Note: Descriptions are shown in the official language in which they were submitted.
CA 02807564 2013-02-26
MICROFLUIDIC DEVICES AND METHODS OF USING SAME
This application is a divisional application of Canadian Patent Application
No.
2,521,171 filed April 5, 2004.
BACKGROUND OF THE INVENTION
[0003] Recently, there have been concerted efforts to develop and. manufacture
microfluidic
systems to perform various chemical and biochemical analyses and syntheses,
both for
preparative and analytical applications. The goal to make such devices arises
because of the
significant benefits that can be realized from miniaturization with respect to
analyses and
syntheses conducted on a macro scale. Such benefits include a substantial
reduction in time,
cost and the space requirements for the devices utilized to conduct the
analysis or synthesis.
Additionally, microfluidic devices have the potential to be adapted for use
with automated
systems, thereby providing the additional benefits of further cost reductions
and decreased
operator errors because of the reduction in human involvement. Microfluidic
devices have
been proposed for use in a variety of applications, including, for instance,
capillary
electrophoresis, gas chromatography and cell separations.
[0004] However, realization of these benefits has often been thwarted because
of various
complications associated with the microfluidic devices that have thus far been
manufactured.
For instance, many of the current microfluidic devices are manufactured from
silica-based
substrates; these materials are difficult and complicated to machine and
devices made from
such materials are fragile. Furthermore, transport of fluid through many
existing microfluidic
CA 02807564 2013-02-26
devices requires regulation of complicated electrical fields to transport
fluids in a controlled
fashion through the device.
[0005] Thus, in view of the foregoing benefits that can be achieved with
microfluidic devices
but the current limitations of existing devices, there remains a need for
microfluidic devices
designed for use in conducting a variety of chemical and biochemical analyses.
Because of
its importance in modern biochemistry, there is a particular need for devices
that can be
utilized to conduct a variety of nucleic acid amplification reactions, while
having sufficient
versatility for use in other types of analyses as well.
[0006] Devices with the ability to conduct nucleic acid amplifications would
have diverse
utilities. For example, such devices could be used as an analytical tool to
determine whether
a particular target nucleic acid of interest is present or absent in a sample.
Thus, the devices
could be utilized to test for the presence of particular pathogens (e.g.,
viruses, bacteria or
fungi), and for identification purposes (e.g., paternity and forensic
applications). Such
devices could also be utilized to detect or characterize specific nucleic
acids previously
correlated with particular diseases or genetic disorders. When used as
analytical tools, the
devices could also be utilized to conduct genotyping analyses and gene
expression analyses
(e.g., differential gene expression studies). Alternatively, the devices can
be used in a
preparative fashion to amplify sufficient nucleic acid for further analysis
such as sequencing
of amplified product, cell-typing, DNA fingerprinting and the like. Amplified
products can
also be used in various genetic engineering applications, such as insertion
into a vector that
can then be used to transform cells for the production of a desired protein
product.
SUMMARY OF THE INVENTION
[0007] A variety of devices and methods for conducting microfluidic analyses
are provided
herein including devices that can be utilized to conduct then-nal cycling
reactions such as
nucleic acid amplification reactions. The devices differ from conventional
microfluidic
devices in that they include elastomeric components; in some instances, much
or all of the
device is composed of elastomeric material.
[0008] Certain devices are designed to conduct thermal cycling reactions
(e.g., PCR) with
devices that include one or more elastomeric valves to regulate solution flow
through the
device. Thus, methods for conducting amplification reactions with devices of
this design are
also provided.
[0009] Some of the devices include blind flow channels which include a region
that functions
as a reaction site. Certain such devices include a flow channel formed within
an elastomeric
2
CA 02807564 2013-02-26
material, and a plurality of blind flow channels in fluid communication with
the flow channel,
with a region of each blind flow channel defining a reaction site. The devices
can also
include one or more control channels overlaying and intersecting each of the
blind flow
channels, wherein an elastomeric membrane separates the one or more control
channels from
the blind flow channels at each intersection. The elastomeric membrane in such
devices is
disposed to be deflected into or withdrawn from the blind flow channel in
response to an
actuation force. The devices can optionally further include a plurality of
guard channels
formed within the elastomeric material and overlaying the flow channel and/or
one or more
of the reaction sites. The guard channels are designed to have fluid flow
therethrough to
reduce evaporation from the flow channels and reaction sites of the device.
Additionally, the
devices can optionally include one or more reagents deposited within each of
the reaction
sites.
[0010] In certain devices, the flow channel is one of a plurality of flow
channels, each of the
flow channels in fluid communication with multiple blind flow channels which
branch
therefrom. Of devices of this design, in some instances the plurality of flow
channels are -
interconnected with one another such that fluid can be introduced into each of
the reaction
sites via a single inlet. In other devices, however, the plurality of flow
channels are isolated
from each other such that fluid introduced into one flow channel cannot flow
to another flow
channel, and each flow channel comprises an inlet at one or both ends into
which-fluid can be
introduced.
[0011] Other devices include an array of reaction sites having a density of at
least 50
sites/cm2, with the reaction sites typically formed within an elastomeric
material. Other
devices have even higher densities such as at least 250, 500 or 1000
sites/cm2, for example.
[0012] Still other device include a reaction site formed within an elastomeric
substrate, at
which a reagent for conducting a reaction is non-covalently immobilized. The
reagent can be
one or more reagents for conducting essentially any type of reaction. The
reagent in some
devices includes one reagents for conducting a nucleic acid amplification
reaction. Thus, in
some devices the reagent comprises a primer, polymerase and one or more
nucleotides. In
other devices, the reagent is a nucleic acid template.
[0013] A variety of matrix or array-based devices are also provided. Certain
of these devices
include: (i) a first plurality of flow channels formed in an elastomeric
substrate, (ii) a second
plurality of flow channels formed in the elastomeric substrate that intersect
the first plurality
of flow channels to define an array of reaction sites, (iii) a plurality of
isolation valves
disposed within the first and second plurality of flow channels that can be
actuated to isolate
3
CA 02807564 2013-02-26
solution within each of the reaction sites from solution at other
reaction=sites, and (iv) a
plurality of guard channels overlaying one or more of the flow channels and/or
one or more
of the reaction sites to prevent evaporation of solution therefrom.
[0014] The foregoing devices can be utilized to conduct a number of different
types of
reactions, including those involving temperature regulation (e.g.,
thermocycling of nucleic
acid analyses). Methods conducted with certain blind channel type devices
involve providing
a microfluidic device that comprises a flow channel formed within an
elastomeric material;
mid a plurality of blind flow channels in fluid communication with the flow
channel, with an
end region of each blind flow channel defining a reaction site. At least one
reagent is
introduced into each of the reaction sites, and then a reaction is detected at
one or more of the
reaction sites. The method can optionally include heating the at least one
reagent within the
reaction site. Thus, for example, a method can involve introducing the
components for a
nucleic acid amplification reaction and then thermocycling the components to
form amplified
product.
[0015] Other methods involve providing a microfluidic device comprising one or
more
reaction sites, each reaction site comprising a first reagent for conducting
an analysis that is
non-covalently deposited on an elastomeric substrate. A second reagent is then
introduced
into the one or more reaction sites, whereby the first and second reagents mix
to form a
reaction mixture. A reaction between the first and second reagents at one or
more of the
reaction sites is subsequently detected.
[0016] Still other methods involve providing a microfluidic device comprising
an array of
reaction sites formed within a substrate and having a density of at least 50
sites/cm2. At least
one reagent is introduced into each of the reaction sites. A reaction at one
or more of the
reaction sites is then detected.
[G017] Yet other methods involve providing a microfluidic device comprising at
least one
reaction site which is formed within an elastomeric substrate and a plurality
of guard
channels also formed within the elastomeric substrate. At least one reagent is
introduced into
each of the reaction sites and then heated within the reaction sites. A fluid
is flowed through
the guard channels before or during heating to reduce evaporation from the at
least one
reaction site. A reaction within the at least one reaction site is
subsequently detected.
[0018] Additional devices designed to reduce evaporation of fluid from the
device are also
provided. In general, such devices comprise a cavity that is part of a
microfluidic network
formed in an elastomeric substrate; and a plurality of guard channels
overlaying the cavity
and separated from the cavity by an elastomeric membrane. The guard channel in
such
4
CA 02807564 2013-02-26
devices is sized (i) to allow solution flow therethrough, and (ii) such that
there is not a
substantial reduction in solution flow in, out or through the cavity due to
deflection of the
membrane(s) upon application of an actuation force to the guard channels.
Other such
devices include (i) one or more flow channels and/or one or more reaction
sites; and (ii) a
plurality of guard channels overlaying the microfluidic system and separated
therefrom by
elastomer, wherein the spacing between guard channels is between 1 pm to 1 mm.
In other
devices the spacing is between 5 pm and 500 pm, in other devices between 10 pm
and 100
pm, and in still other devices between 40 p.m and 75 pm.
[0019] Compositions for conducting nucleic acid analyses in reaction sites of
certain
microfluiclic devices are also provided. Certain such compositions include one
or more of the
following: an agent that blocks protein binding sites on an elastomeric
material and a
detergent. The blocking agent is typically selected from the group consisting
of a protein
(e.g., gelatin or albumin, such as bovine serum albumin (BSA)). The detergent
can be SDS
or Triton, for example.
BRIEF DESCRIPTION OF THE DRAWINGS
[0020] FIG. lA is a schematic representation of an exemplary device with a
matrix design of
intersecting vertical and horizontal flow channels.
[0021] FIGS. 1B-E show enlarged views of a portion of the device shown in FIG.
IA and
illustrates its operation.
[0022] FIG. IF is a schematic representation of another exemplary matrix
design device that
utilizes guard channels to reduce sample evaporation.
[0023] FIG. 2 is a plan view of an exemplary blind channel device.
[0024] FIG. 3A is a plan view of another exemplary blind channel device.
[0025] FIG. 3B is a schematic representation of a more complex blind channel
device based
upon the unit of the general design depicted in FIG. 3A.
[0026] FIG. 3C is an enlarged view of a region of the device shown in FIG. 3B,
and
illustrates the orientation of the guard flow channels in this particular
design.
[0027] FIG. 4 is a plan view of a device utilizing the hybrid design.
[0028] FIG. 5 is a chart showing ramp up and down times to conduct a
thennocycling
reaction.
[0029] FIG. 6 shows the location of spotted reagents within reaction sites in
a blind channel =
type device illustrating proper alignment of the reagents within reaction
sites at the comers of
the device.
5
CA 02807564 2013-02-26
[0030] FIGS. 7A and 7B respectively are a cross-sectional view and a schematic
diagram of
another hybrid type microfluidic device and represents the type of device used
to conduct the
experiments described in Examples 1-4.
[0031] FIG. 8 is a bar graph in which the average FAM/PR1/Control ratios are
plotted for six
different 13-actin TaqMan reactions. The reactions were thermocycled in the
micro fluidic
device (chip) shown in FIG. 7B (solid bars) and Macro TaqMan reactions
(striped bars). The
controls are the first and fourth bar sets that have no DNA. The error bars
are the standard
deviation of the ratios.
[0032] FIG. 9 is a diagram depicting an exemplary pin spotting process.
Reagents are picked
up from a source (e.g., a microtiter plate) and then printed by bringing the
loaded pin into
contact with the substrate. The wash step consists of agitation in deionized
water followed by
vacuum drying.
[0033] FIG. 10 is a bar graph depicting FAM signal strength for the
microfluidic device
(chip) described in Example 1 (see FIG. 7B) based on the experiments described
in Example
2. The data are in the form of (FAM signal / PR1 signal) scaled by the FAM/PR1
ratio for
the reference lanes. Error bars are the standard deviation along a lane. The
"1.3X" and "lX"
designations refer to the concentration of the spotted primers and probes, in
relation to their
nominal values.
[0034] FIG. 11 is a bar graph showing average VIC/PF1/Control ratios for 9-10
wells for
Macro TaqMan (striped bars), and TaqMan reactions in the microfluidic device
(solid bars).
Two negative controls (Control) and two samples with 100 pg/n1 genomic DNA
were
thermocycled with reaction components as described above with 4x the standard
amount of
primer/probe. The error bars represent the standard deviation of the average
ratios.
[0035] FIG. 12 is a bar graph that shows FAM/PR1/Control ratios for each of 10-
1 n1 wells
branching Emu a single flow channel of a microfluidic device (see FIG. 7B).
The amount of
genomic DNA was 0.25 pginl, which results in an average of one target copy per
well.
[0036] FIG. 13 is a bar graph depicting the average VIC/PR1/Control ratios for
CYP2D6
SNP reactions using the microfluidic device shown in FIG. 7B. Allele 1 (A1-1)
is the positive
control for the VIC probe against the reference or wild type allele CYP2D6*1.
Allele 2 (Al-
2) is the positive control for the FAM probe against the variant or mutant
allele, CYP2D6*3.
The control has no DNA template. Genomic DNA was used at either 100 pg/n1 or
20 pg/nl.
The error bars are the standard deviation of the ratios.
6
CA 02807564 2013-02-26
[0037] FIG. 14 is a bar graph showing the average FAM/PR1/Control ratios for
CYP2D6
SNP reactions in the microfluidic device shown in FIG. 7B. The samples are the
same as
described with respect to FIG. 13 and in Example 3.
[0038] FIGS. 15 is a schematic diagram of the microfluidic device used for the
experiments
in Example 4.
[0039] FIG. 16 is a polyacrylamide gel containing PCR product from Macro PCR
and PCR
reactions in the microfluidic device shown in FIG. 7B. The results on the left
show the
approximate migration of different DNA base pair lengths. The lanes containing
interspersed
bands are molecular weight markers. The lanes labeled "Macro" are the PCR
products from
the Macro reactions at different dilutions. The lanes labeled "In chip" are
PCR products
generated in the chip. The lanes containing many bands throughout the gel are
nonspecific
background signals.
[0040] FIGS 17a-17d depict two preferred designs of a partitioning
microfluidic device in a
valve off and valve actuated state.
[00411 FIGS 18a and 18b depict images of a partitioning microfluidic devices
after a
thermocycling reaction was performed. Figure 18a depicts a two color image,
and figure 18b '
depicts the remaining signal after subtraction of the control red signal.
=
[0042] FIG 19 depicts a graph of comparing the average number of copies per
well to the
number of positive wells.
DETAILED DESCRIPTION
[0043] I. Definitions
[0044] Unless defined otherwise, all technical and scientific terms used
herein have the
meaning commonly understood by a person skilled in the art to which this
invention belongs.
The following references provide one of skill with a general definition of
many of the tenns
used in this invention: Singleton et al., DICTIONARY OF MICROBIOLOGY AND
MOLECULAR BIOLOGY (2d ed. 1994); THE CAMBRIDGE DICTIONARY OF
SCIENCE AND TECHNOLOGY (Walker ed., 1988); THE GLOSSARY OF GENETICS,
5TH ED., R. Rieger et al. (eds.), Springer Verlag (1991); and Hale & Marham,
THE
HARPER COLLINS DICTIONARY OF BIOLOGY (1991). As used herein, the following
terms have the meanings ascribed to them unless specified otherwise.
[0045] A "flow channel" refers generally to a flow path through which a
solution can flow.
7
CA 02807564 2013-02-26
[0046] The term "valve" unless otherwise indicted refers to a configuration in
which a flow
channel and a control channel intersect and are separated by an elastomeric
membrane that
can be deflected into or refracted from the flow channel in response to an
actuation force.
[0047] A "blind channel" or a "dead-end channel" refers to a flow channel
which has an
entrance but not a separate exit. Accordingly, solution flow in and out of the
blind channel
occurs at the same location. The process of filling one or more blind channels
is sometimes
simply referred to as "blind fill."
[0048] An "isolated reaction site" generally refers to a reaction site that is
not in fluid
communication with other reactions sites present on the device. When used with
respect to a
blind channel, the isolated reaction site is the region at the end of the
blind channel that can
be blocked off by a valve associated with the blind channel.
[0049] A "via" refers to a channel formed in an elastomeric device to provide
fluid access
between an external port of the device and one or more flow channels. Thus, a
via can serve
as a sample input or output, for example.
[0050] The term "elastomer" and "elastomeric" has its general meaning as used
in the art.
Thus, for example, Allcock et al. (Contemporary Polymer Chemistry, 2nd Ed.)
describes
elastomers in general as polymers existing at a temperature between their
glass transition
temperature and liquefaction temperature. Elastomeric materials exhibit
elastic properties
because the polymer chains readily undergo torsional motion to permit
uncoiling of the
backbone chains in response to a force, with the backbone chains recoiling to
assume the
prior shape in the absence of the force. In general, elastomers deform when
force is applied,
but then return to their original shape when the force is removed. The
elasticity exhibited by
elastomeric materials can be characterized by a Young's modulus. The
elastomeric materials
utilized in the microfluidic devices disclosed herein typically have a Young's
modulus of
betweeii ilbOUt 1 Pa ¨ 1 1:Pa, in other instances between about 10 Pa ¨100
CiPa, in still other
instances between about 20 Pa ¨ 1 GPa, in yet other instances between about 50
Pa 10 MN,
and in certain instances between about 100 Pa ¨ 1 IVit'a. Elastomeric
materials having a
Young's modulus outside of these ranges can also be utilized depending upon
the needs of a
particular application.
