Note: Descriptions are shown in the official language in which they were submitted.
CA 02828954 2013-09-03
WO 2012/121691
PCT/US2011/027180
IMPLANTABLE PRESSURE-ACTUATED DRUG DELIVERY
SYSTEMS AND METHODS OF MANUFACTURE AND USE
CROSS REFERENCE TO RELATED APPLICATIONS
This application is related to prior filed U.S. non-provisional application
No. 12/985,015, filed January 5, 2011 which is incorporated herein by
reference
in its entirety.
BACKGROUND
TECHNICAL FIELD
Mechanically actuated substance delivery methods and systems, including
implantable, pressure-actuated drug delivery methods and systems.
RELATED ART
Coronary stents are commonly used to treat heart attacks. Stents,
however, have been known to abruptly clot. Conventional stent clot treatment
includes emergency angioplasty, which is expensive, difficult to coordinate,
and
accompanied by relatively high mortality rates.
Stents have been coated with drugs ("drug-eluting stents") to reduce the
formation of scar tissue. However, drug-eluting stents have been shown to
increase the likelihood of abrupt in-stent clot formation.
Stents have been textured and coated with antibodies and proteins to
attract native cell coverings in attempts to promote vessel healing and reduce
clot
formation, without much success and, in some cases, with increased clotting.
Furthermore, such passive systems are not able to sense nor adapt to new clots
that may occur.
CA 02828954 2013-09-03
WO 2012/121691
PCT/US2011/027180
Conventional intracoronary drug-delivery systems are passive. Drugs are
released with pre-determined kinetics, and these systems cannot sense nor
respond to new, abrupt environmental changes such as clotting. Active drug
delivery systems deliver drugs in response to an external stimulus, but
conventional systems require a bulky or externalized power source, a separate
sensor and are unsuitable for intracoronary application. For example, World
Intellectual Property Organization (WIP 0) publication number WO
2007/042961, to Johnson et al., teaches a substrate having a chemical
containing
reservoir sealed with a rupturable barrier layer (Johnson, page 2, lines 18-
26),
including a polymer layer and a metal layer (Johnson, page 6, lines 1-10), and
an
"inkjet printer and/or hydraulic system (i.e., a fluid pressure system with
controlled release valves)," or a "piezo-electrical means, e.g. a
piezoactuator
and/or loudspeaker," (Johnson, page 5, lines 21-29), to rupture the barrier
layer.
What are needed are small-scale, implantable, minimally invasive, and
mechanically actuated drug delivery methods and systems that do not require an
external power source or a separate sensor.
SUMMARY
Disclosed herein are implantable pressure-actuated systems to deliver a
drug and/or other substance in response to a pressure difference between a
system
cavity and an exterior environment, and methods of fabrication and use.
A pressure-rupturable membrane or diaphragm may be tuned to rupture at
a desired rupture threshold, rupture site, with a desired rupture pattern,
and/or
within or after a desired rupture time. The diaphragm may be tuned by
selection
of material, controlling a thickness, patterning a surface, patterning an
underlying
substrate support, and/or changing the material composition of the diaphragm.
- 2 -
CA 02828954 2013-09-03
WO 2012/121691
PCT/US2011/027180
The cavity may be pressurized above or evacuated below the rupture
threshold, and a diaphragm-protective layer may be provided to prevent
premature diaphragm rupture in an ambient environment and to dissipate within
an implant environment. The cavity may be pressurized above an implant
environment pressure, such as a blood vessel pressure. Under nollnal
conditions,
diaphragm integrity is maintained by counteracting pressure of the implant
envirnment. When the counteracting pressure of the implant environment falls,
such as loss of blood pressure due to an occlusive blood clot, and the
pressure
difference across the diaphragm falls, the diaphragm ruptures and the drug
and/or
other substance is released or delivered into the implant environment.
The drug and/or other substance may include, without limitation, a
thrombolytic, plasminogen, plasmin, heparin, and/or other anti-thrombotic
agent,
antiplatelet agent, anti-inflammatory agent, immunomodulator, and/or other
enzyme or protein, DNA, RNA, mRNA and/or other genetic material, virus
particles, cells, small molecule, anti-lipid agent, and/or medication.
Methods and systems disclosed herein may be implemented alone and/or
in combination with one or more other implant platforms, such as a stent,
including a coronary and a non-coronary or peripheral stent.
