Note: Descriptions are shown in the official language in which they were submitted.
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
SELECTIVE HISTONE DEACTYLASE 6 INHIBITORS
ACKNOWLEDGEMENT OF GOVERNMENT SUPPORT
This invention was made with government support from the National Institutes
of Health
under Grant number CA134807. The government has certain rights in this
invention.
CROSS REFERENCE TO REALTED APPLICATIONS
This application claims the benefit of priority to U.S. Provisional
Application No.
61/607,083 filed March 7, 2012; U.S. Provisional Application No. 61/618,150,
filed March 30,
2012; U.S. Provisional Application No. 61/648,946, filed May 18, 2012; U.S.
Provisional
Application No. 61/651,595, filed May 25, 2012; U.S. Provisional Application
No. 61/651,896,
filed May 25, 2012; U.S. Provisional Application No. 61/674,942, filed July
24, 2012; and U.S.
Provisional Application No. 61/715,379, filed October 18, 2012, all of which
are incorporated by
reference herein in their entirities.
BACKGROUND
Epigenetic regulation and subsequent gene expression or silencing represents a
tightly
orchestrated interplay among enzymes responsible for modifying the tails of
histones, around
which nuclear DNA is wrapped. Among the various modifiers of the histones, the
cell is capable
of balancing the activity of both histone acetyltransferases (HAT) and histone
deacetylases
(HDAC) to attach or remove the acetyl group, respectively, from the lysine
tails of these histone
barrels. This particular epigenetic marker masks the positive lysine residues
from interacting
closely with the DNA phosphate-backbone, resulting in a more "open" chromatin
state, whereas
the deacetylases remove these acetyl groups, resulting in a more "closed" or
compacted DNA-
histone state.
There are currently no selective HDAC6 inhibitors (HDAC6i) approved for
oncology
purposes. Such molecules would be advantageous as a therapeutic approach for
they can result
in reduced side effects, which is an apparent problem associated with less
selective HDACIs
(Zhang et al., "Mice lacking histone deacetylase 6 have hyperacetylated
tubulin but are viable
and develop normally," Mol Cell Biol 2008, 28(5):1688-1701). Recent pre-
clinical efforts are
being directed toward the use of HDAC6i for certain cancers, specifically in
combination with
known drugs (Santo et al., "Preclinical activity, pharmacodynamic, and
pharmacokinetic
properties of a selective HDAC6i, ACY-1215, in combination with bortezomib in
multiple
myeloma," Blood 2012, 119(11):2579-2589). HDACIs can be useful as possible
therapeutics for
melanoma; however, studies to date have focused on using pan-HDACIs, such as
suberoylanilide hydroxamic acid (SAHA) (Peltonen et al., "Melanoma cell lines
are susceptible
to histone deacetylase inhibitor TSA provoked cell cycle arrest and
apoptosis," Pigment Cell Res
1
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
2005, /8(3):196-202; Facchetti et al., "Modulation of pro- and anti-apoptotic
factors in human
melanoma cells exposed to histone deacetylase inhibitors," Apoptosis 2004,
9(5):573-582).
While SAHA exhibits activity against all Zn-dependant HDAC isozymes, it has
been approved
solely for the treatment of cutaneous T cell lymphoma (Wagner et al., "Histone
deacetylase
(HDAC) inhibitors in recent clinical trials for cancer therapy," Clinical
Epigenetics 2010, 1(3-
4):117-136). It has previously been reported that HDAC6 forms an association
with HDAC11
(Gao et al., "Cloning and functional characterization of HDAC11, a novel
member of the human
histone deacetylase family," J Biol Chem 2002, 277(28):25748-25755). Recent
efforts have
begun to uncover the biological significance of HDAC11 as a participant in
activating the
immune response and targeting one or both of these enzymes is of therapeutic
value (Villagra et
al., "The histone deacetylase HDAC11 regulates the expression of interleukin
10 and immune
tolerance," Nat Immunol 2009, 10(1):92-100; Wang et al., "Histone Deacetylase
Inhibitor
LAQ824 Augments Inflammatory Responses in Macrophages through Transcriptional
Regulation of IL-10," J Immunol 2011,186(7):3986-3996). Thus, HDAC6 has
emerged as a
target in the treatment of melanoma and other cancers. Such an approach can be
devoid of the
cytotoxic properties of the pan-HDACi's and thus of value in the context of
safer cancer
therapeutics (Parmigiani et al., "HDAC6 is a specific deacetylase of
peroxiredoxins and is
involved in redox regulation," Proc Nat Acad Sci USA 2008,105(28):9633-9638).
What are
needed then are new and selective HDAC6 inhibitors and methods of making and
using them to
treat various cancers as well as to augment various tumor immune responses.
The compositions
and methods disclosed herein address these and other needs.
BRIEF DESCRIPTION OF FIGURES
The accompanying figures, which are incorporated in and constitute a part of
this
specification, illustrate several aspects described below.
Figure 1 is an image depicting the grouping ofHDACs.
Figure 2 is an image depicting HDACs are targets for histone deacetylase
inhibitors
(HDACi).
Figure 3 is an image depicting HDAC6 was found to influence the IL-10 gene
expression in APCs.
Figure 4 is an image depicting the genetic or pharmacologic disruption of
HDAC6
inhibits IL-I0.
Figure 5 is an image depicting the genetic disruption of HDAC6 enhances APC
function.
Figure 6 is an image depicting mechanisms as shown by CHIP analysis oflL-10
gene
promotor in macrophages include H3 and H4 acetylation; HDAC6 recruitment; and
binding of
STAT3 and other transcription factors at several timepoints after LPS
stimulation.
2
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
Figure 7 is an image depicting that knocking down HDAC6 results in a decreased
recruitment of the transcriptional activator STAT3 to the IL-10 gene promotor.
Figure 8 is an image depicting disruption of STAT3 binding to the gene
promoter
resulted in decreased recruitment of HDAC6 and diminished IL-10 production.
Figure 9 is an image depicting disruption of HDAC6 inhibits STAT3
phosphorylation.
Figure 10 is a series of images depicting that increased expression of HDAC6
and IL-
10mRNA in human melanoma.
Figure 11 is a series of images that illustrate HDAC6 expression in murine and
human
melanoma cell lines.
Figure 12 is a series of images depicting HDAC protein expression in melanoma.
Figure 13 is a series of images depicting decreased proliferation and cell
cycle arrest in
melanoma cells lacking HDAC6.
Figure 14 is a series of images depicting melanoma cells lacking in HDAC6 are
more
Immunogemc.
Figure 15 is a series of images depicting the pharmacologic inhibition of
HDAC6 in
melanoma cells resulted in cell cycle arrest and increased expression of MHC
molecules.
Figure 16 is an image depicting tubastatin-A inhibits JAK2/STAT3
phosphorylation in
B16 murine melanoma cells in vivo.
Figure 17 is a series of images depicting tubastatin A augments antigen-
specific CD4+T
-cell responses to vaccination in melanoma bearing mice.
Figure 18 is an image depicting tubastatin A, a selective HDAC6 inhibitor
decreased
STAT3 phosphorylation and recruitment to the IL-10 gene promotor in APCs.
Figure 19 is an image depicting the phenotypic and functional changes in APCs
treated
with Tubastatin A.
Figure 20 is an image depicting tubastatin A-treated APCs are better
activators of naIve
T -cells and restore the responsiveness of anergic T cells.
Figure 21 is a graph depicting the antitumor effect of tubastatin A in vivo.
Figure 22 is a series of images depicting that tubastatin A does not affect
PEM.
Figure 23 is a series of images depicting the immunological effects of
Tubastatin A upon
macrophages.
Figure 24 is a series of images depicting Tubastatin A inhibits IL-I0
transcription by
disrupting the JAKISTAT3 pathway in macrophages.
Figure 25 is a graph depicting the inhibitory effect of tubastatin A upon IL-
I0
production is lost in the absence ofHDAC6.
3
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
Figure 26 is a flow chart depicting the experimental design of the in vitro
antigenpresenting studies.
Figure 27 is a series of images depicting that tubastatin A treated
macrophages are better
activators of naIve T -cells and restore function of anergic T -cells.
Figure 28 is a series of images depicting in vivo treatment with tubastatin A
augment the
response of antigen-specific T-cell to vaccination.
Figure 29 is an image depicting HDAC6 expression in human MCL.
Figure 30 is a series of images depicting the disruption ofHDAC6 in human MCL
cell
lines.
Figure 31 is an image depicting the disruption of HDAC6 in murine FC-muMCLI
cells.
Figure 32 is an image depicting the immunological effects of HDAC6 inhibition
in
MCL. Changes in MHC, co-stimulatory molecules and/or cytokine production in
response to
LPS or CpG +/- ST-3-06 or Tubastatin A are show.
Figure 33 is a series of images depicting the antigen-presenting function of
FCmuMCLI
cells treated with ST-3-06.
Figure 34 is a senes of images depicting the antigen-presenting function of
FCmuMCL1
cells treated with tubastatin A.
Figure 35 is an image depicting the antitumor effect of tubas tat in A in
vivo.
Figure 36 is a series of images depicting the disruption of HDAC6 inhibits
STAT3
phosphorylation in APCs.
Figure 37 is a series of images depicting ST -3-06 decreased STAT3
phosphorylation
and recruitment to the IL-10 gene promotor in APCs.
Figure 38 is a Western blot showcasing substrate specificity of 5g.
SUMMARY
In accordance with the purposes of the disclosed materials, compounds,
compositions,
articles, devices, and methods, as embodied and broadly described herein, the
disclosed subject
matter relates to compositions and methods of making and using the
compositions. In other
aspects, the disclosed subject matter relates to compounds having activity as
selective HDAC6
inhibitors, methods of making and using the compounds, and compositions
comprising the
compounds. In certain aspects, the disclosed subject matter relates to
compounds having the
chemical structure shown in Formulas I or II, in particular Formula I-A, I-B,
and I-C, as defined
herein. In still further aspects, the disclosed subject matter relates to
methods for treating
oncological disorders in a patient. For example, disclosed herein are methods
whereby an
effective amount of a compound or composition disclosed herein is administered
to a patient
having an oncological disorder, for example melanoma, and who is in need of
treatment thereof.
4
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
Methods of using the disclosed compounds to inhibit or kill tumor cells, to
inhibit HDAC6, and
to augument tumor inflammatory responses are also disclosed.
Additional advantages of the disclosed subject matter will be set forth in
part in the
description that follows and the Figures, and in part will be obvious from the
description, or can
be learned by practice of the aspects described below. The advantages
described below will be
realized and attained by means of the elements and combinations particularly
pointed out in the
appended claims. It is to be understood that both the foregoing general
description and the
following detailed description are exemplary and explanatory only and are not
restrictive.
DETAILED DESCRIPTION
The materials, compounds, compositions, articles, and methods described herein
may be
understood more readily by reference to the following detailed description of
specific aspects of
the disclosed subject matter and the Examples and Figures included therein.
Before the present materials, compounds, compositions, and methods are
disclosed and
described, it is to be understood that the aspects described below are not
limited to specific
synthetic methods or specific reagents, as such may, of course, vary. It is
also to be understood
that the terminology used herein is for the purpose of describing particular
aspects only and is
not intended to be limiting.
Also, throughout this specification, various publications are referenced. The
disclosures
of these publications in their entireties are hereby incorporated by reference
into this application
in order to more fully describe the state of the art to which the disclosed
matter pertains. The
references disclosed are also individually and specifically incorporated by
reference herein for
the material contained in them that is discussed in the sentence in which the
reference is relied
upon.
General Definitions
In this specification and in the claims that follow, reference will be made to
a number of
terms, which shall be defined to have the following meanings:
Throughout the description and claims of this specification the word
"comprise" and
other forms of the word, such as "comprising" and "comprises," means including
but not limited
to, and is not intended to exclude, for example, other additives, components,
integers, or steps.
As used in the description and the appended claims, the singular forms "a,"
"an," and
"the" include plural referents unless the context clearly dictates otherwise.
Thus, for example,
reference to "a composition" includes mixtures of two or more such
compositions, reference to
"the compound" includes mixtures of two or more such compounds, reference to
"an agent"
includes mixture of two or more such agents, and the like.
5
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
"Optional" or "optionally" means that the subsequently described event or
circumstance
can or cannot occur, and that the description includes instances where the
event or circumstance
occurs and instances where it does not.
Ranges can be expressed herein as from "about" one particular value, and/or to
"about"
another particular value. When such a range is expressed, another aspect
includes from the one
particular value and/or to the other particular value. Similarly, when values
are expressed as
approximations, by use of the antecedent "about," it will be understood that
the particular value
forms another aspect. It will be further understood that the endpoints of each
of the ranges are
significant both in relation to the other endpoint, and independently of the
other endpoint. It is
also understood that there are a number of values disclosed herein, and that
each value is also
herein disclosed as "about" that particular value in addition to the value
itself For example, if
the value "10" is disclosed, then "about 10" is also disclosed. It is also
understood that when a
value is disclosed, then "less than or equal to" the value, "greater than or
equal to the value," and
possible ranges between values are also disclosed, as appropriately understood
by the skilled
artisan. For example, if the value "10" is disclosed, then "less than or equal
to 10" as well as
"greater than or equal to 10" is also disclosed. It is also understood that
throughout the
application data are provided in a number of different formats and that this
data represent
endpoints and starting points and ranges for any combination of the data
points. For example, if
a particular data point "10" and a particular data point "15" are disclosed,
it is understood that
greater than, greater than or equal to, less than, less than or equal to, and
equal to 10 and 15 are
considered disclosed as well as between 10 and 15. It is also understood that
each unit between
two particular units are also disclosed. For example, if 10 and 15 are
disclosed, then 11, 12, 13,
and 14 are also disclosed.
As used herein, by a "subject" is meant an individual. Thus, the "subject" can
include
domesticated animals (e.g., cats, dogs, etc.), livestock (e.g., cattle,
horses, pigs, sheep, goats,
etc.), laboratory animals (e.g., mouse, rabbit, rat, guinea pig, etc.), and
birds. "Subject" can also
include a mammal, such as a primate or a human.
By "reduce" or other forms of the word, such as "reducing" or "reduction," is
meant
lowering of an event or characteristic (e.g., tumor growth). It is understood
that this is typically
in relation to some standard or expected value, in other words it is relative,
but that it is not
always necessary for the standard or relative value to be referred to. For
example, "reduces
tumor growth" means reducing the rate of growth of a tumor relative to a
standard or a control.
By "prevent" or other forms of the word, such as "preventing" or "prevention,"
is meant
to stop a particular event or characteristic, to stabilize or delay the
development or progression of
a particular event or characteristic, or to minimize the chances that a
particular event or
6
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
characteristic will occur. Prevent does not require comparison to a control as
it is typically more
absolute than, for example, reduce. As used herein, something could be reduced
but not
prevented, but something that is reduced could also be prevented. Likewise,
something could be
prevented but not reduced, but something that is prevented could also be
reduced. It is
understood that where reduce or prevent are used, unless specifically
indicated otherwise, the use
of the other word is also expressly disclosed.
By "treat" or other forms of the word, such as "treated" or "treatment," is
meant to
administer a composition or to perform a method in order to reduce, prevent,
inhibit, or eliminate
a particular characteristic or event (e.g., tumor growth or survival). The
term "control" is used
synonymously with the term "treat."
The term "anticancer" refers to the ability to treat or control cellular
proliferation and/or
tumor growth at any concentration.
It is understood that throughout this specification the identifiers "first"
and "second" are
used solely to aid in distinguishing the various components and steps of the
disclosed subject
matter. The identifiers "first" and "second" are not intended to imply any
particular order,
amount, preference, or importance to the components or steps modified by these
terms.
Chemical Definitions
As used herein, the term "substituted" is contemplated to include all
permissible
substituents of organic compounds. In a broad aspect, the permissible
substituents include
acyclic and cyclic, branched and unbranched, carbocyclic and heterocyclic, and
aromatic and
nonaromatic substituents of organic compounds. Illustrative substituents
include, for example,
those described below. The permissible substituents can be one or more and the
same or
different for appropriate organic compounds. For purposes of this disclosure,
the heteroatoms,
such as nitrogen, can have hydrogen substituents and/or any permissible
substituents of organic
compounds described herein which satisfy the valences of the heteroatoms. This
disclosure is
not intended to be limited in any manner by the permissible substituents of
organic compounds.
Also, the terms "substitution" or "substituted with" include the implicit
proviso that such
substitution is in accordance with permitted valence of the substituted atom
and the substituent,
and that the substitution results in a stable compound, e.g., a compound that
does not
spontaneously undergo transformation such as by rearrangement, cyclization,
elimination, etc.
" 1 " " II 44 II
z , z2 4 z3 4 and "Z4" are used herein as generic symbols to
represent various
specific substituents. These symbols can be any substituent, not limited to
those disclosed
herein, and when they are defined to be certain substituents in one instance,
they can, in another
instance, be defined as some other substituents.
7
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
The term "aliphatic" as used herein refers to a non-aromatic hydrocarbon group
and
includes branched and unbranched, alkyl, alkenyl, or alkynyl groups.