[0051] Some of the microfluidic devices described herein are fabricated from
an elastomeric
polymer such as GE RTV 615 (formulation), a vinyl-silane crosslinked (type)
silicone
elastomer (family). However, the present microfluidic systems are not limited
to this one
formulation, type or even this family of polymer; rather, nearly any
elastomeric polymer is
suitable. Given the tremendous diversity of polymer chemistries, precursors,
synthetic
8
CA 02807564 2013-02-26
methods, reaction conditions, and potential additives, there are a large
number of possible
elastomer systems that can be used to make monolithic elastomeric microvalves
and pumps.
The choice of materials typically depends upon the particular material
properties (e.g.,
solvent resistance, stiffness, gas permeability, and/or temperature stability)
required for the
application being conducted. Additional details regarding the type of
elastomeric materials
that can be used in the manufacture of the components of the microfluidic
devices disclosed
herein are set forth in Unger et al. (2000) Science 288:113-116, and PCT
Publications WO
02/43615, and WO 01/01025.
[0052] The terms "nucleic acid," "polynucleotide," and "oligonucleotide" are
used herein to
include a polymeric form of nucleotides of any length, including, but not
limited to,
ribonucleotides or deoxyribonucleotides. There is no intended distinction in
length between
these terms. Further, these terms refer only to the primary structure of the
molecule. Thus, in
certain embodiments these terms can include triple-, double- and single-
stranded DNA, as
well as triple-, double- and single-stranded RNA. They also include
modifications, such as
by methylation and/or by capping, and unmodified forms of the polynucleotide.
More
particularly, the terms "nucleic acid," "polynucleotide," and
"oligonucleotide," include
polydeoxyribonucleotides (containing 2-deoxy-D-ribose), polyribonucleotides
(containing D-
ribose), any other type of polynucleotide which is an N¨ or C-glycoside of a
purine or
pyrimidine base, and other polymers containing nonnucleotidic backbones, for
example,
polyamide (e.g., peptide nucleic acids (PNAs)) and polymorpholino
(commercially available
from the Anti-Virals, Inc., Corvallis, Oregon, as Neugene) polymers, and other
synthetic
sequence-specific nucleic acid polymers providing that the polymers contain
nucleobases in a
configuration which allows for base pairing and base stacking, such as is
found in DNA and
RNA.
[0053] A "probe" is an nucleic acid capable of binding to a target nucleic,
acid of
complementary sequence through one or more types of chemical bonds, usually
through
complementary base pairing, usually through hydrogen bond formation, thus
forming a
duplex structure. The probe binds or hybridizes to a "probe binding site." The
probe can. be
labeled with a detectable label to permit facile detection of the probe,
particularly once the
probe has hybridized to its complementary target. The label attached to the
probe can include
any of a variety of different labels known in the art that can be detected by
chemical or
physical means, for example. Suitable labels that can be attached to probes
include, but are
not limited to, radioisotopes, fluorophores, chromophores, mass labels,
electron dense
9
CA 02807564 2013-02-26
particles, magnetic particles, spin labels, molecules that emit
chemilurainescence,
electrochemically active molecules, enzymes, cofactors, and enzyme substrates.
Probes can
vary significantly in size. Some probes are relatively short. Generally,
probes are at least 7
to 15 nucleotides in length. Other probes are at least 20, 30 or 40
nucleotides long. Still
other probes are somewhat longer, being at least 50, 60, 70, 80, 90
nucleotides long. Yet
other probes are longer still, and are at least 100, 150, 200 or more
nucleotides long. Probes
can be of any specific length that falls within the foregoing ranges as well.
[0054] A "primer" is a single-stranded polynucleotide capable of acting as a
point of
initiation of template-directed DNA synthesis under appropriate conditions
(i.e., in the
presence of four different nucleoside triphosphates and an agent for
polymerization, such as,
DNA or RNA polymerase or reverse transcriptase) in an appropriate buffer and
at a suitable
temperature. The appropriate length of a primer depends on the intended use of
the primer
but typically is at least 7 nucleotides long and, more typically range from 10
to 30 nucleotides
in length. Other primers can be somewhat longer such as 30 to 50 nucleotides
long. Short
primer molecules generally require cooler temperatures to form sufficiently
stable hybrid
complexes with the template. A primer need not reflect the exact sequence of
the template
but must be sufficiently complementary to hybridize with a template. The term
"primer site"
or "primer binding site" refers to the segment of the target DNA to which a
primer
hybridizes. The term "primer pair" means a set of primers including a 5'
"upstream primer"
that hybridizes with the complement of the 5' end of the DNA sequence to be
amplified and a
3' "downstream primer" that hybridizes with the 3' end of the sequence to be
amplified.
[0055] A primer that is "perfectly complementary" has a sequence fully
complementary
= across the entire length of the primer and has no mismatches. The
primer is typically
perfectly complementary to a portion (subsequence) of a target sequence. A
"mismatch"
refers to a site at which the nucleotide in the primer and the nucleotide in
the target nucleic
acid with which it is aligned are not complementary. The term "substantially
complementary" when used in reference to a primer means that a primer is not
perfectly
complementary to its target sequence; instead, the primer is only sufficiently
complementary
= to hybridize selectively to its respective strand at the desired
primer-binding site.
[0056] The term "complementary" means that one nucleic acid is identical to,
or hybridizes
= selectively to, another nucleic acid molecule. Selectivity of
hybridization exists when
hybridization occurs that is more selective than total lack of specificity.
Typically, selective
hybridization will occur when there is at least about 55% identity over a
stretch of at least 14-
25 nucleotides, preferably at least 65%, more preferably at least 75%, and
most preferably at
10
CA 02807564 2013-02-26
least 90%. Preferably, one nucleic acid hybridizes specifically to the other
nucleic acid. See
M. Kanehisa, Nucleic Acids Res. 12:203 (1984).
[0057] The term "label" refers to a molecule or an aspect of a molecule that
can be detected
= by physical, chemical, electromagnetic and other related
analytical techniques. Examples of
detectable labels that can be utilized include, but are not limited to,
radioisotopes,
fluorophores, chromophores, mass labels, electron dense particles, magnetic
particles, spin
labels, molecules that emit chemiluminescence, electrochemically active
molecules, enzymes,
cofactors, enzymes linked to nucleic acid probes and enzyme substrates. The
term
"detectably labeled" means that an agent has been conjugated with a label or
that an agent has
some inherent characteristic (e.g., size, shape or color) that allows it to be
detected without
having to be conjugated to a separate label.
[0058] A "polymorphic marker" or "polymorphic site" is the locus at which
divergence
occurs. Preferred markers have at least two alleles, each occurring at
frequency of greater
than 1%, and more preferably greater than 10% or 20% of a selected population.
A
polymorphic locus may be as small as one base pair. Polymorphic markers
include
restriction fragment length polymorphisms, variable number of tandem repeats
(VNTR's), =
hypervariable regions, minisatellites, dinucleotide repeats, trinucleotide
repeats,
tetranucleotide repeats, simple sequence repeats, and insertion elements such
as Alu. The
first identified allelic form is arbitrarily designated as the reference form
and other allelic
forms are designated as alternative or variant alleles. The allelic form
occurring most
frequently in a selected population is sometimes referred to as the wildtype
form. Diploid
organisms may be homozygous or heterozygous for allelic forms. A diallelic
polymorphism
has two forms. A triallelic polymorphism has three forms.
[0059] A "single nucleotide polymorphism" (SNP) occurs at a polymorphic site
occupied by
a single nucleotide, which is the site of variation between allelic sequences.
The site is
usually preceded by and followed by highly conserved sequences of the allele
(e.g.,
sequences that vary in less than 1/100 or 1/1000 members of the populations).
A siugle
nucleotide polymorphism usually arises due to substitution of one nucleotide
for another at
the polymorphic site. A transition is the replacement of one purine by another
purine or one
pyrimidine by another pyrimidine. A transversion is the replacement of a
purine by a
= pyrimidine or vice versa. Single nucleotide polymorphisms can also
arise from a deletion of
a nucleotide or an insertion of a nucleotide relative to a reference allele.
[0060] A "reagent" refers broadly to any agent used in a reaction. A reagent
can include a =
single agent which itself can be monitored (e.g., a substance that is
monitored as it is heated)
11
CA 02807564 2013-02-26
or a mixture of two or more agents. A reagent may be living (e.g., a cell) or
non-living.
Exemplary reagents for a nucleic acid amplification reaction include, but are
not limited to,
buffer, metal ions, polymerase, primers, template nucleic acid, nucleotides,
labels, dyes,
nucleases and the like. Reagents for enzyme reactions include, for example,
substrates,
cofactors, coupling enzymes, buffer, metal ions, inhibitors and activators.
Reagents for cell-
based reactions include, but are not limited to, cells, cell specific dyes and
ligands (e.g.,
agonists and antagonists) that bind to cellular receptors.
[0061] A "ligand" is any molecule for which there exists another molecule
(i.e., an
"antiligand") that specifically or non-specifically binds to the ligand, owing
to recognition of
some portion of the ligand by the antiligand.
Overview
[0062] A number of different microfluidic devices (also sometimes referred to
as chips)
having unique flow channel architectures are provided herein, as well as
methods for using
such devices to conduct a variety of high throughput analyses. The devices are
designed for
use in analyses requiring temperature control, especially analyses involving
thennocycling
(e.g., nucleic acid amplification reactions). The microfluidic devices
incorporate certain
design features that: give the devices a significantly smaller footprint than
many conventional
microfluidic devices, enable the devices to be readily integrated with other
instrumentation
and provide for automated analysis.
[0063] Some of the microfluidic devices utilize a design typically referred to
herein as "blind
channel" or "blind fill" are characterized in part by having a plurality of
blind channels,
which, as indicated in the definition section, are flow channels having a dead
end or isolated
end such that solution can only enter and exit the blind channel at one end
(i.e., there is not a
separate inlet and outlet for the blind channel). These devices require only a
single valve for
each blind channel to isolate a region of the blind channel to form an
isolated reaction site.
During manufacture of this type of device, one or more reagents for conducting
an analysis
are deposited at the reaction sites, thereby resulting in a significant
reduction in the number of
input and outputs. Additionally, the blind channels are connected to an
interconnected
network of channels such that all the reaction sites can be filled from a
single, or limited
number, of sample inputs. Because of the reduction in complexity in inputs and
outputs and
the use of only a single valve to isolate each reaction site, the space
available for reaction
sites is increased. Thus, the features of these devices means that each device
can include a
large number of reaction sites (e.g., up to tens of thousands) and can achieve
high reaction
12
CA 02807564 2013-02-26
site densities (e.g., over 1,000 ¨4,000 reaction sites/cm2). Individually and
collectively, these
features also directly translate into a significant reduction in the size of
these devices
compared to traditional microfluidic devices.
[0064] Other microfluidic devices that are disclosed herein utilize a matrix
design. In
general, microfluidic devices of this type utilize a plurality of intersecting
horizontal and
vertical flow channels to define an array of reaction sites at the points of
intersection. Thus,
devices of this design also have an array or reaction sites; however, there is
a larger number
of sample inputs and corresponding outputs to accommodate the larger number of
samples
with this design. A valve system referred to as a switchable flow array
architecture enables
solution be flowed selectively through just the horizontal or flow channels,
thus allowing for
switchable isolation of various flow channels in the matrix. Hence, whereas
the blind
channel devices are designed to conduct a large number of analyses under
different
conditions with a limited number of samples, the matrix devices are
constructed to analyze a
large number of sample under a limited number of conditions. Still other
devices are hybrids
of these two general design types.
[0065] The microfluidic devices that are described are further characterized
in part by
utilizing various components such as flow channels, control channels, valves
and/or pumps
from elastomeric materials. In some instances, essentially the entire device
is made of
elastomeric material. Consequently, such devices differ significantly in form
and function
from the majority of conventional microfluidic devices that are formed from
silicon-based
material.
[0066] The design of the devices enables them to be utilized in combination
with a number of
different heating systems. Thus, the devices are useful in conducting diverse
analyses that
require temperature control. Additionally, those microfluidic devices for use
in heating
applications can incorporate a further design feature to minimize evaporation
of sample from
the reaction sites. Devices of this type in general include a number of guard
channels formed
within the elastomeric device through which water can be flowed to increase
the water vapor
pressure within the elastomeric material from which the device is formed,
thereby reducing
evaporation of sample from the reaction sites.
[0067] The array format of certain of the devices means the devices can
achieve high
throughput. Collectively, the high throughput and temperature control
capabilities make the
devices useful for performing large numbers of nucleic acid amplifications
(e.g., polymerase
chain reaction ¨ PCR). Such reactions will be discussed at length herein as
illustrative of the
utility of the devices, especially of their use in any reaction requiring
temperature control.
13
CA 02807564 2013-02-26
However, it should be understood that the devices are not limited to these
particular
applications. The devices can be utilized in a wide variety of other types of
analyses or
reactions. Examples include analyses of protein-ligand interactions and
interactions
between cells and various compounds. Further examples are provided infra.
III. General Structure of Microfluidic Devices
A. Pumps and Valves
[00681 The microfluidic devices disclosed herein are typically constructed at
least in part
from elastomeric materials and constructed by single and multilayer sofl
lithography
(MSL) techniques and/or sacrificial-layer encapsulation methods (see, e.g.,
Unger et al.
(2000) Science 288:113-116, and PCT Publication WO 01/01025. Utilizing such
methods,
micro fluidic devices can be designed in which solution flow through flow
channels of the
device is controlled, at least in part, with one or more control channels that
are separated
from the flow channel by an elastomeric membrane or segment. This membrane or
segment can be deflected into or retracted from the flow channel with which a
control
channel is associated by applying an actuation force to the control channels.
By controlling
the degree to which the membrane is deflected into or retracted out from the
flow channel,
solution flow can be slowed or entirely blocked through the flow channel.
Using
combinations of control and flow channels of this type, one can prepare a
variety of
different types of valves and pumps for regulating solution flow as described
in extensive
detail in Unger et al. (2000) Science 288:113-116, and PCT Publication
WO/02/43615 and
WO 01/01025.
[00691 The devices provided herein incorporate such pumps and valves to
isolate
selectively a reaction site at which reagents are allowed to react. The
reaction sites can be
located at any of a number of different locations within the device. For
example, in some
matrix-type devices, the reaction site is located at the intersection of a set
of flow channels.
In blind channel devices, the reaction site is located at the end of the blind
channel.
100701 If the device is to be utilized in temperature control reactions (e.g.,
thermocycling
reactions), then, as described in greater detail infra, the elastomeric device
is typically
fixed to a support (e.g., a glass slide). The resulting structure can then be
placed on a
temperature control plate, for example, to control the temperature at the
various reaction
sites. In the case of thermocycling reactions, the device can be placed on any
of a number
of thermocycling plates.
14
CA 02807564 2013-02-26
[0071] Because the devices are made of elastomeric materials that are
relatively optically
transparent, reactions can be readily monitored using a variety of different
detection systems
at essentially any location on the microfluidic device. Most typically,
however, detection
occurs at the reaction site itself (e.g., within a region that includes an
intersection of flow
channels or at the blind end of a flow channel). The fact that the device is
manufactured from
substantially transparent materials also means that certain detection systems
can be utilized
with the current devices that are not usable with traditional silicon-based
microfluidic
devices. Detection can be achieved using detectors that are incorporated into
the device or
that are separate from the device but aligned with the region of the device to
be detected.
B. Guard Channels
[0072] To reduce evaporation of sample and reagents from the elastomeric
microfluidic
devices that are provided herein, a plurality of guard channels can be formed
in the devices.
The guard channels are similar to the control channels in that typically they
are formed in a
layer of elastomer that overlays the flow channels and/or reaction site.
Hence, like control
channels, the guard channels are separated from the underlying flow channels
and/or reaction
sites by a membrane or segment of elastomeric material. Unlike control
channels, however, =
the guard channels are considerably smaller in cross-sectional area. In
general, a membrane
with smaller area will deflect less than a membrane with larger area under the
same applied
pressure. The guard channels are designed to be pressurized to allow solution
(typically
water) to be flowed into the guard channel. Water vapor originating from the
guard channel
can diffuse into the pores of the elastomer adjacent a flow channel or
reaction site, thus
increasing the water vapor concentration adjacent the flow channel or reaction
site and
reducing evaporation of solution therefrom.
