Note: Descriptions are shown in the official language in which they were submitted.
CA 02794430 2012-10-31
DEMANDES OU BREVETS VOLUMINEUX
LA PRESENTE PARTIE DE CETTE DEMANDE OU CE BREVETS
COMPREND PLUS D'UN TOME.
CECI EST LE TOME I DE 2
NOTE: Pour les tomes additionels, veillez contacter le Bureau Canadien des
Brevets.
JUMBO APPLICATIONS / PATENTS
THIS SECTION OF THE APPLICATION / PATENT CONTAINS MORE
THAN ONE VOLUME.
THIS IS VOLUME 1 OF 2
NOTE: For additional volumes please contact the Canadian Patent Office
CA 02794430 2014-07-09
76433-30D
1
STABILIZED BIOACTIVE PEPTIDES AND METHODS OF IDENTIFICATION,
SYNTHESIS AND USE
This is a division of Canadian Patent Application Serial No. 2,346,122 filed
on
October 12, 1999.
Background of the Invention
Bioactive peptides are small peptides that elicit a biological activity. Since
the
discovery of secretin in 1902, over 500 of these peptides which average 20
amino acids in size
have been identified and characterized. They have been isolated from a variety
of systems,
exhibit a wide range of actions, and have been utilized as therapeutic agents
in the field of
medicine and as diagnostic tools in both basic and applied research. Tables 1
and 2 list some
of the best known bioactive peptides.
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
2
Table I: Bioactive peptides utilized in medicine
Name Isolated From Size In Therapeutic Use
Amino Acids
Angiotensin II Human Plasma 8 Vasoconstrictor
Bradykinin Human Plasma 9 Vasodilator
Caerulein Frog Skin 10 Choleretic Agent
Calcitonin Human Parathyroid Gland 32 Calcium Regulator
Cholecystokinin Porcine Intestine 33 Choleretic
Agent
Corticotropin Porcine Pituitary Gland 39 Hormone
Eledoisin Octopod Venom 11 Hypotensive Agent
Gastrin Porcine Stomach 17 Gastric Activator
Glucagon Porcine Pancreas 29 Antidiabetic Agent
Gramicidin D Bacillus brevis Bacteria 11 Antibacterial Agent
Insulin Canine Pancreas Antidiabetic Agent
Insulin A 21
Insulin B 30
Kallidin Human Plasma 10 Vasodilator
Luteinizing Bovine Hypothalamus 10 Hormone Stimulator
Hormone-
Releasing Factor
Melittin Bee Venom 26 Antirheumatic Agent
Oxytocin Bovine Pituitary Gland 9 Oxvtocic Agent
Secretin Canine Intestine 27 Hormone
Sermorelin Human Pancreas 29 Hormone Stimulator
Somatostatin Bovine Hypothalamus 14 Hormone Inhibitor
Vasopressin Bovine Pituitary Gland 9 Antidiuretic Agent
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
3
Table 2: Bioactive peptides utilized in applied research
Name Isolated From Size In Biological Activity
Amino Acids
Atrial Natriurctic Rat Atria 28 Natriuretic Agent
Peptide
Bombesin Frog Skin 14 Gastric Activator
Conantokin G Snail Venom 17 Neurotransmitter
Conotoxin Cl Snail Venom 13 Neuromuscular Inhibitor
Defensin HNP-1 Human Neutrophils 30 Antimicrobial
Agent
Delta Sleep- Rabbit Brain 9 Neurological Affector
Inducing
Peptide
Dermascptin Frog Skin 34 Antimicrobial Agent
Dynorphin Porcine Brain 17 Neurotransmitter
EET1 II Ecballium elaterium 29 Protease Inhibitor
seeds
Endorphin Human Brain 30 Neurotransmitter
Enkephalin Human Brain 5 Neurotransmitter
Histatin 5 Human Saliva 24 Antibacterial Agent
Mastoparan Vespid Wasps 14 Mast Cell Degranulator
Magainin 1 Frog Skin 23 Antimicrobial Agent
Melanocyte Porcine Pituitary Gland 13 Hormone Stimulator
Stimulating
Hormone
Motilin Canine Intestine 22 Gastric Activator
Neurotensin Bovine Brain 13 Neurotransmitter
Physalaemin Frog Skin 11 Hypotensive Agent
Substance P Horse Intestine 11 Vasodilator
Vasoactive Porcine Intestine 28 Hormone
Intestinal
Peptide
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
4
Where the mode of action of these peptides has been determined, it has been
found to be due to the interaction of the bioactive peptide with a specific
protein
target. In most of the cases, the bioactive peptide acts by binding to and
inactivating its protein target with extremely high specificities. Binding
constants of these peptides for their protein targets typically have been
determined to be in the nanomolar (nM, 10-9 M) range with binding constants as
high as 10-12 M (picomolar range) having been reported. Table 3 shows target
proteins inactivated by several different bioactive peptides as well as the
binding
constants associated with binding thereto.
Table 3: Binding constants of bioactive peptides
Bioactive Size in Inhibited Binding
Peptide Amino Protein Constant
Acids
a-Conotoxin GIA 15 Nicotinic Acetylcholine 1 oxio- 9
Receptor
EE11 11 29 Trypsin 1.0)(10-12 M
H2 (7-15) 8 HSV Ribonucleotide 3.6x10-5 M
Reductase
Histatin 5 24 Bacteroides gingivalis 5.5)(10-8 m
Protease
Melittin 26 Calmodulin 3.0x10-9 M
Myotoxin (29-42) 14 ATPase 1.9x10-5 M
Neurotensin 13 Ni Regulatory Protein 5 .6x10-11 M
Pituitary Adenylate 38 Calmodulin 1. 5x10 6 M
Cyclase Activating
Polypeptide
PKI (5-24) 20 cAMP-Dependent Protein 2.3x10-9 m
Kinase
SCP (153-180) 27 Calpain 3.0x10-8 M
Secretin 27 HSR G Protein 3 .2x10-9 M
Vasoactive Intestinal 28 GPRN1 G Protein 2.5x10-9 M
Peptide
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
Recently, there has been an increasing interest in employing synthetically
derived bioactive peptides as novel pharmaceutical agents due to the
impressive
ability of the naturally occurring peptides to bind to and inhibit specific
protein
targets. Synthetically derived peptides could be useful in the development of
5 new antibacterial, antiviral, and anticancer agents. Examples of
synthetically
derived antibacterial or antiviral peptide agents would be those capable of
binding to and preventing bacterial or viral surface proteins from interacting
with
their host cell receptors, or preventing the action of specific toxin or
protease
proteins. Examples of anticancer agents would include synthetically derived
peptides that could bind to and prevent the action of specific oncogenic
proteins.
To date, novel bioactive peptides have been engineered through the use
of two different in vitro approaches. The first approach produces candidate
peptides by chemically synthesizing a randomized library of 6-10 amino acid
peptides (J. Eichler et al., Med. Res. Rev. 15:481-496 (1995); K. Lam,
Anticancer Drug Des. 12:145-167 (1996); M. Lebl et at,, Methods Enzymol.
289:336-392 (1997)). In the second approach, candidate peptides are
synthesized by cloning a randomized oligonucleotide library into a Ff
filamentous phage gene, which allows peptides that are much larger in size to
be
expressed on the surface of the bacteriophage (H. Lowman, Ann. Rev. Biophys.
Biomol. Struct. 26:401-424 (1997); G. Smith et al., etal. Meth. Enz. 217:228-
257 (1993)). To date, randomized peptide libraries up to 38 amino acids in
length have been made, and longer peptides are likely achievable using this
system. The peptide libraries that are produced using either of these
strategies
are then typically mixed with a preselected matrix-bound protein target.
Peptides that bind are eluted, and their sequences are determined. From this
information new peptides are synthesized and their inhibitory properties are
determined. This is a tedious process that only screens for one biological
activity at a time.
Although these in vitro approaches show promise, the use of
synthetically derived peptides has not yet become a mainstay in the
pharmaceutical industry. The primary obstacle remaining is that of peptide
instability within the biological system of interest as evidenced by the
unwanted
degradation of potential peptide drugs by proteases and/or pentidases in the
host
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
6
cells. There are three major classes of peptidases which can degrade larger
peptides: amino and carboxy exopeptidases which act at either the amino or the
carboxy terminal end of the peptide, respectively, and endopeptidases which
act
on an internal portion of the peptide. Aminopeptidases, carboxypeptidases, and
endopeptidases have been identified in both prokaryotic and cukaryotic cells.
Many of those that have been extensively characterized were found to function
similarly in both cell types. Interestingly, in both prokaryotic and
eukaryotic
systems, many more aminopeptidases than carboxypeptidases have been
identified to date.
Approaches used to address the problem of peptide degradation have
included the use of D-amino acids or modified amino acids as opposed to the
naturally occurring L-amino acids (e.g., J. Eichler et al., Med Res Rev.
15:481-
496 (1995); L. Sanders, Eur. J. Drug Metabol. Pharmacokinetics 15: 95-102
(1990)), the use of cyclized peptides (e.g., R. Egleton, et al., Peptides
18:1431-
1439 (1997)), and the development of enhanced delivery systems that prevent
degradation of a peptide before it reaches its target in a patient (e.g., L.
Wearley,
Crit. Rev. Thor. Drug Carrier Syst. 8: 331-394 (1991); L. Sanders, Eur. J.
Drug
Metabol. Pharmacokinetics 15: 95-102 (1990)). Although these approaches for
stabilizing peptides and thereby preventing their unwanted degradation in the
biosystem of choice (e.g., a patient) are promising, there remains no way to
routinely and reliably stabilize peptide drugs and drug candidates. Moreover,
many of the existing stabilization and delivery methods cannot be directly
utilized in the screening and development of novel useful bioactive peptides.
A
biological approach that would serve as both a method of stabilizing peptides
and a method for identifying novel bioactive peptides would represent a much
needed advance in the field of peptide drug development.
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
7
Summary of the Invention
The present invention provides an intracellular screening method for
identifying novel bioactive peptides. A host cell is transformed with an
expression vector comprising a tightly regulable control region operably
linked
to a nucleic acid sequence encoding a peptide. The transformed host cell is
first
grown under conditions that repress expression of the peptide and then,
subsequently, expression of the peptide is induced. Phenotypic changes in the
host cell upon expression of the peptide are indicative of bioactivity, and
are
evaluated. If, for example, expression of the peptide is accompanied by
inhibition of host cell growth, the expressed peptide constitutes a bioactive
peptide, in that it functions as an inhibitory peptide.
Intracellular identification of bioactive peptides can be advantageously
carried out in a pathogenic microbial host cell. Bioactive peptides having
antimicrobial activity arc readily identified in a microbial host cell system.
Further, the method can be carried out in a host cell that has not been
modified to
reduce or eliminate the expression of naturally expressed proteases or
peptidases.
When carried out in a host cell comprising proteases and peptides, the
selection
process of the invention is biased in favor of bioactive peptides that are
protease-
and peptidase-resistant.
The tightly regulable control region of the expression vector used to
transform the host cell according to the invention is preferably derived from
the
wild-type Escherichia call lac operon, and the transformed host cell
preferably
comprises an amount of Lac repressor protein effective to repress expression
of
the peptide during host cell growth under repressed conditions. To insure a
sufficient amount of Lac repressor protein, the host cell can be transformed
with
a second vector the overproduces Lac repressor protein.
Optionally, the expression vector used to transform the host cell can be
genetically engineered to encode a stabilized peptide that is resistant to
peptidases and proteases. For example, the coding sequence can be designed to
encode a stabilizing group at either or both of the peptide N-terminus or C-
terminus. As another example, the coding sequence can be designed to encode a
stablizing motif such as an a-helix motif or an opposite charge ending motif,
as
CA 02794430 2016-09-12
60412-4886D
8
described below. The presence of a stabilizing group at a peptide terminus or
a stabilizing
motif can slow down the rate of intracellular degradation of the peptide.
In another aspect, the invention provides a bioactive peptide comprising a
plurality of sequential uniformly charged amino acids comprising the N-
terminus of the
bioactive peptide and a plurality of sequential oppositely charged amino acids
comprising the
C-terminus of the bioactive peptide, wherein the bioactive peptide comprises
at least three
charged amino acids at each end, wherein one terminus of the bioactive peptide
is positively
charged and the other terminus of the bioactive peptide is negatively charged,
wherein each of
the positively charged amino acids is independently selected from the group
consisting of
lysine and arginine, wherein each of the negatively charged amino acids is
independently
selected from the group consisting of aspartate and glutamate, and wherein the
bioactive
peptide is stabilized by the ionic interaction of the oppositely charged
termini.
The invention further provides a bioactive peptide having a first stabilizing
group comprising the N-terminus and a second stabilizing group comprising the
C-terminus.
Preferably, the first stabilizing group is selected from the group consisting
of a small stable
protein, Pro-, Pro-Pro-, Xaa-Pro- and Xaa-Pro-Pro-; and the second stabilizing
group is
selected from the group consisting of a small stable protein, -Pro, -Pro-Pro, -
Pro-Xaa and
-Pro-Pro-Xaa. Suitable small stable proteins include Rop protein, glutathione
sulfotransferase, thioredoxin, maltose binding protein, and glutathione
reductase. In addition,
the invention provides a bioactive peptide stabilized by an opposite charge
ending motif, as
described below. The bioactive peptide is preferably an antimicrobial peptide
or a therapeutic
peptide drug.
Also provided by the invention is a polypeptide that can be cleaved to yield a
bioactive peptide having a stabilizing group at either or both of its N- and C-
termini. The
cleavable polypeptide accordingly comprises a chemical or enzymatic cleavage
site either
immediately preceding the N-terminus of the bioactive peptide or immediately
following the
C-terminus of the bioactive peptide.
CA 02794430 2016-09-12
60412-4886D
9
Thus, in another embodiment, the invention provides a polypeptide
comprising: a bioactive peptide comprising a plurality of sequential uniformly
charged amino
acids comprising the N-terminus of the bioactive peptide and a plurality of
sequential
oppositely charged amino acids comprising the C-terminus of the bioactive
peptide, wherein
the bioaetive peptide comprises at least three charged amino acids at each
end, wherein one
terminus of the bioactive peptide is positively charged and the other terminus
of the bioactive
peptide is negatively charged, wherein each of the positively charged amino
acids is
independently selected from the group consisting of lysine and arginine,
wherein each of the
negatively charged amino acids is independently selected from the group
consisting of
aspartate and glutamate, and wherein the bioactive peptide is stabilized by
the ionic
interaction of the oppositely charged termini; and a cleavage site immediately
preceding the
plurality of sequential uniformly charged amino acids comprising the N-
terminus of the
bioactive peptide.
In another embodiment, the invention provides a polypeptide comprising: a
bioactive peptide comprising a plurality of sequential uniformly charged amino
acids
comprising the N-terminus of the bioactive peptide and a plurality of
sequential oppositely
charged amino acids comprising the C-terminus of the bioactive peptide,
wherein the
bioactive peptide comprises at least three charged amino acids at each end,
wherein one
terminus of the bioactive peptide is positively charged and the other terminus
of the bioactive
peptide is negatively charged, wherein each of the positively charged amino
acids is
independently selected from the group consisting of lysine and arginine,
wherein each of the
negatively charged amino acids is independently selected from the group
consisting of
aspartate and glutamate, and wherein the bioactive peptide is stabilized by
the ionic
interaction of the oppositely charged termini; and a cleavage site immediately
following the
plurality of sequential oppositely charged amino acids comprising the C-
terminus of the
bioactive peptide.
The invention further provides a fusion protein comprising a four-helix bundle
protein, preferably the Rop protein, and a polypeptide. The four-helix bundle
protein is
81685434
9a
positioned at either the N-terminus or the C-terminus of the fusion protein,
and accordingly can be
fused to either the N-terminus or the C-terminus of the polypeptide.
The present invention also provides a method for using an antimicrobial
peptide.
An antimicrobial peptide is stabilized by linking a first stabilizing group to
the N-terminus of an
antimicrobial peptide, and, optionally, a second stabilizing group to the C-
terminus of the
antimicrobial peptide. Alternatively, the antimicrobial peptide is stabilized
by flanking the
peptide sequence with an opposite charge ending motif, as described below. The
resulting
stabilized antimicrobial peptide is brought into contact with a microbe,
preferably a pathogenic
microbe, for example to inhibit the growth or toxicity of the microbe.
In a particular embodiment, the invention provides use of an antimicrobial
peptide
for inhibiting microbial growth, said antimicrobial peptide having an N-
terminus with a plurality
of sequential uniformly charged amino acids covalently linked thereto and
having a C-terminus
with a plurality of sequential oppositely charged amino acids covalently
linked thereto, wherein
the antimicrobial peptide comprises at least three charged amino acids at each
end, wherein one
terminus of the antimicrobial peptide is positively charged and the other
terminus of the
antimicrobial peptide is negatively charged, wherein each of the positively
charged amino acids is
independently selected from the group consisting of lysine and arginine,
wherein each of the
negatively charged amino acids is independently selected from the group
consisting of aspartate
and glutamate, and wherein the antimicrobial peptide is stabilized by the
ionic interaction of the
oppositely charged termini.
The invention also provides a method for treating a patient having a condition
treatable with a peptide drug, comprising administering to the patient a
stabilized peptide drug
having at least one of a first stabilizing group comprising the N-terminus of
the stabilized peptide
drug and a second stabilizing group comprising the C-terminus of the
stabilized peptide drug.
Optionally, prior to administration of the stabilized peptide drug, the first
stabilizing group is
covalently linked to the N-terminus of a peptide drug, and the second
stabilizing group is
covalently linked to the C-terminus of the peptide drug to yield the
stabilized peptide drug.
Alternatively, the method comprises administering to the patient a peptide
drug that has been
stabilized by flanking the peptide sequence with an opposite charge ending
motif, as described
below.
CA 2794430 2017-09-27
CA 02794430 2016-09-12
76433-30D
9b
In a further embodiment, the invention relates to use, for treating a patient
having a
condition treatable with a peptide drug, of a stabilized form of the peptide
drug, wherein the
stabilized form of the peptide drug comprises a plurality of sequential
uniformly charged amino
acids comprising the N-terminus of the peptide drug and a plurality of
sequential oppositely
charged amino acids comprising the C-terminus of the peptide drug, wherein the
peptide drug
comprises at least three charged amino acids at each end, wherein one terminus
of the bioactive
peptide is positively charged and the other terminus of the bioactive peptide
is negatively charged,
wherein each of the positively charged amino acids is independently selected
from the group
consisting of lysine and arginine, wherein each of the negatively charged
amino acids is
independently selected from the group consisting of aspartate and glutamate,
and wherein the
peptide drug is stabilized by the ionic interaction of the oppositely charged
termini.
Brief Description of the Drawings
Figure 1 shows the control region (SEQ ID NO:1) of the wild-type lac operon
from the auxiliary operator 03 through the translational start of the lacZ
gene. DNA binding sites
include the operators 03 and 01 (both underlined), CAP (boxed), the -35 site
(boxed), and the -10
site (boxed), while important RNA and protein sites include the Lad
translation stop site (TGA),
the +1 lacZ transcription start site, the Shine Dalgarno (SD) ribosome binding
site for lacZ, and
the LacZ translation start site (ATG).
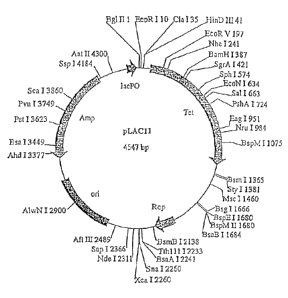
Figure 2 is a map of plasmid pLAC11. The unique restriction sites and the base
pair at which they cut are indicated. Other sites of interest are also shown,
including Tet (98-
1288), Rop (1931-2122), on (2551-3138), Amp (3309-4169), and lacP0 (4424-
4536).