BRIEF DESCRIPTION OF THE DRAWINGS/FIGURES
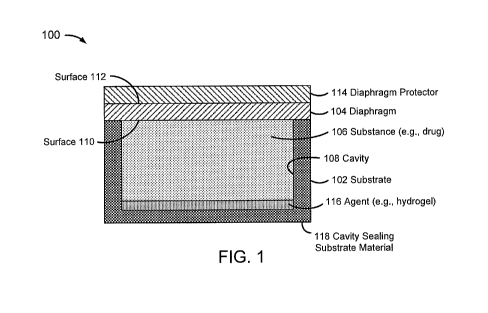
FIG. 1 is a block diagram of a pressure-actuated drug delivery system,
including a substrate and a pressure-rupturable diaphragm to releasably retain
a
drug within a cavity of the substrate.
FIG. 2 is a flowchart of a method of fabricating a pressure-actuated drug
delivery system.
FIG. 3 is a graphic illustration of a fabrication stage of the drug delivery
system of FIG. 1.
FIG. 4 is a graphic illustration of another fabrication stage of the drug
delivery system of FIG. 1.
FIG. 5 is a graphic illustration of another fabrication stage of the drug
delivery system of FIG. 1.
FIG. 6 is a graphic illustration of another fabrication stage of the drug
delivery system of FIG. 1.
-3 -
CA 02828954 2013-09-03
WO 2012/121691
PCT/US2011/027180
FIG. 7 is a flowchart of a method of fabricating a pressure-rupturable
diaphragm.
FIG. 8 is a graphic illustration of a progression of fabrication stages of a
pressure-rupturable diaphragm.
FIG. 9 is a flowchart of a method of fabricating a drug cavity.
FIG. 10 is a graphic illustration of a progression of fabrication stages of a
drug cavity.
FIG. 11 is a flowchart of a method of disposing a diaphragm protector
over a diaphragm, disposing a drug within a cavity, and sealing the cavity.
FIG. 12 is a graphic illustration of a progression of stages of disposing a
diaphragm protector over a diaphragm, disposing a drug within a cavity, and
sealing the cavity.
FIG. 13 is a block diagram of another drug delivery system, including
example dimensions.
FIG. 14 is a flowchart of a method of using an implantable pressure-
actuated substance delivery system.
FIG. 15 is a flowchart of a method of using an implantable pressure-
actuated drug delivery system within a vascular implant environment to treat a
potential blood clot.
FIG. 16 is a flowchart of a method of using an implantable pressure-
actuated substance delivery system that includes one or more of a hydrogel and
a
sensory agent.
In the drawings, the leftmost digit(s) of a reference number identifies the
drawing in which the reference number first appears.
DETAILED DESCRIPTION
FIG. 1 is a block diagram of a pressure-actuated drug delivery system
100, including a substrate 102 and a pressure-rupturable diaphragm 104 to
releasably retain a drug 106 within a cavity 106 of substrate 102.
- 4 -
CA 02828954 2013-09-03
WO 2012/121691
PCT/US2011/027180
Diaphragm 104 may be configured to rupture upon a pre-determined
pressure difference across first and second surfaces or sides 110 and 112,
referred
to herein as a rupture threshold. A pressure on surface 110 may be a pre-
determined pressure, which may be set during fabrication and/or prior to or at
the
time of implantation. A pressure on surface 112 may correspond to an implant
environment pressure, such as a vascular blood pressure.
The rupture threshold may be a function of a material and physical
characteristics of diaphragm 104. Physical characteristics may include
thickness,
and/or other physical features of diaphragm 104. For example, surface 112 may
have ridges, walls, posts, grooves, depressions, and/or other physical
features
formed therein, which may served to anchor and/or stiffen diaphragm 104.
Physical characteristics of diaphragm 104 may be configured to control the
rupture threhsold, and may be designed with assistance of a finite element
analysis (FEA).
The rupture threshold may further be a function of substrate features
within cavity 108, such as described in examples below.
Material and/or physical characteristics of diaphragm 104 and/or substrate
features within cavity 108 may also be configured to control a rupture site,
rupture pattern, and/or rupture time, such as described in examples below.
Diaphragm 104 may include one or more layers or films of one or more
materials to faun a frangible layer.
Substrate 102 and diaphragm 104 may be fabricated from one or more
materials that remain relatively stable within an implant environment for at
least a
pre-determined time, referred to herein as an operating life of system 102,
and
may include a material that will remain relatively stable within the implant
environment beyond the operating life. Stability may be measured in terms of
dissolution, bio-degradation, dissipation, and/or corrosion within the implant
environment.
For example, the American Heart Association recommends treating stent
patients with clopidogrel (a blood thinner) for one year following stent
implantation to provide the blood vessel sufficient time to heal. Several
pathology
studies suggest that, at least with respect to animals, a stented blood vessel
may
need up to two or more 2 years to heal.