[0073] In general, the guard channels are sufficiently small such that when
pressurized the
membrane that separates the guard channel from the underlying flow channel or
reaction site
does not substantially restrict solution flow in, out, or through the flow
channel or reaction
site which the guard channel overlays. When used in this context, the term
"substantially
restrict" or other similar terms means that solution flow is not reduced in,
out or through the
flow channel or reaction site by more than 40%, typically less than 30%,
usually less than
20%, and in some instances less than 10%, as compared to solution flow in, to
or through the
flow channel or reaction site under the same conditions, when the guard
channel is not
pressurized to achieve solution flow therethrough. Usually this means that the
guard
channels have a cross-sectional area of between 100 pm2 and 50, 0001=2, or any
integral or
15
CA 02807564 2013-02-26
non-integral cross-sectional area therebetween. Thus, for example, in some
instances, the
cross-sectional area is less than 50,000 m2, in other instances less than
10,000 tim2, in still
other instances less than 10,00 pm2, and in yet other instances less than 100
pm2. The guard
channels can have any of a variety of shapes including, but not limited to,
circular, elliptical,
square, rectangular, hexagonal and octahedral shapes.
[0074] The guard channels are designed to reduce the evaporation of sample and
reagents
from the device during the time and under the conditions that it takes to
conduct a
thermocycling reaction to less than 50%, in other instance less than 45%, 40%,
35%, 30%,
25%, 20%, 15%, 10%, 5% or 1%. Thus, for example, a typical PCR reaction
involving 40
cycles can be conducted within 120 minutes. The guard channel system is
designed to
reduce evaporation during approximately this time frame to the foregoing set
of limits. To
achieve this level of evaporation reduction, the guard channels are typically
present at a
density of at least 10 lines/cm2 to 1000 lines/cm2, or any integral density
level therebetween.
More specifically, the guard channels are generally at least 25 lines/cm2, in
other instances at
least 50 lines/cm2, in still other instances at least 100 lines/cm2, and in
yet other instances at
least SOO lines/cm2. To achieve this level of evaporation reduction, the guard
channels are
typically present at a spacing between 1 mm to 1 gm as measured from the outer
edge of one
line to the nearest outer edge of adjacent line, or any integral density level
therebetween.
More specifically, the guard channels are generally spaced between 500 p.m to
5 pm, in other
instances between 100 gm to 10 p.m, in still other instances between 75 p.m to
40 pm. Thus,
the spacing is typically at least 1 gm, but is less than 1 mm, in other
instances less than 500
gna, in still other instances less than 400 pm, in yet other instances less
than 300 gm, in other
instances less than 200 p.m, and in still other instances less than 100 gm, 50
gm or 25 pm.
[0075] The guard channels can be formed as a separate network of channels or
can be smaller
channels that branch off of the control channels. The guard channels can
extend across the
device or only a particular region or regions of the device. Typically, the
guard channels are
placed adjacent and over flow channels and reaction sites as these are the
primary locations at
which evaporation is the primary concern. Exemplary locations of guard
channels on certain
matrix devices are illustrated in FIG. 1C, and on certain blind channel
devices in FIGS. 3B
and 3C, and discussed in greater detail infra. =
[0076] The solution flowed through the guard channel includes any substance
that can reduce
evaporation of water. The substance can be one that increases the water vapor
concentration
adjacent a flow line and/or reaction site, or one that while not increasing
the water vapor
16
CA 02807564 2013-02-26
concentration nonetheless blocks evaporation of water from the flow line
and/or reaction site
(blocking agent). Thus, one option is to utilize essentially any aqueous
solution in which
case suitable solutions include, but are not limited to, water and buffered
solution (e.g.,
TaqMan buffer solution, and phosphate buffered saline). Suitable blocking
agents include,
for example, mineral oil.
[0077] Guard channels are typically formed in the elastomer utilizing the MSL
techniques
and/or sacrificial-layer encapsulation methods cited above.
[00781 The following sections describe in greater detail a number of specific
configurations
that can be utilized to conduct a variety of analyses, including analyses
requiring temperature
control (e.g., nucleic acid amplification reactions). It should be understood,
however, that
these configurations are exemplary and that modifications of these systems
will be apparent
to those skilled in the art.
IV. Matrix Design
A. General
[00791 Devices utilizing the matrix design generally have a plurality of
vertical and
horizontal flow channel that intersect to form an array of junctions. Because
a different
sample and reagent (or set of reagents) can be introduced into each of the
flow channels, a
large number of samples can be tested against a relatively large number of
reaction conditions
in a high throughput format. Thus, for example, if a different sample is
introduced into each
of M different vertical flow channels and a different reagent (or set of
reagents) is introduced
into each of N horizontal flow channels, then M x N different reactions can be
conducted at
the same time. Typically, matrix devices include valves that allow for
switchable isolation of
the vertical and horizontal flow channels. Said differently, the valves are
positioned to allow
selective flow just through the vertical flow channels or just through the
horizontal flow
channels. Because devices of this type allow flexibility with respect to the
selection of the
type and number of samples, as well as the number and type of reagents, these
devices are
well-suited for conducting analyses in which one wants to screen a large
number of samples
against a relatively large number of reaction conditions. The matrix devices
can optionally
incorporate guard channels to help prevent evaporation of sample and
reactants.
B. Exemplary Designs and Uses
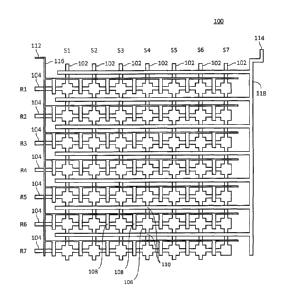
[0030] FIG. 1A provides an illustration of one exemplary matrix device. This
device 100
comprises seven vertical flow channels 102 and seven horizontal flow channels
104 that
17
CA 02807564 2013-02-26
intersect to form an array of 49 different intersections or reaction sites
106. This particular
device thus enables seven samples to be reacted with seven different reagents
or sets of
reagents. Column valves 110 that regulate solution flow in the vertical
direction can be
controlled by control channels 118 that can all be actuated at a single inlet
114.
[0031] Similarly, row valves 108 regulate solution flow in the horizontal
direction; these are
controlled by control channels 116 that are actuated by a single control inlet
112. As shown
in FIG. 1A, the control channels 116 that regulate the row valves 108 vary in
width
depending upon location. When a control channel 116 crosses a vertical flow
channel 102,
the control channel 116 is sufficiently narrow that when it is actuated it
does not deflect into
the vertical flow channel 102 to reduce substantially solution flow
therethrough. However,
the width of the control channel 116 is increased when it overlays one of the
horizontal flow
channels 104; this makes the membrane of the control channel sufficiently
large to block
solution flow through the horizontal flow channel 104.
[0082] In operation, reagents R1-R7 are introduced into their respective
horizontal flow
channels 104 and samples Si-Si are injected into their respective vertical
flow channels 102.
The reagents in each horizontal flow channel 104 thus mix with the samples in
each of the
vertical flow channels 102 at the intersections 106, which in this particular
device are in the
shape of a well or chamber. Thus, in the specific case of a nucleic acid
amplification
reaction, for example, the reagents necessary for the amplification reaction
are introduced
into each of the horizontal flow channels 104. Different nucleic acid
templates are
introduced into the vertical flow channels 102. In certain analyses, the
primer introduced as
part of the reagent mixture that is introduced into each of the horizontal
flow channels 104
might differ between flow channels. This allows each nucleic acid template to
be reacted
with a number of different primers.
[0.083; FIGS. 1.13-13 show enlarged plan views of at-ljaixiii .i.ektution
sites in the device depicted
in FIG. lA to illustrate more specifically how the device operates during an
analysis. For the
purposes of clarity, the intersections 106 are not shown in the form of
reaction wells and
control channels 116, 118 have been omitted, with just the column and row
valves 110, 108
being shown (rectangular boxes). As shown in FIG. 1B, an analysis is commenced
by =
closing column valves 110 and opening row valves 108 to allow solution flow
through
horizontal flow channel 104 while blocking flow through vertical flow channels
102.
Reagent R1 is then introduced into horizontal flow channel 104 and flowed
completely
through the length of the horizontal flow channel 104 such that all the
reaction sites 106 are
filled. Solution flow through horizontal channel 104 can be achieved by an
external pump,
18
CA 02807564 2013-02-26
but more typically is achieved by incorporating a peristaltic pump into the
elastomerie device
itself as described in detail in Unger et al. (2000) Science 288:113-116, and
PCT Publication
WO 01/01025, for example.
[0084] Once R1 has been introduced, row valves 108 are closed and column
valves 102
opened (see FIG. 1C). This allows samples Si and 82 to be introduced into
vertical flow
channels 102 and to flow through their respective flow channels. As the
samples flow
through the vertical flow channels 102, they expel R1 from the reaction sites
106, thus
leaving sample at reaction sites 106. Then, as shown in FIG. 1D, row valves
108 are opened
to allow Si and S2 to diffuse and mix with R1. Thus, a mixture of sample and
reactant
(R1S1 and RI S2) is obtained in the region of each intersection or reaction
site 106. After
allowing a sufficient time for Si and S2 to diffuse with R1, all row and
column valves 108,
110 are closed to isolate Si and S2 within the region of their respective
reaction sites 106 and=
to prevent intermixing of Si and S2 (see FIG. 1E). The mixtures are then
allowed to react
and the reactions detected by monitoring the intersection 106 or the cross-
shaped region that
includes the intersection 106. For analyses requiring heating (e.g.,
thermocycling during
amplification reactions), the device is placed on a heater and heated while
the samples remain
isolated.
[0085] A modified version of the device shown in FIG. lA is shown in FIG. 1F.
The general
structure bears many similarities with that depicted in FIG. 1A, and common
elements in both
figures share the same reference numbers. The device 150 illustrated in FIG.
1F differs in
that pairs of horizontal flow channels 104 are joined to a common inlet 124.
This essentially
enables duplicate sets of reagents to be introduced into two adjacent flow
channels with just a
single injection into inlet 124. The use of a common inlet is further extended
with respect to
the vertical flow channels 102. In this particular example, each sample is
introduced into five
vertical flow channels 102 with a single injection into sample inlet 120.
Thus, with this
particular device, there are essentially ten replicate reactions for each
particular combination
of sample and reagent. Of course, the number of replicate reactions can be
varied as desired
by altering the number of vertical and/or horizontal flow channels 102, 104
that are joined to
a common inlet 120, 124.
[0086] The device shown in FIG. 1F also includes a separate control channel
inlet 128 that
regulates control channel 130 that can be used to govern solution flow toward
outlets 132 and
another control channel inlet 132 that regulates control channel 134 that
regulates solution
flow to outlets 136. Additionally, device 150 incorporates guard channels 138.
In this
particular desigi, the guard channels 138 are formed as part of control
channels 116. As
19
CA 02807564 2013-02-26
indicated supra, the guard channels 138 are smaller than the row valves 108;
consequently,
the membranes of the guard channels 138 are not deflected into the underlying
horizontal
flow channels 104 such that solution flow is disrupted.
[0087] Finally, the design shown in FIG. IF differs in that reaction does not
occur in wells at
5 the intersection of the horizontal and vertical flow lines, but in the
intersection itself.
V. Blind Channel Designs
A. General.
[0083] Devices utilizing a blind channel design have certain features. First,
the devices
include one or more flow channels from which one or more blind channels
branch. As
indicated above, the end region of such channels can serve as a reaction site.
A valve formed
by an overlaying flow channel can be actuated to isolate the reaction site at
the end of the
blind channel. The valves provide a mechanism for switchably isolating the
reaction sites.
[0089] Second, the flow channel network in communication with the blind
channels is
= . configured such that all or most of the reaction sites can
be filled with a single or a limited
-4- 15 number of inlets (e.g., less than 5 or less than 10). The
ability to fill a blind flow channel is
=. . made possible because the devices are made from
elastomeric material. The elastonaeric
material is sufficiently porous such that air within the flow channels and
blind channels can
escape through these pores as solution is introduced into the channels. The
lack of porosity
of materials utilized in other microfluidic devices precludes use of the blind
channel design
because air in a blind channel has no way to escape as solution is injected.
4 [0090] A third characteristic is that one or more reagents
are non-covalently deposited on a
base layer of elastorner during manufacture (see infra for further details on
the fabrication
process) within the reaction sites. The reagent(s) are non-covalently attached
because the
reagents are designed to become dissolved when sample is introduced into the
reaction site.
To maximize the number of analyses, a different reactant or set of reactants
is deposited at
each of the different reaction sites.
[0091] Certain blind channel devices are designed such that the reaction sites
are arranged in
= the form of an array.
[0092] Thus, in those blind channel devices designed for conducting nucleic
acid
amplification reactions, for example, one or more of the reagents required for
conducting the
extension reaction are deposited at each of the reaction sites during
manufacture of the
device. Such reagents include, for example, all or some of the following:
primers,
polymerase, nucleotides, cofactors, metal ions, buffers, intercalating dyes
and the like. To
=
20
CA 02807564 2013-02-26
maximize high throughput analysis, different primers selected to amplify
different regions of
DNA are deposited at each reaction site. Consequently, when a nucleic acid
template is
introduced into the reaction sites via inlet, a large number of extension
reactions can be
performed at different segments of the template. Thermocycling necessary for
an
amplification reaction can be accomplished by placing the device on a
thermocycling plate
and cycling the device between the various required temperatures.
[0093] The reagents can be immobilized in a variety of ways. For example, in
some
instances one or more of the reagents are non-covalently deposited at the
reaction site,
whereas in other instances one or more of the reagents is covalently attached
to the substrate
at the reaction site. If covalently attached, the reagents can be linked to
the substrate via a
linker. A variety of linker types can be utilized such as
photochemical/photolabile linkers,
themolabile linkers, and linkers that can be cleaved enzymatically. Some
linkers are
bifunctional (i.e., the linker contains a functional group at each end that is
reactive with=
groups located on the element to which the linker is to be attached); the
functional groups at
each end can be the same or different. Examples of suitable linkers that can
be used in some
assays include straight or branched-chain carbon linkers, heterocyclic linkers
and peptide
linkers. A variety of types of linkers are available from Pierce Chemical
Company in
Rockford, Elinois and are described in EPA 188,256; U.S. Pat. Nos. 4,671,958;
4,659,839;
4,414,148; 4,669,784; 4,680,338, 4,569, 789 and 4,589,071, and by Eggenweiler,
H.M,
Pharmaceutical Agent Discovery Today 1998, 3, 552. NVOC (6
nitroveratryloxycarbonyl)
linkers and other NVOC-related linkers are examples of suitable photochemical
linkers (see,
e.g., WO 90/15070 and WO 92/10092). Peptides that have protease cleavage sites
are
discussed, for example, in US 5,382,513.
B. Exemplary Designs and Uses
[0094] FIG. 2 is a simplified plan view of one exemplary device utilizing the
blind channel
design. The device 200 includes a flow channel 204 and a set of branch flow
channels 206
branching therefrom that are formed in an elastomeric substrate 202. Each
branch flow
channel 206 terminates in a reaction site 208, thereby forming an array of
reaction sites.
Overlaying the branch flow channels 206 is a control channel 210 that is
separated from the
branch flow channels 206 by membranes 212. Actuation of control channel 210
causes
membranes 212 to deflect into the branch flow channels 206 (i.e., to function
as a valve), thus
enabling each of the reaction sites 208 to be isolated from the other reaction
sites.
21
CA 02807564 2013-02-26
[0095] Operation of such a device involves injecting a test sample into flow
channel 204 with
solution subsequently flowing into each of branch channels 206. Once the
sample has filled
each branch channel 206, control channel 210 is actuated to cause activation
of
valves/membranes 212 to deflect into branch channels 206, thereby sealing off
each of
reaction sites 208. As the sample flows into and remains in reaction sites
208, it dissolves
reagents previously spotted at each of the reaction sites 208. Once dissolved,
the reagents
can react with the sample. Valves 212 prevent the dissolved reagents at each
reaction site
208 from intermixing by diffusion. Reaction between sample and reagents are
then detected,
typically within reaction site 208. Reactions can optionally be heated as
described in the
temperature control section infra.
[0096] FIG. 3A illustrates an example of a somewhat more complex blind flow
channel
design. In this particular design 300, each of a set of horizontal flow
channels 304 are
connected at their ends to two vertical flow channels 302. A plurality of
branch flow
channels 306 extend from each of the horizontal flow channels 304. The branch
flow
channels 304 in this particular design are interleaved such that the branch
channel 306
attached to any given horizontal flow channel 304 is positioned between two
branch channels
306 joined to an immediately adjacent horizontal flow channel 304, or
positioned between a
branch flow channel 306 joined to an immediately adjacent flow channel 304 and
one of the
vertical flow channels 302. As with the design depicted in FIG. 3A, each
branch flow
channel 306 terminates in a reaction site 308. Also consistent with the design
shown in FIG.
3A, a control channel 310 overlays each of the branch channels and is
separated from the
underlying branch channel by membrane 312. The control channel is actuated at
port 316.
