Note: Descriptions are shown in the official language in which they were submitted.
CA 02790433 2012-09-19
DEMANDES OU BREVETS VOLUMINEUX
LA PRESENTE PARTIE DE CETTE DEMANDE OU CE BREVETS
COMPREND PLUS D'UN TOME.
CECI EST LE TOME 1 DE 2
NOTE: Pour les tomes additionels, veillez contacter le Bureau Canadien des
Brevets.
JUMBO APPLICATIONS / PATENTS
THIS SECTION OF THE APPLICATION / PATENT CONTAINS MORE
THAN ONE VOLUME..
THIS IS VOLUME I OF 2
NOTE: For additional volumes please contact the Canadian Patent Office.
CA 02790433 2012-09-19
-1-
ANTI-ADDL ANTIBODIES AND USES THEREOF
15 Background of the Invention
Alzheimer's Disease is a progressive and degenerative
dementia (Terry, et al. (1991) Ann. Neurol,. 30:572-580;
Coyle (1987) In: Encyclopedia of Neuroscience, Adelman
(ed.), Birkhauser, Boston-Basel-Stuttgart, pp 29-31,). In
its early stages, Alzheimer's Disease manifests primarily
as a profound inability to form new memories (Selkoe (2002)
Science 298:789-791), reportedly due to neurotoxins derived
from amyloid beta (AR). AD is an amphipathic peptide whose
abundance is increased by mutations and risk factors linked
to Alzheimer's Disease. Fibrils formed from AD constitute
the core of amyloid plaques, which are hallmarks of an
Alzheimer's Disease brain. Analogous fibrils generated in
vitro are lethal to cultured brain neurons. These findings,
indicate that memory loss is a consequence of neuron death
caused by fibrillar A(3.
Despite strong experimental support for fibrillar AD
and memory loss, a poor correlation exists between dementia
and amyloid plaque burden (Katzman (1988) Ann. Neurol.
23:138-144). Moreover, transgenic hAPP mice (Dodart, et al.
(2002) Nat. Neurosci. 5:452-457; Kotilinek, et al. (2002)
CA 02790433 2012-09-19
-2-
J. Neurosci. 22:6331-6335), which develop age-dependent
amyloid plaques and, most importantly, age-dependent memory
dysfunction, show that within 24 hours of vaccination with
monoclonal antibodies against AR memory loss can be
reversed with no change in plaque levels. Such findings are
not consistent with a mechanism for memory loss dependent
on neuron death caused by amyloid fibrils.
Additional neurologically active molecules formed by
AR self-assembly have been suggested. These molecules
include soluble AR oligomers, also referred to as AR-
derived diffusible ligands or ADDLs. Oligomers are
metastable and form at low concentrations of AR1-42
(Lambert, et al. (1998) Proc. Natl. Acad. Sci. USA 95:6448-
6453) . AR oligomers rapidly inhibit long-term potentiation
(LTP), a classic experimental paradigm for memory and
synaptic plasticity. As such, memory loss stems from
synapse failure, prior to neuron death and synapse failure
by AR oligomers, not fibrils (Hardy & Selkoe (2002) Science
297:353-356) . Soluble oligomers have been found in brain
tissue and are strikingly elevated in Alzheimer's Disease
(Kayed, et al. (2003) Science 300:486-489; Gong, et al.
(2003) Proc. Natl. Acad. Sci. USA 100:10417-10422) and in
hAPP transgenic mice Alzheimer's Disease models (Kotilinek,
et al. (2002) J. Neurosci. 22:6331-6335; Chang, et al.
(2003) J. Mot. Neurosci. 20:305-313).
A variety of Alzheimer's Disease treatment options
have been suggested. Vaccine clinical trials have revealed
that persons mounting a vigorous immune response to the
vaccine exhibit cognitive benefit (Hock, et al. (2003)
Neuron 38:547-554); however, frequency of CNS inflammation
caused early termination of part of the trial (Birmingham &
Frantz (2002) Nat. Med. 8: 199-200) . As an alternative to a
vaccine, therapeutic antibodies that target ADDLs without
binding monomers or fibrils have been suggested (Klein
(2002) Neurochem. In t. 41:345-352). ADDLs are highly
CA 02790433 2012-09-19
-3-
antigenic, generating oligomer-selective polyclonal
antibodies in rabbits at concentration of -50 pg/mL
(Lambert, et al. (2001) .J. Neurochem. 79:595-605). Results
from transgenic mice models also suggest that antibodies
can be successful in reversing memory decline (Dodart, et
al. (2002) Nat. Neurosci. 5:452-457). Accordingly, there is
a need in the art for ADDL-selective therapeutic antibodies
for the prevention and treatment of Alzheimer's Disease.
The present invention meets this need.
Summary of the Invention
The present invention is an isolated antibody, or
fragment thereof, capable of differentially recognizing a
multi-dimensional conformation of one or more AR-derived
diffusible ligands. In particular embodiments, the antibody
of the present invention is in admixture with a
pharmaceutically acceptable carrier. In other embodiments,
the antibody of the present invention is in a kit.
Methods for preventing binding of AR-derived
diffusible ligands to a neuron, inhibiting assembly of AR-
derived diffusible ligands, and blocking the
phosphorylation of tau protein at Ser202/Thr205 employing
an antibody or antibody fragment which binds a multi-
dimensional conformation of one or more AR-derived
diffusible ligands are also provided.
The present invention further embraces a method for
prophylactically or therapeutically treating a disease
associated with AR-derived diffusible ligands using an
antibody of the instant invention. Administration of an
antibody of the invention can prevent binding of AR-derived
diffusible ligands to a neuron thereby preventing or
treating the disease associated with AR-derived diffusible
ligands.
The present invention is also a method for identifying
a therapeutic agent that prevents the binding of AP-derived
CA 02790433 2012-09-19
-4-
diffusible ligands to a neuron. This method of the
invention involves contacting a neuron with A(3-derived
diffusible ligands in the presence of an agent and using an
antibody of the present invention to determine binding of
the A(3-derived diffusible ligands to the neuron in the
presence of the agent.
The present invention also embraces a method for
detecting AP-derived diffusible ligands in a sample and a
method for diagnosing a disease associated with A(3-derived
diffusible ligands. Such methods involve contacting a
sample with an antibody of the instant invention so that
the A(3-derived diffusible ligands can be detected and a
disease associated with A(3-derived diffusible ligands can
be diagnosed.
In one aspect, there is provided an isolated antibody,
or fragment thereof, for differentially recognizing a
multi-dimensional conformation of one or more A(3-derived
diffusible ligands, the antibody having: A) a heavy chain
CDR1 having an amino acid sequence of: i) TSGMGVX (X =
S/G/A) or ii) SFGMH (SEQ ID NO: 28) ; B) a heavy chain CDR2
having an amino acid sequence of: i) HIX1WDDDKX2YNPSLKS (X1 =
F/Y/W, X2 = S/R/Y) or ii) YIXIX2X3SX4TIYYADTVKG (XI/X2 =
R/S/G/T/C/Y/N/Q/K/H, X3 = G/V, X4 = G/S/T/C/Y/N/Q); C) a
heavy chain CDR3 having an amino acid sequence of: i)
RSIXIXZX3X4PEDYFX5Y (X1 = G/S/T/C/Y/N/Q, X2 = SIT, X3/X4 =
A/V/L/I/P, X5 = D/A), ii) RQLGLRSIDAMDY (SEQ ID NO: 43),
iii) YD(3YPYWYFDV, iv) GGNYYGSSRFAY, v) YGNYGYYYGMDY, vi)
SGYGSSYGYGMDY, or vii) GITTALDY; D) a light chain CDR1
having an amino acid sequence of: i) RSSQSXIX2HSNGNTYLX3
(X1/X2 = A/V/L/I/P, X3 = D/E/R/H/K)or ii) KASQDINSYLS; E) a
light chain CDR2 having an amino acid sequence of: i)
KXISNRFX2 (X1 = A/V/L/I/P, X2 = S/F) or ii) RANRFVD; and F) a
light chain CDR3 having an amino acid sequence of: i)
XIQX2X3X4VPX5T (X1 = S/F, X2 = G/S/T, X3 = SIT, X4 = H/Y/L, X5
= A/V/L/I/P) , ii) LQYDEFPLT, or iii) X1QX2TRVPLT (X1 F/L,
X2 = AM.
CA 02790433 2012-09-19
-4a-
Brief Description of the Drawings
Figure 1 shows the results from an alkaline
phosphatase assay, wherein anti-ADDL antibodies
differentially block neurons.
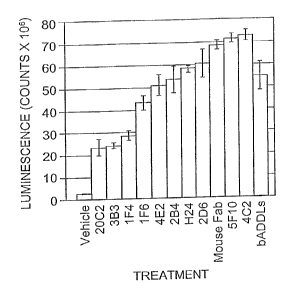
Figure 2 shows a summary of bADDL binding when B103
cells are pre-incubated with anti-ADDL antibodies.
Figure 3 shows a summary of binding characteristics of
antibodies capable of differentially recognizing
multidimensional conformations of ADDLs.
Figure 4 shows a summary of ADDL assembly inhibition
of the antibodies disclosed herein.
Figure 5 shows an N2A binding:kADDL correlation plot.
Figure 6 shows the nucleic acid sequences for the
heavy and light chain variable regions, respectively, for
murine anti-ADDL antibodies, 20C2 (Figures 6A and 6B), 5F10
(Figures 6C and 6D), 2D6 (Figures 6E and 6F), 2B4 (Figures
6G and 6H), 4E2 (Figures 61 and 6J), 2H4 (Figures 6K and
6L) , 2A10 (Figures 6M and 6N) , 3B3 (Figures 60 and 6P) , 1F6
(Figures 6Q and 6R), 1F4" (Figures 6S and 6T), 2E12 (Figure
6U and 6V) and 4C2 (Figures 6W and 6X) . Lower case letters
CA 02790433 2012-09-19
-5-
indicate the antibody leader sequences and uppercase
letters indicate antibody variable region sequences. The
nucleotides coding for the complementary determining
regions (CDRs) are underlined.
Figure 7 shows comparisons of CDR1 (Figure 7A), CDR2
(Figure 7B), CDR3 (Figure 7C) sequences for the heavy chain
variable regions and CDR1 (Figure 7D), CDR2 (Figure 7E),
CDR3 (Figure 7F) sequences for the light chain variable
regions for the mouse anti-ADDL antibodies.
Figure 8 shows the amino acid sequences for the heavy
and light chain variable regions, respectively, for
humanized anti-ADDL antibodies 20C2 (Figures 8A and 8B),
26D6 (Figures 8C and 8D), 4E2 (Figures 8E and 8F), 3B3
(Figures 8G and 8H), 2H4 (Figures 81 and 8J) and 1F6
(Figures 8K) created by CDR grafting. Sequences are
presented as comparisons between the mouse sequence, the
most homologous human sequence obtained from the NCBI
protein database, the most homologous human genomic
sequence and the humanized sequence. Amino acids in the
mouse, human and human genomic sequences that differ from
the humanized sequences are in bold. CDRs are underlined.
Residues important for the maintenance of CDR loop
conformation are indicated with an *. Conserved residues
found at the VL/VH interface are indicated with a #.
Potential glycosylation sites are indicated by italic. For
the 20C2 heavy chain two humanized sequences were generated
(HCVRA and HCVRB) that differ by one amino acid at position
24. In 20C2 HCVRA the human amino acid was used and in 20C2
HCVRB the mouse amino acid was used. No light chain was
designed for 1F6 because it has the same sequence as that
of the light chain for 4E2.
Figure 9 shows the amino acid sequences for the heavy
and light chain variable regions, respectively, for
humanized anti-ADDL antibodies 20C2 (Figures 9A and 9B) and
26D6 (Figures 9C and 9D) created by veneering. Sequences
CA 02790433 2012-09-19
-6-
are presented as comparisons between the mouse sequence,
the most homologous human sequence obtained from the NCBI
protein database, the most homologous human genomic
sequence and the humanized sequence. Amino Acids in the
mouse, human and human genomic sequences that differ from
the humanized sequences are bold. CDRs are underlined.
Residues important for the maintenance of CDR loop
conformation are indicated with an asterisk. Conserved
residues found at the VL/VH interface are indicated with a
pound symbol. Potential glycosylation sites are indicated
by italic. For the 20C2 heavy chain, two humanized
sequences were generated (HCVRVenA and HCVRVenB) that
differ by one amino acid at position 81. In 20C2 HCVRVenA,
the mouse amino acid was used and in 20C2 HCVRVenB, the
human amino acid was used. For the 26D6 heavy chain, three
humanized sequences were designed based on veneering (HCVR
Vent,. Ven 2and Ven3) that differ at amino acids 11, 23, 15,
81, 89 and 118. In HCVR Vent, the mouse amino acid was used
at all positions. In Ven2, the mouse amino acid was used
for residues 81 and 118 and the human amino acid for
residues 11, 13, 15, and 89. In Ven3, the human amino acids
were used at all positions. For the 26D6 light chain, two
veneered humanized sequences were designed (LCVR Venl and
Ven2) that differ at amino acids 88 and 105. In LCVR Venl,
the mouse amino acid was used at both positions and in
Ven2, the human amino acid was used.
Figure 10 shows nucleic acid sequences for the heavy
and light chain variable regions (HCVRs and LCVRs,
respectively) for humanized anti-ADDL antibodies- CDR
grafted HCVRs and LCVRs for 20C2, 2D6, 4E2, 3B3, 2H4, and
IF6, are respectively presented in Figure 10A to Figure
10K. Veneered HCVRs (VenA and VenB) and the LCVR for 20C2
are presented in Figure 1OL to Figure ION, whereas the
veneered HCVRs (Vent, Ven2, Ven3) and LCVRs (Vent, Ven2)
for 26D6 are presented in Figure 100 to Figure lOS.
CA 02790433 2012-09-19
-7-
Uppercase indicates antibody variable region sequences.
CDRs are underlined. Variable region sequences were cloned
into full heavy and light chain antibody expression
vectors.
Figure 11 shows the amino acid sequences for the full
IgG1 and IgG2m4 humanized heavy chains and humanized Kappa
light chains for anti-ADDL antibodies. Figure 11A, CDR
grafted 20C2 HCVRA IgGi; Figure 11B, CDR grafted 20C2 HCVRB
IgGi; Figure IIC, CDR grafted 20C2 HCVRA IgG2m4; Figure
11D, CDR grafted 20C2 HCVRB IgG2m4; Figure IIE, CDR grafted
20C2 LCVR Kappa; Figure 11F, CDR grafted 26D6 HCVR IgGl;
Figure IIG, CDR grafted 26D6 HCVR IgG2m4; Figure 11H, CDR
grafted 26D6 LCVR Kappa; Figure 111, CDR grafted 4E2 HCVR
IgGl; Figure 1IJ, CDR grafted 4E2 LCVR Kappa; Figure 1IK,
CDR grafted 3B3 HCVR IgGi; Figure 11L, CDR grafted 3B3 LCVR
Kappa; Figure 1IM, CDR grafted 2H4 HCVR IgGl; Figure 1IN,
CDR grafted 2H4 LCVR Kappa; Figure 110, CDR grafted IF6
HCVR IgG1; Figure 11P, veneered 20C2 HCVR VenA IgGi; Figure
11Q, veneered 20C2 HCVR VenB IgGl; Figure 11R, veneered
20C2 HCVR VenB IgG2m4; Figure 115, veneered 20C2 LCVR
Kappa; Figure 11T, veneered 26D6 HCVR Vent Ig; Figure 11U,
veneered 26D6 HCVR Venl IgG1; Figure 11V, 26D6 HCVR Ven2
IgGI; Figure 11W, veneered 26D6 HCVR Ven3; Figure 11X,
veneered 26D6 LCVR Venl Kappa; and Figure 11Y, veneered
26D6 LCVR Ven2 Kappa. Underlining indicates variable region
sequences and amino acids corresponding to the CDRs are
double-underlined. The remaining amino acid sequences are
constant region sequences.
Figure 12 shows a comparison of the amino acid
sequence of human antibody constant regions and the
sequence of IgG2m4. The asterisk indicates a glycosylation
site at Asn297. Regions of FcRn binding are indicated.
Sequences in which IgG2m4 is different from IgG2 are
underlined.
CA 02790433 2012-09-19
-8-
Figure 13 shows the annotated amino acid sequence for
heavy (Figure 13A) and light (Figure 13B) chains of 20C2
humanized antibody in Fab phage-display vector pFab3d.
Figure 14 depicts the design and primers employed in
preparing two LC-CDR3 libraries, namely LC3-1 and LC3-2,
for generating an affinity matured 20C2 light chain CDR3.
Restriction endonuclease recognition sites used for cloning
are indicated in italic. Uppercase indicates nucleic acids
encoding antibody variable region sequences. Nucleic acids
encoding.CDRs are underlined.
Detailed Description of the Invention
Monoclonal antibodies, which differentially recognize
multi-dimensional conformations of AR-derived diffusible
ligands (i.e., ADDLs), have now been generated.
Advantageously, the instant monoclonal antibodies can
distinguish between Alzheimer's Disease and control human
brain extracts, and identify endogenous oligomers in
Alzheimer's Disease brain slices and in cultured
hippocampal cells. Further, the instant antibodies
neutralize endogenous and synthetic ADDLs in solution. So-
called "synthetic" ADDLs are produced in vitro by mixing
purified amyloid R1-42 under conditions that generate
ADDLs. See U.S. Patent No. 6,218,506. Particular antibodies
disclosed herein exhibit a high degree of selectivity for
3-24mers, with minimal detection of monomer AR peptides.
Further, recognition of ADDLs by selected antibodies of the
invention is not blocked by short peptides that encompass
the linear sequence of API-42 or ARl-40. However, binding
is blocked by API-28, indicating an epitope based on a
conformationally unique structure also found in ARl-28.
Delineation of epitopes of the instant antibodies indicated
that these antibodies recognize similar core linear
sequences with similar affinity and specificity
characteristics as measured by ELISA. Moreover, the instant
CA 02790433 2012-09-19
-9-
antibodies differentially block the ability of ADDL-
containing preparations to bind primary cultures of rat
hippocampal neurons and immortalized neuroblastoma cell
lines, and also block ADDL assembly. This finding
demonstrates that these antibodies possess a differential
ability to recognize a multi-dimensional conformation of
ADDLs despite similar linear sequence recognition and
affinities. Since ADDLs are known to associate with a
subset of neurons and disrupt normal neuronal function, one
use of this current invention is the development and/or
identification of antibodies that prevent the binding of
ADDLs to neurons. Such antibodies would be useful in the
treatment of ADDL related diseases including Alzheimer's
Disease. A refinement of this use would be to specifically
use humanized and/or affinity-matured versions of these
antibodies for the prevention of ADDL binding to neurons
and assembly of ADDLs_
Accordingly, the present invention is an isolated
antibody that differentially recognizes one or more multi-
dimensional conformations of ADDLs. An antibody of the
instant invention is said to be isolated when it is present
in the substantial absence of other biological
macromolecules of the same type. Thus, an "isolated
antibody" refers to an antibody which is substantially free
of other antibodies; however, the molecule may include some
additional agents or moieties which do not deleteriously
affect the basic characteristics of the antibody (e.g.,
binding specificity, neutralizing activity, etc.)_
Antibodies which are capable of specifically binding
one or more multi-dimensional conformations of ADDLs, bind
particular ADDLs derived from the oligomerization of ARl-
42, but do not cross-react with other AR peptides, namely
API-12, API-28, AR1-40, and AR12-28 as determined by
western blot analyses as disclosed herein; and
preferentially bind ADDLs in solution (see, e.g., Example
CA 02790433 2012-09-19
-10-
21). Specific binding between two entities generally refers
to an affinity of at least 106, 107, 108, 101, or 10" M_'_
Affinities greater than 108 M-' are desired to achieve
specific binding.
In particular embodiments, an antibody that is capable
of specifically binding a multi-dimensional conformation of
one or more ADDLs is also raised against (i.e., an animal
is immunized with) multi-dimensional conformations of
ADDLs. In other embodiments, an antibody-that is capable of
specifically binding a multi-dimensional conformation of
one or more ADDLs is raised against a low n-mer-forming
peptide such as A(31-42[Nle35-Dpro37].
The term "epitope" refers to a site on an antigen to
which B and/or T cells respond or a site on a molecule
against which an antibody will be produced and/or to which
an antibody will bind. For example, an epitope can be
recognized by an antibody defining the epitope.
A linear epitope is an epitope wherein an amino acid
primary sequence comprises the epitope recognized- A linear
epitope typically includes at least 3, and more usually, at
least 5, for example, about 8 to about 10 amino acids in a
unique sequence.
