Note: Descriptions are shown in the official language in which they were submitted.
CA 02805815 2013-01-17
WO 2012/010788 PCT/FR2011/051710
1
PROCEDE DE PREPARATION DE DERIVES D' AMINO-BENZOYL-
BENZOFURANE
La présente invention se rapporte, d'une manière
générale, à la préparation de dérivés d'amino-benzoyl-
benzofurane.
Plus précisément, l'invention concerne un procédé
pour la préparation de dérivés de 5-amino-benzoyl-
benzofurane de formule générale:
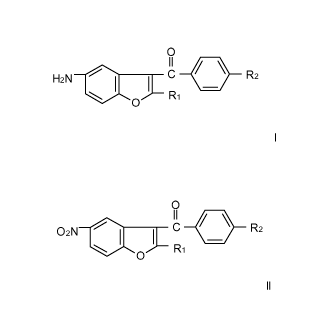
0
H
H2N el 0 I C Ri a R2
I
dans laquelle R1 représente l'hydrogène ou un groupe
alkyle et R2 représente l'hydrogène, un groupe alkyle,
alkoxy ou dialkylaminoalkoxy.
Dans la formule I ci-dessus :
= R1 représente en particulier un groupe alkyle,
linéaire ou ramifié, en C1-C8 notamment un groupe
alkyle, linéaire ou ramifié, en C1-C4 tel que
méthyle, éthyle, n-propyle, isopropyle, n-butyle,
sec-butyle ou tert-butyle,
= R2 représente en particulier un groupe alkyle,
linéaire ou ramifié, en C1-C8 notamment un groupe
WO 2012/010788 CA 02805815 2013-01-17 PCT/FR2011/051710
2
alkyle, linéaire ou ramifié, en Cl-C4 tel que
méthyle, éthyle, n-propyle, isopropyle, n-butyle,
sec-butyle ou tert-butyle ; un groupe alkoxy,
linéaire ou ramifié, en Cl-C8 notamment un groupe
alkoxy, linéaire ou ramifié, en Cl-C4 tel que
méthoxy, éthoxy, n-propoxy, isoproxy, n-butoxy,
sec-butoxy ou tert-butoxy ou encore un groupe
dialkylaminoalkoxy dans lequel chaque groupe
alkyle, linéaire ou ramifié, est en Cl-C8 et le
groupe alkoxy, linéaire ou ramifié, est en C1-C8
notamment dans lequel chaque groupe alkyle,
linéaire ou ramifié, est en Cl-C4 tel que méthyle,
éthyle, n-propyle, isopropyle, n-butyle, sec-butyle
ou tert-butyle et le groupe alkoxy, linéaire ou
ramifié, est en Cl-C4 tel que méthoxy, éthoxy, n-
propoxy, isopropoxy, n-butoxy, sec-butoxy ou tert-
butoxy.
De préférence, R1 représente n-butyle et R2 représente
3-(di-n-butylamino)-propoxy.
Les composés de formule I ci-dessus sont, pour la
plupart, des composés décrits dans le brevet EP 0 471 609
où ils sont présentés comme des produits intermédiaires
pour la préparation finale de dérivés aminoalkoxybenzoyl-
benzofurane utiles pour leurs applications thérapeutiques
dans le domaine cardiovasculaire.
Parmi ces dérivés aminoalkoxybenzoyl-benzofurane, le
2-n-buty1-3-14-[3-(di-n-butylamino)-propoxy]-benzoy11-5-
méthanesulfonamido-benzofurane, communément dénommé
dronédarone, ainsi que ses sels pharmaceutiquement
acceptables, s'est montré particulièrement intéressant
notamment comme agent antiarythmique.
WO 2012/010788 CA 02805815 2013-01-17 PCT/FR2011/051710
3
On a rapporté dans le susdit brevet EP 0 471 609, un
procédé pour la préparation de la dronédarone, procédé
selon lequel le 2-n-buty1-3-14-[3-(di-n-butylamino)-
propoxy]-benzoy11-5-nitro-benzofurane est réduit sous
pression avec l'hydrogène en présence d'oxyde de platine
comme catalyseur pour former le 2-n-buty1-3-14-[3-(di-n-
butylamino)-propoxy]-benzoy11-5-amino-benzofurane (ci-
après Composé A) lequel est ensuite traité avec le
chlorure de méthanesulfonyle, en présence d'un accepteur
d'acide, pour donner le composé désiré. Selon ce procédé,
la dronédarone a pu être obtenue avec un rendement global
de l'ordre de 60% au départ du dérivé 5-nitro-
benzofurane.
