N N 0
ClirN -11&.'
R R'ACI R.-- 0 VI \ 411 140.,N
0 N
R
The compounds in scheme P-4 can be functionalized by the acylating indoles
with
Grignard reagents and zinc chloride.
Scheme P-5
R ¨ H
0 ,
Cmiy1 0, ,,--Th EtrAgB, . ay I,L.2 0
\ N,
sn_ZI 0 1 N 'NHirrN) 4111
t:,, or jv
0j\\I
0 H b --%0 N
Bn H R
R > H
. 0 0
H
0 0 nitrate ).. 02N 0 POBr3 02N Abli
protect 1:31\1 = \
. ______________ ,....
\ Br
CI MP V N Br
N N
H H H N
Bac
1 2 3 4
0 0
-41 OH
Pd(ciPpi)012 02N _ NaCI02, NaH2PO4 02N Am
__________________ a R¨NH2
40 \ 11 NHBoc m ¨y..-. *-
?le 1-4 butene MI \ IP
NHBoc
HR = N N couple *
B NHBoc Boo Boo
,
5 6
0 /, 0 r 0
NH NH H NH
reduce1.TFA s ON is
\ MAWR"
IF
0,N, so
\ . NHBoc ______________________ H2N
k 0 \ ilik NHBoc 2 HAM
N
N N acid, DIPEA R"
H
Boo Boo
7 8 9
The compounds in scheme P-5 can be functionalized by deprotonating the indoles
with a base such as ethylmagnesium bromide and treating the resulting
intermediate with
chlorosulfonyl isocyanate. Alternatively, the indoles 3 can be prepared using
Vilsmeier-Haack
conditions, which can subsequently be protected and coupled under Suzuki
conditions to give
intermediates 5. The aldehydes can be oxidized using standard methodology for
carboxylic acid
- 58 -
:A 02756172 2011 09 21
WO 2010/111483 PCT/US2010/028653
formation. Indole carboxylic acids 6 can be coupled to amines using a reagent,
such as HATU,
to give 7, which can be further functionalized by reduction of the nitro
group, deprotection of the
Boc group and coupling of the anilines to an appropriately substituted
carboxylic acid and an
amide bond-forming reagent, such as HATU.
Scheme P-6
Qyl zB(oH)2
fp NH N
c)
RA, 0 I \ NH N --ko 0 13,
c , N 0
0 c N ,µ Pd(cIppf}C12 R
TFH/HIC)
C-3 halogenated compounds can be coupled to a variety of alkyl and aryl
boronic
acids using standard Suzuki conditions.
Scheme Q
0914 N E7--0 1
(EtO.V00 CI' base
0N
0 lo 0
03-0E1
1 2 3
triflale 02N =Pd couple 02N NHAc reduco
011 011/
¨=-7¨CH,NHAc
EV/
4 5
H2N == basc H2k coo,õ RIN" /Num
4111r. N N
6 00N/0
7
Compounds of scheme Q can be prepared starting from the lactam 1. Reaction
with ethyl chlorofomiate and treatment of the product with a mild base, such
as ammonium
carbonate, provides intermediate 3, which can be activated for coupling by
conversion to the
corresponding vinyl triflate 4. Sonagashira coupling provides compounds 5,
which can be
reduced with iron and ammonium chloride to provide aniline 6. Deprotection of
the indole and
coupling of the aniline to an appropriately substituted carboxylic acid and an
amide bond-
forming reagent, such as HATU, provides the desired targets.
Scheme R
Br Alt
RCOON oot,t_ple Br so
N N
1
Z43(OH)2
.2 02¨H
Amide coupling of the aniline from scheme K-1 with an appropriately
substituted
carboxylic acid and a coupling agent can provide intermediates 2, which can
then be subjected to
Pd-catalyzed cross-coupling reactions to provide the final targets R.
- 59 -
:A 02756172 2011-09-21
WO 2010/111483
PCT/US2010/028653
Scheme S
NH
(NH BrCt4 HO.õ(41.
heat
N
.,..--..-1 ...._./-- H214NO2 ,
Z 3
1
t-BuNO2 02N.,..1.4, Rd(f1)
02N _._..N I ;.)¨Br
Culir2 ._
(BochN #
\ B(OH)2
N
4 5 Boc
(Boc)2N , 0 _ __ (82)0)2N , oy-....,1
deorotect ,
1 - ,,, .., CL. rolquw
AO , I, ,
N NO2 N 1,1-- 'NH,
Boo Roo
6 7
:fly H
H214 ..L..,_ 0
) ,, couple N is..ii
4 RIPR-k 0 \ 0 611
N N NH, RCOOH 0 M '1111-7 NH
8 OR
Compounds in scheme S can be prepared by coupling indole boronic acids with
an appropriately substituted 2-bromobenzoxazoles, such as 5, under standard
Suzuki conditions.
The nitro group in 6 can be reduced under catalytic hydrogenation conditions
and the protecting
groups can be removed with HC1. The penultimate diamine can be coupled with an
appropriately substituted carboxylic acid and an amide bond-forming reagent,
such as BOP,
reagent to give compounds with the targeted central scaffold.
Scheme T
.
Br h
HO
HO at
40 A=CI 0 0 Ana . I. ' 40
NH, AcOH,E1OH
Etiq A. ..,- '
Br e
HO Br Br
1 2 3 4
Z
H functionehze Br App. cyclIze
gli N-1,1, 010 MA \
,
2 10 W Br - 1- LIP N' lik
N B. ..
Br HO Br H HO H HO
5 6 7
Z n_( 1
Br Z PmB , Z
Pd(I)
IP N\ 11 ST '.- iv 4IP BPin __ "1) ,LN io
N ' N
sX--0 \--X X-0
Boo'
8 9 10
N
N
C-" .>_4, 1 Z
N L'I / N
deprotect I , N
1 ¨NH H 10itAi.õ, awgdo ccuOlinO, H ,,.0 - 0 ` it
R N
X-0 H Hri.) .
N
/(:)-c) R
11
Compounds in scheme T can be prepared by starting from a suitably substituted
phenol 3 and a hydrazine reagent, such as 4, using established Fisher indole
conditions. The
indole 3 position can then be functionalized or the indole NH can be cyclized
onto the C-2
aromatic ring using standard conditions to give tetracycles 8, which can
subsequently be
converted to the corresponding boronate esters using standard procedures.
Intermediates 9 can
- 60 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
then be coupled to a heterocyclic halide in the presence of a Pd(II) catalyst
to provide
compounds 10. Deprotection and coupling with an appropriately substituted
carboxylic acid and
an amide bond-forming reagent, such as HATU, can provide the targeted T
scaffold.
The following examples serve only to illustrate the invention and its
practice. The
examples are not to be construed as limitations on the scope or spirit of the
invention.
List of Abbreviations
Ac20 Acetic anhydride
B(O/Pr)3, (iPrO)3B Triisopropyl borate
B(OMe)3 Trimethyl borate
BF3 Boron trifluoride
BOC, Boc, boc tert-Butyloxycarbonyl
BOP Benzotriazole-1-yl-oxy-tris-(dimethylamino)-
phosphonium hexafluorophosphate
BrCN Cyanogen bromide
BuLi, n-BuLi Butyl lithium
CBZ, Cbz, cbz Benzyloxycarbonyl
CDC13 Deuterio-trichloromethane
CH3CN, MeCN Acetonitrile
Cs2CO3 Cesium carbonate
CuBr2 Copper(II) bromide
CuCN Copper(I) cyanide
CuI Copper iodide
DBU 1,8-Diazabicyclo[5.4.0]undec-7-ene
DCE Dichloroethane
DCM, CH2Cl2 Diehloromethane
DIPEA, DIEA Diisopropylethylamine
DMAP 4-Dimethylamino pyridine
DMF Dimethylformamide
DMSO Dimethyl sulfoxide
DPPF, Dppf, dppf 1,1'-bis(Diphenylphosphino)ferrocene
EDC, EDCI N-(3-Dimethylaminopropy1)-N'-
ethylcarbodinnide
Et20 Diethyl ether
- 61 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Et3N, TEA Triethylamine
EtMgBr Bromo(ethyl)magnesium or ethyl magnesium bromide
Et0Ac Ethyl acetate
Et0H Ethanol
FeC13 Ferric chloride or Iron(III) chloride
H2 Hydrogen or hydrogen atmosphere
H20 Water
1-12SO4 Sulfuric acid
HATU 49-(7-Azabenzotriazol-1-y1)-N,N,N',N'-
tetrarnethyluronium hexafluorophosphate
HBr Hydrobromic acid
HC1 Hydrochloric acid
HNO3 Nitric Acid
HOAc, HAc Acetic acid
HOBT, HOBt 1-Hydroxy benzotriazole
HPLC High performance liquid chromatography
InBr3 Indium tribromide
iPr2NH Diisopropylamine
K2CO3 Potassium carbonate
KI Potassium iodide
KI03 Potassium iodate
KOAc, AcOK Potassium acetate
KOH Potassium hydroxide
LDA Lithium diisopropylamide
LHMDS, LiHMDS Lithium hexamethyldisilamide
MeMgBr Bromo(methyl)magnesium or methyl magnesium
bromide
Me0D Methan(2H)ol
Me0H, CH3OH Methanol
MgSO4 Magnesium sulfate
MOC, Moe Methoxy carbonyl
MS Mass spectroscopy
N2 Nitrogen or nitrogen atomosphere
Na2CO3 Sodium carbonate
- 62 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Na2SO4 Sodium sulfate (anhydrous)
NaC102 Sodium perchlorate
NaH2PO4 Dihydrogen sodium phosphate
NaHCO3 Sodium hydrogen carbonate (sodium bicarbonate)
NaNO2 Sodium nitrite
NaOH Sodium hydroxide
NBS N-bromosuccinimide
NCS N-chlorosuccinimide
NH40Ac Ammonium acetate
NMM N-methylmorpholine
NMR, 1H-NMR Proton nuclear magnetic resonance spectroscopy
NXS N-halosuccinimide
P205, P4010. Phosphorus pentoxide
Pd Palladium
Pd(dppf)C12 Dichloro(1,11-bis(Diphenylphosphino)ferrocene)
palladium(II)
Pd(II) Palladium(II)
Pd(PPh3)2C12, PdC12(PPh3)2 Dichlorobis(triphenylphosphine)palladium(II)
Pd(PPh3)4 Tetrakis(triphenylphosphine)palladium(0)
Pd/C, Pd-C Palladium on carbon
Pd2(dba)3 Tris(dibenzylidene acetone)dipalladium(0)
PdC12 Palladium(II) chloride
PE Petroleum ether
Phg Phenylglycine
PhCH3, PhMe Toluene
Piv Pivaloyl
PivC1 Pivaloyl chloride
POBr3 Phosphorus oxybromide
PPA Polyphosphoric acid
PPH3, TPP Triphenylphosphine
Pro Proline
Proc iso-Propylcarbamate
ptau3 Tri-tert-butyl phosphine
- 63 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Py Pyradine
PyBOP (Benzotriazole-1-yl-oxy)-
tripyrrolidinophosphonium
hexafluorophosphate
RPLC Reverse phase liquid chromatography
RT, rt, r.t. Room temperature, approximately 25 C
Si02 Silica or silica gel
SnC12 Stannous chloride or Tin(II) chloride
SOCl2 Thionyl chloride
STP Standard temperature and pressure
t-BuLi tert-Butyl lithium
t-BuNO2 tent -Butyl nitrate
t-BuOH tent -Butanol
t-BuOK, KOt-Bu Potassium tent -butoxide
TFA Trifluoroacetic acid
TFAA Trifluoroacetic anhydride
THF Tetrahydrofuran
TLC Thin layer chromatography
ZnC12 Zinc chloride
EXAMPLES
Example 1 ¨N-14-15-(acetvlamino)-1H-pyrrolo13,2-blpyridin-2-vIlphenv11-1-
(phenvlacetv1)-L-
prolinamide
0
,N N
N )
NH N
0
Step 1
No,
Ac,,o
N
H2N
To a suspension of 2-amino-5-nitropyridine (25.0 g, 0.18 mol) and 0.5 g of
DMAP in 200 mL of pyridine, Ac20 (37 g, 0.36 mol) was added drop wise at 0 C.
The mixture
was stirred at RT for 5 hours. The volatile was removed in vacuo. The residue
was washed with
Et0Ac to yield an off-white solid (28 g, 86%). MS (ES!) mie (M+H+): 182.
- 64 -
CA 02756172 2014-03-03
=
Step 2
/CI =NO2
H2,Pd-C NH,
N
A heterogeneous mixture of 2-acetamido-5-nitropyridine (28 g, 0.15 mol) and
10% Pd/C (2.8 g) in 300 mL of Me0H was stirred in 50 psi of H2 for 6 hours.
The mixture was
filtered through CELliand concentrated in vacuo to yield a solid (20.5 g). MS
(ESI) tn/e
(M+H+): 152.
Step 3
0
A
N NuIJHz 1. HCI,NaNO2 0 -"I NH2
2. SnC12,HCI ANL
NaNO2 (5.4 g, 78.2 mmol) was added slowly to a solution of 2-acetamido-5-
aminopyridine (9.0 g, 60 mmol) in 6 M aqueous HC1 (300 mL) at 0 C and was
stirred for 45
minutes. A solution of SnC12 (40.5 g, 180 mmol) in 15 mL of 6 M aqueous HC1
was added, and
the reaction mixture was allowed to warm to RT slowly while stirring for 16
hours. The reaction
mixture was basified with 40 percent aqueous K.OH, extracted with Et0Ac (3x),
and the organic
layers are combined, dried over Na2S0.4 and concentrated in vacuo to give the
desired compound
(3.2 g). MS (ESI) m/e (M+H+): 167.
Step 4
"
v... IT ..N,
0 Et,N ? N lilt W
N N pH=9 5
A suspension of the product from step 3 (3.32 g, 20 mmol) and N-(4-
acetylphenyl)acetamide (3.54 g, 20 mmol) in 8 mL of Et0H was diluted with TEA
to adjust the
pH to about 9.5. The resulting reaction mixture was refluxed for 3 hours. The
solvent was
removed in vacuo, and the resulting residue was treated with 5 % aqueous
citric acid to form a
precipitate. The precipitate was filtrated, washed with water and dried in
vacuo (3.2 g). MS
(ESI) mle (MA-114): 326.
Step 5
ppA ===,,N N
NH
0 0 0
A mixture of the product from step 4 above (0.6 g, 1.8 mmol) and PPA (5 mL)
was heated to 90 C for 75 minutes under N2. After cooling to RT, the reaction
mixture was
- 65 -
:A 02756172 2011 09 21
WO 2010/111483 PCT/US2010/028653
poured to an ice water, neutralized with solid NaOH, while maintaining the
temperature of the
mixture at or below RT. A solution of iso-propanol and DCM (1:3) was added to
exact the
organic. The combine organic phase was washed with brine, dried over Na2SO4
and
concentrated in vacuo. The residue was purified by preparative HPLC to yield a
solid (280 mg).
MS (ESI) m/e (M+114): 309.
Step 6
õ11-11 N
P \ NH HCI 112N,N,.
1 \ NH2
0 N
N
A mixture of the 4-azaindole (280 mg, 0. 9 mmol) in 10 mL of 3 N HC1 was
refluxed for 2 hours. The solvent was removed in vacuo. The residue was
purified by HPLC to
yield a solid (120 mg). MS (EST) mie (M+H): 225.
Step 7
0 . 0
H2N N H2N N
\ IF NH2 + Ho N HATU, ip, NH N
N
0 41, 0 4It
To a suspension of the product from step 6 (23 mg, 0.1 mmol), acid (23 mg,
0.1 mmol) and D1PEA (20 mg, 0.15 mmol) in 1 mL of CH3CN was added HATU (42 mg,
0.12 mmol). The resulting mixture was stirred at RT overnight. After reaction
completed, the
mixture was purified by pre-HPLC (10 mg). MS (ESI) mire (M+H+): 440.
Step 8
0
0
H2N N s
11\ N Si
N 0
A mixture of product from step 7 (10 mg, 0.023 mmol) and TEA (3 g, 0.03 mmol)
in CH3CN (100 mL) was stirred at 0 C. Acetyl chloride (2 mg, 0.023 mmol) was
added
dropwise, and the resulting mixture was stirred at RT for 0.5 hour. The
solvent was evaporated
in vacuo, and the residue was purified by preparative HPLC to afford the
desired product (5 mg).
MS (ESI) rn/e (M+H+): 482. 1H NMR (Me0D): 6 8.25 (d, 1-8.4 Hz, 1H), 7.74 ¨
7.89 (m, 411),
7.26 ¨ 7.32 (m, 511), 7.00 ¨ 7.02 (m, 2H), 4.63 ¨ 4.64 (m,11-1), 3.72 ¨ 3.84
(m, 4H), 2.18 ¨ 2.33
(m, 211), 2.07 ¨ 2.10 (m, 511).
- 66 -
:A 02756172 2011-09-21
WO 2010/111483
PCT/US2010/028653
Examples 2-3
The compounds of Examples 2 and 3 were prepared in a similar manner starting
from intermediate 7 in step 6.
Example Structure MW Name
2 Fl 654.776 (2,5)-1-
(phenylacety1)-N-{244-
40 N N
I \ IP NHo N
0 (phenylacetyppyrrolidin-
2-
0
ylicarbonyl}amino)phenylj- 11/-
pyrrolo[3,2-b]pyridin-5-
yl}pyrrolidine-2-carboxamide
3 H 0 686.774 benzyl (25)-
24(2-{44({(2,5)-1-
N N.
NH N [(benzyloxy)carbonyljpyn-olidin=-
I
C:A0 0 N2-yl}carbonypamino]pheny1)-
}i
1if-pyrrolo[3,2-bjpyridin-5-
.
yl)carbamoylipyrrolidine-l-
carboxylate
Example 4¨ tert-butyl f(1S)-2-1.(2S)-244-(5-broma-IH-pyrrolol3,2-blpyridin-2-
v1)Phenyllearbamoylipyrrolidin-l-yll-2-oxo-1-phenylethylkarbamate
\ lit
N
goc-14H
Step 1
NH2 NBS NH2
Br"- N Br
NBS (14.9 g, 84 mmol) was added portion wise to a solution of compound 3-
aminopyridine (14.9 g, 84 mmol) in DMS0 (80 mL) and water (20 mL) at 0 C, and
the reaction
was stirred at RT for 3 hours. The mixture was poured into ice-water (250 rnt)
and stirred for 30
minutes. The precipitate was collected and dried to yield a solid (7.0 g). MS
(ESI) mie (M+1-1+):
250. 1H NMR (DMS0): 8, 7.28 (d, J7.6 Hz, 1H), 7.03 (d, J7.6 Hz, 1H), 5.69 (s,
2H).
Step 2
Et,N 0õ so
.2NN
4-Ethynylacetanilide was prepared using the similar method shown in Example 1,
step 1. MS (ESI) m/e (M+H+): 160.
Step 3
NH,
NH2
))t, 101 pcephop,,cui
=- 41. NH
Br ti Br Et3N
Br
- 67 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
To a solution of 3-amino-2,6-dibromopyridine (9.41 g, 37.5 mmol), 4-
ethynylacetanilide (4.77 g, 30 mmol) and Pd(PPh3)2Cl2 (1.31 g, 1.9 mmol) in a
mixture of 150
mL of Et3N and 50 mL of DMF was added CuI (0.71 g, 0.4 mmol) under N2. The
resulting
mixture was stirred at RT overnight. The solvent was removed, and the residue
was purified by
chromatography (8.5 g). MS (ESI) m/e (MAT): 331. 111NMR (DM50): 6 7.59 (d,
J=8.8 Hz,
2H), 7.54 (d, J=8.4 Hz, 2H), 7.21 (d, J=8.8 Hz, 2H), 7.07 (d, J=8.4 Hz, 2H),
2.12 (s, 3H).
Step 4
NH2
TFAA,Py
==
NH I \ tir
1,4-ctioxane
Br
To a 0 C solution of the product from step 3 (8.5 g, 25.7 mmol) and pyridine
(4.0
g, 51.4 mmol) in 50 mL of 1,4-dioxane was added TFAA (10.8 g, 51.4 mmol). The
resulting
mixture was then heated to 100 C overnight. The mixture was cooled and poured
into 200 mL
of water, and the precipitate was filtered and washed by water then dried to
give a solid (1.3 g).
MS (ESI) m/e (M+H+): 331.
Step 5
- (7)3N H01 \
NH
N
The reaction was conducted similar to that describe in Example 1, step 6. MS
(ESI) m/e (M+H+): 288.
Step 6
C)
r
Br N B N
\ NH2 HATU NH N
N 0
Boc-N-H
To a suspension of the product from step 5 (0.1 mmol), N-Boc-L-Phg-L-Pro-OH
(0.1 mmol) and DIPEA (20 mg, 0.15 mmol) in 1 mL of CH3CN was added HATU (42
mg, 0.12
mmol). The resulting mixture was stirred at RT overnight, concentrated and
purified by RPLC
to give the desired compound. MS (ESI) nile (M H ): 619. 1H NMR (Me0D 400) 6:
7.85 -
7.77 (m, 5 H), 7.43 - 7.36 (m, 6 H), 6.89 (s, 1 H), 5.50(s, 1H), 4.54 (d, J-
8.0 Hz 1H), 3.93(t, 1
H), 2.10 - 1.87 (rn, 4 H) 1.41 (s, 9 H).
- 68 -
:A 02756172 2011 09 21
WO 2010/111483 PCT/US2010/028653
Example 5¨ (2S).-N-13-chloro-2-14411(2S)-1-(phenvlacetyl)pyrrolidin-2-
vlIcarbonyllamino)
phenyll-1H-pyrrolo12,3-clppridin-5-v11-1-(phenvlacetvlipyrrolidine-2-
carboxarnide
eti clir,H ,.,.. sOI 0
N , ___ C
\ 40 NH N
0 N ,----
0 H 0 =
Step 1
__ ft NO, H7, PdfC , y
F121,1 N
A heterogeneous mixture of 2-amino-4-methyl-5-nitropyridine (4.15 g, 27 mmol)
and 10% Pd/C (0.4 g) in 50 mL of THF was stirred in 50 psi of H2 for 3 hours.
The mixture was
filtered through CELITE and concentrated to yield a yellow solid (3.20 g). MS
(EST) in/e
(M+H+): 124. III NMR (DMS0): 8 7.38 (s, 1 H), 6.16 (s, 1 H), 4.84 (s, 2 H),
4.06 (s, 2 H), 1.96
(s, 3 H).
Step 2
NH, 0
H2NN''' 0 kl 111><-
,L , . .,\Ac., Et3N xN it, X
N-;--- 0
H
A mixture of diamine from step 1 (3.20 g, 26 mmol), TEA (5.25 g, 52 mmol) and
a catalytic amount of DMAP in THE (100 mL) was stirred at 5-10 C, then treated
with pivaloyl
chloride (3.74 g, 31 mmol). The resulting mixture was stirred at RI for 5
hours, diluted with a
5% solution of citric acid, and extracted with Et0Ac. The combined organic
extracts were
sequentially washed with water and brine, dried, filtered, and the filtrate
was concentrated in
vacuo to yield a residue. The residue was purified by column chromatography on
silica gel to
afford 7.4 g of the desired compound. MS (EST) mie (M-4-1-1+): 292. 11-1 NMR
(CDC13): 8 8.77 (s,
1 H), 8.47 (s, 1 H), 8.20 (s, 1 14), 7.15 (s, 1 H), 2.24 (s, 3 11), 1.30-1.32
(m, 18 H).
Step 3
0 0
OH soci2 0
-..... ,0., ik 0 ri
* (i)
N.,
p- x-
02N 02N ...."'111P.
ON
To a cooled solution of 4-nitrobenzoic acid (12 g, 72 mmol) in 50 mL of PhMe
was added 20 mL of SOC12 drop wise. After the addition, the suspension was
heated to reflux
25 for 4 hours. The solvent was removed, and the residue was azeotroped
with 50 mL of PhMe to
afford 14.5 g of crude acid chloride. To a solution of TEA (101 g, 100 mmol),
a catalytic
amount of DMAP and NO-dimethylhydroxyla.mine (5.3 g, 87 mmol) in 100 mL of DCM
was
-69-
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
added drop wise 14.5 g of the freshly prepared acid chloride in 100 mL of DCM.
The resulting
mixture was stirred at RT for 5 hours, then diluted with a 5 % solution of
citric acid, and
extracted with DCM. The combined organic extracts were sequentially washed
with water and
brine, dried and filtered, and the filtrate was concentrated to yield a
residue. The residue was
purified by column chromatography on silica gel to afford 7.0 g of the Weinreb
amide. MS
(ESI) m/e (M-FH+): 211. 1H NMR (CDCI3): 6 8.25 (d, J=8.8 Hz, 2 H), 7.82 (d, J--
-9.6 Hz, 2 H),
3.52 (s, 3 H), 3.82 (s, 3 H).
Step 4
H2,Pd-C
ON1
1;)
02N H2N
A heterogeneous mixture of the nitro compound above and 10% Pd/C in THF was
stirred at STP with a balloon of H2 for 3 hours. The mixture was filtered
through CELITE, and
concentrated to yield a yellow solid MS (EST) m/e (M+H+): 181.
Step 5
0
0 0
CI 1410
FINEt3N
The product from step 4 was pivaloylated using conditions described in step 2.
MS (PSI) m/e (M+11+): 265.1H NMR (CDCI3): 8 7.66 (d, J=8.4 Hz, 2 H), 7.56 (d,
J=8.8 Hz, 2
H), 3.51 (s, 3 H), 3.32 (s, 3 H).
Step 6
0 )c)
0 jc,r6 _________________________________________________________ \ N
NH
A solution of compound isolated from step 2 above (2.2 g, 7.5 mmol) in 15 mL
of
THF was cooled to below -40 C. t-BuLi in hexane (15 mL, 2.5 M, 37.5 mmol) was
added
dropwise, and the resulting solution was stirred at -40 C for 1 hour. A
solution of compound
from step 5 (2.2 g, 8.25 mmol) in 10 mL of THF was added drop wise, and the
resulting solution
was continued to stir at this temperature for 30 minutes before being warmed
to RT and stirred
for 30 minutes again. An aqueous 5 % citric acid solution was added to quench
the reaction,
which was extracted with DCM (x3), and the combined organic layers were washed
with water
- 70 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
and brine, dried over Na2SO4 and concentrated in yam . The residue was
purified by
preparative HPLC to yield a solid (0.4 g). MS (ESI) mie (M+H ): 495. 111 NMR
(CDCI3): 5 8.85
(s, 1 H), 8.68 (s, 1 H), 8.26 (s, 1 H), 8.03-8.05 (m, 3 H), 7.21 (d, J=8.8 Hz,
2 H), 7.53 (s, 1 H),
4.20 (s, 2 H), 1.33-1,29 (m, 27 H).
Step 7
0 HN
aq HIBr
NH2
haat
NH
01<
A solution of the product from step 6 (400 mg, 0.8 mmol) in 33% aqueous HBr
(15 mL) was refluxed overnight. After cooling, the mixture was concentrated in
vactio. The
residue was purified by preparative HPLC to give a solid (120 mg). MS (ESI)
nile (M-1-114): 225.
1H NMR (Me0D): 5 8.04 (s, 1 H), 7.78 (d, 2 H), 7.04 (d, 2 H), 6.86 (s, 1 H),
6.71 (s, 1 H).
Step 8
II2N NH2 HATUDI
,EA)... =\
NH 0 N N 0 4,
The product from step 7 was coupled to 2 equivalents of N-phenylacetyl-L-
proline using 2 equivalents of HATU and DIEA in a manner similar to that shown
in Example I.
MS (ESI) nee (M+H+): 687. 1H NMR (Me0D): 8 8.49-8.57 (m, 1 H), 7.38-7.45 (m, 2
H), 7.72
¨7.80 (m, 2 H), 5.15-5.20 (m, 411), 4.42-4.50 (m, 2 H), 3.57-2.59 (m, 4 H),
2.31-2.42 (m, 4
H), 1.89-2.15 (m, 611).
Step 9
Ph NCS ay CI ()
0 141 N
,A0 0 0,ph NH
0*-Ph
To a solution of product from step 8 (15 mg, 0.02 mmol) in 2 mL of dry THE was
added NCS (2 mg, 0.015 mmol). The resulting mixture was stirred at RT for 30
minutes. The
solvent was evaporated, and the residue was purified by prep HPLC to give 5 mg
of the desired
product. MS (ESI) rnie (M+H+): 689. 1H NMR (Me0D): 8 8.53 (s, 1 H), 7.91 (d,
J=9.2 Hz, 2
H), 7.77 (d, J-8.8 Hz, 2 H), 7.49 (s, 1 H), 7.20-7.23 (m, 10 H), 4.53-4.59 (m,
2 H), 3.64-3.79
(m, 8 H), 1.98-2.25 (m, 8 H).
-71 -
:A 02756172 2011 09 21
WO 2010/111483 PCT/US2010/028653
Example 6¨ (2S)-N-{3-ehloro-244-({1(2S)-1-(phenvlacetpl)pyrrolidin-2-
yllearbonvIl
amino)phenyll4H-pyrrolo[2,3-blpyridin-5-v114-(phenylacetyl)pyrrolidine-2-
carboxamide
= CN-rri \c'
0
N N 0 fit
Step 1
02N Ki, K103, H2SO4, H20õ.. 02NU I
kr NH2 1000
N NH2
2-Amino-5-nitropyridine (7.00 g, 50.0 mmol) was dissolved in H2SO4 (2 M, 100
mL). Potassium iodate (4.28 g, 20 mmol) was added portion at RT with stirring.
The solution
was heated to 100 C under reflux. Potassium iodide (8.00 g, 48.2 mmol) was
added drop wise
over 1 hour as a solution in water (20 mL). A brown solution resulted, with
solid iodine
collecting in the reflux condenser. Heating at reflux was continued for 30
minutes, and the
mixture was cooled to RT. The mixture was adjusted to pH 7 with the careful
addition of solid
NaHCO3. The mixture was diluted with water (200 mL) and CH2Cl2 (250 mL) was
added. Solid
sodium thiosulfate was added with vigorous stirring until the iodine
coloration had been
disappeared. A significant amount of yellowish solid remained out of solution,
which was
collected by filtration, washed with water and dried to give a yellow solid
(10.5 g). The CH2C12
fraction was filtered through a silicone-treated filter paper and evaporated
to give a yellow solid
(2.4 g). The solids were combined to give the desired iodopyridine (12.7 g).
MS (EST) m/e
(MAO 266. III NMR (DMS0): 5 8.89 (s, 1 H), 8.62 (s, 1 H), 7.75 (bs, 1 H).
Step 2
02N
02N(JI
Cul, PdC)2(PPh3}2, EtaN), \
HN it NH
N NH2
NH,
A solution of the iodide (1.05 g, 4.8 mmol), 4-ethynylacetanilide (636 mg, 4.0
mmol) and Pd(PPh3)2Cl2 (76 mg, 0.4 mmol) in 10 mL of Et3N and 5 mL of DMF was
stirred at
RT over 17 hours under N2. The solvent was removed, and the residue was
purified by column
chromatography to give the product (0.9 g). MS (ESI) mie (M+144): 297. Ili NMR
(DMS0): 5
10.11 (s, 1H), 8.80 (s, 1H), 8.22 (s, 1H), 7.59 (s, 111), 2.01 (s, 3H).
Step 3
02N
02N
= 1-Bu0K, DMF,700 N dip NH
NH
NH2
- 72 -
:A 02756172 2011 09 21
WO 2010/111483 PCT/US2010/028653
To a solution of the product from step 2 (730 mg, 2.5 mmol) in 3 mL of THF and
6 mL of DMF was added t-BuOK (580 mg, 5.25 mmol). The resulting mixture was
heated to
70 C for 6 hours. The solvent was removed and 10 mL of DCM, 5 mL of water was
added, and
the resulting precipitate was filtered to give the desired product as a yellow
solid (680 mg). MS
(ESI) m/e (M+H+): 297. 1H NMR (DMS0): 5 10.11 (s, 1 H), 8.96 (s, 1 H), 8.65
(s, 1 H), 7.88 (d,
J=8.8 Hz, 2 H), 7.63 (d, J=8.8 Hz, 2 H), 6.98 (s, 1 II), 2.02 (s, 3 H).
Step 4
02N 0,N
\ N \H H0I , NH,
Or- N N __
The synthetic method for the removal of the acetyl group was the same as used
in
Example 1, step 6. MS (ESI) m/e (M+H+): 285.
Step 5
ON OaN )H
N\
I NH 2 4- HATU N
N N 0
0 e
The synthetic method used for the coupling of the proline analog to the
aniline
prepared in step 4 was the same as used in Example 1, step 7. MS (ESI) m/e
(M+H+): 470.
Step 6
02N 17/\ H2N
0 %fr¨C
NH N Ha,Pd-C
\irt N
41, N
A heterogeneous mixture of product from step 6 (20 mg, 0.04 mmol) and 10%
Pd/C in 5 mL of Me0H was stirred in 10 psi of H2 for 3 hours. The mixture was
filtered through
CELITE, and concentrated in vacua to yield a yellow solid (17 g). MS (EST) m/e
(M+H ): 440.
Step 7
I-12N = CJY
4. NH N HATU õ / NH N
N N __ 0 e
The product from step 6 was coupled to 1 equivalent of N-phenylacetyl-L-
proline
using 1 equivalent of HATU and DIEA in a manner similar to that shown in
Example 1. MS
(ESI) m/e (M+H+): 655. 1H NMR (Me0D): 6 8.26 (s, 1 H), 8.19 (s, 1 H), 7.30-
7.51 (m, 3 H),
7.18-7.27 (m, 1 H), 6.65 (s, 1 H), 4.51-4,56 (m, 2 H), 3.61-3.76 (m, 8 H),
1.95-2.15 (m, 8 H).
- 73 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Step 8
= F4 = 0 \N CI
CNly N NCS #it (NiirN I \
=0 ' H 0
0 0 * 0 N
0 =
To a solution of product from step 7 (30 mg, 0.04 mmol) in 4 mL of dry THF was
added NCS (4 mg, 0.03 mmol). The resulting mixture was stirred at RT for 30
minutes. The
solvent was evaporated, and the residue was purified by prep HPLC to give the
desired product.
MS (ESI) ink (M+H+): 690. 1H NMR (Me0D): 8.29 (s, 1 H), 8.20 (s, 1 H), 7.81-
7.84 (m, 2
H), 7.66-7.69 (m, 2 H), 7.18-7.29 (m, 10 H), 4.52-4.55 (m, 2H), 3.62-3.78 (m,
8 H), 1.90-2.29
(m, 8 H).
Example 7 ¨N-f4-(3-oxo-7-12-oxo-24(2S)-1-(phenvlacetvl)pyrrolidin-2-yllethyli-
3,7-
dihydroimidazo[1,2-alpyrazin-2-1,0Phenvil-.14Phenylacetp1)-L-prolinamide
Elp C3 ,> \
NH ,m
0 Q
0
Step 1
0
40 HE3r, DM80
110
02N 02N H
To a stirred solution of 4-nitroacetophenone (20 g, 121 mmol) in 100 ml of
DMS0 was added slowly 42 ml of 48% aqueous 11Br (363 mmol). The solution was
stirred in
an open flask at 55 C and the reaction was followed by TLC. When the starting
material was
consumed, the solution was poured into ice. The solid products were filtered,
washed with
water, and dried under vacuum at RT over P205.
Step 2
0
NO2 HNOr \
H LIIIIN
021\1 0
Arylglyoxal hydrate (5g, 27.8mmol) was added in one portion over a slurry of
the
heterocyclic amine (2.773 g, 29.2 mmol) in methylene chloride (10 ml). The
resulting
suspension was treated with 1 drop of freshly distilled 1F3-Et20 and stirred
until most of the
amine was consumed. The reaction products were isolated as hydrates by
filtration of the thick,
intensely colored reaction mixture. The residue obtained by concentration is
allowed to cool,
-74-
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
filtered with suction, washed twice with diethyl ether and dried under reduced
pressure to give
the desired product (4 g). MS (ESI) mile (M-I-H4): 256.
