Note: Descriptions are shown in the official language in which they were submitted.
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
COMPOUNDS AND METHODS FOR INHIBITION OF RENIN, AND INDICATIONS
THEREFOR
FIELD OF THE INVENTION
[0001] Disclosed are novel compounds and uses thereof. In certain embodiments
disclosed
compounds are renin inhibitors.
SUMMARY OF THE INVENTION
[0002] In certain aspects and embodiments disclosed herein, compounds are
provided, as well as
various salts thereof, formulations thereof, conjugates thereof, derivatives
thereof, forms thereof and
uses thereof. Also contemplated in accordance with the present invention are
methods for the use of
the compounds in treating renin-mediated diseases and conditions. Thus, the
use of compounds for
therapeutic methods involving inhibition of renin are provided. In certain
embodiments, the
compounds are used for therapeutic methods involving inhibition of renin
activity, including
treatment of a variety of indications, including, but not limited to,
hypertension, In some
embodiments, compounds are of Formula I, Formula la or Formula Ib, as
described below.
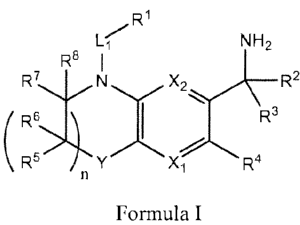
[0003] In a first aspect, compounds having the structure according to the
following Formula I are
provided:
Ri
L~ NH2
Re I
R7 N 2 R2
(::X:4R3
6 Formula I
or a salt, a prodrug, a tautorner or a stereoisomer thereof,
wherein:
XI and X2 are independently -N= or -C(H)=,-
n is0or1;
L1 is a bond, -C(R9R` )-, -C(O)-, -C(R9Rt0) C(R"R`2) , -C(ReR )-C(O)-, -C(0)-
C(R"Rt2)-,
-C(O)-N(R13)-, -N(R'3)-C(O), -S(O)-N(R13) N(R13)-S(O)2, -N(R1)-C(O)-N(Rt3) or
-N(R13)-S(O)z-N(R`) ;
Y is -0- or -C(R14R's) ;
R' is selected from the group consisting of lower alkyl, cycloalkyl,
heterocycloalkyl, aryl and
heteroaryl, wherein lower alkyl is optionally substituted with one or more
substituents R2`,
1
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
and wherein cycloalkyl, heterocycloalkyl, aryl and heteroaryl are optionally
substituted with
one or more substituents R26;
R2 and R3 arc independently hydrogen, lower alkyl, -R'6, or -[C(R17R18)],,-L2-
[C(R'4R20)]e-R16
wherein lower alkyl is optionally substituted with one or more substituents
R27; or
R2 and R' combine with the carbon to which they are bound to form a 3-7
membered cycloalkyl
or a 5-7 membered monocyclic heterocycloalkyl, wherein the 3-7 membered
monocyclic
cycloalkyl or 5-7 membered monocyclic heterocycloalkyl are optionally
substituted with one
or more substituents selected from the group consisting of halogen, -OH, -NH2,
lower alkyl,
fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy,
lower alkylthio,
fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and
cycloalkylamino;
a is1,2,or3;
bis0, 1,2,or3;
L2 is a bond, -0-, -S-, -N(R13)-, -C(O)-N(R13)-, -N(R13)-C(O), -C(S)-N(R'3)-, -
N(R13)-C(S)-,
-S(O)-N(R13)-, -S(O)2-N(R13)-, -N(R13)-S(O), -N(R13)-S(O)2, -N(R13)-C(O)-
N(R13)-,
-N(R13)-C(S)-N(R13)- or -N(R13)-S(O)2-N(R13)-;
R4 is -R21 or -L3-R21:
L3 is -0-, -S-, or -N(R13)-;
R21 2223 24
is -(CRR)p-R, cycloalkyl, heterocycloalkyl, aryl, or heteroaryl, wherein
cycloalkyl,
heterocycloalkyl, aryl or heteroaryl are optionally substituted with one or
more substituents
R29;
pis 1,2,3,4,5or6;
R5, and R6 are independently hydrogen, fluoro, lower alkyl, fluoro substituted
lower alkyl, phenyl,
or benzyl, wherein the phenyl ring of phenyl or benzyl is optionally
substituted with one or
more substituents R30;
R' and R8 are independently hydrogen, fluoro, lower alkyl, luoro substituted
lower alkyl, phenyl,
or benzyl, wherein the phenyl ring of phenyl or benzyl is optionally
substituted with one or
more substituents R31; or
R7 and R8 together form oxo;
R9, R10, R11, R12, and each R22 and R23 are independently hydrogen, fluoro,
lower alkyl, or fluoro
substituted lower alkyl; or
R9 and RS , or Rat and R12, or any two R22 and R23 on the same carbon, combine
with the carbon to
which they are bound to form a 3-7 membered cycloalkyl or a 5-7 membered
monocyclic
heterocycloalkyl, wherein the 3-7 membered monocyclic cycloalkyl or 5-7
membered
monocyclic heterocycloalkyl are optionally substituted with one or more
substituents selected
from the group consisting of halogen, -011, -NII2, lower alkyl, fluoro
substituted lower alkyl,
lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro
substituted lower
alkylthio, mono-alkylamino, di-alkylamino, and cycloalkylamino;
2
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
each R13 is independently hydrogen or lower alkyl;
R14 and R15 are independently hydrogen, fluoro, lower alkyl, or fluoro
substituted lower alkyl;
R16 is cycloalkyl, heterocycloalkyl, aryl or heteroaryl, wherein cycloalkyl,
heterocycloalkyl, aryl
or heteroaryl are optionally substituted with one or more substituents Rib;
R", R's, R19, and R20 are independently hydrogen, fluoro, lower alkyl, or
fluoro substituted lower
alkyl; or
any two R 17 and R18 on the same carbon, or any two R19 and R2D on the same
carbon, combine
with the carbon to which they are bound to form a 3-7 membered cycloalkyl or a
5-7
membered monocyclic hetcrocycloalkyl, wherein the 3-7 membered monocyclic
cycloalkyl or
5-7 membered monocyclic heterocycloalkyl are optionally substituted with one
or more
substituents selected from the group consisting of halogen, -OH, -NH2, lower
alkyl, fluoro
substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy, lower
alkylthio, fluoro
substituted lower alkylthio, mono-alkylamino, di-alkylamino, and
cycloalkylamino; or
any two R" and R'8 on the same carbon, or any two R19 and R20 on the sam
carbon form oxo;
R24 is selected from the group consisting of hydrogen, fluoro, -O-R32, -S-R34,
-N(R32)-R33,
-C(O)-R34, -C(S)-R34, -S(O)-R34, -S(O),-R34, -C(O)-O-R32, -C(O)-N(R322)-R33,
-C(S)-N(R32)-R33, -S(O)-N(R32)-R33, -S(O)2-N(R32)-R33 , -N(R35)-C(O)-R 3ar -
N(R35)-C(S)-R14,
-N(R35)-S(O)-R34 -N(R35)-S(O)2-R34 -N(R35)-C(O)-N(R32)-R33 -N(R35)-C(S)-N(R32)-
R33,
-N(R35)-S(O)2-N(R32)-R33, cycloalkyl, heterocycloalkyl, aryl and heteroaryl,
wherein
cycloalkyl, heterocycloalkyl, aryl or heteroaryl are optionally substituted
with one or more
substituents R29;
each R25 and R2', if present, is independently selected from the group
consisting of fluoro, -O-R36
-S-R 3S, and -N(R36)-R31;
each R26, R78, R29, R30 and R31, if present, is independently selected from
the group consisting of
-CN, -NO7, -O-R39, -S-R41, -N(R39)-R40, -C(O)-R41, -C(S)-R41, -S(O)-R41, -
S(O)2-R41,
-C(O)-O-R39, -C(O)-N(R39)-R40, -C(S)-N(R39)-R40, -S(O)-N(R39)-R40, -S(O)2-
N(R39)-R40,
-N(R35)-C(O)-R41, -N(R35)-C(S)-R41 N(R35)-S(O)-R4i N(R35)-S(O)2-R41,
-N(R35)-C(O)-N(R39)-R40, -N(R35)-C(S)-N(R39)-R40, -N(R35)-S(O)2-N(R39)-R40,
oxo, halogen,
lower alkyl, cycloalkyl, heterocycloalkyl, aryl, and heteroaryl, wherein lower
alkyl is
optionally substituted with one or more substituents selected from the group
consisting of
fluoro, lower alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro
substituted
lower alkylthio, mono-alkylamino, di-alkylamino, cycloalkyl, heterocycloalkyl,
aryl, and
heteroaryl, and wherein cycloalkyl, heterocycloalkyl, aryl, and heteroaryl as
R26, Rea, R29, Rao
or R31, or as a substituent of lower alkyl are optionally substituted with one
or more
substitucnts selected from the group consisting of -CN, -NO2, -O-R42, -S-R44, -
N(R42)-R43,
N(R35)-C(O)-R44, -N(R35)-S(O)2-R44, -C(O)-R44, -S(O)-R44, -S(O)2-R=74, -C(O)-O-
R42,
3
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
-C(O)-N(R -)-R 3, -S(O)2-N(R4`)-R 3, oxo, halogen, lower alkyl, fluoro
substituted lower
alkyl, and cycloalkylamino; or
any two R26, any two R28, any two R29, any two R30, or any two R31, on
adjacent ring atoms,
combine to form a fused ring selected from the group consisting of 3-7
membered cycloalkyl
and 5-7 membered heterocycloalkyl, wherein the 3-7 membered cycloalkyl or 5-7
membered
heterocycloalkyl are optionally substituted with one or more halogen, -OH, -
NP,, oxo, lower
alkyl, fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower
alkoxy, lower
alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino,
and
cycloalkylamino;
each R32, R33, R39 and R4 is independently selected from the group consisting
of hydrogen, lower
alkyl, cycloalkyl, heterocycloalkyl, aryl, and heteroaryl, wherein lower alkyl
is optionally
substituted with one or more substituents selected from the group consisting
of fluoro, lower
alkoxy, fluoro substituted lower alkoxy, lower alkylthio, fluoro substituted
lower alkylthio,
mono-alkylamino, di-alkylamino, cycloalkyl, heterocycloalkyl, aryl, and
heteroaryl, and
wherein cycloalkyl, heterocycloalkyl, aryl, and heteroaryl as R32, R33, R39 or
R40, or as a
substituent of lower alkyl are optionally substituted with one or more
substituents selected
from the group consisting of -CN -NO2, -O-R 42, -S-R44, -N(R 42)-R 43, -N(R
35)-C(O)-R44
,
-N(R35)-S(O)2-R44, -C(O)-R44, -S(O)-R44, -S(O)2-R44, -C(O)-O-R 42, -C(O)-
N(R42)-R43
-S(O)2-N(R42)-R43, oxo, halogen, lower alkyl, fluoro substituted lower alkyl,
and
cycloalkylamino;
each R34 and R41 is independently selected from the group consisting of lower
alkyl, cycloalkyl,
heterocycloalkyl, aryl, and heteroaryl, wherein lower alkyl is optionally
substituted with one
or more substituents selected from the group consisting of fluoro, lower
alkoxy, fluoro
substituted lower alkoxy, lower alkylthio, fluoro substituted lower alkylthio,
mono-
alkylamino, di-alkylamino, cycloalkyl, heterocycloalkyl, aryl, and heteroaryl,
and wherein
cycloalkyl, heterocycloalkyl, aryl, and heteroaryl as R34 or R41, or as a
substituent of lower
alkyl are optionally substituted with one or more substituents selected from
the group
consisting of -CN, -NO2, -O-R42, -S-R44, -N(R42)-R43, -N(R35)-C(O)-R44, -
N(R35)-S(O)2-R44,
_C(O)_R44, 44 44, 42, 4` ' 13, 4`^ 43,
-S(O)-R , -S(O)2-R , -C(O)-O-R , -C(O)-N(R)-R , -S(O)2-N(R )-R oxo,
halogen, lower alkyl, fluoro substituted lower alkyl, and cycloalkylamino;
each R35 is hydrogen or lower alkyl;
each R36 and R3' is independently hydrogen, lower alkyl, or fluoro substituted
lower alkyl;
each R38 is lower alkyl or fluoro substituted lower alkyl;
each R42 and R43 is independently selected from the group consisting of
hydrogen, lower alkyl,
heterocycloalkyl and heteroaryl, wherein lower alkyl is optionally substituted
with one or
more substituents selected from the group consisting of fluoro, lower alkoxy,
fluoro
substituted lower alkoxy, lower alkylthic, fluoro substituted lower alkylthio,
mono-
4
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
alkylamino, di-alkylamino, and cycloalkylamino, and wherein heterocycloalkyl
and
heteroaryl are optionally substituted with one or more substituents selected
from the group
consisting of halogen, -CN, lower alkyl, fluoro substituted lower alkyl, lower
alkoxy and
fluoro substituted lower alkoxy; and
each R44 is independently selected from the group consisting of lower alkyl,
heterocycloalkyl and
heteroaryl, wherein lower alkyl is optionally substituted with one or more
substituents
selected from the group consisting of fluoro, lower alkoxy, fluoro substituted
lower alkoxy,
lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino, di-
alkylamino, and
cycloalkylamino, and wherein heterocycloalkyl and heteroaryl are optionally
substituted with
one or more substituents selected from the group consisting of halogen, -CN,
lower alkyl,
fluoro substituted lower alkyl, lower alkoxy and fluoro substituted lower
alkoxy.
[0004] In some embodiments of compounds of Formula 1, X1 and X2 are both -N=,
n is 0 or 1,
preferably 1, and Y is -0- or -C(R14R15)-, preferably -C(R14R15)-; X1 is -N=
and X2 is -C(H)=, n is 0 or
1, preferably 1, and Y is -0- or -C(R14R'5)-, preferably -C(R14R15)-; X1 is -
C(H)= and X2 is -N=, n is 0
or 1, preferably 1, and Y is -0- or -C(R14R'5)-, preferably -C(R14Ri5)-; X1
and X2 are both -C(H)= ,
n is 0 or 1, preferably 1, and Y is -0- or -C(R14R15)-, preferably -C(R14R15)-
.
[0005] In some embodiments of compounds of Formula I, further to any of the
above embodiments
of compounds of Formula I, L1 is a bond, -C(R9R1D)-, -C(O)-, -C(R9R10)-
C(R"R12)-, -C(R9R'0)-C(O)-,
or -C(O)-C(R11R12), preferably -C(R9R10)- or -C(R9R1))-C(R11R12)-, and R' is
phenyl or monocyclic
heteroaryl, wherein phenyl or monocyclic heteroaryl are optionally substituted
with one or more
substituents R26.
[0006] In some embodiments of compounds of Formula I, further to any of the
above embodiments
of compounds of Formula I, Y is -C(R14R15)- and R14 and R15 are independently
lower alkyl or fluoro
substituted lower alkyl.
[0007] In a second aspect, compounds of Formula I having a structure according
to one of the
following structures, Formulae la or lb, are provided:
R45 R45
L4 NH, NH2
R54 ( L4
N Roe Rah
Rb' R54 N
R47 R47
R52 R53
Rog R48
R51 R50
R5 R49 R49
or
Formula la Formula lb
or a salt, a prodrug, a tautomer or a stercoisomer thereof,
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
wherein:
L4 is a bond, -C R55R56 55 56) ( 57 ( 55 56)_C(O)_, ( ) (R5-'R58);
( ) , -C(O)-, C(R R -C R` R58)_' -C RR or -C O -C R45 is selected from the
group consisting of lower alkyl, cycloalkyl, heterocycloalkyl, aryl and
heteroaryl, wherein lower alkyl is optionally substituted with one or more
substituents R70,
and wherein cycloalkyl, heterocycloalkyl, aryl and heteroaryl are optionally
substituted with
one or more substituents R71:
R46 and R47 are independently hydrogen, lower alkyl, or -[C(R59R60)],-R61,
wherein lower alkyl is
optionally substituted with one or more substituents R' ; or
R46 and R47 combine with the carbon to which they are bound to form a 3-7
membered cycloalkyl
or a 5-7 membered monocyclic heterocycloalkyl, wherein the 3-7 membered
monocyclic
cycloalkyl or 5-7 membered monocyclic heterocycloalkyl are optionally
substituted with one
or more substituents selected from the group consisting of halogen, -OH, -NH2,
lower alkyl,
fluoro substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy,
lower alkylthio,
fluoro substituted lower alkylthio, mono-alkylamino, di-alkylamino, and
cycloalkylamino;
c is 0, 1, 2, 3, 4, or 5;
R41 is -R62 or -L5-R62;
L4 is -0-, -S-, or -N(R63)-;
gis1,2,3,4,5or6;
Rog and R50 arc independently lower alkyl or fluoro substituted lower alkyl;
R51 and R52 are independently hydrogen, fluoro, lower alkyl, or fluoro
substituted lower alkyl;
R53 and R54 are independently hydrogen, fluoro, lower alkyl, or fluoro
substituted lower alkyl; or
R53 and R54 together form oxo;
R55 R56 R57 R58 and each R59 R60 R64 and R65 are independently hydrogen, en,
fluoro, lower alkyl,
Y g >
or fluoro substituted lower alkyl; or
R55 and R56, or R57 and R58, any two R59 and R60 on the same carbon, or any
two R64 and R65 on
the same carbon, combine with the carbon to which they are bound to form a 3-7
membered
cycloalkyl or a 5-7 membered monocyclic heterocycloalkyl, wherein the 3-7
membered
monocyclic cycloalkyl or 5-7 membered monocyclic heterocycloalkyl are
optionally
substituted with one or more substituents selected from the group consisting
of halogen, -OH,
-NH2, lower alkyl, fluoro substituted lower alkyl, lower alkoxy, fluoro
substituted lower
alkoxy, lower alkylthio, fluoro substituted lower alkylthio, mono-alkylamino,
di-alkylamino,
and cycloalkylamino;
R61 is cycloalkyl, heterocycloalkyl, aryl or heteroaryl, wherein cycloalkyl,
heterocycloalkyl, aryl
or heteroaryl are optionally substituted with one or more substituents R73;
R62 is -(CR64R65)q-R66, cycloalkyl, heterocycloalkyl, aryl, or heteroaryl,
wherein cycloalkyl,
heterocycloalkyl, aryl or heteroaryl are optionally substituted with one or
more substituents
R74:
6
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
each R63 is independently hydrogen or lower alkyl;
R66 is independently selected from the group consisting of hydrogen, fluoro, -
O-R67, -S-R61
,
-N(R67)-R68, -C(O)-R69, -C(S)-R69, -S(O)-R69, -S(O)2-R69, -C(O)-N(R67) R68
-S(O)7-N(R67)-R68> -N(R63)-C(O)-R69 -N(R63)-S(O)z-R69, cycloalkyl,
heterocycloalkyl, aryl or
heteroaryl, wherein cycloalkyl, heterocycloalkyl, aryl or heteroaryl are
optionally substituted
with one or more substituents Rr4;
each R7C and R72, if present, is independently selected from the group
consisting of fluoro, -O-R75
-S-R77, and -N(R75)-R7D;
each R71, R73 and R74, if present, is independently selected from the group
consisting of -CN,
-NO2, -O-R78, -S-R$0, -N(R78)-Ri9, -C(O)-R8O, -S(O)-R80, -S(O)2-R80, -C(O)-
N(R7s)-R79,
-S(0)2-N(R7B)-R79, -N(R63)-C(O)-Rft , -N(R63)-S(O)2-R80, oxo, fluoro, chloro,
lower alkyl,
cycloalkyl, heterocycloalkyl, aryl, and heteroaryl, wherein lower alkyl is
optionally
substituted with one or more substituents selected from the group consisting
of fluoro, lower
alkoxy, fluoro substituted lower alkoxy, mono-alkylamino, di-alkylamino, and
cycloalkylamino, and wherein cycloalkyl, heterocycloalkyl, aryl, and
heteroaryl as R", R73 or
R74, or as a substituent of lower alkyl are optionally substituted with one or
more substituents
selected from the group consisting of -CN, -NO,, -O-R", -S-RS3, -N(R")-R82,
-N(R63)-C(O)-R83, -N(R63)-S(O)2-Rs3, -C(O)-R83, -S(O)-R83, -S(O)2-R83, -C(O)-O-
R ,
-C(O)-N(R8')-R82, -S(O),-N(R81)-RB2, oxo, fluoro, chloro, lower alkyl, fluoro
substituted
lower alkyl, and cycloalkylamino; or
any two R71, any two R73, or any two R74, on adjacent ring atoms, combine to
form a fused ring
selected from the group consisting of 3-7 membered cycloalkyl and 5-7 membered
heterocycloalkyl, wherein the 3-7 membered cycloalkyl or 5-7 membered
heterocycloalkyl
are optionally substituted with one or more halogen, -OH, -NH2, oxo, lower
alkyl, fluoro
substituted lower alkyl, lower alkoxy, fluoro substituted lower alkoxy, lower
alkylthio, fluoro
substituted lower alkylthio, mono-alkylamino, di-alkylamino, and
cycloalkylamino;
each R67, R68, R78 and R79 is independently selected from the group consisting
of hydrogen, lower
alkyl, cycloalkyl, heterocycloalkyl, aryl, and heteroaryl, wherein lower alkyl
is optionally
substituted with one or more substituents selected from the group consisting
of fluoro, lower
alkoxy, fluoro substituted lower alkoxy, mono-alkylamino, di-alkylamino, and
cycloalkylamino, and wherein cycloalkyl, heterocycloalkyl, aryl, and
heteroaryl as R6', R63,
R78 79
or R, or as a substituent of lower alkyl are optionally substituted with one
or more
substituents selected from the group consisting of -CN, -NO2, -O-Rn, -S-R s3, -
N(R81)-R82,
-N(R6i)-C(O)-R83, -N(R63)-S(O)2-R53, -C(O)-R s3, -S(O)-R83, -S(O)2-R13, -C(O)-
O-Ru,
-C(O)-N(R8')-R", -S(O)2-N(R8')-R82, oxo, fluoro, chloro, lower alkyl, fluoro
substituted
lower alkyl, and cycloalkylamino;
7
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
each R69 and R& is independently selected from the group consisting of lower
alkyl, cycloalkyl,
heterocycloalkyl, aryl, and heteroaryl, wherein lower alkyl is optionally
substituted with one
or more substituents selected from the group consisting of fluoro, lower
alkoxy, fluoro
substituted lower alkoxy, mono-alkylamino, di-alkylamino, and cycloalkylamino,
and
wherein cycloalkyl, heterocycloalkyl, aryl, and heteroaryl as R69 or RHO, or
as a substituent of
lower alkyl are optionally substituted with one or more substituents selected
from the group
consisting of -CN, -NO2, -O-R$1, -S-R83, -N(R8')-R82, -N(R63)-C(O)-R53, -
N(R63)-S(O),-R$3,
-C(O)-R83, -S(O)-R", -S(O)2-R33, -C(O)-O-Rs', -C(O)-N(R")-R82, -S(O)2-N(R81)-
R82, oxo,
fluoro, chloro, lower alkyl, fluoro substituted lower alkyl, and
cycloalkylamino;
each R75 and R76, if present, is independently hydrogen, lower alkyl, or
fluoro substituted lower
alkyl;
each R77 is lower alkyl or fluoro substituted lower alkyl;
each Ref and RB2 is independently selected from the group consisting of
hydrogen, lower alkyl,
heterocycloalkyl and heteroaryl, wherein lower alkyl is optionally substituted
with one or
more substituents selected from the group consisting of fluoro, lower alkoxy,
fluoro
substituted lower alkoxy, mono-alkylamino, di-alkylamino, and cycloalkylamino,
and
wherein heterocycloalkyl and heteroaryl are optionally substituted with one or
more
substituents selected from the group consisting of halogen, -CN, lower alkyl,
fluoro
substituted lower alkyl, lower alkoxy and fluoro substituted lower alkoxy; and
each RB3 is independently selected from the group consisting of hydrogen,
lower alkyl,
heterocycloalkyl and heteroaryl, wherein lower alkyl is optionally substituted
with one or
more substituents selected from the group consisting of fluoro, lower alkoxy,
fluoro
substituted lower alkoxy, mono-alkylamino, di-alkylamino, and cycloalkylamino,
and
wherein heterocycloalkyl and heteroaryl are optionally substituted with one or
more
substituents selected from the group consisting of halogen, -CN, lower alkyl,
fluoro
substituted lower alkyl, lower alkoxy and fluoro substituted lower alkoxy.
[0008] In reference to compounds herein, unless clearly indicated to the
contrary, specification of a
compound or group of compounds includes salts of such compound(s) (including
pharmaceutically
acceptable salts), formulations of such compound(s) (including
pharmaceutically acceptable
formulations), conjugates thereof, derivatives thereof, forms thereof,
prodrugs thereof, and all
stereoisomers thereof. In reference to compositions, kits, methods of use,
etc, of compounds of
Formula 1 described herein, it is understood (unless indicated otherwise) that
a compound of Formula
I includes all sub-embodiments thereof (e.g. including Formula la, Formula Ib
and all embodiments as
described above).
8
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
100091 In one embodiment of compounds of Formula I, the compound is selected
from the group
consisting of:
1-[7-(l-Amino-ethyl)-6-ethyl-4,4-dimethyl-3,4-dihydro-2H-quinolin-l -yl] -2-
methoxy-ethanone
(P-0001),
1-[6-Ethyl-l -(2-methoxy-ethyl)-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl]-
ethylamine (P-0002),
1-[6-Ethyl-l -(3-methoxy-propyl)-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-
yl]-ethylamine
(P-0003),
1-[6-Ethyl-4,4-dimethyl-l-(3,3,3-trifluoro-propyl)-1,2,3,4-tetrahydro-quinolin-
7-yl]-ethylamine
(P-0004),
1-[6-Ethyl-4,4-dimethyl-l-(4,4,4-trifluoro-butyl)-1,2,3,4-tetrahydro-quinolin-
7-yl]-ethylamine
(P-0005),
N-{2-[7-(1-Amino-ethyl)-6-ethyl-4,4-dimethyl-3,4-dihydro 2H-quinolin-l-yl]-
ethyl}-acetamide
(P-0006),
1-[7-(1-Amino-ethyl)-6-ethyl-4,4-dimethyl-3,4-dihydro-2H-quinolin-1-yl]-
ethanone (P-0007),
C-[6-Ethyl-l-(3 -methoxy-p ropyl)-4,4-dimethyl -1, 2,3,4-tetra hydro-quip o 1
in-7-yl ]-in ethyl amine
(P-0008),
1-[6-Ethyl-4,4-dimethyl-l -(2-pyrazol-l -yl-ethyl)-1,2,3,4-tetrahydro-quinolin-
7-yl]-ethylamine
(P-0009),
1-(6-Ethyl -4,4-dimethyl-l-pyridin-3-ylmethyl-1,2,3,4-tetrahydro-quinolin-7-
yl)-ethylamine (P-0010),
7-(1-Amino-ethyl)-6-bromo-l -(3-methoxy-propyl)-4,4-dimethyl-3,4-dihydro-1 H-
quinolin-2-one
(P-0011),
7-(1-Amino-ethyl)-1-(3-methoxy-propyl)-4,4-dimethyl-3,4-dihydro-111-quinolin-2-
one (P-0012),
7-(1-Amino-ethyl)-6-methoxy-l -(3 -methoxy-propyl)-4,4-dimethyl-3,4-dihydro-1
H-quinolin-2-one
(P-0013),
7-(1-Amino-ethyl)-6-ethyl-l -(3 -methoxy-propyl)-4,4-dimethyl-3,4-dihydro-1 H-
quinolin-2-one
(P-0014),
1-(6-Ethyl-4,4-dimethyl-l-pyridin-4-ylmethyl-1,2,3,4-tetrahydro-quinolin-7-yl)-
ethylamine (P-0015),
1-[6-Ethyl- l -(3 -m ethoxy-propyl)-4,4-dimethyl -1,2,3 ,4-tetrahydro-quinolin-
7-yl]-propylamine
(P-0016),
1-[6-Ethyl-4,4-dimethyl-l -(5-methyl-isoxazol-3 -ylmethyl)-1,2,3,4-tetrahydro-
quinolin-7-yl] -
ethylamine (P-0017),
[7-(1-Amino -ethyl)-6-ethyl-4,4-dimethyl-3,4-dihydro-2H-quinolin-l-yl]-pyridin-
3-yl-methanone
(P-0018),
[7-(1-Amino-ethyl)-6-ethyl-4,4-dimethyl-3,4-dihydro-2H-quinolin-1-yl]-pyridin-
4-yl-methanone
(P-0020),
1-[ 1-(3 .Methoxy-propyl)-4,4-dimethyl-6-phenyl-1,2,3,4-tetrahydro-quinolin-7-
yl]-ethylamine
(P-0021),
9
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
[7-(1-Amino-ethyl)-6-ethyl-4,4-dimethyl-3,4-dihydro-2H-quinolin- l -yl] -
isoxazol-5-yl-methanone
(P-0022),
1-[1-(3-Bromo-isoxazol-5-ylmethyl)-6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-
quinolin-7-yl]-
ethylamine (P-0023),
1-[6-Ethyl -4,4-dimethyl-l -(3-methyl-isoxazol-5-ylmethyl)-1,2,3,4-tetrahydro-
quinolin-7-yl]-
ethylamine (P-0024),
1-(1-Bcnzyl-6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl)-ethylamine
(P-0025),
1-[6-Ethyl-4,4-dimethyl- l -(3-trifluoromethyl-benzyl)-1,2,3,4-tetrahydro-
quinolin-7-yl]-ethylamine
(P-0026),
1-[1-(3,5-Difluoro-benzyl)-6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-
yl]-ethylamine
(P-0027),
1-[6-Ethyl-l-(3-fluoro-benzyl)-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl]-
ethylamine (P-0028),
1-[6-Ethyl- l -(4-methoxy-benzyl)-4,4-dimethyl-1, 2, 3,4-tetrahydro-quinolin-7-
yl]-ethylamine
(P-0029),
1-[ 1-(2,3-Difluoro-benzvl)-6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-
yl]-ethylamine
(P-0030),
1-[6-Ethyl -l-(3-methoxy-benzyl)-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-
yl]-ethylamine
(P-0031),
1-[6-Ethyl-1 -(4-fluoro-benzyl)-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl]-
ethylamine (P-0032),
4-[7-(1-Amino -ethyl)-6-ethyl-4,4-dimethyl-3,4-dihydro-2H-quinolin-l-ylmethyl]-
pyridine-2-
carbonitrile (P-0033),
1-[6-Ethyl- I -(2 -fluoro-pyridin-4-ylmethyl)-4,4-dimethyl- 1,2,3,4-tetrahydro-
quinolin-7-yl]-ethyla mine
(P-0034),
1-(6-Ethyl-4,4-dimethyl-l-phenethyl-1,2,3,4-tetrahydro-quinolin-7-yl)-
ethylamine (P-0035),
1-[6-Ethyl-l-(6-fluoro-pyridin-3-ylmethyl)-4,4-dimethyl-1,2,3,4-tetrahydro-
quinolin-7-yl]-ethylamine
(P-0036),
1-[6-Ethyl-4,4-dimethyl-l-(2-methyl-pyridin-4-ylmethyl)-1,2,3,4-tetrahydro-
quinolin-7-yl]-
ethylamine (P-0037),
1-[ 1-(2,2-Difluoro-benzo [ 1, 3] di oxol-5-ylmethyl)-6-ethyl-4,4-dimethyl-
1,2, 3,4-tetrahydro-quinolin-7-
yl]-ethylamine (P-0038),
1-(1-Benzyl-4,4-dimethyl-6-p-tolyl-1,2,3,4-tetrahydro-quinolin-7-yl)-
ethylamine (P-0039),
7-(1-Amino-ethyl)-1-(3-methoxy-propyl)-4,4-dimethyl-6-p-tolyl-3,4-dihydro-1 H-
quinolin-2-one
(P-0040),
1-[6-Ethyl-l -(2-methoxy-pyridin-4-y lmethyl)-4,4-dimethyl-1,2, 3,4-tetrahydro-
quinolin-7-yl]-
ethylamine (P-0041),
1 -[6-Ethyl-4,4-dimethyl-l -(6-methyl-pyridin-3-ylmethyl)-1,2, 3,4-tetrahydro-
quinolin-7-yl]-
ethylamine (P-0042),
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
1-[1-(3-Methoxy-propyl)-4,4-dimethyl-6-p-tolyl-1,2,3,4-tetrahydro-quinolin-7-
yl]-ethylamine
(P-0043),
1-[1-Benzyl-6-(4-fluoro-phenyl)-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl]-
ethylamine (P-0044),
1-[1-Benzyl-6-(4-chloro-phenyl)-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl]-
ethyl amine (P-0046),
1 -(1 -Benzyl-6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl)-
propylamine (P-0047),
(R)-1-(l -Ben zyl-6-ethyl -4,4-dimethyl- 1,2,3,4-tetrahydro-quinolin-7-yl)-
ethylamine (P-0048),
(S)-1-(1-Benzyl-6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl)-
ethylamine (P-0049),
1-[1-Benzyl-6-(3-methoxy-phenyl)-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-
yl]-ethylamine
(P-0050),
1-(1-Benzo[1,3]dioxol-4-ylmethyl-6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-
quinolin-7-yl)-ethyl amine
(P-0051),
7-(1-Amino -ethyl)-1-benzyl-6-ethyl -4,4-dimethyl-3,4-dihydro-IH-quinolin-2-
one (P-0052),
l -[] -(2,2-Difluoro-bcnzo[1,3]dioxol-4-ylmethyl)-6-ethyl-4,4-dimcthyl-1,2,3,4-
tetrahydro-quinolin-7-
yl]-ethylamine (P-0053),
1-[1-Benzyl-6-(4-methoxy-phenyl)-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-
yl]-ethyl amine
(P-0054),
1-(1-Benzyl-4,4-dimethyl-6-thiophen-2-yl-1,2,3,4-tetrahydro-quinolin-7-yl)-
ethylamine (P-0055),
(R)-1-[6-Ethyl-4,4-dimethyl-l -(2-methyl-pyridin-4-ylmethyl)-1,2,3,4-
tetrahydro-quinolin-7-yl]-
ethylamine (P-0056),
(S)-1-[6-Ethyl-4,4-dimethyl-l -(2-methyl-pyridin-4-ylmethyl)-1,2,3,4-
tetrahydro-quinolin-7-yl]-
ethylamine (P-0057),
1-[6-Ethyl-4,4-dimethyl-1 -(tetrahydro-pyran-4-ylmethyl)-1,2,3,4-tetrahydro-
quinolin-7-yl]-
ethylamine (P-0058),
1-[l-Benzyl-6-(3-ethyl phenyl)-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl]-
ethylamine (P-0059),
1-(l-Benzyl-6-benzytoxy-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl)-
ethylamine (P-0060),
7-(1-Amino-ethyl)-6-ethyl-4,4-dimethyl-l-(2-methyl-pyridin-4-ylmethyl)-3,4-
dihydro-111-quinolin-2-
one (P-0061),
1-[4,4-Dimethyl-l-(2-methyl-pyridin-4-ylmethyl)-6-p-tolyl-1,2,3,4-tetrahydro-
quinolin-7-yl]-
ethylamine (P-0062),
1-[ 1-Benzyl-6-(3 -benzytoxy-4-methoxy-phenyl)-4,4-dimethyl-1,2,3,4-tetrahydro-
quinolin-7-yl]-
ethylamine (P-0063),
1-[ 1-Benzyl-4,4-dimethyl-6-(3 -methyl-3H-imidazol-4-yl)-1,2,3,4-tetrahydro-
quinolin-7-yl]-
ethylamine (P-0064),
5-[7-(l -Amino-ethyl)-l -benzyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-6-yl]
-2-methoxy-pheno 1
(P-0065),
1-[1-Benzyl-6-(3-chloro-phenyl)-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl]-
ethylamine (P-0066),
11
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
1 -(6-B enzo [ 1, 3] dioxol-5 -yl-1-benzyl-4,4-dimethyl-1,2,3,4-tetrahydro-
quinolin-7-yl)-ethyl amine
(P-0067),
1-[1-Benzyl-6-(4-ethyl-phenyl)-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl]-
ethylamine (P-0068),
7-(1-Amino-ethyl)-6-ethyl-4,4-dimethyl-3,4-dihydro-2H-quinoline-l-carboxylic
acid tert-butyl ester
(P-0069),
1-(6-Ethyl-4,4-dimethyl-l-phenyl-1,2,3,4-tetrahydro-quinolin-7-yl)-ethylamine
(P-0070),
6-(1 -Amino -ethyl) -7-ethyl-4-(2-methoxy-ethyl) -4H-benzo [ 1,4] oxazin-3 -
one (P-0071),
3-[7-(1-Amino-ethyl)-1-benzyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-6-yl]-
benzamide (P-0072),
4-[7-(1-Amino -ethyl)-1-benzyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-6-yl]-
benzamide (P-0073),
1-[ 1-Benzyl-6-(3-fluoro-4-methyl-phenyl)-4,4-dimethyl-1,2,3,4-tetrahydro-
quinolin-7-yl]-ethylamine
(P-0074),
1- [1 -B enzyl -6 -(2,4-dimethyl-thiazol-5 -yl) -4,4-dimethyl- 1,2,3 , 4-
tetrahydro-quinolin-7-yl]-ethyl amine
(P-0075),
1-(1-Benzyl-4,4,6-trimethyl-1,2,3,4-tetrahydro-quinolin-7-yl)-ethylamine (P-
0076),
1-[1-Benzyl-6-(1H-indol-5-yl)-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl]-
ethylamine (P-0077),
7-(1-Amino-ethyl)-1-benzyl-4,4-dimethyl-6-p-tolyl-3,4-dihydro-IH-quinolin-2-
one (P-0078),
1-[6 -Ethyl-l-(3 -methoxy-phenyl)-4,4-dimethyl- 1,2,3,4-tetrahydro-quinolin-7 -
yl] -ethylamine
(P-0079),
1-[ 1-Benzyl-4,4-dimethyl-6-(5-methyl-thiophen-2-yl)-1,2,3,4-tetrahydro-
quinolin-7-yl] -ethylamine
(P-0080), and
any salt, prodrug, tautomer, or stereoisomer thereof.
