Note: Descriptions are shown in the official language in which they were submitted.
CA 02809478 2013-03-13
LUMINESCENT COMPOUNDS
.,
AND METHODS OF USING SAME
FIELD OF THE INVENTION
The invention relates to compounds having luminescent (e.g., fluorescent,
phosphorescent) properties, and to methods of using such compounds. The
invention more
particularly relates to compounds having photoluminescent and/or
electroluminescent properties,
and to uses of same. The invention also relates to compounds having photo-
receptor
properties due to their ability to separate charges. The invention also
relates to compounds
having photon harvesting properties.
BACKGROUND OF THE INVENTION
Bright and efficient organic light-emitting diode (OLED) devices and
electroluminescent
(EL) devices have attracted considerable interest due to their potential
application for flat panel
displays (e.g., television and computer monitors) and lighting. OLED based
displays offer
advantages over the traditional liquid crystal displays, such as: wide viewing
angle, fast
response, lower power consumption, and lower cost. However, several challenges
still must be
addressed before OLEDs become truly affordable and attractive next generation
display and
lighting. To realize white lighting and other full color display applications,
it is essential to have
the three fundamental colors of red, green, and blue provided by emitters with
sufficient color
purity and sufficiently high emission efficiency.
Phosphorescent Organic Light-Emitting Diodes (PhOLEDs) have recently received
much
attention because of their high energy efficiency for next generation flat
panel displays and solid
state lighting devices. OLEDs based on phosphorescent emitters can have three
to four-fold
higher device quantum efficiencies than those based on fluorescent emitters.
The key challenge
in PhOLEDs research is the development of phosphorescent metal complexes with
high
quantum efficiency and high stability, especially blue phosphorescent
compounds. Earlier
research efforts on phosphorescent materials for OLEDs focused on
2-phenylpyridine(Hppy)-based Ir(111) complexes because of their high
photoluminescent quantum
efficiencies . Although some efficient PhOLEDs based on Ir(111) emitters have
been achieved,
1
CA 02809478 2013-03-13
stable blue PhOLEDs based on 1r(111) compounds remain elusive. Blue
phosphorescent
compounds are among the most sought-after materials by industry around the
world as one of
the key color components for electroluminescent devices.
SUMMARY OF THE INVENTION
An aspect of the invention provides a compound having general formula (100):
Rh
5
Rk B h _______________ X
1 _________________________________________________ X 2
4
/
Rm ________________________________________
Pt
\
Y
Rp
100
wherein B is sterically sheltered and is located on ring 1 either para or meta
to the C-Pt
bond, R is a non-aromatic carbocycle or heterocycle that is attached as a
fused ring or as a
substituent, an aryl group that is attached as a fused ring or as a
substituent, aliphatic-aryl,
hydroxy, nitro, amino, halo, B(R')2, BR'(ary1), B(aryl)2, aryl-B(aryl)2, 0,
NR', OR', a nitrile group,
C(halo)3which is optionally CF3, or R', where R' is independently an aliphatic
group having 1-24
carbon atoms which may be straight, branched, cyclic, or any combination
thereof, k, p and h are
independently 0 to 5 and m and j are independently 0 to 3, and k, h and m are
not all 0, with the
proviso that there is at least one substituent located ortho to B so that the
boron is sufficiently
sterically sheltered to prevent nucleophilic attack on the B, and wherein if
there is only one
substituent that is ortho to B, then that substituent is branched C3, branched
C4, or linear or
2
CA 02809478 2013-03-13
_
branched C5-or higher, t is 0 or 1, a dotted line in a ring indicates that the
ring can be saturated,
unsaturated, aromatic, or non-aromatic, X is independently C or N and at least
two X are N; and
Y is independently N or 0, wherein a substituent can be further substituted.
In an embodiment of this aspect the compound of general formula 100 comprises
at least
two C1 substituents which are both are located ortho to the boron.
In an embodiment of this aspect, the invention provides compounds that are
photoluminescent or electroluminescent.
In an embodiment of this aspect, the compound of general formula 100 is a
compound of
general formula 101:
Ri
(Mes)2B // ____ AX v
1 _______________________________________________
c ,.T X
\,....,,
RI( \ - X ------ X
Pt7
/ - - -\
Yir 3 %.,Y
, , I
lo
RP
t
101
wherein R, m, j, p, t, X and Y are defined previously, and Mes is mesityl.
In an embodiment of this aspect, Y is oxygen. In an embodiment of this aspect,
the at
least two X that are N are three X that are N, so that ring 2 is a triazole.
In another
embodiment of this aspect, the compound is BC1 or BC2. In another embodiment
of this
aspect, the compound is a Pt(II) complex of Table 1. In an embodiment of this
aspect, the
compound is C5, BC1-acac, BC1-nacnac, BC2-acac, or BC2-nacnac, Pt-12, 51, or
52.
3
CA 02809478 2013-03-13
In another aspect the invention provides, the invention provides a compound of
general formula
200
Rh
Rk
(-4)13/X7- xy,J
______________________________________________ x 2 I
Rnr ____________________________________________ X 200
wherein B is sterically sheltered and is located on ring 1 either para or meta
to the C-Pt
bond, R is a non-aromatic carbocycle or heterocycle that is attached as a
fused ring or as a
substituent, an aryl group that is attached as a fused ring or as a
substituent, aliphatic-aryl,
hydroxy, nitro, amino, halo, B(R')2, BR'(ary1), B(aryl)2, aryl-B(aryl)2, 0,
NR)2, OR', a nitrile group,
C(halo)3which is optionally CF3, or R', where R' is independently an aliphatic
group having 1-24
carbon atoms which may be straight, branched, cyclic, or any combination
thereof, k and h are
independently 0 to 5 and m and j are independently 0 to 3, and k, h and m are
not all 0, with the
proviso that there is at least one substituent located ortho to B so that the
boron is sufficiently
sterically sheltered to prevent nucleophilic attack on the B, and wherein if
there is only one
substituent that is ortho to B, then that substituent is branched C3, branched
C4, or linear or
branched C5-or higher, t is 0 or 1, a dotted line in a ring indicates that the
ring can be saturated,
unsaturated, aromatic, or non-aromatic, X is independently C or N and at least
two X are N; and
Y is independently N or 0. In an embodiment of this aspect, the compound is
photoluminescent
or electroluminescent.
In an embodiment of this aspect, the compound is B-NHC1, B-NHC2, B-triazole1,
B-triazole2, B-Me-triazole1, B-triazole3, B-triazole4, or B-Me-benzimidazole1.
In another aspect the invention provides, the invention provides a composition
comprising
a photoluminescent or electroluminescent compound of general formula 100 or
200, an organic
polymer, and a solvent.
In another aspect the invention provides a photoluminescent product or an
electroluminescent product comprising a compound of the above aspects.
In an embodiment of this aspect, such a product is a flat panel display device
or a lighting
4
CA 02809478 2013-03-13
device. In an embodiment of this aspect, such a product is a luminescent probe
or sensor.
In another aspect the invention provides, a method of producing
electroluminescence,
comprising the steps of: providing an electroluminescent compound of general
formula 100 or
200 and applying a voltage across said compound so that said compound
electroluminesces.
In another aspect the invention provides, an electroluminescent device for use
with an
applied voltage, comprising a first electrode, an emitter which is an
electroluminescent
compound of general formula 100 or 200 optionally in a host layer, and a
second, transparent
electrode, wherein voltage is applied to the two electrodes to produce an
electric field across the
emitter so that the emitter electroluminesces.
In another aspect the invention provides, an electroluminescent device for use
with an
applied voltage, comprising a first electrode, a second, transparent
electrode, an electron
transport layer adjacent the first electrode, a hole transport layer adjacent
the second electrode,
and an emitter which is an electroluminescent compound of general formula 100
or 200
optionally in a host layer, interposed between the electron transport layer
and the hole transport
layer, wherein voltage is applied to the two electrodes to produce an electric
field across the
emitter so that the emitter electroluminesces.
In another aspect the invention provides, a method of harvesting photons
comprising the
steps of: providing a compound of general formula 100 or 200, and providing
light such that
photons strike said compound and charge separation occurs in said compound.
In
some embodiments separated charges recombine and photons are released. In an
embodiment separated charges migrate to respective electrodes to produce a
potential
difference.
In another aspect the invention provides, a method of separating charges
comprising the
steps of: providing a compound of general formula 100 or 200 and providing
light such that
photons strike said compound and charge separation occurs in said compound. In
some
embodiments, separated charges recombine and photons are released.
In an embodiment of this aspect, separated charges migrate to respective
electrodes to
produce a potential difference.
In another aspect the invention provides, a photocopier employing the above
method of
harvesting photons or the above method of separating charges.
In yet another aspect the invention provides, a photovoltaic device employing
the above
method of harvesting photons or the above method of separating charges.
In another aspect the invention provides, a photoreceptor employing the above
method of
CA 02809478 2013-03-13
harvesting photons or the above method of separating charges.
In another aspect the invention provides, a solar cell employing the above
method of
harvesting photons or the above method of separating charges.
In another aspect the invention provides, a semiconductor employing the above
method
of harvesting photons or the above method of separating charges.
In another aspect the invention provides, a light emitting device comprising
an anode, a
cathode, and an emissive layer, disposed between the anode and the cathode,
wherein the
emissive layer comprises a compound of general formula 100 or a compound of
general formula
200. In another aspect the invention provides, a consumer product comprising
such a device.
In another embodiment of this aspect, the device's emissive layer further
comprises a host.
In another aspect the invention provides, the invention provides a method of
synthesizing
a compound of general formula 100, comprising combining in an appropriate
solvent to form a
reaction mixture (i) a cyclometalating ligand comprising two rings joined by
one bond, the first ring
being an aromatic or non-aromatic heterocycle that comprises at least one ring
heteroatom, and
the second ring being an aromatic carbocycle, wherein the first and second
rings may be
substituted or unsubstituted; and (ii) a charge-neutral platinum(II) compound,
wherein at least one
Pt(II) is bonded to four monodentate ligands, optionally, allowing reaction to
proceed for an
appropriate reaction time, adding to the reaction mixture strong acid,
optionally, allowing reaction
to proceed for an appropriate reaction time, adding to the reaction mixture a
stabilizing ligand
comprising a bidentate heteroaryl ligand comprising at least two heteroatoms,
each heteroatom
being available for bonding to the Pt(II), wherein the bidentate heteroaryl
ligand may be
substituted or unsubstituted; and
obtaining a product that is a Pt(II) chelated by two different
bidentate ligands wherein the first bidentate ligand is derived from the
cyclometalating ligand and
the second bidentate ligand is derived from the stabilizing ligand, wherein
substituents may be
further substituted and comprise a non-aromatic carbocycle or heterocycle, an
aryl group that is
attached as a fused ring or as a substituent, aliphatic-aryl, hydroxy, nitro,
amino, halo, BR2,
B(aryl)2, aryl-B(aryl)2, 0, NR', OR', a nitrile group, C(halo)3which includes
CF3, or R', where R' is
independently an aliphatic group having 1-24 carbon atoms which may be
straight, branched,
cyclic, or any combination thereof.
In an embodiment of this aspect, the amount of cyclometalating ligand and
strong acid are
equimolar to the amount of Pt(II), and the amount of bidentate ligand is twice
as much as the
amount of Pt(II). Another embodiment further comprising purifying the product.
In some
embodiments, the strong acid is: HBF4, p-toluenesulfonic acid (Ts0H),
trifluoroacetic acid
6
CA 02809478 2013-03-13
(TFA), picolinic acid (PA), or trifluoromethanesulfonic acid (Tf0H). In some
embodiments, the
-
stabilizing ligand is p-diketonato, 1,3-diketiminato, picolinato, or
N . In some embodiments, substituents comprise aliphatic, aryl,
B(aryl)2, B(aliphatic)(ary1), B(aliphatic)(aliphatic). In certain embodiments,
the substituent
comprises phenyl, isopropyl, n-butyl, t-butyl, or phenyl-BMes2. In certain
embodiments, the
stabilizing ligand is added as a solution formed by dissolving a salt form of
the stabilizing ligand
that comprises a sodium, lithium or potassium counterion. In certain
embodiments, the reaction
mixture is maintained at ambient temperature and/or pressure. In certain
embodiments, the
reaction mixture is maintained at about 55 C. In certain embodiments, the
product is la, lb, lc,
2a, 2b, 2c, 3, 4, 5, 6, 7, 8a, 8b, 8c, 9a, 9b, 9c, 21, 22, 23, 24, or 25. In
certain embodiments, the
stabilizing ligand is the conjugate base of the strong acid. In certain
embodiments, the strong
acid is picolinic acid. Certain embodiments, further comprising adding heat.
In certain
embodiments, one or more steps are performed under an inert atmosphere. In
certain such
embodiments the product is 11. Certain embodiments also comprising cooling. In
certain
embodiments, the product is BC1, BC2, Pt(B-NHC1)(nacnac), or Pt(B-
NHC2)(nacnac).
BRIEF DESCRIPTION OF THE DRAWINGS
For a better understanding of the present invention and to show more clearly
how it may
be carried into effect, reference will now be made by way of example to the
accompanying
drawings, which illustrate aspects and features according to preferred
embodiments of the
present invention, and in which:
Figure 1 shows a preferred embodiment of a three layer electroluminescent (EL)
display
device according to the invention;
Figure 2A shows the absorption (dashed) and emission (solid) spectra of BC1
and BC2
in methylene chloride at a concentration of 1.0 x 10-5 M;
Figure 2B shows a crystal structure schematic for the molecular structure of
BC1 with
50% thermal ellipsoids and labeling;
Figure 2C shows a crystal structure schematic for the molecular structure of
BC2 with
50% thermal ellipsoids and labeling;
Figure 2D graphically shows current efficiencies for OLEDs based on BC1 and
BC2;
Figure 2E graphically shows power efficiencies for OLEDs based on BC1 and BC2;
7
CA 02809478 2013-03-13
Figure 2F graphically shows cyclic voltammetry diagrams of BC1 and BC2
recorded in
t
DMF with NBu4PF6as the electrolyte, scan rate 200 mV/s;
Figure 3A shows the emission spectra of Cl (A); C2 (U); and C3 (0) in PMMA (10
wt%);
Figure 3B shows the emission spectra of C4 (a); C5 (0); and C8 (11I) in PMMA
(10 wt%);
Figure 3C shows the emission spectra of C6 (0); C6 (5%) (A); and C9 (II) in
PMMA (10
wt% and 5 wt%, respectively) ;
Figure 3D shows the emission spectra of C10 (0); and C11 (5%)(U) in PMMA (10
wt%
and 5 wt%, respectively) ;
Figure 3E shows the emission spectra of C27 (5%) (III); and C27(0) in PMMA (10
wt%
and 5 wt%, respectively) ;
Figure 3F shows emission spectra of compound C12 in 1 wt%, 5 wt% and 10 wt%
doped
PMMA films, respectively, after drying for 1 day;
Figure 4A shows the absorption spectrum of Pt-12 in dichloromethane
(concentration=
2.0 x 10-5 M) at room temperature;
Figure 4B shows the emission spectrum of Pt-12 in dichloromethane (c = 2.0x10-
5 M) at
r.t. under nitrogen;
Figure 5A shows the electrolurninescent spectra of compound C6 doped at 5% in
TcTa
(tris(4-carbazoy1-9-ylphenyl)amine) host layer; and
Figure 5B shows a plot of external quantum efficiency (EQE) versus luminance
for
compound C6.