Step 3
so IQ-1'0H 1. CICOOEt C-3--1(C1-1/42
0 2 CH2N2 411 N
0
0 0
The N-protected praline (10 g, 42.8 mmol) in dry ether (60m1) and THF (60m1)
was stirred under argon at -25 C. TEA (42.8 mol, 4.08 ml) and ethyl
chloroformate (42.8 mmol,
2.6 4.14 ml) were added to this solution. The solution was stirred for further
30 minutes, the
temperature then allowed to reach -10 C, and the diazomethane solution in
ether (2-3
equivalents) was added drop wise. The suspension was stirred for an additional
3 hours and
allowed to reach ambient temperature. The triethylamine hydrochloride was then
filtered off,
and the filtrate was evaporated to half of its original volume. The resulting
solution was washed
with saturated aqueous NalIC03 (50 ml) and brine (50 ml). The organic layer
was dried and
evaporated to give a crude product, which was used without further
purification. MS (ESI) mie
(M+H+): 258.
Step 4
CHN2
HOAc =
CVN Br
0 0
0
To a solution of a-diazoketone (2 g, 7.78 mmol) in glacial HOAc (25m1) was
treated with 48% HBr (2.8 ml) drop wise with stirring. After stirring for 1
hour, the reaction
mixture was extracted with DCM and washed with water. Evaporation of the
solvent and
crystallization from ether-pet ether gave the pure product. MS (EST) nile
(M+Fr): 310.
Step 5
(--N3C3rHirr-NI, NO2 EI3N, THF =
=0 NO2
0 0 0
0
The product from step 4 (420 mg, 1.35 mmol) and the heterocycle from step 2
(347 mg, 1.35 mmol) in THF (2m1) were stirred at RT overnight with Et3N (0.3
mL). When the
reaction was completed, the mixture was concentrated, and the residue was
purified by RPLC to
give the product (300 mg). MS (ESI) ink (M-1-11+): 486.
- 75 -
:A 02756172 2011 09 21
WO 2010/111483 PCT/US2010/028653
Step 6
fik N
NO, SnC12
0 = NH2
o N
0 0
0 0
A solution of the product from step 5 (180 mg, 0.371 mmol) in absolute EWE (3
ml) was added to stannous chloride dihydrate (418.6 mg, 1.85 mmol), and the
mixture was
stirred at 70 C for 2 hours. The reaction mixture was cooled to RT and poured
into ice/water (50
ml), and the pH was made strongly alkaline by the addition of saturated NaOH
(100 ml) before
being extracted with Et0Ac (2x). The organic phase was combined and washed
with brine,
dried by MgSO4, filtered, and concentrated to yield the crude product (150mg).
MS (ESI) m/z :
(M+11 ) 456.
Step 7
== (N-11--- r RCOOH} Ott
0 N 2 HATU
0 --c,.õN NH
0 0 0 =
0 0
The mixture of the product from step 6 (30 mg, 0.066 mmol), N-phenylacetyl-L-
proline (46.08 mg, 0.197 mmol), D1PEA (50.1 mg, 0.197 mmol) in CH3CN (2 mL)
was stirred at
RT for 5 minutes, then HATU (74.86 mg, 0.197 mind) was added into the mixture.
The mixture
was stirred at RT overnight, concentrated, and the residue was purified by
RPLC to give the
desired compound (20mg).1}1 NMR (DMSO) 8: 9.26 (s, 1H), 8.84-8.86 (m, 2H),
7.79-8.03 (m,
4H), 7.11-7.31(m, 12H), 4.41-4.63 (m, 2H), 3.52-3.80 (m, 12F1), 2.26-2.12 (m,
2H), 1.87-1.75
(m, 6H).
Example 8 ¨ (2S)-1-(evelabutylcarbonvl)-N-f2-14-61(2S)-1-(Pyridin-3-
ylearbanvOpyrrolidin-2-
v/karbonvilanzino)phenvii-1H-benzimidazoi-5-v/imirro/idine-2-carboxamide
H
CNIIN 110 ;:1H 0
Step I
NH,
H2N 02N II" PPA NH2 02N = = NH2
OH
p-Arninobenzoic acid (0.200 g, 1.45 mmol) and nitrophenylene diamine (0.221 g,
1.45 mmol) were added into PPA (30 mL). The mixture was stirred at 210 C for
20 minutes.
- 76 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Then, it was poured into ice water and extracted with DCM. The organic layer
was washed with
brine, dried (NaSO4), filtered and concentrated to afford 200 mg of the
desired compound. MS
m/z: 255 (M+1).
Step 2
C": OH
02N N\ 411
07N N\ Boo 0 11", N
NH, Fl NN-
0 N
N HOBt, DPEA
Boci
H EDCI, DMF, RI
Compound from step 1 above (1.2 g, 4.7 mmol), N-Boc-proline (1.52 g, 7.07
mmol), EDCI (1.8 g, 9.44 mmol), HOBT (1.27 g, 9.44 mmol) and DIPEA (2.4 g,
18.8 mmol)
were taken in DMF (30 mL) and stirred for overnight at RT. DMF was removed
under reduced
pressure, and the residue was extracted with DCM/water. The organic layer was
washed with
brine, dried (NaSO4), concentrated and purified by column (DCM: Me0H/100:1) to
afford L2 g
of the desired compound. III NMR (Me0D) 6 8.49 (s, 1H), 8.28-8.19 (m, 3H),
7.79-7.76 (d, J =
4.4Hz, 2H), 7.72-7.65 (m, 1H), 4.41-4.29 (t, J = 8.8Hz, 1H), 3.59-3.51 (m,
2H), 2.18-1.95 (m,
4H), 1.49 (s, 9H).
Step 3
02N
fit NH HCl/Me0H 02N NH
N .11r" N
0 N 0 Ne-
BDC
HCI
Compound from step 2 (0.600 g, 1.06 mmol) was stirred in Me0H/HC1 (10 mL)
for 1 hour at RT, and solvent was removed under reduced pressure. The
resulting compound was
dried at high vacuum to afford 370 mg of desired compound.
Step 4
di
02N
fp. NH ss---j 0
11111111" N 02N NN
0
0 N N
H HAM, DIPEA
The product from step 3 (0.370 g, 1.052 mmol), pyridine-3-carboxylic acid
(0.157
g, 1.27 mmol), HATU (1.2 g, 3.18 mmol) and DIPEA (0.814 g, 6.36 mmol) were
taken in DMF
(10 mL) and stirred for overnight at RT. DMF was removed under reduced
pressure, and the
residue was extracted with DCM/water. The organic layer was washed with brine,
dried
(NaSO4), concentrated and purified by column (DCM: Me0H/100:1) to afford 300
mg of desired
compound. III NMR (Me0D) 6 ppm: 0.883 (s, 1H), 0.87-0.85 (d, J = 4.4Hz, 111),
8.49 (s, 1H),
- 77 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
8.21-8.18 (m, 1H), 8.17-8.10 (m, 3H), 7.98-7.95 (d, dr¨ 8.8Hz, 2H), 7.73-7.68
(m, 1H), 7.60-7.51
(m, 1H), 4.79-4.76 (t, J= 6Hz, 1H), 3.77-3.73 (m, 2H), 3.27-3.19 (m, 2H), 2.19-
2.12 (m, 2H).
Step 5
0211 so H2N
N N H Pd/C2, N w
0
The product from step 4 (0.300 g, 0.657 mmol) was taken in Me0H (10 mL) and
Pd/C (0.07 g) was added under N2. The reaction was stirred for overnight at RT
under H2. The
Pd/C was filtered through CELITE, and the filtrate was concentrated under
reduced pressure to
afford 234 mg of desired compound.
Step 6
oH
N (30c 0N
004 la+P1P,ART
0
The product from step 5 (0.370 g, crude), N-Boc-proline (0.157 g, 1.27mmol),
HATU (1.2 g, 3.18 mmol) and DIPEA (0.814 g, 6.36 mmol) were taken in DMF (10
mL) and
stirred for overnight at RT. DMF was removed under reduced pressure, and the
residue was
extracted with DCM/water. The organic layer was washed with brine, dried
(NaSO4),
concentrated and purified by column (DCM: Me0H/100:1) to afford 300 mg of
targeted
compound.
Step 7
=H
ici)___1,11 NN Ha/MOH Au
NH 0 N vir NH 0
Boc
N Ft 0
\J
The product from step 6 (0.600 g, 1.06 mmol) was stirred in Me0H/HC1 (15 mL)
for 1 hour at RT, and the solvent was removed under reduced pressure. The
compound was
dried at high vacuum to afford 370 mg of desired compound. Ili NMR (Me0D) 5:
9.39 (s, 1H),
9.18-8.82 (m, 2H), 8.39-8.38 (m, 1H), 8.32-8.22 (m, 1H), 8.21-8.10 (m, 2H),
8.09-7.98 (m, 211),
7.86-7.72 (m, 1H), 7.71-7.65 (m, 1H), 4.87-4.84 (t, J= 8.8Hz, 2H), 3.51-3.34
(m, 8H), 2.49-2.38
(m, 4H).
-78-
:A 02756172 2011-09-21
WO 2010/111483
PCT/US2010/028653
Step 8
H
CY 0 NIH 4Pt
N HATU,DPEADMF (13-1Erl er
NIN:$11 et 0)r-NN
H 0
0 N. N R-000H crLo 0
0 ,...N
HC1 I I
---
The product from step 7 (0.100 g, 0.191 mina , cyclobutanecarboxylic acid
(0.018 mg, 0.183 mmol), HATU (0.116 g, 0.305 mmol) and DIPEA (0.059 g, 0.416
mmol) were
taken in DMF (5 mL) and stirred for overnight at RT. DMF was removed under
reduced
pressure, and the residue was extracted with DCM/water. The organic layer was
washed with
brine, dried (NaSO4) and concentrated. The residue was purified by HPLC
purification to afford
12 mg of the final product. 1H NMR (Me0D) 6: 8.91 (s, 1H), 8.75 (s, 1H), 8.41
(s, 111), 8.25
(s,1H), 7.96-7.95 (d, J= 5.2Hz, 4H), 7.73-7.67 (d, J= 3.2Hz, 213), 7.52-7.51
(d, J= 3.2Hz, 1H),
4.54-4.82 (t, J = 3.6Hz, 211), 3.63-3.46 (m, 411), 2.48-2.46 (t, J= 2Hz, Hi),
2.34-2.29 (m, 6H),
2.18-2.10 (m, 411), 2.08-2,02 (m, 4H).
Examples 9-15
Compounds of Examples 9-15 were prepared in a similar manner to Example 8.
,
Example Structure MW t Name
9clic H 626321 (2S)-1-
(phenylearbonyi)-N- {4-
N
id1-PN [54
{ [(2S)-1-
I. o 0 ril o
(phenylcarbonyl)pyrrolidin-2-
o 0 Acarbonyl}amino)-
1H-
benzimidazol-2-
yliphenyllpyrrolidine-2-
carboxamicle
10H 682.83 (28)-1-
[(445Z)-4-
crlyllN
SO tvi\ = i'l r-)
- ethylidenehept-5-
enoyll-N-{2 {2 -
0 . 1---N
[4-( { [(28)-1-(3-
14 0
1101 pbenylpropanoyppyn-olidin-2-
yl] carbonyl } amino)pheny1]-1 H-
benzimidazol-5 -yllpyrr olidine-
2- carboxamide
11682.83 (2S)-1-[(4Z,54-
4-
Crjr-^ ethylidenehept-5-
enoyll-N- it c {2-
N N
miliFI 0 0
{3-UR2S)-1-(3-
o \
0 gp phenylpropanoyppyrrolidin-2-
N
H ylic arbonyl)
amino)pheny1)-1H-
0 benzirnidazol-5-
y1}pyrrolidine-
2-carboxamide
12 H N 1 686.774 (2S)-1-
[(2R)-2-hydroxy-2-
5
0,..,.,N
0 = itphenylacetyl]-N-(4-{5-[({(23)-1-
, N
11 11)--<I1
41 6 0 N [(2R)-2-hydroxy-
2-
6H 00,oH
phenylacetylipyrrolidin-2-
yl} carbonyl)amino1-1 1/-
fitbenzimidazol-2-
yllphenyl)pyrrolidine-2-
carboxamide
- 79 -
:A 02756172 2011-09-21
WO 2010/111483
PCT/US2010/028653
Example I Structure MW Name
._
13 cirli N 654.776
(23)- 1-(phenylacety1)-N- {44
5-
o 0 W
t ({[(2S)-1-(phenylacetyl)
pyrrolidin-2-yl] carbonyl }
H 0 40 . amino)-1H-benzimidazol-2-
11 yliphenyllpyn-olidine-2-
earboxamide
14flitA 885.041 tert-butyl { (18)-24(15)-2-U4-
=
iL N butoxycarbouyp
Ni:i
0 111127 amino]-2-
0===.--- NH 0 H OU N.-)
phenylacetyl }pyrrolid in-2 -
CH, 8
yljearbonyl) amino)-1H-
Fii3O
benzimidazol-2-yl]phenyl)
cH3
r -
cH
)k2 earbamoyljpyrrolidin-l-y11-2-
CHs H3
ox0-1-phenylethyl}earbamate
_
15 H 885.041 tert-butyl {(1R)-
2-[(23)-2-( {4 -
. QN1rN 16. N, II
Ni-Lo [5-({[(25)-1-{(2R)-2-[(tert-
o butoxyearbonyl)aminoi -2-
0 _, 0 W. =N
....---NH H
041 N--'
0 IRE
phenylacetyl) pyrrolidin-2-
0 F)1.,<(c)11,13
yl] carbonyl amino)-1 H-
HN
benzimidazo1-2-ylipbenyl)
CH, ,..0
6
carbamoyl)pyrrolidin-l-y11-2-
cH3
l'01-13. exo-l-phenylethyl} earbamate 1
-
Example 16¨ Benzyl OS)-2-114-(4-11(4-methylphenyOsulfonyllaminol-111-
benzimidazol-2-
yOphenylicarbamoyllpyrrolidine-1-carboxylate
. 0 IC N
Op &¨NH Na,... IIW 0 0 .
0 .NH
Step I
NO, NO2 H =NHBac
0
BocHN Ilio + 111 NH2 HATU to N
OH 0
111111-ri 14H2 NH,
To a solution of N-Boc-p-aminobenzoic acid (1.86 g, 7.84 mmol) in DMF, 3-
nitrophenylenediamine (1.0 g, 6.536 mmol), HOBt (0.875 g, 6.536 mmol) and EDCI
(2.5 g,
9.804 mmol) were added, and reaction was stirred for overnight at RT. The
excess of solvent
was removed under reduced pressure, and the residue was diluted with DCM. The
organic layer
was washed with brine, dried (NaSO4), filtered, concentrated and purified by
column to obtain
400 mg of compound. MS miz: 273 (M+1).
Step 2
NO2 H . NHBoc NO,
N
111 N 0 AcOH , (110 \ it
N NI-1
NH, H
- 80 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
The compound from step 1 above (0.600 g, 1.611 mmol) and KOAc (0.158 g,
1.609 mmol) were taken in HOAc (9.3 rnL). The reaction was stirred at 120 C
for overnight,
cooled to RT and poured into ice-water. The aqueous layer was extracted with
DCM. The
organic layer was washed with brine, dried (NaSO4), filtered and concentrated
to obtain 120 mg
of the desired compound. MS m/z: 255 (M+1). 'H NMR (DMSO) 8: 11.44 (s, 111),
8.95-9.01
(m, 211), 8.81-8.83 (d, j¨ 8.0Hz, 2H), 8.22-8.27 (m, 111), 8.22-8.27 (m, 2H),
4.97 (s, 21).
Step 3
NO, NO2
=N\ = N-Bac-prohne N\
H
FcnDITTEA N
0 Bac
To a solution of the aniline (0.200 g, 0.787 mmol) in DMF, N-Boc-proline
(0.186
g, 0.865 mmol), DIPEA (0.302 g, 2.361 mmol) and HATU (0.329 g, 0.865 mmol)
were added.
The reaction was stirred for overnight. The excess of solvent was removed
under reduced
pressure, and the residue was diluted with DCM. The organic layer was washed
with brine,
dried (NaSO4), filtered, concentrated and purified by column to obtain 150 mg
of the desired.
MS miz: 452 (M+1). 11-1 NMR (Me0D) S: 8.70-8.73 (m, 2H), 8.40-8.42 (d, J= 8Hz,
211), 7.87-
7.89 (d, J¨ 8Hz, 1H), 7.50-7.53 (m, 211), 6.78-6.80 (d, J= 8Hz, 1H), 3.83 (s,
111), 3.69-3.76 (m,
211), 3.56-3.60 (m, 411), 1.37-1.43 (m, 9H).
Step 4
NO, NH,
40 Pd/C =H
N W NYY
0 Bac 0 Bac
To a solution of compound from step 3 above (0,200 g, 0.443 mmol), Pd/C (10
mg) was added under argon, and the reaction was stirred for 2 hours in 112.
The Pd/C was
filtered and washed with MeOH for several times. The solvent was evaporated to
obtain 180 mg
of desired compound. MS miz: 422 (M+1). 1H NMR (Me0D) 8: 9.51 (s, 111), 7.95-
8.20 (m,
111), 7.58-7.60 (m, 1H), 7.19-7.48 (m, 211), 6.78-6.87 (m, 211), 6.38 (s, 1H),
4.25-4.42 (m, 1H),
3.34-3.67 (m, 211), 1.79-2.20 (m, 4H), 1.17-1.41 (m, 911).
Step 5
H
NH 2 = NH
N 411k
0 Boc
ipTEA 0
0 Boc
To a solution of the compound from step 4 above (0.196 g, 0.465 mmol) in THF,
-81 -
:A 02756172 2011 09 21
WO 2010/111483 PCT/US2010/028653
TEA (0.070 g, 0.693 mmol) and 4-methylbenzene-1-sulfonyl chloride (0.088 g,
0.461 mrnol)
were added drop wise at 0 C. The reaction was stirred for overnight, and the
solvent was
removed under reduced pressure. The residue was diluted with DCM and washed
with brine.
The organic layer was dried (NaSO4), filtered and concentrated. The residue
was purified by
preparative TLC to afford 110 mg of desired compound. MS m/z: 576 (M+1). 1H
NMR
(Me0D) 6: 7.98-8.00 (dõ f----- 8.0Hz, 211), 7.78-7.80 (d, Jr--= 8.0Hz, 2H),
7.66-7.68 (d, Jr= 8.0Hz,
211), 7.26-7.28 (m, 1111), 7.19-7.21 (d, J= 8.0Hz, 211), 7.07-7.09 (m, 2H),
4.36-4.38 (m, 111),
3.55-3.58 (m, 211), 2.32-2.35 (m, 411), 1.89-2.11 (m, 311), 1.50 (s, 911).
Step 6
H
9 (1) Me0H/Hel
ter N (2)cbzcl ii-NH N 410 N 0 01-0Brt
NH 0
NH
The product from step 5 above (0.180 g, 0.312 mmol) was stirred in Me0H/HCI
(5.0 mL) for 1 hour at RT. The solvent was removed under reduced pressure and
dried at high
vacuum. It was used directly without any further purification. The residue was
taken in DMF,
benzoic acid (0.042 g, 0.344 mmol), DIPEA (0.320 g, 2.504 mmol) and HATU
(0.143 g, 0.375
mmol) were added. The reaction was stirred for overnight at RT. The excess of
solvent was
removed under reduced pressure, and the residue was diluted with DCM. The
organic layer was
washed with brine, dried (NaSO4), filtered, concentrated and purified by
column to afford 40 mg
of the final compound. MS ink: 580 (M+1). 1H NMR (Me0D) 8: 7.92-7.94 (d, J=
8.811z, 211),
7.23-7.68 (m, 1411), 6.81-6.83 (d, J 10Hz, 111), 5.21 (s, 211), 4.65-4.79 (m,
111), 3.52-3.89 (m,
2H), 2.47-2.58 (m, 1H), 2.43 (s, 31I), 1.93-2.23 (m, 3H).
Example 17¨ (2S)-1-(3-phenylpropanov1)-N-14-15-W2S)-1-(3-
phetrylpropanoyl)pyrrolidin-2-
vlicarbonyllamino)-1,3-benzoxazol-2-yllphenyllpyrrolidine-2-carboxamide
fiµ)(11 ith 41-1 NH
0 0 Nmirl
0 W-
. 0
Step 1
it OH
Ask 0 H2N NI-I2 H2N aigt.
H2N ir II0 MP NH2
OH PPA
- 82 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
p-Aminobenzoic acid (1.37 g, 10 mmol) and 2,4-diarninophenol (1.24 g, 10
mmol) were combined under argon and treated with 12 mL of PPA. The resulting
solution was
heated at 200 C for 30 minutes. The black solution was poured onto ice, and
the resulting
yellow solid was collected (1.12 g). 1H-NMR (DMSO) 6: 10.2-10.5 (s, 2H), 8.10-
8.20 (m, 4H),
7.10-7.80 (m, 3H). MS m/z: 226 (M+1).
Step 2
try0H
H2N lab NN it
NH 2 ______________________________ Boc 0 N "ANt H
HATU, DIPEA, DMF' Bac 0 0 wf
"I 0
0 Boc
NTL
The product from step 1 above (0.100 g, 0.236 mmol), N-Boc-proline (0.098 g,
0.355 mmol), HATU (0.135 g, 0.355 mmol), TEA (0.100 g, 0.944 mmol) were taken
in DCM
(10 mL) and stirred overnight at RT. The reaction was diluted with DCM, and
the organic layer
was washed with water, brine, dried (NaSO4), concentrated and purified by
preparative TLC to
afford 100 mg of desired compound. MS miz: 620 (M+1).
Step 3
,H
isN. it HCl/Me0H Q---11-111=
N anik H
Boc 0
0 0 ic;
Q Boc 0
2HCt
The product from step 2 above (0.100 g, 0.161 mmol) was stirred in Me0H/HC1
(3.0 mL) for 1 hour at RT, and the solvent was removed under reduced pressure.
The compound
was dried at high vacuum to afford 80 mg of the desired compound. MS m/z: 420
(M+1).
Step 4
01-1
Cclic-N Alb N\
ip. NH ,
HO
1110 0
H F
N io Auk N
H 0 0 HATU, DIPEA, DM; fit 0 N
0
2HC) 0
Compound from step 3 (0.080 g, 0.191 mmol), 3-phenylpropanoic acid (0.086 g,
0.574 mmol), HATU (0.218 g, 0.574 mmol), DIPEA (0.146 g, 1.146 mmol) were
taken in DMF
(3 mL) and stirred for overnight at RT. DMF was removed under reduced
pressure, and the
residue was extracted with DCM/water. The organic layer was washed with brine,
dried
(NaSO4) and concentrated. The residue was purified by HPLC purification to
afford 108 mg of
target. 1H NMR (DMSO) 5: 10.2-10.5 (s, 21-1), 8.10-8.20 (m, 31-1), 7.10-7.80
(m, 1411), 4,37-4.55
(m, 2H), 3.32-3.58 (m, 4H), 2.67-2.85 (m, 7H), 1.80-2.4 (m, 9H).
- 83 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Example 18¨ (2S)-1-(3-phenylpropanav1)-N-f4-154N2S)-1-(3-
phenylpropanov1)pyrrolidin-2-
yllearbonyliamino)-1-benzolaran-2-yllPhenvlipyrrolidine-2-earboxamide
H
* NHPh ,
ON
Ph
Step 1
00N-0-1Br No,
H0 dab,
H NO2 13IPA= 1=4-dlcxan Si
0 02N H 0
To a solution of 5-nitrosalicylaldehyde (1.0 g, 5.90 mmol) in 1,4-dioxane (10
mL), p-nitrobenzyl bromide (1.33 g, 6.15 mmol) and D1PEA (1.25 g, 9.70 mmol)
were added,
and the reaction was refluxed at 100 C for 2 hours. The reaction was cooled,
and the solids were
filtered, washed with Et0H and dried with high vacuum to afford the desired
compound. MS
miz: 303 (M+1).
Step 2
=1111111 DU 4-diaxane 02N
NO2
0
02N H o
To a solution of the target compound from step 1 (1.5 g, 4.96 mmol) in 1, 4-
dioxane (5 mL), DBU (0.9 g, 6.45 mmol) was added. The reaction was heated to
100 C for 3
hours, then cooled to RT, and the resulting solid was filtered off and
sufficiently washed with
Et0H to afford 927 mg of the desired compound. 11-1 NMR (Me0D) 8: 8.66 (s,
1H), 8.37-8.39
(d, J 8.01-1z, 2H), 8.30-8.32 (d, J 8.0Hz, 1H), 8.18-8.21 (d, J 12Hz, 21-1),
7.78-7.80 (d, J=
8.0Hz, 1H), 7.70 (s, IH).
Step 3
02N rati \ fit
NO2 Fe, HGI H2N NH,
o
To a solution of the product from step 2 above (0.050 g, 0.176 mmol) in 1,4-
dioxane (1.8 mL), water (1.8 mL), Fe (0.054 g) and HCI (1.1 !IL) were added.
The reaction was
heated to 110 C for 3 hours. Then, the solid was filtered, and the organic
layer was concentrated
to afford 40 mg of the desired compound. Ili NMR (Me0D) 6: 8.01-8.05 (m, 2H),
7.90-7.96 (m,
1H), 7.79 (s, 1H), 7.46-7.51 (in, 11-1), 7.34 (s, 1H), 7.17-7.21 (in, 2H). MS
miz: 225 (M+1).
- 84 -
:A 02756172 2011-09-21
WO 2010/111483
PCT/US2010/028653
Step 4
, N
N-Boc-Proline
H2N NH, ___ Boo 0is *
D1PEA, HATU
BoCil
To a solution of the product from step 3 above (0.020 g, 0.089 mmol) in DCM
(10
mL) N-Boc-proline (0.042 g, 0.196 mmol) and DIPEA (0.035 g, 0.267 mmol) were
added. The
reaction was stirred at RT for 5 minutes and then HATU (0.101 g, 0.267 mmol)
was added. The
reaction was stirred overnight and poured into brine and extracted with Et0Ac.
The organic
layer was dried (Na2SO4), filtered and concentrated to afford 30 mg of the
desired compound.
MS rn/z: 619 (M+1).
Step 5
c.õ11,1-1
IV\ HCI-Me0H
00,j 0
0N ['--11 0 iso N
0 1
BoC 2HCI +0
3-phertylpropanaic acid \ NH
DIPEA, HATU 1-µ0
Ph 0 (3,¨K'N:D
Ph
The product from step 4(0.70 g, 1.13 mmol) was stirred in Me0H/HC1 (20 int)
for 1 hour. The solvent was removed at high vacuum to afford the desired
proline compound,
which was used directly to the next step without further purification. 3-
Phenylpropanoic acid
(0.428 g, 2.85 mmol) were taken in DCM (30 mL) was reacted with the proline
compound
(0.500 g, 0.96 mmol) and DIPEA (0.9 g, 7.1 mmol). The reaction was stirred at
RT for 5
minutes, and then HATU (1.0 g, 2.85 mmol) was added. The reaction was stirred
overnight and
poured into brine and extracted with Et0Ac. The organic layer was dried
(Na2SO4),
concentrated and purified by HPLC to afford 30 mg of the desired compound. 1H
NMR (CDCI3)
8: 9.85-9.87 (d, J 8.0Hz, 2H), 7.86 (s, 1H), 7.42-7.44 (d, J 8.8Hz, 211), 7.46-
7.48 (d, J-= 8.8Hz,
211), 7.03-7.31 (m, 9H), 6.90-6.99 (d, J-= 3.6Hz, 1H), 6.58 (s, 1H), 4.71-4.82
(m, 2H), 3.62-3.71
(m, 2H), 3.43-3.50 (m, 2H), 2.95-3.05 (m, 4H), 2.63-2.88 (m, 4H), 2.21-2.43
(m, 4H), 1.87-2.14
(m, 4H). MS m/z: 683 (M+1).
- 85 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Example 19 ¨N-(2-14-(acetylamino)pheny11-1-benzofuran-5-141-1-1(2R)-2-f(tert-
butaxwarbonv1)aminol-2-phenvlaceoll-L-prolinamide
0
N
*- NH
0
Boe4I
Step 1
Br Ai CHO
Br .0 CI+0 Br
CyJ IF' 0 io
OH ______________________________________
No,
K2CO3 (68 g, 0.497 mol) was added to a solution of bromosalicylaldehyde (50 g,
0.248 mol) in DMF (300 ml). The resulting solution was stirred at RT for 1
hour, then to it was
added compound 4-nitrobenzyl bromide (54 g, 0.25 mol). The reaction mixture
was stirred for
30 minutes, filtered, and the filtrate was poured into water and extracted
with Et0Ac (3x). The
combined organic layers were dried and concentrated. The product was
recrystallized from
dioxane to afford a white solid (50 g). 11-INMR (CDC13) 5: 10.43 (s,1H), 8.24
(d, .1 411), 7.51-
7.58 (m, 2H), 7.37-7.42 (m, 4H), 7.31-7.36 (in, 611), 5.52 (s, 111), 4.57 (s,
211), 3.90 (s, 21-1), 3.34
(s, 211), 2.06-2.15 (m, 6H), 1.86-1.88 (m, 111), 1.41 (d, 1811).
Step 2
BrCHO
SDBLI Br mak..
0 40
w 002
No,
DBU (9 ml, 61.58 mmol) was added to a solution of the product from step 1 (10
g, 29.85 mmol) in dioxane (70 m1). The resulting solution was heated to reflux
for 1 hour,
cooled and filtered. The filter cake was washed with Et0Ac and dried in air to
afford a yellow
solid (6.5 g).
Step 3
Br
\ No2 L.,,,mps 112N \
No,
mai 0 mr 0
PtBu3(1.93 ml, 0.32 mmol) was added to a solution of the product from step 2
(2
g, 6.3 mmol) and Pd2(dba)3 (0.29 g, 0.32 mmol) in THE (100 ml) under N2. Then
a solution of
LiHMDS (18.9 ml, 18.9 mmol) was added dropwise. The resulting solution was
heated to reflux
for 3 hours and then cooled to RT. The reaction mixture was adjusted to pH = 1
using 1M HC1,
then stirred for 0.5 hour. The reaction mixture was basified to pH = 8-9 using
aq. saturated
NaHCO3, and extracted with Et0Ac (3x). The combined organic layers were dried
and
concentrated. The residue was recrystallized from Me0H to afford the product
as brown solid.
-86-
CA 02756172 2014-03-03
Step 4
H
\ = _______________________________________
0 NO2 BocHs:
Ph
--Zj 0 \= NO2
DIPEA, HATU BocHN
The mixture of the product from step 3 (500 mg, 2 mmol), R-Boc-Phg-L-Pro-OH
(660 mg, 2.16 mmol), NMM (400 mg, 4 mmol) and DMF (30 ml) was stirred at RT
for 30
minutes, then to it was added HATU (1.13 g, 3 mmol). The resulting mixture was
stirred at RT
overnight. The reaction mixture was diluted with water and filtered. The cake
was washed with
water and dried; the solid was used next step without purification.
Step 5
f (
\J N--1
I
0 0 * NO, H2/ Raney NI
0 ! * NH,
0
BocHN BeeHN
The product from step 4 (0.4 g, 1.3 mmol) in THF (10 ml) was hydrogenated
using RaneTymNi (0.2 mg) as the catalyst. After being stirred under H2
atmosphere at RT
overnight, the reaction slurry was filtered through CELITE) and the filtrate
was concentrated
under reduced pressure to afford 0.33 g of the desired compound.
Step 6
cirri ,
I 0\ = h1H2 Ph)\ 0 'CXo' * NH
\ 0
BocFiN BocHN
Ac20 (18 mg, 0.18 mmol) was added to a solution of the aniline from step 5 (50
mg, 0.09 mmol) in THF (2 ml) at RT. The resulting solution was stirred at RT
overnight,
concentrated, and the residue was purified by RPLC to afford the desired
product. III NMR
(acetone-do) 5: 7.95 (s, 1 II,NH), 7.65-7.74 (m, 7 H, ArH), 7.40 ¨7.43 (m, 3
H, ArH), 7.30 ¨7.34
(m, 2 H, ArH), 7.01 (s, 1 H, ArH), 5.64 (s, 1 H, CH), 4.56-4.59 (m, 1 H, CH),
3.93-.3.99 (n, 1 1-1,
CH2), 3.27-3.42 (m, 2 H, CH2), 3.02-3.17 (in, 311, CH2), 1.98 (s, 311, C113),
1.98- 1.95 (m, 411,
CH2), 1.22 (t, 3 = 7.2 Hz, 6 11, CH3).
Examples 20-36
Compounds of Examples 20-36 were prepared in a similar manner as described in
either Example 18 or Example 19.
- 87 -
:A 02756172 2011-09-21
WO 2010/111483 PCT/US2010/028653
' Example Structure MW Name
(25)-1-(pheny lac ety1)-N- {445-
=
r'l .z-- ( {[(25)-1-
(phenylacetyl)
6.-.L.0 0 0 0= -1-N
654.773 pyrrolidin-2-ylicarbonyl} arnino)-
0)6
1-benzofuran-2-yl)phenyl)
pyrrolidine-2-carboxamide
tert-butyl 10 S)-2-[(25)-24 {445-
H ¶[(25)-1-{(2S)-24(tert-
* CIYN
=
c
o A 1
butoxycarbon yl)amino1-2-
H phenylacetyl} pyrrolidin-2-
21 1r
0 o 885.039 ylicarbonyl} amino)-
1-
CH2 0 8r)(CH$ benzofuran-2-
yl]phenyl)
criph
carbarnoyppyrrolidin-1-y11-2-
oxo- I -phenylethyl) carbamate
_
tert-butyl {(1R)-2-[(15)-2-( {415-
( {[(25)- I - {(2R)-2-[(tert-
[1 --D butoxycarbonyl)amino]-2-
1-0 H Cir \ NH N
22 CHCH3 tN 0 0 ' 0814\ _ 6-013 885.039
0 it
phenylacetyllpyrrolidin-2-yii
tik
carbonyl} am ino)-1-ben zofuran-
2-yilphenyl } carbamoyl)
pyn-olidin-1-y1)-2-oxo-1-
phenylethyl} carbamate
propan-2-y1 [(1R)-2-oxo-1-
,
phenyl-2-1(25)-2 -[(4- {5-[( { (25)-
H
Clia 0 H fly31 so 5--;----3 1-
[(2R)-2-pheny1-2- { Rpropan-2-
0 0 NH N
-i-i, YN 0
yloxy)carbonyljamino) acetyl]
23 0 0 H
81N\ _r, 856.984
411, , --..rc.3
CH3 pyrrolidin-2-yll carb onyl)aminol-
1-benzofuran-2 -y1} phenyl)
carbamoylipyrrolidin-l-
y1} ethyllcarbamate
methyl {(1R)-2-[(25)-2-(1445-
( {[(25)-1-{(2R)-2-
ot ,:s--,
,,Q,,..11 NCIY'l . \ = ,,,i--\,,i3 [(methoxycarbonypaminol -2-
24 O µ0 Q 0 , H
800.876 phenylacetyl } pyrrolidin-2-
tit mi. )ra-cH3
glir ylicarbonyl}amino)-1-
benzofuran-2-yliphenyl)
carbamoyl)pyrrolidin-l-y11-2-
oxo-l-phenylethyl) carbamate
(25)-1 -[(2R)-2-(dimethylamino)-
H 2-phenylacetyl] -N-(4-
{5 -[( {(25)-
pH,Qõ.r.N ....
