Note: Descriptions are shown in the official language in which they were submitted.
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B20081001814
GLYCOSYLATION OF MOLECULES
Technical Field
The invention relates to methods of obtaining glycosylated molecules,
particularly
protein and lipid molecules.
Background
High performance expression systems are required to produce most
biopharmaceuticals
(e.g., recombinant proteins) currently under development. The biological
activity of many of
these biopharmaceuticals is dependent on their modification (e.g.,
phosphorylation or
glycosylation). A yeast-based expression system combines the ease of genetic
manipulation and
fermentation of a microbial organism with the capability to secrete and to
modify proteins.
However, recombinant glycoproteins produced in yeast cells exhibit mainly
heterogeneous high-
mannose and hyper-mannose glycan structures, which can be detrimental to
protein function,
downstream processing, and subsequent therapeutic use, particularly where
glycosylation plays a
biologically significant role.
Summary
The present invention is based, at least in part, on: (a) the discovery that
single gene
deletion (Outer CHain elongation (OCH 1) deletion) in Yarrowia lypolitica
cells resulted in the
substantially homogeneous production of glycosylated proteins having a-1,2-
linked mannose
residues on a Man5GlcNAc2 (structural formula IV; Fig. 1) backbone; (b) the
discovery that
overexpression of an engineered alpha-1,2-mannosidase targeted to the ER of
Yarrowia
lipolytica cells (both with AND without OCH I deletion) resulted in the
substantially
homogenous production of glycosylated proteins carrying the Man5GlcNAc2 N-
glycan structure
(structural formula IV; Fig. 1); (c) the discovery that inactivating the
Asparagine Linked
Glycosylation 3 (ALG3) enzyme activity in Yarrowia lipolytica cells results in
highly increased
levels of glucosylated glycans; and (d) the discovery that overexpression of a
wild-type form of a
Yarrowia lipolytica gene (MNN4) in Yarrowia lipolytica results in
hyperphosphorylation of a-
1,2-linked mannose residues. Thus, the genetically engineered cells (e.g.,
Yarrowia lipolytica,
Arxula adeninivorans, or other related species dimorphic yeast cells) can be
used in methods to
1
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
produce target molecules having an altered N-glycosylation form as compared to
the N-
glycosylation form of the target molecules produced in non-genetically
engineered cells of the
same species. As administration of N-glycosylated target molecules (e.g., N-
glycosylated
proteins) to patients having a metabolic disorder (e.g., a lysosomal storage
disorder) has been
shown to ameliorate the symptoms of the disorder, the methods and cells
described are useful for
the preparation of N-glycosylated target molecules for the treatment of, inter
alia, metabolic
disorders such as lysosomal storage disorders.
The present invention is also based, at least in part, on the discovery of the
spliced form
of the Yarrowia lipolytica and Pichia pastoris HAC1 gene. The protein encoded
by the HAC1
gene, Haclp, is a transcriptional activator that activates transcription of
several target genes by
binding to a DNA sequence motif termed the Unfolded Protein Response (UPR)
element.
Among the Haclp target genes are those that encode chaperones, foldases, and
proteins which
are responsible for lipid-and inositol metabolism. As the spliced form Hacip
is a more potent
transcriptional activator than the form encoded by the unspliced HACI mRNA,
overexpression
of the spliced form of Hac1p transcription factor can lead to an increased
expression of native
and heterologeous proteins as well as an increase in ER membrane. Thus, the
spliced form of
Hac I p can be used to increase the production of membrane and secreted
proteins in a variety of
eukaryotic cells (e.g., fungal cells (e.g., Yarrowia lipolytica or any other
yeast cells described
herein), plant cells, or animal cells (e.g., mammalian cells such as human
cells) by simultaneous
activation of the UPR and expression of target molecules.
The present invention is further based on the discovery of a mutant form of
the MNS 1
mannosidase capable of converting Man5GlcNAc2 (structural formula I; Fig. 4)
structures to
Man5GIcNAc2 (structural formula IV; Fig. 4), Man6GlcNAc2 (structural formula
V; Fig. 4) and
Man7GIcNAc2 (structural formula VI; Fig. 4) when expressed in Yarrowia
lipolytica. Thus,
genetically engineered eukaryotic cells (e.g., fungal cells (e.g., Yarrowia
lipolytica or any other
yeast cells described herein), plant cells, or animal cells (e.g., mammalian
cells such as human
cells)) expressing mutant forms of mannosidase such as MNSI can be used in
methods to
produce target molecules having an altered N-glycosylation form as compared to
the N-
glycosylation form of the target molecules produced in non-genetically
engineered cells of the
same species. Therefore, the cells and methods described are useful for the
preparation of N-
2
CA 02762982 2011-12-22
WO 2008/120107 PCTlIB2008/001814
glycosylated target molecules for the treatment of, inter alia, metabolic
disorders such as
lysosomal storage disorders (see below).
In one aspect, the disclosure features a method of producing an altered N-
glycosylation
form of a target protein. The method includes the step of introducing into a
cell a nucleic acid
encoding a target protein, wherein the cell produces the target protein in an
altered N-
glycosylation form and wherein the cell is a Yarrowia lipolytica or an Arxula
adeninivorans cell
(or a related species dimorphic yeast cell) genetically engineered to contain
at least one modified
N-glycosylation activity. The method can also include the step of providing
the Yarrowia
lipolytica or an Arxula adeninivorans cell (or related species dimorphic yeast
cell) genetically
engineered to contain at least one modified N-glycosylation activity. The
method can also
include the step of isolating the altered N-glycosylation form of the target
protein.
In some embodiments, the target protein can be an endogenous protein or an
exogenous
protein. The target protein can be a mammalian protein such as a human
protein. The target
protein can be, for example, a pathogen protein, a lysosomal protein, a growth
factor, a cytokine,
a chemokine, an antibody or antigen-binding fragment thereof, or a fusion
protein. The fusion
protein can be, for example, a fusion of a pathogen protein, a lysosomal
protein, a growth factor,
a cytokine, or a chemokine with an antibody or an antigen-binding fragment
thereof. The target
protein can be, for example, one associated with a lysosomal storage disorder
(LSD). The target
protein can be, for example, glucocerebrosidase, galactocerebrosidase, alpha-L-
iduronidase,
beta-D-galactosidase, beta-glucosidase, beta-hexosaminidase, beta-D-
mannosidase, alpha-L-
fucosidase, arylsulfatase B, arylsulfatase A, alpha-N-acteylgalactosaminidase,
aspartylglucosaminidase, iduronate-2-sulfatase, alpha-glucosaminide-N-
acetyltransferase, beta-
D-glucoronidase, hyaluronidase, alpha-L-mannosidase, alpha- neuraminidase,
phosphotransferase, acid lipase, acid ceramidase, sphinogmyelinase,
thioesterase, cathepsin K, or
lipoprotein lipase.
In some embodiments, the altered N-glycosylation form can contain one or more
N-
glycan structures such as, e.g., Man5GlcNAc2i Man8GIcNAc2, Man9GIcNAc2,
Man3GlcNAc2,
Glc,Man5GIcNAc2, GIc2Man5GlcNAc2. In some embodiments, the altered
glycosylation can be,
for example, Man5GIcNAc2, Man8GlcNAc2i Man9GIcNAc2, Man3GlcNAc2,
GlcIMan5GIcNAc2,
GIc2Man5GlcNAc2.
3
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
In some embodiments, the altered N-glycosylation form of the target protein
can be
homogenous or substantially homogenous. For example, the fraction of altered
target molecules
that contain the altered glycosylation can be at least about 20%, at least
about 30%, at least about
40%, at least about 45%, at least about 50%, at least about 55%, at least
about 60%, at least
about 65%, at least about 70%, at least about 75%, at least about 80%, at
least about 85%, at
least about 90%, or at least about 95% or more.
In some embodiments, the cell can be genetically engineered to be deficient in
at least
one N-glycosylation activity. The N-glycosylation activity can be, for
example, ALG3 activity,
OCHI activity, MNS1 activity, or MNN9 activity.
In some embodiments, at least one modification can be: (a) deletion of a gene
encoding a
protein having the N-glycosylation activity; (b) expression of a mutant form
of a protein having
the N-glycosylation activity; (c) introduction or expression of an RNA
molecule that interferes
with the functional expression of a protein having the N-glycosylation
activity; (d) expression of
a protein having N-glycosylation activity (such as ALG6 or an alpha-
mannosidase (e.g., an
alpha-mannosidase targeted to the endoplasmic reticulum). The expressed
protein can be a
protein encoded by an exogenous nucleic acid in the cell. The expressed
protein can be an
alpha-mannosidase with a pH optimum below 7.5 (e.g., a pH optimum below 5.1).
The protein
having N-glycosylation activity can be an exogenous protein. The protein
having N-
glycosylation activity can be a mammalian protein (such as a human protein) or
a lower
eukaryotic (e.g., a fungus, a protozoan, or a trypanosome) protein. The lower
eukaryote can be
selected from the group consisting of Typanosoma brucei, Trichoderma
harzianum, an
Aspergillus, and any other lower eukaryote described herein.
In some embodiments, the N-glycosylation activity can be a glucosyltransferase
activity.
In some embodiments, the protein having N-glycosylation activity is ALG6 or an
alpha-
mannosidase. The alpha-mannosidase can be targeted to the endoplasmic
reticulum. For
example, the protein having N-glycosylation activity can be a fusion protein
comprising an
alpha-mannosidase polypeptide and an HDEL endoplasmic reticulum retention
peptide.
In some embodiments, the protein having N-glycosylation activity can be a
protein that is
capable of removing glucose residues from Man5GIcNAc2. For example, the
protein having N-
glycosylation activity can be a protein having a-1,3-glucosidase activity such
as, but not limited
4
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
to, a glucosidase II (e.g., one or both of the alpha and beta subunit of a
glucosidase II) or a
mutanase.
In some embodiments, the cell can be genetically engineered to comprise at
least two
modified N-glycosylation activities such as any of the modified N-
glycosylation activities
described herein. The at least two modified N-glycosylation activities can
comprise, e.g., a
deficiency in an ALG3 activity and an elevated level of an ALG6 activity.
In some embodiments, the cell can be genetically engineered to comprise at
least three
modified N-glycosylation activities such as any of the modified N-
glycosylation activities
described herein. The at least three modified N-glycosylation activities can
comprise, e.g., a
deficiency in an ALG3 activity; an elevated level of an ALG6 activity; and an
elevated level of a
a glucosidase II activity.
In some embodiments, the cell is not genetically engineered to be deficient in
an OCHI
activity.
In some embodiments, modification can comprise expression of a protein or
biologically
active variant thereof capable of effecting mannosyl phosphorylation of the
target protein. The
protein or biologically active variant thereof capable of effecting mannosyl
phosphorylation can
be MNN4, PNOI, or MNN6. In some embodiments, at least about 30% of the
mannosyl
residues of a glycoprotein can be phosphorylated.
In some embodiments, the method can further include additional processing of
the
glycoprotein. The additional processing can occur in vitro or in vivo. The
additional processing
can comprise addition of a heterologous moiety to the modified glycoprotein.
The heterologous
moiety can be a polymer or a carrier. The additional processing can comprise
enzymatic or
chemical treatment of the altered N-glycosylation form of the target protein.
For example, the
additional processing can comprise treatment of the altered N-glycosylation
form of the target
protein with a mannosidase, a mannanase, a phosphodiesterase, a glucosidase,
or a
glycosyltransferase. The additional processing can include treatment of the
altered N-
glycosylation form of the target protein with hydrofluoric acid. The
additional processing can
include phosphorylation of the altered N-glycosylation form of the target
protein.
In another aspect, the disclosure provides a method of producing an altered N-
glycosylation form of a target protein. The method includes the steps of.
providing a eukaryotic
cell (e.g., a fungal cell, a plant cell, or an animal cell) genetically
engineered to comprise at least
5
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
one modified N-glycosylation activity; and introducing into the cell a nucleic
acid encoding a
target protein, wherein the cell produces the target protein in an altered N-
glycosylation form.
In another aspect, the disclosure features a method of producing an altered N-
glycosylation form of a target protein. The method includes the step of
contacting a target
protein with a cell lysate prepared from a Yarrowia lipolytica or an Arxula
adeninivorans cell
genetically engineered to comprise at least one modified N-glycosylation
activity, wherein the
contacting of the target protein with the cell lysate results in an altered N-
glycosylation form of
the target protein.
In yet another aspect, the disclosure features a method of producing an
altered N-
glycosylation form of a target protein, which method includes the step of
contacting a target
protein with one or more proteins having N-glycosylation activity, wherein the
one or more
proteins having N-glycosylation activity are obtained from a Yarrowia
lipolytica or an Arxula
adeninivorans cell genetically engineered to comprise at least one modified N-
glycosylation
activity and wherein contacting the target molecule with the one or more
proteins having N-
glycosylation activity results in an altered N-glycosylation form of the
target protein.
In another aspect, the disclosure provides an isolated protein having altered
N-
glycosylation, wherein the protein is produced by any of the methods described
above.
In yet another aspect, the disclosure provides an isolated Yarrowia lipolytica
or Arxula
adeninivorans cell (or other related species dimorphic yeast cell) genetically
engineered to
comprise at least one modified N-glycosylation activity. The N-glycosylation
activity can be, for
example, ALG3 activity, OCH I activity, MNS I activity, or MNN9 activity. The
modification
can be any of those described herein. For example, the modification can
include: (a) deletion of
a gene encoding a protein having the N-glycosylation activity, (b) expression
of a mutant form of
a protein having the N-glycosylation activity, (c) introduction or expression
of an RNA molecule
that interferes with the functional expression of a protein having the N-
glycosylation activity, or
(d) expression of a protein having N-glycosylation activity. The protein
having N-glycosylation
activity can be, for example, ALG6. The protein having N-glycosylation
activity can be a
mammalian protein such as a human protein. The modification can also include
expression of a
protein (e.g., MNN4 or PNOI) or biologically active variant thereof capable of
promoting
mannosyl phosphorylation of the modified glycoprotein.
6
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
In another aspect, the disclosure provides a method of treating a disorder
treatable by
administration of a protein having altered N-glycosylation. The method
includes the steps of
administering to a subject a protein obtained by any of the methods described
above, wherein the
subject is one having, or suspected of having, a disease treatable by
administration of a protein
having altered N-glycosylation. The method can also include the steps of (a)
providing a subject
and/or (b) determining whether the subject has a disease treatable by
administration of a protein
having altered N-glycosylation. The subject can be mammal such as a human. The
disorder can
be, for example, a cancer, an immunological disorder (e.g., an inflammatory
condition) or a
metabolic disorder. The metabolic disorder can be any of those described
herein, e.g., a
lysosomal storage disorder (LSD) such as Gaucher disease, Tay-Sachs disease,
Pompe disease,
Niemann-Pick disease, or Fabry disease. The protein can be one associated with
an LSD, e.g.,
the protein can be, for example, glucocerebrosidase, alpha-galactosidase. The
protein can be, for
example, alpha-L-iduronidase, beta-D-galactosidase, beta-glucosidase, beta-
hexosaminidase,
beta-D-mannosidase, alpha-L-fucosidase, arylsulfatase B, arylsulfatase A,
alpha-N-
acteylgalactosaminidase, aspartylglucosaminidase, iduronate-2-sulfatase, alpha-
glucosaminide-
N-acetyltransferase, beta-D-glucoronidase, hyaluronidase, alpha-L-mannosidase,
alpha-
neurominidase, phosphotransferase, acid lipase, acid ceramidase,
sphinogmyelinase, thioesterase,
cathepsin K, or lipoprotein lipase.
In another aspect, the disclosure provides a substantially pure culture of
Yarrowia
lipolytica or Arxula adeninivorans cells (or other related species dimorphic
yeast cells), a
substantial number of which being genetically engineered to comprise at least
one modified N-
glycosylation activity (such as any of the modifications described herein).
The culture of cells
can contain one or more subpopulations of cells, each subpopulation comprising
a different
modified glycosylation activity.
In yet another aspect, the disclosure provides: (a) an isolated nucleotide
sequence
comprising SEQ ID NO:1 or SEQ ID NO:2; (b) an isolated nucleotide sequence
comprising a
sequence that is at least 80% identical to SEQ ID NO: I or SEQ ID NO:2; or (c)
a polypeptide
encoded by the isolated nucleotide sequence of (a) or (b). In some
embodiments, the isolated
nucleic acid sequence is SEQ ID NO:1 or SEQ ID NO:2.
7
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
In another aspect, the disclosure features an isolated nucleic acid
containing: (a) a
nucleotide sequence that hybridizes under highly stringent conditions to the
complement of SEQ
ID NO:1 or SEQ ID NO:2; or (b) the complement of the nucleotide sequence.
In yet another aspect, the disclosure provides: (a) an isolated nucleotide
sequence
comprising (or consisting of) any of the nucleic acid sequences depicted
herein; (b) an isolated
nucleotide sequence comprising a sequence that is at least 80% identical to
any of the nucleic
acid sequences depicted herein; or (c) a polypeptide encoded by the isolated
nucleotide sequence
of (a) or (b). In some embodiments, the isolated nucleic acid sequence is any
of the nucleic acid
sequences depicted herein.
In another aspect, the disclosure features an isolated nucleic acid
containing: (a) a
nucleotide sequence that hybridizes under highly stringent conditions to the
complement of any
of the nucleic acid sequences depicted herein; or (b) the complement of the
nucleotide sequence.
In yet another aspect, the disclosure provides: (a) a vector comprising any of
the nucleic
acid sequences described above or (b) a cultured cell containing the vector of
(a). The vector can
be an expression vector. The nucleic acid sequence in the vector can be
operably linked to
expression control sequence.
In another aspect, the disclosure provides a method for producing a protein.
The method
includes the step of culturing any of the cells described above under
conditions permitting the
expression of the polypeptide. The method can also include the step of after
culturing the cell,
isolating the polypeptide from the cell or the medium in which the cell was
cultured. The cell
can be, e.g., a cultured cell containing a vector comprising any of the
nucleic acid sequences
described above.
The target molecules (e.g., target proteins), proteins having N-glycosylation
activity, and
altered N-glycosylation molecules described herein (collectively referred to
as "molecules of the
invention") can, but need not, be isolated. The term "isolated" as applied to
any of the molecules
of the invention described herein refers to a molecule, or a fragment thereof,
that has been
separated or purified from components (e.g., proteins or other naturally-
occurring biological or
organic molecules) which naturally accompany it. It is understood that
recombinant molecules
(e.g., recombinant proteins) will always be "isolated." Typically, a molecule
of the invention is
isolated when it constitutes at least 60%, by weight, of the total molecules
of the same type in a
preparation, e.g., 60% of the total molecules of the same type in a sample.
For example, an
8
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
altered glycosylation protein is isolated when it constitutes at least 60%, by
weight, of the total
protein in a preparation or sample. In some embodiments, a molecule of the
invention in the
preparation consists of at least 75%, at least 90%, or at least 99%, by
weight, of the total
molecules of the same type in a preparation.
As used herein, an "altered N-glycosylation form" of a target molecule is an N-
glycosylation form of a target molecule produced by a genetically engineered
host cell (e.g.,
Yarrowia lipolytica cell, Arxula adeninivorans cell, or a cell of another
related dimorphic yeast
cell species) that differs from the N-glycosylation form of the target
molecule produced in a non-
genetically engineered cell of the same species as the genetically engineered
cell. Thus, an
altered glycosylation form of a target molecule can be, for example, a form of
the target
molecule that is not N-glycosylated. Moreover, an altered glycosylation form
of a target
molecule can be, e.g., a form of the target molecule that has altered
phosphorylation of one or
more N-linked glycans.
As used herein, the term "other related dimorphic yeast cell species" refers
to yeasts
related to Yarrowia lipolytica and Arxula adeninivorans that belong to the
family Dipodascaceae
such as Arxula, Dipodascus (e.g. D. albidus, D. ingens, or D. specifer),
Galactomyces (e.g. G.
reesii or G. geotrichum), Sporopachyderma, Stephanoascus
(e.g., S. cferii), Wickerhamiella, and Zygoascus. Specifically, yeasts in the
Glade Metchnikowia
(e.g., M. pulcherrima or M. agaves) and Stephanoascus (to which Y. lipolytica
is assigned by
analysis of the DI/D2 domain of the 26S-rDNA sequences of species such as
Arxula (e.g. A.
adeninivorans or A. terrestris)) and some Candida species (e.g., C. apicola
but not C. albicans,
C. maltosa, or C. tropicalis).
"Polypeptide" and "protein" are used interchangeably and mean any peptide-
linked chain
of amino acids, regardless of length or post-translational modification.
The disclosure also provides (i) biologically active variants and (ii)
biologically active
fragments or biologically active variants thereof, of the wild-type, full-
length, mature "target
proteins" or "proteins having N-glycosylation activity" described herein.
Biologically active
variants of full-length, mature, wild-type proteins or fragments of the
proteins can contain
additions, deletions, or substitutions. Proteins with substitutions will
generally have not more
than 50 (e.g., not more than one, two, three, four, five, six, seven, eight,
nine, ten, 12, 15, 20, 25,
30, 35, 40, or 50) conservative amino acid substitutions. A conservative
substitution is the
9
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
substitution of one amino acid for another with similar characteristics.
Conservative
substitutions include substitutions within the following groups: valine,
alanine and glycine;
leucine, valine, and isoleucine; aspartic acid and glutamic acid; asparagine
and glutamine; serine,
cysteine, and threonine; lysine and arginine; and phenylalanine and tyrosine.
The non-polar
hydrophobic amino acids include alanine, leucine, isoleucine, valine, proline,
phenylalanine,
tryptophan and methionine. The polar neutral amino acids include glycine,
serine, threonine,
cysteine, tyrosine, asparagine and glutamine. The positively charged (basic)
amino acids include
arginine, lysine and histidine. The negatively charged (acidic) amino acids
include aspartic acid
and glutanvc acid. Any substitution of one member of the above-mentioned
polar, basic or
acidic groups by another member of the same group can be deemed a conservative
substitution.
By contrast, a non-conservative substitution is a substitution of one amino
acid for another with
dissimilar characteristics.
Deletion variants can lack one, two, three, four, five, six, seven, eight,
nine, ten, 11, 12,
13, 14, 15, 16, 17, 18, 19, or 20 amino acid segments (of two or more amino
acids) or non-
contiguous single amino acids.
Additions (addition variants) include fusion proteins containing: (a) full-
length, wild-
type, mature polypeptides or fragments thereof containing at least five amino
acids; and (b)
internal or terminal (C or N) irrelevant or heterologous amino acid sequences.
In the context of
such fusion proteins, the term "heterologous amino acid sequences" refers to
an amino acid
sequence other than (a). A fusion protein containing a peptide described
herein and a
heterologous amino acid sequence thus does not correspond in sequence to all
or part of a
naturally occurring protein. A heterologous sequence can be, for example a
sequence used for
purification of the recombinant protein (e.g., FLAG, polyhistidine (e.g.,
hexahistidine),
hemagluttanin (HA), glutathione-S-transferase (GST), or maltose-binding
protein (MBP)).
Heterologous sequences can also be proteins useful as diagnostic or detectable
markers, for
example, luciferase, green fluorescent protein (GFP), or chloramphenicol
acetyl transferase
(CAT). In some embodiments, the fusion protein contains a signal sequence from
another
protein. In certain host cells (e.g., yeast host cells), expression and/or
secretion of the target
protein can be increased through use of a heterologous signal sequence. In
some embodiments,
the fusion protein can contain a carrier (e.g., KLH) useful, e.g., in
eliciting an immune response
(e.g., for antibody generation; see below) or endoplasmic reticulum or Golgi
apparatus retention
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
signals. Heterologous sequences can be of varying length and in some cases can
be a longer
sequences than the full-length target proteins to which the heterologous
sequences are attached.
A "fragment" as used herein, refers to a segment of the polypeptide that is
shorter than a
full-length, immature protein. Fragments of a protein can have terminal
(carboxy or amino-
terminal) and/or internal deletions. Generally, fragments of a protein will be
at least four (e.g., at
least five, at least six, at least seven, at least eight, at least nine, at
least 10, at least 12, at least 15,
at least 18, at least 25, at least 30, at least 35, at least 40, at least 50,
at least 60, at least 65, at
least 70, at least 75, at least 80, at least 85, at least 90, or at least 100
or more) amino acids in
length.
Biologically active fragments or biologically active variants of the target
proteins or
proteins having N-glycosylation activity have at least 25% (e.g., at least:
30%; 40%; 50%; 60%;
70%; 75%; 80%; 85%; 90%; 95%; 97%; 98%; 99%; 99.5%, or 100% or even greater)
of the
activity of the wild-type, full-length, mature protein. In the case of a
target protein, the relevant
activity is the ability of the target protein to undergo altered N-
glycosylation in a genetically
engineered cell. In the case of a protein having N-glycosylation activity, the
relevant activity is
N-glycosylation activity.
Depending on their intended use, the proteins, biologically active fragments,
or
biologically active variants thereof can be of any species, such as, e.g.,
fungus (including yeast),
nematode, insect, plant, bird, reptile, or mammal (e.g., a mouse, rat, rabbit,
hamster, gerbil, dog,
cat, goat, pig, cow, horse, whale, monkey, or human). In some embodiments,
biologically active
fragments or biologically active variants include immunogenic and antigenic
fragments of the
proteins. An immunogenic fragment is one that has at least 25% (e.g., at
least: 30%; 40%; 50%;
60%; 70%; 75%; 80%; 85%; 90%; 95%; 97%; 98%; 99%; 99.5%, or 100% or even more)
of the
ability of the relevant full-length, immature protein to stimulate an immune
response (e.g., an
antibody response or a cellular immune response) in an animal of interest. An
antigenic
fragment of a protein is one having at least 25% (e.g., at least: 30%; 40%;
50%; 60%; 70%; 75%;
80%; 85%; 90%; 95%; 97%; 98%; 99%; 99.5%, or 100% or even greater) of the
ability of the
relevant full-length, immature protein to be recognized by an antibody
specific for the protein or
a T cell specific to the protein.
"N-glycosylation activity" as used herein refers to any activity that is (1)
capable of
adding N-linked glycans to a target molecule (i.e., an
oligosaccharyltransferase activity); (ii)
11
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
removing N-linked glycans from a target molecule, (iii) modifying one or more
N-linked glycans
on a target molecule, (iv) modifying dolichol-linked oligosaccharides; or (v)
is capable of aiding
the activity of the activities under (i-iv). As such, N-glycosylation activity
includes, e.g., N-
glycosidase activity, glycosidase activity, glycosyltransferase activity,
sugar nucleotide
synthesis, modification, or transporter activity. Modification of one or more
N-linked glycans on
a target molecule includes the action of a mannosylphosphoryltransferase
activity, a kinase
activity, or a phosphatase activity, e.g., a mannosylphosphoryltransferase, a
kinase, or a
phosphatase activity that alters the phosphorylation state of N-linked glycans
on target
molecules.
