Note: Descriptions are shown in the official language in which they were submitted.
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
FUSIDIC ACID DOSING REGIMENS FOR TREATMENT
OF BACTERIAL INFECTIONS
BACKGROUND OF THE INVENTION
[0001] Fusidic acid (FA) is a tetracyclic triterpenoid or fusidane
(steroidal) antibiotic
derived from the fungus Fusidium coccineum that inhibits bacterial protein
synthesis. FA
is effective against gram-positive bacteria such as Staphylococcus species and
Corynebacterium species (L. Verbist, J. Antimicro. Chemo. 25, Suppl. B, 1-5
(1990); A.
Bryskier, Fusidic Acid, Chapter 23, in Antimicrobial Agents: Antibacterials
and
Antifungals (Andre Bryskier, Ed., ASM Press, Washington, USA, 2005)). FA also
has
moderate activity against Group A beta-hemolytic streptococci, or
Streptococcus pyogenes
(L. Verbist, J. Antimicro. Chemo. 25, Suppl. B, 1-5 (1990); A. Bryskier,
Fusidic Acid,
Chapter 23, in Antimicrobial Agents: Antibacterials and Antifungals (Andre
Bryskier, Ed.,
ASM Press, Washington, USA, 2005); Skov et al., Diag. Micro. Infect. Dis.
40:111-116
(2001)).
[0002] FA was developed for clinical use in the 1960s and it is approved
for human
use outside of the United States, such as in the UK, Canada, Europe, Israel,
Australia and
New Zealand. It is typically prescribed at doses of 500 mg TID for treating
skin and skin
structure infections caused by Staphylococcus aureus (A. Bryskier, Fusidic
Acid, Chapter
23, in Antimicrobial Agents: Antibacterials and Antifungals (Andre Bryskier,
Ed., ASM
Press, Washington, USA, 2005); Collignon et al., Int'l J. Antimicrobial Agents
12:S45¨S58
(1999); D. Spelman, Int? J. Antbnicrobial Agents 12:S59¨S66 (1999)), although
some
physicians have routinely prescribed the compound at 500 mg BID for treating
skin and
skin structure infections due to the long half-life of the compound (Fusidic
Acid, in
Principles and Practice of Infectious Diseases, 6th ed. (Mandell et al. eds.,
Elsevier,
2006)).
[0003] Treatment using FA has been well studied and it is generally
regarded as safe
when administered to humans, as evidenced by the fact that the drug has been
in
continuous use for more than 40 years. There are, however, several
characteristics of FA
1
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
that have prevented use of the drug against a wider spectrum of bacteria and
in the
treatment in additional types of infection. For example, approved dosing
regimens have
been shown to select for bacterial resistance, such as in S. aureus. Approved
dosing
regimens provide low multiples of the MIC and as a result, S. aureus resistant
mutants can
be selected after the first day of dosing. Once resistance has developed, FA
is not effective
against the resistant strains. Resistance is reported to occur if FA is used
as a single drug
as the resistance frequency at 4 and 8 times the MIC is in the range of 10-6
or 10-8 (Evans et
al., J. Clin. Path. 19:555-560 (1966); Hansson et al., J. Hol. Biol. 348:939-
949 (2005),
Jensen et al., Acta Pathol Micro biol Scand. 60:271-284 (1964); Besier et al.,
Antimicrob.
Agents Chemo., 49(4):1426-1431 (2005); Gemmell et al., J. Antimicrobial Chemo.
57:589-608 (2006)).
[0004] The dosage of the drug cannot be simply increased as a means of
avoiding
development of resistance. It is difficult to achieve high concentrations of
FA in the blood
due to the substantial protein binding of the drug (approximately 95-97%) (K.
Christiansen, International Journal of Antimicrobial Agents 12:S3¨S9 (1999);
Coutant et
al., Diagn Microbiol Infect Dis 25:9-13 (1996); D. Reeves, J. Antimicrob.
Chemo. 20:467-
476 (1987); J. Tumidge, Intl J. Antimicrobial Agents 12:S23¨S34 (1999);
Rieutord et al.,
Intl J. Pharmaceutics 119:57-64 (1995)). Moreover, high dosages of FA are not
well-
tolerated by patients receiving the drug. High doses of FA (e.g., 1 gram TID)
are required
if the drug is to be used in the treatment of bone and joint infections, less
susceptible
bacteria and other serious infections. However, treatment regimens using high
doses of the
drug induce nausea and vomiting and are rejected by patients (Fusidic Acid, in
Principles
and Practice of Infectious Diseases, 6th ed. (Mandell et al. eds., Elsevier,
2006); K.
Christiansen, International Journal of Antimicrobial Agents 12:S3¨S9 (1999);
Nordin et
al., Eur. I Clin. Res. 5:97-106 (1994)).
[0005] In view of the tremendous costs associated with the de novo
development of
new anti-bacterials, expanding the indications for drugs that have already
been
demonstrated to be safe and effective is strongly needed. Overcoming the
limitations on
the uses of FA would broaden the population of bacterial infections against
which it could
be used and thus meet this need.
2
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
BRIEF SUMMARY OF THE INVENTION
[0006] According to a first embodiment, the present invention is directed
to a method
of treating or preventing a bacterial infection in a subject comprising:
(a) administering at least one loading dose of a pharmaceutical composition
comprising fusidic acid, or a pharmaceutically acceptable salt thereof, to a
subject in need
of treatment or prevention, in an amount sufficient to reach a pharmacokinetic
(PK) profile
for fusidic acid comprising a maximum plasma concentration (C.) of fusidic
acid of not
less than about 70 ug/ml, a time to maximum plasma concentration (Tmax) of
fusidic acid of
no more than about 24 hours and a minimum trough plasma concentration of
fusidic acid
of not less than about 50 ug/ml, and
(b) administering at least one maintenance dose of a pharmaceutical
composition comprising fusidic acid, or a pharmaceutically acceptable salt
thereof, to the
subject of (a) in an amount sufficient to maintain a minimum trough plasma
concentration
of fusidic acid of not less than about 50 ug/ml for at least about 24 hours
after the
administering of (a).
[0007] In a preferred aspect of the first embodiment, the C. is not less
than about 80
ug/ml, the minimum trough plasma concentration of (a) is not less than about
60 ug/ml and
the minimum trough plasma concentration of (b) is not less than about 70
ug/ml. In a
further preferred aspect, the C. is not less than about 100 ug/ml, the minimum
trough
plasma concentration of (a) is not less than about 80 ug/ml and the minimum
trough
plasma concentration of (b) is not less than about 80 ug/ml.
[0008] In preferred aspects with regard to the loading dose, the loading
dose comprises
two loading doses, wherein the second loading dose is administered about 12
hours after
administration of the first loading dose, and wherein each loading dose is
between about
1000 mg and about 1850 mg. In further preferred aspects, the total loading
dose
administered to the subject is between about 2000 mg and about 3600 mg.
[0009] In preferred aspects with regard to the maintenance dose, the
maintenance dose
comprises multiple maintenance doses of independently between about 500 mg and
1000
3
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
mg, administered about 12 hours apart, beginning about 12 hours after
administration of
the loading dose.
[0010] In preferred aspects with regard to the minimum trough plasma
concentration, a
minimum trough plasma concentration of not less than about 50 ug/ml is
maintained for at
least about 48 hours after administering of the loading dose. More preferably,
a minimum
trough plasma concentration of not less than about 50 ug/ml is maintained for
at least about
72 hours. Even more preferably, a minimum trough plasma concentration of not
less than
about 50 ug/ml is maintained for at least about 96 hours.
[0011] In a preferred aspect of the first embodiment, the loading dose
comprises two
loading doses, wherein each loading dose is independently between about 1000
mg and
about 1850 mg, and wherein the second loading dose is administered about 12
hours after
administration of the first loading dose; and the maintenance dose comprises
multiple
maintenance doses of independently between about 500 mg and 1000 mg
administered
about 12 hours apart, beginning about 12 hours after administration of the
second loading
dose. Preferably, each loading dose is independently at least about 1500 mg.
Also
preferably, each maintenance dose is independently at least about 600 mg.
[0012] In preferred aspects with regard to the subject, the subject is a
human.
[0013] In preferred aspects with regard to the bacterial infection, the
bacterial infection
is an infection caused by bacteria selected from the group consisting of
staphylococci,
including coagulase-negative staphylococci and coagulase-positive
staphylococci,
streptococci, including Group A beta hemolytic streptococci, non-Group A beta
hemolytic
streptococci and viridans group streptococci, enterococci, Nesseria species,
Clostridiunz
species, Bordetella species, Bacillus species and Cor.,vnebacterium species.
Preferably, the
bacterial infection is an infection caused by bacteria selected from the group
consisting of
Staphylococcus aureus (methicillin-resistant and -susceptible), Staphylococus
epiclermiclis,
Staphylococus hemolyticus, Staphylococus saprophyticus, Staphylococus
lugdunensis,
Staphylococus capitis, Staphylococus caprae, Staphylococus saccharolyticus,
Staphylococus simulans, Staphylococus warneri, Staphylococus hominis,
Staphylococus
internzedius, Staphylococcus pseudointermedius, Staphylococus lyricus,
Streptococcus
pyogenes, Streptococcus agalactiae, Streptococcus dysgalactiae subspecies
dysgalactiae,
4
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
Streptococcus anginosus, Streptococcus mitis, Streptococcus salivarius,
Streptococcus
bovis, Streptococcus mutans, Neisseria gonorrhoeae, Neisseria men ingitidis,
Bacillus
anthracis, Boraletella pertussis, Clostridium difficile, Enterococcus
faecalis, Enterococcus
faecium and Corynebacterium diphtheriae. In particular aspects, the bacterial
infection is
an infection caused by Enterococcus faecalis or Enterococcus faecium.
[0014] In other preferred aspects with regard to the bacterial infection,
the bacterial
infection is an infection selected from the group consisting of a skin and
soft tissue
infection, a bone infection, a joint infection, pneumonia, a wound infection,
a burn
infection, an infection of the blood, and an infection associated with cystic
fibrosis.
[0015] In preferred aspects with regard to the pharmaceutical composition
comprising
fusidic acid, or a pharmaceutically acceptable salt thereof, the
pharmaceutical composition
is in the form of a tablet, a capsule, an IV solution, an inhalable
formulation, a powder
formulation, or a formulated suspension. Preferably, the pharmaceutical
composition is
administered orally, by injection or by intravenous infusion.
[0016] In another preferred aspect of the first embodiment, the subject
does not
experience an adverse level of nausea.
[0017] According to a second embodiment, the present invention is directed
to a
method of treating or preventing a bacterial infection in a subject
comprising:
(a) administering at least one loading dose of a pharmaceutical composition
comprising fusidic acid, or a pharmaceutically acceptable salt thereof, to a
subject in need
of treatment or prevention in an amount sufficient to achieve a minimum plasma
concentration of not less than about 50 ug/ml fusidic acid within 24 hours,
and
(b) administering at least one maintenance dose of a pharmaceutical
composition comprising fusidic acid, or a pharmaceutically acceptable salt
thereof, to the
subject of (a) in an amount sufficient to maintain a minimum plasma
concentration of not
less than about 50 ug/ml fusidic acid for at least about 24 hours after the
administering of
(a).
[0018] In a preferred aspect of the second embodiment, a minimum plasma
concentration of fusidic acid of not less than about 60 ug/ml is achieved in
(a) and a
minimum plasma concentration of fusidic acid of not less than about 60 ug/ml
is
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
maintained in (b). In a further preferred aspect, a minimum plasma
concentration of
fusidic acid of not less than about 80 ug/ml is achieved in (a) and a minimum
plasma
concentration of fusidic acid of not less than about 80 ug/ml is maintained in
(b).
[0019] In preferred aspects with regard to the loading dose, the loading
dose comprises
two loading doses, wherein the second loading dose is administered about 12
hours after
the first loading dose, and wherein each loading dose is between about 1000 mg
and about
1850 mg. In further preferred aspects, the total loading dose administered to
the subject is
between about 2000 mg and about 3600 mg.
[0020] In preferred aspects with regard to the maintenance dose, the
maintenance dose
comprises multiple maintenance doses of independently between about 500 mg and
1000
mg administered about 12 hours apart, beginning about 12 hours after
administration of the
loading dose.
[0021] In preferred aspects with regard to the minimum plasma
concentration, a
minimum plasma concentration of not less than about 50 ug/ml is maintained for
at least
about 48 hours after administering the loading dose, preferably a minimum
plasma
concentration of not less than about 50 ug/ml is maintained for at least about
72 hours,
more preferably, a minimum plasma concentration of not less than about 50
ug/ml is
maintained for at least about 96 hours.
[0022] In a preferred aspect of the second embodiment, the loading dose
comprises
two loading doses, wherein each loading dose is independently between about
1000 mg
and about 1850 mg, and wherein the second loading dose is administered about
12 hours
after administration of the first loading dose; and multiple maintenance doses
of
independently between about 500 mg and 1000 mg are administered, about 12
hours apart,
beginning about 12 hours after administration of the second loading dose.
Preferably, each
loading dose is independently at least about 1500 mg. Equally preferably, each
maintenance dose is independently at least about 600 mg.
[0023] In preferred aspects with regard to the subject, the subject is a
human.
[0024] In preferred aspects with regard to the bacterial infection, the
bacterial infection
is an infection caused by bacteria selected from the group consisting of
staphylococci,
including coagulase-negative staphylococci and coagulase-positive
staphylococci,
6
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
streptococci, including Group A beta hemolytic streptococci, non-Group A beta
hemolytic
streptococci and viridans group streptococci, enterococci, Nesseria species,
Clostridium
species, Bordetella species, Bacillus species and Corynebacterium species.
Preferably, the
bacterial infection is an infection caused by bacteria selected from the group
consisting of
Staphylococcus aureus (methicillin-resistant and -susceptible), Staphylococus
epidermidis,
Staphylococus hemolyticus, Staphylococus saprophyticus, Staphylococus
lugdunensis,
Staphylococus capitis, Staphylococus caprae, Staphylococus saccharolyticus,
Staphylococus simulans, Staphylococus warneri, Staphylococus hominis,
Staphylococus
internzedius, Staphylococcus pseudointerinedius, Staphylococus lyricus,
Streptococcus
pyogenes, Streptococcus agalactiae, Streptococcus dysgalactiae subspecies
dysgalactiae,
Streptococcus anginosus, Streptococcus mitis, Streptococcus salivarius,
Streptococcus
bovis, Streptococcus nuttans, Neisseria gonorrhoeae, Neisseria meningitidis,
Bacillus
anthracis, Bordetella pertussis, Clostridium difficile, Enterococcus faecalis,
Enterococcus
faeciutn and Corynebacterium diphtheriae. In particular aspects, the bacterial
infection is
an infection caused by Enterococcus .faecalis or Enterococcus faecium.
[0025] In further preferred aspects with regard to the bacterial infection,
the bacterial
infection is an infection selected from the group consisting of a skin and
soft tissue
infection, a bone infection, a joint infection, pneumonia, a wound infection,
a burn
infection, an infection of the blood, and an infection associated with cystic
fibrosis.
[0026] In preferred aspects with regard to the pharmaceutical composition
comprising
fusidic acid, or a pharmaceutically acceptable salt thereof, the
pharmaceutical composition
is in the form of a tablet, a capsule, an IV solution, an inhalable
formulation, a powder
formulation, or a formulated suspension. Preferably, the pharmaceutical
composition is
administered orally, by injection or by intravenous infusion.
