Note: Descriptions are shown in the official language in which they were submitted.
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
ANTI-CD98 ANTIBODIES AND METHODS OF USE THEREOF
FIELD OF THE INVENTION
[0001] The present invention relates generally to anti-CD98 antibodies and
to methods of
using such antibodies.
BACKGROUND
[0002] CD98 (also referred to as CD98 heavy chain; 42F heavy
chain; SLC3A2) is a type II
I 0 transmembrane glycoprotein composed of 529 amino acid residues. The
protein comprises a 75 amino
acid N-terminal intracellular cytoplasmic domain, a single transmembrane
domain, and a 426 amino acid
C-terminal extracellular domain (Parmacek et al., Nucleic Acids Res. 17: 1915-
1931, 1989). CD98
covalently links via a disulfide bond to one of several light chains (SLC7A5,
6, 7, 8, 10, or 11), which are
L-type amino acid transporters. This interaction is required for the cell
surface expression and amino
acid transport function of the light chains. CD98 also associates with
integrin 13 subunits, thereby
regulating integrin signaling that controls cell proliferation, survival,
migration, and epithelial
adhesion/polarity (Cai et al., J. Cell Sci. 118: 889-899, 2005).
[0003] CD98 was originally identified as a cell surface antigen
associated with lymphocyte
activation (Haynes et al., J. Immunol. 126: 1409-1414, 1981). CD98 has since
been identified in all cell
types with the exception of platelets and is expressed at the highest levels
in the gastrointestinal (GI) tract
and the tubules of the kidney (Verrey et al., Pflugers Arch. 440: 503-512,
2000). Upregulation of CD98
has been observed in intestinal inflammation. Recently, intestinal CD98
expression was shown to have a
crucial role in controlling homeostatic and innate immune responses in the
gut. Modulation of CD98
expression in intestinal epethilial cells has therefore been suggested as a
promising therapeutic strategy
for the treatment and prevention of inflammatory intestinal diseases, such as
inflammatory bowel disease
(IBD) and colitis-associated cancer (Nguyen et al., J. Clin. Invest. 121: 1733-
1747, 2011). CD98 is also
overexpressed on the cell surface of almost all tumor cells, regardless of
tissue of origin (Roll et al., Jpn.
J. Cancer Res. 92: 1313-1321, 2001).
100041 Increased expression of one of the light chains that binds
CD98, L-type amino acid
transporter 1 (LAT1; also known as SLC7A5) has also been observed in many
types of human cancer
cells, including breast cancer, colon cancer, oral cancer, ovarian cancer,
esophageal cancer, glioma and
leukemia (Fan et al., Biochem. Pharmacol. 80: 811-818, 2010). Increased amino
acid supply may be
required to support the high growth rate of cancer cells, both by providing
the amino acid building blocks
for protein synthesis, and by stimulating growth via mammalian target of
rapamycin (mTOR) (Fan et al.,
supra; Imai et al., Anticancer Res. 30: 4819-4828, 2010). The expression of
LAT1 and CD98 is
significantly higher in metastatic sites of human cancers than in the primary
sites, suggesting that
overexpression of LAT1/CD98 may be essential for progression and metatstasis
of human cancers. In
1
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
particular, LAT1/CD98 overexpression appears to be required for tumor
metastasis in patients with colon
cancer. (Kaira et al., Cancer Sci. 99: 2380-2386, 2008).
100051 The expression pattern and functions of CD98 and LAT1
suggest these proteins as
promising targets for treatment of a variety of human cancers. Inhibitors of
LAT1 activity have
demonstrated antitumor activity in a number of cancer types, including non-
small cell lung cancers (Imai
et al., supra), colon cancer cells (Oda et al., Cancer Sci. 101: 173-179,
2010), oral cancer cells (Kim et
al., Biol. Pharm. Bull. 33: 1117-1121, 2010), and breast cancer cells (Shennan
and Thomson, Oncol. Rep.
20: 885-889, 2008). LATI has also been suggested as a target for treatment of
ovarian cancer (Fan et al,
supra).
[0006] A murine monoclonal antibody to CD98, identified as HBJ127, was
found to inhibit
lymphocyte proliferation (Yagita and Hashimoto, J. Immunol. 136: 2062-2068,
1986) and to inhibit the
growth of bladder tumor and lymphoma cells (Yagita et al., Cancer Res. 46:
1478-1489, 1986). The
epitope for the HJ127 antibody was found to be residues 442AFS444 of human
CD98 (Itoh et al., 2007). A
different murine monoclonal antibody to CD98 was shown to significantly
inhibit tumor cell growth in
vitro for glioma, prostate and colon cancer cells (Papetti and Herman, Am. J.
Pathol. 159: 165-178,
2001). Additional monoclonal antibodies to human CD98 have been disclosed in
U.S. Publication No.
20100143367. These monoclonal antibodies bind to epitopes within amino acid
regions 372-530 or 104-
371 of CD98. Five of these antibodies were found to inhibit amino acid uptake
in a bladder cancer cell,
and three of these antibodies were shown to suppress tumor growth in a mouse
model.
[0007] As disclosed herein, analysis of fresh primary acute myelogenous
leukemia (AML)
tumor samples from patients using surface tagged antigen profiling (sTAg) of
the cell surface proteome
identified the transmembrane protein CD98 as being present at high density on
the surface of AML tumor
cells. CD98 is therefore a target for the treatment of AML, for example, by
using binding agents such as
antibodies which specifically bind to CD98. Binding agents specific for CD98,
such as anti-CD98
antibodies, were also shown in various in vivo xengraft models to have utility
in treating not only AML
but various cancers, such as sarcoma, lymphoma, non-small cell lung cancer
(NSCLC) and colorectal
cancer.
[0008] The invention provides antibodies to CD98 that are useful
in the diagnosis and
treatment of various types of human cancers.
SUMMARY
[0009] Using in-solution labeling of intact AML tumor cell
surfaces, followed by high-
resolution, solution-based liquid chromatography coupled tandem mass
spectrometry (LC-MS/MS),
CD98 was identified as being present at high density on the surface of a
majority of AML cell subtypes
as compared to normal cells including developing blood cells. Thus, the
invention provides anti-CD98
antibodies and methods of using the such antibodies in the treatment of AML
and other cancers,
including but not limited to lymphoma, sarcoma, non-small cell lung cancer and
colorectal cancer.
2
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
[0010] In an embodiment, the invention provides an isolated
antibody or a functional
fragment thereof that specifically binds to human CD98, wherein the antibody
or functional fragment
binds to an epitope comprising residues A377, D397,1398, G400 and A401 of
human CD98. In some
embodiments, the epitope further comprises residues D374 and L378 of human
CD98. In some
embodiments, the epitope further comprises residues P379 and G380 of human
CD98. In some
embodiments, the epitope further comprises residues F395 and P396 of human
CD98. In some
embodiments, the epitope further comprises residues Q381, P382 and P399 of
human CD98. In some
embodiments, the epitope further comprises any one or more additional residues
selected from the group
consisting of D374, L378, P379, G380, Q381, P382, F395, P396 and P399 of human
CD98.
[0011] In an embodiment, the invention provides an isolated antibody or a
functional
fragment thereof that specifically binds to human CD98, wherein the antibody
binds to an epitope
comprising residues P379, G380, D397 and 1398 of human CD98. In some
embodiments, the epitope
further comprises residues F395 and P396 of human CD98. In some embodiments,
the epitope further
comprises residues Q381, P382, P399, G400 and A401 of human CD98. In some
embodiments, the
epitope further comprises residues D374, A377 and L378 of human CD98. In some
embodiments, the
epitope further comprises any one or more additional residues selected from
the group consisting of
D374, A377, L378, Q381, P382, F395, P396, P399, G400 and A401 of human CD98.
[0012] In some embodiments, the invention provides an isolated
antibody or a functional
fragment thereof, wherein the antibody or functional fragment binds to an
epitope comprising residues
D374, A377, L378, P379, G380, Q381, P382, F395, P396, D397, 1398, P399, G400
and A401 of human
CD98.
[0013] In some embodiments, the invention provides an isolated
antibody or a functional
fragment thereof that specifically binds to human CD98, wherein the antibody
or functional fragment
binds to an epitope comprised within amino acid residues 369-405 of human
CD98. In some
embodiments, the invention provides an isolated antibody or a functional
fragment thereof that
specifically binds to human CD98, wherein the antibody or functional fragment
binds to an epitope
consisting of amino acid residues 369-405 of human CD98.
[0014] In some embodiments, the monoclonal antibody of the
invention is a humanized,
human or chimeric antibody. In some embodiments, the antibody functional
fragment of the invention is
an Fab, F(ab')2, Fv or scFv fragment.
[0015] n an embodiment, the invention provides an isolated
antibody or a functional
fragment thereof comprising all three heavy chain complementarity determining
regions (CDRs) from a
heavy chain variable domain having an amino acid sequence selected from the
group consisting of SEQ
ID NO: 4, SEQ ID NO: 8, SEQ ID NO: 12, SEQ ID NO: 31, and SEQ ID NO: 35,
and/or all three light
chain CDRs from a light chain variable domain having an amino acid sequence
selected from the group
consisting of SEQ ID NO: 6, SEQ ID NO: 10, SEQ ID NO: 14, SEQ ID NO: 33, and
SEQ ID NO: 37.
[0016] In an embodiment, the invention provides an isolated antibody or a
functional fragment
thereof comprising all three heavy chain CDRs from a heavy chain variable
domain having an amino acid
3
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
sequence selected from the group consisting of SEQ ID NO: 4, SEQ ID NO: 8, SEQ
ID NO: 12, SEQ ID
NO: 31, and SEQ ID NO: 35, and all three light chain CDRs from a light chain
variable domain having
an amino acid sequence selected from the group consisting of SEQ ID NO: 6, SEQ
ID NO: 10, SEQ ID
NO: 14, SEQ ID NO: 33, and SEQ ID NO: 37. In some embodiments, the antibody or
functional
fragment thereof comprises all heavy and light chain complementarity
determining regions (CDRs) from:
(a) the antibody designated 8-34B; (b) the antibody designated 18-2A 2.2; (c)
the antibody designated 18-
2A 7.1; (d) the antibody designated 1-47C; or (e) the antibody designated 1-
115A. In some embodiments,
the antibody or functional fragment thereof comprises all heavy and light
chain CDRs from the antibody
designated 8-34B. In some embodiments, the antibody or functional fragment
thereof comprises all all
heavy and light chain CDRs from the antibody designated 18-2A 2.2. In some
embodiments, the antibody
or functional fragment thereof comprises all all heavy and light chain CDRs
from the antibody designated
I 8-2A 7.1. In some embodiments, the antibody or functional fragment thereof
comprises all all heavy and
light chain CDRs from the antibody designated 1-47C. In some embodiments, the
antibody or functional
fragment thereof comprises all all heavy and light chain CDRs from the
antibody designated 1-115A.
I 5 [0017] In some embodiments, the antibody comprises a heavy chain
variable domain
sequence selected from the group consisting of SEQ ID NO: 4, SEQ ID NO: 8, SEQ
ID NO: 12, SEQ ID
NO: 31, and SEQ ID NO: 35. In some embodiments, the antibody comprises a light
chain variable
domain sequence consisting of SEQ ID NO: 6, SEQ ID NO: 10, SEQ ID NO: 14, SEQ
ID NO: 33, and
SEQ ID NO: 37. In some embodiments, the antibody comprises a heavy chain
variable domain sequence
selected from the group consisting of SEQ ID NO: 4, SEQ ID NO: 8, SEQ ID NO:
12, SEQ ID NO: 31,
and SEQ ID NO: 35, and further comprises a light chain variable domain
sequence consisting of SEQ ID
NO: 6, SEQ ID NO: 10, SEQ ID NO: 14, SEQ ID NO: 33, and SEQ ID NO: 37.
[0018] In an embodiment, the antibody comprises the heavy chain
variable domain
sequence of SEQ ID NO: 4 and the light chain variable domain sequence of SEQ
ID NO: 6. In an
embodiment, the antibody comprises the heavy chain variable domain sequence of
SEQ ID NO: 8 and the
light chain variable domain sequence of SEQ ID NO: 10. In an embodiment, the
antibody comprises the
heavy chain variable domain sequence of SEQ ID NO: 12 and the light chain
variable domain sequence
of SEQ ID NO: 14. In an embodiment, the antibody comprises the heavy chain
variable domain sequence
of SEQ ID NO: 31 and the light chain variable domain sequence of SEQ ID NO:
33. In an embodiment,
the antibody comprises the heavy chain variable domain sequence of SEQ ID NO:
35 and the light chain
variable domain sequence of SEQ ID NO: 37.
[0019] In an embodiment, the invention provides humanized
antibodies. In some
embodiments, the humanized antibody comprises a heavy chain variable domain
sequence selected from
SEQ ID NO: 17, SEQ ID NO: 18, SEQ ID NO: 19, SEQ ID NO: 22, and SEQ ID NO: 23.
In some
embodiments, the humanized antibody comprises a light chain variable domain
sequence selected from
SEQ ID NO: 15, SEQ ID NO: 16, SEQ ID NO: 20, and SEQ ID NO: 21. In some
embodiments, the
humanized antibody comprises a heavy chain variable domain sequence selected
from SEQ ID NO: 17,
SEQ ID NO: 18, SEQ ID NO: 19, SEQ ID NO: 22, and SEQ ID NO: 23, and further
comprises a light
4
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
chain variable domain sequence selected from SEQ ID NO: 15, SEQ ID NO: 16, SEQ
ID NO: 20, and
SEQ ID NO: 21. In some embodiments, the humanized antibody comprises a light
chain variable domain
sequence selected from SEQ ID NO: 15 and SEQ ID NO: 16, and a heavy chain
variable domain
sequence selected from SEQ ID NO: 17, SEQ ID NO: 18 and SEQ ID NO: 19. In an
embodiment, the
humanized antibody comprises the light chain variable domain sequence of SEQ
ID NO: 15 and the
heavy chain variable domain sequence of SEQ ID NO: 18.
[0020] In an embodiment, the humanized antibody comprises the
light chain variable
domain sequence of SEQ ID NO: 20 and the heavy chain variable domain sequence
of SEQ ID NO: 22.
In an embodiment, the humanized antibody comprises the light chain variable
domain sequence of SEQ
ID NO: 21 and the heavy chain variable domain sequence of SEQ ID NO: 23. In an
embodiment, the
humanized antibody comprises the light chain variable domain sequence of SEQ
ID NO: 20 and the
heavy chain variable domain sequence of SEQ ID NO: 23. In an embodiment, the
humanized antibody
comprises the light chain variable domain sequence of SEQ ID NO: 21 and the
heavy chain variable
domain sequence of SEQ ID NO: 22. In an embodiment, the invention provides an
antibody that bind to
the same epitope as a humanized antibody comprising the light chain variable
domain sequence of SEQ
ID NO: 21 and the heavy chain variable domain sequence of SEQ ID NO: 22. In an
alternative
embodiment, the invention comprises a binding agent that binds to essentially
the same epitope as an
antibody from bin 1 or bins 3-7 as shown in Fig. 1.
100211 In a further embodiment, the invention comprises a binding
agent that binds to
essentially the same epitope as any of the antibodies disclosed above. In some
embodiments, the binding
agent inhibits the growth of a tumor expressing CD98. In some embodiments, the
binding agent is an
antibody or a functional fragment thereof. In other embodiments, the binding
agent is an anticalin, an
adnectin, an affibody, a DARPin, a fynomer, an affitin, an affilin, an avimer,
a cysteine-rich knottin
peptide, or an engineered Kunitz-type inhibitor.
[0022] In one embodiment, the invention provides a binding agent capable of
binding to
CD98, wherein any one of the antibodies disclosed above displaces the binding
agent in a competitive
binding assay. In some embodiments, the binding agent is an antibody, or a
functional fragment thereof.
In another embodiment, the invention provides a binding agent capable of
binding to CD98, wherein the
binding agent displaces any one of the antibodies disclosed above in a
competitive binding assay. In
some embodiments, the binding agent is an antibody, or a functional fragment
thereof.
[0023] In some embodiments, the invention provides an antibody
that binds to CD98,
wherein the antibody comprises a heavy chain variable domain having at least
90%, at least 91%, at least
92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at
least 98%, or at least 99%
sequence identity to an amino acid sequence selected from SEQ ID NO: 4, SEQ ID
NO: 8, SEQ ID NO:
12, SEQ ID NO: 17, SEQ ID NO: 18; SEQ ID NO: 19, SEQ ID NO: 22, SEQ ID NO: 23,
SEQ ID NO:
31, and SEQ ID NO; 35. In some embodiments, the antibody comprises a light
chain variable domain
having at least 90%, at least 91%, at least 92%, at least 93%, at least 94%,
at least 95%, at least 96%, at
least 97%, at least 98%, or at least 99% sequence identity to an amino acid
sequence selected from the
5
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
group consisting of SEQ ID NO: 6, SEQ ID NO: 10, SEQ ID NO: 14, SEQ ID NO: 15,
SEQ ID NO: 16,
SEQ ID NO: 20, SEQ ID NO: 21, SEQ ID NO: 33, and SEQ ID NO: 37. In some
embodiment, the
antibody comprises a heavy chain variable domain having at least 90%, at least
91%, at least 92%, at
least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least
98%, or at least 99% sequence
identity to an amino acid sequence selected from SEQ ID NO: 4, SEQ ID NO: 8,
SEQ ID NO: 12, SEQ
ID NO: 17, SEQ ID NO: 18; SEQ ID NO: 19, SEQ ID NO: 22, SEQ ID NO: 23, SEQ ID
NO: 31, and
SEQ ID NO: 35, and the antibody further comprises a light chain variable
domain having at least 90%, at
least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least
96%, at least 97%, at least 98%,
or at least 99% sequence identity to an amino acid sequence selected from the
group consisting of SEQ
ID NO: 6, SEQ ID NO: 10, SEQ ID NO: 14, SEQ ID NO: 15, SEQ ID NO: 16, SEQ ID
NO: 20, SEQ ID
NO: 21, SEQ ID NO: 33, and SEQ ID NO: 37.
[0024] In some embodiments, the invention provides an antibody
that is a variant of any of
the above antibodies having one or more amino acid substitutions, deletions,
insertions or modifications,
and which retains a biological function of the antibody. In some embodiments,
the invention provides an
antibody that binds to CD98 expressed on the cell surface and inhibits the
growth of the cell. In some
embodiments, the anti-CD98 antibody binds to CD98 expressed on the cell
surface and inhibits cell
proliferation. In some embodiments, the anti-CD98 antibody binds to CD98
expressed on the cell
surface and induces cell death. In some embodiments, the invention provides an
antibody that is a variant
of any one of the above antibodies having improvements in one or more of a
property such as binding
affinity, specificity, thermostability, expression level, effector function,
glycosylation, reduced
immunogenicity, or solubility as compared to the unmodified antibody.
[0025] In some embodiments, the invention provides any one of the
above antibodies or
functional fragments, wherein theantibody or fragment is conjugated to a
cytotoxic agent. In various
embodiments, the cytotoxic agent is selected from a chemotherapeutic agent, a
drug, a growth inhibitory
agent, a toxin, or a radioactive isotope. In some embodiments, the invention
provides any one of the
above antibodies or functional fragments, wherein theantibody or fragment is
conjugated to a detectable
marker. In various embodiments, the detectable marker is selected from a
radioisotope, a metal chelator,
an enzyme, a fluorescent compound, a bioluminescent compound and a
chemiluminescent compound.
[0026] In an embodiment, the invention provides a hybridoma that
produces a monoclonal
antibody of the invention. In an embodiment, the invention provides a
transgenic animal that produces a
monoclonal antibody of the invention.
[0027] In some embodiments, a polynucleotide encoding any of the
above antibodies is
provided. In an embodiment, a vector comprising the polynucleotide is
provided. In an embodiment, a
host cell comprising the vector is provided. In an embodiment, the host cell
is prokaryotic. In an
embodiment, the host cell is an E. coli cell. In another embodiment, the host
cell is eukaryotic. In an
embodiment, the host cell is a Chinese Hamster Ovary (CHO) cell. In an
embodiment, a method of
making an anti-CD98 antibody is provided, wherein the method comprises
culturing the host cell under
6
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
conditions suitable for expression of the polynucleotide encoding the
antibody, and isolating the
antibody.
[0028] In one embodiment, the invention provides a pharmaceutical
composition
comprising any of the above antibodies or functional fragments thereof,
antibody conjugates, or binding
agents of the invention. In a further embodiment, the invention provides a
method of inhibiting growth of
cancer cells that express CD98, the method comprising exposing the cells to
any one or more of the
above antibodies or functional fragments thereof, antibody conjugates, or
binding agents of the invention.
In various embodiments, the cancer cells are from a cancer selected from
bladder, breast, colon, rectal,
gastric, esophageal, lung, laryx, kidney, oral, ovarian, or prostate cancer,
or a sarcoma, melanoma,
glioma, lymphoma or leukemia, or a metatasis of any of these cancers.
[0029] In an embodiment, the invention provides a method for
treating a cancer in a subject
comprisingadministering to the subject a pharmaceutical composition comprising
any of the above
antibodies or functional fragments thereof, antibody conjugates, or binding
agents of the invention. In
various embodiments, the cancer is selected from bladder, breast, colon,
rectal, gastric, esophageal, lung,
laryx, kidney, oral, ovarian, or prostate cancer, or a sarcoma, melanoma,
glioma, lymphoma or leukemia,
or a metatasis of any of these cancers. In some embodiments, the cancer is
acute myeloid leukemia. In
some embodiments, the subject has relapsed or refractory acute myeloid
leukemia. In some
embodiments, the cancer is associated with increased expression of CD98 on the
surface of a cell.
[0030] In some embodiments, the subject is administered one or
more chemotherapeutic
compound in combination with the antibody or functional fragment, wherein the
chemotherapeutic
compound is selected from bendamustine hydrochloride, cyclophosphamide,
ifosfamide, fludurabine,
cytarabine, gemcitabine, prednisone, prednisolone, methylprednisolone,
paclitaxel, docetaxel,
vinorelbine, vincristine, etoposide, irinotecan, anthracycline, adriamycin,
cisplatin, carboplatin and
rituximab.
[0031] In an embodiment, a method of detecting the presence of CD98 in a
biological
sample is provided, the method comprising contacting the biological sample
with any of the above
antibodies under conditions permissive for binding of the antibody to CD98,
and detecting whether a
complex is formed between the antibody and CD98. In some embodiments, the
biological sample is
from a mammal having or suspected of having a cancer of cells or tissues
including, but not limited to,
bladder, breast, colon, rectal, gastric, esophageal, lung, laryx, kidney,
oral, ovarian, or prostate cancer, or
a sarcoma, melanoma, glioma, lymphoma or leukemia, or a metatasis of any of
these cancers.
[0032] In an embodiment, a method of diagnosing a cancer
associated with increased
expression of CD98 is provided, the method comprising contacting a test cell
with any of the above
antibodies; determining the level of expression of CD98 by detecting binding
of the antibody to CD98;
and comparing the level of expression of CD98 by the test cell with the level
of expression of CD98 by a
control cell, wherein a higher level of expression of CD98 by the test cell as
compared to the control cell
indicates the presence of a cancer associated with increased expression of
CD98. In some embodiments,
the test cell is a cell from a patient suspected of having a cancer selected
from bladder, breast, colon,
7
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
rectal, esophageal, lung, laryx, kidney, oral, ovarian, or prostate cancer, or
a sarcoma, glioma, lymphoma
or leukemiaõ or a metatasis of any of these cancers. In an embodiment, the
method comprises
determining the level of expression of CD98 on the surface of the test cell
and comparing the level of
expression of CD98 on the surface of the test cell with the level of
expression of CD98 on the surface of
the control cell. In some embodiments, the test cell is a cancer cell and the
control cell is a normal cell of
the same tissue type.
[0033] In an embodiment, the invention provides a use of any of
the above antibodies or
functional fragments in the in the manufacture of a medicament, wherein the
medicament is for use in a
method of inhibiting growth of cancer cells that express CD98. In various
embodiments, the cells are
from a cancer is selected from bladder, breast, colon, rectal, gastric,
esophageal, lung, laryx, kidney, oral,
ovarian, or prostate cancer, or a sarcoma, melanoma, glioma, lymphoma or
leukemia, or a metatasis of
any of these cancers.
[0034] In an embodiment, the invention provides any of the above
antibodies or functional
fragments for use in inhibiting the growth of cancer cells that express CD98.
In various embodiments, the
cells are from a cancer is selected from bladder, breast, colon, rectal,
gastric, esophageal, lung, laryx,
kidney, oral, ovarian, or prostate cancer, or a sarcoma, melanoma, glioma,
lymphoma or leukemia, or a
metatasis of any of these cancers.
[0035] In an embodiment, the invention provides a use of a
pharmaceutical composition
comprising any of the above antibodies or functional fragments in the
manufacture of a medicament,
wherein the medicament is for use in a method of treating cancer in a subject.
In various embodiments,
the cancer is selected from bladder, breast, colon, rectal, gastric,
esophageal, lung, laryx, kidney, oral,
ovarian, or prostate cancer, or a sarcoma, melanoma, glioma, lymphoma or
leukemia, or a metatasis of
any of these cancers. In some embodiments, the cancer is acute myeloid
leukemia. In some embodiments,
the subject has relapsed or refractory acute myeloid leukemia. In some
embodiments, the cancer is
associated with increased expression of CD98 on the surface of a cell. In some
embodiments, the subject
is administered one or more chemotherapeutic compound in combination with the
antibody or functional
fragment, wherein the chemotherapeutic compound is selected from bendamustine
hydrochloride,
cyclophosphamide, ifosfamide, fludurabine, cytarabine, gemcitabine,
prednisone, prednisolone,
methylprednisolone, paclitaxel, docetaxel, vinorelbine, vincristine,
etoposide, irinotecan, anthracycline,
adriamycin, cisplatin, carboplatin and rituximab.
[0036] In an embodiment, the invention provides a pharmaceutical
composition comprising
any of the above antibodies or functional fragments and a pharmaceutically
acceptable carrier, for use in
treating cancer in a subject. In various embodiments, the cancer is selected
from bladder, breast, colon,
rectal, gastric, esophageal, lung, laryx, kidney, oral, ovarian, or prostate
cancer, or a sarcoma, melanoma,
glioma, lymphoma or leukemia, or a metatasis of any of these cancers. In some
embodiments, the cancer
is acute myeloid leukemia. In some embodiments, the subject has relapsed or
refractory acute myeloid
leukemia. In some embodiments, the cancer is associated with increased
expression of CD98 on the
surface of a cell. In some embodiments, the subject is administered one or
more chemotherapeutic
8
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
compound in combination with the antibody or functional fragment, wherein the
chemotherapeutic
compound is selected from bendamustine hydrochloride, cyclophosphamide,
ifosfamide, fludurabine,
cytarabine, gemcitabine, prednisone, prednisolone, methylprednisolone,
paclitaxel, docetaxel,
vinorelbine, vincristine, etoposide, irinotecan, anthracycline, adriamycin,
cisplatin, carboplatin and
rituximab.
[0037] In an embodiment, the invention provides the use of any of
the above antibodies or
functional fragments in the manufacture of a medicament, wherein the
medicament is for use in a method
for detecting the presence of of CD98 in a biological sample. In some
embodiments, the method
comprisescontacting the biological sample with any of the above antibodies
under conditions permissive
for binding of the antibody to CD98, and detecting whether a complex is formed
between the antibody
and CD98. In some embodiments, the biological sample is from a mammal having
or suspected of
having a cancer of cells or tissues including, but not limited to, bladder,
breast, colon, rectal, gastric,
esophageal, lung, laryx, kidney, oral, ovarian, or prostate cancer, or a
sarcoma, melanoma, glioma,
lymphoma or leukemia, or a metatasis of any of these cancers.
[0038] In an embodiment, the invention provides any of the above antibodies
or functional
fragments for use in a method of detecting the presence of CD98 in a
biological sample. In some
embodiments, the method comprisescontacting the biological sample with any of
the above antibodies
under conditions permissive for binding of the antibody to CD98, and detecting
whether a complex is
formed between the antibody and CD98. In some embodiments, the biological
sample is from a mammal
having or suspected of having a cancer of cells or tissues including, but not
limited to, bladder, breast,
colon, rectal, gastric, esophageal, lung, laryx, kidney, oral, ovarian, or
prostate cancer, or a sarcoma,
melanoma, glioma, lymphoma or leukemia, or a metatasis of any of these
cancers.
[0039] In an embodiment, the invention provides the use of any of
the above antibodies or
functional fragments in the manufacture of a medicament, wherein the
medicament is for use in a method
of diagnosing a cancer associated with increased expression of CD98. In some
embodiments, the method
comprises contacting a test cell with any of the above antibodies; determining
the level of expression of
CD98 by detecting binding of the antibody to CD98; and comparing the level of
expression of CD98 by
the test cell with the level of expression of CD98 by a control cell, wherein
a higher level of expression
of CD98 by the test cell as compared to the control cell indicates the
presence of a cancer associated with
increased expression of CD98. In some embodiments, the test cell is a cell
from a patient suspected of
having a cancer selected from bladder, breast, colon, rectal, esophageal,
lung, laryx, kidney, oral, ovarian,
or prostate cancer, or a sarcoma, glioma, lymphoma or leukemiaõ or a metatasis
of any of these cancers.
In an embodiment, the method comprises determining the level of expression of
CD98 on the surface of
the test cell and comparing the level of expression of CD98 on the surface of
the test cell with the level of
expression of CD98 on the surface of the control cell. In some embodiments,
the test cell is a cancer cell
and the control cell is a normal cell of the same tissue type.
[0040] In an embodiment, the invention provides any of the above
antibodies or functional
fragments for use in a method of diagnosing a cancer associated with increased
expression of CD98. In
9
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
some embodiments, the method comprises contacting a test cell with any of the
above antibodies;
determining the level of expression of CD98 by detecting binding of the
antibody to CD98; and
comparing the level of expression of CD98 by the test cell with the level of
expression of CD98 by a
control cell, wherein a higher level of expression of CD98 by the test cell as
compared to the control cell
indicates the presence of a cancer associated with increased expression of
CD98. In some embodiments,
the test cell is a cell from a patient suspected of having a cancer selected
from bladder, breast, colon,
rectal, esophageal, lung, laryx, kidney, oral, ovarian, or prostate cancer, or
a sarcoma, glioma, lymphoma
or leukemiaõ or a metatasis of any of these cancers. In an embodiment, the
method comprises
determining the level of expression of CD98 on the surface of the test cell
and comparing the level of
expression of CD98 on the surface of the test cell with the level of
expression of CD98 on the surface of
the control cell. In some embodiments, the test cell is a cancer cell and the
control cell is a normal cell of
the same tissue type.
[0041] In another embodiment of the invention, an article of
manufacture, or -kit",
containing materials useful for the treatment of the disorders described above
is provided. The article of
manufacture comprises a container and a label or package insert on or
associated with the container.
Suitable containers include, for example, bottles, vials, syringes, blister
pack, etc. The containers may be
formed from a variety of materials such as glass or plastic. The container
holds an antibody or an
antibody-drug conjugate (ADC) composition which is effective for treating the
condition, and may have a
sterile access port (for example the container may be an intravenous solution
bag or a vial having a
stopper pierceable by a hypodermic injection needle). At least one active
agent in the composition is an
antibdy or ADC. The label or package insert indicates that the composition is
used for treating the
condition of choice, such as cancer. Alternatively, or additionally, the
article of manufacture may further
comprise a second (or third) container comprising a pharmaceutically-
acceptable buffer, such as
bacteriostatic water for injection (BWFI), phosphate-buffered saline, Ringer's
solution and dextrose
solution. It may further include other materials desirable from a commercial
and user standpoint,
including other buffers, diluents, filters, needles, and syringes.
BRIEF DESCRIPTION OF THE FIGURES
[0042] Fig. 1 shows the protein expression level of CD98 that was
identified and quantified
by sTAg analysis in the AML, CLL, CRC specimens and relevant normal controls.
Lines indicate the
mean of % normalized spectral abundance factor (NSAF) in positive samples.
[0043] Fig. 2 is a graph showing the results of epitope binning
for 39 anti-CD98 antibodies.
[0044] Fig. 3 shows the binding properties of chimeric anti-CD98
monoclonal antibodies 8-
34B, 18-2A 2.1, 18-2A 2.2, and 18-2A 2.7. Fig. 3A is a graph showing the
results of epitope binning for
chimeric anti-CD98 monoclonal antibodies. The four reference antibodies are as
in Fig. 1. "Isotype" is a
control antibody of the same isotype that does not bind CD98. Fig. 3B shows
the Kd of chimeric anti-
CD98 monoclonal antibodies as determined by FACS analysis with colon cancer
cell line DLD1. Fig.
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
3C shows the results of FACS analysis of three AML primary tumor samples and a
cell line expressing
cynomolgus monkey CD98 (cynCD98), stained with chimeric anti-CD98 monoclonal
antibodies.
[0045] Fig. 4 shows the construction of the humanized 8-34B
antibodies. Fig. 4A shows the
sequences of the murine 8-34B light chain variable domain (IGN 34) aligned to
the sequence of the
human acceptor sequence (AC) and the humanized light chains Ll and L2. The
CDRs according to
Kabat numbering are shown in red, and the substitutions in L2 as compared to
Ll are underlined. Fig. 4A
discloses SEQ ID NOS 6, 38, 15-16 and 38, respectively, in order of
appearance. Fig. 4B shows the
sequences of the murine 8-34B heavy chain variable domain (IGN 34) aligned to
the sequence of the
human acceptor sequence (AC) and the humanized heavy chains H1, H2 and H3. The
CDRs according
to Kabat numbering are shown in red, and the substitutions in H2 and H3 as
compared to H3 are
underlined. Fig. 4B discloses SEQ ID NOS 4, 39, 17-19 and 40, respectively, in
order of appearance.
