Note: Descriptions are shown in the official language in which they were submitted.
CA 02783400 2012-06-06
WO 2011/073746 PCT/IB2010/002747
Calibration of electrochemical sensor
Field of the Invention
This invention is concerned with electrochemical analysis of fluids. It may be
applied to
fluids encountered in many locations but it has particular applicability in
connection with
electrochemical determination of hydrogen sulfide and dissolved sulfide anions
at a
location below ground.
Background
The analysis of fluid samples from hydrocarbon wells is a significant step in
the
evaluation of the producibility and economic value of the hydrocarbon
reserves. An
important factor in determining the economic value of gas and liquid
hydrocarbon
reserves is their chemical composition, particularly the concentration of
gaseous
components, such as hydrogen sulfide, carbon dioxide and methane. Therefore,
real
time gas detection is an important process for downhole fluid analysis.
The presence of sulfide species, principally hydrogen sulfide (H2S) but
possibly also
encountered as dissolved sulfide anions, has an important impact on the
economic value
of the produced hydrocarbons and the cost of production operations. Typically,
the
sulfur content of crude oils is in the range 0.3-0.8 weight percent and the
H2S content of
natural gas is in the range 0.01-0.4 weight percent, although concentrations
of 1-12S in
natural gas,of up to 30 weight percent have been reported.
There is a desire to be able to measure concentration of sulfide species in
fluids
downhole, rather than capturing a sample downhole and transporting it to the
surface
for laboratory analysis. One incentive for measurement downhole is to avoid
any effects
of change in temperature and pressure during travel to the surface with the
risk that the
composition analysed at the surface is not representative of the composition
downhole.
Another incentive to carry out measurement downhole is to avoid the delay
between
collecting a sample downhole and receiving analytical results from ,a surface
laboratory
CA 02783400 2012-06-06
WO 2011/073746 PCT/IB2010/002747
2
facility. However, elevated temperature and pressure downhole, possibly
accompanied
by corrosive materials, present a challenging environment for an analytical
sensor.
There have been a number of proposals for the determination of sulfide species
by
electrochemical techniques. These include proposals in which electrochemistry
is
coupled through a mediator compound to sulfide which is the intended analyte.
This
mediator compound is present in an electrochemical cell which is exposed to
the sulfide.
Both in the presence and absence of the sulfide analyte, an electrochemical
oxidation
and reduction of the mediator compound can take place when appropriate
electrical
potential is applied to the electrodes. However, one of the redox reactions of
the
mediator compound can also be brought about through a chemical reaction with
the
sulfide, and when this takes place there is a measurable change to the
electrochemistry.
A number of compounds including ferrocyanide ion and phenylene diamine
derivatives
have been proposed as mediator compounds. More recently ferrocene compounds
have
been used. In the presence of sulfide, the oxidized form of the ferrocene
compound or
other mediator can be reduced by homogeneous chemical reaction with the
sulfide
rather than by electrochemical reduction. In consequence, the presence of the
sulfide
analyte leads to an increase in the observed oxidative current and a reduction
in the
observed reductive current for the electrochemical reactions. The magnitude of
these
changes is dependent on the concentration of the sulfide, which is the
analyte, and can
be used to determine the analyte concentration.
This approach to the electrochemical determination of sulfide was described in
W02001/063094 and W02004/011929. Subsequently, ferrocene carboxylate and
sulfonate have been suggested as as mediator compounds in Electroanalysis Vol
18
pp1658-63 (2006) and in Electrochimica Acta Vol 52 pp499-50 (2006). A number
of
ferrocene sulfonates for possible use in this way have been described in
Journal of
Organometallic Chemistry Vol 692 pp5173-82 (2007). Experimental work in this
area
has, however, generally been confined to laboratory experiments at ambient
room
temperature.
CA 02783400 2012-06-06
WO 2011/073746 PCT/IB2010/002747
3
Determining sulfide downhole by means of this kind of electrochemical
procedure entails
exposing a sensor, including the compound which acts as the mediator, to the
aggressive
environment of the subterranean temperature and fluid. It may be necessary to
accept
that the sensor will have a limited working life, perhaps no more than a day.
The
magnitude of the analytical signal, deriving from reaction between the analyte
and the
mediator compound, is dependent on the concentration of the mediator compound
as
well as on the concentration of the analyte. However, at working temperatures
likely to
be encountered, the mediator compound may undergo significant decomposition
within
the limited working lifetime of an electrochemical sensor located downhole.