A conformational epitope, in contrast to a linear
epitope, is an epitope wherein the primary sequence of the
amino acids comprising the epitope is not the sole defining
component of the epitope recognized (e.g., an epitope
wherein the primary sequence of amino acids is not
necessarily recognized by the antibody defining the
epitope). Typically a conformational epitope encompasses an
increased number of amino acids relative to a linear
epitope. With regard to recognition of conformational
epitopes, the antibody recognizes a three-dimensional
structure of the peptide or protein. For example, when a
protein molecule folds to form a three-dimensional
structure, certain amino acids and/or the polypeptide
CA 02790433 2012-09-19
-ll-
backbone forming the conformational epitope become
juxtaposed enabling the antibody to recognize the epitope.
Methods of determining conformation of epitopes include but
are not limited to, for example, x-ray crystallography,
two-dimensional nuclear magnetic resonance spectroscopy and
site-directed spin labeling and electron paramagnetic
resonance spectroscopy. See, for example, Epitope Mapping
Protocols in Methods in Molecular Biology (1996) Vol. 66,
Morris (Ed.).
A(3-derived diffusible ligands or ADDLs refer to
soluble oligomers of amyloid PI-42 which are desirably
composed of aggregates of less than eight or nine amyloid
(31-42 peptides and are found associated with Alzheimer's
Disease. This is in contrast to high molecular weight
aggregation intermediates, which form stings of micelles
leading to fibril formation.
As exemplified herein, the instant antibody binds or
recognizes at least one multi-dimensional conformation of
an ADDL (see, e.g., Figure 3). In particular embodiments,
the instant antibody binds at least two, at least three, or
at least four multi-dimensional conformations of an ADDL.
Multi-dimensional conformations of ADDLs are intended to
encompass dimers, trimers, tetramers pentamers, hexamers,
heptamers, octamers, nonamers, decamers, etc as defined by
analysis via SDS-PAGE. Because trimer, tetramer, etc.
designations can vary with the assay method employed (see,
e.g., Bitan, et al. (2005) Amyloid 12:88-95) the definition
of trimer, tetramer, and the like, as used herein, is
according to SDS-PAGE analysis. To illustrate the
differentially binding capabilities of the instant
antibodies, it has been found that certain antibodies will
recognize one multi-dimensional conformation, for example,
tetramers of ADDLs (e.g., antibody 2D6 or 4E2), while other
antibodies recognize several multi-dimensional
conformations, for example, trimers and tetramers of ADDLs
CA 02790433 2012-09-19
-12-
(e_g., antibody 2A10, 2B4, 5F10, or 20C2). As such, the
antibodies of the instant invention have oligomer-specific
characteristics. In particular embodiments, a multi-
dimensional conformation of an ADDL is associated with a
specific polypeptide structure which results in a
conformational epitope that is recognized by an antibody of
the present invention. In other embodiments, an antibody of
the invention specifically binds a multi-dimensional
conformation ADDL having a size range of approximately a
trimer or tetramer, which have molecular weights in excess
of >50 kDa.
In certain embodiments, in addition to binding to a
multi-dimensional conformation, the instant antibody binds
to a selected linear epitope of amyloid (31-42. A linear
epitope of an ADDLs is intended as a four, five, six or
more amino acid residue peptide located in the N-terminal
10, 11, 12, 15 or 20 amino acid residues of amyloid (31-42.
In particular embodiments, an antibody of the invention
specifically binds to a linear epitope within residues 1-
10, 1-8, 3-10, or 3-8 of amyloid (31-42. Exemplary linear
epitopes of amyloid (3 1-42 include, but are not limited to,
amino acid residues EFRHDS (SEQ ID NO:177); DAEFRH,DS (SEQ
ID NO:178), and EFRHDSGY (SEQ ID N0:179).
While antibodies of the instant invention may have
similar linear epitopes, such linear epitopes are not
wholly indicative of the binding characteristics of the
instant antibodies (i.e., ability to block ADDL binding to
neurons, prevent tau phosphorylation and inhibit ADDL
assembly) because, as is well known to the skilled artisan,
the linear epitope may only correspond to a portion of the
antigen's epitope (see, e.g., Breitling and Diibel (1999)
In: Recombinant Antibodies, John Wiley & Sons, Inc., NY,
pg. 115) . For example, 20C2 was found to bind assemblies of
charge-inverted, truncated A(37-42 peptide, which lack the
linear epitope for 20C2 (i.e., amino acid residues 3-8) and
CA 02790433 2012-09-19
-13-
contain a very different sequence corresponding to residues
7-16 of A(3. Therefore 20C2 binds to conformational epitopes
that depend upon elements from within residues 17-42 of A(3,
but only when in a multidimensional conformation. The
antibodies of the instant invention can be distinguished
from those of the art as being capable of differentially
recognizing multi-dimensional ADDLs and accordingly
differentially blocking ADDL binding to neurons,
differentially preventing tau phosphorylation and
differentially inhibiting ADDL assembly.
An antibody, as used in accordance with the instant
invention includes, but is not be limited to, polyclonal or
monoclonal antibodies, and chimeric, human (e.g. isolated
from B cells), humanized, neutralizing, bispecific or
single chain antibodies thereof. In one embodiment, an
antibody of the instant invention is monoclonal. For the
production of antibodies, various hosts including goats,
rabbits, chickens, rats, mice, humans, and others, can be
immunized by injection with synthetic or natural ADDLs.
Methods for producing antibodies are well-known in the art.
See, e.g., Kohler and Milstein ((1975) Nature 256:495-497)
and Harlow and Lane (Antibodies: A Laboratory Manual (Cold
Spring Harbor Laboratory, New York (1988)).
Depending on the host species, various adjuvants can
be used to increase the immunological response. Adjuvants
used in accordance with the instant invention desirably
augment the intrinsic response to ADDLs without causing
conformational changes in the immunogen that affect the
qualitative form of the response. Particularly suitable
adjuvants include 3 De-O-acylated monophosphoryl lipid A
(MPL'M; RIBI ImmunoChem Research Inc., Hamilton, MT; see GB
2220211) and oil-in-water emulsions, such as squalene or
peanut oil, optionally in combination with immune
stimulants, such as monophosphoryl lipid A (see Stoute, et
al. (1997) N. Engl. J. Med. 336:86-91), muramyl peptides
CA 02790433 2012-09-19
-14-
(e.g., N-acetylmuramyl-L-threonyl-D-isoglutamine (thr-MDP),
N-acetyl-normuramyl-L-alanyl-D-isoglutamine (nor-MDP), N-
acetylmuramyl-L-alanyl-D-isoglutaminyl-L-alanine-2-(1'-
2 'dipalmitoyl-sn-glycero-3-hydroxyphosphoryloxy)-ethylamine
(E-PE), N-acetylglucsaminyl-N-acetylmuramyl-L-A1-D-isoglu-
L-Ala-dipalmitoxy propylamide (DTP-DPP)), or other
bacterial cell wall components. Specific examples of oil-
in-water emulsions include MF59 (WO 90/14837), containing
5% Squalene, 0.5% TWEENT 80, and 0.5% SPAN 85 (optionally
containing various amounts of MTP-PE) formulated into
submicron particles using a microfluidizer such as Model
110Y microfluidizer (Microfluidics, Newton, MA); SAF
containing 10% Squalene, 0.4% TWEENm 80, 5% PLURONIC -
blocked polymer L121, and thr-MDP, either microfluidized
into a submicron emulsion or vortexed to generate a larger
particle size emulsion; and RIBITM adjuvant system (RAS)
(Ribi ImmunoChem, Hamilton, MT) containing 2% squalene,
0.2% TWEENI 80, and one or more bacterial cell wall
components such as monophosphoryllipid A, trehalose
dimycolate (TDM), and cell wall skeleton (CWS).
Another class of adjuvants is saponin adjuvants, such
as STIMULONT"' (QS-21, Aquila, Framingham, MA) or particles
generated therefrom such as ISCOMs (immunostimulating
complexes) and ISCOMATRIX (CSL Ltd., Parkville,
Australia). Other suitable adjuvants include Complete
Freund's Adjuvant (CFA), Incomplete Freund's Adjuvant
(IFA), mineral gels such as aluminum hydroxide, and
surface-active substances such as lysolecithin, PLURONIC
polyols, polyanions, peptides, CpG (WO 98/40100), keyhole
limpet hemocyanin, dinitrophenol, and cytokines such as
interleukins (IL-1, IL-2, and IL-12), macrophage colony
stimulating factor (M-CSF), and tumor necrosis factor
(TNF). Among adjuvants used in humans, BCG (bacilli
Calmette-Guerin) and Corynebacterium parvum are
particularly suitable.
CA 02790433 2012-09-19
-15-
An antibody to a multi-dimensional conformation ADDL
is generated by immunizing an animal with ADDLs. Generally,
ADDLs can be generated synthetically or by recombinant
fragment expression and purification. Synthetic ADDLs can
be prepared as disclosed herein or in accordance with the
methods disclosed in U.S. Patent No. 6,218,506 or in co-
pending applications USSN 60/621,776, 60/652,538,
60/695,526 and 60/695,528. Further, ADDLs can be fused with
another protein such as keyhole limpet hemocyanin to
generate an antibody against the chimeric molecule. The
ADDLs can be conformationally constrained to form an
epitope useful as described herein and furthermore can be
associated with a surface for example, physically attached
or chemically bonded to a surface in such a manner so as to
allow for the production of a conformation which is
recognized by the antibodies of the present invention.
Monoclonal antibodies to multi-dimensional
conformations of ADDLs can be prepared using any technique
which provides for the production of antibody molecules by
continuous cell lines in culture. These include, but are
not limited to, the hybridoma technique, the human B-cell
hybridoma technique, and the EBV-hybridoma technique
(Kohler, et al. (1975) Nature 256:495-497; Kozbor, et al.
(1985) J. Immunol. Methods 81:31-42; Cote, et al. (1983)
Proc. Natl. Acad. Sci. 80:2026-2030; Cole, et al. (1984)
Mol. Cell Biol. 62:109-120). Exemplary monoclonal
antibodies include murine antibodies designated 2A10, 4C2,
2D6, 4E2, 20C2, 2B4, 5F10, 2H4, 2E12, 1F6, 1F4, 3B3, 5G12,
6B7, 6B11, 11B4, 11B5, 14A11, 15G6, 17G4, 20C2, 3B7, 1E3,
1A9, 1G3, 1A7 and 1E5.
In addition, humanized and chimeric antibodies can be
produced by splicing of mouse antibody genes to human
antibody genes to obtain a molecule with appropriate
antigen specificity and biological activity (see Morrison,
et al. (1984) Proc. Natl. Acad. Sci. 81, 6851-6855;
CA 02790433 2012-09-19
-16-
Neuberger, et al. (1984) Nature 312:604-608; Takeda, et al.
(1985) Nature 314:452-454; Queen, et al. (1989) Proc. Natl.
Acad. Sci. USA 86:10029-10033; WO 90/07861). For example, a
mouse antibody is expressed as the Fv or Fab fragment in a
phage selection vector. The gene for the light chain (and
in a parallel experiment, the gene for the heavy chain) is
exchanged for a library of human antibody genes. Phage
antibodies, which still bind the antigen, are then
identified. This method, commonly known as chain shuffling,
provided humanized antibodies that should bind the same
epitope as the mouse antibody from which it descends
(Jespers, et al. (1994) Biotechnology NY 12:899-903). As an
alternative, chain shuffling can be performed at the
protein level (see, Figini, et al. (1994) J. Mot. Biol.
239:68-78).
Human antibodies can also be obtained using phage-
display methods. See, e.g., WO 91/17271 and WO 92/01047. In
these methods, libraries of phage are produced in which
members display different antibodies on their outer
surfaces. Antibodies are usually displayed as Fv or Fab
fragments. Phage displaying antibodies with a desired
specificity are selected by affinity enrichment to ADDLs.
Human antibodies against ADDLs can also be produced from
non-human transgenic mammals having transgenes encoding at
least a segment of the human immunoglobulin locus and an
inactivated endogenous immunoglobulin locus. See, e.g., WO
93/12227 and WO 91/10741,
Human antibodies can be selected by competitive
binding experiments, or otherwise, to have the same epitope
specificity as a particular mouse antibody. Such antibodies
are particularly likely to share the useful functional
properties of the mouse antibodies. Human polyclonal
antibodies can also be provided in the form of serum from
humans immunized with an immunogenic agent. Optionally,
CA 02790433 2012-09-19
-17-
such polyclonal antibodies can be concentrated by affinity
purification using ADDLs as an affinity reagent.
Humanized antibodies can also be produced by veneering
or resurfacing of murine antibodies. Veneering involves
replacing only the surface fixed region amino acids in the
mouse heavy and light variable regions with those of a
homologous human antibody sequence. Replacing mouse surface
amino acids with human residues in the same position from a
homologous human sequence has been shown to reduce the
immunogenicity of the mouse antibody while preserving its
ligand binding. The replacement of exterior residues
generally has little, or no, effect on the interior
domains, or on the interdomain contacts. (See, e.g., U.S.
Patent No. 6,797,492).
Human or humanized antibodies can be designed to have
IgG, IgD, IgA, IgM or IgE constant regions, and any
isotype, including IgGl, IgG2, IgG3 and IgG4. In particular
embodiments, an antibody of the invention is IgG or IgM, or
a combination thereof. A particular combination embraces a
constant region formed by selective incorporation of human
IgG4 sequences into a standard human IgG2 constant region.
An exemplary mutant IgG2 Fc is IgG2m4, set forth herein as
SEQ ID NO:254. Antibodies can be expressed as tetramers
containing two light and two heavy chains, as separate
heavy chains and light chains or as single chain antibodies
in which heavy and light chain variable domains are linked
through a spacer. Techniques for the production of single
chain antibodies are well-known in the art.
Exemplary humanized antibodies produced. by CDR
grafting and veneering are disclosed herein for antibodies
designated 4E2, 26D6, 20C2, 3B3, 2H4, and 1F6. Amino acid
sequences for IgG1 and IgG2M4 heavy chain variable regions,
as well as kappa light chain variable regions for humanized
4E2, 26D6, 20C2, 3B3, 2H4, and 1F6 generated by CDR
CA 02790433 2012-09-19
-18-
grafting and veneering are presented in Figures 11A to 11Y
and set forth herein as SEQ ID NOs:152 to 176.
Diabodies are also contemplated. A diabody refers to
an engineered antibody construct prepared by isolating the
binding domains (both heavy and light chain) of a binding
antibody, and supplying a linking moiety which joins or
operably links the heavy and light chains on the same
polypeptide chain thereby preserving the binding function
(see, Holliger et al. (1993) Proc. Natl. Acad. Sci. USA
90:6444; Poljak (1994) Structure 2:1121-1123) . This forms,
in essence, a radically abbreviated antibody, having only
the variable domain necessary for binding the antigen. By
using a linker that is too short to allow pairing between
the two domains on the same chain, the domains are forced
to pair with the complementary domains of another chain and
create two antigen-binding sites. These dimeric antibody
fragments, or diabodies, are bivalent and bispecific. The
skilled artisan will appreciate that any method to generate
diabodies can be used. Suitable methods are described by
Holliger, et al. (1993) supra, Poljak (1994) supra, Zhu, et
al. (1996) Biotechnology 14:192-196, and U.S. Patent No.
6,492,123,
Fragments of an isolated antibody of the invention are
also expressly encompassed by the instant invention.
Fragments are intended to include Fab fragments, F(ab')2
fragments, F(ab') fragments, bispecific scFv fragments, Fd
fragments and fragments produced by a Fab expression
library, as well as peptide aptamers. For example, F(ab')2
fragments are produced by pepsin digestion of the antibody
molecule of the invention, whereas Fab fragments are
generated by reducing the disulfide bridges of the F(ab')2
fragments. Alternatively, Fab expression libraries can be
constructed to allow rapid and easy identification of
monoclonal Fab fragments with the desired specificity (see
Huse, et al. (1989) Science 254:1275-1281). In particular
CA 02790433 2012-09-19
-19-
embodiments, antibody fragments of the present invention
are fragments of neutralizing antibodies which retain the
variable region binding site thereof. Exemplary are F(ab')2
fragments, F(ab') fragments, and Fab fragments. See
generally Immunology: Basic Processes (1985) 2d edition, J.
Bellanti (Ed.) pp. 95-97-
Peptide aptamers which differentially recognize multi-
dimensional conformations of ADDLs can be rationally
designed or screened for in a library of aptamers (e.g.,
provided by Aptanomics SA, Lyon, France). In general,
peptide aptamers are synthetic recognition molecules whose
design is based on the structure of antibodies. Peptide
aptamers consist of a variable peptide loop attached at
both ends to a protein scaffold. This double structural
constraint greatly increases the binding affinity of the
peptide aptamer to levels comparable to that of an antibody
(nanomolar range).
Exemplary nucleic acid sequences encoding heavy and
light chain variable regions for use in producing antibody
and antibody fragments of the instant invention are
disclosed herein in Figures 6 and 10 (i.e., SEQ ID NOs:l-24
and SEQ ID NOs:132-151). As will be appreciated by the
skilled artisan, the heavy chain variable regions disclosed
herein can be used in combination with any one of the light
chain variable regions disclosed herein to generate
antibodies with modified affinities, dissociate constants,
epitopes and the like. For example, combining the light
chain variable region of 2H4 (encoded by SEQ ID NO:12) with
the heavy chain variable region of 2A10 (encoded by SEQ ID
NO:13) may provide for recognition of a larger linear
epitope.
Exemplary heavy and light chain CDRs for use in
producing an antibody or antibody fragment of the instant
invention are disclosed in Figures 7A-7F and have amino
acid sequences set forth in SEQ ID NOs:25, 26, and 28
CA 02790433 2012-09-19
-20-
(heavy chain CDR1) ; SEQ ID NOs: 29, 30, 31, 33, 34, 35, and
36 (heavy chain CDR2); SEQ ID NOs:38, 39, 40, 41, 43, 44,
45, 46, 47 and 48 (heavy chain CDR3); SEQ ID NOs:49, 50, 51
and 53 (light chain CDR1); SEQ ID NOs:54, 55, 56, and 58
(light chain CDR2) ; and SEQ ID NOs:59, 60, 61, 62, 63, 64,
and 66 (light chain CDR3). Particular embodiments of the
heavy and light chains of the antibody or antibody
fragments of the instant invention are as follows. A heavy
chain CDR1 having an amino acid sequence of Ser-Phe-Gly-
Met-His (SEQ ID NO:28) or Thr-Ser-Gly-Met-Gly-Val-Xaa (SEQ
ID NO:27), wherein Xaa is an amino acid with no side chain
or a small side chain (e.g., Ser, Gly, or Ala). A heavy
chain CDR2 having an amino acid sequence of His-Ile-Xaal-
Trp-Asp-Asp-Asp-Lys-Xaa2-Tyr-Asn-Pro-Ser-Leu-Lys-Ser (SEQ ID
NO:32), wherein Xaal is an amino acid with an aromatic side
chain group (e. g. , Phe, Tyr or Trp) and Xaa2 is Ser, Arg or
Tyr; or a heavy chain CDR2 having an amino acid sequence of
Tyr-Ile-Xaas-Xaa2-Xaa3-Ser-Xaa4-Thr-Ile-Tyr-Tyr-Ala-Asp-Thr-
Val-Lys-Arg (SEQ ID NO:37), wherein Xaaj and Xaa2 are amino
acids with a polar side chain group (e.g., Arg, Ser, Gly,
Thr, Cys, Tyr, Asn, Gln, Lys, or His); Xaa3 is Gly or Val;
and Xaa4 is an amino acid with a polar and uncharged side
group (e.g., Gly, Ser, Thr, Cys, Tyr, Asn, or Gln) . A heavy
chain CDR3 having an amino acid sequence of Arg-Ser-Ile-
Xaal-Xaa2-Xaa3-Xaa4-Pro-Glu-Asp-Tyr-Phe-Xaa5-Tyr (SEQ ID
NO:42), wherein Xaal is an amino acid with a polar and
uncharged side group (e.g., Gly, Ser, Thr, Cys, Tyr, Asn,
or Gln); Xaa2 is an amino acid with hyroxyl side chain group
(e.g., Ser or Thr) ; Xaa3 and Xaa4 are amino acids with an
aliphatic side chain group (e.g., Ala, Val, Leu, Ile, or
Pro); and Xaas is Asp or Ala. A light chain CDR1 having an
amino acid sequence of Arg-Ser-Ser-Gln-Ser-Xaas-Xaa2-His-
Ser-Asn-Gly-Asn-Thr-Tyr-Leu-Xaa3 (SEQ ID N0:52), wherein
Xaal and Xaa2 are amino acids with an aliphatic side chain
group (e.g., Ala, Val, Leu, Ile, or Pro) and Xaa3 is an
CA 02790433 2012-09-19
-21-
amino acid with a charged side chain group (e.g., Asp, Glu,
Arg, His, or Lys). A light chain CDR2 having an amino acid
sequence of Lys-Xaa1-Ser-Asn-Arg-Phe-Xaa2 (SEQ ID NO: 57) ,
wherein Xaal is an amino acid with an aliphatic side chain
group (e.g., Ala, Val, Leu, Ile, or Pro) and Xaa2 is Ser or
Phe. A light chain CDR3 having an amino acid sequence of
Xaal-Gln-Xaa2-Xaa3-Xaa4-Val -Pro-Xaas-Thr (SEQ ID NO: 65),
wherein Xaal is Ser or Phe; Xaa2 is an amino acid with no
side. chain (e.g., gly) or hyroxyl side chain group (e.g.,
Ser or Thr); Xaa3 is an amino acid with a hyroxyl side chain
group (e. g. , Ser or Thr) ; Xaa4 is His, Tyr or Leu; and Xaa5
is an amino acid with an aliphatic side chain group (e.g.,
Ala, Val, Leu, Ile, or Pro). As will be appreciated by the
skilled artisan, one or more of the CDRs within the heavy
and light chain variable regions of an antibody can be
replaced with one or more CDRs from another antibody to
generate a wholly new antibody or antibody fragment. For
example, replacing CDR3 of the heavy chain of 5F10 with the
CDR3 of the heavy chain from 4E2 (SEQ ID NO:41) may enhance
that ability of 5F10 to block binding of ADDLs to neuronal
cells.