CRY 0 so \ 4. 0õ)._s-D NH N
C H3 1-
[(2R)-2-(dimethylarnino)-2-
o o o,
. \N,CH3 740.91
phenylacetylipynolidin-2-y1}
41Ik* carb onyl)amino}-1 -ben zofuran-2 -
yl} phenyppynolidine-2-
carboxamide
_
(2S)-1-[(2R)-2-(diethylamino)-2-
H 213 WI 1 > 0 phenylacetylj-N-(4-
{54(1(25)-1-
1 \ 40 NH N [(2R)-2-(diethylamino)-2-
o...-- 0
26 o .1NrCH' 797,019 phenylacetylipyffolidin-2-
y1}
*
carbonyl)amino}-1-benzofuran-2-
y1} phenyl)pyrmlidine-2-
carboxamide
- 88 -
:A 02756172 2011-09-21
WO 2010/111483 PCT/US2010/028653
Example Structure MW Name
propan-2 -y1 [(1R)-2- { (25)-2-[(4-
{ 54( {(28)-14(2R)-2-
cHriliA =
o)._ <,- -. 1
(dimethylamino)-2-
c4
' N 0 illt \ 41111 NH N-j
phenylacetyl]pyrrolidin-2-y1)
27 0 -1r-- o `iw
08 H
= 4N\ 798.947
Ift rQ).--cH,
CH3 carbonypaminol-1 -benzofuran-2-
yl I phenyl)carbamoylipyrrolidin-
1-yll -2-oxo-1-
_ phenylethylicarbamate
,p-13(N11.0 aht N12-(4-aminopheny1)-1-
CHV 0 0 RP =' benzofuran-5-y11-14(2R)-2-
28 0 Illj H2 482.587
(dimethylamino)-2-
phenylacety1R-prolinamide
(28)-1- { (2R)-2-[methyl(3-
methylbutypamino] -2-
,T.....õ)0),,,,__.
phenylacetyl) -N- {4454 { [(28)-1-
cHehr 1100 \ . NH 14"-I r_õ, {(2R)-24rnetby1(3-
29 0 0 = = 414, reli3 853.127 methylbutypa,min61-2-
clip-13
phenylacetyn pyrrolidin-2-
ylicarbonylIamino)-1-
benzofuran-2-yljphenyll
' pyn-o1icline-2-
carboxamide
N
N
* cfgr - õ a>-CH3 N-
1244-(acetylamino)pheny1}-1-
benzofuran-5-y1I -14(2R)-2-
30 0 10111 it NH 552.679
- o -' 0 (diethylamino)-2-
phenylacetyli-
cH3 i.:1-
.....--cH3 L-prolinamide
,
(23)-1-acetyl-N-(2- {44( { (28)-1-
o>i
[(2R)-2-(diethylamino)-2-
11
ClyN HN
31 ...\ 0 1 NH N
CH3 .---- 0 a 649.797 phenylacetylipyn-olidin-2-
ylI carbonypaminoiphenyl) -1-
CH3
\----N
benzofuran-5-yl)pyrrolidine-2-
carboxamide
_.
tert-butyl {(1R)-24(28)-2-( {445-
o 0 \ qp, NHc
N ( {
[(2S)-1-acetylpyn-olidin-2-
o cH, a 0 693.807
4it ylicarbonyl} amino)-1-
32
uV_ N,- NH benzofuran-2-Apbenyl)
'
carbamoyl)pyrroliclin-1-y11-2-
cH3cK3 oxo- I -p henylethyl I carbamate
._
(28)-1-acetyl-N-(2- {44( { (28)-1 -
N [(2R)-2-(dimethylamino)-
2-
.ill-i ) \ = NH N phenylacetyl]pyrrolidin-2-
33 0 cH30 ---- a 621.743
yl} carbonyl)aminoipbenyl) -1- .
cnkl...c.H3
benzofiiran-5-yl)pyrrolidine-2-
carboxarnide
N-{4[5-(acetylarnino)-I-
CH3 benzofuran-2-
yl]phenyl} -1-
- o .
34 0 ift
596.689 { (2R)-2 - [(tert-
_ ).õ- NH
'''3 3 butoxycarbonyparnino]-2-
)iln
phenylacety1)-L-prolinamide
01-12. -
-89-
:A 02756172 2011-09-21
WO 2010/111483
PCT/US2010/028653
Example Structure MW Name
methyl {(1R)-24(2,57)-24 {4-[5-
({[(28)-1-{(2R)-2-
¨ Y-rj
[(methoxycarbonypaminoi-2-
35 H, 814.903
H cliNH
N HH N
0 phenylacetyl}
pyrrolidin-2-
o 0
cH3 yflearbonyl}amino)-7-
methy1-1-
benzofuran-2-yl]phenyl)
earbamoyl)pyrrolidin-l-y11-2-
oxo-l-phenylethyl}carbamate
(25)-1- {(2R)-2-[ethyl(propyl)
amino]-2-phenylacetyl) -N- {445-
CN3.,rH dik \o git 0 * [(25)- I - (2R)-2-
iethyl(propyl)
36 825.073 amino]-2-
phenylacetyl)
0 pyrrolidin-2-
ylicarbonyllamino)-
1-benzofuran-2-yllphenyl)
1 pyrrolidine-2-carboxamide
Example 37¨ (2S)-1-(phenylacetvl)-N-14-16-W2S)-1-(phenylacetyl)pyrrolidin-2-
vikarbonvllamino)irnidazoil,2-alpyrimidin-2-vilphenvilpwrolidine-2-carboxamide
0
N
0 ., NH N
0 N N CA
Bn
Step
N e ,,Ny NH2
02N\)---NH2 _____________ }12 H2N¨CS¨NH2 =
THF,pyridtn
CzNN
¨N Pd/C
A suspension of pyrimidine (280 mg, 2 mmol), Pd/C (15 mg, 0.1 mmol) in 40 mL
Et0H was hydrogenated under 30 psi for 1 hour. The mixture was then filtered,
and the filtrate
was then concentrated to give the product (200 mg). The residue was dissolved
in 20 ml THF
and CbzCl (375 mg, 2.19 mmol) and pyridine (1 ml) were added_ The mixture was
stirred at RT
for 1 hour, then the mixture was concentrated in vacua, and the residue was
extracted with
Et0Ac (2x), washed with H20 (30 mL) and brine (30 mL), dried over anhydrous
NaSO4,
concentrated in vacua to give the desired compound as white powder (330 mg).
MS (ES!) m/e
(M+H+): 245.
Step 2
Cbz
(..N NH2 Br + so NO2
,N
1) Acetone, reflux CZZ \
-.. = NO2
2) HBr, methanol, reflux N N
0
A solution of the product from step 1 above (244 mg, 1.00 mmol) and 2-bromo-1-
(4-nitrophenypethanone (244 mg, I mmol) in 40 mL of acetone was heated to
reflux and stirred
for 6 hours. Then, the mixture was cooled to RT and filtered, and the filtrate
was then dissolved
-90-
Ammm722011m21
WO 2010/111483
PCT/US2010/028653
in 30 ml Me0H, and 0.5 ml HBr was added, the mixture was heated to reflux for
another 3
hours; after that, the mixture was concentrated in vaeuo to give the product
as a pale yellow
powder (120 mg). MS (ESI) nile (M+H ): 390.
Step 3
CbzN \ = SnCI, Cbz' NH,
N \
N NO2
To the product from step 2 above (50mg, 0.128mmol) was dissolved in 10 mL
CH3OH was added SnC12(144 mg, 0.64 mmol). The reaction mixture was stirred at
RT for 30
minutes and then heated to reflux for 3 hours. Me0H was removed in vacua, and
the residue
was purified (DCM/Me0H ¨50:1) to afford the desired compound (35mg). MS (EST)
ink
(M+H+): 360.
Step 4
CIDI-NrN \ = NH, AcOH, HBr H2N \ =
NH2
N N N
The compound from step 3 (35 mg, 0.1 mmol) was dissolved in 5 ml of HOAc,
then HBr (1.5 ml) was added. The reaction mixture was heated to reflux and
stirred for 6 hours,
cooled and concentrated. The residue was extracted with Et0Ac (2x), washed
with aq. NaHCO3
and water (30 mL) and brine (30 mL), dried over anhydrous NaSo4. Concentration
afforded the
desired compound as a brown solid (16mg). MS (ESI) rnie (M+1-1+): 226.
Step 5
COOH
H2N,
Bn
NN
'CX =N N IIATU,DIPEA Bn
A mixture of the diamine product from step 4 above (16 mg, 0.071 mmol), N-
phenylacetyl-L-proline (40mg, 0.170 mmol), DIPEA (36.9mg, 0.02 mmol ) in CH3CN
(5 mL)
was stirred at RT for 10 minutes, then HATU (54 mg, 0.142 mmol ) was added.
The mixture
was stirred at RT overnight then the mixture was concentrated, and the residue
was purified to
give compound (15 mg). MS (ESI) m/e (M+H+): 657. NMR (Me0D) 5: 9.45(s, 1H),
8.31(s,
1H), 8.05(s, 1H), 7.61-7.48 (m, 4H), 7.34-7.21 (m, 10H), 4.60-4.52 (m, 2H),
3.83-3.69 (m,
8H), 2.24-1.97(m, 8H).
- 91 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Example 38¨ (2S)-1-(phenylacety1)-N-14-16-WS)-1-(phenylacetv0pyrrolidin-2-
vIkarbonyliamino)imidaza[1,2-alpyridin-2-vIlPheavllpyrrolidine-2-carboxamide
ais
phcHA0 0 W
Step 1
1102 1) Acetone, reilux
Br 02N _______________________________________________ N
02N¨C.,¨NN2
WV 2} HBr, methanol, reflux ). \ 4p,
NO2
The mixture containing 2-amino-5-nitropyridine (1.39 g, 10 mmol) and p-nitro-
alpha-bromoacetophenone (2.42g, 10 mmol) in 100 mL of acetone was heated at
reflux for 12
hours. The solid was collected by filtration and then dissolved in 20 mL of
Me0H and treated
with a trace amount of HBr. The mixture was stirred at reflux for 1 hour,
cooled and the solid
was collected by filtration to give the desired (L4 g). MS (m/z): 285 (M--1-1-
1)+.
Step 2
Pd/C
02N 0 \
NO2 ____________________________________ H2 11P¨NH2
To a suspension of the product from step 1 above (0.7 g, 2.5 mmol) in Me0H (50
mL) was added 0.1 g of Pd/C (20%), and the suspension was stirred under 25 psi
of H2 at RT.
After filtration, the filtrate was concentrated in vacuo to give the desired
compound. MS (m/z):
225 (M+H)+.
Step 3
0,
c.>
N OH
H2N
L
/ Q'yEN-C'¨N \ \
= NH, phoH0 , =NH N
PhCH2A0 (1\
HATU
The mixture of the diamine from step 2 above (225 mg, 1 mmol), N-
phenylacetylproline (1 mmol), DIPEA (5 mmol) and HATU (380 mg, 1 mmol) in 10
mL of
MeCN was stirred at RT for 1 hour. The reaction mixture was concentrated, and
the residue was
purified by column chromatography to give the desired compound. 1HNMR (Me0D)
6: 9.3 (s,
1H), 8.2 (s, 114), 7.5-7.1 (m, 16H), 4.6 (m, 1H), 4.5 (m, 1H), 3.8 (m, 811),
2.3-1.8 (m, 8H).
- 92 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Example 39¨ (2S,2S)-N,N1-1H,111-2,21-bithdole-515'-divlbisil-
(phenylacetyl)pyrrolidine-2-
earboxamidel
=fll H
yN Ny-;"N
N N 0
H H
Step .1
02N io pc,Br3
0 \ Br
To a suspension of the lactarn (10.0 g, 56 mmol) in 200 ml of 1,2-
dichloroethane,
was added POBr3 (15.3g, 53.2 mmol) at RT. The resulting mixture was heated at
reflux
temperature in a 90 C oil bath for 0.5 hour (the reaction formed a copious
amount of precipitate,
and an oil bath was preferred over a heating mantle, as it provided gentle
heating and avoided a
darkening of the precipitate). The reaction was cooled just below reflux
temperature, and
imidazole (4.57 g, 62 mmol) was added in one portion. The resulting gummy
suspension was
heated at reflux temperature in an oil bath for another 2 hours. The reaction
was cooled to RT,
and 100 mL of ice-water was added. Solid NaHCO3 (ca. 50 g) was added to the
mixture until no
further gas was evolved. The suspension was extracted with DCM (4x), and the
combined DCM
extracts were washed with 300 mL of brine. The DCM extracts were filtered
through silica gel
and concentrated to dryness to afford a crude product. The crude product was
recrystallized
from chloroform to give 5.41 g of the desired compound as a white solid. The
filtrate was
concentrated to dryness, and the residue was purified by flash column
chromatography (30%
Et0Ac/Hex) to give an additional 2.6 g of desired product. MS (EST) m/e
242.
Step 2
Bac
02N so
N Pd(dppf)C12 C)2N µ
Br + (H0)2B ip
N N 111r NHBoc
NHBoe
H Boc
The mixture of compound from step 1 above (602.6 mg, 2.5 mmol), the indole
boronic acid (1.034 g, 2.75mmol), Pd(dppf)C12(183 mg, 0.25 rnmol), Na2CO3 (
530 mg, 5.0
mmol) in 5 mL dioxane-1-120 (5:1) was heated to reflux under N2 atmosphere
overnight. When
reaction was complete the mixture was poured into water and extracted with
DCM. The organic
phase was dried over Na2SO4, concentrated, and the residue was purified to
give compound the
desired product. MS (ESI) nile (M+H+): 493.
- 93 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Step 3
02N NH2
HCI-Me0H /
N N
H Bac H H
The product from step 2 (600 mg, 1.3 mmol) was added into HC1 (30 ml, 3M in
Me0H). Then the mixture stirred at RT for 2-3 hours. When reaction was
complete, the mixture
was concentrated to give the crude product (400 mg). MS (ESI) m/e (M+H+): 293.
Step 4
02N lip
401 .H2 NH?
RUC
N N
H H H2 H H
The product from step 3 (400 mg, 1.36 mmol) was dissolved in Et0Ac and treated
with Pd/C (100 mg, 20%). Then the mixture was stirred at RT overnight under H2
atmosphere.
When the reaction was complete, the Pd/C was filtered off, and the resulting
solution was
concentrated to give the crude product (300 mg). MS (ESI) m/e (M+H+): 263.
Step 5
H2N NH2 H H
RCOOH WP
-1\1 0
H H BOP H H
A solution containing (131 mg, 0.5 mmol) of the product from step 4, RCOOH
(256.608 mg, 1.1 mmol), DIPEA (390 mg, 1.5 mmol) in CH3CN (2 mL) was stirred
at RT for 5
minutes, then HATU (418 mg, 1.1 mmol) was added into the mixture. The mixture
was stirred
at RT overnight. When reaction was complete, the mixture was concentrated, and
the residue
was purified to give the desired product. 1HNMR (Me0D) 6: 7.15-7,75 (m, 18H),
4.51-4.59 (in,
2H), 3.56-3.80 (m, 10H), 2.37 (s, 3H), 1.97-2.30 (m, 8H). MS (ESI) m/e (M+H+):
693.
Example 40 ¨ di-tert-bulvl (11-1,111-2,2'-blindole-5,5'-
dirlbisfearbamovI(2S)pwrolidine-2,1-
diplifiR)-2-oxo-1-phenylethane-2,1-diyll1)bisearbamate
H H
NThrN a
0 N N 41111-2-F 0 0
BocHN H H NHBoc
This compound was prepared using the similar method as Example 39, step 5
using N-Boc-R-Phg-L-Pro-OH. 1HNMR (Me0D) 6: 6.78-7.78 (m, 181-1), 5.49 (m,
2H), 4.55-
4.58 (m, 21-1), 3.94-3.97 (m, 311), 2.37 (s, 311), 1.87-2.14 (m, 811), 1.40(s,
18H). MS (EST) mie
(M+H+):924.
- 94 -
:A 02756172 2011-09-21
WO 2010/111483
PCT/US2010/028653
Example 41 ¨ tert-butyl f(1R)--2-oxo-1-phenyl-2-1(2S)-2-(f2-14-(11(2S)-1-
(phenylaceivli
pyrralldin-2-vlicarbonyliamino)phenv11-1H-indo1-5-vlicarbamovbwrolidin-1-
vIlethyllearbamate
cl11
Boc.FIN 0y-' =NH N-
o 0
4It
Step 1
0 No, 02N
Br up,=
asti
.N1-12 = H,N =Br
¨N
To a solution of 4-bromophenylhydrazine (2,5 g, 13.4 mmol) in acetic acid
(19.5
mL) and Et0H (14.5 mL), 4-nitroacetophenone (1.66 g, 10.0 mmol) was added. The
reaction
was refluxed for 5 hours, and water (35 mL) was added. The resulting mixture
was stirred for
another 1 hour, and the resulting solid was filtered, washed with water to
afford 3.1 g of the
desired compound. 1H NMR (Me0D) 6: 8.21-8.23 (d, J= 8.011z, 211), 8.01-8.03
(d,J 8.0Hz,
2H), 7.34-7.36 (d, J= 8.0Hz, 2H), 7.20-7.22 (d, J= 8.0Hz, 211), 2.03 (s, 3H).
Step 2
02N
Hp 41 Br ___________________________________ Br
NO2
The product from step 1 above (2.0 g, 6.0 mmol) was added into PPA (20 mL),
and the mixture was stirred at 80 C for 1 hour before it was cooled in ice-
bath, diluted with
water/Et0Ac (60/20 mL) and stirred for another 1 hour. The mixture was
extracted with Et0Ac
and washed to yield the target. 111 NMR (Me0D) 8: 8.28-8.30 (d, J= 8.0Hz,
211), 7.97-7.99 (d, J
= 8.0Hz, 211), 7,72 (s, 1H), 7.33-7.35 (d, J¨ 8.0Hz, 114), 7.24-7.26 (d, J=
8.0Hz, 1H), 7.02 (s,
1H).
Step 3
Br
AO \
NO2 Pt12(dba)a H2N
NO,
P1Bu3, lithium bis(lrimethylsily1)-
amide, THF, reflux, lh
An oven-dried argon-cooled round-bottom flask was charged with the indole from
step 2 (2.0 g, 6.31 mmol) and 0.05 equivalents of Pd2(dba)3 in THF (100 mL). A
solution of tri-
tert-butyl phosphine (10 wt %) in hexane (1.93 mL, 0.63 mmol) was added
followed by lithium
- 95 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
hexamethyidiSilazane (1.0 M in THF) (18.9 mL, 18.9 mmol). The dark solution
was heated to
reflux overnight then cooled to RT. This mixture was poured into ice-cold aq
1.0 M (70
mL) and stirred vigorously. Hexane was added, and stirring was continued for
30 minutes. The
precipitate was filtered, washed with 20 mL of cold water and then 20 mL of
THF:Hexanes
(5:95) solution. The precipitate was washed with 200 mL of Me0H, and the
filtrate was
concentrated to give 1.5 g of the desired compound. MS trilz: 254 (M+1).
Step 4
H2NOH
BocHN s 40 BocHN * 1402 o \ o 0
00 41k
To a solution of the product from step 3 above (1 g, 3.9 mmol) in acetonitrile
(20
mL) was added R-N-Boc-Phg-S-Pro-OH (1.4 g, 3.9 mmol), HATU (3 g, 7.8 mmol) and
DIPEA
(1 g, 7.8 mmol). The mixture was stirred at RT overnight. The solvent was
distilled, and the
residue was dissolved in Et0Ac and washed with water. The organic layer was
dried and
concentrated in vaeuo, and the residue was purified by column chromatography
to give the
desired compound (1.8 g). MS (ESI) in/e (M+H+): 584.
Step 5
Fi
(Nly *
BocHNfir NO2 N2 BocHN so \ gp NH,
0 0 N
0 0
To a solution of compound from step 4 (300 mg, 0.51 mmol) in Me0H (10 mL)
was added Pd/C (50 mg, 0.28 mmol). The mixture was stirred under H2 atmosphere
at RT for 1
hour. The catalyst was filtered off, and the filtrate was concentrated in
vacuo to give the desired
compound (230 mg) as a yellow oil, which was used directly in next step. MS
(ESI) mie
(M+H)+: 554.
Step 6
BocHt4 (N.IY'l
40 *
NH (14-OH
0 ______________________________________________ BocHN criyH
NH N
41k
To a solution of the compound from step 5 (100 mg, 0.18 mmol) in acetonitrile
(3
mL) was added compound N-phenylacetyl-L-proline (42 mg, 0.18 mmol), HATU (140
mg, 0.36
mmol) and DIPEA (46 mg, 0.36 mmol). The mixture was stirred at RT overnight
then
- 96 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
concentrated, and the residue was purified by RPLC to give (60 mg). 11-1 NMR
(CDC13) 8: 9.62
(s, 1H), 9.08 (s, 1H), 7.84 (s, 111), 7.45-7.09 (m, 15H), 6.52 (s, 1H), 5.67
(s, 1H), 5.49 (m, 1H),
4.72-4.51 (m, 2H), 3.82-3.48 (m, 5H), 3.20 (m, 1H), 2.15-1.72 (in, 8H), 1.43
(s, 9H). MS (EST)
m/e (M+H+): 769.
Example 42¨ (2S)-1-(phenvlacetvl)-N-12-15-W2S)-1-(phenvlacetvl)pwrolidin-2-
vilcarbonyllamino)-.1H-indol-2-vilpyrimidin-5-vlipyrralidine-2-carboxamide
=(19)(N ___________________________________________ ) 0
0 r N
0 N 0 *
Step 1
o2N H2N
\ Fe/NH4CI io
A mixture of 5-nitroindole (5 g, 30.9 mmol), Fe (8.6 g, 154 mmol), NR4C1 (16.5
g, 309 mmol), Et0H (80 mL) and water (20 mL) was refluxed under N2 protection
for 2 hours.
The mixture was cooled to RTand filtered. The filtrate was concentrated and
dissolved in water.
The mixture was basified with Na2CO3 and extracted with CH2C12 two times. The
combined
organic phases were combined, dried over Na2SO4 and filtered. The filtrate was
concentrated to
yield the product (3.6 g). MS (ES1) m/e (M+H+): 133. 1H NMR (DMSO) 6: 10.55
(s, 1H), 7.12-
7.06 (m, 2H), 6.68 (d, J-2.0 Hz, 1H), 6.48 (dd, J=8.4 Hz, 2.0 Hz, 1H), 6.12
(t, J=2.0 Hz, I11),
4.39 (s, 2H).
Step 2
H2N io
Boc20, DMAP Boc2N 0
Boc
A mixture of 5-aminoindole (20 g, 76 mmol), DMAP (9.2 g, 38 mmol) THF (250
mL) and CH3CN (100 mL) was cooled to 0 C Boc20 (132 g, 304 mmol) was slowly
added to the
mixture. The reaction mixture was allowed to warm to RT and stirred over the
weekend. The
mixture was poured into water and exacted with CH2C12 three times. The organic
phase was
combined, dried over Na2SO4 and filtered. The filtrate was concentrated,
dissolved in CH2Cl2
and poured into PE and filtered. The solid was purified by column
chromatography
(PE/EA-20/1) to yield the product (23 g). NMR (CDC13): 8.05 (d, J-8.0 Hz,
1H), 7.56 (d,
J=3.2 Hz, 1H), 7.28 (d, J=2.4 Hz, 1H), 7.03 (dd, J=8.8 Hz, 1.6 Hz, 1H), 1.63
(s, 9H), 1.36 (s,
18H).
- 97 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Step 3
sou2t4 so BocHN ra
\ LDA, {1PrO)kB
\ B(011)2
Boo Boo
A mixture of the product from step 2 above (5.0 g, 11.6 mmol), (iPrO)3B (17.5
mL, 92.8 mmol) and dry THF (100 mL) was cooled to 0 C LDA (prepared from nBuLi
and
iPr2NH in THF, about 116 mmol) was slowly added to the mixture at 0 C. The
mixture was
allowed to warm to RT and stirred for 2 hours. The mixture was quenched by the
addition of 1N
HCI to pH=3 and extracted with CH2C12 three times. The combined organic phases
were
combined, dried over Na2SO4 and filtered. The filtrate was concentrated and
purified by column
chromatography (PE/CH2Cl2=1/1 to pure CH2C12 to CH2C12/acetone=10/1 to pure
acetone) to
afford the product (2.8g). IFI NMR (DMSO) 8: 9.22 (s, 111), 8.10 (s, 211),
7.84 (d, J=9.2 Hz,
1H), 7.65 (s, 111), 7.20 (dd, J=9.2 Hz, 2.0 Hz, 111), 6.48 (d, 1H), 1.51 (s,
911), 1.41 (s, 911).
Step 4
Fo/NH4C1
C1--0¨\ NH2
N¨
A mixture of the pyrimidine compound (0.3 g, 2 mmol), Fe powder (560 mg, 10
mmol), NH4C1 (1.07 g, 20 mmol), Et0H (8 mL) and water (2 mL) was refluxed
under N2
overnight. The mixture was cooled to RT and filtered. The filtrate was
concentrated and
dissolved in water. The mixture was basified with Na2CO3 and extracted with
CH2C12 two times.
The combined organic phases were dried over Na2SO4 and filtered. The filtrate
was
concentrated to yield the product (120 mg). MS (ESI) mie (M+H+): 130. 1H NMR
(DMSO) 8:
7.99 (s, 211), 5.73 (s, 211).
Step 5
BocHN B(01-02 BocHN N
Pd(dpp1)C12
Boo
A
BOO OC
A mixture of the pyrimidine from step 4 (100 mg, 0.77 mmol), indole boronic
acid from step 3 (290 mg, 0.77 mmol), Pd(dppDC12 (56 mg, 0.077 mmol), Na2CO3
(244 mg, 2.3
mmol), THF (20 mL) and H20 (2 mL) was refluxed under N2 overnight. The mixture
was
poured into water and extracted with CH2C12. The organic phase was combined,
dried over
Na2SO4 and filtered. The filtrate was purified by prep TLC (CH2C12/Me0H=20/1)
to afford the
product. MS (EST) m/e (M+H+): 426.
- 98 -
:A 02756172 2011-09-21
WO 2010/111483
PCT/US2010/028653
Step 6
BocHN =,,,
1 \ * NH2 HCIAlle0H_ H2N 0 , 0,,_
NH,
---- N N N¨
Boc H
A mixture of compound from step 5 above was added to a solution HC1 in Me0H
(4M) cooled with ice bath. The mixture was allowed to warm to RT and stirred
overnight. The
mixture was concentrated, dissolved in water, washed by CH2C12 and
concentrated. The residue
was directly used in the next step without further purification. MS (EST) m/e
(M-FH+): 226
Step 7
7-1 HATU
H2N *1<-N-1-1--"
,
so , Ho_o to ,
\ 7p4D¨NH2 +c ' 0 = \Nj-
-1\1 0 " Nit
0
H 0
The product of step 6 above (0.22 mmol), N-phenylacetyl-L-proline (51 mg, 0.22
mmol), DIPEA (100 mg), and DMF (3 mL) was added HATU (84 mg, 0.22 mmol), and
the
mixture was stirred at RT overnight. The mixture was purified by RPLC to
afford the product.
MS (ES1) mie (M+H+): 656. 111 NMR (CDCI3) 8: 10.42 (s, 1H), 10.01 (s, 11-1),
9.77 (s, 111), 8.32
(s, 1H), 7.34-7.27 (m, 11H), 7.05-7.00 (m, 2H), 6.58 (d, .J=8.0 Hz, 11-1),
4.63-4.53 (m, 2H), 3.84-
3.58 (in, 8H), 2.30-1.94 (m, 8H).
Examples 43-87
Compounds of Examples 43-87 were prepared in a similar manner as described in
either Example 41 or Example 42.
1
Example Structure MW Name
tert-butyl , NH N {(1R)-2-[(25)-2-(
{445-
* ri
W 0 N ilk
({[(2,5)-1-{(2R)-2-Rtert-
butoxycarbonypamino]-2-
43 OH 0 0 H
phenylacetyl}pyrrolidin-2
CH H3-
r.õ1,TH
S N 0 = IN
\ro 884.054
yllcarbonyl} amino)-1H-indo1-2-
* 0)c-cH3
yliphenyl} carbamoyl)pyn-olid in-
cHP3 1-
y1)-2-oxo-1-
phenylethyl) carbamate _
methyl {(1R)-2-[(2S)-2-({445-
il
N 0)...,-==-.)
CH5yr,1 Cif 0 N \ it, N' 0 N) H Rmethoxyearbonyl)amino]-2-
0
44 0
H
= µN 799.891
phenylacetyllpyrrolidin-2-
41 it rckcH3
yl] carbonyl} amino)-1H-indo1-2-
yl]phenyl} c arbamoyppyrrolidin-
1-y1}-2-oxo-1-
1
phenylethyl) carbamate
- 99 -
:A 02756172 2011-09-21
WO 2010/111483
PCT/US2010/028653
_
Example Structure MW Name
(2S)-1-[(2R)-2-pheny1-2-
N
(pyn-olidin- 1 -ypacetyli-N-(4- {5-
[( {(25)-1-[(2R)-2-phenyl-2-
,., --- NI
45 o 792.002
(pyrrolidin-l-yl)acetyl)
* pyn-olidin-2-yll
carbonyl)amino] -
Ili-indol-2-y1) phenyl)
pyrrolidine-2-carboxamide
(2S)-1- {(2R)-2-
1 CIH [(cyclopropylacetypamino]-2-
N 0)----3 phenylacety1)-N-{4-15-({[(25)-1-
-10c 1 ,....,' NH N
H
0 ,or 848.023
{(2R)-24(cyclopropylacetyl)
. Ea rv
ix
amino] -2-phenylac etyl)
46 0 0 H
pyrrolidin-2 -ylicarhonyl I amino)-
1H-indo1-2-yljphenyll
pytmlidine-2-carboxamide .
(23)-1- ( (2R)-24(3-
methylbutanoyDaminoi-2-
phenylacetyll -N- (4-[5-({[(28)-1-
CHa 0 0 0 ,H {
(2R)-2-[(3-methylbutanoyl)
47 H 41\1,,,,,,,,,c H 852.055
4It * 6 & 3 amino}-2-
phenylacetyl)
, pyrrolidin-2-ylicarbonyl) amino)-
I H-indo1-2-yllphenyl)
pyrrolidirke-2-carboxamide
-
(23)-1-[(2R)-2-(morpholin-4-y1)-
(5
H
N N _.j ,..õ...
2-phenylacetylj-N-(4- {5-[({(2S)-
\ . NH N 1-[(2R)-2-(morpholin-4 -y1)-2-
0 --- N 0 , INr-ji 824.001
48 0 H
phenylacetyllpyrroliclin-2-
*
Ilk yl)carbonyI)aminol-1H-
indo1-2-
yllphenyl)pyrrolidine-2- '
carboxamide
,
H (25)-1 -(2,3 ( -diphenylpropanoy1)-
N 4.1 o
49 0 N \ lik NH 834,04
diphenylpropanoyl)pyrrolidin-2-
o
H o 834.04
fit * * yl] carbonyl ) amino)-1H-indo1-2-
AphenyI) pyrrol idine-2 -
carboxamide
(2S)-14(2R)-2-{ [(4-
c.H.,
rnethylphenypsulfonyljaminol-2-
H phenylacety1)-N-(4-154( 425)-1-
* õ.1=1 Cl-r-N Ail Ai cNt-c)
01:1 0 0 lir N lirr [(2R)-
2- {[(4-methylphenyl)
50 H 0 , ,I, 4. , 992.195
sulfonyl]amino)-2-phenylacetyli
6A0 pyrrolidin-2-y1)
ca.rbonyl)arninoi-
1H-indo1-2-y1) phenyl)
pyrrolidine-2-carboxamide
(23)-1- {(2R)-2-
O
0 N
[(cyclohexylcarbonyl)amino]-2-
phenylacetyl) -N- {445 -( ( [(2S)-1-
r ,d ciirN 0 , is,- N17 )\N)
{(2/0-24(cyclohexylcarbonyl)
51 o 0 I"
H ,u1>ra 904.132
amino]-2-phenylacetyl)
411t o pyrrolidin-2-yli carbonyl ) amino)-
1H-indo1-2-yljphenyl)
pyrrolidine-2-carboxamide
- 100-
:A 02756172 2011-09-21
WO 2010/111483 PCT/US2010/028653
_
Example Structure MW Name
H methyl f (1R)-24(25)-24
{ 244-
N _.
CH? IrVi lit ' IP :1-)rc"'
(acetylamino)pheny1]-1H-indo I-
52 r , 553.623
5-y1) carbamoyl)pyrro1idin-1-y1]-
* 2-oxo-1-phenylethyl} carbamate
_
H \ _ 0)_....õ---j
tert-butyl {(1R)-24(25)-24{4-[5-
,
tN N ,.., 0 = . NH N (acetylamino)-1H-indo1-2-
' , .,,, H
53 H
CH , y N 595.704 yliphenyl)
carbamoyl)pyrrolidin-
2 t ck)ccH3 1-y1]-2-oxo- 1 -
)
CH=pi3
phenylethyl) carbamate
N-{ 244-(acetylam ino)phenyIj-
_
0 '1,1rN At a
1H-indo1-5-yll -14(2R)-2-
54 o IF :,1 1.1r1 7---cH3 565.678
(morpholin-4-y1)-2-
I. phenylacetylj-L-prolinamide
H
ot-CI43 propan-2-y1 {(1R)-2-
[(2S)-2-( {2-
I-13 0 H .-NIIN
2:3)or N [4-(acetylamino)pheny1]-1H-
55 0 N
H 581.677 indoI-5-y1)
carbamoyl)pyrrolidin-
* 1-y11-2-oxo-l-
phenylethyl) carbamate
_
(25)-1-{(2R)-2-
\ =. N r)
al,t4 Allith- :11--,,,4
[(cyclopentylcarbamoyl)aminol-
2-phenylacetyl} -N-(4- {54( { (25)-
t!1 N
0 ir N
1-[(2R)-2-(dimethylamino)-2-
56 ---- 0 0 .,,N,Ior No 817.953
phenylacetyljpyrrolidin-2-
SI 11 yl} carbonyparnino]-1H-
indo1-2-
yl)phenyl)pyrrolidine-2-
carboxamide
tert-butyl { (2R)-14(25)-24 {445-
( {[(25)-1-{(2R)-24(tert-
butoxycarbonypaminol-3-
CH3 H Clio
*I , 4m.-- 54-3
1 57 cHrt N.rk.0 0 my isimc,
H 816.019
methylbutanoyl)pyrrolidin-2-
CH3 CH3 c$.1 iccH3
_0 ylicarbonyll arnino)-1H-
indo1-2-
f, j \
3 -- cHpH, yl]phenyl)
carbamoyl)pyrrolidin-
1-y11-3 -methyl-l-oxobutan-2-
yl) carbamate
-
H N- 1)-
{244-[4 -
a clirm . lik ii 1H-indol-5-yll -1- [(2R)-2-
phenyl-
58 0
N _.
H Ne CH3 549.679
2-(pyrrolidin-1 -yl)acetyli-L-
I. prolinamide
-,-
(25)-1- [(2R)-2-(dimethylamino)-
4-methylpentanoylj-N-(4 - {5-
H
Cfirm N 0,)
li
- r------ ( )---/ NH ,,, (dimethylamino)-4-
cH3
59 0 0 '-'""----N \ / 0 >-.r13 699.945
methylpentanoyljpyrrolid in-2-
H = 1N
CH3 CH3 µCH3 yl) carbonyl)amino1-1H-indo1-2-
CH yl)phenyppyrrolidine-2-
cH3 carboxamide (non-
preferred
name)
- 101 -
:Ammm72m11-m21
WO 2010/111483 PCT/US2010/028653
......., ____________________________________________________________________
Example Structure MW Name
_
(28)-I -[(2R)-2-phenyl-2-
H
(?)¨\..:4--- (piperidin- 1 -3/1)acetyli -N-(4- { 5-
a CliN 0 \ . NH '11/41"-- R{(28)-1-[(2R)-2-pheny1-2-
0
60 0 N
H 0 . \ NO 820.056 (piperidin-l-y pat etYlip yrrolid in-
*
* 2-y1} carbonyparnino]-1H-indol-
2-y1} phenyppyrrolid ine-2-
carboxarnide
____.N
Q (2S)-1-[(2R)-2-(1H-imidazol-1-
:;----
) \ y1)-2 -phenylacety1}-
N-(4- {5-
\ / NH N"-- R {(25)-1- [(2R)-2 -(1H-
imidazol-
' 0 --...."---1
61 o H 0 .\N) 785.914 1-y1)-2-phenylacetyllpyn-
olidin-
*
. 2-y1) earbonyl)aminol-1H-indol-
2-y1) phenyl)pyrrolidine-2-
earboxamide
.