One way to
overcome this is to provide two sensors, only one of which is exposed to the
analyte
whilst the other is not exposed to the analyte and serves as a reference.
Apart from
requiring two sensors and thereby doubling a cost, a difficulty with this
approach is that
the sensors cannot be exposed to identical conditions and the accuracy of the
reference
is therefore open to question.
The invention
The present invention provides a way to calibrate a sensor whilst it is in
position below
ground, so that the mediator compound and the sensor containing it have a
useful
lifetime even though the concentration of the mediator compound is
progressively
decaying with time.
Although this problem of a decomposing mediator compound has been recognized
in the
context of determining sulfide species - to which the invention does indeed
have
particular applicability - it is also applicable with other analytes. This
invention can be
applied where an electrochemical analytical process entails the utilisation of
a mediator
compound whose concentration needs to be determined.
In a first aspect this invention provides a method of analysing a fluid for an
analyte
species, comprising:
i) providing a sensor comprising electrodes and a mediator compound capable of
undergoing a cycle of oxidation and reduction in response to varying potential
applied to
CA 02783400 2012-06-06
WO 2011/073746 PCT/IB2010/002747
4
the electrodes, where the mediator compound is also able to undergo a portion
of the
cycle through reaction with the analyte;
ii) bringing the sensor and the fluid into contact sufficiently for the
analyte to
react with the mediator,
iii) applying potential to the electrodes and observing current flow with
sufficient
time for reaction between the mediator compound and the analyte, for thereby
enabling
observation of the concentration of the analyte species present,
characterized in that the method also comprises
iv) calibrating the sensor by applying rapidly changing potential to the
electrodes
and observing current flow, the change of this potential and observation of
current being
performed sufficiently quickly to observe electrochemical oxidation and
reduction of the
mediator compound taking place independently of the concentration of analyte,
for
thereby observing the concentration of the mediator compound.
The changing potential required for this calibrating part (iv) of the
invention may be
varied continuously or changed in discrete steps or pulses.
In the above method, point (iv) has the effect of separating baseline
conditions, as would
be observed in the absence of analyte, from the conditions with analyte
present. This
allows calibration of the sensor through observation of the current
concentration of the
mediator compound which enables the concentration of analyte to be derived
from
observations made in point (iii) of the method.
This invention is not limited to a specific mediator chemical nor to a
specific
electrochemical reaction of the mediator. However, the electrochemical change
may be
oxidation and/or reduction and such a redox reaction may be a change in
oxidation state
of the mediator brought about by electron transfer.
The method of this invention may be carried out at a subterranean location,
notably
downhole in a wellbore. Possibilities for the fluid which is subjected to
analysis include
subterranean water or brine and fossil hydrocarbon such as natural gas or
crude oil.
However, the method could also be carried out at the surface, possibly to
analyse
produced hydrocarbon or to analyse an effluent stream.
CA 02783400 2012-06-06
WO 2011/073746 PCT/IB2010/002747
The sensor used in the method of this invention may be of a type which is
already
known. It may be an electrochemical cell of the Clark type in. which the
electrodes are in
contact with a fluid electrolyte and the mediator compound is in solution in
the
electrolyte. In such an arrangement, the electrolyte may be separated from
other fluid
5 by means of a membrane which is permeable to the analyte so as to allow the
analyte to
pass from the subterranean fluid into the electrolyte. A sensor of this
character,
intended for analysis of sulfide downhole, is the subject W02004/063743. A
cell of this
type using microelectrodes with a thin layer of electrolyte separated by a
membrane of
polytetrafluoroethylene (ptfe) from the fluid to be analysed has been
described in
Anal. Chem. vol.75 pp 2499-2503 (2003).
Another possibility for sensor construction is that the mediator compound and
electrolyte may be contained within a porous electrode, as described in
W02004/011929, where the porous electrode may be separated from subterranean
fluid by a membrane permeable to the analyte.
It is also possible that electrodes can be screen printed onto an insulating
support, as has
also been discussed in W02004/011929. Such an arrangement has also been
described
in Anal. Chem. vol.75 pp 2054-2059 (2003) where it is proposed that the
mediator
compound should be an insoluble compound mixed with a carbon-based electrode.
The
electrolyte may then be provided by constituents present in the fluid for
analysis.