[0046] Fig. 5 shows that anti-CD98 antibody treatment induces
strong tumor growth
inhibition in established Ramos tumors. Tumor volumes at which treatment was
initiated increased from
(A) ¨75mm3, (B) ¨150mm3 to (C) ¨250mm3. Dosing of the antibodies was stopped
at day 29 (A) or day
22 (B) and tumor regrowth was measured for the duration of the study.
Rituxirnab (anti-CD20 antibody)
was used as a positive therapeutic control antibody, and antibody HB121 (ATCC)
was used as an IgG2a
isotype negative control.
[0047] Fig. 6 shows that anti-CD98 antibodies prolong
significantly time to progression of
treated RAMOS tumors. Tumor doubling time of previous tumor regrowth data
(Figs. 4A-C) was
calculated and used for further prediction of time to progression (TTP). TTP
was then extrapolated for
each animal within the treatment groups, until 2000mm3 would have been reached
and graphed as a
Kaplan-Meier curve.
[0048] Fig. 7 shows the inhibition of in vivo tumor growth in a
lymphoma xenograft by the
anti-CD98 monoclonal antibody 18-2A as compared to rituxan and a negative
control IgG2a. Arrows
indicate administration of antibody treatment.
[0049] Fig. 8 shows the inhibition of in vivo tumor growth in an
acute myeloid leukemia
xenograft by the anti-CD98 monoclonal antibodies 18-2A and 8-34B as compared
to a negative control
IgG2a. Arrows indicate administration of antibody treatment.
[0050] Fig. 9 shows the inhibition of in vivo tumor growth in a
colorectal cancer xenograft
by the anti-CD98 monoclonal antibody 18-2A as compared to erbitux and a
negative control IgG2a (first
study) and to DC101 + CTX (cyclophosphamide) and a negative control IgG2a
(second study). DC101 is
a rat anti-mouse VEGFR2/KDR IgG1 mAb (ATCC No. HB-11534) and serves as a
positive control.
Arrows indicate administration of antibody treatment.
[0051] Fig. 10 shows the inhibition of in vivo tumor growth in a
non-small cell lung
carcinoma xenograft by the anti-CD98 monoclonal antibody 18-2A as compared to
Erbitux (anti-EGFR)
and a negative control IgG2a. Arrows indicate administration of antibody
treatment.
11
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
[0052] Fig. 11 shows the effect of anti-CD98 monoclonal antibodies
on in vivo tumor
growth of a lymphoma xenograft in mouse strains with different immunodeficient
backgrounds: (A) NSG
mice; (B) NOD.SCID mice, and (C) SCID mice.
[0053] Fig. 12 shows a comparison of the effect of chimeric anti-
CD98 monoclonal
antibodies (18-2A-ch7.1 and 8-34B-ch) as compared to their parent murine
monoclonal antibodies (18-
2A and 8-34B) on in vivo tumor growth of a lymphoma xenograft.
[0054] Fig. 13 illustrates the regions of the mouse CD98 sequence
(SEQ ID NO: 96) that
were substituted into the human CD98 sequence (SEQ ID NO: 1) to form the 13
mouse-human CD98
chimera constructs used to map the epitope on human CD98 bound by humanized
monoclonal antibody
IGN523.
[0055] Fig. 14 shows the binding of humanized monoclonal antibody
IGN523 and a control
antibody to each of the 13 mouse-human CD98 chimera constructs as determined
by FACS analysis.
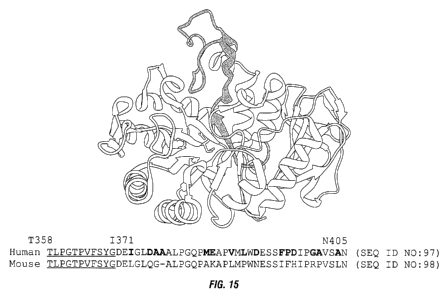
[0056] Fig. 15 shows the sequence of the region of human CD98
within which IGN523
binds, as identified using the mouse-human CD98 chimera constructs, and the
location of this sequence
within the three-dimensional structure of CD98. Amino acids T358-G368
(underlined) are buried in the
crystal structure and are unlikely to be part of the binding interface. Non-
conserved residues between the
human and mouse sequences are shown in bold.
[0057] Fig. 16 shows the binding of IGN523 to four constructs made
by introducing
nonhomologous residues from mouse CD98 into the targeted loop region of the
human sequence.
Construct 4.1 consists of mutations: 1371E, D374Q, A375G and Deletion of A376.
Construct 4.2
consists of mutations: M383A, and E384K. Construct 4.3 consists of mutations:
D391N, F395I, P396F
and D397H. Construct 4.4 consists of mutations: G400R, A401P and A404L.
Binding was detected by
FACS analysis of CHO cells transfected with the respective constructs.
[0058] Fig. 17 shows the binding of IGN523 to single mutation
constructs of hydrophobic
residues in the targeted loop region. Each indicated hydrophobic residue was
substituted with a highly
charged amino acid as shown. Binding was detected by FACS analysis of CHO
cells transfected with the
respective constructs.
[0059] Fig. 18 shows the binding of IGN523 to constructs
containing multiple mutations of
residues in the targeted loop region, as detected by FACS analysis of CHO
cells transfected with the
respective constructs. M1 containts mutations D374Q, D397H, G400R and A401P.
M2 contains
mutations D374E and A375E. M3 contains mutations D3975 and I398T.
[0060] Fig. 19 shows the results of a variable-length peptide
screen for epitope mapping of
humanized monoclonal antibody IGN523. ELISA results for each peptide are shown
as a horizontal line.
Start and end points of the lines indicate which residues are included in the
peptide. The Y-value of the
line shows the ELISA result obtained for that peptide. The results indicated
dominant binding for
395FPDIPGA401 and secondary binding for 379PGQP382 (shaded regions).
[0061] Fig. 20 shows the results of a best-binding single-
positions alanine-replacement
peptide set. Each residue was replaced by A (or G if the orginal amino acid
was A). The height at which
12
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
the replacement letter is plotted in the graph is the obtained ELISA value for
that mutated peptide. The
central line and shaded interval indicate the reference ELISA value.
[0062] Fig. 21 shows heat maps representing the data obtained from
CLIPS conformational
matrix structures that combined two partial sequences of human CD98 (SEQ ID
NOS 45-59 shown on
the X axis, and SEQ ID NOS 60-74 shown on the Y axis).
[0063] Fig. 22 shows the results of a mutagenesis screen of
strongly-binding peptides from
the matrix analyses shown in Fig. 21. SEQ1 shows the sequence of the peptide
and DIF1 indicates where
the mutation is located in the peptide. Grey fields indicate peptides having
non-mutated sequences. The
last column shows the difference in ELISA value between wild-type and mutated
peptide. High values
indicate that the mutation has a strong negative effect on binding.
[0064] Fig. 23 shows the location in the sequence and on the
surface of human CD98 of the
amino acid residues determined to be important for binding of humanized
monoclonal antibody IGN523.
Fig. 23A shows the location in the sequence of residues determined by the
chimera and mutagenesis
studies (bold), by Pepscan analysis (gray) or both (shaded). Fig. 23B shows
the location of the residues
determined by the chimera and mutagenesis studies (dark gray). Fig. 23C shows
the location of residues
determined by Pepscan analysis (light gray). Fig. 23D shows the overlap of
both sets of residues (black).
[0065] Fig. 24 shows the inhibition of in vivo tumor growth in a
RAMOS (RA.1) Burkitt
lymphoma xenograft by the humanized monoclonal antibody IGN523 as compared to
Rituximab and a
negative control IgG. Antibodies were dosed interperitoneally at 10 mg/kg on
days 11, 17 and 25.
Arrows indicate administration of antibody treatment.
[0066] Fig. 25 shows the inhibition of in vivo tumor growth in a
DAU Burkitt lymphoma
xenograft by the humanized monoclonal antibody IGN523 as compared to rituxan
and a negative control
IgG. Antibodies were dosed interperitoneally at 10 mg/kg on days 20 and 26.
Arrows indicate
administration of antibody treatment.
[0067] Fig. 26A shows the inhibition of in vivo tumor growth in a IGN-LNG-
12 lung tumor
xenograft by the humanized monoclonal antibody IGN523 as compared to
carboplatin and a negative
control IgG. IGN523 and carboplatin were dosed interperitoneally on days 17,
24 and 31 at 10 mg/kg or
75 mg/kg, respectively. Arrows indicate administration of treatment. Fig. 26B
shows body weight
measurements corresponding to mice in Fig. 26A treated with the indicated
reagents. Carboplatin was
dosed at its maximum tolerated dose, which induced body weight loss in NOD-
SCID mice.
[0068] Fig. 27 shows the inhibition of in vivo tumor growth in a
KG-1 acute myeloid
leukemia xenograft by the humanized monoclonal antibody IGN523 as compared to
rituxan and a
negative control IgG. Antibodies were dosed interperitoneally at 15 mg/kg on
days 21, 28 and 34.
Arrows indicate administration of antibody treatment.
13
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
[0069] Fig. 28 shows dose dependent inhibition of in vivo tumor
growth in a lung tumor
xenograft by the humanized monoclonal antibody IGN523. The antibody was dosed
intraperitoneally at
the indicated doses on days 12 and 19.Arrows indicate administration of
antibody treatment.
[0070] Fig. 29 shows staining of human and cynomolgus monkey
frozen tissue sections by
humanized monoclonal antibody IGN523. Cryosections of human and cynomolgus
monkey kidney,
cerebrum, and placenta were stained with 10 lig/mL of IGN523. Modifications of
the methods of Tuson,
Fung, and Hierck for immunohistochemistry were used to eliminate the
requirement for labeling of
IGN523 and to preclude nonspecific reactivity between the secondary labeled
anti-human IgG and IgG
endogenous to the tissues to be examined (Fung 1992, Hierck 1994, Tuson 1990).
Sections were cut at
approximately 5 Jim. All slides were initially assessed for the adequacy of
tissue elements and staining,
then evaluated and subjectively graded by the Study Pathologist for intensity
of staining. Representative
images are shown at 40x magnification with the exception of human cerebrum
(20x).
DETAILED DESCRIPTION OF EMBODIMENTS OF THE INVENTION
General Techniques
[0071] The techniques and procedures described or referenced
herein are generally well
understood and commonly employed using conventional methodology by those
skilled in the art, such as,
for example, the widely utilized methodologies described in Sambrook et al.,
Molecular Cloning: A
Laboratory Manual 3rd. edition (2001) Cold Spring Harbor Laboratory Press,
Cold Spring Harbor, N.Y.;
Current Protocols in Molecular Biology (F. M. Ausubel, et al. eds., (2003));
Therapeutic Monoclonal
Antibodies: From Bench to Clinic, Z. An, ed, Wiley, Hoboken N.J. (2009);
Monoclonal Antibodies:
Methods and Protocols, M. Albitar, ed., Humana Press, Totawa, N.J. (2010); and
Antibody Engineering,
2' Ed., Vols 1 and 2, Kontermann and Dubel, eds., Springer-Verlag, Heidelberg,
2010.
DEFINITIONS AND ABBREVIATIONS
Definitions
[0072] For purposes of interpreting this specification, the
following definitions will apply
and whenever appropriate, terms used in the singular will also include the
plural and vice versa. In the
event that any definition set forth conflicts with any document incorporated
herein by reference, the
definition set forth below shall control.
[0073] The term "CD98", as used herein, refers to any native CD98
from any vertebrate
source, including mammals such as primates (e.g. humans, cynomolgus monkey
(cyno)), dogs, and
rodents (e.g., mice and rats), unless otherwise indicated. The amino acid and
encoding nucleic acid
sequences of human CD98 are provided below as SEQ ID NO:1 and SEQ ID NO:2,
respectively.
[0074] MS QDTEVDMKEVELNELE PEKQ PMNAAS GAAMS LAGAEKNGLVKI KVAE DEAEAAA
AAKFTGLSKEELLKVAGS PGWVRTRWALLLLEWLGWLGMLAGAVVI I VRAPRCREL PAQKWWHT GALYRI
GDLQAFQGHGAGNLAGLKGRLDYLS SLKVKGLVLGP I HKNQKDDVAQT DLLQ I DPNEGSKEDEDSLLQSA
KKKS I RVIL DLT PNYRGENSWFS TQVDTVATKVKDALE FWLQAGVDGFQVRDI ENLKDAS S FLAEWQN
I T
14
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
KGFSEDRLL IAGTNS S DLQQILSLLESNKDLLLT S SYLS DS GS TGEHT KS LVT QYLNAT
GNRWCSWSL S Q
ARLLT S FL PAQLLRLYQLML FT L PGT PVFS YGDE I GLDAAAL PGQPMEAPVMLWDES S FPDI
PGAVSANM
TVKGQ SE DPGSLL S L FRRL S DQRSKERSLLHGDFHAFSAGPGL FS YI RHWDQNERFLVVLNFGDVGL
SAG
LQASDL PASASL PAKADLLL STQPGREEGS PLELERLKLEPHEGLLLRFPYAA (SEQ ID NO:1)
[0075] AT GAGCCAGGACACCGAGGT GGATAT GAAGGAGGT GGAGCT GAATGAGTTAGAGCC
CGAGAAGCAGCCGAT GAACGCGGCGT CTGGGGCGGCCAT GTCCCT GGCGGGAGCCGAGAAGAATGGTCTG
GTGAAGATCAAGGTGGCGGAAGACGAGGCGGAGGCGGCAGCCGCGGCTAAGTTCACGGGCCTGTCCAAGG
AGGAGCT GCTGAAGGT GGCAGGCAGCCCCGGCT GGGTACGCACCCGCT GGGCACT GCT GCTGCTCTTCTG
GCTCGGCTGGCTCGGCATGCTTGCTGGTGCCGTGGTCATAATCGTGCGAGCGCCGCGTTGTCGCGAGCTA
CCGGCGCAGAAGTGGTGGCACACGGGCGCCCTCTACCGCATCGGCGACCTTCAGGCCTTCCAGGGCCACG
GCGCGGGCAACCT GGCGGGT CT GAAGGGGCGTCTCGATTACCT GAGCT CT CT GAAGGT GAAGGGCCTTGT
GCTGGGTCCAATT CACAAGAACCAGAAGGAT GATGTCGCTCAGACTGACTT GCT GCAGATCGACCCCAAT
TT T GGCT CCAAGGAAGATT TTGACAGTCT CTT GCAATCGGCTAAAAAAAAGAGCAT CCGTGTCATTCTGG
ACCT TACTCCCAACTACCGGGGTGAGAACT CGTGGTT CTCCACT CAGGT T GACACT GT GGCCACCAAGGT
GAAGGATGCTCT GGAGTT T TGGCTGCAAGCT GGCGTGGAT GGGTT CCAGGTT CGGGACATAGAGAATCT G
AAGGATGCATCCT CAT T CT TGGCT GAGT GGCAAAATAT CACCAAGGGCTT CAGT
GAAGACAGGCTCTTGA
TT GCGGGGACTAACTCCT CCGACCTTCAGCAGATCCT GAGCCTACTCGAATCCAACAAAGACTTGCT GTT
GACTAGCTCATACCTGTCT GAT T CTGGTTCTACTGGGGAGCATACAAAAT CCCTAGT CACACAGTATTT G
AATGCCACTGGCAAT CGCT GGT GCAGCTGGAGTT TGTCTCAGGCAAGGCT CCT GACT TCCT TCTTGCCGG
CTCAACTTCTCCGACT CTACCAGCT GAT GCTCTTCACCCT GCCAGGGACCCCTGTT T TCAGCTACGGGGA
TGAGATTGGCCTGGATGCAGCTGCCCTTCCTGGACAGCCTATGGAGGCTCCAGTCATGCTGTGGGATGAG
TCCAGCTTCCCTGACATCCCAGGGGCTGTAAGTGCCAACATGACT GT GAAGGGCCAGAGTGAAGACCCTG
GCTCCCTCCTTTCCTTGTTCCGGCGGCTGAGTGACCAGCGGAGTAAGGAGCGCTCCCTACTGCATGGGGA
CTTCCACGCGTTCTCCGCT GGGCCT GGACTCTT CT CCTATAT CCGCCACT GGGACCAGAAT GAGCGTTTT
CT GGTAGT GCTTAACT TT GGGGATGTGGGCCT CTCGGCT GGACT GCAGGCCT CCGACCT
GCCTGCCAGCG
CCAGCCTGCCAGCCAAGGCTGACCTCCTGCTCAGCACCCAGCCAGGCCGTGAGGAGGGCTCCCCTCTTGA
GCTGGAACGCCTGAAACTGGAGCCTCACGAAGGGCTGCTGCTCCGCTTCCCCTACGCGGCCTGA (SEQ ID
NO:2)
[0076] The term "CD98" encompasses "full-length," unprocessed CD98
as well as any form
of CD98 that results from processing in the cell. The term also encompasses
naturally occurring variants
or mutations of CD98, e.g., splice variants, allelic variants, SNP variants
and isoforms. The CD98
polypeptides described herein may be isolated from a variety of sources, such
as from human tissue types
or from another source, or prepared by recombinant or synthetic methods. A
"native sequence CD98
polypeptide" comprises a polypeptide having the same amino acid sequence as
the corresponding CD98
polypeptide derived from nature. Such native sequence CD98 polypeptides can be
isolated from nature
or can be produced by recombinant or synthetic means. The term "native
sequence CD98 polypeptide"
specifically encompasses naturally-occurring truncated or secreted forms of
the specific CD98
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
polypeptide (e.g., an extracellular domain sequence), naturally-occurring
variant forms (e.g., alternatively
spliced forms) and naturally-occurring allelic variants of the polypeptide.
[0077] The term "antibody" is used in the broadest sense and
specifically covers, for
example, single anti-CD98 monoclonal antibodies (including agonist,
antagonist, neutralizing antibodies,
full length or intact monoclonal antibodies), anti-CD98 antibody compositions
with polyepitopic
specificity, polyclonal antibodies, multivalent antibodies, multispecific
antibodies (e.g., bispecific
antibodies so long as they exhibit the desired biological activity), formed
from at least two intact
antibodies, single chain anti-CD98 antibodies, and fragments of anti-CD98
antibodies, as defined below.
The term "immunoglobulin" (Ig) is used interchangeable with antibody herein.
An antibody can be
human, humanized and/or affinity matured.
[0078] An "antigen" is a predetermined antigen to which an
antibody can selectively bind.
The target antigen may be a polypeptide, carbohydrate, nucleic acid, lipid,
hapten or other naturally
occurring or synthetic compound. Preferably, the target antigen is a
polypeptide.
[0079] An antibody "which binds" an antigen of interest is one
that binds the antigen with
sufficient affinity such that the antibody is useful as a therapeutic agent in
targeting a cell or tissue
expressing the antigen, and does not significantly cross-react with other
proteins. In such embodiments,
the extent of binding of the antibody to a "non-target" protein will be less
than about 10% of the binding
of the antibody to its particular target protein as determined by fluorescence
activated cell sorting (FACS)
analysis or radioimmunoprecipitation (RIA). With regard to the binding of an
antibody to a target
molecule, the term "specific binding" or "specifically binds to" or is
"specific for" a particular
polypeptide or an epitope on a particular polypeptide target means binding
that is measurably different
from a non-specific interaction. Specific binding can be measured, for
example, by determining binding
of a molecule compared to binding of a control molecule, which generally is a
molecule of similar
structure that does not have binding activity. For example, specific binding
can be determined by
competition with a control molecule that is similar to the target, for
example, an excess of non-labeled
target. In this case, specific binding is indicated if the binding of the
labeled target to a probe is
competitively inhibited by excess unlabeled target. The term "specific
binding" or "specifically binds to"
or is "specific for" a particular polypeptide or an epitope on a particular
polypeptide target as used herein
can be exhibited, for example, by a molecule having a Kd for the target of at
least about 104 M,
alternatively at least about le M, alternatively at least about 10-6 M,
alternatively at least about 10-7 M,
alternatively at least about 10-8 M, alternatively at least about 10-9 M,
alternatively at least about 10-1 M,
alternatively at least about 1041 M, alternatively at least about 10-12 M, or
greater. In one embodiment,
the term "specific binding" refers to binding where a molecule binds to a
particular polypeptide or
epitope on a particular polypeptide without substantially binding to any other
polypeptide or polypeptide
epitope.
[0080] The term "anti-CD98 antibody" or "an antibody that binds to
CD98" refers to an
antibody that is capable of binding CD98 with sufficient affinity such that
the antibody is useful as a
diagnostic and/or therapeutic agent in targeting CD98. Preferably, the extent
of binding of an anti-CD98
16
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
antibody to an unrelated, non-CD98 protein is less than about 10% of the
binding of the antibody to
CD98 as measured, e.g., by fluorescence activated cell sorting (FACS) analysis
or a radioimmunoassay
(RIA). An antibody that "specifically binds to" or is "specific for" CD98 is
defined as above. In certain
embodiments, an antibody that binds to CD98 has a dissociation constant (Kd)
of < 1 tM,< 100 nM,
10 nM, < 1 nM, or < 0.1 nM. In certain embodiments, anti-CD98 antibody binds
to an epitope of CD98
that is conserved among CD98 from different species.
10081] An "isolated antibody" is one that has been identified and
separated and/or recovered
from a component of its natural environment. Contaminant components of its
natural environment
include, but are not limited to, materials that would interfere with
therapeutic uses for the antibody, and
may include enzymes, hormones, and other proteinaceous or nonproteinaceous
solutes. In preferred
embodiments, the antibody will be purified (I) to greater than 95% by weight
of antibody as determined
by the Lowry method (Lowry et al., J. Bio. Chem. 193: 265-275, 1951) and most
preferably more than
99% by weight, (2) to a degree sufficient to obtain at least 15 residues of N-
terminal or internal amino
acid sequence by use of a spinning cup sequenator, or (3) to homogeneity by
SDS-PAGE under reducing
or nonreducing conditions using Coomassie blue or, preferably, silver stain.
Isolated antibody includes
the antibody in situ within recombinant cells since at least one component of
the antibody's natural
environment will not be present. Ordinarily, however, isolated antibody will
be prepared by at least one
purification step.
[0082] The basic 4-chain antibody unit is a heterotetrameric
glycoprotein composed of two
identical light (L) chains and two identical heavy (H) chains. In the case of
IgGs, the 4-chain unit is
generally about 150,000 daltons. Each L chain is linked to a H chain by one
covalent disulfide bond,
while the two H chains are linked to each other by one or more disulfide bonds
depending on the H chain
isotype. Each H and L chain also has regularly spaced intrachain disulfide
bridges. Each H chain has at
the N-terminus, a variable domain (VH) followed by three constant domains (CH)
for each of the a and y
chains and four CH domains for 1.1 and c isotypes. Each L chain has at the N-
terminus, a variable domain
(VL) followed by a constant domain (CL) at its other end. The VL is aligned
with the VH and the CL is
aligned with the first constant domain of the heavy chain (CH1). Particular
amino acid residues are
believed to form an interface between the light chain and heavy chain variable
domains. The pairing of a
VH and VL together forms a single antigen-binding site. For the structure and
properties of the different
classes of antibodies, see, e.g., Basic and Clinical Immunology, 8th edition,
Daniel P. Stites, Abba I. Terr
and Tristram G. Parslow (eds.), Appleton & Lange, Norwalk, CT, 1994, page 71
and Chapter 6.
[0083] The "variable region" or "variable domain" or "V domain" of
an antibody refers to
the amino-terminal domains of the heavy or light chain of the antibody. The
variable domain of the
heavy chain may be referred to as "VH." The variable domain of the light chain
may be referred to as
"VL." The term "variable" refers to the fact that certain segments of the
variable domains differ
extensively in sequence among antibodies. The V domain mediates antigen
binding and defines
specificity of a particular antibody for its particular antigen. However, the
variability is not evenly
distributed across the 110-amino acid span of the variable domains. Instead,
the V regions consist of
17
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
relatively invariant stretches called framework regions (FRs) of 15-30 amino
acids separated by shorter
regions of extreme variability called "hypervariable regions" that are each 9-
12 amino acids long. The
variable domains of native heavy and light chains each comprise four FRs,
largely adopting a13-sheet
configuration, connected by three hypervariable regions, which form loops
connecting, and in some cases
forming part of, the [3-sheet structure. The hypervariable regions in each
chain are held together in close
proximity by the FRs and, with the hypervariable regions from the other chain,
contribute to the
formation of the antigen-binding site of antibodies (see Kabat et al.,
Sequences of Proteins of
Immunological Interest, 5th Ed. Public Health Service, National Institutes of
Health, Bethesda, MD,
1991)). The constant domains are not involved directly in binding an antibody
to an antigen, but exhibit
various effector functions, such as participation of the antibody in antibody
dependent cellular
cytotoxicity (ADCC) and complement dependent cytotoxicity (CDC).
[0084] An "intact" antibody is one comprising an antigen-binding
site as well as a CL and at
least heavy chain constant domains, CHI, CH2 and CH3. The constant domains may
be native sequence
constant domains (e.g. human native sequence constant domains) or amino acid
sequence variant thereof.
Preferably, the intact antibody has one or more effector functions.
[0085] "Antibody fragments" comprise a portion of an intact
antibody, preferably the
antigen binding or variable region of the intact antibody. Examples of
antibody fragments include,
without limitation, Fab, Fab', F(ab')2, and Fv fragments; diabodies and di-
diabodies (see, e.g. Holliger, P.
et al (1993) Proc. Natl. Acad. Sci. 90:6444-8; Lu, D. et al. (2005) J. Biol.
Chem. 280:19665-72; Hudson
et al., Nat. Med. 9:129-134 (2003); WO 93/11161; and U.S. Patent Nos.
5,837,242 and 6,492,123);
single-chain antibody molecules (see, e.g. U.S. Patent Nos. 4,946,778;
5,260,203; 5,482,858 and
5,476,786); dual variable domain antibodies (see, e.g. U.S. Patent No.
7,612,181); single variable domain
antibodies (SdAbs) (see, e.g. Woolven et al., Immunogenetics 50: 98-101, 1999;
Streltsov et al., Proc
Natl Acad Sci USA. 101:12444-12449, 2004); and multispecific antibodies formed
from antibody
fragments.
[0086] A "functional fragment" of a therapeutic antibody will
exhibit at least one if not
some or all of the biological functions attributed to the intact antibody, the
function comprising at least
specific binding to the target antigen.
[0087] The term "monoclonal antibody" as used herein refers to an
antibody obtained from a
population of substantially homogeneous antibodies, i.e., the individual
antibodies comprising the
population are identical except for possible naturally occurring mutations
that may be present in minor
amounts. The modifier "monoclonal" is not to be construed as requiring
production of the antibody by
any particular method. For example, the monoclonal antibodies useful in the
present invention may be
prepared by the hybridoma methodology first described by Kohler et al.,
Nature, 256:495 (1975), or may
be made using recombinant DNA methods in bacterial, eukaryotic animal or plant
cells (see, e.g., U.S.
Patent No. 4,816,567). The "monoclonal antibodies" may also be isolated from
phage antibody libraries
using the techniques described in Clackson et al., Nature, 352:624-628 (1991)
and Marks et al., J. Mol.
Biol., 222:581-597 (1991), for example.
18
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
[0088] The monoclonal antibodies herein include "chimeric"
antibodies in which a portion
of the heavy and/or light chain is identical with or homologous to
corresponding sequences in antibodies
derived from a particular species or belonging to a particular antibody class
or subclass, while the
remainder of the chain(s) is identical with or homologous to corresponding
sequences in antibodies
[0089] "Humanized" forms of non-human (e.g., rodent) antibodies
are chimeric antibodies
that contain minimal sequence derived from the non-human antibody. For the
most part, humanized
[0090] A "human antibody" is one which possesses an amino acid
sequence which
corresponds to that of an antibody produced by a human and/or has been made
using any of the
techniques for making human antibodies as disclosed herein. This definition of
a human antibody
19
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
disabled, e.g., mice (see, e.g., Jakobovits, A., Curr. Opin. Biotechnol. 1995,
6(5):561-6; Brtiggemann and
Taussing, Curr. Opin. Biotechnol. 1997, 8(4):455-8; and U.S. Pat. Nos.
6,075,181 and 6,150,584
regarding XENOMOUSETm technology). See also, for example, Li et al., Proc.
Natl. Acad. Set USA,
103:3557-3562 (2006) regarding human antibodies generated via a human B-cell
hybridoma technology.
[0091] The term "hypervariable region", "HVR", or "IN", when used herein
refers to the
regions of an antibody variable domain that are hypervariable in sequence
and/or form structurally
defined loops. Generally, antibodies comprise six hypervariable regions; three
in the VH (H1, H2, H3),
and three in the VL (L1, L2, L3). A number of hypervariable region
delineations are in use and are
encompassed herein. The Kabat Complementarity Determining Regions (CDRs) are
based on sequence
variability and are the most commonly used (Kabat et al., Sequences of
Proteins of Immunological
Interest, 5th Ed. Public Health Service, National Institutes of Health,
Bethesda, MD. (1991)). Chothia
refers instead to the location of the structural loops (Chothia and Lesk MoL
Biol. 196:901-917 (1987)).
The end of the Chothia CDR-HI loop when numbered using the Kabat numbering
convention varies
between H32 and H34 depending on the length of the loop (this is because the
Kabat numbering scheme
places the insertions at H35A and H35B; if neither 35A nor 35B is present, the
loop ends at 32; if only
35A is present, the loop ends at 33; if both 35A and 35B are present, the loop
ends at 34). The AbM
hypervariable regions represent a compromise between the Kabat CDRs and
Chothia structural loops,
and are used by Oxford Molecular's AbM antibody modeling software. The
"contact" hypervariable
regions are based on an analysis of the available complex crystal structures.
The residues from each of
these hypervariable regions are noted below.
Loop Kabat AbM Chothia Contact
LI L24-L34 L24-L34 L26-L32 L30-L36
L2 L50-L56 L50-L56 L50-L52 L46-L55
L3 L89-L97 L89-L97 L91-L96 L89-L96
H1 H31-H35B H26-H35B H26-H32 H30-H35B (Kabat
Numbering)
H1 H31-H35 H26-H35 H26-H32 H30-H35 (Chothia
Numbering)
H2 H50-H65 H50-H58 H53-H55 H47-H58
H3 H95-H102 H95-H102 H96-H101 H93-H101
[0092] Hypervariable regions may comprise "extended hypervariable
regions" as follows:
24-36 or 24-34 (L1), 46-56 or 50-56 (L2) and 89-97 or 89-96 (L3) in the VL and
26-35 or 26-35A (H1),
50-65 or 49-65 (H2) and 93-102, 94-102, or 95-102 (H3) in the VH. The variable
domain residues are
numbered according to Kabat et al., supra, for each of these definitions. As
used herein, the terms
"HVR" and "CDR" are used interchangeably.
[0093] "Framework" or "FR" residues are those variable domain
residues other than the
hypervariable region residues herein defined.
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
[0094] The term "variable domain residue numbering as in Kabat" or
"amino acid position
numbering as in Kabat", and variations thereof, refers to the numbering system
used for heavy chain
variable domains or light chain variable domains of the compilation of
antibodies in Kabat et al.,
Sequences of Proteins of Immunological Interest, 5th Ed. Public Health
Service, National Institutes of
Health, Bethesda, MD. (1991). Using this numbering system, the actual linear
amino acid sequence may
contain fewer or additional amino acids corresponding to a shortening of, or
insertion into, a FR or CDR
of the variable domain. For example, a heavy chain variable domain may include
a single amino acid
insert (residue 52a according to Kabat) after residue 52 of H2 and inserted
residues (e.g. residues 82a,
82b, and 82c, etc according to Kabat) after heavy chain FR residue 82. The
Kabat numbering of residues
may be determined for a given antibody by alignment at regions of homology of
the sequence of the
antibody with a "standard" Kabat numbered sequence.
[0095] The Kabat numbering system is generally used when referring
to a residue in the
variable domain (approximately residues 1-107 of the light chain and residues
1-113 of the heavy chain)
(e.g, Kabat et al., Sequences of Irnmunological Interest. 5th Ed. Public
Health Service, National Institutes
of Health, Bethesda, Md. (1991)). The "EU numbering system" or "EU index" is
generally used when
referring to a residue in an immunoglobulin heavy chain constant region (e.g.,
the EU index reported in
Kabat et al.. supra). The "EU index as in Kabat" refers to the residue
numbering of the human IgG1 EU
antibody. Unless stated otherwise herein, references to residue numbers in the
variable domain of
antibodies means residue numbering by the Kabat numbering system. Unless
stated otherwise herein,
references to residue numbers in the constant domain of antibodies means
residue numbering by the EU
numbering system.
[0096] An "affinity matured" antibody is one with one or more
alterations in one or more
HVRs thereof which result in an improvement in the affinity of the antibody
for antigen, compared to a
parent antibody which does not possess those alteration(s). Preferred affinity
matured antibodies will
have nanomolar or even picomolar affinities for the target antigen. Affinity
matured antibodies are
produced by procedures known in the art. For review, see Hudson and Souriau,
Nature Medicine 9 :129-
134 (2003); Hoogenboom, Nature Biotechnol. 23 : 1105-1116 (2005); Quiroz and
Sinclair, Revista
Ingeneria Biomedia 4 : 39-51 (2010).
[0097] A "blocking" antibody or an "antagonist" antibody is one
which inhibits or reduces
biological activity of the antigen it binds. Preferred blocking antibodies or
antagonist antibodies
substantially or completely inhibit the biological activity of the antigen.
[0098] An "agonist antibody", as used herein, is an antibody which
mimics at least one of
the functional activities of a polypeptide of interest.
