Note: Descriptions are shown in the official language in which they were submitted.
CA 02762016 2011-12-09
72249-156D
METHODS FOR INCREASING POLYPEPTIDE PRODUCTION
This is a divisional application of Canadian patent application Serial
No. 2,480,121, filed on March 27, 2003.
FIELD OF THE INVENTION
=
The invention is in the field of polypeptide production, particularly
recombinant polypeptide production in cell culture. The subject matter of this
divisional application relates to production methods and culture media
containing
xanthine derivatives, whereas the parent application relates to methods and
media
containing hybrid polar compounds. The expression "the invention" or the like
encompasses the subject matter of both the parent and this divisional
application.
BACKGROUND
Polypeptides are useful in a variety of diagnostic, therapeutic,
agricultural, nutritional., and research applications. Although polypeptides
can be
isolated from natural sources, the isolation of large quantities of a specific
polypeptide
from natural sources may be expensive. Also, the polypeptide may not be of
uniform
quality due to variation in the source material. Recombinant DNA technology
allows
more uniform and cost-effective large-scale production of specific
polypeptides.
One goal of recombinant polypeptide production is the optimization of
culture conditions so as to obtain the greatest possible productivity.
Incremental
increases in productivity can be economically significant. Some of the methods
to
increase productivity in cell culture include using enriched medium,
monitoring
osmolarity during production, decreasing temperatures during specific phases
of a
cell culture, and/or the addition of sodium butyrate (see, e.g., U.S. Patent
No. 5,705,364).
-1-
_
CA 02762016 2011-12-09
72249-156D
However, as more polypeptide-based drugs demonstrate clinical
effectiveness and increased commercial quantities are needed, available
culture
facilities become limited. Accordingly, there remains a need in the art to
continually
improve yields of recombinant polypeptides from each cell culture run.
SUMMARY
As shown by the experimental data reported herein, xanthine
derivatives and/or hybrid polar compounds can dramatically induce the
production of
polypeptides, especially recombinant polypeptides, from mammalian cell lines.
Moreover, xanthine derivatives and/or hybrid polar compounds can be used in
combination with other induction methods to further increase polypeptide
expression.
Thus, in one aspect, the invention provides a method for producing a
polypeptide, which may be a recombinant polypeptide, comprising culturing a
mammalian cell line in a growth phase followed by a production or induction
phase,
which can occur at a temperature
-1a-
,
CA 02762016 2011-12-09
WO 03/083066
PCT/US03/09266
of less than 37 C, and adding to the culture during the production phase a
xanthine derivative.
The addition of the xanthine derivative can increase the production of the
polypeptide. The
mammalian cell line can be a cell line that has been genetically engineered to
produce the
polypeptide or a hybridoma cell line that can produce an antibody. The
xanthine derivative
may be caffeine at a concentration from about 0.01 millimolar to about 5.0
millimolar or from
about 0.01 millimolar to about 3.0 millimolar. In some embodiments, the
mammalian cell
line is a C110 cell line, and it may have been transformed with a recombinant
vector encoding
the recombinant polypeptide. Optionally, the vector can comprise a
cytomegalovirus (CNN)
promoter. Typically, the cell does not naturally express the polypeptide or
only naturally
expresses the polypeptide at very low levels (in the absence of genetic
engineering). The
polypeptide rnay be a recombinant fusion polypeptide or a human or humanized
antibody.
The production or induction phase can occur at a temperature from about 29 C
to about 36 C
or from about 30 C to about 33 C. The growth phase can occut at a temperature
from about
35 C to about 38 C.
Optionally, at least two different xanthine derivatives can be added. The
xanthine
derivative(s) can be selected from the group consisting of caffeine, 3-
isobuty1-1-
methylxanthine, theophylline, theobromine, pentoxyphylline, and arninophylline
or from a
subset of this group. If two different xanthine derivatives are added, they
can be caffeine and
3-isobuty1-1-methylxanthine. Xanthine derivatives can be added multiple times
during the
culturing of the cell line, and the cell tine can be cultured in the presence
of the xanthine
derivative for at least about 5 days. The concentration of each xanthine
derivative added to
the culture can be from about 0.001 millimolar to about 3 millimolar. The
recombinant
polypeptide can be collected from the medium and formulated. The medium may
further
comprise a hybrid polar compound and/or an alkanoic acid. The hybrid polar
compound can
be hexamethylene bisacetatnide, optionally at a concentration from about 0.1
millimolar to
about 5 millimolar. The xanthine derivative can be caffeine, optionally at a
concentration
from about 0.1 millimolar to about 4 millimolar. The alkanoic acid can be a
salt of butyric
acid, optionally at a concentration from about 0.1 millimolar to about 2
millimolar. The
mammalian cells can be cultured at a temperature from about 29 C to about 36 C
or from
about 30 C to about 33 C. The mammalian cells can be cultured in a growth
phase at a first
temperature from about 35 C to about 38 C before they are shifted to a
production phase at a
second temperature from about 29 C to about 36 C, wherein the second
temperature can be
lower than the first temperature. The xanthine can be added at the time of the
shift from the
first temperature to the second temperature and/or before and/or after the
shift.
In another aspect the invention provides a method for producing a recombinant
polypeptide comprising growing in culture a mammalian cell line, optionally a
CHO cell line
that has been genetically engineered to produce the recombinant polypeptide,
and adding to
CA 02762016 2011-12-09
WO 03/083066
PCT/US03/09266
=
the culture medium at least one xanthine derivative selected from the group
consisting of
theobromine and caffeine. The addition of the xanthine derivative can increase
the production
of the recombinant polypeptide. The mammalian cell line may have been
transformed with a
recombinant vector encoding the recombinant polypeptide. Optionally, the
vector can
comprise a cytomegalovirus (CMV) promoter. Typically, the cell line does not
naturally
express the recombinant polypeptide or only naturally expresses the
recombinant polypeptide
at very low levels (in the absence of genetic engine,ering). The recombinant
polypeptide may
be a recombinant fusion polypeptide or a human or humanized antibody. The cell
line can be
cultured in a growth phase, which is distinct from a production or induction
phase. The
production phase can occur at a temperature less than 37 C. The cell line can
be cultured at a
temperature of from about 29 C to about 36 C or from about 30 C to about 33 C.
Optionally, at least two different xanthine derivatives can be added. Xanthine
derivatives can
be added multiple times during the culturing of the cell line. The
concentration of each
xanthine derivative added to the culture can be from about 0.001 millimolar to
about 3
millimolar. The recombinant polypeptide can be collected from the medium and
formulated.
The man-unalian cell line can be cultured at a first temperature from about 35
C to about 38 C
before it is shifted to a second temperature from about 29 C to about 36 C,
and the xanthine
derivative can be added at the time of the shift from the first temperature to
the second
temperature and/or before and/or after the shift The second temperature can be
lower than
the first temperature.
In another aspect, the invention provides a culture comprising a CHO cell
genetically
engineered to produce a polypeptide, a production medium, and at least one
xanthine
derivative selected from the group consisting of caffeine, 3-isobuty1-1-
methylxanthine,
theophylline, theobromine, pentoxyphylline, and aminophylline or from a subset
of this
group. The culture can comprise at least two xanthine derivatives. The
concentration of each
xanthine derivative present can be from about 0.001 millimolar to about 3
millimolar or from
about 0.01 millimolar to about 3 millimolar. The culture can comprise serum-
free medium,
and may comprise no added protein or may comprise insulin or IGF-1.
Additionally, the
culture can comprise dimethylformamide, dimethylsulfoxide, or
dimethylacetamide. The
invention, because of its low cost and convenience, is particularly useful for
large scale
culturing of CHO cells. The culture can be a large scale culture of at least
100 liters, or even
at least 500 liters, in size. The culture can comprise a homogeneous CHO cell
line.
In still another aspect, the invention encompasses a culture comprising a CHO
cell
genetically engineered to produce a polypeptide, a production medium, and at
least one
xanthine derivative, wherein the culture is grown at less than 37 C for at
least part of its life.
The xanthine derivative or derivatives present can be within the concentration
range from.
about 0.001 millimolar to about 3 millimolar or from about 0.01 millimolar to
about 3
-3-
CA 02762016 2011-12-09
WO 03/083066
PCT/US03/09266
rnillimolar, and the culture can contain at least two different xanthine
derivatives. The
xanthine derivatives can be selected from the group consisting of caffeine, 3-
isobuty1-1-
methylxanthine, theophylline, theobromine, pentoxyphylline, and aminophylline
or from a
subset of this group such as caffeine, theobromine and pentoxyphylline. The
size of the
culture can be at least 100 liters, and the production medium can be serum-
free medium and
can comprise either no added protein or insulin or IGF-1. The culture can
comprise a
homogeneous CHO cell line.
In a further aspect, the invention includes a method for producing a
polypeptide in a
culture of mammalian cells comprising incubating the culture at a temperature
of about 37 C
and thereafter incubating the culture at a temperature from about 29 C to 36
C, and adding to
the culture a xanthine derivative during the incubation at a temperature from
about 29 C to
36 C, wherein the polypeptide is a recombinant polypeptide or an antibody. The
xanthine
derivative can be selected from the group consisting of caffeine, 3-isobuty1-1-
methylxanthine,
theophylline, theobromine, pentoxyphylline, and aminophylline or from a subset
of this group
such as caffeine, theobromine, and pentoxyphylline. The mammalian cells can be
hybridoma
cells or CHO cells. The xanthine derivative or derivatives present can be
within the
concentration range from about 0.001 millimolar to about 3 millimolar or from
about 0.01
millimolar to about 3 millimolar, and the culture can contain at least two
different xanthine
derivatives. Xanthine derivatives can be added multiple times during the
culturing of the cell
line.
The invention provides a method for producing a recombinant polypeptide
comprising culturing a mammalian cell line, in some embodiments a CHO cell
line, at a
temperature from about 29 C to about 36 C, optionally at temperatures between
about 29 C
and 35 C or from about 30 C to about 33 C, in a medium comprising a hybrid
polar
compound. The medium can be serum free. The addition of the hybrid polar
compound can
increase the production of the recombinant polypeptide. The hybrid polar
compound can be
hexamethylene bisacetamide, optionally at a concentration from about 0.1
millimolar to about
20 millimolar or from about 0.1 millimolar to about 5 millimolar. Furthermore,
the medium
may comprise an alkanoic acid, such as a salt of butyric acid, at a
concentration, for example,
from about 0.05 millimolar to about 10 raillimolar, optionally from about 0.1
millimolar to
about 2 millimolar. Furthermore, the medium may comprise a xanthine
derivative, for
example, caffeine, at a concentration from about 0.005 millimolar to 10
millimolar, optionally
from about 0.01 millimolar to 4 millimolar or from about 0.1 millimolar to 4
raillimolar. The
mammalian cells can be cultured at a first temperature from about 35 C to
about 38 C before
they are shifted to a second temperature between about 29 C and 36 C, and the
hybrid polar
compound can be added after the shift from the first temperature to the second
temperature.
The manunalian cells may be genetically engineered to produce a polypeptide,
optionally a
-4-
CA 02762016 2011-12-09
WO 03/083066
PCT/US03/09266
secreted polypeptide that can be recovered from the medium, including RANK:Fc,
type II
interleukin-1 receptor, TNFR:Fc, CD40 ligand, TRAIL, flt3-ligand, IL-4
receptor, G-CSF,
erythropoietin, an antibody, or a substantially similar polypeptide, among
others.
In another embodiment, the invention provides an improved method for producing
a
polypeptide by culturing mammalian cells comprising culturing the cells in a
medium
comprising a hybrid polar compound, optionally at temperatures from about 29 C
to about
36 C, between about 29 C and 35 C, or from about 30 C to about 33 C. The
hybrid polar
compound may be hexamethylene bisacetamide, optionally at a concentration
between about
0.1 millimolar and about 5 millimolar. The addition of the hybrid polar
compound can
increase the production of the polypeptide, which may be a recombinant
polypeptide.
Furthermore, the medium may comprise an alkanoic acid, for example, butyric
acid,
optionally at a concentration from about 0.05 millimolar to about 10
millimolar or from about
0.1 millimolar to about 2 millimolar. Furthermore, the medium may comprise a
xanthine,
such as, for example, caffeine, optionally at a concentration from about 0.005
millimolar to 10
millimolar or from about 0.01 millimolar to 5 millimolar. Optionally, the
polypeptide may be
RANK:Fc, type II interleukin-1 receptor, TNFR:Fc, CD40 ligand, TRAIL, flt3-
ligand, IL-4
receptor, GM-CSF, erythropoietin, an antibody, or a substantially similar
polypeptide among
others.
In another aspect, the invention provides a method for obtaining a
polypeptide,
optionally a recombinant polypeptide, comprising recovering the polypeptide
from medium in
which mammalian cells have been grown, wherein the mammalian cells can secrete
the
polypeptide and are grown at temperatures between about 29 C and 35 C,
optionally from
about from about 30 C to about 33 C, in medium comprising hexamethylene
bisacetamide.
The hexamethylene bisacetamide may be present at concentrations between about
0.1
millimolar and about 5 millimolar. Furthermore, the medium may comprise an
alkanoic acid,
for example, butyric acid, optionally at a concentration from about 0.05
millimolar to about
10 rnillimolar or from about 0.1 millimolar to about 2 millimolar.
Furthermore, the medium
may comprise a xantliine, for example, caffeine, optionally at a concentration
from about
0.005 millimolar to 10 millimolar or from about about 0.01 millimolar to 5
millimolar. The
polypeptide may be RANK:Fc, type II interleukin-1 receptor, TNFR:Fc, CD40
ligand,
TRAIL, flt3-ligand, IL-4 receptor, G-CSF, erythropoietin, an antibody, or a
substantially
similar polypeptide, among others.