[0027] In another preferred aspect of the second embodiment, the subject
does not
experience an adverse level of nausea.
[0028] According to a third embodiment, the present invention is directed
to a method
of treating or preventing a bacterial infection in a subject comprising:
(a) administering a first and a second loading dose of a
pharmaceutical
composition comprising fusidic acid, or a pharmaceutically acceptable salt
thereof, to a
7
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
subject in need of treatment or prevention, wherein each loading dose is
independently
between about 1000 mg and about 1850 mg, and wherein the second loading dose
is
administered about 12 hours after administration of the first loading dose;
and
(b) administering two or more maintenance doses of a pharmaceutical
composition comprising fusidic acid, or a pharmaceutically acceptable salt
thereof, to the
subject of (a), wherein each maintenance dose is independently between about
500 mg and
1000 mg, wherein a first maintenance dose is administered about 12 hours after
administration of the second loading dose, and wherein subsequent maintenance
doses are
administered about 12 hours apart.
[0029] In a preferred aspect of the third embodiment, each loading dose is
independently at least about 1500 mg.
[0030] In a further preferred aspect of the third embodiment, each
maintenance dose is
independently at least about 600 mg. In related aspects, at least three, four,
five or six
maintenance doses are administered to the subject.
[0031] In a preferred aspect with regard to the subject, the subject is a
human.
[0032] In a preferred aspect with regard to the bacterial infection, the
bacterial
infection is an infection caused by bacteria selected from the group
consisting of
staphylococci, including coagulase-negative staphylococci and coagulase-
positive
staphylococci, streptococci, including Group A beta hemolytic streptococci,
non-Group A
beta hemolytic streptococci and viridans group streptococci, enterococci,
Nesseria species,
Clostridium species, Bordetella species, Bacillus species and Corynebacterium
species.
Preferably, the bacterial infection is an infection caused by bacteria
selected from the
group consisting of Staphylococcus aureus (methicillin-resistant and -
susceptible),
Staphylococus epidermidis, Staphylococus hemolyticus, Staphylococus
saprophyticus,
Staphylococus lugdunensis, Staphylococus capitis, Staphylococus caprae,
Staphylococus
saccharolyticus, Staphylococus simulans, Staphylococus warneri, Staphylococus
hominis,
Staphylococus intermedius, Staphylococcus pseudointermedius, Staphylococus
lyricus,
Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus dysgalactiae
subspecies
dysgalactiae, Streptococcus anginosus, Streptococcus mitis, Streptococcus
salivarius,
Streptococcus bovis, Streptococcus mutans, Neisseria gonorrhoeae, Neisseria
8
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
meningitidis, Bacillus anthracis, Bordetella pertussis, Clostridium dUjicile,
Enterococcus
faecalis, Enterococcus faeciurn and Corynebacterium diphtheriae. In particular
aspects,
the bacterial infection is an infection caused by Enterococcus faecalis or
Enterococcus
faecium.
[0033] In an additional preferred aspect with regard to the bacterial
infection, the
bacterial infection is an infection selected from the group consisting of a
skin and soft
tissue infection, a bone infection, a joint infection, pneumonia, a wound
infection, a burn
infection, an infection of the blood, and an infection associated with cystic
fibrosis.
[0034] In a preferred aspect with regard to the pharmaceutical composition
comprising
fusidic acid, or a pharmaceutically acceptable salt thereof, the
pharmaceutical composition
is in the form of a tablet, a capsule, an IV solution, an inhalable
formulation, a powder
formulation, or a formulated suspension. In a related aspect, the
pharmaceutical
composition is preferably administered orally, by injection or by intravenous
infusion to
the subject.
[0035] In another preferred aspect of the third embodiment, the subject
does not
experience an adverse level of nausea.
[0036] According to a fourth embodiment, the present invention is directed
to a
method of reducing development of an antibiotic-resistant strain of bacteria
in a subject
having a bacterial infection comprising:
(a) administering at least one loading dose of a pharmaceutical composition
comprising fusidic acid, or a pharmaceutically acceptable salt thereof, to a
subject having a
bacterial infection, in an amount sufficient to reach a pharmacokinetic (PK)
profile for
fusidic acid comprising a maximum plasma concentration (Cmax) of fusidic acid
of not less
than about 70 ug/ml, a time to maximum plasma concentration (Tmax) of fusidic
acid of no
more than about 20 hours and a minimum trough plasma concentration of fusidic
acid of
not less than about 50 ug/ml, and
(b) administering at least one maintenance dose of a pharmaceutical
composition comprising fusidic acid, or a pharmaceutically acceptable salt
thereof, to the
subject of (a) in an amount sufficient to maintain a minimum trough plasma
concentration
9
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
of fusidic acid of not less than about 50 ug/ml for at least about 24 hours
after the
administering of (a).
[0037] In a preferred aspect of the fourth embodiment, the C. is not less
than about
80 ug/ml, the minimum trough plasma concentration of (a) is not less than
about 60 ug/ml,
and the minimum trough plasma concentration of (b) is not less than about 70
ug/ml. In a
further preferred aspect, the C. is not less than about 100 ug/ml, the minimum
trough
plasma concentration of (a) is not less than about 80 ug/ml and the minimum
trough
plasma concentration of (b) is not less than about 80 ug/ml.
[0038] In preferred aspects with regard to the loading dose, the loading
dose comprises
two loading doses, wherein the second loading dose is administered about 12
hours after
the first loading dose, and wherein each loading dose is between about 1000 mg
and about
1850 mg. In further preferred aspects, the total loading dose administered to
the subject is
between about 2000 mg and about 3600 mg.
[0039] In a preferred aspect of the fourth embodiment, the loading dose
comprises two
loading doses, wherein the second loading dose is administered about 12 hours
after the
first loading dose, and wherein each loading dose is between about 1000 mg and
about
1850 mg.
[0040] In another preferred aspect of the fourth embodiment, the
maintenance dose
comprises multiple maintenance doses of independently between about 500 mg and
1000
mg administered about 12 hours apart, beginning about 12 hours after
administration of the
loading dose.
[0041] In preferred aspects with regard to the minimum trough plasma
concentration, a
minimum trough plasma concentration of not less than about 50 ug/ml is
maintained for at
least about 48 hours after administering the loading dose, more preferably a
minimum
trough plasma concentration of not less than about 50 ug/ml is maintained for
at least about
72 hours, even more preferably a minimum trough plasma concentration of not
less than
about 50 ug/ml is maintained for at least about 96 hours.
[0042] In a further preferred aspect of the fourth embodiment, the loading
dose
comprises two loading doses, wherein each loading dose is independently
between about
1000 mg and about 1850 mg, and wherein the second loading dose is administered
about
=
=
12 hours administration of after the first loading dose; and multiple
maintenance doses of
independently between about 500 mg and 1000 mg are administered to the subject
about 12
hours apart, beginning about 12 hours after administration of the second
loading dose.
Preferably, each loading dose is independently at least about 1500 mg. Equally
preferably, each
maintenance dose is independently at least about 600 mg.
[0043] In preferred aspects with regard to the subject, the subject
is a human.
[0044] In preferred aspects with regard to the infection, the
bacterial infection is caused by a
bacteria selected from the group consisting of staphylococci, including
coagulase-negative
staphylococci and coagulase-positive staphylococci, streptococci, including
Group A beta
hemolytic streptococci, non-Group A beta hemolytic streptococci and viridans
group streptococci,
enterococci, Nesseria species, Clostridium species, Bordetella species.
Bacillus species and
Corynebacterium species. Preferably, the infection is caused by a bacteria
selected from the group
consisting of Staphylococcus aureus (methicillin-resistant and -susceptible),
Staphylococus
epidermidis, Staphylococus hemolyticus, Staphylococus saprophyticus,
Staphylococus
lugdunensis, Staphylococus capitis, Staphylococus capraeõS'taphylococus
saccharolyticus,
Staphylococus simulans, Staphylococus warneri, Staphylococus hominis,
Staphylococus
intermedius. Staphylococcus pseudo intermedius, Staphylococus lyricus.
Streptococcus pyogenes.
Streptococcus agalactiae. Streptococcus dysgalactiae subspecies dysgalactiae.
Streptococcus
anginosus. Streptococcus mitis. Streptococcus salivarius. Streptococcus bovis,
Streptococcus
mutans. Neisseria gonorrhoeae. Neisseria meningitidis. Bacillus anthracis,
Bordetella pertussis,
Clostridium difficile, Enterococcus faecalis, Enterococcus faecium and
Corynebacterium
diphtheriae. In particular aspects, the infection is caused by Enterococcus
faecalis or
Enterococcus faeciutn.
[0044a] According to one particular aspect, the invention relates to
the use of a
pharmaceutical composition comprising fusidic acid, or a pharmaceutically
acceptable salt
thereof, for treating or preventing a bacterial infection in a subject in need
of treatment or
prevention, comprising:
(a) two loading doses of 1200 mg; and
(b) two maintenance doses of 600 mg, wherein the second loading dose is
formulated to
be administered 12 hours after administration of the first loading dose,
11
CA 2767614 2018-04-23
wherein the first maintenance dose is formulated to be administered 12 hours
after administration
of the second loading dose, and wherein the second maintenance dose is
formulated to be
administered 12 hours after administration of the first maintenance dose.
[0044b] According to a related aspect, the invention relates to the use of
a pharmaceutical
composition comprising fusidic acid, or a pharmaceutically acceptable salt
thereof, for treating or
preventing a bacterial infection in a subject in need of treatment or
prevention, comprising:
(a) two loading doses of 1500 mg; and
(b) two maintenance doses of 600 mg, wherein the second loading dose is
formulated to be
administered 12 hours after administration of the first loading dose, wherein
the first maintenance
dose is formulated to be administered 12 hours after administration of the
second loading dose, and
wherein the second maintenance dose is formulated to be administered 12 hours
after
administration of the first maintenance dose.
[0044c] According to another particular aspect, the invention relates to
the use of a
pharmaceutical composition comprising fusidic acid, or a pharmaceutically
acceptable salt thereof,
for reducing development of an antibiotic-resistant strain of bacteria in a
subject having a bacterial
infection, comprising:
(a) two loading doses of 1200 mg; and
(b) two maintenance doses of 600 mg, wherein the second loading dose is
formulated to be
administered 12 hours after administration of the first loading dose, wherein
the first maintenance
dose is formulated to be administered 12 hours after administration of the
second loading dose, and
wherein the second maintenance dose is formulated to be administered 12 hours
after
administration of the first maintenance dose.
[0044d] According to a related aspect, the invention relates to the use of
a pharmaceutical
composition comprising flisidic acid, or a pharmaceutically acceptable salt
thereof, for reducing
development of an antibiotic-resistant strain of bacteria in a subject having
a bacterial infection,
comprising:
(a) two loading doses of 1500 mg; and
(b) two maintenance doses of 600 mg, wherein the second loading dose is
formulated to be
administered 12 hours after administration of the first loading dose, wherein
the first maintenance
dose is formulated to be administered 12 hours after administration of the
second loading dose,
ha
CA 2767614 2018-04-23
and wherein the second maintenance dose is formulated to be administered 12
hours after
administration of the first maintenance dose.
[0044e] According to another particular aspect, the invention relates to
the use of a
pharmaceutical composition comprising fusidic acid, or a pharmaceutically
acceptable salt
thereof, for treating or preventing a bacterial infection in a subject in need
of treatment or
prevention, comprising:
(a) two loading doses of 1200 mg; and
(b) three maintenance doses of 600 mg, wherein the second loading dose is
formulated
to be administered 12 hours after administration of the first loading dose,
wherein the first
maintenance dose is formulated to be administered 12 hours after
administration of the second
loading dose, wherein the second maintenance dose is formulated to be
administered 12 hours
after administration of the first maintenance dose, and wherein the third
maintenance dose is
formulated to be administered 12 hours after administration of the second
maintenance dose.
[0044f] According to another particular aspect, the invention relates to
the use of a
pharmaceutical composition comprising fusidic acid, or a pharmaceutically
acceptable
salt thereof, for treating or preventing a bacterial infection in a subject in
need of
treatment or prevention, comprising:
(a) two loading doses of 1500 mg; and
(b) three maintenance doses of 600 mg, wherein the second loading dose is
formulated
to be administered 12 hours after administration of the first loading dose,
wherein the first
maintenance dose is formulated to be administered 12 hours after
administration of the second
loading dose, wherein the second maintenance dose is formulated to be
administered 12 hours
after administration of the first maintenance dose, and wherein the third
maintenance dose is
formulated to be administered 12 hours after administration of the second
maintenance dose.
[0044g] According to another particular aspect, the invention relates to
the use of a
pharmaceutical composition comprising fusidic acid, or a pharmaceutically
acceptable salt
thereof, for reducing development of an antibiotic-resistant strain of
bacteria in a subject
having a bacterial infection, comprising:
lib
CA 2767614 2018-04-23
(a) two loading doses of 1200 mg; and
(b) three maintenance doses of 600 mg, wherein the second loading dose is
formulated
to be administered 12 hours after administration of the first loading dose,
wherein the first
maintenance dose is formulated to be administered 12 hours after
administration of the second
loading dose, wherein the second maintenance dose is formulated to be
administered 12 hours
after administration of the first maintenance dose, and wherein the third
maintenance dose is
formulated to be administered 12 hours after administration of the second
maintenance dose.
[0044h] According to another particular aspect, the invention relates to
the use of a
pharmaceutical composition comprising fusidic acid, or a pharmaceutically
acceptable salt
thereof, for reducing development of an antibiotic-resistant strain of
bacteria in a subject
having a bacterial infection, comprising:
(a) two loading doses of 1500 mg; and
(b)three maintenance doses of 600 mg, wherein the second loading dose is
formulated
to be administered 12 hours after administration of the first loading dose,
wherein the first
maintenance dose is formulated to be administered 12 hours after
administration of the second
loading dose, wherein the second maintenance dose is formulated to be
administered 12 hours
after administration of the first maintenance dose, and wherein the third
maintenance dose is
formulated to be administered 12 hours after administration of the second
maintenance dose.
[0044i] According to another particular aspect, the invention relates to
the use of a
pharmaceutical composition comprising fusidic acid, or a pharmaceutically
acceptable
salt thereof, for treating or preventing a bacterial infection in a subject in
need of
treatment or prevention, comprising:
(a) two loading doses of 1200 mg; and
(b) four maintenance doses of 600 mg, wherein the second loading dose is
formulated to be administered 12 hours after administration of the first
loading dose,
wherein the first maintenance dose is formulated to be administered 12 hours
after
administration of the second loading dose, wherein the second maintenance dose
is
formulated to be administered 12 hours after administration of the first
maintenance
I 1 c
CA 2767614 2018-04-23
dose, wherein the third maintenance dose is formulated to be administered 12
hours after
administration of the second maintenance dose, and wherein the fourth
maintenance dose
is formulated to be administered 12 hours after administration of the third
maintenance
dose.
[0044j] According to another particular aspect, the invention relates to
the use of a
pharmaceutical composition comprising fusidic acid, or a pharmaceutically
acceptable
salt thereof, for treating or preventing a bacterial infection in a subject in
need of
treatment or prevention, comprising:
(a) two loading doses of 1500 mg; and
(b)four maintenance doses of 600 mg, wherein the second loading dose is
formulated to
be administered 12 hours after administration of the first loading dose,
wherein the first
maintenance dose is formulated to be administered 12 hours after
administration of the second
loading dose, wherein the second maintenance dose is formulated to be
administered 12 hours
after administration of the first maintenance dose, wherein the third
maintenance dose is
formulated to be administered 12 hours after administration of the second
maintenance dose,
and wherein the fourth maintenance dose is formulated to be administered 12
hours after
administration of the third maintenance dose.