The term "alkyl" as used herein is a branched or unbranched saturated
hydrocarbon
group of 1 to 24 carbon atoms, such as methyl, ethyl, n-propyl, isopropyl, n-
butyl, isobutyl, t-
butyl, pentyl, hexyl, heptyl, octyl, nonyl, decyl, dodecyl, tetradecyl,
hexadecyl, eicosyl,
tetracosyl, and the like. The alkyl group can also be substituted or
unsubstituted. The alkyl
group can be substituted with one or more groups including, but not limited
to, alkyl,
halogenated alkyl, alkoxy, alkenyl, alkynyl, aryl, heteroaryl, aldehyde,
amino, carboxylic acid,
ester, ether, halide, hydroxy, ketone, nitro, silyl, sulfo-oxo, sulfonyl,
sulfone, sulfoxide, or thiol,
as described below.
Throughout the specification "alkyl" is generally used to refer to both
unsubstituted alkyl
groups and substituted alkyl groups; however, substituted alkyl groups are
also specifically
referred to herein by identifying the specific substituent(s) on the alkyl
group. For example, the
term "halogenated alkyl" specifically refers to an alkyl group that is
substituted with one or more
halide, e.g., fluorine, chlorine, bromine, or iodine. The term "alkoxyalkyl"
specifically refers to
an alkyl group that is substituted with one or more alkoxy groups, as
described below. The term
"alkylamino" specifically refers to an alkyl group that is substituted with
one or more amino
groups, as described below, and the like. When "alkyl" is used in one instance
and a specific
term such as "alkylalcohol" is used in another, it is not meant to imply that
the term "alkyl" does
not also refer to specific terms such as "alkylalcohol" and the like.
This practice is also used for other groups described herein. That is, while a
term such as
"cycloalkyl" refers to both unsubstituted and substituted cycloalkyl moieties,
the substituted
moieties can, in addition, be specifically identified herein; for example, a
particular substituted
cycloalkyl can be referred to as, e.g., an "alkylcycloalkyl." Similarly, a
substituted alkoxy can
be specifically referred to as, e.g., a "halogenated alkoxy," a particular
substituted alkenyl can
be, e.g., an "alkenylalcohol," and the like. Again, the practice of using a
general term, such as
"cycloalkyl," and a specific term, such as "alkylcycloalkyl," is not meant to
imply that the
general term does not also include the specific term.
The term "alkoxy" as used herein is an alkyl group bound through a single,
terminal ether
linkage; that is, an "alkoxy" group can be defined as ¨OZ' where Z1 is alkyl
as defined above.
The term "alkenyl" as used herein is a hydrocarbon group of from 2 to 24
carbon atoms
with a structural formula containing at least one carbon-carbon double bond.
Asymmetric
structures such as (Z1Z2)C=C(Z3Z4) are intended to include both the E and Z
isomers. This can
be presumed in structural formulae herein wherein an asymmetric alkene is
present, or it can be
explicitly indicated by the bond symbol C=C. The alkenyl group can be
substituted with one or
8
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
more groups including, but not limited to, alkyl, halogenated alkyl, alkoxy,
alkenyl, alkynyl,
aryl, heteroaryl, aldehyde, amino, carboxylic acid, ester, ether, halide,
hydroxy, ketone, nitro,
silyl, sulfo-oxo, sulfonyl, sulfone, sulfoxide, or thiol, as described below.
The term "alkynyl" as used herein is a hydrocarbon group of 2 to 24 carbon
atoms with a
structural formula containing at least one carbon-carbon triple bond. The
alkynyl group can be
substituted with one or more groups including, but not limited to, alkyl,
halogenated alkyl,
alkoxy, alkenyl, alkynyl, aryl, heteroaryl, aldehyde, amino, carboxylic acid,
ester, ether, halide,
hydroxy, ketone, nitro, silyl, sulfo-oxo, sulfonyl, sulfone, sulfoxide, or
thiol, as described below.
The term "aryl" as used herein is a group that contains any carbon-based
aromatic group
including, but not limited to, benzene, naphthalene, phenyl, biphenyl,
phenoxybenzene, and the
like. The term "heteroaryl" is defined as a group that contains an aromatic
group that has at least
one heteroatom incorporated within the ring of the aromatic group. Examples of
heteroatoms
include, but are not limited to, nitrogen, oxygen, sulfur, and phosphorus. The
term "non-
heteroaryl," which is included in the term "aryl," defines a group that
contains an aromatic group
that does not contain a heteroatom. The aryl or heteroaryl group can be
substituted or
unsubstituted. The aryl or heteroaryl group can be substituted with one or
more groups
including, but not limited to, alkyl, halogenated alkyl, alkoxy, alkenyl,
alkynyl, aryl, heteroaryl,
aldehyde, amino, carboxylic acid, ester, ether, halide, hydroxy, ketone,
nitro, silyl, sulfo-oxo,
sulfonyl, sulfone, sulfoxide, or thiol as described herein. The term "biaryl"
is a specific type of
aryl group and is included in the definition of aryl. Biaryl refers to two
aryl groups that are
bound together via a fused ring structure, as in naphthalene, or are attached
via one or more
carbon-carbon bonds, as in biphenyl.
The term "cycloalkyl" as used herein is a non-aromatic carbon-based ring
composed of at
least three carbon atoms. Examples of cycloalkyl groups include, but are not
limited to,
cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, etc. The term
"heterocycloalkyl" is a
cycloalkyl group as defined above where at least one of the carbon atoms of
the ring is
substituted with a heteroatom such as, but not limited to, nitrogen, oxygen,
sulfur, or
phosphorus. The cycloalkyl group and heterocycloalkyl group can be substituted
or
unsubstituted. The cycloalkyl group and heterocycloalkyl group can be
substituted with one or
more groups including, but not limited to, alkyl, alkoxy, alkenyl, alkynyl,
aryl, heteroaryl,
aldehyde, amino, carboxylic acid, ester, ether, halide, hydroxy, ketone,
nitro, silyl, sulfo-oxo,
sulfonyl, sulfone, sulfoxide, or thiol as described herein.
The term "cycloalkenyl" as used herein is a non-aromatic carbon-based ring
composed of
at least three carbon atoms and containing at least one double bound, i.e.,
C=C. Examples of
cycloalkenyl groups include, but are not limited to, cyclopropenyl,
cyclobutenyl, cyclopentenyl,
9
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
cyclopentadienyl, cyclohexenyl, cyclohexadienyl, and the like. The term
"heterocycloalkenyl" is
a type of cycloalkenyl group as defined above, and is included within the
meaning of the term
"cycloalkenyl," where at least one of the carbon atoms of the ring is
substituted with a
heteroatom such as, but not limited to, nitrogen, oxygen, sulfur, or
phosphorus. The
cycloalkenyl group and heterocycloalkenyl group can be substituted or
unsubstituted. The
cycloalkenyl group and heterocycloalkenyl group can be substituted with one or
more groups
including, but not limited to, alkyl, alkoxy, alkenyl, alkynyl, aryl,
heteroaryl, aldehyde, amino,
carboxylic acid, ester, ether, halide, hydroxy, ketone, nitro, silyl, sulfo-
oxo, sulfonyl, sulfone,
sulfoxide, or thiol as described herein.
The term "cyclic group" is used herein to refer to either aryl groups, non-
aryl groups (i.e.,
cycloalkyl, heterocycloalkyl, cycloalkenyl, and heterocycloalkenyl groups), or
both. Cyclic
groups have one or more ring systems that can be substituted or unsubstituted.
A cyclic group
can contain one or more aryl groups, one or more non-aryl groups, or one or
more aryl groups
and one or more non-aryl groups.
The term "aldehyde" as used herein is represented by the formula ¨C(0)H.
Throughout
this specification "C(0)" or "CO" is a short hand notation for C=0, which is
also refered to
herein as a "carbonyl."
The terms "amine" or "amino" as used herein are represented by the formula
¨NZ1Z2,
where Z1 and Z2 can each be substitution group as described herein, such as
hydrogen, an alkyl,
halogenated alkyl, alkenyl, alkynyl, aryl, heteroaryl, cycloalkyl,
cycloalkenyl, heterocycloalkyl,
or heterocycloalkenyl group described above. "Amido" is
¨C(0)NZ1Z2.
The term "carboxylic acid" as used herein is represented by the formula
¨C(0)0H. A
"carboxylate" or "carboxyl" group as used herein is represented by the formula
¨C(0)0 =
The term "ester" as used herein is represented by the formula ¨0C(0)Z1 or
¨C(0)0Z1, where Z1 can be an alkyl, halogenated alkyl, alkenyl, alkynyl, aryl,
heteroaryl,
cycloalkyl, cycloalkenyl, heterocycloalkyl, or heterocycloalkenyl group
described above.
The term "ether" as used herein is represented by the formula Zi0Z2, where Z1
and Z2
can be, independently, an alkyl, halogenated alkyl, alkenyl, alkynyl, aryl,
heteroaryl, cycloalkyl,
cycloalkenyl, heterocycloalkyl, or heterocycloalkenyl group described above.
The term "ketone" as used herein is represented by the formula ZiC(0)Z2, where
Z1 and
Z2 can be, independently, an alkyl, halogenated alkyl, alkenyl, alkynyl, aryl,
heteroaryl,
cycloalkyl, cycloalkenyl, heterocycloalkyl, or heterocycloalkenyl group
described above.
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
The term "halide" or "halogen" as used herein refers to the fluorine,
chlorine, bromine,
and iodine.
The term "hydroxyl" as used herein is represented by the formula ¨OH.
The term "nitro" as used herein is represented by the formula ¨NO2.
The term "sily1" as used herein is represented by the formula ¨SiZ1Z2Z3, where
Z1, Z2,
and Z3 can be, independently, hydrogen, alkyl, halogenated alkyl, alkoxy,
alkenyl, alkynyl, aryl,
heteroaryl, cycloalkyl, cycloalkenyl, heterocycloalkyl, or heterocycloalkenyl
group described
above.
The term "sulfonyl" is used herein to refer to the sulfo-oxo group represented
by the
formula ¨S(0)2Z1, where Z1 can be hydrogen, an alkyl, halogenated alkyl,
alkenyl, alkynyl,
aryl, heteroaryl, cycloalkyl, cycloalkenyl, heterocycloalkyl, or
heterocycloalkenyl group
described above.
The term "sulfonylamino" or "sulfonamide" as used herein is represented by the
formula
¨S(0)2NH¨.
The term "thiol" as used herein is represented by the formula ¨SH.
The term "thio" as used herein is represented by the formula ¨S¨.
"R1," "R2," "R3," "Rn," etc., where n is some integer, as used herein can,
independently,
possess one or more of the groups listed above. For example, if R' is a
straight chain alkyl
group, one of the hydrogen atoms of the alkyl group can optionally be
substituted with a
hydroxyl group, an alkoxy group, an amine group, an alkyl group, a halide, and
the like.
Depending upon the groups that are selected, a first group can be incorporated
within second
group or, alternatively, the first group can be pendant (i.e. , attached) to
the second group. For
example, with the phrase "an alkyl group comprising an amino group," the amino
group can be
incorporated within the backbone of the alkyl group. Alternatively, the amino
group can be
attached to the backbone of the alkyl group. The nature of the group(s) that
is (are) selected will
determine if the first group is embedded or attached to the second group.
Unless stated to the contrary, a formula with chemical bonds shown only as
solid lines
and not as wedges or dashed lines contemplates each possible isomer, e.g.,
each enantiomer,
diastereomer, and meso compound, and a mixture of isomers, such as a racemic
or scalemic
mixture.
Reference will now be made in detail to specific aspects of the disclosed
materials,
compounds, compositions, articles, and methods, examples of which are
illustrated in the
accompanying Examples and Figures.
11
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
Compounds
A variety of HDAC6 inhibitors have been investigated (Butler et al., "Rational
Design
and Simple Chemistry Yield a Superior, Neuroprotective HDAC6 Inhibitor,
Tubastatin A," J Am
Chem Soc 2010, 132(30:10842-10846; Kalin et al., "Second-Generation Histone
Deacetylase 6
Inhibitors Enhance the Immunosuppressive Effects of Foxp3+ T-Regulatory
Cells," J Med Chem
2012, 55(2):639-651). A feature of these agents is the presence of a benzylic
linker that is built
into a canonical inhibitor, which comprises a "cap-linker-zinc binding group"
system. There is a
report that discloses HDACi's without the zinc-binding group (ZBG) (Vickers et
al., "Discovery
of HDAC Inhibitors That Lack an Active Site Zn2'-Binding Functional Group,"
ACS Med Chem
Lett 2012, 3(6):505-508). These agents possessed modest activity against the
Class 1 enzymes.
The compounds disclosed herein maintain a ZBG, most prefereably a hydroxamic
acid
group. Futher, the disclosed compounds contain certain urea-based cap groups
that are
incorporated into a benzyl hydroxamic acid scaffold, leading to potent and
selective HDAC6
inhibitors with in vitro anti-melanoma activity. As such, disclosed herein are
compounds having
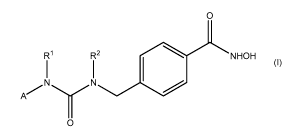
Formula I:
O
R1 R2 NHOH
1 1
N N
A
0
I
wherein
A is aryl, heteroaryl, or C1-C8 alkyl, any of which is optionally substituted
with one or more
groups chosen from acetyl, C1-05 alkyl, amino, -NR6R7, -C(0)NR6R7, C1-C4
alkoxy, C1-
C4 alkylhydroxy, C5-C6 cycloalkyl, C5-C6 heterocycloalkyl, aryl, heteroaryl,
halo,
hydroxy, thiol, cyano, or nitro; and
Rl and R2 are independently chosen from hydrogen, C1-C8 alkyl, C1-C8 alkenyl,
C1-C8 alkynyl,
C1-C8 haloalkyl, C5-C6 cycloalkyl, C5-C6 heterocycloalkyl, C1-C3 alkylaryl,
aryl, C1-C3
alkylheteroaryl, or heteroaryl, any of which is optionally substituted with
acetyl, C1-05
alkyl, amino, -NR6R7, -C(0)NR6R7, C1-C4 alkoxy, C1-C4 alkylhydroxy, C5-C6
cycloalkyl,
C5-C6 heterocycloalkyl, aryl, heteroaryl, carbonyl, halo, hydroxy, thiol,
cyano, or nitro;
Or
Rl and R2 are joined such that together they form an alkylene bridge
comprising 2 atoms so that
a 5-membered ring is formed with the -NC(0)N- moiety, in which case A is as
defined
12
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
above or hydrogen, and which 5-membered ring is optionally substituted with
R1', R2',
Ri", and R2", which are independently, hydrogen, or are C1-C8 alkyl, C5-C6
cycloalkyl,
C5-C6 heterocycloalkyl, Cl-C3 alkylaryl, aryl, Cl-C3 alkylheteroaryl, or
heteroaryl any of
which is optionally substituted with amino, aryl, C1-C4 alkoxy, halo, or
hydroxy; or R1'
and Ri" together or R2" and R2' together form a carbonyl (i.e., =0); or R1'
and R2' are
null and Ri" and R2" together form a fused phenyl group; and
R6 and R7 are independently H, C1-C4 alkyl, or are joined such that together
they form an
alkylene bridge comprising 4 or 5 atoms so that a 5 or 6-membered ring is
formed with
the nitrogen;
or a pharmaceutically acceptable salt or hydrate thereof.
In some examples, when Rl and R2 are both hydrogen, A is not hydroxyphenyl.
In specific examples, A can be a phenyl, pyridyl, oxazolidyl, or pyrimidyl
optionally
substituted with C1-05 alkyl, amino, alkoxy, alkylhydroxy, halo, hydroxy, or
thiol. In still other
examples, A can be phenyl or phenyl substituted with Cl-05 alkyl, Ci-C4
alkoxyl, or halo. In
still other example, A can be pyridyl or pyridyl substituted with C1-05 alkyl,
Ci-C4 alkoxyl, or
halo. In a preferred example, A is phenyl, or methoxyl substituted phenyl, or
halo substituted
phenyl. In further examples, A can be a phenyl. In still further examples, A
can be a phenyl
substituted with one or more methoxyl, ethoxyl, or propoxyl groups, for
example, A can be a
phenyl substituted with one methoxyl group at the ortho-, para-, or meta-
position. In a most
preferred example, A can be a phenyl substituted with a methoxyl group at the
ortho-position.
Still further, A can be a phenyl substituted with one or more halo groups, for
example, A can be
a phenyl with one halo (e.g., Cl, Br, or F) group at the ortho-, para-, or
meta-position. In other
examples, A can be a phenyl group with one or more carboxylic acids group or
an alkyl ester
group (e.g., an acetyl group). A can be a C1-C8 alkyl group. In still other
examples, A can be a
phenyl substituted with one or more C1-C4 alkyl groups. A can be a phenyl
substituted with one
methyl, ethyl, n-propyl, i-propyl, n-butyl, t-butyl, s-butyl, i-butyl group at
the ortho-, para-, or
meta-position. A can be a phenyl substituted with one or more NH2 or N(Ci-C4)2
groups.