As used herein, to "genetically engineer" a cell or a "genetically engineered
cell" and like
terminology refers to any artificially created genetic alteration of a cell
that results in at least one
modified N-glycosylation activity in the cell as compared to a non-genetically
engineered cell
(e.g., a fungal cell such as Yarrowia lipolytica cell, Arxula adeninivorans
cell, or other related
species dimorphic yeast cell, a plant cell, or an animal cell (e.g., a
mammalian cell such as a
human cell)). Thus, it is understood that artificially created genetic
alterations do not include,
e.g., spontaneous mutations. Examples of artificial genetic alterations are
described below (see
"Genetically Engineered Cells").
As used herein, the term "wild-type" as applied to a nucleic acid or
polypeptide refers to
a nucleic acid or a polypeptide that occurs in, or is produced by,
respectively, a biological
organism as that biological organism exists in nature.
The term "heterologous" as applied herein to a nucleic acid in a host cell or
a polypeptide
produced by a host cell refers to any nucleic acid or polypeptide (e.g., an
protein having N-
glycosylation activity) that is not derived from a cell of the same species as
the host cell.
Accordingly, as used herein, "homologous" nucleic acids, or proteins, are
those that occur in, or
are produced by, a cell of the same species as the host cell.
The term "exogenous" as used herein with reference to nucleic acid and a
particular host
cell refers to any nucleic acid that does not occur in (and cannot be obtained
from) that particular
cell as found in nature. Thus, a non-naturally-occurring nucleic acid is
considered to be
exogenous to a host cell once introduced into the host cell. It is important
to note that non-
naturally-occurring nucleic acids can contain nucleic acid subsequences or
fragments of nucleic
acid sequences that are found in nature provided that the nucleic acid as a
whole does not exist in
12
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
nature. For example, a nucleic acid molecule containing a genomic DNA sequence
within an
expression vector is non-naturally-occurring nucleic acid, and thus is
exogenous to a host cell
once introduced into the host cell, since that nucleic acid molecule as a
whole (genomic DNA
plus vector DNA) does not exist in nature. Thus, any vector, autonomously
replicating plasmid,
or virus (e.g., retrovirus, adenovirus, or herpes virus) that as a whole does
not exist in nature is
considered to be non-naturally-occurring nucleic acid. It follows that genomic
DNA fragments
produced by PCR or restriction endonuclease treatment as well as cDNAs are
considered to be
non-naturally-occurring nucleic acid since they exist as separate molecules
not found in nature.
It also follows that any nucleic acid containing a promoter sequence and
polypeptide-encoding
sequence (e.g., cDNA or genomic DNA) in an arrangement not found in nature is
non-naturally-
occurring nucleic acid. A nucleic acid that is naturally-occurring can be
exogenous to a
particular cell. For example, an entire chromosome isolated from a cell of
yeast x is an
exogenous nucleic acid with respect to a cell of yeast y once that chromosome
is introduced into
a cell of yeast y.
It will be clear from the above that "exogenous" nucleic acids can be
"homologous" or
"heterologous" nucleic acids. In contrast, the term "endogenous" as used
herein with reference
to nucleic acids or genes (or proteins encoded by the nucleic acids or genes)
and a particular cell
refers to any nucleic acid or gene that does occur in (and can be obtained
from) that particular
cell as found in nature.
As an illustration of the above concepts, an expression plasmid encoding a Y.
lipolytica
ALG6 protein that is transformed into a Y. lipolytica cell is, with respect to
that cell, an
exogenous nucleic acid. However, the ALG6 protein coding sequence and the ALG6
protein
produced by it are homologous with respect to the cell. Similarly, an
expression plasmid
encoding a Arxula adeninivorans ALG6 protein that is transformed into a Y.
lipolytica cell is,
with respect to that cell, an exogenous nucleic acid. In contrast with the
previous example,
however, the ALG6 protein coding sequence and the ALG6 protein produced by it
are
heterologous with respect to the cell.
As used herein, a "promoter" refers to a DNA sequence that enables a gene to
be
transcribed. The promoter is recognized by RNA polymerase, which then
initiates transcription.
Thus, a promoter contains a DNA sequence that is either bound directly by, or
is involved in the
recruitment, of RNA polymerase. A promoter sequence can also include "enhancer
regions,"
13
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
which are one or more regions of DNA that can be bound with proteins (namely,
the trans-acting
factors, much like a set of transcription factors) to enhance transcription
levels of genes (hence
the name) in a gene-cluster. The enhancer, while typically at the 5' end of a
coding region, can
also be separate from a promoter sequence and can be, e.g., within an intronic
region of a gene or
3' to the coding region of the gene.
As used herein, "operably linked" means incorporated into a genetic construct
so that
expression control sequences effectively control expression of a coding
sequence of interest.
Variants of any of the nucleic acid sequences described herein (e.g., the HACI
sequences
as depicted in SEQ ID NO:1 or SEQ ID NO:2) can have a sequence that is
homologous, e.g., a
sequence bearing at least about 70% (e.g., at least about 75%, at least about
80%, at least about
85%, at least about 90%, at least about 95%, or at least about 99%) homologous
(identical) to the
wild-type nucleic acid sequence. Such wild-type sequences can be isolated from
nature or can be
produced by recombinant or synthetic methods. Thus a wild-type sequence
nucleic acid can
have the nucleic acid sequence of naturally occurring human nucleic acid
sequences, monkey
nucleic acid sequences, murine nucleic acid sequences, or any other species
that contains a
homologue of the wild-type nucleic acid of interest. As used herein, a
"homologous" or
"homologous nucleic acid sequence" or similar term, refers to sequences
characterized by
homology at the nucleotide level of at least a specified percentage and is
used interchangeably
with sequence identity.
Percent homology or identity can be determined by, for example, the Gap
program
(Wisconsin Sequence Analysis Package, Version 8 for UNIX, Genetics Computer
Group,
University Research Park, Madison, WI), using default settings, which uses the
algorithm of
Smith and Waterman ((1981) Adv. Appl. Math. 2:482-489). In some embodiments,
homology
between a probe and target (see below) is between about 50% to about 60%. In
some
embodiments, homology between a probe and target nucleic acid is between about
55% to 65%,
between about 65% to 75%, between about 70% to 80%, between about 75% and 85%,
between
about 80% and 90%, between about 85% and 95%, or between about 90% and 100%.
The term "probe," as used herein, refers to nucleic acid sequences of variable
length. In
some embodiments, probes comprise at least 10 and as many as 6,000
nucleotides. In some
embodiments probes comprise at least 12, at lease 14, at least 16, at least
18, at least 20, at least
25, at least 50 or at least 75 or 100 contiguous nucleotides. Longer length
probes are usually
14
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
obtained from natural or recombinant sources (as opposed to direct, chemical
synthesis), are
highly specific to the target sequence, and are much slower to hybridize to
the target than longer
oligomers. Probes can be single or double stranded nucleic acid molecules.
In some embodiments, a variant nucleic acid described herein can have a
sequence
comprising one or both strands with partial complementary (e.g., at least 50%,
at least 60%, at
least 70%, at least 80%, at least 90%, at least 95%, at least 96%, at least
97%, at least 98%, at
least 99% complementary) to a region, portion, domain, or segment of the wild-
type nucleic acid
of interest (e.g., the HAC I nucleic acid sequences as depicted in SEQ ID NO:
I or SEQ ID
NO:2). In some embodiments, a variant nucleic acid sequence of interest can
have a sequence
comprising one or both strands with full complementary (i.e., 100%
complementary) to a region,
portion, domain, or segment of the wild-type nucleic acid sequence. Sequence
"complementarity" refers to the chemical affinity between specific nitrogenous
bases as a result
of their hydrogen bonding properties (i.e., the property of two nucleic acid
chains having base
sequences such that an antiparallel duplex can form where the adenines and
uracils (or thymine,
in the case of DNA or modified RNA) are apposed to each other, and the
guanines and cytosines
are apposed to each other). Fully complementary sequences, thus, would be two
sequences that
have complete one-to-one correspondence (i.e., adenine to uraciUthymidine and
guanine to
cytosine) of the base sequences when the nucleotide sequences form an
antiparallel duplex.
Hybridization can also be used as a measure of homology between two nucleic
acid
sequences. A nucleic acid sequence described herein, or a fragment or variant
thereof, can be
used as a hybridization probe according to standard hybridization techniques.
The hybridization
of a certain probe of interest (e.g., a probe of a HACI nucleotide sequence,
e.g., the HAC1
nucleotide sequences as depicted in SEQ ID NOS:1 or 2) to DNA or RNA from a
test source
(e.g., a eukaryotic cell) is an indication of the presence of DNA or RNA
(e.g., a HAC 1
nucleotide sequence) corresponding to the probe in the test source.
Hybridization conditions are
known to those skilled in the art and can be found in Current Protocols in
Molecular Biology,
John Wiley & Sons, N.Y., 6.3.1-6.3.6, 1991. Moderate hybridization conditions
are defined as
equivalent to hybridization in 2X sodium chloride/sodium citrate (SSC) at 30
C, followed by a
wash in I X SSC, 0.1% SDS at 50 C. Highly stringent conditions are defined as
equivalent to
hybridization in 6X sodium chloride/sodium citrate (SSC) at 45 C, followed by
a wash in 0.2 X
SSC, 0.1 % SDS at 65 C.
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
Unless otherwise defined, all technical and scientific terms used herein have
the same
meaning as commonly understood by one of ordinary skill in the art to which
this invention
belongs. Although methods and materials similar or equivalent to those
described herein can be
used in the practice or testing of the present invention, the exemplary
methods and materials are
described below. In case of conflict, the present application, including
definitions, will control.
The materials, methods, and examples are illustrative only and not intended to
be limiting.
Other features and advantages of the invention, e.g., methods of producing
altered N-
glycosylation molecules, will be apparent from the following detailed
description, and from the
claims.
Brief Description of the Drawings
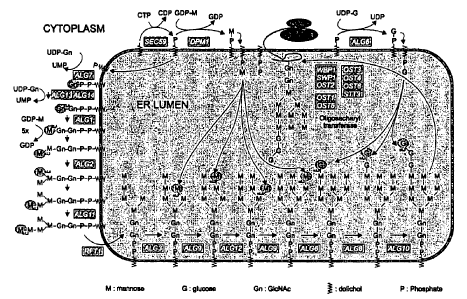
Fig. 1 is a schematic diagram depicting N-glycan precursor synthesis at the
yeast
endoplasmic reticulum. Genes whose encoded protein has an activity mediating
the indicated
enzymatic conversions are in shaded boxes (e.g., ALG7; upper left). "UDP" and
"UMP" refer to
uridine diphosphate and uridine monophosphate, respectively. "GDP" and "GMP"
refer to
guanosine diphosphate and guanosine monophosphate respectively. "Gn" refers to
N-
acetylglucosamine. "M" refers to monomeric mannose, G refers to glucose, Pi
refers to
phosphate
Fig. 2 is a schematic diagram depicting N-glycan processing in the yeast
endoplasmic
reticulum.
Fig. 3 is a schematic diagram depicting N-glycan processing in the S.
cerevisiae Golgi
apparatus. Genes whose encoded protein has an activity mediating the indicated
enzymatic
conversions are in shaded boxes (e.g., OCH 1; middle left).
Fig. 4 is a schematic diagram depicting the structure of the various N-glycan
structures
described herein.
Fig. 5 is a schematic diagram depicting the cloning strategy for OCH1 gene
disruption in
Yarrowia lipolytica. "PCR" refers to polymerase chain reaction.
16
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
Fig. 6 is a schematic diagram depicting the cloning strategy for MNN9 gene
disruption
fragment. "PCR" refers to polymerase chain reaction.
Fig. 7 is a series of electroferograms depicting N-glycan analysis of
mannoproteins
obtained from wild-type Yarrowia lipolytica cells or glycosylation mutant
(e.g., Dochl c19,
Omnn9 I and Aochl Amnn9) cells and MTLY60 strain cells. In some cases, the N-
glycans were
further treated with a-1,2 mannosidase. Analysis was performed using DNA
sequencer-assisted,
fluorophore-assisted carbohydrate electrophoresis (DSA-FACE). "M5," "M6,"
"M7," "M8,"
and "M9," "refer to the number of mannose residues conjugated to the base N-
acetylglucosamine
structure. The Y-axis represents the relative fluorescence units as an
indication of the amount of
each of the mannose structures. The X-axis represents the relative mobility of
each complex
mannose structure through a gel. The top electroferogram is an analysis of
oligomaltose for use
as a mobility standard.
Fig. 8 is a schematic diagram depicting the cloning strategy for S. cerevisiae
MNSI
expression vector. "PCR" refers to polymerase chain reaction.
Fig. 9 is a series of electroferograms depicting N-glycan analysis of secreted
glycoproteins obtained from MTLY60 cells expressing wild-type (WT) Mnslp or
various mutant
forms (i.e., R273G, R273L, or R269S/S272G/R273L) of Mnslp as indicated.
Analysis was
performed using DSA-FACE. "M5," "M6," "M7," "M8," "M9," "refers to the number
of
mannose residues conjugated to the base N-acetylglucosamine structure. The Y-
axis represents
the relative fluorescence units as an indication of the amount of each of the
mannose structures.
The X-axis represents the relative mobility of each complex mannose structure
through a gel.
The top electroferogram is an analysis of oligomaltose for use as a mobility
standard.
Fig. 10 is a schematic diagram depicting the cloning strategy for an MNN4
expression
vector.
Fig. 11 is a series of electroferograms depicting N-glycan analysis of
secreted
glycoproteins obtained from wild-type MTLY60 cells or glycosylation mutant
cells as indicated.
Analysis was performed using DSA-FACE. "M5," "M6," "M7," "M8,"M9," refers to
the
number of mannose residues conjugated to the chitobiose core structure. "P"
refers to
manmoproteins containing one phosphate residue and "PP" refers to
mannoproteins containing
two phosphate residues. The Y-axis represents the relative fluorescence units
as an indication of
the amount of each of the mannose structures. The X-axis represents the
relative mobility of
17
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
each complex mannose structure through a gel. The top electroferogram is an
analysis of
oligomaltose for use as a mobility standard.
Fig. 12 is a schematic diagram depicting the cloning strategy for an a-
galactosidase
expression vector.
Fig. 13 is a series of electroferograms depicting N-glycan analysis of
mannoproteins and
phosphomannoproteins obtained from wild-type MTLY60 cells or various clones of
glycosylation mutant cells as indicated. "alga" indicates that the cell is an
ALG3 knockout.
"ALG6 overexpression" indicates that the protein product of ALG6 is
overexpressed in the cell.
Analysis was performed using DSA-FACE. "M5," "M6," "M7," "M8," and "M9,""
refer to the
number of mannose residues conjugated to the base N-acetylglucosamine
structure, The Y-axis
represents the relative fluorescence units as an indication of the amount of
each of the mannose
structures. The X-axis represents the relative mobility of each complex
mannose structure
through a polyacrylamide gel. The top electroferogram is an analysis of
oligomaltose for use as
a mobility standard.
Fig. 14 is a series of electroferograms depicting N-glycan analysis of
mannoproteins and
phosphomannoproteins obtained from wild-type MTLY60 cells or various clones of
glycosylation mutant cells as indicated. "alga" indicates that the cell is an
ALG3 knockout.
"ALG6 overexpression" indicates that the protein product of ALG6 is
overexpressed in the cell.
One peak runs at the same position as Man5GlcNAc2 of the RNaseB marker and
shifts with two
glucose-units after a-1,2-mannosidase treatment and with 4 glucose-units after
alpha-
mannosidase (JB) digest. This fits with a Man5GlcNAc2 structure as expected.
The additional
two peaks run at a distance of about one and two glyco-units and are not
affected by a-1,2-
mannosidase digestion. Both peaks shift one glucose-unit upon a-mannosidase
(JB) digestion.
Minor shifts are due to the higher salt concentrations of the added enzymes,
e.g. JB mannosidase.
Analysis was performed using DSA-FACE. "M5," "M6," "M7," "M8," and "M9," refer
to the
number of mannose residues conjugated to the chitobiose core structure. The Y-
axis represents
the relative fluorescence units as an indication of the amount of each of the
mannose structures.
The X-axis represents the relative mobility of each complex mannose structure
through a gel.
The top electroferogram is an analysis of oligomaltose for use as a mobility
standard.
Fig. 15 is a sequence alignment of an isolated DNA fragment (SEQ ID NO:1)
sequence
obtained from the unfolded protein response (UPR)-induced strain Yarrowia
lipolylica with a
18
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
genomic HAC1 DNA sequence (SEQ ID NO:5). The boxed sequence corresponds to the
non-
conventionally spliced intron.
Fig. 16 is a series of sequence alignments of the predicted 5' (top) and 3'
(bottom) splice
sites of Pichia pastoris and Saccharomyces cerevisiae. Nucleotides in bold
underlined are
present in the loop structure.
Fig. 17A and 17B are two partial views of a sequence alignment of the HACI
cDNA
obtained from DTT-induced (I) (SEQ ID NO:2) and non-induced (NI) (SEQ ID NO:6)
Pichia
pastoris cultures.
Fig. 18 is a sequence alignment of the 18 amino acid C-terminal regions of
Pichia
pastoris and Saccharomyces cerevisiae. Conserved amino acids are in bold and
underlined.
Fig. 19 is a bar graph depicting the comparison of the relative expression
levels of KAR2
mRNA. Clones 3, 4, and 5 (Pichia pastoris GSM5 cells) were grown on methanol
as carbon
source. "3+," "4+," and "5+" refer to the respective clones grown on methanol
as carbon source,
whereas "3-," "4-," and "5-" refer to the respective clones grown on glucose
as carbon source.
The Y-axis represents the relative expression of the KAR2 gene using real-time
PCR.
Fig. 20 is a bar graph depicting the relative expression level of Kart and HAC
1 mRNA
in two Pichia pastoris clones (clone 6 and 8). "6+" and "8+" refer to the
respective clones grown
on methanol as carbon source, whereas "6-"and "8-" refer to the respective
clones grown on
glucose as carbon source. The Y-axis represents the relative expression of the
KAR2 gene using
real-time PCR.
Fig. 21 is a schematic diagram depicting the cloning strategy for a YIMNN6
expression
vector.
Fig. 22 is a series of electroferograms depicting N-glycan analysis of
glycoproteins
obtained from Dochl Y. lipolytica cells, alone, or various clones (Z3, Z4, Z5,
U5, U6, and U8) of
Aoch1 Y. lipolytica expressing YIMNN6 as indicated. Analysis was performed
using DSA-
FACE. The Y-axis represents the relative fluorescence units as an indication
of the amount of
each of the mannose structures. The X-axis represents the relative mobility of
each complex
mannose structure through a gel. The top electroferogram is an analysis of
oligomaltose for use
as a mobility standard.
Fig. 23 is a schematic diagram depicting the cloning strategy for an MFManHDEL
expression vector.
19
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
Fig. 24 is a series of electroferograms depicting N-glycan analysis of
glycoproteins
obtained from Aochl Y. lipolytica cells, alone, or various clones (9, 11, 10,
3, 5, and 6) of Aochl
Y. lipolytica expressing MFManHDEL as indicated. Analysis was performed using
DSA-
FACE. The Y-axis represents the relative fluorescence units as an indication
of the amount of
each of the mannose structures. The X-axis represents the relative mobility of
each complex
mannose structure through a gel. The top electroferogram is an analysis of
oligomaltose for use
as a mobility standard.
Fig. 25 is a schematic diagram depicting the cloning strategy for an
LIP2preManHDEL
expression vector.
Fig. 26 is a series of electroferograms depicting N-glycan analysis of
glycoproteins
obtained from Aochl Y. lipolytica cells, alone, or various clones (1, 5, 10,
and 11) of Aochl Y.
lipolytica expressing LIP2ManHDEL as indicated. Analysis was performed using
DSA-FACE.
"M5," "M6," "M7," "M8," and "M9," refer to the number of mannose residues
conjugated to the
chitobiose core structure. The Y-axis represents the relative fluorescence
units as an indication
of the amount of each of the mannose structures. The X-axis represents the
relative mobility of
each complex mannose structure through a gel. The top electroferogram is an
analysis of
oligomaltose for use as a mobility standard.
Figs. 27A and 27B are amino acid sequences of HAC I proteins of Yarrowia
lipolytica
(Fig. 27A; SEQ ID NO:3) and Pichia pastoris (Fig. 27B; SEQ ID NO:4).
Fig. 28 is a photograph of a Coomassie blue stained polyacrylamide gel
depicting the
results of Lip2p overexpression in various Yarrowia lipolytica cell (MTLY60,
MTLY60Aa1g3
and MTLY60Aalg3ALG6) cultures. The following samples were resolved in the gel:
Lane 1
("ladder"), a combination of proteins of known molecular weight; Lane 2
("WT"), Lip2p protein
obtained from WT Yarrowia lipolytica cells (MTLY60) overexpressing Lip2p; Lane
3
("WT+PGase F"), Lip2p protein obtained from WT Yarrowia lipolytica cells
overexpressing
Lip2p and treated with PNGase F enzyme; Lane 4 ("alg3-ALG6"), Lip2p protein
obtained from
Yarrowia cells deficient in alg3 and overexpressing both Lip2p and ALG6
(MTLY60.alg3ALG6
); Lane 5 ("alg3-ALG6+PNGase F"), Lip2p protein obtained from Yarrowia cells
deficient in
alg3 and overexpressing both Lip2p and ALG6 (MTLY60talg3ALG6) and treated with
PNGase
F enzyme; Lane 6 ("alga"), Lip2p protein obtained from Yarrowia lipolytica
cells deficient in
alg3 and overexpressing Lip2p (MTLY60Aalg3); Lane 7 ("alg3 + PNGase F"), Lip2p
protein
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
obtained from Yarrowia lipolytica cells deficient in alg3 and overexpressing
Lip2p
(MTLY60Aalg3) treated with PNGase F enzyme; Lane 8 ("WT without Lip2p
overexpression"),
protein obtained from MTLY60 cells; and Lane 9 ("WT without Lip2p
overexpression +
PNGase F"), protein obtained from MTLY60 cells and treated with PNGase F
enzyme.
Fig. 29 is a series of electroferograms depicting N-glycan analysis of
glycoproteins
obtained from various Yarrowia lipolytica cells (WT (MTLY60); zalg3; Aalg3
ALG6
overexpressing; and clones of Aalg3 overexpressing ALG6 along with the alpha
subunit of
glucosidase II from Y. lipolytica (Yl) or Typanosoma brucei (Th)) as
indicated. Analysis was
performed using DSA-FACE. "M5," "M6," "M7," "M8," and "M9," refer to the
number of
mannose residues conjugated to the chitobiose core structure. The Y-axis
represents the relative
fluorescence units as an indication of the amount of each of the mannose
structures. The X-axis
represents the relative mobility of each complex mannose structure through a
gel. The top
electroferogram is an analysis of oligomaltose for use as a mobility standard.
The bottom
electroferogram is an analysis of RNAse B.
Fig. 30 is a series of electroferograms depicting N-glycan analysis of
glycoproteins
obtained from various Yarrowia lipolytica cells (Ealg3; Aalg3 ALG6
overexpressing; and clones
of Aalg3 overexpressing ALG6 along with the alpha subunit of glucosidase II
from Y. lipolytica
(Yl) containing an HDEL sequence as indicated. Analysis was performed using
DSA-FACE.
The Y-axis represents the relative fluorescence units as an indication of the
amount of each of
the mannose structures. The X-axis represents the relative mobility of each
complex mannose
structure through a gel.
Fig. 31 is a series of electroferograms depicting N-glycan analysis of
glycoproteins
obtained from various Yarrowia lipolytica cells (Aalg3; Aalg3 ALG6
overexpressing; and clones
of Aalg3 overexpressing ALG6 along with the alpha subunit of glucosidase II
from Trypanosoma
brucei (Th) containing an HDEL sequence) as indicated. Analysis was performed
using DSA-
FACE. The Y-axis represents the relative fluorescence units as an indication
of the amount of
each of the mannose structures. The X-axis represents the relative mobility of
each complex
mannose structure through a gel.
Fig. 32 is a series of electroferograms depicting N-glycan analysis of
glycoproteins
obtained from alg3ALG6 Yarrowia lipolytica cells treated in vitro with
different concentrations
of mutanase as indicated. Analysis was performed using DSA-FACE. The Y-axis
represents the
21
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
relative fluorescence units as an indication of the amount of each of the
mannose structures. The
X-axis represents the relative mobility of each complex mannose structure
through a gel. The
top electroferogram is an analysis of oligomaltose for use as a mobility
standard. The bottom
electroferogram is an analysis of RNAse B.
Fig. 33 is a series of electroferograms depicting N-glycan analysis of
glycoproteins
obtained from various Yarrowia lipolytica cells (Dalg3; Aalg3 ALG6
overexpressing; and clones
of Dalg3 overexpressing ALG6 along with the alpha subunit of glucosidase II
from Y. lipolytica
(Y.1.) and the beta subunit of glucosidase II from Y.I. expressed under the
control of Hp4d or
TEF promoters) as indicated. The Y-axis represents the relative fluorescence
units as an
indication of the amount of each of the mannose structures. The X-axis
represents the relative
mobility of each complex mannose structure through a gel. The top
electroferogram is an
analysis of oligomaltose for use as a mobility standard. The bottom
electroferogram is an
analysis of RNAse B.
Fig. 34 is a series of electroferograms depicting N-glycan analysis of
glycoproteins
obtained from various Yarrowia lipolytica cells (Dalg3 ALG6 overexpressing;
and clones of
Dalg3 overexpressing ALG6 along with the HDEL-containing alpha subunit of
glucosidase II
from Y. lipolytica (Y.1.) and the beta subunit of glucosidase II from Y.I.
expressed under the
control of Hp4d or TEF promoters) as indicated. Analysis was performed using
DSA-FACE.
The Y-axis represents the relative fluorescence units as an indication of the
amount of each of
the mannose structures. The X-axis represents the relative mobility of each
complex mannose
structure through a gel. The top electroferogram is an analysis of
oligomaltose for use as a
mobility standard. The bottom electroferogram is an analysis of RNAse B.
Fig. 35 is a series of electroferograms depicting N-glycan analysis of
glycoproteins
obtained from various Yarrowia lipolytica cells (Dalg3 and clones of Aalg3
overexpressing the
alpha subunit of glucosidase II from Y. lipolytica (Y.l.) and the beta subunit
of glucosidase II
from Y.I. expressed under the control of a TEF promoter) as indicated. The Y-
axis represents
the relative fluorescence units as an indication of the amount of each of the
mannose structures.
The X-axis represents the relative mobility of each complex mannose structure
through a gel.
The top electroferogram is an analysis of oligomaltose for use as a mobility
standard. The
bottom electroferogram is an analysis of RNAse B.