Toutefois, ce procédé n'est pas dépourvu
d'inconvénients inhérents notamment au type de réaction
utilisée dans la formation du Composé A, à savoir une
hydrogénation sous pression qui comporte un risque
industriel.
D'autre part, cette méthode nécessite d'isoler le
Composé A à partir de son milieu de formation,
l'isolement de ce composé, habituellement sous la forme
de son oxalate, constituant par conséquent une étape
supplémentaire dans la préparation de la dronédarone.
La recherche d'un procédé de préparation industriel
capable de pallier ces inconvénients tout en offrant des
rendements importants en Composé A ainsi qu'une mise en
uvre facilitée de celui-ci, de manière à produire des
rendements significativement supérieurs en dronédarone
par rapport au procédé antérieur, reste par conséquent
d'un intérêt incontestable.
CA 02805815 2013-01-17
WO 2012/010788
PCT/FR2011/051710
4
Or, on a maintenant trouvé que le Composé A peut être
préparé selon un procédé impliquant une réduction
sélective de sa fonction nitro par rapport à sa fonction
cétone. Cette réduction sélective élimine, par
conséquent, la nécessité d'isoler ce Composé A via son
oxalate lors de la synthèse finale de la dronédarone
laquelle peut être obtenue de cette manière avec des
rendements globaux supérieurs à 90% à partir du dérivé
5-nitro-benzofurane de départ.
Les dérivés aminoalkoxybenzoyl-benzofurane du brevet
EP 0 471 609, en particulier la dronédarone, pourront
être synthétisés, par conséquent, dans le milieu même de
formation du composé approprié de formule I.
Selon un premier objet de l'invention, les dérivés de
5-amino-benzofurane de formule I peuvent être préparés en
réduisant un dérivé de 5-nitro-benzofurane de formule
générale :
0
Il
02N el 0 I C Ri 11, R2
H
dans laquelle R1 et R2 ont la même signification que
précédemment, au moyen d'un agent de transfert
d'hydrogène, en présence de charbon palladié comme
catalyseur et dans un éther ou un mélange d'éthers comme
solvant, ce qui forme les composés désirés.
CA 02805815 2013-01-17
WO 2012/010788 PCT/FR2011/051710
Dans la formule II ci-dessus, R1 représente, de
préférence, n-butyle et R2 représente, de préférence, 3-
(di-n-butylamino)-propoxy.
En outre, selon un autre de ses objets, l'invention
5 se rapporte à un procédé de préparation de dérivés de
sulfonamido-benzofurane de formule générale :
0
Il =
R-3 SO¨N . C R2
' H 1Ri
0
M
ainsi que de leurs sels pharmaceutiquement acceptables,
dans laquelle R1 et R2 ont la même signification que
précédemment et R3 représente un groupe alkyle, procédé
selon lequel :
a) on réduit un dérivé de 5-nitro-benzofurane de
formule II au moyen d'un agent de transfert
d'hydrogène, en présence de charbon palladié comme
catalyseur et dans un éther ou un mélange d'éthers
comme solvant, pour former un milieu réactionnel
contenant un dérivé de 5-amino-benzoyl-benzofurane
de formule I, ci-dessus, sous forme de base libre,
b) on traite le milieu réactionnel contenant le dérivé
de 5-amino-benzoyl-benzofurane de formule I sous
forme de base libre obtenu précédemment, avec un
halogénure de formule générale :
Hal-S02-R3 IV
WO 2012/010788 CA 02805815 2013-01-17 PCT/FR2011/051710
6
dans laquelle Hal représente un halogène tel que
chlore et R3 a la même signification que précédemment,
en présence d'un agent basique, pour obtenir les
composés désirés sous forme de base libre que l'on
fait réagir, si nécessaire, avec un acide organique ou
inorganique pour former un sel pharmaceutiquement
acceptable de ce composé désiré.