The application of potential at part (iii) of the method set out above could
be application
of a fixed potential, for example the potential at which the current flow
associated with
oxidation is at a maximum. Alternatively, it may be application of a varying
potential
with observation of the current as the potential is varied. However, the
variation and
observation of current must allow time for reaction of the mediator and the
analyte,
whereas the change of potential in the calibrating part (iv) of the method
must be
considerably faster than any variation of potential at part (iii) so that
electrochemical
oxidation and reduction of the mediator compound take place independently of
the
concentration of analyte.
CA 02783400 2012-06-06
WO 2011/073746 PCT/IB2010/002747
6
More specifically, the application of potential at part (iii) of the method
may be carried
out as cyclic voltammetry in which the potential applied to a working
electrode is cycled
over a sufficient range to bring about the oxidation and reduction reactions
while
recording the current flow as the potential is varied. Such cyclic voltammetry
has been
described and exemplified in Electroanalysis vol 12 page 1453 (2000) and in
later
documents including W02004/063743. The recorded current shows peaks at the
potentials associated with the reduction and oxidation reactions.
Cyclic voltammetry is customarily performed with a continuously varying
potential,
changing sufficiently slowly that electrochemically oxidised mediator compound
is able
to come into contact with analyte within the electrolyte. Potential which
changes in
steps rather than continuously can possibly be employed as an alternative,
provided the
steps are long enough for steady-state conditions to be established before a
subsequent
step in potential.
It is also possible that this variation in potential whilst recording current
flow could be
carried out over only a portion of the reduction and oxidation cycle. This
would be
classed as linear scan voltammetry.
The characterising feature which is the calibration referred to as point (iv)
of this
invention as stated above may be implemented as one of the various forms of
pulse
voltammetry, with square wave voltammetry being the preferred technique. This
will
now be explained further and the invention will be exemplified with reference
to the
following drawings.
Brief Description of the Drawings
Fig. 1 shows the waveform applied in cyclic voltammetry;
Fig. 2 shows the results of cyclic voltammetry applied to solutions containing
varying
concentrations of sulfide and also shows a plot of peak current against
sulfide
concentration;
CA 02783400 2012-06-06
WO 2011/073746 PCT/IB2010/002747
7
Fig. 3 shows sequences of voltammograms taken at intervals at two different
temperatures;
Fig. 4 is a graph in which current flow is plotted against time at various
temperatures;
Figs 5, 6 and 7 are schematic representations of waveforms applied in pulse
voltammetry
together with illustrations of typical observations;
Fig. 8 shows the results of square wave voltammetry applied to solutions
containing
varying concentrations of sulfide;
Fig. 9 shows the results of square wave voltammetry applied to solutions
containing
varying concentrations of t-BuFcSO3 and varying concentrations of sulfide;
Fig. 10 is a schematic representation of a wellbore tool which is positioned
in a wellbore;
Fig. 11 is a schematic cross sectional view of the electrochemical sensor
within the tool
of Fig. 10; and
Fig. 12 shows the electrodes on one face of an electrode assembly within the
sensor
of Fig. 11.
Detailed Description
Cyclic voltammetry is normally carried out using an electrochemical cell with
three
electrodes: a working electrode, a counter electrode and a reference
electrode. A
varying potential relative to the reference electrode is applied to the
working electrode.
This potential is varied, usually linearly, over a period of time in a cycle
from a lower limit
value to an upper limit value and then back again after which the cycle may be
repeated.
This linearly varying waveform, with the cycle being repeated, has the form
shown
schematically in Figure 1.
The direct measurement from the procedure is the current flow as potential is
applied.
The values of particular interest are peak values of current flow together
with the
CA 02783400 2012-06-06
WO 2011/073746 PCT/IB2010/002747
8
applied potentials at which these peaks of current occur. However, it is also
possible for
the data obtained throughout a cyclic voltammetry experiment to be used as
input to a
computer program for modelling the chemical processes which occur.
Figure 2 shows the results of a laboratory experiment in which cyclic
voltammetry was
carried out at room temperature on pH 6.5 aqueous solutions containing 0.5 mM
t-butylferrocene sulfonate (t-BuFcSO3) containing increasing concentrations of
sulfide.
For this laboratory experiment a glassy carbon electrode can be used as the
working
electrode with a standard calomel electrode as the reference electrode.
The left side of Figure 2 shows the voltammetric responses (sometimes termed
voltammograms) obtained as potential applied to the working electrode relative
to the
reference electrode was scanned (i.e varied) linearly between 0.0 and +0.6
volt and back
to O.Ovolt at a scan rate of 0.1 volt/sec. The response in the absence of
sulfide (bottom
curve) revealed an oxidative wave at +0.4volt, and a corresponding reduction
wave at
+0.3volt. This is consistent with the oxidation and reduction of the
ferrocene/ferricenium redox couple. In the presence of sulfide the
voltammograms at
the left show an increase in the oxidation peak current along with a
concomitant
decrease in the reduction wave. The graph at the right shows the oxidation
peak
currents plotted against the concentrations of sulfide, with tielines to the
corresponding
voltammograms.