[0099] "Binding affinity" generally refers to the strength of the
sum total of noncovalent
interactions between a single binding site of a molecule (e.g., an antibody)
and its binding partner (e.g.,
an antigen). Unless indicated otherwise, as used herein, "binding affinity"
refers to intrinsic binding
affinity which reflects a 1:1 interaction between members of a binding pair
(e.g., antibody and antigen).
The affinity of a molecule X for its partner Y can generally be represented by
the dissociation constant
21
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
(Kd). Affinity can be measured by common methods known in the art, including
those described herein.
Low-affinity antibodies generally bind antigen slowly and tend to dissociate
readily, whereas high-
affinity antibodies generally bind antigen faster and tend to remain bound
longer. A variety of methods
of measuring binding affinity are known in the art, any of which can be used
for purposes of the present
invention. Specific illustrative embodiments are described in the following.
[0100] "Or better" when used herein to refer to binding affinity
refers to a stronger binding
between a molecule (e.g. antibody) and its binding partner, and is represented
by a smaller numerical Kd
value. For example, an antibody which has an affinity for an antigen of ".6 nM
or better", the antibody's
affinity for the antigen is <.6 nM, i.e. .59 nM, .58 nM, .57 nM etc. or any
value less than .6 nM.
[0101] In one embodiment, the "Kd" or "Kd value" according to this
invention is measured
by a radiolabeled antigen binding assay (RIA) performed with the Fab version
of an antibody of interest
and its antigen as described by the following assay that measures solution
binding affinity of Fabs for
antigen by equilibrating Fab with a minimal concentration of (1251)-labeled
antigen in the presence of a
titration series of unlabeled antigen, then capturing bound antigen with an
anti-Fab antibody-coated plate
(Chen, et al., (1999) J. Mol Biol 293:865-881). According to another
embodiment the Kd or Kd value is
TM
measured by using surface plasmon resonance assays using, for example, a
BlAcore -2000 or a
TM
BlAcore -3000 (BlAcore, Inc., Piscataway, NJ).
[0102] An "on-rate" or "rate of association" or "association rate"
or "k0
,5 according to this
invention can also be determined with the same surface plasmon resonance
technique described above
TM TM
using, for example, a BlAcore -2000 or a BlAcore -3000 (BlAcore, Inc.,
Piscataway, NJ).
[0103] The phrase "substantially similar," or "substantially the
same", as used herein,
denotes a sufficiently high degree of similarity between two numeric values
(generally one associated
with an antibody of the invention and the other associated with a reference
antibody) such that one of
skill in the art would consider the difference between the two values to be of
little or no biological and/or
statistical significance within the context of the biological characteristic
measured by said values (e.g.,
Kd values). The difference between the two values is preferably less than
about 50%, preferably less
than about 40%, preferably less than about 30%, preferably less than about
20%, preferably less than
about 10% as a function of the value for the reference antibody.
[0104] The phrase "substantially reduced," or "substantially
different", as used herein,
denotes a sufficiently high degree of difference between two numeric values
(generally one associated
with an antibody of the invention and the other associated with a reference
antibody) such that one of
skill in the art would consider the difference between the two values to be of
statistical significance
within the context of the biological characteristic measured by said values
(e.g., Kd values, HAMA
response). The difference between said two values is preferably greater than
about 10%, preferably
greater than about 20%, preferably greater than about 30%, preferably greater
than about 40%, preferably
greater than about 50% as a function of the value for the reference antibody.
22
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
[0105] An antibody that "inhibits the growth of tumor cells
expressing a CD98 polypeptide"
or a "growth inhibitory" antibody is one which results in measurable growth
inhibition of cancer cells
expressing or overexpressing the appropriate CD98 polypeptide. The CD98
polypeptide may be a
transmembrane polypeptide expressed on the surface of a cancer cell or may be
a polypeptide that is
produced and secreted by a cancer cell. Preferred growth inhibitory anti-CD98
antibodies inhibit growth
of CD98-expressing tumor cells by greater than 20%, preferably from about 20%
to about 50%, and even
more preferably, by greater than 50% (e.g., from about 50% to about 100%) as
compared to the
appropriate control, the control typically being tumor cells not treated with
the antibody being tested. In
one embodiment, growth inhibition can be measured at an antibody concentration
of about 0.1 to 30
pg/ml or about 0.5 nM to 200 nM in cell culture, where the growth inhibition
is determined 1-10 days
after exposure of the tumor cells to the antibody. Growth inhibition of tumor
cells in vivo can be
determined in various ways such as is described below. The antibody is growth
inhibitory in vivo if
administration of the anti-CD98 antibody at about 1 pg/kg to about 100 mg/kg
body weight results in
reduction in tumor size or tumor cell proliferation within about 5 days to 3
months from the first
administration of the antibody, preferably within about 5 to 30 days.
[0106] An antibody that "induces apoptosis" is one that induces
programmed cell death as
determined by binding of annexin V, fragmentation of DNA, cell shrinkage,
dilation of endoplasmic
reticulum, cell fragmentation, and/or formation of membrane vesicles (called
apoptotic bodies). The cell
is usually one that overexpresses a CD98 polypeptide. Preferably the cell is a
tumor cell. Various
methods are available for evaluating the cellular events associated with
apoptosis. For example,
phosphatidyl serine (PS) translocation can be measured by annexin binding; DNA
fragmentation can be
evaluated through DNA laddering; and nuclear/chromatin condensation along with
DNA fragmentation
can be evaluated by any increase in hypodiploid cells. Preferably, the
antibody which induces apoptosis
is one which results in about 2 to 50 fold, preferably about 5 to 50 fold, and
most preferably about 10 to
50 fold, induction of annexin binding relative to untreated cell in an annexin
binding assay.
[0107] An antibody that "induces cell death" is one that causes a
viable cell to become
nonviable. The cell is of a cell type that specifically expresses or
overexpresses a CD98 polypeptide.
The cell may be cancerous or a normal cell of the particular cell type. The
CD98 polypeptide may be a
transmembrane polypeptide expressed on the surface of a cancer cell or may be
a polypeptide that is
produced and secreted by a cancer cell. Cell death in vitro may be determined
in the absence of
complement and immune effector cells to distinguish cell death induced by
antibody-dependent cell-
mediated cytotoxicity (ADCC) or complement dependent cytotoxicity (CDC). Thus,
the assay for cell
death may be performed using heat inactivated serum (i.e., in the absence of
complement) and in the
absence of immune effector cells. To determine whether the antibody is able to
induce cell death, loss of
membrane integrity as evaluated by uptake of propidium iodide (PI), trypan
blue (see Moore et al.
Cytotechnology 17:1-11 (1995)) or 7AAD can be assessed relative to untreated
cells. Preferred cell
death-inducing antibodies are those which induce PI uptake in the PI uptake
assay in BT474 cells.
23
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
[0108] Antibody "effector functions" refer to those biological
activities attributable to the
Fc region (a native sequence Fc region or amino acid sequence variant Fc
region) of an antibody, and
vary with the antibody isotype. Examples of antibody effector functions
include: Clq binding and
complement dependent cytotoxicity; Fc receptor binding; antibody-dependent
cell-mediated cytotoxicity
(ADCC); phagocytosis; down regulation of cell surface receptors (e.g., B cell
receptor); and B cell
activation.
[0109] The term "Fc region" herein is used to define a C-terminal
region of an
immunoglobul in heavy chain, including native sequence Fc regions and variant
Fc regions. Although the
boundaries of the Fc region of an immunoglobulin heavy chain might vary, the
human IgG heavy chain
Fc region is usually defined to stretch from an amino acid residue at position
Cys226, or from Pro230, to
the carboxyl-terminus thereof. The C-terminal lysine (residue 447 according to
the EU numbering
system) of the Fc region may be removed, for example, during production or
purification of the antibody,
or by recombinantly engineering the nucleic acid encoding a heavy chain of the
antibody. Accordingly, a
composition of intact antibodies may comprise antibody populations with all
K447 residues removed,
antibody populations with no K447 residues removed, and antibody populations
having a mixture of
antibodies with and without the K447 residue.
[0110] A "functional Fc region" possesses an "effector function"
of a native sequence Fc
region. Exemplary "effector functions" include Clq binding; CDC; Fc receptor
binding; ADCC;
phagocytosis; down regulation of cell surface receptors (e.g. B cell receptor;
BCR), etc. Such effector
functions generally require the Fc region to be combined with a binding domain
(e.g., an antibody
variable domain) and can be assessed using various assays as disclosed, for
example, in definitions herein
[0111] A "native sequence Fc region" comprises an amino acid
sequence identical to the
amino acid sequence of an Fc region found in nature. Native sequence human Fc
regions include a native
sequence human IgG1 Fc region (non-A and A allotypes); native sequence human
IgG2 Fc region; native
sequence human IgG3 Fc region; and native sequence human IgG4 Fc region as
well as naturally
occurring variants thereof.
[0112] A "variant Fc region" comprises an amino acid sequence
which differs from that of a
native sequence Fc region by virtue of at least one amino acid modification,
preferably one or more
amino acid substitution(s). Preferably, the variant Fc region has at least one
amino acid substitution
compared to a native sequence Fc region or to the Fc region of a parent
polypeptide, e.g. from about one
to about ten amino acid substitutions, and preferably from about one to about
five amino acid
substitutions in a native sequence Fc region or in the Fc region of the parent
polypeptide. The variant Fc
region herein will preferably possess at least about 80% homology with a
native sequence Fc region
and/or with an Fc region of a parent polypeptide, and most preferably at least
about 90% homology
therewith, more preferably at least about 95% homology therewith.
[0113] Antibody-dependent cell-mediated cytotoxicity" or "ADCC"
refers to a form of
cytotoxicity in which secreted Ig bound onto Fc receptors (FcRs) present on
certain cytotoxic cells (e.g.,
Natural Killer (NK) cells, neutrophils, and macrophages) enable these
cytotoxic effector cells to bind
24
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
specifically to an antigen-bearing target cell and subsequently kill the
target cell with cytotoxins. The
antibodies "arm" the cytotoxic cells and are absolutely required for such
killing. The primary cells for
mediating ADCC, NK cells, express FcyRIII only, whereas monocytes express
FcyRI, FcyRII and
FcyRIII. FcR expression on hematopoietic cells is summarized in Table 3 on
page 464 of Ravetch and
Kinet, Annu. Rev. Immunol. 9:457-92 (1991). To assess ADCC activity of a
molecule of interest, an in
vitro ADCC assay, such as that described in US Patent No. 5,500,362 or
5,821,337 may be performed.
Useful effector cells for such assays include peripheral blood mononuclear
cells (PBMC) and Natural
Killer (NK) cells. Alternatively, or additionally, ADCC activity of the
molecule of interest may be
assessed in vivo, e.g., in a animal model such as that disclosed in Clynes et
al. (USA) 95:652-656 (1998).
[0114] "Fc receptor" or "FcR" describes a receptor that binds to the Fc
region of an
antibody. The preferred FcR is a native sequence human FcR. Moreover, a
preferred FcR is one that
binds an IgG antibody (a gamma receptor) and includes receptors of the FcyRI,
FcyRII and FcyRIII
subclasses, including allelic variants and alternatively spliced forms of
these receptors. FcyRII receptors
include FcyRIIA (an "activating receptor") and FcyRIIB (an "inhibiting
receptor"), which have similar
amino acid sequences that differ primarily in the cytoplasmic domains thereof
(see review M. in Daeron,
Annu. Rev. hnmunol. 15:203-234 (1997)). FcRs are reviewed in Ravetch and
Kinet, Annu. Rev.
lmmunol. 9:457-492 (1991); Capel et al., Immunomethods 4:25-34 (1994); and de
Haas et al., J. Lab.
Clin. Med. 126:330-41 (1995). Other FcRs, including those to be identified in
the future, are
encompassed by the term "FcR" herein. The term also includes the neonatal
receptor, FcRn, which is
responsible for the transfer of maternal IgGs to the fetus (Guyer et al., J.
Immunol. 117:587 (1976) and
Kim et al., J. Immunol. 24:249 (1994)). Antibody variants with improved or
diminished binding to FcRs
are described, for example, in WO 2000/42072, and U.S. Patent Nos. 7,183,387;
7,332,581; and
7.335,742. See also, e.g., Shields et al. J. Biol. Chem. 9(2):6591-6604
(2001).
[0115] "Complement dependent cytotoxicity" or "CDC" refers to the
lysis of a target cell in
the presence of complement. Activation of the classical complement pathway is
initiated by the binding
of the first component of the complement system (C1q) to antibodies (of the
appropriate subclass) which
are bound to their cognate antigen. To assess complement activation, a CDC
assay, e.g., as described in
Gazzano-Santoro et al., J. Immunol. Methods 202:163 (1996), may be performed.
Polypeptide variants
with altered Fc region amino acid sequences (polypeptides with a variant Fc
region) and increased or
decreased Clq binding capability are described, e.g., in US Patent No.
6,194,551 B1 and WO
1999/51642. See also, e.g., Idusogie et al. J. ImmunoL 164: 4178-4184 (2000).
[0116] The CD98 polypeptide "extracellular domain" or "ECD" refers
to a form of the
CD98 polypeptide that is essentially free of the transmembrane and cytoplasmic
domains. Ordinarily, a
CD98 polypeptide ECD will have less than 1% of such transmembrane and/or
cytoplasmic domains and
preferably, will have less than 0.5% of such domains. The transmembrane domain
of CD98 comprises
amino acid residues 76-103 (Parmacek et al., Nucleic Acids Res. 17: 1915-1931,
1989). The exact
boundaries of a transmembrane domain may vary but most likely by no more than
about 5 amino acids at
either end of the domain as initially identified. Optionally, therefore, an
extracellular domain of a CD98
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
polypeptide may comprise amino acids from about 98-108 to 529 of the sequence
of CD98 as disclosed
in Parmacek et al, supra..
[0117] "Percent (%) amino acid sequence identity" with respect to
a reference polypeptide
sequence is defined as the percentage of amino acid residues in a candidate
sequence that are identical
with the amino acid residues in the reference polypeptide sequence, after
aligning the sequences and
introducing gaps, if necessary, to achieve the maximum percent sequence
identity, and not considering
any conservative substitutions as part of the sequence identity. Alignment for
purposes of determining
percent amino acid sequence identity can be achieved in various ways that are
within the skill in the art,
for instance, using publicly available computer software such as BLAST, BLAST-
2, ALIGN or Megalign
(DNASTAR) software. Those skilled in the art can determine appropriate
parameters for aligning
sequences, including any algorithms needed to achieve maximal alignment over
the full length of the
sequences being compared.
[0118] A "modification" of an amino acid residue/position, as used
herein, refers to a
change of a primary amino acid sequence as compared to a starting amino acid
sequence, wherein the
change results from a sequence alteration involving said amino acid
residue/positions. For example,
typical modifications include substitution of the residue with another amino
acid (e.g., a conservative or
non-conservative substitution), insertion of one or more (generally fewer than
5 or 3) amino acids
adjacent to said residue/position, and deletion of said residue/position.
[0119] An "epitope" is the site on the surface of an antigen
molecule to which a single
antibody molecule binds. Generally an antigen has several or many different
epitopes and reacts with
many different antibodies. The term specifically includes linear epitopes and
conformational epitopes.
[0120] An antibody binds "essentially the same epitope" as a
reference antibody, when the
two antibodies recognize identical or sterically overlapping epitopes. The
most widely used and rapid
methods for determining whether two epitopes bind to identical or sterically
overlapping epitopes are
competition assays, which can be configured in all number of different
formats, using either labeled
antigen or labeled antibody. Usually, the antigen is immobilized on a 96-well
plate, and the ability of
unlabeled antibodies to block the binding of labeled antibodies is measured
using radioactive or enzyme
labels.
[0121] "Epitope mapping" is the process of identifying the binding
sites, or epitopes, of
antibodies on their target antigens. Antibody epitopes may be linear epitopes
or conformational epitopes.
Linear epitopes are formed by a continuous sequence of amino acids in a
protein. Conformational
epitopes are formed of amino acids that are discontinuous in the protein
sequence, but which are brought
together upon folding of the protein into its three-dimensional structure.
[0122] "Epitope binning", as defined herein, is the process of
grouping antibodies based on
the epitopes they recognize. More particularly, epitope binning comprises
methods and systems for
discriminating the epitope recognition properties of different antibodies,
combined with computational
processes for clustering antibodies based on their epitope recognition
properties and identifying
antibodies having distinct binding specificities.
26
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
[0123] A "disorder" is any condition or disease that would benefit
from treatment with an
substance/molecule or method of the invention. This includes chronic and acute
disorders including
those pathological conditions which predispose the mammal to the disorder in
question. Non-limiting
examples of disorders to be treated herein include cancerous conditions such
as bladder, breast, colon,
rectal, gastric, esophageal, lung, laryx, kidney, oral, ovarian, or prostate
cancer, or a sarcoma, melanoma,
glioma, lymphoma or leukemia, as well as metastases of these cancers.
[0124] The terms "cell proliferative disorder" and "proliferative
disorder" refer to disorders
that are associated with some degree of abnormal cell proliferation. In one
embodiment, the cell
proliferative disorder is cancer.
10125] "Tumor," as used herein, refers to all neoplastic cell growth and
proliferation,
whether malignant or benign, and all pre-cancerous and cancerous cells and
tissues. The terms "cancer,"
"cancerous," "cell proliferative disorder," "proliferative disorder" and
"tumor" are not mutually exclusive
as referred to herein.
10126] The terms "cancer" and "cancerous" refer to or describe the
physiological condition
in mammals that is typically characterized by unregulated cell growth.
Examples of cancer include, but
are not limited to, carcinoma, lymphoma, blastoma, sarcoma, and leukemia or
lymphoid malignancies.
More particular examples of such cancers include squamous cell cancer (e.g.
epithelial squamous cell
cancer), lung cancer including small-cell lung cancer, non-small cell lung
cancer, adenocarcinoma of the
lung and squamous carcinoma of the lung, cancer of the peritoneum,
hepatocellular cancer, gastric or
stomach cancer including gastrointestinal cancer, pancreatic cancer,
glioblastoma, cervical cancer,
ovarian cancer, oral cancer, liver cancer, bladder cancer, cancer of the
urinary tract, hepatoma, breast
cancer, colon cancer, rectal cancer, colorectal cancer, endometrial or uterine
carcinoma, salivary gland
carcinoma, kidney or renal cancer, prostate cancer, vulval cancer, thyroid
cancer, hepatic carcinoma, anal
carcinoma, penile carcinoma, melanoma, multiple myeloma and B-cell lymphoma,
brain cancer, as well
as head and neck cancer, and associated metastases.
10127] A "CD98-expressing cell" is a cell that expresses
endogenous or transfected CD98
on the cell surface. A "CD98-expressing cancer" is a cancer comprising cells
that have CD98 protein
present on the cell surface. A "CD98-expressing cancer" produces sufficient
levels of CD98 on the
surface of cells thereof, such that an anti-CD98 antibody can bind thereto and
have a therapeutic effect
with respect to the cancer. A cancer that "overexpresses" CD98 is one that has
significantly higher levels
of CD98 at the cell surface thereof, compared to a noncancerous cell of the
same tissue type. Such
overexpression may be caused by gene amplification or by increased
transcription or translation. CD98
overexpression may be determined in a diagnostic or prognostic assay by
evaluating increased levels of
the CD98 protein present on the surface of a cell (e.g. via an
immunohistochemistry assay; FACS
analysis). Alternatively, or additionally, one may measure levels of CD98-
encoding nucleic acid or
mRNA in the cell, e.g. via fluorescent in situ hybridization; (FISH; see
W098/45479 published October,
1998), Southern blotting, Northern blotting, or polymerase chain reaction
(PCR) techniques, such as real
time quantitative PCR (RT-PCR). Aside from the above assays, various in vivo
assays are available to
27
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
the skilled practitioner. For example, one may expose cells within the body of
the patient to an antibody
which is optionally labeled with a detectable label, e.g. a radioactive
isotope, and binding of the antibody
to cells in the patient can be evaluated, e.g. by external scanning for
radioactivity or by analyzing a
biopsy taken from a patient previously exposed to the antibody. A CD98-
expressing cancer includes, but
[0128] As used herein, "treatment" (and variations such as "treat"
or "treating") refers to
clinical intervention in an attempt to alter the natural course of the
individual or cell being treated, and
[0129] The above parameters for assessing successful treatment and
improvement in the
disease are readily measurable by routine procedures familiar to a physician.
For cancer therapy, efficacy
can be measured, for example, by assessing the time to disease progression
(TTP) and/or by determining
the response rate (RR). Other endpoints for measuring efficacy include, for
example, overall survival
[0130] An "individual" is a vertebrate. In certain embodiments,
the vertebrate is a mammal.
Mammals include, but are not limited to, farm animals (such as cows), sport
animals, pets (such as cats,
dogs, and horses), primates, mice and rats. In certain embodiments, a mammal
is a human.
[0131] An "effective amount" refers to an amount effective, at
dosages and for periods of time
28
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
effective amount of the drug may, for example, reduce the number of cancer
cells; reduce the tumor size;
inhibit (i.e., slow to some extent and preferably stop) cancer cell
infiltration into peripheral organs;
inhibit (i.e., slow to some extent and preferably stop) tumor metastasis;
inhibit, to some extent, tumor
growth; and/or relieve to some extent one or more of the symptoms associated
with the cancer. See
preceding definition of "treating". To the extent the drug may prevent growth
and/or kill existing cancer
cells, it may be cytostatic and/or cytotoxic.
[0132] "Chronic" administration refers to administration of the
agent(s) in a continuous mode
as opposed to an acute mode, so as to maintain the initial therapeutic effect
(activity) for an extended
period of time. "Intermittent" administration is treatment that is not
consecutively done without
interruption, but rather is cyclic in nature.
[0133] Administration "in combination with" one or more further
therapeutic agents includes
simultaneous (concurrent) and consecutive administration in any order.
[0134] "Carriers" as used herein include pharmaceutically
acceptable carriers, excipients, or
stabilizers that are nontoxic to the cell or mammal being exposed thereto at
the dosages and
concentrations employed. Often the physiologically acceptable carrier is an
aqueous pH buffered
solution. Examples of physiologically acceptable carriers include buffers such
as phosphate, citrate, and
other organic acids; antioxidants including ascorbic acid; low molecular
weight (less than about 10
residues) polypeptide; proteins, such as serum albumin, gelatin, or
immunoglobulins; hydrophilic
polymers such as polyvinylpyrrolidone; amino acids such as glycine, glutamine,
asparagine, arginine or
lysine; monosaccharides, disaccharides, and other carbohydrates including
glucose, mannose, or dextrins;
chelating agents such as EDTA; sugar alcohols such as mannitol or sorbitol;
salt-forming counterions
such as sodium; and/or nonionic surfactants such as TWEENTm, polyethylene
glycol (PEG), and
PEURONICSTM.
[0135] The term "pharmaceutical formulation" refers to a
preparation which is in such form as
to permit the biological activity of the active ingredient to be effective,
and which contains no additional
components which are unacceptably toxic to a subject to which the formulation
would be administered.
Such formulation may be sterile.
[0136] A "sterile" formulation is aseptic of free from all living
microorganisms and their
spores. An "effective amount" of an antibody as disclosed herein is an amount
sufficient to carry out a
specifically stated purpose. An "effective amount" may be determined
empirically and in a routine
manner, in relation to the stated purpose.
[0137] The term "therapeutically effective amount" refers to an
amount of an antibody or
other drug effective to "treat" a disease or disorder in a subject or mammal.
In the case of cancer, the
therapeutically effective amount of the drug may reduce the number of cancer
cells; reduce the tumor
size; inhibit (i.e., slow to some extent and preferably stop) cancer cell
infiltration into peripheral organs;
inhibit (i.e., slow to some extent and preferably stop) tumor metastasis;
inhibit, to some extent, tumor
growth; and/or relieve to some extent one or more of the symptoms associated
with the cancer. See the
definition herein of "treating". To the extent the drug may prevent growth
and/or kill existing cancer
29
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
cells, it may be cytostatic and/or cytotoxic. A "prophylactically effective
amount" refers to an amount
effective, at dosages and for periods of time necessary, to achieve the
desired prophylactic result.
Typically but not necessarily, since a prophylactic dose is used in subjects
prior to or at an earlier stage of
disease, the prophylactically effective amount will be less than the
therapeutically effective amount.
[0138] The term "cytotoxic agent" as used herein refers to a substance that
inhibits or prevents
the function of cells and/or causes destruction of cells. The term is intended
to include radioactive
isotopes (e.g., At2i /131, /125, y90, Re186, Re188, smI53, Bi212,
P32 and radioactive isotopes of Lu),
chemotherapeutic agents e.g. methotrexate, adriamicin, vinca alkaloids
(vincristine, vinblastine,
etoposide), doxorubicin, melphalan, mitomycin C, chlorambucil, daunorubicin or
other intercalating
agents, enzymes and fragments thereof such as nucleolytic enzymes,
antibiotics, and toxins such as small
molecule toxins or enzymatically active toxins of bacterial, fungal, plant or
animal origin, including
fragments and/or variants thereof, and the various antitumor or anticancer
agents disclosed below. Other
cytotoxic agents are described below. A tumoricidal agent causes destruction
of tumor cells.
[0139] A "toxin" is any substance capable of having a detrimental
effect on the growth or
proliferation of a cell.
[0140] A "chemotherapeutic agent" is a chemical compound useful in
the treatment of cancer,
regardless of mechanism of action. Chemotherapeutic agents include compounds
used in "targeted
therapy" and conventional chemotherapy. Examples of chemotherapeutic agents
include, but are not
limited to, alkylating agents such as thiotepa and CYTOXAN cyclosphosphamide;
alkyl sulfonates
such as busulfan, improsulfan and piposulfan; aziridines such as benzodopa,
carboquone, meturedopa,
and uredopa; ethylenimines and methylamelamines including altretamine,
triethylenemelamine,
trietylenephosphoramide, triethiylenethiophosphoramide and
trimethylolomelamine; acetogen ins
(especially bullatacin and bullatacinone); delta-9-tetrahydrocannabinol
(dronabinol, MARINOLO); beta-
lapachone; lapachol; colchicines; betulinic acid; a camptothecin (including
the synthetic analogue
topotecan (HYCAMTINO), CPT-11 (irinotecan, CAMPTOSAR8), acetylcamptothecin,
scopolectin, and
9-aminocamptothecin); bryostatin; callystatin; CC-1065 (including its
adozelesin, carzelesin and
bizelesin synthetic analogues); podophyllotoxin; podophyllinic acid;
teniposide; cryptophycins
(particularly cryptophycin 1 and cryptophycin 8); dolastatin; duocarmycin
(including the synthetic
analogues, KW-2189 and CB1-TM1); eleutherobin; pancratistatin; a sarcodictyin;
spongistatin; nitrogen
mustards such as chlorambucil, chlornaphazine, cholophosphamide. estramustine,
ifosfamide,
mechlorethamine, mechlorethamine oxide hydrochloride, melphalan, novembichin,
phenesterine,
prednimustine, trofosfamide, uracil mustard; nitrosureas such as carmustine,
chlorozotocin, fotemustine,
lomustine, nimustine, and ranimnustine; antibiotics such as the enediyne
antibiotics (e.g., calicheamicin,
especially calicheamicin gammal I and calicheamicin omegaIl (see, e.g., Agnew,
Chem Intl. Ed. Engl.,
33: 183-186 (1994)); dynemicin, including dynemicin A; an esperamicin; as well
as neocarzinostatin
chromophore and related chromoprotein enediyne antiobiotic chromophores),
aclacinomysins,
actinomycin, authramycin, azaserine, bleomycins, cactinomycin, carabicin,
caminomycin, carzinophilin,
chromomycinis, dactinomycin, daunorubicin, detorubicin, 6-diazo-5-oxo-L-
norleucine,
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
ADRIAMYCIN , doxorubicin (including morpholino-doxorubicin, cyanomorpholino-
doxorubicin, 2-
pyrrolino-doxorubicin and deoxydoxorubicin), epirubicin, esorubicin,
idarubicin, marcellomycin,
mitomycins such as mitomycin C, mycophenolic acid, nogalamycin, olivomycins,
peplomycin,
potfiromycin, puromycin, quelamycin, rodorubicin, streptonigrin, streptozoc
in, tubercidin, ubenimex,
zinostatin, zorubicin; anti-metabolites such as methotrexate and 5-
fluorouracil (5-FU); folic acid
analogues such as denopterin, methotrexate, pteropterin, trimetrexate; purine
analogs such as fludarabine,
6-mercaptopurine, thiamiprine, thioguanine; pyrimidine analogs such as
ancitabine, azacitidine, 6-
azauridine, carmofur, cytarabine, dideoxyuridine, doxifluridine, enocitabine,
floxuridine; androgens such
as calusterone, dromostanolone propionate, epitiostanol, mepitiostane,
testolactone; anti-adrenals such as
aminoglutethimide, mitotane, trilostane; folic acid replenisher such as
frolinic acid; aceglatone;
aldophosphamide glycoside; aminolevulinic acid; eniluracil; amsacrine;
bestrabucil; bisantrene;
edatraxate; defofamine; demecolcine; diaziquone; elfornithine; elliptinium
acetate; an epothilone;
etoglucid; gallium nitrate; hydroxyurea; lentinan; lonidainine; maytansinoids
such as maytansine and
ansamitocins; mitoguazone; mitoxantrone; mopidanmol; nitraerine; pentostatin;
phenamet; pirarubicin;
losoxantrone; 2-ethylhydrazide; procarbazine; PSK polysaccharide complex (JHS
Natural Products,
Eugene, Oreg.); razoxane; rhizoxin; sizofuran; spirogermanium; tenuazonic
acid; triaziquone; 2,2',2"-
trichlorotriethylamine; trichothecenes (especially T-2 toxin, verracurin A,
roridin A and anguidine);
urethan; vindesine (ELDISINE , FILDESIN ); dacarbazine; mannomustine;
mitobronitol; mitolactol;
pipobroman; gacytosine; arabinoside ("Ara-C"); thiotepa; taxoids, e.g., TAXOL
paclitaxel (Bristol-
Myers Squibb Oncology, Princeton, N.J.), ABRAXANETM Cremophor-free, albumin-
engineered
nanoparticle formulation of paclitaxel (American Pharmaceutical Partners,
Schaumberg, Ill.), and
TAXOTERE doxetaxel (Rhone-Poulenc Rorer, Antony, France); chloranbucil;
gemcitabine
(GEMZAR ); 6-thioguanine; mercaptopurine; methotrexate; platinum analogs such
as cisplatin and
carboplatin; vinblastine (VELBAN ); platinum; etoposide (VP-16); ifosfamide;
mitoxantrone;
vincristine (ONCOVIN ); oxaliplatin; leucovovin; vinorelbine (NAVELBINE );
novantrone;
edatrexate; daunomycin; aminopterin; ibandronate; topoisomerase inhibitor RFS
2000;
difluoromethylornithine (DMF0); retinoids such as retinoic acid; capecitabine
(XELODAR);
pharmaceutically acceptable salts, acids or derivatives of any of the above;
as well as combinations of
two or more of the above such as CHOP, an abbreviation for a combined therapy
of cyclophosphamide,
doxorubicin, vincristine, and prednisolone, and FOLFOX, an abbreviation for a
treatment regimen with
oxaliplatin (ELOXATINTm) combined with 5-FU and leucovovin. Additional
chemotherapeutic agents
include cytotoxic agents useful as antibody drug conjugates, such as
maytansinoids (DM1 and DM4, for
example) and auristatins (MMAE and MMAF, for example).
101411 Also included in the definition of "chemotherapeutic agent"
are: (i) anti-hormonal
agents that act to regulate or inhibit hormone action on tumors such as anti-
estrogens and selective
estrogen receptor modulators (SERMs), including, for example, tamoxifen
(including NOLVADEX ;
tamoxifen citrate), raloxifene, droloxifene, 4-hydroxytamoxifen, trioxifene,
keoxifene, LY117018,
onapristone, and FARESTON (toremifine citrate); (ii) aromatase inhibitors
that inhibit the enzyme
31
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
aromatase, which regulates estrogen production in the adrenal glands, such as,
for example, 4(5)-
imidazoles, aminoglutethimide, MEGASE (megestrol acetate), AROMASIN
(exemestane; Pfizer),
formestanie, fadrozole, RIVISOR (vorozole), FEMARA (letrozole; Novartis),
and ARIMIDEX
(anastrozole; AstraZeneca); (iii) anti-androgens such as flutamide,
nilutamide, bicalutamide, leuprolide,
and goserelin; as well as troxacitabine (a 1,3-dioxolane nucleoside cytosine
analog); (iv) protein kinase
inhibitors such as MEK inhibitors (WO 2007/044515); (v) lipid kinase
inhibitors; (vi) antisense
oligonucleotides, particularly those which inhibit expression of genes in
signaling pathways implicated in
aberrant cell proliferation, for example, PKC-alpha, Raf and H-Ras, such as
oblimersen
(GENASENSER), Genta Inc.); (vii) ribozymes such as VEGF expression inhibitors
(e.g.,
ANGIOZYMEO) and HERZ expression inhibitors; (viii) vaccines such as gene
therapy vaccines, for
example, ALLOVECTIN , LEUVECTIN , and VAXIDO; PROLEUKIN rIL-2; topoisomerase
1
inhibitors such as LURTOTECANO; ABARELIXO rmRH; (ix) anti-angiogenic agents
such as
bevacizumab (AVASTIN , Genentech); and pharmaceutically acceptable salts,
acids and derivatives of
any of the above.