DETAILED DESCRIPTION OF THE INVENTION
Definitions
As used herein, the term "TfOH" means trifluoromethanesulfonic acid, which is
also
known as triflic acid or CF3S03H. The term "Ts0H" means p-toluenesulfonic
acid. The term
"TFA" means trifluoroacetic acid. The term "PA" means picolinic acid.
As used herein, the terms "NAC" chelate, "PAC" chelate and "CAC" chelate are
used to
indicate what atoms are bonded to the metal. That is, "NAC" indicates that a
nitrogen and a
carbon are bonded to the metal, "PAC" indicates that a phosphorus and a carbon
are bonded to
the metal, and "CAC" indicates that a carbon and another carbon are bonded to
the metal.
As used herein, the term "chelation" indicates formation or presence of bonds
(or other
attractive interactions), e.g., coordination bonds, between a single central
atom and two or more
separate binding sites within the same ligand.
8
CA 02809478 2013-03-13
As used herein, the term "cyclometalation" refers to a reaction of transition
metal
a
complexes in which an organic ligand undergoes intramolecular metalation with
formation of a
metal-carbon sigma bond (Bruce, Michael I., Angewandte Chemie Intl Ed. (2003)
16(2): 73-86).
As used herein "aliphatic" includes alkyl, alkenyl and alkynyl. An aliphatic
group may be
substituted or unsubstituted. It may be straight chain, branched chain or
cyclic.
As used herein "aryl" includes aromatic carbocycles and aromatic heterocycles
and may
be substituted or unsubstituted.
As used herein, B means boron.
As used herein the term "Mes" means mesityl, which is also known as
2,4,6-trimethylphenyl.
As used herein the term "acac" refers to 8-diketonato. As used herein the term
"nacnac"
refers to 8-diketimino. As used herein the term picolinato may appear
abbreviated as "pico".
As used herein, the term "unsubstituted" refers to any open valence of an atom
being
occupied by hydrogen. Also, if an occupant of an open valence position on an
atom is not
specified then it is hydrogen.
As used herein "substituted" refers to the structure having one or more
substituents.
As used herein "heteroatom" means a non-carbon, non-hydrogen atom. In some
cases,
a heteroatom may have a lone pair of electrons available to form dative or
coordinate bonds (e.g.,
N, 0, P).
As used herein, the term "dative bond" refers to a coordination bond formed
when one
molecular species serves as a donor and the other as an acceptor of an
electron pair to be
shared in formation of a complex.
As used herein, the term "monodentate ligand" refers to a moiety that has a
single site
that is suitable for binding a metal ion. In general, the stability of a metal
complex correlates
with the denticity of its ligands, where denticity is defined as, "in a
coordination entity the number
of donor groups from a given ligand attached to the same central atom" (IUPAC
Gold Book). This
is thought to be because monodentate ligands are more apt to dissociate from a
metal ion than a
bidentate or multidentate ligand. This phenomenon is considered to be due to
the proximity of
the ligand to the metal ion. For example, in solution, when a monodentate
ligand dissociates
from a metal ion, it drifts away from the metal ion. In contrast, when a
bidentate ligand
dissociates at one of its two binding sites, the other binding site's bond
means that the bidentate
ligand remains in the proximity of the metal ion. For this reason, it is
likely to reform a bond
9
CA 02809478 2013-03-13
_
between the available binding site and the metal ion. Thus a bidentate metal
complex is more
stable than a monodentate metal complex.
Embodiments
Compared with Ir(111) complexes, Pt(II) compounds that have a high
phosphorescent
quantum efficiency are scarce. This scarcity is due to strong intermolecular
rc-it stacking
interactions caused by Pt(11) complexes' square planar geometry. Such
interactions lead to
excimer formation and decreases in emission quantum efficiency and color
purity in the solid
state. The square planar geometry of Pt(II) may have one advantage over
Ir(111), namely the
access to a higher triplet state. Such access is due to greater ligand field
splitting for a given
set of chelate ligands, which greatly increases the energy of the d-d state.
Thus, Pt(11)
compounds are good candidates for development of blue phosphorescent emitters,
if low
emission efficiency and intermolecular interaction issues can be addressed.
Several examples
of green, orange or red phosphorescent Pt(11) compounds have been demonstrated
recently for
successful use in PhOLEDs (A. F. Rausch etal., Proc. SPIE 2007, 6655,
66550F/1; B. Ma etal.,
Adv. Funct. Mater. 2006, 16, 2438; V. Adamovich etal., New J. Chem. 2002, 26,
1171; B. W.
D'Andrade et al.,Adv. Mater. 2002, 14, 1032; J. A. G. Williams etal., Coord.
Chem. Rev. 2008,
252, 2596; W. -Y. Wong etal., Organometallics, 2005, 24, 4079; G. Zhou etal.,
J. Mater. Chem.,
2010, 20, 7472; J. Kavitha et al., Adv. Funct. Mater., 2005, /5, 223; S.-Y.
Chang etal., lnorg.
Chem., 2007, 46, 7064; S.-Y. Chang etal., Dalton Trans., 2008, 6901; Z. M.
Hudson etal., Adv.
Funct Mater. 2010, 20, 3426; Z. M. Hudson etal., Dalton Trans., 2011, 40,
7805; Z. Wang etal.,
App!. Phys. Lett., 2011, 98, 213301; Z. M. Hudson etal., Chem. Commun., 2011,
47, 755).
Blue PhOLEDs based on Pt(II) compounds are very rare and only a few examples
are
known in the literature (K. Li, X. Guan etal., Chem. Commun., 2011, 47, 9075;
Y. Unger, etal.,
Angew. Chem. mt. Ed., 2010, 49, 10214; E. L. Williams etal., Adv. Mater.2007,
19,197; M.
Cocchi, etal., App!. Phys. Lett. 2009, 94, 073309; M. Cocchi, etal., Adv.
Funct. Mater., 2007, 17,
285; X. Yang et al., Adv. Mater. 2008, 20, 2405; S.-Y. Chang et al., Inorg.
Chem. 2007, 46,
11202).
The inventors of this discovery have found that cyclometalated Pt(II) li-
diketonate,
cyclometalated Pt(II) p-diketiminate, and cyclometalated Pt(II) picolinate
complexes, as
described herein, have promising PhOLED properties such as photoluminescent
quantum
efficiencies and may offer one or more of the key color components for
electroluminescent
devices. General formula 100 representing such cyclometalated Pt(II) P-
diketonate,
CA 02809478 2013-03-13
_
cyclometalated Pt(I1)p-diketiminate, and cyclometalated Pt(II) picolinate
complexes is shown
below:
Rh
1 5
R.
Rk B 4 ____________ X 1 1
1
1 4
X --
RmN ___________ X ------- X
Pt/
Y= 'Y
, I
,,, .. _ , , ............
RP
t
100
wherein B is sterically sheltered and is located on ring 1 either para or meta
to the C-Pt
bond, R is a non-aromatic carbocycle or heterocycle that is attached as a
fused ring or as a
substituent, an aryl group that is attached as a fused ring or as a
substituent, aliphatic-aryl,
hydroxy, nitro, amino, halo, B(R)2, BR'(ary1), B(aryl)2, aryl-B(aryl)2, 0,
NR', OR', a nitrile group,
C(halo)3which is optionally CF3, or R', where R' is independently an aliphatic
group having 1-24
carbon atoms which may be straight, branched, cyclic, or any combination
thereof (e.g.,
adamantyl), k, p and h are independently 0 to 5 and m and j are independently
0 to 3, and k, h
and m are not all 0, with the proviso that there is at least one substituent
located ortho to B so
that the boron is sufficiently sterically sheltered to prevent nucleophilic
attack on the B, and
wherein if there is only one substituent that is ortho to B, then that
substituent is branched C3,
branched Ca, or linear or branched C5-or higher, t is 0 or 1, a dotted line in
a ring indicates that
11
CA 02809478 2013-03-13
_
the ring can be saturated, unsaturated, aromatic, or non-aromatic, X is
independently C or N and
at least two X are N, and Y is independently N or 0. A substituent may be
further substituted.
The term "cyclometalation" refers to a reaction of a transition metal complex
in which an
organic ligand undergoes intramolecular metalation with formation of a metal-
carbon sigma bond
(Bruce, Michael I., Angewandte Chemie Intl Ed. (2003) 16(2): 73-86). A
cyclometalating ligand
is one component of the compounds described and characterized herein.
General structure 200, showing structural features of cyclometalating ligands
described
herein, is provided below:
Rh
1 5
Rk
15<- B __________________________ -
1>
______
Rm4 _________________________________________________________________
X 'X
200
wherein R, k, h, m and j are as defined for general formula 100.
Notably, B (boron) is sterically sheltered to protect it from nucleophilic
attack, so there is
at least one substituent located ortho to B so that the boron is sufficiently
sterically sheltered to
prevent nucleophilic attack by, for example, water. If there is only one
substituent ortho to B
then that substituent is branched C3, branched C4, or linear or branched C5-or
higher. A dotted
line in a ring indicates that it can be saturated, unsaturated, aromatic or
non-aromatic. A
substituent can be further substituted.
In certain embodiments, compounds of general formula 200 are luminescent
(i.e.,
fluorescent).
12
CA 02809478 2013-03-13
_
The cyclometalating ligand has two rings bonded together through one bond so
that
when this ligand chelates a metal ion, the metal atom becomes part of a newly-
formed five- or
six-membered ring (see below). The first ring (shown as ring 1 in general
formulas 100 and
200) is a six-membered aromatic carbocycle. The second ring (shown as ring 2
in general
formulas 100 and 200) is an aromatic or non-aromatic 5- membered heterocycle
that has at least
two ring heteroatoms. Both the first and second rings may be substituted or
unsubstituted.
The cyclometalating ligand is a bidentate ligand, and as such, two atoms form
bonds with the
Pt(II). The first metal-bonding atom is a carbon atom of the ring 1, and the
second is an atom of
the ring 2. In some embodiments, the atom of ring 2 is one of the at least two
ring heteroatoms.
For clarity, schematics of an example cyclometalating ligand and a Pt complex,
which is an
intermediate product of Scheme 1, including the same cyclometalating ligand,
are shown below.
. \ /
N
Cyclometalating ligand
Newly formed ring =
\/
,N
Pt
Tf0/ \SMe2
The working examples provide details regarding synthesis and characterization
of
compounds of general formula 200.
Acceptable substituents include any chemical moiety that does not interfere
with the
desired reaction or desired property such as luminescence, and may include,
for example: R is a
non-aromatic carbocycle or heterocycle, an aryl group that is attached as a
fused ring or as a
substituent, aliphatic-aryl, hydroxy, nitro, amino, halo, BR'2, BR'(ary1),
B(aryl)2, aryl-B(aryl)2, 0,
NR'2, OR, a nitrile group, C(halo)3which is optionally CF3, or R', where R' is
independently an
aliphatic group having 1-24 carbon atoms which may be straight, branched or
cyclic or any
combination thereof;
A substituent may be further substituted.
In certain embodiments, boron disubstituted by respective substituted aryl
carbocyclic
moities (e.g., BMes2) is a substituent of the cyclometalating ligand either at
the ring 1, at ring 2, or
13
CA 02809478 2013-03-13
at both ring 1 and at ring 2. In some embodiments, this type of substituted
cyclometalated
ligand has phosphorescent properties. In certain embodiments, this type of
substituted ligand is
a blue phosphorescent compound. Also, when this type of substituted
cyclometalating ligand is
used to form a compound of general formula 100, a highly efficient
phosphorescent Pt(II)
compound can be achieved. In some embodiments, the phosphorescence is blue.
In some embodiments of general formula 100, the invention provides a compound
wherein the boron moiety is substituted by two mesityl groups, see general
formula 101 below.
R-
(Mes)2B ii __________________________________________________
c 1 ______________________________________________ / X
Rrir \ -
Pt7
/- - \
Y
'- - \ ----- RP
t
101
The terms of general formula 101 are as defined for general formula 100. This
embodiment is similar to general formula 100, except it specifies the
substituents on boron.
The effect of the presence of such substituents (i.e., boron disubstituted by
respective
substituted aryl carbocyclic moities (e.g., BMes2)) on the N,C-chelate
backbone plays several
important roles in the high performance of the resulting Pt(II) compounds of
general formula 100
in PhOLEDs. It facilitates the mixing of the 3LC and the MLCT state, thus
enhancing the
intrinsic phosphorescent efficiency of the molecule. It minimizes
intermolecular interactions,
thus enhancing emission efficiency in the solid state. Also, it facilitates
electron injection into
the emissive layer/dopant, thus improving the device efficiency. In certain
embodiments of the
invention, a compound of general formula 101 exhibits intense luminescence,
which may be
photoluminescence and/or electroluminescence.
Notably, compounds of general formula 101 comprise a phenyl ring (ring 1) that
is
bonded to the Pt. Ring 1 can be singly-substituted or multi-substituted. Ring
1 is substituted
14
CA 02809478 2013-03-13
by a B(Mes)2 moiety in either the para or meta positions relative to Pt.
Optionally, it is also
substituted by one or more R moieties. Suitable R substituents include any
moiety that does
not interfere with the luminescence of such compounds and may be fused rings
(i.e, bound to
ring 1 at two locations).
Importantly, when a B(Mes)2 moiety is bound to ring 1 at the para position
relative to Pt,
the compound's luminescence is blue in colour. When a B(Mes)2 moiety is bound
to ring 1 at
the meta position relative to Pt, the compound's luminescence is green, or
greenish blue in
colour. When a B(Mes)2 moiety is bound to ring 1 at the meta position relative
to Pt, and ring 2
contains two fused aryl rings, then the compound's luminescence is yellow,
orange or red in
colour.
Compounds of general formula 101 further comprise a five-membered
heteroaromatic
ring (ring 2) that has at least two heteroatoms. Ring 2 can be singly-
substituted or
multi-substituted. Suitable R substituents include any moiety that does not
interfere with the
luminescence of such compounds. Optionally, ring 2 may be part of a fused ring
system. The
rings of the fused ring system may be substituted.
Another component of compounds described and characterized herein, is Pt(II).