These changes in the presence of sulfide are attributed to at least some of
the oxidised
ferricenium species undergoing reduction by homogenous chemical reaction with
sulfide
in solution, instead of electrochemical reduction. Because this alternative
reductive
pathway is available, the oxidative current is increased and the reductive
current is
reduced.
The homogenous chemical reduction requires interaction between molecules of
the
oxidised mediator (ie the ferrocinium compound) and molecules of the analyte.
Consequently the rate of this reduction will depend on both the concentrations
of the
mediator compound and on the concentration of the sulfide which is the
analyte. The
scan rate, i.e. the rate at which applied electrical potential is varied, it
is chosen to be
CA 02783400 2012-06-06
WO 2011/073746 PCT/IB2010/002747
9
sufficiently slow that mediator compound which has undergone electrochemical
oxidation will have time to diffuse and collide with analyte, in order to be
reduced by
reaction in solution.
As mentioned previously, such cyclic voltammetry in the presence of a mediator
can be
used to observe the concentration of the sulfite analyte, implementing point
(iii) of the
invention as set out above. However, obtaining a meaningful result requires
the
concentration of the mediator compound present during the voltammetry
experiment to
be known. The present inventor has found that this becomes problematic at
elevated
temperatures.
Figure 3 shows the cyclic voltammetric responses of 0.5mM t-BuFcSO3 solutions
(pH 7)
obtained in the absence of sulfide over a period of 24 hours at 87 C and 133
C
respectively. In each case the voltammograms shown were measured at hourly
intervals. At 87 C, while some shift in the peak potential can be seen, only a
small decay
(about 15 %) in the oxidative peak current was observed over the 24 hour
period. This
can be attributed to thermal degradation of the mediator compound. When the
temperature was raised to 133 C, a much larger decrease (about 87 %) in the
oxidative
peak current was observed. This shows that the t-BuFcSO3- mediator compound
decays
at these temperatures - confirming a hypothesis that the decay process is
thermally
activated. The concentration of the t-BuFcSO3 mediator compound thus ceases to
have
a known value.
The decay of the mediator compound at various temperatures is also shown by
Figure 4.
This plots the oxidative peak current against time for each of five
temperatures. The
values are all shown as the remaining percentage of an initial amount at the
temperature
concerned. It can be clearly seen that as the temperature is increased from 87
to 133 C,
the loss of mediator compound, leading to a loss of analytical signal, becomes
progressively quicker.
To overcome this problem, in accordance with this invention, the concentration
of the
mediator is observed with rapid change to the applied potential. In
conventional cyclic
voltammetry the variation of electrical potential is sufficiently slow that
mediator
CA 02783400 2012-06-06
WO 2011/073746 PCT/IB2010/002747
compound which has undergone electrochemical oxidation will have time to
diffuse and
collide with analyte, so as to be reduced by the chemical reaction with the
analyte. In
contrast with this practice of cyclic voltammetry with a slow variation of
electrical
potential, when a rapidly changing potential is applied as required for point
(iv) of the
5 invention, there is insufficient time for diffusion of the mediator compound
and so little
or no chemical reduction by the sulfide analyte can take place. This chemical
reaction
cannot now compete effectively with the electrochemical reduction, or to put
it another
way the electrochemical reduction outruns the chemical reaction. As will now
be shown,
the consequence is that the electrochemical process becomes independent of the
10 presence of analyte.
There are several possibilities for providing the rapid change of potential
required for the
calibrating part (iv) of the invention. One possibility is to use cyclic
voltammetry with a
fast scan rate so that the variation of the applied potential becomes rapid.
Calculation,
carried out by use of a simulator program, has indicated that a scan rate of
15 volt/sec
would be suitable. Other possibilities are to adopt one of the various forms
of pulse
voltammetry. There are three main pulse techniques available, normal pulse
voltammetry (NPV), differential pulse voltammetry (DPV) and square wave
voltammetry
(SWV). All three entail changing the applied potential in steps and making a
brief
measurement of current flow between changes. The following description of
these
techniques will assume for the purposes of explanation that the applied
potential is
positive and causes electrochemical oxidation. However, it should be
appreciated that
these techniques could also be carried out with negative potential causing
electrochemical reduction.