- 5 -
CA 02828954 2013-09-03
WO 2012/121691
PCT/US2011/027180
Where system 100 is implemented within a stent, substrate 102 and
diaphragm 104 may thus be fabricated from one or more materials that will
remain relatively stable within a blood vessel for up to one year, up to two
years,
or longer. Methods and systems disclosed herein are not, however, limited to a
pre-determined period of time or an operating life.
Substrate 102 and diaphragm 104 may be fabricated, for example, from
one or more metals and/or non-metal materials. A metal may include, without
limitation, one or more of chromium, titanium, nickel, cobalt, gold, tungsten,
and/or combinations and/or alloys thereof A non-metal material may include,
without limitation, silcon dioxide and/or other a silicon compound, and/or a
polymer that remains stable over the operating life, which may include a
polylactic or polyglycolic acid.
Substrate 102 may include a silicon substrate, and diaphragm 104, or
portions thereof, may be spin-coated and/or deposited by electron-beam
evaporation onto a surface of substrate 102, such as described in one or more
examples below. Physical features may be imparted to diaphragm 104, such as
with photo-lithographic etching, such as described in one or more examples
below.
Cavity 108 may be pressurized and/or evacuated based on an implant
environment, and diaphragm 104 may be configured with a rupture threshold
based on a potential adverse pressure change within the implant environment.
Cavity 108 may be pressurized to a pressure substantially equal to a sum of a
desired rupture pressure and a rupture threshold of diaphragm 104. Conversely,
the rupture pressure may be defined as a difference between the cavity
pressure
and the rupture threshold.
Where system 100 is implanted within a blood vessel, for example,
normal physiological blood pressure maintains sufficient pressure against
diaphragm surface 112 to prevent rupture. In the event of an occlusive blood
clot,
counteracting physiological blood pressure at diaphragm surface 112 may fall
below the pre-determined rupture pressure, which may cause diaphragm 104 to
rupture and relase drug 106 and/or other substance into the blood vessel to
contact the clot. Drug 106 may include one or more drugs to mechanically
disrupt
and/or enzymatically degrade the clot, which may re-establish blood flow.
- 6 -
CA 02828954 2013-09-03
WO 2012/121691
PCT/US2011/027180
Example pressure values are provided below with respect to a blood
vessel implant environment and an example rupture threshold. Methods and
systems disclosed herein are not, however, limited to any of the pressure
values
or the implant environment of the examples below. In the examples below,
presure values are provided below in millimeters of mercury (mmHg), relative
to
one (1) atmosphere.
A normal systolic blood pressure may be within a range of approximately
100-140 mmHg.
A normal diastolic blood pressure may be within a range of approximately
70-90 mmHg.
A lowered blood pressure indicative of a distal clot may be within a range
of approximately 5-20 mmHg.
Cavity 108 may be pressurized based on a desired rupture pressure (e.g., a
lowered blood pressure), and a diaphragm rupture threshold.
The desired rupture pressure may be patient specific and/or implant
location specific, and may be determined in consultation with a medical
professional. A desired rupture pressure may be, for example, approximately 40
mmHg.
A rupture threshold of an example chromium based diaphragm may be
approximately 300 mmHg.
Cavity 108 may thus be pressurized to approximately at 340 mmHg to
cause diaphragm 104 to rupture when the blood pressure falls below 40 mmHg.
In other examples, cavity 108 may be pressurized to less than or greater
than 340 mmHg, including less than or equal to 300 mmHg, greater or equal to
1000 mmHg, and may be within a range of approximately 300 mmHg to 1000
mmHg, within a range of approximately 200 mmHg to 1200 mmHg, or may be
evacuated, depending upon the desired rupture pressure and the rupture
threshold.
In other examples, diaphragm 104 may be fabricated and/or subsequently
configured to provide a rupture threshold less than or greater than 300 mmHg,
including less than or equal to 200 mmHg, less than or equal to 1200 mmHg, and
may be within a range of approximately 200 mmHg to 1200 mmHg.
In other examples, a rupture pressure may be greater or less than 40
mmHg, including less than or equal to 20 mmHg, less than or equal to 50 mmHg,
and may be within a range of approximately 20 mmHg to 50 mmHg.
- 7 -
CA 02828954 2013-09-03
WO 2012/121691
PCT/US2011/027180
Diaphragm 104 may be fabricated and/or subsequently configured to
rupture substantially immediately upon exposure to a rupture pressure.
Alternatively, diaphragm 104 may be fabricated and/or subsequently configured
to maintain integrity when exposed to the rupture pressure for less than a pre-
determined period of time, and to rupture after exposure to the rupture
pressure
for at least a pre-determined period of time. Such a pre-detemiined period of
time
is referred to herein as a rupture time. Delayed rupture may be useful, for
example, to prevent rupture due to a spurious condition, such as a spurious
drop
in blood flow that may not be indicative of a blood clot. Diaphragm 104 may be
fabricated and/or subsequently configured to rupture upon exposure to a
rupture
pressure of, for example, within 5 seconds, after at least 5 seconds, within
15
second, after at least 15, within 60 seconds, or after at least 60 seconds or
more.
