Note: Descriptions are shown in the official language in which they were submitted.
CA 02796884 2012-11-27
IMPROVED CONTROLLED RELEASE ORAL DOSAGE FORM
FIELD OF THE INVENTION
The present invention relates to oral controlled release dosage formulations
containing bupropion hydrochloride.
BACKGROUND OF THE INVENTION
The compound designated bupropion hydrochloride is described in United States
Patent Nos. 3,819,706 and 3,885,046. It is marketed as an anti-depressant and
an aid to smoking
cessation. Bupropion is an aminoketone-derivative chemically unrelated to
other currently
available antidepressants (e.g., selective serotonin-reuptake inhibitors,
tricyclics, tetracyclics).
While the neurochemical mechanisms of the antidepressant and smoking
cessation effects are unknown, noradrenergic pathways and/or dopaminergic
effects appear to be
primarily involved. Bupropion does not inhibit monoamine oxidase and is a
weak blocker of serotonin and norepinephrine uptake.
1
CA 02796884 2012-11-27
WO 021062299 PCT/US02103523
The drug is useful in the treatment of depressive affective disorders
(e.g., major depression) at dosages of 75 to 600 mg daily. Bupropion may be
preferable to other agents because of its minimal anticholinergic,
cardiovascular, and
antihistaminic effects or in those patients who have experienced weight gain
or sexual
dysfunction with another antidepressant. Bupropion, as extended-release
tablets, is
used in the cessation of smoking at dosages of 100-300 mg daily. Withdrawal
symptamns and cigarette craving are reduced with bupropion. Other uses include
patients with b.polar depression, attention-deficit hyperactivity in both
adult and
pediatric patients, and panic symptoms superimposed on depression.
Immediate release bupropion tablets pro vide more than 75% of
bupropion release into the dissolution media in 45 minutes. In studies to
date, the risk
of seizures appears to be strongly associated, in part, with the use of
instant release
tablets.
Numerous techniques exist in the prior art for preparing sustained or
controlled release pharmaceutical formulations. One common technique involves
surrounding an osmotically active drug core with a semipermeable membrane. The
drug is released from the core over time by allowing a fluid such as gastric
or
intestinal fluid to permeate the coating membrane and dissolve the drug so the
dissolved drug can permeate through the membrane. In some cases a hvdrogel is
employed to push the active ingredient through the passageway of the membrane.
Another common technique for preparing controlled-release
pharmaceutical formulations is to encapsulate a plurality of beads, pellets or
tablets
that are coated with varying levels of diffusion barriers. The barriers can be
of the
same or different chemical composition. Release of the pharmaceutical may
occur by
leaching, erosion, rupture, diffusion or similar actions depending on the
nature and
2
CA 02796884 2012-11-27
WO 02/062299 PCTiU, S02/03523
thickness of the coating material. These products require multi-layered
coating,
sometimes as much as 30 to 90 coats.
Film coating techniques are characterized by the deposition of a
uniform film onto the surface of a substrate. Because of the capability of
depositing a
variety of coating materials onto solid cores, this process has been used to
make
controlled release dosage forms starring from different formulations, such as
tablets,
granules, pellets and capsules. Cores are usually prepared using one of the
following
processes. compaction, surface layering, or agglomeration.
One limitation associated with these dosage forms consists in their
failure o delay drug delivery. Many o the m ilti-walled preparations described
above
do not provide prolonged delayed release of the drag prior to initiation of
sustained
release, which. is important when biphasic release profiles are desired. Other
systems
are essentially "delayed releases mechanisms. There is delay of drug release
in the
stomach. but once the coated drug reaches the i~rtestines, the release of
medication is
rapid. There is no sustained release in the intestines.
Bupropion hydrochloride is highly soluble in water with a high
permeability characterized by rapid and almost complete absorption. Peak
plasma
concentrations occur within 2 hours for bupropion and 3 hours for bupropion
sustained-release. Its biphasic pharniacokinetics is characterized by a two-
compartment model; the distributive phase has a mean half-life of 3 to 4 hours
with a
biphasic decline and a terminal T Vz of about 14 hours following single doses.
A
major drawback is extensive first-pass metabolism. It appears that only a
small
portion of any oral dosage reaches the systemic circulation intact. Immediate-
release
tablets are dosed three times a day, preferably with 6 or more hours
separating the
doses. For those patients requiring doses greater than 300 mg daily, each
divided
3
CA 02796884 2012-11-27
WO 02/062299 PCTiUS02/03523
dose should not exceed 150 mg each. This necessitates administration of the
tablets 4
times daily with at least 4 hours between successive doses. Commercially
available
sustained-release products are available in film-coated tablets marketed by
Glaxo
Wellcome under the tradenames Welibutrin SR and Zyban . These are dosed
Mice daily. For those patients requiring above 300 mg daily, the regimen
remains
twice daily dosing. No currently available product provides a sustained
release
profile suitable for once daily dosing.