22
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
Figs. 36A and 36B is the depiction of a nucleotide sequence of a cDNA encoding
a
mature form of Aspergillus niger (lacking signal peptide) glucosidase II a,
which is codon-
optimized cDNA for expression in Yarrowia lipolytica. (SEQ ID NO:7).
Fig. 37 is the depiction of a nucleotide sequence of a cDNA encoding a mature
form of
Aspergillus niger (lacking signal peptide) glucosidase lI (3, which is codon-
optimized cDNA for
expression in Yarrowia lipolytica. (SEQ ID NO:8).
Fig. 38 is a series of electroferograms depicting N-glycan analysis of
glycoproteins
obtained from various Yarrowia lipolytica cells (Aalg3 and ALG6 overexpressing
along with the
alpha subunit of glucosidase II from Aspergillus niger (An) expressed under
the control of a TEF
or hp4d promoter) as indicated. The Y-axis represents the relative
fluorescence units as an
indication of the amount of each of the mannose structures. The X-axis
represents the relative
mobility of each complex mannose structure through a gel. The top
electroferogram is an
analysis of oligomaltose for use as a mobility standard. The bottom
electroferogram is an
analysis of RNAse B.
Figs. 39A and 3916 are a pair of bar graphs depicting the relative expression
level (Y-
axis) of the HAC1 (39A) or KAR (39B) gene in WT (MTLY60) Yarrowia lipolytica
cells or in
two clones (clone 7 and clone 2) of Yarrowia lipolytica cells containing a
spliced form of HAC I
cDNA under the expression control of the hp4d promoter.
Fig. 40 is line graph depicting the growth of wild type Pichia pastoris GS115
cells
transformed with an empty vector as compared to the growth of Pichia pastoris
GS 115 cells
expressing the HacIp protein.
Fig. 41 is a photograph of a Coomassie blue stained polyacrylamide gel
comparing the
expression level of the murine IL-10 (mIL-10) protein from a culture of Pichia
pastoris GS 115
cell cells expressing mIL- 10 protein with the expression of the mIL-10
protein obtained from a
culture of GS 115 cells expressing mIL-10 and the spliced HACI protein from
Pichia pastoris
under the control of an inducible promoter, AOX 1. The following samples were
resolved in the
gel: Lane I ("ladder"), a combination of proteins of known molecular weight;
Lane 2
("Reference"), protein obtained from the reference mIL-10 expressing Pichia
pastoris strain
(GS 115); Lane 3 ("Reference"), protein obtained from the reference mIL-10
expressing Pichia
pastoris strain after PNGase F enzyme treatment of the proteins; Lane 4
("Clone I"), protein
obtained from a mIL-l0 expressing Pichia pastoris cells inducibly expressing
HACI protein;
23
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
Lane 5 ("Clone 1"), protein obtained from a mIL-l0 expressing Pichia pastoris
cells inducibly
expressing HACI protein after treatment of the protein with PNGase F enzyme;
Lane 6 ("Clone
2"), protein obtained from a mIL-l0 expressing Pichia pastoris cells inducibly
expressing
HACI protein I; Lane 7 ("Clone 2"), protein obtained from a mIL-10 expressing
Pichia pastoris
cells inducibly expressing HACI protein after treatment of the proteins with
PNGase F enzyme.
Fig. 42 is the depiction of a nucleotide sequence of an exemplary cDNA
sequence
encoding a Trichoderma reesei a-1,2 mannosidase, codon optimized for
expression in Yarrowia
lipolytica (SEQ ID NO:9) containing the LIP2 pre signal sequence.
Fig. 43 is the depiction of a nucleotide sequence of an exemplary nucleotide
sequence for
the GAP promoter of Yarrowia lipolytica. (SEQ ID NO: 10).
Figs. 44A-44C are the depiction of a nucleotide sequence of an exemplary
nucleic acid
sequence (SEQ ID NO: 11) for the expression vector pYLHUXdL2preManHDEL, which
contains a cDNA sequence encoding a Trichoderma reesei a-1,2 mannosidase,
codon optimized
for expression in Yarrowia lipolytica and containing the LIP2 pre signal
sequence.
Figs. 45A-45C are the depiction of a nucleotide sequence of an exemplary
nucleic acid
sequence (SEQ ID NO: 12) for the expression vector pYLGUXdL2preManHDEL, which
contains a cDNA sequence encoding a Trichoderma reesei a-1,2 mannosidase,
codon optimized
for expression in Yarrowia lipolytica and containing the LIP2 pre signal
sequence.
Figs. 46A-46C are the depiction of a nucleotide sequence of an exemplary
nucleic acid
sequence (SEQ ID NO: 13) for the expression vector pYLPUXdL2preManHDEL, which
contains
a cDNA sequence encoding a Trichoderma reesei a-1,2 mannosidase, codon
optimized for
expression in Yarrowia lipolytica and containing the LIP2 pre signal sequence.
Figs. 47A-47C are the depiction of a nucleotide sequence of an exemplary
nucleic acid
sequence (SEQ ID NO: 14) for the expression vector pYLTUXdL2preManHDEL, which
contains
a cDNA sequence encoding a Trichoderma reesei a-1,2 mannosidase, codon
optimized for
expression in Yarrowia lipolytica and containing the LIP2 pre signal sequence.
Fig. 48 is a series of electroferograms depicting N-glycan analysis of
glycoproteins
obtained from Yarrowia lipolytica cells transformed with different expression
vectors as
indicated: "hp4dL2ManHDEL" (pYLHUXdL2preManHDEL, Figs. 44A-44C);
"GAPL2ManHDEL" (pYLGUXdL2preManHDEL, Figs. 45A-45C); "TEFI L2ManHDEL"
(pYLTUXdL2preManHDEL, Figs. 47A-47C). The Y-axis represents the relative
fluorescence
24
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
units as an indication of the amount of each of the mannose structures. The X-
axis represents the
relative mobility of each complex mannose structure through a gel. The top
electroferogram is
an analysis of dextran for use as a mobility standard. The second
electroferogram in the series is
an analysis of RNAse B.
Fig. 49 is a series of electroferograms depicting N-glycan analysis of
glycoproteins
obtained from Yarrowia lipolytica MTLY60 DochI cells containing a stably
integrated
expression vector pYLTUXdL2preManHDEL (Figs. 47A-47C). Glycoprotein samples
were
obtained from cell cultures at 24, 48, 72, and 96 hours. The top
electroferogram is an analysis of
dextran for use as a mobility standard. The second electroferogram in the
series is an analysis of
RNAse B.
Fig. 50 is an exemplary nucleic acid sequence for human glucocerebrosidase
(GLCM,
Swiss Prot entry nr: P04062; SEQ ID NO: 15), which was chemically synthesized
as a codon-
optimized cDNA for expression in Yarrowia lipolytica.
Fig. 51 is a photograph of an immunoblot depicting the mobility pattern of
human
glucocerebrosidase expressed in Yarrowia lipolytica strains MTLY60 (WT; lanes
4 and 6) and
MTLY600och1 (t ochl; first three lanes). The molecular weight (kDa) of the
proteins is
depicted, by way of molecular weight markers, at the far right of the
immunoblot.
Fig. 52 is an exemplary nucleic acid sequence for human erythropoietin (Epo,
Swiss Prot
entry nr: P01588; SEQ ID NO: 16), which was chemically synthesized as a codon-
optimized
cDNA for expression in Yarrowia lipolytica.
Fig. 53 is an exemplary nucleic acid sequence for human a-galactosidase A
(AGAL,
Swiss Prot entry nr: P06280; SEQ ID NO:17), which was chemically synthesized
as a codon-
optimized cDNA for expression in Yarrowia lipolytica.
Fig. 54 is a series of electron micrographs of wild type Pichia pastoris cells
or Pichia
pastoris cells overexpressing the spliced form of Haclp protein. Discrete
regions of stacked
lipid membranes in the cells are boxed.
Fig. 55 is a series of electroferograms depicting N-glycan analysis of
glycoproteins
obtained from WT Yarrowia lipolytica cells (polld) and Yarrowia lipolytica
cells expressing a
fusion protein of alpha-l,2-mannosidase and a HDEL sequence as indicated.
Analysis was
performed using DSA-FACE. "M5," "M6," "M7," "M8," and "M9," refer to the
number of
mannose residues conjugated to the chitobiose core structure. The Y-axis
represents the relative
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
fluorescence units as an indication of the amount of each of the mannose
structures. The X-axis
represents the relative mobility of each complex mannose structure through a
gel. The top
electroferogram is an analysis of RNAse B. The bottom electroferogram is an
analysis of
oligomaltose for use as a mobility standard.
Detailed Description
The methods and genetically engineered cells described herein can be used to
produce
target molecules (e.g., target protein or target dolichol) having an altered N-
glycosylation form
as compared to the N-glycosylation form of the target molecules produced in
non-genetically
engineered cells. Administration of glycosylated target molecules (e.g.,
glycosylated proteins) to
patients having metabolic disorders (e.g., lysosomal storage disorders) has
been shown to
ameliorate the symptoms of the disorders. Thus, the methods and cells
described are useful for
the preparation of altered N-glycosylated target molecules for, inter alia,
the treatment of
metabolic disorders such as lysosomal storage disorders. Such altered N-
glycosylation
molecules are also useful in a wide-variety of other fields, e.g., the food
and beverage industries;
the pharmaceutical industry (e.g., as vaccines); the agriculture industry; and
the chemical
industry, to name a few.
Altered N-Glycosylation Molecule
Target molecules, as used herein, refer to any molecules that undergo altered
N-
glycosylation by one or more N-glycosylation activities from a genetically
engineered cell (e.g.,
a fungal cell such as Yarrowia lipolvtica or Arxula adeninivorans (or other
related species
dimorphic yeast) cell; a plant cell, or an animal cell). In some embodiments,
the target
molecules are capable of being trafficked through one or more steps of the
Yarrowia lipolytica or
Arxxula adeninivorans (or other related species dimorphic yeast) secretory
pathway, resulting in
their altered N-glycosylation by the host cell machinery. The target molecules
can be
endogenous or exogenous.
Target proteins, their biologically active fragments, or biologically active
variants
thereof, can include proteins containing additions, deletions, or
substitutions as described above.
Suitable target proteins include pathogen proteins (e.g., tetanus toxoid;
diptheria toxoid; viral
26
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
surface proteins (e.g., cytomegalovirus (CMV) glycoproteins B, H and gCIII;
human
immunodeficiency virus I (HIV-1) envelope glycoproteins; Rous sarcoma virus
(RSV) envelope
glycoproteins; herpes simplex virus (HSV) envelope glycoproteins; Epstein Barr
virus (EBV)
envelope glycoproteins; varicella-zoster virus (VZV) envelope glycoproteins;
human papilloma
virus (HPV) envelope glycoproteins; Influenza virus glycoproteins; and
Hepatitis family surface
antigens), lysosomal proteins (e.g., glucocerebrosidase, cerebrosidase, or
galactocerebrosidase),
insulin, glucagon, growth factors, cytokines, chemokines, antibodies or
fragments thereof, or
fusions of any of the proteins to antibodies or fragments of antibodies (e.g.,
protein-Fc). Growth
factors include, e.g., vascular endothelial growth factor (VEGF), Insulin-like
growth factor
(IGF), bone morphogenic protein (BMP), Granulocyte-colony stimulating factor
(G-CSF),
Granulocyte-macrophage colony stimulating factor (GM-CSF), Nerve growth factor
(NGF); a
Neurotrophin, Platelet-derived growth factor (PDGF), Erythropoietin (EPO),
Thrombopoietin
(TPO), Myostatin (GDF-8), Growth Differentiation factor-9 (GDF9), basic
fibroblast growth
factor (bFGF or FGF2), Epidermal growth factor (EGF), Hepatocyte growth factor
(HGF).
Cytokines include, e.g., interleukins (e.g., IL-I to IL-33 (e.g., IL-1, IL-2,
IL-3, IL-4, IL-5, IL-6,
IL-7, IL-8, IL-9, IL-10, IL-12, IL-13, or IL-15)). Chemokines include, e.g., 1-
309, TCA-3,
MCP-1, MIP-1 a, MIP-10, RANTES, C 10, MRP-2, MARC, MCP-3, MCP-2, MRP-2, CCF
18,
MIP-ly, Eotaxin, MCP-5, MCP-4, NCC-1, CkR10, HCC-1, Leukotactin-l, LEC, NCC-4,
TARC,
PARC, or Eotaxin-2. Also included are tumor glycoproteins (e.g., tumor-
associated antigens),
for example, carcinoembryonic antigen (CEA), human mucins, HER-2/neu, and
prostate-specific
antigen (PSA) [R. A. Henderson and O. J. Finn, Advances in Immunology, 62, pp.
217-56
(1996)]. In some embodiments, the target protein can be one associated with a
lysosomal storage
disorder, which target proteins include, e.g., alpha-L-iduronidase, beta-D-
galactosidase, beta-
glucosidase, beta-hexosaminidase, beta-D-mannosidase, alpha-L-fucosidase,
arylsulfatase B,
arylsulfatase A, alpha-N-acetylgalactosaminidase, aspartylglucosaminidase,
iduronate-2-
sulfatase, alpha-glucosaminide-N-acetyltransferase, beta-D-glucoronidase,
hyaluronidase, alpha-
L-mannosidase, alpha-neuraminidase, phosphotransferase, acid lipase, acid
ceramidase,
sphingomyelinase, thioesterase, cathepsin K, and lipoprotein lipase.
Target proteins can also be fusion proteins. Fusions proteins include, e.g., a
fusion of (i)
any protein described herein or fragment thereof with (ii) an antibody or
fragment thereof. As
used herein, the term "antibody fragment" refers to an antigen-binding
fragment, e.g., Fab,
27
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
F(ab')2, Fv, and single chain Fv (scFv) fragments. An scFv fragment is a
single polypeptide
chain that includes both the heavy and light chain variable regions of the
antibody from which
the scFv is derived. In addition, diabodies [Poljak (1994) Structure
2(12):1121-1123; Hudson et
al. (1999) J. Immunol. Methods 23(102):177-189, and intrabodies [Huston et al.
(2001) Hum.
Antibodies 10(3-4):127-142; Wheeler et al. (2003) Mol. Ther. 8(3):355-366;
Stocks (2004) Drug
Discov. Today 9(22): 960-966, can be used in the methods of the invention.
Target proteins can also be joined to one or more of a polymer, a carrier, an
adjuvant, an
immunotoxin, or a detectable (e.g., fluorescent, luminescent, or radioactive)
moiety. For
example, a target protein can be joined to polyethyleneglycol, which polymer
moiety can be
used, e.g., to increase the molecular weight of small proteins and/or increase
circulation
residence time.
In some embodiments, the target molecule can be, or contain, dolichol.
Genetically Engineered Cells
Described herein are genetically engineered cells having at least one modified
N-
glycosylation activity, which cells are useful for the production of one or
more target molecules
having an altered N-glycosylation form. Cells suitable for genetic engineering
include, e.g.,
fungal cells (e.g., Yarrowia lipolylica or any other related dimorphic yeast
cells described
herein), plant cells, or animal cells (e.g., (nematode, insect, plant, bird,
reptile, or mammal (e.g.,
a mouse, rat, rabbit, hamster, gerbil, dog, cat, goat, pig, cow, horse, whale,
monkey, or human)).
The cells can be primary cells, immortalized cells, or transformed cells. The
cells can be those in
an animal, e.g., a non-human mammal. Such cells, prior to the genetic
engineering as specified
herein, can be obtained from a variety of commercial sources and research
resource facilities,
such as, for example, the American Type Culture Collection (Rockville, MD).
Target molecules
include proteins such as any of the target proteins described herein (see
above). Target
molecules also include dolichol.
Genetic engineering of a cell includes genetic modifications such as: (i)
deletion of an
endogenous gene encoding a protein having N-glycosylation activity; (ii)
introduction of a
28
CA 02762982 2011-12-22
WO 2008/120107 PCT/182008/001814
recombinant nucleic acid encoding a mutant form of a protein (e.g., endogenous
or exogenous
protein) having N-glycosylation activity (i.e., expressing a mutant protein
having an N-
glycosylation activity); (iii) introduction or expression of an RNA molecule
that interferes with
the functional expression of a protein having the N-glycosylation activity;
(iv) introduction of a
recombinant nucleic acid encoding a wild-type (e.g., endogenous or exogenous)
protein having
N-glycosylation activity (i.e., expressing a protein having an N-glycosylation
activity); or (v)
altering the promoter or enhancer elements of one or more endogenous genes
encoding proteins
having N-glycosylation activity to thus alter the expression of their encoded
proteins. RNA
molecules include, e.g., small-interfering RNA (siRNA), short hairpin RNA
(shRNA), anti-sense
RNA, or micro RNA (miRNA). It is understood that item (ii) includes, e.g.,
replacement of an
endogenous gene (e.g., by homologous recombination) with a gene encoding a
protein having
greater N-glycosylation activity relative to the endogenous gene so replaced.
Genetic
engineering also includes altering an endogenous gene encoding a protein
having an N-
glycosylation activity to produce a protein having additions (e.g., a
heterologous sequence),
deletions, or substitutions (e.g., mutations such as point mutations;
conservative or non-
conservative mutations). Mutations can be introduced specifically (e.g., site-
directed
mutagenesis or homologous recombination; see accompanying Examples) or can be
introduced
randomly (for example, cells can be chemically mutagenized as described in,
e.g., Newman and
Ferro-Novick (1987) J. Cell Biol. 105(4):1587.
The genetic modifications described herein can result in one or more of (i) an
increase in
one or more N-glycosylation activities in the genetically modified cell, (ii)
a decrease in one or
more N-glycosylation activities in the genetically modified cell, (iii) a
change in the localization
or intracellular distribution of one or more N-glycosylation activities in the
genetically modified
cell, or (iv) a change in the ratio of one or more N-glycosylation activities
in the genetically
modified cell. It is understood that an increase in the amount of an N-
glycosylation activity can
be due to overexpression of one or more proteins having N-glycosylation
activity, an increase in
copy number of an endogenous gene (e.g., gene duplication), or an alteration
in the promoter or
enhancer of an endogenous gene that stimulates an increase in expression of
the protein encoded
by the gene. A decrease in one or more N-glycosylation activities can be due
to overexpression
of a mutant form (e.g., a dominant negative form) of one or more proteins
having N-
29
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
glysosylation altering activities, introduction or expression of one or more
interfering RNA
molecules that reduce the expression of one or more proteins having an N-
glycosylation activity,
or deletion of one or more endogenous genes that encode a protein having N-
glycosylation
activity.
Methods of deleting or disrupting one or more endogenous genes are described
in the
accompanying Examples. For example, to disrupt a gene by homologous
recombination, a "gene
replacement" vector can be constructed in such a way to include a selectable
marker gene. The
selectable marker gene can be operably linked, at both 5' and 3' end, to
portions of the gene of
sufficient length to mediate homologous recombination. The selectable marker
can be one of
any number of genes which either complement host cell auxotrophy or provide
antibiotic
resistance, including URA3, LEU2 and HIS3 genes. Other suitable selectable
markers include
the CAT gene, which confers chloramphenicol resistance to yeast cells, or the
lacZ gene, which
results in blue colonies due to the expression of R-galactosidase. Linearized
DNA fragments of
the gene replacement vector are then introduced into the cells using methods
well known in the
art (see below). Integration of the linear fragments into the genome and the
disruption of the
gene can be determined based on the selection marker and can be verified by,
for example,
Southern blot analysis.
As detailed in the accompanying examples, subsequent to its use in selection,
a selectable
marker can be removed from the genome of the host cell by, e.g., Cre-loxP
systems (see below).
Alternatively, a gene replacement vector can be constructed in such a way as
to include a
portion of the gene to be disrupted, which portion is devoid of any endogenous
gene promoter
sequence and encodes none or an inactive fragment of the coding sequence of
the gene. An
"inactive fragment" is a fragment of the gene that encodes a protein having,
e.g., less than about
10% (e.g., less than about 9%, less than about 8%, less than about 7%, less
than about 6%, less
than about 5%, less than about 4%, less than about 3%, less than about 2%,
less than about I%,
or 0%) of the activity of the protein produced from the full-length coding
sequence of the gene.
Such a portion of the gene is inserted in a vector in such a way that no known
promoter sequence
is operably linked to the gene sequence, but that a stop codon and a
transcription termination
sequence are operably linked to the portion of the gene sequence. This vector
can be
subsequently linearized in the portion of the gene sequence and transformed
into a cell. By way
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
of single homologous recombination, this linearized vector is then integrated
in the endogenous
counterpart of the gene.
Expression vectors can be autonomous or integrative.
A recombinant nucleic acid (e.g., one encoding a wild-type or mutant form of a
protein
having N-glycosylation activity) can be in introduced into the cell in the
form of an expression
vector such as a plasmid, phage, transposon, cosmid or virus particle. The
recombinant nucleic
acid can be maintained extrachromosomally or it can be integrated into the
yeast cell
chromosomal DNA. Expression vectors can contain selection marker genes
encoding proteins
required for cell viability under selected conditions (e.g., URA3, which
encodes an enzyme
necessary for uracil biosynthesis or TRP I, which encodes an enzyme required
for tryptophan
biosynthesis) to permit detection and/or selection of those cells transformed
with the desired
nucleic acids (see, e.g., U.S. Pat. No. 4,704,362). Expression vectors can
also include an
autonomous replication sequence (ARS). For example, U.S. Pat. No. 4,837,148
describes
autonomous replication sequences which provide a suitable means for
maintaining plasmids in
Pichia pastoris
Integrative vectors are disclosed, e.g., in U.S. Pat. No.
4,882,279. Integrative vectors generally include a
serially arranged sequence of at least a first insertable DNA fragment, a
selectable marker gene,
and a second insertable DNA fragment. The first and second insertable DNA
fragments are each
about 200 (e.g., about 250, about 300, about 350, about 400, about 450, about
500, or about 1000
or more) nucleotides in length and have nucleotide sequences which are
homologous to portions
of the genomic DNA of the species to be transformed. A nucleotide sequence
containing a gene
of interest (e.g., a gene encoding a protein having N-glycosylation activity)
for expression is
inserted in this vector between the first and second insertable DNA fragments
whether before or
after the marker gene. Integrative vectors can be linearized prior to yeast
transformation to
facilitate the integration of the nucleotide sequence of interest into the
host cell genome.
An expression vector can feature a recombinant nucleic acid under the control
of a yeast
(e.g., Yarrowia lipolytica, Arxula adeninivorans, or other related dimorphic
yeast species)
promoter, which enables them to be expressed in yeast. Suitable yeast
promoters include, e.g.,
ADC 1, TP11, ADH2, hp4d, POX, and Gal10 (see, e.g., Guarente et al. (1982)
Proc. Natl. Acad.
31
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
Sci. USA 79(23):7410) promoters. Additional suitable promoters are described
in, e.g., Zhu and
Zhang (1999) Bioinformatics 15(7-8):608-611 and U.S. Patent No. 6,265,185.
Where the expression vector
is to be introduced into an animal cell, such as a mammalian cell, the
expression vector can
feature a recombinant nucleic acid under the control of an animal cell
promoter suitable for
expression in the host cell of interest. Examples of mammalian promoters
include, e.g., SV40 or
cytomegalovirus (CMV) promoters.
A promoter can be constitutive or inducible (conditional). A constitutive
promoter is
understood to be a promoter whose expression is constant under the standard
culturing
conditions. Inducible promoters are promoters that are responsive to one or
more induction cues.
For example, an inducible promoter can be chemically regulated (e.g., a
promoter whose
transcriptional activity is regulated by the presence or absence of a chemical
inducing agent such
as an alcohol, tetracycline, a steroid, a metal, or other small molecule) or
physically regulated
(e.g., a promoter whose transcriptional activity is regulated by the presence
or absence of a
physical inducer such as light or high or low temperatures). An inducible
promoter can also be
indirectly regulated by one or more transcription factors that are themselves
directly regulated by
chemical or physical cues.
Genetic engineering of a cell also includes activating an endogenous gene
(e.g., a gene
encoding a protein having N-glycosylation activity) that is present in the
host cell, but is
normally not expressed in the cells or is not expressed at significant levels
in the cells. For
example, a regulatory sequence (e.g., a gene promoter or an enhancer) of a
endogenous gene can
be modified such that the operably-linked coding sequence exhibits increased
expression.
Homologous recombination or targeting can be used to replace or disable the
regulatory region
normally associated with the gene with a regulatory sequence which causes the
gene to be
expressed at levels higher than evident in the corresponding non-genetically
engineered cell, or
causes the gene to display a pattern of regulation or induction that is
different than evident in the
corresponding non-genetically engineered cell. Suitable methods for
introducing alterations of a
regulatory sequence (e.g., a promoter or enhancer) of a gene are described in,
e.g., U.S.
Application Publication No. 20030147868.
32
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
It is understood that other genetically engineered modifications can also be
conditional.
For example, a gene can be conditionally deleted using, e.g., a site-specific
DNA recombinase
such as the Cre-loxP system (see, e.g., Gossen et al. (2002) Ann. Rev.
Genetics 36:153-173 and
U.S. Application Publication No. 20060014264).
A recombinant nucleic acid can be introduced into a cell described herein
using a variety
of methods such as the spheroplast technique or the whole-cell lithium
chloride yeast
transformation method. Other methods useful for transformation of plasmids or
linear nucleic
acid vectors into cells are described in, for example, U.S. Patent No.
4,929,555; Hinnen et al.
(1978) Proc. Nat. Acad. Sci. USA 75:1929; Ito et al. (1983) J. Bacteriol.
153:163; U.S. Patent
No. 4,879,231; and Sreekrishna et al. (1987) Gene 59:115.
Electroporation and PEG 1000 whole cell
transformation procedures may also be used, as described by Cregg and Russel,
Methods in
Molecular Biology: Pichia Protocols, Chapter 3, Humana Press, Totowa, N.J.,
pp. 27-39 (1998).
Transfection of animal
cells can feature, for example, the introduction of a vector to the cells
using calcium phosphate,
electroporation, heat shock, liposomes, or transfection reagents such as
FUGENE or
LIPOFECTAMINE , or by contacting naked nucleic acid vectors with the cells in
solution (see,
e.g., Sambrook et al., Molecular Cloning: A Laboratory Manual Second Edition
vol. 1, 2 and 3.
Cold Spring Harbor Laboratory Press: Cold Spring Harbor, New York, USA, Nov.
1989).
Transformed yeast cells can be selected for by using appropriate techniques
including,
but not limited to, culturing auxotrophic cells after transformation in the
absence of the
biochemical product required (due to the cell's auxotrophy), selection for and
detection of a new
phenotype, or culturing in the presence of an antibiotic which is toxic to the
yeast in the absence
of a resistance gene contained in the transformants. Transformants can also be
selected and/or
verified by integration of the expression cassette into the genome, which can
be assessed by, e.g.,
Southern blot or PCR analysis.