Par la suite, le sel pharmaceutiquement acceptable du
composé de formule III peut être récupéré de son milieu
de formation, par exemple par cristallisation.
Dans la formule III ci-dessus, R3 représente en
particulier un groupe alkyle, linéaire ou ramifié, en Cl-
Cg notamment un groupe alkyle, linéaire ou ramifié, en Cl-
C4 tel que méthyle, éthyle, n-propyle, isopropyle, n-
butyle ou tert-butyle.
De préférence, R1 représente n-butyle, R2 représente
3-(di-n-butylamino)-propoxy et R3 représente méthyle dans
la formule III ci-dessus.
La réduction par transfert d'hydrogène selon
l'invention est réalisée habituellement dans un éther ou
un mélange d'éthers comme solvant contrairement à l'état
de la technique où ce type de réaction s'opère
généralement dans un alcool. Cette réduction dans un
éther ou un mélange d'éthers permet notamment une
importante chimio-sélectivité de la fonction nitro au
détriment de la fonction cétone également présente et qui
elle aussi est susceptible d'une réduction en alcool.
Cette réduction sélective de la fonction nitro évite, par
conséquent, l'isolement du composé de formule I de
quelque manière que ce soit notamment par transformation
WO 2012/010788 CA 02805815 2013-01-17 PCT/FR2011/051710
7
de ce composé, obtenu sous forme basique, en un sel
aisément séparable de son milieu de formation.
L'éther utilisé comme solvant est habituellement un
dialkyléther tel que le méthyl tert-butyl éther ou encore
un éther cyclique par exemple le tétrahydrofurane tandis
que le mélange d'éthers correspond généralement à un
mélange de dialkyléther et d'éther cyclique par exemple
un mélange de méthyl tert-butyléther et de
tétrahydrofurane.
Le méthyl tert-butyl éther représente un solvant
particulièrement préféré dans le cadre de la présente
invention, en particulier pour la préparation du Composé
A et subséquemment de la dronédarone.
Habituellement, l'agent de transfert d'hydrogène est
un formiate, de préférence le formiate d'ammonium ou un
phosphinate, en particulier le phosphinate de sodium. Cet
agent de transfert d'hydrogène est utilisé en excès par
rapport au composé de formule II, cet excès pouvant
atteindre de 3 à 5 équivalents d'agent de transfert
d'hydrogène par équivalent de composé de formule II ou
davantage. Préférentiellement, on utilise 5 ou environ 5
équivalents d'agent de transfert d'hydrogène par
équivalent de composé de formule II, par exemple 5 ou
environ 5 équivalents d'agent de transfert d'hydrogène
dissous par exemple dans un volume d'eau. En particulier,
on utilise 5 équivalents de formiate d'ammonium dissous
par exemple dans un volume d'eau.
La réduction peut avoir lieu à température ambiante.
Toutefois, celle-ci est généralement entreprise par
chauffage du milieu réactionnel à une température allant
WO 2012/010788 CA 02805815 2013-01-17 PCT/FR2011/051710
8
jusqu'à par exemple 50 C à 60 C, de préférence à une
température de l'ordre de 40 C, en particulier à 40 C.
Selon un de ses aspects particuliers, l'invention se
rapporte, en outre, à un procédé de préparation du 2-n-
buty1-3-14-[3-(di-n-butylamino)-propoxy]-benzoy11-5-
amino-benzofurane, procédé selon lequel on réduit le 2-n-
buty1-3-14-[3-(di-n-butylamino)-propoxy]-benzoy11-5-
nitro-benzofurane au moyen de formiate d'ammonium ou de
phosphinate de sodium comme agent de transfert
d'hydrogène, en présence de charbon palladié comme
catalyseur et dans le méthyl tert-butyl éther ou un
mélange de méthyl tert-butyl éther et de tétrahydrofurane
comme solvant, pour former un milieu réactionnel
contenant le 2-n-buty1-3-14-[3-(di-n-butylamino)-
propoxy]-benzoy11-5-amino-benzofurane sous forme de base
libre.