Figure 3 is a map of plasmid pLAC22. The unique restriction sites and the base
pair at which they cut are indicated. Other sites of interest are also shown,
including Tet (98-
1288), Rop (1927-2118), on (2547-3134), Amp (3305-4165), /acicl (4452-5536),
and lacP0
(5529-5641).
Figure 4 is a map of plasmid pLAC33. The unique restriction sites and the base
pair at which they cut are indicated. Other sites of interest are also shown,
including Tet (98-
1288), on (1746-2333), Amp (2504-3364), and lacP0 (3619-3731).
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
Figure 5 shows the response of the pLAC I I -lacZ construct (open circles)
to varying amounts of isopropyl 13-D-thiogalactoside (IPTG). A filled square
indicates the f3-galactosidase activity that was obtained when MG1655 or
CSH27 cells were grown in rich media induced with 1 mM IPTG. while a filled
5 diamond indicates the Vgalactosidase activity that was obtained when
MG1655
or CSH27 cells were grown in M9 minimal lactose media.
Figure 6 shows growth curves depicting the inhibitory effects of a two
day inhibitor (pPep12) versus a one day inhibitor (pPepl). Data points for the
control, pLAC11, for pPepl, and for pPep12, are indicated by squares, circles,
10 and triangles, respectively.
Figure 7 is a map of the p-Rop(C) fusion vector. The unique restriction
sites and the base pair at which they cut are indicated. Other sites of
interest are
also shown, including Rop (7-198), on (627-1214), Amp (2245-1385), lacP0
(2500-2612).
Figure 8 is a map of the p(N)Rop-fusion vector. The unique restriction
sites and the base pair at which they cut are indicated. Other sites of
interest are
also shown; Rop (7-204), on (266-853), Amp (1024-1884), lacP0 (2139-2251).
Figure 9 illustrates a peptide (SEQ ID NO:2) having the opposite charge
ending motif, wherein the amino and carboxy termini of the peptide are
stabilized
by the interactions of the opposite charge ending amino acids.
Detailed Description
The present invention represents a significant advance in the art of
peptide drug development by allowing concurrent screening for peptide
bioactivity and stability. Randomized recombinant peptides are screened for
bioactivity in a tightly regulated inducible expression system, preferably
derived
from the wild-type lac operon, that permits essentially complete repression of
peptide expression in the host cell. Subsequent induction of peptide
expression
can then be used to identify peptides that inhibit host cell growth or possess
other bioactivitics.
Intracellular screening of randomized peptides has many advantages over
existing methods. Bioactivity is readily apm.1:_lai, many di-, else
bioactivities can
CA 02794430 2012-10-31
WO 00/22112 PCPUS99/23731
11
be screened for simultaneously, very large numbers of peptides can be screened
using easily generated peptide libraries, and the host cell, if desired, can
be
genetically manipulated to elucidate an affected protein target.
Advantageously,
randomized peptides can be screened in a host cell that is identical to or
closely
resembles the eventual target cell for antimicrobial applications. An
additional
and very important feature of this system is that selection is naturally
biased in
favor of peptides that arc stable in an intracellular environment; i.e., that
are
resistant to proteases and peptidases. Fortuitously, bacterial peptidases are
very
similar to eukaryotic peptidases. Peptides that are stable in a bacterial host
are
thus likely to be stable in a eukaryotie cell as well, allowing bacterial
cells to be
used in initial screens to identify drugs that may eventually prove useful as
human or animal therapeutics.
The invention is directed to the identification and use of bioactive
peptides. A bioactive peptide is a peptide having a biological activity. The
term
"bioactivity" as used herein includes, but is not limited to, any type of
interaction
with another biomolecule, such as a protein, glycoprotein, carbohydrate, for
example an oligosaccharide or polysaccharide, nucleotide, polynucleotide,
fatty
acid, hormone, enzyme, cofactor or the like, whether the interactions involve
covalent or noncovalent binding. Bioactivity further includes interactions of
any type with other cellular components or constituents including salts, ions,
metals, nutrients, foreign or exogenous agents present in a cell such as
viruses,
phage and the like, for example binding, sequestration or transport-related
interactions. Bioactivity of a peptide can be detected, for example, by
observing
phenotypic effects in a host cell in which it is expressed, or by performing
an in
vitro assay for a particular bioactivity, such as affinity binding to a target
molecule, alteration of an enzymatic activity, or the like. Examples of
bioactive
peptides include antimicrobial peptides and peptide drugs. Antimicrobial
peptides are peptides that adversely affect a microbe such as a bacterium,
virus,
protozoan, or the like. Antimicrobial peptides include, for example,
inhibitory
peptides that slow the growth of a microbe, microbiocidal peptides that are
effective to kill a microbe (e.g., bacteriocidal and virocidal peptide drugs,
sterilants, and disinfectants), and peptides effective to interfere with
microbial
reproduction, host toxicity, or the like. Peptide drugs for therapeutic use in
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
12
humans or other animals include, for example, antimicrobial peptides that are
not
prohibitively toxic to the patient, peptides designed to elicit, speed up,
slow
down, or prevent various metabolic processes in the host such as insulin,
oxytocin, calcitonin, gastrin, somatostatin, anticancer peptides, and the
like.
The term "peptide" as used herein refers to a plurality of amino acids
joined together in a linear chain via peptide bonds. Accordingly, the term
"peptide" as used herein includes a dipeptide, tripeptide. oligopeptide and
polypeptide. A dipeptide contains two amino acids: a tripeptide contains three
amino acids; and the term oligopeptide is typically used to describe peptides
having between 2 and about 50 or more amino acids. Peptides larger than about
50 are often referred to polypeptides or proteins. For purposes of the present
invention, a "peptide" is not limited to any particular number of amino acids.
Preferably, however, the peptide contains about 2 to about 50 amino acids,
more
preferably about 5 to about 40 amino acids, most preferably about 5 to about
20
amino acids.
The library used to transform the host cell is formed by cloning a
randomized, peptide-encoding oligonucleotide into a nucleic acid construct
having a tightly regulable expression control region. An expression control
region can be readily evaluated to determine whether it is "tightly
regulable," as
the term is used herein, by bioassay in a host cell engineered to contain a
mutant
nonfunctional gene "X." Transforming the engineered host cell with an
expression vector containing a tightly regulable expression control region
operably linked to a cloned wild-type gene "X" will preserve the phenotype of
the engineered host cell under repressed conditions. Under induced conditions,
however, the expression vector containing the tightly regulable expression
control region that is operably linked to the cloned wild-type gene "X" will
complement the mutant nonfunctional gene X to yield the wild-type phenotype.
In other words, a host cell containing a null mutation which is transformed
with
a tightly regulable expression vector capable of expressing the chromosomally
inactivated gene will exhibit the null phenotype under repressed conditions;
but
when expression is induced the cell will exhibit a phenotype indistinguishable
from the wild-type cell. It should be understood that the expression control
region in the tightly regulable es71Jression vect.r of the present invention
can be
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
13
readily modified to produce higher levels of an encoded biopeptidc, if desired
(see, e.g., Example I, below). Such modification may unavoidably introduce
some "leakiness" into expression control, resulting in a low level of peptide
expression under repressed conditions.
In a preferred embodiment, the expression control region of the inducible
expression vector is derived from the wild-type E. coil lac promoter/operator
region. In a particularly preferred form, the expression vector contains a
regulatory region that includes the auxiliary operator 03, the CAP binding
region, the ¨35 promoter site, the -10 promoter site, the operator 01, the
Shine-
Dalgamo sequence for lacZ, and a spacer region between the end of the Shine-
Dalgarno sequence and the ATG start of the iticZ coding sequence (see Fig. 1).
It is to be understood that variations in the wild-type nucleic acid
sequence of the lac promoter/operator region can be tolerated in the
expression
control region of the preferred expression vector and are encompassed by the
invention, provided that the expression control region remains tightly
regulable
as defined herein. For example, the ¨10 site of the wild-type lac operon
(TATGTT) is weak compared to the bacterial consensus ¨10 site sequence
TATAAT, sharing four out of six positions. It is contemplated that other
comparably weak promoters are equally effective at the ¨10 site in the
expression control region; a strong promoter is to be avoided in order to
insure
complete repression in the uninduced state. With respect to the ¨35 region,
the
sequence of the wild-type lac operon, TTTACA, is one base removed from the
consensus ¨35 sequence TTGACA. It is contemplated that a tightly regulable
lac operon-derived expression control region could be constructed using a
weaker ¨35 sequence (i.e., one having less identity with the consensus ¨35
sequence) and a wild-type ¨10 sequence (TATAAT), yielding a weak promoter
that needs the assistance of the CAP activator protein. Similarly, it is to be
understood that the nucleic acid sequence of the CAP binding region can be
altered as long as the CAP protein binds to it with essentially the same
affinity.
The spacer region between the end of the Shine-Dalgarno sequence and the ATG
start of the lacZ coding sequence is typically between about 5 and about 10
nucleotides in length, preferably about 5 to about 8 nucleotides in length,
more
preferably about 7-9 nucleotide o length. Th most preferred composition and
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
14
length of the spacer region depends on the composition and length of Shine-
Dalgarno sequence with which it is operably linked as well as the translation
start codon employed (i.e., AUG, GUG, or UUG), and can be determined
accordingly by one of skill in the art. Preferably the nucleotide composition
of
the spacer region is "AT rich"; that is, it contains more A's and Ts than it
does
G's and C's.
In a preferred embodiment of the method of the invention, the expression
vector has the identifying characteristics of pLAC11 (ATCC No. 207108). More
preferably, the expression vector is pLAC11 (ATCC No. 207108).
As used in the present invention, the term "vector" is to be broadly
interpreted as including a plasmid, including an episome. a viral vector, a
cosmid, or the like. A vector can be circular or linear, single-stranded or
double-
stranded, and can comprise RNA, DNA, or modifications and combinations
thereof. Selection of a vector or plasmid backbone depends upon a variety of
characteristics desired in the resulting construct, such as selection
marker(s),
plasmid copy number, and the like. A nucleic acid sequence is "operably
linked" to an expression control sequence in the regulatory region of a
vector,
such as a promoter, when the expression control sequence controls or regulates
the transcription and/or the translation of that nucleic acid sequence. A
nucleic
acid that is "operably linked" to an expression control sequence includes, for
example, an appropriate start signal (e.g., ATG) at the beginning of the
nucleic
acid sequence to be expressed and a reading frame that permits expression of
the
nucleic acid sequence under control of the expression control sequence to
yield
production of the encoded peptide. The regulatory region of the expression
vector optionally includes a termination sequence, such as a codon for which
there is no corresponding aminoacetyl-tRNA, thus ending peptide synthesis.
Typically, when the ribosome reaches a termination sequence or codon during
translation of the mRNA, the polypeptide is released and the ribosome-mRNA-
tRNA complex dissociates.
An expression vector optionally includes one or more selection or marker
sequences, which typically encode an enzyme capable of inactivating a
compound in the growth medium. The inclusion of a marker sequence can, for
example, render the host resistant to an antibiotic, or it can confer a
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
compound-specific metabolic advantage on the host cell. Cells can be
transformed with the expression vector using any convenient method known in
the art, including chemical transformation, e.g., whereby cells are made
competent by treatment with reagents such as CaCl2, electroporation and other
5 electrical techniques: microinjection and the like.
In embodiments of the method that make use of a tightly regulable
expression system derived from the lac operon, the host cell is or has been
genetically engineered or otherwise altered to contain a source of Lac
repressor
protein in excess of the amount produced in wild-type E. coll. A host cell
that
10 contains an excess source of Lac repressor protein is one that expresses
an
amount of Lac repressor protein sufficient to repress expression of the
peptide
under repressed conditions, i.e., in the absence of an inducing agent, such as
isopropyl p-D-thiogalactoside (IPTG). Preferably, expression of Lac repressor
protein is constitutive. For example, the host cell can be transformed with a
15 second vector comprising a gene encoding Lac repressor protein,
preferably lad,
more preferably laclq, to provide an excess source of Lac repressor protein in
trans, i.e., extraneous to the tightly regulable expression vector. An episome
can
also serve as a trans source of Lac repressor. Another option for providing a
trans source of Lac repressor protein is the host chromosome itself, which can
be
genetically engineered to express excess Lac repressor protein. Alternatively,
a
gene encoding Lac repressor protein can be included on the tightly regulable
expression vector that contains the peptide-encoding oligonucleotide so that
Lac
repressor protein is provided in cis. The gene encoding the Lac repressor
protein
is preferably under the control of a constitutive promoter.
The invention is not intended to be limited in any way by the type of host
cell used for screening. The host cell can be a prokaryotic or a eukaryotic
cell.
Preferred mammalian cells include human cells, of any tissue type, and can
include cancer cells or hybridomas, without limitation. Preferred bacterial
host
cells include gram negative bacteria, such as E. coli and various Salmonella
spp.,
and gram positive bacteria, such as bacteria from the genera Staphylococcus,
Streptococcus and Enterococcus. Protozoan cells are also suitable host cells.
In
clear contrast to conventional recombinant protein expression systems, it is
preferable that the ho-,,t cell contaii,s proteascs and/or peptidases, since
the
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
16
selection will, as a result, be advantageously biased in favor of peptides
that are
protease- and peptidase-rcsistant. More preferably. the host cell has not been
modified, genetically or otherwise, to reduce or eliminate the expression of
any
naturally expressed proteases or peptidases. The host cell can be selected
with a
particular purpose in mind. For example, if it is desired to obtain peptide
drugs
specific to inhibit Staphylococcus, peptides can be advantageously expressed
and
screened in Staphylococcus.
There is, accordingly, tremendous potential for the application of this
technology in the development of new antibacterial peptides useful to treat
various pathogenic bacteria. Of particular interest are pathogenic
Staphylococci,
Streptococci, and Enterococci, which are the primary causes of nosocomial
infections. Many of these strains are becoming increasingly drug-resistant at
an
alarming rate. The technology of the present invention can be practiced in a
pathogenic host cell to isolate inhibitor peptides that specifically target
the
pathogenic strain of choice. Inhibitory peptides identified using pathogenic
microbial host cells in accordance with the invention may have direct
therapeutic
utility; based on what is known about peptide import, it is very likely that
small
peptides are rapidly taken up by Staphylococci õStreptococci, and Enterococci.
Once internalized, the inhibitory peptides identified according to the
invention
would be expected to inhibit the growth of the bacteria in question. It is
therefore contemplated that novel inhibitor peptides so identified can be used
in
medical treatments and therapies directed against microbial infection. It is
further contemplated that these novel inhibitor peptides can be used, in turn,
to
identify additional novel antibacterial peptides using a synthetic approach.
The
coding sequence of the inhibitory peptides is determined, and peptides are
then
chemically synthesized and tested in the host cell for their inhibitory
properties.
Novel inhibitor peptides identified in a pathogenic microbial host cell
according to the invention can also be used to elucidate potential new drug
targets. The protein target that the inhibitor peptide inactivated is
identified
using reverse genetics by isolating mutants that are no longer inhibited by
the
peptide. These mutants are then mapped in order to precisely determine the
protein target that is inhibited. New antibacterial drugs can then be
developed
using various known or yet t:: be discover;--d pharmaceutical strategies.
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
17
Following transformation of the host cell, the transformed host cell is
initially grown under conditions that repress expression of the peptide.
Expression of the peptide is then induced. For example, when a lac
promoter/operator system is used for expression. IPTG is added to the culture
medium. A determination is subsequently made as to whether the peptide is
inhibitory to host cell growth, wherein inhibition of host cell growth under
induced but not repressed conditions is indicative of the expression of a
hioactive peptide.
Notably, the bioactive peptides identified according to the method of the
invention are, by reason of the method itself, stable in the intracellular
environment of the host cell. The method of the invention thus preferably
identifies bioactive peptides that are resistant to proteases and peptidases.
Resistance to proteases and peptidases can be evaluated by measuring peptide
degradation when in contact with appropriate cell extracts or purified
peptidases
and/or proteases, employing methods well-known in the art. A protease- or
peptidase-resistant peptide is evidenced by a longer half-life in the presence
of
proteases or peptidases compared to a control peptide.
Randomized peptides used in the screening method of the invention can
be optionally engineered according to the method of the invention in a biased
synthesis to increase their stability by making one or both of the N-terminal
or
C-terminal ends more resistant to proteases and peptidases, and/or by
engineering into the peptides a stabilizing motif.
In one embodiment of the screening method of the invention, the putative
bioactive peptide is stabilized by adding a stabilizing group to the N-
terminus,
the C-terminus, or to both termini. To this end, the nucleic acid sequence
that
encodes the randomized peptide in the expression vector or the expression
vector
itself is preferably modified to encode a first stabilizing group comprising
the N-
terminus of the peptide, and a second stabilizing group comprising the C-
terminus of the peptide.
The stabilizing group can be a stable protein, preferably a small stable
protein such as thioredoxin, glutathione sulfotransferase, maltose binding
protein, glutathione reductasc, or a four-helix bundle protein such as Rop
protein, although no specific size limitation on the protein anchor is
intended.
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
18
Proteins suitable for use as a stabilizing group can be either naturally
occurring
or non-naturally occurring. They can be isolated from an endogenous source.
chemically or enzymatically synthesized, or produced using recombinant DNA
technology. Proteins that are particularly well-suited for use as a
stabilizing
group are those that are relatively short in length and form very stable
structures
in solution. Proteins having molecular weights of less than about 50 kD are
preferred for use as a stabilizing group; more preferably the molecular weight
of
the small stable protein is less than about 25 kD, most preferably less than
about
12 kD. For example, E. coil thioredoxin has a molecular weight about 11.7 kD;
E. coil glutathione sulfotransferase has a molecular weight of about 22.9 kD,
and
Rop from the ColE1 replicon has a molecular weight of about 7.2 kD; and
maltose binding protein (without its signal sequence) is about 40.7 kD. The
small size of the Rop protein makes it especially useful as a stabilizing
group,
fusion partner, or peptide anchor, in that it is less likely than larger
proteins to
interfere with the accessibility of the linked peptide, thus preserving its
bioactivity. Rop's highly ordered anti-parallel four-helix bundle topology
(after
dimerization) and slow unfolding kinetics ( see, e.g., Betz et al,
Biochemistry 36,
2450-2458 (1997)) also contribute to its usefulness as a peptide anchor
according
to the invention. Other proteins with similar folding kinetics and/or
thermodynamic stability (e.g., Rop has a midpoint temperature of denaturation,
T,,õ of about 71 C, Steif et al., Biochemistry 32, 3867-3876 (1993)) are also
preferred peptide anchors. Peptides or proteins having highly stable tertiary
motifs, such as a four-helix bundle topology, are particularly preferred.
Alternatively, the stabilizing group can constitute one or more prolines
(Pro). Preferably, a proline dipeptide (Pro-Pro) is used as a stabilizing
group,
however additional prolines may be included. The encoded proline(s) are
typically naturally occurring amino acids, however if and to the extent a
proline
derivative, for example a hydroxyproline or a methyl- or ethyl- proline
derivative, can be encoded or otherwise incorporated into the peptide, those
proline derivatives are also useful as stabilizing groups.
At the N-terminus of the peptide, the stabilizing group can alternatively
include an oligopeptide having the sLquence Xaa-Pro,-. wherein Xaa is any
amino acid .1.t.1 m is gre- t than 0. Preferably, m is about 1 to about 5:
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
19
preferably m = 2 or 3, more preferably, m = 2. Likewise, at the C-terminus of
the peptide, the stabilizing group can alternatively include an oligopeptide
having the sequence -Pro,õ-Xaa, wherein Xaa is any amino acid, and m is
greater
than 0. Preferably, n is about 1 to about 5; preferably a = 2 or 3, more
preferably, m = 2. In a particularly preferred embodiment of the method of the
invention, the nucleic acid sequence that encodes the randomized peptide in
the
expression vector is modified to encode each of a first stabilizing group
comprising the N-terminus of the peptide, the first stabilizing group being
selected from the group consisting of small stable protein, Pro-, Pro-Pro-,
Xaa-
Pro- and Xaa-Pro-Pro-, and a second stabilizing group comprising the C-
terminus of the peptide, the second stabilizing group being selected from the
group consisting of a small stable protein, -Pro, -Pro-Pro, Pro-Xaa and Pro-
Pro-
Xaa. The resulting peptide has enhanced stability in the intracellular
environment relative to a peptide lacking the terminal stabilizing groups.