In a further embodiment, the invention comprises method for producing a
recombinant polypeptide comprising culturing mammalian cells in a medium
comprising a
hybrid polar compound and a xanthine, wherein the mammalian cells have been
genetically
engineered to express the recombinant polypeptide. The medium may further
comprise an
alkanoic acid, such as, for example, a salt of butyric acid, which may be at a
concentration
-5-
CA 02762016 2011-12-09
WO 03/083066
PCT/US03/09266
=
from about 0.1 millimolar to about 2 millimolar. The hybrid polar compound can
be
hexamethylene bisacetamide, which may be at a concentration from about 0.1
millimolar to
about 5 millimolar, and/or the xanthine can be caffeine, which may be at a
concentration from
about 0.1 millimolar to about 4 millimolar. The cells can be cultured at a
temperature from
about 29 C to about 36 C or from about 30 C to about 33 C. The mammalian cells
can be
cultured at a first temperature from about 35 C to about 38 C before they are
shifted to a
second temperature from about 29 C to about 36 C, and the hybrid polar
compound and the
xanthine can be added at the time of the shift from the first temperature to
the second
temperature and/or before and/or after the shift. The can be medium can be
serum free.
In a further embodiment, the invention encompasses a method for producing a
polypeptide, optionally a recombinant polypeptide, comprising culturing
mammalian cells in
a medium comprising a hybrid polar compound and an alkanoic acid, wherein the
mammalian
cells may have been genetically engineered to express the rr.cf=rnbinnt
polypeptide The
hybrid polar compound can be hexamethylene bisacetamide, and the hybrid polar
compound
can be present at a concentration of from about 0.5 millimolar to about 10
millimolar or at a
concentration between about 0.5 millimolar and 2.5 millimolar. The alkanoic
acid can be a
salt of butyric acid, and the alkanoic acid can be present at a concentration
from about 0.1
millimolar to about 5 millimolar or at a concentration between about 0.1
millimolar and about
2.0 millimolar. The mammalian cells can be cultured at a temperature from
about 29 C to
about 36 C, and the medium can be serum free. The mammalian cell line can be
cultured at a
first temperature from about 35 C to about 38 C before they are shifted to a
second
temperature from about 29 C to about 36 C, and the hybrid polar compound and
the alkanoic
acid may be added after the shift from the first temperature to the second
temperature. The
medium can further comprise a xanthine derivative at a concentration from
about 0.001
millimolar to about 5.0 tni llimolar. The mammalian cell line can be a
hybridoma cell line or a
CHO cell line.
In still another embodiment, the invention provides a method for producing a
polypeptide comprising culturing a mammalian cell line in a production phase
at a second
temperature from about 30 C to 34 C in a medium comprising a hybrid polar
compound,
wherein the production phase follows a growth phase at a first temperature
from about 35 C
to about 38 C. The polypeptide can be a recombinant polypeptide or an
antibody. The
hybrid polar compound can be hexamethylene bisacetamide, optionally at a
concentration
from about 0.1 millimolar to about 5 millimolar. The hybrid polar compound may
be added
after the shift from the first temperature to the second temperature. The
medium can further
comprise an alkanoic acid, which can be a salt of butyric acid, optionally at
a concentration
from about 0.05 millimolar to about 10.0 millimolar. The medium can also
comprise a
xanthine derivative, optionally at a concentration from about 0.001 millimolar
to about 5.0
-6-
CA 02762016 2014-11-21
72249-156D
millimolar. The medium can be serum free. The mammalian cell line can be a
hybridoma cell line or a CHO cell.
The invention also provides a method for producing a polypeptide
comprising culturing a mammalian cell line in a medium comprising a hybrid
polar
compound at a concentration between about 0.5 millimolar and 2.5 millimolar,
an
alkanoic acid at a concentration from about 0.1 millimolar and 2.0 millimolar,
and a
xanthine derivative at a concentration from about 0.001 millimolar to about
4 millimolar.
In still another embodiment, the invention provides a method for
producing a polypeptide, optionally RANK:Fc, type II interleukin-1 receptor,
TNFR:Fc,
CD40 ligand, TRAIL, flt3-ligand, IL-4 receptor, G-CSF, erythropoietin, an
antibody, or
a substantially similar polypeptide, comprising culturing mammalian cells,
which may
have been genetically engineered to produce any of these polypeptides, in a
medium
comprising between about 0.1 millimolar and about 5 millimolar HMBA, from
about 0.1 millimolar to about 2 millimolar butyric acid, and from about 0.1
millimolar to
about 4 millimolar caffeine at a temperature from about 29 C to about 36 C or
from
about 30 C to about 33 C.
The present invention as claimed relates to:
- a method for producing a polypeptide comprising: culturing a CHO
cell line in a production phase, wherein the production phase occurs at a
temperature
of less than 37 C; and adding to the culture medium a xanthine derivative
selected
from the group consisting of caffeine, 3-isobuty1-1-methylxanthine, and
pentoxyphylline; wherein the CHO cell line has been genetically engineered to
produce the polypeptide;
- a method for producing a recombinant antibody comprising: culturing
a CHO cell line that has been genetically engineered to produce the
recombinant
antibody at a temperature from about 29 C to about 36 C; and adding caffeine
to the
culture medium; wherein the recombinant antibody binds to EGF receptor; and
-7-
CA 02762016 2014-11-21
72249-156D
- a culture comprising a CHO cell genetically engineered to produce a
polypeptide, a production medium, and a xanthine derivative selected from the
group
consisting of caffeine, 3-isobutyl-1-methylxanthine, and pentoxyphylline.
-7a-
CA 02762016 2012-05-25
72249-156D
' BRIEF DESCRIPTION OF THE FIGURES
Figure 1 shows the percentage of the total cells that are viable under the
indicated
20 conditions at 31 C for the #9 CHO cell line as a function of days in
culture.
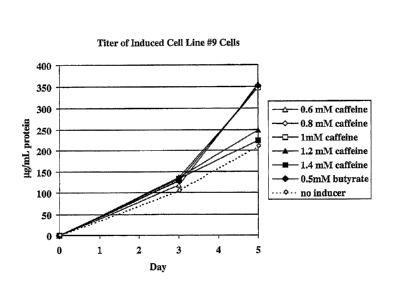
Figure 2 shows the micrograms of protein per milliliter of cell culture, i.e.,
protein
titer, under the indicated conditions at 31 C for the #9 CHO cell line as a
function of days in
culture.
Figure 3 shows the micrograms of protein per 106 cells per day under the
indicated
25 conditions at 31 C for #9 CHO cell line as a function of days in
culture.
Figure 4 shows a graph displaying the concentration of an antibody against
murine
IL-4 receptor recovered from medium as a function of days of growth of a CHO
cell line
comprising a vector encoding the antibody at the stated temperatures in the
presence or
absence of HMBA or sodium butyrate. Markings are as follows: --A- , no inducer
37 C;
30 ¨111-- , no inducer 34 C; ----, 0.5 millimolar sodium butyrate 34 C;
, 2.0
millimolar HMBA 34 C; ¨0¨, no inducer 31 C; ¨IV¨, 0.5 millimolar sodium
butyrate
31 C; and ¨0¨ , 2.0 millimolar HMBA 31 C.
DETAILED DESCRIPTION OF THE INVENTION
An "antibody" is a polypeptide or complex of polypeptides, each of which
comprises at least one variable antibody immunoglobulin domain and at least
one constant
-7b-
CA 02762016 2011-12-09
WO 03/083066 PCT/US03/09266
antibody immunoglobulin domain. Antibodies may be single chain antibodies,
dimeric
antibodies, or some higher order complex of polypeptides including, but not
limited to,
heterodimeric antibodies. A "human antibody" is an antibody encoded by nucleic
acids that
are ultimately human in origin. Such an antibody can be expressed in a non-
human cell or
organism. For example, DNA encoding a human antibody can be introduced into
tissue
culture cells and expressed in transformed cell lines. Alternatively, human
antibodies can be
Q
expressed in transgenic animals such as, for example, the transgenic mice
described in
Mendez et al. ((1997), Nature Genetics 16(4): 146-56). Such transgenic mice
are utilized in
making the fully human antibodies in US Patent No. 6,235,883 Bl. Human
antibodies can
also be expressed in hybridoma cells. A "humanized antibody" is a chimeric-
antibody
comprising complementarity determining regions (CDR1, CDR2, and CDR3) from a
non-
human source and other regions that conform to sequences in human antibodies
(and may be
of human origin) as explained in, e.g., US Patent Nos. 5,558,864 arid
5,693,761 and
International Patent Application WO 92/11018.
A "constant antibody immunoglobulin domain" is an immunoglobulin domain
that is identical to or substantially similar to a CL, CH 1 , C112, C113, or
CH4, domain of human
or animal origin. See e.g. Hasemann and Capra, Lmmunoglobulins: Structure and
Function, in
William E. Paul, ed., Fundamental Immunology, Second Edition, 209, 210-218
(1989);
Kabat et al., Sequences of Proteins of Immunological Interest, U.S. Dept of
Health and
Human Services (1991).
An "Fc portion of an antibody" includes human or animal itnmunoglobulin
=
domains C112 and CH3 or immunoglobufm domains substantially similar to these.
For
discussion, see Hasemann and Capra, supra, at 212-213 and Kabat et al., supra.
Cells have been "genetically engineered" to express a specific polypeptide
when
recombinant nucleic acid sequences that allow expression of the polypeptide
have been
introduced into the cells using methods of "genetic engineering," such as
viral infection with
a recombinant virus, transfection, transformation, or electroporation. See
e.g. Kaufman et al.
(1990), Meth. Enzymol. 185: 487-511; Current Protocols in Molecular Biology,
Ausubel et
al., eds. (Wiley & Sons, New York, 1988, and quarterly updates). Infection
with an
unaltered, naturally-occurring virus, such as, for example, hepatitis B virus,
human
immunodeficiency virus, adenovirus,-etc., does not constitute genetic
engineering as meant
herein. The term "genetic engineering" refers to a recombinant DNA or RNA
method used
to create a host cell that expresses a gene at elevated levels or at lowered
levels, or expresses a
mutant form of the gene. In other words, the cell has been transfected,
transformed or
transduced with a recombinant polynucleotide molecule, and thereby altered so
as to cause the
cell to alter expression of a desired polypeptide_ For the purposes of the
invention, the
antibodies produced by a hybridoma cell line resulting from a cell fusion are
not
-8-
CA 02762016 2011-12-09
,
WO 03/083066
PCT/US03/09266
"recombinant polypeptides." Further, viral polypeptides produced by a cell as
a result of viral
infection are also not "recombinant polypeptides" as meant herein unless the
viral nucleic acid
has been altered by genetic engineering prior to infecting the cell. The
methods of "genetic
engineering" also encompass numerous methods including, but not limited to,
amplifying
nucleic acids using polymerase chain reaction, assembling recombinant DNA
molecules by
cloning them in Escherichia coli, restriction enzyme digestion of nucleic
acids, ligation of
nucleic acids, and transfer of bases to the ends of nucleic acids, among
numerous other
methods that are well-la-town in the art. See e.g. Sambrook et al., Molecular
Cloning: A
Laboratory Manual, 2.11d ed_, vol. 1-3, Cold Spring Harbor Laboratory, 1989.
Methods and
vectors for genetically engineering cells and/or cell lines to express a
polypeptide of interest
are well known to those skilled in the art_ Genetic engineering techniques
include but are not
limited to expression vectors, targeted homologous recombination and gene
activation (see,
for example, US Patent No. 5,272,071 to Chappel) and trans activation by
engineered
transcription factors (see e.g., Segal et al., 1999, Proc. Natl. Acad. Sci.
USA 96(6):2758-63).
Optionally, the polypeptides are expressed under the control of a heterologous
control
element such as, for example, a promoter that does not in nature direct the
production of that
polypeptide. For example, the promoter can be a strong viral promoter (e.g.,
CMV, SV40)
that directs the expression of a mammalian polypeptide. The host cell may or
may not
normally produce the polypeptide. For example, the host cell can be a CHO cell
that has been
genetically engineered to produce a human polypeptide, meaning that nucleic
acid encoding
the human polypeptide has been introduced into the CHO cell. Alternatively,
the host cell can
be a human cell that has been genetically engineered to produce increased
levels of a human
polypeptide normally present only at very low levels (e.g., by replacing the
endogenous
promoter with a strong viral promoter).
95 "Growth phase" means a period during which cultured cells are rapidly
dividing and
increasing in number. During growth phase, cells are generally cultured in a
medium and
under conditions designed to maximize cell proliferation.
A "hybrid polar compound" is compound having two polar groups separated by an
apolar carbon chain. This includes hexamethylene bisacetamide (HM13A) and the
other
molecules discussed below and in the following references: Richon et al.
(1998), Proc. Natl.
Acad. Sci. 95: 3003-07; Marks et al. (1994), Proc. Natl. Acad. Sci. 91: 10251-
54; and US
Patent Nos. 5,055,608 and 6,087,367.
The production of a polypeptide is "increased" by the addition of an inducing
agent,
such as hexamethylene bisacetamide (H2MBA) or caffeine, if the amount the
polypeptide
produced in a culture containing the inducing agent is more than the amount of
the
polypeptide produced in an otherwise identical culture that does not contain
the inducing
agent. Similarly, the production of a polypeptide is "increased" by growth at
a temperature
-9-
CA 02762016 2011-12-09
õ.õ
WO 03/083066
PCT/US03/09266
_other than 37 C if the amount of polypeptide produced in a culture incubated
at a temperature
other than 37 C is more than the amount of the polypeptide produced in an
otherwise
identical culture incubated at 37 C.