System 100 may include a dissolvable and/or bio-degradable diaphragm
protector 114 disposed over second surface 112 of diaphragm 104, which may be
configured to prevent premature rupture of diaphragm 104. Diaphragm protector
114 may be useful to protect diaphragm 104 from rupture prior to implantation,
such as where an ambient or pre-implant environment pressure is below the
rupture pressure.
Diaphragm protector 114 may include one or more water-soluble and/or
bio-degradable materials, such that diaphragm protector 114 substantially
dissolves, bio-degrades, or otherwise dissipates within an implant
environment.
Diaphragm protector 114 may include, for example, a water-soluble polymer, and
may include one or more relatively long chain polyethylene glycols of varying
molecular weights. Alternatively, or additionally, diaphragm protector 114 may
include a biodegradable polymer, such as a polylactic and/or polylactic-co-
glycolic acid.
The material of diaphragm protector 114 may be spun onto diaphragm
surface 112 to a thickness sufficient to protect diaphragm 104 from mechanical
and pressure-related damage during fabrication, storage, shipping, and/or
implantation. Diaphragm 104 may have a thickness of, for example, greater than
10 micrometers (um).
When system 100 is implanted, diaphragm protector 114 dissolves,
degrades, and/or otherwise dissipates to expose diaphragm surface 112 to the
implant environment.
- 8 -
CA 02828954 2013-09-03
WO 2012/121691
PCT/US2011/027180
Drug 106 within cavity 108 may include, without limitation, a
thrombolytic, plasminogen, plasmin, heparin, and/or other anti-thrombotic
agent,
antiplatelet agent, anti-inflammatory agent, immunomodulator, and/or other
enzyme or protein, DNA, RNA, mRNA and/or other genetic material, virus
particles, cells, small molecule, anti-lipid agent, and/or medication.
Drug 106 and/or cavity 108 may include one or more compounds or
agents 116 applied to a surface of cavity 108 and/or within an interior
portion of
cavity 108.
Agent 116 may include a swelling agent to expand upon contact with a
fluid, which may help to expel drug 106 from cavity 108 into the implant
environment. The swelling agent may expand to occupy all or substantially all
of
cavity 108. The swelling agent may include a hydrogel, such as a drug-eluting
hydrogel, which may include one or more of an anti-coagulant, such as heparin,
a
thrombolytic, and/or other drug.
Agent 116 and/or drug 106 may include an indicator to alert the patient
when membrane 104 releases drug 106. The indicator may include, for example,
allyl methyl sulfide, a compound that provides a garlic sensation.
Drug delivery system 100 may be implemented as a stand-alone
implantable platform, and/or may be affixed to, embedded with, and/or
integrated
within another implantable platform. Drug delivery system 100 may, for
example, be embedded within wells of a metal ring and/or a stent to be
implanted
within a vascular passage.
Drug delivery system 100 may be implemented, for example, as part of a
coronary and/or non-coronary stent. Non-coronary stents may be implemented to
treat non-coronary vessels, such as carotid, renal, subclavian, aorta,
femoral,
popliteal, iliac and other arteries. System 100 may be implemented, for
example,
to treat peripheral artery disease (PAD), which may affect femoral, popliteal,
iliac
and other arteries. Non-coronary stents are also referred to herein as
peripheral
stents and PAD stents. Peripheral stents may need more flexibility than
coronary
stents because lesions may be longer and may be located between muscle and
bone in the legs, which may place greater stress and torque on a stent. To
reduce
the chance of displacement, fracture, and/or crushing of stents, a peripheral
stent
- 9 -
CA 02828954 2013-09-03
WO 2012/121691
PCT/US2011/027180
may be implemented as a self-expanding stent, and may be manufactured from a
relatively flexible nickel-titanium alloy developed by the U.S. Naval
Ordinance
Lab, commercially known as Nitinol, an acronym for Nickel Titanium Naval
Ordinance Laboratory.
Where drug delivery system 100 is incorporated within a stent, the stent
may be implanted within an vessel, such as a coronary artery, using
conventional
percutaneous techniques. Once implanted, drug delivery system 100 may operate
without external sensory or control intervention.
Drug delivery system 100 may be implemented to treat heart attacks
caused by occlusive coronary clots, and may be retroactively placed in a
patient
who has already received a stent. Other example implementations are provided
below.