In the
synthesis of compounds of general formula 100, Pt(II) is obtained from a
reactant that is a
charge-neutral platinum(II) compound. In such reactants, a Pt(II) is bonded to
four
monodentate ligands. By being charge-neutral, this starting material is
soluble in a variety of
non-aqueous solvents (e.g., tetrahydrofuran (THF)). By having monodentate
ligands occupying
the four coordination sites of the Pt(II), this starting material is a good
source of Pt(II) that is
readily able to form bonds with a cyclometalating bidentate ligand. Thus, when
treated with
stoichiometric quantities of the cyclometalating ligand (e.g., 2-
phenylpyridine ("ppy")) in an
appropriate solvent at ambient temperature, the charge-neutral platinum(II)
starting material
reacts and affords a cyclometalated Pt(II) complex. Specifically, the first
reaction product,
Pt(ppy)Me(SMe2), is obtained through irreversible loss of a monodentate ligand
(e.g., CH4).
An example of such a starting material for the synthesis of such
cyclometalated Pt(II)
diketonate or diketiminate complexes, is [PtMe2(SMe2)]2, which has been widely
used as a
precursor in C-H activation chemistry and can be easily prepared on a multi-
gram scale from
K2PtC14 (Scott, J. D.; Puddephatt, R. J. Organometallics 1983, 2, 1643-1648).
Other examples of
suitable Pt starting materials include Pt(pheny1)2(DMS0)2 (Klein, A. et al.,
Organometallics,
2005,17, 4125) and [Pt(pheny1)2(SMe2)1, ( where n = 2,3) (Song, D.et al., J.
Organomet Chem.
2002, 648, 302-305).
CA 02809478 2013-03-13
Another starting material for the synthesis of such compounds of general
formula 100 is a
strong acid. Such an acid is able, for example, to protonate an alkanyl moiety
(e.g., -CH3 is
protonated to CH4). An example of a strong acid is HBF4. In some embodiments
the strong acid
is a strong organic acid. Examples of strong organic acids include: p-
toluenesulfonic acid
(Ts0H), trifluoroacetic acid (TEA), or picolinic acid (PA). For certain
embodiments
trifluoromethanesulfonic acid (Tf0H) may be used; however, for other
embodiments this choice of
acid may lead to unwanted side reactions.
In some cases, the acid not only protonates but also can act as the
stabilizing ligand). As
shown in the second step of Scheme 1, treatment of this reaction mixture with
one equivalent of a
solution of a strong organic acid (e.g., trifluoromethane sulfonic acid
("TfOH")) leads to rapid loss
of a second equivalent of monodentate ligand (e.g., CH4) giving the
corresponding complex (e.g.,
Pt(PPY)(OTO(SMe2)), which incorporates two labile ligands that can be replaced
by a stabilizing
ligand (see ring 3 of general formula 100). In some embodiments, the conjugate
base of the
strong acid acts as the stabilizing ligand (see compound 9, as an example
where picolinate acts
stabilizing ligand.)
As shown in the third step of Scheme 1, another component of general formula
100 is
known herein as a stabilizing ligand (see ring 3 of general formula 100). When
introduced in the
synthesis of compounds of general formula 100, this ligand is a negatively-
charged bidentate
chelate ligand, with a cationic counterion. This chelate has at least two
heteroatoms (e.g., N, 0)
wherein the at least two heteroatoms are each available for bonding to the
Pt(II). The stabilizing
ligand may be substituted or unsubstituted. The stabilizing ligand forms a 5-
or 6-membered
metallocycle with the Pt, which may be aromatic or non-aromatic, saturated or
unsaturated, and
may have fused rings bonded to the metallocycle. As shown in Scheme 1,
exemplary
stabilizing ligands include P-diketonato ("acac"), and p-diketiminato
("nacnac"). Other
exemplary stabilizing ligands include picolinate, and
N> HO
N .
The stabilizing ligand can be unsubstituted, singly-substituted, or multi-
substituted. A
substituent may form fused rings with the metal-bonding heteroatoms (e.g.
pyridyl, triazole,
pyrazole, imidazole, etc). Suitable substituents include any moiety that does
not interfere with
the phosphorescence of such compounds. Ring 3 may be part of a fused ring
system. Such a
16
CA 02809478 2013-03-13
fused ring system may be substituted.
A one-pot, two- or three-step reaction (number of steps depends whether the
acid
provides the stabilizing ligand) provides a product with general formula 100,
which has P1(11)
chelated by two different bidentate ligands wherein the first bidentate ligand
is derived from the
cyclonnetalating ligand and the second bidentate ligand is derived from the
stabilizing ligand. As
shown in the example reaction shown in Scheme 1, the reaction proceeds fairly
quickly, at
ambient temperature, and for this particular example, the product was isolated
as analytically
pure material in 87% yield following column chromatography.
1 [PtMe2(SMe2)]2
2 CF3S03H
*
3 Na(acac)
Pt N
THF/Me0H, 25 C, 3h
one pot ,))c
Scheme 1. One-pot synthesis of cyclometalated Pt(II) p-diketonates
The working examples provide detailed descriptions of syntheses of specific
compounds
of general formulas 100 and 200, whose structures are shown in Table 1. As
would be
apparent to a person of ordinary skill in the art, other structural variations
may be used according
to the invention. Starting materials may be modified to include moieties that
confer desirable
physical or chemical properties, such as increased stability or luminescence.
Structural formulae of compounds of general formula 100 are shown in Table 1,
together
with data regarding their luminescence. Such compounds of general formula 100
are
photoluminescent or electroluminescent. Thus, embodiments of the invention
provide
compounds that are photoluminescent and, in at least some embodiments of the
invention,
electroluminescent; they may produce intense light. In embodiments of the
invention, a
composition is provided which comprises a photoluminescent or
electroluminescent compound
of general formula 100, an organic polymer, and a solvent. In other
embodiments of the
invention, a composition is provided which comprises a photoluminescent or
electroluminescent
compound of general formula 200, an organic polymer, and a solvent.
The invention also provides a method of producing photoluminescence comprising
the
steps of: providing a photoluminescent compound of the invention having
general formula 100
or general formula 200; and irradiating said photoluminescent compound with
radiation of a
wavelength suitable for exciting the compound to photoluminesce.
17
CA 02809478 2013-03-13
-
The invention further provides a method of producing electroluminescence
comprising
the steps of: providing an electroluminescent compound of the invention having
general
formula 100 or general formula 200; and applying a voltage across said
electroluminescent
compound.
The invention further provides an electroluminescent device for use with an
applied
voltage, comprising: a first electrode, an emitter (e.g., phosphor) which is
an
electroluminescent compound of the invention optionally doped in a host
material, and a second,
transparent electrode, wherein a voltage is applied between the two electrodes
to produce an
electric field across the emitter.
The invention further provides an electroluminescent device for use with an
applied
voltage, comprising: a first electrode, an electron transport layer, an
emitter (e.g., phosphor)
which is an electroluminescent compound of the invention doped in a host
material, a hole
transport material, and a second, transparent electrode, wherein a voltage is
applied between
the two electrodes to produce an electric field across the emitter.
The emitter consequently electroluminesces. In some embodiments of the
invention,
the device includes one or more charge transport layers interposed between the
emitter and one
or both of the electrodes. For example, spacing of an embodiment of the
device, called for the
purposes of the present specification, a "three layer EL device", is: first
electrode, first charge
transport layer, emitter in a host layer, second charge transport layer, and
second transparent
electrode.
An advantage of certain embodiments of the invention is that they compounds
that are
soluble in common solvents such as toluene, diethyl ether, tetrahydrofuran
(THF), and
dichloromethane. This permits the compounds to be blended easily and
conveniently with
polymers. The role of the polymer in such a mixture is at least two-fold.
First, a polymer can
provide protection for the compound from air degradation. Second, a polymer
host matrix
permits use of a solution-based process (e.g., ink-jet printing), a spin-
coating process, or a
dip-coating process as an alternative way to make films. Although spin-coating
and dip-coating
processes may not produce as high quality films as those produced by chemical
vapor
deposition (e.g., ink-jet printing) or vacuum deposition, they are often much
faster and more
economical.
Other embodiments of the invention provide compounds that are water-soluble.
Accordingly, the invention further provides methods of applying compounds as
described
above to a surface. These methods include solvent cast from solution,
electrochemical
18
CA 02809478 2013-03-13
deposition, vacuum vapor deposition, chemical vapor deposition, spin coating
and dip coating.
The compounds may be applied alone or with a carrier. In some embodiments of
the invention,
they are applied in a composition including an organic polymer. Such
compositions are also
encompassed by the invention. As an example of this application, compounds of
general
formula 100 form a clear transparent solution with the weakly-luminescent
polymer PMMA.
This can be converted to a transparent film by evaporating the toluene solvent
via either a
dip-coating or spin-coating process. Films obtained in this way are stable.
Certain polymers
such as, for example, PVK, are expected to further enhance the luminescence of
an emitter in
the film. Conveniently, spin coating may be performed using a Chemat
Technology spin-coater
KW-4A; and vacuum deposition may be performed using a modified Edwards manual
diffusion
pump.
Certain compounds of the invention have high chemical and/or thermal
stability. As a
result, they are suitable for vacuum deposition methods used in fabricating
single- or multi-layer
OLED devices.
The invention provides a method of producing electroluminescence comprising
the steps
of: providing an electroluminescent compound of the invention having
general formula 100 or
general formula 200; and applying a voltage across said electroluminescent
compound so that
the compound electroluminesces.
According to the invention, electroluminescent devices for use with an applied
voltage are
provided. In general, such a device has a first electrode, an emitter which is
an
electroluminescent compound of the invention, and a second, transparent
electrode, wherein a
voltage is applied between the two electrodes to produce an electric field
across the emitter of
sufficient strength to cause the emitter to electroluminesce. Preferably, the
first electrode is of a
metal, such as, for example, aluminum, which reflects light emitted by the
compound; whereas
the second, transparent electrode permits passage of emitted light
therethrough. The
transparent electrode is preferably of indium tin oxide (ITO) glass, flexible
polymer, or an
equivalent known in the art. Here, the first electrode is the cathode and the
second electrode is
the anode.
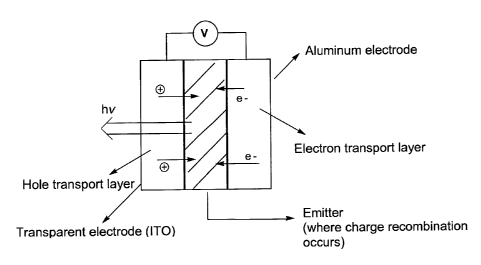
Referring to Figure 1, an embodiment of an electroluminescent device of the
invention is
shown. In general, when a potential is applied across an OLED, holes are said
to be injected
from an anode into a hole transporting layer (HTL) while electrons are
injected from a cathode
into an electron transporting layer (ETL). The holes and electrons migrate to
an ETUHTL
interface. Materials for these transporting layers are chosen so that holes
are preferentially
19
CA 02809478 2013-03-13
transported by the HTL, and electrons are preferentially transported by the
ETL. At the
ETL/HTL interface, the holes and electrons recombine to give excited molecules
which
radiatively relax, producing an EL emission that can range from blue to near-
infrared (Koene, B.;
Loy, D.; and Thompson, M. Unsymmetrical Triaryldiamines as Thermally Stable
Hole
Transporting Layers for Organic Light-Emitting Devices. Chemistry of
Materials. (1998) 10(8):
2235-2250).
As shown in Figure 1, the electron transport material is adjacent to the first
electrode (the
cathode, which can be, for example, LiF/Aluminum,). The emitter is doped in a
host layer, which
can be, for example, 4,4'-N,ff-dicarbazolebiphenyl (CBP) or tris(4-carbazoy1-9-
ylphenyl)amine
(TcTa). The hole transport material (for example,
N,A1-61(1-naphthaleny1)-N,IT-dipheny1]-(1,1'-biphenyl)-4,4'-diamine (NPB),) is
placed between
the ITO electrode (the anode) and the emitting layer. The choice of the
materials employed as
charge transport layers and host layers will depend upon the specific
properties of the particular
emitter employed. The hole transport layer or the electron transport layer may
also function as
a host layer. The device is connected to a voltage source such that an
electric field of sufficient
strength is applied across the emitter. Light, preferably blue light,
consequently emitted from
the compound of the invention passes through the transparent electrode.
Referring to Figure 2A, absorption and emission spectra of BC1 and BC2 are
shown.
Referring to Figures 2B and 2C, single crystals of both BC1 and BC2 were
successfully
obtained and were examined by X-ray diffraction analyses. The resultant
crystal structures of
BC1 and BC2 are shown (see Figures 2B and 2C, respectively). Both molecules
display highly
planar geometries about the Pt(II) centre with minimal strain apparent in
either structure,
important for the maximization of phosphorescent quantum yields. Strength of
the carbene donor
(carbene is ring 2 of general formula 100 for BC1 and BC2) is evident in both
cases, exhibiting C
¨ Pt bond lengths shorter than those observed between the Pt(II) centre and
the phenyl ring (ring
1 of general formula 100). The considerable trans influence of the carbene can
also be
observed, with the Pt-0 bond trans to the carbene lengthened by as much as
0.05 A relative to
more common nitrogen donors in similar NAC chelate cyclometalated systems (see
Hudson, Z.
M.; etal. Adv. Fund. Mater. 2010, 20, 3426). The crystal structures of both
BC1 and BC2 show
discrete dimeric Pt-Pt stacking, with Pt ¨ Pt distances of 3.389(2) and
3.505(2) A, respectively (for
more detailed information see Supporting Materials of Hudson, Z.M.; et al. J.
Am. Chem. Soc.
(2012) 134: 13930-13933).
In Figure 2D, current efficiencies for OLEDs based on BC1 and BC2 are
graphically
CA 02809478 2013-03-13
displayed. In Figure 2E, power efficiencies for OLEDs based on BC1 and BC2 are
shown. As
_
shown, compounds of the invention display excellence current and power
efficiencies. In Figure
2F, cyclic voltammetry diagrams of BC1 and BC2 are shown, which provide
insight into the
HOMO and LUMO levels.
Referring to Figures 3A-3F, emission spectra are shown for compounds Cl, C2,
C3, C4,
C5, C6 at 5% and 10% in PMMA, C8, C9, C10, C11 at 5% in PMMA, C12 at 1%, 5%
and 10%
doping level in PMMA, and C27 at 5% and 10% in PMMA.
Referring to Figures 4A-4B, absorption and emission spectra of compound Pt-12
are
shown, respectively.
Referring to Figure 5A, an electroluminescence spectrum is shown for compound
C6
doped at 5% in a TcTa (tris(4-carbazoy1-9-ylphenyl)amine) host layer.
Referring to Figure 5B, a plot is shown presenting external quantum efficiency
(EQE)
versus luminance from compound C6. The C.I.E. coordinate for compound C6 was
found to be
(0.178, 0.197).