A "multimerization domain" is a domain within a polypeptide molecule that
confers upon it a propensity to associate with other polypeptide molecules
through covalent or
non-covalent interactions.
A "naturally-occurring polypeptide" is a polypeptide that occurs in nature,
that is,
a polypeptide that can be produced by cells that have not been genetically
engineered. Such a
polypeptide may also be produced by cells genetically engineered to produce
it.
"Polypeptide" means a chain of at least 6 amino acids linked by peptide bonds.
Optionally, a polypeptide can comprise at least 10, 20, 30, 40, 50, 60 , 70,
80, 90, 100, 150,
200, 250, or 300 amino acids linked by peptide bonds.
-Production tnedium" means a cell culture medium designed to be used to
culture
cells during a production phase.
"Production phase" means a period during which cells are producing maximal
amounts of recombinant polypeptide. A production phase is characterized by
less cell
division than during a growth phase and by the use of medium and culture
conditions
designed to maximize polypeptide production.
A "recombinant fusion polypeptide" is a fusion of all or part of at least two
polypeptides, which is made using the methods of genetic engineering.
A "recombinant polypeptide" is a polypeptide resulting from the process of
genetic
engineering. For the purposes of the invention, the antibodies produced by a
hybridoma cell
line resulting from a cell fusion are not "recombinant polypeptides." Further,
viral proteins
produced by a cell as a result of viral infeCtion with a naturally-occurring
virus are also not
"recombinant polypeptides" as meant herein unless the viral nucleic acid has
been altered by
genetic engineering prior to infecting the cell.
"Substantially similar" polypeptides are at least 80%, optionally at least
90%,
identical to each other in amino acid sequence and maintain or alter in a
desirable manner the
biological activity of the unaltered polypeptide. Conservative amino acid
substitutions,
unlikely to affect biological activity, include, without limitation, the
following: Ala for Ser,
Val for Ile, Asp for Glu, Thr for Ser, Ala for Gly, Ala for Thr, Ser for Asn,
Ala for Val, Ser
for Gly, Tyr for Phe, Ala for Pro, Lys for Arg, Asp for Asn, L,eu for Ile, Leu
for Val, Ma for
Glu, Asp for Gly, and these changes in the reverse. See e.g. Neurath et al.,
The Proteins,
Academic Press, New York (1979). In addition exchanges of amino acids among
members of
the following six groups of amino acids will be considered to be conservative
substitutions for
the purposes of the invention. The groups are: 1) methionine, alanine, valine,
leucine, and
isoleucine; 2) cysteine, serine, threonine, asparagine, and glutamine; 3)
aspartate and
-10-
CA 02762016 2011-12-09
WO ()3/083066
PCT/US03/09266
glutamate; 4) histidine, lysine, and arginine; 5) glycine and proline; and 6)
tryptophan,
tyrosine, and phenylalanine. The percent identity of two amino sequences can
be determined
by visual inspection and mathematical calculation, or more preferably, the
comparison is done
by comparing sequence information using a computer program such as the
Genetics
Computer Group (GCG; Madison, WI) Wisconsin package version 10.0 program,
'GAP'
(Devereux et al. (1984), Nucl. Acids Res. 12: 387) or other comparable
computer programs.
The preferred default parameters for the 'GAP' program includes: (1) the
weighted amino acid
comparison matrix of.Gribskov and Burgess (1986), Nucl. Acids Res. 14: 6745,
as described
by Schwartz and Dayhoff, eds., Atlas of Polypeptide Sequence and Structure,
National
Biomedical Research Foundation, pp. 353-358 (1979), or other comparable
comparison
matrices; (2) a penalty of 30 for each gap and an additional penalty of 1 for
each symbol in
each gap for amino acid sequences; (3) no penalty for end gaps; and (4) no
maximum penalty
for long gaps. Other programs used by those skilled in the art of sequence
comparison can
also be used.
"Transition phase" means a period of cell culture between a "growth phase" and
a
If
production phase." During transition phase, the medium and environmental
conditions are
typically shifted from those designed to maximize proliferation to those
designed to maximize
polypeptide production.
A "variable antibody immunoglobulin domain" is an immunoglobulin domain that
is identical or substantially similar to a VL or a VH domain of human or
animal origin.
The present invention is directed towards improved methods for culturing
mammalian
cells, which may have been genetically engineered to produce a particular
polypeptide. In
particular, the invention is directed towards culture methods that maximize
the production of
specific polypeptides. It is also directed towards methods of producing and
obtaining such
polypeptides from cultured mammalian cells_ Polypeptides are useful in a large
variety of
diagnostic, therapeutic, agricultural, nutritional, and research applications.
As shown by the experimental data reported herein, it has been discovered that
xanthine derivatives and hybrid polar compounds used separately or together
can dramatically
induce the production of recombinant polypeptide from CHO cell lines. In
particular,
addition of the xanthine derivative caffeine to the production phase of a cell
culture enhances
recombinant polypeptide production. The hybrid polar compound hexamethylene
bisacetamide is also shown to be an effective inducer of recombinant
polypeptide production.
Further, other inducers, such as, for example, alkanoic acids, can also be
added to either a
xanthine derivative, a hybrid polar compound, or both. Other methods, such as,
for example,
culturing the cells at temperatures from about 29 C to about 36 C, between
about 29 C and
35 C, and/or from about 30 C to about 33 C can also be used. Thus, the
invention relates to
-11-
CA 02762016 2011-12-09
/ 2 2 4 9 - 1 5 6
inducing increased production of a recombinant polypeptide from a cell grown
in culture by
exposing the cell to chemical inducers, including hybrid polar compounds
and/or xanthine
derivatives.
The methods of the invention include culturing marrunalian cells in medium
comprising a hybrid polar compound, for example, hexamethylene bisacetamide
(aMBA),
optionally at temperatures between about 29 C and 35 C or from about 30 C to
about 33 C.
Other embodiments of the invention encompass culture conditions in which an
alkanoic acid
and/or a xanthine, in addition to the hybrid polar compound, are added to the
culture medium.
In one embodiment, a xanthine and a hybrid polar compound and culture
temperatures
between about 29 C and 36 C are used. Another embodiment comprises the
addition of an
alkanoic acid and a hybrid polar compound plus culture temperatures between
about 29 C and
36 C. Still another embodiment comprises addition of a xanthine, an alkanoic
acid, and a
hybrid polar compound plus culture temperatures between about 29 C and 36 C.
Optionally,
cell culture using the methods of the invention can take place during a
production phase, as
distinguished from a growth phase. A growth phase can be distinguished from a
production
phase by, for example, a temperature shift and/or a change in medium such as,
for example,
the addition of one or more inducers.
In one aspect, the invention provides a method comprising growing in culture a
marrunalian cell that has been genetically engineered to produce a
polypeptide; and adding to
the culture a xanthine derivative. A genetically engineered cell may be a cell
that has been
transformed with a recombinant vector encoding the polypeptide. In addition,
the polypeptide
can be expressed under the control of a heterologous promoter such as, for
example, a CMV
promoter. Typically, the cell does not naturally express the polypeptide or
only naturally
expresses the polypeptide at very low levels (in the absence of genetic
engineering). In
another aspect, the invention provides a culture containing a cell genetically
engineered to
produce a polypeptide, a production medium, and the xanthine derivative.
In addition, the methods and compositions of the invention c,an be used in
combination with any other known or yet to be discovered methods of inducing
the
production of recombinant polypeptides. Such techniques include cold
temperature shift,
alkanoic acid additions (as described in U.S. Patent No. 5,705,364 to
Etcheverry et al),
MAE, and DMSO, to name just a few examples, as well as
any yet to be described and/or discovered induction techniques. As used
herein, "inducing"
polypeptide production or "induction" refers to culturing cells under a set of
conditions
designed to maximize the total amount of a desired polypeptide made by the
cells. An
3,5 "inducer" is an agent that, when added to culture medium, can increase
the production of a
desired polypeptide in at least some cell lines. C,orribining the addition of
xanthine
derivatives with other protein induction techniques can have a synergistic
effect on
-12-
CA 02762016 2011-12-09
WO 03/083066
PCT/US03/09266
polypeptide induction, allowing for lower additions of xanthine derivatives
and/or lower
additions of other inducing agents and/or more conservative temperature
shifts. The other
methods of induction can take place at around the same time as xanthine
addition, and/or
before and/or after xanthine addition. For example, one can shift the
temperature of the
culture at day 0, and then add a xanthine derivative and/or a hybrid polar
compound, and
optionally other chemical inducers, later, e.g. one to several hours or days
later. Such a
protocol allows some additional growth of a seeded culture before full
induction.
Furthermore, multiple additions of xanthine derivatives and/or hybrid polar
compounds can
be added to the culture during the production phase, separated by about 12,
24, 48, and/or 72
hours or more, with or without additions of other inducing agents or changes
in culture
conditions. For example, an inducer can be added at day 0 and again at day 4.
Alternatively,
an inducer can be added for the first time one, two, three, or four days after
a temperature
shift.
In one aspect, the invention entails performing a low temperature shift
(shifting the
temperature of the medium from the optimal growth temperature, usually around
37 C, to a
lower temperature, usually from about 29 C to about 36 C, and optionally about
30 C to
about 34 C at the time of, before, and/or after adding the xanthine derivative
or the hybrid
polar compound. Alternatively, or in addition, an alkanoic acid or salt
thereof (e.g. sodium
butyrate) can be added to the culture at around the same time as the xanthine
derivative and/or
hybrid polar compound is added. Alkanoic acid can be added at concentrations
typically used
for induction, or even at lower concentrations than would typically be used.
Thus, by
manipulating both transcriptional and post-transcriptional controls, higher
levels of
productivity may be achieved.
There are individual differences between cell lines in the effectiveness of
various
inducers. For example, although sodium butyrate is a widely-used inducer, it
can have no
effect or an adverse effect on polypeptide production in some cell lines. See
Table 5.
Different inducers or different concentrations of the same inducers may be
appropriate for
different cell lines. Furthermore, different temperatures may be appropriate
for different cell
lines. In spite of this variability, some inducers, such as, for example,
caffeine,
hexamethylene bisacetamide, and sodium butyrate, can be useful in a wide
variety, though
perhaps not all, cell lines.
Generally, xanthine derivatives have the structure illustrated below.
-13-
CA 02762016 2011-12-09
/2249-156
0
XN
N'11
0).N
X, Y, and Z can be independently selected from a straight or branched chain
alkyl radical
having from 1 to 12 carbons, a straight or branched chain allcynyl radical
having from 1 to 12
carbons (including a propynyl radical), a straight or branched chain acyl
radical having from 1
to 12 carbons, a straight or branched chain radical. with the structure -R-
acyl containing from
1 to 12 carbons where R is a saturated or unsaturated aliphatic group, a
straight or branched
chain allenyl radical having from I to 12 carbons, a straight or branched
chain hydroxyalk-yl
radical having from 1 to 12 carbons, a straight or branched chain
hydroxyallenyl radical
having from 1 to 12 carbons, a straight or branched chain radical with the
structure -allenyl-
.
halogen having from 1 to 12 carbons, a cyclohexyl radical, and hydrogen. In
some
embodiments, at least one of X, Y and Z is a methyl group_ In some
embodiments, each of X
and Y independently represents a hydrogen atom, a linear or branched alkyl
radical having up
to 5 carbon atoms, an ally( radical, a propynyl radical or a cyclohexyl
radical, with the proviso
that X and Y do not simultaneously represent a hydrogen atom, and Z represents
a hydrogen,
methyl, ethyl, hydroxymethyl, hydroxyethyl or heterocyclo radical. These
xanthines can be
obtained using conventional processes and/or purchased. A number of different
xanthine
derivatives that can be used are described in Beavo et al. (1970), Molec.
Pharm. 6:597-603,
Illustrative examples of xanthine derivatives that can be used in the methods
and
compositions of the invention include, but are not limited to, caffeine (1,3,7-
trimethylxanthine), theophylline (1,3-dimethylxanthine), theobroinine (3,7-
dimethylxanthine), 3-isobuty1-1-methylxanthine, 3-buty1-1-methylxanthine,
1,3,7-
triethylxanthine, 3-cyclohexy1-1-ethylxanthine, 3-ethy1-1-propyny1xanthine, 3-
ethyl-I-
pentylxanthine, pentoxifylline, and aminophylline. Aminophyltine is
theophylline compound
with 1,2-ethylenediamine (2:1) dihydrate. Generally, the xanthine derivative
is added at a
concentration in the culture from about 0.0005 to about 25 millimolar,
optionally from about
0.001 to about 10 millimolar, from about 0.005 to about 5 millimolar, or from
about 0.01 to
-14-
CA 02762016 2011-12-09
7 22 4 9-15 6
about 3 millimolar. The optimal concentration of the xanthine derivative will
vary depending
upon its activity and the cell line, and can be determined by those skilled in
the art using the
guidance provided herein_
The xanthine derivative can be dissolved in any appropriate solvent. For
example, 3-
isobutyl-l-methylxanthine (ISX) can be dissolved in water, but must to be
heated to almost
the boiling point_ Alternatively, LBX can be dissolved in the solvents DMSO
(dimethylsulfoxide), DMF (dimethylformamide), or DMA (dirnethylacetamide).
113X can
also be easily dissolved as a 100 millirnolar stock solution in 0.5 M NaOH.