[0044k] According to another particular aspect, the invention relates to
the use of a
pharmaceutical composition comprising fusidic acid, or a pharmaceutically
acceptable salt
thereof, for reducing development of an antibiotic-resistant strain of
bacteria in a subject
having a bacterial infection, comprising:
(a)two loading doses of 1200 mg; and
(b) four maintenance doses of 600 mg, wherein the second loading dose is
formulated to
be administered 12 hours after administration of the first loading dose,
wherein the first
maintenance dose is formulated to be administered 12 hours after
administration of the second
loading dose, wherein the second maintenance dose is formulated to be
administered 12 hours
after administration of the first maintenance dose, wherein the third
maintenance dose is
formulated to be administered 12 hours after administration of the second
maintenance dose,
and wherein the fourth maintenance dose is formulated to be administered 12
hours after
administration of the third maintenance dose.
1 1 d
CA 2767614 2018-04-23
[00441] According to another particular aspect, the invention relates to
the use of a
pharmaceutical composition comprising fusidic acid, or a pharmaceutically
acceptable salt
thereof, for reducing development of an antibiotic-resistant strain of
bacteria in a subject
having a bacterial infection, comprising:
(a) two loading doses of 1500 mg; and
(b) four maintenance doses of 600 mg, wherein the second loading dose is
formulated to
be administered 12 hours after administration of the first loading dose,
wherein the first
maintenance dose is formulated to be administered 12 hours after
administration of the second
loading dose, wherein the second maintenance dose is formulated to be
administered 12 hours
after administration of the first maintenance dose, wherein the third
maintenance dose is
formulated to be administered 12 hours after administration of the second
maintenance dose,
and wherein the fourth maintenance dose is formulated to be administered 12
hours after
administration of the third maintenance dose.
[0044m] According to another particular aspect, the invention relates to the
use of a
pharmaceutical composition comprising fusidic acid, or a pharmaceutically
acceptable
salt thereof, for treating or preventing a bacterial infection in a subject in
need of
treatment or prevention, comprising:
(a) two loading doses of 1200 mg; and
(b) five maintenance doses of 600 mg, wherein the second loading dose is
formulated
to be administered 12 hours after administration of the first loading dose,
wherein the first
maintenance dose is formulated to be administered 12 hours after
administration of the second
loading dose, wherein the second maintenance dose is formulated to be
administered 12 hours
after administration of the first maintenance dose, wherein the third
maintenance dose is
formulated to be administered 12 hours after administration of the second
maintenance dose,
wherein the fourth maintenance dose is formulated to be administered 12 hours
after
administration of the third maintenance dose, and wherein the fifth
maintenance dose is
formulated to be administered 12 hours after administration of the fourth
maintenance dose.
[0044n] According to another particular aspect, the invention relates to
the use of a
pharmaceutical composition comprising fusidic acid, or a pharmaceutically
acceptable salt
lie
CA 2767614 2018-04-23
thereof, for treating or preventing a bacterial infection in a subject in need
of treatment or
prevention, comprising:
(a) two loading doses of 1500 mg; and
(b)five maintenance doses of 600 mg, wherein the second loading dose is
formulated to
be administered 12 hours after administration of the first loading dose,
wherein the first
maintenance dose is formulated to be administered 12 hours after
administration of the second
loading dose, wherein the second maintenance dose is formulated to be
administered 12 hours
after administration of the first maintenance dose, wherein the third
maintenance dose is
formulated to be administered 12 hours after administration of the second
maintenance dose,
wherein the fourth maintenance dose is formulated to be administered 12 hours
after
administration of the third maintenance dose, and wherein the fifth
maintenance dose is
formulated to be administered 12 hours after administration of the fourth
maintenance dose.
[0044o] According to another particular aspect, the invention relates to
the use of a
pharmaceutical composition comprising fusidic acid, or a pharmaceutically
acceptable salt
thereof, for reducing development of an antibiotic-resistant strain of
bacteria in a subject
having a bacterial infection, comprising:
(a) two loading doses of 1200 mg; and
(b) five maintenance doses of 600 mg, wherein the second loading dose is
formulated to
be administered 12 hours after administration of the first loading dose,
wherein the first
maintenance dose is formulated to be administered 12 hours after
administration of the second
loading dose, wherein the second maintenance dose is formulated to be
administered 12 hours
after administration of the first maintenance dose, wherein the third
maintenance dose is
formulated to be administered 12 hours after administration of the second
maintenance dose,
wherein the fourth maintenance dose is formulated to be administered 12 hours
after
administration of the third maintenance dose, and wherein the fifth
maintenance dose is
formulated to be administered 12 hours after administration of the fourth
maintenance dose.
[0044p] According to another particular aspect, the invention relates to
the use of a
pharmaceutical composition comprising fusidic acid, or a pharmaceutically
acceptable salt
llf
CA 2767614 2018-04-23
thereof, for reducing development of an antibiotic-resistant strain of
bacteria in a subject
having a bacterial infection, comprising:
(a) two loading doses of 1500 mg; and
(b) five maintenance doses of 600 mg, wherein the second loading dose is
formulated to
be administered 12 hours after administration of the first loading dose,
wherein the first
maintenance dose is formulated to be administered 12 hours after
administration of the second
loading dose, wherein the second maintenance dose is formulated to be
administered 12 hours
after administration of the first maintenance dose, wherein the third
maintenance dose is
formulated to be administered 12 hours after administration of the second
maintenance dose,
wherein the fourth maintenance dose is formulated to be administered 12 hours
after
administration of the third maintenance dose, and wherein the fifth
maintenance dose is
formulated to be administered 12 hours after administration of the fourth
maintenance dose.
[0044q] According to another particular aspect, the invention relates to
the use of a
pharmaceutical composition comprising fusidic acid, or a pharmaceutically
acceptable
salt thereof, for treating or preventing a bacterial infection in a subject in
need of
treatment or prevention, comprising:
(a) two loading doses of 1200 mg; and
(b) six maintenance doses of 600 mg, wherein the second loading dose is
formulated to
be administered 12 hours after administration of the first loading dose,
wherein the first
maintenance dose is formulated to be administered 12 hours after
administration of the second
loading dose, wherein the second maintenance dose is formulated to be
administered 12 hours
after administration of the first maintenance dose, wherein the third
maintenance dose is
formulated to be administered 12 hours after administration of the second
maintenance dose,
wherein the fourth maintenance dose is formulated to be administered 12 hours
after
administration of the third maintenance dose, wherein the fifth maintenance
dose is formulated
to be administered 12 hours after administration of the fourth maintenance
dose, and wherein
the sixth maintenance dose is formulated to be administered 12 hours after
administration of
the fifth maintenance dose.
[0044r] According to another particular aspect, the invention relates to
the use of
pharmaceutical composition comprising fusidic acid, or a pharmaceutically
acceptable
Jig
CA 2767614 2018-04-23
salt thereof, for treating or preventing a bacterial infection in a subject in
need of
treatment or prevention, comprising:
(a) two loading doses of 1500 mg; and
(b) six maintenance doses of 600 mg, wherein the second loading dose is
formulated to
be administered 12 hours after administration of the first loading dose,
wherein the first
maintenance dose is formulated to be administered 12 hours after
administration of the second
loading dose, wherein the second maintenance dose is formulated to be
administered 12 hours
after administration of the first maintenance dose, wherein the third
maintenance dose is
formulated to be administered 12 hours after administration of the second
maintenance dose,
wherein the fourth maintenance dose is formulated to be administered 12 hours
after
administration of the third maintenance dose, wherein the fifth maintenance
dose is formulated
to be administered 12 hours after administration of the fourth maintenance
dose, and wherein
the sixth maintenance dose is formulated to be administered 12 hours after
administration of the
fifth maintenance dose.
[0044s] According to another particular aspect, the invention relates to
the use of a
pharmaceutical composition comprising fusidic acid, or a pharmaceutically
acceptable salt
thereof, for reducing development of an antibiotic-resistant strain of
bacteria in a subject having
a bacterial infection, comprising:
(a) two loading doses of 1200 mg; and
(b) six maintenance doses of 600 mg, wherein the second loading dose is
formulated to be administered 12 hours after administration of the first
loading dose,
wherein the first maintenance dose is formulated to be administered 12 hours
after
administration of the second loading dose, wherein the second maintenance dose
is
formulated to be administered 12 hours after administration of the first
maintenance
dose, wherein the third maintenance dose is formulated to be administered 12
hours after
administration of the second maintenance dose, wherein the fourth maintenance
dose is
formulated to be administered 12 hours after administration of the third
maintenance
dose, wherein the fifth maintenance dose is formulated to be administered 12
hours after
administration of the fourth maintenance dose. and
11h
CA 2767614 2018-04-23
wherein the sixth maintenance dose is formulated to be administered 12 hours
after administration of the fifth maintenance dose.
[0044t] According to another particular aspect, the invention relates to
the use of a
pharmaceutical composition comprising fusidic acid, or a pharmaceutically
acceptable salt
thereof, for reducing development of an antibiotic-resistant strain of
bacteria in a subject having a
bacterial infection, comprising:
(a) two loading doses of 1500 mg; and
(b) six maintenance doses of 600 mg, wherein the second loading dose is
formulated to be
administered 12 hours after administration of the first loading dose, wherein
the first maintenance
dose is formulated to be administered 12 hours after administration of the
second loading dose,
wherein the second maintenance dose is formulated to be administered 12 hours
after
administration of the first maintenance dose, wherein the third maintenance
dose is formulated to
be administered 12 hours after administration of the second maintenance dose,
wherein the fourth
maintenance dose is formulated to be administered 12 hours after
administration of the third
maintenance dose, wherein the fifth maintenance dose is formulated to be
administered 12 hours
after administration of the fourth maintenance dose, and wherein the sixth
maintenance dose is
formulated to be administered 12 hours after administration of the fifth
maintenance dose.
BRIEF DESCRIPTION OF THE DRAWINGS
[0045] Figure 1 - Mean plasma concentrations of fusidic acid after a single
dose of sodium
fusidate (semi-log plot).
[0046] Figure 2 - Mean plasma concentrations of fusidic acid after a single
dose of sodium
fusidate (linear plot).
1 1 i
CA 2767614 2018-04-23
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
[0047] Figure 3 ¨ Mean plasma concentrations of CEM-102 (Sodium fusidate)
after 13
doses - semi-log plot.
[0048] Figure 4 - Mean plasma concentrations of CEM-102 (Sodium fusidate)
after 13
doses - linear plot.
[0049] Figure 5 - Mean CEM-102 (Sodium fusidate) single and multiple dose
plasma
concentrations (linear scale). Cohort 1 (550 mg), cohort 2 (1100 mg), cohort 3
(1650 mg),
and cohort 4 (2200 mg - single dose only).
[0050] Figure 6 - Mean CEM-102 (Sodium fusidate) plasma concentrations in
loading
dose regimens (linear scale).
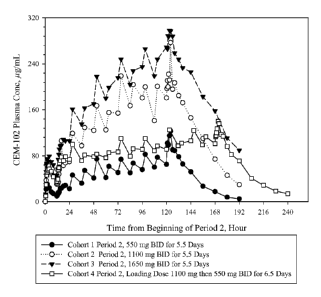
[0051] Figure 7 ¨ Mean CEM-102 (Sodium fusidate) multiple dose plasma
concentrations (linear scale) which is a compilation of the Period 2 data of
Figure 5 and the
Cohort 4, Period 2, data of Figure 6.
[0052] Figure 8 ¨ CEM-102 pharmacodynamics in hollow fiber. CEM-102 (Sodium
fusidate) 600 mg ql2h vs. USA 300.
[0053] Figure 9 ¨ CEM-102 pharmacodynamics in hollow fiber. CEM-102 (Sodium
fusidate) 1200 mg x 2 then 600 mg ql2h vs. USA 300.
[0054] Figure 10 ¨ CEM-102 pharmacodynamics in hollow fiber. CEM-102
(Sodium
fusidate) 1500 mg x 2 then 600 mg ql2h vs. USA 300.
DETAILED DESCRIPTION OF THE INVENTION
[0055] Conventional treatment using FA is problematic due to the fact that
while low
doses of the drug result in the selection of drug-resistant organisms, higher
optimal doses
are not possible due to severe nausea and vomiting induced in subjects. This
has resulted
in the dogma that monotherapy results in resistance and combination therapy is
required to
defeat resistance selection.
[0056] Through numerous studies and the diligent efforts of the inventors,
and as
disclosed herein, it has been discovered that both problems associated with
conventional
FA therapy can be avoided by administering to a subject a high (loading) dose
of FA
during the first day of treatment, and a moderate (maintenance) dose of FA on
succeeding
days of treatment. The loading dose is of sufficient amount that at least a 2-
fold excess,
12
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
and in some cases at least a 10-fold excess, of the drug above the MIC of the
bacterial
infection being treated or inhibited, is administered to the subject on the
first day of
treatment. By administering a large amount of the drug to the subject within
the first day
of treatment, a high plasma concentration of the drug can be reached within a
short period
of time. Quickly reaching a maximum plasma concentration (Cõ.) that is well
above the
MIC for the bacterial infection that is being treated or prevented prevents
development of
FA-resistance in the bacteria.
[0057] Limiting the administration of high doses of FA to the loading phase
(the first
day of treatment), and decreasing the dosage(s) given to the subject on the
second and
succeeding days of treatment (the maintenance phase), avoids the severe nausea
and
vomiting seen when large amounts of FA are administered for prolonged periods
of time.
The doses given during the maintenance phase are sufficient to maintain a
minimum
trough plasma concentration of FA for sufficient time in which to treat or
prevent the
bacterial infection, but not so great as to cause an unacceptable degree of
nausea or to
induce vomiting.
[0058] As an example, and as further discussed in the Examples below, a
loading dose
regimen of 1100 mg BID FA for the first day, followed by 550 mg BID FA for
five days as
the maintenance dose, showed that the drug was well tolerated and that a
trough level of at
least 74 ug/ml in the plasma was achieved, much higher than the MIC of 2.5
ug/ml of S.
aureus in humans.
[0059] The use of a high loading dose and moderate maintenance doses has
the added
benefit of expanding the number species of bacteria against which FA can be
used in
treatment.
[0060] According, the present invention is directed to methods for treating
or
preventing bacterial infections in a subject. The methods are based on the
novel dosing
regimens disclosed herein.
[0061] In each of the methods of the present invention, the novel dosing
regimens
comprise two phases, a loading dose phase followed by a maintenance dose
phase.
Loading Dose
13
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
[0062] The loading dose phase is the period of time over which a high dose
of FA is
administered to a subject. By administering a high dose of FA to the subject
over a short
period of time, the development of drug-resistant strains of bacteria can be
avoided.
