Note: Descriptions are shown in the official language in which they were submitted.
DESCRIPTION
2,2-DIFLUOROPROPIONAMIDE DERIVATIVES OF BARDOXOLONE
METHYL, POLYMORPHIC FORMS AND METHODS OF USE THEREOF
BACKGROUND OF THE INVENTION
Pursuant to 37 C.F.R. 1.821(c), a sequence listing is submitted herewith as an
ASCII compliant text file named "REATP0073WO_ST25," created on April 24, 2013
and having a size of ¨6 kilobytes.
I. Field of the Invention
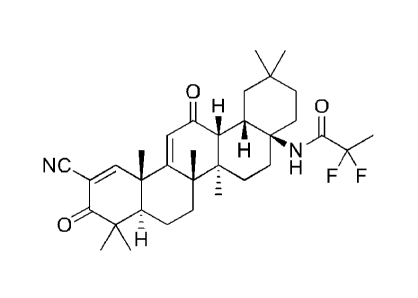
The present invention relates generally to the compound:
N-((4aS, 6aR,6bS,8aR,12aS,14aR,14b5)-11 -cyano-2,2,6a,6b,9,9,12a-
heptamethy1-10,14-dioxo-
1,2,3 ,4,4a,5,6,6a,6b,7,8,8a,9,10,12a,14,14a,14b-octadecahydropieen-
4a-y1)-2,2-difluoropropanamide,
also referred to herein as RTA 408, 63415, or PP415. The present invention
also
relates to polymorphic forms thereof, methods for preparation and use thereof,
pharmaceutical compositions thereof, and kits and articles of manufacture
thereof
II. Description of Related Art
The anti-inflammatory and anti-proliferative activity of the naturally
occurring
triterpenoid, oleanolic acid, has been improved by chemical modifications. For
example, 2-cyano-3,12-diooxoolcana-1,9(11)-dien-28-oic acid (CDDO) and related
compounds have been developed. See Honda et al., 1997; Honda etal., 1998;
Honda
et al., 1999; Honda el al., 2000a; Honda et al., 2000b; Honda et al., 2002;
Suh et al.,
1998; Suh et al., 1999; Place et al., 2003; Liby et al., 2005; and U.S.
Patents
8,129,429, 7,915,402, 8,124,799, and 7,943,778.
The methyl ester, bardoxolone methyl (CDDO¨Me), has been
1
CA 2869783 2019-09-05
evaluated in phase II and III clinical trials for the treatment and prevention
of diabetic
nephropathy and chronic kidney disease. See Pergola et al., 2011.
Synthetic triterpenoid analogs of oleanolic acid have also been shown to be
inhibitors of cellular inflammatory processes, such as the induction by IFN-y
of
inducible nitric oxide synthase (iNOS) and of COX-2 in mouse macrophages. See
Honda et al, (2000a), Honda et al. (2000b), Honda et al. (2002), and U.S.
Patents
8,129,429, 7,915,402, 8,124,799, and 7,943,778,
Compounds derived from oleanolic acid have been shown to affect the
function of multiple protein targets and thereby modulate the activity of
several
important cellular signaling pathways related to oxidative stress, cell cycle
control,
and inflammation (e.g., Dinkova-Kostova et al., 2005; Ahmad et al., 2006;
Ahmad el
al., 2008; Liby et al., 2007a, and U.S. Patents 8,129,429, 7,915,402,
8,124,799, and
7,943,778).
Given that the biological activity profiles of known triterpenoid derivatives
vary, and in view of the wide variety of diseases that may be treated or
prevented with
compounds having potent antioxidant and anti-inflammatory effects, and the
high
degree of unmet medical need represented within this variety of diseases, it
is
desirable to synthesize new compounds 'with different biological activity
profiles for
the treatment or prevention of one or more indications.
=
2
CA 2869783 2020-04-08
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
SUMMARY OF THE INVENTION
In some aspects of the present invention, there is provided a compound of the
formula (also referred to herein as RTA 408, 63415, or PP415):
0
0
N
NC F F
0
I:1
or a pharmaceutically acceptable salt thereof
In some embodiments, the compound is in the form of a pharmaceutically
acceptable salt. In some embodiments, the compound is not in the form of a
salt.
In another aspect of the present invention, there are provided polymorphic
forms of the above compound. In some embodiments, the polymorphic form has an
X-ray powder diffraction pattern (CuKa) comprising a halo peak at about 14
20. In
some embodiments, the X-ray powder diffraction pattern (CuKa) further
comprises a
shoulder peak at about 8 020. In some embodiments, the X-ray powder
diffraction
pattern (CuKa) is substantially as shown in FIG. 59. In some embodiments, the
polymorphic form has a Ts from about 150 C to about 155 C, including for
example, a Ts of about 153 C or a Ts of about 150 C. In some embodiments,
the
polymorphic form has a differential scanning calorimetry (DSC) curve
comprising an
endotherm centered from about 150 C to about 155 C. In some embodiments, the
endotherm is centered at about 153 C. In some embodiments, the endotherm is
centered at about 150 C. In some embodiments, the differential scanning
calorimetry
(DSC) curve is substantially as shown in FIG. 62.
In some embodiments, the polymorphic form is a solvate having an X-ray
powder diffraction pattern (CuKa) comprising peaks at about 5.6, 7.0, 10.6,
12.7, and
14.6 020. In some embodiments, the X-ray powder diffraction pattern (CuKa) is
substantially as shown in FIG. 75, top pattern.
In some embodiments, the polymorphic form is a solvate having an X-ray
powder diffraction pattern (CuKa) comprising peaks at about 7.0, 7.8, 8.6,
11.9, 13.9
(double peak), 14.2, and 16.0 020. In some embodiments, the X-ray diffraction
pattern (CuKa) is substantially as shown in FIG. 75, second pattern from top.
3
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
In some embodiments, the polymorphic form is an acetonitrile hemisolvate
having an X-ray powder diffraction pattern (CuKa) comprising peaks at about
7.5,
11.4, 15.6 and 16.6 020. In some embodiments, the X-ray diffraction pattern
(CuKa)
is substantially as shown in FIG. 75, second pattern from bottom. In some
embodiments, the polymorphic form has a Tg of about 196 C. In some
embodiments,
the polymorphic form has a differential scanning calorimetry (DSC) curve
comprising
an endotherm centered at about 196 C. In some embodiments, the differential
scanning calorimetry (DSC) curve is substantially as shown in FIG. 116.
In some embodiments, the polymorphic form is a solvate having an X-ray
powder diffraction pattern (CuKa) comprising peaks at about 6.8, 9.3, 9.5,
10.5, 13.6,
and 15.6 020. In some embodiments, the X-ray diffraction pattern (CuKa) is
substantially as shown in FIG. 75, bottom pattern.
In another aspect of the present invention, there are provided pharmaceutical
compositions comprising an active ingredient consisting of the above compound
or a
polymorphic form thereof (such as, e.g., any one of the polymorphic forms
described
herein above and below), and a pharmaceutically acceptable carrier. In some
embodiments, the pharmaceutical composition is formulated for administration:
orally, intraadiposally, intraarterially, intraarticularly, intracranially,
intradermally,
intral es i onal ly, intramuscularly, intranasally, intraocularly, intraperi
cardi al ly,
intraperitoneally, intrapleurally, intraprostatically, intrarectally,
intrathecally,
intratracheally, intratumorally, intraumbilically, intravaginally,
intravenously,
intravesicularlly, intravitreally, liposomally, locally, mucosally,
parenterally, rectally,
subconjunctival, subcutaneously, sublingually, topically, transbuccally,
transdermally,
vaginally, in cremes, in lipid compositions, via a catheter, via a lavage, via
continuous
infusion, via infusion, via inhalation, via injection, via local delivery, or
via localized
perfusion. In some embodiments, the pharmaceutical composition is formulated
for
oral, intraarterial, intravenous, or topical administration. In some
embodiments, the
pharmaceutical composition is formulated for oral administration.
In some embodiments, the pharmaceutical composition is formulated as a hard
or soft capsule, a tablet, a syrup, a suspension, an emulsion, a solution, a
solid
dispersion, a wafer, or an elixir. In some embodiments, the pharmaceutical
composition according to the invention further comprises an agent that
enhances
solubility and dispersibility. (For example, agents that enhance solubility
and
dispersibility include, but are not limited to, PEGs, cyclodextrans, and
cellulose
4
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
derivatives.) In some embodiments, the compound or polymorphic form is
suspended
in sesame oil.
In other embodiments, the pharmaceutical composition is formulated for
topical administration. In other embodiments, the pharmaceutical composition
is
formulated as a lotion, a cream, a gel, an oil, an ointment, a salve, an
emulsion, a
solution, or a suspension. In some embodiments, the pharmaceutical composition
is
formulated as a lotion, as a cream, or as a gel. In some embodiments, the
amount of
the active ingredient is from about 0.01% to about 5% by weight, about 0.01%
to
about 3% by weight, or 0.01%, 0.1%, 1%, or 3% by weight.
In another aspect of the present invention there are provided methods of
treating or preventing a condition associated with inflammation or oxidative
stress in
a patient in need thereof, comprising administering to the patient a
therapeutically
effective amount of the pharmaceutical composition as described above or
below.
The invention likewise relates to the compound
N-
((4aS,6aR,6bS,8aR,12aS,14aR,14bS)-11-cyano-2,2,6a,6b,9,9,12a-heptamethy1-10,14-
dioxo-1,2,3,4,4a,5,6,6a,6b,7,8,8a,9,10,12a,14,14a,14b-octadecahydropicen-4a-
y1)-2,2-
difluoropropanamide (or RTA 408) or a pharmaceutically acceptable salt
thereof, or a
polymorphic form of that compound (such as, e.g., any one of the polymorphic
forms
described herein above or below), or a pharmaceutical composition comprising
any of
the aforementioned entities and a pharmaceutically acceptable carrier
(including, e.g.,
the pharmaceutical compositions described above), for use in treating or
preventing a
condition associated with inflammation or oxidative stress. The invention also
relates
to the use of the aforementioned compound, polymorphic form or pharmaceutical
composition for the preparation of a medicament for the treatment or
prevention of a
condition associated with inflammation or oxidative stress. In some
embodiments,
the condition is associated with inflammation. In other embodiments, the
condition is
associated with oxidative stress. In some embodiments, the condition is a skin
disease
or disorder, sepsis, dermatitis, osteoarthritis, cancer, inflammation, an
autoimmune
disease, inflammatory bowel disease, a complication from localized or total-
body
exposure to ionizing radiation, mucositis, acute or chronic organ failure,
liver disease,
pancreatitis, an eye disorder, a lung disease or diabetes.
The present invention furthermore relates to the compound N-
((4 aS,6aR,6bS,8aR,12 aS,14aR ,14bS)-11-cyano-2,2,6a,6b,9,9,12 a-heptamethyl-
10,14 -
dioxo-1,2,3,4,4a,5,6,6a,6b,7,8,8a,9,10,12a,14,14a,14b-octadecahydropicen-4a-
y1)-2,2-
5
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
difluoropropanamide (or RTA 408) or a pharmaceutically acceptable salt
thereof, or a
polymorphic form of that compound (such as, e.g., any one of the polymorphic
forms
described herein above or below), or a pharmaceutical composition comprising
any of
the aforementioned entities and a pharmaceutically acceptable carrier
(including, e.g.,
the pharmaceutical compositions described above), for use in treating or
preventing a
condition selected from a skin disease or disorder, sepsis, dermatitis,
osteoarthritis,
cancer, inflammation, an autoimmune disease, inflammatory bowel disease, a
complication from localized or total-body exposure to ionizing radiation,
mucositis,
acute or chronic organ failure, liver disease, pancreatitis, an eye disorder,
a lung
disease, or diabetes. Accordingly, the invention relates to the use of the
aforementioned compound, polymorphic form or pharmaceutical composition for
the
preparation of a medicament for the treatment or prevention of a condition
selected
from a skin disease or disorder, sepsis, dermatitis, osteoarthritis, cancer,
inflammation, an autoimmune disease, inflammatory bowel disease, a
complication
from localized or total-body exposure to ionizing radiation, mucositis, acute
or
chronic organ failure, liver disease, pancreatitis, an eye disorder, a lung
disease, or
diabetes. The invention also relates to a method of treating or preventing a
condition
selected from a skin disease or disorder, sepsis, dermatitis, ostcoarthritis,
cancer,
inflammation, an autoimmune disease, inflammatory bowel disease, a
complication
from localized or total-body exposure to ionizing radiation, mucositis, acute
or
chronic organ failure, liver disease, pancreatitis, an eye disorder, a lung
disease, or
diabetes in a patient in need thereof, the method comprising administering to
the
patient a therapeutically effective amount of the aforementioned compound,
polymorphic form or pharmaceutical composition. In some embodiments, the
condition is a skin disease or disorder such as dermatitis, a thermal or
chemical burn,
a chronic wound, acne, alopecia, other disorders of the hair follicle,
epidermolysis
bullosa, sunburn, complications of sunburn, disorders of skin pigmentation, an
aging-
related skin condition; a post-surgical wound, a scar from a skin injury or
burn,
psoriasis, a dermatological manifestation of an autoimmune disease or a graft-
versus
host disease, skin cancer, or a disorder involving hyperproliferation of skin
cells. In
some embodiments, the skin disease or disorder is dermatitis. In some
embodiments,
the dermatitis is allergic dermatitis, atopic dermatitis, dermatitis due to
chemical
exposure, or radiation-induced dermatitis. In other embodiments, the skin
disease or
disorder is a chronic wound. In some embodiments, the chronic wound is a
diabetic
6
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
ulcer, a pressure sore, or a venous ulcer. In other embodiments, the skin
disease or
disorder is alopecia. In some embodiments, the alopecia is selected from
baldness or
drug-induced alopecia. In other embodiments, the skin disease or disorder is a
disorder of skin pigmentation. In some embodiments, the disorder of skin
pigmentation is vitiligo. In other embodiments, the skin disease or disorder
is a
disorder involving hyperproliferation of skin cells. In some embodiments, the
disorder involving hyperproliferation of skin cells is hyperkeratosis.
In other embodiments, the condition is an autoimmune disease, such as
rheumatoid arthritis, lupus, Crohn's disease, or psoriasis. In other
embodiments, the
condition is a liver disease, such as fatty liver disease or hepatitis.
In other embodiments, the condition is an eye disorder, such as uveitis,
macular degeneration, glaucoma, diabetic macular edema, blepharitis, diabetic
retinopathy, a disease or disorder of the corneal endothelium, post-surgical
inflammation, dry eye, allergic conjunctivitis or a form of conjunctivitis. In
some
embodiments, the eye disorder is macular degeneration. In some embodiments,
the
macular degeneration is the dry form. In other embodiments, the macular
degeneration is the wet form. In some embodiments, the disease or disorder of
the
corneal endothelium is Fuchs endothelial corneal dystrophy.
In other embodiments, the condition is a lung disease, such as pulmonary
inflammation, pulmonary fibrosis, COPD, asthma, cystic fibrosis, or idiopathic
pulmonary fibrosis. In some embodiments, the COPD is induced by cigarette
smoke.
In other embodiments, the condition is sepsis. In other embodiments, the
condition is mucositis resulting from radiation therapy or chemotherapy. In
some
embodiments, the mucositis presents orally. In other embodiments, the
condition is
associated with exposure to radiation. In some embodiments, the radiation
exposure
leads to dermatitis. In some embodiments, the radiation exposure is acute. In
other
embodiments, the radiation exposure is fractionated.
In other embodiments, the condition is cancer. In some embodiments, the
cancer is a carcinoma, sarcoma, lymphoma, leukemia, melanoma, mesothelioma,
multiple myeloma, or seminoma. In other embodiments, the cancer is of the
bladder,
blood, bone, brain, breast, central nervous system, cervix, colon,
endometrium,
esophagus, gall bladder, genitalia, genitourinary tract, head, kidney, larynx,
liver,
lung, muscle tissue, neck, oral or nasal mucosa, ovary, pancreas, prostate,
skin,
spleen, small intestine, large intestine, stomach, testicle, or thyroid.
7
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
In some embodiments, the pharmaceutical composition is administered before
or immediately after a subject is treated with a radiation therapy or a
chemotherapy
wherein the chemotherapy does not comprise RTA 408 or its polymorphic forms.
In
some embodiments, the pharmaceutical composition is administered both before
and
after the subject is treated with radiation therapy, chemotherapy or both. In
some
embodiments, the treatment reduces a side effect of the radiation therapy or
the
chemotherapy. In some embodiments, the side effect is mucositis and
dermatitis. In
some embodiments, the treatment enhances the efficacy of the radiation therapy
or the
chemotherapy. In some embodiments, the chemotherapy comprises administering to
the patient a therapeutically effective amount of 5-fluorouracil or docetaxel.
Additional combination treatment therapy is also contemplated by the present
disclosure. For example, in some embodiments, the methods of treating cancer
in a
subject, comprising administering to the subject a pharmaceutically effective
amount
of a compound of the present disclosure, the methods may further comprise one
or
more treatments selected from the group consisting of administering a
pharmaceutically effective amount of a second drug, radiotherapy,
immunotherapy,
gene therapy, and surgery. In some embodiments, the methods may further
comprise
(1) contacting a tumor cell with the compound prior to contacting the tumor
cell with
the second drug, (2) contacting a tumor cell with the second drug prior to
contacting
the tumor cell with the compound, or (3) contacting a tumor cell with the
compound
and the second drug at the same time. The second drug may, in certain
embodiments,
be an antibiotic, anti-inflammatory, anti-neoplastic, anti-proliferative, anti-
viral,
immunomodulatory, or immunosuppressive. In other embodiments, the second drug
may be an alkylating agent, androgen receptor modulator, cytoskeletal
disruptor,
.. estrogen receptor modulator, histone-deacetylase inhibitor, HMG-CoA
reductase
inhibitor, prenyl-protein trans ferase inhibitor, retinoid receptor modulator,
topoisomerase inhibitor, or tyrosine kinase inhibitor. In certain embodiments,
the
second drug is 5-azacitidine, 5-fluorouracil, 9-cis-retinoic acid, actinomycin
D,
alitretinoin, all-trans-retinoic acid, annamycin, axitinib, belinostat,
bevacizumab,
bexarotene, bosutinib, busulfan, capecitabine, carboplatin, carmustine, CD437,
cediranib, cetuximab, chlorambucil, cisplatin, cyclophosphamide, cytarabine,
dacarbazine, dasatinib, daunorub ic
in, dec itabine, docetaxel, do lastatin-10,
doxifluridine, doxorubicin, doxorubicin, epirubicin, erlotinib, etoposide,
gefitinib,
gemcitabine, gemtuzumab ozogamicin, hexamethylmelamine, idarubicin,
ifosfamide,
8
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
imatinib, irinotecan, isotretinoin, ixabepilone, lapatinib, LBH589, lomustine,
mechlorethamine, melphalan, mercaptopurine, methotrexate, mitomycin,
mitoxantrone, MS-275, neratinib, nilotinib, nitrosourea, oxaliplatin,
paclitaxel,
plicamycin, procarbazine, semaxanib, semustine, sodium butyrate, sodium
phenylacetate, streptozotocin, suberoylanilide hydroxamic acid, sunitinib,
tamoxifen,
teniposide, thiopeta, tioguanine, topotecan, TRAIL, trastuzumab, tretinoin,
trichostatin A, valproic acid, valrubicin, vandetanib, vinblas tine,
vincristine,
vindesine, or vinorelbine.
Methods of treating or preventing a disease with an inflammatory component
in a subject, comprising administering to the subject a pharmaceutically
effective
amount of a compound of the present disclosure are also contemplated. In some
embodiments, the disease may be, for example, lupus or rheumatoid arthritis.
In other
embodiments, the disease may be an inflammatory bowel disease, such as Crohn's
disease or ulcerative colitis. In other embodiments, the disease with an
inflammatory
component may be a cardiovascular disease. In other embodiments, the disease
with
an inflammatory component may be diabetes, such as type 1 or type 2 diabetes.
In
other embodiments, RTA 408, its polymorphs, and pharmaceutical compositions
may
also be used to treat complications associated with diabetes. Such
complications are
well-known to a person of skill in the art and include but are not limited to,
for
example, obesity, hypertension, atherosclerosis, coronary heart disease,
stroke,
peripheral vascular disease, hypertension, nephropathy, neuropathy,
myonecrosis,
retinopathy and metabolic syndrome (syndrome X). In other embodiments, the
disease with an inflammatory component may be a skin disease, such as
psoriasis,
acne, or atopic dermatitis. Administration of RTA 408, its polymorphs, and
pharmaceutical compositions in treatment methods of such skin diseases may be
but
are not limited to, for example, topical or oral.
In other embodiments, the disease with an inflammatory component may be
metabolic syndrome (syndrome X). A patient having this syndrome is
characterized
as having three or more symptoms selected from the following group of five
symptoms: (1) abdominal obesity; (2) hypertriglyceridemia; (3) low high-
density
lipoprotein cholesterol (HDL); (4) high blood pressure; and (5) elevated
fasting
glucose, which may be in the range characteristic of Type 2 diabetes if the
patient is
also diabetic. Each of these symptoms is defined in the Third Report of the
National
Cholesterol Education Program Expert Panel on Detection, Evaluation and
Treatment
9
WO 2013/163344
PCT/US2013/038064
of High Blood Cholesterol in Adults (Adult Treatment Panel III, or ATP III),
National
Institutes of Health, 2001, NIH Publication No. 01-3670,
Patients with metabolic syndrome, whether or not they have or develop
overt diabetes mellitus, have an increased risk of developing the
macrovascular and
microvascular complications that are listed above that occur with type 2
diabetes,
such as atherosclerosis and coronary heart disease.
Another general method of the present disclosure entails a method of treating
or preventing a cardiovascular disease in a subject, comprising administering
to the
subject a pharmaceutically effective amount of a compound of the present
disclosure.
In some embodiments, the cardiovascular disease may be but not limited to, for
example, atherosclerosis, cardiomyopathy, congenital heart disease, congestive
heart
failure, myocarditis, rheumatic heart disease, valve disease, coronary artery
disease,
endocarditis, or myocardial infarction. Combination therapy is also
contemplated for
methods of treating or preventing a cardiovascular disease in a subject. For
example,
such methods may further comprise administering a pharmaceutically effective
amount of one or more cardiovascular drugs. The cardiovascular drug may be but
not
limited to, for example, a cholesterol lowering drug, an anti-hyperlipidemic,
a calcium
channel blocker, an anti-hypertensive, or an HMG-CoA reductase inhibitor. In
some
embodiments, non-limiting examples of cardiovascular drugs include amlodipine,
aspirin, ezetimibe, felodipine, lacidipine, lercanidipine, nicardipine,
nifedipine,
nimodipine, nisoldipine or nitrendipine. In other embodiments, other non-
limiting
examples of cardiovascular drugs include atenolol, bucindolol, carvedilol,
clonidine,
doxazosin, indoramin, labetalol, methyldopa, metoprolol, nadolol, oxprenolol,
phenoxybenzamine, phentolamine, pindolol, prazosin, propranolol, terazosin,
timolol
or tolazoline. In other embodiments, the cardiovascular drug may be, for
example, a
statin, such as atorvastatin, cerivastatin, fluvastatin, lovastatin,
mevastatin,
pitavastatin, pravastatin, rosuvastatin or simvastatin.
Methods of treating or preventing a neurodegenerative disease in a subject,
comprising administering to the subject a pharmaceutically effective amount of
a
compound of the present disclosure are also contemplated. In some embodiments,
the
neurodegenerative disease maybe selected, for example, from the group
consisting of
Parkinson's disease, Alzheimer's disease, multiple sclerosis (MS),
Huntington's
disease and amyotrophic lateral sclerosis. In
particular embodiments, the
neurodegenerative disease is Alzheimer's disease. In particular embodiments,
the
CA 2869783 2020-04-08
neurodegenerative disease is MS, such as primary progressive, relapsing-
remitting
secondary progressive or progressive relapsing MS. In some embodiments, the
subject may be, for example, a primate. In some embodiments, the subject may
be a
human.
In particular embodiments of methods of treating or preventing a
neurodegenerative disease in a subject, comprising administering to the
subject a
pharmaceutically effective amount of a compound of the present disclosure, the
treatment suppresses the demyelination of neurons in the subject's brain or
spinal
cord. In certain embodiments, the treatment suppresses inflammatory
demyelination.
In certain embodiments, the treatment suppresses the transection of neuron
axons in
the subject's brain or spinal cord. In certain embodiments, the treatment
suppresses
the transection of neurites in the subject's brain or spinal cord. In
certain
embodiments, the treatment suppresses neuronal apoptosis in the subject's
brain or
spinal cord. In certain embodiments, the treatment stimulates the
remyelination of
neuron axons in the subject's brain or spinal cord. In certain embodiments,
the
treatment restores lost function after an MS attack. In certain embodiments,
the
treatment prevents a new MS attack. In certain embodiments, the treatment
prevents
a disability resulting from an MS attack.
One general aspect of the present disclosure contemplates a method of treating
or preventing a disorder characterized by overexpression of iNOS genes in a
subject,
comprising administering to the subject a pharmaceutically effective amount of
RTA
408, polymorphic forms, or a pharmaceutical composition of the present
disclosure.
Another general aspect of the present disclosure contemplates a method of
inhibiting IFN-y-induced nitric oxide production in cells of a subject,
comprising
administering to said subject a pharmaceutically effective amount of RTA 408,
polymorphic forms, or a pharmaceutical composition of the present disclosure.
Yet another general method of the present disclosure contemplates a method
of treating or preventing a disorder characterized by overexpression of COX-2
genes
in a subject, comprising administering to the subject a pharmaceutically
effective
amount of RTA 408, polymorphic forms, or a pharmaceutical composition of the
present disclosure.
Methods of treating renal/kidney disease (RKD) in a subject, comprising
administering to the subject a pharmaceutically effective amount of a compound
of
the present disclosure are also contemplated. See U.S. Patent 8,129,429,
11
CA 2869783 2019-09-05
The RKD may result from, for example, a toxic
insult. The toxic insult may result from but not limited to, for example, an
imaging
agent or a drug. The drug may be a chemotherapeutic, for example. The RKD may
result from ischemia/reperfusion injury, in certain embodiments. In certain
embodiments, the RKD results from diabetes or hypertension. In some
embodiments,
the RKD may result from an autoimmune disease. The RKD may be further defined
as chronic RKD or acute RKD.
In certain methods of treating renal/kidney disease (RKD) in a subject,
comprising administering to the subject a pharmaceutically effective amount of
a
compound of the present disclosure, the subject has undergone or is undergoing
dialysis. In certain embodiments, the subject has undergone or is a candidate
to
undergo kidney transplant. The subject may be a primate. The primate may be a
human. The subject in this or any other method may be, for example, a cow,
horse,
dog, cat, pig, mouse, rat or guinea pig.
Also contemplated by the present disclosure is a method for improving
glomerular filtration rate or creatinine clearance in a subject, comprising
administering to the subject a pharmaceutically effective amount of RTA 408,
polymorphic forms, or a pharmaceutical composition of the present disclosure.
In some embodiments, the pharmaceutical composition is administered in a
single dose per day. In other embodiments, the pharmaceutical composition is
administered in more than one dose per day. In some embodiments, the
pharmaceutical composition is administered in a pharmaceutically effective
amount.
In some embodiments, the dose is from about 1 mg/kg to about 2000 mg/kg.
In other embodiments, the dose is from about 3 mg/kg to about 100 mg/kg. In
other
embodiments, the dose is about 3, 10, 30, or 100 mg/kg.
In other embodiments, the pharmaceutical composition is administered
topically. In some embodiments, the topical administration is administered to
the
skin. In other embodiments, the topical administration is administered to the
eye.
In other embodiments, the pharmaceutical composition is administered orally.
In other embodiments, the pharmaceutical composition is administered
intraocularly.
Other objects, features and advantages of the present disclosure will become
apparent from the following detailed description. It should be understood,
however,
that the detailed description and the specific examples, while indicating
specific
embodiments of the invention, are given by way of illustration only, since
various
12
CA 2869783 2019-09-05
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
changes and modifications within the spirit and scope of the invention will
become
apparent to those skilled in the art from this detailed description. Note that
simply
because a particular compound is ascribed to one particular generic formula
does not
mean that it cannot also belong to another generic formula.
13
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
BRIEF DESCRIPTION OF THE DRAWINGS
The following drawings form part of the present specification and are included
to further demonstrate certain aspects of the present disclosure. One or more
German
words can be found in the drawings including "Masseanderung" and "temperatur",
which mean "change in mass" and "temperature", respectively. The invention may
be
better understood by reference to one of these drawings in combination with
the
detailed description of specific embodiments presented herein.
FIG. 1 ¨ Effect of RTA 408 on IFN7-induced nitric oxide production and cell
viability in RAW264.7 cells.
FIGS. 2a & b ¨ Effect of RTA 408 on antioxidant response element (ARE)
activation: (a) NQ01-ARE luciferase activity; (b) GSTA2-ARE luciferase
activity.
FIGS. 3a-f ¨ Relative Nrf2 GST ARE fold increase after cellular treatment
with (a) RTA 402; (b) 63415 (RTA 408); (c) 63170; (d) 63171; (e) 63179; and
(f)
63189. The graphs also show the viability of the cells as assayed using the
WST1 cell
proliferation reagent and measuring the absorbance after 1 hour. All drugs
were
administered in DMSO and cells were grown at 10,000 cells/well in 384-well
plates in
DMEM low glucose supplemented with 10% FBS, 1% Penicillin Streptomycin, and
0.8 mg/mL Geneticin.
FIGS. 4a¨d ¨ Effect of RTA 408 on Nrf2 target gene expression in HFLI
lung fibroblasts. (a) NQ01; (b) HMOX1; (c) GCLM; (d) TXNRD1.
FIGS. 5a¨d ¨ Effect of RTA 408 on Nrf2 target gene expression in BEAS-2B
bronchial epithelial cells. (a) NQ01; (b) HMOX1; (c) GCLM; (d) TXNRD1.
FIGS. 6a & b ¨ Effect of RTA 408 on Nrf2 target protein levels. (a) SH-
SY5Y cells; (b) BV2 cells.
FIG. 7 ¨ Effect of RTA 408 on NQ01 enzymatic activity in RAW264.7 cells.
FIG. 8 ¨ Effect of RTA 408 on total glutathione levels in the AML-12
hepatocyte cell line.
FIG. 9 ¨ Effect of RTA 408 on WST-1 absorbance as a marker of NADPH.
14
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
FIGS. 10a¨d ¨ Effect of RTA 408 on expression of genes involved in
NADPH synthesis. (a) H6PD; (b) PGD; (c) TKT; (d) ME1.
FIGS. ha & b ¨ (a) Effect of RTA 408 on TNF-a-induced activation of an
NF-KB luciferase reporter in the mouse NIH3T3 cell line with WST1 viability
and
WST1/2 viability overlaid. (b) TNF-a-induced activation of an NF-KB luciferase
reporter in the mouse NIH3T3 cell line. The graph shows relative fold change
as a
function of log change in RTA 408 concentration.
FIG. 12 ¨ Effect of RTA 408 on TNF-a-induced activation of a NF-KB
luciferase reporter construct.
FIGS. 13a & b ¨ (a) Effect of RTA 408 on TNF-a-induced activation of an
NF-KB luciferase reporter in the human A549 cell line with WST1 viability and
WST1/2 viability overlaid. (b) TNF-a-induced activation of an NF-KB luciferase
reporter in the human A549 cell line. The graph shows relative fold change as
a
function of log change in RTA 408 concentration.
FIG. 14 ¨ Effect of RTA 408 on TNF-a-induced phosphorylation of IxBa.
FIGS. 15a-d ¨ Effect of RTA 408 on transaminase gene expression: (a) ALT1
(GPT1); (b) ALT2 (GPT2); (c) ASTI (GOT1); (d) ASTI (GOT2). Asterisks indicate
a statistically-significant difference from the control group (*P < 0.05; **P
< 0.01).
FIG. 16 ¨ Effect of RTA 408 on pyruvate levels in cultured muscle cells (*P <
0.05).
FIG. 17 ¨ Effect of 63415 in a model of pulmonary LPS-mediated
inflammation (% change in pro-inflammatory cytokines relative to LPS
treatment).
Compound 63415 was administered QDx3 at Time 0, 24, and 48 h followed by LPS
one h after the last dose of 63415 in female BALB/c mice. Animals were
sacrificed
20 h after LPS administration. BALF was examined for pro-inflammatory cytokine
expression. Compoound 63415 reduced pro-inflammatory cytokines in a dose-
dependent manner, with peak reductions ranging from 50%-80% in TNF¨a, IL-6,
and
IL-12.
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
FIGS. 18a & b ¨ Effect of RTA 408 on LPS-induced pulmonary
inflammation in mice. (a) inflammatory cytokines; (b) Nrf2 targets. RTA 408
was
administered to female BALB/c mice (n = 10) QDx6 at Time 0, 24, 48, 72, 96,
and
120 h followed by LPS at 121 h with animals sacrificed at 141 h. Pro-
inflammatory
cytokine protein expression assayed in BALF. Nrf2 biomarkers assayed in lung.
Asterisks indicate a statistically significant difference from the saline
control group
(*P < 0.05; **P < 0.01; ***P <0.001).
FIGS. 19a & b ¨ Effect of 63415 on BALF infiltrates in bleomycin-induced
pulmonary inflammation: (a) BAL fluid cell count; (b) body weight. Compound
63415 was administered QDx39 on Days -10 to 28 in C57BL/6 mice. Bleomycin was
given on Day 0. Daily weights were measured. BALF cell counts were obtained at
sacrifice. A notable reduction in inflammatory infiltrate was observed. No
significant
improvements in chronic inflammation score, interstitial fibrosis, or number
of
fibrotic foci were observed.
FIGS. 20a & b ¨ Effect of RTA 408 on bleomycin-induced pulmonary
fibrosis in rats: (a) PMN; (b) Hydroxyproline. Asterisks indicate a
statistically
significant difference from the bleomycin control group (*P < 0.05).
FIG. 21 ¨ Effect of RTA 408 on Nrf2 target enzymes in lungs from rats with
bleomycin-induced pulmonary fibrosis. Asterisks indicate a statistically
significant
difference from the saline control group (*P <0.05; **P < 0.01; ***P < 0.001).
FIGS. 22a¨e ¨ Effect of RTA 408 on cigarette smoke-induced COPD in mice.
(a) KC; (b) IL-6; (c) TNF-a; (d) TN-7; (e) RANTES. RTA 408 (63415) was tested
at
dose levels of 3 mg/kg (low), 10 mg/kg (mid), and 30 mg/kg (high). An AIM
analog
(63355) was tested in the same study for comparison. Asterisks indicate a
statistically
significant difference form the CS control group.
FIG. 23 ¨ Effect of RTA 408 on Nrf2 target enzymes in lungs from mice with
cigarette smoke-induced COPD. Asterisks
indicate a statistically significant
difference from the saline control group (*P < 0.05; **P < 0.01; ***P <
0.001).
Daggers represent a statistically significant difference from mice expose to
cigarette
smoke and administered vehicle (-l-P < 0.05).
16
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
FIGS. 24a¨d ¨ Effects of 63415 (RTA 408) on body weight in a BALB/c
mouse model of sepsis. LPS was administered to all animals on Day 0. (a) Body
Weight: 63415 (RTA 408); (b) Body Weight: RTA 405; (c) Systemic LPS: %
Survival: 63415 (RTA 408); (d) Systemic LPS: % Survival: RTA 405. Both RTA
405 and 63415 (RTA 408) were administered QDx5 on Days -2 to 2. Compound
63415 (RTA 408) improved survival. Body weight as a function of time in 63415-
treated BALB/c mice serves as a model for sepsis.
FIG. 25 ¨ Effect of 63415 in a model of radiation-induced oral mucositis.
RTA 405 or 63415 (RTA 408) was administered BIDx20 on Days -5 to -1 and Days 1
to 15 to male Syrian Golden Hamsters. Radiation occurred on Day 0. Mucositis
scores range from 0 to 5 based on clinical manifestations (0: completely
healthy; 1-2:
light to severe erythema; 3-5: varying degrees of ulceration). 63415 improved
mucositis at 30 mg/kg and 100 mg/kg with up to a 36% reduction in ulceration.
FIG. 26 ¨ Effect of 63415 on Nrf2 target gene induction in a 14-day mouse
toxicity study in C57BL/6 mice. mRNAs of Nrf2 target genes were assessed in
livers
of mice treated PO QDx14. Substantial increases in mRNA expression for
multiple
Nrf2 target genes were observed and were consistent with tissue exposure.
FIGS. 27a & b ¨ Effect of 63415 on Nrf2 target gene induction in rat livers:
(a) Target genes; (b) Negative regulators. mRNAs of Nrf2 target genes were
assessed
in livers of rats treated PO QDx14.
FIGS. 28a & b ¨ Effect of 63415 on Nrf2 target genes in monkey tissues: (a)
Liver; (b) Lung. mRNAs of Nrf2 target genes were assessed in monkeys treated
PO
QDx14 using Panomics QuantiGenet 2.0 Plex technology.
FIGS. 29a & b ¨ Effect of 63415 on Nrf2 target enzyme activity in mouse
liver: (a) NQ01 activity; (b) GST activity. Nr12 target enzyme activity was
assessed
in livers of mice treated PO QDx14. NQ01 and GST enzyme activities were
induced
in a dose-dependent manner.
FIGS. 30a & b ¨ Effect of 63415 on Nrf2 target enzyme activity in rat liver:
(a) NQ01 activity; (b) GST activity. Nrf2 target enzyme activity was assessed
in
17
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
livers of rats treated PO QDx14. NQ01 and GST enzyme activities were induced
in a
dose dependent manner.
FIGS. Ma & b ¨ Effects of 63415 on Nrf2 target enzyme activity induction in
various tissues of cynomolgus monkeys: (a) NQ01 activity; (b) GSR activity.
FIGS. 32a & b ¨ RTA 408 concentration in mouse liver, lung, and brain, and
NQ01 activity in mouse liver after 14 days of daily oral administration. (a)
Tissue
distribution of RTA 408 in mice after 14 days of daily oral administration.
Data
represent the mean SD of RTA 408 concentrations in tissue collected 4 h
after the
final dose of the study. Numbers above the error bars are representative of
the mean.
(b) Correlation of mouse liver RTA 408 content with NQ01 enzyme activity.
Individual mouse liver RTA 408 liver content was plotted against individual
enzyme
activity from this report.
FIGS. 33a & b ¨ RTA 408 concentration in rat plasma, liver, lung, and brain,
and NQ01 activity in rat liver after 14 days of daily oral administration. (a)
Tissue
distribution of RTA 408 in rats after 14 days of daily oral administration.
Data
represent the mean SD of RTA 408 concentrations in tissue collected 4 h
after the
final dose of the study. Numbers above the error bars are representative of
the mean.
*Two values were excluded from the mean calculation due to being outliers,
defined
as values causing the set of data to fail the Shapiro-Wilk normality test. (b)
Correlation of rat liver RTA 408 content with NQ01 enzyme activity. Individual
rat
liver RTA 408 content was plotted against individual enzyme activity from this
report. The tissues from the 100 mg/kg RTA 408 dose group were collected on
Day 6, and the observed toxicities in this group precluded liver NQ01 enzyme
activity evaluations.