The vertical and horizontal flow channels 302, 304 are interconnected such
that injection of
sample into inlet 314 allows solution to flow throughout the horizontal and
vertical flow
uhaunel neiwork and ultimately into each of the reaction sites 308 via the
branch flow
channels 306.
[0097] Hence, in operation, sample is injected into inlet to introduce
solution into each of the
reaction sites. Once the reaction sites are filled, valves/membranes are
actuated to trap
solution within the reaction sites by pressurizing the control channels at
port. Reagents
previously deposited in the reaction sites become resuspended within the
reaction sites,
thereby allowing reaction between the deposited reagents and sample within
each reaction
site. Reactions within the reaction sites are monitored by a detector. Again,
reactions can
optionally be controllably heated according to the methods set forth in the
temperature
control section below.
22
CA 02807564 2013-02-26
[00981 An even more complicated version of the general design illustrated in
FIG. 3A is
shown in FIG. 3B. The device shown in FIG. 3B is one in which the unit
organization of the
horizontal and branch flow channels 302 shown in FIG. 3A is repeated multiple
times. The
device shown in FIG. 3B further illustrates the inclusion of guard channels
320 in those
devices to be utilized in applications that involve heating (e.g.,
thermocycling). An
exemplary orientation of the guard channels 320 with respect to the flow
channels 304 and
branch channels 306 is shown in the enlarged view depicted in FIG. 3C. The
guard channels
320 overlay the branch flow channels 306 and reaction sites 308. As discussed
above, water
is flowed through the guard channels 320 during heating of the device 300 to
increase the
local concentration of water in the device, thereby reducing evaporation of
water from
solution in the flow channels 306 and reaction sites 308.
[00991 The features of blind channel devices discussed at the outset of this
section minimizes
the footprint of the device and enable a large number of reaction sites to be
formed on the
device and for high densities to be obtained. For example, devices of this
type having 2500
reaction sites can readily be manufactured to fit on a standard microscope
slides (25 mm x 75
mm). The foregoing features also enable very high densities of reaction sites
to be obtained
with devices utilizing the blind channel design. For example, densities of at
least 50, 60, 70,
80, 90 or 100 reaction sites/cm2 or any integral density value therebetween
can be readily
obtained. However, certain devices have even higher densities ranging, for
example, between
100 to 4000 reaction sites/ cm2, or any integral density value therebetween.
For instance,
some devices have densities of at least 100, 150, 200, 250, 300, 400, 500,
600, 700, 800, 900
or 1000 sites/ cm2. Devices with very high densities of at least, 2000, 3000,
or 4000 sites/
cm2 are also obtainable. Such high densities directly translate into a very
large number of
reaction sites on the device. Devices utilizing the blind channel architecture
typically have at
least 10400 reaction sites, or any integral number of sites therebetween. More
typically, the
devices have at least 100-1,000 reaction sites, or any integral number of
sites therebetween.
Higher density devices can have even more reaction sites, such as at least
1,000- 100,000
reaction sites, or any integral number of sites therebetween. Thus, certain
devices have at
least 100; 500; 1,000; 2,000; 3,000; 4,000; 5,000; 6,000; 7,000; 8,000; 9,000;
10,000; 20,000;
30,000; 40,000; 50,000; or 100,000 reaction sites depending upon the overall
size of the
device.
[01001 The large number of reaction sites and densities that can be obtained
is also a
consequence of the ability to fabricate very small wells or cavities. For
example, the cavities
or wells typically have a volume of less than 50 nL; in other instances less
than 40 nL, 30
23
CA 02807564 2013-02-26
nL, 20 nL or 10 nL; and in still other instances less than 5 nL or lnL. As a
specific example,
certain devices have wells that are 300 microns long, 300 microns wide and 10
microns deep.
[0101] The blind channel devices provided herein can utilize certain design
features and
methodologies discussed in PCT Applications PCT/US01/44549 (published as WO =
02/43615) and PCT/US02/10875 (published as WO 02/082047), including, for
example,
strategies for filling dead-ended channels, liquid priming, pressurized outgas
priming, as well
as various strategies for displacing gas during the filling of microfluidic
channels.
VI. Hybrid Designs
[0102] Still other devices are hybrids of the matrix and blind fill designs.
The design of
devices of this type is similar to the blind channel device shown in FIG. 3A,
except that each
horizontal flow channel is connected to its own sample inlet port(s) and the
horizontal flow
channels are not interconnected via vertical flow channels. Consequently,
sample introduced
into any given horizontal flow channel fills only that horizontal flow channel
and reaction
sites attached thereto. Whereas, in the blind flow channel device shown in
FIG. 3A, sample
can flow between the horizontal flow channels 304 via vertical flow channels
302_
[0103] An example of devices of this general device is shown in FIG. 4. Device
400
= comprises a plurality of horizontal flow channels 404, each of which has a
plurality of branch
flow channels 406 extending from it and its own sample inlet 414. A control
channel 410
overlays each of the branch flow channels 406 and membrane (valve) 412
separates the
. control channel 410 from the underlying branch flow channel 406. As with
the blind flow
channel design, actuation of the control channel at inlet 416 causes
deflection of membranes
412 into the branch flow channels 406 and isolation of reaction sites 408. In
a variation of
this design, each horizontal flow channel 404 can include an inlet 414 at each
end, thus
allowing sample to be introduced from both ends.
[0104] In some instances, reagents are deposited at the reaction sites during
manufacture of
the device. This enables a large number of samples to be tested under a
relatively large
number of reaction conditions in a short period of time without requiring time-
consuming
additions of reagents as required with the matrix devices. Alternatively,
reaction mixtures
can be prepared prior to injection on the chip. Once the mixtures are
injected, they can be
analyzed or further treated (e.g., heated).
101051 By injecting different samples into each of the horizontal flow
channels, a large
number of samples can be rapidly analyzed. Assuming reagents have been
previously
24
CA 02807564 2013-02-26
deposited at the reaction sites, the presence of the same reagent at each
reaction site
associated with any given horizontal flow channel provides a facile way to
conduct a number
of replicate reactions with each sample. If instead, the reagent at the
reaction sites differ for
any given flow channel, then each sample is essentially simultaneously exposed
to a variety
. 5 of different reaction conditions.
[0106] Thus, the devices provided herein are tailored for a variety of
different types of
investigations. If an investigation involves screening of a relatively large
number of different
- -
samples under user controlled conditions (e.g., 100 samples against 100 user
selected
reagents), then the matrix devices provide a useful solution. If, however, the
investigation
involves analyzing one or a limited number of samples under a wide variety of
reaction
conditions (e.g., one sample against 10,000 reaction conditions), then the
blind channel
design is useful. Finally, if one wants to examine a relatively large number
of samples
against defined reaction conditions without having to inject reagents (e.g.,
100 samples
against 100 previously defined reagents), then the hybrid devices are useful.
VII. Temperature Control
A. Devices and Components
[0107] A number of different options of varying sophistication are available
for controlling
temperature within selected regions of the microfluidic device or the entire
device. Thus, as
used herein, the term temperature controller is meant broadly to refer to a
device or element
that can regulate temperature of the entire microfluidic device or within a
portion of the
microfluidic device (e.g., within a particular temperature region or at one or
more junctions in
a matrix of blind channel-type microfluidic device).
[0108] Generally, the devices are placed on a thermal cycling plate to thermal
cycle the
device. A variety of such plates are readily available from commercial
sources, including for
example the ThermoHybaid Px2 (Franklin, MA), MI Research PTC-200 (South San
Francisco, CA), Eppendorf Part# E5331 (Westbury, NY), Techne Part# 205330
(Princeton,
NJ).
[01091 To ensure the accuracy of thermal cycling steps, in certain devices it
is useful to
incorporate sensors detecting temperature at various regions of the device.
One structure for
- 30 detecting temperature is a thermocouple. Such a thermocouple could be
created as thin film
wires patterned on the underlying substrate material, or as wires incorporated
directly into the
microfabricated elastomer material itself.
25
CA 02807564 2013-02-26
[0110] Temperature can also be sensed through a change in electrical
resistance. For
example, change in resistance of a thermistor fabricated on an underlying
semiconductor
substrate utilizing conventional techniques can be calibrated to a given
temperature change.
Alternatively, a thermistor could be inserted directly into the
microfabricated elastonaer
material. Still another approach to detection of temperature by resistance is
described in
Wu et al. in "MEMS Flow Sensors for Nano-fluidic Applications", Sensors and
Actuators
A 89 152-158 (2001). This paper describes the use of doped polysilicon
structures to both
control and sense temperature. For polysilicon and other semiconductor
materials, the
temperature coefficient of resistance can be precisely controlled by the
identity and amount
of dopant, thereby optimizing performance of the sensor for a given
application.
[0111] Thermo-chromatic materials are another type of structure available to
detect
temperature on regions of an amplification device. Specifically, certain
materials
= dramatically and reproducibly change color as they pass through
different temperatures.
Such a material could be added to the solution as they pass through different
temperatures.
Thenno-chromatic materials could be formed on the underlying substrate or
incorporated
within the elastomer material. Alternatively, thermo-chromatic materials could
be added to
the sample solution in the form of particles.
[0112] Another approach to detecting temperature is through the use of an
infrared camera.
An infrared camera in conjunction with a microscope could be utilized to
determine the
temperature profile of the entire amplification structure. Permeability of the
elastomer
material to radiation would facilitate this analysis.
[0113] Yet another approach to temperature detection is through the use of
pyroelectric
sensors. Specifically, some crystalline materials, particularly those
materials also
exhibiting piezoelectric behavior, exhibit the pyroelectric effect. This
effect describes the
phenomena by which the polarization of the material's crystal lattice, and
hence the voltage
across the material, is highly dependent upon temperature. Such materials
could be
= incorporated onto the substrate or elastomer and utilized to detect
temperature.
[0114] Other electrical phenomena, such as capacitance and inductance, can be
exploited
to detect temperature in accordance with embodiments of the present invention.
B. Verification of Accurate Thennocycling
[0115] As described in greater detail in the fabrication section infra, blind
channel devices
have a base layer onto which reagents are placed. The structure comprising the
two layers
26
CA 02807564 2013-02-26
containing the flow channels and control channels is overlayed on the base
layer such that the =
flow channels are aligned with the deposited reagents. The other side of the
base layer is then
placed upon a substrate (e.g., glass). Usually, the reaction site at which
reaction occurs is
about 100-150 microns above the substrate/glass interface. Using known
equations for
thermal diffusivity and appropriate values for the elastomers and glass
utilized in the device,
one can calculate the time required for the temperature within the reaction
site to reach the
temperature the controller seeks to maintain. The calculated values shown in
Table 1
demonstrate that temperature can rapidly be reached, even using elastomer and
glass layers
considerably thicker than utilized in devices in which the reaction site is
approximately 100-
150 microns (i.e., the typical distance for the devices described herein).
[0116] Table 1: Calculated heat diffusion lengths through PDMS and glass
layers at the
indicated time periods.
1 second 10 seconds , 100 seconds .=
PDMS 400 um 1.26 nun 4.0 mm
Glass 640 um 2.0 mm 6.4 mm
[0117] FIG. 5 illustrates the rapidity at which the desired temperature is
achieved using a
blind channel device.
VIII. Detection
A. General
[0118] A number of different detection strategies can be utilized with the
microfluidic
devices that are provided herein. Selection of the appropriate system is
informed in part on
the type of event and/or agent being detected. The detectors can be designed
to detect a
number of different signal types including, but not limited to, signals from
radioisotopes,
fluorophores, chromophores, electron dense particles, magnetic particles, spin
labels,
molecules that emit chemiluminescence, electrochemically active molecules,
enzymes,
= cofactors, enzymes linked to nucleic acid probes and enzyme
substrates.
[0119] Illustrative detection methodologies suitable for use with the present
microfluidic
devices include, but are not limited to, light scattering, multichannel
fluorescence detection,
UV and visible wavelength absorption, luminescence, differential reflectivity,
and confocal
laser scanning. Additional detection methods that can be used in certain
application include
scintillation proximity assay techniques, radiochemical detection,
fluorescence polarization,
27
CA 02807564 2013-02-26
fluorescence correlation spectroscopy (FCS), time-resolved energy transfer
(TRET),
fluorescence resonance energy transfer (FRET) and variations such as
bioluminescence
resonance energy transfer (BREI). Additional detection options include
electrical resistance,
resistivity, impedance, and voltage sensing.
[0120] Detection occurs at a "detection section," or "detection region." These
terms and
other related terms refer to the portion of the microfluidic device at which
detection occurs.
= As indicated above, with devices utilizing the blind channel design,
the detection section is
generally the reaction site as isolated by the valve associated with each
reaction site. The
detection section for matrix-based devices is usually within regions of flow
channels that are
adjacent an intersection, the intersection itself, or a region that
encompasses the intersection
and a surrounding region.
[0121] The detection section can be in communication with one or more
microscopes, diodes,
light stimulating devices (e.g., lasers), photomultiplier tubes, processors
and combinations of
the foregoing, which cooperate to detect a signal associated with a particular
event and/or
^,- 15 agent. Often the signal being detected is an optical signal that is
detected in the detection
section by an optical detector. The optical detector can include one or more
photodiodes
(e.g., avalanche photodiodes), a fiber-optic light guide leading, for example,
to a
- photomultiplier tube, a microscope, and/or a video camera (e.g., a
CCD camera).
101221 Detectors can be microfabricated within the microfluidic device, or can
be a separate
element. If the detector exists as a separate element and the microfluidic
device. includes a
plurality of detection sections, detection can occur within a single detection
section at any
given moment. Alternatively, scanning systems can be used. For instance,
certain automated
systems scan the light source relative to the microfluidic device; other
systems scan the
emitted light over a detector, or include a multichannel detector. As a
specific illustrative
example, the microfluidic device can be attached to a translatable stage and
scanned under a
microscope objective. A signal so acquired is then routed to a processor for
signal
interpretation and processing. Arrays of photomultiplier tubes can also be
utilized.
Additionally, optical systems that have the capability of collecting signals
from all the
different detection sections simultaneously while determining the signal from
each section
can be utilized.
10123] External detectors are usable because the devices that are provided are
completely or
= largely manufactured of materials that are optically transparent at
the wavelength being
monitored. This feature enables the devices described herein to utilize a
number of optical
detection systems that are not possible with conventional silicon-based
microfluidic devices.
.1
28
CA 02807564 2013-02-26
[0124] A detector can include a light source for stimulating a reporter that
generates a
detectable signal. The type of light source utilized depends in part on the
nature of the
reporter being activated. Suitable light sources include, but are not limited
to, lasers, laser
diodes and high intensity lamps. If a laser is utilized, the laser can be
utilized to scan across a
set of detection sections or a single detection section. Laser diodes can be
microfabricated
into the microfluidic device itself. Alternatively, laser diodes can be
fabricated into another
device that is placed adjacent to the microfluidic device being utilized to
conduct a thermal
cycling reaction such that the laser light from the diode is directed into the
detection section.
[0125] Detection can involve a number of non-optical approaches as well. For
example, the
detector can also include, for example, a temperature sensor, a conductivity
sensor, a
potentiometric sensor (e.g., pH electrode) and/or an amperometric sensor
(e.g., to monitor
oxidation and reduction reactions).
[0126] A number of commercially-available external detectors can be utilized.
Many of
these are fluorescent detectors because of the ease in preparing fluorescently
labeled reagents.
Specific examples of detectors that are available include, but are not limited
to, Applied
Precision ArrayWoRx (Applied Precision, Issaquah, WA)). =
B. Detection of Amplified Nucleic Acids
1. Intercalation Dyes
[0127] Certain intercalation dyes that only fluoresce upon binding to double-
stranded DNA
can be used to detect double-stranded amplified DNA. Examples of suitable dyes
include,
but are not limited to, SYBRI'm and Pico Green (from Molecular Probes, Inc. of
Eugene, OR),
ethidium bromide, propidium iodide, chromomycin, acridine orange, Hoechst
33258, Toto-1,
Yoyo-1, and DAPI (4',6-diamidino-2-phenylindole hydrochloride). Additional
discussion
regarding the use of intercalation dyes is provided by Zhu et al., Anal. Chem.
66:1941-1948
(1994),
2. FRET Based Detection Methods
[0128] Detection methods of this type involve detecting a change in
fluorescence from a
donor (reporter) and/or acceptor (quencher) fluorophore in a donor/acceptor
fluorophore pair.
The donor and acceptor fluorophore pair are selected such that the emission
spectrum of the
donor overlaps the excitation spectrum of the acceptor. Thus, when the pair of
fluorophores
are brought within sufficiently close proximity to one another, energy
transfer from the donor
to the acceptor can occur. This energy transfer can be detected.