[0010] In a third aspect, methods arc provided for treating or prophylaxis of
a renin-mediated
disease or condition in an animal subject in need thereof, wherein the method
involves administering
to the subject an effective amount of any one or more compound(s) of Formula
I. The terms "treat,"
"therapy," and like terms refer to the administration of material, e.g., one
or more compound(s) of
Formula I in an amount effective to prevent, alleviate, or ameliorate one or
more symptoms of a
disease or condition, i.e., indication, and/or to prolong the survival of the
subject being treated, The
term "renin-mediated disease or condition" refers to a disease or condition in
which the biological
function of renin affects the development, course, and/or symptoms of the
disease or condition, and/or
in which inhibition of the renin activity alters the development, course,
anchor symptoms of the
disease or condition. A renin-mediated disease or condition includes a disease
or condition for which
inhibition of renin provides a therapeutic benefit, e.g. wherein treatment
with renin inhbitors,
including compounds described herein, provides a therapeutic benefit to the
subject suffering from or
at risk of the disease or condition. In one aspect, the method involves
administering to the subject an
effective amount of a compound of Formula I in combination with one or more
other therapies for the
disease or condition.
12
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
[0011] Ina fourth aspect, compositions are provided that include a
therapeutically effective amount
of any one or more compound(s) of Formula I and at least one pharmaceutically
acceptable carrier,
excipient, and/or diluent, including combinations of any two or more compounds
of Formula I. The
composition can further include a plurality of different pharmacologically
active compounds, which
can include a plurality of compounds of Formula I. In certain embodiments, the
composition can
include any one or more compound(s) of Formula I along with one or more
compound(s) that are
therapeutically effective for the same disease indication. In one embodiment,
the composition
includes any one or more compound(s) of Formula I along with one or more
compound(s) that are
therapeutically effective for the same disease indication, wherein the
compounds have a synergistic
effect on the disease indication.
[0012] In a fifth aspect, methods are provided for treating or prophylaxis of
a renin-mediated
disease or condition in an animal subject in need thereof, wherein the method
involves administering
to the subject an effective amount of a composition including any one or more
compound(s) of
Formula I.
[0013] In a sixth aspect, the invention provides a kit that includes any one
or more compound(s) of
Formula I, or a composition thereof as described herein. In some embodiments,
the composition is
packaged, e.g., in a vial, bottle, flask, which may be further packaged, e.g.,
within a box, envelope, or
bag; the composition is approved by the U.S. Food and Drug Administration or
similar regulatory
agency for administration to a mammal, e.g., a human; the composition is
approved for administration
to a mammal, e.g., a human, for a renin-mediated disease or condition; the
invention kit includes
written instructions for use and/or other indication that the composition is
suitable or approved for
administration to a mammal, e.g., a human, for a renin-mediated disease or
condition; and the
composition is packaged in unit dose or single dose form, e.g., single dose
pills, capsules, or the like.
[0014] In a seventh aspect, the invention provides methods for treating or
prophylaxis of a renin-
mediated disease or condition in an animal subject in need thereof, wherein
the method involves
administering to the subject an effective amount of any one or more
compound(s) of Formula 1, or an
effective amount of a composition including any one or more compound(s) of
Formula I, wherein the
disease or condition is, for example, without limitation:
Cardiovascular and cardiovascular-related diseases, including, but not limited
to, hypertension,
essential hypertension, malignant hypertension, pulmonary hypertension,
atherosclerosis,
congestive heart failure, coronary artery disease, myocardial infarction,
cardiac hypertrophy,
cardiac fibrosis, ischemic or hemorrhagic stroke, stenosis of arteries,
restcnosis after
angioplasty or other interventions, myocarditis, cardiomyopathy (e.g. diabetic
cardiac
myopathy, post-infarction cardiac myopathy), arrhythmia (e.g. supraventricular
and
ventricular arrhythmia, atrial fibrillation and atrial flutter), myocardial
ischemia, cardiac
13
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
insufficiency, unstable coronary syndrome, detrimental vascular remodeling,
angina (stable or
unstable), diastolic dysfunction, left ventricular dysfunction, and abnormal
vascular growth;
Pulmonary-related diseases, including, but not limited to, chronic obstructive
pulmonary disease,
cor pulmonale, bronchiectasis, acute respiratory distress syndrome, acute lung
injury, asthma,
bronchiolitis obliterans, bronchiolitis obliterans with organizing pneumonia,
cystic fibrosis,
idiopathic pulmonary fibrosis, connective tissue or autoirmmune disease-
related pulmonary
fibrosis (e.g. resulting from scleroderma, systemic lupus erythematosus,
rheumatoid arthritis,
or polymyositis), hypersensitivity pneumonitis, eosinophilic granuloma
(Langerhan's cell
histiocytosis), chronic eosinophilic pneumonia, Wegener's granulomatosis,
idiopathic
pulmonary hemosiderosis, lymphangioleiomyomatosis, silicosis, asbestosis,
berylliosis,
sarcoidosis, systemic sclerosis, atypical pneumonia, pneumocystis pneumonia,
Hamman-Rich
syndrome, histiocytosis X, collagen vascular disease, granulomatous
vasculitis,
Goodpasture's syndrome, Hermansky-Pudlak syndrome, pulmonary alveolar
proteinosis, lung
cancer, and tuberculosis;
Vasculitis and autoimmune disease and related tissue fibrosis, including, but
not limited to,
fibrosis caused by sclerodactyly, scleroderma, amyloidosis, systemic
sclerosis,
dermatomyositis, dupytren's contracture, peyronie's disease, polymyositis,
amyloidosis,
atrophoderma of Pasini and Pierini, Raynaud's phenomenon, Still's disease,
eosinophilic
fasciitis, Hutchinson-Gilford progeria syndrome, Lichen myxedematosus, mixed
connective
tissue disease, morphoea, porphyria cutanea tarda type 1, scleredema
adultorum, systemic
sclerosis or lymphangitis carcinomatosis;
Nephropathies, including, but not limited to, diabetic nephropathy, IgA
nephropathy, Fabry
nephropathy, renal fibrosis, nephritis, lupus nephritis, glomerulonephritis,
glomcrular
sclerosis, renal vascular hypertension, hyperaldosteronism, chronic kidney
disease, renal
insufficiency, renal ischemia, renal failure, and renal colic;
Liver diseases, including, but not limited to, hepatocellular carcinoma, liver
fibrosis, liver
cirrhosis, and hepatitis;
Diabetes and complications resulting from diabetes, including, but not limited
to, nephropathy,
vasculopathy, neuropathy, and diabetic retinopathy; and
Other diseases, including metabolic syndrome, obesity, thyroiditis, gastritis,
peptic ulcer,
inflammatory bowel syndrome (e.g. ulcerative colitis, Crohn's disease),
multiple sclerosis,
rheumatoid arthritis, osteoarthritis, osteoporosis, endometriosis,
preeclampsia, brain
infarction, interstitial cystitis, pancreatitis, pancreatic cancer, erectile
dysfunction, proteinuria,
albuminuria, cognitive impairment, alzheimer's disease, dementia, anxiety
states, cognitive
disorders, increased intraocular pressure, glaucoma, angiogenesis related
disorders (e.g.
macular degeneration), carcinoid tumor, graft versus host disease
(transplant), Sjogren's
disease, uveitis, pemphigus, bullous pemphigoid, epidermolysis bullosa,
aphthous ulcers,
14
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
stomatitis, Bchcet's disease, and other diseases known to be related to the
the Renin
Angiotensin System.
10015] In an eighth aspect, the invention provides methods for treating or
prophylaxis of a renin-
mediated disease or condition in an animal subject in need thereof, wherein
the method involves
administering to the subject an effective amount of any one or more
compound(s) of Formula I, or an
effective amount of a composition including any one or more compound(s) of
Formula I, in
combination with one or more other therapies for the disease or condition. In
some embodiments, the
one or more other therapies include, for example, without limitation:
angiotensin converting enzyme (ACE) inhibitors, including, but not limited to,
benazepril,
cilazapril, captopril, delapril, enalapril, fosinopril, indolapril,
lisinopril, meoxipril,
perindopril, pivopril, quinapril, ramipril, rescinnamine, spirapril,
trandolapril, and zofenopil;
angiotensin II receptor antagonists, including, but not limited to,
candesartan, eprosartan,
irbesartan, losartan, olmesartan, saprisartan, tasosartan, telmisartan, and
valsartan;
endothelin receptor antagonists, including, but not limited to, ambrisentan,
avosentan, bosentan,
clazosentan, sitaxsentan, tezosentan),
alpha- and beta-adrenergic antoginists, including, but not limited to,
acebutolol, albutcrol,
alfuzosin, alprenolol, atenolol, betaxolol, bisoprolol, butaxamine, carteolol,
carvedilol,
celiprolol, dilevalol, doxazosin, esmolol, labetalol, levobunolol, mepindolol,
metipranolol,
metoprolol, nadolol, nebivolol, oxprenolol, penbutolol, phentolamine,
phenoxybenzamine,
pindolol, prazosin, propranolol, salmeterol, sotalol, tamsulosin, terazosin,
timolol, and
tolazine;
vasodilators, including, but not limited to, diazoxide, dipyridamole,
fenoldopam, flosequinan,
hydralazine, iloprost, isosorbide mononitrate, minoxidil, nitroprusside, and
treprostinil;
calcium channel blockers such as amlodipine, azelnidipine, bencyclane,
cilnidipine, diltiazem,
felodipine, fendiline, flunarizine, gallopamil, lacidipine, lercanidipine,
manidipine,
nicardipine, nifedipine, nimodipine, nisoldipine, nitrendipine, pencexilcnc,
tcludipinc, and
verapamil;
potassium activators, including, but not limited to, nicorandil and pinacidil;
diuretics, including, but not limited to, acetazolamide, amiloride,
bendroflumethiazide,
benzthiazide, bumetanide, canrenone, chlorothiazide, chlortalidone,
eplerenone, ethacrynic
acid, furosemide, hydrochlorothiazide, indacrinone, indapamide, mefruside,
metolazone,
spironolactone, torasemide, tricrynafen, and triamterene;
sympatholytics, including, but not limited to, clonidine, dobutamine,
guanabenz, guanethidine,
b anfacine, indoramin, lofexidine, mecamylamine, methyldopa, moxonidine,
reserpine,
rilmenidine, trirnethaphan, and urapidil;
serotonin receptor antagonists, including, but not limited to,?(see below);
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
thromboxane-synthetase inhibitors;
neutral endopeptidase inhibitors (NEP inhibitors);
renin inhibitors, including, but not limited to, aliskerin;
phosphodiesterase inhibitors, including, but not limited to, amrinone,
pentoxifylline, sildenafil,
tadalafil, and vardenafil;
anti-inflammatory agents, such as aceclofenac, acemetacin, aiclofenac,
algestone, alminoprofen,
amcinafal, amcinafide, amfenac, amiprilose, ampyrone, anakinra, anirolac,
apazone,
azapropazone, balsalazide, bendazac, benoxaprofen, benzydamine, bromelains,
bromfenac,
broperamole, carprofen, cicloprofen, cintazone, cliprofen, clobetasol,
clobetasone, clofezone,
clopirac, cloticasone, cortodoxone, deflazacort, desonide, desoximetasonc,
dexibuprofen,
dexketoprofen, diclofenac, diflumidone, diflunisal, difluprednate, diftalone,
drocinonide,
droxicam, enlimomab, enolicam, epirizole, etodolac, etofenamate, felbinac,
fenamole,
fenbufen, fenclofenac, fenclorac, fcndosal, fenpipalonc, fentiazac, flazalone,
fluazacort,
flufenamic acid, flumizole, flunisolide acetate, flunixin, flunoxaprofen,
fluocortin butyl,
fluorometholone, fluquazone, flurbiprofen, fluretofen, furaprofen, furobufen,
ibufenac,
ibuprofen, ibuproxam, ilonidap, indomethacin, indoprofen, indoxole, intrazole,
isoflupredone,
isoxepac, isoxicam, kebuzone, ketorolac, ketoprofen, lofemizole, lornoxicam,
loxoprofen,
meclofenamic acid, mefenamic acid, meloxicam, meseclazone, metamizole,
mofebutazone,
morniflumate, nabumetone, naproxen, naproxol, nimazone, olsalazine sodium,
orgotein,
orpanoxin, oxametacin, oxaprozin, oxyphenbutazone, paranyline, pentosan,
phenazone,
phenylbutazone, pirfenidone, piroxicam, pirprofen, prednazate, prifelone,
prodolic acid,
proglumetacin, proquazone, proxazole, rimexolone, romazarit, salcolex,
salsalate, salycilates,
sanguinarium chloride, seclazone, sermetacin, sudoxicam, sulfinpyrazonc,
sulindac, suprofen,
talmetacin, talniflumate, talosalate, tebufelone, tenidap, tenoxicam, tesicam,
tesimide,
tetrydamine, tiaprofenic acid, tiopinac, tolfenamic acid, tolmetin,
triclonide, triflumidate,
zidometacin, and zomepirac;
agents suitable for treating inflammatory bowel syndrome, including, but not
limited to,
corticosteroids, 6-mercaptopurine, mesalamine, and TNF anatogonists
(including, but not
limited to, adalimumab, etanercept, and infliximab);
agents suitable for treating diabetes and complications associated with
diabetes, including, but not
limited to, benzafibrate, ciprofibrate, clofibrate, fenofibrate, gemfibrozil,
metformin,
pioglitazone, repaglinide, and rosiglitazone;
agents suitable for treating cystic fibrosis, including, but not limited to,
alendronate, dornase alfa,
denufosol, fluticasone, inhaled sodium bicarbonate, miglustat, and
perfenidone;
agents suitable for treating pulmonary fibrosis, including, but not limited
to, azathioprine,
budesonide, cyclophosphamide, dapsonc, dexamethasone, etanercept, imatinib,
interferon
16
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
gamma-lb, interferon alpha 2b, minocycline, prednisone, methylprednisolone,
and
thalidomide;
agents suitable for treating COPD, including, but not limited to, advair and
Covent;
agents suitable for treating liver fibrosis, including, but not limited to,
adefovir dipivoxil,
colchicine, conivaptan, cyclosporine, everolimus, gabapentin, methotrexate,
satavaptan,
tacrolimus, and zilcuton;
agents suitable for treating hepatitis, including, but not limited to, 4-
methylumbelliferone and
cystadane;
agents suitable for treating other nephropathies, including, but not limited
to, rapamycin,
rituximab, and daclizumab;
agents suitable for treating pancreatitis, including, but not limited to,
pancrelipase;
agents suitable for treating scleroderma, including, but not limited to,
abatacept;
agents suitable for treating macular degeneration, including, but not limited
to, ranibizumab;
agents suitable for treating infections that may be result in fibrosis,
including, but not limited to,
adefovir, azithrornycin, aztreonarn, ceftazidime, ciprofloxacin,
clarithromycin, efavirenz,
itraconazole, lamivudine, lopinavir, ritonavir, nelfinavir, tefibazumab,
telbivudine, tcnofovir,
tobramycin, and zidovudine;
other agents suitable for treatment of, for example, hypertension,
cardiovascular diseases, or
fibrotic diseases, including, but not limited to, amitriptyline, L-arginine,
depclestat,
hydroxychloroquine, ketanserin, Nitric oxide, nitroglycerin, octreotide,
perhexiline,
ranolazine, statins (including, but not limited to, atorvastatin,
cirivastatin, fluvastatin,
lovastatin, pravastatin, and simvastatin), and tetrahydrobiopterin.
[0016] In a ninth aspect, the invention provides a method of treating or
prophylaxis of a disease or
condition in an animal subject in need thereof, by administering to the
subject a therapeutically
effective amount of any one or more compound(s) of Formula I, a prodrug of
such compound, a
pharmaceutically acceptable salt of such compound or prodrug, or a
pharmaceutically acceptable
formulation of such compound or prodrug. The compound can be alone or can be
part of a
composition. In some embodiments, the invention provides a method of treating
or prophylaxis of a
disease or condition in an animal subject in need thereof, by administering to
the subject a
therapeutically effective amount of any one or more compound(s) of Formula I,
a prodrug of such
compound, a pharmaceutically acceptable salt of such compound or prodrug, or a
pharmaceutically
acceptable formulation of such compound or prodrug in combination with one or
more other suitable
therapies for the disease or condition.
[0017] In a tenth aspect, a compound of Formula I will have an IC50 of less
than 500 nm, less than
100 nM, less than 50 nM, less than 20 nM, less than 10 nM, less than 5 nM. or
less than 1 nM as
17
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
determined in a generally accepted renin activity assay. In some embodiments,
the compound
selectively inhibits renin relative to other enzymes, including, but not
limited to, selectivity of the
compound relative to other proteases, whether a protease discussed herein, or
other proteases. In
some embodiments, the selectivity is such that the compound is at least 2-
fold, 5-fold, 10-fold, or 100-
fold more active on renin than any other aspartyl protease, including, but not
limited to, any of pepsin,
cathepsin-D or BACE. Selective inhibition of renin relative to another
protease is such that the IC6o
for the renin may be at least about 2-fold, also 5-fold, also 10-fold, also 20-
fold, also 50-fold, or at
least about 100-fold less than the IC50 for any of the other proteases as
determined in a generally
accepted activity assay for said protease.
10018] Additional aspects and embodiments will be apparent from the following
Detailed
Description of the Invention and from the claims.
DETAILED DESCRIPTION OF THE INVENTION
[00191 As used herein the following definitions apply unless clearly indicated
otherwise:
[00201 All atoms designated within a Formula described herein, either within a
structure provided,
or within the definitions of variables related to the structure, is intended
to include any isotope
thereof, unless clearly indicated to the contrary. It is understood that for
any given atom, the isotopes
may be present essentially in ratios according to their natural occurrence, or
one or more particular
atoms may be enhanced with respect to one or more isotopes using synthetic
methods known to one
skilled in the art. Thus, hydrogen includes for example 'H, 2H, 3H; carbon
includes for example "C,
"-C, 13C,14C; oxygen includes for example 160, 170, 180; nitrogen includes for
example 13N, 14N, 15N;
sulfur includes for example 32S, 33S, 34S,35S, 36S, 37S, 38S; fluoro includes
for example17F, 18F, 19F;
chloro includes for example 35C1, 36C1, 37C1, 38C1, 39C1; and the like.
[00211 "Halogen" refer to all halogens, that is, chloro (Cl), fluoro (F),
bromo (Br), or iodo (I).
100221 "Oxo" refers to =0, e.g. an oxo substituted carbon atom provides a
carbonyl (C=O) group.
[00231 "Lower alkyl" alone or in combination means an alkane-derived radical
containing from I
to 6 carbon atoms (unless specifically defined) that includes a straight chain
alkyl or branched alkyl.
The straight chain or branched lower alkyl group is chemically feasible and
attached at any available
point to provide a stable compound. In many embodiments, a lower alkyl is a
straight or branched
alkyl group containing from 1-6, 1-4, or 1-2, carbon atoms, such as methyl,
ethyl, propyl, isopropyl,
butyl, t-butyl, and the like. Lower alkyl that is "optionally substituted"
denotes lower alkyl that is
optionally independently substituted, unless indicated otherwise, with one or
more, preferably 1, 2, 3,
4 or 5, also 1, 2, or 3 substituents as indicated, for example, in the
description of compounds of
18
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
Formula I. Furthermore, possible substitutions include subsets of these
substitutions, such as are
indicated herein. For example, fluoro substituted lower alkyl denotes a lower
alkyl group substituted
with one or more fluoro atoms, such as perfluoroalkyl, where preferably the
lower alkyl is substituted
with 1, 2, 3, 4 or 5 fluoro atoms, also 1, 2, or 3 fluoro atoms. It is
understood that any substitutions
are chemically feasible and attached at any available atom to provide a stable
compound.
[0024] "'Cycloalkyl" refers to saturated or unsaturated, non-aromatic
monocyclic, bicyclic or
tricyclic carbon ring systems of 3-10, also 3-8, more preferably 3-6, ring
members per ring, such as
cyclopropyl, cyclopentyl, cyclohexyl, adamantyl, and the like. Cycloalkyl that
is "optionally
substituted" denotes cycloalkyl that is optionally independently substituted,
unless indicated
otherwise, with one or more, preferably 1, 2, 3, 4 or 5, also 1, 2, or 3
substituents as indicated, for
example, in the description of compounds of Formula I. It is understood that
any substitutions are
chemically feasible and attached at any available atom to provide a stable
compound.
[0025] "Heterocycloalkyl" refers to a saturated or unsaturated non-aromatic
cycloalkyl group having
from 5 to 10 atoms in which from 1 to 3 carbon atoms in the ring are replaced
by heteroatoms of 0, S
or N, and are optionally fused with benzo or lieteroaryl of 5-6 ring members.
Heterocycloalkyl is also
intended to include oxidized S or N, such as sulfinyl, sulfonyl and N-oxide of
a tertiary ring nitrogen.
Heterocycloalkyl is also intended to include compounds in which a ring carbon
may be oxo
substituted, i.e. the ring carbon is a carbonyl group, for example as found in
lactones and lactams.
The point of attachment of the heterocycloalkyl ring is chemically feasible,
at a carbon or nitrogen
atom, such that a stable compound is provided. Examples of heterocycloalkyl
groups include, but are
not limited to, morpholino, tetrahydrofuranyl, dihydropyridinyl, piperidinyl,
pyrrolidinyl,
pyrrolidonyl, piperazinyl, dihydrobenzofuryl, and dihydroindoly].
Heterocycloalkyl that is
"optionally substituted" denotes heterocycloalkyl that is optionally
independently substituted, unless
indicated otherwise, with one or more, preferably 1, 2, 3, 4 or 5, also 1, 2,
or 3 substituents as
indicated, for example, in the description of compounds of Formula I. It is
understood that any
substitutions are chemically feasible and attached at any available atom to
provide a stable compound.
[0026] "Aryl" alone or in combination refers to a monocyclic or bicyclic ring
system containing
aromatic hydrocarbons such as phenyl or naphthyl, which may be optionally
fused with a cycloalkyl
of preferably 5-7, more preferably 5-6, ring members. Aryl that is "optionally
substituted" denotes
aryl that is optionally independently substituted, unless indicated otherwise,
with one or more,
preferably 1, 2, 3, 4 or 5, also 1, 2, or 3 substituents as indicated, for
example, in the description of
compounds of Formula I. It is understood that any substitutions are chemically
feasible and attached
at any available atom to provide a stable compound.
19
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
[00271 "Heteroaryl" alone or in combination refers to a monocyclic aromatic
ring structure
containing 5 or 6 ring atoms, or a bicyclic aromatic group having 8 to 10
atoms, containing one or
more, preferably 1-4, more preferably 1-3, even more preferably 1-2,
heteroatoms independently
selected from the group consisting of 0, S, and N. Heteroaryl is also intended
to include oxidized S
or N, such as sulfmyl, sulfonyl and N-oxide of a tertiary ring nitrogen. The
point of attachment of the
heteroaryl ring is chemically feasible, at a carbon or nitrogen atom, such
that a stable compound is
provided. Examples of heteroaryl groups include, but are not limited to,
pyridinyl, pyridazinyl,
pyrazinyl, quinaoxalyl, indolizinyl, benzo[b]thienyl, quinazolinyl, purinyl,
indolyl, quinolinyl,
pyrimidinyl, pyrrolyl, pyrazolyl, oxazolyl, thiazolyl, thienyl, isoxazolyl,
oxathiadiazolyl, isothiazolyl,
tetrazolyl, imidazolyl, triazolyl, furanyl, benzofuryl, and indolyl.
Hetcroaryl that is "optionally
substituted" denotes heteroaryl that is optionally independently substituted,
unless indicated
otherwise, with one or more, preferably 1, 2, 3, 4 or 5, also 1, 2, or 3
substituents as indicated, for
example, in the description of compounds of Formula I. It is understood that
substitutions are
chemically feasible and attached at any available atom to provide a stable
compound.
[0028] "Lower alkoxy" denotes the group -OR', where R' is lower alkyl. Lower
alkoxy that is
"optionally substituted" denotes lower alkoxy in which R' is lower alkyl
optionally independently
substituted with one or more substituents as indicated herein, for example, in
the description of
compounds of Formula I. Preferably, substitution of lower alkoxy is with 1, 2,
3, 4, or 5 substituents,
also 1, 2, or 3 substituents, For example "fluoro substituted lower alkoxy"
denotes lower alkoxy in
which the lower alkyl is substituted with one or more fluoro atoms, where
preferably the lower alkoxy
is substituted with 1, 2, 3, 4 or 5 fluoro atoms, also 1, 2, or 3 fluoro
atoms. It is understood that any
substitutions on alkoxy are chemically feasible and attached at any available
atom to provide a stable
compound.
10029] "Lower alkylthio" denotes the group -SR", where Re is lower alkyl.
Lower alkylthio that is
'optionally substituted" denotes lower alkylthio in which Rh is lower alkyl
substituted with one or
more substituents as indicated herein, for example, in the description of
compounds of Formula I.
Preferably, substitution of lower alkylthio is with 1, 2, 3, 4, or 5
substituents, also 1, 2, or 3
substituents. For example "fluoro substituted lower alkylthio" denotes lower
alkylthio in which the
lower alkyl is substituted with one or more fluoro atoms, where preferably the
lower alkylthio is
substituted with 1, 2, 3, 4 or 5 fluoro atoms, also 1, 2, or 3 fluoro atoms.
It is understood that any
substitutions on alkylthio are chemically feasible and attached at any
available atom to provide a
stable compound.
[0030] "Amino" denotes the group -NH2. "Mono-alkylamino" denotes the group -
NHR where R` is
lower alkyl. "Di-alkylamino" denotes the group -NR`Rd, where R` and Rd are
independently lower
alkyl, "Cycloalkylaminco" denotes the group -NReR', where R' and Rf combine
with the nitrogen to
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
form a 5-7 membered heterocycloalkyl, where the heterocycloalkyl may contain
an additional
heteroatom within the ring, such as 0, N, or S, and may also be further
substituted with lower alkyl.
Examples of 5-7 membered heterocycloalkyl include, but are not limited to,
piperidine, piperazine,
4-methylpiperazine, morpholine, and thiomorpholine. It is understood that when
mono-alkylamino,
di-alkylamino, or cycloalkylamino are substituents on other moieties, these
substitutions are
chemically feasible and attached at any available atom to provide a stable
compound.
[0031] The terms "animal subject", "subject" and the like refer to human and
non-human
vertebrates, e.g. mammals, such as non-human primates, sports and commercial
animals, e.g.,
equines, bovines, porcines, ovines, rodents, and pets, e.g., canines and
felines.
[0032] As used herein, the term "solid form" refers to a solid preparation
(i.e. a preparation that is
neither gas nor liquid) of a pharmaceutically active compound that is suitable
for administration to an
intended animal subject for therapeutic purposes. The solid form includes any
complex, such as a
salt, co-crystal or an amorphous complex, as well as any polymorph of the
compound. The solid form
may be substantially crystalline, semi-crystalline or substantially amorphous.
The solid form may be
administered directly or used in the preparation of a suitable composition
having improved
pharmaceutical properties. For example, the solid form may be used in a
formulation comprising at
least one pharmaceutically acceptable carrier or excipient.
[0033] As used herein, the term "substantially crystalline" material embraces
material which has
greater than about 90% crystallinity; and "crystalline" material embraces
material which has greater
than about 98% crystallinity.
[0034] As used herein, the term "substantially amorphous" material embraces
material which has no
more than about 10% crystallinity; and "amorphous" material embraces material
which has no more
than about 2% crystallinity.
[0035] As used herein, the term "semi-crystalline" material embraces material
which is greater than
10% crystallinity, but no greater than 90% crystallinity; preferably "semi-
crystalline" material
embraces material which is greater than 20% crystallinity, but no greater than
80% crystallinity. In
one aspect of the present invention, a mixture of solid forms of a compound
may be prepared, for
example, a mixture of amorphous and crystalline solid forms, e.g. to provide a
"semi-crystalline"
solid form. Such a "semi-crystalline" solid form may be prepared by methods
known in the art, for
example by mixing an amorphous solid form with a crystalline solid form in the
desired ratio. In
some instances, a compound mixed with acid or base forms an amorphous complex;
a semi-crystalline
solid can be prepared employing an amount of compound component in excess of
the stoichiometry of
the compound and acid or base in the amorphous complex, thereby resulting in
an amount of the
21
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
amorphous complex that is based on the stoichiometry thereof, with excess
compound in a crystalline
form. The amount of excess compound used in the preparation of the complex can
be adjusted to
provide the desired ratio of amorphous complex to crystalline compound in the
resulting mixture of
solid forms. For example, where the amorphous complex of acid or base and
compound has a 1:1
stoichiometry, preparing said complex with a 2:1 mole ratio of compound to
acid or base will result in
a solid form of 50% amorphous complex and 50% crystalline compound. Such a
mixture of solid
forms may be beneficial as a drug product, for example, by providing an
amorphous component
having improved biopharmaceutical properties along with the crystalline
component. The amorphous
component would be more readily bioavailable while the crystalline component
would have a delayed
bioavailablity. Such a mixture may provide both rapid and extended exposure to
the active
compound.
[0036] As used herein, the tern "complex" refers to a combination of a
pharmaceutically active
compound and an additional molecular species that forms or produces a new
chemical species in a
solid form. In some instances, the complex may be a salt, i.e. where the
additional molecular species
provides an acid/base counter ion to an acid/base group of the compound
resulting in an acid:base
interaction that forms a typical salt. While such salt forms are typically
substantially crystalline, they
can also be partially crystalline, substantially amorphous, or amorphous
forms. In some instances, the
additional molecular species, in combination with the pharmaceutically active
compound, forms a
non-salt co-crystal, i.e. the compound and molecular species do not interact
by way of a typical
acid:base interaction, but still form a substantially crystalline structure.
Co-crystals may also be
formed from a salt of the compound and an additional molecular species. In
some instances, the
complex is a substantially amorphous complex, which may contain salt-like
acid:base interactions that
do not form typical salt crystals, but instead form a substantially amorphous
solid, i.e. a solid whose
X-ray powder diffraction pattern exhibits no sharp peaks (e.g. exhibits an
amorphous halo),
[0037] As used herein, the term "stoichiometry" refers to the molar ratio of
two or more reactants
that combine to form a complex, for example, the molar ratio of acid or base
to compound that form
an amorphous complex. For example, a 1:1 mixture of acid or base with compound
(i.e. 1 mole acid
or base per mole of compound) resulting in an amorphous solid form has a 1:1
stoichiometry.
10038] As used herein, the term "composition" refers to a pharmaceutical
preparation suitable for
administration to an intended animal subject for therapeutic purposes that
contains at least one
pharmaceutically active compound, including any solid form thereof, The
composition may include
at least one pharmaceutically acceptable component to provide an improved
formulation of the
compound, such as a suitable carrier or excipient.
22
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
100391 The term "pharmaceutically acceptable" indicates that the indicated
material does not have
properties that would cause a reasonably prudent medical practitioner to avoid
administration of the
material to a patient, taking into consideration the disease or conditions to
be treated and the
respective route of administration. For example, it is commonly required that
such a material be
essentially sterile, e.g., for injectibles.
100401 In the present context, the term "therapeutically effective" or
"effective amount" indicates
that the materials or amount of material is effective to prevent, alleviate,
or ameliorate one or more
symptoms of a disease or medical condition, and/or to prolong the survival of
the subject being
treated.
10041] In the present context, the terms "synergistically effective" or
"synergistic effect" indicate
that two or more compounds that are therapeutically effective, when used in
combination, provide
improved therapeutic effects greater than the additive effect that would be
expected based on the
effect of each compound used by itself.
10042] The term "inhibit" is used to refer to a decrease the activity of a
target biomolecule, e.g., an
enzyme such as a protease. Generally an inhibitor will be a small molecule,
i.e. a compound with a
molecular weight of 1500 daltons or less, or preferably 1000 daltons or less,
800 daltons or less, or
600 daltons or less. As such, a compound that inhibits renin (a renin
inhibitor) will decrease the
activity of renin, for example, the compound will reduce the conversion by
renin of angiotensinogen
to angiotensin I.
Renin Angiotensin System
[0043] The renin-angiotensin system (RAS) is involved in the regulation of
blood pressure and fluid
homeostasis. Renin is an endopeptidase of approximately 40 kDa that is
produced and secreted by the
juxtaglomerular cells of the kidney, and circulates through the blood. It is
also produced locally in
tissues, including, but not limited to brain, heart, vasculature, adipose
tissue, adrenal glands, pancreas,
placenta and kidney, as well as in mast cells, and thus is active in a variety
of tissues. Renin
specifically cleaves angiotensinogen, a naturally occurring plasma
glycoprotein, to form angiotensin I.