In another preferred embodiment of the screening method of the
invention, the expression vector encodes a four-helix bundle protein fused, at
either the C-terminus or the N-terminus, to the randomized peptide.
Preferably,
the four-helix bundle protein is E. coli Rop protein or a homolog thereof. The
non-fused terminus of the randomized peptide can, but need not, comprise a
stabilizing group. The resulting fusion protein is predicted to be more stable
than the randomized peptide itself in the host intracellular environment.
Where
the four-helix bundle protein is fused to the N-terminus, the randomized
peptide
can optionally be further stabilized by engineering one or more prolines, with
or
without a following undefined amino acid (e.g., -Pro, -Pro-Pro, -Pro-Xaa, -Pro-
Pro-Xaa, etc.) at the C-terminus of the peptide sequence; likewise, when the
four-helix bundle protein is fused to the C-terminus, the randomized peptide
can
be further stabilized by engineering one or more prolines, with or without a
preceding undefined amino acid (e.g., Pro-, Pro-Pro-. Xaa-Pro-, Xaa-Pro-Pro-,
etc.) at the N-terminus of the peptide sequence.
In yet another embodiment of the screening method of the invention, the
putative bioactive peptide is stabilized by engintering into the peptide a
stabilizing motif such as an a-helix motif or an opposite charge ending motif.
ChemicP' nthesis oligonucleotide according to the scheme
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
[(CAG)A(TCAG)] yields an oligonucleotide encoding a peptide consisting of a
random mixture of the hydrophilic amino acids His. GM, Asn, Lys, Asp. and Glu
(see Table 14). Except for Asp, these amino acids are most often associated
with
a-helical secondary structural motifs; the resulting oligonucleotides are thus
biased
5 in favor of oligonucleotides that encode peptides that are likely to form
a-helices in
solution. Alternatively, the putative bioactive peptide is stabilized by
flanking a
randomized region with a region of uniform charge (e.g., positive charge) on
one
end and a region of opposite charge (e.g., negative) on the other end, to form
an
opposite charge ending motif. To this end, the nucleic acid sequence that
encodes
10 the randomized peptide in the expression vector or the expression vector
itself is
preferably modified to encode a plurality of sequential uniformly charged
amino
acids comprising the N-terminus of the peptide. and a plurality of sequential
oppositely charged amino acids comprising the C-terminus of the peptide. The
positive charges are supplied by a plurality of positively charged amino acids
15 consisting of lysine, histidine, arginine or a combination thereof; and
the negative
charges are supplied by a plurality of negatively charged amino acids
consisting of
aspartate, glutamate or a combination thereof. It is expected that such a
peptide
will be stabilized by the ionic interaction of the two oppositely charges
ends.
Preferably, the putative bioactive peptide contains at least three charged
amino
20 acids at each end. More preferably, it contains at least four charged
amino acids at
each end. In a particularly preferred embodiment, the larger acidic amino acid
glutamate is paired with the smaller basic amino acid lysine, and the smaller
acidic
amino acid aspartate is paired with the larger basic amino acid arginine.
It is to be understood that novel bioactive peptides identified using the
method for identification of bioactive peptides described herein are also
included
in the present invention.
The present invention further provides a bioactive peptide containing one
or more structural features or motifs selected to enhance the stability of the
bioactive peptide in an intracellular environment. During development and
testing of the intracellular screening method of the present invention, it was
surprisingly discovered that several bioactive peptides identified from the
randomized peptide library shared particular structural features. For example,
a
dist- portionatel- .agh number of bioactive peptides identified nsing the
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
'71
intracellular screening method contained one or more proline residues at or
near
a peptide terminus. A disproportionate number also contained sequences
predicted, using structure prediction algorithms well-known in the art, to
form
secondary structures such as a helices or 13 sheets; or a hydrophobic membrane
spanning domain. Bioactive fusion proteins comprising the randomized peptide
sequence fused to the Rop protein, due to a deletion event in the expression
vector, were also identified.
Accordingly, the invention provides a bioactive peptide having a
stabilizing group at its N-terminus, its C-terminus, or at both termini. In a
bioactive peptide stabilized at only one terminus (i.e., at either the N- or
the C-
terminus) the stabilizing group is preferably either a four-helix bundle
protein,
such as Rop protein, a proline (Pro), or a proline dipeptide (Pro-Pro). It
should
be understood that in any synthetic peptide having a stabilizing group that
includes one or more prolines according to the present invention, the proline
is
preferably a naturally occurring amino acid; alternatively, it can be a
synthetic
derivative of proline, for example a hydroxyproline or a methyl- or ethyl-
proline
derivative. Accordingly, where the abbreviation "Pro" is used herein in
connection with a stabilizing group that is part of a synthetic peptide, it is
meant
to include proline derivatives in addition to a naturally occurring proline.
A peptide stabilized at both termini includes a first stabilizing group
comprising the N-terminus, and a second stabilizing group stabilizing the C-
terminus, where the first and second stabilizing groups are as defined
previously
in connection with the method for identifying bioactive peptides. The
stabilizing
group is covalently attached to the peptide. The bioactive peptide of the
invention includes a bioactive peptide that has been detectably labeled,
derivatized, or modified in any manner desired prior to use, provided it
contains
one or more terminal stabilizing groups as provided herein. In one preferred
embodiment of the bioactive peptide of the invention, the first stabilizing
group,
comprising the N-terminus, is Xaa-Pro-Pro-, Xaa-Pro-, Pro- or Pro-Pro-; and
second stabilizing group, comprising the C-terminus, is Pro-Pro-Xaa, -Pro-Xaa,
-
Pro or -Pro-Pro; preferably ¨Pro-Pro. In another preferred embodiment, the
first
(N-terminal) stabilizing prc ap is a small stable protein, preferably a four-
helix
bu-1 protein F. as Rop protein; and the
second (C-terminal) stabilizing
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
27
group is Pro-Pro-Xaa, -Pro-Xaa, -Pro or -Pro-Pro; preferably ¨Pro-Pro. In yet
another preferred embodiment, the second (C-terminal) stabilizing group is a
small stable protein, preferably a four-helix bundle protein such as Rop
protein,
and the first (N-terminal) stabilizing group is Pro-, Pro-Pro-, Xaa-Pro- or
Xaa-
Pro-Pro-.
The invention further provides a peptide stabilized by flanking the amino
acid sequence of a bioactive peptide with an opposite charge ending motif, as
described herein. Preferably, the resulting stabilized peptide retains at
least a
portion of the biological activity of the bioactive protein. The stabilized
peptide
includes a peptide that has been detectably labeled, derivatized, or modified
in
any manner desired prior to use.
It should be understood that any bioactive peptide, without limitation,
can be stabilized according to the invention by attaching a stabilizing group
to
either or both of the N- and C-termini, or by attaching oppositely charged
groups
to the N- and C-termini to form an opposite charge ending motif. Included in
the
present invention are any and various antimicrobial peptides, inhibitory
peptides,
therapeutic peptide drugs, and the like, as, for example and without
limitation,
those listed in Tables 1 and 2, that have been modified at one or both peptide
termini to include a stabilizing group, for example a four-helix bundle
protein
such as Rop protein, proline (Pro-), a proline-proline dipeptide (Pro-Pro-),
an
Xaa-Pro- dipeptide, or an Xaa-Pro-Pro-tripeptide at the N-terminus, and/or a
four-helix bundle protein such as Rop protein, proline (-Pro), or a proline-
proline
dipeptide (-Pro-Pro), a Pro-Xaa dipeptide, or a Pro-Pro-X tripeptide at the C-
terminus; or that have been modified to contain an opposite charge ending
motif
according to the invention. In this aspect the invention is exemplified by
peptides such as Pro-Pro-Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His-Ile-Pro-Pro
(SEQ ID NO: 3) and Glu-Asp-Glu-Asp-Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His-
lie-Arg-Lys-Arg-Lys (SEQ ID NO: 4), wherein the middle nine amino acids (-
Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His-I le-; SEQ ID NO: 5) constitute the
sequence of angiotensin.
Modification of a bioactive ne,ptide to yield a stabilized bioactive peptide
according to the invention can be achieved by standard techniques well-known
- -the arts of iietics and peptide synthesis. For example. where the peptide
is
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
23
synthesized de novo, as in solid state peptide synthesis. one or more prolines
can
be added at the beginning and the end of the peptide chain during the
synthetic
reaction. In recombinant synthesis, for example as described in Example Ill
herein, one or more codons encoding proline can be inserted into the peptide
coding sequence at the beginning and/or the end of the sequence. as desired.
Preferably, codons encoding N-terminal prolines are inserted after (i.e., 3'
to) the
initiation site ATG (which encodes for methionine). Analogous techniques are
used to synthesize bioactive peptides having an opposite charge ending motif.
When a known bioactive peptide is modified to yield a stabilized bioactivc
peptide according to the invention, the unmodified peptide can conveniently be
used as a control in a protease- or peptidase-resistance assay as described
hereinabove to confirm, if desired, that the modified peptide exhibits
increased
stability.
The present invention also provides a cleavable polypeptide comprising a
stabilized, bioactive peptide either immediately preceded by (i.e., adjacent
to the
N-terminus of the bioactive peptide) a cleavage site, or immediately followed
by
(i.e., adjacent to the C-terminus of the bioactive peptide) a cleavage site.
Thus, a
bioactive peptide as contemplated by the invention can be part of a cleavable
polypeptide. The cleavable polypeptide is cleavable, either chemically, as
with
cyanogen bromide, or enzymatically, to yield the bioactive peptide. The
resulting bioactive peptide either includes a first stabilizing group
comprising its
N-terminus and a second stabilizing group comprising its C-terminus, or it
includes an opposite charge ending motif, both as described hereinabove. The
cleavage site immediately precedes the N-terminal stabilizing group or
immediately follows the C-terminal stabilizing group. In the case of a
bioactive
peptide having an opposite charge ending motif, the cleavage site immediately
precedes the first charged region or immediately follows the second charged
region. The cleavage site makes it possible to administer a bioactive peptide
in a
form that could allow intracellular targeting and/or activation.
Alternatively, a bioactive peptide of the invention can be fused to a
noncleavable N-terminal or C-terminal targeting sequence wherein the targeting
sequence allows targ.;ed delivery of the bioactive peptide, e.g.,
intracellular
targeting r- Assue-specific targeting of the bioactive peptide. In one
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
24
embodiment of this aspect of the invention, the free terminus of the bioactive
peptide comprises a stabilizing group as described hereinabove in connection
with the screening method for identifying bioactive peptides, for example one
or
more prolines. The targeting sequence forming the other peptide terminus can,
but need not, contain a small stable protein such as Rop or one or more
prolines
comprising its terminus, as long as the targeting function of the targeting
sequence is preserved. In another embodiment of this aspect of the invention,
the bioactive peptide comprises a charge ending motif as described
hereinabove,
wherein one charged region occupies the free terminus of the bioactive
peptide,
and the other charged region is disposed between the targeting sequence and
the
active sequence of the bioactive peptide.
The invention further includes a method for using an antimicrobial
peptide that includes covalently linking a stabilizing group, as described
hereinabove, to the N-terminus, the C-terminus, or to both termini, to yield a
stabilized antimicrobial peptide, then contacting a microbe with the
stabilized
antimicrobial peptide. Alternatively, the stabilized antimicrobial peptide
used in
this aspect of the invention is made by covalently linking oppositely charged
regions, as described hereinabove, to each end of the antimicrobial peptide to
form an opposite charge ending motif. An antimicrobial peptide is to be
broadly
understood as including any bioactive peptide that adversely affects a microbe
such as a bacterium, virus, protozoan, or the like, as described in more
detail
hereinabove. An example of an antimicrobial peptide is an inhibitory peptide
that inhibits the growth of a microbe. When the antimicrobial peptide is
covalently linked to a stabilizing group at only one peptide terminus, any of
the
stabilizing groups described hereinabove can be utilized. When the
antimicrobial peptide is covalently linked to a stabilizing group at both
peptide
termini, the method includes covalently linking a first stabilizing group to
the N-
terminus of the antimicrobial peptide and a second stabilizing group to the C-
terminus of the antimicrobial peptide, where the first and second stabilizing
groups are as defined previously in connection with the method for identifying
bioactive peptides. In a preferred embodiment of the method for using an
antimicrobial per,ide, one or more prolines, more preferably a proline-proline
dipept'' _, is attached to at least one, preferably both, termini of the
antimicro1:'..1
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
peptide. Alternatively, or in addition, an Xaa-Pro- or an Xaa-Pro-Pro sequence
can be attached to the N-terminus of a microbial peptide, and/or a Pro-Xaa or
a
Pro-Pro-Xaa sequence can be attached to the C-terminus, to yield a stabilized
antimicrobial peptide.
5 The antimicrobial peptide thus modified in accordance with the invention
has enhanced stability in the intracellular environment relative to an
unmodified
antimicrobial peptide. As noted earlier, the unmodified peptide can
conveniently
be used as a control in a protease- or peptidase-resistance assay as described
hereinabove to confirm, if desired, that the modified peptide exhibits
increased
10 stability. Further, the antimicrobial activity of the antimicrobial
peptide is
preferably preserved or enhanced in the modified antimicrobial peptide;
modifications that reduce or eliminate the antimicrobial activity of the
antimicrobial peptide are easily detected and are to be avoided.
The invention further provides a method for inhibiting the growth of a
15 microbe comprising contacting the microbe with a stabilized inhibitory
peptide.
In one embodiment of this aspect of the invention, the stabilized inhibitory
peptide has a stabilizing group at the N-terminus, the C-terminus, or to both.
Preferably, the inhibitory peptide has a first stabilizing group comprising
the N-
terminus of the inhibitory peptide, and a second stabilizing group comprising
the
20 C-terminus of the inhibitory peptide; the first and second stabilizing
groups are
as defined previously in connection with the method for identifying bioactive
peptides. In another embodiment of this aspect of the invention, the
inhibitory
peptide is stabilized by the addition of oppositely charged regions to each
end to
form an opposite charge ending motif, as described hereinabove.
25 Also included in the present invention is a method for treating a
patient
having a condition treatable with a peptide drug, comprising administering to
the
patient a peptide drug that has been stabilized as described herein. Peptide
drugs
for use in therapeutic treatments are well-known (see Table 1). However, they
are often easily degraded in biological systems, which affects their efficacy.
In
on embodiment of the present method, the patient is treated with a stabilized
drug comprising the peptide drug of choice and a stabilizing group attached at
either the N-te:minus, the C-terminus of, or at both termini of the peptide
drug.
In _,Lrier embodiment of the present method, the patient is treated with a
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
26
stabilized drug comprising the peptide drug of choice and stabilized by
attachment of oppositely charged regions to both termini of the peptide drug.
Because the peptide drug is thereby stabilized against proteolytic
degradation,
greater amounts of the drug should reach the intended target in the patient.
In embodiments of the method involving administration of a peptide drug
that is covalently linked to a stabilizing group at only one peptide terminus,
the
stabilizing group is preferably a four-helix bundle protein such as Rop
protein,
provided that attachment of the four-helix bundle protein to the peptide
terminus
preserves a sufficient amount of efficacy for the drug. It is to be
nonetheless
understood that group or groups used to stabilize the peptide drug are as
defined
hereinabove, without limitation. In embodiments involving administration of a
peptide drug covalently linked to a stabilizing group at both peptide termini,
the
peptide drug includes a first stabilizing group comprising the N-terminus of
the
peptide drug and a second stabilizing group comprising the C-terminus of the
peptide drug. Thus, in another preferred embodiment of the treatment method of
the invention, the stabilized peptide drug comprises one or more prolines,
more
preferably a proline-proline dipeptide, attached to one or both termini of the
peptide drug. For example, the peptide drug can be stabilized by covalent
attachment of a Rop protein at one terminus, and by a proline or proline
dipeptide at the other terminus; in another preferred embodiment, the peptide
drug can be stabilized by proline dipeptides at each of the N-terminus and C-
terminus. Alternatively, or in addition, the stabilized peptide drug used in
the
treatment method comprises an Xaa-Pro- or an Xaa-Pro-Pro- sequence at the N-
terminus of the peptide drug, and/or a -Pro-Xaa or a -Pro-Pro-Xaa sequence at
the C-terminus. Optionally, prior to administering the stabilized peptide
drug,
the treatment method can include a step comprising covalently linking a
stabilizing group to one or both termini of the peptide drug to yield the
stabilized peptide drug.
If desired, the unmodified peptide drug can conveniently be used as a
control in a protease- or peptidase-resistance assay as described hereinabove
to
confirm that the stabilized peptide drug exhibits increased stability.
Further, the
therapeutic ':;fiicacy of the peptide drug is preferably preserved or enhanced
in
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
27
the stabilized peptide drug; modifications that reduce or eliminate the
therapeutic
efficacy of the peptide drug are easily detected and are to be avoided.
The present invention further includes a fusion protein comprising a four-
helix bundle protein, preferably Rop protein, and a polypeptide. Preferably
the
polypeptide is bioactive; more preferably it is a bioactive peptide. The
fusion
protein of the invention can be used in any convenient expression vector known
in the art for expression or overexpression of a peptide or protein of
interest.
Optionally, a cleavage site is present between four helix bundle protein and
the
polypeptide to allow cleavage, isolation and purification of the polypeptide.
In
one embodiment of the fusion protein, the four helix bundle protein is
covalently
linked at its C-terminus to the N-terminus of the polypeptidci in an
alternative
embodiment, the four helix bundle protein is covalcntly linked at its N-
terminus
to the C-terminus of the polypeptide. Fusion proteins of the invention, and
expression vectors comprising nucleic acid sequences encoding fusion proteins
wherein the nucleic acid sequences are operably linked to a regulatory control
element such as a promoter, are useful for producing or overproducing any
peptide or protein of interest.
EXAMPLES
The present invention is illustrated by the following examples. It is to be
understood that the particular examples, materials, amounts, and procedures
are
to be interpreted broadly in accordance with the scope and spirit of the
invention
as set forth herein.
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
28
Example I
Construction and characterization of a highly regulable expression vector,
pLAC11, and its multipurpose derivatives, pLAC22 and pLAC33
A number of different expression vectors have been developed over the
years to facilitate the production of proteins in E. coli and related
bacteria. Most
of the routinely employed expression vectors rely on lac control in order to
overproduce a gene of choice. While these vectors allow for overexpression of
the gene product of interest. they are leaky due to changes that have been
introduced into the lac control region and gene expression can never be shut
off
under repressed conditions, as described in more detail below. Numerous
researchers have noticed this problem with the more popular expression vectors
pKK223-3 (G. Posfai et al. Gene. 50: 63-67 (1986); N. Scrutton et al., Biochem
J.245: 875-880 (1987)), pKK233-2 (P. Beremand et al., Arch Biochem
Biophys. 256: 90-100 (1987); K. Ooki et al., Biochemie. 76: 398-403 (1994)),
pTrc99A (S. Ghosh, Protein Expr. Purif. 10: 100-106 (1997); J. Ranie et al.,
Mol. Biochem. Parasitol. 61: 159-169 (1993)), as well as the pET series (M.
Eren eta!,, J. Biol. Chem. 264: 14874-14879 (1989): G. Godson, Gene 100: 59-
64 (1991)).