In some embodiments of the invention, an EL device includes one or more charge
transport layers interposed between the emitter and one or both of the
electrodes. Such charge
transport layer(s) are employed in prior art systems with inorganic salt
emitters to reduce the
voltage drop across the emitter. In a first example of such a device, layers
are arranged in a
sandwich in the following order: first electrode, charge transport layer,
emitter and host, second
charge transport layer, and second transparent electrode. In anembodiment of
this type, a
substrate of glass, quartz or the like is employed. A reflective metal layer
(corresponding to the
first electrode) is deposited on one side of the substrate, and an insulating
charge transport layer
is deposited on the other side. The emitter layer which is a compound of the
invention is
deposited on the charge transport layer, preferably by vacuum vapor
deposition, though other
methods may be equally effective. A transparent conducting electrode (e.g.,
ITO) is then
deposited on the emitter layer. An effective voltage is applied to produce
electroluminescence
of the emitter.
In a second example of an EL device of the invention, a second charge
transport layer is
employed, and the sandwich layers are arranged in the following order: first
electrode, first
charge transport layer, emitter and host, second charge transport layer and
second, transparent
electrode.
Electroluminescent devices of the invention may include one or more of the
emitting
compounds described herein. In some embodiments of the invention, an
electroluminescent
21
CA 02809478 2013-03-13
device such as a flat panel display device may include not only a blue- or
green-emitting
phosphor as described herein, but may be a multiple-color display device
including one or more
other phosphors. The other phosphors may emit in other light ranges, e.g.,
red, green, and/or
be "stacked" relative to each other. Convenient materials, structures and uses
of
electroluminescent display devices are described in Rack, P.D.; Naman, A.;
Holloway, P.H.; Sun,
S.-S.; and Tuenge, R. T. Materials used in electroluminescent displays." MRS
Bulletin (1996)
21(3): 49-58.
For photoluminescence, the compounds absorb energy from ultraviolet radiation
and emit
visible light near the ultraviolet end of the visible spectrum, e.g., in the
blue region. For
electroluminescence, the absorbed energy is from an applied electric field.
The invention further provides methods employing compounds of the invention to
harvest
photons, and corresponding devices for such use. Spectroscopic studies have
demonstrated
that compounds of the invention have high efficiency to harvest photons and
produce highly
polarized electronic transitions. In general, when such compounds are excited
by light, a charge
separation occurs within the molecule; a first portion of the molecule has a
negative charge and a
second portion has a positive charge. Thus the first portion acts as an
electron donor and the
second portion as an electron acceptor. If recombination of the charge
separation occurs, a
photon is produced and luminescence is observed. In photovoltaic devices,
recombination of
the charge separation does not occur; instead the charges move toward an anode
and a cathode
to produce a potential difference, from which current can be produced.
Molecules with the ability to separate charges upon light initiation are
useful for
applications such as photocopiers, photovoltaic devices and photoreceptors.
Photoconductors
provided by the present invention are expected to be useful in such
applications, due to their
stability and ability to be spread into thin films. Related methods are
encompassed by the
invention.
Organic semiconducting materials can be used in the manufacture of
photovoltaic cells
that harvest light by photo-induced charge separation. To realize an efficient
photovoltaic
device, a large interfacial area at which effective dissociation of excitons
occurs must be created;
thus an electron donor material is mixed with an electron acceptor material.
(Here, an exciton is
a mobile combination of an electron and a hole in an excited crystal, e.g., a
semiconductor.)
Luminescent compounds as semiconductors are advantageous due to their long
lifetime,
efficiency, low operating voltage and low cost.
Photocopiers use a light-initiated charge separation to attract positively-
charged
22
CA 02809478 2013-03-13
molecules of toner powder onto a drum that is negatively charged.
The molecular design of compounds of general formula 100 was intended to
achieve
high-energy blue phosphorescence with maximum quantum yield (Op). The CAC or
NAC chelate
backbone presents a strong ligand field to the Pt(II) centre, raising the
energy of non-radiative d-d
excited states and reducing thermal quenching. An acetylacetonate (acac)
stabilizing ring (ring 3)
provides good solubility as well as solution- and solid-state stability, while
its rigid structure and
high triplet energy level help to increase (I)p. The BMes2 group and related
BAr2 groups on ring 1
serve to greatly enhance metal-to-ligand charge-transfer phosphorescence.
As show in Table 2, doped PMMA films (10 wt%) of BC1 and BC2 exhibit good
quantum
yields of 90 and 86%, respectively, compared to only 13% for an analogous
control compound
lacking the BMes2 group. The solid-state quantum yield of BC2 represents the
highest observed
for a blue phosphorescent Pt(II) complex. BC1 exhibits blue-green
phosphorescence in the solid
state and solution, with an emission maximum of 478 nm in CH2Cl2. This
emission is blue-shifted
by 20 nm in BC2, resulting in sky-blue emission from the complex at Amõ = 462
nm (see Figures
2A-2F and Tables 2 and 3).
As shown in Table 3, BMes2-functionalized triazole chelate Pt(II) compounds
and
BMes2-functionalized benzimidazolyl chelate Pt(II) compounds display bright
phosphorescence
with emission colors ranging from blue to yellow or orange.
As shown in Figure 5, preliminary electroluminescent property evaluation
indicated that
BMes2-functionalized triazole chelate Pt(II) compounds are very promising as
phosphorescent
emitters in OLEDs.
Certain embodiments of the invention provide compounds suitable for use in
biological
and/or medical imaging. For example, for use in cells (in vivo or in vitro) to
use the compounds'
luminescent properties for visualizing structures such as tumours or other
anomaly.
As described herein, triarylboron-functionalized metal-carbene and
triarylboron-functionalized metal-triazole complexes have been prepared and
tested. It has been
shown that the boron moiety greatly increases the phosphorescent quantum yield
of such
complexes.
WORKING EXAMPLES
All reactions were carried out under air unless otherwise noted. Reagents were
purchased
from Aldrich chemical company (Oakville, ON, Canada) and used without further
purification.
Solvents were freshly distilled over appropriate drying reagents. Thin Layer
Chromatography
23
CA 02809478 2013-03-13
_
was carried out on Si02 (silica gel F254, Whatman). Flash chromatography was
carried out on
silica (silica gel 60, 70-230 mesh). 1H and 13C spectra were recorded on a
Bruker Avance 300
spectrometer () operating at 300 and 75.3 MHz respectively. Deuterated
solvents were
purchased from Cambridge Isotopes (St. Leonard, QC, Canada) and used without
further drying.
Excitation and emission spectra were recorded using a Photon Technologies
International
QuantaMaster Model 2 spectrometer (Anaheim, California, USA) UV-visible
absorbance spectra
were recorded using a Varian Cary 50 UV-visible absorbance spectrophotometer
(Varian, Inc. of
Agilent Technologies, Mississauga, ON, Canada). Solution quantum yields were
calculated using
optically dilute solutions (A = 0.1) relative to Ir(ppy)3(T. Sajoto, P. I.
Djurovich, A. B. Tamayo, J.
Oxgaard, W. A. Goddard, M. E. Thompson, J. Am. Chem. Soc. 2009, 131, 9813-
9822). Data
collection for the X-ray crystal structural determinations were performed on a
Bruker SMART
CCD 1000 X-ray diffractometer with graphite-monochromated Mo Ka radiation (A =
0.71073 A) at
298K and the data were processed on a Pentium PC using the Bruker AXS Windows
NT
SHELXTL software package (version 5.10). Elemental analyses were performed by
the
University of Montreal Elemental Analysis Laboratory (Montreal, Canada).
Melting points were
determined on a Fisher-Johns melting point apparatus. Conveniently EL spectra
may be
obtained using Ocean Optics HR2000; and data involving current, voltage and
luminosity may be
obtained using a Keithley 238 high current source measure unit.
Example 1. Fabrication on EL Device
Devices were fabricated in a Kurt J. Lesker LUMINOS cluster tool with a base
pressure
of --10-8 Torr without breaking vacuum. The ITO anode is commercially
patterned and coated on
glass substrates 50 x 50 mm2 with a sheet resistance less than 15 0./square.
Substrates were
ultrasonically cleaned with a standard regiment of Alconox , acetone, and
methanol followed by
UV ozone treatment for 15 min. The active area for all devices was 2 mm2. The
film thicknesses
were monitored by a calibrated quartz crystal microbalance. Current-Voltage
characteristics
were measured using a HP4140B picoammeter in ambient air. Luminance
measurements and
EL spectra were taken using a Minolta LS-110 luminance meter and an Ocean
Optics USB200
spectrometer with bare fiber, respectively. The external quantum efficiency of
EL devices was
calculated following standard procedure. Additional details regarding device
fabrication and
characterization measurements have been described elsewhere (Hudson, Z. of aL
J. Am. Chem.
Soc. (2012) 134, 13930-13933).
Devices were fabricated by vacuum vapor deposition on ITO-coated glass
substrates.
24
CA 02809478 2013-03-13
Due to the wide bandgaps of these materials, care was taken to ensure that the
HOMO and
LUMO energy levels of both emitters were contained within the bandgap of the
host material, to
ensure efficient trapping of both holes and electrons. Furthermore, it was
necessary to employ a
host material with a sufficiently high triplet level to ensure that excitons
within the device were
confined to the dopant. Based on these considerations, preliminary devices
were fabricated using
4,4'-N,1\f-dicarbazolylbiphenyl (CBP) as the hole-transport
layer,
1,3,5-tris(N-phenylbenzimidazole-2-yl)benzene (TPBI) as the electron-transport
layer, and
N,Nr-dicarbazoly1-3,5-benzene (mCP) as host. These devices had a structure of
ITO/Mo03 (1
nm)/CBP (35 nm)/mCP (5 nm)/mCP:emitter (12%, 15 nm)ITPBI (65 nm)/LiF (1
nm)/Al.
Example 2. One-Pot Synthesis of Cyclometalated Pt(I1)13-Diketonates
pt,N
0"0
[PtMe2(SMe2)]2
(0.5 equiv) Na(acac)
THF (2 equiv)
Me0H
cF3so3H (1 equiv)
õN ,N
Pt THF Pt
Me/ NSMe2
Tf0 SMe2
General Synthesis To a 20 mL screw-cap vial equipped with a magnetic stir bar
was added
one equivalent of a cyclometalating ligand (0.35 mmol), [PtMe2(SMe2)]2 dimer
(100 mg, 0.17
mmol), and 3 mL of THF. The resulting mixture was allowed to stir 1 hr at
ambient temperature,
then a solution of CF3S03H organic acid (1 mL, 0.35 M in THF) was added
dropwise. The
mixture was stirred for 30 minutes, then a solution of Na(acac) (0.70 mmol in
2 mL Me0H) was
added. The mixture was stirred for 1.5 hours, then partitioned between water
and CH2Cl2. The
hydrophobic layer was washed with brine, dried over MgSO4, filtered, and
concentrated under
reduced pressure. The resulting residue was then purified using a plug of
silica gel, with
hexanes and CH2Cl2 as eluent, to give analytically pure material.
CA 02809478 2013-03-13
Characterization Data for Compounds Prepared Using the above general synthesis
with
the appropriate cyclometalating ligand (structural formulae shown in Table 1).
1 [ptme2(sme2)]2
2 TfOH
3 Na(R0(0)CHC(0)R N'Pt
THF/Me0H, rt 0-0
la: 1H NMR (400 MHz, Chloroform-d) 59.00 (d, sat, Jpt_H = 41.8 Hz, J = 5.8 Hz,
1H), 7.80 (t, J =
7.8 Hz, 1H), 7.71 -7.55 (m, 2H), 7.45 (d, J = 7.6 Hz, 1H), 7.21 (t, J = 7.4
Hz, 1H), 7.15 - 7.06 (m,
2H), 5.48 (s, 1H), 2.01 (s, 6H) ppm, Anal. calc'd for C16H15NO2Pt: C 42.86, H
3.37, N 3.12; found
C 43.56, H 3.39, N 2.98.
lb: 1H NMR (400 MHz, Chloroform-d) 6 9.00 (d, sat, Jpt_H = 40.9 Hz, J = 5.8
Hz, 1H), 7.81 (t, J =
7.7 Hz, 1H), 7.67 (d, J = 7.6 Hz, 1H), 7.63 (d, J = 8.1 Hz, 1H), 7.45 (d, J =
7.6 Hz, 1H), 7.22 (t, J =
7.4 Hz, 1H), 7.16 - 7.07 (m, 2H), 5.82 (s, 1H), 1.29 (s, 9H), 1.28 (s, 9H)
ppm, Anal. calc'd for
C22H27NO2Pt: C 49.62, H 5.11, N 2.63; found C 49.86, H 5.05, N 2.59.
1c: 1H NMR (400 MHz, Chloroform-d) 6 9.15(d, sat, Jpt-H = 39.0 Hz, J = 5.8 Hz,
1H), 8.11 (d, J =
7.3 Hz, 2H) 8.08 (d, J = 7.1 Hz, 2H), 8.01 (d, J = 7.1 Hz, 1H), 7.85 (t, J =
7.7 Hz, 1H), 7.80 (d, J =
7.3 Hz, 1H), 7.69 (d, J = 8A Hz, 1H), 7.57 (t, J = 7.3 Hz, 2H), 7.51 (t, J =
7.8 Hz, 4H), 7.29 (t, J =
8.3 Hz, 1H), 7.20 (t, J = 6.1 Hz, 1H), 7.15 (t, J = 7.5 Hz, 1H), 6.79 (s, 1H)
ppm, Anal. calc'd for
C26H19NO2Pt: C 54.54, H 3.35, N 2.45, found C 55.05, H 2.92, N 2.40.
2a: 1H NMR (400 MHz, Chloroform-d) 68.97 (d, sat, Jpt-H = 42.3 Hz, J = 5.8 Hz,
1H), 7.77 (t, J =
7.7 Hz, 1H), 7.57 (d, J = 8.0 Hz, 1H), 7.42 (s, 1H), 7.34 (d, J = 7.8 Hz, 1H),
7.07 (dd, J = 7.4, 5.8
Hz, 1H), 6.92 (d, J = 7.8 Hz, 1H), 5.48 (s, 1H), 2.41 (s, 3H), 2.03 (s, 3H),
2.01 (s, 3H) ppm; Anal.
calc'd for C17H17NO2Pt: C 44.16, H 3.71, N 3.03; found C 44.99, H 3.61, N
2.98.