29
CA 02807564 2013-02-26
[0129] FRET and template extension reactions. These methods generally utilize
a primer
labeled with one member of a donor/acceptor pair and a nucleotide labeled with
the other
member of the donor/acceptor pair. Prior to incorporation of the labeled
nucleotide into the
primer during an template-dependent extension reaction, the donor and acceptor
are spaced
far enough apart that energy transfer cannot occur. However, if the labeled
nucleotide is
incorporated into the primer and the spacing is sufficiently close, then
energy transfer occurs
and can be detected. These methods are particularly useful in conducting
single base pair
extension reactions in the detection of single nucleotide polymotphisms (see
infra) and are
described in U.S. Patent No. 5,945,283 and PCT Publication WO 97/22719.
[0130] Ouantitative RT-PCR. A variety of so-called "real time amplification"
methods or
"real time quantitative PCR" methods can also be utilized to determine the
quantity of a
target nucleic acid present in a sample by measuring the amount of
amplification product
formed during or after the amplification process itself. Fluorogenic nuclease
assays are one
specific example of a real time quantitation method which can be used
successfully with the
devices described herein. This method of monitoring the formation of
amplification product
involves the continuous measurement of PCR product accumulation using a dual-
labeled
fluorogenic oligonucleotide probe -- an approach frequently referred to in the
literature as the
"TaqMan" method.
[0131] The probe used in such assays is typically a short (ca. 20-25 bases)
polynucleotide
that is labeled with two different fluorescent dyes. The 5' terminus of the
probe is typically
attached to a reporter dye and the 3' terminus is attached to a quenching dye,
although the
dyes can be attached at other locations on the probe as well. The probe is
designed to have at
least substantial sequence complementarity with the probe binding site on the
target nucleic
acid. Upstream and downstream PCR primers that bind to regions that flank the
probe
binding site are also included in the reaction mixture.
[0132] When the probe is intact, energy transfer between the two fluorophors
occurs and the
quencher quenches emission from the reporter. During the extension phase of
PCR, the
probe is cleaved by the 5' nuclease activity of a nucleic acid polymerase such
as Tag
polyrnerase, thereby releasing the reporter from the polynucleotide-quencher
and resulting in
an increase of reporter emission intensity which can be measured by an
appropriate detector.
[0133] One detector which is specifically adapted for measuring fluorescence
emissions such
as those created during a fluorogenic assay is the ABI 7700 manufactured by
Applied
Biosystems, Inc. in Foster City, CA. Computer software provided with the
instrument is
capable of recording the fluorescence intensity of reporter and quencher over
the course of
30
CA 02807564 2013-02-26
the amplification. These recorded values can then be used to calculate the
increase in
normalized reporter emission intensity on a continuous basis and ultimately
quantify the
amount of the mRNA being amplified.
[0134] Additional details regarding the theory and operation of
fluorogenic.methods for
making real time determinations of the concentration of amplification products
are described,
for example, in U.S. Pat Nos. 5,210,015 to Gelfand, 5,538,848 to Livak, etal.,
and 5,863,736
to Haaland, as well as Heid, C.A., et al., Genome Research, 6:986-994 (1996);
Gibson,
U.E.M, et al., Genome Research 6:995-1001 (1996); Holland, P. M., et al.,
Proc. Natl. Acad.
Sci. USA 88:7276-7280, (1991); and Livak, K.J., et al., PCR Methods and
Applications 357-
362 (1995). =
[0135] Thus, as the amplification reaction progresses, an increasing amount of
dye becomes
bound and is accompanied by a concomitant increase in signal.
[0136] Intercalation dyes such as described above can also be utilized in a
different approach
to quantitative PCR methods. As noted above, these dyes preferentially bind to
double
stranded DNA (e.g., SYDR GREEN) and only generate signal once bound. Thus, as
an
amplification reaction progresses, an increasing amount of dye becomes bound
and is
accompanied by a concomitant increase in signal that can be detected..
[0137] Molecular Beacons: With molecular beacons, a change in conformation of
the probe
as it hybridizes to a complementary region of the amplified product results in
the formation
of a detectable signal. The probe itself includes two sections: one section at
the 5' end and
the other section at the 3' end. These sections flank the section of the probe
that anneals to
the probe binding site and are complementary to one another. One end section
is typically
attached to a reporter dye and the other end section is usually attached to a
quencher dye.
[0138] In solution, the two end sections can hybridize with each other to form
a hairpin loop.
In this conformation, the reporter and quencher dye are in sufficiently close
proximity that
fluorescence from the reporter dye is effectively quenched by the quencher
dye. Hybridized
probe, in contrast, results in a linearized conformation in which the extent
of quenching is
decreased. Thus, by monitoring emission changes for the two dyes, it is
possible to indirectly
monitor the formation of amplification product. Probes of this type and
methods of their use
is described further, for example, by Piatek, A.S., et al., Nat. Biotechnol.
16:359-63 (1998);
Tyagi, S. and Kramer, F.R., Nature Biotechnology 14:303-308 (1996); and Tyagi,
S. et al.,
Nat. Biotechuol. 16:49-53 (1998).
31 =
CA 02807564 2013-02-26
[0139] Invader: Invader assays (Third Wave Technologies, (Madison, WI)) are
used for SNP
genotyping and utilize an oligonucleotide, designated the signal probe, that
is complementary
to the target nucleic acid (DNA or RNA) or polymorphism site. A second
oligonucleotide,
designated the Invader Oligo, contains the same 5' nucleotide sequence, but
the 3' nucleotide
sequence contains a nucleotide polymorphism. The Invader Oligo interferes with
the binding of
the signal probe to the target nucleic acid such that the 5' end of the signal
probe forms a "flap"
at the nucleotide containing the polymorphism. This complex is recognized by a
structure
specific endonuclease, called the Cleavase enzyme. Cleavase cleaves the 5'
flap of the
nucleotides. The released flap binds with a third probe bearing FRET labels,
thereby forming
another duplex structure recognized by the Cleavase enzyme. This time the
Cleavase enzyme
cleaves a fluorophore away from a quencher and produces a fluorescent signal.
For SNP
genotyping, the signal probe will be designed to hybridize with either the
reference (wild type)
allele or the variant (mutant) allele. Unlike PCR, there is a linear
amplification of signal with
no amplification of the nucleic acid. Further details sufficient to guide one
of ordinary skill in
the art is provided by, for example, Neri, B.P., et al., Advances in Nucleic
Acid and Protein
Analysis 3826:117-125, 2000).
[0140] Nasba: Nucleic Acid Sequence Based Amplification (NASBA) is a detection
method
using RNA as the template. A primer complementary to the RNA contains the
sequence for
the T7 promoter site. This primer is allowed to bind with the template RNA and
Reverse
Transcriptase (RT) added to generate the complementary strand from 3' to 5'.
RNase H is
subsequently added to digest away the RNA, leaving single stranded cDNA
behind. A second
copy of the primer can then bind the single stranded cDNA and make double
stranded cDNA.
T7 RNA polymerase is added to generate many copies of the RNA from the T7
promoter site
that was incorporated into the cDNA sequence by the first primer. All the
enzymes
mentioned are capable of functioning at 41 C. (See, e.g., Compton, J. Nucleic
Acid
Sequence-based Amplification, Nature 350: 91-91, 1991.)
[0141] Scorpion. This method is described, for example, by Thelwell N., et al.
Nucleic Acids
Research, 28:3752-3761, 2000, and which figure 20 depicts the scheme thereof,
wherein
Scorpion probing mechanism is as follows. Step 1: initial denaturation of
target and Scorpion
stem sequence. Step 2: annealing of Scorpion primer to target. Step 3:
extension of Scorpion
primer produces double-stranded DNA. Step 4: denaturation of double-stranded
DNA
produced in step 3. This gives a single-stranded target molecule with the
Scorpion primer
attached. Step 5: on cooling, the Scorpion probe sequence binds to its target
in an
intramolecular manner. This is favoured
32
CA 02807564 2013-02-26
=
over the intermolecular binding of the complementary target strand. A Scorpion
(as shown in
Fig. ) consists of a specific probe sequence that is held in a hairpin loop
configuration by
complementary stem sequences on the 5' and 3' sides of the probe. The
fluorophore attached
to the 5'-end is quenched by a moiety (normally methyl red) joined to the 3'-
end of the loop.
= 5 The hairpin loop is linked to the 5'-end of a primer via a PCR stopping
sequence (stopper).
After extension of the primer during PCR amplification, the specific probe
sequence is able to
bind to its complement within the same strand of DNA. This hybridization event
opens the
hairpin loop so that fluorescence is no longer quenched and an increase in
signal is observed.
The PCR stopping sequence prevents read-through, that could lead to opening of
the hairpin
loop in the absence of the specific target sequence. Such read-through would
lead to the
detection of non-specific PCR products, e.g. primer dimers or mispriming
events.
3. Capacitive DNA Detection
101421 There is a linear relationship between DNA concentration and the change
in
capacitance that is evoked by the passage of nucleic acids across a 1-kHz
electric field. This
relationship has been found to be species independent. (See, e.g., Sohn, et
al. (2000) Proc.
Natl. Acad. Sci. U.S.A. 97:10687-10690). Thus, in certain devices, nucleic
acids within the
flow channel (e.g., the substantially circular flow channel of FIG. 1 or the
reaction chambers
of FIG. 2) are subjected to such a field to determine concentration of
amplified product.
Alternatively, solution containing amplified product is withdrawn and then
subjected to the
electric field.
Composition of Mixtures for Conducting Reactions
[0143] Reactions conducted with the microfluidic devices disclosed herein are
typically
conducted with certain additives to enhance the reactions. So, for example, in
the case of
devices in which reagents are deposited, these additives can be spotted with
one or more
reactants at a reaction site, for instance. One set of additives are blocking
reagents that block
protein binding sites on the elastomeric substrate. A wide variety of such
compounds can be
utilized including a number of different proteins (e.g., gelatin and various
albumin proteins,
= such as bovine serum albumin) and glycerol.
[01.44] A detergent additive can also be useful. Any of a number of different
detergents can
be utilized. Examples include, but are not limited to SDS and the various
Triton detergents.
[0145] In the specific case of nucleic acid amplification reactions, a number
of different types
of additives can be included. One category are enhancers that promote the
amplification
33
CA 02807564 2013-02-26
reaction. Such additives include, but are not limited to, reagents that reduce
secondary
structure in the nucleic acid (e.g., betaine), and agents that reduce
naispriming events (e.g.,
. = tetramethylammonium chloride).
[0146] It has also been found in conducting certain amplification reactions
that some
5 polymerases give enhanced results. For example, while good results
were obtained with
AmpliTaq Gold polymerase (Applied Biosystenas, Foster City, CA) from Thennus
aquaticus,
improved reactions were in some instances obtained using DyNAzyme polymerase
from
' Finnzyme, Espoo, Finland. This polymerase is from the
thermophilic bacterium, Therms
brockianus. Other exemplary polymerases that can be utilized include, but are
not limited to,
10 rTH polymerase XL, which is a combination of Thermus thermophilus
(Tth) and
nermococcus litoralis (Tli), hyperthermo-philic archaebacterium Pyrosoccus
woesei (Pwo),
= and Tgo DNA Polymerase.
[0147] Further details regarding additives useful in conducting reactions with
certain of the
devices disclosed herein, including nucleic acid amplification reactions, are
provided in
15 Example 1 infra.
X. Exemplary Applications
[0148] Because the microfiuidic devices provided herein can be manufactured to
include a
large number of reaction sites, the devices are useful in a wide variety of
screening and
analytical methods. In general, the devices can be utilized to detect
reactions between species
20 that react to form a detectable signal, or a product that upon
interaction with another species
generates a detectable signal. In view of their use with various types of
temperature control
systems, the devices can also be utilized in a number of different types of
analyses or
reactions requiring temperature control.
A. Nucleic Acid Amplification Reactions
25 [0149] The devices disclosed herein can be utilized to conduct
essentially any type of nucleic
acid amplification reaction. Thus, for example, amplification reactions can be
linear
amplifications, (amplifications with a single primer), as well as exponential
amplifications
(i.e., amplifications conducted with a forward and reverse primer set).
[0150] When the blind channel type devices are utilized to perform nucleic
acid amplification
30 reactions, the reagents that are typically deposited within the
reaction sites are those reagents
necessary to perform the desired type of amplification reaction. Usually this
means that some
or all of the following are deposited, primers, polymerase, nucleotides, metal
ions, buffer, and
34
CA 02807564 2013-02-26
cofactors, for example. The sample introduced into the reaction site in such
cases is the
nucleic acid template. Alternatively, however, the template can be deposited
and the
amplification reagents flowed into the reaction sites. As discussed supra,
when the matrix
device is utilized to conduct an amplification reaction, samples containing
nucleic acid
template are flowed through the vertical flow channels and the amplification
reagents through
the horizontal flow channels or vice versa. =
[0151] While PCR is perhaps the best known amplification technique. The
devices are not
limited to conducting PCR amplifications. Other types of amplification
reactions that can be
conducted include, but are not limited to, (i) ligase chain reaction (LCR)
(see Wu and
Wallace, Genomics 4:560 (1989) and Landegren et al., Science 241:1077 (1988));
(ii)
transcription amplification (see Kwoh et al., Proc. Natl. Acad. Sci. USA
86:1173 (1989)); (iii)
self-sustained sequence replication (see Guatelli et al., Proc. Nat. Acad.
Sci. USA, 87:1874
(1990)); and (iv) nucleic acid based sequence amplification (NASBA) (see,
SooknEman, R.
and Malek, L., BioTechnology 13: 563-65 (1995)).
[01521 Detection of the resulting amplified product can be accomplished using
any of the
detection methods described supra for detecting amplified DNA.
B. SNP Analysis and Genotyping
1. General
[01531 Many diseases linked to genome modifications, either of the host
organism or of
infectious organisms, are the consequence of a change in a small number of
nucleotides,
frequently involving a change in a single nucleotide. Such single nucleotide
changes are
referred to as single nucleotide polymorphisms or simply SNPs, and the site at
which the SNP
occurs is typically referred to as a polymorphic site. The devices described
herein can be
utilized to determine the identify of a nucleotide present at such polymorphic
sites. As an
extension of this capability, the devices can be utilized in genotyping
analyses. Genotyping
involves the determination of whether a diploid organism (i.e., an organism
with two copies
of each gene) contains two copiei of a reference allele (a reference-type
homozygote), one
copy each of the reference and a variant allele (i.e., a heterozygote), or
contains two copies of
the variant allele (i.e., a variant-type homozygote). When conducting a
genotyping analysis,
the methods of the invention can be utilized to interrogate a single variant
site. However, as
described fiuther below in the section on multiplexing, the methods can also
be used to
35
CA 02807564 2013-02-26
determine the genotype of an individual in many different DNA loci, either on
the same gene,
. = different genes or combinations thereof.
= [0154] Devices to be utilized for conducting genotyping analyses
are designed to utilize
reaction sites of appropriate size to ensure from a statistical standpoint
that a copy of each of
the two alleles for a diploid subject are present in the reaction site at a
workable DNA
= concentrations. Otherwise, an analysis could yield results
suggesting that a heterozygote is a
homozygote simply because a copy of the second allele is not present at the
reaction site.
Table 2 below indicates the number of copies of the genome present in a 1 111
reaction volume
at various exemplary DNA concentrations that can be utilized with the devices
described
herein.
[0155] Table 2: Number of genome copies present in a 1 riL volume at the
indicated DNA
= concentration.
Volume (nL) [DNA] (ug/uL)
1 0.33 100
1 0.10 32
1 0.05 16
1 0.01 3
1 0.003 1
,
[0156] As a general matter, due to stochastic proportioning of the sample, the
copy number
present before an amplification reaction is commenced determines the likely
error in the
meamirinT1Mt_ Clenfltypng analyses using certain devices arc typically
conducted with
samples having a DNA concentration of approximately 0.10 ug/uL, although the
current
inventors have run successful TaqMan reactions at concentrations in which
there is a single
genome per reaction site.
2. Methods
[0157] Genotyping analyses can be conducted using a variety of different
approaches. In
these methods, it is generally sufficient to obtain a "yes" or "no" result,
i.e., detection need
= only be able to answer the question whether a given allele is
present. Thus, analyses can be
conducted only with the primers or nucleotides necessary to detect the
presence of one allele
36
CA 02807564 2013-02-26
potentially at a polymorphic site. However, more typically, primers and
nucleotides to detect -
the presence of each allele potentially at the polymorphic site are included.
Examples of
suitable approaches follow. =,.
[0158] Sinkle Base Pair Extension (SBPE) Reactions. SBPE reactions are one
technique
specifically developed for conducting genotyping analyses. Although a number
of SPBE
assays have been developed, the general approach is quite similar. Typically,
these assays
involve hybridizing a primer that is complementary to a target nucleic acid
such that the 3'
end of the primer is immediately 5' of the variant site or is adjacent
thereto. Extension is
conducted in the presence of one or more labeled non-extendible nucleotides
that are
complementary to the nucleotide(s) that occupy the variant site and a
polymerase. The non-
extendible nucleotide is a nucleotide analog that prevents further extension
by the polymerase
once incorporated into the primer. If the added non-extendible nucleotide(s)
is(are)
complementary to the nucleotide at the variant site, then a labeled non-
extendible nucleotide
is incorporated onto the 3' end of the primer to generate a labeled extension
product. Hence,
extended primers provide an indication of which nucleotide is present at the
variant site of a
target nucleic acid. Such methods and related methods are discussed, for
example, in U.S.