The expression vector described in this example, pLAC1I, was designed
to be more regulable and thus more tightly repressible when grown under
repressed conditions. This allows better regulation of cloned genes in order
to
conduct physiological experiments. pLAC11 can be used to conduct
physiologically relevant studies in which the cloned gene is expressed at
levels
equal to that obtainable from the chromosomal copy of the gene in question.
The expression vectors described here were designed utilizing the wild-type
lac
promoter/operator in order to accomplish this purpose and include all of the
lac
control region, without modification, that is contained between the start of
the
03 auxiliary operator through the end of the 01 operator. As with all lac
based
vectors, the pLAC1I expression vector described herein can be turned on or off
by presence or absence of the gratuitous inducer IPTG. In experiments in
which a bacterial cell contained both a null allele in the ehromosi;_.e and a
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
29
second copy of the wild-type allele on pLAC11. cells grown under repressed
conditions exhibited the null phenotype while cells grown under induced
conditions exhibited the wild-type phenotype. Thus the pLAC11 vector truly
allows for the gene of interest to be grown under either completely repressed
or
fully induced conditions. Two multipurpose derivatives of pLAC11, pLAC22
and pLAC33, were also constructed to fulfill different experimental needs.
The vectors pLAC1 I, pLAC22 and pLAC33 were deposited with the
American Type Culture Collection (ATCC), 10801 University Blvd., Manassas,
VA, 20110-2209, USA, on February 16, 1999, and assigned ATCC deposit
numbers ATCC 207108, ATCC 207110 and ATCC 207109, respectively. It is
nonetheless to be understood that the written description herein is considered
sufficient to enable one skilled in the art to fully practice the present
invention.
Moreover, the deposited embodiment is intended as a single illustration of one
aspect of the invention and is not lobe construed as limiting the scope of the
claims in any way.
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
MATERIALS AND METHODS
Media. Minimal M9 media (6 g disodium phosphate, 3 g potassium phosphate,
1 g ammonium chloride, 0.5 g sodium chloride, distilled water to 1L;
autoclave;
5 add 1 mL magnesium sulfate (1M) and 0.1 ml, calcium chloride (1M); a
sugar
added to a final concentration of 0.2%; vitamins and amino acids as required
for
non-prototrophic strains) and rich LB media (10 g tryptone, 5 g yeast extract,
10
g sodium chloride, distilled water to 1L; autoclave) were prepared as
described
by Miller (J. Miller, "Experiments in molecular genetics" Cold Spring Harbor
10 Laboratory, Cold Spring Harbor, N.Y. (1972). The antibiotics ampicillin.
kanamycin, streptomycin, and tetracycline (Sigma Chemical Company. St.
Louis, MO) were used in rich media at a final concentration of 100, 40, 200,
and
20 tig/ml, respectively. When used in minimal media, tetracycline was added at
a final concentration of 10 ug/ml. 5-bromo-4-chloro-3-indoyl p-D-
15 galactopyranoside (Xgal) was added to media at a final concentration of
40
ug/ml and unless otherwise noted. IPTG was added to media at a final
concentration of 1 mM.
Chemicals and Reagents. When amplified DNA was used to construct the
20 plasmids that were generated in this study, the PCR reaction was carried
out
using native Pfu polymcrase from Stratagene (Cat. No. 600135). Xgal and IPTG
were purchased from Diagnostic Chemicals Limited.
Bacterial Strains and Plasmids. Bacterial strains and plasmids are listed in
25 Table 4. To construct ALS225, ALS224 was mated with ALS216 and
streptomycin resistant, blue recombinants were selected on a Rich LB plate
that
contained streptomycin, Xgal. and IPTG. To construct ALS226. ALS224 was
mated with ALS217 and streptomycin resistant, kanomycin resistant
recombinants were selected on a Rich LB plate that contained streptomycin and
30 kanamycin. To construct ALS515, ALS514 was mated with ALS2I 6 and
streptomycin resistant, blue recombinants were selected on a Rich LB plate
that
contained streptomycin, Xgal, and IPTG. To construct ALS527, ALS524 was
mated with ALS224 and streptomycin resistant, tetracyclinf. .esistant
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
31
recombinants were selected on a Rich LB plate that contained streptomycin and
tetracycline. To construct ALS535õALS533 was mated with ALS498 and
tetracycline resistant recombinants were selected on a Minimal M9 Glucose
plate that contained tetracycline, leucine and thiamine (Bi ) (Sigma Chemical
Company). To construct ALS533, a P1 lysate prepared from E. coil strain
K5076 (H. Miller et al., Cell 20: 711-719 (1980)) was used to transduce ALS224
and tetracycline resistant transductants were selected.
0
TaWe 4: Bacterial strains and plasmids used in Example 1
E. coil Strains
).)
Laboratou Original Name Genotype Source
Name
ALS216 SE9100 araD139 A(1001.1169 thi J-11)135301 deoC7 E. Altman
et al., J. Biol. Chem.265:18148-18153 (1990)
ptsF25 rpsE I Flac] Z 14-1 A1
ALS217 SE9100.1 araD139 A(lac)U169 thi fl bB5301 deoC7 S. Emr
ptsF25 rpsE I F /ac/q1 Z::Tn5 Y+ A+ (Univ. of California,
San Diego)
A LS221 BL21(DE3) ompT hsdS(b) (R-M-)gal dcnt F. Studier et al., J.
Mol. Biol. 189: 113-130 (1986) 0
N.)
-.1
ALS224 MC1061 araD139 A(araABOIC-leu)7679 A(lac)X74
M. Casadaban et al., J. Mol. Bio1.138:179-207
(1980). 1/40
14.
galLI galK rpsL hsr- hstu+
JALS225 MC 1061 / F'laclq I Z+4 - A+ This example
N.)
ALS226 MC1061 / Flacill Z::Tn5 Y-1- A-4- This example
N.)
ALS269 CSH27 F- trpA33 thi J. Miller, -Experiments
in molecular geneticS- Cold Spring
Harbor Laboratory, Cold Spring Harbor, N.Y. (1972).
1-)
ALS413 MG1655 E. colt wild-type F-;).- M. Guyer et al.,Cold
Spring Harbor Symp. Quant. Biol.
45:135-140(1980).
ALS498 JM 101 supE thi A(lac-proAB) I F' 1raD36 prok C. Yanisch-
Perron et al., Gene. 33: 103-119 (1985)
/ac/q A(lacZ)/1115
ALS514 NM554 MC1061 recA13 E. Raleigh et al., Nucl.
Acids Res. 16: 1563-1575
(1988).
,\ES515 MC1061 recA13 I F'lacicil Z+ y-+ This example
AI,S524 XL I -Blue recAl endA1 gyrA96 thi-1 hsdR17 supE44 Stratag,ene
(Cat. No. 200268)
relAl lac I F'proAB laclq A(lacZ)M15
Tn10
N.)
ALS527 MC1061 / F'proAB laclq A(lacZ)MI5 This example
Tn10
ALS533 MC1061 proAB::Tn10 This example
ALS535 MCI 061 proAB::Tn10 / F'lac/q This example
AflacZ)/1/11 5 proA+B+
ALS598 CAG18615 zjb-3179::TnlOdKan lambda- rph-1 M. Singer etal..
Microbiol. Rev. 53: 1-24 (1989).
Plasmids
NJ
Plasmicl Relevant Characteristics Source
1/40
Name
0
pBH20 wild-type lac promoter! operator, AmpR, TetR,
K. Itakura etal., Science. 198:1056-1063 (1977)
N.)
0
1-`
N.)
colE1 replicon
0
012322 AmpR, TetR, colE1 replicon F. Bolivar et al., Gene. 2:95-
113(1977)
7ET-21(+) T7 promoter //ac operator, laclq, AmpR, co1E1 Novagen
(Cat No. 69770-1)
replicon
pGE226 wild-type recA gene, AmpR J. Weisemann, et al., J.
Bacteriol. 163:748-755 (1985)
KK223-3 lac promoter, / operator, AmpR, colE1 replicon J. Brosius et
al., Proc. Natl. Acad. Sci. USA. 81:6929-6933 (1984)
pKK233-2 Irc promoter / operator, AmpR, colE1 replicon F. Amann etal.,
Gene. 40:183-190 (1985)
pLysE T7 lysozyme, CamR, PISA replicon F. Studier, J. Mol. Biol. 219:37-
44 (1991)
pLysS 17 lysozyme, Camp-, PISA replicon F. Studier, J. Mol. Biol.
219:37-44 (1991)
174
pMS421 wild-type lac promoter / operator. laclq, StrcpR, D. Grafia et
al., Genetics. 120:319-327 (1988)
SpecR, SC101 replicon
pTer7 wild-type /acZ coding region, AmpR R. Young
(Texas A&M University)
pTre99A trc promoter / operator, /ac/q, AmpR, colE1 E. Amann etal., Gene.
69:301-315 (1988)
0
replicon
1/40
pUC8 lac promoter / operator, AmpR, colE1 replicon J. Vieira et al.,
Gene, 19: 259-268 (1982)
0
pXE60 wild-type TOL pWW0 xylE gene, AmpR J. Westpheling
N.)
(Univ. of Georgia)
NJ
1-`
0
lA)
(44
(4.!
=-=
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
Construction of the pLAC11, pLAC22, and pLAC33 expression vectors. To
construct pLAC11, primers #1 and #2 (see Table 5) were used to PCR amplify a
952 base pair (bp fragment from the plasmid pBH20 which contains the wild-
type lac operon. Primer #2 introduced two different base pair mutations into
the
5 seven base spacer region between the Shine Dalgamo site and the ATG start
site
of the lacZ which converted it from AACAGCT to AAGATCT thus placing a
Bgl 11 site in between the Shine Dalgamo and the start codon of the lacZ gene.
The resulting fragment was gel isolated, digested with Pst I and EcoR I. and
then
ligated into the 3614 bp fragment from the plasmid pBR322z\Aval which had
10 been digested with the same two restriction enzymes. To construct
pBR322.6AvaI, pBr322 was digested with AvaI, filled-in using Klenow, and
then religated. To construct pLAC22, a 1291 bp Nco I. EcoR I fragment was gel
isolated from pLAC21 and ligated to a 4361 bp Nco I. EcoR I fragment which
was gel isolated from pBR322/Neol. To construct pLAC21, primers #2 and #3
15 (see Tables) were used to PCR amplify a 1310 bp fragment from the
plasmid
pMS421 which contains the wild-type /ac operon as well as the lacici
repressor.
The resulting fragment was gel isolated, digested with EcoR 1, and then
ligated
into pBR322 which had also been digested with EcoR I. To construct
pBR322/Nco I, primers 44 and #5 (sec Table 5) were used to PCR amplify a 788
20 bp fragment from the plasmid pBR322. The resulting fragment was gel
isolated,
digested with Pst I and EcoR I, and then ligated into the 3606 bp fragment
from
the plasmid pBR322 which had been digested with the same two restriction
enzymes. The pBR322/Nco I vector also contains added Kpn I and Sma I sites
in addition to the new Nco I site. To construct pLAC33, a 2778 bp fragment was
25 gel isolated from pLAC12 which had been digested with BsaB I and Bsa 1
and
ligated to a 960 bp fragment from pUC8 which had been digested with Afl III,
filled-in with Klenow, and then digested with Bsa I. To construct pLAC12, a
1310 bp Pst I, Bamll I fragment was gel isolated from pLAC11 and ligated to a
3232 bp Pst I, BamH I fragment which was gel isolated from pBR322.
CA 02794430 2012-10-31
N4)011(22112 PCT/US99/23731
36
Table 5. Primers employed to PCR amplify DNA fragments that were
used in the construction of the various plasmids described in
Example I
pLAC11 and pLAC22
1 (for) alT_GCC_ATT_GCT GCA GGC_AT (SEQ ID NO:6)
2 (rev) ATT GAA TTC ATA AGA TOT TTC CTG .TOT GAA ATT GTE ATC.
(SEQ ID 140:7)
3 (for) ATT GAA TTC ACC ATG 3AC ACC ATC GAA
(SEQ ID NO:8)
p1312322/Nco I
4 (for) G7T_GTI_GCC_ATT_C=CCA_C (SEQ ID NO:9)
5 (rev) TGT ATG AAT TQC CGG GTA CCA TOG TTG AAG ACG. AAA_GGG
C.C.T...C(SEQ ID NO:10)
Bgl II - lacZ - Hind III
6 (for) TAO TAT AGA TOT ATG_ACC_ATG_ATT_ACG_GAT_TCA_CTO (SEQ
ID NO:11)
7 (rev) TAO ATA AAG CTT GGC CTG_ CCC.GGT TAT_TAT_TAT_ITT (SEQ
ID NO:12)
Pst I - lacZ - Hind III
8 (for) TAT CAT CTG EL3 AGG AAA CAD CTA TGA CCA.TGA_TTA_C.GG
ATT_CAC_TG (SEQ ID NO:13)
9 (rev) TAC ATA CTC GAG CAG GAA AGC TTG_GCC_TGQ_CCG_GTT_ATT
NIT_ATT_TT (SEQ ID 140:14)
BamH 1 - lacZ - Hind III (also uses primer #9)
10 (for) TAT CAT GGA TOO AGG AAA CAD CTA_TGA CCA_TGA_TTAC.GG
ATT_CAC_TG (SEQ ID NO:15)
Bgl II - recA - Hind III
11 (for) TAO TAT AGA TOT ATGACTIATC_OAC_GA.k_AAC_AAA_CAG (SEQ
ID 140:16)
12 (rev) ATA TAT AAG OTT TIiAATQ.TTOGTLAGTTECTDOTAC
G (SEQ ID NO:17)
BamH 1 - xylE - EcoR I
13 (for) TAO TAT AGA TOT ATa_21.M_LA_GGT_GTA_ATG...CA¨Q (SEQ
ID NO:18)
14 (rev) ATT AGT GAA TTC GCA. CAA TCT.CTG CA& TAA CTC_GT (SEQ
ID NO:19)
CA 02794430 2012-10-31
WO 00/22112 PCT/1JS99/23731
$7
In Table 5 the regions of the primers that are homologous to the DNA target
template are indicated with a dotted underline, while the relevant restriction
sites
are indicated with a solid underline. All primers are listed in the 5' 3'
orientation.
Compilation of the DNA sequences for the pLAC11, pLAC22, and pLAC33
expression vectors. All of the DNA that is contained in the pLAC11, pLAC22,
and, pLAC33 vectors has been sequenced.
The sequence for the pLAC11 vector which is 4547 bp can be compiled
as follows: bp 1-15 are AGATCTTATGAATTC (SEQ ID NO:20) from primer
42 (Table 5); bp 16-1434 are bp 4-1422 from pBR322 (GenBank Accession 4
J01749); bp 1435-1442 are TCGCITCGG. caused by filling in the Ava I site in
pBR3226,AvaI; bp 1443-4375 is bp 1427-4359 from pBR322 (GenBank
Accession # J01749); and bp 4376-4547 are bp 1106-1277 from the wild-type E.
coli lac operon (GenBank Accession 4 J01636).
The sequence for the pLAC22 vector which is 5652 bp can be compiled
as follows: bp 1-15 are AGATCTTATGAATTC (SEQ ID NO:21) from primer
42 (Table 5); bp 16-4370 are bp 4-4358 from pBR322 (GenBank Accession 4
J01749); bp 4371-4376 is CCATGG which is the Nco I site from pl3R322/Nco I;
and bp 4377-5652 are bp 2-1277 from the wild-type E. coli lac operon
(GenBank Accession 4 J01636), except that bp 44391 of the pLAC22 sequence
or bp#16 from the wild-type E. coli lac operon sequence has been changed from
a "C" to a "T" to reflect the presence of the lacIci mutation (J. Brosius et
al.,
Proc. Natl. Acad. Sci. USA. 81:6929-6933 (1984)).
The sequence for the pLAC33 vector which is 3742 bp can be compiled
as follows: bp 1-15 is AGATCTTATGAATTC (SEQ ID N0:22) from primer
42 (Table 5); bp 16-1684 are bp 4-1672 from pBR322 (GenBank Accession
J01749); bp 1685-2638 are bp 786-1739 from pUC8 (GenBank Accession #
L09132); bp 2639-3570 are bp 3428-4359 from pBR322 (GenBank Accession 4
J01749); and bp 3571-3742 are bp 1106-1277 from the wild-type E. coli lac
operon (GenBank Accession 4 J01636). In the maps for these vectors the on is
identified as per Balbas (P. Balbas et al., Gene. 50:3-40 (1980) while the
lacP0
is indicated btarting with the 03 a- ',Lary opera. and ending with the 01
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
38
operator as per Muller-Hill (B. Muller-Hill, The lac Operon: A Short History
of
a Genetic Paradigm, Walter de Gruyter, Berlin, Germany (1996)).
Construction of the pLAC11-, pLAC22-, pLAC33-, pKK223-3-, 1)KK233-2-,
pTrc99A-, and pET-21(+)- lacZ constructs. To construct pLAC11-lacZ,
pLAC22-/acZ, and pLAC33-/acZ, primers 46 and 47 (see Table 5) were used to
PCR amplify a 3115 bp fragment from the plasmid pTer7 which contains the
wild-type lacZ gene. The resulting fragment was gel isolated, digested with
Bgl
II and Hind III, and then ligated into the pLAC11. pLAC22. or pLAC33 vectors
which had been digested with the same two restriction enzymes. To construct
pKK223-3-/acZ and pKK233-2-/acZ, primers #8 and #9 (see Table 5) were used
to PCR amplify a 3137 bp fragment from the plasmid pTer7. The resulting
fragment was gel isolated, digested with Pst I and Hind III, and then ligated
into
the pK1(223-3 or pKK233-2 vectors which had been digested with the same two
restriction enzymes. To construct pTrc99A-/acZ and pET-21(+)-lacZ, primers
#9 and #10 (see Table 5) were used to PCR amplify a 3137 bp fragment from the
plasmid pTer7. The resulting fragment was gel isolated, digested with BamH I
and Hind III, and then ligated into the pTrc99A or pET-21(+) vectors which had
been digested with the same two restriction enzymes.
Construction of the pLAC11- recA and xylE constructs. To construct
pLAC11-recA, primers #11 and #12 (see Table 5) were used to PCR amplify a
1085 bp fragment from the plasmid p0E226 which contains the wild-type recA
gene. The resulting fragment was gel isolated, digested with Bgl 11 and Hind
III,
and then ligated into the pLAC11 vector which had been digested with the same
two restriction enzymes. To construct pLAC11-xylE, primers #13 and #14 (see
Table 5) were used to PCR amplify a 979 bp fragment from the plasmid pXE60
which contains the wild-type Pseudonionas putida xylE gene isolated from the
TOL pWW0 plasmid. The resulting fragment was gel isolated, digested with
Bgl II and EcoR 1, and then ligated into the pLAC11 vector which had been
digested with the same two restriction enzymes.
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
39
Assays. 13-galactosidase assays were performed as described by Miller (I.
Miller, "Experiments in molecular genetics" Cold Spring Harbor Laboratory.
Cold Spring Harbor, N.Y. (1972)), while catechol 2.3-dioxygenase (cat02ase)
assays were performed as described by Zukowski, et. al. (M. Zukowski et al.,
Proc. Natl. Acad. Sci. U.S.A. 80:1101-1105(1983)).
RESULTS
Construction and features of pLAC11, pLAC22, and pLAC33. Plasmid
1 0 maps that indicate the unique restriction sites, drug resistances,
origin of
replication, and other relevant regions that are contained in pLAC11, pLAC22,
and pLAC33 are shown in Figs. 2, 3 and 4, respectively. pLAC1 I was designed
to be the most tightly regulable of these vectors. It utilizes the ColE1
origin of
replication from pBR322 and Lad I repressor is provided in trans from either
an
episome or another compatible plasmid. pLAC22 is very similar to pLAC11,
however, it also contains /ac/q, thus a source of Lad does not have to be
provided in trans. pLAC33 is a derivative of pLAC I 1 which utilizes the
mutated
ColE1 origin of replication from pUC8 (S. Lin-Chao et al., Mol. Micro. 6:3385-
3393 (1992)) and thus pLAC33's copy number is significantly higher than
pLAC11 and is comparable to that of other pUC vectors. Because the cloning
regions of these three vectors are identical, cloned genes can be trivially
shuffled
between and among them depending on the expression demands of the
experiment in question.
