Note: Descriptions are shown in the official language in which they were submitted.
WO 2012/009217 CA 02804970 2013-01-09
PCT/US2011/043330
SPIROCYCLIC COMPOUNDS
TECHNICAL FIELD
The present invention is directed to novel spirocyclic compounds.
Specifically,
the compounds act as a diacylglycerol 0-acyltransferase type linhibitors
(hereinafter also
referred to as "DGAT1"), and can be useful in preventing, treating or acting
as a remedial agent
for hyperlipidemia, diabetes mellitus and obesity.
BACKGROUND
Metabolic syndrome is associated with obesity and is recognized as an upstream
risk factor for many conditions such as diabetes mellitus, lipidosis,
hypertension (Journal of
Japan Society for the Study of Obesity, Vol. 12, Extra Edition, 2006). Since
metabolic syndrome
is associated with an increase in the risks of arteriosclerosis,
cardiovascular disorder and
cerebrovascular disorder, treatment of obesity has been recognized to be
important for preventing
these diseases. Although the need to treat obesity is recognized to be
important, there are
extremely-limited drug therapies for obesity that are currently available, and
thus, the advent of
novel anti-obesity drugs having more definite action and few side-effects is
desired.
In general, obesity is caused by the accumulation triacylglycerol (TO) in
adipose
tissue which is a result of lack of exercise, intake of excessive calories and
ageing. In the body
there are two TO synthesis pathways, a glycerol phosphate pathway, which is
present in most
organs and causes de novo TG synthesis, and a monoacylglyeerol pathway, which
is involved
principally in absorption of aliphatic acid from the small intestine.
Diacylglycerol
acyitransferases (DGATs, EC 2.3.1.20), which are membrane-bound enzymes
present in the
endoplasmic reticulum, catalyze the final step of the TO synthesis common to
the two TO
synthesis pathways. The final reaction consists of transferring an acyl group
of aeyl-coenzyme A
to the 3-position of 1,2-diacylglyeerol to generate TO (Frog.' Lipid Res.,
43.134-176. 2004 and
Ann. Med., 36, 252-261, 2004). There are two subtypes of DGATs: DGAT I and
DGAT2.
There is no significant homology at the generic or amino acid level between
the DGAT1 and
DGAT2, which are encoded by different genes (ProcNatl.Acad.Sci.USA.,95,13018-
13023,1998
and JBC,276,38870-38876,2001). DGAT1 is present in the small intestine,
adipose tissue and
liver and is believed to be involved in lipid absorption in the small
intestine; lipid accumulation
in the fated!; and VLDL secretion and lipid accumulation in the liver
(Ann.Med.,36,252-
261,2004 and JBC,280,21506-21514,2005). In consideration of these functions, a
DGAT1
inhibitor is expected to be an effective obesity treatment through inhibition
of lipid absorption in
the small intestine, lipid accumulation in the adipose tissue and the liver,
and the lipid secretion
from the liver.
- 1 -
WO 2012/009217 CA 02804970 2013-01-09
PCT/US2011/043330
In order to carry out in vivo examination of the physiological function(s) of
DGAT1 and inhibitory activity against DGAT 1, DGAT1-knockout mice deficient in
DGAT1 at
the generic level was produced, and analyses thereof were conducted. The DGAT
1-knockout
mice have been found to have smaller fat masses than those of wild-type mice
and became
resistant to obesity, abnormal glucose tolerance, insulin resistance and fatty
liver due when fed a
high-fat diet (Nature Genetics,25,87-90,2000 and JCI,109,1049-1055,2002). In
addition, energy
expense has been reported to be accelerated in the DGAT1-knockout mice; and
transplantation of
the adipose tissues of DGAT1-knockout mice into wild-type mice has been
reported to make the
wild-type mice resistant to obesity and abnormal glucose tolerance, induced by
a high-fat diet
(JCI,111,1715-1722,2003 and Diabetes,53,1445-1451,2004). In contrast, obesity
and diabetes
mellitus due to a high-fat diet have been reported to worsen in mice with
overexpression of
DGAT1 in adipose tissue (Diabetes,51,3189-3195,2002 and Diabetes,54,3379-
3386,2005).
From the results, DGAT1 inhibitors are likely to be therapeutic drugs with
efficacy for obesity, type 2 diabetes mellitus, lipidosis, hypertension, fatty
liver, arteriosclerosis,
cerebrovascular disorder, coronary artery disease and metabolic syndrome,
associated with the
obesity.
SUMMARY OF THE INVENTION
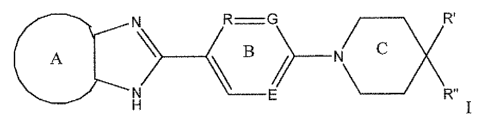
The present invention is directed to compounds of structural formula
A B C X
N) R"
or a pharmaceutically acceptable salt thereof, wherein A is independently
selected from the group
consisting of benzene, pyridine, pyrazine and pyrimidine;
R, G and E are independently selected from the group consisting of ¨N- and ¨CH-
,
wherein if one of R, G and E is ¨N-, the remaining two are ¨CH-;
Ri and R" together form ring D, wherein D is selected from the group
consisting of
cycloalkyl and heterocycloalkyl wherein A, B, C and D are independently
unsubstituted or
substituted with one or more substituents selected from the group a, CI-
C6alkyl, C3-
Cmcycloalkyl, aryl, heteroaryl, cycloheteroalkyl, Ci-C6alky1C3-Ciocycloalkyl,
C1-C6alkylaryl, C1-
C6alkylheteroaryl and C1-C6alkylcycloheteroalkyl, wherein CI-C6a1kyl, C3-
C1ocycloalkyl, aryl,
heteroaryl, cycloheteroalkyl, CI-C6alky1C3-Clocycloalkyl, CI-C6alkylaryl, Ci-
C6alkylheteroaryl
- 2 -
WO 2012/009217
CA 02804970 2013-01-09
PCT/US2011/043330
and CI-C6alkylcycloheteroalkyl are independently unsubstituted or substituted
with one or more
substituents selected from the group consisting of a;a is selected from the
group consisting of halogen, C1-C6alkyl, halogen-
substitutedE1-C6alkyl, COC1-E6alkyl, oxo, -OH, halogen-substitutedCi-
C6alkylOH, -0C1-
C6alkyl, -Ohalogen-substitutedCi-C6alkyl, -COOH, -COOCI-C6alkyl, -C1-
C6alkylCOOCi-
C6alkyl, -C1-C6alky1COOH, -0C1-C6alkylCOOH, -EN, Ci-C6alkylCN, -NO2, NH2, NHC1-
C6alkyl, N(C1-E6alky1)2, -NHCOOH, -NHCOOC1-C6alkyl, -CONH2, -CONHC 1-C6alkyl, -
CON(Ci-C6alky1)2, -CONHCI-C6alkyl-N(Ci-C6alky1)2, -NEISO2C1-C6alkyl, -SO2NH2, -
S02C1-
C6alkyl, C3-C10eycloalkyl, aryl, heteroaryl, cycloheteroalkyl,
cycloheteroalkylCOOH, C-
C6alky1C3-C10cycloalkyl, C1-C6alkylaryl, CI-C6allcylheteroaryl and C1-
C6alkylcycloheteroalkyl,
DETAILED DESCRIPTION OF THE INVENTION
Compounds The present invention
is directed to compounds of structural formula I:
A
B N/ C
R"
or a pharmaceutically acceptable salt thereof, wherein A is independently
selected from the group
consisting of benzene, pyridine, pyrazine and pyrimidine;
R, G and E are independently selected from the group consisting of -N- and -CH-
,
wherein if one of R, G and E is -N-, the remaining two are -CH-;
R and R" together form ring D, wherein D is selected from the group consisting
of
cycloalkyl and heterocycloalkyl wherein A, B, C and D are independently
unsubstituted or
substituted with one or more substituents selected from the group a, C1-
C6alkyl, C3-
Ciocycloalkyl, aryl, heteroaryl, cycloheteroalkyl, CI-C6alky1C3-C1ocycloalkyl,
C1-C6alkylaryl, C1-
C6alkylheteroaryl and C1-C6alkylcycloheteroalkyl, wherein C1-C6alkyl, C3-
C10cycloalkyl, aryl,
heteroaryl, cycloheteroalkyl, C1-C6alky1C3-C1ocycloalkyl, C1-C6alkylaryl, E1-
C6alkylheteroaryl
and C1-C6alkylcycloheteroalkyl are independently unsubstituted or substituted
with one or more
substituents selected from the group consisting of a;
substitutedE1-C6alkyl, COC1-C6alkyl, oxo, a is selected from the
group consisting of halogen, C1-C6alkyl, halogen-
halogen-substitutedQ-C6alkylOH, -0C1-
C6alkyl, -Ohalogen-substitutedC1-C6alkyl, -E0OH, -COOC1-C6alkyl, -C1-
C6alkylCOOCI-
C6alkyl, -C1-C6alkylCOOH, -0C1-C6a1ky1COOH, -CN, C1-C6alkylCN, -NO2, NH2, NHCI-
C6alkyl, -C6alkyl)2, -NHCOOH, -
NHCOOC 1-C6alkyl, -CONH2, -CONHC -C6alkyl,
-3-
WO 2012/009217 CA 02804970 2013-01-09
PCT/US2011/043330
CONCI-C6alicY1)2, -CONHC1-C6aikyi-N(C1-C6alkyl)2, -NBISO2CI-C6aikyi, -SO2N1-
12, -S02C1-
C6alkyl, C3-C1ocycloalkyl, aryl, heteroaryl, cycloheteroalkyl,
cycloheteroalkylCOOH, Ci-
C6alky1C3-Cioeycloalkyl, C1-C6alkylaryl, Ci-C6alkylheteroaryl and C1-
C6alkylcycloheteroalkyl.
In certain embodiments of the compounds described herein, A is selected from
the
group consisting of benzene, pyridine, pyrazine and pyrimidine. In some
embodiments A is
selected from the group consisting of benzene and pyridine. In other
embodiments, A is
benzene. In still other embodiments, A is pyridine. In yet other embodiments,
A is pyrazine. In
still other embodiments, A is pyrimidine. In some embodiments A is
unsubstituted. In other
embodiments, A is substituted.
In some embodiments, A is substituted with one or more substituents selected
from a. In certain embodiments, A is further substituted with one or more
substituents selected
from the group consisting of halogen, C1-C6alkyl,
-CN, SO2Me and halogen-
substitutedCI-C6alkyl. In some embodiments, A is further substituted with one
substituents
selected from the group consisting of halogen, CI-C6alkyl, -OCI-C6alkyl, -CN,
SO2Me and
halogen-substitutedC1-C6alkyl. In some embodiments, A is further substituted
with two
substituents selected from the group consisting of halogen, CI-C6alkyl, -OCI-
C6alkyl, -CN,
SO2Me and halogen-substitutedC1-C6alkyl. For example, A can be substituted
with one or more
halogens. Examples of halogens include, but are not limited to, chlorine,
bromine and fluorine.
In another example, A can be substituted with one or more Ci-C6alkyls.
Examples of C1-C6alkyl
include but are not limited to methyl and ethyl. A can also be substituted
with halogen-
substitutedCi-Colkyl. Examples of halogen-substituted Ci-C6alkyl, include but
are not limited
to, trifluoromethyl. In still other examples, A can be substituted with one or
more substituents
selected from the group consisting of methoxy, trifluoromethoxy, ¨OH, CN,
CH2CN, NHSO2Me
and SO2NH2. In yet other examples A is substituted with (C21-14)NHCOCH3 or
CONH(C2R4)N(C21-14)2.
In some embodiments, A is substituted with one or more substituents selected
from aryl, heteroaryl or cycloheteroalkyl, wherein aryl, heteroaryl or
cycloheteroalkyl are
unsubstituted or substituted with one or more substituents selected from a. In
certain
embodiments, A is substituted with phenyl. In other embodiments, A is
substituted with
pyridine. The pyridine can be unsubstituted or substituted with one or more
substituents selected
from the group consisting of halogen or halogen-substitutedCI-C6alkyl.
Suitable examples
include, but are not limited to, fluorine or trifluoromethyl. In other
embodiments, A is
substituted with pyrimidine. The pyrimidine can be unsubstituted or
substituted with one or
more substituents selected from the group consisting of halogen or halogen-
substitutedCi-
C6alkyl. Suitable examples include, but are not limited to, fluorine and
trifluoromethyl.
In still other embodiments, A is substituted with one or more substituents
selected
from aryl, heteroaryl or cycloheteroalkyl, wherein aryl, heteroaryl or
cycloheteroalkyl are fused to
- 4 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
A, forming a polycyclic ring structure with ring A and the pyrrole of formula
I. Suitable aryls,
heteroaryls or cycloheteroalkyls include phenyl, pyridine, pyrimidine and
triazole. Examples
include but are not limited to:
ei N N
N)
N
N
L
N\ )
N N
In certain embodiments, G is ¨N- and E is ¨CH-. In other embodiments, G is ¨
CH- and E is ¨N-. In other embodiments, G and E are both ¨CH-. In other
embodiments, R is ¨
N-, G and E are both ¨CH-. In certain embodiments, R is ¨CH-, G is ¨N- and E
is ¨CH-. In
other embodiments, R is ¨CH-, G is ¨CH- and E is ¨N-. In other embodiments, R,
G and E are
all ¨CH-. In still other embodiments, R is ¨CH- and G or E is ¨CH- and the
other is ¨N-.
In certain embodiments of the compounds described herein, B is selected from
the
group consisting of benzene, pyridine and pyrimidine. In some embodiments B is
selected from
the group consisting of benzene and pyridine. In other embodiments, B is
benzene. In still other
embodiments, B is pyridine. In still other embodiments, B is pyrimidine. In
some embodiments
B is unsubstituted. In other embodiments, B is substituted.In certain
embodiments of the compounds described herein, B is substituted with
one or more substituents selected from the group consisting of a. In some
embodiments, B is
substituted with one or more substituents selected from the group consisting
of halogen, CI-
C6alkyl and halogen-substitutedCi-Coalicyl. Examples of suitable halogens
include, but are not
limited to chlorine- and fluorine. Examples of suitable C1-C6alkyl include,
but are not limited to
methyl and ethyl and examples of suitable halogen-substitutedC1-C6alkyl
include, but are not
limited to trifluoromethyl.
Suitable examples of ring B and its substituents include, but are not limited
to,
CI CI
CI CI
111 and
/
Ring C as shown in formula I can be substituted or unsubstituted. In certain
embodiments, C is unsubstituted. In certain embodiments of the compounds
described herein, C
- 5 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
is substituted with one or more substituents selected from the group
consisting of a. In some
embodiments, C is substituted with one or more substituents selected from the
group consisting
of halogen, C1-C6alkyl and halogen-substitutedCi-C6alkyl. Examples of suitable
halogens
include, but are not limited to chlorine and fluorine. Examples of suitable Ci-
C6alkyls include,
but are not limited to methyl and ethyl and examples of suitable halogen-
substitutedC1-C6alkyls
include, but are not limited to trifluoromethyl.
As shown in formula I, R' and R" together form ring D. In certain embodiments
of the compounds described herein, D is cycloalkyl or heterocycloalkyl. In one
embodiment, D
is cycloalkyl, Suitable examples of cycloalkyl include but are not limited to
cyclohexane and
bycyclic cycloalkyls, such as,
S.
In another embodiment, D is heterocycloalkyl. For example, in one embodiment,
D is
\1õAN 11
wherein T is selected from the group consisting of¨O-, -CH2-, -NH and ¨NCI-
C6alkyl-; and V,
U, Q and W are independently selected from the group consisting of ¨N-, -C-
and ¨CH-. In one
embodiment, T is ¨0- and V, U, Q and W are ¨CH-. In another embodiment, T is 0
and one of
V, U, Q and W is N and the remaining variables are ¨CH-. In yet another
embodiment, T is
N(CH3) and V, U, Q and W are ¨CH-. In yet another embodiment, T is ¨CI-12- and
V , U, Q and
W are ¨CH-.
In certain embodiments of the compounds described herein, D can be selected
from the group consisting of:
0 0 0 0 0
Ra N
NM÷
/P
fb, eRas and Vir
- 6 -
WO 2012/009217 CA 02804970
2013-01-09
PCT/US2011/043330
wherein Ra is selected from the group consisting of H and CI-C6alkyl. In
certain embodiments,
D, when selected from the group above can be unsubstituted. In more particular
embodiments,
D, when selected from the group above, can be further substituted with an oxo
group, such as
=O. In other embodiment, when D is selected from the group above, D can be
substituted with
one or more substituents selected from the group consisting of oxo, -OH, -
COOH, -COOCI-
C6alkyl, halogen, C1-C6alkyl, C1-C6alkylCOOH, Ci-C6alkylCN, cyclopropyl,
halogen-
substitutedCi-C6alkyl and C1-C6alkyltriazole, wherein the triazole is
substituted with methyl.
In particular embodiments, D, when selected from the group above, can be
further
substituted with a C1-C6alkylaryl group. In certain embodiments, D is
substituted with a ¨CH2-
phenyl, wherein the ¨CI-12-phenyl is unsubstituted. In certain embodiments, D
is substituted with
¨CH2-phenyl, wherein the ¨CH2-phenyl is substituted with one or more
substituents selected
from the group consisting of halogen, Ci-C6alkyl and halogen-substitutedCi-
C6alkyl.
In other embodiments of the compounds described herein, D is selected from the
group consisting of:
X
wherein X, Y and Z are independently selected from the group consisting of ¨C-
, -CH-, -CH2-, -
N-, -NH- and ¨0-. In one embodiment, Z is N, X is CH2 and Y is ¨CO. In another
embodiment,
X is 0, Z is N and Y is ¨CO. In yet another embodiment, X and Z are both ¨NH.
In still another
embodiment, Z is ¨N- and X is NH.
In certain embodiments of the compounds described herein, D is selected from
the
group consisting of:
0
> ' j and\c)
In certain embodiments, D is substituted with one or more oxo groups. For
example, D can be
substituted with one =0. In particular embodiments, D, when selected
from the group above, can be farther
substituted with an aryl or heteroaryl group. In certain embodiments, D is
substituted with a
phenyl ring, wherein the phenyl is unsubstituted. In certain embodiments, D is
substituted with a
phenyl ring, wherein the phenyl is substituted with one or more substituents
selected from the
group consisting of ¨COOH, COOCI-C6alkyl, heterocycloalkyl-COOH, halogen, Ci-
C6alkyl,
ha1ogen-substitutedC1-C6alkyl, -OCI-C6alky1C001-1, NO2, -CN, C1-C6alkylCOOH,
C1-
C6alkylCOOC i-C6alkyl and -CF3OH. In certain embodiments, D is substituted
with a pyridine
ring, wherein the pyridine is unsubstituted. In certain embodiments, D is
substituted with a
- 7 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
pyridine ring, wherein the pyridine is substituted with one or more
substituents selected from the
group consisting of halogen, C1-C6alkyl and halogen-substitutedC1-C6alkyl.
In still other embodiments of the compounds described herein D is selected
from
the group consisting of:
1110 and 101
P is selected from the group consisting of-0- or -CH2-. In one embodiment, P
is -0-. In
another embodiment, P is -CH2-. In particular embodiments, wherein D is
selected from the
group above, D is substituted with one or more substituents selected from the
group consisting of
-COOH, oxo, -C1-C6alkylCOOH and NHSO2Me. In one embodiment, D is cyclohexane,
wherein the cyclohexane is substituted with -COOH. In another embodiment, D is
cyclohexane,
wherein the cyclohexane is substituted with -CI-C6alkylCOOH.
In yet another embodiment of the compounds described herein, D is selected
from
the group consisting of:
0 0
cs-05 cs=SSO cs-5S cs-550
I
, 0 0
and -"0"-
In particular embodiments, wherein D is selected from the group above, D is
substituted with one or more substituents selected from the group consisting
of -COOH and -C1-
C6alkylCOOH. Examples of D and its substituents include, but are not limited
to,
0 R\
OR /\--'0H
artd [XI)
0 0 0
In of the above described embodiments of D, D can be substituted with one or
more substituents selected from the group consisting of a. In certain
embodiment, any of the
above described embodiments of D, D can be substituted with one or more
substituents selected
from the group consisting of halogen, C1-C6alkyl, halogen-substitutedCI-
C6alkyl, oxo, -OH, -
COOH, -COOCI-C6alkyl, -C1-C6alkylCOOCI-C6alkyl, -C1-C6alkylCOOH, -0C1-
C6alkylCOOH,
-CN, Ci-C6alkylCN and -NHSO2CI-C6alkyl.
Also described herein are compounds of formula Ia:
Ri
R2
--NH)
Ia
- 8 -
WO 2012/009217 CA 02804970 2013-01-09
PCT/US2011/043330
or a pharmaceutically acceptable salt thereof, wherein G and E are
independently selected from
the group consisting of ¨N- and ¨CH-, wherein if one of G and E is ¨N-, the
remaining one is ¨
CH-;
RI is selected from the group consisting of halogen or halogen-substitutedCi-
C6alkyl;
D is a C3-Cl0cycloa1kyl; and
R2 is selected from the group consisting of hydrogen, C1-C6alkyl, halogen-
substitutedCi -C6alkyl, COCI-C6alkyl, -OH, oxo, halogen-substitutedCI-
C6alkylOH,
-Ohalogen-substitutedC1-C6alkyl, -COOH, -COOC1 -C6alkyl, -C1-C6alkylCOOCi-
C6alkyl, -C1-C6alkylCOOH and -OCI-C6alkylCOOH.
In certain embodiments, G is ¨N- and E is ¨CH-. In other embodiments, G is ¨
CH- and E is ¨N-. In other embodiments, G and E are both ¨CH-.
In other embodiments of the compounds described herein, D is cycloalkyl
wherein
the cycloalkyl is selected from the group consisting of:
ONO land
In certain embodiments, D is cyclohexane.
In some embodiments, R1 is halogen. Suitable examples of halogen include, but
are not limited to, chlorine, fluorine and bromine. In some embodiments, RI is
halogen-
substitutedCI-C6alkyl. Suitable examples of halogen-substitutedCi-C6alkyl
include, but are not
limited to, trifluoromethyl.
In certain embodiments, R2 is selected from the group consisting of -COOH, -
COOCI-C6alkyl, -C1-C6alkylCOOC1-C6allcyl and -C1-C6alkylCOOH. For example, in
some
embodiments R2 is selected from the group consisting of ¨COOH and -C1-
C6alkylCOOH.
Examples of -C1-C6alkylCOOH include, but are not limited to, -CH2COOH.