[0142] The term "prodrug" as used in this application refers to a precursor
or derivative form
of a compound of the invention that may be less cytotoxic to cells compared to
the parent compound or
drug and is capable of being enzymatically or hydrolytically activated or
converted into the more active
parent form. See, e.g., Wilman, "Prodrugs in Cancer Chemotherapy" Biochemical
Society Transactions,
14, pp. 375-382, 615th Meeting Belfast (1986) and Stella et al., "Prodrugs: A
Chemical Approach to
Targeted Drug Delivery," Directed Drug Delivery, Borchardt et al., (ed.), pp.
247-267, Humana Press
(1985). The prodrugs of this invention include, but are not limited to,
phosphate-containing prodrugs,
thiophosphate-containing prodrugs, sulfate-containing prodrugs, peptide-
containing prodrugs, D-amino
acid-modified prodrugs, glycosylated prodrugs, 13-lactam-containing prodrugs,
optionally substituted
phenoxyacetamide-containing prodrugs, optionally substituted phenylacetamide-
containing prodrugs, 5-
fluorocytosine and other 5-fluorouridine prodrugs which can be converted into
the more active cytotoxic
free drug. Examples of cytotoxic drugs that can be derivatized into a prodrug
form for use in this
invention include, but are not limited to, compounds of the invention and
chemotherapeutic agents such
as described above.
[0143] A "small molecule" is defined herein to have a molecular
weight below about 500
Daltons.
[0144] An "isolated nucleic acid" is a nucleic acid, e.g., an RNA,
DNA, or a mixed polymer,
which is substantially separated from other genome DNA sequences as well as
proteins or complexes
such as ribosomes and polymerases, which naturally accompany a native
sequence. The term embraces a
nucleic acid sequence that has been removed from its naturally occurring
environment, and includes
recombinant or cloned DNA isolates and chemically synthesized analogues or
analogues biologically
synthesized by heterologous systems. A substantially pure molecule includes
isolated forms of the
molecule.
32
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
[0145] "Polynucleotide," or "nucleic acid," as used interchangeably
herein, refer to polymers
of nucleotides of any length, and include DNA and RNA. The nucleotides can be
deoxyribonucleotides,
ribonucleotides, modified nucleotides or bases, and/or their analogs, or any
substrate that can be
incorporated into a polymer by DNA or RNA polymerase or by a synthetic
reaction. A polynucleotide
may comprise modified nucleotides, such as methylated nucleotides and their
analogs.
"Oligonucleotide," as used herein, generally refers to short, generally single-
stranded, generally synthetic
polynucleotides that are generally, but not necessarily, less than about 200
nucleotides in length. The
terms "oligonucleotide" and "polynucleotide" are not mutually exclusive. The
description above for
polynucleotides is equally and fully applicable to oligonucleotides.
[0146] The cell that produces an anti-CD98 antibody of the invention will
include the parent
hybridoma cell e.g., the hybridomas that are deposited with the ATCC, as well
as bacterial and
eukaryotic host cells into which nucleic acid encoding the antibodies have
been introduced. Suitable host
cells are disclosed below.
[0147] The term "package insert" is used to refer to instructions
customarily included in
commercial packages of therapeutic products, that contain information about
the indications, usage,
dosage, administration, contraindications and/or warnings concerning the use
of such therapeutic
products.
COMPOSITIONS AND METHODS OF MAKING THE SAME
[0148] Antibodies that bind to CD98 are provided. Immunoconjugates
comprising anti-CD98
antibodies are provided. Antibodies and immunoconjugates of the invention are
useful, e.g., for the
diagnosis or treatment of disorders associated with altered expression, e.g.,
increased expression, of
CD98. In certain embodiments, antibodies or immunoconjugates of the invention
are useful for the
diagnosis or treatment of a cell proliferative disorder, such as cancer.
Anti-CD98 Antibodies
[0149] In one embodiment, the present invention provides anti-CD98
antibodies that may find
use herein as therapeutic agents. Exemplary antibodies include polyclonal,
monoclonal, humanized,
human, bispecific, and heteroconjugate antibodies, as well as variants thereof
having improved affinity or
other properties.
I. Polyclonal Antibodies
[0150] The antibodies of the invention may comprise polyclonal
antibodies. Methods of
preparing polyclonal antibodies are known to the skilled artisan. Polyclonal
antibodies can be raised in a
mammal, for example, by one or more injections of an immunizing agent and, if
desired, an adjuvant.
Typically, the immunizing agent and/or adjuvant will be injected in the mammal
by multiple
subcutaneous or intraperitoneal injections. The immunizing agent may include
the CD98 polypeptide or
a fusion protein thereof. It may be useful to conjugate the immunizing agent
to a protein known to be
immunogenic in the mammal being immunized. Examples of such immunogenic
proteins include but are
33
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
not limited to keyhole limpet hemocyan in, serum albumin, bovine
thyroglobulin, and soybean trypsin
inhibitor. Examples of adjuvants which may be employed include Freund's
complete adjuvant and MPL-
TDM adjuvant (monophosphoryl Lipid A, synthetic trehalose dicorynomycolate).
The immunization
protocol may be selected by one skilled in the art without undue
experimentation. The mammal can then
be bled, and the serum assayed for CD98 antibody titer. If desired, the mammal
can be boosted until the
antibody titer increases or plateaus.
2. Monoclonal Antibodies
[0151] The antibodies of the invention may alternatively be
monoclonal antibodies. Monoclonal
antibodies may be made using the hybridoma method first described by Kohler et
al., Nature, 256:495
(1975), or may be made by recombinant DNA methods (see, e.g., U.S. Patent No.
4,816,567).
[0152] In the hybridoma method, a mouse or other appropriate host
animal, such as a hamster,
is immunized as described above to elicit lymphocytes that produce or are
capable of producing
antibodies that will specifically bind to the protein used for immunization.
Alternatively, lymphocytes
may be immunized in vitro. After immunization, lymphocytes are isolated and
then fused with a
myeloma cell line using a suitable fusing agent, such as polyethylene glycol,
to form a hybridoma cell
(Goding, Monoclonal Antibodies: Principles and Practice, pp.59-103 (Academic
Press, 1986)).
[0153] The hybridoma cells thus prepared are seeded and grown in a
suitable culture medium
which medium preferably contains one or more substances that inhibit the
growth or survival of the
unfused, parental myeloma cells (also referred to as fusion partner). For
example, if the parental
myeloma cells lack the enzyme hypoxanthine guanine phosphoribosyl transferase
(HGPRT or HPRT),
the selective culture medium for the hybridomas typically will include
hypoxanthine, aminopterin, and
thymidine (HAT medium), which substances prevent the growth of HGPRT-deficient
cells.
[0154] Preferred fusion partner myeloma cells are those that fuse
efficiently, support stable
high-level production of antibody by the selected antibody-producing cells,
and are sensitive to a
selective medium that selects against the unfused parental cells. Preferred
myeloma cell lines are murine
myeloma lines, such as those derived from MOPC-21 and MPC-11 mouse tumors
available from the Salk
Institute Cell Distribution Center, San Diego, California USA, and SP-2 and
derivatives e.g., X63-Ag8-
653 cells available from the American Type Culture Collection, Manassas,
Virginia, USA. Human
myeloma and mouse-human heteromyeloma cell lines also have been described for
the production of
human monoclonal antibodies (Kozbor, J. Immunol., 133:3001 (1984); and Brodeur
et al., Monoclonal
Antibody Production Techniques and Applications, pp. 51-63 (Marcel Dekker,
Inc., New York, 1987)).
[0155] Culture medium in which hybridoma cells are growing is
assayed for production of
monoclonal antibodies directed against the antigen. Preferably, the binding
specificity of monoclonal
antibodies produced by hybridoma cells is determined by immunoprecipitation or
by an in vitro binding
assay, such as radioimmunoassay (RIA) or enzyme-linked immunosorbent assay
(ELISA). The binding
affinity of the monoclonal antibody can, for example, be determined by the
Scatchard analysis described
in Munson et al., Anal. Biochem., 107:220 (1980).
34
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
[0156] Once hybridoma cells that produce antibodies of the desired
specificity, affinity, and/or
activity are identified, the clones may be subcloned by limiting dilution
procedures and grown by
standard methods (Goding, Monoclonal Antibodies: Principles and Practice,
pp.59-103 (Academic Press,
1986)). Suitable culture media for this purpose include, for example, D-MEM or
RPMI-1640 medium.
[0157] The monoclonal antibodies secreted by the subclones are
suitably separated from the
culture medium, ascites fluid, or serum by conventional antibody purification
procedures such as, for
example, affinity chromatography (e.g., using protein A or protein G-
Sepharose) or ion-exchange
[0158] DNA encoding the monoclonal antibodies is readily isolated
and sequenced using
conventional procedures (e.g., by using oligonucleotide probes that are
capable of binding specifically to
genes encoding the heavy and light chains of murine antibodies). The hybridoma
cells serve as a
preferred source of such DNA. Once isolated, the DNA may be placed into
expression vectors, which
20 [0159] In a further embodiment, monoclonal antibodies or antibody
fragments can be isolated
from antibody phage libraries generated using the techniques described in,
e.g, Antibody Phage Display:
Methods and Protocols, P.M. O'Brien and R. Aitken, eds, Humana Press, Totawa
N.J., 2002. In
principle, synthetic antibody clones are selected by screening phage libraries
containing phage that
display various fragments of antibody variable region (Fv) fused to phage coat
protein. Such phage
[0160] Variable domains can be displayed functionally on phage,
either as single-chain Fv
[0161] Repertoires of VH and VL genes can be separately cloned by polymerase
chain
reaction (PCR) and recombined randomly in phage libraries, which can then be
searched for antigen-
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
734 (1993). Finally, naive libraries can also be made synthetically by cloning
the unrearranged V-gene
segments from stem cells, and using PCR primers containing random sequence to
encode the highly
variable CDR3 regions and to accomplish rearrangement in vitro as described by
Hoogenboom and
Winter, J. Mol. Biol., 227: 381-388 (1992).
[0162] Screening of the libraries can be accomplished by various techniques
known in the art.
For example, CD98 can be used to coat the wells of adsorption plates,
expressed on host cells affixed to
adsorption plates or used in cell sorting, or conjugated to biotin for capture
with streptavidin-coated
beads, or used in any other method for panning display libraries. The
selection of antibodies with slow
dissociation kinetics (and good binding affinities) can be promoted by use of
long washes and
monovalent phage display as described in Bass et al., Proteins, 8: 309-314
(1990) and in WO 92/09690,
and a low coating density of antigen as described in Marks et al.,
Biotechnol., 10: 779-783 (1992).
[0163] Any of the anti-CD98 antibodies of the invention can be
obtained by designing a
suitable antigen screening procedure to select for the phage clone of interest
followed by construction of
a full length anti-CD98 antibody clone using the Fv sequences from the phage
clone of interest and
suitable constant region (Fc) sequences described in Kabat et al., Sequences
of Proteins of
Immunological Interest, Fifth Edition, NIH Publication 91-3242, Bethesda MD
(1991), vols. 1-3.
3. Antibody Fragments
[0164] The present invention encompasses antibody fragments. In
certain circumstances there
are advantages of using antibody fragments, rather than whole antibodies. The
smaller size of the
fragments allows for rapid clearance, and may lead to improved access to solid
tumors. For a review of
certain antibody fragments, see Hudson et al. (2003) Nat. Med. 9:129-134.
[0165] Various techniques have been developed for the production of
antibody fragments.
Traditionally, these fragments were derived via proteolytic digestion of
intact antibodies (see, e.g.,
Morimoto et al., Journal of Biochemical and Biophysical Methods 24:107-117
(1992); and Brennan et
al., Science, 229:81 (1985)). However, these fragments can now be produced
directly by recombinant
host cells. Fab, Fv and ScEv antibody fragments can all be expressed in and
secreted from E. coli or yeast
cells, thus allowing the facile production of large amounts of these
fragments. Antibody fragments can be
isolated from the antibody phage libraries discussed above. Alternatively,
Fab'-SH fragments can be
directly recovered from E. coli and chemically coupled to form
F(abt)2fragments (Carter et al.,
Bio/Technology 10:163-167 (1992)). According to another approach, F(ab')2
fragments can be isolated
directly from recombinant host cell culture. Fab and F(a1302 fragment with
increased in vivo half-life
comprising salvage receptor binding epitope residues are described in U.S.
Pat. No. 5,869,046. Other
techniques for the production of antibody fragments will be apparent to the
skilled practitioner. In certain
embodiments, an antibody is a single chain Fv fragment (scFv). See WO
93/16185; U.S. Pat. Nos.
5,571,894; and 5,587,458. Fv and scFv are the only species with intact
combining sites that are devoid of
constant regions; thus, they may be suitable for reduced nonspecific binding
during in vivo use. scFv
fusion proteins may be constructed to yield fusion of an effector protein at
either the amino or the
carboxy terminus of an scFv. See Antibody Engineering, ed. Borrebaeck, supra.
The antibody fragment
36
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
may also be a "linear antibody", e.g., as described for example, in the
references cited before. Such linear
antibodies may be monospecific or multi-specific, such as bispecific.
[0166] The smallest antibody-derived binding structures are the
separate variable domains (V
domains) also termed single variable domain antibodies (SdAbs). Certain types
of organisms, the
camelids and cartilaginous fish, possess high affinity single V-like domains
mounted on an Fc equivalent
domain structure as part of their immune system. (Woolven et al.,
Immunogenetics 50: 98-101, 1999;
Streltsov et al., Proc Natl Acad Sci USA. 101:12444-12449, 2004). The V-like
domains (called VhH in
camelids and V-NAR in sharks) typically display long surface loops, which
allow penetration of cavities
of target antigens. They also stabilize isolated VH domains by masking
hydrophobic surface patches.
These VhH and V-NAR domains have been used to engineer sdAbs. Human V domain
variants have
been designed using selection from phage libraries and other approaches that
have resulted in stable, high
binding VL- and VH-derived domains.
4. Humanized Antibodies
[0167] The invention encompasses humanized antibodies. Various methods for
humanizing
non-human antibodies are known in the art. For example, a humanized antibody
can have one or more
amino acid residues introduced into it from a source that is non-human. These
non-human amino acid
residues are often referred to as "import" residues, which are typically taken
from an "import" variable
domain. Humanization can be essentially performed following the method of
Winter and co-workers
(Jones et al. (1986) Nature 321:522-525; Riechmann et al. (1988) Nature
332:323-327; Verhoeyen et al.
(1988) Science 239:1534-1536), by substituting hypervariable region sequences
for the corresponding
sequences of a human antibody.
[0168] In some cases, the humanized antibodies are constructed by
CDR grafting, in which
the amino acid sequences of the six complementarity determining regions (CDRs)
of the parent rodent
antibody are grafted onto a human antibody framework. Padlan et al. (FASEB J.
9:133-139, 1995)
determined that only about one third of the residues in the CDRs actually
contact the antigen, and termed
these the "specificity determining residues," or SDRs. In the technique of SDR
grafting, only the SDR
residues are grafted onto the human antibody framework (Kashmiri et al.,
Methods 36: 25-34, 2005).
[0169] The choice of human variable domains, both light and heavy,
to be used in making the
humanized antibodies can be important to reduce antigenicity. According to the
so-called "best-fit"
method, the sequence of the variable domain of a rodent antibody is screened
against the entire library of
known human variable-domain sequences. The human sequence which is closest to
that of the rodent is
then accepted as the human framework for the humanized antibody (Sims et al.
(1993)J. linntunoL
151:2296; Chothia et al. (1987) J. MoL Biol. 196:901. Another method uses a
particular framework
derived from the consensus sequence of all human antibodies of a particular
subgroup of light or heavy
chains. The same framework may be used for several different humanized
antibodies (Carter et al.
(1992) Proc. Natl. Acad. Sci. USA, 89:4285; Presta et al. (1993)J. ImmunoL,
151:2623. In some cases,
the framework is derived from the consensus sequences of the most abundant
human subclasses, Voc.
37
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
subgroup I (VocI) and VH subgroup III (VIII). In another method, human
germline genes are used
at the source of the framework regions.
[0170] In an alternative paradigm based on comparison of CDRs,
called Superhumanization,
FR homology is irrelevant. The method consists of comparison of the non-human
sequence with the
functional human germline gene repertoire. Those genes encoding the same or
closely related canonical
structures to the murine sequences are then selected. Next, within the genes
sharing the canonical
structures with the non-human antibody, those with highest homology within the
CDRs are chosen as FR
donors. Finally, the non-human CDRs are grafted onto these FRs. (Tan et al.,
J. Immunol. 169: I 119-
1125, 2002).
[0171] It is further generally desirable that antibodies be humanized with
retention of high
affinity for the antigen and other favorable biological properties. To achieve
this goal, according to one
method, humanized antibodies are prepared by a process of analysis of the
parental sequences and
various conceptual humanized products using three-dimensional models of the
parental and humanized
sequences. Three-dimensional immunoglobulin models are commonly available and
are familiar to those
skilled in the art. Computer programs are available which illustrate and
display probable three-
dimensional conformational structures of selected candidate immunoglobulin
sequences. These include,
for example, WAM (Whitelegg and Rees, Protein Eng. 13: 819-824, 2000),
Modeller (Sali and Blundell,
Mol. Biol. 234: 779-815, 1993), and Swiss PDB Viewer (Guex and Peitsch,
Electrophoresis 18: 2714-
2713, 1997). Inspection of these displays permits analysis of the likely role
of the residues in the
functioning of the candidate immunoglobulin sequence, L e., the analysis of
residues that influence the
ability of the candidate immunoglobulin to bind its antigen. In this way, FR
residues can be selected and
combined from the recipient and import sequences so that the desired antibody
characteristic, such as
increased affinity for the target antigen(s), is achieved. In general, the
hypervariable region residues are
directly and most substantially involved in influencing antigen binding.
[0172] Another method for antibody humanization is based on a metric of
antibody
humanness termed Human String Content (HSC). This method compares the mouse
sequence with the
repertoire of human germline genes and the differences are scored as HSC. The
target sequence is then
humanized by maximizing its HSC rather than using a global identity measure to
generate multiple
diverse humanized variants. (Lazar et al., Mol. Immunol. 44: 1986-1998, 2007).
[0173] In contrast to the methods described above, empirical methods may be
used to generate
and select humanized antibodies. These methods are based upon the generation
of large libraries of
humanized variants and selection of the best clones using enrichment
technologies or high throughput
screening techniques. Antibody variants may be isolated from phage, ribosome
and yeast display
libraries as well as by bacterial colony screening. (Hoogenboom, Nat.
Biotechnol. 23: 1105-1116, 2005;
Dufner et al., Trends Biotechnol. 24: 523-529, 2006; Feldhaus et al., Nat.
Biotechnol. 21: 163-70, 2003;
Schlapschy et al., Protein Eng. Des. Sel. 17: 847-60, 2004).
[0174] In the FR library approach, a collection of residue variants
are introduced at specific
positions in the FR followed by selection of the library to select the FR that
best supports the grafted
38
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
CDR. The residues to be substituted may include some or all of the "Vernier"
residues identified as
potentially contributing to CDR structure (Foote and Winter, J. Mol. Biol.
224: 487-499, 1992), or from
the more limited set of target residues identied by Baca et al. (J. Biol.
Chem. 272: 10678-10684, 1997).
[0175] In FR shuffling, whole FRs are combined with the non-human CDRs instead
of
creating combinatorial libraries of selected residue variants. (Dal l'Acqua et
al., Methods 36: 43-60,
2005). The libraries may be screened for binding in a two-step selection
process, first humanizing VL,
followed by V14. Alternatively, a one-step FR shuffling process may be used.
Such a process has been
shown to be more efficient than the two-step screening, as the resulting
antibodies exhibited improved
biochemical and physico-chemical properties including enhanced expression,
increased affinity and
thermal stability (Damschroder et al, Mol. Immunol. 44: 3049-60, 2007)
[0176] The "hurnaneering" method is based on experimental
identification of essential
minimum specificity determinants (MSDs) and is based on sequential replacement
of non-human
fragments into libraries of human FRs and assessment of binding. It begins
with regions of the CDR-3 of
non-human VH and VL chains and progressively replaces other regions of the non-
human antibody into
the human FRs, including the CDR-1 and CDR-2 of both VH and VL. This
methodology typically results
in epitope retention and identification of antibodies from multiple sub-
classes with distinct human V-
segment CDRs. Humaneering allows for isolation of antibodies that are 91-96 %
homologous to human
germline gene antibodies. (Alfenito, Cambridge Healthtech Institute's Third
Annual PEGS, The Protein
Engineering Summit, 2007).
5. Human Antibodies
[0177] Human anti-CD98 antibodies of the invention can be constructed by
combining Fv
clone variable domain sequence(s) selected from human-derived phage display
libraries with known
human constant domain sequences(s) as described above. Alternatively, human
monoclonal anti-CD98
antibodies of the invention can be made by the hybridoma method. Human myeloma
and mouse-human
heteromyeloma cell lines for the production of human monoclonal antibodies
have been described, for
example, by Kozbor J. Immunol., 133: 3001 (1984); Brodeur et al., Monoclonal
Antibody Production
Techniques and Applications, pp. 51-63 (Marcel Dekker, Inc., New York, 1987);
and Boemer et al., J.
Immunol., 147: 86 (1991).
[0178] It is also possible to produce transgenic animals (e.g. mice)
that are capable, upon
immunization, of producing a full repertoire of human antibodies in the
absence of endogenous
immunoglobul in production. Transgenic mice that express human antibody
repertoires have been used to
generate high-affinity human sequence monoclonal antibodies against a wide
variety of potential drug
targets. See, e.g., Jakobovits, A., Curr. Opin. Biotechnol. 1995, 6(5):561-6;
Brtiggemann and Taussing,
Curr. Opin. Biotechnol. 1997, 8(4):455-8; U.S. Pat. Nos. 6,075,181 and
6,150,584; and Lonberg et al.,
Nature Biotechnol. 23: 1117-1125, 2005).
[0179] Alternatively, the human antibody may be prepared via immortalization
of human B
lymphocytes producing an antibody directed against a target antigen (such B
lymphocytes may be
recovered from an individual or may have been immunized in vitro). See, e.g.,
Cole et al., Monoclonal
39
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
Antibodies and Cancer Therapy, Alan R. Liss, p. 77 (1985); Boerner et al., J.
Immunol., 147 (1):86-95
(1991); and US Pat No. 5,750,373.
[01801 Gene shuffling can also be used to derive human antibodies from non-
human, e.g.
rodent, antibodies, where the human antibody has similar affinities and
specificities to the starting non-
human antibody. According to this method, which is also called "epitope
imprinting" or "guided
selection", either the heavy or light chain variable region of a non-human
antibody fragment obtained by
phage display techniques as described herein is replaced with a repertoire of
human V domain genes,
creating a population of non-human chain/human chain scFv or Fab chimeras.
Selection with antigen
results in isolation of a non-human chain/human chain chimeric scFv or Fab
wherein the human chain
restores the antigen binding site destroyed upon removal of the corresponding
non-human chain in the
primary phage display clone, i.e. the epitope guides (imprints) the choice of
the human chain partner.
When the process is repeated in order to replace the remaining non-human
chain, a human antibody is
obtained (see PCT WO 93/06213; and Osbourn et al, Methods., 36, 61-68, 2005).
Unlike traditional
humanization of non-human antibodies by CDR grafting, this technique provides
completely human
antibodies, which have no FR or CDR residues of non-human origin. Examples of
guided selection to
humanize mouse antibodies towards cell surface antigens include the folate-
binding protein present on
ovarian cancer cells (Figini et al., Cancer Res., 58, 991-996, 1998) and
CD147, which is highly expressed
on hepatocellular carcinoma (Bao et al., Cancer Biol. Then, 4, 1374-1380,
2005).
101811 A potential disadvantage of the guided selection approach is
that shuffling of one
antibody chain while keeping the other constant could result in epitope drift.
In order to maintain the
epitope recognized by the non-human antibody, CDR retention can be applied
(Klimka et al., Br. J.
Cancer., 83, 252-260, 2000; Beiboer et al., J. Mol. Biol., 296, 833-49, 2000)
In this method, the non-
human CDR-H3 is commonly retained, as this CDR is at the center of the antigen-
binding site and has
proven to be the most important region of the antibody for antigen
recognition. In some instances,
however, CDR-H3 and CDR-L3, as well as CDR-H3, CDR-L3 and CDR-L2 of the non-
human antibody
may be retained.
6. Bispecific Antibodies
101821 Bispecific antibodies are monoclonal antibodies that have
binding specificities for at
least two different antigens. In certain embodiments, bispecific antibodies
are human or humanized
antibodies. In certain embodiments, one of the binding specificities is for
CD98 and the other is for any
other antigen. In certain embodiments, bispecific antibodies may bind to two
different epitopes of CD98.
Bispecific antibodies may also be used to localize cytotoxic agents to cells
that express CD98. These
antibodies possess a CD98-binding arm and an arm which binds a cytotoxic
agent, such as, e.g., saporin,
anti-interferon-a, vinca alkaloid, ricin A chain, methotrexate or radioactive
isotope hapten. Bispecific
antibodies can be prepared as full length antibodies or antibody fragments
(e.g. F(ab1)2bispecific
antibodies).
101831 Methods for making bispecific antibodies are known in the
art, such as, for example,
by co-expression of two immunoglobulin heavy chain-light chain pairs, where
the two heavy chains have
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
different specificities (Milstein and Cuello, Nature, 305: 537 (1983)). For
further details of generating
bispecific antibodies see, for example, Bis I ecific Antibodies, Kontermann,
ed., Springer-Verlag,
Hiedelberg (2011).
Multivalent Antibodies
[0184] A multivalent antibody may be internalized (and/or catabolized)
faster than a bivalent
antibody by a cell expressing an antigen to which the antibodies bind. The
antibodies of the present
invention can be multivalent antibodies (which are other than of the IgM
class) with three or more
antigen binding sites (e.g. tetravalent antibodies), which can be readily
produced by recombinant
expression of nucleic acid encoding the polypeptide chains of the antibody.
The multivalent antibody can
comprise a dimerization domain and three or more antigen binding sites. In
certain embodiments, the
dimerization domain comprises (or consists of) an Fc region or a hinge region.
In this scenario, the
antibody will comprise an Fc region and three or more antigen binding sites
amino-terminal to the Fc
region. In certain embodiments, a multivalent antibody comprises (or consists
of) three to about eight
antigen binding sites. In one such embodiment, a multivalent antibody
comprises (or consists of) four
antigen binding sites. The multivalent antibody comprises at least one
polypeptide chain (for example,
two polypeptide chains), wherein the polypeptide chain(s) comprise two or more
variable domains. For
instance, the polypeptide chain(s) may comprise VD1-(X1)11 -VD2-(X2)n -Fc,
wherein VD1 is a first
variable domain, VD2 is a second variable domain, Fc is one polypeptide chain
of an Fc region, X1 and
X2 represent an amino acid or polypeptide, and n is 0 or 1. For instance, the
polypeptide chain(s) may
comprise: VH-CH1-flexible linker-VH-CH I -Fc region chain; or VH-CHI-VH-CHI-Fc
region chain. The
multivalent antibody herein may further comprise at least two (for example,
four) light chain variable
domain polypeptides. The multivalent antibody herein may, for instance,
comprise from about two to
about eight light chain variable domain polypeptides. The light chain variable
domain polypeptides
contemplated here comprise a light chain variable domain and, optionally,
further comprise a CL domain.
8. Effector Function Engineering
[0185] It may be desirable to modify the antibody of the invention
with respect to effector
function, e.g., so as to enhance antigen-dependent cell-mediated cyotoxicity
(ADCC) and/or complement
dependent cytotoxicity (CDC) of the antibody. This may be achieved by
introducing one or more amino
acid substitutions in an Fc region of the antibody. See, e.g. Lazar et al.,
Proc. Natl. Acad. Sci. USA
2006, 103(11):4005-4010; Presta, L.G., Curr. Opin. Immunol. 2008, 20(4):460-
70; and U.S. Patent Nos.
7,183,387; 7,332,581; and 7.335,742.
[0186] Alternatively or additionally, cysteine residue(s) may be
introduced in the Fc region,
thereby allowing interchain disulfide bond formation in this region. The
homodimeric antibody thus
generated may have improved internalization capability and/or increased
complement-mediated cell
killing and antibody-dependent cellular cytotoxicity (ADCC). See Caron et al.,
J. Exp Med. 176:1191-
1195 (1992) and Shopes, B. J. Immunol. 148:2918-2922 (1992). Homodimeric
antibodies with enhanced
anti-tumor activity may also be prepared using heterobifunctional cross-
linkers as described in Wolff et
al., Cancer Research 53:2560-2565 (1993). Alternatively, an antibody can be
engineered which has dual
41
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
Fc regions and may thereby have enhanced complement lysis and ADCC
capabilities. See Stevenson et
al., Anti-Cancer Drug Design 3:219-230 (1989). To increase the serum half life
of the antibody, one may
incorporate a salvage receptor binding epitope into the antibody (especially
an antibody fragment) as
described in U.S. Patent 5,739,277, for example. As used herein, the term
"salvage receptor binding
epitope" refers to an epitope of the Fc region of an IgG molecule (e.g., IgG,
IgG2, IgG3, or IgG4) that is
responsible for increasing the in vivo serum half-life of the IgG molecule.
9. Alternative Binding Agents
[01871 The invention encompasses non-immunoglobulin binding agents
that specifically bind
to the same epitope as an anti-CD98 antibody disclosed herein. In some
embodiments, a binding agent is
identified an agent that displaces or is displaced by an anti-CD98 antibody of
the invention in a
competive binding assay. These alternative binding agents may include, for
example, any of the
engineered protein scaffolds known in the art. Such scaffolds include, for
example, anticalins, which are
based upon the lipocalin scaffold, a protein structure characterized by a
rigid beta-barrel that supports
four hypervariable loops which form the ligand binding site. Novel binding
specificities are engineered
by targeted random mutagenesis in the loop regions, in combination with
functional display and guided
selection (Skerra (2008) FEBS J. 275: 2677-2683). Other suitable scaffolds may
include, for example,
adnectins, or monobodies, based on the tenth extracellular domain of human
fibronectin III (Koide and
Koide (2007) Methods Mol. Biol. 352: 95-109); affibodies, based on the Z
domain of staphylococcal
protein A (Nygren et al. (2008) FEBS J. 275: 2668-2676)); DARPins, based on
ankyrin repeat proteins
(Stumpp et al. (2008) Drug. Discov. Today 13: 695-701); fynomers, based on the
SH3 domain of the
human Fyn protein kinase Grabulovski et al. (2007) J. Biol. Chem. 282: 3196-
3204); affitins, based on
Sac7d from Sulfolobus acidolarius (Krehenbrink et al. (2008) J. Mol. Biol.
383: 1058-1068); affilins,
based on human y-B-crystall in (Ebersbach et al. (2007) J. Mol. Biol. 372: 172-
185); avimers, based on
the A domains of membrane receptor proteins (Silverman et al. (2005)
Biotechnol. 23: 1556-1561);
cysteine-rich knottin peptides (Kolmar (2008) FEBS J. 275: 2684-2690); and
engineered Kunitz-type
inhibitors (Nixon and Wood (2006) CUIT. Opin. Drug. Discov. Dev. 9: 261-268)
For a review, see
Gebauer and Skerra (2009) Curr. Opin. Chem. Biol. 13: 245-255.
Antibody Variants
101881 In some embodiments, amino acid sequence modification(s) of the
antibodies
described herein are contemplated. For example, it may be desirable to improve
the binding affinity
and/or other biological properties of the antibody, including but not limited
to specificity, thermostability,
expression level, effector functions, glycosylation, reduced immunogenicity or
solubility. In addition to
the anti-CD98 antibodies described herein, it is contemplated that anti-CD98
antibody variants can be
prepared. Anti-CD98 antibody variants can be prepared by introducing
appropriate nucleotide changes
into the encoding DNA, and/or by synthesis of the desired antibody or
polypeptide. Those skilled in the
art will appreciate that amino acid changes may alter post-translational
processes of the anti-CD98
42
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
antibody, such as changing the number or position of glycosylation sites or
altering the membrane
anchoring characteristics.
[0189] Variations may be a substitution, deletion or insertion of
one or more codons encoding
the antibody or polypeptide that results in a change in the amino acid
sequence as compared with the
[0190] Amino acid sequence insertions include amino- and/or carboxyl-
terminal fusions
ranging in length from one residue to polypeptides containing a hundred or
more residues, as well as
intrasequence insertions of single or multiple amino acid residues. Examples
of terminal insertions
include an antibody with an N-terminal methionyl residue. Other insertional
variants of the antibody
[0191] Substantial modifications in the biological properties of the
antibody are accomplished
by selecting substitutions that differ significantly in their effect on
maintaining (a) the structure of the
polypeptide backbone in the area of the substitution, for example, as a sheet
or helical conformation, (b)
(l) non-polar: Ala (A), Val (V), Leu (L), Ile (I), Pro (P), Phe (F), Trp (W),
Met (M)
(2) uncharged polar: Gly (G), Ser (S), Thr (T), Cys (C), Tyr (Y), Asn (N), Gln
(Q)
25 (3) acidic: Asp (D), Glu (E)
(4) basic: Lys (K), Arg (R), His(H)
[0192] Alternatively, naturally occurring residues may be divided
into groups based on
common side-chain properties:
(1) hydrophobic: Norleucine, Met, Ala, Val, Leu, Ile;
30 (2) neutral hydrophilic: Cys, Ser, Thr, Asn, Gln;
(3) acidic: Asp, Glu;
(4) basic: His, Lys, Arg;
(5) residues that influence chain orientation: Gly, Pro;
(6) aromatic: Trp, Tyr, Phe.
35 [0193] Non-conservative substitutions will entail exchanging a member
of one of these classes
for another class. Such substituted residues also may be introduced into the
conservative substitution
sites or, into the remaining (non-conserved) sites.