Antibodies with particular characteristics are
contemplated. In one embodiment, an antibody which binds
the 3-8 amino acid epitope of A(31-42 has a heavy chain CDR1
amino acid sequence of Thr-Ser-Gly-Met-Gly-Val-Xaa (SEQ ID
NO:27), wherein Xaa is an amino acid with no side chain or
a small side chain (e.g., Ser, Gly, or Ala); or a heavy
chain CDR2 amino acid sequence of His-Ile-Xaas-Trp-Asp-Asp-
Asp-Lys-Xaa2-Tyr-Asn -Pro- Ser-Leu-Lys-Ser (SEQ ID NO:32),
wherein Xaal is an amino acid with an aromatic side chain
group (e.g., Phe, Tyr or Trp) and Xaa2 is Ser, Arg or Tyr.
In another embodiment, an antibody with a moderate affinity
for large (>50 kDa) ADDL aggregates over small (<30 kDa)
aggregates (i.e. SEC Peak 1 and Peak 2, respectively), has
a heavy chain CDR3 amino acid sequence of Arg-Ser-Ile-Xaal-
CA 02790433 2012-09-19
-22-
Xaa2-Xaa3-Xaa4-Pro-Glu-Asp-Tyr-Phe-Xaa5-Tyr (SEQ ID NO:42),
wherein Xaay is an. amino acid with a polar and uncharged
side group (e.g., Gly, Ser, Thr, Cys, Tyr, Asn, or Gln),
Xaa2 is an amino acid with hyroxyl side chain group (e.g.,
Ser or Thr), Xaa3 and Xaa4 are amino acids with an aliphatic
side chain group (e.g., Ala, Val, Leu, Ile, or Pro), and
Xaa5 is Asp or Ala.
Antibodies or antibody fragments of the present
invention can have additional moieties attached thereto.
For example, a microsphere or microparticle can be attached
to the antibody or antibody fragment, as described in U.S.
Patent No. 4,493,825, the disclosure of which is
incorporated. herein by reference.
Moreover, antibody or antibody fragments of the
invention can be mutated and selected for increased antigen
affinity, neutralizing activity (i.e., the ability to block
binding of ADDLs to neuronal cells or the ability to block
ADDL assembly), or a modified dissociation constant.
Mutator strains of E. coli (Low, et al. (1996) J_ Mot.
Biol. 260:359-368), chain shuffling (Figini, et al. (1994)
supra), and PCR mutagenesis are established methods for
mutating nucleic acid molecules encoding antibodies. By way
of illustration, increased affinity can be selected for by
contacting a large number of phage antibodies with a low
amount of biotinylated antigen so that the antibodies
compete for binding- In this case, the number of antigen
molecules should exceed the number of phage antibodies, but
the concentration of antigen should be somewhat below the
dissociation constant. Thus, predominantly mutated phage
antibodies with increased affinity bind to the biotinylated
antigen, while the larger part of the weaker affinity phage
antibodies remains unbound. Streptavidin can then assist in
the enrichment of the higher affinity, mutated phage
antibodies from the mixture (Schier, et al. (1996) J_ Mot.
Biol. 255:28-43). Exemplary affinity-maturated light chain
CA 02790433 2012-09-19
-23-
CDR3 amino acid sequences are disclosed herein (see Tables
11 and 12), with particular embodiments embracing a light
chain CDR3 amino acid sequence of Xaa1-Gln-Xaa2-Thr-Arg-Val-
Pro-Leu-Thr (SEQ ID NO:316), wherein Xaal is Phe or Leu, and
Xaa1 is Ala or Thr.
For some therapeutic applications it may be desirable
to reduce the dissociation of the antibody from the
antigen. To achieve this, the phage antibodies are bound to
biotinylated antigen and an excess of unbiotinylated
antigen is added. After a period of time, predominantly the
phage antibodies with the lower dissociation constant can
be harvested with streptavidin (Hawkins, et al. (1992) J.
Mol. Biol. 226:889-96).
Various immunoassays including those disclosed herein
can be used for screening to identify antibodies, or
fragments thereof, having the desired specificity for
multi-dimensional conformations of ADDLs. Numerous
protocols for competitive binding (e.g, ELISA), latex
agglutination assays, immunoradiometric assays, kinetics
(e.g., BIACORETM analysis) using either polyclonal or
monoclonal antibodies, or fragments thereof, are well-known
in the art. Such immunoassays typically involve the
measurement of complex formation between a specific
antibody and its cognate antigen. A two-site, monoclonal-
based immunoassay utilizing monoclonal antibodies reactive
to two non-interfering epitopes is suitable, but a
competitive binding assay can also be employed. Such assays
can also be used in the detection of multi-dimensional
conformations of ADDLs in a sample.
An antibody or antibody fragment can also be subjected
to other biological activity assays, e.g., displacement of
ADDL binding to neurons or cultured hippocampal cells or
blockade of ADDL assembly, in order to evaluate
neutralizing or pharmacological activity and potential
CA 02790433 2012-09-19
-24-
efficacy as a prophylactic or- therapeutic agent. Such
assays are described herein and are well-known in the art.
Antibodies and fragments of antibodies can be produced
and maintained as hydridomas or alternatively recombinantly
produced in any well-established expression system
including, but not limited to, E. coli, yeast (e.g.,
Saccharomyces spp. and Pichia spp.), baculovirus, mammalian
cells (e.g., myeloma, CHO, COS), plants, or transgenic
animals (Breitling and Diabel (1999) In: Recombinant
Antibodies, John Wiley & Sons, Inc., NY, pp_ 119-132).
Exemplary nucleic acid sequences of IgG1 and IgG2m4 heavy
chain variable regions, as well as kappa light chain
variable regions for humanized 4E2, 26D6, 20C2, 3B3, 2H4,
and 1F6 generated by CDR grafting and veneering are
presented in Figures 10A to lOS and set forth herein as SEQ
ID NOs:132 to 151. For antibodies and fragments of
antibodies can be isolated using any appropriate methods
including, but not limited to, affinity chromatography,
immunoglobulins-binding molecules (e.g., proteins A, L, G
or H), tags operatively linked to the antibody or antibody
fragment (e.g., His-tag, FLAG -tag, Strep tag, c-myc tag)
and the like. See, Breitling and Diibel (1999) supra.
Antibodies and antibody fragments of the instant
invention have a variety of uses including, diagnosis of
diseases associated with-accumulation of ADDLs, blocking or
inhibiting binding of ADDLs to neuronal cells, blocking
ADDL assembly, prophylactically or therapeutically treating
a disease associated with ADDLs, identifying therapeutic
agents that prevent binding of ADDLs to neurons, and
preventing the phosphorylation of tau protein at
Ser202/Thr205.
Antibody and antibody fragments of the instant
invention are also useful in a method for blocking or
inhibiting binding of ADDLs to neuronal cells. This method
of the invention is carried out by contacting a neuron, in
CA 02790433 2012-09-19
-25-
vitro or in vivo, with an antibody or antibody fragment of
the present invention so that binding of ADDLs to the
neuron is blocked. In particular embodiments, an antibody
or antibody fragment of the instant invention achieves at
least a 15%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%,
or 97% decrease in the binding of ADDLs as compared to
binding of ADDLs in the absence of the antibody or antibody
fragment. The degree to which an antibody can block thee
binding of ADDLs to a neuron can be determined in
accordance with the methods disclosed herein, i.e.,
immunocytochemistry or cell-based alkaline phosphatase
assay or any other suitable assay. Antibodies particularly
useful for decreasing binding of ADDLs to neuronal cells
include the exemplary 20C2, 3B3, 1F4, 1F6, 4E2, 2B4, 2D6,
and 2H4 monoclonal antibodies.
Antibody and antibody fragments of the instant
invention are further useful in a method for blocking or
inhibiting assembly of ADDLs. This method involves
contacting a sample containing amyloid (3 i-42 peptides with
an antibody or antibody fragment of the instant invention
so that ADDL assembly is inhibited. The degree to which an
antibody can block the assembly of ADDLs can be determined
in accordance with the methods disclosed herein, i.e., FRET
or fluorescence polarization or any other suitable assay.
Antibodies particularly useful for blocking the assembly of
ADDLs include the exemplary 1F4, 20C2, 4C2, 1F6, 2B4, 5F10,
2A10, and 2D6 antibodies.
Antibodies disclosed herein are also useful in methods
for preventing the phosphorylation of tau protein at
Ser202/Thr205. This method involves contacting a sample
containing tau protein with an antibody or antibody
fragment of the instant invention so that binding of ADDLs
to neurons is blocked thereby preventing phosphorylation of
tau protein. The degree to which an antibody can prevent
the phosphorylation of tau protein at Ser202/Thr205 can be
CA 02790433 2012-09-19
-26-
determined in accordance with the methods disclosed herein
or any other suitable assay.
Blocking or decreasing binding of ADDLs to neurons,
inhibiting assembly of ADDLs, and preventing the
phosphorylation of tau protein at Ser202/Thr205 all find
application in methods of prophylactically or
therapeutically treating a disease associated with the
accumulation of ADDLs. Accordingly, the present invention
also embraces the use of an antibody or antibody fragment
of the instant invention to prevent or treat a disease
associated with the accumulation of ADDLs (e.g. Alzheimer's
or similar memory-related disorders. Patients amenable to
treatment include individuals at risk of disease but not
exhibiting symptoms, as well as patients presently
exhibiting symptoms. In the case of Alzheimer's Disease,
virtually anyone is at risk of suffering from Alzheimer's
Disease if he or she lives long enough. Therefore, the
antibody or antibody fragments of the present invention can
be administered prophylactically to the general population
without the need for any assessment of the risk of the
subject patient- The present methods are especially useful
for individuals who have a known genetic risk of
Alzheimer's Disease. Such individuals include those having
relatives who have been diagnosed with the disease, and
those whose risk is determined by analysis of genetic or
biochemical markers. Genetic markers of risk for
Alzheimer's Disease include mutations in the APP gene,
particularly mutations at position 717 and positions 670
and 671 referred to as the Hardy and Swedish mutations
respectively. Other markers of risk are mutations in the
presenilin genes, PSI and PS2, and ApoE4, family history of
Alzheimer's Disease, hypercholesterolemia or
atherosclerosis. Individuals presently suffering from
Alzheimer's Disease can be recognized from characteristic
dementia, as well as the presence of risk factors described
CA 02790433 2012-09-19
-27-
above. In addition, a number of diagnostic tests are
available for identifying individuals who have Alzheimer's
Disease. These include measurement of CSF tau and A(31-42
levels. Individuals suffering from Alzheimer's Disease can
also be diagnosed by ADRDA criteria or the method disclosed
herein.
In asymptomatic patients, treatment can begin at any
age (e.g., 10, 20, 30 years of age). Usually, however, it
is not necessary to begin treatment until a patient reaches
40, 50, 60 or 70 years of age. Treatment typically entails
multiple dosages over a period of time. Treatment can be
monitored by assaying for the presence of ADDLs over time.
In therapeutic applications, a pharmaceutical
composition or medicament containing an antibody or
antibody fragment of the invention is administered to a
patient suspected of, or already suffering from such a
disease associated with the accumulation of ADDLs in an
amount sufficient to cure, or at least partially arrest,
the symptoms of the disease (biochemical, histologic and/or
behavioral), including its complications and intermediate
pathological phenotypes in development of the disease. In
prophylactic applications, a pharmaceutical composition or
medicament containing an antibody or antibody fragment of
the invention is administered to a patient susceptible to,
or otherwise at risk of, a disease associated with the
accumulation of ADDLs in an amount sufficient to achieve
passive immunity in the patient thereby eliminating or
reducing the risk, lessening the severity, or delaying the
outset of the disease, including biochemical, histologic
and/or behavioral symptoms of the disease, its
complications and intermediate pathological phenotypes
presenting during development of the disease. In some
methods, administration of agent reduces or eliminates
myocognitive impairment in patients that have not yet
developed characteristic Alzheimer's pathology. In
CA 02790433 2012-09-19
-28-
particular embodiments, an effective amount of an antibody
or antibody fragment of the invention is an amount which
achieves at least a 15%, 20%, 30%, 40%, 50%, 60%, 70%, 80%,
90%, 95%, or 97% decrease in the binding of ADDLs to
neurons in the patient as compared to binding of ADDLs in
the absence of treatment. As such, impairment of long-term
potentiation/memory formation is decreased-
Effective doses of the compositions of the present
invention, for the treatment of the above described
conditions vary depending upon many different factors,
including means of administration, physiological state of
the patient, whether the patient is human or an animal,
other medications administered, and whether treatment is
prophylactic or therapeutic. Usually, the patient is a
human but nonhuman mammals such as dogs or transgenic
mammals can also be treated.
.Treatment dosages are generally titrated to optimize
safety and efficacy. For passive immunization with an
antibody or antibody fragment, dosage ranges from about
0.0001 to 100 mg/kg, and more usually 0.01 to 5 mg/kg, of
the host body weight are suitable. For example, dosages can
be 1 mg/kg body weight or 10 mg/kg body weight or within
the range of 1-10 mg/kg. An exemplary treatment regime
entails administration once per every two weeks or once a
month or once every 3 to 6 months. In some methods, two or
more antibodies of the invention with different binding
specificities are administered simultaneously, in which
case the dosage of each antibody administered falls within
the ranges indicated. Antibodies are usually administered
on multiple occasions, wherein intervals between single
dosages can be weekly, monthly or yearly. Intervals can
also be irregular as indicated by measuring blood levels of
antibody to ADDLs in the patient. In some methods, dosage
is adjusted to achieve a plasma antibody concentration of
1-1000 pg/mL and in some methods 25-300 pg/mL.
CA 02790433 2012-09-19
-29-
Alternatively, the antibody or antibody fragment can be
administered as a sustained-release formulation, in which
case less frequent administration is required- Dosage and
frequency vary depending on the half-life of the antibody
in the patient- In general, human and humanized antibodies
have longer half-lives than chimeric antibodies and
nonhuman antibodies. As indicated above, dosage and
frequency of administration can vary depending on whether
the treatment is prophylactic or therapeutic. In
prophylactic applications, a relatively low dosage is
administered at relatively infrequent intervals over a long
period of time. Some patients continue to receive treatment
for the rest of their lives. In therapeutic applications, a
relatively high dosage at relatively short intervals is
sometimes required until progression of the disease is
reduced or terminated, and preferably until the patient
shows partial or complete amelioration of symptoms of
disease. Thereafter, the patient can be administered a
prophylactic regime.
Antibody and antibody fragments of the instant
invention can be administered as a component of a
pharmaceutical composition or medicament- Pharmaceutical
compositions or medicaments generally contain the active
therapeutic agent and a variety of other pharmaceutically
acceptable components. See Remington: The Science and
Practice of Pharmacy, Alfonso R_ Gennaro, editor, 20th ed_
Lippincott Williams & Wilkins: Philadelphia, PA, 2000. The
preferred form depends on the intended mode of
administration and therapeutic application. Pharmaceutical
compositions can contain, depending on the formulation
desired, pharmaceutically-acceptable, non-toxic carriers or
diluents, which are defined as vehicles commonly used to
formulate pharmaceutical compositions for animal or human
administration. Diluents are selected so as not to affect
the biological activity of the combination. Examples of
CA 02790433 2012-09-19
-30-
such diluents are distilled water, physiological phosphate-
buffered saline, Ringer's solutions, dextrose solution, and
Hank's solution.
Pharmaceutical compositions can also contain large,
slowly metabolized macromolecules such as proteins,
polysaccharides such as chitosan, polylactic acids,
polyglycolic acids and copolymers (such as latex-
functionalized SEPHAROSET", agarose, cellulose, and the
like), polymeric amino acids, amino acid copolymers, and
lipid aggregates (such as oil droplets or liposomes).
Administration of a pharmaceutical composition or
medicament of the invention can be carried out via a
variety of routes including, but not limited to, oral,
topical, pulmonary, rectal, subcutaneous, intradermal,
intranasal, intracranial, intramuscular, intraocular, or
intra-articular injection, and the like. The most typical
route. of administration is intravenous followed by
subcutaneous, although other routes can be equally
effective. Intramuscular injection can also be performed in
the arm or leg muscles. In some methods, agents are
injected directly into a particular tissue where deposits
have accumulated, for example, intracranial injection. In
some embodiments, an antibody or antibody fragment is
injected directly into the cranium. In other embodiments,
antibody or antibody fragment is administered as a
sustained-release composition or device, such as a MEDIPADT"
device.
For parenteral administration, antibody or antibody
fragments of the invention can be administered as
injectable dosages of a solution or suspension of the
substance in a physiologically acceptable diluent with a
pharmaceutical carrier that can be a sterile liquid such as
water, oils, saline, glycerol, or ethanol. Additionally,
auxiliary substances, such as wetting or emulsifying
agents, surfactants, pH buffering substances and the like
CA 02790433 2012-09-19
-31-
can be present in compositions. Other components of
pharmaceutical compositions are those of petroleum, animal,
vegetable, or synthetic origin, for example, peanut oil,
soybean oil, and mineral oil. In general, glycols such as
propylene glycol or polyethylene glycol are suitable liquid
carriers, particularly for injectable solutions. Antibodies
can be administered in the form of a depot injection or
implant preparation which can be formulated in such a
manner as to permit a sustained-release of the active
ingredient An exemplary composition contains an antibody at
5 mg/mL, formulated in aqueous buffer composed of 50 mM L-
histidine, 150 mM NaCl, adjusted to pH 6.0 with HC1.
Typically, compositions are prepared as injectables,
either as liquid solutions or suspensions; solid forms
suitable for solution in, or suspension in, liquid vehicles
prior to injection can also be prepared. The preparation
also can be emulsified or encapsulated in liposomes or
micro particles such as polylactide, polyglycolide, or
copolymer for enhanced delivery.
For suppositories, binders and carriers include, for
example, polyalkylene glycols or triglycerides; such
suppositories can be formed from mixtures containing the
active ingredient in the range of 0.5% to 10%, or more
desirably 1%-2%.
Oral formulations include excipients, such as
pharmaceutical grades of mannitol, lactose, starch,
magnesium stearate, sodium saccharine, cellulose, and
magnesium carbonate- These compositions take the form of
solutions, suspensions, tablets, pills, capsules,
sustained-release formulations or powders and contain 10%-
95% of active ingredient, or more suitably 25%-70%.
Topical application can result in transdermal or
intradermal delivery- Topical administration can be
facilitated by co-administration of the agent with cholera
toxin or detoxified derivatives or subunits thereof or
CA 02790433 2012-09-19
-32-
other similar bacterial toxins (see Glenn, et al. (1998)
Nature 391:851) . Co-administration can be achieved by using
the components as a mixture or as linked molecules obtained
by chemical crosslinking or expression as a fusion protein-
Alternatively, transdermal delivery can be achieved
using a skin path or using transferosomes (Paul, et al.