[0063] In one embodiment of the invention, the loading dose phase is
defined by the
administration of fusidic acid to a subject in an amount sufficient to reach a
pharmacokinetic (PK) profile for fusidic acid defined by three variables: (i)
a maximum
plasma concentration (Cmax), (ii) a time to maximum plasma concentration (T.)
of fusidic
acid, and (iii) a minimum trough plasma concentration of fusidic acid. With
regard to the
first variable, the maximum plasma concentration (C.) of fusidic acid is not
less than
about 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 105, 110, 115 or 120 ug/ml,
or a value
within this range, preferably not less than about 80 ug/ml, more preferably
not less than
about 100 ug/ml. With regard to the second variable, the time to maximum
plasma
concentration (T.) of fusidic acid is no more than about 12, 13, 14, 15, 16,
17, 18, 19, 20,
21, 22, 23, 24, 25, 26, 27, 28, 29 or 30 hours, preferably no more than about
24 hours.
With regard to the third variable, the minimum trough plasma concentration of
fusidic acid
is not less than about 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95 or 100
ug/ml, or a value
within this range, preferably not less than about 60 ug/ml, more preferably
not less than
about 80 ug/ml.
[0064] The loading dose administered to the subject may also be defined
based on the
amount of FA sufficient to achieve a minimum plasma concentration within a
particular
period of time. The minimum plasma concentration of fusidic acid is not less
than about
40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95 or 100 ug/ml, or a value within
this range,
preferably not less than about 60 ug/ml, more preferably not less than about
80 ug/ml. The
period of time in which the minimum plasma concentration of fusidic acid is
achieved is
within about 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27,
28, 29 or 30
hours, preferably within about 24 hours. In preferred aspects, the loading
dose of fusidic
acid is an amount sufficient to achieve a minimum plasma concentration of not
less than
about 50 ug/ml fusidic acid within about 24 hours of administration to the
subject.
[0065] The skilled artisan will understand that as long as the particular
PK profile or
minimum plasma concentration is achieved, the loading dose may be split into
discrete
14
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
loading doses that are administered to a subject over a particular period of
time. Therefore,
in addition to a single loading dose, the loading dose may comprise two,
three, four, five,
six or more discrete loading doses. The total amount of FA administered to a
subject as
the loading dose is about 1500, 1600, 1700, 1800, 1900, 2000, 2100, 2200,
2300, 2400,
2500, 2600, 2700, 2800, 2900, 3000, 3100, 3200, 3300, 3400, 3500, 3600, 3700,
3800,
3900, 4000 mg or more of FA, or a value within this range. Preferably, the
total loading
dose is between about 2000 mg and 3600 mg of FA. Where two or more discrete
loading
doses are administered to a subject, the skilled artisan will understand that
any fraction of
the total loading dose may be used in each discrete loading dose, whether the
discrete
doses are equal in concentration or unequal (e.g., two-thirds of the total
loading dose in a
first dosage, and one-third of the total loading dose in a second dosage). In
a preferred
aspect, the loading dose is administered to a subject as two discrete loading
doses, where
each discrete loading dose is about 1000, 1100, 1200, 1300, 1400, 1500, 1600,
1700, 1800
or 1850 mg, or a value within this range. Preferably, each loading dose is
between about
1000 mg and 1850 mg of FA
[0066] With regard to the period of time over which the loading dose is
administered
to the subject, the skilled artisan will understand that quickly reaching a
desired C. or a
particular minimum plasma concentration is important to avoid development of
FA-
resistant strains of bacteria. However, the entirety of the loading dose will
be administered
to the subject within about 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18,
19, 20, 21, 22, 23,
24 or more hours. In a preferred aspect, the entirety of the loading dose will
be
administered to the subject within about 20 hours. When the loading dose is
split into two
or more discrete loading doses, the time periods between administrations of
the discrete
loading doses may be equal or unequal. Preferably, the time periods are equal.
[0067] The loading dose administered to the subject may further be defined
simply
based on the amount of FA administered to the subject. The total amount of FA
administered to a subject as the loading dose may be about 1500, 1600, 1700,
1800, 1900,
2000, 2100, 2200, 2300, 2400, 2500, 2600, 2700, 2800, 2900, 3000, 3100, 3200,
3300,
3400, 3500, 3600, 3700, 3800, 3900, 4000 mg or more of FA, or a value within
this range.
Preferably, the total loading dose is between about 2000 mg and 3600 mg of FA.
As
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
above, the loading dose may be split into discrete loading doses that are
administered to a
subject over a particular period of time, such as two, three, four, five, six
or more discrete
loading doses. Where two or more discrete loading doses are administered to a
subject,
any fraction of the total loading dose may be used in each discrete loading
dose, whether
the discrete doses are equal in concentration or unequal (e.g., two-thirds of
the total
loading dose in a first dosage, and one-third of the total loading dose in a
second dosage).
In a preferred aspect, the loading dose is administered to a subject as two
discrete loading
doses, where each discrete loading dose is about 1000, 1100, 1200, 1300, 1400,
1500,
1600, 1700, 1800 or 1850 mg, or a value within this range.
[0068] In preferred aspects, the loading dose is administered to a subject
as two
discrete loading doses, where each discrete loading dose is about 1000 mg, and
where in
the second loading dose is administered about 12 hours after administration of
the first
loading dose. In additionally preferred aspects, the loading dose is
administered to a
subject as two discrete loading doses, where each discrete loading dose is
about 1500 mg,
and where in the second loading dose is administered about 12 hours after
administration
of the first loading dose.
Maintenance Dose
[0069] The maintenance dose phase is the period of time following the
loading dose
phase where a particular minimum trough plasma concentration or plasma
concentration of
the FA is maintained in the subject. The skilled artisan will understand that
the particular
concentration of FA in the plasma, and the length of time needed to maintain
the desired
concentration, will depend on the condition that is being treated or
prevented. While the
methods of the invention may be practiced through the use of a single
maintenance dose,
the methods of the present invention will generally require that two or more
maintenance
doses be administered to a subject in order to maintain the desired minimum
trough plasma
concentration or plasma concentration of fusidic acid for a particular period
of time.
[0070] In one embodiment of the invention, the maintenance dose is defined
by the
administration of fusidic acid to the subject in an amount sufficient to
maintain a minimum
trough plasma concentration of fusidic acid for a particular period of time. A
minimum
trough plasma concentration of not less than about 30, 35, 40, 45, 50, 55, 60,
65, 70, 75,
16
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
80, 85, 90, 95 or 100 ug/ml, or a value within this range, is maintained in
the subject
during the maintenance dose phase, preferably not less than about 70 ug/ml,
more
preferably not less than about 80 ug/ml. The minimum trough plasma
concentration is
maintained for a sufficient period of time to achieve the goal of the method
being practiced
(treatment or prevention) and will vary depending on the identity of the
bacterial infection.
However, it is considered that the minimum trough plasma concentration will
need to be
maintained at least about 12, 18, 24, 36, 48, 60, 72, 84, 96, 108, 120, 132,
144, 156, 168,
180, 192, 204, 216, 228, 240 or more hours, beginning from the time at which
the last
loading dose was administered to the subject. The first maintenance dose may
be
administered about 6,7, 8,9, 10, 11, 12, 13, 14, 15, 16, 17, 18 or more hours
after the last
loading dose.
[0071] In order to achieve particular minimum trough plasma concentrations,
the
specific amount of FA administered to a subject will depend on the
characteristics of the
subject, including age, weight, and general health. However, discrete
maintenance doses
will generally be about 200, 300, 400, 500, 600, 700, 800, 900, 1000, 1100,
1200 mg or
more, or a value within this range. Preferably, each maintenance dose is
between about
500 mg and 1000 mg. The amount of FA in each maintenance dose may be the same,
or
the doses may increase or decrease with time. The maintenance doses will be
administered to the subject on a course of therapy determined by a physician,
but in
general the maintenance doses may be administered to the subject 1, 2, 3, 4,
5, 6 or more
times per day, or a value within this range.
[0072] In an aspect, a minimum trough plasma concentration of not less than
about 50
ug/ml is maintained for at least about 12, 18, 24, 36, 48, 60, 72, 84, 96,
108, 120, 132, 144,
156, 168, 180, 192, 204, 216, 228, 240 or more hours, or a value within this
range, in
particular at least about 24, 48 or 96 hours, after the last loading dose was
administered to
the subject.
[0073] The maintenance dose in the methods of the present invention may
also be
simply defined by the amount of fusidic acid in a discrete dose that is
administered to a
subject. Each discrete maintenance dose is individually about 200, 300, 400,
500, 600,
700, 800, 900, 1000, 1100, 1200 mg or more, or a value within this range.
Preferably, each
17
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
maintenance dose is between about 500 mg and 1000 mg. The discrete maintenance
doses
of FA may be administered 1, 2, 3, 4, 5, 6 or more times per day, or a value
within this
range. The first maintenance dose may be administered about 6, 7, 8, 9, 10,
11, 12, 13, 14,
15, 16, 17, 18 or more hours after the last loading dose. The maintenance dose
phase is
maintained for at least about 12, 18, 24, 36, 48, 60, 72, 84, 96, 108, 120,
132, 144, 156,
168, 180, 192, 204, 216, 228, 240 or more hours, or a value within this range,
beginning
from the time at which the last loading dose was administered to the subject.
In a preferred
aspect, at least three maintenance doses of 500 mg or 600 mg are administered
to a subject
during the maintenance dose phase, more preferably at least four doses, even
more
preferably at least five doses, wherein the maintenance doses arc administered
about 12
hours apart beginning about 12 hours after the last loading dose.
Combination of LD and MD
[0074] The skilled artisan will understand that different combinations of
values for the
three variables comprising the PK during the loading phase and the minimum
trough
plasma concentration during maintenance phase can be used, and that the exact
values will
depend on the condition being treated or prevented in the subject. However, in
one
example, the method of treating or preventing a bacterial infection in a
subject comprises:
(a) administering at least one loading dose of fusidic acid, or a
pharmaceutically acceptable salt thereof, to a subject in need of treatment or
prevention, in
an amount sufficient to reach a pharmacokinetic (PK) profile for fusidic acid
comprising a
maximum plasma concentration (C.) of fusidic acid of not less than about 70
ug/ml, a
time to maximum plasma concentration (T.) of fusidic acid of no more than
about 20
hours and a minimum trough plasma concentration of fusidic acid of not less
than about 50
ug/ml, and
(b) administering at least one maintenance dose of fusidic acid, or a
pharmaceutically acceptable salt thereof, to the subject of (a) in an amount
sufficient to
maintain a minimum trough plasma concentration of fusidic acid of not less
than about 50
ug/ml for at least about 24 hours after the administering of (a).
[0075] In preferred aspects, the PK profile comprises (i) a C. of not less
than about
80 ug/ml, (ii) a minimum trough plasma concentration of not less than about 60
ug/ml and
18
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
(iii) a T. of no more than 20 hours, with the minimum trough plasma
concentration being
maintained at not less than about 70 ug/ml. In further preferred aspects, the
PK profile
comprises (i) a C. of not less than about 100 ug/ml, (ii) a minimum trough
plasma
concentration of not less than about 80 ug/ml and (iii) a T. of no more than
20 hours,
with the minimum trough plasma concentration being maintained at not less than
about 80
ug/ml.
[0076] The skilled artisan will also understand that different combinations
of values for
the minimum plasma concentration during the loading phase and the minimum
trough
plasma concentration during maintenance phase can be used, and that the exact
values will
depend on the condition being treated or prevented in the subject. However, in
one
example, the method of treating or preventing a bacterial infection in a
subject comprises:
(a) administering at least one loading dose of fusidic acid, or a
pharmaceutically acceptable salt thereof, to a subject in need of treatment or
prevention in
an amount sufficient to achieve a minimum plasma concentration of not less
than about 50
ug/ml fusidic acid within 24 hours, and
(b) administering at least one maintenance dose of fusidic acid, or a
pharmaceutically acceptable salt thereof, to the subject of (a) in an amount
sufficient to
maintain a minimum plasma concentration of not less than about 50 ug/ml
fusidic acid for
at least about 24 hours after the administering of (a).
[0077] In a preferred aspect, a minimum plasma concentration of fusidic
acid of not
less than about 60 ug/ml is achieved in (a) and a minimum plasma concentration
of fusidic
acid of not less than about 60 ug/ml is maintained in (b). In a further
preferred aspect, a
minimum plasma concentration of fusidic acid of not less than about 80 ug/ml
is achieved
in (a) and a minimum plasma concentration of fusidic acid of not less than
about 80 ug/ml
is maintained in (b).
[0078] The skilled artisan will further understand that different
combinations of values
for loading phase doses and maintenance phase doses can be used, and that the
exact
values will depend on the condition being treated or prevented in the subject.
However, in
one example, the method of treating or preventing a bacterial infection in a
subject
comprises:
19
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
(a) administering a first and a second loading dose of fusidic acid, or a
pharmaceutically acceptable salt thereof, to a subject in need of treatment or
prevention,
wherein each loading dose is independently between about 1000 mg and about
1850 mg,
and wherein the second loading dose is administered about 12 hours after the
first loading
dose; and
(b) administering two or more maintenance doses of fusidie acid, or a
pharmaceutically acceptable salt thereof, to the subject of (a), wherein each
maintenance
dose is independently between about 500 mg and 1000 mg, wherein a first
maintenance
dose is administered about 12 hours after administration of the second loading
dose, and
wherein subsequent maintenance doses arc administered about 12 hours apart.
[0079] In a preferred aspect, at least three, four, five or six maintenance
doses are
administered to the subject. In a further preferred aspect, each loading dose
is
independently at least about 1500 mg and each maintenance dose is
independently at least
about 600 mg.
Preventing Development of Resistant Strains of Bacteria
[0080] The present invention is also directed to methods for reducing
development of
antibiotic-resistant strains of bacteria in a subject undergoing antibiotic
therapy. These
methods are also based on the novel dosing regimens disclosed herein. As in
the methods
of treating or preventing bacterial infections described herein, the methods
for reducing
development of antibiotic-resistant strains of bacteria are based on the novel
dosing
regimens comprising a loading dose phase and a maintenance dose phase. All
aspects of
the loading dose phase and the maintenance dose phase described above for the
methods of
treating or preventing bacterial infections are the same for the loading dose
phase and the
maintenance dose phase of the methods for reducing development of antibiotic-
resistant
strains of bacteria and are specifically incorporated herein.
[0081] The skilled artisan will understand that different combinations of
values for the
three variables comprising the PK during the loading phase and the minimum
trough
plasma concentration during maintenance phase can be used, and that the exact
values will
depend on the bacterial infection for which a subject is undergoing treatment.
However, in
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
one example, the method for reducing development of antibiotic-resistant
strains of
bacteria in a subject having a bacterial infection comprises:
(a) administering at least one loading dose of fusidic acid, or a
pharmaceutically acceptable salt thereof, to a subject having a bacterial
infection, in an
amount sufficient to reach a pharmacokinetic (PK) profile for fusidic acid
comprising a
maximum plasma concentration (C.) of fusidic acid of not less than about 70
ug/ml, a
time to maximum plasma concentration (T.) of fusidic acid of no more than
about 24
hours and a minimum trough plasma concentration of fusidic acid of not less
than about 50
ug/ml, and
(b) administering at least one maintenance dose of fusidic acid, or a
pharmaceutically acceptable salt thereof, to the subject of (a) in an amount
sufficient to
maintain a minimum trough plasma concentration of fusidic acid of not less
than about 50
ug/ml for at least about 24 hours after the administering of (a).
[0082] In a preferred aspect, the C. is not less than about 80 ug/ml, the
minimum
trough plasma concentration of (a) is not less than about 60 ug/ml, and the
minimum
trough plasma concentration of (b) is not less than about 70 ug/ml. In a
further preferred
aspect, the Cmax is not less than about 100 ug/ml, the minimum trough plasma
concentration of (a) is not less than about 80 ug/ml and the minimum trough
plasma
concentration of (b) is not less than about 80 ug/ml.