Ri Aldo described herein are compounds of formula Ia:
R2
\CM
N> Ia
or a pharmaceutically acceptable salt thereof, wherein G and E are
independently selected from
the group consisting of ¨N- and ¨CH-, wherein if one of G and E is ¨N-, the
remaining one is ¨
CH-; Rl is selected from the group consisting of halogen or
halogen-substitutedCI-
C6alkyl;
- 9 -
WO 2012/009217
CA 02804970 2013-01-09
PCT/US2011/043330
D is a heterocycloalkyl; and
R2 is selected from the group consisting of hydrogen, halogen, C1-C6alky1,
halogen-substitutedC1-C6alkyl, COCI-C6alkyl, oxo, -OH, halogen-substitutedCi-
C6alkylOH, -
0C1-C6alkyl, -Ohalogen-substitutedCi-C6alkyl, -COOH, -COOC1-C6alkyl, -C1-
C6alkylCOOC1-
C6alkyl, -C1-C6alkylCOOH, -0C1-C6alkylCOOH, -CN, C1-C6alkylCN, -NO2, NH2, NHCI-
C6alkyl, N(C1-C6alky1)2, -NHCOOH, -NHCOOCI-C6alkyl, -CONH2, -CONHCE-C6alkyl, -
CON(C1-C6alky1)2, -NHSO2C1-C6alkyl, -S02C1-C6alkyl, C3-Ciocycloalkyl, aryl,
heteroaryl,
cycloheteroalkyl, cycloheteroalkylCOOH, C1-C6alky1C3-Ciocycloalkyl, C1-
C6alkylaryl, CI-
Coalkylheteromyl and CI-Colkylcycloheteroalkyl.
In certain embodiments, G is ¨N- and E is ¨CH-. In other embodiments, G is ¨
CH- and E is ¨N-. In other embodiments, G and E are both ¨CH-.
In other embodiments of the compounds described herein, D is heterocycloalkyl
wherein the heterocycloalkyl is selected from the group consisting of: o
0
0 0
0
0
>
4itt, = =
'
0 and
In certain embodiments, D is selected from the group consisting of:=
0 0
0 0
\\ N
and \
In certain embodiments, D is selected from the group consisting of:
fa and
In certain embodiments, D is selected from the group consisting of:
- 10 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
, >0 and
In some embodiments, RI is halogen. Suitable examples of halogen include, but
are not limited to, chlorine, fluorine and bromine. In some embodiments, RI is
halogen-
substitutedCi-C6allcyl. Suitable examples of halogen-substitutedCI-C6alkyl
include, but are not
limited to, trifluoromethyl.
In certain embodiments, R2 is selected from the group consisting of halogen,
C1-
halogen-substitutedCi-C6alkyl, oxo, -COOH, -COOCI-C6alkyl, -C1-C6alkylCOOCi-
C6alkyl, -Ci-C6alkylCOOH, -0C1-C6alkylCOOH, -CN, C1-C6alkylCN and C1-
C6alkylcyclopropyl. In other embodiments R2 is selected from the group
consisting of hydrogen,
halogen, CI -C6alkyl, halogen-substitutedCi-C6alkyl, oxo, -OH, halogen-
substitutedCi-
C6alkylOH, -COOH, -COOCI-C6alkyl, -C1-C6alkylCOOCI-C6alkyl, -Ci-C6alkylCOOH, -
0C1-
C6alkylCOOH, -CN, C1-C6alkylCN, -NO2, -NHSO2C1-C6alkyl, -S02C1-C6alkyl, C3-
Ciocycloalkyl and cycloheteroalkylCOOH. In another embodiment, R2 is hydrogen.
In still other
embodiments, R2 is selected from the group consisting of phenyl or pyridine,
wherein the phenyl
or pyridine is unsubstituted or substituted. In one embodiment, R2 is phenyl.
In one
embodiment, R2 is pyridine. In certain embodiments, the phenyl or pyridine can
be substituted
with one or more substituents selected from the group consisting of halogen,
Ci-C6alkyl,
halogen-substitutedC1-C6alkyl, -COOH, -COOC1-C6alkyl, -CI-C6alkylCOOCI-
C6alkyl, -C1-
C6alkylCOOH, -0C1-C6alkylCOOH and ¨CN.
Also described herein are compounds of formula Ib:
R3
1/,\,/ // 'R4
R5 U " lb
or a pharmaceutically acceptable salt thereof, wherein G and E are
independently selected from
the group consisting of ¨N- and ¨CH-, wherein if one of G and E is ¨N-, the
remaining one is -
CH-;
T is selected from the group consisting of¨O-, -CH2-, -NH- and ¨NC1-C6alkyl-;
V, U, Q and W are independently selected from the group consisting of ¨N-, -C-
and ¨CH-;
-11-
WO 2012/009217 CA 02804970 2013-01-09 PCT/US2011/043330
R1 is selected from the group consisting of halogen or halogen-substitutedC1-
C6alkyl; and
R3, R4 and R5 are independently selected from the group consisting of
hydrogen,
halogen, C1-C6alkyl, halogen-substitutedCi-C6alkyl, COCI-C6alkyl, oxo, -OH,
halogen-
substitutedC1-C6alkylOH, -OCI-C6alkyl, -Ohalogen-substitutedCi-C6alkyl, -COOH,
-COOCI-
C6alkyl, -C1-C6alkylCOOCI-C6alkyl, -C1-C6alkylCOOH, -0C1-C6alkylCOOH, -CN, C1-
C6alkylCN, -NO2, NH2, NHC1-C6alkyl, N(Ci-C6alkyl)2, -NFICOOH, -NHCO0C1-
C6alkyl, -
CONH2, -CONHC -C6alkyl, -CON(C -C6alky1)2, -NHSO2C -C6alkyl, -S02C1-C6alkyl,
C3-
C locycloalkyl, aiyl, heteroaryl, cycloheteroalkyl, cycloheteroalkylCOOH, Ci-
C6alkyl C3-
1 0 Ciocycloalkyl, C1-C6alkylatyl, Ci-C6alkylheteroaryl and C1-
C6alkyleycloheteroalkyl, wherein the
C3-C10cycloalkyl, aryl, heteroaryl, cycloheteroalkyl, cyclobeteroalky1C001-1,
C1-C6alky1C3-
C10cycloalkyl, Ci-C6alkylatyl, C1-C6alkylheteroaly1 and C1-
C6alkylcycloheteroalkyl are
unsubstituted or substituted with at least one substituent selected from the
group consisting of
halogen.
In certain embodiments, G is -N- and E is -CH-. In other embodiments, G is -
CH- and E is -N-. In other embodiments, G and E are both -CH-.
In certain embodiment, T is -0-. In other embodiments, T is -C112-. In still
other
embodiments, T is -NH-. In yet other embodiment, T is -NCH3.
In some embodiments, RI is halogen. Suitable examples of halogen include, but
are not limited to, chlorine, fluorine and bromine. In some embodiments, R1 is
halogen-
substitutedC1-C6alkyl. Suitable examples of halogen-substitutedCi-C6alkyl
include, but are not
limited to, trifluoromethyl.
In certain embodiments, one of V, U, Q or W is -N- and three of V, U, Q or W
are selected from the group consisting of -C- and -CH-. In other embodiments,
all of V, U, Q
and W are -CH-.
R3 is independently selected from the group consisting of hydrogen, halogen,
C1-
C6alkyl, halogen-substitutedCi-C6alkyl, COCI-C6alkyl, oxo, -OH, halogen-
substitutedC1-
C6alkylOH, -OCI-C6alkyl, -Ohalogen-substitutedCi-C6alkyl, -COOFI, -COOC1-
C6alkyl, -C1-
C6alkylCOOCI-C6alkyl, -C1-C6alkylCOOH, -0C1-C6alky1C0011, -CN, C1-C6alkyleN, -
NO2,
NH2, NHCI-C6alkyl, N(C1-C6alky1)2, -NHCOOH, -NHCOOC1-C6alkyl, -CONH2, -CONHCI-
C6alkyl, -CON(C1-C6alky1)2, -NHSO2C1-C6alkyl, -S02CI-C6alkyl, C3-
C10cycloalkyl, aryl,
heteroaryl, cycloheteroalkyl, cycloheteroalkylCOOH, C1-C6alky1C3-
C1ocycloalkyl, C1-
C6alkylaryl, CI-C6alkylheteroaryl and CI-C6alkyleycloheteroalkyl, wherein the
C3-C10cycloalkyl,
aryl, heteroaryl, cycloheteroalkyl, cycloheteroalkylCOOH, CI -C6alky1C3-
Ciocycloalkyl, C1-
C6alkylaryl, CI-C6alkylheteroaryl and Ci-C6alkylcycloheteroalkyl are
unsubstituted or substituted
with at least one sub stituent selected from the group consisting of halogen.
In certain
embodiments, R3 is selected from the group consisting of hydrogen, oxo, CI-
C6alkyl, -COOH, -
-12-
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
CI-C6alkylCOOH, CI-C6alkyleN, -S02C1-C6alkyl, cyclopropyl, C1-C6alkylaryl, and
C1-
C6alkylcycloheteroalkyl, wherein the C1-C6alkylaryl and Ci-
C6alkylcycloheteroalkyl are
unsubstituted or substituted with at least one substituent selected from the
group consisting of
halogen. In other embodiments, R3 is independently selected from the group
consisting of
hydrogen, halogen, C1-C6alkyl, halogen-substitutedCi-C6alkyl, COC)-C6alkyl, -
OH, -COOH, -
COOC1-C6alkyl, -C1-C6alkylCOOCi-C6alkyl, Ci-C6alkylCN and -C1-C6alkylCOOH. In
one
embodiment, R3 is oxo. In another embodiment R3 is hydrogen. In still another
embodiment, R3
is C1-C6alkylphenyl, wherein the phenyl is unsubstituted or substituted with a
halogen or -
COOH.
R4 is independently selected from the group consisting of hydrogen, halogen,
C1-
C6alkyl, halogen-substitutedCI-C6alkyl, COC1-C6alkyl, oxo, -OH, halogen-
substitutedCi-
C6alkylOH, -0 Ci-C6alkyl, -Ohalogen-substitutedCI-C6alkyl, -COOH, -COOCI-
C6alkyl, -C 1-
C6alkylCOOCI-C6alkyl, -Ci-C6alkylCOOH, -0C1-C6alkylCOOH, -CN, C1-C6alkylCN, -
NO2,
NH2, NHC1-C6alkyl, N(C1-C6alky1)2, -NICOOH, -NHCOOCI-C6alkyl, -CONH2, -CONHC1-
C6alkyl, -CON(CI-C6alkyl)2, -NHSO2CI-C6alkyl, -S02C1-C6alkyl, C3-
Ciocycloalkyl, aryl,
heteroaryl, cycloheteroalkyl, cycloheteroalkylCOOH, C1-C6alky1C3-
C1ocycloalkyl, C1-
C6alkylatyl, CI-C6alkylheteroaryl and CI -C6allcylcyeloheteroalkyl, wherein
the C3-C10cycloalkyl,
aryl, heteroaryl, cycloheteroalkyl, cycloheteroalkylCOOH, C1-C6alky1C3-
C1ocycloalkyl, C1-
C6alkylaryl, Ci-C6alkylheteroaryl and C1-C6alkyleyeloheteroalkyl are
unsubstituted or substituted
with at least one substituent selected from the group consisting of halogen.
In other
embodiments, R4 is independently selected from the group consisting of
halogen, C1-C6alkyl,
halogen-substitutedC1-C6alkyl, COC1-C6alkyl, -OH, -COOH, -COOCI-C6alkyl, -C1-
C6alkylCOOCI-C6alkyl and -C1-C6a1kylCOOH. In another embodiment, R4 is
hydrogen.
R5 is independently selected from the group consisting of hydrogen, halogen,
C1-
C6alkyl, halogen-substitutedC1-C6alkyl, COC1-C6alkyl, oxo, -OH, halogen-
substitutedCI-
C6alkylOH, -0C1-C6alkyl, -Ohalogen-substitutedCi-Colkyl, -COOH, -COOCI-
C6alkyl, -C1-
C6alkylCOOCI-C6alkyl, -C1-C6alkylCOOH, -0C1-C6alkylCOOH, -CN, Ci-C6alkylCN, -
NO2,
NH2, NHC1-C6alkyl, N(C1-C6alky1)2, -NHCOOH, -NHCOOCI-C6alkyl, -CONH2, -CONHC1-
C6alkyl, -CON(C1-C6alky1)2, -NHSO2C1-C6alkyl, -S02C1-C6alkyl, C3-
C1ocycloalkyl, aryl,
heteroaryl, cycloheteroalkyl, cycloheteroalkylCOOH, C1-C6alky1C3-
C1ocycloalkyl, C1-
C6alkylaryl, CI-C6alkylheteroaryl and C1-C6alkylcycloheteroalkyl, wherein the
C3-C10cycloalkyl,
aryl, heteroaryl, cycloheteroalkyl, cycloheteroakICOOH, CI-C6alky1C3-
C1ocycloalkyl, C1-
C6alkylatyl, C1-C6alkylheteroaryl and C1-C6alkyleyeloheteroalkyl are
unsubstituted or substituted
with at least one substituent selected from the group consisting of halogen.
In certain
embodiments, R5 is selected from the group consisting of hydrogen, oxo, C1-
C6alkyl, -COOH, -
C1-C6alkylCOOH, CI-C6alkylEN, -S02C1-C6alkyl, cyclopropyl, CI-Colkylaryl, and
C1-
C6alkyleyeloheteroalkyl, wherein the C1-C6alkylaryl and C1-
C6alkylcycloheteroalkyl are
- 13 -
WO 2012/009217 CA 02804970 2013-01-09
PCT/US2011/043330
unsubstituted or substituted with at least one substituent selected from the
group consisting of
halogen. In other embodiments, R5 is independently selected from the group
consisting of
halogen, C1-C6alkyl, halogen-substitutedCi-C6alkyl, COCi-C6alkyl, -OH, -COOH, -
COOCi-
C6alkyl, -Cl-C6alkylCOOCI-C6alkyl and -C1-C6alkylCOOH. In one embodiment, R5
is
hydrogen. Also described herein are compounds formula Ic:
R1
R 6
Xm R7 " R8 IC
or a pharmaceutically acceptable salt thereof, wherein G and E are
independently selected from
the group consisting of -N- and -CH-, wherein if one of G. and E is -N-, the
remaining one is -
CH-;
K, L and M are independently selected from the group consisting of-O-, -CH2-,
-CH-, -C- and -N -;RI is selected from the group consisting of halogen or
halogen-substitutedCi-
C6alkyl; and
R6, R7 and R8 are independently selected from the group consisting of
hydrogen,
halogen, C1-C6alkyl, halogen-substitutedCi-C6alkyl, COCI-C6alkyl, oxo, -OH,
halogen-
substitutedC1-C6alkylOH, -0C1-C6alkyl, -Ohalogen-substitutedC1-C6alkyl, -COOH,
-COOC1-
-C1-C6alkylCOOCI-C6alkyl, -C1-C6alkylCOOH, -0C1-C6alkylCOOH, -CN,
ColkylCN, -NO2, NH2, NHCI-C6alkyl, N(C1-C6alky1)2, -NHCOOH, -NHCOOC1-C6alkyl, -
CONH2, -CONHC1-C6alkyl, -CON(C1-C6alky1)2, -NHSO2C1-C6alkyl, -S02C1-C6alkyl,
C3-
C1ocycloalkyl, aryl, heteroaryl, cycloheteroalkyl, cycloheteroalkylCOOH, CI-
C6alkylC3-
CI ocycloalkyl, C1-C6alkylaryl, C1-C6alkylheteroaryl and C1-
C6alkylcycloheteroalk-yl, wherein the
C3-C1ocycloalkyl, aryl, heteroaryl, cycloheteroalkyl, cycloheteroalkylCOOH, C1-
C6alkylC3-
C1 ocycloalkyl, C1-C6alkylaryl, C1-C6alkylheteroaryl and CI-
Colkylcycloheteroalkyl are
unsubstituted or substituted with at least one substituent selected from the
group consisting of
halogen.
In certain embodiments, G is -N- and E is -CH-. In other embodiments, G is -
CH- and E is -N-. In other embodiments, G and E are both -CH-.
In some embodiments, RI is halogen. Suitable examples of halogen include, but
are not limited to, chlorine, fluorine and bromine. In some embodiments, RI is
halogen-
- 14 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
substitutedCi-C6alkyl. Suitable examples of halogen-substitutedCi-C6alkyl
include, but are not
limited to, trifluoromethyl.
In certain embodiments, J is ¨CH2-. In other embodiments, J is ¨0-. In still
other
embodiments, K is CO. In yet another embodiment, K is ¨N-. In one embodiment,
M is ¨NH-.
In anther embodiment, N is ¨CHR8-. In other embodiments, L is N(R7). In
certain embodiments,
J is ¨0-; K is ¨C(R6), wherein R6 is oxo; L is N(R7), wherein R7 is defined
above; and M is ¨
CH(R8), wherein R8 is hydrogen. In other embodiments, J is ¨0-; K is ¨N-; L is
C(R6); and M is
¨C(R7)( R8), wherein R6, R7 and R8 are hydrogen.
In certain embodiments, R6 is oxo. In other embodiments, R7 is hydrogen. In
still
other embodiments R7 is selected from the group consisting of phenyl or
pyridine, wherein the
phenyl or pyridine is unsubstituted or substituted. In one embodiment, R7 is
phenyl. In one
embodiment, R7 is pyridine. In certain embodiments, the phenyl or pyridine can
be substituted
with one or more substituents selected from the group consisting of halogen,
C1-C6alkyl,
halogen-substitutedCi-C6alkyl, heterocyclicalkylCOOH, -COOH, -COOCI-C6alkyl, -
C1-
C6alkylCOOCi-C6alkyl, -CI-C6alkylCOOH, -0C1-C6alkylCOOH, NO2 and ¨CN. In still
other
embodiments, R8 is hydrogen.
Also described herein are compounds of formula Id:
RI N)
N
110
R9 Id
or a pharmaceutically acceptable salt thereof, wherein G and E are
independently selected from
the group consisting of ¨N- and ¨CH-, wherein if one of G and E is ¨N-, the
remaining one is ¨
CH-;
J selected from the group consisting of¨O- or ¨CH2-;
RI is selected from the group consisting of halogen or halogen-substitutedCi-
C6alkyl; and
R9 is selected from the group consisting of hydrogen, Ci-C6alkyl, halogen-
substitutedC1-C6alkyl, COCI-C6alkyl, -OH, halogen-substitutedCi-C6alkylOH, -
0C1-C6alkyl, -
Ohalogen-substitutedCI-C6alkyl, -CO OH, -COOC -C6alkyl, -C -C6alkylCOOC -
C6alkyl, -C 1-
C6alkylCOOH, -0C1-C6alkylCOOH, -CN, CI-C6alkylCN, -NO2, -S02C1-C6alkyl, C3-
C ocycloalkyl, aryl, heteroaryl, cycloheteroalkyl, cycloheteroalkylCOOH, Ci-
C6alkyIC3-
- 15 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
Caocycloalkyl, Ci-C6alkylaryl, Ci-Colkylheteroaryl and C1-
C6alkylcycloheteroalkyl, wherein the
C3-C1ocycloalkyl, aryl, heteroaryl, cycloheteroalkyl, cycloheteroalkylCOOH, C1-
C6alky1C3-
Ciocycloalkyl, C1-C6alkylaryl, C1-C6alkylheteroaryl and C1-
C6alky1cycloheteroa1ky1 are
unsubstituted or substituted with at least one substituent selected from the
group consisting of -
COOH.
In certain embodiments, G is -N- and E is -CH-. In other embodiments, G is -
CH- and E is -N-. In other embodiments, G and E are both -CH-.
In certain embodiments, J is -0-. In other embodiments, J is -CH-.
In some embodiments, Rl is halogen. Suitable examples of halogen include, but
are not limited to, chlorine, fluorine and bromine. In some embodiments, RI is
halogen-
substitutedC1-C6alkyl. Suitable examples of halogen-substitutedC1-C6alkyl
include, but are not
limited to, trifluoromethyl.
In certain embodiments, R9 is Ci-C6alkyl, halogen-substitutedCi-C6alkyl, -OH,
halogen-substitutedC1-C6alkylOH, -COOH, -COOCI-C6alkyl, -Cj-C6alkylCOOCj-
C6alkyl, -C1-
C6a1kylCOOH, -0C1-C6alkylCOOH, -CN, Ci-C6alkylCN, -NO2 and cycloheteroalkyl,
wherein
the cycloheteroalkyl is substituted with -COOH. In other embodiments, R9 is -
COOH or -C1-
C6alkylCOOH.
Also described herein are compounds of founula le: Rio
Ri G>
N)
Ie
or a pharmaceutically acceptable salt thereof, wherein G and E are
independently selected from
the group consisting of -N- and -CH-, wherein if one of G and E is -N-, the
remaining one is -
CH-;
P selected from the group consisting of-O- or -CH2-;
RI is selected from the group consisting of halogen or halogen-substitutedCr
C6alkyl;
RI is selected from the group consisting of hydrogen, Ci-C6alkyl, halogen-
substitutedCi-C6alkyl, -OH, oxo, halogen-substitutedCi-C6alkylOH, -COOH, -
COOC1-C6alkyl, -
C1-C6alkylCOOCI-C6alkyl, -C1-C6alky1COOH and -0C1-C6alkylCOOH; and
Ri1 is selected from the group consisting of halogen, Ci-C6alkyl, halogen-
substitutedC1-C6alkyl, COCI-C6alkyl, oxo, -OH, halogen-substitutedCi-
C6alkylOH, -0C1-
C6alkyl, -Ohalogen-substitutedCi-C6alkyl, -COOH, -COOCI-C6alkyl, -C1-
C6alkylCOOC1-
C6alkyl, -C1-C6alkylCOOH, -0C1-C6alky1C001-1, -CN, C1-C6alkylCN, -NO2, NH2,
NHCi-
- 16 -
WO 2012/009217
CA 02804970 2013-01-09
PCT/US2011/043330
C6alkyl, N(CI-C6alky1)2, -NHCOOH, -NHCOOCI-C6alkyl, -CONH2, -CONHCI-C6alkyl, -
CON(C1-C6alky1)2, -NHSO2CI-C6alkyl, -S02C1-C6a1ky1, C3-C10cycloalkyl, aryl,
heteroaryl,
cycloheteroalkyl, cycloheteroalkylCOOH, -C6alkyIC3-Ciocycloalkyl, Ci-
C6alkylaryl, C1-
C6alkylheteroaryl and C1-C6alkyleycloheteroalkyl.In certain embodiments, G is -
N- and E is -CH-. In other embodiments, G is -
CH- and E is -N-. In other embodiments, G and E are both -CH-.
In certain embodiments, P is -0-. In other embodiments, P is -C1-12-.
In some embodiments, R1 is halogen. Suitable examples of halogen include, but
are not limited to, chlorine, fluorine and bromine. In some embodiments, R1 is
halogen-
substitutedCi-C6alkyl. Suitable examples of halogen-substitutedC1-C6alkyl
include, but are not
limited to, trifluoromethyl.
In certain embodiments, RI is selected from the group consisting of oxo and -
COOH. In other embodiments, RI is hydrogen.