Prior to introducing the vectors into a target cell of interest, the vectors
can be grown
(e.g., amplified) in bacterial cells such as Escherichia coli (E. coli). The
vector DNA can be
isolated from bacterial cells by any of the methods known in the art which
result in the
33
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
purification of vector DNA from the bacterial milieu. The purified vector DNA
can be extracted
extensively with phenol, chloroform, and ether, to ensure that no E. coli
proteins are present in
the plasmid DNA preparation, since these proteins can be toxic to mammalian
cells.
Genetic engineering, as described herein, can be used to express (e.g.,
overexpress),
introduce modifications into, or delete any number of genes, e.g., genes
encoding proteins having
N-glycosylation activity. Such genes include, e.g., ALG7, ALG13, ALG14, ALGI,
ALG2,
ALG11, RFT1, ALG3, ALG9, ALG12, ALG6, ALG8, ANL1, ALG10, ALG5, OST3, OST4,
OST6, STT3, OSTI, OST5, WBP1, SWP1, OST2, DPMI, SEC59, OCHI, MNN9, VAN1,
MNN8, MNN 10, MNN 11, HOC 1, MNN2, MNN5, MNN6, KTR 1, YUR 1, MNN4, KRE2,
KTR2, KTR3, MNNI, MNSI, MNN4, PNOI, MNN9, glucosidase I, glucosidase II, or
endomannosidase. The genes encoding proteins having N-glycosylation activity
can be from any
species (e.g., lower eukaryotes (e.g., fungus (including yeasts) or
trypanosomes), plant, or animal
(e.g., insect, bird, reptile, or mammal (e.g., a rodent such as mouse or rat,
dog, cat, horse, goat,
cow, pig, non-human primate, or human)) containing such genes. Exemplary
fungal species
from which genes encoding proteins having N-glycosylation activity can be
obtained include,
without limitation, Pichia anomala, Pichia bovis, Pichia canadensis, Pichia
carsonii, Pichia
farinose, Pichia fermentans, Pichiafluxuum, Pichia membranaefaciens, Pichia
membranaefaciens, Candida valida, Candida albicans, Candida ascalaphidarum,
Candida
amphixiae, Candida Antarctica, Candida atlantica, Candida atmosphaerica,
Candida blattae,
Candida carpophila, Candida cerambycidarum, Candida chauliodes, Candida
corydalis,
Candida dosseyi, Candida dubliniensis, Candida ergatensis, Candidafructus,
Candida glabrata,
Candidafermentati, Candida guilliermondii, Candida haemulonii, Candida
insectamens,
Candida insectorum, Candida intermedia, Candida jeffresii, Candida kefyr,
Candida krusei,
Candida lusitaniae, Candida lyxosophila, Candida maltosa, Candida
membranifaciens, Candida
milleri, Candida oleophila, Candida oregonensis, Candida parapsilosis, Candida
quercitrusa,
Candida shehatea, Candida temnochilae, Candida tenuis, Candida tropicalis,
Candida
tsuchiyae, Candida sinolaborantium, Candida sojae, Candida viswanathii,
Candida utilis,
Pichia membranaefaciens, Pichia silvestris, Pichia membranaefaciens, Pichia
chodati, Pichia
membranaefaciens, Pichia menbranaefaciens, Pichia minuscule, Pichia pastoris,
Pichia
pseudopolymorpha, Pichia quercuum, Pichia robertsii, Pichia saitoi, Pichia
silvestrisi, Pichia
strasburgensis, Pichia terricola, Pichia vanriji, Pseudozyma Antarctica,
Rhodosporidium
34
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
toruloides, Rhodotorula glutinis, Saccharomyces bayanus, Saccharomyces
bayanus,
Saccharomyces tnomdshuricus, Saccharomyces uvarum, Saccharomyces bayanus,
Saccharomyces cerevisiae, Saccharomyces bisporus, Saccharomyces chevaliers,
Saccharomyces
delbrueckii, Saccharomyces exiguous, Saccharomycesfermentati,
Saccharomycesfragilis,
Saccharomyces marxianus, Saccharomyces mellis, Saccharomyces rosei,
Saccharomyces rouxii,
Saccharomyces uvarum, Saccharomyces willianus, Saccharomycodes ludwigii,
Saccharomycopsis capsularis, Saccharomycopsisfibuligera,
Saccharomycopsisfibuligera,
Endomyces hordei, Endomycopsisfobuligera. Saturnispora saitoi,
Schizosaccharomyces
octosporus, Schizosaccharomyces pombe, Schwanniomyces occidentalis,
Torulaspora
delbrueckii, Torulaspora delbrueckii, Saccharomyces dairensis, Torulaspora
delbrueckii,
Torulasporafermentati, Saccharomycesfermentati, Torulaspora delbrueckii,
Torulaspora rosei,
Saccharomyces rosei, Torulaspora delbrueckii, Saccharomyces rosei, Torulaspora
delbrueckii,
Saccharomyces delbrueckii, Torulaspora delbrueckii, Saccharomyces delbrueckii,
Zygosaccharomyces mongolicus, Dorulaspora globosa, Debaryomyces globosus,
Torulopsis
globosa, Trichosporon cutaneum, Trigonopsis variabilis, Williopsis
californica, Williopsis
saturnus, Zygosaccharomyces bisporus, Zygosaccharomyces bisporus, Debaryomyces
disporua.
Saccharomyces bisporas, Zygosaccharomyces bisporus, Saccharomyces bisporus,
Zygosaccharomyces mellis, Zygosaccharomyces priorianus, Zygosaccharomyces
rou.xiim,
Zygosaccharomyces rouxii, Zygosaccharomyces barkeri, Saccharomyces rouxii,
Zygosaccharomyces rouxii, Zygosaccharomyces major, Saccharomyces rousii,
Pichia anomala,
Pichia bovis, Pichia Canadensis, Pichia carsonii, Pichia farinose,
Pichiafermentans, Pichia
fluxuum, Pichia membranaefaciens, Pichia pseudopolymorpha, Pichia quercuum,
Pichia
robertsii, Pseudozyma Antarctica, Rhodosporidium toruloides, Rhodosporidium
toruloides,
Rhodotorula glutinis, Saccharomyces bayanus, Saccharomyces bayanus,
Saccharomyces
bisporus, Saccharomyces cerevisiae, Saccharomyces chevalieri, Saccharomyces
delbrueckii,
Saccharomyces fermentati, Saccharomycesfragilis, Saccharomycodes ludwigii,
Schizosaccharomyces pombe, Schwanniomyces occidenialis, Torulaspora
delbrueckii,
Torulaspora globosa, Trigonopsis variabilis, Williopsis californica,
Williopsis saturnus,
Zygosaccharomyces bisporus, Zygosaccharomyces mellis, Zygosaccharomyces
rouxii, or any
other fungi (e.g., yeast) known in the art or described herein. Exemplary
lower eukaryotes also
include various species of Aspergillus including, but not limited to,
Aspergillus caesiellus,
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
Aspergillus candidus, Aspergillus carneus, Aspergillus clavatus, Aspergillus
deflectus,
Aspergillus flavus, Aspergillusfumigatus, Aspergillus glaucus, Aspergillus
nidulans, Aspergillus
niger, Aspergillus ochraceus, Aspergillus oryzae, Aspergillus parasiticus,
Aspergillus
penicilloides, Aspergillus restrictus, Aspergillus sojae, Aspergillus sydowi,
Aspergillus tamari,
Aspergillus terreus, Aspergillus ustus, or Aspergillus versicolor. Exemplary
protozoal genera
from which genes encoding proteins having N-glycosylation activity can be
obtained include,
without limitation, Blastocrithidia, Crithidia, Endotrypanum, Herpetomonas,
Leishmania,
Leptomonas, Phytomonas, Trypanosoma (e.g., T. bruceii, T gambiense, T
rhodesiense, and T.
cruzi), and Wallaceina.
It is understood that genetic engineering, as described herein, can be used to
express (e.g.,
overexpress), introduce modifications into, or delete any number of genes
(e.g., genes encoding
proteins having N-glycosylation activity) and/or any combination of one or
more (e.g., two,
three, four, five, six, seven, eight, nine, 10, 11, 12, 15, or 20 or more) of
any of the genes recited
herein.
In some embodiments, the genetically engineered cell lacks the ALG3 (Genbank(D
Accession Nos: XM_503488, Genolevures Ref: YALIOE03190g) gene or gene product
(e.g.,
mRNA or protein) thereof. In some embodiments, the genetically engineered cell
expresses
(e.g., overexpresses) the ALG6 (Genbank Acccession Nos: XM_502922,
Genolevures Ref:
YALIOD17028g) protein. In some embodiments, the genetically engineered cell
expresses the
MNN4 gene (Genbank(& Acccession Nos: XM_503217, Genolevures Ref: YALIOD24101
g). In
some embodiments, the genetically engineered cell lacks the OCH I and/or MNN9
gene or gene
products (e.g., mRNA or protein) thereof. In some embodiments, the genetically
engineered cell
does not lack the OCHI gene or a gene product (e.g., mRNA or protein) thereof.
In some
embodiments, the genetically engineered cell expresses an alpha or beta
subunit (or both the
alpha and the beta subunit) of a glucosidase II such as the glucosidase II of
Yarrowia lipolytica
or Trypanosoma brucei. In some embodiments, the genetically engineered cell
expresses a
mutantase such as the mutanase of T. harzianum. In some embodiments, the
genetically
engineered cell can have any combination of these modifications.
For example, in some embodiments, the genetically engineered cell can lack the
ALG3
(e.g., the ALG3 gene exemplified by Genbank Accession Nos: XM_503488,
Genolevures Ref:
YALIOE03I90g) gene or gene product (e.g., mRNA or protein) thereof; can
overexpress the
36
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
ALG6 (e.g., the ALG6 as exemplified by Genbank Acccession Nos: XM_502922,
Genolevures
Ref: YALIODI7028g) protein; can overexpress one or both of the alpha and the
beta subunit of a
glucosidase II (such as the glucosidase II of Yarrowia lipolytica, Trypanosoma
brucei, or any
other species described herein); can overexpress an alpha-1,2-mannosidase; and
overexpress one
or more (and any combination) of the following: a glycosidase, a
glycosyltransferase, a sugar-
nucleotide transporter, a sugar-nucleotide modifying enzyme. In some
embodiments, the
genetically engineered cell does not lack the OCH I gene or a gene product
(e.g., mRNA or
protein) thereof.
In some embodiments, the genetically modified cell can contain a mannosidase
activity
(e.g., an (x-mannosidase activity). The mannosidase activity can be targeted
to the endoplasmic
reticulum. The mannosidase can have a pH optimum at least below 7.5 (e.g., at
least below 7.4,
at least below 7.3, at least below 7.2, at least below 7.1, at least below
7.0, at least below 6.9, at
least below 6.8, at least 6.7, at least below 6.6, at least below 6.5, at
least 6.4, at least below 6.3,
at least below 6.2, at least below 6.1, at least below 6.0, at least below
5.9, at least below 5.8, at
least below 5.7, at least below 5.6, at least below 5.5, at least below 5.4,
at least below 5.3, at
least below 5.2, at least below 5.1, at least below 5.0, at least below 4.9,
at least below 4.8, or at
least below 4.7).
The mannosidase can be MNSI.
For example, the genetically engineered cell can overexpress a mannosidase
(e.g., an
alpha- 1,2-mannosidase or any other mannosidase described herein), but not
lack the OCH I gene
or a gene product (e.g., mRNA or protein) thereof. The mannosidase can be a
wild-type form of
the protein or can be a mutant form such as a fusion protein containing a
mannosidase and an
HDEL ER-retention amino acid sequence (see Examples). (It is understood that
any protein
having N-glycosylation activity can be engineered into a fusion protein
comprising an HDEL
sequence).
In some embodiments, the genetically modified cell can contain an activity
capable of
promoting mannosyl phosphorylation of the altered N-glycosylation form of the
target molecule.
For example, a nucleic acid encoding an activity that promotes phosphorylation
of N-glycans
(e.g. MNN4, MNN6, PNOI) can be introduced in the genetically engineered cell,
which cell is
capable of increasing phosphorylating the N-glycosylation of the target
molecule.
37
CA 02762982 2011-12-22
WO 2008/120107 PCTlIB2008/001814
In some embodiments, the genetically modified cell can contain an activity
capable of
removing mannose residues that cap phosphorylation (e.g., a mannosidase such
as the one from
Jack Bean) from the altered N-glycosylation molecules.
In some embodiments, the genetically modified cell is capable of removing
glucose
residues from Man5GIcNAc2. For example, the genetically modified cell can
overexpress a
protein having a-1,3-glucosidase activity such as, but not limited to, a
mutanase or one or both
of the alpha and beta subunit of a glucosidase II (such as the glucosidase II
of Yarrowia
lipolytica, Trypanosoma brucei, or any other fungal species described herein).
In embodiments where a protein having N-glycosylation activity is derived from
a cell
that is of a different type (e.g., of a different species) than the cell into
which the protein is to be
expressed, a nucleic acid encoding the protein can be codon-optimized for
expression in the
particular cell of interest. For example, a nucleic acid encoding a protein
having N-glycosylation
from Trypanosoma brucei can be codon-optimized for expression in a yeast cell
such as
Yarrowia lipolytica. Such codon-optimization can be useful for increasing
expression of the
protein in the cell of interest. Methods for codon-optimizing a nucleic acid
encoding a protein
are known in the art and described in, e.g., Gao et al. (Biotechnol. Prog.
(2004) 20(2): 443 -448),
Kotula et al. (Nat. Biotechn. (1991) 9, 1386 - 1389), and Bennetzen et al. (J.
Biol. Chem. (1982)
257(6):2036-3031).
A cell can also be genetically engineered to produce predominantly N-glycans
that are
intermediates of a mammalian (e.g., human) glycosylation pathway. For example,
one or more
nucleic acids encoding human proteins having N-glycosylation activity can be
introduced into
the cell. In some embodiments, human proteins can be introduced into the cell
and one or more
endogenous yeast proteins having N-glycosylation activity can be suppressed
(e.g., deleted or
mutated). Techniques for "humanizing" a fungal glycosylation pathway are
described in, e.g.,
Choi et al. (2003) Proc. Natl. Acad. Sci. USA 100(9):5022-5027; Verveken et
al. (2004) Appl.
Environ. Microb. 70(5):2639-2646; and Gerngross (2004) Nature Biotech.
22(11):1410-1414.
Where the genetic engineering involves, e.g., changes in the expression of a
protein or
expression of an exogenous protein (including a mutant form of an endogenous
protein), a
variety of techniques can be used to determine if the genetically engineered
cells express the
protein. For example, the presence of mRNA encoding the protein or the protein
itself can be
38
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
detected using, e.g., Northern Blot or RT-PCR analysis or Western Blot
analysis, respectively.
The intracellular localization of a protein having N-glycosylation activity
can be analyzed by
using a variety of techniques, including subcellular fractionation and
immunofluorescence.
Additional genetic modifications and methods for introducing them into any of
the cells
described herein can be adapted from the disclosures of, e.g., U.S. Patent
Nos. 7,029,872;
5,272,070; and 6,803,225; and U.S. Application Publication Nos. 20050265988,
20050064539,
20050170452, and 20040018588.
While the engineering steps performed in dimorphic yeast species to achieve in
vivo
production of the Man5GIcNAc2 and Man3GlcNAc2 can be different from the
engineering steps
performed in other yeast species, it will be clear to those skilled in the art
that the engineering
techniques to produce modified glycoproteins (with the Man5GIcNAc2 and
Man3GlcNAc2 core
N-glycan structures) in dimorphic yeasts in vivo can be adapted by routine
experimentation from
the methods disclosed in, inter alia, U.S. Patent No. 7,326,681 and U.S.
Publication Nos.
20040018590, 20060040353, and 20060286637.
The adapted methods can thus be used to achieve production of
glycoproteins modified with human-type hybrid and complex N-glycans. These
complex N-
glycans can have 2 to 5 branches initiated with a GIcNAc residue onto the
above-named core
glycans, which can be further extended, e.g., with galactose, fucose and
sialic acid residues.
In some embodiments, the mutant or wild-type proteins having N-glycosylation
activity
can be isolated from the genetically engineered cells using standard
techniques. For example,
following the expression of a mutant or wild-type protein in the genetically
engineered cell, the
protein can be isolated from the cell itself or from the media in which the
cell was cultured.
Methods of isolating proteins are known in the art and include, e.g., liquid
chromatography (e.g.,
HPLC), affinity chromatography (e.g., metal chelation or immunoaffinity
chromatography), ion-
exchange chromatography, hydrophobic-interaction chromatography,
precipitation, or
differential solubilization.
In some embodiments, the isolated proteins having N-glycosylation activity can
be
frozen, lyophilized, or immobilized and stored under appropriate conditions,
which allow the
proteins to retain activity.
39
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
The disclosure also provides a substantially pure culture of any of the
genetically
engineered cells described herein. As used herein, a "substantially pure
culture" of a genetically
engineered cell is a culture of that cell in which less than about 40% (i.e.,
less than about : 35%;
30%; 25%; 20%; 15%; 10%; 5%; 2%; 1%; 0.5%; 0.25%; 0.1%; 0.01%; 0.001%;
0.0001%; or
even less) of the total number of viable cells in the culture are viable cells
other than the
genetically engineered cell, e.g., bacterial, fungal (including yeast),
mycoplasmal, or protozoan
cells. The term "about" in this context means that the relevant percentage can
be 15% percent of
the specified percentage above or below the specified percentage. Thus, for
example, about 20%
can be 17% to 23%. Such a culture of genetically engineered cells includes the
cells and a
growth, storage, or transport medium. Media can be liquid, semi-solid (e.g.,
gelatinous media),
or frozen. The culture includes the cells growing in the liquid or in/on the
semi-solid medium or
being stored or transported in a storage or transport medium, including a
frozen storage or
transport medium. The cultures are in a culture vessel or storage vessel or
substrate (e.g., a
culture dish, flask, or tube or a storage vial or tube).
The genetically engineered cells described herein can be stored, for example,
as frozen
cell suspensions, e.g., in buffer containing a cryoprotectant such as glycerol
or sucrose, as
lyophilized cells. Alternatively, they can be stored, for example, as dried
cell preparations
obtained, e.g., by fluidized bed drying or spray drying, or any other suitable
drying method.
Methods of Producing Altered N-Glycosylation Molecules
Described herein are methods of producing an altered N-glycosylation form of a
target
molecule. The methods generally involve the step of contacting a target
molecule with one or
more N-glycosylation activities from a genetically engineered cell (e.g., a
fungal cell (e.g.,
Yarrowia lipolyrica, Arxula adeninivorans, or any other related dimorphic
yeast cells described
herein), a plant cell, or an animal cell (e.g., nematode, insect, plant, bird,
reptile, or mammal
(e.g., a mouse, rat, rabbit, hamster, gerbil, dog, cat, goat, pig, cow, horse,
whale, monkey, or
human)). The methods can be cell-based or non-cell based.
Cell based methods can include the steps of introducing into a cell (e.g., a
fungal cell
(e.g., Yarrowia lipolylica, Arxula adeninivorans, or any other related
dimorphic yeast cells
described herein), a plant cell, or an animal cell) genetically engineered to
have at least one
modified N-glycosylation activity a nucleic acid encoding a target molecule
subject to N-
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
glycosylation in the cell, wherein the cell produces the target molecule in an
altered N-
glycosylation form. The target molecule can be, e.g., a protein such as any of
the target proteins
described herein. In embodiments where the target protein is a lipid, the
nucleic acid can be one
encoding one or more enzymes which promote the synthesis of the lipid.
The types of modifications produced by the genetic engineering of the cells
are described
herein (see the accompanying Examples and "Genetically Engineered Cells"
above).
Methods for introducing a nucleic acid are known in the art and are described
in the
accompanying Examples and above.
Introduction or expression of a target molecule (e.g., a target protein) into
a genetically
engineered cell can result in the trafficking of the target molecule through
the endoplasmic
reticulum and/or Golgi apparatus of the cell, thereby producing an altered N-
glycosylation form
of the target molecule.
Following the processing of the target molecule (e.g., in the genetically
modified cell),
the altered N-glycosylation form of the target molecule (e.g., the target
protein) can contain one
or more N-glycan structures. For example, the altered form of the target
molecule can contain
one or more specific N-glycan structures such as Man5GlcNAc2 (structural
formula I or VII; Fig.
4), Man3GIcNAc2 (structural formula I; Fig. 4), Man9GlcNAc2 (structural
formula II; Fig. 4),
Man3GlcNAc2 (structural formula XIV; Fig. 4), G1c,Man5GIcNAc2 (structural
formula VIII; Fig.
4), or GIc2Man5GlcNAc2 (structural formula IX; Fig. 4) ("Man" is mannose;
"Glc" is glucose;
and "GlcNAc" is N-acetylglucosamine).
The target molecules having altered N-glycosylation produced from the
genetically
engineered cells can be homogeneous (i.e., all altered N-glycosylation
molecules containing the
same specific N-glycan structure) or can be substantially homogeneous. By
"substantially
homogeneous" is meant that the altered target molecules are at least about 25%
(e.g., at least
about 27%, at least about 30%, at least about 35%, at least about 40%, at
least about 45%, at
least about 50%, at least about 55%, at least about 60%, at least about 65%,
at least about 70%,
at least about 75%, at least about 80%, at least about 85%, at least about
90%, or at least about
95%, or at least about 99%) of the target molecules having altered N-
glycosylation produced by
the genetically engineered cell.
Where the genetically engineered cell includes one or more N-glycosylation
activities
that effect the phosphorylation of an N-glycan, an altered N-glycosylation
form of a target
41
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
molecule can have at least about 25% (e.g., at least about 27%, at least about
30%, at least about
35%, at least about 40%, at least about 45%, at least about 50%, at least
about 55%, at least
about 60%, at least about 65%, at least about 70%, at least about 75%, or at
least about 80%) of
its mannosyl residues phosphorylated.
Where any of the genetic modifications of the genetically engineered cell are
inducible or
conditional on the presence of an inducing cue (e.g., a chemical or physical
cue), the genetically
engineered cell can, optionally, be cultured in the presence of an inducing
agent before, during,
or subsequent to the introduction of the nucleic acid. For example, following
introduction of the
nucleic acid encoding a target protein, the cell can be exposed to a chemical
inducing agent that
is capable of promoting the expression of one or more proteins having N-
glycosylation activity.
Where multiple inducing cues induce conditional expression of one or more
proteins having N-
glycosylation activity, a cell can be contacted with multiple inducing agents.
Following processing by one or more N-glycosylation activities, the altered
target
molecule can be isolated. The altered target molecule can be maintained within
the yeast cell
and released upon cell lysis or the altered target molecule can be secreted
into the culture
medium via a mechanism provided by a coding sequence (either native to the
exogenous nucleic
acid or engineered into the expression vector), which directs secretion of the
molecule from the
cell. The presence of the altered target molecule in the cell lysate or
culture medium can be
verified by a variety of standard protocols for detecting the presence of the
molecule. For
example, where the altered target molecule is a protein, such protocols can
include, but are not
limited to, immunoblotting or radioinununoprecipitation with an antibody
specific for the altered
target protein (or the target protein itself), binding of a ligand specific
for the altered target
protein (or the target protein itself), or testing for a specific enzyme
activity of the altered target
protein (or the target protein itself).
In some embodiments, the isolated altered target molecules can be frozen,
lyophilized, or
immobilized and stored under appropriate conditions, e.g., which allow the
altered target
molecules to retain biological activity.
The altered N-glycosylation form of the target molecule can be further
processed in vivo
(e.g., in the genetically engineered cell) or can be processed in vitro
following isolation from the
genetically engineered cell or cell medium. The further processing can include
modifications of
one or more N-glycan residues of the altered target molecule or modifications
to the altered
42
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
target molecule other than to its N-glycan residues. The additional processing
of the altered
target molecule can include the addition (covalent or non-covalent joining) of
a heterologous
moiety such as a polymer or a carrier. The further processing can also involve
enzymatic or
chemical treatment of the altered target molecule. Enzymatic treatment can
involve contacting
the altered target molecule with one or more of a glycosidase (e.g.,
mannosidase or mannanase),
a phosphodiesterase, a phospholipase, a glycosyltransferase, or a protease for
a time sufficient to
induce modification of the altered target molecule. Enzymatic treatment can
also involve
contacting the altered target molecule with an enzyme capable of removing one
or more glucose
residues from Man5GlcNAc2 such as, but not limited to, a mannosidase or one or
both of the
alpha and beta subunit of a glucosidase II. Chemical treatment can, for
example, involve
contacting the altered target molecule with an acid such as hydrofluoric acid
for a time sufficient
to induce modification of the altered target molecule. Hydrofluoric acid
treatment under certain
conditions specifically removes the mannose residues that are phosphodiester-
linked to glycans,
while leaving the phosphate on the glycan. An altered target molecule can be
further processed
by addition or removal of a phosphate group from one or more N-glycans. For
example, a
altered target molecule can be contacted with a mannosyl kinase or a mannosyl
phosphatase.
In some embodiments, any of the altered target molecules described herein,
following
isolation, can be attached to a heterologous moiety, e.g., using enzymatic or
chemical means. A
"heterologous moiety" refers to any constituent that is joined (e.g.,
covalently or non-covalently)
to the altered target molecule, which constituent is different from a
constituent originally present
on the altered target molecule. Heterologous moieties include, e.g., polymers,
carriers,
adjuvants, immunotoxins, or detectable (e.g., fluorescent, luminescent, or
radioactive) moieties.
In some embodiments, an additional N-glycan can be added to the altered target
molecule.
It is understood that a target molecule can be, but need not be, processed in
a genetically
engineered cell. For example, the disclosure also features cell-free methods
of producing a target
molecule having an altered N-glycosylation form, which methods include the
step of contacting a
target molecule under N-glycosylation conditions with a cell lysate prepared
from a cell (e.g., a
fungal cell (e.g., Yarrowia lipolylica, Arxula adeninivorans, or any other
related dimorphic yeast
cells described herein), a plant cell, or an animal cell (e.g., nematode,
insect, plant, bird, reptile,
or mammal (e.g., a mouse, rat, rabbit, hamster, gerbil, dog, cat, goat, pig,
cow, horse, whale,
monkey, or human)) genetically engineered to have at least one modified N-
glycosylation
43
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
activity, wherein the contacting of the target molecule to the cell lysate
produces an altered N-
glycosylation form of the target molecule.
By "N-glycosylation conditions" is meant that a mixture (e.g., of target
molecule and cell
lysate) is incubated under conditions that allow for altered N-glycosylation
(as described above).