FIGS. 34a & b ¨ Effect of 63415 treatment on Nrf2 activation in monkey
PBMC: (a) PBMC NQ01 vs. Plasma Concentration; (b) Lung NQ01 vs. PBMC
NQ01.
FIG. 35 ¨ Summary of 63415 14-day monkey toxicity study. All doses were
well-tolerated without adverse clinical signs. Clinical chemistry data
suggested no
obvious toxicity.
18
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
FIG. 36 ¨ Effect of dosing time on plasma concentration of RTA 408 after
topical ocular and oral administration. The plasma concentration of RTA 408
was
also measured after 5 days of daily topical ocular administration of RTA 408
and
determined to remain relatively consistent from the measurements taken after
the first
day.
FIGS. 37a & b ¨ Con-elation of exposure to RTA 408 in monkey plasma with
NQ01 and SRXN1 mRNA expression in PBMCs: (a) NQ01; (b) SRXN1.
FIG. 38 ¨ Concentration of RTA 408 in various tissues or fluids within the
eye as a function of time after 5 days of topical ocular dosing. RTA 408
concentration in plasma was also measured after topical ocular administration.
FIG. 39 ¨ Effect of RTA 408 on the incidence of grade 3 dermatitis caused by
acute radiation exposure for different concentrations of topically
administered
RTA 408.
FIG. 40 ¨ Effect of RTA 408 on the incidence of grade 2 dermatitis caused by
acute radiation exposure over a 30 day treatment course for different
concentrations of
topically administered RTA 408.
FIG. 41 ¨ Effect of RTA 408 on the incidence of grade 2 dermatitis caused by
acute radiation exposure over a 28 day treatment course for different
concentrations of
orally administered RTA 408.
FIGS. 42a & b ¨ (a) Area under the curve analysis of clinical score of the
dermatitis as a function of time for each of the different control groups
including all
of the animals used in the test. (b) Area under the curve analysis of the
clinical score
of the dermatitis as a function of the duration of that score for each of the
different
control groups including only animals that completed the entire 30 days in the
trial.
FIG. 43 ¨ Average l st blind score of acute radiation dermatitis as a function
of
time for untreated, untreated with no radiation exposure, vehicle only and
three oral
amounts of RTA 408 at 3, 10, and 30 mg/kg. The dermatitis score was based upon
a
scale that 0 was completely healthy, 1-2 exhibited mild to moderate erythema
with
19
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
minimal to slight desquamation, 3-4 exhibited moderate to severe erythema and
desquamation, and 5 exhibited a frank ulcer.
FIG. 44 ¨ Mean score of the acute radiation dermatitis as a function of time
for untreated, untreated with no radiation exposure, vehicle only, and three
oral
amounts of RTA 408 at 3, 10, and 30 mg/kg measured every other day from day 4
to
day 30. The dermatitis score was based upon a scale that 0 was completely
healthy,
1-2 exhibited mild to moderate erythema with minimal to slight desquamation, 3-
4
exhibited moderate to severe erythema and desquamation, and 5 exhibited a
frank
ulcer.
FIG. 45 ¨ Mean score of the acute radiation dermatitis as a function of time
for untreated, untreated with no radiation exposure, vehicle only, and three
topical
amounts of RTA 408 at 0.01%, 0.1%, and 1% measured every other day from day 4
to
day 30. The dermatitis score was based upon a scale that 0 was completely
healthy,
1-2 exhibited mild to moderate erythema with minimal to slight desquamation, 3-
4
exhibited moderate to severe erythema and desquamation, and 5 exhibited a
frank
ulcer.
FIG. 46 ¨ Clinical scores of fractional radiation dermatitis plotted versus
time
and changes in dermatitis score for each testing group. The dermatitis score
was
based upon a scale that 0 was completely healthy, 1-2 exhibited mild to
moderate
erythema with minimal to slight desquamation, 3-4 exhibited moderate to severe
erythema and desquamation, and 5 exhibited a frank ulcer.
FIG. 47 ¨ Graph of the AUC analysis showing the dermatitis score (severity x
days) for each of the testing groups over the entire observation period. The
dermatitis
scores were assessed every two days from day 4 to day 30 of the study.
FIGS. 48a & b ¨ (a) Graph of the absorbance at 595 nm for treated prostate
cancer cell line LNCaP showing relative cytotoxic effect on cells treated with
a
chemotherapeutic agent and RTA 408 versus RTA 408 alone. (b) Graph of the
absorbance at 595 nm for treated prostate cancer cell line DU-145 showing
relative
cytotoxic effect on cells treated with a chemotherapeutic agent and RTA 408
versus
RTA 408 alone.
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
FIG. 49 ¨ Black and white versions of color photographs of imaged mice
showing the luciferase activity of tumors for three mice: a control animal
with no
treatment, an animal subjected only to ionizing radiation (single dose, 18
Gy), and an
animal given both ionizing radiation (single dose, 18 Gy, day 0) and RTA 408
(17.5
mg/kg i.p., once daily on days -3 to -1, then single doses on days 1, 3, and
5). The
colors indicated by the arrows are indicative of intensity with the
intensities being
represented by red, yellow, green, and blue in order from highest to lowest
intensity.
FIG. 50 ¨ Reduction of aqueous humor protein concentrations for different
formulations of RTA 408 (dark bars) compared to literature values for MaxiDex0
(0.1% dexamethasone) and mapracorat (light bars) after induction of
paracentesis.
FIG. 51 ¨ Dose-dependent suppression of NO in vivo by 63415. CD-1 mice
(n = 6) were dosed with DMSO or AIM by oral gavage. LPS (5 mg/kg) was
administered 24 h later. Twenty-four hours after LPS administration, whole
blood
was collected for NO assay. NO inhibition was determined by Griess Reaction
from
reduced, de-proteinated plasma.
FIG. 52 ¨ Extensive distribution of 63415 (RTA 408) into mouse tissues.
Mice were dosed PO QDx3 with either 25 mg/kg 63415 (RTA 408) or 25 mg/kg
RTA 405. Blood (plasma and whole blood) and tissues (brain, liver, lung, and
kidney) were collected 6 h after the last dose. Semi-quantitative analysis of
drug
content was performed. Notable levels were observed in the CNS.
FIG. 53 ¨ NQ01 activity induction in mouse liver, lung, and kidney by
63415. Mice were dosed PO QDx3 with 25 mg/kg. Tissues were collected 6 11
after
the last dose, and analysis of NQ01 activity was performed. Meaningful
activation of
NQ01 was observed in multiple tissues.
FIG. 54 ¨ Summary of 63415 14-day mouse toxicity study. C57BL/6 mice
were dosed PO QDx14. Endpoints
included survival, weight, and clinical
chemistries. All animals survived to day 14. No significant weight changes
occurred
compared to the vehicle group, and there was no evidence of toxicity at any
dose
based on clinical chemistries.
21
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
FIG. 55 ¨ Tissue distribution of 63415 from 14-day mouse toxicity study in
C57BL/6 mice. Brain, lung, and liver samples were collected 4 h after final
dose and
quantified for 63415 content using sensitive LC/MS/MS method. Exposures at 10
and 100 mg/kg in lung exceeded the in vitro IC50 for NO induction by 55- and
1138-
fold, respectively. Exposure at 10 and 100 mg/kg in brain exceeded the in
vitro IC50
for NO induction by 29- and 541-fold, respectively.
FIG. 56 ¨ Tissue distribution of 63415 in Sprague Dawley rats. Tissues were
collected four hours after final dosing on Day 14 or Day 6 (100 mg/kg),
extracted, and
quantified for 63415 content using a sensitive LC/MS/MS method. Compound 63415
distributes well into target tissues. Exposures at 10 mg/kg in lung and brain
exceed
the in vitro IC50 for NO inhibition by 294- and 240-fold, respectively.
FIG. 57 ¨ Target tissue distribution of compound 63415 in cynomolgus
monkeys. Tissues were collected four hours after final dosing on Day 14.
Compound
63415 content was extracted and quantified using a sensitive LC/MS/MS method.
FIG. 58 ¨ FT-Raman spectrum (3400-50 cm-1) of the sample PP415-P1,
which corresponds to the amorphous form (Class 1).
FIG. 59 ¨ PXRD (1.5-55.5 020) pattern of the sample PP415-P1, which
corresponds to the amorphous form (Class 1).
FIG. 60 ¨ TG-FTIR thermogram (25-350 C) of the sample PP415-P1, which
corresponds to the amorphous form (Class 1).
FIG. 61 ¨ 11-I-NMR spectrum in DMSO-d6 of the sample PP415-P1, which
corresponds to the amorphous form (Class 1).
FIG. 62 ¨ DSC thermogram of the sample PP415-P1, which corresponds to
the amorphous form (Class 1).
FIG. 63 ¨ DVS isotherm of the sample PP415-P1, which corresponds to the
amorphous form (Class 1).
FIG. 64 ¨ FT-Raman spectrum of the sample PP415-P1, which corresponds to
the amorphous form (Class 1), after DVS measurement (top) is unchanged
compared
22
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
to the material before the DVS measurement (bottom). The spectra have been
scaled
and offset in the y-direction for the purpose of comparison.
FIG. 65 ¨ PXRD pattern of the sample PP415-P I, which corresponds to the
amorphous form (Class 1), after DVS measurement (top) is unchanged compared to
the material before the DVS measurement (bottom). The patterns have not been
scaled but are offset in the y-direction for the purpose of comparison.
FIG. 66 ¨ PXRD pattern of the sample PP415-P40 (top) corresponds to the
pattern of the solvate form (Class 2) (bottom, sample PP415-P19). The patterns
have
been scaled and offset in the y-direction for the purpose of comparison.
FIG. 67 ¨ PXRD patterns of the stability samples PP415-P2a (top),
PP415-P3a (21d from top), PP415-P4a (middle), and PP415-P5a (2nd from bottom),
which corresponds to the amorphous form (Class 1), after one week show no
differences compared to the starting material at time point to (bottom, sample
PP415-P1). The patterns are not scaled but are offset in the y-direction for
the
purpose of comparison.
FIG. 68 ¨ PXRD patterns of the stability samples PP415-P2b (top),
PP415-P3b (2nd from stop), PP415-P4b (middle), and PP415-P5b (211d from
bottom),
which corresponds to the amorphous form (Class 1), after two weeks show no
differences compared to the starting material at time point to (bottom, sample
PP415-P1). The patterns are not scaled but are offset in the y-direction for
the
purpose of comparison.
FIG. 69 ¨ PXRD patterns of the stability samples PP415-P2c (top),
PP415-P3c (2nd from top), PP415-P4c (middle), and PP415-P5c (211d from
bottom),
which corresponds to the amorphous form (Class 1), after four weeks show no
differences compared to the starting material at time point to (bottom, sample
PP415-P1). The patterns are not scaled but are offset in the y-direction for
the
purpose of comparison.
FIG. 70 ¨ FT-Raman spectra (2400-50 cm-1) of samples of the solvate form
(Class 2) (PP415-P7: top; PP415-P8: 2'd from top; PP415-P9: 3rd from top;
PP415-P10: 4th from top; PP415-P11: middle; PP415-P15: 4th from bottom; PP415-
23
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
P17: 3'd from bottom; PP415-P21: 2nd from bottom; PP415-P24: bottom). The
spectra
have been scaled and offset in the y-direction for the purpose of comparison.
FIG. 71 ¨ FT-Raman spectrum (1750-1000 cm4) of the solvate form (Class 2)
(PP415-P7: top) clearly differs from the spectrum of the amorphous form (Class
1)
(PP415-P1: bottom). The spectra have been scaled and offset in the y-direction
for
the purpose of comparison.
FIG. 72 ¨FT-Raman spectra (1750-1000 cm-1) of class 2 (sample PP415-P19:
top), class 3 (sample PP415-P6: 2nd from top), class 4 (sample PP415-P13: 2nd
from
bottom), and class 5 (sample PP415-P14: bottom) differ significantly from each
other.
The spectra have been scaled and offset in the y-direction for the purpose of
comparison.
FIG. 73 ¨ PXRD patterns (2-32 028) of samples of the solvate form (Class 2)
(PP415-P7: top; PP415-P8: 2nd from top; PP415-P10: 3rd from top; PP415-P15:
4th
from top; PP415-P17: middle; PP415-P18: 4th from bottom; PP415-P19: 31d from
bottom; PP415-P21: 2nd from bottom; PP415-P24: bottom). The patterns have been
scaled and offset in the y-direction for the purpose of comparison.
FIG. 74 ¨ PXRD patterns (11-21 020) of some samples of the solvate form
(Class 2) (PP415-P7: top; PP415-P8: 2nd from top; PP415-P10: middle; PP415-
P21:
2nd from bottom; PP415-P24: bottom). The patterns have been scaled and offset
in
the y-direction for the purpose of comparison.
FIG. 75 ¨ PXRD patterns (2-32 020) of class 2 (sample PP415-P19: top),
class 3 (sample PP415-P6: 2nd from top), class 4 (sample PP415-P13: 2nd from
bottom), and class 5 (sample PP415-P14: bottom) are distinctly different. The
patterns have been scaled and offset in the y-direction for the purpose of
comparison.
FIG. 76 ¨ TG-FTIR thermogram of the sample PP415-P7, which corresponds
to a solvate form (Class 2).
FIG. 77 ¨ TG-FTIR thermogram of the sample PP415-P21, which
corresponds to a solvate form (Class 2).
24
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
FIG. 78 ¨ TG-FTIR thermogram of the sample PP415-P24, which
corresponds to a solvate form (Class 2).
FIG. 79 - TG-FTIR thermogram of the sample PP415-P29, which corresponds
to a solvate form (Class 2).
FIG. 80 ¨ TG-FTIR thermogram of the sample PP415-P47, which
corresponds to a solvate form (Class 2).
FIG. 81 ¨ TG-FTIR thermogram of the sample PP415-P48, which
corresponds to a solvate form (Class 2).
FIG. 82 ¨ FT-Raman spectra (1800-700 cm-1) of the solvate form (Class 2)
(bottom, sample PP415-P7) and of the dried solvate form (Class 2) (top, sample
PP415-P30) are similar and show only small differences which can hardly be
distinguished within the graph. The spectra are scaled for the purpose of
comparison.
FIG. 83 - PXRD pattern of the dried solvate form (Class 2), sample
PP415-P30 (top) in comparison to the pattern of the solvate form (Class 2),
sample
PP415-P7 (bottom). The patterns are not scaled but are offset in the y-
direction for
the purpose of comparison.
FIG. 84 ¨ TG-FTIR thermogram of the dried sample PP415-P30, which
corresponds to a solvate form (Class 2).
FIG. 85 ¨ FT-Raman spectrum of the dried sample PP415-P18 (light grey) is
identical to the spectrum of the original sample PP415-P15 (dark grey) are
similar and
show only small differences which can hardly be distinguished within the
graph. The
spectra have been scaled for the purpose of comparison.
FIG. 86 ¨ PXRD pattern of the dried sample PP415-P18 (top) shows small
differences from the pattern of the original sample PP415-P15 (bottom),
although
both solvate forms (Class 2). The patterns have been scaled and offset in the
y-
direction for the purpose of comparison.
FIG. 87 ¨ TG-FTIR thermogram of the sample PP415-P18, which
corresponds to a solvate form (Class 2).
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
FIG. 88 ¨ FT-Raman spectrum of the sample PP415-P17 (top) is nearly
identical to the spectra of the dried samples PP415-P19 (middle) and PP415-P32
(bottom) and show only small differences which can hardly be distinguished
within
the graph. The spectra have been scaled and offset in the y-direction for the
purpose
of comparison.
FIG. 89 ¨ PXRD pattern of the dried sample PP415-P19 (middle) is different
from the pattern of the original sample PP415-P17 (top) but still corresponds
to
class 2 form. The pattern of the further dried sample PP415-P32 (bottom) shows
broader peaks with a lower SN ratio. The material is less crystalline, but
still
corresponds to class 2 form. The patterns have been scaled and offset in the y-
direction for the purpose of comparison.
FIG. 90 ¨ TG-FTIR thermogram of the sample PP415-P19, which
corresponds to a solvate form (Class 2).
FIG. 91 ¨ TG-FTIR thermogram of the sample PP415-P32A, which
corresponds to a solvate form (Class 2).
FIG. 92 ¨ FT-Raman spectrum of the sample PP415-P21 (top) is identical to
the spectra of the dried samples PP415-P28 (middle) and PP415-P34 (bottom).
The
spectra have been scaled and offset in the y-direction for the purpose of
comparison.
FIG. 93 ¨ PXRD patterns of the dried samples PP415-P28 (middle) and
PP415-P34 (bottom) show broader peaks with a lower S/N ratio, indicating a
lower
crystallinity of the samples compared to the pattern of the original sample
PP415-P21
(top). The patterns are somewhat different but still correspond to class 2
form. They
have been scaled and offset in the y-direction for the purpose of comparison.
FIG. 94 ¨ TG-FTIR thermogram of the dried sample PP415-P28, which
corresponds to a solvate form (Class 2).
FIG. 95 ¨ TG-FTIR thermogram of the dried sample PP415-P34, which
corresponds to a solvate form (Class 2).
26
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
FIG. 96 ¨ FT-Raman spectra (2400-50 cm-1) of the samples of the solvate
form (Class 3) (PP415-P6: top; PP415-P12: middle; PP415-P20: bottom). The
spectra
have been scaled and offset in the y-direction for the purpose of comparison.
FIG. 97 ¨ FT-Raman spectra (1750-1000 cm-1) of the samples of the solvate
form (Class 3) (PP415-P6: top; PP415-P12: 211d from top; PP415-P20: 211d from
bottom) are very similar to each other with only small differences, e.g., at
¨1690 cm-1,
but are clearly different from class 1 (PP415-P1: bottom). The spectra have
been
scaled and offset in the y-direction for the purpose of comparison.
FIG. 98 ¨ PXRD patterns (2-32 '20) of the samples of the solvate form
(Class 3) (PP415-P6: top; PP415-P12: middle; PP415-P20: bottom). The patterns
have been scaled and offset in the y-direction for the purpose of comparison.
FIG. 99 ¨ PXRD patterns (13.5-18.5 '20) of the samples of solvate form
(Class 3) (PP415-P6: top; PP415-P12: middle; PP415-P20: bottom) show small
differences. The patterns have been scaled and offset in the y-direction for
the
purpose of comparison.
FIG. 100 ¨ TG-FTIR thermogram of the sample PP415-P6, which
corresponds to the solvate form (Class 3).
FIG. 101 ¨ TG-FTIR thermogram of the sample PP415-P12, which
corresponds to the solvate form (Class 3).
FIG. 102 ¨ TG-FTIR thermogram of the dried solvate form (Class 3), sample
PP415-P25.
FIG. 103 ¨ TG-FTIK thermogram of the further dried solvate form (Class 3),
sample PP415-P33.
FIG. 104 ¨ FT-Raman spectra (1800-700 cm-1) of the solvate form (Class 3)
(top, sample PP415-P6), of the dried solvate form (Class 3) (middle, sample
PP415-P25), and of the further dried solvate form (Class 3) (bottom, sample
PP415-
P33) are identical. The spectra have been scaled and offset in the y-direction
for the
purpose of comparison.
27
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
FIG. 105 ¨ PXRD patterns (4-24 '20) of the solvate form (Class 3) (top,
sample PP415-P6), of the dried solvate form (Class 3) (middle, sample PP415-
P25),
and of the further dried solvate form (Class 3) (bottom, sample PP415-P33).
The
patterns have been scaled and offset in the y-direction for the purpose of
comparison.
FIG. 106 ¨ TG-FTIR thermogram of the sample PP415-P13, which
corresponds to an acetonitrile solvate form (Class 4).
FIG. 107 ¨ FT-Raman spectra (1800-700 cm-1) of the acetonitrile solvate form
(Class 4) (dark grey, sample PP415-P13) and of the dried material of an
acetonitrile
solvate form (Class 4) (light grey, sample PP415-P26) are identical and
overlay
perfectly. The spectra have been scaled for purposes of comparison.
FIG. 108 ¨ PXRD pattern of the dried acetonitrile solvate form (Class 4),
sample PP415-P26 (bottom), in comparison to the reference pattern of the
acetonitrile
solvate form (Class 4), sample PP415-P13 (top). The patterns have not been
scaled
but were offset in the y-direction for purposes of comparison.
FIG. 109 ¨ TG-FTIR thermogram of the dried acetonitrile solvate form
(Class 4), sample PP415-P26.
FIG. 110¨ FT-Raman spectra (1800-700 cm-1) of the acetonitrile solvate form
(Class 4) (top, sample PP415-P35), and of the dried acetonitrile solvate form
(Class 4)
(middle, sample PP415-P36 and bottom, sample PP415-P37) correspond to each
other. The spectra have been scaled and offset in the y-direction for purposes
of
comparison.
FIG. 111 ¨ PXRD patterns (4-24 '20) of the acetonitrile solvate form
(Class 4) (top, sample PP415-P35) and of the dried acetonitrile solvate form
(Class 4)
(middle, sample PP415-P36 and bottom, sample PP415-P37) agree with each other.
The patterns have been scaled and offset in the y-direction for the purpose of
comparison.
FIG. 112 ¨ TG-FTIR thermogram of the dried acetronitrile solvate form
(Class 4), sample PP415-P36.
28
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
FIG. 113 ¨ TG-FTIR thermogram of the dried acetonitrile solvate form
(Class 4), sample PP415-P37.
FIG. 114 ¨ DVS isotherm of the desolvated acetronitrile solvate form
(Class 4) sample PP415-P37).
FIG. 115 ¨ PXRD pattern of the sample PP415-P37, an acetonitrile solvate
form (Class 4) after the DVS measurement (bottom) is unchanged compared to the
material before the DVS measurement (top). The patterns have not been scaled
but
are offset in the y-direction for the purpose of comparison.
FIG. 116 ¨ DSC thermogram of the desolvated acetonitrile solvate form
(Class 4) (sample PP415-P37).
FIG. 117 ¨ DSC thermogram of a ¨1:1 mixture of the amorphous form
(Class 1), sample PP415-P1, with the desolvated acetonitrile solvate form
(Class 4),
sample PP415-P36.
FIG. 118 ¨ DSC thermogram of a ¨1:1 mixture of the amorphous form
(Class 1), sample PP415-P1, with the desolvated acetonitrile solvate form
(Class 4),
sample PP415-P36 (experiment number: PP415-P39). The heating scan (Step 1) was
stopped for 30 min at 173 C (Step 2) and then resumed (Step 3).
FIG. 119 ¨ TG-FTIR thermogram of the sample PP415-P14, which
corresponds to a THF solvate form (Class 5).
FIG. 120 ¨ FT-Raman spectra (1800-1100 cm-I) of a THF solvate form
(Class 5) (dashed line, sample PP415-P14), dried material of a THF solvate
form
(Class 5) (dotted line, sample PP415-P27), and of the amorphous form (Class 1)
(solid
line, sample PP415-P1). The spectra have been scaled for the purpose of
comparison
and show small changes in magnitude but little corresponding change in
spectral
shape.
FIG. 121 ¨ PXRD pattern of the dried THF solvate form (Class 5), sample
PP415-P27 (top) in comparison to the pattern of the THF solvate form (Class
5),
sample PP415-P14 (bottom). The patterns have not been scaled but are offset in
the
y-direction for the purpose of comparison.
29
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
FIG. 122 ¨ TG-FTIR thermogram of the sample PP415-P27, which
corresponds to a dried THF solvate (Class 5).
FIG. 123 ¨ PXRD pattern of sample PP415-P41 (top) corresponds to the
pattern of the THF solvate form (Class 5) (middle, sample PP415-P14) and not
to the
pattern of the heptane solvate form, (Class 2) (bottom, sample PP415-P19). The
patterns have been scaled and offset in the y-direction for purposes of
comparison.
FIG. 124 ¨ PXRD pattern of sample PP415-P45 (top) corresponds to the
pattern of the THF solvate form (Class 5) (middle, sample PP415-P14) and not
to the
pattern of the heptane solvate form (Class 2) (bottom, sample PP415-P19). The
patterns have been scaled and offset in the y-direction for the purpose of
comparison.
FIG. 125 ¨ PXRD pattern of sample PP415-P41 (top) corresponds to a THF
solvate form (Class 5). After drying sample PP415-P41 for 1 day (211d from
top,
sample: PP415-P44), the material is mainly amorphous. Some broad peaks with
low
intensity remain. After
further drying overnight (21d from bottom, sample
PP415-P44a) the intensity of these broad peaks is further reduced. The
amorphous
form (Class 1) is shown as a reference (bottom, sample: PP415-P42). The
patterns
have not been scaled but are offset in the y-direction for the purpose of
comparison.
FIG. 126 ¨ TG-FTIR thermogram of the sample PP415-P44a, which
corresponds to the amorphous form (Class 1).
FIG. 127 ¨ PXRD pattern of sample PP415-P45 (top) corresponds to a THF
solvate form (Class 5). After drying sample PP415-P45 for 1 day (211d from
top,
sample PP415-P46), the material is mainly amorphous. Some broad peaks with low
intensity remain. After a total of 4 days of drying (211d from bottom, sample
PP415-P46a), the pattern remains unchanged. The amorphous form (Class 1) is
shown as reference (bottom, sample PP415-P42). The patterns have not been
scaled
but are offset in the y-direction for the purpose of comparison.
FIG. 128 ¨ TG-FTIR thermogram of the sample PP415-P46a, which
corresponds to the amorphous form (Class 1).
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
FIG. 129 ¨ PXRD pattern of sample PP415-P42 (top) corresponds to the
pattern of the amorphous form (Class 1) (bottom, sample PP415-P1). The
patterns
have been scaled and offset in the y-direction for the purpose of comparison.
FIG. 130 ¨ PXRD pattern of sample PP415-P43 (top) corresponds to the
pattern of the isostructural solvate form (Class 2) (bottom, sample PP415-P19)
and
not to the pattern of the THF solvate form (Class 5) (middle, sample PP415-
P14).
The patterns have been scaled and offset in the y-direction for the purpose of
comparison.
FIG. 131 ¨ PXRD patterns of samples PP415-P47 (top) and PP415-P48
(middle) correspond essentially to the pattern of the isostructural solvate
forms
(Class 2) (bottom, sample PP415-P19), although there are some differences. The
patterns have been scaled and offset in the y-direction for the purpose of
comparison.
FIG. 132 ¨ PXRD pattern of sample PP415-P49 (top) corresponds to the
pattern of the amorphous form (Class 1) (bottom, sample PP415-P1). The
patterns
.. have been scaled and offset in the y-direction for the purpose of
comparison.
31
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
DESCRIPTION OF ILLUSTRATIVE EMBODIMENTS
The present invention provides in one aspect the compound:
N-((4aS,6aR,6bS,8aR,12aS,14aR,14bS)-11-cyano-2,2,6a,6b,9,9,12a-
heptamethyl- 10,14 -di oxo-
1,2,3 ,4,4a,5,6,6a,6b,7,8,8a,9,10,12 a,14,14 a,14b-octadecahydropicen-
4a-y1)-2,2-difluoropropanamide,
which is also referred to herein as RTA 408, 63415, or PP415. In other non-
limiting
aspects, the present invention also provides polymorphic forms thereof,
including
solvates thereof. In other non-
limiting aspects, the invention also provides
pharmaceutically acceptable salts thereof. In other non-limiting aspects,
there are also
provided methods for preparation, pharmaceutical compositions, and kits and
articles
of manufacture of these compounds and polymorphic forms thereof.
I. Definitions
When used in the context of a chemical group: "hydrogen" means ¨H;
"hydroxy" means ¨OH; "oxo" means =0; "carbonyl" means ¨C(=0)¨; "carboxy"
means ¨C(=0)0H (also written as ¨COOH or ¨CO2H); "halo" means independently
¨F, ¨Cl, ¨Br or ¨I; "amino" means ¨NH2; "hydroxyamino" means ¨NHOH; "nitro"
means ¨NO2; imino means =NH; "cyano" means ¨CN; "isocyanate" means
¨N=C=O; "azido" means ¨N3; in a monovalent context "phosphate" means
¨0P(0)(OH)2 or a deprotonated form thereof; in a divalent context "phosphate"
means ¨0P(0)(OH)0¨ or a deprotonated form thereoff, "mercapto" means ¨SH; and
"thio" means =S; "sulfonyl" means ¨S(0)2¨; and "sulfinyl" means ¨S(0)¨. Any
undefined valency on an atom of a structure shown in this application
implicitly
represents a hydrogen atom bonded to the atom.
In the context of this disclosure, the formulas:
0 0
0 0
N)2(
-L
NC F F NC F F
0 0 a
and
32
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
represent the same structures. When a dot is drawn on a carbon, the dot
indicates that
the hydrogen atom attached to that carbon is coming out of the plane of the
page.
The use of the word "a" or "an," when used in conjunction with the term
"comprising" in the claims and/or the specification may mean "one," but it is
also
consistent with the meaning of "one or more," "at least one," and "one or more
than
one."
Throughout this application, the term "about" is used to indicate that a value
includes the inherent variation of error for the device, the method being
employed to
determine the value, or the variation that exists among the study subjects.
When used
in the context of X-ray powder diffraction, the term "about" is used to
indicate a value
of 0.2 020 from the reported value, preferably a value of 0.1 020 from the
reported
value. When used in the context of differential scanning calorimetry or glass
transition temperatures, the term "about" is used to indicate a value of +10
C relative
to the maximum of the peak, preferably a value of 2 C relative to the
maximum of
the peak. When used in other contexts, the term "about" is used to indicate a
value of
10% of the reported value, preferably a value of 5% of the reported value. It
is to
be understood that, whenever the term "about" is used, a specific reference to
the
exact numerical value indicated is also included.
The terms "comprise," "have" and "include" are open-ended linking verbs.
Any forms or tenses of one or more of these verbs, such as "comprises,"
"comprising," "has," "having," "includes" and "including," are also open-
ended. For
example, any method that "comprises," "has" or "includes" one or more steps is
not
limited to possessing only those one or more steps and also covers other
unlisted
steps.
The term "effective," as that term is used in the specification and/or claims,
means adequate to accomplish a desired, expected, or intended result.
"Effective
amount," "Therapeutically effective amount" or "pharmaceutically effective
amount"
when used in the context of treating a patient or subject with a compound
means that
amount of the compound which, when administered to a subject or patient for
treating
a disease, is sufficient to effect such treatment for the disease.
The term "halo peak" in the context of X-ray powder diffraction would mean a
broad peak, often spanning >10 20 in an X-ray powder diffractogram, typically
characteristic of an amorphous solid or system.
33
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
The term "hydrate" when used as a modifier to a compound means that the
compound has less than one (e.g., hemihydrate), one (e.g., monohydrate), or
more
than one (e.g., dihydrate) water molecules associated with each compound
molecule,
such as in solid forms of the compound.
As used herein, the term "IC50" refers to an inhibitory dose that is 50% of
the
maximum response obtained. This quantitative measure indicates how much of a
particular drug or other substance (inhibitor) is needed to inhibit a given
biological,
biochemical, or chemical process (or component of a process, i.e. an enzyme,
cell,
cell receptor or microorganism) by half
An "isomer" of a first compound is a separate compound in which each
molecule contains the same constituent atoms as the first compound, but where
the
configuration of those atoms in three dimensions differs.
As used herein, the term "patient" or "subject" refers to a living mammalian
organism, such as a human, monkey, cow, sheep, goat, dog, cat, mouse, rat,
guinea
pig, or transgenic species thereof. In certain embodiments, the patient or
subject is a
non-human animal. In certain embodiments, the patient or subject is a primate.
In
certain embodiments, the patient or subject is a human. Non-limiting examples
of
human subjects are adults, juveniles, infants and fetuses.
As generally used herein "pharmaceutically acceptable" refers to those
compounds, materials, compositions, and/or dosage forms which are, within the
scope
of sound medical judgment, suitable for use in contact with the tissues,
organs, and/or
bodily fluids of human beings and animals without excessive toxicity,
irritation,
allergic response, or other problems or complications commensurate with a
reasonable benefit/risk ratio.
"Pharmaceutically acceptable salts" means salts of compounds of the present
invention which are pharmaceutically acceptable, as defined above, and which
possess the desired pharmacological activity. Such salts include acid addition
salts
formed with inorganic acids such as hydrochloric acid, hydrobromic acid,
sulfuric
acid, nitric acid, phosphoric acid, and the like; or with organic acids such
as
1,2-ethanedisulfonic acid, 2-hydroxyethanesulfonic acid, 2-naphthalenesulfonic
acid,
3 -ph enylpropionic acid, 4,4'-methylenebis(3-hydroxy-2-ene-l-carboxylic
acid),
4-methylbicyclo[2.2.2]oct-2-ene-1-carboxylic acid, acetic acid, aliphatic mono-
and
dicarboxylic acids, aliphatic sulfuric acids, aromatic sulfuric acids,
benzenesulfonic
acid, benzoic acid, camphorsulfonic acid, carbonic acid, cinnamic acid, citric
acid,
34
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
cyclopentanepropionic acid, ethanesulfonic acid, fumaric acid, glucoheptonic
acid,
gluconic acid, glutamic acid, glycolic acid, heptanoic acid, hexanoic acid,
hydroxynaphthoic acid, lactic acid, laurylsulfuric acid, maleic acid, malic
acid,
malonic acid, mandelic acid, methanesulfonic acid, muconic acid,
o-(4-hydroxybenzoyl)benzoic acid, oxalic acid, p-chlorobenzenesulfonic acid,
phenyl-
substituted alkanoic acids, propionic acid, p-toluenesulfonic acid, pyruvic
acid,
salicylic acid, stearic acid, succinic acid, tartaric acid,
tertiarybutylacetic acid,
trimethylacetic acid, and the like. Pharmaceutically acceptable salts also
include base
addition salts which may be formed when acidic protons present are capable of
reacting with inorganic or organic bases. Acceptable inorganic bases include
sodium
hydroxide, sodium carbonate, potassium hydroxide, aluminum hydroxide and
calcium
hydroxide. Acceptable
organic bases include ethan o 1 ami ne, di eth an o I ami ne,
triethanolamine, tromethamine, N-methylglucamine and the like. It should be
recognized that the particular anion or cation forming a part of any salt of
this
invention is not critical, so long as the salt, as a whole, is
pharmacologically
acceptable. Additional examples of pharmaceutically acceptable salts and their
methods of preparation and use are presented in Handbook of Pharmaceutical
Salts:
Properties, and Use (P. H. Stahl & C. G. Wermuth eds., Verlag Helvetica
Chimica
Acta, 2002).
"Prevention" or "preventing" includes: (1) inhibiting the onset of a disease
in a
subject or patient which may be at risk and/or predisposed to the disease but
does not
yet experience or display any or all of the pathology or symptomatology of the
disease, and/or (2) slowing the onset of the pathology or symptomatology of a
disease
in a subject or patient which may be at risk and/or predisposed to the disease
but does
not yet experience or display any or all of the pathology or symptomatology of
the
disease.
"Prodrug" means a compound that is convertible in vivo metabolically into an
inhibitor according to the present invention. The prodrug itself may or may
not also
have activity with respect to a given target protein. For example, a compound
comprising a hydroxy group may be administered as an ester that is converted
by
hydrolysis in vivo to the hydroxy compound. Suitable esters that may be
converted in
vivo into hydroxy compounds include acetates, citrates, lactates, phosphates,
tartrates,
malonates, oxalates, salicylates, propionates, succinates, fumarates,
maleates,
methylene-bis-13-hydroxynaphthoate, gentisates, isethionates, di-p-
toluoyltartrates,
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
methanesulfonates, ethanesulfonates, benzenesulfonates, p-toluenesulfonates,
cyclohexylsulfamates, quinates, esters of amino acids, and the like.
Similarly, a
compound comprising an amine group may be administered as an amide that is
converted by hydrolysis in vivo to the amine compound.
A "stereoisomer" or "optical isomer" is an isomer of a given compound in
which the same atoms are bonded to the same other atoms, but where the
configuration of those atoms in three dimensions differs. "Enantiomers" are
stereoisomers of a given compound that are mirror images of each other, like
left and
right hands. "Diastereomers" are stereoisomers of a given compound that are
not
enantiomers. Chiral molecules contain a chiral center, also referred to as a
stereocenter or stereogenic center, which is any point, though not necessarily
an atom,
in a molecule bearing groups such that an interchanging of any two groups
leads to a
stereoisomer. In organic compounds, the chiral center is typically a carbon,
phosphorus or sulfur atom, though it is also possible for other atoms to be
stereocenters in organic and inorganic compounds. A molecule can have multiple
stereocenters, giving it many stereoisomers. In compounds whose
stereoisomerism is
due to tetrahedral stereogenic centers (e.g., tetrahedral carbon), the total
number of
hypothetically possible stereoisomers will not exceed 2n, where n is the
number of
tetrahedral stereocenters. Molecules with symmetry frequently have fewer than
the
maximum possible number of stereoisomers. A 50:50 mixture of enantiomers is
referred to as a racemic mixture. Alternatively, a mixture of enantiomers can
be
enantiomerically enriched so that one enantiomer is present in an amount
greater than
50%. Typically, enantiomers and/or diastereomers can be resolved or separated
using
techniques known in the art. It is contemplated that for any stereocenter or
axis of
chirality for which stereochemistry has not been defined, that stereocenter or
axis of
chirality can be present in its R form, S form, or as a mixture of the R and S
forms,
including racemic and non-racemic mixtures. As used
herein, the phrase
"substantially free from other stereoisomers" means that the composition
contains
<15%, more preferably <10%, even more preferably <5%, or most preferably <1%
of
another stereoisomer(s).
"Treatment" or "treating" includes (1) inhibiting a disease in a subject or
patient experiencing or displaying the pathology or symptomatology of the
disease
(e.g., arresting further development of the pathology and/or symptomatology),
(2)
ameliorating a disease in a subject or patient that is experiencing or
displaying the
36
, õ.. .. õ.
pathology or symptomatology of the disease (e.g., reversing the pathology
and/or
symptomatology), and/or (3) effecting any measurable decrease in a disease in
a
subject or patient that is experiencing or displaying the pathology or
symptomatology
of the disease.
Thc above definitions supersede any conflicting definition in any of the
reference that is referenced herein. The fact
that certain terms are
defined, however, should not be considered as indicative that any term that is
undefined is indefinite. Rather, all terms used are believed to describe the
invention
in terms such that one of ordinary skill can appreciate the scope and practice
the
present invention.
U. RTA 408 and Synthetic Methods
RTA 408 can be prepared according to the methods described in the Examples
section below. These methods can be further modified and optimized using the
principles and techniques of organic chemistry as applied by a person skilled
in the
art. Such principles and techniques are taught, for example, in March's
Advanced
Organic Chemisoy: Reactions, Mechanisms, and Structure (2007) .
It should be recognized that the particular anion or cation forming a part of
any salt of this invention is not critical, so long as the salt, as a whole,
is
pharmacologically acceptable. Additional examples of pharmaceutically
acceptable
salts and their methods of preparation and use are presented in Handbook of
Pharmaceutical Salts: Properties, and Use (2002).