To clone into pLAC I 1, pLAC22, or pLAC33, PCR amplification is
performed with primers that are designed to introduce unique restriction sites
just upstream and downstream of the gene of interest. Usually a Bgl II site is
introduced immediately in front of the ATG start codon and an EcoR I site is
introduced immediately following the stop codon. An additional 6 bases is
added
to both ends of the oligonucleotide in order to ensure that complete digestion
of
the amplified PCR product will occur. After amplification the double-stranded
(ds) DNA is restricted with Bgl II and EcoR I, an t.,ioned into the vector
which
has also been restricted with the same '.:wo enzymes. If the gene of interest
contains a Bp-' -- site, then iiH I or 13cl I can be used instead since
they
CA 02794430 2012-10-31
WO 00/22112 PCTTUS99/23731
generate overhangs which are compatible with BO II. If the acne of interest
contains an EcoR I site, then a site downstream of EcoR I in the vector (such
as
Hind III) can be substituted.
5 Comparison of pLAC11, pLAC22, and pLAC33, to other expression
vectors. In order to demonstrate how regulable the pLAC11, pLAC22, and
pLAC33 expression vectors were, the wild-type lacZ gene was cloned into
pLAC I 1, pLAC22, pLAC33, pKK223-3, pKK233-2, pTrc99A. and pET-21(+).
Constructs which required an extraneous source of Lac I for their repression
were
10 transformed into ALS225, while constructs which contained a source of
Lacl on
the vector were transformed into ALS224. pET-21(+) constructs were
transformed into BL21 because they require T7 RNA polymerase for their
expression. Four clones were chosen for each of these seven constructs and p-
galactosidase assays were performed under repressed and induced conditions.
15 Rich Amp overnights were diluted I to 200 in either Rich Amp Glucose or
Rich
Amp IPTG media and grown until they reached mid-log (0D550 0.5). In the
case of pET-21(+) the pLysE and pLysS plasmids, which make T7 lysozyme and
thus lower the amount of available T7 polymerase, were also transformed into
each of the constructs. Table 6 shows the results of these studies and also
lists
20 the induction ratio that was determined for each of the expression
vectors. As
the data clearly indicates, pLAC11 is the most regulable of these expression
vectors and its induction ratio is close to that which can be achieved with
the
wild-type lac operon. The vector which yielded the lowest level of expression
under repressed conditions was pLAC1 I, while the vector which yielded the
25 highest level of expression under induced conditions was pLAC33.
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
4
Table 6: P-galactosidase levels obtained in different expression vectors
grown under either repressed or induced conditions
Vector Source # of Miller Units Observed
Repressed Induced Fold
Conditions Conditions Induction
pLAC11 F 19 11209 590X
pLAC22 Plasmid 152 13315 88X
pLAC33 F' 322 23443 73X
pK1(223-3 F' 92 11037 120X
pKK233-2 F' 85 10371 122X
pTrc99A Plasmid 261 21381 82X
pET-21(+) Plasmid 2929 16803 6X
pET-21(+) / pLysE Plasmid 4085 19558 5X
pET-21(+) / pLysS Plasmid 1598 20268 13X
The average values obtained for the four clones that were tested from each
vector are listed in the table. Standard deviation is not shown but was less
than
5% in each case. Induction ratios are expressed as the ratio of enzymatic
activity
observed at fully induced conditions versus fully repressed conditions. The
plasmid pLysE yielded unexpected results; it was expected to cause lower
amounts of lacZ to be expressed from pET-2l (-0 under repressed conditions
and, instead, higher amounts were observed. As a result, both pLysE and pLysS
were restriction mapped to make sure that they were correct.
Demonstrating that pLAC11 constructs can be tightly regulated. pLAC I I
was designed to provide researchers with an expression vector that could be
utilized to conduct physiological experiments in which a cloned gene is
studied
under completely repressed conditions where it is off or partially induced
conditions where it is expressed at physiologically relevant levels. Figure 5
demonstrates how a pLAC I 1 -lacZ construct can be utilized to mimic
chromosomally expressed /arZ that occurs under various physiological
conditions by var rig the amount of IPTCI inducer that is added. ALS226 cells
containin, pLAC11-/acZ were grown to mid-log in rich inedia that contained
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
42
varying amounts of IPTG and then P-galactosidase activity was assayed. Also
indicated in the graph are the average p-galactosidase activities obtained for
strains with a single chromosomal copy of the wild-type lacZ gene that were
grown under different conditions.
To demonstrate just how regulable pLAC11 is, the recA gene was cloned
into the pLAC11 vector and transformed into cells which contained a null recA
allele in the chromosome. As the results in Table 7 clearly shows,
recombination can not occur in a host strain which contains a nonfunctional
RecA protein and thus P1 lysates which provide a TnlOdKan transposon can not
be used to transduce the strain to KanR at a high frequency. recA- cells which
also contain the pLAC11-recA construct can be transduced to KanR at a high
frequency when grown under induced conditions but can not be transduced to
KanR when grown under repressed conditions.
Table 7: The recombination (-) phenotype of a recA null mutant strain
can be preserved with a pLAC11-recA (wild-type) construct
under repressed conditions
Repressed Conditions Induced Conditions
Strain Number of KanR Number of KanR
transductants transductants
ALS225 (recA ) 178,000 182,000
ALS514 (recil-) 4
ALS515 (recA- pCyt-3-recA) 4 174,000
The data presented in Table 7 are the number of KanR transductants that were
obtained from the different MCI 061 derivative strains when they were
transduced with a PI lysate prepared from strain ALS598 which harbored a
Tnl OdKan transposon insertion. Overnights were prepared from each of these
strains using either rich medium to which glucose was added at a final
concentration of 0.2% (repressed conditions) or rich medium to which IPTG was
added at a iinal concentration of 1 mM (induced conditions). The overnights
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
43
were then diluted 1 to 10 into the same medium which contained CaCl2 added to
a final concentration of 10 mM and aerated for two hours to make them
competent for transducti On with PI phage. Cells were then
spectrophotometrically normalized and aliquots off OD550 cell equivalents in a
volume of approximately 0.1 ml were transduced with 0.1 ml of concentrated P1
Iysate as well as 0.1 ml of PI lysates that had been diluted to 10-1, 10-2, or
10-3.
0.2 ml of 0.1M Sodium Citrate was added to the celliphage mixtures and 0.2 ml
of the final mixtures were plated onto Rich Kanamycin plates and incubated
overnight at 37 C. The total number of KanR colonies were then counted.
ALS225 recil+ data points were taken from the transductions which used the 10-
3 diluted phage, while ALS514 reci4- data points were taken from the
transductions which used the concentrated phage. The data points for ALS515
recA pCyt-3-recil grown under repressed conditions were taken from the
transductions which used the concentrated phage, while the data points for
ALS515 recA pCyt-3-recA grown under induced conditions were taken from
the transductions which used the 10-3 diluted phage.
Testing various sources of Lacl for trans repression of pLAC11. Because
pLAC11 was designed to be used with an extraneous source of Lad I repressor,
different episomal or plasmid sources of Lad l which are routinely employed by
researchers were tested. Since one of the Lad I sources also contained the
lacZ
gene, a reporter construct other than pLAC11-lacZ was required and thus a
pLAC11 -xylE construct was engineered. Table 8 shows the results of this
study.
All of the Lad sources that were tested proved to be adequate to repress
expression from pLAC11, however, some were better than others. The basal
level of expression that was observed with F's which provided lac1c11 or with
the
plasmiid pMS421 which provided /ac/'l at approximately six copies per cell was
lower than the basal level of expression that was observed with F's which
provided laclq all three times that the assay was run. Unfortunately, however,
the xylE gene could not be induced as high when /aciql on a F' or /ac/q on a
plarilid was used as the source of Lac repressor.
CA 02794430 2012-10-31
WO 00/22112
PCT/US99/23731
44
Table 8: Catechol 2,3-dioxygenase levels obtained for a pLAC11-xy/E
construct when Lac repressor is provided by various sources
Strain Source of Lad Catechol 2,3-dioxygenase activity in
milliunits/mg
Repressed Induced
Conditions Conditions
ALS224 None 32.7 432.8
ALS535 ziaaczoim .3 204.4
proA+B+ In10
ALS527 Fiac/1 d(7acZ)M15 .3 243.3
proA+B+
ALS227 pMS421 IacN .2 90.9
ALS225 pvacici 1 z+171- A+ .2 107.4
ALS226 Fri0c/(11 Z::Tn5 PA4- .2 85.1
The wild-type xylE gene was cloned into the pLACI 1 vector and the resulting
pLAC1 1-xylE construct was then transformed into each of the MC1061
derivative strains listed in the table. Rich overnights were diluted 1 to 200
in
either Rich Glucose or Rich IPTG media and grown until they reached mid-log
(0D550 = 0.5). Cell extracts were then prepared and catechol 2,3-dioxygenase
assays were performed as described by Zukowski, et. al (Proc. Natl. Acad. Sci.
U.S.A. 80:1101-1105 (1983)). The average values obtained in three different
experiments are listed in the table. Standard deviation is not shown but was
less
than 10% in each case.
DISCUSSION
Most of the routinely employed expression vectors rely on lac control in
order to overproduce a gene of choice. The lac promoter/operator functions as
it
does due to the interplay of three main components. First, the wild-t:, ix;
hie -10
CA 02794430 2012-10-31
WO 00/22112
PCT/US99/23731
region (TATGTT) is very weak, c-AMP activated CAP protein is able to bind to
the CAP site just upstream of the -35 region which stimulates binding of RNA
polymerase to the weak -10 site. Repression of the lac promoter is observed
when glucose is the main carbon source because very little c-AMP is present
5 which results in low amounts of available c-AMP activated CAP
protein. When
poor carbon sources such as lactose or glycerol are used, c-AMP levels rise
and
large amounts of c-AMP activated CAP protein become available. Thus
induction of the lac promoter can occur. Second, Lac repressor binds to the
lac
operator. Lac repressor can be overcome by allolactose which is a natural
10 byproduct of lactose utilization in the cell, or by the gratuitous
inducer, IPTG.
Third, the lac operator can form stable loop structures which prevents the
initiation of transcription due to the interaction of the Lac repressor with
the lac
operator (01) and one of two auxiliary operators, 02 which is located
downstream in the coding region of the lacZ gene, or 03 which is located just
15 upstream of the CAP binding site.
While binding of Lac repressor to the lac operator is the major effector of
lac regulation, the other two components are not dispensable. However, most of
the routinely used lac regulable vectors either contain mutations or deletions
which alter the affect of the other two components. The pKI(.223-3 (J. Brosius
et
20 al., Proc. Natl. Acad. Sci. USA. 81:6929-6933 (1984)), pKK233-2
(E. Amann et
al., Gene. 40:183-190 (1985)), pTrc99A (E. Amann et al., Gene. 69:301-315
(1988)), and pET family of vectors (F. Studier, Method Enzymol. 185:60-89
(1990)) contain only the /ac operator (01) and lack both the CAP binding site
as
well as the 03 auxiliary operator. 0(1(223-3, pKK233-2, and pTrc99 use a trp-
25 lac hybrid promoter that contains the trp -35 region and the
lacUV5 -10 region
which contains a strong TATAAT site instead of the weak TATGTT site. The
pET family of vectors use the strong T7 promoter. Given this information,
perhaps it is not so surprising researchers have found it is not possible to
tightly
shut off genes that are cloned into these vectors.
30 The purpose of the studies described in Example I was to
design a vector
which would allow researchers to better regulate their cloned genes in order
to
condu6t physiological experiments. The expression vectors described herein
= were designed utilizing the wild-type lac promoter/operator in ord,:r to
CA 02794430 2012-10-31
WO 00/22112
PCT/US99/23731
46
accomplish this purpose and include all of the lac control region, without
modification, that is contained between the start of the 03 auxiliary operator
through the end of the 01 operator. As with all lac based vectors, the pLAC11,
pLAC22, and pLAC33 expression vectors can be turned on or off by the
presence or absence of the gratuitous inducer IPTG.
Because the new vector, pLAC11, relies on the wild-type lac control
region from the auxiliary lac 03 operator through the lac 01 operator, it can
be
more tightly regulated than the other available expression vectors. In direct
comparison studies with pKK223-3, pKK233-2, pTre99A, and pET-21(+), the
lowest level of expression under repressed conditions was achievable with the
pLAC 11 expression vector. Under fully induced conditions, pLAC11 expressed
lacZ protein that was comparable to the levels achievable with the other
expression vectors. Induction ratios of 1000x have been observed with the wild-
type lac operon. Of all the expression vectors that were tested, only pLAC11
yielded induction ratios which were comparable to what has been observed with
the wild-type lac operon. It should be noted that the regulation achievable by
pLAC 11 may be even better than the data in Table 6 indicates. Because lacZ
was used in this test, the auxiliary lac 02 operator which resides in the
coding
region of the lacZ gene was provided to the pKK223-3, pK1(233-2, pTre99A,
and pET-21(+) vectors which do not normally contain either the 02 or 03
auxiliary operators. Thus the repressed states that were observed in the study
in
Table 6 are probably lower than one would normally observe with the pKK223-
3, pKK233-2, pTrc99A, and pET-21(+) vectors.
To meet the expression needs required under different experimental
circumstances, two additional expression vectors which are derivatives of
pLAC11 were designed. pLAC22 provides laclq on the vector and thus unlike
pLAC11 does not require an extraneous source of Lad for its repression.
pLAC33 contains the mutated ColE1 replicon from pIJC8 and thus allows
proteins to be expressed at much higher levels due to the increase in the copy
number of the vector. Of all the expressions that were evaluated in direct
comparison studies, the highest level of protein expression under fully
induced
= conditions was achieved using the pLAC33 vector. Because the cloning
regions
are identical in pLAC11, pLAC22, and pLAC33, genes that crc cloned into one
CA 02794430 2012-10-31
WO 00/22112
PCT/US99/23731
47
of these vectors can bc trivially subcloned into either of the other two
vectors
depending on experimental circumstances. For physiological studies, pLAC I I
is the best suited of the three vectors. If, however, the bacterial strain of
choice
can not be modified to introduce elevated levels of Lac repressor protein
which
can be achieved by F's or compatible plasmids that provide laclq or /ac/[11,
the
pLAC22 vector can be utilized. If maximal overexpression of a gene product is
the goal, then the pLAC33 vector can be utilized.
Numerous experiments call for expression of a cloned gene product at
physiological levels; i.e., at expression levels that are equivalent to the
expression levels observed for the chromosomal copy of the gene. While this is
not easily achievable with any of the commonly utilized expression vectors,
these kinds of experiments can be done with the pLAC11 expression vector. By
varying the IPTG concentrations, expression from the pLAC11 vector can be
adjusted to match the expression levels that occur under different
physiological
conditions for the chromosomal copy of the gene. In fact, strains which
contain
both a chromosomal null mutation of the gene in question and a pLAC11
construct of the gene preserve the physiological phenotype of the null
mutation
under repressed conditions.
Because the use of Lac repressor is an essential component of any
expression vector that utilizes the lac operon for its regulation; the ability
of
different source of Lad I to repress the pLAC11 vector was also investigated.
Researchers have historically utilized either Lac/c1 constructs which make 10
fold
more Lac repressor than wild-type lad l or /acitli constructs which make 100
fold
more Lac repressor than wild-type lad (B. Muller-Hill, Prog. Biophys. Mol.
Biol. 30:227-252 (1975)). The greatest level of repression of pLAC11
constructs
could be achieved using F's which provided approximately one copy of the
laciq I gene or a multicopy compatible plasmid which provided approximately
six copies of the /ac/q gene. However, the induction that was achievable using
these lad I sources was significantly lower than what could be achieved when
F's
which provided approximately one copy of the laclq gene were used to repress
the pLAC11 construct. Thus if physiological studies are the goal of an
investigation, then Fs which provide approximately one copy of the /ac/cli
gene
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
48
or a multicopy compatible plasnaid which provides approximately six copies of
the laclq gene can be used to regulate the pLAC I I vector. However, if
maximal
expression is desired, then F's which provide approximately one copy of the
tac/c1 gene can be utilized. Alternatively, if a bacterial strain can tolerate
prolonged overexpression of an expressed gene and overexpression of a gene
product is the desired goal, then maximal expression under induced conditions
is
obtained when a bacteria strain lacks any source of Lac repressor.
Example II
An in vivo approach for generating novel bioactive peptides that inhibit the
growth of E. coil
A randomized oligonucleotide library containing sequences capable of
encoding peptides containing up to 20 amino acids was cloned into pLAC11
(Example I) which allowed the peptides to either be tightly turned off or
overproduced in the cytoplasm of E. coll. The randomized library was prepared
using a [1\1NN] codon design instead of either the [NN(G,T)] or [NN(G,C))
codon design used by most fusion-phage technology researchers. [NN(G,T)1 or
INN(G,C)1 codons have been widely used instead of [NNN] cOcions to eliminate
two out of the three stop codons, thus increasing the amount of full-length
peptides that can be synthesized without a stop codon (J. Scott et al.,
Science
249:386-390 (1990); J. Delvin et al., Science 249:404-406 (1990); S. Cwirla et
al., Proc. Nat'l. Acad. Sci. U.S.A. 87:6378-6382 (1990)). However, the
[1\11\1(G,T)) and [NN(G,C)] oligonucleotide codon schemes eliminate half of
the
otherwise available codons and, as a direct result, biases the distribution of
amino acids that are generated. Moreover, the [NN(G,T)] and [NN(G,C)] codon
schemes drastically affect the preferential codon usage of highly expressed
genes
and removes a number of the codons which are utilized by the abundant tRNAs
that are present in .E. coli (H. Grosjean et al., Gene. 18: 199-209 (1982); T.
Ikemura, J. Mol. Biol. 151: 389-409 (1981)).
Of the 20,000 peptides screened in this Example, 21 inhibitors of cell
growth were found which could prevent the growth of E coil on mini,nal media.
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
49
The top twenty inhibitor peptides were evaluated for strength of inhibition,
and
the putative amino acid sequences of the top 10 "anchorless" inhibitor
peptides
were examined for commonly shared features or motifs.
MATERIALS AND METHODS
Media. Rich LB and minimal M9 media used in this study was prepared as in
Example I. Ampicillin was used in rich media at a final concentration of 100
ug/ml and in minimal media at a final concentration of 50 ug/ml. IPTG was
added to media at a final concentration of 1 mM.
Chemicals and Reagents. Extension reactions were carried out using Klenow
from New England Biolabs while ligation reactions were performed using 14
DNA Ligase from Life Sciences. IPTG was obtained from Diagnostic
Chemicals Limited.
Bacterial Strains and Plasmids. ALS225, which is MC1061IF'lacici1.Z+Y+A+
(see Example I), was the E. coli bacterial strain used in this Example. The
genotype for MC1061 is araD139 A(araABOIC-lett)7679 A(lac)X74 galU galK
rpsL hsr- hsm+ (M. Casadaban et al., J. Mol. Biol. 138:179-207 (1980)).
pLAC11, a highly regulable expression vector, is described in Example I.
