Note: Descriptions are shown in the official language in which they were submitted.
CA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
1
ARYLCYCLOPROPYLAMINE BASED DEMETHYLASE INHIBITORS OF LSD1 AND THEIR MEDICAL
USE
The invention relates to (hetero)aryl cyclopropylamine compounds, particularly
the
compounds of formula (I), (Ia), (1b), OD or (III) as described and defined
herein, and their
use in therapy, including e.g., in the treatment or prevention of cancer, a
neurological
disease or condition, or a viral infection.
Aberrant gene expression in affected tissue as compared to normal tissue is a
common
characteristic of many human diseases. This is true for cancer and many
neurological
diseases which are characterized by changes in gene expression patterns. Gene
expression
patterns are controlled at multiple levels in the cell. Control of gene
expression can occur
through modifications of DNA: DNA promoter methylation is associated with
suppression
of gene expression. Several inhibitors of DNA methylation are approved for
clinical use
including the blockbuster VidazaTM. Another class of modifications involve
histones which
form the protein scaffold that DNA is nothially associated with (coiled
around) in
eukaryotic cells. Histones play a crucial role in organizing DNA and the
regulated coiling
and uncoiling of DNA around the histones is critical in controlling gene
expression ¨ coiled
DNA is typically not accessible for gene transcription. A number of histone
modification
have been discovered including histone acetylation, histone lysine
methylation, histone
arginine methylation, histone ubiquinylation, and histone sumoylation, many of
which
modify accessibility to the associated DNA by the cells transcriptional
machinery. These
histone marks serve to recruit various protein complexes involved in
transcription and
repression. An increasing number of studies are painting an intricate picture
of how various
combinations of histone marks control gene expression in cell-type specific
manner and a
new term has been coined to capture this concept: the histone code.
The prototypical histone mark is histone acetylation. Histone acetyl
transferase and histone
deacctylases are the catalytic machines involved in modulation of this histone
mark
although typically these enzymes are parts of multiprotcin complexes
containing other
2
proteins involved in reading and modifying histone marks. The components of
these protein
complexes are typically cell type and typically comprise transcriptional
regulators,
repressors, co-repressors, receptors associated with gene expression
modulation (e.g.,
estrogen or androgen receptor). Histone deacetylase inhibitors alter the
histone acetylation
profile of chromatin. Accordingly, histone deacetylase inhibitors like SAHA,
TSA, and
many others have been shown to alter gene expression in various in vitro and
in vivo animal
models. Clinically, histone deacetylase inhibitors have demonstrated activity
in the cancer
setting and are being investigated for oncology indications as well as for
neurological
conditions and other diseases.
Another modification that is involved in regulating gene expression is histone
methylation
including lysine and arginine methylation. The methylation status of histone
lysines has
recently been shown to be important in dynamically regulating gene expression.
A group of enzymes known as histone lysine methyl transferases and histone
lysine
demethylases are involved in histone lysine modifications. One particular
human histone
lysine demethylase enzyme called Lysine Specific Demethylase-1 (LSD1) was
recently
discovered (Shi et al. (2004) Cell 119:941) to be involved in this crucial
histone
modification. LSD1
has a fair degree of structural similarity, and amino acid
identity/homology to polyamine oxidases and monoamine oxidases, all of which
(i.e.,
MAO-A, MAO-B and LSD1) are flavin dependent amine oxidases which catalyze the
oxidation of nitrogen-hydrogen bonds and/or nitrogen carbon bonds.
Several groups have reported LSD1 inhibitors in the literature. Sharma et al.
recently
reported a new series of urea and thiourea analogs based on an earlier series
of polyamines
which were shown to inhibit LSD1 and modulate histone methylation and gene
expression
in cells ((2010) J. Med Chem. PMID: 20568780). Sharma et aL note that "To
date, only a
few existing compounds have been shown to inhibit LSD1." Some efforts were
made to
make analogs of the histone peptide that is methylated by the enzyme, other
efforts have
focused on smaller molecule like molecules based on known MAO inhibitors.
Cyclopropylamine containing compounds are known to inhibit a number of
medically
important targets including amine oxidases like Monoamine Oxidase A (MAO-A; or
CA 2806008 2018-02-26
3
MAOA), Monoamine Oxidase B (MAO-B; or MA0B), and Lysine Specific Demethylase-1
(LSD1). Tranylcypromine (also known as 2-phenylcyclopropylamine), which is the
active
ingredient of Parnatet and one of the best-known examples of a
cyclopropylamine, is
known to inhibit all of these enzymes.
Gooden et al. reported trans-2-arylcyclopropylamine analogues that inhibit
LSD1 with Ki
values is the range of 188-566 micromolar (Gooden et al. ((2008) Bioorg. Med.
Chem. Let.
18:3047-3051)). Most of these compounds were more potent against MAO-A as
compared
to MAO-B. Ueda et al. ((2009) J. Am. Chem Soc. 131(48):17536-17537) reported
cyclopropylamine analogs selective for LSD1 over MAO-A and MAO-B that were
designed
based on reported X-ray crystal structures of these enzymes with a
phenylcyclopropylamine-FAD adduct and a FAD-N-propargyl lysine peptide. The
reported
1050 value for phenylcyclopropylamine was about 32 micromolar for LSD1 whereas
compounds 1 and 2 had values of 2.5 and 1.9 micromolar respectively.
Mimasu et al. disclose a series of phenylcyclopropylamine derivatives having
benzoyl
substitutions at the ortho-position (2010) Biochemistry PMID: 20568732. Ortho-
substituted
compounds from this series without a benzoyl group in the ortho-position e.g.,
phenyl,
alkoxy, or having a combination of ortho- and para- substitution appeared to
be less potent
inhibitors of LSD1 than those compounds having benzoyl substituents in the
ortho-position.
The most active compounds from this series had a benzoyl group at the ortho-
position and
one or two meta- fluor substitutions: biphenyls like S1310 and compounds
having large
groups in the para- position were less effective LSD1 inhibitors.
The phenylcyclopropylamines have been the subject of many studies designed to
elucidate a
SAR for MAO inhibition. Kaiser et al. ((1962)1. Med. Chem, 5:1243-1265);
Zirkle et al.
((1962) J. Med. Chem. 1265-1284; US patent nos. 3,365,458; 3,471,522;
3,532,749) have
disclosed the synthesis and activity of a number of phenylcyclopropylamine
related
compounds. Other phenylcyclopropylamine type compounds are disclosed in
Bolesov et al.
((1974) Zhurnal Organicheskoi Khimii 10:8 1661-1669) and Russian Patent No.
230169
(19681030).
Studies have been conducted with phenylcyclopropylamine related compounds to
determine
selectivity for MAO-A versus MAO-B since MAO-A inhibitors can cause dangerous
side-
CA 2806008 2018-02-26
. .
4
effects (see e.g., Yoshida et at. (2004) Bioorg. Med Chem. 12(10):2645-2652;
Hruschka et
al. (2008) Biorg Med Chem. (16):7148-7166; Folks et at. (1983)1 Clin.
Psychopharmacol.
(3)249; and Youdim et al. (1983) Mod. Probl. Pharmacopsychiatry (19):63).
Binda et al. examined a series of phenylcyclopropylamine derivatives in
relation to their
inhibitory activity against LSD1 and LSD2 as well as examining stereochemical
issues in
relation to the cyclopropyl ring (J. Am. Chem. Soc. (2010) May 19;132(19):6827-
33).
Binda et al. reported that their para substituted phenylcyclopropylamine
derivatives are
non-selective which as a group appear to be better MAO-A inhibitors than MAO-B
inhibitors. Furthermore, their inhibitory activities against MAO-A and LSD1
were roughly
the same.
Substituted cyclopropylamines can be chiral. Chiral compounds are often
characterized by
their ability to rotate plane polarized light and are typically referred to as
(+) or (-)
depending on the direction they rotate the light. Another nomenclature is the
d- and 1-
which are short for dextrorotatory and levorotatory. R and S designation are
used to specify
absolute configurations since the ability of chiral molecules to rotate plane
polarized light,
e.g., the direction of rotation, does not correlate always with absolute
configurations.
Tranylcypromine has two stereocenters corresponding to the carbons of the
cyclopropyl ring
that bear the amino substituent and the phenyl substituent. Theoretically, a
compound
having the phenylcyclopropylamine structure of tranylcypromine can have four
stereochemical configurations: two corresponding to the cis (1S,2S or 1R,2R)
and two
corresponding to the trans (1S,2R or 1R,2S). Tranylcypromine corresponds to
the trans
isomer of 2-phenylcyclopropylamine and is a racemate of the (-) and (+)
enantiomers (i.e., a
50:50 mixture of the 1S,2R and 1R,2S enantiomers) and, thus. is optically
inactive.
The (-) enantiomer of tranylcypromine was synthesized, characterized, and
absolute
configuration determined. Riley et al. (1972) 1 Med. Chem. 15(11):1187-1188.
The (-)
stereoisomer was determined to have the 1R,2S absolute configuration by
synthesis from
1R,2R-2-phenylcyclopropanecarboxylic acid.
Later studies using other techniques
confirmed this assignment (Binda et at. (2010) JACS 132:6827-6833) by
determination of
the X-ray structure of the para-bromo derivative of a stereoisomer of 2-
CA 2806008 2018-02-26
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
phenylcyclopropylamine and comparison to the CD spectra of the tranylcypromine
stereoisomers. These results were confilined by stereoselective synthesis of
the enantiomers
of tranylcypromine.
5 It is reported that the (+) isomer (1S,2R) of trans-2-
phenylcyclopropylamine is more potent
against MAO than the (-) isomer (1R,2S) (Riley et al. (1972) J. Med. Chem.
15(11):1187-
1188) using an in vivo tryptamine convulsion model (Zirkle et al. (1962) J.
Med. Pharm.
Chem. 5:1265).
Binda et al. reported a significant difference in the ability of the
stereoisomers of
tranylcypromine to inhibit MAO-B with the (+) stereoisomer being much more
potent using
in vitro biochemical assays. The (-) stereoisomer (1R,2S) was a slightly
stronger inhibitor
of LSD1 than the (+) stereoisomer (Ki of 168 uM versus a Ki of 284 uM) but
these
differences were considered marginal by the authors (Binda et al. ((2010) JACS
132:6827-
6833 see page 6828).
Recently, a group reported another investigation of the stereoisomers of
tranylcypromine
with LSD1 (Benelkebir et al. (2011) Bioorg Med. Chem.doi: 10.1016/j
.bmc.2011.02.017).
They found that the stereoisomers of tranylcypromine were about equipotent for
LSD1:
Ki(inact) for the (+) stereoisomer was of 26.6 uM, 28.1 uM for the (-)
stereoisomer and 25.0
uM for the racemate.
Another group studying the effect of the stereochemical configuration around
the
cyclopropylamine group of substituted 2-phenylcyclopropylamine compounds
reported the
stereoisomer having the (1S,2R) absolute configuration (as determined by NMR
using chiral
shift reagents) was a more potent LSD1 inhibitor as compared to its
enantiomer, both in in
vitro biochemical assays and cell based growth inhibition assays in Hela and
HEK293,
whereas the enantiomers behaved equally in the neuroblastoma line, SH-SY5Y
(Ogasawara
et al. (2011) Bioorg. Med. Chem. Doi:10.1016/j.bmc. 2010.12.024).
Given the differences in assays/experimental protocols used in the different
studies
referenced above, it is difficult to compare results between studies.
Regardless, it is not
clear from these data how derivatives or analogs of compounds having a
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
6
phenylcyclopropylamine core can be optimized to provide potent inhibitors of
LSD1, and
LSD1 and MAO-B, based on the stereochemical configuration of the carbons of
the
cyclopropyl moiety. Furtheimore, it is not clear from these studies how the
selectivity of N-
substituted aryl- and hetero- cyclopropylamine compounds for both LSD1 and MAO-
B can
be modulated to provide compounds that inhibit these enzymes to a greater
extent than
MAO-A. Such compounds are expected to have beneficial safety windows by
avoiding
MAOA inhibition and the so-called "cheese effect".
In view of the lack of adequate treatments for conditions such as cancer and
neurodegeneration, there is a desperate need for disease modifying drugs and
drugs that
work by inhibiting novel targets. There is a need for the development of
better LSD1
selective inhibitors particularly those which selectively inhibit LSD1 or LSD1
in
combination with MAO-B.
SUMMARY OF THE INVENTION
The present invention relates to the identification of compounds and their use
in treating or
preventing diseases. The invention provides (hetero)cyclopropylamine
compounds,
including the compounds of Formula (1), (II) or (Ill) as described and defined
herein. The
present invention particularly provides a compound of Formula (1) or a
pharmaceutically
acceptable salt or solvate thereof, pharmaceutical compositions comprising a
compound of
Formula (1) or a pharmaceutically acceptable salt or solvate thereof, and a
pharmaceutically
acceptable carrier, and their uses for treating diseases. One use of the
compound of Formula
(I) is for treating or preventing cancer. Another use for the compound of
Foimula (I) is to
inhibit LSD1. The present invention thus relates to a compound of Formula (I)
or an
enantiomer, a diastereomer, or a mixture thereof, or a pharmaceutically
acceptable salt or
solvate thereof for use in treating or preventing cancer. Thus, the invention
provides a
compound of Formula (I), or a pharmaceutically acceptable salt or solvate
thereof, and
further relates to its use in treating or preventing human disease.
Accordingly, the present invention provides a compound of Formula (I) or a
pharmaceutically acceptable salt or solvate thereof:
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
7
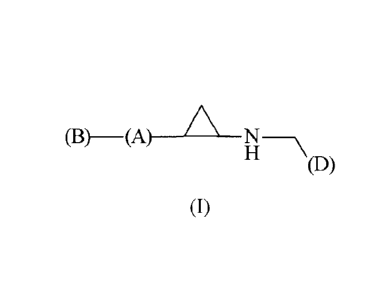
(B)
HD)
(1)
(A) is a cyclyl group having n substituents (R3).
(B) is a cyclyl group or an -(L1)-cyclyl group, wherein said cyclyl group or
the cyclyl
moiety comprised in said -(L1)-cyclyl group has n substituents (R2).
(L1) is -0-, -NH-, -N(alkyl)-, alkylene or heteroalkylene.
(D) is a heteroaryl group or an -(L2)-heteroaryl group, wherein said
heteroaryl group or the
heteroaryl moiety comprised in said -(L2)-heteroaryl group has one substituent
(R1), and
further wherein said heteroaryl group is covalently bonded to the remainder of
the molecule
through a ring carbon atom or the heteroaryl moiety comprised in said -(L2)-
heteroaryl
group is covalently bonded to the (L2) moiety through a ring carbon atom.
(L2) is -0-, -NH-, -N(alkyl)-, alkylene or heteroalkylene.
(R1) is a hydrogen bonding group.
Each (R2) is independently selected from alkyl, alkenyl, alkynyl, cyclyl,
amino, amido, C-
amido, alkylamino, hydroxyl, nitro, halo, haloalkyl, haloalkoxy, cyano,
sulfinyl, sulfonyl,
sulfonamide, alkoxy, acyl, carboxyl, carbamate or urea.
Each (R3) is independently selected from alkyl, alkenyl, alkynyl, cyclyl,
amino, amido, C-
amido, alkylamino, hydroxyl, nitro, halo, haloalkyl, haloalkoxy, cyano,
sulfinyl, sulfonyl,
sulfonamide, alkoxy, acyl, carboxyl, carbamate or urea.
n is independently 0, 1, 2, 3 or 4.
GA 02806008 2013-01-18
WO 2012/013728 PCT/EP2011/062949
8
The substituents of the cyclopropyl moiety, i.e., the group (A) and the group
¨NH-CH2-(D),
are preferably in the trans- configuration.
In a related aspect, the invention provides a phaimaceutical composition
comprising a
compound of Formula (I) or an enantiomer, a diastereomer, or a mixture
thereof, or a
phaimaceutically acceptable salt or solvate thereof as defined above and a
pharmaceutically
acceptable carrier. Preferred embodiments of the compound of Formula (I), e.
g., for use in
the composition of the invention are defined and described herein below in
more detail.
In another aspect, the invention provides a method of treating or preventing a
disease or
condition comprising administering, to a patient (preferably a human) in need
of treatment
or prevention, a therapeutically effective amount of a pharmaceutical
composition
comprising a compound of Fotinula (I) as described above or as in the
embodiments thereof
as described below, or a pharmaceutically acceptable salt thereof and a
pharmaceutically
acceptable carrier. This aspect can be refaimulated as a compound of Formula
(I) as
defined above in the first aspect of the invention for use as a medicine. In a
related aspect,
the invention provides a pharmaceutical composition for use in treating or
preventing a
disease or condition wherein said composition comprises a therapeutically
effective amount
of a compound of Formula (I) sufficient for treating or preventing said
disease or condition.
In a more specific embodiment, the invention provides a compound of Formula
(I) for use in
the treatment of a disease associated with I,SD1.
In yet another aspect, the invention provides a method of inhibiting LSD1
activity
comprising administering, to a patient in need of treatment, a therapeutically
effective
amount of a composition comprising a compound of Formula (I) or a
pharmaceutically
acceptable salt thereof, and a pharmaceutically acceptable carrier sufficient
to inhibit LSD1
activity. Preferably the patient is a human. This aspect can be refoimulated
as a compound
of Foimula (I) as herein defined for use as a LSD1 inhibitor. In a related
aspect, a method
for treating an individual is provided, said method comprising identifying an
individual in
need of treatment and administering to said individual a therapeutically
effective amount of
a compound of Formula (I). In a preferred aspect, the therapeutically
effective amount of a
compound of Formula (I) is an amount sufficient to inhibit LSD1. Preferred
embodiments
GA 02806008 2013-01-18
WO 2012/013728 PCT/EP2011/062949
9
of the compounds of Formula (I) for use in the composition and method of this
aspect of the
invention are as described in more detail herein.
In again another aspect, the invention provides a method of treating or
preventing cancer
comprising administering, to a patient in need of treatment or prevention, a
therapeutically
effective amount of a composition comprising a compound of Formula (I) as
defined above
or as the embodiments described in more detail herein, and a pharmaceutically
acceptable
carrier. This aspect can be reformulated as a compound of For ____________
nula (I) as defined above in
the first aspect of the invention for use in the treatment or prevention of
cancer. In a related
aspect, the invention provides a pharmaceutical composition for use in
treating or
preventing cancer wherein said composition comprises a therapeutically
effective amount of
a compound of Formula (I) sufficient for treating or preventing cancer. In
another related
aspect, the invention provides a compound of Formula (I) or a pharmaceutical
composition
for the treatment or prevention of a cancer wherein said cancer is chosen from
breast cancer,
lung cancer, prostate cancer, colorectal cancer, brain cancer, skin cancer,
blood cancer (e.g.,
leukemia, including, for example, acute myelogenous leukemia (AML), chronic
myelogenous leukemia (CML), chronic neutrophilic leukemia, chronic
eosinophilic
leukemia, chronic lymphocytic leukemia (CLL), acute lymphoblastic leukemia
(ALL), or
hairy cell leukemia), lymphoma and myeloma. Said composition preferably
comprises a
therapeutically effective amount of a compound of Formula (I) sufficient for
treating or
preventing said cancer. In a preferred aspect, the therapeutically effective
amount of a
compound of Formula (I) is an amount sufficient to inhibit LSD I . In another
preferred
aspect, the therapeutically effective amount is an amount sufficient to
modulate histone
methylation levels. In another preferred aspect, the therapeutically effective
amount is an
amount sufficient to modulate histone-3 lysine-4 methylation levels. In
another preferred
aspect, the therapeutically effective amount is an amount sufficient to
modulate histone-3
lysine-9 methylation levels.
In again another aspect, the invention provides a method of treating or
preventing a
neurological disease or condition comprising administering, to a patient in
need of treatment
or prevention, a therapeutically effective amount of a composition comprising
a compound
of Formula (I) as defined above or in the embodiments described in more detail
herein, and
a pharmaceutically acceptable carrier. This aspect can be reformulated as a
compound of
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
Formula (I) as defined above for use in the treatment or prevention of a
neurological
condition or disease. In a related aspect, the invention provides a
pharmaceutical
composition for use in treating or preventing a neurological condition or
disease wherein
said composition preferably comprises a therapeutically effective amount of a
compound of
5 Formula (I) sufficient for treating or preventing said neurological
disease or condition. In
another related aspect, the invention provides a compound of Formula (I) or a
pharmaceutical composition for the treatment or prevention of a neurological
disease or
condition wherein said neurological disease or condition is chosen from
depression,
Alzheimer's disease, Huntington disease, Parkinson's disease, Amyotrophic
Lateral
10 Sclerosis, Dementia with Lewy Bodies, or Frontotemporal Dementia,
particularly from
depression, Alzheimer's disease, Huntington disease, Parkinson's disease, or
Dementia with
Lewy Bodies. Said composition preferably comprises a therapeutically effective
amount of
a compound of Formula (I) sufficient for treating or preventing said disease
or condition. In
a preferred aspect, the therapeutically effective amount of a compound of
Foimula (I) is an
amount sufficient to inhibit LSD1. In another preferred aspect, the
therapeutically effective
amount is an amount sufficient to modulate histone methylation levels. In
another preferred
aspect, the therapeutically effective amount is an amount sufficient to
modulate histone-3
lysine-4 methylation levels. In another preferred aspect, the therapeutically
effective
amount is an amount sufficient to modulate histone-3 lysine-4 methylation
levels. In
another preferred aspect, the therapeutically effective amount is an amount
sufficient to
modulate histone-3 lysine-9 methylation levels.
In still another aspect, the invention provides a method for identifying a
compound which is
a selective inhibitor of LSD1, the method comprising selecting or providing a
compound of
Formula (I) as defined herein, and determining the ability of the compound to
inhibit LSD1
and MAO-A and/or MAO-B, wherein a compound that inhibits LSD1 to a greater
extent
than MAO-A and/or MAO-B is identified as a LSD1 selective inhibitor. The
compound of
this aspect that is an LSD1 inhibitor can be used to treat disease,
particularly human disease.
In still another aspect, the invention provides a method for identifying a
compound which is
a dual inhibitor of LSD1 and MAO-B, the method comprising selecting or
providing a
compound of Formula (I) as defined herein, and determining the ability of the
compound to
inhibit LSD1, MAO-A, and MAO-B, wherein a compound that inhibits LSD I and MAO-
B
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
11
to a greater extent than MAO-A is identified as a LSD1 MAO-B dual inhibitor.
The
compound of this aspect that is an LSD1 MAO-B inhibitor can be used to treat
disease,
particularly human disease.
Thus, in one embodiment of the invention, the phalmaceutical composition
comprising a
LSD1 selective inhibitor of Formula (I), or a pharmaceutically acceptable salt
or solvate
thereof is useful for treating and/or preventing a disease in an individual.
In one aspect, a
therapeutically effective amount of the composition is administered to an
individual in an
amount sufficient to prevent or treat a disease. In a more specific aspect,
the disease is
cancer. In an even more specific aspect, the disease is a cancer chosen from
prostate, brain,
colorectal, lung, breast, skin, and blood cancer. In one specific aspect, the
cancer is prostate
cancer. In one specific aspect, the cancer is lung cancer. In one specific
aspect, the cancer
is brain cancer. In one specific aspect, the cancer is blood cancer (e.g.,
leukemia, including,
for example, acute myelogenous leukemia (AML), chronic myelogenous leukemia
(CML),
chronic neutrophilic leukemia, chronic eosinophilic leukemia, chronic
lymphocytic
leukemia (CLL), acute lymphoblastic leukemia (ALL), or hairy cell leukemia).
In one
specific aspect, the cancer is breast cancer. In one specific aspect, the
cancer is colorectal
cancer. In one specific aspect, the cancer is lymphoma. In one specific
aspect, the cancer is
myeloma. In another preferred aspect, the therapeutically effective amount is
an amount
sufficient to inhibit LSD1. In another preferred aspect, the therapeutically
effective amount
is an amount sufficient to modulate histone methylation levels. In another
preferred aspect,
the therapeutically effective amount is an amount sufficient to modulate
histone-3 lysine-4
methylation levels. In another preferred aspect, the therapeutically effective
amount is an
amount sufficient to modulate histone-3 lysine-9 rnethylation levels.
Furthermore, the inventors unexpectedly found that the stereochemical
configuration of the
cyclopropyl carbons of N-substituted arylcyclopropylamine compounds
substantially affects
the potency of LSD1 inhibition, MAO-B inhibition and MAO-A inhibition. The
inventors
have shown that the (-) stereoisomer of 5-
(((trans)-2-(4-
(benzyloxy)phenyl)cyclopropylamino)methyl)-1,3,4-oxadiazol-2-amine is about 20-
fold
more potent against LSD1 than the corresponding (+) stereoisomer. Furthermore,
the (-)
stereoisomer retained substantial MAO-B inhibitory activity. Notably, the
selectivity for
LSD I /MAO-A for the (-)/(+) stereoisomer was over 100 fold as judged by
kinact/KI values.
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
12
Thus, (-) stereoisomers of N-substituted (hetero)arylcyclopropylamine
compounds are
unexpectedly potent and selective LSD1 inhibitors compared to their respective
enantiomers. Furtheiniore, the compounds of the invention have improved
selectivity
against MAO-A, preferentially inhibiting MAO-B and LSD1. The invention
therefore
relates to optically active (hetero)arylcyclopropylamine compounds, in
particular optically
active N-substituted aryl- or heteroaryl- cyclopropylamines, and their use for
treating or
preventing a disease or a disorder.
Thus, in one specific aspect the invention relates to a substantially pure
stereoisomer of an
N-substituted aryl- or heteroaryl- cyclopropylamine (e.g., a compound of
Formula (II) or
(III) as described and defined herein below) for use in a method of treating
or preventing a
disease or disorder. Desirably, the disease or disorder is one that is
treatable or preventable
by LSD1 inhibition, LSD1 inhibition and MAO-B inhibition, or MAO-B inhibition.
In a
specific aspect, a substantially pure stereoisomer of an N-substituted aryl-
or heteroaryl-
cyclopropylamine refers to an N-substituted aryl- or heteroaryl-
cyclopropylamine which is
90% or greater (-) stereoisomer and 10% or less (+) stereoisomer. In a more
specific aspect,
a substantially pure stereoisomer of an N-substituted aryl- or heteroaryl-
cyclopropylamine
refers to an N-substituted aryl- or heteroaryl- cyclopropylamine which is 95%
or greater (-)
stereoisomer and 5% or less (+) stereoisomer. In yet a more specific aspect, a
substantially
pure stereoisomer of an N-substituted aryl- or heteroaryl- cyclopropylamine
refers to an N-
substituted aryl- or heteroaryl- cyclopropylamine which is 98% or greater (-)
stereoisomer
and 2% or less (+) stereoisomer. In an even more specific aspect, a
substantially pure
stereoisomer of an N-substituted aryl- or heteroaryl- cyclopropylamine refers
to an N-
substituted aryl- or heteroaryl- cyclopropylamine which is 99% or greater (-)
stereoisomer
and 1% or less (+) stereoisomer. In yet an even more specific aspect, a
substantially pure
stereoisomer of an N-substituted aryl- or heteroaryl- cyclopropylamine refers
to an N-
substituted aryl- or heteroaryl- cyclopropylamine which is 99.5% or greater (-
) stereoisomer
and 0.5% or less (+) stereoisomer. In one embodiment, the above-described
percentages
refer to mole-%. The substantially pure stereoisomer of an N-substituted aryl-
or heteroaryl-
cyclopropylamine, in one aspect, is for use in a method of treating or
preventing cancer,
depression, a neurodegenerative disease or disorder, or a viral infection.
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
13
Furthermore, in another aspect, the invention is a composition comprising a
stereoisomer of
an N-substituted aryl- or heteroaryl- cyclopropylamine (e.g., a compound of
Formula (II) or
(III) as described and defined herein below) wherein said composition has a
90% or more
enantiomeric excess of the (-) stereoisomer of the N-substituted aryl- or
heteroaryl-
cyclopropylamine. In a specific aspect said composition has a 95% or more
enantiomeric
excess of the (-) stereoisomer of the N-substituted aryl- or heteroaryl-
cyclopropylamine. In
a more specific aspect said composition has a 98% or more enantiomeric excess
of the (-)
stereoisomer of the N-substituted aryl- or heteroaryl- cyclopropylamine. In an
even more
specific aspect said composition has a 99% or more enantiomeric excess of the
(-)
stereoisomer of the N-substituted aryl- or heteroaryl- cyclopropylamine. The
composition in
one aspect of the invention is for use in a method of treating or preventing
cancer,
depression, a neurodegenerative disease or disorder, or a viral infection.
Furthennore, in another aspect, the invention is a phaimaceutical composition
comprising a
stereoisomer of an N-substituted aryl- or heteroaryl cyclopropylamine (e.g., a
compound of
Formula (II) or (III) as described and defined herein below) and a
pharmaceutically
acceptable carrier wherein said composition has a 90% or more enantiomeric
excess of the
(-) stereoisomer of the N-substituted aryl- or heteroaryl- cyclopropylamine.
In a specific
aspect said composition has a 95% or more enantiomeric excess of the (-)
stereoisomer of
the N-substituted aryl- or heteroaryl- cyclopropylamine. In a
specific aspect said
composition has a 99% or more enantiomeric excess of the (-) stercoisomer of
the N-
substituted aryl- or heteroaryl- cyclopropylamine. The phainiaceutical
composition of this
paragraph is for use in a method of treating or preventing cancer, depression,
a
neurodegenerative disease or disorder, or a viral infection.
In one aspect of the invention, the optically active N-substituted aryl- or
heteroaryl-
cyclopropylamine or phaimaceutically acceptable salt or solvate thereof, for
use in a method
of treating or preventing a disease or disorder, as described herein, is of
Foi mula (II):
RI "¨(A'1)¨R2"
(II)
wherein:
14
(A") is an aryl or heteroaryl group having 2 substituents, RI" and R2", and 1,
2, or 3
optional substituents wherein said optional substituents are independently
chosen from halo,
CI-C3 alkyl, or CI-C3 alkoxy;
R111 is an II)-(R3 II) group;
R3" is an aryl or heteroaryl group having 1, 2, 3, 4, or 5 optional
substituents independently
chosen from halo, -OH, -NHSO2RA, alkyl, alkoxy, cyano, -CF3, or -0CF3, wherein
RA is a
CI-C6 alkyl or phenyl;
LI" is chosen from a bond, -CH20-, -CH2CH20-, -OCH2-, - OCH2CH2-, - CH2CH2-, -
CH2-,
-CH2CH2CH2-, or -0-;
R2" is ¨Cyclopropyl-NII-(L211)-(R4") wherein said cyclopropyl group has two
chiral centers
substituted in the trans orientation corresponding to the carbons to which (A)
and ¨NII-
(L2")-(R411) are covalently attached;
R411 is a 5 or 6 membered heteroaryl ring having 1, 2, or 3 optional
substituents chosen from
alkyl, -NHRB, -ORB, or halo wherein RB is hydrogen, C1-C3 alkyl, or -C(=0)CH3;
L2Il is a branched or unbranched C,-C4 alkylene group
and wherein said compound of Formula (II) is optically active.
The invention also is a pharmaceutical composition comprising an optically
active N-
substituted aryl- or heteroaryl- cyclopropylamine of Formula (II) as defined
above, or a
pharmaceutically acceptable salt or solvate thereof, and a pharmaceutically
acceptable
carrier.
The invention also is a pharmaceutical composition comprising an optically
active N-
substituted aryl- or heteroaryl cyclopropylamine of Formula (II) as defined
above, or a
pharmaceutically acceptable salt or solvate thereof, and a pharmaceutically
acceptable
carrier, for use in treating or preventing a disease or disorder. Preferably,
the disease or
CA 2806008 2018-02-26
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
disorder is mediated through an amine oxidase. In one aspect, the amine
oxidase is LSD1 or
MAO-B.
Furthermore, the inventors found a subset of optically active compounds of
Formula (II) as
5 shown in Formula (III) which are inhibitors of LSD 1 or of LSD 1 and MAO-
B.
The invention thus further relates to an optically active compound of Formula
(III) or a
pharmaceutically acceptable salt or solvate thereof:
10 R2HI
(III)
wherein:
(Am) is an aryl or heteroaryl group having 2 substituents, ROI and R2111, and
1, 2, or 3
15 optional substituents independently chosen from halo, C1-C3 alkyl, or CI-
C3 alkoxy;
R1 111 is an ¨(L1111)4R3111) group;
R3" .5 is a phenyl, pyridyl, thiazolyl, or thienyl group having 1, 2, 3, 4, or
5 optional
substituents independently chosen from halo, -OH, -NHSO2RA, alkyl, alkoxy,
cyano,
or -0CF3, wherein RA is a CI-C6 alkyl or phenyl;
L1111 is chosen from a bond, -OCH2-, or -CH20-;
R2111 is ¨Cyclopropyl-NH-(L2111)-(R4111) wherein said cyclopropyl group has
two chiral
centers substituted in the trans orientation corresponding to the carbons to
which (A111) and
-NH-(L2111)-(R = 4Ills ) are covalently attached:
R4111 is a 5-membered heteroaryl ring having 1, 2, or 3 optional substituents
wherein said
optional substituents are independently chosen from ¨NH2 or ¨NH(Ci-C3) alkyl;
L2111 is _c-2_
H. Or -CH2CH2-;
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
16
and wherein said compound of Formula (III) is optically active.
Additionally, the invention is a pharmaceutical composition comprising an
optically active
N-substituted aryl- or heteroaryl- cyclopropylamine of Formula (II) or (III)
as defined
above, or a pharmaceutically acceptable salt or solvate thereof, and a
pharmaceutically
acceptable carrier.
The invention also is a pharmaceutical composition comprising an optically
active N-
substituted aryl- or heteroaryl- cyclopropylamine of Formula (II) or (III) as
defined above,
or a pharmaceutically acceptable salt or solvate thereof, and a
pharmaceutically acceptable
carrier, for use in treating or preventing a disease or disorder.
In one aspect, the invention is a method of treating or preventing a disease
or disorder
comprising administering, to an individual in need of treatment, a
therapeutically effective
amount of an optically active N-substituted aryl- or heteroaryl-
cyclopropylamine,
particularly a compound of Formula (II) or (III), or a pharmaceutically
acceptable salt or
solvate thereof. In a more specific aspect, the disease or disorder is human
disease or
disorder chosen from cancer, depression, a neurodegenerative disease or
disorder, or a viral
infection. In one aspect, the neurodegenerative disease or disorder is
Huntington disease,
Parkinson disease, Alzheimer disease, Amyotrophic Lateral Sclerosis,
Frontotemporal
Dementia, or Dementia with Levvy Bodies.
In one aspect, the invention is a method of treating or preventing a disease
or disorder
comprising identifying an individual in need of treating or preventing and
administering to
said individual a therapeutically effective amount of an optically active N-
substituted aryl-
or heteroaryl- cyclopropylamine, particularly a compound of Formula (II) or
(III), or a
pharmaceutically acceptable salt or solvate thereof. In a more specific
aspect, the disease or
disorder is human disease or disorder chosen from cancer, depression, a
neurodegenerative
disease or disorder, or a viral infection. In one aspect, the
neurodegenerative disease or
disorder is Huntington disease, Parkinson disease, Alzheimer disease,
Amyotrophic Lateral
Sclerosis, Frontotemporal Dementia, or Dementia with Lcwy Bodies.
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
17
In one aspect, the invention provides a method for enriching an enantiomer of
a trans N-
substituted cyclopropylarnine (Jarticularly of a compound of Formula (II) or
(III), or of a
compound of Foimula (I) wherein the substituents on the cyclopropyl moiety are
in trans-
orientation), the method comprising:
Contacting a trans-substituted cyclopropylamine with a chiral
recrystallization agent in a
solvent (particularly under conditions that are sufficient for the
crystallization of the salt of
the chiral recrystallization agent and the trans substituted cylopropylamine);
and isolating
the crystallized salt of the chiral recrystallization agent and the trans
substituted
cyclopropylamine. In one aspect, the trans cyclopropylamine is an N-
substituted aryl- or
heteroaryl-cylopropylamine. In one aspect, the trans cyclopropylamine is 4
benzoxy-2-
phenylcyclopropylamine or a derivative thereof wherein the amine is protected
with a
protecting group.