Patient compiiance is especially problematic in depressed patients.
There is a need for improved patient compliance. One of the means employed
clinica'I_- to improve p tint adherence _ .
o , s
es r; simplification or the dosing
regi~ Een. Thus, need exists for a once daily bupropion formulation.
Sustained release tablet fours of bupropion are described in United
States Patent No. 5,427,798. comprising a . ustained release tablet which
provides
peals bupropion blood levels at approximately 2-3 hours, thereby requiring
twice daily
dosing. Controlled release is achieved by combining bupropion particles with
microcrvstalline cellulose and hy-drogel-forming hydroxv~propyl
methyicelluiose.
Another sustained release bupropion tablet or caplet formulation
disclosed in united States 4,687,660, comprises a difficult manufacturing
process and
limited shelf life. United States Patent No. 5,358,970 discloses a formulation
of
bupropion hydrochloride that contains an acid stabilizer.
United States Reissue Patent No. 33,994 discloses a tablet formulation
of a water insoluble, water-permeable film coating surrounding the drug core
and a
particulate, water-soluble, pore-forming material dispersed within the film
coating;
this osmotic gradient and channel forming system is applicable for tablet
dosage
forms. However, here also at least twice daily dosing is necessitated by the
release
3
CA 02796884 2012-11-27
WO 021062299 PCT/US02103523
profile of 25-70% of bupropion within 4 hours, and 40-90% within 6 hours.
Wellbutrin SR is a commercially available twice a day dosage form of
bupropion
which contains carnauba wax, cy steire hydrochloride, hydroxypropyl
methylcellulose, magnesium stearate, microcrystalline cellulose, polyethylene
glycol
and titanium dioxide.
There is no capsule form of bupropion commercially available.
Capsules are advantageous in those patients who have difficulty swallowing
where
the contents of the capsule may be sprinkled on food.
Immediate release tablets must be stored at a temperature above 15-
C a _d protected from light and moist ere. Extendeu release tablets should be
stored in tight, light-resistant containers at a temperature of 20-25 C.
The need exists for a relayed, sustained release pharmaceutical
preparation that provides a longer delay of drag dissolution thereby allowing
greater
fiexibili in designing sustained release profiles, provides improved plasma
levels
wherein the maximum plasma concentration (Caw,) can be substantially reduced
without a concomitant reduction in A1. C, and is simply and economically
produced.
Such a delayed delivery dosage form has a practical application, and it
represents a
valuable contribution to the medical arts. The present invention provides such
a
composition and offers an efficient and cost effective method of preparation.
Accordingly, it is an object of this invention to provide a sustained
release formulation of bupropion suitable for once daily administration.
Another object of the present invention is to provide a capsule dosage
form comprising means for delaying delivery of the drug in gastric fluids for
6 hours
up to 12 hours, usually 4 hours to 8 hours.
CA 02796884 2012-11-27
WO 02/062299 PCT/US02/03523
It is also an object of this invention to provide a controlled and
extended release bupropion capsule formulation that is easy to manufacture and
can
be used to prepare a range of dosing levels suitable for once daily
administration.
It is a further object of the present invention to provide 24-hour control
of symptoms of depression or tobacco dependence withdrawal.
Seizures result more commonly by single dosages of bupropion over
150, mg, hence the need for twice to four times dally dosing regimens. Another
object
of this i vention is to provide simplified once daily dosing regimen with the
potential
to prevent or reduce the incidence of seizures caused by bupropion.
The present in v'ention also relates to a new sustained release bupropion
pharmaceutical composition producing novel blood plasma levels after ingestion
over
24 ` o r^ t n n or rep tss
ho hat is not disclosed; ; nc_ . dead obvious by, the prior art. Other abJec
f L-tunes and advantages of the invention are not taught in the prior art but
will be
more apparent to those versed in the art from the follo~. ng specification;
taken in
conjunction with the d_rawirigs.
SUNENIARY OF THE PRESENT INVENTION
The present invention meets the unfulfilled needs of the
pharmaceutical industry.
The current invention involves a new pelletization process, typified by
the application of a bupropioncellulose ether suspension to inert spheres and
two
unique formulations of sustained release coatings that are applied to separate
active
pellets. The formulation functions by membrane-controlled extended-release in
a pH
dependent manner. The bupropion release rate has been improved by the
introduction
6
CA 02796884 2012-11-27
WO 02/062299 PCT/US02/03523
of two types of film coated active pellets that release the drug at different
pH resulting
in novel dissolution profiles.
Inert spheres are initially coated with bupropion and hydroxypropyl
methylceilulose. T% -e active pellets containing bupropion comprise 70-75
weight % of
the dosage form. An enteric coating, applied to about one third of the active
drug
pellets, is comprised of a film insoluble at low pH, such as hydroX'Vpropyl
.methylceilulose phthalate. The second coating applied to the other two thirds
of
active drug pellets is comprised of a combination of a hydrophobic coating
agent and
methyl acrylic acid copolymer. The two pellet types are then combined in a
capsule.