In other examples, A can be a n-propyl, i-propyl, n-butyl, t-butyl, s-butyl, i-
butyl, n-
pentyl, i-pentyl, or s-pentyl group.
In specific examples, Rl can be hydrogen, C1-C8 alkyl, C5-C6 cycloalkyl, C5-C6
heterocycloalkyl, aryl, or heteroaryl, any of which is optionally substituted
with C1-C3 alkyl,
amino, -NR6R7, C1-C4 alkoxy, C1-C4 alkylhydroxy, carbonyl, hydroxy, thiol, or
cyano. In
specific examples, Rl can be C1-C8 alkyl, for example a C1-C4 alkyl. In other
examples, Rl can
be a C1-C8 alkyl which is optionally substituted with acetyl, NH2, N(Ci-C4)2
C1-C4 alkoxy, C1-C4
C5-C6 heterocycloalkyl, carbonyl, halo, or hydroxy. In preferred examples, Rl
is hydrogen.
13
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
In specific examples, R2 can be hydrogen, C1-C8 alkyl, C5-C6 cycloalkyl, C5-C6
heterocycloalkyl, aryl, or heteroaryl, any of which is optionally substituted
with C1-05 alkyl,
amino, -NR6R7, C1-C4 alkoxy, C1-C4 alkylhydroxy, carbonyl, hydroxy, thiol, or
cyano. In
specific examples, R2 can be C1-05 alkyl, or C1-05 alkyl substituted with a
methoxy, amino, -
NR6R7, alkylhydroxy, carbonyl, hydroxy, cyano. In other examples, R2 can be a
C1-C4alkyl
substituted with a heteroaryl, such as imidazole or indole. In other examples
R2 can be a C1-C4
alkyl substituted with a phenyl, hydroxy substituted phenyl, methoxy substited
phenyl, halo
substituted phenyl, or amino substituted phenyl.
In further examples, the disclosed compounds can have Formula I-A
O
R2 NHOH
H 1
0
N N
II
0
w'''===õ,....X.%) W
R5
I-A
where R2 is as noted herein; each W is independent of the others CH or N; and
R5 is hydrogen,
C1-C8 alkyl, C1-C8 alkenyl, Ci-C 8 alkynyl, Ci-C 8 haloalkyl, C5-C6
cycloalkyl, C5-C6
heterocycloalkyl, aryl, or heteroaryl, any of which is optionally substituted
with acetyl, C1-05
alkyl, amino, -NR6R7, -C(0)NR6R7, C1-C4 alkoxy, C1-C4 alkylhydroxy, C5-C6
cycloalkyl, C5-C6
heterocycloalkyl, aryl, heteroaryl, carbonyl, halo, hydroxy, thiol, cyano, or
nitro, or a
pharmaceutically acceptable salt or hydrate thereof
In further examples, the disclosed compounds can have Formula I-B
O
R1 NHOH
1 H
0
N N
(W%
W, 0
\IV
R5
I-B
where Rl is as noted herein; each W is independent of the others CH or N; and
R5 is hydrogen,
C1-C8 alkyl, C1-C8 alkenyl, C1-C8 alkynyl, C1-C8 haloalkyl, C5-C6 cycloalkyl,
C5-C6
heterocycloalkyl, aryl, or heteroaryl, any of which is optionally substituted
with acetyl, C1-05
alkyl, amino, -NR6R7, -C(0)NR6R7, C1-C4 alkoxy, C1-C4 alkylhydroxy, C5-C6
cycloalkyl, C5-C6
14
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
heterocycloalkyl, aryl, heteroaryl, carbonyl, halo, hydroxy, thiol, cyano, or
nitro, or a
pharmaceutically acceptable salt or hydrate thereof
The disclosed compounds can be selective HDAC6i's. A homology model (Butler et
al.,
J Am Chem Soc 2010, 132(30:10842-10846) shows the entrance to the binding site
is wider and
shallower for HDAC6 than that of HDAC1. This model also shows a lipophillic
cavity. As
such, the disclosed compounds can contain cetain branched-elements
incorporated into the aryl
urea cap group, primarily at A, Rl and/or R2, to enhance both the potency and
selectivity by
accessing this side cavity and leading to better interactions with the surface
of HDAC6.
Accessing this side cavity can be, in one aspect, accomplished through
substituting the
proximal nitrogen atom of the urea linker relative to the benzyl linker, i.e.,
R2 in Formula I. The
synthesis of these branched acyclic ureas is accomplished as outlined in
Scheme 1. As an
example, a variety of amines undergo reductive amination with methyl 4-
formylbenzoate 2 to
form the desired secondary amines 3a-h. Subsequent reaction of 3a-h with the
approprate
isocyanates affords the branched urea esters 4a-h. Aryl isocyanates are shown
in Scheme 1;
however, other isocyanates can be used to vary the "A" group in Formula I
(e.g., heteroaryl, or
alkyl). This chemistry generates a series of ureas displaying branched
substitutions on the
nitrogen proximal to the benzylic linker (R2). The hydroxamic acid group is
installed using
hydroxylamine under basic conditions to provide the hydroxamic acids 5a-h.
CA 02866707 2014-09-08
WO 2013/134467 PCT/US2013/029521
Scheme 1: Synthesis of proximal N-substituted hydroxamic acids
0
,OH
OMe ki
1.1HH H
N N
100 0 1
0
0 0
0
0 e a HN R2 lel e ___________________________ b Rlo H I2 0 Z
N N
lel 0
2 3a-h 4a-g Z = -OCH3
e L
Rlo = -0Me 5a-g Z = -NHOH
NH
R2 = N 14' I 'µOH 0 OH I .
a b c d
R10 = _Fi
N(0 ,õ,(.0 '',.(\/\ I.
R2=
e f g h
Reagents and conditions: (a) R2-NH2, NaCNBH3, rt, 5% AcOH/DCM, 16 h; (b) Aryl-
NCO,
DCM, rt, 16 h; (c) 50% wt NH2OH, NaOH, THF/Me0H (1:1), 0 C to rt, 30 min.
Accessing this side cavity can be, in another aspect, accomplished through
substituting
the distal nitrogen atom of the urea linker relative to the benzyl linker,
i.e., Rl in Formula I. The
synthesis of these branched acyclic ureas is accomplished as outlined in
Scheme 2. As an
example, a copper-mediated Buchwald coupling reaction is used in order to
assemble anilines
6a-b from iodobenzene, as these intermediates are not commercially available
(Kwong et al.,
"Copper-catalyzed coupling of alkylamines and aryl iodides: An efficient
system even in an air
atmosphere" Org Lett 2002, 4(4):581-584). Triphosgene chemistry is implemented
to convert
methyl 4-(aminomethyl)benzoate into the corresponding isocyanate, which
undergoes reaction
with seconday amines 6a-c to afford the penultimate esters 7a-c. Final
conversion to the
hydroxamic acids is accomplished as in Scheme 1 to complete the synthesis of
8a-c.
16
CA 02866707 2014-09-08
WO 2013/134467 PCT/US2013/029521
Scheme 2: Synthesis of distal N-substituted hydroxamic acids
0
R1 R1 0 Z N . .
1 1 H
I NH N
0 1) ,... .
0 n 7a -c Z OCH3
0 c L.
6a-b 8a-c Z = -NHOH
a b c
Reagents and conditions: (a) R1-NH2, CuI, K3PO4, ethylene glycol, iPrOH, 80
C, 18 h; (b) i.
triphosgene, sat. aq bicarbonate/DCM (1:1), 0 C, 30 min; ii. methyl 4-
(aminomethyl)benzoate-
HC1, Et3N, DCM, rt, 16 h; (c) 50% wt NH2OH, NaOH, THF/Me0H (1:1), 0 C to rt,
30 min.
Specific compounds according to Formula I are as follows.
0 I
_OH N
,-= 0
0
110
H H
0
N-OH
NT N 0
110 H
O 1 0 11
0 5a
HCD.
0 HN 1110
N_OH N
0 0
0 H
0
_OH
0 Y 0
H
0 N N
5b 0 T
0 5c
OH I
0
0 0
N_OH
0 H 1110 H
N N
,OH
0
1.1 "
H 110 0
SNT N 5e
O 5d
O 0
0
N_OH 0
N-OH
H 1
H H 0 H H
N N 0 NN
401 0 0
5f 5g
17
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
SI / 0
N,OH
0
,OH Ic NI 110 H
H I. H
N{N 0 8
8a
lel 0 5h
I 0
0
N,OH
0
H 1.1N,OH NrN H
c FN I. H 11 0 I 8 8c
11 0 I 8
8b
0 0
m-OH _OH
H H 0 P H H 40 hi
N N N N N
f ' T
0 8 o
H3C0 9a
9b
0 0
,OH N,OH
H H (101 H H
N NN NT =1101 H
0;fy A
9c Br 0 N
0 0
N-OH õ.õ----...., 0
N,OH
H H
NI yN 1.1 H
0 8 I 10 I Or
F
0 0
H 1101 N,OH
H
FN1.(N 0 N,OH
1\1 N N
H
I y
I II
0 0
0 0
H 10 N,OH
H H N,OH
H
NyNTN =0 1\1 N I.
I II
N 0 0
or pharmaceutically acceptable salts or hydrates thereof.
18
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
Accessing the side cavity of HDAC6 can also be, in still another aspect,
accomplished
through linking the distal and proximal nitrogen atoms of the urea linker via
an alkylene bridge,
i.e., joining Rl and R2 together to form a 5-membered ring with the
¨NC(0)N¨ moiety in Formula I. Thus, in further examples the disclosed
compounds can have
Formula I-C
O
R1" R2"R2.
R1\'
/ _______________________________ /
\
0 NHOH
N N
A
0
I-C
wherein
A is hydrogen, or A is aryl, heteroaryl, or C1-C8 alkyl, any of which is
optionally substituted
with one or more groups chosen from acetyl, C1-05 alkyl, amino, -NR6R7, -
C(0)NR6R7,
C1-C4 alkoxy, C1-C4 alkylhydroxy, C5-C6 cycloalkyl, C5-C6 heterocycloalkyl,
aryl,
heteroaryl, halo, hydroxy, thiol, cyano, or nitro, with R6 and R7 as defined
above; and
R1', R2', Ri", and R2", are independently, hydrogen, or are C1-C8 alkyl, C5-C6
cycloalkyl, C5-C6
heterocycloalkyl, Cl-C3 alkylaryl, aryl, Cl-C3 alkylheteroaryl, or heteroaryl
any of which
is optionally substituted with amino, aryl, C1-C4 alkoxy, halo, or hydroxy; or
R1' and Rill
together or R2" and R2' together form a carbonyl (i . e. , =0); or R1' and R2'
are null and
Ri" and R2" together form a fused phenyl group.
In certain examples, R2' and R2" are both methyl. In other examples R2' is
hydrogen and
R2" is methyl, ethyl, propyl, i-propyl, n-butyl, i-butyl, benzyl, tosyl,
hydroxyphenyl, C1-C4
alkoxyphenyl, or aminophenyl. In other examples R2' and R2" form a carboxyl
(i. e ., =0) group,
or a pharmaceutically acceptable salt or hydrate thereof.
In certain examples, R1' and Ri" are both methyl. In other examples R1' is
hydrogen and
Ri" is methyl, ethyl, propyl, i-propyl, n-butyl, i-butyl, benzyl, tosyl,
hydroxyphenyl, C1-C4
alkoxyphenyl, or aminophenyl. In other examples R1' and Ri" form a carboxyl
(i.e., =0) group.
The synthesis of these cyclic ureas is accomplished as outlined in Scheme 3.
19
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
Scheme 3: Synthesis of cyclic urea hydroxamic acids
O
R2
R OR2
2 OCH3
NCO 1 bP
0
¨ON- -D.
R1 a NH
,
H3CO2C NH2 N ....< NN -........<
R1
R1
0
0
0
R2
0 . 0............<
I. NHOH
c
N
NI ....,,\(
R1/
0
0
0
0
tta
0
eI
OCH3 d R1t
I. OCH3
H
HO N HN....,....<
R1 0
Reagents and conditions: (a) i. aq NaOH, ii. H+ 60 C lh; (b) KOtBu, DMF, 0 C-
RT; (c) aq
NH2OH, NaOH, THF/Me0H; (d) triphosgene, Et3N.
Specific examples of compounds of Formula I-C are as follows.
\ 0 0
0
fa 0___ \ 0)_.......õ...
N,OH fit ,OH
I. N
s Ni\\l/ 0 H N N
0 10a 0 1 Ob
1110
0
0 0
-OH HN 0
No
HN \,..N 0 il 4Ik N 0 H
ii
0 1 1 a
HO HO
1104
/ 0 , 0 00 0
¨N
el
N-OH
. N is N-OH
H
iyil\I
0=
0
or pharmaceutically acceptable salts or hydrates thereof.
CA 02866707 2014-09-08
WO 2013/134467 PCT/US2013/029521
The disclosed compounds comprise, in one aspect, branched aryl or alkyl urea
cap
groups, or cyclic urea cap groups, introduced into the canonical HDACi
platform. Introduction
of branching elements, particularly to the proximal nitrogen atom of the urea
motif, has led to
the discovery of potent inhibitors that show excellent selectivity for HDAC6
versus the full
panel of HDACs and are capable of inducing selective hyperacetylation of a-
tubulin compared to
histone protein. The SAR developed to this point indicates the branched urea
scaffold imparts
substantial gains in the desired biochemical activity. These compounds were
also screened in
cell systems, and both 5g and 5h were found to be capable of inhibiting the
growth of B16
melanoma cell line.
Also disclosed are hydroxamate compounds that are devoid of the urea motif
Such
compounds have Formula II.
0
R2
1
10 NHOH
N
/
R8
II
wherein R2 is as defined above and R8 is acetyl, C1-05 alkyloxycarbonyl,
carbobenzyloxy,
methoxybenzyl carbonyl, benzoyl, benzyl, methoxybenzyl, dimethoxybenzyl,
methoxyphenyl,
C1-05 alkylcarbamate, or aryl sulfonyl, i.e.,R9 (SO2), where R9 is aryl
optionally substituted with
C1-05 alkyl, amino, methoxyl, halo, or hydroxy, or a pharmaceutically
acceptable salt or hydrate
thereof In preferred examples, R8 can be C1-05 alkyloxycarbonyl or
arylsulfonyl. Specific
examples of compounds of Formula II are as follows.
el
0
N,OH
0
101 1111 H
m,OH ,N
0 121S
ON dflO
1 II
0
0
0
N,OH
e
N,OH l H 40 H
0 N
y 0 N
y
0 0
or pharmaceutically acceptable salts or hydrates thereof
21
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
Also disclosed herein are pharmaceutically-acceptable salts and hydrates of
the disclosed
compounds. Pharmaceutically-acceptable salts include salts of the disclosed
compounds that are
prepared with acids or bases, depending on the particular substituents found
on the compounds.
Under conditions where the compounds disclosed herein are sufficiently basic
or acidic to form
stable nontoxic acid or base salts, administration of the compounds as salts
can be appropriate.
Examples of pharmaceutically-acceptable base addition salts include sodium,
potassium,
calcium, ammonium, or magnesium salt. Examples of physiologically-acceptable
acid addition
salts include hydrochloric, hydrobromic, nitric, phosphoric, carbonic,
sulphuric, and organic
acids like acetic, propionic, benzoic, succinic, fumaric, mandelic, oxalic,
citric, tartaric, malonic,
ascorbic, alpha-ketoglutaric, alpha-glycophosphoric, maleic, tosyl acid,
methanesulfonic, and the
like. Thus, disclosed herein are the hydrochloride, nitrate, phosphate,
carbonate, bicarbonate,
sulfate, acetate, propionate, benzoate, succinate, fumarate, mandelate,
oxalate, citrate, tartarate,
malonate, ascorbate, alpha-ketoglutarate, alpha-glycophosphate, maleate,
tosylate, and mesylate
salts. Pharmaceutically acceptable salts of a compound can be obtained using
standard
procedures well known in the art, for example, by reacting a sufficiently
basic compound such as
an amine with a suitable acid affording a physiologically acceptable anion.
Alkali metal (for
example, sodium, potassium or lithium) or alkaline earth metal (for example
calcium) salts of
carboxylic acids can also be made.
Methods of Use
Further provided herein are methods of treating or preventing cancer in a
subject,
comprising administering to the subject an effective amount of a compound or
composition as
disclosed herein. Additionally, the method can further comprise administering
an effective
amount of ionizing radiation to the subject.
Methods of killing a tumor cell are also provided herein. The methods comprise
contacting a tumor cell with an effective amount of a compound or composition
as disclosed
herein. The methods can further include administering a second compound or
composition (e.g.,
an anticancer agent) or administering an effective amount of ionizing
radiation to the subject.
Also provided herein are methods of radiotherapy of tumors, comprising
contacting the
tumor with an effective amount of a compound or composition as disclosed
herein and
irradiating the tumor with an effective amount of ionizing radiation. Methods
of treating
inflammation in a subject are further provided herein, the methods comprising
administering to
the subject an effective amount of a compound or composition as described
herein. Optionally,
the methods can further include administering a second compound or composition
(e.g., an anti-
inflammatory agent).