Angiotensin-converting enzyme (ACE) acts in the lungs, kidneys, or other
tissues to cleave
angiotensin 1, forming angiotensin 11. Angiotensin II constricts blood vessels
and acts to release
aldostcrone from the adrenal gland, resulting in a significant increase in
blood pressure. Thus, any
mechanism to block the activity of angiotensin II, for example, by inhibiting
the formation of
angiotensin II or by blocking the receptors of angiotensin II may be effective
in treating
cardiovascular diseases such as hypertension and congestive heart failure.
While ACE inhibitors may
be useful in such treatment, these inhibitors interfere with other functions
of the enzyme as well,
23
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
including degradation of bradykinin, enkcphalins, and substance P, which may
result in undesired side
effects. Additional concerns in using ACE inhibitors or angiotensin II
receptors involve feedback
whereby renin increases, resulting in increased angiotensin 1, which can
overwhelm the ACE
inhibitor, or increase in angiotensin II, which can overwhelm the angiotensin
II receptor antagonists.
Renin, by contrast, is very specific to cleaving angiotensinogen, and
inhibitors of renin are expected
to have few side effects. The renin inhibitor aliskerin was approved for use
in treating hypertension in
March of 2007.
[0044] In addition to hypertension and other cardiovascular diseases, blocking
of the renin-
angiotensin system, for example with renin inhibitor, can provide therapeutic
benefits in a variety of
diseases. Segall et al. (Nephrol Dial Transplant, 2007, 22: 2435-2439)
discusses the use of renin
inhibitors in treating nephropathies, including chronic kidney disease,
diabetic nephropathy and
proteinuria, as well as treating cardiovascular diseases, including heart
failure and myocardial
infarction. Discussion of renin-angiotensin system inhibition in renal
diseases is also found in
Remuzzi ct al. (Annals of Internal Medicine, 2002, 136(8): 604-615), MacKinnon
et al. (American
Journal of Kidney Diseases, 2006, 48(1): 8-20), Russo et al. (American Journal
of Kidney Diseases,
2001, 38(1): 18-25), and Kelly et al. (Diabetologia, 2007, 50: 2398-2404).
[0045] Renin inhibitors may also be used to treat fibrosis, for example
fibrotic diseases in various
tissues, including liver (e.g. cirrhosis, as caused, for example, by viral
hepatitis, schistosomiasis or
alcoholism), lung (e.g. interstitial lung diseases, such as idiopathic
pulmonary disease, sarcoidosis,
silicosis, drug reactions and infections, as well as collagen vascular
diseases, such as rheumatoid
arthritis and systemic sclerosis), kidney (e.g. diabetic nephropathy), heart
and vascular (e.g. post
myocardial infarction, hypertension, atherosclerosis and restenosis), eye
(e.g. macular degeneration,
retinal and vitreal retinopathy), skin (e.g. keloids, hypcrtrophic scars, and
sclerodcrma), pancreas (e.g.
pancreatitis), intestine (e.g. inflammatory bowel disease), and brain (e,g.
Alzheimer's disease and
AIDS). Fibrosis results from, for example, chronic inflammation, which is
induced by a variety of
stimuli, such as infection, autoimmune reaction, allergic reaction, chemical
insults, radiation, or tissue
injury, Discussion of the role of the renin-angiotensin system in fibrosis is
found in T.A. Wynn
(Journal of Pathology, 2008, 214: 199-210), Kisseleva et al. (Journal of
Gastroenterology and
Hepatology, 2007, 22 (Suppl. 1): S73-S78), Zhang et al. (Current Bye Research,
2007, 32(10): 883-
889), Nagai et al. (Investigative Ophthalmology & Visual Science, 2007, 48(5):
2321-2326;
Investigative Ophthalmology & Visual Science, 2005, 46(8): 2925-2931: and
Investigative
Ophthalmology & Visual Science, 2005, 46(3): 1078-1084), Yoshiji et al.
(Journal of
Gastroenterology and Ilepatology, 2007, 22(Suppl. 1): S93-S95), Yao et al.
(Respiration, 2006, 73:
236-242), Kuba et al. (Current Opinion in Pharmacology, 2006, 6:271-276), and
Kurihara et al.
(Investigative Ophthalmology & Visual Science, 2006, 47(12): 5545-5552),
Inhibition of renin
24
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
produced by mast cells in various tissues may also be used to treat a variety
of diseases, as discussed
in Veerappan ct al. (PNAS, 2008, 105(4): 1315-1320) and Reid et al.
(Immunological Reviews, 2007,
217: 123-140).
[00461 Renin inhibitors may be useful in treating cardiovascular and
cardiovascular-related diseases,
such as hypertension, essential hypertension, malignant hypertension,
pulmonary hypertension,
atherosclerosis, congestive heart failure, coronary artery disease, myocardial
infarction, cardiac
hypertrophy, cardiac fibrosis, ischemic or hemorrhagic stroke, stenosis of
arteries, restenosis after
angioplasty or other interventions, myocarditis, cardiomyopathy (e.g. diabetic
cardiac myopathy, post-
infarction cardiac myopathy), arrhythmia (e.g. supraventricular and
ventricular arrhythmia, atrial
fibrillation and atrial flutter), myocardial ischemia, cardiac insufficiency,
unstable coronary syndrome,
detrimental vascular remodeling, angina (stable or unstable), diastolic
dysfunction, left ventricular
dysfunction, and abnormal vascular growth; pulmonary-related diseases, such as
chronic obstructive
pulmonary disease, cor pulmonale, bronchiectasis, acute respiratory distress
syndrome, acute lung
injury, asthma, bronchiolitis obliterans, bronchiolitis obliterans with
organizing pneumonia, cystic
fibrosis, idiopathic pulmonary fibrosis, connective tissue or autoimmune
disease-related pulmonary
fibrosis (e.g. resulting from scleroderma, systemic lupus erythematosus,
rheumatoid arthritis, or
polymyositis), hypersensitivity pneumonitis, eosinophilic granuloma
(Langerhan's cell histiocytosis),
chronic cosinophilic pneumonia, Wegener's granulomatosis, idiopathic pulmonary
hemosiderosis,
lymphangioleiomyomatosis, silicosis, asbestosis, berylliosis, sarcoidosis,
systemic sclerosis, atypical
pneumonia, pneumocystis pneumonia, Hamman-Rich syndrome, histiocytosis X,
collagen vascular
disease, granulomatous vasculitis, Goodpasture's syndrome, Hermansky-Pudlak
syndrome,
pulmonary alveolar proteinosis, lung cancer, and tuberculosis; vasculitis and
autoimmune disease and
related tissue fibrosis, such as fibrosis caused by sclerodactyly,
scleroderma, amyloidosis, systemic
sclerosis, dermatomyositis, dupytren's contracture, peyronie's disease,
polymyositis, amyloidosis,
atrophoderma of Pasini and Pierini, Raynaud's phenomenon, Still's disease,
eosinophilic fasciitis,
Hutchinson-Gilford progeria syndrome, Lichen myxedematosus, mixed connective
tissue disease,
morphoea, porphyria cutanea tarda type 1, scleredema adultorum, systemic
sclerosis or lymphangitis
carcinomatosis; Nephropathies, such as diabetic nephropathy, IgA nephropathy,
Fabry nephropathy,
renal fibrosis, nephritis, lupus nephritis, glomerulonephritis, glomerular
sclerosis, renal vascular
hypertension, hyperaldosteronism, chronic kidney disease, renal insufficiency,
renal ischemia, renal
failure, and renal colic; liver diseases, such as hepatocellular carcinoma,
liver fibrosis, liver cirrhosis,
and hepatitis; diabetes and complications resulting from diabetes, such as
nephropathy, vasculopathy,
neuropathy, and diabetic retinopathy; and other diseases, including metabolic
syndrome, obesity,
thyroiditis, gastritis, peptic ulcer, inflammatory bowel syndrome (e.g.
ulcerative colitis, Crohn's
disease), multiple sclerosis, rheumatoid arthritis, osteoarthritis,
osteoporosis, endometriosis,
preeclampsia, brain infarction, interstitial cystitis, pancreatitis,
pancreatic cancer, erectile dysfunction,
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
proteinuria, albuminuria, cognitive impairment, Alzheimer's disease, dementia,
anxiety states,
cognitive disorders, increased intraocular pressure, glaucoma, angiogenesis
related disorders (e.g.
macular degeneration), carcinoid tumor, graft versus host disease
(transplant), Sjogren's disease,
uveitis, pemphigus, bullous pemphigoid, epidermolysis bullosa, aphthous
ulcers, stomatitis, Beheet's
disease, and other diseases known to be related to the the Renin Angiotensin
System.
Aspartyl Protease Activity Assays
100471 A number of different assays for renin activity can be utilized for
assaying for the ability of a
compound to inhibit renin. Such assays can be formatted either in a
fluorescence resonance energy
transfer (FRET) format, or using an AlphaScreen (amplified luminescent
proximity homogeneous
assay) format by varying the donor and acceptor reagents accordingly. Similar
assays may be utilized
for assaying the activity against other aspartyl proteases, or other
proteases, in order to assess the
selectivity of the compound for inhibition of renin. One of ordinary skill in
the art will know of
assays that can be utilized or modified for a particular application,
including, for example, the assays
described in the Examples below. Further, animal models (e.g. rat, marmoset)
known in the art may
be used to test the activity of renin inhibitors in vivo.
Organic Synthetic Techniques
100481 A wide array of organic synthetic techniques exist in the art to
facilitate the preparation of
potential inhibitors. Many of these organic synthetic methods are described in
detail in standard
reference sources utilized by those skilled in the art. One example of such a
reference is March, 1994,
Advanced Or anic Chemistry; Reactions, Mechanisms and Structure, New York,
McGraw Hill.
Thus, the techniques useful to synthesize a potential renin inhibitor are
readily available to those
skilled in the art of organic chemical synthesis.
Alternative Compound Forms or Derivatives
100491 Compounds contemplated herein are described with reference to both
generic formulae and
specific compounds. In addition, invention compounds may exist in a number of
different forms or
derivatives, all within the scope of the present invention. Alternative forms
or derivatives, include,
for example, (a) prodrugs, and active metabolites (b) tautomers, isomers
(including stereoisomers and
regioisomers), and racemic mixtures (c) pharmaceutically acceptable salts and
(d) solid forms,
including different crystal forms, polymorphic or amorphous solids, including
hydrates and solvates
thereof. and other forms.
26
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
(a) Prodrugs and Metabolites
[00501 In addition to the present formulae and compounds described herein, the
invention also
includes prodrugs (generally pharmaceutically acceptable prodrugs), active
metabolic derivatives
(active metabolites), and their pharmaceutically acceptable salts.
[0051] Prodrugs are compounds or pharmaceutically acceptable salts thereof
which, when
metabolized under physiological conditions or when converted by solvolysis,
yield the desired active
compound. Prodrugs include, without limitation, esters, amides, carbamates,
carbonates, ureides,
solvates, or hydrates of the active compound. Typically, the prodrug is
inactive, or less active than
the active compound, but may provide one or more advantageous handling,
administration, and/or
metabolic properties. For example, some prodrugs are esters of the active
compound; during
metabolysis, the ester group is cleaved to yield the active drug. Esters
include, for example, esters of
a carboxylic acid group, or S-acyl or O-acyl derivatives of thiol, alcohol, or
phenol groups. In this
context, a common example is an alkyl ester of a carboxylic acid. Some
prodrugs are activated
enzymatically to yield the active compound, or a compound may undergo further
chemical reaction to
yield the active compound. Prodrugs may proceed from prodrug form to active
form in a single step
or may have one or more intermediate forms which may themselves have activity
or may be inactive.
Prodrug of a compound of Formula I includes, for example, a carbamate
derivative of the primary
amine substitution at the 7 position, i.e. a prodrug includes where the amine
at this position is in the
form of NH-C(O)-OR, where R, for example, is alkyl, such that the compound is
metabolized to give
the free NH2 at this position.
[0052] As described in The Practice of Medicinal Chemistry, Ch, 31-32 (Ed.
Wermuth, Academic
Press, San Diego, CA, 2001), prodrugs can be conceptually divided into two non-
exclusive categories,
bioprecursor prodrugs and carrier prodrugs. Generally, bioprecursor prodrugs
are compounds that are
inactive or have low activity compared to the corresponding active drug
compound, that contain one
or more protective groups and are converted to an active form by metabolism or
solvolysis. Both the
active drug form and any released metabolic products should have acceptably
low toxicity. Typically,
the formation of active drug compound involves a metabolic process or reaction
that is one of the
following types:
[0053] Oxidative reactions Oxidative reactions are exemplified without
limitation by reactions
such as oxidation of alcohol, carbonyl, and acid functionalities,
hydroxylation of aliphatic carbons,
hydroxylation of alicyclic carbon atoms, oxidation of aromatic carbon atoms,
oxidation of carbon-
carbon double bonds, oxidation of nitrogen-containing functional groups,
oxidation of silicon,
phosphorus, arsenic, and sulfur, oxidative N-dealkylation, oxidative 0- and S-
dealkylation, oxidative
deamination, as well as other oxidative reactions.
27
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
[0054] Reductive reactions: Reductive reactions are exemplified without
limitation by reactions
such as reduction of carbonyl functionalitites, reduction of alcohol
functionalities and carbon-carbon
double bonds, reduction of nitrogen-containing functional groups, and other
reduction reactions.
[00551 Reactions without change in the oxidation state: Reactions without
change in the state of
oxidation arc exemplified without limitation to reactions such as hydrolysis
of esters and ethers,
hydrolytic cleavage of carbon-nitrogen single bonds, hydrolytic cleavage of
non-aromatic
heterocycles, hydration and dehydration at multiple bonds, new atomic linkages
resulting from
dehydration reactions, hydrolytic dehalogenation, removal of hydrogen halide
molecule, and other
such reactions.
[0056] Carrier prodrugs are drug compounds that contain a transport moiety,
e.g., that improves
uptake and/or localized delivery to a site(s) of action. Desirably for such a
carrier prodrug, the
linkage between the drug moiety and the transport moiety is a covalent bond,
the prodrug is inactive
or less active than the drug compound, the prodrug and any release transport
moiety are acceptably
non-toxic. For prodrugs where the transport moiety is intended to enhance
uptake, typically the
release of the transport moiety should be rapid. In other cases, it is
desirable to utilize a moiety that
provides slow release, e.g., certain polymers or other moieties, such as
cyclodextrins. (See, e.g.,
Cheng et al., U.S. Patent Publ. No. 20040077595, App. No. 10/656,838,
incorporated herein by
reference.) Such carrier prodrugs are often advantageous for orally
administered drugs. In some
instances, the transport moiety provides targeted delivery of the drug, for
example the drug maybe
conjugated to an antibody or antibody fragment. Carrier prodrugs can, for
example, be used to
improve one or more of the following properties: increased lipophilicity,
increased duration of
pharmacological effects, increased site-specificity, decreased toxicity and
adverse reactions, and/or
improvement in drug formulation (e.g., stability, water solubility,
suppression of an undesirable
organoleptic or physiochemical property). For example, lipophilicity can be
increased by
esterification of hydroxyl groups with lipophilic carboxylic acids, or of
carboxylic acid groups with
alcohols, e.g., aliphatic alcohols. Wermuth, supra.
[0057] Metabolites, e.g., active metabolites, overlap with prodrugs as
described above, e.g.,
bioprecursor prodrugs. Thus, such metabolites are pharmacologically active
compounds or
compounds that further metabolize to pharmacologically active compounds that
are derivatives
resulting from metabolic processes in the body of a subject. Of these, active
metabolites are such
pharmacologically active derivative compounds. For prodrugs, the prodrug
compound is generally
inactive or of lower activity than the metabolic product. For active
metabolites, the parent compound
may be either an active compound or may be an inactive prodrug. For example,
in some compounds,
one or more alkoxy groups can be metabolized to hydroxyl groups while
retaining pharmacologic
activity and/or carboxyl groups can be esterified, e.g., glueuronidation. In
some cases, there can be
28
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
more than one metabolite, where an intermediate metabolite(s) is further
metabolized to provide an
active metabolite. For example, in some cases a derivative compound resulting
from metabolic
glucuronidation may be inactive or of low activity, and can be further
metabolized to provide an
active metabolite.
[00581 Metabolites of a compound may be identified using routine techniques
known in the art, and
their activities determined using tests such as those described herein. See,
e.g., Bertolini et al., 1997,
J. Med. Chem., 40:2011-2016; Shan et al., 1997, JPharm Sci 86(7):756-757;
Bagshawe, 1995, Drug
Dcv. Res., 34:220-230; Wermuth, supra.
(b) Tautomers, Stereoisomers, and Regloisomers
[00591 It is understood that some compounds may exhibit tautomerism. In such
cases, the formulae
provided herein expressly depict only one of the possible tautomeric forms. It
is therefore to be
understood that the formulae provided herein are intended to represent any
tautomeric form of the
depicted compounds and are not to be limited merely to the specific tautomeric
form depicted by the
drawings of the formulae.
[00601 Likewise, some of the compounds according to the present invention may
exist as
stereoisomers, i.e, having the same atomic connectivity of covalently bonded
atoms yet differing in
the spatial orientation of the atoms. For example, compounds may be optical
stereoisomers, which
contain one or more chiral centers, and therefore, may exist in two or more
stereoisomeric forms (e.g.
cnantiomers or diastereomers). Thus, such compounds may be present as single
stereoisomers (i.e.,
essentially free of other stereoisomers), racemates, and/or mixtures of
enantiomers and/or
diastereomers. As another example, stercoisomers include geometric isomers,
such as cis- or trans-
orientation of substituents on adjacent carbons of a double bond. All such
single stereoisomers,
racemates and mixtures thereof are intended to be within the scope of the
present invention. Unless
specified to the contrary, all such steroisomeric forms are included within
the formulae provided
herein.
[00611 In some embodiments, a chiral compound of the present invention is in a
form that contains
at least 80% of a single isomer (60% enantiomeric excess ("e.e.") or
diastereomeric excess ("d.e.")),
or at least 85% (70% c.c. or d.c.), 90 ,/0 (80% e.e. or d.e.), 95% (90% e.e.
or d.c,), 97.5% (95% e.e. or
d.e.), or 99% (98% e.e, or d.e.). As generally understood by those skilled in
the art, an optically pure
compound having one chiral center is one that consists essentially of one of
the two possible
enantiomers (i.e., is enantiomerically pure), and an optically pure compound
having more than one
chiral center is one that is both diastereomerically pure and enantiomerically
pure. In some
embodiments, the compound is present in optically pure form, such optically
pure form being
29
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
prepared and/or isolated by methods known in the art (e.g. by
recrystallization techniques, chiral
synthetic techniques (including synthesis from optically pure starting
materials), and chromatographic
separation using a chiral column.
(c) Pharmaceutically acceptable salts
[00621 Unless specified to the contrary, specification of a compound herein
includes
pharmaceutically acceptable salts of such compound. Thus, compounds of Formula
I can be in the
form of pharmaceutically acceptable salts, or can be formulated as
pharmaceutically acceptable salts.
Contemplated pharmaceutically acceptable salt forms include, without
limitation, mono, bis, tris,
tctrakis, and so on. Pharmaceutically acceptable salts are non-toxic in the
amounts and concentrations
at which they are administered. The preparation of such salts can facilitate
the pharmacological use
by altering the physical characteristics of a compound without preventing it
from exerting its
physiological effect. Useful alterations in physical properties include
lowering the melting point to
facilitate transmucosal administration and increasing the solubility to
facilitate administering higher
concentrations of the drug. A compound of the invention may possess a
sufficiently acidic, a
sufficiently basic, or both functional groups, and accordingly can react with
any of a number of
inorganic or organic bases, and inorganic and organic acids, to form a
pharmaceutically acceptable
salt.
100631 Pharmaceutically acceptable salts include acid addition salts such as
those containing
chloride, bromide, iodide, hydrochloride, acetate, dichloroacetate,
phenylacetate, acrylate, ascorbate,
aspartate, benzoate, 2-phenoxybenzoate, 2-acetoxybenzoate, dinitrobenzoate,
hydroxybenzoate,
methoxybenzoate, methylbenzoate, bicarbonate, butyne-1,4 dioate, hexyne-l,6-
dioate, caproate,
caprylate, chlorobenzoate, cinnamate, citrate, decanoate, formate, fumarate,
glycolate, gluconate,
glucarate, glucuronate, glucose-6-phosphate, glutamate, heptanoate, hexanoate,
isethionate,
isobutyrate, gamma-hydroxybutyrate, phenylbutyrate, lactate, malate, maleate,
hydroxymaleate,
methylmaleate, malonate, mandelate, nicotinate, nitrate, isonicotinate,
octanoate, oleate, oxalate,
pamoate, phosphate, monohydrogenphosphate, dihydrogenphosphate,
orthophosphate,
metaphosphate, pyrophosphate, 2-phosphoglycerate, 3-phosphoglyceratc,
phthalate, propionate,
phenylpropionatc, propiolate, pyruvate, quinate, salicylate, 4-amino
salicylate, sebacate, stearate,
suberate, succinate, sulfate, pyrosulfate, bisulfate, sulfite, bisulfite,
sulfamate, sulfonate,
benzenesulfonate (i.e, besylate), ethanesulfonate (i.e. esylate), ethane-1,2-
disulfonate,
2-hydroxyethanesulfonatc (i.e. isethionate), methanesulfonate (i.e. mesylate),
naphthalene-l -
sulfonate, naphthalene-2-sulfonate (i,e. napsylate), propanesulfonate, p-
toluenesulfonate (i.e.
tosylate), xylenesulfonates, eyelohexylsulfain ate, tartrate, and
trifluoroacetate. These
pharmaceutically acceptable acid addition salts can be prepared using the
appropriate corresponding
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
[0064] When acidic functional groups, such as carboxylic acid or phenol are
present,
pharmaceutically acceptable salts also include basic addition salts such as
those containing
bcnzathine, chloroprocaine, choline, ethanolamine, diethanolamine,
triethanolamine, t-butylamine,
dicyclohexylamine, ethylenediamine, N,N'-d ibenzylethylenediamine, megluminc,
hydroxyethylpyrrolidine, piperidine, morpholine, piperazine, procaine,
aluminum, calcium, copper,
iron, lithium, magnesium, manganese, potassium, sodium, zinc, ammonium, and
mono-, di-, or tri-
alkylamines (e.g. diethylamine), or salts derived from amino acids such as L-
histidine, L-glycine,
L-lysine, and L-arginine. For example, see Rernington's Pharmaceutical
Sciences, 19th ed., Mack
Publishing Co., Easton, PA, Vol. 2, p. 1457, 1995. These pharmaceutically
acceptable base addition
salts can be prepared using the appropriate corresponding base.
[0065] Pharmaceutically acceptable salts can be prepared by standard
techniques. For example, the
free-base form of a compound can be dissolved in a suitable solvent, such as
an aqueous or aqueous-
alcohol solution containing the appropriate acid and then isolated by
evaporating the solution. In
another example, a salt can be prepared by reacting the free base and acid in
an organic solvent. If the
particular compound is an acid, the desired pharmaceutically acceptable salt
may be prepared by any
suitable method, for example, treatment of the free acid with an appropriate
inorganic or organic base.
[0066] The pharmaceutically acceptable salt of the different compounds may be
present as a
complex. Examples of complexes include 8-chlorotheophylline complex (analogous
to, e.g.,
dimenhydrinate: diphenhydramine 8-chlorotheophylline (1:1) complex; Dramamine)
and various
cyclodextrin inclusion complexes.
(d) Other compound forms
[0067] In the case of agents that are solids, it is understood by those
skilled in the art that the
compounds and salts may exist in different crystal or polymorphic forms, or
may be formulated as co-
crystals, or may be in an amorphous form, or may be any combination thereof
(e.g. partially
crystalline, partially amorphous, or mixtures of polymorphs) all of which are
intended to be within the
scope of the present invention and specified formulae. Whereas salts are
formed by acid/base
addition, i.e. a free base or free acid of the compound of interest forms an
acid/base reaction with a
corresponding addition base or addition acid, respectively, resulting in an
ionic charge interaction, co-
crystals are a new chemical species that is formed between neutral compounds,
resulting in the
compound and an additional molecular species in the same crystal structure,
[0068] In some instances, compounds of the invention are complexed with an
acid or a base,
including base addition salts such as ammonium, diethylamine, ethanolamine,
ethyl enediamine,
diethanolamine, t-butylamine, piperazine, meglumine; acid addition salts, such
as acetate,
31
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
acetylsalicylate, besylate, camsylate, citrate, formate, fumarate, glutarate,
hydrochlorate, maleatc,
mesylate, nitrate, oxalate, phosphate, succinate, sulfate, tartrate,
thiocyanate and tosylate; and amino
acids such as alanine, arginine, asparagine, aspartic acid, cysteine,
glutamine, glutamic acid, glycine,
histidine, isoleucine, leucine, lysine, methionine, phenylalanine, proline,
serine, threonine, tryptophan,
tyrosine or valine. In combining the compound of the invention with the acid
or base, an amorphous
complex is preferably formed rather than a crystalline material such as a
typical salt or co crystal. In
some instances, the amorphous form of the complex is facilitated by additional
processing, such as by
spray-drying, mechanochcmical methods such as roller compaction, or microwave
irradiation of the
parent compound mixed with the acid or base. Such amorphous complexes provide
several
advantages. For example, lowering of the melting temperature relative to the
free base facilitiates
additional processing, such as hot melt extrusion, to further improve the
biopharmaceutical properties
of the compound. Also, the amorphous complex is readily friable, which
provides improved
compression for loading of the solid into capsule or tablet form.
[00691 Additionally, the formulae are intended to cover hydrated or solvated
as well as unhydrated
or unsolvated forms of the identified structures. For example, the indicated
compounds include both
hydrated and non-hydrated forms. Other examples of solvates include the
structures in combination
with a suitable solvent, such as isopropanol, ethanol, methanol, DMSO, ethyl
acetate, acetic acid, or
ethanolamine.
Formulations and Administration
[00701 The methods and compounds will typically be used in therapy for human
subjects.
However, they may also be used to treat similar or identical indications in
other animal subjects.
Compounds of Formula I can be administered by different routes, including
injection (i.e. parenteral,
including intravenous, intraperitoneal, subcutaneous, and intramuscular),
oral, transdermal,
transmucosal, rectal, or inhalant. Such dosage forms should allow the compound
to reach target cells.
Other factors are well known in the art, and include considerations such as
toxicity and dosage forms
that retard the compound or composition from exerting its effects. Techniques
and formulations
generally may be found in Remington: The Science and Practice of Pharmacy, 210
edition,
Lippincott, Williams and Wilkins, Philadelphia, PA, 2005 (hereby incorporated
by reference herein).
[00711 In some embodiments, compositions will comprise pharmaceutically
acceptable carriers or
excipients, such as fillers, binders, disintegrants, glidants, lubricants,
complexing agents, solubilizers,
and surfactants, which may be chosen to facilitate administration of the
compound by a particular
route. Examples of carriers include calcium carbonate, calcium phosphate,
various sugars such as
lactose, glucose, or sucrose, types of starch, cellulose derivatives, gelatin,
lipids, liposomes,
nanopartieles, and the like. Carriers also include physiologically compatible
liquids as solvents or for
32
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
suspensions, including, for example, sterile solutions of water for injection
(WFI), saline solution,
dextrose solution, Hank's solution, Ringer's solution, vegetable oils, mineral
oils, animal oils,
polyethylene glycols, liquid paraffin, and the like. Excipients may also
include, for example, colloidal
silicon dioxide, silica gel, talc, magnesium silicate, calcium silicate,
sodium aluminosilicate,
magnesium trisilicate, powdered cellulose, macrocrystalline cellulose,
carboxymethyl cellulose, cross-
linked sodium carboxymethylcellulose, sodium benzoate, calcium carbonate,
magnesium carbonate,
stearic acid, aluminum stearate, calcium stearate, magnesium stearate, zinc
stearate, sodium stearyl
fumarale, syloid, stearowet C, magnesium oxide, starch, sodium starch
glycolate, glyceryl
monostearate, glyceryl dibehenate, glyceryl palmitostearate, hydrogenated
vegetable oil,
hydrogenated cotton seed oil, castor seed oil mineral oil, polyethylene glycol
(e.g, PEG 4000-8000),
polyoxyethylene glycol, poloxamers, povidone, crospovidonc, croscarmellose
sodium, alginic acid,
casein, methacrylic acid divinylbenzene copolymer, sodium docusate,
cyclodextrins (e.g. 2-
hydroxypropyl-.delta.-cyclodextrin), polysorbates (e.g. polysorbate 80),
cetrimide, TPGS (d-alpha-
tocopheryl polyethylene glycol 1000 succinate), magnesium lauryl sulfate,
sodium lauryl sulfate,
polyethylene glycol ethers, di-fatty acid ester of polyethylene glycols, or a
polyoxyalkylene sorbitan
fatty acid ester (e.g., polyoxyethylene sorbitan ester Tween"),
polyoxyethylene sorbitan fatty acid
esters, sorbitan fatty acid ester, e.g. a sorbitan fatty acid ester from a
fatty acid such as oleic, stearic or
palmitic acid, mannitol, xylitol, sorbitol, maltose, lactose, lactose
monohydrate or lactose spray dried,
sucrose, fructose, calcium phosphate, dibasic calcium phosphate, tribasic
calcium phosphate, calcium
sulfate, dextrates, dextran, dextrin, dextrose, cellulose acetate,
maltodextrin, simethicone,
polydextrosem, chitosan, gelatin, HPMC (hydroxypropyl methyl celluloses), HPC
(hydroxypropyl
cellulose), hydroxycthyl cellulose, hypromellose, and the like.
10072] In some embodiments, oral administration may be used. Pharmaceutical
preparations for
oral use can be formulated into conventional oral dosage forms such as
capsules, tablets, and liquid
preparations such as syrups, elixirs, and concentrated drops. Compounds of
Formula I may be
combined with solid excipients, optionally grinding a resulting mixture, and
processing the mixture of
granules, after adding suitable auxiliaries, if desired, to obtain, for
example, tablets, coated tablets,
hard capsules, soft capsules, solutions (e.g. aqueous, alcoholic, or oily
solutions) and the like.
Suitable excipients are, in particular, fillers such as sugars, including
lactose, glucose, sucrose,
mannitol, or sorbitol; cellulose preparations, for example, core starch, wheat
starch, rice starch, potato
starch, gelatin, gum tragacanth, methyl cellulose, hydroxypropylmethyl-
cellulose, sodium
carboxymethylcellulose (CMC), and/or polyvinylpyrrolidone (PVP: povidone);
oily excipients,
including vegetable and animal oils, such as sunflower oil, olive oil, or
codliver oil. The oral dosage
formulations may also contain disintegrating agents, such as the cross-linked
polyvinylpyrrolidone,
agar, or alginic acid, or a salt thereof such as sodium alginate; a lubricant,
such as talc or magnesium
stearate; a plasticizer, such as glycerol or sorbitol; a sweetening such as
sucrose, fructose, lactose, or
33
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
aspartame; a natural or artificial flavoring agent, such as peppermint, oil of
wintergreen, or cherry
flavoring; or dye-stuffs or pigments, which may be used for identification or
characterization of
different doses or combinations. Also provided are dragee cores with suitable
coatings. For this
purpose, concentrated sugar solutions may be used, which may optionally
contain, for example, gum
arabic, talc, poly-vinylpyrrolidone, carbopol gel, polyethylene glycol, and/or
titanium dioxide.. lacquer
solutions, and suitable organic solvents or solvent mixtures,
[0073] Pharmaceutical preparations that can be used orally include push-fit
capsules made of gelatin
("gelcaps"), as well as soft, scaled capsules made of gelatin, and a
plasticizer, such as glycerol or
sorbitol. The push-fit capsules can contain the active ingredients in
admixture with filler such as
lactose, binders such as starches, and/or lubricants such as talc or magnesium
stcaratc and, optionally,
stabilizers. In soft capsules, the active compounds may be dissolved or
suspended in suitable liquids,
such as fatty oils, liquid paraffin, or liquid polyethylene glycols.
[0074] In some embodiments, injection (parenteral administration) may be used,
e.g., intramuscular,
intravenous, intraperitoneal, and/or subcutaneous. Compounds of Formula I for
injection may be
formulated in sterile liquid solutions, preferably in physiologically
compatible buffers or solutions,
such as saline solution, Hank's solution, or Ringer's solution. Dispersions
may also be prepared in
non-aqueous solutions, such as glycerol, propylene glycol, ethanol, liquid
polyethylene glycols,
triacetin, and vegetable oils. Solutions may also contain a preservative, such
as methylparaben,
propylparaben, chlorobutanol, phenol, sorbic acid, thimerosal, and the like.
In addition, the
compounds may be formulated in solid form, including, for example, lyophilized
forms, and
redissolved or suspended prior to use.
[0075] In some embodiments, transmucosal, topical or transdermal
administration may be used. In
such formulations of compounds of Formula I, penetrants appropriate to the
barrier to be permeated
are used. Such penetrants are generally known in the art, and include, for
example, for transmucosal
administration, bile salts and fusidic acid derivatives. In addition,
detergents may be used to facilitate
permeation. Transmucosal administration, for example, may be through nasal
sprays or suppositories
(recta] or vaginal). Compositions of compounds of Formula I for topical
administration may be
formulated as oils, creams, lotions, ointments, and the like by choice of
appropriate carriers known in
the art. Suitable carriers include vegetable or mineral oils, white petrolatum
(white soft paraffin),
branched chain fats or oils, animal fats and high molecular weight alcohol
(greater than CIA). In some
embodiments, carriers are selected such that the active ingredient is soluble,
Emulsifiers, stabilizers,
humectants and antioxidants may also be included as well as agents imparting
color or fragrance, if
desired. Creams for topical application are preferably formulated from a
mixture of mineral oil, self-
emulsifying beeswax and water in which mixture the active ingredient,
dissolved in a small amount of
solvent (e.g., an oil), is admixed. Additionally, administration by
transderral means may comprise a
34
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
transdermal patch or dressing such as a bandage impregnated with an active
ingredient and optionally
one or more carriers or diluents known in the art. To be administered in the
form of a transdermal
delivery system, the dosage administration will be continuous rather than
intermittent throughout the
dosage regimen.
[00761 In some embodiments, compounds are administered as inhalants. Compounds
of Formula I
may be formulated as dry powder or a suitable solution, suspension, or
aerosol. Powders and
solutions may be formulated with suitable additives known in the art. For
example, powders may
include a suitable powder base such as lactose or starch, and solutions may
comprise propylene
glycol, sterile water, ethanol, sodium chloride and other additives, such as
acid, alkali and buffer salts.
Such solutions or suspensions may be administered by inhaling via spray, pump,
atomizer, or
nebulizer, and the like. The compounds of Formula I may also be used in
combination with other
inhaled therapies, for example corticosteroids such as fluticasone
proprionate, beclomethasone
dipropionate, triamcinolone acetonide, budesonide, and mometasone furoate;
beta agonists such as
albuterol, salmeterol, and formoterol; anticholinergic agents such as
ipratroprium bromide or
tiotropium; vasodilators such as treprostinal and iloprost; enzymes such as
DNAase; therapeutic
proteins; immunoglobulin antibodies; an oligonucleotide, such as single or
double stranded DNA or
RNA, siRNA; antibiotics such as tobramycin; muscarinic receptor antagonists;
leukotriene
antagonists; cytokine antagonists; protcasc inhibitors; cromolyn sodium;
nedocril sodium; and sodium
cromoglycate.