RTA 408 may also exist in prodrug form. Since prodrugs are known to
enhance numerous desirable qualities of pharmaceuticals, e.g., solubility,
bioavailability, manufacturing, etc., the compounds employed in some methods
of the
invention may, if desired, be delivered in prodrug form. Thus, the invention
contemplates prodrugs of compounds of the present invention as well as methods
of
delivering prodrugs. Prodrugs of the compounds employed in the invention may
be
prepared by modifying functional groups present in the compound in such a way
that
the modifications are cleaved, either in routine manipulation or in vivo, to
the parent
compound. Accordingly, prodrugs include, for example, compounds described
herein
in which a hydroxy, amino, or carboxy group is bonded to any group that, when
the
37
CA 2869783 2020-04-08
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
prodrug is administered to a patient, cleaves to form a hydroxy, amino, or
carboxylic
acid, respectively.
RTA 408 may contain one or more asymmetrically-substituted carbon or
nitrogen atoms, and may be isolated in optically active or racemic form. Thus,
all
chiral, diastereomeric, racemic form, epimeric form, and all geometric
isomeric forms
of a structure are intended, unless the specific stereochemistry or isomeric
form is
specifically indicated. RTA 408 may occur as racemates and racemic mixtures,
single
enantiomers, diastereomeric mixtures and individual diastereomers. In some
embodiments, a single diastereomer is obtained. The chiral centers of RTA 408
according to the present invention can have the S or the R configuration.
In addition, atoms making up RTA 408 of the present invention are intended
to include all isotopic forms of such atoms. Isotopes, as used herein, include
those
atoms having the same atomic number but different mass numbers. By way of
general example and without limitation, isotopes of hydrogen include tritium
and
deuterium, and isotopes of carbon include i'C and 14C. Similarly, it is
contemplated
that one or more carbon atom(s) of a compound of the present invention may be
replaced by a silicon atom(s). Furthermore, it is contemplated that one or
more
oxygen atom(s) of RTA 408 may be replaced by a sulfur or selenium atom(s).
RTA 408 and polymorphic form thereof may also have the advantage that they
may be more efficacious than, be less toxic than, be longer acting than, be
more
potent than, produce fewer side effects than, be more easily absorbed than,
and/or
have a better pharmacokinetic profile (e.g., higher oral bioavailability
and/or lower
clearance) than, and/or have other useful pharmacological, physical, or
chemical
advantages over, compounds known in the prior art for use in the indications
stated
herein.
III. Polymorphic Forms of RTA 408
In some embodiments, the present invention provides different solid forms of
RTA 408, including solvates thereof. A
preformulation and preliminary
polymorphism study was performed, and RTA 408 was found to have a high
tendency
for solvate formation. Crystalline forms of classes 2, 3, 4, and 5 are
consistent with
solvates. For a description of the classes, see Table 1 below. Attempts to dry
classes 2 and 3 (two groups of isostructural solvates) were not successful,
which is
consistent with tightly bound solvent molecules. In some embodiments, drying
of a
38
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
class 4 solid (acetonitrile solvate) led to an isostructural desolvated form.
In some
embodiments, drying of a class 5 solid (THF solvate) resulted in the amorphous
form
class 1. Non-solvated forms of RTA 408 include the amorphous form (class 1)
and
the crystalline desolvated solvate of class 4 (isostructural to the class 4
acetonitrile
.. solvate). In some embodiments, the amorphous form has a high glass
transition with
Ts 153 C (ACp = 0.72 J/g C) and is only slightly hygroscopic (Am = +0.4%
50%¨>85% r.h.). In some embodiments, the amorphous form is stable for at least
four weeks under elevated temperature and humidity conditions (i.e., open at
40
C/-75% r.h. or closed at 80 C). In some embodiments, the amorphous form
(class 1) was successfully prepared from class 2 material in a two-step
process
(transformation into class 5 and subsequent drying of class 5 to obtain the
amorphous
form), as well as in a direct one-step method (precipitation from an acetone
solution
in a cold water bath). The crystalline desolvated solvate of class 4
(isostructural to the
class 4 solvate) is slightly hygroscopic (mass gain of ¨0.7 wt.-% from 50%
r.h. to
.. 85% r.h.) and has a possible melting point at 196.1 C (AH = 29.31 J/g).
A sample of the amorphous form of 63415, class 1, was characterized by
FT-Raman spectroscopy, PXRD, TG-FTIR, Karl Fischer titration, 11-1-NMR, DSC,
and DVS (see Examples section for additional details). The sample was found to
contain ¨0.9 wt.-% Et0H with traces of H20 (according to the TG-FTIR). A water
content of 0.5 wt.-% was determined by Karl Fischer titration. DSC shows a
high
glass transition temperature with Ts 153 C (ACp = 0.72 .1/g C). According to
DVS,
the material is slightly hygroscopic (Am = +0.4% 500/0z,
85% r.h.). No crystallization
was observed in the DSC or DVS experiments.
The chemical stability of the amorphous form was investigated in organic
.. solvents, including acetone, Et0Ac, Me0H, and MeCN, as well as different
aqueous
media (e.g., 1% aq. Tween 80, 1% aq. SDS, 1% aq. CTAB) at a concentration of
1 mg/mL at time points 6 h, 24 h, 2 d, and 7 d. Decomposition >1% was observed
only for solutions in MeCN after 7 days and for suspensions in the 1% aqueous
Tween 80 medium (at all times points at 254 nm and after 24 h, 2 d, and 7 d at
.. 242 nm).
In addition, the stability of the amorphous form was investigated by storage
under elevated temperature and humidity conditions (open at 25 C/62% r.h. and
C/75% r.h. and closed at 60 C and 80 C). After one week, two weeks, and four
39
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
weeks, the stored samples were analyzed by PXRD. None of the samples differed
from the amorphous starting material.
More than 30 crystallization and drying experiments were carried out,
including suspension equilibration, slow cooling, evaporation, and
precipitation. Four
new crystalline forms were obtained (classes 2, 3, 4, and 5) in addition to
the
amorphous form (class 1).
The four new forms (classes 2, 3, 4, and 5) were characterized by FT-Raman
spectroscopy, PXRD, and TG-FTIR. All forms correspond to solvates (Table 1).
Drying experiments under vacuum or N2 flow were carried out with the aim to
obtain
a crystalline, non-solvated form of 63415.
Table 1. Summary of Obtained Classes
Class Characteristics Result of Drying Experiments
Class 1 amorphous form
isostructural solvates (e.g.,
Class 2 drying not successful
heptane)
Class 3 isostructural solvates (e.g., ethanol) drying not successful
Class 4 MeCN solvate & desolvated drying successful, structure
solvate unchanged
Class 5 THF solvate drying
resulted in amorphous form
Class 2: Most crystallization experiments that were conducted resulted in
solid material of class 2 (see Examples section below). Its members may
correspond
to isostructural, non-stoichiometric (<0.5 eq.) solvates (of heptane,
cyclohexane,
isopropyl ether, 1-butanol, triethylamine, and possibly other solvents, such
as hexane,
other ethers, etc.) with tightly bound solvent molecules. The Raman spectra
and
PXRD patterns within this class are very similar to each other, thus the
structures
might be essentially identical with only small differences due to the
different solvents
that were incorporated.
Drying experiments on class 2 samples have not resulted in a crystalline, non-
solvated form. Even elevated temperatures (80 C) and a high vacuum (<1x10-3
mbar)
could not remove the tightly bound solvent molecules completely; a solvent
content of
>2 wt.-% always remained. The crystallinity of these partially dried samples
is
reduced, but neither transformation into a different structure nor substantial
amorphization was observed.
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
Class 3: Solid
material of class 3 may be obtained from several
crystallizations (see Examples section below). The samples of class 3 are
likely
isostructural solvates of 2PrOH, Et0H, and probably acetone with tightly bound
solvent molecules. They could correspond to either stoichiometric hemisolvates
or
non-stoichiometric solvates with a solvent content of ¨0.5 eq. As with class
2, the
Raman spectra and PXRD patterns within this class are very similar to each
other,
indicating similar structures that incorporate different solvents.
Similar to class 2, drying experiments were not successful. The very tightly
bound solvent molecules could only partially be removed (i.e., ¨5.4 wt.-% to
¨4.8 wt.-% after up to 3 d at 1x103 mbar and 80 C). The PXRD patterns
remained
unchanged.
Class 4 may be obtained from a 7:3 MeCI\14120 solvent system (see Examples
section below). It most likely corresponds to a crystalline acetonitrile
hemisolvate.
By drying (under vacuum or N2 flow at elevated temperatures) most of the
solvent
molecules could be removed without changing or destroying the crystal
structure
(PXRD remained unchanged). Thus, a crystalline, non-solvated form (or rather
desolvated solvate) was obtained. It is slightly hygroscopic (mass gain of
¨0.7 wt.-%
from 50% r.h. to 85% r.h.) and has a possible melting point at 196.1 C (AH =
29.31 J/g).
Class 5 may be obtained from an ¨1:1 THF/H20 solvent system. Class 5
contains bound THF (and maybe H20). As the content of the two components
cannot
be readily quantified separately, the exact nature of this crystalline solvate
has not
been determined.
Drying of class 5 resulted in significant desolvation and transformation in
the
direction of the amorphous form (class 1). In some embodiments, the amorphous
form of RTA 408 may be prepared by suspending class 2 heptane solvate in 1:1
THF/H20 to form a class 5 solid, followed by drying and amorphization.
Experiments with the aim of preparing the amorphous form (class 1) were
carried out using class 2 starting material. Mainly amorphous material (class
1) was
prepared starting from class 2 material in a two-step process via class 5 on a
100-mg
and 3-g scale (drying at 100 mbar, 80 C, several days). The preparation of
fully
amorphous material (class 1) was found to be possible in a one-step process
avoiding
41
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
the solvent THF by direct precipitation of the amorphous form (class 1) from
an
acetone solution of class 2 material in a cold water bath.
IV. Diseases Associated with Inflammation and/or Oxidative Stress
Inflammation is a biological process that provides resistance to infectious or
.. parasitic organisms and the repair of damaged tissue. Inflammation is
commonly
characterized by localized vasodilation, redness, swelling, and pain, the
recruitment of
leukocytes to the site of infection or injury, production of inflammatory
cytokines,
such as TNF-ct and IL-1, and production of reactive oxygen or nitrogen
species, such
as hydrogen peroxide, superoxide, and peroxynitrite. In later stages of
inflammation,
.. tissue remodeling, angiogenesis, and scar formation (fibrosis) may occur as
part of the
wound healing process. Under normal circumstances, the inflammatory response
is
regulated, temporary, and is resolved in an orchestrated fashion once the
infection or
injury has been dealt with adequately. However, acute inflammation can become
excessive and life-threatening if regulatory mechanisms fail.
Alternatively,
inflammation can become chronic and cause cumulative tissue damage or systemic
complications. Based at least on the evidence presented herein, RTA 408 can be
used
in the treatment or prevention of inflammation or diseases associated with
inflammation.
Many serious and intractable human diseases involve dysregulation of
inflammatory processes, including diseases such as cancer, atherosclerosis,
and
diabetes, which were not traditionally viewed as inflammatory conditions. In
the case
of cancer, the inflammatory processes are associated with processes that
include
tumor formation, progression, metastasis, and resistance to therapy. In some
embodiments, RTA 408 may be used in the treatment or prevention of cancers
including a carcinoma, sarcoma, lymphoma, leukemia, melanoma, mesothelioma,
multiple myeloma, or seminoma, or cancer of the bladder, blood, bone, brain,
breast,
central nervous system, cervix, colon, endometrium, esophagus, gall bladder,
genitalia, genitourinary tract, head, kidney, larynx, liver, lung, muscle
tissue, neck,
oral or nasal mucosa, ovary, pancreas, prostate, skin, spleen, small
intestine, large
intestine, stomach, testicle, or thyroid. Atherosclerosis, long viewed as a
disorder of
lipid metabolism, is now understood to be primarily an inflammatory condition,
with
activated macrophages playing an important role in the formation and eventual
rupture of atherosclerotic plaques. Activation of inflammatory signaling
pathways
42
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
has also been shown to play a role in the development of insulin resistance,
as well as
in the peripheral tissue damage associated with diabetic hyperglycemia.
Excessive
production of reactive oxygen species and reactive nitrogen species, such as
superoxide, hydrogen peroxide, nitric oxide, and peroxynitrite, is a hallmark
of
inflammatory conditions. Evidence of dysregulated peroxynitrite production has
been
reported in a wide variety of diseases (Szabo et al., 2007; Schulz et al.,
2008;
Forstermann, 2006; Pall, 2007).
Autoimmune diseases such as rheumatoid arthritis, lupus, psoriasis, and
multiple sclerosis involve inappropriate and chronic activation of
inflammatory
processes in affected tissues, arising from dysfunction of self vs. non-self
recognition
and response mechanisms in the immune system. In neurodegenerative diseases
such
as Alzheimer's and Parkinson's diseases, neural damage is correlated with
activation
of microglia and elevated levels of pro-inflammatory proteins, such as
inducible nitric
oxide synthase (iNOS). Chronic organ failure, such as renal failure, heart
failure,
liver failure, and chronic obstructive pulmonary disease, is closely
associated with the
presence of chronic oxidative stress and inflammation, leading to the
development of
fibrosis and eventual loss of organ function. Oxidative stress in vascular
endothelial
cells, which line major and minor blood vessels, can lead to endothelial
dysfunction
and is believed to be an important contributing factor in the development of
systemic
cardiovascular disease, complications of diabetes, chronic kidney disease and
other
forms of organ failure, and a number of other aging-related diseases,
including
degenerative diseases of the central nervous system and the retina.
Many other disorders involve oxidative stress and inflammation in affected
tissues, including inflammatory bowel disease; inflammatory skin diseases;
mucositis
and dermatitis related to radiation therapy and chemotherapy; eye diseases,
such as
uveitis, glaucoma, macular degeneration, and various forms of retinopathy;
transplant
failure and rejection; ischemia-reperfusion injury; chronic pain; degenerative
conditions of the bones and joints, including osteoarthritis and osteoporosis;
asthma
and cystic fibrosis; seizure disorders; and neuropsychiatric conditions,
including
schizophrenia, depression, bipolar disorder, post-traumatic stress disorder,
attention
deficit disorders, autism-spectrum disorders, and eating disorders, such as
anorexia
nervosa. Dysregulation of inflammatory signaling pathways is believed to be a
major
factor in the pathology of muscle wasting diseases, including muscular
dystrophy and
various forms of cachexia.
43
WO 2013/163344
PCT/US2013/038064
A variety of life-threatening acute disorders also involve dysregulated
inflammatory signaling, including acute organ failure involving the pancreas,
kidneys,
liver, or lungs, myocardial infarction or acute coronary syndrome, stroke,
septic
shock, trauma, severe burns, and anaphylaxis.
Many complications of infectious diseases also involve dysregulation of
inflammatory responses, Although an inflammatory response can kill invading
pathogens, an excessive inflammatory response can also be quite destructive
and in
some cases can be a primary source of damage in infected tissues. Furthermore,
an
excessive inflammatory response can also lead to systemic complications due to
= 10 overproduction of inflammatory cytolcines, such as TNF-cc and
IL-1. This is believed
to be a factor in mortality arising from severe influenza, severe acute
respiratory
syndrome, and sepsis.
The aberrant or excessive expression of either iNOS or cyclooxygenase-2
(COX-2) has been implicated in the pathogenesis of many disease processes. For
example, it is clear that NO is a potent mutagen (Tamir and Tannebaum, 1996),
and
that nitric oxide can also activate COX-2 (Salvemini et al., 1994).
Furthermore, there
is a marked increase in iNOS in rat colon tumors induced by the carcinogen,
azoxymethane (Takahashi et aL, 1997). A series of synthetic triterpenoid
analogs of
olcanolic acid have been shown to be powerful inhibitors of cellular
inflammatory
processes, such as the induction by IFN-y of inducible nitric oxide synthase
(iNOS)
and of COX-2 in mouse macrophages. See Honda et al. (2000a), Honda et al.
(2000b), and Honda et al. (2002).
In one aspect, RTA 408 disclosed herein is in part characterized by its
ability
to inhibit the production of nitric oxide in macrophage-derived RAW 264.7
cells
induced by exposure to 7-interferon. RTA 408 is further characterized by the
ability
to induce the expression .of antioxidant proteins, such as NQ01, and reduce
the
expression of pro-inflammatory proteins, such as COX-2 and inducible nitric
oxide
synthase (iNOS). These properties are relevant to the treatment of a wide
array of
diseases and disorders involving oxidative stress and dysregulation of
inflammatory
processes, including cancer, complications from localized or total-body
exposure to
ionizing radiation, mucositis and dermatitis resulting from radiation therapy
or
chemotherapy, autoimmune diseases, cardiovascular diseases, including
atherosclerosis, ischemia-reperfusion injury, acute and chronic organ failure,
44 .
CA 2869783 2020-04-08
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
including renal failure and heart failure, respiratory diseases, diabetes and
complications of diabetes, severe allergies, transplant rejection, graft-
versus-host
disease, neurodegenerative diseases, diseases of the eye and retina, acute and
chronic
pain, degenerative bone diseases, including osteoarthritis and osteoporosis,
inflammatory bowel diseases, dermatitis and other skin diseases, sepsis, bums,
seizure
disorders, and neuropsychiatric disorders.
In another aspect, RTA 408 may be used for treating a subject having a
condition such as eye diseases. For example, uveitis, macular degeneration
(both the
dry form and wet form), glaucoma, diabetic macular edema, blepharitis,
diabetic
retinopathy, diseases and disorders of the corneal endothelium such as Fuchs
endothelial corneal dystrophy, post-surgical inflammation, dry eye, allergic
conjunctivitis and other forms of conjunctivitis are non-limiting examples of
eye
diseases that could be treated with RTA 408.
In another aspect, RTA 408 may be used for treating a subject having a
condition such as skin diseases or disorders. For example, dermatitis,
including
allergic dermatitis, atopic dermatitis, dermatitis due to chemical exposure,
and
radiation-induced dermatitis; thermal or chemical burns; chronic wounds
including
diabetic ulcers, pressure sores, and venous ulcers; acne; alopecia including
baldness
and drug-induced alopecia; other disorders of the hair follicle; epidermolysis
bullosa;
sunburn and its complications; disorders of skin pigmentation including
vitiligo;
aging-related skin conditions; post-surgical wound healing; prevention or
reduction of
scarring from skin injury, surgery, or burns; psoriasis; dermatological
manifestations
of autoimmune diseases or graft-versus host disease; prevention or treatment
of skin
cancer; disorders involving hyperproliferation of skin cells such as
hyperkeratosis is a
non-limiting example of skin diseases that could be treated with RTA 408.
Without being bound by theory, the activation of the antioxidant/anti-
inflammatory Keapl/Nrf2/ARE pathway is believed to be implicated in both the
anti-
inflammatory and anti-carcinogenic properties of the compound disclosed
herein.
In another aspect, RTA 408 may be used for treating a subject having a
condition caused by elevated levels of oxidative stress in one or more
tissues.
Oxidative stress results from abnormally high or prolonged levels of reactive
oxygen
species, such as superoxide, hydrogen peroxide, nitric oxide, and
peroxynitrite
(formed by the reaction of nitric oxide and superoxide). The oxidative stress
may be
accompanied by either acute or chronic inflammation. The oxidative stress may
be
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
caused by mitochondrial dysfunction, by activation of immune cells, such as
macrophages and neutrophils, by acute exposure to an external agent, such as
ionizing
radiation or a cytotoxic chemotherapeutic agent (e.g., doxorubicin), by trauma
or
other acute tissue injury, by ischemia/reperfusion, by poor circulation or
anemia, by
localized or systemic hypoxia or hyperoxia, by elevated levels of inflammatory
cytokines and other inflammation-related proteins, and/or by other abnormal
physiological states, such as hyperglycemia or hypoglycemia.
In animal models of many such conditions, stimulating expression of inducible
heme oxygenase (H0-1), a target gene of the Nrf2 pathway, has been shown to
have a
significant therapeutic effect including in models of myocardial infarction,
renal
failure, transplant failure and rejection, stroke, cardiovascular disease, and
autoimmune disease (e.g., Sacerdoti et al., 2005; Abraham & Kappas, 2005;
Bach,
2006; Araujo et al., 2003; Liu et al., 2006; Ishikawa et al., 2001; Kruger et
al., 2006;
Satoh et al., 2006; Zhou et al., 2005; Morse and Choi, 2005; Morse and Choi,
2002).
This enzyme breaks free heme down into iron, carbon monoxide (CO), and
biliverdin
(which is subsequently converted to the potent antioxidant molecule,
bilirubin).
In another aspect, RTA 408 may be used in preventing or treating tissue
damage or organ failure, acute and chronic, resulting from oxidative stress
exacerbated by inflammation. Examples of diseases that fall in this category
include
heart failure, liver failure, transplant failure and rejection, renal failure,
pancreatitis,
fibrotic lung diseases (cystic fibrosis, COPD, and idiopathic pulmonary
fibrosis,
among others), diabetes (including complications), atherosclerosis, ischemia-
reperfusion injury, glaucoma, stroke, autoimmune disease, autism, macular
degeneration, and muscular dystrophy. For example, in the case of autism,
studies
suggest that increased oxidative stress in the central nervous system may
contribute to
the development of the disease (Chauhan and Chauhan, 2006).
Evidence also links oxidative stress and inflammation to the development and
pathology of many other disorders of the central nervous system, including
psychiatric disorders, such as psychosis, major depression, and bipolar
disorder;
seizure disorders, such as epilepsy; pain and sensory syndromes, such as
migraine,
neuropathic pain, or tinnitus; and behavioral syndromes, such as the attention
deficit
disorders. See, e.g., Dickerson et al., 2007; Hanson et al., 2005; Kendall-
Tackett,
2007; Lencz et al., 2007; Dudhgaonkar et al., 2006; Lee et al., 2007; Morris
et al.,
2002; Ruster et al., 2005; McIver et al., 2005; Sarchielli et al., 2006;
Kawakami et al.,
46
2006; Ross et aL, 2003. For example,
elevated levels of inflammatory cytokines, including TNF-a, interferon-y, and
IL-6,
are associated with major mental illness (Dickerson et al., 2007). Microglial
activation has also been linked to major mental illness. Therefore,
downregulating
inflammatory cytolcines and inhibiting excessive activation of microglia could
be
beneficial in patients with schizophrenia, major depression, bipolar disorder,
autism-
spectrum disorders, and other neuropsychiatric disorders.
Accordingly, in pathologies involving oxidative stress alone or oxidative
stress
exacerbated by inflammation, treatment may comprise administering to a subject
a
therapeutically effective amount of a compound of this invention, such as
those
described above or throughout this specification. Treatment may be
administered
preventively, in advance of a predictable state of oxidative stress (e.g.,
organ
transplantation or the administration of radiation therapy to a cancer
patient), or it
may be administered therapeutically in settings involving established
oxidative stress
and inflammation. In some embodiments, when a compound of the present
invention
is used for treating a patient receiving radiation therapy and/or
chemotherapy, the
compound of the invention may be administered before, at the same time, and/or
after
the radiation or chemotherapy, or the compound may be administered in
combination
with the other therapies. In some embodiments, the compound of the invention
may
prevent and/or reduce the severity of side effects associated with the
radiation therapy
or chemotherapy (using a different agent) without reducing the anticancer
effects of
the radiation therapy or chemotherapy. Because such side effects may be dose-
limiting for the radiation therapy and/or chemotherapy, in some embodiments,
the
compound of the present invention may be used to allow for higher and/or more
frequent dosing of the radiation therapy and/or chemotherapy, for example,
resulting
in greater treatment efficacy. In some embodiments, the compound of the
invention
when administered in combination with the radiation therapy and/or
chemotherapy
may enhance the efficacy of a given dose of radiation and/or chemotherapy. In
some
embodiments, the compound of the invention when administered in combination
with
the radiation therapy and/or chemotherapy may enhance the efficacy of a given
dose
of radiation and/or chemotherapy and reduce (or, at a minimum, not add to) the
side
effects of the radiation and/or chemotherapy. In some embodiments, and without
being bound by theory, this combinatorial efficacy may result from inhibition
of the
47
CA 2869783 2019-09-05
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
activity of the pro-inflammatory transcription factor NF-KB by the compound of
the
invention. NF-KB is often chronically activated in cancer cells, and such
activation is
associated with resistance to therapy and promotion of tumor progression
(e.g., Karin,
2006; Aghajan et al., 2012). Other transcription factors that promote
inflammation
and cancer, such as STAT3 (e.g., He and Karin 2011; Grivennikov and Karin,
2010),
may also be inhibited by the compound of the invention in some embodiments.
RTA 408 may be used to treat or prevent inflammatory conditions, such as
sepsis, dermatitis, autoimmune disease, and osteoarthritis. RTA 408 may also
be used
to treat or prevent inflammatory pain and/or neuropathic pain, for example, by
inducing Nrf2 and/or inhibiting NF-KB.
RTA 408 may also be used to treat or prevent diseases, such as cancer,
inflammation, Alzheimer's disease, Parkinson's disease, multiple sclerosis,
autism,
amyotrophic lateral sclerosis, Huntington's disease, autoimmune diseases, such
as
rheumatoid arthritis, lupus, Crohn's disease, and psoriasis, inflammatory
bowel
.. disease, all other diseases whose pathogenesis is believed to involve
excessive
production of either nitric oxide or prostaglandins, and pathologies involving
oxidative stress alone or oxidative stress exacerbated by inflammation. RTA
408 may
be used in the treatment or prevention of cancers include a carcinoma,
sarcoma,
lymphoma, leukemia, melanoma, mesothelioma, multiple myeloma, or seminoma, or
cancer of the bladder, blood, bone, brain, breast, central nervous system,
cervix,
colon, endometrium, esophagus, gall bladder, genitalia, genitourinary tract,
head,
kidney, larynx, liver, lung, muscle tissue, neck, oral or nasal mucosa, ovary,
pancreas,
prostate, skin, spleen, small intestine, large intestine, stomach, testicle,
or thyroid.
Another aspect of inflammation is the production of inflammatory
prostaglandins, such as prostaglandin E. RTA 408 may be used to promote
vasodilation, plasma extravasation, localized pain, elevated temperature, and
other
symptoms of inflammation. The inducible form of the enzyme COX-2 is associated
with their production, and high levels of COX-2 are found in inflamed tissues.
Consequently, inhibition of COX-2 may relieve many symptoms of inflammation
and
a number of important anti-inflammatory drugs (e.g., ibuprofen and celecoxib)
act by
inhibiting COX-2 activity. It has been demonstrated that a class of
cyclopentenone
prostaglandins (cyPGs) (e.g., 15-deoxy prostaglandin J2, a.k.a. PGJ2) plays a
role in
stimulating the orchestrated resolution of inflammation (e.g., Rajakariar et
al., 2007).
48
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
COX-2 is also associated with the production of cyclopentenone prostaglandins.
Consequently, inhibition of COX-2 may interfere with the full resolution of
inflammation, potentially promoting the persistence of activated immune cells
in
tissues and leading to chronic, "smoldering" inflammation. This effect may be
responsible for the increased incidence of cardiovascular disease in patients
using
selective COX-2 inhibitors for long periods of time.
In one aspect, RTA 408 may be used to control the production of pro-
inflammatory cytokines within the cell by selectively activating regulatory
cysteine
residues (RCRs) on proteins that regulate the activity of redox-sensitive
transcription
factors. Activation of RCRs by cyPGs has been shown to initiate a pro-
resolution
program in which the activity of the antioxidant and cytoprotective
transcription factor
Nrf2 is potently induced and the activities of the pro-oxidant and pro-
inflammatory
transcription factors NF-03 and the STATs are suppressed. In some embodiments,
RTA 408 may be used to increase the production of antioxidant and reductive
molecules
(NQ01, HO-1, SOD1, y-GCS) and decrease oxidative stress and the production of
pro-
oxidant and pro-inflammatory molecules (iNOS, COX-2, 'TNF-a). In some
embodiments, RTA 408 may be used to cause the cells that host the inflammatory
event
to revert to a non-inflammatory state by promoting the resolution of
inflammation and
limiting excessive tissue damage to the host.
A. Cancer
In some embodiments, RTA 408, the polymorphic forms, and methods of the
present disclosure may be used to induce apoptosis in tumor cells, to induce
cell
differentiation, to inhibit cancer cell proliferation, to inhibit an
inflammatory
response, and/or to function in a chemopreventative capacity. For example, the
invention provides new polymorphic forms that have one or more of the
following
properties: (1) an ability to induce apoptosis and differentiate both
malignant and non-
malignant cells, (2) an activity at sub-micromolar or nanomolar levels as an
inhibitor
of proliferation of many malignant or premalignant cells, (3) an ability to
suppress the
de novo synthesis of the inflammatory enzyme inducible nitric oxide synthase
(iNOS),
(4) an ability to inhibit NF-KB activation, and (5) an ability to induce the
expression
of heme oxygenase-1 (H0-1).
49
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
The levels of iNOS and COX-2 are elevated in certain cancers and have been
implicated in carcinogenesis and COX-2 inhibitors have been shown to reduce
the
incidence of primary colonic adenomas in humans (Rostom et al., 2007; Brown
and
DuBois, 2005; Crowel et al., 2003). iNOS is expressed in myeloid-derived
suppressor cells (MDSCs) (Angulo et al., 2000) and COX-2 activity in cancer
cells
has been shown to result in the production of prostaglandin E2 (PGE2), which
has
been shown to induce the expression of arginase in MDSCs (Sinha et al., 2007).
Arginase and iNOS are enzymes that utilize L-arginine as a substrate and
produce
L-omithine and urea, and L-citrulline and NO, respectively. The depletion of
arginine
from the tumor microenvironment by MDSCs, combined with the production of NO
and peroxynitrite has been shown to inhibit proliferation and induce apoptosis
of
T cells (Bronte et al., 2003). Inhibition of COX-2 and iNOS has been shown to
reduce the accumulation of MDSCs, restore cytotoxic activity of tumor-
associated T
cells, and delay tumor growth (Sinha et al., 2007; Mazzoni et al., 2002; Zhou
et al.,
2007).
Inhibition of the NF-KB and JAK/STAT signaling pathways has been
implicated as a strategy to inhibit proliferation of cancer epithelial cells
and induce
their apoptosis. Activation of STAT3 and NF-KB has been shown to result in
suppression of apoptosis in cancer cells, and promotion of proliferation,
invasion, and
metastasis. Many of the target genes involved in these processes have been
shown to
be transcriptionally regulated by both NF-KB and STAT3 (Yu et al., 2007).
In addition to their direct roles in cancer epithelial cells, NF-KB and STAT3
also have important roles in other cells found within the tumor
microenvironment.
Experiments in animal models have demonstrated that NF-KB is required in both
cancer cells and hematopoeitic cells to propagate the effects of inflammation
on
cancer initiation and progression (Greten et al., 2004). NF-KB inhibition in
cancer
and myeloid cells reduces the number and size, respectively, of the resultant
tumors.
Activation of STAT3 in cancer cells results in the production of several
cytokines (IL-
6, IL-10) which suppress the maturation of tumor-associated dendritic cells
(DC).
.. Furthermore, STAT3 is activated by these cytokines in the dendritic cells
themselves.
Inhibition of STAT3 in mouse models of cancer restores DC maturation, promotes
antitumor immunity, and inhibits tumor growth (Kortylewski et al., 2005). In
some
embodiments, RTA 408 and its polymorphic forms can be used to treat cancer,
including, for example, prostate cancer. In some embodiments, RTA 408 and its
polymorphic forms can be used in a combination therapy to treat cancer
including, for
example, prostate cancer. See, e.g., Example H below.
B. Multiple Sclerosis and Other Neurodegenerative Conditions
In some embodiments, RTA 408, the polymorphic forms, and the methods of
this invention may be used for treating patients for multiple sclerosis (MS)
or other
neurodegenerative conditions such as Alzheimer's disease, Parkinson's disease,
or
amyotrophic lateral sclerosis. MS is known to be an inflammatory condition of
the
central nervous system (Williams et al., 1994; Merrill and Benvenist, 1996;
Genain
and Nauser, 1997). Based on several
investigations, evidence suggests that
inflammatory, oxidative, and/or immune mechanisms are involved in the
pathogenesis
of Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral
sclerosis
(ALS), and MS (Bagasra et aL, 1995; McGeer and McGeer, 1995; Simonian and
Coyle, 1996; Kaltschmidt et al., 1997). Epidemiologic data indicate that
chronic use
of NSAIDs which block synthesis of prostaglandins from arachidonate, markedly
lowers the risk for development of AD (McGeer et al., 1996; Stewart et al.,
1997).
Thus, agents that block formation of NO and prostaglandins, may be used in
approaches to prevent and treat neurodegenerative diseases. Successful
therapeutic
candidates for treating such a disease typically require an ability to
penetrate the
blood-brain barrier. See, for example, U.S. Patent Publication 2009/0060873.
C. Neuroinflammation
In some embodiments, RTA 408, the polymorphic forms, and methods of this
invention may be used for treating patients with neuroinflammation.
Neuroinflammation encapsulates the idea that microglial and astrocytic
responses and
actions in the central nervous system have a fundamentally inflammation-like
character, and that these responses are central to the pathogenesis and
progression of a
wide variety of neurological disorders. These ideas have been extended from
Alzheimer's disease to other neurodegenerative diseases (Eikelenboom et al.,
2002;
Ishizawa and Dickson, 2001), to ischemic/toxic diseases (Gehrmann et al.,
1995;
Touzani et al., 1999), to tumor biology (Graeber et al., 2002) and even to
normal
brain development. Neuroinflammation incorporates a wide spectrum of complex
51
CA 2869783 2019-09-05
=
cellular responses that include activation of microglia and astrocytes and
induction of
cytokines, chemokines, complement proteins, acute phase proteins, oxidative
injury,
and related molecular processes, and the events may have detrimental effects
on
neuronal function, leading to neuronal injury, further glial activation, and
ultimately
neuro degeneration.
D. Renal Diseases
In some embodiments, RTA 408, as well as polymorphic forms thereof, may
be used for treating patients with renal diseases and disorders, including
renal failure
and chronic kidney disease (CKD), based, for example, on the methods taught by
US 8,129,429. Renal failure, resulting
in
inadequate clearance of metabolic waste products from the blood and abnormal
concentrations of electrolytes in the blood, is a significant medical problem
throughout the world, especially in developed countries. Diabetes and
hypertension
are among the most important causes of chronic renal failure, also known as
chronic
kidney disease (CKD), but it is also associated with other conditions such as
lupus.
Acute renal failure may arise from exposure to certain drugs (e.g.,
acetaminophen) or
toxic chemicals, or from ischernia-reperfusion injury associated with shock or
surgical
procedures such as transplantation, and may result in chronic renal failure.
In many
patients, renal failure advances to a stage in which the patient requires
regular dialysis
or kidney transplantation to continue living. Both of these procedures are
highly
invasive and associated with significant side effects and quality of life
issues.
Although there are effective treatments for some complications of renal
failure, such
as hyperparathyroidism and hyperphosphatemia, no available treatment has been
shown to halt or reverse the underlying progression of renal failure. Thus,
agents that
can improve compromised renal function would represent a significant advance
in the
treatment of renal failure.
Inflammation contributes significantly to the pathology of CKD. There is also
a strong mechanistic link between oxidative stress and renal dysfunction. The
NF-x13
signaling pathway plays an important role in the progression of CKD as NF-KB
regulates the transcription of MCP-1, a chemolcine that is responsible for the
recruitment of monocytes/macrophages resulting in an inflammatory response
that
ultimately injures the kidney (Wardle, 2001). The Keapl /Nrf2/ARE pathway
controls
the transcription of several genes encoding antioxidant enzymes, including
home
52
CA 2869783 2020-04-08
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
oxygenase-1 (H0-1). Ablation of the Nrf2 gene in female mice results in the
development of lupus-like glomerular nephritis (Yoh et al., 2001).
Furthermore,
several studies have demonstrated that HO-1 expression is induced in response
to
renal damage and inflammation and that this enzyme and its products ¨
bilirubin and
carbon monoxide ¨ play a protective role in the kidney (Nath et al., 2006).
Acute kidney injury (AK1) can occur following ischemia-reperfusion,
treatment with certain pharmacological agents, such as cisplatin and
rapamycin, and
intravenous injection of radiocontrast media used in medical imaging. As in
CKD,
inflammation and oxidative stress contribute to the pathology of AKI. The
molecular
mechanisms underlying radiocontrast-induced nephropathy (RCN) are not well
understood; however, it is likely that a combination of events including
prolonged
vasoconstriction, impaired kidney autoregulation, and direct toxicity of the
contrast
media all contribute to renal failure (Tumlin et al., 2006). Vasoconstriction
results in
decreased renal blood flow and causes ischemia-reperfusion and the production
of
reactive oxygen species. HO-1 is strongly induced under these conditions and
has
been demonstrated to prevent ischemia-reperfusion injury in several different
organs,
including the kidney (Nath et al., 2006). Specifically, induction of HO-1 has
been
shown to be protective in a rat model of RCN (Goodman et al., 2007).
Reperfusion
also induces an inflammatory response, in part though activation of NF-KB
signaling
(Nichols, 2004). Targeting NF-KB has been proposed as a therapeutic strategy
to
prevent organ damage (Zingarelli et al., 2003).
E. Cardiovascular Disease
In some embodiments, RTA 408, the polymorphic forms and methods of this
invention may be used for treating patients with cardiovascular disease. The
etiology
of CV disease is complex, but the majority of causes are related to inadequate
or
completely disrupted supply of blood to a critical organ or tissue. Frequently
such a
condition arises from the rupture of one or more atherosclerotic plaques,
which leads
to the formation of a thrombus that blocks blood flow in a critical vessel.
In some incidences, atherosclerosis may be so extensive in critical blood
vessels that stenosis (narrowing of the arteries) develops and blood flow to
critical
organs (including the heart) is chronically insufficient. Such chronic
ischemia can
lead to end-organ damage of many kinds, including the cardiac hypertrophy
associated with congestive heart failure.
53
Atherosclerosis, the underlying defect leading to many forms of
cardiovascular disease, occurs when a physical defect or injury to the lining
(endothelium) of an artery triggers an inflammatory response involving the
proliferation of vascular smooth muscle cells and the infiltration of
leukocytes into the
affected area. Ultimately, a complicated lesion known as an atherosclerotic
plaque
may form, composed of the above-mentioned cells combined with deposits of
cholesterol-bearing lipoproteins and other materials (e.g., Hansson et al.,
2006).
Despite the significant benefits offered by current therapeutic treatments,
mortality
from cardiovascular disease remains high and significant unmet needs in the
treatment
of cardiovascular disease remain.
Induction of HO-1 has been shown to be beneficial in a variety of models of
cardiovascular disease, and low levels of HO-1 expression have been clinically
correlated with elevated risk of CV disease. RTA 408, the polymorphic forms
and
methods of the invention, therefore, may be used in treating or preventing a
variety of
cardiovascular disorders including but= not limited to atherosclerosis,
hypertension,
myocardial infarction, chronic heart failure, stroke, subarachnoid hemorrhage,
and
restenosis. In some embodiments, RTA 408, the polymorphic forms and methods of
the invention may be used as a combination therapy with other known
cardiovascular
therapies such as but not limited to anticoagulants, thrombolytics,
streptokinase, tissue
plasminogen activators, surgery, coronary artery bypass grafting, balloon
angioplasty,
the use of stents, drugs which inhibit cell proliferation, or drugs which
lower
cholesterol levels.
F. Diabetes
In some embodiments, RTA 408, as well as polymorphic forms thereof, may
be used for treating patients with diabetes, based, for example, on the
methods taught
by US 8,129,429.. Diabetes
is a complex
disease characterized by the body's failure to regulate circulating levels of
glucose.