Generation of the Randomized Peptide Library. The 93 base oligonucleotide
5' TAC TAT AGA TCT ATG (N1'N)2() TAA TAA GAA TIC TCG ACA 3'
(SEQ ID NO: 23), where N denotes an equimolar mixture of the nucleotides A,
C, G, or T, was synthesized with the trityl group and subsequently purified
with
an OPC cartridge using standard procedures. The complementary strand of the
93 base oligonucleotide was generated by an extension/fill-in reaction with
Klenow using an equimolar amount of the 18 base oligonucleotide primer 5'
TGT CGA GAA TTC TTA TTA 3' (SEQ ID NO: 24). After extension, the
resulting ds-DNA was purified using a Promega DNA clean-up kit and restricted
with EcoR 1 and Bgl II (Promega, Madison, WI). The digested DNA was again
purified using a Promega DNA clean-up kit and ligatc,i to pLAC 1 1 vector
which
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
had been digested with the same two restriction enzymes. The resulting library
was transformed into electrocompetent ALS225 E coli cells under repressed
conditions (LB, ampicillin, plus glucose added to 0.2%).
Screening of Transformants to Identify Inhibitor Clones. Transformants
5 were screened to identify any that could not grow on minimal media when
the
peptides were overproduced. Using this scheme, any transforrnant bacterial
colony that overproduces a peptide that inhibits the production or function of
a
protein necessary for growth of that transfonnant on minimal media will be
identified. Screening on minimal media, which imposes more stringent growth
10 demands on the cell, will facilitate the isolation of potential
inhibitors from the
library. It is well known that growth in minimal media pins more demands on a
bacterial cell than growth in rich media as evidenced by the drastically
reduced
growth rate; thus a peptide that adversely affects cell growth is more likely
to be
detected on minimal media. Screening was carried out using a grid-patching
15 technique. Fifty clones at a time were patched onto both a rich
repressing plate
(LB Amp glucose) and a minimal inducing plate (M9 glycerol Amp IPTG) using
an ordered grid. Patches that do not grow are sought because presumably these
represent bacteria that are being inhibited by the expressed bioactive
peptide. To
verify that all of the inhibitors were legitimate, plasmid DNA was made from
20 each inhibitory clone (QIA Prep Spin Miniprep kit; Qiagen Cat. No.
27104) and
transformed into a fresh background (ALS225 cells), then checked to confirm
that they were still inhibitory on plates and that their inhibition was
dependent
on the presence of the inducer, IPTCi.
25 Growth Rate Analysis in Liquid Media. Inhibition strength of the
peptides
was assessed by subjecting the inhibitory clones to a growth rate analysis in
liquid media. To determine the growth rate inhibition, starting cultures of
both
the peptides to be tested and a control strain which contains pLAC11 were
diluted from a saturated overnight culture to an initial 01)550 of ¨.0L All
30 cultures were then induced with 1 niM IPTG and 0D550 readings were taken
until the control culture reached an 0D550 of'-0.5. The hypothetical data in
Table 9 shows that when the control strain reaches an OD550 of about 0.64 (at
CA 02794430 2012-10-31
WO 00/22112
PCT/US99/23731
51
about 15 hours), a strain which contains a peptide that inhibits the growth
rate at
50% will only have reached an 0D550 of only about 0.08. Thus, the growth of a
50% inhibited culture at 15 hours (i.e., the 013550 at 15 hours, which is
proportional to the number of cells in a given volume of culture) is only
about
12.5% (that is, 0.08/0.64 x 100) of that of a control strain after the same
amount
of time, and the inhibitor peptide would thus have effectively inhibited the
growth of the culture (as measured by the 0D550 at the endpoint) by 87.5%
(=100% - 12.5%).
Table 9: Hypothetical data from a peptide that inhibits growth rate at
30%, 50% and 70%
Time in hours 0D550 readings on a 0D550 readings on a culture which contains a
control culture which peptide that inhibits the growth rate at...
contains pLAC I 1
25% 50% 75%
0 .010 .010 .010 .010
2.5 .020 .017 .015 .012
5 .040 .028 .020 .014
7.5 .080 .047 .030 .017
10 .160 .079 .040 .020
12.5 .320 .133 .060 .024
.640 .226 .080 .028
15 An example is shown in Fig. 6, wherein ALS225 cells containing the
pLAC I 1 vector (control), and either the one day inhibitor pPepl or the two
day
inhibitor pPep12 (see below), were grown in minimal M9 glycerol media with
IPTG added to 1 mM. OD55() readings were then taken hourly until the cultures
had passed log phase. Growth rates were determined by measuring the
spectrophotometric change in OD550 per unit time within the log phase of
growth. The inhibition of the growth rate was then calculated for the
inhibitors
using pLAC11 as a control.
Sequencing the Coding Regions of the Inhibitor Peptide Clones. The
forward primer 5' TCA TTA ATG CAG CTG GCA CG 3' (SEQ ID NO: 25) and
the reverse primer 5' TTC ATA CAC GOT GCC TGA CT 3' (SEQID NO: 26)
CA 02794430 2012-10-31
WO 00122112 PCT/US99/23731
52
were used to sequence both strands of the top ten "anchorless- inhibitor
peptide
clones identified by the grid-patching technique. If an error-free consensus
sequence could not be deduced from these two sequencing runs, both strands of
the inhibitor peptide clones in question were resequenced using the forward
primer 5' TAG CTC ACT CAT TAG GCA CC 3' (SEQ ID NO: 27) and the
reverse primer 5' GAT GAC GAT GAG CGC ATT GT 3' (SEQ ID NO: 28).
The second set of primers were designed to anneal downstream of the first set
of
primers in the pLAC11 vector.
Generating Antisense Derivatives of the Top Five "Anehorless" Inhibitor
Clones. Oligonucleotides were synthesized which duplicated the DNA insert
contained between the Bgl II and EcoR I restriction sites for the top five
"anchorless" inhibitor peptides as shown in Table 12 with one major nucleotide
change. The "T" of the ATG start codon was changed to a "C" which resulted in
a ACG which can not be used as a start codon. The oligonucleotides were
extended using the same 18 base oligonucleotide primer that was used to build
the original peptide library. The resulting ds-DNA was then restricted, and
cloned into pLAC11 exactly as described in the preceding section "Generating
the randomized peptide library". The antisense oligonucleotides that were used
are as follows:
pPepl(antisense): 5' TAC TAT AGA TCT ACG GTC ACT GAA ITT TGT
GGC TTG TTG GAC CAA CTG CCT TAG TAA TAG TOG AAG OCT GAA
ATT AAT AAG AAT TCT CGA CA 3' (SEQ ID NO: 29);
pPep5(antisense): 5' TAC TAT AGA TCT ACG TGG CGG GAC TCA TOG
ATT AAG GGT AUG GAC GTG OGG TTT ATG GGT TAA AAT AGT TTG
ATA ATA AGA ATT CTC GAC A 3' (SEQ ID NO: 30)
pPep12(antisense): 5' TAC TAT AGA TCT ACG AAC GGC CGA ACC AAA
CGA ATC COG GAC CCA CCA GCC GCC TAA ACA OCT ACC AGC TGT
GGT AAT AAG AAT TCT CGA CA 3' (SEQ ID NO: 31)
CA 02794430 2012-10-31
WO 00/22112
PCT/US99/23731
53
pPep13(antisense): 5' TAC TAT AGA TCT ACG GAC CGT GAA GTG ATG
TOT GCG GCA AAA CACi GAA TOG AAG GAA CGA ACG CCA TAG
GCC GCG TAA TAA GAA TTC TCG ACA 3 (SEQ ID NO: 32)
pPep19(antisense): 5' TAC TAT AGA TCT ACG AGG GGC GCC AAC TAA
GGG GGG GGG AAG GTA TTT GTC CCG TGC ATA ATC TCG GGT GTT
GTC TAA TAA GAA TTC TCG ACA 3' (SEQ ID NO: 33)
RESULTS
Identifying and Characterizing Inhibitor Peptides from the Library.
Approximately 20,000 potential candidates were screened as described
hereinabove, and 21 IPTG-dependent growth inhibitors were isolated. All the
inhibitors so identified were able to prevent the growth of the E. coil
bacteria at
24 hours, and three of the 21 inhibitors were able to prevent the growth of
the E.
coli bacteria at 48 hours, using the grid patching technique. These three
inhibitors were classified as "two day" inhibitors; the other 18 were
classified as
"one-day" inhibitors.
Results from the growth rate analysis for candidate peptide inhibitors are
shown in Table 10. The % inhibition of the growth rate was calculated by
comparing the growth rates of cells that contained induced peptides with the
growth rate of cells that contained the induced pLAC 11 vector. Averaged
values
of three independent determinations arc shown.
CA 02794430 2012-10-31
WO 00/22112
PCT/US99/23731
54
Table 10: Ability of the Inhibitor Peptides to Inhibit Cell Growth
Inhibitor Type % Inhibition Inhibitor Type % Inhibition
pLAC 11 0 Ppepli 1 Day 22
(control)
pPepl 1 Day 25 Ppep12 2 Day 82
pPep2 1 Day 23 Ppep13 1 Day 28
pPep3 2 Day 80 Ppep14 2 Day 71
pPep4 1 Day 21 Ppep15 1 Day 23
pPep5 1 Day 24 Ppep16 1 Day 24
pPep6 1 Day 27 Ppep17 1 Day 28
pPep7 1 Day 26 PPepl 8 1 Day 24
pPep8 1 Day 29 pPep19 I Day 29
pPep9 1 Day 22 pPep20 1 Day 19
pPep10 1 Day 24 pPep21 I Day 23
Of the 21 peptides that were tested, the one-day inhibitor peptides
inhibited the bacterial growth rate at a level of approximately 25%, while the
two-day inhibitor peptides inhibited the bacterial growth rate at levels
greater
than 75%. As can be seen from the hypothetical data in Table 9, a one-day
inhibitor which inhibited the growth rate at 25% would have only reached an
0D550 of 0.226 when the control strain reached an 0D550 of 0.64. At that point
in time, the growth of the culture that is inhibited by a one-day inhibitor
(as
measured by the end-point 0D550) will only be only 35.3% of that of a control
strain at that point; thus the inhibitor peptide would have effectively
inhibited the
growth of the culture by 64.7%. A two-day inhibitor which inhibited the growth
rate at 75% would have only reached an 0D550 of 0.028 when the control strain
reached an 0D550 of 0.64. Thus the growth of the culture that is being
inhibited
by a two-day inhibitor will only be 4.4% of that of the control strain at this
point,
and the inhibitor peptide would have effectively inhibited the growth of the
culture by 95.6%. These calculations are consistent with the observation that
two-day inhibitors prevent the of bacteria on plates for a full 48 hours
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
while the one-day inhibitors only prevent the growth of bacteria on plates for
24
hours.
All 21 candidates were examined using restriction analysis to determine
whether they contained 66 bp inserts as expected. While most of them did, the
5 two day inhibitors pPep3 and pPep14 were found to contain a huge
deletion.
Sequence analysis of these clones revealed that the deletion had caused the
carboxy-terminal end of the inhibitor peptides to become fused to the amino-
terminal end of the short 63 amino acid Rop protein. The rap gene which is
part
of the ColE1 replieon is located downstream from where the oligonucleotide
10 library is inserted into the pLAC11 vector.
Sequence Analysis of the Top 10 "Anchorless" Inhibitor Peptides. The DNA
fragments comprising the sequences encoding the top 10 "anchorless" inhibitor
peptides (i.e., excluding the two Rop fusion peptides) were sequenced, and
their
15 coding regions are shown in Table 11. Stop codons are represented by
stars, and
the landmark Bgl Ii and EcoR I restriction sites for the insert region are
underlined. Since the ends of the oligonucleotide from which these inhibitors
were constructed contained these restriction sites, the oligonucleotide was
not
gel isolated when the libraries were prepared in order to maximize the
20 oligonucleotide yields. Because of this, several of the inhibitory
clones were
found to contain one (n-1) or two (n-2) base deletions in the randomized
portion
of the oligonucleotide.
CA 02794430 2012-10-31
WC)00/22112
PCT/US99/23731
56
Table 11: Sequence analysis of the insert region from the top 10
inhibitory clones and the peptides that they are predicted to
encode
pPepl - 13 aa
GAG GAO AGO TOT ATG GTC ACT GAA TTT TOT GGC TTG TTG GAG CAA CTG CCT TAG TAO
TAG TGG MG OCT
MVTEFCGLL(3OLP. . = (SEQ ID NO:
34)
GAO AAT AM PAT TO (SEQ ID NO: 35)
pPep5 - 16 as
COG GPO AGO TCT ATG TOG CGG GAC TCA TOG OTT AAG GOT AGG GAC GIG GGG ITT ATG
GGT TAO OAT OAT
MWRDSWIKGRDVGFMG. (550 10
NO: 36)
TTG MA ATA AGO OTT C (SEQ ID NO: 31)
pPep5 - 42 as - las': 25 aa could form a hydrophobic membrane-
spanning domain
COG GAO AGO TOT ATG TCA GGG GGA CAT GTG ACG AGG GAG TGC MG TCG GCG ATG TCC OAT
COT TGG ATC
MSGGHVIRECKSANSNRWI
'SAC GTA ATA AGO ATT CTC ATG TTT GAC AGC TTA TCA TCG 0Th AGC ITT OAT GCG GTA
OTT TAT CAC AOT
YvIRILMFDSLSSISFNAVVYNS
TAP (SEQ ID NO: 38)
= (SEQ ID NO: 39)
pPep7 - 6 aa
COG GAO AGA TOT ATG TAT TTG TTC RTC GGA TAA TAC TTA ATG GTC CGC TOG AGO ACT
TCA OTT TAO TAO
MYLFIG= (SEQ ID NO: 40)
GOO TTC (SEQ ID NO: 41)
pPep8 - 21 aa
CAG GAA AGO TOT ATG CTT CTA TI'S GGO GGG GAC TGC COG COG AAA GCC GGA TAO TOT
ACT GTG CIA CCG
MLLFGG(3CGOKAGYFTVLP
TCA AGG TPA TOO GAA TIC (SEQ ID NO: 42)
S R = (SEQ ID NO: 43)
pPep10 - 20 as - predicted to be 45% 0-sheet -amino acids 6-14
COG GAO AGO OCT ATG OTT 000 GGA TCG TTG AGC TTC GCC TOG GE:A ATA OTT TOT OAT
AAG PAT TOT CAT
M IGGSLSFAWAIVCNKNSH
GTT TOO (SEQ ID NO: 44)
V . (SEQ ID NO: 45)
CA 02794430 2012-10-31
N/000/22112 PCT/US99/23731
57
pPep12 - 14 aa
CAG GM AGA TCT ATG MC CCC CGA ACC AAA CGA ATC CCC MC CCA CCA CCC CCC TAA ACA
GCT ACC AGC
MNGRTKNIRDPPAA. ISCQ ID NO: 46)
TGT GOT PAT MG MT TC_(SEQ ID NO: 47)
pPep13 -18 aa - predicted to be 72% a-helical - amino acids
3-15
CAG GM AGA TCT ATG GAC CGT GM GIG ATG TOT CCC GCA AAA CAG GM TGG AAG GAP CGA
ACS CCA TAG
MDREVMCAAKQEWKERTP.
(GC) ID NO: 40)
CCC CCC TAA TPA GM TTC :SEQ ID NO: 40)'
pPep17 - 12 aa
CAG GM AGA TCT ATG TAG CCC MT GCA CTG GOP GCA CGC GIG TTA GGT CM GAP CCC ACG
TAC CCA ITT
MLGLEATYPF
APT CCA TAA 1AA GM TTC ,SEQ ID NO: 50)
N P (SEC ID NO: 51)
pPep19 - 5 aa
CAG GM AGA TCT ATG AGG GGC CCC PAD TAA GGG GGG GGG MG GTA TTT CTC CCC TCC ATA
ATC TCG COT
WAN. (GED ID NO: 52)
OTT GTC TAA TAA GM TIC (SEQ ID NO: 53)
Eight out of the top 10 inhibitors were predicted to encode peptides
which terminate before the double TAA TAA termination site which was
engineered into the oligonucleotide. Two of the inhibitors, pPep6 and pPep10,
which contain deletions within the randomized portion of the oligonucleotide,
are terminated beyond the EcoR I site. One of the inhibitors, pPepl 7,
contains a
termination signal just after the ATG start codon. However, just downstream
from this is a Shine Dalgarno site and a GIG codon which should function as
the start codon. Interestingly, the start sites of several proteins such as
Rop are
identical to that proposed for the pPep17 peptide (G. Cesarcni at al., Proc.
Natl.
Acad. Sci. USA. 79:6313-6317 (1982)). The average and median length for the
8 peptides whose termination signals occurred before or at the double TAA TAA
termination site was 13 amino acids.
The characteristics of the predicted coding regions of the inhibitor
peptides proved to be quite interesting. Three out of the top 10 peptides,
pPepl,
pPep13, and pPep17, contained a proline residue as their last (C-terminal)
amino
acid. Additionally, one of the peptides, pPep12, contained 2 proline residues
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
58
near the C-terminus, at the n-2 and n-3 positions. Thus there appears to be a
bias
for the placement of proline residues at or near the end of several of the
inhibitory peptides. Secondary structure analysis predicted that 3 out of the
10
peptides contained a known motif that could potentially form a very stable
structure. pPep13, a peptide containing a C-terminal proline, is predicted to
be
72% a-helical, pPepl 0 is predicted to be 45%n-sheet, and pPep6 is predicted
to
form a hydrophobic membrane spanning domain.
Verifying that the Inhibitory Clones do not Function as Antisense. To rule
out the possibility that the bioactivity of the inhibitory clones resulted
from their
functioning as antisense RNA or DNA (thus hybridizing to host DNA or RNA)
rather than by way of the encoded peptides, the insert regions between the Bgl
II
and EcoR I sites for the top five inhibitors from Table 10 were recloned into
the
pLAC I I vector using oligonucleotides which converted the ATG start codon to
an ACG codon thus abolishing the start site. In all five cases the new
constructs
were no longer inhibitory (see Table 12), thus confirming that it is the
encoded
peptides that causes the inhibition and not the DNA or transcribed mRNA.
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
59
Table 12: Antisense test of the top 5 "anchorless" inhibitory peptides from
Inhibitory peptide % inhibition versus Antisense construct %
inhibition
pLAC1 I control versus pLAC I I
control
pPepl 26 pPepl-anti 0
pPep5 23 pPep5-anti 0
pPep12 80 pPep12-anti 0
pPepI3 28 pPep13-anti 0
pPepl 9 29 pPepl9-anti 0
Growth rates for cells containing the induced inhibitors or antisense
constructs
were determined and then the % inhibition was calculated by comparing these
values to the growth rate of cells that contained the induced pCyt-3 vector.
DISCUSSION
Use of the tightly regulable pLAC11 expression vector made possible the
identification of novel bioactive peptides. The bioactive peptides identified
using the system described in this Example inhibit the growth of the host
organism (E. coli) on minimal media. Moreover, bioactive peptides thus
identified are, by reason of the selection process itself, stable in the
host's
cellular environment. Peptides that are unstable in the host cell, whether or
not
bioactive, will be degraded; those that have short half-lives are, as a
result, not
part of the selectable pool. The selection system thus makes it possible to
identify and characterize novel, stable, degradation-resistant bioactive
peptides
in essentially a single experiment.
The stability of the inhibitory peptides identified in this Example may be
related to the presence of certain shared structural features. For example,
three
out of the top 10 inhibitory "anehorless" (i.e., non-Rop fusion) peptides
contained a proline residue as their last amino acid. According to the genetic
code, a randomly generated cligonucfeoti.le such as the one used in this
Example
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
has only a 6% chance of encoding a proline at a given position, yet the
frequency
of a C-terminal proline among the top ten inhibitory peptides is a full 30%.