[0083] In each of the embodiments of the present invention, the following
common
aspects and preferred common aspects are encompassed within the scope of the
invention.
[0084] Fusidic Acid (FA) has the following structure.
21
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
26 27
"
24
23
22 20 COOH
(21)
H 0/11 E OAc
'4., 1^ 17
11 13 16
(19) CH3
14 15
9 CH 3
1
2 10 H
CH3
3 4 6 (18)
HO\ . =
17I
(30)
Fusidic Acid
The skilled artisan will understand that for the sake of brevity alone, all
references herein
to "fusidic acid" or "FA", unless otherwise stated, also refers to the
hemihydrate form of
the compound, as well as pharmaceutically acceptable salts, other hydrates,
solvates, or
mixtures thereof
[0085] The term "pharmaceutically acceptable salt" refers to non-toxic base
addition
salts derived from inorganic and organic bases. Base addition salts include
those derived
from inorganic bases, such as ammonium or alkali or alkaline earth metal
hydroxides,
carbonates, bicarbonates, and the like, as well as alkylamine and organic
amino salts, such
as an ethanolamine salt. Such bases useful in preparing the salts of this
invention thus
include sodium hydroxide, potassium hydroxide, ammonium hydroxide, potassium
carbonate, sodium carbonate, sodium bicarbonate, potassium bicarbonate,
calcium
hydroxide, calcium carbonate, and the like. The potassium and sodium salt
forms are
particularly preferred. In preferred embodiments, sodium fusidate is a
pharmaceutically
acceptable salt that is used in the methods of the present invention. Sodium
fusidate, also
termed CEM-102 herein, has the following structure.
22
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
26 27
24
23
22 20 COO Na
(21)
HOiõ, 12 9 E 17 OAc
11 13 16
(19) CH3
9 CF-i314 19
1
2 10 H 8
Cl-I3
3 5 7 (18)
4 6
HO\ . =
H
eH3
(30)
Sodium Fusidate
[0086] It should be recognized that the particular counter-ion forming a
part of any salt
of this invention is not of a critical nature, so long as the salt as a whole
is
pharmacologically acceptable and as long as the counter-ion does not
contribute undesired
qualities to the salt as a whole.
[0087] In each of the embodiments of the present invention, the subject
being
subjected to treatment or prevention is a human, non-human primate, bird,
horse, cow,
goat, sheep, a companion animal, such as a dog, cat or rodent, or other
mammal. The
subject may have a bacterial infection, such as where the present invention is
directed to
methods for treating a bacterial infection in a subject. The subject may also
be at risk for
developing a bacterial infection, such as where the present invention is
directed to methods
for preventing a bacterial infection in a subject. Examples of subjects at
risk for
developing bacterial infections include patients undergoing treatment for
bacterial
infections whereby normal gut flora is inhibited by antimicrobial therapy,
patients with
impaired immune function (e.g., immunoglobulin deficiency, splenic
dysfunction,
splenectomy, HIV infection, impaired leukocyte function, hemoglobinopathies),
the
elderly, children, people with certain malignancies (e. g., multiple myeloma,
chronic
lympocytic leukemia, lymphoma), people at increased occupational risk (e.g.,
public
services workers, such a fire, water, sanitary, police, medical, and
laboratory workers,
23
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
hospital workers), people in closed populations (e.g., hospitals and medial
clinics, prisons,
military, nursing homes), people having cystic fibrosis, people that have
immunological
deficiencies that might enhance their susceptibility to bacterial infection,
people entering
an emergency room, such as those with wounds or cellulitis, and patients
leaving a hospital
on step-down therapy after having been on intravenous therapy
[0088] In each of the embodiments of the present invention, the bacterial
infection
being treated or prevented is an infection caused by bacteria selected from
the group
consisting of staphylococci, including coagulase-negative staphylococci and
coagulase-
positive staphylococci, streptococci, including Group A beta hemolytic
streptococci, non-
Group A beta hemolytic streptococci and viridans group streptococci,
enterococci,
Nesseria species, Clostridium species, Bordetella species, Bacillus species
and
Corynebacterium species. In particular aspects, the bacterial infection is an
infection
caused by bacteria selected from the group consisting of Staphylococcus aureus
(methicillin-resistant and -susceptible), Staphylococus epidermidi s,
Staphylococus
hemolyticus, Staphylococus saprophyticus, Staphylococus lugdunensis,
Staphylococus
capitis, Staphylococus caprae, Staphylococus saccharolyticus, Staphylococus
simulans,
Staphylococus warneri, Staphylococus hominis, Staphylococus internzedius,
Staphylococcus pseudointermedius, Staphylococus lyricus, Streptococcus
pyogenes,
Streptococcus agalactiae, Streptococcus dysgalactiae subspecies dysgalactiae,
Streptococcus anginosus, Streptococcus mitis, Streptococcus salivarius,
Streptococcus
bovis, Streptococcus mutans, Neisseria gonorrhoeae, Neisseria meningitidis,
Bacillus
anthracis, Bordetella pertussi s, Clostridiunz difficile, Enterococcus
faecalis, Enterococcus
filecium and Cozynebacterium diphtheriae. In particular aspects, the bacterial
infection is
an infection caused by Enterococcus faecalis or Enterococcus faecium.
[0089] In each of the embodiments of the present invention, the bacterial
infection may
also be defined based on the type of infection that it causes. For example,
each of the
embodiments of the present invention may be used to treat or prevent an
infection selected
from the group consisting of a skin and soft tissue infection, a bone
infection, a joint
infection, pneumonia, a wound infection, a bum infection, an infection of the
blood, and an
infection associated with cystic fibrosis.
24
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
[0090] In each of the embodiments of the present invention, the fusidic
acid may be
administered to a subject in conjunction with a second therapeutic agent, such
as a second
antibiotic. The second therapeutic agent may be administered before,
concurrent with or
after administration of the fusidic acid, whether in the same formulation or
in a separate
formulation. Suitable second therapeutic agents include rifampin, rifamycin, a
sulfonamide, a beta-lactam, a tetracycline, a chloramphenicol, an
aminoglycoside, a
macrolide, a streptogramin, a quinolone, a fluoroquinolone, an oxazolidinone
and a
lipopeptide. In particular, tetracycline, tetracycline derived antibacterial
agents,
glycylcycline, glycylcycline derived antibacterial agents, minocycline,
minocycline
derived antibacterial agents, oxazolidinone antibacterial agents,
aminoglycoside
antibacterial agents, quinolone antibacterial agents, vancomycin, vancomycin
derived
antibacterial agents, teicoplanin, teicoplanin derived antibacterial agents,
eremomycin,
eremomycin derived antibacterial agents, chloroeremomycin, chloroeremomycin
derived
antibacterial agents, daptomycin, and daptomycin derived antibacterial agents
are
preferred. In a preferred aspect of the embodiments, rifampin is administered
concurrently
with the fusidic acid.
[0091] In each of the embodiments of the present invention, the subject
does not
experience an adverse level of nausea over the entire course of treatment or
prevention. As
used herein, an adverse level of nausea is considered to be a level of nausea
severe enough
that at least 15% of subjects in a population of subjects being treated with
fusidic acid
discontinue treatment.
[0092] The pharmaceutical compositions of the present invention comprise
fusidic
acid, a hemihydrate form thereof, or pharmaceutically acceptable salts, other
hydrates,
solvates, or mixtures thereof, and one or more of a carrier, diluent and
excipient. The
terms specifically exclude cell culture medium. Suitable diluents (for both
dry and liquid
pharmaceutical formulations) are well known to those skilled in the art and
include saline,
buffered saline, dextrose (e.g., 5% dextrose in water), water, glycerol,
ethanol, propylene
glycol, polysorbate 80 (Tween-80Tm), poly(ethylene)glycol 300 and 400 (PEG 300
and
400), PEGylated castor oil (e.g. Cremophor EL), poloxamer 407 and 188, a
cyclodextrin or
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
a cyclodextrin derivative (including HPCD ((2-hydroxypropy1)-cyclodextrin) and
(2-
hydroxyethyl)-cyclodextrim see, e.g., U.S. patent application publication
20060194717).
[0093] Carriers are compounds and substances that improve and/or prolong
the
delivery of an active ingredient to a subject in the context of a
pharmaceutical formulation.
Carrier may serve to prolong the in vivo activity of a drug or slow the
release of the drug in
a subject, using controlled-release technologies. Carriers may also decrease
drug
metabolism in a subject and/or reduce the toxicity of the drug. Carrier can
also be used to
target the delivery of the drug to particular cells or tissues in a subject.
Common carriers
(both hydrophilic and hydrophobic carriers) include fat emulsions, lipids,
PEGylated
phospholids, liposomcs and lipospheres, microsphcres (including those made of
biodegradable polymers or albumin), polymer matrices, biocompatible polymers,
protein-
DNA complexes, protein conjugates, erythrocytes, vesicles and particles.
[0094] Excipients included in a pharmaceutical composition have different
purposes
depending, for example on the nature of the drug, and the mode of
administration.
Examples of generally used excipients include, without limitation: stabilizing
agents,
solubilizing agents and surfactants, buffers and preservatives, tonicity
agents, bulking
agents, lubricating agents (such as talc or silica, and fats, such as
vegetable stearin,
magnesium stearate or stearic acid), emulsifiers, suspending or viscosity
agents, inert
diluents, fillers (such as cellulose, dibasic calcium phosphate, vegetable
fats and oils,
lactose, sucrose, glucose, mannitol, sorbitol, calcium carbonate, and
magnesium stearate),
disintegrating agents (such as crosslinked polyvinyl pyrrolidonc, sodium
starch glycolatc,
cross-linked sodium carboxymethyl cellulose), binding agents (such as
starches, gelatin,
cellulose, methyl cellulose or modified cellulose such as microcrystalline
cellulose,
hydroxypropyl cellulose, sugars such as sucrose and lactose, or sugar alcohols
such as
xylitol, sorbitol or maltitol, polyvinylpyrrolidone and polyethylene glycol),
wetting agents,
antibacterials, chelating agents, coatings (such as a cellulose film coating,
synthetic
polymers, shellac, corn protein zein or other polysaccharides, and gelatin),
preservatives
(including vitamin A, vitamin E, vitamin C, retinyl palmitate, and selenium,
cysteine,
methionine, citric acid and sodium citrate, and synthetic preservatives,
including methyl
26
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
paraben and propyl paraben), sweeteners, perfuming agents, flavoring agents,
coloring
agents, administration aids, and combinations thereof.
[0095] In particular, the pharmaceutical compositions may contain common
carriers
and excipients, such as microcrystalline cellulose, crospovidone,
hypromellose, lactose
monohydrate, magnesium stearate, silica, all-rac-a-tocopherol, talc and
titanium dioxide.
[0096] Pharmaceutically acceptable excipients also include tonicity agents
that make
the composition compatible with blood. Tonicity agents are particularly
desirable in
injectable formulations.
[0097] The particular carrier, diluent or excipient used will depend upon
the means and
purpose for which the active ingredient is being applied.
[0098] The pharmaceutical compositions of the present invention may be
formulated,
for example, for oral, sublingual, intranasal, intraocular, rectal,
transdermal, mucosa',
pulmonary, topical or parenteral administration. Parenteral modes of
administration
include without limitation, intradermal, subcutaneous (s.c., s.q., sub-Q,
Hypo),
intramuscular (i.m.), intravenous (i.v.), intraperitoneal (i.p.), intra-
arterial, intramedulary,
intracardiac, intra-articular (joint), intrasynovial (joint fluid area),
intracranial, intraspinal,
and intrathecal (spinal fluids). Any known device useful for parenteral
injection or
infusion of drug formulations can be used to effect such administration.
[0099] Formulations for parenteral administration can be in the form of
aqueous or
non-aqueous isotonic sterile injection solutions, suspensions or fat
emulsions. The
parenteral form used for injection must be fluid to the extent that easy
syringability exists.
These solutions or suspensions can be prepared from sterile concentrated
liquids, powders
or granules.
[00100] Excipients used in parenteral preparations also include, without
limitation,
stabilizing agents (e.g. carbohydrates, amino acids and polysorbates, such as
5% dextrose),
solubilizing agents (e.g. cetrimide, sodium docusate, glyceryl monooleate,
polyvinylpyrolidone (PVP) and polyethylene glycol (PEG)), surfactants (e.g.
polysorbates,
tocopherol PEG succinate, poloxamer and CremophorTm), buffers (e.g. acetates,
citrates,
phosphates, tartrates, lactates, succinates, amino acids and the like),
preservatives (e.g.
BHA, BHT, gentisic acids, vitamin E, ascorbic acid, sodium ascorbate and
sulfur
27
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
containing agents such as sulfites, bisulfites, metabisulfites, thioglycerols,
thioglycolates
and the like), tonicity agents (for adjusting physiological compatibility),
suspending or
viscosity agents, antibacterials (e.g. thimersol, benzethonium chloride,
benzalkonium
chloride, phenol, cresol and chlorobutanol), chelating agents, and
administration aids (e.g.
local anesthetics, anti-inflammatory agents, anti-clotting agents,
vasoconstrictors for
prolongation and agents that increase tissue permeability), and combinations
thereof
[00101] Parenteral formulations using hydrophobic carriers include, for
example, fat
emulsions and formulations containing lipids, lipospheres, vesicles, particles
and
liposomes. Fat emulsions include in addition to the above-mentioned
excipients, a lipid
and an aqueous phase, and additives such as emulsifiers (e.g. phospholipids,
poloxamers,
polysorbates, and polyoxyethylene castor oil), and osmotic agents (e.g. sodium
chloride,
glycerol, sorbitol, xylitol and glucose). Liposomes include natural or derived
phospholipids
and optionally stabilizing agents such as cholesterol.
[00102] In another embodiment, the parenteral unit dosage form can be a ready-
to-use
solution of the active ingredient in a suitable carrier in sterile,
hermetically sealed
ampoules or in sterile pre-loaded syringes. The suitable carrier optionally
comprises any of
the above-mentioned excipients.
[00103] Alternatively, the unit dosage of the pharmaceutical composition can
be in a
concentrated liquid, powder or granular form for ex tempore reconstitution in
the
appropriate pharmaceutically acceptable carrier, such as sterile water, at the
time of
delivery. In addition to the above-mentioned excipients, powder forms
optionally include
bulking agents (e.g. mannitol, glycine, lactose, sucrose, trehalose, dextran,
hydroxyethyl
starch, ficoll and gelatin), and cryo or lyoprotectants.
[00104] In intravenous (IV) use, a sterile formulation of the pharmaceutical
compositions of the present invention and optionally one or more additives,
including
solubilizers or surfactants, can be dissolved or suspended in any of the
commonly used
intravenous fluids and administered by infusion. Intravenous fluids include,
without
limitation, physiological saline, phosphate buffered saline, 5% dextrose in
water or
Ringer'STM solution.