43
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
[0194] The variations can be made using methods known in the art such as
oligonucleotide-
mediated (site-directed) mutagenesis, alanine scanning, and PCR mutagenesis.
Site-directed mutagenesis
(Carter et al., Nucl. Acids Res., 13:4331 (1986); Zoller et al., Nucl. Acids
Res., 10:6487 (1987)), cassette
mutagenesis (Wells et al., Gene, 34:315 (1985)), restriction selection
mutagenesis (Wells et al., Philos.
Trans. R. Soc. London SerA, 317:415 (1986)) or other known techniques can be
performed on the cloned
DNA to produce the anti-CD98 antibody variant DNA.
[0195] Any cysteine residue not involved in maintaining the proper
conformation of the anti-
CD98 antibody also may be substituted, generally with serine, to improve the
oxidative stability of the
molecule and prevent aberrant crosslinking. Conversely, cysteine bond(s) may
be added to the anti-
CD98 antibody to improve its stability (particularly where the antibody is an
antibody fragment such as
an Fv fragment). Cysteine-engineered antibodies, which can be used to generate
antibody-drug
conjugates, are described, for example, in WO 2006/034488.
[0196] In an embodiment, an anti-CD98 antibody molecule of the invention is
a "de-
immunized" antibody. A "de-immunized" anti-CD98 antibody is an antibody
derived from a humanized
or chimeric anti-CD98 antibody, that has one or more alterations in its amino
acid sequence resulting in a
reduction of immunogenicity of the antibody, compared to the respective
original non-de-immunized
antibody. One of the procedures for generating such antibody mutants involves
the identification and
removal of T-cell epitopes of the antibody molecule. In a first step, the
immunogenicity of the antibody
molecule can be determined by several methods, e.g. by in vitro determination
of T-cel I epitopes or in
silico prediction of such epitopes, as known in the art. Once the critical
residues for T-cell epitope
function have been identified, mutations can be made to remove immunogenicity
and retain antibody
activity. For review, see, e.g., Jones et al., Methods in Molecular Biology
525: 405-423, 2009.
In vitro affinity maturation
[0197] In an embodiment, antibody variants having an improved property such
as affinity,
stability, or expression level as compared to a parent antibody is in vitro
affinity maturation. Like the
natural prototype, in vitro affinity maturation is based on the principles of
mutation and selection.
Libraries of antibodies are displayed as Fab, scFv or V domain fragments
either on the surface of an
organism (e.g., phage, bacteria or yeast) or in association (covalently or non-
covalently) with their
encoding mRNA or DNA. Affinity selection of the displayed antibodies allows
isolation of organisms or
complexes carrying the genetic information encoding the antibodies. Two or
three rounds of mutation
and selection using display methods such as phage display usually results in
antibody fragments with
affinities in the low nanomolar range. Preferred affinity matured antibodies
will have nanomolar or even
picomolar affinities for the target antigen.
[0198] Phage display
is the most widepread method for display and selection of antibodies.
The antibodies are displayed on the surface of Fd or M13 bacteriophages as
fusions to the bacteriophage
coat protein. Selection involves exposure to antigen to allow phage-displayed
antibodies to bind their
targets, a process referred to as "panning." Phage bound to antigen are
recovered and infected in bacteria
44
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
to produce phage for further rounds of selection. For review, see Hoogenboom,
Methods. Mol. Biol.
178: 1-37, 2002; Bradbury and Marks, J. Immuno. Methods 290: 29-49, 2004).
[0199] In the yeast display system (Boder et al., Nat. Biotech. 15:
553-57, 1997; Chao et al.,
Nat. Protocols 1:755-768, 2006), the antibody is displayed as single-chain
variable fusions (scFv) in
which the heavy and light chains are connected by a flexible linker. The scFv
is fused to the adhesion
subunit of the yeast agglutinin protein Aga2p, which attaches to the yeast
cell wall through disulfide
bonds to Agalp. Display of a protein via Aga2p projects the protein away from
the cell surface,
minimizing potential interactions with other molecules on the yeast cell wall.
Magnetic separation and
flow cytometry are used to screen the library to select for antibodies with
improved affinity or stability.
Binding to a soluble antigen of interest is determined by labeling of yeast
with biotinylated antigen and a
secondary reagent such as streptavidin conjugated to a fluorophore. Variations
in surface expression of
the antibody can be measured through immunofluorescence labeling of either the
hemagglutinin or c-
Myc epitope tag flanking the scFv. Expression has been shown to correlate with
the stability of the
displayed protein, and thus antibodies can be selected for improved stability
as well as affinity (Shusta et
al., J. Mol. Biol. 292: 949-956, 1999). An additional advantage of yeast
display is that displayed proteins
are folded in the endoplasmic reticulum of the eukaryotic yeast cells, taking
advantage of endoplasmic
reticulum chaperones and quality-control machinery. Once maturation is
complete, antibody affinity can
be conveniently `titrated' while displayed on the surface of the yeast,
eliminating the need for expression
and purification of each clone. A theoretical limitation of yeast surface
display is the potentially smaller
functional library size than that of other display methods; however, a recent
approach uses the yeast cells'
mating system to create combinatorial diversity estimated to be 1014 in size
(US Patent Publication
2003/0186,374; Blaise et al., Gene 342: 211-218, 2004).
[0200] In ribosome display, antibody-ribosome-mRNA (ARM) complexes are
generated for
selection in a cell-free system. The DNA library coding for a particular
library of antibodies is
genetically fused to a spacer sequence lacking a stop codon. This spacer
sequence, when translated, is
still attached to the peptidyl tRNA and occupies the ribosomal tunnel, and
thus allows the protein of
interest to protrude out of the ribosome and fold. The resulting complex of
mRNA, ribosome, and
protein can bind to surface-bound ligand, allowing simultaneous isolation of
the antibody and its
encoding mRNA through affinity capture with the ligand. The ribosome-bound
mRNA is then reversed
transcribed back into cDNA, which can then undergo mutagenesis and be used in
the next round of
selection. (Fukuda et al., Nucleic Acids Res. 34, e127, 2006). In mRNA
display, a covalent bond
between antibody and mRNA is established using puromycin as an adaptor
molecule (Wilson et al., Proc.
Natl. Acad. Sci. USA 98, 3750-3755, 2001).
[0201] As these methods are performed entirely in vitro, they
provide two main advantages
over other selection technologies. First, the diversity of the library is not
limited by the transformation
efficiency of bacterial cells, but only by the number of ribosomes and
different mRNA molecules present
in the test tube. Second, random mutations can be introduced easily after each
selection round, for
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
example, by non-proofreading polymerases, as no library must be transformed
after any diversification
step.
[0202] Diversity may be introduced into the CDRs or the whole V genes of the
antibody
libraries in a targeted manner or via random introduction. The former approach
includes sequentially
[0203] Screening of the libraries can be accomplished by various
techniques known in the art.
For example, CD98 can be immobilized onto solid supports, columns, pins or
cellulose/poly(vinylidene
fluoride) membranes/ other filters, expressed on host cells affixed to
adsorption plates or used in cell
sorting, or conjugated to biotin for capture with streptavidin-coated beads,
or used in any other method
[0204] For review of in vitro affinity maturation methods, see
Hoogenboom, Nature
Biotechnology 23: 1105-1116, 2005 and Quiroz and Sinclair, Revista Ingeneria
Biomedia 4: 39-5 I, 2010
and references therein.
25 Modifications of Anti-CD98 Antibodies
[0205] Covalent modifications of anti-CD98 antibodies are included
within the scope of this
invention. Covalent modifications include reacting targeted amino acid
residues of an anti-CD98
antibody with an organic derivatizing agent that is capable of reacting with
selected side chains or the N-
or C- terminal residues of the anti-CD98 antibody. Other modifications include
deamidation of
35 [0206] Other types of covalent modification of the anti-CD98 antibody
included within the
scope of this invention include altering the native glycosylation pattern of
the antibody or polypeptide
(Beck et al., Curr. Pharm. Biotechnol. 9: 482-501, 2008; Walsh, Drug Discov.
Today 15: 773-780, 2010),
and linking the antibody to one of a variety of nonproteinaceous polymers,
e.g., polyethylene glycol
46
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
(PEG), polypropylene glycol, or polyoxyalkylenes, in the manner set forth in
U.S. Patent Nos. 4,640,835;
4,496,689; 4,301,144; 4,670,417; 4,791,192 or 4,179,337.
[0207] The anti-CD98 antibody of the present invention may also be modified to
form
chimeric molecules comprising an anti-CD98 antibody fused to another,
heterologous polypeptide or
amino acid sequence, e.g., an epitope tag (Terpe, Appl. Microbiol. Biotechnol.
60: 523-533, 2003) or the
Fc region of an IgG molecule (Aruffo, "Immunoglobulin fusion proteins" in
Antibody Fusion Proteins,
S.M. Chamow and A. Ashkenazi, eds., Wiley-Liss, New York, 1999, pp. 221-242).
Preparation of Anti-CD98 Antibodies
[0208] Anti-CD98 antibodies may be produced by culturing cells transformed
or transfected
with a vector containing anti-CD98 antibody-encoding nucleic acid.
Polynucleotide sequences encoding
polypeptide components of the antibody of the invention can be obtained using
standard recombinant
techniques. Desired polynucleotide sequences may be isolated and sequenced
from antibody producing
cells such as hybridoma cells. Alternatively, polynucleotides can be
synthesized using nucleotide
synthesizer or PCR techniques. Once obtained, sequences encoding the
polypeptides are inserted into a
recombinant vector capable of replicating and expressing heterologous
polynucleotides in host cells.
Many vectors that are available and known in the art can be used for the
purpose of the present invention.
Selection of an appropriate vector will depend mainly on the size of the
nucleic acids to be inserted into
the vector and the particular host cell to be transformed with the vector.
Host cells suitable for
expressing antibodies of the invention include prokaryotes such as
Archaebacteria and Eubacteria,
including Gram-negative or Gram-positive organisms, eukaryotic microbes such
as filamentous fungi or
yeast, invertebrate cells such as insect or plant cells, and vertebrate cells
such as mammalian host cell
lines. Host cells are transformed with the above-described expression vectors
and cultured in
conventional nutrient media modified as appropriate for inducing promoters,
selecting transformants, or
amplifying the genes encoding the desired sequences. Antibodies produced by
the host cells are purified
using standard protein purification methods as known in the art.
[0209] Methods for antibody production including vector
construction, expression and
purification are further described in PlOckthun et al., (1996) in Antibod En.
ineerin.: Producin=
antibodies in Escherichia coli: From PCR to fermentation (McCafferty, J.,
Hoogenboom, H. R., and
Chiswell, D. J., eds), 1 Ed., pp. 203-252, IRL Press, Oxford; Kwong, K. &
Rader, C. E. coli expression
and purification of Fab antibody fragments. Current protocols in protein
science editorial board John E
Coligan et al., Chapter 6, Unit 6.10 (2009); Tachibana and Takekoshi,
"Production of Antibody Fab
Fragments in Escherischia coli," in Antibody Expression and Production, M. Al-
Rubeai, Ed., Springer,
New York, 2011; Therapeutic Monoclonal Antibodies: From Bench to Clinic (ed Z.
An), John Wiley &
Sons, Inc., Hoboken, NJ, USA.
[0210] It is, of course, contemplated that alternative methods,
which are well known in the art,
may be employed to prepare anti-CD98 antibodies. For instance, the appropriate
amino acid sequence, or
portions thereof, may be produced by direct peptide synthesis using solid-
phase techniques (see, e.g.,
47
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
Stewart et al., Solid-Phase Peptide Synthesis, W.H. Freeman Co., San
Francisco, CA (1969); Merrifield,
J. Am. Chem. Soc., 85:2149-2154 (1963)). In vitro protein synthesis may be
performed using manual
techniques or by automation. Various portions of the anti-CD98 antibody may be
chemically synthesized
separately and combined using chemical or enzymatic methods to produce the
desired anti-CD98
antibody. Alternatively, antibodies may be purified from cells or bodily
fluids, such as milk, of a
transgenic animal engineered to express the antibody, as disclosed, for
example, in US Pat. No.
5,545,807 and US Pat. No. 5,827,690.
Immunocon'u=ates
[0211] The invention also provides immunoconjugates (interchangably
referred to as
"antibody drug conjugates," or "ADCs") comprising any one of the anti-CD98
antibodies of the invention
covalently bound by a synthetic linker to one or more cytotoxic agents. ADCs
combine the high
specificity of monoclonal antibodies with the pharmacological potency of
cytotoxic molecules, allowing
specific targeting of cytotoxic agents to tumor cells and avoiding the
nonspecific toxicity of most anti-
cancer drugs. For review, see, e.g. Carter and Senter,_Cancer J. 14: 154-169
(2008); Ducry and Stump,
Bioconjugate Chem. 21:5-13 (2010); Beck et al., Discov. Med. 10: 329-339
(2010).
[0212] Cytotoxic agents for use in the immunoconjugates of the
invention may include
chemotherapeutic agents, drugs or growth inhibitory agents as described above,
toxins (e.g., an
enzymatically active toxin of bacterial, fungal, plant or animal origin, or
fragments thereof) or
radioisotopes. In some embodiments, the immunoconjugate comprises a DNA binder
(e.g.,
calicheamycin) or a tubulin depolymerization agent (e.g., a maytansinoid or an
auristatin). The present
invention further contemplates an immunoconjugate formed between an antibody
and a compound with
nucleolytic activity (e.g., a ribonuclease or a DNA endonuclease such as a
deoxyribonuclease; DNase).
[0213] Enzymatically active toxins and fragments thereof that can be
used in the
immunoconjugates of the invention include diphtheria A chain, nonbinding
active fragments of
diphtheria toxin, exotoxin A chain (from Pseudomonas aeruginosa), ricin A
chain, abrin A chain,
modeccin A chain, alpha-sarcin, Aleurites fordii proteins, dianthin proteins,
Phytolaca americana proteins
(PAP', PAPIL and PAP-S), momordica charantia inhibitor, curcin, crotin,
sapaonaria officinalis inhibitor,
gelonin, mitogellin, restrictocin, phenomycin, enomycin, and the
tricothecenes. See, e.g., WO 93/21232.
[0214] A variety of radioactive isotopes are available for the production
of radioconjugated
antibodies. Examples include At211, 1131, /125, y90, Re186, Re188, sm153,
Bi212, F,32, p+ 212
and radioactive
isotopes of Lu. When the conjugate is used for detection, it may comprise a
radioactive atom for
scintigraphic studies, for example tc99mor 1123, or a spin label for nuclear
magnetic resonance (NMR)
imaging (also known as magnetic resonance imaging, MRI), such as iodine-123
again, iodine-131,
indium-111, fluorine-19, carbon-13, nitrogen-15, oxygen-17, gadolinium,
manganese or iron. The
radioisotopes may be incorporated in the conjugate in known ways as described,
e.g., in Reilly, "The
radiochemistry of monoclonal antibodies and peptides," in Monoclonal Antibody
and Peptide-Targeted
Radiotherapy of Cancer, R.M. Reilly, ed., Wiley, Hoboken N.J., 2010.
48
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
[0215] The linker may be a "cleavable linker" facilitating release
of the cytotoxic drug in the
cell, but non-cleavable linkers are also contemplated herein. Linkers for use
in the immunoconjugates of
the invention include without limitation acid labile linkers (e.g., hydrazone
linkers), disulfide-containing
linkers, peptidase-sensitive linkers (e.g, peptide linkers such as citrulline-
valine or phenylalanine-lysine),
photolabile linkers, dimethyl linkers (Chari et al., Cancer Research 52:127-
131 (1992); U.S. Patent No.
5,208,020), thioether linkers, or hydrophilic linkers designed to evade
multidrug transporter-mediated
resistance (Kovtun et al., Cancer Res. 70: 2528-2537, 2010).
[0216] Conjugates of the antibody and cytotoxic agent may be made
using a variety of
bifunctional protein coupling agents such as BMPS, EMCS, GMBS, HBVS, LC-SMCC,
MBS, MPBH,
SBAP, SIA, SIAB, SMCC, SMPB, SMPH, sulfo-EMCS, sulfo-GMBS, sulfo-KMUS, sulfo-
MBS, sulfo-
SIAB, sulfo-SMCC, and sulfo-SMPB, and SVSB (succinimidy1-(4-
vinylsulfone)benzoate)). For
example, a ricin immunotoxin can be prepared as described in Vitetta et al.,
Science 238:1098 (1987).
The invention further contemplates that conjugates of antibodies and cytotoxic
agents may be prepared
using any suitable methods as disclosed in the art, e.g., in Bioconjugate
Techniques, 2nd Ed., G.T.
Hermanson, ed., Elsevier, San Francisco, 2008.
[0217] Conventional antibody-drug conjugation strategies have been
based on random
conjugation chemistries involving the c-amino group of Lys residues or the
thiol group of Cys residues,
which results in heterogenous conjugates. Recently developed techniques allow
site-specific conjugation
to antibodies, resulting in homogeneous drug loading and avoiding ADC
subpopulations with altered
antigen-binding or pharmacokinetics. These include engineering of "thiomabs"
comprising cysteine
substitutions at positions on the heavy and light chains that provide reactive
thiol groups and do not
disrupt immunoglobul in folding and assembly or alter antigen binding
(Junutula et al., J. Immunol. Meth.
332: 41-52 (2008); Junutula et al., Nat. Biotechnol. 26: 925-932, 2008). In
another method,
selenocysteine is cotranslationally inserted into an antibody sequence by
recoding the stop codon UGA
from termination to selenocysteine insertion, allowing site specific covalent
conjugation at the
nucleophilic selenol group of selenocysteine in the presence of the other
natural amino acids (Hofer et al.,
Proc. Natl. Acad. Sci. USA 105: 12451-12456 (2008); Hofer et al, Biochemistry
48(50): 12047-12057,
2009).
Pharmaceutical Formulations
[0218] The antibodies or antibody-drug conjugates (ADC) of the invention
may be
administered by any route appropriate to the condition to be treated. The
antibody or ADC will typically
be administered parenterally, i.e., infusion, subcutaneous, intramuscular,
intravenous, intradermal,
intrathecal and epidural.
[0219] For treating cancers, in one embodiment, the antibody or
antibody-drug conjugate is
administered via intravenous infusion. The dosage administered via infusion is
in the range of about 1
ug/m2 to about 10,000 jig,/m2 per dose, generally one dose per week for a
total of one, two, three or four
doses. Alternatively, the dosage range is of about 1 ug/m2 to about 1000
ug/m2, about 1 ug/m2 to about
800 ug/m2, about 1 jig/m2 to about 600 ug/m2, about 1 ug/m2 to about 400
ug/m2, about 10 ug/m2 to
49
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
about 500 t1g/m2, about 10 lig/m2 to about 300 tg/m2, about 10 tg/m2 to about
200 fig/m2, and about 1
jtg/m2 to about 200 ig/m2. The dose may be administered once per day, once per
week, multiple times
per week, but less than once per day, multiple times per month but less than
once per day, multiple times
per month but less than once per week, once per month or intermittently to
relieve or alleviate symptoms
of the disease. Administration may continue at any of the disclosed intervals
until remission of the tumor
or symptoms of the cancer being treated. Administration may continue after
remission or relief of
symptoms is achieved where such remission or relief is prolonged by such
continued administration.
[0220] In one aspect, the invention further provides pharmaceutical
formulations comprising
at least one anti-CD98 antibody of the invention and/or at least one
immunoconjugate thereof and/or at
least one anti-CD98 antibody-drug conjugate of the invention. In some
embodiments, a pharmaceutical
formulation comprises 1) an anti-CD98 antibody and/or an anti-CD98 antibody-
drug conjugate and/or an
immunoconjugate thereof, and 2) a pharmaceutically acceptable carrier. In some
embodiments, a
pharmaceutical formulation comprises 1) an anti-CD98 antibody and/or an
immunoconjugate thereof,
and optionally, 2) at least one additional therapeutic agent.
I 5 [0221] Pharmaceutical formulations comprising an antibody or
immunoconjugate of the
invention or the antibody-drug conjugate of the invention are prepared for
storage by mixing the antibody
or antibody-drug conjugate having the desired degree of purity with optional
physiologically acceptable
carriers, excipients or stabilizers (Remington's Pharmaceutical Sciences 16th
edition, Osol, A. Ed.
(1980)) in the form of aqueous solutions or lyophilized or other dried
formulations. The formulations
herein may also contain more than one active compound as necessary for the
particular indication being
treated, preferably those with complementary activities that do not adversely
affect each other. For
example, in addition to an anti-CD98 antibody, it may be desirable to include
in the one formulation, an
additional antibody, e.g., a second anti-CD98 antibody which binds a different
epitope on the CD98
polypeptide, or an antibody to some other target such as a growth factor that
affects the growth of the
particular cancer. Alternatively, or additionally, the composition may further
comprise a
chemotherapeutic agent, cytotoxic agent, cytokine, growth inhibitory agent,
anti-hormonal agent, and/or
cardioprotectant. Such molecules are suitably present in combination in
amounts that are effective for the
purpose intended.
[0222] The antibodies or immunoconjugates of the invention may be formulated
in any
suitable form for delivery to a target cell/tissue, e.g, as microcapsules or
macroemulsions (Remington's
Pharmaceutical Sciences, 16th edition, Osol, A. Ed. (1980); Park et al.,
Molecules 10: 146-161 (2005);
Malik et al., Curr. Drug. Deliv. 4: 141-151 (2007)); as sustained release
formulations (Putney and Burke,
Nature Biotechnol. 16: 153-157, (1998)) or in liposomes (Maclean et al., Int.
J. Oncol. 11: 235-332
(1997); Kontermann, Curr. Opin. Mol. Ther. 8: 39-45 (2006)).
Therapeutic methods
[0223] An antibody or immunoconjugate of the invention may be used in, for
example, in
vitro, ex vivo, and in vivo therapeutic methods. In one aspect, the invention
provides methods for
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
inhibiting cell growth or proliferation, either in vivo or in vitro, the
method comprising exposing a cell to
an anti-CD98 antibody or immunoconjugate thereof under conditions permissive
for binding of the
immunoconjugate to CD98. "Inhibiting cell growth or proliferation" means
decreasing a cell's growth or
proliferation by at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, or
100%, and includes
inducing cell death. In certain embodiments, the cell is a tumor cell. In
certain embodiments, the cell is
a bladder, breast, colon, rectal, gastric, esophageal, lung, laryx, kidney,
oral, ovarian, or prostate tumor
cell, or a sarcoma, melanoma, glioma, lymphoma or leukemia cell.
[0224] In one aspect, an antibody or immunoconjugate of the
invention is used to treat or
prevent a cell proliferative disorder, such as cancer. In certain embodiments,
the cell proliferative
disorder is associated with increased expression and/or activity of CD98. For
example, in certain
embodiments, the cell proliferative disorder is associated with increased
expression of CD98 on the
surface of a cancer cell. Examples of cell proliferative disorders to be
treated by the antibodies or
immunoconjugates of the invention include, but are not limited to, bladder,
breast, colon, rectal, gastric,
esophageal, lung, laryx, kidney, oral, ovarian, or prostate cancers, or
sarcomas, melanomas, gliomas,
lymphomas or leukemias, or metatases of any of these cancers.
[0225] In one aspect, the invention provides methods for treating a
cell proliferative disorder
comprising administering to an individual an effective amount of an anti-CD98
antibody or
immunoconjugate thereof. In certain embodiments, a method for treating a cell
proliferative disorder
comprises administering to an individual an effective amount of a
pharmaceutical formulation
comprising an anti-CD98 antibody or anti-CD98 immunoconjugate and, optionally,
at least one
additional therapeutic agent, such as those provided below. In one embodiment,
an anti-CD98 antibody
or immunoconjugate can be used for targeting CD98 on cancer cells by
contacting the antibody or
immunoconjugate with CD98 to form an antibody or immunoconjugate-antigen
complex such that a
conjugated cytotoxic agent of the immunoconjugate accesses the interior of the
cell. In one embodiment,
the bound antibody or immunoconjugate is internalized into the cancer cell
expressing CD98.
[0226] An anti-CD98 antibody or immunoconjugate can be administered to a human
for
therapeutic purposes. Moreover, an anti-CD98 antibody or immunoconjugate can
be administered to a
non-human mammal expressing CD98 with which the antibody cross-reacts (e.g., a
primate, pig, rat, or
mouse) for veterinary purposes or as an animal model of human disease.
Regarding the latter, such
animal models may be useful for evaluating the therapeutic efficacy of
antibodies or immunoconjugates
of the invention (e.g., testing of dosages and time courses of
administration).
[0227] Antibodies or immunoconjugates of the invention can be used
either alone or in
combination with other compositions in a therapy. For instance, an antibody or
immunoconjugate of the
invention may be co-administered with at least one additional therapeutic
agent and/or adjuvant. In
certain embodiments, an additional therapeutic agent is a cytotoxic agent, a
chemotherapeutic agent, or a
growth inhibitory agent. In some embodiments, a chemotherapeutic agent is an
agent or a combination
of agents such as an alkylating agent (for example, bendamustine
hydrochloride, cyclophosphamide or
ifosfamide) a nucleoside analog (for example, fludurabine, cytarabine or
gemcitabine) a corticosteroid
51
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
(for example, prednisone, prednisolone or methylprednisolone), an anti-mitotic
agent (for example,
paclitaxel, docetaxel or vinorelbine), a vinca alkaloid (for example,
vincristine or etoposide), a
topoisomerase inhibitor (for example, irinotecan), an antibiotic (for example,
anthracycline or
adriamycin), a platinum analog (for example, cisplatin or carboplatin), a
therapeutic antibody (for
example, rituximab) or a combination of agents (for example CHOP or CVP)
wherein the combination
therapy is useful in the treatment of cancers. In some embodiments, the
additional compound is a
therapeutic antibody other than an anti-CD98 antibody (for example,
rituximab).
[0228] Such combination therapies noted above encompass combined
administration (where
two or more therapeutic agents are included in the same or separate
formulations), and separate
administration, in which case, administration of the antibody or
immunoconjugate of the invention can
occur prior to, simultaneously, and/or following, administration of the
additional therapeutic agent and/or
adjuvant. Antibodies or immunoconjugates of the invention can also be used in
combination with
additional therapeutic regimens including without limitation radiation therapy
and/or bone marrow and
peripheral blood transplants.
[0229] An antibody or immunoconjugate of the invention (and any additional
therapeutic
agent or adjuvant) can be administered by any suitable means, including
parenteral, subcutaneous,
intraperitoneal, intrapulmonary, and intranasal, and, if desired for local
treatment, intralesional
administration. Parenteral infusions include intramuscular, intravenous,
intraarterial, intraperitoneal, or
subcutaneous administration. In addition, the antibody or immunoconjugate is
suitably administered by
pulse infusion, particularly with declining doses of the antibody or
immunoconjugate. Dosing can be by
any suitable route, e.g. by injections, such as intravenous or subcutaneous
injections, depending in part
on whether the administration is brief or chronic.
[0230] Antibodies or immunoconjugates of the invention would be
formulated, dosed, and
administered in a fashion consistent with good medical practice. Factors for
consideration in this context
include the particular disorder being treated, the particular mammal being
treated, the clinical condition
of the individual patient, the cause of the disorder, the site of delivery of
the agent, the method of
administration, the scheduling of administration, and other factors known to
medical practitioners. The
antibody or immunoconjugate need not be, but is optionally formulated with one
or more agents currently
used to prevent or treat the disorder in question. The effective amount of
such other agents depends on
the amount of antibody or immunoconjugate present in the formulation, the type
of disorder or treatment,
and other factors discussed above. These are generally used in the same
dosages and with administration
routes as described herein, or about from 1 to 99% of the dosages described
herein, or in any dosage and
by any route that is empirically/clinically determined to be appropriate.
[0231] For the prevention or treatment of disease, the appropriate
dosage of an antibody or
immunoconjugate of the invention (when used alone or in combination with one
or more other additional
therapeutic agents, such as chemotherapeutic agents) will depend on the type
of disease to be treated, the
type of antibody or immunoconjugate, the severity and course of the disease,
whether the antibody or
immunoconjugate is administered for preventive or therapeutic purposes,
previous therapy, the patient's
52
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
clinical history and response to the antibody or immunoconjugate, and the
discretion of the attending
physician. The antibody or immunoconjugate is suitably administered to the
patient at one time or over a
series of treatments. Depending on the type and severity of the disease, about
1 lig/kg to 100 mg/kg (e.g.
0.1mg/kg-20ing/kg) of antibody or immunoconjugate can be an initial candidate
dosage for
administration to the patient, whether, for example, by one or more separate
administrations, or by
continuous infusion. One typical daily dosage might range from about 1 ig/kg
to 100 mg/kg or more,
depending on the factors mentioned above. For repeated administrations over
several days or longer,
depending on the condition, the treatment would generally be sustained until a
desired suppression of
disease symptoms occurs. One exemplary dosage of the antibody or
immunoconjugate would be in the
range from about 0.05 mg/kg to about 10 mg/kg. Thus, one or more doses of
about 0.5 mg/kg, 2.0
mg/kg, 4.0 mg/kg or 10 mg/kg (or any combination thereof) of antibody or
immunoconjugate inay be
administered to the patient. Such doses may be administered intermittently,
e.g. every week or every
three weeks (e.g. such that the patient receives from about two to about
twenty, or e.g. about six doses of
the antibody or immunoconjugate). An initial higher loading dose, followed by
one or more lower doses
may be administered. An exemplary dosing regimen comprises administering an
initial loading dose of
about 4 mg/kg, followed by a weekly maintenance dose of about 2 mg/kg of the
antibody. However,
other dosage regimens may be useful. The progress of this therapy is easily
monitored by conventional
techniques and assays.
Diagnostic methods and methods of detection
[0232] In one aspect, anti-CD98 antibodies and immunoconjugates of
the invention are useful
for detecting the presence of CD98 in a biological sample. The term
"detecting" as used herein
encompasses quantitative or qualitative detection. In certain embodiments, a
biological sample
comprises a cell or tissue. In certain embodiments, such tissues include
normal and/or cancerous tissues
that express CD98 at higher levels relative to other tissues, for example,
bladder, breast, colon, rectal,
gastric, esophageal, lung, latyx, kidney, oral, ovarian, or prostate cancer,
or a sarcoma, melanoma,
glioma, lymphoma or leukemia, or a metatasis of any of these cancers.
[0233] In one aspect, the invention provides a method of detecting
the presence of CD98 in a
biological sample. In certain embodiments, the method comprises contacting the
biological sample with
an anti- CD98 antibody under conditions permissive for binding of the anti-
CD98 antibody to CD98, and
detecting whether a complex is formed between the anti- CD98 antibody and
CD98.
[0234] In one aspect, the invention provides a method of diagnosing
a disorder associated
with increased expression of CD98. In certain embodiments, the method
comprises contacting a test cell
with an anti-CD98 antibody; determining the level of expression (either
quantitatively or qualitatively) of
CD98 by the test cell by detecting binding of the anti-CD98 antibody to CD98;
and comparing the level
of expression of CD98 by the test cell with the level of expression of CD98 by
a control cell (e.g., a
normal cell of the same tissue origin as the test cell or a cell that
expresses CD98 at levels comparable to
such a normal cell), wherein a higher level of expression of CD98 by the test
cell as compared to the
53
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
control cell indicates the presence of a disorder associated with increased
expression of CD98. In certain
embodiments, the increased expression corresponds to higher density of CD98
expression on the surface
of a tumor cell as compared toa normal cell. In certain embodiments, the test
cell is obtained from an
individual suspected of having a disorder associated with increased expression
of CD98. In certain
embodiments, the disorder is a cell proliferative disorder, such as a cancer
or a tumor. Exemplary cell
proliferative disorders that may be diagnosed using an antibody of the
invention include bladder, breast,
colon, rectal, gastric, esophageal, lung, laryx, kidney, oral, ovarian, or
prostate cancer, or a sarcoma,
melanoma, glioma, lymphoma or leukemia, or a metatasis of any of these
cancers.
[0235] In certain embodiments, a method of diagnosis or detection,
such as those described
above, comprises detecting binding of an anti-CD98 antibody to CD98 expressed
on the surface of a cell
or in a membrane preparation obtained from a cell expressing CD98 on its
surface. In certain
embodiments, the method comprises contacting a cell with an anti-CD98 antibody
under conditions
permissive for binding of the anti-CD98 antibody to CD98, and detecting
whether a complex is formed
between the anti-CD98 antibody and CD98 on the cell surface. An exemplary
assay for detecting
binding of an anti-CD98 antibody to CD98 expressed CD98 on the surface of a
cell is a "FACS" assay.
[0236] Certain other methods can be used to detect binding of anti-
CD98 antibodies to CD98.
Such methods include, but are not limited to, antigen-binding assays that are
well known in the art, such
as western blots, radioimmunoassays, ELISA (enzyme linked immunosorbent
assay), "sandwich"
immunoassays, immunoprecipitation assays, fluorescent immunoassays, protein A
immunoassays, and
immunohistochemistry (IHC).
[0237] In certain embodiments, anti-CD98 antibodies are labeled.
Labels include, but are not
limited to, labels or moieties that are detected directly (such as
fluorescent, chromophoric, electron-
dense, chemiluminescent, and radioactive labels), as well as moieties, such as
enzymes or ligands, that
are detected indirectly, e.g., through an enzymatic reaction or molecular
interaction. Exemplary labels
include, but are not limited to, the radioisotopes 32P, 14C, 125.,
31-1, and 1311, fluorophores such as rare earth
chelates or fluorescein and its derivatives, rhodamine and its derivatives,
dansyl, umbel liferone,
luceriferases, e.g., firefly luciferase and bacterial luciferase (U.S. Pat.