Figure 5A shows the waveform used for normal pulse voltammetry.. There is a
baseline
potential applied to the reference electrode and at intervals the potential is
stepped up
to a higher value, held at that value for a short duration and then returned
to the
baseline. The steps in potential are increased progressively. The pulses, i.e.
the steps up
to a higher potential, are of short duration separated by much longer
intervals at the
baseline potential. The measurements of current flow are taken towards the end
of a
CA 02783400 2012-06-06
WO 2011/073746 PCT/IB2010/002747
11
pulse as indicted by the points tin Figure 5A. The data obtained consists of a
succession
of values of applied potential paired with the measured current at that
potential. This
data can be plotted in similar manner to the oxidative portion of a
voltammogram
obtained in cyclic voltammetry with continuously varying potential. However,
because
the pulses are short each measurement of current is made before the
electrochemical
process becomes influenced by any homogenous chemical reduction. As shown
schematically in Figure 513, the plot typically shows a rise in current as the
pulses reach
the potential required to effect oxidation.
Figure 6A shows the waveform used for differential pulse voltammetry. Again
the
applied potential is stepped up in short pulses separated by much longer
intervals, but
this is superimposed on a baseline which is of a staircase form so that the
baseline
potential rises after each pulse. Current flow is measured just before each
pulse begins,
as indicated by the points a in Figure 6A and also measured just before each
pulse ends,
as indicated by the points b in Figure 6A. The difference between the current
measured
at each point b and that measured at an adjacent point a is determined and
this
difference between the two measured values is recorded as the output from the
procedure.
Figure 6B shows schematically this difference in current plotted against the
applied
potential. When the baseline potential is well below that required for
oxidation, the step
in potential has little effect on the current flow and the difference in
current before and
during a pulse of raised potential is negligible, as seen at the left of
Figure 6B. When
baseline potential is well above the value required for oxidation the
electrochemical
process is proceeding at a rate limited by diffusion and the increase in
potential during a
pulse does not increase the rate of reaction very much, so that the difference
in current
during a pulse of higher potential is fairly small as seen at the right hand
side of Figure
6B. However, in between these extremes, the increase in potential during a
pulse
significantly increases the current and the plot of current difference rises
to a peak at a
potential associated with the electrochemical oxidation, as seen in Figure 6B.
CA 02783400 2012-06-06
WO 2011/073746 PCT/IB2010/002747
12
Figure 7A shows the waveform used in square wave voltammetry which is the
preferred
technique for the characterizing part (iv) of this invention. A square wave is
combined
with a staircase baseline, so that the potentials applied at the peaks and
troughs of the
square wave increase progressively. Current flow is measured close to the end
of each
peak and trough of the square wave as indicated by the points f and r in
Figure 7A. The
amplitude of the square wave is chosen to be sufficiently large that, in at
least part of the
scan, the potential at the peak of the square wave causes oxidation and the
measured
current is a forward current while the potential at the trough of the square
wave
reverses the oxidation and the measured current is a reverse current. Both the
peaks
and troughs of the square wave are pulses of short duration (usually the
troughs and
peaks are of equal duration) and so, as with the other forms of pulse
voltammetry each
measurement of current is made before the electrochemical process becomes
influenced
by any homogenous chemical reduction.
Three outputs can be obtained from a square wave voltammetry experiment. One
is the
applied potentials and associated current flow at the points f, i.e at the
peaks of the
square wave, plotted as the'curve W(f) in Figure 7B. Another is the applied
potentials
and associated current flow at the points r, i.e at the troughs of the square
wave, plotted
as the curve W(r). The third is the applied potentials and the associated
current
difference. This is plotted as the curve OW in Figure 7B and with differential
pulse
voltammetry this rises to a peak at a potential associated with the
electrochemical
oxidation.
One advantage of square wave voltammetry is that the peak in the curve OW is
larger
than the peaks in the plots of forward and reverse current W(f) and W(r) so
that the
technique has good sensitivity. A second advantage is that the technique can
be carried
out with a more rapid scan rate than other forms of pulse voltammetry so that
measurements can be made quickly.