Dilutions of this
stock solution can be added to the induction media as it is being prepared
(pre-sterile) and the
effects of the NaOH should be inconsequential since base must often be added
to raise the pH
of the medium to 7Ø
Many of the xanthine derivatives for use in the invention are cAMP
phosphodiesterase inhibitors. Thus, in addition to using xanthine derivatives
that are cAMP
phosphodiesterase inhibitors, it is believed that cAlv1P phosphodiesterase
inhibitors that are
not xanthine derivatives could also be used to induce. polypeptide production
in alternative
methods of the invention. Examples of such inducers include but are not
limited to
imidazopyrimidine, pyrazolopyridine, etazotate, pyiazoloquinoline, and
triazoloquinazoline
(Pflugers Archly 407: S31, 1936). Other examples cAMP phosphodiesterase
inhibitors can be
found in US Patent No. RE37,234.
The hybrid polar compounds, the use of which is encompassed by the invention,
can
have two polar groups separated by a non-polar carbon chain, such as those
described in
Richon et al_ (1998), Proc. Natl. Acad. Sci. 95: 3003-07, Marks et al_ (1994),
Proc. Natl.
Acad. Sci. 91: 10251-54, US Patent Nos. 5,055,608 and 6,087,367. The hybrid
polar
compounds of the invention may have the property of inducing one or more
changes
characteristic of a terminally differentiated state of the host cells. These
compounds include
those with the structure:
RI \
N -(CH2L---N
R2
R1 and R2 can be the same as or different from each other. R1 and R2 can each
be a carbonyl
group to which a hydrogen atom, a hydroxyl group, a substituted or
unsubstituted, branched
or unbranched alkyl, alkenyl, cycloalkyl, aryl, alkyrd, allenyl, allyl,
alkyloxy, aryloxy,
arylallcyloxy, which contains 12 or fewer carbon atoms, or pyridine group, may
also be
attached. The "n" can be an integer from about four to about eight.
Specifically, HMBA is
included within this class of hybrid polar compounds, and its structure is:
-15-
CA 02762016 2011-12-09
WO 03/083066
PCT/US03/09266
O
C /H
CH
CH3 N ¨(CH2)6¨ N
/ 3
O
The present invention further encompasses the use of hybrid polar compounds
with
the following structure:
R3\
C ¨(CH2).¨ C
R4
R3 and R4 can be the same as or different from each other. When R3 and R4 are
the same,
each is a substituted or unsubstituted arylamino, cycloallcylamino,
pyridineamino, piperidino,
9-purine-6-amine, or thiozoleamino group containing 12 or fewer carbon atoms.
Where R3
and R4 are different, R3 is equal to R6, where R5 and R6 are the same as or
different
from each other and are a hydrogen atom, a hydroxyl group, a substituted or
unsubstituted,
branched or unbranched alkyl, alkenyl, cycloalkyl, aryl, alkynl, allenyl,
allyl, allcyloxy,
aryloxy, arylalk-yloxy, or pyridine group, which contains 12 or fewer carbon
atoms, or R5 and
R6 bond together to form a piperidine group, and R4 is a hydroxylarnino,
hydroxyl, amino,
alkylamino, diallcylamino, or alkyloxy group, which contains 12 or fewer
carbon atoms. The
"n" is an integer from about four to about eight.
The invention further embraces the use of all compounds disclosed in US Patent
No.
6,087,367, US Patent No. 5,055,608, Richon et al., supra, and Marks et al.,
supra. In some of
these, the apolar carbon chain may be shorter than 4 carbons and longer than 8
carbons, and it
may be interrupted by aromatic groups, apolar groups, and/or polar groups.
If LIMB A is used, it can be added at concentrations from about 0.1 millimolar
to
about 20 millimolar, optionally, between about 0.1 millimolar and about 5
millimolar. Other
hybrid polar compounds may be active at lower or higher concentrations. The
optimal
concentration for a particular hybrid polar compound will vary depending on
its activity and
the cell line in which it is used and can be determined by one of skill in the
art using routine
methods and the guidance provided herein. For example, compounds such as
suberoylanilide
hydroxamic acid or m-carboxycinnarnic acid bishydroxarnide can be used at
concentrations
about one thousand fold lower than those required for 1-11MBA, from about 0.01
micromolar to
about 10 micromolar. See Richon et al., supra. Concentrations of hybrid polar
compounds
required to induce cell differentiation as disclosed in Marks et al. (supra)
and Richon et al.
(supra) can be used as a guide for determining the concentration of a hybrid
polar compound
-16-
CA 02762016 2011-12-09
6-Th
1 WO 03/()83066
PCT/1JS03/09266
required to enhance polypeptide production. Deterrnination of the
concentration needed for a
specific hybrid polar compound used in a specific cell line can be done using
routine methods
as described herein and the guidance provided in Richon et al. (supra) and
Marks et al.
(supra).
The alkanoic acids for use in the invention include the selected acid and/or a
corresponding salt. The acids include straight or branched chain, saturated or
unsaturated
alkanoic acids or salts thereof. An alkanoic acid generally comprises from one
to ten carbon
atoms. Examples of alkanoic acids contemplated by the invention are pentanoic
acid, butyric
acid, isobutyric acid, propionic acid, and acetic acid. Concentrations for
alkanoic acids
encompassed by the invention range from about 0.05 millimolar to about 10
millimolar,
optionally from about 0.1 millimolar to about 2 millimolar. Appropriate
concentrations of
alkanoic acids will vary depending upon their activity and the cell line and
can be determined
by one of skill in the art using routine methods and the guidance provided
herein. An
exemplary salt of butyric acid is sodium butyrate. Appropriate salts of the
alkanoic acids
described above include those comprising sodium, potassium, or ammonium
groups, among
others.
Particularly preferred polypeptides for expression are polypeptide-based
drugs, also
known as biologics. Preferably, the polypeptides are secreted as extracellular
products. The
polypeptide being produced can comprise part or all of a polypeptide that is
identical or
substantially similar to a naturally-occurring polypeptide, and/or it may, or
may not, be a
recombinant fusion polypeptide. Optionally, the polypeptide may be a human
polypeptide, a
fragment thereof, or a substantially similar polypeptide that is at least 15
amino acids in
length. It may comprise a non-antibody polypeptide and/or an antibody_ It may
be produced
intracellularly or be secreted into the culture medium from which it can be
recovered. It may
or may not be a soluble polypeptide.
The polypeptide being produced cart comprise part or all of a polypeptide that
is
identical or substantially similar to a naturally-occurring polypeptide,
and/or it may, or may
not, be a recombinant fusion polypeptide. It may comprise a non-antibody
polypeptide and/or
an antibody. It may be produced intracellularly or be secreted into the
culture medium from
which it can be recovered.
The invention can be used to induce the production of just about any
polypeptide, and
is particularly advantageous for polypeptides whose expression is under the
control of a
strong promoter, such as for example, a viral promoter, and/or polypeptides
that are encoded
on a message that has an adenoviral tripartite leader element. Examples of
useful expression
vectors that can be used to produce proteins are disclosed in International
Application
WO 01/27299 and in McMahan et al., (1991), EMBO J. 10: 2821, which describes
the
-17-
CA 02762016 2011-12-09
72249-156
pDC409 vector. A protein is generally understood to be a polypeptide of at
least about 10
amino acids, optionally about 25, 75, or 100 amino acids.
Generally, the methods of the invention are useful for inducing the production
of
recombinant polypeptides. Some polypeptides that can be produced with the
methods of the
invention include polypeptides comprising amino acid sequences identical to or
substantially
similar to all or part of one of the following polypeptides: a f113 ligand (as
described in
International Application WO 94/28391), a CD40 ligand
(as described in US Patent No. 6,087,329), erythropoeitin,
thrombopoeitin, calcitonin, leptin, angiopoietin-2 (as described by
Maisonpierre et al_
(1997), Science 277(5322): 55-60), Fas ligand, ligand for
receptor activator of NF-kappa B (RANKL, as described in International
Application
WO 01/36637), tumor necrosis factor (TNF)-related
apoptosis-inducing ligand (TRAIL, as described in International Application WO
97/01633),
thymic stroma-derived lymphopoietin, granulocyte colony
stimulating factor, granulocyte-macrophage colony stimulating factor (GM-CSF,
as described
in Australian Patent No. 588819 ), mast cell growth factor,
stem cell growth factor (described in e.g. US Patent No.6,204,363),
epidermal growth factor, keratinocyte growth factor, megakaryote growth and
development factor, RANTES, growth hormone, insulin, insulinotropin, insulin-
like growth
factors, parathyroid hormone, interferons including a interferons, y
interferon, and consensus
interferons (such as those described in US Patent Nos. 4,695,623 and
4,897471),
=
nerve growth factor, brain-derived neurotrophic factor,
synaptotagmin-like proteins (SLP 1-5), neurotrophin-3, glucdgon, interleuldns
1 through 18,
colony stimulating factors, lymphotoxin-13, tumor necrosis factor (TNF),
leukemia inhibitory
factor, oncostatin-M, and various ligands for cell surface molecules ELK and
Hek (such as the
ligands for eph-related lcinases or LERKS). Descriptions of polypeptides that
can be
produced according to the inventive methods may be found in, for example,
Human
Cytokines: Handbook for Basic and Clinical Research, Vol. II (Aggarwal and
Gutterman, eds.
Blackwell Sciences, Cambridge, MA, 1998); Growth Factors: A Practical Approach
(McKay
and Leigh, eds., Oxford University Press Inc., New York, 1993); and The C
tolcine Handbook
(A.W. Thompson, ed., Academic Press, San Diego, CA, 1991).
Other polypeptides that can be produced using the methods of the invention
include
polypeptides comprising all or part of the amino acid sequence of a receptor
for any of the
above-mentioned polypeptides, an antagonist to such a receptor or any of the
above-
mentioned polypeptides, and/or polypeptides substantially similar to such
receptors or
antagonists. These receptors and antagonists include: both forms of tumor
necrosis factor
-18-
CA 02762016 2011-12-09
/2249-156
=
= receptor (TNFR, referred to as p55 and p75, as described in US Patent No.
5,395,760 and US
Patent No. 5,610,279), Interleukin-1
(IL-1) receptors (types I and II; described in EP Patent No. 0 460 846, US
Patent
No. 4,968,607, and US Patent No. 5,767,064),
IL-1 receptor antagonists (such as those described in US Patent No.
6,337,072),
IL-1 antagonists or inhibitors (such as those described in
US Patent Nos. 5,981,713, 6,096,728, and 5,075,222)
IL-2 receptors, IL-4 receptors (as described in EP Patent No. 0 367 566 and US
Patent No. 5,856,296), 1L-15 receptors, IL-17
receptors, IL-18 receptors, granulocyte-macrophage colony stimulating factor
receptor,
granulocyte colony stimulating factor receptor, receptors for oncostatin-M and
leukemia
inhibitory factor, receptor activator of NF-kappa B (RANK, described in WO
01/36637 and
US Patent No. 6,271,349), osteoprotegerin
(described in e.g. US. Patent No. 6,015,938), receptors for TRAIL
(including TRAIL receptors 1, 2, 3, and 4), and receptors that comprise death
domains, such
as Fas or Apoptosis-Inducing-Receptor (AIR).
Other polypeptides that can be produced using the process of the inve,ntion
include
polypeptides comprising all or part of the amino acid sequences of
differentiation antigens
(referred to as CD polypeptides) or their ligands or polypeptides
substantially similar to either
of these. Such antigens are disclosed in Leukocyte Typing VI (Proceedings of
the VIM
Intentational Workshop and Conference, Kishimoto, Kikutani et al., eds., Kobe,
Japan. 1996).
Similar CD polypeptides are disclosed in subsequent
worlcshops. Examples of such antigens include CD22, CD27, CD30, CD39, CD40,
and
ligands thereto (CD27 ligand, CD30 ligand, etc.). Several of the CD antigens
are members of
the TNF receptor family, which also includes 41BB and 0X40. The ligands are
often
members of the TNF family, as are 41BB ligand and 0X40 ligand. Accordingly,
members of
the TNF and TNFR families can also be purified using the present invention.
Enzymatically active polypeptides or their ligands can also be produced
according to
the methods of the invention. Examples include polypeptides comprising all or
part of one of
the following polypeptides or their ligands or a polypeptide substantially
similar to one of
these: metalloproteinase-disintegrin family members, various lcinases,
glucocerebrosidase,
superoxide dismutase, tissue plasminogen activator, Factor V111, Factor IX,
apolipoprotein E,
apolipoprotein A-I, globins, an IL-2 antagonist, alpha-1 antitrypsin, TNF-
alpha Converting
Enzyme, ligands for any of the above-mentioned enzymes, and numerous other
enzymes and
their ligands.
The methods of the invention can also be used to produce antibodies or
portions
thereof and chimeric antibodies, i.e. antibodies having human constant
antibody
-19-
CA 02762016 2011-12-09
7 2 2 4 9-1 5 6
immunoglobulin domains coupled to one or more murine variable antibody
immunoglobulin
domain, fragments thereof, or substantially similar proteins_ The method of
the invention
may also be used to produce conjugates comprising an antibody and a cytotoxic
or
luminescent substance. Such substances include: maytansine derivatives (such
as DM1);
enterotoxins (such as a Staphlyococcal enterotoxin); iodine isotopes (such as
iodine-125);
technium isotopes (such as Tc-99m); cyanine fluorochromes (such as Cy5.5.18);
and
ribosome-inactivating polypeptides (such as bouganin, gelonin, or saporin-S6).