Drugs and/or endocrine conditions may lead to a critical rise in blood
pressure. Drug delivery system 100 may be implemented to release anti-
hypertensive drugs when blood pressure exceeds a pre-determined threshold.
Fluid overload in a patient suffering from heart failure may experience an
increase in central venous pressure (CVP), which may be treated with
diuretics.
Drug delivery system 100 may be implemented to release diuretics when the CVP
exceeds a pre-determined threshold.
Patients with end-stage renal disease and who are on dialysis may
experience clotting of an arteriovenous conduit (fistula or graft). Drug
delivery
system 100 may be incorporated within the arteriovenous conduit to release
thrombolytics to clear the clot from the conduit.
Anaphylaxis is a sudden allergic reaction that can cause a fatal drop in
blood pressure. Drug delivery system 100 may be implemented to relese
epinephrine, corticosteroids and/or anti-histamines to prevent and/or
ameliorate
anaphylactic shock.
An acute rise in intracranial pressure may cause fatal brain herniation
before emergency neurosurgical intervention can be performed. Drug delivery
system 100 may be implemented to repond to intracranial pressure by releasing
steroids and/or diuretics to provide additional time for neurosurgical
decompression.
- 10 -
CA 02828954 2013-09-03
WO 2012/121691
PCT/US2011/027180
System 100 may be implemented to release vasopressants when blood
pressure falls as a result of trauma, which may temporarily sustain blood
pressure
until medical assistance is provided.
System 100 may be implemented to repair a non-biological system, such
as to release a liquid sealant to seal a puncture in reponse to a pressure
change.
System 100 may include a layer or membrane laced with a high-resistivity
heating wire, such as nickel-chromium alloy wire. The coil may be inductively
heated with a magnetic resonance coil to melt the membrane and expose
diaphragm 104 and/or diaphragm protector 114. For example, with multiple
instances of System 100 on a stent each with membranes of slightly different
melting points, the stent will be able to function for multiple different
episodes of
stent clotting. Alternatively, a wire-laced membrane may be implemented as
diaphragm 104.
Multiple insances of system 100 may be fabricated on a shared substrate,
which may be cut into portions of one or more instances of system 100.
Subsets of instances of system 100 may be configured with different
rupture thresholds, which may permit delivery of drugs in response to
different
environmental pressures.
Subsets of instances of system 100 may also be provisioned with different
drugs and/or combinations of drugs, which may permit delivery of different
drugs
in response to different environmental pressures.
Subsets of instances of system 100 may include membranes having
disparate melting points, such as different formulations of ethylene vinyl
acetate
copolymer, and the membranes may be laced with the high-resistivity heating
wire described above to permit selective activation of the the instances of
system
100.
The shared substrate may be cut to provide an implantable substrate that
includes multiple instances of system 100, which may include multiple rupture
thresholds, multiple drugs and/or combinations of drugs, and/or membranes
having disparate melting temperatures.
- 11 -
CA 02828954 2013-09-03
WO 2012/121691
PCT/US2011/027180
An ultrasound transducer may be implanted physically proximate to
system 100 to emit signals at a resonnant wavelength of cavity 108 to rupture
diaphragm 104 with relatively minimal energy. The acousitic signals may have a
wavelength substantially equal to integer multiple of 4L, where L is depth of
cavity 106. The ultrasound transducer may be used as actuate drug delivery
system 100 independently of an implant environment pressure, and/or may be
used as a back-up actuator. The ultrasound transducer may be coupled to an ECG
sensor to actuate drug delivery system 100 when the ECG sensor detects ST
segment elevations.
FIG. 2 is a flowchart of a method 200 of fabricating a pressure-actuated
drug delivery system. For illustrative purposes, method 200 is described below
with reference to FIGS. 1 and 3 through 6. FIGS. 3-6 are graphic illustrations
of
stages of fabrication of drug delivery system 100. Method 200 is not however,
limited to any of the examples of FIGS. 1 and 3 through 6.
At 202, diaphragm 104 is disposed over a surface of substrate 102, as
illustrated FIG. 3, such as by spin-coating and/or electron-beam deposition or
evaporation.
At 204, diaphragm protector 114 is disposed over diaphragm 104, as
illustrated FIG. 4.
As described in examples below, diaphragm protector 114 may be
disposed over diaphragm 104 subsequent to formation of cavity 108 at 206.
At 206, cavity 108 is formed within substrate 102, to expose at least a
portion of diaphragm surface 110 to cavity 108, as illustrated FIG. 5.
At 208, drug 106 is disposed within cavity 108, and cavity 108
pressurized and hermetically sealed. In FIG. 6, cavity 108 is sealed with a
material 602, which may be substantially similar to a material of substrate
102.