This
5-fold bias in favor of a C-terminal proline is quite surprising, because
although
the presence of proline in a polypeptide chain generally protects biologically
5 active proteins against nonspecific enzymatic degradation, a group of
enzymes
exists that specifically recognize proline at or near the N- and C-termini of
peptide substrates. Indeed, praline-specific peptidases have been discovered
that
cover practically all situations where a proline residue might occur in a
potential
substrate (D.F. Cunningham at al., Biochimica at Biophysica Acta 1343:160-186
10 (1997)). For example, although the N-terminal sequences Xaa-Pro-Yaa- and
Xaa-Pro-Pro-Yaa (SEQ ID NO: 54) have been identified as being protective
against nonspecific N-terminal degradation, the former sequence is cleaved by
aminopeptidase P (at the Xaa-Pro bond) and dipeptidyl peptidases IV and II (at
the ¨Pro-Yaa-bond) ) (Table 5, G. Vanhoof et al., FASEB J. 9:736-44 (1995);
15 D.F. Cunningham at al., Biochimica et Biophysica Acta 1343:160-186
(1997));
and the latter sequence, present in bradykinin, interleukin 6, factor XII and
erytluopoietin, is possibly cleaved by consecutive action of aminopeptidase P
and dipeptidyl peptidase IV (DPPIV), or by proly1 oligopeptidase (post Pro-Pro
bond) (Table 5, G. Vanhoof et al., FASEB J. 9:736-44 (1995)). Prolyl
20 oligopeptidase is also known to cleave Pro-Xaa bonds in peptides that
contain an
N-terminal acyl-Yaa-Pro-Xaa sequence (D.F. Cunningham et al., Biochimica et
Biophysica Acta 1343:160-186(1997)). Other proline specific peptidases acting
on the N-terminus of substrates include prolidase, proline iminopeptidase and
prolinase. Prolyl carboxypeptidase and carboxypeptidase P, on the other hand,
25 cleave C-terminal residues from peptides with proline being the
preferred P1
residue (D.F. Cunningham et al., Biochimica et Biophysica Acta 1343:160-186
(1997).
Also of interest with respect to the stability of the inhibitory peptides,
three of the top ten (30%) contained motifs that were predicted, using
standard
30 protein structure prediction algorithms, to form stable secondary
structures. One
of the peptides (which also has a C-terminal proline) was predicted to be 72%
o:-
helical. Another was predicted to be 45% I3-sheet; this peptide may dimerize
in
order to effect the hydrogen bonding necessary to form the 13-sheet. A third
was
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
61
predicted to possess a hydrophobic membrane spanning domain. According to
these algorithms (see, e.g., P. Chou et al., Adv. Enzymol. 47:45-148 (1978);
J.
Gamier et al., J. Mol. Biol. 120:97-120 (1978); P. Chou, "Prediction of
protein
structural classes from amino acid composition," In Prediction of Protein
Structure and the Principles of Protein Conformation (Fasman, G.D. ed.).
Plenum Press, New York, N.Y. 549-586 (1990); P. Klein et al., Biochim.
Biophys. Acta 815:468-476 (1985)), a randomly generated oligonucleotide such
as the one used in our studies would have had no better than a 1 in a 1000
chance
of generating the motifs that occurred in these peptides.
Finally, two of the three two-day inhibitors proved to be fusion peptides
in which the carboxyl terminus of the peptides was fused to the amino terminus
of the Rop protein. Rop is a small 63 amino acid protein that consists of two
antiparallel a-helices connected by a sharp hairpin loop. It is a dispensable
part
of the ColE1 replieon which is used by plasmids such as pBr322, and it can be
deleted without causing any ill-effects on the replication, partitioning, or
copy
numbers of plasmids that contain a ColE1 on (X. Soberon, Gene. 9: 287-305
(1980). Rop is known to possess a highly stable structure (W. Eberle et al.,
Biochem. 29:7402-7407 (1990); S. Betz et al., Biochemistry 36:2450-2458
(1997)), and thus it could be serving as a stable protein anchor for these two
peptides.
Table 13 lists naturally occurring bioactivc peptides whose structures
have been determined. Most of these peptides contain ordered structures,
further
highlighting the importance of structural stabilization. Research on
developing
novel synthetic inhibitory peptides for use as potential therapeutic agents
over
the last few years has shown that peptide stability is a major problem that
must
be solved if designer synthetic peptides are to become a mainstay in the
pharmaceutical industry (J. Bai et al., Crit. Rev. Ther. Drug. 12:339-371
(1995);
R. Egleton Peptides. 18:1431-1439 (1997); L. Wearley, Crit Rev Ther Drug
Carrier Syst. 8: 331-394 (1991). The system described in this Example
represents a major advance in the art of peptide drug development by biasing
the
selection process in favor of bioactive peptides that exhibit a high degree of
stability in an intracellular environment.
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
62
Table 13: Structural motifs observed in naturally occurring bioactive
peptides
Bioactive Size in Structural Reference
Peptide Amino acids Motif
Dermaseptin 34 a-helix 34
Endorphin 30 a-helix 7
Glucagon 29 a-helix 6
_
Magainins a 23 a-helix 5
Mastoparan 14 a-helix 11
Melittin 26 a-helix 44
Motilin 22 a-helix 25
PKI (5-24) 20 a-helix 38
Secretin 27 a-helix 8
Atrial Natriuretic Peptide 28 disulfide bonds 33
_
Caleitonin 32 disulfide bonds 4
Conotoxins a 10-30 disulfide bonds 37
Defensins a 29-34 disulfide bonds 30
EETI II 29 disulfide bonds 23
Oxytocin 9 disulfide bonds - 45
_
Somatostatin 14 disulfide bonds 35
Vasopressin 9 disulfide bonds 20
Bombesin 14 disordered 12
Histatin 24 disordered 51
_
Substance P 11 disordered 50
a These peptides belong to multi-member families.
CA 02794430 2012-10-31
WO 00/22112 PCT/1JS99/23731
63
Example III
Directed synthesis of stable synthetically engineered inhibitor peptides
These experiments were directed toward increasing the number of
bioactive peptides produced by the selection method described in Example II.
In
the initial experiment, randomized peptides fused to the Rop protein, at
either the
N- or C- terminus, were evaluated. In the second experiment, nucleic acid
sequences encoding peptides containing a randomized internal amino acid
sequence flanked by terminal prolines were evaluated. Other experiments
included engineering into the peptides an a-helical structural motif, and
engineering in a cluster of opposite charges at the N- and C-termini of the
peptide.
MATERIALS AND METHODS
Media. Rich LB and minimal M9 media used in this study was prepared as
described by Miller (see Example 1). Ampicillin was used in rich media at a
final concentration of 100 ug/ml and in minimal media at a final concentration
of
50 ug/ml. IPTG was added to media at a final concentration of 1 mM.
Chemicals and Reagents. Extension reactions were carried out using Klenow
from New England Biolabs (Bedford, MA) while ligation reactions were
performed using T4 DNA ligase from Life Sciences (Gaithersburg, MD)
Alkaline phosphatase (calf intestinal mucosa) from Pharmacia (Piscataway, NJ)
was used for dephosphorylation. IPTG was obtained from Diagnostic Chemicals
Limited (Oxford, CT).
Bacterial Strains and Plasmids. ALS225, which is MC1061/P/ac/q1Z+Y+A+,
was the E. coli bacterial strain used in this study (see Example I). The
genotype
for MCI 061 is araDI 39 A(araABO1C-leu)7679 A(lac)X74 galU galK rpsl, hsr-
hsm+ as previously described. pLAC11 (Example I), a highly regulable
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
64
expression vector, was used to make p-Rop(C) and p(N)Rop- fusion vectors as
well as the other randomized peptide libraries which are described below.
Construction of the p-Rop(C) Fusion Vector. The forward primer 5' TAC
TAT AGA TCT ATG ACC AAA CAG GAA AAA ACC GCC 3' (SEQ ID NO:
55) and the reverse primer 5' TAT ACG TAT TCA GTT OCT CAC ATG TTC
TTT CCT GCG 3' (SEQ ID NO: 56) were used to PCR amplify a 558 bp DNA
fragment using pBR322 as a template. This fragment contained a Bgl II
restriction site which was incorporated into the forward primer followed by an
ATG start codon and the Rop coding region. The fragment extended beyond the
Rop stop codon through the All III restriction site in pBR322. The amplified
dsDNA was gel isolated, restricted with Bgl II and Afi III, and then ligated
into
the pLAC11 expression vector which had been digested with the same two
restriction enzymes. The resulting p-Rop(C) fusion vector is 2623 bp in size
(Fig. 7).
Construction of the p(N)Rop- Fusion Vector. The forward primer 5' AAT
TCA TAC TAT AGA TCT ATG ACC AAA CAG GAA AAA ACC GC 3'
(SEQ ID NO: 57) and the reverse primer 5' TAT ATA ATA CAT GTC AGA
AT f CGA GGT TTT CAC COT CAT CAC 3' (SEQ ID NO: 58) were used to
PCR amplify a 201 bp DNA fragment using pBR322 as a template. This
fragment contained a Bgl II restriction site which was incorporated into the
forward primer followed by an ATG start codon and the Rop coding region. The
reverse primer placed an EcoR I restriction site just before the Rop TGA stop
codon and an All III restriction site immediately after the Rop TGA stop
codon.
Thc amplified dsDNA was gel isolated, restricted with Bgl II and All III, and
then ligated into the pLAC11 expression vector which had been digested with
the same two restriction enzymes. The resulting p(N)Rop- fusion vector is 2262
bp in size (Fig. 8).
Generation of Rop Fusion Randomized Peptide Libraries. Peptide
libraries were constructed as described in Example H. The synthetic
oligoriucleotide 5' TAC TAT AGA TCT ATG (NNN)20 CAT AGA TCT GCG
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
TGC TOT GAT 3' (SEQ ID NO: 59) was used to construct the randomized
peptide libraries for use with the p-Rop(C) fusion vector, substantially as
described in Example II. The complementary strand of this oligonucleotide was
generated by a fill-in reaction with Klenow using an equimolar amount of the
5 oligonucleotide primer 5' ATC ACA GCA CGC AGA TCT ATG 3' were used
(SEQ ID NO: 60). After extension the resulting dsDNA was digested with Bgl
II and ligated into the pLAC11 expression vector which had been digested with
the same restriction enzyme and subsequently dephosphorylated using alkaline
phosphatase. Because of the way the oligonucleotide library has been
10 engineered, either orientation of the incoming digested double-stranded
DNA
fragment results in a fusion product.
To construct the randomized peptide libraries for use with the p(N)Rop-
fusion vector, the randomized oligonucleotide 5' TAC TAT GAA TTC (NNN)2()
GAA TTC TGC CAC CAC TAC TAT 3' (SEQ ID NO: 61), and the primer 5'
15 ALA GTA GTCf GTG GCA GAA TTC 3' (SEQ ID NO: 62) were used. After
extension the resulting dsDNA was digested with EcoRI and ligated into the
pLAC11 expression vector which had been digested with the same restriction
enzyme and subsequently dephosphorylated using alkaline phosphatase. Because
of the way the oligonucleotide library has been engineered, either orientation
of
20 the incoming digested double-stranded DNA fragment results in a fusion
product.
Generation of a Randomized Peptide Library Containing Terminal
Prolines. Randomized 20 amino acid peptide libraries containing two proline
25 residues at both the amino and the carboxy terminal ends of the peptides
were
constructed using the synthetic oligonucleotide 5' TAC TAT AGA TCT ATG
CCG CCG (NNN)16 CCG CCG TAA TAA GAA TTC GTA CAT 3' (SEQ ID
NO: 63). The complementary strand of the 93 base randomized oligonucleotide
was generated by filling-in with Klenow using the oligonucleotide primer 5'
30 ATG TAC GAA TTC TTA TTA CGG CGG 3' (SEQ ID NO: 64). After
extension the resulting dsDNA was digested with Bgl II and EcoR I and ligated
into the pLAC11 expression vector which had been digested with the same two
rt.::,triction enzymes. Because the initiating methionine of the peptides
coded by
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
66
this library is followed by a proline residue, the initiating methionine will
be
removed (F. Sherman et al, Bioessays 3: 27-31 (1985)). Thus the peptide
libraries encoded by this scheme are 20 amino acids in length.
Generation of a Randomized Hydrophilic a-Helical Peptide Library. Table
14 shows the genetic code highlighted to indicate certain amino acid
properties.
Table 14: Genetic Code Highlighted to Indicate Amino Acid Properties
phe ha TCT ser TAT tYr ha TGT cys
TTC phc ha TCC ser TAC tYr ha TGC cys
TrA leu Ha TCA ser TAA OCH TGA OPA
TTG leu Ha TCG ser TAG AMB TGG trP
CTT leu Ha CCT pro B. CAT his ha CGT arg
CTC leu Ha CCC Pro Ba CAC his ha CGC arg
CFA ten Ha CCA pro Ba CAA gin ha CGA arg
CTG leu Ha CCG pro Ba CAG gin ha CGG arg
ATT He ha ACT thr AAT am n ba AGT ser
ATC He ha ACC thr AAC asn ba AGC ser
ATA lie ha ACA thr AAA lys ha AGA arg
ATG met Ha ACG thr AAG ZYS ha AUG arg
G1-1 val ha OCT ala Ha GAT asp ha GUT gly Ba
GTC val ha GCC ala Ha GAC asp ha GGC gly Ba
GTA val ha GCA ala H. GAA giu Ha GGA gly Ba
GTG val ha GCG ala Ha GAG gin Ha GGG gly BR
Boldface amino acids are hydrophobic while italicized amino acids are
hydrophilic. The propensity for various amino acids to form a-helical
structures is
also indicated in this table using the conventions first described by Chou and
Fasman (P. Chou et al., Adv. Enzytnol. 47:45-148 (1978)). Ha= strong a-helix
former, ha = a-helix former, Ba = strong a-helix breaker, ba = a-helix
breaker. The
assignments given in this table are the consensus agreement from several
different
sources. Hydrophilic versus hydrophobic assignments for the amino acids were
made from data found in Wolfenclen et. at. (Biochemistry. 20:849-55 (1981));
CA 02794430 2012-10-31
WO 00/22112 PCIMS99/23731
67
Miller et. al. (J. Mol. Biol. 196:641-656 (1987)); and Roseman (J. Mol. Biol,
200:513-22(1988)). The propensity for amino acids to form a-helical structures
were obtained from consensus agreements of the Chou and Fasman (P. Chou et
at.,
Adv. Enzyinol. 47:45-148 (1978); P. Chou, "Prediction of protein structural
classes from amino acid compositions," in Prediction ofprotein structure and
the principles of protein conformation (G. Fasman, G.D. ed.). Plenum Press,
New York, N.Y. 549-586 (1990)); Gamier, Osguthorpe, and Robson (J. Mol.
Biol. 120:97-120 (1978)); and O'Neill and DeGrado (Science. 250:646-651
(1990)) methods for predicting secondary structure.
By analyzing the distribution pattern of single nucleotides in the genetic
code relative to the properties of the amino acids encoded by each nucleotide
triplet, a novel synthetic approach was identified that would yield randomized
18
amino acid hydrophilic peptide libraries with a propensity to form a-helices.
According to Table 14, the use of a [(CAG)A(TCAG)J codon mixture yields the
hydrophilic amino acids His, Gln, Asn, Lys, Asp, and Glu. These amino acids
are
most often associated with a-helical motifs except for asparagine, which is
classified as a weak a-helical breaker. If this codon mixture was used to
build an
a-helical peptide, asparagine would be expected to occur in about 17% of the
positions, which is acceptable in an a-helical structure according to the
secondary
structure prediction rules of either Chou and Fasman (P. Chou et al., Adv.
Enzymol. 47:45-148 (1978); P. Chou, "Prediction of protein structural classes
from amino acid compositions," in Prediction ofprotein structure and the
principles ofprotein conformation (G. Fasman, G.D. ed.). Plenum Press, New
York, N.Y. 549-586 (1990)) or Gamier, Osguthorpe, and Robson (J. Gamier et
at., J. Mol. Biol. 120:97-120 (1978)). Additionally, several well-
characterized
proteins have been observed to contain up to three ba breaker amino acids
within a
similarly sized a-helical region of the protein (T. Creighton, "Conformational
properties of polypeptide chains," in Proteins: structures and molecular
properties, W.H. Freeman and Company, N.Y., 182-186 (1993)). Since in most
a-helices there are 3.6 amino acids per complete turn, the 18 amino acid
length was
chosen in order to generate a-helical peptides which contained 5 complete
turns.
Moreover, the use of hydrophilic amino acids would be expected to yield
peptides
which are soluble in the cellular cytosol.
Randomized 18 amino acid hydrophilic a-helical peptide libraries were
synthesized using the synthetic oligonucleotide 5' TAC TAT AGA TCT ATG
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
68
(VAN)I7 TAA TAA GAA TTC TGC C.AG CAC TAT 3' (SEQ ID NO: 65). The
complementary strand of the 90 base randomized oligonucleotide was generated
by filling-in with Klenow using the oligonucleotide primer 5' ATA GIG CTG
GCA CAA TIC TTA TTA 3' (SEQ ID NO: 66). After extension the resulting
dsDNA was digested with Bgl II and EcoR I and ligated into the pLAC11
expression vector which had been digested with the same two restriction
enzymes.
Generating a Randomized Peptide Library Containing the +1- Charge
Ending Motif. Randomized peptide libraries stabilized by the interaction of
oppositely charge amino acids at the amino and carboxy termini were generated
according to the scheme shown in Fig. 9. To maximize the potential
interactions
of the charged amino acids, the larger acidic amino acid glutamate was paired
with
the smaller basic amino acid lysine, while the smaller acidic amino acid
aspartate
was paired with the larger basic amino acid arginine. To construct the
randomized
peptide libraries, the synthetic oligonucleotide 5' TAC TAT AGA TCT ATG
GAA GAC GAA GAC (N1N)16CGT AAA CUT AAA TAA TAA GAA TIC
GTA CAT 3' (SEQ ID NO: 67) and the oligonucleotide primer 5' ATG TAG
GAA TIC TTA TTA TTT ACG TTT ACG 3' (SEQ ID NO: 68) were used.
After extension the resulting dsDNA was digested with Bgl II and EcoR I and
ligated into the pLAC11 expression vector which had been digested with the
same two restriction enzymes.
For all libraries of randomized oligonucleotides, N denotes that an
equimolar mixture of the four nucleotides A, C, G, and T was used, and V
denotes that an equimolar mixture of the three nucleotides A, C and G was
used.
The resulting libraries were transformed into electrocompetent ALS225 E. colt
cells (Example I) under repressed conditions as described in Example II.
Screening of Transformants to Identify Inhibitor Clones. Transformants
were initially screened using the grid-patching technique to identify any that
could not grow on minimal media as described in Example II when the peptides
were overproduced. To verify that all the inhibitors were legitimate. plasmid
CA 02794430 2012-10-31
WO 00122112 PCT/US99/23731
69
DNA was made from each inhibitory clone, transformed into a fresh
background, then checked to make sure that they were still inhibitory on
plates
and that their inhibition was dependent on the presence of the inducer, IPTG,
as
in Example 11.
Growth Rate Analysis in Liquid Media. Inhibition strength of the peptides
was assessed by subjecting the inhibitory clones to a growth rate analysis in
liquid media. Minimal or rich cultures containing either the inhibitor to he
tested
or the relevant vector as a control were diluted to an initial OD 550 of
approximately 0.01 using new media and induced with I mM 1PTG. 0D550
readings were then taken hourly until the cultures had passed log phase.
Growth
rates were determined as the spectrophotornetric change in 0D55() per unit
time
within the log phase of growth, and inhibition of the growth rate was
calculated
for the inhibitors using the appropriate vector as a control.
RESULTS
Isolation and Characterization of Inhibitor Peptides that are Fused at Their
Carboxy Terminal End to the Amino Terminal End of the Rop Protein.