Unless otherwise defined, all technical and scientific terms used herein have
the same
meaning as commonly understood by one of ordinary skill in the art to which
this invention
pertains. Although methods and materials similar or equivalent to those
described herein
can be used in the practice or testing of the present invention, suitable
methods and
materials are described below. In case of conflict, the present specification,
including
definitions, will control. In addition, the materials, methods, and examples
are illustrative
only and not intended to be limiting.
Other features and advantages of the invention will be apparent from the
following detailed
description, and from the claims.
DETAILED DESCRIPTION OF THE INVENTION
The present invention relates to the identification of compounds and their use
in treating and
preventing diseases. The present invention provides compounds of Formula
(I),
phaimaceutical compositions comprising a compound of Folinula (I) or a
pharmaceutically
acceptable salt or solvate thereof and a pharmaceutically acceptable carrier,
and their use for
treating diseases. One use of the compounds of Formula (1) is for treating
cancer. The
compounds of Formula (I) can be used as LSD1 selective inhibitors that inhibit
LSD1 to a
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
18
greater extent than MAO-A and MAO-B or as LSD1/MAO-B dual inhibitors that
inhibit
LSD1 and MAO-B to a greater extent than MAO-A. The compounds of Foimula (I) as
described herein are generally better inhibitors of LSD1 by a factor of more
than 10 to 20 or
more as compared to tranylcypromine, with improved selectivity against MAO-A.
Thus,
these compounds are LSD1 selective in that they inhibit LSD1 to an extent
greater than
MAO-A and MAO-B or are LSD1/MAO-B duals inhibitors that inhibit LSD1 and MAO-B
to a greater extent than MAO-A.
The present invention provides a compound of Formula (I) or a pharmaceutically
acceptable
salt or solvate thereof:
(B) N
H \(D)
(I)
(A) is a cyclyl group having n substituents (R3). Preferably, (A) is an aryl
group or a
heteroaryl group, wherein said aryl group or said heteroaryl group has n
substituents (R3).
More preferably, (A) is phenyl, pyridinyl, thiophenyl, pyrrolyl, furanyl, or
thiazolyl,
wherein (A) has n substituents (R3). Even more preferably, (A) is phenyl or
pyridyl,
wherein said phenyl or said pyridyl has n substituents (R3). In one
embodiment, (A) has 0
or 1 substituent (R3). In a further embodiment, (A) has 0 substituents (R3).
In a further
embodiment, (A) has 1 substituent (R3). It is to be understood that, if n is
0, the cyclyl
group is not substituted with any substituents (R3) but may instead be
substituted with
hydrogen.
(B) is a cyclyl group or an -(L1)-cyclyl group, wherein said cyclyl group or
the cyclyl
moiety comprised in said -(Li )-cyclyl group has n substituents (R2). Said
cyclyl group or
the cyclyl moiety comprised in said -(L1)-cyclyl group may, for example, be an
aryl group
(e.g., phenyl, naphthyl or anthracenyl) or a heteroaryl group (e.g.,
pyridinyl, thiophenyl,
pyrrolyl, furanyl, thiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, thiadiazolyl,
triazinyl,
pyridazinyl, pyrazinyl, or pyrimidinyl). Preferably, (B) is -0-CH2-phenyl or
phenyl, wherein
(B) has n substituents (R2). In one embodiment, (B) is phenyl having n
substituents (R2).
In a further embodiment, (B) is -0-CH2-phenyl having n substituents (R2). In
one
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
19
embodiment, (B) has 0, 1 or 2 substituents (R2). In a further embodiment, (B)
has 0 or 1
substituent (R2). In a further embodiment, (B) has 0 substituents (R2). In a
further
embodiment, (B) has 1 substituent (R2).
(L1) is -0-, -NH-, -N(alkyl)-, alkylene or heteroalkylene. Said alkylene may,
e.g., be a
straight-chain or branched chain alkylene having from 1 to 6 carbon atoms.
Said
heteroalkylene may, e.g., be a straight-chain or branched chain alkylene
having from 1 to 6
carbon atoms, wherein 1, 2 (if present) or 3 (if present) carbon atoms are
each replaced by a
heteroatom selected independently from 0, N or S; accordingly, said
heteroalkylene may,
e.g., be a straight-chain or branched chain alkylene haying from 1 to 4 carbon
atoms,
wherein 1 or 2 non-adjacent carbon atoms are each replaced by 0.
(D) is a heteroaryl group or a -(L2)-heteroaryl group, wherein said heteroaryl
group or the
heteroaryl moiety comprised in said -(L2)-heteroaryl group has one substituent
(R1), and
further wherein said heteroaryl group is covalently bonded to the remainder of
the molecule
through a ring carbon atom or the heteroaryl moiety comprised in said -(L2)-
heteroaryl
group is covalently bonded to the (L2) moiety through a ring carbon atom.
Preferably, (D)
is thiazolyl, oxadiazolyl, oxazolyl, isoxazolyl, thiadiazolyl, triazinyl,
pyridazinyl, pyrazinyl,
pyridinyl or pyrimidinyl, wherein said thiazolyl, oxadiazolyl, oxazolyl,
isoxazolyl,
thiadiazolyl, triazinyl, pyridazinyl, pyrazinyl, pyridinyl or pyrimidinyl has
one substituent
(RI). In particular, (D) may be thiazolyl, oxadiazolyl or pyrimidinyl, wherein
said thiazolyl,
said oxadiazolyl or said pyrimidinyl has one substituent (R1). Most
preferably, (D) is
oxadiazolyl.
(L2) is -0-, -NH-, -N(alkyl)-, alkylene or heteroalkylene. Said alkylene may,
e.g., be a
straight-chain or branched chain alkylene having from 1 to 6 carbon atoms.
Said
heteroalkylene may, e.g., be a straight-chain or branched chain alkylene
having from 1 to 6
carbon atoms, wherein 1, 2 (if present) or 3 (if present) carbon atoms are
each replaced by a
heteroatom selected independently from 0, N or S; accordingly, said
heteroalkylene may,
e.g., be a straight-chain or branched chain alkylene having from 1 to 4 carbon
atoms,
wherein 1 or 2 non-adjacent carbon atoms are each replaced by 0.
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
(R1) is a hydrogen bonding group. For example, (R1) may be -OH, -0(alkyl), -
NH2,
-NH(alkyl) (e.g., -NHCH3), -N(alkyl)(alkyl) (e.g., -N(CH3)2), amido, -SO-NH2,
-SO-NH(alkyl), -SO-N(alkyl)(alkyl), -S(0)2NH2, -S(0)2NH(alkyl), -
S(0)2N(alkyl)(alkyl),
-C(0)NH2, -C(=0)NH(alkyl), -C(=0)N(alkyl)(alkyl), -alkylene-C(=0)NH2 (e.g.,
5 -CH2-C(=0)NH2), -alkylene-C(=0)NH(alkyl) (e.g.,
-CH2-C(=0)NH(alkyl)),
-alkylene-C(=0)N(alkyl)(alkyl) (e.g., -CH2-C(=0)N(alkyl)(alkyl)), -NHC(=0)-
alkyl (e.g.,
-NHC(=0)CH3), -N(alkyl)-C(=0)-alkyl (e.g., -N(-CH3)-C(=0)CH3), -alkylene-NH2
(e.g.,
-CH2-NH2), -alkylene-NH(alkyl), or -alkylene-N(alkyl)(alkyl), wherein it is
preferred that
the aforementioned alkyl and alkylene groups each independently have from 1 to
6 carbon
10 atoms. Preferably, (R1) is -OH, -NH2, amido, -S(0)2NH2, -C(0)NH2, -CH2-
C(=0)NH2, -
NHC(---0)CH3, -NHCH3, -N(CH3)2 or -CH2-NH2, particularly -OH, -
NHCH3, amido,
-S(0)21\1112, -C(=0)NH2, -CH2-C(=0)NH2, or -CH2-NH2. More preferably, (RI) is -
NH2 or
-NHCH3. Even more preferably, (R1) is -NH2.
15 Each (R2) is independently selected from alkyl, alkenyl, alkynyl,
cyclyl, amino, amido, C-
amido, alkylamino, hydroxyl, nitro, halo, haloalkyl, haloalkoxy, cyano,
sulfinyl, sulfonyl,
sulfonamide, alkoxy, acyl, carboxyl, carbamate or urea. For example, each (R2)
may be
independently selected from hydroxyl, halo (e.g., -Cl or -F) or haloalkyl
(e.g., -CF3).
Accordingly, each (R2) may, for example, be selected independently from
hydroxyl or
20 haloalkyl (e.g., -CF3). It is preferred that each (R2) is halo, more
preferably -F.
Each (R3) is independently selected from alkyl, alkenyl, alkynyl, cyclyl,
amino, amido, C-
amido, alkylamino, hydroxyl, nitro, halo, haloalkyl, haloalkoxy, cyano,
sulfinyl, sulfonyl,
sulfonamide, alkoxy, acyl, carboxyl, carbamate, or urea. For example, each
(R3) may be
independently selected from alkyl, cyclyl, amino, amido, alkylamino, hydroxyl,
halo,
haloalkyl, haloalkoxy, cyano, sulfonamide, alkoxy, acyl, carboxyl, carbamate,
or urea.
n is independently 0, 1, 2, 3 or 4. For example, each n may be independently
0. 1 or 2. In
particular, each n may be independently 0 or 1.
The substituents of the cyclopropyl moiety, i.e., the (A) group and the -NH-
CH2-(D) group,
are preferably in trans-configuration.
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
21
In one preferred embodiment of the first aspect, the invention provides a
compound of
Formula (I) wherein (A) is an aryl or heterocyclyl. In a more preferred
embodiment (A) is
phenyl, pyridinyl, thiophenyl, pyrrolyl, furanyl, or thiazolyl. In an even
more preferred
embodiment (A) is phenyl or pyridinyl.
In one preferred embodiment of the first aspect, the invention provides a
compound of
Formula (I) wherein, (B) is a -L2-cyclyl. In a more preferred embodiment (B)
is -0-phenyl
or -0-CH2-phenyl. In an even more preferred embodiment (B) is -0-CH2-phenyl.
In one
specific embodiment the phenyl group of said (B) group has I, 2, 3, or 4
optional
substituents (R2) independently chosen from alkyl, alkenyl, alkynyl, cyclyl,
amino, amido,
C-amido, alkylamino, hydroxyl, nitro, halo, haloalkyl, haloalkoxy, cyano,
sulfinyl, sulfonyl,
sulfonamide, alkoxy, acyl, carboxyl, carbamate or urea.
In one preferred embodiment of the first aspect, the invention provides a
compound of
Formula (I) wherein, (B) is cyclyl. In a more preferred embodiment (B) is
phenyl. In one
specific embodiment, the phenyl group of said (B) group has 1, 2, 3, or 4
optional
substituents (R2) independently chosen from alkyl, alkenyl, alkynyl, cyclyl,
amino, amido,
C-amido, alkylamino, hydroxyl, nitro, halo, haloalkyl, haloalkoxy, cyano,
sulfinyl, sulfonyl,
sulfonamide, alkoxy, acyl, carboxyl, carbamate or urea.
In one preferred embodiment of the first aspect, the invention provides a
compound of
Formula (I), wherein (R2) is hydroxyl, halo or haloalkyl. In one preferred
embodiment (R2)
is ¨OH or ¨CF3. In another preferred embodiment (R2) is fluoro or chloro.
In one preferred embodiment of the first aspect, the invention provides a
compound of
Formula (I), wherein (D) is a monocyclic heteroaryl. In a more preferred
embodiment (D)
is thiazolyl, oxadiazolyl, oxazolyl, isoxazolyl, thiadiazolyl, triazinyl,
pyridazinyl, pyrazinyl,
pyridinyl or pyrimidinyl. In one specific embodiment, said cyclyl (D) has one
substituent
(R1).
In one preferred embodiment of the first aspect, the invention provides a
compound of
Foimula (1) wherein (R1) is a hydrogen bonding group. For example, (RI) may be
-OH, -
0(alkyl), -NH2, -NH(alkyl) (e.g., -NHCH3), -N(alkyl)(alkyl), amido, -SO-NH2, -
SO-
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
22
NH(alkyl), -SO-N(alkyl)(alkyl), -S(0)2NH2, -S(0)2NH(alkyl), -
S(0)2N(alkyl)(alkyl),
-C(=0)NH2, -C(=0)NH(alkyl), -C(=0)N(alkyl)(alkyl), -alkylene-C(=0)NH2 (e.g., -
CH2-
C(=0)NH2), -alkylene-C(=0)NH(alkyl) (e. g. , -
CH2-C(=0)NH(alkyl)),
-alkylene-C(=0)N(alkyl)(alkyl) (e.g., -CH2-C(=0)N(alkyl)(alkyl)), -NHC(=0)-
alkyl (e.g.,
-NHC(----0)CH3), -N(alkyl)-C(=0)-alkyl (e.g., -N(-CH3)-C(=0)CH3), -alkylene-
NH2 (e.g., -
CH2-NH2), -alkylene-NH(alkyl), or -alkylene-N(alkyl)(alkyl), wherein it is
preferred that
the aforementioned alkyl and alkylene groups each independently have from 1 to
6 carbon
atoms. In a more preferred embodiment (R1) is a -NH2, -OH, amido, -NHC(=0)CH3,
-
NHCH3 or -S(0)2NI-12. In an even more preferred embodiment (R1) is -NFI2.
The compound of Folinula (I) as described and defined herein may, for example,
be a
compound of the following Formula (Ia) or a pharmaceutically acceptable salt
or solvate
thereof:
(B) __________________________ (A)
/NH (D)
(RAI
(RAI
(R1)
(Ia)
wherein (A), (B), (D), (R1), (R2), (R3) and n have the meanings or the
preferred meanings
described herein for the compound of Formula (I).
Preferably, the compounds of the invention, including in particular the
compounds of
Formula (I), (Ia) or (lb) as described herein, are used to treat a disease in
a mammal and
more preferably a human. More preferably, the human disease is chosen from
cancer (e.g.,
breast cancer, lung cancer, prostate cancer, colorectal cancer, brain cancer,
skin cancer,
blood cancer (e.g., leukemia, including, for example, acute myelogenous
leukemia (AML),
chronic myelogenous leukemia (CML), chronic neutrophilic leukemia, chronic
eosinophilic
leukemia, chronic lymphocytic leukemia (CLL), acute lymphoblastic leukemia
(ALL), or
hairy cell leukemia), lymphoma, or myeloma), a neurological condition or
disease (e.g.,
depression, Alzheimer's disease, Huntington disease, Parkinson's disease,
Amyotrophic
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
23
Lateral Sclerosis, Frontotemporal Dementia, or Dementia with Lewy Bodies), or
a viral
infection.
In one preferred embodiment of the first aspect, the invention provides a
compound of
Formula (Ia) wherein (A) is aryl or heterocyclyl. In a more preferred
embodiment (A) is
phenyl, pyridinyl, thiophenyl, pyrrolyl, furanyl, and thiazolyl. In an even
more preferred
embodiment (A) is a phenyl or a pyridinyl.
In one preferred embodiment of the first aspect, the invention provides a
compound of
Formula (la) wherein (B) is -L2-cyclyl. In a more preferred embodiment (B) is -
0-phenyl
or -0-CH2-phenyl. In an even more preferred embodiment (B) is -0-CH2-phenyl.
In one
specific embodiment the phenyl group of said (B) group has 1, 2, 3, or 4
optional
substituents (R2) independently chosen from alkyl, alkenyl, alkynyl, cyclyl,
amino, amido,
C-amido, alkylamino, hydroxyl, nitro, halo, haloalkyl, haloalkoxy, cyano,
sulfinyl, sulfonyl,
sulfonamide, alkoxy, acyl, carboxyl, carbamate or urea.
In one preferred embodiment of the first aspect, the invention provides a
compound of
Formula (Ia) wherein (B) is cyclyl. In a more preferred embodiment (B) is
phenyl. In one
specific embodiment the phenyl group of said (B) group has 1, 2, 3, or 4
optional
substituents (R2) independently chosen from alkyl, alkenyl, alkynyl, cyclyl,
amino, amido,
C-amido, alkylamino, hydroxyl, nitro, halo, haloalkyl, haloalkoxy, cyano,
sulfinyl, sulfonyl,
sulfonamide, alkoxy, acyl, carboxyl, carbamate or urea.
In one preferred embodiment of the first aspect, the invention provides a
compound of
Formula (Ia) wherein (R2) is hydroxyl, halo or haloalkyl. In one preferred
embodiment
(R2) is ¨OH or ¨CF3. In another preferred embodiment (R2) is fluoro or chloro.
In one preferred embodiment of the first aspect, the invention provides a
compound of
Foimula (Ia.), wherein (D) is a monocyclic heteroaryl. In a more preferred
embodiment (D)
is thiazolyl, oxadiazolyl, oxazolyl, isoxazolyl, thiadiazolyl, triazinyl,
pyridazinyl, pyrazinyl,
pyridinyl or pyrimidinyl. In one specific embodiment said cyclyl (D) has one
substituent
(R1).
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
24
In one preferred embodiment of the first aspect, the invention provides a
compound of
Formula (Ia) wherein (R1) is a hydrogen bonding group. For example, (R1) may
be -OH, -
0(alkyl), -NH2, -NH(alkyl) (e.g., -NHCH3), -N(alkyl)(alkyl), amido, -SO-NH2, -
SO-
NH(alkyl), -SO-N(alkyl)(alkyl), -S(0)2NH2, -S(0)2NH(alkyl), -
S(0)2N(alkyl)(alkyl),
-C(=0)NH2, -C(=0)NH(alkyl), -C(=0)N(alkyl)(alkyl), -alkylene-C(=0)NH2 (e.g., -
CH2-
C(=0)NH2), -alkylene-C(=0)NH(alkyl)
(e.g., -CH2-C(=0)NH(alkyl)),
-alkylene-C(=0)N(alkyl)(alkyl) (e.g., -CH2-C(=0)N(alkyl)(alkyl)), -NFIC(=0)-
alkyl (e.g.,
-NHC(=0)CH3), -N(alkyl)-C(=0)-alkyl (e.g., -N(-CH3)-C(=0)CH3), -alkylene-NH2
(e.g., -
CH2-NH2), -alkylene-NH(alkyl), or -alkylene-N(alkyl)(alkyl), wherein it is
preferred that
the aforementioned alkyl and alkylene groups each independently have from 1 to
6 carbon
atoms. In a more preferred embodiment (R1) is a -NH2, -OH, amido, -NHC(=0)CH3,
-
NHCH3 or -S(0)2NI-12. In a more preferred embodiment (R1) is a -NH2, -OH,
amido, or -
S(0)2NH2. In an even more preferred embodiment (R1) is -NH2.
Thus in a preferred aspect, the invention provides a compound of Fount'la (lb)
or a
pharmaceutically acceptable salt or solvate thereof or its use in treating or
preventing a
disease or disorder:
(B) ____________________________________ (A)."4
/NH(D)
(R3)n
(R2)n
(R1)
(Ib)
wherein:
(A) is a phenyl, pyridinyl, thiophenyl, pyrrolyl, furanyl, or thiazolyl group
having n optional
substituents (R3);
(B) is ¨0-CH2-phenyl or phenyl, wherein the phenyl group has n optional
substituents (R2);
(D) is thiazolyl, oxadiazolyl or pyrimidinyl wherein said (D) has one
substituent (R.1);
(RI) is -NH2, -OH, amido, -NHC(=0)CH3, -NHCH3 or -S(0)2NH2;
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
each (R2) is independently chosen from alkyl, alkenyl, alkynyl, cyclyl, amino,
amido, C-
amido, alkylamino, hydroxyl, nitro, halo, haloalkyl, haloalkoxy, cyano,
sulfinyl, sulfonyl,
sulfonamide, alkoxy, acyl, carboxyl, carbamate or urea;
each (R3) is independently chosen from alkyl, alkenyl, alkynyl, cyclyl, amino,
amido, C-
5 amido, alkylamino, hydroxyl, nitro, halo, haloalkyl, haloalkoxy, cyano,
sulfinyl, sulfonyl,
sulfonamide, alkoxy, acyl, carboxyl, carbamate, or urea; and
n is independently 1, 2, 3 or 4.
In one embodiment of this aspect, the compound of Formula (lb) is used to
treat a disease in
10 a mammal and more preferably a human. In another embodiment, the disease
or disorder is
chosen from cancer, a neurological condition or disease, or a viral infection.
In one
embodiment, the neurological disease or disorder is Huntington disease,
Parkinson disease,
Alzheimer disease, Amyotrophic Lateral Sclerosis, or Frontotemporal Dementia.
15 In another embodiment of this aspect, the disease or disorder is cancer.
In another
embodiment the cancer is prostate cancer. In another specific embodiment of
this aspect the
cancer is breast cancer. In another yet specific embodiment of this aspect the
cancer is lung
cancer. In another yet specific embodiment of this aspect the cancer is
colorectal cancer. In
another yet specific embodiment of this aspect the cancer is brain cancer. In
another yet
20 specific embodiment of this aspect the cancer is skin cancer. In another
yet specific
embodiment of this aspect the cancer is blood cancer (e.g., leukemia,
including, for
example, acute myelogenous leukemia (AML), chronic myelogenous leukemia (CML),
chronic neutrophilic leukemia, chronic eosinophilic leukemia, chronic
lyrnphocytic
leukemia (CLL), acute lymphoblastic leukemia (ALL), or hairy cell leukemia), a
lymphoma,
25 or myeloma.
In another specific embodiment of this aspect, the invention provides a
compound of
Formula (lb) for use in treating or preventing a disease or disorder wherein
(A) is aryl or
heterocyclyl. In a more preferred embodiment (A) is phenyl, pyridinyl,
thiophenyl,
pyrrolyl, furanyl, or thiazolyl. In an even more preferred embodiment (A) is
phenyl or
pyTidinyl.
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
26
In another specific embodiment of this aspect, the invention provides a
compound of
Formula (lb) for use in treating or preventing a disease or disorder wherein
(B) is -L2-
cyclyl. In a more preferred embodiment (B) is -0-phenyl or -0-CH2-phenyl. In
an even
more preferred embodiment (B) is -0-CH2-phenyl. In one specific embodiment the
phenyl
group of said (B) group has 1, 2, 3, or 4 optional substituents (R2)
independently chosen
from alkyl, alkenyl, alkynyl, cyclyl, amino, amido, C-amido, alkylamino,
hydroxyl, nitro,
halo, haloalkyl, haloalkoxy, cyano, sulfinyl, sulfonyl, sulfonamide, alkoxy,
acyl, carboxyl,
carbamate or urea.
In another specific embodiment of this aspect, the invention provides a
compound of
Formula (lb) for use in treating or preventing a disease or disorder wherein
(B) is cyclyl. In
a more preferred embodiment (B) is phenyl. In one specific embodiment the
phenyl group
of said (B) group has 1, 2, 3, or 4 optional substituents (R2) independently
chosen from
alkyl, alkenyl, alkynyl, cyclyl, amino, amido, C-amido, alkylamino, hydroxyl,
nitro, halo,
haloalkyl, haloalkoxy, cyano, sulfinyl, sulfonyl, sulfonamide, alkoxy, acyl,
carboxyl,
carbamate or urea.
In another specific embodiment of this aspect, the invention provides a
compound of
Formula (lb) for use in treating or preventing a disease or disorder wherein
(R2) is
hydroxyl, halo or haloalkyl. In one preferred embodiment (R2) is ¨OH or ¨CF3.
In another
preferred embodiment (R2) is fluor or chloro.
In another specific embodiment of this aspect, the invention provides a
compound of
Formula (Ib) for use in treating or preventing a disease or disorder wherein
(D) is a
monocyclic heteroaryl. In a more preferred embodiment (D) is thiazolyl,
oxadiazolyl,
oxazolyl, isoxazolyl, thiadiazolyl, triazinyl, pyridazinyl, pyrazinyl,
pyridinyl or pyrimidinyl.
In one specific embodiment said cyclyl (D) has one substituent (RI).
In another specific embodiment of this aspect, the invention provides a
compound of
Foimula (lb) for use in treating or preventing a disease or disorder wherein
(R1) is a
hydrogen bonding group. For example, (R1) may be -OH, -0(alkyl), -NH2, -N1-
1(alkyl)
(e.g., -NHCH3), -N(alkyl)(alkyl), amido, -SO-NH2, -SO-NI-1(a] kyl), -SO-
N(alkyl)(alkyl),
-S(0)2N1-12, -S(0)2NH(alkyl), -S(0)2N(allcyl)(alkyl), -C(=0)NH2, -
C(=0)NH(alkyl),
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
27
-C(=0)N(alkyl)(alkyl), -alkylene-C(=0)NH2 (e.g., -
CH2-C(=0)NH2), -alkyl ene-
C(=0)NH(alkyl) (e.g., -CH2-C(-----0)NR(alkyl)), -alkylene-C(=0)N(alkyl)(alkyl)
(e.g.,
-CH2-C(=0)N(alkyl)(alkyl)), -NHC(=0)-alkyl (e.g., -NHC(=0)CH3), -N(alkyl)-
C(=0)-
alkyl (e.g., -N(-CH3)-C(-0)CH3), -alkylene-NH2 (e.g., -CH2-NH2), -alkylene-
NH(alkyl), or
-alkylene-N(alkyl)(alkyl), wherein it is preferred that the aforementioned
alkyl and alkylene
groups each independently have from 1 to 6 carbon atoms. In a more preferred
embodiment
(R1) is -NH2, -OH, amido, -NHC(=0)CH3, -NHCH3 or -S(0)2NH2. In an even more
preferred embodiment (R1) is -NI-12.
In one aspect, the invention provides a stereoisomer or a mixture thereof, of
a compound of
Formula (1), (la) or (lb).
In another aspect, the invention relates to a derivative or analog of a
compound of Formula
(I), (Ia) or (lb).
In yet another aspect, the invention relates to a solvate or polymorph of a
compound of
For ____ nula (I), (Ia) or (lb).
In yet another aspect, the invention relates to a prodrug of a compound of
Formula (I), (la)
or (Ib).
In yet another aspect, the invention relates to a metabolite of a compound of
Formula (I),
(Ia) or (lb).
In another aspect, the invention provides a method of treating or preventing a
disease or
condition comprising administering, to a patient (preferable human) in need of
treatment or
prevention, a therapeutically effective amount of a phairnaceutical
composition comprising
a compound of Formula (I), (Ia) or (lb) as defined above, or a
pharmaceutically acceptable
salt or solvate thereof, and a pharmaceutically acceptable carrier. This
aspect can be
reformulated as a compound of Formula (I), (Ia) or (Ib) for use as a medicine.
In a related
aspect, the invention provides a pharmaceutical composition for use in
treating or
preventing a disease or condition wherein said composition comprises a
therapeutically
effective amount of a compound of Formula (I), (Ia) or (Ib) sufficient for
treating or
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
28
preventing said disease or condition. In a more specific embodiment the
invention provides
a compound of Foimula (I), (Pa) or (lb) for use in the treatment of a disease
associated with
LSD1. In another preferred aspect, the therapeutically effective amount is an
amount
sufficient to modulate histone methylation levels. In another preferred
aspect, the
therapeutically effective amount of a compound of Formula (I), (Ia) or (lb) is
an amount
sufficient to modulate the level of histone-3 lysine-4 methylation. In another
preferred
aspect, the therapeutically effective amount is an amount sufficient to
modulate histone-3
lysine-9 methylation levels.
In yet another aspect, the invention provides a phaimaceutical composition
comprising a
compound of Formula (I), (Ia) or (Ib) and a pharmaceutically acceptable
carrier. In a more
specific aspect, the pharmaceutical composition comprises a therapeutically
effective
amount of a compound of Formula (I), (Ia) or (lb). In an even more specific
aspect, the
therapeutically effective amount of a compound of Foimula (I), (Ia) or (lb) is
an amount
effective to inhibit LSD1. In another preferred aspect, the therapeutically
effective amount
is an amount sufficient to modulate histone methylation levels. In another
preferred aspect,
the therapeutically effective amount of a compound of Formula (I), (Ia) or
(lb) is an amount
sufficient to modulate the level of histone 3 lysine 4 methylation. In another
preferred
aspect, the therapeutically effective amount is an amount sufficient to
modulate histone-3
lysine-9 methylation levels.
In again another aspect, the invention provides a method of inhibiting LSD1
activity
comprising administering, to a patient in need of treatment, a therapeutically
effective
amount of a composition comprising a compound of Formula (I), (Ia) or (lb) or
a
pharmaceutically acceptable salt or solvate thereof, and a pharmaceutically
acceptable
carrier sufficient to inhibit LSD1 activity. This aspect can be reformulated
as a compound
of Formula (I), (Ia) or (Ib) as herein defined for use as a LSD1 inhibitor.
This aspect can
also be refoimulated as a compound of Formula (I), (Ia) or (lb) for the
manufacture of a
medicament for the treatment of a disease associated to LSD1. In a related
aspect, a method
for treating an individual is provided, said method comprising identifying an
individual in
need of treatment and administering to said individual a therapeutically
effective amount of
a compound of Formula (I), (Ia) or (lb). In a preferred aspect, the
therapeutically effective
amount of a compound of Formula (I), (Ia) or (Ib) is an amount sufficient to
inhibit LSD1.
:
29
In another preferred aspect, the therapeutically effective amount is an amount
sufficient to
modulate histone methylation levels. In another preferred aspect, the
therapeutically
effective amount of a compound of Formula (I), (Ia) or (Ib) is an amount
sufficient to
modulate the level of histone 4 lysine 3 methylation. In another preferred
aspect, the
therapeutically effective amount is an amount sufficient to modulate histone-3
lysine-9
methylation levels.
Preferred embodiments of the compounds of Formula (I), (Ia) or (Ib) for use in
the
composition and method of this aspect of the invention are as defined herein
above in the
first aspect of the invention.
In still another aspect, the invention provides a method of treating or
preventing cancer
comprising administering, to a patient in need of treatment, a therapeutically
effective
amount of a composition comprising a compound of Formula (I), (Ia) or (Ib) as
defined
above in the first aspect of the invention, and a pharmaceutically acceptable
carrier. This
aspect can be reformulated as a compound of Formula (I), (Ia) or (Ib) as
defined above in
the first aspect of the invention for use in the treatment or prevention of
cancer. In a related
aspect, the invention provides a pharmaceutical composition for use in
treating or
preventing cancer wherein said composition comprises a therapeutically
effective amount of
a compound of Formula (I), (Ia) or (lb) sufficient for treating or preventing
cancer. In
another related aspect, the invention provides a compound of Formula (I), (Ia)
or (lb) or a
pharmaceutical composition for the treatment or prevention of a cancer wherein
said cancer
is chosen from testicular cancer, breast cancer, lung cancer, prostate cancer,
colorectal
cancer, brain cancer, skin cancer, blood cancer (e.g., leukemia, including,
for example, acute
myelogenous leukemia (AML), chronic myelogenous leukemia (CML), chronic
neutrophilic
leukemia, chronic eosinophilic leukemia, chronic lymphocytic leukemia (CLL),
acute
lymphoblastic leukemia (ALL), or hairy cell leukemia), lymphoma and myeloma,
wherein
said composition comprises a therapeutically effective amount of a compound of
Formula
(I), (Ia) or (lb) sufficient for treating or preventing the said cancer. In a
preferred aspect, the
therapeutically effective amount of a compound of Formula (1), (Ia) or (Ib) is
an amount
sufficient to inhibit LSD1. In another preferred aspect, the therapeutically
effective amount
is an amount sufficient to modulate histone methylation levels. In another
preferred aspect,
the therapeutically effective amount of a compound of Formula (I), (Ia) or
(Ib) is an amount
CA 2806008 2018-02-26
30
sufficient to modulate the level of histone-3 lysine-4 methylation. In another
preferred
aspect, the therapeutically effective amount is an amount sufficient to
modulate histone-3
lysine-9 methylation levels.
In still another aspect, the invention provides a method of treating or
preventing a
neurological disease or disorder comprising administering, to a patient in
need of treatment
or prevention, a therapeutically effective amount of a composition comprising
a compound
of Formula (I), (Ia) or (Ib) as defined above in the first aspect of the
invention, and a
pharmaceutically acceptable carrier. This aspect can be reformulated as a
compound of
Formula (I), (Ia) or (Ib) as defined above in the first aspect of the
invention for use in the
treatment or prevention of a neurological disease or disorder. In a related
aspect, the
invention provides a pharmaceutical composition for use in treating or
preventing a
neurological disease or disorder wherein said composition comprises a
therapeutically
effective amount of a compound of Formula (I), (la) or (Ib) sufficient for
treating or
preventing a neurological disease or disorder. In another related aspect, the
invention
provides a compound of Formula (I), (Ia) or (Ib) or a pharmaceutical
composition for the
treatment or prevention of a neurological disease or disorder wherein said
neurological
disease or disorder chosen from Huntington disease, Parkinson disease,
Alzheimer disease,
Amyotrophic Lateral Sclerosis, or Frontotemporal Dementia, or Dementia with
Lewy
Bodies, and further wherein said composition preferably comprises a
therapeutically
effective amount of the compound of Formula (I), (la) or (lb) sufficient for
treating or
preventing the said neurological disease or disorder. In a preferred
aspect, the
therapeutically effective amount of a compound of Formula (I), (Ia) or (Ib) is
an amount
sufficient to inhibit LSD1. In another preferred aspect, the therapeutically
effective amount
of a compound of Formula (1), (Ia) or (lb) is an amount sufficient to modulate
the level of
hi stone-3 lysine-4 methylation.
In a still yet aspect, the invention provides a method for identifying a
compound which is a
selective inhibitor of LSD1, the method comprising selecting or providing a
compound of
Formula (I) and determining the ability of the said compound to inhibit LSD1
and MAO-A
and/or MAO-B, wherein a compound that inhibits I,SD1 to a greater extent than
MAO-A
and/or MAO-B is identified as a LSD1 selective inhibitor. Thus, the invention
provides a
pharmaceutical composition comprising a pharniaceutically acceptable carrier
and a
CA 2806008 2018-09-26
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
31
compound of Formula (I), (Ia) or (Ib) which is a selective inhibitor of LSD1.
LSD1
selective inhibitors have Ki (IC50) values for LSD1 which are lower than the
Ki (IC50)
value for MAO-A and/or MAO-B. Preferably, the Ki (IC50) values for LSD1 are
two-fold
lower than for MAO-A and/or MAO-B. In one aspect of this embodiment, the LSD1
Ki
value is at least 5-fold lower than the Ki (IC50) value for MAO-A and/or MAO-
B. In one
aspect of this embodiment, the LSD1 Ki (IC50) value is at least 10-fold lower
than the Ki
(IC50) value for MAO-A and/or MAO-B. In one embodiment of this aspect of the
invention, the pharmaceutical composition comprising a LSD1 selective
inhibitor of
Formula (I), (la) or (lb) or a pharmaceutically acceptable salt or solvate
thereof is useful for
treating and/or preventing a disease in an individual. In one specific
embodiment, a
therapeutically effective amount of the composition is administered to an
individual in an
amount sufficient to prevent or treat a disease or disorder. In a more
specific, the disease is
cancer, a neurological disease or condition, or a viral infection. In an even
more specific
aspect, the disease is a cancer chosen from prostate, testicular, brain,
colorectal, lung, breast,
skin, and blood cancer. In another aspect, the neurological disease or
disorder is Huntington
disease, Parkinson disease, Alzheimer disease, Amyotrophic Lateral Sclerosis,
Frontotemporal Dementia, or Dementia with Lowy Bodies.