No. 4,737,456), luciferin, 2,3-
dihydrophthalazinediones, horseradish peroxidase (HRP), alkaline phosphatase,
p-galactosidase,
glucoamylase, lysozyme, saccharide oxidases, e.g., glucose oxidase, galactose
oxidase, and glucose-6-
phosphate dehydrogenase, heterocyclic oxidases such as uricase and xanthine
oxidase, coupled with an
enzyme that employs hydrogen peroxide to oxidize a dye precursor such as HRP,
lactoperoxidase, or
microperoxidase, biotin/avidin, spin labels, bacteriophage labels, stable free
radicals, and the like.
[0238] In certain embodiments, anti-CD98 antibodies are immobilized
on an insoluble matrix.
Immobilization entails separating the anti-CD98 antibody from any CD98 that
remains free in solution.
This conventionally is accomplished by either insolubilizing the anti-CD98
antibody before the assay
procedure, as by adsorption to a water-insoluble matrix or surface (Bennich et
al.., U.S. 3,720,760), or by
covalent coupling (for example, using glutaraldehyde cross-linking), or by
insolubilizing the anti-CD98
54
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
antibody after formation of a complex between the anti-CD98 antibody and CD98,
e.g., by
immunoprecipitation.
[0239] Any of the above embodiments of diagnosis or detection may be carried
out using an
immunoconjugate of the invention in place of or in addition to an anti-CD98
antibody.
Assays
[0240] Anti-CD98 antibodies and immunoconjugates of the invention may be
characterized
for their physical/chemical properties and/or biological activities by various
assays known in the art.
Activity assays
[0241] In one aspect, assays are provided for identifying anti-CD98
antibodies or
immunoconjugates thereof having biological activity. Biological activity may
include, e.g., the ability to
inhibit cell growth or proliferation (e.g., "cell killing" activity), or the
ability to induce cell death,
including programmed cell death (apoptosis). Antibodies or immunoconjugates
having such biological
activity in vivo and/or in vitro are also provided.
[0242] In certain embodiments, an anti-CD98 antibody or immunoconjugate
thereof is tested
for its ability to inhibit cell growth or proliferation in vitro. Assays for
inhibition of cell growth or
proliferation are well known in the art. Certain assays for cell
proliferation, exemplified by the "cell
killing" assays described herein, measure cell viability. One such assay is
the CellTiter-GlOrm
Luminescent Cell Viability Assay, which is commercially available from Promega
(Madison, WI). That
assay determines the number of viable cells in culture based on quantitation
of ATP present, which is an
indication of metabolically active cells. See Crouch et al (1993) J. Immunol.
Meth, 160:81-88, US Pat.
No. 6602677. The assay may be conducted in 96- or 384-well format, making it
amenable to automated
high-throughput screening (HTS). See Cree et al (1995) AntiCancer Drugs 6:398-
404.
[0243] Another assay for cell proliferation is the "MTT" assay, a
colorimetric assay that
measures the oxidation of 3-(4,5-dimethylthiazol-2-y1)-2,5-diphenyltetrazolium
bromide to formazan by
mitochondrial reductase. Like the CellTiter-GloTm assay, this assay indicates
the number of
metabolically active cells present in a cell culture. See, e.g., Mosmann
(1983) J. Immunol. Meth. 65:55-
63, and Zhang et al. (2005) Cancer Res. 65:3877-3882.
[0244] In one aspect, an anti-CD98 antibody is tested for its
ability to induce cell death in
vitro. Assays for induction of cell death are well known in the art. In some
embodiments, such assays
measure, e.g., loss of membrane integrity as indicated by uptake of propidium
iodide (PI), trypan blue
(see Moore et al. (1995) Cytotechnology, 17:1-11), or 7AAD. In an exemplary PI
uptake assay, cells are
cultured in Dulbecco's Modified Eagle Medium (D-MEM): Ham's F-12 (50:50)
supplemented with 10%
heat-inactivated FBS (Hyclone) and 2 mM L-glutamine. Thus, the assay is
performed in the absence of
complement and immune effector cells. Cells are seeded at a density of 3 x 106
per dish in 100 x 20 mm
dishes and allowed to attach overnight. The medium is removed and replaced
with fresh medium alone or
medium containing various concentrations of the antibody or immunoconjugate.
The cells are incubated
for a 3-day time period. Following treatment, monolayers are washed with PBS
and detached by
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
trypsinization. Cells are then centrifuged at 1200 rpm for 5 minutes at 4 C,
the pellet resuspended in 3
ml cold Ca2+ binding buffer (10 mM Hepes, pH 7.4, 140 mM NaCI, 2.5 mM CaC12)
and aliquoted into 35
mm strainer-capped 12 x 75 mm tubes (1 ml per tube, 3 tubes per treatment
group) for removal of cell
clumps. Tubes then receive PI (101.1g/m1). Samples are analyzed using a
FACSCANTM flow cytometer
and FACSCONVERTTm CellQuest software (Becton Dickinson). Antibodies or
immunoconjugates that
induce statistically significant levels of cell death as determined by PI
uptake are thus identified.
[0245] In one aspect, an anti-CD98 antibody or immunoconjugate is
tested for its ability to
induce apoptosis (programmed cell death) in vitro. An exemplary assay for
antibodies or
immunconjugates that induce apoptosis is an annexin binding assay, for
example, as in Zhang et al.
(BioTechniques 23: 525-531, 1997). Another exemplary assay for antibodies or
immunconjugates that
induce apoptosis is a histone DNA ELISA colorimetric assay for detecting
internucleosomal degradation
of genomic DNA. Such an assay can be performed using, e.g., the Cell Death
Detection ELISA kit
(Roche, Palo Alto, CA).
[0246] Cells for use in any of the above in vitro assays include
cells or cell lines that naturally
express CD98 or that have been engineered to express CD98. Such cells include
tumor cells that
overexpress CD98 relative to normal cells of the same tissue origin. Such
cells also include cell lines
(including tumor cell lines) that express CD98 and cell lines that do not
normally express CD98 but have
been transfected with nucleic acid encoding CD98.
[0247] In one aspect, an anti-CD98 antibody or immunoconjugate
thereof is tested for its
ability to inhibit cell growth or proliferation in vivo. In certain
embodiments, an anti-CD98 antibody or
immunoconjugate thereof is tested for its ability to inhibit tumor growth in
vivo. In vivo model systems,
such as xenograft models, can be used for such testing. In an exemplary
xenograft system, human tumor
cells are introduced into a suitably immunocompromised non-human animal, e.g.,
a SCID mouse. An
antibody or immunoconjugate of the invention is administered to the animal.
The ability of the antibody
or immunoconjugate to inhibit or decrease tumor growth is measured. In certain
embodiments of the
above xenograft system, the human tumor cells are tumor cells from a human
patient. Such cells useful
for preparing xenograft models include without limitation cells expressing
exogenous CD98, and cells
naturally expressing CD98. In certain embodiments, the human tumor cells are
introduced into a suitably
immunocompromised non-human animal by subcutaneous injection or by
transplantation into a suitable
site, such as a mammary fat pad.
Binding assays and other assays
[0248] In one aspect, an anti-CD98 antibody is tested for its
antigen binding activity. For
example, in certain embodiments, an anti-CD98 antibody is tested for its
ability to bind to exogenous or
endogenous CD98 expressed on the surface of a cell. A FACS assay may be used
for such testing.
[0249] A panel of monoclonal antibodies raised against CD98 may be grouped
based upon the
epitiopes they recognize, a process known as epitope binning. Epitope binning
is typically carried out
using competition assays, which evaluate an antibody's ability to bind to an
antigen in the presence of
another antibody. In an exemplary competition assay, immobilized CD98 is
incubated in a solution
56
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
comprising a first labeled antibody that binds to CD98 and a second unlabeled
antibody that is being
tested for its ability to compete with the first antibody for binding to CD98.
The second antibody may be
present in a hybridoma supernatant. As a control, immobilized CD98 is
incubated in a solution
comprising the first labeled antibody but not the second unlabeled antibody.
After incubation under
conditions permissive for binding of the first antibody to CD98, excess
unbound antibody is removed,
and the amount of label associated with immobilized CD98 is measured. If the
amount of label
associated with immobilized CD98 is substantially reduced in the test sample
relative to the control
sample, then that indicates that the second antibody is competing with the
first antibody for binding to
CD98. In certain embodiments, immobilized CD98 is present on the surface of a
cell or in a membrane
preparation obtained from a cell expressing CD98 on its surface.
[0250] High-throughput methods of epitope binning are also known in
the art. See, for
example, Jia et al., J. Immunol. Methods 2004, 288(1-2):91-98, describing a
method of multiplexed
competitive antibody binning for the characterization of monoclonal
antibodies; and Miller et al., J.
Immunol. Methods 2011, 365(1-2):118-25, describing epitope binning of murine
monoclonal antibodies
by a multiplexed pairing assay.
[0251] Epitope mapping
[0252] Epitope mapping is the process of identifying the binding
sites, or epitopes, of an
antibody on its target protein antigen. Antibody epitopes may be linear
epitopes or conformational
epitopes. Linear epitopes are formed by a continuous sequence of amino acids
in a protein.
Conformational epitopes are formed of amino acids that are discontinuous in
the protein sequence, but
which are brought together upon folding of the protein into its three-
dimensional structure.
[0253] A variety of methods are known in the art for mapping antibody epitopes
on target
protein antigens. These include mutagenesis methods, peptide scanning methods,
display methods,
methods involving and mass spectroscopy, and structural determination.
[0254] The site directed mutagenesis method involves targeted site-directed
mutagenesis
where critical amino acids are identified by systematically introducing
substitutions along the protein
sequence and then determining the effects of each substitution on antibody
binding. This may be done by
"alanine scanning mutagenesis," as described by Cunningham and Wells (1989)
Science 244: 1081-1085,
or some other form of point mutagenesis of amino acid residues in human CD98.
Mutagenesis studies,
however, may also reveal amino acid residues that are crucial to the overall
three-dimensional structure
of CD98 but that are not directly involved in antibody-antigen contacts, and
thus other methods may be
necessary to confirm a functional epitope determined using this method.
[0255] Shotgun mutagenesis mapping utilizes a comprehensive plasmid-
mutation library for
the target gene, with each clone in the library bearing a unique amino acid
mutation and the entire library
covering every amino acid in the target protein. The clones that constitute
the mutation library are
individually arranged in microplates, expressed within living mammalian cells,
and tested for
immunoreactivity with antibodies of interest. Amino acids critical for
antibody epitopes are identified by
a loss of reactivity and are then mapped onto a protein structure to visualize
epitopes. By automating the
57
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
analysis, new epitope maps can be derived within days to weeks. Because it
uses the native structure of
proteins within mammalian cells, the technique allows both linear and
conformational epitope structures
to be mapped on complex proteins. (Paes et al., J. Am. Chem. Soc. 131(20):
6952-6954 (2009); Banik
and Doranz, Genetic Engineering and Biotechnology News 3(2): 25-28 (2010)).
[0256] The epitope bound by an anti-CD98 antibody may also be determined using
peptide
scanning methods. In peptide scanning, libraries of short peptide sequences
from overlapping segments
of the target protein, CD98 are tested for their ability to bind antibodies of
interest. The peptides are
synthesized and screened for binding, e.g. using ELISA or BIACORE, or on a
chip, by any of the
multiple methods for solid-phase screening (Reineke et al, Curr. Opin.
Biotechnol. 12: 59-64, 2001) as in
the "pepscan" methodology (WO 84/03564; WO 93/09872). Such peptide screening
methods may not be
capable of detecting some discontinuous functional epitopes, i.e. functional
epitopes that involve amino
acid residues that are not contiguous along the primary sequence of the CD98
polypeptide chain.
[0257] A recently developed technology termed CLIPS (chemical linkage of
peptides onto
scaffolds) may be used to map conformational epitopes. The loose ends of the
peptides are affixed onto
synthetic scaffolds, so that the scaffolded peptide may be able to adopt the
same spatial structure as the
corresponding sequence in the intact protein. CLIPS technology is used to fix
linear peptides into cyclic
structures ('single-loop' format), and to bring together different parts of a
protein binding site (double-
loop', 'triple-loop', etc. format), so as to create conformational epitopes
that may be assayed for antibody
binding. (US Pat. No. 7,972,993).
[0258] The epitopes bound by antibodies of the invention may also be mapped
using display
techniques, including, for example, phage display, microbial display, and
ribosome/mRNA display as
described above. In these methods, libraries of peptide fragments are
displayed on the surface of the
phage or cell. Epitopes are then mapped by screening mAbs against these
fragments using selective
binding assays. A number of computational tools have been developed which
allow the prediction of
conformational epitopes based upon linear affinity-selected peptides obtained
using phage display.
(Mayrose et al., Bioinformatics 23: 3244-3246 , 2007). Methods are also
available for the detection of
conformational epitopes by phage display. Microbial display systems may also
be used to express
properly folded antigenic fragments on the cell surface for identification of
conformational epitopes
(Cochran et al., J. Immunol. Meth. 287: 147-158, 2004; Rockberg et al., Nature
Methods 5: 1039-1045,
2008).
102591 Methods involving proteolysis and mass spectroscopy may also
be used to determine
antibody epitopes (Baerga-Ortiz et al., Protein Sci. 2002 June; 11(6): 1300-
1308). In limited proteolysis,
the antigen is cleaved by different proteases, in the presence and in the
absence of the antibody, and the
fragments are identified by mass spectrometry. The epitope is the region of
the antigen that becomes
protected from proteolysis upon binding of the antibody (Suckau et al., Proc.
Natl. Acad. Sci. USA 87:
:9848-9852, 1990). Additional proteolysis based methods include, for example,
selective chemical
modification (Fiedler et al., Bioconjugate Chemistry 1998, 9(2): 236-234,
1998), epitope excision (Van
de Water et al., Clin. Immunol. Immunopathol. 1997, 85(3): 229-235, 1997), and
the recently developed
58
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
method of hydrogen-deuterium (H/D) exchange (Flanagan, N., Genetic Engineering
and Biotechnology
News 3(2): 25-28, 2010).
[0260] The epitope bound by antibodies of the present invention may also be
determined by
structural methods, such as X-ray crystal structure determination (e.g., WO
2005/044853), molecular
modeling and nuclear magnetic resonance (NMR) spectroscopy, including NMR
determination of the H-
D exchange rates of labile amide hydrogens in IL-23R when free and when bound
in a complex with an
antibody of interest (Zinn-Justin et al. (1992) Biochemistry 31:11335-11347;
Zinn-Justin et al. (1993)
Biochemistry 32:6884-6891).
[0261] Additional antibodies binding to the same epitope as an
antibody of the present
invention may be obtained, for example, by screening of antibodies raised
against CD98 for binding to
the epitope, by immunization of an animal with a peptide comprising a fragment
of human CD98
comprising the epitope sequence, or by selection of antibodies using phage
display for binding to the
epitope sequence. Antibodies that bind to the same functional epitope might be
expected to exhibit
similar biological activities, such as blocking a biological activity of CD98,
and such activities can be
confirmed by functional assays of the antibodies.
Additional Activity Assays
[0262] In one embodiment, an anti-CD98 antibody of the invention is
an antagonist antibody
that inhibits a biological activity of CD98. The anti-CD98 antibodies of the
invention may be assayed to
determine if they inhibit a biological activity of CD98, for example, binding
to light chains. In order to
determine whether CD98 antibodies of the invention inhibit binding to light
chains, the ability of the
CD98 antibodies to inhibit amino acid uptake in cancer cell lines is conducted
in accordance with the
method described in Kim et al., Biochim. Biophys. Acta 1565: 112-122, 2002.
[0263] In one aspect, purified anti-CD98 antibodies can be further
characterized by a series of
assays including, but not limited to, N-terminal sequencing, amino acid
analysis, non-denaturing size
exclusion high pressure liquid chromatography (HPLC), mass spectrometry, ion
exchange
chromatography and papain digestion.
[0264] In one embodiment, the invention contemplates an altered
antibody that possesses
some but not all effector functions, which make it a desirable candidate for
many applications in which
the half life of the antibody in vivo is important yet certain effector
functions (such as complement and
ADCC) are unnecessary or deleterious. In certain embodiments, the Fc
activities of the antibody are
measured to ensure that only the desired properties are maintained. In vitro
and/or in vivo cytotoxicity
assays can be conducted to confirm the reduction/depletion of CDC and/or ADCC
activities. For
example, Fc receptor (FcR) binding assays can be conducted to ensure that the
antibody lacks FcyR
binding (hence likely lacking ADCC activity), but retains FcRn binding
ability. An example of an in
vitro assay to assess ADCC activity of a molecule of interest is described in
U.S. Patent No. 5,500,362 or
5,821,337. Useful effector cells for such assays include peripheral blood
mononuclear cells (PBMC) and
Natural Killer (NK) cells. Alternatively, or additionally, ADCC activity of
the molecule of interest may
be assessed in vivo, e.g., in a animal model such as that disclosed in Clynes
et al. PNAS (USA) 95:652-
59
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
656 (1998). Clq binding assays may also be carried out to confirm that the
antibody is unable to bind
Clq and hence lacks CDC activity. To assess complement activation, a CDC
assay, e.g. as described in
Gazzano-Santoro et al., J. Immunol. Methods 202:163 (1996), may be performed.
FcRn binding and in
vivo clearance/half life determinations can also be performed using methods
known in the art.
[0265] Although the foregoing invention has been described in some detail
by way of
illustration and example for purposes of clarity of understanding, the
descriptions and examples should
not be construed as limiting the scope of the invention. The disclosures of
all patent and scientific
literatures cited herein are expressly incorporated in their entirety by
reference.
EXAMPLES
[0266] The following are examples of methods and compositions of the
invention. It is
understood that various other embodiments may be practiced, given the general
description provided
above.
EXAMPLE 1: IDENTIFICATION OF C 98 ON THE SURFACE OF ACUTE
MYELOGENOUS LEUKEMIA (AML) TUMOR CELLS
[0267] A total of 16 primary AML samples were obtained from Fred Hutchinson
Cancer
Research Center (FHCRC). Eleven samples from healthy donors were analyzed. To
monitor the quality
of individual AML samples, hematoxylin and eosin staining of AML blasts were
performed. Only
samples containing at least 75% tumor cells were analyzed. Additionally,
analysis was performed on 23
primary chronic lymphocytic leukemia (CLL) samples obtained from Billings
Clinic or the University of
Florida, and 27 primary colorectal carcinomas (CRC) with 22 normal adjacent
colon control samples
obtained from The Cooperative Human Tissue Network (CHTN) or the National
Disease Research
Interchange (NDRI). Sample handling was optimized so as to maximally maintain
cell viability during
sample isolation. Optimal labeling times for AML, CLL and CRC samples were
determined to allow for
efficient labeling without compromise of cellular integrity.
[0268] Surface tagged antigen profiling (sTAg) was used to
identify and quantitatively
profile the repertoire of surface proteins on cells in sixteen core AML
samples, five bone marrow
mononuclear cell (BMMC) control and six peripheral blood mononuclear cell
(PBMC) control samples,
20 core CLL samples, 27 CRC samples, and 22 normal adjacent colon samples. The
extracellular
domains of proteins associated with the AML tumor cell membranes of intact
primary tumor cells were
chemically tagged and then chromatographically enriched for tagged proteins
using a solid-phase affinity
resin. Eluted proteins were stored at -80 C prior to mass spectrometry
analysis as described below.
[0269] Proteins enriched by the sTAg method were identified and
quantified using high-
resolution, shotgun liquid chromatography tandem mass spectrometry (MS). A
hybrid ThermoFisher
LTQ-Orbitrap Velos mass spectrometer, which combines the sensitivity of a
linear ion trap with the high-
resolution and mass accuracy afforded by the revolutionary orbitrap mass
analyzer (Olsen et al., Mol.
Cell Proteomics 8:2759-2769, 2009) coupled to a nanoflow liquid chromatography
apparatus was
employed for shotgun-based, bottoms-up proteomics to determine the identities
and quantitative
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
abundance measurements of proteins in the AML cell surface enrichment
fractions (Yates et al., Annu.
Rev. Biomed. Eng. 11: 49-79, 2009). Tryptic digests from enriched surface
proteins were separated by
hydrophobicity via online, nanoflow liquid chromatography as peptide masses
and fragmentation patterns
were recorded dynamically by the mass spectrometer. To determine peptide and
protein identities, the
raw MS data was processed using the SEQUEST algorithm executed on a fast-
processing Sorcerer 2
platform (Lundgren et al., Curr. Protoc. Bioinformatics, Chapter 13: Unit
13.3, 2009), to determine best-
fit matches between experimental fragmentation patterns and those determined
in-silico from the human
proteome. Resulting matches were statistically validated using the
PeptideProphet (Keller et al., Anal.
Chem. 74: 5383-5392, 2002) and ProteinProphet (Nesvizhskii et al., Anal. Chem.
75: 4646-4658, 2003)
software tools to ensure the lowest possible false discovery rates (FDR) and
thus inclusion of only
robustly identified proteins in the candidate pool.
[0270] The relative quantitative levels of identified proteins in
the sTAg samples were
determined using the spectral counting method (Neilson et al., Proteomics 11:
535-553, 2011). Spectral
counting is based on the empirical demonstration that the number of assigned
(positively identified)
spectra associated with peptides from each protein correlates strongly with
that protein's relative
abundance in the original mixture (Liu et al.,Anal. Chem. 76:4193-4201, 2004).
Spectral counts of
identified peptides were obtained from proteomics analytical software
platforms including Scaffold
(Proteome Software) and ProteolQ (NuSep) that display, sort and filter the
results of SEQUEST-searched
mass spectrometry data. Raw spectral counts were transformed to percent
Normalized Spectral
Abundance Factor (%NASF) values (Zybailov et al., J. Proteome Res. 5: 2339-
2347, 2006) to account for
differences in protein length and variability in sample size. Selected
monoclonal antibodies were used to
validate the proteomic measurements using quantitative FACS as an independent,
external confirmatory
measure of the sTAg mass spectrometry-based proteomic profiling of the primary
tumor cell surface
expression.
[0271] Using sTAg, the heterodimeric type H transmembrane glycoprotein CD98
having the
amino acid sequence of SEQ ID NO.: 1 was identified as being present at high
density on the surface of
AML, CLL and CRC tumor cells. As shown in Fig. 1, using sTAg CD98 was
identified in 7 of 16
primary AML samples with a mean %NSAF of 0.11 and in 20 of 20 primary CLL
samples with a mean
%NSAF of 0.15. CD98 was identified in 5 out of 5 bone marrow mononuclear cells
(BMMC) and 5 out
of 6 peripheral blood mononuclear cells (PBMC) samples with mean %NSAFs of
0.05 and 0.06
respectively. Furthermore, CD98 was identified in 11 of 27 primary CRC samples
with a mean %NSAF
of 0.10 and in only one normal adjacent colon sample with a mean %NSAF less
than 0.01. Based on this
analysis, CD98 is substantially enriched on a significant portion of patient-
derived AML, CLL, and CRC
primary tumor specimens relative to relevant normal controls.
EXAMPLE 2: IDENTIFICATION OF CD98 IN TUMORS
61
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
[0272] Antibody titration experiments were conducted with anti-
CD98 monoclonal antibody
8-34B (see Example 3) and the isotype control antibody HB-121 to establish
dilutions that would result
in minimal background and maximal detection of signal. Serial dilutions were
performed using steam-
based antigen retrieval (pH 6.0 citrate buffer) at 1:50, 1:100, 1:200, and
1:400 on formalin-fixed,
paraffin-embedded (FFPE) tissues or on fresh frozen tissues. Frozen control
cell lines (F244 and F244-P)
and formalin fixed control cell lines (F244, RM, and F244-P) were provided by
Igenica and prepared by
LifeSpan. The dilutions of 8-34B of 1:20 and 1:50 were selected for the study
on formalin-fixed,
paraffin-embedded tissues, whereas 8-34B at a dilution of 1:400 was selected
for the study on fresh
frozen tissues.
[0273] The principal detection system consisted of a Vector anti-mouse
secondary antibody
(BA-2000) and a Vector ABC-AP kit (AK-5000) with a Vector Red substrate kit
(SK-5100), which was
used to produce a fuchsia-colored deposit. Tissues were also stained with
positive control antibodies (to
CD31 and vimentin) to ensure that the tissue antigens were preserved and
accessible for
immunohistochemical analysis. Only tissues that were positive for CD31 and
vimentin staining were
selected for the remainder of the study.
[0274] Antibody 8-34B, at dilutions of 1:20 and 1:50, showed
positive staining within ten
out of 15 malignant melanomas and four out of 18 lung carcinomas on formalin-
fixed, paraffin-
embedded tissues. In addition, at a dilution of 1:400, antibody 8-34B showed
positive staining of six of
the six frozen lung carcinoma samples and also of two of the two frozen
melanoma samples.
[0275] Table 1: Frequency of positive C 98 staining in lung carcinomas and
melanomas
Carcinoma Frequency
Non-small cell lung 4/18 (FFPE); 6/6 (Frozen)
Melanoma 10/15 (FFPE); 2/2 (Frozen)
EXAMPLE 3: PREPARATION OF MONOCLONAL ANTIBODIES TO C 98
[0276] Monoclonal antibodies were prepared in accordance with a
general method as
described in "Antibodies A Laboratory Manual" (Harlow and Lane 1988 CSH
Press). Male 12956/SvEv
mice purchased from Taconic Farms were used for immunization. Mice were
immunized via
subcutaneous injection in the flank with 106 human CD98 (huCD98) expressing
tumor cells. On day 39
post immunization, mice were boosted intraperitoneally with 5 million huCD98
expressing tumor cells.
Spleens were harvested on day 42. Individual splenocytes were prepared and
fused with CRL-2016
myeloma cells (ATCC) using a PEG based method as generally described in
"Antibodies A Laboratory
Manual" (Harlow and Lane 1988 CSH Press) to establish hybridomas.
[0277] Hybridomas were grown in 384 well tissue culture plates and
supernatants from
individual wells were screened by ELISA for production of antibodies
recognizing huCD98. Positive
wells were then transferred to 48 well plates, expanded, and supernatants were
collected for huCD98
binding confirmation by ELISA. Individual hybridomas producing anti-huCD98
antibodies were
62
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
established as confirmed unique clones producing monoclonal anti-huCD98
antibodies by plating single
hybridoma cells in wells of 96 well plates. These cells were grown into
colonies and the supernatant from
these individual colonies was screened by ELISA to confirm monoclonal antibody
binding to huCD98.
Clonal hybridomas were injected into pristane treated Balb/C mice to produce
ascites. Ascites was
collected and purified using Gammabind sepharose (GE Healthcare product code
17-0885-01), Protein A
IgG binding buffer (Thermo Scientific part number 21001), and IgG elution
buffer (Thermo Scientific
part number 21004) following the general antibody purification protocol
published by Thermo Scientific
(Product Instructions #21001).
102781 The nucleic acid and amino acid sequences for the heavy chain
and light chain variable
regions of the antibody 8-34B are shown below:
102791 8-34B heavy chain variable region
102801 CAGGTGCAGCTGAAGGAGTCCGGCCCCGGCCTGGTGGCCCCCTCCCAGTCCCT
GTCCATCACCTGCACCGTGTCCGGCTTCTCCCTGACCTCCTACGGCGTGCACTGGATCCGCCAG
CCCCCCGGCAAGGGCCTGGAGTGGCTGGGCCTGATCTGGGCCGGCGGCTCCATCAACTACAACT
CCGCCCTGATGTCCCGCCTGTCCATCTCCAAGGACAACTCCAAGTCCCAGGTGTTCCTGAAGAT
GAACTCCCTGGAGACCGAGGACACCGCCATGTACTACTGCGCCCGCAAGGGCCACATGTACTCC
TACGCCATGGACTACTGGGGCCAGGGCACCTCCGTGACCGTGTCCTCC ( SEQ ID NO: 3)
10281] QVQLKESGPGLVAPSQSLS ITCTVSGFSLTSYGVHWIRQPPGKGLEWLGLIWA
GGS INYNSALMSRLS I SKDNSKSQVFLKMNSLETEDTAMYYCARKGHMYSYAMDYWGQGTSVTV
SS (SEQ ID NO:4 ; CDRs are underlined)
102821 8-34B light chain variable region
102831 GACATCGTGATGACCCAGTCCCCCTCCTCCCTGACCGTGACCGCCGGCGAGAA
GGTGACCATGTCCTGCAAGTCCTCCCAGTCCCTGCTGAACTCCGGCAACCAGAAGACCTACCTG
ACCTGGTACCAGCAGAAGCCCGGCCAGCCCCCCAAGCTGCTGATCTACTGGGCCTCCACCCGCG
AGTCCGGCGTGCCCGACCGCTTCACCGGCTCCGGCTCCGGCACCGAGTTCACCCTGACCATCTC
CTCCGTGCAGGCCGAGGACCTGGCCGTGTACTACTGCCAGAACGACTACTCCTACCCCCCCTGG
ACCTTCGGCGGCGGCACCAAGCTGGAGATCAAG ( SEQ ID NO : 5 )
102841 DIVMTQSPSSLTVTAGEKVTMSCKSSQSLLNSGNQKTYLTWYQQKPGQPPKLL
IYWASTRESGVPDRFTGSGSGTEFTLTISSVQAEDLAVYYCQNDYSYPPWTFGGGTKLEIK
(SEQ ID NO: 6; CDRs are underlined)
102851 The nucleic acid and amino acid sequences for the heavy chain
and light chain variable
regions of the antibody 18-2A 2.2 are shown below:
102861 18-2A 2.2 heavy chain variable region
102871 CAGGTGCAGCTGCAGCAGTCCGGCGCCGAGCTGGTGAAGCCCGGCGCCTCCGT
GAAGCTGTCCTGCAAGGCCTCCGGCTACACCTTCACCTCCTACTACATGTACTGGGTGAAGCAG
CGCCCCGGCCAGGGCCIGGAGIGGATCGGCGTGATCAACCCCGGCTCCGGCATCACCAACTACA
63
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
ACGAGAAGTTCAAGGGCAAGGCCACCCTGACCGCCGACAAGTCCTCCAACACCGCCTACATGCA
GCTGTCCTCCCTGTCCTCCGACGACTCCGCCGTGTACTTCTGCTCCGGCTCCGCCAACTGGTTC
GCCTACTGGGGCCAGGGCACCCTGGTGACCGTGTCCGCC (SEQ ID NO: /)
[0288] QVQLQQSGAELVKPGASVKLSCKASGYTFTSYYMYWVKQRPGQGLEWIGVINP
GSGITNYNEKFKGKATLTADKSSNTAYMQLSSLSSDDSAVYFCSGSANWFAYWGQGTLVTVSA
(SEQ ID NO:8; CDRs are underlined)
[0289] 18-2A 2.2 light chain variable region
[0290] GACATCGTGATGTCCCAGTCCCCCTCCTCCCTGGCCGTGTCCGTGGGCGAGAA
GGTGACCATGTCCTGCAAGTCCTCCCAGTCCCTGCTGTACTCCTCCAACCAGAAGAACTACCTG
GCCTGGTACCAGCAGAAGCCCGGCCAGTCCCCCAAGCTGCTGATCTACTGGGCCTCCACCCGCG
ACTCCGGCGTGCCCGACCGCTTCACCGGCTCCGGCTCCGGCACCGACTTCACCCTGACCATCTC
CTCCGTGAAGGCCGAGGACCTGGCCGTGTACTACTGCCAGCGCTACTACGGCTACCCCTGGACC
TTCGGCGGCGGCACCAAGCTGGAGATCAAG (SEQ ID NO: 9)
102911 DIVMSQSPSSLAVSVGEKVTMSCKSSQSLLYSSNQKNYLAWYQQKPGQSPKLL
IYWASTRDSGVPDRFTGSGSGTDFTLTISSVKAEDLAVYYCQRYYGYPWTFGGGTKLEIK
(SEQ ID NO:10; CDRs are underlined)
[0292] The nucleic acid and amino acid sequences for the heavy
chain and light chain variable
regions of the antibody 18-2A 7.1 are shown below:
[0293] 18-2A 7.1 heavy chain variable region
102941 CAGGTGCAGCTGCAGCAGTCCGGCGCCGAGCTGGTGCGCCCCGGCACCTCCGT
GAAGGTGTCCTGCAAGGCCTCCGGCAACGCCTTCACCAACTACCTGATCGAGTGGATCAAGCAG
CGCCCCGGCCAGGGCCTGGAGTGGATCGGCGTGATCAACCCCGGCTCCGGCATCACCAACTACA
ACGAGAAGTTCAAGGGCAAGGCCACCCTGACCGCCGACAAGTCCTCCAACACCGCCTACATGCA
GCTGTCCTCCCTGTCCTCCGACGACTCCGCCGTGTACTTCTGCTCCGGCTCCGCCAACTGGTTC
GCCTACTGGGGCCAGGGCACCCTGGTGACCGTGTCCGCC (SEQ ID NO:11)
[0295] QVQLQQSGAELVRPGTSVKVSCKASGNAFTNYLIEWIKQRPGQGLEWIGVINP
GSGITNYNEKFKGKATLTADKSSNTAYMQLSSLSSDDSAVYFCSGSANWFAYWGQGTLVTVSA
(SEQ ID NO:12; CDRs are underlined)
102961 18-2A 7.1 light chain variable region
102971 GACATCGTGATGTCCCAGTCCCCCTCCTCCCTGGCCGTGTCCGTGGGCGAGAA
GGTGACCATGTCCTGCAAGTCCTCCCAGTCCCTGCTGTACTCCTCCAACCAGAAGAACTACCTG
GCCTGGTACCAGCAGAAGCCCGGCCAGTCCCCCAAGCTGCTGATCTACTGGGCCTCCACCCGCG
ACTCCGGCGTGCCCGACCGCTTCACCGGCTCCGGCTCCGGCACCGACTTCACCCTGACCATCTC
CTCCGTGAAGGCCGAGGACCTGGCCGTGTACTACTGCCAGCGCTACTACGGCTACCCCTGGACC
TTCGGCGGCGGCACCAAGCTGGAGATCAAG (SEQ ID NO:13)
64
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
[0298] DIVMSQSPSSLAVSVGEKVTMSCKSSQSLLYSSNQKNYLAWYQQKPGQSPKLL
TYWASTRDSGVPDRFTGSGSGTDFTLTISSVKAEDLAVYYCQRYYGYPWTEGGGTKLEIK
(SEQ ID NO:14; CDRs are underlined)
[0299] The nucleic acid and amino acid sequences for the heavy chain
and light chain variable
regions of the antibody 1-47C are shown below:
[0300] 1-47C heavy chain variable region
103011 CAGGTGCAGTTGAAGGAGTCAGGACCTGGCCTGGTGGCGCCCTCACAGAGCCT
GTCCATCACTTGCACTGTCTCTGGGTTTTCATTAACCACCTATGGTGTACACTGGGTTCGCCAG
CCTCCAGGAAAGGGTCTGGAGTGGCTGGGAGTGATGTGGACTAATGGAATCACAAATTATAATT
CGGCTCTCATGTCCAGACTGAGCATCAGCAAAGACAACTCCAAGAGCCAAGTTTTCTTAAAAAT
GAACAGTCTGCAAACTGATGACACAGCCATGTACTACTGTGCCAGAGGAGGACACTACGGTAGT
ACCTCCTATGCTATGGACTTCTGGAGTCAAGGA (SEQ ID NO: 30)
[0302] QVQLKESGPGLVAPSQSLSITCTVSGFSLTTYGVHWVRQPPGKGLEWLGVMWT
NGITNYNSALMSRLSISKDNSKSQVFLKMNSLQTDDTAMYYCARGGHYGSTSYAMDFWSQG
(SEQ ID NO:31; CDRs are underlined)
106031 1-47C light chain variable region
[0304] GACATCCAGATGACTCAGTCTCCAGCCTCCCTATCTGCATCTGTGGGAGAAAC
TGTCACCATCACATGTCGAGCAAGTGGGAATATTCACAATTATTTAACATGGTATCAGCAGAAA
CAGGGAAAATCTCCTCAGCTCCTGGTCTATACTGCAAAAACCTTAGCAGATGGTGTGCCATCAA
GGTTCAGTGGCAGTGGATCAGGAACACAATATTCTCTCAAGATCAACAGCCTGCAGCCTGAAGA
TTTTGGGAGTTATTACTGTCAACATTTTTGGAATACTCCTTACACGTTCGGAGGGGGGACCAAG
CTGGAAATAAAACGGGCTGATGCTGCACCAACTGTATCCATC (SEQ ID NO: 32)
[0305] DIQMTQSPASLSASVGETVTITCRASGNIHNYLTWYQQKQGKSPQLLVYTAKT
LADGVPSRFSGSGSGTQYSLKINSLQPEDEGSYYCQHFWNTPYTEGGGTKLEIK (SEQ ID
NO: 33; CDRs are underlined)
[0306] The nucleic acid and amino acid sequences for the heavy chain
and light chain variable
regions of the antibody 1-115A are shown below:
[0307] 1-115A heavy chain variable region
[0308] CAGGTGCAGCTGGAGGAGTCAGGACCTGGCCTGGTGGCGACCTCACAGAGCCT
GTCCATCACTTGCACTGTCTCTGGGTTTTCATTAACCAACTGTGGTGTACACTGGGTTCGCCAG
CCTCAAGGAAAGGGTCTAGAGTGGCTGGGAGTGATATGGCCTAATGGAATCACAATTTATAATT
CGGGTCTCATGTCCAGACTGAGTATCAGCAAAGACAACTCCAAGAGCCAAGTT T TCTTAAAAAA
GAACAGTCTGCAAACTGATGACACAGCCATGTACTACTGTGCCAGAGGAGGACATTACGGTAGT
AGCTCCTATGCTATGGACTACTGGAGTCAAGGA (SEQ ID NO: 34)
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
[0309] QVQLEESGPGLVATSQSLSITCTVSGESLTNCGVHWVRQPQGKGLEWLGVIWP
NGITIYNSGLMSRLSISKDNSKSQVFLKKNSLQTDDTAMYYCARGGHYGSSSYANDYWSQG
(SEQ ID NO:35; CDRs are underlined)
[0310] 1-115A light chain variable region
[0311] GACATCCAGATGACTCAGTCTCCAGCCTCCCTATCTGCATCTGTGGGAGAAAC
TGTCACCATCACATGTCGAGCAAGTGGGAATATTCACAATTATTTAACATGGTATCAGCAGAAA
CCOGGAAAATCTCCTCAACTCCTGGTCTATACTGCAAAAACCTTAGCAGATGGTGTGCCATCAA
GGTTCAGTGGCAGTGGATCAGGAACACAATATTCTCTCAAGATCAACAGCCTGCAGCCTGAAGA
TTTTGGGAGTTATTACTGTCAACATTTTTGGAATACTCCTTACACATTCGGAGGGGGGACCAAG
CTGGAAATAAAACGGGCTGATGCTGCACCAACTGTATCCATCTTCCCACCATCCAGTAAGC
(SEQ ID NO: 36)
[0312] DIQMTQSPASLSASVGETVTITCRASGNIHNYLTWYQQKPGKSPQLLVYTAKT
LADGVPSRFSGSGSGTQYSLKINSLQPEDEGSYYCQHFWNTPYTEGGGTKLEIK (SEQ ID
NO:37; CDRs are underlined)
EXAMPLE 4: ISOTYPING AND BINNING OF MONOCLONAL ANTIBODIES
[0313] Individual hybridoma supernatants containing antibodies that
recognize huCD98 were
assessed for isotype by detection on ELISA with isotype specific secondary
antibodies purchased from
Jackson lmmunologicals (Goat x IgG1 HRP ¨ Product# 115-035-206, Goat x IgG2a
HRP - Product#
115-035-206, Goat x IgG2b HRP ¨ Product# 115-035-207, Goat x IgG3 HRP ¨
Product# 115-035-209).