22
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
The disclosed subject matter also concerns methods for treating a subject
having an
oncological disorder or condition. In one embodiment, an effective amount of
one or more
compounds or compositions disclosed herein is administered to a subject having
an oncological
disorder and who is in need of treatment thereof The disclosed methods can
optionally include
identifying a subject who is or can be in need of treatment of an oncological
disorder. The
subject can be a human or other mammal, such as a primate (monkey, chimpanzee,
ape, etc.),
dog, cat, cow, pig, or horse, or other animals having an oncological disorder.
Means for
administering and formulating compounds for administration to a subject are
known in the art,
examples of which are described herein. Oncological disorders include, but are
not limited to,
cancer and/or tumors of the anus, bile duct, bladder, bone, bone marrow, bowel
(including colon
and rectum), breast, eye, gall bladder, kidney, mouth, larynx, esophagus,
stomach, testis, cervix,
head, neck, ovary, lung, mesothelioma, neuroendocrine, penis, skin, spinal
cord, thyroid, vagina,
vulva, uterus, liver, muscle, pancreas, prostate, blood cells (including
lymphocytes and other
immune system cells), and brain. Specific cancers contemplated for treatment
include
carcinomas, Karposi's sarcoma, melanoma, mesothelioma, soft tissue sarcoma,
pancreatic
cancer, lung cancer, leukemia (acute lymphoblastic, acute myeloid, chronic
lymphocytic, chronic
myeloid, and other), and lymphoma (Hodgkin's and non-Hodgkin's), and multiple
myeloma.
Other examples of cancers that can be treated according to the methods
disclosed herein
are adrenocortical carcinoma, adrenocortical carcinoma, cerebellar
astrocytoma, basal cell
carcinoma, bile duct cancer, bladder cancer, bone cancer, brain tumor, breast
cancer, Burkitt's
lymphoma, carcinoid tumor, central nervous system lymphoma, cervical cancer,
chronic
myeloproliferative disorders, colon cancer, cutaneous T-cell lymphoma,
endometrial cancer,
ependymoma, esophageal cancer, gallbladder cancer, gastric (stomach) cancer,
gastrointestinal
carcinoid tumor, germ cell tumor, gliomaõ hairy cell leukemia, head and neck
cancer,
hepatocellular (liver) cancer, hypopharyngeal cancer, hypothalamic and visual
pathway glioma,
intraocular melanoma, retinoblastoma, islet cell carcinoma (endocrine
pancreas), laryngeal
cancer, lip and oral cavity cancer, liver cancer, medulloblastoma, Merkel cell
carcinoma,
squamous neck cancer with occult mycosis fungoides, myelodysplastic syndromes,
myelogenous
leukemia, nasal cavity and paranasal sinus cancer, nasopharyngeal cancer,
neuroblastoma, non-
small cell lungcancer, oral cancer, oropharyngeal cancer, osteosarcoma,
ovarian cancer,
pancreatic cancer, paranasal sinus and nasal cavity cancer, parathyroid
cancer, penile cancer,
pheochromocytoma, pineoblastoma and supratentorial primitive neuroectodermal
tumor,
pituitary tumor, plasma cell neoplasm/multiple myeloma, pleuropulmonary
blastoma, prostate
cancer, rectal cancer, renal cell (kidney) cancer, retinoblastoma,
rhabdomyosarcoma, salivary
gland cancer, Ewing's sarcoma, soft tissue sarcoma, Sezary syndrome, skin
cancer, small cell
23
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
lung cancer, small intestine cancer, supratentorial primitive neuroectodermal
tumors, testicular
cancer, thymic carcinoma, thymoma, thyroid cancer, transitional cell cancer of
the renal pelvis
and ureter, trophoblastic tumor, urethral cancer, uterine cancer, vaginal
cancer, vulvar cancer,
Waldenstrom's macroglobulinemia, and Wilms' tumor.
Melanoma and mantle cell lymphoma
In a preferred embodiment, disclosed herein is a method of treating a subject
with
melanoma by administering an effective amount of a compound of Formula 1 or
II. Melanoma
is currently the fastest growing cancer in incidence according to the World
Health Organization.
Currently, few therapies provide significant prolongation of survival for
metastatic melanoma.
Immunotherapy is an attractive modality with potentially few side effects due
to the antigen
specificity of adaptive immunity. The latest therapy approved by the FDA for
the treatment of
melanoma was ipilimumab, an antibody against CTLA-4, a key regulator of T-cell
activity;
however, this therapy offers modest improvements in overall survival.
Overcoming mechanisms of tumor-mediated immune suppression requires targeting
multiple pathways. One strategy that has gained attention has been the use of
histone
deacetylase inhibitors (HDACi). Indeed, HDACi treatment has been shown to
augment the
expression of immunologically relevant genes such as MHC and co-stimulatory
molecules.
Inhibition of IL-10 is a potent anti-inflammatory cytokine upon treatment of
macrophages with
an HDACi. However; most studies to date have used pan-HDACi, which inhibit all
11 zinc-
dependent HDACs. Therefore, the use of more selective HDACi is preferable in
order to
minimize side effects.
As demonstrated herein, HDAC6 is a molecular target in at least melanoma. Both
pharmacologic and genetic disruption of HDAC6 in B16 murine melanoma cells'
using
HDAC6-selective inhibitors (HDAC6i) and targeted shRNA (HDAC6KD),
respectively, led to
inhibition of proliferation, characterized by G1 arrest measured by propidium
iodine staining for
DNA content. Furthermore, treatment with the HDAC6i led to enhanced expression
of
immunologically relevant receptors including MHC-I and MHC-II. In vivo,
subcutaneous
injection in wild type mice of HDAC6KD B16 cells led to delayed tumor growth
as compared
with control cells. However, this effect was abrogated in experiments using
SCID mice, which
lack T- and B-cells, suggesting a critical immune component for tumor control
in vivo.
The mechanism(s) by which HDAC6 regulates tumor immunogenicity are yet to be
defined. One possible mechanism arises from protein immunoprecipitation
studies which
demonstrate that HDAC6 interacts with, and potentially regulates of STAT3, an
important
survival and pathogenic factor in melanoma, which also has implications for
immune tolerance.
24
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
The expression HDAC6 was found to be upregulated in a majority of melanoma
patient
tumor biopsies by gene microarray analysis, as compared with normal skin. This
observation
was supported by immunohistochemically-stained patient melanoma tissue
microarray.
Taken together, HDAC6 inhibition is an attractive therapeutic target in
melanoma and
mantle cell lymphoma by both delaying tumor growth and conferring a more
attractive immune
target, providing rationale for the development and use of selective HDAC6i.
Inflammamatory responses
It has previously been shown that tumor antigen specific CD8+ T cells are
unresponsive
in patients with melanoma (Lee et al., Nat. Med. 1999, 5:677-85). T cells
infiltrating the bone
marrow of patients with multiple myeloma are also unresponsive (Noonan et al.
Cancer Res.
65:2026-34, 2005). The conclusion of these studies, along with several other
reports in the
literature, is that CD4+ T cells are rendered tolerant to tumor antigens early
in tumor
progression. This presents a significant barrier to the development of
effective cancer
immunotherapy.
The role of histone deacetylases (HDACs) in the epigenetic regulation of
inflammatory
responses in APCs is disclosed herein. HDACs are a group of enzymes that
remove an acetyl
group from lysine residues on histones to regulate chromatin architecture and
gene expression.
HDACs are grouped into four distinct classes as depicted in Figure 1.
HDACs are targets for histone deacetylase inhibitors (HDACi) as depicted in
Figure 2.
HDACi's are structurally diverse compounds that are capable of targeting
several HDACs.
HDACi's induce differentiation, cell cycle and growth arrest in cancer cells.
There is an
emerging role for HDACi's as modulators of inflammation and antitumor
responses.
It was previously found that Pan-HDACi LAQ824 augments inflammatory responses
in
macrophages through transcriptional regulation of IL-10. Pan-HDACI LAQ824 was
also found
to restore the responsiveness of tolerant T cells (Wang et al. J lmmunol 2011,
186:3986-96).
The mechanisms and relevant targets of Pan-HDACIs are difficult to elucidate
given their
multiple effects. Understanding the expression and function of specific HDACs
in APCs may
unveil novel targets to influence immune activation versus immune tolerance.
The identified
target(s) may then be subject to pharmacologic inhibition with isotype-
selective HDACi's.
HDAC6 was found to influence the IL-10 gene expression in APCs as shown in
Figure 3.
HDAC6 is a 131kDa protein encoded on the X chromosome that is mainly
cytoplasmic;
however, recent data suggests that HDAC6 may also be present in the nucleus).
HDAC6 has
tubulin deacetylase activity related to cell motility and T cell/APC synapse.
There are isotype-
selective HDAC6 inhibitors available. Figure 4 illustrates the genetic or
pharmacologic
disruption of HDAC6 inhibits IL-10. Figure 5 illustrates the genetic
disruption of HDAC6
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
enhances APC function. Mechanisms as shown by CHIP analysis of IL-I0 gene
promotor in
macrophages include H3 and H4 acetylation; HDAC6 recruitment; and binding of
STAT3 and
other transcription factors at several timepoints after LPS stimulation as
shown in Figure 6.
Figure 7 illustrates that knocking down HDAC6 results in a decreased
recruitment of the
transcriptional activator STAT3 to the IL-10 gene promotor. The C-terminus of
HDAC6 is
required for interaction with HDAC11. Figure 8 illustrates that disruption of
STAT3 binding to
the gene promotor resulted in decreased recruitment of HDAC6 and diminished IL-
10
production. Figure 9 illustrates that the disruption of HDAC6 inhibits STAT3
phosphorylation.
Figure 10 illustrates that there is increased expression of HDAC6 and IL-
10mRNA in
human melanoma. Figure 11 is a series of images that illustrate HDAC6
expression in murine
and human melanoma cell lines. Figure 12 is a series of images depicting HDAC
protein
expression in melanoma. Figure 13 is a series of images depicting decreased
proliferation and
cell cycle arrest in melanoma cells lacking HDAC6. Figure 14 is a series of
images depicting
melanoma cells lacking in HDAC6 are more immunogenic.
As shown in Figure 14A and 14B, B16 cells treated with the HDAC6i ST-2-92
displayed
an elevated expression of MHC-I and -II molecules relative to untreated B16
cells. Similar
changes in MHC expression were observed in B16 cells in which HDAC6 was
knocked down.
Of note, a delay in tumor growth was observed in C57BL16 mice challenged in
vivo with B16-
KDHDAC6 cells (Figure 14C). This delay in tumor growth in KDHDAC6 melanoma
cells
could be a reflection of their diminished proliferation (FigureS 12-13) and/or
an increase in their
immunogenicity leading to improved immune recognition and clearance. To
address this
question, C57BL16 SCID mice were challenged with either KDHDAC6 or WTB16
melanoma
cells. Unlike immune competent mice in which a delay in KDHDAC6 tumor growth
was
observed (Figure 14C); such an effect was not observed in SCID mice challenged
with the same
KDHDAC6 cells (Figure 14D). These results suggest that the immunological
effects triggered
by disruption of HDAC6 in melanoma cells make these cells "better seen" by the
immune
system.
Figure 15 is a series of images depicting the pharmacologic inhibition of
HDAC6 in
melanoma cells resulted in cell cycle arrest and increased expression of MHC
molecules. It was
also found that melanoma cells treated with HDAC6 specific inhibitors are
better activators of T-
cells (CD4 and/or CD8). The procedures for this finding include loading OVA-
peptide into
melanoma cells (treated or not with Tubastatin A) and adding OT ¨lor OT -II
transgenic T -cells
(naive or tolerized) and determining their production of IL-2 and IFN-gamma.
Figure 16 is an image depicting Tubastatin-A inhibits JAK2/STAT3
phosphorylation in
B16 murine melanoma cells in vivo. Figure 17 is a series of images depicting
Tubastatin A
26
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
augments antigen-specific CD4+ T-cell responses to vaccination in melanoma
bearing mice.
There is an anti-melanoma effect after administration of tubastatin A in vivo
(alone or in
combination with anti-CLTA4).
Figure 18 illustrates the Tubastatin A, a selective HDAC6 inhibitor decreased
STAT3
phosphorylation and recruitment to the 1L-10 gene promotor in APCs. Figure 19
illustrates the
phenotypic and functional changes in APCs treated with Tubastatin A. Figure 20
illustrates that
tubastatin A-treated APCs are better activators of naive T cells and restore
the responsiveness of
anergic T cells. Figure 21 is a graph depicting the antitumor effect of
Tubastatin A in vivo.
Figure 22 depicts that Tubastatin A does not affect PEM. Figure 23 is a series
of images
depicting the immunological effects of Tubastatin A upon macrophages. Figure
24 is a series of
images depicting Tubastatin A inhibits IL-10 transcription by disrupting the
JAKISTAT3
pathway in macrophages. Figure 25 is a graph depicting the inhibitory effect
of Tubastatin A
upon IL-10 production is lost in the absence of HDAC6. Figure 26 is a flow
chart depicting the
experimental design of the in vitro antigen-presenting studies. Figure 27 is a
series of images
depicting that Tubastatin A treated macrophages are better activators of naIve
Tcells and restore
function of anergic T-cells. Figure 28 is a series of images depicting in vivo
treatment with
tubastatin A augment the response of antigen-specific T-cell to vaccination.
The experiments with Tubastatin A in APCs above indicate that treatment of
macrophages with Tubastatin-A increased the expression of co-stimulatory
molecules and
inhibits IL-10 production by these cells. Tubastatin A-treated macrophages are
better activators
of naIve T-cells and restore the responsiveness of anergic T -cells in vitro.
In vivo treatment
with Tubastatin-A enhances antigen-specific T-cell responses to vaccination.
Mechanistically,
Tubastatin-A disrupt JAKISTAT3/IL-i0 pathway and tip the balance towards
immunogenic
rather than tolerogenic macrophages.
Figure 29 is an image depicting HDAC6 expression in human MCL. Figure 30 is a
series
of images depicting the disruption of HDAC6 in human MCL cell lines. Figure 31
is an image
depicting the disruption of HDAC6 in murine FC-muMCLI cells. Figure 32 is an
image
depicting the immunological effects of HDAC6 inhibition in MCL. Changes in
MHC,
costimulatory molecules and/or cytokine production in response to LPS or CpG
+/- ST-3- 06 or
Tubastatin A are show. Figure 33 is a series of images depicting the
antigenpresenting function
of FC-muMCLI cells treated with ST-3-06. Figure 34 is a series of images
depicting the
antigen-presenting function of FC-muMCLI cells treated with Tubastatin A.
Figure 35 is an
image depicting the antitumor effect of tubastatin A in vivo. The data showed
that HDAC6
inhibition augments the immunogenicity of MCL cells. HDAC6 is required for
STAT3
activation in APCs and STAT3 diminishes the immunogenicity of tumor cells.
27
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
Figure 36 is a series of images depicting the disruption of HDAC6 inhibits
STAT3
phosphorylation in APCs. The C-terminus of HDAC6 is required for interaction
with HDAC 11.
(A) Constructs of HDAC6 coding for different lengths of the proteins and
carrying the FLAG
epitope. (B) HDAC6 constructs were over-expressed in HeLa cells and their
expression was
evaluated by western blot using an anti-FLAG antibody or (C)
immunoprecipatated to evaluate
their interaction with HDAC 11.
Figure 37 is a series of images depicting ST-3-06 decreased STAT3
phosphorylation and
recruitment to the IL-10 gene promotor in APCs. Human MCL cells display an
enhanced
expression of HDAC6. Disruption of HDAC6 in malignant B-cells inhibits their
proliferation
and is associated with induction of apoptosis. Pharmacologic or genetic
disruption of HDAC6 in
MCL cells augment their antigen-presenting capabilities leading to better T-
cell activation and
restoration of function of anergic Tcells in vitro. In vivo treatment of MCL-
bearing mice with
Tubastatin-A is associated with a strong antitumor effect. Mechanistically, is
has been have
found that HDAC6 interacts with STAT3 in APCs. Disclosed herein is the
rationale to use
HDAC6 specific inhibitor(s) alone or in combination with STAT3 inhibitors in
MCL.
Compositions, Formulations and Methods of Administration
In vivo application of the disclosed compounds, and compositions containing
them, can
be accomplished by any suitable method and technique presently or
prospectively known to
those skilled in the art. For example, the disclosed compounds can be
formulated in a
physiologically- or pharmaceutically-acceptable form and administered by any
suitable route
known in the art including, for example, oral, nasal, rectal, topical, and
parenteral routes of
administration. As used herein, the term parenteral includes subcutaneous,
intradermal,
intravenous, intramuscular, intraperitoneal, and intrasternal administration,
such as by injection.
Administration of the disclosed compounds or compositions can be a single
administration, or at
continuous or distinct intervals as can be readily determined by a person
skilled in the art.