The invention
can also be used to produce chimeric proteins selected in vitro to bind to a
specific target
protein and modify its activity such as those described in International
Applications
W0,01/83525 and WO 00124782.. Examples of
antibodies, in vitro-selected chimeric proteins, or antibody/cytotoxin or
antibody/luminophore
conjugates that cart be produced by the methods of the invention include those
that recognize
any one or a combination of polypeptides including, but not limited to, the
above-mentioned
proteins and/or the following antigens: CD2, CD3, CD4, CD8, CD I la, CD14,
CD18, CD20,
CD22, C1)23, CD25, CD33, CD40, CD14, CD52, CD80 (B7.1), CD86 (B7.2), CD147, IL-
la,
IL-1(3, IL-2, 1L-3, LL-7, 1L4, IL-5, 1L-8, IL-10, 1L-2 receptor, 1T.-4
receptor, 1L-6 receptor,
1L-13 receptor, IL-18 receptor subunits, PDGF-I3 and analogs thereof (such as
those described
in US Patent Nos. 5,272,064 and 5,149,792), VEGF, TGF, TGF-132, TGF-131, EGF
receptor
(including those described in US Patent No. 6,235,883 B1) VEGF
receptor, hepatocyte growth factor, osteoprotegerin ligand, interferon gamma,
B lymphocyte
stimulator (BlyS, also blown as BAFF, THANK, TALL-1, and zTNF4; see Do and
Chen-
Kiang (2002), Cytokine Growth Factor Rev_ 13(1): 19-25), C5 complement, IgE,
turnor
antigen CA125, tumor antigen MUCI, PEM antigen, LCG (which is a gene product
that is
expressed in association with lung cancer), HER-2, a tumor-associated
glycoprotein TAG-72,
the SK-1 antigen, tumor-associated epitopes that are present in elevated
levels in the sera of
patients with colon ancUor pancreatic cancer, cancer-associated epitopes or
polypeptides
expressed on breast, colon, squamous cell, prostate, pancreatic, lung, and/or
kidney cancer
cells and/or on melanoma, glioma, or neuroblastoma cells, the necrotic core of
a tumor,
integrin alpha 4 beta 7, the integrin VLA-4, B2 integrins, TRAIL receptors I,
2, 3, and 4,
RANK, RANK ligand, TNF-a, the adhesion molecule VAP-1, epithelial cell
adhesion
molecule (EpCAM), intercellular adhesion molecule-3 (ICA1v1-3), leukointegrin
adhesin, the
platelet glycoprotein gp 11b/Ifla, cardiac myosin heavy chain, parathyroid
hormone, rNAPc2
(which is an inhibitor of factor Vna-tissue factor), MHC I, carcinoembryonic
antigen (CEA),
alpha-fetoprotein (AFP), tumor necrosis factor (TNF), CTLA-4 (which is a
cytotoxic T
lymphocyte-associated antigen), Fc-y-1 receptor, HLA-DR 10 beta, HLA-DR
antigen, L-
selectin, Respiratory Syncitial Virus, human inununodeficiency virus (HIV),
hepatitis B virus
(HBV), Streptococcus inutans, and Staphlvcoccus entreus.
-20-
CA 02762016 2011-12-09
32249-156
The invention may also be used to produce all or part of an anti-idiotypic
antibody or
a substantially similar polypeptide, including anti-idiotypic antibodies
against: an antibody
targeted to the tumor antigen gp72; an antibody against the ganglioside GD3;
an antibody
against the ganglioside GD2; or antibodies substantially similar to these.
The methods of the invention can also be used to produce recombinant fusion
polypeptides comprising any of the above-mentioned polypeptides. For example,
recombinant fusion polypeptides comprising one of the above-mentioned
polypeptides plus a
multimerization domain, such as a leucine zipper, a coiled coil, an Fc portion
of an antibody,
or a substantially similar protein, can be produced using the methods of the
invention. See
e.g. W094/10308; Lovejoy et al. (1993), Science 259:1238-1293; Harbury et al.
(1993),
'Science 262:1401-05; Harbury et al. (1994), Nature 371:80-83; Wakansson et
al.(1999),
Structure 7:255-64. Specifically included among
such recombinant fusion polypeptides are polypeptides in which a portion of
TNFR or RANK
is fused to an Fc portion of an antibody (TNFR:Fc or RANK:Fc). TNFR:Fc
comprises the Fc
IS portion of an antibody fused to an extrac,ellular domain of TN141{,
which includes amino acid
sequences substantially similar to amino acids 1-163, 1-185, or 1-235 of
Figure 2A of US
Patent No. 5,395, 760. RAN L:Fc is described in
International Application WO 01/36637,.
Preferably, the polypeptides are expressed under the control of a heterologous
control
element such as, for example, a promoter that does not in nature direct the
production of that
polypeptide. For example, the promoter can be a strong viral promoter (e.g.,
CMV, SV40)
that directs the expression of a mammalian polypeptide. The host cell may or
may not
normally produce the polypeptide_ For example, the host cell can be a CHO cell
that has been
genetically engineered to produce a human polypeptide, meaning that nucleic
acid encoding
the human polypeptide has been introduced into the CHO cell. Alternatively,
the host cell can
be a human cell that has been genetically engineered to produce increased
levels of a human
polypeptide normally present only at very low levels (e.g., by replacing the
endogenous
promoter with a strong viral promoter). For the production of recombinant
polypeptides, an
expression vector encoding the recombinant polypeptide can be transferred, for
example by
transfection or viral infection, into a substantially homogeneous culture of
host cells. The
expression vector, which can be constructed using the methods of genetic
engineering, can
include nucleic acids encoding the polypeptide of interest operably linked to
suitable
regulatory sequences.
The regulatory sequences are typically derived from mammalian, microbial,
viral,
and/or insect genes. Examples of regulatory sequences include transcriptional
promoters,
operators, and enhancers, a ribosomal binding site (see e.g. Kozak (1991), J.
Biol. Chem.
266:19867-19870), appropriate sequences to control transcriptional and
translational initiation
-21-
CA 02762016 2011-12-09
WO 03/083066
PCT/US03/09266
and termination, polyadenylation signals (see e.g. McLauchlan et al. (1988),
Nucleic Acids
Res. 16:5323-33), and matrix and scaffold attachment sites (see Phi-Van et al.
(1988), Mol.
Cell. Biol. 10:2302-07; Stief et al. (1989), Nature 341:342-35; Bonifer et al.
(1990), EMBO
J. 9:2843-38). Nucleotide sequences are operably linked when the regulatory
sequence
functionally relates to the polypeptide coding sequence. Thus, a promoter
nucleotide
sequence is operably linked to a polypeptide coding sequence if the promoter
nucleotide
sequence controls the transcription of the coding sequence. A gene encoding a
selectable
marker is generally incorporated into the expression vector to facilitate the
identification of
recombinant cells.
Transcriptional and translational control sequences for mammalian host cell
-- -
expression vectors can be excised from viral genomes. Commonly used promoter
and
= enhancer sequences are derived from polyoma virus, adenovirus 2, simian
virus 40 (SV40),
and human cytomegalovirus (CMV). For example, the human GMAT promoter/enhancer
of
immediate early gene 1 may be used. See e.g. Patterson et al. (1994), Applied
Microbiol.
Biotechnol. 40:691-98. DNA sequences derived from the SV40 viral genome, for
example,
SV40 origin, early and late promoter, enhancer, splice, and polyadenylation
sites can be used
to provide other genetic elements for expression of a structural gene sequence
in a
mammalian host cell. Viral early and late promoters are particularly useful
because both are
easily obtained from a viral genome as a fragment, which can also contain a
viral origin of
replication (Fiers et al. (1978), Nature 273:113; Kaufman (1990), Meth. in
Enzymol.
185:487-511). Smaller or larger SV40 fragments can also be used, provided the
= approximately 250 bp sequence extending from the Hind ILI site toward the
Bgl 1 site located
in the SV40 viral origin of replication site is included.
In addition, a sequence encoding an appropriate native or heterologous signal
peptide
(leader sequence) can be incorporated into the expression vector, to promote
extracellular
secretion of the recombinant polypeptide. The signal peptide wilt be cleaved
from the
recombinant polypeptide upon secretion from the cell. The choice of signal
peptide or leader
depends on the type of host cells in which the recombinant polypeptide is to
be produced.
Examples of signal peptides that are functional in manunalian host cells
include the signal
sequence for interleukin-7 (IL-7) described in US Patent No. 4,965,195, the
signal sequence
for interleukin-2 receptor described in Cosman et al. (1984), Nature 312:768;
the interleukin-4
receptor signal peptide described in EP Patent No. 367,566; the type I
interleukin-1 receptor
signal peptide described in US Patent No. 4,968,607; and the type II
interleukin-1 receptor
signal peptide described in EP Patent No. 0 460 846.
Established methods for introducing DNA into marrunalian cells have been
described.
Kaufman, R.J., Large Scale Mammalian Cell Culture, 1990, pp. 15-69. Additional
protocols
using commercially available reagents, such as the cationic lipid reagents
= -22-
CA 02762016 2011-12-09
(Th
WO 03/033066
PCT/US03/09266
L1POFF,C1 L1POFECTAMINElm-2000, or LIPOFECf A/v11NETm-PLUS (which
can be purchased from Invitrogen), can be used to transfect cells. Feigner et
al. (1987)., Proc.
Natl. Acad. Sci. USA 84:7413-7417. In addition, electroporation or bombardment
with
microprojectiles coated with nucleic acids can be used to transfect mammalian
cells using
procedures, such as those in Sambrook et al., Molecular Cloning: A Laboratory
Manual, 2nd
ed. Vol. 1-3, Cold Spring Harbor Laboratory Press (1989) and Fitzpatrick-
McElligott (1992),
Biotechnology (NY) 10(9):1036-40. Selection of stable transformants can be
performed
using methods known in the art, such as, for example, resistance to cytotoxic
drugs. Kaufman
et al. ((1990), Meth. in Enzymology 185:487-511), describes several selection
schemes, such
as dihydrofolate reductase (DHFR) resistance. A suitable host strain for DHFR
selection can
be CHO strain DX-B11, which is deficient in DHFR. Urlaub and Chasin (1980),
Proc. Natl.
Acad. Sci. USA 77:4216-4220. A plasmid expressing the DHFR cDNA can be
introduced
into strain DX-B 11, and only cells that contain the plasmid can grow in the
appropriate
selective media. Other examples of selectable markers that can be incorporated
into an
expression vector include cDNAs conferring resistance to antibiotics, such as
G418 and
hygromycin B. Cells harboring the vector can be selected on the basis of
resistance to these
compounds.
Additional control sequences shown to improve expression of heterologous genes
from mammalian expression vectors include such elements as the expression
augmenting
sequence element (EASE) derived from CHO cells (Morris et al., in Animal Cell
Technology,
pp. 529-534 (1997); US Patent Nos. 6,312,951 Bl, 6,027,915, and 6,309,841 B I)
and the
tripartite leader (TPL) and VA gene RNAs from Adenovirus 2 (Gingeras et al.
(1982), J. Biol.
Chem. 257:13475-13491). The internal ribosome entry site (IRES) sequences of
viral origin
allows dicistronic iriRNAs to be translated efficiently (Oh and Sarnow (1993),
Current
Opinion in Genetics and Developinein 3:295-300; Ramesh et al. (1996). Nucleic
Acids
Research 24:2697-2700). Expression of a heterologous cDNA as part of a
dicistronic mRNA
followed by the gene for a selectable marker (e.g. DI-]FR) has been shown to
improve
transfectability of the host and expression of the heterologous cDNA (Kaufman
et al_ (1990),
Methods in Enzymol. 185:487-511). Exemplary expression vectors that employ
dicistronic
mRNAs are pTR-DC/GFP described by Mosser et al., Biotechniques 22:150-161
(1997), and
p2A5I described by Morris et al., in Aninzal Cell Technology, pp. 529-534
(1997).
A useful high expression vector, pCAVNOT, has been described by Mosley et al.
((1989), Cell 59:335-348). Other expression vectors for use in mammalian host
cells can be
constructed as disclosed by Okayama and Berg ((1983), Mol. Cell. Biol. 3:280).
A useful
system for stable high level expression of manunalian cDNAs in C127 murine
mammary
epithelial cells can be constructed substantially as described by Cosman et
al. ((1986), Mol.
Immunol. 23:935). A useful high expression vector, PMLSV N I/N4, described by
-23-
CA 02762016 2011-12-09
72249-156
=
Cosman et at. ((1984), Nature 312:768), has been deposited as ATCC 39890.
Additional
useful-mammalian expression vectors are described in EP Patent No.-A-0 367 566
and
WO 01/27299 Al. The vectors can be derived from retroviruses. In place of the
native signal
sequence, a heterologous signal sequence can be added, such as one of the
following
sequences: the signal sequence for 1L-7 described in US Patent No. 4,965,195;
the signal
sequence for IL-2 receptor described in Cosman et al. (Nature 312:768 (1984));
the IL-4
signal_ peptide described in EP Patent No. 0 367 566; the type I LL-1 receptor
signal peptide
described in US Patent No. 4,968,607; and the type LE IL-1 receptor signal
peptide described
in EP Patent No. 0 460 846.
The polypeptides can be produced recombinantly in eukaryotic cells and are
preferably secreted by host cells adapted to grow in cell culture. Optionally,
host cells for use
in the invention are preferably mammalian cells. The cells can be also
genetically engineered
to express a gene of interest, can be mammalian production cells adapted to
grow in cell
culture, and/or can be homogenous cell lines. Examples of such cells commonly
used in the
industry are VERO, MIK, HeLa, CV1 (including Cos),MDC.K, 9.93, 3T3, myeloma
cell lines
(e.g., NSO, NS1), PC12, WI38 cells, and Chinese hamster ovary (CHO) cells,
which are
widely used for the production of several cotnplex recombinant polypeptides,
e.g. cytokines,
clotting factors, and antibodies (Brasel et al_ (1996), Blood 88:2004-2012;
Kaufman et al.