2b: 1H NMR (400 MHz, Chloroform-d) 69.00 (d, sat, Jpt-H = 40.5 Hz, J = 5.8 Hz,
1H), 7.82 (t, J =
7.8 Hz, 1H), 7.64 - 7.47 (m, 2H), 7.36 (d, J = 8.3 Hz, 1H), 7.14 (t, J = 6.6
Hz, 1H), 7.10 (dd, J = 8.2,
2.1 Hz, 1H) 5.50 (s, 1H), 2.04 (s, 3H), 2.02 (s, 3H) ppm; 13C NMR (100 MHz,
Chloroform-d) 6
185.8, 184.4, 167.3, 147.3, 143.1, 141.0, 138.3, 134.8, 130.0, 124.0, 123.6,
121.4, 118.5, 102.6,
28.2,27.1 ppm; Anal. calc'd for C16H14CIN02Pt: C 39.80, H 2.92, N 2.90; found
C 40.29, H 2.91, N
2.68; m.p. > 300 C.
26
CA 02809478 2013-03-13
2c:1H NMR (400 MHz, Chloroform-d) 6 8.99 (d, sat, Jpt-H = 39.6 Hz, J = 5.8 Hz,
1H), 7.82 (t, J = 7.8
Hz, 1H), 7.72 (d, J = 1.8 Hz, 1H), 7.58 (d, J = 8.1 Hz, 1H), 7.32-7.23 (m,
2H), 7.15 (t, J = 6.5 Hz,
1H), 5.49 (s, 1H), 2.04 (s, 3H), 2.02 (s, 3H) ppm; 13C NMR (100 MHz,
Chloroform-d) 6 185.8,
184.3, 167.3, 147.3, 143.5, 141.4, 138.3, 132.8, 126.5, 124.2, 123.9, 121.5,
118.5, 102.6, 28.2,
27.1 ppm, Anal. calc'd for C161-114BrNO2Pt: C 36.45, H 2.68, N 2.66; found C
36.89, H 2.63, N 2.56;
m.p. > 300 C.
3: 1H NMR (400 MHz, Chloroform-d) 6 9.07 (d, sat, Jpt-H = 40.7 Hz, J = 5.8 Hz,
1H), 8.04 (d, J = 8.1
Hz, 1H), 7.88(t, J= 8.1 Hz, 1E1), 7.32 (dd, J = 8.4, 4.9 Hz, 1H), 7.24 ¨ 7.17
(m, 1H), 7.14 ¨ 7.04 (dt,
J = 10.9, 8.3 Hz, 1H), 5.49 (s, 1H), 2.02 (s, 3H), 2.01 (s, 3H) ppm; Anal.
calc'd for C161-113NO2FPt:
C 39.68, H 2.71, N 2.89; found C 40.11, H 2.70, N 2.78.
4: 1H NMR (400 MHz, Chloroform-d) 69.00 (d, sat, Jpt-H = 39.8 Hz, J = 5.8 Hz,
1H), 7.80 (t, J = 7.8
Hz, 1H), 7.58 (d, J = 8.1 Hz, 1H), 7.51 (d, J = 8.4 Hz, 1H), 7.10 (dd, J =
7.3, 5.6 Hz, 1H), 7.04 (d,
J = 2.7 Hz, 1H), 6.91 (dd, J = 8.3, 2.7 Hz, 1H), 5.47 (s, 1H), 3.85 (s, 3H),
2.00 (s, 6H) ppm; 13C
NMR (100 MHz, Chloroform-d) 6 185.6, 183.9, 168.0, 157.1, 147.3, 145.0, 138.1,
131.1, 128.9,
121.3,118.3, 115.6,108.8, 102.5, 55.4, 28.3, 27.1 ppm, Anal. calc'd for
C17H17NO3Pt: C 42.68, H
3.58, N 2.93; found C 43.19, H 3.55, N 2.79; m.p. 227-228 C.
5: 1H NMR (400 MHz, Chloroform-d) 58.41 (d, J = 6.9 Hz, 1H), 7.58 (d, J = 7.3
Hz, 1H), 7.37 (d, J
= 7.6 Hz, 1H), 7.15 (t, J = 7.4 Hz, 1H), 7.05 (t, J = 7.4 Hz, 1H), 6.74 (d, J
= 3.0 Hz, 1H), 6.34 (dd,
J = 7.0, 3.0 Hz, 1H), 5.43 (s, 1H), 3.11 (s, 6H), 1.97 (s, 3H), 1.96 (s, 3H)
ppm; 13C NMR (100 MHz,
Chloroform-d) 6 185.1, 183.8, 166.7, 155.1, 146.0, 145.8, 138.1, 130.5, 128.3,
123.0, 121.9,
103.8, 102.3, 100.2, 39.4, 28.2, 27.2 ppm; Anal. calc'd for C18H20N202Pt: C
43.99, H 4.10, N 5.70;
found C 44.99, H 4.15, N 5.68; m.p. 265-266 C.
6: 1H NMR (400 MHz, Chloroform-d) 6 9.14 (d, J = 5.4 Hz, 1H), 8.26 (d, J = 8.0
Hz, 1H), 7.85 ¨
7.73 (m, 2H), 7.63 ¨ 7.57 (m, 2H), 7.53 (d, J = 8.8 Hz, 1H), 7.44 (dd, J =
8.0, 5.4 Hz, 1H), 5.54 (s,
1H), 2.07 (s, 6H) ppm; Anal. calc'd for C18H15NO2Pt: C 45.76, H 3.20, N 2.97;
found C 46.11, H
3.12, N 2.92.
7: 1H NMR (400 MHz, Chloroform-d) 69.57 (d, J = 8.9 Hz, 1H), 8.26 (d, J = 8.7
Hz, 1H), 7.85 ¨
27
CA 02809478 2013-03-13
7.72 (m, 4H), 7.59 (d, J = 7.7 Hz, 1H), 7.55 (dd, J = 8.1, 6.9 Hz, 1H), 7.23
(d, J = 7.5 Hz, 1H), 7.17
(t, J = 7.5 Hz, 1H), 5.58 (s, 1H), 2.06 (s, 3H), 2.05 (s, 3H) ppm; Anal.
calc'd for C201-117NO2Pt: C
48.19, H 3.44, N 2.81; found C 48.47, H 3.28, N 2.67.
8a: 1H NMR (400 MHz, Chloroform-d) 6 9.04 (s, sat, Jpt_H = 38.4 Hz, 1H), 7.88
(d, J = 8.0 Hz, 1H),
7.63 (d, J = 7.6 Hz, 1H), 7.57 (d, J = 8.1 Hz, 1H), 7.50 (d, J = 7.8 Hz, 1H),
7.22 (t, J = 7.4 Hz, 1H),
7.10 (t J = 7.5 Hz, 1H), 6.87 (s, 4H), 5.40 (s, 1H), 2.32 (s, 6H), 2.09 (s,
12H), 1.98 (s, 3H), 1.66 (s,
3H) ppm; Anal. calc'd. for C34H3613NO2Pt: C 58.63, H 5.21, N 2.01; found C
57.63, H 5.23, N 1.83.
8b: 1H NMR (400 MHz, Chloroform-d) 6 8.89 (d, sat, Jpt-H = 39.6 Hz, J = 5.3
Hz, 1H), 7.72 (t, J =
7.6 Hz, 1H), 7.47 (d, J = 7.8 Hz, 1H), 7.32-7.24 (m, 6H), 7.24-7.18 (m, 4H),
7.04 (t, J = 7.2 Hz, 2H),
7.00 (d, J = 6.5 Hz, 1H), 6.69 (dd, J = 8.4, 2.4 Hz, 1H), 5.39 (s, 1H), 1.97
(s, 3H), 1.73 (s, 3H) ppm,
Anal. calc'd for C28H24N202Pt: C 54.63, H 3.93, N 4.55; found C 55.31, H 3.94,
N 4.35.
8c: 1H NMR (400 MHz, Chloroform-d) 68.90 (s, sat, Jpt_H = 35.3 Hz, 1H), 8.01
(d, J = 8.5 Hz, 1H),
7.88 (d, J = 8.2 Hz, 1H), 7.79 (d, J = 8.2 Hz, 1H), 7.75 (d, J = 8.1 Hz, 1H),
7.52 ¨ 7.40 (m, 3H), 7.40
¨ 7.31 (m, 2H), 7.23 (m, 6H), 6.99 (m, 1H), 6.85 (m, 5H), 6.55 (d, J = 8.5 Hz,
1H), 5.31 (s, 1H) 2.30
(s, 6H), 2.08 (s, 12H), 1.62 (s, 6H) ppm; Anal. calc'd for C50H47BN202Pt: C
65.72, H 5.18, N 3.07;
found C 66.09, H 5.07, N 3.08.
9a: 1H NMR (400 MHz, Chloroform-d) 6 8.86 (d, sat, Jpt_H = 42.8 Hz, J = 5.6
Hz, 1H), 8.08 (d, J =
7.8 Hz, 1H), 7.73 (t, J = 7.8 Hz, 1H), 7.44 (d, J = 8.3 Hz, 1H), 7.37-7.30 (m,
2H), 7.24 (t, J = 7.3 Hz,
1H), 7.03 (dd, J = 7.4, 5.8 Hz, 1H), 5.54 (s, 1H), 2.06 (s, 3H), 2.03 (s, 3H)
ppm 13C NMR (100 MHz,
Chloroform-d) 6 185.1, 183.6, 159.5, 156.6, 147.8, 138.9, 133.3, 125.4, 123.7,
122.8, 119.1,
116.6, 116.3, 111.1, 102.5, 28.1, 26.4 ppm; Anal. calc'd for C18H15NO3Pt: C
44.27, H 3.10, N 2.87;
found C 44.68, H 2.72, N 2.73; m.p. 247-248 C.
9b: 1H NMR (400 MHz, Chloroform-d) 6 8.92 (d, sat, Jpt-H = 40.0 Hz, J = 5.8
Hz, 1H), 8.83 ¨8.76
(m, 1H), 7.86 ¨ 7.78 (m, 1H), 7.72 (t, J = 7.8 Hz, 1H), 7.40 ¨ 7.28 (m, 4H),
6.96 (dd, J = 7.3, 5.8 Hz,
1H), 5.56 (s, 1H), 2.10 (s, 3H), 2.03 (s, 3H) ppm; Anal. calc'd. for
C18F115NO2PtS: C 42.97, H 3.00,
N 2.78; found C 42.97, H 2.71, N 2.76.
9c: 1H NMR (400 MHz, Chloroform-d) 6 8.92 (d, sat, Jpt_H = 40.7 Hz, J = 5.8
Hz, 1H), 8.30 (d, J =
7.9 Hz, 1H), 7.57 (t, J = 7.3 Hz, 2H), 7.49 (t, J = 7.3 Hz, 1H), 7.43 (d, J =
7.3 Hz, 2H), 7.36 (t, J =
28
CA 02809478 2013-03-13
. 7.9 Hz, 1H), 7.19(t, J = 7.5 Hz, 1H), 7.11 (t, J = 7.3 Hz, 1H), 7.02(d,
J = 8.3 Hz, 1H), 6.79 (dd, J
= 7.3, 5.7 Hz, 1H), 6.38 (d, J = 8.2 Hz, 1H), 5.54 (s, 1H), 2.09 (s, 3H), 2.02
(s, 3H) ppm; 13C NMR
(100 MHz, Chloroform-d) 6 184.9, 183.5, 159.7, 148.2, 142.6, 142.3, 138.6,
138.1, 132.7, 129.6,
128.2, 127.9, 124.2, 123.6, 120.3, 118.2, 117.2, 116.5, 110.0, 102.4, 28.3,
26.4 ppm, Anal. calc'd
for C24H20N202Pt: C 51.15, H 3.58, N 4.97; found C 51.67, H 3.51, N 4.73; m.p.
> 300 C.
Pt-Bppy (21): Yield: 92%. 1H NMR (400 MHz, CD2Cl2, 25 C, TMS) 6 = 9.24 (d,
sat, 3J = 5.6 Hz,
JPt-H = 36 Hz, 1H), 7.81 (td, 3J = 8.0, 4J= 1.6 Hz, 1H), 7.65-7.53 (m, 3H),
7.28 (dd, 3J = 7.6, 4J = 1.2
Hz, 1H), 7.18 (td, 3J= 6.4, 4J = 1.2 Hz, 1H), 6.87(s, 4H), 5.55 (s, 1H), 2.35
(s, 6H), 2.07 (s, 12H),
2.06 (s, 3H), 2.02 (s, 3H); 13C NMR (100 MHz, CD2Cl2, 25 C, TMS) 6=186.2,
184.4, 167.9, 147.9,
147.2, 144.9, 140.7, 138.2, 131.2, 130.3, 128.2, 128.1, 121.5, 118.6, 102.3,
27.9, 26.9, 23.2,
20.9. Anal. Calcd (%) for C33H35BNO2Pt: C, 57.99; H, 5.16; N, 2.05. Found: C,
57.89; H, 5.19; N,
2.08. This product was also obtained when Pt(pheny1)2(DMS0)2was used in place
of
[PtMe2(SMe2)]2=
Pt-Bmppy (22): Yield: 89%. 1H NMR (400 MHz, CD2Cl2, 25 C, TMS) 6 = 9.10 (dd,
sat, 3J = 5.6
Hz, 4J = 1.2 Hz, Jpt-H = 42 Hz, 1H), 7.79-7.74 (m, 2H), 7.45 (d, sat, 3J = 7.7
Hz, Jpt-H = 28 Hz, 1H),
7.05 (td, 3J = 7.2 Hz, 4J =1.2 Hz, 1H), 6.84(d, 3J = 7.6 Hz, 1H), 6.70 (s,
4H), 5.41 (s, 1H), 2.40 (s,
3H), 2.20 (s, 6H), 1.93 (s, 12H), 1.87 (s, 3H), 1.45 (s, 3H); 13C NMR (100
MHz, CD2Cl2, 25 C,
TMS) 6 = 186.3, 184.4, 168.4, 147.8, 146.2, 143.8, 143.4, 140.2, 138.5, 137.9,
134.8, 128.2,
128.0, 123.2, 120.8, 102.2, 28.0, 26.9, 22.8, 20.9. Anal. Calcd (%) for
C34H376NO2Pt : C, 58.54; H,
5.35; N, 2.01. Found: C, 58.52; H, 5.38; N, 2.05. This product was also
obtained when
Pt(pheny1)2(DMS0)2was used in place of [PtMe2(SMe2)]2.
Pt-Bfppy (23): Bright yellow crystals were obtained after recrystallization
from DCM/hexane. 1H
NMR (400 MHz, CD2Cl2, 25 C, TMS): 6= 9.11 (d, 4J = 0.5 Hz, 1H), 8.02 (d, 3J =
8.5 Hz, 1H), 7.86
(t, 3J = 8.0 Hz, 1H), 7.45 (d, 3J= 8.5 Hz, 1H), 7.20 (t, 3J = 7.5 Hz, 1H),
7.06 (t, 3J = 7.5 Hz, 1H), 6.85
(s, 4H), 5.54 (s, 1H), 2.33 (s, 6H), 2.10 (s, 12H), 1.75 (s, 3H), 1.15 (s,
3H). 13C NMR (100 MHz,
CD2Cl2, 25 C, TMS): 6 = 186.4, 184.5, 147.4, 140.2, 138.8, 138.5, 137.6,
137.5, 128.1, 126.4,
123.4, 123.2, 121.6, 102.4, 27.9, 26.8, 22.8, 20.9. Anal. Calcd (%) for
C34H35BFNO2Pt: C, 57.15;
H, 4.94; N, 1.96. Found: C, 56.97; H, 4.95; N, 1.87. This product was also
obtained when
Pt(pheny1)2(DMS0)2was used in place of [PtMe2(SMe2)12.