This failure may result from a lack of insulin, a peptide hormone that
regulates both
the production and absorption of glucose in various tissues. Deficient insulin
compromises the ability of muscle, fat, and other tissues to absorb glucose
properly,
leading to hyperglycemia (abnormally high levels of glucose in the blood).
Most
commonly, such insulin deficiency results from inadequate production in the
islet
cells of the pancreas. In the majority of cases this arises from autoimmune
54
CA 2869783 2020-04-08
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
destruction of these cells, a condition known as type 1 or juvenile-onset
diabetes, but
may also be due to physical trauma or some other cause.
Diabetes may also arise when muscle and fat cells become less responsive to
insulin and do not absorb glucose properly, resulting in hyperglycemia. This
phenomenon is known as insulin resistance, and the resulting condition is
known as
type 2 diabetes. Type 2 diabetes, the most common type, is highly associated
with
obesity and hypertension. Obesity is associated with an inflammatory state of
adipose
tissue that is thought to play a major role in the development of insulin
resistance
(e.g., Hotamisligil, 2006; Guilherme et al., 2008).
Diabetes is associated with damage to many tissues, largely because
hyperglycemia (and hypoglycemia, which can result from excessive or poorly
timed
doses of insulin) is a significant source of oxidative stress. Because of
their ability to
protect against oxidative stress, particularly by the induction of HO-1
expression,
RTA 408, the polymorphic forms, and methods of the current invention may be
used
in treatments for many complications of diabetes. As noted above (Cai et al.,
2005),
chronic inflammation and oxidative stress in the liver are suspected to be
primary
contributing factors in the development of type 2 diabetes. Furthermore, PPAR7
agonists such as thiazolidinediones are capable of reducing insulin resistance
and are
known to be effective treatments for type 2 diabetes. In some embodiments,
RTA 408, the polymorphic forms, and methods of the current invention may be
used
as combination therapies with PPARy agonists such as thiazolidinediones.
G. Arthritis
In some embodiments, RTA 408, the polymorphic forms, and methods of this
invention may be used for treating patients with a form of arthritis. In some
embodiments, the forms of arthritis that could be treated with RTA 408 and the
polymorphic forms of this invention are rheumatoid arthritis (RA), psoriatic
arthritis
(F'sA), spondyloarthropathies (SpAs) including ankylosing spondylitis (AS),
reactive
arthritis (ReA), and enteropathic arthritis (EA), juvenile rheumatoid
arthritis (JRA),
and early inflammatory arthritis.
For rheumatoid arthritis, the first signs typically appear in the synovial
lining
layer, with proliferation of synovial fibroblasts and their attachment to the
articular
surface at the joint margin (Lipsky, 1998). Subsequently, macrophages, T cells
and
other inflammatory cells are recruited into the joint, where they produce a
number of
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
mediators, including the cytokines interleukin-1 (IL-1), which contributes to
the
chronic sequelae leading to bone and cartilage destruction, and tumor necrosis
factor
(TNE-a), which plays a role in inflammation (Dinarello, 1998; Arend and Dayer,
1995; van den Berg, 2001). The concentration of IL-1 in plasma is
significantly
higher in patients with RA than in healthy individuals and, notably, plasma 1L-
1 levels
correlate with RA disease activity (Eastgate et al., 1988). Moreover, synovial
fluid
levels of IL-1 are correlated with various radiographic and histologic
features of RA
(Kahle et al., 1992; Rooney et al., 1990).
Other forms of arthritis include psoriatic arthritis (PsA), which is a chronic
inflammatory arthropathy characterized by the association of arthritis and
psoriasis.
Studies have revealed that PsA shares a number of genetic, pathogenic and
clinical
features with other spondyloarthropathies (SpAs), a group of diseases that
comprise
ankylosing spondylitis, reactive arthritis and enteropathic arthritis (Wright,
1979).
The notion that PsA belongs to the SpA group has recently gained further
support
from imaging studies demonstrating widespread enthesitis in PsA but not RA
(McGonagle et al., 1999; McGonagle et al., 1998). More specifically,
enthesitis has
been postulated to be one of the earliest events occurring in the SpAs,
leading to bone
remodeling and ankylosis in the spine, as well as to articular synovitis when
the
inflamed entheses are close to peripheral joints. Increased amounts of TNE-a.
have
been reported in both psoriatic skin (Ettehadi et al., 1994) and synovial
fluid (Partsch
et al., 1997). Recent trials have shown a positive benefit of anti-TNF
treatment in
both PsA (Mease et al., 2000) and ankylosing spondylitis (Brandt et al.,
2000).
Juvenile rheumatoid arthritis (JRA), a term for the most prevalent form of
arthritis in children, is applied to a family of illnesses characterized by
chronic
inflammation and hypertrophy of the synovial membranes. The term overlaps, but
is
not completely synonymous, with the family of illnesses referred to as
juvenile
chronic arthritis and/or juvenile idiopathic arthritis in Europe.
Polyarticular JRA is a distinct clinical subtype characterized by inflammation
and synovial proliferation in multiple joints (four or more), including the
small joints
of the hands (Jarvis, 2002). This subtype of JRA may be severe, because of
both its
multiple joint involvement and its capacity to progress rapidly over time.
Although
clinically distinct, polyarticular JRA is not homogeneous, and patients vary
in disease
manifestations, age of onset, prognosis, and therapeutic response. These
differences
56
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
very likely reflect a spectrum of variation in the nature of the immune and
inflammatory attack that can occur in this disease (Jarvis, 1998).
Ankylosing spondylitis (AS) is a disease subset within a broader disease
classification of spondyloarthropathy. Patients affected with the various
subsets of
spondyloarthropathy have disease etiologies that are often very different,
ranging
from bacterial infections to inheritance. Yet, in all subgroups, the end
result of the
disease process is axial arthritis.
AS is a chronic systemic inflammatory rheumatic disorder of the axial
skeleton with or without extraskeletal manifestations. Sacroiliac joints and
the spine
are primarily affected, but hip and shoulder joints, and less commonly
peripheral
joints or certain extra-articular structures such as the eye, vasculature,
nervous system,
and gastrointestinal system may also be involved. The disease's etiology is
not yet
fully understood (Wordsworth, 1995; Calin and Taurog, 1998). The etiology is
strongly associated with the major histocompatibility class I (MHC I) HLA-B27
allele
(Calin and Taurog, 1998). AS affects individuals in the prime of their life
and is
feared because of its potential to cause chronic pain and irreversible damage
of
tendons, ligaments, joints, and bones (Brewerton et al., 1973a; Brewerton et
al.,
1973b; Schlosstein et al., 1973).
H. Ulcerative Colitis
In some embodiments, RTA 408, the polymorphic forms and methods of this
invention may be used for treating patients with ulcerative colitis.
Ulcerative colitis is
a disease that causes inflammation and sores, called ulcers, in the lining of
the large
intestine. The inflammation usually occurs in the rectum and lower part of the
colon,
but it may affect the entire colon. Ulcerative colitis may also be called
colitis or
proctitis. The inflammation makes the colon empty frequently, causing
diarrhea.
Ulcers form in places where the inflammation has killed the cells lining the
colon and
the ulcers bleed and produce pus.
Ulcerative colitis is an inflammatory bowel disease (IBD), the general name
for diseases that cause inflammation in the small intestine and colon.
Ulcerative
colitis can be difficult to diagnose because its symptoms are similar to other
intestinal
disorders and to another type of IBD, Crohn's disease. Crohn's disease differs
from
ulcerative colitis because it causes inflammation deeper within the intestinal
wall.
Also, Crohn's disease usually occurs in the small intestine, although the
disease can
57
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
also occur in the mouth, esophagus, stomach, duodenum, large intestine,
appendix,
and anus.
I. Crohn's Disease
In some embodiments, RTA 408, the polymorphic forms, and methods of this
invention may be used for treating patients with Crohn's disease. Crohn's
disease
symptoms include intestinal inflammation and the development of intestinal
stenosis
and fistulas; neuropathy often accompanies these symptoms. Anti-inflammatory
drugs, such as 5-aminosalicylates (e.g., mesalamine) or corticosteroids, are
typically
prescribed, but are not always effective (reviewed in Botoman et al., 1998).
Immunosuppression with cyclosporine is sometimes beneficial for patients
resistant to
or intolerant of corticostcroids (Brynskov et al., 1989).
In active cases of Crohn's disease, elevated concentrations of INF-a and IL-6
are secreted into the blood circulation, and INF-a, IL-1, IL-6, and IL-8 are
produced
in excess locally by mucosal cells (id.; Funakoshi et al., 1998). These
cytokincs can
have far-ranging effects on physiological systems including bone development,
hematopoiesis, and liver, thyroid, and neuropsychiatric function. Also, an
imbalance
of the IL-113/1L-lra ratio, in favor of pro-inflammatory IL-113, has been
observed in
patients with Crohn's disease (Rogler and Andus, 1998; Saiki et al., 1998;
Dionne et
al., 1998; but see Kuboyama, 1998).
Treatments that have been proposed for Crohn's disease include the use of
various cytokine antagonists (e.g., IL-lra), inhibitors (e.g., of 1L-113
converting
enzyme and antioxidants) and anti-cytokine antibodies (Rogler and Andus, 1998;
van
Hogezand and Verspaget, 1998; Reimund et al., 1998; Lugering et al., 1998;
McAlindon et al., 1998). In particular, monoclonal antibodies against TNF-ct
have
been tried with some success in the treatment of Crohn's disease (Targan et
al., 1997;
Stack et al., 1997; van Dullemen et al., 1995). These compounds may be used in
combination therapy with RTA 408, the polymorphic forms, and methods of the
present disclosure.
J. Systemic Lupus Erythematosus
In some embodiments, RTA 408, the polymorphic forms and methods of this
invention may be used for treating patients with SLE. Systemic lupus
erythematosus
(SLE) is an autoimmune rheumatic disease characterized by deposition in
tissues of
58
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
autoantibodies and immune complexes leading to tissue injury (Kotzin, 1996).
In
contrast to autoimmune diseases, such as MS and type 1 diabetes mellitus, SLE
potentially involves multiple organ systems directly, and its clinical
manifestations
are diverse and variable (reviewed by Kotzin and O'Dell, 1995). For example,
some
patients may demonstrate primarily skin rash and joint pain, show spontaneous
remissions, and require little medication. At the other end of the spectrum
are patients
who demonstrate severe and progressive kidney involvement that requires
therapy
with high doses of steroids and cytotoxic drugs such as cyclophosphamide
(Kotzin,
1996).
One of the antibodies produced by SLE, IgG anti-dsDNA, plays a major role
in the development of lupus glomerulonephritis (GN) (Hahn and Tsao, 1993;
Ohnishi
et al., 1994). Glomerulonephritis is a serious condition in which the
capillary walls of
the kidney's blood purifying glomeruli become thickened by accretions on the
epithelial side of glomerular basement membranes. The disease is often chronic
and
.. progressive and may lead to eventual renal failure.
K. Irritable Bowel Syndrome
In some embodiments, RTA 408, the polymorphic forms, and methods of this
invention may be used for treating patients with irritable bowel syndrome
(IBS). IBS
is a functional disorder characterized by abdominal pain and altered bowel
habits.
This syndrome may begin in young adulthood and can be associated with
significant
disability. This syndrome is not a homogeneous disorder. Rather, subtypes of
IBS
have been described on the basis of the predominant symptom--diarrhea,
constipation,
or pain. In the absence of "alarm" symptoms, such as fever, weight loss, and
gastrointestinal bleeding, a limited worlcup is needed.
Increasingly, evidence for the origins of IBS suggests a relationship between
infectious enteritis and subsequent development of IBS. Inflammatory cytokines
may
play a role. In a survey of patients with a history of confirmed bacterial
gastroenteritis (Neal et al., 1997), 25% reported persistent alteration of
bowel habits.
Persistence of symptoms may be due to psychological stress at the time of
acute
infection (Gwee et al., 1999).
Recent data suggest that bacterial overgrowth in the small intestine may also
have a role in IBS symptoms. In one study (Pimentel et al., 2000), 157 (78%)
of 202
IBS patients referred for hydrogen breath testing had test findings that were
positive
59
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
for bacterial overgrowth. Of the 47 subjects who had follow-up testing, 25
(53%)
reported improvement in symptoms (i.e., abdominal pain and diarrhea) with
antibiotic
treatment.
L. Sjogren's Syndrome
In some embodiments, RTA 408, the polymorphic forms, and methods of this
invention may be used for treating patients with Sjogren's syndrome. Primary
Sjogren's syndrome (SS) is a chronic, slowly progressive, systemic autoimmune
disease, which affects predominantly middle-aged women (female-to-male ratio
9:1),
although it can be seen in all ages including childhood (Jonsson et aL, 2002).
The
disease is characterized by lymphocytic infiltration and destruction of the
exocrine
glands, which are infiltrated by mononuclear cells including CD4+, CD8+
lymphocytes, and B-cells (Jonsson et al., 2002). In addition,
extraglandular
(systemic) manifestations are seen in one-third of patients (Jonsson et al.,
2001).
In other systemic autoimmune diseases, such as RA, factors critical for
ectopic
germinal centers (GCs) have been identified. Rheumatoid synovial tissues with
GCs
were shown to produce chemokines CXCL13, CCL21, and lymphotoxin (LT)-13
(detected on follicular center and mantle zone B cells). Multivariate
regression
analysis of these analytes identified CXCL13 and LT-(3 as the solitary
cytokines
predicting GCs in rheumatoid synovitis (Weyand and Goronzy, 2003). Recently
CXCL13 and CXCR5 in salivary glands has been shown to play an essential role
in
the inflammatory process by recruiting B and T cells, therefore contributing
to
lymphoid neogenesis and ectopic GC formation in SS (Salomonsson et al., 2002).
M. Psoriasis
In some embodiments, RTA 408, the polymorphic forms, and methods of this
invention may be used for treating patients with psoriasis. Psoriasis is a
chronic skin
disease of scaling and inflammation that affects 2 to 2.6 percent of the
United States
population, or between 5.8 and 7.5 million people. Psoriasis occurs when skin
cells
quickly rise from their origin below the surface of the skin and pile up on
the surface
before they have a chance to mature. Usually this movement (also called
turnover)
takes about a month, but in psoriasis turnover may occur in only a few days.
In its
typical form, psoriasis results in patches of thick, red (inflamed) skin
covered with
silvery scales. These patches, which are sometimes referred to as plaques,
usually
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
itch or feel sore. The plaques most often occur on the elbows, knees, other
parts of
the legs, scalp, lower back, face, palms, and soles of the feet, but they can
occur on
skin anywhere on the body. The disease may also affect the fingernails, the
toenails,
and the soft tissues of the genitals and inside the mouth.
Psoriasis is a skin disorder driven by the immune system, especially involving
a type of white blood cell called a T cell. Normally, T cells help protect the
body
against infection and disease. In the case of psoriasis, T cells are put into
action by
mistake and become so active that they trigger other immune responses, which
lead to
inflammation and to rapid turnover of skin cells.
N. Infectious diseases
In some embodiments, RTA 408, the polymorphic forms, and methods of the
present disclosure may be useful in the treatment of infectious diseases,
including
viral and bacterial infections. As noted above, such infections may be
associated with
severe localized or systemic inflammatory responses. For example, influenza
may
cause severe inflammation of the lung and bacterial infection can cause the
systemic
hyperinfiammatory response, including the excessive production of multiple
inflammatory cytokines, which is the hallmark of sepsis. In addition,
compounds of
the invention may be useful in directly inhibiting the replication of viral
pathogens.
Previous studies have demonstrated that related compounds such as CDDO can
inhibit the replication of HIV in macrophages (Vazquez et al., 2005). Other
studies
have indicated that inhibition of NF-kB signaling may inhibit influenza virus
replication, and that cyclopentenone prostaglandins may inhibit viral
replication (e.g.,
Mazur et al., 2007; Pica et al., 2000).
The present invention relates to the treatment or prevention of each of the
diseases/disorders/conditions referred to above in section IV using the
compound
RTA 408 or a pharmaceutically acceptable salt thereof, or a polymorphic form
of that
compound (such as, e.g., any one of the polymorphic forms described herein
above or
below), or a pharmaceutical composition comprising any of the aforementioned
entities and a pharmaceutically acceptable carrier (including, e.g., the
pharmaceutical
compositions described above).
61
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
V. Pharmaceutical Formulations and Routes of Administration
RTA 408 may be administered by a variety of methods, e.g., orally or by
injection (e.g., subcutaneous, intravenous, intraperitoneal, etc.). Depending
on the
route of administration, the active compounds may be coated in a material to
protect
the compound from the action of acids and other natural conditions which may
inactivate the compound. They may
also be administered by continuous
perfusion/infusion of a disease or wound site.
To administer RTA 408 by other than parenteral administration, it may be
necessary to coat the compound with, or co-administer the compound with, a
material
to prevent its inactivation. For example, the therapeutic compound may be
administered to a patient in an appropriate carrier, for example, liposomes,
or a
diluent. Pharmaceutically acceptable diluents include saline and aqueous
buffer
solutions. Liposomes include water-in-oil-in-water CGF emulsions as well as
conventional liposomes (Strejan et al., 1984).
RTA 408 may also be administered parenterally, intraperitoneally,
intraspinally, or intracerebrally. Dispersions can be prepared in glycerol,
liquid
polyethylene glycols, and mixtures thereof and in oils. Under ordinary
conditions of
storage and use, these preparations may contain a preservative to prevent the
growth
of microorganisms.
Sterile injectable solutions can be prepared by incorporating RTA 408 in the
required amount in an appropriate solvent with one or a combination of
ingredients
enumerated above, as required, followed by filtered sterilization.
Generally,
dispersions are prepared by incorporating the therapeutic compound into a
sterile
carrier that contains a basic dispersion medium and the required other
ingredients
from those enumerated above. In the case of sterile powders for the
preparation of
sterile injectable solutions, the preferred methods of preparation are vacuum
drying
and freeze-drying, which yields a powder of the active ingredient (i.e., the
therapeutic
compound) plus any additional desired ingredient from a previously sterile-
filtered
solution thereof.
RTA 408 may be rendered fully amorphous using a direct spray drying
procedure. RTA 408 can be orally administered, for example, with an inert
diluent or
an assimilable edible carrier. The therapeutic compound and other ingredients
may
also be enclosed in a hard or soft shell gelatin capsule, compressed into
tablets, or
62
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
incorporated directly into the patient's diet. For oral therapeutic
administration, the
therapeutic compound may be incorporated, for example, with excipients and
used in
the form of ingestible tablets, buccal tablets, troches, capsules including
hard or soft
capsules, elixirs, emulsions, solid dispersions, suspensions, syrups, wafers,
and the
like. The percentage of the therapeutic compound in the compositions and
preparations may, of course, be varied. The amount of the therapeutic compound
in
such therapeutically useful compositions is such that a suitable dosage will
be
obtained.
It is especially advantageous to formulate parenteral compositions in dosage
unit form for ease of administration and uniformity of dosage. Dosage unit
form as
used herein refers to physically discrete units suited as unitary dosages for
the patients
to be treated, each unit containing a predetermined quantity of therapeutic
compound
calculated to produce the desired therapeutic effect in association with the
required
pharmaceutical carrier. The specification for the dosage unit forms of the
invention
are dictated by and directly dependent on (a) the unique characteristics of
the
therapeutic compound and the particular therapeutic effect to be achieved, and
(b) the
limitations inherent in the art of compounding such a therapeutic compound for
the
treatment of a selected condition in a patient.
RTA 408 may also be administered topically to the skin, eye, or mucosa. In
some embodiments, the compound may be prepared in a lotion, cream, gel, oil,
ointment, salve, solution, suspension, or emulsion. Alternatively, if local
delivery to
the lungs is desired the therapeutic compound may be administered by
inhalation in a
dry-powder or aerosol formulation.
RTA 408 will typically be administered at a therapeutically effective dosage
sufficient to treat a condition associated with a given patient. For example,
the
efficacy of a compound can be evaluated in an animal model system that may be
predictive of efficacy in treating the disease in humans, such as the model
systems
shown in the examples and drawings.
The actual dosage amount of RTA 408 or composition comprising RTA 408
administered to a patient may be determined by physical and physiological
factors,
such as age, sex, body weight, severity of condition, the type of disease
being treated,
previous or concurrent therapeutic interventions, idiopathy of the patient,
and the
route of administration. These factors may be determined by a skilled artisan.
The
practitioner responsible for administration will typically determine the
concentration
63
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
of active ingredient(s) in a composition and appropriate dose(s) for the
individual
patient. The dosage may be adjusted by the individual physician in the event
of any
complication.
An effective amount typically will vary from about 0.001 mg/kg to about
1000 mg/kg, from about 0.01 mg/kg to about 750 mg/kg, from about 100 mg/kg to
about 500 mg/kg, from about 1.0 mg/kg to about 250 mg/kg, from about 10.0
mg/kg
to about 150 mg/kg in one or more dose administrations daily, for one or
several days
(depending of course of the mode of administration and the factors discussed
above).
Other suitable dose ranges include 1 mg to 10000 mg per day, 100 mg to 10000
mg
per day, 500 mg to 10000 mg per day, and 500 mg to 1000 mg per day. In some
particular embodiments, the amount is less than 10,000 mg per day with a range
of
750 mg to 9000 mg per day.
The effective amount may be less than 1 mg/kg/day, less than 500 mg/kg/day,
less than 250 mg/kg/day, less than 100 mg/kg/day, less than 50 mg/kg/day, less
than
25 mg/kg/day, or less than 10 mg/kg/day. It may alternatively be in the range
of
1 mg/kg/day to 200 mg/kg/day. In some embodiments, the amount could be 10, 30,
100, or 150 mg/kg formulated as a suspension in sesame oil as described below
in
Example Cl. In some embodiments, the amount could be 3, 10, 30 or 100 mg/kg
administered daily via oral gavage as described below in Examples C2 and C3.
In
.. some embodiments, the amount could be 10, 30, or 100 mg/kg administered
orally as
described below in Example C6. For example, regarding treatment of diabetic
patients, the unit dosage may be an amount that reduces blood glucose by at
least 40%
as compared to an untreated patient. In another embodiment, the unit dosage is
an
amount that reduces blood glucose to a level that is 10% of the blood glucose
level
of a non-diabetic patient.
In other non-limiting examples, a dose may also comprise from about 1 vg/kg
body weight, about 5 1..tg/kg body weight, about 10 pg/kg body weight, about
50 jig/kg body weight, about 100 mg/kg body weight, about 200 jig/kg body
weight,
about 350 jig/kg body weight, about 500 jig/kg body weight, about 1 mg/kg body
weight, about 5 mg/kg body weight, about 10 mg/kg body weight, about 50 mg/kg
body weight, about 100 mg/kg body weight, about 200 mg/kg body weight, about
350 mg/kg body weight, about 500 mg/kg body weight, to about 1000 mg/kg body
weight or more per administration, and any range derivable therein. In non-
limiting
examples of a derivable range from the numbers listed herein, a range of about
64
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
mg/kg body weight to about 100 mg/kg body weight, about 5 [ig/kg body weight
to
about 500 mg/kg body weight, etc., can be administered, based on the numbers
described above.
In certain embodiments, a pharmaceutical composition of the present
5 disclosure may comprise, for example, at least about 0.01% of RTA 408. In
other
embodiments, RTA 408 may comprise between about 0.01% to about 75% of the
weight of the unit, or between about 0.01% to about 5%, for example, and any
range
derivable therein. In some embodiments, RTA 408 may be used in a formulation
such
as a suspension in sesame oil of 0.01%, 0.1%, or 1% as described below in
Examples
F and G. In some embodiments, RTA 408 may be formulated for topical
administration to the skin or eye, using a pharmaceutically suitable carrier
or as a
suspension, emulsion, or solution in concentrations ranging from about 0.01%
to
10%. In some embodiments the concentration ranges from about 0.1% to about 5%.
The optimal concentration may vary depending upon the target organ, the
specific
preparation, and the condition to be treated.
Single or multiple doses of the agent comprising RTA 408 are contemplated.
Desired time intervals for delivery of multiple doses can be determined by one
of
ordinary skill in the art employing no more than routine experimentation. As
an
example, patients may be administered two doses daily at approximately 12 hour
intervals. In some embodiments, the agent is administered once a day. The
agent(s)
may be administered on a routine schedule. As used herein a routine schedule
refers
to a predetermined designated period of time. The routine schedule may
encompass
periods of time that are identical or that differ in length, as long as the
schedule is
predetermined. For instance, the routine schedule may involve administration
twice a
day, every day, every two days, every three days, every four days, every five
days,
every six days, a weekly basis, a monthly basis, or any set number of days or
weeks
there-between. Alternatively, the predetermined routine schedule may involve
administration on a twice daily basis for the first week, followed by a daily
basis for
several months, etc. In other embodiments, the invention provides that the
agent(s)
may be taken orally and that the timing of which is or is not dependent upon
food
intake. Thus, for example, the agent can be taken every morning and/or every
evening, regardless of when the patient has eaten or will eat.
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
VI. Combination Therapy
In addition to being used as a monotherapy, RTA 408 and the polymorphic
forms described in the present invention may also find use in combination
therapies.
Effective combination therapy may be achieved with a single composition or
pharmacological formulation that includes both agents, or with two distinct
compositions or formulations, administered at the same time, wherein one
composition includes RTA 408 or its polymorphic forms, and the other includes
the
second agent(s). The other therapeutic modality may be administered before,
concurrently with, or following administration of RTA 408 or its polymorphic
forms.
The therapy using RTA 408 or its polymorphic forms may precede or follow
administration of the other agent(s) by intervals ranging from minutes to
weeks. In
embodiments where the other agent and RTA 408 or its polymorphic forms are
administered separately, one would generally ensure that a significant period
of time
did not expire between the time of each delivery, such that each agent would
still be
able to exert an advantageously combined effect. In such instances, it is
contemplated
that one would typically administer RTA 408 or the polymorphic forms and the
other
therapeutic agent within about 12-24 hours of each other and, more preferably,
within
about 6-12 hours of each other, with a delay time of only about 12 hours being
most
preferred. In some situations, it may be desirable to extend the time period
for
treatment significantly, however, where several days (2, 3, 4, 5, 6 or 7) to
several
weeks (1, 2, 3, 4, 5, 6, 7 or 8) lapse between the respective administrations.
It also is conceivable that more than one administration of RTA 408 or its
polymorphic forms, or the other agent will be desired. In this regard, various
combinations may be employed. By way of illustration, where RTA 408 or its
polymorphic forms is "A" and the other agent is "B", the following
permutations
based on 3 and 4 total administrations are exemplary:
A/B/A B/A/B B/B/A A/A/B B/A/A A/B/B B/B/B/A B/B/A/B
A/A/B/B A/B/A/B A/B/B/A B/B/A/A B/A/B/A B/A/A/B B/B/B/A
A/A/A/B B/A/A/A A/B/A/A A/A/B/A A/B/B/B B/A/B/B B/B/A/B
Other combinations are likewise contemplated. Non-limiting examples of
pharmacological agents that may be used in the present invention include any
pharmacological agent known to be of benefit in the treatment of a cancer. In
some
embodiments, combinations of RTA 408 or its polymorphic forms with a cancer
66
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
targeting immunotherapy, gene therapy, radiotherapy, chemotherapeutic agent,
or
surgery are contemplated. Also contemplated is a combination of RTA 408 or its
polymorphic forms with more than one of the above mentioned methods including
more than one type of a specific therapy. In some embodiments, the
immunotherapy
can be other cancer targeting antibodies such as but not limited to
trastuzumab
(Herceptint), alemtuzumab (Campathg)), bevacizumab (Avastin0), cetuximab
(Eribituxt), and panitumumab (Vectibixt) or conjugated antibodies such as
ibritumomab tiuxetan (Zevaling)), tositumomab (Bexxarg), brentuximab vedotin
(Adcetrisg), ado-trastuzumab emtansine (KadcylaTm), or denileukin dititox
(Ontakt).
Furthermore, in some embodiments, RTA 408 or its polymorphic forms are
envisioned to be used in combination therapies with dendritic cell-based
irnmunotherapies such as Sipuleucel-T (Provengeg)) or adoptive T-cell
immunotherapies.
Furthermore, it is contemplated that RTA 408 or its polymorphic forms are
used in combination with a chemotherapeutic agent such as but not limited to
anthracyclines, taxanes, methotrexate, mitoxantrone, estramustine,
doxorubicin,
etoposide, vinblastine, carboplatin, vinorelbine, 5-fluorouraci1, cisplatin,
topotecan,
ifosfamidc, cyclophosphamide, epirubicin, gemcitabinc, vinorelbine,
irinotecan,
etoposide, vinblastine, pemetrexed, melphalan, capecitabine, and oxaliplatin.
In some
embodiments, RTA 408 or its polymorphic forms are used in combination with
radiation therapy including but not limited to the use of ionizing radiation.
In some
embodiments, the effects of the cancer therapeutic agent are synergistically
enhanced
through administration with RTA 408 and its polymorphic forms. In some
embodiments, combination therapies which included RTA 408 are used to treat
cancer, including, for example, prostate cancer. See, e.g., Example H below.
In some embodiments, the methods may further comprise (1) contacting a
tumor cell with the compound prior to contacting the tumor cell with the
second
chemotherapeutic agent, (2) contacting a tumor cell with the second
chemotherapeutic
agent prior to contacting the tumor cell with the compound, or (3) contacting
a tumor
cell with the compound and the second chemotherapeutic agent at the same time.
The
second chemotherapeutic agent may, in certain embodiments, be an antibiotic,
anti-
inflammatory, anti-neoplastic, anti-proliferative, anti-viral,
immunomodulatory, or
immunosuppressive. In other embodiments, the second chemotherapeutic agent may
be an alkylating agent, androgen receptor modulator, eytoskeletal disruptor,
estrogen
67
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
receptor modulator, histone-deacetylase inhibitor, HMG-CoA reductase
inhibitor,
prenyl-protein trans ferase inhibitor, retinoid receptor modulator,
topoisomerase
inhibitor, or tyrosine kinase inhibitor. In certain
embodiments, the second
chemotherapeutic agent is 5 -azac itidine, 5 -fluorouracil, 9-c is-retinoic
acid,
actinomycin D, alitretinoin, all-trans-retinoic acid, annamycin, axitinib,
belinostat,
bevacizumab, bexarotene, bosutinib, busulfan, capecitabine, carboplatin,
carmustine,
CD437, cediranib, cetuximab, chlorambucil, cisplatin, cyclophosphamide,
cytarabine,
dacarbazine, dasatinib, daunorub ic
in, decitabine, docetaxel, do lastatin-10,
doxifluridine, doxorubicin, doxorubicin, epirubicin, erlotinib, etoposide,
gefitinib,
gemcitabine, gemtuzumab ozogamicin, hexamethylmelamine, idarubicin,
ifosfamide,
imatinib, irinotecan, isotretinoin, ixabepilone, lapatinib, LBH589, lomustine,
mechloreth amine, melphal an, mercaptopurine, meth
otrexate, mitomycin,
mitoxantronc, MS-275, ncratinib, nilotinib, nitrosourea, oxaliplatin,
paclitaxcl,
plicamycin, procarbazine, semaxanib, semustine, sodium butyrate, sodium
phenylacetate, streptozotocin, suberoylanilide hydroxamic acid, sunitinib,
tamoxifen,
teniposide, thiopeta, tioguanine, topotecan, TRAIL, trastuzumab, tretinoin,
trichostatin A, valproic acid, valrubicin, vandetanib, vinblastine,
vincristine,
vindesine, or vinorclbine.
Additionally, combination therapies for the treatment of cardiovascular
disease utilizing RTA 408, polymorphic forms, and pharmaceutical compositions
of
the present disclosure are contemplated. For example, such methods may further
comprise administering a pharmaceutically effective amount of one or more
cardiovascular drugs in addition to RTA 408, polymorphic forms, and
pharmaceutical
compositions of the present disclosure. The cardiovascular drug may be but not
limited to, for example, a cholesterol lowering drug, an anti-hyperlipidemic,
a calcium
channel blocker, an anti-hypertensive, or an HMG-CoA reductase inhibitor. In
some
embodiments, non-limiting examples of cardiovascular drugs include amlodipine,
aspirin, ezetimibe, felodipine, lacidipine, lercanidipine, nicardipine,
nifedipine,
nimodipine, nisoldipine or nitrendipine. In other embodiments, other non-
limiting
examples of cardiovascular drugs include atenolol, bucindolol, carvedilol,
clonidine,
doxazosin, indoramin, labetalol, methyldopa, metoprolol, nadolol, oxprenolol,
phenoxybenzamine, phentolamine, pindolol, prazosin, propranolol, terazosin,
timolol
or tolazoline. In other embodiments, the cardiovascular drug may be, for
example, a
68
statin, such as atorvastatin, cerivastatin, fluvastatin, lovastatin,
mevastatin,
pitavastatin, pravastatin, rosuvastatin or simvastatin.
VII. Examples
The following examples are included to demonstrate preferred embodiments
of the invention. It should be appreciated by those of skill in the art that
the
techniques disclosed in the examples which follow represent techniques
discovered by
the inventor to function well in the practice of the invention, and thus can
be
considered to constitute preferred modes for its practice. However, those of
skill in
the art should, in light of the present disclosure, appreciate that many
changes can be
made in the specific embodiments which arc disclosed and still obtain a like
or similar
result without departing from the spirit and scope of the invention.
A. Synthesis of RTA 408 (63415)
0 H 0
0 H
OH a N3 b N,
NC NC
0
0
0 0 -
H
RTA 401 1 2
0 0
0
N1...ki<CH3
NC
NC H F F
NH2 d_
0 0
3 RTA 406 (63415)
Reagents and conditions: (a) (Ph0)2P0N3(DPPA), Et3N, toluene, 0 C for 5 min,
then
r.t. overnight, ¨94%; (b) benzene, 80 C for 2 h; (c) HC1, CH3CN, r.t. for 1
h; (d)
CH1CF2CO2H, DCC, DMAP, CH2C12, r.t. overnight, 73% from RTA 401 (4 steps).
Compound 1: To a solution of toluene (400 mL), RTA 401 (which can be
prepared according to the methods taught, for example, by Honda, etal., 1998;
Honda
et al., 2000b; Honda et al., 2002; Yates et al., 2007; and U.S. Patents
6,326,507 and
6,974,801), (20.0 g, 40.6 mmol) and
Et3N (17.0 mL, 122.0 mmol) were added into a reactor and cooled to 0 C with
stirring. Diphenyl phosphoryl azide (DPPA) (13.2 mL, 61.0 mmol) was added with
stirring at 0 C over 5 min and the mixture was continually stirred at room
temperature overnight (HPLC-MS check shows no RTA 401 left). The reaction
69
CA 2869783 2019-09-05
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
mixture was directly loaded on a silica gel column and purified by column
chromatography (silica gel, 0% to 5% Et0Ac in CH2C12) to give compound 1 (19.7
g,
¨94%, partially converted into compound 2) as a white foam.
Compound 2: Compound 1 (19.7 g, ¨38.1 mmol) and benzene (250 mL) were
added into a reactor and heated to 80 C with stirring for 2 h (HPLC-MS check
shows
no compound 1 left). The reaction mixture was concentrated at reduced pressure
to
afford crude compound 2 as a solid residue, which was used for the next step
without
purification.
Compound 3: Crude compound 2 (<38.1 mmol) and CRICN (200 mL) were
added into a reactor and cooled to 0 C with stirring. HC1 (12 N, 90 mL) was
added
at 0 C over 1 min and the mixture was continually stirred at room temperature
for 1 h
(HPLC-MS check shows no compound 2 left). The reaction mixture was cooled to
0 C and 10% NaOH (-500 mL) was added with stirring. Then, saturated NaHCO3
(1 L) was added with stirring. The aqueous phase was extracted by Et0Ac
(2x500 mL). The combined organic phase was washed by H20 (200 mL), saturated
NaCl (200 mL), dried over Na2SO4, and concentrated to afford crude compound 3
(16.62 g) as a light yellow foam, which was used for the next step without
purification.
RTA 408: Crude amine 3 (16.62 g, 35.9 mmol), CH3CF2CO2H (4.7388 g, 43.1
mmol), and CH2C12 (360 mL) were added into a reactor with stirring at room
temperature. Then, dicyclohexylcarbodiimide (DCC) (11.129 g, 53.9 mmol) and 4-
(dimethylamino)-pyridine (DMAP) (1.65 g, 13.64 mmol) were added and the
mixture
was continually stirred at room temperature overnight (HPLC-MS check shows no
compound 3 left). The reaction mixture was filtered to remove solid by-
products and
the filtrate was directly loaded on a silica gel column and purified by column
chromatography (silica gel, 0% to 20% Et0Ac in Hexanes) twice to give compound
RTA 408 (16.347 g, 73% from RTA 401 over 4 steps) as a white foam: IFI NMR
(400 MHz, CD3C1) 6 8.04 (s, 1H), 6.00 (s, 1H), 5.94 (s, br, 1H), 3.01 (d, 1H,
J =
4.8 Hz), 2.75-2.82 (m, 1H), 1.92-2.18 (m, 4H), 1.69-1.85 (m, 7H), 1.53-1.64
(m, 1H),
1.60 (s, 3H), 1.50 (s, 3H), 1.42 (s, 3H), 1.11-1.38 (m, 3H), 1.27 (s, 3H),
1.18 (s, 3H),
1.06 (s, 3H), 1.04 (s, 3H), 0.92 (s, 3H); rniz 555 (M+1).
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
B. Pharmacodynamics
A summary of the in vitro and in vivo studies to evaluate the primary
pharmacodynamic effects of RTA 408 is provided below.
1. Effects of RTA 408 on Keapl-Nrf2 and NF-KB in Vitro
Inhibition of IFNy-induced NO production by AIMs is Nrf2-dependent
(Dinkova-Kostova, 2005). RAW264.7 mouse macrophages were plated in 96-well
plates at 30,000 cells/well in triplicated in RPM' 1640 supplemented with 0.5%
FBS
and incubated at 37 C with 5% CO2. On the next day, cells were pre-treated
with
DMSO (vehicle) or RTA 408 for 2 h, followed by treatment with 20 ng/mL of
mouse
IFNy for 24 h. Nitrite NO2) levels in the media were measured as a surrogate
for
nitric oxide using the Griess Reagent System (cat # G2930, Promega), according
to
the manufacturer's instructions, since nitrite is a primary, stable breakdown
product of
NO. Cell viability was assessed using the WST-1 Cell Proliferation Reagent
(cat #
11644807001, Roche Applied Science) according to the manufacturer's
instructions.
IC50 values were determined based on the suppression of IFN7-induced nitric
oxide
production normalized to cell viability. Treatment with RTA 408 resulted in a
dose-
dependent suppression of IFNy-induced NO production, with an average IC50
value of
3.8 1.2 nM. Results from a representative experiment are shown in FIG. 1.
The
IC50 value for RTA 408 was found to be 45%-65% lower than the IC50 values for
compounds 63170 (8 3 nM), 63171 (6.9 0.6 nM), 63179 (11 2 nm), and 63189
(7 + 2 nM). 63170, 63171, 63179, and 63189 are compounds of the formulas:
0
0
NC
0
63170
0
0
0
63171
71
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
0
0 F
NC
z
0
63179
0
0
NA
NC
z
0
63189.