(1995) Eur. J. Immunol. 25:3521-24; Cevc, et al. (1998)
Bi ochem. Biophys. Acta 1368:201-15).
An antibody or antibody fragment of the invention can
optionally be administered in combination with other agents
that are at least partly effective in treatment of
amyloidogenic disease.
Antibody and antibody fragments of the instant
invention also find application in the identification of
therapeutic agents that prevent the binding of ADDLs to
neurons (e.g. a hippocampal cell) thereby preventing
downstream events attributed to ADDLs. Such an assay is
carried out by contacting a neuron with ADDLs in the
presence of an agent and using an antibody of antibody
fragment of the invention to determine binding of the ADDLs
to the neuron in the presence of the agent. As will be
appreciated by the skilled artisan, an agent that blocks
binding of ADDLs to a neuron will decrease the amount of
ADDLs bound to the neuron as compared to a neuron which has
not been contacted with the agent; an amount which is
detectable in an immunoassay employing an antibody or
antibody fragment of the instant invention. Suitable
immunoassays for detecting neuronal-bound ADDLs are
disclosed herein.
Agents which can be screened using the method provided
herein encompass numerous chemical classes, although
typically they are organic molecules, preferably small
organic compounds having a molecular weight of more than
100 and less than about 2,500 daltons. Agents encompass
functional groups necessary for structural interaction with
CA 02790433 2012-09-19
-33-
proteins, particularly hydrogen bonding, and typically
include at least an amine, carbonyl, hydroxyl or carboxyl
group, preferably at least two of the functional chemical
groups. The agents often contain cyclical carbon or
heterocyclic structures and/or aromatic or polyaromatic
structures substituted with one or more of the above
functional groups. Agents can also be found among
biomolecules including peptides, antibodies, saccharides,
fatty acids, steroids, purines, pyrimidines, derivatives,
structural analogs or combinations thereof. Agents are
obtained from a wide variety of sources including libraries
of natural or synthetic compounds-
A variety of other reagents such as salts and neutral
proteins can be included in. the screening assays. Also,
reagents that otherwise improve the efficiency of the
assay, such as protease inhibitors, nuclease inhibitors,
anti-microbial agents, and the like can be used. The
mixture of components can be added in any order that
provides for the requisite binding.
Agents identified by the screening assay of the
present invention will be beneficial for the treatment of
amyloidogenic diseases and/or tauopathies. In addition, it
is contemplated that the experimental systems used to
exemplify these concepts represent research tools for the
evaluation, identification and screening of novel drug
targets associated with amyloid beta induction of tau
phosphorylation.
The present invention also provides methods for
detecting ADDLs and diagnosing a disease associated with
accumulation of ADDLs using an antibody or antibody
fragment of the instant invention. A disease associated
with accumulation of ADDLs is intended to include any
disease wherein the accumulation of ADDLs results in
physiological impairment of long-term potentiation/memory
formation. Diseases of this type include, but are not
CA 02790433 2012-09-19
-34-
limited to, Alzheimer's Disease and similar memory-related
disorders.
In accordance with these methods, a sample from a
patient is contacted with an antibody or antibody fragment
of the invention and binding of the antibody or antibody
fragment to the sample is indicative of the presence of
ADDLs in the sample. As used in the context of the present
invention, a sample is intended to mean any bodily fluid or
tissue which is amenable to analysis using immunoassays.
Suitable samples which can be analyzed in accordance with
the methods of the invention include, but are not limited
to, biopsy samples and fluid samples of the brain from a
patient (e.g., a mammal such as a human). For in vitro
purposes (e.g., in assays monitoring oligomer formation), a
sample can be a neuronal cell line or tissue sample. For
diagnostic purposes, it is contemplated that the sample can
be from an individual suspected of having a disease
associated with accumulation of ADDLs or from an individual
at risk of having a disease associated with accumulation of
ADDLs, e.g., an individual with a family history which
predisposes the individual to a disease associated with
accumulation of ADDLs.
Detection of binding of the antibody or antibody
fragment to ADDLs in the sample can be carried out using
any standard immunoassay (e.g., as disclosed herein), or
alternatively when the antibody fragment is, e.g., a
peptide aptamer, binding can be directly detected by, for
example, a detectable marker protein (e.g., j3-
galactosidase, GFP or luciferase) fused to the aptamer.
Subsequently, the presence or absence of the ADDL-antibody
complex is correlated with the presence or absence,
respectively, of ADDLs in the sample and therefore the
presence or absence, respectively, of a disease associated
with accumulation of ADDLs_ It is contemplated that one or
more antibodies or antibody fragments of the present
CA 02790433 2012-09-19
-35-
invention can be used in conjunction with current non-
invasive immuno-based imaging techniques to greatly enhance
detection and early diagnosis of a disease associated with
accumulation of ADDLs.
To facilitate diagnosis the present invention also
pertains to a kit for containing an antibody or antibody
fragment of the instant invention. The kit includes a
container holding one or more antibody or antibody
fragments which recognizes multi-dimensional conformation
of ADDLs and instructions for using the antibody for the
purpose of binding to ADDLs to form an antibody-antigen
complex and detecting the formation of the antibody-antigen
complex such that the presence or absence of the antibody-
antigen complex correlates with presence or absence of
ADDLs in the sample. Examples of containers include
multiwell plates which allow simultaneous detection of
ADDLs,in multiple samples.
The invention is described in greater detail by the
following non-limiting examples.
Example 1: General Materials and Methods
ADDL Preparation. ADDLs in F12 medium (Biosource,
Camarillo, CA) were prepared from AR1-42 in accordance with
established methods (Lambert, et al. (2001) supra).
Briefly, A31-42 peptide (American Peptide Co., Sunnyvale,
CA or California Peptide Research, Inc., Napa, CA) was
weighed and placed in a glass vial capable of holding a
sufficient quantity of HFIP (1,1,1,3,3,3-hexafluoro-2-
propanol) to achieve a peptide concentration of 10 mg/mL.
HFIP was added to the dry peptide, the vial was capped and
gently swirl to mix, and the peptide/HFIP solution was
stored at room temperature for at least one hour- Aliquots
(50 or 100 pL, 0.5 or 1.0 mg, respectively) of peptide
solution was dispensed into a series of 1.5 mL conical
centrifuge tubes. The tubes were placed in a speedvac
CA 02790433 2012-09-19
-36-
overnight to remove the HFIP. Tubes containing the dried
peptide film were capped and stored at -70 C in a sealed
container with dessicant.
Prior to use, the A~1-42 peptide film was removed from
-70 C storage and allowed to warm to room temperature.
Fresh DMSO (44 jiL/mg of peptide film; 5 mM) was added and
the peptide/DMSO mixture was incubated on a vortex mixer at
the lowest possible speed for ten minutes. F12 media (2
mL/mg peptide) was dispensed into each tube of DMSO/peptide
and the tube was capped and mixed by inversion. The 100 pM
preparation was stored at 2-8 C for eighteen to twenty four
hours. The samples were centrifuged at 14,000 x g for ten
minutes at 2-8 C. The supernatant was transferred to a
fresh tube and stored at 2-8 C until used.
Blotinylated ADDL preparations (bADDLs) were prepared
in the same manner as described above for ADDL preparations
using 100% N-terminal biotinylated amyloid beta peptide
(American Peptide Company, Sunnyvale, CA).
ADDL Fibril Preparation. To room temperature ADDL
peptide film was added 2 mL of 10 mM hydrochloric acid per
mg peptide. The solution was mixed on a vortex mixer at the
lowest possible speed for five to ten minutes and the
resulting preparation was stored at 37 C for eighteen to
twenty four hours before use.
Monomer Preparation. HFIP dry down preparations of
amyloid beta (1-40) peptide (A01-40) were prepared as
outlined for A(3(1-42) peptide. The peptide film was
dissolved in 2 mL of 25 mM borate buffer (pH 8.5) per mg of
peptide, divided into aliquots, and frozen at -70 C until
used.
Human Fibril Preparation. Samples obtained from frozen
human cortex were homogenized in 20X cold F12 medium with
protease inhibitors (COMPLETE , Roche Diagnostics
Corporation, Indianapolis, IN) for 1 minute. The sample was
then centrifuged at 10,000 x g for 1 hour at 4 C. After
CA 02790433 2012-09-19
-37-
washing twice with F12, the pellet was resuspended in 2%
SDS/F12 and incubated on ice for 30 minutes. The sample was
subsequently centrifuged at 220,000 x g for 1 hour at 4 C.
The pellet was resuspended in cold F12 and sonicated for 1
minute in 15-second bursts- Protein was determined using
COOMASSIE PLUSTM kit (Pierce Biotechnology, Rockford, IL) .
Immunization. The resulting soluble A(3 oligomers,
referred to herein as "synthetic" ADDLs, were mixed 1:1
with complete Freund's adjuvant (first and second
vaccination) or incomplete Freund's adjuvant (all
subsequent vaccinations) and injected subcutaneously (first
two vaccinations) or intraperitoneally into three mice in a
total volume of ^-1 mL/mouse. Each injection consisted of
purified ADDLs equivalent to 194 25 pg total protein.
Mice were injected approximately every three weeks. After
six injections, one mouse died and its spleen was frozen.
The spleen from the mouse with the highest titer serum was
then fused with SP2/0 myeloma cells in the presence of
polyethylene glycol and plated out into six 96-well plates.
The cells were cultured at 37 C with 5% CO2 for ten days in
200 pL of HAT selection medium, which is composed of ISCOV
medium supplemented with 10% fetal bovine serum (FBS), 1
pg/mL HYBRIMAX (azaserine-hypoxanthine; Sigma-Aldrich, St.
Louis, MO), and 30% conditioned media collected from SP2/0
cell culture. The cultures were fed once with ISCOV medium
supplemented with 10% FBS on day 10, and the culture
supernatants were removed on day 14 to screen for positive
wells in ELISA_ The positive cultures were further cloned
by limiting dilutions with probability of 0.3 cells per
well. The positive clones were confirmed in ELISA and
further expanded.
Screening of supernates involved five assays: a dot
blot and western immunoblot (Lambert, et al. (2001) supra) ,
a native immunoblot using synthetic ADDLs, and a dot blot
and western blot using endogenous fibrils obtained from
CA 02790433 2012-09-19
-38-
human tissue. These assays tested the binding of antibodies
to ADDLs (the dot blot) and identified the oligomer(s) that
had the greatest affinity (western). All antibodies were
tested in the dot blot using 5 pmole ADDLs (576 supernates
in the first fusion and 1920 supernates in the second).
Those supernatants that tested positive were then screened
further using western blot at 10-20 pmole ADDLs. The screen
was repeated to identify low positives or false positives.
Teri wells supernatants expanded for the first mouse and
forty-five wells were expanded for the second mouse. The
expanded cells were then frozen or subcloned.
Monoclonal antibody-containing ascites were produced
in female balb/c mice using standard protocols (Current
Protocol of Molecular Biology)_- Briefly, mice were primed
by intraperitoneal injection of 0.5 mL of pristane. One
week after the priming, mice were injected
intraperitoneally with approximately 5 x 106 hybridoma cells
in 1 mL phosphate-buffered saline (PBS). Ascites were
collected ten to fourteen days later. IgG purification was
carried out by using BIO-RAD AFFI-GEL Protein A MAPS II
kit, according to manufacturer's protocol. For each run, 3
mL ascites were desalted by passage through a desalting
column and elution in 4 mL binding buffer. The sample was
then applied to the Protein A column. After washing with 40
mL binding buffer, the column was eluted with elution
buffer and the 5 mL fractions were collected. Samples were
neutralized by addition of 60 pL of 10 N NaOH. To exchange
the buffer to PBS, the samples were applied to a second
desalting column and eluted with PBS.
Control Antibodies. Polyclonal antibodies M71/2 and
M90/1 were obtained from Bethyl Laboratories, Inc.
(Montgomery, TX). Anti-A3 monoclonal antibodies 6E10
(raised against residues 1-17) and 4G8 (raised against
residues 17-24) were obtained from Signet Labs (Dedham,
MA). Monoclonal antibody WO-2 is known in the art for its
CA 02790433 2012-09-19
-39-
ability to recognize both 1-40 and 1-42 via western blot
analysis (Ida, et al. (1996) J. Biol. Chem. 271: 22908-
22914. Monoclonal antibody BAM-10 (raised against API-40)
was obtained from ABCAM (Cambridge, MA). Monoclonal
antibody 26D6 is well-known in the art for its ability to
recognize amino acids 1-12 of A(3 sequence (Lu, et al.
(2000) Nat. Pled. 6:397-404).
Immunoblot Analysis. Sodium dodecyl sulfate
polyacrylamide gel electrophoresis (SDS-PAGE) was performed
using established methods (Lambert, et al. (2001) supra),
except that 10-20% Tris-Tricine gels (BIO-RAD , Hercules,
CA) were used and the separation was performed at 120 V.
Gels were transferred according to standard methods and
secondary antibody was routinely used at a 1:40,000
dilution.
For initial screening, 2.7 pg ADDLs, equivalent to
-16-20 pmol/lane, were separated on two-dimensional (2D) 4-
20% gels. Electrophoresis and transfer were as above- Using
the tracking dye as a guide, the nitrocellulose was placed
into a Surf-blot apparatus (Idea Scientific, Minneapolis,
MN) and 200 pL of hybridoma supernate mixed with blocking
buffer, composed of 5% nonfat dry milk in tris-buffered
saline with TWEEN 1 20 (TBS-T; Lambert et al. (2001) supra),
was added to each of 20-21 wells. After incubation at room
temperature for 1.5 hour with rocking, the supernatants
were removed and the wells were washed with 200 pL blocking
buffer. The membrane was then removed from the Surf-blot
apparatus and washed 3 x 15 minutes in TBS-T. The secondary
antibody (anti-mouse, IgG conjugated-HRP, 1:40,000;
Molecular Probes, Eugene, OR) was then incubated with the
membrane for 1 hour at room temperature. After washing (3 x
15 minutes), the oligomers were visualized with half-
strength SUPERSIGNAL (Pierce, Rockland, IL) . The western
immunoblot using human fibrils was performed in the same
CA 02790433 2012-09-19
-40-
manner using approximately 64 pg of human fibrillar tissue
in each 2D SDS-PAGE immunoblot.
Native polyacrylamide gel electrophoresis was
performed according established methods (Chromy, et al.
(2003) Biochemistry 42:12749-12760) except that the
separation was performed at 120 V_
Western Blot. Separated proteins were transferred to
nitrocellulose. Blots were blocked with 5% non-fat dry milk
or 1% bovine serum albumin (BSA) in TBS-T (TBS with 0.1%
TWEENT1" 20) overnight, incubated with primary antibody(ies)
for 1.5 hours, washed, and incubated the horseradish
peroxidase (HRP) -conjugated secondary antibody (Amersham
Biosciences Corp., Piscataway, NJ) for I hour. After final
washing, proteins were visualized with a West Femto
chemiluminescence kit (Pierce Biotechnology, Rockford, IL)
and a KODAK Image Station 440 CF or with film (HYPERFILMT",
Amersham Biosciences Corp., Piscataway, NJ).
Hippocampal Cultures. Cultures were prepared from E18
embryos according to standard methods (Brewer (1997) J.
Neurosci. Methods 71:143-155; Stevens, et al. (1996) J.
Neurosci. Res. 46:445-455). Viable cells were counted and
plated on coverslips coated with polylysine (200 pg/mL) at
densities from 1.5 x 104_106 cells/cm2. The medium was
changed by removing half of the medium and replacing it
with supplemented NEUROBASALT" media.
Primary Neurons. Primary hippocampal cultures were
prepared from frozen, dissociated neonatal rat hippocampal
cells (Cambrex, Corp., East Rutherford, NJ) that were
thawed and plated in 96-well COSTAR plates at a
concentration of 20,000 cells per well. The cells were
maintained in NEUROBASALT" media without L-glutamine (GIBCO-
BRLT"', Gaithersburg, MD) and supplemented with B27 (GIBCO-
BRL''", Gaithersburg, MD) for a period of two weeks and then
used for binding studies.
CA 02790433 2012-09-19
-41-
B103 Cells. The B103 neuroblastoma cell line (Schubert
and Behl (1993) Brain Res. 629:275-82) was grown in DMEM
without phenol red (GIBCO-BRLT"', Gaithersburg, MD), in the
presence of 10% FBS (Hyclone, Logan, UT) and 1% Pen-Strep
(GIBCO-BRLTM, Gaithersburg, MD) . Exponentially growing B103
cells--were dissociated and plated in 96-well CORNING&
plates at a concentration of 5,000 cells/well. Twenty-four
hours after plating, the cells were used to assess ADDL and
bADDL binding as well as characterize commercial and novel
anti-ADDL monoclonal antibodies.
Dot Blot Analysis- Dot blots were performed according
to Lambert, et al. ((2001) supra) applying either ADDLs (5
pmole/dot) or fibrils to the nitrocellulose. For later dot
blots, ADDLs were applied to dry nitrocellulose in
duplicate at various pmolar concentrations in 0.5 jiL using
a template derived from the Surf-blot apparatus. Samples
were then dried for 15 minutes, blocked with blocking
buffer for 1 hour, and incubated for 1.5 hour with antibody
plus or minus peptide, which had been pre-incubated for at
least 1 hour at room temperature. The solution was removed
from the Surf-blot apparatus, the wells were washed with
blocking buffer, and the membrane was removed from the
apparatus. The nitrocellulose was washed, treated with
secondary antibody, and visualized as indicated above.
Immunocytochemistry. Immunocytochemistry was performed
according to established methods (Lambert, et al. (2001)
supra), except the secondary antibodies were conjugated to
ALEXAFLUOR 588 (Molecular Probes, Eugene, OR). Antibodies
and ADDLs were preincubated for 1 hour at room temperature,
at a molar ratio of 1:4 antibody:ADDL before application to
the 21-day hippocampal cell culture. For endogenous ADDLs,
human brain protein (prepared as in Lambert, et al. (2001)
supra) was incubated with cells for 1 hour before the cells
were washed, fixed, and visualized as above.
CA 02790433 2012-09-19
-42-
Lightly fixed frozen sections (4% paraformaldehyde at
4 C for 30 hours and cryoprotected in 40 pm sucrose) from
Alzheimer's Disease and control hippocampus were incubated
with antibody (1:1000 in phosphate-buffered saline (PBS))
overnight at 4 C. After removal of antibody, sections were
washed 3 times with PBS and incubated with secondary
antibody at room temperature. Binding was then visualized
with DAB (SIGMA", St. Louis, MO). Sections were then
counterstained with hematoxylin, mounted, and imaged on a
NIKON ECLIPSE E600 light microscope with a SPOTTh INSIGHTTM
digital video camera (v. 3.2).
Quantitative Immunocytochemistry_ Cultured hippcampal
cells were incubated with 500 nM ADDLs for 1 hour at 37 C.
ADDLs were removed by washing and cells were fixed with
3.7% formaldehyde. Cells were incubated with 0.1% TRITON''
X-100 in PBS-NGS (PBS with 10% normal goat serum) for 30
minutes, washed once, and incubated with the desired
primary antibody(ies) (diluted in PBS-NGS) overnight at
4 C. Samples were washed and incubated with the appropriate
secondary antibody(ies), e.g., ALEXAFLUOR 488 or 594 anti-
mouse and anti-rabbit IgGs (Molecular Probes, Inc., Eugene,
OR), for 2 hours at 37 C. Coverslips were washed and
mounted in PROLONG anti-fade mounting medium (Molecular
Probes, Inc., Eugene, OR) and imaged using a LEICA TCS SP2
confocal Scanner DMRXE7 microscope.
ELISA. Polyclonal anti-ADDL5 IgG (M90/1; Bethyl
Laboratories, Inc., Montgomery, TX) was plated at 0.25
mg/well on IMMULONTM 3 REMOVAWELLTM strips (Dynatech Labs,
Chantilly, VA) for 2 hours at room temperature and the
wells blocked with 2% BSA in TBS. Samples diluted with 1%
BSA in F12 were added to the wells, allowed to bind for 2
hours at 4 C, and washed 3X with BSA/TBS at room
temperature. Monoclonal antibodies diluted in BSA/TBS were
incubated for 90 minutes at room temperature and detected
with a VECTASTAIN ABC kit to mouse IgG_ The HRP label was
CA 02790433 2012-09-19
-43-
visualized with BIO-RAD peroxidase substrate and read at
405 nm on a Dynex MRX-TC microplate reader.
Example 2: Development and Characterization of Anti-ADDL
Antibodies
Three mice were inoculated with ADDLs (194 25 pg
protein/injection) every three weeks for a total of six
inoculations- Hybridomas made from the fusion of these mice
spleens with SP2 cells were grown in 96-well plates.