(1988), J.B iol Chem 263:6352-6362; McKinnon et al_ (1991), J Mol Endocrinol
6:231-239;
Wood et al. (1990), J. Immunol. 145:3011-3016). The dihydrofolate reductase
(DHFR)-
deficient mutant cell lines (Urlaub et al. (1980), Proc Natl Acad Sci USA 77:
4216-4220),
DXB11 and DG-44, are desirable CHO host cell lines
because the efficient DHFR selectable and amplifiable gene expression system
allows high
level recombinant polypeptide expression in these cells (Kaufman R.I. (1990),
Meth Enzymol
185:537-566.). In addition, these cells are easy to
manipulate as adherent or suspension cultures and exhibit relatively good
genetic stability.
CHO cells and recombinant polypeptides expressed in then-t have been
extensively
characterized and have been approved for use in clinical commercial
manufacturing by
regulatory agencies. The methods of the invention can also be practiced using
hybridoma cell
lines that produce an antibody. Methods for making hybridoma lines are well
known in the
art. See e.g. Berzofsky et al. in Paul, ed., Fundamental Mununology, Second
Edition,
pp.315-356, at 347-350, Raven Press Ltd., New York (1989). Cell lines derived
from the
above-mentioned lines are also suita6le for practicing the invention.
According to the present invention, a man-unalian host cell is cultured under
conditions that promote the production of the polypeptide of interest, which
can be an
- antibody or a recombinant polypeptide. Basal cell culture medium-
formulations are well
known in the art. To these basal culture medium formulations the skilled
artisan will add
-24-
CA 02762016 2011-12-09
WO 03/083066
PCT/US03/09266
components such as amino acids, salts, sugars, vitamins, hormones, growth
factors, buffers,
antibiotics, lipids, trace elements and the like, depending on the
requirements of the host cells
to be cultured. The culture medium may or may not contain serurn and/or
protein. Various
tissue culture media, including serum-free and/or defined culture media, are
commercially
available for cell culture. Tissue culture media is defined, for purposes of
the invention, as a
media suitable for growth of animal cells, and preferably mammalian cells, in
in vitro cell
culture. Typically, tissue culture media contains a buffer, salts, energy
source, amino acids,
vitamins and trace essential elements. Any media capable of supporting growth
of the
appropriate eukaryotic cell in culture can be used; the invention is broadly
applicable to
eukaryotic cells in culture, particularly mammalian cells, and the choice of
media is not
crucial to the invention. Tissue culture media suitable for use in the
invention are
commercially available from, e.g., ATCC (Manassas, VA). For example, any one
or
combination of the following media can be used: RPM1-1640 Medium, R_PlvII-1641
Medium,
Dulbecco's Modified Eagle's Medium (DMEM), Minimum Essential Medium Eagle, F-
12K
Medium, Ham's F12 Medium, Iscove's Modified Dulbecco's Medium, McCoy's 5A
Medium,
Leibovitz's L-15 Medium, and serum-free media such as EX-CELLTm 300 Series
(available
from JRH Biosciences, Lenexa, Kansas, USA), among others, which can be
obtained from the
American Type Culture Collection or TRH Biosciences, as well as other vendors.
When
defined medium that is serum-free and/or peptone-free is used, the medium is
usually highly
enriched for amino acids and trace elements. See, for example, US Patent Nos.
5,122,469 to
Mather et al. and 5.633,162 to Keen et al.
In the methods and compositions of the invention, cells can be grown in serum-
free,
protein-free, growth factor-free, and/or peptone-free media. The term "serum-
free" as applied
to media includes any mammalian cell culture medium that does not contain
serum, such as
fetal bovine serum. The term "insulin-free" as applied to media includes any
medium to
which no exogenous insulin has been added. By exogenous is meant, in this
context, other
than that produced by the culturing of the cells themselves. The term "IGF-1-
free" as applied
to media includes any medium to which no exogenous Insulin-like growth factor-
1 (IGF-1) or
analog (such as, for example, LongR3, [A1a31], or [Leu241[A1a31] IGF-1 analogs
available
from GroPep Ltd. of Thebarton, South Australia) has been added. The term
"growth-factor
free" as applied to media includes any medium to which no exogenous growth
factor (e.g.,
insulin, IGF-1) has been added. The term "protein-free" as applied to media
includes medium
free from exogenously added protein, such as, for example, transferrin and the
protein growth
factors IGF-1 and insulin. Protein-free media may or may not have peptones.
The term
"peptone-free" as applied to media includes any medium to which no exogenous
protein
hydrolysates have been added such as, for example, animal and/or plant protein
hydrolysates.
Eliminating peptone from media has the advantages of reducing lot to lot
variability and
CA 02762016 2011-12-09
(71
WO 03/083066
PCT/US03/09266
enhancing processing such as filtration. Chemically defined media are media in
which every
component is defined and obtained from a pure source, preferably a non-animal
source.
The skilled artisan may also choose to use one of the.many individualized
media
formulations that have been developed to maximize cell growth, cell viability,
and/or
recombinant polypeptide production in a particular cultured host cell. The
methods according
to the current invention may be used in combination with commercially
available cell culture
media or with a cell culture medium that has been individually formulated for
use with a
particular cell line. For example, an enriched medium that could support
increased
polypeptide production may comprise a mixture of two or more courunercial
media, such as,
for instance, DMEM and Ham's F12 media combined in ratios such as, for
example, 1:1, 1:2,
1:3, 1:4, 1:5, 1:6, 1:7, 1:8, or even up to 1:15 or higher. Alternatively or
in addition, a
medium can be enriched by the addition of nutrients, such as amino acids or
peptone, and/or a
medium (or most of its components with the exceptions noted below) can be used
at greater
than its usual, recommended concentration, for example at 2X, 3X, 4X, 5X, 6X,
7X, 8X, or
even higher concentrations. As used herein, "IX" means the standard
concentration, "2X"
means twice the standard concentration, etc. In any of these embodiments,
medium
components that can substantially affect osmolarity, such as salts, cannot be
increased in
concentration so that the osmolarity of the medium falls outside of an
acceptable range. Thus,
a medium may, for example, be 8X with respect to all components except salts,
which can be
present at only IX. An enriched medium may be serum free and/or protein free.
Further, a
medium may be supplemented periodically during the time a culture is
maintained to
replenish medium components that can become depleted such as, for example,
vitamins,
amino acids, and metabolic precursors. As is known in the art, different media
and
temperatures may have somewhat different effects on different cell lines, and
the same
medium and temperature may not be suitable for all cell lines.
Suitable culture conditions for mammalian cells are known in the art. See e.g.
Animal cell culture: A Practical Approach, D. Rickwood, ed., Oxford university
press, New
York (1992). Mammalian cells may be cultured in suspension or while attached
to a solid
substrate. Furthermore, mammalian cells may be cultured, for example, in
fluidized bed
bioreactors, hollow fiber bioreactors, roller bottles, shake flasks, or
stirred tank bioreactors,
with or without microcarriers, and operated in a batch, fed batch, continuous,
semi-
continuous, or perfusion mode.
The methods according to the present invention may be used to improve the
production of recombinant polypeptides in both single phase and multiple phase
culture
processes. In a single phase process, cells are inoculated into a culture
environment and the
disclosed methods are employed during the single production phase. In a
multiple stage
process, cells are cultured in two or more distinct phases. For example cells
may be cultured
-26-
CA 02762016 2011-12-09
wo 03/083066
PCT/US03/09266
first in a growth phase, under environmental conditions that maximize cell
proliferation and
viability, then transferred to a production phase, under conditions that
maximize polypeptide
production. The growth and production phases may be preceded by, or separated
by, one or
more transition phases. In multiple phase processes the methods according to
the present
invention are employed at least during the production phase. A growth phase
may occur at a
higher temperature than a production phase. For example, a growth phase may
occur at a
first temperature from about 35 C to about 38 C, and a production phase may
occur at a
second temperature from about 29 C to about 36 C, optionally from about 30 C
to about
33 C. Chemical inducers of polypeptide production, such as, for example,
caffeine, butyrate,
and HMBA, may be added at the same time as, before, and/or after a temperature
shift. If
inducers are added after a temperature shift, they can be added from one hour
to five days
after the temperature shift, optionally from one to two days after the
temperature shift.
After induction using the niethods of the invention, the resulting expressed
polypeptide can then be collected. In addition, the polypeptide can purified,
or partially
purified, from such culture or component (e.g., from culture medium or cell
extracts or bodily
fluid) using known processes. By "partially purified" means that some
fractionation
procedure, or procedures, have been carried out, but that more polypeptide
species (at least
10%) than the desired polypeptide is present. By "purified" is meant that the
polypeptide is
= essentially homogeneous, i.e., less than 1% contaminating polypeptides
are present.
Fractionation procedures can include but are not limited to one or more steps
of filtration,
centrifugation, precipitation, phase separation, affinity purification, gel
filtration, ion
exchange chromatography, hydrophobic interaction chromatography (H1C; using
such resins
as phenyl ether, butyl ether, or propyl ether), HPLC, or some combination of
above.
For example, the purification of the polypeptide can include an affinity
column
containing agents which will bind to the polypeptide; one or more column steps
over such
affinity resins as concanavalin A-agarose, heparinTOYOPEARL (Toyo Soda
Manufacturing Co., Ltd., Japan) or Cibacrom blue 3GA SEPHAROSE (Pharmacia
Fine
Chemicals, Inc., New York); one or more steps involving elution; and/or
immunoaffinity
chromatography. The polypeptide can be expressed in a form that facilitates
purification. For
example, it may be expressed as a fusion polypeptide, such as those of maltose
binding
polypeptide (MBP), glutathione-S-transferase (GST), or thioredoxin (TRX). Kits
for
expression and purification of such fusion polypeptides are commercially
available from New
England BioLab (Beverly, Mass.), Pharmacia (Piscataway, N.J.) and InVitrogen,
respectively.
The polypeptide can be tagged with an epitope and subsequently purified by
using a specific
antibody directed to such epitope. One such epitope (FLAG ) is commercially
available from
Kodak (New Haven, Conn.). It is also possible to utilize an affinity column
comprising a
-27-
CA 02762016 2011-12-09
WO 03/083066
PCT/US03/09266
polypeptide-binding protein, such as a monoclonal antibody to the recombinant
polypeptide,
to affinity-purify expressed polypeptides. Other types of affinity
purification steps can be a
Protein A or a Protein G column, which affinity agents bind to proteins that
contain Fc
domains. Polypeptides can be removed from an affinity column using
conventional
techniques, e.g., in a high salt elution buffer and then dialyzed into a lower
salt buffer for use
or by changing pH or other components depending on the affinity matrix
utilized, or can be
competitively removed using the naturally occurring substrate of the affinity
moiety.
The desired degree of final purity depends on the intended use of the
polypeptide. A
relatively high degree of purity is desired when the polypeptide is to be
administered in vivo,
for example_ In such a case, the polypeptides are purified such that no
polypeptide bands
corresponding to other polypeptides are detectable upon analysis by SDS-
polyacrylamide gel
electrophoresis (SDS-PAGE). It will be recognized by one skilled in the
pertinent field that
multiple bands corresponding to the polypeptide can be visualized by SDS-PAGE,
due to
differential glycosylation, differential post-translational processing, and
the like. Optionally,
the polypeptide of the invention is purified to substantial homogeneity, as
indicated by a
single polypeptide band upon analysis by SDS-PAGE. The polypeptide band can be
= visualized by silver staining, Coomassie blue staining, or (if the
polypeptide is radiolabeled)
by autoradiography.
The invention also optionally encompasses further formulating the
polypeptides. By
the term "formulating" is meant that the polypeptides can be buffer exchanged,
sterilized,
bulk-packaged, and/or packaged for a final user. For purposes of the
invention, the term
"sterile bulk form" means that a formulation is free, or essentially free, of
microbial
contamination (to such an extent as is acceptable for food and/or drug
purposes), and is of
defined composition and concentration. The term "sterile unit dose form" means
a form that
is appropriate for the customer and/or patient administration or consumption.
Such
compositions can comprise an effective amount of the polypeptide, in
combination with other
components such as a physiologically acceptable diluent, carrier, or
excipient. The term
"physiologically acceptable" means a non-toxic material that does not
interfere with the
effectiveness of the biological activity of the active ingredient(s).
Formulations suitable for administration include aqueous and non-aqueous
sterile
injection solutions which may contain anti-oxidants, buffers, bacteriostats,
and solutes which
render the fonnulation isotonic with the blood of the recipient; and aqueous
and non-aqueous
sterile suspensions which may include suspending agents or thickening agents.
The
polypeptides can be forrnulated according to known methods used to prepare
pharmaceutically useful compositions. They can be combined in admixture,
either as the sole
active material or with other known active materials suitable for a given
indication, with
pharmaceutically acceptable diluents (e.g., saline, Tris-HCI, acetate, and
phosphate buffered
CA 02762016 2011-12-09
WO (13/083066
PCT/US03/09266
solutions), preservatives (e.g., thimerosal, benzyl alcohol, parabens),
emulsifiers, solubilizers,
adjuvants, and/or caniers. Suitable formulations for pharmaceutical
compositions include
those described in Remington's Pluznnaceutical Sciences, 16th ed. 1980, Mack
Publishing
Company, Easton, PA. In addition, such compositions can be complexed with
polyethylene
glycol (PEG), metal ions, or incorporated into polymeric compounds such as
polyacetic acid,
polyglycolic acid, hydrogels, dextran, etc., or incorporated into liposomes,
rnicroemulsions,
micelles, unilamellar or multilamellar vesicles, erythrocyte ghosts or
spheroblasts. Suitable
lipids for liposomal formulation include, without limitation, monoglycerides,
diglycerides,
sulfatides, lysolecithin, phospholipids, saponin, bile acids, and the like.