28
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
[00105] In a preferred intravenous (IV) formulation, fusidic acid is dissolved
in a
buffered solution (pH 7.4 - 7.6) containing disodium hydrogen phosphate,
citric acid,
disodium edetate and water for injections. The buffered solution is then added
to a suitable
infusion, such as a sodium chloride intravenous infusion, a dextrose
intravenous infusion, a
compound sodium lactate intravenous infusion ("Ringer-lactate solution"), a
sodium
lactate intravenous infusion, sodium chloride and dextrose intravenous
infusion, or
potassium chloride and dextrose intravenous infusion. Suitable amounts of
fusidic acid to
be dissolved in the buffered solution range from about 10 to about 4000 mg,
with preferred
amounts including about 250, 300, 350, 400, 450, 500, 550, 600, 650, 700, 750,
800, 850,
900, 950, or 1000 mg of fusidic acid, or a value within this range.
Preferably, the final
concentration of fusidic acid in the intravenous infusion is about 1, 2, 3, 4,
5, 6, 7, 8, 9, 10,
15, 20, 25, 30, 35 or 40 mg/ml or more. Suitable periods of time over which
the fusidic
acid-containing intravenous infusion may be administered include 15, 30, 45 or
60
minutes, or 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5 or 5 or more hours.
[00106] In intramuscular preparations, a sterile formulation of the
pharmaceutical
composition of the present invention can be dissolved and administered in a
pharmaceutical diluent such as Water-for-Injection (WFI), physiological saline
or 5%
dextrose in water. A suitable insoluble form of the pharmaceutical
compositions may be
prepared and administered as a suspension in an aqueous base or a
pharmaceutically
acceptable oil base, e.g. an ester of a long chain fatty acid such as ethyl
oleate. Suitable
amounts of fusidic acid to be administered in a formulation for injection
range from about
to about 4000 mg, with preferred amounts including about 10, 25, 50, 100, 150,
200,
250, 300, 350, 400, 450, 500, 550, 600, 650, 700, 750, 800, 850, 900, 950, or
1000 mg of
fusidic acid, or a value within this range.
[00107] For oral use, the oral pharmaceutical composition may be made in the
form of a
unit dosage containing a therapeutically-effective amount of the
pharmaceutical
composition. Solid formulations such as tablets and capsules are particularly
useful.
Sustained released or enterically coated preparations may also be devised. For
pediatric
and geriatric applications, suspension, syrups, slow release and chewable
tablets are
especially suitable. For oral administration, the pharmaceutical compositions
are in the
29
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
form of, for example, tablets, capsules, suspensions or liquid syrups or
elixirs, wafers and
the like. For general oral administration, excipient or additives include, but
are not limited
to inert diluents, fillers, disintegrating agents, binding agents, wetting
agents, lubricating
agents, sweetening agents, flavoring agents, coloring agents and
preservatives.
[00108] For therapeutic purposes, the tablets and capsules can contain, in
addition to
fusidic acid, inert diluents (e.g., sodium and calcium carbonate, sodium and
calcium
phosphate, and lactose), binding agents (e.g., acacia gum, starch, gelatin,
sucrose,
polyvinylpyrrolidone (Povidone), sorbitol, tragacanth methylcellulose, sodium
carboxymethylcellulose, hydroxypropyl methylcellulose, and ethylcellulose),
fillers (e.g.,
calcium phosphate, glycinc, lactose, maize-starch, sorbitol, or sucrose),
wetting agents,
lubricating agents (e.g., metallic stearates, stearic acid, polyethylene
glycol, waxes, oils,
silica and colloical silica, silicon fluid or talc), disintegrating agents
(e.g., potato starch,
corn starch and alginic acid), flavoring (e.g. peppermint, oil of wintergreen,
fruit flavoring,
cherry, grape, bubblegum, and the like), and coloring agents. The tablets and
capsules may
also include coating excipients such as glyceryl monostearate or glyceryl
distearate, to
delay absorption in the gastrointestinal tract.
[00109] In a particular oral formulation, the pharmaceutical compositions of
the present
invention may be in the form of a tablet containing microcrystalline
cellulose,
crospovidone, hypromellose, lactose monohydrate, magnesium stearate, silica,
all-rac-a-
tocopherol, talc and titanium dioxide, and optionally one or more other
inactive
ingredients. Suitable amounts of fusidic acid in a tablet may range from about
10 to about
4000 mg, with preferred amounts including about 250, 300, 350, 400, 450, 500,
550, 600,
650, 700, 750, 800, 850, 900, 950, or 1000 mg of fusidic acid per tablet, or a
value within
this range.
[00110] Oral liquid preparations, generally in the form of aqueous or oily
solutions,
suspensions, emulsions or elixirs, may contain conventional additives such as
suspending
agents, emulsifying agents, non-aqueous agents, preservatives, coloring agents
and
flavoring agents. Examples of additives for liquid preparations include
acacia, almond oil,
ethyl alcohol, fractionated coconut oil, gelatin, glucose syrup, glycerin,
hydrogenated
edible fats, lecithin, methyl cellulose, microcrystalline cellulose, methyl or
propyl para-
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
hydroxybenzoate, propylene glycol, sorbitol, or sorbic acid. In a particular
oral
formulation, the pharmaceutical composition comprises fusidic acid and the
following
inactive ingredients: acesulfame potassium, flavor, citric acid, disodium
phosphate
dihydrate, hydroxyethylcellulose, glucose liquid, methylcellulose, sodium
benzoate,
sorbitol, and purified water. Suitable amounts of fusidic acid in an oral
formulation may
range from about 10 to about 4000 mg, with preferred amounts including about
250, 300,
350, 400, 450, 500, 550, 600, 650, 700, 750, 800, 850, 900, 950, or 1000 mg of
fusidic acid
in the oral formulation, or a value within this range.
[00111] For topical use, the pharmaceutical compositions of present invention
can also
be prepared in suitable forms to be applied to the skin, or mucus membranes of
the nose
and throat, and can take the form of creams, ointments, nasal drops, liquid
sprays or
inhalants, lozenges, or throat paints. Such formulations further can include
chemical
compounds such as dimethylsulfoxide (DMSO) to facilitate surface penetration
of the
active ingredient. For application to the eyes or ears, the pharmaceutical
compositions can
be presented in liquid or semi-liquid form formulated in hydrophobic or
hydrophilic bases
as ointments, creams, lotions, paints or powders. For rectal administration
the
pharmaceutical compositions can be administered in the form of suppositories
admixed
with conventional carriers such as cocoa butter, wax or other glyceride. In
particular
embodiment, a cream may be prepared comprising fusidic acid and the following
inactive
ingredients: steareth-21, cetostearyl alcohol, white soft paraffin, liquid
paraffin,
hypromellose, citric acid monohydrate, methyl parahydroxybenzoate, propyl
parahydroxybenzoate, potassium sorbate, and purified water, or the following
inactive
ingredients: butylated hydroxyanisole, cetanol, glycerol, liquid paraffin,
potassium sorbate,
Tween 60, white soft paraffin, and purified water. In another particular
formulation, an eye
drop may be prepared comprising fusidic acid and the following inactive
ingredients:
benzalkonium chloride, disodium edetate, mannitol, carbomer, sodium hydroxide,
and
water. Suitable amounts of fusidic acid in an eye drop formulation may range
from about
1 to about 100 mg, with preferred amounts including about 5, 10, 15, 20, 25,
30, 35, 40,
45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, or 100 mg of fusidic acid in the
formulation, or a
value within this range.
31
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
[00112] For pulmonary administration, the pharmaceutical compositions of
present
invention can be prepared in suitable forms for inhalation (an inhalable
formulation; for
nasal or buccal inhalation). The compounds may be conveniently delivered in
the form of
an aerosol spray presentation from pressurized packs or nebulisers. The
compounds may
also be delivered as powders which may be formulated and the powder
composition may
be inhaled with the aid of an insufflation powder inhaler device. The
preferred delivery
system for inhalation is a metered dose inhalation (MDI) aerosol, which may be
formulated as a suspension or solution of a compound of this invention in
suitable
propellants, such as fluorocarbons or hydrocarbons.
[00113] As used herein, the terms "dose", "dosage", "unit dose", "unit
dosage",
"effective dose" and related terms refer to physically discrete units that
contain a
predetermined quantity of active ingredient calculated to produce a desired
therapeutic
effect. These terms are synonymous with the therapeutically effective amounts
and
amounts sufficient to achieve the stated goals of the methods disclosed
herein. The skilled
artisan will understand that the term "maintenance dose" can refer to single
discrete dose,
as well as more than one discrete dose. The maintenance dose is the total
amount of FA
administered during the maintenance dose phase. Similarly, the term "loading
dose" can
refer to single discrete dose, as well as more than one discrete dose. The
loading dose is
the total amount of FA administered during the loading dose phase.
[00114] As used herein, the terms "treating" and "treatment" have their
ordinary and
customary meanings, and include one or more of, ameliorating a symptom of a
bacterial
infection in a subject, blocking or ameliorating a recurrence of a symptom of
a bacterial
infection in a subject, decreasing in severity and/or frequency a symptom of a
bacterial
infection in a subject, stasis, decreasing, or inhibiting growth of bacteria
in a subject,
killing bacteria in a subject, inhibiting bacterial sporulation, inhibiting
activation of a
bacterial spore in a subject, inhibiting germination of a bacterial spore in a
subject, and
inhibiting outgrowth of a bacterial spore in a subject. Treatment means
ameliorating,
blocking, reducing, decreasing or inhibiting by about 1% to about 100% versus
a subject to
which a pharmaceutical composition has not been administered. Preferably, the
ameliorating, blocking, reducing, decreasing or inhibiting is about 100%, 99%,
98%, 97%,
32
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
96%, 95%, 90%, 80%, 70%, 60%, 50%, 40%, 30%, 20%, 10%, 5% or 1% versus a
subject
to which a pharmaceutical composition has not been administered.
[00115] As used herein, the terms "preventing" and "prevention" have their
ordinary
and customary meanings, and include one or more of preventing bacterial
colonization in a
subject, preventing an increase in the growth of a bacterial population in a
subject,
preventing activation, germination or outgrowth of bacterial spores in a
subject, preventing
bacterial sporulation in a subject, preventing development of a disease caused
by a
bacterial infection in a subject, and preventing symptoms of a disease caused
by a bacterial
infection in a subject. As used herein, the prevention lasts at least about
0.5, 1, 2, 3, 4, 5,
6, 7, 8, 9, 10, 12, 14, 16, 18, 20, 25, 30, 35, 40, 45, 50 or more days after
administration of
a pharmaceutical composition.
EXAMPLES
Example 1 - Study of the Spectrum of CEM-102 (Fusidic Acid) Against
Contemporary Wildtype (WT) Bacterial Species Including Mutational Resistance
(R)
Analysis, and Synergy Testing
[00116] Methods: A collection of 114 WT isolates (>80 species) was used to
define the
contemporary limits of CEM-102 (fusidic acid; FA) spectrum against Gram-
positive (GP)
and -negative (GN) species. CLSI broth microdilution (BMD) and anaerobic agar
dilution
(AD) methods were performed. Modifications of standard test methods included
adding
10% human scrum, adjusting the medium pH to 5, 6, and 8, and synergy was
assessed by
the checkerboard method. Mutational rates to R were determined at 4X, 8X and
16X MIC.
[00117] Results: Against GP, FA MIC values ranged from 0.06-32 pg/m1 with
greatest
potency against S. aureus, Corynebacterium spp. and M luteus (MIC results
0.25, <0.12
and <0.5 !..ig/ml, respectively). Enterococci and streptococci were less
susceptible (S; MIC
ranges of 2-8 and 16-32 pg/ml, respectively).
[00118] When tested against 217 S. aureus Canadian isolates, the MIC50 for CEM-
102
was 0.12 gg/mL and the MIC90was 0.25 ,Li.g/mL, which is only slightly higher
than strains
in the US (MIC90 0.12 i.tg/mL). The MIC population distribution study showed
that only
6.5% of the Canadian S. aureus isolates had MICs of 2..0 g/mL and would be
classified
33
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
as resistant. By comparison, ciprofloxacin, clindamycin, and erythromycin
resistance rates
were 41.5, 30.9, and 52.1% respectively). This is based upon established
breakpoints of
greater than or equal to 2 ug/ml (more strains are expected to be classified
as susceptible in
view of the dosing regimes disclosed in the present application due to the
much higher
blood levels of FA; many resistant strains, with MICs of 4-32 ug/ml, are
expected to be
susceptible under the new dosing regimens).
[00119] FA activity against GN species was limited (all MIC values >2 ilg/m1)
except
for E. brevis, M. catarrhalis and N. meningitidis (MICs, 0.12-0.5 g/m1).
[00120] A 4-fold increase in FA MIC results was observed when 10% serum was
added.
[00121] Decreasing medium pH to 5.0-6.0 negated the protein binding effects.
[00122] Among the 8 combinations of drugs tested, gentamicin (GEN) and
rifampin
(RIF) showed the greatest enhanced activity combined with FA (No antagonism;
Table 1).
Single-step mutational rates ranged from 1.2 x 10-6 for 4X MIC to 9.8 x 10-8
for 16X MIC.
TABLE 1
Synergy
FA/co-drug Complete Partial Additive Indifferent Antagonism Indeterminate
Rifampin 0 5 1 0 0 0
Levofloxacin 0 0 0 4 0 2
Gentamicm 1 1 3 1 0 0
Oxacillin 0 1 1 3 0 1
Vancomycin 0 0 2 4 0 0
All agents tested 1 7 9 24 0 7
[00123] Conclusions: FA demonstrated potent GP activity, especially against
the
staphylococci. A more limited activity was observed against GN species
isolates. Added
serum proteins adversely influenced MIC values; however lower media pH like
seen at
infection sites decreased negative protein binding effects. FA in vitro
activity was most
improved when combined with RIF.
Example 2 - Antimicrobial Activity of CEM-102 (Fusidic Acid) Against Canadian
Isolates of Staphylococci and Streptococci.
34
CA 02767614 2012-01-09
WO 2011/008193
PCT/US2009/050353
[00124] Methods: To determine a contemporary susceptibility (S) spectrum
pattern,
153 GP isolates (123 S. aureus, 15 coagulase-negative staphylococci [CoNS] and
15 S.
pyogenes [SPYO]) were collected from 5 Canadian medical centers between 2001
and
2006. Reference broth microdilution (BMD) S testing was performed by CLSI M07-
A8,
2009 methods for FA and 13 comparator antimicrobials.
[00125] Results: FA MIC results for S. aureus had MIC50 and MIC90 values of
0.12
[tg/mL for the 2001-2002 and 2003-2004 time periods, however, for 2005-2006
the MIC90
increased to > 2 pg/m1 (Table 2). Applying an international breakpoint from
literature
reviews at < 0.5 pg/m1 (S) and > 2 pg/m1 (R), the S. aureus isolates showed a
small
increase in the R rate over time (5.0-12.2%), now confirmed by 2007-2008
results. The
overall S. aureus population had a MIC90 of 0.25 jig/ml and R rate of 8.1%.
Some
comparator agents showed higher R rates that remained stable over the period
tested with
highest R noted for erythromycin (ERY, 52.0%), ciprofloxacin (43.9%), and
clindamycin
(CLI, 28.5%). CoNS isolates had FA MIC50 and MIC90 values at 0.12 and 16
tg/ml,
respectively. SPYO isolates were only moderately S to FA with all values at 4
or 8 [tg/ml.
Among the comparator agents, ERY had an R rate of 20.0% and CLI at 13.3% for
SPYO.