Patent Nos. 5,846,710; 6,004,744; 5,888,819; 5,856,092; and 5,710,028; and in
WO
92/16657.
[0159] Detection of the extended products can be detected utilizing the FRET
detection
approach described for extension reactions in the detection section supra.
Thus, for example,
using the devices described herein, a reagent mixture containing a primer
labeled with one
member of a donor/acceptor fluorophore, one to four labeled non-extendible
nucleotides
(differentially labeled if more than one non-extendible nucleotide is
included), and
polymerase are introduced (or previously spotted) at a reaction site. A sample
containing
template DNA is then introduced into the reaction site to allow template
extension to occur.
Any extension product formed is detected by the formation of a FRET signal
(see, e.g., U.S.
Patent No. 5,945,283 and PCT Publication WO 97/22719.). The reactions can
optionally be
thermocycled to increase signal using the temperature control methods and
apparatus
described above.
[0160] Quantitative PCR. Genotyping analyses can also be conducted using the
quantitative
PCR methods described earlier. In this case, differentially labeled probes
complementary to
each of the allelic forms are included as reagents, together with primers,
nucleotides and
polymerase. However, reactions can be conducted with only a single probe,
although this can
create ambiguity as to whether lack of signal is due to absence of a
particular allele or simply
37
CA 02807564 2013-02-26
a failed reaction. For the typical biallelic case in which two alleles are
possible for a
= - polymorphic site, two differentially labeled probes, each
perfectly complementary to one of
the alleles are usually included in the reagent mixture, together with
amplification primers,
nucleotides and polymerase. Sample containing the target DNA is introduced
into the
-= ' 5 reaction site. If the allele to which a probe is
complementary is present in the target DNA,
then amplification occurs, thereby resulting in a detectable signal as
described in the
. detection above. Based upon which of the differential signal
is obtained, the identity of the
nucleotide at the polymorphic site can be determined. If both signals are
detected, then both
alleles are present. Thermocycling during the reaction is performed as
described in the
temperature control section supra.
C. Gene Expression Analysis
- 1. General
10161] Gene expression analysis involves determining the level at which one or
more genes
is expressed in a particular cell. The determination can be qualitative, but
generally is
quantitative. In a differential gene expression analysis, the levels of the
gene(s) in one cell
(e.g., a test cell) are compared to the expression levels of the same genes in
another cell
(control cell). A wide variety of such comparisons can be made. Examples
include, but are
not limited to, a comparison between healthy and diseased cells, between cells
from an P.
individual treated with one drug and cells from another untreated individual,
between cells
exposed to a particular toxicant and cells not exposed, and so on. Genes whose
expression
levels vary between the test and control cells can serve as markers and/or
targets for therapy.
For example, if a certain group of genes is found to be up-regulated in
diseased cells rather
than healthy cells, such genes can serve as markers of the disease and can
potentially be
utilized as the basis for diagnostic tests. These genes could also he tprgets_
A strategy for
treating the disease might include procedures that result in a reduction of
expression of the
up-regulated genes.
[0162] The design of the devices disclosed herein is helpful in facilitating a
variety of gene
expression analyses. Because the devices contain a large number of reaction
sites, a large
number of genes and/or samples can be tested at the same time. Using the blind
flow channel
devices, for instance, the expression levels of hundreds or thousands of genes
can be
determined at the same time. The devices also facilitate differential gene
expression
analyses. With the matrix design, for example, a sample obtained from a
healthy cell can be
tested in one flow channel, with a sample from a diseased cell run in an
immediately adjacent
38
CA 02807564 2013-02-26
channel. This feature enhances the ease of detection and the accuracy of the
results because
the two samples are run on the same device at the same time and under the same
conditions.
2. Sample Preparation and Concentration
[0163] To measure the transcription level (and thereby the expression level)
of a gene or
= 5 genes, a nucleic acid sample comprising mRNA transcript(s) of the gene(s)
or gene
fragments, or nucleic acids derived from the naRNA transcript(s) is obtained.
A nucleic acid
derived from an mRNA transcript refers to a nucleic acid for whose synthesis
the mRNA
transcript or a subsequence thereof has ultimately served as a template. Thus,
a cDNA
reverse transcribed from an mRNA, an RNA transcribed from that cDNA, a DNA
amplified
from the cDNA, an RNA transcribed from the amplified DNA, are all derived from
the
mRNA transcript and detection of such derived products is indicative of the
presence and/or
abundance of the original transcript in a sample. Thus, suitable samples
include, but are not
limited to, mRNA transcripts of the gene or genes, cDNA reverse transcribed
from the
mRNA, cRNA transcribed from the cDNA, DNA amplified from the genes, RNA
transcribed
from amplified DNA..
[0164] In some methods, a nucleic acid sample is the total mRNA isolated from
a biological
sample; in other instances, the nucleic acid sample is the total RNA from a
biological sample.
The term "biological sample", as used herein, refers to a sample obtained from
an organism
or from components of an organism, such as cells, biological tissues and
fluids. In some
methods, the sample is from a human patient. Such samples include sputum,
blood, blood
cells (e.g., white cells), tissue or fine needle biopsy samples, urine,
peritoneal fluid, and
fleural fluid, or cells therefrom. Biological samples can also include
sections of tissues such
as frozen sections taken for histological purposes. Often two samples are
provided for
purposes of comparison. The samples can be, for example, from different cell
or tissue types,
from different individuals or from the same original sample subjected to two
different
treatments (e.g., drug-treated and control).
[0165] Any RNA isolation technique that does not select against the isolation
of mRNA can
be utilized for the purification of such RNA samples. For example, methods of
isolation and
purification of nucleic acids are described in detail in WO 97/10365, WO
97/27317, Chapter
3 of Laboratory Techniques in Biochemistry and Molecular Biology:
Hybridization With
Nucleic Acid Probes, Part I. Theory and Nucleic Acid Preparation, (P. Tijssen,
ed.) Elsevier,
N.Y. (1993); Chapter 3 of Laboratory Techniques in Biochemistry and Molecular
Biology:
Hybridization With Nucleic Acid Probes, Part 1. Theory and Nucleic Acid
Preparation, (P.
39
CA 02807564 2013-02-26
Tijssen, ed.) Elsevier, N.Y. (1993); and Sambrook et al., Molecular Cloning: A
Laboratory
r Manual, Cold Spring Harbor Press, N.Y., (1989);
Current Protocols in Molecular Biology,
(Ausubel, F.M. et al., eds.) John Wiley & Sons, Inc., New York (1987-1993).
Large numbers
of tissue samples can be readily processed using techniques known in the art,
including, for
5 example, the single-step RNA isolation process of Chomczynski, P.
described in U.S. Pat.
No. 4,843,155.
101661 In gene expression analyses utilizing the devices that are described, a
significant
factor affecting the results is the concentration of the nucleic acid in the
sample. At low copy
number, noise is related to the square root of copy number. Thus, the level of
error that is
10 deemed acceptable governs the copy number required. The required copy
number in the
particular sample volume gives the required DNA concentration. Although not
necessarily
= optimal, quantitation reactions can be conducted with an
error level of up to 50%, but
= preferably is less. Assuming a 1 nanoliter volume, the DNA
concentrations required to
= .= achieve a particular error level are shown in Table
3. As can be seen, 1 nanoliter volumes
15 such as used with certain of the devices have sufficient copies of
gene expression products at
concentrations that are workable with inicrofluidic devices.
[0167] Table 3: Gene Expression DNA Quantity
=
En-or (%) N (Copy No) Volume (nL)
[DNA] (10-12
2 2500 1
4.2
, =
10 100 1
0.17
25 16 1
0.027
50 4 1
0.0066
[0168] A further calculation demonstrates that the certain of the devices
provided herein
which utilize a 1 nanoliter reaction site contain sufficient DNA to achieve
accurate
20 expression results. Specifically, a typical mRNA preparation
procedure yields approximately
.>. 10 ug of mRNA. It has been demonstrated that typically
there are 1 to 10,000 copies of each
mRNA per cell. Of the mRNAs that are expressed within any given cell,
approximately the
four most common messages comprise about 13% of the total mRNA levels. Thus,
such
highly expressed messages comprise 1.3 ug of mRNA (each is 4 x 1042 mole or
25 approximately 2.4 x 1012 copies). In view of the foregoing expression
ranges, rare messages
40
CA 02807564 2013-02-26
are expeetea to oe present at a level ot about 2 x HY- copies. If in a
standard analysis the
mRNA sample is dissolved in 10 ul, the concentration of a rare message is
approximately 2 x
107 copies/u1; this concentration corresponds to 20,000 copies per 1 nl well
(or 4 x 1011 M).
3. Methods
[0169] Because expression analysis typically involves a quantitative analysis,
detection is
typically achieved using one of the quantitative real time PCR methods
described above.
Thus, if a TaqMan approach is utilized, the reagents that are introduced (or
previously
spotted) in the reaction sites can include one or all of the following:
primer, labeled probe,
nucleotides and polymerase. If an intercalation dye is utilized, the reagent
mixture typically
includes one or all of the following: primer, nucleotides, polymerase, and
intercalation dye.
D. Multiplexing
[0170] The array-based devices described herein (see, e.g., FIGS. 1A, 1F, 2,
3A and 3B and
accompanying text) are inherently designed to conduct a large number of
amplification
reactions at the same time. This feature, however, can readily be further
expanded. upon by
conducting multiple analyses (e.g., genotyping and expression analyses) within
each reaction
site.
[0171] Multiplex amplifications can even be performed within a single reaction
site by, for
example, utilizing a plurality of primers, each specific for a particular
target nucleic acid of
interest, during the thermal cycling process. The presence of the different
amplified products
can be detected using differentially labeled probes to conduct a quantitative
RT-PCR reaction
or by using differentially labeled molecular beacons (see supra). In such
approaches, each
differentially labeled probes is designed to hybridize only to a particular
amplified target. By
judicious choice of the different labels that are utilized, analyses can be
conducted in which
the different labels are excited and/or detected at different wavelengths in a
single reaction.
Further guidance regarding the selection of appropriate fluorescent labels
that are suitable in
such approaches include: Fluorescence Spectroscopy (Pesce et al., Eds.) Marcel
Dekker,
New York, (1971); White et al., Fluorescence Analysis: A Practical Approach,
Marcel
= Dekker, New York, (1970); Berlman, Handbook of Fluorescence Spectra of
Aromatic
Molecules, 211d ed., Academic Press, New York, (1971); Griffiths, Colour and
Constitution of
Organic Molecules, Academic Press, New York, (1976); Indicators (Bishop, Ed.).
Pergamon
Press, Oxford, 19723; and Haugland, Handbook of Fluorescent Probes and
Research
Chemicals, Molecular Probes, Eugene (1992).
41
CA 02807564 2013-02-26
[0172] Multiple genotyping and expression analyses can optionally be conducted
at each
reaction site. If quantitative PCR methods such as TaqMan is utilized, then
primers for
amplifying different regions of a target DNA of interest are included within a
single
reaction site. Differentially labeled probes for each region are utilized to
distinguish
product that is formed.
E. Non-Nucleic Acid Analyses
[0173] While useful for conducting a wide variety of nucleic acid analyses,
the devices can
also be utilized in a number of other applications as well. As indicated
earlier, the devices
can be utilized to analyze essentially any interaction between two or more
species that
generates a detectable signal or a reaction product that can be reacted with a
detection
reagent that generates a signal upon interaction with the reaction product.
[0174] Thus, for example, the devices can be utilized in a number of screening
applications to identify test agents that have a particular desired activity.
As a specific
example, the devices can be utilized to screen compounds for activity as a
substrate or
inhibitor of one or more enzymes. In such analyses, test compound and other
necessary
enzymatic assay reagents (e.g., buffer, metal ions, cofactors and substrates)
are introduced
(if not previously deposited) in the reaction site. The enzyme sample is then
introduced and
reaction (if the test compound is a substrate) or inhibition of the reaction
(if the test
compound is an inhibitor) is detected. Such reactions or inhibition can be
accomplished by
standard techniques, such as directly or indirectly monitoring the loss of
substrate and/or
appearance of product.
[0175] Devices with sufficiently large flow channels and reaction sites can
also be utilized
to conduct cellular assays to detect interaction between a cell and one or
more reagents. For
instance, certain analyses involve determination of whether a particular cell
type is present
in a sample. One example for accomplishing this is to utilize cell-specific
dyes that
preferentially react with certain cell types. Thus, such dyes can be
introduced into the
reaction sites and then cells added. Staining of cells can be detected using
standard
microscopic techniques. As another illustration, test compounds can be
screened for ability
to trigger or inhibit a cellular response, such as a signal transduction
pathway. In such an
analysis, test compound is introduced into a site and then the cell added. The
reaction site is
then checked to detect formation of the cellular response.
[0176] Further discussion of related devices and applications of such devices
is set forth in
copending and commonly owned U.S. Provisional application No. 60/335,292,
filed
November 30, 2001, which relates to U.S. published application No.
20030138829.
42
CA 02807564 2013-02-26
XI. Fabrication
A. General Aspects
[0177] As alluded to earlier, the micro fluidic devices that are provided are
generally =
constructed utilizing single and multilayer soft lithography (MSL) techniques
and/or
sacrificial-layer encapsulation methods. The basic MSL approach involves
casting a
series of elastomeric layers on a micro-machined mold, removing the layers
from the
mold and then fusing the layers together. In the sacrificial-layer
encapsulation
approach, patterns of photoresist are deposited wherever a channel is desired.
These
techniques and their use in producing micro fluidic devices is discussed in
detail, for
example, by Unger et al. (2000) Science 288:113-116, by Chou, et al. (2000)
"Integrated Elastomer Fluidic Lab-on-a-chip-Surface Patterning and DNA
Diagnostics,
in Proceedings of the Solid State Actuator and Sensor Workshop, Hilton Head,
S.C.;
and in PCT Publication WO 01/01025.
[0178] In brief, the foregoing fabrication methods initially involve
fabricating mother '
molds for top layers (e.g., the elastomeric layer with the control channels)
and bottom
layers (e.g., the elastomeric layer with the flow channels) on silicon wafers
by
photolithography with photoresist (Shipley SJR 5740). Channel heights can be
controlled precisely by the spin coating rate. Photoresist channels are formed
by
exposing the photoresist to UV light followed by development. Heat reflow
process
and protection treatment is typically achieved as described by M.A. Unger, H.-
P.
Chou, T. Thorsen, A. Scherer and S.R. Quake, Science (2000) 288:113. A mixed
two-
part-silicone elastomer (GE RTV 615) is then spun into the bottom mold and
poured
onto the top mold, respectively. Here, too, spin coating can be utilized to
control the
thickness of bottom polymeric fluid layer. The partially cured top layer is
peeled off
from its mold after baking in the oven at 80 C for 25 minutes, aligned and
assembled
with the bottom layer. A 1.5-hour final bake at 80 C is used to bind these two
layers
irreversibly. Once peeled off from the bottom silicon mother mold, this RTV
device is
typically treated with HCL (0.1N, 30 min at 80 C). This treatment acts to
cleave some
of the Si-O-Si bonds, thereby exposing hydroxy groups that make the channels
more
hydrophilic.
[0179] The device can then optionally be hermetically sealed to a support. The
support
can be manufactured of essentially any material, although the surface should
be flat to
ensure a
43
CA 02807564 2013-02-26
good seal, as the seal formed is primarily due to adhesive forces. Examples of
suitable
supports include glass, plastics and the like.
[0180] The devices formed according to the foregoing method result in the
substrate (e.g.,
glass slide) forming one wall of the flow channel. Alternatively, the device
once removed
from the mother mold is sealed to a thin elastomeric membrane such that the
flow channel is
totally enclosed in elastomeric material. The resulting elastomeric device can
then optionally
be joined to a substrate support.
B. Devices Utilizing Blind Channel Design
1. Layer Formation
[01811 Microfluidic devices based on the blind channel design in which
reagents are
deposited at the reaction sites during manufacture are typically formed of
three layers. The
bottom layer is the layer upon which reagents are deposited. The bottom layer
can be formed
from various elastomeric materials as described in the references cited above
on MLS
methods. Typically, the material is polydirnethylsiloxane (PlVfDS) elastomer.