The compounds disclosed herein, and compositions comprising them, can also be
administered utilizing liposome technology, slow release capsules, implantable
pumps, and
biodegradable containers. These delivery methods can, advantageously, provide
a uniform
dosage over an extended period of time. The compounds can also be administered
in their salt
derivative forms or crystalline forms.
The compounds disclosed herein can be formulated according to known methods
for
preparing pharmaceutically acceptable compositions. Formulations are described
in detail in a
number of sources which are well known and readily available to those skilled
in the art. For
example, Remington 's Pharmaceutical Science by E.W. Martin (1995) describes
formulations
that can be used in connection with the disclosed methods. In general, the
compounds disclosed
28
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
herein can be formulated such that an effective amount of the compound is
combined with a
suitable carrier in order to facilitate effective administration of the
compound. The compositions
used can also be in a variety of forms. These include, for example, solid,
semi-solid, and liquid
dosage forms, such as tablets, pills, powders, liquid solutions or suspension,
suppositories,
injectable and infusible solutions, and sprays. The preferred form depends on
the intended mode
of administration and therapeutic application. The compositions also
preferably include
conventional pharmaceutically-acceptable carriers and diluents which are known
to those skilled
in the art. Examples of carriers or diluents for use with the compounds
include ethanol, dimethyl
sulfoxide, glycerol, alumina, starch, saline, and equivalent carriers and
diluents. To provide for
the administration of such dosages for the desired therapeutic treatment,
compositions disclosed
herein can advantageously comprise between about 0.1% and 99%, and especially,
1 and 15% by
weight of the total of one or more of the subject compounds based on the
weight of the total
composition including carrier or diluent.
Formulations suitable for administration include, for example, aqueous sterile
injection
solutions, which can contain antioxidants, buffers, bacteriostats, and solutes
that render the
formulation isotonic with the blood of the intended recipient; and aqueous and
nonaqueous
sterile suspensions, which can include suspending agents and thickening
agents. The
formulations can be presented in unit-dose or multi-dose containers, for
example sealed
ampoules and vials, and can be stored in a freeze dried (lyophilized)
condition requiring only the
condition of the sterile liquid carrier, for example, water for injections,
prior to use.
Extemporaneous injection solutions and suspensions can be prepared from
sterile powder,
granules, tablets, etc. It should be understood that in addition to the
ingredients particularly
mentioned above, the compositions disclosed herein can include other agents
conventional in the
art having regard to the type of formulation in question.
Compounds disclosed herein, and compositions comprising them, can be delivered
to a
cell either through direct contact with the cell or via a carrier means.
Carrier means for
delivering compounds and compositions to cells are known in the art and
include, for example,
encapsulating the composition in a liposome moiety. Another means for delivery
of compounds
and compositions disclosed herein to a cell comprises attaching the compounds
to a protein or
nucleic acid that is targeted for delivery to the target cell. U.S. Patent No.
6,960,648 and U.S.
Application Publication Nos. 20030032594 and 20020120100 disclose amino acid
sequences
that can be coupled to another composition and that allows the composition to
be translocated
across biological membranes. U.S. Application Publiation No. 20020035243 also
describes
compositions for transporting biological moieties across cell membranes for
intracellular
delivery. Compounds can also be incorporated into polymers, examples of which
include poly
29
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
(D-L lactide-co-glycolide) polymer for intracranial tumors; poly[bis(p-
carboxyphenoxy)
propane:sebacic acid] in a 20:80 molar ratio (as used in GLIADEL);
chondroitin; chitin; and
chitosan.
For the treatment of oncological disorders, the compounds disclosed herein can
be
administered to a patient in need of treatment in combination with other
antitumor or anticancer
substances and/or with radiation and/or photodynamic therapy and/or with
surgical treatment to
remove a tumor. These other substances or treatments can be given at the same
as or at different
times from the compounds disclosed herein. For example, the compounds
disclosed herein can
be used in combination with mitotic inhibitors such as taxol or vinblastine,
alkylating agents
such as cyclophosamide or ifosfamide, antimetabolites such as 5-fluorouracil
or hydroxyurea,
DNA intercalators such as adriamycin or bleomycin, topoisomerase inhibitors
such as etoposide
or camptothecin, antiangiogenic agents such as angiostatin, antiestrogens such
as tamoxifen,
and/or other anti-cancer drugs or antibodies, such as, for example, GLEEVEC
(Novartis
Pharmaceuticals Corporation) and HERCEPTIN (Genentech, Inc.), respectively, or
an
immunotherapeutic such as ipilimumab and bortezomib. In other aspect, the
disclosed
compounds are coadministered with other HDAC inhibitors like ACY-1215,
Tubacin, Tubastatin
A, ST-3-06, OR ST-2-92.
In certain examples, compounds and compositions disclosed herein can be
locally
administered at one or more anatomical sites, such as sites of unwanted cell
growth (such as a
tumor site or benign skin growth, e.g., injected or topically applied to the
tumor or skin growth),
optionally in combination with a pharmaceutically acceptable carrier such as
an inert diluent.
Compounds and compositions disclosed herein can be systemically administered,
such as
intravenously or orally, optionally in combination with a pharmaceutically
acceptable carrier
such as an inert diluent, or an assimilable edible carrier for oral delivery.
They can be enclosed
in hard or soft shell gelatin capsules, can be compressed into tablets, or can
be incorporated
directly with the food of the patient's diet. For oral therapeutic
administration, the active
compound can be combined with one or more excipients and used in the form of
ingestible
tablets, buccal tablets, troches, capsules, elixirs, suspensions, syrups,
wafers, aerosol sprays, and
the like.
The tablets, troches, pills, capsules, and the like can also contain the
following: binders
such as gum tragacanth, acacia, corn starch or gelatin; excipients such as
dicalcium phosphate; a
disintegrating agent such as corn starch, potato starch, alginic acid and the
like; a lubricant such
as magnesium stearate; and a sweetening agent such as sucrose, fructose,
lactose or aspartame or
a flavoring agent such as peppermint, oil of wintergreen, or cherry flavoring
can be added.
When the unit dosage form is a capsule, it can contain, in addition to
materials of the above type,
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
a liquid carrier, such as a vegetable oil or a polyethylene glycol. Various
other materials can be
present as coatings or to otherwise modify the physical form of the solid unit
dosage form. For
instance, tablets, pills, or capsules can be coated with gelatin, wax,
shellac, or sugar and the like.
A syrup or elixir can contain the active compound, sucrose or fructose as a
sweetening agent,
methyl and propylparabens as preservatives, a dye and flavoring such as cherry
or orange flavor.
Of course, any material used in preparing any unit dosage form should be
pharmaceutically
acceptable and substantially non-toxic in the amounts employed. In addition,
the active
compound can be incorporated into sustained-release preparations and devices.
Compounds and compositions disclosed herein, including pharmaceutically
acceptable
salts, or hydrates thereof, can be administered intravenously,
intramuscularly, or
intraperitoneally by infusion or injection. Solutions of the active agent or
its salts can be
prepared in water, optionally mixed with a nontoxic surfactant. Dispersions
can also be prepared
in glycerol, liquid polyethylene glycols, triacetin, and mixtures thereof and
in oils. Under
ordinary conditions of storage and use, these preparations can contain a
preservative to prevent
the growth of microorganisms.
The pharmaceutical dosage forms suitable for injection or infusion can include
sterile
aqueous solutions or dispersions or sterile powders comprising the active
ingredient, which are
adapted for the extemporaneous preparation of sterile injectable or infusible
solutions or
dispersions, optionally encapsulated in liposomes. The ultimate dosage form
should be sterile,
fluid and stable under the conditions of manufacture and storage. The liquid
carrier or vehicle
can be a solvent or liquid dispersion medium comprising, for example, water,
ethanol, a polyol
(for example, glycerol, propylene glycol, liquid polyethylene glycols, and the
like), vegetable
oils, nontoxic glyceryl esters, and suitable mixtures thereof The proper
fluidity can be
maintained, for example, by the formation of liposomes, by the maintenance of
the required
particle size in the case of dispersions or by the use of surfactants.
Optionally, the prevention of
the action of microorganisms can be brought about by various other
antibacterial and antifungal
agents, for example, parabens, chlorobutanol, phenol, sorbic acid, thimerosal,
and the like. In
many cases, it will be preferable to include isotonic agents, for example,
sugars, buffers or
sodium chloride. Prolonged absorption of the injectable compositions can be
brought about by
the inclusion of agents that delay absorption, for example, aluminum
monostearate and gelatin.
Sterile injectable solutions are prepared by incorporating a compound and/or
agent
disclosed herein in the required amount in the appropriate solvent with
various other ingredients
enumerated above, as required, followed by filter sterilization. In the case
of sterile powders for
the preparation of sterile injectable solutions, the preferred methods of
preparation are vacuum
31
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
drying and the freeze drying techniques, which yield a powder of the active
ingredient plus any
additional desired ingredient present in the previously sterile-filtered
solutions.
For topical administration, compounds and agents disclosed herein can be
applied in as a
liquid or solid. However, it will generally be desirable to administer them
topically to the skin as
compositions, in combination with a dermatologically acceptable carrier, which
can be a solid or
a liquid. Compounds and agents and compositions disclosed herein can be
applied topically to a
subject's skin to reduce the size (and can include complete removal) of
malignant or benign
growths, or to treat an infection site. Compounds and agents disclosed herein
can be applied
directly to the growth or infection site. Preferably, the compounds and agents
are applied to the
growth or infection site in a formulation such as an ointment, cream, lotion,
solution, tincture, or
the like.
Useful solid carriers include finely divided solids such as talc, clay,
microcrystalline
cellulose, silica, alumina and the like. Useful liquid carriers include water,
alcohols or glycols or
water-alcohol/glycol blends, in which the compounds can be dissolved or
dispersed at effective
levels, optionally with the aid of non-toxic surfactants. Adjuvants such as
fragrances and
additional antimicrobial agents can be added to optimize the properties for a
given use. The
resultant liquid compositions can be applied from absorbent pads, used to
impregnate bandages
and other dressings, or sprayed onto the affected area using pump-type or
aerosol sprayers, for
example.
Thickeners such as synthetic polymers, fatty acids, fatty acid salts and
esters, fatty
alcohols, modified celluloses or modified mineral materials can also be
employed with liquid
carriers to form spreadable pastes, gels, ointments, soaps, and the like, for
application directly to
the skin of the user.
Useful dosages of the compounds and agents and pharmaceutical compositions
disclosed
herein can be determined by comparing their in vitro activity, and in vivo
activity in animal
models. Methods for the extrapolation of effective dosages in mice, and other
animals, to
humans are known to the art.
Also disclosed are pharmaceutical compositions that comprise a compound
disclosed
herein in combination with a pharmaceutically acceptable carrier.
Pharmaceutical compositions
adapted for oral, topical or parenteral administration, comprising an amount
of a compound
constitute a preferred aspect. The dose administered to a patient,
particularly a human, should be
sufficient to achieve a therapeutic response in the patient over a reasonable
time frame, without
lethal toxicity, and preferably causing no more than an acceptable level of
side effects or
morbidity. One skilled in the art will recognize that dosage will depend upon
a variety of factors
including the condition (health) of the subject, the body weight of the
subject, kind of concurrent
32
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
treatment, if any, frequency of treatment, therapeutic ratio, as well as the
severity and stage of
the pathological condition.
Kits
The disclosed subject matter also concerns a packaged dosage formulation
comprising in
one or more containers at least one inhibitor compound or composition
disclosed herein, e.g.,
any compound of Formulas I through II. A packaged dosage formulation can
optionally
comprise in one or more containers a pharmaceutically acceptable carrier or
diluent. A packaged
dosage formulation can also optionally comprise, in addition to an inhibitor
compound or
composition disclosed herein, other HDAC inhibitors, or an immunotherapeutic
such as
ipilimumab.
Depending upon the disorder or disease condition to be treated, a suitable
dose(s) can be
that amount that will reduce proliferation or growth of the target cell(s). In
the context of cancer,
a suitable dose(s) is that which will result in a concentration of the active
agent in cancer tissue,
such as a malignant tumor, which is known to achieve the desired response. The
preferred
dosage is the amount which results in maximum inhibition of cancer cell
growth, without
unmanageable side effects. Administration of a compound and/or agent can be
continuous or at
distinct intervals, as can be determined by a person of ordinary skill in the
art.
To provide for the administration of such dosages for the desired therapeutic
treatment,
in some embodiments, pharmaceutical compositions disclosed herein can comprise
between
about 0.1% and 45%, and especially, 1 and 15%, by weight of the total of one
or more of the
compounds based on the weight of the total composition including carrier or
diluents.
Illustratively, dosage levels of the administered active ingredients can be:
intravenous, 0.01 to
about 20 mg/kg; intraperitoneal, 0.01 to about 100 mg/kg; subcutaneous, 0.01
to about 100
mg/kg; intramuscular, 0.01 to about 100 mg/kg; orally 0.01 to about 200 mg/kg,
and preferably
about 1 to 100 mg/kg; intranasal instillation, 0.01 to about 20 mg/kg; and
aerosol, 0.01 to about
20 mg/kg of animal (body) weight.
Also disclosed are kits that comprise a composition comprising a compound
disclosed
herein in one or more containers. The disclosed kits can optionally include
pharmaceutically
acceptable carriers and/or diluents. In one embodiment, a kit includes one or
more other
components, adjuncts, or adjuvants as described herein. In another embodiment,
a kit includes
one or more anti-cancer agents, such as those agents described herein. In one
embodiment, a kit
includes instructions or packaging materials that describe how to administer a
compound or
composition of the kit. Containers of the kit can be of any suitable material,
e.g., glass, plastic,
metal, etc., and of any suitable size, shape, or configuration. In one
embodiment, a compound
and/or agent disclosed herein is provided in the kit as a solid, such as a
tablet, pill, or powder
33
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
form. In another embodiment, a compound and/or agent disclosed herein is
provided in the kit
as a liquid or solution. In one embodiment, the kit comprises an ampoule or
syringe containing a
compound and/or agent disclosed herein in liquid or solution form.
EXAMPLES
The following examples are set forth below to illustrate the methods,
compositions, and
results according to the disclosed subject matter. These examples are not
intended to be
inclusive of all aspects of the subject matter disclosed herein, but rather to
illustrate
representative methods, compositions, and results. These examples are not
intended to exclude
equivalents and variations of the present invention, which are apparent to one
skilled in the art.
Efforts have been made to ensure accuracy with respect to numbers (e.g.,
amounts,
temperature, etc.) but some errors and deviations should be accounted for.
Unless indicated
otherwise, parts are parts by weight, temperature is in C or is at ambient
temperature, and
pressure is at or near atmospheric. There are numerous variations and
combinations of reaction
conditions, e.g., component concentrations, temperatures, pressures, and other
reaction ranges
and conditions that can be used to optimize the product purity and yield
obtained from the
described process. Only reasonable and routine experimentation will be
required to optimize
such process conditions.
1H and 13C spectra were obtained on a Bruker spectrometer with TMS as an
internal
standard. The following abbreviations for multiplicity were used: s = singlet,
d = doublet, t =
triplet, m = multiplet, dd = double doublet, br = broad. Reactions were
monitored by TLC using
precoated silica gel plates (Merck silica gel 60 F254, 250 gm thickness) and
visualized under UV
light. LRMS experiments were carried out using an Agilent 1946A LC-MSD with
MeCN and
H20 spiked with 0.1% formic acid as mobile phase. HRMS determinations were
done with a
Shimadzu IT-TOF instrument with MeCN and H20 spiked with 0.1% formic acid as
mobile
phase. Flash chromatography was accomplished using the automated CombiFlash Rf
system
from Teledyne ISCO and prepacked silica gel cartridges according to the
recommended loading
capacity.
Preparatory HPLC was used in purification of all final compounds using a
Shimadzu
preparative liquid chromatograph with the following specifications: Column:
ACE 5AQ (150 x
21.2 mm) with 5 gm particle size. Method 1 ¨ 25-100% Me0H/H20, 30 min; 100%
Me0H, 5
min; 100-25% Me0H/H20, 4 min. Method 2 ¨ 8-100% Me0H/H20, 30 min; 100% Me0H, 5
min; 100-8% Me0H/H20, 4 min. Method 3 ¨ 0% Me0H, 5 min; 0-100% Me0H/H20, 25
min;
100% Me0H, 5 min; 100-0% Me0H/H20, 4 min. Flow rate = 17 mL/min with
monitoring at
254 and 280 nm. Both solvents were spiked with 0.05% TFA. Analytical HPLC was
carried out
using an Agilent 1100 series instrument with the following specifications:
column: Luna 5
34
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
C18(2) 100A (150 x 4.60 mm) 5 gm particle size; gradient¨ 10-100% Me0H/H20, 18
min,
100% Me0H, 3 min; 100-10% Me0H/H20, 3 min; 10% Me0H/H20, 5 min. Both solvents
were
spiked with 0.05% TFA. The purity of all tested compounds was >95%, as
determined by
analytical HPLC.