Suitable methods for obtaining cell lysates that preserve the activity or
integrity of one or
more N-glycosylation activities in the lysate can include the use of
appropriate buffers and/or
inhibitors, including nuclease, protease and phosphatase inhibitors that
preserve or minimize
changes in N-glycosylation activities in the cell lysate. Such inhibitors
include, for example,
chelators such as ethylenediamine tetraacetic acid (EDTA), ethylene glycol
bis(P-aminoethyl
ether) N,N,NI,Nl-tetraacetic acid (EGTA), protease inhibitors such as
phenylmethylsulfonyl
fluoride (PMSF), aprotinin, leupeptin, antipain and the like, and phosphatase
inhibitors such as
phosphate, sodium fluoride, vanadate and the like. Inhibitors can be chosen
such that they do not
interfere with or only minimally adversely affect the N-glycosylation
activity, or activities, of
interest. Appropriate buffers and conditions for obtaining lysates containing
enzymatic activities
are described in, e.g., Ausubel et al. Current Protocols in Molecular Biology
(Supplement 47),
John Wiley & Sons, New York (1999); Harlow and Lane, Antibodies: A Laboratory
Manual
Cold Spring Harbor Laboratory Press (1988); Harlow and Lane, Using Antibodies:
A Laboratory
Manual, Cold Spring Harbor Press (1999); Tietz Textbook of Clinical Chemistry,
3rd ed. Burtis
and Ashwood, eds. W.B. Saunders, Philadelphia, (1999).
A cell lysate can be further processed to eliminate or minimize the presence
of interfering
substances, as appropriate. If desired, a cell lysate can be fractionated by a
variety of methods
well known to those skilled in the art, including subcellular fractionation,
and chromatographic
techniques such as ion exchange, hydrophobic and reverse phase, size
exclusion, affinity,
hydrophobic charge-induction chromatography, and the like (see, e.g., Scopes,
Protein
Purification: Principles and Practice, third edition, Springer-Verlag, New
York (1993); Burton
and Harding, J. Chromatogr. A 814:71-81 (1998)).
In some embodiments, a cell lysate can be prepared in which whole cellular
organelles
remain intact and/or functional. For example, a lysate can contain one or more
of intact rough
endoplasmic reticulum, intact smooth endoplasmic reticulum, or intact Golgi
apparatus. Suitable
methods for preparing lysates containing intact cellular organelles and
testing for the
functionality of the organelles are described in, e.g., Moreau et al. (1991)
J. Biol. Chem.
44
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
266(7):4329-4333; Moreau et al. (1991) J. Biol. Chem. 266(7):4322-4328; Rexach
et al. (1991)
J. Cell Biol. 114(2):219-229; and Paulik et al. (1999) Arch. Biochem. Biophys.
367(2): 265-273.
The disclosure also provides methods of producing a target molecule having an
altered
N-glycosylation form that includes the step of contacting a target molecule
under N-
glycosylation conditions with one or more isolated proteins having N-
glycosylation activity,
wherein contacting the target molecule with the one or more proteins having N-
glycosylation
activity produces an altered N-glycosylation form of the target molecule and
wherein the one or
more proteins having N-glycosylation activity are prepared from a cell (e.g.,
a fungal cell (e.g.,
Yarrowia lipolytica, Arxula adeninivorans, or any other related dimorphic
yeast cells described
herein), a plant cell, or an animal cell (e.g., nematode, insect, plant, bird,
reptile, or mammal
(e.g., a mouse, rat, rabbit, hamster, gerbil, dog, cat, goat, pig, cow, horse,
whale, monkey, or
human)) genetically engineered to have at least one modified N-glycosylation
activity.
One of more proteins having N-glycosylation activity can be purified using
standard
techniques as described above. A target molecule can be contacted with one or
more proteins in
a suitable buffer for a time sufficient to induce modification of the target
molecule as described
in, e.g., Lee and Park (2002) 30(6):716-720 and Fujita and Takegawa (2001)
Biochem. Bionhvs.
Res. Commun. 282(3):678-682.
In some embodiments, the target molecule can be contacted with just one
protein having
N-glycosylation activity. In some embodiments, the target molecule can be
contacted with more
than one protein having N-glycosylation activity. The target molecule can be
contacted with
more than one protein at the same time or sequentially. Where the target
molecule is contacted
sequentially to more than one protein having N-glycosylation activity, the
target molecule can,
but need not, be purified after one or more steps. That is, a target molecule
can be contacted
with protein activity A, then purified before contacting the molecule to
protein activity B, and so
on.
It some embodiments of the cell free methods, it can be advantageous to link
the target
molecule to a solid-phase support prior to contacting the target molecule with
one or more N-
glycosylation activities. Such linkage can allow for easier purification
following the N-
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
glycosylation modifications. Suitable solid-phase supports include, but are
not limited to, multi-
well assay plates, particles (e.g., magnetic or encoded particles), a column,
or a membrane.
Methods for detecting N-glycosylation (e.g., altered N-glycosylation) of a
target
molecule include DNA sequencer-assisted (DSA), fluorophore-assisted
carbohydrate
electrophoresis (FACE) (as described in the accompanying Examples) or surface-
enhanced laser
desorption/ionization time-of-flight mass spectrometry (SELDI-TOF MS) and. For
example, an
analysis can utilize DSA-FACE in which, for example, glycoproteins are
denatured followed by
immobilization on, e.g., a membrane. The glycoproteins can then be reduced
with a suitable
reducing agent such as dithiothreitol (DTT) or (3-mercaptoethanol. The
sulfhydryl groups of the
proteins can be carboxylated using an acid such as iodoacetic acid. Next, the
N-glycans can be
released from the protein using an enzyme such as N-glycosidase F. N-glycans,
optionally, can
be reconstituted and derivatized by reductive amination. The derivatized N-
glycans can then be
concentrated. Instrumentation suitable for N-glycan analysis includes, e.g.,
the ABI PRISM
377 DNA sequencer (Applied Biosystems). Data analysis can be performed using,
e.g.,
GENESCAN 3.1 software (Applied Biosystems). Optionally, isolated
mannoproteins can be
further treated with one or more enzymes to confirm their N-glycan status.
Exemplary enzymes
include, e.g., a-mannosidase or a-1,2 mannosidase, as described in the
accompanying Examples.
Additional methods of N-glycan analysis include, e.g., mass spectrometry
(e.g., MALDI-TOF-
MS), high-pressure liquid chromatography (HPLC) on normal phase, reversed
phase and ion
exchange chromatography (e.g., with pulsed amperometric detection when glycans
are not
labeled and with UV absorbance or fluorescence if glycans are appropriately
labeled). See also
Callewaert et al. (2001) Glycobiology 11(4):275-281 and Freire et al. (2006)
Bioconjug. Chem.
17(2): 559-564.
Disorders Treatable by Altered N-Glycosylation Molecules
The isolated, altered N-glycosylation molecules (e.g., the altered N-
glycosylation
proteins or dolichol) described herein can be used to treat a variety of
disorders, which disorders
are treatable by administration of one or more altered N-glycosylation
molecules (e.g., a protein
having altered N-glycosylation). Examples of some specific medical conditions
that can be
46
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
treated or prevented by administration of an altered N-glycosylation molecule
(e.g., an altered N-
glycoprotein or an altered N-glycosylated dolichol) are reviewed in the
following sections.
(i) Metabolic Disorders
A metabolic disorder is one that affects the production of energy within
individual human
(or animal) cells. Most metabolic disorders are genetic, though some can be
"acquired" as a
result of diet, toxins, infections, etc. Genetic metabolic disorders are also
known as inborn errors
of metabolism. In general, the genetic metabolic disorders are caused by
genetic defects that
result in missing or improperly constructed enzymes necessary for some step in
the metabolic
process of the cell. The largest classes of metabolic disorders are disorders
of carbohydrate
metabolism, disorders of amino acid metabolism, disorders of organic acid
metabolism (organic
acidurias), disorders of fatty acid oxidation and mitochondrial metabolism,
disorders of
porphyrin metabolism,
disorders of purine or pyrimidine metabolism, disorders of steroid metabolism
disorders of mitochondrial function, disorders of peroxisomal function, and
lysosomal storage disorders (LSDs).
Examples of metabolic disorders that can be treated through the administration
of one or
more altered N-glycosylation molecules (or pharmaceutical compositions of the
same) described
herein can include, e.g., hereditary hemochromatosis, oculocutaneous albinism,
protein C
deficiency, type I hereditary angioedema, congenital sucrase-isomaltase
deficiency, Crigler-
Najjar type II, Laron syndrome, hereditary Myeloperoxidase, primary
hypothyroidism,
congenital long QT syndrome, tyroxine binding globulin deficiency, familial
hypercholesterolemia, familial chylomicronemia, abeta-lipoproteinema, low
plasma lipoprotein
A levels, hereditary emphysema with liver injury, congenital hypothyroidism,
osteogenesis
imperfecta, hereditary hypofibrinogenemia, alpha-l antichymotrypsin
deficiency, nephrogenic
diabetes insipidus, neurohypophyseal diabetes insipidus, adenosine deaminase
deficiency,
Pelizaeus Merzbacher disease, von Willebrand disease type IIA, combined
factors V and VIII
deficiency, spondylo-epiphyseal dysplasia tarda, choroideremia, I cell
disease, Batten disease,
ataxia telangiectasias, ADPKD-autosomal dominant polycystic kidney disease,
microvillus
inclusion disease, tuberous sclerosis, oculocerebro-renal syndrome of Lowe,
amyotrophic lateral
sclerosis, myelodysplastic syndrome, Bare lymphocyte syndrome, Tangier
disease, familial
47
CA 02762982 2011-12-22
WO 2008/120107 PCT/182008/001814
intrahepatic cholestasis, X-linked adreno-leukodystrophy, Scott syndrome,
Hermansky-Pudlak
syndrome types I and 2, Zellweger syndrome, rhizomelic chondrodysplasia
puncta, autosomal
recessive primary hyperoxaluria, Mohr Tranebjaerg syndrome, spinal and bullar
muscular
atrophy, primary ciliary diskenesia (Kartagener's syndrome), giantism and
acromegaly,
galactorrhea, Addison's disease, adrenal virilism, Cushing's syndrome,
ketoacidosis, primary or
secondary aldosteronism, Miller Dieker syndrome, lissencephaly, motor neuron
disease, Usher's
syndrome, Wiskott-Aldrich syndrome, Optiz syndrome, Huntington's disease,
hereditary
pancreatitis, anti-phospholipid syndrome, overlap connective tissue disease,
Sjbgren's syndrome,
stiff-man syndrome, Brugada syndrome, congenital nephritic syndrome of the
Finnish type,
Dubin-Johnson syndrome, X-liriked hypophosphosphatemia, Pendred syndrome,
persistent
hyperinsulinemic hypoglycemia of infancy, hereditary spherocytosis,
aceruloplasminemia,
infantile neuronal ceroid lipofuscinosis, pseudoachondroplasia and multiple
epiphyseal,
Stargardt-like macular dystrophy, X-linked Charcot-Marie-Tooth disease,
autosomal dominant
retinitis pigmentosa, Wolcott-Rallison syndrome, Cushing's disease, limb-
girdle muscular
dystrophy, mucoploy-saccharidosis type IV, hereditary familial amyloidosis of
Finish, Anderson
disease, sarcoma, chronic myelomonocytic leukemia, cardiomyopathy,
faciogenital dysplasia,
Torsion disease, Huntington and spinocerebellar ataxias, hereditary
hyperhomosyteinemia,
polyneuropathy, lower motor neuron disease, pigmented retinitis, seronegative
polyarthritis,
interstitial pulmonary fibrosis, Raynaud's phenomenon, Wegner's
granulomatosis, preoteinuria,
CDG-Ia, CDG-Ib, CDG-Ic, CDG-Id, CDG-Ie, CDG-If, CDG-Ila, CDG-11b, CDG-IIc, CDG-
IId,
Ehlers-Danlos syndrome, multiple exostoses, Griscelli syndrome (type 1 or type
2), or X-linked
non-specific mental retardation. In addition, metabolic disorders can also
include lysosomal
storage disorders such as, but not limited to, Fabry disease, Farber disease,
Gaucher disease,
GMi-gangliosidosis, Tay-Sachs disease, Sandhoff disease, GM2 activator
disease, Krabbe
disease, metachromatic leukodystrophy, Niemann-Pick disease (types A, B, and
C), Hurler
disease, Scheie disease, Hunter disease, Sanfilippo disease, Morquio disease,
Maroteaux-Lamy
disease, hyaluronidase deficiency, aspartylglucosaminuria, fucosidosis,
mannosidosis, Schindler
disease, sialidosis type 1, Pompe disease, Pycnodysostosis, ceroid
lipofuscinosis, cholesterol
ester storage disease, Wolman disease, Multiple sulfatase deficiency,
galactosialidosis,
mucolipidosis (types II 11II, and IV), cystinosis, sialic acid storage
disorder, chylomicron
48
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
retention disease with Marinesco-Sjogren syndrome, Hermansky-Pudlak syndrome,
Chediak-
Higashi syndrome, Danon disease, or Geleophysic dysplasia.
Symptoms of a metabolic disorder are numerous and diverse and can include one
or more
of, e.g., anemia, fatigue, bruising easily, low blood platelets, liver
enlargement, spleen
enlargement, skeletal weakening, lung impairment, infections (e.g., chest
infections or
pneumonias), kidney impairment, progressive brain damage, seizures, extra
thick meconium,
coughing, wheezing, excess saliva or mucous production, shortness of breath,
abdominal pain,
occluded bowel or gut, fertility problems, polyps in the nose, clubbing of the
finger/toe nails and .
skin, pain in the hands or feet, angiokeratoma, decreased perspiration,
corneal and lenticular
opacities, cataracts, mitral valve prolapse and/or regurgitation,
cardiomegaly, temperature
intolerance, difficulty walking, difficulty swallowing, progressive vision
loss, progressive
hearing loss, hypotonia, macroglossia, areflexia, lower back pain, sleep
apnea, orthopnea,
somnolence, lordosis, or scoliosis. It is understood that due to the diverse
nature of the defective
or absent proteins and the resulting disease phenotypes (e.g., symptomatic
presentation of a
metabolic disorder), a given disorder will generally present only symptoms
characteristic to that
particular disorder. For example, a patient with Fabry disease can present a
particular subset of
the above-mentioned symptoms such as, but not limited to, temperature
intolerance, corneal
whirling, pain, skin rashes, nausea, or dirarrhea. A patient with Gaucher
syndrome can present
with splenomegaly, cirrhosis, convulsions, hypertonia, apnea, osteoporosis, or
skin discoloration.
In addition to the administration of one or more altered N-glycosylation
molecules
described herein, a metabolic disorder can also be treated by proper nutrition
and vitamins (e.g.,
cofactor therapy), physical therapy, and pain medications.
Depending on the specific nature of a given metabolic disorder, a patient can
present
these symptoms at any age. In many cases, symptoms can present in childhood or
in early
adulthood. For example, symptoms of Fabry disease can present at an early age,
e.g., at 10 or 11
years of age.
As used herein, a subject "at risk of developing a metabolic disorder" (such
as one
described herein) is a subject that has a predisposition to develop a
disorder, i.e., a genetic
predisposition to develop metabolic disorder as a result of a mutation in a
enzyme such as alpha-
L-iduronidase, beta-D-galactosidase, beta-glucosidase, beta-hexosaminidase,
beta-D-
mannosidase, alpha-L-fucosidase, arylsulfatase B, aryisulfatase A, alpha-N-
49
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
acteylgalactosaminidase, aspartylglucosaminidase, iduronate-2-sulfatase, alpha-
glucosaminide-
N-acetyltransferase, beta-D-glucoronidase, hyaluronidase, alpha-L-mannosidase,
alpha-
neurominidase, phosphotransferase, acid lipase, acid ceramidase,
sphinogmyelinase, thioesterase,
cathepsin K, or lipoprotein lipase. Clearly, subjects "at risk of developing a
metabolic disorder"
are not all the subjects within a species of interest.
A subject "suspected of having a disorder" is one having one or more symptoms
of a
disorder (e.g., a metabolic disorder or any other disorder described herein)
such as any of those
described herein.
(ii) Cancer
Cancer is a class of diseases or disorders characterized by uncontrolled
division of cells
and the ability of these to spread, either by direct growth into adjacent
tissue through invasion, or
by implantation into distant sites by metastasis (where cancer cells are
transported through the
bloodstream or lymphatic system). Cancer can affect people at all ages, but
risk tends to increase
with age. Types of cancers can include, e.g., lung cancer, breast cancer,
colon cancer, pancreatic
cancer, renal cancer, stomach cancer, liver cancer, bone cancer, hematological
cancer, neural
tissue cancer, melanoma, thyroid cancer, ovarian cancer, testicular cancer,
prostate cancer,
cervical cancer, vaginal cancer, or bladder cancer.
As used herein, a subject "at risk of developing a cancer" is a subject that
has a
predisposition to develop a cancer, i.e., a genetic predisposition to develop
cancer such as a
mutation in a tumor suppressor gene (e.g., mutation in BRCAI, p53, RB, or APC)
or has been
exposed to conditions that can result in cancer. Thus, a subject can also be
one "at risk of
developing a cancer" when the subject has been exposed to mutagenic or
carcinogenic levels of
certain compounds (e.g., carcinogenic compounds in cigarette smoke such as
Acrolein, Arsenic,
Benzene, Benz (a)anthracene, Benzo{a}pyrene, Polonium-210 (Radon), Urethane,
or Vinyl
Chloride). Moreover, the subject can be "at risk of developing a cancer" when
the subject has
been exposed to, e.g., large doses of ultraviolet light or X-irradiation, or
exposed (e.g., infected)
to a tumor-causing/associated virus such as papillomavirus, Epstein-Barr
virus, hepatitis B virus,
or human T-cell leukemia-lymphoma virus. From the above it will be clear that
subjects "at risk
of developing a cancer" are not all the subjects within a species of interest.
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
A subject "suspected of having a cancer" is one having one or more symptoms of
a
cancer. Symptoms of cancer are well-known to those of skill in the art and
include, without
limitation, breast lumps, nipple changes, breast cysts, breast pain, weight
loss, weakness,
excessive fatigue, difficulty eating, loss of appetite, chronic cough,
worsening breathlessness,
coughing up blood, blood in the urine, blood in stool, nausea, vomiting, liver
metastases, lung
metastases, bone metastases, abdominal fullness, bloating, fluid in peritoneal
cavity, vaginal
bleeding, constipation, abdominal distension, perforation of colon, acute
peritonitis (infection,
fever, pain), pain, vomiting blood, heavy sweating, fever, high blood
pressure, anemia, diarrhea,
jaundice, dizziness, chills, muscle spasms, colon metastases, lung metastases,
bladder
metastases, liver metastases, bone metastases, kidney metastases, and pancreas
metastases,
difficulty swallowing, and the like.
In addition to the administration of one or more altered N-glycosylation
molecules
described herein, a cancer can also be treated by chemotherapeutic agents,
ionizing radiation,
immunotherapy agents, or hyperthermotherapy agents. Chemotherapeutic agents
include, e.g.,
cisplatin, carboplatin, procarbazine, mechlorethamine, cyclophosphamide,
camptothecin,
adriamycin, ifosfamide, melphalan, chlorambucil, bisulfan, nitrosurea,
dactinomycin,
daunorubicin, doxorubicin, bleomycin, plicomycin, mitomycin, etoposide,
verampil,
podophyllotoxin, tamoxifen, taxol, transplatinum, 5-flurouracil, vincristin,
vinblastin, and
methotrexate.
(iii) Inflammatory Disorders
An "inflammatory disorder," as used herein, refers to a process in which one
or more
substances (e.g., substances not naturally occurring in the subject), via the
action of white blood
cells (e.g., B cells, T cells, macrophages, monocytes, or dendritic cells)
inappropriately trigger a
pathological response, e.g., a pathological immune response. Accordingly, such
cells involved in
the inflammatory response are referred to as "inflammatory cells." The
inappropriately triggered
inflammatory response can be one where no foreign substance (e.g., an antigen,
a virus, a
bacterium, a fungus) is present in or on the subject. The inappropriately
triggered response can
be one where a self-component (e.g., a self-antigen) is targeted (e.g., an
autoimmune disorder
such as multiple sclerosis) by the inflammatory cells. The inappropriately
triggered response can
also be a response that is inappropriate in magnitude or duration, e.g.,
anaphylaxis. Thus, the
51
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
inappropriately targeted response can be due to the presence of a microbial
infection (e.g., viral,
bacterial, or fungal). Types of inflammatory disorders (e.g., autoimmune
disease) can include,
but are not limited to, osteoarthritis, rheumatoid arthritis (RA),
spondyloarthropathies, POEMS
syndrome, Crohn's disease, multicentric Castleman's disease, systemic lupus
erythematosus
(SLE), multiple sclerosis (MS), muscular dystrophy (MD), insulin-dependent
diabetes mellitus
(IDDM), dermatomyositis, polymyositis, inflammatory neuropathies such as
Guillain Barre
syndrome, vasculitis such as Wegener's granulomatosus, polyarteritis nodosa,
polymyalgia
rheumatica, temporal arteritis, Sjogren's syndrome, Bechet's disease, Churg-
Strauss syndrome,
or Takayasu's arteritis. Also included in inflammatory disorders are certain
types of allergies
such as rhinitis, sinusitis, urticaria, hives, angioedema, atopic dermatitis,
food allergies (e.g., a
nut allergy), drug allergies (e.g., penicillin), insect allergies (e.g.,
allergy to a bee sting), or
mastocytosis. Inflammatory disorders can also include ulcerative colitis and
asthma.
A subject "at risk of developing an inflammatory disorder" refers to a subject
with a
family history of one or more inflammatory disorders (e.g., a genetic
predisposition to one or
more inflammatory disorders) or one exposed to one or more inflammation-
inducing conditions.
For example, a subject can have been exposed to a viral or bacterial
superantigen such as, but not
limited to, staphylococcal enterotoxins (SEs), a streptococcus pyogenes
exotoxin (SPE), a
staphylococcus aureus toxic shock-syndrome toxin (TSST-1), a streptococcal
mitogenic exotoxin
(SME) and a streptococcal superantigen (SSA). From the above it will be clear
that subjects "at
risk of developing an inflammatory disorder" are not all the subjects within a
species of interest.
A subject "suspected of having an inflammatory disorder" is one who presents
with one
or more symptoms of an inflammatory disorder. Symptoms of inflammatory
disorders are well
known in the art and include, but are not limited to, redness, swelling (e.g.,
swollen joints), joints
that are warm to the touch, joint pain, stiffness, loss of joint function,
fever, chills, fatigue, loss
of energy, headaches, loss of appetite, muscle stiffness, insomnia, itchiness,
stuffy nose,
sneezing, coughing, one or more neurologic symptoms such as dizziness,
seizures, or pain.
In addition to the administration of one or more altered N-glycosylation
molecules
described herein, an inflammatory disorder can also be treated by non-
steroidal anti-
inflammatory drug (NSAID), a disease-modifying anti-rheumatic drug (DMARD), a
biological
response modifier, or a corticosteroid. Biological response modifiers include,
e.g., an anti-TNF
52
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
agent (e.g., a soluble TNF receptor or an antibody specific for TNF such as
adulimumab,
infliximab, or etanercept).
Methods suitable for treating (e.g., preventing or ameliorating one or more
symptoms of)
any of the disorders described herein using any of the altered N-glycosylation
molecules (or
pharmaceutical compositions thereof) are set forth in the following section.
Pharmaceutical Compositions and Methods of Treatment
An altered N-glycosylation molecule (e.g., an altered N-glycosylation form of
a target
molecule such as a target protein) can be incorporated into a pharmaceutical
composition
containing a therapeutically effective amount of the molecule and one or more
adjuvants,
excipients, carriers, and/or diluents. Acceptable diluents, carriers and
excipients typically do not
adversely affect a recipient's homeostasis (e.g., electrolyte balance).
Acceptable carriers include
biocompatible, inert or bioabsorbable salts, buffering agents, oligo- or
polysaccharides,
polymers, viscosity-improving agents, preservatives and the like. One
exemplary carrier is
physiologic saline (0.15 M NaCl, pH 7.0 to 7.4). Another exemplary carrier is
50 mM sodium
phosphate, 100 mM sodium chloride. Supplementary active compounds can also be
incorporated
into the compositions.
Administration of a pharmaceutical composition containing an altered N-
glycosylation
molecule can be systemic or local. Pharmaceutical compositions can be
formulated such that
they are suitable for parenteral and/or non-parenteral administration.
Specific administration
modalities include subcutaneous, intravenous, intramuscular, intraperitoneal,
transdermal,
intrathecal, oral, rectal, buccal, topical, nasal, ophthalmic, intra-
articular, intra-arterial,
sub-arachnoid, bronchial, lymphatic, vaginal, and intra-uterine
administration.
Administration can be by periodic injections of a bolus of the pharmaceutical
composition or can be uninterrupted or continuous by intravenous or
intraperitoneal
administration from a reservoir which is external (e.g., an IV bag) or
internal (e.g., a bioerodable
implant, a bioartificial organ, or a colony of implanted altered N-
glycosylation molecule
production cells). See, e.g., U.S. Pat. Nos. 4,407,957, 5,798,113, and
5,800,828.
Administration of a pharmaceutical
53
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
composition can be achieved using suitable delivery means such as: a pump
(see, e.g., Annals of
Pharmacotherapy, 27:912 (1993); Cancer, 41:1270 (1993); Cancer Research,
44:1698 (1984),
microencapsulation (see, e.g., U.S. Pat. Nos. 5,284,761; 4,352,883; 4,353,888;
and 5,084,350;
continuous release polymer implants (see, e.g., Sabel, U.S. Pat. No.
4,883,666;
macroencapsulation (see, e.g. U.S. Pat. Nos. 5,284,761, 5,158,881, 4,976,859
and 4,968,733 and
published PCT patent applications W092/19195, WO 95/05452; injection, either
subcutaneously,
intravenously, intra-arterially, intramuscularly, or to other suitable site;
or oral administration, in
capsule, liquid, tablet, pill, or prolonged release formulation.
Examples of parenteral delivery systems include ethylene-vinyl acetate
copolymer
particles, osmotic pumps, implantable infusion systems, pump delivery,
encapsulated cell
delivery, liposomal delivery, needle-delivered injection, needle-less
injection, nebulizer,
aerosolizer, electroporation, and transdermal patch.
Formulations suitable for parenteral administration conveniently contain a
sterile aqueous
preparation of the altered N-glycosylation molecule, which preferably is
isotonic with the blood
of the recipient (e.g., physiological saline solution). Formulations can be
presented in unit-dose
or multi-dose form.
Formulations suitable for oral administration can be presented as discrete
units such as
capsules, cachets, tablets, or lozenges, each containing a predetermined
amount of the altered N-
glycosylation molecule; or a suspension in an aqueous liquor or a non-aqueous
liquid, such as a
syrup, an elixir, an emulsion, or a draught.