[0314] A competition ELISA was performed to establish competitive
binding bins. Individual
anti-huCD98 isotyped antibody containing hybridoma supernatants were allowed
to bind to huCD98 in
individual wells of an ELISA plate. After 1 hour, the wells were washed and
fixed using 4%
paraformaldehyde. Then individual anti-huCD98 isotyped antibody (of a
different isotype) containing
hybridoma supernatants were allowed to bind to huCD98 in individual wells of
an ELISA plate for an
hour. After washing, the wells were incubated with a specific secondary
antibody (Jackson
Immunologicals Goat x IgG2a HRP - Product# 115-035-206) and detected with
Supersignal ELISA Pico
Chemiluminescent substrate (Thermo Scientific ¨ Product# 37069). Individual
IgG2a isotype antibodies
that were able to bind in the presence of an IgG1 are considered to be in a
unique epitope bin from that
particular IgGl. Individual IgG2a isotype antibodies that were unable to bind
in the presence of an IgG1
are considered to be in the same epitope bin as that particular IgGl. In this
way multiple epitope bins
were defined for huCD98 binding antibodies, as illustrated in Fig. 2.
EXAMPLE 5: BINDING AFFINITY
[0315] Purified anti-CD98 monoclonal antibodies are tested for affinity by
the general method
published by Carderelli et al. (2002) Cancer Immunol Immunother 51; 15-24.
Briefly, CD98 expressing
cells are incubated with different amounts of anti-CD98 monoclonal antibodies
overnight, then assessed
66
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
by FACS with a goat anti-human Fc specific or anti-mouse Fc specific PE-
conjugated secondary
antibody (Jackson Immunologicals). FACS data is analyzed using Graphpad Prism
software and Kds are
determined using the Graphpad Prism Kd calculation tool.
EXAMPLE 6: PRODUCTION AND CHARACTERIZATION OF CHIMERIC ANTIBODIES
[0316] Total RNA was extracted from hybridomas producing anti-CD98 monoclonal
antibodies using the Qiagen RNeasy Mini kit (Cat No. 74104), followed by the
Qiagen OneStep RT-PCR
Kit (Cat No. 210210). RT-PCR was performed with primer sets specific for
murine heavy and light chain
sequences. For each RNA sample, 12 individual heavy chain and 11 light chain
RT-PCR reactions were
set up using degenerate forward primer mixtures covering the leader sequences
of murine variable
regions. Reverse primers were located in the constant regions of murine heavy
and light chains. The RT-
PCR products from the first-round reactions were further amplified in the
second-round PCR. 12
individual heavy chain and 11 light chain RT-PCR reactions were set up using
semi-nested primer sets
specific for antibody variable regions. PCR reactions were run on agarose gels
and heavy and light chain
PCR products were cut from the gel and cloned into sequencing vectors. 10-20
clones per hybridoma
were sequenced to determine the anti-CD98 monoclonal antibody variable region.
Heavy chain variable
regions were then cloned in-frame into a vector containing human IgG1 heavy
chain constant region
sequence and light chain variable regions were cloned in-frame into a vector
containing human kappa
light chain constant region sequence. Chimeric antibodies were generated from
transient transfection of
HEK293 Freestyle cells and purified using the methodology described in Example
3.
[0317] Purified chimeric antibodies were subjected to binding
analysis as described in
Example 4 for the mouse monoclonal anti-CD98 antibodies. Fig. 3A shows the
results of competitive
binding assays for the 8-34B, 18-2A 2.1, 18-2A 2.2 and 18-2A 7.1 chimeric
antibodies. Reference
antibodies 1-4 are as in Fig. 2, while "isotype" is a control IgG2a antibody
that does not bind to CD98.
The results shown in Fig. 3A demonstrate that the chimeric antibodies retain
the epitope binding
specificities of the murine antibodies from which they were derived. The
binding affinities of the murine
and chimeric anti-CD98 monoclonal antibodies were determined by FACS analysis
as described in
Example 5 with colon cancer cell line DLD1. The Kd values (ranging from 0.9nM
to 4.5 nM) are shown
in Fig. 3B, indicating that all these recombinant antibodies retain high-
affinity binding to CD98,
comparable to their parental murine antibodies. Purified chimeric monoclonal
antibodies were also
subjected to FACS analysis as described in Example 5 with three AML primary
tumor samples and a cell
line expressing cynomolgus monkey CD98 (cynCD98). The results shown in Fig. 3C
demonstrate that all
chimeric antibodies retained the binding to human CD98 on AML cells and
cynCD98.
[0318] The binding affinities of the murine and chimeric anti-CD98
monoclonal antibodies
were determined by on/off rate determination using a BIACORE system (reviewed
in Lipschultz et al.,
Methods 20: 310-318, 2000). As shown in Table 2, the affinities of the
chimeric antibodies were similar
to those of the parent murine antibodies. Data for the humanized antibody 8-
34B H2 Ll (see Example 7)
is also shown.
67
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
[0319] Table 2
Antibody ka kd Kd [nM]
(on-rate) (off-rate)
[M-1.s-1] [s-1]
18-2A 6.2 x 105 3.5 x10-5 0.56
18-2A ch7.1 1.2 x 105 2.6 x10-5 0.22
18-2A ch2.2 2.5 x 105 3.5 x10-4 1.4
8-34B 8.2 x 104 1.6 x10-4 2.0
8 -34B ch 1.7x 105 1.6 x10-4 0.94
8-34B H2 L1 2.5 x 105 3.2 x10-4 1.3
EXAMPLE 7: PREPARATION OF HUMANIZED ANTIBODIES
[0320] A humanized form of the murine monoclonal antibody 8-34B was made by
grafting the CDRs of the murine heavy chain and light chain variable domains
into the human
acceptor framework regions as shown in Fig. 4. The humanized 8-34B light chain
variable
domain Ll (SEQ ID NO:15) was constructed by grafting the CDRs of the murine
light chain
into the human acceptor sequence (GenBank Accession No. ACJ71709.1). The 8-34B
humanized light chain variable domain L2 (SEQ ID NO:16) was constructed by
replacing two
residues in FR3 of the human acceptor light chain with the corresponding
residues of the murine
monoclonal antibody (amino acid substitutions S63T andD7OE by Kabat numbering;
see Fig.
4A). The 8-34B humanized heavy chain HI (SEQ ID NO:17) was constructed by
grafting the
CDRs of the murine heavy chain into the human acceptor sequence (GenBank
Accession No.
137782). The 8-34B humanized heavy chain variable domain H2 (SEQ ID NO:18) was
constructed by replacing one residue in FR2 and two residues in FR3 of the
human acceptor
heavy chain with the corresponding residues of the murine monoclonal antibody,
resulting in the
amino acid substitutions I48L, V71K and F78V by Kabat numbering (see Fig. 4B).
The 8-34B
humanized heavy chain variable domain H3 (SEQ ID NO:19) added two additional
substitutions
back to the murine residue in FR3, V67L and T73N, as shown in Fig. 4B.
[0321]
The nucleic acid and amino acid sequences for the H2 heavy chain and the Ll
light
chain variable regions of the humanized antibody 8-34B are shown below:
[0322] Humanized 8-34B heavy chain variable region H2
[0323] CAGGTGCAGCTGCAGGAGTCCGGCCCCGGCCTGGTGAAGCCCTCCGAGACCCT
GTCCCTGACCTGCACCGTGTCCGGCTTCTCCCTGACCTCCTACGGCGTGCACTGGATCCGCCAG
CCCCCCGGCAAGGGCCTGGAGTGGCTGGGCCTGATCTGGGCCGGCGGCTCCATCAACTACAACT
CCGCCCTGATGTCCCGCGTGACCATCTCCAAGGACACCTCCAAGAACCAGGTGTCCCTGAAGCT
68
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
GTCCTCCGTGACCGCCGCCGACACCGCCGTGTACTACTGCGCCCGCAAGGGCCACATGTACTCC
TACGCCATGGACTACTGGGGCCAGGGCACCCTGGTGACCGTGTCCTCC (SEQ ID NO: 25)
[0324] QVQLQESGPGLVKPSETLSLTCTVSGFSLTSYGVHWIRQPPGKGLEWLGLIWA
GGSTNYNSALMSRVTISKDTSKNQVSLKLSSVTAADTAVYYCARKGHMYSYAMDYWGQGTLVTV
SS (SEQ ID NO: 18; CDRs are underlined)
[0325] Humanized 8-34B light chain variable region Ll
[0326] GACATCGTGATGACCCAGTCCCCCGACTCCCTGGCCGTGTCCCTGGGCGAGCG
CGCCACCATCAACTGCAAGTCCTCCCAGTCCCTGCTGAACTCCGGCAACCAGAAGACCTACCTG
ACCTGGTACCAGCAGAAGCCCGGCCAGCCCCCCAAGCTGCTGATCTACTGGGCCTCCACCCGCG
AGTCCGGCGTGCCCGACCGCTTCTCCGGCTCCGGCTCCGGCACCGACTTCACCCTGACCATCTC
CTCCCTGCAGGCCGAGGACGTGGCCGTGTACTACTGCCAGAACGACTACTCCTACCCCCCCTGG
ACCTTCGGCCAGGGCACCAAGGTGGAGATCAAG (SEQ ID NO: 24)
[0327] DIVMTQSPDSLAVSLGERATINCKSSQSLLNSGNQKTYLTWYQQKPGQPPKLL
TYWASTRESGVPDRFSGSGSGTDFTLTISSLQAEDVAVYYCQNDYSYPPWTFGQGTKVEIK
(SEQ ID NO: 15; CDRs are underlined)
[0328] A humanized form of the murine monoclonal antibody 18-2A was made by
grafting the CDRs of the murine heavy chain and light chain variable domains
from the chimeric
antibody 18-2A 7.1 into the human acceptor framework regions. The humanized 18-
2A 7.1 light
chain variable domain Ll (SEQ ID NO:20) was constructed by grafting the CDRs
of the murine
light chain into the human acceptor sequence (GenBank Accession No.
ACJ71709.1). The 18-
2A 7.1 light chain variable domain L2 (SEQ ID NO:21) was constructed by
replacing certain
human framework residues with the corresponding residues from the murine
monoclonal
antibody. The 18-2A 7.1 humanized heavy chain variable domain H1 (SEQ ID NO:
22) was
constructed by grafting the CDRs of the murine heavy chain into the human
acceptor sequence.
The 18-2A 7.1 humanized heavy chain variable domain H2 (SEQ ID NO: 23) was
constructed
by replacing certain human framework residues with the corresponding residues
from the murine
monoclonal antibody.
[0329] The nucleic acid and amino acid sequences for the heavy
chain and light chain variable
regions of the humanized antibody 18-2A 7.1 are shown below:
[0330] Humanized 18-2A 7.1 heavy chain variable region H1
[0331] CAGGTGCAGCTGGTGCAGTCCGGCGCCGAGGTGAAGAAGCCCGGCTCCTCCGT
GAAGGIGTCCTGCAAGGCCTCCGGCAACGCCTTCACCAACTACCTGATCGAGTGGGTGCGCCAG
GCCOCCGGCCAGGGCCTGGAGTGGATGGGCGTGATCAACCCCGGCTCCGGCATCACCAACTACA
ACGAGAAGITCAAGGGCAAGGCCACCATCACCGCCGACAAGTCCACCTCCACCGCCTACATGGA
GCTGTCCTCCCTGCGCTCCGAGGACACCGCCGTGTACTACTGCTCCGGCTCCGCCAACTGGTTC
GCCTACTGGGGCCAGGGCACCCTGGTGACCGTGTCCTCC (SEQ ID NO: 26)
69
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
[0332] QVQLVQSGAEVKKPGSSVKVSCKASGNAFTNYLIEWVRQAPGQGLEWMGVINP
GSGITNYNEKFKGKATITADKSTSTAYMELSSLRSEDTAVYYCSGSANWFAYWGQGTLVTVSS
[0333] (SEQ ID NO: 22; CDRs are underlined)
[0334] Humanized 18-2A 7.1 light chain variable region L1
[0335] GACATCGTGATGACCCAGTCCCCCGACTCCCTGGCCGTGTCCCTGGGCGAGCG
CGCCACCATCAACTGCAAGTCCTCCCAGTCCCTGCTGTACTCCTCCAACCAGAAGAACTACCTG
GCCTGGTACCAGCAGAAGCCCGGCCAGCCCCCCAAGCTGCTGATCTACTGGGCCTCCACCCGCG
ACTCCGGCGTGCCCGACCGCTTCTCCGGCTCCGGCTCCGGCACCGACTTCACCCTGACCATCTC
CTCCCTGCAGGCCGAGGACGTGGCCGTGTACTACTGCCAGCGCTACTACGGCTACCCCTGGACC
TTCGGCGGCGGCACCAAGGTGGAGATCAAG (SEQ ID NO: 27)
[0336] DIVMTQSPDSLAVSLGERATINCKSSQSLLYSSNQKNYLAWYQQKPGQPPKLL
IYWASTRDSGVPDRFSGSGSGTDFTLTISSLQAEDVAVYYCQRYYGYPWTFGGGTKVEIK
(SEQ ID NO: 20; CDRs are underlined)
[0337] Humanized 18-2A 7.1 heavy chain variable region H2
[0338] CAGGTGCAGCTGGTGCAGTCCGGCGCCGAGGTGAAGAAGCCCGGCTCCTCCGT
GAAGGTGTCCTGCAAGGCCTCCGGCAACGCCTTCACCAACTACCTGATCGAGTGGATCCGCCAG
GCCCCCGGCCAGGGCCTGGAGTGGATCGGCGTGATCAACCCCGGCTCCGGCATCACCAACTACA
ACGAGAAGTTCAAGGGCAAGGCCACCCTGACCGCCGACAAGTCCACCTCCACCGCCTACATGGA
GCTGTCCTCCCTGCGCTCCGAGGACACCGCCGTGTACTACTGCTCCGGCTCCGCCAACTGGTTC
GCCTACTGGGGCCAGGGCACCCTGGTGACCGTGTCCTCC (SEQ ID NO: 28)
[0339] QVQLVQSGAEVKKPGSSVKVSCKASGNAFTNYLIEWIRQAPGQGLEWIGVINP
GSGITNYNEKFKGKATLTADKSTSTAYMELSSLRSEDTAVYYCSGSANWFAYWGQGTLVTVSS
(SEQ ID NO: 23; CDRs are underlined)
[0340] Humanized 18-2A 7.1 light chain variable region L2
[0341] GACATCGTGATGACCCAGTCCCCCGACTCCCTGGCCGTGTCCCTGGGCGAGCG
CGCCACCATCAACTGCAAGTCCTCCCAGTCCCTGCTGTACTCCTCCAACCAGAAGAACTACCTG
GCCTGGTACCAGCAGAAGCCCGGCCAGCCCCCCAAGCTGCTGATCTACTGGGCCTCCACCCGCG
ACTCCGGCGTGCCCGACCGCTTCACCGGCTCCGGCTCCGGCACCGACTTCACCCTGACCATCTC
CTCCCTGCAGGCCGAGGACGTGGCCGTGTACTACTGCCAGCGCTACTACGGCTACCCCTGGACC
TTCGGCGGCGGCACCAAGGTGGAGATCAAG (SEQ ID NO: 29)
[0342] DIVMTQSPDSLAVSLGERATINCKSSQSLLYSSNQKNYLAWYQQKPGQPPKLL
IYWASTRDSGVPDRFTGSGSGTDFTLTISSLQAEDVAVYYCQRYYGYPWTEGGGTKVEIK
(SEQ ID NO: 21; CDRs are underlined)
70
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
EXAMPLE 8: ANTI-CD-98 MONOCLONAL ANTIBODY-MEDIATED INHIBITION OF
TUMORS IN VIVO
Antibody efficacy on tumor growth and metastasis formation is studied, e.g.,
in mouse
subcutaneous or orthotopic cancer xenograft models. The antibodies can be
unconjugated, or can be
conjugated to a therapeutic agent, as appreciated in the art.
Monoclonal antibodies are raised against CD-98 as described in Example 1, and
purified and
characterized as described above. Chimeric or humanized antibodies as
described above may also be
used. A therapeutic monoclonal antibody or a cocktail comprising a mixture of
individual monoclonal
antibodies is prepared and used for the treatment of mice receiving
subcutaneous or orthotopic injections
of tumor xenografts.
Subcutaneous tumors are generated by injection of 1 x 107 cancer cells in a
mixture of PBS
(without magnesium or calcium) and BD Matrigel (BD Biosciences) at a 1:1 ratio
in the right flank of
female SCID or nu "1" mice. The injected total volume per mouse is 200m1 with
50% being Matrigel (BD
Biosciences). Mice are randomized once tumors reach a size between 65-200mm3.
Antibodies are
administered weekly, and body weights and tumors are measured once and twice
weekly, respectively.
Tumor volume is calculated as described (van der Horst et al. (2009) Neoplasia
11: 355-364). As a
negative control, mice are injected with either purified mouse IgG or PBS; or
a purified monoclonal
antibody that recognizes an antigen other than CD98.
EXAMPLE 9: EFFECT OF CD-98 MONOCLONAL ANTIBODIES ON THE GROWTH OF B-
CELL LYMPHOMA XENOGRAFTS IN MICE
[0343] The Ramos (B-cell lymphoma) cell line was obtained from ATCC and
cultured
according to the suppliers' protocols. Animals were obtained from Charles
River Laboratories.
103441 4-6 week-old immunodeficient SCID female mice on a CB.17 background
were
subcutaneously injected on the right flank with 1x107 viable cells in a
mixture of PBS (without
magnesium or calcium) and BD Matrigel (BD Biosciences) at a 1:1 ratio. The
injected total volume per
mouse was 200 1 with 50% being Matrigel (BD Biosciences). Once the tumor
reached a size between
65-200mm3 mice were randomized. Antibodies were administered weekly, and body
weights and tumors
were measured once and twice weekly, respectively. Tumor volume was calculated
as described (van der
Horst et al. (2009) Neoplasia 11: 355-364). All the experiments were performed
on groups of at least 7
animals per experimental point. Animal experiments were performed in
accordance with protocols
approved by the Igenica Inc. Institutional Review Board - Animal Care and Use
Committee.
[0345]
Statistical significance between treatment and control groups was calculated
using the
Graphpad Prism software package and applying Student's two-tailed t-test. A p-
value of less than 0.05
was considered significant. Doubling time and time to progression analysis was
calculated as described
in Daniel et al. (2007) Blood 110:4037-4046.
71
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
[0346] Rituximab (anti-CD20 antibody) was used as a positive
therapeutic control antibody.
Antibody HB121 was an IgG2a negative control. Rituximab and the anti-CD98
antibodies 8-93A and 18-
3A are IgG1 antibodies, whereas all other anti-CD98 antibodies are IgG2a
antibodies.
[0347] Treatment with anti-CD98 antibodies was shown to induce strong tumor
growth
inhibitiion in established B cell lymphoma (Ramos) tumors. Notably, anti-CD98
antibody treatment was
superior to rituximab (see Fig. SA, Fig. 5B) in inducing tumor growth
inhibition.
[0348] Anti-CD98 antibodies were also shown to significantly prolong
time to progression of
treated RAMOS tumors. The tumor doubling time of the tumor regrowth data from
Figs. 5A-C was
calculated as described above and used for further prediction of time to
progression (TTP). TTP was then
extrapolated for each animal within the treatment groups, until 2000mm3 would
have been reached and
graphed as a Kaplan-Meier curve, as shown in Figs. 6A-C. As shown in Figs. 6A-
C, various anti-CD98
antibodies are superior to rituximab in prolonging time to progression in
Ramos tumors.
[0349] The starting tumor volumes of established Ramos tumors were increased
in order to
assess the potential maximum therapeutic efficacy of anti-CD98 antibodies.
Table 3 shows the
therapeutic efficacy of anti-CD98 antibodies in Ramos xenograft tumor models
with increasing tumor
volume starting size. As can be seen, anti-CD98 antibodies retain tumor growth
inhibition (TGI) even at
increasing tumor volumes.
Table 3
Antibody [Vo=75.6mm3] [V0=144.3mm3]
[V0=249.6mm31
TGI [%] p-Value TGI ['A] p-Value TGI [%1 p-
Value
18-2A -96.74 0.000127 -93.23 0.000009 -76.08%
0.000022
8-34B -95.13 0.000121 -86.3 0.000004
8-101A - -86.73
0.000009 -65.26% 0.000046
1-115A - -85.8 0.000006
-51.29% 0.000665
8-213A - -85.2 0.000015
8-32A - -83.96 0.000022
8-300B - -
63.52% 0.000105
18-4A - -
62.14% 0.000072
8-361A6 - -
60.59% 0.000141
8-25C _ - -
57.94% 0.000140
-
456-83A - -
57.08% 0.000151
8-162A4 - -
55.89% 0.000236
1-47C - -
54.23% 0.000874
456-26A - -
53.07% 0.000277
8-319A2 - -
52.55% 0.000307
8-120A - -
51.97% 0.000576
72
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
Rituximab -87.65 0.005564
8-93A -68.76 0.016095 _
18-3A -48.42 0.066085
EXAMPLE 10: EFFECT OF ANTI-CD-98 MONOCLONAL ANTIBODIES ON THE
INHIBITION OF TUMOR GROWTH IN VIVO
[0350] The effects of the anti-CD98 monoclonal antibodies 8-34B and 18-2A were
tested in
several xenograft models , using the protocol described in Example 7.
[0351] The DLD-1 (colorectal carcinoma), A549 (non small cell lung
carcinoma), Ramos (B-
cell lymphoma), and OCI-AML-3 (acute myeloid leukemia) cell lines were
obtained from ATCC and
cultured according to the suppliers' protocols. Animals were obtained from
Charles River Laboratories.
4-6 week-old immunodeficient NOD.SCID female mice on a CB.17 background were
used for the
sarcoma tumor model, 4-6 week-old immunodeficient SCID female mice on a CB.17
background were
used for the Ramos and DLD-1 tumor models, and 4-6 week-old immunodeficient nu
-I- female mice
were used for the A549 and the OCI-AML-3 tumor model. Either rituximab (an
IgG1 anti-CD20
antibody) or Erbitux (an IgG1 anti-EGFR antibody) was used as a positive
control antibody, and an IgG2a
antibody to an irrelevant antigen was used as a negative control. DC101 is a
rat anti-mouse
VEGFR2/KDR IgGI mAb (ATCC No. HB-11534) and serves as a positive control.
Injections, antibody
treatment and statistical calculations were performed as described in Example
9.
[0352] As shown in Figs. 6-9, the anti-CD98 monoclonal antibody 18-2A is a
potent inhibitor
of tumor growth in the colorectal cancer (DLD-1), non-small cell lung cancer
(A549), Burkitts
lymphoma (Ramos) andAML (OCI-AML-3) xenografts. The effect of 18-2A compared
favorably to that
of Rituxan (Fig. 7) and Erbitux (Fig. 9 and Fig. 10).
[0353] The anti-CD98 monoclonal antibody 8-34B inhibited tumor growth in the
Ramos and
AML (OCI-AML-3) xenografts, as shown in Fig. 7 and Fig. 8.
EXAMPLE 11: ANTI-CD98 MONOCLONAL ANTIBODIES IN MOUSE STRAINS WITH
DIFFERENT IMMUNODEFICIENT BACKGROUNDS
[0354] Three anti-CD98 monoclonal antibodies, 8-300B, 8-34B and 18-2A, were
tested in the
RAMOS xenograft model as described in Example 9, using different mouse
strains. An IgG2a antibody
was used as a negative control. The three immunodeficient mouse strains ranged
from less to highly
immunocompromised. SCID mice lack functional B and T cells, but retain natural
killer (NK) cell
function and some complement function. NOD.SCID mice lack complement function
and have only
partial NK function, while NSG (NOD/SCID/gamma) mice lack NK function.
[0355] As shown in Fig. 11, the tumor-growth inhibitory effect of anti-CD98
antibodies in the
RAMOS xenograft model increases when assessed in more immuno-competent mouse
strains, indicating
that the in vivo anti-tumor activity of the anti-CD98 antibodies is due to a
combination of
immunoeffector function and inhibition of CD98 activity. The results in the
NSG mice, which lack all
73
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
ADCC and CDC function, suggests that anti-CD98 antibodies may also inhibit the
biological function of
CD98 in the RAMOS xenograft, and that CD98 may be critical for RAMOS tumor
growth and/or
maintenance in vivo.
EXAMPLE 12: IN VIVO EFFICACY OF MURINE AND CHIMERIC ANTI-CD98
MONCLONAL ANTIBODIES
[03561 The in vivo efficacy of the murine anti-CD98 antibodies 8-34B and 18-2A
were
compared to that of the chimeric antibodies 8-34B-ch and 18-2A-ch7.1 in the
RAMOS xenograft model,
as described in Example 9. The antibodies were tested at 0.5 mg/kg dose (-30
nm). The results shown in
Fig. 12 confirm that the chimeric anti-CD98 antibodies inhibit in vivo tumor
growth at an effectiveness
similar to their parental murine counterparts.
EXAMPLE 13: EPITOPE MAPPING OF HUMANIZED MONOCLONAL ANTIBODY IGN523
Materials and methods
[03571 Reagents. FREESTYLETm CHO-S cells (Invitrogen) were maintained in
FREESTYLE"' CHO expression medium (Invitrogen) supplemented with GlutaMAXTm.
Control
antibodies 4F2 and MEM108 were obtained from Santa Cruz Biotechnology and
Thermo Scientific,
respectively. Antibodies were labeled with Alexa Fluor 647 using the
appropriate Zenon Antibody
Labeling Kit (Invitrogen).
[03581 Plasmid constructions. Full length CD98, CD98 point mutants and mouse
and human
CD98 chimeras were constructed by gene synthesis (GeneWiz) and cloned into the
pCDNA3.1 vector
(Invitrogen). CD98 ECD, CD98 ECD point mutants and CD98 ECD chimeras were
constructed by gene
synthesis and cloned into the pDisplay vector (Invitrogen). All constructs
were confirmed by DNA
sequencing.
[03591 Fluorescence-activated cell sorter (FACS) analysis of IGN523 binding to
CD98.
CD98 chimeras, CD98 point mutation constructs and human wild type control
constructs were
transfected by electroporation with a Nucleofector 4D unit (Lonza). Constructs
were mixed with the
transfection solution and then transiently transfected into FREESTYLE"' CHO-S
cells. Transfected
CHO-S cells were harvested 24 hours after transfection. Cells were quantitated
and then stained with
IGN523, MEM108 or 4F2 antibodies. Before staining, the antibodies were labeled
with Alexa Fluor 647
using the appropriate Zenon Antibody Labeling Kit (Invitrogen). Flow data were
acquired on a Miltenyi
MACSQuant Analyzer (Miltenyi Biotec) and data analysis was performed using
FlowJo software version
9.5.3 (Tree Star, Inc.).
Determining the IGN523 binding region on CD98
[0360] To determine the region of CD98 that is involved in IGN523 binding,
regions of
approximately 40 contiguous amino acids of human CD98 (SEQ ID NO: 1) were
substituted with the
corresponding regions of mouse CD98 (SEQ ID NO: 96), and the effects of these
substitutions on the
overall binding of IGN523were monitored using FACS analysis. Fig. 13 shows the
regions of the mouse
74
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
sequence that were substituted into the human sequence to form the 13 mouse-
human CD98 chimera
constructs. Fig. 14 shows the binding of IGN523 and a control antibody to
murine CD98 (Mu), human
CD98 (Hu) and the 13 mouse-human chimeras. The results demonstrate that the
region substituted in
chimera construct 10 is required for IGN523 binding to CD98. In contrast, it
can be seen that the region
responsible for binding of the control antibody is found in the regions
substituted in chimeras 11 and 12.