[00771 The amounts of various compounds to be administered can be determined
by standard
procedures taking into account factors such as the compound activity (in
vitro, e.g. the compound IC50
vs. target, or in vivo activity in animal efficacy models), pharmacokinetic
results in animal models
(e.g. biological half-life or bioavailability), the age, size, and weight of
the subject, and the disorder
associated with the subject. The importance of these and other factors are
well known to those of
ordinary skill in the art. Generally, a dose will be in the range of about
0.01 to 50 mg/kg, also about
01 to 20 mg/kg of the subject being treated. Multiple doses may be used.
[00781 The compounds of Formula I may also be used in combination with other
therapies for
treating the same disease. Such combination use includes administration of the
compounds and one
or more other therapeutics at different times, or co-administration of the
compound and one or more
other therapies. In some embodiments, dosage may be modified for one or more
of the compounds
of the invention or other therapeutics used in combination, e.g., reduction in
the amount dosed relative
to a compound or therapy used alone, by methods well known to those of
ordinary skill in the art.
[00791 It is understood that use in combination includes use with other
therapies, drugs, medical
procedures etc., where the other therapy or procedure may be administered at
different times (e.g.
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
within a short time, such as within hours (e.g. 1, 2, 3, 4-24 hours), or
within a longer time (e.g. 1-2
days, 2-4 days, 4-7 days, 1-4 weeks)) than a compound of Formula I, or at the
same time as a
compound of Formula 1. Use in combination also includes use with a therapy or
medical procedure
that is administered once or infrequently, such as surgery, along with a
compound of Formula I
administered within a short time or longer time before or after the other
therapy or procedure. In
some embodiments, the present invention provides for delivery of a compound of
Formula I and one
or more other drug therapeutics delivered by a different route of
administration or by the same route
of administration. The use in combination for any route of administration
includes delivery of a
compound of Formula I and one or more other drug therapeutics delivered by the
same route of
administration together in any formulation, including formulations where the
two compounds are
chemically linked in such a way that they maintain their therapeutic activity
when administered. In
one aspect, the other drug therapy may be co-administered with a compound of
Formula 1. Use in
combination by co-administration includes administration of co-formulations or
formulations of
chemically joined compounds, or administration of two or more compounds in
separate formulations
within a short time of each other (e.g. within an hour, 2 hours, 3 hours, up
to 24 hours), administered
by the same or different routes. Co-administration of separate formulations
includes co-
administration by delivery via one device, for example the same inhalant
device, the same syringe,
etc., or administration from separate devices within a short time of each
other. Co-formulations of a
compound of Formula I and one or more additional drug therapies delivered by
the same route
includes preparation of the materials together such that they can be
administered by one device,
including the separate compounds combined in one formulation, or compounds
that are modified such
that they are chemically joined, yet still maintain their biological activity.
Such chemically joined
compounds may have a linkage that is substantially maintained in vivo, or the
linkage may break
down in vivo, separating the two active components.
Examples
100801 Examples related to the present invention are described below. In most
cases, alternative
techniques can be used. The examples are intended to be illustrative and are
not limiting or restrictive
to the scope of the invention. In some examples, the mass spectrometry result
indicated for a
compound may have more than one value due to the isotope distribution of an
atom in the molecule,
such as a compound having a bromo or chloro substituent.
[00811 Unless specifically indicated otherwise, the Formula enumeration and R
group enumeration
used in the following examples is not related to such enumeration in other
sections of this application.
The reagents and solvents used in these examples can be readily substituted
with appropriate
alternatives as are known in the art and isolation of products is readily
achieved by methods known in
the art, including, but not limited to, extraction, crystaIli- i; i - rn. ,knd
chromatographic methods.
36
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
100821 Ring numbering for the 1,2,3,4-tetrahydroquinoline and 3,4-dihydro-IH-
quinolin-2-one
comopunds in the following Examples is as follows:
I I
H & H
2 N 7 O 2 N 7
3 6 3 6
4 5 and 4 5 , respectively.
Example 1: Preparation of 1-(6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-
7-yl)-ethanone
9.
100831 1-(6-Ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl)-ethanone 9
was prepared in six
steps from 4-ethylaniline I as shown in Scheme 1.
Scheme 1
NHz H H H
+ O CI Step 1 O N Step 2 0 T;P4 Step 3 2 3 5
C CF3 F3C\/0 F3CY0 O H O
+ 0 O Step 4 Step 5 Step 6 N O=< 1
CF3 7 8 9
6
Step I - Preparation of 3-metlvl-but-2-enoic acid (4-ethyl phenyl)-amide (3).-
[00841 To a mixture of 4-ethylaniline (1, 100 mL, 0.809 mol) and 730 mL of 10%
aqueous sodium
hydroxide in 454 mL of dichloromethane, 3,3-dimethylacryloyl chloride (2, 115
g, 0.970 mol) in 454
mL of dichloromethanc was slowly added. The reaction mixture was stirred at
room temperature
overnight, poured into 200 mL of water and extracted with 3 x 500 mL of
dichloromethane. The
combined organic layers were dried over sodium sulfate, filtered and the
filtrate concentrated under
vacuum. The resulting material was crystallized using heptane to provide the
desired compound as a
light brown solid that was used without further purification in the next step
(3, 175 g, >100%).
Step 2 - Preparation of 6-ethyl-4, 4-dimethyl-3, 4-dihydro-I H-quinolin-2-one
(4).-
[00851 To a solution of 3-methyl-but-2-enoic acid (4-ethyl-phenyl)-amide (3,
164.4 g, 0.809 mol
theoretical) in 1.6 L of 1,2-dichlorobenzene, aluminum trichloride (162 g,
1.21 mol) was added. The
reaction mixture was stirred at 100 C for 2 hours, allowed to cool to room
temperature and poured
into 7 L of ice water. To this, 1 L of 2M hydrochloric acid was added and the
suspension was
37
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
extracted with 2 x 4 L of heptanes. The organic layers were pooled, washed
with saturated sodium
bicarbonate solution, washed with brine, dried over sodium sulfate, filtered
and the filtrate
concentrated under vacuum. The residue was purified by silica gel column
chromatography eluting
with a gradient of heptane and ethyl acetate. Appropriate fractions were
combined and concentrated
under vacuum, and the resulting material was triturated with heptane to
provide the desired compound
as a white solid (4, 136 g, 83% for steps 1 and 2).
Step 3-Preparation of 6-ethyl-4, 4-dirnet/tyl-1 2,3, 4-tetrahydroquinoline
(5):
[0086] To a solution of 6-ethyl-4,4-dimethyl-3,4-dihydro-1H-quinolin-2-one (4,
70 g, 0.344 mol) in
700 mL of toluene, 140 ml- of borane-dimethyl sulfide complex (1.48 mol) was
slowly added at 0 C.
The reaction mixture was stirred at 0 'C for 15 minutes and then stirred at
100 C for 2 hours. The
reaction mixture was cooled to room temperature and poured slowly into 1 L of
10% aqueous sodium
carbonate. After stirring at room temperature for 30 minutes, the organic
layer was collected. The
aqueous layer was extracted with 1 L of ethyl acetate and the pooled organic
layers were dried over
sodium sulfate, filtered and the filtrate concentrated under vacuum, The
resulting material was
purified by silica gel column chromatography, eluting with a gradient of 0-50%
ethyl acetate in
heptane. Appropriate fractions were combined and concentrated under vacuum to
provide the desired
compound as a clear oil (5, 50 g, 77%).
Step 4 - Preparatoin of 1-(6-ethyl-4,4-dimethyl-3, 4-dihydro-2H-quinolin-I y1)-
2,2,2-trifluoro-
ethanone (7):
[0087] To a solution of 6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydroquinoline (5,
50 g, 0.264 mol) in
500 ml, of dichloromethane, triethylamine (93 mL, 0.660 mol), 4-
dimethylaminopyri dine (3.2 g,
0.026 mol), and trifluoroacetic anhydride (6, 66 mL, 0.475 mol) were added.
The reaction mixture
was stirred at 40 C for 2 hours, allowed to cool to room temperature and
poured into saturated
aqueous sodium bicarbonate. The organic layer was collected and the aqueous
layer was back-
extracted with ethyl acetate. The organic layers were combined and dried over
sodium sulfate,
filtered and the filtrate concentrated under vacuum. The resulting material
was purified by silica gel
column chromatography eluting with a gradient of 0-50% ethyl acetate in
heptane. Appropriate
fractions were combined and concentrated under vacuum to provide the desired
compound as a clear
oil (7, 70 g, 92%).
Step 5 -Preparation of I-(7-(i.cetyl-6-ethyl-4,4-dimethyl-3,4-dihydro-2H
quinolin-1 y!)-2,2,2-
trifluoro-ethanone (8):
[0088] A mixture of 1-(6-ethyl-4,4-dimethyl-3,4-dihydro-2H-quinolin-1-yl)-
2,2,2-trifluoro-
ethanone (7, 100 mg, 0.4 mmol), acetyl chloride (78 mg, 1.0 mmol) and aluminum
trichloride (100
mg, 1.0 mmol) in 5 ml- of nitromethane was warmed to reflux for 5 hours. The
reaction mixture was
cooled to room temperature, poured into water and extracted with ethyl
acetate, The layer
38
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
was washed with saturated sodium bicarbonate solution and brine, dried over
magnesium sulfate,
filtered and the filtrate concentrated under vacuum. The resulting crude
material was purified by
silica gel chromatography to provide the desired compound as a colorless oil
(8, 0.11 g, 90%). MS
(ESI) [M+H-]` = 328.05.
Step 6-Preparation of 1-(6-eth))l-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-
yl)-ethmzone (9):
[0089] To a solution of 10% potassium hydroxide in 5 mL of methanol, 1-(7-
acetyl-6-ethyl-4,4-
dimethyl-3,4-dihydro-2H-quinolin-1-yl)-2,2,2-trifluoro-ethanone (8, 107 mg,
0.327 mol) was added.
The reaction mixture was stirred at room temperature for 1.5 hours, then
concentrated under reduced
pressure and the residue was partitioned between ethyl acetate and saturated
sodium bicarbonate
solution. The organic layer was washed with brine, dried over magnesium
sulfate, filtered and the
filtrate concentrated under vacuum. The resulting crude material was purified
by silica gel
chromatography to provide the desired compound as a colorless oil (9, 61 mg,
81 %). MS (ESI)
[M+H+]+ = 232.40.
Example 2: Alternative preparation of 1-(6-ethyl-4,4-dimethyl-1,2,3,4-
tetrahydro-quinolin-7-
yl)-ethanone 9.
10090] 1-(6-Ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl)-ethanone 9
was prepared in five
steps from 1-(6-ethyl-4,4-dimethyl-3,4-dihydro-2H-quinolin-1-yl)-2,2,2-
trifluoro-ethanone 7 as
shown in Scheme 2.
Scheme 2
F3CY0 F3CY0 H
O 0
N Step 1 N Br Step 2 N Br + 0
7 10 11 7 0 12
00 O O H O
Step 3 Br Step 4 N Step 5 VN
13 1 9
Step I -Preparation ofl-(7-bromo-6-ethyl-4,4-dimethyl-3,4-dihydro-2Hquinolin-1-
yl)-2,2,2-
trifluoroethanone (10):
[00911 1-(6-ethyl-4,4-dimethyl-3,4-dihydro-2H-quinolin-1-yl)-2,2,2-trifluoro-
ethanone (7, 65 g,
0.224 mol) was stirred in 2 L of nitromethane at room temperature. Bromine
(15.2 mL, 0.296 mol)
was added dropwise followed by the addition of aluminum trichloride (33 g,
0,244 mol). The reaction
39
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
mixture was stirred overnight at room temperature under an inert atmosphere.
The reaction mixture
was concentrated under reduced pressure and was poured into aqueous sodium
thiosulfate and
extracted with ethyl acetate. The organic fraction was washed with brine,
dried over sodium sulfate,
filtered and the filtrate concentrated under vacuum to provide the desired
compound (10, 79 g, 95%).
MS (ESI) [M+H+]+ = 364.1, 366Ø
Step 2 - Preparation of 7-bromo-6-ethyl-4,4-dinmethvl-1,2,3,4-
tetrahvdroquinoline (11):
[0092] A solution of 1-(7-bromo-6-ethyl-4,4-dimethyl-3,4-dihydro-2H-quinolin-l-
yl)-2,2,2-
trifluoroethanone (10, 79 g, 0.217 mol) in 800 mL of 10% potassium hydroxide
in methanol was
stirred at room temperature overnight. The reaction mixture was concentrated
and partitioned
between ethyl acetate and water. The organic layer was collected and the
aqueous layer was back-
extracted with ethyl acetate. The organic layers were combined, dried over
sodium sulfate, filtered,
and the filtrate concentrated under vacuum. The resulting material was
purified by silica gel column
chromatography eluting with a gradient of 0-20% ethyl acetate in hexane.
Appropriate fractions were
combined and concentrated under vacuum to provide the desired compound as a
light orange solid
(11, 70 g, 52%). MS (ESI) [M+H+]+= 267.9, 270Ø
Step 3 - Preparation of 7-bromo-6-ethyl-4,4-dimethyl-3,4-dihydro-2H-quinoline-
l-carboxylic acid
tert-butyl ester (13):
[00931 To 7-bromo-6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydroquinoline (11, 26 g,
97 mmol) in 300
mL of tetrahydrofuran, 1 M sodium hexamethyldisilazide in 107 mL
tetrahydrofuran was added at
room temperature and was allowed to stir for 30 minutes. Di-tert-
butyldicarbonate (12, 32 g, 146
mmol) was added as a solution in tetrahydrofuran. The reaction mixture was
stirred for 1 hour and
then quenched with the addition of 300 mL of brine. The mixture was extracted
with ethyl acetate
and the organic layer was separated and concentrated under reduced pressure.
The crude product was
purified by silica gel column chromatography eluting with a gradient of 0-20%
ethyl acetate in
hexane. Appropriate fractions were combined and concentrated under vacuum to
provide the desired
compound as a clear oil (13, 25 g, 70%).
Step 4 - Preparation of 7-acetyl-6-ethyl-4,4-dimethyl-3,4-dihydro-2H-quinoline-
l-carboxylic acid
tert-butyl ester (14):
[0094] A mixture of 7-bromo-6-ethyl-4,4-dimethyl-3,4-dihydro-2H-quinoline-l-
carboxylic acid
tert-butyl ester (13, 25 g, 68 mmol), tri-n-butyl tin-vinyl ether (31 mL, 238
mmol), palladium(II)
acetate (0.47 g, 2.1 mmol), 1,3-bis(diphenylphosphino)propaue (1.74 g, 4.2
mmol), and potassium
carbonate (9.7 g, 70 mmol) in 125 mL of dimethylformamide and 7.5 mL of water
under nitrogen was
heated to 80 C overnight. The mixture was cooled to room temperature and
quenched by adding 250
mL of 10% aqueous hydrochloric acid. This mixture was transferred to a
separatory funnel
containing saturated aqueous potassium carbonate and extracted with methyl
tent-butyl ether. The
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
organic layer was dried over sodium sulfate, filtered and the filtrate
concentrated under vacuum. The
resulting crude material was purified by silica gel column chromatography
eluting with a gradient of
0-200%o ethyl acetate in hexane. Appropriate fractions were combined and
concentrated under vacuum
to provide the desired compound as a light brown oil (14, 6 g, 27 %). MS (ESI)
[M+H-`]* = 354.3.
Step 5 - Preparation of 1-(6-ethyl-4,4-dimethyl-I, 2,3,4-tetrahydro-quinolin-7-
yl)-ethanone (9):
[0095] To a solution of 7-acetyl-6-ethyl-4,4-dimethyl-3,4-dihydro-2H-quinoline-
l-carboxylic acid
tcrt-butyl ester (14, 1.40 g, 0.00422 mol) in 35 mL of diehloromethane, 8.0 mL
of 4 M hydrogen
chloride in 1,4-dioxane was added. The reaction mixture was stirred at room
temperature for 0.5
hours, then concentrated under vacuum. The residue was partitioned between
ethyl acetate and
saturated sodium bicarbonate solution, The organic layer was washed with
brine, dried over
magnesium sulfate, filtered and the filtrate concentrated under vacuum. The
resulting material was
purified by silica gel chromatography to provide the desired compound as a
pale yellow solid (9, 0.88
g, 88%). MS (ESI) [M+H+]+ = 232,2,
Example 3: Preparation of [1-(6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-
7-yl)-ethyl]-
carbamic acid tert-butyl ester 17.
[0096] [1 -(6-Ethyl-4,4-dimethyl- 1,2,3,4-tetrahydro-quinol in-7-yl) -ethyl] -
carb amic acid tert-butyl
ester 17 was prepared in three steps from 1-(6-ethyl-4,4-dimethyl-1,2,3,4-
tetrahydro-quinolin-7-yl)-
ethanone 9 as shown in Scheme 3.
Scheme 3
H O H HO,N H NHz Step 3 H HN'' O
Step 1 N Step 2 O+ O
1
17
9 15 16 12
Step 1 - Preparation of 1-(6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7
yl)-ethanone oxime
(15):
[0097] A solution of 1-(6-ethyl-4,4-dimethyl-1.2,3,4-tetrahydro-quinolin-7-yl)-
ethanone (9, 3 g,
10.0 mmol), hydroxylamine (1 g, 40.0 mmol), and pyridine (1 mL, 10.0 mmol) in
50 mL of ethanol
was stirred at 80 C overnight. The reaction mixture was concentrated under
vacuum and the
resulting material was partitioned between ethyl acetate and water. The
organic layer was washed
with brine, dried over magnesium sulfate, filtered and the filtrate
concentrated under vacuum. The
resulting material was purified by silica gel chromatography to provide the
desired compound as a
yellow solid (15, 1.65 g, 50%). MS (ESI) [M I lI+]+ = 247.15.
41
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
Step 2 - Preparation of I-(6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7
yl)-ethylamine (16):
[0098] A mixture of 1-(6-ethyl -4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl)-
ethanone oxime (15,
0.8 g, 3.0 mmol) and Raney nickel (50%, aqueous slurry) in 40 mL of methanol
and 8 mL of
ammonium hydroxide was agitated under hydrogen (55 psi) in a Parr shaker for
3.5 hours. The
mixture was filtered and the filtrate was concentrated under vacuum to provide
the desired compound
as a brownish oil (16, 0.8 g, 80%). MS (ESI) [M+H']' = 234.05,
Step 3 - Preparation of[]-(6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-gtuinolin-7-
yl)-ethyiJ-carbamic
acid tert-butyl ester (17):
[0099] To a solution of 1-(6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-
yl)-ethylamine (16,
1.2 g, 2.8 mmol) in 100 mL of dichloromethane, N,N-diisopropylethylamine (1.5
mL, 8.6 mmol) and
di-tert-butyldicarbonate (12, 0.4 g, 2.0 mmol) were added. The reaction
mixture was stirred at room
temperature overnight and concentrated under vacuum. The residue was
partitioned between ethyl
acetate and water, The organic layer was washed with sodium bicarbonate and
brine, dried over
magnesium sulfate, filtered and the filtrate concentrated under vacuum. The
crude material was
purified by silica gel chromatography to provide the desired compound as a
light yellow solid (17,
645 mg, yield 90 ./o). MS(ESI): [M+H']' =333.05.
Example 4: Preparation of 1-[6-ethyl-l-(2-methoxy-ethyl)-4,4-dimethyl-1,2,3,4-
tetrahydro-
quinolin-7-yl]-ethylamine P-0002.
[0100] 1-[6-Ethyl-l-(2-methoxy-ethyl)-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-
7-yl]-ethylamine
P-0002 was prepared in three steps from 1-(6-ethyl-4,4-dimethyl-1,2,3,4-
tetrahydro-quinolin-7-yl)-
ethanone 9 as shown in Scheme 4.
Scheme 4
_O -O -O
H O H O rJ HON rJ NH2
O I
N (~ Step 1 N Step 2 N Step 3 N
9 18 19 20
P-0002
Step I - Preparation of 1 [6 ethyl l-(2 methox v ethyl) 4,4-dimethyl 1, 2,3,4
tetrahti~dro quinolin 7 vlJ
ethanone (19).:
[0101] To a solution of 1-(6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-
yl)-ethanone (9, 55
mg, 0.17 mmol) in 10 mL of acetonitrile, 1-bromo-2-methoxy-ethane (18, 0.043
mL, 0.45 mmol),
potassium iodide (25 mg, 0.15 mmol) and potassium carbonate (84 mg, 0.60 mmol)
were added. The
reaction mixture was stirred at 100 'C for 20 hours. The mixture was poured
into water, extracted
with eth c dried over magnesium "ltered and the filtrate concentrated under
vacuum,
42
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
The crude material was purified by silica gel chromatography to provide the
desired compound as a
yellow oil (19, 13 mg, 27%). MS (ESI) [M+H]' = 290.50.
Step 2 - Preparation of 1-[6-ethyl-]-(2-methoxy-ethyl)-4,4-dirnethv!-1,2,3,4-
tetrahydro-quinolin-7-ylJ-
ethanone oxime (20).-
[01021 A solution of 1-[6-ethyl-l-(2-methoxy-ethyl)-4,4-dimethyl-1,2,3,4-
tetrahydro-quinolin-7-
yl]-ethanonc (19, 13 mg, 0.045 mmol), hydroxylamine hydrochloride (6.2 mg,
0.090 mmol), and
pyridine (0.018 mL, 0.22 mmol) in 5 mL of ethanol was stirred at 80 C for 2
hours. The reaction
mixture was concentrated under vacuum and the residue was partitioned between
ethyl acetate and
water. The organic layer was washed with brine, dried over magnesium sulfate,
filtered and the
filtrate concentrated under vacuum. The crude material was used for the next
step without further
purification (20, 10 mg, 70%). MS (ESI) [M+Hf]+= 305.65.
Step 3 - Preparation of I-[6-ethyl-I-(2-methoxy-ethyl)-4,4-dimethyl-1,2,3,4-
tetrahydro-quinolin-7 yl]-
ethylamine (P-0002):
[0103] A mixture of 1-[6-ethyl-l-(2-methoxy-ethyl)-4,4-dimethyl-1,2,3,4-
tetrahydro-quinolin-7-
yl]-ethanone oxime (20, 10 mg, 0.03 mmol) and Raney nickel (50%, aqueous
slurry) in 5 mL of
methanol and 2 mL of ammonium hydroxide was agitated under hydrogen (55 psi)
in a Parr shaker for
4 hours. The mixture was filtered and the filtrate was concentrated under
vacuum. The resulting
material was purified by silica gel chromatography eluting with
dichloromethane and methanol to
provide the desired compound as a pale yellow viscous oil (P-0002, 4 mg, 40%).
MS (ESI) [M+H+]+
= 291.00.
[0104] 1-[7-(1-Amino-ethyl)-6-ethyl-4,4-dimethyl-3,4-dihydro-2H-quinolin-l-yl]-
ethanone P-0007,
'--f- O NH2
N
was prepared similarly to the protocol of Scheme 4, where 1-(6-ethyl-4,4-
dimethyl-1,2,3,4-tetrahydro-
quinolin-7-yl)-ethanone (9, 20 mg, 0.09 mmol) in 10 ml. of acetonitrile is
mixed with methyl iodide
(0.016 mL, 0.26 mmol) in step 1, where the 1-acetyl derivative was formed
instead of the desired
1-methyl derivative and carried through the last two steps. MS (ESI) [M+H]+ =
275.95.
[0105] Additional compounds maybe prepared similarly to the protocol of Scheme
4, replacing 1-
bromo-2-methoxy-ethane 18 with a suitably substituted alkyl halide in Step 1.
The following
compounds were made following this procedure:
1-[7-(1-Amino-ethyl)-6-ethyl-4,4-dimethyl-3,4-dihydro-2H-quinolin-l-yl]-2-
methoxy-ethanone
(P-0001),
1-[6-Ethyl-l-(3-,.u:i!~-_) iropyl)-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-
yl]-ethylainine
43
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
(P-0003),
1-[6-Ethyl-4,4-dimethyl-l -(3,3,3-trifluoro-propyl)-1,2,3,4-tetrahydro-
quinolin-7-yl]-ethylamine
(P-0004),
1-[6-Ethyl-4,4-dimethyl- l -(4,4,4-trifluoro-butyl)-1,2, 3, 4-tetrahydro-
quinolin-7-yl] -ethylamine
(P-0005),
N- {2-[7-(1-Amino-ethyl)-6-ethyl-4,4-dimethyl-3,4-dihydro-2H-quinolin-l-yl]-
ethyl} -acetamide
(P-0006), and
I -[6-Ethyl-4,4-dimethyl-l -(2-pyrazol-l-yl-ethyl)-1,2,3,4-tetrahydro-quinolin-
7-yl]-ethylamine
(P-0009).
The following table shows compounds that were prepared similarly to the
protocol of Scheme 4.
where optimal reaction conditions may have varied, for example, in terms of
solvents, time and
temperature of the reaction, and in chromatography conditions for purification
of the desired
compounds, with the compound number in Column 1, the alkyl halide used in Step
1 in Column 2, the
resulting compound in Column 3 and the experimental mass spectrometry result
in Column 4.
Com ound number Alkyl halide Compound structure MS ESI M-H1
O Y 0 NH2
P-0001 N 304.50
NHZ
P-0003 Bra, ~,-Q~ N 305.60
CF3
H NH2
P-0004 Br ,-,_,CF3 N 330.65
NH2
F3C~
P-0005 Bra ~,CF3 N 334.65
0
AN'-"I NH2
P-0006 Cl" H 31805
H
N N NH2
P-0009 CND Br vv N 328.05
(/~
N
44
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
Example 5: Preparation of 1-(6-ethyl-4,4-dimethyl-l-pyridin-3-yhnethyl-1,2,3,4-
tetrahydro-
quinolin-7-yl)-ethylamine P-0010 and related compounds.
[0106] 1-(6-Ethyl-4,4-dimethyl-l-pyridin-3-ylmethyl-1,2,3,4-tetrahydro-
quinolin-7-yl)-ethyl amine
P-0010 was prepared in two steps from [1-(6-ethyl-4,4-dimethyl-1,2,3,4-
tetrahydro-quinolin-7-yl)-
ethyl]-carbamic acid tert-butyl ester 17 as shown in Scheme 5. Compounds such
as 1-[6-Ethyl-4,4-
dimethyl-l-(3-trifluoromethyl-benzyl)-1,2,3,4-tetrahydro-quinolin-7-yl]-
ethylamine P-0026 were
prepared similarly according to Scheme 5a. Compounds such as 1-[6-Ethyl-4,4-
dimethyl-] -(2-
methyl-pyridin-4-ylmethyl)-1,2,3,4-tetrahydro-quinolin-7-yl]-ethylamine P-0037
were prepared
similary, using an aldehyde instead of bromomethyl compound in step 1,
according to Scheme 5b.
Scheme 5
N N
Boc Boc
HN N HN/ NH2
N YB S tep 1 Step 2 r
17 21 22 P-0010
Step 1 - Preparation of[] -(6-ethyl-4,4-dimethyl-1pyridin-3-yimethyl-1,2,3,4-
tetrahydroquinolin-7-
yl)-ethyl]-carbamic acid tert-butyl ester (22):
[0107] To a solution of [1-(6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-
yl)-ethyl]-
carbamic acid tert-butyl ester (17, 42.3 mg, 0.127 mmol) in 10 mL of
acetonitrile,
3-bromomethyl-pyridine (21, 96 mg, 0.38 mmol), potassium iodide (21 mg, 0.13
mmol) and sodium
bicarbonate (107 mg, 1.27 mmol) were added. The reaction mixture was stirred
at 80 C overnight,
then cooled to room temperature and poured into water, extracted with ethyl
acetate, dried over
magnesium sulfate, filtered and the filtrate concentrated under vacuum. The
crude material was
purified by silica gel chromatography to provide the desired compound as a
yellow solid (22, 20 mg,
40%). MS (ESI) [M+H+]-` = 424.20.
Step 2 - Preparation of 1-(6-ethyl-4,4-dimethyl-1 pyridin-3-ylmethyl- 1,2,3,4-
tetrahydroquinolin-7 yl)-
ethylamine (P-0010):
[0108] To a solution of [1-(6-ethyl-4.4-dimethyl-l-pyridin-3-ylmethyl-1,2,3,4-
tetrahydro-quinolin-
7-yl)-ethyl]-carbamic acid tent-butyl ester (22, 16 mg, 0.038 mmol) in 0.5 mL
of dichloromethane,
hydrochloric acid (3 mL, 100 mmol) was added. The mixture was stirred at room
temperature for 1.5
hours and concentrated under vacuum to provide the desired compound as a
yellow solid (P-0010,
15.1 mg, 88%). MS (ESI): [M+H']' = 325.05.
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
Scheme 5a
CF3 CF3
f'
Boc Boc
HN/ CF3 \ HN/ \ NH2
N \ ( f Step 1 a N Step 2a
Br
17 21a 22a P-0026
Step la - Preparation of (1-[6-ethyl-4,4-dimethyl-l-(3-trifluoromethyl-benzyl)-
1,2,3,4-tetrahydro-
quinolin-7-yl}-ethyl}-carbamic acid tert-butyl ester (22a):
[0109] To a solution of [1-(6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-
yl)-ethyl]-
carbamic acid tert-butyl ester (17, 15 mg, 0.045 mmol) in 500 sL of N-
methylpyrrolidone,
1-bromomethyl-3-trifluoromethyl-benzene (21a, 32.3 mg, 0.135 mmol), and N,N-
diisopropylethylamine (32 .iL, 0.18 mmol) were added. The reaction mixture was
irradiated 5
minutes at 150 C in microwave, then extracted with ethyl acetate. The organic
layer was washed
with sodium bicarbonate, then brine, dried over magnesium sulfate, filtered
and the filtrate
concentrated under vacuum. The residue was used in the next step without
further purification.
Step 2a - Preparation of 1-[6-ethyl-4,4-dimethyl-l-(3-trifluoromethyl-benzyl)-
1,2,3,4-tetrahydro-
quinolin-7-yl}-ethylamine (P-0026):
[0110] {1-[6-Ethyl-4,4-dimethyl-l-(3-trifluoromethyl-benzyl)-1,2,3,4-
tetrahydro-quinolin-7-yl]-
ethyl}-carbamic acid tert-butyl ester (22a, Step la) was dissolved in 500 4L
of dioxanc and 250 L of
4M hydrochloric acid in dioxane. The mixture was stirred at room temperature
for 30 minutes and
concentrated under vacuum. The residue was dissolved in 1 mL of dimethyl
sulfoxide and purified by
HPLC using a Hamilton PRP-3 column eluting with 10%-80% solvent B at a flow
rate of 7
mL/minute, where solvent A was 10 mM ammonium hydroxide in water and solvent B
was 10 MM
ammonium hydroxide in acctonitrile. The appropriate fractions were combined
and the solvents
removed under vacuum to provide the desired compound (P-0026). MS (ESI): [M-
NH2-]+ = 374.3.
Scheme 5b
N N
HNBoc N HN Boc
NH2
H
N \ / Step 1 b N Step 2b N
o H
17 21b 22b P-0037
Ib Preparation of t-E t; ethyl 4,4 d~;~~ 12 mail,, ~`in 4-'lmethylr .',2,3,4
_.
Step
46
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
quinolin-7-ylJ-ethyl}-carbamic acid tert-butyl ester (22b):
[0111] [1-(6-Ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl)-ethyl]-
carbamic acid tert-butyl
ester (17, 12 mg, 0.036 mmol) and 2-methyl-pyridine-4-carbaldehyde (21b, 13.2
mg, 0.12 mmol)
were dissolved in 600 .tL of 95:5 ethanol:acetic acid, and cyanoborohydride on
silica (55 mg, 0.05
mmol) was added and the mixture irradiated for 15 minutes at 120 C in the
microwave. The vial was
centrifuged to remove the silica, and the supernatant transferred to a new
vial. The silica residue was
washed with 500 L of ethanol, centrifuged and the supernatants combined. The
solvents were
removed under vacuum and the residue used in the next step without further
purification.
Step 2b - Preparation of 1-[6-ethyl-4,4-dimethyl-l-(2-methyl pyridin-4 ylmeth
i)-],2,3,4-tetrahydro-
quinolin-7ylJ-ethylamine (P-0037):
[0112] {1-[6-Ethyl-4,4-dimethyl- l-(2-methyl-pyridin-4-ylmethyl)-1,2,3,4-
tetrahydro-quinolin-7-
yl]-ethyl }-carbamic acid tert-butyl ester (22b, 16 mg, 0.038 mmol) was
dissolved in 0,3 mL of
dioxane and 0.3 mL of 4M hydrochloric acid in dioxane and stirred for 30
minutes. The solvents
were removed under vacuum and the residue purified by HPLC as per step 2a of
Scheme 5a. MS
(ESI) [M+H-]'= 339.00,
[0113] 1-[6-Ethyl-4,4-dimethyl-l-(2-methyl-pyridin-4-ylmethyl)-1,2,3,4-
tetrahydro-quinolin-7-yl]-
ethylamine P-0037 was further purified by chiral HPLC to provide (R)-1-[6-
Ethyl-4,4-dimethyl-l-(2-
methyl-pyridin-4-ylmethyl)-1,2,3,4-tctrahydro-quinolin-7-yl]-ethylamine P-0056
and (S)-1-[6-Ethyl-
4,4-dimethyl-l-(2-methyl-pyridin-4-ylmethyl)-1,2,3,4-tetrahydro-quinolin-7-yl]-
ethylamine P-0057,
N' I N~
NH2 NH2
N ~ N \
P-0056 and P-0057
using a ChiralCel OD-H column, with isocratic 6% Buffer A (Isopropanol with
0.1 % trifluoroacetic
acid), and 94% Buffer B (hexane with 0.1 % trifluoroacetic acid).