In a still yet aspect, the invention provides a method for identifying a
compound which is a
dual inhibitor of LSD1 and MAO-B, the method comprising selecting or providing
a
compound of Formula (I) and determining the ability of the said compound to
inhibit LSD1
and MAO-A and/or MAO-B, wherein a compound that inhibits LSD1 and MAO_B to a
greater extent than MAO-A is identified as a LSD1/MAO-B dual inhibitor. Thus,
the
invention provides a pharmaceutical composition comprising a pharmaceutically
acceptable
carrier and a compound of Formula (I), (Ia) or (Ib) which is a dual inhibitor
of LSD1/MAO-
B. LSD1/MAO-B dual inhibitors have Ki (IC50) values for LSD1 and MAO-B which
are
lower than the Ki (IC50) value for MAO-A. Preferably, the Ki (IC50) values for
LSD1 and
MAO-B are two-fold lower than for MAO-A. In one aspect of this embodiment, the
LSD1/MAO-B dual inhibitors have Ki (IC50) values at least 5-fold lower than
the Ki (IC50)
value for MAO-A. In one aspect of this embodiment, the LSD1/MAO-B Ki (IC50)
values
are at least 10-fold lower than the Ki (IC50) value for MAO-A. In one
embodiment of this
aspect of the invention, the pharmaceutical composition comprising a LSD1/MAO-
B dual
inhibitor of Formula (I), (Ia) or (Ib) or a pharmaceutically acceptable salt
or solvate thereof
32
is useful for treating and/or preventing a disease in an individual. In one
specific
embodiment, a therapeutically effective amount of the composition is
administered to an
individual in an amount sufficient to prevent or treat a disease or disorder.
In a more
specific, the disease is cancer, a neurological disease or condition, or a
viral infection. In an
even more specific aspect, the disease is a cancer chosen from prostate,
testicular, brain,
colorectal, lung, breast, skin, and blood cancer. In another aspect, the
neurological disease
or disorder is Huntington disease, Parkinson disease, Alzheimer disease,
Amyotrophic
Lateral Sclerosis, Frontotemporal Dementia, or Dementia with Lewy Bodies.
Recent studies have implicated LSD1 in viral infection and reactivation. In
particular it was
shown that pharmacological inhibitors of LSD1 like pamate and siRNA knock down
of
LSD1 caused reduced viral infectivity and reduced reactivation after latency
(Liang et al.
(2009) Nat. Med. 15:1312-1317). Therefore, it is believed that the compounds
of the
invention can be used for treating or preventing viral infection. Furthermore,
it is believed
that the compounds of the invention can treat or prevent viral reactivation
after latency.
Thus, in another aspect, the invention provides a method for treating or
preventing a viral
infection, the method comprising administering to an individual (preferably a
human) a
compound of Formula (I), (Ia) or (Ib) as defined above in any of the aspects
and
embodiments of the invention or a pharmaceutically acceptable salt or solvate
thereof.
Accordingly, the invention also provides a compound of Formula (I), (Ia) or
(Ib) as defined
above in any of the aspects and embodiments of the invention or a
pharmaceutically
acceptable salt or solvate thereof for use in treating or preventing a viral
infection. In one
specific embodiment, the viral infection is a herpesvirus infection. In a more
specific
embodiment, the herpesvirus infection is caused by and/or associated with a
herpesvirus
chosen from HSV-1, HSV-2, and Epstein-Barr virus. In another embodiment of
this
seventh aspect, the viral infection is caused by and/or associated with HIV.
In an even more
specific embodiment, the invention provides a method for treating or
preventing viral
reactivation after latency, the method comprising administering to an
individual (preferably
a human) a compound of Formula (I), (Ia) or (lb) as defined above in any of
the aspects and
embodiments of the invention or a pharmaceutically acceptable salt or solvate
thereof.
Accordingly, the invention also provides a compound of Formula (I), (Ia) or
(Ib) as defined
above in any of the aspects and embodiments of the invention or a
pharmaceutically
CA 2806008 2018-02-26
33
acceptable salt or solvate thereof for use in treating or preventing viral
reactivation after
latency. The invention also provides a compound of the invention, for use in
treating or
preventing viral reactivation after latency. The invention also provides a
pharmaceutical
composition comprising a compound of the invention and a pharmaceutically
acceptable
carrier, for use in treating or preventing viral reactivation after latency.
The invention also
provides a use of a compound of the invention, for the preparation of a
medicament for
treating or preventing viral reactivation after latency. The invention also
provides a use of a
compound of the invention, or of a pharmaceutical composition comprising a
compound of
the invention and a pharmaceutically acceptable carrier, for treating or
preventing viral
reactivation after latency. In a specific embodiment, the virus that is
reactivating is a
herpesvirus. In a more specific embodiment, the herpesvirus that is
reactivating is chosen
from HSV-1, HSV-2, and Epstein-Barr virus. In an even more specific
embodiment, the
virus that is reactivating is HSV.
In another aspect, the invention also provides a compound of the invention,
for use in
inhibiting histone-3 lysine-4 demethylation. The invention also provides a
pharmaceutical
composition comprising a compound of the invention and a pharmaceutically
acceptable
carrier, for use in inhibiting histone-3 lysine-4 demethylation. The invention
also provides a
use of a compound of the invention, for the preparation of a medicament for
inhibiting
histone-3 lysine-4 demethylation. The invention also provides a use of a
compound of the
invention, or of a pharmaceutical composition comprising a compound of the
invention and
a pharmaceutically acceptable carrier, for inhibiting histone-3 lysine-4
demethylation.
In yet another aspect, the invention also provides a compound of the
invention, for use in
inhibiting histone-3 lysine-9 demethylation. The invention also provides a
pharmaceutical
composition comprising a compound of the invention and a pharmaceutically
acceptable
carrier, for use in inhibiting histone-3 lysine-9 demethylation. The invention
also provides a
use of a compound of the invention, for the preparation of a medicament for
inhibiting
histone-3 lysine-9 demethylation. The invention also provides a use of a
compound of the
invention, or of a pharmaceutical composition comprising a compound of the
invention and
a pharmaceutically acceptable carrier, for inhibiting histone-3 lysine-9
demethylation.
CA 2806008 2018-02-26
33a
During the inventors' investigation of amine oxidases like LSD], MAO-B and MAO-
A, it
was unexpectedly found that the stereochemical configuration of the
cyclopropyl carbons of
N-substituted arylcyclopropylamine compounds substantially affects the potency
of LSD1
inhibition. The inventors have shown that the (-) stereoisomer of 5-(((trans)-
2-(4-
(benzyloxy)phenypcyclopropylamino)methyl)-1,3,4-oxadiazol-2-amine is about 20-
fold
more active against LSD1 than the corresponding (+) stercoisomer. Furthermore,
the (-)
stereoisomer retained substantial MAO-B inhibitory activity. Notably, the
selectivity for
LSD1/MAO-A for the (-)/(+) stereoisomer was over 100 fold as judged by
kinact/Ki values.
Thus, (-) stereoisomers of the compounds of the present invention, including
particularly the
compounds of Formula (I), (Ia) and (Ib), are unexpectedly potent LSDI
inhibitors compared
to their respective enantiomers. The invention thus relates to a compound of
Formula (I),
(Ia) or (Ib) as described and defined herein, wherein the substituents on the
cyclopropyl
moiety (i.e., the group (A) and the group ¨NH-CH2-(D)) are in trans-
configuration and
further wherein the compound is optically active.
The invention, in one aspect, relates to a substantially pure, optically
active stereoisomer of
a compound of Formula (I), (Ia) or (Ib) as described and defined herein,
wherein the
substituents on the cyclopropyl moiety (i.e., the group (A) and the group ¨NH-
CH2-(D)) are
in trans-configuration, or a pharmaceutically acceptable salt or solvate
thereof, as well as its
use in as a medicament. In a specific aspect, the substantially pure,
optically active
stereoisomer of a compound of Formula (I), (Ia) or (Ib), wherein the
substituents on the
cyclopropyl moiety (i.e., the group (A) and the group ¨NH-CH2-(D)) are in
trans-
configuration, is 90 mole-% or greater (-) stereoisomer and 10 mole-% or less
(+)
stereoisomer. In a more specific aspect, the substantially pure, optically
active stereoisomer
is 95 mole-% or greater (-) stereoisomer and 5 mole-% or less (+)
stereoisomer. In yet a
more specific aspect, the substantially pure, optically active stereoisomer is
98 mole-% or
greater (-) stereoisomer and 2 mole-% or less (+) stereoisomer. In an even
more specific
CA 2806008 2018-02-26
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
34
aspect, the substantially pure, optically active stereoisomer is 99 mole-% or
greater (-)
stereoisomer and 1 mole-% or less (+) stereoisomer. In yet an even more
specific aspect,
the substantially pure stereoisomer is 99.5 mole-% or greater (-) stereoisomer
and 0.5 mole-
% or less (+) stereoisomer. The substantially pure, optically active
stereoisomer of a
compound of Formula (I), (Ia) or (lb), wherein the substituents on the
cyclopropyl moiety
(i.e., the group (A) and the group ¨NH-Cif,-(D)) are in trans-configuration,
is useful in
treating or preventing a disease or disorder, particularly cancer, depression,
a
neurodegenerative disease or disorder, or a viral infection.
The invention also relates to a composition comprising a stereoisomer of a
compound of
Formula (I), (Ia) or (lb), wherein the substituents on the cyclopropyl moiety
(i.e., the group
(A) and the group ¨NH-CH2-(D)) are in trans-configuration, or a
phaimaceutically
acceptable salt or solvate thereof, wherein said composition has a 90% or more
enantiomeric excess of the (-) stereoisomer of the compound. In a specific
aspect said
composition has a 95% or more enantiomeric excess of the (-) stereoisomer of
the
compound. In a more specific aspect said composition has a 98% or more
enantiomeric
excess of the (-) stereoisomer of the compound. In an even more specific
aspect said
composition has a 99% or more enantiomeric excess of the (-) stereoisomer of
the
compound. The composition, in one aspect, is for use in treating or preventing
a disease or
disorder, particularly cancer, depression, or a neurodegenerative disease or
disorder, or a
viral infection.
Accordingly, the invention relates to a compound of Formula (I), (Ia) or (lb),
wherein the
substituents on the cyclopropyl moiety (i.e., the group (A) and the group -NH-
CH2-(D)) are
in trans-configuration, or a pharmaceutically acceptable salt or solvate
thereof, wherein the
(-) stereoisomer of said compound is present at an enantiomeric excess of 90%
or more,
preferably of 95% or more, more preferably of 98% or more, and even more
preferably of
99% or more.
In a further aspect, the invention relates to a pharmaceutical composition
comprising a
stereoisomer of a compound of Formula (I), (Ia) or (Ib), wherein the
substituents on the
cyclopropyl moiety (i.e., the group (A) and the group ¨NH-CH2-(D)) are in
trans-
configuration, or a pharmaceutically acceptable salt or solvate thereof, and a
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
phamiaceutically acceptable carrier, wherein said composition has a 90% or
more
enantiomeric excess of the (-) stereoisomer of the compound. In a specific
aspect said
composition has a 95% or more enantiomeric excess of the (-) stereoisomer of
the
compound. In a specific aspect said composition has a 99% or more enantiomeric
excess of
5 the (-) stereoisomer of the compound. The pharmaceutical composition of
this aspect is
particularly useful in treating or preventing cancer, depression, a
neurodegenerative disease
or disorder, or a viral infection.
In one aspect, the invention relates to an optically active compound of
Formula (I), (Ia) or
10 (Ib), wherein the substituents on the cyclopropyl moiety (i.e., the
group (A) and the group
-NH-CH2-(D)) are in trans-configuration, or a pharmaceutically acceptable salt
or solvate
thereof, or a composition comprising any of the aforementioned compounds,
wherein said
compound is the (1 R, 2S) enantiomer (in respect to the substituents on the
cyclopropyl ring)
substantially free of the (1S, 2R) enantiomer. Preferably, the compound is
more than 90
15 mole-% (1R. 2S) enantiomer and less than 10 mole-% (1S, 2R) enantiomer.
More
preferably, the compound is more than 95 mole-% (1R, 2S) enantiomer and less
than 5
mole-% (I S, 2R) enantiomer. Yet more preferably, the compound is more than 98
mole-%
(IR, 2S) enantiomer and less than 2 mole-% (IS, 2R) enantiomer. Even yet more
preferably, the compound is more than 99 mole- /0 (IR, 2S) enantiomer and less
than I
20 mole-% (1S, 2R) enantiomer. Still even more preferably, the compound is
more than 99.5
mole-% (1R, 2S) enantiomer and less than 0.5 mole-% (1S, 2R) enantiomer. The
enantiomeric content can be determined, for example, by chiral HPLC (e.g., as
described in
Example 36).
25 In one aspect, the invention relates to an optically active compound of
Formula (I), (Ia) or
(lb), wherein the substituents on the cyclopropyl moiety (i.e., the group (A)
and the group ¨
NH-CH2-(D)) are in trans-configuration, or a pharmaceutically acceptable salt
or solvate
thereof, or a composition comprising any of the aforementioned compounds,
wherein said
compound is the (is, 2R) enantiomer (in respect to the substituents on the
cyclopropyl ring)
30 substantially free of the (1R, 2S) enantiomer. Preferably, the compound
is more than 90
mole-% (1S, 2R) enantiomer and less than 10 mole-% (1R, 2S) enantiomer. More
preferably, the compound is more than 95 mole-% (15, 2R) enantiomer and less
than 5
mole-% (1R, 2S) enantiomer. Yet more preferably, the compound is more than 98
mole-%
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
36
(1S, 2R) enantiomer and less than 2 mole-% (1R, 2S) enantiomer. Even yet more
preferably, the compound is more than 99 mole-% (1S, 2R) enantiomer and less
than 1
mole-% (1R, 2S) enantiomer. Still even more preferably, the compound is more
than 99.5
mole-% (1S, 2R) enantiomer and less than 0.5 mole-% (IR, 2S) enantiomer. The
enantiomeric content can be determined, for example, by chiral HPLC (e.g., as
described in
Example 36).
In one aspect, the invention relates to an optically active compound of
Formula (I), (Ia) or
(Ib), wherein the substituents on the cyclopropyl moiety (i.e., the group (A)
and the group
-NH-CH2-(D)) are in trans-configuration, or a pharmaceutically acceptable salt
or solvate
thereof, wherein the cyclopropyl ring carbon atom which is bound to the amino
group of the
compound has the (S)-configuration and the cyclopropyl ring carbon atom which
is bound
to the cyclic group (A) attached to the cyclopropyl ring of the compound has
the
(R)-configuration. Preferably, the compound is provided in an enantiomeric
excess of at
least 90%. Even more preferably the compound is provided in an enantiomeric
excess of at
least 95%. Yet still more preferably the compound is provided in an
enantiomeric excess of
at least 98%. Still more preferably the compound is provided in an
enantiomeric excess of
at least 99%. The enantiomeric excess can be determined, for example, by
chiral HPLC
(e.g., as described in Example 36).
In one aspect, the invention relates to an optically active compound of
Formula (I), (la) or
(Ib), wherein the substituents on the cyclopropyl moiety (i.e., the group (A)
and the group
-NI-1-CH2-(D)) are in trans-configuration, or a pharmaceutically acceptable
salt or solvate
thereof, wherein the cyclopropyl ring carbon atom which is bound to the amino
group of the
compound has the (R)-configuration and the cyclopropyl ring carbon atom which
is bound
to the cyclic group (A) attached to the cyclopropyl ring of the compound has
the
(S)-configuration. Preferably, the compound is provided in an enantiomeric
excess of at
least 90%. Even more preferably the compound is provided in an enantiomeric
excess of at
least 95%. Yet still more preferably the compound is provided in an
enantiomeric excess of
at least 98%. Still more preferably the compound is provided in an
cnantiomeric excess of
at least 99%. The enantiomeric excess can be determined, for example, by
chiral HPLC
(e.g., as described in Example 36).
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
37
In one aspect, the invention relates to an optically active compound of
Formula (I), (Ia) or
(Ib), wherein the substituents on the cyclopropyl moiety (i.e., the group (A)
and the group
-NH-CH2-(D)) are in trans-configuration, or a pharmaceutically acceptable salt
or solvate
thereof, for use in treating or preventing a disease or disorder, such as,
e.g., cancer, a
neurological disease, disorder or condition, or a viral infection. In one
aspect, the
neurological disease, disorder or condition is depression, Huntington disease,
Parkinson
disease, Alzheimer disease, Arnyotrophic Lateral Sclerosis, Frontotemporal
Dementia, or
Dementia with Lewy Bodies. In one specific aspect, the cancer is prostate
cancer. In
another specific, the cancer is breast cancer. In another aspect, the cancer
is lung cancer. In
another aspect, the cancer is colorectal cancer. in another specific aspect,
the cancer is brain
cancer. In another specific aspect, the cancer is skin cancer. In another
specific aspect, the
cancer is blood cancer (e.g., a leukemia or a lymphoma; the leukemia to be
treated or
prevented includes, for example, acute myelogenous leukemia (AML), chronic
myelogenous leukemia (CML), chronic neutrophilie leukemia, chronic
eosinophilic
leukemia, chronic lymphocytic leukemia (CLL), acute lymphoblastic leukemia
(ALL), or
hairy cell leukemia). In one aspect, the neurological disease, disorder or
condition is
depression, Huntington disease, Parkinson disease, or Alzheimer disease. In
one aspect, the
viral infection is an infection with HSV1 or HSV2. In one aspect, the disease
or disorder is
depression. In one aspect, the neurological disease, disorder or
condition is a
neurodegenerative disease, disorder or condition. In one aspect, the
neurodegenerative
disease. disorder or condition is Huntington disease, Parkinson disease,
Alzheimer disease,
Amyotrophic Lateral Sclerosis, or Frontotemporal Dementia.
The invention further relates to the optically active compound of Formula (I),
(Ia) or (Ib),
wherein the substituents on the cyclopropyl moiety (i.e., the group (A) and
the group
-NH-CH2-(D)) are in trans-configuration, or a phaimaceutically acceptable salt
or solvate
thereof, as defined in any of the above embodiments or aspects, for use in the
treatment or
prevention of a disease or disorder, in particular cancer (e.g., breast
cancer, lung cancer,
prostate cancer, colorectal cancer, brain cancer, skin cancer, blood cancer,
leukemia
(including, for example, acute myelogenous leukemia (AML), chronic myelogenous
leukemia (CML), chronic neutrophilic leukemia, chronic eosinophilic leukemia,
chronic
lymphocytic leukemia (CLL), acute lymphoblastic leukemia (ALL), or hairy cell
leukemia),
lymphoma, or myeloma), a neurological disease or condition (e.g., depression,
Alzheimer's
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
38
disease, Huntington disease, Parkinson's disease, or Dementia with Lewy
Bodies), or a viral
infection (e.g., a viral infection is caused by and/or associated with HIV, or
a herpesvirus
infection, such as a herpesvirus infection caused by and/or associated with a
herpesvirus
chosen from HSV-1, HSV-2, or Epstein-Barr virus) in a subject (preferably a
mammal,
more preferably a human).
The invention further relates to the optically active compound of Formula (I),
(Ia) or (Ib),
wherein the substituents on the cyclopropyl moiety (i.e., the group (A) and
the group
-NH-CH2-(D)) are in trans-configuration, or a pharmaceutically acceptable salt
or solvate
thereof, as defined in any of the above embodiments or aspects, for use in the
treatment or
prevention of a disease or disorder wherein said disease or disorder is a
neurodegenerative
disease or disorder. In one aspect, the neurodegenerative disease or disorder
is Huntington
disease, Parkinson disease, Alzheimer disease, Amyotrophic Lateral Sclerosis,
or
Frontotemporal Dementia.
In another preferred aspect, the invention is an optically active N-
substituted aryl- or
hetcroaryl cyclopropylamine, or pharmaceutically acceptable salt or solvate
thereof, for use
in a method of treating or preventing a disease or disorder. Preferably, the
optically active
N-substituted aryl- or heteroaryl cyclopropylamine, as described herein, is a
compound of
Foiniula (II) or a pharmaceutically acceptable salt or solvate thereof:
R1 11.¨(Aii) R2 I I
( II)
wherein:
(A") is an aryl or heteroaryl group having 2 substituents, R111 and R211, and
1, 2, or 3
optional substituents independently chosen from halo, Cl-C3 alkyl, or Cl-C3
alkoxy;
Reis anu-R311 group;
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
39
R311 is an aryl or heterocyclyl group having 1, 2, 3, 4, or 5 optional
substituents
independently chosen from halo, -OH, -NHSO,RA, alkyl, alkoxy, cyano, -CF3, or -
0CF3
wherein RA is a Cl-C6 alkyl or phenyl;
Lill is chosen from a bond, -CH20-, -CH2CH20-, - OCH2-, - OCH2CH2-, -CH2-, -
CH2CH2-,
-CH2CH2CH2-, or -0-;
R2B is ¨Cyclopropyl-NH-L2-R4 wherein said cyclopropyl group has two chiral
centers
substituted in the trans orientation corresponding to the carbons to which
(AB) and ¨NH-
L211-R4B are covalently attached;
R4B is a 5 or 6 membered heteroaryl ring having 1, 2, or 3 optional
substituents
independently chosen from alkyl, -NH-RB, -ORB, or halo wherein RB is hydrogen,
Cl -C3
alkyl, or ¨C(=0)CH3;
L21' is a branched or unbranched C1-C4 alkylene group
and wherein said compound of Fonnula (II) is optically active.
Optically active compounds for use in the methods of the invention and
optically active
compounds of the invention refer to enriched stereoisomers of compounds
wherein the
enrichment is in reference to the chiral centers corresponding to the
enantiomers of the
trans-substituted cyclopropyl moiety. Other chiral centers may or may not be
present in the
molecule and the configuration or optical rotation attributable to these
centers are not
intended to be addressed by this invention e.g., their effect on LSD1 , MAO-A,
or MAO-B.
The invention is also is a pharmaceutical composition comprising an optically
active N-
substituted aryl- or heteroaryl cyclopropylamine of Formula (II) as defined
above, or a
pharmaceutically acceptable salt or solvate thereof, and a phannaceutically
acceptable
carrier.
The invention is also is a pharmaceutical composition comprising an optically
active N-
substituted aryl- or heteroaryl cyclopropylamine of Fonnula (H) as defined
above, or a
40
pharmaceutically acceptable salt or solvate thereof, and a pharmaceutically
acceptable
carrier, for use in treating or preventing a disease or disorder.
Preferably, the optically active N-substituted aryl- or heteroaryl
cyclopropylamine, or
pharmaceutically acceptable salt or solvate thereof, for use in a method of
treating or
preventing a disease or disorder, as described herein, is of Foimula (II):
R1"¨(A")--R2"
(II)
wherein:
(A") is an aryl or heteroaryl group having 2 substituents, R 1 " and R2". and
1. 2, or 3
optional substituents independently chosen from halo, Cl -C3 alkyl, or Cl -C3
alkoxy;
Rl" is an ¨L1"-R3" group;
R3" is a phenyl, pyridyl, thiazolyl, or thienyl group having 1, 2, or 3
optional substituents
independently chosen from halo, -OH, -NHSO2RA, alkyl, alkoxy, cyano, -CF3, or -
0CF3
wherein RA is Cl-C6 alkyl or phenyl;
is chosen from a bond, -CH20-, or -CH20- ;
R2" is ¨Cyclopropyl-NH-L2"-R4" wherein said cyclopropyl group has two chiral
centers
substituted in the trans orientation corresponding to the carbons to which
(An) and ¨NH-
are covalently attached;
R4" is a 5-membered heteroaryl ring having 1, 2, or 3 optional substituents
independently
chosen from ¨NH2 or ¨NH(C1-C3) alkyl;
L2I1 is -CH2- or ¨CH2CH2-;
and wherein said compound of Formula (II) is optically active.
CA 2806008 2018-02-26
CA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
41
The invention is also is a pharmaceutical composition comprising an optically
active N-
substituted aryl- or heteroaryl-cyclopropylamine of Formula (II) as defined
above, or a
pharmaceutically acceptable salt or solvate thereof; and a pharmaceutically
acceptable
carrier.
The invention is also is a pharmaceutical composition comprising an optically
active N-
substituted aryl- or heteroaryl cyclopropylamine of Formula (II) as defined
above, or a
phaimaceutically acceptable salt or solvate thereof, and a pharmaceutically
acceptable
carrier, for use in treating or preventing a disease or disorder.
Furthermore, the inventors have unexpectedly found that a subset of optically
active
compounds of Founula (II) as shown in Formula (III) are potent and selective
inhibitors of
LSD1 or LSD1 and MAO-B.
The invention therefore is an optically active compound of Formula (III) or a
pharmaceutically acceptable salt or solvate thereof:
Rim¨(km) R2ai
(III)
wherein:
(Am) is an aryl or heteroaryl group having 2 substituents, R1111 and R2111,
and 1, 2, or 3
optional substituents wherein said optional substituents are independently
chosen from halo,
Cl-C3 alkyl, or Cl-C3 alkoxy;
Ri in is an group;
R3111 is a phenyl, pyridyl, thiazolyl, or thienyl group having 1, 2, 3, 4, or
5 optional
substituents independently chosen from halo, -OH, -NHSO2RA, alkyl, alkoxy,
cyano, -CF3,
or -0CF3 wherein RA is a CI-C6 alkyl or phenyl;
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
42
LIIllis chosen from a bond, - OCE12-, or -CH20-;
R21n is ¨Cyclopropyl-NH-L2111-R4111 wherein said cyclopropyl group has two
chiral centers
substituted in the trans orientation corresponding to the carbons to which
(A111) and ¨NH-
L2"-R4''
are covalently attached;
R4111 is a 5-membered heteroaryl ring having 1, 2, or 3 optional substituents
independently
chosen from ¨NH2 or ¨NH(C I -C3) alkyl;
Lillis -CH2- or ¨CH7CH2-;
and wherein said compound of Formula (III) is optically active.
Preferably, the optically active compound of Formula (III) is as follows:
R1111¨(AIII) R.2111
(III)
wherein:
(Ani) is a phenyl or pyridyl group having 2 substituents, R1111 and R2111;
R1111is an ¨L1111-R3III group;
.5 is a phenyl having 0, 1, 2, or 3 substituents independently chosen from ¨F,
-Cl, -OH, -
NHSO,RA, Cl-C3 alkyl, C1-C3 alkoxy, cyano, -CF3, or ¨OCF3 wherein RA is Cl -C6
alkyl
or phenyl;
Lim is chosen from a bond, - OCH2-, or -CH20-;
iu
is ¨Cyclopropyl-NH 4 wherein said cyclopropyl group has two chiral
centers
corresponding to the carbons to which (A111) and ¨NH-L2111-R4III are
covalently attached;
43
R4 is a 5-membered heteroaryl ring wherein said the chain of atoms comprising
said 5-
membered heteroaryl ring has 2 or 3 hetero atoms independently chosen from N,
S, or 0,
wherein said 5 membered heteroaryl has 1 optional substituent wherein said
optional
substituent is ¨NH2 or ¨NH(C1-C3) alkyl;
L2111 is -CH2- Or -CH2CH2-;
or a pharmaceutically acceptable salt or solvate thereof,
and wherein said compound of Formula (III) is optically active.
The invention is also a pharmaceutical composition comprising an optically
active N-
substituted aryl- or heteroaryl cyclopropylamine of Formula (III) as defined
above, or
pharmaceutically acceptable salt or solvate thereof, and a pharmaceutically
acceptable
carrier.
The invention is also a pharmaceutical composition comprising an optically
active N-
substituted aryl- or heteroaryl cyclopropylamine of Formula (III) as defined
above, or
pharmaceutically acceptable salt or solvate thereof, and a pharmaceutically
acceptable
carrier, for use in treating or preventing a disease or disorder.
Even more preferably, the optically active compound of Formula (III) is as
follows:
R1I" ¨(A"1)¨R2"1
(III)
wherein:
(AIll) is a phenyl or pyridyl group having 2 substituents, R1 lli and R2III;
ROI is an ¨Lim-R3"1 group;
CA 2806008 2018-02-26
. :
44
R3111 is a phenyl having 1, 2, or 3 optional substituents independently chosen
from ¨F, -Cl, -
OH, -NHSO2CH3, methyl, methoxy, cyano, -CF3, or ¨0CF3;
Lim is chosen from a bond, - OCH2-, or -CH20-;
R2111 is ¨Cyclopropyl-NH-L)m-Re wherein said cyclopropyl group has two chiral
centers
substituted in the trans orientation corresponding to the carbons to which
(Am) and ¨NH-
L2111-R4" are covalently attached;
R4111 is an oxadiazolyl, thiadiazolyl, or thiazolyl ring having 1 optional
substituent wherein
said optional substituent is ¨NH2 or ¨NH(C1-C3) alkyl;
L2111 is -CH2- or ¨CH2C1I2-;
or a pharmaceutically acceptable salt or solvate thereof,
and wherein said compound of Formula (III) is optically active.
The invention is also a pharmaceutical composition comprising an optically
active N-
substituted aryl- or heteroaryl cyclopropylamine of Formula (III) as defined
above, or
pharmaceutically acceptable salt or solvate thereof, and a pharmaceutically
acceptable
carrier.
The invention is also a pharmaceutical composition comprising an optically
active N-
substituted aryl- or heteroaryl cyclopropylamine of Formula (III) as defined
above, or
pharmaceutically acceptable salt or solvate thereof, and a pharmaceutically
acceptable
carrier, for use in treating or preventing a disease or disorder.
Preferably, the optically active compound of Formula (III) is as follows:
R1 111¨(Am)¨R2m
(III)
CA 2806008 2018-02-26
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
wherein:
(An1) is a phenyl or pyridyl group having 2 substituents, R1111 and R2111;
5 ROI is an __L1in-R3:11 group;
R3H1 is a phenyl having 0, I, 2, or 3 substituents independently chosen from
¨F, -Cl, -OH, -
NHSO2RA, C1-C3 alkyl, Cl-C3 alkoxy, cyano, -CF3, or ¨0CF3 wherein RA is C1-C6
alkyl
or phenyl;
Li is chosen from a bond, - 0CH2-, or -CH20- ;
R2111 _NH-L2Ili-R
is ¨Cyclopropyl 4 wherein said cyclopropyl group has two chiral
centers
substituted in the trans orientation corresponding to the carbons to which
(Am) and ¨NH-
L2111-R4111 are covalently attached;
R4111 is a 5-membered heteroaryl ring wherein said the chain of atoms
comprising said 5-
membered heteroaryl ring has 2 or 3 hetero atoms independently chosen from N,
S, or 0,
having 1 optional substituent wherein said optional substituent is ¨NH2 or
¨NH(C I -C3)
alkyl;
L2111 is -CH2- or ¨CH2CH2-;
or a pharmaceutically acceptable salt or solvate thereof,
and wherein said compound of Formula (III) is optically active.
The invention is also is a pharmaceutical composition comprising an optically
active N-
substituted aryl- or heteroaryl-cyclopropylamine of Formula (III) as defined
above, or
pharmaceutically acceptable salt or solvate thereof, and a pharmaceutically
acceptable
carrier.
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
46
The invention is also is a phamiaceutical composition comprising an optically
active N-
substituted aryl- or heteroaryl-cyclopropylamine of Formula (III) as defined
above, or
pharmaceutically acceptable salt or solvate thereof, and a pharmaceutically
acceptable
carrier, for use in treating or preventing a disease or disorder.
Even more preferably, the optically active compound of Formula (III) is as
follows:
R R2iti
(III)
wherein:
(Alit) is a phenyl or pyridyl group having 2 substituents, R1111 and R2I11;
R1111 is an 3 group;
.5 is a phenyl having 0, 1, 2, or 3 substituents independently chosen from ¨F,
-Cl, -OH, -
NHSO2CH3, methyl, methoxy, cyano, -CF3, or ¨0CF3;
LI" is chosen from a bond, -OCH2-, or -CH20-;
is ¨Cyclopropyl-NH-L2'11 wherein said cyclopropyl group has two
chiral centers
substituted in the trans orientation corresponding to the carbons to which
(A111) and ¨NH-
L2"-R4' are covalently attached;
R41" is a heteroaryl group which is amide isostere having 1 optional
substituent wherein
said optional substituent is ¨NH2 or ¨NH(C1-C3) alkyl;
L2I11 is -CH2- or ¨CH2CH2-;
or a pharmaceutically acceptable salt or solvate thereof,
and wherein said compound of Formula (III) is optically active.
:
47
The invention is also a pharmaceutical composition comprising an optically
active N-
substituted aryl- or heteroaryl cyclopropylamine of Formula (III) as defined
above, or
pharmaceutically acceptable salt or solvate thereof, and a pharmaceutically
acceptable
carrier.
The invention is also a pharmaceutical composition comprising an optically
active N-
substituted aryl- or heteroaryl cyclopropylamine of Formula (III) as defined
above, or
pharmaceutically acceptable salt or solvate thereof, and a pharmaceutically
acceptable
carrier, for use in treating or preventing a disease or disorder.
In one aspect, the invention is an optically active N-substituted aryl- or
heteroaryl-
cyclopropylamine for use in a method of treating or preventing a disease or
disorder. In a
specific aspect, the disease or disorder is a human disease or disorder chosen
from cancer,
depression, a neurodegenerative disease or condition, or a viral infection. In
one aspect, the
neurodegenerative disease or disorder is Huntington disease, Parkinson
disease, Alzheimer
disease, Amyotrophic Lateral Sclerosis, Frontotemporal Dementia, or Dementia
with Lewy
Bodies. In a specific aspect, the optically active N-substituted aryl- or
heteroaryl-
cyclopropylamine is as defined in Formula (II) or (III).
The invention, in one aspect, is a substantially pure stereoisomer of an N-
substituted aryl- or
heteroaryl- cyclopropylamine (e.g., a compound of Formula (II) or (III) as
described herein)
or a pharmaceutically acceptable salt or solvate thereof, for use in a method
of treating or
preventing a disease or disorder. In a related aspect, the method comprises
administering to
an individual a therapeutically effective amount of a substantially pure
stereoisomer of an
N-substituted aryl- or heteroaryl- cyclopropylamine. Desirably, the disease or
disorder is
one that is treatable or preventable by LSD1 inhibition, LSD1 inhibition and
MAO-B
inhibition, or MAO-B inhibition. In a specific aspect, a substantially pure
stereoisomer of
an N-substituted aryl- or heteroaryl- cyclopropylamine refers to an N-
substituted aryl- or
heteroaryl- cyclopropylamine which is 90% or greater (-) stereoisomer and 10%
or less (+)
stereoisomer. In a more specific aspect, a substantially pure stereoisomer of
an N-
substituted aryl- or heteroaryl- cyclopropylamine refers to an N-substituted
aryl- or
heteroaryl- cyclopropylamine which is 95% or greater (-) stereoisomer and 5%
or less (+)
CA 2806008 2018-02-26
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
48
stereoisomer. In yet a more specific aspect, a substantially pure stereoisomer
of an N-
substituted aryl- or heteroaryl- cyclopropylamine refers to an N-substituted
aryl- or
heteroaryl- cyclopropylamine which is 98% or greater (-) stereoisomer and 2%
or less (+)
stereoisomer. In an even more specific aspect, a substantially pure
stereoisomer of an N-
substituted aryl- or heteroaryl- cyclopropylamine refers to an N-substituted
aryl- or
heteroaryl- cyclopropylamine which is 99% or greater (-) stereoisomer and 1%
or less (+)
stereoisomer. In yet an even more specific aspect, a substantially pure
stereoisomer of an
N-substituted aryl- or heteroaryl- cyclopropylamine refers to an N-substituted
aryl- or
heteroaryl- cyclopropylamine which is 99.5% or greater (-) stereoisomer and
0.5% or less
(+)stereoisomer. In one embodiment, the above-described percentages refer to
mole-%. The
substantially pure stereoisomer of an N-substituted aryl- or heteroaryl-
cyclopropylamine of
this aspect is useful in treating or preventing cancer, depression, a
neurodegenerative
disease or disorder, or a viral infection. In a specific aspect, the optically
active N-
substituted aryl- or heteroaryl- cyclopropyl amine is as defined in Formula
(11) or (III).