(2,3)-14(2R)-2-(diethylamino)-2-
QLll
cH3 40 0)_,,,,.,,_,
,--...
phenylacetyll-N-(4- {54( { (2S)- 1-
..,
[(2R)-2-(diethylamino)-2-
o
62 0 N N.-- -I
rcH3
H 0 = µN 796.034 phenylacetylipyrrolidin-2- ,
Ili
* .._.3 y4}carbonyparninol-1H-
indol-2-
y1) phenyppyrrolidine-2-
carboxamide
O -
tert-butyl {(18)-21(28)-24 {445-
Z CI , 0H
f
-
( { [(28-1- a cetylpy-rroldin-2--
63 0CH- , 692.822ylicarbonyl1amino)1H-
indo12-
)c-cH3 yllphenyl }
carbamoyppyrroldin-
4,0 cl.e.13 1-y1]-2-oxo-l-
phenylethyl) carbamate
.
tert-butyl {(1R)-21(28)-24 {445-
({[(25)- I -acetylpyrrolidin-2-
J 40 . NH N
yl] carbonyl} amino)-1H-indo1-2-
64 0=-- "\cH30 N
H H
1N 692.822
, r )c-cH, yl]phenyl}
carbamoyppyrrolidin-
qAt cHP3 1-y1]-2-oxo-1-
phenylethyl carbarnate
tert-butyl {(1R)-2-oxo-l-phenyl-
cH3 H
41-111 CILrN 24(2S)-2-({244-
({[(2S)-1-
CF r
* \ IP 5-\ \
65 o 692.866<., (Propan-2-
yl)pyrrolidin-2-
yti carbonyl } amino)pheny11-1H-
4,N N -
H H IN
CH( indo1-5-
y1}carbamoyl)pyffolidin-
cH3 1-yliethyll
carbamate
CHO,-,-/'-i- N-{244-[4
11 iiii .
66 C4 0 "
NH 1H-indo1-5-y1} -1-
[(2R)-2-
ur
0 ' '
H t"' 523.64
(dimethylamino)-2-
at phenylacety1I-L-
prolinamide
HN-{214-(acetylamino)phenyli-
CNi 0 . CIINCH3
1H-indo1-5-y1}-14(2R)-2-phenyl-
67 o N
H 563.706
2-(piper idin-l-yDacetyli -L-
O. prolinamide
- 102 -
:Ammm72m11-m21
WO 2010/111483
PCT/US2010/028653
Example Structure MW Name
tert-butyl-2- {(2S)-24(2- {4-
cH3 0 H fir 11 40 , o)__,
.õ--j R {(2S)-1-1(2R)-2-
(dimethylarnino)-2-
cHti, y N
N * NH N
0 CH 3 phenylacetyl]pyrrolidin-2-
68 0
H 0'
, 811.99
ft
* c1-13 yl} carbonypamino]phenyl} -1 H-
indo1-5-yl)carbamoyl]pyn-olidin-
1-y1} -2-oxo-1-
phenylethylicarbatnate
,
tert-butyl f(1R)-2- {(2S)-2-[(4- {5-
0) D
cH3fly. Fri
01.i31 0 0 N\ * NH N (dimethylainino)-2-
phenylacetyll
69 o H 08 cHH3 carb H
N 0
,1 811.99 pyrrolidin-2-
yl}carbony1)amino]-
\iccH3
P 1H-indo1-2-yll
phenyl)
amoyli pyrrolidin-1-y1) -2-
oxo-l-phenylethyllearbamate _
(28)-1-[(2 R)-2-(diniethy1amino)-
1
H 2-phenylacetyI]-N- {4454
{ [(28)-
N
H N \ 1-{(2R)-2-[(3,3-
CriN Cif . H N li, N
CH3 dirnethylbutanoyDamino]-
2-
70 CH õ , 0 0 ' 810.017
,cH3 .4N
r-13 v phenylacetyl} pyrrolidin-2-
lir
*
/ ylicarbonyl} amino)-1H-
indol-2-
yl]phenyl}pyrrolidine-2-
carboxamide
.,.
(2S)-1-(cyc1opropylacetyI)-N-(4-
H
cr\N
NH (dirnethylamino)-2-
71 ci4 0 ca., 660.823
phenylacetylipyrrolidin-2-
yl} carbonyl)aminoi-IH-indol-2-
4110
y1}phenyppyrrolidine-2-
carboxamide
_
(25)-1-[(2R)-2-(d1methylamino)-
H
0 \ fa, NcN) 2-phenylacetyll-N-(2- {4-
[({(25)-
N
NH 1- [(2R)-
tetrahydrofuran-2-
72 cas1 o 0 o'N=10.4 676.823
ylearbonylipmolidin-2-
40 yl}
carbonyl)aminolphenyl) -1 H-
indo1-5-yl)pyrrolidine-2 -
carboxamide
tert-butyl [(1R)-2- {(2S)-2-[(4- {5-
H [({(13)-1-[(2R)-2-[(tert-
Ctl" H '..111.( N Aiii..,6 \ la 02i--,;,
$ND
butoxycarbonypamino}-2-(4-
ct-i,r1 o 0 1,1 N 'W n H
73
4. Ft ' .µN, , 920 035
fluorophenyl)acetyl]pyrrolidin-2-
r -xcii3 - yl } carbonyl)amino)-111-
indol-2-
* cHpa
yl}phenyl)carbamoyljpyiTolidin-
P
F 1-3/1) -1-(4-
fluoropheny1)-2-
oxoethyl]carbamate
(25)-1- {(2R)-2-[methyl(3-
methylbutyparnino)-2-
H , ,z=
phenylacetyl) -N - {4454 { [(2S)-1 -
CH PH3fifN AIN le Nin -ND
\
0 11, CH3 {(2R)-2-[methyl(3-
methyIbutyl)
74 cH8 0 N ' 0 .µNi 852.142
#gt "Th--cHa
amino]-2-phenylacetyl }
pyrrolidin-2-yllcarbonyl} amino)-
eHa
1H-indo1-2-yliphenyl)
pyrrolidine-2-carboxamide
- 103 -
:A 02756172 2011-09-21
WO 2010/111483 PCT/US2010/028653
Example _ Structure MW Name
(28)-1-[(25)-tetrahydrofuran-2-
is_ H ylcarbonylj-N-(4- {5- [(
{ (28)-1 -
[-I H
Ai, __LNN'ThfN ip \NH ix NrC75 [(25)-tetrahydrofuran-
2-
613.72 y1carbonyllpyn-olidin-2-
o
c-or o oNi= 6 \ yl} carbonyl)amino]-1H-
indo1-2-
0,/ yl}phenyl)pyrrolidine-2-
carboxamide
N-(tert-butoxyearbony1)-D-valyl-
N-{445-({1-[(2R)-2-
Z3-1,N =N;J hi rm H (dimethylarnino)-2-
76 c1-13/ o - im 0., \ Nõ J.., (GH3 777.972
phenylacety1]-1,-prolyll amino)-
40 CH3 CH? ce-13. 1H-indo1-2-yliphenyl)-
1,-
prolinarnide
, .
(28)-1- [(2R)-2-(dimethy lam ino)-
H
cHPH AdiFk \ . N _Z-7-3 2-pheny lac etyll-N- {2144 { [(28)-
--IN " ipt, NH cr \ N I -( 1,3 -
oxazol-2-
77 0141 0 (:).,.-:- N \
673.778 ylcarbonyl)pyn-olidin-2-
o-../ yflcarbonyl} am ino)pheny 1]-1H-
0 indo1-5-yllpyr-
rolidine-2-
carboxamide
tert-butyl [(1R)-2- { [(28)-14 {4-
ci-t3 , 0_cii, [5-({(25)-2-[{(2R)-2-
[(tert-
cH, 1
\ [4 NH N-CH3 butoxycarbonypamino}-2-
CH3 0 H N' y ip .
78 01K rN 0 0
N 0 .di 860.032 phenylacetyl}
(rnethyl)aminolpro
et)r x-cria
CH,panoyl) am ino)-1H-indo1-2-
çj yliphenyl} amino)-1 -oxoprop an-
2-ylj (rn ethyl)amino} -2-oxo-1-
phenylethylicarbamate
oCHO
p::
tert-butyl {(1R)-2-[(25)-2-( {214-
1----NH (1.-r.0 (methylam ino)pheny11-1H-
ind ol-
79 567.694
= a 0 * \ . 5-y1)
carbamoyl)pyn-olidin- 1 -y11-
.. 11,F1 2-oxo-1-pheny 'ethyl}
carbamate
cH,
H 0 (28)-1-[(2R)-2-(d im
ethy lam ino)-
80 0
. 0 NH N pH3c1,11, N N--- -D 2-
phenylacetyli-N- {2444 { [(25)-
N = \ =
CH3 1 -
(phenylacetyl)pyiTolidin-2-
H
o8 696.857
ft yllearbonyl} am
ino)phenyl] -1H-
Md ol-5-y1) pyrrolidine-2-
carboxamide
,
H (2S)-1-{ [(3R)-1-
benzylpyrrolidin-
H \ 40 t4)._-) :z--
3-yli carbonyl} -N-(4- {54( {(2.8)-
CH3N IP NH 0 J1
1- [(2R)-2-(dimethylam ino)-2-
81 c1 o 0
l- o ,...--) 765.964
phenyIacetylipyrrolidin-2-
411 ----- N
1110 yl} carbonyl)aminol-1H-
indo1-2-
y1} phenyppyuolid in e-2-
carboxamide
H (28)-1- { [(38)-1-
benzylpyrrolidin-
ri hi
N illa \ . Ni------N) 3-yl] carbonyl } -N-(4- ( 5- [( {(25)-
wir NH 1-[(2R)-2-
(climethylamino)-2-
82 oda' o 0 oAn 765.964
phenylaeetyllpyrrolidin-2-
=
* N
* yl} carbonyparninol-1H-
indo1-2-
yllphenyppyrro1idine-2-
carboxamide
- 104 -
:A 02756172 2011-09-21
WO 2010/111483
PCT/US2010/028653
Example Structure MW Name
NN-dimethyl-D-leucyl-N- {445-
C%
83
N N N 0 \ ip NH PI) ,õ,..
( {1-R2R)-2-(dimethylarnino)-2-
0
0 N
H (21), ,,,i73 719.935
phenylacetyli-L-proly1) amino)-
* CH3 CI i3 11/-indol-2-
y1ipheny1 } -I,-
CHa prolinamide
¨
c'H/nH
-IlN\ 0,.__a 577733
N-(2- (4-Rcyclopentylcarbonyl)
84
CHti 0 gp = If mi
aminoThhenyl} -1H-indo1-5-y1)-1-
o
1.1 .
[(2R)-2-(dimethy1amino)-2-
0.,_,fH3 tpehret-butyl
-L--1p4ro{41757{d1e,_
_ .
,
CHN3p-t3(1,1 [(2R)-2-(dimethylarnino)-2-
0 \ I. NH HN--< CH3
85 0 0 N 0õ \--CH3 652.8
pheny1acetylj-L-proly1) amino)-
II
lilt CH3
1H-indo1-2-yllphenyl}amino)-1-
oxopropan-2-yl]carbamate
0 tert-butyl [(1S)-
24 {4454 { 1-
Fl
pH [(2R)-2-
(dimethylamino)-2-
3cliN , \)4
86 chy 0 a N\ ii NH HN---< CH3cH 714.872
phenylacetyli-L-prolyll amino)-
0 H 0--1/4-cii 3
111-indo1-2-yl]phenyll amino)-2-
41, oxo-l-
phenylethylicarbamate
CH3 tert-butyl [(2R)-1-
( {4454 { 1-
o)ot.ot3 [(2R)-2-
(dimethylamino)-2-
pH3ciH
N
87 CHII 0r io \ . NH HN----( CH3680.855
phenylacetyli-L-proly1) amino)-
ic-
o N
ocH3 11/-indol-2-yl]phenyl) amino)-3-
OH,
methy1-1-oxobutan-2-
ylicarbamate
Example 88¨ (2S,2S)-N,M-5,6,7,12-tetrahydrobenzo16,71eyeloheptafl,2-blindole-
3,9-
divlbisil-(phenvlacetyl)pyrrolidine-2-earboxamidel
di 4 i OR NH -N-J
CkrkCoN iir NH 0 ,O,
0
Step 1
SO HNO3,H7s.4 0
___________________________________________ 0,N C001 i
COOH
0
0
To a mixture of HNO3 (4 mL) and H2SO4 (2 mL) at 0 C was slowly added the
above carboxylic acid (2 g, 10.4 mmol). The mixture was stirred under 0 C for
30 minutes. The
resulting solution was poured into 20 mL of H20 at 0 C, and the precipitate
was filtered to give
compound (2 g) as a yellow solid. MS (ESI) mie (M+H+): 238.
Step 2
0 H2, Pd, HAc, Ac.20 0 Illi
02N 000H ' AN COOK
0 H
To a mixture of HOAc (10 mL) and Ac20 (3 mL) was added the nitro compound
from step 1 (1 g, 4.3 mmol) and Pd/C (100 mg, 0.6 mmol). The mixture was
stirred under 112 for
- 105 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
6 hours. The catalyst was filtered, and the filtrate was concentrated in vacuo
to give the desired
compound (1 g) as a brown solid. MS (ESI) m/e (MAI): 236.
Step 3
0
0 PPA
N,'"W-000H
The compound from step 2 above (150 mg, 0.64 mmol) was slowly added to PPA
(6 mL) at 100 C. The mixture was stirred for 3 hours. After cooling, the
resulting solution was
poured into 40 mL mixture of water and ice and extracted with DCM. The organic
layer was
concentrated to give the cyclic product (70 mg) as a brown solid. MS (EST) ink
(M+H+): 218.
Step 4
0
11 10% Ac Et0H
)10. * ) L=
NH ______________________________________________ , W1P
41V NH
To a solution of the ketone from step 3 (140 mg, 0.65 mmol) in 10% HOAc/Et0H
(10 mL) was added 4-acetamidophenylhydrazine (144 mg, 0.72 mmol). The mixture
was stirred
at re-flux for 4 hours. After cooling, the resulting solution was concentrated
in vacua, washed
with water and extracted by Et0Ac. The organic layer was concentrated in vacuo
to give the
desired compound (200 mg) as a brown solid. MS (EST) (M+H+): 348.
Step 5
io 021
MCI
. H2N NH2
NH
To a solution of the product from step 4 above (200 mg, 0.57 mmol) in Et011
(10
mL) was added 6N HC1 (2 mL, 12 mmol). The mixture was stirred at reflux
overnight and
cooled, and the resulting solution was concentrated then purified by silica
gel flash
chromatography (petroleum ether! ethyl acetate-5:1) to give the desired
compound (150 mg) as
a brown solid. MS (EST) m/e (M+H+): 264.
Step 6
OH HATU, DIPEA
H2N O. NH, 0 NH N
0
SO c")*'H 11
NH
NH 0
0
To a solution of the aniline from step 5 (40 mg, 0.15 mmol) in MeCN (5 mL) was
added the proline analog (70 mg, 0.3 mol), HATU (250 mg, 0.6 mmol) and DIPEA
(80 mg, 0.6
- 106 -
:A 02756172 2011-09-21
WO 2010/111483
PCT/US2010/028653
rnmol). The mixture was stirred overnight. The resulting solution was purified
by pre-HPLC to
give the desired compound (10 mg) as a brown solid. 111 NMR 8: 7.62-7.20 (m,
1611), 4.58-4.54
(m, 211), 3.79-3.60 (m, 611), 2.95 (m, 211), 2.80 (m, 211), 2.26-1.94 (m, 8H).
MS (EST) inie
(M+114): 694.
Examples 89-98
Compounds of Examples 89-98 were prepared in a similar manner as described in
Example 88.
Example , Structure _ MW Name
(2S, 2 15)- N,N`-6 ,11 -dihydro-5H -
89 0 0 . N
. 14111.,
679.826benzo[a]carbazole-3,8-diylbis[1-
(phenylacetyppyrTolidine-2-
H 0
carboxamide]
cill Am (:),, ,..,...-Th dibenzyl (2S,2'S)-
2,2'-(6,11-
0 o...L. 0 00 ilk NH'N-i
N
w o)---o 10 711.825 dihydro-5H-
benzo[a]carbazole-
H 3 ,8-diyldicarbamoyl)
dipyrrolidine- I -carboxylate
N-(8-bromo-6511-dihydro-5H-
.-
91 13r Ali O. )\H N,Nj :S---)
N 0 528.453
benzo ralcarbazol-3-y1)-1-
H
lir N (phenylacety1)-L-prolinamide
=
di-tert-butyl (6,11 -dihydro-5H-
- H benzo[alcarbazole-3,8-diyibis
92 0 " N
H 0 Nyo 910.092 {carbamoy1(25)pyrrolidine-2,1-
>õ01 NH
= o
diy1[(1S)-2-oxo- l -phenylethane-
2,1-diy1]) )biscarbarnate
_
BY rib, iiii o )--s
NH N -..j
tert-butyl
N
bromo-6,11-dihydro-5H-
93 IF
H =
01-ir 643.586
benzoiajcarbazol-3-y1)
carbamoylipyrrolidin- I -yll -2-
oxo-l-phenylethyli carbamate
= <
(2,5,2' S)-N, N r-6 711-dihydro-5H-
,µ Cilir" ip= )¨.--J
o \ it N benzo[a]carbazole-378-
diyIbis {1-
94 765.942 [(2R)-2-
(dimethylarnino)-2-
phenylacetyllpyrrolidine-2-
carboxamide)
- -,-
(2.5,215)-N,N1-5,6,7,12-
-Ni Ci)(' dp = .
tetrahydrobenzo[6,7]cyclohepta
ark = 0 * 14)'' [1,2-b]indole-3,9-
diylbis {1-
II, 779.968
[(2R)-2-(dimethylamino)-2-
phenylacetyl]pyrradine-2-
carboxamide)
w N0
N -(6,11-dihyd -ro-5H-
benzo[a]carbazol-3y1)-1-{(2S)-
4r 562.718
0 24(3 ,3-
dimethylbutanoyl)amino]96
-2-phenylacetyl} -L-prolinamide
A
tert-butyl {(1 R)-2-[(2S)-2-(6,11 -
.....--
H
dihydro-5H-benzo[a] carbazol-3-
97 N '
H
564.69
yIcarbamoyppyrrolidin- I -yl] -2-
oxo-l-phenylethyl} carbarnate
- 107-
:A 02756172 2011-09-21
WO 2010/111483
PCT/US2010/028653
Example Structure MW Name
di-tert-butyl (5,6,7,12-
N 11 NH N-j
tetrahydrobenzo [6,7] cycIohepta[
"
98 =-...,(.0yN 00 40 =
0 ANY() 924.119
I
{carbamoy1(2.3)pyrrolidine-2,1-
0 0 0 diyl 1 /0-2-oxo-l-
phenylethane-
t
2,1-diy11})laiscarbamate
Example 99¨ tert-butyl f(1R)-2-1(2S)-2-(54445-a1(2S)-14(2R)-2-f(tert-butoxv
carbonyl)aminol-2-phenylacetyllpyrrolidin-2-ylkarbonyllamino)-1H-indol-2-
yllpheny11-1H-
imidazol-2-yOpyrrolidin-.1-yll-2-oxa-1-phenylethyllearbamate
N ,
BOCHN
NHBoc
Step I
HATU 0 0
NH2
MI + HO L io NH Cbz
Br
Br
HATU (20 g, 52.3 mmol) was added to a heterogeneous mixture of the amino
ketone (12 g, 48.5 mmol) and L-Cbz-Pro (12.4 g, 50 mmol) in MeCN (156 mL). The
mixture
was cooled in an ice-water bath, and immediately afterward DIPEA (27 mL, 155
mmol) was
added dropwise. After the addition of the base, the cooling bath was removed,
and the reaction
mixture was stirred for an additional 50 minutes. The volatile component was
removed, and
water (125 mL) was added to the resulting crude solid and stirred for about 1
hour. The off-
white solid was filtered and washed with copious water, and dried in vacuo to
provide the
desired compound as a white solid (20.68 g). MS (EST) rnie (MAO 446.
Step 2
0 cy Nfl4oAc
Br 4111111)kir
irk NH Cbz ____ 111
""" sr Cbz
A mixture of the product from step 1 above (12.8 g, 31.12 mmol) and NH40Ac
(12.0 g, 155.7 mmol) in xylenes (155 mL) was heated in a sealed tube at 160 C
for 2 hours. The
volatile component was removed in vacua, and the residue was partitioned
carefully between
Et0Ac and water, where by enough saturated NaHCO3 solution was added so as to
make the pH
of the aqueous phase slightly basic after the shaking of the biphasic system.
The layers were
- 108 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
separated, and the aqueous layer was extracted with an additional Et0Ac. The
combined organic
phase was washed with brine, dried, filtered, and concentrated in vacuo to
yield a yellow solid.
MS (ESI) m/e (M+H4): 426. 1H NMR (CDCI3) 6: 7.31-7.52 (m, 9H), 7.17 (s, 1H),
5.12 - 5.20
(m, 2H), 5.00 - 5.01 (m, 1H), 3.50 - 3.52 (m, 2H), 2.96 - 2.97 (m, IH), 1.97 -
2.17 (m, 3H).
Step 3
,N
B
Boo'N B(OH)2 Pd(clopf)C12 13 c 001
111PIS "
Na2C0 loc
Br Boa
Cbz"N
A mixture of the product from step 2 above (327 mg, 0.77 mmol), indole boronic
acid from Example 42 (290 mg, 0.77 mmol), Pd(dppf)C12 (56 mg, 0.077 mmol),
Na2CO3 (244
mg, 2.3 mmol), THF (20 mL) and H20 (2 mL) was refluxed under N2 overnight. The
mixture
was poured into water and extracted with CH2C12. The organic phase was
combined, dried over
Na2SO4 and filtered to give the desired compound, which was used directly in
the next step. MS
(ESI) m/e (M+H+): 678.
Step 4
Bac,. H2N
\ 4111 $-ÃC
I
N
Bac
Cbz
A solution of the product of step 3 in FIC1/CH3OH (5 N) was stirred for 3
hours.
Concentration in vacua afforded the crude product. MS (ESI) m/e (M+H+): 478.
Step 5
H2N
1 \ Boo
c)).(1IN
0 OH Bac 0 \ 41, /
F'11-1\1 D
Cbl-N
,N
HATU Cbz
This reaction was carried out using the standard HATU-mediated coupling
procedure described in step 1 between Boc-L-Pro-OH and the product from step 4
above. MS
(ESI) m/e (M+H+): 808. 1H NMR (Me0D) 6: 8.95 (bs, 11-1), 6.82 - 7.56 (m, 17H),
6.50 - 6.62
(m, 1H), 5.74 (bs, 1H), 5.38 - 5.39 (m, 1H), 4.91 - 5.08 (m, 211), 4.66 (bs,
111), 3.79 (bs, 111),
3.40 -3.54 (m, 211), 3.19 (bs, 1H), 1.93 - 2.25 (m, 411), 1.75 - 1.88 (m, 4H),
1.35 - 1.32 (m,
9H).
- 109 -
:A 02756172 2011-09-21
WO 2010/111483
PCT/US2010/028653
Step 6
=/--- _______________________________________ HE3r,
N N
\ N
El ' 0 C/N--11) 80C 0 I N
H
HN
cbz--
To a solution of the product from step 5 (220mg, 0.3 mmol) in 20 mL of AcOH
was added 3 mL of 48% HBr. The solution was heated to 80 C for 6 hours. The
volatiles were
removed in yam , and the residue was dissolved in DCM/i-PrOH (3:1), washed
with saturated
Na2CO3 and brine, dried and concentrated in vaeuo to give a solid, which was
used in the next
step directly. MS (ESI) nile (M+H+): 441.
Step 7
,
CNAyN= 't
H I /1 HAM BocHN 0 N
N NNy ______
= 0 14-1"
NI-1Boc
A cooled solution containing HATU (0.6 mmol), the diamine from step 6 above
(132 mg, 0.3 mmol) and R-Boe-Pro (129 mg, 0.6 mmol) in MeCN (3 mL), was
treated with
DIPEA (2.4 mmol), added dropwise over 13 minutes. After the addition of the
base was
completed, the cooling bath was removed, and the reaction mixture was stirred
for an additional
30 minutes. The volatile component was removed in vaeuo; water was added to
the resulting
crude solid and stirred for about 1 hour. The off-white solid was filtered,
washed with water, and
dried in vacuo to provide the desired compound as a white solid. MS (ESI) m/e
(M+H+): 908.
1HNMR (Me0D) b: 7.66 ¨ 7.84 (m, 6 H), 7.28 ¨ 7.40 (m, 12H), 6.80 (s, 1H),
5.40'-- 5.45 (m,
2H), 5.18 --5.20 (m, 1H), 3.70 --4.02 (m, 4 H), 1.80 -2.l2 (m, 8 H), 1.35 ¨
1.37 (in, 18 H).
Examples 100-116
Compounds of Examples 100-116 were prepared in a similar manner as described
in Example 99.
Example Structure MW Name
benzyl C (2S)-245-(4-
(54(1-
r"1 {(2R)-2-[(tert-
butoxycarbonyl) II)r) aminoi-2-phenylacety1}-1,-
100 807_958
prolyl)amino]-1H-indol-2-
* 1110 yllpheny0-1H-
iruidazoi-2-
L yllpyn-olidine-l-
carboxylate
-110-
:A 02756172 2011-09-21
WO 2010/111483
PCT/US2010/028653
¨ _________________________________________________________________________
Example Structure MW Name
c-IH = N 1-[(2R)-2-
(dimethylamino)-2-
1$
cH3N 11 \ ,
. µ1.1.-,,\ phenylacetyli-N-{2-[4-(2-{(2S)-
uh 0 0 NH H
N 1-[(2R)-2-
(dimethylamino)-2-
101 o 762.963
411 101 phenylacety1]pyrrolidin-
2-y1)-
cHp
1H4nnidazol-5-y1)pheny1]-1H-
C1-1.$ indo1-5-y1)-L-
pro1inamide
_ . _____________________________
H tert-butyl {(1R)-2-
[(2S)-2-( {2-
ceEi.3-- [I Cie ip , . ,p.,,II [4-(1H-imidazol-4-
y1)phenylj-
102 chi,.
NH 604.715 1H-indo1-5-
y1)carbamoyl)
H
4It pyrrolidin-l-y1]-2-oxo-l-
phenylethyl}carbamate
cHNH / N 1-[(28)-2-(dimethylamino)-2-
N cili \ . 'õ,-1 ,\ phenylacetyll-N-{2-[4-(2-{(25)-
cf. 0 0 'Ur" NH k
N 1-[(2S)-2-(dimethylamino)-2-
103 0 762.963
411 R 110
phenylacetylipyrrolidin-2-y1)-
CHK ,
lli-imidazol-5-y0phenyll-I H-
0H3 _ indo1-5-y1)-L-
prolinamide
.,
H / N tert-butyl {(1R)-2-oxo-l-
phenyl-
cHl cH3
G4( H CNIN 40 \ * ' \ 2-[(2S)-2- { [2-(4- (2-[(2S)-1-
104 rN 0 0 NH ri 1)
0 N 791.958 (pheny1acety1)pyrro1idirk-
2-y11-
1H-imidazol-5-yllpheny1)-11-1-
4111 . indo1-5-
ylicarbamoyl)pyrrolidin-
l-yliethyl)carba.mate
H / N tert-butyl{(1R)-2-[(25)-
2-([2-(4-
cHo cH3
ciV, H )f N ilik \ . ' )- \ {2-[(25)-1-
acetylpyrrolidin-2-
0 W- NH ri il.>
0,(N 715.86 y11-1H-imidazol-5-
yl)pheny1)-
1H-indol-5-y1)carbarnoyl)
cH3 pyn-olidin-1-y1]-2-oxo-
1-
phenylethy1)carbarnate
-
ccHF1' WI benzyl (2S)-2-(5-{4-[5-
( {1-
0,7r(3..c.. [(2R)-2-phenyl-2-
{[(propan-2-
N - 11 106 0.1,.,N..., 793.931
yloxy)earbonyl]amino) acetyli-L-
proly1) amino)-1H-indo1-2-
6 ylipheny1}-1H-imidazol-
2-
y1)pyn-olidine-1-carboxylate
tert-butyl { (1R)-2-[(2S)-2-( {2-
(IYI
:t% r'l N a 1 , [4-(2-
{(25)-1-[(2R)-2-
(dimetbylamino)-2-
o 00 a 3-1 VI 1.
1111, 0 835.027
107 N> phenylacetyljpyrrolidin-
2-y1)-
1H-imidazol-5-yl)pheny1)-1H-
./wC1-6
indo1-5-yl)carbarnoyl)pyrrolidin-
40 eH3 1-y1J-2-oxo-1-
phenylethyl)carbamate
,CH3. propan-2-y1 [(1R)-2-
{(28)-241-
H / N methy1-4-(4- {54( {(25)-
1-[(2R)-
(N-1)(1\I 11 \ =
CHt_arH 0 N
0 NH 2-
pheny1-2-{[(propan-2-
N ,,N yloxy)carbonyli amino )
acetyl]
108 cH, 0 0
4111 .
.
0) 893.064
pyn-olidin-2-yl)carbonypamino)-
. -0
1H-indol-2-yl)pheny1)-1H-
.A--rw imidazol-2-yllpyrrolidin-
1 -y11-2-
CH3 ¨3 oxo-l-
phenylethylicarbamate
-111-
:A 02756172 2011-09-21
WO 2010/111483 PCT/US2010/028653
-
Example , Structure MW Name
propan-2-y1 {(1R)-2 4(19)-2- { [2-
CH
tcHH3 (Nil( 11
(4- {2-[(25)-1-acetylpyrrolidin-2-
109 .-k).%,
tN . 0 a 11 / '
\) 701.833
H H yI]-1H-Unidazol-5 -y1) pheny1)-
1H-indo1-5-yllcarbamoyl)
filk cr---/
pyrrolidin- 1 -y11-2-oxo-1-
phenylethyl) carbamate
* Ny.....0
/ µ propan-2-y1 {(1R)-2-
oxo-l-
ri 1-1
At \ pheny1-2-[(2S)-2- { [244- {2-
CHr 111! NINIfN ir NH 3-1 HN [(2S)-pyrrolidin-2-y1]-11/-
110 o...(Nto 0 659.795
imidazol-5-y1) pheny1)-1H-indol-
o 5-yl]carbamoyl}pyrrolidin-1 -
yll ethyl} carbamate
111 -) H 0---CH' propan-2-y1 {(1R)-2 4(28)-2 -
{5-
[4-(1H-inclo1-2-yl)pheny11-1H-
411, N W'
I-1 f-
õ \ IA ' CH3
0 0 547.663
imidazo1-2-y1) pyrrolidin- 1 -y1]-2-
H
di ..0-1-phenylethyl}
carbamate
H 7"--) c,H3 (2R)-2-(dimetby1amino)-1-[(2S)-
0
N n 489.626 2- { 5 44-(1H-indo1-2-yl)phenyll-
112 0
. 1H-imidazol-2-y1} pyn-olidin- 1 -
H
y1}-2-pheny1ethanone
propan-2-y1 [(1R)-2-oxo-1-
0kl0H3 Cl 0 N pheny1-2 - {(25)-245-(4- {5 -
,..i, NH Ali / 1..)
Ft N '' \ R {(15)-1-[(2R)-2-
phenyl-2
113 0 0 N 879.037 -
0 N lir NH H {[(propan-2-
yloxy)carbonyl]
,
NI(
o ---/
0 iciak amino}
acetyl]pyrrolidin-2-
lir cHz
..L )¨N110 yl} carbonyparnino]-1H-
indol-2-
0 Hz H yl}pheny1)-1H-imidazol-
2-yl]
pyrrolidin-l-y1}ethyl}carbamate
propan-2-y1 {(1R)-24(2S)-2-( {2-
H N * 1 iiµ\)
H 'N [4-(2-{(2S)-1-[(2R)-
2-
N
CHt-CHF2i (-AI 10 NH (dimethylamino)-2-phenylacetyll
114 0 [cH3 821 pyrrolidin-2-y1}-1H-
imidazol-5-
0,,r Nto ,1/N
CH3 yl)pheny11-1H-indo1-
5-
yl} carbamoyl)pyrrolidin-l-y1]-2-
o
oxo-l-phenylethyljcarbarnate .
0
ci _.(cH3 propan-2-y1 {(1R)-2 1(25)-2- {4-
115 \
N IIP \N----N?" CH3 582.108
0 H 0
:\N---<0 [4-(3-chloro-1H-
indol-2-
Aphenyll- 1H-imdazol-2-
i
H
0 yl} pyrrolidin-l-y1]-2-oxo-1-
phenyJethyll carbamate
-
/ .)._'\\' ___.
H . N- \ s\...
irk ..... {24442 - { (28)-14N-
H N-(methoxycarbony1)-1,-valyl-N-
116 91-13 H lir NH 0,N
754.894 (methoxycarbonyI)-L-
olE.ko 0
CHN)El-o imi valylipyrrolidin-2 -
y1} -1H-
dazol-5 -yl)phenyI]-1H-indol-
0
..N, cH3 cn3 CI--1 %
CH3 5-y1)-L-pro1inamide
'
- 112 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Example 117¨ Propan-2-yl l(1R)-2-exo-l-pheny1-242S)-2-f344-f5-11[(2S)-1-1(2R)-
2-phenvl-
24(propan-2-vloxy)carbonyllaminolacetyllpyrrolidin-2-yllearbonyl)aminol-11-1-
indol-2-
vliatenv0-1H-pyrazol-5-vIlpyrrolidin-l-yllethvllearbarnate
0-1\
.11H
= :),1 so \ \ H N.14
0 _
0 Elo 0 r N
"Li H N =
Step]
eta
EtMg8r
7= = Br THF N
Cbz" = Br
OC, 2 h
0¨ 0
A solution of 4-bromophenylacetylene (5.0g, 27.6mmoD in THF (100mL) at 0 C
was treated a solution of EtMgBr (3M in THF, 9.84mL, 29.5mmol). After 10
minutes, the
cooling bath was removed, and the mixture was allowed to stir at RT for 3
hours. The reaction
mixture was then cooled to 0 C and added to the Weinreb amide of Z-proline
(6.10g, 20.9 mmol)
in THF (50mL). The reaction mixture was warmed to RT for 48 hours. The
reaction mixture
was quenched with saturated NH4C1 and diluted with Et0Ac/H20. The aqueous
phase was back-
extracted with Et0Ac (2x), and the combined organic layers were washed (H20,
brine), dried
(Na2SO4), and filtered. The solvent was removed, and the residue was purified
by silica gel (PE:
EA=10:1-4:1) to give the product (7.0g) as a cream-colored solid. 1HNMR
(CDC13) 5: 7.37 ¨
7.47 (m, 2 H), 7.10 ¨ 7.30 (m, 7 H), 4.98 ¨ 5.32 (m, 2 H), 4.32 ¨ 4.47 (m, 1
H), 3.45 ¨ 3.61 (m, 2
H), 2.15 ¨ 2.27 (m, 1 H), 2.03 ¨ 2.12 (m, 1 H), 1.76 ¨ 193 (m, 2 H). MS (ESI)
rrile (M+H4):
413.
Step 2
NH2NH2
ethano$ N
Cbz7 = Br Clo; \ Br
0
A mixture of the product from step 1 (7.0g, 17 mmol) and hydrazine hydrate
(85%, 1.6 mL) in Et0H (50 mL) was heated at 80 C for 16 hours. The reaction
mixture was
cooled and concentrated to afford the desired product (6.7g). MS (ESI) mie
(M+H+): 426.
Step 3
Na2CO3
.,(E5Qc)2N \ Pagicoi2
rki , :oc
cbr d_\ Br Chz . \ 401
Boc reflux, 24 h "¨N
N(13002
- 113 -
:A 02756172 2011-09-21
WO 2010/111483 PCT/US2010/028653
A mixture of the product from step 2 above (0.77 mmol), indole boronic acid
from Example 42 (290 mg, 0.77 mmol), Pd(dppf)C12 (56 mg, 0.077 mmol), Na2CO3
(244 mg, 2.3
mmol), THF (20 mL) and H20 (2 mL) was refluxed under N2 overnight. The mixture
was
poured into water and extracted with CH2C12, dried over Na2SO4 and filtered to
give the desired
compound, which was used directly in the next step. MS (EST) mie (M-FH+): 678.