[0114] Additional compounds maybe prepared similarly to the protocols of
Scheme 5, 5a, and 5b,
replacing the bromomethyl compound or aldehyde compound with an appropriate
aryl/heteroaryl-
alkyl halide or aryl/heteroaryl-aldehyde in Step 1/la/lb. The following
compounds were made
following this procedure:
1-(6-Ethyl-4,4-dimethyl-l-pyridin-4-ylmethyl-1,2,3,4-tetrahydro-quinolin-7-yl)-
ethylamine (P-0015),
1-[6-Ethyl-4,4-dimethyl-l -(5-methyl-i soxazol-3-ylmethyl)-1,2,3,4-tetrahydro-
quinolin-7-yl]-
ethylamine (P-0017),
1-[1-(3-Bromo-isoxazol-5-ylmethyl)-6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-
quinolin-7-yl]-
ethylamine (P-0023),
47
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
l -[6-Ethyl-4,4-dimethyl-l-(3-methyl-isoxazol-5-ylmethyl)-1,2,3,4-tetrahydro-
quinolin-7-yl]-
ethylamine (P-0024),
l -(1-Benzyl-6-ethyl-4,4-dimethvl-1,2,3,4-tetrahydro-quinolin-7-yl)-ethylamine
(P-0025),
l -[6-Ethyl-4,4-dimethyl-1-(3-trifluoromethyl-benzyl)-1,2,3,4-tetrahydro-
quinolin-7-yl]-ethyl amine
(P-0026),
1-[ 1-(3,5-Difluoro-benzyl)-6-ethyl-4,4-dimethvl-1,2,3,4-tetrahydro-quinolin-7-
yl]-ethylamine
(P-0027),
1-[6-Ethyl-l-(3-fluoro-benzyl)-4,4-dimethyl- 1,2,3,4-tetrahydro-quinolin-7-yl]-
ethylamine (P-0028),
1-[6-Ethyl-l-(4-methoxy-benzyl)-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl]
-ethylamine
(P-0029),
1-[ 1-(2,3-Difluoro-benzyl)-6-ethyl-4,4-dimethvl-1 ,2,3,4-tetrahydro-quinolin-
7-yl]-ethylamine
(P-0030),
1-[6-Ethyl-l -(3-methoxy-benzyl)-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7 -
yl]-ethylamine
(P-0031),
1-[6-Ethyl-l-(4-fluoro-benzyl)-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl]-
ethylamine (P-0032),
4-[7-(1-Amino-ethyl)-6-ethyl-4,4-dimethyl-3,4-dihydro-2H-quinolin-l-ylmethyl]-
pyridine-2-
carbonitrile (P-0033),
1-[6-Ethyl-l -(2-fluoro-pyridin-4-ylmethyl)-4,4-dimethyl-1,2, 3,4-tetrahydro-
quinolin- 7 -yl]-ethylamine
(P-0034),
1-(6-Ethyl-4,4-dimethyl-l-phenethyl-1,2,3,4-tetrahydro-quinolin-7-yl)-
ethylamine (P-0035),
1-[6-Ethyl-l -(6 -flu oro-pyri din-3-ylmethyl)-4,4-dimethyl-1,2,3,4-tetrahydro-
quinolin-7-yl]-ethylamine
(P-0036),
1-[6-Ethyl-l -(2-methoxy-pyridin-4-ylmethyl)-4,4-dimethyl-1,2,3,4-tetrahydro-
quinolin-7-yl]-
ethylamine (P-0041),
1-[6-Ethyl-4,4-dimethyl-l -(6-methyl-pyridin-3-ylmethyl)-1,2,3,4-tetrahydro-
quinolin-7-yl]-
ethylamine (P-0042),
1-(1-Benzo[ 1,3] dioxol-4-ylmethyl-6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-
quinolin-7-yl)-ethylamine
(P-0051), and
1-[I -(2,2-Difluoro-benzo[ 1 ,3] dioxol-4-ylmethyl)-6-ethyl-4,4-dimethyl-
1,2,3,4-tetrahydro-quinolin-7-
yl]-ethylammine (P-0053).
The following table shows compounds that were prepared similarly to the
protocol of Scheme
5/5a/5b, or suitable variations thereof, where optimal reaction conditions may
have varied, for
example, in terms of solvents used, time and temperature of the reaction, and
in chromatography
conditions for purification of the desired compounds, with the compound number
in Column 1, the
aryl-alkyl halide or aryl-aldehyde used in Step 1/ l a/l b in Column 2, the
resulting compound in
Column 3 and the experimental mass spectrometry result in Column 4,
48
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
Compound Aryl/heteroaryl- Compound structure MS (ESI)
number alkyl halide or aldehyde [M+H+]+
N,~ r
N NH2
P-0015 1 , Br 325.25
P-0017 -No O'Nl-j NH2 329.05
Br
Br
\ N OU~ NH2 329.95
P-0023 N 394.95
Br I e
N'
P-0024 \,N '0 NH' 329.05
o NIA
ci 1
/I
~ NH2
P-0025 / Br 324.25
F
F
F ( NH, 342.3
P-0027 N [M-NH,]
F e Br
I
F NH2 324.3
P-0028 F Br N o [M-NH2-1'
'0
e0 NH2 3363
P-0029 Br N (~ [M NH2 ]"
' \ F NHZ 342,3
P-0030 F e Br F N [h'f NIH,
F
49
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
NHo z 336.3
P-0031
~p (, Br N [M-NHz
F
F NH2 324.3
P-0032 [M NH2 ]
Br N
-_-
N
N NC NHz 332.3
P-0033
[M-NH2]e
NC Br
-----------
N'
N F NH,
P-0034 F I, Br N 343.05
O (/ \ NH2
N 338.25
P-0035
H
F
N NHz
P-0036 N Br 343.0
N
N o NH2 337.5
P-0041 O I , Br [M-NHz-]'
\ N '
NHz 321.1
P-0042 N i H
[M-NHS -]"
O
\ I / NHz
P-0051 p o `-o 368.0
\-p H
j , O p \ NHz
P-0053 F-\-o 404.0
FFO H F
[01151 1-(1-Ben zyl-6-ethyl -4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl)-
ethylamine P-0025 was
further purified by chiral HPLC to provide (R)-1-(1-Benzyl-6-ethyl-4,4-
dimethyl-1,2,3,4-tetrahydro-
quinolin-7-y1)-ethylamine P-0048 and (S)-1-(1-Benzyl-6-ethyl-4,4-dimmethyl-
1,2,3,4-tctrahydro-
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
quinolin-7-yl)-ethylamine P-0049,
NH2 ' NH2
N
P-0048 and P-0049
using a ChiralCel OD-H column, with isocratic 6% Buffer A (Isopropanol with
0.1% trifluoroacetic
acid), and 94% Buffer B (hexane with 0.1 % trifluoroacetic acid).
Example 6: Preparation of 7-(1-amino-ethyl)-1-benzyl-6-ethyl-4,4-dimethyl-3,4-
dihydro-lH-
quinolin-2-one P-0052.
10116] 7-(1-Amino-ethyl)-1-benzyl-6-ethyl-4,4-dimethyl-3,4-dihydro-IH-quinolin-
2-one P-0052
was prepared in four steps from 1-(6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-
quinolin-7-yl)-ethanone 9
as shown in Scheme 6.
Scheme 6
O 0 0
N + Step 1 Step 2 0 N
Br
9 23 24 25
N~OH 0-) NH2
2
Step 3 O N' Step 4 0 N'
i i
26 P-0052
Step I -Preparation oft -(1-benzyl-6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-
quinolin-7-yl)-ethanone
(24):
10117] A mixture of 1-(6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl)-
ethanone (9, 500 mg,
2 mmol), benzyl bromide (23, 0.43 mL, 3.6 mmol) and N,N-diisopropylethylamine
(0,92 mL, 5.3
mmol) were combined with 0.4 mL of N-methylpyrrolidone. The reaction was
irradiated in a
microwave at 100 C for 40 minutes. The mixture was diluted with ethyl
acetate, and the organic
layer was washed with brine, dried with magnesium sulfate, filtered and the
filtrate concentrated
under vacuum. The crude material was purified by silica gel chromatography to
provide the desired
compound as a light yellow oil (24, 590 mg, 90%). MS (ESI) [M+H,], = 322.30,
51
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
Step 2 - Preparation of 7-acetyl-l-benzyl-6-ethyl-4,4-dimethyl-3,4-dihydro-IH-
quinolin-2-one (25):
[0118) To a solution of 1-(1-benzyl-6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-
quinolin-7-yl)-ethanone
(24, 55.0 mg, 0.17 mmol) in 10 mL of dichloromethane, a premixed fine powder
of potassium
permanganate (140 mg, 0.86 mmol) and copper(II) sulfate pentahydrate (210 mg,
0.86 mmol) was
added. The reaction mixture was gently refluxed for 20 hours, then filtered
through a Celite pad and
the pad was washed with dichloromethane and ether. The organic portions were
collected and
combined, the solvent was removed under vacuum, and the residue was purified
by silica gel
chromatography to provide the desired compound as a light yellow oil (25, 8
mg, 14%). MS (ESI)
[M+H+j*= 335.95.
Step 3 - Preparation ofd-benzyl-6-ethyl-7-(1-hy(iroxvinzino-ethyl)-4,4-
dimethyl-3,4-dihydro-IH-
quinolin-2-one (26):
[01191 A solution of 7 -acetyl- I -benzyl-6-ethyl-4,4-dimethyl-3,4-dihydro- I
H-quinolin-2 -one (25,
10.0 mg, 0.028 mmol) , hydroxylamine hydrochloride (9.8 mg, 0.14 mmol), and
pyridine (0.014 mL,
0.17 mmol) in 5 mL of ethanol was stirred at 80 C for 4 hours. The reaction
mixture was
concentrated and the residue was dissolved in ethyl acetate, the organic layer
washed with water,
brine, and dried over magnesium sulfate, filtered and the filtrate
concentrated under vacuum. The
residue was purified by silica gel chromatography to provide the desired
compound as a light yellow
solid (26, 10 mg, 70%). MS (ESI) [M+H ]`= 350.95.
Step 4- Preparation of 7-(1-arnino-ethyl)-1-benzyl-6-ethyl-4,4-dimethyl-3,4-
dihydro-III-quinolin-2-
one (P-0052):
[012011 A mixture of 1-benzyl-6-ethyl-7-(1-hydroxyimino-ethyl)-4,4-dimethyl-
3,4-dihydro-lH-
quinolin-2-one (26, 10 mg, 0.03 mmol) and Raney nickel (50%, aqueous slurry)
in 10 mL of methanol
and 3 mL of ammonium hydroxide was stirred under an hydrogen balloon for 5
hours. The catalyst
was removed by filtration and the filtrate was concentrated under vacuum. The
residue was purified
by silica gel chromatography to provide the desired compound as a colorless
viscous liquid (P-0052,
4.6 mg, 40%). MS (ESI) [M+H+] '= 337.95.
Example 7: Preparation of 1-(1-benzyl-6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-
quinolin-7-yl)-
propylamine P-0047.
[01211 1-(1-Benzyl-6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl)-
propylamine P-0047
was prepared in three steps from 1-(1-benzyl-6-ethyl -4,4-dimethyl-1,2,3,4-
tetrahydro-quinolin-7-yl)-
ethanone 24 as shown in Scheme 7.
52
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
Scheme 7
__ I ~I I off ~I
'
O O N NH2
N Step 1 N I Step 2 N Step 3
24 27 28
P-0047
Step 1 - Preparation of 1-(1-benzyl-6-ethyl-4,4-dimethyl-1, 2,3,4-tetrahydro-
quinolin-7 yl) propan-l-
one (27):
[0122] 1-(1-Benzyl-6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl)-
ethanone (24, 0.115 g,
0.358 mmol) was dissloved in 10 mL of tetrahydrofuran and chilled to -78 T.
Lithium
hexamethyldisilazide in tetrahydrofuran (1.0 M, 0.36 mL) was added dropwise
and the reaction
mixture was stirred for 15 minutes at -78 T. Methyl iodide (0.0223 mL, 0.358
mmol) was added in
one portion and the reaction mixture was allowed to warm to room temperature.
Ammonium chloride
solution was added to quench the reaction and the mixture was washed with
ethyl acetate. The
organic layer was washed with brine, dried over magnesium sulfate, filtered
and the filtrate
concentrated under vacuum. The crude material was purified by silica gel
chromatography to provide
the desired compound as a pale yellow waxy solid (27, 40 mg). MS (ESI) [M+H']'
= 336.05.
Step 2 - Preparation of]-(] -benzyl-6-ethyl-4,4-dime(hyl-1,2,3,4-tetrahydro-
quiyrolin-7 yl) propan-l-
one oxime (28):
[0123] 1-(1-Benzyl-6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl)-
propan-l-onc (27,
0.040 g, 0.12 mmol), and hydroxylamine hydrochloride (0.043 g, 0.62 mmol) were
added to a
solution of 0.5 mL of pyridine in 10 mL of ethanol. The reaction mixture was
warmed to reflux for 4
hours and then concentrated under vacuum. The residue was partitioned between
ethyl acetate and
ammonium chloride solution. The organic layer was washed with water and brine,
then dried over
magnesium sulfate, filtered and filtrate concentrated under vacuum. The
resulting material was
purified by silica gel chromatography to provide the desired compound (28, 20
mg). MS (ESI)
[M+H+]+= 37 7Ø
Step 3-Preparation of 1-(I-benzyl-6-ethyl-4,4-dime(hyl-1,2,3,4-tetrahydro-
quinolin-7yl)-
propylarnine (P-0047):
[0124] 1-(1-Benzyl-6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl)-
propan-l-one oximc
(28, 0.0055 g, 0.016 mmol) was dissolved in 10 mL of ethanol in a Parr vessel.
Raney nickel (0.05 g,
0.8 mmol) was added, and the vessel was evacuated and charged with hydrogen
(10 g, 5 mol, 55 PSI),
The reaction mixture was agitated for 20 minutes. The Raney nickel was removed
via filtration and
the filtrate concentrated under vacuum to provide the desired compound as a
pale, yellow, waxy solid
53
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
(P-0047, 3.3 mg). MS (ESI) [M+H+]+= 338.05.
[0125] 1-[6-Ethyl-l-(3-methoxy-propyl)-4,4-dimethyl-1,2,3,4-tetrahydro-
quinolin-7-yl]-
propylamine P-0016
NH,
N
was prepared similarly to the protocol of Scheme 7, replacing 1-(1-benzyl-6-
ethyl-4,4-dimethyl-
1,2,3,4-tetrahydro-quinolin-7-yl)-ethanone 24 with 1-[6-ethyl-l-(3-methoxy-
propyl)-4,4-dimethyl-
1,2,3,4-tetrahydro-quinolin-7-yl]-ethanone (isolated after Step I of Scheme 4
in preparation of P-0003
per Example 4). MS (ESI) [M'1-H+]' = 338.05.
Example 8: Preparation of trifluoro-methanesulfonic acid 7-acetyl-4,4-dimethyl-
l-(2,2,2-
trifluoro-acetyl)-1,2,3,4-tetrahydro-quinolin-6-yl ester 35.
[0126] Trifluoro-methanesulfonic acid 7-acetyl-4,4-dimethyl- l-(2,2,2-
trifluoro-acetyl)-1,2,3,4-
tetrahydro-quinolin-6-yl ester 35 was prepared in six steps from 4-
methoxybenzenamine 29 as shown
in Scheme S.
Scheme 8
O CI H H H
H2N + / Step 1 N Step 2 O Step 3 111
29 2 30 31 1 32 I
F3CO F3CYO O F3Clf~ O O
Step 4 N I Step 5 N I Step 6
O - OH OTf
33 1 34 35
Step 1 - Preparation of 3-methyl-but-2-enoic acid (4-tnethoxy-phenyl)-amide
(30):
101271 To a chilled (0 C) mixture of 4-methoxybenzenamine (29, 2.50 g, 20.3
mmol) and sodium
hydroxide (2.0 g, 51.0 mmol) in 20 mL of water and 30 mL of dichloromethane,
3,3-dimethylacryloyl
chloride (2, 2.7 mL, 24.0 mmol) was added over a period of n=5 minutes. The
reaction mixture was
allowed to warm to room temperature and stirred overnight. The reaction
mixture was partitioned
between ethyl acetate and saturated sodium bicarbonate solution. The aqueous
layer was washed 3x
with ethyl acetate and the pooled organic layer was washed with ammonium
chloride solution and
brine, then dried over anhydrous magnesium sulfate, filtered and the filtrate
concentrated under
vacuum. The resulting brown oil was purified by silica gel column
chromatography eluting with a
54
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
gradient of 0-7% methanol in dichloromethane. Appropriate fractions were
combined and the
solvents removed under vacuum to provide the desired compound as a tan waxy
solid (30, 3.78g,
91%).
Step 2 - Preparation of 6-methoxyy-4,4-dimethyl-3, 4-dihydro-IH-quinolin-2-one
(31):
[0128] Aluminum trichloride (19.1 g, 143 mmol) was suspended in 300 mL of
nitromethane and
3-methyl-but-2-enoic acid (4-mcthoxy-phenyl)-amide (30, 9.5 g, 46.0 mmol) was
added dropwise
over a period of -5 minutes. The reaction mixture was stirred under an inert
atmosphere overnight.
Ice was added piece-by-peice to quench the reaction and the mixture was
partitioned between ethyl
acetate and water. The organic layer was collected and the aqueous layer was
washed 3x with ethyl
acetate. The organic fractions were pooled and washed with water, sodium
bicarbonate solution and
brine, then concentrated under vacuum. The resulting material was crystalized
from an ethyl acetate
and hexane solution to provide the desired compound as long colorless needles
(31, 7.8 g, 82%).
Step 3 - Preparation of 6-methoxy-4,4-dimethyl-1,2,3,4-tetrahydro-quinoline
(32):
[0129] To a solution of 6-methoxy-4,4-dimethyl-3,4-dihydro-I H-quinolin-2-one
(31, 2.0 g, 9.7
mmol) in 150 ml- of toluene, borane-dimethyl sulfide complex (1.7 mL, 19.0
mmol) was slowly
added at 0 C. The reaction mixture was stirred at 0 C for 15 minutes and
then stirred at 100 C for
2 hours. The reaction mixture was cooled to room temperature and poured into
150 mL of 10%
sodium carbonate solution. The mixture was stirred at room temperature for 30
minutes. The organic
layer was collected and the aqueous layer was extracted with 2 x 60 mL of
ethyl acetate. The organic
layers were combined and dried over magnesium sulfate, filtered and the
filtrate concentrated under
vacuum. The crude material was purified by silica gel chromatography to
provide the desired
compound as a light brown liquid (32, 1.2g, 64%).
Step 4 - Preparation oft,2,2-trifluoro-l-(6-methoxy-4,4-dimethyl-3,4-dihydro-
2H-quinolin-1 yl)-
ethanone (33):
[0130] To a solution of 6-methoxy-4,4-dimethyl-1,2,3,4-tetrahydro-quinoline
(32, 4.49 g, 23.5
mmol) in 25 mL of dichloromethane, triethylamine (65.0 mL, 47.0 mmol) and
trifluoroacetic
anhydride (6.6 mL, 47.0 mmol) were added. The reaction mixture was stirred
overnight, then
partitioned between dichloromethane and water. The organic layer was washed
with brine and dried
over magnesium sulfate, filtered and the filtrate concentrated under vacuum.
The resulting material
was purified by silica gel chromatography eluting with 10% ethyl acetate in
hexanes to provide the
desired compound (33, 5.4g, 80%).
Step 5 - Preparation oft-(7-acetyl-6-hydroxy-4,4-dimethyl-3,4-dihydro-2H-
quinolin-1 yl)-2,2,2-
trifluoro-ethanone (34):
[0131] To a suspension of aluminum trichloride (2.6 g, 19.0 mmol) in 8 mL of
nitromethane,
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
chilled to at 0 C, a solution of acetyl chloride (1.1 mL, 16.0 mmol) and
2,2,2-trifluoro-l -(6-methoxy-
4,4-dimethyl-3,4-dihydro-2H-quinolin-l-yl)-ethanone (33, 1.24 g, 4.32 mmol) in
10 mL of
nitromethane was slowly added. Once addition was complete, the ice bath was
removed and the
reaction mixture was stirred at room temperature for 5 hours. The reaction
mixture was chilled to at
0 C and water was added dropwise, resulting in heat and foaming, until all
solids were dissolved.
The mixture was extracted with 100 mL dichloromethane. The organic layer was
washed with water,
ammonium chloride solution and brine, dried over magnesium sulfate, filtered
and the filtrate
concentrated under vacuum. The resulting golden colored oil was purified by
silica gel column
chromatography eluting with a gradient of 0-5% methanol in dichloromethane
over 40 minutes.
Appropriate fractions were combined and the solvents removed to provide the
desired compound (34,
656 mg, 48%). MS (ESI) [M+H+]+_ 316.2.
Step 6 - Preparation of trifluoro-nmethanesulfonic acid 7-acetyl-4,4-dimethyl-
l-(2,2,2-trifluoro-
acetyl)-1,2,3,4-tetrahydro-quinolin-6yl ester (35):
[0132] To a chilled (-78 C) solution of 1-(7-acetyl-6-hydroxy-4,4-dimethyl-
3,4-dihydro-2H-
quinolin-1-yl)-2,2,2-trifluoro-ethanone (34, 2.1 g, 6.7 mmol) in 10 mL of
dichloromethane, pyridine
(1.2 mL, 15.0 mmol) and trifluoromethane sulfonyl anhydride (1.4 mL, 8.4 mmol)
were added. The
reaction mixture was stirred at -78 C for 30 minutes and at room temperature
for 2 hours. The
reaction mixture was poured into 1M aqueous hydrochloric acid, then extracted
with
dichloromethane. The organic layer was collected and washed with 1M
hydrochloric acid, sodium
bicarbonate and brine, dried over magnesium sulfate, filtered and the filtrate
concentrated under
vacuum, The resulting material was purified by silica gel chromatography to
provide the desired
compound (35, 2.4 g, 84%). MS (ESI) [M+H+]+= 448.2.
Example 9: Preparation of 1-[ 1-benzyl-6-(4-fluoro-phenyl)-4,4-dimethyl-
1,2,3,4-tetrahydro-
quinolin-7-yl]-ethylamine P-0044.
[0133] 1-[1-Benzyl-6-(4-fluoro-phenyl)-4,4-dimethyl-1,2,3,4-tetrahydro-
quinolin-7-yl]-ethylamine
P-0044 was prepared in five steps from trifluoro-methanesulfonic acid 7-acetyl-
4,4-dimethyl-1-(2,2,2-
trifluoro-acetyl)-1,2,3,4-tetrahydro-quinolin-6-yl ester 35 as shown in Scheme
9.
56
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
Scheme 9
F3C.0 O B(OH)2 F3CYO 0 H 0
N OTf + Step 1 N Step 2 N ~=
35 F 36 37 F 38 F
Ia
Br NfOH ~ NH2
+ 6 Step 3 N Step 4 N Step 5 N
39 F 40 F P-0044 F
23 3
Step I - Preparation of I-[7-acetyl-6-(4 fluoro phenyl)-4,4-dimethyl-3,4-
dihydro-2H-quinolin-]yll-
2,2,2-trifluoro-ethanone (37):
[0134] Into a solution of trifluoromethanesulfonic acid 7-acetyl-4,4-dimethyl-
l-(2,2,2-trifluoro-
acetyl)-1,2,3,4-tetrahydro-quinolin-6-yl ester (35, 50.0 mg, 0112 mmol) in
2.00 mL tetrahydrofuran
and 0.12 mL of dimethyl sulfoxide in a microwave reaction vial, 4-
fluorophenylboronic acid (36, 36.0
mg, 0.257 mmol), palladium acetate (25.1 mg, 0.112 mmol),
tricyclohexylphosphine (37.6 mg, 0.134
mmol) and potassium fluoride (23 mg, 0.39 mmol) were added. The reaction
mixture was warmed to
70 C for 80 minutes via microware irradiation. The reaction mixture was
diluted with ethyl acetate
and the organic layer washed with brine, dried over magnesium sulfate,
filtered, and purified by silica
gel chromatography to provide the desired compound (37, 46 mg, 70%). MS (ESI)
[M+H']4=
393.95.
Step 2 Preparation of ]-[6-(4- l]-
_
(38):
[0135] Into a 1-neck round-bottom flask was added 1-[7-acetyl-6-(4-fluoro-
phenyl)-4,4-dimethyl-
3,4-dihydro-2H-quinolin-l-yl]-2,2,2-trifluoro-ethanone (37, 87 mg, 0.16 mmol),
potassium hydroxide
(26 mg, 0.46 mmol), 15.0 mL of methanol and 2.0 mL of water. The reaction
mixture was stirred at
room temperature for 2 hours and concentrated under reduced pressure. The
residue was partitioned
between ethyl acetate and brine, The organic layer was dried over magnesium
sulfate, filtered and
purified by silica gel chromatography to provide the desired compound as a
pale yellow solid (38, 35
mg, 57%). MS (ESI) [M+H"]`- 298.46
Step 3 - Preparation oft-[I-benzyl-6-(4-fluoro phenyl)-4,4-dimethyl-1,2,3,4-
tetrahydro-quinolin-7-
yl/-ethanone (39):
[0136] A solution of 1-[6-(4-fluoro-phenyl)-4,4-dimethyl-1,2,3,4-tetrahydro-
quinolin-7-yl]-
ethanonc (38, 35 mg. 0.088 mmol), benzyl bromide (23, 0.026 mL, 022 mmol) and
0.038 mL of N,N-
,1;;s,pr;;pylethylamine in 0.1 mL of N-methylpyrrolidone was warmed to 100
`'C" for 40 minutes via
57
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
microwave irradiation. The reaction mixture was diluted with ethyl acetate and
the organic phase
washed 2x with brine, dried over magnesium sulfate, filtered and purified by
silica gel
chromatography to provide the desired compound as a pale yellow oil (39, 45
mg, 99%). MS (ESI)
[M+H-]-= 388Ø
Step 4 - Preparation of 1-[1-benzyl-6-(4 fluorophenyl)-4,4-dimethyl-1,2,3,4-
tetrahydro-quinolin-7-
y1J-ethanone oxime (40):
[01371 A solution of 1-[1-benzyl-6-(4-fluoro-phenyl)-4,4-dimethyl-1,2,3,4-
tetrahydro-quinolin-7-
yl]-ethanone (39, 45 mg, 0.087 mmol), hydroxylamine hydrochloride (50 mg, 0.7
mmol), and
pyridine (0.1 mL, 1.0 mmol) in 15 mL of ethanol was stirred at 80 C for 4
hours. The reaction
mixture was concentrated under vacuum and the residue was partitioned between
ethyl acetate and
water. The organic layer was washed with brine, dried over magnesium sulfate,
filtered and the
filtrate concentrated under vacuum. The residue was purified by silica gel
chromatography to provide
the desired compound as a pale yellow solid (40, 29 mg). MS (ESI) [M+H+]' =
403.45
Step 5 - Preparation of] -[I-benzyl 6 (4 fluorophenyl)-4,4-dimethyl-1,2,3,4-
tetrahydro-quinolin-7-
yl]-ethylarnine (P-0044):
[01381 A mixture of 1-[1-benzyl-6-(4-fluoro-phenyl)-4,4-dimethyl-1,2,3,4-
tetrahydro-quinolin-7-
yl]-ethanone oxime (40, 29 mg, 0.070 mmol) and Raney nickel (50%, aqueous
slurry) in 20 mL of
methanol and 3 mL of ammonium hydroxide was agitated in a Parr shaker under
hydrogen (55 psi) for
1.5 hours. The mixture was filtered and the filtrate purified by silica gel
chromatography to provide
the desired compound as a yellow oil (P-0044, 4.0 mg, 14%). MS (ESI) [M-H]-
=387.00.
[01391 Additional compounds maybe prepared similarly to the protocol of Scheme
9, replacing 4-
fluorophenylboronic acid 36 with an appropriate boronic acid or boronic acid
ester in Step 1,
Purification for the last step was alternatively done by HPLC, using
Phenomenex C18 column with
20-100% solvent B gradient over 40 minutes, with flow rate of 20 mL/minute,
Solvent A was water
with 0.1% trifluoroacetic acid and solvent B was acetonitrile with 0.1%
trifluoroacetic acid. For
compounds P-0059, P-0063, P-0064, P-0065, P-0066, P-0067, P-0068, P-0072-P-
0075, P-0077 and
P-0080 the first two steps of the reaction were performed in one step (per the
following Step I a for
compound P-0059):
F3C0 0 B(OH)2 H 0
Step 1 a N
OTf r'C~
35 36a 38a
Step la - Preparation ofl-[6-(3-ethylphenyl)-4,4-dimethyl-1,2,3,4-tetrahydro-
quinolin-7-yl]-
ethanone (38a):
[01401 Into a solution of trifluo.romethanesulfonic acid 7-acetyl-4,4-dimethyl-
l-(2,2,2-trifluoro-
58
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
acetyl)-1,2,3,4-tetrahydro-quinolin-6-yl ester (35, 80 mg, 0.18 mmol) and 3-
ethylphenylboronic acid
(36a, 54 mg, 0.36 mmol) in 800 l of acetonitrile, 400 l of lM aqueous
potassium carbonate and
[1,1'-bis(diphcnylphosphino)ferrocene] dichloropalladium(II) (-0.02 mmol) as
catalysis were added
and the reaction mixture was irradiated under microwave conditions for 10
minutes at 155 T. The
reaction mixture was diluted with water and extracted 2x with ethyl acetate.
The combined organic
extracts were washed with water, brine, dried over anhydrous magnesium
sulfate, filtered and the
filtrate concentrated under vacuum. The residue was dissolved in 1 ml of
dichloromethane and excess
of hexane was added. A precipitate was removed by filtration and washed with
hexane. The filtrate
solution was concentrated under vacuum to provide the desired compound as
yellow oil (38a, 67 mg),
which was used in the next step without further purification.
The following compounds were made similarly to the protocol of Scheme 9:
1-(1-Benzyl-4,4-dimetliyl-6-p-tolyl-1,2,3,4-tetrahydro-quinolin-7-yl)-
ethylamine (P-0039),
1 -[1 -B enzyl-6-(4-chloro-phcnyl)-4,4-dimethyl- 1,2,3,4-tetrahydro-quinolin-7-
yl] -ethyl amine (P-0046),
1-[ 1-Benzyl-6-(3-methoxy-phenyl)-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-
yl] -ethyl arnine
(P-0050),
1-[ 1-Benzyl-6-(4-methoxy-phenyl)-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-
yl]-ethylamine
(P-0054),
l-(]-Benzyl-4,4-dimethyl-6-thiophen-2-yl-1,2,3,4-tetrahydro-quinolin-7-yl)-
ethylamine (P-0055),
1-[l-Benzyl-6-(3-ethyl-phenyl)-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl]-
ethylamine (P-0059),
1-[ 1-Benzyl-6-(3 -benzyloxy-4-methoxy-phcnyl)-4,4-dimethyl-1,2, 3,4-
tetrahydro-quinolin-7-yl] -
ethylamine (P-0063),
1-[ 1-Benzyl-4,4-dimethyl -6-(3 -methyl-3 H-imidazol-4-yl)-l, 2,3,4-tetrahydro-
quinolin-7-yl] -
ethylamine (P-0064),
5-[7-(1-Amino-ethyl)-1-benzyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-6-yl]-2-
methoxy-phenol
(P-0065),
1-[1-Benzyl-6-(3-chloro-phenyl)-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl]-
ethylamine (P-0066),
1-(6-B enzo [ 1,3 ] dioxol-5 -yl- l -benzyl-4,4-dimethyl-1,2, 3,4-tetrahydro-
quinolin -7-yl)-ethylamine
(P-0067),
1-[1-Benzyl-6-(4-ethyl-phenyl)-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl]-
ethylamine (P-0068),
3-[7-(1-Amino-ethyl)-1-benzyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-6-yl]-
benzamide (P-0072),
4-[7-(1-Amino-ethyl)-1-benzyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-6-yl]-
benzamide (P-0073),
1-[ 1-B cnzyl-6-(3 -fluoro-4-methyl-phenyl)-4,4-dimethyl-1,2,3,4-tetrahydro-
quinolin-7-yl]-ethylamine
(P-0074),
1-[ 1-Benzyl -6-(2,4-dimethyl-thiazol-5 -yl) -4,4 -dimethyl-1,2, 3,4-
tetrahydro-quinolin-7-yl] -ethylamine
(P-0075),
1-[1-Benzyl-6-(1H-indol-5-yl)-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl]-
ethylamine (P-0077),
and
59
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
1-[1-Benzyl-4,4-dimethyl-6-(5-methyl-thiophen-2-yl)-1,2,3,4-tetrahydro-
quinolin-7-yl]-ethylamine
(P-0080).
The following table shows compounds that were prepared similarly to the
protocol of Scheme 9,
where optimal reaction conditions may have varied, for example, in terms of
solvents, time and
temperature of the reaction, and in chromatography conditions for purification
of the desired
compounds, with the compound number in Column 1, the boronic acid or ester
used in Step 1 or 1a in
Column 2, the resulting compound in Column 3 and the experimental mass
spectrometry result in
Column 4.
Compound number Boronic acid/ester Compound structure MS (ESI)
[M+H]'
P-0039 QBOH N 368.0
off [M-NH2 ] 399.9
P-0046 i B'OH N NHZ [M NH,-]'
OH
CI
i
NH2
P-0050 O 'a % B-OH N 400.20
OH
~O \ NH2 354.0
P-0054 Q8OH N [M NH ] `
OH
O
NH2
P-0055 S OH 376.95
OH
~I
NHZ
P-0059 (~ B.oH N 352.3
I i [M-NH,-]+
OH
NH2
P-0063` 'O N 490.3
B.O o [MNIh]+
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
~I
\ NH2
P-0064 B0 N I 375.8
NJN
O
\s-/
NH2
P-0065* l0 N 400.3
(~ B.0 OH [M-NH2_]
NHz 388.3
P-0066 cis ..B-OH N I M NH,
OH
0 NHZ
P-0067 <O BOH 1N 398.3
OH o [M-NH,
Flo
NHZ
P-0068 CBOH N ( 382.3
OH
I
NH2
P-0072 H2N (/ B,0 H N NH2
0 OH 0
i I
O NH2
P-0073 H2N OH
B' r
OH NHz
NH2
P-0074 nL N 386.3
F XBOH H F [M NH,
N ,
/ 1 1 \ NH2
N 389,5
P-0075 S B'
0 s LM NH2]T
N
61
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
NHL----
OH
P-0077 HOB W'I:ZIMI
OH NH2
P-0080 HO' B S f N 374.3
s
* P-0063 and P-0065 isolated from the same reaction.
Example 10: Preparation of 1-(1-Benzyl-6-benzyloxy-4,4-dimethyl- 1,2,3,4-
tetrahydro-quinolin-
7-yl)-ethylamine P-0060.
101411 1 -(1 -Benzyl-6-benzyloxy-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-
yl)-cthylamine P-0060
was prepared in five steps from 1-(7-acetyl-6-hydroxy-4,4-dimethyl-3,4-dihydro-
2H-quinolin-1-yl)-
2,2,2-trifluoro-ethanone 34 as shown in Scheme 10.
Scheme 10
OyCF3 O Br OYCF3 O H O Step 3
N ~ Step 2 + OH
6Br
34 23 41 ' 42
23
SOH
p N NH2
r1---J I
N' Step 4 N Step 5 N
O 1 O O
43 44 P-0060
Step 1-Preparation of 1-(7-acetyl-6-benzyloxy-4,4-dirnethvl-3,4-dihydro-2H-
quinolin-1 yl)-2,2,2-
trifuoro-ethanone (41):
[0142] To a solution of 1-(7-acetyl-6-hydroxy-4,4-dimethyl-3,4-dihydro-2H-
quinolin-l-yl)-2,2,2-
trifluoro-etha none (34, 500.0 mg, 1.58 mmol) in 30 mL of acetone, potassium
carbonate (600 mg. 4,8
mmol), potassium iodide (260 mg, 1.6 mmol) and benzyl bromide (23, 0.21 mL,
1.7 mmol) were
added. The reaction mixture was stirred at room temperature for 4 hours, then
partitioned between
ethyl acetate and water. The organic layer was washed with brine, dried over
magnesium sulfate,
filtered and the filtrate purified by silica gel chromatography to provide the
desired compound (41,
422 mg, 65%).
62
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
Step 2-Preparation of 1-(6-be),izyloxy-4,4-dime(hyl-1,2,3,4-tetrahydro-
quinolin-7-yl)-ethanone (42):
[01431 Into a 1-neck round-bottom flask, 1-(7-acetyl-6-benzyloxy-4,4-dimethyl-
3,4-dihydro-21I-
quinolin-1-yl)-2,2,2-trifluoro-ethanone (41, 550 mg, 1.35 mmol) and 1 N
potassium hydroxide
(1.5 mL, 1.5 mmol), were mixed in 25 mL of methanol. The reaction mixture was
stirred at room
temperature for 2 hours and concentrated under vacuum. The residue was
partitioned between ethyl
acetate and brine. The organic layer was dried over magnesium sulfate,
filtered and the filtrate
purified by silica gel chromatography to provide the desired compound as pale
yellow solid (42,
400 mg, 95 %). MS (ESI) [M+H+]+= 310.2.