Furthetinore, in another aspect, the invention is a composition comprising a
stereoisomer of
an N-substituted aryl- or heteroaryl cyclopropylamine (e.g., a compound of
Fotmula (II) or
(III) as described herein), or a pharmaceutically acceptable salt or solvate
thereof, wherein
said composition has a 90% or more enantiomeric excess of the (-) stereoisomer
of the N-
substituted aryl- or heteroaryl- cyclopropylamine. In a specific aspect said
composition has
a 95% or more enantiomeric excess of the (-) stereoisomer of the N-substituted
aryl- or
heteroaryl- cyclopropylamine. In a more specific aspect said composition has a
98% or
more enantiomeric excess of the (-) stereoisomer of the N-substituted aryl- or
heteroaryl-
cyclopropylamine. In an even more specific aspect said composition has a 99%
or more
enantiomeric excess of the (-) stereoisomer of the N-substituted aryl- or
heteroaryl-
cyclopropylamine. The composition, in one aspect, is for use in treating or
preventing
cancer, depression, or a neurodegenerative disease or disorder, or a viral
infection. In a
specific aspect, the optically active N-substituted aryl- or heteroaryl-
cyclopropylamine is as
defined in Formula (II) or (III).
Furthermore, in another aspect, the invention is a pharmaceutical composition
comprising a
stereoisomer of an N-substituted aryl- or heteroaryl cyclopropylamine (e.g., a
compound of
Foimula (11) or (III) as described herein), or a phannaceutically acceptable
salt or solvate
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
49
thereof, and a pharmaceutically acceptable carrier wherein said composition
has a 90% or
more enantiomeric excess of the (-) stereoisomer of the N-substituted aryl- or
heteroaryl-
cyclopropylamine. In a specific aspect said composition has a 95% or more
enantiomeric
excess of the (-) stereoisomer of the N-substituted aryl- or heteroaryl-
cyclopropylamine. In
a specific aspect said composition has a 99% or more enantiomeric excess of
the (-)
stereoisomer of the N-substituted aryl- or heteroaryl- cyclopropylamine.
The
pharmaceutical composition of this aspect is useful in treating or preventing
cancer,
depression, a neurodegenerative disease or disorder, or a viral infection. In
a specific
aspect, the optically active N-substituted aryl- or heteroaryl-
cyclopropylamine is as defined
in Formula (II) or (III).
In one aspect, the invention is a composition as defined herein comprising an
optically
active N-substituted aryl- or heteroaryl- cyclopropylamine of Founula (11) or
(111), as
described in any one of the above embodiments or aspects, or a solvate or a
phannaceutically acceptable salt thereof, wherein said N-substituted aryl- or
heteroaryl-
cyclopropylamine is the (IR, 2S) enantiomer (in respect to the substituents on
the
cyclopropyl ring) substantially free of the (15, 2R) enantiomer. Preferably,
the N-substituted
aryl- or heteroaryl- cyclopropylamine is more than 90% (1R, 2S) enantiomer and
less than
100/ (15, 2R) enantiomer. More preferably, the N-substituted aryl- or
heteroaryl-
cyclopropylamine is more than 95% (IR, 2S) enantiomer and less than 5% (15,
2R)
enantiomer. Yet more preferably, the N-substituted aryl- or heteroaryl-
cyclopropylamine is
more than 98% (1R, 2S) enantiomer and less than 2% (15, 2R) enantiomer. Even
yet more
preferably, the N-substituted aryl- or heteroaryl- cyclopropylamine is more
than 99% (1R,
2S) enantiomer and less than 1% (1S, 2R) enantiomer. Still even more
preferably, the N-
substituted aryl- or heteroaryl- cyclopropylamine is more than 99.5% (1R, 2S)
enantiomer
and less than 0.5% (1S, 2R) enantiomer. The enantiomeric content can be
determined, for
example, by chiral HPLC e.g., (as described in Example 36).
In one aspect, the invention is a composition as defined herein comprising an
optically
active N-substituted aryl- or heteroaryl- cyclopropylamine of Formula (II) or
(III), as
described in any one of the above embodiments or aspects, or a solvate or a
phatmaceutically acceptable salt thereof, wherein said N-substituted aryl- or
heteroaryl-
cyclopropylamine is the (15, 2R) enantiomer (in respect to the substitucnts on
the
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
cyclopropyl ring) substantially free of the (IR, 2S) enantiomer. Preferably,
the N-
substituted aryl- or heteroaryl- cyclopropylamine is more than 90% (1S, 2R)
enantiomer and
less than 10% (1R, 2S) enantiomer. More preferably, the N-substituted aryl- or
heteroaryl-
cyclopropylamine is more than 95% (1S, 2R) enantiomer and less than 5% (1R,
2S)
5 enantiomer. Yet more preferably, the N-substituted aryl- or heteroaryl-
cyclopropylamine is
more than 98% (1S, 2R) enantiomer and less than 2% (1R, 2S) enantiomer. Even
yet more
preferably, the N-substituted aryl- or heteroaryl- cyclopropylamine is more
than 99% (1S,
2R) enantiomer and less than 1% (1R, 2S) enantiomer. Still even more
preferably, the N-
substituted aryl- or heteroaryl- cyclopropylamine is more than 99.5% (1S, 2R)
enantiomer
10 and less than 0.5% (1R, 2S) enantiomer. The enantiomeric content can be
determined, for
example, by chiral HPLC e.g., (as described in Example 36).
In one aspect, the invention is an optically active N-substituted aryl- or
heteroaryl
cyclopropylamine (e.g., a compound of Formula (II) or (III)), as defined in
any one of the
15 above embodiments or aspects, or a solvate or a pharmaceutically
acceptable salt or solvate
thereof, wherein the cyclopropyl ring carbon atom which is bound to the amino
group of the
N-substituted aryl- or heteroaryl- cyclopropylamine has the (S)-absolute
configuration and
the cyclopropyl ring carbon atom which is bound to the cyclic group adjacent
to the
cyclopropyl ring of the N-substituted aryl- or heteroaryl- cyclopropylamine
has the (R)-
20 absolute configuration. Preferably, said N-substituted aryl- or
heteroaryl- cyclopropylamine
is provided in an enantiomeric excess of at least 90%. Even more preferably
said N-
substituted aryl- or heteroaryl- cyclopropylamine is provided in an
enantiomeric excess of at
least 95%. Yet still more preferably said compound is provided in an
enantiomeric excess of
at least 98%. Still more preferably said N-substituted aryl- or heteroaryl-
cyclopropylamine
25 is provided in an enantiomeric excess of at least 99%. The enantiomeric
excess can be
determined, for example, by chiral HPLC e.g., (as described in Example 36).
In one aspect, the invention provides an optically active N-substituted aryl-
or heteroaryl
cyclopropylamine (e.g., a compound of Formula (II) or (III)), as defined in
any one of the
30 the above embodiments or aspects, or a pharmaceutically acceptable salt
or solvate thereof,
wherein the cyclopropyl ring carbon atom which is bound to the amino group of
the
compound has the (R) absolute configuration and the cyclopropyl ring carbon
atom which is
bound to the cyclic group adjacent to the cyclopropyl ring of the compound has
the (S)
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
51
absolute configuration. Preferably. said compound is provided in an
enantiomeric excess of
at least 90%. Even more preferably said compound is provided in an
enantiomeric excess of
at least 95%. Yet still more preferably said compound is provided in an
enantiomeric
excess of at least 98%. Still more preferably said compound is provided in an
enantiomeric
excess of at least 99%. The enantiorneric excess can be determined, for
example, by chiral
HPLC e.g., (as described in Example 36).
In one aspect, the invention is an optically active N-substituted aryl- or
heteroaryl-
cyclopropylamine (e.g., a compound of Formula (II) or (III) as described
herein), or a
pharmaceutically acceptable salt or solvate thereof, for use in treating or
preventing a
disease or disorder. In a related aspect, the method comprises administering
to an individual
in need of treatment a therapeutically effective amount of an optically active
N-substituted
aryl- or heteroaryl-cyclopropylamine or a pharmaceutically acceptable salt or
solvate
thereof In one aspect, the disease or disorder is a human disease or disorder
chosen from
cancer, a neurological disease or condition, or a viral infection. In one
aspect, the
neurological disease or disorder is depression, Huntington disease, Parkinson
disease,
Alzheimer disease, Amyotrophic Lateral Sclerosis, Frontotemporal Dementia, or
Dementia
with Lewy Bodies. In one specific aspect, the cancer is prostate cancer. In
another specific,
the cancer is breast cancer. In another aspect, the cancer is lung cancer. In
another aspect,
the cancer is colorectal cancer. In another specific aspect, the cancer is
brain cancer. In
another specific aspect, the cancer is skin cancer. In another specific
aspect, the cancer is
blood cancer (e.g., a leukemia (including, for example, acute myelogenous
leukemia
(AML), chronic myelogenous leukemia (CML), chronic neutrophilic leukemia,
chronic
eosinophilic leukemia, chronic lymphocytic leukemia (CLL), acute lymphoblastic
leukemia
(ALL), or hairy cell leukemia) or a lymphoma). In another specific aspect, the
cancer is a
inyeloma. In one aspect, the neurological disease or condition is depression,
Huntington
disease, Parkinson disease, or Alzheimer disease. In one aspect, the viral
infection is HSV1
or HSV2. In one aspect, the disease or condition is depression. In one aspect,
the
neurological disease or condition is a neurodegenerative disease or condition.
In one aspect,
the neurodegenerative disease or disorder is Huntington disease, Parkinson
disease,
Alzheimer disease, Amyotrophic Lateral Sclerosis, or Frontotemporal Dementia.
In one
aspect, the neurodegenerative disease or disorder is Huntington disease. In
one aspect, the
ncurodegenerative disease or disorder is Parkinson disease. In
one aspect, the
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
52
neurodegenerative disease or disorder is Alzheimer disease. In
one aspect, the
neurodegenerative disease or disorder is Amyotrophic Lateral Sclerosis. In one
aspect, the
neurodegenerative disease or disorder is Frontotemporal Dementia.
The invention further relates to the optically active N-substituted aryl- or
heteroaryl
cyclopropylamine of Foimula (II) or (III), or a phaimaceutically acceptable
salt or solvate
thereof, as defined in the above embodiments or aspects, for use in the
treatment or
prevention of a disease or disorder, in particular cancer (e.g., breast
cancer, lung cancer,
prostate cancer, colorectal cancer, brain cancer, skin cancer, blood cancer,
leukemia
(including, for example, acute myelogenous leukemia (AML), chronic myelogenous
leukemia (CML), chronic neutrophilic leukemia, chronic eosinophilic leukemia,
chronic
lymphocytic leukemia (CLL), acute lymphoblastic leukemia (ALL), or hairy cell
leukemia),
lymphoma, or myeloma), a neurological disease or condition (e.g., depression,
Alzheimer's
disease, Huntington disease, Parkinson's disease, or Dementia with Lewy
Bodies), or a viral
infection (e.g., a viral infection is caused by and/or associated with HIV, or
a herpesvirus
infection, such as a herpesvirus infection caused by and/or associated with a
herpesvirus
chosen from HSV-1, HSV-2, or Epstein-Barr virus) in a subject (preferably a
mammal,
more preferably a human).
The invention further relates to the optically active N-substituted aryl- or
heteroaryl
cyclopropylamine of Formula (II) or (III), or a pharmaceutically acceptable
salt or solvate
thereof, as defined in anyone of the above embodiment, for use in the
treatment or
prevention of a disease or disorder wherein said disease or disorder is a
neurodegenerative
disease or disorder. In one aspect, the neurodegenerative disease or disorder
is Huntington
disease, Parkinson disease, Alzheimer disease, Amyotrophic Lateral Sclerosis,
or
Frontotemporal Dementia.
In one embodiment the invention provides a compound Formula (I), (Ia) or (lb)
or a solvate
or a pharmaceutically acceptable salt thereof, wherein the compound is:
5 -(((trans)-2 -(4-(benzyl oxy)phenyl)cyclopropylamino)methyl)p yrimidin
-2-amine;
5-(((trans)-2-(4-(benzyloxy)phenyl)cyclopropylamino)methypthiazol-2-amine;
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
53
-(((trans)-2-(6-(3 -(trifluoromethyl)phenyl)pyridin-3 -
yl)cyclopropylamino)methyl)pyrimidin-2-amine;
5-(((trans)-2-(6-(3 -(trifluoromethyl)phenyl)pyridin-3 -yl)cycloprop
ylamino)methyl)thiazol-
2-amine;
5 3 -(5-((trans)-242-arninopyrimi din-5-ypmethylamino)cyclopropyl)pyridin-2-
yl)phenol;
3-(5-((trans)-24(2-aminothiazol-5-yOmethylamino)cyclopropyl)pyridin-2-
ypphenol;
4?-((trans)-2-42-aminopyrimidin-5-yl)methylamino)cyclopropyl)bipheny1-3-ol;
4'-((trans)-242-aminothiazol-5-yl)methylamino)cyclopropyl)bipheny1-3-ol;
5-(((trans)-2-(4-(benzyloxy)phenyl)cyclopropylamino)methyl)-1 ,2,4-oxadiazol-3-
amine;
5-(((trans)-2-(4-(benzyloxy)phenyl)cyclopropylamino)methyl)-1,3,4-oxadiazol-2-
amine;
5-((((trans)-2-(444- fluorobenzyl) oxy)phenyl)cyclopropyl)amino)methyl)- 1,3
,4-oxadiazol-
2-amine;
5-((((trans)-2-(44(3-fluorobenzyl)oxy)phenyl)cyclopropypamino)methyl)- 1,3 ,4-
oxadiazol-
2-amine;
54((trans)-2-(4-((3,5-difluorobenzyl)oxy)phenyl)cyclopropyl)amino)methyl)-
1,3,4-
oxadiazol-2-amine;
5-((((trans)-2-(4-((4-chlorobenzyl)oxy)phenyl)cyclopropyl)amino)metb y1)- 1,3
,4-ox adi azol-
2-amine;
5-((((trans)-2-(443 -chlorobenzyl)oxy)phenypc yc lopropyl)amino)methyl)- 1,3
,4-oxadiazol-
2-amine;
54((trans)-2-(442-fluorobenzypoxy)phenyl)cyclopropyl)amino)methyl)- 1,3 ,4-
oxadiazol-
2-amine;
5-((((trans)-2-(4-(benzyloxy)phenyl)cycloprop yeamino)methyl)-N-methyl-1,3 ,4-
oxadiazol-
2-amine;
N-(5-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)mcthyl)-1,3,4-
oxadiazol-2-
ypacetamide;
4'-((trans)-2-(((5-amino- 1,3 ,4-oxadiazol-2-yl)m ethyDarnino)cycl prop y1)41
,1'-bipheny1]-3-
ol;
5-((((trans)-2-(6-(3-(trifluoromethyl)phenyl)pyridin-3 -yl)cycloprop
yl)amino)methyl)- 1,3,4-
oxadiazol-2-amine;
5-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)-1,3,4-thiadiazol-
2-amine;
2-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyeamino)methyl)thiazol-5-amine;
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
54
4-((((trans)-2-(3'-(trifluoromethy1)41,1'-biphenyl]-4-
y0cyclopropyl)amino)methyl)thiazol-
2-amine;
2-4((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methypoxazol-5-amine;
3-4((trans)-2-(4-(benzyloxy)phenyl)cyclopropyeamino)methypisoxazol-5-amine;
5-((((trans)-2-(4-(benzyl oxy)phenyl )cycl opropyl)am ino)methyl)-1 ,2,4-
oxadiazol-3 -amine;
3-((((trans)-2-(4-(benzy1oxy)phenyl)cyc1opropy1)amino)methyl)-1,2,4-oxadiazol-
5-amine;
5-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)- 1 ,2,4-
thiadiazol -3-amine;
5-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)pyridin-2-amine;
6-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)pyridazin-3-
amine;
54((trans)-2-(4-(benzyloxy)phenyl)cyclopropypamino)methyl)pyrazin-2-amine;
2-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)pyrimidin-5-
amine;
64((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)-1,2,4-triazin-3-
amine; or
3-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)-1,2,4-triazin-6-
amine;.
In one embodiment of the invention provides a pharmaceutical composition
comprising a
pharmaceutically carrier and a compound, or a solvate or a pharmaceutically
acceptable salt
thereof, wherein said compound is:
5-(((trans)-2-(4-(benzyloxy)phenyl)cyclopropylamino)methyl)pyrimidin
-2-amine;
5-(((trans)-2-(4-(benzyloxy)phenyl)cyclopropylamino)methyl)thiazol-2-amine;
5-(((trans)-2-(6-(3-(trifluoromethyl)phenyppyridin-3-
ypcyclopropylamino)methyppyrimidin-2-amine;
5-(((trans)-2-(6-(3-(trifluoromethyl)phenyppyridin-3-
y0cyclopropylamino)methypthiazol-
2-amine;
3-(5-((trans)-24(2-aminomimidin-5-yl)methylamino)cyclopropyl)pyridin-2-
yl)phenol;
3-(5-((trans)-2-((2-aminothiazol-5-y1)methylamino)cyclopropyljpyridin-2-
y1)phenol;
4'-((trans)-242-aminopyrimidin-5-yl)methylamino)cyclopropyl)bipheny1-3-ol;
4'-((trans)-2((2-aminothiazol-5-yl)methylamino)cyclopropyl)bipheny1-3-ol;
5-(((trans)-2-(4-(benzyloxy)phenyl)cyclopropylamino)methyl)-1,2,4-oxadiazol-3-
amine;
5-4(trans)-2-(4-(benzyloxy)phenyl)cyclopropylamino)methyl)-1,3,4-oxadiazol-2-
amine;
5-((((trans)-2-(444-fluorobenzyl)oxy)phenypeyclopropyl)amino)methyl)-1,3,4-
oxadiazol-
2-amine;
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
5-((((trans)-2-(44(3 -fluorob enzyl)oxy)phenyl)cyclopropypamino)methyl)-1,3,4-
oxadiazol-
2-amine;
5 -((((trans)-2-(4-((3 ,5-difluorob cnzypoxy)phenyl)cyclopropyeamino)methyl)-
1,3,4-
ox adi azol-2-amine;
5 5-((((trans)-2-(4-((4-chlorob enzypoxy)phenyl)cyclopropyl)amino)methyl)-
1,3,4-oxadiazol-
2-amine;
5-((((trans)-2-(4-((3 -chlorob enzyl)oxy)phenyl)cyc lopropyl)amino)methyl)-
1,3,4-oxadi azol -
2-amine;
54((trans)-2-(4-((2-fluorobenzypoxy)phenyl)cyclopropyl)amino)methyl)-1,3,4-
oxadiazol-
10 2-amine;
5-4((trans)-2-(4-(benzyloxy)phenyl)cycloprop yl)amino)methyl)-N-methy1-1,3,4-
oxadiazol-
2-amine ;
N-(54((trans)-2-(4-(benzy1oxy)phenyl)cyclopropypamino)methyl)-1,3,4-oxadiazol-
2-
ypacetamide;
15 4'-((trans)-2-(((5-am ino-1,3,4-oxadiazol-2-
yl)methypamino)cyclopropy1)41,1'-biphenyl] -3 -
ol;
5-((((trans)-2-(6-(3-(trifluoromethyl)phenyl)p yridin-3 -yl)cyclopropyl)ami
no)meth y1)-1,3,4-
oxadiazol-2-amine;
54((trans)-2-(4-(benzyloxy)phenyecyclopropyl)amino)methyl)-1,3,4-thiadiazol-2-
amine;
20 2-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)thiazol-5-
amine;
4-((((trans)-2-(3'-(trifluoromethy1)41,1'-biphenyl] -4-
yecyclopropyl)amino)methyl)thiazol-
2-amine;
2-((((trans)-2-(4-(benzyloxy)phenyl)cycloprop yl)amino)methyl)oxazol-5 -amine;
3-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)isoxazol-5-amine;
25 5-((((trans)-2-(4-(benzyl oxy)ph en yl)cycl prop yl)amino)methyl)-1,2,4-
oxadiazol-3-amine;
3-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyparnino)methyl)-1,2,4-oxadiazol-
5-amine;
5-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)m ethyl)-1,2,4-thiadi
azol-3-amine;
5-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)py-ridin-2-amine;
6-4((trans)-2-(4-(benzyloxy)phenyl)cycloprop yl)amino)methyl)pyridazin-3 -
amine;
30 5-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)pyrazin-2-
amine;
2-((((trans)-2-(4-(benzyl oxy)phenyl)cyclopropyl)amino)methyl)pyriraidin-5 -
amine;
6-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)-1,2,4-triazin-3-
amine; or
3-4((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)-1,2,4-triazin-6-
amine;
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
56
In one embodiment of the invention relates to a compound, or a solvate or a
pharmaceutically acceptable salt thereof. wherein said compound is:
5-(((trans)-2-(4-(benzyloxy)phenyl)cyclopropylamino)methyppyrimidin
-2-amine;
5-(((trans)-2-(4-(benzyloxy)phenyl)cyclopropylamino)methypthiazol-2-amine;
5-(((trans)-2-(6-(3-(trifluoromethyl)phenyppyridin-3-
y0cyclopropylamino)methyl)pyrimidin-2-amine;
5-(((trans)-2-(6-(3-(trifluoromethyl)phenyl)pyridin-3-yl)cycloprop
ylamino)methyl)thiazol-
I 0 2-amine;
3-(5-((trans)-242-aminopyrimidin-5-yl)methylamino)cyclopropyppyridin-2-
yDphenol;
3-(5-((trans)-2-((2-aminothiazol-5-yl)methylamino)cyclopropyl)pyridin-2-
yl)phenol;
4'-((trans)-2((2-aminopyrimidin-5-yl)methylamino)cyclopropyl)bipheny1-3-ol;
41-((trans)-2((2-aminothiazol-5-yOmethylamino)cyclopropyl)bipheny1-3-ol;
5-(((trans)-2-(4-(benzyloxy)phenyl)cyclopropylamino)methyl)-1,2,4-oxadiazol-3-
amine;
5-(((trans)-2-(4-(benzyloxy)phen yl)c ycloprop ylamino)m ethyl)- 1,3,4-
oxadiazol-2-amine;
54((trans)-2-(44(4-fluorobenzyl)oxy)phenyl)cyclopropyl)amino)methyl)- I ,3,4-
oxadiazol-
2-amine;
54((trans)-2-(4-((3-fluorobenzyl)oxy)phenyl)cyclopropyl)amino)methyl)-1,3,4-
oxadiazol-
2-amine;
54((trans)-2-(4-((3,5-difluorobenzyl)oxy)phenyl)cyclopropyl)amino)methyl)-
1,3,4-
oxadiazol-2-amine;
5-((((trans)-2-(44(4-chlorobenzypoxy)phenyl)cyclopropyl)amino)methyl)-1,3 ,4-
oxadiazol-
2-amine;
5-((((trans)-2-(4-((3 -chi orobenzyl)oxy)phenyt)cyclopropyl)amino)methyl)-
1,3,4-oxadiazol-
2-amine;
54((trans)-2-(442-fluorobenzyl)oxy)phenyecyclopropyl)amino)methyl)-1 ,3,4-ox
adiazol-
2-amine;
5-((((trans)-2-(4-(benzyloxy)phenyl)cycloprop yl)amino)methyl)-N-methy1-1,3,4-
oxadiazol-
2-amine;
N-(54((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)-1,3,4-oxadiazol-
2-
y1)acetamide;
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
57
4'-((trans)-2-(((5-amino-1,3,4-oxadiazol-2-yl)methypamino)cyclopropy1)41,1'-
bipheny1]-3-
01;
54((trans)-2-(6-(3-(trifluoromethyl)phenyppyridin-3-
y1)cyclopropypamino)methyl)-1,3,4-
oxadiazol-2-amine;
5-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)-1,3,4-thiadiazol-
2-amine;
2-((((trans)-2-(4-(benzylox y)phenyl)cyclopropyl)amino)methyl)thiazol-5-amine;
4-4((trans)-2-(3'-(trifluoromethy1)41,1'-biphenyll-4-
y1)cyclopropyl)amino)methyl)thiazol-
2-amine;
2-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)oxazol-5-amine;
3-4((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)isoxazol-5-amine;
5-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)-1,2,4-oxadiazol-
3-amine;
34((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)-1,2,4-oxadiazol-5-
amine;
54((trans)-2-(4-(benzyloxy)phenyecyclopropyeamino)methyl)-1,2,4-thiadiazol-3-
amine;
5-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)pyridin-2-amine;
6-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)pyridazin-3-
amine;
5 -((((tran s)-2-(4-(benzyl oxy)ph en yfleyclopropyl)amino)methyl)mazin-2-
amine;
24((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)pyrimidin-5-amine;
6-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)-1,2,4-triazin-3-
amine; or
34((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)-1,2,4-triazin-6-
amine;
for use in a method of treating or preventing a neurological disease or
condition. In one
aspect, the neurological disease or condition is chosen from Alzheimer
Disease, Parkinson
Disease, Huntington Disease, Amyotrophic Lateral Sclerosis, Frontotemporal
Dementia, or
Dementia with Lewy Bodies.
In one embodiment the invention relates to a compound, or a solvate or a
pharmaceutically
acceptable salt thereof, wherein said compound is:
5-(((trans)-2-(4-(benzyloxy)phenyl)cyclopropylamino)methyl)pyrimidin
-2-amine;
5-(((trans)-2-(4-(benzyloxy)phenyl)cyclopropylamino)methyl)thiazol-2-amine;
5-(((trans)-2-(6-(3-(trifluoromethyl)phenyl)pyridin-3-
yl)cyclopropylamino)methyl)pyrimidin-2-amine;
5-(((trans)-2-(6-(3-(trifluoromethyl)phenyl)pyridin-3-
yl)cyclopropylamino)methyethiazol-
2-amine;
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
58
345 -((trans)-242-aminopyrimidin-5-yOmethylamino)cyclopropy1)pyridin-2-
yl)pheno1;
3-(5-((trans)-2-((2-aminothiazol-5-yl)methylamino)cyclopropyl)pyridin-2-
y1)phenol;
4'-((trans)-2((2-aminopyrimidin-5-yOmethylamino)cyclopropyl)bipheny1-3-ol;
4'-((trans)-2-((2-aminothiazol-5-yl)methylamino)cyclopropyl)bipheny1-3-ol;
5-(((trans)-2-(4-(b enzyl oxy)phen yl)cyclopropylamino)methyl)- 1 ,2,4-
oxadiazol-3 -amine;
5-(((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl amino)rnethyl)-1 ,3 ,4-
oxadiazol-2-amine;
5 -((((trans)-2-(444- fluorob enzyl)oxy)phenyl)cyclopropyl)amino)meth y1)- 1,3
,4-ox adi azol-
2-amine;
54((trans)-2-(4-((3-fluorobenzypoxy)phenyecyclopropyl)amino)methyl)-1 ,3 ,4-
oxadiazol-
1 0 2-amine;
5-((((trans)-2-(4-((3 ,5-difluorobenzypoxy)phenyl)cycloprop yl)amino)methyl)-
1 ,3,4-
oxadiazol-2-amine;
5-((((trans)-2-(4-((4-chl orobenzyl)oxy)phenyl)cyclopropypamino)methyl)- 1,3
,4-oxadiazol-
2-amine;
5-((((tran s)-2-(44(3-chlorobenzyl)oxy)phenyl)cyc lopropypamino)methyl)- 1,3
,4-oxadi azol-
2-amine;
5-((((trans)-2-(4-((2-fluorobenzyl)oxy)ph enyl)cycl opropyl)am i no)m eth y1)-
1 ,3 ,4-oxadiazol-
2-amine;
5-((((trans)-2-(4-(benzyl oxy)phenyl)cycloprop yl)amino)methyl)-N-methyl- 1,3
,4-oxa diazol-
2 0 2-amine;
N-(5-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)-1,3 ,4-
oxadiazol-2-
yl)acetamide;
4'-((trans)-2-(((5 -amino- 1,3 ,4-oxadiazol-2-yl)methyl)amino)cyclopropy1)-[
1,1 '-biphenyl] -3-
01;
5 -((((trans)-2-(6-(3-(tri fl uoromethyl)phenyppyrid in-3-
yl)cyclopropyl)amino)methyl)- 1,3,4-
oxadiazol -2-amine;
5 -((((trans)-2-(4-(benzyl oxy)phenyl)cycl oprop yl)ami no)m ethyl)-1,3,,4-thi
adiazol-2-amine ;
2-0((trans)-2-(4-(benzyloxy)phenyecyclopropyl)amino)methyl)thiazol-5 -amine;
4-((((trans)-2-(3'-(trifluoromethyl)-{ 1 , 1 '-biphenyl] -4-
ypcyclopropyl)amino)methypthia zol-
3 0 2-amine;
2-4((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)oxazol-5 -amine;
3-((((trans)-2-(4-(benzy1 oxy)phenyl)cycloprop yl)amino)methyl)is oxazol-5 -
amine;
5 -((((trans)-2-(4-(benzytox y)phenyl)cycloprop yl)amino)methyl)- 1,2,4-
oxadiazol-3 -amine;
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
59
3 -((((trans)-2-(4-(benzyl oxy)phenyl)cycloprop yl)aminonnethyl)-1,2,4-
oxadiazol-5-amine ;
5-4((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)-1,2,4-thiadiazol-
3-amine;
5-((((trans)-2-(4-(b enzyloxy)phenyl)c ycloprop yl)amino)methyl)p yridin-2-
amine ;
6-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)midazin-3-amine;
5 -((((trans)-2-(4-(b enzyl oxy)phenyl)cyclopropyl)amino)methyl)mazin-2-amine;
2-((((trans)-2-(4-(b enzyloxy)phenyl)cycloprop yl)amino)methyl)pyrimidin-5 -
amine;
6-((((trans)-2-(4-(b enz yloxy)phenyl)cycloprop yl)amino)methyl)-1,2,4-triazin-
3-amine ; or
3 -((((trans)-2-(4-(benz yloxy)phenyl)cyclopropyl)amino)methyl)-1,2,4-triazin-
6-amine ;
for use in a method of treating or preventing cancer. In one aspect, the
cancer is chosen
from prostate, testicular, brain, colorectal, lung, breast, lymphoma, skin, or
blood cancer.
In one embodiment the invention relates to a compound, or a solvate or a
pharmaceutically
acceptable salt thereof, wherein said compound is:
4'-((trans)-24(2-aminothiazol-5-yl)methylamino)cyclopropyl)biphcny1-3-ol;
5 -(((trans)-2-(4-(b enzyl oxy)ph enyl )cycl opropyl ami noim ethyl )-1,2,4-
oxadi azol-3-am ine;
5-(((trans)-2-(4-(b enzyl oxy)phenyl)cyclopropylamino)methyl)-1,3,4-ox adiazol-
2-amin e ;
5 -((((trans)-2-(4-((4-fluorobenzyl)oxy)phenyl)cycl opropyl)amino)methyl)- 1,3
,4-oxadiazol-
2-amine; or
5 -((((trans)-2-(443 - fluorobenzyl)oxy)phenyl)cyclopropyl)amino)methyl)- 1,3
,4-oxadiazol -
2-amine;
for use in a method of treating or preventing a neurological disease or
condition. In one
aspect, the neurological disease or condition is chosen from Alzheimer
Disease, Parkinson
Disease, Huntington Disease, Amyotrophic Lateral Sclerosis, Frontotemporal
Dementia, or
Dementia with Lcwy Bodies.
In one embodiment the invention relates to a compound, or a solvate or a
pharmaceutically
acceptable salt thereof, wherein said compound is
4'-((trans)-242-aminothiazol-5-ylimethylamino)cyclopropyl)bipheny1-3-ol,
for use in a method of treating or preventing a neurological disease or
condition. In one
aspect, the neurological disease or condition is chosen from Alzheimer
Disease, Parkinson
Disease, Huntington Disease, Amyotrophic Lateral Sclerosis, Frontotemporal
Dementia, or
Dementia with Lewy Bodies.
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
In one embodiment the invention relates to a compound, or a solvate or a
phainiaceutically
acceptable salt thereof, wherein said compound is
5-(((trans)-2-(4-(benzyloxy)phenyl)cycloprop ylamino)methyl)-1 ,2 ,4-oxadiazol-
3 -amine, for
use in a method of treating or preventing a neurological disease or condition.
In one aspect,
5 the neurological disease or condition is chosen from Alzheimer Disease,
Parkinson Disease,
Huntington Disease, Amyotrophic Lateral Sclerosis, Frontotemporal Dementia, or
Dementia
with Lewy Bodies.
In one embodiment the invention relates to a compound, or a solvate or a
pharmaceutically
10 acceptable salt thereof, wherein said compound is
5 -(((trans)-2-(4-(b enz ylo xy)phenyl)c ycloprop ylam ino)m ethyl)-1,3 ,4-
oxadiazol-2-amine, for
use in a method of treating or preventing a neurological disease or condition.
In one aspect,
the neurological disease or condition is chosen from Alzheimer Disease,
Parkinson Disease,
Huntington Disease, Amyotrophic Lateral Sclerosis, Frontotemporal Dementia, or
Dementia
15 with Lewy Bodies.
In one embodiment the invention relates to a compound, or a solvate or a
pharmaceutically
acceptable salt thereof wherein said compound is
5 -((((trans)-2-(4-((4- fluorob enz yl)ox y)phenyl)c ycloprop yl)amino)methyl)-
1 ,3 ,4-oxadiaz ol-
20 2-amine, for use in a method of treating or preventing a neurological
disease or condition.
In one aspect, the neurological disease or condition is chosen from Alzheimer
Disease,
Parkinson Disease, Huntington Disease, Amyotrophic Lateral Sclerosis,
Frontotemporal
Dementia, or Dementia with Lewy Bodies.
25 In one embodiment the invention relates to a compound, or a solvate or a
pharmaceutically
acceptable salt thereof, wherein said compound is
5-((((trans)-2-(4-((3-fluorobenzyl)oxy)phenyl)cyclopropyl)amino)m ethyl)- 1,3
,4-ox a diazol-
2-amine, for use in a method of treating or preventing a neurological disease
or condition.
In one aspect, the neurological disease or condition is chosen from Alzheimer
Disease,
30 Parkinson Disease, Huntington Disease, Amyotrophic Lateral Sclerosis,
Frontotemporal
Dementia, or Dementia with Lewy Bodies.
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
61
In one embodiment of the invention provides an optically active N-substituted
aryl- or
heteroaryl cyclopropylamine or a phattnaceutically acceptable salt or solvate
thereof,
wherein said optically active N-substituted aryl- or heteroaryl-
cyclopropylamine is chosen
from
(-) 5-((((trans)-
2-(44(3-fluorobenzyl)oxy)phenyl)cyclopropyl)amino)methyl)-1,3,4-
oxadiazol-2-amin e;
(-) 5-
((((trans)-2-(4-(benzyl oxy)phenyl)cyclopropyeamino)m eth yl )-N-methy1-1,3 ,4-
oxadiazol-2-amine;
(-) N-
(5-((((trans)-2-(4-(benzyloxy)phenyl)cycl opropyl)amino)methyl)-1 ,3 ,4-
oxadiazol -2-
yl)acetamide;
(-) 5-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)pyrimidin-2-
amine;
(-) 5-
((((trans)-2-(4-(benzyloxy)phenyl)c yc lopropyl)amino)methyl)-1 ,3 ,4-thiadiaz
01-2-
amine; or
(-) 5-
4((trans)-2-(442-fluorob enzyl)oxy)phenyl)cycloprop yl)amino)m ethyl)-1,3,4-
oxadiazol-2-amine.