Drug 106 may be disposed within cavity 108 by direct microinjection
using one or more conventional micro-injection techniques.
- 12 -
CA 02828954 2013-09-03
WO 2012/121691
PCT/US2011/027180
Alternatively, or additionally, drug 106 may drug may be disposed within
cavity 108 with a nebulization process. Referring to FIG. 5, the nebulization
process may include masking exposed portions 118 of substrate 102 with a
protective shadow mask. A drug solution may be supersaturated or concentrated
at a solubility limit, and aerosolized into relatively fine droplets using a
jet
nebulizer. As the droplets accumulate on the shadow mask and within cavity
108,
solvent within the droplets evaporates, leaving behind a film of drug
crystals. The
nebulizing process may continue until a desired drug mass is provided within
cavity 108. The shadow mask may then be removed.
One or more agents 116 (FIG. 1) may be applied to an inner surface of
material 602 prior to sealing cavity 108.
Cavity 108 may be pressurized prior to sealing cavity 108. For example,
pressurization of cavity 108 may be achived by hermetically sealing cavity 108
in
a pressurized atmosphere.
Cavity 108 may be pressurized subsequent to sealing of cavity 108. For
example, pressurization of cavity 108 can be achieved by hermetically sealing
cavity 108 under relatively low-temperature conditions. Pressurization then
ensues with expansion of gas within cavity 108 when warmed to ambient and/or
body temperature. A specific pressure and/or temperature at which cavity 108
is
hermetically sealed may be selected based on a desired rupture pressure and
rupture threshold.
Air transport through metal is relatively negligible and may be further
reduced and/or minimized by sealing cavity 108 within a high molecular weight
inert gas atmosphere, such as argon or xenon.
Cavity 108 may be sealed using one or more of a variety of types of
sealants, which may include a conventional silicon adhesive, such as an epoxy
and/or a crystal-bonding sealant, and/or a low-temperature ultrasound-assisted
bonding technique.
Method 200 may be implemented to form a plurality of cavities within a
shared substrate, and may include cutting or dicing the substrate into
multiple
substrates, each including one or more drug-containing cavities, such as
described
above.
- 13 -
CA 02828954 2013-09-03
WO 2012/121691
PCT/US2011/027180
Where method 200 is implemented to form a plurality of cavities,
different drugs and/or combinations of drugs may be disposed within different
cavities at 208, such as with one or more masking procedures. Similarly,
diaphragms having different rupture thresholds may be disposed over different
cavities. The substrate may be cut to provide a substrate having multiple
cavities,
which may include different drugs or combinations of drugs, and/or different
rupture thresholds.
FIG. 7 is a flowchart of a method 700 of fabricating a pressure-rupturable
diaphragm. Method 700 is described below with reference to example materials
for illustrative purposes. Method 700 is not, however, limited to the example
materials described below.
Method 700 is further described with reference to FIG. 8, which is a
graphic illustration of a progression of fabrication stages of a pressure-
rupturable
diaphragm. Method 700 is not, however, limited to the example of FIG. 8.
At 702, a polished silicon wafer 802 is thermally oxidized to create a
silicon dioxide layer 804.
At 704, an adhesive titanium layer 806 is deposited over silicon dioxide
layer 804. Titanium layer 806 may be deposited by electron-beam evaporation
under high vacuum, and may have a thickness within a range of approximately 5-
30 nanometers (nm).
At 706, an elemental chromium layer 808 is deposited onto titanium layer
806. Chromium layer 808 may be deposited by electron-beam evaporation under
high vacuum, and may have a thickness within a range of approximately 50-120
nm.
At 708, a negative photoresist 810 is deposited onto chromium layer 808.
Negative photoresist 810 may be spin deposited, and may have a thickness in
excess of 1000 nm.
At 710, portions of negative photoresist 810 are masked (see mask 812 in
FIG. 8).
At 712, one or more features and/or patterns of features are
photolithographically etched in negative photoresist 810, based on the masking
at
710. The features may include walls 814 extending from chromium layer 808
defining trenches 815 therebetween.
- 14 -
CA 02828954 2013-09-03
WO 2012/121691
PCT/US2011/027180
At 714, a second chromium layer 816 is deposited over chromium layer
808. Second chromium layer 816 may be deposited by electron-beam
evaporation, and may have a thickness within a range of approximately of 30-50
nm. Chromium layers 808 and 816, together, may have a minimum thickness of
approximately 80 nm.
At 716, remaining portions of photoresist 810, such as walls 814, are
removed from chromium layers 808 and 816, leaving a rupturable diaphragm
818, including titanium layer 806 and chromium layers 808 and 816, and having
a pattern of grooves 820 formed therein.