Ge nerail= , -1__ eig _ratio of the airs' pellet to the second pellet will be
from about
.
9E:1 0 to about 10:90 y;; atho ugl'i a ~~~ elg__. ratio of from about 71v:70
to about -10-1:30 is
preferred- Especially prof rred is a weight ratio of about 33.3:66.7.
This formulation can provide 24-hour efficacy with once daily dosing,
with less than 50% of the drug released at 10 hours. Therapeutic plasma levels
are
maintained from 12 to 24 hours. The usual dosage range is 75-450 mg.
In another embodiment of the present, an uncoated bupropion
component is also employed. In this embodiment, bupropion powder or granules,
or
the uncoated active pellets (bupropion and hydroxypropyl methylcellulose
sprayed
onto an ine sphere.) may be used directly (first co(mponent). The bupropion
release
rate is further modified and improved by the introduction of uncoated
bupropion and
the two types of film coated active pellets that release the drug at different
pH
resulting in further novel dissolution profiles.
In. this embodiment, the enteric coating (hydroxypropyl
methylceilulose phthalate) is applied to from about 10 to about 90 weight
percent of
the active drug pellets (second component). The second coating (hydrophobic
and
1
CA 02796884 2012-11-27
methyl acrylic acid copolymer) is applied to from about 90 to about 10 weight
percent of active
drug pellets (third component). The three components are then combined in a
capsule. Generally,
the weight ratio of the first component to the second component may vary from
about 1: 50 to
about 50: 1, the weight ratio of the first component to the third component
may vary from about
1: 50 to about 50: 1, and the weight ratio of the second component to the
third component may
vary from about 10: 90 to about 90: 10, although a weight ratio of from about
30: 70 to about
70:30 is preferred. Especially preferred is a weight ratio of three components
of about 10: 30 : 60.
In a broad aspect, the present invention relates to a pharmaceutical
composition
comprising: (A) at least one pellet consisting of. (i) a core which comprises
an inert carrier,
wherein said inert carrier is coated with bupropion or a pharmaceutically
acceptable salt or
stereoisomer thereof, and (ii) a sustained release coating surrounding said
core wherein said
sustained release coating comprises at least one controlled release polymer
for controlled release
delivery of said bupropion or a pharmaceutically acceptable salt or
stereoisomer thereof, (B) said
pharmaceutical composition further comprising bupropion or a pharmaceutically
acceptable salt
or stereoisomer thereof in an immediate release form, and wherein the
pharmaceutical
composition is designed for once daily dosing.
In another broad aspect, the present invention relates to a pharmaceutical
composition
comprising: (A) at least one pellet comprising: (i) a core which comprises an
inert carrier.
wherein said inert carrier is coated with bupropion or a pharmaceutically
acceptable salt or
stereoisomer thereof, and (ii) a sustained release coating surrounding said
core wherein said
sustained release coating comprises at least one controlled release polymer
for controlled release
delivery of said bupropion or a pharmaceutically acceptable salt or
stereoisomer thereof, (B) said
pharmaceutical composition further comprising bupropion or a pharmaceutically
acceptable salt
or stereoisomer thereof in an immediate release form, and wherein the
composition provides
therapeutic plasma levels of bupropion for 12 to 24 hours with once daily
dosing and less than
50% of the bupropion released at 10 hours.
R
CA 02796884 2012-11-27
In another broad aspect, the present invention relates to a once-a-day
composition
comprising: (a) an immediate release component comprising bupropion or a
pharmaceutically
acceptable salt thereof; (b) a first pellet comprising an enteric release
component comprising
bupropion or a pharmaceutically acceptable salt thereof and a pH dependent
coating polymer; and
(c) a second pellet comprising a sustained release component comprising
bupropion or a
pharmaceutically acceptable salt thereof and a water insoluble coating
polymer, wherein said
composition contains 75 to 450 mg of bupropion or a pharmaceutically
acceptable salt thereof
and provides an in vivo plasma profile selected from: (a) a mean Cmar of at
least 50.0 ng/ml; (b) a
mean AUCo_;,,f of greater than approximately 500.0 ng=hr/ml; and (c) a mean
Tmax of between
approximately 5.0 hours and 8.5 hours based upon a single dose administration
of a composition
containing 150 mg of bupropion or a pharmaceutically acceptable salt.