An altered N-glycosylation molecule (e.g., an altered N-glycosylation form of
a target
molecule such as a target protein) suitable for topical administration can be
administered to a
mammal (e.g., a human patient) as, e.g., a cream, a spray, a foam, a gel, an
ointment, a salve, or a
dry rub. A dry rub can be rehydrated at the site of administration. An altered
N-glycosylation
molecule can also be infused directly into (e.g., soaked into and dried) a
bandage, gauze, or
patch, which can then be applied topically. Altered N-glycosylation molecules
can also be
maintained in a semi-liquid, gelled, or fully-liquid state in a bandage,
gauze, or patch for topical
54
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
administration (see, e.g., U.S. Patent No. 4,307,717).
Therapeutically effective amounts of a pharmaceutical composition can be
administered
to a subject in need thereof in a dosage regimen ascertainable by one of skill
in the art. For
example, a composition can be administered to the subject, e.g., systemically
at a dosage from
0.01.tg/kg to 10,000 pg/kg body weight of the subject, per dose. In another
example, the dosage
is from I gg/kg to 100 gg/kg body weight of the subject, per dose. In another
example, the
dosage is from 1 .tg/kg to 30 gg/kg body weight of the subject, per dose,
e.g., from 3 pg/kg to 10
pg/kg body weight of the subject, per dose.
In order to optimize therapeutic efficacy, an altered N-glycosylation molecule
can be first
administered at different dosing regimens. The unit dose and regimen depend on
factors that
include, e.g., the species of mammal, its immune status, the body weight of
the mammal.
Typically, levels of an altered N-glycosylation molecule in a tissue can be
monitored using
appropriate screening assays as part of a clinical testing procedure, e.g., to
determine the efficacy
of a given treatment regimen.
The frequency of dosing for an altered N-glycosylation molecule is within the
skills and
clinical judgement of medical practitioners (e.g., doctors or nurses).
Typically, the
administration regime is established by clinical trials which may establish
optimal administration
parameters. However, the practitioner may vary such administration regimes
according to the
subject's age, health, weight, sex and medical status. The frequency of dosing
can be varied
depending on whether the treatment is prophylactic or therapeutic.
Toxicity and therapeutic efficacy of such altered N-glycosylation molecules
(e.g., an
altered N-glycosylation form of target molecules such as target proteins) or
pharmaceutical
compositions thereof can be determined by known pharmaceutical procedures in,
for example,
cell cultures or experimental animals. These procedures can be used, e.g., for
determining the
LD50 (the dose lethal to 50% of the population) and the ED50 (the dose
therapeutically effective
in 50% of the population). The dose ratio between toxic and therapeutic
effects is the therapeutic
index and it can be expressed as the ratio LD50/ED50. Pharmaceutical
compositions that exhibit
high therapeutic indices are preferred. While pharmaceutical compositions that
exhibit toxic side
effects can be used, care should be taken to design a delivery system that
targets such
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
compounds to the site of affected tissue in order to minimize potential damage
to normal cells
(e.g., non-target cells) and, thereby, reduce side effects.
The data obtained from the cell culture assays and animal studies can be used
in
formulating a range of dosage for use in appropriate subjects (e.g., human
patients). The dosage
of such pharmaceutical compositions lies generally within a range of
circulating concentrations
that include the ED50 with little or no toxicity. The dosage may vary within
this range
depending upon the dosage form employed and the route of administration
utilized. For a
pharmaceutical composition used as described herein (e.g., for treating a
metabolic disorder in a
subject), the therapeutically effective dose can be estimated initially from
cell culture assays. A
dose can be formulated in animal models to achieve a circulating plasma
concentration range that
includes the IC50 (i.e., the concentration of the pharmaceutical composition
which achieves a
half-maximal inhibition of symptoms) as determined in cell culture. Such
information can be
used to more accurately determine useful doses in humans. Levels in plasma can
be measured,
for example, by high performance liquid chromatography.
As defined herein, a "therapeutically effective amount" of an altered N-
glycosylation
molecule is an amount of the molecule that is capable of producing a medically
desirable result
(e.g., amelioration of one or more symptoms of a metabolic disorder or
decreased proliferation of
cancer cells) in a treated subject. A therapeutically effective amount of an
altered N-
glycosylation molecule (i.e., an effective dosage) includes milligram or
microgram amounts of
the compound per kilogram of subject or sample weight (e.g., about 1 microgram
per kilogram to
about 500 milligrams per kilogram, about 100 micrograms per kilogram to about
5 milligrams
per kilogram, or about I microgram per kilogram to about 50 micrograms per
kilogram).
The subject can be any mammal, e.g., a human (e.g., a human patient) or a non-
human
primate (e.g., chimpanzee, baboon, or monkey), a mouse, a rat, a rabbit, a
guinea pig, a gerbil, a
hamster, a horse, a type of livestock (e.g., cow, pig, sheep, or goat), a dog,
a cat, or a whale.
An altered N-glycosylation molecule or pharmaceutical composition thereof
described
herein can be administered to a subject as a combination therapy with another
treatment, e.g., a
treatment for a metabolic disorder (e.g., a lysosomal storage disorder). For
example, the
combination therapy can include administering to the subject (e.g., a human
patient) one or more
additional agents that provide a therapeutic benefit to the subject who has,
or is at risk of
developing, (or suspected of having) a metabolic disorder (e.g., a lysosomal
storage disorder).
56
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
Thus, the compound or pharmaceutical composition and the one or more
additional agents are
administered at the same time. Alternatively, the altered N-glycosylation
molecule (e.g., protein
or dolichol) can be administered first in time and the one or more additional
agents administered
second in time. The one or more additional agents can be administered first in
time and the
altered N-glycosylation molecule (e.g., protein or dolichol) administered
second in time. The
altered N-glycosylation molecule can replace or augment a previously or
currently administered
therapy. For example, upon treating with an altered N-glycosylation molecule
of the invention,
administration of the one or more additional agents can cease or diminish,
e.g., be administered
at lower levels. Administration of the previous therapy can also be
maintained. In some
instances, a previous therapy can be maintained until the level of the altered
N-glycosylation
molecule (e.g., the dosage or schedule) reaches a level sufficient to provide
a therapeutic effect.
The two therapies can be administered in combination.
It will be appreciated that in instances where a previous therapy is
particularly toxic (e.g.,
a treatment for a metabolic disorder with significant side-effect profiles),
administration of the
altered N-glycosylation molecule (e.g., protein or dolichol) can be used to
offset and/or lessen
the amount of the previously therapy to a level sufficient to give the same or
improved
therapeutic benefit, but without the toxicity.
In some instances, when the subject is administered an altered N-glycosylation
molecule
(e.g., protein, dolichol, or a dolichol-linked lipid) or pharmaceutical
composition of the invention
the first therapy is halted. The subject can be monitored for a first pre-
selected result, e.g., an
improvement in one or more symptoms of a metabolic disorder such as any of
those described
herein (e.g., see above). In some cases, where the first pre-selected result
is observed, treatment
with the altered N-glycosylation molecule (e.g., an altered N-glycosylation
protein or an altered
N-glycosylation dolichol) is decreased or halted. The subject can then be
monitored for a second
pre-selected result after treatment with the altered N-glycosylation molecule
(e.g., protein or
dolichol) is halted, e.g., a worsening of a symptom of a metabolic disorder.
When the second
pre-selected result is observed, administration of the altered N-glycosylation
molecule (e.g.,
protein or dolichol) to the subject can be reinstated or increased, or
administration of the first
therapy is reinstated, or the subject is administered both an altered N-
glycosylation molecule
(e.g., protein, dolichol, or a dolichol-linked lipid) and first therapy, or an
increased amount of the
altered N-glycosylation molecule (e.g., protein or dolichol) and the first
therapeutic regimen.
57
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
The altered N-glycosylation molecule (e.g., protein or dolichol) can also be
administered
with a treatment for one or more symptoms of a disease (e.g., a metabolic
disorder). For
example, the altered N-glycosylation molecule (e.g., protein, dolichol, or a
dolichol-linked lipid)
can be co-administered (e.g., at the same time or by any combination regimen
described above)
with, e.g., a pain medication.
It is understood that in some embodiments, an altered N-glycosylation molecule
is one in
which the altered glycosylation increases the ability of the molecule to
produce a medically
relevant product. For example, an altered N-glycosylation molecule can be an
enzyme capable
of producing a therapeutic product (e.g., a small molecule or therapeutic
peptide), which
enzyme's activity is increased or optimized by glycosylation. Such products
and methods of
using the products are within the scope of the present disclosure.
Any of the pharmaceutical compositions described herein can be included in a
container,
pack, or dispenser together with instructions for administration.
The following are examples of the practice of the invention. They are not to
be construed
as limiting the scope of the invention in any way.
Examples
Example 1. Plasmids, Primers and Strains
Table I contains a list of all of the plasmids used in the construction of
vectors (e.g.,
expression vectors) and deletion cassettes used in the experiments described
herein. The
MTLY60 strain of Yarrowia lipolytica was used in the experiments.
Table 2 contains a list of primers (the names of the primers) and the utility
of the primers
used in the following examples.
Table 1.
Plasmids:
JMP62
pYLTsA
58
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
pYLHmL
pYLHmA
JMP113
JMP114
pRRQ2
JME 507
JME 509
JME 461
KS-LPR-
URA3
KS-LPR-LEU2
Cre ARS68
LEU2
Table 2.
Primers: Name: Use:
TCGCTATCACGTCTCTAGC YlochI prom fw Amplification YIOCHI
(SEQ 1D NO: 18) Amplification YIOCH l P fragment
TCTCTGTATACTTGTATGT YlochI ter rev Amplification YIOCHI
ACTG (SEQ ID NO:19 Amplification YIOCH I T fragment
CTAGGGATAACAGGGTAA Y1OCH I Pfrag rev Amplification P fragment incl I-Sce
TGGTGTGACGAAGTATCG I site
AG (SEQ ID NO:20)
CATTACCCTGTTATCCCTA Y1OCH1 Tfrag fw Amplification T fragment incl I-Sce
GCGAGATCATGGACTGG I site
(SEQ ID NO:21)
GACGCGGCCGCATGAGCT YIMNSI ORF+Ter Amplification of YIMNS I P frag.
TCAACATTCCCAAAAC (Pfrag) S (ORF + terminator)
(SEQ ID NO:22
CTAGGGATAACAGGGTAA Y1MNS1 ORF+Ter Amplification of YIMNSI P frag.
TACAAAATTCAGAAATAA (Pfrag) AS (ORF + termin.) + I-SceI
AAATACTTTACAG (SEQ ID
NO:23)
CATTACCCTGTTATCCCTA YIMNS 1 Tfrag S Amplification of YIMNS I T frag.
AGTAACATGAGTGCTATG (downstream terminator.) + I-SceI
AG SE ID NO:24)
CGCTTAATTAAATGCATGG YIMNSI Tfrag AS Amplification of YIMNSI,T frag.
AGGTATTGCTG (SEQ ID (downstream terminator.)
NO:25)
59
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
Primers: Name: Use:
GGTGCTTCGACTATCAGTT ScMNSI mut 269-273 S ScMNSI mutation primer to shift to
TCGGAGGATTGGGTGATTC mam. Golgi type mannase => proof
TTTTTATG (SEQ ID NO:26) of concept in Sc
CATAAAAAGAATCACCCA ScMNSI mut 269-273 YIMNSI mutation primer to shift to
ATCCTCCGAAACTGATAGT AS mam. Golgi type mannase => proof
CGAAGCACC (SEQ ID of concept in Sc
NO:27)
TGAGCGGCCGCTTTTCTAC Y1MNN9 P fw YIMNN9 KO primer
TTCAGAGCTGGAG (SEQ ID
NO:28
GGCTTAATTAATTGGTAGT Y1MNN9 T rv YIMNN9 KO primer
GATATAATGTAACGC (SEQ
ID NO:29
TAGGGATAACAGGGTAAT YIMNN9 P rv YIMNN9 KO primer
CACGACACATACTCATTCA
AG (SEQ ID NO:30)
ATTACCCTGTTATCCCTAG YIMNN9 T fw YIMNN9 KO primer
AAGGAGATGTAGCGTAAG
(SEQ ID NO:31
TGATAAATAGCTTAGATAC LIP2 rv Reverse primer used for sequencing
CACAG (SEQ ID NO:32)
ACATACAACCACACACAT 5' hp4d Forward primer used for sequencing
C (SEQ ID NO:33)
GGCGGATCCATGGTGCTGC YIMNN4 BamHI fw Forward primer for amplification of
ACCCGTTTC (SEQ ID NO:34) YIMNN4
GGCCCTAGGCTACTCAAAC Y1MNN4 AvrII rv Reverse primer for amplification of
TCCTCGCGAATC (SEQ ID YIMNN4
NO:35)
GGTCTCGCCAGCGCGCCCA HAC 1 FW06-003 Forward primer region around
CCCTCTTC (SEQ ID NO:36)
HAC 1 splice site
CTAGATCAGCAATAAAGT HAC 1 Rv06-001 Reverse primer region around HAC I
CGTGCTGGGC (SEQ ID
NO:37) splice site
GGATCCATGTCTATCAAGC HAC I Fw06-002 Amplification of HACI gene
GAGAAGAG TCC (SEQ ID includes start codon and BamHI
NO:38)
restriction site
CCTAGGCTAGATCAGCAAT HACIRV06-006 Amplification of HACI gene
AAAGTCGTGCTGGGC (SEQ includes stop codon and AvrlI
ID NO:39)
restriction site
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
Example 2, Yarrowia Iipolytica OCH1 and MNN9 disruption
A strategy to knock out both OCHI (GenBank Accession No: AJ563920) and MNN9
(GenBank(& Accession No: AF441127) genes in Yarrowia lipolytica was set up as
described in
Fickers et al. ((2003) J Microbiol Methods. 55(3):727-37) for the LIP2 gene.
The gene
construction strategy that was followed for the OCHI gene is depicted in Fig,
5.
The OCH I KO fragment was isolated from the plasmid YIOCH I PUT TOPO by
restriction digest and by PCR and was transformed to Yarrowia lipolytica
strain MTLY60. 20
uracil prototrophic strains were obtained and screened by PCR on genomoic DNA
(gDNA) using
primers Ylochl promfw (SEQ ID NO: 18) and YlochI ter rev (SEQ ID NO: 19) to
analyse the
genomic integration of the plasmid. A fragment of the correct size (i.e., 2618
bp vs. 1894 bp in
the wild type) was amplified in 2 of the 20 clones tested. Several clones
contained a random
integrated copy of the construct and therefore both fragments were amplified.
To remove the URA3 gene, the two positive clones were transformed with the
episomal
plasmid pRRQ2 that contains an expression cassette for the Cre recombinase.
Removal of the
URA3 gene was screened for by PCR on gDNA using primers Ylochl prom fw and
Ylochl ter
rev (see above). The 2328 bp fragment (incl. URA3) was absent from, and a 1075
bp (excl.
URA3) fragment of 1075 bp was present in, the positive clones.
A Southern blot analysis was performed on the 2 positive clones to check
whether
aberrant DNA integration had occurred. Genomic DNA (gDNA) was double digested
with
EcoRV/HindI1I, subjected to agarose-gel electrophoresis, and transferred to
nitrocellulose
membrane. The membrane was probed with a 500 bp Spel/I-Scel fragment from
plasmid
YIOCHI PT TOPO. A fragment of 1456 bp was present in Aoch1 PUT, whereas a
fragment of
2066 bp in Aochl PT and a fragment of 2893 bp in the wild type strain was
present.
A construction strategy to inactivate MNN9 was set up and is depicted in Fig.
6.
The disruption fragment was cut out of plasmid Y1MNN9PUT TOPO by a Notl/Pacl
double digest and transformed to MTLY60 and Aochl PT clone 9. Several URA3
positive clones
were obtained for both strains and they were screened for correct integration
of the construct by
PCR on gDNA after single clones were isolated. A fragment of 2349 bp was
amplified in the
disruptant strains, whereas in the non-transformants, a fragment of 2056 bp
was amplified using
primers YIMNN9 P f v and Y1MNN9 T rv. (Table 2).
61
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
To analyze the N-glycan structures that were synthesized by the mutant
strains, DSA-
FACE was performed on glycans derived from mannoproteins (Fig. 7). The wild-
type
(MTLY60) strain has as main core type glycan structures mainly Man8GlcNAc2
(structural
formula I; Fig. 4) and a substantial amount of Man9GlcNAc2 (structural formula
II; Fig. 4) the
latter most probably containing an additional mannose as a result of Och I p
activity.
Furthermore, some larger structures can be seen. The ioch1 strain has mainly
Man8GlcNAc2
(structure formula I) and a small portion of Man9GlcNAc2 (structural formula
II; Fig. 4), both of
which are sensitive to a-1,2-mannosidase treatment (indicated Dochl a-l,2-man)
resulting in
trimming to Man5GlcNAc2 (structural formula IV; Fig. 4). The Emnn9 strain
accumulates more
Man9GlcNAc2 (structural formula II; Fig. 4) than the Dochl strain, which
indicates that Mnn9p is
involved in the elongation of the glycan structure subsequent to Ochlp
activity. The double
mutant Eoch1 Omnn9 displays a glycosylation phenotype that resembles the one
from the Aoch1
strain.
Example 3. Mutagenesis of MNSI
MNSJ (ER a-1,2-mannosidase) is involved in the trimming of the Man9GlcNAc2 to
Man8GlcNAc2 and has a strict substrate specificity in the sense that it is
only able to trim the a-
1,2-mannose that is linked to the a-1,3-mannose of the central arm (Fig. 2).
To determine
where the ANSI gene could be mutagenized in order to shift its substrate
specificity towards a
Golgi type a-I,2-mannosidase, the primary sequences of several ER type
mannosidases were
compared with Golgi type mannosidases. One region that is different between
the two classes
was identified. In addition, an oligosaccharide that was crystallised in the
catalytic site of the
Golgi type mannosidase into the yeast MNSI was also analyzed to identify
possible interactions
between sugar and protein. Surprisingly, the same sites were identified using
both methods.
The MNSI gene from Saccharomyces cerevisiae (GenBank Accession No: Z49631,
sgd: YJR131 W) was mutated in order to change its substrate specificity. Three
mutated versions
were made: two with one mutation (R273L and R273G) and one with 3 mutations
(R269S/S272G/R273L) in the same region:
A) R273L (arginine 273 to leucine)
B) R273G (arginine 273 to glycine)
C) R269S/S272G/R273L (arginine 269 to serine/serine 272 to glycine/arginine
273 to leucine).
62
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
All mutations were made using the Quick Change (Stratagene) mutagenesis kit.
Constructs were made to express the 3 different mutant genes under control of
the strong
constitutive TPI1 promoter. Oligonucleotides
CGACTATCCGGTTCGGATCATTGGGTGATTCTTTTTATGAG (SEQ ID NO:40).
and CTCATAAAAAGAATCACCCAATGATCCGAACCGGATAGTCG (SEQ ID NO:41)
were used to generate mutant R273L, and oligonucleotides
CGACTATCCGGTTCGGATCAGGTGGTGATTCTTTTTATGAG (SEQ ID NO:42)
and CTCATAAAAAGAATCACCACCTGATCCGAACCGGATAGTCG (SEQ ID NO:43)
were used to obtain mutant R273G using the wild type gene as a template.
Oligonucleotides
GGTGCTTCGACTATCAGTTTCGGAGGATTGGGTGATTCTTTTTATG (SEQ ID NO:44)
and CATAAAAAGAATCACCCAATCCTCCGAAACTGATAGTCGAAGCACC (SEQ ID
NO:45) were used to obtain mutant R269S/S272G/R273L using mutant R273L as
template
DNA. Via PCR reaction using oligonucleotides
CCCGATATCGGATCCATGAAGAACTCTGTCGGTATTTC (SEQ ID NO:46) and
GGGAAGCTTAACGCGGTTCCAGCGGGTCCGGATACGGCACCGGCGCACCCAACGAC
CAACCTGTGGTCAG (SEQ ID NO:47) the coding sequence of an E-tag was added at the
3'
end of the mutant and the wild type MNSIopen reading frames to allow protein
detection after
expression. An overview of the construction strategy is presented in Fig. 8.
The three constructs, as well as the non-mutated gene (as a negative control),
were
transformed to S. cerevisiae strain XW27 (MATa leu2 ura3 trpl his3 ade2 Iys2
och1::LEU2
mnn1::URA3 mnn6::ADE2) using TRPI as a selection marker after digestion of the
plasmids
with XbaI to direct the construct to the TRPI locus in the S. cerevisiae
genome. The latter strain
is able to synthesize uniform MangGlcNAc2 (on its glycoproteins. If the
mutated enzyme is
active this Man8GlcNAc2 (structural formula I; Fig. 4) should be trimmed to
Man5GIcNAc2
(structural formula IV; Fig. 4), Man6GlcNAc2 (structural formula V; Fig. 4)
and/or
Man7GlcNAc2 (structural formula VI; Fig. 4).
Tryptophan prototrophic strains were isolated, grown in liquid SDC-trp medium
and
mannoproteins were prepared. N-glycans derived from mannoproteins were
analysed via DSA-
FACE. As can be appreciated from Fig. 9, a small amount of Man8GlcNAc2
(structural formula
I; Fig. 4) from the strains that contain the R273G and R269S/S272G/R273L
mutations are
converted to Man5GlcNAc2 (structural formula IV; Fig. 4), Man6GlcNAc2
(structural formula V;
63
*Trademark
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
Fig. 4) and Man7GlcNAc2 (structural formula VI; Fig. 4). The expression of the
other mutant or
the wild type gene cause an altered N-glycosylation phenotype. To evaluate
whether all mutants
are equally well expressed, a Western blot analysis was performed using an
antibody specific for
an E-tag (a 13 amino acid epitope added to the MNS I proteins). All mutant
proteins, as well as
the wild-type MNS1 protein, were expressed equally well.
Example 4. Increasing Phosphorylation.
Expression of Yarrowia lipolvtica MNN4
To increase the phosphorylation of Man8GIcNAc2, Yarrowia liplytica MNN4 (a
homologue of the P. pastoris PNOI) was overexpressed in Yarrowia lipolvtica to
promote the
core type phosphorylation of N-glycans.
The coding sequence of the Yarrowia lipolytica MNN4 (XM_503217, YALIOD24101g)
gene was amplified using primers GGCGGATCCATGGTGCTGCACCCGTTTC (Y1MNN4
BamHI fw; SEQ ID NO:34) and GGCCCTAGGCTACTCAAACTCCTCGCGAATC (Y1MNN4
AvrII rv; SEQ ID NO:35). This open reading frame (ORF) was cloned into the
plasmid using
BamHI and AvrII sites, which placed the ORF under control of the hp4d promoter
of plasmid
pY1HURA3 that contains the URA3dI gene as a selection marker and the zeta
sequences for
improving random integration (Fig. 10).
Prior to transformation in the MTLY60 Aoch1 strain, the plasmid containing the
MNN4
expression cassette was digested either with Eco47111 for integration in the
URA3 locus, Pvul for
integration in the M7VN4 locus, or RsrIIIBstBI for random integration.
Transformants targeted to
the URA3 and MNN4 locus were analysed by PCR using a primer in the hp4d
promoter and one
in the LIP2 terminator. Transformants with random integration of the construct
were evaluated
by Southern blot analysis.
To evaluate whether manno-phosphorylation was increased we analysed N-glycans
derived from secreted glycoproteins after 48 hours culture in YPD medium by
DSA-FACE
capillary electrophoresis (Fig. 11). The amount of Man8GIcNAc2 (structural
formula I) was
drastically reduced in favour of two structures that migrate faster (compared
to Man8GIcNAc2
(structural formula I; Fig. 4)) and that are likely to contain one (P)
(structural formula X or XI;
Fig. 4) and two (PP) (structural formula XII; Fig. 4) phosphate residues,
respectively (Fig. 11).
Thus, it can be concluded that the random integrated expression cassettes
perform better than the
64
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
cassettes integrated in the URA3 locus or the MNN4 locus, in that order. The
MZ2 exhibited the
highest level of phosphorylation.
Assuming that both peaks derive from the Man8GlcNAc2 (structural formula I;
Fig. 4)
peak, the amount of MangGlcNAc2 converted to phosphorylated glycans was
quantitated (Table
3).
Table 3.
Phosph-
N I can struct. height area % signal Status
Strain Aochl
M82P (struct. form. XII) 18 302 1,0282618 18.91045*
M8P (struct. form. X or XI) 261 5252 17,88219
M8 (struct. form. I) 928 23816 81,08955 81.08955*
29370 100 100
Strain MU5 % signal
M82P (struct. form. XII) 1319 19736 27,1677381 17283=
M8P (struct. form. X or XI) 2025 39232 54,00509
M8 (struct. form. 1) 539 13677 18,82717 18.82717*
72645 100 100
Strain MZ2 % signal
M82P (struct. form. XII) 1182 17662 27,7529983.11282*
M8P (struct. form. X or XI) 1803 35231 55,35984
M8 (struct. form. 1) 419 10747 16,88718 16.88718*
63640 100 100
Table 3 Legend: Height and area refer to the peak height and peak area as
determined from electropherograms, "%
signal" refers to the proportion of each glycan in the N-glycan mixture, The
numbers identified by asterisk depict
the proportion of phosphorylated MangGn2 (top) and the proportion of non-
phosphorylated Man3Gn2 (bottom).
These results indicated that more than 80% of MangGlcNAc2 (structural formula
I; Fig. 4)
that is present in the parent Mochl is phosphorylated in the strain that over
expresses the Y1MNN4
gene.
Example 5. Modifying Glycosylation by Lipid-linked Oligosaccharide
Modification in the
Endoplasmic Reticulum
Materials and Methods
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
Strains, culture conditions and reagents. Escherichia coli strains MC 1061 or
TOP 10 or DH5Q
were used for the amplification of recombinant plasmid DNA and grown in a
thermal shaker at
37 C in Luria-Broth (LB) medium supplemented with 100 pg/ml of carbenicillin
or 50 g/ml of
kanamycin depending on the plasmids used.
Yarrowia lipolytica MTLY60 (ura3 leu2) strain was used as parent strain. All
yeast strains were
cultured in a 28 C incubator. They were grown on YPD medium (2% dextrose, 2%
bacto-
peptone and 1% yeast extract) or synthetic dextrose complete (SDC) medium
(0.17% YNB w/o
amino acids and without ammonium sulphate, 1% glucose, 0.5% NH4CI, 50mM K/Na
phosphate
buffer pH 6.8 and 0.077% Complete Supplement Mixture (Qbiogene Inc, Morgan
Irvine, CA)).
For selection of Ura+ and Leu+ transformants 0.077% CSM -ura or CSM -leu was
added
respectively.
Standard genetic techniques. Transformation competent cells of Yarrowia
lipolytica were
prepared as described in Boisrame et al. (1996) J. Biol. Chem. 271(20):11668-
75.