[03611 Fig. 15 shows the sequence of the region of human CD98 within which
IGN523 binds,
as identified using the mouse-human CD98 chimera constructs, and the location
of this sequence within
the three-dimensional structure of CD98. The region defined by chimera 10
consists of amino acid
residues T358-N405 of human CD98. Amino acids T358-G368 (underlined) are
buried in the crystal
structure and are unlikely to be part of the binding interface. Non-conserved
residues between the human
and mouse sequences are shown in bold. The substitution of N at site D391
results in an extra
glycosylation site in the mouse sequence as compared to human.
[03621 Fine mapping of IGN523 with single and multiple mutations in CD98
[0363] To further define the IGN523 epitope, single or multiple
amino acid changes were
introduced in the region defined by mouse-human chimera 10. Four constructs
were made, each
introducing the nonhomologous residues from a portion of the mouse CD98
sequence into the human
CD98 sequence. Construct 4.1 contained mutations I371L, D374Q, A375G and a
deletion of A376.
Construct 4.2 contained mutations M383A and E384K. Construct 4.3 contained
mutations D391N,
F395I, P396F and D397H. Construct 4.4 contained mutations G400R, A401P and
A404L. FACS analysis
of CHO cells transfected with the respective constructs showed that the
mutations contained in construct
4.1 prevented IGN523 from binding to CD98 (Fig. 16). The mutations contained
in construct 4.4 also
affected binding of 1GN523 to CD98, to a lesser degree (Fig. 16).
[0364] To determine which hydrophobic residues are involved in the
binding interface of
IGN523 to human CD98, single mutation constructs of hydrophobic residues in
the targeted loop region
were created by substituting these hydrophobic residues with highly charged
amino acids such as aspartic
acid or asparagine. The single mutant constructs were individually transfected
into CHO cells and
analyzed by FACS to determine binding to 1GN523. As shown in Fig. 17, mutation
of hydrophobic
residues A377 and L378 to a charged amino acid negatively affected IGN523
binding. To a lesser extent,
mutation of residues 1398 and A401 showed a similar negative effect on binding
of IGN523.
[0365] Further constructs containing multiple mutations were made to
identify additional
important residues for IGN523 binding to CD98. As shown in Fig. 18, the
multiple mutations in
construct M1 (D374Q, D397H, G400R and A401P) completely prevented IGN523 from
binding,
indicating the significance of these residues. Construct M2 (D374E and A375E)
and M3 (D397S and
I398T) also resulted in reduced binding. Additional experiments with
constructs comprising a deletion of
A376 (not shown) indicated that the presence of this residue appears to be
required for correct folding of
the epitope loop region.
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
[0366] Based upon the binding studies using the mouse-human chimeras,
mutations of
hydrophobic residues, and multiple mutations, the following amino acids were
determined to be part of
the IGN523 epitope: D374, A377, L378, D397,1398, G400 and A401.
Peptide scanning analysis
[0367] As a complement to these mutational studies, the binding epitope of
IGN523 was
further analyzed using a peptide scanning analysis (PepScan).
[0368] Synthesis of peptides: To reconstruct discontinuous epitopes of the
target molecule, a
library of structured peptides was synthesized using Pepscan's proprietary
Chemically Linked Peptides
on Scaffolds (CLIPS) technology (Pepscan). Chemical linkage of peptides onto
scaffolds was carried out
essentially as follows: The side-chains of multiple cysteines in the peptides
were coupled to one or two
CLIPS templates. A 0.5 mM solution of the T2 CLIPS template 1,3-bis
(bromomethyl) benzene was
dissolved in ammonium bicarbonate (20 mM, pH 7.9)/acetonitrile (1:1(v/v). This
solution was added
onto the peptide arrays, causing the CLIPS template to bind to side-chains of
two cysteines as present in
the solid-phase bound peptides of the peptide-arrays (455 wells plate with 3
)1.1 wells). The peptide arrays
were gently shaken in the solution for 30 to 60 minutes while completely
covered in solution. Finally, the
peptide arrayswere washed extensively with excess of H20 and sonicated in
disrupt-buffer containing 1
percent SDS/0.1 percent beta-mercaptoethanol in PBS (pH 7.2) at 70 C for 30
minutes, followed by
sonication in H20 for another 45 minutes. See also the methods described in
Timmerman et al. (2007), J.
Mol. Recognit. 20:283-99; and Slootstra et al. (1996), Molecular Diversity 1:
87-96.
[0369] ELISA screening: The binding of antibody to each of the synthesized
peptides was
tested in a PEPSCAN-based ELISA. The peptide arrays were incubated with
primary antibody solution
(overnight at 4 C). After washing, the peptide arrays were incubated with a
1/1000 dilution of an
antibody peroxidase conjugate (SBA, catalog no. 2010-05) for one hour at 25 C.
After washing, the
peroxidase substrate 2,2'-azino-di-3-ethylbenzthiazoline sulfonate (ABTS) and
2 [t1/m1 of 3 percent H702
were added. After one hour, the color development was measured. The color
development was quantified
with a charge coupled device (CCD)-camera and an image processing system.
[0370] Data processing: The values obtained from the CCD camera range from 0
to 3000
mAU, similar to a standard 96-well plate ELISA-reader. The results were
quantified and stored into the
Peplab database. Occasionally a well contains an air-bubble resulting in a
false-positive value. The cards
were manually inspected and any values caused by an air-bubble are scored as
O.
[0371] Synthesis quality control: To verify the quality of the
synthesized peptides, a separate
set of positive and negative control peptides was synthesized in parallel.
These were screened with
antibody 57.9 (Posthumus et al., J. Virology, 1990, 64:3304-3309).
[0372] The results of a variable-length peptide screen are shown in
Fig. 19. ELISA results for
each peptide are shown as a horizontal line. Start and end points of the lines
indicate which residues are
included in the peptide, and the Y-value of the line shows the ELISA result
obtained for that peptide. The
ELISA results for the peptides show dominant binding for 395FPDIPGA401 (SEQ ID
NO: 42) and
secondary binding for 379PGQP382(SEQ ID NO: 43). A global analysis of 29
single-positions alanine
76
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
replacement sets showed strongest binding for 394SFDIPGAVASANMTV407 (SEQ ID
NO: 44). Fig. 20
shows the results of a best-binding single-positions alanine-replacement
peptide set, in which each
residue of peptide SEQ ID NO:44 was replaced by alanine, or by glycine if the
original residue was
alanine. The analysis shows strongest dependency for residues F395, P396, D397
and 1398, which appear
to form the core of this epitope.
[0373] Next, CLIPS conformational matrix structures were used to to bring
together pairs of
peptides from the two regions identified in the peptide screen, so as to assay
antibody binding to
conformational epitopes. Fig. 21 shows heat maps representing the data
obtained from CLIPS
conformational matrix structures that combined two partial sequences of human
CD98 (shown on X axis
and Y axis). The results indicated a high dependency on secondary structure.
The best binding was
observed for peptides that combined 395FPDIPGAVSAN405 (SEQ ID NO: 70) and
372GLDAAALPGQp382
(SEQ ID NO: 50). These two peptides were used as the basis for a mutagenesis
screen as shown in Fig.
22. SEQ1 shows the sequence of the peptide and D1F1 indicates where the
mutation is located in the
peptide. Grey fields indicate peptides having non-mutated sequences. The last
column shows the
difference in ELISA value between wild-type and mutated peptide. High values
indicate that the mutation
has a strong negative effect on binding. The mutagenesis screen identified
P379, G380, D397 and 1398 as
important binding residues.
[0374] Fig. 23 shows the location on the surface of human CD98 of the amino
acid residues
determined to be important for binding of humanized monoclonal antibody
IGN523. Fig. 23A shows the
location of the residues identified by the chimera and mutagenesis studies,
while Fig. 23B shows the
location of residues determined by Pepscan analysis. Fig. 23C shows that both
sets of residues
substantially overlap, confirming that the highlighted loop region is the
binding epitope for IGN523.
EXAMPLE 14: IN VIVO ANTI-TUMOR ACTIVITY OF HUMANIZED ANTI-CD98
MONOCLONAL ANTIBODY IGN523
[0375] The effects of the anti-CD98 humanized monoclonal antibody IGN523 were
tested in
several xenograft models , using the protocol described in Example 8.
Injections, antibody treatment and
statistical calculations were performed as described in Example 9. IGN523 was
compared to the standard
of care drug rituximab in the RAMOS (RA.1) and DAU Burkitt lymphoma models
(Fig. 24 and Fig. 25).
IGN523 was then compared to carboplatin, at its maximum tolerated dose, in the
patient-derived NSCLC
xenograft model IGN-LNG-12 (Fig. 26). IGN523 was also tested in the AML
xenograft model KG-1
(Figure 27). The tumors used were minimally passaged in NOD/SCID mice, without
any intervening cell
culture, in order to preserve the heterogeneity of the original tumors.
[0376] In all cases IGN523 showed significant tumor growth
inhibition. Interestingly, IGN-
LNG-12 patient-derived tumors lead to weight-loss in NOD-SCID mice, which
correlated with tumor
burden (Fig. 26A and 26B). Although carboplatin at its maximal tolerated dose
induced a significant
tumor growth inhibition, it also displayed an increase in body weight loss. On
the other hand, IGN523
treatment exerted a similar anti-tumor effect as carboplatin, but reduced IGN-
LNG-12 induced body
77
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
weight loss. These data demonstrate that IGN523 treatment is as effective as
carboplatin in this NOD-
SCID model without inducing body weight loss.
[0377] The data demonstrate significant tumor growth inhibition in Burkitt
lymphoma models
RAMOS (RA.1) and DAU, in the patient-derived lung tumor xenograftIGN-LNG-12,
and in the AML
model KG-1. Moreover, the tumor growth inhibition of IGN523 is comparable to
the anti-cancer agents
carboplatin or rituximab, respectively (Table 4).
Table 1
TGI
RAMOS
Treatment (RA.1) DAU IGN-LNG-12 KG-1
IGN523 92 76 44 53
Rituximab 84 67 N/A N/A
Carboplatin N/A N/A 55 N/A
[0378] The dose response relationship of1GN523 in IGN-LNG-12 tumors was also
investigated. 1GN-LNG-12 was chosen to determine the dose response to the
antibody using a
"therapeutic dosing regimen". IGN523 was dosed on days 12 and 19 between 1
mg/kg and 30 mg/kg
(Figure 15). A dose of 10 and 30 mg/kg produced the maximum tumor growth
reduction of 50-66%
relative to the control group in IGN-LNG-12 lung tumors.
[0379] Taken together, in vivo efficacy data demonstrate that
IGN523 induces significant
tumor growth inhibition in various xenograft models, which is at least
comparable to that of standard
clinical agents.
EXAMPLE 15: RECEPTOR BINDING SPECIFICITY FOR 111,11VIANIZED ANTI-CD98
MONOCLONAL ANTIBODY IGN523
[0380] Table 5 shows the percentage of sequence homology of the extracellular
domain
(ECD) of CD98 between the indicated species and of the epitope of CD98 to
which IGN523 binds. As
shown in Table 5, the homology between the human and cynomolgus monkey epitope
of CD98 to which
IGN523 binds is 96%. Receptor binding specificity studies utilizing various
methodologies, such as
surface plasmon resonance (SPR, Biacore), bio-layer interferometry (Octet) or
ELISA, determined that
IGN523 binds with high affinity to human and cynomolgus monkey CD98, but does
not bind to other
species including the murine, rat, rabbit, dog, and pig homologs of CD98, due
to decreasing homology
(Table 5). Biacore and Octet data demonstrate that the KD of IGN523 ranges
between 2 and 6 nM for
human CD98, and between 8 and 14 nM for cynomolgus monkey CD98. ELISA binding
data show that
the EC50 for IGN523 is 9 ng for human and 39 ng for cynomolgus monkey CD98.
78
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
Table 5
% Identity to `)/0 Identity to IGN523 Biacore ELISA Octet
Human Epitope Region of KD (nM) EC50 (ng) KD
(nM)
Species CD98 ECD Human CD98
Human 100 100 2 - 6 9 2
Cynomolgus monkey 96 95 13 - 14 39
8
Rabbit 80 74
negative negative ND
Dog 78 74 negative
negative = ND
Rat 73 66
negative negative ND
Mouse 70 60
negative negative ND
Pig 79 63
negative negative ND
ND = Not Determined.
[0381] In order to corroborate that cynomolgus monkey is the
relevant species for toxicology
studies, human and cynomolgus frozen tissue sections of kidney, placenta and
cerebrum were stained
with IGN523 (Fig. 29). In cryosections of human kidney and placenta (positive
control), moderate to
intense membrane and cytoplasmic staining of renal tubular epithelial (kidney)
and trophoblastic
epithelium cells (placenta) was observed, which was similar, if not identical,
to the staining intensity in
counterpart cynomolgus monkey tissue sections. In the case of human and
cynomolgus monkey
cerebrum, similar weak-to-moderate cytoplasmic staining of neuropil and
cytoplasmic granule epithelium
was observed. Taken together, it appears that staining in cynomolgus monkey
tissue is comparable to
that in human tissues. Based on the affinity and staining data the cynomolgus
monkey is considered to be
the appropriate species to evaluate the safety of IGN523.
103821 EXAMPLE 16: SINGLE-DOSE PHARMACOKINETIC STUDY IN
CYNOMOLGUS MONKEYS
103831 An exploratory non-GLP single dose intravenous pharmacokinetic study of
IGN523
was performed in male and female cynomolgus monkeys at doses of 1, 3, 10, and
100 mg/kg (N=-- 2 per
sex/group). Pronounced dose-dependent kinetics of IGN523 was observed
following a single 1-h IV
infusion of 1, 3, 10 or 100 mg of IGN523 per kg of body weight in the monkey.
Therefore, the basic
assumption regarding linearity that is implicit in the application of the non-
compartmental analysis
methods used does not apply to IGN523 over the entire dose-range studied, and
the parameters are
displayed for making descriptive comparisons among the dose groups. Although
only 2 animals per sex
were evaluated at each dose level, there were no apparent differences in PK
profiles of male versus
female animals. The mean concentration-time profiles of male and female
animals were similar within
each dose group at doses ranging from 1 to 100 mg of IGN523 per kg of body
weight (Table 6).
79
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
Table 6: PK Parameters of IGN523
Dose T112A (hours) CL, (mL/h/kg) Võ, (mL/kg)
(mg/kg) Mean Range Mean Range Mean
Range
1 4.7 3.7-5.9 4.30 3.54-4.82
32.1 27.4-39.8
3 12.4 10.2-13.5 1.7 1.58-1.88
31.5 28.2-35.1
12.0 6.4-15.8 1.03 1.00-1.10 29.3 25.7-31.7
100 60.9 44.9-96.9 0.44 0.34-0.49
41.1 37.8-44
T1,2A.; terminal half-life, CL; clearance, Vss; Volume of distribution
103841 Taken together, there was a 10-fold reduction in the "apparent" plasma
CL of IGN523
over the dose range of I to 100 mg/kg. Mean "apparent" MRT and mean "apparent"
T1/2 increased 12 to
5 13-fold over the dose range of 1 to 100 mg/kg. There were no differences
in mean "apparent" Võ at doses
of 1, 3 or 10 mg/kg, but mean "apparent" Võ in the 100 mg/kg dose cohort was
approximately 30 to 40%
higher compared to the 1, 3 and 10 mg/kg cohorts. All of these findings
suggest that the nonlinear
disposition characteristics of IGN523 in the monkey may be due to target-
mediated drug disposition
(TMDD). TMDD models have previously been used to describe the nonlinear
disposition of other
10 monoclonal antibodies. TMDD arises when the antibody has specificity for
densely populated cell-
surface targets that are abundantly expressed so that the target-antibody
interactions represent a
quantitatively important clearance pathway at low doses (Mager 2001, Mager
2003, Luu 2012).
EXAMPLE 17: REPEAT DOSE GLP STUDY IN CYNOMOLGUS MONKEY
103851 The toxicology, PK and immunogenicity of IGN523 are studied in a GLP
multi-dose
IV administration toxicology study in cynomolgus monkeys. This study provides
comprehensive data on
clinical endpoints, toxicokinetics, immunogenicity (development of anti-IGN523
antibodies), and
histopathology involving a broad list of tissues (including the injection
site) and is conducted with
formulated material representative of that which will be administered in the
clinical trial. In addition,
selected safety pharmacology endpoints (neurobehavioral, electrocardiography,
respiratory behavior) are
also evaluated.
103861 The dosing regimen is a once per week intravenous 60-min infusion for 8
weeks. The
doses employed are 10, 30, and 100 mg/kg of IGN523 once weekly (total of 9
doses) followed by a 4-
week treatment-free recovery period (Table 5). The 4-week recovery period is
considered sufficient to
allow for complete clearance of IGN523 and to assess the reversibility of any
potential toxicity. The
highest dose is anticipated to approximate a MTD and provide for a significant
exposure multiple beyond
that anticipated in patients. Immunogenicity (anti-IGN523 antibodies) and
toxicokinetics are monitored.
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
Table 7: Treatment Groups for Proposed Cynomolgus GLP Toxicology Study
Group Treatment in mg/kg No. of Males No. of Females Treatment Schedule
1 0 5 5 Once/VVeek for 9
doses
2 10 5 5 Once/1Neek for 9
doses
3 30 5 5 Once/Week for 9 doses
4 100 5 5 OnceNVeek for 9 doses
103871 Table 8 contains a detailed summary of the study design for the multi-
dose
cynomolgus GLP toxicology study. Selected safety pharmacology endpoints
(neurobehavioral,
electrocardiography, respiratory behavior) will be evaluated in the context of
the GLP repeat dose study
in cynomolgus monkeys.
81
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
Table 8: Tabular Overview of Cynomolgus GLP Toxicology Study
Eight Week Intermittent (9 Weekly Doses) Intravenous Toxicity and
Toxicokinetic Study with
IGN523 in Cynomolgus Monkeys with a 4-Week Recovery Period
No. of Animals: 40 (20M, 20F): 3 animals per sex per Main Group
(0, 10, 30, 100
mg/kg) plus 2M and 2F per Recovery Group (all dose groups)
Dose: 0, 10, 30, 100 mg/kg, once weekly
Weight: 2-4 kg, 2 to 4 years of age
Route: Intravenous 60 min infusion
Recovery: 4 weeks (2M and 2F Control; 2M and 2F 10, 30, 100
mg/kg)
Toxicokinetic (TK) Sampling: Days 0, 56: pre-dose, 0.083, 1, 3, 6, 12, 24,
48, 72, 96, 120 hr post
dose.
Days 7, 14, 21, 28, 35, 42, and 49: pre and 0.083 hr post dose.
Days 56: pre-dose, 0.083, 1, 3, 6, 12, 24, 48 hr post dose.
Additional (TK) Recovery Relative to dosing on Day 56, at approximately
0.083, 1, 3, 6, 12, 24,
Samples: and 48 hr following dosing.
During the recovery period, 3, 4, 5, 7, 14, and 28 days following the
final dose.
Immunogenicity: Days 0, 14, 28, 42, 56: pre-dose
(Anti-drug antibody) Days 70, 84 (recovery animals)
Complement Activation Day 56: Approximately 0.25 hr following dosing.
Evaluation
Clinical Pathology: Samples collected from all animals during pretest
and on days 2, 58
and 84 are evaluated for hematology, coagulation (including
fibrinogen), serum chemistry (including C-reactive protein) and
urinalysis.
Morbidity/Mortality: Twice daily
Peripheral Blood Phenotyping: Blood collected and analyzed from all animals
once during pretest and
on Days 7, 14, 58 and once near the end of the recovery period
Detailed Observations: Once during pre-dose phase, Pre-dose on Day 1,
weekly during the
dosing and recovery phase, and on day of scheduled sacrifice
including neurobehaviour (e.g tremor, convulsions, hyper-,
hypoactivity)
Cageside Observations: 1-2 hours post each dose (time recorded) and once
daily during non-
dose days and recovery period including neurobehaviour (e.g tremor,
convulsions, hyper-, hypoactivity)
Qualitative Food Consumption: Once daily during dosing phase and recovery
phase
Physical Examination: Once prior to treatment, on the days of dosing,
once weekly during the
remainder of the study period
Ophthalmic Examination: Once prior to treatment, during the last week of
the treatment period
and during the last week of recovery phase
ECGs, Respiratory Rate and Once prior to treatment, 2-3 hr post-end
infusion on day 0 and day 56,
Blood pressure measurements: near the end of the recovery period
EXAMPLE 18: HEMOLYTIC POTENTIAL EVALUATION OF IGN523
103881 The hemolytic potential of IGN523 will be assessed in vitro using
cynomolgus whole
blood. The results of this test will be used to identify any potential effects
on hemoglobin. The study
82
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
will be performed using non-human primate whole blood on the day of whole
blood collection. Table 8
contains a summary of the GLP study design for the hemolytic potential
evaluation of IGN523.
Table 2: Outline of Proposed Hemolytic Potential Evaluation GLP Study
Number of Blood Treatment
Group Treatment Formulation Samples Volume Volume
1 Test Article 1 x Projected 3 0.5 mL 0.5 mL
Concentration
2 Test Article 2 x Projected 3 0.5 mL 0.5 mL
Concentration
3 Test Article 4 x Projected 3 0.5 mL 0.5 mL
Concentration
4 Negative Saline 3 0.5 mL 0.5 mL
Control
Positive Control Distilled Water 3 0.5 mL 0.5 mL
5 EXAMPLE 19:ACUTE INTRAVENOUS AND PERIVASCULAR IRRITATION
[0389] The irritation and local tissue tolerance study is designed
to assess the short-term
toxicities of compounds in the immediate area of injection at high
concentrations. As part of this study
the compound is administered intravenously and perivascularly to identify
effects on tissues expected to
encounter the initial exposure. The rabbit is the standard animal model for
this evaluation.
EXAMPLE 20:TISSUE CROSS-REACTIVITY STUDY OF IGN523
103901 The tissue cross-reactivity profile of IGN523 is
characterized using a full range of
human tissues as detailed in the FDA guidance, Points to Consider in the
Manufacture and Testing of
Monoclonal Antibody Products for Human Use (February, 1997). A comprehensive
list of tissues to be
studied is included in Table 9.
83
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
Table 3: Outline of Proposed GLP Tissue Cross-Reactivity Study
1. Adrenal 17. Lymph Node
2. Bladder 18. Ovary
3. Blood Cells 19. Pancreas
4. Bone Marrow 20. Parathyroid
5. Breast 21. Pituitary
6. Cerebellum 22. Placenta (if available)
7. Cerebral Cortex 23. Prostate
8. Colon 24. Skin
9. Endothelium. _ 25. Spinal Cord
10. Eye 26. Spleen
11. Fallopian Tube 27. Striated Muscle
12. Gastrointestinal Tract 28. Testis
13. Heart 29. Thymus
14. Kidney (glomerulus, tubule) 30. Thyroid
15. Liver 31. Ureter
16. Lung 32. Uterus (cervix, endometrium)
EXAMPLE 21: PHASE I CLINICAL STUDY TO EVALUATE THE SAFETY AND
PHARNIACOKINETICS OF IGN523 IN PATIENTS WITH RELAPSED OR
REFRACTORY ACUTE MYELOID LEUKEMIA
103911 Primary objectives
The primary objectives of this study are
= To evaluate the safety and tolerability of IGN523 administered on a
schedule starting with
weekly dosing to patients with relapsed or refractory acute myeloid leukemia
(AML)
= To determine the maximum tolerated dose (MTD) and dose-limiting toxicities
(DLTs) of
IGN523 when administered weekly x 4
= To identify a recommended Phase 2 dose (RP2D) of IGN523 on the basis of
safety,
pharmacokinetic, and pharmacodynamics data
103921 Secondary objectives
= To assess the incidence of antibody formation to IGN523
= To characterize the pharmacokinetics of IGN523 in patients with relapsed
or refractory AML
= To make a preliminary assessment of the anti-leukemic activity of IGN523
in patients with
relapsed or refractory AML
= To make a preliminary assessment of biologic markers that might predict
IGN523 anti-leukemic
activity
[0393] Methodology
Open-label, dose-escalation study of approximately 6 dose cohorts using
standard 3+3 design, plus
expansion cohort at MTD or RP2D
84
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
[0394] Number of Subjects
Dose Escalation: approximately 21-30
Dose Expansion: 20
[0395] Investigational drug
103961 IGN523 drug product will be supplied as 20 mL of a 10 mg/mL solution in
a 25-mL
single-use glass vial. The appropriate volume of IGN523 drug product will be
diluted to 250 mL and
infused intravenously (IV) over 1 hour. In the Phase I dose-escalation study
(see Protocol Synopsis in
Section 10.2.1), cohorts of patients may be treated at escalating doses up to
30 mg/kg weekly for 8 doses.
Continued treatment beyond 8 weeks will be offered to patients with ongoing
clinical benefit (i.e., lack of
disease progression and no unacceptable toxicity). Intra-patient dose
escalation may be permitted under
specific conditions (described in Section 10.2.1) in order to maximize the
accumulation of data at
relevant doses and to minimize treatment of patients at potentially sub-
therapeutic doses.
[0397] Diagnosis and Main Criteria for Inclusion
Relapsed or Treatment Refractory AML for which no effective standard therapy
exists.
= Must have measurable disease
= Age >18 years,
= Eastern Cooperative Oncology Group (ECOG) performance status 0-2
= Life expectancy of at least 12 weeks
= Platelet count > 25,000/mm3 (may be maintained by transfusion)
= AST(SGOT)/ALT(SGPT) 5 2.5x institutional upper limit of normal (ULN)
= Total bilirubin < 1.5x institutional ULN
= Creatinine < 2x institutional ULN or calculated or measured creatinine
clearance > 50 mL/min
= For women of childbearing potential and men, agreement to use adequate
contraception
(hormonal or barrier method of birth control; abstinence) prior to study entry
and for the
duration of study participation.
= Ability to understand and the willingness to sign a written informed
consent document
103981 Exclusion Criteria
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
= Use of monoclonal antibody therapy within 4 weeks, or chemotherapy or
radiotherapy within 2
weeks, before Cycle 1 Day 1 (hydroxyurea given to control peripheral blast
counts up to 72 hours
before Cycle 1, Day 1 is allowed)
= Unresolved acute toxicity of NCI CTCAE v4.0 Grade >1 from prior anti-
cancer therapy
= Prior allogeneic stern cell transplant currently requiring
immunosuppressive therapy
= History of severe allergic or anaphylactic reactions to monoclonal
antibody therapy
= Known leptomeningeal or CNS involvement of leukemia
= Uncontrolled intercurrent illness, including, but not limited to, ongoing
or active infection,
symptomatic congestive heart failure, unstable angina pectoris, cardiac
arrhythmia, or psychiatric
illness/social situations that would limit compliance with study requirements
= Recent major surgery within 4 weeks prior to Cycle 1, Day 1
= Pregnant or lactating women
103991 Diagnostic Plan
104001 Patient AML samples from peripheral blood (or bone marrow) will be
analyzed
retrospectively after study enrollment for expression of CD98 (and not
analyzed as part of patient
screening and used as a condition for study eligibility), based upon the
following rationale:
= CD98 is differentially overexpressed in the CD34+/CD33+ and CD34+/CD33-
subpopulations of
the majority (-94%) of AML patients compared to similarly "gated" cells from
normal bone
marrow samples.
= There is no clinically validated method of detecting and quantitating CD98
expression on patient
samples that would be adequate to select patients for participation (or
exclude patients from
participating) in the proposed Phase I study.
= Qualification of assays and analysis of Phase I results will be used to
inform the design of future
clinical studies that could require patient selection.
104011 Test Product, Mode of Administration, Starting Dose
IGN523
Intravenous Infusion
Starting Dose: No greater than 1/6 the human-equivalent dose (HED) of the No
Observed
Adverse Effect Level (NOAEL) observed in the GLP multi-dose cynomolgus
toxicity study
104021 Cohort Initiation and Duration of Treatment
104031 The first patient in each new dose cohort will be dosed at
least 1 day prior to any other
patients in that cohort, to allow for observation of possible severe and/or
serious acute (e.g. infusion-
related) toxicities that might affect subsequent patient enrollment or dosing
decisions.
104041 Patients will receive weekly intravenous doses of IGN523 for 8 weeks
(two 28-day
cycles). Dosing beyond 8 weeks will be permitted for patients meeting criteria
for ongoing clinical
benefit (i.e. lack of disease progression) and acceptable safety for up to 1
year.
104051 Intra-Patient Dose Escalation
86
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
104061 To maximize the collection of information at relevant doses
and to minimize the
exposure of patients to potentially sub-optimal doses, intra-patient dose
escalation may be permitted
under the following conditions:
= Patients must complete at least two 28-day cycles at their originally
assigned dose level prior
to any dose escalation
= Patients may only escalate their dose to the highest dose level cleared
by a completed 3-6
patient dose cohort through at least one 28-day cycle of IGN523 administration
= All intra-patient dose escalation decisions will be based on the
investigator's judgment of
whether it is felt to be in the best interest of the patient, in coordination
with the Medical
Monitor
104071 Definition of DLT
04081 A DLT will be any of the following adverse events considered by the
investigator to be
related to 1GN523 (and not attributable to another clearly identifiable cause)
occurring during Days 1-28
of Cycle 1:
= Grade 3 or 4 non-hematologic toxicity, except for
- Reversible Grade 3 non-allergic infusion toxicities (including
symptoms such as fever,
chills/rigors, nausea, vomiting, pruritis, headache, rhinitis, rash, asthenia,
and/or hypoxia (in
the absence of signs/symptoms of respiratory distress) occurring during or
within 24 hours
after completing an infusion and resolving within 24 hours with a reduced
infusion rate,
supportive care, and/or administration or corticosteroids
- Grade 3 or 4 hyperuricemia, hyperphosphatemia, or hypocalcemia,
or Grade 3 hyperkalemia,
if transient (i.e. lasting (48 hours) and without manifestations of clinical
tumor lysis
syndrome (i.e. creatinine > 1.5 x ULN, cardiac arrhythmias, sudden death, or
seizures)
= Grade 3 or 4 thrombocytopenia (in a patient without pre-existing
thrombocytopenia requiring
transfusion support) that either results in bleeding, or does not improve to >
80% of baseline
value within 2 weeks without platelet transfusion
= Grade 3 or 4 neutropenia (in a patient without pre-existing neutropenia
requiring growth factor
support) that either is associated with a fever (oral or tympanic temperature
of 100.4 F/38 C) or
does not improve to > 80% of baseline value within 2 weeks without growth
factor support
104091 DLT Window
Days 1-28 of Cycle 1
104101 Dose Escalation Scheme
104111 Dose escalation may only take place after each individual in
a given cohort has reached
Day 28. Patients who experience disease progression and withdraw from the
study prior to Day 28
without DLT will not be evaluable for DLT and will be replaced. Dose
escalation will proceed between
cohorts at up to 100% increments (or less if significant AEs are observed)
according to the following
scheme:
87
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
= If 0/3 patients have DLT at a given dose level, 3 patients may be
enrolled at the next dose level
= If > 2/3 patients experience a DLT at a given dose level, dose escalation
will be stopped, and this
dose will be declared to exceed the MTD.
= If 1/3 patients experience DLT at a given dose level, at least 3 more
patients will be enrolled at
the same dose level. If 0 of these 3 patients experience DLT, proceed to the
next dose level
(which may be at a (100% dose increment). If 1 or more of this group
experience DLT, then
dose escalation is stopped, and this dose will be declared to exceed the MTD.
= Once MTD has been exceeded, if the preceding dose escalation increment
was <30%, then a
minimum of 6 evaluable patients may be enrolled at the previous dose level to
evaluate it as an
MTD. If the preceding dose escalation increment was > 30%, then at least one
dose level
intermediate between the two highest dose levels may be evaluated.
= The highest dose level resulting in DLTs in less than one-third of a
minimum of 6 patients will
be declared the MTD.
104121 Dose Escalation Committee
104131 Agreement to proceed with dose escalation, modify dose escalation
scheme, or stop the
study will be made by an internal Dose Escalation Committee made up of the
Medical Monitor, Drug
Safety representative, and additional ad hoc study team members, in
consultation with the study
investigators at the conclusion of each study cohort. This committee will
review all available study data
from the current cohort and all available safety data in the previous cohorts
before deciding on dose
escalation for the subsequent cohort. All study subjects will receive study
therapy until disease
progression, the development of unacceptable toxicity, noncompliance, or
withdrawal of consent by the
subject, or by investigator decision.
104141 Criteria for Evaluation Efficacy
Peripheral blood counts
Bone marrow aspirate and biopsy
Safety
Safety outcome measures are as follows:
Incidence and nature of DLTs,
Incidence and severity of adverse events
104151 PK Sampling
PK assessment will be done on the following schedule:
Day 1 dose: pre-dose, and 30 minutes, 4 hours, 24 hours, and 48 (or 72 hours)
post-dose
Days 8, 15, and 21 doses: pre-dose, and 30 minutes, 4 hours, and 48 (or 72
hours) post-dose
Subsequent doses: pre-dose and 30 minutes post-dose
104161 Expansion Cohort
104171 In order to obtain additional safety, tolerability, and
pharmacokinetic data, and
preliminary evidence of clinical activity, up to an additional 20 patients
with relapsed or refractory AML
will be enrolled into an expansion cohort at the MTD or RP2D. While the MTD
will be determined
88
CA 02856873 2014-05-23
WO 2013/078377
PCT/US2012/066347
primarily by the safety (DLT) data observed during the DLT observation periods
of Phase I, the RP2D
will also take into account additional safety data beyond the DLT windows, and
may also include
information gathered during Phase I dose escalation, including PK and target
occupancy data.
10
89