Step 3 - Preparation of 1-(1-benzyl-6-benzyloxy-4,4-dime(hyl-1, 2, 3,4-
tetrahydro-quinolin-7-yl)-
ethanone (43):
101441 A solution of 1-(6-benzyloxy-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-
yl)-ethanone (42,
422 mg, 1.36 mmol), benzyl bromide (23, 0.49 mL, 4.1 mmol) and N,N-
diisopropylethylamine
(0.71 mL) in 20 mL of N-methylpyrrolidone was warmed to 1 00 C for 40 minutes
via microwave
irradiation. The reaction mixture was diluted with ethyl acetate and washed 2x
with brine. The
organic phase was dried with magnesium sulfate, filtered and the filtrate
purified by silica gel
chromatography to provide the desired compound as pale yellow oil (43, 266 mg,
41 %). MS (ESI)
[M--H+]+ = 400.2.
Step 4 - Preparation of 1-(1-benzyl-6-benzyloxy-4, 4-dimethyl-1,2,3,4-
tetrahydro-quinolin-7wl)-
ethanone oxime (44):
[01451 A solution of 1-(1-benzyl-6-benzyloxy-4,4-dimethyl-1,2,3,4-tetrahydro-
quinolin-7-yl)-
ethanone (43, 100 mg, 0.25 mmol), hydroxylamine hydrochloride (35 mg, 0.50
mmol), and pyridine
(0.04 mL, 0.5 mol) in 15 mL of ethanol was stirred at at 80 C for 4 hours.
The reaction mixture was
concentrated under reduced pressure and the residue was partitioned between
ethyl acetate and water.
The organic layer was washed with brine, dried over magnesium sulfate,
filtered and the filtrate
concentrated under vacuum. The residue was purified by silica gel
chromatography to provide the
desired compound as a pale yellow solid (44, 60 mg, 57 %). MS (ESI) [M+H+]+=
415.2,
Step 5 - Preparation of1-(1-benzyl-6-benzyloxy-4,4-dimethyl-1,2,3,4-te(rahydro-
quinolin-7-yl)-
ethylamine (P-#06#),-
[01461 A mixture of 1-(1-benzyl-6-benzyloxy-4,4-dimethyl-1,2,3,4-tetrahydro-
quinolin-7-yl)-
ethanone oxime (44, 30 mg, 0.072 mmol) and Raney nickel (50%, aqueous slurry)
in 5 mL of
methanol and 2 mL of ammonium hydroxide was agitated in a Parr shaker under
hydrogen (55 psi) for
1.5 hours. The mixture was filtered and the filtrate purified by silica gel
chromatography to provide
the desired compound as a yellow oil (P-0060, 7.2 mg, 25 %). MS (ESI) [M+H+]+_
400.8.
63
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
Example 11: Preparation of 2-methyl-pyridine-4-carbaldehyde 21 b.
[0147] 2-Methyl-pyridine-4-carbaldehyde 21b (used e.g. in Scheme 5b) was
prepared in three steps
from 2-methyl-isonicotinic acid 45 as shown in Scheme 11.
Scheme 11
O OH
F F Ste 1 N`!~/O F F Step 2 Step 3 H
N\ / OH + F F P / O \ / F OH N\ O
45 F 46 47 F F 48 21b
Step I - Preparation of 2-methyl-isonicotinic acid pentafluorophenvl ester
(47):
[0148] To a suspension of 2-methyl-isonicotinic acid (45, 2,74 g, 20 mmol) and
pcntafluorophenol
(46, 3.7 g, 20 mmol) in 100 mL of anhydrous tetrahydrofuran, N,N'-
diisopropylcarbodiimide (3.1 mL,
20 mmol) was added. The mixture was stirred at room temperature overnight,
then filtered through a
pad of Celite. The filtrate was collected and used in the next step without
further purification.
Step 2 - Preparation of (2-znetlzyl pyridin-4 yl)-methanol (48):
[0149] To a solution of 2-methyl-isonicotinic acid pentafluorophenyl ester
(47), lithium borohydride
(12 rL, 2.0 M in tetrahydrofuran) was added slowly. The reaction mixture was
stirred at room
temperature overnight, then cooled with ice water bath and 40 mL of IN sodium
hydroxide was
slowly added. The mixture was then extracted with chloroform and the combined
organic layers were
washed with brine, dried over sodium sulfate, filtered and the filtrate
concentrated under vacuum.
The residue was purified by silica gel chromatography eluting with hexanes and
ethyl acetate to
provide the desired compound as a white solid (48, 2.38 g, 80%). MS (ESI)
[M+H']' = 123.85.
Step 3 - Preparation of 2-n7ethylpyridine-4-carbaldehyde (21b):
[0150] A solution of chromium (VI) oxide (2 g, 20 mmol), pyridine (4 mL, 50
mmol) and 20 mL of
dichloromethane was stirred at room temperature for 30 minutes. To this
mixture, cooled with an ice
water bath, (2-methyl-pyridin-4-yl) -methanol (48, 0.5 g, 4.0 mmol) in 5 mL of
dichloromethane was
added. The reaction mixture was stirred at room temperature for 30 minutes.
The reaction mixture
was cooled with an ice water bath and diluted with ethyl acetate, then
filtered through a pad of Celitc.
The filtrate was concentrated under vacuum and the residue was purified by
silica gel chromatography
eluting with hexanes and ethyl acetate to provide the desired compound as a
colorless liquid (21b, 0.1
g, 20%).
64
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
Example 12: Preparation of 1-[4,4-dimethyl-l-(2-methyl-pyridin-4-ylmethyl)-6-p-
tolyl-1,2,3,4-
tetrahydro-quinolin-7-yl]-ethylamine P-0062.
[0151] 1-[4,4-Dimethyl-1-(2-methyl-pyridin-4-ylmethyl)-6-p-toly1-1,2,3,4-
tetrahydro-quinolin-7-
yl]-ethylamine P-0062 was prepared in four steps from trifluoromethanesulfonic
acid 7-acetyl-4,4-
dimethyl-l-(2,2,2-trifluoro-acetyl)-1,2,3,4-tetrahydro-quinolin-6-yl ester 35
as shown in Scheme 12.
Scheme 12
F30Y00 B(OH)2 H 0 Step 2
NY Y i
Step 1 p H
N
OTf
35 49 50 + k-N
21b
4C,~,N \N OH 0 N NH2
N Step 3 N I Step 4
51 52
P-0062
Step I - Preparation of 1-(4,4-dimethyl-6 p-tolyl-1,2,3,4-tetrahydro-quinolin-
7 yl)-ethanone (50):
101521 Into a solution of trifluoromethanesulfonic acid 7-acetyl-4,4-dimethyl-
l-(2,2,2-trifluoro-
acetyl)-1,2,3,4-tetrahydro-quinolin-6-yl ester (35, 100 mg, 0.20 mmol) in 3 mL
of tetrahydrofuran in
a microwave reaction vial, 4-toluene boronic acid (49, 61 mg, 0.45 mmol) and
tetrakis(triphenylphosphine)palladium (0) (-2 mg, catalyst) were added. The
reaction mixture was
warmed to 70 C for 20 minutes via microware irradiation. The reaction mixture
was diluted with
ethyl acetate and the organic layer washed with brine, dried over magnesium
sulfate, filtered, and the
filtrate purified by silica gel chromatography to provide the desired compound
(50, 28 mg, 42%). MS
(ESI) [M+H+] `= 294.3.
Step 2 - Preparation ofl-[4,4-dimethyl-]-(2-rneth_ylpyridin-4-ylmethyl)-6p-
tolyl-1,2,3,4-tetrahydro-
quinolin-7 yl]-ethanone (51):
[0153] To a mixture of 2-methylpyridine-4-carbaldehyde (21b, 14 mg, 0.11 mmol)
and 1-(4,4-
dimethyl-6-p-tolyl-1,2,3,4-tetrahydro-quinolin-7-yl)-ethanone (50, 100 mg, 0.4
mmol) in 5 mL of
acetonitrile, triethylsilane (0.2 mL, 1.2 mmol) and trifluoroacetic acid (0.4
mL, 5.0 mmol) were
added. The reaction mixture was stirred at 100 C for 2 hours. The reaction
mixture was
concentrated under vacuum and partitioned between aqueous potassium carbonate
and ethyl acetate,
The organic layer was dried over anhydrous sodium sulfate, filtered and the
filtrate purified by silica
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
gel chromatography to provide the desired compound as a yellow solid (51, 18
mg, 47%). MS (ESI)
[M+H-]+= 399.2.
Step 3 Preparation ofl-[4,4-dimethyl-l-(2-methylpyridin-4ylme/hyl)-6 p-tolyi-
1,2,3,4-tetrahydro-
quinolin-7ylJ-ethanone oxime (52):
[0154] A solution of 1-[4,4-dimethyl-l-(2-methyl-pyridin-4-ylmethyl)-6-p-tolyl-
1,2,3,4-tetrahydro-
quinolin-7-yl]-ethanone (51, 22.7 mg, 0.057 mmol), hydroxylamine hydrochloride
(7.9 mg, 0,11
mmol), and 0.03 mL of pyridine in 15 mL of ethanol was stirred at 80 C for 4
hours. The reaction
mixture was concentrated under vacuum and the residue was partitioned between
ethyl acetate and
water. The organic layer was washed with brine, dried over magenesium sulfate,
filtered and the
filtrate concentrated under vacuum. The residue was purified by silica gel
chromatography to provide
the desired compound as a pale yellow solid (52, 10 mg, 43 %). MS (ESI) [M+H+]
414.2.
Step 4 -Preparation of 1-[4,4-dimethyl-]-(2-methyl pyridin-4ylmethyl)-6p-tolyi-
1,2,3,4-tetra/iydro-
quinolin-7 ylJ-ethylamine (P-0062):
[0155] A mixture of 1-[4,4-dimethyl-l-(2-methyl-pyridin-4-ylmethyl)-6-p-tolyl-
1,2,3,4-tetrahydro-
quinolin-7-yl]-ethanone oxime (52, 10 mg, 0.024 mmol) and Raney nickel (50%,
aqueous slurry) in 5
mL of methanol and 1 mL of ammonium hydroxide was agitated in a Parr shaker
under hydrogen (55
psi) for 1.5 hours. Filtration and silica gel chromatography provided the
desired compound as a
yellow oil (P-0062, 0.9 mg, 9%). MS (ESI) [M+H+]+= 400.3.
Example 13: Preparation of 1-[7-(1-amino-ethyl)-6-ethyl-4,4-dimethyl-3,4-
dihydro-2H-
quinolin-1-yl]-2-methoxy-ethanone P-0001
[0156] 1-[7-(1-Amin o-ethyl)-6-ethyl-4,4-dimethyl -3,4-dihydro-2H-quinolin-l-
yl]-2-methoxy-
ethanone P-0001 was prepared in three steps from 1-(6-ethyl-4,4-dimethyl-
1,2,3,4-tetrahydro-
quinolin-7-yl)-ethanone 9 as shown in Scheme 13.
Scheme 13
O OH O
H O O O~ O O O N N/ 0---f:- NHZ
N e Step 1 N Step 2 Step 3
9 53 Ot
54 55 P-0001
Step I - Preparation of I -(7-acetyl-6-ethyl-4, 4-dimethyl-3, 4-dihydro-2H-
quinolin-1-yl)-2-methoxy-
ethanone (54):
[0157] To a mixture of 1-(6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-
yl)-ethanone (9, 110
mg, 0.43 mmol), N,N-diisopropylethylamine (0.38 mL, ?.2 mmol), and 4-
dimethylaminopyridine (5
mg, 0 " t ;H) in 20 mL oftetril :.._ir, u:nricth i, ._ ty 1 chloride (53, 0.12
mL, 1.3 mmol) in 2
66
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
mL of tetrahydrofuran was added dropwisc at 0 C. The reaction mixture was
stirred at room
temperature overnight. The reaction mixture was poured into water and
extracted with
dichloromethane. The organic layer was collected, washed with saturated sodium
bicarbonate
solution and brine, dried over magnesium sulfate, filtered and the filtrate
concentrated under vacuum.
The residue was purified by silica gel chromatography eluting with ethyl
acetate and hexanes to
provide the desired compound as a light brown oil (54, 81 mg, 62%). MS (ESI)
[M+1T]`= 304.50.
Step 2 -Preparation of ]-(6-ethyl-7-(1-hydroxyimino-ethyl)-4, 4-dimethyl-3,4-
dihydro-2H-quinolin-l -
yl)-2-methoxy-ethanone (55):
[01581 A solution of 1-(7-acetyl-6-ethyl-4,4-dimethyl-3,4-dihydro-2H-quinolin-
l-yl)-2-methoxy-
ethanone (54, 75 mg, 0.25 mmol), hydroxylamine hydrochloride (30 mg, 0.5
mmol), and pyridine (0.1
mL, 1 mmol) in 10 mL of ethanol was stirred at 80 C for 2 hours. The reaction
mixture was
concentrated and the residue was dissolved in ethyl acetate, washed with water
and brine, dried over
magnesium sulfate, filtered and the filtrate concentrated under vacuum. The
residue was purified by
silica gel chromatography eluting with ethyl acetate and hexanes to provide
the desired compound as
a colorless oil (55, 40 mg, 50%). MS (ESI) [MfH+]-= 319.45.
Step 3 - Preparation of 1-[7-(1-amino-ethyl)-6-ethyl-4, 4-dimethyl-3,4-dihydro-
2H-quinolin-l-ylJ-2-
methovy-ethanone (P-0001):
10159] A mixture of 1-(6-ethyl-7-(1-hydroxyimino-ethyl)-4,4-dimethyl-3,4-
dihydro-2H-quinolin-l-
yl)-2-methoxy-ethanone (55, 40 mg, 0,1 mmol) and Raney nickel (50%, aqueous
slurry) in 6 mL of
methanol and 3 mL of ammonium hydroxide was shaken under hydrogen (55 psi) for
5 hours. The
reaction mixture was filtered and the filtrate concentrated under vacuum. The
residue was purified by
silica gel chromatography eluting with methanol and dichloromethane to provide
the desired
compound as a colorless oil (P-0001, 22 mg, 60%). MS (ESI) [MfH+]+= 305.95.
Example 14: Preparation of 7-(1-amino-ethyl)-1-(3-methoxy-propyl)-4,4-dimethyl-
6-p-tolyl-3,4-
dihydro-1Il-quinolin-2-one P-0040 and 1-[1-(3-methoxy-propyl)-4,4-dimethyl-6-p-
tolyl-1,2,3,4-
tetrahydro-quinolin-7-yl]-ethylamine P-0043.
[0160] 7-(I-amino-ethyl)-1-(3-methoxy-propyl)-4,4-dimethyl-6-p-tolyl-3,4-
dihydro-IH-quinolin-2-
one P-0040 and 1-[1-(3-methoxy-propyl)-4,4-dimethyl-6-p-tolyl-1,2,3,4-
tctrahydro-quinolin-7-yl]-
ethylamine P-0043 were prepared in eight steps from 3-ethylaniline 56 as shown
in Scheme 14,
67
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
Scheme 14
H
O O N Step 4
0
H2N Step 1 0 N Step 2 I Step 3
+ ~CIi 9 Br
2 57 58
56
0, 0,
Step 6
H 0 0 0 Step 7
O N Step 5 0 N B(OH)2 O TN
Br + Br'v 0 I Br
+
60 0 61 62 63
49
(J( NH2
O N ~
'OH Step 8a
N P-0040
O N
NH2
64 Step 8b N
P-0043
Step I Preparation of 3-methyl-but-2-enoic acid (3-ethyl-phenyl)-amide (57):
[0161] To a mixture of 3-ethylaniline (56, 20.00 g, 0.16 mol) and sodium
hydroxide (16 g, 0.41
mol) in 160 mL of water and 100 mL of dichloromethane, a solution of 3,3-
dimcthylacryloyl chloride
(2, 22 mL, 0.20 mol) in 50 mL of dichloromethane was slowly added at C. The
reaction mixture
was then stirred at room temperature for 16 hours, then poured into 400 mL of
water and extracted
with dichlorornethane. The combined organic layers were collected, washed with
brine, dried over
magnesium sulfate, filtered and the filtrate concentrated under vacuum. The
resulting material was
purified by recrystallization from ethyl acetate and hexanes to provide the
desired compound as a
white solid (57, 31.1 g, 93%). MS (ESI) [M+H4]' = 204.15.
Step 2 - Preparation of 7-ethyl-4, 4-dimethyl-3, 4-dihydro-1 H quinolin-2-one
(58):
[01621 To a suspension of aluminum trichloride (13.1 g, 98.4 mmol) in 50 mL of
1,2-
dichlorobenzene, a solution of 3-methyl-but-2-enoic acid (3-ethyl-phenyl)-
amide (57, 10 g, 49.19
mmol) in chlorobcnzcnc was added dropwisc at 0-10 C. After completion of
addition, the mixture
was stirred at 110 C for 2 hours, then cooled down to room temperature and
poured into crushed ice.
To this mixture was added 60 mL of 2 M hydrochloric acid solution, followed by
extraction with
68
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
dichloromethane. The organic layers were collected, washed with saturated
sodium bicarbonate and
brine, dried over magnesium sulfate, filtered and the filtrate concentrated
under vacuum, The
resulting material was purified by recrystallization from ethyl acetate to
provide the desired
compound as a white solid (58, 9.9 g, 89%).
Step 3 -Preparation of 6-bromo--7-ethyl-4,4-dimethyl-3,4-dihydro-IH-quinolin-2-
one (59):
[0163] To a solution of 7-ethyl-4,4-dimethyl-3,4-dihydro-lH-quinolin-2-one
(58, 3.0 g, 15.0 mmol)
in 100 mL of nitromethane, bromine (0.84 mL, 16.0 mmol) was added dropwise,
followed by
aluminum trichloride (100 mg, 0.7 mmol). The reaction mixture was stirred at
room temperature
overnight, then poured into ice, extracted with dichloromethane, washed with
sodium bicarbonate
solution and water, dried over magnesium sulfate, filtered and the filtrate
concentrated under vacuum.
The resulting material was purified by silica gel chromatography, eluting with
hexanes and
dichloromethane and ethyl acetate to provide the desired compound as a white
solid (59, 2.4 g, 58%).
MS (ESI) [M+H+]+= 283.90.
Step 4-Preparation of 7-acetyl-6-bromo-4,4-dimethyl-3,4-dihydro-]H-quinolin-2-
one (60):
[0164] To a mixture of 6-bromo-7-ethyl-4,4-dimethyl-3,4-dihydro-1 H-quinolin-2-
one (59, 1 g, 3
mmol) in 50 mL of dichloromethane, a premixed fine powder of potassium
permanganate (4 g, 20.0
mmol) and copper(II) sulfate pentahydratc (6 g, 20.0 mmol) were added. The
reaction mixture was
gently refluxed for 72 hours, and then filtered through a Celite pad. The
Celite pad was washed with
dichloromethane and ether. The organic portions were collected and combined
and solvents removed
under vacuum. The resulting material was purified by silica gel
chromatography, eluting with
hexanes and dichloromethane and ethyl acetate to provide the desired compound
as a white solid (60,
0.13 g, 20%). MS (ESI) [M+H+]+= 295/297.87.
Step 5 -Preparation of 7-acetyl-6-bromo-l-(2-rnethoxy-ethyl)-4,4-dimethyl-3,4-
dihydro-IH-quinolin-
2-one (62):
[0165] To a solution of 7-acetyl-6-bromo-4,4-dimethyl-3,4-dihydro- I H-
quinolin-2-one (60, 170
mg, 0.57 mmol) in 10 mL of acetonitrile, 1-bromo-3-methoxy-propane (61, 120
mg, 0.75 mmol),
potassium iodide (30 mg, 0.2 mmol) and potassium carbonate (240 mg, 1.7 mmol)
were added. The
reaction mixture was stirred at 80 C for 20 hours, then cooled down and
poured into water and
extracted with ethyl acetate. The organic layers were dried over magnesium
sulfate, filtered and the
filtrate concentrated under vacuum, The resulting material was purified by
silica gel chromatography,
eluting with hexanes%dichloromethane/ethyl acetate to provide the desired
compound as a colorless oil
(62, 0.11 g, 42%).
69
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
Step 6-Preparation of 7-acetyl-1-(3-methoxy-propyl)-4,4-dimethyl- 6 p-tolyl-
3,4-dihydro-IH-
quinolin-2-one (63):
[0166] To a mixture ofp-tolyl boronic acid (49, 18 mg, 0.13 mmol), potassium
phosphate (37 mg,
0.17 mmol), palladium acetate (4 mg, 0.02 mmol), and diphenyl(t-Bu)2 (10 mg,
0.03 mmol) in 1 mL
of tetrahydrofuran in a sealed tube, 7-acetyl-6-bromo-1-(2-methoxy-ethyl)-4,4-
dimethyl-3,4-dihydro-
I H-quinolin-2-one (62, 32 mg, 0.087 mmol) in 1 mL of tetrahydrofuran was
added. The reaction
mixture was stirred at 65 C for 20 hours, then quenched with aqueous
saturated sodium bicarbonate
and extracted with ethyl acetate. The organic layer was washed with brine,
dried over sodium sulfate,
filtered and the filtrate concentrated under vacuum. The resulting material
was purified by silica gel
chromatography, eluting with dichloromethane and methanol to provide the
desired compound as a
colorless oil (63, 15 mg, 44%). MS (ESI) [M+H+]+= 380.05.
Step 7 - Preparation of 7-(I -[(E)-hydroxyimino]-ethyl)-1-(3-methoxy propyl)-
4,4-dimethyl-6 p-tolyl-
3, 4-dihydro-IH-quinolin-2-one (64):
[0167] A solution of 7-acetyl-l -(3-methoxy-propyl)-4,4-dimethyl-6-p-tolyl-3,4-
dihydro-1 H-
quinolin-2-one (63, 19 mg, 0.04 mmol), hydroxylaminc (3 mg, 0.09 mmol), and a
drop of pyridine in
4 mL of ethanol was stirred at 80 C for 2 hours. The reaction mixture was
concentrated under
vacuum and the residue was dissolved in ethyl acetate, washed with water and
brine, dried over
magnesium sulfate, filtered and the filtrate concentrated under vacuum, The
resulting material was
used in next step without further purification.
Step 8a Preparation of 7-(1-amino-ethyl)-1-(3-methoxy-propyl)-4,4-dimethyl-6p-
tolyl-3,4-dihydro-
IH-quinolin-2-one (P-0040):
[0168] A mixture of 7-{1-[(E)-hydroxyimino]-ethyl}-1-(3-methoxy-propyl)-4,4-
dimethyl-6-p-tolyl-
3,4-dihydro-IH-quinolin-2-one (64, 12 mg, 0.02 mmol) and Raney nickel (50%,
aqueous slurry) in 5
mL of methanol and 1 mL of ammonium hydroxide was shaken under hydrogen (55
psi) for 4 hours.
The catalyst was removed by filtration and the filtrate was concentrated. The
residue was purified by
preparative reverse phase HPLC, mobile phase A, 5% of acetonitrile, 95% water,
0.1 % acetic acid,
and mobile phase B, 95% acetonitrile, 5% of water, 0.1% of acetic acid, to
provide the desired
compound as a white solid (P-0040, 6 mg, 70%). MS (ESI) [M-NH3]*- 364.00.
Step 8b - Preparation of I -(I -butyl-4, 4-dimethyl-6p-tolyl-1,2, 3, 4-
tetrahydro-quinolin-7 -yl)-
et vlamine (P-0043):
[0169] To a solution of 7-{1-[(E)-hydroxyimino]-ethyl }-1-(3-methoxy-propyl)-
4,4-dimethyl-6-p-
tolyl-3,4-dihydro-1 H-quinolin-2-one (64, 10 mg, 0.02 mmol) in 5 mL of
toluene, borane-dimethyl
sulfide complex (0.05 mL, 0.6 mmol) was added. The reaction mixture was
stirred at 100 C for 3
hours, then cooled down and poured into 20 mL of 10% aqueous sodium carbonate
solution. The
mixtu stirred at room temperature for 30 minutes, The organic layer was
collected, and the
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
aqueous layer was extracted with ethyl acetate. The combined organic layers
were washed with brine,
dried over magnesium sulfate, filtered and the filtrate concentrated under
vacuum. The resulting
material was separated by preparative TLC, eluting with dichloromethane and
methanol to provide the
desired compound as a colorless oil (P-0043, 1.1 mg, 20%). MS (ESI) [M+H*]- =
367.00.
Example 15: Preparation of 7-(1-amino-ethyl)-6-ethyl-l-(3-methoxy-propyl)-4,4-
dimethyl-3,4-
dihydro-1H-quinolin-2-one P-0014.
101701 7-(1-Amino-ethyl)-6-ethyl- l -(3-methoxy-propyl)-4,4-dimethyl-3,4-
dihydro- I H-quinolin-2-
one P-0014 was prepared in four steps from 1-(6-ethyl-4,4-dimethyl-1,2,3,4-
tetrahydro-quinolin-7-
yl)-ethanone 9 as shown in Scheme 15.
Scheme 15
O O,
H O O
H
O Step 1 N Step 2 0 N
9 Br 61 65 66
O O,
NOH NH2
Step 3 0 N Step 4
67 P-0014
Step 1 -Preparation f 1-[6-ethyl-]-(3-methoxy-propyl)-4,4-dimethyl-1,2,3,4-
tetrahydro-quinolin-7-
ylJ-ethanone (65):
[0171] To a solution of 1-(6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-
yl)-ethanone (9, 190
mg, 0.82 mmol) in 10 mL of aectonitrile, 1-bromo-3-methoxy-propane (61, 184
mg, 1.2 mmol),
potassium iodide (140 mg, 0.82 mmol) and potassium carbonate (450 mg, 3.3
mmol) were added,
The reaction mixture was stirred at 100 C for 20 hours, then cooled down and
poured into water and
extracted with ethyl acetate, The organic layer was dried over magnesium
sulfate, filtered and the
filtrate concentrated under vacuum. The resulting material was purified by
silica gel chromatography,
eluting with hexanes and ethyl acetate to provide the desired compound as
yellow oil (65, 84 mg,
34%).
Step 2 - Preparation of 7-acetyl-6-ethyl-l-(3-methoxy-propy I)-4,4-dimethyl-
3,4-dihydro-IH-
quinolin-2-one (66):
101721 To a solution of 1-[6-ethyl-l-(3-methoxy-propyl)-4,4-dimethyl-1,2,3,4-
tetrahydro-quinolin-
7-yl]-ethanone (65, 81 mg, 0,27 mmol) in 10 mL of dichloromethane, a premixed
fine powder of
71
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
potassium permanganate (210 mg, 1.3 mmol) and copper(11) sulfate pentahydrate
(330 mg, 1.3 mmol)
was added. The reaction mixture was gently refluxed for 3 days. The reaction
mixture was filtered
through a Celite pad and the pad was washed with dichloromethane and ether.
The organic portions
were collected and combined, the solvent removed under vacuum, and the residue
was purified by
silica gel chromatography, eluting with dichloromethane and methanol to
provide the desired
compound as light yellow oil (66, 50 mg, 601,10). MS (ESI) [M+H`]' = 318.05.
Step 3 - Preparation of 6-ethyl-7-(1-[(E)-hydroxyimino]-ethyl)-1-(3-methoxy
propyl)-4,4-dimethyl-
3,4-dihydro-IH-quinolin-2-one (67):
[0173] A solution of 7-acetyl-6-ethyl-l-(3-methoxy-propy 1)-4,4-dimethyl -3,4-
dihydro-lH-quinolin-
2-one (66, 0.08 g, 0.1 mmol), hydroxylamine hydrochloride (0.04 g, 0.5 mmol),
and pyridine (0.1 mL,
I mol) in 5 mL of ethanol was stirred at 80 C for 24 hours. The reaction
mixture was concentrated
under vacuum and the residue was dissolved in ethyl acetate, washed with water
and brine, dried over
magnesium sulfate, filtered and the filtrated concentrated under vacuum. The
resulting material was
used in the next step without purification.
Step 4 - Preparation of 7-(1-amino-ethyl)-6-ethyl-1-(3-methoxy propyl)-4,4-
dimethyl-3,4-dihydro-
IH-quinolin-2-one (P-0014):
[0174] Amixture of6-ethyl-7-{1-[(E)-hydroxyimino]-ethyl }-1 -(3-methoxy-
propyl)-4,4-dimethyl-
3,4-dihydro-1H-quinolin-2-one (67, 32 mg, 0.048 mmol) and Raney nickel (50%,
aqueous slurry) in 5
mL of methanol and 2 mL of ammonium hydroxide was shaken under hydrogen (55
psi) for 4 hours.
The catalyst was filtered off and the filtrate was concentrated under vacuum.
The resulting material
was purified by preparative reverse phase HPLC, mobile phase A, 5%
acetonitrile, 95% water, 0.1%
acetic acid and mobile phase B, 95% acetonitrile, 5% water, 0.1% of acetic
acid, to provide the
desired compound as a colorless oil (P-0014, 4.3 mg, 28%). MS (ESI) [M-fiH+]+-
319.2.
Example 16: Preparation of [7-(1-amino-ethyl)-6-ethyl-4,4-dimethyl-3,4-dihydro-
2H-quinolin-
1-yl]-pyridin-3-yl-methanone P-0018.
101751 [7-(1-Amino-ethyl)-6-ethy]-4,4-dimethyl-3,4-dihydro-2H-quinolin-l-yl]-
pyridin-3-yl-
methanone P-0018 was prepared in two steps from nicotinic acid 68 and [1-(6-
ethyl-4,4-dimethyl-
1,2,3,4-tetrahydro-quinolin-7-yl)-ethyl]-carbamic acid tert-butyl ester 17 as
shown in Scheme 16.
72
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
Scheme 16
11 Boc
H HN N \ ( HN' Boc N ( NH2
/
N
N. ( N,Step 1 Step 2
68 OH
17 69
P-0018
Step 1 -Preparation of (1-[6-ethyl-4,4-dimetlzvl-]-(pyridine-3-carbonyl)-
1,2,3,4-tetrahydro-quinolin-
7 yl]-ethyl}-carbamic acid tert-butyl ester (69):
[0176] A suspension of nicotinic acid (68, 100 mg, 0.8 mmol) in thionyl
chloride (5 mL, 70.0
mmol) was stirred at 85 C for 2 hours. The excess of thionyl chloride was
removed under vacuum,
and the residue was dissolved in 5 mL of dichloromethane, to which a mixture
of [1-(6-ethyl-4,4-
dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl)-ethyl]-carbamic acid tert-butyl
ester (17, 60 mg, 0.2
mmol) and N,N-diisopropylethylamine (0.10 mL, 0.6 mmol) in 2 mL of
dichloromethane was added.
The reaction mixture was at room temperature for 3 hours, then poured into
water and extracted with
dichloromethane. The organic layer was washed with brine, dried over magnesium
sulfate, filtered
and the filtrate concentrated under vacuum. The resulting material was
purified by silica gel
chromatography, eluting with dichloromethane and methanol to provide the
desired compound as a
light yellow solid (69, 64 mg, 8 0%). MS (ESI) [M+H]= 438.70.
Step 2 - Preparation of [7-(1-amino-ethyl)-6-ethyl-4, 4-dimethyl-3,4-dihydro-
2H-quinolin-1-yl]-
pyridin-3 yl-methan one (P-0018):
[0177] To a solution of {1-[6-ethyl-4,4-dimethyl-l-(pyridine-3-carbonyl)-
1,2,3,4-tetrahydro-
quinolin-7-yl]-ethyl}-carbamic acid tert-butyl ester (69) in 3 mL of
dichloromethane, a solution of
hydrochloric acid (0.2 mL, 4 M in dioxane) in 1 mL of dichloromethane was
added. The reaction
mixture was at room temperature for 2 hours, then poured into water and
extracted with
dichloromethane. The organic layer was washed with brine, dried over magnesium
sulfate, filtered
and the filtrate concentrated and dried under vacuum to provide the
hydrochloric acid salt of the
desired compound as a light yellow viscous oil (P-0018, 7 mg, 90%). MS (ESI)
[hi+H]-'= 339.00.
[0178] [7-(I-Amino-ethyl)-6-ethyl-4,4-dimethyl-3,4-dihydro-2H-quinolin-l-yl]-
pyridin-4-yl-
methanone P-0020, and [7-(1-amino-ethyl)-6-ethyl -4,4-dimethyl-3,4-dihydro-2H-
quinolin-l-yl]-
isoxazol-5-yl-methanone P-0022,
O N/ 0 ` o
NH2 NH2
NN
P-0020,and P-0022,
73
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
were prepared similarly to the protocol of Scheme 16, where optimal reaction
conditions may have
varied, for example, any of time and temperature of the reaction or
chromatography conditions for
purification of the desired compounds, replacing nicotinic acid 68 with
isonicotinic acid and
isoxazole-5-carboxylic acid, respectively, in step 1. MS (ESI) [M+H+]+_ 338.95
(P-0020); 329.15
(P-0022).
Example 17: Preparation of 1-[1-(2,2-difluoro-benzo[1,3]dioxol-5-,ylmethyl)-6-
ethyl-4,4-
dimethyl- 1,2,3,4-tetrahydro-quinolin-7-yl]-ethylamine P-0038.
[0179] 1-[1-(2,2-difluoro-benzo[1,3]dioxol-5-ylmethyl)-6-ethyl-4,4-dimethyl-
1,2,3,4-tetrahydro-
quinolin-7-yl]-ethylamine P-0038 was prepared in two steps from [1-(6-ethyl-
4,4-dimethyl-1,2,3,4-
tetrahydro -quinolin-7 -yl) -ethyl] -carb amic acid tert-butyl ester 17 as
shown in Scheme 17.
Scheme 17
O FX \
HN~Boc F F FX HN_Boc F O NH2
2
H y-O N
N Step 1 Step 2
17 70 H 71 P-0038
Step I Preparation of (I-[I-(2,2-difluoro-benzo[l,3Jdioxol-5-ylmethyl)-6-ethyl-
4,4-dimethyl-
1,2,3,4-tetrahydro-quinolin-7 y1J-ethyl)-carbamic acid tert-butyl ester (71):
[0180] To a mixture of 2,2-difluoro-benzo[1,3]dioxole-5-carbaldehyde (70, 48
mg, 0.26 mmol) and
sodium cyanoborohydride (8.1 mg, 0.13 mmol) in 1 mL of tetrahydrofuran in a
sealed tube, [1 -(6-
ethyl-4,4-dimethyl- 1,2,3,4-tetrahydro-quino lin-7 -yl) -ethyl] -carbamic acid
tert-butyl ester (17, 29 mg,
0.086 mmol) in 1 mL of tetrahydrofuran was added. The reaction mixture was
stirred at 65 C for 20
hours, then quenched with aqueous saturated sodium bicarbonate and extracted
with ethyl acetate.
The organic layer was washed with brine, dried over sodium sulfate, filtered
and the filtrate
concentrated under vacuum. The resulting material was purified by silica gel
chromatography, eluting
with hexanes and ethyl acetate to provide the desired compound as a colorless
oil (71, 32 mg, 74%).
MS (ESI) [M+H']+= 503.10.
Step 2-Preparation of 1-[] -(2,2-difluoro-benzo[],3Jdioxol-5ylmethyl)-6-ethyl-
4,4-rlirneihvl-I.2,3,4-
tetrahydro-quinolin-7-yl]-ethylamine (P-0038).