TABLE 2
No. inhibited at MIC (i.tgInaL) of:
S. aureus (years tested) <0.03 0.06 0.12 0.25 0.5 1
>2 % at <0.5'
2001-2002 29 95.0
2003-2004 6 33 3 92.9
2005-2006 2 32 2 5 87.8
a6.0% R for 2007-2008
[00126] Conclusions: FA demonstrated potent activity against Canadian
staphylococci
isolates with a low overall R rate (8.1%) among S. aureus. CoNS isolates had a
greater R
rate than S. aureus. FA had a narrow range of MIC results (4-8 tg/m1) and was
less active
against SPYO.
Example 3 - In Vitro Activity of CEM-102 (Fusidic acid) Against Resistant
Strains of
Staphylococcus aureus
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
[00127] The activity of CEM-102 (fusidic acid) against a variety of resistant
strains of
Staphylococcus aureus was investigated.
[00128] Methods: The in vitro activity of CEM-102 was compared with that of
telithromycin, azithromycin, erythromycin, levofloxacin, linezolid, and
doxycycline
against a total of 145 resistant S. aureus by agar dilution procedures (CLSI,
M7-A7, M100-
S18). The tested strains included S. aureus MRSA (Mec A genotype; 100
isolates),
macrolide-resistant (ermA, B, C genotype or MLSb-resistant; 25), and
ciprofloxacin-
resistant (gyrA and parC genotype; 20).
[00129] Results: Against S. aureus MRSA (MecA), CEM-102 (MIC90 0.25 mg/L) and
telithromycin (MIC90 0.06 mg/L) were more active than doxycycline (MIC90 1
mg/L),
linezolid (MIC90 2 mg/L), levofloxacin (MIC90 16 mg/L), azithromycin (MIC90
>32 mg/L)
and erythromycin (MIC90 >32 mg/L). CEM-102 (MIC90 0.25 mg/L) was significantly
superior to linezolid (MIC90 2 mg/L), levofloxacin (MIC90 4 mg/L),
telithromycin (MIC90
4 mg/L), azithromycin (MIC90 >32 mg/L), and erythromycin (MIC90 >32 mg/L)
against
macrolide-resistant S. aureus (ertnA, B, C genotype or MLSb-resistant).
Against
ciprofloxacin-resistant (gyrA and parC genotype) S. aureus, erythromycin
(MIC90 >32
mg/L), levofloxacin (MIC90 >32 mg/L), azithromycin (MIC90 16 mg/L), linezolid
(MIC90 2
mg/L), and doxycycline (MIC90 1 mg/L) were less active than CEM-102 (MIC90
0.25
mg/L) and telithromycin (MIC90 0.06 mg/L).
[00130] Conclusions: These data confirm the activity of CEM-102 against
resistant S.
aureus and show the promise of this unique antibiotic that has no cross
resistance with
other classes of antibiotics.
Example 4 ¨ Single oral dose bioavailability study of CEM-102 (sodium
fusidate)
[00131] The primary objective of the study was to evaluate the relative
bioavailability
of single oral doses of CEM-102 (sodium fusidate) 500 mg (2 x 250 mg) tablets
and
Fucidin0 (sodium fusidate) 500 mg (2 x 250 mg) tablets in healthy subjects in
a fed or
fasted state.
[00132] This was a single-center, Phase 1, double-blind, randomized, 3 period,
fed,/fasted crossover bioequivalence study designed to evaluate the relative
bioavailability
36
CA 02767614 2012-01-09
WO 2011/008193
PCT/US2009/050353
and safety and tolerability of a single oral dose of CEM-102 500 mg compared
to Fucidin
500 mg in healthy subjects.
[00133] Subjects were randomized in equal numbers to the 4 treatment sequences
(Table 3).
Table 3 Treatment Sequences
Treatment Subjects
Sequence Randomized Study Period 1 Study Period 2 Study
Period 3
1 7 Fucidin 500 mg CEM-102 500 mg Fucidin
500 mg
(2 x 250 mg) (2 x 250 mg) (2 x 250 mg)
Fasting Fasting Fed
2 7 Fucidin 500 mg CEM-102 500mg CEM-102 500
mg
(2 x 250 mg) (2 x 250 mg) (2 x 250 mg)
Fasting Fasting Fed
3 7 CEM-102 500 mg Fucidin 500 mg Fucidin
500 mg
(2 x 250 mg) (2 x 250 mg) (2 x 250 mg)
Fasting Fasting Fed
4 7 CEM-102 500 mg Fucidin 500 mg CEM-102
500 mg
(2 x 250 mg) (2 x 250 mg) (2 x 250 mg)
Fasting Fasting Fed
[00134] Study Period 1, 2, and 3 (Days 1, 15, and 29): One dose of study drug
500 mg
(2 x 250 mg) of CEM-102 or Fucidin was given at 8:00 AM (-30 minutes). On Day
1 of
Study Periods 1 and 2 (Days 1 and 15), subjects fasted overnight for at least
10 hours prior
to dosing. On Day 1 of Study Period 3 (Day 29), subjects were given a high fat
caloric
meal which was to be entirely consumed within 30 minutes prior to study drug
administration.
[00135] Blood samples for assay of plasma concentrations were collected on
Days 1, 15
and 29 at pre-dose and 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 6, 8, 12, 16, 24, 36,
and 48 hours after
the dose. Plasma concentrations were assayed at MicroConstants, Inc. in San
Diego, CA
using a validated liquid chromatography with dual tandem mass spectrometry
(LC,/MS/MS) method with a lower limit of quantitation (LLOQ) of 20.0 ng/mL
[00136] Mean plasma concentrations of fusidic acid are depicted in Figures 1
and 2 on
semi-log and linear scales. The median (minimum-maximum) trnax values for
fusidic acid
37
CA 02767614 2012-01-09
WO 2011/008193
PCT/US2009/050353
for the CEM-102 and Fucidin products under fasting conditions were 3.00 hours
(1.00 to
4.00 hours) and 2.52 hours (1.00 to 6.00 hours), respectively.
[00137] Descriptive statistics of pharmacokinetic results for fusidic acid in
plasma are
presented in Table 4.
Table 4 Descriptive
Statistics for the Assessment of Food-effect and Fed
Pharmacokinetics
CEM-102 CEM-102 Fuci . .
ding
Fed Fasting Fed
Parameter (C) (A) (D)
Geometric Mean (CV%) AUC04 (ng=h/mL) 268561
(31.5) 300352 (30.9) 264713 (40.5)
AUCinf (ng-h/mL) 285056 (32.7) 318391 (32.0) 280680
(40.5)
Cmax(flg/mL) 21175 (25.2) 27413 (23.8) 21541
(34.0)
Arithmetic Mean SD t% (h) 11.5 2.75 11.8 2.20 11.5 2.62
Median (Min-Max) 3.50 3.00 4.00
tmax (h)
(1.50- 6.00) (1.00- 4.00) (1.00-
8.25)
[00138] Food appeared to decrease the Cmax of CEM-102 by approximately 23%.
However, the total exposure (AUC), the time to peak plasma concentrations and
the half-
life were comparable when the CEM-102 500 mg dose was administered under fed
and
fasting conditions (approximately 285 vs. 318 [tg=h/mL, 3.50 vs. 3.00 hours
and 11.5 vs.
11.8 hours, respectively)
Example 5 - Multi-dose bioavailability study using FA at 500 mg TID
[00139] The pharmacokinctics of multiple oral doses of CEM-102 (sodium
fusidatc)
500 mg tablets in healthy subjects was evaluated. Subjects were randomized in
a 3:1 ratio
and received either CEM-102 500 mg (2 x 250 mg; n = 18) or placebo tablets TID
(n = 6)
for 4.5 days for a total of 13 doses.
[00140] Blood samples for assay of plasma concentrations of CEM-102 and
placebo
were collected pre-dose on Day 1 and at 24, 48, 72, and 96 hours. Samples were
also
collected at the following times after the morning dose on Day 5: 0.5, 1, 1.5,
2, 2.5, 3, 3.5,
4, 6, 8, 12, 16, 24, 36, and 48 hours.
38
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
[00141] Plasma concentrations of CEM-102 were assayed at MicroConstants, Inc.
in
San Diego, CA, USA using a validated LC/MS/MS method with a lower limit of
quantitation (LLOQ) of 20.0 ng/mL.
[00142] Mean plasma concentrations of CEM-102 on Day 5 (13 doses) are depicted
on
semi-log (Figure 3) and linear scales (Figure 4).
[00143] Descriptive statistics of pharmacokinetic results for CEM-102 in
plasma are
presented in Table 5.
Table 5
CEM-102 in Plasma
Parameter (Day 5)
Geometric Mean (CV%) AUCO-tss (ng.h/mL) 3562344 (34.7)
AUCO-Tss (ng-h/mL) 1030827 (26.3)
Cmaxss (ng/mL) 145680 (25.3)
Cmin (ng/mL) 106600 (25.5)
Arithmetic Mean SD t1/2ss (h) 18.7 5.30
Median (Min-Max) tmaxss (h) 3.00 (0.50 ¨ 6.00)
[00144] On Day 5, the PK profile of CEM-102 in plasma following repeated oral
doses
of 500 mg T1D was well characterized. Mean plasma concentrations of CEM-102
remained above the LLOQ (20.0 ng/mL) throughout the sampling schedule. Mean
CEM-
102 concentrations rose rapidly and peaked at approximately 3 hours (range 0.5-
6 hours)
post-dose and then declined slowly with an apparent half-life of approximately
19 hours.
Maximum plasma concentrations ranged between 87.6 and 245 ug/mL. Trough levels
rose
steadily from Days 1 through 5 attaining 105 ug/mL prior to dosing on Day 5
and 120
ug/mL at 8 hours post-dose. This continuing rise in mean trough concentrations
following
the last dose may indicate that steady-state was not yet reached by Day 5.
[00145] The pharmacokinetic profile of CEM-102 in plasma following multiple
oral
doses of 500 mg administered TID for 13 doses over 4.5 consecutive days was
well
characterized. However, the continuing rise in mean trough concentrations
following the
last dose may indicate that steady-state was not yet reached by Day 5.
39
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
[00146] The results of this study supported the published reports that a
steady state high
level of FA is not reached until after the 4th day using the standard dosing
regimen of 500
mg TID.
Example 6 ¨ Dose Escalation Study Using Single- and Multi-Dose FA
[00147] The pharmacokinetics (PK), safety, and tolerability of single and
multiple doses
of CEM-102 (sodium fusidate) were evaluated in a single-center, Phase 1,
double-blind,
randomized, placebo-controlled, dose-escalating study in 32 healthy adult
subjects enrolled
in 4 dosage groups (550, 1100, 1650, and 2200 mg). In each cohort, six
subjects were to
receive CEM-102 and two were to receive placebo in a single dose (Period 1)
and then
multiple doses BID for a total of 11 doses over 5.5 days (Period 2). The two
dosing
periods were separated by a seven-day washout period between the single dose
in Period 1
and the first dose in Period 2.
[00148] Dosing proceeded as planned for Cohorts 1 to 3; however, Cohort 4
received
only the single dose of 2200 mg (Period 1), because dose-limiting
gastrointestinal
intolerance was observed after multiple doses of 1650 mg BID in Cohort 3.
Period 2 for
Cohort 4 evaluated an initial loading dose regimen, and a second loading dose
regimen was
evaluated in an additional cohort (Cohort 5) per a protocol amendment.
[00149] CEM-102 was considered safe and generally well tolerated. No serious
or life-
threatening adverse events (AEs) occurred. Seventy-four AEs were reported in
19 of the
32 study subjects, all of which were mild or moderate in severity. Of the 70
treatment-
emergent AEs reported all except 7 were in Period 2, in which subjects
received multiple
daily doses over 5.5 days. Most of the AEs were considered possibly related to
study drug.
[00150] Single doses of CEM-102 up to 2200 mg were well tolerated. The only
AEs
reported in the 550 mg and 1100 mg cohorts were in subjects on placebo and no
AEs were
reported in the 1650 mg cohort. As described in product information documents
from
countries in which sodium fusidate is approved and from published reports,
gastrointestinal
AEs are the most commonly reported AEs associated with administration of oral
sodium
fusidate. In the present study, no gastrointestinal AEs were reported in the
550, 1100, or
1650 mg cohorts (Table 6). Nausea and vomiting occurred in 1 subject after the
single
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
dose of 2200 mg and nausea alone occurred in 2 others; therefore,
gastrointestinal
intolerance appeared to be dose-related with a threshold between 1650 and 2200
mg.
There were no clinically significant changes in physical examinations, vital
signs,
electrocardiograms (ECGs), or laboratory parameters after single doses of CEM-
102.
[00151] As with single doses, the most common AE after multiple doses of CEM-
102
was nausea, reported in 9 subjects (550 mg, 2 subjects; 1100 mg, 2 subjects;
1650 mg, 5
subjects) (Table 7). Nausea occurred most frequently after 3 to 5 days of
dosing with 1650
mg BID. No pharmacologic treatment was required for most of the AEs.
[00152] Four of the subjects at the 1650 mg multiple dose level also
experienced
vomiting (Grade 1 in 3 subjects and Grade 2 in 1 subject). Although the
vomiting was
considered mild in 3 of the 4 subjects, the decision was made to limit
evaluation of
multiple doses to the 1650 mg dose level. Consequently, escalation to the 2200
mg dose in
Cohort 4 involved only Period 1 with a single dose of study drug.
41
0
N
Table 6
CEM-102 Single-Dose Treatment-emergent Adverse Events =
Cohort/ Subject Gender Severity
Treatment # Age (Y)
-C"
Day Adverse Event Frequency
Grade- Relationship Treatment Outcome c
cc
1--,
550 mg 12 M/48 1 BP elevation x1 1
Possibly None Recovered w
1100 mg 37 F/46 3 Headache Constant 1
Possibly None Recovered
1650 mg - - None - -
- - -
2200 mg 67 F/54 2 Abdomen distention Constant 1
Unlikely None Recovered
80 F/21 1 Nausea x1 1
Probably None Recovered
1 Vomiting x1 1
Probably None Recovered
81 F/54 1 Nausea x1 1
Probably None Recovered
a
85 M/51 1 Nausea x1 1
Probably None Recovered
0
IV
-.1
01
--.1
01
I-I
N
IV
0
I-.
IV
I
0
I-.
I
0
l0
JI
*I:
n
1-
--C
cA
w
=
=
---=
=
f...)
un
f...)
o
Table 7 CEM-102 Multiple-Dose Treatment-Emergent
Adverse Events (Blinded) IJ
C
I--,
Cohort/ Subject Gender Severity
1¨
Day Adverse Event Frequency
Relationship Treatment Outcome
Treatment # Age (Y) Grade
c
oe
550 mg 01 F/48 2 Hoarseness Constant 1
Unrelated None Recovered 1--.
3 Sore throat Constant 1
Unrelated None Recovered w
02 F/31 1 Nausea x1 1
Possibly None Recovered
2 Nausea x1 1
Possibly None Recovered
3 Nausea x1 1
Possibly None Recovered
6 Nausea xl 1
Possibly None Recovered
7 Epigastric pain x1 1
Possibly None Recovered
17 M/21 5 Dizziness Constant 1
Possibly None Recovered
Elevated BP Intermittent 1 Possibly None
Recovered a
5 Headache Constant 1
Possibly None Recovered 0
5 Tachycardia Intermittent 1
Possibly None Recovered n,
-.1
5 Pharyngitis Constant 1
Unrelated None Recovered 01
--.1
.6. 5 Nausea x1 1
Possibly None Recovered 0,
1-.
1100 mg 24 F/33 1 Heartburn Constant 1
Possibly None Recovered n)
3 Constipation Constant 1
Possibly None Recovered 0
H.