29
CA 02809478 2013-03-13
Example 3: Synthesis of boron-functionalized CAC-chelate carbene complexes BC1
and
BC2
=I, II
Ek iii=
B
N
n-NH2
=
p: la, 90% p: lb, 85%
m: 2a, 83% m: 2b, 81%
O
E3
iv i, ix-xi
-vii viii igpir
B ______________________________ ;=-N,
Pt
¨ ____________________________________________ ¨
0 p
1-
p: lc, 38% p: Id, 80%
p: BC1, 25%
m: 2c, 73% m: 2d, 70% m: BC2, 17%
Scheme 2. Synthesis of boron-functionalized CAC-chelate carbene complexes.
Reagents and
conditions: i) n-BuLi, THF, -78 C; ii) FBMes2, THF, -78 C to RT; iii) Pd/C,
Ts0H, Et0H, 25 C; iv)
glyoxal THF/Me0H, 25 C; v) H2CO, NH4CI, 25 C; vi) H3PO4, reflux; vii)
Na0H(aq), 0 C; viii) Mel,
THF, 25 C; ix) [PtMe2(SMe2)]2, -78 C to 55 C; x) Ts0H, 25 C; xi) Na(acac),
THF/Me0H, -78 C.
N,N-dibenzy1-4-(dimesitylboryl)aniline (la): To a 250 mL Schlenk flask was
added
N,N-dibenzy1-4-bromoaniline (1.8 g, 5.1 mmol) and 80 mL dry THF. The resulting
mixture was
cooled to -78 C, then n-BuLi (3.5 mL, 5.6 mmol, 1.6 M in hexanes) was added
dropwise with
stirring. The reaction was stirred for 1 h at -78 C, and then FBMes2 (1.6 g,
6.1 mmol) was added.
The reaction mixture was stirred at -78 C for 1 h, then allowed to warm slowly
to room
temperature and stirred for 16 h. After removal of the solvent in vacuo, the
mixture was washed
with saturated aqueous NH4CI, then extracted with CH2Cl2 and water. The
combined organic
layers were dried using MgSO4, filtered, and purified using flash
chromatography on silica gel (4:1
hexanes:CH2Cl2 as eluent) to afford 2.4 g la as a white solid (90% yield). 1H
NMR (400 MHz,
CA 02809478 2013-03-13
sc
_ CDCI3) (57.43 (d, J = 8.2 Hz, 2H, -C6H4-), 7.35(t, J = 7.1 Hz, 4H, -
Ph), 7.32-7.22 (m, 6H, -Ph), 6.83
(s, 4H, Mes), 6.73 (d, J = 8.2 Hz, 2H, -C6H4-), 4.72 (s, 4H, -CH2-), 2.32 (s,
6H, Mes), 2.12 (s, 12H,
Mes) ppm; 13C {1H} NMR (100 MHz, CDCI3) 5 152.5, 142.1, 140.6, 140.0, 137.8,
137.5, 133.0,
128.7, 127.9, 127.1, 126.7, 111.2, 53.8, 23.5, 21.1 ppm; HRMS (High Resolution
Mass
Spectrometry) calc'd for C38H40BN: 521.3254, found 321.3246.
4-dimesitylborylaniline (lb): To a 500 mL round-bottomed flask equipped with a
magnetic stir
bar was added la (2.37 g, 4.5 mmol), palladium on carbon (0.50 g, 5 wt% Pd), p-
toluenesulfonic
acid (0.50 g, 2.9 mmol) and ethanol (200 mL). The reaction was bubbled with
hydrogen gas for 16
h at room temperature, then passed through a pad of celite and concentrated in
vacuo. The
residue was washed with 1M aq. NaOH, then extracted with CH2Cl2 and water. The
combined
organic layers were dried using MgSO4, filtered, and purified using flash
chromatography on silica
gel (1:1 hexanes:CH2Cl2 as eluent) to afford 1.31 g lb as a white solid (85%
yield). 1H NMR (300
MHz, CDCI3) (57.39 (d, J = 8.4 Hz, 2H, -C6H4-), 6.82 (s, 4H, Mes), 6.61 (d, J
= 8.4 Hz, 2H, -C61-14-),
4.03 (s, br, 2H, -NH2), 2.32 (s, 6H, Mes), 2.07 (s, 12H, Mes) ppm; 13C {1F1}
NMR (75 MHz, CDCI3)
(5 150.5, 141.9, 140.7, 139.9, 137.7, 135.1, 128.0, 113.8, 23.4, 21.1 ppm;
HRMS calc'd for
C24H28BN: 341.2315, found 341.2309.
N-(4-dimesitylborylphenyl)imidazote (lc): To a 100 mL round-bottomed flask
with stir bar was
added lb (1.12 g, 3.3 mmol), glyoxal (0.375 mL, 40 wt.%, 3.28 mmol) and 10 mL
1:1 THF:Me0H.
The reaction was stirred for 16 h at room temperature, then NH4CI (0.35 g, 6.6
mmol),
formaldehyde (0.45 mL, 37 wt.%, 6.6 mmol) and 25 mL Me0H were added. The
reaction was
heated to reflux for 1 h, then 0.5 mL 85% H3PO4 was added. The mixture was
heated to reflux for
an additional 8h, then poured over ice (25 g), washed with 2M aq. NaOH, and
extracted with
CH2Cl2 and water. The combined organic layers were dried using MgSO4,
filtered, and purified
using flash chromatography on silica gel (4:1 ethyl acetate:hexanes as eluent)
to afford 488 mg
lc as a white solid (38% yield). 1H NMR (400 MHz, CDCI3) 67.94 (s, br, 1H, -
Im), 7.63 (d, J = 8.2
Hz, 2H, -C6H.4-), 7.37 (d, J = 8.2 Hz, 2H, -C6H4-), 7.35 (s, br, 1H, -Im),
7.21 (s, br, 1H, -Im), 6.84 (s,
4H, Mes), 2.31 (s, 6H, Mes), 2.02 (s, 12H, Mes) ppm; 13C {1F1) NMR (100 MHz,
CDCI3) (5144.8,
141.3, 140.7, 139.8, 139.0, 138.1, 135.4, 130.7, 128.3, 120.1, 117.7, 23.4,
21.2 ppm; HRMS
calc'd for C27H2913N2: 392.2424, found 392.2429.
31
CA 02809478 2013-03-13
t
,. N-(4-dimesitylborylphenyI)-N'-methylimidazolium iodide (1d): To a 25 mL
round-bottomed
flask with stir bar was added 1c (400 mg, 1.01 mmol), methyl iodide (0.32 mL,
5.1 mmol) and 10
mL THF. After stirring at room temperature for 40 h under air, the white
precipitate was filtered,
washed with THE and dried to afford 433 mg Id (80% yield). 1H NMR (300 MHz,
Me0H-d4) 6
9.59 (s, 1H, Im), 8.15 (d, J = 2.1 Hz, 1H, Im), 7.81 (d, J = 2.1 Hz, 1H, Im),
7.76 (d, J = 8.6 Hz, 2H,
-C6H4-), 7.69 (d, J = 8.6 Hz, 2H, -C6H4-), 6.86 (s, 4H, Mes), 4.06 (s, 3H, -
CH3), 2.30 (s, 6H, Mes),
1.99 (s, 12H, Mes) ppm; Anal. Calc'd for C28H33131N2: C 62.83, H 6.21, N 5.23,
found C 62.82, H
5.99, N 5.12.
N,N-dibenzy1-3-(dimesitylborypaniline (2a): Prepared in analogy with la (83%
yield). 1H NMR
(400 MHz, CDCI3) 6 7.37 (t, J = 7.4 Hz, 1H, -C6H4-), 7.34-7.25 (m, 7H, -Ph, -
C6H4-), 7.21-7.15
(m, 5H, -Ph, -C6H4-), 6.92 (s, 1H, -06H4-), 6.77 (s, 4H, Mes), 4.59 (s, 4H, -
CH2-), 2.33 (s, 6H, Mes),
1.99 (s, 12H, Mes) 13C {1H} NMR (100 MHz, CDCI3) 6148.4, 146.4, 141.9, 140.6,
138.8, 138.0,
128.6, 128.5, 128.0, 126.9, 125.2, 121.0, 116.8, 112.5, 55.0, 23.2, 21.2 ppm;
HRMS calc'd for
C38H40E3N: 521.3254, found 521.3260.
3-dimesitylborylaniline (2b): Prepared in analogy with lb (81% yield). 1H NMR
(400 MHz,
CDCI3) 67.15 (t, J = 7.5 Hz, 1H, -C6H4-), 6.94 (d, J = 7.2 Hz, 1H, -C6H4-),
6.86-6.78 (m, 6H, -C6H4-,
Mes), 3.52 (s, br, 2H, -NH2), 2.32 (s, 6H, Mes), 2.04 (s, 12H, Mes) ppm; 13C
{1H} NMR (100 MHz,
CDCI3) 6 147.2, 145.9, 141.9, 140.8, 138.5, 128.8, 128.1, 126.8, 122.1, 118.7,
23.3, 21.2 ppm;
HRMS calc'd for C24H28BN: 341.2315, found 341.2319.
N-(3-dimesitylborylphenyl)imidazole (2c): Prepared in analogy with 1c (73%
yield). 1H NMR
(400 MHz, CDCI3) 67.76 (s, br, 1H, -Im), 7.52-7.42 (m, 4H, -C6H4-), 7.21 (s,
br, 1H, -Im), 7.15 (s,
br, 1H, -Inn), 6.83(s, 4H, Mes), 2.31 (s, 6H, Mes), 2.00 (s, 12H, Mes) ppm;
130 {1F1} NMR (75 MHz,
CDCI3) 5 148.2, 141.1, 140.8, 139.3, 137.2, 135.6, 135.1, 130.1, 129.5, 128.4,
128.2, 124.6,
118.4, 23.4, 21.2 ppm; HRMS calc'd for C27F129BN2: 392.2424, found 392.2411.
N-(3-dimesitylborylphenyI)-N'-methylimidazolium iodide (2d): Prepared in
analogy with 7.1
(70% yield). 1H NMR (400 MHz, Me0H-d4) 69.43 (s, 1H, Im), 7.98 (d, J = 2.1 Hz,
1H, Im), 7.87 (dt,
J = 7.6 Hz, J = 1.7 Hz, 1H, -06H4-), 7.74 (d, J = 2.1 Hz, 1H, Im), 7.68 (t, J
= 7.7 Hz, 1H, -C6H4-),
7.66 (d, J = 1.7 Hz, 1H, -C6H4-), 7.65 (d, J = 7.9 Hz, 1H, -C6H4-), 6.86 (s,
4H, Mes), 4.00 (s, 3H,
-CH3), 2.29 (s, 6H, Mes), 1.99 (s, 12H, Mes) ppm; 130 {1H} NMR (100 MHz, Me0H-
d4) 6 150.5,
32
CA 02809478 2013-03-13
=
. 142.4, 142.2, 141.2, 138.5, 137.2, 136.7, 131.7, 129.7, 129.6, 126.9,
125.9, 123.0, 37.1, 23.9,
21.5 ppm; Anal. Calc'd for C28H33BIN2: C 62.83, H 6.21, N 5.23, found C 62.92,
H 6.53, N 4.58.
As shown in Scheme 2 above, compounds 1d and 2d were reacted using the general
synthesis provided in Example 5 to form their Pt complexes BC1 and BC2 with
the addition of
cooling to the indicated temperatures. An analogous reaction with sodium
nacnac in place of
sodium acac would form Pt(B-NHC1)(nacnac) and Pt(B-NHC2)(nacnac).
Example 4. The following schematic shows a synthetic pathway for a organoboron
ligand that
comprises a BMes2functionalized phenyl ring and a triazole ring.
Scheme 3
Br Mes26µ
Mes26 Mes26 R i, ii
iii, iv V /
---ill. ( ) _______________________________________________________________ l
N
I
...............õ..,. --). ,.........õ7õ..7.
N-----N
Br Br
.õCH2Ph
Mes2B I/ / N
1 1
3a
i) n-BuLi, -78 C, Et20, 1h; ii) BMes2F, RT, overnight; iii) TMS acetylene,
Pd(PPh3).4, Cul, Et3N,
80 C, overnight; iv) NaOH, THF/Me0H, RT, 2h; v) RN3, Cu(CH3CN)4PF6, DIPEA,
TBTA, DCM,
RT.
Synthesis of (4-bromophenyl)dimesitylborane see steps (i) and (ii) of scheme
3: To a 100
mL Schlenk flask equipped with a magnetic stir bar was added para-
dibromobenzene (1.0 g,
4.24mmol) and 30 mL of dry diethyl ether (Et20). The resulting solution was
cooled to -78 C and
stirred for 30 minutes. At that time, 2.9 mL of 1.6 M n-butyllithium (n-BuLi)
(4.64mmol) was
slowly added. The mixture was maintained at -78 C for 1h, and dimesitylboron
fluoride (1.36g,
5.07mmol) was added. The resulting mixture was stirred at -78 C for another
hour. It was then
slowly warmed to room temperature (RT) and stirred overnight. The following
morning, the
33
CA 02809478 2013-03-13
_ solvent was removed under reduced pressure. A crude product was
dissolved using
dichloromethane solvent. The hydrophobic solvent solution was washed with
brine and water.
The combined hydrophobic phase was dried over MgSO4 and filtered through
filter paper. The
product was further purified using flash chromatography through silica using
hexane as eluent to
afford 1.2 g of (4-bromophenyl)dimesitylborane, as a white solid (70% yield).
Notably, the above
synthetic procedure could also be used to synthesize (3-
bromophenyl)dimesitylborane when
meta-dibromobenzene is used in place of para-dibromobenzene. Also,
(2-bromophenyl)dimesitylborane can be synthesized when ortho-dibromobenzene is
used in
place of para-dibromobenzene.