In a related aspect, the invention relates to a compound selected from:
(-) 5-((((trans)-2-(4-(benzyloxy)phen yl)cycl opropyl ino)methyl )-1,3 ,4-
oxadi azol -2-amine ;
(-) 5 -
((((trans)-2-(443-fluorobenzyl)oxy)phenyl)cycloprop yl)amino)methyl)-1 ,3,4-
oxadiazol-2-amine;
(-) 5 -
((((trans)-2-(4-(b enzyloxy)phenyl)cyclopropyl)amino)methyl)-N-methyl-1 ,3,4-
oxadiazol-2-amine;
(-) N-
(5-((((trans)-2-(4-(benzyloxy)phenyl)cycl opropyl)amino)methyl)-1 ,3 ,4-
oxadiazol-2-
yl)acetamide;
(-) 5-4((trans)-2-(4-(benzyloxy)phenyl)cyclopropyeamino)methyl)pyrimidin-2-
amine;
(-) 5-
((((trans)-2-(4-(benzyloxy)phenyl)cyc lopropyl)amino)methyl)-1 ,3 ,4-thiadiaz
ol-2-
amine;
(-) 5 -
((((trans)-2-(442-fl u orobenzyl)oxy)phen yl)cycl prop yl)amino)methyl)-1
,3,4-
oxadiazol-2-amine; or
a pharmaceutically acceptable salt or solvate thereof;
for use in a method of treating or preventing a neurological disease or
condition (e.g.,
depression). Preferably, the neurological disease or condition is chosen from
depression,
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
62
Alzheimer disease, Huntington disease, Parkinson disease, Frontotemporal
Dementia,
Dementia with Lewy Bodies, or Amyotrophic Lateral Sclerosis.
The invention thus is a compound or composition having an optically active N-
substituted
aryl- or heteroaryl- cyclopropylamine. Preferably, the optically active N-
substituted aryl- or
heteroaryl-cyclopropylamine is as defined herein in any one of the embodiments
or aspects
of Formula (II) or (III). More preferably, the optically active N-substituted
aryl- or
heteroaryl-cyclopropylamine has a kinact/KI value for LSD1 which is at least
50 fold
higher than the kinact/KI value for MAO-A. Still more preferably, the
kinact/Ki value for
LSD1 is at least 100-fold higher than kinact/KI for MAO-A. Even more
preferably, the
LSD1 kinact/KI value is at least 250-fold higher than the kinact/KI value for
MAO-A. Yet
even more preferably, the LSD1 kinact/KI value is at least 500-fold higher
than the
kinact/KI value for MAO-A.
Preferably, the optically active N-substituted aryl- or heteroaryl-
cyclopropylamine is as
defined herein in any one of the embodiments or aspects of Formula (II) or
(III). More
preferably, the optically active N-substituted aryl- or heteroaryl-
cyclopropylamine has
kinact/KI value for LSD1 and MAO-B which are at least 50-fold higher than the
kinact/Ki
value for MAO-A. Preferably, the kinact/KI value for LSD1 and MAO-B are at
least 100-
fold higher than kinact/KI for MAO-A. Even more preferably, the LSD I and MAO-
B
kinact/KI values are at least 250-fold higher than the kinact/KI value for MAO-
A. Yet even
more preferably, the LSD1 and MAO-B kinact/KI values are at least 500-fold
higher than
the kinact/KI value for MAO-A.
Preferably, the optically active N-substituted aryl- or heteroaryl-
cyclopropylamine is as
defined herein in any of the embodiments of Formula (1) or (Ti). More
preferably, the
optically active N-substituted aryl- or heteroaryl-cyclopropylamine has a
kinact/KI value for
MAO-B which is at least 50-fold higher than the kinact/KI value for MAO-A.
Preferably,
the kinact/KI value for MAO-B is at least 100-fold higher than kinact/KI for
MAO-A. Even
more preferably, the MAO-B kinact/KI value is at least 500-fold higher than
the kinact/KI
value for MAO-A. Yet even more preferably, the MAO-B kinact/KI is at least
1000-fold
higher than the kinact/KI value for MAO-A. Yet even still more preferably, the
MAO-B
kinact/KI value is at least 2000-fold higher than the kinact/KI value for MAO-
A.
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
63
The optically active N-substituted aryl- or hetcroaryl-cyclopropylamine,
phainiaceutical
composition comprising the optically active N-substituted aryl- or hetcroaryl-
cyclopropylamine, or a phaimaceutically acceptable salt thereof, and a
pharmaceutically
acceptable carrier, and methods of their use have unexpected selectivity for
LSD1 andior
MA0B. The (-) stereoisomers of N-substituted aryl- or heteroaryl-
cyclopropylamines (e.g.,
of the compounds of Formula (II) or (III) as described herein) are
unexpectedly potent and
selective inhibitors of LSD I and/or MAO-B. Avoiding inhibition of "off-
targets" can avoid
unwanted or undesirable side-effects like the cheese effect associated with
MAO-A.
The optically active compounds of the invention can be prepared by chiral HPLC
from e.g.,
racemates, chiral synthesis with compounds of known chirality, or chiral
recrystallization
using chiral salts.
In one aspect, the invention provides a method for enriching an enantiomer of
a trans N-
substituted cyclopropylamine (e.g., an enantiomer of a compound of Formula
(II) or (III) or
an enantiomer of a compound of Folinula (I), wherein the substituents on the
cyclopropyl
moiety comprised in Formula (I) (i.e., the substituent (A) and the substituent
-NH-CH2-(D))
are in trans-configuration, the method comprising: contacting a trans-
substituted
cyclopropylamine with a chiral recrystallization agent in a solvent
(particularly under
conditions that are sufficient for the crystallization of the salt of the
chiral recrystallization
agent and the trans substituted cylopropylamine); and isolating the
crystallized salt of the
chiral recrystallization agent and the trans substituted cyclopropylamine. In
another
preferred aspect, the trans cyclopropylamine is trans 4-benzoxy-2-
phcnylcyclopropylamine
or a protected derivative thereof. In a preferred aspect, the trans N-
substituted
cyclopropylamine is of Formula (II) or (III) as described above or a
derivative thereof
wherein the -L211-R411 group or the the -L2111-R4111 group is absent or
substituted with a
protecting group. In a preferred aspect, the chiral recrystallization agent is
chosen from S
(+) mandelic acid, D (-) tartaric acid, L (¨) di-p-toluoyl tartaric acid, or R
(-) mandelic acid.
In one preferred aspect, the chiral recrystallization agent is R (-) mandelic
acid. In one
aspect, the solvent is THF and H20.
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
64
In one aspect, the invention provides a method for preparing an enantiomer of
a trans N-
substituted cyclopropylamine comprising: contacting a trans-substituted
cyclopropylamine
with a chiral recrystallization agent in a solvent (particularly under
conditions are sufficient
for the crystallization for the salt of the chiral recrystallization agent and
the trans
substituted cyclopropylamine); and isolating the crystallized salt of the
chiral
recrystallization agent and the trans substituted cyclopropylamine, thereby
preparing an
enatiomer of a trans N-substituted cyclopropylamine. In a preferred aspect,
the trans N-
substituted cyclopropylamine is of Formula (II) or (III) as defined above or a
derivative
thereof wherein the -L2"-R4" group or the the -L2m-R4111 group is absent or
substituted with
a protecting group. In another preferred aspect, the trans cyclopropylamine is
trans 4-
benzoxy-2-phenylcyclopropylamine or a protected derivative thereof. In a
preferred aspect,
the chiral recrystallization agent is chosen from S (+) mandelic acid, D (-)
tartaric acid, L (¨
) di-p-toluoyl tartaric acid, or R (-) mandelic acid. In one preferred aspect,
the chiral
recrystallization agent is R (-) mandelic acid. In one aspect, the solvent is
THF and H20.
Additionally, the invention relates to the (+) enantiomer of a trans N-
substituted aryl- or
heteroaryl-cyclopropylamine, including the compounds of Foinaula (II) or (III)
and the
compounds of Foimula (I), in which the substituents on the cyclopropylamine
moiety are in
trans-orientation. For example, a corresponding optically active (+)
enantiomer may be
selected from:
( ) 5 -
((((trans)-2-(44(3 -fluorob enzyl)oxy)phenyl)c yc loprop yl)amino)methyl)-1,3
,4-
oxadiazol-2-amine;
(-0 5-
4((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)-N-methyl-1,3,4-
oxadiazol-2-amine;
(+) N-(5-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)-1,3,4-
oxadiazol-2-
ypacetamide;
(+) 5-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)pyrimidin-2-
amine;
(m) 5 -
((((trans)-2-(4-(benzyloxy)phenyl)c yc lopropyl)amino)methyl)-1 ,3 ,4-
thiadiazol-2-
amine;
(+) 5-((((trans)-
2-(44(2-fluorobenzyl)oxy)phenyl)cyclopropyl)amino)methyl)-1,3,4-
oxadiazol-2-amine;
or a pharmaceutically acceptable salt or solvate thereof.
The present invention furthermore relates to the following items:
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
1. An optically active N-substituted aryl- or heteroaryl-
cyclopropylamine or a
pharmaceutically acceptable salt or solvate thereof for use in a method for
treating or
preventing a disease.
5 2. The optically active N-substituted aryl- or heteroaryl-
cyclopropylamine of item 1
wherein said N-substituted aryl- or heteroaryl cyclopropylamine is a trans N-
substituted aryl- or heteroaryl cyclopropylamine that rotates plane polarized
light in
the (-) sense or is the (-) stereoisomer.
3. The optically active N-substituted aryl- or heteroaryl-cyclopropylamine
of item 1 or
10 2 wherein said N-substituted aryl- or heteroaryl cyclopropylamine is
90% or greater
(-) stereoisomer and 10% or less (+) steroisomer.
4. The optically active N-substituted aryl- or heteroaryl-cyclopropylamine
of item 1 or
2 wherein said N-substituted aryl- or heteroary.1 cyclopropylamine is 95% or
greater
(-) stereoisomer and 5% or less (+).
15 5. The optically active N-substituted aryl- or heteroaryl-
cyclopropylamine of item 1 or
2 wherein said N-substituted aryl- or heteroaryl. cyclopropylamine is 98% or
greater
(-) stereoisomer and 2% or less (+).
6. The optically active N-substituted aryl- or heteroaryl- cyclopropylamine
of item 1 or
2 wherein said N-substituted aryl- or heteroaryl cyclopropylamine is 99% or
greater
20 (-) stereoisomer and 1% or less (¨).
7. The optically active N-substituted aryl- or heteroaryl- cyclopropylamine
of item 1 or
2 wherein said N-substituted aryl- or heteroaryl- cyclopropylamine is 99.5% or
greater (-) stereoisomer and 0.5% or less (+).
8. The optically active N-substituted aryl- or heteroaryl- cyclopropylamine
of item 1 or
25 2 having a 90% or more enantiomeric excess of the (-) stereoisomer of
the N-
substituted aryl- or heteroaryl- cyclopropyi amine.
9. The optically active N-substituted aryl- or heteroaryl- cyclopropylamine
of item 1 or
2 having a 95% or more enantiomeric excess of the (-) stereoisomer of the N-
substituted aryl- or heteroaryl- cyclopropylamine.
30 10. The optically active N-substituted aryl- or heteroaryl
cyclopropylamine of item 1 or
2 having a 98% or more enantiomeric excess of the (-) stereoisomer of the N-
substituted aryl- or heteroaryl- cyclopropylamine.
CA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
66
11. The optically active N-substituted aryl- or heteroaryl cyclopropylamine
of item 1 or
2 having a 99% or more enantiomcric excess of the (-) stereoisomer of the N-
substituted aryl- or heteroaryl- cyclopropylamine.
12. The optically active N-substituted aryl- or heteroaryl cyclopropylamine
of any of of
items 1-11, or a pharmaceutically acceptable salt thereof, wherein said N-
substituted
aryl- or heteroaryl cyclopropylamine is of Formula (II):
RI "-(A")-R2"
wherein (A11) is an aryl or heteroaryl group having 2 substituents, R1 D and
R2D, and
1 to 3 optional substituents wherein said optional substituents are
independently
chosen from halo, Cl-C3 alkyl, or CI-C3 alkoxy;
R111 is an¨Li-R3 group;
R311 is a aryl or heteroaryl group having 1, 2, 3, 4, or 5 optional
substituents
independently chosen from halo, -OH, -NITS02RA, alkyl, alkoxy, cyano, -CF3, or
-0CF3 wherein RA is a C1-C6 alkyl or phenyl;
LID is chosen from a bond, -CF120-, -CH2CH20-, -0 CH2-, -OCH2CH2-, -CH2-,
-CH2CH2-, -CH2CH2CH2-, or -0-.
R211 is ¨Cyclopropyl-NH-L211-R411 wherein said cyclopropyl group has two
chiral
centers substituted in the trans orientation corresponding to the carbons to
which
(AD) and ¨NH-L211-R411 are covalently attached;
R411 is a 5 or 6 membered heteroaryl ring having 1, 2, or 3 optional
substituents
wherein said optional substituents are independently chosen from alkyl, NEIRD,
-
ORB, or halo wherein RD is a hydrogen, Cl-C3 alkyl, or ¨C(=0)CH3;
L21' =
is a branched or unbranched Cl-C4 alkylene group.
13. The optically active N-substituted aryl- or heteroaryl-cyclopropylamine
of item 12
wherein (AII) is an aryl or heteroaryl group having 2 substituents, R111 and
R211, and
1, 2, or 3 optional substituents wherein said optional substituents are
independently
chosen from halo, CI-C3 alkyl, or Cl-C3 alkoxy.
14. The optically active N-substituted aryl- or heteroaryl cyclopropylamine
of item 12
wherein R311 is a phenyl, pyridyl, thiazolyl, or thienyl group having 1, 2, or
3
optional substituents independently chosen from halo, -OH, -NHSO2RA, alkyl,
alkoxy, cyano, -CF3, or -0CF3 wherein RA is C1-C6 alkyl or phenyl.
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
67
15. The optically active N-substituted aryl- or heteroaryl cyclopropylamine
of item 12
wherein L11' is chosen from a bond, - OCH2-, or -CH20-.
16. The optically active N-substituted aryl- or heteroaryl cyclopropylamine
of item 12
wherein R411 is a 5-membered heteroaryl ring having 1, 2, or 3 optional
substituents
independently chosen from ¨NH2 or ¨NH(CI-C3) alkyl.
17. The optically active N-substituted aryl- or heteroaryl cyclopropylamine
of item 12
wherein L211 is -CH2- or ¨CH2CF12-=
18. The use of any one of items 1-17 wherein said method of treating or
preventing is a
method of treating or preventing cancer, depression, a neurodegenerative
disease or
disorder, or a viral infection.
19. The use of item 17 wherein said neurodegenerative disease or disorder
is chosen
from Alzheimer Disease, Parkinson Disease, Huntington Disease, Frontotemporal
Dementia or Amytrophic Lateral Sclerosis.
20. The use of item 17 wherein said neurodegenerative disease or disorder
is chosen
from Alzheimer Disease, Parkinson Disease, Huntington Disease, Frontotemporal
Dementia or Amytrophic Lateral Sclerosis.
21. The use of item 17 wherein said neurodegenerative disease or disorder
is Alzheimer
Disease.
22. The use of item 17 wherein said neurodegenerative disease or disorder
is Parkinson
Disease.
23. The use of item 17 wherein said neurodegenerative disease or disorder
is Huntington
Disease.
24. The use of item 17 wherein said neurodegenerative disease or disorder
is
Frontotemporal Dementia.
25. The use of item 17 wherein said neurodegenerative disease or disorder
is
Amytrophic Lateral Sclerosis.
26. An optically active compound, or a pharmaceutically acceptable salt or
solvate
thereof, of Formula (III):
R111t4A111)-R2111
wherein (A111) is an aryl or heteroaryl group having 2 substituents, R1111 and
R2111,
and 1 to 3 optional substituents wherein said optional substituents are
independently
chosen from halo, CI-C3 alkyl, or C1-C3 alkoxy;
CA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
68
Ri is an ¨L1111-R3 group;
R3111 is a phenyl, pyridyl, thiazolyl, or thienyl group having 0, 1, 2, or 3
substituents
independently chosen from ¨F, -Cl, -OH, -NHSO7RA, C1-C3 alkyl, C 1 -C3 alkoxy,
cyano, -CF3, or ¨0CF3 wherein RA is Cl -C6 alkyl or phenyl;
LI" is chosen from a bond, -CH20-, or -CH20-,
R2111 is ¨Cyclopropyl-NH-L2111-R4111 wherein said cyclopropyl group has two
chiral
centers substituted in the trans orientation corresponding to the carbons to
which
(Ain) and ¨NH-L2111-R4" are covalently attached;
R4111 is a 5-membered heteroaryl ring having 1, 2, or 3 optional substituents
wherein
said optional substituents are independently chosen from ¨NH2 or ¨NH(C1-C3)
alkyl; and
L2111 is -CH2- or ¨CH2CH2-.
27. The compound of item 26 wherein (Am) is a phenyl or pyridyl group.
28. The compound of item 26 wherein R3111 is a phenyl having 0, 1, 2, or 3
substituents
independently chosen from ¨F, -Cl, -OH, -NHSO2RA, C1-C3 alkyl, CI-C3 alkoxy,
cyano, -CF3, or ¨0CF3 wherein RA is C1-C6 alkyl or phenyl;
29. The compound of item 26 wherein L1111 is chosen from a bond, -OCH2-, or
-CH20-,
30. The compound of item 26 wherein R4111 is a 5-membered heteroaryl ring
wherein the
chain of atoms comprising said 5-membered heteroaryl ring has 2 or 3 hetero
atoms
independently chosen from N, S, or 0 and said heteroaryl ring has 1 optional
substituent wherein said optional substituent, if present, is ¨NH2 or ¨NH(C1-
C3)
alkyl.
31. The compound of item 26 wherein L2111 is -CH2- or ¨CH2CH2-=
32. The compound of item 26 wherein R3111 is a phenyl having 0, 1, 2, or 3
substituents
independently chosen from ¨F, -Cl, -OH, -NHSO/CH3, methyl, methoxy, cyano, -
CF3, or ¨0CF3.
33. The compound of item 26 wherein R4111 is an oxadiazolyl, thiadiazolyl,
or thiazolyl
ring having 1 optional substituent wherein said optional substituent, if
present, is
-NH, or ¨NH(C1-C3) alkyl.
34. The compound of item 26 wherein R4111 is an oxadiazolyl ring having 1
optional
substituent chosen from ¨NH2 or ¨NH(C1-C3) alkyl.
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
69
35. The optically active N-substituted aryl- or heteroaryl-cyclopropylamine
of any one
of items 26-34 wherein said N-substituted aryl- or heteroaryl cyclopropylamine
rotates plane polarized light in the (-) sense or is the (-) enantiomer.
36. The optically active N-substituted aryl- or heteroaryl cyclopropylamine
of any one
of items 26-34 wherein said N-substituted aryl- or heteroaryl-
cyclopropylamine is
90% or greater (-) stereoisomer and 10% or less (+) steroisomer.
37. The optically active N-substituted aryl- or heteroaryl cyclopropylamine
of item 1
any one of items 26-34 wherein said N-substituted aryl- or heteroaryl-
cyclopropylamine is 95% or greater (-) stereoisomer and 5% or less (+).
38. The optically active N-substituted aryl- or heteroaryl cyclopropylamine
of any one
of items 26-34 wherein said N-substituted aryl- or heteroaryl-cyclopropylamine
98%
or greater (-) stereoisomer and 2% or less (+).
39. The optically active N-substituted aryl- or heteroaryl-
cyclopropylamine of any one
of items 26-34 wherein said N-substituted aryl- or heteroaryl-cyclopropylamine
99%
or greater (-) stereoisomer and 1% or less (+).
40. The optically active N-substituted aryl- or heteroaryl-
cyclopropylamine of any one
of items 26-34 wherein said N-substituted aryl- or heteroaryl-
cyclopropylamine
99.5% or greater (-) stereoisomer and 0.5% or less (+).
41. The optically active N-substituted aryl- or heteroaryl-
cyclopropylamine of any one
of items 26-34 wherein said N-substituted aryl- or heteroaryl-
cyclopropylamine has
a 90% or more enantiomeric excess of the (-) stereoisomer of the N-substituted
aryl-
or heteroaryl- cyclopropylamine.
42. The optically active N-substituted aryl- or hcteroaryl cyclopropylamine
of any one
of items 26-34 wherein said N-substituted aryl- or heteroaryl-
cyclopropylamine has
a 95% or more enantiomeric excess of the (-) stereoisomer of the N-substituted
aryl-
or heteroaryl- cyclopropylamine.
43. The optically active N-substituted aryl- or heteroaryl cyclopropylamine
of any one
of items 26-34 wherein said N-substituted aryl- or heteroaryl-
cyclopropylamine has
a 98% or more enantiomeric excess of the (-) stereoisomer of the N-substituted
aryl-
or heteroaryl- cyclopropylamine.
44. The optically active N-substituted aryl- or heteroaryl cyclopropylamine
of any one
of items 26-34 wherein said N-substituted aryl- or heteroaryl-
cyclopropylamine has
GA 02806008 2013-01-18
WO 2012/013728 PCT/EP2011/062949
a 99% or more enantiomeric excess of the (-) stereoisomer of the N-substituted
aryl-
or heteroaryl- cyclopropylamine.
45. A method of treatment or prevention of a disease or disorder said
method comprising
administering to an individual in need of said treatment or prevention an
effective
5 amount of an optically active N-substituted aryl- or heteroaryl-
cyclopropylamine or
a pharmaceutically acceptable salt or solvate thereof.
46. The method of item 45 wherein said optically active N-substituted aryl-
or
heteroaryl-cyclopropylamine is as in any one of items 26-44.
47. The method of item 45 or 46 wherein said disease or disorder is chosen
from cancer,
10 a neurodegenerative disease or disorder, viral infection, or
depression.
48. The method of item 45 or 46 wherein said disease or disorder is a
neurodegenerative
disease or disorder chosen from Alzheimer disease, Parkinson disease,
Huntington
Disease, Frontotemporal Dementia, or Amytrophic Lateral Sclerosis.
49. A optically active compound or a pharmaceutically acceptable salt or
solvate thereof
15 wherein said optically active compound is chosen from:
(-) 5 -
((((trans)-2 -(4-(b enzyloxy)phenyl)cyclopropylamino)methyl)-1,3,4-oxadiazol-
2-amine;
(-) 5-4((trans)-2-(44(3-fluorobenzypoxy)phenyl)cyclopropyl)amino)methyl)-1,3,4-
oxadiazol-2-amine;
20 (-) 5 -((((trans)-2-(4-(b enzyloxy)phenyl)cyclopropyl)amino)methyl)-N-
methy1-1,3,4-
oxadiazol-2-amine;
N-(54((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)-1,3,4-
oxadiazol-2-ypacetamide;
(-) 5-
((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyppyrimidin-2-
25 amine; or
(-) 5 -((((trans)-2-(4-(b enzyloxy)phenyl)cyclopropyl)amino)methyl)- 1 ,3,4-
thiadiazol-
2-amine.
50. The compound of item 49 chosen from (-) N -(5
4(244-
(benzyl ox y)phenyl )cyclopropyl)amino)methyl)-1,3,4-oxadiazol -2-
yl)acetamide; (-)
30 5-(((2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)-N-methyl- 1,3 ,4-
oxaci iazol-
2- amine ; or (-) 5-(((2-(4-(b enzyloxy)phenyl)cyclopropylamino)methyl)- 1
,3,4-
oxadiazol-2-amine.
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
71
51. The compound of item 49 which is (-) 5-(((2-(4-
(benzy1oxy)pheny1)cyclopropyl)amino)methy1)-N-methyl-1,3,4-oxadiazol-2-amine.
52. The compound of item 49 which is (-) 5-(((2-(4-
(benzyloxy)phenyl)cyclopropylamino)methyl)-1,3,4-oxadiazol-2-amine.
53. The compound of item 49 which is (-) N-(5-(((2-(4-
(benzyloxy)phenyl)cyclopropyl)amino)methyl)-1,3,4-oxadiazol-2-y1)acetamide.
54. A pharmaceutical composition comprising an optically active compound as
in any
one of items 26-44 or 49 or a pharmaceutically acceptable salt thereof, and a
pharmaceutically acceptable carrier.
55. The pharmaceutical composition of 54 for use in a method of treating or
preventing
a disease or disorder.
56. The pharmaceutical composition of item 55 wherein said disease or
disorder is a
human disease or disorder chosen from cancer, a neurological disease or
disorder, or
a viral infection.
57. The pharmaceutical composition of item 56 where n said neurological
disease or
disorder is depression or a neurodegenerative disease or disorder.
58. The pharmaceutical composition of item 57 wherein said
neurodegenerative disease
or disorder is Alzheimer disease, Parkinson disease, Huntington Disease,
Frontotemporal Dementia, or Amytrophic Lateral Sclerosis.
59. An optically active compound chosen from:
(+) 54((trans)-2-(4-((3-fluorobenzypoxy)phenyl)cyclopropyl)amino)methyl)-1,3,4-
oxadiazol-2-amine;
(+) 5-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyeamino)methyl)-N-methyl-
1,3,4-
oxadiazol-2-amine;
N-(54((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)-1,3,4-
oxadiazol-2-ypacetarnide;
(-9 5-
((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyppyrimidin-2-
amine;
or
( ) 5-((((trans)-
2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)-1,3,4-
thiadiazol-2-amine; or a pharmaceutical acceptable salt or solvate thereof.
60. A method for enriching an enantiomer of a trans N-substituted
cyclopropylamine
comprising: contacting a trans-substituted cyclopropylarnine with a chiral
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
72
recrystallization agent in a solvent and under conditions are sufficient for
the
crystallization for the salt of the chiral recrystallization agent and the
trans
substituted cylopropylamine; and isolating the crystallized salt of the chiral
recrystallization agent and the trans substituted cyclopropylamine.
61. The method of item 60 wherein the trans cyclopropylamine is trans 4-
benzoxy-2-
phenylcyclopropylamine or a protected derivative thereof.
62. The method of item 60 wherein the chiral recrystallization agent is
chosen from S
(+) mandelic acid, D (-) tartaric acid, L (¨) di-p-toly1 tartaric acid, or R (-
) mandelic
acid.
63. The method of item 60 or 61 wherein the chiral recrystallization agent
is R (-)
mandelic acid.
64. The method of item 60, 61, 62, or 63 wherein the solvent is THF and
HA/
Definitions:
Any definition herein may be used in combination with any other definition to
describe a
composite structural group. By convention, the trailing element of any such
definition is that
which attaches to the parent moiety. For example, the composite group
alkylamido would
represent an alkyl group attached to the parent molecule through an amido
group, and the
teini alkoxyalkyl would represent an alkoxy group attached to the parent
molecule through
an alkyl group.
As used herein, the term "acyl," refers to a carbonyl attached to an alkenyl,
alkyl, aryl,
cycloalkyl, heteroaryl, heterocyclyl, or any other moiety where the atom
attached to the
carbonyl is carbon. An "acetyl" group refers to a -C(=0)CH3 group. An
"alkylcarbonyl" or
"alkanoyl" group refers to an alkyl group attached to the parent molecular
moiety through a
carbonyl group. Examples of such groups include, but are not limited to,
methylearbonyl or
ethylcarbonyl. Examples of acyl groups include, but are not limited to,
formyl, alkanoyl or
aroyl.
As used herein, the term "alkenyl," refers to a straight-chain or branched-
chain hydrocarbon
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
73
group having one or more double bonds and containing from 2 to 20 carbon
atoms. A (C2-
C6)alkenyl has from 2 to 6 carbon atoms.
As used herein, the tem). "alkoxy," refers to an alkyl ether group, wherein
the term alkyl is
as defined below. Examples of suitable alkyl ether groups include, but are not
limited to,
methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, iso-butoxy, sec-butoxy, tert-
butoxy, or
n-pentoxy.
As used herein, the term "alkyl," refers to a straight-chain or branched-chain
alkyl group
containing from 1 to 20 carbon atoms. A (C I -C10)alkyl has from 1 to 10
carbon atoms and
a (C1-C6)alkyl has from 1 to 6 carbon atoms and a (C1-C4)alkyl has from 1 to 4
carbon
atoms. Examples of alkyl groups include, but are not limited to, methyl,
ethyl, n-propyl,
isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, pentyl, isopentyl, neo-
pentyl, iso-amyl,
hexyl, heptyl, octyl, or nonyl.
As used herein, the term "alkylene" refers to an alkyl group attached at two
positions, i.e. an
alkanediyl group. Examples include, but are not limited to, methylene,
ethylene, propylene,
butylene, pentylene, hexylene, heptylene, octylene, or nonylene. Accordingly,
the term
"alkylene" may, e.g., refer to a straight-chain or branched-chain alkylene
group having from
1 to 6 carbon atoms.
As used herein, the term "alkylamino," refers to an alkyl group attached to
the parent
molecular moiety through an amino group. Suitable alkylamino groups may be
mono- or
dialkylated, forming groups including, but not limited to N-methylamino, N-
ethylamino,
N,N-dimethylamino, N,N-ethylmethylamino, N,N-diethylamino, N-propylamino, and
N,N-
methylprop ylamino.
As used herein, the term "alkynyl," refers to a straight-chain or branched-
chain hydrocarbon
group having one or more triple bonds and containing from 2 to 20 carbon
atoms. A (C2-
C6)alkynyl has from 2 to 6 carbon atoms. A (C2-C4)alkynyl has from from 2 to 4
carbon
atoms. Examples of alkynyl groups include, but are not limited to, ethynyl,
propynyl,
hydroxypropynyl, butyn-l-yl, butyn-2-yl, pentyn-l-yl, 3-methylbutyn-1-yl, or
hexyn-2-yl.
:
74
As used herein, the terms "amido" and "carbamoyl," refer to an amino group as
described
below attached to the parent molecular moiety through a carbonyl group (e.g., -
C(-0)NRR') , or vice versa (-N(R)C(=0)R'). "Amido" and ''carbamoyl" encompass
"C-
amido", "N-amido" and "acylamino" as defined herein. R and R' are as defined
herein.
As used herein, the term "C-amido," refers to a -C(=0)NRR' group with R and R'
as
defined herein.
As used herein, the term "amino," refers to -NRR', wherein R and R' are
independently
selected from the group consisting of hydrogen, alkyl, heteroalkyl, aryl,
carbocycly 1, and
heterocyclyl.
Additionally, R and R' may be combined to form a heterocyclyl.
As used herein, the term "aryl," refers a carbocyclic aromatic system
containing one ring, or
two or three rings fused together where in the ring atoms are all carbon. The
term "aryl"
includes, but is not limited to groups such as phenyl, naphthyl, or
anthracenyl.
As used herein, the term ''arylalkoxy" or "aralkoxy," refers to an aryl group
attached to the
parent molecular moiety through an alkoxy group. Examples of arylalkoxy groups
include,
but are not limited to, benzyloxy or phenethoxy.
As used herein, the term "arylalkyl" or "aralkyl," refers to an aryl group
attached to the
parent molecular moiety through an alkyl group.
As used herein, the term "aryloxy," refers to an aryl group attached to the
parent molecular
moiety through an oxy (-0-).
As used herein, the term "carbamate," refers to an 0-carbamyl or N-carbamyl
group as
defined herein.
As used herein, the term "carbonyl," when alone includes formyl -C(=0)H and in
combination is a -C(=0)- group.
As used herein, the term "carboxyl" or "carboxy" refers to -C(0)OH or the
corresponding
CA 2806008 2018-02-26
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
"carboxylate" anion, such as is in a carboxylic acid salt. An "O-carboxy"
group refers to a
RC(=0)0- group, where R is as defined herein. A "C-carboxy" group refers to a -
C(=0)OR
groups where R is as defined herein.
5 As used herein, the teini "cyano" refers to -CN.
As used herein, the term "carbocyclyl" refers to a saturated or partially
saturated monocyclic
or a fused bicyclic or tricyclic group wherein the ring atoms of the cyclic
system are all
carbon and wherein each cyclic moiety contains from 3 to 12 carbon atom ring
members.
10 "Carbocycly1" encompasses benzo fused to a carbocyclyl ring system. One
group of
carbocyclyls have from 5 to 7 carbon atoms. Examples of carbocyclyl groups
include, but
are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and
cycloheptyl,
tetrahydronapthyl, indanyl, octahydronaphthyl, 2,3-dihydro-1H-indenyl, or
adamantyl.
15 As used herein, the Willi "cycloalkyl" refers to a saturated monocyclic,
bicyclic or tricyclic
group wherein the ring atoms of the cyclic system are all carbon and wherein
each cyclic
moiety contains from 3 to 12 carbon atom ring members. One group of
cycloalkyls has from
5 to 7 carbon atoms. Examples of cycloalkyl groups include, but are not
limited to,
cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, or adamantyl.
As used herein, the term "cycloalkenyl" refers to a partially saturated
monocyclic, bicyclic
or tricyclic group wherein the ring atoms of the cyclic system are all carbon
and wherein
each cyclic moiety contains from 3 to 12 carbon atom ring members. One group
of
carboalkenyls have from 5 to 7 carbon atoms. Examples of cycloalkenyl groups
include, but
are not limited to, cyclobutenyl, cyclopentenyl, or cyclohexenyl.
As used herein, the term "cycly1" refers to an aryl, heterocyclyl, or
carbocyclyl group as
defined herein.
As used herein, the teini "halo" or "halogen" refers to fluorine, chlorine,
bromine, or iodine.
As used herein, the term "haloalkoxy" refers to a haloalkyl group attached to
the parent
molecular moiety through an oxygen atom. Examples of haloalkoxy groups
include, but are
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
76
not limited to, trifluoromethoxy, 2-
fluoroethoxy, or 3-chloropropoxy.
As used herein, the teini "haloalkyl" refers to an alkyl group having the
meaning as defined
above wherein one or more hydrogens are replaced with a halogen. Specifically
embraced
are monohaloalkyl, dihaloalkyl or polyhaloalkyl groups. A monohaloalkyl group,
for one
example, may have an iodo, bromo, chloro or fluoro atom within the group.
Dihalo or
polyhaloalkyl groups may have two or more of the same halo atoms or a
combination of
different halo groups. Examples of haloalkyl groups include, but are not
limited to,
fl uoromethyl, difl uorom ethyl, trifluoromethyl,
chloromethyl, dichloromethyl,
trichloromethyl, pentafluoroethyl,
heptafluoropropyl , difluorochloromethyl,
dichlorofluoromethyl, difluoroethyl, difluoropropyl, dichloroethyl or
dichloropropyl.
As used herein, the term "heteroalkyl" refers to a straight or branched alkyl
chain, wherein
one, two, or three carbons forming the alkyl chain are each replaced by a
heteroatom
independently selected from the group consisting of 0, N, and S, and wherein
the nitrogen
and/or sulfur heteroatom(s) (if present) may optionally be oxidized and the
nitrogen
heteroatom(s) (if present) may optionally be quaternized. The heteroatom(s) 0,
N and S
may, for example, be placed at an interior position of the heteroalkyl group,
i.e., the
heteroalkyl may be bound to the remainder of the molecule via a carbon atom.
Up to two
heteroatoms may be consecutive, such as, for example, -CH2-NH-OCH3.
Accordingly, a
further example for a "heteroalkyl" group is a straight or branched alkyl
group, in which
two consecutive carbon atoms are replaced by the heteroatoms S and N,
respectively, and
the sulfur heteroatom is furthermore oxidized, resulting in moieties such as,
e.g.,
-S(=0)2-NH2, -S(=0)2-NH(alkyl) or -S(=0)2-N(alkyl)(alkyl).
As used herein, the term "heteroalkylene" refers to a heteroalkyl group
attached at two
positions. Examples include, but are not limited to, -CH2OCH2-, -CH2SCH2-. and
-
CH2NHCH2-, -CH2S-, or -CH2NHCH(CH3)CH2-. Accordingly, the term
"heteroalkylene"
may, e.g., refer to a straight or branched alkylene group (i.e., a straight or
branched
alkanediyl group) having from 1 to 6 carbon atoms, wherein 1, 2 (if present)
or 3 (if present)
of said carbon atoms are each replaced by a heteroatom independently selected
from 0, N or
S. It is to be understood that the presence of hydrogen atoms will depend on
the valence of
the heteroatom replacing the respective carbon atom. If, for example, the
carbon atom in a
. .
77
-CH2- group is replaced by 0 or S, the resulting group will be -0- or -S-,
respectively, while
it will be -N(H)- when the carbon atom replaced by N. Likewise, if the central
carbon atom
in a group -CH2-CH(-CH3)-CH2- is replaced by N, the resulting group will be -
CH2-
N(-CH3)-CH2-. An example for a "heteroalkylene" group is a straight or
branched alkylene
group, in which two consecutive carbon atoms are replaced by the heteroatoms S
and N,
respectively, and the sulfur heteroatom is furthermore oxidized, resulting in
moieties such
as, e.g., -S(=0)2-N(H)- or -S(=0)2-N(alkyl)-. Accordingly, the groups -S(=0)2.-
N(H)- and
-S(=0)2-N(alkyl)- (e.g., -S(=0)2-N(C1-C6 alkyl)-) are exemplary
"heteroalkylene" groups.