In certain embodiments, R" is selected from the group consisting of hydrogen.
In
other embodiments, R" is selected from the group consisting of -COOH, -COOCI-
C6alkyl, -C1-
C6alkylCOOCI-C6alkyl, -Ci-CaalkylCOOH, -0C1-C6alkylCOOH, -NHCOOH, -NHCOOCI-
C6alkyl, -CONH2, -CONHCI-C6alkyl, -CON(Ci-C6alky1)2, -NHSO2C1-C6alkyl and -
S02C1-
C6alkyl.
Also described herein are compounds of formula I(f):
N
A )
1\14\
H
/)p12
Kt)
or a pharmaceutically acceptable salt thereof, wherein G and E are
independently selected from
the group consisting of -N- and -CH-, wherein if one of G and E is -N-, the
remaining one is -
CH-;
A is selected from the group consisting of benzene and pyridine, wherein A is
unsubstituted or substituted with one or more substituents selected from the
group a, C1-C6alkyl,
C3-Ciocycloalkyl, aryl, heteroaryl, cycloheteroalkyl, CI-C6alkyIC3-
Ciocycloalkyl, C1-C6alkylaryl,
C1-C6alkylheteroaryl and C1-C6alkylcycloheteroalkyl, wherein Ci-C6alkyl, C3-
C10cycloalkyl,
aryl, heteroaryl, cycloheteroalkyl, C1-C6alkylC3-C1ocycloalkyl, Ci-
C6alkylaryl, C1-
C6alkylheteroaryl and C1-C6alkylcycloheteroalkyl are independently
unsubstituted or substituted
with one or more substituents selected from the group consisting of a;
a is selected from the group consisting of halogen, CI-C6alkyl, halogen-
substitutedC1-C6alkyl, COC1-C6alkyl, oxo, -OH, halogen-substitutedCi-ColkylOH,
-0C1-
C6alkyl, -Ohalogen-substitutedCi-C6alkyl, -COOH, -COOC1-C6alkyl, -C1-
C6alkylCOOC1-
C6alkyl, -C1-C6alkylCOOH, -OCI-C6alkylCOOH, -CN, C1-C6alkylCN, -NO2, NH2, NHC1-
- 17 -
WO 2012/009217 CA 02804970 2013-01-09 PCT/US2011/043330
C6alkyl, N(C1-C6alkyl)2, -NHCOOH, -NHCOOC1-C6alkyl, -CONH2, -CONHCI-C6alkyl, -
CON(C1-C6alky1)2, -CONHCI-C6alkyl-N(C1-C6alkyl)2, -NHSO2C1-C6alkyl, -SO2NH2, -
S02C1-
C6alkyl, C3-C10cycloalkyl, aryl, heteroaryl, cycloheteroalkyl,
cycloheteroalkylCOOH, C1-
C6alkyIC3-C10cycloalkyl, C1-C6alkylalyl, Ci-C6alkylheteroatyl and C1-
C6alkylcycloheteroalkyl;
D selected from the group consisting of: csss. 0
0 and
;
R12 is selected from the group consisting of -COOH, -COOC1-C6alkyl, -C1-
C6alkylCOOC1-C6alkyl, -C1-C6alkylCOOH and -0C1-C6alkylCOOR
In certain embodiments, G is -N- and E is -CH-. In other embodiments, G is -
CH- and E is -N-. In other embodiments, G and E are both -CH-.
Also described herein are compounds of formula Ig:
A ) N
H \ R12Ig
or a pharmaceutically acceptable salt thereof, wherein 0 and E are
independently selected from
the group consisting of -N- and -CH-, wherein if one of G and E is -N-, the
remaining one is -
CH-;
A is selected from the group consisting of benzene and pyridine, wherein A is
unsubstituted or substituted with one or more substituents selected from the
group a, C1-C6alkyl,
C3-C1ocycloalkyl, aryl, heteroaryl, cycloheteroalkyl, C1-C6alkylC3-
CI0cycloalkyl, C1-C6alkylaryl,
C1-C6alkylheteroaryl and C1-C6alkyleycloheteroalkyl, wherein C1-C6alkyl, C3-
C1ocycloalkyl,
aryl, heteroaryl, cycloheteroalkyl, C1-C6alky1C3-C10cycloalkyl, C1-
C6alkylaryl, C1-
C6alkylheteroaryl and C1-C6alkylcycloheteroalkyl are independently
unsubstituted or substituted
with one or more substituents selected from the group consisting of a;
a is selected from the group consisting of halogen, C1-C6alkyl, halogen-
substitutedCI-C6alkyl, COCI-C6alkyl, oxo, -OH, halogen-substitutedCI-C6alky101-
1, -0C1-
C6alkyl, -Ohalogen-substitutedCi-C6alkyl, -COOH, -COOC1-C6alkyl, -Ci-
C6alkylCOOCi-
C6alkyl, -C1-C6alkylCOOH, -0C1-C6alkylCOOH, -CN, C1-C6alkylCN, -NO2, NH2,
NFICI-
C6alkyl, N(C1-C6alky1)2, -NHCOOH, -NHCOOC1-C6alkyl, -CONH2, -CONHCI-C6alkyl, -
CON(CI-C6alky1)2, -CONHC1-C6alkyl-N(Ci-C6alkyl)2, -NHSO2CI-C6alkyl, -SO2NH2, -
S02Ci-
C6alkyl, C3-Clocycloalkyl, aryl, heteroaryl, cycloheteroalkyl,
cycloheteroalkylCOOH, CI-
C6alky1C3-C10cycloalkyl, C1-C6alkylaryl, C1-C6alkylheteroaxyl and C1-
C6alkylcycloheteroalkyl;
- is -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
R12 is selected from the group consisting of -COOH, -COOCi-C6alkyl, -C1-
C6alkylCOOCI-C6alkyl, -C1-C6alkylCOOH and -0C1-C6alkylCOOH.
Examples of compounds or pharmaceutically acceptable salt thereof include, but
are not limited to:
H 0
F F o
0 r/
0 )--- N
la K-7)--- OC
N
" n
CI N N
1100 "
41V 40
'
C11-0".-0(--=
CI
\ i
N
, r
./¨ . 0
0
10 4'---C)
a 0--(-)---N N
1110 .
40
_
H 30\ r F
Ci 0 õ_____ N õ0
, 40 K)-0Crõ
1 ...___N
.
ip
N7 \--\\ N
H
_
H
F
/ 40 F
0 N N
HN
H
.
k
F
. 0
N µ N
ih
H
11t OH
, OH
,
0
0
t___0.,_ / cõ..,.0
F F
,
*MOD
= SO / \ / Fk \ N N
40 ':?---0-/, --CIX-L
\ /
* 0
OH
_
0
i'li 6 0
F 0
IlkN\ I
0
..
F F
k - r-- \-10Cr
Wil 11-((
1 ,
N N /
F 6
- 19 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
F
F
. 0 ti ri
.0
F 0 =Nei>...0_,/4 0(0.1..f H
N
N
*
CH5
F
r r
. =
r
F 0 1 \ \ /
01 I 1 la Crib
H
ill
N \ /
044
r F
"""r"" 6
a Ol , N4-11
N N
H
v
.
F
0 =F
0
H
N \---14
=
i I 1
F =
a
144
F
-
F F
F F
. cHz
H
/ X.....,),,/za
F 11101 \ \¨,,. N
F OP _
PI N
N
H *
N N \\===
1 'N...
f
...--
_
F
F
= F
if = \ \f
/1
I 4 . -
"...
F.t.c.
Pr-- CY-
8
0
FF
F = u$.--Q--: -
F
H
0 õ,.)::)
F fall
/X .
N ........ N
0O
r'.... C 1-i3
F F
a 0 F
F H
F . N\ ¨
N
0..
/
N \ / N N ----
H
\ 1
110 N/>----C)--N \ A---- N
B r
.
=
.
-." N
- 20 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
-
r F
F f
e,...,..,0
F * \ ¨0.-=-
. \ .
ii
.
ON
, F
F
F
0 iiith II, r"-'--" \ 7---- \ /01
N.\ \ ..._/"=" ,,, \ ___\ /
F
F N 11111. \ VP ''',17"-
\ ___.A.,51
fi
N"--N7
H
. i
*
r
, aN '¨'11
F 0 \>¨'0-- =
N N Oil
F F
0
¨
F r
F H
0
; * t4,,. 0
F = 15-0-0ech
/,4
'1111111P OH
11
F
" F '
F
' * i \"--/ -- I\ .._
NII ==='' .
OH
Br
IS
F
F
F el N\>< N\:µ\ Ni/ \ ,..... 0 0
111.
11111 aq,
N /1- \ k:r
til N
ti
0
411
¨
F
r, F
0
il
N \
H
0
' =
a
0
II
F
Ft N
0 I II
- -
ON
F F
-F>r=VN l',õ.... )., ,...... F =1 N), .._.,
0.,...00 =
= *
o
= t4 --,4 o
N ¨ ii
_ ori
\ /
r
- 21 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
I
0 0i.,
* 0.,
F
14
* '-'0-= -
F alp Ns> c )...._, N
q 0
--N
NA CH, N
0 *
H
0
F
r
F
, * , r ti*,___
N
F
ta \
is/ ¨
o = 8.--:.
H
*
F
F
,
At
el F = \ \--/
VI OH
N N
IA
=
H
F 0
H ,=
CH
F
F
' . N\. \ .¨N
Ait.
ii---- I-r"\)_ 0 .
F= >--c_."--)--
H
VI ON
14 N
H
110- F
0
CH)
F F
OH
F
1
0 0
N
i "r=-/- CH
H,
CH,
N.--
vN¨cm4
F
F
N
\
\ N
11/
0
H
HO
Cl H
0--- N
. cN3
F . 0.....
1. >. \----/ I
N N
H0 CH,
t
* 0
II,C OH)
MO
CH3
H ,O 01 N\
---r4 HO
N HO
0
0 \ * 0
H
F
N
F F
/ I
N HO
..-...N
..
1 \
H .
e 141---cl-'- 0(4 a
0' \ \-../ 14 N
H
- 22 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
F
N HG
--N
---,--- N HO
0 I `= 0
0
N
H =
..,
= H
ON
HO N 411 HO
0 1
¨0 H30
0
ei \
= 4 .
\ / N
H
_
0 H3
H=
i \ 43
1 N, N HO
¨N
N N
H = \
..---- \ / N
N 0 N
H =
H&c.,1 H
Oy...-N
-- N HO
' HsC
II f \ 0
H 111)
0
0
7.-1,1.--- N =
I. g
0
0,J
N---- N HO
FizN-- 0 --- / N
_N
HO
/ \ 0
I N \\
N
N 14 N
H =
H =
CHs H3C N
HO
¨N HO \
i 0
1 \
0 .------'
H,C N'--- N \ / N
H . H
411
H
N r.-N 1
N HO =
/
N I 0 \ 1 \ . 0
\\ N
N N N ..õ
/ \
0
N
N
H N
=
1
N.
I I
/
N HO
N N ¨N HO
N 0 it \
H =
N
H III
N
riC
1 N, N ¨ N HO
F j
\
0
HO .-,--- \ / N
. \..._/N .
N
o
H
H = 1
II 110 N * ,
,
- 23 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
F N (
= ---N HO ----'-'
0 N HO
--N
.
F el \ 0
H
H II
,
N HO
I -N
F
.,"*
HO0 = \
F \ / 111
H C
' a N
N H I)
H = . HO
- N ------ N
4111 \ 0 40 \
0
N
H H3C
H =
H3C H3C
It
HO N HO
- N
el \ 0 \
P=
III 0
= N \ \ /
H C 1111 N
H
F
N - N HO N
.-^,--, N HO
4111 I \
0
\ / N
H . 0 F ill N\
H
1111
H
.
/ 0 N HO
1110 \
0 N 'N=
0 \ / 1
0
N N
H =
H30 N
H N
=
==-- N HO N H =
0 el \
et \
0
----
N \ /
H = I N ----
HO H =
.. I e F
N 0
H.
N HO
0
N N
H N H
=
. a
_
F F
III N HO
-N
\
0 40, ,
\ / t4 111 HO
- N
N
H
0
\ / N
F
N
H
=
F40
HO
HO
0I \ o
-0
o N \ /=
H
1 N\ \ /
: F
H =
F
- 24 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
= H.
0 \ * .
0 ,
N
o'r H
=
HP
Examples of compounds or pharmaceutically acceptable salt thereof described
herein include, but are not limited to:
F30, N õN ,
\ 7 F3c 0 N /
0___.--. õ
\ 7 \ K)
õ \ N\___X I V
N -N
H CO2H H
OH
,
F30, N) c \ 0 F3C 00 N õ
\ 4 \)-Nr-Y:fo
N ip
N -N
CO2H H OH
H
F305 N\\___27 ,\,,__Na.).,.__(.. 0
FaC la .\ ____FN-N
N * 0 VP N/ \=N7
N/ \\,,,,
1-4
H \--CO2H
1OH
0
F30 r,L1, ,,,)....i, 0 cl
40 N,--A-N 0 - N
I
Nf____\--:--N1
CO0 2Et - H
H
111 OH
0
,
FaC 0 N FaC
40 N / \\
OH \ 'L \)¨ --N = 0H O
\ / \ Nil: k-r
ill N -N =
H \--i
H CFa
0
FaC 0 N/ 0
XD:r F
N t CN
N -N CI
N 0 0
H 40 ' II N
H
4Ik
F 0
F3C is -N / ) .1-1-
I-1
FaC 0 IA OH
, ip, N ii,
r1.4 gp, NO2
HN
0
F is a N , , , ....,0*
F3C 5 N N __. =, \ OH
\ /C N N.\ _jc j 1
N -N 1 .---CF3
---N it
N N
H N H
F3 C lis N ru 0
0
\ / \ / \ \ i.,--f'
.--N OH
N
I N it
N -N \ l\----N *
H Nzzr---
8
41.0 NH
FaC
FaC 40 N A / 0.,_.,[0
0
,--N OH
\ ¨ \)----14\_X,
N 111
N -N Nr1 S'-
-- N =
H
Facs = NH
..
I:
- 25 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
, 0
F3c 0 N o-f
....-N OH
\ / \ N
N = CF3 I ,--N 411
N -N
H N_.--,C-S
. NH
CI
Me
_
F3C\ / \ 0 N NI/ CF3
F 0
F3C 0 NC OH
\>--------Ni it
.
N -N \ N N -N
H H
F3C, N /
0,-,
,
\ \ N/DUL K---N p3n0
n 0
\ 1 . _
N -N N\ / \ Ni
''--
-
H
N ¨N
1
HO
_
0
FaC 40 N
:,.._)--treco2me
OH
nN\ \---/ N it
N -N
H Me0 N1
N
Me 0
CI 40 N, /0 0
\-__.N OH
\--% ---N
N N NI \---N
HH Me gill Am
110
....
0
Cl 0 N\>_< ____NI 0
me 0 NN,/ \_\_____/__\ N>,.,õ ii OH
N -N
H H
I.
CI 0
Cl. N 0,-
N -\ OH
">---- --N
cl 0 \>_i,,,,i_N .
N N
H
0
Cl 410 N 0 0
0 N_ N it OH
\>¨< '---N
01 N \ N
N -N H
H
\., /
N
o
Cl I. N- .
N\v_o_N . OH
--- ----.N/
-- F Ni \--N
N aN
H
H I
\ N
-
0
F3C si tsj,> /,,,,,,, \\ / 0,_,4-.0
OH
go N.___/-1_,N 410
\ \)-----N
FaCO IµIIIP FiN"-N
N \--N
H N \ I
_
- 26 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
CI I. N / 0-__,/,
Cl o
= OH
\ / N
N -N ---
H- N F3c * FIN\/ \------
7--
\ i
N F
0
OH
N\)-------0,,
F 0 N N
H H
l
F
CI si N\>___L/ \\\?____N 0 0 F
0
0 N_____K-I___N * OH
N -N
H F3C Ni \--N
H
11.
OH
0
F
0
CI 0 N\>< cN 0 0 cliN - \
OH
= ''N 1 FiN\>*14/2-N =
N -N
H
.
_
0
F3C 0 N\> g / 0-=__-
OH
\ ) N ---N e
N \-N N\) C N
NN (----
H
N \ /
Br
0
F3C to õ ,õõ .\.) 0 0
, N OH
, I\>-{)--N it
N \---Th
H H
CO2H
...
0
F3C . N 0 õN An N\\
47-'>_. N 41, OH
\>-- ------N a
'PP Ni \-1,4'
1 N -N I
H
H . ---.1-,N
CF3
0 .
F3C so N 0 Me
0 N\ \--/ N OH
\)-----< ---N
N -NI 10 N N N
H H
c02H
F3C = g / 0,- F3c
\ \? N\>--=C \)---N
N \ -N \
H
H
=
N \ /
Me
-27 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
,
I
F3C,i\I N \--N"
OH
CI F 0
Me N m 0
11
Me0..,_,NN)___(_ HFaC 0 0 .
N1\4/
N' \'-'---N 0
H OH
'----õ------"----N N \ ---
N ,,, I
F3C iso N\.j,/,, \_ )____N 0 CO2H
,
d \-N
H F3C is N
\ / \ N 0
N ¨N
H OH
a CO2H H
F3C, N 0
N/:,_(,_,,,,N,
\)-4 --.-N
N -N ill CI N ---IN
OH
H
0 H
F3C, N , \\ OH 41 /)___. N
N 0, 0
\>---C \)---N =
N ¨N
N -1\1
OH
H
0 Fsc 0 N / \ H
00 ii, OH
\ / \ Nr-rssThro
N ¨N
H
N ¨N
H
H F2C
\ / õ N \ 111 ell
F3C 0N N -N O
H
H
0 F3C
\ OH
N -NI OH
F3C 0 N
H
---< ----N
N ¨N O=
H
_
Me02C 0 Nai/0
F3C 411 N,,_____F, VN CI
N".=--N OH
H
Ni \-----NI 0 lit CO2Me
H .
- 28 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
HO2C
N.,>
0
0
H
0 ___c,,,,._N
F3C 0N õ
F
N -N
OH
\
N
-N
0 40 CO2Me
H
,
,
HO2C
F3C 0 N>< \\
0
p
N -N
--_ )----N
'"%H
F3C
N
si
\ /õ \ N
H
N
-N
0 411 CO2H
H
o
F3C 0 N , \\
0
o
\>--___ \)---N
F3C 40 N>
\
OH
N -N
H
OH
\
/ )- N
N
¨N
0 .
H
o
11
0
o
F3c 0N õ
0
N
-IV
0 . NHSO2Me Et
N
-N
OH
H
-
0
H
ci
F3C, N0 N
0 ,53
/ \ N
40H
04
/
'N
NH Et N -N
N ¨N
H
.
'
Me
H
F30
0
5Ni\>-<_---N
FCS 4111 jia 0
3
N
i / \ N
N -N 0 /<0
OH
N
-N
H
OH
H,
F3C 0 N
0 0
\ / \ N
NSO2Me
0
.,
N -N
le
F3C0 N -N
OH
H
H
F3C 0 c
ENt
/ \
0
0
N
/
N-,o
/ ,---N
.r_Nl
Or ,>--G-N
\
Et0 N -N
N
- N
OH
H
0 116
CN
FTC, \/ N ,N
0
/< \
kli
0
0
\
i\>--0---
-
IP
N -N
0 0
OH
F
H
0
H
F3C 0> N ,
N
0
ii0
\
.<---NF3C 0
0
/>----N
N
N -N
N -N
OH
H
III
iv
002H
- 29 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
F3C 0 0
\ N
-N OH
40 c02,_
F3c N 0 0
r
N -N \ NH Me0 N N -N OH
-N OH
Definitions
Examples of "halogen" include a fluorine atom, a chlorine atom, a bromine
atom, and an
iodine atom.
The term "C1-C 6alkyl" encompasses straight alkyl having a carbon number of 1
to 6 and
branched alkyl having a carbon number of 3 to 6. Specific examples thereof
include methyl,
ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, n-
pentyl, isopentyl, neopentyl,
tert-pentyl, 1-methylbutyl, 2-methylbutyl, 1,2-dimethylpropyl, 1-ethylpropyl,
n-hexyl, isohexyl,
1-methylpentyl, 2-methylpentyl, 3-methylpentyl, 1,1-dimethylbutyl, 1,2-
dimethylbutyl, 2,2-
dimethylbutyl, 1-ethylbutyl, 1,1,2-trimethylpropyl, 1,2,2-trimethylpropyl, 1-
ethy1-2-
methylpropyl, 1-ethyl-l-methylpropyl, and the like.
The term "-OCI-C 6alkyl " refers to an alkyl group having 1 to 6 carbons
linked to
oxygen, also known as an alkoxy group. Examples include methoxy, ethoxy,
butoxy and
propoxy.
The term "halogen-substitutedCi-C6 alkyl" encompasses C1-C6 alkyl with the
hydrogen
atoms thereof being partially or completely substituted with halogen, examples
thereof including
fluoromethyl, difluoromethyl, trifluoromethyl, 2-fluoroethyl, 1,2-
difluoroethyl, 2,2-difluoroethyl
and the like.
The term "-Ohalogen-substitutedCI-C6alkyl" means a -0C1-C6alkyl as defined
above,
which is substituted with 1-3 halogen atoms which are identical or different,
and specifically
includes, for example, a trifluoromethoxy group.
The term "CI-C6alkylCOOH" means a C1-C6alkyl as defined above substituted with
a
carboxylic acid group (COOH).
The term "COOCi-C6alkyl" means a -COOH group wherein the ¨OH is replaced with
an
alkoxy group as defined above. Examples include methoxycarbonyl,
ethoxycarbonyl and
butoxyearbonyl.
The term "C3-C10cycloalkyl" means a monocyclic or polycyclic, saturated or
partially-
unsaturated carbocyclic group having from 3 to 10 carbon atoms, for example,
cyclopropyl,
- 30 -
WO 2012/009217 CA 02804970 2013-01-09
PCT/US2011/043330
cyclobutenyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooetyl, cyclononyl,
cyclodecyl,
bicyclohexyl, bycyclodecyl, bicyclononyl, tetrahydronaphthyl,
decahydronaphthyl, indanyl and
adamantly.
The term "S02C1-C6alkyl" means a group having C1,6 alkyl bonded to sulfonyl (-
SO2-).
Specific examples thereof include methanesulfonyl, ethanesulfonyl, n-
propanesulfonyl,
isopropanesulfonyl, n-butanesulfonyl, sec-butanesulfonyl, tert-butanesulfonyl,
and the like.
The term "COC1.6 alkyl" means groups having C1-6 alkyl bonded to carbonyl, and
encompasses alkylcarbonyl having a carbon number of 1 to 6. Specific examples
thereof include
acetyl, propionyl, butyryl, isobutyryl, valeryl, isovaleryl, pivaloyl, and the
like.