BREW DESCRIPTION OF THE DRAWINGS
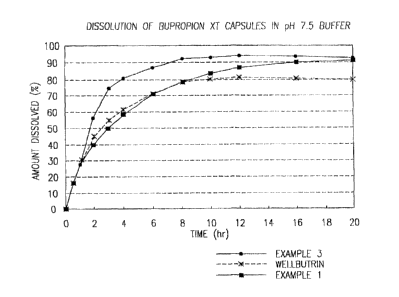
FIG. I is a graph depicting the dissolution profile in a pH 7.5 buffer of the
formulations
as described in Examples 1 and 3 versus the dissolution of the commercially
available sustained
release form of bupropion (Wallbutrin(k SR).
FIG. 2 is a graph depicting the dissolution profile in simulated gastric fluid
(pH 1.5) of
the formulations as described in Examples 1 and 3 versus the dissolution of
the commercially
available sustained release form of bupropion (Wellbutrin SR).
FIG. 3 is a graph depicting the mean plasma concentration-time profiles of
bupropion in
seven healthy subjects (smokers) following a single oral dose of the
formulation in Example 2
versus 150 mg of the commercially available sustained release product (Zyban
).
FIG. 4 is a graph depicting the mean plasma concentration-time profiles of
bupropion in
seven healthy subjects (smokers) following a single oral dose of the
formulation in Example 4
versus 150 mg of the commercially available sustained release product
(Zyban(k).
da
CA 02796884 2012-11-27
WO 02/062299 PCT/ S02i03523
DETAILED DESCRIPTION OF THE PRESENT INVENTION
The present invention, in a first embodiment provides a two
component controlled release bupropion formulation for oral administration the
formulation comprising:
(1) a first pellet comprising:
(i) a core comprising:
(a) bupropion and its salts, isomers, or a pharmaceuticall
acceptable aminoketone antidepressant agent;
(b} an inert pellet as a stating material; and
(c) a binder; and;
(ii) a coating comprising:
(a) a pH dependent coating agent;
(b) a plasticizer; and
(c) a lubricant; and
(2) a second pellet comprising:
(i) a core comprising:
(a) bupropion and its salts, isomers, or a pharmaceutically
acceptable aminoketone antidepressant agent;
(b) an inert pellet as a starting material; and
(c) a binder; and
(ii) a coating comprising:
(a) a methyl acrylic acid copolymer;
(b) a water insoluble polymer;
(c) a plasticizer; and
9
CA 02796884 2012-11-27
WO 02/062299 PCT! tS02,'03523
(d) an antisticking agent.
In other embodiments of the present invention, there may also be
present another component, a form of immediate release bupropion.
The immediate release bupropion component may comprise any form
of immediate release bupropion. This may take the form of uncoated bupropion
granules or powders, may comprise bupropion active pellets (as described
hereinbelow), may include bupropion granules or active pellets coated with a
highly
soluble immediate r such as 2I' ad '^ r. n ; ?' t ,
ui at.elease coa_ r.~, s L Op y tyke coating, as are ko.
those skilled in the art (see generally, United States Patent No. 5,098,715),
or a
combination of any tof the Ibregoing
t
The active pellets of bupropion hydro-6 ,lori'de usef=ul in the practice of
the present invention are preferably bas d on active pellets having a core
formic g
inert col ponent that may comprise any type of comm- on ' known pellet sta. in
material, which may be water insoluble, such as, but not limited to, cellulose
spheres
or silicon dioxide. or may be water soluble, such as, but not limited to,
starch or sugar
spheres having a diameter ranging from about 15 to. about 50 mesh, preferably
ranging from about 30 to about 35 mesh. The preferred pellet starting material
is
sugar spheres, NF, containing not less than about 62.5 percent and not more
than
about 91.5 percent of sucrose. The spheres should have consistent bulk
density, low
friability, and low dust generation properties.
The inert core is preferably coated with an aminoketone antidepressant
agent or a pharmaceutically acceptable salt or stereoisomer thereof Most
preferably;
the core drug is bupropion hydrochloride.
The core forming inert component is coated with a formulation that
comprises bupropion hydrochloride and a binding agent. The binding agent
should be
Iv
CA 02796884 2012-11-27
WO 02/062299 PCTitJS02/03523
water soluble, and should possess high adhesivity and an appropriate
viscosity, to
guarantee good adhesion between the sugar cores and bupropion particles,
resulting in
a high concentration of drug in the pellets. The binding agents employed can
be any
type of binding agent commonly known in the art such as polyvinyl pyrrolidone
hydroxyethyI cellulose, hydroxypropyl cellulose, low molecular weight
hydroxyprop,'I methylcellulose (HPMC}, polyinethacrylate or ethyl cellulose-
In a
preferred embodiment of the present invention, the binding agent is a ,rater-
soluble
polymer such as Iiydroxypr=opyl metylcei!uiose having a viscosity in the range
o 2-
12 cps at 20 C, preferably= 4-6 cps, such as the material sold as MIethocel
E5. A
preferred ocn~.posltion of the binder for b?uproplo_ is about 2-10"=10 ev.W
and most
preferably 3-5'%,
The active pellets of the present invention will preferably comprise the
following ingredients:
E GREDIENT PREFERRED MOST
PREFERRED
Bupropion HCI 40-80%% 60-%{0
%
HPIC 2-1 03% 2.5-5ilo
starting pellets 10-35% 15-30%
All the percentages in the above-table are based on the total weight of the
core.