28 M/32 7 Skin Irritation Constant 1
Unlikely None Recovered n)
1
0
31 F/50 5 Constipation Constant 1
Possibly None Recovered
I
6 Heartburn Constant 1
Possibly None Recovered 0
QD
38 F/34 1 Nausea x1 1
Unlikely None Recovered
5 Nausea Constant 1
Possibly None Recovered
6 Nausea Constant 1
Possibly None Recovered
6 Dizziness Constant 1
Possibly None Recovered
39 M/38 1 Viral URI Constant 1
Unlikely None Recovered
1 Dizziness x1 1
Possibly None Recovered
1 Nausea x1 1
Possibly None Recovered *I:
n
1650 mg 44 F/49 1 Heartburn Constant 1
Possibly Ranitidine Recovered 1-3
C.
2 Constipation Constant 1
Unlikely None Recovered cn
3 Headache Transient 1
Possibly None Recovered n.)
c
c
3 Nausea x1 1
Possibly None Recovered
4 Nausea Constant 1
Possibly None Recovered --C-
cA
c
4 Headache Constant 1
Possibly None Recovered w
cA
5 Dizziness Constant 1
Possibly None Recovered w
Cohort/ Subject Gender Severity
0
Day Adverse Event Frequency
Relationship Treatment Outcome
Treatment # Age (Y) Grade
IJ
C
I--I
Vomiting x1 1
Possibly None Recovered
6 Dysuria Intermittent 1
Unrelated None Recovered -C-
c
oe
6 Nausea Constant 2
Possibly None Recovered 1--,
6 Vomiting xl 2
Possibly IV Fluids Recovered w
47 M/30 1 Nausea x1 1
Possibly None Recovered
50 M/43 3 Constipation Constant 1
Unlikely None Recovered
3 Nausea Constant 1
Possibly None Recovered
4 Nausea Constant 1
Possibly None Recovered
6 Heartburn Constant 1
Possibly Ranitidine Recovered
6 Dizziness Constant 1
Possibly None Recovered
6 Nausea Constant 1
Possibly None Recovered a
6 Vomiting x1 1
Possibly None Recovered
0
59 F/31 3 Paresthesia Constant 1
Possibly None Recovered n,
-.1
4 Constipation Constant 1
Unlikely None Recovered 0,
--.1
5 Vomiting xl 1
Possibly None Recovered 0,
.6.
1-
.6, 60 M/46 3 Dizziness Constant 1
Possibly None Recovered
3 Headache Constant 1
Possibly None Recovered n)
0
3 Loose stools x1 1
Possibly None Recovered
IV
I
3 Nausea Constant 1
Possibly None Recovered 0
I-.
I 3 Paresthesia
Transient 1 Possibly None Recovered 0
3 Weakness Constant 1
Possibly None Recovered QD
4 Headache Constant 1
Possibly None Recovered
5 Nausea Constant 1
Possibly None Recovered
5 Paresthesia Constant 1
Possibly None Recovered
5 Vomiting x1 1
Possibly None Recovered
6 Loose stools Multiple 1
Possibly None Recovered
6 Nausea Constant 1
Possibly None Recovered
6 Paresthesia Constant 1
Possibly None Recovered n
1-
6 Vomiting X1 1
Possibly None Recovered C.
7 Nausea Constant 1
Possibly None Recovered cn
n.)
7 Paresthesia Constant 1
Possibly None Recovered c
c
--C-
cA
c
w
cA
w
CA 02767614 2012-01-09
WO 2011/008193
PCT/US2009/050353
[00153] PK results from single and multiple dose administration of CEM-102 are
shown
in Table 8 and Figure 5.
Table 8 CEM-102 Single and Multiple Dose Pharmacokinetic Parameters
Single and Multiple Dose Groups Single Dose Only
Cohort 1 Cohort 2 Cohort 3 Cohort 4
550 mg 1100 mg 1650 mg 2200 mg
Parameter Mean SD Mean SD Mean SD Mean SD
Period 1 Day 1
C., Pgirril- 33.4 12.2 72.2 10.8 102 25.8 128 28.3
Tax, ha 2.00 (2-3) 3.50 (1-4) 3.00 (2-4)
6.00 (3-8)
Kel, h 0.0564 0.0564 0.0130 0.0617 0.0120 0.0498
0.0151 0.0500 0.0159
T112, hc 12.3 2.98 11.2 2.17 13.9 4.29 13.9
4.52
AUC(o_24), pg-h/mL 242 102 844 115 1,260 386 1,690
427
AUC(04,0, pg=h/mL 441 269 1,100 247 1,800 689 2,650
978
CL/F, L/h 1.69 1.00 1.04 0.202 1.07 0.519 0.924
0.434
Vd/F, L 28.4 10.8 16.9 1.20 21.1 4.96 19.2
6.38
Period 2 Day 6
C., pg/mL 130 30.5 281 52.5 324 26.8
T., ha 3.00 (1.5-4) 4.00 (4-8) 4.00 (1.5-6)
Kel, h 0.0554 0.0131 0.0404 0.0162 0.0199 0.0118
T112, hc 12.5 3.05 17.1 6.79 31.6 18.9
AUC(0_12), pg=h/mL 1,150 433 2,530 417 3290 146
CL/F, L/h 0.553 0.255 0.449 0.100 0.0503 0.0223
Vd/F, L 9.88 3.05 12.7 5.66 34.8 22.9
C. accumulation ratio d 3.89 3.89 3.18
AUC(0_12) accumulation
2.61 2.30 1.83
ratio e
a Expressed as median and range
Apparent first-order terminal elimination rate constant
Expressed as harmonic mean and pseudo SD
Mean Cm. Day 6/Mean C. Day 1
Mean AUC(0_12) Day 6/Mean AUC(o-inf) Day 1
[00154] As shown in Figure 5, the increases in Cnia), and AUC appear to be
more than
dose proportional from the 550 mg to the 1100 mg dose, but then approximately
dose
proportional from the 1100 mg to the 2200 mg dose (only single doses). In the
550 to
1650 mg dose groups CEM-102 demonstrated higher PK exposures after 5.5 days of
dosing in Period 2 compared to a single dose on Day 1 in Period 1, indicating
that
accumulation occurs over the dosing period.
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
[00155] The protocol was amended for assessment of the safety, tolerability,
and PK of
front-loaded dose regimens comprised of a higher dose of CEM-102 on Day 1
followed by
lower doses for the subsequent 6.5 days. All loading dose regimens employed
multiple
doses lower than the maximum tolerated dose (MTD) established in the first
part of the
study (1650 mg administered as multiple doses).
[00156] The loading dose regimens assessed were 1100 mg BID on Day 1 and 550
mg
BID on the following 6.5 days in Period 2 of Cohort 4 and 1650 mg BID on Day 1
and 825
mg BID on the following 6.5 days in Cohort 5.
[00157] Four AEs, all mild in severity, were reported by 3 subjects that
received loading
dose regimens (Table 9). One subject in the 1100/550 mg group reported nausea
on the
first day of dosing only. No gastrointestinal AEs were reported in the
1650/825 mg group.
Table 9 GEM-102 Loading-Dose Regimens Treatment-emergent Adverse
Events
Dose Treatment Subj S MJF Adverse Sev
Group Group Age (Y) Day Event Freq. Grade
Relation Treatment Outcome
1100/550 mg CEM-102 080 F/21 1 Urinary Constant 1
Possibly None Recovered
frequency
1 Nausea Constant 1 Possibly
None Recovered
CEM-102 081 F/54 2 Constipation Constant 1
Unlikely Fruit juice Recovered
1650/825 mg CEM-102 087 F/24 6 Redness to Constant
1 Unrelated None Recovered
face
[00158] There were no clinically significant changes in physical examinations,
vital
signs, ECGs, or laboratory parameters after either loading dose regimen.
[00159] PK results of the loading dose regimens are shown in Figure 6. The
1100 mg
BID loading dose followed by 550 mg BID for 6.5 days resulted in a mean trough
plasma
concentration of approximately 74 i_tg/mL at 24 hours and approximately 105
iug/mL after
7 days of dosing. The 1650 mg BID loading dose followed by 825 mg BID for 6.5
days
resulted in a mean trough plasma concentration of approximately 150 iug/mL at
24 hours
and approximately 200 g/mL after 7 days of dosing. Figure 7 is a compilation
of the
period 2 portion of Figure 5 and the 1100/550 mg BID loading dose regimen
data. Figure
7 demonstrates that, in contrast with the conventional BID regimens, near
steady state
plasma concentrations are reached at 24 hours after the first dose on Day 1
and these
46
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
plasma concentrations are maintained at or above this level throughout the
remainder of
the 7 day treatment course.
[00160] The results demonstrated that CEM-102 was safe and well tolerated in
single
doses up to 2200 mg, with a threshold for symptoms of gastrointestinal
intolerance
between 1650 mg and 2200 mg. CEM-102 administered in doses up to 1650 mg BID
for
5.5 days was safe and generally well tolerated; however, as 4 subjects had
vomiting at the
1650 mg dose level, escalation to the 2200 mg dose level was limited to single
doses. AEs
appeared to be related to dose and duration of exposure, with gastrointestinal
symptoms
reported more frequently after 3-5 days of dosing and at higher doses. Loading
dose
regimens of CEM-102 1100/550 mg and 1650/825 mg for a total of 7.5 days were
very
well tolerated. Loading dose regimens appeared to be generally better
tolerated than the
conventional BID dosing regimens for similar levels of overall plasma
exposure. There
were no clinically significant changes in physical examinations, vital signs,
ECGs, or
laboratory parameters after single doses of CEM-102 up to 2200 mg, multiple
doses up to
1650 mg for 5.5 days, or loading dose regimens of 1100/500 mg or 1650/825 mg
for a total
of 7.5 days.
[00161] The study revealed the following information concerning
pharmacokinetics
associated with FA administration to humans using the noted dosing regimens.
C. and
AUC showed more than dose proportional increases from 550 mg to 1100 mg, then
approximately dose proportional increases from 1100 to 2200 mg (single dose
only for
2200 mg). Accumulation of CEM-102 occurred from Day 1 to Day 6 of dosing at
all dose
levels. The 1100 mg BID loading dose followed by a 550 mg BID maintenance dose
resulted in mean trough plasma concentrations of approximately 74 g/mL at 24
hours and
approximately 105 iag/mL after 7 days of dosing. The 1650 mg BID loading dose
followed by an 825 mg BID maintenance dose resulted in mean trough plasma
concentrations of approximately 150 iug/mL at 24 hours and approximately 200
tig/mL
after 7 days of dosing. The loading dose regimens produced plasma
concentrations that
approached steady-state levels at 24 hours and appeared to be better tolerated
than the
conventional BID regimens for comparable levels of overall exposure.
47
CA 02767614 2012-01-09
WO 2011/008193 PCT/US2009/050353
[00162] The data shows that a loading dose between 1100 mg BID - 1650 mg BID
is
sufficient to give a substantial blood level to provide multiples of the MIC
against S.
aureus and beta hemolytic streptococci. These high blood levels are sufficient
to inhibit
the bacterium as well as to prevent the selection of resistant bacteria.
Keeping the growth
to below detectable levels for a period of 72 hours without selection of
resistant strains has
been noted to be sufficient time in which to allow for clearance by
neutrophils and
macrophages, and it is sufficient to prevent the selection of resistant
strains (Louie et al.,
Antimicrob. Agents Chemother. 52:2486-2496 (2008)). Thus, after the loading
dose, the
600 mg as the maintenance dose was sufficient to maintain a steady state level
of 80
micrograms per ml throughout the dosing period.
Example 7 ¨ Pharmacodynamics of CEM-102 against Methicillin-Resistant
Staphylococcus aureus Using In vitro Models
[00163] A hollow fiber model was used to evaluate CEM-102 resistance potential
in
methicillin-resistant S. aureus (MRSA) strain USA 300 (Network of
Antimicrobial
Resistance in Staphylococcus aureus (NARSA), Chantilly, VA). USA 300 is a
highly
virulent strain of MRSA and is the most common community-associated MRSA
isolate in
the USA. MIC values were determined in accordance with Clinical and Laboratory
Standards Institute (CLSI).
[00164] The hollow fiber model comprised a two-compartment hollow fiber model
(Louie et al. Antimicrob Agents Chernother 52:2486-2496(2008)) consisting of a
volume of
15 mL in the central compartment, with multiple ports for the removal of
broth, delivery of
antibiotics, and collection of bacterial and antimicrobial samples. A
peristaltic pump was
used to continually replace antibiotic-containing medium with fresh media
(Mueller
Hinton Broth, supplemented with calcium, magnesium, and human albumin to a
final
concentration of 4 g/dL, simulating human physiologic levels) at a rate to
simulate the
half-life of CEM-102 based on human PK data. All experiments were performed in
duplicate.
[00165] Hollow fibers contain 15 ml of media was prepared and inoculated with
106
colony forming units (CFU)/mL USA 300 bacteria. One of three different dosing
48
CA 02767614 2015-12-04
regimens of CEM-120 was then applied to the fibers: (1) 600 mg/ml ql2h; (2)
1200 mg/ml
x 2, followed by 600 mg/ml, q12h; (3) 1500 mg/ml x 2, followed by 600 mg/ml,
q12h.
[00166] On Days 0, 1, 2, 3, 4, 5, 6, 7, 8, 9 and 10 samples were withdrawn
from the
fibers and plated on media with no-drug (TSA II with 5% sheep blood) or drug-
containing
agar (brain heart infusion), consisting of 4, 8, 16 x MIC of CEM-102 (1 ug/ml,
2 ug/ml, 4.0
ug/ml. respectively; MIC = 0.25 ug/ml).
[00167] Figure 8 shows the growth of samples withdrawn from 600 mg/ml ql2h
fibers
at the different time points on plates without CEM-102 (solid circles), with 1
ug/ml CEM-
102 (open circles), with 2 ug/ml CEM-102 (solid triangle), and with 4 ug/ml
CEM-102
(open triangle). The results in Figure 8 show that treatment using 600mg FA
ql2h resulted
in regrowth and development of resistance by 24h and 48h.
[00168] Figure 9 shows the growth of samples withdrawn from 1200 mg/ml x 2,
600
mg/ml, ql2h, fibers at the different time points on plates without CEM-1 02
(solid circles),
with 1 ug/ml CEM-102 (open circles), with 2 ug/ml CEM-102 (solid triangle),
and with 4
ug/ml CEM-102 (open triangle). The results in Figure 9 show that treatment
using 1200
mg x2 then 600 mg ql2h suppressed bacterial counts and reduced bacterial
counts by ¨2
logs, with rebound at 72h.
[00169] Figure 10 shows the growth of samples withdrawn from 1500 mg/ml x 2,
600
mg/ml, ql2h, fibers at the different time points on plates without CEM-102
(solid circles),
with 1 ug/ml CEM-102 (open circles), with 2 ug/ml CEM-102 (solid triangle),
and with 4
ug/ml CEM-102 (open triangle). The results in Figure 10 show that treatment
using 1500
mg x2 then 600 mg q12h suppressed resistance until > 72h and resulted in
killing over 2.5
log and even approached the threshold of bactericidal activity, and rebounded
at 144h.
[00170] While the foregoing specification teaches the principles of the
present
invention, with examples provided for the purpose of illustration, it will be
appreciated by
one skilled in the art from reading this disclosure that various changes in
form and detail
can be made without departing from the true scope of the invention.
49