Synthesis of (4-ethynylphenyl)dimesitylborane see steps (iii) and (iv) of
scheme 3: A 100
mL three-necked round bottomed flask, equipped with a magnetic stir bar and
condenser, was
charged with ligand (4-bromophenyl)dimesitylborane (1.22 g, 3.03mmol),
trimethylsilylacetylene
(0.45mL, 3.44mmol), tetrakis(triphenylphosphine)palladium(0) (0.175g,
0.15mmol), copper iodide
(0.03g, 0.15mmol) and 30 mL of degassed triethylamine. The mixture was stirred
at 80 C for 20
hours, and then concentrated under reduced pressure. The product was dissolved
in
dichloromethane solvent. The hydrophobic solvent solution was then
sequentially washed with
saturated ammonium chloride solution, brine and water. The combined
hydrophobic phase was
dried over MgSO4 and filtered through a filter paper. The product was then
purified using flash
chromatography through silica using hexane as eluent. After removal of eluent
solvent under
reduced pressure, the resulting white solid was dissolved in 10 mL of
tetrahydrofuran solvent and
treated with sodium hydroxide in methanol (20 mL of a 2.0 M solution). After
stirring for 2 hour,
the resulting mixture was concentrated under reduced pressure. After
extraction with
dichloromethane, the hydrophobic solution was dried over MgSO4, filtered and
the solvent was
removed under reduced pressure to give the product (4-
ethynylphenyl)dimesitylborane as a white
solid (0.67g, 65%).
Notably, the above synthetic procedure could also be used to synthesize
(2-ethynylphenyl)dimesitylborane or 3--ethynylphenyl)dimesitylborane when 2-
and 3-
-bromophenyl)dimesitylborane are used instead of (4-
bromophenyl)dimesitylborane,
respectively.
Synthesis of 4-(4-(dimesitylboryl)pheny1)-1-benzyl-1,2,3-triazole (3a) see
step (v) of
scheme 3: To a 50 mL Schlenk flask equipped with a magnetic stir bar was added
34
CA 02809478 2013-03-13
(4-ethynylphenyl)dimesitylborane (0.64g, 1.84mmol), benzyl azide (0.245g,
1.84mmol),
diisopropylethylamine (0.475g, 3.68mmol), tris[(1-benzy1-1H-1,2,3-triazol-4-
yl)methyl]amine (1
mol %) and 30 mL of dichloromethane. The resulting solution was bubbled with
nitrogen gas for
20 minutes. [Cu(CH3CN)4]PF6 (1 mol %) was added as a catalyst. The resulting
mixture was
stirred overnight, after which the solvent was removed under reduced pressure.
The crude
product was dissolved in dichloromethane. The solution was washed with
saturated ammonium
chloride solution, brine and water. Following isolation, the non-aqueous phase
was dried over
MgS0.4 and filtered through a filter paper. The product was then purified
using flash
chromatography through silica (4:1 hexanes:ethyl acetate as eluent) to afford
0.64 g
4-(4-(dimesitylboryl)pheny1)-1-benzy1-1,2,3-triazole (3a) as white solid (72%
yield).
Notably, the above synthetic procedure could also be used to synthesize
4-(3-(dimesitylboryl)phenyI)-1-benzyl-1H-1,2,3-triazole when (3-
ethynylphenyl)dimesitylborane is
used instead of (4-ethynylphenyl)dimesitylborane.
Example 5. Synthesis of Platinum complexes
R Mes2
(
I.
v
Mes2B Mes2B
Pt Pt
/ No /
N--N
R/
R/
C1-05 C6-C11
30H2Ph H2Ph
m/C
/C
Mes2B /
Mes2B
P/
/
Pt Pt
N __________________________________________________________________
0 0
o)
35-
CA 02809478 2013-03-13
,
Scheme 4 i) [PtMe2(u-SMe2)]2, acetone, 80 C, 2-3h; ii) p-toluenesulfonic acid,
THF, 1h; iii)
Na(acac), Me0H, overnight; iv) picolinic acid, Me0H, overnight.
Synthesis of Pt(4-(4-BMes2-phenyl)-1-benzyl-1,2,3-triazoly1)(acac) (C5), see
scheme 4:
BMes2-functionalized phenyl-triazole (3a) ligand (0.10 mmol) and [PtMe2(u-
SMe2)]2 (0.032
g, 0.055 mmol) were added to a 20 mL screw-cap vial with of acetone (5 mL).
The resulting
mixture was heated to and maintained at 75 C for 2 hours. Then, a 0.10 M
solution of Ts0H in
THF (1 mL) was added. The resulting solution was stirred for 1 hour. Next, 0.1
M solution of
Na(acetylacetonate) in methanol (2 mL) was added and the mixture was stirred
overnight. The
solvent was then removed under reduced pressure. The crude product was
dissolved in
dichloromethane and washed with with brine and water. The combined non-aqueous
phase was
dried over MgS0.4 and filtered through a filter paper. The product was
purified through silica
using dichloromethane as the eluent to to afford 0.0195g C5 as yellow solid
(24% yield). 1H NMR
(400 MHz, CD2Cl2):67.51 (s, 1H), 7.47(s, 1H), 7.40-7.28(m, 5H), 7.11 (d,
3J=7.6Hz, 1H), 7.07(d,
3J=7.6Hz, 1H), 6.74 (s, 4H), 5.49 (s, 2H), 5.37 (s,1H), 2.20 (s, 6H), 1.97 (s,
12H), 1.88 (s, 3H),
1.60 (s, 3H) ppm; elemental analysis calcd (%) for C38H40BN302Pt: C 58.77, H
5.19, N 5.41,
found: C 58.76, H 5.21, N 5.39.
Synthesis of Pt-(4-(4-BMes2-phenyl)-1-benzy1-1,2,3-triazoly1)(picolinate)
(C10): Ligand 3a
(0.05g, 0.10mmol) and [PtMe2(u-SMe2)]2 (0.032g, 0.055mmol) were added to a
20mL screw-cap
vial with 5mL of acetone. The mixture was heated at 75 C for 2 hours before
2mL of 0.10 M
solution of picolinic acid in methanol was added. The resulting solution was
stirred overnight. After
the solvent was removed under reduced pressure, the product was extracted with
dichloromethane, and then washed with brine and water. The combined organic
phase was dried
over MgSO4, filtered and purified on silica (2:1 dichloromethane:ethyl acetate
as eluent) to afford
0.021g of C10 as yellow solid (26% yield). 1H NMR (500 MHz, CD2C12):69.52 (d,
3J=5.6Hz, 1H),
8.14 (m, 1H), 7.80 (s, 1H), 7.70 (m, 1H), 7.62 (s, 1H), 7.50-7.42 (m, 5H),
7.27 (d, 3J=7.5Hz, 1H),
7.16 (d, 3J=7.5Hz, 1H), 6.88 (s, 4H), 5.64 (s, 2H), 2.34 (s, 6H), 2.06 (s,
12H); elemental analysis
calcd (%) for C39H37BN402Pt: C 58.58, H 4.68, N 7.01, found: C 59.83, H 5.18,
N 6.44.
36
CA 02809478 2013-03-13
-
Synthesis of C11
A BMes2-functionalized phenyl-triazole ligand (0.10mmol) and [PtMe2(u-SMe2)]2
(0.032g,
0.055mmol) were added to a 20mL screw-cap vial with acetone (5mL). The
resulting mixture was
heated to and maintained at 75oC for 2 hours. Then, 0.1M solution of the
1,5-dimethy1-1H-pyrazole-3-carboxylic acid in methanol (2mL) was added. The
resulting solution
was stirred overnight. A precipitated solid was collected on a filter paper
and washed with
methanol, hexane and acetone (3 x 5 mL each) and dried in air.
Synthesis of C12
A BMes2-functionalized phenyl-triazole (3a) ligand (0.10 mmol) and [PtMe2(u-
SMe2)]2 (0.032
g, 0.055 mmol) were added to a 20 mL screw-cap vial with of acetone (5 mL).
The resulting
mixture was heated to and maintained at 75 C for 2 hours. Then, a 0.10 M
solution of Ts0H in
THE (1 mL) was added. The resulting solution was stirred for 1 hour. Next, 0.1
M solution of
2-(1H-1,2,4-triazol-3-yl)pyridine in methanol (2 mL) was added and the mixture
was stirred
overnight. The solvent was then removed under reduced pressure. The crude
product was
dissolved in methanol and purified on TLC plate using acetone as the eluent.
Synthesis of Pt(BMes2-triazolyI)(picolinate)
See C6-C11 of Scheme 4: The BMes2-functionalized phenyl-triazole ligand
(0.10mmol)
and [PtMe2(u-SMe2)]2 (0.032g, 0.055mmol) were added to a 20mL screw-cap vial
with acetone
(5mL). The resulting mixture was heated to and maintained at 75 C for 2 hours.
Then, 0.1M
solution of the corresponding picolinic acid or substituted picolinic acid in
methanol (2mL) was
added. The resulting solution was stirred overnight. A precipitated solid was
collected on a filter
paper and washed with methanol, hexane and acetone (3 x 5 mL each) and dried
in air.
30
37
CA 02809478 2013-03-13
Synthesis of 2-(3-bromo-phenyl)-benzimidazole (see first step of scheme 5)
COOH
Br
Br 1. KOt-Bu
Br
1. n-BuLi
40 NH2
PPA 2. CH3I N
2. BIVIes2F
1110 "= __________________________________________________
150 C
NH2 THF, r.t.
THF, -78 C
B
B 1. PtMe2ISMe2)2
2. Ts0H, Na(acac)
N/
11/ = THF, r.t. Pt
Pt-12
imidazole 1
Scheme 5
3-bromobenzoic acid (2.0g, 9.9 mmol) was added to the solution of 1,2-
phenylenediamine
(1.07 g, 9.9 mmol) in polyphsphoric acid (PPA) (40 mL) at 120 C. The
resulting solution was
heated to and maintained at 150 C and stirred for 3 hrs. Upon cooling of the
solution, it was
poured into water. A resulting precipitate was filtered off. 10% NaOH aqueous
solution was added
to the filtrate until the pH was 10. In this process, a large quantity of
precipitate was produced,
which was then filtrated off using a filter paper. This filtrate was extracted
with diethyl ether 3
times. 2-(3-bromo-phenyl)-benzimidazole was obtained as a white solid after
the solvent was
removed under reduced pressure (yield, 55%). 1H NMR (ppm, 300 M in d6-DMS0):
13.03 (1H,
s), 8.37 (1H, s), 8.18 (1H, d, J= 8.18 Hz), 7.70 (2H, m), 7.53 (2H, m), 7.25
(2H, m).
Synthesis of N-Me-2-(3-bromo-phenyl)-benzimidazole (see second step of scheme
5)
K-Ot-Bu (0.23 g, 0.2 mmol) was added to a stirred solution of
2-(3-bromo-phenyl)benzimidazole (0.45 g, 0.2 mmol) in THF for 20 min. Excess
methyl iodide
was added to the solution, which was then stirred overnight. After filtering
off the precipitate and
removal of the solvent under reduced pressure, N-Me-2-(3-bromo-
phenyl)benzimidazole was
obtained quantitatively. 1H NMR (ppm, 300 M in CDCI3): 7.97 (1H, s), 7.84 (1H,
m), 7.69 (1H, d,
38
CA 02809478 2013-03-13
J = 7.5 Hz), 7.64 (1H, J = 8.1 Hz), 7.38 (4H, m), 3.87 (3H, m).
Synthesis of N-Me-2-(3-BMes2-phenyl)-benzimidazole (see imidazole 1 in scheme
5)
n-BuLi (0.8 mL, 1.3 mmol) was added slowly to a solution of
N-Me-2-(3-bromo-phenyl)-benzimidazole (0.29 g, 1.0 mmol) in THF (30 mL) at -78
C and the
resulting solution was stirred for about 1 hour at -78 C. BMes2F (0.37 g, 1.4
mmol) was then
added under a stream of nitrogen and the solution was stirred at the same
temperature for about
2 hours and then stirred overnight at ambient temperature. The solvents were
removed under
reduced pressure. The residue was purified over silica gel by flash column
chromatography using
a CH2Cl2/hexanes (1:1) mixture give a white powder of N-Me-2-(3-BMes2-phenyl)-
benzimidazole
("imidazole 1") (0.23 g, 50%). 1H NMR (400 MHz, CDCI3, ppm): 8.08 (1H, broad),
7.90 (1H, b),
7.76 (1H, s), 7.73 (1H, m), 7.62 (1H, m), 7.41 (3H, b), 6.85 (4H, s), 3.78
(3H, s), 2.33 (3H, s), 2.05
(12H, s).
Synthesis of Pt complexes Pt-12 as shown in scheme 5
Pt complexs Pt-12 were synthesized using procedures similar to that reported
in the
literature procedure (Z. M. Hudson etal., Org. Lett. 2012, 14, 1700-1703). 1
eq of ligand imidazole
1(1 mmol) and 1 equivalent PtMe2(SMe2)2 (1 mmol) were combined and stirred at
RT in 3mL THF
for 2hrs. 1 equivalent of p-tolunenesulfonic acid was added to the solution
and stirred for another
0.5 h, which was then followed by the addition of 2 equivalents of Na(acac) in
2 mL Me0H. The
mixture was stirred for 2 hrs. The solvent was then removed under vacuum and
the residue was
purified using column chromatograph on silica (dichlormethane /hexane: 1/1 v),
producing Pt-12
in good yield. 1H NMR (400 MHz, CD2Cl2, ppm): 8.84 (1H, m), 7.77 (2H, m), 7.39
(3H, m), 7.32
(1H, dd, J= 8.0 Hz, J = 1.2 Hz), 6.89(4H, s), 5.06(1H, s), 2.35 (6H, s),
2.12(12H, s), 2.11 (3H, s),
2.02 (3H, s). 13C NMR (125.6 MHz, CD2Cl2, ppm): 185.4, 183.6, 141.7, 140.6,
138.3, 138.1, 132.4,
130.6, 128.0, 124.0, 123.0, 116.4, 109.6, 101.9, 31.2, 27.6, 27.0, 23.3, 21Ø
Absorption and
emission spectra are shown for Pt-12 in Figures 4A and 4B. The solution
luminescence
quantum efficiency of Pt-12 compared to that of Ir(ppy)3 is 0.5. Compounds 51
and 52 are
synthesized in a similar way to the synthesis of Pt-12, by replacing Na(acac)
with Na(nacnac) in
the reaction.
Syntheses of Pt complexes with nacnac as a stabilizing ligand are procedurally
the same
as the corresponding acac complex, except that sodium nacnac would be used
instead of
sodium acac. A person with skill in the art of the invention would recognize
that this ligand with
39
CA 02809478 2013-03-13
another counterion would be equivalent (e.g., potassium nacnac).
Example 6. Synthesis of 10, a PAC chelate Pt(II)P-diketonate complex
Scheme 6. Synthesis of PAC chelate Pt(II) p-diketonate complex.