Genomic DNA from all yeast strains
was isolated using a published protocol (Epicenter Kit catologue No. MPY80200;
Epicenter
Biotechnologies, Madison, WI). The protocol involves non-enzymatic cell lysis
at 65 C,
followed by removal of protein by precipitation and nucleic acid precipitation
and resuspension.
PCR amplification was performed in a final volume of 50 l containing 5M1 of
lOx buffer
(200mM Tris-HC1 pH8.4 and 500 mM KCJ), a variable quantity of MgC12i 2.5 M
dNTP, Song
of template, 50 pmol of the proper primers and 2.5 units of either Taq or Pfu
DNA polymerase.
Cycling conditions used were as follows: denaturation at 94 C for 10 minutes
followed by hot
start and 30 cycles of 94 C for 45 seconds, suitable annealing temperature for
45 seconds and
extension at 72 C for 1 minute per kb followed by 10min of extension at 72 C.
DNA fragments
(PCR products or fragments) recovered from gel were purified using
NucleoSpin*extract II.
(Macherey-Nagel). DNA sequencing was performed by VD3 Genetic Service Facility
(Antwerp,
Belgium).
Vector construction.
(i) Knock-out (gene-replacement) of the ALG3 gene. The promoter fragment (P)
of the
ALG3 gene (GenBank Accession No: XM_503488, Genolevures: YALIOE03I90g) was
amplified from genomic DNA of the Yarrowia lipolytica MTLY60 strain by PCR
with
66
*Trademark
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
5'CAGTGCGGCCGCACTCCCTCTTTTCACTCACTATTG3' (SEQ ID NO:48) and
5'CATTACCCTGTTATCCCTACGCTCAGATCCAATTGTTTTGGTGGTC3' (SEQ ID
NO:49) as the forward and reverse primers, respectively, using Taq polymerase
(Invitrogen). The
overhanging A nucleotide was removed with T4 DNA polymerase (Fermentas,
Ontario, Canada).
The terminator fragment (T) of the ALG3 gene was amplified from genomic DNA of
the
Yarrowia lipolytica MTLY60 strain by PCR with
5'GTAGGGATAACAGGGTAATGCTCTCAAGGACGGACCAGATGAGACTGTTATCG3'
(SEQ ID NO:50) and
5'GACTTTAATTAAACCCTATGTGGCACCTCAACCCACATCTCCCGTC3' (SEQ ID
NO:51)
as the forward and reverse primers, respectively, using the proofreading Pfu
DNA polymerase
(Fermentas). Because of overlapping primer sequences containing an IScel
restriction site, both
fragments could be linked by PCR with the P-forward primer and the T-reverse
primer. This co-
amplicon was then subcloned in a pCR-2.1 TOPO TA (Invitrogen) vector and the
correctness of
the co-amplicon's sequence was confirmed by sequencing. The co-amplicon was
then cloned
using the Notl-PacI sites into an intermediate vector.
(ii) Overexpression of the ALG6 gene. The ALG6 ORF (1725bp) together with the
terminator (415bp downstream) of the ALG6 gene (GenBank(g Accession No:
XM_502922,
Genolevures: YALIOD17028g) were cloned from genomic DNA of the Yarrowia
lipolytica
MTLY60 strain by PCR with 5'CAGTGGATCCATGAACTCTCCTATTTTCACTACCG3'
(SEQ ID NO:52) and 5'GACTCCTAGGAAGCTTCCAGGTTACAAGTTGTTAC3'(SEQ ID
NO:53) as the forward and reverse primers, respectively, using the
proofreading Pfu DNA
polymerase (Fermentas). The sequence was cloned in pCR-Blunt II-TOPO
(Invitrogen) and the
correctness of the ALG6 ORF sequence was confirmed by sequencing (as above).
Next, the
ALG6 ORF was cloned in a vector (pYLHmA) containing the hp4d promoter via
BamHI and
AvrII and subsequently cloned in the intermediate vector via the unique
restriction sites Clal and
HindIII present in the terminator fragment of ALG3.
(iii) Selection marker cassette. To remove the selectable marker URA3 from the
host
genornic DNA, the Cre-lox recombination system was used, e.g., as described by
Fickers et al.
67
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
((2003) J. Microbiol. Methods 55(3):727-737.
Upon expression of the Cre recombinase from the plasmid pRRQ2
(hp4d-cre, LEU2) (a gift from the Institut National de Recherche Agronomique
(INRA)), the
marker gets excised by recombination between the two lox sites. In both
constructs, with and
without the ALG6 overexpression cassette, the URA3 selection marker flanked by
lox sites, was
inserted in the introduced I-SceI site between P and T fragments of the
vector, resulting in a
"PUT" construct.
Preparation of mannoproteins. Yeast strains were grown overnight in 10 ml
standard YPD
medium in 50 ml falcon tubes, rotating at 250 rpm in a 28 C incubator. The
cells were then
pelleted by centrifugation at 4000 rpm at 4 C. The supernatants were removed,
and the cells
were first washed with 2 ml of 0.9% NaCl solution followed by two washes with
2 ml of water
and subsequently resuspended in 1.5 ml of 0.02 M sodium citrate pH 7 in a
microcentrifuge tube.
After autoclaving the tubes for 90 minutes at 121 C, the tubes were vortexed
and the cellular
debris was pelleted by centrifugation. The supernatants were collected and the
mannoproteins
were precipitated overnight with 4 volumes of methanol at 4 C with rotary
motion. The
precipitate was then obtained by centrifugation of the alcohol precipitated
material. The pellets
were allowed to dry and dissolved in 50 l of water.
Sugar analysis. DNA sequencer-assisted (DSA), fluorophore-assisted
carbohydrate
electrophoresis (FACE) was performed with an ABI 3130 DNA sequencer as
described by
Callewaert et a!. (2001; supra). Briefly, glycoproteins were denatured for 1
hour in RCM buffer
(8M urea, 360mM Tris pH 8.6 and 3.2 mM EDTA) at 50 C followed by
immobilization on a
prewetted PVDF membrane of a IP plate containing 15 I RCM. Prewetting of the
membrane
was done with 300 I MeOH, 3 times washed with 300 I water and 50 1 RCM,
followed by
vacuum removal. The glycoproteins were reduced for 1 hour with 50 l 0.1 M
dithiothreitol and
washed 3 times with 300 I water. A 30 minute incubation in the dark with 50 I
0.IM iodoacetic
acid was used to carboxymethylate the SH groups, followed by 3 washes with 300
I water. The
plates were subsequently incubated for 1 hour with 100 I I%
polyvinylpyrrolidone 360 to
saturate the unoccupied binding sites on the membrane, again followed by 3
washes with 300 I
water. Next, the N-glycans were released by 3 hours treatment with peptide: N-
glycosidase F
68
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
(PNGase F) x U in 50 l of 10mM Tris-acetate pH 8.3. N-glycans were recuperated
and
derivatized with the fluorophore 8-aninopyrene-1,3,6-trisulfonate (APTS) by
reductive
amination. This was accomplished with an overnight (ON) incubation at 37 C
with I.tl of 1:1
mixture of 20 mM APTS in 1.2M citric acid and I M NaCNBH3 in DMSO and
quenching by
addition of 4 1 water. Excess label was removed by size fractionation on
Sephadex G-10 resin.
The remaining labeled N-glycans were then concentrated by evaporation. The N-
glycans of
RNase B and an oligomaltose ladder were included as size markers. Data
analysis was
performed using Genemapper software (Applied Biosystems). Glycosidase digests
on the
labeled sugars were performed ON at 37 C in 100mM NH4AC pH5.. Additional Jack
bean (JB)
mannosidase was added after ON digestion and left for another 24 hours at 37
C.
Disruption of the ALG3 gene in Yarrowia lipo! tY ica
To disrupt the ALG3 gene, a vector was generated that includes parts of the
promoter and
terminator of ALG3 and has a URA3 selection marker cassette and was designated
pYLalg3PUT. A Nod and PacI site were integrated to linearize the vector and
thereby remove
the E. coli related DNA elements. Double homologous recombination at the
promoter and
terminator site was used to replace ALG3 with the URA3 selectable marker,
which resulted in an
alg3::URA3 mutant strain. The knockout strategy applied was described by
Fickers et al. (2003;
supra) and makes use of the Cre-lox recombination system, that facilitates
efficient marker
rescue. Upon integration in the genomic ALG3 contig the Alg3p a-1,6-
mannosyltransferase
activity should be lost. This was monitored by analyzing the glycosylation
pattern of the
mannoproteins of several transformants. The N-glycans derived from
mannoproteins were
analysed by DSA-FACE (capillary electrophoresis) and treated with a selection
of
exoglycosidases to reveal the structures. Seven out of 24 transformants gave a
change in
glycosylation profile (three of which are depicted in Fig. 13). In all seven
transformants, correct
integration of the knockout cassette in the genome could be confirmed by PCR.
Three main
glycan structures were found by analyzing the profiles: (i) one (structural
formula VII; Fig. 4)
that runs at the same size as the Man5G1cNAc2 structure of RNase B (the latter
being structural
formula IV; Fig. 4); (ii) one at a distance of one glucose-unit extra; and
(iii) one at the distance of
two extra glucose-units. (Fig. 13). These results indicate that ALG3 was
disrupted in these cells.
69
*Trademark
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
Overexpression of a-1,6-mannosyltransferase Alg6p
A strategy was developed in which a constitutively active overexpression
cassette for the
first glucosyltransferase, i.e., Alg6p, was incorporated into the alg3 gene
replacement vector.
This vector was designated pYLa1g3PUT-ALG6. A NotI/Pacl fragment of this
vector was
transformed into the Yarrowia lipolytica MTLY60 strain. In this way,
disruption of ALG3 and
overexpression of ALG6 under control of the hp4d promoter is achieved. Correct
integration in
the genome was again confirmed by PCR. DSA-FACE analysis of the N-glycans
derived from
mannoproteins showed that half of the transformants, i.e., 12 out of 24,
exhibited a change in
glycosylation pattern comparing to the WT strain. Overexpression of ALG6 led
to a mild clonal
variation (Fig. 13).
Identification of the N-glycan structures
To further elucidate the nature of the glycan structures from the experiments
described
above, in vitro digests of glycans derived from the mannoproteins (as above)
were performed
with a selection of exoglycosidases. The mannoprotein glycans were analyzed
with the
following enzymes: a-1,2-mannosidase; a-mannosidase (JB) and glucosidase II.
Three observed
glycanstructures represent Man5GlcNAc2 (structural formula VII; Fig. 4),
GlcMan5GIcNAc2
(structural formula VIII; Fig. 4) and Glc2Man5GlcNAc2 (structural formula IX;
Fig. 4) (Fig. 14).
These results indicate that there is very little to no high mannose elongation
by a-1,6-
mannosyltranferases (e.g., Ochlp).
To determine if ALG6 overexpression is necessary for promoting N-glycosylation
site-
occupancy, Lipase 2 (LIP2) from Yarrowia lipolytica was expressed in three
different strains of
Yarrowia: MTLY60, MTLY60Aalg3 and MTLY60AaIg3ALG6. A construct for the
Yarrowia
lipolytica LIP2, under control of a TEF constitutive promoter was obtained
from INRA. The
expression cassette was transformed to the above-mentioned strains and the
expression of the
protein was verified by subjecting the supernatant prepared from the
transformed cells to SDS-
PAGE analysis (Fig. 28). The Lip2p protein has 2 glycosylation sites. Lip2p
protein derived
from the alga-deficient ("knockout") yeast strain was resolved by SDS-PAGE
into three distinct
bands that were visualized using Coomassie blue staining of the gel (Fig. 28).
To confirm that
all three forms of protein in the gel were different glycosylation forms of
the Lip2p protein,
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
Lip2p protein obtained from the alg3-deficient ("knockout") yeast strain was
subject to treatment
with PNGase F (an enzyme that removes oligosaccharide residues from
glycoproteins) and then
subjected to SDS-PAGE analysis as described above. Treatment of the Lip2p
protein with
PNGase F resulted in a single band (which had the same molecular weight as non-
glycosylated
Lip2p) on the gel following Coomassie blue staining and indicated that all
three forms of protein
previously observed were different glycosylation forms of the same Lip2p
molecule. The same is
true for the Lip2p derived from the alg3ALG6 strain. However, the amount of
protein in a
reduced glycosylation form is decreased. Thus, it can be concluded that
overexpression of ALG6
can (at least partially) restore N-glycosylation site-occupancy, which is
reduced in the a1g3
knockout mutant yeast strain.
Removing Capping Glucose Structures
Next, to eliminate mono (structural formula VIII; Fig. 4) and bi-glucosylated
(structural
formula IX; Fig. 4) Man5GlcNAc2 (structural formula VII; Fig. 4) structures in
vivo, cells were
genetically engineered to overexpress the a-subunit of the enzyme glucosidase
II. The a subunit
of glucosidase II of Yarrowia (GenBank(K Accession No: XM_500574) and the a
subunit of
glucosidase II Trypanosoma brucei (GenBank Accession No: AJ865333) were
independently
cloned as two strategies to overexpress the protein. The a subunit of
glucosidase II
Trypanosoma brucei was chosen since its natural substrate is GlcMan5GlcNAc2
(structural
formula VIII; Fig. 4). Both genes were cloned under control of the
constitutive hp4d promoter
and their plasmids contain the URA3 marker. These constructs were transformed
into a1g3
mutant yeast strains, both with and without ALG6 overexpression.
Oligosaccharides were prepared from secreted proteins derived from cultured
cells
containing the constructs and the profile of the oligosaccharides was
determined by DSA-FACE
analysis. All transformants gave the same DSA-FACE profile, two different
clones of each
glucosidasell a are depicted in Fig. 29. From these results it was concluded
that the
overexpression of either the Yarrowia or the Trypanosoma glucosidase II a
subunit has only a
minor effect on the amount of mono (structural formula VIII; Fig. 4) and bi-
glucosylated
(structural formula IX; Fig. 4) Man5GIcNAc2 (structural formula VII; Fig. 4)
structures.
71
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
Expression of glucosidase II a-subunits of Yarrowia lipolvtica and Trypanosoma
brucei tagged
with an HDEL sequence
To improve the effect of the expression of Yarrowia or the Trypanosoma
glucosidase 11 a
subunit on removing glucose residues from Man5GlcNAc2 in vivo, a nucleic acid
encoding an
HDEL tag was added using molecular biology techniques in frame to the 3'end of
the nucleic
acid encoding each of the two GIsII a enzymes. The HDEL tag was meant to serve
as a retrieval
mechanism from the Golgi to the ER. Plasmids encoding HDEL-tagged glucosidase
II
sequences from both Yarrowia lipolytica (Y.1.) and Trypanosoma brucei (Tb.)
under control of
the hp4d promoter were transformed to the alga KO strain with and without
overexpression of
the ALG6 gene. As can be seen in Fig. 30, overexpression of the Yarrowia
lipolytica
glucosidase II a subunit had only a minor effect on the amount of glucosylated
structures. In
contrast, overexpressing the a-Glucosidase II of Typanosoma brucei a subunit
with an extra
HDEL tag leads to a reduction of the mono-glucose peak (see Fig. 31).
Treatment of glucosylated glycans with Mutanase
The above-described results demonstrate one exemplary means of reducing mono-
glucosylated forms of Man5GlcNAc2. To reduce bi-glucosylated forms of
Man5GlcNAc2 from
glycoproteins, the mutanase of T harzianum was investigated as one potential
solution. An
enzyme preparation was obtained from Novozymes (Novozyme 234; Bagsvaerd,
Denmark) and
was used to digest oligosaccharides in vitro. That is, mutanase was added in
different
concentrations to the oligosaccharides derived from a alg3ALG6 strain
(glycans: Man5GlcNAc2,
GlcMan5GlcNAc2 and Glc2Man5GlcNAc2). As shown in the DSA-FACE profile of Fig.
32, the
bi-glucose peak observed in the oligosaccharides was effectively reduced.
Next, the mutanase of T harzianum was overexpressed in vivo. An HDEL-sequence
containing mutanase was synthesized as a codon-optimized cDNA for expression
in Yarrowia
lipolytica. The mature protein was cloned in frame with the LIP2 pre signal
sequence under
control of the TEF1 promoter (Fig. 33). This construct is transformed into
a1g3 mutant yeast
strains, both with and without ALG6 overexpression. Oligosaccharides are
prepared from
cultured cells containing the construct and the profile of the
oligosaccharides is determined by
DSA-FACE analysis. It is expected that the DSA-FACE profile will show a
reduction in the bi-
glucose peak observed in the oligosaccharides. From these results it will be
concluded that the
72
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
overexpression of mutanase in vivo is effective at reducing the bi-glucose
peak observed in
oligosaccharides as compared to cells not overexpressing the mutanase.
Co-expression of Yl GIsIl a- and 0 subunits
It is known that the a- and (3-subunits of glucosidase II form a heterodimeric
complex
whereby the (3-subunit is responsible for retrieval of the complex to the ER
and is also involved
in substrate recognition, whereas the a-subunit contains the catalytic
activity. Since the
overexpression of only the a-subunit of glucosidase II had a small effect on
bi-glucose
oligosaccharide structures, the a- and (3- subunits were co-expressed.
The open reading frame of the (3-subunit (YALIOB03652g) was amplified from
genomic
DNA that was isolated from the MTLY60 strain using PCR and was cloned under
control of the
TEF1 and hp4d promoter. The constructs were made with LEU2 as a selection
marker and with
the glucosidase II R-subunit under control of the TEFL and the hp4d promoter.
These were
transformed to the alga knockout strains with and without ALG6 overexpression
and
overexpressing the Yarrowia lipolytica Glucosidase II a subunit with and
without an HDEL
sequence tag. N-glycans were prepared from proteins secreted from the cells
and the DSA-
FACE profiles of the N-glycans are depicted in Figs. 33 and 34 (alg3 knockout
with
overexpression of ALG6). It can be concluded from these profiles that
overexpressing the R
subunit of glucosidase II from Yarrowia lipolytica did have a positive effect
on the trimming of
the glucosylated sugars. In general, the efficacy of the R subunit of
glucosidase II was improved
when expressed under the TEF1 promoter. The glucosylated structures were even
more reduced
when the Yarrowia lipolytica glucosidase II a subunit contained an HDEL tag
(Figs. 33 and 34).
For a1g3-deficient cells without ALG6 overexpression, similar results
regarding reduction
of glucosylated structures were observed for each of the different cell
populations (Fig. 35).
Expression of Aspergillus GIsII a and b subunit
In order for the glucose residues to be removed from the glucose bearing
structures that
occur in alg3-deficient background, the Aspergillus niger mature (lacking
signal peptide)
glucosidase II a and a were synthesized as codon-optimized cDNA for expression
in Yarrowia
lipolytica (a-subunit (SEQ ID NO:7; Figs. 36A-36B) R-subunit: (SEQ ID NO:8;
Fig. 37).
73
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
Aspergillus niger (An) glucosidase a subunit was cloned under control of the
constitutive TEF1
and hp4d promoters and had URA3 gene as a selection marker. The expression
cassettes (ORFs
under control of TEF1 and hp4d) were transformed to Yarrowia lipolytica
alg3ALG6 strain.
Transformant candidates were grown in YPD and glycans from secreted proteins
were analysed
by DSA-FACE. It can be deduced from Fig. 38 that the two glucosylated
structures are less
abundant in the transformant strains compared to the non-transformant
(alg3ALG6).
To further reduce the glucosylated glycan structures a construct is made with
(3-subunit of
the Aspergillus niger glucosidase II under control of TEF I promoter or hp4d
promoter with
LEU2 as a selection marker. This construct is transformed to Yarrowia
lipolytica alg3ALG6
strain expressing the An glucosidase II a-subunit. It is expected that
expression of the a-subunit
of the Aspergillus niger glucosidase II will result in a decrease in
glucosylated structures in
Yarrowia lipolytica cells.
Example 6. Identification of the-HA C1 Intron and Cloning and Isolation of the
HACI Gene
Y. lipolytica HACI splice site. On the basis of sequence homology between the
intronic
regions of HAC I in Yarrowia lipolytica and the fungi Trichoderma reesei and
Aspergillus
nidulans, a potential splice site of the Yarrowia lipolytica HAC I (Genbank:
XM_500811,
Genolevures: YaliOB 12716g) was identified. The 5' and 3' splice sites were
predicted to be
localized in a characteristic loop structure and the intron was calculated to
be 29 bp long.
Primers were developed around the splice site in order to identify the intron.
First strand
cDNA was synthesized from the isolated mRNA from an UPR (unfolded protein
response)
induced (by means of growth in dithiothreitol (DTT)) and non-induced culture
(negative control)
with gene specific primers. PCR was then performed on first strand using
primers HACIFW06-
003 and HACIRv06-001. Amplification products were analyzed on a 1.5% agarose
gel.
A fragment of +/- 400 bp was expected to be amplified for the non-induced
cells; a 29 bp
smaller fragment was expected to be amplified for the induced cells. Fragments
of the correct
size were obtained from the non-induced cells and the UPR induced cells. Two
more
amplification products were obtained for the UPR induced culture. The middle
fragment was the
same size as the band obtained for the non-induced culture and was interpreted
as being
unspliced HACI. The lower, most prominent band was purified from the gel and
cloned into a
sequencing vector. After sequencing the construct, a sequence alignment was
performed in order
74
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
to identify the splice site (Fig. 15). From the sequence alignment it can be
seen that the splice
site is located at the position that was predicted from the comparison of the
Yarrowia lipolytica
and the fungal (Trichoderma reesei and Aspergillus nidulans) HACI sequences.
The splice site
is 29 bp long.
In order to isolate the active full length HACI sequence, primers were
engineered to have
restriction sites suitable for cloning into an expression vector. Primer
sequences were as follows:
Hac 1 ylRv07-018:
CCTAGGTCACTCCAATCCCCCAAACAGGTTGCTGACGCTCGACTCATAGTGAGCTAG
ATCAGCAATAAAGTCG (SEQ ID NO:54) and HAC1 Fw06-002 :GGA TCC ATG TCT ATC
AAG CGA GAA GAG TCC (SEQ ID NO:55). A 10 ml culture of yeast cells was
incubated for
1.5 hours in the presence of 5 mM DTT to induce the UPR response. Following
the incubation,
RNA was isolated from the DTT-treated cells and first strand cDNA was prepared
from the
isolated RNA using reverse transcriptase and PCR using the cDNA as a template
and the above
primers. The PCR-amplified sequence containing the spliced HACI was inserted
into the pCR-
blunt-TOPO cloning vector using standard molecular biology techniques and
sequenced.
Pichia pastoris HACI splice site. On the basis of sequence homology of the
intronic
regions of the Pichia pastoris and Saccharomyces cerevisiae HAC I genes, a
potential splice site
in the Pichia pastoris HAC1 gene was identified (Fig. 16). The 5' and 3'splice
sites were
predicted to be localized in a characteristic loop structure and the intron
was calculated to be 322
bp in length.
Primers (HAC 1 Fw06-004 and HAC 1 Rv06-005) were developed around the
predicted
splice site in order to identify the intron (see Table 4). A fragment of 257
nucleotides was
expected to be amplified when the intron is removed and a 579 bp fragment if
intron is still
present. First strand cDNA was synthesized from the isolated mRNA from an UPR
induced and
non-induced culture. The UPR was induced by adding 5mM DTT to a 10 ml culture
of
exponentially growing cells. The cells were cultured in the presence of DTT
for 1.5 hours. The
amplification product was analyzed by 1.5 % agarose gel electrophoresis. A
fragment of
approximately 257 bp was obtained from cDNA from both non-induced and induced
cells.
Table 4. Primers
Primer code sequence 5'--> 3' Information
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
Primer code sequence 5'--> 3' Information
GAATTCATGCCCGTAGATTC Forward primer Hacl gene+ start codon and
HAC1-Karl TTCTC (SEQ ID NO:56) EcoRl site
GAGTCTTCCGGAGGATTCA Forward primer Hac1 gene region around 5'
s lice site
HAC1 Fw06-004 G (SEQ ID NO:57)
CCTGGAAGAATACAAAGTC
HAC1 Rv06-005 (SEQ ID NO:58 Reverse primer Had gene gene near stop codon
CCTAGGCTATTCCTGGAAG
AATACAAAGTC (SEQ ID reverse primer Hacl gene + stop codon and
HAC1RvO6-009 NO:59) Avrll site
GGTATTGCTGAGCGTATGC
ACT Fw07-007 AAA (SEQ ID NO:60) Act1 forward primer for QPCR
CCACCGATCCATACGGAGT
ACT Rv07-003 ACT (SEQ ID NO:61) Actl reverse primer for QPCR
CGACCTGGAATCTGCACTT
HAC1 Fw07-008 CAA (SEQ ID NO:62) Hacl forward primer QPCR
CGGTACCACCTAAGGCTTC
HAC1 RV07-004 CAA (SEQ ID NO:63) Hacl reverse primer QPCR
CCAGCCAACTGTGTTGATTC
Kar2ppFwO7-009 AA (SEQ ID NO:64) Kar2 forward primer QPCR
GGAGCTGGTGGAATACCAG
Kar2ppRvO7-005 TCA (SEQ ID NO:65) Kar2 reverse primer QPCR
To verify the length of the unspliced P. pastoris HACI gene, PCR was performed
on
genomic DNA using primers HAC1-Karl and HACIRvO6-005. The length of the
obtained
fragment was compared with the length of a PCR product obtained from the cDNA
from an
induced cell culture. The amplified fragment from the genomic DNA is about 300
bp longer
than the amplicon derived from the cDNA using the same primers indicating that
the intron is
present in the genomic DNA sequence and absent from the spliced mRNA.
The cDNA fragment of 257 bp was isolated from the gel and cloned in a
sequencing
vector. The fragment was sequenced and an alignment was performed in order to
identify the
splice site (Fig. 17). To isolate and clone the spliced P. pastoris HAC I
gene, PCR primers were
developed with restriction enzyme sites for cloning into an expression vector
(HAC1-Karl and
HAC I RvO6-009). A 10 ml culture was UPR-induced with 5 mM DTT for 1.5 hours.
First strand
cDNA was prepared from the isolated RNA using reverse transcriptase and PCR
was
subsequently performed on the cDNA template DNA using the above primers. The
spliced
HACI was isolated and cloned in pCR-blunt-TOPO cloning vector for sequencing.
The spliced
gene was also cloned under the control of the methanol inducible AOXI promoter
in the
expression vector pBLHIS LX to obtain the vector pBLHIS LX ppHACI spliced. The
correct
insertion of the HAC I gene into the expression vector was confirmed using PCR
and restriction
enzyme analysis.
76
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
In Saccharomyces cerevisiae, upon splicing, the coding sequence of the C-
terminal 10
amino acids in the non-spliced mRNA is replaced with the coding sequence of 18
amino acids.