[0181] To {1-[1-(2,2-difluoro-benzo[1,3]dioxol-5-ylmethyl)-6-ethyl-4,4-
dimethyl-1,2,3,4-
tetrahydro-quinolin-7-yl]-ethyl}-carbamic acid tert-butyl ester (71, 32 mg,
0.064 mmol) dissolved in 1
ml, of dichloromethane, hydrochloric acid (0.2 mL, 4.0 M in dioxane) was added
and the mixture was
stirred at room temperature for 4 hours. The mixture was concentrated under
vacuum and the
resulting material was purified by silica gel chromatography, eluting with
dichloromethane and
74
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
methanol to provide the desired compound as a pale yellow solid (P-0038, 20
mg, 74%). MS (ESI)
[M+H `] } = 404.45.
Example 18: Preparation of 1-[6-ethyl-4,4-dimethyl-l-(tetrahvdro-pyran-4-
ylmethyl)-1,2,3,4-
tetrahvdro-quinolin-7-yl]-ethylamine P-0058.
101821 1-[6-Ethyl-4,4-dimethyl-l -(tetrahvdro-pyran-4-ylmethyl)-1,2,3,4-
tetrahydro-quinolin-7-yl]-
ethylamine P-0058 was prepared in two steps from [1-(6-ethyl-4,4-dimethyl-
1,2,3,4-tetrahydro-
quinolin-7-yl)-ethyl]-carbamic acid tert-butyl ester 17 as shown in Scheme 18.
Scheme 18
H HN Boc r/ HN Boc O 61 NHZ
+ Step 1 N Step 2
17 72 73
P-0058
Step I -Preparation of (I-[6-ethyl-4,4-dimethyl-I-(tetrahydro pyran-4
ylme(hyl)-1,2,3,4-tetrahvdro-
quinolin-7-ylJ-e(hyl)-carbamic acid tert-butyl ester (73):
10183] A solution of[ I -(6-cthyl-4,4-dimethyl- 1,2,3,4-tetrahydro-quinolin-7 -
yl) -ethyl] -carbamic
acid ter(-butyl ester (17, 16 mg, 0.048 mmol), iodomethyl tetrahydropyran (72,
13 mg, 0.058 mmol),
and diisopropylethylamine in 3 mL of acetonitrile was irradiated in a
microwave at 180 C for 30
minutes. The reaction mixture was concentrated under vacuum and the residue
was dissolved in ethyl
acetate, washed with brine, dried over magnesium sulfate, filtered and the
filtrate concentrated under
vacuum. The resulting material was purified by silica gel chromatography,
eluting with hexanes and
ethyl acetate to provide the desired compound as a colorless oil (73, 14 mg,
68%), MS (ES1) [M+H']4
= 431.06.
Step 2 -Preparation of 1-[6-ethyl-4,4-dimethyl-]-(tetrahydro pyran-4ylmethyl)-
1,2,3,4-tetrahydro-
quinolin-7ylJ-ethylamine (P-0058):
10184] To a solution of {1-[6-ethyl-4,4-dimethyl-l-(tetrahydro-pyran-4-
ylmethyl)-1,2,3,4-
tetrahydro-quinolin-7-yl]-ethyl ]-carbamic acid tert-butyl ester (73, 85 mg,
0.2 mmol) in 5 mL of
acetonitrile, hydrochloric acid (0.2 mL, 4.0 M in dioxane) was added and the
mixture was stirred at
room temperature for 4 hours. The reaction mixture was concentrated under
vacuum and the residue
was dissolved in ethyl acetate, washed with brine, dried over magnesium
sulfate, filtered and the
filtrate concentrated under vacuum. The resulting material was purified by
silica gel
chromatography, eluting with dichloromcthane and methanol to provide the
desired compound as a
yellow viscous liquid (P-0058, 52 mg, 74%). MS (ESI) [M+H]+= 330.95.
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
Example 19: Preparation of 7-(1-amino-ethyl)-6-ethyl-4,4-dimethyl-l-(2-methyl-
pyridin-4-
ylmethyl)-3,4-dihydro-1H-quinolin-2-one P-0061.
101851 7-(1-Amino-ethyl)-6-ethyl-4,4-dimethyl-l -(2-methyl-pyridin-4-ylmethyl)-
3,4-dihydro-lH-
quinolin-2-one P-0061 was prepared in three steps from [1-(6-ethyl-4,4-
dimethyl-1,2,3,4-tetrahydro-
quinolin-7-yl)-ethyl]-carbamic acid tert-butyl ester 17 as shown in Scheme 19.
Scheme 19
Boc 61- Boc -N _BoC N
HN HN/ HN NH2
N + N 'H Step 1 N Step 2 0 N O N
Step 3
17 49 74 75 P-0061
Step 1 - Preparation of (1-[6-ethyl-4,4-dimethyl-1-(2-methyl-pyridin-4
ylmethyl)-1,2,3,4-tetrahydro-
quinolin-7ylJ-ethyl)-carbamic acid tert-butyl ester (74):
[01861 To a mixture of 2-methylpyridine-4-carbaldehyde (49, 100 mg, 0.8 mmol)
and [1-(6-ethyl-
4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl)-ethyl]-carbamic acid tert-butyl
ester (17, 100 mg, 0.3
mmol) in 3 mL ethanol:acetic acid (95:5), sodium cyanoborohydride (50 mg, 0.8
mmol) was added.
The reaction mixture was irradiated at 120 C for 30 minutes, then
concentrated under vacuum. The
residue was dissolved in ethyl acetate, washed with saturated sodium
bicarbonate and brine, dried
over sodium sulfate, filtered and the filtrate concentrated under vacuum. The
resulting material was
purified by silica gel chromatography, eluting with dichloromethane and
methanol to provide the
desired compound as a colorless oil (74, 40 mg, 30%). MS (ESI) [M+H`]' =
439.00.
Step 2-Preparation of{1-[6-eth l-4,4-dimethyl-]-(2-metliy l pyridin-4ylmethyl)-
2-oxo-1,2,3,4-
tetrahydro-quinolin-7-ylJ-ethyl]-carbamic acid tert-butyl ester (75):
[01871 To a solution of {1-[6-ethyl-4,4-dimethyl-l-(2-methyl-p),ridin-4-
ylmethyl)-l,2,3,4-
tetrahydro-quinolin-7-yl]-ethyl }-carbamic acid tert-butyl ester (74, 20 mg,
0.044 mmol) in 10 mL of
dichloromethane, a fine powder pre-mixture of potassium permanganate (25 mg,
0.16 mmol) and
copper(II) sulfate pentahydrate (39 mg, 0.16 mmol) was added. The reaction
mixture was gently
refluxed for 2.5 hours, then filtered through a Celite pad and the Celite
residue was washed with
dichloromethane and ether. The organic portions were collected and combined,
the solvents removed
under vacuum, and the residue purified by silica gel chromatography, eluting
with dichloromethane
and methanol to provide the desired compound as a light yellow oil (75, 7 mg,
35%). MS (ESI)
[M+H'] = 452.15.
76
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
Step 3 - Preparation of 7-(1-amino-ethyl)-6-ethyl-4,4-dimethyl-I-(2-methyl-
pyridin-4-ylmethyl)-3,4-
dihydro-111-quinolin-2-one (P-0061):
101881 A solution of {1-[6-ethyl-4,4-dimethyl-l-(2-methy 1-pyridin-4-ylmethyl)-
2-oxo-1,2,3,4 -
tetrahydro-quinolin-7-yl]-ethyl}-carbamic acid tert-butyl ester (75, 7 mg,
0.016 mmol) in 5 mL of
dichloromethane, hydrogen chloride (0.1 mL, 3 mmol) was added. The mixture was
stirred at room
temperature for 1.5 hours, then concentrated under vacuum and the residue was
purified by reverse
phase HPLC, mobile phase A, 5% acetonitrile, 95% water, and mobile phase B,
95% acetonitrile, 5%
water, to provide the desired compound as a light yellow solid (P-0061, 1.6
mg, 28%). MS (ESI)
[M+H+]+ = 352.3.
Example 20: Preparation of 6-(1-amino-ethyl)-7-ethyl-4-(2-methoxy-ethyl)-4H-
benzo [1,4] oxazin-3-one P-0071.
[0189] 6-(1-Amino-ethyl)-7-ethyl -4-(2-methoxy-ethyl)-4H-benzo[1,4] oxazin-3 -
one P-0071 was
prepared in seven steps from N-(4-acetyl-2-hydroxyphenyl) acetarnide 80 as
shown in Scheme 20.
Scheme 20
-YO -YO Step 3 O N Step 4 OyNH 0
HN Step 1 HN Step 2 H2N +
HO I i O HO HO CI 0 0 80 0 81
76 77 78 79
O H
Step 5 O N" NH2
OyN I j Step 6 OyN I j Step 7 0 N
0 Br O 0 0 i
61 82 83 P-0071
Step 1 Preparation ofN-(4-ethyl-2-hydroxy phenyl)-acetamide (77):
[0190] A mixture of N-(4-acetyl-2-hydroxyphenyl) acetarnide (76, 12.00 g; 62.0
mmol) and
trifluoroacetic acid (46.8 mL, 0.62 mol) was cooled down to 0 C in an ice
water bath. To this
suspension, triethylsilane (22.4 mL, 014 mol) was added. The reaction mixture
was stirred at room
temperature for 64 hours, then poured into 750 mL of ice water. The
precipitated solid was collected
by filtration, washed with water and dissolved in 150 mL of ethyl acetate. The
organic solution was
washed with 50 mL of water and 50 mL of brine, then dried over magnesium
sulfate, filtered and the
filtrate concentrated under vacuum to provide crude compound as a beige solid,
which was used in the
next step without further purification.
Step 2 - Preparation of 2-amino-5-ethyl phenol (78):
[0191] To N-(4-ethyl-2-hydroxy-phenyl)-acetamide (77, 10 g, 34.0 mmol) in 70
mL of ethyl
acetate, 12,5 mL of water was added. The mixture was stirred vigorously and
hydrochloric acid (30
77
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
mL, 0.99 mol) was added and the mixture was heated at reflux for 17 hours. The
reaction mixture
was cooled down to 0 C in an ice water bath, and the pH of the solution was
adjusted to 7 by addition
of aqueous sodium bicarbonate. The organic phase was separated and the aqueous
phase was washed
with 2x 100 mL of ethyl acetate. The organic extracts were combined, dried
over magnesium sulfate,
filtered and the filtrate concentrated under vacuum. The resulting material
was purified by silica gel
chromatography, eluting with ethyl acetate to provide the desired compound as
a dark beige solid.
Step 3 - Preparation of 7-ethyl-4H-benzo[I, 4]oxazin-3-one (80):
[0192] To a solution of 2-amino-5-ethyl-phenol (78, 2.40 g, 17.5 mmol) in 145
mL of acetonitrile,
2-chloroacetyl chloride (79, 1.5 mL, 19.2 mmol) was added dropwise, followed
by potassium
carbonate (6.28 g, 45.5 mmol). The reaction mixture was heated at reflux for 2
hours, then cooled
down to room temperature. The mixture was filtered and the solid washed with
50 mL of acetonitrile,
The filtrates were combined and concentrated under vacuum. The residue was
dissolved in 120 mL of
dichloromethane, washed with 150 mL of water, dried over magnesium sulfate,
filtered and the
filtrated concentrated under vacuum. The resulting material was purified by
flash silica gel column
chromatography, eluting with hexane and ethyl acetate. Appropriate fractions
were combined and
concentrated under vacuum to provide the desired compound as a beige solid
(80, 1.2 g, 40%).
Step 4-Preparation of 6-acetyl-7-ethyl-41f-benzo[I,4]oxazin-3-one (81):
[0193] To aluminium chloride (229 mg, 1.7 mmol), cooled in an ice water bath,
52 L of
dimethylformamide was added dropwise. The mixture was stirred at 10 C for 1
hour, then heated to
50 C. To this mixture, 7-ethyl-4H-benzo[l,4]oxazin-3-one (80, 50 mg, 0.28
mmol) was added,
followed by 28 L of acetyl chloride. The mixture was stirred at 50-70 'C for
20 minutes, then
cooled down to room temperature and the melt was taken up in 1 mL of toluene
and quenched with 3
g of ice. The mixture was stirred for 30 minutes until all of the ice melted.
The mixture was extracted
2 x 5 mL with dichloromethane. The combined organic extracts were dried over
magnesium sulfate,
filtered and the filtrate concentrated under vacuum. The resulting material
was purified by flash silica
gel column chromatography, eluting with dichloromethane and hexane.
Appropriate fractions were
combined and concentrated under vacuum to provide the desired compound as a
pale yellow solid
(81, 6 mg, 10%)
Step 5 - Preparation of 6-acetyl-7-ethyl-4-(3-mnethoxy pro ?yl)-4H-benzo[1,
4]oxazin-3-one (82):
[0194] To 6-acetyl-7-ethyl-4H-benzo [ 1,4]oxazin-3 -one (81, 120 mg, 0.55
mmol) in 6 mL of
acetonitrile, 1-bromo-3-methoxy-propane (61, 210 mg, 1.37 mmol), potassium
fluoride on alumina
(412 mg, 2.84 mmol), and potassium iodide (5 mg, 0.03 mmol) were added. The
mixture was stirred
at 80 C for 3 hours, then cooled down to room temperature. The inorganic
material was filtered off
and the filtrate was concentrated under vacuum, The residue was purified by
flash silica gel column
78
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
chromatography, eluting with ethyl acetate and hexane. Appropriate fractions
were combined and
concentrated under vacuum to provide the desired compound as a colorless oil
(82, 120 mg, 78 ./o).
Step 6 - Preparation of 7-ethyl-6-(I-[(E)-hydroxyimino]-ethyl]-4-(3-methoxy
propyl)-4H-
benzo[I,4]oxazin-3-one (83):
101951 To 6-acetyl-7-ethyl-4-(3 -methoxy-propyl)-4H-benzo[ 1,4]oxazin-3 -one
(82, 122 mg, 0.42
mmol) in 8 mL of ethanol, hydroxylamine hydrochloride (58 mg, 0.84 mmol) and
pyridine (73 mg,
0.92 mmol) were added. The mixture was stirred at 80 C for 2 hours, then
cooled down to room
temperature and concentrated. The residue was purified by flash silica gel
column chromatography,
eluting with ethyl acetate and hexane. Appropriate fractions were combined and
concentrated under
vacuum to provide the desired compound as a white solid (83, 30 mg, 23%).
Step 7 - Preparation of 6-(1-amino-ethyl)-7-ethyl-4-(2-methoxy-ethyl)-4H-
benzo[1,4]oxazin-3-one
(P-0071):
101961 A mixture of 7-ethyl-6-{1-[(E)-hydroxyimino]-ethyl}-4-(3-methoxy-
propyl)-4H-
bcnzo[1,4]oxazin-3-one (83, 7 mg, 0.02 mmol) and Raney nickel (50%, aqueous
slurry) in 5 mL of
methanol and 2 mL of ammonium hydroxide was shaken under hydrogen (55 psi) for
4 hours. The
catalyst was filtered off and the filtrate was concentrated under vacuum. The
residue was purified by
silica gel chromatography, eluting with dichloromethane and methanol to
provide the desired
compound as a colorless oil (P-0071, 4 mg, 70%). MS (ESI) [M+H+], = 280.00.
Example 21: Preparation of 1-(6-ethyl-4,4-dimethyl- 1-phenyl-1,2,3,4-
tetrahydro-quinolin-7-yl)-
ethylamine P-0070.
101971 1-(6-Ethyl-4,4-dimethyl-l-phenyl-1,2,3,4-tetrahydro-quinolin-7-yl)-
ethylamine P-0070 was
prepared in three steps from 1-(6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-
quinolin-7-yl)-ethanone 9 as
shown in Scheme 21.
Scheme 21
H O NOH I / NH2
B(OH)2 Step 1 Step 2 Step 3 N
9 84 B5 86 P-0070
Step I - Preparation of 1-(6-ethyl-4,4-dimethyl-Iphenyl-1,2,3,4-(etrahydro-
quinolin-7-yl)-ethanone
(85):
101981 A mixture of 1-(6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl)-
ethanone (9, 62 mg,
0.27 rnmol), phenylboronic acid (84, 49 mg, 0.4 mmol), cupric acetate (80 mg,
0.4 mmol),
79
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
triethylaniine (0.06 mL, 0.4 mmol), and molecular sieves (powder) in 10 mL of
dichloromethane was
stirred at 35 C for 22 hours. The residue was dissolved in ethyl acetate,
washed with water and
brine, and dried over magnesium sulfate. The mixture was filtered, the
filtrated concentrated under
vacuum and the residue was purified by silica gel chromatography, eluting with
hcxanes and ethyl
acetate to provide the desired compound as a pale yellow oil (85, 20 mg, 20%).
MS (ESI) [M+H+]+=
308.52
Step 2 - Preparation of ]-(6-ethyl-4, 4-dimethyl-1 phenyl-1,2, 3,4-tetrahydro-
quinolin-7-yl)-ethanone
oxinie (86):
[0199] A solution of 1-(6-ethyl-4,4-dimethyl-I-phenyl-1,2,3,4-tetrahydro-
quinolin-7-yl)-ethanone
(85, 51 mg, 0.13 mmol), hydroxylamine hydrochloride (30 mg, 0.4 mmol), and
pyridine (0.1 mL, 1
mmol) in 10 mL of ethanol was stirred at 90 C for 5 hours. The reaction
mixture was concentrated
and the residue was dissolved in ethyl acetate, washed with water and brine,
and dried over
magnesium sulfate. The mixture was filtered, the filtrated concentrated and
dried under vacuum. The
resulting material was used in the next step without further purification.
Step 3 - Preparation of l-(6-ethyl-4,4-dimethyl-l-phenyl-1,2,3,4-tetrahydro-
quinolin-7 yl)-ethylamine
(P-0070):
[0200] A mixture of ]-(6-ethyl-4,4-dimethyl-l-phenyl-1,2,3,4-tetrahydro-
quinolin-7-yl)-ethanone
oxime (86, 40 mg, 0.1 mmol) and Raney nickel (50%, aqueous slurry) in 5 mL of
methanol and 2 mL
of ammonium hydroxide was stirred under an hydrogen balloon for 3 hours. The
mixture was filtere
and the filtrate was concentrated under vacuum. The resulting material was
purified by silica gel
chromatography, eluting with dichloromethane and methanol to provide the
desired compound as a
pale yellow oil (P-0070, 25 mg, 80%). MS (ESI) [M+H+]+ = 309.90.
[0201] 1-[6-Ethyl-l-(3-methoxy-phenyl)-4,4-dimethyl-1,2,3,4-tetrahydro-
quinolin-7-yl]-ethylamine
P-0079,
Y NH,
c
was prepared similarly to the protocol of Scheme 21, replacing phenylboronic
acid 84 with 3-
methoxy-phenylboronic acid in step 1, where optimal reaction conditions may
have varied, for
example, any of time and temperature of the reaction, or chromatography
conditions for purification
of the desired compound, MS (ES1) [M+H+]+- 339.25.
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
Example 22: Preparation of C-[6-ethyl-l-(3-methoxy-propyl)-4,4-dimethyl-
1,2,3,4-tetrahydro-
quinolin-7-yl]-methylamine P-0008.
[0202] C-[6-Ethyl-l-(3-methoxy-propyl)-4,4-dimethyl-1,2,3,4-tetrahydro-
quinolin-7-yl]-
methylamine P-0008 was prepared in three steps from 7-bromo-6-ethyl-4,4-
dimethyl-1,2,3,4-
tetrahydroquinoline 11 (see Scheme 2) as shown in Scheme 22.
Scheme 22
Br
N Br
C X + Step 1 ~jBr Step 2 ~(CN Step 3 NHZ
O
11 61
87 88 P-0008
Step I -Preparation of7-bromo-6-ethyl-l-(3-methoxy propyl)-4,4-dimethyl-
1,2,3,4-tetrahydro-
quinoline (87):
[0203] To 7-bromo-6-ethyl-4,4-dimethyl-1,2,3, 4-tetrahydro-quinoline (11,
500.00 mg, 1.86 mmol)
in 5.00 mL of acetonitrile, potassium carbonate (260 mg, 1.9 mmol) and 1-bromo-
3-methoxy-propane
(61, 0.86 g, 5.6 mmol) were added and the reaction stirred for 2 days at 100
T. The reaction was
diluted with water and extracted with ethyl acetate. The organic layer was
washed with brine, dried,
filtered and the filtrate concentrated under vacuum. The resulting material
was purified by silica gel
column chromatography eluting with I% ethyl acetate in hexane. Appropriate
fractions were
combined and solvents removed under vacuum to provide the desired compound
(87, 513 mg) as a
light orange oil. MS (EST) [M+H']'= 342.
Step 2 - Preparation of 6-ethyl-l -(3-meth oxy-propyl)-4, 4-dimethyl-1, 2,3, 4-
tetrahydro-quinoline-7-
carbonitrile (88):
[0204] To 7-bromo-6-ethyl-l-(3-methoxy-propyl )-4,4-dimethyl-1,2,3,4-
tetrahydro-quinoline (87,
25.00 mg, 0.073 mmol) in 1.2 mL of N-methylpyrrolidone, zinc cyanide (9.5 mg,
0.081 mmol) and a
catalytic amount of 4% tetrakis(triphenylphosphine)palladium(0) were added.
The reaction was
microwaved on 60 watts, 175 C for 3 minutes. The organic layer was washed
with brine, dried,
filtered and the filtrate concentrated under vacuum. The resulting material
was purified by silica gel
column chromatography eluting with 5% ethyl acetate in hexane. Appropriate
fractions were
combined and solvents removed under vacuum to provide the desired compound
(88, 16 mg). MS
(EST) [M+H+]*= 287,
81
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
Step 3-Preparation ofC-[6-ethyl-l-(3-rnethoxy-propyl)-4,4-dirnethyl- 1,2,3,4-
tetra hydro-quinolin-7-
yl]-inetlrylarnine (P-0008):
[0205] 6-Ethyl-l-(3-methoxy-propyl)-4,4-di methyl-1,2,3,4-tetrahydro-quinoline-
7-carbonitrile (88,
21.0 mg, 0.073 rnmol) in 5.00 mL of methanol was degassed with nitrogen.
Hydrochloric acid
(0.0045 mL, 0.15 mmol) and a catalytic amount of palladium on carbon were
added and the reaction
was stirred in a hydrogenation apparatus at 50 psi for 3 hours. The reaction
was filtered through celite
and the filter rinsed with methanol. The filtrate was combined and
concentrated under vacuum to
solid, which was purified by silica gel column chromatography eluting with 5%
methanol in
dichloromethane. Appropriate fractions were combined and solvents removed
under vacuum to
provide the desired compound (P-0008, 8 mg). MS (ESI) [M+H]+- 291.
Example 23: Preparation of 7-(1-amino-ethyl)-6-methoxy-l-(3-methoxy-propyl)-
4,4-dimethyl-
3,4-dihydro-1H-quinolin-2-one P-0013.
[0206] 7-(1-Amino-ethyl)-6-methoxy-1-(3-methoxy-propyl)-4,4-dimethyl-3,4-
dihydro-lH-
quinolin-2-one P-0013 was prepared in five steps from 3-methyl-but-2-enoic
acid (4-methoxy-
phenyl)-amide 93 as shown in Scheme 23.
Scheme 23
H O H O Step3
O NH Step 1 O N Step 2 +
O~ Oi Br/\
89 / 61
90 91
011
O O O
0 N' OH NH2
O Step 4 O N I Step 5 O
92 93 P-0013
Step I -Preparation of 6-rnetho.xv-4,4-dimethyl-3,4-dihydro-]H-quinolin-2-one
(90):
[0207] Aluminum trichloride (10.4 g, 77.9 mmol) was suspended in 100 mL of 1,2-
dichloroethane
and 3-methyl-but-2-enoic acid (4-methoxy-phenyl)-amide (89, 5,16 g, 25.1 mmol)
was added
dropwise over a period of -5 minutes. The reaction mixture was stirred under
an inert atmosphere
for 4 hours and partitioned between ethyl acetate and water. The organic layer
was collected and the
aqueous layer was washed 3x with ethyl acetate. The organic fractions were
pooled and washed with
water, sodium bicarbonate solution and brine, then dried over magnesium
sulfate, filtered and the
82
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
filtrate concentrated under vacuum. The resulting yellow oil was purified by
silica gel column
chromatography, eluting with ethyl acetate and hexane. Appropriate fractions
were collected and the
solvents removed under vacuum to provide the desired compound as a light pink
solid (90, 4.05 g).
'H NMR was consistent with the compound structure.
Step 2 Preparation of 7-acetyl-6-methoxy--4,4-dimethvi-3,4-dihydro-IH-quinolin-
2-one (91):
[0208] To a suspension of aluminum trichloride (2.9 g, 22.0 mmol) in 8 mL of
nitromethane,
chilled to 0 C, a solution of acetyl chloride (0.48 mL, 6.8 mmol) and 6-
methoxy-4,4-dimethyl-3,4-
dihydro-1H-quinolin-2-one (90, 1,00 g, 4.87 mmol) in 10 mL of dichloromethane
was added
dropwise. Once addition was complete, the reaction was removed from the ice
bath and stirred at
room temperature for 4-5 hours. The reaction mixture was re-cooled to 0 C and
water was added
dropwise, resulting in heat and foaming, until all solids were dissolved. The
mixture was extracted
with 100 mL of dichloromethane, and the organic layer was washed with water,
ammonium chloride
solution and brine, then dried over magnesium sulfate, filtered and the
filtrate concentrated under
vacuum. The resulting oil was purified by silica gel column chromatography,
eluting with gradient of
0-5% methanol in dichloromethane over 40 minutes, Appropriate fractions were
combined and the
solvents removed under vacuum to provide the desired compound. 'H NMR was
consistent with the
compound structure.
Step 3 - Preparation of 7-acetyl-6-methoxy-l-(3-methoxy propyl)-4,4-dimethyl-
3,4-dihydro-IH-
quinolin-2-one (92):
[0209] To a solution of 7-acetyl-6-methoxy-4,4-dimethyl-3,4-dihydro-1H-
quinolin-2-one (91,
261.00 mg, 1.055 mmol) in 3.0 mL of N,N-dimethylformamide, 1-bromo-3-methoxy-
propane (61,
190 mg, 13 mmol) was added, followed by sodium hydride (33 mg, 1.4 mmol). The
reaction mixture
was heated in the microwave at 107 C for 20 minutes. The reaction mixture was
quenched with
water and extracted 2x with dichloromethane. The combined organic layer was
washed 2x with
water, then with brine, dried over sodium sulfate, filtered and the filtrate
concentrated under vacuum.
The resulting material was purified by silica gel column chromatography
eluting with 5-40% ethyl
acetate in hexane. Appropriate fractions were combined and the solvents
removed under vacuum to
provide the desired compound (92, 156 mg). 'H NMR was consistent with the
compound structure.
Step 4 - Preparation of 7-(1-[(E)-hydrotyimino]-ethyl}-6-methoxy-l -(3-methoxy
propyl)-4, 4-
dimethyl-3,4-dihydro-1H-quinolin-2-one (93):
[0210] A solution of 7-acetyl-6-methoxy-l-(3-methoxy-propyl)-4,4-dimethyl-3,4-
dihydro-lH-
quinolin-2-one (92, 38 mg, 0.12 mmol), hydroxylamine hydrochloride (16.9 mg,
0.243 mmol), and
pyridine (0.046 mL, 0.57 mmol) in 5 mL of ethanol was stirred at 80 C for 2
hours. The reaction
mixture was concentrated under vacuum and the residue was dissolved in ethyl
acetate, washed with
water and brine, dried over magnesium sulfate, filtered and the filtrate
concentrated under vacuum,
83
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
The resulting material was used for the next step without further purification
(93, 45 mg). MS (ESI)
[M+H']f = 335.2.
Step 5 - Preparation of 7-(1-atnino-ethyl)-6-methoxy-l -(3-methoxy propyl)-4,4-
dirnethyl-3,4-dihydro-
IH-quinolin-2-one (P-0013):
[02111 To a solution of7-{ 1-[(E)-hydroxyimino]-ethyl}-6-methoxy-l-(3-methoxy-
propyl)-4,4-
dimethyl-3,4-dihydro-1H-quinolin-2-one (93, 63.00 mg, 0.188 mmol) in 5.0 mL of
toluene, borane-
dimethyl sulfide complex (0.026 mL, 0.30 mmol) was added slowly at 0 T. The
reaction mixture
was stirred at 0 C for 15 minutes and then stirred at 100 C for 1.5 hours.
The reaction mixture was
cooled down and poured into 150 mL of 10% aqueous sodium carbonate solution.
The mixture was
stirred at room temperature for 30 minutes. The organic layer was removed and
the aqueous layer
extracted with 2 x 60 mL of ethyl acetate, The organic layers were combined
and dried over
magnesium sulfate, filtered and the filtrate concentrated under vacuum to
provide the desired
compound (P-0013, 12 mg).
Example 24: Preparation of 1-[1-(3-methoxy-propyl)-4,4-dimethyl-6-phenyl-
1,2,3,4-tetrahydro-
quinoiiin-7-yl]-ethylamine P-0021.
[0212] 1-[1-(3-Methoxy-propyl)-4,4-dirnethyl-6-phenyl-1,2,3,4-tetrahydro-
quinolin-7-yl]-
ethylamine P-0021 was prepared in four steps from trifluoro-methanesulfonic
acid 7-acetyl-4,4-
dimethyl-1-(2,2,2-trifluoro-acetyl)-1,2,3,4-tetrahydro-quinolin-6-yl ester 35
(see Scheme 8) as shown
in Scheme 24.
Scheme 24
F3CY0 0
p B(OH)2 H 0
Step 1 N Step 2 0
N
OTf +
35 84 94 I i Bra /~0~
61
I I
0 0
N'OH NH2
Step 3 N E Step 4 N
96 P-0021
Step I - Preparation of 1-(4,4-dirnethyl-6phenyl-1,2,3,4-tetrahydro-quinolin-7-
y1)-ethanone (94):
[0213] Into a round bottom flask, trifluoro-methanesulfonic acid 7-acetyl-4,4-
dimethyl-l-(2,2,2-
trifluoro-acetyl)-1,2.3,4-tetrahydro : ua 6 .1 != (34 iu ( Y6 mmol), was
combined with
84
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
potassium carbonate (44 mg, 0.32 mmol), phenylboronic acid (84, 20 mg, 0.0002
mol),
tetrakis(triphenylphosphine)palladium(0) (5 mg, 0.004 mmol), and 1.5 mL of
water. The reaction was
microwaved on 60 watts, 100 C for 10 minutes. Solvents were removed under
vacuum and the
residue extracted with ethyl acetate and saturated bicarbonate, then washed
with brine, The solvents
were removed from the organic layer and the residue purified by silica gel
column chromatography
eluting with hexanes:ethyl acetate 80:20. The appropriate fractions were
combined and the solvents
removed under vacuum to provide the desired compound (98, 19.5 mg). MS (ESI)
[M+HT]' = 280.1.
Steps 2-4
[0214] 1-(4,4-Dimethyl-6-phenyl-1,2,3,4-tetrahydro-quinolin-7-yl)-ethanone 94
was reacted
similarly to steps 5-7 of Scheme 14 above to provide the desired compound 1-[1-
(3-Methoxy-propyl)-
4,4-dmethyl-6-phenyl-1,2,3,4-tetrahydro-quinolin-7-yl]-ethylamine P-0021. MS
(ESI) [M+H*]+=
353.1.
Example 25: Preparation of 1-(1-benzyl-4,4,6-trimethyl- 1,2,3,4-tetrahydro-
quinolin-7-yl)-
ethylamine P-0076.
[0215] 1-(1-Benzyl-4,4,6-trimethyl-1,2,3,4-tetrahydro-quinolin-7-yl)-
ethylamine P-0076 was
prepared in nine steps from 3-methyl-but-2-enoic acid p-tolylamide 101 as
shown in Scheme 25.
Scheme 25
H H F3CYO
O N Step 1 O N Step 2 N Step 3 N
98 99 100
97
F3C O H Step 6
Step 4 N Br Step 5 N 1Br N Br Step 7
102
101 Br 23 103
O OH NH
N f\ Step 8 N Step 9 N 2
104 105 P-0076
Step I - Preparation of 4,4, 6-Trimethyl-3, 4-dihydro-1 H quinolin-2-one (98).
[0216] Aluminum trichloride (11 g, 84.0 mmol) was suspended in 30 mL of
nitromcthane and 3-
methyl-but-2-enoie acid p-tolylamide (97, 5.3 g, 28.0 mmol, prepared similarly
to step 1 of Scheme 1,
substituting 4-ethylaniline 1 with p-tolylamine) was added portionwise over a
period of -5 minutes.
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
The reaction mixture was stirred under an inert atmosphere at 80 C for 12
hours, then partitioned
between ethyl acetate and water. The organic layer was collected and the
aqueous layer was washed
additional 3x with ethyl acetate. The organic fractions were pooled and washed
with water, aqueous
sodium bicarbonate solution and brine, dried over magnesium sulfate, filtered
and the filtrate
concentrated under vacuum. The resulting yellow oil was purified by silica gel
column
chromatography eluting with ethyl acetate and hexane. The appropriate
fractions were combined and
solvents removed under vacuum to provide the desired compound as an off-white
solid (98, 705 mg).
'H NMR was consistent with the compound structure. Additional material was
prepared similarly for
the next step.
Step 2 - Preparation of 4,4,6-Trmmethvl-1,2,3,4-tetrahydro-quinoline (99):
102171 To a solution of 4,4,6-trimethyl-3,4-dihydro- I H-quinolin-2-one (98,
3.86 g, 20.4 mmol) in
150 mL of toluene, borane-dimethyl sulfide complex (3.6 mL, 41.0 mmol) was
added slowly at 0 T.
The reaction mixture was stirred at 0 C for 15 minutes and then stirred at
100 C for 2 hours. The
reaction mixture was cooled down and poured into 150 mL of aqueous 10% sodium
carbonate
solution. The mixture was stirred at room temperature for 30 minutes and the
organic layer was
collected. The aquous layer was extracted with 2 x 60 mL of ethyl acetate and
the organic layers were
combined and dried over magnesium sulfate, filtered and the filtrate
concentrated under vacuum. The
residue was purified by silica gel chromatography to provide the desired
compound as a light brown
liquid (99, 2.3 g). 'H NMR was consistent with the compound structure.
Step 3 - Preparation of2,2,2-Trifluoro-I-(4,4,6-trimethyl-3,4-dihydro-
2Hquinolin-1 yl)-ethanone
(100):
[0218] In a reaction vessel, 4,4,6-trimethyl-1,2,3,4-tetrahydro-quinoline (99,
2.25 g, 12.8 mmol)
was dissolved in 25.0 mL of dichloromethane and triethylamine (2.70 mL, 19.0
mmol) was added and
the reaction stirred for a couple of minutes. Trifluoroacetic anhydride (2.20
mL, 15.0 mmol) in 5 mL
of dichloromethane was then added and the reacion stirred for 3-4 hours. The
reaction was diluted
with additional dichloromethane, washed with water, then brine, and the
organic solvents removed
under vacuum. The resulting residue was purified by silica gel column
chromatography eluting with
10% ethyl acetate in hexane. Appropriate fractions were combined and the
solvents removed to
provide the desired compound (100, 2.6 g).