The term "oxo" means the functional group '1=0", such as, for example, (1) "C--
-(0)", that
is a carbonyl group; (2) "S=(0)", that is, a sulfoxide group; and (3) 11\1----
(0)", that is, an N-oxide
group, such as pyridyl-N-oxide.
The term "NHCI-C6alkyl" means a group with one of the hydrogen atoms of amino
(-
NH2) being substituted with a C1_6 alkyl group. Specific examples thereof
include methylamino,
ethylamino, n-propylamino, isopropylamino, n-butylamino, sec-butylamino, tert-
butylamino, and
the like.
The term "N(CI-C6alky1)2" means a group with the two amino hydrogen atoms each
being substituted with a Ci_6 alkyl group. Specific examples thereof include
dirnethylamino,
diethylamino, ethylmethylamino, di(n-propyl)amino, methyl(n-propyl)amino,
diisopropylamino,
and the like.
The term "NHCO2C1-C6alkyl" means a group with one of the amino hydrogen atoms
being substituted with Ci_6 alkoxycarbonyl and encompasses alkoxycarbonylamino
having a
carbon number of 1 to 6. Specific examples thereof include
methoxycarbonylamino,
ethoxycarbonylamino, n-propyloxycarbonylamino, isopropyloxycarbonylamino, n-
butoxycarbonylamino, isobutoxycarbonylamino, tert-butoxycarbonylamino, n-
pentyloxycarbonylamino, and the like.
The term "CONHCI-C6alkyl" means a group with one of the hydrogen atoms of
carbamoyl (-CONH2) being substituted with C1_6 alkyl. Specific examples
thereof include
methylcarbamoyl, ethylcarbamoyl, n-propylcarbamoyl, isopropylcarbamoyl, n-
butylcarbamoyl,
sec-butylcarbamoyl, tert-butylcarbamoyl, and the like.
The term "CON(C1-C6alky1)2" means a group with the two carbamoyl hydrogen
atoms
each being substituted with C1_6 alkyl. Specific examples thereof include
dimethylcarbamoyl,
diethylcarbamoyl, ethylmethylcarbarnoyl, di(n-propyl)carbarnoyl, methyl(n-
propyl)carbamoyl,
diisopropylcarbamoyl, and the like.
Examples of "aryl" include phenyl, naphthyl, tolyl, and the like.
The term "heteroaryl" means 5-membered or 6-membered monocyclic heteroaryl
containing one or more, preferably one to three, same or different heteroatoms
selected from the
- 31 -
WO 2012/009217 CA 02804970 2013-01-09PCT/US2011/043330
group consisting of an oxygen atom, a nitrogen atom, and a sulfur atom, or
otherwise means
condensed-ring heteroaryl formed by condensation of such monocyclic heteroaryl
and the above-
mentioned heteroaryl or alternatively by mutual condensation of the same or
different
monocyclic heteroaryl groups. Examples thereof include pyrrolyl, furyl,
thienyl, imidazolyl,
pyrazolyl, thiazolyl, isothiazolyl, oxazolyl, isoxazolyl, triazolyl,
tetrazolyl, oxadiazolyl, 1,2,3-
thiadiazolyl, 1,2,4-thiadiazolyl, 1,3,4-thiadiazolyl, pyridyl, pyrazinyl,
pyrimidinyl, pyridazinyl,
1,2,4-triazinyl, 1,3,5-triazinyl, indolyl, benzofuranyl, benzothienyl,
benzimidazolyl,
benzopyrazolyl, benzoxazolyl, benzisoxazolyl, benzothiazolyl,
benzisothiazolyl, indazolyl,
purinyl, quinolyl, isoquinolyl, phthalazinyl, naphthyridinyl, quinoxalinyl,
quinazolinyl,
cinnolinyl, pteridinyl, pyrido[3,2-b]pyridyl, and the like.
"Cycloheteroalkyl" means mono- or bicyclic or bridged saturated rings
containing at least
one heteroatom selected from N, S and 0, each of said ring having from 3 to 10
atoms in which
the point of attachment may be carbon or nitrogen. The term also includes
monocyclic
heterocycle fused to an aryl or heteroaryl group in which the point of
attachment is on the non-
aromatic portion. Examples of "cycloheteroalkyl" include tetrahydropyranyl,
tetrahydrofuranyl,
piperidinyl, piperazinyl, dioxanyl, imidazolidinyl, 2,3-dihydrofuro(2,3-
b)pyridyl,
benzoxazinyl, benzoxazolinyl, 2-H-phthalazinyl, isoindolinyl, benzoxazepinyl,
5,6-
dihydroimidazo[2,1-bjthiazolyl, tetrahydroquinolinyl, morpholinyl,
tetrahydroisoquinolinyl,
dihydroindolyl, and the like. The term also includes partially unsaturated
monocyclic rings that
are not aromatic, such as 2- or 4-pyridones attached through the nitrogen or N-
substituted-(1H,
311)-pyrimidine-2,4-diones (N-substituted uracils). The temi also includes
bridged rings such as
5-azabicyclo[2.2.1]heptyl, 2,5-diazabicyclo[2.2.1]heptyl, 2-
a.zabicyclo[2.2.11heptyl, 7-
azabicyclo[2.2.1]heptyl, 2,5-diazabicyclo[2.2.2]octyl, 2-
azabicyclo[2.2.2joctyl, and 3-
azabicyclo[3.2.2]nonyl, and azabicyclo[2.2.11heptanyl. The cycloheteroalkyl
ring may be
substituted on the ring carbons and/or the ring nitrogens.
"S02C1-C6alkyl" group means a group in which a Ci-C6alkyl group is attached to
a sulfonyl (-SO2-) group. Specific examples thereof include methanesulfonyl,
ethanesulfonyl, n-
propylsulfonyl, isopropanesulfonyl, n-butanesulfonyl, sec-butanesulfonyl and
tert-butanesulfonyl
groups and the like.
The term "pharmaceutically acceptable salt" refers to salts prepared from
pharmaceutically acceptable non-toxic bases or acids including inorganic or
organic bases and
inorganic or organic acids. Salts of basic compounds encompassed within the
term
"pharmaceutically acceptable salt" refer to non-toxic salts of the compounds
of this invention
which are generally prepared by reacting the free base with a suitable organic
or inorganic acid.
Representative salts of basic compounds of the present invention include, but
are not limited to,
the following: acetate, benzenesulfonate, benzoate, bicarbonate, bisulfate,
bitartrate, borate,
bromide, camsylate, carbonate, chloride, clavulanate, citrate,
dihydrochloride, edetate, edisylate,
-32-
WO 2012/009217 CA 02804970 2013-01-09PCT/US2011/043330
estolate, esylate, fumarate, gluceptate, gluconate, glutamate,
glycollylarsanilate, hexylresorcinate,
hydrabamine, hydrobromide, hydrochloride, hydroxynaphthoate, iodide,
isothionate, lactate,
lactobionate, laurate, inalate, maleate, mandelate, mesylate, methylbromide,
methylnitrate,
methylsulfate, mucate, napsylate, nitrate, N-methylglucarnine ammonium salt,
oleate, oxalate,
palmate (embonate), palmitate, pantothenate, phosphate/diphosphate,
polygalacturonate,
salicylate, stearate, sulfate, subacetate, succinate, tannate, tartrate,
teoclate, tosylate, triethiodide
and valerate. Furthermore, where the compounds of the invention carry an
acidic moiety,
suitable pharmaceutically acceptable salts thereof include, but are not
limited to, salts derived
from inorganic bases including aluminum, ammonium, calcium, copper, ferric,
ferrous, lithium,
magnesium, manganic, mangamous, potassium, sodium, zinc, and the like.
Particularly preferred
are the ammonium, calcium, magnesium, potassium, and sodium salts. Salts
derived from
pharmaceutically acceptable organic non-toxic bases include salts of primary,
secondary, and
tertiary amines, cyclic amines, and basic ion-exchange resins, such as
arginine, betaine, caffeine,
choline, N,N-dibenzylethylenediamine, diethylamine, 2-diethylarninoethanol, 2-
dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethylmorpholine, N-
ethylpiperidine,
glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine,
methylglucamine,
morpholine, piperazine, piperidine, polyamine resins, procaine, purines,
theobromine,
triethylamine, trimethylamine, tripropylamine, tromethamine, and the like.
If desired, racemic mixtures of the compounds may be separated so that the
individual
enantiomers are isolated. The separation can be carried out by methods well
known in the art,
such as the coupling of a racemic mixture of compounds to an enantiomerically
pure compound
to form a diastereomeric mixture, followed by separation of the individual
diastereomers by
standard methods, such as fractional crystallization or chromatography. The
coupling reaction is
often the formation of salts using an enantiomerically pure acid or base. The
diasteromeric
derivatives may then be converted to the pure enantiomers by cleavage of the
added chiral
residue. The racemic mixture of the compounds can also be separated directly
by
chromatographic methods utilizing chiral stationary phases, which methods are
well known in
the art.
Alternatively, any enantiomer of a compound may be obtained by stereoselective
synthesis using optically pure starting materials or reagents of known
configuration by methods
well known in the art.
It will be understood that, as used herein, references to the compounds of the
structural
foimulas described herein are meant to also include the pharmaceutically
acceptable salts, and
also salts that are not pharmaceutically acceptable when they are used as
precursors to the free
compounds or their pharmaceutically acceptable salts or in other synthetic
manipulations.
Solvates, and in particular, the hydrates of the compounds of the structural
formulas
described herein are included in the present invention as well.
-33 -
CA 02804970 2013-01-09
WO 2012/009217 PCT/US2011/043330
Some of the compounds described herein may exist as tautomers, which have
different
points of attachment of hydrogen accompanied by one or more double bond
shifts. For example,
a ketone and its enol form are keto-enol tautomers. The individual tautomers
as well as mixtures
thereof are encompassed with compounds of the present invention.
In the compounds of the formulas described herein, the atoms may exhibit their
natural
isotopic abundances, or one or more of the atoms may be artificially enriched
in a particular
isotope having the same atomic number, but an atomic mass or mass number
different from the
atomic mass or mass number predominantly found in nature. The present
invention is meant to
include all suitable isotopic variations of the compounds of the formulas
described herein. For
example, different isotopic forms of hydrogen (H) include protium (1H) and
deuterium (2H).
Protium is the predominant hydrogen isotope found in nature. Enriching for
deuterium may
afford certain therapeutic advantages, such as increasing in vivo half-life or
reducing dosage
requirements, or may provide a compound useful as a standard for
characterization of biological
samples. Isotopically-enriched compounds within generic formula can be
prepared without
undue experimentation by conventional techniques well known to those skilled
in the art or by
processes analogous to those described in the Schemes and Examples herein
using appropriate
isotopically-enriched reagents and/or intermediates.
Methods of Treatment
Also encompassed by the present invention are methods of treating DGAT1-
related
diseases. The compounds described herein are effective in preventing or
treating various
DGAT1-related diseases, such as metabolic diseases such as obesity, diabetes,
hormone secretion
disorder, hyperlipemia, gout, fatty liver, and the like; circulatory diseases
such as angina pectoris,
acute/congestive cardiac insufficiency, myocardial infarction, coronary
arteriosclerosis,
hypertension, nephropathy, electrolyte abnormality, and the like; central and
peripheral nervous
system diseases such as bulimia, affective disorder, depression, anxiety,
epilepsy, delirium,
dementia, schizophrenia, attention deficit/hyperactivity disorder, dysmnesia,
somnipathy,
cognitive impairment, dyskinesia, dysesthesia, dysosmia, morphine resistance,
drug dependence,
alcohol dependence, and the like; reproductive system diseases such as
infertility, premature
delivery, sexual dysfunction, and the like; and other conditions including
digestive diseases,
respiratory diseases, cancer, and ehromatosis. The compound of the invention
is especially
useful as a preventive or a remedy for obesity, diabetes, fatty liver,
bulimia, depression, or
anxiety.
One aspect of the invention described herein provides a method for the
treatment and
control of obesity or metabolic syndrome, which comprises administering to a
patient in need of
such treatment a therapeutically effective amount of a compound having the
formulas described
herein or a pharmaceutically acceptable salt thereof. For example, the
compounds described
-34-
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
herein are useful for treating or preventing obesity by administering to a
subject in need thereof a
composition comprising a compound of formula I or formula Ia.
Methods of treating or preventing obesity and conditions associated with
obesity refer to
the administration of the pharmaceutical formulations described herein to
reduce or maintain the
body weight of an obese subject or to reduce or maintain the body weight of an
individual at risk
of becoming obese. One outcome of treatment may be reducing the body weight of
an obese
subject relative to that subject's body weight immediately before the
administration of the
compounds or combinations of the present invention. Another outcome of
treatment may be
preventing body weight, regain of body weight previously lost as a result of
diet, exercise, or
pharmacotherapy and preventing weight gain from cessation of smoking. Another
outcome of
treatment may be decreasing the occurrence of and/or the severity of obesity-
related diseases.
Yet another outcome of treatment may be decreasing the risk of developing
diabetes in an
overweight or obese subject. The treatment may suitably result in a reduction
in food or calorie
intake by the subject, including a reduction in total food intake, or a
reduction of intake of
specific components of the diet such as carbohydrates or fats; andior the
inhibition of nutrient
absorption; and/or the inhibition of the reduction of metabolic rate; and in
weight reduction in
patients in need thereof. The treatment may also result in an alteration of
metabolic rate, such as
an increase in metabolic rate, rather than or in addition to an inhibition of
the reduction of
metabolic rate; and/or in minimization of the metabolic resistance that
normally results from
weight loss.
Prevention of obesity and obesity-related disorders refers to the
administration of the
pharmaceutical formulations described herein to reduce or maintain the body
weight of a subject
at risk of obesity. One outcome of prevention may be reducing the body weight
of a subject at
risk of obesity relative to that subject's body weight immediately before the
administration of the
compounds or combinations of the present invention. Another outcome of
prevention may be
preventing body weight regain of body weight previously lost as a result of
diet, exercise, or
pharrnacotherapy. Another outcome of prevention may be preventing obesity from
occurring if
the treatment is administered prior to the onset of obesity in a subject at
risk of obesity. Another
outcome of prevention may be decreasing the occurrence and/or severity of
obesity-related
disorders if the treatment is administered prior to the onset of obesity in a
subject at risk of
obesity. Moreover, if treatment is commenced in already obese subjects, such
treatment may
prevent the occurrence, progression or severity of obesity-related disorders,
such as, but not
limited to, arteriosclerosis, type 2 diabetes, polycystic ovary disease,
cardiovascular diseases,
osteoarthritis, dermatological disorders, hypertension, insulin resistance,
hypercholesterolemia,
hypertriglyceridemia, and cholelithiasis.
Another aspect of the invention that is of interest relates to a method of
treating
hyperglycemia, diabetes or insulin resistance in a mammalian patient in need
of such treatment
- 35 -
WO 2012/009217 CA 02804970 2013-01-09PCT/US2011/043330
which comprises administering to said patient a compound in accordance with
the formulas
described herein or a pharmaceutically acceptable salt thereof in an amount
that is effective to
treat hyperglycemia, diabetes or insulin resistance.
More particularly, another aspect of the invention that is of interest relates
to a method of
treating type 2 diabetes in a mammalian patient in need of such treatment
comprising
administering to the patient a compound in accordance with the formulas
described herein or a
pharmaceutically acceptable salt thereof in an amount that is effective to
treat type 2 diabetes.
Yet another aspect of the invention that is of interest relates to a method of
treating non-
insulin dependent diabetes mellitus in a mammalian patient in need of such
treatment comprising
administering to the patient a compound in accordance with the formulas
described herein or a
pharmaceutically acceptable salt thereof in an amount that is effective to
treat non-insulin
dependent diabetes mellitus.
The present invention is also directed to the use of a compound of any of the
formulas
described herein in the manufacture of a medicament for use in treating
various DGAT1-related
diseases, such as metabolic diseases such as obesity, diabetes, hormone
secretion disorder,
hyperlipemia, gout, fatty liver, and the like; circulatory diseases such as
angina pectoris,
acute/congestive cardiac insufficiency, myocardial infarction, coronary
arteriosclerosis,
hypertension, nephropathy, electrolyte abnouuality, and the like; central and
peripheral nervous
system diseases such as bulimia, affective disorder, depression, anxiety,
epilepsy, delirium,
dementia, schizophrenia, attention deficit/hyperactivity disorder, dysmnesia,
somnipathy,
cognitive impairment, dyskinesia, dysesthesia, dysosmia, morphine resistance,
drug dependence,
alcohol dependence, and the like; reproductive system diseases such as
infertility, premature
delivery, sexual dysfunction, and the like; and other conditions including
digestive diseases,
respiratory diseases, cancer, and chromatosis. The compounds described herein
are especially
useful as a preventive or a remedy for obesity, diabetes, fatty liver,
bulimia, depression, or
anxiety.
For example, the present invention is directed to the use of a compound of any
of the
formulas described herein in the manufacture of a medicament for use in
treating obesity,
diabetes, hormone secretion disorder, hyperlipemia, gout and fatty liver.
Additionally, the present invention is directed to the use of a compound of
any of the
formulas described herein in the manufacture of a medicament for use in
treating obesity.
Pharmaceutical Compositions
Compounds of the invention may be administered orally or parenterally. As
formulated
into a dosage form suitable for the administration route, the compound of the
invention can be
used as a pharmaceutical composition for the prevention, treatment, or remedy
of the above
diseases.
-36-
CA 02804970 2013-01-09
WO 2012/009217 PCT/US2011/043330
In clinical use of the compound of the invention, usually, the compound is
formulated
into various preparations together with pharmaceutically acceptable additives
according to the
dosage form, and may then be administered. By "pharmaceutically acceptable" it
is meant the
additive, carrier, diluent or excipient must be compatible with the other
ingredients of the
formulation and not deleterious to the recipient thereof. As such additives,
various additives
ordinarily used in the field of pharmaceutical preparations are usable.
Specific examples thereof
include gelatin, lactose, sucrose, titanium oxide, starch, crystalline
cellulose, hydroxypropyl
methylcellulose, carboxymethylcellulose, corn starch, microcrystalline wax,
white petrolatum,
magnesium metasilicate aluminate, anhydrous calcium phosphate, citric acid,
trisodium citrate,
hydroxypropylcellulose, sorbitol, sorbitan fatty acid ester, polysorbate,
sucrose fatty acid ester,
polyoxyethylene, hardened castor oil, polyvinylpyrrolidone, magnesium
stearate, light silicic acid
anhydride, talc, vegetable oil, benzyl alcohol, gum arabic, propylene glycol,
polyalkylene glycol,
cyclodextrin, hydroxypropyl cyclodextrin, and the like.
Preparations to be formed with those additives include, for example, solid
preparations
such as tablets, capsules, granules, powders, suppositories; and liquid
preparations such as
syrups, elixirs, injections. These may be formulated according to conventional
methods known
in the field of pharmaceutical preparations. The liquid preparations may also
be in such a form
that may be dissolved or suspended in water or in any other suitable medium in
their use.
Especially for injections, if desired, the preparations may be dissolved or
suspended in
physiological saline or glucose liquid, and a buffer or a preservative may be
optionally added
thereto.
The pharmaceutical compositions may contain the compound of the invention in
an
amount of from to 99.9 % by weight, preferably from 1 to 60 % by weight of the
composition.
The compositions may further contain any other therapeutically-effective
compounds.
In case where the compounds of the invention are used for prevention or
treatment
for the above-mentioned diseases, the dose and the dosing frequency may be
varied, depending
on the sex, the age, the body weight and the disease condition of the patient
and on the type and
the range of the intended remedial effect. In general, when orally
administered, the dose may be
from 0.001 to 50 mg/kg of body weight/day, and it may be administered at a
time or in several
times. The dose is preferably from about 0.01 to about 25 mg/kg/day, more
preferably from
about 0.05 to about 10 mg/kg/day. For oral administration, the compositions
are preferably
provided in the form of tablets or capsules containing from 0.01 mg to 1,000
mg, preferably 0.01,
0.05, 0.1, 0.2, 0.5, 1.0, 2.5, 5, 10, 15, 20, 25, 30, 40, 50, 75, 100, 125,
150, 175, 200, 225, 250,
500, 750, 850 and 1,000 milligrams of a compound described herein. This dosage
regimen may
be adjusted to provide the optimal therapeutic response.
Combination Therapy
-37-
WO 2012/009217 CA 02804970 2013-01-09 PCT/US2011/043330
The compounds of the present invention are further useful in methods for the
prevention or
treatment of the aforementioned diseases, disorders and conditions in
combination with other therapeutic
agents.
The compounds of the present invention may be used in combination with one or
more other
drugs in the treatment, prevention, suppression or amelioration of diseases or
conditions for which
compounds of the formulas described herein or the other drugs may have
utility, where the combination
of the drugs together are safer or more effective than either drug alone. Such
other drug(s) may be
administered, by a route and in an amount commonly used therefore,
contemporaneously or sequentially
with a compound of any of the formulas described herein. When a compound of
any of the formulas
described herein is used contemporaneously with one or more other drugs, a
pharmaceutical composition
in unit dosage form containing such other drugs and the compound of any of the
formulas described
herein is preferred. However, the combination therapy may also include
therapies in which the
compound of any of the fothiulas described herein and one or more other drugs
are administered on
different overlapping schedules. It is also contemplated that when used in
combination with one or more
other active ingredients, the compounds of the present invention and the other
active ingredients may be
used in lower doses than when each is used singly. Accordingly, the
pharmaceutical compositions of the
present invention include those that contain one or more other active
ingredients, in addition to a
compound of any of the formulas described herein.