Compound Synthesis: making reference to Scheme 1
Methyl 4-(03-(dimethylamino)propyl)amino)methyl)benzoate (3a) The synthesis of
3a is representative, General Procedure A: A round-bottom flask charged with
methyl 4-formyl
benzoate (328 mg, 2 mmol) and 3-dimethylamino propylamine (0.252 mL, 2 mmol)
was taken
up in a solution of 5% AcOH in DCM (10 mL). After 5 minutes NaCNBH3 (126 mg, 2
mmol)
was added in portions and the resulting mixture was allowed to stir at room
temperature under an
atmosphere of Ar overnight. The reaction was quenched with 1N NaOH (10 mL) and
the
aqueous layer extracted with DCM (3x 10mL). The combined organic extracts were
washed
with brine, dried over sodium sulfate, concentrated in vacuo and purified via
flash
chromatography affording the product as a waxy solid (313 mg, 63%). 1H NMR
(400 MHz,
CDC13) 6 7.98 (d, J= 8.0 Hz, 2H), 7.38 (d, J= 8.0 Hz, 2H), 3.89 (s, 3H), 3.84
(s, 2H), 2.79 (br s,
1H), 2.68 (t, J= 6.8 Hz, 2H), 2.35 (t, J= 6.8 Hz, 2H), 2.23 (s, 6H), 1.70
(quint, J= 6.8 Hz, 2H).
13C NMR (100 MHz, CDC13) 6 166.96, 145.27, 129.71, 128.87, 127.95, 58.04,
53.42, 51.99,
47.87, 45.36, 27.41. LRMS ESI: [M+H] ' = 251.1
Methyl 4-(((3-hydroxypropyl)amino)methyl)benzoate (3b) Made according to
General
Procedure A affording a waxy solid (89 mg, 29%). 1H NMR (400 MHz, CDC13) 6
8.00 (d, J=
8.0 Hz, 2H), 7.39 (d, J= 8.0 Hz, 2H), 3.91 (s, 3H), 3.90 (s, 2H), 3.78 (broad,
4H), 2.93 (t, J= 5.6
Hz, 2H), 1.78-1.73 (m, 2H). 13C NMR (100 MHz, CDC13) 6 166.79, 143.16, 129.77,
129.44,
128.26, 63.55, 53.14, 52.10, 48.88, 30.18. LRMS ESI: [M+H] = 224.2.
Methyl 4-(02-(1H-indo1-3-y1)ethyl)amino)methyl)benzoate (3c) Made according to
General Procedure A affording a waxy solid (446 mg, 72%). 1H NMR (400 MHz,
CDC13) 6 8.28
(br s, 1H), 7.98 (d, J= 8.0 Hz, 2H), 7.62 (d, J= 7.6 Hz, 1H), 7.36-7.34 (m,
3H), 7.21 (t, J= 7.6
Hz, 1H), 7.13 (t, J= 7.6 Hz, 1H), 7.00 (s, 1H), 3.92 (s, 3H), 3.87 (s, 2H),
3.02-2.98 (m, 4H), 1.68
(br s, 1H). 13C NMR (100 MHz, CDC13) 6 167.06, 145.66, 136.34, 129.60, 128.62,
127.85,
127.31, 122.02, 121.89, 119.13, 118.74, 113.47, 111.15, 53.36, 51.97, 49.28,
25.64. LRMS ESI:
[M+H]' = 309.1.
Methyl 4-(((4-hydroxyphenethyl)amino)methyl)benzoate (3d) Made according to
General Procedure A affording a waxy solid (130 mg, 46%). 1H NMR (400 MHz,
CDC13) 6 7.97
(d, J= 8.0 Hz, 2H), 7.32 (d, J= 8.0 Hz, 2H), 7.01 (d, J= 8.4 Hz, 2H), 6.71 (d,
J= 8.4 Hz, 2H),
3.90 (s, 3H), 3.86 (s, 2H), 2.86 (t, J= 6.8 Hz, 2H), 2.76 (t, J= 6.8 Hz, 2H).
13C NMR (100 MHz,
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
CDC13) 6 167.07, 154.60, 145.02, 131.05129.78, 129.75, 128.91, 128.04, 115.55,
53.29, 52.08,
50.37, 35.02. LRMS ESI: [M+H] = 268.1.
Methyl 4-(((3-methoxypropyl)amino)methyl)benzoate (3e) Made according to
General
Procedure A affording a colorless oil (127 mg, 54%). 1H NMR (400 MHz, CDC13) 6
7.94 (d, J =
8.0 Hz, 2H), 7.35 (d, J = 8.0 Hz, 2H), 3.86 (s, 3H), 3.80 (s, 2H), 3.41 (t, J=
6.4 Hz, 2H), 3.28 (s,
3H), 2.67 (t, J= 6.4 Hz, 2H), 1.72 (m, 2H). 13C NMR (100 MHz, CDC13) 6 166.88,
145.78,
129.54, 128.58, 127.76, 71.17, 58.48, 53.50, 51.85, 46.72, 29.83. LRMS ESI:
[M+H]' = 238.2.
Methyl 4-(((2-methoxyethyl)amino)methyl)benzoate (3f) Made according to
General
Procedure A affording a colorless oil (29 mg, 13%). 1H NMR (400 MHz, CDC13) 6
7.97 (d, J =
8.0 Hz, 2H), 7.38 (d, J = 8.0 Hz, 2H), 3.88 (s, 3H), 3.84 (s, 2H), 3.49 (t, J=
5.2 Hz, 2H), 3.33 (s,
3H), 2.77 (t, J= 5.2 Hz, 2H), 1.88 (br, 1H). 13C NMR (100 MHz, CDC13) 6
166.98, 145.58,
129.65, 128.74, 127.90, 71.85, 58.74, 53.47, 51.94, 48.69. LRMS ESI: [M+H]' =
224.2.
Methyl 4-((butylamino)methyl)benzoate (3g) Made according to General Procedure
A
affording colorless oil (150 mg, 68%). 1H NMR (400 MHz, CDC13) 6 7.99 (d, J =
8.0 Hz, 2H),
7.39 (d, J= 8.0 Hz, 2H), 3.91 (s, 3H), 3.84 (s, 2H), 2.62 (t, J= 7.2 Hz, 2H),
1.49 (m, 2H), 1.34
(m, 3H), 0.91 (t, J= 7.2 Hz, 3H). 13C NMR (100 MHz, CDC13) 6 167.00, 145.89,
129.65,
128.70, 127.87, 53.63, 51.95, 49.14, 32.14, 20.38, 13.93. LRMS ESI: [M+H]' =
222.1.
Methyl 4-((phenethylamino)methyl)benzoate (3h) Made according to General
Procedure A affording a colorless oil (203 mg, 75%). 1H NMR (400 MHz, CDC13) 6
8.01 (m,
2H), 7.35 (m, 4H), 7.22 (m, 3H), 3.93 (s, 3H), 3.88 (s, 2H), 2.89 (m, 4H). 13C
NMR (100 MHz,
CDC13) 6 166.97, 145.66, 139.81, 129.65, 128.65, 128.43, 127.82, 126.15,
53.38, 51.96, 50.46,
36.29. LRMS ESI: [M+H]' = 270.1.
Methyl 4-01-(3-(dimethylamino)propy1)-3-(2-methoxyphenyl)ureido)methyl)-
benzoate (4a) The synthesis of 4a is representative, General Procedure B. A
solution of 3a (99
mg, 0.395 mmol) in DCM (5 mL) was added the appropriate isocyante (0.053 mL,
0.395 mmol)
at room temperature under and atmosphere of Ar and the resulting solution was
allowed to stir
overnight. The reaction was quenched with saturated bicarbonate (10 mL) and
extracted with
DCM (3x 10 mL). The combined organics were washed with brine (15 mL), dried
over sodium
sulfate, concentrated in vacuo and purified via flash chromatography affording
the urea ester as a
waxy solid (156 mg, 98%). 1H NMR (400 MHz, CDC13) 6 8.64 (br s, 1H), 8.18 (d,
J= 7.2 Hz,
1H), 7.99 (d, J= 8.0 Hz, 2H), 7.40 (d, J= 8.0 Hz, 2H), 6.97-6.94 (m, 2H), 6.84
(d, J = 7.2 Hz,
1H), 4.64 (s, 2H), 3.91 (s, 3H), 3.81 (s, 3H), 3.42 (t, J= 6.0 Hz, 2H), 2.34
(t, J = 6.0 Hz, 2H),
2.20 (s, 6H), 1.74 (quint, J= 6.0 Hz, 2H). 13C NMR (100 MHz, CDC13) 6 166.85,
156.64,
148.86, 143.75, 129.77, 129.43, 129.02, 127.53, 122.25, 120.94, 120.36,
109.92, 55.52, 54.60,
51.96, 49.19, 44.64, 44.18, 24.98. LRMS ESI: [M+H]' = 400.2.
36
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
Methyl 4-01-(3-hydroxypropy1)-3-(2-methoxyphenyl)ureido)methyl)benzoate (4b)
Made according to General Procedure B affording a waxy solid (113 mg, 74%).
1FINMR (400
MHz, CDC13) 6 8.07-8.03 (m, 3H), 7.39 (d, J= 8.0 Hz, 2H), 7.20 (br s, 1H),
6.93-6.90 (m, 2H),
6.77-6.47 (m, 1H), 4.59 (s, 2H), 3.91 (s, 3H), 3.67-3.62 (m, 7H), 1.80-1.77
(m, 2H). 13C NMR
(100 MHz, CDC13) 6 166.65, 156.31, 147.85, 142.24, 130.14, 129.70, 128.43,
126.77, 122.44,
121.04, 119.48, 109.88, 58.25, 55.53, 52.12, 50.41, 44.00, 30.32. LRMS ESI:
[M+H]' = 373.2.
Methyl 4-01-(2-(1H-indo1-3-yl)ethyl)-3-(2-methoxyphenyl)ureido)methyl)-
benzoate
(4c) Made according to General Procedure B affording a solid (200 mg, 95%).
1FINMR (400
MHz, CDC13) 6 8.23-8.21 (m, 1H), 8.15 (br s, 1H), 7.99 (d, J= 8.0 Hz, 2H),
7.60 (d, J= 7.6 Hz,
1H), 7.36-7.34 (m, 3H), 7.22-7.10 (m, 3H), 7.02 (s, 1H), 6.97-6.94 (m, 2H),
6.81-6.79 (m, 1H),
4.59 (s, 2H), 3.91 (s, 3H), 3.70-3.67 (m, 5H), 3.12 (t, J= 7.6 Hz, 2H). 13C
NMR (100 MHz,
CDC13) 6 166.83, 155.17, 147.60, 143.28, 136.29, 129.98, 129.34, 128.79,
127.30, 127.13,
122.18, 122.08, 121.15, 119.51, 118.96, 118.46, 112.53, 111.29, 109.29, 55.56,
52.10, 50.89,
48.98. LRMS ESI: [M+H] = 458.2.
Methyl 4-01-(4-hydroxyphenethyl)-3-(2-methoxyphenyl)ureido)methyl)-benzoate
(4d) Made according to General Procedure B affording a solid (178 mg, 90%).
1FINMR (400
MHz, CDC13) 6 8.15 (d, J= 4.8 Hz, 1H), 8.01 (d, J= 7.6 Hz, 2H), 7.35 (d, J=
8.0 Hz, 2H), 7.11
(s, 1H), 7.02 (d, J= 8.0 Hz, 2H), 6.95-6.93 (m, 2H), 6.80-6.77 (m, 3H), 6.43
(br s, 1H), 4.53 (s,
2H), 3.91 (s, 3H), 3.73 (s, 3H), 3.56 (t, J= 6.8 Hz, 2H), 2.87 (t, J= 6.8 Hz,
2H). 13C NMR (100
MHz, CDC13) 6 166.89, 155.25, 154.99, 147.69, 142.93, 130.04, 129.83, 129.76,
129.40, 128.50,
127.21, 122.35, 121.18, 119.10, 115.67, 109.78, 55.60, 52.15, 50.97, 50.33,
33.88. LRMS:
[M+H]' = 435.2.
Methyl 4-01-(3-methoxypropy1)-3-phenylureido)methyl)benzoate (4e) Made
according to General Procedure B affording a colorless oil (70 mg, 73%).
1FINMR (400 MHz,
CDC13) 6 8.00 (d, J= 8.4 Hz, 2H), 7.84 (s, 1H), 7.45 (d, J= 8.4. Hz, 2H), 7.40
(d, J= 8.0 Hz,
2H), 7.30 (m, 2H), 7.02 (t, J= 7.6 Hz, 1H), 4.64 (s, 2H), 3.92 (s, 3H), 3.49
(t, J= 5.2 Hz, 2H),
3.44 (s, 3H), 3.415 (t, J= 6.4 Hz, 2H), 1.77 (m, 2H). 13C NMR (100 MHz, CDC13)
6 166.87,
156.36, 143.76, 139.85, 129.88, 129.18, 128.81, 127.72, 122.45, 119.17, 68.16,
58.63, 52.05,
49.41, 43.05, 27.55. LRMS ESI: [M+H]' = 358.2.
Methyl 4-01-(2-methoxyethyl)-3-phenylureido)methyl)benzoate (4f) Made
according
to General Procedure B affording a colorless oil (40 mg, 91%). 1FINMR (400
MHz, CDC13) 6
8.33 (br, 1H), 8.02 (d, J= 8.0 Hz, 2H), 7.35 (m, 6H), 7.02 (t, J= 7.2 Hz, 1H),
4.68 (s, 2H), 3.93
(s, 3H), 3.50 (s, 3H), 3.46 (s, 4H). 13C NMR (100 MHz, CDC13) 6 166.88,
157.07, 143.81,
139.37, 129.92, 129.28, 128.81, 127.73, 122.34, 119.15, 72.59, 59.28, 52.07,
50.90, 48.44.
LRMS ESI: [M+H]' = 343.2.
37
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
Methyl 4-((1-butyl-3-phenylureido)methyl)benzoate (4g) Made according to
General
Procedure B affording colorless oil (59 mg, 98%). 1H NMR (400 MHz, CDC13)
68.04 (d, J = 8.0
Hz, 2H), 7.39 (d, J= 8.0 Hz, 2H), 7.27 (m, 4H), 7.03 (t, J= 7.2 Hz, 1H), 6.32
(s, 1H), 4.65 (s,
2H), 3.93 (s, 3H), 3.36 (t, J= 7.2 Hz, 2H), 1.64 (m, 2H), 1.37 (m, 2H), 0.96
(t, J= 7.2 Hz, 3H).
13C NMR (100 MHz, CDC13) 6 166.74, 155.30, 143.11, 138.83, 130.14, 129.50,
128.85, 127.04,
123.16, 119.90, 52.12, 50.49, 47.74, 30.52, 20.18, 13.81. LRMS ESI: [M+H] =
341.1
Methyl 4-((1-phenethy1-3-phenylureido)methyl)benzoate (4h) Made according to
General Procedure B affording a colorless oil (113 mg, 92%). 1H NMR (400 MHz,
CDC13) 6
8.02 (d, J = 8.4 Hz, 2H), 7.36 (m, 4H), 7.30 (d, J = 7.2 Hz, 1H), 7.21 (m,
4H), 7.08 (d, J= 8.0
Hz, 2H), 6.99 (t, J= 7.2 Hz, 1H), 6.00 (s, 1H), 4.58 (s, 2H), 3.91 (s, 3H),
3.59 (t, J = 6.8 Hz,
2H), 2.90 (t, J= 6.8 Hz, 2H). 13C NMR (100 MHz, CDC13) 6 166.71, 155.61,
142.99, 138.86,
138.72, 130.06, 129.43, 129.00, 128.86, 128.65, 127.28, 126.92, 122.95,
119.83, 52.08, 50.38,
49.94, 34.74. LRMS ESI: [M+H]' = 389.2.
4-01-(3-(dimethylamino)propy1)-3-(2-methoxyphenyl)ureido)methyl)-N-
hydroxybenzamide (5a) The synthesis of 5a is representative, General Procedure
C. Solid
NaOH (125 mg, 3.12 mmol) was dissolved in an aq. solution (50% wt, 1 mL) at 0
C. Then a
solution of 4a (156 mg, 0.390 mmol) in THF/Me0H (1:1, 6 mL total) was added
dropwise where
the biphasic solution became homogenous upon compete addition. The resulting
solution was
allowed to stir 30 min at room temperature. The reaction was quenched with
AcOH (0.223 mL,
3.90 mmol) and concentrated in vacuo, and the crude product was purified via
HPLC Method 2
and neutralized with bicarbonate wash affording the title compound (20 mg,
13%). 1H NMR
(400 MHz, DMSO-d6) 6 11.22 (br s, 1H), 9.02 (br s, 1H), 7.80-7.76 (m, 3H),
7.39 (d, J= 8.4 Hz,
2H), 7.00-6.94 (m, 2H), 6.88-6.84 (m, 2H), 4.62 (s, 2H), 3.72 (s, 3H), 3.43-
3.40 (m, 2H), 2.82
(br s, 2H), 2.56 (s, 6H), 1.91-1.86 (m, 2H). 13C NMR (100 MHz, DMSO-d6) 6
155.35, 149.62,
141.56, 131.69, 128.60, 127.24, 127.15, 127.02, 123.14, 121.40, 120.23,
110.78, 55.62, 54.33,
49.32, 44.24, 42.76, 23.45. HRMS ESI: calc. for C2iH28N404 [M+H]' m/z =
401.2183; found
401.2164.