As used herein, the term "heteroaryl," refers to a 3 to 7 membered unsaturated
monocyclic
ring, or a fused bicyclic or tricyclic ring system in which the rings are
aromatic and which at
least one ring contains at least one atom selected from the group consisting
of 0, S. and N.
One group of heteroaryls has from 5 to 7 carbon atoms. Examples of heteroaryl
groups
include, but are not limited to, pyridinyl, imidazolyl, imidazopyridinyl,
pyrimidinyl,
pyrazolyl, triazolyl, pyrazinyl, tetrazolyl, furyl, thienyl. isoxazolyl,
thiazolyl, oxadiazolyl,
oxazolyl, isothiazolyl, pyrrolyl, quinolinyl, isoquinolinyl, indolyl,
benzimidazolyl,
benzofuranyl, cinnolinyl, indazolyl, indolizinyl, phthalazinyl, pyridazinyl,
triazinyl,
isoindolyl, pteridinyl, purinyl, oxadiazolyl, triazolyl, thiadiazolyl,
thiadiazolyl, furazanyl,
benzofurazanyl, benzothiophenyl, benzothiazolyl, benzoxazolyl, quinazolinyl,
quinoxalinyl,
naphthyridinyl, or furopyridinyl.
As used herein, the term "heterocycly1" or "hetercycle," each refer to a
saturated, partially
unsaturated, or fully unsaturated monocyclic, bicyclic, or tricyclic
heterocyclic group
containing at least one heteroatom as a ring member, wherein each said
heteroatom may be
independently selected from the group consisting of nitrogen, oxygen, and
sulfur wherein
the nitrogen or sulfur atoms may be oxidized (e.g., -N=0, -S(=0)-, or -S(=0)2-
). Additionally,
1, 2, or 3 of the carbon atoms of the heterocyclyl may be optionally oxidized
(e.g., to give
an oxo group or =0). One group of heterocyclyls has from 1 to 4 heteroatoms as
ring
members. Another group of heterocyclyls has from 1 to 2 heteroatoms as ring
members.
One group of heterocyclyls has from 3 to 8 ring members in each ring. Yet
another group of
heterocyclyls has from 3 to 7 ring members in each ring. Again, another group
of
heterocyclyls has from 5 to 6 ring members in each ring. "Heterocycly1" is
intended to
encompass a heterocyclyl group fused to a carbocyclyl or benzo ring systems.
Examples of
CA 2806008 2018-02-26
CA 02806008 2016-07-27
78
heterocyclyl groups include, but are not limited to, pyrrolidinyl,
tetrahydrofuranyl,
dihydrofuranyl, tetrahydrothienyl, tetrahydropyranyl, dihydropyranyl,
tetrahydrothiopyranyl, piperidino, morpholino, thiomorpholino, thioxanyl,
piperazinyl,
homopiperazinyl, azetidinyl, oxetanyl, thietanyl, homopiperidinyl, oxepanyl,
thiepanyl,
oxazepinyl, diazepinyl, thiazepinyl, 2-pyrrolinyl, 3-pyrrolinyl, indolinyl, 2H-
pyranyl, 4H-
pyranyl, dioxanyl, 1,3-dioxolanyl, pyrazolinyl, dithianyl, dithiolanyl,
dihydropyranyl,
dihydrothienyl, dihydrofuranyl, pyrazolidinylimidazolinyl, or imidazolidinyl.
Examples of
heteroaryls that are heterocyclyls include, but are not limited to, pyridinyl,
imidazolyl,
imidazopyridinyl, pyrimidinyl, pyrazolyl, triazolyl, pyrazinyl, tetrazolyl,
fury!, thienyl,
isoxazolyl, thiazolyl, oxadiazolyl, oxazolyl, isothiazolyl, pyrrolyl,
quinolinyl, isoquinolinyl,
indolyl, benzimidazolyl, benzofuranyl, cinnolinyl, indazolyl, indolizinyl,
phthalazinyl,
pyridazinyl, triazinyl, isoindolyl, pteridinyl, purinyl, oxadiazolyl,
triazolyl, thiadiazolyl,
thiadiazolyl, furazanyl, benzofurazanyl, benzothiophenyl, benzothiazolyl,
benzoxazolyl,
quinazolinyl, quinoxalinyl, naphthyridinyl, or furopyridinyl.
As used herein, the term "heterocycloalkyl," refers to a heterocyclyl group
that is not fully
unsaturated e.g., one or more of the rings systems of a heterocycloalkyl is
not aromatic.
Examples of heterocycloalkyls include piperazinyl, morpholinyl, piperidinyl,
or
pyrrolidinyl.
As used herein, the term "hydroxyl," as used herein, refers to -OH.
As used herein, the term "hydroxyalkyl," as used herein, refers to a hydroxyl
group attached
to the parent molecular moiety through an alkyl group.
As used herein, the phrase ''in the main chain," refers to the longest
contiguous or adjacent
chain of carbon atoms starting at the point of attachment of a group to the
compounds of any
one of the formulas disclosed herein.
As used herein, the term phrase "linear chain of atoms" refers to the longest
straight chain of
atoms independently selected from carbon, nitrogen, oxygen and sulfur.
As used herein, the teini ''lower," where not otherwise specifically defined,
means
CA 02806008 2016-07-27
79
containing from 1 to and including 6 carbon atoms.
As used herein, the term "lower aryl," means phenyl or naphthyl.
As used herein, the term ''lower heteroaryl," means either 1) monocyclic
heteroaryl
comprising five or six ring members, of which between one and four said
members may be
heteroatoms selected from 0, S, or N.
As used herein, the term "nitro," refers to -NO2.
As used herein, the terms "sulfonate," "sulfonic acid" and "sulfonic," refer
to the -S03H
group and its anion as the sulfonic acid is used in salt formation.
As used herein, the term "sulfanyl," refers to -S-.
As used herein, the term "sulfinyl," refers to -S(=0)R, with R as defined
herein.
As used herein, the term "sulfonyl," refers to -S(=0)2R, with R as defined
herein.
As used herein, the term "sulfonamide", refers to an N-sulfonamido or S-
sulfonamido group
as defined herein.
As used herein, the term "N-sulfonamido," refers to a RS(=0)2N(R')- group with
R and R'
as defined herein. Exemplary, non-limiting N-sulfonamido groups are ¨NHSO2CH3,
-NHSO2CH2CH3, -NHS02(phenyl), or -NHS02(isopropy1).
As used herein, the term "S-sulfonamido," refers to a -S(=0)2NRR', group, with
R and R' as
defined herein.
As used herein, the term "urea," refers to a ¨N(R)C(=0)N(R) group wherein R
and R' are as
defined herein.
CA 02806008 2016-07-27
As used herein, "hydrogen bonding group" refers to a substituent group, which
is capable of
taking part in a non-covalent bonding between hydrogen and another atom
(usually nitrogen
or oxygen). Examples include, but are not limited to, -NH2, -OH, amido, -
S(0)2NH2, -
C(=0)NH2, -CH2-C(=0)NH2, - and -CH2-NH2. Other non-limiting examples include
5 NHC(=0)CH3 or -NHCH3.
As used herein, the term "amide isostere" refers to a monocyclic or bicyclic
ring system that
is isosteric or bioisosteric with an amide moiety. Examples of amide isoteres
include but
are not limited to those disclosed in, e.g., Meanwell (2011) J. Med. Chem.
PM1D:
10 21413808,
As used herein, the term "optionally substituted" means the preceding or
anteceding group
may be substituted or unsubstituted. When substituted, the substituents of an
"optionally
substituted" group may include, without limitation, one or more substituents
independently
15 selected from the following groups or a particular designated set of
groups, alone or in
combination: lower alkyl, lower alkenyl, lower alkynyl, lower alkanoyl, lower
heteroalkyl,
lower heterocycloalkyl, lower haloalkyl, lower cycloalkyl, phenyl, aryl,
aryloxy, lower
alkoxy, lower haloalkoxy, oxo, lower acyloxy, carbonyl, carboxyl, lower
alkylcarbonyl,
lower carboxyester, lower carboxamido, cyano, hydrogen, halogen, hydroxyl,
amino, lower
20 alkylamino, arylamino, aminoalkyl, amido, nitro, thiol, lower alkylthio,
lower haloalkylthio,
lower perhaloalkylthio, arylthio, sulfonate, sulfonic acid, trisubstituted
silyl, N3, SH, SCH3,
C(0)CH3, CO2CH3, CO2H, pyridinyl, thiophene, furanyl, carbamate, and urea. Two
substituents may be joined together to form a fused five-, six-, or seven-
membered
carbocyclie or heterocyclic ring consisting of zero to three heteroatoms, for
example
25 forming methylenedioxy or ethylenedioxy. An optionally substituted group
may be
unsubstituted (e.g -CH2CH3), fully substituted (e.g., -CF2CF3),
monosubstituted (e.g., -
CH2CH2F) or substituted at a level anywhere in-between fully substituted and
monosubstituted (e.g., -CH2CF3). Where substituents are recited without
qualification as to
substitution, both substituted and unsubstituted forms are encompassed. Where
a
30 substituent is qualified as "substituted," the substituted form is
specifically intended.
Additionally, different sets of optional substituents to a particular moiety
may be defined as
needed; in these cases, the optional substitution will be as defined, often
immediately
following the phrase, "optionally substituted with." In one specific
definition, the optional
CA 02806008 2016-07-27
81
substituents are chosen from hydroxyl, halo, alkyl, alkoxy, haloalkyl,
haloalkoxy, -N((C I -
C3)alky1)2, -NH((C1-C3)alkyl), -NHC(=0)((C1-C3)alkyl), -C(=0)0H, -C(=0)0((C1-
C3)alkyl), -C(=0)(C1-C3)alkyl), -C(=0)NH2, -C(=0)NH(C1-C3)alkyl),
C(=0)NH(cycloalkyl), -C(=0)N(C1-C3)alky1)2, -S(=0)2((C1-C3)alkyl), -S(=0)2NH2,
-
S(=0)2N((C1-C3)alkyl)2, - S(=0)2NH((C1-C3)alkyl), -CHF2, -0CF3, -OCHF2, -SCF3,
-CF3,
-CN, -NH2, -NO2, or tetrazolyl.
As used herein, the term "optional substituent" denotes that the corresponding
substituent
may be present or may be absent. Accordingly, a compound having 1, 2 or 3
optional
substituents may be unsubstituted or may be substituted with 1, 2 or 3
substituents.
The term R or the term R', appearing by itself and without a number
designation, unless
otherwise defined, refers to a moiety selected from the group consisting of
hydrogen, alkyl,
cycloalkyl, heteroalkyl, aryl, heteroaryl and heterocycloalkyl. Whether an R
group has a
number designation or not, every R group, including R, R' and RP where p=(1,
2, 3, . . . p),
every substituent, and every term should be understood to be independent of
every other in
terms of selection from a group. Should any variable, substituent, or term
(e.g., aryl,
heterocycle, R, etc.) occur more than one time in a formula or generic
structure, its
definition at each occurrence is independent of the definition at every other
occurrence.
Those of skill in the art will further recognize that certain groups may be
attached to a
parent molecule or may occupy a position in a chain of elements from either
end as written.
Thus, by way of example only, an unsymmetrical group such as -C(=0)N(R)- may
be
attached to the parent moiety at either the carbon or the nitrogen.
Asymmetric centers exist in the compounds disclosed herein. These centers are
designated
by the symbols "R" or "S," depending on the configuration of substituents
around the chiral
carbon atom. It should be understood that the invention encompasses all
stereochemical
isomeric forms, including diastereomeric, enantiomeric, and epimeric forms, as
well as d-
isomers and 1-isomers, and mixtures thereof Individual stereoisomers of
compounds can
be prepared synthetically from commercially available starting materials which
contain
chiral centers or by preparation of mixtures of enantiomeric products followed
by separation
such as conversion to a mixture of diastereomers followed by separation or
recrystallization,
chromatographic techniques, direct separation of enantiomers on chiral
chromatographic
. :
=
82
columns, or any other appropriate method known in the art. Starting compounds
of
particular stereochemistry are either commercially available or can be made
and resolved by
techniques known in the art. Additionally, the compounds disclosed herein may
exist as
geometric isomers. The present invention includes all cis, trans, syn, anti,
entgegen (E), and
zusammen (Z) isomers as well as the appropriate mixtures thereof.
Additionally,
compounds may exist as tautomers; all tautomeric isomers are provided by this
invention.
Additionally, the compounds disclosed herein can exist in unsolvated as well
as solvated
forms with pharmaceutically acceptable solvents such as water, ethanol, and
the like. In
general, the solvated forms are considered equivalent to the unsolvated forms.
As used herein, the term "optically active," refers to the ability of a
compound to rotate
plane polarized light. In the context of the invention, the term refers to
mixtures of
enantiomers which are not racemic mixtures; that is to say, not a 50:50
mixture of a (+)
enantiomer and the corresponding (-) enantiomer.
As used herein, the term "N-substituted aryl or heteroarylcyclopropylamine"
(or, likewise,
"N-substituted aryl- or heteroaryl- cyclopropylamine"), refers to a compound
having a 1,2
disubstituted cyclopropyl core wherein the 1 and 2 positions are substituted
with a
substituted amine group and a substituted aryl or heteroaryl group. Compounds
of Formula
(II) and Formula (III) as described herein are examples of N-substituted aryl-
or heteroaryl-
cyclopropylamines.
As used herein, the term "enantiomeric excess" or "cc" or "percent
enantiomeric excess"
refers to the difference between the mole fraction of one specific enantiomer
(i e., the
specified enantiomer) and the mole fraction of the other enantiomer in
relation to the sum of
the mole fractions of both enantiomers, expressed as a percent value, and thus
describes the
extent of the excess of one specific enantiomer in relation to the other
enantiomer. If, for
example, a specific enantiomer is provided in the absence of the other
enantiomer, the
enantiomeric excess will be 100%, while a racemate comprising equal molar
amounts of the
two enantiomers will have an enantiomeric excess of 0%. Accordingly, the
"enantiomeric
excess" or "ee" or "percent enantiomeric excess" is defined by the following
formula:
CA 2806008 2018-02-26
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
83
(mole fraction of the specified enantiomer) - (mole fraction of the other
enantiorner)
= 100
(mole fraction of the specified enantiomer) + (mole fraction of the other
enantiomer)
As used herein, the term "preventing an increase in a symptom," refers to both
not allowing
a symptom to increase or worsen, as well as reducing the rate of increase in
the symptom.
For example, a symptom can be measured as the amount of particular disease
marker, i.e., a
protein (e.g., cancer biomarker). In another example the symptom can be
cognitive decline.
Preventing an increase, according to the definition provided herein, means
that the amount
of symptom (e.g., protein or cognitive decline) does not increase or that the
rate at which it
increases is reduced.
1 0
As used herein, the term "treating a disease or disorder," refers to a slowing
of or a reversal
of the progress of the disease. Treating a disease or disorder includes
treating a symptom
andior reducing the symptoms of the disease.
As used herein, the teim "preventing a disease or disorder," refers to a
slowing of the
disease or of the onset of the disease or the symptoms thereof. Preventing a
disease or
disorder can include stopping the onset of the disease or symptoms thereof As
used herein,
the term "unit dosage form" refers to a physically discrete unit, such as a
capsule or tablet
suitable as a unitary dosage for a human patient. Each unit contains a
predetelinined
quantity of a compound of Formula (I), (Ia), (Ib), (II) or (III) which was
discovered or
believed to produce the desired pharmacokinetic profile which yields the
desired therapeutic
effect. The dosage unit is composed of a compound of Formula (I), (Ia), (lb),
(II) or (III) in
association with at least one pharmaceutically acceptable carrier, salt,
excipient, or
combination thereof.
As used herein, the term "subject" or "patient" or "individual", such as the
subject in need of
treatment or prevention, may be a eukaryote, an animal, a vertebrate animal, a
mammal, a
rodent (e.g., a guinea pig, a hamster, a rat, a mouse), a murine (e.g., a
mouse), a canine (e.g.,
a dog), a feline (e.g., a cat), an equine (e.g. a horse), a primate, a simian
(e.g., a monkey or
ape), a monkey (e.g., a marmoset, a baboon), an ape (e.g., gorilla,
chimpanzee, orangutang,
gibbon), or a human. The meaning of the terms "eukaryote", "animal", "mammal",
etc. is
well known in the art and can, for example, be deduced from Wehner und Gehring
(1995;
84
Thieme Verlag). In the context of this invention, it is particularly envisaged
that animals are
to be treated which are economically, agronomically or scientifically
important.
Scientifically important organisms include, but are not limited to, mice,
rats, and rabbits.
Lower organisms such as, e.g., fruit flies like Drosophila tnelagonaster and
nematodes like
Caenorhabditis elegans may also be used in scientific approaches. Non-limiting
examples
of agronomically important animals are sheep, cattle and pig, while, for
example, cats and
dogs may be considered as economically important animals.
Preferably, the
subject/patient/individual is a mammal; more preferably, the
subject/patient/individual is a
human or a non-human mammal (such as, e.g., a guinea pig, a hamster, a rat, a
mouse, a
rabbit, a dog, a cat, a horse, a monkey, an ape, a marmoset, a baboon, a
gorilla, a
chimpanzee, an orangutang, a gibbon, a sheep, cattle, or a pig); even more
preferably, the
subject/patient/individual is a human.
As used herein, the term "dose" or "dosage," refers the amount of active
ingredient that an
individual takes or is administered at one time. For example, a 40-mg dose of
a compound
of Formula (I), (Ia), (Ib), (II) or (III) refers to, in the case of a twice-
daily dosage regimen, a
situation where the individual takes 40 mg of a compound of Formula (I) twice
a day, e.g..
40 mg in the morning and 40 mg in the evening. The 40 mg of a compound of
Formula (I).
(Ia), (Ib), (II) or (III) dose can be divided into two or more dosage units,
e.g., two 20 mg
dosage units of a compound of Formula (I), (Ia), (Ib), (II) or (III) in tablet
form or two 20
mg dosage units of a compound of Formula (I), (Ia), (lb), (II) or (III) in
capsule form.
As used herein, a "pharmaceutically acceptable prodrug" is a compound that may
be
converted under physiological conditions or by solvolysis to the specified
compound or to a
pharmaceutically acceptable salt of such compound.
As used herein, a "pharmaceutically active metabolite" is intended to mean a
pharmacologically active product produced through metabolism in the body of a
specified
compound or salt thereof. Metabolites of a compound may be identified using
routine
techniques known in the art and their activities determined using tests such
as those
described herein.
CA 2806008 2018-02-26
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
As used herein, a "pharmaceutically acceptable salt" is intended to mean a
salt that retains
the biological effectiveness of the free acids and bases of the specified
compound and that is
not biologically or otherwise undesirable. A compound for use in the invention
may possess
a sufficiently acidic, a sufficiently basic, or both functional groups, and
accordingly react
5 with any of a number of inorganic or organic bases, and inorganic and
organic acids, to
form a pharmaceutically acceptable salt. Exemplary pharmaceutically acceptable
salts
include those salts prepared by reaction of the compounds of the present
invention with a
mineral or organic acid or an inorganic base, such as salts including
sulfates, pyrosulfates,
bisulfates, sulfites, bisulfites, phosphates, monohydrophosphates,
dihydrophosphates,
10 metaphosphates, pyrophosphates, chlorides, bromides, iodides, acetates,
propionates,
decanoates, caprylates, acrylates, foiniates, isobutyrates, caproates,
heptanoates, propiolates,
oxalates, malonates, succinates, suberates, sebacates, fumarates, maleates,
butyne-1,4
dioates, hexyne-1,6-dioates, benzoates, chlorobenzoates, methylbenzoates,
dinitrobenzoates,
hydroxybenzoates, methoxybenzoates, phthalates, sulfonates, xylenesulfonates,
15 phenylacetates, phenylpropionates, phenylbutyrates, citrates, lactates,
gamma-
hydroxybutyrates, glycollates, tartrates, methane-sulfonates,
propanesulfonates,
naphthalene-l-sulfonates, naphthalene-2-sulfonates, or mandelates.
As used herein, a "pharmaceutically acceptable carrier" refers to a non-API
(API refers to
20 Active Pharmaceutical Ingredient) substances such as disintegrators,
binders, fillers, and
lubricants used in formulating pharmaceutical products. They are generally
safe for
administering to humans according to established governmental standards,
including those
promulgated by the United States Food and Drug Administration and the European
Medical
Agency.
As is understood by the skilled artisan, certain variables in the list of
substituents are
repetitive (different name for the same substituent), generic to other teinis
in the list, and/or
partially overlap in content with other terms. In the compounds of the
invention, the skilled
artisan recognizes that substituents may be attached to the remainder of the
molecule via a
number of positions and the preferred positions are as illustrated in the
Examples.
As described herein above, the compound of Formula (I), (Ia) or (lb) contains
asymmetric
carbon atoms and can therefore exist in racemic and optically active forms.
Thus, optical
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
86
isomers or enantiomers, racemates, tautomers, and diastereomers are also
encompassed by
the compounds of Formula (I), (Ia) or (lb). The methods of the present
invention include
the use of all such isomers and mixtures thereof. Methods of separation of
enantiomeric and
diastereomeric mixtures are well known to one skilled in the art and are
furthermore
described in the appended examples. The present invention encompasses any
isolated
racemic or optically active form of compounds according to Foimula (I), (Ia)
or (lb), or any
mixture thereof. In one aspect, the compounds of the invention have a trans
configuration
around the cyclopropyl ring as in trans-phenylcyclopropylamine. In one aspect,
the
compounds of the invention have a cis configuration around the cyclopropyl
ring as in cis-
phenylcyclopropylamine. In a preferred aspect, the compounds of Formula (I),
(Ia) or (lb)
have the trans configuration. In a more preferred aspect, the compounds of
Formula (I), (Ia)
or (Ib) are (-) stereoisomers having the trans configuration around the
cyclopropyl ring.
Typically, compounds according to Formula (I), (Ia), (lb), (II) or (III) can
be effective at an
amount of from about 0.01 ig/kg to about 100 mg/kg per day based on total body
weight.
The active ingredient may be administered at once, or may be divided into a
number of
smaller doses to be administered at predetermined intervals of time. The
suitable dosage
unit for each administration can be, e.g., from about 11.1.g to about 2000 mg,
preferably from
about 5 1.ig to about 1000 mg. Even more preferably, the amount of active
ingredient
administered is from about 5 lug to about 100 mg per day. These doses will
depend of the
pharmacokinetic parameters of the particular compound and other ADME
properties as well
as the efficacy of the compound in a particular disease setting.
It should be understood that the dosage ranges set forth above are exemplary
only and are
not intended to limit the scope of this invention. The therapeutically
effective amount for
each active compound can vary with factors including but not limited to the
activity of the
compound used, stability of the active compound in the patient's body, the
severity of the
conditions to be alleviated, the total weight of the patient treated, the
route of
administration, the ease of absorption, distribution, and excretion of the
active compound by
the body, the age and sensitivity of the patient to be treated, and the like,
as will be apparent
to a skilled artisan. The amount of administration can be adjusted as the
various factors
change over time.
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
87
For oral delivery, the active compounds can be incorporated into a formulation
that includes
pharmaceutically acceptable carriers such as binders (e.g., gelatin,
cellulose, gum
tragacanth), excipients (e.g., starch, lactose), lubricants (e.g., magnesium
stearate, silicon
dioxide), disintegrating agents (e.g., alginate, Primogel, and corn starch),
and sweetening or
flavoring agents (e.g., glucose, sucrose, saccharin, methyl salicylate, and
peppermint). The
formulation can be orally delivered in the form of enclosed gelatin capsules
or compressed
tablets. Capsules and tablets can be prepared in any conventional techniques.
The capsules
and tablets can also be coated with various coatings known in the art to
modify the flavors,
tastes, colors, and shapes of the capsules and tablets. In addition, liquid
carriers such as
fatty oil can also be included in capsules.
Suitable oral formulations can also be in the form of suspension, syrup,
chewing gum,
wafer, elixir, and the like. If desired, conventional agents for modifying
flavors, tastes,
colors, and shapes of the special forms can also be included. In addition, for
convenient
administration by enteral feeding tube in patients unable to swallow, the
active compounds
can be dissolved in an acceptable lipophilic vegetable oil vehicle such as
olive oil, corn oil
and safflower oil.
The active compounds can also be administered parenterally in the form of
solution or
suspension, or in lyophilized form capable of conversion into a solution or
suspension form
before use. In such formulations, diluents or pharmaceutically acceptable
carriers such as
sterile water and physiological saline buffer can be used. Other conventional
solvents, pH
buffers, stabilizers, anti-bacteria agents, surfactants, and antioxidants can
all be included.
For example, useful components include sodium chloride, acetates, citrates or
phosphates
buffers, glycerin, dextrose, fixed oils, methyl parabens, polyethylene glycol,
propylene
glycol, sodium bisulfate, bcnzyl alcohol, ascorbic acid, and the like. The
parenteral
formulations can be stored in any conventional containers such as vials and
ampoules.
Routes of topical administration include nasal, bucal, mucosal, rectal, or
vaginal
applications. For topical administration, the active compounds can be
formulated into
lotions, creams, ointments, gels, powders, pastes, sprays, suspensions, drops
and aerosols.
Thus, one or more thickening agents, humectants, and stabilizing agents can be
included in
the formulations. Examples of such agents include, but are not limited to,
polyethylene
=
88
glycol, sorbitol, xanthan gum, petrolatum, beeswax, or mineral oil, lanolin,
squalene, and
the like. A special form of topical administration is delivery by a
transdermal patch.
Methods for preparing transdermal patches are disclosed, e.g., in Brown, et
al. (1988) Ann.
Rev, Med. 39:221-229.
Subcutaneous implantation for sustained release of the active compounds may
also be a
suitable route of administration. This entails surgical procedures for
implanting an active
compound in any suitable formulation into a subcutaneous space, e.g., beneath
the anterior
abdominal wall. See, e.g., Wilson et al. (1984) J. Clin. Psych. 45:242-247.
Hydrogels can
be used as a carrier for the sustained release of the active compounds.
Hydrogels are
generally known in the art. They are typically made by crosslinking high
molecular weight
biocompatible polymers into a network, which swells in water to form a gel
like material.
Preferably, hydrogels are biodegradable or biosorbable. For purposes of this
invention,
hydrogels made of polyethylene glycols, collagen, or poly(glycolic-co-L-lactic
acid) may be
useful. See, e.g., Phillips et al. (1984).1 Pharmaceut. Sci., 73: 1718-1720.
The active compounds can also be conjugated, to a water soluble non-
immunogenic non-
peptidic high molecular weight polymer to form a polymer conjugate. For
example, an
active compound is covalently linked to polyethylene glycol to form a
conjugate. Typically,
such a conjugate exhibits improved solubility, stability, and reduced toxicity
and
immunogenicity. Thus, when administered to a patient, the active compound in
the
conjugate can have a longer half-life in the body, and exhibit better
efficacy. See generally,
Burnham (1994) Am. J. Hosp. Pharm. 15:210-218. PEGylated proteins are
currently being
used in protein replacement therapies and for other therapeutic uses. For
example,
PEGylated interferon (PEG-INTRON AC) is clinically used for treating Hepatitis
B.
PEGylated adenosine deaminase (ADAGENS) is being used to treat severe combined
immunodeficiency disease (SCIDS). PEGylated L-asparaginase (ONCAPSPARt) is
being
used to treat acute lymphoblastic leukemia (ALL). It is preferred that the
covalent linkage
between the polymer and the active compound and/or the polymer itself is
hydrolytically
degradable under physiological conditions. Such conjugates known as "prodrugs"
can
readily release the active compound inside the body. Controlled release of an
active
compound can also be achieved by incorporating the active ingredient into
microcapsules,
nanocapsules, or hydrogels generally known in the art. Other pharmaceutically
acceptable
CA 2806008 2018-02-26
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
89
prodrugs of the compounds of this invention include, but are not limited to,
esters,
carbonates, thiocarbonates, N-acyl derivatives, N-acyloxyalkyl derivatives,
quaternary
derivatives of tertiary amines, N-Mannich bases, Schiff bases, amino acid
conjugates,
phosphate esters, metal salts and sulfonate esters.
Liposomes can also be used as carriers for the active compounds of the present
invention.
Liposomes are micelles made of various lipids such as cholesterol,
phospholipids, fatty
acids, and derivatives thereof Various modified lipids can also be used.
Liposomes can
reduce the toxicity of the active compounds, and increase their stability.
Methods for
preparing liposomal suspensions containing active ingredients therein are
generally known
in the art. See, e.g., U.S. Patent No. 4,522,81 1; Prescott, Ed., Methods in
Cell Biology,
Volume XIV, Academic Press, New York, N. Y. (1976).
The active compounds can also be administered in combination with another
active agent
that synergistically treats or prevents the same symptoms or is effective for
another disease
or symptom in the patient treated so long as the other active agent does not
interfere with or
adversely affect the effects of the active compounds of this invention. Such
other active
agents include but are not limited to anti-inflammation agents, antiviral
agents, antibiotics,
antifungal agents, antithrombotic agents, cardiovascular drugs, cholesterol
lowering agents,
anti-cancer drugs, hypertension drugs, and the like.
Examples of antineoplastic agents that can be used in combination with the
compounds and
methods of the present invention include, in general, and as appropriate,
alkylating agents,
anti-metabolites, epidophyllotoxins, antineoplastic enzymes, topoisomerase
inhibitors,
procarbazines, mitoxantrones, platinum coordination complexes, biological
response
modifiers and growth inhibitors, hormonal/anti-hormonal therapeutic agents and
haematopoietic growth factors. Exemplary classes of antineoplastic include
the
anthracyclines, vinca drugs, mitomycins, bleomycins, cytotoxic nucleosides,
epothilones,
discodermolides, ptericlines, diynenes and podophyllotoxins. Particularly
useful members
of those classes include, for example, caiminomycin, daunorubicin,
aminopterin,
methotrexate, methopterin, dichloromethotrexate, mitomycin C, porfiromycin, 5-
fiuorouracil, 6-mercaptopurine, gemcitabine, cytosine arabinoside,
podophyllotoxin or
podo-phyllotoxin derivatives such as etoposide, etoposide phosphate or
teniposide,
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
melphalan, vinblastine, vincristine, leurosidine, vindesine, leurosine,
paclitaxel and the like.
Other useful antineoplastic agents include estramustine, carboplatin,
cyclophosphamide,
bleomycin, gemcitibine, ifosamide, melphalan, hexamethyl melamine, thiotepa,
cytarabin,
idatrexate, trimetrexate, dacarbazine, L-asparaginase, camptothecin, CPT-11,
topotecan,
5 ara-C, bicalutamide, flutamide, leuprolide, pyridobenzoindole
derivatives, interferons and
interleukins.
CA 02806008 2013-01-18
WO 2012/013728 PCT/EP2011/062949
91
General Synthetic Route Description
Compounds of Formula (I), (Ia), (lb), (II) or (III) can be synthesized in
accordance with or
in analogy to the general routes described in Schemes 1, 2 and 3. Other routes
known by
the ordinary skilled artisan, as well as other reactants and intermediates,
can also be used to
arrive at the compounds of Formula (I), (Ia), (lb), (II) or (III).
No2
Me3S(0)-1, t-BuOK 010A, yo.
n(R2) (A) (A) NO2
DMSO
(R3)n (R3)n
(1) (2)
Zn, HCI (aq.)
0 i-PrOH
(D)
(4)
(R ) NaBH(OAc)3
n(R2) (A) NH2
(D)
(R3)n (R3)n
(5) (3)
SCHEME 1: DMSO (Dimethyl sulfoxide)
Commercially available nitrostyrenes of formula (1) have been subjected to a
cyclopropanation reaction using trimetilsulfoxonium iodide and potassium
tertbutylate. The
nitro group of the resulted trans nitrocyclopropyl derivatives of folinula (2)
(being trans
S, 2R), (1R, 2S)) mixture although the individual diastereoisomers
corresponding to (1S,
2R) and (1R, 2S) can be used) has been then reduced using zinc in hydrochloric
acid to
afford the cyclopropylamino derivatives of formula (3). Later reductive
alkylation with
commercially available aldehydes of formula (4) using sodium
triacetoxyborohydride as
reducing agent leads to the formation of cyclopropylamino derivatives of
formula (5) which
are subjects of the present invention.
GA 02806008 2013-01-18
WO 2012/013728 PCT/EP2011/062949
92
0
BrN ,CHO (E10)2P(0)CH2002Et
(A) t-BuOK,THF. 0 C, 2 h BrN
(A) OEt
(R3)n
(R3)n
(6) (7)
Me3S(0)-1, NaH
DMSO, t a , 2 h
NaOH (ac)
Me0H
BrN(A)-c&õ OH t.a., 16 h
BrN ,,A.õ, OEt
(A)
0 0
(9)
(8)
1. CICO2Et, Et3N,
acetone, -15 C, 1 h
2. NaN3, water
-10 C, 20 min
0
BrN. BrN )L.
(A) õIrN3
, (A) 0"D'`
0 tBuOH
(R3)n 75 C, 20 h (R3)n
(11) n(R2)
(10)
(B)
B(OH)2
(12)
ACN, K2003, H20,
Pd(PPh3)4
0
'NH2 ,HCI / Et20
n(R2) 0
HCI
(R3)n (R3)n
(3) (13)
0 ,..(Ri)
AR-1) Br,C1 (D)
(D) (14)
(4)
NaH / DMF
NaBH(OAc)3
0
n(rN2)(A)
,HCI / Et20 n(R2),(B),A)
j==_
- 0
H
(R3)n (5) (R3)n L'(D)¨(R1)
(15)
SCHEME 2: ACN (acetonitrile), DMSO (Dimethyl sulfoxide), THF
(Tetrahydrofuran).
=
93
Commercially availables aldehydes of formula (6) were subjected to a Homer-
Wadsworth-
Emmons reaction using triethyl phosphono acetate and potassium tert-butoxide
in
tetrahydrofuran at 0 C to get the ethyl acrylate derivatives of formula (7)
which is subjected
to cyclopropanation using trimetilsulfoxonium iodide and sodium hydride in
dimethy 1
sulfoxide as a solvent, leading to (trans)-ethyl cyclopropanecarboxylate
derivatives of
formula (8) (being trans ((i S, 2R), (1R, 2S)) mixture although the individual
diastereoisomers corresponding to (1S, 2R) and (1R, 2S) can be used).
Hydrolysis to the
corresponding (trans)-cyclopropanecarboxylic acid derivatives of formula (9)
was
performed using NaOH in Me0H. Reaction, first with ethyl chloroformate and
triethylamine in acetone and later with sodium azide in water leads to the
formation of
(trans)-cyclopropanecarbonyl azide derivatives of formula (10). Reaction with
tert-butanol
results in the formation of tert-butyl (trans)-cyclopropylcarbamate
derivatives of formula
(11). The reaction with commercially available boronic acid or boronate ester
derivatives of
formula (12) using acetonitrile and water as a solvent, potassium carbonate as
a base and
Tetrakis(triphenylphospine) Paladium (0) as a catalyst leads to the formation
of tert-butyl
(trans)-cyclopropylcarbamate derivatives of formula (13).
Deprotection of the Boc-group using HC1 2M in diethyl ether using diethyl
ether as a
solvent, leads to the formation of the corresponding hydrochloride salt of the
(trans)-
cyclopropanamine derivatives of formula (9). Reductive alkylation with
commercially
available aldehides of formula (4) using sodium triacetoxyborohydride as
reducing agent
leads to the formation of cyclopropylamino derivatives of formula (5) which
are subjects of
the present invention.