Figure 8 shows current difference (i.e. OW) curves obtained by square wave
voltammetry applied to pH 7 solutions containing 0.5mM t-BuFCSO3- and
increasing
sulfide concentrations at room temperature. The sulfide concentrations ranged
from 0
CA 02783400 2012-06-06
WO 2011/073746 PCT/IB2010/002747
13
to 1mM by 0.2mM steps. It can be seen that the curves are virtually
superimposed thus
showing that with this square wave voltammetry the electrochemical oxidation
and
reduction of the t-BuFcSO3- had been successfully isolated from its reduction
by sulfide.
This was further confirmed by the data shown in Figure 9. Square wave
voltammetry
was applied to pH 7 solutions at room temperature containing varying
concentrations of
t-BuFcSO3- and varying concentrations of sulfide.. In Figure 9A the oxidative
peak current
(ie the maximum of the W(f) curve) is plotted as a function of sulfide
concentration for
various concentrations of t-BuFcSO3 and it can be seen that the recorded
values are
effectively independent of the sulfide concentration. In Figure 9B the same
data is
presented as a plot of the oxidative peak currents against t-BuFcS03
concentration. The
points lay on a straight line, showing that the measured peak currents were
proportional
to the concentration of the t-BuFcSO3 mediator compound.
Figures 10 to 12 illustrate equipment used to perform the method of the
invention
below ground, within a wellbore. The tool 10 comprises an elongate
substantially
cylindrical body which is suspended on a wireline 14 in the wellbore 16,
adjacent an
earth formation 18 believed to contain recoverable hydrocarbons. The tool is
provided
with a radially projecting sampling probe 20. The sampling probe 20 is placed
into firm
contact with the formation 18 by hydraulically operated rams 22 projecting
radially from
the tool on the opposite side from the sampling probe 20 and is connected to a
conduit
26 within the tool. A pump 28 within the tool 10 can be used to draw a sample
of the
hydrocarbons into the conduit 26. The pump 28 is controlled from the surface
at the
top of the wellbore via the wireline 14 and control circuitry (not shown)
within the tool.
The conduit 26 leads through an electrochemical sensor 30 located close to the
sampling
probe 20.
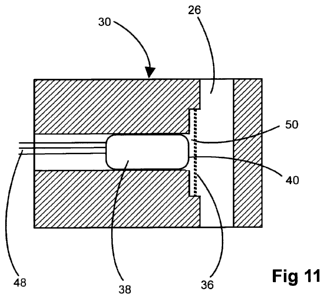
The sensor 30 is shown rather schematically in cross section in Figure 11. It
may be
constructed as described in greater detail in W02004/063743 and/or
W02005/066618.
The sensor 30 is generally cylindrical. A flowpath for the fluid whose sulfide
content is to
be determined extends through the sensor 30 and forms part of the conduit 26.
A gas
permeable membrane 36 separates this flow path from an axial bore through the
sensor,
CA 02783400 2012-06-06
WO 2011/073746 PCT/IB2010/002747
14
within which an electrode assembly 38 is located. This assembly 38 comprises
an
insulating body, having three electrodes on its face 40, namely a working
electrode 42
made from boron-doped diamond, a reference electrode 44 in the form of a
silver dot
coated with silver chloride or silver iodide, and a counter electrode 46
comprising a
printed platinum track. The electrodes 42, 44, 46 are connected via respective
electrical
conductors moulded into and extending axially through the body of the
electrode
assembly 38 to respective electrical leads 48, which connect the sensor 30 to
control
circuitry 32 within the tool. The space 50 between the face 40 of the
electrode assembly
and the membrane 36 is filled with a polar electrolyte which may be an aqueous
solution
in which the mediator compound t-BuFcSO3 is dissolved.
Once the tool is in place, fluid is drawn through the conduit 26 by the pump
28.
Hydrogen sulfide in the fluid can pass through the membrane 36 into the
electrolyte in
the space 50. After a time for equilibrium to be reached, the control unit 32
(possibly on
command received via the wireline 14) applies varying potential to the
electrodes and
meters the current flowing. This is done as cyclic voltammetry with a scan
rate which is
slow enough to allow time for reaction between the mediator compound and the
sulfide
which has entered the electrolyte. The current flowing and the applied
potential may be
communicated to the surface in real time via the wireline 14 or may be
recorded until
the tool is retrieved to the surface. Separately, either before or after this
voltammetric
measurement, the control unit 32 applies a square wave to the electrodes to
calibrate
the sensor 30 by square wave voltammetry. The current flowing is recorded
during the
peaks and troughs of the square wave as described above with reference to
Figure 7.
The recorded current and/or the values of current difference together with the
associated values of applied potential may be communicated to the surface via
the
wireline 14 or recorded until the tool is retrieved.