N-hydroxy-4-01-(3-hydroxypropy1)-3-(2-methoxyphenyl)ureido)methyl)-benzamide
(5b) Made according to General Procedure C and purified via Method 3 affording
the title
compound (95 mg, 84 %). 1H NMR (400 MHz, DMSO-d6) 6 11.19 (br s, 1H), 7.82 (d,
J= 7.6
Hz, 1H), 7.76-7.71 (m, 3H), 7.37 (d, J= 8.0 Hz, 2H), 6.95 (d, J = 4.0 Hz, 2H),
6.88-06.85 (m,
1H), 4. 58 (s, 2H), 3.74 (s, 3H), 3.48 (t, J= 5.6 Hz, 2H), 3.40 (t, J= 6.8 Hz,
2H), 1.73-1.69 (m,
2H). 13C NMR (100 MHz, DMSO-d6) 6 164.10, 155.18, 149.08, 141.99, 131.56,
128.91, 127.18,
127.10, 126.93, 122.61, 120.49, 120.29, 110.76, 57.59, 55.69, 49.28, 43.97,
30.61. HRMS ESI:
calc. for Ci9H23N305 [M+H]' m/z = 374.1710; found 374.1693.
38
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
4-01-(2-(1H-indo1-3-yl)ethyl)-3-(2-methoxyphenyl)ureido)methyl)-N-
hydroxybenzamide (5c) Made according to General Procedure C and purified via
Method 1
affording the title compound (62 mg, 57%). 1H NMR (400 MHz, DMSO-d6) 6 11.21
(br s, 1H),
10.86 (s, 1H), 10.12 (br s, 1H), 7.86 (t, J = 7.6 Hz, 1H), 7.75 (d, J = 8.0
Hz, 2H), 7.56 (d, J = 8.0
Hz, 1H), 7.42-7.40 (m, 3H), 7.34 (d, J = 8.0 Hz, 1H), 7.18 (s, 1H), 7.07 (t, J
= 7.2 Hz, 1H), 7.01-
6.96 (m, 3H), 6.90-6.86 (m, 1H), 4.64 (s, 2H), 3.71 (s, 3H), 3.62 (t, J= 7.6
Hz, 2H), 3.00 (t, J=
7.6 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) 6 163.97, 158.37, 154.72, 148.88,
141.90, 136.21,
131.65, 128.70, 127.12, 122.98, 122.65, 120.97, 120.35, 118.31, 118.18,
111.41, 111.11, 110.66,
55.66, 49.77, 48.47, 23.89. HRMS ESI: calc. for C26H26N404 [M+H] m/z =
459.2027; found
459.2030.
N-hydroxy-4-01-(4-hydroxyphenethyl)-3-(2-methoxyphenyl)ureido)methyl)-
benzamide (5d) Made according to General Procedure C and purified via Method 2
affording
the title compound (63 mg, 63%). 1H NMR (400 MHz, DMSO-d6) 6 11.20 (br s, 1H),
9.20 (br s,
1H), 7.82 (d, J= 8.0 Hz, 2H), 7.74 (d, J= 8.0 Hz, 2H), 7.38 (d, J= 8.4 Hz,
2H), 7.37 (s, 1H),
7.04 (d, J = 8.4 Hz, 2H), 6.98 (d, J = 8.0 Hz, 2H), 6.89-6.85 (m, 1H), 6.69
(d, J= 8.4 Hz, 2H),
4.57 (s, 2H), 3.75 (s, 3H), 3.49 (t, J = 7.6 Hz, 2H), 2.75 (t, J= 7.6 Hz, 2H).
13C NMR (100 MHz,
DMSO-d6) 6 164.00, 155.76, 154.62, 148.97, 141.88, 131.63, 129.63, 128.83,
127.12, 122.72,
120.44, 120.33, 115.22, 110.69, 55.69, 49.68, 49.49, 33.21. HRMS ESI: calc.
for C24H25N305
[M+H] ' m/z =436.1867; found 436.1858.
N-hydroxy-4-01-(3-methoxypropy1)-3-phenylureido)methyl)benzamide (5e) Made
according to General Procedure C and purified via Method 2 affording the title
compound (48
mg, 76%). 1H NMR (400 MHz, DMSO-d6) 6 11.19 (br, 1H), 8.36 (s, 1H), 7.72 (d, J
= 8.4 Hz,
2H), 7.45 (d, J= 7.6 Hz, 2H), 7.32 (d, J= 8.4 Hz, 2H), 7.23 (m, 2H), 6.94 (t,
J= 7.2 Hz, 1H),
4.61 (s, 2H), 3.34 (m, 4H), 3.21 (s, 3H), 1.74 (m, 2H). 13C NMR (100 MHz, DMSO-
d6) 6164.04,
155.30, 142.25, 140.42, 131.49, 128.31, 127.06, 121.87, 119.88, 69.16, 57.88,
49.08, 43.56,
27.81. HRMS ESI: calc. for Ci9H23N304 [M+H]' m/z = 358.1761; found 358.1785.
N-hydroxy-4-01-(2-methoxyethyl)-3-phenylureido)methyl)benzamide (5f) Made
according to General Procedure C and purified Method 2 affording the title
compound (20 mg,
50%). 1H NMR (400 MHz, DMSO-d6) 6 11.17 (s, 1H), 9.01 (br, 1H), 8.44 (s, 1H),
7.72 (d, J=
8.0 Hz, 2H), 7.42 (d, J = 7.6 Hz, 2H), 7.32 (d, J = 8.0 Hz, 2H), 7.23 (t, J=
7.6 Hz, 2H)H, 6.94 (t,
J = 7.2 Hz, 1H), 4.64 (s, 2H), 3.49 (s, 4H), 3.28 (s, 3H). 13C NMR (100 MHz,
DMSO-d6) 6
164.06, 155.46, 142.17, 140.30, 131.43, 128.32, 126.97, 121.83, 119.64, 70.88,
58.31, 49.82,
46.35. HRMS ESI: calc. for Ci8H2iN304 [M+H]' m/z = 344.1605; found 344.1601.
4-((1-butyl-3-phenylureido)methyl)-N-hydroxybenzamide (5g) Made according to
General Procedure C and purified by Method 2 affording the title compound (40
mg, 68%). 1H
39
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
NMR (400 MHz, DMSO-d6) 6 11.17 (br, 1H), 8.36 (s,1H), 7.72 (d, J= 8.0 Hz, 2H),
7.46 (d, J=
7.6 Hz, 2H), 7.32 (d, J = 8.0 Hz, 2H), 7.22 (t, J= 7.6 Hz, 2H), 6.94 (t, J=
7.2 Hz, 1H), 4.62 (s,
2H), 3.30 (m, 1H), 1.48 (m, 2H), 1.27 (m, 2H), 0.86 (t, J = 7.6 Hz, 3H). 13C
NMR (100 MHz,
DMSO-d6) 6 164.01, 155.22, 142.30, 140.45, 131.42, 128.19, 127.00, 126.96,
121.80, 120.04,
49.01, 46.11, 29.64, 19.43, 13.77. HRMS ESI: calc. for Ci9H23N303 [M+H] m/z =
342.1812;
found 342.1802.
N-hydroxy-4-((1-phenethy1-3-phenylureido)methyl)benzamide (5h) Made according
to General Procedure C and purified by Method 2 affording the title compound
(71 mg, 63%).
1H NMR (400 MHz, DMSO-d6) 6 11.15 (br, 1H), 7.72 (d, J= 8.0 Hz, 2H), 7.45 (d,
J= 8.0 Hz,
2H), 7.31 (d, J= 8.0 Hz, 2H), 7.24 (m, 7H), 6.95 (t, J= 7.2 Hz, 1H), 4.60 (s,
2H), 3.54 (t, J = 7.6
Hz, 2H), 2.82 (t, J= 7.6 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) 6 164.04, 155.12,
142.12,
140.35, 139.04, 131.50, 128.77, 128.31, 128.20, 127.09, 127.05, 126.16,
121.91, 120.13, 49.20,
48.04, 33.91. HRMS ESI: calc. for C23H23N303 [M+H]' m/z = 390.1812; found
390.1793.
Compund Synthesis: makin reference to Scheme 2
N-butylaniline (6a) Was synthesized in an analogous manner previously reported
(Org
Lett, 4, 581)." Briefly, CuI (19 mg, 0.1 mmol) and freshly ground K3PO4 (849
mg, 4 mmol)
were placed in a sealed tube followed by sequential addition of isopropanol (2
mL), ethylene
glycol (0.222 mL, 4.0 mL), phenyl-iodide (0.224 mL, 2.0 mmol) and n-butylamine
(0.237 mL,
2.4 mmol). The tube was then sealed and stirring commenced at 80 C for 18 h.
After cooling
to room temperature the reaction was diluted with water: ethyl ether (1:1, 10
mL). The aqueous
layer was extracted with ether (3x 5mL), washed with brine (15 mL), dried over
sodium sulfate
and concentrated in vacuo. Purification via flash chromatography afforded the
title compound as
a yellow oil (235 mg, 79%). 1H NMR (400 MHz, CDC13) 6 7.19 (m, 2H), 6.70 (t, J
= 7.2 Hz,
1H), 6.61 (d, J= 8.4 Hz, 2H), 3.60 (br, 1H), 3.12 (t, J= 7.2 Hz, 2H), 1.62 (m,
2H), 1.44 (m, 2H),
0.98 (t, J= 7.2 Hz, 3H). Spectra matches that reported in Okano et al.,
"Synthesis of secondary
arylamines through copper-mediated intermolecular aryl amination," Org Lett
2003, 5(26):4987-
4990.
N-(3-methoxypropyl)aniline (6b) Made following the same procedure for 6a
affording
a light yellow oil (282 mg, 85%). 1H NMR (400 MHz, CDC13) 6 7.18 (m, 2H), 6.69
(t, J = 7.2
Hz, 1H), 6.61 (d, J = 8.4 Hz, 2H), 3.92 (br, 1H), 3.52 (tJ= 6.0 Hz, 2H), 3.60
(s, 3H), 3.23 (t, J=
6.8 Hz, 2H), 1.90 (m, 2H). Spectra matches that reported in Guo et al.,
Efficient Iron-Catalyzed
N-Arylation of Aryl Halides with Amines," Org Lett 2008,10(20):4513-4516.
Methyl 4-((3-butyl-3-phenylureido)methyl)benzoate (7a) Methyl 4-
(aminomethyl)benzoate hydrochloride (101 mg, 0.5 mmol) was taken up in a
biphasic solution
of DCM:sat. bicarbonate (1:1, 4 mL) and added triphosgene (49 mg, 0.17 mmol)
at 0 C. After
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
30 min, the aqueous layer was extracted with DCM (3x 5mL), washed with brine
(15 mL) and
concentrated in vacuo. The crude isocyante was taken up in DCM (2 mL) and
added 6a (75 mg,
0.5 mmol) and Et3N (0.209 mL, 1.5 mmol) and resulting solution allowed to stir
overnight at
room temperature. The reaction was quenched with with sat. bicarbonate (5 mL)
and extracted
with DCM (3x 5mL). The combined organics were washed with brine (15 mL), dried
over
sodium sulfate, concentrated in vacuo. The crude material was purified via
flash
chromatography affording the title compound as an off-white waxy solid (93 mg,
55%). 1H
NMR (400 MHz, CDC13) 6 7.95 (d, J= 8.0 Hz, 2H), 7.42 (t, J= 7.6 Hz, 2H), 7.32
(m, 1H), 7.25
(m, 4H), 4.45 (t, J= 5.6 Hz, 1H), 4.41 (d, J= 6.0 Hz, 2H), 3.89 (s, 3H), 3.70
(t, J= 7.6 Hz, 2H),
1.48 (m, 2H), 1.31 (m, 2H), 0.89 (t, 7.2 Hz, 3H). 13C NMR (100 MHz, CDC13) 6
166.89, 156.87,
145.14, 141.61, 130.08, 129.78, 128.85, 128.73, 127.81, 126.96, 52.01, 49.21,
44.25, 30.68,
19.92, 13.82. LRMS ESI: [M+H] = 341.1.
Methyl 4-03-(3-methoxypropy1)-3-phenylureido)methyl)benzoate (7b) Made
according to that of 7a except using 6b as the secondary amine affording the
title compound as
an off-white waxy solid (65 mg, 36%). 1H NMR (400 MHz, CDC13) 6 7.96 (d, J=
8.0 Hz, 2H),
7.43 (t, J= 7.6 Hz, 2H), 7.33 (m, 1H), 7.27 (m, 4H), 4.69 (br, 1H), 4.42 (6.0
Hz, 2H), 3.90 (s,
3H), 3.80 (t, J= 7.2 Hz, 2H), 3.43 (t, J= 6.4 Hz, 2H), 3.27 (s, 3H), 1.83 (m,
2H). 13C NMR (100
MHz, CDC13) 6 166.88, 156.95, 145.05, 141.63, 130.10, 129.80, 128.90, 127.79,
127.00, 70.23,
58.54, 52.03, 46.86, 44.29, 28.82. LRMS ESI: [M+H]' = 357.1.
Methyl 4-((3-ethyl-3-phenylureido)methyl)benzoate (7c) Made according to that
of 7a
except using commercially available N-ethylaniline as the secondary amine
affording the title
compound as an off-white waxy solid (197 mg, 63%). 1H NMR (400 MHz, CDC13) 6
7.94 (d, J=
8.4 Hz, 2H), 7.24 (t, J= 7.6 Hz, 2H), 7.32 (t, J= 7.2 Hz, 1H), 7.26 (m, 4H),
4.57 (br, 1H), 4.41
(d, J= 5.6 Hz, 1H), 3.88 (s, 3H), 3.76 (dd, J= 14, 7.2 Hz, 2H), 1.12 (t, J=
7.2 Hz, 3H). 13C
NMR (100 MHz, CDC13) 6 166.82, 156.64, 145.09, 141.22, 130.03, 129.72, 128.79,
127.82,
126.92, 51.95, 44.17, 44.09, 13.82. LRMS ESI: [M+H]' = 313.1.
4-((3-butyl-3-phenylureido)methyl)-N-hydroxybenzamide (8a) Made according to
General Procedure C and purified by Method 2 affording the title compound as
an off-white
solid (74 mg, 80%). 1H NMR (400 MHz, DMSO-d6) 6 11.13 (br, 1H), 8.72 (br, 1H),
7.66 (d, J
=8.0 Hz, 2H), 7.43 (t, J= 7.6 Hz, 2H), 7.27 (m, 5H), 6.23 (t, J= 5.6 Hz, 1H),
4.20 (d, J= 5.6 Hz,
2H), 3.57 (t, J= 6.8 Hz, 2H), 1.35 (m, 2H), 1.23 (m, 2H), 0.82 (t, J= 6.8 Hz,
3H). 13C NMR
(100 MHz, DMSO-d6) 6 164.17, 156.48, 144.50, 142.12, 130.129.55, 128.22,
126.67, 126.61,
48.44, 43.38, 30.21, 19.35, 13.72. HRMS ESI: calc. for Ci9H23N303 [M+H]' m/z =
342.1812;
found 342.1825.
41
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
N-hydroxy-4-03-(3-methoxypropy1)-3-phenylureido)methyl)benzamide (8b) Made
according to General Procedure C and purified by Method 2 affording an off-
white solid (59 mg,
91%). 1H NMR (400 MHz, DMSO-d6) 6 11.14 (br, 1H), 7.66 (d, J= 8.0 Hz, 2H),
7.43 (t, J = 7.6
Hz, 2H), 7.27 (m, 5H), 6.27 (t, J = 6.0 Hz, 1H), 4.21 (d, J = 5.8 Hz, 2H),
3.62 (t, J = 7.2 Hz, 2H),
3.28 (t, J= 6.4 Hz, 2H), 3.14 (s, 3H), 1.63 (m, 2H). 13C NMR (100 MHz, DMSO-
d6) 6 164.13,
156.51, 144.44, 142.19, 130.89, 129.57, 128.18, 126.70, 126.64, 69.49, 57.80,
46.35, 43.39,
28.32. HRMS ESI: calc. for Ci9H23N304 [M+H] ' m/z = 358.1761; found 358.1749.
4-((3-ethyl-3-phenylureido)methyl)-N-hydroxybenzamide (8c) Made according to
General Procedure C and purified by Method 2 affording an off-white solid (91
mg, 96%). 1H
NMR (400 MHz, DMSO-d6) 6 11.13 (br, 1H), 7.67 (d, J = 8.0 Hz, 2H), 7.43 (t, J=
7.6 Hz, 2H),
7.26 (m, 5H), 6.30 (t, J = 6.0 Hz, 1H), 4.21 (d, J = 5.6 Hz, 2H), 3.61 (dd, J=
14, 7.2 Hz, 2H),
0.99 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) 6 164.21, 156.34, 144.54,
142.00,
130.92, 129.57, 128.32, 126.75, 126.67, 43.70, 43.39, 13.76. HRMS ESI: calc.
for Ci7Hi9N303
[M+H] ' m/z = 314.1499; found 314.1489.