Step 4
Boc
N H
Cbzi I \ ilik \N 4 TFA/DCM(11), N
N.-. N(Boc) Cbz \ is
N 2
H N
H NH,
A solution of the product from step 3 above (339 mg, 0.5 mmol) was dissolved
in
3 mL of DCM and cooled to 0 C. After the addition of 3 mL of TFA, the reaction
mixture was
warmed to RT and stirred for 3 hours. Removal of the solvent left the desired
product as an oil,
which was used directly in the next reaction. MS (ESI) pole (M-1-H+): 378.
Step 5
H N-BocPro,PH H /, \N-
N PYBOB , fl
CI4 NI-\ 411 N II
-- NH bir NMM/DMF G j\ .... , It
\ õN ilk X Bc.c
N 11 N 11(111F N 0
H H H
A solution containing PyBOP (0.3 mmol), the amine from step 4 above (132 mg,
0.3 mmol) and N-Boc-L-Pro-OH (62 mg, 0.3 mmol) in DMF (2 mL) was treated with
N-
methylmorpholine (1.2 mmol). The reaction mixture was stirred for 3 hours,
diluted with Et0Ac
and washed with water (5x). The organic phase was dried and concentrated then
chromatographed by RPLC to afford the desired compound. MS (EST) m/e (M+H+):
675.
Step 6
H T--
,, N_ Pd/C,Me0H H /
N ,,,Iiin ,./ Boo __ N 0 =
e --õ N Boc
CbZ . \ IP ,L ip ,,N. 40 .1
il =tl 0
H $1,05ps0 N N
H N'''''0
H
The product from step 5 (100 mg, 0.15 mmol) was dissolved in Me0H and treated
with 20 mg of 20% Pd(OH)2 then hydrogenated at 45 psi for 4 hours. The
catalyst was removed
by filtration through CELITE, and the filtrate was evaporated to leave the
desired product. MS
(EST) mile (MAT): 541.
Step 7
H ITN- n
N N air ',, z B H oo
N ,, NH
H .1 \ /I
N'N \ imp
N __ N O X
H H N N
HN 0
H
- 114-
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
A solution of the product from step 6 above was dissolved in 2 mL of DCM and 2
mL of TFA. The reaction mixture was stirred for 3 hours before the solvent was
evaporated to
give the desired product as an oil, which was used directly in the next
reaction. MS (ESI) m/e
(M+H): 441.
Step 8
N 11 OZ"(
NH
N N
H ....JA H PyBOP 0 MMLDME, 0.__f 0
io
HN
N
A solution containing PyBOP (0.6 nunol), the diamine from step 7 above (132
mg, 0.3 mmol) and R-i-Proe-Phg-OH (125 mg, 0.6 mmol) in DMF (5 mL) was treated
with N-
methylmorpholine (2.4 mmol). The reaction mixture was stirred for 3 hours,
diluted with 20 mL
of Et0Ac and washed with water (5x). The organic phase was dried and
concentrated then
chromatographed by RPLC to afford the desired compound. 1H NMR (Me0D) 5: 7.70
¨ 7.80(m,
4H), 7.05 ¨ 7.55(m, 14H), 6.80 ¨ 7.00(m, 1H), 5.10 ¨ 5.50(m, 3H), 4.40 ¨
4.65(m, 2H), 3.25 ¨
4.00(m, 4H), 1.70 ¨ 2.40(m, 9H), 1.05 ¨ 1.20(m, 12H). MS (EST) m / e (M+H ):
880.
Example 118 ¨ Propan-2-y1 f(JR)-2-oxo-1-pheny1-2-0S)-2-15-(4-15-N(2S)-14(2R)-2-
phenv1-
2-fl(propan-2-vlaxy)carbonyllaminolacetyllpyrralidin-2-ylkarbonvnaminol-1H-
indol-2-
0,Pheny1)-1,3-thiazol-2-vIlpyrrolidin-1-vilethyllearbamate
0 N CANN
0 -
411
Step 1
0
/0H i) c\74H2
\ 2) NH3, H20 N
Cbz Cbz
20 Ethyl chloroformate (12 mL, 125 mmol) in 180 mL of THF was added
drop-wise
to a cooled solution (-5 C) of compound Z-Pro-OH (13.8 g, 55.5 mmol), TEA
(7.71 mL, 55.5
mmol). The resulting slurry was stirred for 20 minutes at -5 C before
saturated NR4OH (15 mL)
was added. The solution was stirred at RT for 18 hours, volatiles were
removed, and the residue
was taken up in Et0Ac (180 mL). The undissoloved white precipitate was
filtered off and rinsed
- 115-
:A 02756172 2011 09 21
WO 2010/111483 PCT/US2010/028653
with Et0Ac (100 mL). The organic layers were dried over Na2SO4 and
concentrated in vaeuo to
give the desired product (13.5 g) as off-white amorphous solid. MS (EST) m/e
(M+H+): 249.
Step 2
NH2 Lawessen's reagent 7H2
0
Cbz
Cbz
Lawesson's reagent (16.1 g, 39.9 mmol) was added to a stirred slurry of the
amide
(18 g, 72.6 mmol) in PhMe (200 mL) at RT. The reaction mixture was heated to
100 C for 3
hours before the solvent was removed. The residue was purified by flash Si02
chromatography
(DCM/Me0H=1:0-20:1) to afford the product (18 g). MS (EST) m/e (M+H+): 265.
Step 3
õ.N1-12 0
Br = Br ethanol,reflux
S N, S ip
\Cbz Ctlz
1 0 Br
A mixture of the thioamide from step 2 (10.0 g, 37.8 mmol) and the
bromoacetophenone (10.0 g, 35.9 mmol) in DOH (100 mL) was heated at 90 C for
150 minutes.
The reaction mixture was cooled and concentrated, and the residue was purified
by Si02
chromatography to afford the product (11 g). MS (EST) m/e (M+H+): 444.
Step 4
t\IH
s N
0 N\ mks.
Cafl 14y 41, Br __________________
411
The product from step 3 above can be converted to the final compounds using
the
same procedure as described in Example 117, steps 4-8. III NMR (Me0D) 8: 7.00
¨ 8.10(m,
19H), 5.40 ¨ 5.60(m, 31-1), 4.50 ¨ 4.70(m, 1H), 3.45 ¨ 4.10(m, 4H), 3.35 ¨
3.40(m, 1H), 1.80 ¨
2.6(m, 9H), 1.05 ¨ 1.30(m, 12H). MS (EST) m / e (M+H4): 897.
- 116 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Example 119 ¨ proPam-2-vi 1(1R)-2-oxo-1-phenv1-2-1(2S)-2-12-(4-15-1-a0S1-
14(2R)-2-phenv1-
2-1[(propan-2-vloxv)earbonyllaminolacetyllpyrrolidin-2-yllearbonyl)aminol-11-1-
indol-2-
vliPhenyl)-11-1-imidazol-5-41Pwrolidin-1-yllethyllearbamate
NH
0 7 0 F
0
NH 'y N N
H H
Step 1
NH
N LIHMDS di NH,
___________________________________________ 7..
Br HO Br
To a solution of 4-bromobenzonitrile (1.82 g, 10 mmol) in anhydrous THF (50
mL) was added LiHMDS (2N, 15 mmol) under N2 atmosphere at RT, and the mixture
was stirred
for 1 hour. After quenching with 1N HC1, the reaction mixture was heated at
reflux for 5
minutes. The precipitate was collected by filtration and then dried in vaeuo
to give the desired
compound (L9 g). 1H NMR (DMSO) 6: 7.74 (d, 211, J=8.2 Hz), 7.62 (d, 211, J-8.2
Hz) 6.43 (br,
311).
Step 2
,o
cri-o
N Br
Cbz OH Cbz
To a solution of Cbz-Pro-OH (2.9986 g, 12.0 mmol) in THF (100 mL) was added
TEA (1.7 mL, 12.2 mmol). The solution was cooled to -25 C and ethyl
chloroformate (1.6 mL,
12.3 mmol) was added. The resulting solution was stirred at RT for lhour. The
precipitate was
removed by filtration, and the filtrate was used in next step without
purification. A solution of
0.5 M diazomethane was added to the above reaction mixture. The sample was
stin-ed at -10 C
for 1 hour. The reaction mixture was concentrated to one half of its original
volume and washed
once with saturated NaHCO3 (50 mL). The organic layer was dried over MgSO4 and
filtered.
The crude material was adsorbed onto silica gel and purified by flash
chromatography (40 g
SiO2, 0-50% ethyl acetate in hexanes) to give the diazoketone (2.29 g). 1H NMR
(CDC13) 6:
7.32 (m, 5H) 5.13 (m, 211), 4.61 (m, 111), 3.81, 4.03, 4.17 (s, AB quartet,
211, J=4.0 Hz), 3.58 (m,
2H), 1.88-2.09, 2.17-2.38 (2, br m, 4H).
- 117 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
To a solution of the N-carbobenzyloxy-L-proline diazoketone (1.0 g, 3.6 mmol)
in
anhydrous diethyl ether (10 mL) was added a saturated solution of HBr in
diethyl ether until N2
evolution ceased. The solution was stirred for about 1 hour at about 25 C,
then washed with
saturated NaHCO3, water and brine. The crude material was purified by silica
gel column
chromatography and eluted with 40% ethyl acetate in pentane to obtain the
bromoketone (0.49 g)
as clear oil. 1H NMR (CDC13; mixture of cis-trans amide rotamers) 5: 7.35 (in,
511), 5.28 (t, 1H),
5.17 (m, 211), 4.32 (in, 111), 3.58 (m, 2H), 1.84-2.30 (br m, 411).
Step 3
NH
io NH2 NaHcos Br H
c..1\10
=Br
Cbz Br
To a mixture of bromoketone (3.25 g, 10 mmol) and the amidine (1.97 g, 10 mol)
in THF (100 mL) was added NaHCO3 (1.7 g, 20 mmol), and the suspension was
stirred at reflux
for 12 hours. The reaction was cooled, concentrated and chromatographed to
give compound 7
(0.425 g). MS (ESI) mie (M+11 ): 426, 428.
Step 4
0\
(N)N¨ri
0 N
014 = Br _________
HN irrN01 =
The product from step 3 above can be converted to the final compounds using
the
same procedure as described in Example 117, steps 4-8. 1H NMR (Me0D) 5: 7.7-
8.0 (in,
5H), 7.3-7.5 (m, 10H), 6.9-7.1 (m, 3H), 6.8 (d, J=4.8Hz, 1011), 5.4-5.6 (in,
211), 5.2-5.3 (m,
1H), 4.8 (s, 211), 4.5-4.7 (m, 1011)6 4.0 (d, J-2.411z, 111), 3.7 (d, J----
4.84Hz, 1H), 3.1-3.3 (in,
111), 2.3-2.5 (m, 111), 1.8-2.2 (m, 111)6 1.1-1.4 (in, 12H). MS (ESI) mie
(M+H+): 880.
Example 120 ¨ 14(2R)-2-phenv1-2-il(Prapan-2-vlaxy)carbonvllaminolacetyll-N44-
15-(2-
1(2S)-14(2R)-2-phenv1-2-111Propan-2-vloxvkarbonvllaminalaceollwrolidin-2-v11-
1H-
imidazol-5-171)-1-benzofuran-2-yllphenyll-L-prolinamide
,..8N io NH N
HN 0 31
t0
- 118 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Step I
0
HyL.H
H20 0L0 N
Glyoxal (2.0 rriL of 40% in water) was added dropwise to a Me0H solution of
NH4OH (32 mL) and (S)-Boc-prolinal (8.564 g, 42.98 mmol), then the whole was
stirred at
ambient temperature for 19 hours. The volatile component was removed in vaeuo,
and the
residue was purified by a flash chromatography (silica gel, ethyl acetate)
followed by a
recrystallization (ethyl acetate) to provide compound as a white fluffy solid
(4.43 g). 1H NMR
(DMSO) 6: 11.68/11.59 (br s, 111), 6.94 (s, 111), 6.76 (s, 1H), 4.76 (m, 1H),
3.48 (m, 111), 3.35-
3.29 (m, 111), 2.23-1.73 (m, 4H), 1.39/1.15 (s, 911).
Step 2
NB8,..11_1.
N N Br
NBS (838.4 mg, 4.71 mmol) was added in batches over 15 minutes to a cooled
(ice/water) CH2C12 (20 inL) solution of imidazole (1.06 g, 4.50 mmol). The
reaction mixture
was stirred for 75 minutes and concentrated. The crude material was purified
by RPLC to
separate the mono bromide from its dibromo analog and the starting material.
The HPLC elute
was neutralized with excess NH3/Me0H, and the volatile component was removed
in vacuo.
The residue was partitioned between C112C12 and water, and the aqueous layer
was extracted
with water. The combined organic phase was dried (MgSO4), filtered, and
concentrated to
provide compound as a white solid (374 mg). 1H NMR (DMSO) 6: 12.12 (br s,
111), 7.10 (m,
1H), 4.70 (m, 111), 3.31 (m, 111; overlapped with water signal), 2.25-1.73 (m,
4H), 1.39/1.17 (s,
3.8H + 5.211).
Step 3
Pd(cIppt)C12, KOM B
th
1402
0 400_300:: 0
To a mixture of the benzofuran from Example 19, step 1 (15 g, 0.05 mol),
bis(pinacolato)diboron (25.4 g, 0.1 mol), Pd(dppf)C12 (1 g), KOAc (0.1 mol) in
dioxane (500
rriL) was stirred at reflux under N2 atmosphere for 2 hours. Concentration of
the reaction
mixture left a residue that was chromatographed to give the desired compound
(12 g). 111NMR
- 119 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
(DMSO) 6: 8.28 (d, J=8.8 Hz, 2H), 8.10 (s, 1H), 7.95 (d, J=8.8 Hz, 2H), 7.72
(d, J=8.8 Hz, 1H),
7.43 (d, J=8.8 Hz, 1H), 7.05 (s, 114).
Step 4
N1)¨ti Br
Pd(dppf)Ci2
L-Ni8oc 411&. = , * NO2
NO2
NBoc
0 Na2CO3 0
This reaction was conducted in a similar manner to that described in Example
117. MS (ESI) m/e (M+11+): 475.
Step 5
L-1,18oe io it NO2 112 1 14 so \
NH2
0 Pd/C 0
The product from step 4 (475 mg, 1.0 mmol) was dissolved in Et0H and treated
with 20 mg of 10% Pd/C then hydrogenated over 5 hours. The catalyst was
removed by
filtration through CELITE, and the filtrate was evaporated to leave the
desired product. MS (ESI)
nile (M-41 ): 445.
Step 6
HATU C))
L-14B.
NH,
Boo L,pro' H 110=
BNoc
0
A solution containing HATU (1.0 mmol), the amine from step 5 above (445 mg,
1.0 mmol) and N-Boc-L-Pro-OH (215 mg, 1.0 mmol) in MeCN (10mL) was treated
with DIPEA
(1.2 mmol). The reaction mixture was stirred for 3 hours, diluted with Et0Ac
and washed with
water (5x). The organic phase was dried and concentrated then chromatographed
by silica gel
chromatography (Et0Ac) to afford the desired compound. MS (EST) m/e (M+11+):
642.
Step 7
C1/4 TFA CItH
L¨Ni3oc
\ H N
8oe
0 0
A solution of the product from step 6 above was dissolved in 2 mL of DCM and 2
mL of TFA. The reaction mixture was stirred for 3 hours before the solvent was
evaporated to
give the desired product as an oil, which was used directly in the next
reaction. MS (EST) rn/e
(M-111+): 442.
- 120 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Step 8
)
NH N"--
0-2
NH ti
NH N 1N 0 111 \
4W... 0 0 H
0
=
fi 0
A solution containing BOP reagent (222 mg, 0.5 mmol), the diamine from step 7
above (112 mg, 0.25 mmol) and R-i-Proc-Phg-OH (125 mg, 0.6 mmol) in DMF (5 mL)
was
treated with N-methylmorpholine (2.4 mmol). The reaction mixture was stirred
for 3 hours,
diluted with 20 mL of Et0Ac and washed with water (5x). The organic phase was
dried and
concentrated, then chromatographed by RPLC to afford the desired compound.
111NMR
(Me0D) 8: 6.8-7.9 (m, 1911), 5.1-5.5 (m, 311), 4.5 (m, 111), 3.5-4.04 (m, 2H),
1.6-2.5 (m, 9H),
0.9-1.3(m, 1211). MS (ESI) rrile (M+H+): 880.
Example 121 ¨ propan-2-y/ f(1R).-2-1(2S)-2-(5-1244-(acetylamino)phenv11-111-
indol-5-v11-
1,3,4-oxadiazo1-2-0)pyrrolidin-1-v11-2-oxo-1-phenylethyllearbamatNH
40 wf
õ = NH
c--a
Step 1
HO so NaH003
MeWDMF ? 40
A mixture of 5-carboxyindole (32.2 g, 0.2 mol), NaHCO3 (53.36 g, 0.64 mol),
methyl iodide (122.22 g, 0.86 mol) in 60 mL of DMF were stirred at RT for 2
days. Water and
Et0Ac are added, and the organic layer was washed with bicarbonate solution,
dried and
concentrated to obtain 5-(methoxycarbonyl)indole. 114 NMR (DMSO) 6: 11.44 (s,
111), 8.22 (s,
111), 7.68(d, J = 8.4 Hz, 1H), 7.37 ¨ 7.47 (m, 2 H), 6.56(d, J = 2.0 Hz, 1H),
3.80 (s, 311). MS
(ESI)m/e (M+H+): 176.
Step 2
o 0
NH2NH2(85%) H
? so \ ________________________________________ N
A mixture of the ester (28 g, 0.16 mmol) and NH2NH2 (85%, 50 mL) in ethanol
(200 mL) was heated at reflux for 48 hours. The reaction mixture was
concentrated, and the
- 121 -
A02756172201109-21
WO 2010/111483
PCT/US2010/028653
residue was purified by column chromatography (5% Me0H / DCM) to give compound
(25 g).
1H NMR (DMS0) 5: 1L27 (s, 1H), 9.54 (s, 1H), 8.06 (s, 1H), 7.57 (d, J = 8.8
Hz, 1H), 7.37 (d, J
= 8.8 Hz, 2H), 4.42 (s, 1H), 6.48 (s, 2H), 3.32 (s, 1H). MS (ESI) m/e (M+H+):
176.
Step 3
0 0
1N2=
\ Trl-AllAjelt = 0 N,11
N t OH
Cbz N-Cbz
The product from step 3 above was coupled using a standard HATU amide bond
forming procedure. MS (ESI) m/e (M+11 ): 407.
Step 4
0
0 N. PRI13,02Cie
=ONT-0 N\ CHON,IAPEA / 1. NH
N-N
To a suspension of the product from step 3 above (100 g, 0.25 mol), PPh3 (98.4
g,
0.375 mol) and DIPEA (96.7 g, 0.75mo1) in CH3CN (500 mL) at RT was added
C2C16(82.8 g,
0.35 mol). The reaction was stirred at RT for 1.5 hours, and the solvent was
removed, and the
residue was portioned with Et0Ac/H20. The layers were separated, the aqueous
phase was re"
extracted with Et0Ac (2X), and the combined organic layers were removed in
vacuo, and the
residue purified by column chromatography (5% Me0H/DCM) to give compound 4 (55
g). MS
(ESI) m/e (M+H+): 389
Step 5
B&VkDDICNIAP
Cbz
f j
NI=== " RT, 14 h Cbz 411 N.
-N
Di-tert-butyl dicarbonate (30.7 g, 142 mmol) was added drop wise to a solution
of
indole (55.0 g, 142 mmol), DMAP (2.0 g) and DIPEA (18.3 g, 142 mmol) in 50 mL
of DCM at
0 C. The reaction was allowed to stir to RT overnight before it was
concentrated, and the
residue purified by prep TLC (PE/EA=2:1). MS (ESI) m/e (M+Fl+): 489.
Step 6
OH
LOA
c
13(01-Pr)1
Cb N'Boc -5C, 3 hBoc
N-1,4
A mixture of compound indole from step 5 (977 mg, 2 mmol), (iPrO)3B (3.0 g, 16
mmol) and dry THF (100 mL) was cooled to 0 C. LDA (prepared from n-BuLi and
iPr2NH in
THF, about 8 mmol) was slowly added and the mixture was allowed to warm to RT
over 2 hours.
- 122 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
The mixture was quenched by 1N HC1 to p3 and extracted with CH2Cl2 three
times. The
combined organic phases were combined, dried over Na2SO4 and filtered. The
filtrate was
concentrated and purified by column chromatography (PE/DCM=1/1 to pure CH2C12
to
CH2C12/acetone=10/1 to pure acetone) to afford the product 8 (0.5 g).
Step 7
9H
B, 40 4=1 Br
Pcrlicla N
it)2g12 T
(1,1)).-0 =
Cbz
'-N'BocOH 4
-N reflux, 24 h
CbZ / = N'Bo,
A mixture of the product from step 6 (0.38 mmol), indole boronic acid from
Example 42 (145 mg, 0.38 mmol), Pd(dppf)C12 (28 mg, 0.038 mmol), Na2CO3 (122
mg, 1.15
mmol), THF (10 mL) and H20 (1 mL) was refluxed under N2 overnight. The mixture
was
poured into water and extracted with CH2C12. The organic phase was combined,
dried over
Na2SO4 and filtered to give the desired compound, which was used directly in
the next step. MS
(ESI) m/e (M+H ): 622.
Step 8
Pd/C,N100H
NT
H2(45psi) so
9Nr leo
Cbz
then TFA H / = NH
N-N Boc "-N
The product from step 7 (0.15 mmol) was dissolved in Me0H and treated with 20
mg of 20% Pd(OH)2 then hydrogenated at 45 psi for 4 hours. The catalyst was
removed by
filtration through CELITE, and the filtrate was evaporated then dissolved in 1
mL of DCM then
treated with 1 mL of TFA. After stirring for 2 hours, the mixture was
evaporated and the residue
was used directly in the next reaction without further purification. MS (EST)
mie (M+H+): 488.
Step 9
(0) PyBOP 0 .44
NMWOMF 0 NH
NH 11111
RT, 14 h
N,t4
A solution containing PyBOP (44 mg, 0.1 mrnol), the amine from step 8 above
(49 mg, 0.1 mmol) and R-i-Proc-Phg-OH (21 mg, 0.1 mmol) in DMF (1 mL) was
treated with N-
methylmorpholine (0,6 mmol). The reaction mixture was stirred for 3 hours,
diluted with Et0Ac
and washed with water (five times). The organic phase was dried and
concentrated, then
chromatographed by RPLC to afford the desired compound. 1H NMR (Me0D): 67.90 ¨
8.35(m,
- 123 -
:A 02756172 2011 09 21
WO 2010/111483 PCT/US2010/028653
111), 7.70 ¨ 7.85(m, 2H), 7.60 ¨ 7.70(m, 211), 720 ¨ 7.52(m, 71-1), 6.55 ¨
7.20(m, 1H), 5.50 ¨
5.60(m, 111), 5.30 ¨ 5.50(m, 111), 4.75 ¨ 4.85(m, 111), 3.70 ¨ 4.10(m, 111),
3.35 ¨ 3.50(m, 1H),
1.95 ¨ 2.50(m, 711), 1.10 ¨ 1.30(m, 611). MS (ES1) m / e (M+H+): 607.
Example 122 ¨ (2S)-1-(phenvlacetyl)-N-13-15411(2S)-1-(phenplacetpOpyrroliditi-
2-
vilcarbonvliamino)-111-indol-2-vilpheavlimrrolidine-2-carboxamide
NH 0
PhJsijo /IN \ *
Step 1
0 HN4
N paiop N:Pc;ow14a0C 0ve Mk'
NNHOHCI 1r
A solution of the hydrazine (1 g, 5 mmol) and 3-acetylacetanilide (0.88 g, 5
mmol) in NMP (5 ml) was heated at 150 C under microwave for 10 minutes. The
solution was
poured into water and extracted with Et0Ac three times. The organic layer was
washed with
water, dried over sodium sulfate and then concentrated in vacua. The residue
was purified by
RPLC to give the desired compound. MS (m/z): 308 (M+H)+.
Step 2
tiN4 INN2
"lorN 0 Fici HON,
N
To aqueous HC1 (4N, 5 mL) was added the product from step 1 above (300 mg, 1
mmol), and the mixture was heated at reflux for 1 hour. The reaction mixture
was cooled and
concentrated, and the residue was purified by RPLC to give compound (200 mg).
MS (rn/z): 224
(M+H)+.
Step 3
,((p
N =H, t--,7 OH
H2t4 NH 0 PhCH; k(111-11
\ ipHATU
0
To a solution of the compound from step 2 above (35 mg, 0.148 mmol) in MeCN
(5 mL) were added N-phenylacetyl-L-proline (15 mg, 0.0673 mmol), DIPEA (26 mg,
0.202
mmol) and HATU (56 mg, 0.148 mmol). The reaction was stirred overnight and
concentrated,
and the residue was purified by RPLC to give the desired product (15 mg). MS
(ESI) aile
- 124 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
(M+H ): 654. Ili NMR (Me0D) 6: 8.0 (in, 1H), 7.7(m, 111), 7.6-7.1 (m, 14H),
6.7 (m, 1H), 4.6
(m521-1), 3.9-3.5 (m, 9H)2.4-1.7 (m, 8H).
Example 123 ¨ (2S)-1-(Phenylacetyl)-N-1146411(2S)-1-(phenvlacetvl)pyrrolidin-2-
vlicarbonylfamino)-11-1-indol-2-vlbthenvlipwrolidine-2-carboxamide
=NH N--1
N H 0
N
0
=
Step]
Ct
NI,
0CM. TEA
To a stirred solution of 4-ethynylaniline (1 g, 8.5 mmol) in DCM (60 ml) was
added acetyl chloride (0.8 g, 10 mmol) and TEA (1.7 g, 17 mmol). The mixture
was stirred for 3
hours. The resulting solution was washed with water, 1 N HC1 and brine. The
organic layer was
concentrated in vacuo to give the desired product (900 mg), which was used in
next step without
purification. MS (ESI) m/e (M H+): 160.
Step 2
Pd(PPhi)2Ciz
IP 8
- 02. .H2
02. .H2
To a stirred solution of 4-ethynylacetanilide (800 mg, 3.1 mmol) in anhydrous
THF (6 ml) was added compound 2 (0.5 g, 3.1 mmol), PdC12(PPh)3 (33 mg, 0.05
mmol), CuI (10
mg, 0.05 mmol) and TEA (2 mL). The mixture was protected from light and
stirred at RT
overnight. The resulting solution was concentrated in vacuo, and the residue
was washed with
DCM to give the desired compound (300 mg) as a yellow solid. MS (ESI) m/e
(M+H+): 296.
Step 3
1011 N MI3r3
lo \ it NH
1.1 11
02N NH2 02N
To a stirred solution of compound from step 2 above (200 mg, 0.68 mmol) in
toluene (2 ml) was added InBr3 (2 mg, 0.004 mmol). The mixture was stirred at
reflux for 3
hours. The resulting solution was washed with water and extracted with Et0Ac.
The combined
- 125 -
:A 02756172 2011 09 21
WO 2010/111483 PCT/US2010/028653
organic layers were dried over sodium sulfate, concentrated in vacuo to give
the desired indole
(170 mg) as a brown solid. MS (ESI) m/e (M+H+): 296.
Step 4
07N
0 \ ft 1:"-- HOI
02N
N4-1 ______________________________________________ \ 41, NH2
N )" - H
H
To a stirred solution of the product from step 3 above (100 mg, 0.34 mmol) in
EtOH (5 ml) was added 3N HC1 (1 mg). The mixture was stirred at reflux
overnight. The
resulting solution was concentrated in vacuo to give the desired aniline (80
mg) as a brown solid.
MS (BSI) mie (M+H+): 254.
Step 5
) 0 . 0
__________________________________ ) 0 It NH2 +
HO N--
0 HATU, D1PEA
* 02N 0 \ .
N NH N
02N H N H
fk =
To a solution of the aniline from step 4 (50 mg, 0.2 mmol) in acetonitrile (5
inL)
was added N-phenylacetyl-L-proline (56 mg, 0.2 mol), HATU (167 mg, 0.4 mmol)
and DIPEA
(100 mg, 0.8 mmol), The mixture was stirred overnight. The resulting solution
was purified by
RPLC to give the desired compound (40 mg) as a brown solid. MS (EST) m/e
(M+H+): 469.
Step 6
07N N )--
) ________________________________________________________________ j
o ,
iso\ 4., NH N-- H2, PcIle
40 ' IIP NH N
H 0 ). H2N
N
H 0
= 41,
To a solution of the nitro compound (40 mg, 0.08 mmol) in THF (2 mL) was
added Pd/C (20 mg, 0.1 mmol), The mixture was stirred under H2 atmosphere for
1 hour. After
replacement of 112 with N2, the Pd/C was filtered off, and the filtrate was
evaporated in vacuo to
give the desired aminoindole (40 mg) as a brown solid. MS (ESI) m/e (M+H+):
439.
Step 7
0 . 41,
.0 , itõ ,sõ, 1,4 t HO HATU, D1PEA H 0
N2N N
H 08 0 ------"" eh, N
0
. * Or
- 126 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
To a solution of the product from step 6 above (40 mg, 0.1 mmol) in
acetonitrile
(5 mL) was added N-phenylacetyl-L-proline (23 mg, 0.1 mmol), HATU (70 mg, 0.2
mmol) and
DTPEA (25 mg, 0.2 mmol). The mixture was stirred overnight. The resulting
solution was
purified by RPLC to give the desired compound (15 mg) as a brown solid. III
NMR (Me0D) a:
7.81 (m, 1H), 7.70 (d, J=8.6 Hz, 2H), 7.60 (d, J=8.4 Hz, 2H), 7.39 (m, 1H),
7.30-7.28 (m, 10H),
6.96 (m, 1H), 6.69 (s, 1H), 4.63-4.50 (m, 2H), 3.78-3.60 (m, 8H), 2.23-1.92
(m, 8H). MS (EST)
m/e (M+H+): 654.
Example 124 ¨ (2S)-1-(phenvlacetpl)-N-13-16411(2S)-1-(phenylacelyppwrolidin-2-
yllearbonyllamin0-1H-indol-2-vl1Pheavilpyrrolidine-2-carboxamide
)0t,
0
0
I.
=
Step 1
cH,c1,
EtaN N 0
To a solution of 3-ethynylaniline (1.17 g, 10 mmol) and 1.5 ml Et3N in 40 mL
DCM was added dropwise acetyl chloride (1 g, 13 mmol). The reaction mixture
was stirred at
RT for 1 hour. After that, the solvents were evaporated, and the residue was
extracted with
Et0Ac (100 mL), washed with water (50 mL) and brine (50 mL), dried over
anhydrous NaSO4,
and concentrated in vacuo to afford 3-ethynylacetanilide (1.5 g). MS (EST) ink
(M+H+): 160.
Step 2
02N NH2
0 id& 2.
C H OH 2N
2 5
RT W
3-nitroaniline (6.9 g, 0.05 mmol) was dissolved in 150m1 ethanol, iodine
chloride
(8.1 g, 0.05 mmol) added with dropwise. The reaction mixture was stirred at RT
for 4 hours.
After that, the solvents were evaporated, and the residue was extracted with
Et0Ac (100 mL),
washed with water (50 mL) and brine (50 mL), dried over anhydrous NaSO4. After
concentrated
in vaeuo, the residue was purified by column chromatography (PE/Et0Ac
=40:1=420:1) to
afford the desired product (8.9 g). MS (EST) aile (M+H ): 265.
Step 3
0
02N 40 NH2
PdCI7(RPN2, H2N
NO Cul,EtN =NO2
- 127-
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
3-Ethynylacetanilide (480 mg, 3 mmol) and 2-iodo-5-introaniline (800 mg, 3
mmol) were dissolved in anhydrous THF (30 mL), PdC12(PPh3)2 (105 mg,0.15 mmol)
and CuI
(28.5 mg,0.15 mmol) Et3N(1 ml) was added sequentially. The reaction mixture
was protected by
N2 and stirred at RT for overnight, After that, the solvents were evaporated,
and the residue was
extracted with Et0Ac (50x2 mL), washed with water (40 mL) and brine (30 mL),
dried over
anhydrous NaSO4. After concentrated in vacuo, the residue was purified by
column
chromatography (DCM/Me0H =50:1-420:1) to afford the desired product (620 mg).
MS (EST)
m/e (M+11 ): 296.
Step 4
0 HN-4
)--NH 1-17N
AR1/4 - PdC12, FeCl2 N
1 0 lir \ NO, DCE8OC 02N
To a solution of the product from step 3 (295 mg, 1.0 mop in DCE (15 mL) was
added PdC12 (9 mg, 0.05 mmol) and FeCl3 (8 mg, 0.05 mmol). The reaction
mixture was heated
at 80 C for 2 hours. The reaction was cooled, and the solvents were
evaporated, and the residue
was extracted with Et0Ac (2x), washed with water (30 mL) and brine (30 mL),
dried over
anhydrous Na2SO4. After concentrated in vacuo, the residue was purified by
Prep-TLC
(DCM/Me0H =50:1) to afford the desired product (240 mg). MS (EST) m/e (M+H+):
296. 1H
NMR (DMSO) 8: 0.12 (s, 1 H), 8.25 (d, J= 8.0 Hz, 2 H), 7.45-7.94 (m, 6 H),
7.03 (s, 1 H), 2.10
(s, 3 H).
Step 5
Hts1¨µ 0
NH2
\
02N ri H2N 1-1,N p
A suspension of the product from step 4 (200 mg, 0.67 mmol), Pd/C (10 mg,
0.034 mmol) in 40 mL Et0H was under H2 protection and stirred for 1 hour. The
mixture was
then filtered, and the filtrate was then concentrated to give the product (160
mg). The residue
was dissolved in 20 ml 3N HCI, the mixture was stin-ed at 80 C for 1 hour. It
was cooled to RT,
concentrated in vacuo and the residue was purified to give desired compound
(120 mg) as a
brown solid. MS (m/z) (Milt): 224
- 128 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Step 6
NH, 0õ.
i= N
H H
\ Hr'N o ATU lPEA
11/
H2N 0
0
1110
The mixture of compound 10 (50 mg, 0.224 mmol), N-phenylacetyl-L-proline
(110 mg, 0.45 nanaol), D1PEA (88mg, 0.7 mmol) in CH3CN (5 mL) was stirred at
RT for 5
minutes, then HATU (82 mg, 0.54 mmol) was added to it. The mixture was stirred
at RT
overnight. When reaction completed, the mixture was concentrated in vactio,
the residue was
purified by chromatography on silica gel to give the desired target (70 mg).
MS (ESI) nt/e
(M+H+): 654 1H NMR (Me0D): 6 7.95 (d, J= 8.0 Hz, 2 H), 7.86-7.21 (m, 13 H),
6.98 (d, J= 8.0
Hz, 1 H), 6.72 (s, 1 H), 4.57(m,2H), 3.53 (in, 3 H), 2.02-2.31 (m, 8 H).
Example 125 - tert-butyl 1(1R)-2-1(2S)-2-112-12-10S1-142R)-2-1(tert-
butoxyearbonyl)
amino1-2-pbenylacetyllpyrrolidin-2-yll-1H-benzinzidazol-5-y11-111-indo1-5-
yl)carbamoyll
pwrolidin4-y11-2-oxo-1-phenylethvilearbamate
BocHN
N N NHBoc
N\ 41).111
0
0
Step I
Br NH2 n H
µP -FicY--11 HA" :r siNT-Lz
iii
NH2 0 Cbz
NH2
The mixture of 4-brorno-1,2-phenylenediamine (3.1 g, 16 mmol), L-proline (4.3
g, 16 mmol), DIPEA (3 ml) in MeCN (100 mL) was stirred at RT for 5 minutes,
then HATU (6
g, 17 mmol ) was added. The mixture was stirred at RT overnight. When reaction
completed,
the mixture was concentrated, the residue was washed with water (100 mL) and
extracted with
Et0Ac (three times), washed with brine (50 mL), dried over anhydrous Na2SO4.