Approximately 10,000 peptides protected by the Rop protein af their carboxy
terminal end were screened using the grid-patching technique described in
Example II, and 16 two day inhibitors were isolated. The inhibitory effects
were
determined as described in the Example II, using pRop(C) as a control. Unlike
the anehorless inhibitors identified in Example H that were only inhibitory on
minimal media, many of the Rop fusion inhibitors were also inhibitory on rich
media as well, which reflects increased potency. As indicated in Table 15, the
inhibitors inhibited the bacterial growth rate at levels that averaged 90% in
minimal media and at levels that averaged 50% in rich media. The data in Table
15 is the average of duplicate experiments.
CA 02794430 2012-10-31
WO 00/22112 PCTAIS99/23731
Table 15: Inhibitory effects of peptide inhibitors stabilized by fusing the
carboxy terminal end of the peptide to the amino terminal
end of the Rop protein (Rop(C) fusion peptide inhibitors
Inhibitor % inhibition in % inhibition in
minimal media rich media
PRop(C)1 87 47
?Rop(C)2 99 58
PRop(C)3 85 54
PRop(C)4 98 49
PRop(C)5 95 54
PRop(C)6 99 46
PRop(C)7 91 59
PRop(C)8 86 51
PRop(C)9 93 57
PRop(C)10 91 35
5
Isolation and Characterization of Inhibitor Peptides that are Fused at Their
Amino Terminal End to the Carboxy Terminal End of the Rop Protein.
Approximately 6000 peptides protected at their amino terminal end by Rop
10 protein were screened using the grid-patching technique described in
Example II,
and 14 two day inhibitors were isolated. As observed for the Rop fusion
peptides isolated using the p-Rop(C) vector, most of the inhibitor peptides
isolated using the p(N)Rop- vector were inhibitory on rich media as well as
minimal media. The inhibitors were verified as described hereinabove and
15 subjected to growth rate analysis using p(N)Rop- as a control in order
to
determine their potency. As indicated in Table 16, the inhibitors inhibited
the
bacterial growth rate at levels that averaged 90% in minimal media and at
levels
that averaged 40% in rich media. The data in Table 16 is the average of
duplicate experiments.
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
71
Table 16: Inhibitory effects of peptide inhibitors stabilized by fusing the
amino terminal end of the peptide to the carboxy terminal
end of the Rop protein (Rop(N) fusion peptide inhibitors)
Inhibitor % inhibition in % inhibition in
minimal media rich media
pRop(N)1 81 30
pRop(N)2 96 53
pRop(N)3 95 43
pRop(N)4 92 38
pRop(N)5 99 33
pRop(N)6 93 38
pRop(N)7 87 34
pRop(N)8 91 44
pRop(N)9 95 37
pRop(N)10 96 40
Isolation and Characterization of Anchorless Inhibitor Peptides Containing
Two Prolines at Both Their Amino Terminal and Carboxy Terminal Ends.
Approximately 7500 peptides were screened using the grid-patching technique
described in Example II, and 12 two day inhibitors were isolated. As indicated
in Table 17, the top ten inhibitors inhibited the bacterial growth rate at
levels that
averaged 50% in minimal media. The inhibitory effects were determined as
described in the text using pLAC11 as a control. The data in Table 17 is the
. average of duplicate experiments.
CA 02794430 2012-10-31
WO 00/22112
72 PCT/US99/23731
Table 17: Inhibitory effects of peptide inhibitors stabilized by two
proline residues at both the amino and earboxy terminal ends of the peptide
Inhibitor % inhibition in
minimal media
pProl 50
pPro2 49
pPro3 50
pPro4 59
pPro5 52
pPro6 93
pPro7 54
pPro8 42
pPro9 41
pProl0 42
Sequence analysis of the coding regions for the top ten inhibitors is
shown in Table 19. The landmark Bgl II and EcoR I restriction sites for the
insert
region are underlined, as are the proline residues.
Since the ends of the oligonucleotide from which these inhibitors were
constructed contained Bgl II and EcoR I restriction sites, the oligonucleotide
was
not gel isolated when the libraries were prepared in order to maximize the
oligonucleotide yields. Because of this, three of the inhibitory clones,
pPro2,
Ppro5, and pPro6 were found to contain deletions in the randomized portion of
the
oligonucleotide.
Table 18: Sequence analysis of the insert region from the proline
peptides
pProl ¨ 21-aa
,
ACA TCT ATG CCG CCG ATT CTA TOG GGC GAA GCG AGA AAG CCC TOG TOG GOT COG GAT
CAT ACA CCG CCG TAA TAA
2U MPPILWGEARERLWGGEHIPP.
(SEQ ID NO: Th(
GAA TOG (SEC: ID NO: 69)
pPro2 27aa
MA TOT MG Ca; CCG CCG TOG GAT ATT GIG TOG GOT ATT GAG CIA GGG GGO CAT TOG TGG
TGC CGC COT ATT AM
CA 02794430 2012-10-31
W000/22112
73 PCT/US99/23731
MPPPLDIVSGIEVGGEFFICREIN
MT TCT CAT GTT YON (SEQ ID NO: 711
N S EV. (SEQ ID NO: 72)
pPro3 - 8aa
AGA TOT ATG CCG CCG GAC AM CCG GTC CTG SOP TM AGO GM GOT CGA CM AGO GOP TAT
CAD CCG CCG TAP TM
MPI,DNPVF. (SEQ ID NO: 74)
GAP TTC (SEQ ID NO: 73)
pPro4 - 9aa
AGA TCT ATG CCG CCG CTA TTG GAG MA GAT GAO AAA TAG PTA TAT GCG TOG TTG TTT TTC
TGT CCG CCG TAP TAP
7<PPFLDGDDIC* (SEQ ID NO: 76)
GAP TTC (SEQ ID NO: 75)
pPro5 - 1 Oaa
AGA TCT RIG CCG COG AGO TGG APP ATG TTG PTA AGA CAG TOP CM ATG CGT 'FCC ATT
ACT CCC GCC GOP PTA AGA
MFPRWKIIFIRQ* (SEQ ID NO: 78)
AFT . C (SEQ YD NO: 77)
pPro6 - 7aa
r AGA TCT ATG ATG AGA MA GCC CCC CCG 'FAA TAP GAP TTC (SEQ ID NO: 79)
23 --- MMEVAPP* = (SEQ ID NO: 60)
pPro7 - 14aa
AGA TCT ATG CCG COG TTG CCC COG GCA 'FCC GAT GTA TAT COG CTA PAT TOP ATG TCT
TGT COG CCG CCG TAP TAP
NPPLKOACDVYGVN. (SEQ ID NO: 82)
GAP TIC (SEQ ID HO: el)
pPro8 - 21aa
AGA TCT ATG CCG CCG GGG AGA GGG GAP COG GTG GOP GTG ACA TOO TTG AGC COG AAC
GTG TAC CCG CCG TAN TAP
HPFGEIGEAVGVTCLSANVY77* =
(SEQ ID NO: 8-4)
GAP TTC (SEQ ID NO: 81)
pPro9 - 2 laa
AGA OCT ATG CCG CCG GGA AGG GTA GTG TTC 'ITT GTC OCT ATC TIT GTT TCC GCA ATA
TGC CTC CCG CCG TAP FAA
¨ MPPGAVVFEVAIFVSAICLPP* *
(SEC? ID NO: -8-6)
GAP TTC (SEQ ID NO: e5)
pProl0 - 21aa
415 AGA TCT ATG CCG CCG AGO TTC COT CAT GAG ACT GTT AAA tGG CTG COG MC GTT
ACA AAA OCT CCG CCG TAP TAP
=MPPRFAHESVKGFGDVTKAPP* *
(SEQ ID NO: 88)
GAP TTC (SEQ ID NO: 87)
All the inhibitors were found to contain two proline residues at either their
amino
or carboxy termini as expected. Four inhibitors contained two proline residues
at
both their amino and carboxy termini, five inhibitors contained two proline
residues at only their amino termini, and one inhibitor contained two proline
residues at only its carboxy terminus.
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
74
Isolation and Characterization of Anchorless Hydrophilic Inhibitor
Peptides Stabilized by an a-Helical Motif. Approximately 12,000 peptides were
screened using the grid-patching technique and 5 two day inhibitors were
isolated.
The inhibitors were verified as already described for the Rop-peptide fusion
studies
and subjected to growth rate analysis using pLAC I I as a control in order to
determine their potency. As indicated in Table 19, the inhibitor peptides
inhibited
the bacterial growth rate at levels that averaged 50% in minimal media. The
averaged values of two independent determinations are shown.
Table 19: Inhibitory effects of the hydrophilic a-helical peptides
Inhibitor % inhibition in
minimal media
pHelixl 67
pHelix2 46
pHelix3 48
pHelix4 45
pHelix5 42
Sequence analysis of the coding regions for the 5 inhibitors is shown in
Table 20. The landmark Bgl II and EcoR I restriction sites for the insert
region are
underlined. Since the ends of the oligonucleotide from which these inhibitors
were
constructed contained these restriction sites, the oligonucleotide was not gel
isolated when the libraries were prepared in order to maximize the
oligonucleotide
yields. Because of this, two of the inhibitory clones, pHelix2 and pHelix3,
were
found to contain deletions in the randomized portion of the oligonucleotide.
The
predicted a-helical content of these peptides is indicated in Table 20
according to
the secondary structure prediction rules of Gamier, Osguthorpe, and Robson (J.
Gamier et al., J. Mol. Biol. 120:97-120 (1978)) prediction rules.
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
Table 20: Sequence analysis of the insert region from the hydrophilic a-
helical peptides
5 pHelixl - 18aa, 83% a-helical
AGO TCT ATG CAT GAG GAO CAA GAG GAG GAG CAC MT AAA MG GAT AAC CPA AAA CPA CAC
TAP TAP GAO
MHDEGEEENNKKONEKER* * (SEQ
ID NO: 90)
10 TTC (SEQ ID NO: 89)
pHelix2 - 22aa, 68% a-helical
15 AGO TCT ATG CAN GAG GAG CAC GAG CAA GGC AGG ATG AGC AAG AGO ATG AAG MT
PAT MG OAT TCT CAT
MQQEHEQGRMSKRMKNNKNSH
GTT TGA (SEQ ID NO: 91)
V 4 (SEQ ID NO: 92)
pHelix3 - 22aa, 55% a-helical
r
AGO TCT ATG AAC CAT CAT OAT GAG GCC ATG ATC MC ACA ATG AAA ACG AGG ART PAT MG
OAT TCT CAT
---MNHHNEAMINTMKTRNNK-NT H
GTT TOP (SEQ ID NO: 93)
V * (SEQ ID NO: 94)
pHelix4 - 18aa, 17% a-helical
AGO TCT ATG AAC GAC GAC OAT GAG CAA GAG GAT PAT CAT GAT CAG CAT RAG GAT AAC
AAA TAR TAP GAP
MNDDNUOEDNRDQHKDNK. * (SEQ
"I'D NO:'OSZID NO: 95)
pHelix5 - 18aa, 50% a-helical
LIO AGO TCT ATG CAA GAG COG GAT GAG CAT OAT GAT MC CAT CAC GAG GAT AAA CAT
RAG MG TAR TAP. GM
M OEQDQUN DNHREDNFIKK` * (SEQ
ID NO: 98)
TTC (SEQ ID NO: 97)
According to Gamier, Osguthorpe, and Robson secondary structure prediction,
all
of the encoded peptides are expected to be largely a-helical except for
pHelix4.
Interestingly, pHelix I which had the highest degree of a-helical content was
also
the most potent inhibitory peptide that was isolated in this study.
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
76
Isolation and Characterization of Anchorless Inhibitor Peptides Stabilized
by an Opposite Charge Ending Motif. Approximately 20,000 peptides were
screened using the grid-patching technique and 6 two day inhibitors were
isolated.
The inhibitors were verified as already described for the Rop-peptide fusion
studies
and subjected to growth rate analysis using pLAC I 1 as a control in order to
determine their potency. As indicated in Table 21, the inhibitor peptides
inhibited
the bacterial growth rate at levels that averaged 50% in minimal media. The
averaged values of two independent determinations are shown.
Table 21: Inhibitory effects of peptide inhibitors that are stabilized by the
opposite charge ending motif
Inhibitor % inhibition in
minimal media
p+/-1 41
p+/-2 43
p+/-3 48
p+/-4 60
p+/-5 54
p+/-6 85
Sequence analysis of the coding regions for the six inhibitors is shown in
Table 22. The landmark Bgl II and EcoR I restriction sites for the insert
region
are underlined. With the exception of p+/-4 which was terminated prematurely,
the coding regions for the inhibitors were as expected based on the motif that
was used to generate the peptide libraries.
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
77
Table 22: Sequence analysis of the insert region from the opposite charge
ending peptides
p+/-1 - 25aa
MA TCT ATG GAA GAG CAA aAc GAG GOT GCG TM GCG TOG GM GM GM CTT TOG TCG TGG MG
TCG GTG
NEDEDEGASAWGAEINSWOSV
, COT AAA CGT AAA TMTMGMTTC (SEQ ID NO: 99)
1U RKRK*" (SEQ ID NO: 100)
p+/-2 - 25aa
AM TCT ATG GAA GAO GAA GAC GOT CTA GGC ATG GGG GOT GGG TIC GTC AOC CTC ACT TTA
TTA TOG TTC
MEDEDGIGMGGGIVRITLIFF
CGT AAA CGT AAA TM TM GAA TT: (SEQ ID NC: 101)
RKRK. " (SEQ ID NO: 102)
p+/-3 - 25aa
AGA TCT AIG GAA GAC GM aAc GGG GAG AGG ATC MG OGG GCC CGC TOT CM Gil. COG CTG
GTA GAT AGA
MEDEDGERIQGARCEVALVDR
CGT AAA CGT AAA TA?. TA?. GA?. TTC (SEQ ID NO: 103)
RKEK* (SEQ ID NO: 104)
p+/-4 - 11aa
AGA TCT MG GA?. MC GM MC GAO AGG GGG CGT GCG COG TAG CTT TM GTT GGG CIA SCOT
TOO GAG ATA
MEDFDDRGRGR= (SEG ID NO: 105)
COT AAA CGT AAA TN'. TA?. GSA TTC (SEQ ID NO: 106)
p+/-5 - 25aa
AGA TCT ATG GM GA: GA?. MC GGG GGG GCC GGG AGG AGG GCC TOT CTT TOT TCC (CG CTT
OTT GGG GA?.
MEDEDGGAGRRACLCSALVGE
, COT AAA CGT AAA TA.?. TA?. GM TTC (SEQ ID NO: 107)
LtURKRE. " (SEQ ID NO: 100)
p+/-6 - 25aa
AGA TCT ATG GAS'. GAC GAA GAC AAG CGT CGC GAG AGG ACT CCA AAA GGG CGT CAT GTC
GOT COG TCG ATG
NEDEDKRAFRSAKGRHVGRSM
CGT AAA CGT PAA TM MC TOT (SEQ ID NO: 109)
R KR K (SEQ ID NO: 110)
DISCUSSION
In Example II, where fully randomized peptides were screened for
inhibitory effect, only three peptides (one "anchorless" and two unanticipated
Rop fusions resulting from deletion) were identified out of 20,000 potential
candidates as a potent (i.e., two day) inhibitor of E. coli bacteria. Using a
biased
CA 02794430 2012-10-31
WO 00/22112 PCT/US99/23731
78
synthesis as in this Example, it was possible to significantly increase the
frequency of isolating potent growth inhibitors (see Table 23).
Table 23: Summary of the frequency at which the different types of
inhibitor peptides can be isolated
Type of inhibitor peptide Frequency at which a two
day Reference
inhibitor peptide can be isolated
anchorless 1 in 20,000 Example 11
protected at the C-terminal 1 in 625 This example
end via Rop
protected at the N-terminal 1 in 429 This example
end via Rop
protected at both the C- 1 in 625 This example
terminal and N-terminal end
via two pro] ines
protected with an a-helix 1 in 2,400 This example
structural motif
protected with an opposite 1 in 3,333 This example
charge ending motif
Many more aminopeptidases have been identified than carboxypeptidases
in both prokaryotic and eukaryotic cells (J. Bai, et al., Pharm. Res. 9:969-
978
(1992); J. Brownlees et al., J. Neurochem. 60:793-803 (1993); C. Miller, In
Escherichia coli and Salmonella typhimurium cellular and molecular biology,
2nd edition (Neidhardt, F.C. ed.), ASM Press, Washington, D.C. 1:938-954
(1996)). In the Rop fusion studies, it might have therefore been expected that
stabilizing the amino terminal end of the peptide would have been more
effective
at preventing the action of exopeptidases than stabilizing the carboxy end of
the
peptides. Surprisingly, it was found that stabilizing either end of the
peptide
caused about the same effect.
Peptides could also be stabilized by the addition of two proline residues
at the amino and/or carboxy termini, the incorporation of opposite charge
ending
amino acids at the amino and carboxy termini, or the use of helix-generating
hydrophilic amino acids. As shown in Table 23, the frequency at which potent
inhibitor peptides could be isolated increased significantly over that of the
anchorless peptides characterized in -Example II.
CA 02794430 2012-10-31
WO 00/22112
PCT/US99/23731
79
These findings can be directly implemented to design more effective
peptide drugs that are resistant to degradation by peptidases. In this
example,
several strategies were shown to stabilize peptides in a bacterial host.
Because
the aminopeptidases and carboxypeptidases that have been characterized in
prokaryotic and eukaryotic systems appear to function quite similarly (C.
Miller,
In Escherichia coil and Salmonella typhimurium cellular and molecular biology,
2nd edition (Neidhardt, F.C. ed.), ASM Press, Washington, D.C. 1:938-954
(1996); N. Rawlings et al., Biochem J. 290:205-218 (1993)), the incorporation
of
on or more of these motifs into new or known peptide drugs should slow or
prevent the action of exopeptidases in a eukaryotic host cell as well.
Sequence Listing Free Text
SEQ ID NO:2
peptide sequence having opposite charge ending motif
SEQ ID NOs:3-4
stabilized angiotensin
SEQ ID NOs:6-19, 24-28, 55-58, 60, 62, 64, 66, 68
printer
SEQ ID NOs:20-22
primer fragment
SEQ ID NOs:23, 59, 61, 63, 65, 67
randomized oligonucleotide
SEQ ID NOs:29-33
antisense oligonucleotide
CA 02794430 2012-10-31
WO 00/22112 PCT1US99/23731
SEQ ID NOs:34, 36, 39, 40, 43, 45, 46, 48, 51, 52, 70, 72, 74, 76, 78, 80, 82,
84,
86, 88, 90, 92, 94, 96, 98, 100, 102, 104, 105, 108, 110
stabilized peptide
5 SEQ ID NOs:35, 37, 38, 41,42, 44, 47, 49, 50, 53, 69, 71, 73, 75, 77, 79,
81, 83,
85, 87, 89, 91, 93, 95, 97, 99, 101, 103, 106, 107, 109
nucleic acid encoding stabilized peptide
SEQ ID NO:54
10 N-terminal protective sequence
The foregoing detailed description and examples have been given for
clarity of understanding only. No unnecessary limitations are to be understood
therefrom. The invention is not limited to the exact details shown and
described,
15 for variations obvious to one skilled in the art will be included within
the
invention defined by the claim.
CA 02794430 2012-10-31
DEMANDES OU BREVETS VOLUMINEUX
LA PRESENTE PARTIE DE CETTE DEMANDE Ot CE BREVETS
COMPREND PLUS D'UN TOME.
CEC1 EST LE TOME 1 ____________________ DE 2
NOTE: Pour les tomes additionels, veillez contacter le Bureau Canadien des
Brevets.
JUMBO APPLICATIONS / PATENTS
THIS SECTION OF THE APPLICATION / PATENT CONTAINS MORE
THAN ONE VOLUME.
THIS IS VOLUME 1 OF 2
NOTE: For additional volumes please contact the Canadian Patent Office.