FIG. 9 is a flowchart of a method 900 of fabricating a drug cavity. Method
900 is described below with reference to example materials for illustrative
purposes. Method 900 is not, however, limited to the example materials
described
below.
Method 900 is further described with reference to FIGS. 8 and 10. FIG. 10
is a graphic illustration of a progression of fabrication stages of a drug
cavity.
Method 900 is not, however, limited to the examples of FIGS. 8 and 10.
At 902, a positive photoresist 1002 is spun onto a surface of substrate 802.
At 904, positive photoresist 1002 is masked with a mask 1004.
At 906, positive photoresist 1002 is eteched in accordance with mask
1004 to expose a portion 1006 of the surface of substrate 802, and mask 1004
is
removed.
At 908, a cavity 1008 is etched into substrate 802 through exposed
portion 1006 of substrate 802.
Up to this point, silicon dioxide layer 804 may structurally support
diaphragm 818.
At 910, exposed portions of silicon dioxide layer 804 are removed, such
as with a wet-etching process, which may utilize hydrofluoric acid to remove
release diaphragm 804 from silicon dioxide layer 804.
- 15 -
CA 02828954 2013-09-03
WO 2012/121691
PCT/US2011/027180
Referring back to 906, positive photoresist 1002 may be eteched to create
one or more physical features or shapes such as, for example, lines, circles,
ellipses, and/or one or more polygons. At 908, substrate 802 may be etched in
accordance with the one or more physical features or shapes of positive
photoresist 1002 until silicon dioxide layer 804 is reached, such as with an
STS-
DRIE silicon etching technique, to leave one or more "residual posts" of
substrate
material within cavity 1008. At 910, silicon dioxide layer 804 may be wet-
etched
to retain corresponding residual posts.
The residual posts may serve to anchor and/or stiffen diaphragm 818. A
rupture threshold, rupture site, rupture pattern, and/or rupture time of
diaphragm
818 may be defined, at least in part, by the residual posts.
In the example of FIG. 10, cavity 1008 is formed in substrate 802 through
a surface of substrate 802 that is opposite diaphragm 818. Alternatively,
cavity
1008 may be formed through another surface of substrate 802, such as a surface
adjacent to diaphragm 818.
FIG. 11 is a flowchart of a method 1100 of disposing a diaphragm
protector over a diaphragm, disposing a drug within a cavity, and sealing the
cavity. Method 1100 is described below with reference to example materials for
illustrative purposes. Method 1100 is not, however, limited to the example
materials described below.
Method 1100 is further described with reference to FIGS. 8, 10, and 12.
FIG. 12 is a graphic illustration of a progression of stages of disposing a
diaphragm protector over a diaphragm, disposing a drug within a cavity, and
sealing the cavity. Method 1100 is not, however, limited to the examples of
FIGS. 8, 10, and 12.
At 1102, a layer 1202 of a diaphragm protector material, such as
polyethylene glycol (PEG), is spun onto diaphragm 818. Layer 1202 may be spun
to a thickness of greater than 10 um.
At 1104, a shadow mask 1204 is disposed over exposed portions of
substrate 802.
At 1106, a drug 1210 is disposed within cavity 1008, such as by
nebulization. Shadow mask 1204 is then removed.
-16-
CA 02828954 2013-09-03
WO 2012/121691
PCT/US2011/027180
At 1108, cavity 1008 is hermetically sealed with a substrate material
1212, such as described in one or more examples above. Substrate material 1212
may include an agent 1214, applied and/or affixed to an inner surface of
substrate
material 1212. Agent 1214 may include a hydrogel, such as described in one or
more examples above.
Cavity 1008 may be pressurized and/or evacuated, such as described in
one or more examples herein.
At 1110, substrate 802, including diaphragm 818, diaphragm protector
layer 1202, drug 1210, substrate material 1212, and agent 1214, is mounted
within a well of an implantable platform 1216, which may include a stent.
Where substrate 802 includes a plurality of drug-containing cavities 1008,
the substrate may be cut into a plurality of substrates, each of which may
include
one or more cavities 1008, and which may mounted within a well of implantable
platform 1216.
At 1112, platform 1216 is implanted and diaphragm protector layer 1202
dissolves, degrades, and/or otherwise dissipates within the implant
environment.
FIG. 13 is a block diagram of a drug delivery system 1300 to illustrate an
example pattern formed within a surface of a rupturable diaphragm 1302. For
illustrative purposes, FIG. 13 includes example dimensions. Methods and
systems
disclosed herein are not, however, limited to example dimensions disclosed
herein.
FIG. 14 is a flowchart of a method 1400 of using an implantable pressure-
actuated substance delivery system.