The alkylation of tert-butyl (trans)-cyclopropylcarbamate derivatives of
formula (13) with
commercial available alkyl halides of formula (14) using sodium hydride as a
base and
DMF as a solvent, leads to the formation of the tert-butyl (trans)-
cyclopropylcarbamate
derivatives of formula (15). Deprotection of the Boc-group using HC1 2M in
diethyl ether
using diethyl ether as a solvent, results in the formation of the
corresponding hydrochloride
salt of the (trans)-cyclopropanamine derivatives of formula (5), which are
subject of the
present invention as defined above.
CA 2806008 2018-02-26
GA 02806008 2013-01-18
WO 2012/013728 PCT/EP2011/062949
94
(R2)ri
Br, CI¨(B1)
HO,, /CHO (17)
(R2)riN, õCHO
(A) (B) (A)
1 K2003/DMF
/ DMF ,1
(R3)0 (N3)
(16) (18)
(Et0)2P(0)CH2CO2Et
t-BuOK,THF. 0 C, 2 h
Me3S(0)-1, NaH 0
(R2)r(B)õ0(A),,Ay.0Et DMSO, t.a., 2 h (R2)n-, /0II
(B) (A) OEt
, 0 1
(R3)3
(R3i n (20)
NaOH (ac) (19)
Me0H 1. CICO2Et, Et3N,
t.a., 16 h acetone, -15 C, 1 h
2. NaN3, water
(R2)n a ' , . H __ -10 C, 20 min (R2)n,A.N3
,1
(m3)n 0 (R3)n 0
(21) (22)
tBuOH
75 C, 20 h
0
HCI / Et20 (R2)n=-,,, /0,,, A, 1
(B) (A) 'NH2 ___________ (B) (A) `-=
1 1
(R3)n HCI (R3)n
(24) (23)
0
ARt)
H (D CI
)
(4) (14)
NaBH(OAc)3 K2CO3 / DMF
,1
(R3)n
(25)
SCHEME 3: DMF (N,N-dimethylformarnide), DMSO (Dimethyl sulfoxide), THF
(Tetrahydrofuran).
The alkylation of commercially available aldehydes of fonnula (16) using
commercially
available alkyl halides of formula (17), potassium carbonate in N,N-
dimethylfon-namide
leads to the formation of the aldehyde derivatives of fonnula (18). A Homer-
Wadsworth-
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
Emmons reaction using triethyl phosphono acetate and potassium tert-butoxide
in
tetrahydrofuran at 0 C gives the ethyl acrylate derivatives of formula (19)
which are
subjected to cyclopropanation using trimetilsulfoxonium iodide and sodium
hydride in
dimethyl sulfoxidc as a solvent leading to (trans)-ethyl
cyclopropanecarboxylate derivatives
5 of formula (20). Hydrolysis to the corresponding (trans)-
cyclopropanecarboxylic acid
derivatives of formula (21) was performed using NaOH in Me0H. Reaction, first
with ethyl
chloroformate and triethylamine in acetone and later with sodium azide in
water leads to the
formation of (trans)-cyclopropanecarbonyl azide derivatives of formula (22).
Reaction with
tert-butanol results in the foimation of tert-butyl (trans)-
cyclopropylcarbamate derivatives
10 of formula (23). Boc-group deprotection using HC1 2M in diethyl ether
and diethyl ether as
a solvent leads to the formation of the corresponding hydrochloride salt of
the (trans)-
cyclopropanamine derivatives of formula (24).
Reductive alkylation with commercially available aldehydes of fonnula (4)
using sodium
15 triacctoxyborohydride as a reducing agent leads to the foimation of
(trans)-
cyclopropylamino derivatives of formula (25) which are also subjects of the
present
invention.
Alternatively, alkylation of (trans)-cyclopropanamine derivatives of fonnula
(24) with
20 commercial available alkyl halides of formula (14) using potassium
carbonate as a base and
N,N-dimethylfonnamide as a solvent also leads to the formation of (trans)-
cyclopropylamino derivatives of formula (25), which are subject of the present
invention as
defined above.
25 As known by those skilled in the art, (trans)-cyclopropylamino
derivatives of formula (5)
and (25) can also be obtained from the (trans)-cyclopropanamine derivatives of
formula (3)
and (24), respectively, by a well-known reactions (i.e, cyclization).
Optically pure or enantiomerically enriched compounds can be isolated at
various stages of
30 the synthetic procedure and can be used in subsequent steps.
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
96
Examples
The program used to generate the names corresponding to the structures in the
Example
compounds below was ChemBioDraw Ultra 11Ø1. This program named the molecules
as
the (1S,2R) configuration due to the configuration of the input structure and
the "trans" term
has been substituted in the place of the (1S,2R) term specified by the
program. The
structures depicted below for the Example compounds below are shown as having
one
particular stereochemical configuration around the cyclopropyl carbon atoms of
the
phenylcyclopropylamine core (1S, 2R). Unless stated otherwise, the compounds
synthesized in the Examples are mixtures having both configurations (1 R, 2S)
and (1S, 2R),
that is to say they are "trans" in respect to the substituents on the
cyclopropyl ring system.
This is due to the fact the cyclopropyl derivatives used as starting material
are "trans". It is
contemplated that the cis configuration starting material or the individual
diastereomers
could be used as starting material, all of which are either commercially or
synthetically
available. Thus, the invention relates to compounds of Formula (I), (Ia),
(lb), (II) or (III),
including those of the examples, that have specific stereochemical
configurations around the
cyclopropyl ring e.g., trans ((IR, 2S) and (1S, 2R)) and cis ((1R, 2R) and
(1S, 2S)). A
preferred stereochemical configuration around the cyclopropyl ring is trans.
The compounds of the examples can also be synthesized or provided in a salt
form. The
skilled artisan is aware and capable of making salt forms and/or converting
salt forms of the
compounds of the invention, e.g., compounds of Formula (I), (Ia), (lb), (H) or
(III) and those
of the Examples. In some cases the compounds of Foimula (I), (Ia), (lb), (II)
or (III) and the
Examples can be more stable as salt forms as compared to free base.
In reference to the synthetic schemes described herein the following intei __
niediates (and
analogous intermediates or derivatives thereof) can be made using the
following procedures.
. :
. =
97
Intermediate A: 1-(benzyloxy)-4-[(trans)-2-nitrocyclopropyllbenzene
NO2
0
Trimethylsulfoxonium iodide (0.62 g, 2.82 mmol) was added in portions to a
solution of t-
BuOK (0.32 g, 2.82 mmol) in dry DMSO (5 mL). After 10 min a solution of 1-
(benzyloxy)-
4-[(E)-2-nitrovinyl]benzene (0.60 g, 2.35 mmol) in DMSO (5 mL) was transferred
via
canula and the mixture was stirred at room temperature for 6 h. The reaction
was poured
over water (10 mL) and extracted with Et20 (3x10 mL); the organic layers were
washed
with brine (2x15 mL), dried over anhydrous Na2SO4 and filtered. After removal
of the
solvent, the residual orange oil was purified by column chromatography on
silica gel (5%
Et0Ac/hexanes) affording 0.16 g of 1-(benzyloxy)-4-[(trans)-2-
nitrocyclopropyl]benzene
[Rf--- 0.5 (20% Et0Ac/hexanes), white solid, 26% yield].
Intermediate B: Trans-2-[4-(benzyloxy)phenyl]cyclopropanamine
iNH2
cr
Zn dust (1.97 g, 30 mol) was added in small portions, over a period of 30 min,
to a
vigorously stirred solution of 1-(benzyloxy)-4-[(trans)-2-
nitrocyclopropyllbenzene
(Intennediate A, 0.81 g, 3.0 mmol) in i-PrOH (25 mL) and HCl (11 mL of aqueous
solution
2.7 N, 30 mmol). After 17 h the mixture was filtered through a pad of
CeliteTM, that was
washed with 10 mL of methanol. The filtrate was concentrated and 10 mL of
water were
added, washing with CH2C12 (3x15 mL). The organic layers were dried over
anhydrous
Na2SO4 and filtered. After removal of the solvent, the crude product was
purified by
column chromatography on silica gel (10% Me0H/CH2C12) affording 0.50 g of
(trans)-2-[4-
(benzyloxy)phenyl]cyclopropanamine [Rf= 0.2 (10% Me0H/CH2C12), white solid,
70%
CA 2806008 2018-02-26
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
98
yield]. 1H¨NMR (Me0H, 250 MHz, 8): 7.45-7.27 (m, 5HõAsH); 6.96 (d, ./--= 8.5
Hz, 2H,
ArH); 6.86 (d, .1= 8.5 Hz, 2H, ArH); 5.03 (s, 2H, CH2); 2.41-2.34 (m, 1H, CH);
1.86-1.76
(m, 1H, CH); 0.98-0.85 (m, 2H, CH2).
Intermediate C: (E)-ethyl 3-(6-bromopyridin-3-yl)acrylate
0
0 B Et
r N
Triethyl phosphonoacetate (26.6g, 118.8 mmol) was added slowly dropwise to a
mixture of
Potassium-tert-butoxide (14.5g, 129.6 mmol) in dry THE (200 mL) at -5 C,
stirred for 20
mm and then a solution of 6-bromopyridine-3-carboxaldehyde (20 g, 108 mmol) in
dry THF
(100 mL) was added slowly dropwise at -5 C and stirred for 30 min. After
completion, the
reaction mixture was poured into ice water (350 mL) and extracted with Et0Ac
(2 x 300
mL). The combined organic extracts were washed with saturated NaHCO3 solution
(250
mL), water (250 mL) and brine (250 mL) and dried over anhydrous Na2SO4,
filtered and
evaporated to get (E)-ethyl 3-(6-bromopyridin-3-y1) acrylate (20 g, 72.9 %) as
brown color
liquid. This is carried to next step without further purification.
Intermediate D: (Trans)-ethyl-2-(6-bromopyridin-3 -ypcycloprop anecarb oxyl
ate
Br 0
N
Trimethyl sulfoxonium iodide (20.8g, 94.7 mmol) was added in small portions to
a
suspension of sodium hydride (4g, 170.6 mmol) in dry DMSO (400 mL) at rt.,
stirred for 1 h
until clear solution was obtained. A solution of (E)-ethyl 3-(6-bromopyridin-3-
y1) acrylate
(Intermediate C. 20 g, 78.7 mmol) in dry DMSO (20 mL) was added and stirred
for 4 h.
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
99
After completion, the reaction mixture was poured into ice water (700 mL),
extracted with
Et0Ac (2 x 350 mL). The combined organic extracts were washed with water ( 250
mL),
brine (250 mL) and dried over anhydrous Na2SO4, filtered and evaporated to
give (trans)-
ethy1-2-(6-bromopyridin-3-yl)cyclopropanecarboxylate (10g, 47 %) as brown
liquid.
Intermediate E: (Trans)-2-(6-bromopyridin-3-yl)cyclopropanecarboxylic acid
hydrochloride
,r0H
.-/.A. 0
Br 1N
.HCI
NaOH 4N solution (60 mL) was added to a solution of (trans)-ethy1-2-(6-
bromopyridin-3-
yl)cyclopropanecarboxylate (Intermediate D, 10 g, 37.1 mmol) in methanol (100
mL) and
the reaction mixture was stirred at RT for 4 h. After completion, the solvent
was evaporated
and the residue was diluted with ice water (250 mL) and acidified with 4 N HCI
solution,
the aqueous layer was extracted with Et0Ac (2 x 350 mL). The combined organic
extracts
were washed with water ( 250 mL), brine (250 mL) and dried over anhydrous
Na2SO4,
filtered and evaporated to give (trans)-2-(6-bromopyridin-3-
yl)cyclopropanecarboxylic acid
hydrochloride (5g, 55.8 %) as a light brown color solid.
Intermediate F: (Trans)-2-(6-bromopyridin-3-yl)cyclopropanecarbonyl azide
I Br IrN3
..
,,,CTA
0
N
Ethyl chloroformate (5.8 mL, 62 mmol) was added to a solution of (trans)-2-(6-
bromopyridin-3-yl)cyclopropanecarboxylic acid hydrochloride (Intermediate E, 5
g, 20.7
mmol) and Etdsl (14,2 mL, 103.7 mmol) in Acetone (100 mL) at -5 C, then
reaction
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
100
mixture was stirred at -5 C for 1 h, then a solution of NaN3 (2.7g, 41.4
mmol) in water (10
mL) was added and stirred for 30 mins at RT. After completion the solvent was
evaporated
under vacuum. The crude residue was dissolved in ethyl acetate (200 mL),
washed with
water (80 mL), brine (80 mL), dried over anhydrous Na2SO4, filtered and
evaporated to get
(trans)-2-(6-bromopyridin-3-yl)cyclopropaneearbonyl azide (2.5 g, 45.5 %) as a
brown
color gummy liquid.
Intermediate G: tert-butyl (trans)-2-(6-bromopyridin-3-yl)cyclopropylcarbamate
0
N 0
lo Br
A solution of (trans)-2-(6-bromopyridin-3-yl)cyclopropanecarbonyl azide
(Intermediate F,
2.5 g, 9.36 mmol) in tert-butanol (80 mL) was heated at 90 C for 16 h. After
completion,
the solvent was evaporated under vacuum and the residue was taken in water
(100 mL) and
extracted with Et0Ac (2 x 100 mL). The combined organic extracts were washed
with water
(100 mL), brine (100 mL) and dried over anhydrous Na2SO4, filtered and
evaporated. The
crude residue was purified by flash column chromatography (SiO2) by eluting
with Et0Ac:
Hexane (2: 8) to get tert-butyl (trans)-2-(6-bromopyriclin-3-
yl)cyclopropylcarbamate (1.1g,
37.5 %) as a light yellow solid. 1H-NMR (CDC13) 6 (ppm): 1.16 (q, 1H), 1.23
(quin, 1H),
1.45 (s, 9H), 2.01 (m, 1H), 2.69 (m, 1H), 4.88 (br, 1H), 7.36 (s, 2H), 8.20
(s, 1H).
Intemediate H: (E)-ethyl 3-(4-bromophenyl)acrylate
0
OEt
Br
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
101
A solution of triethyl phosphonoacetate (13.1 g, 0.0589 mol) was added slowly
(dropwise) to a solution of Potassium-tert-butoxide (6.59 g, 0.0589 mol), in
dry
THF (150 mL) at -5 C, stirred for 30-45 mins at the same temperature, then a
solution of 4-Bromo benzaldehyde (10 g, 0.054 mol), in dry THF (50 inL) was
slowly added dropwise at -5 C over a period of 15 mins, stirred the reaction
mixture for 30 mins at the same temperature. After completion of reaction by
TLC,
the reaction mixture was poured into ice water (300 mL), extracted with Et0Ac
(2
x 200 mL). The combined organic extracts were washed with sat NaHCO3 solution
(200 mL), water (200 mL), brine (200 mL) and dried over anhydrous Na2SO4,
filtered and evaporated to get crude (E)-ethyl 3-(4-bromophenyl) acrylate (10
g, 72
%) as pale green liquid. This is carried to next step without further
purification.
Intermediate I: (Trans)-ethyl 2-(4-bromophenyl)cyclopropanecarboxylate
Ir0Et
0
Br
Trimethyl sulfoxonium iodide (5.19 g, 0.0236 mol) was added slowly in small
portions
over a period of 20 min. to a suspension of sodium hydride (0.44 g, 0.0236
mol) in dry
DMSO (80 mL) at rt, stirred for 1 h, till the formation of clear solution.
Then a solution of
(E)-ethyl 3(4-bromophenyl) acrylate (Intermediate H, 5 g, 0.01968), in dry
DMSO (20 mL)
was added slowly dropwise, stirred at rt for 30 mins. After completion of
reaction, checked
by TLC, the reaction mixture was poured into ice water (200 mL), extracted
with Et0Ac (2
x 150 mL). Combined organic extracts were washed with ice water (2 x 150 mL),
brine (150
mL), dried over anhydrous Na2SO4, filtered and evaporated to get (trans)-ethyl
2-(4-
bromophenypcyclopropanecarboxylate (4 g, 75.9 %) as a green liquid. The crude
is carried
to next step without further purification.
Intennediate J: (Trans)-2-(4-bromophenyl)cyclopropanecarboxylic acid
CA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
102
A
Br' '0õir.0 FI
Br
0
Na OH 4N (20 mL) was added to a solution of (trans)-ethyl 2-(4-
bromophenyl)cyclopropanecarboxylate (Intermediate I, 4 g, 0.0149 mol), in
Methanol (40
mL) and stirred at rt for 2 h. After completion of reaction, checked by TLC,
the solvent was
evaporated and the residue was diluted with water (50 mL), acidified with HC1
4 N solution,
the solid formed was filtered and dried to get (trans)-2-(4-
bromophenyl)cyclopropanecarboxylic acid (2.59 g, 72 %), as a white solid.
Intermediate K: (Trans)-2-(4-bromophenyl)cyclopropaneearbonyl azide
A
Br0
0
Ethyl chloroformate (1.9 mL) was added to a solution of (trans)-2-(4-
bromophenyl)
cyclopropanecarboxylic acid (Intermediate J, 4 g, 0.0165 mol) and Et3N (2.51
mL, 0.0199
mol) in acetone (60 mL) at -20 C, stirred at same temperature for 1 h, then a
solution of
NaN3 (1.3 g, 0.0199 mol) in water (5 mL), was added and stirred for 30 mins at
rt. After
completion of reaction, checked by TLC, the solvent was evaporated and crude
residue was
dissolved in ethyl acetate (100 mL), washed with water (40 mL), dried over
anhydrous
Na2SO4, filtered and evaporated to get (trans)-2-(4-
bromophenyl)cyclopropanecarbonyl
azide (4 g). The crude residue is carried to next step without further
purification.
Intermediate L: tert-butyl (trans)-2-(4-bromophenyl)cyclopropylcarbamate
A o
-A
0 ,
gq*N0-i'=
H
Br
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
103
A solution of (trans)-2-(4-bromophenyl) cyclopropanecarbonyl azide
(Intermediate K, 4 g)
in tert-Butanol (40 mL) was heated at 90 C for 16 h. After completion of
reaction, checked
by TLC, the solvent was evaporated residue was poured into water (50 mL),
extracted with
Et0Ac (2 x 50 mL). The combined organic extracts were washed with water (50
mL), brine
(50 mL), dried over anhydrous Na2S0.4, filtered and evaporated. The crude
residue was
purified by column chromatography (SiO2) by eluting with Et0Ac: Pethroleum
ether (2:
98), to get tert-butyl (trans)-2-(4-bromophenyl)cyclopropylcarbamate (2.5 g,
48 % overall 2
steps) as a white solid. 1H-NMR (CDC13, 250 MHz) 6 (ppm): 1.07-1.19 (m, 2H),
1.44 (s,
9H); 2.05-1.94 (m, 1H); 2.72-2.62 (m, 1H); 4.85 (br, 1H); 7.09-6.96 (m, 2H);
7.44-7.33 (m,
2H).
Intermediate M: Ethyl 5-((tert-b utoxyc arbonyl)amino)-1,3,4-ox adiazole-2-
carboxylate
N¨N
N,Boc
0
r
Sodium hydride (280 mg, 0.007 mol) in DMF (10 mL) was added to a suspension of
Ethyl
5-amino-1,3,4-oxadiazole-2-carboxylate (1 g, 0.006 mol) in DMF (2 mL) at 0 C,
stirred for
10 mins, then Di tert-butyl dicarbonate (1.65 g, 0.0076 mol) was added and
stirred at RI for
16 h. After completion, the reaction mixture was poured into ice water (25 mL)
and
extracted with Et0Ac (3 x 25 mL). The combined extracts were washed with cold
water (2
x 25 mL), brine (25 mL), dried over anhydrous Na2SO4, filtered and evaporated.
The crude
residue was purified by column chromatography (SiO2) using Et0Ac: Petroleum
ether (1:3)
as eluent to get Ethyl 5-((tert-butoxycarbonyeamino)-1,3,4-oxadiazole-2-
carboxylate (900
mg, 56.2 %) as a white solid.
Intermediate N: Tert-butyl (5-(hydroxymethyl)-1,3,4-oxadiazol-2-yl)carbamate
:
104
N¨N
HO
NaBH4 (330 mg, 0.0087 mol) was added to a solution of Ethyl 5-((tert-
butoxycarbonyl)amino)-1,3,4-oxadiazole-2-carboxylate (Intermediate M, 900 mg,
0.0035
mol) in THF (18 mL) at 0 C and stirred at RT for 16 h. After completion, the
solvent was
evaporated and the residue was taken in water (15 mL) and extracted with Et0Ac
(3 x 20
mL). The combined extracts were washed with water (20 mL), brine (20 mL),
dried over
anhydrous Na2SO4, filtered and evaporated. The crude residue was purified by
column
chromatography (SiO2) using Et0Ac: Petroleum ether (8: 2) as eluent to get
tert-butyl (5-
(hydroxymethyl)-1,3,4-oxadiazol-2-y1) carbamate (450 mg, 54.2 %) as a white
solid.
Intermediate 0: Tert-butyl (5-formy1-1,3,4-oxadiazol-2-yl)carbamate
N¨N
Boc
0
0
Mn02 (500 mg) was added to a solution of tert-butyl (5-(hydroxymethyl)-1,3,4-
oxadiazol-2-
y1) carbamate (Intermediate N, 450 mg, 0.0021 mol) in THF (9 mL) at RT and
stirred for 16
h. After completion, the reaction mixture was filtered through a pad of
CeliteTM and the
filtrate was evaporated to get crude tert-butyl (5-formy1-1,3,4-oxadiazol-2-
y1)carbamate
(250 mg). This crude was carried to next step without further purification.
Intermediate P: 4-(benzyloxy)benzaldehyde
CHO
Bn0
Potassium Carbonate (678 g, 4.91 mol) was added to a solution of 4-
hydroxybenzaldehyde
(200 g, 1.63 mol) in DMF (2 L) followed to the addition of benzyl bromide (214
mL, 1.80
CA 2806008 2018-02-26
GA 02806008 2013-01-18
WO 2012/013728 PCT/EP2011/062949
105
mol) at 0 C and stirred for 18 h at RT. After completion, the reaction
mixture was poured
into ice water (3 L), filtered the solid and dried to get 4-
(benzyloxy)benzaldehyde (230 g, 66
%).
Intermediate Q: (E)-ethyl 3-(4-(benzyloxy)phenyeacrylate
0
0 Et
Bn0
Triethyl phosphonoacetate (259 mL, 1.3 mol) was added slowly drop wise to a
solution of
Potassium-tert-butoxide (145 g, 1.29 mol) in dry THF (2 L) at -5 C and
stirred for 30-45
mins. Then a solution of 4-(benzyloxy)benzaldehyde (Intermediate P, 230 g,
1.08 mol) in
dry THF ( 1.5 L) was added slowly drop wise at -10 C over a period of 15 mins
and stirred
for 30 mins. After completion, the reaction mixture was poured into ice water
(1 L) and
extracted with Et0Ac (2 x 1.5 L). The combined organic extracts were washed
with sat
NaHCO3 solution (1 L), water (1 L), brine (1 L), dried over anhydrous Na2SO4,
filtered and
evaporated to get crude (E)-ethyl 3-(4-(benzyloxy)phenyl)acrylate (290 g, 95
%). The crude
was carried to next step without further purification.
Intermediate R: (Trans)-ethyl 2-(4-(benzyloxy)phenyl)cyclopropanecarboxylate
11
Bn0 0
Trimethyl sulfoxonium iodide (224 g, 1.02 mol) was added portion wise to a
suspension of
Nall (40.8 g, 1.02 mol) in dry DMSO (2 L) at RT over a period of 20 min and
stirred for 1 h
till the formation of a clear solution. A solution of (E)-ethyl 3-(4-
(benzyloxy) phenyl)
acrylate (Intermediate Q, 240 g, 0.85 mol) in dry DMSO (2 L) was added drop
wise and
stirred at RT for 30 mins. After completion, the reaction mixture was poured
into ice water
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
106
(2 L), extracted with Et0Ac (2 x 1 L). Combined organic extracts were washed
with ice
water (1 L), brine (1 L), dried over anhydrous Na2SO4, filtered and evaporated
to afford
(Trans)-ethyl 2-(4-(benzyloxy)phenyl)cyclopropanecarboxylate (142 g, 58.6 %)
as an off
white solid. The crude was carried to next step without further purification.
Intermediate S: (Trans)-2-(4-(benzyloxy)phenyl)cyclopropanecarboxylic acid
OH
Bn0 0
4N NaOH solution (4 L) was added to a solution of (trans)-ethyl 2-(4-
(benzyloxy)phenyl)cyclopropanecarboxylate (Intermediate R, 250 g, 0.844 mol)
in
Methanol (1.2 L) at 0 C and stirred at RT for 4 h. After completion, the
solvent was
evaporated, the residue was diluted with water (1 L), acidified with 4 N 1-IC1
solution,
extracted with Et0Ac (2 x 2 L). Combined organic extracts were washed with
water (1 L),
brine ( 1 L), dried over anhydrous Na2SO4, filtered and evaporated to afford
(trans)-2-(4-
(benzyloxy)phenyl)cyclopropanecarboxylie acid (190 g, 84 %) as off white
solid. The crude
was carried to next step without further purification.
Intermediate T: (Trans)-2-(4-(benzyloxy)phenyl)cyclopropanecarbonyl azide
N3
Bn0
Ethyl chloroformate (143 mL, 1.48 mol) was added to a solution of (trans)- 2-
(4-
(benzyloxy) phenyl) cyclopropanecarboxylic acid (Intermediate S, 190 g, 0.70
mol),
Triethyl amine (229 mL, 1.63 mol) in acetone ( 2.8 L) at -20 C and stirred
for 1 h, then a
solution of NaN3 ( 138 g, 2.1 mol) in water (200 mL) was added and stirred at
RT for 30
mins. After completion, the solvent was evaporated, residue was dissolved in
Et0Ac (2 L),
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
107
washed with water (2 L), brine ( 1 L), dried over anhydrous Na2SO4, filtered
and evaporated
to afford (trans)-2-(4-(benzyloxy)phenyl)cyclopropanecarbonyl azide ( 178 g,
85.9 %).
Intermediate U: Tert-butyl ((trans)-2-(4-
(benzyloxy)phenyl)cyclopropyl)carbamate
0
IN 0
Bn0
A solution of (trans)-2-(4-(benzyloxy)phenyl)cyclopropanecarbonyl azide
(Intermediate T,
178 g, 0.64 mol) in tert-butanol (2.6 L) was heated at 90 C for 16 h. After
completion, the
solvent was evaporated and the crude residue was purified by column
chromatography by
using (SiO2) Et0Ac: Pet ether (4: 96) to get tert-butyl ((trans)-2-(4-
(benzyloxy)phenyl)cyclopropyl)carbamate (78 g, 37.8 %) as off-white solid.
Intermediate V: (Trans)-2-(4-(benzyloxy)phenyl)cyclopropanamine hydrochloride
'''NH2
Bn0 HCI
HC1 in Dioxane (390 ml) was added to a solution of tert-butyl ((trans)-2-(4-
(benzyloxy)phenyl)cyclopropyl)carbamate (Intermediate U, 78 g, 0.23 tnol) in
1,4-dioxane
(780 mL) at 0 C and stirred at RT for 12 h. After completion, the solvent was
evaporated
and the residue was triturated with diethyl ether (1 L) followed by hexane (1
L) to give
(trans)-2-(4-(benzyloxy)phenyl)cyclopropanamine hydrochloride (55 g, 87 %) as
off-white
solid.
Intermediate W: ethyl 2-amino-2-thioxoacetate
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
108
0
N H2
P2S5 (28.5 g, 128 mmol) was added portion wise to a solution of ethyl 2-amino-
2-oxoacetate
(30 g, 25.6 mmol) in pyridine (300 mL) over a period of 30 mins, and stirred
at 90 C for 3
h. After completion, the solvent was evaporated, the residue was diluted with
water (300
mL) and extracted with Et0Ac (2 x 300 mL). The combined extracts were washed
with
water (2 x 200 mL), brine (200 mL) and dried over anhydrous Na2SO4, filtered
and
evaporated. The crude was purified by column chromatography (SiO2) by eluting
(1:9)
Et0Ac:Hexane to afford ethyl 2-amino-2-thioxoacetate (18 g, 52.9 %) as white
solid.
Intermediate X: 2-(ethoxycarbonyl)thiazole-5-carboxylic acid
N
0 OH
Bromopyruvic acid (22.7 g, 135.33 mmol) was added to a solution of ethyl 2-
amino-2-
thioxoacetate (Intermediate W, 18 g, 135.3 mmol) in dioxane (200 mL) and
refluxed for 5 h.
After completion the reaction mixture was poured into water (200 mL), the
residue was
basified with sat NaHCO3 and extracted with Et0Ac (2 x 250 mL). The aqueous
layer was
acidified with 2N HC1 and extracted with Et0Ac (2 x 250 mL). The combined
extracts were
washed with water (250 mL), brine (250 mL), dried over anhydrous Na2SO4,
filtered and
evaporated to afford 2-(ethoxycarbonyl)thiazole-5-carboxylic acid (13 g
crude). The crude
was carried to next step without further purification.
Intermediate Y: Ethyl 5-(azidocarbonyl)thiazole-2-carboxylate
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
109
N
0 N3
Ethyl chloroformate (9.8 g, 83.6 mmol) was added to a solution of 2-
(ethoxycarbonyl)thiazole-5-carboxylic acid (Intermediate X, 13 g, 64.67 mmol),
TEA (9.79
g, 97.01 mmol) in acetone (130 mL) at -20 C, stirred for 1 h, then a
solution of NaN3 (5.4
g, 83.6 mmol) in water (15 mL) was added and stirred at RT for 30 mins. After
completion,
the solvent was evaporated, the crude residue was diluted with water (150 mL)
and
extracted with Et0Ac (2 x 150 mL). The combined extracts were washed with
water (100
mL), brine (100 mL), dried over anhydrous Na2SO4, filtered and evaporated to
afford Ethyl
5-(azidocarbonyl)thiazole-2-carboxylate (11 g crude) as brown liquid. The
crude was
carried to next step without further purification.
Intermediate Z: Ethyl 5-((tert-butoxycarbonyl)amino)thiazole-2-carboxylate
0
A solution of ethyl 5-(azidocarbonyl)thiazole-2-carboxylate (Intermediate Y,
11 g, 48.6
mmol) in tert-butanol (150 mL) was refluxed at 90 C for 16 h. After
completion, the
solvent was evaporated. The crude residue was purified by column
chromatography by
using (SiO2), eluting with Et0Ac: Petroleum ether (2:98) to afford Ethyl 5-
((tert-
butoxycarbonyl)amino)thiazole-2-carboxylate (4 g, 30.23 %) as white solid.
Intermediate AA: Tert-butyl (2-(hydroxymethyl)thiazol-5-yl)carbamate
NHBoc
HO
:
_
110
NaBH4 (1.1 g, 29.2 mmol) was added portion wise to a solution of Ethyl 5-(tert-
butoxycarbonyl amino)thiazole-2-carboxylate (Intermediate Z, 4 g. 14.6 mmol)
in Me0H
(40 mL) at 0 C over a period of 30 mins and stirred at RT for 16 h. After
completion,
solvent was evaporated, the solid residue was dissolved in ice water (50 mL)
and extracted
with Et0Ac (2 x 50 mL). The combined extracts were washed with water (50 mL),
brine
(50 mL), dried over anhydrous Na2SO4, filtered and evaporated. The crude was
purified by
column chromatography by using SiO2, eluting with Et0Ac:Petroleum ether (2:8)
to afford
Tert-butyl (2-(hydroxymethyl)thiazol-5-yl)carbamate (2.9 g, 84.8 %) as white
solid.
Intermediate AB: Tert-butyl (2-formylthiazol-5-yl)carbamate
HJ
NHBoc
0
Mn02 (1.5 g, 18.2 mmol) was added to a solution of Tert-butyl (2-
(hydroxymethyl)thiazol-
5-yl)carbamate (Intermediate AA, 700 mg, 3.04 mmol) in DCM (15 mL) and stirred
at RT
for 16 h. After completion reaction mixture was diluted with DCM, filtered
through
CeliteTM. The filtrate was concentrated under vacuum to afford Tert-butyl (2-
formylthiazol-
5-yecarbamate (500 mg crude). The crude was carried to next step without
further
purification.
The compounds described in examples 1-24 are racemic, that is to say a 50:50
mixture of
the enantiomers corresponding the trans racemate.
Example 1: 5-(((trans)-2-(4-
(benzyloxy)phenyl)cyclopropylamino)methyl)pyrimidin
-2-amine
N
0 NH2
CA 2806008 2018-02-26
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
1 1 1
Sodium triacetoxy borohydride (883 mg, 4.166 mmol) was added slowly at 0 C to
a
solution of (trans)-2-(4-(benzyloxy)phenyl)cyclopropanamine (Inteimediate B,
500 mg,
2.083 mmol), 2-aminopyrimidine-5-carbaldehyde (256 mg, 2.083 mmol) in DCE (10
mL)
and stirred for 20 h. After completion, the solvent was evaporated. The
residue was
dissolved in Methanol (15 mL), NaBH4 (237 mg, 6.249 mmol) was added slowly at
0 C
and stirt-ed for 3 h. After completion, the solvent was evaporated, the
residue was dissolved
in ice water (20 mL) and extracted with Et0Ac (2 x 20 mL). The combined
organic layers
were washed with brine (20 mL) and dried over anhydrous Na2SO4, filtered and
evaporated.
The crude residue was purified by prep HPLC to afford 5-(((trans)-2-(4-
(benzyloxy)phenyl)cyclopropylamino)methyl)pyrimidin-2-amine (180 mg, 25 %) as
white
solid.1H-NMR (400 MHz, DMSO-d6) 6 (ppm): 0.85 (q, 1H), 0.90 (quin, 1H), 1.73
(m, 1H),
2.07 (m, 1H), 2.75 (brs, 1H), 3.53 (s, 2H), 5.04 (s, 2H), 6.46 (s, 2H), 6.85
(d, 2H), 6.92 (d,
2H), 7.33 (m, 1H), 7.42 (m, 4H), 8.11 (s, 2H). Mass (M+H): 347.3
Following example has been synthesized using the procedure described for
Example 1 and
the corresponding starting materials.
Example 2: 5-(((tran s)-2-(4-(benzyl oxy)ph
enyl)cyclopropylamino)methypthiazol-2 -amine
hydrochloride
S-2(
0 HCI
NH2
1H-NMR (400 MHz, DMSO-d6) 6 (ppm): 1.22 (q, 1H), 1.48 (quin, 1H), 2.46 (in,
1H), 2.80
(br, 1H), 4.35 (s, 2H), 5.08 (s, 2H), 6.93 (d, 2H), 7.06 (d, 2H), 7.32 (m,
2H), 7.40 (m, 4H),
8.98 (br, 1H), 9.90 (br, 2H). Mass (M+H): 351.9
The following compounds can be synthesized following the methodology described
in
Scheme 1 and 2 or other synthetic routes known to the ordinary skilled
artisan.
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
112
Example 3: 5-(((trans)-2-(6-(3-(trifluoromethyl)phenyl)pyridin-3-
yl)cyclopropylamino)methyl)pyrimidin-2-amine
A
I H
S
N N N H2
C F3
Example 4: 5-(((trans)-2-(6-(3-
(trifluoromethyl)phenyl)pyridin-3-
yl)cyclopropylamino)methypthiazol-2-amine
A,
...,... ,,
I 5 _,õ H
S ---.1/
N-
N H2
CF3
Example 5: 3-(5-((trans)-24(2-aminopyrimidin-5-yemethylaminoleyclopropyl)midin-
2-
y1)phenol
=/,
- N
f2\
5
OH
Example 6: 3-(5-((trans)-24(2-aminothiazol-5-
yl)methylamino)cyclopropyl)pyridin-2-
yl)phenol
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
113
N N
S
101 \ (-1
N H 2
OH
Example 7: 4'-((trans)-2((2-aminopyrimidin-5-
yl)methylamino)cyclopropyl)bipheny1-3-ol
/10 N
1110
N N H2
0 H
Example 8: 4'-((trans)-24(2-aminothiazol-5-yl)methylamino)cyclopropyl)bipheny1-
3-ol
,,
S
41101 N H2
LI
Lin
Example 9: 5-(((trans)-2-(4-(benzyloxy)phenyl)cyclopropylamino)methyl)-1,2,4-
oxadiazol-
3-amine
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
114
0 '//1\1"--%syN NH2
Example 10: 5-(((trans)-2-(4-(benzyloxy)phenyecyclopropylamino)meth
y1)-1,3,4-
oxadi azol-2-amine
',ill .'"=...õ.r.:3N,
11110
NH2
This compound can be synthesized following scheme 1 or scheme 3, or other
synthetic
routes known to the ordinary skilled artisan.