Preparation of such
liposomal formulations is within the level of skill in the art, as disclosed,
for example, in US
Patent Nos. 4,235,871, 4,501,728, 4,837,028, and 4,737,323. Such compositions
will
influence the physical state, solubility, stability, rate of in vivo release,
and rate of in vivo
clearance, and are thus chosen according to the intended application, so that
the
characteristics of the carrier will depend on the selected route of
administration. Sustained-
release forms suitable for use include, but are not limited to, polypeptides
that are
encapsulated in a slowly-dissolving biocompatible polymer (such as the
alginate
microparticles described in US Patent No. 6,036,978), admixed with such a
polymer
(including topically applied hydrogels), and or encased in a biocompatible
semi-permeable
implant.
The invention having been described, the following examples are offered by way
of
illustration, and not limitation.
EXAMPLE 1
Comparison of the Inducing Activity of Caffeine and Butyrate at 31 C
In this experiment, caffeine (at concentrations from 0.5 to 2.0 mIV1) was
compared to
sodium butyrate for its ability to induce expression of a recombinant
polypeptide. A CHO
cell production line genetically engineered to express TNFR:Fc (cell line 115)
was used to test
the effectiveness of caffeine as an inducing agent. CHO cells were grown in
spinner flasks at
37 C using serum-free growth medium containing methotrexate. When the
appropriate cell
mass was obtained, the cells were placed into induction conditions by a five
minute
centrifugation at 1000 x g, followed by replacement of the growth medium with
serum-free
medium without methotrexate. The cells, at initial cell densities of 2 x 106
cells/ml in 20 ml,
were placed in 125 ml plastic Erlenmeyer flasks with plug seal caps and placed
on shaker
platforms in incubators set to the appropriate temperatures. Cell viability
and number were
monitored by haemocytometer counting using trypan blue dye. Recombinant
polypeptide
titers were assessed by ELISA-based assays.
CA 02762016 2011-12-09
r-2.)
WO 03/083066
PCT/US03/09266
For this cell line, 0.2 raM was known to be the optimal concentration of
sodium
butyrate for induction_ Accordingly, 0.2 mM sodium butyrate was compared
against the
inducing effects of 0.5, 1.0, and 2.0 niM caffeine. A flask containing no
inducing compound
was also included. The shaker flasks were incubated in this induction phase
for 5 days at
31oC in incubators without carbon dioxide control.
After 5 days in culture, cell viabilities for all of the tested conditions
were very
similar and ranged between 75 and 85 %. The highest relative protein titer (in
ug/m1), which
was about 1.15 times the titer of the control culture without inducers, and
relative productivity
(in pg protein/106 cells/day), which was about 1.3 times the productivity of
the control
culture, was exhibited by the cells that were induced with 1 raM caffeine.
Cells induced with
0.2 mM butyrate produced about 1.11 times the total protein (in Rival)
produced by the
control culture at a rate (in ug protein/106 cells/day) that was about 1.07
times the rate of
control cuitures Similar protein titers were observed in the cells induced
with 0.5 mM
caffeine, although these cultures had slightly higher rates of production. At
caffeine
concentrations of 2 mM, protein titer was similar to that observed with no
inducing agent,
although the rate of productivity per cell was higher.
These results indicate that caffeine can be used as an inducing agent and can
induce
product titers equal or exceeding those observed using sodium butyrate as an
inducing agent.
In addition, further experimental data was obtained which indicated that
recombinant
polypeptide produced using caffeine was equal in product quality (e.g.,
glycosylation,
folding, and amino acid composition) to that produced using sodium butyrate.
EXAMPLE 2
Induction of Recombinant Polypcptide Expression in Cell Line #9
In this experiment, the effect of caffeine (at concentrations from 0 to 1.4
inM) on the
induction of expression of a different recombinant polypeptide, a soluble form
of the 1L-1
receptor type II, in a second CLIO cell line (cell line #9) was examined. -
CHO cells were grown in spinner flasks at 37 C using serum-free growth medium
containing methotrexate. When the appropriate cell mass was obtained, spent
medium was
removed by a five minute centrifugation at 1000 x g and replaced with
production medium
without methotrexate. The cells, with initial cell densities of 2 x 106
cells/ml in 20 ml, were
placed in 125 ml plastic Erlenmeyer flasks with plug seal caps. The following
caffeine
concentrations were tested: 0, 0.6, 0_8, 1.0, 1.2, and 1.4 mM caffeine_ The
flasks were then
incubated in this induction phase for 5 days at 31 C in incubators without
carbon dioxide
control. Cell viability and number were monitored by haemocytometer counting
using trypan
blue dye. Recombinant polypeptide titers were assessed by ELISA-based assays.
Each
induction assessment experiment was carried out for 5 days.
-30-
CA 02762016 2011-12-09
rTh
WO 03/083066
PCT/US03/09266
After 5 days in culture, cell viability for most of the tested conditions was
similar and
averaged around 85%. Figure 1. For the flask induced with 1.4 mM caffeine, 67%
cell
viability was observed after 5 days. Similar protein titers were observed
using 0.6 mM,
0.8 mM, and 1.0 mM caffeine, that is, about 350 p.g/mL, which is equal to the
titer observed
for 0.5 mIVI butyrate. Figure 2. Since 0.6 mM is the lowest caffeine
concentration tested,
these data do not exclude the possibility that even lower concentrations of
caffeine might give
equal or better results. The highest productivity (in pg protein/106
cells/day) observed for a
caffeine-induced culture was in the 0.8 mlvl caffeine culture. Figure 3. At
higher levels of
caffeine, i.e., 1.2 and 1.4 mM, protein titers were comparable to the negative
control (no
inducing agent), although productivity on a per cell basis was somewhat
higher. Figures 2
and 3.
EXAMPLE 3
Induction Of Recombinant Polypeptide Expression In Cell Line #60
In this experiment, the use of caffeine to induce recombinant production from
a third
CHO cell line (cell line #60) expressing a third recombinant product, a human
antibody that
recognizes epidermal growth factor receptor, was analyzed. For this cell line,
the inducing
effects of 0, 0.5, 1.0, 1.5, and 2.0 mM caffeine were tested, and the
experiment was conducted
as in the previous experiment except that the induction phase was performed at
36 C.
At day 5, the flask of cells with no inducer and the flask of cells induced
with 0.5 m/v1
caffeine exhibited the highest cell viabilities (about 76%) of all the
conditions. Viabilities of
cultures containing 1.0 mM and 1.5 tnM caffeine were about 68% and 60%,
respectively.
Cultures containing 0.75 mM butyrate or 2.0 mM caffeine were about 51% viable.
Thus
viability, overall, was lower than that seen in cell line 49 at 5 days, an
effect that might be
attributed to a variety of factors including the difference in temperature
and/or cell line
differences. A clear dose-Tesponse was observed with higher caffeine
concentrations leading
to lower cell viabilities.
The highest day 5 protein titer was observed in cells induced by 0.5 triM
caffeine
(305 lig/m1), which was about 111% of the titer of the control culture with no
inducer.
Generally, the titer of recombinant polypeptide was less as caffeine
concentrations increased
above 0.5 mM. Productivity (in pg protein/ 106 cells/day) appeared to be
linked to caffeine
concentration, with the highest productivity obtained from cells induced with
2.0 m_M caffeine
and a lower level of productivity obtained from the cells induced with lower
caffeine
concentrations. Since a 0_5 triM was the lowest caffeine concentration tested
as well as the
most effective concentration tested for the induction of protein production,
these data do not
exclude the possibility that a lower concentration of caffeine might be
equally or more
effective as an inducer of cell line 1#60 incubated at 36 C_
-31-
CA 02762016 2011-12-09
(7)
WO 03/083066
PCT/US03/09266
This experiment, along with those described in Examples 1 and 2, demonstrates
that
the ability of caffeine to induce recombinant polypeptide expression is not
cell line-specific
and that favorable cell viability is maintained in caffeine's presence. In
addition, caffeine can
be used in an induction or production phase implemented at temperatures from
31 C to 36 C.
However, these data also indicate differences between cell lines in how
effectively caffeine
induces the synthesis of a recombinant protein. For example, induction of cell
line #9 with
caffeine is more effective than induction of cell line #60. Compare Example 2
and Figure 2
to Example 3.
EXAMPLE 4
Optimization of Induction for Cell Line #60
The purpose of this experiment was to test ranges of temperature and caffeine
concentrations in shake flasks in order to optimize the induction conditions
for the cell line
#60.
Materials and Methods. Twelve shaker flasks were set up under the conditions
described in Table 1.
TABLE 1: Caffeine Concentrations and Temperatures of Samples
Flask Temperature Caffeine
Number ( C) (rn1V1)
1 36 0
2 36 0.5
3. 36 1.0
4 36 1.5
5 36 2.0
6 36 2.5
7 37 0
8 37 0.5
9 37 1.0
10 37 1_5
11 37 2.0
12 37 2.5
Cells were collected via centrifugation from a spinner culture of cell line
#60 (26.85 x 105
cells/ml, 95.2% viable) and inoculated into a 575 ml spinner flask at 2 x 106
cells/ml in
serum-free production medium_ The culture was then aliquoted into twelve shake
flasks.
Caffeine was added according to the experimental plan described in Table 1.
The shake
flasks were incubated at the designated temperatures for 7 days_ Samples were
taken on days
3, 5 and 7. Cell density and viability were measured using an automated system
of cell
counting that employs trypan blue staining to determine viability (the Cell
Density
Examination System or Cedex, developed by innovatis GmbH, Bielefeld, Germany).
-32-
CA 02762016 2011-12-09
WO 03/083066
PCT/US03/09266
Glucose and lactate measurements were taken with the Yellow Springs
Instruments 2700
Select (available from Yellow Springs Instruments, Yellow Springs, Ohio, USA).
Glucose
was added on demand to maintain a concentration of > 2g/1. CO2 and external pH
were
measured using the Ciba-Corning 248 blood gas analyzer (available from Bayer
Diagnostics,
Tarryton, New York, USA). Protein titers were determined via a pre-
purification of the
= antibody on a Protein A column followed by a measurement of the
absorbance of the protein
bound and eluted from the column at 280 nanometers. Cumulative viable cell
densities
(CVCDs) were calculated as follows: the CVCD for day 1 is the number of viable
cells per
milliliter of culture as measured on day 1; the CVCD for day 2 is the number
of viable cells
_ per milliliter of culture as measured on day 2 plus the number of viable
cells per milliliter of
culture as measured on day 1; the CVCD for day 3 is the number of viable cells
per milliliter
of culture as measured on day 3 plus the numbers of viable cells per
milliliter of culture
measured on days 1 and 2; and CVCDs for subsequent days are calculated in a
similar
manner.
Results. Higher CVCDs were achieved in the presence of little or no caffeine.
Lower temperature, i.e., 36 C rather than 37 C, and lower levels of caffeine
resulted in higher
final viability. Caffeine at 2.5 mN1 resulted in cell death and termination of
the cultures.
Over the rest of the concentration range tested, increased levels of caffeine
resulted in
increased cumulative specific productivity (Cum Qp), with the highest level
being almost
30 pg/106 cells/day. Cultures containing the highest levels of caffeine
resulting in viable
cultures (2 rnM), while having a high Cum Qp, had a low CVCD, indicating that
2 inNI
caffeine decreased cell viability but increased the productivity of remaining
viable cells.
However, protein titers of cultures induced with 2 mIVI caffeine were lower
than for
uninduce,d cultures at 7 days at both temperatures.
The highest protein titers resulted at the low to intermediate levels of
caffeine for both
temperatures. The highest day 7 titer was observed in the culture grown at 36
C in the
presence of 0.5 mM caffeine, and its titer was about 124% of the titer seen in
a control culture
grown at 36 C for 7 days without inducers. Day 7 titers of 36 C cultures grown
in the
presence of 1.0 m1V1 and 1.5 mM caffeine were about 116% and 111% of control
levels,
respectively. The day 7 titers of cultures grown at 37 C in the presence of
0.5 rnM, 1.0 rnM,
and 1.5 nilVl caffeine were about 110%, 112%, and 109%, respectively, of the
37 C no
inducer control culture_ Together, these data indicate that induction of cell
line #60 was more
effective at 36 C than it was at 37 C. Day 7 titers of control cultures
without inducers grown
at 36 C and 37 C were comparable. Thus, as in Example 3, the lowest
concentration of
caffeine tested led to the highest protein titers at 36 C, suggesting the
possibility that even
lower concentrations might produce equal or higher titers.
-33-
CA 02762016 2011-12-09
wo 03/083066
PCT/US03/09266
In summary, induction with caffeine increased specific productivity and titer
at both
36 C and 37 C. Titers were modestly higher at 36 C than at 37 C, despite lower
Cum Qp
values because of higher CVCDs and viability at the lower temperature. Based
on cell
performance and productivity, caffeine can be used to induce production from
this cell line.
EXAMPLE 5
Induction Effects for Compounds Related to Caffeine in Cell Line #9
Since the above experiments showed that caffeine as an inducing agent
increased
titers of recombinant polypeptide between about 9% and about 67%, additional
experiments
were performed with other xanthine derixatives to test their inducing ability.
Based upon the
structure of xanthine, a variety of compounds were modeled and chosen for
testing. These
include 3-isobuty1-1-methylxanthine, theophylline, theobromine,
pentoxifylline, and
aminophylline, the structures of which are illustrated below.
TABLE 2: Xanthine Derivatives
0
Z
X N
I ____________________________________________ I I
0
Compound
X
caffeine methyl methyl methyl
3-isobuty1-1-
methylxanthine methyl isobutyl hydrogen
(IBX)
methyl methyl hydrogen
theophylline
theobromine hydrogen methyl methyl
pentoxifylline 5-oxyhexyl methyl methyl
Arninophylline is theophylline compound with 1,2-ethylenediamine (2:1)
dihydrate.