Supernates from these wells were screened in dot blots with
synthetic ADDLs to identify positive clones, which were
compared with dot blots of endogenous fibrils to identify
differences. Hybridomas that bound only synthetic ADDLs and
not endogenous fibrils were sought. To further refine what
the products of the hybridomas bound to and under what
conditions binding occurred, three western blots of each
positive clone were performed: SDS-PAGE of ADDLs, native
gels of ADDLs, and SDS-PAGE with endogenous fibrils.
Approximately 40 clones were selected for further
examination. Each clone was tested for recognition of
soluble Alzheimer`s Disease brain extract, for
identification of ADDLs bound to cultured hippocampal
cells, and for the ability to block ADDL binding under
various conditions. Selected antibodies were collected from
culture medium and further purified using Protein G
SEPHAROSE~.
Each time a set of hybridomas was screened via dot
blot, approximately -30% yielded positive supernates. Of
these, only one or two hybridomas bound synthetic ADDLs and
not endogenous fibrils. Approximately 2% of the original
number of clones bound synthetic ADDLs and not monomer at
low ADDL concentrations, as determined by western blot
analysis- Clone 3B7, which bound synthetic ADDLs and not
fibrils on western blots, was kept for further analysis.
CA 02790433 2012-09-19
-44-
One to two clones were identified that bound higher
molecular weight material (12-24 mer) better than
trimer/tetramer oligomers. Two to three clones were
identified which could bind to native ADDLs under native
conditions, but failed to bind ADDLs in the presence of
SDS.
The results of this analysis indicated that ADDLs are
good antigens in mice and monoclonal antibodies can be
developed that bind to synthetic ADDLs with much greater
affinity than to monomers.
Example 3: Immunohistochemical Analysis of Endogenous and
Synthetic ADDLs Bound to Cultured Hippocampal Cells
Cultured hippocampal cells were also analyzed to
determine whether monoclonal antibodies that distinguish
between Alzheimer's Disease and control brain extracts
could identify ADDLs (either endogenous or synthetic) bound
to cultured cells. Hippocampal cultures were prepared
according to established protocols and allowed to grow for
3-4 weeks. Synthetic ADDLs were prepared according to
standard protocols (e.g., U.S. Patent No. 6,218,506).
Endogenous ADDLs were extracted from Alzheimer's Disease
brain according to Gong, et al. ( (2003) supra) . ADDLs (100
nM in F12, or 2 mg total protein in F12) were incubated
with the cells for 1 hour and then washed and fixed
according to standard methods- Following washing, the cells
were incubated with 20C2, 3B7,. M94, 2A10, 4E2, 2D6, 4C2,
2B4, 5F10, or 5G12 monoclonal antibody and subsequently
with anti-mouse secondary conjugated to ALEXAFLUOR 488-
Images were taken on a NIKON DIAPHOTTM epifluorescent
microscope with COOLSNAPT" HQ camera and analyzed using
METAMORPHTM software (Universal Imaging, Downingtown, PA).
Both endogenous and synthetic ADDLs exhibited the
standard hot spot pattern in cultured cells when visualized
by 20C2. Thus, monoclonal antibody 20C2 identifies both
CA 02790433 2012-09-19
-45-
synthetic and endogenous ADDLs bound to cultured
hippocampal cells. As 3B7 did not bind to fibrils, higher
molecular weight oligomers, and monomers, hot spot binding
of ADDLs by 3B7 was attributed to oligomeric ADDLs. The
other antibodies appeared to recognize a variety of
epitopes on ADDLs bound to cells, ranging from hot spots on
processes (M94, 2A10) to cell body specific attachment
(4E2) and other states in between (2D6, 4C2, 2B4, 5F10,
5G12).
Example 4: Inhibition of ADDL Binding to Neurons Using
Marine Anti-ADDL Antibodies
To determine whether monoclonal antibodies that
distinguish between Alzheimer's Disease and control brain
extracts could also block binding of ADDLs to cultured
cells, cultured hippocampal cells were preincubated with
20C2. antibody and ADDL binding was determined by
immunocytochemistry. Hippocampal cultures were prepared
according to established methods and allowed to grow for 3-
4 weeks. Synthetic ADDLs were prepared according to
standard protocols (e.g., see U.S. Patent No. 6,218,506 and
the like). Endogenous ADDLs were extracted from Alzheimer's
Disease brain according to Gong, et al. ((2003) supra).
ADDLs (100 nM in F12, or 2 mg total protein in F12) were
preincubated with 20C2 antibody for 1 hour and subsequently
added to cells for 1 hour at 37 C. Cells were washed,
fixed, and incubated with anti-mouse secondary conjugated
to ALEXAFLUOR 488.
Both endogenous and synthetic ADDL binding to cultured
cells was blocked by preincubation with 20C2. Vehicle and
no-secondary antibody control images were black.
CA 02790433 2012-09-19
-46-
Example 5: Detection of ADDL Binding to Neurons Using
Biotinylated ADDLs
The binding of ADDLs or bADDLs (biotinylated ADDLs) to
neurons was detected using standard immunofluorescence
procedures. Primary hippocampal neurons (cultured for
fourteen days) or B103 cells (plated for twenty-four hours)
were incubated with 5-25 pm ADDLs or bADDLs for one hour at
37 C and the cells were subsequently washed three to four
times with warm culture medium to remove unbound ADDLs or
bADDLs. The cells were then fixed for ten minutes at room
temperature with 4% paraformaldehyde prepared from 16%
paraformaldehyde (Electron Microscopy Sciences, Fort
Washington, PA) diluted in PBS. Subsequently, the solution
was removed and fresh fixative added for an additional ten
minutes at room temperature. The cells were permeabilized
(4% paraformaldehyde solution with 0.1% TRITON'-X 100;
SIGMA, St. Louis, MO) for ten minutes, washed six times
with PBS and incubated for one hour at 37 C with blocking
buffer (PBS with 10% BSA; Sigma, St. Louis, MO). At this
point, the protocols for the detection of bound ADDLs and
bADDLs diverge. To detect ADDL binding, the cells were
incubated overnight at 37 C with 4G8 (diluted 1:1,000 in
PBS containing 1% BSA; Signet Labs, Dedham, MA), 6E10
(1:1,000; Signet Labs, Dedham, MA), or one of the anti-ADDL
monoclonal antibodies disclosed herein (diluted 1:1,000).
In addition, a polyclonal antiserum raised against tau
(1:1,000; Sigma, St. Louis, MO) was used to visualize the
cell processes. The next day, the cells were washed three
times with PBS, incubated for one hour at room temperature
with an ALEXA 594-labeled anti-mouse secondary (diluted
1:500 in PBS with 1% BSA; Molecular Probes, Eugene, OR,)
and an ALEXAO 488-labeled anti-rabbit secondary (diluted
1:1,000; Molecular Probes, Eugene, OR), washed three times
in PBS and the binding observed using a microscope with
fluorescence capabilities. For the detection of bADDL
CA 02790433 2012-09-19
-47-
binding, the cells were incubated overnight with the tau
antibody. Subsequently, the cells were washed three times
with PBS, incubated for one hour at room temperature with
an ALEXA 488-labeled anti-rabbit secondary (as above) and
an ALEXA 594-labeled streptavidin, 1:500 dilution
(Molecular Probes, Eugene, OR), washed 5-6 times in PBS and
the binding visualized with a fluorescence microscope. If
the staining of the cell nuclei was desired, the nuclei
were labeled with DAPI (1:1000) according to standard
protocols.
For immunocytochemical analysis of ADDLs using an
ADDL-specific monoclonal antibody, cells were washed,
fixed, permeabilized and blocked after incubation with
ADDLs. To detect the bound bADDLs with monoclonal
antibodies, the cells were incubated overnight with 4G8,
6E10 or one of the instant anti-ADDL monoclonal antibodies
and immunoreactivity was subsequently detected with an
ALEXA 488-labeled anti-mouse secondary antibody. The bound
bADDLs were visualized with an ALEXA 594-labeled
streptavidin and the nuclei stained with DAPI. After
staining, the colocalization of bADDL binding and ADDL
immunoreactivity was detected with a fluorescence
microscope.
Specific immunoreactivity with primary hippocampal
cells incubated with ADDLs was seen with each of the
monoclonal antibodies evaluated (.i.e., 20C2, 2H4, 2B4, and
2A10). The bound ADDLs appeared as punctate staining along
the neuronal processes and cell soma. This pattern was only
seen on a subset of neurons, a pattern that is consistent
with previous reports describing ADDL binding to primary
neurons using both commercial and non-commercial
antibodies. The pattern of staining and the results of a
number of control studies demonstrated the specificity of
these antibodies.
CA 02790433 2012-09-19
-48-
The use of bADDLs offered a simplified method to
detect bound ADDLs and evaluate the blockade of ADDL
binding with the monoclonal antibodies. When bADDLs were
added to primary hippocampal cells and the binding
evaluated with a fluorescent-labeled streptavidin, specific
binding was seen along the neuronal processes of a subset
of cells in culture. If the cells were then fixed,
processed for immunocytochemistry and an anti-ADDL antibody
used to visualize binding, a similar pattern of staining
was observed. Furthermore, the superimposition of these
staining patterns revealed a perfect overlap of the
antibody staining and bound bADDLs, thus demonstrating that
bADDLs and ADDLs are functionally equivalent and the use of
bADDLs in binding assays.
Example 6: Detecting and Measuring Murine anti-ADDL
Monoclonal Antibody Differential Displacement of bADDL
Binding to Neurons
The ability of antibodies to block the binding of
ADDLs or bADDLs to neuronal cultures (primary neurons or
B103 cells) was characterized using the immunocytochemical
methods described herein with a few modifications.
Monoclonal antibodies were mixed with 1-10 pm bADDLs at a
molar ratio of 1:1, 1:5 or 1:10 (antibody:bADDLs) and
incubated in a siliconized microcentrifuge tube for one
hour at 37 C on a slow rotator (Miltenyi Biotec, Auburn,
CA). Subsequently, the antibody/bADDL mixture was added to
cells and allowed to further incubate for one hour at 37 C.
After incubation, the cells were washed, fixed,
permeabilized, blocked and incubated overnight with a
polyclonal antiserum raised against tau to visualize the
cell processes. The next day, the cells were washed,
incubated with an ALEXA 488-labeled anti-rabbit secondary
antibody and an ALEXA 594-labeled streptavidin and the
cells were stained with DAPI to allow detection of nuclei.
CA 02790433 2012-09-19
-49-
Once stained, the degree of"binding was assessed visually
with a fluorescence microscope.
To quantitatively assess the degree of bADDL binding
and the ability of anti-ADDL antibodies to abate this
interaction, a cell-based alkaline phosphatase assay was
developed. Monoclonal antibodies or PBS were mixed at a 1:1
(B103 cells) or 1:5 (primary neurons) molar ratio with 2.5-
pm (final concentration) of bADDLs and incubated for one
hour at 37 C on a slow rotator. After preincubation, the
10 antibody/bADDL preparations were added to the B103 or
primary neuron cultures and incubated for an additional one
hour at 37 C. At the end of the incubation period, the
bADDLs/antibody mixture was removed and the plates washed
six times with media. The cells were fixed in 4%
paraformaldehyde for ten minutes at room temperature, the
solution removed, fresh fixative added and the cells fixed
for an additional ten minutes. The cells were permeabilized
with 4% paraformaldehyde containing 0.1% TRITON""' X-100 (2
times, each for ten minutes at room temperature), washed
six times in PBS and treated with 10% BSA in PBS for one
hour at 37 C. Alkaline phosphatase-conjugated streptavidin
(1:1,500 in 1% BSA; Molecular Probes, Eugene, OR) was added
to the cells for one hour at room temperature. The cells
were rinsed six times with PBS, the alkaline phosphatase
substrate (CDP-STAR with SAPPHIRE-II'r'; Applied Biosystems,
Foster City, CA) added to the cells and incubated for
thirty minutes prior to determining the luminescence on a
LJL Luminometer (Analyst AD; LJL BioSystems, Sunnyvale,
CA).
When the binding of bADDLs to the neurons was
evaluated, an antibody-dependant pattern of staining was
observed. Some of the antibodies investigated markedly
reduced the binding of bADDLs, while others were less
effective. Unexpectedly, a third group of antibodies
appeared to enhance the binding of bADDLs to neurons. While
CA 02790433 2012-09-19
-50-
the results of these studies were qualitative and not
quantitative in nature, they indicated that the antibodies
differentially blocked bADDL binding to neurons.
Quantitative assessment demonstrated a similar trend
(Figure 1). That is, some antibodies abated the binding of
bADDLs to neurons, some were weak or had little effect and
a few enhanced the binding (i.e., 5F10 and 4C2). Moreover,
a mouse Fab was unable to block the binding of bADDLs,
further demonstrating the specificity of the monoclonal
antibodies in this assay.
Analysis of bADDL binding and blockade with monoclonal
antibodies in the neuroblastoma cell line B103 demonstrated
specific bADDL binding to B103 cells, but not to an ovarian
cell line (CHO) . Moreover, the binding was dramatically
attenuated when bADDLs were pre-incubated with an anti-ADDL
monoclonal antibody prior to the addition to B103 cells.
Quantitative assessment of the blockade of bADDL binding to
B103 cells with monoclonal antibodies indicated that the
monoclonal antibodies were not equal in their ability to
block bADDL binding to cells (Figure 2). As seen with the
primary hippocampal cells, some antibodies were quite good
at blocking binding, while others were less effective.
Furthermore, the antibody 4C2 also enhanced the ability of
bADDLs to bind to B103 cells in culture.
- To show that bADDLs also bind to regions of the
hippocampus that are involved in learning and memory, a
series of binding studies were conducted using rat
hippocampal slice cultures. Binding studies showed that
neurons in the CAI-3 and dentate gyrus regions of the
hippocampus were capable of binding bADDLs, while neurons
in other regions did not. When the bADDLs were pre-
incubated with an anti-ADDL monoclonal antibody, the degree
of bADDL binding was attenuated in a dose-dependant manner.
These results showed that monoclonal antibodies can also
CA 02790433 2012-09-19
-51-
abate the binding of bADDLs to a subset of hippocampal
neurons, neurons that a critical for learning and memory.
Example 7: Binding of Anti-ADDL Antibodies to Endogenous
ADDLs from Alzheimer's and Control Brain
To further characterize the monoclonal antibodies
disclosed herein, it was determined whether the monoclonal
antibodies could identify ADDLs from soluble extracts of
human Alzheimer's Disease brain (endogenous ADDLs) and
distinguish that from extracts of control brain. Synthetic
ADDLs and human brain extracts prepared in F12 were diluted
in F12 and spotted (1 pmole ADDLs; 0.5 pg brain extract) in
duplicate onto dry HYBONDTm ECLT" nitrocellulose. Brain
tissue, with corresponding CERAD grades (Consortium to
Establish a Registry for Alzheimer's Disease) and Braak
stages, was obtained from NU Brain Bank Core. The blot was
allowed to dry 20 minutes and then incubated in 3% H202 in
TBS (20 mM Tris-HC1, pH 7.5, 0.8% NaCl) for 20 minutes at
room temperature. The blot was cut into strips and blocked
with 5% milk in TBS-T (0.1% TWEENT"-20 in TBS) for 1 hour at
room temperature. Rabbit polyclonal antibody M71/2 (1:2500,
0.4 pg; Bethyl Laboratories, Inc., Montgomery, TX);
monoclonal antibody 6E10 (1:500, 3 pg; Signet Labs, Dedham,
MA); and monoclonal antibodies 20C2 (1.52 mg/mL, 5 pg),
11B5 (2.54 mg/ml, 5 pg), 2B4 (1.71 mg/mL, 5 pg), and 2A10
(1.93 mg/mL, 7.5 pg) as disclosed herein (Figure 3) were
diluted in 1.5 mL of milk/TBS-T and incubated for 1 hour at
room temperature. The blots were washed 3 x 10 minutes with
TBS-T. The blots were incubated with horseradish peroxidase
(HRP)-linked secondary antibody (1:40,000 in milk/TBS-T;
Amersham Life Science, Inc., Arlington Heights, IL) for 1
hour at room temperature. The blots were washed 3 x 10
minutes with TBS-T, rinsed 3 times with dH2O, developed with
SUPERSIGNALT ' substrate (1:1 dilution with ddH2O; Pierce,
CA 02790433 2012-09-19
-52-
Rockland, IL) and exposed to HYPERFILM`" ECL71' (Amersham Life
Science, Inc., Arlington Heights, IL).
All antibodies tested identified synthetic ADDLs with
robust binding, except 2A10, which had weaker binding, even
though it was tested at higher protein concentration.
Polyclonal antibody M71/2 and monoclonal antibodies 20C2
and 11B5 bound strongly to both Alzheimer's Disease
samples, but showed only very faint binding, similar to
background in control brain. In contrast, monoclonal
antibodies 6E10, 2B4, and 2A10 showed weak binding to
Alzheimer's Disease brain.
The results of this analysis indicated that two of the
monoclonal antibodies tested could distinguish between
Alzheimer's Disease and control brain, wherein binding to
endogenous oligomers was with a high degree of specificity.
In addition, these data indicate that detection can be
accomplished in early stages of Alzheimer's Disease.
Example 8: Immunohistochemical Analysis of Alzheimer's
Disease and Control Brain Slices
Immunohistochemical analysis using the monoclonal
antibodies disclosed herein was carried out to determine
whether ADDLs can be visualized in brain slices using
monoclonal antibodies that distinguish between Alzheimer's
Disease and control brain extracts, and to demonstrate the
nature of ADDL labeling (e.g., diffuse, perineuronal,
plaque-like, etc.) and its distribution in human tissue.
Sections (40 pm) of fixed Alzheimer's Disease and control
brain were prepared in accordance with standard methods.
The slices were labeled with several monoclonal and one
polyclonal antibody and subsequently counterstained with
hematoxylin to identify cell nuclei. Images were obtained
using a NIKON ECLIPSE E600 light microscope with a SPOTTM
INSIGHTT11 digital video camera (v. 3.2).
CA 02790433 2012-09-19
-53-
Immunohistochemical analysis indicated that ADDL
staining was manifest in Alzheimer's Disease brain in the
hippocampus, entorhinal cortex, and middle frontal gyrus.
In a severe Alzheimer's Disease case, there was abundant
light ADDLs staining in what appeared predominantly as a
plaque-type distribution. Some light ADDL staining was
observed as peri-neuronal in one Alzheimer's Disease case.
In contrast, there is no staining using either antibody in
any regions of control samples, not even a rare neuron
surrounded by dot-like immunostaining.
These data indicate that polyclonal and monoclonal
antibodies can be used to identify ADDLs in fixed human
tissue, wherein labeling is varied, consisting of plaque-
like regions, vascular regions, and peri-neuronal labeling
of individual cells and some clusters. Further, labeling of
ADDLs in Alzheimer's Disease, but not control, brain was
observed in at least three brain regions: hippocampus,
entorhinal cortex, and middle frontal gyros.
Example 9: Immunostaining of API-40 Monomer-Like Control
API-40 oligomerizes slowly in DMSO/F12 compared to
ADDLs. Thus, it was determined whether API-40 could serve
as a monomer-like control. ADDLs were subjected to size
exclusion chromatography (SEC) on a SUPERDEX 75 column
(ADDL063), which resolved into two peaks. API-40 was
prepared in DMSO/F12 (45.5 mM), frozen and thawed. Samples
were diluted with F12 and mixed -2:1 with Tricine sample
buffer (BIO-RAD , Waltham, MA). SDS-PAGE was carried out on
10-20% Tris-Tricine gels (BIO-RAD , Waltham, MA) with
Tris/Tricine/SDS buffer (BIO-RAD , Waltham, MA) at 120V at
room temperature for 80 minutes. The gel was silver stained
(60 pmoles Af31-40 or ADDLs; 40 pmoles Peaks 1 or 2) with
SILVERXPRESSTM (INVITROGENTM, Carlsbad, CA) . Alternatively,
the gels (20 pmoles A131-40 or ADDLs; 30 pmoles Peaks 1 or
2) were electroblotted onto HYBONDTM ECLTM nitrocellulose
CA 02790433 2012-09-19
-54-
using 25 mM Tris-192 mM glycine, 20% v/v methanol, pH 8.3,
0.02% SDS at 100V for 1 hour at 8 C_ The blots were blocked
with 5% milk in TBS-T (0.1% TWEENn1-20 in 20 mM Tris-HC1, pH
7.5, 0.8% NaC1) overnight at 8 C_ Monoclonal antibody 6E10
(1:2000; Signet Labs, Dedham, MA), monoclonal antibody 20C2
(1.52 mg/mL, 1:2000; Figure 3), or polyclonal antibody
M71/2 (1:4000, Bethyl Laboratories, Inc-, Montgomery, TX)
was diluted in milk/TBS-T and incubated with the blots for
90 minutes at room temperature. The blots were washed 3 x
10 minutes with TBS-T and subsequently incubated with HRP-
conjugated secondary antibody (1:40,000 in TBS-T; Amersham
Life Science, Inc., Arlington Heights, IL) for 1 hour at
room temperature. After three washes with TBS-T, 10 minutes
per wash, the blots were rinsed 3X with dH2O, developed with
SUPERSIGNAL West Femto Maximum Sensitivity substrate (1:1
dilution with ddH2O; Pierce, Rockland, IL) and exposed to
HYPERFILMTM ECL'r" (Amersham Life Science, Inc-, Arlington
Heights, IL).