Scheme 1 Procedure
Step 1:
Tert-butyl (5-formy1-1,3,4-oxadiazol-2-yl)carbamate (Intermediate 0, 220 mg,
1.041 mmol)
and sodium triacetoxy borohyride (441 mg, 2.08 mmol) was added to a solution
of Trans-2-
[4-(benzyloxy)phenyl]cyclopropanamine (Intermediate B, 250 mg,1.041 mmol) in
dry
Dichloro ethane (2.5 mL) at 0 C and stirred at RT for 24 h, then the solvent
was
evaporated. The residue was taken in Me0H (2.5 mL) and NaBH4 (116 mg, 3.138
mmol)
was added at 0 'C and stirred for 2 h at RT. After completion, the solvent was
evaporated,
the residue was taken in water (10 mL) and extracted with Et0Ac (4 x 10 mL).
Combined
extracts were washed with water (10 mL), brine (10 mL), dried over anhydrous
Na2SO4,
filtered and evaporated. The residue was purified by column chromatography
(Si02) using
MeOH:CHC13 (1:99) to get tert-butyl (5-((((trans)-2-(4-
(benzyloxy)phenyl)cyclopropyl)aminolmethyl)-1,3,4-oxadiazol-2-ypearbamate (70
mg,
15.3 %) as pale green liquid.
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
115
Step 2:
HC1 in 1, 4 dioxane (1 mL) was added to a solution of tert-butyl (54((trans)-2-
(4-
(benzyloxy)phenyl)cyclopropyl)amino)methyl)-1,3,4-oxadiazol-2-yl)carbamate
(100 mg) in
1, 4 dioxane (1 mL) at 0 C and stirred for 18 h. After completion, the
solvent was
evaporated and residue was dissolved in water (10 mL), basified with Na2CO3
solution,
extracted with Et0Ac (3 X 5 mL). The combined extracts were washed with water
(5 mL),
brine (5 mL), dried over anhydrous Na2SO4, filtered and evaporated. The crude
residue was
purified by column chromatography using MeOH: CHC13 (5:95) as eluent to afford
5-
(((trans)-2-(4-(benzyloxy)phenyl)cyclopropylamino)methyl)-1,3,4-oxadiazol-2-
amine (40
mg, 52 %) as a white solid.
3F1-NMR (400 MHz, DMSO-d6) 8 (ppm): 0.85 (m, 2H), 1.72 (m, 1H), 2.2 (m, 1H),
3.0 (m,
1H), 3.75 (s, 2H), 5.08 (s, 2H), 6.8-7.0 (m, 6H), 7.4 (m, 5H); Mass (M+H):
337.1
Scheme 3 Procedure
This compound can be synthesized following the same method as described in the
Scheme 1
procedure but, in Step 1, the intermediate V is used instead of intermediate
B.
The following compounds can be synthesized following the method described for
example
10 using Scheme 3 procedure and the corresponding commercial available alkyl
halides to
get the suitable Intermediate P derivatives.
Example 11: 5-((((trans)-2-(444-
fluorobenzypoxy)phenyl)cyclopropyl)amino)methyl)-
1,3,4-oxadiazol-2-amine
0 110 \N
NH2
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
116
'fINMR (400 MHz, DMSO d6) 6 (ppm): 0.84 (m, 2H), 1.79 (m, 1H), 2.20 (m, 1H),
3.12 (m,
111), 3.78 (s, 111), 5.02 (s, 211), 6.85-7.00 (m, 611), 7.2 (t, 2H), 7.46 (t,
2H); Mass (M-H):
353.3
Example 12: 5-((((trans)-2-(44(3-
fluorobenzypoxy)phenyl)cyclopropyl)amino)methyl)-
1,3,4-oxadiazol-2-amine
0-4
0
NH2
'HNMR (400 MHz, DMSO d6) 6 (ppm): 0.85 (m, 211), 1.73 (m, 1H), 2.19 (m, 111),
3.00 (m,
1H), 3.75 (s, 2H), 5.07 (s, 2H), 6.85-7.00 (m, 611), 7.14 (t, 1H), 7.25 (t,
2H), 7.41 (m, 111);
Mass (M-H): 353.3
Example 13: 5-((((trans)-2-(4-((3,5-
difluorobenzyl)oxy)phenyecyclopropyeamino)methyl)-
1,3,4-oxadiazol-2-amine
\N
0
N H2
1117'MR (400 MHz, DMSO d6) 6 (ppm): 0.87 (m, 2H), 1.75 (m, 1H), 2.20(m, 1H),
3.04 (m,
1H), 3.75 (s, 211), 5.13 (s, 2H), 6.80-7.05 (m, 6H), 7.16 (m, 311); Mass (M-
41): 373.0
117
Example 14: 5-((((trans)-2-(44(4-
chlorobenzyl)oxy)phenyl)cyclopropypamino)methyl)-
1,3,4-oxadiazol-2-amine
N
0
NH2
CI
1HNMR (400 MHz, DMSO d6) ö (ppm): 0.86 (m, 2H), 1.73 (m, 1H), 2.18 (m, 1H),
2.98 (m,
1H), 3.75 (s, 2H), 5.05 (s, 2H), 6.82-6.95 (m, 6H), 7.44 (m, 4H); Mass (M-H):
369.0
Example 15: 5-((((trans)-2-(44(3-
chlorobenzyl)oxy)phenyl)cyclopropyl)amino)methyl)-
1,3,4-oxadiazol-2-amine
N
0
NH2
CI
1HNMR (400 MHz, DMSO d6) 6 (ppm): 0.85 (m, 2H), 1.73 (m, 1H), 2.19 (m, 1H),
3.00 (m,
1H), 3.75 (s, 2H), 5.07 (s, 2H), 6.84-7.02 (m, 6H), 7.39-7.54 (m, 4H); Mass
(M+H): 371.0
The following compounds can be synthesized following the methodologhy
described in
Scheme 1, 2 and 3. Alternatively, as known by those skilled in the art, the
following
compounds can also be obtained from the (trans)-cyclopropanamine derivatives
of formula
(3) and (24), respectively, by a well-known reaction (i.e., heterocycle
formation or
cyclization)
CA 2806008 2018-02-26
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
1 1 8
Example 16: 5-((((trans)-2-(44(2-
fluorobenzyl)oxy)phenyl)eyelopropyl)amino)methyl)-
1,3,4-oxadiazol-2-amine
0-1(
0
NH2
Example 17: 54((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)-N-
methyl-
1,3,4-oxadiazol-2-amine
0 /
)--NH
NN
0
Example I 8: N-(5-((((trans)-2-(4-(benzyl ox y)phenyl)cycloprop
ylIamino)methyl)- 1 ,3 ,4-
oxadiazol-2-ypacetamide
0\
0
Example 19: 4'-((trans)-2-(((5-amino-1,3,4-oxadiazol-2-
yl)methyl)amino)eyelopropyl)-
[1,1'-biphenyl]-3-ol
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
119
pir0
c )--N H2
N-N
OH
Example 20: 5-((((trans)-2-(6-(3-(trifluoromethyl)phenyl)pyridin-3-
yl)eyclopropyl)amino)methyl)-1,3,4-oxadiazo1-2-amine
0
N-N
C F 3
Example 21: 5-(qtrans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)-1,3,4-
thiadiazol-2-amine
N
/10 0
NH2
Example 22 2-((((trans)-2-(4-
(benzyloxy)phenyl)cyclopropyl)amino)methyl)thiazol-5-
amine
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
120
NH
N
Example 23: 4-((((trans)-2-(3'-(trifluoromethyl)-{1,1'-biphenyl]-4-
y1)cyclopropyl)amino)methyl)thiazol-2-amine
5
=
CF3
Example 24: 2-((((trans)-2-(4-(benzyloxy)phenypeyelopropyl)amino)methyl)oxazol-
5-
amine
0
H2
N
0
Example 25: 3-((((trans)-2-(4-
(benzyloxy)phenyl)cyclopropyl)amino)methyl)isoxazol-5-
amine
HN \ NH2
. .
0
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
121
Example 26: 5-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)-N,N-
dimethyl-1,3,4-oxadiazol-2-amine
o
N¨
/
Example 27: 3-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyparnino)methyl)-
1,2,4-
oxadiazol-5-amine
iI10 N-0
Example 28: 5-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)-
1,2,4-
thiadiazol-3-amine
H 2
Example 29: 5-((((trans)-2-(4-
(benzyloxy)phenyl)cyclopropyl)amino)methyl)pyridin-2-
amine
101 0
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
122
Example 30: 6-((((trans)-2-(4-
(benzyloxy)phenyl)cyclopropypamino)methyl)pyridazin-3-
amine
0
Example 31: 5-0((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyppyrazin-
2-
amine
H NI
0 NH2
Example 32: 2-((((trans)-2-(4-
(benzyloxy)phenyl)cyclopropyeamino)methyl)pyrimidin-5-
amine
H
0 NH2
Example 33: 6-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)-
1,2,4-triazin-
3-amine
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
123
Example 34: 3 -((((trans)-2 -(4-(benz yloxy)phenyl)cycl opropyl)amino)methyl)-
1,2,4-triazin-
6-amine
N H2
Example 35: Preparation of enantiomerically enriched or optically active
compounds
5-(((trans)-2-(4-(benzyloxy)phenyl)cyc1opropy1amino)methyl)-1,3,4-oxadiazol-2-
amine
can be synthesized according to the procedure describe in example 10.
Alternatively,
enantiomerically enriched or pure intermediates can be prepared and then used
in
subsequent reactions in order to synthesize the corresponding (-) or (+)
enantiomer, e.g., of
5-(((trans)-2-(4-(b enzyloxy)phenyl)cyclopropyl amino)methyl)-1 ,3 ,4-
oxadiazol-2-amine.
Step 1
R-(-)-Mandelic acid (22.2 g, 0.14 mol) was added to a solution of (trans)-2-(4-
(benzyloxy)phenyl)cyclopropanamine hydrochloride (intermediate V) (35 g, 0.14
mol) in a
mixture of THF and H20 (6: 4) (650 mL) and refluxed for 1 h. After formation
of a clear
solution the reaction mixture was cooled to RT. The solid precipitated formed
was filtered,
basificd with sat. NaHCO3 solution and extracted with ethyl acetate (3 x 500
mL). The
combined organic extracts were washed with water (500 mL), brine (500 mL),
dried over
anhydrous Na2SO4, filtered and concentrated under vacuum to afford trans 244-
(benzyloxy)phenyl)cyclopropanamine (enantiomer-(-) ) (14 g, 46.6 %) as an off
white solid.
Step 2
Tert-butyl (5-(ehloromethyl)-1,3,4-oxadiazol-2-y1)earbamate (141 mg, 0.606
mmol) was
added to a solution of 2-(4-(benzyloxy)phenyl)cyclopropanamine (enantiomer (-
)) (145 mg,
0.606 mmol) and K2CO3 (166 mg, 1.213 mmol) in dry DMF (1.5 mL) and stirred at
RT for
2 h. After completion, the reaction mixture was poured into ice water (10 mL)
and extracted
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
124
with Et0Ac (4 x 10 mL). The combined organic extracts were washed with water
(3 x 10
mL), brine (10 mL), dried over anhydrous Na2SO4., filtered and concentrated
under vacuum.
The residue obtained was purified by column chromatography (SiO2) using
MeOH:CHC13
(1:99) as eluent to afford tert-butyl (5-
(((2-(4-
(benzyloxy)phenyl)cyclopropyl) amino)methyl)-1 ,3 ,4-oxadiazol-2-yl)carbamate
(enantiomer-(-)) (100 mg, 37.7 %) as a pale green liquid.
Step 3
To a solution of (-) tert-butyl 5-
(((trans)-2-(4-
(benzyloxy)phenyl)cyclopropylamino)methyl)-1,3,4-oxadiazol-2-y1 carbamate (100
mg,
0.229 mmol ) in 1, 4 dioxane (1 mL) at 0 C was added HC1 in 1, 4 dioxane (1
mL) and
stirred for 18 h. After completion, the solvent was evaporated and residue was
dissolved in
water (10 mL), basified with Na2CO3 solution, extracted with Et0Ac (3 X 5 mL).
The
combined extracts were washed with water (5 mL), brine (5 mL), dried over
anhydrous
Na2SO4, filtered and evaporated. The crude residue was purified by column
chromatography using MeOH: CHC13 (5: 95) as the eluent to afford (-) 5-
(((trans)-2-(4-
(benzyloxy)phenyl)cyclopropylamino)methyl)-1,3,4-oxadiazol-2-amine (40 mg,
52%) as a
white solid.
1HNMR (400 MHz, DMSO d6) 5: 7.46 (t, 2H), 7.2 (t, 2H), 6.98 (q, 6H), 5.0 (s,
2H), 3.78 (s,
1H), 3.1 (brs, 1H), 2.2 (brs, 1H), 1.79 (brs, 1H), 0.92 (m, 2H)
Mass (M+H): 337.1
HPLC Purity: 96.02 %
Chiral HPLC Purity: 95.12 %
Specific optical rotation [4)272 (c=0.5 % in DMSO): - 37.76
The corresponding enantiomer-(+) can be synthesized by following the same
procedure but
using S-(+)-Mandelic acid in Step 1.
1H-NMR (400 MHz, DMSO d6) 6: 7.46 (t, 2H), 7.2 (t, 2H), 6.98 (q, 6H), 5.0 (s,
2H), 3.78
(s, 1H), 3.1 (brs, 1H), 2.2 (brs, 1H), 1.79 (brs, 1H), 0.92 (m, 2H);
Mass (M+H): 337.1
HPLC Purity: 98.16 %
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
125
Chiral HPLC Purity: 98.34 %
Specific optical rotation [a] D26.9 (C4.5 % in DMS0): + 37.76
Salts for chiral recrystallization include S (+) Mandelic acid,
D (-) tartaric acid,
L (¨) di-p-toluoyl tartaric acid, or
R (-) Mandelic acid.
The following compounds can be prepared according to the synthetic description
provided
herein and the skill of an ordinary skilled artisan, wherein the absolute
configuration is as
specified in the drawn structure:
5-((((trans)-2-(44(3-fluorobenzypoxy)phenyl)cyclopropyl)amino)methyl)-1,3,4-
oxadiazol-
2-amine
H N
0
0
NH2
5-((((trans)-2-(44(3-fluorobenzyl)oxy)phenyl)cyclopropyl)amino)methyl)-1,3,4-
oxadiazol-
2-amine
*
H \ N
0
0
NH2
5-((((trans)-2-(442-fluorob enzyl)oxy)phenyl)cyclopropyl)amino)methyl)-1 ,3 ,4-
oxadiazol-
2-amine
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
126
N
0
di 0
F NH2
5-((((trans)-2-(44(2-fluorobenzyl)oxy)phenyl)cyclopropypamino)methyl)-1,3,4-
oxadiazol-
2-amine
IN\ /\*FN2nN N
Si 0 o-'
NH2
5-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)-N-methyl-1,3,4-
oxadiazol-
2-amine
,
H
0 =-=-(
0
/NH
54((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)-N-methyl-1,3,4-
oxadiazol-
2-amine
110 0
/NH
N-(54((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)-1,3,4-oxadiazol-
2-
y1)acetamide
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
127
H N
0
0
NH
0
N-(5-((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)-1,3,4-
oxadiazol-2-
y1)acetamide
rah\ __
H N
110 0 111111
(NH
0
5-((((trans)-2-(4-(benzyloxy)phenyl)cyc lopropyeamino)m ethyl )pyrimidin-2-
amine
0 N NH2
5 -((((trans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)
pyrimidin-2-amine
0 N NH2
Is
5-((qtrans)-2-(4-(benzyloxy)phenyl)cyclopropyl)amino)methyl)-1,3,4-thiadiazol-
2-amine
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
128
a A
0
NH2
5-((((trans)-2-(4-(benzyloxy)phenyl)cycloprop yl)amino)m etliy1)- 1 ,3,4-
thiadiazol-2-amine
N N
s
=NH2
5-((((trans)-2-(4-(benzyloxy)phenyl) cyclopropyl)amino)methyl)-N,N-dimethyl-
1,3 ,4-
oxadiazol-2-amine
o
0
N¨
/
5 -((((trans)-2-(4-(benzyloxy)phenyl)eycl oprop yl )amino)methyl)-N,N-dimethyl
-1,3,4-
oxadiazol-2-amine
sN
04
0
N¨
/
GA 02806008 2013-01-18
WO 2012/013728 PCT/EP2011/062949
129
Example 36: Isolation of single enantiomers of (trans) racemic N-substituted
aryl- or
heteroaryl-cyclopropylamine compounds
Chiral HPLC: Conditions to perform the chiral separation of compounds or
intermediates of
the invention can be similar to the following:
Separation by chiral preparative HPLC: Every injection is prepared from e.g.,
about 15 mg
of the N-substituted aryl- or heteroaryl- trans-cyclopropylamine compound
dissolved in a
mixture of Et0H, n-pentane and HFIPA (1,1,1,3,3,3-Hexafluoro-2-propanol). The
optically
active N-substituted aryl- or heteroaryl- trans-cyclopropylamine compound
(e.g., (-) 5-
(((trans)-2-(4-(benz ylox y)phenyl)c yeloprop yl ami no)methyl)- 1,3,4-
oxadiazol-2-amine) can
be separated on e.g., a ChiralPak- IA (250 X 20 mm ID) 5 firn at ambient
temperature
eluting with 0.1% DEA in 70/30 hexane/Et0H at 18 mL/min. The solutions from
the chiral
separation can be concentrated under vacuum (15 psi, 35 C) to afford the
resolved
enantiomers.
Analytical determination of enantiomeric excess (ee): ChiralPak IA 250 x 4.6
mm ID, 51.tm,
0.1 % DEA in 80/20 hexane/Et0H at 1 mL/min at ambient temperature, with UV
analysis at
230 nm. Enantiomers eluted at 11.35 and 16.51 min, each with > 90%
enantiomeric excess.
Analytical purity: Acquity UPLC BEH C18 100 x 2.1 mm ID, 1.7um, 0.025 "A) TFA
in a
gradient H20:ACN (T/%B, 0/30, 4/80, 6/80, 6.1/30) at 0.4 mL/min at ambient
temperature,
with UV analysis at 229 nm. Elution at 1.64 min, each with > 95.0% purity.
Without being
bound by theory, it is believed that mixtures, e.g., racemates corresponding
to a compound
of Formula (I), (Ia), (lb), (II) or (III) can be resolved in the individual
enantiomers or an
enantiomer substantially free of the other enantiomer. Thus, the skilled
artisan, in view of
the disclosure described herein can isolate or purify enantiomers from
racemates or mixtures
of enantiomers in view of the disclosure herein utilizing standard organic
chemistry
techniques for separating cnantiomers.
Enantiomer 1, the (-) optical stereoisomer of
5 -(((trans)-2-(4-
(benzyloxy)phenyl)cyclopropylamino)methyl)-1,3,4-oxadiazol-2-amine is
characterized as
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
130
follows: 111NMR (400 MHz, DMSO d6) 6: 7.46 (t, 2H), 7.2 (t, 2H), 6.98 (q, 6H),
5.0 (s,
2H), 3.78 (s, 1H), 3.1 (brs, 1H), 2.2 (brs, 1H), 1.79 (brs, 1H), 0.92 (m, 2H);
Mass (M+H):
337.1; HPLC Purity: 96.02 %; Chiral HPLC Purity: 95.18 %; Specific optical
rotation
[a]027=2 (c=0.5 % in DMSO): - 37.76 .
Enantiomer 2, the (+) optical stereoisomer of
5-(((trans)-2-(4-
(benzyloxy)phenyl)cyclopropylamino)methyl)-1,3,4-oxadiazol-2-amine is
characterized as
follows: 1HNMR (400 MHz, DMSO d6) 6: 7.46 (t, 2H), 7.2 (t, 2H), 6.98 (q, 6H),
5.0 (s,
2H), 3.78 (s, 1H), 3.1 (brs, 1H), 2.2 (brs, 1H), 1.79 (brs, 1H), 0.92 (m, 2H);
Mass (M+H):
337.1; HPLC Purity: 98.16 %; Chiral HPLC Purity: 98.34 %; Specific optical
rotation
[a]026=9 (c=0.5 % in DMSO): + 37.76 .
The optical activity determination experiment was performed with a Jasco-P-
1030
Polarimeter at a temperature of about 26.9 and 27.2 and a compound
concentration (0.5%)
and solvent of choice e.g., (DMSO).
Example 37: Determination of Kinetic Parameters for Optically Active Compounds
of the
Invention
The kinetic parameters of LSD1 demethylase inhibition were obtained using the
peroxidase-
coupled reaction method. In this assay the demethylase reaction was initiated
by
simultaneously mixing LSD1 protein (either 31 nM or 10 nM) (BPS), 31.25 i_tM
H3-K4me2
peptide (Millipore), increasing concentrations of test compound (e.g., an
optically active
stereoisomer of 5 -(((Trans)-2 -(4-(b enzyloxy)phenyec
yclopropylamino)methyl)-1,3,4-
ox adi azol -2-amine or an optically active N -substituted aryl- or heteroaryl-
cyclopropylamine compound) and 501.tM Amplex Red and 0,11J/m1 horseradish
peroxidase (HPR) (Invitrogen) in a buffer containing 50mM sodium phosphate
buffer
pH=7.4. The final DMSO concentration was 0.7% and constant in all assay wells.
The conversion of the Amplex Red reagent to resorufin due to generation of
H202 was
continuously monitored by fluorescence (excitation at 540 rim, emission at 590
nm) using a
microplate reader (Infinite 200, Tecan). A solution of 11..t,M H202 was used
to calibrate the
fluorescence signal and the temperature was kept constant at 25 C.
GA 02806008 2013-01-18
WO 2012/013728 PCT/EP2011/062949
131
Kinetic parameters were obtained following the method described by Szewczuk et
al
((2007) Biochemistry, 46, 6892-6902).
Briefly, progress curves obtained in the presence of test compound were fit to
derive kobs
(k) based on the following equation:
Va (1 ¨
prodixt = __________________ + o f f set
The kobs values were then used to derive the kinetic constant by using the
following
equations (Kitz and Wilson analysis):
r 1
ktritaC 1.1,) /(Ricapp) [-11
K1 (app)
K
[Si
-I- at
with Km = 24 M.
The determination of the kinetic inhibition constants for MAOs was done
following the
same protocol as for LSD1 with the following modifications:
For MAO-A, the protein was kept at 1,8ng/u1 (Sigma M7316) and kynuramine
(Sigma) at
64uM was used as substrate. In this case the Km was 64 uM.
For MAO-B, the protein was kept between 1.8 and 3.6ng/u1 (Sigma M7441) and
kynuramine (Sigma) at 50uM was used as substrate. In this case the Km was 32
uM.
For both MAO assays, the final DMSO concentrations were 0.54%.
Selegine hydrochloride and rasagiline mesylate were obtained from Sigma-
Aldrich and
Carbone Scientific Co. Ltd respectively.
These studies were used to calculate the values obtained in Table 1.
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
132
Table 1: The Catalytic Efficiency, kinact/Kb Obtained for the Enantiomers of 5-
(qTrans)-2-
(4-(benzyloxy)phenyl)cyclopropylamino)methyl)-1,3,4-oxadiazol-2-amine
ki n actIKI (M-1 s-1)
Enantiomer -1 Enantiomer -2 Selegiline Rasagiline
(-) optical (+) optical
antipode antipode
LSD1 15,516 767 Inactive Inactive
MAO-A 17 182 <100 62
MAO-B 38,298 34,940 32,500 7,463
The results described herein show that the (-) stereoisomer of 5-(((trans)-2-
(4-
(benzyloxy)phenyl)cyclopropylamino)methyl)-1,3,4-oxadiazol-2-amine is a
potent, highly
selective inhibitor of LSD1 and MAOB. The selectivity of the (-) stereoisomer
of 5-
(((trans)-2-(4-(b enzyloxy)phenyecyclopropylamino)methyl)-1,3 ,4-oxadi azol-2-
amine for
LSD', MAO-A, and MAO-B as judged by the selectivity index kinact/Kt indicates
that the
compound is highly selective for both LSD1 and MAO-B. In particular, the
selectivity
index of the (-) stereoisomer of 5 -(((trans)-
2-(4-
(benzyloxy)phenyl)cyclopropylamino)m ethyl)-1,3,4-oxadiazol-2-amine for MAO-
B/MA0-
A is about 2253 and is thus more advantageous than the corresponding values
for Rasagiline
and Selegiline which are 120 and < 325, respectively. The selectivity index of
the (+)
stereoisomer of 5 -
(((trans)-2-(4-(b enzyloxy)phenyl)cycloprop ylamino)methyl)-1,3,4-
oxadiazol-2-amine for MAO-B/MAO-A is about 192. Furthermore, other
irreversible
monoamine oxidase inhibitors like Rasagiline and Selegiline are not active
against LSD1 in
these assays. Notably, the ratio of kinact/KT for LSD1/MAO-A was over 100-
larger for the (-
) stereoisomer as compared to the (+) stereoisomer of 5-(((trans)-2-(4-
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
133
(benzyloxy)phenyl)cyclopropylamino)m ethyl )- 1 ,3,4-oxadiazol-2-amine.
Thus, the
inventors have unexpectedly found that optically active N-substituted aryl- or
lieteroaryl-
trans-cyclopropylamine compounds, including the compounds of Formula (I),
wherein the
substituents on the cyclopropyl moiety are in trans orientation, as well as
the compounds of
Foiniula (II) or (III), have unexpected selectivity for inhibiting LSD1 and
for inhibiting
LSD1 and MAO-B.
Example 38: Biological Assays
The compounds of the invention can be tested for their ability to inhibit
LSD1. The ability
of the compounds of the invention to inhibit LSD1 can be tested as follows.
Human
recombinant LSD1 protein was purchased from BPS Bioscience Inc. In order to
monitor
LSD1 enzymatic activity and/or its inhibition rate by our inhibitor(s) of
interest, di-
methylated H3-K4 peptide (Millipore) was chosen as a substrate. The
demethylase activity
was estimated, under aerobic conditions, by measuring the release of H202
produced during
the catalytic process, using the Amplex Red peroxide/peroxidase-coupled assay
kit
(Invitrogen, Carlsbad, CA).
Briefly, a fixed amount of LSD1 was incubated on ice for 15 minutes, in the
absence and/or
in the presence of various concentrations of inhibitor (e.g., from 0 to 75 M,
dependina, on
the inhibitor strength). Tranylcypromine (Biomol International) was used as a
control for
inhibition. Within the experiment, each concentration of inhibitor was tested
in duplicate.
After leaving the enzyme interacting with the inhibitor, 12.5 uM of di-
methylated 1-13-K4
peptide was added to each reaction and the experiment was left for 30 minutes
(or e.g., an
hour) at 37 C in the dark. The enzymatic reactions were set up in a 50 mM
sodium
phosphate, pH 7.4 buffer. At the end of the incubation, Amplex Red reagent
and
horseradish peroxidase (HPR) solution were added to the reaction according to
the
recommendations provided by the supplier (Invitrogen), and mixed well for 5
minutes (e.g.,
or alternatively 30 mintues) at room temperature in the dark. A 1 jiM H202
solution was
used as a control of the kit efficiency. The conversion of the Amplex Red
reagent to
resorufm due to the presence of H202 in the assay, was monitored by
fluorescence
(excitation at 540 nm, emission at 590 ran) using a microplate reader
(Infinite 200, Tecan).
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
134
Arbitrary units were used to measure level of F1202 produced in the absence
and/or in the
presence of inhibitor.
The maximum demethylase activity of LSD1 was obtained in the absence of
inhibitor and
corrected for background fluorescence in the absence of LSD1. The Ki (IC50) of
each
inhibitor was estimated at half of the maximum activity.
The results presented in Table 2 below show the results of the LSD1 inhibition
studies for a
number of the Example compounds. Parnate (2-trans phenylcyclopropylamine) was
found
to have a Ki (IC50) of from about 15 to 35 micromolar depending on the enzyme
preparation. The studies show that the compounds of the invention have
unexpectedly
potent LSD1 inhibition.
Example 39: Biological Assays - Monoamine Oxidase Assays for determining the
selectivity of the compounds of the invention for LSD1
Human recombinant monoamine oxidase proteins MAO-A and MAO-B were purchased
from Sigma Aldrich. MAOs catalyze the oxidative deamination of primary,
secondary and
tertiary amines. In order to monitor MAO enzymatic activities and/or their
inhibition rate
by inhibitor(s) of interest, a fluorescent-based (inhibitor)-screening assay
was set up. 3-(2-
Aminopheny1)-3-oxopropanamine (kynuramine dihydrobromide, Sigma Aldrich), a
non
fluorescent compound was chosen as a substrate. Kynuramine is a non-specific
substrate
for both MAOs activities. While undergoing oxidative deamination by MAO
activities,
kynuramine is converted into 4-hydroxyquinoline (4-HQ), a resulting
fluorescent product.
The monoamine oxidase activity was estimated by measuring the conversion of
kynuramine
into 4-hydroxyquinoline. Assays were conducted in 96-well black plates with
clear bottom
(Corning) in a final volume of 100 L. The assay buffer was 100 mM HEPES, pH
7.5.
Each experiment was performed in triplicate within the same experiment.
Briefly, a fixed amount of MAO (0.25 )...tg for MAO-A and 0.5 jig for MAO-B)
was
incubated on ice for 15 minutes in the reaction buffer, in the absence and/or
in the presence
135
of various concentrations of inhibitor (e.g., from 0 to 50 uM, depending on
the inhibitor
strength). Tranylcypromine (Biomol International) was used as a control for
inhibition.
After leaving the enzyme(s) interacting with the inhibitor, 60 to 90 uM of
kynuramine was
added to each reaction for MAO-B and MAO-A assay respectively, and the
reaction was left
for 1 hour at 37 C in the dark. The oxidative deamination of the substrate was
stopped by
adding 50 pt (v/v) of NaOH 2N. The conversion of kynuramine to 4-
hydroxyquinoline,
was monitored by fluorescence (excitation at 320 nm, emission at 360 nm) using
a
microplate reader (Infinite 200, Tecan). Arbitrary units were used to measure
levels of
fluorescence produced in the absence and/or in the presence of inhibitor.
The maximum of oxidative deamination activity was obtained by measuring the
amount of
4-hydroxyquinoline formed from kynuramine deamination in the absence of
inhibitor and
corrected for background fluorescence in the absence of MAO enzymes. The Ki
(IC50) of
each inhibitor was determined at Vmax/2.
Example MAO-A MAO-B LSD1
No. (Ki) (Ki) (Ki)
1 I II III
2 ..
11 I 11 II
12 I II II
13 I II TI
14 I II II
I II IT-Ill
Table 2: Summary of Data from MAO-A, MAO-B, and LSD1 Inhibition Studies
The ranges for the Ki value reported in Table 2 are for MAO-A, MAO-B and LSD1 -
I =
between 1 uM and 40 uM; II = between 0.1 uM and 1 uM; III between 0.001 uM and
0.1
Generally, the compounds of the Examples were found to have Ki (IC50) values
for
MAO-A and MAO-B greater than the LSD I Ki values, whereas LSD1 Ki values were
lower
than 0.6 M.
CA 2806008 2018-02-26
GA 02806008 2013-01-18
WO 2012/013728
PCT/EP2011/062949
136
Thus the compounds of the invention are unexpectedly potent LSD1 inhibitors
and
unexpectedly selective for LSD1 as compared to MAO-A and MAO-B, or the
compounds
are dual inhibitors of LSD1 and MAO-B.
Some compounds of the Examples have been tested for
antiproliferative/eytotoxic activity
and been found to have activity in the micromolar to low micromolar range
against cancer
cell lines including HCT-116.
Previous reports of LSD1 have found that it is involved in cell proliferation
and growth.
Some studies have implicated LSD1 as a therapeutic target for cancer. Huang et
al. (2007)
PIUS 104:8023-8028 found that polyamine inhibitors of LSD1 modestly cause the
reexpression of genes aberrantly silenced in cancer cells and particularly
colorectal cancer
(Huang et at. Clin Cancer Res. (2009) Dec 1;15(23):7217-28. Epub 2009 Nov 24.
PMID:
19934284). Scoumanne et al. ((2007) J. Biol. Chem. May 25;282(21):15471-5)
found that
deficiency in LSD1 leads to a partial cell cycle arrest in G2/M and sensitizes
cells to growth
suppression induced by DNA damage. Kahl et at. ((2006) Cancer Res.
66(23):11341-7.)
found that LSD1 expression is correlated with prostate cancer aggressiveness.
Metzger et
at. reported that LSD1 modulation by siRNA and pargyline regulates androgen
receptor
(AR) and may have therapeutic potential in cancers where AR plays a role, like
prostate,
testis, and brain cancers. Lee et at. ((2006) (hem. Biol. 13:563-567) reported
that
tranylcypromine derepresses Egr-1 gene expression in some cancer lines. A body
of
evidence is accumulating that Egr-1 is a tumor suppressor gene in many
contexts (see e.g.,
Calogero et at. (2004) Cancer Cell International 4:1 exogenous expression of
EGR-1
resulted in growth arrest and eventual cell death in primary cancer cell
lines; Lucema et al.
(2006) Cancer Research 66, 6708-6713 show that sustained expression of Egr-1
causes
antiangiogeneic effects and inhibits tumor growth in some models; Ferraro et
at. ((2005)
Clin. Oncol. Mar 20;23(9):1921-6) reported that Egr-1 is downregulated in lung
cancer
patients with a higher risk of recurrence and may be more resistant to
therapy. Thus,
increasing Egr-1 expression via inhibition of LSD1 is a therapeutic approach
for some
cancers. Recent studies have also implicated LSD1 in brain cancer (Schulte et
al. (2009)
Cancer Res. Mar 1;69(5):2065-71). Other studies have implicated LSD1 in breast
cancer
(Lims et al. Carcinogenesis. 2009 Dec 30. [Epub ahead of print] PMID:
20042638).
137
Thus, a body of evidence has implicated LSD1 in a number of cancers, which
suggests that
LSD1 is a therapeutic target for cancer. The instant inventors have discovered
a class of
LSD1 inhibitors that can be used to treat diseases where LSD1 is implicated as
a therapeutic
target like cancer. Accordingly, the phenylcyclopropylamine compounds of the
invention
can be used to treat such diseases.
Recent studies have also implicated LSD1 in viral infection and reactivation.
In particular it
was shown that pharmacological inhibitors of LSD1 like parnate and siRNA knock
down of
LSD1 caused reduced viral infectivity and reduced reactivation after latency
(Liang et al.
(2009) Nat. Med. 15:1312-1317). Therefore, it is believed that the compounds
of the
invention can be used for treating or preventing viral infection. Furthermore,
it is believed
that the compounds of the invention can treat or prevent viral reactivation
after latency.
Thus, without being bound by theory, the inventors have identified a new class
of
cyclopropanamine derivatives containing LSD1 inhibitors with unexpected
potency and
selectivity for LSD1 a biologically relevant target in oncology and other
diseases and/or
LSD1/MAO-B.
All publications and patent applications mentioned in the specification are
indicative of the
level of those skilled in the art to which this invention pertains. The mere
mentioning of the
publications and patent applications does not necessarily constitute an
admission that they
are prior art to the instant application.
Although the foregoing invention has been described in some detail by way of
illustration
and example for purposes of clarity of understanding, it will be obvious that
certain changes
and modifications may be practiced within the scope of the appended claims.
CA 2806008 2018-02-26