2. Effect of RTA 408 on Nrf2 Target Genes
RTA 408 was tested in two different luciferase reporter assays to assess
activation of the ARE. The first luciferase reporter tested was under the
control of a
single ARE derived from the promoter of the human NQ01 gene, which allows for
quantitative assessment of the endogenous activity of the Nrf2 transcription
factor in
cultured mammalian cells. Expression of Firefly luciferase from NQ01-ARE
luciferase reporter plasmid is controlled by binding of Nrf2 to a specific
enhancer
sequence corresponding to the antioxidant response element (ARE) that was
identified
in the promoter region of the human NADPH:quinone oxidoreductase 1 (NQ01) gene
(Xie et al., 1995). The NQ01-ARE-luciferase reporter plasmid was constructed
by
inserting the human NQ01-ARE (5'-CAGTCACAGTGACTCAGCAGAATCTG-3')
into the pLuc-MCS vector using HindIII/Xhof cloning sites (GenScript Corp.,
Piscataway, NJ). The HuH-7 human hepatoma cell line, maintained in DMEM
(Invitrogen) supplemented with 10% FBS and 100 U/mL (each) of penicillin and
streptomycin, was transiently transfected using Lipofectamine 2000
(Invitrogen) with
the NQ01-ARE luciferase reporter plasmid and the pRL-TK plasmid, which
constitutively expresses Renilla luciferase and is used as an internal control
for
normalization of transfection levels. Thirty hours of transfection, cells were
treated
with RTA 408 for 18 h. Firefly and Renilla luciferase activity was assayed by
Dual-
Glo Luciferase Assay (cat # E2920, Promega), and the luminescence signal was
measured on an L-Max II luminometer (Molecular Devices). Firefly luciferase
activity was normalized to the Renilla activity, and fold induction over a
vehicle
72
control (DMSO) of normalized Firefly activity was calculated. FIG. 2a shows a
dose-
dependent induction of luciferase activity by RTA 408 in this cell line.
Values
represent the average of three independent experiments, Twenty percent less
RTA 408 (12 nM) than 63189 (14.9 nM) was required to increase transcription
from
the NQ01 ARE in HuH-7 cells by 2-fold. Likewise, 2.1-2.4 fold less RTA 408
than
63170 (25.2 nM) and 63179 (29.1 nM), respectively, was required to increase
transcription from the NQ01 ARE in HuH-7 cells by 2-fold.
The effect of RTA 408 on luciferase reporter activation was also assessed in
the AREc32 reporter cell line. This cell line is derived from human breast
carcinoma
MCF-7 cells and is stably transfected with a Firefly luciferase reporter gene
under the
transcriptional control of eight copies of the rat GSTA2 ARE sequence (Wang,
et al.,
2006). Following
treatment with RTA 408
for 18 h, Firefly lucifcrase activity was measured using the ONE-Glo
Luciferase
Assay System (Promega, Catalog #E6110) according to the manufacturer's
instructions. A dose-dependent response was observed in the AREc32 reporter
cell
line (FIG. 2b). A ¨2-fold induction of luciferase activity was evident
following
treatment with 15.6 nM RTA 408 in both the NQ01-ARE and GSTA2-ARE reporter
assay system. When looking at the results from the GSTA2-ARE (AREc32)
luciferase activity study,.the effects of 63415 (RTA 408) on GSTA2-ARE
induction
can be directly compared to that of RTA 402, 63170, 63171, 63179, and 63189
along
with WST1 viability studies (FIGS. 3a-f). Compared to the values of RTA 402,
63415 showed the quickest induction of GSTA2-ARE-mediated transcription of the
five comparison compounds with a concentration of 93 nM needed to reach 4-fold
induction in the luciferase reporter assay. All other compounds showed a
similar
induction only with much higher concentrations with 63170 needing a
concentration
of 171 nM, 63171 needing a concentration of 133 nM, 63179 needing a
concentration
of 303 nM and 63189 needing a concentration of 174 nM to achieve a 4 fold
induction
of luciferase activity. These values correspond to a 1.86 (63415), 3.40
(63170), 2.65
(63171), 6.05 (63179) and 3.47 (63189) fold increase in the amount of the
active
compound needed compared to RTA 402 to lead to the same amount of activity.
73
CA 2869783 2020-04-08
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
0
ON.
NC 0
0 z
RTA 402
RTA 408 was also shown to increase transcript levels of known Nrf2 target
genes in the HFL1 human fetal lung fibroblast and BEAS-2B human bronchial
epithelial cell lines. HFL1 cells were cultured in F-12K media supplemented
with
10% fetal bovine serum and 1% penicillin-streptomycin. BEAS-2B cells were
cultured in DMEM/F-12 media supplemented with 10% fetal bovine serum and 1%
penicillin-streptomycin. Cells were plated in 6-well dishes at a density of
2.5 x 105
cells/well. The following day, cells were treated with DMSO (vehicle) or RTA
408
(7.8, 15.6, 31.3, 62.5, or 125 nM) for 18 h. Each well received the same
amount of
vehicle. Following treatment, media was removed and cells were harvested using
RLT buffer (Qiagen). Lysates were homogenized using QIASIredder columns
(Qiagen, Catalog #79654) and RNA was isolated using RNeasy Mini kits (Qiagen,
Catalog #74104). For reverse transcription, RNA (1 p,g) was combined with
Oligo(dT)12-18 primer and H20 in a final volume of 23.25 p,L. The mixture was
heated to 70 C for 10 min and then placed on ice. A master mix containing 8 pL
5X
et strand buffer, 2 pL 1 mg/mL BSA, 2 pL 20 mM DTI, 4 p,L 5 mM dNTP mix, 0.25
pL RNaseOUTTm and 0.5 !AL Superscript II reverse transcriptase was added to
the
RNA mixture and incubated at 42 C for 1 h. The reaction was inactivated by
heating
to 70 C for 10 min. The reaction mixture was diluted 1:3 with H20 prior to use
in
qPCR. 2.5 pi., of the diluted reverse transcription reaction was combined with
one set
of PCR primers (0.36 !LIM final concentration), 2X iQn`i SYBR Green Supermix
(Bio-Rad, Catalog #170-8885) and H20 to a final volume of 20 !AL Sequences for
PCR primers are as follows: Glutamate-eysteine ligase, modifier subunit
(GCLAI),
forward primer 5'-GCTGTGGCTACTGCGGTATT-3' (SEQ ID NO: 1) reverse
primer 5'-ATCTGCCTCAATGACACCAT-3' (SEQ ID NO: 2); Henze oxygenase-1
(HMOX1) forward primer 5`-TCCGATGGGTCCITACACTC-3' (SEQ ID NO: 3),
reverse primer 5'-TAGGCTCCTTCCTCCTTTCC-3' (SEQ ID NO: 4); NAD(P)H
dehydrogenase, quinone 1 (NQ01) forward primer 5'-
AAAACACTGCCCTCTTGTGG-3' (SEQ ID NO: 5), reverse primer 5'-
74
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
GTGCCAGTCAGCATCTGGTA-3' (SEQ ID NO: 6); Ribosomal protein S9 (RPS9)
forward primer 5'-GATGAGAAGGACCCCACGGCGTCTG-3' (SEQ ID NO: 7),
reverse primer 5'-GAGACAATCCAGCAGCCCAGGAGGG-3' (SEQ ID NO: 8);
Thioredoxin Reductase 1 (TXNRDi) forward primer 5'-
ATTGCCACTGGTGAAAGACC-3' (SEQ ID NO: 9) , reverse primer 5'-
ACCAATTTTGTTGGCCATGT-3' (SEQ ID NO: 10). All primers had previously
been validated for specificity and amplification efficiency. cDNA was
amplified
using the following cycle conditions: (95 C for 3 min, 44 cycles of 95 C for
30 sec,
60 C for 15 sec, 72 C for 15 sec, followed by a melt curve of 55 C to 95 C
in
increments of 0.5 C). The relative abundance of each Nrf2 target gene was
determined using the comparative CT method (AACT). PCR reactions were run in
triplicate wells for each sample. Two independent experiments were performed
using
the conditions described above. Treatment of HFL1 lung fibroblasts with RTA
408
for 18 h resulted in increased expression of several Nrf2 target genes,
including
NQ01, HMOX1, GCLM, and TXNRD1, as measured by quantitative PCR (FIGS. 4a-
d). For all genes tested, induction by RTA 408 was dose-dependent and evident
at
concentrations as low as 15.6 nM. Treatment of BEAS-2B bronchial epithelial
cells
with RTA 408 for 18 h resulted in a similar dose-dependent increase of all
Nrf2 target
genes evaluated (FIGS. 5a¨d). RTA 408 also increased expression of Nrf2 target
genes in normal human mesangial cells (nHMC), the mouse BV2 microglial cell
line,
and the human SH-SY5Y neuroblastoma cell line at similar concentrations.
Protein levels of Nrf2 targets NQ01 and HMOX1 were measured in
SH-5Y5Y and BV-2 cells by Western blot following treatment with RTA 408. SH-
SY5Y cells were plated in 6-well plates at a density of 4 x 105 cells per
well. BV-2
cells were plated in 6-well plates at a density of 2.5 x 104 cells per well.
Twenty-four
(BV-2) or 48 (SH-SY5Y) 11 after plating, cells were treated with RTA 408 for
24
hours. Following treatment, cells were washed twice with cold PBS and
harvested in
lysis buffer. Cells were sonicated and debris was cleared by centrifugation
(10 min g
18,000 rcf, Beckman Coulter, microfuge 18 centrifuge). Total protein in
supernatant
was quantified using Bio-Rad protein reagent with BSA as a standard. Equal
amounts
of total cellular protein were separated on SDS-PAGE, and proteins were
transferred
to nitrocellulose membrane. Membranes were blocked for 1 h in TBST (lx TBS
with
0.1% Tween-20) containing 5% milk, washed 3 times with TBST, and incubated
with
primary antibodies overnight at 4 C. NQ01 antibody was from Abeam (#AB2346);
HMOX1 (H0-1) antibody was from Santa Cruz (#sc-10789); actin antibody was from
Millipore (#MAB 1501). After washing with TBST, secondary antibodies were
added
in TBST + 5% milk for 1 h at room temperature. AffiniPure goat anti-rabbit or
anti-
mouse IgG secondary antibodies were from Jackson ImmunoResearch (catalog #111-
035-144 and #115-035-146, respectively). Membranes were washed in TBST,
developed using ECL, and exposed to X-ray film. Treatment with RTA 408 also
increased NQ01 protein levels in SH-SY5Y cells in a dose-dependent manner
(FIG.
6a). HMOX1 protein was not detected in untreated or RTA 408-treated SH-SY5Y
cells. In BV2 cells, treatment with RTA 408 increased NQ01 and HMOX1 protein
levels at concentrations up to 125 nM (FIG. 6b). The EC50 value for induction
of
Nrf2 protein expression in SK-N-SH cells by RTA 408 (56.4 nM) was 45%-65%
lower than the EC50 values for 63171 (122 nM), 63189 (102 nM), and 63179 (126
nM). The same amount of 63170 (54.6 nM) was required.
The EC50 was measured using an in-cell western NQ01 assay where the cells
were incubated with the compound under evaluation for three days. After
incubation
with the compound of interest, the cells were reacted with mouse NQ01 antibody
and
then the next day the cells were reacted with IRDye-800CW-anti-mouse IgG
antibody. The target signals were visualized and then analyzed.
Consistent with induction of Nrf2 target genes and corresponding protein
products, treatment of RAW264.7 mouse macrophage cells for 24 h increased NQ01
enzymatic activity in a dose-dependent manner, with increases evident at 7.8
nM
(FIG. 7). NQ01 enzymatic activity was measured by a modified Prochaska assay
(Prochaska and Santamaria, Anal Biochem. 169:328-336, 1988.
Taken together, these data from multiple cell lines demonstrate that treatment
with RTA 408 increases transcriptional activity controlled by antioxidant
response
elements, increases expression of Nrf2 target genes, and increases the
activity of
NQOI, an Nrf2 target gene product.
3. Effect of RTA 408 on Markers of Cellular Redox Capacity
Glutathione and NADPH are critical factors required for the maintenance of
cellular redox capacity. Several genes involved in the synthesis of
glutathione (e.g.,
GCLC and GLCM) and NADPH [e.g., hexose-6-phosphate dehydrogenase (H6PD)
and malic enzyme 1 (MEI)] have been demonstrated to be regulated by Nrf2 (Wu,
76
CA 2869783 2019-09-05
2011). The effect of RTA 408 treatment on total glutathione levels was
evaluated in
the mouse AML-12 hepatocyte cell line using the GSH-GloTm Glutathione Assay
kit
(Promega, Catalog 0/6912) according to the manufacturer's instructions.
Treatment
of AML-12 cells for 24 h with RTA 408 increased total cellular glutathione
levels, in a
dose-dependent manner (FIG. 8). Data shown are representative of two
independent
experiments. A >2-fold increase in total glutathione was observed at RTA 408
concentrations as low as 15.6 nM. The ECK, value using a RAW264.7 mouse model
for induction of glutathione levels by RTA 408 (9.9 nM) was 22%-57% lower than
the ECso values for 63170 (12.1 nM), 63171 (23.2 nM), and 63189 (16 nM).
The effect of RTA 408 treatment on the levels of NADPH, as measured by the
absorbance of a redox-sensitive dye, WST-1 (Roche Applied Science, Catalog
#11644807001), was evaluated in HCT-116 cells. WST-1 absorbance is commonly
used to assess cell viability by measuring glycolytic production of NAD(P)H by
viable cells. Therefore, in situations where NADPH production increases in the
absence of any effect on cell viability, WST-1 absorbance also increases
(Berridge et
al., 1996). Several
key genes involved in
NADPH production have also been shown to be regulated by Nrf2 (Thimmulappa et
aL, 2002; Wu, et aL, 2011).
RTA 408 treatment for 24 h increased WST-1 absorbance in a dose-dependent
manner (FIG. 9), suggesting that NADPH levels were increased.
The effect of RTA 408 on the expression of genes involved in NADPH
synthesis pathways was also evaluated in this study. HCT-116 cells were
treated with
RTA 408 for 24 h, and mRNA levels of H6PD, phosphogluconatc dehydrogcnase
(POD), transketolase (TKT), and MEI were measured using quantitative PCR.
HCT-116 cells were plated in 6-well dishes at a density of 3 x 105 cells/
well. The
following day, cells were treated with DMSO (vehicle), 10 nM RTA 408, or 50 nM
RTA 408 for 24 h. Each well received the same amount of vehicle. Following
treatment, media was removed and cells were harvested using RLT buffer
(Qiagen).
Lysates were homogenized using QIAShredder columns (Qiagen, Catalog #79654)
and RNA was isolated using RNeasy Mini kits (Qiagen, Catalog #74104). For
reverse
transcription, RNA (1 1..tg) was combined with Oligo(dT)12-18 primer and H20
in a
final volume of 23.25 L. The mixture was heated to 70 C for 10 min and then
placed on ice. A master mix containing 8 I 5X 1st strand buffer, 2 L I mg/mL
BSA, 2 !AL 20 mM DTT, 4 L 5 mM dNTP mix, 0.25 L RNaseOUTTm and 0.5 L
77
CA 2869783 2020-04-08
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
Superscript II reverse transcriptase was added to the RNA mixture and
incubated at
42 C for 1 h. The reaction was inactivated by heating to 70 C for 10 min.
The
reaction mixture was diluted 1:3 with H20 prior to use in qPCR. 2.5 L of the
diluted
reverse transcription reaction was combined with one set of PCR primers (0.36
M
final concentration), 2X iQTM SYBRO Green Supermix (Bio-Rad, Catalog #170-
8885) and H20 to a final volume of 20 L. Sequences for PCR primers are as
follows: Ribosomal protein S9 (RPS9) forward primer 5'-
GATGAGAAGGACCCCACGGCGTCTG-3' (SEQ ID NO: 7), reverse primer 5'-
GAGACAATCCAGCAGCCCAGGAGGG-3' (SEQ ID NO: 8); Hexose-6-phosphate
dehydrogenase (H6PD) forward primer 5'-GAGGCCGTGTACACCAAGAT-3' (SEQ
ID NO: 11), reverse primer 5'-AGCAGTGGGGTGAAAATACG-3' (SEQ ID NO:
12), Phosphogluconate dehydrogenase (PGD) forward primer
5 '-
AAGGCACTCTACGCTTCCAA-3' (SEQ ID NO: 13), reverse primer 5'-
AGGAGTCCTGGCAGTTTTCA-3' (SEQ ID NO: 14), Transketolase (TKT) forward
primer 5'-CATCTCCGAGAGCAACATCA-3' (SEQ ID NO: 15), reverse primer 5'-
TTGTATTGGCGGCTAGTTCC-3' (SEQ ID NO: 16); Malie enzyme I (MEI)
forward primer 5'-TATATCCTGGCCAAGGCAAC-3' (SEQ ID NO: 17) reverse
primer 5'-GGATAAAGCCGACCCICTTC-3' (SEQ ID NO: 18). All primers had
previously been validated for specificity and amplification efficiency. cDNA
was
amplified using the following cycle conditions: (95 C for 3 min, 44 cycles of
95 C
for 30 sec, 60 C for 15 sec, 72 C for 15 sec, followed by a melt curve of 55
C to 95
C in increments of 0.5 C). The relative abundance of each target gene was
determined using the comparative CT method (AACT). PCR reactions were run in
triplicate wells for each sample. Two independent experiments were performed
using
the conditions described above. Treatment with RTA 408 resulted in a dose-
dependent increase in expression of genes involved in NADPH synthesis (FIGS.
10a-
d).
In summary, treatment with RTA 408 increased total glutathione levels in
AML-12 hepatocytes and increased WST-1 absorbance, a marker of NADPH
production, in HCT-116 cells. This observation correlated with an increase in
the
expression of several key genes encoding enzymes involved in NADPH synthesis.
78
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
4. Effect of RTA 408 on TNFa-induced NF-KB Signaling
NF-x13 is a transcription factor that plays a central role in the regulation
of
many immune and inflammatory responses. RTA 402 and other AIMs have been
shown to inhibit pro-inflammatory NF-x13 signaling in a variety of cell lines
(Shishodia, 2006; Ahmad, 2006; Yore, 2006). Using the mouse NIH3T3/NF-i<13-luc
cell line (Panomics), the effects of RTA 408 and the compounds 63171, 63179,
63170, and 63189 on the NF-x13-Luc reporter were explored. The NIH3T3/NF-KB-
luc cell line maintains a chromosomal integration of a Firefly luciferase
reporter
construct regulated by eight copies of the NF-x13 response element. The
effects of
these compounds can be quantified by measuring the value of the NF-KB IC5o.
RTA 408 showed a 1.2 M IC50, which when normalized for viability showed an
IC50
of 1.4 M. The other four compounds showed NIH3T3/1\1F-KB IC50 values of 1.7,
0.2, 1.1, and 1.1 M, which when viability normalized showed IC50 values of
1.8, 0.6,
1.1, and 1.0 M, respectively. RTA 408 and its effects on NF-x13 are plotted
as a
function of dosing and relative fold change as well as WST1 and WST1/2 are
shown
in FIGS. 1 la & b. The effect of RTA 408 on TNFa-induced NF-KB signaling was
evaluated in HeLa/NF-KB-Luc cells, a human cervical adenocarcinoma cell line
stably
transfected with a luciferase reporter construct under the control of multiple
NF-03
transcriptional response elements. HeLaINF-KB-Luc cells were pretreated for 1
h
with RTA 408, followed by treatment with TNF-a (10 ng/mL) for an additional 5
h.
After treatment, luminescence was measured, and the effect of RTA 408
pretreatment
on TNF-a-induced luciferase activity was determined. The average results and
standard deviations from three independent experiments are shown in FIG. 12.
RTA 408 dose-dependently inhibited TNF-a-induced NF-KB activation with an ICo
value of 517 83 nM. Similar results were observed in another NF-KB reporter
cell
line (A549/NF-KB-Luc) where RTA 408 inhibited TNF-a-induced NF-KB activation
with an 1050 value of 627 nM (range 614-649 nM). RTA 408 was 1.6-1.8 fold more
efficient at reducing expression from the NF-K13 promoter reporter in HeLaINF-
03-
Luc cells than 63189 (854 nM) and 63170 (953 nM), respectively. Further
experimentation with the human A549 cell line showed an IC50 for RTA 408 as
1.7iuM and a value that has been viability normalized to 1.7iuM. The IC50 of
RTA
408 showed similar activity to 63189, 63179, 63171, and 63170 which showed
1050
values of 1.1, 1.4, 2.0, and 1.0, respectively. When those values were
viability
79
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
normalized, the assay showed 1.2, 1.5, 2.1 and 1.1 uM IC50, respectively. The
fold
change for NF-KB as a function of RTA 408 concentration along with WST1 and
WST1/2 curves were plotted and are shown in FIGS. 13a & b.
The effect of RTA 408 on TNF-a-induced phosphorylation of IxBa, a key step
in activation of the NF-KB pathway, was also evaluated in HeLa cells. HeLa
cells
were pretreated with RTA 408 for 6 h, followed by treatment with TNF-a (20
ng/mL)
for 5 min. Total and phosphorylated levels of IkBa were evaluated by Western
blot.
Primary IxBa antibodies were from Santa-Cruz (sc-371), pIxBa antibody was from
Cell Signaling (9246), actin antibody was from Millipore (MAB 1501).
Peroxidase-
conjugated affini-pure Goat anti-Rabbit (IgG) and peroxidase-conjugated affini-
pure
Goat anti-Mouse IgG secondary antibodies were purchased from Jackson
ImmunoResearch. Protein blots were developed using ECL, and exposed to X-ray
film. Consistent with the results from the luciferase reporter assay, RTA 408
inhibited INF-a-induced phosphorylation of IxBa in a dose-dependent manner
(FIG. 14).
RTA 408 has also been demonstrated to inhibit other pro-inflammatory
signaling pathways, such as IL-6-induced signal transducer and activator of
transcription 3 (STAT3) phosphorylation and receptor activator of NF-KB ligand
(RANKL)-induced osteoclastogenesis. In HeLa cells, pretreatment with 1 !LEM
RTA
408 for 6 h inhibited phosphorylation of STAT3 induced by IL-6. STAT3 (124H6)
and phospho-STAT3 (Tyr705) monoclonal antibodies were from Cell Signaling
Technology. Peroxidase-conjugated Affini-pure Goat anti-Rabbit IgG and
Peroxidase-
conjugated Affini-pure Goat anti-Mouse IgG were from Jackson ImmunoResearch.
Osteoclastogenesis is a multi-step differentiation process that results from
the binding
of RANKL to its receptor, RANK, on cells of hematopoietic origin. This results
in
the activation of NF-KB and MAPK, which in turn increase transcription of
osteoclast-specific target genes, including tartrate-resistant acid
phosphatase (TRAP).
The effect of RTA 408 on RANKL-induced osteoclastogenesis was evaluated in the
mouse macrophage cell line RAW264.7. RAW 264.7 cells were plated in 24-well
plates at a density of 5,000 cells/well. The next day, cells were treated with
RTA 408
for 2 Ii and then treated with 50 ng/mL recombinant mouse RANKL (R&D systems).
The treated cells were incubated for four days to allow differentiation into
osteoclasts.
Differentiation into osteoclasts was assessed by measuring TRAP activity. In
brief,
90 uL of conditioned cell culture media was removed from each test well and
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
aliquoted into triplicate wells (30 L/well) of a 96-well plate. 170 L of
TRAP Assay
buffer (Kamiya Biomedical) was then added to each well and the plate was
incubated
at 37 C for 3 hours. Following the incubation, absorbance at 540 nm was
determined
using a Spectramax M2 plate reading spectrophotometer. RTA 408 dose-
dependently
inhibited RANKL-induced TRAP activity and the formation of osteoclasts, with
an
1050 of ¨5-10 nM.
5. Effect of RTA 408 on Expression of Genes Encoding
Transaminase Enzymes
Transaminase elevations were observed in the 28-day toxicity studies with
RTA 408 in rats and, to a much lower extent, in monkeys. Similar findings have
been
observed following oral administration of a related AIM (bardoxolone methyl)
in
humans (Pergola, 2011). One hypothesis for this effect is that AIMs directly
or
indirectly increase transaminasc gene expression in the absence of cellular
toxicity.
To assess whether treatment with RTA 408 affects transaminase mRNA levels,
mouse
AML-12 hepatocytes were treated with RTA 408 for 18 h, and the mRNA levels of
genes encoding transaminases were measured using quantitative PCR. AML-12
cells
were plated in 6-well culture dishes at 3 x 105 cells per well using 2 mL of
media per
well. The following day cells were treated with DMSO (vehicle) or 250 nM and
500 nM RTA 408 for 18 h at 37 C. Each well received 0.1% DMSO. Three
independent replicate experiments were performed. Following treatment, media
was
removed and cells were harvested using RLT buffer (Qiagen). Lysates were
homogenized using QIAShredder columns (Qiagen, Catalog #79654) and RNA was
isolated using RNeasy Mini kits (Qiagen, Catalog #74104). For reverse
transcription,
RNA (1 litg) was combined with Oligo(dT)12-18 primer and H2O in a final volume
of
23.25 L. The mixture was heated to 70 C for 10 min and then placed on ice. A
master mix containing 8 pL 5X 1st strand buffer, 2 litL 1 mg/mL BSA, 2 1_, 20
mM
DTT, 4 L 5 mM dNTP mix, 0.25 L RNaseOUTTm and 0.5 pi. Superscript II
reverse transcriptase was added to the RNA mixture and incubated at 42 C for
1 h.
The reaction was inactivated by heating to 70 C for 10 min. The reaction
mixture
was diluted 1:3 with H20 prior to use in qPCR. 2.5 L of the diluted reverse
transcription reaction was combined with one set of PCR primers (0.36 M final
concentration), 2X iQ'M SYBR Green Supermix (Bio-Rad, Catalog #170-8885) and
H2O to a final volume of 20 L. Sequences for PCR primers are as follows:
81
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
Ribosomal protein L19 (Rp119) forward primer 5'-
TCAGGCTACAGAAGAGGCTTGC-3' (SEQ ID NO: 19), reverse primer 5'-
ACAGTCACAGGCTTGCGGATG-3' (SEQ ID NO: 20); NAD(P)H dehydrogenase,
quinone 1 (Nqol) forward primer 5'-TCGGGCTAGTCCCAGTTAGA-3' (SEQ ID
NO: 21), reverse primer 5'-AAAGAGCTGGAGAGCCAACC-3' (SEQ ID NO: 22);
Glutamie pyruvie transaminase 1 (Gpt1 or Alt1) forward primer 5'-
CACGGAGCAGGTCTTCAACG-3 (SEQ ID NO: 23), reverse primer 5'-
AGAATGGTCATCCGGAAATG-3' (SEQ ID NO: 24); Glutamic pyruvie
transaminase 2 (Gpt2 or Alt2) forward primer 5'-CGCGGTGCAGGTCAACTACT-3'
(SEQ ID NO: 25), reverse primer 5'-CCTCATCAGCCAGGAGAAAA-3' (SEQ ID
NO: 26); Glutamate oxaloaeetate transaminase 1 (Gotl or Asti) forward primer
5'-
GGCTATTCGCTATTTTGTGT-3' (SEQ ID NO: 27), reverse primer 5'-
GACCAGGTGATTCGTACAAT-3' (SEQ ID NO: 28); Glutamate oxaloacetate
transaminase 2 (Got2 or Ast2) forward primer 5'-AGAGTCCTCTTCAGTCATTG-3'
(SEQ ID NO: 29), reverse primer 5'-ATGATTAGAGCAGATGGTGG-3' (SEQ ID
NO: 30). All primers had previously been validated for specificity and
amplification
efficiency. cDNA was amplified using the following cycle conditions: (95 C
for 3
min, 44 cycles of 95 C for 30 sec, 60 C for 15 sec, 72 C for 15 sec,
followed by a
melt curve of 55 C to 95 C in increments of 0.5 C). The relative abundance
of
each target gene was determined using the comparative CT method (AACT). PCR
reactions were run in triplicate wells for each sample. Treatment with RTA 408
increased mRNA levels of alanine transaminase 1 (Altl or Gptl) and aspartate
transaminase 1 (Astl or Gotl) (FIGS. 15a,c). RTA 408 had no effect on alanine
transaminase 2 (Alt2 or Gpt2) mRNA levels and reduced mRNA levels of aspartate
transaminase 2 (Ast2 or Got2) (FIGS. 15b,d). These results demonstrate that
RTA
408, at the concentrations tested (250 nM or 500 nM), affects transaminase
gene
expression in vitro.
6. Effect of RTA 408 on Levels of Glycolytic Intermediates
Studies in diabetic mice have demonstrated that bardoxolone methyl increases
muscle-specific insulin-stimulated glucose uptake (Saha, 2010). In humans, a
higher
percentage of patients receiving bardoxolone methyl reported experiencing
muscle
cramps compared with patients receiving placebo (Pergola, 2011). Muscle spasms
have also been reported in diabetic patients following insulin administration,
82
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
suggesting a possible association with muscle glucose metabolism. The effect
of
RTA 408 on glycolytic metabolism was evaluated through the assessment of
lactate
and pyruvate levels in cultured rodent C2C12 muscle cells. To measure lactate
levels,
differentiated C2C12 myotubes were treated with 1 jiM or 2 iuM RTA 408 or
insulin
for 3 h at 37 C. Buffer was removed and saved for measurement of
extracellular
lactate levels. Cell debris was pelleted by centrifugation (10 min at 14,000
rpm) prior
to measurement of lactate. To measure intracellular lactate, cells were
suspended in
0.1% Triton X-100 in PBS and lysed by shearing with a 25 gauge needle. Cell
lysate
was centrifuged (10 min at 14,000 rpm, 4 C) and lactate was measured in the
supernatant. Intracellular and extracellular lactate was measured using the
Lactate
Assay Kit (BioVision, Catalog # K607-100). Similar to treatment with insulin,
treatment of differentiated C2C12 myotubes with 1 !LEM or 2 ILLM RTA 408 for 3
h
significantly increased intracellular and extracellular lactate levels in a
dose-
dependent manner.
To measure pyruvate levels, differentiated C2C12 myotubes were treated with
250 or 500 nM RTA 408 or 100 nM insulin for 18 h. Following drug treatment,
media
was removed and cells were washed with PBS. Cells were lysed in Pyruvate Assay
Buffer (Pyruvate Assay Kit, BioVision, Catalog # K609-100). Cell lysates were
centrifuged (10 min at 14,000 rpm, 4 C) and pyruvate levels were measured in
the
supernatant. Treatment of C2C12 differentiated myotubes with 250 nM or 500 nM
RTA 408 for 18 h also significantly (P < 0.0001, noted by asterisks) increased
intracellular pyruvate levels in a dose-dependent manner (FIG. 16). Together,
these
results demonstrate that RTA 408, at the concentrations tested, can affect
muscle
glycolytic intermediates in vitro; however, it is unclear how the results from
this in
vitro system at the RTA 408 concentrations tested relate to the potential
effects on
glucose metabolism at clinically-relevant dose levels in humans.
7. In Vitro Evaluation of RTA 408 Efflux by MRP-1
One of characteristic of a drug candidate is the compound's efflux ratio. The
efflux ratio measures how easily the compound is transported across a
membrane.
The MRP-1 protein, or the multidrug resistance-assistance protein 1, is one of
a
family of proteins which help to facilitate the transport of organic anions
and other
small molecules through cellular membranes. A larger efflux ratio typically
means
that the drug candidate is more readily transported out of the membrane and
less
83
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
available to modulate intracellular processes. Similar proteins also regulate
the
transport of compounds across the blood-brain barrier. The efflux ratio MRP-1
for
RTA 408 (1.3) was experimentally determined to be approximately ten-fold lower
than 63170 (10) and 63171 (11.2) and over 40-fold lower than 63179 (56.5) and
63189 (57.1). Without being bound by theory, RTA 408 may not be a good
substrate
for MRP-1 and/or a candidate for p-glycoprotein mediated efflux at the blood-
brain
barrier. In some embodiments, RTA 408 may be used for treating disorders of
the
central nervous system (CNS).
C. Protective Effects of RTA 408 in Animal Models of Lung Disease
RTA 408 was tested in several animal models of pulmonary disease to
evaluate its potential efficacy in the lung. For all studies, RTA 408 was
orally
administered daily in sesame oil at dose levels in the range of 3 to 150
mg/kg. In
most cases, RTA 408 was administered starting several days prior to the
induction of
the lung injury response.
1. LPS-induced Pulmonary Inflammation in Mice
RTA 408 was tested in two studies of LPS-induced pulmonary inflammation
in mice. In the first study, intended to be a preliminary dose-range finder,
RTA 408
(30, 100, or 150 mg/kg) was administered orally once daily for three days,
followed
by LPS administration 1 h after the final dose. Bronchoalveolar lavage fluid
(BALF)
was collected 20 h after LPS administration (21 h after the final dose of RTA
408)
and evaluated for levels of pro-inflammatory markers (i.e., IL-6, IL-12p40,
TNF-a,
and RANTES) using Luminexim technology. RTA 408 treatment resulted in a
significant reduction in IL-12p40 at all doses and in TNF-a at the 100 and 150
mg/kg
doses (FIG. 17). In the second study, RTA 408 (10, 30, or 100 mg/kg) was
administered daily for six days, followed by LPS administration 1 h after the
final
dose. In this study, significant decreases in body weight were observed at the
100 mg/kg dose level starting on Day 3. Significant reductions in TNF-a were
observed at the 10 mg/kg dose, and significant reductions in IL-12p40, TNF-a,
and
RANTES were observed at the 30 mg/kg dose (FIG. 18a). Further evaluation of
lungs
from mice in this study revealed meaningful engagement of relevant Nrf2 target
genes, including significant induction of NQ01 enzyme activity (by measurement
of
84
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
rate of reduction of 2,6-dicholorphenol-indophenol) and increases in total GSH
(GSH-
GloTM, Promega, Madison, WI) at 10 and 30 mg/kg (FIG. 18b).
2. Bleomycin-induced Pulmonary Fibrosis
The effect of RTA 408 was also evaluated in models of bleomycin-induced
pulmonary fibrosis in mice and rats. In the first preliminary study, RTA 408
(10, 30,
or 100 mg/kg) was administered to mice daily via oral gavage for 39 days, with
bleomycin challenge (intranasal) on day 10. On the last day of dosing, lung
tissue
was collected and histology was performed to evaluate the extent of
inflammation and
interstitial fibrosis. In this model, no statistically significant effects
were observed at
the RTA 408 doses tested (FIGS. 19a & b). Additional evaluation was performed
using a rat model of pulmonary fibrosis that has been extensively
characterized at the
Lovelace Respiratory Research Institute. In this study, rats were challenged
with
bleomycin or saline by intratracheal administration on day 0. Following the
challenge, animals received RTA 408 (3, 10, or 30 mg/kg) daily via oral gavage
for
28 days. Administration of the 30-mg/kg dose was stopped on day 14 due to
excessive dehydration and diarrhea in the animals. For the remaining animals,
bronchoalveolar lavage fluid was collected on day 28 for assessment of pro-
inflammatory infiltrates by flow cytometry, and lung tissue was analyzed for
hydroxyproline levels by LC-MS and histopathology. Challenge with bleomycin
sulfate induced a substantial release of neutrophils and an increase in
soluble collagen
in the BALF, as well as an increase in hydroxyproline in the lung. Treatment
with 3
and 10 mg/kg RTA 408 significantly suppressed polymorphonuclear (PMN) cell
infiltration into the lungs and also produced a meaningful reduction (-10%-
20%) in
hydroxyproline deposition (FIGS. 20a & b).
Importantly, histopathological evaluation revealed a significant decrease in
collagen deposition, as assessed by trichrome staining, in rats treated with
RTA 408.
Whereas bleomycin control animals primarily exhibited moderate staining,
animals
treated with 10 mg/kg RTA 408 had predominantly minimal to mild staining
(Table 2).
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
Table 2: Effect of RTA 408 on collagen deposition in rat lung as assessed by
intensity of trichrome staining
RTA 408 RTA 408 (10
Staining Intensity' Bleomycin Control
(3 mg,/kg) mg/kg)
Minimal 0 0 3
Mild 1 0 4
Moderate 7 7 1
a Values represent intensity of staining in animals with interstitial
trichrome staining
in areas of bleomycin-mduced lung alterations.
Further evaluation of lungs from rats in this study also revealed meaningful
engagement of relevant Nrf2 target genes as assayed by Quantigene Plex 2.0
Multiplex assay (Affymetrix, Santa Clara, CA) (FIG. 21). RTA 408 significantly
and
dose-dependently increased NQ01, Txnrd, Gsr, and Gst enzyme activity in the
lungs
of rats exposed to bleomycin, demonstrating Nrf2 activation by RTA 408 in this
disease setting. NQ01 enzyme activity was assessed by measuring the rate of
reduction of DCPIP. Txnrd, Gst, and Gst enzyme activities were measured using
commercially available kits from Cayman Chemical (Ann Arbor, MI).
3. Cigarette Smoke-induced COPD in Mice
RTA 408 was also tested in a mouse model of cigarette smoke-induced
COPD. Mice received RTA 408 (3, 10, or 30 mg/kg) daily via oral gavage for two
weeks and were exposed to cigarette smoke five days per week during the RTA
408
dosing period. At the end of the study, lung tissue and BALF were collected
for
analysis of inflammatory infiltrates and cytokines. In this experiment,
multiple-dose
administration of RTA 408 at doses as low as 3 mg/kg RTA 408 resulted in
significant suppression of pro-inflammatory cytokines, including KC
(functional
mouse homolog of human IL-8) and TNF-a as measured using LuminexTM
Technology. A summary of results from this study is presented in FIGS. 22a-e.
An
AIM analog (63355) was tested in the same study for comparison. 63355 is a
compound of the formula:
0
OH
NC
z
0
86
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
Further evaluation of lungs from mice in this study also revealed meaningful
engagement of relevant Nrf2 target genes (FIG. 23). NQ01 enzyme activity in
the
lung, measured as the rate of reduction of DCPIP, was significantly decreased
by
cigarette smoke exposure; administration of RTA 408 rescued this loss. Txnrd
enzyme activity was also induced by the 30 mg/kg dose of RTA 408. In general
Gsr
enzyme activity was not altered, and Gst enzyme activity was decreased with
treatment ¨ both of which were likely the consequence of a temporal response
for
these enzymes. Txnrd, Gst, and Gst enzyme activities were measured using
commercially available kits from Cayman Chemical (Ann Arbor, MI).
4. Ovalbumin-induced Asthma in Mice
The potential activity of RTA 408 was also evaluated in a pilot study in a
mouse model of ovalbumin-induced asthma. Mice were sensitized with an IP
injection of ovalbumin and aluminum hydroxide on Day 0 and Day 14 and
challenged
intranasally with ovalbumin in saline on Days 14, 25, 26, and 27. Mice
received
RTA 408 (3, 10, or 30 mg/kg) daily via oral gavage on Days 1-13 and 15-27.
Following sensitization and challenge with ovalbumin, vehicle-treated mice had
a
significant increase in the total number of leukocytes compared with positive
control
(dexamethasone)-treated mice. An increase in the number of T cells and B cells
was
also observed in the vehicle-treated mice. Treatment with RTA 408 at 30 mg/kg
significantly reduced the number and percentage of B cells within the airways.