Examples of other active ingredients that may be administered in combination
with a compound
of any of the formulas described herein, and either administered separately or
in the same
pharmaceutical composition, include, but are not limited to:
(1) dipeptidyl peptidase-IV (DPP-4) inhibitors;
(2) insulin sensitizers, including (i) PPARy agonists, such as the glitazones
(e.g. pioglitazone,
rosiglitazone, netoglitazone, rivoglitazone, and balaglitazone) and other PPAR
ligands, including (1)
PPARoVy dual agonists, such as muraglitazar, aleglitazar, sodelglitazar, and
naveglitazar, (2) PPARa
agonists, such as fenofibric acid derivatives (gemfibrozil, clofibrate,
ciprofibrate, fenofibrate and
bezafibrate), (3) selective PPARy modulators (SPPARyM's), such as those
disclosed in WO 02/060388,
WO 02/08188, WO 2004/019869, WO 2004/020409, WO 2004/020408, and WO
2004/066963, and (4)
PPARy partial agonists; (ii) biguanides, such as metformin and its
pharmaceutically acceptable salts, in
particular, rnetformin hydrochloride, and extended-release formulations
thereof, such as Glumetza ,
Fortamet , and GlucophageXR0; (iii) protein tyrosine phosphatase-1B (PTP-1B)
inhibitors;
(3) insulin or insulin analogs, such as insulin lispro, insulin detemir,
insulin glargine, insulin
glulisine, and inhalable formulations of each thereof;
(4) leptin and leptin derivatives and agonists;
(5) arnylin and amylin analogs, such as pramlintide;
(6) sulfonylurea and non-sulfonylurea insulin secretagogues, such as
tolbutamide, glyburide,
glipizide, glimepiride, mitiglinide, and meglitinides, such as nateglinide and
repaglinide;
- 38 -
WO 2012/009217 CA 02804970 2013-01-09PCT/US2011/043330
(7) a-glucosidase inhibitors (such as acarbose, voglibose and miglitol);
(8) glucagon receptor antagonists, such as those disclosed in WO 98/04528, WO
99/01423, WO
00/39088, and WO 00/69810;
(9) incretin mimetics, such as GLP-1, GLP-1 analogs, derivatives, and
mimetics; and GLP-1
receptor agonists, such as exenatide, liraglutide, taspoglutide, AVE0010, CJC-
1131, and B1M-51077,
including intranasal, transdermal, and once-weekly formulations thereof;
(10) LDL cholesterol lowering agents such as (i) HMG-CoA reductase inhibitors
(lovastatin,
simvastatin, pravastatin, cerivastatin, fluvastatin, atorvastatin,
pitavastatin, and rosuvastatin), (ii) bile
acid sequestering agents (such as cholestyramine, colestimide, colesevelam
hydrochloride, colestipol,
and dialkylaminoalkyl derivatives of a cross-linked dextran, (iii) inhibitors
of cholesterol absorption,
such as ezetimibe, and (iv) acyl CoA:cholesterol acyltransferase inhibitors,
such as avasimibe;
(11) HDL-raising drugs, such as niacin or a salt thereof and extended-release
versions thereof;
MK-524A, which is a combination of niacin extended-release and the DP-1
antagonist MK-524; and
nicotinic acid receptor agonists;
(12) antiobesity compounds;
(13) agents intended for use in inflammatory conditions, such as aspirin, non-
steroidal anti-
inflammatory drugs (NSAIDs), glucocorticoids, and selective cyclooxygenase-2
(COX-2) inhibitors;
(14) antihypertensive agents, such as ACE inhibitors (such as enalapril,
lisinopril, ramipril,
captopril, quinapril, and tandolapril), A-II receptor blockers (such as
losartan, candesartan, irbesartan,
olmesartan medoxomil, valsartan, telmisartan, and eprosartan), renin
inhibitors (such as aliskiren), beta
blockers (such as and calcium channel blockers).
(15) glucokinase activators (GKAs), such as LY2599506;
(16) inhibitors of 1113-hydroxysteroid dehydrogenase type 1, such as those
disclosed in U.S.
Patent No. 6,730,690; WO 03/104207; and WO 04/058741;
(17) inhibitors of cholesteryl ester transfer protein (CETP), such as
torcetrapib and MK-0859;
(18) inhibitors of fructose 1,6-bisphosphatase, such as those disclosed in
U.S. Patent Nos.
6,054,587; 6,110,903; 6,284,748; 6,399,782; and 6,489,476;
(19) inhibitors of acetyl CoA carboxylase-1 or 2 (ACC1 or ACC2);
(20) AMP-activated Protein Kinase (AMPK) activators;
(21) agonists of the 0-protein-coupled receptors: GPR-109, GPR-119, and GPR-
40;
(22) SSTR3 antagonists, such as those disclosed in WO 2009/011836;
(23) neuromedin U receptor agonists, such as those disclosed in W02009/042053,
including, but
not limited to, neuromedin S (NMS);
(24) inhibitors of stearoyl-coenzyme A delta-9 desaturase (S CD);
(25) GPR-105 antagonists, such as those disclosed in WO 2009/000087;
(26) inhibitors of glucose uptake, such as sodium-glucose transporter (SGLT)
inhibitors and its
various isoforms, such as SGLT-1; SGLT-2, such as dapagliflozin and
remogliflozin; and SGLT-3;
-39-
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
(27) inhibitors of acyl coenzyme A:diacylglycerol acyltransferase 1 and 2
(DGAT-1 and DGAT-
2);
(28) inhibitors of fatty acid synthase;
(29) inhibitors of acyl coenzyme A:monoacylglycerol acyltransferase 1 and 2
(MOAT-1 and
MGAT-2);
(30) agonists of the TGR5 receptor (also known as GPBAR1, BG37, GPCR19,
GPR131, and M-
BAR); and
(31) bromocriptine mesylate and rapid-release formulations thereof.
Dipeptidyl peptidase-IV (DPP-4) inhibitors that can be used in combination
with compounds of
the formulas described herein include, but are not limited to, sitagliptin
(disclosed in US Patent No.
6,699,871), vildagliptin, saxagliptin, alogliptin, denagliptin, carrnegliptin,
dutogliptin, melogliptin,
linagliptin, and pharmaceutically acceptable salts thereof, and fixed-dose
combinations of these
compounds with metforrnin hydrochloride, pioglitazone, rosiglitazone,
simvastatin, atorvastatin, or a
sulfonylurea.
Other dipeptidyl peptidase-1V (DPP-4) inhibitors that can be used in
combination with
compounds of any of the formulas described herein include, but are not limited
to:
(2R,3S,5R)-5-(1-methy1-4,6-dihydropyrrolo[3,4-c]pyrazol-5(111)-y1)-2-(2,4,5-
trifluorophenyl)tetrahydro-2H-pyran-3-amine;
(2R,3S,5R)-5-(1-methy1-4,6-dihydropyrrolo[3,4-elpyrazol-5(111)-y1)-2-(2,4,5-
trifluorophenyl)tetrahydro-2H-pyran-3-amine;
= (2R,3S,5R)-2-(2,5-difluorophenyptetrahydro)-5-(4,6-
dihydropyrrolo[3,4-c]pyrazol-5(11/)-y1)
tetrahydro-2H-pyran-3-amine;
(3R)-4-[(3R)-3-amino-4-(2,4,5-trifluorophenyl)butanoy1]-hexahydro-3-methyl-211-
1,4-diazepin-
2-one;
= 25 4-[(3R)-3-amino-4-(2,5-difluorophenyl)butanoyllhexahydro-I-methyl-2H-
1,4-diazepin-2-one
hydrochloride; and
(3R)-4-[(3R)-3-amino-4-(2,4,5-trifluorophenyl)butanoyfl-hexahydro-3-(2,2,2-
trifluoroethyl)-2H-1,4-
diazepin-2-one; and
pharmaceutically acceptable salts thereof.
Antiobesity compounds that can be combined with compounds of any of the
formulas described
herein include topiramate; zonisamide;,naltrexone; phentermine; bupropion; the
combination of
bupropion and naltrexone; the combination of bupropion and zonisamide; the
combination of topiramate
and phenteimine; fenfluramine; dexfenfluramine; sibutramine; lipase
inhibitors, such as orlistat and
cetilistat; melanocortin receptor agonists, in particular, melanocortin-4
receptor agonists; CCK-1
agonists; melanin-concentrating hormone (MCH) receptor antagonists;
neuropeptide Yi or Y5
antagonists (such as MK-0557); CB1 receptor inverse agonists and antagonists
(such as rimonabant and
taranabant); f33 adrenergic receptor agonists; ghrelin antagonists; bombesin
receptor agonists (such as
- 40 -
WO 2012/009217 CA 02804970 2013-01-09
PCT/US2011/043330
bombesin receptor subtype-3 agonists); and 5-hydroxytryptamine-2c (5-HT2c)
agonists, such as
lorcaserin. For a review of anti-obesity compounds that can be combined with
compounds of the present
invention, see S. Chaki et al., "Recent advances in feeding suppressing
agents: potential therapeutic
strategy for the treatment of obesity," Expert Opin. Ther. Patents, 11; 1677-
1692 (2001); D. Spanswick
and K. Lee, "Emerging antiobesity drugs," Expert Opin. Emerging Drugs, 8: 217-
237 (2003); J.A.
Fernandez-Lopez, et al., "Pharmacological Approaches for the Treatment of
Obesity," Drugs, 62: 915-
944 (2002); and K.M. Gadde, et al., "Combination pharmaceutical therapies for
obesity," Exp. Opin.
Pharmacother., 10: 921-925 (2009).
Glucagon receptor antagonists that can be used in combination with the
compounds of any of the
formulas described herein include, but are not limited to:
N- [4415)-1 - 3-(3 ,5-dichloropheny1)-5- [6-(trifluoromethoxy)-2-naphthyl] -1H-
pyrazol-1-
yllethyl)benzoy111-13-alanine;
N-[4-((1R)-1-{3-(3,5-dichloropheny1)-5-[6-(trifluoromethoxy)-2-naphthyl]-1H-
pyrazol-1-
y1) ethyl)benzoyli-p-alanine;
N-(4- {1- [3-(2,5-dichtoropheny1)-5-(6-methoxy-2-naphthyl)- I H-pyrazol-1-
yllethyl benzoy1)-13-
alanine;
N-(4- { (15)-143 -(3,5 -dichloropheny1)-5-(6-methoxy-2-naphthyl)-1H-pyrazol-1-
yl] ethyllbenzoy1)-13-alanine;
N-(4- {(1S)-1- [(R)-(4-chlorophenyl)(7-fluoro-5-methyl-1H-indo1-3-yOmethyll
butyl } benzoy1)-13-
alanine; and
N-(4- { (15)-1 -{(4-chlorophenyl)(6-chloro-8-methylquiriolin-4-
y1)methylibutyllbenzoy1)-13-
alanine; and
phatinaceutically acceptable salts thereof.
Inhibitors of ste,aroyl-coenzyme A delta-9 desaturase (SCD) that can be used
in
combination with the compounds of any of the formulas described herein
include, but are not
limited to:
[5-(5-{442-(trifluoromethyl)phenoxy]piperidin-1-y1}-1,3,4-thiadiazol-2 -y1)-2H-
tetrazol-2-
yll acetic acid;
(2'-{4-[2-(trifluoromethyl)phenoxy]piperidin-1-y1}-2,5r-bi-1,3-thiazol-4-
y1)acetic acid;
(5- {3-[4-(2-bromo-5-fluorophenoxy)piperidin-1-Aisoxazol-5-y1}-2H-tetrazol-2-
yl)acetic acid;
(3- {3- [4-(2-bromo-5-fluorophenoxy)piperidin- I -y1]-1,2,4-oxadiazol-5-y1}-1H-
pyrrol- -yl)acetic
acid;
(5-{5-[4-(2-bromo-5-fluorophenoxy)piperidin-1-y1ipyrazin-2-y1}-2H-tetrazol-2-
y1)acetic acid;
and
(5- {2-[4-(5-bromo-2-chlorophenoxy)piperidin-1-ylipyrimidin-5-y1}-2H-tetrazol-
2-yl)acetic acid;
and pharmaceutically acceptable salts thereof.
- 41 -
WO 2012/009217 CA 02804970 2013-01-09
PCT/US2011/043330
Glucokinase activators that can be used in combination with the compounds of
any of the
formulas described herein include, but are not limited to:
3-(6-ethanesulfonylpyri din-3-yloxy)-5-(2-hydroxy- 1-methyl-ethoxy)-N-(1-
methy1-1H-pyrazol-3-
yl)benzamide;
5-(2-hydroxy-1-methyl-ethoxy)-3-(6-methanesulfonylpyridin-3-yloxy)-N-(1-methyl-
1H-pyrazol-
3-yObenzamide;
5-(1-hydroxymethyl-propoxy)-3-(6-methanesulfonylpyri din-3-yloxy)-N-(1-methy1-
1H-pyrazol-3
yl)benzamide;
3-(6-methanesulfonylpyri din-3-yloxy)-5-(1-methoxymethyl-propoxy)-N-(1 -methy1-
1H-pyrazol-
3-yl)benzamide;
5-isopropoxy-3-(6-methanesulfonylpyridin-3-yloxy)-N-(1-methy1-1H-pyrazol-3-
y1)benzamide;
5-(2-fluoro- I -fluoromethyl-ethoxy)-3 -(6-methanesulfonylpyri din-3 -yloxy)-N-
(1 -methyl-1H-
pyrazol-3-yl)benzamide;
3 -( 442-(dimethylamino)ethoxy] phenyl ) thio)-N-(3 -methy1-1,2,4-thiadiazol-5
-y1)-6- [(4-methyl -
4H-1,2,4-triazol-3-ypthio]pyridine-2-carboxamide;
3 -({ 4-[(1-methylazetidin-3 -yl)oxyl phenyl) thio)-N-(3 -methy1-1,2,4-
thiadiazol-5-y1)-6-[(4-methyl-
4H-1,2,4-triazol-3-ypthio]pyridine-2-carboxamide;
N-(3-methyl-1,2,4-thiadiazol-5-y1)-6-[(4-methyl-4H-1,2,4-triazol-3-y1)thio] -3-
{ [4-(2-pyiTolidin-
1-ylethoxy)phenyl]thiolpyridine-2-carboxamide; and
3-[(4- { 2- [(2R)-2-methylpyrro lidin- 1-yl] ethoxy phenyl)thio-N-(3-methy1-
1,2,4-thiadiazo1-5-y1)-6- [(4-
methy1-4H-1,2,4-triazol-3-y1)thiolpyridine-2-carboxamide; and pharmaceutically
acceptable salts
thereof.
Agonists of the GPR-119 receptor that can be used in combination with the
compounds of any of
the formulas described herein include, but are not limited to:
rac-cis 5-chloro-2- {44242- { [5-(methylsulfonyl)pyridin-2-yl]oxyl
ethyl)cyclopropyl] piperidin-l-
yllpyrimidine;
5-chloro-2- {4- [(1R,2S)-2-(2- { [5-(methylsulfonyl)pyridin-2-yl]oxy)
ethyl)cyclopropylipiperi din-
1-yllpyrimidine;
rac cis-5-chloro-2- [4-(2- {2- [4-(rnethylsulfonyl)phenoxy]
ethylIcyclopropyppiperidin-1
Apyrimidine;
5-chloro-2- [44(1S,2R)-2- {2- [4-(methylsulfonyl)phenoxy]ethyl) cyclopropyl)
piperidin-l-
yl]pyrimidine;
5-chloro-2-[4-((lR,2S)-2-1244-(methylsu1fonyl)phenoxyl ethyl) cyclopropyl)
piperidin-l-
yllpyrimidine;
rac cis-5-chloro-244-(2- {243 -(methylsulfon.yl)phenoxy] ethyl )
cyclopropyl)piperi din-1-
yll pyrimidine ; and
- 42 -
WO 2012/009217 CA 02804970 2013-01-09PCT/US2011/043330
rac cis -5-chloro-2- [442- {2- [3 -(5-methy1-1,3 ,4-oxadiazol-2-yl)phenoxyl
ethyl ) cyclopropyl)
piperidin-l-yl]pyrimidine; and pharmaceutically acceptable salts thereof.
Selective PPARy modulators (SPPARyM's) that can be used in combination with
the compounds
of any of the fotmulas described herein include, but are not limited to:
(25)-24 { 6-ohloro-3- [6-(4-chlorophenoxy)-2-propylpyrid in-3 -y1]-1,2-
benzisoxazol-5 -
yll oxy)propanoic acid;
(25)-2-({6-chloro-346-(4-fluorophenoxy)-2-propylpyridin-3-y1]-1,2-benzisoxazol-
5-
yl)oxy)propanoic acid;
(28)-2- [6-chloro-3 -(6-phenoxy-2-propylpyridin-3 -y1)-1 ,2-benzisoxazol-5-yll
oxy propanoic
acid;
(2R)-2-( {6-ch1oro-3-[6-(4-chlorophenoxy)-2-propylpyridin-3-y1]-1,2-
benzisoxazol-5-
yl)oxy)propanoic acid;
(2R)-2-{3-[3-(4-methoxy)benzoyl-2-methyl-6-(trifluoromethoxy)-1H-indol-1-
ylliphenoxy)butanoic acid;
(2S)-2-{3-[3-(4-methoxy)benzoy1-2-methy1-6-(trifluoromethoxy)-1H-indo1-1-
yljphenoxy)butanoic acid;
2- {3- [3 -(4-methoxy)benzoy1-2-methyl-6-(trifluoromethoxy)-1H-indol-1 -yll
phenoxyl -2-
methylpropanoic acid; and
(2R)-2- { 3 43 -(4-chloro)benzoy1-2-methyl-6-(trifluoromethoxy)-1H-indol -1-
yliphenoxy)propanoic acid; and pharmaceutically acceptable salts thereof.
Inhibitors of 1113-hydroxysteroid dehydrogenase type 1 that can be used in
combination
with the compounds of any of the formulas described herein include, but are
not limited to:
3- [1 -(4-chloropheny1)-trans-3-fluorocyclobutyl] -4,5 -dicyclopropyl-r-4H-
1,2,4-triazole ;3 - [1 -(4-
chloropheny1)-trans-3-fluorocyclobuty1]-4-cyclopropy1-5-(1-methylcyclopropy1)-
r-4H-1,2,4-
triazole;
341-(4-chloropheny1)-trans-3-fluoroeyclobutyl]-4-methyl-5-[2-
(trifluoromethoxy)phenyl]-r-4H-
1,2,4-triazole;
3- [1-(4-chlorophenyl)cyclobutyl] -4-methyl-5[2-(trifluoromethyl)phenyll -4H-
1,2,4-triazo le;
3- {4- [3-(ethylsulfonyl)propyl]bicyclo [2.2.2]oct-l-y1) -4-methyl-5- [2-
(trifluoromethyl)phenyl] -4H
-1,2,4-triazole;
4-methyl-3- 444-(m ethylsulfonyl)phenyl]bicycl o [2.2.2] oct-1 -y1) -5- [2-
(trifluoromethyl)phenyl] -
4H-1,2,4-triazole;
3-(4- {4-methy1-5-[2-(trifluoromethyl)phenyll-4H-1,2,4-triazol-3-
y1)bicyclo[2,2.2]oct-1-y1)-5-
(3,3,3 -trifluoropropy1)-1,2,4-oxadiazole;
3-(4-{4-methy1-5-[2-(trifluoromethyl)phenyl]-4H-1,2,4-triazol-3-
yllbicyclo[2.2.2]oct-1-y1)-5-
(3,3,3-trifluoroethyl)-1,2,4-oxadiazole,
-43 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
5-(3,3-difluorocyclobuty1)-3-(4-14-methy1-5-[2-(trifluoromethyl)pheny11-41-I-
1,2,4-triazol-3-
yl}bicycloP2.2]oct-l-y1)-1,2,4-oxadiazole;
5-(1-fluoro-1-methylethyl)-3-(4-{4-methyl-5-[2-(trifluoromethyl)phenyl]-411-
1,2,4-triazol-3-
yllbicyclo[2.2.2]oct-1-y1)-1,2,4-oxadiazole;
241,1 -difluoroethyl)-5 -(4- {4-methyl-5- [2-(trifluoromethyl)phenyl] -4I-1-
1,2,4-triazol-3 -
yl 1 bicyclo [2.2.2jjoet-l-y1)-1,3,4-oxadiazole;
2-(3,3-difluorocyclobuty1)-5-(4-14-methy1-5-[2-(trifluoromethyl)pheny1]-4H-
1,2,4-triazol-3-
y1) bicyclo [2.2.2]oct-1 -y1)-1,3 ,4-oxadiazole; and
5-(1,1-difluoroethyl)-3-(4-14-methy1-5-[2-(trifluoromethyl)pheny1]-4H-1,2,4-
triazol-3-
ylIbicyclo[2.2.2]oct-1-y1)-1,2,4-oxadiazole; and pharmaceutically acceptable
salts thereof.
Somatostatin subtype receptor 3 (SSTR3) antagonists that can be used in
combination
with the compounds of any of the formulas described herein include, but are
not limited to;
,,,OLN \ / F
=s\\\\LN \ / F
N
NH
141111 I NH
N
N
H N \----
HN
--- \
7 N¨
7
N¨H
/o/0
/N N -----<
5 -------/ N¨N 0 ----<0
,
HN \ ---_
HN \
---
s\\\LN \ / HO i N
0\\\\LN \ / F
N ,
0 1 NH
NH
N
N
----- \
/ N
/
\
N/
-----/ ---N ----< N¨ 0
, /
0 5
--
. N \ / F
'`µ\\\LN N
N
0 I NH
NH
N
N
H N
H
.......__N\
/ \NI (
N/ N
N
0 ,
0
,and
- 44 -
CA 02804970 2013-01-09
WO 2012/009217 PCT/US2011/043330
F
/ \
---N
N \
' N
I H
I
NH
N
H
0
N
( \ j >----0
/ 0 N -----N ,
and pharmaceutically acceptable salts thereof.
AMP-activated Protein Kinase (AMPK) activators that can be used in combination
with the
compounds of any of the formulas described herein include, but are not limited
to:
5
0
HO 10)
0 .,-O 0 00 NI)_.0 I.
CO2H CO2H
CI N CI N
H H
OH
0N SI N \ 0 0 N)-o 5
CO2H CO2H
CI N CI N
H H
,
5 OH
\
N
\ 411 to N)_0 Ill
0 0 N)-0 4111
C 02 H CO2H
N
CI FN
H H
\
N
0 F 1 F
õ..-k.,
N)___ ell \ . 0 N)0 411]
CO2H CO2H
CI N F N
H H
-45 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
1-13C0 F a
N)_.0 4111 CO2H
co2H
CI CI =N>_(:)
HO2C
411k
HO
40 N)__.
1\1___O 401 CO2H
, and NH =
and pharmaceutically acceptable salts thereof.