At 1402, a substance delivery system is received within an implant
environment. The substance delivery system may include:
a substrate;
a substance within a pressurized cavity of the
substrate;
a frangible layer of one or more materials disposed
over a surface of the substrate, including over an
opening to the cavity; and
a substantially non-frangible layer of an implant-
environment-dissipative material disposed over the
frangible layer.
-17-
CA 02828954 2013-09-03
WO 2012/121691
PCT/US2011/027180
At 1404, an implant environment fluid is contacted to the substantially
non-frangible layer within the implant environment.
At 1406, the substantially non-frangible layer is dissipated within the
implant environment in response to the contacting of the implant environment
fluid.
At 1408, the frangible layer of material is exposed to an implant
environment pressure in response to the dissipating of the substantially non-
frangible layer.
At 1410, when a difference between the implant environment pressure
and the cavity pressure does not exceeds a rupture threshold of the frangible
layer, integrity of the frangible layer is maintained at 1408. When the
rupture
threshold is exeeded, processing proceeds to 1412.
At 1412, the frangible layer is ruptured.
At 1414, the substance is released from the cavity into the implant
environment through the substrate opening.
The substance delivery system may include a drug, such as described in
one or more examples herein, which may include an anti-clotting agent. A
method of using a drug delivery system is described below with reference to
FIG.
15.
The substance delivery system may include one or more a sensory agent
to invoke or elicit a sensory perception in patient, and a hydrogel, such as
described in one or more examples herein. A method of using such a substance
delivery system is described further below with reference to FIG. 16.
FIG. 15 is a flowchart of a method 1500 of using an implantable pressure-
actuated drug delivery system within a vascular implant environment to treat a
potential blood clot.
At 1502, a drug delivery system is received within a vascular implant
environment.
The vascular implant environment may include one or more of a cornary
vascular implant environment and a non-coronary or peripherial vascular
implant
environment.
The drug delivery system may be received as an implantable platform,
which may include a stent and/or a stand-lone drug delivery platform.
The drug delivery system may include include:
- 18 -
CA 02828954 2013-09-03
WO 2012/121691
PCT/US2011/027180
a substrate;
a thrombolytic drug or agent within a pressurized
cavity of the substrate;
a frangible layer of one or more materials disposed
over a surface of the substrate, including over an
opening to the cavity; and
a substantially non-frangible layer of an implant-
environment-dissipative material disposed over the
frangible layer.
At 1504, a vascular implant environment fluid is contacted to the
substantially non-frangible layer within the vascular implant environment.
At 1506, the substantially non-frangible layer is dissipated within the
vascular implant environment in response to the contacting of the vascular
implant environment fluid.
At 1508, the frangible layer of material is exposed to an vascular implant
environment pressure (e.g., vascular blood pressure), in response to the
dissipating of the substantially non-frangible layer.
At 1510, when a difference between the vascular implant environment
pressure and the cavity pressure does not exceeds a rupture threshold of the
frangible layer, integrity of the frangible layer is maintained at 1408. When
the
rupture threshold is exeeded, processing proceeds to 1512.
At 1512, the frangible layer is ruptured.
At 1514, the thrombolytic agent is released from the cavity into the
vascular implant environment through the substrate opening.
FIG. 16 is a flowchart of a method 1600 of using an implantable pressure-
actuated substance delivery system that includes one or more of a hydrogel and
a
sensory agent.
Method 1600 includes features 1402 through 1414 of method 1400, and
may include one or more of features 1602 through 1606.
At 1602, an implant environment fluid is contact to a hydrogel within the
substrate cavity. The implant environment fluids of 1404 and 1604 may be the
same fluid or different fluids within the implant environment.
At 1604, the implant environment fluid of 1604 is absorbed by the
hydrogel to expand the hydrogel within the cavity.
- 19 -
CA 02828954 2013-09-03
WO 2012/121691
PCT/US2011/027180
At 1606, a sensory agent is released from the substrate cavity into the
implant environment to invoke or elicit a sensory perception in the patient,
such
as described in one or more examples herein.
Methods and systems are disclosed herein with the aid of functional
building blocks illustrating the functions, features, and relationships
thereof. At
least some of the boundaries of these functional building blocks have been
arbitrarily defined herein for the convenience of the description. Alternate
boundaries may be defined so long as the specified functions and relationships
thereof are appropriately performed.
While various embodiments are disclosed herein, it should be understood
that they have been presented by way of example only, and not limitation. It
will
be apparent to persons skilled in the relevant art that various changes in
form and
detail may be made therein without departing from the spirit and scope of the
methods and systems disclosed herein. Thus, the breadth and scope of the
claims
should not be limited by any of the example embodiments disclosed herein.
-20-