Silver stain analysis showed API-40 as a heavy monomer
band. In contrast, ADDLs and Peak 1 showed monomer, trimer
and tetramer, although there was less tetramer_ Silver
stain analysis of Peak 2 showed heavy monomer with a
lighter trimer and very light tetramer band-
Immunostaining of A(31-40 with 6E10 showed only a light
monomer band. Immunostaining of ADDLs and Peak 1 with 6E10
showed monomer, trimer, tetramer and 12-24mer. Peak 2
showed heavy monomer staining with 6E10 and some light
trimer and tetramer with no 12-24mer. There was no monomer
staining of A131-40 with 20C2 or M71/2. While both 20C2 and
M71/2 showed minimal or no monomer staining of ADDLs and
Peak 1, these samples had trimer, tetramer, and 12-24mer
staining with 20C2 and M71/2. Peak 2 immunostaining with
20C2 and M71/2 showed light monomer, trimer and tetramer
with no 12-24mer observed. API-40 immunostained lighter
CA 02790433 2012-09-19
-55-
with 6E10 than did the ADDL monomer, despite heavier silver
staining.
These results indicated that, in contrast to the 6E10
antibody which shows good recognition of monomer, gels
transferred with 0.02% SDS in the transfer buffer showed
minimal monomer detection with the oligomer-specific
antibodies. Immunostaining of SEC fractions showed Peak 2
composed mostly of monomer with small amounts of trimer and
tetramer and no 12-24mer, while Peak 1 has monomer, trimer,
tetramer and the 12-24mers.
To further characterize the monoclonal antibodies with
respect to binding to Peak 1 and Peak 2, a sandwich ELISA
was developed using polyclonal antibody M90 to ADDLs as the
capture antibody. SEC peak 1 and peak 2 fractions referred
to herein are the two major peaks of ADDLs that were
fractionated on a SEPHADEXTM 75 column to distinguish
between potentially bioactive and inactive oligomers. Non-
denaturing gel electrophoresis confirmed the separation
into large (>50 kDa) and small (<30 kDa) aggregates that
were stable at 37 C. These peaks were used separately as
the detection substance for clone supernates. Binding was
visualized with a VECTASTAIN kit. Differences between
recognition of the two peaks was observed for all
antibodies. For example, compare the ratio of peak 1 to
peak 2 for antibodies 2B4 and 20C2 (Figure 3). Only one
antibody reflects the control antibody (6E10) preference
for peak 2.
Example 10: Detection of ADDL Formation from API-42
Polyclonal antibodies have been used in dot-blots to
show time-dependent ADDL formation from AJ31-42. Thus, it
was demonstrated that monoclonal 20C2 antibody, which
preferentially binds to oligomers, could also show
increased signal with time as ADDLs form from A(31-42. A(31-
42, -750 pmoles HFIP film, was dissolved in 1.5 mL DMSO
CA 02790433 2012-09-19
-56-
(0.5 mM) and 2 pL aliquots diluted to a final volume of 100
pL with F12 (10 nM) and incubated on ice. Two pL (20 fmol)
of reaction mixture was spotted on dry HYBONDI ECLTM
nitrocellulose (Amersham Life Science, Inc., Arlington
Heights, IL) at specified time points. The nitrocellulose
was blocked with 5% non-fat dry milk in TBS-T (20 mM Tris-
HC1, pH 7-5, 0-8% NaCl, 0-1% TWEEN'a'-20) for 1 hour at room
temperature- Polyclonal antibody M90/1 (Bethyl
Laboratories, Inc-, Montgomery, TX) or monoclonal antibody
20C2 (1.52 mg/mL) was diluted 1:2000 in milk/TBS-T and
incubated with the blot for 90 minutes at room temperature
followed by washing 3 x 10 minutes with TBS-T. HRP-
conjugated secondary antibodies (Amersham Life Science,
Inc., Arlington Heights, IL) were diluted 1:40,000 in
milk/TBS-T and the blot incubated for 60 minutes at room
temperature followed by washing as above. After a brief
rinse. with dH2O, the blot was incubated for 60 seconds with
SUPERSIGNAL West Femto Maximum Sensitivity substrate
(diluted 1:1 with ddH2O; Pierce, Rockland, IL) and exposed
to HYPERFILM'M ECLTM (Amersham Life Science, Inc., Arlington
Heights, IL). Dot blots were scanned and intensity of spots
was determined with ADOBE PHOTOSHOP .
Both antibodies detected time-dependent ADDL formation
from A(31-42, wherein the results for 20C2 showed better
signal and consistency. Neither antibody could detect API-
40 at a concentration equivalent to ADDLs_ These data
further demonstrate the oligomer-specificity of this
antibody, since monomers are present all the time and
oligomers form with time. In addition, both M90/1 and 20C2
showed minimal recognition of A[31-40 monomers even at a
100-fold higher concentration than ADDLs_
Example 11: Competition Dot Blot Assays
To determine whether the monoclonal antibodies
disclosed herein could bind monomers, a competition dot
CA 02790433 2012-09-19
-57-
blot assay was performed with synthetic ADDLs, 20C2, and
API-40. ADDLs were applied to dry nitrocellulose at 10
pmol/0.5 pL. While the nitrocellulose was being blocked in
5% NDM/TBS-T for one hour, ADDLs and fresh API-40 at
various concentrations were incubated with 200 pL each of
20C2 (1.5 pg/mL final concentration) in 5% NDM/TBS-T for 1
hour. These solutions were then applied to the
nitrocellulose using the SURF-BLOT apparatus and incubated
at room temperature for 1.5 hours with rocking. The blot
was subsequently visualized with anti-mouse IgG-HRP and
chemiluminescence. Quantitation was performed using the
KODAK IMAGESTATION 440 and EXCEL .
Results of this analysis indicated that synthetic
ADDLs in solution could effectively and specifically block
20C2 binding to ADDLs immobilized on nitrocellulose with a
half maximal inhibition observed at <50 nM for ADDLs. In
contrast, AR1-40 in solution did not block binding of 20C2
to immobilized ADDLs.
To determine which portions constitute the binding
epitope of the API-42 molecule, a competition dot blot
assay was performed with ADDLs, 20C2, and peptides. ADDLs
were spotted on nitrocellulose at four concentrations (1,
0.5, 0.25, and 0.125 pmole) each in 0.5 pL. While the
nitrocellulose was being blocked in 5% NDM/TBS-T for two
hours, the peptides at 50, 100 and 200 pmol were added to
200 pL of 20C2 (1.52 pg/mL final concentration = 1.9 pmol,
in 5% NDM/TBS-T) and rocked at room temperature- The
solutions were subsequently incubated with the
nitrocellulose using the SURF-BLOT apparatus for 1.5 hours
at room temperature. Binding was visualized with anti-mouse
IgG-HRP using chemiluminescence.
The results of this analysis indicated that binding to
ADDLs was blocked by the ADDLs themselves and by A31-28,
but no other combination of peptides. Thus, the binding
epitope required some conformation that ARl-28 could
CA 02790433 2012-09-19
-58-
attain, but that was not available on A(31-12 and A(312-28 or
their combination. Alternatively, A(31-28 forms a dimer that
blocks binding of ADDLs by steric hindrance.
To determine whether A131-28 aggregates (similar to
A(31-42) or folds such that it blocks the binding epitope
for 20C2, SDS-PAGE gels were silver stained and western
blot analysis was performed. ADDLs and A131-28 (60 pmol in
each of two lanes used for silver stain and 20 pmol
otherwise) were separated using a 10-20% Tris-Tricine SDS-
PAGE. The 60 pmol lanes were excised and stained with
SILVERXPRESSTM (INVITROGENT", Carlsbad, CA); alternatively,
the gels (20 pmoles ADDLs and A(31-28) were electroblotted
onto HYBONDT ' ECLTM nitrocellulose using 25 mM Tris-192 mM
glycine, 20% v/v methanol, pH 8.3, 0-02% SDS at IOOV for 1
hour at 8 C. The blots were blocked with 5% milk in TBS-T
(0.1% TWEENT"-20 in 20 mM Tris-HC1, pH 7.5, 0.8% NaCl).
Samples were incubated with 20C2 (1:1000, 1.52 mg/mL) or
20C2 + API-28 (2 nmol, preincubated for 2 hour) for 1.5
hour at room temperature in the above blocking buffer.
Binding was visualized with anti-mouse IgG-HRP (1:40,000 in
TBS-T) and chemiluminescence.
Silver staining showed monomer, trimer and tetramer in
the ADDL lane, whereas the A(31-28 lane had one species,
which ran at about a dimer. ADDLs, but not A(31-28, were
visualized by 20C2 and binding to all ADDL species by 20C2
was blocked by A(31-28. Moreover, while the 20C2 binding
epitope is blocked by A131-28, 20C2 does not recognize the
A131-28 peptide in a western blot.
Example 12: Isotype Analysis of Anti-ADDL Antibodies
To further characterize the monoclonal antibodies
disclosed herein, isotype analysis was performed using the
SIGMA IMMUNOTYPET Kit with the Mouse Monoclonal Antibody
Isotyping Reagents, following the manufacturer's directions
CA 02790433 2012-09-19
-59-
(Sigma-Aldrich Co., St. Louis, MO). Results of this
analysis are presented in Figure 3.
Example 13: Core Linear Epitope Mapping of Anti-ADDL
Antibodies
Specific interaction of the anti-ADDL monoclonal
antibodies with amyloid beta peptide was detected in
standard ELISA assays. Briefly, synthetic peptides, or ADDL
or fibril in some cases, were used as antigen to coat on
NUNCT" MAXISORBT" plate at concentration of 4 pg/mL (about
800 to 1200 nM). Unless specified, the peptides were coated
in 5 mM sodium bicarbonate buffer, pH 9.6, overnight at
4 C. After blocking the plates with PBS containing 0.050
TWEENT"' 20 and 3% (w/v) nonfat dry milk for one hour, the
monoclonal antibody was titrated in blocking buffer at a
determined concentration and the plates were incubated for
one hour at ambient temperature with gentle rocking. After
washing, HRP-conjugated goat anti-mouse IgG (H+L), diluted
in blocking buffer, was added to the plates. The
colorimetric substrate, TMB, was added to the plates after
extensive washes to remove unbound HRP-conjugate. The
absorbance was measured at wavelength of 450 nm on a plate
reader.
To map the core linear epitope for the anti-ADDL
monoclonal antibodies, a set of overlapping, ten amino acid
peptides was synthesized to cover A(31-42 (Table 1). Three
peptides of fourteen amino acids, with reversed amino acid
sequence of A[31-42 were also synthesized as nonspecific
control peptides.
TABLE 1
SEQ
N- C- Peptide Sequence Mol_ ID
Wt. NO:
1 42 DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA 180
1 7 DAEFRHD 929.8 181
1 8 DAEFRHDS 975.4 178
1 9 DAEFRHDSG 10352.5 182
CA 02790433 2012-09-19
-60-
1 10 DAEFRHDSGY 1195.4 183
2 11 AEFRHDSGYE 1209.3 184
3 12 EFRHDSGYEV 1237.4 185
4 13 FRHDSGYEVH 1245.2 186
14 RHDSGYEVHH 1235.7 187
6 15 HDSGYEVHHQ 1207.4 188
7 16 DSGYEVHHQK 1198.5 189
8 17 SGYEVHHQKL 1196.8 190
9 18 GYEVHHQKLV 1208.3 191
19 YEVHHQKLVF 1298.6 192
11 20 EVHHQKLVFF 1282.9 193
12 21 VHHQKLVFFA 1224.4 194
13 22 HHQKLVFFAE 1254.5 195
14 23 HQKLVFFAED 1232.5 196
24 QKLVFFAEDV 1177.3 197
16 25 KLVFFAEDVG 1123.8 198-
17 26 LVFFAEDVGS 1082.3 199
18 27 VFFAEDVGSN 1083.0 200
19 28 FFAEDVGSNK 1112.2 201
29 FAEDVGSNKG 1022.6 202
21 30 AEDVGSNKGA 946.5 203
22 31 EDVGSNKGAI 988.1 204
23 32 DVGSNKGAII 972.2 205
24 33 VGSNKGAIIG 914.4 206
34 GSNKGAIIGL 928.5 207
26 35 SNKGAIIGLM 1002.2 208
27 36 NKGAIIGLMV 1014.7 209
28 37 KGAIIGLMVG 957.4 210
29 38 GAIIGLMVGG 886.3 211
-39 AIIGLMVGGV 928.3 212
31 40 IIGLMVGGVV 956.5 213
32 41 IGLMVGGVVI 956.4 214
33 42 GLMVGGVVIA 914.2 215
14 1 HHVEYGSDHRFEAD 1923.8 216
28 15 KNSGVDEAFFVLKQ 1806.9 217
42 29 AIVVGGVMLGIIAGKK 1751.5 218
All peptides were dissolved in DMSO at about 400 to
500 }iM (1 mg/mL) and stored in multiple aliquots at -20 C.
The peptides were used in an ELISA assay for determination
5 of the core epitope of the anti-ADDL monoclonal antibodies.
Each monoclonal antibody was tested at four concentrations
(3, 1, 0.3 and 0.1 .ig/mL) against either an N-terminal
peptide set (from residues 1 to 25) or a C-terminal peptide
set (from residues 17 to 42), with control peptides. The
10 core linear epitopes for the panel of monoclonal antibodies
are listed in Table 2. Several commercial monoclonal
antibodies (6E10, BAM-10, 4G8 and WO-2) were included in
the experiment to validate the assay format, and the
results confirmed their core linear epitopes as reported in
15 published literature.
CA 02790433 2012-09-19
-61-
TABLE 2
Epitope Sequence within A(31-42 SEQ
Core ID
Antibody Epitope* NO.
DAF.FRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA 180
6E10 5-11 RHDSGYE 219
BAM-10 3-8 EFRHDS 220
4G8 xx-21 EVHHQKI,VFFA 221
WO-2 3-8 EFRHDS 220
26D6 3-8 EFRHDS 220
2AlOa 3-8 EFRHDS 220
2B4b 3-8 EFRHDS 220
4C2a 3-8 EFRHDS 220
4E2a 3-8 EFRHDS 220
2H4` 1-8 DAEFRHDS 178
20C2a 3-8 EFRHDS 220
2D6a 3-8 EFRHDS 220
5F10" 3-8 EFRHDS 220
1F4a nd
IF6a nd
2E12a 3-10 EFRHDSGY 222
3B3a nd
*Position within A(31-42. a1gGl, IgG2b, cIgG2a. nd, not
determined.
Nine out of twelve ADDL-specific monoclonal antibodies
evaluated were mapped to the N-terminal region of A(31-42,
and seven of these mapped to amino acid residues 3 to 8.
Two monoclonal antibodies, 2H4 and 2E12, prefer slightly
bigger epitopes. Three monoclonal antibodies, 1F4, 1F6 and
3B3, failed to bind the overlapping peptide set, even at
high concentration of 3 pg/mL, but their epitopes were
estimated to be located at the N-terminus of A131-42, as
they could bind to A(31-20 peptide, which was used as a
positive control in the experiments.
Example 14: Affinity and Specificity of Mouse Anti-ADDL
Antibodies
A solution-based binding assay was developed to
determine the specificity and affinity of anti-ADDL
antibodies to different amyloid beta peptide preparations
(ADDL, fibril, A(31-40, A(31-20). A quantitative ELISA was
established that was capable of capturing the linear range
of dose-response of monoclonal antibodies against ADDL
CA 02790433 2012-09-19
-62-
coated on NUNCT" plates. Based on this information, a fixed
concentration of monoclonal antibody was selected that
could give consistent OD signals in ELISA just above assay
noise (OD 450 nm reading around 0.2 to 0.5). IgG at this
fixed concentration was then incubated with different
amyloid beta peptide substrates (ADDL, fibril, A(31-40, A131-
20) in 20 point titrations in solution at room temperature
overnight to reach equilibrium. The quantity of free IgG
within the mixture was determined the next day in a
quantitative ELISA with a one hour incubation on regular
ELISA plates. The fraction of bound IgG was calculated and
the correlations of bound IgG to titration of free ligand
(substrates) were used to derive KD, using the GraFit
program (Erithacus Software, Surrey, UK). Thus, the
substrate preference for each antibody to different amyloid
beta peptide preparations was presented as the intrinsic
affinity values (KD).
There were several advantages of using this assay
format. First, the interaction of the antibody and
substrate was in solution phase, thus, there was no
constraint from any solid surface such as in regular ELISA
assay or BIACORE''" experiment, where potential influence of
solid surface from ELISA plates or sensor chip on
monoclonal antibody and substrate interaction has to be
taken into consideration for interpretation of data.
Second, the interactions were allowed to reach equilibrium.
Therefore, the interaction of IgG and substrate occurred at
limiting concentrations of both components with no concerns
for precipitation of IgG or additional amyloid beta peptide
oligomerization due to high experimental concentration.
Third, the assay readout was independent of antigen in the
solution; thus, any heterology of amyloid beta in different
peptide preparations (e.g., ADDL or fibril) would not
interfere with data interpretation and mathematical
modeling. The assay sensitivity was limited to ELISA assay
CA 02790433 2012-09-19
-63-
detection limits which allowed this assay to evaluate
monoclonal antibodies with KD values in the nanomolar range.
Alternative substrates such fluorescent reagents are
contemplated to improve the sensitivity range. It is
believed that the immune complex was minimally disrupted
during the one hour incubation to capture the free IgG in
quantitative ELISA.
The quantities of free IgG were determined by a
standard curve and plotted against titrations of different
substrate. The quantities of bound IgG with different
substrates were plotted and the information was used in
GraFit for curve fitting with appropriate mathematic
models. The summary of KD, expressed in nM ranges, for the
panel of monoclonal antibodies disclosed herein is
presented in Table 3.
TABLE 3
ADDL Fibril A(31-40 A(31-20
Antibody*
KD SE KD SE KD SE KD SE
20C2 0.92 0.09 3.62 0.47 30.48 5.05 71.35 24.41
2A10 2.29 0.25 6.72 0.99 14.69 2.64 22.40 2.43
2B4 2.09 0.24 10.50 1.26 27.57 4.88 1.63 0.26
2D6 5.05 0.52 14.41 2.40 25.66 5.84 30.17 7.07
5F10 11.90 1.63 28.95 5.78 23.54 6.21 6.10 4.39
4E2 4.26 0.42 9.40 1.60 20.24 2.07 28.40 3.23
4C2 8.08 1.03 19.17 3.69 21.89 4.14 28.40 3.23
1F4 9.24 0.84 12.52 1.66 IC IC IC IC
1F6 N/T NIT N/T NIT N/T N/T N/T N/T
3B3 10.02 0.74 7.21 0.59 104.68 21.86 IC IC
2E12 IC IC IC IC IC IC IC IC
WO-2 0.57 0.042 1.15 0.12 6.15 0.62 19.26 3.53
*All antibodies were IgG.
Values listed in italic are high SE and poor fitting.
IC: inconclusive data
N/T: not tested.
Example 15: Detecting and Measuring tau Phosphorylation
Hyperphosporylated Tau (pTau) is a hallmark of
Alzheimer's Disease, although little is known about the
events that cause this hyperphosphorylation. Without
wishing to be bound by any theory, it is believed that
CA 02790433 2012-09-19
-64-
ADDLs may play a role in this phosphorylation event. To
investigate this, neuronal cultures (primary neurons and
B103 cells) were grown as described above, 1 }gym bADDLs or
vehicle was added to the media and the cultures were
maintained for an additional one, six or twenty four hours.
At the end of each incubation, the cells were washed,
fixed, permeabilized, blocked and incubated overnight with
a monoclonal antiserum raised against pTau (AT8, 1:500;
Pierce, Rockland, IL) . The next day, the cells were washed,
incubated with an ALEXA 488-labeled anti-mouse secondary
antibody and an ALEXA 594-labeled streptavidin and the
cells were stained with DAPI to allow detection of nuclei.