Based upon .=
the arrangement and location of the reaction sites that is desired for the
particular device, one
can determine the locations on the bottom layer at which the appropriate
reagents should be
spotted. Because PMDS is hydrophobic, the deposited aqueous spot shrinks to
form a very
small spot. The deposited reagents are deposited such that a covalent bond is
not formed
between the reagent and the surface of the elastomer because, as described
earlier, the
20. reagents are intended to dissolve in the sample solution once it is
introduced into the reaction 7
site.
[0182] The other two layers of the device are the layer in which the flow
channels are formed
and the layer in which the control and optionally guard channels are formed.
These two
layers are prepared according to the general methods set forth earlier in this
section. The
resulting two layer structure is then placed on top of the first layer onto
which the reagents
have been deposited. A specific example of the composition of the three layers
is as follows
(ration of component A to component B): first layer (sample layer) 30:1 (by
weight); second
layer (flow channel layer) 30:1; and third layer (control layer) 4:1. It is
anticipated, however,
that other compositions and ratios of the elastomeric components can be
utilized as well.
[0133] During this process, the reaction sites are aligned with the deposited
reagents such
that the reagents are positioned within the appropriate reaction site. FIG. 6
is a set of
photographs taken from the four corners of a device; these photographs
demonstrate that the
deposited reagents can be accurately aligned within the reaction sites
utilizing the foregoing
44
CA 02807564 2013-02-26
approach. These photographs show guard channels and reaction site located at
the end of
branch flow channel. The white circle indicates the location of the deposited
reagent
relative to the reaction site. As indicated, each reagent spot is well within
the confines of
the reaction site.
2. Spotting
[0184] The reagents can be deposited utilizing any of a number of commercially
available
reagent spotters and using a variety of established spotting techniques.
Examples of
suitable spotter that can be utilized in the preparation of the devices
include pin spotters,
acoustic spotters, automated micropipettors, electrophoretic pumps, ink jet
printer devices,
ink drop printers, and certain osmotic pumps. Examples of commercially
available spotters
include: Cartesian Technologies MicroSys 5100 (Irvine, CA), Hitach SPBIO
(Alameda,
CA), Genetix Q-Array (United Kingdom), Affymetrix 417 (Santa Clara, CA) and
Packard
Bioscience SpotArray (Meriden, CT). In general, very small spots of reagents
are
deposited; usually spots of less than 10 nl are deposited, in other instances
less than 5 nl, 2
n1 or ml, and in still other instances, less than 0.5 nl, 0.25 nl, or 0.1 nl.
[0185] Arrays of materials may also be formed by the methods described in
Foder, et al.,
US Patent No.5,445,934: titled "Array of oligonucleotides on a solid
substrate", wherein
oligonucleotide probes, such as SNP probes, are synthesized in situ using
spatial light
directed photolithography. Such arrays would be used as the substrate or base
of the micro
fluidic devices of the present invention such that the regions of the
substrate corresponding
to the reaction sites, for example, blind fill chambers, would contain one, or
preferably
more than one, oligonucleotide probes arrayed in known locations on the
substrate. In the
case of a partitioning micro fluidic structure, such as the one depicted in
figure 15 herein,
the reaction sites, depicted as square boxes along the serpentine, fluid
channel, would
contain a plurality of different SNP probes, preferably a collection of SNP
probes suitable
for identifying an individual from a population of individuals, and preferably
wherein a
plurality of reaction sites along the serpentine fluid channel, such that if a
fluid sample
containing nucleic acid sequences from a plurality of individuals where
introduced into the
serpentine flow channel, and a plurality of valve in communication with the
serpentine
flow channel such that when actuated causes the serpentine flow channel to be
partitioned
thereby isolating each reaction site from one another to contain a fraction of
the fluid
sample in each reaction site. Amplification of the components of the sample
may be
performed to increase the number of molecules, for example nucleic acid
molecules, for
binding to the array of SNP probes located within each reaction site. In some
embodiments,
45
CA 02807564 2013-02-26
each of the reaction sites along the serpentine fluid channel would be the
same array, that is,
have the same SNP probes arrayed, and in other embodiments, two or more of the
reaction
sites along the serpentine t fluid channel would have a different set of SNP
probes. Other
partitioning fluid channel architectures could also be used, for example,
branched and/or
branched branch systems, and so forth. Other arraying techniques, such as
spotting described
herein, may likewise be used to form the arrays located within the
partitionable reactions sites
along a serpentine or common, such as in branched, fluid channel(s).
[0186] The following examples are presented to further illustrate certain
aspects of the
devices and methods that are disclosed herein. The examples are not to be
considered as
limiting the invention.
EXAMPLES
A. Example 1: Signal Strength Evaluations
1. Introduction
[0187] The purpose of this set of experiments was to demonstrate that
successful PCR
reactions can be conducted with a microfluidic device of the design set forth
herein with
signal strength greater than 50% of the Macro TaqMan reaction.
2. Microfluidic Device
[0188] A three layer microfluidic device, fabricated using the MSL process,
was designed
and fabricated for conducting the experiments described in the following
example. FIG. 7A
shows a cross-sectional view of the device. As shown, the device 700 includes
a layer 722
into which is formed the flow channels. This fluid layer 722 is sandwiched
between an
overlaying layer 720 that includes the control and guard layers and an
underlying sealing
layer 724. The sealing layer 724 forms one side of the flow channels. The
resulting three-
layer structure is affixed to a substrate 726 (in this example, a slide or
coverslip), which
provides structural stiffness, increases thermal conductivity, and helps to
prevent evaporation
from the bottom of microfluidic device 700.
[0189] FIG. 7B shows a schematic view of the design of the flow channels in
flow layer 722
and of the control channels and guard channel in control/guard layer 720.
Device 700
consists of ten independent flow channels 702, each with its own inlet 708,
and branching
blind channels 704, each blind channel 704 having a 1 n1 reaction site 706.
Device 700
contains a network of control lines 712, which isolate the reaction sites 706
when sufficient
46
CA 02807564 2013-02-26
pressure is applied. A series of guard channels 716 are also included to
prevent liquid from
evaporating out of the reaction sites 706; fluid is introduced via inlet 718.
= 3. Experimental Setup
[0190] A PCR reaction using P-actin primers and TaqMan probe to amplify exon 3
of the P-
. 5 actin gene from human male genomic DNA (Promega, Madison WI) was
conducted in device
700. The TaqMan reaction consists of the following components: lx TaqMan
Buffer A (50
mM KC1, 10 mM Tris-HC1, 0.01M EDTA, 60nM Passive Referencel (PR1), pH 8.3);
3.5-4.0 >
mM MgCl; 200 nM dATP, dCTP, dGTP, 400 nM dUTP; 300 nM 13-actin forward primer
and
reverse primer; 200 nM FAM-labeled 13-actin probe; 0.01U/u1 AmpEraseUNG
(Applied
Biosystems, Foster City, CA); 0.1-0.2U/u1DyNAzyme (Finnzyme, Espoo, Finland);
0.5%Triton-x-100 ( Sigma, St. Louis, MO); 0.8ug/u1 Gelatin (Calbiochem, San
Diego, CA);
5.0% Glycerol (Sigma, St. Louis, MO); deionized H20 and male genomic DNA. The
components of the reaction were added to produce a total reaction volume of 25
1. Negative
controls (Control) composed of all the TaqMan reaction components, except
target DNA
were included in each set of PCR reactions.
[0191] Once the TaqMan reaction samples and Control were prepared, they were
injected
into microfluidic device 700 by using a gel loading pipet tip attached to a 1
ml syringe. The
pipet tip was filled with the reaction samples and then inserted into the
fluid via 708. The
flow channels 702 were filled by manually applying backpressure to the syringe
until all the
entire blind channels 704 and reaction sites 706 were filled. Control lines
712 were filled with
deionized water and pressurized to 15-20 psi after all of the samples were
loaded into the
flow lines 702, 704. The pressurized control lines 712 were actuated to close
the valves and
isolate the samples in the 1 nl wells 706. The guard channels 716 were then
filled with
deionized water and pressurized to 5-7 psi. Mineral oil (15 ul) (Sigma) was
placed on the
flatplate of a thermocycler and then the microfluidic device/coverglass 700
was placed on the
thermocycler. Micro fluidic device 700 was then thermocycled using an initial
ramp and
either a three-step or two-step thermocycling profile:
[0192] 1. Initial ramp to 95 C and maintain for 1 minute (1.0 C/s to 75 C,
0.1 C /sec to 95
C).
[0193] 2. Three step thermocycling for 40 cycles (92 C for 30 sec., 54 C for
30 sec., and
72 C for 1 min) or;
[0194] 3. Two step thermocycling for 40 cycles (92 C for 30 seconds and 60 C
for 60 sec.)
47
CA 02807564 2013-02-26
[0195] MicroAmp tubes (Applied Biosystems, Foster City, CA) with the remaining
reaction
mixture, designated Macro TaqMan reactions to distinguish them from reactions
performed
in the microfluidic device, were placed in the GeneAmp PCR System 9700
(Applied
Biosystems, Foster City, CA) and thermocycled in the 9600 mode. The Macro
TaqMan
5 reactions served as macroscopic controls for the reactions performed
in the micro fluidic
device. The thermocycling protocol was set to match that of the microfluidic
device, except
that the initial ramp rate was not controlled for the Macro TaqMan reactions.
. . = [0196] Once thermocycling was completed, the
control and guard lines were depressurized
and the chip was transferred onto a glass slide (VWR, West Chester, PA). The
chip was then
10 placed into an Array WoRx Scanner (Applied Precision, Issaquah, WA)
with a modified
carrier. The fluorescence intensity was measured for three different
excitation/emission
wavelengths: 475/510 nm (PAM), 510/560 nm (VIC), and 580/640nm (Passive
Referencel
= = (PR1)). The Array Works Software was used to image
the fluorescence in the micro fluidic
device and to measure the signal and background intensities of each 1n1 well.
The results
15 were then analyzed using a Microsoft Excel file to calculate the
FA_M/PR1 ratio for 0-actin
TaqMan reactions. For conventional Macro TaqMan, positive samples for target
DNA were "
determined using calculations described in the protocol provided by the
manufacturer
(TaqMan PCR Reagent Kit Protocol). The signal strength was calculated by
dividing the
PAM! PR1 ratio of the samples by the FAM/ PR1 ratio of the controls. A
successful reaction
20 was defined as a sample ratio above the 99% confidence threshold
level.
= r
4. Results
[0197] Initially, AmpliTaq Gold (Applied Biosystems, Foster City, CA) was used
in TaqMan
reactions and PAM! PR1/Control ratios of 1.5-2.0 were produced, compared to
Macro
TaciiM rea &inn ratinc nf S.11-1 4,11. Althrui ah rpciiitc N.211.1.Ft pc%
ClthIP, lnorpacer1 eicrn al etrength
= 25 was desired. Therefore, the AmpliTaq Gold polymerase was
substituted with DyNAzyme
= polymerase due to its increased thermostability,
proofreading, and resistance to impurities.
The standard Macro TaqMan DyNAzyme concentration of 0.025U/u1 was used in the
microfluidic experiments. This polymerase change to DyNAzyme produced
FAM/ROX/Control ratios of 3.5-5.8. The signal strength was improved, but it
was difficult
. = 30 to achieve consistent results. Because it is know that some
proteins stick to PDMS, the
= -
concentration of the polymerase was increased and surface modifying additives
were
included. Two increased concentrations of DyNAzyme were tested, 8x (0.2U/up
and 4x
(0.1U/u1) the standard concentration for Macro TaqMan, with 100 pg or 10 pg of
genomic
48
CA 02807564 2013-02-26
DNA per n1 in the micro fluidic device. Gelatin, Glycerol, and 0.5%Triton-x-
100 were added
to prevent the polymerase from attaching to the PDMS. The results of the
reactions in the
micro fluidic device (chip) and the Macro TaqMan controls are shown in FIG. 8.
101981 The microfluidic TaqMan reaction ratios range from 4.9-8.3, while the
Macro
TaqMan reactions range from 7.7-9.7. Therefore, the signal strength of the
TaqMan reactions
in chip is up to 87% of the Macro TaqMan reactions. There was no significant
difference
between 4x or 8x DyNAzyme. The results demonstrate that PCR reactions can be
done with
greater than 50% signal strength, when compared to the Macro TaqMan reactions,
in the
microfluidic devices. The results have been consistent through at least four
attempts. =
B. EXAMPLE 2- Spotting Reagents
1. Introduction
[0199] The purpose of the experiment was to demonstrate successful spotted PCR
reactions
in a microfluidic device. The term "spotted" in this context, refers to the
placement of small
droplets of reagents (spots) on a substrate that is then assembled to become
part of a
microfluidic device. The spotted reagents are generally a subset of the
reagent mixture
required for performing PCR.
2. Procedure
i. Spotting of Reagents
[0200] Routine spotting of reagents was performed, via a contact printing
process. Reagents
were picked up from a set of source wells on metal pins, and deposited by
contacting the pins :µ
to a target substrate. This printing process is further outlined in FIG. 9. As
shown, reagents
were picked up from a source (e.g., microtiter plates), and then printed by
bringing the loaded
pin into contact with the substrate. The wash step consists of agitation in
deionized water
followed by vacuum drying. The system used to print the reagent spots is a
Cartesian
Technologies MicroSys 5100 (Irvine, CA), employing TeleChem "ChipMaker" brand
pins,
although other systems can be used as described supra.
[0201] Pins employed are Telechcm ChipMaker 4 pins, which incorporate an
electro-milled
slot (see FIG. 9) to increase the uptake volume (and hence the number of
printable spots).
Under the operating conditions employed (typically, 75% relative humidity and
temperature
approximately 25 C), in excess of one hundred spots were printed per pin, per
loading cycle.
Under the conditions above, the volume of reagents spotted onto the PDMS
substrate is on
the order of 0.1 nL.
49
CA 02807564 2013-02-26
[0202] The dimensions of the pin tip are 125x125 gm. The final spot of dried
reagent is
substantially smaller than this (as small as 7 fin-i in diameter), yet the pin
size defines a lower
limit to the readily achievable spot spacing. The achievable spacing
determines the smallest
well-to-well pitch in the final device. Using such a device and the foregoing
methods, arrays
with spacings of 180 gm have been achieved. Arrays built into working chips
tend to have
spacings from 600 to 1300 microns.
[0203] Spotting was done using only one pin at a time. The system in use,
however, has a
pin head which can accommodate up to 32 pins. Printing a standard-size chip
(array
dimensions of order 20x25 mm) takes under 5 minutes.
ii. Assembly of Spotted Chips
= [0204] The flow and control layers of the PCR devices are assembled
according to the
normal MSL process described above. The microfluidic device design is the same
as the one
' = described in Example 1. In parallel, a substrate layer composed of
150 gm ¨thick PDMS
with component ratio A:B of 30:1 is formed via spin-coating a blank silicon
wafer, and then
cured for 90 minutes at 80 C.
[02051 The cured blank substrate layer of PDMS (sealing/substrate layer 724 of
FIG. 7A)
, serves as the target for reagent spotting. Patterns of spots are
printed onto the substrate, =
, which is still on the blank wafer. The reagents spotted for PCR
reactions were primers and
= probes, specific to the particular gene to be amplified. The spotted
reagent included a 1:1:1
volume ratio of 300 nM 3-actin forward primer (FP), 300 nM 13-actin reverse
primer (RP),
and 200 nM j3-actin probe (Prb). In some cases, it is useful to further tune
the chemistry via
concentrating the spotted mixture. It has been found that adjusting the
concentrations such
that primer and probe concentrations are equal to, or slightly higher than,
the normal
.tuaLa upt...vpit, recipe value yields consistently good results. Therefore,
the spotted reagent is
concentrated to be 3 times and 4 times the concentration of the macro
reaction. Concentration
of the reagents is performed in a Centrivap heated and evacuated centrifuge
and does not alter
relative FP:RP:Prb ratios. The increased spot concentration results in the
correct final
concentration when the reagents are resuspended in a 1 nL reaction volume.
Spotted reagents
need not be limited to primers and probes; nor must all three (FP, RP and Prb)
be spotted.
Applications where only the probe, or even one of the primers, is spotted can
be performed.
-= . Experiments have been conducted in which the sample primer/probe sets
spotted were
TaqMan [3-actin and TaqMan RNAse-P.
50
CA 02807564 2013-02-26
102061 Following the spotting process onto the substrate layer, the combined
flow and
control layers (i.e., layers 720 and 722 of FIG. 7A) were aligned with the
spot pattern and
brought into contact. A further bake at 80 C, for 60-90 minutes, was used to
bond the
= substrate to the rest of the chip. After the chip has been assembled,
the remaining components
of the PCR reaction (described in Example 1) are injected into the flow
channels of the chip
and the chip is thermocycled as described in Example 1.
3. Results
[0207] PCR reactions have been successfully and repeatably performed using
devices where
primer (forward and reverse primers) and probe molecules are spotted. An
example of data
from a chip in which a reaction has been successfully performed is shown in
FIG. 10.
The spotted reagents have resulted in successful PCR reactions as defined in
Example 1.