Inhibitors of acetyl-CoA carboxylase-1 and 2 (ACC-1 and ACC-2) that can be
used in
combination with the compounds of any of the formulas described herein
include, but are not limited to:
3-{1'-[(1-cyclopropy1-4-methoxy-1H-indo1-6-yl)carbonyl]-4-oxospiro[chroman-
2,4'-piperidin]-
6-yl}benzoic acid;
5- fl'-{(1-cyclopropyl-4-methoxy-1H-indo1-6-yl)carbonyli -4-oxospiro [chroman-
2,4'-piperidin] -6 -
yl}nicotinic acid;
l'-[(1-cyclopropy1-4-methoxy-1H-indol-6-yl)carbonyl]-6-(1H-tetrazol-5-
yl)spiro[chroman-2,4'-
piperidin]-4-one;
1'-[(1-cyclopropy1-4-ethoxy-3-methy1-1H-indo1-6-y1)carbony11-6-(1H-tetrazol-5-
yl)spiro[chroman-2,4'-piperidin]-4-one; and
5- [(1-cyclopropy1-4-methoxy-3-methy1-1H-indo1-6-ypcarbonyl]-4-oxo-
spiro[chroman-2,4'-
piperiditi]-6-y1}nicotinic acid; and
pharmaceutically acceptable salts thereof
In another aspect of the invention, a pharmaceutical composition is disclosed
which comprises
one or more of the following agents:
(a) a compound of any of the structural formulas described herein;
(b) one or more compounds selected from the group consisting of:
(1) dipeptidyl peptidase-1V (DPP-4) inhibitors;
(2) insulin sensitizers, including (i) PPARy agonists, such as the glitazones
(e.g.
pioglitazone, rosiglitazone, netoglitazone, rivoglitazone, and balaglitazone)
and other PPAR ligands,
including (1) PPARa/y dual agonists, such as muraglitazar, aleglitazar,
sodelglitazar, and naveglitazar,
(2) PPARa agonists, such as fenofibric acid derivatives (gemfibrozil,
clofibrate, ciprofibrate, fenofibrate
and bezafibrate), (3) selective PPARy modulators (SPPARTM's), and (4) PPARy
partial agonists; (ii)
biguanides, such as metformin and its pharmaceutically acceptable salts, in
particular, metformin
- 46 -
CA 02804970 2013-01-09
WO 2012/009217 PCT/US2011/043330
hydrochloride, and extended-release formulations thereof, such as Glumetza ,
Fortamet , and
GlucophageXR0; (iii) protein tyrosine phosphatase-1B (PTP-1B) inhibitors;
(3) sulfonylurea and non-sulfonylurea insulin secretagogues, such as
tolbutamide,
glyburide, glipizide, gliniepiride, mitiglinide, and meglitinides, such as
nateglinide and repaglinide;
(4) a-glucosidase inhibitors (such as acarbose, voglibose and miglitol);
(5) glucagon receptor antagonists;
(6) LDL cholesterol lowering agents such as (i) HMG-CoA reductase inhibitors
(lovastatin, simvastatin, pravastatin, cerivastatin, fluvastatin,
atorvastatin, pitavastatin, and rosuvastatin),
(ii) bile acid sequestering agents (such as cholestyramine, colestimide,
colesevelam hydrochloride,
colestipol, and dialkylaminoalkyl derivatives of a cross-linked dextran, (iii)
inhibitors of cholesterol
absorption, such as ezetimibe, and (iv) acyl CoA:cholesterol acyltransferase
inhibitors, such as
avasimibe;
(7) HDL-raising drugs, such as niacin or a salt thereof and extended-release
versions
thereof; MK-524A, which is a combination of niacin extended-release and the DP-
1 antagonist MK-524;
and nicotinic acid receptor agonists;
(8) antiobesity compounds;
(9) agents intended for use in inflammatory conditions, such as aspirin, non-
steroidal anti-
inflammatory drugs (NSAIDs), glucocorticoids, and selective cyclooxygenase-2
(COX-2) inhibitors;
(10) antihypertensive agents, such as ACE inhibitors (such as enalapril,
lisinopril,
ramipril, captopril, quinapril, and tandolapril), A-II receptor blockers (such
as losartan, candesartan,
irbesartan, olmesartan medoxomil, valsartan, telmisartan, and eprosartan),
renin inhibitors (such as
alisldren), beta blockers (such as and calcium channel blockers (such as;
(11) glucokinase activators (GKAs), such as LY2599506;
(12) inhibitors of 1 113-hydroxysteroid dehydrogenase type 1;
(13) inhibitors of cholesteryl ester transfer protein (CETP), such as
torcetrapib and MK-
0859;
(14) inhibitors of fructose 1,6-bisphosphatase;
(15) inhibitors of acetyl CoA carboxylase-1 or 2 (ACC1 or ACC2);
(16) AMP-activated Protein Kinase (AMPK) activators;
(17) agonists of the 6-protein-coupled receptors: GPR-109, GPR-119, and GPR-
40;
(18) SSTR3 antagonists;
(19) neuromedin U receptor agonists, including, but not limited to, neuromedin
S (NMS);
(20) inhibitors of stearoyl-coenzyme A delta-9 desaturase (SCD);
(21) (}PR-105 antagonists;
(22) inhibitors of glucose uptake, such as sodium-glucose transporter (SGLT)
inhibitors
and its various isoforms, such as SGLT-1; SGLT-2, such as dapagliflozin and
remogliflozin; and SGLT-
3;
-47-
WO 2012/009217 CA 02804970 2013-01-09
PCT/US2011/043330
(23) inhibitors of acyl coenzyme A:diacylglycerol acyltransferase 1 and 2
(DGAT-1 and
DGAT-2);
(24) inhibitors of fatty acid synthase;
(25) inhibitors of acetyl-CoA carboxylase-1 and 2 (ACC-1 and
ACC-2);
(26) inhibitors of acyl coenzyme A:monoacylglycerol acyltransferase 1 and 2
(MGAT-1
and MGAT-2);
(27) agonists of the TGR5 receptor (also known as GPBAR1, B037, GPCR19,
GPR131,
and M-BAR); and
(28) bromocriptine mesylate and rapid-release formulations thereof; and
(c) a pharmaceutically acceptable carrier.
When a compound of the present invention is used contemporaneously with one or
more other
drugs, a pharmaceutical composition containing such other drugs in addition to
the compound of the
present invention is preferred. Accordingly, the pharmaceutical compositions
of the present invention
include those that also contain one or more other active ingredients, in
addition to a compound of the
present invention.
The weight ratio of the compound of the present invention to the second active
ingredient may be
varied and will depend upon the effective dose of each ingredient. Generally,
an effective dose of each
will be used. Thus, for example, when a compound of the present invention is
combined with another
agent, the weight ratio of the compound of the present invention to the other
agent will generally range
from about 1000:1 to about 1:1000, preferably about 200:1 to about 1:200.
Combinations of a
compound of the present invention and other active ingredients will generally
also be within the
aforementioned range, but in each case, an effective dose of each active
ingredient should be used.
In such combinations the compound of the present invention and other active
agents may be
administered separately or in conjunction. In addition, the administration of
one element may be prior
to, concurrent to, or subsequent to the administration of other agent(s).
Examples
Intermediates and Examples are shown below.
Scheme 1
- 48 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
Oxone
2
H
X = F or CI
HN / )(R1 base
\ R"
')<R'R---
:
C% ¨NI
H KIN R"
The following is a list of abbreviations used in the description of the
synthesis of the
Inteimediates and Examples shown below.
List of Abbreviations:
Alk
= alkyl
Ar
= aryl
Boc
= tert-butoxycarbonyl
Cbz
= carbobenzyloxy
CH2C12
= dichloromethane
CoA
= Coenzyme A
dba
= dibenzylidineacetone
DIPEA
= diisopropylethylamine
DMA
= dimethylacetamide
DMF
= dimethylformamide
DMSO
= dimethyl sulfoxide
ESI
= electrospray ionization
Et
= ethyl
Et0Ac
= ethyl acetate
HOAc
= acetic acid
LC-MS
= liquid chromatography-mass spectroscopy
LiOH
= lithium hydroxide
Me
= methyl
Me0H
= methyl alcohol
MgSO4
= magnesium sulfate
MS
= mass spectroscopy
- 49 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
MTBE = methyl tert-butyl ether
NaOH = sodium hydroxide
Na2SO4 sodium sulfate
NMR nuclear magnetic resonance spectroscopy
Ph = phenyl
it or RT = room temperature
TEA triethylamine
TFA = trifluoroacetic acid
THF tetrahydrofuran
Intermediate I.
FaC N
N N
2-(6-fluoropyridin-3-y1)-5-(trifluoromethyl)- I H-benzimidazo le
Step 1. Following the recommendations presented in Beaulieu, P. L.; Hache, B.
and von Moos,
E. Synthesis, 2003, 11, 1683: a solution of 2-fluoropyridine-5-carboxaldehyde
(2.0 g) in DMF
was added dropwise to a suspension of 4-(trifluoromethyl)phenylene-1,2-
diarnine (2.8 g) and
Oxone (6.39 g) in DMF and water. The mixture was cooled in an ice bath during
the addition
and allowed to warm to reaction once the addition of aldehyde was complete.
The reaction was
stirred open to air at room temperature for 45 minutes. To the reaction was
added water and the
mixture was brought to pH 7 by the addition of solid potassium carbonate. The
resulting light
brown solid was filtered and dried in vacuo. [MH] m/z 282.8.
Intermediate 2.
CI r\i>
N N ¨CI
5-chloro-2-(6-chloropyridin-3-y1)-1H-benzimidazole
Step 1. Following the recommendations presented in Beaulieu, P. L.; Hache, B.
and von Moos,
E. Synthesis, 2003, /1, 1683: a solution of 2-chloropyridine-5-carboxaldehyde
(0.60 g) in DMF
was added clropwise to a suspension of 4-chlorophenylene-1,2-diamine (0.57 g)
and Oxonee (1.6
g) in DMF and water. The mixture was cooled in an ice bath during the addition
and allowed to
warm to reaction once the addition of aldehyde was complete. The reaction was
stirred open to
air at room temperature for 45 minutes. To the reaction was added water and
the mixture was
- 50 -
WO 2012/009217
CA 02804970 2013-01-09
PCT/US2011/043330
brought to pH 7 by the addition of solid potassium carbonate. The resulting
light brown solid
was filtered and dried in vacua. [ME] m/z 264Ø
Intermediate 3.
=el N
5-chloro-2-(6-chloro-5-fluoropyridin-3-y1)-1H-benzimidazole
This compound was prepared using the same protocols as described for
Intermediate 1.
Intermediate 4.
meo^e'N N
2-(6-fluoropyridin-3-y1)-5-methoxy-3114midazo [4,5 -bl pyridine
This compound was prepared using the same protocols as described for
Inteimediate 1.
Intermediate 5
F3C ¨4) ¨Br ¨N
2-(6-bromo-5-fluoropyridin-3 -y1)-5-(trifluoromethy1)-1H-benzo [d] imidazole
Step I . 4-(trifluoromethyl)benzene-1,2-diamine (44.0 mg, 1.1 equiv), 6-bromo-
5-
fluoronicotinaldehyde (50 mg, 1 equiv) and (benzotriazol-1-
yloxy)tripyrrolidinophosphonium
hexafluorophosphate (130 mg, 1 equiv) were combined in anhydrous DMF. To the
solution,
DMA (0.119 mL, 3 equiv) was added and the mixture was stirred at room
temperature for 2
hours. The solution was diluted with ethyl acetate and washed with saturated
ammonium
chloride, then water then brine. The organic layer was dried over anhydrous
sodium sulfate,
filtered and evaporated to give N-(2-amino-4-(trifluoromethyl)pheny1)-6-bromo-
5-
fluoronicotinamide as a viscous brown oil: [Miff' m/z 379.
Step 2. Acetic acid was added to N-(2-amino-4-
(trifluoromethyl)pheny1)-6-bromo-5-
fluoronicotinamide obtained in step 1, and the solution was irradiated in a
microwave reactor at
180 C for 40 min. The solution was diluted with 40% acetonitrile-water (4 mL)
and purified by
- 51 -
CA 02804970 2013-01-09
WO 2012/009217 PCT/US2011/043330
reverse phase HPLC to give 2-(6-bromo-5-fluoropyridin-3-y1)-5-
(trifluoromethyl)-1H-
benzo[dlimidazole as a tan solid: [MFI] nilz 361.
Intermediate 6.
0
0
HN CO2H
3-oxo-3H-spiro[2-benzofuran-1,41-piperidinej-5-carboxy1ic acid
Step 1. 4-bromobenzene-1,3-dicarboxylic acid (600 mg, 1 equiv) was taken up in
THF (12 mL)
and cooled to -78 C. A 2.5 M solution of n-butyl lithium in hexanes (3.92 mL,
4 equiv) was
added dropwise over 15 minutes forming a red precipitate. After 2 hours, tert-
butyl 4-
oxopiperidine-l-carboxylate (488 mg, 1 equiv) was added dropwise over 10
minutes as a
solution in THF. The final concentration was 0.15 M. After 2 hours, the
reaction mixture was
warmed to room temperature, acidified to pH ¨ 0 with 1M HCI and stirred
vigorously for 16
hours. Volatiles were removed in vacuo and the residue poured into water then
extracted with
dichloromethane. The organic layer was dried on sodium sulfate and
concentrated to give 1'-
(tert-butoxycarbony1)-3-oxo-3H-spiro[2-benzofaran-1,41-piperidine]-5-
carboxylic acid as a white
oil: [MH¨Bocr m/z 248.
Step 2. The residue from Step 1 (79 mg, 1 equiv) was reconstituted in
dichlorometha.ne (40 mL)
and trifluoroacetic acid (3:1) and stirred for 18 hours at room temperature.
Concentration and
purification by reverse phase HPLC (0% acetonitrile:water with 0.05% TFA to
50%
acetonitrile:water with 0.05% TFA) to give pure 3-oxo-3H-spiro[2-benzofuran-
1,4'-piperidine]-
5-carboxylic acid as a white solid: [MFI] m/z 248.
Intermediate 7.
0
0
/
N_ Br
3-bromo-5H-spiro[furo[3,4-blpyridine-7,41-piperidin]-5-one
Step 1. 5-bromo-2-iodopyridine-3-carbonitrile (6.44 g, 1 equiv) and tert-butyl
4-oxopiperidine-
1-carboxylate (5.82 g, 1.4 equiv) taken up in toluene (65 mL) and cooled in -
78 C bath. A 1.3M
solution of isopropylmagnesium chloride-lithium chloride complex in THF (22.5
mL, 1.4 equiv)
was added rapidly forming a brown-black gel. After 10 minutes, the reaction
was quenched by
addition of methanol (5.9 mL, 7 equiv) then 50% aqueous acetic acid (16.7 mL,
7 equiv) and
warmed to room temperature and stirred for 24 hours. The mixture was poured
into toluene,
washed with water, 1N HC1 then IN NaOH. The organic layer was dried on sodium
sulfate and
- 52 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
concentrated to a brown oil. Purification by silica gel chromatography
(hexanes to ethyl acetate)
gave tert-butyl 3-bromo-5-oxo-1'H,5H-spiro[furo[3,4-b]pyridine-7,4'-
piperidine]-1'-carboxylate
as an oil.
Step 2. The oil from Step 1 (204 mg) was reconstituted in 4 mL dichloromethane-
trifluoroacetic
acid (3:1) and stirred for 5 hours at room temperature to effect Boc-removal.
Concentration gave
the hydrochloride salt which was used without further purification [MI-Ir m/z
283.
Intermediate 8.
0
OH
0 ¨N
2-(3-(5-formylpyridin-2-y1)-3-azaspirof5.5jundecan-9-y1)acetic acid
Sodium bicarbonate (2.70 g, 5 equiv), 2-fluoropyridine-6-carboxaldehyde (0.80
g, 1 equiv) and
the hydrochloride salt of 3-azaspiro[5.5]undec-9-ylacetic acid (1.59 g, 1
equiv) were stirred at 80
C in NMP (12 mL) for 5 hours. The mixture was cooled to room temperature,
acidified with
1M HC1 (25 mL, 4 equiv), diluted with water, and extracted with
dichloromethane. The
combined organic layers were concentrated in vacuo and purified by silica gel
chromatography
(0% acetone:dichloromethane to 50% acetone:dichloromethane) to give the pure
aldehyde as a
white solid: [MHr rn/z 317.
Intermediate 9.
Me02C
0 CO2Me
methyl 4-(2-methoxy-2-oxoethyl)-3,4-dihydrospiro[chromene-2,4'-niperidinej-6-
earboxy1ate
Step 1. Trimethyl phosophonoacetate (680 pL, 2.1 equiv) was added dropwise to
a stirring
suspension of sodium hydride (160 mg, 2 equiv, 60% dispersion in mineral oil)
in THF (8 mL) at
room termperature and the slurry stirred for 30 minutes. The reaction mixture
was cooled to 0 C
and 1'-tert-buty1-6-methyl 4-oxo-3,4-dihydro-1'H-spiro[ehromene-2,41-
piperidine]-dicarboxylate
(750 mg, 1 equiv) was added dropwise as a solution in THF (4 mL). The reaction
mixture was
slowly warmed to room temperature and stirred for 16 hours. Volatiles were
removed in vacuo
and the residue was transferred to a separatory funnel containing 1M
hydrochloric acid.
Extraction with diehloromethane and concentration gave an oil that was
purified by silica gel
column chromatography (hexanes to ethyl acetate) to give the desired product
as an oil.
- 53 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
Step 2. Material from Step 1 (226 mg, 1 equiv) was dissolved in ethanol (2 mL)
and added to
30% Pd/C (125 mg, 35 mol%) then stirred at room temperature under an
atmosphere of hydrogen
for 4 hours, Filtration through Celite, concentration in vacua, and
purification by silica gel
column chromatography (hexanes to ethyl acetate) gave the desired product as a
clear oil.
Step 3. The oil from Step 2 (154 mg) was treated with hydrochloric acid (2
equiv) in 1,4-dioxane
(1.7 mL) at 40 'C. After 2 hours, an additional equivalent of 4M hydrochloric
acid in 1,4-
dioxane (89 RL) was added and the temperature raised to 60 C for 1 hour.
Removal of volatiles
in vacuo gave the desired hydrochloride salt as a white solid: [M1-1]+ in/z
334.
Intermediate 10.
0 0 0
) N 41) OMe
tert-butyl 9- (2-methoxy-2-oxoethoxy)-3 -azaspiro 15 .51undecane-3 -
carboxylate
Step 1. Sodium borohydride (139 mg, 1 equiv) was added to a methanol solution
of tert-butyl 9-
oxo-3-azaspiro[5.51undecane-3-carboxylate (980 mg, 1 equiv) and the mixture
was stirred at
room temperature for 1 hour. Saturated aqueous sodium bicarbonate was added to
the solution
and the mixture was diluted with dichloromethane. The organic layer was
separated and washed
with water then brine then dried over sodium sulfate, filtered and
concentrated in vacuo to give
950 mg of tert-butyl 9-hydroxy-3-azaspiro[5.5]undecane-3-carboxylate: [Mlir
m/z 270.
Step 2. Potassium tert-butoxide (66.5 mg, 1.2 equiv) was added to tert-butyl 9-
hydroxy-3-
azaspiro[5.5]undecane-3-carboxylate (133 mg, 1.0 equiv) in anhydrous DMF and
stirred at 0 C
for 1 hour. To the stirred pale yellow solution was added methyl 2-
bromoaeetate (0.070 mL, 1.5
equiv) and the solution was warmed to room temperature over 1 hour and then
heated at 50 C
for 1 hour. The solution was cooled to room temperature, diluted with 40%
acetonitrile in water
then directly purified by preparative HPLC (20% to 70% acetonitrile in water
with 0.05% TFA
as eluant) to give the product as an off-white solid: [MH1+ tn/z 342.
Example 1.
F3 N
r
N ¨N
CO2H
- 54 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
Step 1. Copper (1) iodide (19 mg, 1 equiv), cesium carbonate (98 mg, 3 equiv)
and commercially
available tert-butyl 2-oxo-1-oxa-3,8-diazaspiro[4.5]decane-8-carboxylate (26
mg, 1 equiv) were
weighed into a vial. To this was added ethyl p-bromophenylacetate (32 mg, 1.3
equiv) and 1,4-
dioxane (0.25 mL) and the mixture stirred at 100 C for 18 hours. After
cooling, the reaction was
quenched by addition of 1M HCI (1 rniõ 10 equiv) and extracted twice with
MTBE. The
combined organic layers were concentrated in vacuo.
Step 2. The residue from Step 1 was dissolved in 1M solution of HC1 in 1,4-
dioxane (400 pL, 4
equiv) and the mixture stirred at room temperature for 4 hours to effect
removal of the Boc
group. Concentration in vacuo gave a crude residue which was carried forward
without further
purification.
Step 3. To the residue from Step 2 was added sodium bicarbonate (42 mg, 5
equiv), 2-(6-
fluoropyridin-3-y1)-5-(trifluoromethyl)-1H-benzimidazole (28 mg, 1 equiv), and
NMP (330 4).
The mixture was stirred at 120 C for 22 hours. The reaction was neutralized
by addition of
acetic acid (231,tL, 4 equiv), diluted with DMSO, filtered and purified by
reverse phase HPLC.
Examples 2-14 were prepared according to a similar method. For examples where
the aryl group
was a pyridine, the aqueous phase from the workup in Step 1 was basified with
1M NaOH and
further extracted with ethyl acetate.
Table 1. F3c N
N ¨N sR
[M111 iymr 1111Hr [M11]+
Example R in/z rn/z Example R
nez /Fez
cale'd found ealc'd found
1 CO2H 552 552 8
563 563
0
2 111 CO2H 594 594 9 = Me Me 508
508
3 co2n 568 568 10
508 508
4 CO2Et 580 580 11 CF3 562
562
- 55 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
it OH F3C
CF3 592 592 12 562
562
6 411 CN 519 519 13
N 495 495
MeO2C
7 11 NO2 539 539 14
411 552 552
Example 15.
cl N 0 0
N N
5 5-chloro-2-(6-chloroppidin-3-y1)-1H-benzimidazole (123 mg, 1 equiv),
Hunig's base (408 uL, 5
equiv) and 3H-spiro[2-benzofuran-1,4'-piperidin]-3-one (95 mg, 1 equiv) were
dissolved in
DMA (3 mL) and heated at 140 C in a microwave for 12 hours. The mixture was
poured into
water and extracted with MTBE thrice. The combined organic fractions were
dried on
magnesium sulfate, filtered, concentrated, then purified by column
chromatography on silica gel
using gradient elution (20% ethyl acetate:hexanes to 100% ethyl acetate). The
title compound
was obtained as a pale yellow solid: [M1-1]+ m/z 431.
Examples 16-56 (Table 2) were prepared according to the procedure described
above or one of
the variations described below.
Procedure A
A 5-substituted-2-(6-chloropyridin-3-y1)-1H-benzirnidazole (1 equiv), Hunig's
base (5 equiv) and
the amine (1 equiv), as a hydrochloride or trifluoroacetate salt, were
dissolved in DMA (0.3 M)
and heated at 140 C in a microwave until complete as judged by LCMS analysis
(10-60 hours).
If the conversion of starting materials to products was low within 3 hours
(LCMS analysis), the
reaction temperature was increased to 200 C. If decomposition products were
observed to form
at a comparable rate to the consumption of starting material, the reaction was
stopped prior to
complete conversion. The mixture was cooled to room temperature, poured into
water and
extracted with MTBE thrice. The combined organic fractions were dried on
magnesium sulfate,
filtered, concentrated, then purified by column chromatography on silica gel
using gradient
elution (generally 20% ethyl acetate:hexanes to 100% ethyl acetate) to give
the pure product.
Procedure B
- 56 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
Performed as in Procedure A except potassium carbonate (5 equiv) was used as
base and DMSO
(0.3 M) was used as solvent. The reaction was heated at 140 C in a microwave
until complete
as judged by LCMS analysis (10-60 hours). The mixture was cooled to room
temperature,
diluted with a small amount of acetonitrile, filtered and purified by
preparative reverse phase
HPLC (generally 30% acetonitrile:water with 0.05% TFA to 95%
acetonitrile:water with 0.05%
TFA) to give the pure product.