The cells were then assessed using a fluorescence
microscope, with the degree of pTau staining and
correlation with bADDL binding being noted at each time-
point.
The results of this analysis indicated that bADDL
binding to B103 cells increased the level of pTau in the
cellular processes, when compared with vehicle-treated
cells. A similar change was also noted in primary
hippocampal cells. When cells were exposed to bADDLs for
six hours, an increase in pTau staining was observed in a
subpopulation of cells, cells that also bound bADDL. A
time-course study with B103 cells further investigated the
modulation of pTau by bADDLs. The addition of bADDLs
resulted in a marginal increase in pTau at one hour.
However, pTau staining was dramatically increased six hours
after the addition of bADDLs and remained elevated up to 24
hours later. Thus, these data indicate that ADDL binding to
neurons can initiate a cascade of intracellular events that
results in the hyperphosphorylation of tau, the
accumulation of neurofibrillary tangles and eventual cell
death- To this end, one skilled in the art can appreciate
that blocking the binding of ADDLs to neurons, would in
turn prevent such downstream events and be beneficial for
CA 02790433 2012-09-19
-65-
the treatment of amyloidogenic diseases and/or tauopathies.
Moreover, a better understanding of the signaling events
that are triggered by ADDL binding and result in pTau
production may also elucidate additional pathways that are
suitable targets for the development of novel therapeutics.
Example 16: AP Peptide/ADDL-Antibody Interaction and
Assembly Inhibition
.Changes in ADDL assembly kinetics and oligomeric size,
in the presence of selected monoclonal antibodies disclosed
herein were observed by fluorescence resonance energy
transfer (FRET) and fluorescence polarization (FP) using a
1:4 mixture of fluorescein-labeled A[31-42 monomers to
native peptide monomers. The auto-quenching of flourescein
emission upon monomer incorporation into ADDLs results in a
three- to five-fold reduction of fluorescence intensity
over the short hour timescale due to FRET. In addition, the
increase in size when monomers assemble into oligomeric
ADDL species results in a two-fold FP increase. The FRET
and FP kinetic progress curves of ADDL assembly, in the
presence of various novel and commercial anti-ADDL and
anti-Ap peptide antibodies, showed differences in the
ability of the antibodies to inhibit ADDL assembly and/or
bind peptide oligomers (Figure 4).
Assays were performed in 384-well CORNING Non-Binding
Surface black, opaque microtiter plates. The assay buffer
was composed of 50 mM MOPS-Tris (pH 8.0) with 100 mM MgC17.
The assay volume, containing 0.2 pM FITC-A(31-42 and 0.8 pM
API-42, was 50 pL and the assay temperature was 37 C. ADDL
assembly was monitored with a Tecan GENios Pro plate
reader, exciting at a wavelength of 485 nm and detecting
emission at a wavelength of 515 nm. Kinetic traces were
collected by recording fluorescence intensity and
polarization readings every five minutes over a six-hour
time course. Negative control reactions, which did not
CA 02790433 2012-09-19
-66-
appreciably assemble into ADDLs during this time, lacked
MgC12 but contained all other buffer and peptide components.
Positive control reactions contained all buffer components
in the absence of added monoclonal antibody reagents. To
test for ADDL binding and assembly inhibition, antibodies
were incubated with the peptide mixture at eight
concentrations from 500 nM decreasing to 5 nM.
This assay was useful for classifying different
profiles of ADDL binding behavior and ADDL assembly
inhibition. The binding and neutralization of larger ADDL
species, through interaction with ADDL-specific and/or
conformational epitopes, serves as a viable therapeutic
strategy. In addition, the inhibition of oligomerization
into large ADDLs by binding an ADDL-specific and/or
conformational epitope present in transient, intermediate
ADDL assembly species (non-monomer) provides an alternative
strategy for anti-ADDL therapy. The FP progress curves,
which demonstrated striking differences between antibodies,
denotes such intermediate or stable species binding.
Correlating the FP/FRET behavior of monoclonal antibodies
with other functional, cellular and in vivo effects allows
for the selection of desired immunotherapy modes of action.
The results of the analyses disclosed herein indicates
that 1F6, 2A10, 5F10, 2D6, and 2B4 exhibit potent assembly
inhibition, whereas 20C2, 1F4, and 4C2 exhibit intermediate
assembly inhibition and 2H4, 3B3 and 4E2 show weak assembly
inhibition (Figure 4). As summarized in Table 4 and
illustrated in Figure 5, 20C2, 4E2, 3B3 and 5FIO show a
variety of biochemical behaviors.
TABLE 4
Potent Assembly Weak or no
Inhibition by Assembly
FP/FRET Inhibition by
FP/FRET
FP _laddering at 30 20C2 4E2
minutes
Low or no FP 5F10, IA9 3B3
CA 02790433 2012-09-19
-67-
laddering at 30
j-
minutes
Further, antibody 1A9, one of five purified antibodies
(i.e., 1A9, 1E3, 1G3, 1A7, and 1E5) generated against a low
n-mer-forming peptide A(31-42[Nle35-Dpro37], segregates with
5F10 in terms of its assembly inhibition and FP behavior.
Moreover, 20C2 was found to bind to assemblies of
charge-inverted, truncated A[37-42 peptide assemblies as
determined by SEC/ICC, indicating a lack of conventional
linear epitope binding to the A(37-42 charge-inverted
peptide, which has a very different sequence corresponding
to residues 7-16 of A(3, i.e., A[3 (7-
42) [Orn-,Orn11D13D14E16N1e35] . Therefore, 20C2 binds to
conformational epitopes that depend upon elements from
within residues 17-42 of AE3, but only when assembled.
Example 17: Isolation of Mouse Antibody Variable Region
Sequences
The cDNAs coding for the variable domains of the mouse
antibody were cloned and sequenced following a polymerase
chain reaction (PCR) using specially designed primers that
hybridize to the 5'-ends of the mouse constant regions and
to the murine leader sequences upstream of the V regions.
This ensured that the mouse variable region sequences
obtained were complete and accurate. In short, mRNA was
extracted from mouse hybridoma cell lines using the QIAGEN
OLIGOTEX Direct mRNA Mini Kit and subsequently converted
to cDNA using a first-strand cDNA synthesis kit. The cDNA
was then used as template in PCR reactions to obtain the
antibody variable region sequences.
To obtain the light chain variable region sequence,
eleven independent PCR reactions were set up using each of
the eleven light chain 5' PCR primers (MKV-1 to MKV-11) and
the 3' PCR primer MKC-1 (Table 5).
CA 02790433 2012-09-19
-68-
TABLE 5
SEQ ID
Primer Sequence NO:
MKV-1 GAT CTC TAG ATG AAG ATT GCC TGT TAG GCT GTT GGT GCT G 223
MKV-2 GAT CTC TAG ATG GAG WCA GAC ACA CTC CTG YTA TGG GTG 224
MKV-3 GAT CTC TAG ATG AGT GTG CTC ACT CAG GTC CTG GSG TTG 225
MKV-4 GAT CTC TAG ATG AGG RCC CCT GCT CAG WTT YTT GGM WTC TTG 226
MKV-5 GAT CTC TAG ATG GAT TTW CAG GTG CAG ATT WTC AGC TTC 227
MKV-6 -GAT CTC TAG ATG AGG TKC YYT GYT SAY CTY CTC TGR GG 228
MKV-7 GAT CTC TAG ATG GGC WTC AAA GAT GGA GTC ACA KWY YCW GG 229
MKV-8 GAT CTC TAG ATG TGG GGA YCT KTT TYC MMT TTT TCA ATG 230
MKV-9 GAT CTC TAG ATG GTR TCC WCA SCT CAG TTC CTT G 231
MKV-10 GAT CTC TAG ATG TAT ATA TGT TTG TTG TCT ATT TCT 232
MKV-11 GAT CTC TAG ATG GAA GCC CCA GCT CAG CTT CTC TTC C 333
Sequence SNQO ID
31 Primer
MKC-1 GAT CGA GCT CAC TGG ATG GTG GGA AGA TGG 234
Underlined and italic sequences denote Xbal and Sacl
restriction sites, respectively. W = A or T, M = A or C, K
= G or T, Y = C or T, and R = A or G.
To obtain the heavy chain variable region sequences
twelve independent PCR reactions were set up using each of
the twelve heavy chain 5' PCR primers (MHV-1 to MHV-12) and
the appropriate isotype specific 3' primer (MHCG-1, MHCG-
2A, MHCG-2B, MHCG-3) (Table 6).
TABLE 6
Sequence SEQ ID
51 Primer NO:
MHV-1 GAT CTC TAG ATG AAA TGC AGC TGG GGC ATS TTC TTC 235
MHV-2 GAT CTC TAG ATG GGA TGG AGC TRT ATC ATS YTC TT 236
MHV-3 GAT CTC TAG ATG AAG WTG TGG TTA AAC TGG GTT TTT 237
MHV-4 GAT CTC TAG ATG RAC TTT GGG YTC AGC TTG RTT T 238
MHV-5 GAT CTC TAG ATG GGA CTC CAG GCT TCA ATT TAG TTT TCC TT 239
MHV-6 GAT CTC TAG ATG GCT TGT CYT TRG SGC TRC TCT TCT GC 240
MI-IV-7 GAT CTC TAG ATG GRA TGG AGC KGG RGT CTT TMT CTT 241
MHV-8 GAT CTC TAG ATG AGA GTG CTG ATT CTT TTG TG 242
MHV-9 GAT CTC TAG ATG GMT TGG GTG TGG AMC TTG CTT ATT CCT G 243
MHV-10 GAT CTC TAG ATG GGC AGA CTT ACC ATT CTC ATT CCT G 244
MHV-11 GAT CTC TAG ATG GAT TTT GGG CTG ATT TTT TTT ATT G 245
MHV-12 GAT CTC TAG ATG ATG GTG TTA AGT CTT CTG TAC CTG 246
SEQ ID
Sequence
Primer NO:
MHCG-1 GCATC GAG CTC CAG TGG ATA GAC AGA TGG GGG 247
MHCG-2A GCATC GAG CTC CAG TGG ATA GAC CGA TGG GGG 248
MHCG-2B GCATC GAG CTC CAG TGG ATG AGC TGA TGG GGG 249
MHCG-3 GCATC GAG CTC CAA GGG ATA GAC AGA TGG GGC 250
Underlined and italic sequences denote Xbal and Sacl
restriction sites, respectively- W = A or T, M = A or C, K
G or T, Y = C or T, and R = A or G-
CA 02790433 2012-09-19
-69-
Each of the light chain PCR reactions contained 46 pL
INVITROGENT" PLATINUM PCR Super Mix, 1.0 pL of one of the
100 pM 5' primers (MKV-1 to MKV-11), 1.0 pL of the 100 pM
3' primer (MKC-1), and 2.0 pL of hybridoma cDNA. Similar
PCR reactions were employed to clone the mouse heavy chain
variable region sequences. Reactions were placed in a DNA
thermal cycler and, after an initial denaturation step at
97 C for 2.0 minutes, subjected to 30 cycles of: 95 C for
30 seconds, 55 C for 45 seconds, and 72 C for 90 seconds.
Following the last cycle, a final extension step at 72 C
for 10 minutes was employed. To determine which PCR
reactions yielded product, 5 pL aliquots from each reaction
were separated on 1.5% (w/v) agarose/1X TAE buffer gels,
containing 0.5 pg/mL ethidium bromide. PCR products from
reactions that produced fragments of the expected size (420
to 500 bp) were then gel purified, digested with Xbal and
Sad - and ligated into the XbaI and SacI sites in the
multicloning region of plasmid pNEB193 (New England
Biolabs, Beverly, MA). Alternatively, PCR products were
ligated directly into plasmid pCR 2.1 using the INVITROGENT`''
TA CLONING kit. Ligation products were then transformed
into XL-1 cells and aliquots of the transformed E. coli
were plated onto LB agar plates containing 50 pg/mL
ampicillin and overlaid with 40 pL of X-Gal stock (50
mg/mL) and 40 pL IPTG (100 mM) solution for blue/white
selection. Plates were incubated overnight at 37 C and
potential clones were identified as white colonies. DNA
from at least 24 independent clones for each PCR product
were sequenced on both strands using universal forward and
reverse primers for pNEB193 and pCR 2.1. The resulting
sequences were then assembled into a contig to generate a
consensus sequence for each antibody light and heavy chain
variable region. Using this approach the sequences were
determined for the light and heavy antibody variable
CA 02790433 2012-09-19
-70-
regions of hybridoma's 20C2, 5F10, 2D6, 2B4, 4E2, 2H4,
2A10, 3B3, 1F6, 1F4, 2E12 and 4C2 (Figures 6A-6X) .
The six complementarity-determining regions (CDRs),
which form the structure complementary to the antigen, are
underlined in Figures 6A-6X. Upon analysis of the CDRs and
corresponding antigen epitopes (Table 2), sequence
similarities were observed. Antibodies sharing the 3-8
amino acid epitope of A[31-42 (i.e., 2A10, 4C2, 2D6, 4E2,
20C2, 2B4, and 5F10) shared highly homologous CDR1 (Figure
7A) and CDR2 (Figure 7B) sequences of the heavy chain.
Antibody 2H4, which was found to recognize the 1-8 amino
acid epitope of A(31-42, appeared to have unique CDR3
(Figure 7C) sequences of the heavy chain and unique CDR1
(Figure 7D), CDR2 (Figure 7E), and CDR3 (Figure 7F)
sequences of the light chain. Similarly, antibody 2E12,
which was found to recognize the 3-10 amino acid epitope of
A(31-42, had unique CDR3 sequences of the heavy chain
(Figure 7C). Further, antibodies 2A10, 2B4, 4C2 and 4E2,
having similar affinities for SEC Peak 1 and Peak 2 ADDLs
(see Figure 3), shared highly homologous CDR3 sequences of
the heavy chain (Figure 7C). Moreover, amino acid
substitutions in CDR3 of the heavy chain of antibody 4E2
appeared to enhance blockage of binding of ADDLs to
neuronal cells, as 4E2 is more effective than antibody 2D6
at blocking ADDL binding to neurons and the sequences of
the heavy and light chains of 4E2 and 2D6 were identical
except for three amino acid residues of CDR3 of the heavy
chain; Ser vs. Asn, Thr vs. Ser, and Ile vs. Val for 2D6
and 4E2, respectively (Figure 7C).
Example 18: Humanization of Mouse Anti-ADDL Antibody
Variable Region Sequences
Mouse antibody heavy and light variable domains
nucleic acids obtained from mouse hybridoma cell lines
20C2, 26D6, 4E2, 3B3, 2H4 and 1F6 were humanized using a
CA 02790433 2012-09-19
-71-
CDR grafting approach and in the case of 20C2 and 26D6 a
veneering strategy. It will be appreciated by those skilled
in the art that humanization of mouse antibody sequences
can maximize the therapeutic potential of an antibody by
improving its serum half-life and Fc effector functions
thereby reducing the anti-globulin response.
Humanization by CDR grafting was carried out by
selecting the human light and heavy chain variable regions
from the NCBI protein database with the highest homology to
the mouse variable domains. The mouse variable region
sequences were compared to all human variable region
sequences in the database using the protein-protein Basic
Local Alignment Search Tool (BLAST). Subsequently, mouse
CDRs were joined to the human framework regions and the
preliminary amino acid sequence was analyzed. All
differences between the mouse and human sequences in the
framework regions were evaluated particularly if they were
part of the canonical sequences for loop structure or were
residues located at the VL/VH interface (O'Brien and Jones
(2001) In: Antibody Engineering, Kontermann and Dubel
(Eds.), Springer Laboratory Manuals). Framework regions
were also scanned for unusual or rare amino acids in
comparison to the consensus sequences for the human
subgroup and for potential glycosylation sites. Wherein
amino acid sequence differences existed between the mouse
and human framework region sequences that were not found to
be involved in canonical sequences, or located at the VL/VH
interface, the human residue was selected at that position.
Wherein a difference in a key residue existed, two versions
of the variable region sequence were generated for
evaluation. The CDR grafting strategy made the minimum:
number of changes to the human framework region so that
good antigen binding was achieved while maintaining human
framework regions that closely matched the sequence from a
CA 02790433 2012-09-19
-72-
natural human antibody. The design of humanized amino acid
sequences using CDR grafting is shown in Figure 8.
Humanized sequences for 20C2 and 26D6 were also
designed using a veneering strategy (See, e.g., U.S. Patent
No. 6,797,492). Humanization was carried out by selecting
the human light and heavy chain variable regions from the
NCBI protein database with the highest homology to the
mouse variable domains, as well as to the closest human
antibody germline family or families (see, Kabat, eta 1.
(1991) Sequences of proteins of immunological interest, 5th
ed., U.S. Dept. Health and Human Services, NIH, Washington
DC)_ The mouse variable region sequences were compared to
all human variable region sequences in the database using
protein-protein BLAST. The murine variable sequences and
their closest human homologues were modeled to the closest
crystallized human antibody as determined by computer
modeling as practiced in the art. From the model of the
murine VH and VL sequences, a surface area map was
constructed, which dictated the solvent accessibility of
the amino acids in the mouse heavy and light variable
regions. To confirm the modeling, these exposed residues
were compared position-by-position with known surface
accessible residues (see, e.g_, Padlan (1994) Mol. immunol.
31(3):169-217). A score was assigned for each residue in the
sequence designating it as exposed, mostly exposed, partly
buried, mostly buried and buried according to established
methods (see, U.S. Patent No. 6,797,492) . Mouse framework
residues that scored as exposed or mostly exposed and
differed from the homologous human sequence were changed to
the human residue at that position. The designed veneered
sequences retained the mouse CDRs, residues neighboring the
CDRs, residues known be involved in canonical sequences,
residues located at the VL/VH interface, and residues at
the N-terminal sequences of the mouse heavy and light
chain. The N-terminal sequences are known to be contiguous
CA 02790433 2012-09-19
-73-
with the CDR surface and are potentially involved in ligand
binding. Likewise, care was taken to limit changes in Pro,
Gly, or charged residues. Once the veneered sequences were
finalized they were remodeled to look for are any potential
obvious structural issues. In some instances, more then one
veneered sequence was generated for analysis. The design of
humanized amino acid sequences using the veneering approach
is shown in Figure 9.
Once the humanized amino acid sequences were selected
the sequences were reverse-translated to obtain the
corresponding DNA sequence. The DNA sequences were codon-
optimized using art-established methods (Lathe (1985) J.
Mot. Biol. 183(1):1-12) and designed with flanking
restriction enzyme sites for cloning into human antibody
expression vectors. The DNA sequences synthesized are
presented in Figures 10A-10S. For the 20C2 humanized
antibodies designed by CDR grafting and veneering, both
human IgG1/kappa and IgG2m4/kappa versions were
constructed, wherein IgG2m4 represents selective
incorporation of human IgG4 sequences into a standard human
IgG2 constant region. IgG1/kappa and IgG2m4/kappa versions
were also made for the 26D6 CDR grafted antibody. For all
other antibodies only the IgG1/kappa versions were made.
The complete amino acid sequence of the resulting
antibodies is shown in Figures 11A-11Y.
Antibodies were expressed by co-transient transfection
of separate light and heavy chain expression plasmids into
293 EBNA cells. In cases where more then one humanized
heavy or light chain sequence was designed for a given
antibody, all combinations of heavy and light chains were
combined to generate the corresponding antibodies.
Antibodies were purified from culture supernatant 7-10 days
post-transfection using protein A columns and used in
subsequent analysis.
CA 02790433 2012-09-19
-74
Example 19: Generation of IgG2m4 Antibodies
IgG2m4 antibody derivatives were prepared to decrease
Fc receptor engagement, C1q binding, unwanted cytotoxicity
or immunocomplex formation while maintaining both the long
half-life and pharmacokinetic properties of a typical human
antibody. The basic antibody format of IgG2m4 is that of
IgG2, which has been shown to possess a superior half-life
in experimental models (Zuckier, et al. (1994) Cancer
Suppl. 73:794-799) . The structure of IgG2 was modified to
eliminate Clq binding, through selective incorporation of
IgG4 sequences, while maintaining the typical low level of
FcyR binding (Canfield and Morrison (1991) J. Exp. Med.
173:1483-1491). This was achieved by using cross-over
points wherein sequences of IgG2 and IgG4 were identical,
thereby producing an antibody containing natural Fe
sequences rather than any artificial mutational sequences.