RTA 408 (3 and 30 mg/kg) also significantly reduced the number of macrophages,
but
not the mean percentage of macrophages, detected in the airways. These
observations
are suggestive of potential efficacy in this model.
5. Effects of RTA 408 on LPS-induced Sepsis in Mice
Sepsis was induced on Day 0 with an IP injection of LPS (21 mg/kg), and
survival was followed until Day 4. RTA 408 (10, 30, or 100 mg/kg) was
administered
daily via oral gavage from Day -2 to Day 2. In the vehicle control group, 60%
of the
animals survived until Day 4 (higher than the ¨40% survival rate expected in
this
model). In the RTA 408 treatment groups, 80% of the animals in the 10 mg/kg
dose
group and 90% of the animals in the 30 mg/kg dose group survived until Day 4
(FIGS. 24c & d). For the 100 mg/kg dose group, 90% of the animals survived
until
Day 4, with only a single death occurring on Day 4. Although these RTA 408-
87
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
induced effects are indicative of profound efficacy in this model, the
relatively high
survival rate in the vehicle control group precluded a statistically-
significant
difference between the control and RTA 408-treated groups. Results obtained
using
the compound RTA 405 are also presented (FIGS. 24a & b). RTA 405 is a compound
.. of the formula:
0
NC
0
0 7_
6. Effects of RTA 408 against Radiation-Induced Oral Mucositis
Exposure to acute radiation directed to the buccal cheek pouch of hamsters
produces effects similar to those observed in oral ulcerative mucositis in
humans.
These effects include moderate to severe mucositis characterized by severe
erythema
and vasodilation, erosion of the superficial mucosa, and formation of ulcers.
A single
study was conducted to evaluate the effects of RTA 408 in this model. On Day
0,
each hamster was given an acute radiation dose of 40 Gy directed to the left
buccal
cheek pouch. RTA 408 (10, 30, or 100 mg/kg) was orally administered twice
daily
from Day -5 to Day -1, and Day 1 to Day 15. Beginning on Day 6 and continuing
until Day 28 on alternate days, oral mucositis was evaluated using a standard
6-point
scoring scale. Both the 30 and 100 mg/kg doses of RTA 408 caused a significant
reduction in the duration of ulcerative mucositis (FIG. 25). Furthermore, a
dose-
dependent decrease in the percentage of animals with mucositis scores >3 was
also
observed. However, administration of RTA 408 at 30 or 100 mg/kg caused
significant dose-dependent reductions in weight gain in irradiated hamsters.
Due to
weight loss in excess of 20%, two out of eight hamsters in the 100 mg/kg dose
group
were euthanized on Day 2.
7. Effect of RTA 408 on the Induction of Nrf2 Biornarkers in Vivo
As described above, a key molecular target of RTA 408 is Nrf2, a central
transcriptional regulator of antioxidative cellular protection. Activation of
Nri2
induces upregulation of a battery of cytoprotective genes, including NQ01,
enzymes
involved in GSH synthesis [i.e., glutamate-cysteine ligase catalytic and
modifier
88
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
subunits (Gcic and Gclm)], enzymes involved in detoxification (i.e.,
glutathione S-
transferases [Gsts]), and efflux transporters [i.e., multidrug
resistance¨associated
proteins (Mrps)]. Induction of these genes results in a coordinated cellular
effort to
protect against oxidative insult, highlighted by increased antioxidative
capacity,
induction of glutathione synthesis, and conjugation and export of potentially
harmful
molecules from the cell. In addition to the efficacy endpoints and Nrf2 target
gene
expression evaluated in the various animal models described above, the ability
of
RTA 408 to induce expression of Nrf2 target genes was also assessed using
tissues
collected from healthy RTA 408-treated mice, rats, and monkeys.
As part of the non-GLP 14-day toxicity studies of RTA 408 in mice, rats, and
monkeys, tissues were collected for the purposes of measuring mRNA and enzyme
activity levels of selected Nrf2 target genes. For mice and rats, liver
samples were
collected 4 h after the final dose on Day 14. For monkeys, blood (for PBMC
isolation), liver, lung, and brain tissue were collected 24 h after the final
dose on
Day 14. Enzyme activity for NQ01, Gst, and glutathione reductase (Gsr), as
described above, were measured in tissue homogenates. Levels of mRNA were
determined using Quantigene Plex 2.0 technology according to the
manufacturer's
protocol, which involves a hybridization-based assay using xMAP Luminex
magnetic beads for direct quantification of mRNA targets. In addition, RTA 408
concentrations were measured in plasma and tissues by LC/MS/MS methods on a
TQD mass spectrometer (Waters, Milford, MA).
RTA 408 generally increased the expression of various Nrf2 target genes in a
dose-dependent manner at doses of 10, 30, and 100 mg/kg (FIG. 26, FIG. 27a,
FIGS. 28a & b). Transcriptional upregulation of Nrf2 target genes by RTA 408
also
resulted in functional increases in the antioxidant response, as manifested by
dose-
dependent increases in NQ01, Gst, and Gsr enzyme activity in rodent liver, as
well as
monkey liver and lung (FIGS. 29a & b, FIGS. 30a & b, FIGS. 31a & b).
Furthermore,
in rodents, liver exposure of RTA 408 correlated with the level of enzyme
activity of
NQ01, the prototypical target gene for Nrf2 (FIG. 32b, FIG. 33b). In monkeys,
the
level of mRNA expression in PBMCs of both NQ01 and sulfiredoxin 1 (SRXN1)
correlated with plasma exposure to RTA 408 (FIGS. 37a & b). Overall, RTA 408
increased mRNA levels and activity of Nrf2 targets, and such increases
generally
correlated with tissue and plasma exposures, suggesting Nrf2 targets may serve
as
89
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
feasible biomarkers for Nrf2 activation (FIGS. 34a & b) and may be useful for
assessing pharmacological activity of RTA 408 in healthy human subjects.
D. Safety Pharmacology
A GLP-compliant safety pharmacology program was completed using
RTA 408. This included in vitro and in vivo (monkey) studies on the
cardiovascular
system, as well as studies on the respiratory system and central nervous
system in rats.
1. Evaluation of the Effects of RTA 408 on Cloned hERG
Channels Expressed in 11EK293 cells
This study was conducted to assess the effects of RTA 408 on the rapidly
activating inward rectifying potassium current (IK,) conducted by hERG (human
ether-a-go-go-related gene) channels stably expressed in the human embryonic
kidney
(HEK293) cell line. The effects of RTA 408 on the hERG-related potassium
current
were assessed using whole-cell patch clamp electrophysiology methods. RTA 408
was determined to have IC50 value of 12.4 M in a hERG QPatch_Kv11.1 assay.
This value was 2.5-3 fold higher than the values for 63170 (4.9 uM) and 63189
(3.8 uM), respectively. The RTA 408 IC50 value was similar to the 63171 value
(15.7 tiM).
2. Cardiovascular Evaluation of RTA 408 in the Cynomolgus
Monkey
A single study was conducted to evaluate the potential cardiovascular effects
of RTA 408 in conscious freely moving cynomolgus monkeys. The same four male
and four female cynomolgus monkeys were administered the vehicle (sesame oil)
and
RTA 408 at dose levels of 10, 30, and 100 mg/kg according to a Latin square
design,
with one animal/sex/treatment dosed each week followed by a 14-day washout
period
between administrations, until each animal received all treatments. Vehicle
and
RTA 408 were administered to all animals via oral gavage at a dose volume of
5 mL/kg.
Animals were instrumented with telemetry transmitters for measurement of
body temperature, blood pressure, heart rate, and electrocardiogram (ECG)
evaluation. Body temperature, systolic, diastolic, and mean arterial blood
pressure,
heart rate, and ECG parameters (QRS duration and RR, PR, and QT intervals)
were
monitored continuously from at least 2 h pre-dose until at least 24 h post-
dose. ECG
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
tracings were printed at designated time points from the cardiovascular
monitoring
data and were qualitatively evaluated by a board-certified veterinary
cardiologist.
Prior to the first administration on study, untreated animals were
continuously
monitored for cardiovascular endpoints for at least 24 h, and these data were
used in
the calculation of the corrected QT interval throughout the study.
Observations for morbidity, mortality, injury, and availability of food and
water were conducted at least twice daily for all animals. Clinical
observations were
conducted pre-dose, approximately 4 h post-dose, and following completion of
the
cardiovascular monitoring period. Body weights were measured and recorded on
the
day prior to each treatment administration.
RTA 408 at dose levels of 10, 30, and 100 mg/kg did not produce mortality,
adverse clinical signs, or result in meaningful changes in body weight, body
temperature, blood pressure, or qualitative or quantitative (PR, RR, QRS, QT
intervals) ECG parameters (FIG. 35; Table 45). In the 100 mg/kg dose group, a
small
(1.6% on average) but statistically significant increase in the corrected QT
interval
was observed; however, individual animal data did not show consistent
increases in
QTc that would indicate a test article related effect. Consequently, due to
the small
magnitude of change and lack of a consistent response in individual animals,
these
slight increases in QTc were not considered to be related to RTA 408
treatment.
.. Therefore, oral administration of RTA 408 produced no effects on
cardiovascular
function in cynomolgus monkeys at doses up to and including 100 mg/kg.
3. Neurobehavioral Evaluation of RTA 408 in Rats
The potential acute neurobehavioral toxicity of RTA 408 was evaluated in
rats. Three treatment groups of 10 male and 10 female CD [Crl:CD (SD)] rats
received RTA 408 at dose levels of 3, 10, or 30 mg/kg. One additional group of
10 animals/sex served as the control and received vehicle (sesame oil).
Vehicle or
RTA 408 was administered to all groups via oral gavage once on Day 1 at a dose
volume of 10 mL/kg.
Observations for morbidity, mortality, injury, and availability of food and
water were conducted twice daily for all animals. Observations for clinical
signs were
conducted prior to dosing on Day 1 and following each functional observational
battery (FOB) evaluation. FOB evaluations were conducted pre-dose (Day -1) and
at
91
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
approximately 4 and 24 h post-dose. Body weights were measured and recorded
pre-
dose on Day 1.
RTA 408 at doses of 3, 10, and 30 mg/kg did not produce mortality, adverse
clinical observations, or effects on any of the neurobehavioral measures
tested. Slight
decreases in body weight gain were observed approximately 24 h after dosing in
the
30 mg/kg group that may potentially be test article-related. With respect to
the basic
neurobehavioral endpoints evaluated in this study, RTA 408 did not produce any
adverse effects in rats at doses up to and including 30 mg/kg.
4. Pulmonary Evaluation of RTA 408 in Rats
The potential effect of RTA 408 on pulmonary function was evaluated in rats.
Three treatment groups of eight male and eight female CD [Crl:CD (SD)] rats
received RTA 408 at dose levels of 3, 10, or 30 mg/kg. One additional group of
8 animals/sex served as the control and received vehicle (sesame oil). Vehicle
or
RTA 408 was administered to all groups via oral gavage once on Day 1 at a dose
volume of 10 mL/kg.
Observations for mortality, morbidity, injury, and availability of food and
water were conducted twice daily for all animals. Clinical observations were
conducted prior to dosing, approximately 4 h post-dose, and following
completion of
the 8-h pulmonary monitoring period. Body weights were measured and recorded
on
the day of RTA 408 administration. Pulmonary function (respiratory rate, tidal
volume, and minute volume) was monitored for at least 1 h prior to dosing to
establish
a baseline and for at least 8 h post-dose.
RTA 408 at doses of 3, 10, and 30 mg/kg did not produce mortality, adverse
clinical observations, or effects on any of the pulmonary parameters
evaluated.
Therefore, with respect to the basic pulmonary endpoints evaluated in this
study,
RTA 408 did not produce any adverse effects in rats at doses up to and
including
mg/kg.
E. Nonclinical Overview
1. Pharmacokinetics
30 RTA 408 has been
investigated both in vitro and in vivo to assess its PK and
metabolism properties. In vitro studies have been conducted to determine RTA
408
plasma protein binding and blood/plasma partitioning, cytochrome P450 (CYP450)
92
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
inhibition and induction, and to identify metabolites formed by liver
microsomes of
mice, rats, monkeys, and humans. Data pertaining to the in vivo absorption and
distribution following repeated administration of RTA 408 has been obtained
primarily through monitoring of drug levels in plasma and select tissues from
toxicology studies. Sensitive and
selective liquid chromatography-mass
spectrometry-based bioanalytical methods (LC/MS/MS) have been used to measure
concentrations of RTA 408 in plasma, blood, and tissues with appropriate
accuracy
and precision. Measurements were performed on TQD and QToF mass spectrometers
(Waters).
a. Absorption
The absorption and systemic pharmacokinetic behavior of RTA 408 was
studied in mice, rats, and monkeys following single and repeated (daily) oral
administration. Following oral administration of a suspension formulation at
doses of
10 to 100 mg/kg, maximal concentrations were observed within 1 to 2 h in mice,
and
within 1 to 24 h in rats and monkeys. Systemic exposure to RTA 408 tended to
be
highest in rats, with lower levels observed in mice and monkeys. Estimates of
the
apparent terminal half-life of RTA 408 observed after oral administration were
generally in the 6- to 26-h range, though the apparent prolonged absorption
phase in
some instances precluded calculation of a definitive half-life estimate.
Systemic exposure to RTA 408 was generally similar in males and females.
Exposure to RTA 408 following repeated daily oral administration tended to be
slightly higher (<2-fold) than the exposure observed after a single dose.
Administration of RTA 408 over a dose range from 3 to 100 mg/kg in a
suspension
formulation generally resulted in dose-proportional increases in systemic
exposure.
However, administration of higher doses (100 to 800 mg/kg in monkeys; 500 to
2000 mg/kg in rats) did not result in similar increases in exposure,
suggesting
saturation of absorption at doses above 100 mg/kg. Following oral
administration of
an unoptimized (loose-filled) capsule formulation of RTA 408 (3 mg/kg) to
monkeys,
dose-normalized systemic exposure tended to be somewhat lower than that
observed
with a suspension formulation.
The absorption and systemic pharmacokinetic behavior of RTA 408 was
studied in rats using single and repeated topical administration. The
administration of
RTA 408 over a range of 0.01% to 3% showed lower plasma concentrations
relative
93
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
to similar oral dosing. The systemic exposure to RTA 408 generally increased
in a
dose dependent manner. The topical administration was formulated as a
suspension
in sesame oil.
Using rabbits, the ocular absorption and systemic pharmacokinetic behavior of
RTA 408 was evaluated. RTA 408 was administered topically to the eye once per
day
for five days. The ocular administration showed lower plasma concentration of
RTA
408 relative to when RTA 408 is administered orally (FIG. 36). The amount of
RTA
408 in the plasma even after five consecutive days showed only a small change
compared to the concentration after the first dose relative to when RTA 408
was
administered orally, where plasma concentrations were almost 100-fold higher
(FIG. 36).
h. Distribution
Plasma protein binding of RTA 408 was evaluated in mouse, rat, rabbit, dog,
minipig, monkey, and human plasma at RTA 408 concentrations of 10-2000 ng/mL
using ultracentrifugation methodology. RTA 408 was extensively bound to plasma
proteins. Plasma protein binding in the nonclinical species ranged from 93%
(mouse)
to >99% (minipig), with binding of 95% in the toxicology species (rat and
monkey)
and 97% in human. There was no evidence of concentration-dependent protein
binding in any species tested. Results from blood-to-plasma partitioning
experiments
indicate that RTA 408 tended to distribute primarily in the plasma fraction of
blood in
a linear manner, with blood:plasma ratios <1.0 for all species and all
concentrations
tested.
The distribution of RTA 408 into tissues has been investigated after oral
administration to mice, rats, and monkeys. In the 14-day non-GLP toxicity
studies,
select tissues (liver, lung, and brain) were collected at a single time point
(4 h for rat
and mouse; 24 h for monkey) after the final dose of the study was administered
and
were analyzed for RTA 408 content using LC/MS/MS. RTA 408 readily distributes
into lung, liver, and brain. In lung, RTA 408 concentrations at 4 h in mice
and rats
were similar to or slightly higher (<2-fold) than concentrations in plasma,
while at
24 h in monkeys, RTA 408 concentrations in lung were 6- to 16-fold higher than
plasma concentrations. A similar pattern was observed for brain. In contrast,
RTA 408 concentrations in liver were 5- to 17-fold higher than plasma for mice
and
rats at 4 h, and 2- to 5-fold higher than plasma at 24 h in monkeys.
94
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
The pharmacodynamic effects of RTA 408 in tissues were assessed in mice,
rats, and monkeys, by monitoring the induction of Nrf2 target genes in the
same
tissues collected for drug exposure from the 14-day toxicity studies.
Induction of
Nrf2 target genes by RTA 408 resulted in increases in the antioxidant response
as
manifested by dose-dependent increases in NQ01, glutathione S-transferase
(Gst),
and glutathione reductase (Gsr) enzyme activity in the examined tissues.
Enzyme
activities were measured as described above. Furthermore, in rodents, RTA 408
liver
content correlated with the level of enzyme activity for NQOI, the
prototypical target
gene for Nrf2. In monkeys, the level of mRNA expression in peripheral blood
mononuclear cells (PBMCs) for both NQ01 and sulfiredoxin 1 (SRXN1) correlated
with plasma exposure of RTA 408 (FIGS. 37a & b). Overall, RTA 408 induced
biomarkers of Nrf2 in rodents and monkeys, and such inductions generally
correlated
well with tissue and plasma exposure to RTA 408.
When RTA 408 was administered to rabbits via ocular topical administration,
the highest concentrations of the compound were found in the cornea, retina,
or iris
while the vitreous humor, aqueous humor, and plasma showed significantly lower
concentrations of RTA 408 (FIG. 38).
c. Metabolism
The metabolism of RTA 408 has been investigated after in vitro incubation of
RTA 408 for 60 min with liver microsomes from mice, rats, monkeys, and humans
in
the presence of a nicotinamide adenine dinucleotide phosphate (NADPH)-
regenerating system and a uridine diphosphate glucuronosyltransferase (UGT)
reaction mixture. Extensive turnover of RTA 408 was observed with primate
microsomes, with <10% of the parent molecule remaining at the end of the 60-
min
incubation in both monkey and human microsomes. In contrast, the extent of
metabolism was lower in rodent microsomes, with >65% of the parent molecule
remaining at the end of the incubation. The lack of available authentic
standards for
the various potential metabolites of RTA 408 precluded quantitative evaluation
of the
observed metabolites. From a qualitative perspective, a similar pattern of RTA
408
metabolites was observed across species, and included peaks with masses
consistent
with reduction and hydroxylation of RTA 408 as well as glucuronidation of RTA
408
or of its reduction/hydroxylation metabolites. No unique human metabolites
were
observed, with all peaks in the human microsome incubations also being
observed in
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
one or more of the preclinical species. In particular, based on in vitro
microsome
data, all human metabolites were present in rat or monkey, the selected rodent
and
non-rodent toxicity species.
d. Pharmacokinetic Drug Interactions
The potential for RTA 408 to inhibit cytochrome P450 (CYP450)-mediated
metabolism was evaluated using pooled human liver microsomes and standard
substrates for specific CYP450 enzymes. RTA 408 directly inhibited CYP2C8 and
CYP3A4/5 with K, values of approximately 0.5 !LIM for each enzyme. No
meaningful
inhibition was observed for the other enzymes tested (CYP1A2, CYP2B6, CYP2C9,
CYP2C19, or CYP2D6), with inhibition <50% at the highest concentration tested
(3 ttM). In addition, there was little or no evidence of metabolism-dependent
inhibition of any of the enzymes tested. Future studies investigating the
potential for
CYP3A4/5-mediated drug-drug interactions may be warranted based on these data,
and the potentially high concentrations that may be achieved locally in the
gastrointestinal (GI) tract after oral administration.
The potential for RTA 408 to induce CYP450 enzyme expression was
evaluated using cultured human hepatocytes. Under conditions where
prototypical
inducers caused the expected increases in CYP activity, RTA 408 (up to 3 pM)
was
not an inducer of CYP 1A2, CYP2B6, or CYP3A4 enzyme activity in cultured human
hepatocytes. Enzyme activity was measured by monitoring substrate conversion
of
phenacetin, bupropion, and testosterone for CYP1A2, CYP2B6, and CYP3A4,
respectively, in isolated microsomes.
F. Effects of RTA 408 on Acute Radiation Dermatitis
The effects of RTA 408 as a topical or oral preventative for acute radiation
dermatitis have been examined. Using male BALB/c mice, a 30 Gy dose of
radiation
was administered on day 0 (Table 3). The sesame oil vehicle or RTA 408 was
administered to the rats on day -5 to -1 and days 1 to 30. RTA 408 was
administered
both orally in 3, 10, and 30 mg/kg in sesame oil and topically in percentage
composition of 0.01%, 0.1%, and 1% in sesame oil. The dermatitis was blindly
evaluated every other day from day 4 to day 30. On day 12, the typical peak of
dermatitis was observed and 4 mice were sacrificed 4 hours after
administration of the
dose. The remaining mice were sacrificed on day 30 at 4 h post-dose. Plasma
was
96
CA 02869783 2014-10-06
WO 2013/163344 PCT/US2013/038064
collected on days 12 and 30 as well as irradiated skin samples for mRNA and
histological examination.
Table 3: Study Design for Acute Radiation Dermatitis Model
Number Radiation
Group of Treatment Treatment Schedule
(Day 0)
Animals
1 9 males Untreated
2 10 males 30 Gy Untreated
Vehicle Control
3 14 males 30 Gy Day -5 to -1 &
Day 1 to 30
(sesame oil)
RTA 408¨ 0.01% or
4 14 males 30 Gy Day -5 to -1 &
Day 1 to 30
3 mg/kg
RTA 408 ¨ 0.1% or
14 males 30 Gy Day -5 to -1 & Day 1 to 30
mg/kg
6 14 males 30 Gy RTA 408¨ 1%
orDay -5 to -1 & Day 1 to 30
30 mg/kg
In the test groups where the mice were treated with RTA 408, the incidence of
5 dermatitis
appeared to be slightly diminished in severity when RTA 408 was given in
either an oral or topical administration (FIGS. 39-42). Furthermore, curves
plotting
the average dermatitis clinic score for the test groups as a function of time
show some
change with the administration of RTA 408 either in oral or topical form from
the
untreated test groups (FIGS. 43-45) particularly in the case where RTA 408 was
given
10 through an oral
administration. Furthermore, as can be seen in Tables 4 and 5 below,
the percentage of mice suffering from dermatitis with a clinical score above 3
was
significantly lower for mice treated with RTA 408 through an oral
administration
while the percentage of mice suffering from dermatitis with a clinical score
above 2
was slightly lower for test groups who were given a topical administration of
RTA 408.
97
N
0
0.,
Table 4: Percentage of mice per testing group which scored above 2 in their
clinical dermatitis exam and given a topical treatment w
,
1--,
containing RTA 408
o,
w
Day Day Day Day Day Day
Day Day Day Day % animal- % animal-
4-
12 14 16 18 20 22 24 26
28 30 days >=2 days >=3
1 no radiation, untreated 0.0 0.0 0.0 0.0 0.0 0.0
0.0 0.0 0.0 0.0 0.0 0.0
2 irradiated, untreated 0.0 50.0 83.3 83.3 83.3
100.0 66.7 50.0 50.0 50.0 35.6 0.0
3 irradiated, sesame oil 21.4 45.0 60.0 50.0 40.0
40.0 0.0 0.0 0.0 0.0 16.6 0.0
4 irradiated, RTA 408- 0.01% 0.0 0.0 20.0
50.0 10.0 40.0 40.0 40.0 20.0 10.0 14.4 0.0
irradiated, RTA 408-0.1 % 7.1 10.0 20.0 80.0 60.0 40.0
30.0 10.0 0.0 0.0 16.3 0.0
0
6 irradiated, RTA 408-1.0% 10.7 20.0 10.0 70.0 30.0
10.0 0.0 0.0 0.0 0.0 9.7 0.0 2
2,1
.4
0
.
Table 5: Percentage of mice per testing group which scored above 3 in their
clinical dermatitis exam and given an oral treatment
c,
containing RTA 408
.
i,
% animal-
% Day 16 16 Day 18 Day 20 Day 22 Day 24 Day 26 Day 28
days >=2 days >=3
1 no radiation, untreated 0 0 0
0 0 0 0 0.0 0.0
2 irradiated, untreated 20 40 20 20 20 20
20 39.0 8.8
3 irradiated, sesame oil 35 50 40 30 20 0
0 45.6 10.9
4 irradiated, RTA 408-3 mg/kg 10 10 0 0 0
0 0 32.5 1.3 It
n
5 irradiated, RTA 408-10 mg/kg 10 25 30 0 0
0 0 33.8 4.1 1-3
6 irradiated, RTA 408-30 mg/kg 10 20 10 0 0
0 0 28.8 2.5 c7)
5
o
1-,
w
,
o
w
of:
0
4-
98
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
G. Effects of RTA 408 on Fractionated Radiation Dermatitis
Utilizing RTA 408 through topical administration, the effects of RTA 408
towards ameliorating the effects of fractionated radiation dermatitis were
measured.
Using Balb/c mice, RTA 408 in a topical preparation was administered to the
mice
daily from day -5 to day 30 in three doses ranging from 0.01% to 1%. The mice
were
irradiated on days 0-2 and 5-7 with six 10-Gy doses per day. Clinical
dermatitis
scores for the mice were evaluated blindly every two days from day 4 until the
end of
the study. In FIG. 46, the graph shows the change in the average clinical
score for
each group were plotted as a function of time. The graph shows a statistically
significant improvement in the scores for mice treated with 0.1% to 1% topical
formulations of RTA 408. Study and treatment parameters can be found in Table
6.
Table 6: Study Conditions for Fractionated Radiation-Induced Dermatitis
Number of Radiation Treatment
Group Treatment
Animals (Days 0-2, 5-7) Schedule
1 9 males Untreated
2 14 males 6x 10 Gy Untreated
Vehicle Control QD Days -5
3 18 males 6x 10 Gy
(sesame oil) to 30
4 18 males 6x 10 Gy RTA 408 ¨0.01% QD Days -5
to 30
5 18 males 6x 10 Gy RTA 408 ¨ 0.1% QD Days -5to 30
6 18 males 6x 10 Gy RTA 408¨ 1% QD Days -5to 30
By analyzing the average clinical scores that were shown in FIG. 46, an area
under
the curve (AUC) analysis was performed, which yielded the severity of the
dermatitis
relative to how long the dermatitis persisted. This AUC analysis allowed for
direct
comparison between the different groups of mice and the effect of the
different
percentage compositions of RTA 408 (FIG. 47 and Table 7). Administration of
topical RTA 408 formulations reduced Grade 2 and Grade 3 lesions from 60% and
33% when the mice were only exposed to the vehicle to 21% and 6% with RTA 408
at 1%, concentration, respectively. The other RTA composition showed some
activity
but was not as significant as that shown by the 1% formulation.
99
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
Table 7: Percentage of Dermatitis Score for Each Treatment Group
Group %Days 2 %Days > 3
No Rad, No Tx 0% 0%
Rad, No Tx 66% 31%
Rad, Sesame Oil 60% 33%
Rad, RTA 408 (0.01%) 54% 29%
Rad, RTA 408 (0.1%) 40% 13%
Rad, RTA 408 (1%) 21% 6%
H. Synergistic Effects of RTA 408 and Cancer Therapeutic Agents on Tumor
Growth
A study of the effects of RTA 408 used in combination with traditional
chemotherapeutic agents was carried out to determine the efficacy of the
potential
treatment. In vitro studies were carried out to determine the effects of RTA
408 on
two different prostate cancer cell lines, LNCaP and DU-145. As can be seen in
FIG. 48a, the treatment of the prostate cancer cell lines (LNCaP) in vitro
with
5-fluorouracil shows a statistically significant increase in cytotoxicity when
combined
with RTA 408 at doses from ranging from 0.125 to 0.5 M. Using the prostate
cell
line DU-145 and docetaxel, RTA 408 amplified the cytotoxicity of the
chemotherapeutic agent in a statistically significant fashion for dosing of
RTA 408
from 0.125 to 0.75 viM as shown in FIG. 48b. This evidence supports the
concept that
RTA 408 could act synergistically with cancer therapeutic agents and may be
used in
some embodiments to provide greater efficacy in treating cancer patients.
After the successful results of the in vitro assay, a pilot in vivo assay was
carried out using LNCaP/C4-2B and DU145 human prostate cancer engineered to
express firefly luciferase (hereafter referred to as C4-2B-Luc and DUI 45-Luc,
respectively). Of note, both of these cell lines grow in an androgen-
independent
fashion. Cells were cultured in RPMI 1640 supplemented with 10% FBS. Cells
were
harvested using TrypLE Express (Invitrogen) and washed in PBS and counted.
Cells
were reconstituted in PBS to arrive at a final concentration of 3 x 106 cells
per 30 p,L
(unless otherwise stated) and aliquoted in separate tubes. Growth factor-
reduced
Matrigel (BD Bioscience) was thawed overnight at +4 C and transferred into
the
tubes in 30 1õ1,1_, aliquots. The cell/Matrigel solutions were transferred to
the vivarium
and mixed right before injection at a 1:1 ratio. Each mouse (n = 1 per group
for a
total of three animals) received a single subcutaneous injection of the tumor
cells.
Tumors were pre-established for 4 weeks. Then, one animal was treated with RTA
100
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
408 (17.5 mg/kg, i.p.) once a day for 3 days (Days -3 to -1). On the following
day
(Day 0), the RTA 408 treated animals and one other animal were treated with a
single
dose of 18 Gy IR, localized to the pelvic region where the tumors were
implanted.
The mouse that was pre-treated with RTA 408 received three additional doses of
RTA 408 (17.5 mg/kg, i.p.), once every other day, over the following week. The
third
animal received no treatment and served as a positive control. Tumor
progression
was monitored weekly via live imaging. To detect luciferase-expressing tumor
cells,
mice were IP injected with D-Luciferin 5 min prior to imaging according to the
manufacturer's protocol (Caliper LifeScience). Prior to
imaging mice were
anesthetized by isoflurane inhalation and imaged on the IVIS Lumina XR system
(Caliper LifeScience). For standardization, minimal exposure time necessary to
image control tumor was determined and all animals were imaged under these
conditions. On Day 7, no apparent reduction in tumor size was visible in the
IR
treated animal compared to the control, whereas the animal receiving both RTA
408
and IR showed a smaller tumor image. On Day 14 and Day 21, the control animal
showed continued tumor development and growth while the animal treated with
ionizing radiation showed some improvement, most notably at Day 21. On the
other
hand, the animal treated with RTA 408 and ionizing radiation showed no
progression
from Day 7 to Day 14 and had no visible tumor on Day 21. The progress of the
tumor
per week can be seen in FIG. 49. Both the in vitro and in vivo data show that
RTA 408 appears to complement the activity of different cancer therapeutic
agents
thus increasing the agent's efficacy.
I. Effects of RTA 408 on a Model of Ocular Inflammation
A study of the effects of RTA 408 on ocular inflammation was carried out
using rabbits of the New Zealand albino strain. The rabbits were divided into
5
groups of 12 rabbits which were given three different concentrations of RTA
408
(0.01%, 0.1%, and 1%), Voltarene collyre at 0.1% and the vehicle (sesame
oil).
Each rabbit was given three instillations within 60 min before induction of
paracentesis and two instillations within 30 min after induction of
paracentesis. Each
instillation was 50 ILIL and given in both eyes. Aqueous humor for 6 animals
per time-
point was collected 30 min and again 2 h after induction of paracentesis. The
amount
of inflammation was determined by protein concentration in the aqueous humor.
As
shown in FIG. 50, RTA 408 showed a reduction in aqueous humor protein similar
to
101
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
that of the highest concentration of any of the other reference compounds
(MaxiDex
or mapracorat) at only 0.01% RTA 408 in the formulation. The effects of
increasing
concentration of RTA 408 appeared to be negligible as all concentrations of
RTA 408
appeared to show relatively similar effects within error in reducing aqueous
humor
protein concentration.
J. Polymorph Screen
A preformulation and polymorphism study was performed for compound
63415. As part of this study, a preliminary polymorphism program was carried
out
with the aim to identify the most stable anhydrous form at room temperature
and
possible hydrates with a reasonably high probability. A total of 30
crystallization
experiments, including phase equilibrations, drying experiments and other
techniques,
were carried out. All obtained solids were characterized by FT-Raman
spectroscopy.
All new forms were characterized by PXRD and TG-FTIR, and optionally by DSC
and DVS.
In addition, the amorphous form was prepared and characterized. Several
experiments using different techniques and approaches were carried out to
prepare the
amorphous form. The
amorphous form was characterized by FT-Raman
spectroscopy, PXRD, TG-FTIR, DSC, DVS, and Karl-Fischer titration. The
stability
of the amorphous form was tested at elevated humidity and temperature
conditions
over the course of four weeks.
1. Starting Material and Nomenclature
Two batches of 63415 were used as starting materials (Table 8). 63415 is also
referred to as PP415 in this disclosure. All samples received or generated
during this
project received a unique identification code of the form PP415-Px, where Px
refers to
the sample/experiment number (x = 1, 2, ..., n).
102
CA 02869783 2014-10-06
WO 2013/163344 PCT/US2013/038064
Table 8: Starting materials
Sample Material Amount Received
63415, batch #: 0141-66-1;
PP415-P1 5.0g March 25, 2011
MW = 554.7 g/mol, C33H44F2N203
63415, batch #: 2083-69-DC;
PP415-P40 5.0g May 27, 2011
MW = 554.7 g/mol, C331-144F2N203
2. Compound 63415, batch # 0414-66-1 (PP415-P1):
The Amorphous Form
The 63415, batch # 0414-66-1, starting material was characterized by
FT-Raman spectroscopy, PXRD, TG-FTIR, Karl-Fischer titration, 1-14-NMR, DSC,
DVS, and approximate solubility measurements. The results are summarized in
Table 9.
Table 9: Characterization of the 63415 starting material (PP415-P1)
Method Results
FT-
will be used as the reference
Raman
PXRD no sharp peak pattern, material is amorphous
TG-FTIR ¨0.9 wt.-% (-0.1 eq.) Et0H with traces of H20 from 25 C to 200 C,
decomposition at T > 290 C
Karl-
0.5 wt.-% H20
Fischer
1-1-1-NMR agrees with structure, ¨0.08 eq. Et0H
DSC heating scan: glass transition Tg = 152.7 C (ACp = 0.72 J/g C);
2nd heating scan: glass transition T, = 149.7 C (ACp = 0.45 J/g C)
DVS slightly hygroscopic; Am = +0.4% (50%¨*85% r.h.);
FT-Raman and PXRD unchanged
The FT-Raman spectrum (FIG. 58) will be used as the reference spectrum for
the starling material. PXRD (FIG. 59) shows no sharp peak pattern. The broad
halo
at ¨10-20 20 is characteristic for amorphous materials.
The TG-FTIR thermogram (FIG. 60) shows the gradual loss of ¨0.9 wt.-%
Et0H (i.e., ¨0.1 eq.) with traces of H20 between 25 and 200 C. Decomposition
starts at T > 290 C.
A water content of 0.5 wt.-% was determined by Karl-Fischer titration.
The 1H-NMR spectrum (FIG. 61) agrees with the structure and shows
¨0.08 eq. Et0H, in agreement with the TG-FTIR thermogram.
103
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
The DSC thermogram (FIG. 62) shows in a first heating scan a glass transition
of the amorphous material at Tg = 152.7 C (ACp = 0.72 J/g C). In a second
scan after
quench cooling, the glass transition occurs at Tg = 149.7 C (ACp = 0.45 J/g
C).
The DVS isotherm (FIG. 63) shows that a gradual mass loss of 1.0 wt.-%
occurred upon lowering the relative humidity from 50% r.h. to 0% r.h.;
equilibrium
was reached at 0% r.h. Upon increasing the relative humidity to 95% r.h. a
gradual
mass gain of 2.1 wt.-% (relative to the mass at 0% r.h.) occurred; equilibrium
was
reached at 95% r.h. Upon lowering the relative humidity from 95% r.h. to 50%
r.h.
the final mass was 0.2 wt-% below the starting mass. The mass increase of 0.4
wt-%
at 85% r.h. (relative to the starting mass) classifies the sample as slightly
hygroscopic.
The FT-Raman spectrum (FIG. 64) and PXRD pattern (FIG. 65) of the sample
after the DVS measurement are unchanged compared to the spectrum and pattern
of
the sample before the measurement.
The approximate solubility of the PP415-131 starting material was measured in
twelve solvents and four solvent mixtures at r.t. by manual dilution combined
with
visual observation (Table 10). Due to the experimental error inherent in this
method,
the solubility values are intended to be regarded as rough estimates and are
to be used
solely for the design of crystallization experiments. All solvent mixtures are
listed as
ratios by volume (v/v).
Table 10: Approximate solubility of the PP415-P1 (amorphous) starting
material
Solvent Solubility S Img/mL]
toluene S > 200
DCM S > 200
Et0Ac S > 210
acetone S > 230
MeCN S > 230
DMF S > 210
Me0H S < 210
Et0Ha 105 < S < 210
2PrOH 16 < S < 19
DEE S > 1 d
heptane S < 1
H20 S < 1
2PrOH / H20 (9:1)b 7.9 <S < 8.5
104
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
Solvent Solubility S Img/mLl
MeCN / FLO (2:3)c S< 1
Et0Ac / heptane (1:1)a 100 <S <200
toluene / DEE (1:1)a S >220
a observed precipitation after ¨ Id;
water activity a(H20) ¨ 0.7 at 25 C;
a water activity a(H20) > 0.9 at 50 C; d incomplete dissolution at first (S <
1), but solid
residue dissolved completely overnight (S> 1).
3. Compound 63415, batch # 2083-69-DC (PP415-P40): Class 2
63415, batch # 2083-69-DC, is a heptane solvate. This material (PP415-P40)
was characterized by PXRD and found to correspond to class 2 (FIG. 66).
Class 2 likely corresponds to isostructural, non-stoichiometric (<0.5 eq.)
solvates (of heptane, cyclohexane, isopropyl ether, 1-butanol, triethyl amine,
and
possibly other solvents, such as hexane and other ethers) with tightly bound
solvent.
The small peaks visible in the pattern of PP415-P40 at 7.9 020 and 13.8 020 do
not correspond to peaks of classes 3, 4, or 5. Their origin is not clear at
this point.
4. Chemical Stability of the Amorphous Form
The chemical stability of the amorphous form was investigated in different
solvents over the course of seven days.
Solutions/suspensions with a concentration of 1 mg/mL were prepared in four
organic solvents (acetone, Me0H, MeCN, Et0Ac) and three aqueous surfactant
media (1% aq. SDS, 1% aq. Tween 80, 1% aq. CTAB).
Four separate solutions/suspensions were prepared for each solvent,
equilibrated for 6 h, 24 h, 2 d, and 7 d and subsequently analyzed by HPLC.
The relative area-% obtained from the HPLC chromatograms are given in
Table 11. The compound seems to be somewhat unstable in the diluent (0.1%
formic
acid in Me(N); over the course of the sequence (i.e., within ¨24 hours) the
area-% of
a reference sample (PP415-P1, ran at the beginning and the end of the
sequence)
decreased from 99.9% to 99.3% at 254 nm and from 99.9% to 99.5% at 242 nm. Due
to this effect, the samples measured towards the end of the sequence (set up
in the
following order: 7 d, 2 d, 24 h, 6 h), might be affected and the obtained area-
% might
be underestimated.