HDAC inhibition assays
HDAC inhibition assays were performed by Reaction Biology Corp. (Malvern, PA)
using
isolated human, recombinant full-length HDAC1 and -6 from a baculovirus
expression system in
5f9 cells. An acetylated fluorogenic peptide, RHKKAc, derived from residues
379-382 of p53
was used as substrate. The reaction buffer was made up of 50 mM Tris-HC1 pH
8.0, 127 mM
NaC1, 2.7 mM KC1, 1 mM MgC12, 1 mg/mL BSA, and a final concentration of 1%
DMSO.
Compounds were delivered in DMSO and delivered to enzyme mixture with
preincubation of 5-
10 min followed by substrate addition and incubation for 2 h at 30 C.
Trichostatin A and
developer were added to quench the reaction and generate fluorescence,
respectively. Dose-
response curves were generated starting at 30 ILIM compound with three-fold
serial dilutions to
generate a 10-dose plot. ICso values were then generated from the resulting
plots, and the values
expressed are the average of duplicate trials standard deviation.
Compound 1 was identified to possess submicromolar HDAC inhibitory activity;
however, it was not selective against representative members of the Zn2'-
dependant Classes 1
and 2 (HDAC1 and HDAC6, respectively). It was discovered that activity and
selectivity could
be improved for HDAC6 by accessing a unique cavity on the surface of HDAC6.
This was
accomplished substitutions on the urea nitrogens. There is a shorter substrate
channel on
HDAC6 relative to HDAC1 and this feature represented an excellent strategy to
impart critical
isoform selectivity (Butler et al., J Am Chem Soc 2010, 132(30:10842-10846;
Kalin et al., J
Med Chem 2012, 55(2):639-651). By incorporating substitutions on the urea
motif, the
additional branched molecular surface could form valuable contacts with the
subtle differences at
42
CA 02866707 2014-09-08
WO 2013/134467 PCT/US2013/029521
the HDAC6 surface while the benzyl linker would give a shorter linker that
would favor away
from HDAC1 inhibition. A summary of the HDAC1 and HDAC6 inhibitory data
obtained is
presented in Table 1.
Table 1. HDAC inhibition screen of substituted urea compounds'
Compound Structure HDAC1 IC50 HDAC6 IC50 Fold
Selective
(nM) (nM) (HDAC1/HDAC6)
1 o 265 59 139 27 2
0
N _OH
H H
Si E1
N N
SI TO
5a l 2550 540 458 64 6
N
--- -... 0
0 H 401 N-OH
H
N N
0 8
5b HO 0 1910 570 2.38 0.38 803
No 0 0 =H
0 TO
5c
HN 8950 770 468 130 19
N 0
(:) NOH
H 0
INI{N1
0 8
5d OH 1690 120 5.80 0.50 292
O0
so0 _OH
H
N N
0 1r
0
5e
l 5180 130 11.7 1.7 443
O
0
N _OH
SI H
IS TO
5f 0 0 2250 420 9.26 0.66 243
H H isN -OH
H
N N
01 8
43
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
5g
0 3020 740 5.02 0.060 600
H is 11,0H
0N{N
8
5h
lei 3120 640 14.0 0.75 222
0
-OH
H 101
NN
110 8
8a 0 8120 600 25.2 2.5 322
N-OH
( H=H
1.1 TO
8b l 1360 450 60.3 15.8 226
0 0
r H is 11,0H
NN
0 8
8c 0 1360 650 41.1 0.40 330
= N-OH
NY kl 0 H
0
0
9a o 1060 8.3 128
N_OH
H H 0 H
r& NyN
0
H3C0
9b 0 579 2.2 263
0 irzi-OH
H H
N' NT N
f
0
9c 0 1000 6.9 145
_OH
H H 0 11
N
N N,
A
44
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
Tubastatin Ab \ 16400 260 15.0 0.01
1903
N 0
--
N 101 NHOH
0
TSAb'' 0 0 5 1.0 1.2 0.30
4
1 1 11NHOH
MGCD0103 . 0 102 38 >10,000d --
rN
(NN =
aic50 displayed are the mean of two experiments standard deviation obtained
from curve fitting
of 10-point enzyme assay starting from 30 iuM analog with 3-fold serial
dilution. Values are
extracted from fitting dose-response curves to the data points, b Butler et
al., J Am Chem Soc
2010, 132(30:10842-10846; C Trichostatin A; d Fournel et al., "MGCD0103, a
novel isotype-
selective histone deacetylase inhibitor, has broad spectrum antitumor activity
in vitro and in
vivo," Mol Cancer Ther 2008, 7(4):759-768.
The analogs based on 1 maintained the same 2-methoxyphenyl cap group but
contained
varied substitutions on the proximal linking nitrogen of the urea (5a-d).
Introducing the
branching element at this position had a dramatic impact on decreasing
activity at HDAC1.
Interestingly, inhibition at HDAC6 was found to be dependent upon the nature
of this
substitution. The dimethylamino substitution as in 5a, and the 3-indoyl
substitution as in Sc,
both proved detrimental to HDAC6 inhibition as they were over three times less
potent
compared to compound 1. They did however, maintain low micromolar inhibitory
activity at
HDAC1; but the activity against HDAC6 was only in the submicromolar range. As
the tertiary
amine in 5a would be protonated at physiological pH, it is possible that a
positive charge is
unfavorable for proper target binding. Likewise, the larger indole group of Sc
may simply
present too much steric bulk to be properly accommodated by the active site.
However, the 3-
hydroxypropyl derivative, 5b, and the 4-hydroxyphenylethyl derivative, 5d,
resulted in a
significant increase in the inhibition of HDAC6. These substitutions had only
a marginal effect
on HDAC1 activity. It is possible the hydroxyl groups of 5b and 5d are able to
serve as H-bond
acceptors or donors and possess favorable interactions with key amino acid
residues on the
HDAC6 surface, thus improving binding affinity.
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
The first series of compounds based on 1 maintained the 2-methoxy group in the
aryl
urea cap. The oxidative potential of phenols presents a substantial hurdle for
in vivo efficacy
thus the structure activity relationship (SAR) investigation was furthered by
synthesizing a series
with a phenyl cap using the same chemistry. To demonstrate the influence of an
H-bond
acceptor the the free hydroxyl moeity was masked. Capping the free hydroxyl
with a methyl
group resulted in 5e, and was also found to be a low nanomolar inhibitor with
>400 fold
selectivity for HDAC6. Shortening to an ethylene bridge in 5f did not
significantly dissuade
HDAC6 inhibition, but did slightly increase activity against HDAC1 ultimately
lowering the
selectivity for HDAC6. The general trend established was that an H-bond donor
and large
aromatic groups deterred activity, whereas smaller groups with H-bond
acceptors were favored
for selective HDAC6 activity. Interestingly, the n-butyl 5g and phenethyl 5h
were proficient
HDAC6i in the low nanomolar range with 5g possessing excellent selectivity
over HDAC1 (600
fold). These data refute the notion that a specific H-bond interaction is
required for activity.
Shifting the branching element to the distal urea nitrogen resulted in analogs
8a-c. The
most potent of this series, 8a, possessed the same n-butyl substitution as 5e.
While 8a is a
nanomolar HDAC6I, it is five-times less potent and more importantly, is less
selective than the
proximally substituted homolog 5e. The methoxy variant 8b suffered a dramatic
decrease in
potency towards HDAC6. Whereas the alkyl to heteroalkyl switch on the proximal
nitrogen
resulted in equipotent inhibition on the distal nitrogen, this modification
was detrimental to
furthering potent HDAC6I development. Decreasing the length of the alkyl
branch in 8c also
resulted in decreased HDAC6 inhibition. These data point to specific
requirements for inhibitors
decorated with a cap groups comprising of an acyclic urea and that potent and
selective
inhibition comes most from urea substitutions on the proximal nitrogen to
generate a branched
cap group.
Evaluating the disclosed compounds against other HDACi's developed by others
reveals
5g, termed "Nexturastat A," is in fact a potent and selective HDAC6i.
Comparing for example,
5g to Tubastatin A, another HDAC6i (Butler et al., J Am Chem Soc 2010,
132(30:10842-
10846), reveals that the inhibition of HDAC6 has been improved while
maintaining excellent
selectivity relative to HDAC1. 5g also demonstrates comparable HDAC6 potency
to
Trichostatin A (TSA) (see Table 1). Additionally, the amino-benzamide ZBG has
been
incorporated into the HDACi's, and its introduction reduces Class 2 inhibition
resulting in Class
1 selectivity; this is typified by MGCD0103, an HDACI that possesses
antiproliferative activity
and that has recently entered clinical trials (Zhou et al., "Discovery of N-(2-
aminopheny1)4- 4-
pyridin-3-ylpyrimidin-2-ylamino)methyl benzamide (MGCD0103), an orally active
histone
46
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
deacetylase inhibitor," J Med Chem 2008, 51(14):4072-4075). Compared to
MGCD0103, 5g
leads to a 30-fold reduction in activity at HDAC1.
Similar experiments were done with compounds 10a, 10b, and lla , which are
cyclic
ureas of Formula I-C (Table 2).
Table 2. HDAC scree of cyclic urea compounds
Compound HDAC1 1050 HDAC6 1050 Fold Selective
(nM) (nM) (HDAC1/HDAC6)
10a 28600 74.2 386
10b 11800 22.2 531
lla 7560 25.6 295
Since the HDAC isoforms are highly homologous obtaining selectivity is
critical for
avoiding off-target effects and is paramount for the development of the
disclosed HDAC6i's. It
is well known that Class 1 inhibition is responsible for the cytotoxicity
associated with pan-
selective HDACi; thus, 5g was screened against all 11 isoforms (Table 3). In
the similar Class 1
and Class 4 isoforms, 5g displayed low micromolar activity compared to the low
nanomolar
activity of HDAC6. Moreover, 5g demonstrated high levels of selective
inhibition against
members of the related Class 2 HDAC isforms reaching >1000-fold selective in
some cases.
These data establish 5g, and similar analogs, to be potent and isoform
selective HDAC6i's.
Table 3. Inhibitory profile of 5g against HDAC1-11a
Isoform ICso Fold selective for
(M) HDAC6
HDAC1 3.02 1.04 600
HDAC2 6.92 0.763 1380
HDAC3 6.68 1.75 1330
HDAC4 9.39 0.863 1870
HDAC5 11.7 0.141 2330
HDAC6 0.00502 0.00060 --
HDAC7 4.46 0.665 888
HDAC8 0.954 0.0799 190
HDAC9 6.72 1.15 1340
HDAC10 7.57 0.481 1510
HDAC11 5.14 0.686 1020
47
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
aIC50 displayed are the mean of two experiments standard deviation obtained
from curve fitting
of 10-point enzyme assay starting from 30 ILIM analog with 3-fold serial
dilution. Values are
extracted from fitting dose-response curves to the data points.
Tubulin and histone acetylation western blot assay
The ability of 5g to induce hyperacetylation of a-tubulin, a hallmark of HDAC6
inhibition, without elevating levels of acetylated histones was evaluated. B16
melanoma cells
were plated at 105 cells/well in 12 well plates and allowed to adhere
overnight. A 50 mM stock
of compound was then added by serial dilutions in complete medium to the
indicated
concentrations. Cells were incubated for 24 h under humidified conditions (37
C, 5% CO2).
Wells were then washed with cold PBS, and cells were lysed in a buffer
containing 10 mM Tris-
HC1 pH 8.0, 10% SDS, 4 mM urea, 100 mM DTT, and lx protease inhibitor (Roche).
Cells
were lysed for 30 min on ice and then sonicated for 8 min (8 cycles of 30 s
on/ 30 s rest). Cells
were then boiled for 10 min with 6x gel loading buffer and resolved on 4-15%
gradient gels and
subsequently transferred onto nitrocellulose membranes. Membranes were blocked
with 5%
milk in PBS-T and detection of specific antigens using antibodies against
acetyl-H3 and H3
(Cell Signaling), and acetyl- a-tubulin and a-tubulin (Sigma). Bands were
detected by scanning
blots with LI-COR Odyssey imaging system using both 700 and 800 channels.
HDAC6 contains two catalytic domains. Its C-terminus domain is the functional
domain
for both synthetic and physiological substrates, whereas the N-terminal domain
is devoid of
enzymatic activity (Zou et al., "Characterization of the two catalytic domains
in histone
deacetylase 6," Biochem Biophys Res Commun 2006, 341(1):45-50). Low nanomolar
treatement
of 5g on B16 murinemelanoma cells led to a dose-dependant increase of acetyl a-
tubulin levels
without a concamanent elevation of histone H3 acetylation (Figure 39)
indicating binding to the
second, enzymatically-active catalytic domain. Not until concentrations of 1
and 10 [iM were
used was an observable increase in histone H3 acetylation found. This was
expected as the
biochemical IC50 of 5g against the Class 1 HDACs, those responsible for
histone acetylation, is
in the micromolar range. There is a clear preference for activity of 5g in a
cellular environment
that corresponds to selective HDAC6 inhibition.
B16 melanoma cell growth inhibition assay
Compounds were evaluated in an MTS assay to determine the ability of selective
HDAC6i to exert an antiproliferative effect on B16 murine melanoma cells. B16
murine
melanoma cells were plated at 5x103/well in 96 well flat bottom plates. The
following day,
media was changed to that containing various concentrations of HDACi or
matched DMSO
vehicle concentrations diluted in complete medium done in triplicate. Cells
were incubated for
48
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
48 hours at 37 C and 5% CO2. Density of viable, metabolically active cells was
quantified using
a standard MTS assay (CellTiter 96TM AO
,ueous One, Promega, Madison, WI) as per
manufacturer's instructions. Briefly, 201AL of reagent were added per well and
incubated at 37 C
for 3 hours. Absorbances at 490 nM were measured spectrophotometrically with
background
subtraction at 690nM. All values were then normalized and expressed as a
percentage of
medium control (100%).
Treatment with the compounds for 48 h resulted in dose-dependent growth
inhibition of
the oncogenic melanoma cells summarized in Table 4. The general trend for
inhibiting cell
growth correlates with potency for HDAC6. However, the potent and selective
5b, performed
very poorly in the cellular assay possibly due to it being highly polar and
lacking efficient cell
permeability. Comparing the most active compounds 5d and 5f-h in this whole-
cells assay
reveals that the most selective HDAC6i's have the greatest efficacy in
inhibiting cell growth. It
should also be noted that they also have higher cLogP values, possibly
contributing to improved
cell permeability. As the cLogP is adjusted to more optimal levels, as
exemplified for 5g (cLogP
= 2.20), cellular efficacy is restored, demonstrating that a proper balance of
physiochemical
parameters must be maintained.
Table 4. Antiproliferative activity against B16 murine melanoma cells
Compound GIso (11-1M)
5a 75.3 1.23
5b >100
Sc 30.4 1.32
5d 18.4 1.23
5e 22.2 1.41
5f 19.1 1.19
5g 14.3 1.15
5h 15.4 1.20
8a 65.8 1.19
8b >100
8c >100
Tubastatin 40.5 1.21
LBH589 0.150
49
CA 02866707 2014-09-08
WO 2013/134467
PCT/US2013/029521
Compared to the pan-selective HDACi LBH589, 5g is approximately 100-fold less
potent in inducing murine B16 melanoma cell death. This decreased efficacy is
unlikely due to
poor cell permeability, for as shown above, the treatment of B16 cells with
nanomolar doses of
5g results in increased acetyl-tubulin levels (Figure 39). Additionally, both
compounds possess
similar cLogP values (2.64 vs 2.20 for LBH589 and 5g, respectively). Rather,
the effects of
nonselective HDAC inhibition with LBH589 treatment are likely contributing to
its increased
potency, and in particular, its Class 1 activity. It is also of interest to
note that 5g has increased
potency against the B16 cell line in comparison to Tubastatin A (Table 4).
While a definitive
explanation for this difference in cellular activity is lacking, it is
possible that this is due to the
improved HDAC6 activity of 5g. While HDAC6-selective inhibitors have not
played a role in
cancer therapy to date, the data indicate that they have utility in this area.
This work thus
constitutes the first report of HDAC6 selective inhibitors that possess
antiproliferative effects
against melanoma cells.
The materials and methods of the appended claims are not limited in scope by
the
specific materials and methods described herein, which are intended as
illustrations of a few
aspects of the claims and any materials and methods that are functionally
equivalent are within
the scope of this disclosure. Various modifications of the materials and
methods in addition to
those shown and described herein are intended to fall within the scope of the
appended claims.
Further, while only certain representative materials, methods, and aspects of
these materials and
methods are specifically described, other materials and methods and
combinations of various
features of the materials and methods are intended to fall within the scope of
the appended
claims, even if not specifically recited. Thus a combination of steps,
elements, components, or
constituents can be explicitly mentioned herein; however, all other
combinations of steps,
elements, components, and constituents are included, even though not
explicitly stated.
50