The active pellets for use in the practice of the present invention that
comprise the bupropion are typically prepared by forming a suspension of the
binder
and the drag and then layering the suspension onto the starting pellet using
any of the
layering techniques known in the industry, such as fluidized bed coating,
rotor
granulation or pan coating. The suspension medium may comprise any low
viscosity
solvent, such as isopropyl alcohol, ethanol, water, mixtures thereof and the
like. A
II
CA 02796884 2012-11-27
WO 02/062299 POT/US02/03523
sufficient amount of coating is applied to provide the desired level of
bupropion.
These active pellets may be used directly as the first component of the three
component formulations of the present invention.
The active pellets are also useful in preparing the other two
components of the present invention (both the two component and three
component
formulations). The active pellets intended for such use are divided into two
groups,
each group receiving a film coax ng that releases the drug at a different pH.
One
group of là is coated to release d tg at a pH correspond a 1 St nd
~
r r _ in-
lower. about ~. an
lower, which is likely to occur the upper gastrointestinal (GI) tract; the
other group
E pellets is Pcoated release drug a pH o_
47 7 and above, which is likely to
occur _ the lower Gi ii act. Thus, the entire does is released from this
product for an -x-ended Period of time its transition through the GI tract.
n e re~ embodiment, one oup of pellets (enteric component) is
coated v h a .91m comprising a pH dependent coating polymer, a plasticizer and
a
lubricant. This group of pellets preferably comprises from about 10 to about
90
weight percent of the total pellets, preferably from about 30 to about 70
weight
percent, and most preferably from about 33 to about 60 weight percent.
The pH dependent coating polymer may be selected from those enteric
coatings known to those skilled in the art. Preferably, the pH dependent
coating is
selected from the group consisting of shellac; methacrylic acid copolymers
(such as,
but not limited to Eudragit E100 (a cationic copolymer of dimethyl aminoethyl
methacrviate and neutral methacrylic acid esters)), cellulose acetate
phthalate,
hydroxypropyl methyleellulose phthalate, hydroxypropyl methylcellulose acetate
succinate, cellulose acetate trimellitate, polyvinyl acetate phthalate or
mixtures
thereof Hvdroxypropyl methyicefulose phthalate (HP ICP) is preferred. The
12
CA 02796884 2012-11-27
FVO 02/062299 PCT/US42/03523
preferred concentration is 2-10% -'AV of the total dosage form, and most
preferably 3-
5%_
The coating preferably also contains plasticizers. Plasticizers that may
be used in the practice of the present invention include any of those known to
those
skilled in the art, including, but not limited to, acetyltributyl citrate,
triacetin,
acetylated monoglyceride, rage oil, olive oil, sesame oil, acetyltriethyl
citrate,
ply ceri sorbitol, d_et'_esvloxalate, diet _yimalate, diethylfi r Brate,
dibutylsuccinate,
diethylmalo <a e. dioc :lphthai :e. dibutylphthaiate, d'sbut isebaCate.
ietliyi citrate
tribuE Gitrate_ gl;-:cerohribut rate, polyethylene glycol propylene glycol and
I es .}_ L T'he preferre cize_ i ace - 5ributYl citrate in an amount
.g .g ^ r a ^ f . 4 t au~ u: out i_ercent based or, t i., t Glat -LO rar.~In P
a~ _: iC t weight Ci tlse final coating
or D. -3 % `,v.., of the totad dosage forrn
T bly includes a lubricant Such as, t::.t not
limited to. hoe selected from the group consisting of giycer;-l m~onostearate;
Myvap ex 600P , ca ci 3tearate or stearic acid. The preferred lubricant
giyceryl
mono tearare in an amount ranging from about 1 to about 15 percent, and most
preferably 1-2.5 ,9 based on the total weight of the coating.
A preferred enteric coating for use in the present invention therefore
comprises the following ingredients:
INGREDIENT PREFERRED OS T
PREFERRED
HPMCP 2-10% 3-5%
Acetyltributyl citrate 0.1-3% 0.5-1%
Glyceryl monostearate 1-3% 1-2.5%
13
CA 02796884 2012-11-27
WO 02/062299 PCT/US02103523
Additional active drug pellets for forming the second coated
component of Cite present invention, preferably from about 90 to about 10
weight
percent of the total pellets, more preferably from about 70 to about 30 weight
percent,
and most preferably from about 67 to about 40 weight percent, are coated with
a
coating that comprises a polymer such as a methacrylic copolymer, water
insoluble
polymer, a plasticizer and an anti-sticking agent.