105
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
Table 11: Chemical stability experiments with the amorphous form of 63415
(PP415-Plr
at 254 nm at 242 nm
Solvent 7d 2d 24h 6h 7d 2d 24h 6h
acetone 99.6% 99.6% 99.6% 99.6% 99.7% 99.7% 99.6% 99.6%
Et0Ac 99.8% 99.8% 99.8% 99.7% 99.8% 99.9% 99.8% 99.7%
Me0H 99.8% 99.8% 99.7% 99.8% 99.8% 99.9% 99.7% 99.8%
MeCN 97.7% 99.5% 99.3% 99.4% 97.4% 99.5% 99.3% 99.3%
Tween
1%b 97.7% 97.1% 95.1% 97.6% 98.7% 98.7% 96.2% 99.1%
SDS 1% 99.7% 99.6% 99.7% 99.7% 99.8% 99.7% 99.7% 99.7%
CTAB 1% 99.3% 99.4% 99.4% 99.6% 99.3% 99.4% 99.4% 99.7%
a at the third wavelength (210 nm), the signal intensity was weak and the
signal-to-noise ratio
large, thus integration was not carried out
b
suspensions, not all material dissolved for all time points
suspensions, not all solid dissolved for time points 24 h and 6 h
Decomposition >1% was observed for solutions in MeCN after seven days and
for suspensions in the 1% aqueous Tween 80 media (at all time points at 254 nm
and
after 24 h, 2 d, and 7 d at 242 nm).
5. Storage Stability of the Amorphous Form
To learn more about its fundamental properties and physical stability, the
amorphous form of 63415 was stressed by storage at elevated temperatures and
relative humidities.
Samples of the amorphous form (the PP415-P1 starting material) were stored
open at 25 C/-62% r.h. (over saturated aqueous solution of NH4NO3) and
40 C/-75% r.h. (over saturated aqueous solution of NaCl) and closed at 60 C
and
80 C (Table 12). At time points 0 w, 1 w, 2 w, and 4 w the samples were
examined
by PXRD and compared to the starting material, PP415-Pl.
Table 12. Storage stability experiments with the amorphous form of 63415
(PP415-P1)
Sample Conditions Time Point PXRD Result
PP415-P2a open, 25 'V / -62% r.h. 1 w amorphous
PP415-P2b open, 25 C / -62% r.h. 2 w amorphous
PP415-P2c open, 25 C / -62% r.h. 4 w amorphous
PP415-P3a open, 40 C / -75% r.h. 1 w amorphous
PP415-P3b open, 40 C / -75% r.h. 2 w amorphous
PP415-P3c open, 40 C / -75% r.h. 4 w amorphous
106
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
Sample Conditions Time Point PXRD Result
PP415-P4a closed, 60 C 1 w amorphous
PP415-P4b closed, 60 C 2 w amorphous
PP415-P4c closed, 60 C 4 w amorphous
PP415-P5a closed, 80 C 1 w amorphous
PP415-P5b closed, 80 C 2 w amorphous
PP415-P5c closed, 80 C 4 w amorphous
After one week (time point 1 w, FIG. 67), two weeks (time point 2 w,
FIG. 68), and four weeks (time point 4 w, FIG. 69) all four samples were still
amorphous, as the powder X-ray diffractograms show no differences compared to
the
starting material at time point 0 w.
6. Crystallization and Drying Experiments
a. Crystallization Experiments
Phase equilibrations, crystallizations from hot solutions, and evaporation
experiments were carried out starting from the amorphous form in order to
identify
with reasonably high probability the most stable anhydrous form at r.t. and
possible
hydrates. All obtained materials were characterized by FT-Raman spectroscopy;
selected samples were also characterized by PXRD.
The FT-Raman spectra were grouped into classes according to the similarity
of their peak positions. The original sample (PP415-P1, see Table 8) was
classified
along with the crystallization products. The spectra within a class, however,
are not
strictly identical, but similar. Small differences and peak shifts might
exist.
Considering the FT-Raman spectra alone, it is difficult to determine if the
spectra of
one class belong to the same polymorphic form.
The peaks in the PXRD patterns were determined and the patterns then
classified into clusters using the PANalytical X'Pert (Highscore Plus)
software.
These clusters identify patterns of a high similarity. However, small but
significant
differences exist within a cluster. Thus, the patterns within a cluster do not
necessarily correspond to the same polymorphs, but represent different forms
with
very similar molecular structures. The FT-Raman classes correspond in all
cases to
the PXRD clusters.
107
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
b. Suspension Equilibration Experiments
Suspension equilibration experiments were carried out in one solvent and
eleven solvent mixtures (Table 13). Suspensions of ¨100 mg of PP415-P1 in
0.2-2.0 mL of the selected solvents were prepared and shaken for 4-15 days at
22-24
C. The solids were recovered and characterized by FT-Raman spectroscopy; most
were characterized also by PXRD.
Table 13. Suspension equilibration experiments starting from the amorphous
form (PP415-P1)
Sample Solvent/Mixture FT-Raman class PXRD cluster
PP415-P6 2PrOH 3 3
PP415-P7 1:2 Et0Ac/heptane 2 2
PP415-P8 1:2 acetone/hexane 2 2
PP415-P9 1:3 toluene/DEE 2d
PP415-P10 1:3 Me0H/TBME 2 2
PP415-P11 1:2 MEK/cyclohexane 2d
PP415-P12 9:1 Et0H/H20a 3 3
PP415-P13 7:3 MeCI\T/H2Ob 4d
4
PP415-P14 ¨1:1 THF/H20e 5d
5
PP415-P29 1:2 Et0Ac/TEA 2 2
PP415-P31 9:1 PEG/H20 1 1
PP415-P35 7:3 MeCN/H20b 4d 4
water activities: a a(H20) ¨ 0.5 at 50 C; a(H20) ¨ 0.85 at 50 C; C a(H20)>
0.99 at 64 C;
d the spectrum contains solvent signals
e. Crystallizations from Hot Solutions
Hot solutions of PP415-P1 were prepared in one solvent and four solvent
mixtures (Table 14). Upon slow cooling to 5 C at a rate of ¨0.2 Kimin,
precipitation
was observed in three cases (-P20, -P21, -P24). In two cases (-P22, -P23) no
solid
precipitated, even after storage at 4-5 C for two days. Here, the solvent was
evaporated under N2 flow at r.t. The solids were recovered and characterized
by FT-
Raman spectroscopy and for those with spectra different from the amorphous
starting
material, FT-Raman class 1, also by PXRD.
108
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
Table 14: Slow cooling experiments starting from the amorphous form
(PP415-P1)
FT-Raman PXRD
Sample Solvent/Mixture Conditions
class cluster
PP415-P20 ¨2:1 acetone/H 0
2 55 C ¨> 5 C 3h
3
PP415-P21 ¨1:5 75 C ¨> 5 C 2 2
Et0H/cyclohexane
PP415-P22 ¨1:3 MeCN/toluene 75 C ¨> 5 Ca lb
PP415-P23 1:3 Et0Ac/dioxane 75 C ¨> 5 Ca lb
PP415-P24 1BuOH 75 C --> 5 C 2b 2
a no precipitation after slow cooling and stirring at 5 C for 2 days;
evaporated solvent under
N2 flow at r.t.
the spectrum contains solvent signals
d. Evaporation/Precipitation Experiments
Clear solutions of PP415-P1 were prepared in three solvent mixtures (Table
15). The solvents were then slowly evaporated at r.t. under ambient
conditions.
However, in two of the three experiments (-P15 and -P17) white solid
precipitated
before evaporation began. The obtained solids were examined by FT-Raman
spectroscopy and PXRD.
Table 15. Slow evaporation experiments with the amorphous form (PP415-P1)
Sample Solvent/Mixture FT-Raman class PXRD cluster
PP415-P15 1:2 DCM/1PE 2a 2
PP415-P16 1:2 Me0H/toluene 1'
PP415-P17 1:3 Et0Ac/heptane 2a 2
a the spectrum contains solvent signals
e. Drying Experiments
At least one sample of each class was dried under vacuum with the aim to
desolvate the solvates and to obtain non-solvated crystalline forms of 63415
(Table 16). The dried materials were characterized further by FT-Raman, PXRD,
and
TG-FTIR.
109
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
Table 16. Drying experiments carried out on samples obtained from the
crystallization experiments
Sample Starting Material (Class) Conditions Result
PP415-P18 PP415-P15 (2) r.t., 2-10 mbar, ¨2 h 2
PP415-P19 PP415-P17 (2) r.t., 2-10 mbar, ¨2 h 2
PP415-P25 PP415-P6 (3) r.t., ¨3 mbar, ¨5 d; 3
60 C, 5-10 mbar, 2x1 h;
40-50 C, 5-20 mbar, ¨1 d
PP415-P26 PP415-P13 (4) r.t., ¨3 mbar, ¨5 d; 4
60 C, 5-10 mbar, 2x1 h;
40-50 C, 5-20 mbar, ¨1 d
PP415-P27 PP415-P14 (5) r.t., ¨3 mbar, ¨5 d; la
60 C, 5-10 mbar, 2x1 h;
40-50 C, 5-20 mbar, ¨1 d
PP415-P28 PP415-P21 (2) r.t., ¨3 mbar, ¨5 d; 2b
60 C, 5-10 mbar, 2x1 h;
40-50 C, 5-20 mbar, ¨1 d
PP415-P30 PP415-P7 (2) 50-70 C, 1-10 mbar, 3 d 2
PP415-P32 PP415-P19 (2) 80 C, <1x10-3mbar,3 d 2b
PP415-P33 PP415-P25 (3) 80 C, <1x10-3mbar,3 d 3
PP415-P34 PP415-P28 (2) 80 C, <1x10-3mbar,3 d 2"
PP415-P36 PP415-P35 (4) 80 C, <1 x10_3 mbar, 3 d
PP415-P37 PP415-P35 (4) 80 C, N2 flow, 3 d
PP415-P44a PP415-P41 (5) 80 C, 100 mbar, 2 d la
PP415-P46a PP415-P45 (6) 80 C, 100 mbar, 4 d la
desolvation successful, significant reduction of solvent content, sample
mainly amorphous;
only few, broad peaks in PXRD
sample less crystalline, as indicated by broader peaks in PXRD
desolvation successful, significant reduction of solvent content, sample still
crystalline; no
change in structure
7. Characterization of New Forms (Classes)
a. Summary of New Classes
In addition to the amorphous form of 63415, four new crystalline forms were
obtained in this study (Table 17).
110
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
Table 17: Summary of obtained classes
Class Characteristics Result of Drying Experiments
Class 1 amorphous form
Class 2 isostructural solvates (e.g., heptane) drying unsuccessful
Class 3 isostructural solvates (e.g., ethanol) drying unsuccessful
drying successful, structure
Class 4 MeCN solvate & desolvated solvate
unchanged
Class 5 THF solvate drying
resulted in amorphous form
Class 2: Most crystallization experiments resulted in solid material of class
2.
These samples likely correspond to isostructural, non-stoichiometric (<0.5
eq.)
solvates (of heptane, cyclohexane, isopropyl ether, 1-butanol, triethylamine,
and
possibly hexane and other ethers, etc.) with tightly bound solvent molecules.
The
Raman spectra and PXRD patterns within this class are very similar to each
other,
thus the structures might be essentially identical with only small differences
due to the
different solvents that were incorporated.
Drying experiments on class 2 samples have not resulted in a crystalline, non-
solvated form. Even elevated
temperatures (80 C) and a high vacuum
(<1x10-3 mbar) could not remove the tightly bound solvent molecules
completely; a
solvent content of =2 wt.-% always remained. The crystallinity of these
samples is
reduced, but neither transformation into a different structure nor substantial
amorphization was observed.
Class 3: Solid material of class 3 was obtained from several crystallization
experiments. The samples of class 3 are likely isostructural solvates of
2PrOH, Et0H,
and probably acetone with tightly bound solvent molecules. They could
correspond
to either stoichiometric hemisolvates or non-stoichiometric solvates with a
solvent
content of ¨0.5 eq. As with class 2, the Raman spectra and PXRD patterns
within this
class are very similar to each other, indicating similar structures that
incorporate
different solvents.
Similar to class 2, drying experiments were also unsuccessful. The very
tightly bound solvent molecules could only partially be removed (-5.4 wt.-% to
¨4.8 wt.-%, up to 3 d at 1x103 mbar and 80 C). The PXRD pattern remained
unchanged.
Class 4: Material of class 4 was only obtained from a 7:3 MeCN/H20 solvent
system. It most likely corresponds to a crystalline acetonitrile hemisolvate.
111
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
By drying (under vacuum or N2 flow at elevated temperatures) most of the
solvent could be removed without changing or destroying the crystal structure
(PXRD
remained unchanged). Thus, a crystalline, non-solvated form (or rather
desolvated
solvate) was obtained. It is slightly hygroscopic (mass gain of ¨0.7 wt.-%
from
.. 50% r.h. to 85% r.h.) and has a possible melting point at 196.1 C (AH =
29.31 J/g).
Class 5: Class 5 was also obtained from only one solvent system
(-1:1 THF/H20) and contains bound THF (and maybe H20). As the content of the
two components cannot be quantified separately, the exact nature of this
crystalline
solvate cannot be determined.
Drying of class 5 resulted in complete desolvation and transformation into the
amorphous form (class 1). One possible process to prepare the amorphous form
from
class 2 material is a transformation of class 2 to class 5, followed by drying
and
amorphization.
h. Class 1 - The Amorphous Form
Class 1, the amorphous form of 63415, was obtained from a few
crystallization experiments (Table 18). Most crystallization experiments
resulted in
crystalline material of classes 2, 3, 4, or 5.
The starting material, PP415-P1, is amorphous and belongs to class 1. Further
experiments, exclusively aimed at preparing the amorphous form (class 1), were
carried out.
Table 18. Crystallization experiments resulting in solid material of class 1
Sample Method Solvent Characterization Drying
PP415-P31 susp. equil. 9:1 PEG/F20 Raman PXRD
PP415-P22 slow a ¨1:3 MeCNitoluene vis. obs.,
Raman
cooling
PP415-P23 slow cooling 1:3 Et0Acklioxanc vis. obs.,
Raman
PP415-P 16 evap./precip. 1:2 Me0H/toluene vis. obs., Raman
a no precipitation after slow cooling and stirring at 5 C for 2 days;
evaporated solvent under
N2 flow at r.t.
c. Class 2-
Isostructural Solvates (e.g., Heptane)
Most crystallization experiments resulted in solid material of class 2
(Table 19). In addition, one batch of a class 2 heptane solvate, PP415-P40,
was used
as starting material (see Table 8).
112
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
The FT-Raman spectra of class 2 are clearly similar to each other (FIG. 70)
but show small differences. They differ significantly from the spectrum of the
amorphous starting material, class 1 (FIG. 71) and from the spectra of classes
3, 4,
and 5 (FIG. 72).
The PXRD patterns of class 2 (FIG. 73) confirm the crystallinity of the
materials. The patterns of the samples are very similar to each other but show
small
differences (FIG. 74). The class 2 patterns differ clearly from the patterns
of
classes 3, 4, and 5 (FIG. 75).
The TG-FTIR thermogram of sample PP415-P7 (FIG. 76) shows the loss of
¨7.5 wt.-% Et0Ac and heptane in two steps from ¨100 C to 290 C and
decomposition at temperatures T > 290 C. Before the TG-FTIR experiments, the
samples were dried briefly (for ¨5 min) under vacuum (10-20 mbar) to remove
excess, unbound solvent. The loss of both Et0Ac and heptane occur together in
the
same temperature range; both solvents seem tightly bound within the structure.
The
theoretical Et0Ac content (b.p. = 76 C) of a hemisolvate is 7.4 wt.-%, the
theoretical
heptane content (b.p. = 98 C) of a hemisolvate is 8.3 wt.-%. Unfortunately,
the
content of the two components cannot be quantified separately.
The TG-FTIR thermogram of sample PP415-P21 (FIG. 77) shows the loss of
¨5.8 wt.-% cyclohexane in two steps from ¨140 C to ¨250 C and decomposition
at
temperatures T > 250 C. With the boiling point of cyclohexane at 81 C, the
solvent
seems tightly bound within the structure. The theoretical cyclohexane content
of a
hemisolvate is 7.1 wt.-%. Thus, sample PP415-P18 possibly corresponds to a non-
stoichiometric cyclohexane solvate (with <0.5 eq. solvent content).
The TG-FTIR thermogram of sample PP415-P24 (FIG. 78) shows the loss of
¨16.6 wt.-% 1BuOH in a step from ¨50 C to ¨160 C, further loss of 1BuOH
(6.6 wt.-%) in a second step from 160 C to 230 C and decomposition at
temperatures T > 230 C. With the boiling point of 1BuOH at 117 C, the
solvent of
at least the second step seems tightly bound within the structure. The
theoretical
1BuOH content of a hemisolvate is 6.3 wt.-%.
The TG-FTIR thermogram of sample PP415-P29 (FIG. 79) shows the loss of
¨5.1 wt.-% Et0Ac and TEA from -'-50 C to -'-220 C, most of it in a step from
180 C
to 210 C. Decomposition occurs at temperatures T > 220 C. The loss of both
Et0Ac and TEA occur together in the same temperature range; both solvents seem
113
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
tightly bound within the structure (with the boiling point of Et0Ac at 77 C
and of
TEA at 89 C).
The TG-FTIR thermogram of sample PP415-P47 (FIG. 80) shows the typical
two-step mass loss for class 2 (total of ¨7.9 wt.-% Et0Ac) at temperatures up
to
240 C, indicating very tightly bound solvent molecules.
The TG-FTIR thermogram of sample PP415-P48 (FIG. 81) shows the mass
loss of ¨3.5 wt.-% ethyl formate and water, at first gradually and then in a
clear step
between 180 C and 200 C. There might be further loss of ethyl formate
concomitant with the decomposition at T > 240 C.
Thus, the samples of class 2 might all correspond to non-stoichiometric
(<0.5 eq.), isostructural solvates with tightly bound solvent molecules. As
the Raman
spectra and PXRD patterns within this class are very similar to each other,
the
structures might be essentially identical to each other with only small
distortions of
the unit cell dimensions or small changes of atomic positions within the unit
cell, due
to the different sizes and shapes of the incorporated solvent molecules.
Table 19: Crystallization experiments resulting in solid material of class 2
Sample Method Solvent Characterization Drying
PP415-P7 susp. equil. 1:2 Et0Ac/heptane
Raman, PXRD,
Tti-FTIR
PP415-P8 susp. equil. 1:2 acetone/hexane
Raman, PXRD
PP415-P9 susp. equil. 1:3 toluene/DEE
Raman, PXRD
PP415-P10 susp. equil. 1:3 Me0H/TBIVIE
Raman, PXRD
PP415-P11 susp. equil. 1:2 Raman
MEK/cyclohexane
PP415-P29 susp. equil. Et0AciTEA Raman,
PXRD,
Tti-FTIR
PP415-P15 evap./precip. 1:2 DCM/1PE Raman, PXRD
PP415-P17 evap./precip. 1:3 Et0Ac/heptane
Raman, PXRD
Raman, PXRD,
PP415-P21 slow cooling ¨1:5
Et0H/cyclohexane TO-FT1R
Raman, PXRD,
PP415-P24 evap./precip. 1BuOH
TO-FT1R
PP415-F'43a evaporation (8:2) THF/hexane PXRD
PP415-P47a evaporation Et0Ac PXRD, TO-FT1R
PP415-P48a evaporation ethyl formate PXRD, TO-FT1R
a starting material: PP415-P40, class 2, in all other experiments PP415-P1,
class 1, was used
as starting material.
114
CA 02869783 2014-10-06
WO 2013/163344 PCT/US2013/038064
d. Diying Experiments on Samples of Class 2
Several samples of class 2 were dried under vacuum (and some at elevated
temperatures) and in an attempt to desolvate them with the aim to obtain an
anhydrous
form of 63415. Details and characterizations of the dried samples are provided
below
in Table 20.
However, even drying for three days at 80 C and a vacuum <1x10-3 mbar
could not remove the tightly bound solvent molecules completely; a solvent
content of
>2 wt.-% remained (see samples -P32 and -P34). The PXRD patterns show a
reduced
crystallinity of these samples, but no transformation into a different
structure was
observed.
Table 20. Drying experiments on class 2 samples
Starting Material Drying Dried Material
Solvent
Sample Solvent Content Conditions Sample Class
Content
PP415 PP415 7 P30 Et0Ac/heptane 50-70 C, heptane 2
-P (_7.5%) 1-10 mbar, 3d - (-2.5%)
IPE r.t., 2-10 mbar, IPE
PP415-P15 PP415-P18 2
(unknown) -2 h (-7.0%)
Et0Ac(?)/heptane r.t., 2-10 mbar, heptane
PP415-P17 PP415-P19 2
(unknown) -2 h (-7.6%)
heptane 80 C, <1 xle heptane
PP415-P19 PP415-P32
r.t., -3 mbar,
-5 d;
PP415-P21 PP415-P28
cyclohexane 60 C, 5-10 mbar, cyclohexane
(_5.8%) 2 x111; (-3.0%)
40-50 C,
5-20 mbar, -1 d
cyclohexane 80 C,<1x10- cyclohexane
PP415-P28 PP415-P34 2
(-3.0%) mbar,3 d (-2.3%)
a according to PXRD somewhat less crystalline
Thus, the class 2 solvates seem to have very tightly bound solvent molecules.
They are difficult to desolvate or transform/amorphize.
e. PP415-P7 ¨> PP415-P30
The solid material of sample PP415-P7, class 2, obtained from a suspension
equilibration experiment in 1:2 Et0Ac/heptane was dried (as PP415-P30) under
vacuum for several days (1-10 mbar, 50-70 C).
115
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
The FT-Raman spectrum of the dried class 2 material (PP415-P30) shows
small differences from the original spectrum (sample PP415-P7, FIG. 82) but
still
corresponds to class 2.
The PXRD pattern of the dried class 2 material (PP415-P30) shows slightly
broader, less intense peaks (FIG. 83) but still corresponds to class 2.
The TG-FTIR thermogram of the dried sample PP415-P30 (FIG. 84) shows
the loss of ¨2.5 wt.-% heptane (and some Et0Ac) in two steps from ¨50 C to
¨250 C and decomposition at temperatures T> 250 C. Compared to the TG-FTIR
of sample PP415-P7 (FIG. 76), the two steps of solvent loss are preserved, but
the
total amount of solvent in the sample has decreased from ¨7.5 wt.-% in PP415-
P7 to
¨2.5 wt.-% in PP415-P30.
Thus, the attempt to desolvate this solvate at elevated temperatures (50-70
C)
and a vacuum of 1-10 mbar) has caused only a partial loss of solvent.
J PP415-P15 PP415-P18
The solid material of sample PP415-P15, class 2, obtained from a precipitation
experiment in 1:2 DCM/IPE was dried (as PP415-P18) under vacuum (-2-20 mbar)
at
r.t. for ¨2 h.
The FT-Raman spectrum of PP415-P18 is identical to the spectrum of sample
PP415-P15 (FIG. 85), both correspond to class 2.
The PXRD pattern of PP415-P18 shows small differences to the pattern of
PP415-P15 (FIG. 86). PP415-P18 still corresponds to class 2.
The TG-FTIR thermogram (FIG. 87) shows the loss of'-7.0 wt.-% IPE in two
steps from ¨140 C to ¨250 C and decomposition at temperatures T> 250 C.
With
the boiling point of IPE being 67 C, the solvent seems tightly bound within
the
structure. The theoretical IPE content of a hemisolvate is 8.4 wt.-%.
Unfortunately, no TG-FTIR was recorded of the material before the drying
step. However, as the solvent seems so tightly bound into the structure and no
(or
only small) changes are observed in the FT-Raman spectra and PXRD patterns, it
is
assumed that the drying has had no significant effect on structure or solvent
content.
g. PP415-P17 PP415-P19 PP415-P32
The solid material of sample PP415-P17, class 2, obtained from a precipitation
experiment in 1:3 Et0Aciheptane was dried (as PP415-P19) under vacuum
(-2-20 mbar) at r.t. for ¨2 h.
116
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
The FT-Raman spectrum of PP415-P19 is identical to the spectrum of sample
PP415-P17 (FIG. 88); no changes can be observed, and both correspond to class
2.
The PXRD pattern of PP415-P19 differs slightly from the pattern of
PP415-P17 (FIG. 89) but still corresponds to class 2.
The TG-FTIR thermogram (FIG. 90) shows the loss of -7.6 wt.-% heptane in
two steps from -140 C to -270 C and decomposition at temperatures T > 270
C.
With the boiling point of heptane being 98 C, the solvent seems tightly bound
in the
structure. The theoretical heptane content of a hemisolvate is 8.3 wt.-%.
A further drying experiment (80 C, <1 >< 10-3 mbar, 3 days) was carried out
on
the same sample as PP415-P32.
The FT-Raman spectrum remained unchanged (FIG. 88). The PXRD pattern
still corresponded to class 2 (FIG. 89), but the sample was less crystalline
(as the
peaks were broader and had a lower S/1\1 ratio).
The TG-FTIR thermogram (FIG. 90) shows the loss of -2.2 wt.-% heptane,
most of it in a step from 170 C to 200 C and decomposition at temperatures T
>
250 'C.
Thus, the heptane content was reduced only from 7.6 wt.-% to 2.2 wt.-%,
confirming the tight binding of the solvent molecules.
It. PP415-P21 ¨> PP415-P28 ¨> PP415-P34
The solid material of sample PP415-P21, class 2, obtained from a slow cooling
experiment in -1:5 Et0H/cyclohexane was dried (as PP415-P28) under vacuum for
several days (2-20 mbar, r.t. to 60 C).
The FT-Raman spectrum of the dried class 2 material (PP415-P28) shows
small differences to the spectrum of class 2 (sample PP415-P21, FIG. 92), but
still
corresponds to class 2.
The PXRD pattern of the dried class 2 material (PP415-P28) shows broader,
less intense peaks compared to the pattern of PP415-P21 (FIG. 93), indicating
that the
dried sample is less crystalline. However, the pattern still corresponds to
class 2.
The TG-FTIR thermogram of the dried sample PP415-P28 (FIG. 94) shows
the loss of -3.0 wt.-% cyclohexane in two steps from -140 C to -250 C and
decomposition at temperatures T > 250 C. Compared to the TG-FTIR of sample
PP415-P21 (FIG. 77), the two steps of solvent loss are preserved, but the
total amount
117
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
of solvent in the sample has decreased from ¨5.8 wt.-% in PP415-P21 to ¨3.0
wt.-%
in PP415-P28.
Thus, the desolvation of this solvate seems to have caused only a partial loss
of solvent, parallel to a partial loss of crystallinity.
Further drying of this sample (at 80 C, <1 x10-3 mbar, 3 days) was carried
out
as PP415-P34.
The FT-Raman spectrum remained unchanged (FIG. 92). The PXRD pattern
still corresponded to class 2 (FIG. 93), but the sample was less crystalline
(as the
peaks were broader and had a lower S/1\1 ratio).
The TG-FTIR thermogram (FIG. 95) shows the loss of ¨2.3 wt.-%
cyclohexane, in two steps from 25 C to 270 C and decomposition at
temperatures
T > 270 C.
Thus, the cyclohexane content was reduced only from 3.0 wt.-% to 2.3 wt.-%
confirming the tight binding of the solvent molecules.
L Class 3 - Isostructural Solvates (e.g., Ethanol)
Several crystallization experiments resulted in solid material of class 3 and
were characterized by FT-Raman spectroscopy, PXRD, and TG-FTIR (Table 21).
The FT-Raman spectra of class 3 are clearly similar to each other (FIG. 96)
but show small differences (FIG. 97). The spectra of class 3 differ
significantly from
the spectrum of the amorphous starting material, class 1 (FIG. 98), and from
the
spectra of classes 2, 4, and 5 (FIG. 72).
The PXRD patterns of class 3 (FIG. 99) confirm the crystallinity of the
materials. The patterns of the three samples are similar to each other but
show small
but significant differences (FIG. 100). The class 3 pattern clearly differs
from the
crystalline patterns of classes 2, 4, and 5 (FIG. 75).
The TG-FTIR thermogram of sample PP415-P6 (FIG. 100) shows the loss of
¨5.4 wt.-% 2PrOH from 25 C to 250 C, most of it in a step from ¨170 C to
190 C.
Decomposition starts at temperatures T > 250 C. Before the TG-FTIR
experiments,
the samples were dried briefly (for ¨5 min) under vacuum (10-20 mbar) to
remove
excess, unbound solvent. The theoretical 2PrOH (b.p. = 82 C) content of a
hemisolvate is 5.1 wt.-%.
The TG-FTIR thermogram of sample PP415-P12 (FIG. 101) shows the loss of
¨4.9 wt.-% Et0H (with traces of water) from 25 C to 250 C, most of it in a
step
118
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
from ¨160 C to 190 C. Decomposition starts at temperatures T > 250 C. The
theoretical Et0H (b.p. = 78 C) content of a hemisolvate is 4.0 wt-%.
Thus, the samples of class 3 seem to be isostructural solvates of 2PrOH,
Et0H, and probably acetone with tightly bound solvent content. They could
correspond to stoichiometric hemisolvates. It cannot be ruled out, however,
that these
forms are non-stoichiometric solvates.
As the Raman spectra and PXRD patterns within this class are very similar to
each other, the structures might be essentially identical with only small
distortions of
the unit cell dimensions or small changes of atomic positions within the unit
cell due
to the incorporation of different solvent molecules.
Table 21. Crystallization experiments resulting in solid material of class 3
Solvent!
Sample Method
Characterization Drying
Mixture
Raman, PXRD,
PP415-P6 suspension equil. 2PrOH X
TG-FTIR
PP415-P12 suspension equil. 9:1 Et0H/H20 Raman, PXRD,
TG-FTIR
PP415-P20 slow cooling ¨2:1 acetone/H20
Raman, PXRD
j. Diying Experiments on Samples of Class 3
One of the samples of class 3 (PP415-P6), obtained from a suspension
equilibration experiment in 2PrOH, was dried (as PP415-P25) under vacuum for
several days (2-20 mbar, r.t. to 60 C, Table 22).
The TG-FTIR thermogram of this dried class 3 material, sample PP415-P25
(FIG. 102), shows the loss of ¨5.4 wt.-% 2PrOH from 50 C to 250 C, most of
it in a
step from 170 C to 190 C, another loss of ¨1.0 wt.-% 2PrOH from 290 C to
320 C, and decomposition at temperatures T > 320 C. Compared to the TG-FTIR
of
the original class 3 sample PP415-P6 (FIG. 103), with a solvent content of
¨5.4 wt.-%
2PrOH, the solvent content does not seem to have decreased significantly.
This material was dried further (as PP415-P33, Table 22) for three days under
high vacuum and elevated temperatures (<1x10-3 mbar, 80 C) with the aim to
desolvate the solvate and to obtain a non-solvated, anhydrous form of 63415.
The TG-FTIR thermogram of this further dried class 3 material, sample
PP415-P33 (FIG. 103) shows the loss of ¨4.2 wt.-% 2PrOH from 50 C to 210 C,
119
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
most of it in a step from 160 C to 190 C, another loss of ¨0.5 wt.-% 2PrOH
from
210 C to 290 C, and decomposition at temperatures T > 290 C.
Compared to the solvent content of the samples PP415-P6 and PP415-P25, the
solvent content has decreased only from ¨5.4 wt.-% to ¨4.8 wt.-%.
Table 22. Drying experiments on samples of class 3
Starting Material Drying Dried Material
Solvent Solvent
Sample Conditions Sample Class
Content Content
r.t., ¨3 mbar, ¨5 d;
60 C 5-10 mbar,
2PrOH 2PrOH
PP415-P6 2x1 h; PP415-P25 3
(-5.4%)
40-50 C, 5-20 (-5.4%)
mbar, ¨1 d
2PrOH PP415-P25 ( 80 C, <1x10-3 2PrOH
PP415-P33 3
¨5.4%) mbar, 3 d (-4.8%)
The FT-Raman spectra of class 3 (sample PP415-P6), of the dried material of
class 3 (sample PP415-P25), and of the further dried material of class 3
(sample
PP415-P33) are identical and show no changes (FIG. 104).
The PXRD patterns of class 3 (sample PP415-P6) and of the further dried
material of class 3 (sample PP415-P33) do not show any significant
differences, while
there are few small shifts and differences from the pattern of the initially
dried
material of class 3 (sample PP415-P25, FIG. 105). All patterns correspond to
class 3.
As the drying had no major effect on the solvent content, it is not surprising
that the FT-Raman spectra and PXRD patterns of the dried materials do not show
differences compared to the non-dried material.
Thus, class 3 is a class of isostructural solvates (2PrOH, Et0H, and probably
acetone) with very tightly bound solvent molecules that could be removed only
partially (-5.4 wt.-% to ¨4.8 wt.-%) by the drying conditions applied here (up
to 3 d
at 1x10-3 mbar and 80 C).
k. Class 4¨ Acetonitrile Solvate
Class 4 was obtained only from a 7:3 MeCN/H20 solvent mixture (Table 23).
The experiment resulting in class 4 (PP415-P13) was repeated as PP415-P35 to
prepare more material for further drying studies.
120
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
The FT-Raman spectrum (FIG. 72) and PXRD pattern (FIG. 75) of class 4
(sample PP415-P13) differ significantly from the spectra and patterns of
classes 2, 3,
and 5.
The TG-FTIR thermogram of class 4 (sample PP415-P13, FIG. 106) shows
the loss of ¨3.4 wt.-% MeCN (with traces of water) from 25 C to 270 C, most
of it
in a step from ¨180 C to 210 C. Decomposition starts at temperatures T>270
C.
Before the TG-FTIR experiments, the samples were dried briefly (for ¨5 min)
under
vacuum (10-20 mbar) to remove excess, unbound solvent. The theoretical MeCN
(b.p. = 81 C) content of a hemisolvate is 3.6 wt.-%.
Table 23. Crystallization experiments resulting in solid material of class 4
Sample Method Solvent
Characterization Drying
PP41 suspension 7:3 Raman, PXRD,
5-P13 X
equilibration MeCN/H20 TG-FTIR
PP415 35 suspension 7:3 Raman, PXRD,
-P X
equilibration MeCN/1-120 TG-FTIR
I. Drying Experiments on Class 4
The samples of class 4 obtained from suspension equilibration experiments in
¨7:3 MeCN/H20 were dried under vacuum for several days or under N2 flow
(Table 24).
Table 24. Drying experiments on samples of class 4
Starting Material Drying Dried Material
Solvent Solvent
Sample Conditions Sample Class
Content Content
r.t., ¨3 mbar, ¨5 d;
60 C, 5-10 mbar' PP415- MeCN MeCN
PP415-P13 2 xl h; 4
(-3.4%) 40-50 P26 (-2.8%)
C,
5-20 mbar, ¨1 d
PP415P35 MeCN 80 C' <1x10 -
mbar, 3 d PP415- MeCN/H203
- 4
(-2.9%) P36 (-0.6%)
MeCN PP415- MeCN/H203
PP415-P35 80 C, N2 flow, 3 d 4
(%)
solvent content possibly MeCN and H20, but difficult to determine as amounts
arc small
The FT-Raman spectrum of the dried class 4 material (PP415-P26) is identical
to the spectrum of class 4 (PP415-P13, FIG. 107).
121
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
The PXRD pattern of the dried class 4 material (PP415-P26) shows only very
small differences from the pattern of class 4, sample PP415-P13 (FIG. 108).
Some
peaks seem better resolved, and peak intensities have shifted. No
amorphization is
observed. The pattern of PP415-P26 corresponds to class 4.
The TG-FTIR thermogram of the dried class 4 material, sample PP415-P26
(FIG. 109) shows the loss of ¨2.8 wt.-% MeCN from 170 C to 250 C and
decomposition at temperatures T > 300 C. Compared to the TG-FTIR of sample
PP415-P13 (FIG. 106), the solvent content of the sample has decreased from
3.4 wt.-% to 2.8 wt.-%.
Thus, the sample seems to be a partially desolvated solvate. As not sufficient
material remained for a second drying experiment with subsequent
characterization,
experiment PP415-P13 was repeated (as PP415-P35). More material of class 4 was
prepared and two drying experiments were carried out with this freshly
prepared
material:
= PP415-P36: drying under vacuum (<1x10-3 mbar) at 80 C for three days
= PP415-P37: drying under N2 flow at 80 C for three days
The FT-Raman spectra of these dried class 4 samples (PP415-P36 and -P37)
correspond to the spectrum of class 4 (i.e., PP415-P35, FIG. 110).
The PXRD patterns (FIG. 111) of the class 4 material (sample PP415-P35)
and the dried samples of class 4 (samples PP415-P36 and PP415-P37) are
identical.
The dried samples are crystalline.
The TG-FTIR thermograms of these dried samples of class 4 (FIG. 112 for
PP415-P36 and FIG. 113 for PP415-P37) show only a small solvent content (MeCN
and/or H20) of ¨0.6 wt.-% and ¨0.9 wt.-% for PP415-P36 and PP415-P37,
respectively, in two steps from 25 C to 280 C. Solvent content is possibly
MeCN
and H20, but is difficult to determine as amounts are small. Decomposition
starts at
temperatures T > 280 C.
Thus, most of the solvent of this solvate could be removed without destroying
the crystal structure. A crystalline, non-solvated form (or rather desolvated
solvate)
was obtained.
m. Further Characterization of the Dried and Desolvated
Class 4
Drying of class 4 (MeCN solvate) resulted in a desolvated solvate with the
solvent content reduced to <1 wt.-% (TG-FTIR).
122
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
No change in the structure occurred upon desolvation (FT-Raman and PXRD).
No significant loss of the crystallinity was observed.
Thus, a non-solvated, crystalline form of 63415 was obtained, the only one
known to date.
This desolvated class 4 material was characterized further by DVS and DSC.
The DVS isotherm (FIG. 114) shows that during initial equilibration time at
50% r.h. a mass gain of ¨0.4 wt.-% occurred. During the measurement, a
gradual,
reversible mass loss of ¨1.3 wt.-')/0 occurred upon lowering the relative
humidity from
50% r.h. to 0% r.h. Equilibrium was reached. Upon increasing the relative
humidity
to 95% r.h., a gradual mass gain of ¨0.8 wt.-% was observed (relative to the
equilibration mass at 50% r.h.). Equilibrium was reached. After lowering the
relative
humidity to 50% r.h., the final mass remained 0.1 wt.-% below the equilibrated
starting mass. The mass gain of ¨0.7 wt.-% upon increasing the relative
humidity
from 50% r.h. to 85% r.h. classified the sample as slightly hygroscopic.
The PXRD pattern of the sample after the measurement is unchanged
compared to the pattern before the measurement (FIG. 115).
The DSC thermogram of a sample of desolvated class 4 material (FIG. 116)
shows no glass transition attributable to the amorphous form, which would have
been
expected at ¨150 C, but instead a sharp endothermic peak with a maximum at T
=
196.1 C (AH = 29.31 J/g), probably corresponding to melting, and no
decomposition
up to 270 C.
In addition, a DSC experiment was carried out with a ¨1:1 mixture of the
amorphous material, class 1, with the desolvated class 4 material to
investigate if the
amorphous material would transform and crystallize into the desolvated class
4, an
event expected to occur (if at all) above the glass transition temperature of
the
amorphous form (Tg 150 C) and
below the melting of the desolvated class 4
(Tm z196 C).