D'autre part, selon un autre de ses aspects
particuliers, l'invention se rapporte à un procédé de
préparation du 2-n-buty1-3-14-[3-(di-n-butylamino)-
propoxy]-benzoy11-5-méthanesulfonamido-benzofurane ou
dronédarone ainsi que de ses sels pharmaceutiquement
acceptables, procédé selon lequel :
a) on réduit le 2-n-buty1-3-14-[3-(di-n-butylamino)-
propoxy]-benzoy11-5-nitro-benzofurane, au moyen de
formiate d'ammonium ou de phosphinate de sodium
comme agent de transfert d'hydrogène, en présence de
charbon palladié comme catalyseur et dans le méthyl
tert-butyl éther ou un mélange de méthyl tert-butyl
éther et de tétrahydrofurane comme solvant, pour
former un milieu réactionnel contenant le 2-n-butyl-
WO 2012/010788 CA 02805815 2013-01-17 PCT/FR2011/051710
9
3-14-[3-(di-n-butylamino)-propoxy]-benzoy11-5-amino-
benzofurane sous forme de base libre,
b) on traite le milieu réactionnel contenant le 2-n-
buty1-3-14-[3-(di-n-butylamino)-propoxy]-benzoy11-5-
amino-benzofurane sous forme de base libre obtenu
précédemment, avec un halogénure de méthanesulfonyle
en présence d'un agent basique, pour obtenir la
dronédarone sous forme basique que l'on fait réagir,
si nécessaire, avec un acide organique ou
inorganique pour former un sel pharmaceutiquement
acceptable de dronédarone.
Par la suite, le sel pharmaceutiquement acceptable de
dronédarone peut être récupéré de son milieu de
formation, par exemple par cristallisation.
A la lumière de la description qui précède,
l'ensemble formé par un dérivé de 5-nitro-benzofurane de
formule II, un agent de transfert d'hydrogène, le charbon
palladié et un éther ou un mélange d'éthers comme
solvant, se révèle particulièrement intéressant comme
milieu réactionnel pour la préparation de divers composés
notamment les composés de formule I et ceux de formule
III ci-dessus.
En conséquence, un autre objet de l'invention
concerne un milieu réactionnel, caractérisé en ce qu'il
est formé :
a) d'un dérivé de 5-nitro-benzofurane de formule II,
en particulier un dérivé de formule II dans
laquelle R1 représente n-butyle et R2 représente 3-
(di-n-butylamino)-propoxy,
b) d'un agent de transfert d'hydrogène tel que le
formiate d'ammonium ou le phosphinate de sodium,
WO 2012/010788 CA 02805815 2013-01-17PCT/FR2011/051710
10
c) de charbon palladié,
d) d'un éther tel que le méthyl tert-butyl éther ou d'
un mélange d'éthers tel qu'un mélange de méthyl
tert-butyl éther et de tétrahydrofurane, comme
solvant.
L'exemple non limitatif suivant illustre la
préparation d'un composé de formule I selon le procédé de
l'invention ainsi que sa mise en uvre dans la synthèse
de la dronédarone.
EXEMPLE
a) 2-n-Buty1-3-14-[3-(di-n-butylamino)-propoxy]-benzoyll-
5-amino-benzofurane (Composé A ou composé de formule I:
R1 = n-C4H9 ; R2 = 3-(di-n-butylamino)-prop0xY)
Dans un réacteur de 5 L, on charge, à température
ambiante, 3,33 kg d'une solution à 30% de 2-n-buty1-3-14-
[3-(di-n-butylamino)-propoxy]-benzoy11-5-nitro-
benzofurane (composé de formule II) et 0,05 kg de charbon
palladié (Pd/C) sec. Sous agitation, on chauffe
l'ensemble à 40 C puis on ajoute, en 2 H environ, une
solution de 0,62 kg (5 équivalents) de formiate
d'ammonium dans 0,62 kg d'eau. On maintient alors la
température du milieu réactionnel à 40 C (+/-2 C) pendant
15 H tout en contrôlant l'évolution de la réaction par
chromatographie en phase liquide. Dès que la réduction
est terminée, on refroidit à 23 C (+/-2 C) puis on filtre
le charbon palladié qui subit alors un lavage avec du
méthyl tert-butyl éther et de l'eau. On décante puis, à
la température ambiante, on lave la phase organique avec
WO 2012/010788 CA 02805815 2013-01-17PCT/FR2011/051710
11
de l'eau. On renouvelle ensuite une fois ces opérations
de décantation et de lavage. On procède à une nouvelle
décantation et on concentre la solution à 40 C sous vide.