Procedure C
Performed as in Procedure A except a 5-substituted-2-(6-fluoropyridin-3-y1)-1H-
benzirnidazole
(1 equiv) was used. The reaction was heated at 140 "C in a microwave until
complete as judged
by LCMS analysis (10-60 hours). The mixture was cooled to room temperature,
diluted with a
small amount of acetonitrile, filtered and purified by preparative reverse
phase HPLC (generally
30% acetonitrile:water with 0.05% TFA to 95% acetonitrile:water with 0.05%
TFA) to give the
pure product.
Procedure D
Performed as in Procedure A except sodium bicarbonate (5 equiv) was used as
base and NMP
(0.3 M) was used as solvent. The reaction was heated at 110 C until complete
as judged by
LCMS analysis (10-60 hours). The mixture was cooled to room temperature,
diluted with a
small amount of DMSO, filtered and purified by preparative reverse phase HPLC
(generally 30%
acetonitrile:water with 0.05% TFA to 95% acetonitrile:water with 0.05% TFA) to
give the pure
product.
Procedure E
Performed as in Procedure A except A 5-chloro-2-(6-chloro-5-fluoropyridin-3-
y1)-1H-
benzimidazole (1 equiv) was used as electrophile.
Procedure F
Perfatmed as in Procedure D except the product was saponified with aqueous
sodium hydroxide
or lithium hydroxide in a water miscible organic solvent such as THF,
methanol, dioxane or
DMA, or a mixture thereof, at room temperature or 60 C. Carboxylic acid
products were
subjected to preparative reverse phase HPLC (generally 30% acetonitrile:water
with 0.05% TFA
to 95% acetonitrile:water with 0.05% TFA) to give the pure product.
Table 2.
Example Structure
1111H1+ twir procedure
m/z m/z
- 57 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
calc'd found
0 0
H \)--N
16 N -N .
417 417 A
H
_
_
CI 0 N,..) g ,,,,,) N/ 0,7-0
17 N \¨N
432 432 B
H
N \ /
CI. N\> , N/
18 N \ ¨N
, 432 432 A
H
/
N
. CI 40 I\1 i N/ 0----,----0
19 N \------N
432 432 A
H /
\ N
,
F3C 0 N
, e d 'o
20 N \¨N \
466 466 D
H
N \ i
CI 0 N\\_// ____N 0--_. 0
21 N"¨N --
432 432 A
H N
\ /
CI 0
= N )--< --1\1
N -N 435 435 B
22
H
.
F
CI \\ 0 0
SIN \>, ---- \)--N
N -N
23 H
475 475 B
OH
0
F
CI N i \ 0 0
0 , . ---1\1
24
449 449 E
N -N
H
_
_<
F3C so N , õ Nir 0-4% 0
25 N \ -N
544 544 C
H
N \ i
Br
,
- 58 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
F3 0 N "
0 0
"------C ----N
N -N
26
H
509 509 C
CO2H
_
0
F3 40N õ
\ / \ N =
27
N -N
.
463 463 C
H
,
F3C 0 N õ
0 Me
\28
N"/ -N \ N
465
465
C
H
=
.
0
F3C 401 I \1'$ N/ X
N
(-N
N \ /
29
\ 12
480 480 C
Me
,
F3C0N, i ..-rµ17
N \--N \
30
H
N\ /
514
514
C
a
Me
-
H
Me0,....,-N
1
31
/ .N
)1_,
\ / N
429 429 D
"-----------^N
N
NI µ /
F3C 40 N
(.,:\----
/ "N a
CO2H
32
N -N
.
493 493 D
H
0
CO2H
499 499 D
,
F3C
N N (,,,, \\
)___N a
33
-N
ilk
H
0
F3C 40 NI,
OH
34
\)---N 11,
473 473 D
N -N
H
0
35
0 INI/ \'-N J ___1\1
it
0H
405 405 D
N
H
-59-
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
-
F3C 40 N , \\
I "¨N it 0
36
459 459 D
N ¨N OH
H
0\
\--OH
37 F3C ,N \\
509 509 D
7 \)---N
N ¨N 0 40
H
Me02C ,
F3C0 N , \\
38
595 595 D
\>--- )---- N
N ¨N 0 104 CO2Me
H
HO2C
F3C 0 N , \\
39
581 581 F
\>----C \----N
N ¨N 0 40 CO2Me
H
HO2C
F3C 0 N , \\
40
567 567 F
\>---- )----N
N ¨N 0 404 CO2H
H
=
, 1,0
\
F3C 010 N, ,,,, õ OH
41
523 523 D
\ N
N ¨N 0 =
H
0
F3C 0 N õ
42 \ / \ N
572 572 D
N ¨N 0 4111 NHSO2Me
H
0
F3C 40 N> , \\ 0 /7 \
--- --N NH
43
480 480 D
N --N
H
. ,
Me
44 F3C 40N \\ 410
521 521 D
`i¨N a 0
N ¨N
_ H
OH
F3C 0 N
, NSO2Me
-----< ----r\1
45 N ¨N
528 528 D
H
1 4Ik
'
- 60 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
_
,
H r,
F3C 0 N / N-.f-
\>-----( ---N
N
46 N -N CN
532 532 D
H 0 1110
,.
F3C 0 N , \\
----- "---N 1110 F 525 525
D
0--N-0 -
H
F3C . N , \\
\hNr-)Cr
N
48 N -N
536 536 C
H
1410
CO2H
F3C si N , ,\ / .,._0
\)----- )---N k 1 =
49 N -N \ N
538 538 C
H
110 CO2H
, F3C 0 N / ,0
50 ---t., \---N' X,C._
418 418 D
N -N \ NH
H
F3C 0 \ ,
K._ -o 0
51 Nk
476 476 F
N -N
H OH
F3C . N , c \\ / \\./0--..õ-:-
7 \)--N 0
52
' 504 504 F
NA---NN-----\A
H OH _
0- N
\)--< --N I
F3C 0N N -N
53 H 11 OH 522 522 D
0
0
Cl, r\i.___j N 0-N
I
54 H 40 OH 488 488
D
, 0
F3C . N / \\
\>--(,_ ---N 111 0 OH
55 \489 489 F
N -N
H
Example 56.
- 61 -
WO 2012/009217 CA 02804970 2013-01-09
PCT/US2011/043330
C N 0 0
4Ik
Step 1. Cesium carbonate (337 mg, 1.4 equiv), Pd2(dba)3 (17 mg, 2.5 mol%) and
RuPhos (69
mg, 20 mol%) were transferred to a vial which was sealed and flushed with
nitrogen. Degassed
THF (3 mL) was added followed by a solution of 3H-spiro[2-benzofuran-1,41-
piperidini-3-one
(165 mg, 1.1 equiv) and 4-bromo-3-fluorobenzaldehyde (150 mg, 1 equiv) in
degassed THF (1
mL). The reaction mixture was heated at 70 C for 5 hours then cooled to room
temperature.
The mixture was poured into water, extracted with ethyl acetate, then purified
by silica gel
chromatography (0% to 40% ethyl acetate:hexanes) to give 3-fluoro-4-(3-oxo-
l'H,3H-spiro[2-
benzofuran-1,4r-piperidin]-1t-yl)benzaldehyde: m/z 326.
Step 2. Next, a solution of 3-fluoro-4-(3-oxo-11-1,3H-spiro[2-benzofuran-1,4'-
piperidini-l'-
yObertzaldehyde (11 mg, 1 equiv), Oxone (13 mg, 0.65 equiv), and 4-
chlorophenylene-1,2-
diamine (5 mg, 1 equiv) in 1.4 mL DMF-water (30:1) was stirred open to air at
room
temperature. After 2 hours, water was added and the mixture was extracted with
ethyl acetate.
The organic layer was concentrated and the residue purified by preparative
reverse phase HPLC
(30% acetonitrile:water with 0.05% TFA to 95% acetonitrile:water with 0.05%
TFA) to give the
pure product as a solid: [M1-1]- m/z 448.
Example 57.
F3C N OH
=N =0
To a vial containing ethyl 2-(3-azaspiro[5.51undecan-9-yeacetate (50 mg, 1
equiv) was added
(BrettPhos)palladium(II) phenethylamine chloride (CAS: 1148148-01-9, 33.4 mg,
20 mol%) and
2-(4-bromo-2-fluoropheny1)-6-(trifluoromethyl)-1H-benzo[d]imidazole (75 mg, 1
equiv). The
vial was capped under nitrogen then THF (1 mL) was added through the septum
and the
suspension was sparged with nitrogen for 5 minutes. Then, a 1.7 M solution of
potassium tert-
butoxide in THF (381111,, 3.1 equiv) was added dropwise inducing a deep, wine-
red color. After
stirring for 20 hours at room temperature, water (1 mL) and methanol (1 mL)
were added and the
mixture stirred for a further two hours. Volatiles were then removed in vacua.
The residue was
dissolved in a mixture of acetic acid and DMSO and purified by preparative
reverse phase HPLC
(10% acetonitrile:water with 0.05% TFA to 90% acetonitrile:water with 0.05%
TFA) to give the
pure product as a solid: [wir- nilz 490.
- 62 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
Example 58.
0
F3C
N v
OH
N
N
Step 1. 2-(3-(tert-butoxycarbony1)-3-azaspiro[5.5]undecan-9-yl)acetic acid
(750 mg) was taken
up in dioxane (6 mL) to which was added a 4 M solution of hydrochloric acid in
dioxane (2.4
mL, 4 equiv). The mixture was heated at 60 C for 3.5 hours then concentrated
in vacuo.
Step 2. To the residue from Step 1 was added solid sodium bicarbonate (1012
mg, 5 equiv) and
5-chloropyrazine-2-carbaldehyde (400 mg, 1.2 equiv) and DMF (8 mL). The
mixture was stirred
at 60 C for 18 hours then cooled and poured into water and the mixture
neutralized with 1 M
HC1. Extraction first with ether then with ethyl acetate gave an organic
fraction that was washed
four times with water, then dried on anhydrous sodium sulfate, filtered and
concentrated in vacuo
to give an orange solid that was carried forward crude: [MH] m/z 318.
Step 3. The aldehyde from Step 2 (50 mg, 1 equiv), 4-(trifluoromethypbenzene-
1,2-diamine (28
mg, 1 equiv) and Oxone (63 mg, 0.65 equiv) were weighed to a vial. To this was
added DMF
(254 !AL) and water (10 fit) and the mixture stirred at room temperature for
90 minutes. The
mixture was poured into water and neutralized with solid potassium carbonate
then the brown
precipitate was collected by filtration. The brown solid was dissolved with
DMSO and the
residue purified by preparative reverse phase HPLC (20% acetonitrile:water
with 0.05% TFA to
70% acetonitrile:water with 0.05% TFA) to give the pure product as a brown
solid: [MI11+ rn/z
474.
Example 59.
0
,N
OH
NH
F3C
Step 1. 2-(3-(tert-butoxycarbony1)-3-azaspiro[5.5]andecan-9-ypacetic acid (208
mg) was taken
up in dioxane (2 mL) to which was added a 4 M solution of hydrochloric acid in
dioxane (1 mL,
6 equiv). The mixture was heated at 60 C for 1.5 hours then concentrated in
vacua.
Step 2. To the residue from Step 1 was added solid sodium bicarbonate (281 mg,
5 equiv) and 2-
chlorothiazole-5-carbaldehyde (99 mg, 1.0 equiv) and DMF (2.2 mL). The mixture
was stirred at
60 C for 15 hours then cooled and filtered through a plug of cotton and
diluted with DMF to a
- 63 -
WO 2012/009217 CA 02804970 2013-01-09
PCT/US2011/043330
final volume of 6 mL. The resulting orange solution was carried forward to
subsequent reactions
assuming a concentration of 0.11 M: [MH]t mtz 323.
Step 3. The aldehyde solution from Step 2 (1 mL, 1 equiv), 4-
(trifluoromethyl)henzene-1,2-
diamine (19.4 mg, 1 equiv) and Oxone (43 mg, 0.64 equiv) were weighed to a
vial. To this was
and water (32 4) and the mixture stirred at room temperature for 16 hours. The
reaction was
quenched by the addition of a few drops of saturated sodium thiosulfate then
the mixture was
diluted with DMSO, filtered and purified by preparative reverse phase HPLC
(20%
aeetonitrile:water with 0.05% TFA to 70% aeetonitrile:water with 0.05% TFA) to
give the pure
product as a pale yellow solid: [mfir 479.
The compounds presented in Table 3 were prepared by a similar method.
Table 3.
[Mff] imnr
Example Structure
neZ nilZ
0 OH caled found
60 r\lj N 111
511 511
F3CS NH ,N 111 0 OH
61 Cl = NH
459 459
Example 62. Me
,,c N 0 OH
=\>¨(3--NN
Step 1. 4-(trifluoromethyl)benzene4,2-diamine (44.0 mg, 1.1 equiv), 6-
bromo-5-
fluoronicotinaldehyde (50 mg, 1 equiv) and (benzotriazol-1-
yloxy)tripyrrolidinophosphonium
hexafluorophosphate (130 mg, I_ equiv) were combined in anhydrous DMF. To the
solution,
DIEA (0.119 mL, 3 equiv) was added and the mixture was stirred at room
temperature for 2
- 64 -
CA 02804970 2013-01-09
WO 2012/009217 PCT/US2011/043330
hours. The solution was diluted with ethyl acetate and washed with saturated
ammonium
chloride, then water then brine. The organic layer was dried over anhydrous
sodium sulfate,
filtered and evaporated to give N-(2-amino-4-(trifluoromethyl)pheny1)-6-bromo-
5-
fluoronicotinamide as a viscous brown oil: [MH]+ m/z 379.
Step 2. Acetic acid was added to N-(2-amino-4-(trifluoromethyl)pheny1)-6-
brorno-5-
fluoronicotinamide obtained in step 1, and the solution was irradiated in a
microwave reactor at
180 C for 40 minutes. The solution was diluted with 40% acetonitrile-water (4
mL) and purified
by reverse phase HPLC to give 2-(6-bromo-5-fluoropyridin-3-y1)-5-
(trifluoromethyl)-1H-
benzo[djimidazole as a tan solid: [MHI" m/z 361.
2-(6-brorno-5-fluoropyridin-3-y1)-5-(trifluoromethyl)-1H-benzo[d]imidazole (9
mg, 1 equiv),
methyl 2-(3-azaspiro[5.5]undecan-9-yl)acetate (5.6 mg, 1 equiv) and solid
sodium bicarbonate
(14.7 mg, 7 equiv) were suspended in NMP and stirred at 110 C for 2.5 hours.
The mixture was
cooled, diluted with a mixture of DMSO:acetonitrile:water and purified by
preparative HPLC
(20% acetonitrile:water with 0.05% TFA to 70% acetonitrile:water with 0.05%
TFA). Fractions
containing miz 491 or 505 by :LCMS analysis were pooled and concentrated. The
residue was
dissolved in a mixture of 1:1:1 THF-methanol-water to which was added solid
lithium hydroxide
(1.8 mg, 10 equiv). The mixture was stirred at room temperature for 30 minutes
at which point
LCMS indicates complete conversion to m/z 491. The mixture was diluted with a
mixture of
DMSO:acetonitrile:water and purified by preparative HPLC (20%
acetonitrile:water with 0.05%
TFA to 70% acetonitrile:water with 0.05% TFA) to give the product as a white
solid: [MIA+ m/z
491.
Example 63.
F3C 40N v0
"Th
N \
HO
A 54rifluoromethy1-2-(6-fluoropyridin-3-y1)-1H-benzimidazole (30 mg, 1 equiv),
Hunig's base
(93 tut, 5 equiv) and the hydrochloride salt of 2-methoxy-5H-spiro[furo[3,4-
b]pyridine-7,4'-
piperidi]-5-one (29 mg, 1 equiv) were dissolved in DMA (0.3 M) and heated at
150 C in a
microwave until for 8 hours. The mixture was cooled to room temperature, put
under vacuum to
remove residual Hunig's base, diluted with DMSO and purified by preparative
reverse phase
HPLC (30% acetonitrile:water with 0.05% TFA to 95% acetonitrile:water with
0.05% TFA) to
give the pure demethylated product: [MI-1] m/z 482.
- 65 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
Example 64.
0
Ns OH
N
[3-(tert-butoxycarbony1)-3-azaspiro{5.5]undec-9-y1iacetic acid (120 mg, 4.5
equiv) was
dissolved in dioxane (2 mL) and a 4 M solution of hydrochloric acid (300 [AL,
14 equiv) in
dioxane was added. The mixture was heated at 60 C for 6 hours at which point
a white
precipitate had formed and analysis of an aliquot by Ili NMR indicated
complete removal of the
Boe group. Volatiles were removed in vacuo and to the white solid was added 2-
(6-
fluoropyridin-3-y1)-5-methoxy-3H-imidazo[4,5-b]pyridine (25 mg, 1 equiv),
sodium bicarbonate
(36 mg, 5 equiv) and NMP (330 pL). The reaction mixture was heated at 110 C
for 18 hours,
neutralized with acetic acid (15 [IL, 3 equiv), diluted with DMSO and purified
by preparative
reverse phase HPLC (10% acetonitrile:water with 0.05% TFA to 50%
acetonitrile:water with
0.05% TFA) to give the pure product: [MEW m/z 436.
Examples 65-77.
Me 0
OH
Me N N
To a mixture of Oxone (9 mg, 1 equiv) and 3,5-dimethy1pheny1enediamine (10
mg, 1 equiv)
was added the hydrochloride salt of [3-(5-formy1pyridin-2-y1)-3-
azaspiro[5.5]undec-9-y1lacetic
acid (30 mg, 1 equiv), as a solution in 3% acetic acid-DMF (1 mL). The mixture
was stirred at
100 C for 16 hours, neutralized with potassium carbonate, filtered and
purified by preparative
reverse phase HPLC.
The compounds presented in Table 4 were prepared according to a similar
method.
Table 4
0
OH
R \ N
=[MH1+ [Mir 1M111+ [MH]
Example R m/z m/z Example R
ni/z nilz
eale'd found eale'd found
- 66 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
,
Me ,
F
N
433 72 01 N....-.. 441
441
Me N
F =N
H
H
N
.,..,..
F 4 N 0
66 419
419 73
491 491
Me H N
N
_
,
H ! ,
CI
F3C 0 ),....,...Me
....)-----õ,-N
67 411, N ¨.... 473
473 74 1
-*-- 420 420
Ci N
1\(----N
H
H ,
Me
0 r\l..._
...)---õ--N
68 CI N 439 439
75 , 1 '-- 434
434
H
Me'N.-----N
, H
N
õ--<----\_-N
69 N 41 .--- 423
423 76 MeN1-----N õ I ,--- 420
420
F H
H
N
,T\I OP NN---
1
70 489
489 77 I .....õ N
H 551 551
F3C0 0 N--"
H
CF3
CI
40 N__.,
0 ....... 507 507 78
N N 456 456
71 F3C 01N
H
Intermediate 11.
CO2H
Boc-N
0
methyl {9-(tert-butoxycarbony1)-1-oxa-9-azaspiro45.5]undec-4-yljacetate
Step 1. N,N-dimethy1-1-amino-3-tert-butyldimethylsilyloxy-1,3-butadiene (1.5
g, 1 equiv) was
added dropwise to a stirring suspension of tert-butyl 4-oxopiperidine-1-
carboxylate in 2-butanol
(11 mL) at room temperature. After 2.5 hours, volatiles were removed in vacua
and the residue
dissolved in diethyl ether (40 mL) then cooled to -78 C. A solution of acetyl
chloride (0.56 mL,
1.2 equiv) in diethyl ether (10 mL) was added slowly and the reaction stirred
for 10 minutes then
quenched by addition of saturated sodium bicarbonate (25 mL) and warmed to Mom
temperature.
The biphasic mixture was transferred to a separatory funnel with enough water
to dissolve all
solids and extracted with ethyl acetate thrice. The combined organic layers
were dried on
- 67 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
magnesium sulfate, filtered, concentrated and purified by flash column
chromatography on silica
gel (0% to 100% ethyl acetate in hexanes) to give tert-butyl 4-oxo-l-oxa-9-
azaspiro[5.5jundec-2-
ene-9-carboxylate as a colorless oil.
Step 2. The residue from Step 1 (1.62 g, 1 equiv) and 30% (w/w) palladium on
carbon (440 mg,
20 mol%) were stirred together at room temperature in methanol (25 mL) under
an atmosphere
of hydrogen for 6 hours. The mixture was filtered through a pad of Celite,
concentrated and
purified by flash column chromatography on silica gel (0% to 100% ethyl
acetate in hexanes) to
give tert-butyl 4-oxo-1-oxa-9-azaspiro[5.5]undecane-9-carboxylate as an oil.
Step 3. Trimethyl phosphonoacetate (149 mg, 1.1 equiv) was added dropwise to a
stirring
suspension of hodium hydride (31 mg, 60% dispersion in mineral oil, 1,05
equiv) in THF (2 mL)
at 0 C. The mixture was warmed to room temperature and stirred for 2 hours.
tert-Butyl 4-oxo-
1 -oxa-9-azaspiro[5.5Iundecane-9-caxboxylate from Step 2 (200 mg) was added
dropwise as a
5 solution in THF (1.4 mL). The reaction was stirred at room temperature
for 2 hours at which
point volatiles were removed in vacua. The mixture was poured in 1 M
hydrochloric acid and
extracted with dichloromethane. The combined organic layers were washed with
brine, dried on
magnesium sulfate, filtered, concentrated and purified by flash column
chromatography on silica
gel (0% to 100% ethyl acetate in hexanes) to give tert-buty1-4-(2-methoxy-2-
oxoethylidene)-1-
oxa-9-azaspiro[5.51undecane-9-carboxy1ate as a clear oil.
Step 4. tert-Buty1-4-(2-methoxy-2-oxoethylidene)-1-oxa-9-azaspiro[5.5]undecane-
9-carboxylate
from Step 3 (246 mg, 1 equiv) and palladium hydroxide (75 mg, 14 mol%) were
stirred together
in ethanol (5 mL) at room temperature under an atmosphere of hydrogen for 6
hours. The
reaction mixture was filtered, concentrated and concentrated and purified by
flash colunm
chromatography on silica gel (0% to 100% ethyl acetate in hexanes) to give
tert-butyl 4-(2-
methoxy-2-oxoethy1)-1-oxa-9-azaspiro[5.51undecane-9-carboxylate as an oil.
Example 79.
CO2H
õc N
¨N 0
Step 1. Intermediate 11(137 mg, 1 equiv) was dissolved in 1,4-dioxane (4 mL)
to which was
added a 4M solution of hydrochloric acid in dioxane (418 pt, 4 equiv) and the
mixture heated at
heated at 60 C for 5 hours. Volatiles were removed under a stream of nitrogen
and to the
remaining white solid was added 2-(6-fluoropyridin-3-y1)-5-(trifluoromethyl)-
1H-benzimidazole
(141 mg, 1.2 equiv), sodium bicarbonate (176 mg, 5 equiv) and NMP (1.5 mL).