The residue
purified by was purified by column chromatography (DCM/Me0H = 100:1-450:1) to
afford the
desired compound (5.0 g). MS (ESI) m/e (M+H+): (418,420).
Step 2
H
Br Mc õBr io
cba
NH? HCbz
- 129 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
The product from step 1 (5 g, 7.2 mmol) was dissolved in 50 mL acetic acid
.The
reaction mixture was stirred at 100 C for 4 hours. The mixture was cooled, and
the acetic acid
was removed in vacuo. The residue was purified by column chromatography
(DCM/Me0H =
150:1¨ 100:1) to afford the desired compound (3.8 g). MS (ESI) m/e
(400,402).
Step 3
Br Ali BocHN
Pd(PPU4 B')CHN io NH Z
\ I3(OH)2
P 4" '1 CH3OH,Na2t-0
Cbz Roe
Boo
A suspension of the product from step 3 (1.2 g, 3 mmol),1,5-bis-Boc-5-
aminoindole-2-boronic acid (1.2 g, 3 mmol), Pd(PPh3)4 (240 mg), Na2CO3 (1 g, 9
mmol) and
H20 (3 mL) in 30 mL of THF under N2 protection was reacted with refluxed at 75
C overnight.
The mixture was filtered, and the filtrate was washed with 50 mL of water and
extracted with
100m1 Et0Ac and dried over anhydrous Na2SO4. Removal of the solvent and column
chromatography (CH2C12/Me0H = 250:1200:1) afforded the desired compound (500
mg).
MS (EST) rn/e (M+H+): 652
Step 4
)
Ny"---1)1 iNinCLH ¨
8-RN HC
NH Cbz
Boo
The product from step 3 (500 mg, 0.9 mmol) was stirred in Me0H/HCI (20 mL)
for 16 hours. The solvent was removed under reduced pressure and the residue
was dried at high
vacuum. MS (ESI) m/e (M+H+): 452.
Step 5
I-1 =NH bz 2N Ny^N HATIJ
b Boo 0C7:1(
40, NH bbz
.--'
The mixture of aniline from step 4 (450 mg, 1 mmol), (51)-N-Boc proline (215mg
I mmol), DIPEA (0.4 mL ) in CH3CN (10 mL) was stirred at RT for 10 minutes,
then HATU
(400 mg, 1.1 mmol) was added. The mixture was stirred at RT overnight,
concentrated, and the
residue was purified by column chromatography (CH2C12 / Me0H = 250:1¨)-200:1).
MS (ESI)
nile (M+H+): 649.
- 130 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Step 6
N
fly
Bol 0 I. * NH 01>z HAc,HBr ("X
0 \ ilk NH
10-80C
The product from step 5 (290 mg, 0.45 mmol) was dissolved in 5 mL of acetic
acid and HBr (imp was added. The reaction mixture was heated to 70-80 C and
stirred for 4
hours. The mixture was cooled to RT and concentrated in -wen . The residue was
extracted
with Et0Ac (2x), washed with aq NaHCO3and water (30 mL) and brine (30 mL),
dried over
anhydrous sodium sulfate. Evaporation of the solvent afforded the desired
compound as brown
solid (160 mg). MS (ESI) mie (M+H+): 415.
Step 7
Nye-, N
0 NH _ HATU BocNN CNIYN
I \ NH
NHBoc.
-N 0
0
qk
A mixture of the product from step 6 (100 mg, 0.24 mmol), (R)-N-Boc-Phg (120
mg, 0.48 mmol), DIPEA (0.4 mL) in CH3CN (10 mL) was stirred at RT for 10
minutes, then
HATU (200 mg, 0.5 mmol) was added. The mixture was stirred at RT overnight
then
concentrated, and the residue was purified by RPLC to afford the desired
compound (54 mg).
MS (ES1) ink (WIT): 882. 111 NMR (Me0D) 8: 7.96-7.69(m, 4H), 7.49-6.84(m,
13H),
5.50-5.40(m, 2H), 4.06-3.94 (in, 2H), 2.27-1.88 (in, 811), 1.37 (s, 18H).
Example 126 ¨ (2S)-N-14-13-bromo-5-(N2S)-1-(phenylacetyl)pwrolidin-2-
vllearbonWamino)-111-Mdol-2-vliphenyll-1-(PhenvlacetOpyrrolidine-2-carboxamide
Br
N N
NBs (NIT )
phZ 110 \ it NH N 0 N\ =Nil
0
o*¨Ph Ph Ph
To a solution of the indole (1 equiv) in 5 mL of THF was added NBS (278 mg, 1
mmol) at RT, and the mixture was stirred for 1 hour. Concentration of the
solvent and
purification of the residue by RPLC afforded the targeted halogenated
compounds. Ill NMR
(Me0D) 8: 7.9-7.5 (m, 51-1), 7.4-7.0 (m, 12H), 5.2-4.9 (m, 211), 4.4 (m, 2H),
3.8-3.5 (m, 611), 2.5-
1.8 (m, 811).
- 131 -
:A 02756172 2011-09-21
WO 2010/111483
PCT/US2010/028653
Example 127¨ tert-butyl {(1S)-24(2S)-2-(1244-(acetylamino)phenyll-3-fluoro-1H-
indo1-5-
vilearbamoyl)pyrrolidin-1-yll-2-oxo-1-phenvlethyllearbamate
H
Alt, F
Kit-41r dvi
CifH14
SocHN
WI 1\2; NW NH p SELECTFLuoRo GocHN 0 a \
= N/H \ N
N
0
(:)'\\. OK
0 H 0 H
= 14,
To a solution of the indole (1 equiv.) in acetonitrile/DMSO (5 ml, 1:1) was
added
SELECTFLUORO (1 equiv.) at 0 C. The mixture was stirred at RT for 3 3 hours
before it was
concentrated, and the residue purified with RPLC. 111 NMR (Me0D) 6: 7.9-7.c8
(m, 311), 7.8-7.7
(m, 211), 7.5-7.3 (m, 611), 7.3 (in, 111), 5.5 (s, 1H), 4.6-4.5 (m, 211), 4.0-
3.9 (m, 1H), 3.8 (m, 111),
3.7 (m, 111), 2.4-2.3 (m, 111), 2.2-2.1 (m, 711), 2.1-2.0 (in, 3H), 2.0-1.9
(in, 111), 1.4 (in, 914).
MS (m/z): 711 (M-I-H)4
.
Examples 128-154
Compounds of Examples 128-154 can be prepared by direct halogenation of the
indole or benzofuran compounds in a similar manner as described in either
Example 126 or
Example 127.
_______________________________________________________________________________
_ _
Example Structure 1 MW Name
-D
ei 0 (2S)-N-{443-chloro-5-({[(2,5)-
1-
(phenyIacetyl)p3rrrolidin-2-
128 N
= Cif" 0 \ = NH N 688.233 yl]carbonyl} amino)-1H-indo1-2-
o N
0 H 0 .
yliphenyl}-1-(phenylacetyl)
_ pyrrolidine-2-carboxamide
-
clyH F (2,5)-N- {4-113-fluoro-5-({[(2S)-1-
N 0
0 . \ 4. NH )___<..,--j N (phenylacetyppyrradin-2-
129 0 N
H
o8 671.778
yllcarbonyli amino)-1H-indo1-2-
lit
yllphenyl} -1-(phenylacetyl)
pyrrolidine-2-carboxamide
tert-butyl {(18)-2-[(25)-24 {445-
F({ [(2,9-1.-{(2S)-21(tert-
* CNIYI . \ ip ,)1,-0
butoxycarbonyl)am ino]-2-
130 cF1 H
henylacetyllpyrrolidin-2-
,,o NH 0 H 0
N
CH .c:, 902.044
ylicParbonyl} amino)-3-fluoro-1 H-
; &13-i-
* 0>cui, indo1-2-yllphenyl}
carbamoyl)
cH3Cf-13
pyrrolidin-1-y11-2-oxo-1-
. phenylethyl} carbatnate
* cAH CI
0 tert-butyl .R1,5)-24(2,9-24{244-
iN -,, \ = N)ti---CH3
(acetylatnino)pheny1]-3 -chloro-
131 C1-
630.15 1H-indo1-5-y1} carbamoyl)
>i()NH 0 H
pyrrolidin-1-y11-2-oxo-1-
* cH3cHt-
phenylethyl) carbamate
i (2S)-N- {443 -iodo-5 -({[(28)-
I-
* <Ji 40 ll a \ strn
NH N
(phenylacetyppyrrolidin-2-yll
o
132 0 N
H 08 779.684 carbonyl} amino)-1H-indo1-2-
yl]
phenyl} -1-(phenylacetyl)
pyrrolidine-2-carboxamide
- 132 -
:A 02756172 2011-09-21
WO 2010/111483 PCT/US2010/028653
- .
Example _ Structure MW Name
tert-butyl ((1S)-2-[(2S)-2-( (244-
N ¨ Y-"<- (
{[(2S)-1-acetylpyn=olidin-2-
, -... \
tit Cli: , , )---NH N"--
yl]earbonyl ) am ino)pheny11-3 -
13 3 ----- N \ 710.813
CH \3_,,.., cEi3 fluoro-1H-indo1-5 -
H
CH3 i I/
yl) carbamoyl)pyrrolidin-l-y1]-2-
c H30 oxo-
1-phenylethyll carbamate
(25)-1- ( (25)-2 - [(3 ,3 -
r
. 14'-lr'il a \ == iptit-C
dimethylbutanoyl)amino]-2-
phenylaeetyll-N- {4454 ( [(25)-1-
el-13 0 N ' 0 H {
(25)-2 -[(3,3-dimethylbutanoyl)
134 cH,--)-Thr 1,41-1 H N 898.1
amino}-2-phenylacetyl)
"3 o
#1 ccH3
pyrro1idin-2-yllearbony1) amino).-
cH, cH3 3-fluoro-1H-indo1-2-yliphenyl)
pyrrolidine-2-carboxamide
tert-butyl ((I/)-24(25)-24 (4-
[5-( ( [(25)-1- {(2R)-2-[(tert-
r1
H F _.--::---i
cHC143-" ,1r-INI N- I NO--S-0-7N4µH \N") butoxycarbonypamino]-2-
135 3 CH, 6 0 - 11
08 H
AN 902.044
phenylacetyl) pyrrolidin -2-
41P ricsH3
yli carbonyl ) amino)-3-11uoro-1 H-
indol-2-yliphenyll earbamoyl)
CH3 CHI,
pyrrolidin-l-y1]-2-oxo-1-
phenylethyllearbarnate
H F 0 tert-butyl {(1R)-2-[(25)-24
{2-
cH, 0 H (NlyN
II N,.._ CH3
H [4-
(aeetylamino)pheny1]-3-
136 cie-- ),...- N
cH$ 6 0 0 N
H 613.695 fluoro-1 H-indo1-5-
i
yl) carbamoyl)pyn-olidin-l-y11-2-
. oxo-l-
phenylethyl)earbarnate
H Ftert-butyl ( (1R)-2-[(2S)-
2-( {2-
0 1
CH3 N N ,
[44 {[(2S)-1-acetylpyrrolidin-2-
-)¨ Cilr I \ lik NH .1\1"-j
yll carbonyl) amino)pheny11-3-
137 t- õ, H ,3 cH3
0 0 ---- . \ 0 \,,H3 710'813
H fluoro-1H-indo1-5-
41k
yl)earbamoyl)pyrrolidin-l-y11-2-
oxo-1-phenylethyl) carbamate
1-
cH3 F 0
ter t -butyl {(1R)-2-[(2S)-2-( {3-
0 H CITN N1
,w \ ,\----, Nb
fluoro-244-(2-oxopyrrolidin-1-
q)-- o
138 cv ..),--N N 639.733 yl)pheny1]-1H-indol-
5-
o
cH3 0 H
lir yl)carbamoyppyrrolidin-
1-y11-2-
oxo-l-phenylethyl) carbamate
H F N-
{244-(acetylamino)pheny11-3-
ON N
(1)r )(:-O-NH fluoro-1H-indo1-5-y1) -1- [(2R)-2 -
139 . 0
0 N
H 1-0H3 567.669
pheny1-2-(pyrrolidin-1-
. ypacety1]-1,prolinami
de
H F 0, N-
(tert-butoxyearbony1)-D-
0143 N ¨ )\-CH3 alanyl-N- (244-
140 cHs--)- )r11.õ 1 ' \ 551.623
0 ...", N \ / NH
(ac etylamino)phenyll-3 411.101-0-
CH3 0 / '0
H
CH3 1H-indo1-5-y1)-L-
prolinamide
_
H F
C\113 1,-Ilif,N N-
{244-(acetylaniino)pheny]]-3-
cH,-N , 0 N . NH
fluoro-1H-indo1-5 -y1) -1- [(2R)-2-
141 o ' (5)--cu3 541.631
H (dimethylamino)-2-
1St phenylacetylj-L-
prolinarnide
- 133 -
:A 02756172 2011-09-21
WO 2010/111483 PCT/US2010/028653
Example Structure MW Name
H F
)---CH3 N-(tert-
butoxyearbony1)-D-
N
c- 0 H (--rN \ leucyl-N-1244-
cm
142 3 cH \orNõõcco . so N . NH
593.705
H (acety1amino)pheny1]-3-
fluoro-
CH3
11/-indo1-5-y1) -L-prolinarnide
cH,
_
A
propan-2-y1 [(1R)-2-{ (2S)-2-[(4-
F {54( { (2S)-1-[(2R)-2-
C H30.1 rl
CH3-4 N r 0. \ NiVr) (dimethylarnino)-2-
143 0 H H
o / 815.953 phenyiacetyl]pyrrolidin-2-y1}
N =
0
. INr carbonyparnino]-3 -
fluoro-1 H -
El \r-CH3
indo1-2-yl)phenypearbarnoyl]
. cH,
pyrrolidin- 1 -y1} -2-oxo-l-
phenylethylicarbamate )
CH3H F tert-butyl (2S)-2-[(3-fluoro-2- {4-
cH,,r? )(N 1 '--- \ /1 NH [( {(25)-1-[(2R)-2-
phenyl-2-
cH, o --- " ----0H {[(propan-2-
yloxy)carbonyll
144 0 N CH
0804Hry3 cH., 754.866 amino}
acetylipyrrolidin-2-
yll carbonypaminolphenyl} - 1 H-
indo1-5-y1)carbamoyll
pyrroli dine- 1 -earboxylate
,
propan-2-y1 [(1R)-2-{(2S)-2-[(4-
H Br {3-bromo-54({(25)-1-
[(2R)-2-
1.1..0 vt clyN 46 \ ip 15.---j phenyl-2- { [(propan-
2-
145 ,,,,r 0 H 935.88
NH N
yloxy)carbonyl]amino} acetyl]
0
, IN
la rosr cH, cH3 pyffolidin-2-y1}
carbonypaminol-
.1-benzofuran-2-y1} phenyl)
earbamoyIl pyrradin- 1 -y1} -2-
oxo-l-phenylethyl) carbamate .
(2S)-N-(4- { 3-brorno-54( { (25)-1 -
11
N Br 0
% .>
1 3 (N-Iy , .-"=-= \ ¨ / :---3 [(2R)-2-
(dimethylamino)-2-
CH
phenylacetyllpyn-olidin-2-
CH3¨N 0 NH N n H3
yl} carbonyl)amino]-1-
146 0 0, \NI: 819.806
41Ik
. CH benzofuran-2-y1)
pheny1)-1-
[(2R)-2-(dirnethylamino)-2-
phenylacetylipyrrolidine-2-
carboxamide
-
methyl {(1 R)-2-[(25)-2-({413-
1-1 Br bromo-5-({[(2S)-1-
{(2R)-2-
,cp H (NlyNC6_0_1-ci
[(methoxycarbonypaminol-2-
cH3 ),-N 0 0
, H phenylacetyl}
prTolidin-2-
147 o 879.772
lik milk,. )orckcH,
yljearbonyl) amino)-1-
benzofuran-2-yl]phenyl}
carbamoyppyrrolidin-l-y11-2-
oxo-l-phenylethyl} carbamate ,
_ -
methyl {(25)-1-[(28)-24 {443-
F fluoro-5-({ [(25)-1-
{(2S)-2-
-H 0s
--r
N 0NH\
NH0 ,4-r
749.847
y[(nehoxyearbo nyl)amin ol-3-
N
148 cH-0-Nrko0 methylbutanoyl) pyrrolidin-
2-
2c13 cH, cH, N llcarbonyl) amino)-1H-indo1-2-
cH, yl]phenyl) earbamoyl)pyrrolidin-
cH, 1-y11-3 -methyl-l-
oxobutan-2-
yl } earbamate
149 0,....ct.6
;!NH3Q.yttccar
CH
0 0 NH
603.521 N- (244-[4-3-
bromo-l-benzofuran-5-y1) -1-
[(2R)-2-(dimethyIamino)-2-
4It phenylacetyli-L-prolinamide
_
- 134 -
:Ammm72m11-m21
WO 2010/111483
PCT/US2010/028653
Example Structure 1 MW Name
(2S)-N-(4- {3 -brorno-54( { (28)-1-
CH CJI1rH Br
[(2R)-2-(diethylamino)-2-
Cf-1,__3 1\13 N N i .."--- \ St CIN,H .'-µ:-Nrj
pheny1acety1lpyrrolidin-2-y1}
0 ...-- 0 rCH3
150 o o . iN 875.915
carbonyl)amino1-1-benzofuran-
I.
. \--CHa 2-
yllpheny1)-1-[(2R)-2-
(diethylamino)-2-phenylacetylj
pyrrolidine-2-earboxamide
OH0I,N H Br 0 :-------
(2S)-1-acetyl-N-(4- {3 -bromo-5-
1 ,
NH N---
151 tari3 0 \
(dimethylamino)-2-phenylacetyli
/
0 700.639
0 00H3 pyrrolidin-2-
y1}carbonyljaminol-
,
1-benzofirart-2-y1) phenyl)
pyrmlidine-2-carboxamide
tert-butyl {(1S)-2-1(28)-2-( {443-
H srbromo-5-( {[(28)-1 - {(2S)-2-
. cl oef ,. \ 4., 1:4VN-D
[(tert-butoxycarbonyl)amino]-2-
CH3 0 up 0 mr7 gH
phenylacetyI) pyrrolidin-2-
152 00.;--0kr,Nu '..' Nro 963.935
yl] carbonyl} amino)-1-
cH3 o
benzofuran-2-yl]phenyl}
cHc CcHH:
carbarnoyljpyrrolidirt-1-y11-2-
_ oxo-l-phenylethyl }
carbamate ) ,
Br 0
4It (N-Ir H N )-0
401 \ it NH N
(phenylacetyl)pyrrolidin-2-yl]
153 o o o 08 733.669
carbonyl} amino)-1-benzofuran-
2-yliphenyl} -1-(p heny lacetyl)
pyrrolidine-2-carboxamide
tert-butyl {(1R)-2-[(2S)-2-( {4-
H Br
\ =[3-bromo-5-( { [(2S)-1-{(2R)-
2-
N 0, ....-,
[(tert-btit0XycarbOnyDaMin01-2-
I
154 3 0 963.935
HQ 0( * Z-KNj
phenylaeetyl } p yrrolidin-2 -
0 0
A
11. r 5ccH3
yli carbonyl} amino)-1-
CH3 C
benzofuran-2-yliphenyl }
H3
carbamoyl)pyrrolidin-l-yll -2-
oxo-1-phenyleth yll earbamate
Example 155 ¨ (2S)-N-1443-eyano-5-([1-(2S)-1-(phenvlacetyl)pyrrolidin-2-
vllearbonyliamino)-
1H-indol-2-vIlphenyli-1-(phenviacetv1)pyrrolidine-2-earboxamide
cly." Br H i.,
`.`----(3
1110 N
\ . C'Y C Cu "\---
CN/DMF Cie iiihõ,. õõ.
- k. 0 ir .
Ph NH N
KI
0 ' H Nmo N ptl
H
A mixture of the bromo compound from Example 126 (150 mg, 0.2 mmol),
CuCN (50 mg, 0.6 mmol) and DMF (3 mL) was refluxed under N2 protection
overnight. The
mixture was purified by RPLC to afford the product. MS (EST) mie (M H+): 679.
1H NMR
(CDC13) 8: 7.73-7.70 (m, 4H), 7.38-7.29 (m, 4H), 7.21-7.04 (in, 6H), 4.63-4.60
(m, 1H), 4.49-
4.47 (m, 1H), 3.81-3.59 (m, 4H), 2.48-1.97 (m, 8H).
- 135 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Example 156 ¨ (2S)-N-1443-(2,2-dimethylpropanay1)-5-(11(2S)-1-(phenvlacetv1)
pyrrolidin-2-
ylkarbonvliamino)-1H-indol-2-ylipheny11-1-(phenylacetyl)pyrrolidine-2-
earboxamide
NZnC12/MeMgEr ClyN czy_(-3
õ,õ___co 0 so N
Ph Ph
To a stirred solution of the indole (50 mg, 0.076 mmol) in CH2C12 (5 mL) was
added anhydrous ZnC12 (54 mg, 0.4mmol) then MeMgBr (3.0 M in Et20, 0.4 mL, 0.4
mmol).
The resulting suspension was stirred for 10 minutes at RT and then cooled to 0
C at an ice bath.
A solution of pivaloyl chloride (14 mg) in CH2C12 (0.2 la) was added to the
mixture. The
reaction mixture was allowed to warm to RT and stirred overnight. The reaction
mixture was
quenched by saturated aqueous NH4C1 and exacted with CH2C12 3 times. The
organic layers
were combined, dried over anhydrous Na2SO4 and filtered. The filtrate was
concentrated and
purified by RPLC to yield the product. MS (ESI) mile (M+H+): 738. 11-1 NMR
(Me0D) 8: 7.69-
7.63 (m, 314), 7.44-7.41 (m, 211), 7.31-7.21 (m, 12H), 4.55-4.52 (m, 2H), 3.76-
3.59 (m, 8H),
2.26-1.94 (m, 8H).
Example 157¨ 5-(11(2S)-1-(phenvlacetv1)pyrrolidin-2-yllearbonyliamino)-244-MS)-
1-
(phenvlaeetvi)pwrolidin-2-vllearbonyllamino)phenvll-1H-indole-3-carboxamide
0
EIMg = roi2
NH N
.ZeiNH go \ NH N __ Bt 7 R0 0
Njc,
Bn \`
0
To a solution of the indole (653 mg, 1 mmol) in THF 10 (mL) was added EtMgBr
(2 mL, 6 mmol), and the mixture was stirred at RT for 30 minutes. Thereto was
added
chlorosulfonyl isocyanate (140 mg, 1 mmol), and the mixture was stirred at RT
for 20 minutes.
Then, DMF (146 mg, 2 mmol) was added to the above mixture, and the stirring
continued for 20
minutes. After adding aqueous NaOH (2N, imL), the resulting solution was
heated at reflux for
5 minutes. Concentration in vacuo, the residue was purified with RPLC to give
(67 mg). III
NMR (Me0D) 8: 8.0 (s, 111), 7.6 (m, 411), 7.1-7.4 (m, 1211), 4.5 (m, 4H), 3.5-
3.7 (m, 8H), 2.5-
2.0 (m, 611).
- 136 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Example 158 - tert-butyl l(1R)-2-[(2S)-2-6445-(11(2S)-14(2R)-24(tert-
butoxvcarbonyl)aminol-2-phenvlacetyllpyrrolidin-2--plkarbonvIlamino)-3-
(eyelopropykarbamoy1)-1H-indol-2-vIlphenyllearbamoyl)pwrolidin-l-vil-2-oxo-1-
phenvlethplicarbamate
0 E
0
c N)
BocHN \
0 go it NH
Step .1
02N is POBrz 0214
DWIF N\ Br
To a solution of POBr3 (113.2 g, 0.4 mol) in DCE (1 L) was added DMF (14.6 g,
0.2 mol) dropwise under ice bath, and the mixture was stirred at RT for 30
minutes. Thereto was
added nitro compound (17.8 g, 0.1 mol), and the mixture was stirred at reflux
for 4 hours. The
precipitate was collected by filtration and then washed with water and Me0H.
The solid was
dried in vacuo to give the desired compound (13.5 g). 1HNMR (DMSO) 6: 13.6 (s,
1H), 9.8 (s,
1H), 8.8 (s, 1H), 8.1 (d, J-9.2 Hz, 1H), 7.6 (d, J=9.2 Hz, 1H).
Step 2
0
02N (Bo.)20 02N
15 IIBac
To a solution of the aldehyde from step 1 (13.5 g, 0.05 mol) in DCM (100 mL)
was added DMAP (0.6 g, 0.005 mol), TEA (10.1 g, Imo]) and (Boc)20 (21.8 g,
0.1 mol), and
the mixture was stirred at RT overnight, The mixture was concentrated, and the
residue was
purified by column chromatography to give the desired compound (14.7 g). 1H
NMR (CDCI3) 8:
20 9.8 (s,111), 8.8 (s, 111), 8.1 (d, J-9.2 Hz, 1H), 7.6 (d, J=9.2 Hz, 1H),
1.4 (s, 911).
Step 3
0 0
HSB
02N NHBoo
Br PrcPf):12 N H it NHBac
HO
BOO 80C
The Suzuki coupling procedure was the same as described in Example 117,
step 3. MS (m/z): 482 (M+H)+.
- 137 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Step 4
0 =
OH
OzN
No0102, NaH2PO4 dot
NHBoc methy1-2-bu 07N tene "\ NHE3c'
Boo Boo
To a solution of compound from step 3 (2.4 g, 5 mmol) in pH 3.5 phosphate
buffer (24 mL) and t-BuOH (30 mL) was added 2-methy1-2-butene (10 mL) and
sodium chlorate
(0.89 g, 10 rnrnol). The reaction was stirred at RT for 16 hours and then
extracted with DCM
(3x). The combined organic extracts were washed with brine, dried over
anhydrous MgSO4, and
concentrated in vacua to give the desired carboxylic acid (2.3 g). MS (m/z):
498 (1\44-H)+.
Step 5
o 0 Y7
OH NH
lj¨NH2
02N abh
NHBoo HATU, DIPEA 02N 7
______________________________________________________ ost ip NHBoc
N N
Boo Boo
The mixture of compound from step 4 (1 mmol), cyclopropyl amine (1 mmol),
HATU (1 mmol) and DIPEA (5 mmol) in DCM was stirred at RT overnight.
Concentration and
purification of the residue by RPLC gave the desired compound (0.8 mmol). MS
(m/z): 538
(M+H)+.
Step 6
0
Nt180 Pd: H2N 0
NH NH
02N
\
\
irioc
Boo Boo
To a solution of the amide from step 5 (0.8 mmol) in Me0H (10 mL) was added
Pd/C (100 mg) and the mixture was stirred under H2 at RT for 1 hour. The Pd/C
was removed
by filtration, and the filtrate was concentrated to give the desired compound
(0.7 mmol). MS
(m/z): 507 (M411) .
Step 7
NH NH
H2N \
NHBoc TFA H211
\
N
Boo
To a solution of compound from step 6 (0.7 mmol) in DCM (5 mL) was added
TFA (2 mL), and the mixture was stirred at RT overnight. The solution was
concentrated, and
the residue was used in next step without purification. MS (m/z): 307 (M+H)+.
- 138-
:A 02756172 2011-09-21
WO 2010/111483 PCT/US2010/028653
Step 8
0X:6_
HATIJ Bad IN 0 0 ' NH
r
NH2 Acid, OIREA 0 11 ,,NI-il3oc
N
=
The coupling procedure was the same as used in Example 72, step 7. 1H NMR
(Me0D) 6: 6.9-7.9 (m, 17H), 5.2-5.5 (m, 211), 4.4-4.5 (m, 211), 3.5-3.9 (m,
3H), 2.7-2.8 (m, 111),
1.7-2.2 (m, 8H), 1.4 (s, 18H), 1.2 (m, 111), 0.4-0.8 (m, 411). MS (m/z): 967
(M+H)+.
Example 159 ¨ tert-butp/ {(1R)-24(2S)-2-(f4-15411(2S)-1-1(2R)-2-I(tert-butaxv-
carbonvl)aminal-2-phenylacetvlIpyrrolidin-2-vllearbanyllamino)-3-(4-
methoxyphenv1)-1H-
indol-2-yllphenylicarbamovl)pprrolidin-l-vll-2-oxo-1-phenvlethylicarbamate
. \ BocHN
BocH
NI 0 . .*
i j io IN' a \ it
(,:_J)
H
Step]
H
01 BocHN
C---4'
NI.
BocHN
11,
C-"''
N 0
A...õ,,, 4NHBo iii Br
c
NHBoo
NI, \ 0
WI N
H NH
NBS (103 mg, 0.5769 mmol) was added in portions to the solution of the indole
(510 mg, 0.5769 mmol) in 20 mL of THF, the mixture was stirred at RT for 1
hour, then
concentrated. The residue was purified by Prep-HPLC to afford the desired
compound (500 mg).
MS (ESI) m/e (M+H+): 962.
Step 2
00 BocHN di \,)
Bociio
0,)- 44 BocHN =.`` BocHN
Ci H Br 0, ,K1 --.." Pd(apP)012
"x " 0 \ = ,,,,,,- , H
Na,2003rdoxanad-420 N N OkJ)
N I 1101
H N
H
The mixture of the product from step 1 above (100 mg, 0.104 mmol), 4-methoxy-
phenylboronic acid (24 mg, 0.1558 mmol), Pd(dppf)C12 (7.6 mg, 0.0104 mmol),
Na2CO3 (3.3
mg, 0.0312 mmol) in 10 mL of dioxane and 2 mL of water was heated to reflux
under N2
atmosphere overnight. The mixture was cooled and concentrated, then the
residue was purified
- 139 -
:A 02756172 2011-09-21
WO 2010/111483
PCT/US2010/028653
by RPLC to give the desired product (30 mg). 111 NMR (Me0D) 6: 7.71-7.54 (m,
311),
7.42-7.26 (m, 1611), 6.96-6.92 (m, 311), 5.45 (s, 211), 4.53-4.50 (m, 211),
3.92-3.81 (m, 511),
2.08-1.84 (m, 811), 1.42-1.32 (m, 1811). MS (ES1) m/e (M+11 ): 991.
Examples 1604 77
Compounds of Examples 160-177 were prepared in a similar manner as described
in Examples 155-159.
Example Structure MW Name
. (2S)-1-
(phenylacety1)-N- {4- [3-
pheny1-54 { [(19-1-
i
D 729.887
(phenylacetyl)pyn-olid in-2-
160 fit ciiiN Ali_
0 ip \ iP NH N
yIl carbonyl I amino)-1H-indo1-2-
14
H A
phertyll pyn-olidine-2-
0 o git
carboxamide
CH3 0 (25)-N- {443 -ethyl-
5-( { [(25)-1-
161 #it ciirmH di \
NH N
(phenylacetyl)pyrrolidin-2-
681.842 ylicarbonyl} amino)-
1H-indo1-2-
o
0 =
o µ11111P N
H ylipheny1)-1-
(phenylacetyl)
pyrrolidine-2-carboxamide
CHa (25)-N- {443 -acetyl-54 ( [(28)-1-
0
H (1
(phenylacetyl)pyrrolidin-2-
162 4111P IK-111-14 0 \ it 7
,,......3
NH N 695.826 yl]carbonyl }
amino)-1H-indo1-2-
0
H N yliphenyl } -1-
(phenylacetyl)
0 0 .
pyrrolidine-2-carboxamide
ft (25)-1-(phenylacety1)-N- {4-[5 -
( {[(25)-1-(phenylacetyl)
0
H 0) N.--3 757.897 pyn-olidin-2-
yl]carbonyl) amino)-
163 *ClirN
- 40 \ . NH N 3-(phenylcarbony1)-
1H-indol-2-
o N
H 0 yllphenyl}pyrrolidine-2-
o tit
carboxamide
N
/ \ (2 S )-1-(phenylac
ety1)-N- {2 - [4-
* C-1 r 1 ( {[(2S)-1-
(phenylacetyl)
= NH ill
pyrrolidin-2-yl] carbonyl}
164
0 0 \ N
730.874
li 0 amino)pheny1)-3-
(pyridin-4-y1)-
1H-Indol-5-y1}pyrrolidine-2-
carboxarnide
411# benzyl (25)-24(4-
{5-[( {(2S)-1-
I 9"-0
[(benzyloxy)carbonyl]pyrrol idin-
165 0-Zo o * ii 761.886 2 -y1) c
arbonyl)amino]-3-phenyl-
40 H
1H-indo1-2-y1) phenyl)
c arbamoyl]pyrrolidine-1-
carboxylate
r¨ N 0 (25)-N- {443-cyano-
54 {[(25)-1-
fAiNH
0 411\ Amlik >-0
1 ti w NH N (phenylacetyl)pytTolidin-2-
166 0 H 08 678.798 ylicarbonyl }
amino)-1H-indo1-2-
yl}pheny1}-1-(phenylacetyI)
0.
pyrrolidine-2-carboxamide
11P. (28)-N- (443-
cycIopropy1-5-
(1 14
s1,1Y.1 iiii \ di :,D ({[(2.9- I -(phenylacetyl)
pyrrolidin-2-yljcarbonyl) amino)-
167 71111P N W 08 693.853
1H-indo1-2-yl]phenyl) -1-
(phenylacety1)pyrrolidine-2-
carboxamide
..