The methacrylic acid copolymer is selected from the known group of
meth acrylic acid cot ivrers, preferably' Eudragt' S (methacrylic acid
copolymer
Type B). and most preferably Eudragit' , 5100. The preferred concentration is
1-15%
in"
of the no-sal wig=___ of ' dosage form, preferably. 4-7%%.
The water insoluble polymer in the preferred embodiments y of the
presen Invent it s formed from a cellulose ester, or a cellulose esLe-,
et+her.
Represei_ta e r _aterials include a member selected from the group consisting
of
ethyl cellulose, cellulose acylate, cellulose diacylate, cellulose triacylate,
cellulose
acetate, cellulose diacetate, cellulose triacetate, cellulose acetate
butyrate, mono-, di-
and t -cellulose aryiates, and the like. Preferred is ethyl cellulose in a
concentration
ranging from about 1 to about 20%, preferably from about 2 to about 13%.
The preferred plasticizer additive for the second coating may be
selected from a :y of those mentioned above. Acetyltributyl citrate is
preferred.
The anti-sticking agents can be chosen from any of the known agents,
such as, but not limited to, those selected from the group consisting of an
alkaline
earth metal stearate, such as magnesium stearate or calcium stearate, or talc.
The anti-
sticking agents can be used alone or in combination in effective amounts. The
preferred anti-sticking agent is talc.
14
CA 02796884 2012-11-27
WO 021062299 PCTrUS02103523
The coating for the active pellet for this (second coated) component of
the present invention is applied to the active pellets by forming a solution
of the
respective coating components in a solvent or a mixture of solvents, such as,
but not
limited to, acetone and isopropyl alcohol, and employing any of the
application
techniques known to those skilled in the art, such as fluidized bed coating,
rotor
granulation or pan coating.
The components. either the two coated or the two coated and the
re ease, of tine present invert ion are blended together in the desired ratio
and placed in gelatin capsule to obtain a finished product. By va~-ing the
ratio of the
used of t e i tL le io n
-onToren. rzc: .~ L_~ rease at 0 o, novel ,iss ilut :._ _.
pro riles and p asrna profiles may be obtained in acco rdanc with he pre-sent
i- ret t_õ__. lter~a`iveiy, the dosage formulation may be made into tablets by
firs..
t 'id pharrliaLeuticair ly acceptable tablet
leet
adding ~d from 25 " percent of a 5~o_
... 40 0 _ [-ei?
excipient that w -ill form a compressible mixture without crushing the
pellets, and then
tabletting the mixture in a suitable tablet press.
The f =llovt-ing examples are intended to illustrate the present invention
but are not intended to limit the scope of the appended claims.
EXAMPLE 1
A batch of controlled release bupropion was manufacture using all
materials that comply with the current USP NF compendia specifications.
A controlled release 150 mg oral bupropion dosage form is prepared
by forming active core pellets having the following composition:
1. ACTIVE CORE PELLETS
Bupropion HCI 70.0%
Sugar sphere 30!35 26.5%
CA 02796884 2012-11-27
WO 02/062299 PCTJUS02/03523
Methocel ES 3.5%
Active pellets of bupropion are formed by dissolving 2.8 kg of
bupropion HCI and 0.140 kg of hydroxypropyl methylcellulose (Methocel E5) in
a
mixture of water and isopropyl alcohol. The active drug solution is then
sprayed onto
1.06 kg of sugar spheres 30135 in a fluidized bed processor with a W 'urster
insert. The
active core pellets are then dried in a fluidized bed processor until the loss
on drying
is below 1`',= . The bupropion pellets are than passed through a 16 mesh
screen and a
3(j! mesh screen and pellets are colectedd that are smaller than 16 mesh and
larger than
30 mesh.
11 EtiTEPJC COATED PELLETS
Bupropion active peflez 75.0%
HPMCP 50 16.9%
Acetvltrfbutvl citrate 2.5%
It vaplex 600P 5.6%
one-third , tJ_ 0.270 k,., of
For a group of about one-third of the pe1le ,
hydroxvpropyl methylcellulose phthalate and 0.040 kg of acetyltributyl citrate
are
dissolved in a mixture of purified water and isopropyl alcohol, USP. Then
0.090 kg
of glyceryI monostearate (1vIyvaplex 600P) is dissolved into the solution
above. The
solution is then sprayed onto 1.2 kg of the bupropion core pellets in a
fluidized bed
processor with a `I'urster insert. The pellets are then dried until the loss
on drying
(LOD) is less than 1%. The pellets are then mixed with 2% (w'w) talk for 10
minutes
in a V"-blender. The pellets are then passed through a 14 mesh screen and a 24
mesh
screen and pellets that are smaller than 14 mesh and larger than 24 mesh are
collected.