The IgG2m4 form of the human antibody constant region
was formed by selective incorporation of human IgG4
sequences into a standard human IgG2 constant region, as
shown in Figure 12. Conceptually, IgG2m4 resulted from a
pair of chain-swaps within the CH2 domain as shown in
Figure 12. Four single mutations were made corresponding to
sequences from IgG4. The Fc residues mutated in IgG2
included His268GIn, Va1309Leu, Ala33OSer, and Pro33lSer,
which minimized the potential for neoepitopes. The specific
IgG4 amino acid residues placed into the IgG2 constant
region are shown in Table 7, along with other alternatives
from the basic structure.
TABLE 7
Residue
(Kabat Residue Residue Residue Alternative
number- in IgG2 in IgG4 in residue in Comment
ing) IgG2m4 IgG2m4
Key polymorphism of IgG2;
Pro or Pro residue present in
189 Thr* Pro Thr Pro IGHG*01 allotype and Thr
residue present in
IGHG2*02 allotypea,e
268 His Gin Gin -- Change in the B/C loop
CA 02790433 2012-09-19
-75-
known to be involved in
FcyRII binding'.
309 Val L Vao or Leu Val FcRn binding domain
Key residue for Clq
bindingd; also
330 Fula Ser Ser -- potentially involved in
binding FcyRII and
FcyRllle.
Key residue for Clq
bindingd'f and FcyRl
331 Pro Ser Ser binding9; also
potentially involved in
binding FcyRII and
FcyRI I I.e
Val residue present in
Met or IGHG*01 allotype and Met
397 Val* Val Met Val residue present in
IGHG2*02 allotypea.
*Positions marked with an asterisk are subject to allelic
variations.
aHougs, et al. (2001) Immunogenetics 52(3-4):242-8.
bWO 97/11971.
cMedgyesi, et al. (2004) Eur. J_ Immunol. 34:1127-1135,
dTao, et al. (1991) J. Exp. Med. 173:1025-1028.
eArmour, et al. (1999) Eur. J. Immunol. 29:2613.
fXu, et al. (1994) J. Biol. Chem_ 269:3469-3474.
9Canfield and Morrison (1991) J. Exp. Med. 173:1483-
Example 20: Binding Affinity of Humanized Anti-ADDL
Antibodies
To evaluate ADDL binding affinity of the humanized
antibodies, titration ELISAs were conducted as disclosed
herein. Streptavidin-coated, 96-well microtiter plates
(Sigma, St. Louis, MO) were coated with 10% biotinylated
ADDL antigen (1 pM). A series of 2-fold dilutions of
purified antibody, starting at 500 ng/mL was added to the
ADDL captured plates and the plates were incubated for 2
hours at 25 C. After washing five times with PBS solution
using a plate washer (Bio-Tek, Winooski, VA), polyclonal
goat anti-human kappa light chain antibody (Biomeda, Foster
City, CA) was added at a 1/2000 dilution in 3% non-fat milk
blocker and incubated at room temperature for 1 hour. A
rabbit anti-goat IgG (H+L) HRP-conjugated (Bethyl
Laboratories, Inc., Montgomery, TX) detection antibody was
then added at a 1/ 2000 dilution in blocking solution and
CA 02790433 2012-09-19
-76-
incubated for 1 hour at room temperature. After washing
with PBS, HRP substrate, 3,3',5'5-tetramethylbenzidine
(ready-to-use TMB; Sigma, St. Louis, MO) was added and the
reaction was stopped after 10 minutes with 0.5 N H2SO4.
Absorbance at wavelength of 450 nm was read in a plate
reader (model VICTOR V; Perkin Elmer, Boston, MA) and data
were processed using EXCEL work sheet. Assay variations
between plates were estimated within 20%.
Different groups of humanized antibodies were compared
in different experiments. A comparison of IgGi antibodies
20C2A, 20C2B, 3B3, 4E2, 1F6 and 2H4 humanized by CDR
grafting indicated that all antibodies could bind to ADDLs,
wherein binding with IF6 was weaker than the majority and
20C2A was the strongest. The four different humanized
versions of 20C2 IgG1 antibodies (two CDR grafted versions
and two veneered versions) were also compared and found to
exhibit very similar ADDL binding curves with all binding
slightly better then a chimeric 20C2 antibody. The seven
different humanized versions of 26D6 IgGi (one CDR grafted
versions and six veneered versions) were also compared. All
were found to have ADDL binding curves similar to the
chimeric form of 26D6. The IgGl and IgG2m4 antibodies for
the two 20C2 versions humanized by CDR grafting were also
analyzed and found to have comparable binding curves as did
the IgGl and IgG2m4 isotypes of 26D6 humanized by CDR
grafting.
Example 21: Inhibition of ADDL Binding to Neurons Using
Humanized Anti-ADDL Antibodies
The humanized anti-ADDL antibodies were further
evaluated for their ability to block ADDL binding to
primary hippocampal neurons using the methods disclosed
herein. The relevant antibodies, or PBS as a control, were
mixed at a 1:1 (B103 neuroblastoma cells) or 1:5 (primary
hippocampal neurons) molar ratio with 2.5-10 pm (final
CA 02790433 2012-09-19
-77-
concentration) of bADDLs and incubated for one hour at 37 C
on a slow rotator. After the preincubation, the
antibody/bADDL preparations were added to the B103 or
primary neuron cultures and incubated for an additional
hour at 37 C. At the end of the incubation period, the
bADDLs/antibody mixture was removed and the plates washed
six times with media. The cells were then fixed in 40
paraformaldehyde for ten minutes at room temperature, the
solution removed, fresh fixative added, and the cells fixed
for an additional ten minutes. The cells were permeabilized
with 4% paraformaldehyde containing 0.1% TRITONT" X-100 (2
times, each for ten minutes at room temperature), washed
six times in PBS and then treated with 10% BSA in PBS for
one hour at 37 C. Alkaline phosphatase-conjugated
streptavidin (1:1,500 in 1% BSA; Molecular Probes, Eugene,
OR) was then added to the cells for one hour at room
tempe-rature. The cells were rinsed six times with PBS, the
alkaline phosphatase substrate (CDP-STAR with SAPPHIRE-
III"; Applied Biosystems, Foster City, CA) added to the
cells and incubated for thirty minutes prior to determining
the luminescence on a LJL Luminometer (Analyst AD; LJL
Biosystems, Sunnyvale, CA). As with the murine antibodies,
the humanized versions of 26D6, 20C2, 4E2, 3B3, 2H4 and 1F6
were capable of inhibiting the binding of ADDL preparations
to B103 neuroblastoma cells and to primary neurons.
Example 22: Affinity Maturation of a Humanized Anti-ADDL
Antibody
Nucleic acid molecules encoding humanized 20C2 version
A variable heavy chain only, light chain only, or heavy
chain and light chain together were cloned in the Fab
phage-display vector pFab3d. Nucleic acid sequence analysis
confirmed sequence and orientation in pFab3d. The annotated
20C2 Fab sequences in pFab3d are presented in Figure 13 and
set forth herein as SEQ ID NO:255 for the heavy chain and
CA 02790433 2012-09-19
SEQ ID NO:256 for the light chain. The three constructs
were used in the 20C2 maturation program using art-
established phage-displayed Fab library methods.
Briefly, two libraries were designed to mutate the
nine wild-type amino acids of CDR3 of the light (kappa)
chain of 20C2 (i.e., Phe-Gln-Gly-Ser-Leu-Val-Pro-Leu-Thr;
SEQ ID NO:60). These libraries were designated LC3-1 and
LC3-2 representing light chain CDR3 sequences of Xaa-Xaa-
Xaa-Xaa-Xaa-Val-Pro-Leu-Thr (SEQ ID NO:257) and Phe-Gln-
Gly-Ser-Xaa-Xaa-Xaa-Xaa-Xaa (SEQ ID NO:258), respectively.
Biotinylated reverse primers, 20C2LC3-1 (SEQ ID NO:259) and
20C2LC3-2 (SEQ ID N0:260), were used in combination with
forward primer 20C2LC3F (SEQ ID NO:261) to generate the
LC3-1 and LC3-2 libraries (see Figure 14). Primers were
purified by polyacrylamide gel electrophoresis, whereas the
vector DNA was purified by gel electrophoresis and
electroelution. The two light chain libraries were designed
to be randomly mutated. The final diversities of the three
IOG5H6 LC3 libraries were 4.76 x 108 and 7.45 x 108,
respectively (Table 8). Sequence analysis of approximately
100 clones from the libraries showed 100% diversity of
mutant clones at the designed amino acid positions.
TABLE 8
20C2 Library
Characteristic
LC3-1 LC3-2
Vector pFab3d20C2HS pFab3d2OC2HS
Number of 4.76 x 108 7.45 x 100
Transformants
Library Diversity 4.76 x 108x 0.89 = 7.45 x 108x 0.90 =
4.24 10 6.71 10
Primary Library 2 mL 2 mL
Volume
Primary Library 2.13 x 1011 *9.3 x 1010
Titer
*Higher titers are achieved by concentration or phage
rescue.
CA 02790433 2012-09-19
-79-
Soluble panning of the two 20C2 light chain libraries
against high molecular weight bADDL was completed- Briefly,
four rounds of panning were carried out using biotinylated
high molecular weight ADDL (bADDL). The first three rounds
were carried out using approximately 1.5 }1M antigen
concentration (input = 1 x 1010 to I x 1011) . Upon completion
of the third round, the outputs of the two libraries were
combined and divided into three groups for analysis with 10
nM, 100 nM and approximately 1.5 pM antigen to increase
panning stringency. As such, a total of 58 output plates
were tested in phage ELISA assays, i.e., two plates per
library in the first round (a total of four plates), six
plates per library in the second round (a total of 12
plates), eight plates for LC3-1 and 10 plates for LC3-2
libraries in the third round (a total of 18 plates) and
eight plates for each antigen concentration in the fourth
round-(a total of 24 plates)-
Panning resulted in 1000 hits, 436 of which were
sequenced (Table 9).
TABLE 9
Round Antigen Input Output o Recovery ELISA Sequenced
Screen*
la 1.6 pM 2.13x1010 7.3x104 3.42x10-6 0
(0/176)
2a 2.0 pM 1.55x1011 1.88x105 1.21x10'6 1.50 8
(8/528)
3a 1. 1 pM 1. 80x 10") 7.8X104 4.3x10-6 5.8 0 41
(41/704)
4
1b 1.6 pM 9.30x109 5.7x104 6.13x10-6 (7/176)
2b 2.0 pM 1.23x1011 1.07x105 8.7x10-7 4.5 24
(24/528)
3b 1.1 pM 1.37x1010 3.32x105 2.42x10-5 15 134
(134/880)
4C 1.1 pM 3.0x1012 1.37x105 4.6x10-7 390 _
(274/704)
4C 100 nM 3.0x1011 3.88x105 1.29x10-6 010
(290/704)
4C 10 nM 3.0x1011 1.6x105 5.3x10-7 225
(225/704)
Total 1000/5104 436
a20C2 LC3-1 versus high molecular weight 10% bADDL.
b2 0C2 LC3-2 versus high molecular weight 10% bADDL.
CA 02790433 2012-09-19
-80-
c20C2_ LC3-1 + 20C2 LC3-2 versus high molecular weight 10%
bADDL.
*Hits per total number of colonies.
Sequence and frequency of highly enriched clones are
presented in Table 10.
TABLE 10
Clone CDR3 SEQ ID Round 2 Round 3 Round 4 Total
Designation NO-
Hu20C2LC FQGSLVPLT 60 6 15 14 35
SJ-pl-31 ADTTHVPLT 262 1 2 3
SJ-pl-14 AQSTFVPLT 263 1 1 2 4
4P2-12-E3 AQASFVPLT 264 2 2
SJ-pl-38 AQATKVPLT 265 1 1 2
4P3-59 AQSSKVPLT 266 2 2
SJ-p2-14 AQSTLVPLT 267 1 2 3
4P3-11 FAASSVPLT 268 2 2
4P3-1 FESTYVPLT 269 2 2
SJ-p2-10 FESSRVPLT 270 1 1 2
SJ-p2-11 FNATWVPLT 271 2 2
SJ-p2-60 FQASRVPLT 272 1 5 6
SJ-PI-18 FQATRVPLT 273 1 5 6
SJ-p3-51 FQGSFIGLS 274 1 1 2
SJ-p3-16 FQGSFIPGT 275 2 3 5
SJ-p8-8F FQGSFLPPS 276 1 1 2
SJ-p3-26 FQGSFLPQL 277 1 2 3
SJ-p3-15 FQGSLFPPV 278 1 2 3
SJ-p2-70 FQGSFFSPS 279 1 5 6
SJ-p3-24 FQGSRLPVS 280 1 1 2
SJ-p3-33 FQGSRLPVS 281 2 3 5
SJ-p3-14 FQGSRVPLV 282 2 1 3
SJ-p2-IF FQSSFVPLT 283 6 8 14
4P1-22 FQSSRVPLT 284 15 15
SJ-p2-44 GQTTLVPLT 285 1 3 4
SJ-pl-56 HESTLVPLT 286 2 1 3
4P1-40 HQSSKVPLT 287 4 4
Si-p2-20 IQTSLVPLT 288 2 2
SJ-pl-41 IQAALVPLT 289 1 1 2
SJ-p2-13 LQSSFVPLT 290 1 4 5
4P1-26 LETSRVPLT 291 3 3
SJ-pl-33 LASTHVPLT 292 2 1 3
SJ-p2-27 LNSTTVPLT 293 2 4 6
SJ-p2-62 LQSKSVPLT 294 2 2
4P2-26-E5 LQSVRVPLT 295 3 3
4P1-32 LQSSFVPLT 296 5 5
SJ-p2-37 LQTGRVPLT 297 2 2 4
SJ-p2-64 LQTSFVPLT 298 3 3
4P1-20 LQTSNVPLT 299 5 5
SJ-p2-39 LQTTRVPLT 300 2 6 8
SJ-p2-52 LSSTFVPLT 301 3 1 4
SJ-p2-6L LSSTHVPLT 302 2 1 3
4P1-77 LTSSAVPLT 303 2 2
SJ-p1-59 LVSSLVPLT 304 2 2
Si-p2-23 METANVPLT 305 2 2
CA 02790433 2012-09-19
-81-
SJ-p1-9M MQSSLVPLT 306 1 3 4
SJ-p2-28 MQSSLVPLT 307 1 2 3
SJ-pl-21 MQTSKVPLT 308 1 1 2
4P1-17 SQARMVPLT 309 3 3
SJ-p2-66 SQASRVPLT 310 1 2 3
SJ-pl-49 TQSTQVPLT 311 2 1 3
SJ-p2-24 VCATFVPLT 312 1 1 2
4P1-41 VQSSAVPLT 313 2 2
SJ-p2-51 VQTSLVPLT 314 12 31 43
4P1-64 VQTSVVPLT 315 3 3
SJ-p2-55 VQTTAVPLT 316 2 2
SJ-pl-25 LQTARVPLT 317 1 3 4
Fab fragments from the 10 top clones based on
enrichment frequency were prepared and a total of 15 clones
were converted into IgGl humanized A version and two
clones, 20C2-6 and 20C2-8, were converted to IgG1 humanized
B version- KD values for these clones were measured by
BIACORETM using biotin-A(31-20 (Table 11) and bADDL (Table
12) as antigens- Dramatic improvements in affinity were
observed as compared to parental humanized 20C2A and 20C2B,
as well as mouse 20C2 antibodies. In particular, low
nanomolar to sub-picomolar KDs were achieved with a light
chain CDR3 of the sequence Xaal-Gln-Xaa2-Thr-Arg-Val-Pro-
Leu-Thr (SEQ ID NO:318), wherein Xaal is Phe or Leu, and
Xaal is Ala or Thr. Moreover, a comparison between KD values
obtained with BIACORETM using biotin-A(31-20 and bADDL
further demonstrates that anti-ADDL antibodies such as 20C2
preferentially bind multi-dimensional conformations of
ADDLs over monomeric Aj3 peptides.
TABLE 11
Name Clone LC-CDR3 SEQ ID KD (Biotin-A(31-20)
NO: Fab IgG11 IgGl#2
0C 2 --1A SJ-p2-60 FQASRVPLT 262 91 nM 1.2 nM
20C2-2A SJ-pl-18 FQATRVPLT 273 28 nM 686 pM 2 nM
20C2-3A SJ-p3-16 FQGSFIPGT 275 -- 1.7 nM
20C2-5A SJ-p2-1F FQSSFVPLT 283 41 nM 912 pM 1.5 nM
20C2-6A 4P1-22 FQSSRVPLT 284 18 nM 544 pM 714 pM
20C2-6B 4P1-22 FQSSRVPLT 284 -- 53 pM --
20C2-7A SJ-p2-27 LNSTTVPLT 293 128 nM -- --
20C2-8A Si-p2-39 LQTTRVPLT 300 14 nM 140 pM 376 pM
20C2-8B SJ-p2-39 LQTTRVPLT 300 -- 46 pM 64 pM
20C2-9A SJ-p2-51 VQTSLVPLT 314 36 nM 241 pM 420 pM
2002-10A Si-p3-33 FQGSRLPVS 281 -- 84 nM
CA 02790433 2012-09-19
-82-
20C2-11A SJ-p3-6 FQGSLLPLS 319 -- -- --
20C2-12A 4P1-32 LQSSLVPLT 296 617 nM 1.5 nM
20C2-13A 4pl-20 LQTSNVPLT 299 94 nM 3 nM --
20C2-18A SJ-pl-9M MQSSFVPLT 306 126 nM 1.8 nM --
20C2-20A SJ-p3-15 FQGSLFPPV 278 21 nM
20C2-22A SJ-p2-66 SQASRVPLT 310 2.3 nM
20C2-23A 4P1-40 HQSSKVPLT 287 649 pM 1.5 nM
20C2-24A Si-p2-44 GQTTLVPLT 285 1.9 nM
20C2A FQGSLVPLT 60 27 nM
20C2B FQGSLVPLT 60 5.4 nM
Mouse- FQGSLVPLT 60 83 nM 3.4 nM
20C2
TABLE 12
Name Clone LC-CDR3 SEQ ID KD (bADDL)
NO: Fab IgGl#1 IgGl#2
20C2-1A SJ-p2-60 FQASRVPLT 262 85 nM 75 pM --
20C2-2A SJ-pl-18 FQATRVPLT 273 28 nM 15 pM 0.3 pM
20C2-3A SJ-p3-16 FQGSFIPGT 275 -- 3.7 nM --
20C2-5A SJ-p2-1F FQSSFVPLT 283 41 nM 317 pM 68 pM
20C2-6A 4P1-22 FQSSRVPLT 284 42 nM 4.3 pM 24 pM
20C2-6B 4P1-22 FQSSRVPLT 284 -- 53 pM --
20C2-7A SJ-p2-27 LNSTTVPLT 293 435 nM --
20C2-8A SJ-p2-39 LQTTRVPLT 300 13 nM 3 pM 0.7 pM
20C2-8B SJ-p2-39 LQTTRVPLT 300 -- 13 pM 0.8 pM
20C2-9A SJ-p2-51 VQTSLVPLT 314 40 nM -- 2 pM
20C2-10A SJ-p3-33 FQGSRLPVS 281 -- 7.7 nM.
20C2-11A SJ-p3-6 FQGSLLPLS 319 -- --
20C2-12A 4P1-32 LQSSLVPLT 296 238 nM 15.pM --
20C2-13A 4p1-20 LQTSNVPLT 299 567 nM 764 pM
20C2-18A SJ-pl-9M MQSSFVPLT 306 85 nM 149 pM
20C2-20A SJ-p3-15 FQGSLFPPV 278 6.9 nM
20C2-22A SJ-p2-66 SQASRVPLT 310 198 pM
20C2-23A 4PI-40 HQSSKVPLT 287 85 pM 66 pM
20C2-24A SJ-p2-44 GQTTLVPLT 285 114 pM
20C2A FQGSLVPLT 60
20C2B FQGSLVPLT 60
Mouse- FQGSLVPLT 60 62 nM 4.1 nM
20C2
CA 02790433 2012-09-19
DEMANDES OU BREVETS VOLUMINEUX
LA PRESENTE PARTIE DE CETTE DEMANDE OU CE BREVETS
COMPREND PLUS D'UN TOME.
CECI EST LE TOME 1 DE 2
NOTE: Pour les tomes additionels, veillez contacter le Bureau Canadien des
Brevets.
JUMBO APPLICATIONS / PATENTS
THIS SECTION OF THE APPLICATION / PATENT CONTAINS MORE
THAN ONE VOLUME..
THIS IS VOLUME 1 OF 2
NOTE: For additional volumes please contact the Canadian Patent Office.