- 140 -
:Ammm72m11-m21
WO 2010/111483 PCT/US2010/028653
Example Structure MW Name
4* 0¨CH3 benzyl (25)-24(4- {54(
{(25)-1-
QH [(benzyloxy)carbonyl}pyrrolidin-
.'? 2-y1) carbonyl)amino]-3-(3-
168 \
"I 0--kip e 01
NH N 791.912 N .
methoxypheny1)-1H-indo1-2-
H () \0"")__.µ
yIlphenyl)carbamoylipp-rolidine
-1-carboxylate
tert-butyl {(1R)-2-[(2S)-2-( {4-
* CH [54 { [(25)-1- {(2R)-2-
[(tert-
, butoxycarbonyl)amino1-2-
H
phenylacetyl) pyrrolidin-2-
169 õerr, Clior 10 \ = NH N 974.18 yl]carbonyl)amino)-3-(3-
cH3 o o 1 o ,
1. )r ,,,-cH,
O c/ H;cr1 yll
methylpheny1)-1H-indo1-2-
phenyl) carbamoyppyrrolidin-
1-yli-2-oxo-l-
phenylethyl)carbamate _
tert-butyl {(1R)-24(28)-24 {4-
[54 { [(25)-1-{(2R)-2-[(tert-
cH,*,)' butoxycarbonypamino]-2-
170 cH H vs
CIAõ ...,
o H cif 0 \ ANI HF;---J
\t,j_ phenylacetyl) pyrrolid in-2-
g,.,a ,c,,,,--N 0 0 - N W 08 H 974.18 yl] carbonyl) amino)-3-
(2-
H . .1N,1/ _0 methylpheny1)-1H-ind 01-
2-
`===,-CH3
O 0/1213cH, yliphenyl)
carbamoyl)pyrrolidin-
1-y1]-2-oxo-1-
phenylethyl)carbamate
,
tert-butyl {(1R)-2-[(2S)-2-( {2-
/ N,,,
[4-({[(2S)-1-{(2R)-2-[(tert-
H
(:\ --0 butoxycarbonyl)am
0 ino]-2-
CH3 H 14 N
phenylacetyll pyrrolidin-2-
171 cH, r
c-p:3`N Cit 110 \ gp, NH H N
961.14
N
o 0 4 = yli carbonyl 1
amino)phonyll-3-
0,3
O cf,3, (pyridin-4-y1)-1H-
indo1-5-
y1) carbarnoyl)pyrrolidin-1-y1}-2-
, oxo-l-phenylethyl)carbamate
__.
-
CH3 )----D btouxtyylca{r(bI
oRn)y-21)12S:no-2112({-4-
terbtu-
N [5 -( {[(2S)-1-{(2R)-2-1(tert-
\_ H (111\1 111 fi
CH3-7- '.1.--N 4111 Ilk NH N phenylacetyl )
pyrrolidin-2-
172 cH3 6 0o N ' H
08 H 909.064
yl] carbonyl) am ino)-3-cyano-1H-
tilt indo1-2-yliphenyl)carbamoyl)
= CH3 CH3
pyrrolidin- 1 -y11-2-oxo- 1 -
phenylethyl) carbamate
N (25)-N-(4- {3-cyano-54(
{(2S)-1-
0
H
c [(2R)-2-(dimethylamino)-
2-
c
11 ,----j
NH N phenylacetyljpyrmlidin-
2-
0113---N
173 0 0 H \ 1
0 N
0 /CH3 764.936 yl} carbonyl)amino]-1H-indo1-2-
. 4N\CHs
* yl)pheny1)-1-[(2R)-2-
(dimethylamino)-2-phenylacetyl]
pyrrolidine-2-carboxamicle ,
N
ii tert-butyl {(1R)-2-[(2S)-
2-( {2-
H
CH3 N Aik , A., C-CH3 [4-
(acetylamino)pheny1]-3-
(1 H N
174 cl-r,)-
-)¨II Clill IF N\ lir NH 620.714 cyano-11/-indo1-5-
cH3 0 0
H yl) carbamoyl)pyrrolidin-
1 -yI]-2-
* oxo-l-phenylethyl)carbarnate
- 141 -
:A 02756172 2011-09-21
WO 2010/111483
PCT/US2010/028653
Example Structure MW Name
_ ......
tert-butyl { (1R)-2-[(2S)-2-( {4-
cH, [5-( { [(2S)-1- {
(2/0-24(tert-
0 /
butoxycarbonyl)aminol-2-
CH3 <-"rli N-CH3 1:)µ___., phenylacetyl}
pyrrolidin-2-
175
cH3-0,,,11 N , NH N
Ok ` Ill ¨
0 N
1 955.133 yl]carbonyl)
amino)-3-
8
cH3 6 0 H 07,1
40 )--`),,x0H3 (dimethylcarbamoy1)-
111-indol-
CH3 oH, 2-yl]phenyl }
carbamoyl)
pyrrolidin-l-y1]-2-oxo-l-
phenylethyl) carbamate
methyl 5-( ( [(2S)-1- ( (2R)-2-
CH, [(tert-
butoxycarbonyl)amino1-2-
CH3 H
o I phenylacetyll pyrrolidin-2-
H 0
clirN .- C))----D
yl] carbonyl j amino)-244-[4 ( [(25)-
176 0143)raNtrN õ 0 N\ . NH N
942.091 1- {(2R)-2-
[(tert-
cu5 0 0 ' H .111
4ft )_0;:lc-.H3
butoxycarbonyl)amino1-2-
ci 0õ, cH3
phenylacetyl}pyrrolidin-2-
yllearbonyl } amino)pheny1]-111-
indole-3-carboxylate
5-( ( [(25)-1- {(2R)-2-Rtert-
butoxycarbony1)amino)-2-
OH H 0
H OH 0 phenylacetyl}
pyrrolidin-2-
,
177 OH. N CliN aih. NH N')
ylicarbonyl}amino)-244-[4 [(2S)-
,H3 .õ.11 0 111.1 \ lep 0 0 N
H , Sill 928.064 1- 42R)-2-[(tert-butoxycarbonyl)
* )r- )CH3
(:)µ amino]-2-
phenylacetyl)
0tH3 CH3
pyrrolidin-2-yl] carbonyl) amino)
pheny1]-1H-indole-3-carboxylie
acid
1
....
Example 178¨ tert-buivl falt)-2-1(2S)-2-(f2-13-(acetplamino)prop-1-vn-1-v11-
111-indo1-5-
yllearbamov1)pyrrolidin-1-v11-2-oxo-1-phenvlethvlkarbamate
H
TrN HN4
BocHN , \ = j o
0 N
0 H
41.
Step 1
0,)
02N 401 02N so -0Et 02N
0 ethyl carbonochlorklaIe 0 {NH4)2CO3 0 .
ti
N N
c,out
0...-'0E1
Ethyl carbonochloridate (3 mL) was added to the nitro lactam (2.0 g, 11.2
mmol),
and the mixture was stirred for 3 hours before being concentrated to give the
crude product. MS
(ESI) m/e (M+H+): 325. The crude product was dissolved in DMF (25 mL), then
(N114)2CO3
(1.5 g) was added, and the mixture was stirred overnight. The mixture was
evaporated and the
residue was poured into ice-water and extracted with DCM, and the organics
were dried. The
solvent was removed, and the residue was purified by chromatography on silica
gel to give the
desired compound as a white solid. MS (ESI) mie (M+1-1 ): 251.
- 142 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Step 2
02N io
OTf 02N .0 HN--(
Pd(PP113)4 _______________________________________________ J 0
o
Cr-
Et
Ei00
The mixture of the triflate (382 mg, 1.0 mmol), N-(prop-2-ynyl)acetamide (97
mg, 1.0 mmol), Et3N (3 mL) in CH3CN(3 mL) was stirred at RT for 3 hours. The
mixture was
concentrated, and the residue was purified by chromatography on silica gel to
give compound
target compound (254 mg). MS (ESI) m/e (M+14+): 330.
Step 3
H2N
02N if& Fe/NH4C1 io __
EtON/
Et0
A solution of the nitro compound from step 2 above (165 mg, 0.50 mmol) in
absolute Et0H (3 mL) was added Fe power (280 mg, 2.5 mmol) and NH4C1 (535 mg,
5.0 mmol).
The mixture was stirred at 70 C for 2 hours, cooled and poured into ice/water
(50m1). The
mixture was extracted with Et0Ac (200 ml), and the organic phase was combined
and washed
with brine, dried and concentrated to yield the crude product (150mg). MS
(ESI) m/z :(M+H)
300.
Step 4
H2N Ht4¨(- / H2N
k2CO3
"WI HN--(
_____________________________________________________________ 0
¨ 0
1\1/
Et
To a solution of the aniline from step 3 (150 mg, 0.50 mmo) in absolute Et0I-1
(3
mL) was added K2CO3 (138 mg, 1.0 mmol), and the mixture was stirred at RT for
12 hours. The
reaction mixture was poured into water (10 mL), extracted with Et0Ac (20 mL),
and the organic
phases were combined and washed with brine, dried over MgSO4 and concentrated
to yield the
crude product (113mg). MS (EST) miz :(M+H+) 228.
Step 5
N
H2N io HATU/E13N BocHN HN4
(1-1(
0 -- 0
R-Boc-Phg-L,Pro
The mixture of the indole (113 mg, 0.5 mmol), R-Boc-Phg-L-Pro-OH (175 mg,
0.5 mmol), DIPEA (115 mg, 1.0mrnol) in MeCN (2 mL) was stirred at RT for 5
minutes, then
HATU (190 mg, 0.5 mmol) was added into the mixture. The mixture was stirred at
RT overnight
- 143 -
/1027561722011-09-21
WO 2010/111483
PCT/US2010/028653
then concentrated. The residue was purified by RPLC to give the desired
compound (110 mg).
1H NMR (Me0D) 8: 1.37 (s, 9H), 1.96 - 2.14 (in, 7H), 3.92 - 3.94 (m, 211),
4.51 - 4.54 (in, 1H),
5.41 (s, 111), 6.56 (s, 1H), 7.20 - 7.43 (m, 714), 7.72 (s, 111). MS (ESI)
:(M+H+) 576.
Example 179 -N-1445-(furan-3-yl)-1H-indo1-2-yllphenv11-1-(phenylacety1)-L-
pralinamide
/ = 13)\--0
1110 ' Ilk NH N
0 *
Step 1
t).õ,c)
Br Q
Nt-I2 "
HATU,DWIF H 0 *
The mixture of the indole from Example 41(1.6 mg, 5.575 mmol), 1-phenylacetyl
pyrrolidine-2-carboxylic acid (1.3 g, 5.575 mmol), DIPEA (1.45 g, 11.15 mmol)
in DMF (50
10 mL) was stirred at RT for 30 minutes, then HATU (2.54 g, 6.689 mmol )
was added. The
mixture was stirred at RT overnight, concentrated in vacuo, and the residue
was purified by
chromatography on silica gel to give the desired product (2.3 g). MS (ES1) m/e
(M+H+): 504.
Step 2
91-/
\-D
Br \ -
NH N 0,0 B.OH /
54'D
NVf N
N=n
Pd(PP113)2C-12,No/COsIGH3CH/H20 H 0 *
A suspension of the product from step 1 above (18 mg, 0.03583 mmol), furan-2-
boronic acid (6 mg, 0.05374 mmol), Pd(PPh3)2C12 (1.4 mg), Na2CO3 (7.6 mg,
0.07166 mmol)
and 1120 (0.15 mL) in 0.5 mL of acetonitrile under N2 protection was heated at
150 C for 10
mintues in a microwave reactor. The mixture was cooled, filtered and washed
with 10 mL of
DCM. The solvents were removed, and the residue was purified by HPLC to give
the desired
product. 1H NMR (Me0D) 8: 7.79-7_74 (in, 311), 7.67-7.62 (m, 3H), 7.50 (s,
1H), 7.37-7.22
(m, 6H), 6.78-6.75 (m, 2H), 4.57-4.55 (m, 111), 3.78-3.61 (m, 411), 2.24-1.99
(m, 411).
Examples 180-189b
Compounds of Examples 180-189b were prepared in a similar manner as
described in Example 179.
Example Structure MW Name
CH3
,N N, N-(4- {546-
(dimethylarnino)-4-
cH,
180 557.701
10 N\ () yl}pheny1}-1-(phenylacetyl)-1,-
H 41, prolinamide
- 144 -
/1027561722011419-21
WO 2010/111483
PCT/US2010/028653
Example StructureMW Name
N- {445-(1-benzothiophen-3 -y1)-
181elt04 --0---NH N 555.704 1H-indo1-2-yl]phenyI)-1-
H 0 *
(phenylacety1)-L-prolinamide
-
PN- (445-(1-benzy1-1H-pyrazol-4-
182o
579.708
y1)-1H-indol-2-ylipheny11-1-
1 .
11o>_,...(phenylacety1)-L-prolinamide
11 0 *
, ___________________________________________________________________________
Co N-
(44542,3 -dihydro-1,4-
' 0
183 10 )-j 557655
benzodioxin-6-y1)-1H-indol-2-
N 9+ NH N yl]pheny11-1-
(phenylaeety1)-L-
H 0 . prolinamide
1-(phenylacety1)-N- (445-
184 N .
ylipheny1)-L-prolinamide
0¨ N 550.666
(quinolin-8-y1)-11/-indo1-2-
fiH
..._.......
S I
5---<"--D 1-
(phenylacety1)-N- (445-
..
185 40\ =
N NH N 505.644
(thiophen-3-y1)-1H-indoI-2-
H 0 *
yl]phenyl) -L-prolinamide
_
411" \ . NH N tert-butyl
N = '
cyano-111-indo1-2-y1)phenyll
186 H 0 tql
563.662
earbamoyllpyrrolidin- 1-y11-2-
= \rcH3
oxo-1-phenylethyl)earbamate
cH9H,
I HN
n--14 1 51---D
propan-2-y1 [(1R)-2-oxo-1-
pheny1-2- ( (25)-24442- (4-
[( {(2S)-1-[(2R)-2-pheny1-2-
H3 :ft ), gib
187 01-13¨<C N o '111111-rir N I"
H {[(propan-2-yloxy)carb0nyl]
o 879.037
H .01
amino} acetyljpyrrolidin-2-
oi ro\rcH3
yll carbonyparnino]pheny1)-1H-
. cn,
indo1-5-y1)-1H-imidazol-2-yl]
pyrro1idin-1-yl)ethyl]carbamate
"
, c__...14-T0 propan-2-y1 {(1R)-
21(28)-2-(5-
mi.
o 40 \ 11 NH 606 {244-
(acetylamino)pheny1]-1 H-
188
o HN'18 N
H .687
indo1-5-y1)-1,3,4-oxadiazol-2-
0113_< -lb
yl)pyrro1idin-1-y11-2-oxo-1-
cH3
phenylethyl) carbamate
N-N N-
{44545- ((2S)-14(2R)-2-
\
o )-- H
N 0 0 * NH C 3 (diethylamino)-2-
189 cH3---,õ 0
N N 576.704
phenylacetylipyrrolidin-2-y1)-
cH3-1 1,3,4-oxadiazol-2-y1)-
1H-indo I-
2-Aphenyl} acetamide
methyl [(28)-1-{(2S)-2-[5-(10-
N (2-[(2S)-1-((2S)-2-
[(methoxycarbonyl)amino]-3-
n * N\ * / 1:J
methylbutanoyl ) pyrrolid in-2-ylj-
189a \ Ht". \ ti, ..," 806.9 1H-imidazol-5-
y1) indolo[1,2-c)
o-io 0 ./
[1,3]benzoxazin-3-y1)-11 / -
3
imidazol-2-ylipyrro1idin-l-y1) -3-
---,0 q
methyl-1 -oxobutan-2-
yflearbamate
..
- 145 -
:A 02756172 2011-09-21
WO 2010/111483
PCT/US2010/028653
Example Structure MW Name
methyl [(28)-1-{(2S)-245-(12-
fluoro-10-{2-[(25)-1-{(2,5)-2-
F
methoxycarbonyl)amino1-3-
Ld \ A -IL
Rmethylbutanoyl}pyrrolidin-2-yli-
Pi
1891) Lc, = 824.9
Q
[1,3]benzoxazin-3-y1)-1H-
imidazo1-2-ylipyrrolidin-l-y1}-3-
methy1-1-oxobutan-2-
ylicarbamate
Example 189b (Alternative Procedure: Methyll(2S)-11(2S)-2-15-(12-fluoro-10-12-
1(2S)-1-
f(2s)-24(methoxycarbonyl)aminol-3-methylbutanoyepyrrolidin-2-yll-lH-imidazol-5-
yllindolof1,2-01,31benzoxazin-3-v1)-1H-imidazol-2-yllpyrrolidin-1-yll-3-methyl-
1-oxobutan-
2-ylkarbamate
C\r¨N
N'LH'`
H
/C)--\(
0 01
Step 1
.0 B AcC1 9 lo
HO Et3N
To a solution of compound 3-bromophenol (51 g, 0.3 mol) and Et3N (36 g, 0.36
mol) in 500 mL of DCM was added dropwise acetyl chloride (26 g, 0.33 mol) in
an ice-water
bath. The mixture was stirred at RT for 30 minutes. The mixture was washed
with 1 N HC1,
saturated Na2CO3 and brine, dried over Na2SO4 and concentrated in vacua to
give a oil (62 g).
Step 2
9 40 A3
21'0 Br 140 C IP
HO Br
A1C13 (40 g, 0.3 mol) was slowly added to the product from step 1 (21.5 g, 0.1
mol) in an ice-water bath. The mixture was stirred at 140 C for 2 hours. After
cooling to 60-
70 C, the mixture was slowly poured into an ice water. The resulting solution
was extracted
with DCM. The combined organic phases were washed with brine, dried over
Na2SO4 and
concentrated in vacua. The residue was purified by column chromatography to
give the desired
compound (14 g). MS (EST) m / e (M+H+): 214.
Step 3
- 146 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
H HCI
-N,2 AOHEOH N(
HO Br Br Br HO P Br
A mixture of the ketone obtained in step 2 (4.2 g, 20 mmol) and 4-bromophenyl
hydrazine hydrochloride (4.4 g, 20 mmol) in AcOH and Et0H (1:10, 100 mL) was
heated to
reflux for 6 hours. The solvent was removed in vacuo to give a solid, which
was used in the next
step without further purification (9.2 g crude). MS (EST) m / e (M+H+): 383.
Step 4
Br Ail \
Br
N,
N PPA 7 N
Br .11r".. HO Br HHO
A mixture of product from step 3 (9.2 g) in PPA was heated to 80 C for 2
hours.
After cooling to RI, the mixture was poured into ice water. The resulting
solution was extracted
with DCM. The combined organic phases were washed with brine, dried over
Na2SO4, and
concentrated in vacuo. The residue was purified by column chromatography to
give the desired
indole (4.8 g). MS (EST) m / e (M+H+): 368.
Step 5
Br
* Br select F Br
s-
111 Br
HO DIVISO,CHaCN N
HO
To a mixture of the indole from step 4(6 g, 16.3 mmol) in DMSO/CH3CN (1:1,
24 mL) was added SELECTFLUOle (5.8 g, 16.3 mmol) in portion at RT. The mixture
was stirred
for an additional 1 hour at RT, and the mixture was purified by HPLC to give a
solid (1.0 g). MS
(EST) m / e (M+H+):386.
Step 6
Br
Br
\ 41, Br GN2Br2 =
Br
N k2CO3,DMF
HO 80 C 0
A mixture of the compound from step 5 (650 mg, 1.63 mmol), CH2Br2 (1.5 g,
8.62 mmol) and K2CO3 (1.2 g, 8.7 mmol) in DMF (32.5 mL) was stirred for 5
hours at 80 C.
Then the mixture was evaporated in vacuo. The residue was diluted with EA and
water. The
organic layer was separated, dried over Na2SO4 and concentrated in vacuo to
give a solid, which
was directly used to next step without further purification (610 mg). MS (EST)
m / e
(M+H):396.
Step 7
- 147 -
:A 02756172 2011-09-21
WO 2010/111483
PCT/US2010/028653
Br
Pin8
=N\ Br PctOpPf/C12 =\
N BPin
KOAc
\--0 \--0
To a solution of the product from step 6 (1 mmol) in 1,4-dioxane was added bis
pinacol borate (1.1 mmol) and Pd(dppf)C12 (0.02 mmol) and KOAc (2 mmol). The
reaction
mixture was stirred under N2 and heated to 110 C for 3 hours. After that, the
solvent was
removed under vacuum, and the residue was purified by column chromatography to
afford the
product. MS (ESI) m / e (M+11+):492.
Step 8
PinB C44
apin Pd(dppf)C12 NN \ , N
Na2COs Eic' N
Boc
A suspension of the boronate from above (2 mmol), tert-butyl 2-(2-bromo-1H-
imidazol-5-y1) pyrrolidine-l-carboxylate (2.4 mmol), Pd(dppf) C12 (200 mg),
Na2CO3 (3 mmol)
and in THF/H20 (10:1, 33 mL) was refluxed at 75 C overnight under N2
protection. The
mixture was cooled and filtered, and the filtrate was washed with water (50
mL) and extracted
with Et0Ac (100 mL), washed with brine and dried over anhydrous sodium
sulfate. After
concentrated in vacua, the residue was purified by column chromatography to
afford the desired
compound. MS (ESI) m / e (M+1-14):710.
Step 9
(-Nisi
N HO c\r_6:,N
Bac 410 \ =
,N-D NH H
Boc H0
The protected proline from above (1.3 mmol) was added to HC1/CH30H (10 mL,
3M). The mixture was stirred at RT for 2-3 hours before the mixture was
concentrated to give
the crude product, which was used in the next step without further
purification. MS (ESI) m / e
(M-1-1-0:510
Step 10
NH H
N
0 0
- 148-
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
To a mixture of the crude product from step 9 (1.0 mmol), (5)-2-
(methoxycarbonylamino)-3-methylbutanoie acid (2.0 mmol) and DIPEA (8 mmol) in
CH3CN
(10 mL) was added BOP (2.2 mmol). The resulting mixture was stirred at RT.
After LCMS
showed the starting material to be consumed, the mixture was filtered, and the
filtrate was
purified by HPLC to give the desired compound as a white solid. MS (ESI) m / e
(M+11 ):825.
1H NMR (Me0D): 6 7.83 - 7.85 (in, 3 H), 7.72 (s, 1 H), 7.53 (s, 2 H), 7.46 -
7.48 (m, 1 H), 7.42
(s, 1 H), 5.92 (s, 2 H), 5.20 - 5.22 (m, 2 H), 4.20 - 4.23 (m, 2 H), 4.06 -
4.09 (m, 2 H), 3.86 - 3.88
(m, 2 H), 3.61 (s, 6 H), 2.50 - 2.52 (m, 2 H), 1.96 - 2.20 (m, 8 H), 0.90 -
0.98 (m, 12 H).
Example 190: (2S)-1-1(2R)-2-(dimethylamino)-2-phenylacetyll-N-(2-15-NOS)-1-
1(2R)-2-
(dimethylamina)-2-phenylacetplipwrolidin-2-vlicarbonyl)aminol-1,3-benzaxazal-2-
1,11-1H-
indol-5-v1)pyrrolidine-2-earboxamide
TE,1 N NH 110
Me2N
NMB2
Step .1
NH
CNN BrCtl
To a solution of imidazole (13.6 g, 0.2 mol) in IL of DCM was added BrCN (7.4
g, 66 mmol), and the mixture was heated at reflux for 30 minutes. The mixture
was cooled to
RI', and the white precipitate removed by filtration, and the filtrate
concentrated to 100 mL then
cooled to 0 C for 2 days. The crystallized solid was filtered and washed with
cold DCM, then
dried in vacua to give the desired product (8.8 g) as a white solid.
Step 2
NH
NNNN
+ HO .0
heat 02N --
401 INH2
H2N NO2 0
A solution containing the product from step 1 (8.36g, 54.2 mmol) and 2-amino-4-
nitrophenol (8.74g, 54.2 mmol) in anhydrous THE (200 mL) was allowed to reflux
under N2 for
14 hours. The mixture was cooled to RI', filtered, and the precipitate was
washed with THF
(cold) then dried in vacua, to afford the desired product (9.0g), as a yellow
solid. MS (ESI) m/e
(M+H4): 180. 1H NMR (DMSO) 6: 7.85 - 7.96(m, 3 H), 7.52(d, J = 8.8 Hz, 1H).
- 149 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Step 3
02N 0
t-BuNo2 02N =
N
N
CuBr2
To a suspension of the product from step 2 (3.58 g, 20 mmol) in acetonitrile
(300
mL) was added CuBr2 (8.96 g, 40 mmol). The solution became dark green and t-
butyl nitrite
(4.12 g, 40 mmol) was added RT over 5 minutes, whereupon the mixture heated at
45 C for 2
hours. The reaction mixture was poured into water (800 mL) and DCM (800 mL),
and the
phases were separated. The aqueous phase was extracted with DCM (3 X 800 mL),
dried with
Na2SO4 and evaporated to afford the crude product. Purification by column
chromatography
afforded the desired product. MS (ESI) na/e (M+H+): 243/245. 111 NMR (DMSO) 8:
8.71 (s, 1
H), 8.42(d, J = 9.2 Hz, 111), 8.10 (d, J = 9.2 Hz, 111).
Step 4
Pd(11)
02N N (Boc)2N __________________ 0
ij lip
(Boc),N ,
0 N N N
Boo NO2
Boo
The mixture of compound from step 3 above (603 mg, 2.5 mmol), the indole
boronic acid from Example 42 (1.0 g, 2.75mmol), Pd(dppl)C12(183 mg, 0.25
mmol), Na2CO3
(530 mg, 5.0 mmol) in 5 mL dioxane4H20 (5:1) was heated to reflux under N2
atmosphere
overnight. When reaction was complete, the mixture was poured into water and
extracted with
DCM. The organic phase was dried over Na2SO4 and concentrated, and the residue
was purified
to give compound the desired product. MS (ESI) m/e (M+H ): 596.
Step 5
03002N is
1. Pd(C) / H2 H2N
N N 1111111"1
N.-,2 2 HHON N NH2
Boc
The product from step 4 (596 mg, 1.0 mmol) was dissolved in Et0Ac and treated
with Pd/C (100 mg, 20%). Then, the mixture was stirred at RT overnight under
H2 atmosphere.
When the reaction was complete, the Pd/C was filtered off, and the resulting
solution was
concentrated to give the crude product MS (ESI) rn/e (M+H+): 565. This
material was covered
with 5 mL of 3M FICI, and the mixture was stirred at RT for 2 hours.
Evaporation of the solvent
afforded the desired product, which was used directly without further
purification. MS (ESI) m/e
(MAO 265.
- 150 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Step 6
H2N \ PyBOP = (Nil/A \ 010
N N NH2 Ai, N
me:f---0 0 WI 1,1
:OH
NH Is
0
NMe2
Me2N
The compound was coupled using the procedure similar to that which was
described in Example 40 starting from 265 mg (1.0 mmol) of the product from
step 5. IH NMR
(Me0D) 8: 8.12(s, 1H), 7.97(d, J=2Hz, 1H), 7.30 - 7.70 (m, 15H), 5.30 - 5.35
(m, 2H), 4.51 -
4.60 (m, 2H), 3.85 - 3.95(m, 2H), 3.15 - 3.25(m, 2H), 3.06(s, 3H), 2.54(s,
6H), 1.80 - 2.30(m,
8H). MS (ESI) m / e (M+H+): 781.
Example 191: 1-1(2R)-2-(diethylamino)-2-phenylacetyll-N-f244-(5-1(2S)-1-112R)-
2-
(diethvlamin0-2-phenvlacetyllpvrrolidin-2-vll-1,3,4-oxadiazol-2-v1)Pheavl1-1H-
indo1-5-vli-L-
prolinarnide
NN
0 up
ift
Step 1
0 o H
-N
NHNI-12 + Ce OH 0 ri
Cbz EtsN tirr
To a solution of N-Cbz-L-Pro (14.9 g, 0.06 mol) and TEA (8.08 g, 0.08 mol) in
100 mL of DCM added dropwise isopropyl chloroformate (8.05 g, 0.066 mol) at 0
C. After
addition, the solution was continued to stir for 1 hour before the hydrazide
(13.0 g, 0.05 mol)
was added, and the mixture was continued to stir for another 1 hour. The
solvent was evaporated
in vacuo, and the residue was recrystallized from Et0H to give a white solid
(22.1 g). IH NMR
(DMSO) 5: 10.47 (s, 114), 10.03 (s, 1H), 7.86 (d, J = 8.0 Hz, 2H), 7.62 (d, J
= 8.0 Hz, 2H), 7.31 -
7.61 (m, 5H), 4.91 - 5.14 (m, 2 H), 4.26 - 4.35 (m, 1 H), 3.30 - 3.4 (m, 2 H),
1.95 - 2.19 (s, 4
H). MS (ESI) m/e (M+H+): 494.
Step 2
F N-N
I
110
N(
IZ D
PPh;EAC2C161 So
- 151 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
To a solution of the product from step 1 (2.1 g, 4.26 mmol), DIPEA (2.3 mL,
17.7
mmol) and PPh3 (1.71 g, 6.5 mmol) in 20 mL of MeCN was added hexchloroethane
(1.41 g, 5.97
mmol), and the mixture was stirred at RT for 1.5 hours. The solvent was
evaporated, and the
residue was purified by chromatography to give a white solid (1.75 g). MS
(EST) mie
494.
Step 3
40 0>-J Bac ,
Pc1(dppt)C1 B c
Cbz Na2CO3 Bab
1 Bob
Cbz
A mixture of the product from step 2 above (494 mg, 1.0 rnmol), indole boronic
acid from Example 42 (377 mg, 1.0 mmol), Pd(dpp0C12 (73 mg, 0.10 mmol), Na2CO3
(318 mg,
3.0 mmol), THE (25 mL) and 1420 (5 mL) was refluxed under N2 overnight. The
mixture was
poured into water and extracted with CH2C12. The organic phase was combined,
dried over
Na2SO4 and filtered to give the desired compound, which was used directly in
the next step. MS
(EST) ink (M+14+): 680.
Step 4
N-
(Bc)c)2N *j1-1`i ciaz Example 99 0 0 1 =
C11"-C--1 H
t...) steps 4-7
i3oc
0 N
Following the procedure described in Example 99, steps 4-7, the oxadiazole
from
step 3 above was converted to the desired product. 1H NMR (Me0D) 6: 8.09 (d, J
- 8.8 Hz, 2H),
7.99 (d, .1- 8.8 Hz, 2H), 7.83 (s, 1H), 7.66 - 7.68 (m, 4H), 7.55 - 7.58 (m,
611), 7.38 - 7.40 (m,
Hi), 7.22 - 7.24 (m, 111), 6.98 (s, 111), 5.39 (s, 111), 5.37 - 5.39 (m, 2H),
4.52 - 4.54 (m, 114),
4.12 - 4.14 (m, IH), 3.94 - 3.96 (m, 1H), 3.10 - 3.41 (m, 811), 2.72 - 2.76
(m, 2H), 1.84 - 2.24
(m, 8H), 1.34 - 1.41 (m, 6H), 1.16 - 1.19 (m, 614). MS (ESI) mic (M+H+): 821.
Example 192: Methyl f(2S)-14(2S)-2-15-14-(5-124(2S)-1-1(2S)-24(methoxy-
carbony0amth01-
3-methylbutanoyilpyrrolidin-2-yll-1H-imidazol-5-y11-1-benzofuran-2-yhphenyll-
111-imidazol-
2-ylipyrrolidin-1-yll-3-methyl-1-oxobutan-2-yllearbamate
õ
crc
=
0
0
- 152 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Step 1
Oxaly$ Chloride QThcH
BaC DMSCVNEt3
Bac t,
A 2 L, 3-necked round bottomed flask equipped with an overhead stir and a N2
inlet was charged with a solution of oxalyl chloride (130 mL, 0.26 mol) in DCM
(250 mL). The
solution was cooled to -78 C, and a solution of DMSO (20 mL, 0.28 mol) in DCM
(30 mL) was
added dropwise. After 30 minutes, a solution of (S)-N-Boc-prolinol (40 g, 0.20
mol) in DCM
(200 mL) was added dropwise. After 30 minutes, TEA (140 mL, 1.00 mol) was
added to the
solution, and the flask was transferred to an ice/water bath and stirred for
another 30 minutes.
The reaction mixture was diluted with DCM (200 mL) and washed successively
with H20, 1M
HC1, saturated NaHCO3, and brine. The DCM layer was dried over Na2SO4,
filtered, and
concentrated to afford crude (S)-2-formyl-pyrrolidine- 1-carboxylic acid tert-
butyl ester (40 g) as
an oil, which was used without further purification.
Step 2
0
HJ
11 4'
NH,, H20 NBoo Boo
Glyoxal (2.0 mL of 40% in water) was added dropwise over 11 minutes to a
methanol solution of NH4OH (32 mL) and (S)-Boc-prolinal (8.564 g, 42.98 mmol)
and stirred at
ambient temperature for 19 hours. The volatile components were removed in
vacua, and the
residue was purified by a flash silica gel chromatography (Et0Ac) followed by
a
recrystallization (Et0Ac) to provide the desired compound as a white fluffy
solid (4.43 g). 1H
NMR (DMSO) 5: 11.68, 11.59 (br s, 1H), 6.94 (s, 1H), 6.76 (s, 1H), 4.76 (m,
1H), 3.48 (m, 1H),
3.35-3.29 (m, 11-1), 2.23-1.73 (m, 4H), 1.39/1.15 (s, 9H). MS (ESI) m/e
(M+11+): 238.
Step 3
Br
NS
NBoc NBoc N Br
To a suspension of the compound from step 2 (140 g, 0.59 mol) in THF (2000 ml)
was added NBS (200 g, 1.1 mol). The mixture was stirred at RT under N2
protection overnight
before the solvent was removed, and the residue was purified by chromatography
on silica gel to
give 230 g of the desired dibromo compound. MS (ESI) m/e (M+H+): 396.
Step 4
- 153 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Br
Nr\S'
Na2S03
NBoc \.--r6oc
To a suspension of compound from step 3 (230 g, 0.58 mol) in Et0H/H20 (3000
ml) was added Na2S03 (733 g, 5.8 mol). The resulting mixture was stirred under
reflux
overnight. After cooling to RT, the mixture was extracted by DCM and
concentrated under
vacuum. The resulting residue was purified by chromatography on silica gel to
give the desired
bromo imidazole target. MS (ESI) m/e (M+11+): 317.
Step 5
COOEt NBS/C04= COOEt
Br * y Br
Br
To a stirred solution of ethyl 4-bromopheny1acetate (50 g, 205.8 mmol) in CC14
(500 mL) was added NBS (38 g, 214.7 mmol), then 48% aqueous HBr (4 drops).
After the
addition, the solution was stirred overnight at 80 C under argon. Then the
reaction was cooled
to RT, filtered, and concentrated. The resulting oil was directly used the
next step.
Step 6
0
B 0
Br H Cs2CO2/DMFSr 10/ \
0 reflux IW? 411111"- OH
Br
To a solution of the compound from step 5 (2 g, 6.2 mmol) in DMF (20 mL) was
added 5-bromosalicylaldehyde (1.21 g, 6.0 mmol) and Cs2CO3 (2 g, 12.3 mmol)
under N2
protection. The resulting suspension was stirred for 5 hours at 160 C, then
cooled and treated
with water. The resulting precipitate was filtered, and the filtrate cake was
dried in vacuo to give
the desired compound, which was used directly in next step.
Step 7
pd(dppi)cliKomid.ne
er
40= LBBçi I;'
0 Br
416, p
--- 0 lir B\c,
A suspension of the product from step 6 above (4.43 g, 12.58 mmol),
bis(pincolato)diboron (8.31 g, 32.72 mmol), AcOK (3.72 g, 37.7 mmol) and
Pd(dppf)C12(921
mg, 1.26 mmol) in dioxane (100 mL) was heated to reflux for 4 hours under N.
The mixture
was concentrated, the residue was partitioned between H20 and DCM, and the
aqueous phase
was extracted with DCM. The combined organic layers were washed with brine,
dried over
Na2SO4, concentrated. The residue was purified by chromatography on silica gel
to afford the
desired compound (5 g).
- 154-
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Step 8
Br
foLõ. PoKtippl)C1,! NBoc
= 0
6 3\ NaGO 0
0-=IN
=
H Boctj,-)
A suspension of the product from step 4 (5 mmol), the boronate ester from step
7
(2 mmol), Pd(dppf)Cl2 (146 mg, 0.2 mmol), and Na2CO3 (636 mg, 6 mmol) were
refluxed in
THF/H20 (10:1, 33 rut) overnight under N2 protection. The mixture was cooled
and filtered,
and the filtrate was washed with water (50 mL) then extracted with Et0Ac (100
mL), washed
with brine and dried over anhydrous sodium sulfate. The solution was
concentrated and the
resulting residue was purified by column chromatography (PE / EA = 8:1¨>5:1)
to afford the
desired compound. MS (ESI) rniz (M+H)+:641).
Step 9
H011Nief01 Vi =
/N3
= 11)N.),,,\ 0
HN--)
80cN--/
The product from step 8 (1.3 mmol) was added into 3M HC1/CH3OH (20 mL) and
the mixture was stirred at RT for 2 to 3 hours. The mixture was concentrated,
and the crude
product was used directly in the next step without further purification. MS
(ES!) m/z (M H)+:
441.
Step 10
\ 41
\ ))y, _______
HNie Bor, C-r1N.N
N H 0 0
L-14H H = 0
HN oo
HN 0
To a mixture of the product from step 9 (1 mmol), N-Moc-L-valine (2.1 mmol)
and DIPEA (0.4 mL) in DME (3 mL) was added BOP reagent (2.2 mmol). The
resulting mixture
was stirred at RT for 16 hours. The solution was subjected directly to RPLC to
afford the
desired compound. NMR (Me0D) 5: 7.7-8.1 (m, 10 H), 7.4 (m, 1 H), 5.3 (m, 2
H), 4.3 (m, 2
H), 4.1 (d, J4.81-1z, 2 H), 3.9 (m, 2 H), 3.7 (m, 6 H), 2.6 (d, J4.8 Hz, 2 H),
2.0-2.4 (m, 8 H),
1.3-1.4 (m, 2 H), 0.9-1.0 (m, 12 H). MS (ES!) na/z (M+H)+: 780.
Examples 193-202
Compounds of Examples 193-202 were prepared in a similar manner as described
in Example 192.