The suspension
- 68 -
WO 2012/009217 CA 02804970 2013-01-09
PCT/US2011/043330
was stirred vigorously at 110 C for 16 hours, cooled, diluted with DMSO (-3
mL), neutralized
with acetic acid (350 ItL), filtered then purified by reverse phase HPLC (20%
to 70% acetonitrile
in water with 0.05% TFA as eluant) to give methyl (9-{545-(trifluoromethyl)-1H-
benzimidazol-
2-yllpyridin-2-y1}-1-oxa-9-azaspiro [5 .51undec-4-yl)acetate as a yellow oil.
Step 2. methyl (9- { 5 -[5 -(trifluoromethyl)-1H-benzimidazol-2-
yljpyridin-2-y1 -1-oxa-9-
azaspiro[5.5]undec-4-yl)acetate from Step 1 (240 mg) was dissolved in a
mixture of 1:1:1
THF:methanol:water (1.33 mL) and lithium hydroxide hydrate (167 mg, 10 equiv)
was added.
After 3.5 hours of stirring at room temperature, volatiles were removed in
vacuo, the residue was
diluted with DMSO (-3 mL) and water (0.1 inL), neutralized with acetic acid
(400 ILL), filtered
then purified by reverse phase HPLC (20% to 70% acetonitrile in water with
0.05% TFA as
eluant) to give (9- {5- [5-(trifluoromethyl)-1H-benzimidazol-2-
yl]pyridin-2-y1}-1-oxa-9-
azaspiro [5 .5]undec-4-yl)acetic acid as a yellow oil: [MEW m/z 475.
Enantiomers were separated by preparative HPLC using a Chiral Technologies 4.6
x 250 mm
Chiralcel AD-H column using 25% isopropanol in supercritical carbon dioxide
with an operating
pressure of 100 bar, flow rate of 2.4 mL/min and temperature of 40 'C. Each
enantiomer was
obtained as a pale yellow solid with [MF1J+ m/z 475.
Example 80. 0
F N\>._,
Step 1. BH3-THF complex (5.8 mL, 3.5 equiv, 1.0 M in THF) was added dropwise
to a stirred
THF solution of tert-butyl 1-oxa-9-azaspiro[5.5jundec-3-ene-9-carboxylate (420
mg, 1 equiv)
(Walters, M. A.; La, F.; Deshmukh, P.; Omeeinsky, D. 0. J. Comb. Chem. 2002,
4(2), 125-130)
over 15 minutes, under an atmosphere of nitrogen. After 4 hours, the mixture
was chilled to 0 C
and 30% H202 (3.39 mL, 20 equiv) was added dropwise, followed by a solution of
4M NaOH
(8.3 mL, 20 equiv). The mixture was warmed to room temperature and stirred for
1 hour until
gas evolution ceased. The mixture was partitioned between ethyl acetate and
water and the
organic phase was washed twice with brine and dried over sodium sulfate. The
resulting crude
residue was then purified by silica gel chromatography (0 to 100% ethyl
acetate: hexanes) to give
tert-buty1-3-hydroxy-1-oxa-9-azaspiro[5.5]undecane-9-carboxylate as an amber
oil: [MNar m/z
294.
Step 2. To a solution of tert-butyl-3-hydroxy-l-oxa-9-azaspiro[5.5]undecane-9-
carboxylate
(180 mg, I equiv) obtained from Step 1 in anhydrous THF (3 mL), under an
atmosphere of
- 69 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
nitrogen was added N-methylmorpholine N-oxide (75.4 mg, 1 equiv) followed by
tetrapropylammonium perruthenate (23.1 mg, 0.1 equiv). After stirring for 40
minutes at room
temperature, the reaction was quenched with water (1.0 mL) and poured into
ethyl acetate. The
ethyl acetate extract was washed with water and brine and dried over sodium
sulfate. The
resulting crude residue was purified by silica gel chromatography (0 to 100%
ethyl acetate:
hexanes) to give tert-bu1y1-3-oxo4-oxa-9-azaspiro[5.5]undecane-9-carboxylate
as a waxy solid:
[MH¨C4H9] m/z 214.
Step 3. Performed as in Intermediate 11, Step 3, to afford tert-buty1-3-(2-
methoxy-2-
oxoethylidene)-1-oxa-9-azaspiro [5 .5] undecane-9-carboxylate : [MNar m/z 348
Step 4. Performed as in Intermediate 11, Step 4, except 10% Pd/C was used as
the catalyst and
the reaction was stirred for 18 hours at room temperature, under atmospheric
hydrogen and then
purified by reverse phase HPLC (10% to 50% acetonitrile in water with 0.05%
TFA as eluant) to
give 9-(tert-butoxycarbony1)-1-oxa-9-azaspiro [5 .51undec-3-yl] methyl
acetate: [MNaj+ m/z 350.
Step 5. Performed as in Example 71, Step 1, except TFA was used as the acid
and DCM was
used as the solvent for the Boc-deprotection. Purification by reverse phase
HPLC (20% to 70%
acetonitrile in water with 0.05% TFA as eluant) gave methyl (9-{545-
(tiifluoromethyl)-1H-
benzimidazol-2-yl]pyridin-2-y1}-1-oxa-9-azaspiro[5.5]undec-3-y1) as an amber
oil: [MH] 4 in/Z
489.
Step 6. Performed as in Example 71, Step 2 to give (9-1545-(trifluorornethyl)-
1H-benzimidazol-
2-Apyridin-2-y1}-1-oxa-9-azaspiro(5.51undec-3-y1)acetic acid as a tan solid:
[MIT]+ nilz 475.
Example 81.
F3C N
N ¨N \-OH
tert-butyl 2-(2-methoxy-2-oxoethyl)-1-oxa-8-azaspiro f4.51decane-8-carboxylate
Step 1.
A 1.5 M solution of DIBAL-H in dichloromethane (2.85 mL, 0.97 equiv) was added
slowly to a
solution of tert-butyl 2-oxo-1-oxa-8-azaspiro[4.5]decane-8-carboxylate (1.125
g, 1.0 equiv) in
anhydrous dichloromethane and the mixture chilled to -30f C. After 2 hours,
the mixture was
allowed to warm to 0 _ C and aged for 2 hours. Saturated aqueous sodium
bicarbonate (1 mL)
was added followed by water (2 mL) forming a slurry. The slurry was filtered
through a pad of
Celite via buchner funnel, rinsing twice with 5 mL of dichloromethane. The
rinses were
- 70 -
WO 2012/009217 CA 02804970 2013-01-09PCT/US2011/043330
combined with the filtrate and concentrated in vacuo to give tert-butyl 2-
hydroxy-l-oxa-8-
azaspiro[4.5]decane-8-carboxylate as a colorless oil: [MNal+ tn/z 280.
Step 2.
Methyl phosphonoacetate (217 mg, 1.1 equiv) was added to a solution of tert-
butyl 2-hydroxy-l.
oxa-8-azaspiro[4.5]decane-8-carboxylate (279 mg, 1.0 equiv) from Step 1 in
anhydrous THF.
The mixture was chilled to 0 C then potassium tert-butoxide (134 mg, 1.1
equiv) was added in
one portion. The mixture was warmed to room temperature and stirred for 18
hours. The
solution was treated with aqueous ammonium chloride (2 mL) and diluted with
ethyl acetate.
The organic phase was separated and washed twice with water then once with
brine. The organic
phase was dried over sodium sulfate, filtered and concentrated in vacuo. The
crude residue was
purified using column chromatography on silica gel (0% to 100% ethyl
acetate:hexanes) to give
tert-butyl 2-(2-methoxy-2-oxoethyl)-1-oxa-8-azaspiro14.51decane-8-carboxylate
as a colorless
oil: [MNa]+ m/z 336.
Step 3.
The carbamate from Step 2 (213 mg, 1.0 equiv) was dissolved in dichloromethane
(1.0 mL) and
TFA (1.0 mL) and stirred at room temperature for 1 hour. Volatiles were
removed in vacua then
the residue was reconstituted in dichloromethane and concentrated in vacuo
again. The tan oil
thus obtained was carried forward to the next step.
Step 4.
The amine salt from Step 3 (54 mg, 1.0 equiv), 2-(6-fluoropyridin-3-y1)-5-
(trifluoromethyl)-1H-
benzo[d]imidazole (47 mg, 1.0 equiv) and sodium bicarbonate (139 mg, 10 equiv)
was
suspended in NMP (0.75 mL) and stirred at 110 C for 4 hours. The mixture was
cooled and
diluted with 40% acetonitrile in water then directly purified by preparative
HPLC (20% to 70%
acetonitrile in water with 0.05% TFA as eluant) to give the product as an off-
white solid: [MH]
nitz 475.
Step 5.
The ester from Step 4 (29 mg, 1 equiv) was dissolved in a 1:1:1 mixture of THF-
water-methanol
to which was added solid lithium hydroxide (14.6 mg, 10 equiv). After stirring
at room
temperature for 3 hours, LCMS showed clean conversion to product. The reaction
mixture was
-71 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
acidified to pH 2 with 2 M hydrochloric acid. The mixture was diluted with 40%
acetonitrile in
water then directly purified by preparative HPLC (20% to 70% acetonitrile in
water with 0.05%
TFA as eluant) to give the product as an off-white solid: [MHT m/z 461.
Enantiomers were separated by preparative HPLC using a Chiral Technologies 4.6
x 250 mm
ChiralPak IA column using 35% ethanol in heptane. Each enantiomer was obtained
as a white
solid with [Mlir m/z 461.
F3 Example 82. N \)--=N 0
N -N OH
Step 1. 2-(8-(tert-butoxycarbony1)-1-oxa-8-azaspiro[4.5]decan-3-yl)acetic acid
(203 mg, 1 equiv)
was dissolved in dioxane (2 mL) then treated with a 4 M solution of
hydrochloric acid in dioxane
(1 mL) at 50 C for 90 minutes. The mixture was then concentrated in vacuo to
yield a white
powder that was carried forward in subsequent steps.
Step 2. The amine salt from Step 1 (80 mg, I equiv), 2-(6-fluoropyridin-3-y1)-
5-
(trifluoromethyl)-1H-benzo[djimidazole (95 mg, 1 equiv) and sodium bicarbonate
(199 mg, 7
equiv) were dissolved in NMP (1 mL) and stirred at 110 C for 16 hours. The
mixture was
diluted with DMSO, filtered and purified by preparative HPLC (20% to 70%
acetonitrile in water
with 0.05% TFA as eluant) to give the product as a yellow solid: [MH} m/z
475.
The compounds presented in Table 7 were prepared according to a similar
method.
Table 7.
0
-N OH
[m11.]+ 1111111+
Example R in& nez
calct d found
83 1110 427 427
Ci
84 393 393
- 72 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
Example 85-86.
F3 N(OH F3C N
OH
N \ 0 0 N -N \ 0
0
Step I. 2-(8-(tert-butoxycarbony1)-1-oxa-8-azaspiro[4.51decan-3-ypacetic acid
(800 mg, I equiv)
was stirred in a 1.25 M solution of hydrochloric acid in methanol (5 mL) at 50
C for 2 hours.
Then additional hydrochloric acid was added as a 4M solution in dioxane (1 mL)
followed by an
additional 90 minutes of stirring at 50 C. The mixture was then concentrated
in vacuo to yield a
white powder that was carried forward in subsequent steps.
Step 2. The amine salt from Step 1 (617 mg, 1 equiv), 2-(6-fluoropyridin-3-y1)-
5-
(trifluoromethyl)-1H-benzo[d]imidazole (736 mg, I equiv) and sodium
bicarbonate (1100 mg, 5
equiv) were dissolved in NMP (8.7 mL) and stirred at 110 C for 18 hours. The
mixture was
poured into ice water and ethyl acetate and saturated aqueous ammonium
chloride were added
until complete dissolution of solids occurred. The layers were shaken and
separated. The
organic layer was then washed five times with water, once with brine then
dried on anhydrous
sodium sulfate, filtered and concentrated in vacua to yield a brown solid:
[M11]4. m/z 475.
Purification was performed by preparative HPLC on a Chiral Technologies 4.6 x
250 mm
Chiralcel OD column using 40% methanol in supercritical carbon dioxide. This
yielded two
separated enantiomers: A and B. Each enantiomer was obtained as an off-white
solid with
[MH]F m/z 475.
Step 4. Enantiomer A of the ester from Step 3 was dissolved in methanol (300
p.L) and THF (300
L) to which was added 2.5 M lithium hydroxide. The reaction was stirred at 50
C for 2 hours
then quenched by the addition of glacial acetic acid (150 pL) and concentrated
in vacua. The
mixture was diluted with DMSO, filtered and purified by preparative HPLC (20%
to 70%
acetonitrile in water with 0.05% TFA as eluant) to give the carboxylic acid
(Enantiomer A) as a
white solid: [M1114- m/z 461.
Step 5. Enantiomer B of the ester from Step 3 was dissolved in methanol (300
pt) and THF (300
1,LL) to which was added 2.5 M lithium hydroxide. The reaction was stirred at
50 C for 2 hours
then quenched by the addition of glacial acetic acid (150 ',it) and
concentrated in vaeuo. The
mixture was diluted with DMSO, filtered and purified by preparative HPLC (20%
to 70%
acetonitrile in water with 0.05% TFA as eluant) to give the carboxylic acid
(Enantiomer B) as a
white solid: [MHir m/z 461.
Intermediate 12.
- 73 -
CA 02804970 2013-01-09
WO 2012/009217 PCT/US2011/043330
HN 0 OMe
methyl 1-oxa-9-azaspiro[5.5]undecane-3-carboxylate (TFA salt)
Step 1. A 60% oil dispersion of sodium hydride (0.438 g, 1.2 equiv) was added
to a solution of
tert-butyl 4-ally1-4-hydroxypiperidine-l-carboxylate (2.2 g, 1 equiv)
(Walters, M. A.; La, F.;
Deshmukh, P.; Omecinsky, D. 0. J. Comb. Chem. 2002, 4(2), 125-130) in
anhydrous DMF (170
mL) and the mixture cooled to 0 C. The mixture was warmed to room temperature
over 1 hour
and methyl 2-(bromomethyl)acrylate (1.63 g, 1 equiv) was added dropwise to the
solution over 5
minutes. The mixture was aged for 72 hours. A saturated solution of ammonium
chloride was
added to the reaction mixture and the mixture was diluted with ethyl acetate.
The organic phase
was separated and washed twice with water then brine, then dried over sodium
sulfate, filtered
and concentrated in vacuo. The crude mixture was purified using column
chromatography on
silica gel (0% to 100% ethyl acetate in hexanes) to give 920 mg of tert-butyl
(methoxycarbonyl)prop-2-en-1-yl] oxy} -4-(prop-2-en-1 -yppiperidine-l-
carboxylate as a colorless
oil: [MNa] m/z 362.
Step 2. tert-butyl 4- { [2-(methoxycarbonyl)prop-2-en-l-yl]oxy}-4-(prop-2-en-1-
y1)piperidine-1-
caiboxylate from Step 1 (340 mg, 1 equiv) in anhydrous 1,2-dichloroethane (75
mL) was
combined with benzylidene [1,3 -bi s(2,4,6-trimethylpheny1)-2-
imidazolidinylidene] dichloro-
(tricyclohexylphosphine)ruthenium (85 mg, 10 mol%) and the mixture was heated
at 85 'C for 18
hours. The mixture was cooled to room temperature, -then diluted with ethyl
acetate and washed
with water twice with brine. The separated organic layer was dried over
anhydrous sodium
sulfate, filtered and evaporated to give tert-butyl 3-oxo-1-oxa-9-
azaspiro[5.5]undecane-9-
carboxylate as an oil [MNar m/z 334.
Step 3. An ethanol solution of the product from Step 2 was subjected to
hydrogenation by a
single pass through a Thales H-Cube Flow Hydrogenation Reactor (Jones, R. V.;
Godorhazy, L.;
Varga, N.; Szalay, D.; Urge, L.; Darvas, F. J. Comb. Chem. 2006, 8, 110-116)
using a palladium
hydroxide cartridge at 50 C and 60 bar. Volatiles were then removed in vacuo
and the residue
purified by preparative HPLC (10% to 98% aeetonitrile in water with 0.05% TFA
as eluant) to
give the product as an oil: [MNa] m/z 336.
- 74 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
Step 4. The residue from Step 3 was dissolved in dichloromethane (4 mL) and
TFA (4 mL) and
stirred at room temperature for 90 minutes at which point volatiles were
removed in vacuo. The
residue was reconstituted in dichloromethane and concentrated again to afford
an amber oil
which was purified by preparative HPLC (10% to 98% acetonitrile in water with
0.05% TFA as
eluant) to give the TFA salt of methyl 1-oxa-9-azaspiro[5.5]undecane-3-
carboxylate as an oil:
[MH]+ Wz 214.
Example 87.
F3 N> 0 0
N ¨N OH
Step I. To the amine salt from Step 4 above (60 mg, 1 equiv), 2-(6-
fluoropyridin-3-y1)-5-
(trifluoromethyl)-1H-benzimidazole (79 mg, 1 equiv) and sodium bicarbonate
(236 mg, 10
equiv) was added NMP (2 mL) and the mixture stirred at 110 C for 2.5 hours.
The mixture was
cooled to room temperature, diluted with 40% acetonitrile in water, filtered
and purified by
preparative HPLC (20% to 70% acetonitrile in water with 0.05% TFA as eluant)
to give methyl
945 -(5-(trifluoromethyl)-1H-benzo [d] imidazol-2-yl)pyridin-2-y1)-1-oxa-9-
azaspiro [5. 5jundecane-3-carboxylate as a tan solid: [M11] 4- in/z 475.
Step 2. The ester from Step 1 (130 mg, 1 equiv) was dissolved in a 1:1:1
mixture of THF-water-
methanol and treated with lithium hydroxide (65.6 mg, 10 equiv) at room
temperature for 2
hours, The reaction mixture was cooled in an ice bath and acidified to pH 2
with 2M
hydrochloric acid. The mixture was diluted with 40% acetonitrile in water,
filtered and purified
by preparative HPLC (20% to 70% acetonitrile in water with 0.05% TFA as
eluant) to give the
product as an off-white solid: [MEW rn/z 461.
The compounds presented in Table 5 were prepared according to a similar
method.
Table 5 R 0 OH
0
[MHI+ [Mif]'
Example R m/z
ealeid found
- 75 -
WO 2012/009217 CA 02804970 2013-01-09
PCT/US2011/043330
88 Ci 427 427
89 1.1 411 411
Examples 90-91.
,3c. N 0 0 õc N
0 0
=N -N OH N
Enantiomers of Example 86 were separated by preparative HPLC using a Chiral
Technologies
4.6 x 250 mm ChiralPak IA column using 50% ethanol in heptane. Enantiorner A
was obtained
as a white solid with [MEW 222/z 475 and Enantiomer B was obtained as a white
solid with [ya]
n2/z 475. Absolute stereochemisty was arbitrarily assigned.
Intermediate 13.
0 0
0 -N OMe
The TFA salt of methyl 1-oxa-9-azaspiro[5.5jundecane-3-carboxylate (876 mg, I
equiv) and 2-
fluoropyridine-5-carboxaldehyde (352 mg, 1.05 equiv) were added to sodium
bicarbonate (1124
mg, 5 equiv) in DMF (10.5 mL) and the mixture stirred overnight at 80 C. The
mixture was
then cooled and filtered through a 0.45 micron membrane and this solution used
crude in
subsequent steps: [MH] nilz 319.
Examples 92-101 in Table 6 were prepared from Intermediate 13 in two steps.
First,
Intermediate 13 and the appropriate diamine were allowed to react at room
temperature under
influence of Oxone in DMF and water as previously described. For examples 100-
101, the
diamine and aldehyde were heated at 65 C for 2 hours prior to addition of
Oxone . The
resulting benzimidazoles were purified by preparative HPLC then subjected to
saponification
with lithium hydroxide then purified as previously described.
Table 6.
0 OH
0
- 76 -
CA 02804970 2013-01-09
WO 2012/009217
PCT/US2011/043330
_
[Mir [Ma]
[Mli] [mH]
Example R
nilz in/z Example
R rn/z In&
eale'd found
eale'd found
H
H
N
001 N......
92
421 421 97 Me
449 449
I. t\l/
Et N
Me
,..
Me
CI 40 N HH
.
N.....
93 ==¨= 455 455 98
F3C
N 501 501
... Et N
A
0 H
H
Nõ..-------N
-*-- 493 493 99
''"¨ 408 408
94 N
F3CS
M eft----N
H
H
N
F C3 =_-,.,¨N
95 110 ...¨ 477 477 100
i ---' 462 462
---.. --;,-----
F3C0 N
N N
. .
H
H
H
N
1 96 1/>¨ 422 422 101
CC, '"'"' 424 424
Et0 N
Me07.14- N
-
Potency of selected DGAT1 inhibitors.
To a 384 well assay plate was added 1 pt of a 400 [tM solution of the test
compound in DMSO and 20 uL of a substrate mix that is 300 JAM in diolein and
40 uM in oleoyl-
CoA in 10% ethanol. To this was added 19 ill, of 1.05 pg/mL human DGAT1-
expressed yeast
membrane fraction in a buffer of the following composition: 200 mM Tris, pH 7,
200 mM
sucrose, 200 mM magnesium chloride, and 20 ItglmL N-ethylmaleimide-treated
bovine serum
albumin. The solution is incubated at room temperature for 1 hour after which
20 ILL of a 90 ttM
7-diethy1amino-3-(4'-ma1eimidy1pheny1)-4-methy1cournarin solution in 90%
ethanol was added.
After incubation in the dark for 30 minutes at room temperature, fluorescence
was measured on a
Perkin Elmer Envision multilabel reader.
The 1050 is detetwined from a 4 parameter fit of the plot of %Inhibition vs.
Concentration of Test Compound in the reaction and is defined as the
concentration at which the
curve crosses the 50% inhibition line.
The inhibitory activity was calculated from the following formula:
% inhibition = [1-(fluorescence counts from test compound- average
fluorescence counts from
LC)/(average fluorescence counts from HC-average fluorescence counts from LC)]
x 100%
- 77 -
WO 2012/009217 CA 02804970 2013-01-09
PCT/US2011/043330
LC = low control = maximal inhibition by excess amount of a Merck DGAT1
inhibitor
HC = high control = DMSO = uninhibited control
Table 7.
Example IC50 (nM) Example I IC50 (nM)
1 3 37 <25
2 11 41 <25
3 6 44 <100
13 49 49 <25
17 11 54 <25
18 70 56 <25
20 3 63 <100
23 4 66 <100
25 27 72 <100
26 3 80 <100
29 12 82 <25
30 42 85 <25
31 <100 86 <25
34 <25 87 <25
36 <25 95 <100
- 78 -