1 [PtMe2(SMe2)12, 55 C
2 TfOH, rt 65%
0-Pt'0
3 -7Failaet, rt
10
To a 20 mL screw-cap vial equipped with a magnetic stir bar was added
1-naphthyldiphenylphosphine (97 mg, 0.35 mmol), [PtMe2(SMe2)]2 dimer (100 mg,
0.17 mmol)
and 3 mL degassed THF. The resulting reaction mixture was stirred for 4 hours
at 55 C under an
N2 atmosphere. Then, HOTf (1 mL, 0.35 M in THF) was added dropwise. The
mixture was stirred
for 30 minutes at room temperature. A solution of Na(acac)-1-120 (98 mg, 0.70
mmol in 2 mL
Me0H) was then added. The mixture was stirred for 1.5 hours. The reaction
mixture was then
partitioned between water and CH2Cl2. The hydrophobic layer was washed with
brine, dried over
MgSO4, filtered, and concentrated under reduced pressure. The residue was then
purified using a
plug of silica gel (hexanes and CH2Cl2 as eluent) to give 10 as a white solid
in 65% yield.
10: 1H NMR (400 MHz, Chloroform-d) 6 8.24(d, sat, Jpt-H = 44.6 Hz, J = 7.1 Hz,
1H), 7.91-7.80 (m,
5H), 7.67 (dd, J = 10.5 Hz, 7.1 Hz, 1H), 7.58 (dd, J = 8.1, 1.8 Hz, 1H), 7.51-
7.36 (m, 8H), 5.52 (s,
1H), 2.16 (s, 3H), 1.93 (s, 3H) ppm; 13C NMR (100 MHz, Chloroform-d) 6186.10,
184.8 (d, JP-C =
3.7 Hz), 151.3 (d, Jr-c = 30.4 Hz), 134.12 (d, Jp_c = 52.7 Hz), 133.81 (d, d,
Jp_c = 16.8 Hz), 133.24
(d, d, Jp_c = 15.0 Hz), 132.93 (d, Jp_c= 11.7 Hz), 130.9(d, Jp_c = 62.9 Hz),
131.1 (d, Jp_c = 2.6 Hz),
130.7 (d, Jp-c = 32.2 Hz), 128.8 (d, Jp-c = 1.8 Hz), 128.5, 128.4, 126.5,
125.0 (d, Jp-c = 10.3 Hz),
122.7, 101.6, 28.2, 28.1 (d, Jp_c = 6.6 Hz) ppm; 31P NMR (169 MHz, Chloroform-
d) 28.27(s, sat,
JIDt-P = 4671 Hz) ppm; Anal. calc'd for C27C2302PPt: C 53.55, H 3.83, found C
53.59, H 3.70; m.p.
222-223 C.
CA 02809478 2013-03-13
Example 7. Synthesis of 11, a CAC chelate Pt(II) /3-diketonate complex
Scheme 7. Synthesis of a CAC chelate Pt(II) 0-diketonate complex.
N'l1= [PtMe2(SMe2)12, 55 C =
N
NN 2 TfOH, rt
-Pt
Ag 3 Na(acac), 0 0 61%
THF/Me0H, -40 C
11
To a 20 mL screw-cap vial with stir bar was added
1-methyl-3-phenylimidazol-2-ylidine)silver chloride (100 mg, 0.35 mmol),
[PtMe2(SMe2)]2 dimer
(100 mg, 0.17 mmol) and 3 mL degassed THF. The reaction was stirred for 1
hour, and then was
filtered to remove Agl. The resulting mixture was then heated to and
maintained at 55 C for two
hours, and then was cooled to room temperature. HOTs (1 mL, 0.35 M in THF) was
then added
dropwise, and the mixture was stirred for 30 minutes at room temperature.
After cooling the
reaction to -40 C, a solution of Na(acac).1-120 (49 mg, 0.35 mmol in 1 mL
Me0H) was added
dropwise. The mixture was stirred for 2 hours, and then was allowed to warm to
room
temperature. After partitioning between water and CH2Cl2, the hydrophobic
layer was washed
with brine, and the combined extracts were dried over MgSO4. The solution was
filtered and
concentrated under reduced pressure. The resulting residue was purified using
a plug of silica
gel, with CH2Cl2 as eluent, to give 11 as a yellow solid in 61% yield.
11: 1H NMR (400 MHz, Chloroform-d) 6 7.78 (dd, sat, Jpt-H = 52.1 Hz, J = 5.3,
2.0 Hz, 1H), 7.24 (d,
J = 2.0 Hz, 1H), 7.01 (m, 2H), 6.93 (dd, J = 6.8, 2.0 Hz, 1H), 6.80 (d, J =
2.0 Hz, 1H), 5.49 (s, 1H),
4.07 (s, 3H), 2.05 (s, 3H), 1.96 (s, 3H) ppm; Anal. calc'd for C15H16N202Pt: C
39.91, H 3.57, N
6.21, found C 40.34, H 3.60, N 6.08.
All scientific and patent publications cited herein are hereby incorporated in
their entirety
by reference.
Although this invention is described in detail with reference to embodiments
thereof,
these embodiments are offered to illustrate but not to limit the invention. It
is possible to make
other embodiments that employ the principles of the invention and that fall
within its spirit and
scope as defined by the claims appended hereto.
41
CA 02809478 2013-03-13
Table 1. Structural Formulae
Compound Compound
110
B 111 N1/
=
B-NHC1
B-NHC2
110
B
Pt
= N<
\\O N
Pt
Pt-(B-NHC1)(acac), BC1d ;
Pt-(B-NHC2)(acac), BC2d
B
-N\ * = N{
Pt
Pt
\.N/
/K)
Pt(B-NHC1)(nacnac)
Pt-(B-NHC2)(nacnac)
42
CA 02809478 2013-03-13
* 401 la
B
1 . 8 = / N
1
---N
N---
B-triazole1
B-triazole2
0
* 0 *
B
pezN
zN-_----N
Pt
/ \
Pt(B-triazole2)(acac)
Pt(B-triazole1)(acac), Cl C5
.
e 0 .
8
1
-- = / N
/ N---
N
-_----N Pt"
/ \ /
PtVN N. N
Pt(B-triazole2)(nacnac)
Pt(B-triazole1)(nacnac)
0
. 10 0
. J B = / N
z/ /
N--:---N
Pt,
Pt
/ NN
Pt(B-triazole1 )(t-bu-acac), C2 Pt(B-triazole2)(pic), C10
43
CA 02809478 2013-03-13
SO 0 40
11 : = B .
/
---N
N----.
N---,----N
Pt7
Pt
0/ NN
0'
NN
o> 0> <
Pt(B-triazole1 )(p-Me-pic), C6
Pt(B-triazole2)(p-Me-pic), C10A
10 0 10
. B
/ N
= B li _____ rs
__/ 7
7,N----
Pt
Pt OZ N
0; ),\I
)
0 ¨
Pt(B-triazole2)(o-Me-pic), C1OB
Pt(B-triazole1 )(o-Me-pic), C7
SO lei 10
= B. /1 . B .0 /
N
1
PtZ
0/
C/ N NN > N
0> ) 0
Pt(B-triazole1 )(pic), C8 Pt(B-triazole2)(Me2-pyrazole-carboxylate),
C11A
44
CA 02809478 2013-03-13
%
._
0
0 . S_
a
B / N---
--
B =
. / Nil
--- N Pt/N---
)1X
VNI--
OZNN--N/
0 Pt(B-Me-triazole2)(acac)
Pt(B-triazole1)(Me2-pyrazole-carboxylate) C11 C5A
0 0
41 B
=5) /
. B >.
NN. /N
NI-----":N
B-Me-triazole1
B-triazole3
1.1 le
111 B. /X I
N---
P/1 PtV
Pt(B-Me-triazole1)(acac), C4 Pt(B-triazole3)(acac), C3
CA 02809478 2013-03-13
4
._
0
0
. B
5
=
B >.
. /j
N 4I /j
7 "Pt
N¨N/ NN Pt
(N) 0/ \
Trans- <¨)
Pt(B-triazole3)(pic), C9
*
. B
/ii
Pt,
N/ N¨
O CN 3
Cis-
Pt(B-triazole1)(py-1,2,4-triazole), C12
=0 401
fit B
41
-
/ . B
,Ni --- . / 7
,..,
N¨N/ \N zN=_=.!--N
411 1 tst) 0 Pt
07 N
Cis and trans <
Pt(B-triazole1)(5-py-3-ph-1,2,4-triazole), C13
Pt(B-triazole3)(p-Me-pic), C27
46
CA 02809478 2013-03-13
0 5.
140 / N
B . r,
=1
0 N
7
N'"----"N
Pt B-triazole4
N-/ \ NJ
F3C )
N -
Cis and trans
Pt(B-triazole1)(5-py-3-CF3 1,2,4-triazole), C14
0
0 *
-,z,----N
1
. B
0
Pf-VN
0O
4I / 7 /\
NN-----"
Pt
7
N-N/ \ N Pt(B-triazole4)(acac), C28
) C>N
Cis and trans
Pt(B-triazole1)(5-py-3-Me-1 ,2,4-triazole), C15
SI
101 liP
B 11 / N
/ Pt
,N ,,,N-:...--N
1 5'
. B
1111
41 J4
N a
N-N/ NN
Cis, trans
Pt(B-triazole4)(5-py-3-Me 1,2,4-triazole),
Cis and trans
C29
Pt(B-triazole1)(3,5-py- 1,2,4-triazole), C16
47
CA 02809478 2013-03-13
-
I
0
0lit
1 \(
/j
=8
0
.
PtVN N
N-N/ NN ?)
V K
Cis and trans
Cis and trans Pt(B-triazole2)(2-py-
pyrazole), C30
Pt(B-triazole1)(2-py-pyrazole), C17
0
0 401
41 B
411 B
5>'
= / it
41 7
ptõ,N
N-N/ NN Pt
/ \ NV NN
40 V
N) )
Cis and trans
Cis and trans
Pt(B-triazole3)(3-Me-5-py- 1,2,4-triazole),
Pt(B-triazole1)(2-py-4-ph-pyrazole), C18
C27A
0
= B. \
11 \N 0
111 /J1
6-Me-benzimidazole1 (imidazole 1)
Pt
N-N/ NN
F3C )
A. <
Cis and trans
Pt(B-triazole1)(2-py-4-CF3 -pyrazole), C19
48
CA 02809478 2013-03-13
-
_
0 I/ Bli \N
41I B lit \ N =
= / hi
N-_----N Pt
7
/
/
0 0
Pt
N¨N/ \ N )
Pt(B-Me-benzimidazole1)(acac), C26
Cis and trans (Pt-12)
Pt(B-triazole1)(2-py-4-Me -pyrazole), C20
140
101 . B
111
4 .
11 \N 40
N---::-
Pt --
./ Pt `,N---
N¨N/ \
Q )
Pt(B-Me-benzimidazole1 )(nacnac), 51
Cis, trans
Pt(B-triazole1)(2-py-1 ,2,3-triazole), C21
11101
O *
B \
41 B
* pt N
/
. /N---
Z
Pt'
N/ XN Pt(B-Me-
benzimidazole2)(nacnac), 52
) < )
N ¨
Cis and trans
Pt(B-triazole1)(2-py-imidazole), C22
49
CA 02809478 2013-03-13
*lei 1
* \
B
N
B
N--'" = N lei
Pt
N/ NN * Pt
pt,N) ) V 0
cis and trans
Pt(B-triazole1)(2-py-4-ph-imidazole), C23 Pt(B-
Me-benzimidazole2)(acac), (Pt-12A)
10 4. : V
le 841/ /1 = \ /
N N
Pt Pt
N/ Nhj 0/ \O
õC N)
cis and trans
Pt(B-triazole1)(2-py-4-CF3 -imidazole), C24 21b
. le V
B
= B
= iii
z,N = \ /
Pt N
N/ \N Pt
) 0 / \
0 0
N -
)L)
Cis and trans
Pt(B-triazole1)(2-py-4-Me -imidazole), C25
22b
CA 02809478 2013-03-13
F
Pt/
)0at
Ck0o
23b
24c
V
Pt,
N
H3C
25'
P.,
411 P"
q P
N
N/
Pt
Ag
)1
11
bY.-L. Rao, Chem. Eur. J., 2012, 18, 11306-11316.
cSoo-Byung Ko etal., Organometaffics, 2013, 32(2), 599-608.
dZ. M. Hudson etal., J. Am. Chem. Soc., 2012, 134, 13930-13933.
5
51
CA 02809478 2013-03-13
Table 2. Photophysical Properties of BC1 and BC2
Absorption, A. Amõõ (nm) Tpa CDpc Eided HOMO
LUMO
E (104 CM-1 Mla Solutiona/Sol (us) Solutiona/Solid (V)d (eV)e
(eV) f
idb
BC1 381 (0.38), 344 478 / 482 6.9 0.87 / 0.90 -2.50 -
5.73 -2.64
(1.16), 316 (2.15)
BC2 371 (0.76), 356 462 / 464 3.4 0.41 / 0.86 -2.49 -
5.86 -2.65
(0.80), 324 (1.48)
[a] Measured in degassed CH2Cl2 at 1x10-5 M,
rbrDoped into PMMA at 10 wt%.
rcr Solution quantum efficiencies were measured in CH2Cl2 relative to Ir(PPY)3
= 0.97.17 Solid state
quantum yields (QY) were measured using an integration sphere. All QYs are
10%.
Ed) In DMF relative to FeCpr.
Eel Measured by UV photoelectron spectroscopy. rfi Calculated from the HOMO
level and the
optical energy gap.
52
CA 02809478 2013-03-13
Table 3. Photophysical properties of BMes2-phenyl-triazolyl/imidazolyl-Pt
compounds
-
Compound AmAnm] (1) PL
Cl (10 wt% PMMA) 471 0.10
C2 (10 wt% PMMA) 453 0.11
C3 (10 wt% PMMA) 455 0.09
C4 (10 wt% PMMA) 453 0.06
C5 (10 wt% PMMA) 493 0.63
C6 (5 wt% PMMA) 456 0.34
C6 (10 wt% PMMA) 456/540(br) 0.24
C7 (10 wt% PMMA) - -
C8 (10 wt% PMMA) 454/563(br) 0.18
C9 (10 wt% PMMA) 457/544(br) 0.20
C10 (10 wt% PMMA) 487 0.54
C11 (10 wt% PMMA) 456 0.17
C12 (5 wt% PMMA)) 456, 544 0.60
C26 (in CH2C12) 485 nm 0.50
C27 (5 wt% PMMA) 454 0.21
C27 (10 wt% PMMA) 455/550(br) 0.15
Table 4. Electroluminescent Device Data of BC1 and BC2
Device BC1 BC2
Von (V) 4.0 3.6
Luminance . (cd m-2) 4165 2098
Current efficiency, max. (cd A-1) 53.0 25.8
Power efficiency, max. (Im W-1) 41.6 22.5
External Quantum efficiency, max (%) 17.9 9.8
C.I.E.a (x, y) (0.34, 0.53)
(0.27, 0.50)
a The International Commission on Illumination (abbreviated CIE for its French
name) is the
international authority on light, illumination, color, and color spaces.
53