- 155 -
:A 02756172 2011-09-21
WO 2010/111483 PCT/US2010/028653
Example Structure .11INMR M+1 Name
-I (2R)-2-(diethylamino)-1-[(25)-2-
(Me0D) 6: 7.2-8.1 (m, 20 (5-{4-[5-(2-{(2S1-1-[(2R)-2-
H), 5.2-5.6 (m, 4 H), 3.9- (diethylamino)-2-pheny1acety1i
193 _.., _02) l .,,.ç- 4.2 (m, 2 H), 3.1 (m, 2 H), 844
pyrrolidin-2-y1}-1H-imidazol-5-
_7 o µ.-- 2.6 (m, 8 H), 1.9-2.5 (m, 8
y1)-1-benzofuran-2-yllpheny1)-
H), 1.0-1.5 (m, 12 H). 1H-imidazol-2-
yl)pyrrolidin-1-
11-2-=hen lethanone
(Me0D) 6: 8.0-8.1 (m, 3 Methyl {(1S)-1-cyelopropy1-2-
H), 7.7-7.9 (in, 6 H), 7.4
(d, 3=2.4Hz, 1 H), 5.3 (d, 2-cyclopropy1-2-
194 3=5.6 Hz, 2 11), 4.1 (m, 2 775
[(rnethoxyearbonyDamino]acetyl)
II), 3.9 (m, 4 H), 3.7 (m, 6 pyrrolidin-2-y1]-1H-imidazol-5-
' 1'0 H), 2.5-2.6 (m, 2 H), 2.1-
y1}-1-benzofuran-2-y1)pheny1l-
2.3 (m, 6 H), 1.1-1.2 (m, 2 1H-imidazol-2-yl}pyrrolidin-1-
H), 0.4-0.6 (in, 9 H). y11-2-
oxoethyl}carbamate
i
Methyl {(1R)-1-cyclopropy1-2-
(Me0D) 6: 7.7-8.1 (in, 9
H), 7 {54445- (2-[(2S)-
1- {(2R)-
2-cyclopropy1-2-
fo_tI,J1)..., (m), 74(m, 2. H),, 16 H), 5 3-
5 4 3,5-4.1 (M, 12
195
Rmethoxycarbonypaminojacetyl)
H), 2.6 (d, J=4.8 Hz, 26 775
2
H), 2.2 (d, J--4.8 Hz, 6
pyrroliclin-2-A-1H-imidazol-5-
-v,
H), 1.1-1.2 (m, 2 H), 0.4-
yl} -1-benzolzuran-2-yl)phenyli-
C 1
1H-imidazol-2-y1}pyrro1idin-1-
0.7 (m, 8 H).
y1)-2-oxoethyl}carbamate
(Me0D) 6: 7.8-7.9 (in, 5
Methyl {(2R)-14(2S)-2-{542-(4-
1-1), 7.1-7.6 (m, 5 H), 5.6-
5.7 (in, 1 H), 5.2 (d, .1
.1.1.,.,\ =4.8 Hz, 1 H), 4.0-4.2 (m,
196 /0-e. 0..c14- 3 H), 3.6-3.8 (m, 8 H),
{24(29-1- {(2R)-2-
[(methoxycarbonyl)amino]-3-
780 methylbutanoyll pyrrolidin-2-yll-
1H-imidazol-5-yl)pheny1)-1-
2.0-2.5 (m, 10 H), 1.6 (d,
benzofuran-5-y1]-1H-imidazol-2-
1. J =4.8 Hz, 1 H), 1.3 (n, 1
H), 0.8-1.1 (m, 11 H), 0.4
yl}pyrrolidin-1-y11-3-methyl-1-
oxobutan-2-y1) carbamate
(m, 26 H).
_
(Me0D), 5 8.10(d, J =4
Hz, 2 H), 8.01(s, 1 H), Methyl {(25)-1-[(2S1-
2-{544-(5-
7.94(s, 1 H), 7.87(d , J =2 (2-{(15)-1-((2,9-2-
Hz, 2 H), 7.84(m, 1 H), {(methoxycarbonyl)amino]-3,3-
0 :
7.73(d, .1 =4 Hz, 1 H), dimethylbutanoyl}
pyrrolidin-2-
")
197
/40 ..1 .--/' 7.44(m, 1 H), 5.25(m, 2 806
y1}-11/-imidazoI-5-y1}-1-
>CL H), 4.33(m, 2 H), 4.16(m,
benzofuran-2-yl)phenyl]-1 H-
e- ? 2 H), 3.89(m, 2 H), imidazol-2-
yl}pyrrolidin-1-y1}-
3.67(s, 6 H), 2.58(m, 2 3,3 -dimethyl-l-
oxobutan-2-
H), 2.20(m, 6 H), 0.97(m, yl}carbamate
18 H)
,
(Me0D) 5: 7.6-8.1 (m, 9
Methyl {(1R)-24(28)-2-{544-(5-
H), 7.3-7.5 (m, 11 H), 7.2 {2-[(2S)-1-{(2R)-2-
e-r (m, 1 II), 5.4-5.5 (m, 2
[(methoxycarbonyl)amino]-2-
phenylaeetyl } pyrrolid in-2-y1]-1 H-
198 j7,:-1::111C0-40-Ctosji H), 5.3 (m, 2 H), 4.1 (d, .1 848
1'
unidazol-4-y1}-1-benzofuran-2-
=4.8 Hz, 2 II), 3.7 (d, .1 0 0
=2.4 Hz, 6 H), 1.9-2.4 (m, yl)pheny1]-1H-imidazol-2-
8 H)
yllpyn-olidin-1-y1]-2-oxo-1-
phenylethyl}carbamate
- 156 -
:A 02756172 2011-09-21
WO 2010/111483 PCT/US2010/028653
Example _ Structure M+1 Name
(Me0D) & 8.08(d, I =4
Hz, 2 H), 8.04(s, 1 H),
7.89(d, 3 H), 7.78(m, 1
H), 7.77 (rn, 2 H),
Methyl {(2S,3R)-1-[(25)-2-(5-{2-
7.42(m,1 H), 5.34(t, 3=4 [4-(2-{(2S)4-[N-
Hz, 2 H), 4.42(d, J 4Hz, (methoxycarbony1)-1,-
199 2 H), 4.10(s, 2 H), 806
alloisoleucyl}pyrrolidin-2-y1}-1 H-
3 .82(m, 2 H), 3.79(m, 3 imidazol-5-
yl)phenyli-1-
H), 3.64(s, 3 H), 2.57(m,
benzoluran-5-y1}-1H-imidazol-2-
I
2 H), 2.20(m, 6 H),
yl)pyrroliclin-l-y1]-3 -methyl-1-
1.88(m, 2 H), 1.48(m, 2 oxopentan-2-
yl}carbarnate
H), 1.32(m, 2 H),
0.97(m, 12 H).
Methyl {(28)-3-hydroxy-14(25)-
2-{544-(5-{2-[(2S)-1-{(25)-3-
(Me0D) 5: 7.6-8.1 (m, 9
hydroxy-2-Rmethoxycarbonyl)
200i-c II) 7.4 (m 6 H) 5.4 (m 2
aoy': 4), 4.6 (M,
, 2 1-1), 3.5-4:1 755
amino]propanoyl}pyrTolidin-2-
(m, 13 H), 2.5-2.7 (m, 6 y1]-1H-imidazol-5-y1}-1-
1-? H), 2.3 (m, 5 1-1). benzofuran-2-yl)pheny11-1 H-
imidazol-2-yl)pyrrolidin-l-y11-1-
oxopropan-2-ylicarbamate
(Me0D), 5 8.04(d, .1 =4
Hz, 2 H), 7.97(s, 1I-I),
7.84(d, 1 =2 Hz, 2 H),
Methyl {(2,5,3R)-3-hydroxy-1-
7.81(m, I H), 7.76(m, 1
[(2,C)-2-(544-(5-{2-[(2,S)-1-
H), 7.64(d, 3=4 Hz, 2 H),
4* TIE
= m 7.38(m, I H),
5.29(m, 2 i {(28,3R)-3-hydroxy-2-
[(methoxycarbonypamino]butano
201 r.c.. .1C-1 H), 4.49(m, 2 H), 4.15(m, I 783
01-4 2 H), 3.97(m, 1 H), yllpyn-olidin-2-y11-1H-
imidazol-
5-y1}-1-benzofuran-2-yl)phenyll-
' 3.92(m, 1 H), 3.66(m, 6
1H-imidazol-2-y1)pyrrolidin-1-
H), 2.63(m, 1 H), 2.60(m,
y11-1-oxobutan-2-yI}carbamate
1 H), 2.54(m, 2 H), 2.16-
2.22(m, 6 H), 1.16 (d, J
=2 Hz, 6 H).
(Me0D), 6 8.05(d, J =4
Hz, 2 H), 7.97(s, 1 H),
7.84(d, 1 H), 7.81(d , 3 =2 Methyl {(26)-14(2,5)-2-
{544-(5-
Hz, 2 H), 7.76(m, 1 H), {2-[(28)-1-{(28)-2-
7.64(d, .1 =4Hz, 2 H),
[(methoxycarbonyl)aminol-4-
7.39(m, 1 H), 5.25(m, 2 methylpentanoyl)
pyrrolidin-2-
202 H), 4.45(m, 2 H), 4.03(m, 806
2 H), 3.84(m, 2 H), benzofuran-2-
yl)pherty1i-11-/-
3.85(m, 2 H), 3.64(s, 6 irnidazol-2-
yl}pyrrolidin-1-y1]-4-
H), 2.55(m, 2 H), 2.22(m, methyl-1-oxopentan-2-
6 H), 1.70(m, 2 H), yl}carbamate
1.51(m, 4 H), 0.98(m, 12
H)
- 157
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Example 203: Methyl f(2S)-14(2S)-2-1543-fluoro-4-(5424(2S)-1-(0S)-2-
ffmethoxycarbanyl)aminol-3-methylbutanoylipyrrolidin-2-y11-1H-imidazol-5-v11-1-
benzofuran-2-yl)phenv11-111-imidazol-2-yliPyrroliditi-1-yll-3-methyl-1-
oxobutan-2-yll
carbarnate
\ \c, 4, tirl)
N F 0
N\ 0
Step]
Br WA
Br 130$1
Ali
+ 130-PrO)3 _________________________________
WI 0 4111111" 0 OH
To a solution of 5-bromobenzofuran (3.9 g, 20 mmol) in dry THF (30 mL) cooled
to -78 C under N2-protected LDA (prepared from n-BuLi and iPr2NH in THF (-30
mmol)) was
slowly added. The mixture was stirred at the same temperature for 30 minutes,
then
triisopropylborate (5.64 g, 30 mmol) was added to the mixture. The mixture was
allowed to
warm to RT and stirred for 2 hours. The mixture was then quenched with IN HC1
to pH=3 and
extracted with Et0Ac. The combined organic phases were combined, dried and
filtered. The
filtrate was concentrated to afford the desired product (4.3 g). MS (ESI) m/e
(M+H+): 241.
Step 2
Br 1110 \,OH + Br Pd(cIpP Brf)C12 \
IF Br
I ig"
A suspension of the boronic acid from step 1 (1.44 mg, 6.0 mmol), 2-fluoro-4-
iodobromobenzene (1.8 g, 6.0 mmol), Pd(dppt)C12 (600 mg), Na2CO3 (954 mg, 9.0
mmol) and in
THF/H20 (9:1, 100 mL) was refluxed at 75 C overnight under N2 protection. The
mixture was
cooled and filtered. The filtrate was washed with water (150 mL) and extracted
with Et0Ac
(200 mL), washed with brine and dried over anhydrous sodium sulfate. The
solution was
evaporated and the residue was purified by column chromatography (PE! EA = 8:1-
45:1) to
afford the desired compound. MS (ESI) m/e (M+H+): 370.
Step 3
Br
.` Br io 0 0 0
Pd(cippOCl2
- 158 -
:A 02756172 2011 09 21
WO 2010/111483 PCT/US2010/028653
To a solution of the product from step 2 (1.85 g, 5 mmol),
bis(pinaeolato)diboron
(2.54 g, 10 mmol) and Pd(dppf)C12 (80 mg) and KOAc (0.98 g, 10 mmol) were
dissolved in 1,4-
dioxane (30 mL), and the reaction mixture was heated at 110 C for 16 hours.
The solvent was
evaporated, and the residue was purified by column chromatography with silica
gel elution with
PE to afford the desired product as white solid (1.95 g). MS (ESI) m/e (M+H+):
465.
Step 4
0_
PDC1
0 B [4,0 2
0 W `c> Pd(dpNa2CO3 Boc 0 \ Boc
A suspension of the bromoimidazole from Example 192 (5 mmol), the boronate
ester from step 3 (2 mmol), Pd(dppf)Cl2 (146 mg, 0.2 mmol) and Na2CO3 (636 mg,
6 mmol) was
refluxed in THF/H20 (10:1, 33 mL) overnight under N2 protection. The mixture
was cooled and
filtered, and the filtrate was washed with water (50 mL) and extracted with
Et0Ac (100 mL),
washed with brine and dried over anhydrous sodium sulfate. The solution was
concentrated, and
the resulting residue was purified by column chromatography (PE / Et0Ac ----
8:1) to afford the
desired compound. MS (ESI) m/e (MAO 683.
Step 5
FICl/Me011
NH H \
pl dip \ 0,
Boc LW 0 \ Bc'c 0 \ NH
The product from step 4 (682 mg, 1.0 mmol) was treated with 3M HC1/CH3OH
(10 mL) and the mixture was stirred at RT for 3 hours. The reaction mixture
was concentrated,
and the crude product was used directly in the next step without further
purification. MS (ESI)
'tile (M+H+): 483.
Step 6
N---
\ NH \
C-F4i 411bN
0 F 0
0
0
1-11\11/
To a mixture of the product from step 5 (482 mg, 1.0 mmol), N-Moc-L-valine
(2.1
mmol) and DIPEA (0.4 mL) in DMF (3 mL) was added BOP reagent (977 mg, 2.2
mmol). The
resulting mixture was stirred at RT for 16 hours. The solution was subjected
directly to RPLC to
afford the desired compound as white solid (40 mg). Ili NMR (Me0D) 6: 7.99
(s,1 H), 7.89-
7.80 (m, 5 H), 7.72-7.67 (in, 2 H), 7.47 (s, 1 H), 5.27-5.22 (m, 2 H), 4.22
(d, 2 H), 4.09 (d, 2 H),
- 159 -
:A 02756172 2011-09-21
WO 2010/111483
PCT/US2010/028653
3.89-3.84 (m, 2 H), 3.64 (s, 6 H), 2.55-2.02 (m, 10 H), 0.92 (d, 6 H), 0.88
(d, 6 H). MS (ESI)
mie (M+H+): 797.
Examples 204-212
Compounds of Examples 204-212 were prepared in a similar manner as described
in Example 203.
Example _ Structure M+1 Name
F
/ \ j \ Methyl {(1R)-2-[(25)-
2-{543-fluoro-4-(5-12-
SI \ 4. /NJ 1 1:\. 866 [(2S)-1- {(2R)-2-
Rtnethoxycarbonyl)arninol-2-
204
0
0 phenylacetyl}
pyrrolidin-2-y1j-1H-imidazol-5-
= --/
yl} -1-benzofuran-2-yl)pheny11-1H-imidazol-
= \
/ O= ill 2-yl}pyrro1idin-1-ylj-2-
oxo- I _
0,0
1 phenylethyl}carbamate
N F 0 c Methyl { (28)-
14(25)-2- {543-fluoro-4-(5- {2-
l'I -)1' 'D
[(2S)-1- { (2,S)-2-[(methoxycarbonyl)amino}-3-
205 A 797 methylbutanoyl}pyrrolidin-
2-y11-1H- 1
10-- \..' 0__ N
irnidazol-5-yll -1-benzofuran-2-yl)phenyIj-
0
--"A7 -.)---
1H-imidazol-2-y1) pyrrolidin-1-yI]-3-methyl-
0 \ I -oxobutan-2-y1) carbamate
/1 \ Methyl {(2S)-1-[(25)-
2-{542-(256-difluoro-4-
\ Ilk 'N {24(25)-1- {(25)-2-
Rmethoxycarbonyl)
amino1-3 -methylbutanoyl) pyrrolidin-2-y11-
206 F :7:D 816
P--i 1H-imidazol-5-y1}pheny1)- I
-benzoftwan-5-
, o t, y11-1H-imidazol-2-y1)
pyrrolidin- I -y1]-3-
e- \ methyl-l-oxobutan-2-y1}carbamate
_
...
0.4 = Methyl { (1R)-2-
[(2S)-2-{543-methoxy-4-(5-
0 N 0' 1.'
= N1) {2-1(2S)-1-{(2R)-2-
[(methoxycarbonyl)
207 0 878
amino1-2-phenylacetyl} pyffolidin-2-yI]-1H-
= N-../
/ 0 it imidazol-5-y1) - l -
benzofuran-2-yl)phenyll-
'N 1H-imidazol-2-y1) pyrrolidin-1
..
-yll -2-oxo-1-
IP pbenylethyl)
earbamate
Methyl {(25)-1-[(25)-2- {542-(2-ehloro-4- {2-
i 0 m
eth{ylbutan oyl) pyrrolidin-2 -y11-1H-
208 r 814
[(2S)-1-(2S)-2-(methoxycarbonyl)aminol-3-
imidazol-4-y1}phenyI)-1-benzofuran-5-ylj-
IH-imidazol-2-yl}pyrrolidin-l-y11-3-methyl-
I -oxobutan-2-y1) earbamate
Methyl {(1R)-24(2S)-2- {443 -cyano-4-(5- {2-
[(2S)-1- {(2R)-2-Kmethoxycarbonyparnin61-2-
209 ra . 60
= 0_
J1 873 phenylacetyl}pmolidin-2-y1]-1H-
imidazol-5-
, i 0 is 411
yl} -1-benzofuran-2-yl)pheny11-1H-imidazol-
2-y1) pyrrolidin-l-y1]-2-oxo-1-
phenylethyl) carbarnate
_
Methyl {(28)-1-[(251l-2-{5-[2-(4-{2-[(28)-1-
{(2S)-2-[(tnethoxycarbonyl)arnino]-3-
methylbutanoyl}pyffolidin-2-yIHH-
0 --
210
0 imidazol-4-y1} -2-methylpheny1)-1-22--c benzofuran-5-y1]-1H-
imidazol-2-
yllpyrrolidin-1-34-3-methyl-l-oxobutan-2-
1 yl} carbamate
- 160 -
:A 02756172 2011-09-21
WO 2010/111483
PCT/US2010/028653
Example ¨ Structure M+1 Name
c..74 Methyl K15)-24(2,9)-245- (3-fluoro-445-(2-
{(2,9-14(25)-24(methoxycarbonypaminol-2-
N 0 N 40 (tetrahydro-2H-pyran-4-ypacetApyffolidin-
211
hõ..N
2-Y1} -1H-imidazol-5-y1)-1-benzofiran-2-
yliphenyl -1H-imidazol-2 -yl)pyrrolidin-1-yll-
0 0),N 0, 2-oxo-1-(tetrahydro-2H-
pyran-4-
yl)ethylicarbarnate
Methyl { (25)-1-[(25)-2- {443-fluoro-4-(5- {2-
[(25)-1- (2R)-24(methoxycarbonypamino}-2
212 :(0 832 phenylacetyl)pyrrolidin-
2-y1]-1H-imidazol-5-
?-< y1}-1-benzofuran-2-
yl)phenyli-IH-imidazol-
2-yllpyrrolidin-1-y1}-3-methyl-1-oxobutan-2-
y11 carbamate
Example 213: Methyl f(110-2-1-(2S)-2-1544-(5-12-1(2S)-142R)-2 [(methoxy-
earbanyl)aminol-2-phenylacetpllpyrrolidin-2-y11-1H-hnidazol-5-01-1,3-
benzoxazol-2-
ybphenyll-1H-imidazol-2-yllpyrrolidin-1-01-2-oxo-1-phenylethylkarbamate
\
,N
N H
0 H
0 N
NHMoc
MocHN
Step
0 it
Br 4 Br NI-12
PPA Br
Br
At,
1.11)
4-Bromobenzoic acid (20 g, 0.1 mol) and 2-amino-4-bromophenol (18.8 g, 0.1
mol) were added into polyphosphoric acid (250 mL), and the mixture was stirred
at 140 C for 90
minutes. After cooling in an ice-bath, the reaction mixture was diluted with
water (4000 mL)
and neutralized with NaOH. The resulting solid was filtered off and dried to
afford the desired
benzoxazole. MS (EST) m/e (M+111): 354.
Step 2
0
Br N8 ________ 0 * ¨0 0
r _____________ 0
WI 0 Pd(5)&10012
A suspension of the product from step 1 above (10.6 g, 30 mmol),
bis(pinacolato)diboron (30.3 g, 120 mmol), KOAc (7.6 g, 78 mmol) and
Pd(dppf)C12 (1.1 g, 1.5
mmol) in dioxane (300 ml) was stirred at 100 C under N2 protection overnight.
The reaction
mixture was cooled and concentrated, then chromatographed on silica gel gave
the product
compound. MS (ES1) mie (M+H+): 366.
- 161 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
Step 3
N 1
0 du N\ r Pd(tippf)C12 NBoc so N
¨
\ N
1. 0 NBoc nia2CO3 0
N BocN--)
A suspension of the product from step 2 (1.2 g, 2.6 mmol), bromoimidazole from
Example 192 (2 g, 6.3 mmol), Na2CO3 (1.3 g, 12 mmol) and Pd(dpp0C12 (220 mg,
0.3 mmol) in
THF/H20 (36 ml) was stirred at 100 C under N2 protection overnight. The
reaction mixture was
concentrated and purified by chromatography on silica gel to give the desired
compound. MS
(ESI) mie (M+1-14): 666.
Step 4
N\ He!! Me01.2 =
-II so ¨
\ N
WI 0 0 __
BocNi> NHN
A solution of the product from step 3 (400 mg, 0.6 mmol) in HCl/Me0H (20 ml)
was stirred at ambient temperature for 3 hours, then concentrated and dried
under high vacuum
to give to desired product. MS (ESI) rn/e (M+1{4): 466.
Step 5
/ 1Y/
1
H Vi .µ)
L-1.1 N N>.¨(1)¨(3) BOP
H 110 0
_______________________________________________ NHMoc
HN¨J MocHN
To a mixture of the product from step 4 (233 mg, 0.5 mmol), N-Moc-D-Phg (1.1
mmol) and DIPEA (0.2 mL) in DMF (3 mL) was added BOP reagent (488 mg, 1.1
mmol). The
resulting mixture was stirred at RT for 16 hours before the solution was
subjected directly to
RPLC to afford the desired compound. 1HNMR (Me0D) 8: 8.4 (d, J=8.4 Hz, 2 H),
8.2 (s, 1 H),
8.0 (m, 3 H), 7.9 (m, 3 H.), 7.5-7.4 (m, 10 H), 5.5 (s, 2 H), 5.3 (m, 2 H),
4.1-4.0 (m, 2 H), 3.6 (d,
J=2.8 Hz, 6 H), 3.3 (m, 1 Fl), 3.3-3.1 (m, 1 H), 2.5-2.3 (m, 2 H), 2.2-2.1 (m,
4 H), 2.0 (m, 2 H).
MS (ESI) m / e (M-f-H+): 780.
Examples 214-215
Compounds of Examples 214-215 were prepared in a similar manner as described
in Example 213.
- 162-
:A 02756172 2011-09-21
WO 2010/111483 PCT/US2010/028653
Example Structure 1H NMR M+1 Name
,
Methyl {(25)-1-[(2S)-2-
(Me0D) 8: 8.4 (d, J=8.4
Hz, 2 H), 8.1 (s, 1 H), 7.9
N 8 2-[(methoxyearbonyl)
(m, 3 H), 7. (m, 2 1-1),
Q¨S, 1 fik N amino]
-3-
0
ip H), 4.2 (m, 2 /Ns, ..,, 7.7 (m, 1 H), 5.3 (m, 2
214
methylbutanoyl)
1.) 14), 4.1-4.0
0 N 848
pyn-olidin-2-y1]-1 H-
/ o (m, 2 H), 3.9-3.8 (m, 2
H), 3.6 (s, 2 H), 2.6 (in, 2
imidazol-5-yl}pheny1)-
1,3-benzoxazol-5-yli-1H-
0'? H), 2.3 (m, 2 H), 2.2 (m,
imidazol-2-yl)pyn-olidin-
4 H), 2.0 (m, 2 H), 0.9
1-y1j-3-methyl-I-
(m, 12 H).
oxobutan-2-yl}carbamate
¨
(Me0D) 8: 8.4 (dõ/=6.8 '
Methyl {(1 R)-1 -
Hz, 2 H), 8.2-8.1 (s, 1
cyclopropy1-2-[(25)-2-{5-
N
H), 8.0 (m, 3 H), 7.9-7.8 [4-(5-{2-[(2,5)-1-{(2R)-2-
r.,4 1
= I* ")_,40-..._(-14 (in,
3 H), 5.3 (m, 2 H), cyclopropy1-2-
0 N-1)...,\
4.1-4.0 (m, 2 H), 4.0 (m, [(methoxyearbonyl)
215 10.-iN
.3,1--/ 2 H), 3.9-3.8 (m, 2 H),
776 amino]acetyl) pyrrolidin-
3.7 (m, 6 H), 2.6 (m, 2
2-y1]-1H-imidazol-5-y1) -
..'N
H), 2.4-2.1 (m, 6 H), 1.3-
1,3-benzoxazol-2-
\ 1.1 (m, 2 H), 0.7-0.6 (m,
yl)pheny11-1H-imidazol-
3 H), 0.6-0.5 (m, 3 H), 2-yl}pyrroliclin-
1-y11-2-
0.4 (m, 2 H). oxoethylicarbamate
Examples 216-227
Compounds of Examples 216-227 were prepared in a similar manner as described
in Example 189b (Alternative Procedure),
Example _ Structure 1 M-I-1 Name
41
/
NH
dimethyl (indolo[1,2-
CN 0 0,ro
11H,,, e c]
[1,3]benzoxazine-3,I0-diylb is {1 H-
216 875 imidazole-5,2-diy1(2S)pyrrolidine-
NH
0
2,1-diy1R1R)-2-oxo-l-phenylethane-
M
N
..---
2,1-diy1biscarbamate
--... \ 1r".-"/
N,L.0
N
.
N methyl 1-
{2-
N[(25)-1-{(25)-245-(10_</. 1
N 4 [(2S)-1-{(2S)-2-
Rmethoxyearbonyl)
7:_cao 11 0
amino]-3-methylbutanoylipyrrolidin-
No
0 " 2-y11-1H-imidazol-5-y1) -
6,7-
217821
N1-1
dihydroindolo[1,2-d][1,4]
CH 3 0 l'o cH,
benzoxazepin-3-y1)-1H-imidazol-2-
ylipyrrolidin-1-y1)-3-methyl-l-
CH,
oxobutan-2-Acarbamate
.
rctõ)_.61H,
CH
i 3
methyl j(25)-1-{(25)-2-[5-(3-{2-
CH,roNro
[(25)-1-{(25)-2-Rmethoxycarbonyl)
amino]-3-methylbutanoyllpyrrolidin-
218
D¨C
0-X 2-y11-1H-imidazol-5-y1}-
6,6-
-iir-NH 835 dimethylindolo[1,2-
c][1,3]
N
411i N\ lip \Hehi
benzoxazin-10-y1)-1H-Mndazol-2-
yllpyrrolidin-l-y1}-3-methyl-1-
rn
1 )7
CH, cHõ oxobutan-2-yl]carbaate
- 163 - .
:A 02756172 2011-09-21
WO 2010/111483 PCT/US2010/028653
Example Structure M+1 Name
0 CH5
)--1 methyl [(2 5)-1- 425)-
245412-fluoro-
7z..(13 cr,
10- {21(25)-1- 425)-2-
C7, cH3 0 cH3
Rmethoxycarbonypaminoi-3 ^
0 HNxi,õ
methylbutanoyl}pyrrolidin-2-y1]-1 H-
219 -: 839
17--NH CH,
imiclazol-5 -y1) -6-methylindolo[1,2-
F 0
\
C1[193}benzoxazin-3-y1)-1H-imidazol-
2-Apyrrolidin-1-y1) -3 -methyl-1-
---0 \ N oxobutan-2-ylicarbamate
0E6
r"
.i,c"' \ methyl [(25)-1- 425)-24543- {2-
(7\ [(25)-1-425)-24(methoxycarbonyl)
amino]-3-methylbutanoyl} pyrrolidin-
1 220 N 13 N
804 2-3,1]-1H-imidazol-5-y1) IMO [1,2-
e]quinazolin-10-y1)-1H-imidazol-2-
CH, thl
4
yl]pyrroliclin-l-y1) -3 -methyl- I -
<'Th70 oxebutan-2-yl]earbamate
crõ
1 30 methyl [(2S)-1-{(25)-245-(12-{2-
[(2S)-1-42S)-24(methoxyearbonyl)
N N ......Ahõ
\,0 H
qp \ . N 0
amino]-3-methylbutanoyl}pyrrolidin-
221
N /0 835 2-
y1]-1H-imidazol-5-y1) -7,8-dihydro-
---?-
01
=,t11 ("3
61/-indolo[ I ,2-e][1,5]benzoxazocin-
,0 CH3
HN \
cH3 CH, 3-
y1)-1H-imidazol-2-yl]pyrrolidin- 1 -
\
yl} -3 -methyl-l-oxobutan-2-
0
\
OH2 ylicarbamate .
¨
4 t methyl 425)-1- 425)-24543-
{2-
. H 425)-1 -{(25)-
24(methoxycarbonyl)
011
eSrio N \ N
i amino1-3-methylbutanoyl) pyrrolidin-
222 CH NH
)-- N 820 H 11 2-
y11-1H-imidazol-5-y1) -6-oxo-5,6-
0
dihydroindolo [1,2-c]quinazolin- I 0-
oc,
.;1 cH3Npi y1)-1H-imidazol-2-
ylipyrrolidin-l-
C1-130_ yl} -3-methyl-l-oxobutan-2-
1
.H,
- ylicarbamate
_
methyl [(25)-1-1(25)-245410- {2 -
[(25)-1-425)-24(methoxycarbonyl)
-I\ O N * ./. -#-',
amino1-3 -methylbutanoyl} pyrrolid in-
ir ,,,
0 2-y1]-11/-imidazol-5-
y1) -6-
0
223 7 --c.
./0 CI-1 t.,
883
phenylindolo[1,2-c][1,3}benzoxazin-
0
3-y1)-IH-imidazol-2-yljpyn-ol idin-1-
LA
y1}-3-methyl- I -oxobutan-2-
CH, , yljcarbamate
_
_
methyl 425)-1-425)-24543- {2-
' - - = 4 ri * \,, IN
[(28)-1-{(25)-24(methoxycarbonyl)
amino]-3-methylbutanoyl} pyrrolidin-
AI N
0-cH4 2-y1]-1H-imidazol-5-y1}
-6-
CF-,.._.=0
224 )---='' 0
cH, CH, 818
CH,
methylindolo[1,2-c]quinazolin-10-
CF.6 NH
y1)-1H-imidazoI-2-yl]pyrrolidin-1-
0
0
yl} -3-methyl-I -oxobutan-2-
cii-b yl]carbarnate
- 164 -
:A 02756172 2011-09-21
WO 2010/111483
PCT/US2010/028653
_______________________________________________________________________________
i
Example i Structure M+1 Name
11 H N H
methyl R2S)-1-{(2,5)-215-(10'-{2-
rN1 40 , go, cif,
(2S)-1-{(2S)-2-[(methoxycarbonyl)
1,
\¨
amino1-3-methylbutanoyljpyrrolidin-
225 mr,)--. d 0
CH3 875.1 2-y11-11/-imidazol-5-yl}spiro
[cyclohexane-1,6'-indolo{1,2-
0 CH3
CI [1,31benzoxazin)-3'-y1)-1H-
/
OH, 0 imidazol-2-
ylipyrrolidin-l-y1}-3-
\CH, , methyl-l-oxobutan-2-
yllcarbamate
F F
methyl [(25)-1-{(2S)-245-(1,12-
0-44N \ 41 \ . /N,I,_
difluoro-10-{24(25)-1-{(25)-2-
II
N H [(methoxycarbonyl)amino]-3-
226
__N \--0 0 ' LI
842
methylbutanoyl}pyrrolidin-2-y11-11/-
Ir.,,, CH3
/0
Hl (
imidazol-5-yl}indolo[1,2-
c][1,3]benzoxazin-3-y1)-11/-imidazol-
CH,
CH3 a 0 2-yl]pynolidin-l-y1}-3-
methyl-1-
1
oxobutan-2-yl]carbamate
Clis
N
methyl R2S)-1-{(2S)-245-(12-eyano-
N \ * 11 /1 10- (24(28)-1-
{(25)-2-
0 \-0 Pi "r--
[(methoxyearbonyl)amino]-3-
methylbutanoyl}pyrrolidin-2-y1]-1 H-
227 0 N---/ 832
''' All,
imidazol-5-yl}indolo[1,2-
C] [ 1,3]benzoxazin-3-y1)-1H-imidazol-
4 cl 0c. c' 2-yllpyrrolidin-l-y1}-
3-methyl-1 -
oxobutan-2-yljcarbamate
Example 228 ¨ Measurinz Compound Inhibitory Potency
Measurement of inhibition by compounds was performed using the HCV replicon
system. Several different replicons encoding different HCV genotypes or
mutations were used.
In addition, potency measurements were made using different formats of the
replicon assay,
including different ways of measurements and different plating formats. See
Jan M. Vrolijk et
al., A replicons-based bioassay for the measurement of interferons in patients
with chronic
hepatitis C, 110 J, VIROLOGICAL METHODS 201 (2003); Steven S. Carroll et al.,
Inhibition of
Hepatitis C Virus RNA Replication by T-Modified Nucleoside Analogs, 278(14)3.
BIOLOGICAL
CHEMISTRY 11979 (2003). However, the underlying principles are common to all
of these
determinations, and are outlined below.
Stable neomycin phosphotransferase encoding replicon-harboring cell lines were
used, so all cell lines were maintained under G418 selection prior to the
assay. In some cases,
the cell lines encoded a luciferase:Neor fusion and could be assayed either
directly by
determination of RNA copy number, or indirectly through measurement of the
luciferase
activity.
To initiate an assay, replicon cells were plated in the presence of a dilution
series
of test compound in the absence of G418. Typically, the assays were performed
in a 96-well
- 165 -
:A 02756172 2011-09-21
WO 2010/111483
PCT/US2010/028653
plate format for manual operation, or a 384-well plate in an automated assay.
Replicon cells and
compound were incubated for 24 to 72 hours. At the end of the assay, cells are
washed free of
media and compound and then lysed. Luciferase activity was measured using a
conventional
luciferase assay. EC50 determinations were calculated as a percent of a DMSO
control by fitting
the data to a four parameter fit function.
The activity table below provides representative data illustrating observed
activity
against genotype lb.
Activity Table
Example ECso (nM) Example EC50 (nM)
2 9 105 0.08
6 200 107 0.04
14 10 116 0.065
0.045 119 0.013
19 25 125 0.016
26 0.063 129 0,7
30 26 130 0.05
39 0.24 131 17
40 0.026 137 0.009
41 0.05 138 8.5
42 14 144 0.036
45 0.02 155 0,9
49 0.072 158 0.5
58 0.97 159 0.002
60 0.13 169 0.004
62 0.067 178 317
72 0.17 186 0.015
94 0.006 189a 0.15
95 0.01 189b 0.001
96 0.015 190 0.067
99 0.038 191 0.02
100 0.031 192 0.002
101 0.5 193 0.05
102 8.3 203 0.004
103 5.7 213 0.009
- 166 -
:A 02756172 2011 09 21
WO 2010/111483
PCT/US2010/028653
It will be appreciated that various of the above-discussed and other features
and
functions, or alternatives thereof, may be desirably combined into many other
different systems
or applications. It will also be appreciated that various presently unforeseen
or unanticipated
alternatives, modifications, variations or improvements therein, also intended
to be encompassed
by the following claims, may be subsequently made by those skilled in the art,
- 167 -