III. SUSTAINED RELEASE (SR) COATED ACTIVE PELLETS
Bupropion active pellets 80.0%
Eudragit .DR S100 12.6%
Ethocel 10 cps 1.4%
16
CA 02796884 2012-11-27
WO 02/062299 PCTitS02103523
Acetyltributyi citrate 2.0%
Talc 4.0%
For another group of about two-thirds of the pellets, a coating is
prepared where the ratio of the methacrylic acid copolymer to ethylcellulose
is about
9:1. The coating is made as follows: 0.378 kg of methacrylic acid copolymer
(Eudragit S 100), 0.42 kg of ethylcellulose (Ethocel 10 cps), and 0.060 kg
of
acet yltributvl citrate are dissolved in a i ixture of 0.690 kg acetone and
6.210 kg
isopropsi alcohol. 0. 12 kv of talcl is then dispersed into to the solution
above.
.. _0 y~ The
suspension is then sprayed onto 2.40 kg of the active bupropion core pellets
in a
fluidized bed v r p rocessor with a W!-{ "t uster insert. The bupropion
pellets are is ar: cried in a
fluidized met pr'_essor unt the LOD is less than 1%. The pe lets are mixed
With k4rw) talc for 10 m mutes in a V-blender and passed through a 14 mesh
screen and -2-L
mesh sCr _>_ --- Pe, iet.. . rt ah. 1.=r t mesh larger t aan 24 mesh are Ci..
ected
_ . -- i ?a.ll and iar, than ~:s_., ct4
.
These pellets have the following coating composition.
INGREDIENT MG`CAPSULE % TOTAL
WEIGHT
Eu agity S,100 22. 5 6.4
Ethocel 10 cps 2.5 0.7
Acetyltributyl citrate 3.6 1.0
Talc 2.0
The enteric coated pellets and the SR pellets are mixed after loading
each group into dosators. The strength of the final product is 150 mg of
bupropion
with 50 mg of active drug in the first group of pellets and 100 mg of active
in the
second group. The pellets are then encapsulated into size "1" light turquoise
blue/light turquoise blue capsules. The total weight of the formulation
(capsule +
pellets) is 350 mg.
17
CA 02796884 2012-11-27
1%VO 02/062299 PCT/US02/03523
The resulting bupropion capsules of Example I were then tested
according to the USP XXIII dissolution test (type 2, basket) at 50 rpm, at 37
C in pH
7.5 buffer and found to have the following release profile:
TABLE I
Time (hours) % Released
1 25
2 60
3 75
4 80
y6 88
8 93
1v 93
12 94
14 94
YT
The release profile of the controlled release product shown in thus
Example is sho n in FIG I by the line filled with circles.
The bupropicn capsules of Example 1 were then tested according to
the liSP XXIII dissolution test (type 2, basket), at 50 rpm, at 3', C in SGF
(pH 1.5) to
determine the percentage of drug dissolved versus time-
TABLE 2
Time (hours) % Released
1 0
2 1
3 2
18
CA 02796884 2012-11-27
WO 02/062299 PCT,I IS02/03523
4 3
6 7
8 15
30
12 45
14 S0
16 56
G' 60
The release profile of the controlled release product shown: in ti s
Example sn -ri sin FIG 2 by the line -,nh the tilled circles.
T e bupr_pion capsules of Example I were ten evaluated in seven
patients us in.- Ãa dard techniques mown in the art. Buprortv pion was 1 ~~ s
frstL detected
.
the plasma at about ~''. hours after adstTutioi_, and showed sustained G` ur
2=1
hours.
Two panels of seven patients were randomly assigned to receive either
tae bupropion formulation described herein or ZS1B AN 3) an open; randomized
single dose study. Blood samples were collected over a ?2-hour period and
anal, zed
for bupropion concentrations with a LCIMSiNIS method.
For the blood levels carried out Cam,:; is the maximum blood level
concentration of bupropion, Tm., is the time at which the maximum blood level
concentration occurs, Tt,g is the sampling point preceding the one at which
concentrations first become quantifiable. AUC is the "area under the curve" of
time
versus blood concentration. The results provided are given in Table 3 and FIG.
3
show that the mean plasma concentration time profiles of bupropion were
different
for the Example I formulation and ZybanS. Following oral administration, the
I
CA 02796884 2012-11-27
VVO 021062299 PCTfUS02J03523
Example 1 formulation had a delayed absorption with a Tig value of 1.9 hours.
The
mean Coax value of the Example 1 bupropion formulation was about one-half of
that
for Zybar: The time to reach (T,.,,, ) maximum plasma concentration occurred
about
8 hours after administration of the Example 1 formulation. The relative
bioavailability of the Example 1 formL:lation to Zyban was 40% o in terms of
C,
and 80 r% in terms of ALTCO_;f ratio.