In accordance, in Pichia pastoris it was revealed that the coding sequence of
the C-terminal 45
amino acids in the non-spliced HAC 1 are replaced upon splicing by the coding
sequence of again
18 amino acids which are homologous to the ones from the S. cerevisiae
sequence (Fig. 18).
Example 7. Transformation and induction of spliced HAC1 gene into Yarrowia
lipolytica
Yarrowia lipolytica cells (MTLY60 strain) were transformed with the vector
"PYHMAXHACIylspliced" containing the spliced HACI cDNA (above) under the
expression
control of the hp4d promoter and the URA3 gene as a selection marker.
Integration of the vector
into the yeast genome was verified using PCR. The MTLY60 strain transformed
with
PYHMAXHACIylspliced was grown in a 2 ml culture in YPG at 28 C for 24 hours,
The
cultured cells were washed twice with YNB, then diluted to OD600 0.6 and grown
for 24 hours in
YTG buffered with 50 mM phosphate buffer pH: 6.8. The cells were then diluted
to OD60o 0.2
and grown for 3 more generations in order to harvest the cells in the mid-
exponential phase. To
the pellet, 1 ml of RNApureTM solution was added to the cells along with I g
of glass beads.
Cells were broken by vigorous shaking. RNA was extracted from the broken cells
by adding 150
l chloroform and precipitating the RNA with isopropanol. The extracted RNA was
also treated
with DNAse to remove any coprecipitated DNA impurities.
First strand cDNA was prepared from 800 ng of the RNA using the iScriptTMcDNA
Synthesis Kit (Bio-Rad Laboratories, Hercules, CA) in a 20 pl total volume
reaction. The
equivalent of 20 ng RNA was used for real time PCR analysis to determine the
amount of HACI
mRNA in the cells. Real time PCR was run using SYBR green as the detection
reagent
(fluorescent) (Eurogentec). In addition to designing primers for detecting the
amount of HAC1
mRNA in the cells, primers were also designed to quantify the amount of ACTI
(household
gene) and KAR2 (UPR responsive gene) genes as controls for the real time PCR.
The relative
amount of mRNA of each gene in the cells was calculated from the comparative
threshold cycle
values using Actin (a housekeeping gene) as the expression control. Induction
of the UPR
response by the cells was confirmed by measuring the expression of UPR. The
expression levels
of KAR2 as well as HAC1 are higher in the strains expressing HACI under
control of a
constitutive promoter compared to the wild type strain MTLY60 (Fig. 39).
77
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
Example 8. Transformation and induction of spliced HACI gene into Pichia
pastoris
Media: For the following experiments, three types of media were used: BMY
(Buffered
Medium for Yeast: 100 mM potassium phosphate pH:6.0/1.34% YNB without amino
acids/l %
Yeast extract/2% peptone); BGMY (Buffered Glycerol-complex Medium for Yeast:
100 mM
potassium phosphate pH:6.0/1.34% YNB without amino acids/1% Yeast extract/2%
peptone/1%
glycerol); and
BMMY (Buffered Methanol-complex Medium for Yeast: 100 mM potassium phosphate
pH:6.0/1.34% YNB without amino acids/l% Yeast extract/2% peptone/0,5%
glycerol).
Pichia pastoris cells were transformed according to the electroporation
protocol from the
Pichia Expression kit (Invitrogen Cat. No. K1710-01). The vector pBLHIS IX
ppHAC1 spliced
was linearized in the HIS4 gene to target the construct to the HIS4 locus for
integration. Ten
micrograms of DNA was transformed into the yeast cells. The correct
integration of the
construct was validated using PCR on genomic DNA after isolation of single
colonies (primers
HAC 1-Karl and HAC I Rv06-005). Fragments of 915 kb and 1237 kb were amplified
from DNA
obtained from the transformed cells, whereas in the non-transformants (cells
without integration
of the construct) a fragment of 1237 kb was amplified. Clones so identified as
positive for
integration of the plasnvd were grown in 10 ml BMGY medium for 24 hours before
induction.
Cells were washed once with BMY. BMGY was added to non-induced cultures while
BMY was
added to the induced cultures. Every 12 hours, induced cultures were fed with
0.5% methanol
(final concentration). Induction was performed for 24 hours after which cells
were harvested by
centrifugation. To prepare RNA, cells were combined with I nil RNApureTM
(Genhunter
Corporation, Nashville, NY) and I g of glass beads, and lysed by vigorous
shaking. RNA was
extracted by the addition of 150 gl chloroform and precipitated with
isopropanol. The extracted
and precipitated RNA was DNAse treated with RNAse-free DNAse obtained from
Qiagen (Cat
No. 79254). 400 ng of total RNA was subjected to reverse transcriptase
reaction using an
oligodT primer and the Superscript II reverse transcriptase (Invitrogen, Cat.
No. 18064-014).
The equivalent of 20 ng RNA was used in a real-time PCR reaction. Primer
sequences were
designed by Primer Express software (Applied Biosystems) (see primer table for
sequence).
Real time PCR utilizing SYBR green fluorescent reagent (Eurogentec) was run in
the iCycler*.
machine from BioRad. The relative amounts of mRNA were calculated from the
comparative
78
*Trademark
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
threshold cycle values using the housekeeping gene actin as a control.
Quantification of UPR is
performed through expression analysis of the UPR-target gene KAR2. A 3 to 7
fold higher
expression of KAR2 was obtained when comparing clones that were not induced as
compared to
the same clones that were induced with methanol (Fig. 19).
The relative amount of HACI mRNA from two additional clones 6 and clone 8 was
determined by quantitative PCR and compared with the relative amount of mRNA
of Kar2. A
strong induction of HAC I was observed in both clones. The relative amount of
KAR2 mRNA
appeared to correlate with the relative amount of HACI mRNA, higher expression
levels of
HAC 1 lead to higher expression level of KAR2 (Fig. 20).
Cell death studies of the methanol-induced cultures were performed using
fluorescence
flow cytometry (FFC) and compared to cell death of non-induced cultures. Ten
thousand cells
were measured per analysis. Cells were analyzed on the FACScaliburTM (Becton
Dickinson) after
12, 36 and 48 hours of induction. No cell death was observed. The
GlycoSwitchM5 (GSM5)
strain has as main core type glycan structures mainly Man5GlcNAc2 (structural
formula IV; Fig.
4). In order to check if Haclp induction has an influence on the N-glycan
structure a DSA-
FACE analysis was performed of I ml of the culture medium. The glycan profiles
obtained after
48 hours of induction of spliced Haclp are similar to the profile of the
parental GSM5 strain.
A growth curve was made in order to check if the induction of Haclp impairs
the growth
of P. pastoris. No growth defect was seen of the Hac ip induced strain
compared to the empty
vector transformed strain (Fig. 22).
Example 9. Expression of YIMNN6
In S. cerevisiae, MNN6 transfers phosphomannose residues to N-glycans.
Therefore,
overexpression of YIMNN6 in Y. lipolytica could lead to increased
phosphorylation. Moreover,
an additional effect on phosphorylation Y. lipolytica be obtained by over
expressing YIMNN4
and YIMNN6. The YIMNN6 coding region (Genbank Accession No. XM_499811,
Genolevures Ref: YALIOA06589g) was PCR amplified from the genome using PCR
primers
YIMNN6 BamHI fw (GCGGGATCCATGCACAACGTGCACGAAGC (SEQ ID NO:34)) and
YIMNN6 Avrll rv (GCGCCTAGGCTACCAGTCACTATAGTTCTCC (SEQ ID NO:35)) and
cloned in the pYHmAX expression vector for expression under control of the
hp4d promoter
(Fig. 21). The plasmid was transformed to the Y. lipolytica strain MTLY60
using zeta sequences
79
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
to improve random integration. Secreted glycoproteins were collected from cell
clones that grew
on medium without uracil and the composition of the glycans synthesized the
glycoproteins was
analyzed using DSA-FACE. However, no increased phosphorylation was observed
(Fig. 22).
Example 10. Effects of Haclp Expression
Evaluation of Haclp overexpression on the secretion of heterologous proteins.
Vectors containing the hygromycin resistance marker and the spliced HACI cDNA
under control
of the inducible AOXI promoter (pPlChygppHACl spliced) or under control of the
constitutive
GAP promoter ( pGAPhygHAC I ppspl iced) were transformed to a GS115 strain
expressing a
mIL-10 protein under the control of the inducible AOX1 promoter. P. pastoris
cells were
transformed according to the electroporation protocol from the Pichia
Expression kit (Invitrogen
Cat. No. K1710-01, Invitrogen, Carlsbad, CA). The vectors were linearized in
the AOXI or
GAP promoter to target the integration of the Haclp gene to respectively the
AOX1 or GAP
locus. Integration of the plasmid into the host genome was confirmed using
PCR.
Precultures (5 ml) from positive identified clones were grown in YPD for 24
hours. The
concentration (OD) at a wavelength of 600 rim (OD600) of the cells in the
cultures was measured
and cultures were diluted to an OD600 of 1 in 2 ml of BMGY media in each well
of a 24 well
plate. Cultures were grown in BMGY for 48 hours, washed twice with BMY, and
then induced
for 24 hours in BMMY. Every 8 to 12 hours, cultures were re-fed with medium
containing 1%
methanol (final concentration). After induction, the supernatant of the cells
was harvested and
the protein from 1 ml of the supernatant was precipitated using
trichloroacetic acid (TCA). The
precipitated protein was subjected to 15% SDS-PAGE.
From the SDS-PAGE, clonal variation in the expression of at least one protein -
mIL-l0
- was observed between the different clones. For example, for the clones
expressing the Haclp
protein constitutively (under control of GAP promoter), no improvement in
expression level was
observed, whereas for the clones expressing the Haclp inducibly (AOX1
promoter), two clones
could be identified that exhibited higher expression levels of the mIL-10
protein (Fig. 40 and
Fig. 41). Expression of m1L-10 by each of the clones was compared to the
expression of mIL-10
produced by a reference GSI 15 mIL-10 expressing strain.
A new induction was performed for these clones. A preculture grown for 24
hours was
diluted to OD I in 20 ml BMGY in a baffled flask. Cells were grown for 48
hours in BMGY,
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
washed twice, and then induced in BMMY. Cultures were re-fed with medium
containing 1%
methanol every 8-12 hours. After induction, the supernatant of the cells was
harvested and the
protein from I ml of the supernatant was precipitated using TCA. Prior to
subjecting the
precipitated protein to 15% SDS-PAGE, the protein was treated with PNGase F
(or not) to
remove all glycosylation (Fig. 41). SDS-PAGE resolved proteins from the
supernatant of
Hac 1 p-expressing strains contained a prominent band of 75 kDa, which is not
present in the
reference strain. This band was identified by means of mass spectrometry as
being Kar2p, which
is the most prominent UPR target gene. It could be shown using the cytokine
bead array (CBA)
that simultaneous inducible expression of the Haclp and the MIL-10 protein can
lead to a 2 fold
higher expression of the mIL-10 protein (clone 1, Fig. 41). CBA was performed
on endoH
treated mIL-10 protein.
Evaluation of Haclp overexpression on the surface expression of heterolgous
proteins. Vectors containing the hygromycin resistance marker and the spliced
HACI cDNA
under control of the inducible AOX1 promoter (pPIChygppHACI spliced) or under
control of the
constitutive GAP promoter ( pGAPhygHACIppspliced) were transformed to
GlycoswitchMan5
strains expressing a mature human interferon-beta/alpha-agglutinin fusion
protein, a mature
mouse interferon gamma/alpha-agglutinin fusion protein, a mature human
erythropoietin/alpha-
agglutinin fusion protein, or a fusion protein of alpha-agglutinin and the
lectin-like domain of
mouse thrombomodulin, each of which were under the control of the inducible
AOX1 promoter.
P. pastoris cells were transformed according to the electroporation protocol
from the Pichia
Expression kit (Invitrogen Cat. No. K1710-01). The vectors were linearized in
the AOXI or
GAP promoter to target the HacIp gene to respectively the AOXI or GAP locus
for integration.
Integration of the plasmid into the host genome was confirmed using PCR.
Precultures (5 ml) from positive identified clones were grown in YPD for 24
hours. The
OD600 was measured and cultures were diluted to ODwO of 1 in 2 ml BMGY in each
well of a 24
well plate. The cultures were grown in BMGY for 24 hours, washed twice with
dionized water,
and then induced (using culture medium containing 1% methanol) for 24 hours in
BMMY.
Surface expression was demonstrated by indirect immunostaining with an
antibody specific for
the V5-epitope, which is fused C-terminally to the VHH coding sequence. After
induction, 10'
cells in 1 ml PBS (pH 7.2), supplemented with 0.1 % bovine serum albumin
(PBS/BSA), were
81
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
incubated with l pl/ml of the anti-V5 antibody (I g/pl; Invitrogen), washed
with PBS/BSA,
and incubated with I pl/ml Alexa fluor 488-labeled goat anti-mouse lgG (t
pg/pl; Molecular
Probes). After washing twice with PBS/BSA, the cells were analyzed by flow
cytometry (Table
5).
Table 5. MFI values determined by flow cytometry
Expressed Protein Wild-type Pichia Pichia+AOX GAP Pichia+GAP RAC
mouse Interferon- 36.6 19.9 42.8
gamma
human EPO 59.5 45.8 66.5
interferon-beta 22.6 12.4 14.4
human 95.5 184.1 67.8
thrombomodulin
MFI=Mean Fluorescence Intensity obtained from the flow cytometry analysis.
For the strains expressing the Hacip protein constitutively no improvement, or
very
minor differences, could be observed in surface expression levels for all four
proteins compared
to reference strains expressing the surface protein alone. In cells expressing
human interferon-
beta, a significant reduction of surface expression levels was observed. For
the strains
overexpressing the inducible Hac Ip (Table 5) the following could be observed:
1) in the human
interferon-gamma surface expressing strain, a 1.8-fold lowering of the surface
expression levels
could be observed compared to the reference strain expressing alone the human
interferon-beta
a-agglutinin fusion; 2) for the strain surface-expressing human erythropoietin
a-agglutinin fusion
protein, a 1.3-fold lowering of the surface expression levels could be
observed compared to the
reference strain; 3) in the strain surface expressing human interferon-beta,
no difference of the
surface expression levels could be observed compared to the reference strain;
and 4) in the strain
surface expressing mouse thrombomodulin lectin-like domain, an 1.9-fold
increase of the surface
expression levels could be observed compared to the reference strain.
Effect of overexpression of Haclp on phosholipid synthesis. To determine
whether
overexpression of the Hac I p product (produced from the spliced HAC 1 cDNA)
had an effect of
lipid metabolism in P. pastoris, cells were transformed with the above-
described spliced HAC I
cDNA and the effect of Hac lp on lipid metabolism in the cells was determined
by electron
82
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
microscopy analysis. Cells were grown for 48 hours on BMGY, washed once with
PBS, and
then grown for another 48 hours on BMMY. The cells were next cultured in
medium containing
1% methanol every 8 to 12 hours. The cells were then prepared for electron
microscopy
according to the method of Baharaeen (Baharaeen et al. (2004) Mycopathologia).
Briefly, a
primary fixative containing glutaraldehyde (3%) and para-formaldehyde (1.5%
buffered in 0.05
M sodium cacodylate at pH 7.2) was contacted with the cells for 2 hours on
ice. The cells were
then washed three times for 20 minutes with 0.05 M sodium cacodylate. After
washing, the cells
were contacted with a 6% potassium permanganate solution for one hour at room
temperature
and then washed with 0.05 M sodium cacodylate three times for 20 minutes. The
results of the
experiment are presented in Fig. 54. Overexpression of the Haclp product
(produced from the
spliced HAC I cDNA) in P. pastoris lead to the formation of discrete regions
of stacked
membranes as can be shown in the electron micrograph (EM) depicted in Fig. 54.
These results
demonstrate that overexpression of Haclp, by way of its transcriptional
activation of genes
involved in lipid metabolism, indeed has a strong effect on lipid metabolism
in P. pastoris.
Example 11. Expression of ManHDEL
For Man5GlcNAc2 to be bound to glycoproteins expressed by the AochI strain, an
a-1,2-mannosidase can be expressed to cleave Man8GlcNAc2 to Man5GlcNAc2 (i.e.,
Golgi type
a-1,2-mannosidase activity). This mannosidase should be targeted to the
secretion system.
Trichoderma reesei a-1,2-mannosidase (Genbank(& accession no. AF212153), fused
to the S.
cerevisiae prepro mating factor and tagged with a HDEL sequence, is able to
trim Man8GIcNAc2
to Man5GlcNAc2 in vivo in Pichia pastoris as well as in Trichoderma reesei and
Aspergillus
niger. An expression construct was made to overexpress MFManHDEL (S.
cerevisiae a-mating
factor prepro fused to Trichoderma reesei a-1,2-mannosidase tagged with an
HDEL sequence)
in Y. lypolytica under control of the constitutive hp4d promoter (Fig. 23).
The expression
cassette was transformed into the cells after digestion of the plasmid
pYHmAXManHDEL with
the restriction enzyme Notl, followed by isolation of the desired fragment
using agarose-gel
electrophoresis.
Glycans derived from mannoproteins from the transformed cells were analysed
using
DSA-FACE. Only A minor fraction of Man8GlcNAc2 was converted to Man5GlcNAc2
(Fig. 24).
Incomplete conversion of Man8GIcNAc2 to Man5GlcNAc2 could have been due to a
non-optimal
83
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
secretion signal. Therefore, the Saccharomyces cerevisiae secretion signal was
replaced with the
secretion signal derived from the well expressed Yarrowia lipolytica LIP2
(LIP2pre). The
LIP2pre sequence was made by hybridizing the synthetic oligonucleotides
LIP2pre fw
GATCCATGAAGCTTTCCACCATCCTCTTCACAGCCTGCGCTACCCTGGCCGCGGTAC
(SEQ ID NO:66) and Lip2prepro rv
GTACCGGCCGGCCGCTTCTGGAGAACTGCGGCCTCAGAAGGAGTGATGGGGGAAGG
GAGGGCGGC (SEQ ID NO:67) and cloning the DNA into pYLHmA vector (at the
BamHUAvrII sites) resulting in the following construct: pYLHUdL2pre. The
ManHDEL coding
sequence was PCR amplified from pGAPZMFManHDEL using oligonucleotides ManHDEL
Eco47111 fw (GGCAGCGCTACAAAACGTGGATCTCCCAAC (SEQ ID NO:68)) and
ManHDEL AvrIl rv (GGCCCTAGGTTACAACTCGTCGTGAGCAAG (SEQ ID NO:69)) and
cloned in pYLHUdL2pre, The construction strategy is depicted in Fig. 25. The
expression
cassette (with L2preManHDEL under control of the constitutive promoter hp4d)
was
transformed to Yarrowia lipolytica Aochl strain after digestion of the plasmid
with Notl and
isolation of the correct fragment (see above). Glycans derived from secreted
proteins were
analysed via DSA FACE. Some conversion of MangGlcNAc2 to Man5GlcNAc2 occurred,
but the
reaction was incomplete (MangGlcNAc2 was present as well as intermediate
products
Man7GlcNAc2 and Man6GIcNAc2i Fig. 26).
To further improve the trimming of Man8GlcNAc2, Man7GlcNAc2, and Man6GIcNAc2
to
Man5GlcNAc2i the Trichoderma reesei a-1,2 mannosidase was codon optimized for
expression
in Yarrowia lipolytica (SEQ ID NO:9; Fig. 42) and fused to the LIP2 pre signal
sequence. This
fusion construct was expressed under control of 4 different promoters: (i)
hp4d, (ii) GAP(SEQ
ID NO:10; Fig. 43), (iii) POX2, and (iv) TEFI. Final expression plasmids were
named
pYLHUXdL2preManHDEL (SEQ ID NO: 11; Figs. 44A-C) pYLGUXdL2preManHDEL (SEQ
ID NO:12; Figs.45A-) pYLPUXdL2preManHDEL (SEQ ID NO: 13; Figs. 46A-C)
pYLTUXdL2preManHDEL (SEQ ID NO: 14; Figs. 47A-C). All 4 plasmids were
transformed to
Yarrowia lipolytica MTLY60 AochI strain (described in example2) after cutting
the plasmid
with Notl and isolation of the fragment containing the ManHDEL expression
cassette.
Transformed strains with the ManHDEL under control of the hp4d, GAP and TEF
promoter
(plasmids pYLHUXdL2preManHDEL, pYLGUXdL2preManHDEL and
pYLTUXdL2preManHDEL) were grown in YPD.
84
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
Glycans derived from secreted proteins of transformed strains were analyzed by
DSA
FACE. Results are represented in Fig. 48. Alternatively, transformants
(including a
transformant that had integrated the pYLPUXdL2preManHDEL plasmid) were grown
in
medium containing oleic acid (protein production conditions) and glycans were
analysed via
DSA-FACE. Data for one of the vectors, pYLTUXdL2preManHDEL, are presented in
Fig. 49.
As can be concluded from the data, by 48 hours of culture, almost all glycans
are converted to
Man5GlcNAc2.
Example 12: Culturingsconditions for POX2 promoter controlled gene expression
Cultures were started from a single colony of a fresh plate and grown
overnight in 10 mL
YPD at 28 C in a 50 mL tube in an orbital shaker at 250 rpm. Next, a 250 mL
shake flask
containing 22 mL of production medium (including 2.5 mL oleic acid emulsion)
was inoculated
with the preculture at a final OD600 of 0.2. This culture was incubated at 28
C in an orbital
shaker at 250 rpm. Samples of the culture were taken at various time points
over a 96 hour
culture.
The oleic acid emulsion (20%) was made the method as follows:
Add to a sterile 50 ml vessel;
ml sterile water;
20 5 ml oleic acid; and
125 l Tween 40.
Sonication resulting in the formation of the emulsion was performed for one
minute at 75Hz.
The production medium consisted of the following:
1 % yeast extract;
2% trypton;
1% glucose; and
50 mM phosphate pH 6.8.
Example 13: Expression of human glucocerebrosidase:
*Trademark
CA 02762982 2011-12-22
WO 2008/120107 PCT/IB2008/001814
Human glucocerebrosidase (GLCM, Swiss Prot entry nr: P04062) was chemically
synthesized as a codon-optimized cDNA for expression in Yarrowia lipolytica
(SEQ ID NO:15;
Fig. 50).
The coding sequence for the mature protein was fused to the coding sequence of
the LIP2
pre signal sequence. This fusion construct was cloned under control of the
oleic acid inducible
POX2 promoter. The resulting plasmid was named pYLPUXL2preGLCM (= pRAN21)).
Before
transformation, the plasmid was digested with Not! and the fragment containing
the expression
cassette was isolated and transformed to Yarrowia lipolytica strain MTLY60,
MTLY60AochI
(described in Example 2 above), and MTLY60Aoch]ManHDEL (described in Exampled
1).
Transformants obtained in these three strains were grown as described in
Example 12. Proteins
were precipitated from the supernatant as described above, subjected to SDS-
PAGE, and
immunoblotted using a rat monoclonal anti- glucocerebrosidase antibody
(Alessandrini et al.
(2004) J. Invest. Dermatol 23(6):1030-6). An exemplary immunoblot analysis is
depicted in Fig.
51. It can be appreciated from Fig. 51 that in a och 1 disrupted strain no
smearing occurs (lanes
1, 2, and 3), whereas heterogeneity of the protein is seen as a smear in WT
cells (lanes 4 and 6).
No smearing of protein was observed in protein obtained from a strain of yeast
expressing
ManHDEL. These results demonstrate that a more homogeneous population of a
target protein
can be obtained using the genetically engineered Yarrowia lipolytica cells
MTLY60toch1 and
MTLY60Aoch I ManHDEL.
Example 14: Expression of human eropoietin:
Human erythropoietin (Epo, Swiss Prot entry nr: P01588) encoding cDNA was
chemically synthesized codon optimized for expression in Yarrowia lipolytica
(SEQ ID NO: 16;
Fig. 52). The cDNA coding sequence for the mature protein was fused to the
coding sequence of
the LIP2 pre signal sequence. This fusion construct was cloned under control
of the oleic acid
inducible POX2 promoter. The resulting plasmid was named pYLPUXL2prehuEPO.
Before
transformation the plasmid was cut Nod and the fragment containing the
expression cassette was
isolated and transformed to Yarrowia lipolytica strain MTLY60Aochl (described
in Example 2).
Transformant candidates were grown as described in Example 12 and secreted
proteins were
analysed by western blot after SDS PAGE using a monoclonal mouse anti human
Epo antibody
86
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
obtained from R&D systems (clone AE7A5). The EPO product obtained from the
cells
exhibited very homogenous glycosylation.
Example 15: Expression of human a-galactosidase A:
Human a-galactosidase A (AGAL, Swiss Prot entry nr: P06280) encoding cDNA was
chemically synthesized as a codon-optimized cDNA for expression in Yarrowia
lipolytica (SEQ
ID NO:17; Fig. 53).
The cDNA coding sequence for the mature protein was fused to the coding
sequence of
the LIP2 pre signal sequence. This fusion construct was cloned under control
of the oleic acid
inducible POX2 promoter. The resulting plasmid was named pYLPUXL2preaGalase).
Before
transformation the plasmid was cut Nod and the fragment containing the
expression cassette was
isolated and transformed to Yarrowia lipolytica strain MTLY60 and
MTLY60Aoch1MNN4
(described in Example 4). Transformants obtained in these two strains were
grown as described
in Example 12. Extracellular proteins obtained from transformants were
analyzed by
immunoblot after SDS-PAGE analysis. Two antibodies specific for a-
galactosidase A (a
chicken polyclonal antibody obtained from Abcam (ab28962) and a rabbit
polyclonal antibody
obtained from Santa Cruz Biotechnology (sc-25823)) were used to detect the
expressed human
a-galactosidase A protein.
Example 16. Expression of Mannosidase in WT Yarrowia lipolytica
To determine whether expression of MannosidaseHDEL alone (that is in cells
containing
a functional OCHI gene) could lead to a more homogenous glycosylation of
proteins expressed
by fungal cells, an expression cassette containing a nucleic acid encoding
MannosidaseHDEL
(see Example 11) was transformed into wild-type Yarrowia lipolytica pold
cells. Glycans
derived from secreted proteins obtained from the cells were analysed by DSA-
FACE (Fig. 55).
The analyzed glycans consisted mainly of Man5GlcNAc2 and a minor part
Man6GlcNAc2. These
results demonstrate that expression of MannosidaseHDEL alone, in the absence
of any disruption
of the OCHI gene, leads to a more homogenous glycosylation of proteins
expressed by Yarrowia
lipolytica.
87
CA 02762982 2011-12-22
WO 2008/120107 PCT/1B2008/001814
Other Embodiments
While the invention has been described in conjunction with the detailed
description
thereof, the foregoing description is intended to illustrate the invention,
which is defined by the
scope of the appended claims.
10
20
30
88