On dilue ensuite le concentrat avec du tétrahydrofurane,
ce qui fournit 3,47 kg d'une solution du composé désiré
dans un mélange de méthyl tert-butyl éther et de
tétrahydrofurane.
Rendement estimé : 99%
b) Chlorhydrate de dronédarone (chlorhydrate du composé
de formule III : R1= n-C4H9 ; R2= 3-(di-n-butylamino)-
propoxy ; R3= CI-13)
Dans un réacteur de 5 L, on charge, à température
ambiante, les 3,47 kg de solution du Composé A dans un
mélange de méthyl tert-butyl éther et de tétrahydrofurane
obtenue précédemment. Sous agitation, on ajoute ensuite,
en 1H, le chlorure de méthanesulfonyle en maintenant la
température du milieu réactionnel inférieure à 30 C. On
refroidit à 25 C puis on coule une solution d'ammoniaque,
la température du milieu réactionnel étant maintenue à
C. On contrôle la fin de la réaction par
chromatographie en phase liquide. Au milieu réactionnel
maintenu à 30 C, on additionne alors de 1"eau et du
25 méthyl tert-butyl éther et on maintient une agitation
durant 15 min. Après décantation, on procède à un lavage
de celle-ci d'abord avec une solution d'eau saline. On
maintient sous agitation durant 10 min. à 28 C, puis on
décante et on concentre à 45 C sous vide. On ajoute alors
de l'isopropanol et on concentre à 50 C sous vide. On
ajoute à nouveau de l'isopropanol et on concentre à
WO 2012/010788 CA 02805815 2013-01-17PCT/FR2011/051710
12
nouveau à 50 C sous vide. On ajuste le milieu réactionnel
par ajout de 2,03 kg d'isopropanol de façon à obtenir
3,62 kg d'une solution, dans l'isopropanol, du composé
désiré sous forme de base. On chauffe cette solution à
50 C, puis on ajoute 0,225 kg d'acide chlorhydrique au
milieu réactionnel maintenu à la température de 50 C à
55 C, puis on amorce la cristallisation du chlorhydrate
désiré par ajout de chlorhydrate de dronédarone au milieu
réactionnel. On filtre ensuite et on lave le gâteau de
filtration avec de l'isopropanol, ce qui fournit le
chlorhydrate désiré que l'on sèche à 45 C sous vide pour
obtenir 1,12 kg de chlorhydrate de dronédarone sec.
Rendement global (par rapport au composé I): 96%
Le procédé selon l'invention présente des avantages
incontestables par rapport à la méthode décrite dans le
brevet EP 0 471 609 ou la demande de brevet
WO 2002/048078.
En effet, la fonction nitro du composé de formule II
peut être réduite dans un réacteur standard, ce qui évite
la nécessité d'opérer avec l'hydrogène sous pression dans
un appareil à hydrogénation. D'autre part, la qualité du
composé de formule I sous forme de base se trouve
significativement améliorée puisque l'on enregistre une
formation d'impuretés différentes en moindre nombre et en
teneurs plus faibles. Cet avantage permet d'éviter la
préparation et l'isolement de l'oxalate du composé de
formule I, opération qui pose de nombreux problèmes à
l'échelle industrielle.
En outre, la mise en uvre des composés non isolés de
formule I dans un procédé de préparation des dérivés
WO 2012/010788 CA 02805815 2013-01-17PCT/FR2011/051710
13
aminoalkoxybenzoyl-benzofurane pharmacologiquement actifs
du brevet EP 0 471 609 et notamment dans un procédé de
préparation des composés de formule III ci-dessus, permet
d'améliorer très significativement le rendement de ce
procédé. Dans le cas particulier de la dronédarone, le
rendement global de sa synthèse, à partir de son dérivé
5-nitro-benzofurane correspondant, s'élève de 60% selon
l'état de la technique à 95% par mise en uvre du procédé
chimio-sélectif de l'invention. Cette amélioration est
liée notamment à l'absence d'isolement de l'oxalate du
composé de formule I et aux pertes y associées.