TABLE 3
Variable Example I Mean Zvban Mean G-Mean
Ratio
i - 1 - C129. 0.40
AL Ce _; ,n g-h~r nu1 321. 0- 998.0 0.8=
(lir) 1 9 T,ax(lrj 8.1 3_a
T112 thr) 17.0 20.3
Thus. it can be seen from the data above that although the Cma of
Example I is significantly lower than, the C a of the. Zyban formulation,
the AUK
has only been slightly reduced.
EXAMPLE 2
The pellets from Example 1 are taken as the second and third
components. These pellets are loaded into the dosator along with active
pellets and
are filled into capsules in a ratio of 10:30:60 while maintaining the dosage
at 150 mg.
The blood profiles from this example will show a Cna,, that is the same as
shown in
Table 3. but will show a slightly increase AUC, thereby rendering the G-Mean
ratio at
about 1.00. The amount of active pellets may be adjusted as is known in the an
CA 02796884 2012-11-27
WW 0 02/062299 PCT/ SO2/03S23
pellets are xmixed with 2`9% (wtw) talc for 10 minutes in a V-blender and
passed
through a 14 mesh screen and a 24 mesh screen. Pellets smaller than 14 mesh
and
larger than 24 mesh are collected.
The pellets have the following coating composi on:
Ingredient mg Capsule % Total Wt.
Methacrylic acid copolymer 12.5 3.6
Ethocel 10 cps 12.5 3.6
Acet sltrihutti-l citrate 3.6 1.0
Talc 7.1 2.0
The {-. an d r^e `:1 flu
Tr )t group of pellets the 1 above pellets are nosed a r
load-n In-Lo o
v: ch group cos~xtoy- Tir Fe stI..a x. e~'gt v t e f -h- G ii
r 3 _ ~l i
prGc:. s 150 ~g of
~iupropi` n with mg. of-act-i-iie drug in the first group of pelts and 1 00
ling of active
=1;. bl
drug inn to se-fond groupThe pellet.. a:-- then aps:l enc ateu .7 lns.i, t"
Size aF
:
r ~~
opaque'light blue opaque capsules. The :oral weight of the formulation
(capsule
pellets) is 352 ME
The resulting bumropion capsules were then tested according - the
raccording to
U SP Xix1i dissolution test (tvp e 2. ba ket), at 50 rpm. at 3 7 C; in pH ?. ~
buf far and
found to have the following release profile:
Time (hours) % Released
1 28
2 40
3 501
4 58
6 70
8 80
2
CA 02796884 2012-11-27
%N O (121062299 PCT/US012103523
IO 83
I2 87
14 88
16 90
I8 90
20 92
The release profile of the con rolled release product of Example 3 is
shown r by the line ~zith the filled in squares, I'll. FIB. . ''
The resulting bupropion capsules were then tested according to USP
II st
e 2. :: :at 50 rpm at 37cC. i SG ip 1-1) an d f and
to have the following release
Times hours) % Released
2
3 4
4 6
6 12
8 21
40
12 57
13 64
16 68
17 71
74
23
CA 02796884 2012-11-27
BYO 021062299 PCTfUS02,03523
The release profile of the controlled release product shown in Example 3 is
shown in
FIG. 2 by the line with the filled in squares.
The bupropion capsules of Example 3 were then analyzed in a seven
patient test using techniques known in the art. Bupropion was first detected
in the
plasma about 1.4 hours after administration and showed a sustained release
over 24
hours.
The testing procedure is as described in Example 1. The results
provided are given in Table 4 and FIG. 4 and show that the mean plasma time
profile
of the bupropion formulation differs om that of Zyban . Bupropion had a
delayed
absorption, the relative 'ahabili-tv abill 0 bupropior2 to ZY can S was 48%
and 55` in
tern's Cma, and AUC Values, respectivei . The terminal elimination ha:11-lives
were
similar
TABLE _4
Parameter Example 3 Zvban G-Mean
Ratio
Cmax (ng rn) `6.9 114.8 0.48
-ALLFC2 - (ng=heml) 531.7 8895 0.59
Ti.. Chr: 1.4 0.1
Tura. (hr) 5.1 4.1
Ti,2 (hr) 12.6 14.1
Thus, again, the C,,,a, of the Example 3 product was reduced
significantly more than the AUC compared to the reference product,
demonstrating
that an effective once-a-day product has been provided.
24
CA 02796884 2012-11-27
EXAMPLE 4
The pellets from Ex- le 3 are taken as the second and third
componen hose pellets are loaded into the dosator along with active pellets
and
are filled into capsules in a ratio of 10:30:60 while maintaining the dosage
at 150 tug
bjsprcpion. The blood profiles from this example will show a C the same as in
Table 3, but wil= show a slightly increased AUC, thereby rendering the G-mean
ratio
at about LOO_ The amount of active pellets may be adjusted as is known in the
art
ithcut undue expenme :tom on based on ne teachings of the present disclosure
in
c- suos_ar_ ah pr_ iae a C- 'eai for AL or approximately _ O.