Step 4-Preparation oft-(7-bromo-4,4,6-trimethyl-3,4-dihydro-2H-quinolin-1-yl)-
2,2,2-trifluoro-
ethanone (101):
[0219] Into a reaction vessel, 2,2,2-trifluoro-l-(4,4,6-trimethyl- 3,4-dihydro-
2H-quinolin-l-yl)-
ethanone (100, 0.500 g, 1.84 mmol) and aluminum trichloride (0.24 g, 1.8 mmol)
were combined in
10.0 mL of nitromethane, Bromine (0.125 mL, 2.43 mmol) was added dropwise at 0
C and the
reaction went overnight. The reaction was diluted with water, extracted with
ether, and the organic
86
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
layer was washed with brine, then concentrated under vacuum. The residue was
purified by silica gel
column chromatography, eluting with 0-5% ethyl acetate in hexane. Appropriate
fractions were
combined and the solvents removed under vacuum to provide the desired compound
(101, 410.0 mg).
Step 5 - Preparation of 7-bromo-4,4,6-trimethyl-1,2,3,4-tetrahydro-quinoline
(102):
[0220] 1-(7-bromo-4,4,6-trimethyl-3,4-dihydro-2H-quinolin-1-yl)-2,2,2-
trifluoro-ethanone 101 was
reacted similarly to step 6 of Scheme 1 to provide the desired compound.
Step 6 - Preparation of 1-benzyl-7-bromo-4, 4, 6-trimethyl-1, 2,3, 4-
tetrahydro-quinoline (103):
[0221] A solution of 7-bromo-4,4,6-trimethyl-1,2,3,4-tetrahydro-quinoline
(102, 249.1 mg, 0.98
mmol), benzyl bromide (23) and N,N-diisoprupylethylamine (0.34 mL, 2.0 mmol)
in 2 mL of N-
methylpyrrolidonc was heated at 80 C overnight. The reaction mixture was
partitioned between
water and ethyl acetate and the organic layer was washed with brine, dried
over magnesium sulfate,
filtered and the filtrate concentrated under vacuum. The residue was purified
by silica gel column
chromataography, eluting with 0-25% ethyl acetate in hexanes. Appropriate
fractions were combined
and the solvents removed under vacuum to provide the desired compound (103,
177 mg).
Step 7- Preparation of 1-(1-benzyl-4,4,6-trimethyl-1,2,3,4-tetrahydro-quinolin-
7 y1)-ethanone (104).
[0222] 1-Benzyl-7-bromo-4,4,6-trimethyl-1, 2,3,4-tetrahydro-quinoline (103,
177.0 mg, 0.514
mmol), was mixed with 1,3-bis(diphenylphosphino)propane (13 mg, 0.032 mmol),
potassium
carbonate (73.2 mg, 0.530 mmol), tributyl-(l-ethoxy-vinyl)-stannane (0.61 mL,
1.8 mmol), and
palladium acetate (3.0 mg, 0.013 mmol) in 1.0 mL of water and 3.0 mL of N,N-
dimethylfonnamide
under nitrogen and heated at 80 C overnight. The reaction was cooled to room
temperature and
diluted with ethyl acetate, and the organic layer then washed with water,
brine and concentrated under
vacuum. The residue was purified by silica gel chromatography to provide the
desired compound
(104, 35 mg). 'H NMR was consistent with the compound structure.
Step 8 - Preparation of 1-(I-benzyl-4, 4, 6-trimethyl-1, 2, 3,4-tetrahydro-
quinolin-7-yl)-ethanone oxiine
(105):
[0223] 1-(1-Benzyl-4,4,6-trimethyl-1,2,3,4 -tetrahydro-quinolin-7-yl)-ethanone
(104, 35.00 mg,
0.114 mmol), hydroxylamine hydrochloride (16 mg, 0.23 mmol), and pyridine
(0.018 mL, 0.23 mmol)
in 15 mL of ethanol was stirred at 80 C for 3 hours. The reaction mixture was
concentrated under
vacuum and the residue dissolved in ethyl acetate, washed with water and
brine, dried over
magnesium sulfate and filtered, The filtrate was concentrated under vacuum and
the residue was
purified by silica gel chromatography to provide the desired compound as a
light yellow glue (105, 19
mg). 'H NMR was consistent with the compound structure.
87
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
Step 9 Preparation ofl-(I-benzyl-4,4,6-trimethyl-1,2,3,4-tetrahydro-quinolin-7-
yl)-ethvlamine
(P-0076):
102241 1 -(1 -Benzyl-4,4,6-trimethyl-1,2,3,4 -tetrahydro-quinolin-7-yl)-
ethanone oxime (105, 19.0
mg, 0.059 mmol) was dissolved in 10 mL of ethanol in a parr vessel. Raney
nickel (0,05 g, 0.8 mmol)
was added and the vessel evacuated and charged with hydrogen (10 g, 5 mol, 55
PSI). The reaction
mixture was shaken for 120 minutes. The raney nickel was removed via
filtration and the filtrate
concentrated under vacuum to provide the desired compound as a yellow, waxy
solid (P-0076, 3.0
mg). 'H NMR was consistent with the compound structure. MS (ESI) [M+H-]-'=
308.2.
Example 26: Preparation of 7-(1-amino-ethyl)-1-benzyl-4,4-dimethyl-6-p-tolyl-
3,4-dihydro-lH-
quinolin-2-one P-0078.
[0225] 7-(l-Amino-ethyl)-l-benzyl-4,4-dimethyl-6-p-tolyl-3,4-dihydro-lH-
quinolin-2-one P-0078
was prepared in four steps from 7-acetyl-6-bromo-4,4-dimethyl-3,4-dihydro-1 H-
quinolin-2-one 60 as
shown in Scheme 26.
Scheme 26
H 0 Step 1 \ 0 Step 2 \ 0
O
O O N
I I
Br Br / I I \
60 Br 23 106 ~ 108
107
HO' B OH
OH
N NH2
Step 3 0 N Step 4 O N
I ( i
109
P-0078
Step I -Preparation of7-acetyl-I-benzyl-6-bronco-4,4-dimethyl-3,4-dihydro-IH-
quinolin-2-one
(706):
[02261 Into a quartz vial 7-acetyl-6-bromo-4,4-dimethyl-3,4-dihydro-1 H-
quinolin-2-one (60, 25
mg, 0.083 mmol, see Scheme 14) was dissolved in S mL of acetonitrile (5 mL,
0.1 mol) and benzyl
bromide (23, 0.015 mL, 0.12 mmol), potassium carbonate (40.0 mg, 0.29 mmol)
and potassium iodide
(4 mg, 0.02 mmol) were added. The reaction was stirred at 80 C over the
weekend, then poured into
water and extracted 2x with ethyl acetate. All organic layers were combined,
washed with brine,
dried over magnesium sulfate, filtered and the filtrate concentrated under
vacuum. The residue was
purified by silica gel column chromatography eluting with 10-35% ethyl acetate
in hexane,
88
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
Appropriate fractions were combined and the solvents removed under vacuum to
provide the desired
compound as a light yellow solid (106, 33 mg, purity 98%, yield 98%).
Step 2 - Preparation of 7-acetyl-I-benzyl-4,4-dimethyl-6 p-tolyl-3,4-dihydro-
1H-quinolin-2-one
(108):
102271 Into a quartz vial, 7-acetyl-l-benzyl-6-bromo-4,4-dimethyl-3,4-dihydro-
1H-quinolin-2-one
(106, 28.0 mg, 0.0725 mmol) was mixed with 2.00 mL. of tetrahydrofuran and
0.08 mL of dimethyl
sulfoxide, then p-tolyl boronic acid (107, 15 mg, 0.11 mmol), palladium
acetate (3 Ong, 0.01 mmol),
diphenyl(t-Bu)2 (9 mg, 0.03 mmol) and potassium phosphate (31 mg, 0.14 mmol)
were added. The
reaction was microwaved at 150 watts, 70 C for 80 minutes. The reaction was
diluted with ethyl
acetate, extracted with sodium chloride solution, the organic layer dried over
magnesium sulfate,
filtered and the filtrate concentrated under vacuum. The residue was purified
by silica gel
chromatography to provide the desired compound (108, 10 mg, purity 50%, yield
20%). MS (ESI)
[M+W]' = 398.8.
Step 3 - Preparation of ' 1-benzyl-7-(1-[(E)-h_ydroxyimino]-ethyl)-4, 4-
dimethyl-6-p-tolyl-3, 4-dihydro-
]H-quinolin-2-one (109).'
[02281 A solution of 7-acetyl-l -benzyl-4,4-dimethyl-6-p- tolyl-3,4-dihydro-1H-
quinolin-2-one
(108, 24 mg, 0.024 mmol), hydroxylaminc hydrochloride (50 mg, 0.7 mmol), and
pyridine (0.5 mL,
6.0 mmol) in 5 mL of ethanol was stirred at 80 C overnight. The reaction
mixture was concentrated
under vacuum. The residue was dissolved in ethyl acetate, washed with water
and brine, dried over
magnesium sulfate, filtered and the filtrate concentrated under vacuum to
provide the desired
compound as a white solid (109, 24 mg, purity 40%), which was used in the next
step without further
purification. MS(ES1) [M+H]+= 413.89, 413.13.
Step 4 - Preparation of'7-(1-amino-ethy1)-1-benzyl-4,4-dimethyl-6p-tolyl-3,4-
dihydro-IH-quinolin-2-
one (P-0078):
[02291 A mixture of 1-benzyl-7-{1-[(E)-hydroxyimino]-ethyl }-4,4-dimethyl-6-p-
tolyl-3,4-dihydro-
IH-quinolin-2-one (109, 55 mg, 0.053 mmol) and Raney nickel (50%, aqueous
slurry) in 15 mL
methanol and 2.0 mL of ammonium hydroxide was stirred under an atmosphere of
hydrogen for 2
hours. The reaction mixture was filtered and the filtrate was concentrated
under vacuum. The residue
was purified by HPLC to provide the desired compound as a white solid (P-0078,
7.5 mg, purity
>95%, yield 34%). MS(ESI) [M-NH2'] I= 383.05.
89
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
Example 27: Preparation of 7-(1-amino-ethyl)-6-ethyl-4,4-dimethyl-3,4-dihydro-
2H-quinoline-
1-carboxylic acid tert-butyl ester P-0069.
10230] 7-(1 -Amino-ethyl)-6-ethyl-4,4-dimethyl-3,4-dihydro-2H-quinoline-l -
carboxylic acid tert-
butyl ester P-0069 was prepared in two steps from 7-acetyl-6-ethyl -4,4-
dimethyl-3,4-dihydro-2H-
quinoline-l-carboxylic acid tert-butyl ester 14 as shown in Scheme 27.
Scheme 27
O 0 O 0 N OH O 0 NH2
z
Step 1 Step 2
14
110 P-0069
Step 1 Preparation of 6-ethyl-7-(1-hydroxyiniino-ethyl)-4,4-dinmetlryl-3,4-
dihydro-2H-quinoline-l-
carboxylic acid tert-butyl ester (110):
102311 A solution of 7-acetyl-6-ethyl-4,4-dimethyl-3,4-dihydro-2H-quinoline-l-
carboxylic acid
tert-butyl ester (14, 500.00 mg, 1.509 mmol, sec Scheme 2), hydroxylamine
hydrochloride (300 mg,
4.0 mmol), and pyridine (0.6 mL, 8.0 mmol) in 35 mL of ethanol was stirred at
80 C for 3 hours. The
reaction mixture was concentrated under vacuum and the residue was dissolved
in ethyl acetate,
washed with water and brine, dried over magnesium sulfate, filtered and the
filtrate removed under
vacuum, The resulting material was purified by silica gel chromatography to
provide the desired
compound as a white solid (110, 484 mg, purity >95%, yield 88%). MS (ESI)
[M+H]* = 347.05.
Step 2 -Preparation of 7-(1-annino-ethyl)-6-ethyl-4,4-dimethyl-3,4-dihydro-2H-
quinoline-l-
carboxylic acid tert-butyl ester (P-0069):
10232] A mixture of 6-ethyl-7-(1-hydroxyimino-ethyl)-4,4-dimethyl-3,4-dihydro-
2H-quinoline- l-
carboxylic acid tert-butyl ester (110, 390.0 mg, 1.126 mmol) and Raney nickel
(50%, aqueous slurry)
in 50 mL of methanol and 15 mL of ammonium hydroxide was shaken under hydrogen
(55 psi) for 4
hours. The reaction was filtered and the filtrate was concentrated under
vacuum. The resulting
material was purified by silica gel chromatography eluting with
dichloromethane and methanol to
provide the desired compound as a colorless oil (P-0069, 317 mg, yield 80.5%).
MS (ESI) [M+Ht]+ _
334.05.
Example 28: Synthesis of Compounds where Li is -S(O)z- or -S(O)2NH-.
[0233] Compounds of Formula IV having a sulfone linker (or aminosulfone) at
the 1-position can
be prepared as exemplified from [1-(6-cthyl-4,4-dimethyl-1,2,3,4-tetrahydro-
quinolin-7-yl)-ethyl]-
carbamic acid tert-butyl ester 17. Compounds are pf ,;, in two steps as shown
in Scheme A.
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
Scheme A
R O Boc R9
HNB c ~S=O HN' ~S=O NH2
H + R~ S' Step I Step 2 N 11 17 III
IV
Step I - Preparation of Compounds of Formula III;
[0234] A mixture of sulfonyl chloride of Formula II (R is e.g. NHR' or R',
where R' is as defined
in paragraph [0003]), [1-(6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-
yl)-ethyl]-carbamic acid
tert-butyl ester (17), and an appropriate base, such as triethyl amine in an
appropriate solvent, such as
dichloromethane is stirred at 20-80 C for 2-24 hours. The reaction is
quenched with an aqueous
work-up and purified by silica gel chromatography or crystallization to
provide Compounds of
Formula III.
Step 2 - Preparation of Compounds of Formula IV,
[0235] A compound of Formula III is stirred in a suitable solvent, such as
dioxane, tetrahydrofuran
or dichloromethane with a suitable acid, such as hydrochloric acid,
trifluoroacetic acid, or tosic acid at
0-80 C for 0,25-24 hours. The reaction mixture is quenched with aqueous base
and purified by silica
gel chromatography or crystallization to provide a compound of Formula IV.
Example 29: Synthesis of Compounds where L, is -NH-, -NHS(O)Z- or -NHC(O)-.
[0236] Compounds of Formula VIII having either -NHS(O)2- or -NHC(O)- linker at
the 1-position
can be prepared, for example, in five steps from 1-(6-ethyl-4,4-dimethyl-
1,2,3,4-tetrahydro-quinolin-
7-yl)-ethanone 9 as shown in Scheme B.
Scheme B
H O NO O NHZ O Step 3
Step 1 Step 2 N --
9 111 112 + V
R~ OH R`a
N~ NH N` NH NH2
Step 4 N Step 5 N 11
VI
VII Vlll
91
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
Step] Preparation ofl-(6-ethyl-4,4-dimethyl-l-nitroso-1,2,3,4-tetrahvdro-
quinolin-7-yl)-ethanone
(111):
[0237] To a mixture of 1-(6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-
yl)-ethanone (9) and
a solution hydrochloric acid, cooled with an ice water bath, a solution of
sodium nitrite is slowly
added. The reaction mixture is then allowed to warm to room temperature and
warmed for a few
hours as needed. The reaction mixture is extracted with an appropriate solvent
(such as ethyl acetate
or toluene). The organic layer is collected, washed with water and dried over
sodium sulfate. After
removal of drying agent and solvent, the residue is dried under vacuum to
provide the desired
compound 111 that can be used without purification in the next step.
Step 2- Preparation of t-(1-amino-6-ethyl-4, 4-dimethyl-1,2, 3, 4-tetrahydro-
quinolin-7 yl)-ethanone
(112):
[0238] To a solution of 1-(6-ethyl-4,4-dimethyl-l-nitroso-1,2,3,4-tetrahydro-
quinolin-7-yl)-
ethanone (111) in an appropriate solvent (such as tetrahydrofuran), a
suspension of lithium aluminum
hydride in an appropriate solvent (such as tetrahydrofuran) is added. The
temperature can be
maintained by use of an ice water bath. The reaction mixture is poured into
water, extracted with an
appropriate solvent (such as ethyl acetate or dichloromethane), and the
organic layer is collected,
washed with water and dried over sodium sulfate. After removal of drying agent
and solvent, the
residue is dried under vacuum to provide the desired compound 112 that can be
used without
purification in the next step.
Step 3 - Preparation of Compounds ofFormula VI:
[0239] To a mixture of 1-(1-amino-6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-
quinolin-7-yl)-ethanone
(112) and a base (such as triethylamine or potassium carbonate) in an
appropriate solvent (such as
dichloromethane or acetonitrile), organo-halide, acid halide, or sulfonyl
halide of Formula V (R' is
e.g. R'-, R'-C(O)- or R'-S(O)z-, where R' is as defined in paragraph [0003])
is slowly added. The
reaction mixture is stirred at room temperature or heated in an oil bath for 2
to 24 hours. The reaction
mixture is poured into water and extracted with an appropriate solvent (such
as dichloromethane or
ethyl acetate), The organic layer is collected, washed with saturated sodium
bicarbonate solution and
brine, and dried over magnesium sulfate. After removal of drying agent and
solvent, the residue can
be purified by silica gel chromatography to provide the desired compound of
Formula VI.
Step 4 - Preparation of Compounds of Formula VII:
[0240] A mixture of a compound of Formula VI, hydroxylamine hydrochloride, and
pyridine in
ethanol is stirred at 80-100 C for 2-24 hours, The reaction mixture is
concentrated and the residue
dissolved in an appropriate solvent such ethyl acetate, washed with water and
brine, and dried over
magnesium sulfate, After removal of drying agent and solvent, the residue can
be purified by silica
gel chromatography to provide the desired compound of Formula VII.
92
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
Step 5 - Preparation of Compounds of Formula VIII:
102411 A mixture of a compound of Formula VII, Raney nickel, and ammonium
hydroxide in
methanol is stirred under an atmosphere of hydrogen gas for 0.5-20 hours. The
catalyst is removed by
filtration and the filtrate concentrated under reduced pressure. The residue
can be purified by silica
gel chromatography to provide the desired compound of Formula VIII.
[0242] Similar compounds with substituents other than ethyl at the 6 position,
e.g. optionally
substituted aryl, can be prepared similarly, for example, by replacing 1-(6-
ethyl-4,4-dimethyl-1,2,3,4-
tetrahydro-quinolin-7-yl)-ethanone 9 in step 1 with a suitable compound,
including, but not limited to,
1-[6-(4-fluoro-phenyl)-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl]-
ethanone 38 and similar
compounds formed in the reaction Scheme 9 of Example 9; 1-(6-benzyloxy-4,4-
dimethyl-1,2,3,4-
tetrahydro-quinolin-7-yl)-ethanone 42 formed in the reaction Scheme 10 of
Example 10; 1-(4,4-
dimethyl-6-p-tolyl-1,2,3,4-tetrahydro-quinolin-7-yl)-ethanone 50 formed in the
reaction Scheme 12 of
Example 12; 6-acctyl-7-ethyl-4H-benzo[1,4]oxazin-3-one 81 formed in the
reaction Scheme 21 of
Example 21; 7-acetyl-6-methoxy-4,4-dimethyl-3,4-dihydro-1H-quinolin-2-one 91
formed in the
reaction Scheme 23 of Example 23; and 1-(4,4-dimethyl-6-phenyl-1,2,3,4-
tetrahydro-quinolin-7-yl)-
ethanone 94 formed in the reaction Scheme 24 of Example 24.
Example 30: Synthesis of Compounds where LI is -NHC(O)NH-.
102431 Compounds of Formula XII where L1 is -NHC(O)NH- are prepared, for
example, in three
steps from 1-(1-amino-6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl)-
ethanone 112 as shown
in Scheme C.
Scheme C
R NH R NH NH
NH2 D O~NH 0 0~NH N0OH O~NH NHz
R", NCO Step 1 N I Step 2 N Step 3 N
IX
112 X XI XII
Step 1 - Preparation of Compounds of Formula X.-
[02441 To a mixture of 1-(1-amino-6-ethyl-4,4-dimethyl-1,2,3,4-tetrahydro-
quinolin-7-yl)-ethanone
(112) and a base (such as triethylamine, potassium carbonate) in an
appropriate solvent (such as
dichloromethane or acetonitrile) an isocyanate of Formula IX (R" is R' as
defined in paragraph
[0003]) is slowly added. The reaction mixture is stirred at room temperature
or heated in an oil bath
for 2 to 24 hours. The reaction mixture is poured into water and extracted
with an appropriate solvent
(such as dichloromethane or ethyl acetate), The organic layer is collected,
washed with saturated
93
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
sodium bicarbonate solution and brine, and dried over magnesium sulfate. After
removal of drying
agent and solvent, the residue can be purified by chromatography to provide
the desired compound of
Formula X.
Step 2 - Preparation of Compounds of Formula XI:
[0245] A solution of a compound of Formula X, hydroxylamine hydrochloride, and
pyridine in
ethanol is stirred at 80-100 C for 2-24 hours. The reaction mixture is
concentrated and the residue
dissolved in an appropriate solvent such as ethyl acetate, washed with water
and brine, and dried over
magnesium sulfate. After removal of drying agent and solvent, the residue can
be purified by silica
gel chromatography to provide the desired compound of Formula XI.
Step 3 - Preparation of Compounds of Formula XII:
[0246] A mixture of a compound of Formula XI, Raney nickel, and ammonium
hydroxide in
methanol is stirred under a hydrogen balloon for 0.5-20 hours. The catalyst is
removed by filtration
and the filtrate is concentrated. The residue can be purified by
chromatography to provide the desired
compound of Formula XII.
[0247] Similar compounds with substituents other than ethyl at the 6 position,
e.g. optionally
substituted aryl, can be prepared similarly, for example, by replacing 1-(1-
amino-6-ethyl-4,4-
dimethyl-1,2,3,4-tetrahydro-quinolin-7-yl)-ethanone 112 in step 1 with the
product of step 1 of
Scheme B made by replacing 1-(6 -ethyl -4,4-dimethyl- 1,2,3,4-tetrahydro-
quinolin-7-yl) -ethanone 9
with a suitable compound as described in Example 30.
Example 31: Synthesis of Compounds where -R4 is alkyl.
[0248] Compounds of Formula XVI where R"' is alkyl (i.e. R4 of Formula I,
paragraph [0003] is
alkyl) are prepared, for example, in five steps from 7-acctyl-4,4-dimethyl-l-
(2,2,2-trifluoro-acetyl)-
1,2,3,4-tetrahydro-quinolin-6-yl ester 35 as shown in Scheme IV.
Scheme IV
F3CO
0 H 1 0
N ` Step 1 N Step N
0Tf OTf OTf
r l R'-CI
35 113 X111
I 0 R, HO.N R, NH2
Step 3a Step 4 Step 5
or R
Step 3b X11/ XV XV1
94
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
Step 1 -Preparation of trifluoro-methanesulfonic acid 7-acetyl-4,4-dimethyl-
1,2,3,4-tetrahydro-
quinolin-6 yl ester (113):
102491 To a solution of 7-acetyl-4,4-dimethyl-l-(2,2,2-trifluoro-acetyl)-
1,2,3,4-tctrahydro-quinolin-
6-yl ester (35, 110 mg, 0.25 mmol) in 3 mL of acetonitrile was added 1.5 mL of
2 M potassium
carbonate in water. The reaction mixture was stirred at room temperature for
10 h and was
concentrated under reduced pressure. The residue was partioned between ethyl
acetate and water and
the organic layer was dried over sodium sulfate. The solution was filtered and
the filtrate
concentrated under vacuum to provide the desired compound as a colorless oil
(113, 57 mg, 65%).
Step 2 - Preparation of Compounds of Formula XIIL:
10250] A mixture of trifluoro-methanesulfonic acid 7-acetyl-.4,4-dimethyl-
1,2,3,4-tetrahydro-
quinolin-6-yl ester (113), a halide such as R'-Cl (R' as defined in paragraph
[0003]), a suitable base
such as potassium carbonate, pyridine or diisopropylethylamine in a suitable
solvent such as
acetonitrile or tetrahydrofuran is stirred at a temperature below 80 C for 1-
36 hours. The reaction
mixture is concentrated under reduced pressure and partitioned between an
organic solvent such as
ethyl acetate and an aqueous solution. The organic layer is dried over an
anhydrous salt, filtered,
concentrated and subjected to silica gel chromatography to provide the desired
compound of Formula
XIII.
Step 3a - Preparation of Compounds of Formula XIV:
[0251] A mixture of a compound of Formula XIII, a terminal alkync or olcfin, a
suitable ligated
palladium catalyst (CuI and an amine solvent such as triethylamine for alkyne)
in a suitable solvent
such or tetrahydrofuran is stirred at a temperature below 80 C for 1-36
hours. The reaction mixture is
concentrated under reduced pressure and partitioned between an organic solvent
such as ethyl acetate
and an aqueous solution. The organic layer is dried over an anhydrous salt,
filtered, concentrated and
subjected to silica gel chromatography to provide the alkyne or olefin
intermediate, which can be
reduced to the corresponding saturated alkyl compound of Formula XIV by
treatment with a
transition metal catalyst such as palladium, nickel or platinum under a
hydrogen atmosphere.
Step 3b - Preparation of Compounds of Formula XIV.:
[0252] A mixture of a compound of Formula XIII, an alkyl tin compound for
Stille cross-coupling,
or an alkyl boronic acid, ester or alkyl trifluoroborate salt for Suzuki cross-
couplings, and a suitable
ligated palladium catalyst in a suitable solvent such or tetrahydrofuran is
stirred at a temperature
below 80 C for 1-36 hours. The reaction mixture is concentrated under reduced
pressure and
partitioned between an organic solvent such as ethyl acetate and an aqueous
solution. The organic
layer is dried over an anhydrous salt, filtered, concentrated and subjected to
chromatography to
provide the desired compound of Formula XIV. Note, vinyl tin and vinyl
boronate reagents can also
be employed to form olefin compounds, These olcfin products can be reduced to
the corresponding
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
saturated alkyl compound of Formula XIV by treatment with a transition metal
catalyst such as
palladium, nickel or platinum under a hydrogen atmosphere.
Step 4- Preparation of Compounds of'Formula XV:
[02531 A solution of a compound of Formula XIV, hydroxylamine hydrochloride,
and pyridine in
ethanol is stirred at 80-100 C for 2-24 hours. The reaction mixture is
concentrated and the residue
dissolved in an appropriate solvent such as ethyl acetate, washed with water
and brine, and dried over
magnesium sulfate. After removal of drying agent and solvent, the residue can
be purified by silica
gel chromatography to provide the desired compound of Formula XV.
Step 5 - Preparation of Compounds of Formula X67.
[02541 A mixture of a compound of Formula XV, Raney nickel, and ammonium
hydroxide in
methanol is stirred under a hydrogen balloon for 0.5-20 hours. The catalyst is
removed by filtration
and the filtrate is concentrated. The residue can be purified by
chromatography to provide the desired
compound of Formula XVI.
Example 32: Enzyme Activity Assays
[02551 Assays for the activity of proteases, including, but not limited to,
renin, pepsin, cathepsinD
and BACE are known in the art. Exemplary biochemical assays used to assess the
activity of
compounds of the invention are described as follows.
[02561 Renin protease activity is assessed in a FRET (Fluorescent resonance
energy transfer) screen.
The substrate peptide is linked to a Fluorescent probe (Hilyte Fluor 488) on
one end and a quencher
on the other end (QXL). In the absence of an inhibitor, protease such as renin
will cleave the peptide
and the fluorescence signal will increase. IC50 values are determined with
respect to inhibition of
renin activity, where inhibition of cleavage of the peptide substrate is
measured as a function of
compound concentration. Compounds to be tested were dissolved in DMSO to a
concentration of 20
mM. These were diluted 30 L into 120 L of DMSO (4 mM) and 1 L was added to
an assay plate.
These were then serially diluted 1:3 (50 gL to 100 L DMSO) for a total of 8
points. Plates were
prepared such that each protease reaction is 20 .tL in 1 x protease buffer (25
mM Hepes, pH 7.4, 100
mM Nacl, 0.01%BSA, 0,01%Tween-20), 5% DMSO. Substrate was 2 pM Hilyte Fluor
488 Arg-
Ile-IIis-Pro-Phe-His-Leu-dial-Ile-His-Thr-Lys (QXL-520)Arg-NH2 (Anaspec,
Inc.), Renin was at 4
ng per sample. The protease reaction was incubated for 210 minutes at 30 C,
and the signal per well
was read on a Tecan Saffire reader. The fluorescence signal was measured and
the signal vs.
compound concentration was used to determine the IC50.
[02571 Porcine pepsin protease activity is assessed in a FRET screen similarly
to that for renin.
Compounds to be tested were dissolved in DMSO to a concentration of 20 mM,
These were diluted
96
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
30 pL into 120 pL of DMSO (4 mM) and 1 L was added to an assay plate. These
were then serially
diluted 1:3 (50 pL to 100 L DMSO) for a total of 8 points. Plates were
prepared such that each
protease reaction is 20 L in 1 x protease buffer (50 mM Sodium Acetate, pH
3.5, 150 mM Nacl,
0.01 %BSA, 0.01 % Tween-20), 5% DMSO, Substrate was 4 pM Hilyte Fluor 488 -Glu-
Dap-Lys-Pro-
Ile-Leu-Phe-Phe-Arg-Leu-Gly-Lys--Glu (QXL-520)Glu-NH2 (Anaspec, Inc.). Porcine
pepsin was at
0.5 ng per sample. The protcasc reaction was incubated for 30 minutes at 30 C
and the signal per
well was read on a Tecan Saffire reader. The fluorescence signal was measured
and the signal vs.
compound concentration was used to determine the IC50.
[0258] CathepsinD protcasc activity is assessed in a FRET screen similarly to
that for renin.
Compounds to be tested were dissolved in DMSO to a concentration of 20 mM.
These were diluted
30 pL into 120 pL of DMSO (4 mM) and 1 pL was added to an assay plate. These
were then serially
diluted 1:3 (50 L to 100 L DMSO) for a total of 8 points. Plates were
prepared such that each
protease reaction is 20 L in 1 x protease buffer (50 mM Sodium Acetate, pH
3.5, 150 mM Nacl,
0.01 %BSA, 0.01 % Tween-20), 5% DMSO. Substrate was 4 .oM Hilyte Fluor 488 -
Glu-Dap-Lys-Pro-
Ile-Leu-Phe-Phe-Arg-Leu-Gly-Lys--Glu (QXL-520)Glu-NH2 (Anaspec, Inc.).
CathepsinD was at
0.15 ng per sample. The protease reaction was incubated for 60 minutes at 25
C and the signal per
well was read on a Tecan Saffire reader. The fluorescence signal was measured
and the signal vs.
compound concentration was used to determine the IC50.
[0259] BACE protease activity is assessed in a FRET screen similarly to that
for renin. Compounds
to be tested were dissolved in DMSO to a concentration of 20 mM. These were
diluted 30 L into
120 L of DMSO (4 mM) and I L was added to an assay plate. These were then
serially diluted 1:3
(50 L to 100 L DMSO) for a total of 8 points. Plates were prepared such that
each protease
reaction is 20.oL in lx protease buffer (50 mM Sodium Acetate, pH 4.5, 150 mM
Nacl, 0.01%BSA,
0.0 1 % Tween-20), 5% DMSO. Substrate was 4 pM Hilyte Fluor 488 -Ser-Glu-Val-
Asn-Leu-Asp-
Ala-Glu-Phe-Lys (QXL-520)Glu-NH2 (Anaspec, Inc.). BACE was at 8 ng per sample.
The protease
reaction was incubated for 210 minutes at 30 C and the signal per well was
read on a Tecan Saffire
reader. The fluorescence signal was measured and the signal vs. compound
concentration was used to
determine the IC50.
[0260] Compounds having ICs of less than 10 pM in a renin biochemical assay
as described above
are listed in the following table:
P-0002, P-0003, P-0005, P-0006, P-0009, P-0010, P-0014, P-0015, P-0016,
P-0017, P-0019, P-0021, P-0023, P-0025, P-0026, P-0027, P-0028, P-0029,
Renin <10 M P-0030, P-0031, P-0032, P-0033, P-0034, P-0035, P-0036, P-0037, P-
0039,
P-0040, P-0041, P-0042, P-0043, P-0044, P-0045, P-0046, P-0047. P-0048,
P-0049, P-0050, P-0051, P-0052. P-0053, P-0054, P-0055, P-0056, P-0057,
P-0059, P-0060, P-0061, P 006 . P 0063, P-0065, P-0066, P-0067, P-0068
97
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
[02611 Compounds were selective with respect to other proteases, as none of
the compounds
screened in a BACE or porcine pepsin assay as described above showed
measureable inhibition up to
200 .tM. Compounds having IC50 between 10 .tM and 100 M in a cathepsinll
biochemical assay as
described above are listed in the following table:
B Raf ! P-0015, P-0023, P-0025, P-0032, P-0034, P-0039, P-0048, P-0059, P-
0060, P-0066,
P-0067, P-0068
[02621 All patents and other references cited in the specification are
indicative of the level of skill of
those skilled in the art to which the invention pertains, and are incorporated
by reference in their
entireties, including any tables and figures, to the same extent as if each
reference had been
incorporated by reference in its entirety individually.
[0263] One skilled in the art would readily appreciate that the present
invention is well adapted to
obtain the ends and advantages mentioned, as well as those inherent therein.
The methods, variances,
and compositions described herein as presently representative of preferred
embodiments are
exemplary and are not intended as limitations on the scope of the invention.
Changes therein and
other uses will occur to those skilled in the art, which are encompassed
within the spirit of the
invention, are defined by the scope of the claims.
[02641 The invention illustratively described herein suitably may be practiced
in the absence of any
element or elements, limitation or limitations which is not specifically
disclosed herein. Thus, for
example, in each instance herein any of the terms "comprising", "consisting
essentially of" and
"consisting of may be replaced with either of the other two terms. Thus, for
an embodiment of the
invention using one of the terms, the invention also includes another
embodiment wherein one of
these terms is replaced with another of these terms. In each embodiment, the
terms have their
established meaning. Thus, for example, one embodiment may encompass a method
"comprising" a
series of steps, another embodiment would encompass a method "consisting
essentially of the same
steps, and a third embodiment would encompass a method "consisting of the same
steps. The terms
and expressions which have been employed are used as terms of description and
not of limitation, and
there is no intention that in the use of such terms and expressions of
excluding any equivalents of the
features shown and described or portions thereof, but it is recognized that
various modifications are
possible within the scope of the invention claimed. Thus, it should be
understood that although the
present invention has been specifically disclosed by preferred embodiments and
optional features,
modification and variation of the concepts herein disclosed may be resorted to
by those skilled in the
art, and that such modifications and variations are considered to be within
the scope of this invention
as defined by the appended claims.
98
CA 02761009 2011-11-04
WO 2010/129467 PCT/US2010/033385
[0265] In addition, where features or aspects of the invention are described
in terms of Markush
groups or other grouping of alternatives, those skilled in the art will
recognize that the invention is
also thereby described in terms of any individual member or subgroup of
members of the Markush
group or other group.
[0266] Also, unless indicated to the contrary, where various numerical values
are provided for
embodiments, additional embodiments are described by taking any 2 different
values as the endpoints
of a range. Such ranges are also within the scope of the described invention.
[0267] Thus, additional embodiments are within the scope of the invention and
within the following
claims.
99