Successful reactions have been performed using 2-stage and 3-stage
thermocycling protocols.
C. EXAMPLE 3: Genotyping
1. Introduction
102081 The purpose of the following experiments was to demonstrate that
genotyping
experiments can be conducted utilizing a microfluidic device or chip such as
described
herein. Specifically, these experiments were designed to determine if
reactions conducted in
the device have sufficient sensitivity and to ensure that other primer/probe
sets, besides 13-
actin, can be performed in the microfluidic device.
2. Methods/Results
1.. RNase P Experiment
[0209] RNase P TaqMan reactions (Applied Biosystems; Foster City, CA) were
performed in
a microfluidic device as described in Example 1 to demonstrate that other
primer/probe sets
produce detectable results. RNaseP reactions also require a higher level of
sensitivity
because the RNaseP primer/probe set detects a single copy gene (2
copies/genome) in
contrast to the 13-actin primer/ probe set. The 13-actin set detects a single
copy p-actin gene
and several pseudogenes, which collectively total approximately 17 copies per
genome. The
RNase P reactions were run with the same components as described in Example 1,
with the
exception that the 13-actin primer/probe set was replaced with the RNase P
primer/probe set.
Further, the RnaseP primer/probe set was used at 4x the manufacturer's
recommended value
to enhance the fluorescence signal. The VIC dye was conjugated to the probe
for RNase P
51
CA 02807564 2013-02-26
and the analysis focused on VIC/PR1 ratios. The results of one of four
experiments are
shown in FIG. 11.
. = [0210] The VIC/PR1/Control ratios for the Macro TaqMan reactions
are 1.23. The
corresponding ratios for the TaqMan reactions in the microfluidic device are
1.11 and 1.21.
The ratios of the genomic DNA samples in the microfluidic device are above the
99%
confidence threshold level. Further, the signal strength of the TaqMan
reactions in the
microfluidic device is 50% and 93.7% of the Macro TaqMan reactions. The
control TaqMan
= reactions in the microfluidic device have standard deviations of
.006 and .012, demonstrating
consistency in the reactions across the micro fluidic device. Therefore, it is
determined that
the TaqMan reactions in the chip are sensitive enough to detect 2 copies per
genome.
DNA Dilution Experiment
= [0211] To further determine the sensitivity of TaqMan reactions in
the microfluidic device,
dilutions of genomic DNA were tested using the 13-actin primer/probe set.
Reaction
compositions were generally composed as described in Example 1 using 4x
DyNAzyme and
dilutions of genomic DNA. The genomic DNA was diluted down to 0.25
pg/n1,=which
corresponds to approximately 1 copy per nl. The result of one dilution series
is shown in =
FIG. 12.
[0212] According to a Poisson distribution, 37% of the total number of wells
should be
negative if the average target number is one. Well numbers 5, 6 and 7 are
below the
calculated threshold and, therefore, negative. This suggests that the 13-actin
TaqMan
reactions in micro fluidic chip can detect an average of one copy per nl.
Therefore, the
sensitivity of the reactions in the microfluidic device is sufficient to
perform genotyping
experiments.
Genotyping Experiment
[0213] Because TaqMan in the microfluidic device is capable of detecting low
target
numbers, preliminary testing of SNP (Single Nucleotide Polymorphism)
genotyping was
performed using the Predetermined Allelic Discrimination kit (Applied
Biosystems; Foster
City, CA) against the CYP2D6 P450 cytochrome gene. The kit contains one primer
set and
two probes; FAM labeled for the wildtype or reference allele, CYP2D6*1, and
VIC labeled
for the CYP2D6*3 mutant or variant allele. Positive controls, PCR products,
for each allele
along with genomic DNA were tested in the device using the same conditions as
described in
52
CA 02807564 2013-02-26
Example 1. The results from one experiment are shown in FIGS. 13 and 14. The
experiment
has been repeated at least three times to validate the results and to
demonstrate reliability.
10214] As shown in FIG. 13, the A1-1 (Allele 1, CYP2D6*1 wild type allele) and
genomic
DNA (100 pg/nl) produced an average VIC/PR1/Control ratio of 3.5 and 2.2,
respectively,
indicating that the genomic DNA was positive for the CYP2D6*1, wild type
allele. These
values are above the threshold limit for the reactions. The signal strength of
the TaqMan
reactions in the microfluidic device is 59% and 40% of the Macro TaqMan
controls,
respectively. A1-2 (Allele 2, CYP2D6*3 mutant or variant allele), which should
be negative
in the VIC channel, showed some signal over control (1.5), possibly due to FAM
fluorescence leaking into the VIC channel of the detector. The leakage can be
minimized
with an improved detection process.
[0215] The A1-2 positive control gave an average FAM/PR1/Control ratio of 3.0,
which was
37% of the Macro TaqMan signal and above the calculated threshold limit (see
FIG. 14). The
genomic samples were negative for the CYP2D6*3 mutant allele, an expected
result since the
frequency of the CYP2D6*3 allele is low. Again, it appears that there is some
leakage of the
A1-1, VIC probe into the FAM channel of the detector. Overall, the SNP
detection reactions
were successful in the microfluidic device.
D. EXAMPLE 4: Verification of PCR by Gel Electrophoresis
1. Introduction
[0216] As an alternative method to prove amplification of DNA was occurring in
the
microfluidic device, an experiment to detect PCR product by gel
electrophoresis was
performed. PCR reactions compositions were as described in Example 1, except
the TaqMan
probe was omitted and the 13-actin forward primer was conjugated to PAM.
2. Procedure
i. Microfluidic Device
= [0217] A three layer microfluidic device, fabricated using the MSL
process, was designed
and fabricated for conducting the experiments described in this example; FIG.
15 shows a
schematic view of the design. The device 1500 generally consists of a sample
region 1502
and a control region 1504. Sample region 1502 contains three hundred and forty-
one 1 nl
reaction sites 1508 represented by the rectangles arrayed along flow channel
1506, which
includes inlet via 1510 and outlet via 1512. Control region 1504 contains
three control flow
channels 1514 each containing ten 1 nl reaction sites 1518, also represented
by the rectangles
53
CA 02807564 2013-02-26
and an inlet via 1516. A network of control lines 1522 isolate each reaction
site 1508, 1518
when sufficient pressure is applied to inlet via 1524. A series of guard
channels 1520 are
included to prevent liquid from evaporating out of the reaction sites 1508,
1518. The device
is a three- layer device as described in Example 1 (see FIG. 7A). The entire
chip is placed
onto a coverslip.
ii.. Experimental Setup
[0218] Microfluidic device 1500 was loaded and thermocycled using the 3
temperature
profile described in Example 1. The remaining reaction sample was thermocycled
in the
GeneAmp 9700 with the same thermocycling profile as for microfluidic device
1500. The
reaction products were recovered after thermocycling was completed. To recover
the
amplified DNA, 3 1 of water was injected into sample input via 1506 and 3-4
j4 of product
were removed from outlet via 1512. The reaction products from device 1500 and
the Macro
reaction were treated with 2 p1 of ExoSAP-IT (USB, Cleveland, OH), which is
composed of
DNA Exonuclease I and Shrimp Alkaline Phosphatase, to remove excess
nucleotides and
primers. The Macro product was diluted from 1:10 to1:106. The product from
device 1500
was dehydrated and resuspended in 4 I of formamide.
3. Results
[0219] Both products, along with negative controls were analyzed, on a
polyacrylamide gel.
FIG. 15 shows the gel electrophoresis results. The appropriate size DNA band
of 294 base
pairs in length is observed in FIG. 16.
[0220] The products from the Macro reactions are shown on the left hand side
of the gel and
correspond to about 294 base pairs, the expected size of the 13-actin PCR
product. The
negative controls lack the PCR product. Similarly, the product derived from
the device gave
the expected 13-actin PCR product. Therefore, target DNA was amplified in the
micro fluidic
device.
E. EXAMPLE 5: Massive Partitioning
[0221] The polymerase chain reaction (PCR) has become an essential tool in
molecular
biology. Its combination of sensitivity (amplification of single molecules of
DNA),
specificity (distinguishing single base mismatches) and dynamic range (105
with realtime
instrumentation) make it one of the most powerful analytical tools in
existence. We
demonstrate here that PCR performance improves as the reaction volume is
reduced: we have
54
CA 02807564 2013-02-26
performed 21,000 simultaneous PCR reactions in a single microfluidic chip, in
a volume of
90 pL per reaction and with single template molecule sensitivity.
[02221 Figures 17a-17d depict a single bank and dual bank partitioning
microfluidic device.
where multilayer soft lithography (MSL) (1),was used to create elastomeric
microfluidic
chips which use active valves to massively partition each of several liquid
samples into a
multitude of isolated reaction volumes. After injection of the samples into
inlet 1703 which
is in communication with branched partitioning channel system 1705 of
microfluidic device
1701 (Fig. 17b), 2400 90 pL volumes 1709 of each sample are isolated by
closing valves
1707 spaced along (fig. 174) simple microfluidic channels. The chip device is
then
themaocycled on a flat plate thermocycler and imaged in a commercially
available
fluorescence reader.
[02231 We assessed the performance of PCR in the chips by varying the
concentration of
template DNA and measuring the number of wells that gave a positive Taqmantni.
signal. We
found that a digital amplification is observed when the average number of
copies per well is
low (Figures 18a and 18b). A mixture of robust positive and clearly negative
signals is
observed even when the average number of copies per well is below 1; this
implies that even
a single copy of target can give good amplification. The number of positive
wells was
consistent with the number of wells calculated to have 1 copy of target by the
Poisson
distribution (Figure 19.). This result validates that this system gives
amplification
consistently even from a single copy of target. Fluorescent signal strengths
from microfluidic
Taqmantm PCR were comparable to macroscopic PCR reactions with the same DNA
concentration ¨ even though the macroscopic reactions contained >104 more
template copies
per reaction.
[0224] We believe that the primary source of this remarkable fidelity is the
effective
concentration of the target: a single molecule in a 90 pL volume is 55,000
times more
concentrated than a single molecule in a 5 uL volume. Since the number of
molecules of
target, nt, does not change (i.e. nt = 1) and the number of molecules that can
produce side
reactions, nõ (i.e. primer-dimers and non-complementary DNA sequences in the
sample) is
linearly proportional to volume (i.e. ns cc V), the ratio of target to side
reactions is inversely
proportional to volume: nthas cc VV. Since side reactions are a primary cause
of PCR failure
(4), the advantage to reducing the volume of the reaction is clear.
[0225] PCR amplification from single copies of template has been previously
reported (5).
However, current methods that achieve reliable amplification from single
copies in a
55
CA 02807564 2013-02-26
macroscopic volumes often require altered thermo cycling protocols (e.g. long
extension
times, many cycles), precautions against mispriming and non-specific
amplification (e.g. "hot
start" PCR (thermal activation of the polymerase), "booster" PCR, additives to
reduce
nonspecific hybridization, etc), and are almost always done with two rounds of
PCR, where
an aliquot of the first PCR is used as template in the second reaction. In
contrast, this system
achieves reliable amplification from single copies using standard conditions -
off-the-shelf
primers and probes and a single-round, standard thermo cycling protocol. Being
completely
enclosed, it is also nearly invulnerable to environmental contamination. The
ability to do
massive numbers of PCR reactions simultaneously provides definite logistical,
cost and time
advantages compared to macroscopic volumes (1 chip with 21,000 reactions vs.
219 separate
96 well plates, and the associated time, equipment, and tracking
infrastructure).
[0226] This principle of massive partitioning with a digital PCR readout may
be used for
absolute quantification of the concentration of target in a sample. It can be
used, for
example, to genotype a pooled sample of genomic DNA simply by counting the
numbers of
wells that give a positive for a particular allele, or plurality of alleles as
described above.
Due to the enhanced resistance to side reactions, it should also be useful in
quantifying =
mutants in a background of wild-type DNA - a problem relevant in cancer
detection. The
general principle of concentration by partitioning may also be useful in other
reactions
where detection of single molecules, bacteria, viruses or cells is of interest
(e. g. ELISA
reactions for protein detection). Digital PCR is described by Brown, et al.,
US Patent No.
6,143,496, titled "Method of sampling, amplifying and quantifying segment of
nucleic acid,
polymerase chain reaction assembly having nanoliter-sized chambers and methods
of
filling chambers", and by Vogelstein, et al, US Patent No. 6,446,706, titled
"Digital PCR".
The small volumes achievable using microfluidics allow both a massive degree
of
parallelization and very high target-to-background concentration ratios. High
target-to-
background ratios allow single-molecule amplification fidelity. These factors
suggest that
for PCR, smaller really is better.
[02271 The invention provides for methods and devices for conducting digital
PCR in a
microfluidic environment comprising the steps of: providing a micro fluidic
device having a
fluid channel therein, said fluid channel having two or more valves associated
therewith, the
valves, when actuated, being capable of partitioning the fluid channel into
two or more
reaction sites or chambers; introducing a sample containing at least one
target nucleic acid
polymer, actuating the valves to partition the fluid sample into two or more
portions, wherein
at least one portion contains a target nucleic acid polymer and another
portion does not
56
CA 02807564 2013-02-26
contain a target nucleic acid polymer, amplifying the target nucleic acid
polymer, and,
determining the number of portions of the fluid channel that contained the
target molecule.
In preferred embodiments, the micro fluidic device comprises an elastomeric
material, and
more preferably, comprises at least one layer comprising an elastomeric
material. In certain
preferred embodiments, the micro fluidic device further comprises a
deflectable membrane
wherein the deflectable membrane is deflectable into and out of the fluid
channel to control
fluid flow within the fluid channel and/or to partition one portion of the
fluid channel from
another, preferably wherein the deflectable membrane is integral to a layer of
the micro
fluidic device having a channel or recess formed therein, and preferably
wherein the
deflectable membrane is formed where a first channel in a first layer is
overlapped by a
second channel in a second layer of the micro fluidic device. In some
embodiments, the
sample fluid contains all of the components needed for conducting an
amplification
reaction, while in other embodiments, the micro fluidic device contains at
least one
component of an amplification reaction prior to the introduction of the sample
fluid. In
some embodiments, the micro fluidic device further comprises a detection
reagent,
preferably one or more nucleic acid polymers complimentary to a least a
portion of the
target nucleic acid polymer, preferably a plurality of different nucleic acid
polymers
spatially arrayed within a reaction site or chamber of the micro fluidic
device.
[0228] Amplification may be achieved by thermo cycling reactions such as PCR,
or by
isothermal reactions, such as described by Van Ness et al., in US Patent
Application Serial
No. 10/196,740 which has published as US 2003/0138800 Al, and teaches an
isothermic
amplification scheme.
[0229] See also Unger et al, Science 288, 113-116 (2000).
[0230] The sample channels and control lines are loaded by "blind filling" -
PDMS is
sufficiently gas permeable that liquid pressurized at a few psi drives the gas
out of the
channels, leaving them completely filled with liquid. See Hansen et al, PNAS
99, 16531-
16536 (2002).
[0231] A 294 bp segment of the human 8-actin gene was amplified using a 5'-
exonuclease
assay (Taqman). Forward and reverse primer sequences were 5'
. TCACCCACACTGTGCCCATCTACGA3' and 5'-
CAGCGGAACCGCTCATTGCCAATGG3', respectively. TAMRA-based FRET probe,
sequence 5' -(FAM)ATGCCC-X(TAMRA)-CCCCCATGCCATCCTGCGTp-3'. Data was
57
CA 02807564 2013-02-26
taken with a dark-quencher based probe, as large numbers of these primer-probe
sets are
becoming commercially available. Reactions contained Ix Taqman buffer A (50 mM
KC1, 10
mM Tris-HC1, 0.01 M EDTA, 60 nM Passive Reference 1, pH 8.3), 4 mM MgC12, 200
nM
dATP, dCTP, dTTP, 400 nM dUTP, 300 nM forward primer, 300 nM reverse primer,
200 nM
probe, 0.01 U/uL Amperase UNG (all from Applied Biosystems, Foster City, CA),
0.2 U/uL
DyNAzyme (Finnzyme, Espoo, Finland), 0.5% Triton-x-100, 0.8 ug/ul Gelatin
(Calbiochem,
San Diego, CA), 5.0% Glycerol, deionized 1120 and human male genomic DNA
(promega).
[0232] See also: Quantitative PCR Technology, Chapter on "Gene
Quantification", L.T
McBride, K Livak, M Lucero, et al, Editor, Francois Ferre, Birkauser, Boston,
MA p 97
110, 1998.See E.T. Lagally, 1. Medintz, R.A. Mathies, Anal Chem 73(3), 565-570
(2001), as
well as B. Vogelstein, K.W. Kinzler, PNAS 96, 9236-9241 (1999).
[0233] The scope of the claims should not be limited by the embodiments set
forth in the
examples, but should be given the broadest interpretation consistent with the
description as a whole.
58