These xanthine derivatives, including some combinations, were tested on cell
line #9
in a shake-flask forrnat (20 rn1 in 125 ml shake flasks) as described above
for Examples 1
and 2. To dissolve 3-isobuty1-1-methylxanthine (LB X), it was solubilized in
water heated to
25 almost the boiling point, and quickly added to the flasks before it
precipitated. Alternatively,
-34-
CA 02762016 2011-12-09
WO 03/083066
PCT/US03/09266
IBX was dissolved in DMF. The induction phase of the cell culture was carried
out for 6 days
at 31 C, and samples were removed for analysis at 3 day and 6 day timepoints.
The protein
titers from each shake flask are shown in Table 3.
TABLE 3: Recombinant Polypeptide Titer Under Various Inducing Conditions
Condition Titer gig/rap
Titer (pg/m1)
Day 3 Day 6
0.6 HIM caffeine + 0.5 m1v1 butyrate 120 280
0.1 rnM theobromine 90 270
0.5 niM theobromine 100 260
1 in1V1 theobrotnine 100 260
0.1 m1V1 aminophylline 110 260
0.5 mIVI aminophylline 120 250
1 mIVI aminophylline 100 200
0.1 rniq pentoxyphylline 110 320
_0.5 m/V1 pentoxYphylline 130 380
1 ni/v1 pentoxyphylline 130 400
0.3% DMF + 0.5 mM IBX not determined 340
0.3% DMF 120 330
0.5 m1VI IBX 150 420
0.5 m1v1 rBx+ 0.6 raM caffeine 130 370
0.1 mM IBX + 0.6 ruM caffeine . 140 390
0.1 in1V1 IBX + 0.6n-iM caffeine + 0.5m1V1 butyrate _ 130 280
0.1 mM IBX + 0.6mM caffeine + 0.3% MP 150 390
0.2 mIVI caffeine 150 400
0.6 mM caffeine 120 320
0.5 mM butyrate 100 220
NO INDUCER 100 290
Several conclusions can be made from this data. Production from the flask
induced with 0.5
mM IBX was even better than caffeine, and the 3 flasks containing
pentoxyphylline also gave
promising results. Titers of pentoxyphylline-induced cultures increased-with
increasing dose
and were higher than the tier of the no inducer control culture. Additionally,
some
combinations of different xanthine derivatives, as well as different xanthine
derivatives with
other inducing agents (e.g., butyrate and/or DMF) yielded protein titers above
control levels.
Theobromine and aminophylline did not induce protein titers above that seen in
the no
inducer control culture. The highest protein titers obtained when caffeine was
used as an
inducer were obtained at the lowest concentration tested, that is, 0.2 mM
caffeine. As
explained above, such a result leaves open the possibility that even lower
concentrations of
= caffeine may be effective. Finally, unlike in Example 2 (Figure 2),
butyrate does not induce
increased protein titer over that seen in a culture with no inducer.
-35 -
CA 02762016 2011-12-09
(5,7)
WO 03/083066
PCT/US03/09266
EXAMPLE 6
Induction of Cell Line #60 by Various Inducing Agents at 37 C
This experiment was done in shaken Erlenmeyer flasks as described above in
Examples 1 and 2, except that the flasks were incubated for 5 days at 37 C,
rather than at
31 C, and cell line #60 was used. As a control, one flask without inducers was
grown at
31 C. The titer of recombinant polypeptide in the medium was assayed after 5
days.
Xanthine derivatives tested included caffeine (at 0.5 mM), theobromine (at 0.1
mM, 0.5 mM,
and 1.0 mM), 3-isobuty1-1-methylxanthine (1BX, at 0.05 niVI, 0.1 mM, and 0.15
rnM), and
pentoxyphylline (at 0.1 mM, 0.5 mM, and 1.0 mM). In addition, butyrate, some
combinations
of inducers, and the non-xanthine compound papaverine were tested.
The 31 C control culture yielded low protein titers compared to the 37 C
control
culture, probably due to the preference of cell line #60 for higher
temperatures. Theobromine
at a concentration of 0.1 mM increased protein titer over that seen in the 37
C control culture,
but was counterproductive at higher concentrations (0.5 mM and 1.0 mM).
Neither caffeine,
rBx, or pentoxyphylline increased protein titers above that seen in a control
culture with no
inducers. Protein titer was inversely proportional to theobromine, LBX, and
pentoxyphylline
concentrations in the ranges tested. Interestingly, cell line #9 (Table 3,
Example 5) showed
increased protein titers with increasing pentoxyphylline concentrations within
this same
range, highlighting the variability in the responses of different cell lines
incubated at different
temperatures to inducing agents. As in other experiments (see Example 3), 0.5
mIVI caffeine
appears to be a better inducer than 0.5 it-LW' butyrate for cell line #60,
although both failed to
increase protein titer over that seen in the control culture with no inducing
agent in this
experiment. In a previous experiment, caffeine had induced slightly higher
protein production
than that seen in a control culture at 37 C at day 7 (about 110% of the titer
seen in the control
culture), although a greater induction was observed at 36 C. Example 4. The
failure of
caffeine to induce increased protein production in this experiment may be
explained by a
variety of factors such as experimental variability, the small size of the
positive effect at 37 C
in cell line #60, and/or the possibility that 0.5 mM may not be an optimum
caffeine
concentration for induction of cell line 1160 at 37 C.
EXAMPLE 7
Production of RANK:Fc in the Presence of Varying Amounts of HIVILBA
Nucleic acids encoding RANK:Fc inserted into a suitable vector (as described
in
International Application WO 01/36637) were introduced into CHO cells. About 2
million
cells from a stably transformed line propagated at 37 C were inoculated into
20 milliliters of
medium at 31 C, either without FEMBA or in the presence of varying
concentrations of
I-MBA, as indicated in Table 4. Cells were grown for a total of 5 days in
shaker flasks.
-36-
CA 02762016 2011-12-09
.4,7)
:4-C)
WO 03/083066
PCT/US03/09266
Thereafter, all medium was harvested. The number of cells present in the
culture was
determined by staining with trypan blue and counting the cells in a
hemocytometer. The titer
of RANK:Fc per milliliter of harvested medium was determined by purifying
RANK:Fc by
Protein A high performance liquid chromatography (HPLC) and subsequently
measuring
absorbance at 280 nanorneters. An average number of cells in the culture was
calculated by
averaging the starting and ending cell numbers. Specific productivity was
determined from
the total number of micrograms of RANK:Fc produced, an average cell number
(calculated as
described above), and the number of days of growth. Data from this experiment
are shown in
Table 4.
TABLE 4: Effects of Varying Concentrations of
IIMBA on Protein Titer and Specific Productivity
1-IMBA Specific Titer of
concentration productivity RANK:Fc
(mM) (pg/106 cells/day)
(pg/ml)
0 17.1 272
0.1 19.1 243
0.5 19.5 347
2.0 23.9 444
These data indicate that the addition of HMBA at concentrations of 0.5 or 2.0
rnIVI
had positive effects on polypeptide production and specific productivity.
EXAMPLE 8
Production of RANK:Fc in the Presence of FINLBA, Caffeine, and/or Butyric Acid
Nucleic acids encoding RANK:Fc inserted into a suitable vector (as described
in
International Application WO 01/36637) were introduced into CHO cells. About 2
million
cells from a stably transformed line prop-agated at 37 C were inoculated into
20 milliliters of
serum-free medium at 31 C without inducers or in the presence of of HIMBA
and/or caffeine
and/or butyric acid, as indicated in Table 5. Cells were grown for a total of
5 days in a shaker
.25 flask. Thereafter, all medium was harvested. The number of cells
present in the culture, the
titer of RANK:Fc per milliliter of harvested medium, and specific productivity
were
determined as described above in Example 7. Data from this experiment are
shown in
Table 5.
-37-
CA 02762016 2011-12-09
r-Th
W. 03/083066
PCT/US03/09266
TABLE 5: Effects of Caffeine, IIMBA, and Butyric Acid Singly
and in Combination on Protein Titer and Specific Productivity
Inducer Specific Titer of I
productivity RANK:Fc
(pg/106 cells/day) (pg/m1)
None 14.8 261
HIVIBA (2 mM) 22.8 410
Caffeine (1 mM) 23.6 362
Butyric acid (0.5 mM) 31.6 476
HMBA (2 niM)+ 46.7 553
Caffeine (1 miV1)-F
butyric acid (0.5 mIVI)
These data indicate that the addition either caffeine (at 1 rnM), butyric acid
(at
0.5 mM), or HMBA (at 2 !I:1M) had positive effects on both polypeptide
production and
specific productivity and that the combination of butyric acid, caffeine, and
HMBA (at the
concentrations mentioned above) had greater positive effects than any of these
compounds
alone.
EXAMPLE 9
Production of Type 11 IL-1 Receptor in the Presence of HIVIBA in a Bioreactor
Nucleic acids encoding a type 111 IL-1 receptor inserted into a suitable
vector were
introduced into CHO cells. About 500 thousand cells from a stably transformed
line were
inoculated into a one liter of serum-free medium in a bioreactor. Cells were
grown for two
days at 37 C. Thereafter, cells were shifted to 31 C, either without filvD3A
or in the presence
of 2 mM HMBA, and grown for 12 more days. Thereafter, all medium was
harvested. The
titer of type II lL-1 receptor per milliliter of harvested medium was
determined by
purification by reverse phase ILPLC followed by the measurement of absorbance
at 280
nanometers. Data from this experiment are shown in Table 6 as a percentage of
the average
of the protein titers obtained from the two samples without HMBA rounded to
the nearest
whole number.
TABLE 6: Effects of 2 mM LIMBA on Protein Titer
Inducer Relative Titer of
type H IL-1
Receptor
(percent of average of
samples without FIMBA)
None 99%
None 101%
_ HIVIBA (2 niM) 120%
HME A (2 mM) 125%
-38-
CA 02762016 2011-12-09
= WO
03/083066 PCT/US03/09266
These data show that bioreactor cultures shifted to 31 C after an initial 37 C
growth
phase produced more type II FL-1 receptor if IIMBA was added at the time of
temperature
shift than if it wasn't. These data further suggest that the invention can be
useful for
producing a variety of polypeptides in a variety of cell lines and that the
mechanics of how
the cells are grown, for example, in a shaker flask versus in a bioreactor,
are not critical.
EXAMPLE 10
Production of an Antibody Against Murine IL-4 Receptor in CHO Cells
The experiment described below tests the effects of using either sodium
butyrate or
IIMBA as an inducer in still another cell Line at various temperatures.
Nucleic acids encoding an antibody against a murine IL-4 receptor inserted
into a
suitable vector were introduced into CHO cells. About two million cells from a
stably
transformed line propagated at 37 C were inoculated into 20 milliliters of
medium at the
temperatures indicated in Figure 4 and in the presence or absence of HMBA (2
mM) or
sodium butyrate (0.5 mM), as indicated in Figure 4. Cells were grown for a
maximum of 14
days in a shaker flask. Aliquots were removed at the times indicated in Figure
4, and the titer
of the antibody (in micrograms per milliliter of harvested medium) was
determined by
enzyme-linked immunosorbent assay (ELISA), a method well known in the art. See
e.g. Reen
(1994), Enzyme-Linked Immunosorbent Assay (ELISA), in Basic Protein and
Peptide
Protocols, Methods Mol. Biol. 32:461-466. The results are shown in Figure 4.
These data
indicate that growth at 31 C resulted in the production of more antibody for a
longer time
than growth at either 34 C or 37 C when medium was harvested at 7 days or
later. These
data also indicate that both HMBA and sodium butyrate, individually, enhanced
production of
the antibody and that HMBA did so to a greater extent than did sodium butyrate
at 31 C.
EXAMPLE 11
Production of TNFR:Fc in CHO Cells
Nucleic acids encoding human TNFR:Fc in a suitable vector were introduced into
CHO cells. About 3 0.5 x 106 cells from a stably transformed cell line
propagated at 37 C
were introduced into each of three 1 liter bioreactors and cultured at 32.5 C
in an enriched,
serum-free medium. Sodium butyrate (0.5 mM) was added to all three cultures,
and MBA
(2 mM) was added to two of the cultures ("day 1 + HMBA") one day after the
shift to 32.5 C.
Cells were incubated for a total of 11 days at 32.5 C. Medium was harvested,
and protein
titer was determined by measuring optical density at 280 nanometers following
a
prepurification using Protein A POROS Perfusion Chromatographyrm (Applied
Biosystems,
Foster City, California, USA). These results are shown in Table 7 as a
percentage of the titer
obtained from the sample with no HMBA ("day 1") rounded to the nearest whole
number.
-39-
CA 02762016 2011-12-09
/ 2 2 4 9 - 1 5 6
TABLE 7: Effects of the Timing of Addition of Inducers
Relative Titer
TNFR:Fc
(percent of the
day 0 titer)
day 1 100%
day 1 + HMEA 131%
day 1 + HMBA 110%
These data indicate that the addition of HMBA increased the titer of TNFR:Fc
produced by these cultures when added one day after a temperature shift to
32.5 C. These
data, together with the data in previous examples, indicate that addition of
HMBA can
increase protein titer when it is added at the time of or after a shift to a
lower temperature.
The foregoing description of specific embodiments reveals the general nature
of the
invention so that others can readily modify and /or adapt such embodiments for
various
applications without departing from the generic concepts presented herein. Any
such
adaptions or modifications arc intended to be embraced within the meaning and
range of
equivalents of the disclosed embodiments. Phraseology and terminology employed
herein are
for the puipose ofclesciiption and not of limitation.
-40-
=