The DSC thermogram of the mixture (FIG. 117) shows an endothermic event
with a peak at T = 156.7 C (AH = 1.47 J/g) and a second endothermic event
with a
peak at 197.0 C (AH = 14.1 J/g). The first event could be attributable to the
amorphous material (glass transition at Tg z-; 150 C). The second event could
correspond to the melting of the &solvated class 4 at Tm z196 C. The heat of
fusion
123
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
(AH = 14.1 Jig) of the mixture correlates well to half of the heat of fusion
(AH =
29.3 J/g) of the pure desolvated class 4.
No exothermic event in the temperature range between the glass transition and
the melting corresponding to a possible crystallization of the amorphous
material can
be observed. Thus, no transformation of the amorphous form into the desolvated
class 4 form seemed to have occurred on this timescale.
In yet another DSC experiment with a ¨1:1 mixture of the amorphous
material, class 1, with the desolvated class 4 material, the heating was
stopped at
173 C (in between the glass transition and the melting) to allow time for a
possible
crystallization.
The DSC thermogram of the mixture (FIG. 118) shows an endothermic event
with a peak at T = 161.4 C (AH = 0.31 J/g) and a second endothermic event
with a
peak at 201.4 C (AH = 11.4 J/g). As in the first experiment, the heat of
fusion of the
second peak did not increase; no indications for a transformation of the
amorphous
form into the desolvated class 4 form are visible.
The curved baseline (-50 C to 150 C) is most likely an artifact (due to a
bent
sample holder lid).
n. Class 5- THF Solvate
Class 5 was obtained only from a 1:1 THF/I-120 solvent mixture (Table 25).
The FT-Raman spectrum (FIG. 71) and PXRD pattern (FIG. 75) of class 5
differs significantly from the spectra and patterns of classes 2, 3, and 4.
The TG-FTIR thermogram of class 5 (sample PP415-P14, FIG. 119) shows
the loss of ¨36.1 wt.-% THF and H70 from 25 to 200 C, most of it in a step
from
¨100 C to 130 C. Before the TG-FTIR experiments, the samples were dried
briefly
(for ¨5 min) under vacuum (10-20 mbar) to remove excess, unbound solvent. The
loss of both THF and F20 occur together in the same temperature range.
Decomposition starts at temperatures T> 300 C. The theoretical THF (b.p. = 66
C)
content of a trisolvate is 28.1 wt.-%. Unfortunately, as the content of the
two
components cannot be quantified separately, the exact solvation state cannot
be
determined.
Details on the experiments and characterizations of samples PP415-P41 and
PP415-P45 are provided.
124
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
Table 25. Crystallization experiments resulting in solid material of class 5
Sample Method Solvent Characterization Drying
PP415-P14
suspension 1:1 Raman, PXRD,
X
equilibration THF/H20 TG-FTIR
suspension 1:1
12'13415-P4lb PXRD X
equilibration THF/H20
PP415- suspension 1:1
PXRD X
p45b.c equilibration THF/H20
b
starting material: PP415-P40, class 2; in all other experiments in this table
PP415-P1, class
1, was used as the starting material
C 3-g scale experiment instead of 100-mg scale
o. Drying Experiments on Samples of Class 5
The sample of class 5 (PP415-P14), obtained from a suspension equilibration
experiment in ¨1:1 THF/H20, was dried (as PP415-P27) under vacuum for several
days (2-20 mbar, r.t. to 60 C, Table 26).
Table 26. Drying experiments of samples of Class 5
Starting Material Drying Dried Material
Solvent Solvent
Sample Conditions Sample Class
Content Content
r.t., ¨3 mbar,
THF & ¨5 d;
H20 60 C,5-
PP415-P14PP415-P27 (-0.3 wt.- 1(+5)a
(-36 10 mbar, 2x1 h;
)
wt.-%) 40-50 C, %
5-20 mbar, ¨1 d
a mainly amorphous, only few broad peaks with low SiN ratio
The FT-Raman spectrum of the dried material (PP415-P27) is different from
the spectrum of class 5 (PP415-P14, FIG. 120) and, with its broadened peaks,
resembles more the spectrum of class 1, the amorphous starting material, PP415-
P1.
The PXRD pattern of the dried class 5 material (PP415-P27) shows only some
broad, low intensity peaks with a low S/N ratio, indicating the poor
crystallinity of the
sample (FIG. 121). Some of the peaks could correspond to class 5, while
others, i.e.,
at 7.35 '20, are new or shifted.
The TG-FTIR thermogram of the dried class 5 material (FIG. 122) shows a
mass loss of ¨0.3 wt.-% from 25 C to 290 C and decomposition at temperatures
T>
290 C. The sample is anhydrous.
125
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
Thus, by drying under vacuum, the material has lost its solvent content and
also much of its crystallinity.
8. Experiments to Prepare the Amorphous Form
Experiments with the aim to prepare the amorphous form, class 1, were carried
out using class 2 material (PP415-P40, Table 8) as the starting material.
Several
strategies and methods were attempted:
= Transformation of class 2 into class 5, followed by drying of class 5 to
obtain
the amorphous form, class 1.
= Preparation of the amorphous form, class 1, directly from class 2, if
possible
using ICH class 3 solvents.
Mainly amorphous material was prepared starting from class 2 material in a two-
step
process via class 5 on a 100-mg and 3-g scale.
Further experiments were carried out with the aim to simplify the procedure to
a one-step process, to avoid the ICH class 2 solvent THF, and to obtain fully
amorphous material. The most promising method was found to be the
precipitation
from an acetone solution in a cold water bath. This direct method gives much
better
results than the two-step method via class 5.
a. Preparation of the Amorphous Form via Class 5
Crystallization experiments using class 2, PP415-P40, as the starting material
were carried out with the aim to transform this heptane solvate into class 5
(likely
THF solvate), followed by drying of class 5 to obtain the amorphous material
(Table 27).
Class 5 is thought to be a good intermediate step, as it is easier to
desolvate
and amorphize than classes 2 or 3.
126
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
Table 27. Summary of experiments aimed at preparation of amorphous form,
class I, via class 5 material
Step Sample Method Conditions Results
suspension 1:1 THF/I-120, 24 C,
1 PP415-P41 class 5
equil. 3 d
suspension
" PP415-P45 1:1 THF/I-120, r.t. 1 d class 5
cquil.
2 PP415-P44a drying 100 mbar, 80 C, 2 d class la; 0.9
wt.-%
THF
" PP415-P46a drying 100 mbar, 80 C, 4 d class la; 0.4
wt.-%
H20
mainly amorphous, only few broad peaks with low SiN ratio
b. Step 1: Transformation of Class 2 into Class 5
Transformation of the heptane solvate, class 2, into the THF solvate, class 5,
was successfully carried out by suspending the PP415-P40 (heptane solvate)
material
in a (1:1) THF/H20 mixture and equilibrating the suspension at r.t. (PP415-
P41,
100 mg-scale). The resulting solid material corresponds to the THF solvate,
class 5
(FIG. 123).
A first scale-up experiment from the mg-scale to the g-scale (x30, i.e., 3-g
scale) was carried out analogous to PP415-P41: the class 2 heptane solvate
starting
material (PP415-P40) was equilibrated in THF/WO (1:1) for one day and
successfully
transformed into class 5, the THF solvate (PP415-P45, FIG. 124).
c. Step 2: Amorphization of Class 5 Material by Drying
The class 5 material (THF solvate) was dried at elevated temperature (80 C)
under vacuum (-100 mbar) taking into account the conditions that can be used
at the
API MFG site.
After drying the material of the 100-mg scale experiment, PP415-P41, for one
day at 80 C and 100 mbar it transformed into mainly amorphous material
(PP415-P44, FIG. 125). The PXRD pattern shows only some broad peaks with low
intensity. After additional drying (80 C, 100 mbar) overnight, the intensity
of these
broad peaks is further reduced (PP415-P44a). The TG-FTIR of this material
shows
the loss of ¨0.9 wt.-% THF (with traces of water) gradually from 25 C to 280
C and
decomposition at temperatures T > 300 C (FIG. 126).
The material of the 3-g scale experiment, PP415-P45, was also dried at 80 C
and 100 mbar (as PP415-P46). It transformed overnight into mainly amorphous
127
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
material with only some broad peaks with low intensity (FIG. 127). After a
total of
four days of drying (80 C, 100 mbar), these broad peaks are still present (-
P46a,
FIG. 128). The TG-FTIR of this material shows no THF content, but the loss of
¨0.4
wt.-% water gradually from 25 C to 250 C and decomposition at temperatures
T>
250 C (FIG. 129).
d. Obtaining the Amorphous Form Directly
The preparation of the amorphous form starting from class 2 material in the
two-step process via class 5 was largely, but not fully, successful. Thus,
further
experiments were carried out with the aim to simplify the procedure to a one-
step
process, to avoid the use of ICH class 2 solvent THF, and to obtain fully
amorphous
material (Table 28).
The amorphous form, class 1, was prepared directly from the class 2 material
in an evaporation experiment of a class 2 solution in THF under N2 flow (PP415-
P42,
FIG. 129).
In an attempt to simulate an incompletely dried heptane/hexane solvate with a
significant amount of remaining solvent, an evaporation of a class 2 solution
in an 8:2
THF/hexane solution was carried out (hexane was used instead of heptane in
order to
have similar boiling points in the solvent mixture). However, the resulting
solid
corresponds to class 2, the class of the isostructural solvates, not to class
5
(PP415-P43, FIG. 130).
In order to avoid the ICH class 2 solvent THF, evaporation experiments were
carried out in ICH class 3 solvents.
Evaporation of a class 2 solution in Et0Ac under N2 flow resulted in
crystalline material with a PXRD pattern corresponding to class 2 (PP415-P47,
FIG. 130). The TG-FTIR (FIG. 80) shows the class 2-typical two-step mass loss
(total of ¨7.9 wt.-% Et0Ac) at temperatures up to 240 C, indicating very
tightly
bound solvent molecules.
Evaporation in ethyl formate also gave crystalline class 2 material and not
the
amorphous form (PP415-P48, FIG. 131). The TG-FTIR (FIG. 78) shows the mass
loss of ¨3.5 wt.-% ethyl formate, at first gradually and then in a clear step
between
180 C and 200 C. There might be further loss of ethyl formate concomitant
with the
decomposition at T> 240 C.
128
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
However, class 2 material was successfully transformed into the amorphous
form, class 1, by adding an acetone solution to a cold (5 C) water bath
(PP415-P49,
FIG. 132).
This direct method for the preparation of the amorphous form gives better
results and is a more promising route than the two-step process.
Table 28. Summary experiments aimed at obtaining the amorphous form
directly from class 2 starting material
Sample Method Solvent Condition Result
PP415-P42 evaporation THF N2 flow, 1 d class 1
8:2
PP415-P43 evaporation THF/hexane N2 flow, 1 d class 2
PP415-P47 evaporation Et0Ac N) flow, 1 d class 2
PP415-P48 evaporation ethyl formate N2 flow, 1 d class 2
H20 bath at 5
PP415-P49 precipitation acetone class 1
C
9. Instrumental - Typical Measurement Conditions
FT-Raman Spectroscopy: Bruker RFS100 with OPUS 6.5 software;
Nd:YAG 1064-nm excitation, Ge detector, 3500-100 cm-I range; typical
measurement
conditions: 100-300 mW nominal laser power, 64-128 scans, 2 cm' resolution.
PXRD: Stoe Stadi P; Mythen1K Detector; Cu-Ka radiation; standard
measurement conditions: transmission; 40 kV and 40 mA tube power; curved Ge
monochromator; 0.02 020 step size, 12 s or 60 s step time, 1.5-50.5 020 or 1.0-
55 020
scanning range; detector mode: step scan; 1 020 or 6 020 detector step;
standard
sample preparation: 10 to 20 mg sample was placed between two acetate foils;
sample
holder: Stoe transmission sample holder; the sample was rotated during the
measurement.
TG-FTIR: Netzsch Thermo-Microbalance TG 209 with Bruker FT-1R
Spectrometer Vector 22; aluminum crucible (with microhole), N? atmosphere,
10 K/min heating rate, 25-250 C or 25-350 C range.
DSC: Perkin Elmer DSC 7; gold crucibles (closed or with microhole), sample
filled in an N2 environment, 10 K/min heating rate, -50 to 250 C or 350 C
range, at
times quench cooling (at -200 K/min) to -50 C between scans.
DVS: Projekt Messtechnik Sorptions Prufsystem S'PS II - 100n or Surface
Measurement Systems DVS-1. The sample was placed on an aluminum or platinum
129
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
holder on top of a microbalance and allowed to equilibrate for 2 h at 50% r.h.
before
starting the pre-defined humidity program:
(1) 50 ¨> 0% r.h. (5%/h); 5 h at 0% r.h.
(2) 0 ¨> 95% r.h. (5%/h); 5 h at 95% r.h.
(3) 95 ¨> 50% r.h. (5%/h); 2 h at 50% r.h.
The hygroscopicity was classified based on the mass gain at 85% r.h. relative
to the initial mass as follows: deliquescent (sufficient water adsorbed to
form a
liquid), very hygroscopic (mass increase of >15')/0), hygroscopic (mass
increase <15%
but >2%), slightly hygroscopic (mass increase <2% but >0.2%), or non-
hygroscopic
(mass increase <0.2%).
Solvents: For all experiments, Fluka, Merck, or ABCR analytical grade
solvents were used.
Approximate Solubility Determination: Approximate solubilities were
determined by a stepwise dilution of a suspension of about 10 mg of substance
in
.. 0.05 mL of solvent. If the substance was not dissolved by addition of a
total of >10
mL solvent, the solubility is indicated as <1 mg/mL. Due to the experimental
error
inherent in this method, the solubility values are intended to be regarded as
rough
estimates and arc to be used solely for the design of crystallization
experiments.
Chemical Stability Determination: Four samples of 1.0 mg of the PP415-P1
material in 1.0 mL of the respective solvent were prepared. The resulting
suspensions/solutions were equilibrated in a temperature-controlled Eppendorf
Thermomixer Condbrt shaker for 7 d, 2 d, 24 h, and 6 h at 25 C at a shaking
rate of
500 rpm. If necessary, the solid phase was separated by filter centrifugation
(0.5-ptm
PVDF membrane). The filtrates were diluted in the diluent (0.1% formic acid in
MeCN) to concentrations <0.2 mg/mL (unknown and likely lower for suspensions)
and examined using the HPLC method given in Table 29. As reference, the PP415-
P1
material was diluted in the diluent to a concentration of 0.25 mg/mL and added
to the
beginning and end of the HPLC sequence.
130
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
HPLC Results
Table 29. HPLC method used for chemical stability determinations
Column Zorbax Eclipse XDB-C18, 3x150 mm, 5 !um (CC19)
Eluent A H20 + 0.1% formic acid
Eluent B MeCN + 0.1% formic acid
0 min 50%A 50%B
10.0 min 10%A 90%B
Gradient 15.0 min 0% A 100% B
15.1 min 50%A 50%B
20.0 min 50% A 50% B
Flow 0.75 mL/min
Injection Volume 10 [IL
Wavelength 254 nm, 242 nm, 210 nm
Acquisition time 20 min
Run time 20 min
Column
25 C
temperature
Retention time 8.9-9.0 min
10. Abbreviations
Methods:
AUC area under the curve analysis
DSC differential scanning calorimetry
DVS dynamic vapor sorption
FT Raman Fourier-transform Raman spectroscopy
1H-NMR proton nuclear magnetic resonance spectroscopy
HPLC high-performance liquid chromatography
PXRD powder X-ray diffraction
TG-FTIR thermogravimetry coupled to Fourier transform
infrared
spectroscopy
Chemicals:
1BuOH 1-butanol
CTAB cetyl trimethylammonium bromide
DCM dichloromethane
DEE diethyl ether
131
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
DMF N,N-dimethylformamide
Et0Ac ethyl acetate
Et0H ethanol
IPE isopropyl ether
MeCN acetonitrile
MEK methyl ethyl ketone
Me0H methanol
PEG propylene glycol
PTFE polytetrafluoroethylene, Teflon
2PrOH 2-propanol, isopropanol
SDS sodium dodecyl sulfate
TBME tert-butyl methyl ether
TEA triethylamine
THF tetrahydrofuran
Tween 80 polyoxyethylene (80)
sorbitan monooleate or polysorbate
Genes, Proteins, and Biological Parameters:
AIM antioxidant inflammation modulator
20 Akr 1 cl aldo-keto reductase family 1, member cl
ALP alkaline phosphatase
ALT alanine transaminase
ARE antioxidant response element
AST aspartate transaminase
25 AUC area under the curve
BAL bronchoalveolar lavage
BALF bronchoalveolar lavage fluid
Bil bilirubin
BUN blood urea nitrogen
30 COPD chronic obstructive pulmonary disease
COX-2 cyclooxygenase-2
Cr creatine
CYP450 cytochrome P450
Eh-1 epoxide hydrolase 1
132
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
G6PD glucose-6 phosphate dehydrogenase
Gcic glutamate-cysteine ligase, catalytic subunit
Gclm glutamate-cysteine ligase, modifier subunit
Ggtl gamma-glutamyltransferase
Glrx glutaredoxin-1
Glu glucose
GOT glutamic-oxaloacetic transaminase
GPT1 glutamic-pyruvate transaminase
Gpx3 glutathione peroxidase 3
GSH glutathione
GSR glutathione reductase
GSs glutathione synthetase
GST glutathione S-transferase
GSTal glutathione S-transferase alpha 1
GSTpl glutathione S-transferase pi 1
Gy Gray
H6PD hexose-6-phosphate dehydrogenase
hERG human ether a-go-go-related gene
HMOX1 heme oxygenase (decycling) 1
HO-1 heme oxygenase
IFN7 interferon-gamma
IL interleukin
iNOS inducible nitric oxide synthase
ItcBa nuclear factor of kappa light polypeptide gene
enhancer
in B-cells inhibitor, alpha
KC mouse 1L-8 related protein
Keapl Kelch-like ECH associated protein-1
LP S lipopolysaccharide
ME1 malic enzyme 1
MPCE micronucleated polychromatic erythrocytes
Mrp metG-related protein
Mrps multidrug resistance-related proteins
NADPH nicotinamide adenine dinucleotide phosphate, reduced
133
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
NFKB nuclear factor of kappa-light-chain-enhancer of
activated B cells
NO nitric oxide
NQ01 NAD(P)H quinone oxidoreductase 1
Nrf2 nuclear factor (erythroid-derived)-like 2
p- IicBa phosphorylated IKBa
PBMC peripheral blood mononuclear cell
PCE polychromatic erythrocytes
PGD phosphogluconate dehydrogenase
PMN polymorphonuclear
RANTES regulated and normal T cell expressed and secreted
SOD 1 superoxide dismutase 1
SRXN1 sulfiredoxin-1
TG total glycerides
TKT transketolase
INFict tumor necrosis factor alpha
Txn thioredoxin
TXNRD1 thioredoxin reductase 1
xCT solute carrier family7, member 11
Misc:
API active pharmaceutical ingredient
aq. aqueous
b.p. boiling point
cryst. crystalline
decomp. decomposition
day(s)
eq. equivalent
equil. equilibration
evap. evaporation
h hour(s)
mat. material
min minute(s)
m.p. melting point
134
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
MS molecular sieves
part. partially
precip. precipitation
r.h. relative humidity
rpm revolutions per minute
r.t. room temperature (-25 C)
S/N signal-to-noise (ratio)
solv. solvent
susp. suspension
1 temperature
Ts glass transition temperature
theo. theoretical
vis. obs. visual observation
week(s)
wt.-% weight percent
135
K. Further Tables
Table 30: List of Samples and Performed Experiments
Sample Experimental Description Test Methods
Result / Remarks
PP415-P1 received ¨5 g of 63415, batch #0141-66-1, on March 25, FT-
Raman: FT-Raman: used as reference for P1
2011; MW ¨ 554.7 gimol, C33H44F2N203 PP415P1.0
PXRD: amorphous, no crystalline peak
PP415P1.1 pattern
PXRD: 117a TG-
FT1R: loss of ¨0.9 wt.-% (-0.1 eq.)
TG-FTIR: a4285 Et0H
with traces of H20 from 25 C to
1H-NMR: 200 C, decomposition at T > 290 C
Mar30-2011-ktr/30 1H-NMR: agrees with structure, ¨0.08 eq.
DSC: d 9840
Et0H
DVS: #0305_02 DSC:
1st scan: Tg = 152.7 C (ACp =
post-DVS Raman: 0.72
Jig C); 2nd scan: Tg = 149.7 C
PP415P1_aDVS
(ACp = 0.45 J/g C)
post-DVS PXRD: DVS: slightly hygroscopic; Am = +0.4%
179a
(50%¨>85% r.h.); total mass gain of 2.1
wt.-% from 0% r.11. to 95% r.h.
post-DVS Raman and PXRD: unchanged
PP415-P2 stored material of PP415-P1 at 25 C open over a saturated
PXRD: PXRD: all amorphous, correspond to PI
solution of NH4NO3 (i.e., at ¨62% r.h.); examined samples 132a (-P2a)
after 1 w (PP415-P2a), 2 w (PP415-P2b), and 4 w 191a (-P2b)
(PP415-P2c). 262a (-P2c)
c7)
oc
136
Sample Experimental Description Test Methods
Result! Remarks
PP415-P3 stored material of PP415-
P1 at 40 C open over a saturated PXRD: PXRD: all amorphous, correspond
to PI
solution of NaCl (i.e., at ¨75% r.h.); examined samples after 133a (-P3 a)
(P P4 15-P3a), 2 w (PP415-P3b), and 4 w (PP415-P3c). 192a (-P3b)
263a (-P3c)
PP415-P4 stored material of PP415-
P1 at 60 C in a closed container; PXRD: PXRD: all amorphous, correspond
to PI
examined samples after I w (PP415-P4a), 2 w (PP4I5-P4b), .. 134a (-P4a)
and 4 w (PP415-P4c). 193a (-P4b)
264a (-P4c)
PP415-P5 stored material of PP415-
P1 at 80 C in a closed container; PXRD: PXRD: all amorphous, correspond
to PI
examined samples after 1 w (PP415-P5a), 2 w (PP415-P5b), 135a (-P5a)
and 4 w (PP415-P5c). 194a (-P5b)
265a (-P5c)
PP415-P6 suspended 97.7 mg of PP415-P1 in 0.4 mL of 2PrOH; FT-
Raman: Raman: corresponds to class 3
obtained white suspension; equilibrated suspension at 22 C PP415P6.0
PXRD: corresponds to class 3
shaking at 400 rpm; added stepwise a total of 0.5 mL of the PXRD: 225a
TG-FTIR: loss of ¨5.4 wt.-% 2PrOH
solvent over the next couple of days; after 15 d recovered TG-FTIR: a4323
from 25 C to 250 C, most of it in a step
solid material by filter centrifugation (0.20- m PTFE from
¨170 C to 190 C; decomposition
membrane); examined material by FT-Raman spectroscopy starts at T>250 C
and PXRD; dried material for 5 min under vacuum (10-
20 mbar); examined material by TG-FTIR.
CID
oc
137
Sample Experimental Description Test Methods
Result! Remarks
PP415-P7 suspended 104.3 mg of PP415-P1
in 0.6 mL of FT-Raman: Raman: corresponds to class 2
1:2 Et0Ac/heptane; obtained white suspension; equilibrated
PP415P7.0 PXRD: corresponds to class 2
suspension at 22 C shaking at 400 rpm; added stepwise a PXRD: 227a TG-
FTIR: loss of ¨7.5 wt.-% Et0Ac and
total of 0.2 mL of the solvent mixture over the next couple TG-
FTIR: a4338 heptane in two steps from ¨100 C to
of days; after 15 d recovered solid material by filter 290 C;
decomposition starts at T > 290
centrifugation (0.20- m PTFE membrane); examined
C
material by FT-Raman spectroscopy and PXRD; dried
material for 5 mm under vacuum (10-20 mbar); examined
material by TG-FTIR.
PP415-P8 suspended 102.0 mg of PP415-P1 in 0.4 mL of 1:2 FT-
Raman: Raman: corresponds to class 2
acetone/hexane; obtained white suspension; equilibrated PP415P8.0 PXRD:
corresponds to class 2
suspension at 22 C shaking at 400 rpm; added stepwise a PXRD: 228a
total of 0.2 mL of the solvent mixture over the next couple
of days; after 15 d recovered solid material by filter
centrifugation (0.20-um PTFE membrane); examined
material by FT-Raman spectroscopy and PXRD.
PP415-P9 suspended 102.6 mg of PP415-P1 in 0.4 mL of 1:3 FT-
Raman: Raman: corresponds to class 2, contains
toluene/diethyl ether; obtained white suspension; PP415P9.0 solvent
signals
equilibrated suspension at 22 C shaking at 400 rpm; added
stepwise a total of 0.2 mL of the solvent mixture over the
next couple of days; after 15 d recovered solid material by
filter centrifugation (0.20-ium PTFE membrane); examined
material by FT-Raman spectroscopy.
k=J
oc
138
Sample Experimental Description Test Methods
Result! Remarks
PP415-P10 suspended 102.5 mg of PP415-P1 in 0.2 mL of FT-
Raman: Raman: corresponds to class 2
1:3 Me0H/TBME; obtained clear solution; equilibrated PP415P10.0
PXRD: corresponds to class 2
solution at 22 C shaking at 400 rpm; after 1 d observed PXRD: 229a
thick suspension; added 0.2 mL of the solvent mixture;
continued equilibration of suspension at 22 C shaking at
400 rpm; after a total of 15 d recovered solid material by
filter centrifugation (0.20- m PTFE membrane); examined
material by FT-Raman spectroscopy and PXRD.
PP415-P11 suspended 97.1 mg of PP415-P1 in 0.4 mL of 1:2 FT-
Raman: Raman: corresponds to class 2, contains
MEK/cyclohexane; obtained white suspension; equilibrated PP415P11.0
solvent signals
suspension at 22 C shaking at 400 rpm; after 15 d recovered
solid material by filter centrifugation (0.20- m PTFE
membrane); examined material by FT-Raman spectroscopy.
PP415-P12 suspended 98.6 mg of PP415-P1 in 0.2 mL of 9:1 FT-
Raman: Raman: corresponds to class 3
Et0H/H20; obtained white suspension; equilibrated PP415P12.0
PXRD: corresponds to class 3
suspension at 22 C shaking at 400 rpm; added stepwise a PXRD: 230a TG-
FTIR: loss of ¨4.9 wt.-% Et0H (with
total of 0.2 mL of the solvent mixture over the next couple TG-FTIR:
a4324 traces of water) from 25 C to 250 C,
of days; after 15 d recovered solid material by filter most
of it in a step from ¨160 C to
centrifugation (0.20-litm PTFE membrane); examined 190 C;
decomposition starts at 1>250 C
material by FT-Raman spectroscopy and PXRD; dried
material for 5 mm under vacuum (10-20 mbar); examined
material by TG-FT1R.
CID
oc
139
CD
N
0
0.,
w
Sample Experimental Description Test Methods
Result! Remarks ,
1-,
o,
PP415-P13 suspended 95.9 mg of PP415-P1 in 0.2 mL of 7.3 FT-
Raman: Raman: corresponds to class 4, contains
t..)
A
MeCN/H20; obtained two clear, separated phases; PP415P13.0
solvent signals A
equilibrated solution at 22 C shaking at 400 rpm; after 1 d PXRD: 231a
PXRD: corresponds to class 4
observed thick suspension; added 0.2 mL of the solvent TG-FTIR: a4321 TG-
FTIR: loss of -3.4 wt.-% MeCN
mixture; continued equilibration of suspension at 22 C (with
traces of water) from 25 C to
shaking at 400 rpm; after a total of 15 d recovered solid 270 C,
most of it in a step from -180 C
material by filter centrifugation (0.20-um PTFE membrane); to 210 C;
decomposition starts at 1>270
examined material by FT-Raman spectroscopy and PXRD; C
dried material for 5 min under vacuum (10-20 mbar);
examined material by TG-FTIR.
0
2
0
PP415-P14 suspended 95.8 mg of PP415-P1 in 0.2 mL of 9:1 THF/H20; FT-Raman:
Raman: corresponds to class 5, contains
..,
obtained two clear, separated phases; equilibrated solution at PP415P14.0
solvent signals ,,,
22 C shaking at 400 rpm; after 1 d observed one clear PXRD: 232a
PXRD: corresponds to class 5 .
phase; added 0.2 mL of H20; observed white precipitate; TG-FTIR: a4322
TG-FTIR: loss of'-36.1 wt.-% THF and
equilibrated suspension at 22 C shaking at 400 rpm; after a H20 from 25 C
to 200 C, most of it in a .
total of 15 d recovered solid material by filter centrifugation step from -
100 C to 130 C;
(0.20-um PTFE membrane); examined material by
decomposition starts at T> 300 C
FT-Raman spectroscopy and PXRD; dried material for 5 min
under vacuum (10-20 mbar); examined material by
TG-FTIR.
PP415-P15 dissolved 100.6 mg of PP415-P1 in 0.2 mL of 1:2 DCM/IPE; FT-
Raman: Raman: corresponds to class 2, contains
obtained clear solution; observed precipitation of white solid PP415P15.0
solvent signals od
n
in < 1 min; added 0.2 mL of solvent mixture; covered vial PXRD: 137a
PXRD: corresponds to class 2
with single-layer tissue and let solvent evaporate under
cp
k=J
ambient conditions; obtained wet, white solid material after
1-
c..J
several hours; examined solid by FT-Raman spectroscopy ,
o
and PXRD.
c,.)
o,
A
140
Sample Experimental Description Test Methods
Result! Remarks
PP415-P16 dissolved 100.3 mg of PP415-P1 in 0.2 mL of 1:2 FT-
Raman: FT-Raman: corresponds to class 1,
Me01-1/toluene; obtained clear solution; covered vial with PP415P16.0
contains toluene solvent peaks
single-layer tissue and let solvent evaporate under ambient
conditions; obtained glassy material after several days;
examined material by FT-Raman spectroscopy.
PP415-P17 dissolved 101.0 mg of PP415-P1 in 0.3 mL of 1:3 FT-
Raman: Raman: corresponds to class 2, contains
Et0Ac/heptane; obtained clear solution; observed PP415P17.0
solvent signals
precipitation of white solid in < l mill; added 0.2 mL of PXRD: 138a
PXRD: corresponds to class 2
solvent mixture; covered vial with single-layer tissue and let
solvent evaporate under ambient conditions; obtained wet,
white solid material after several hours; examined solid by
FT-Raman spectroscopy and PXRD.
PP415-P18 dried material of PP415-
P15 under vacuum (2-20 mbar) at FT-Raman: Raman: corresponds to class 2
r.t. for ¨2 h; examined dry, white solid by FT-Raman PP415P18.0
PXRD: corresponds to class 2
spectroscopy, PXRD, and TG-FTIR. PXRD: 149a TG-FTIR:
loss of ¨7.0 wt.-% IPE in two
TG-FTIR: a4301
steps from ¨140 C to ¨250 C;
decomposition at T > 250 C
PP415-P19 dried material of PP415-
P17 under vacuum (2-20 mbar) at FT-Raman: Raman: corresponds to class 2
r.t. for ¨2 h; examined dry, white solid by FT-Raman PP415P19.0
PXRD: corresponds to class 2
spectroscopy, PXRD, and TG-FTIR. PXRD: 150a TG-FTIR:
loss of ¨7.6 wt.-% heptane in
TG-FTIR: a4302 two
steps from ¨140 C to ¨270 C;
decomposition at T > 270 C
CID
k=J
oc
141
Sample Experimental Description Test Methods
Result! Remarks
PP415-P20 suspended 98.8 mg of PP415-P1 in 2.0 mL of H90; heated FT-
Raman: Raman: corresponds to class 3, contains
suspension to 50 C; added slowly and stepwise 4.0 mL of PP415P20.0
solvent signals
acetone; obtained clear solution; heated solution to 55 C
PXRD: 226a .. PXRD: corresponds to class 3
and held at 55 C for 30 min; slowly cooled in 4 h 10 min to
C (at ¨0.2 Kimin); recovered solid by vacuum filtration
(P4 pore size); examined solid by FT-Raman spectroscopy
and PXRD.
PP415-P21 suspended 100.9 mg of PP415-P1 in 2.0 mL of cyclohexane; FT-
Raman: .. Raman: corresponds to class 2
heated suspension to 70 C; added slowly and stepwise PP415P21.0
PXRD: corresponds to class 2
0.5 mL of cyclohexane and 0.5 mL of Et0H; thin suspension
PXRD: 218a TG-FTIR: loss of ¨5.8 wt.-%
became thicker due to additional precipitation over course of TG-FTIR: a4326
cyclohexane in two steps from ¨140 C
solvent addition; heated suspension to 75 C and held at to ¨250
C; decomposition at T > 250 C
75 C for 30 min; slowly cooled in 5 h to 5 C (at
¨0.23 K/min); recovered solid by vacuum filtration (P4 pore
size); examined solid by FT-Raman spectroscopy and
PXRD; dried material for 5 min under vacuum (10-20
mbar); examined material by TG-FT1R.
PP415-P22 suspended 151.1 mg of PP415-P1 in 1.5 mL of toluene; FT-
Raman: Raman: corresponds to class 1, contains
heated suspension to 70 C; obtained clear solution; added
PP415P22.0 solvent signals
0.5 mL of MeCN; heated solution to 75 C and held at 75 C
for 30 min; slowly cooled in 5 h to 5 C (at ¨0.23 K/min);
observed clear solution and no precipitation; stirred clear
solution at 5 C for 2 d; observed no precipitation;
evaporated solvent under N2 flow at r.t.; obtained glassy
k=J
substance; examined it by FT-Raman spectroscopy.
oc
142
Sample Experimental Description Test Methods
Result! Remarks
PP415-P23 suspended 150.6 mg of PP415-P1 in 1.5 mL of dioxane; FT-
Raman: Raman: corresponds to class 1, contains
heated suspension to 70 C; obtained clear solution; added
PP415P23.0 solvent signals
0.5 mL of Et0Ac; heated solution to 75 C and held at 75 C
for 30 min; slowly cooled in 5 h to 5 C (at ¨0.23 K/min);
observed clear solution and no precipitation; stirred clear
solution at 5 C for 2 d; observed no precipitation;
evaporated solvent under N2 flow at r.t.; obtained glassy
substance; examined it by FT-Raman spectroscopy.
PP415-P24 suspended 99.4 mg of PP415-P1 in 0.3 mL of 1BuOH; FT-
Raman: Raman: corresponds to class 2, contains
heated suspension to 70 C; obtained clear solution; PP415P24.0
solvent signals
observed shortly thereafter precipitation of white solid;
PXRD: 219a PXRD: corresponds to class 2
added 0.5 mL 1BuOH; still suspension; heated suspension to TG-FTIR: a4325
TG-FTIR: loss of ¨16.6 wt.-% 1BuOH in
75 C and held at 75 C for 30 min; slowly cooled in 5 h to a step
from ¨50 C to ¨160 C, further
C (at ¨0.23 K/min); recovered solid by vacuum filtration loss of
1BuOH (6.6 wt.-%) in a second
(P4 pore size); examined solid by FT-Raman spectroscopy
step from 160 C to 230 C;
and PXRD; dried material for 5 min under vacuum (10-20
decomposition at T > 230 C
mbar); examined material by TG-FTIR.
PP415-P25 dried material of PP415-P25 under vacuum: at 60 C and FT-
Raman: Raman: corresponds to class 3
¨5 mbar for ¨1 h; at r.t. and ¨3 mbar for 4.5 d; at 60 C and
PP415P25.0 PXRD: corresponds to class 3
¨10 mbar for 1 h; at 40-50 C and 5-20 mbar for ¨20 h, PXRD: 258a TG-
FTIR: loss of ¨5.4 wt.-% 2PrOH
examined solid by FT-Raman spectroscopy, PXRD, and TG-FTIR:
a4337 from 50 C to 250 C, most of it in a step
TG-FTIR. from 170
C to 190 C, another loss of
¨1.0 wt.-% 2PrOH from 290 C to
320 C; decomposition at T > 320 C
i=J
oc
143
Sample Experimental Description Test Methods
Result! Remarks
PP415-P26 dried material of PP415-P13 under vacuum: at 60 C and FT-
Raman: Raman: corresponds to class 4
¨5 mbar for ¨1 h; at r.t. and ¨3 mbar for 4.5 d; at 60 C and
PP415P26.0 PXRD: corresponds to class 4
¨10 mbar for 1 h; at 40-50 C and 5-20 mbar for ¨2011, PXRD: 259a TG-
FTIR: loss of ¨2.8 wt.-% MeCN
examined solid by FT-Raman spectroscopy, PXRD, and TG-FTIR: a4335 from
170 C to 250 C; decomposition at
TG-FTIR.
T > 300 C
PP415-P27 dried material of PP415-P14 under vacuum: at 60 C and FT-
Raman: Raman: seems to corresponds to a
¨5 mbar for ¨1 h; at r.t. and ¨3 mbar for 4.5 d; at 60 C and
PP415P27.0 mixture of class 1 and class 5
¨10 mbar for 1 h; at 40-50 C and 5-20 mbar for ¨20 h, PXRD: 260a
PXRD: sample is only partially
examined solid by FT-Raman spectroscopy, PXRD, and TG-FTIR: a4336
crystalline; the few, broad peaks
TG-FTIR.
correspond to class 5; thus corresponds to
a mixture of the amorphous class 1 and
class 5
TG-FTIR: loss of ¨0.3 wt.-% from 25 C
to 290 C; decomposition at T > 290 C
PP415-P28 dried material of PP415-P21 under vacuum: at 60 C and FT-
Raman: Raman: corresponds to class 2
¨5 mbar for ¨1 h; at r.t. and ¨3 mbar for 4.5 d; at 60 C and
PP415P28.0 PXRD: corresponds to class 2, sample
¨10 mbar for 1 h; at 40-50 C and 5-20 mbar for ¨20 h, PXRD: 261a less
crystalline, as indicated by broader
examined solid by FT-Raman spectroscopy, PXRD, and TG-FT1R: a4334
peaks
TG-FTIR. TG-
FTIR: loss of ¨3.0 wt.-%
cyclohexane in two steps from ¨140 C
to ¨250 C; decomposition at T > 250 C
CID
oc
144
Sample Experimental Description Test Methods
Result! Remarks
PP415-P29 suspended 132.2 mg of PP415-P1 in 0.8 mL of 1:2 FT-
Raman: Raman: corresponds to class 2
Et0Ac/TEA; observed change in appearance of solid phase; PP415P29.0
PXRD: corresponds to class 2
agitated and sonicated; equilibrated suspension at 24 C PXRD: 282a TG-
FTIR: loss of ¨5.1 wt.-% Et0Ac and
shaking at 500 rpm; after 4 d recovered solid material by TG-FTIR:
a4346 TEA from ¨50 C to ¨220 C, most of it
filter centrifugation (0.20-pm PTFE membrane); examined in a step from 180
C to 210 C;
material by FT-Raman spectroscopy and PXRD; dried
decomposition at T > 220 C
material for 5 min under vacuum (10-20 mbar); examined
material by TG-FTIR.
PP415-P30 dried material of PP415-P7
under vacuum at 50-70 C and FT-Raman: Raman: corresponds to class 2
1-10 mbar for 3 days; examined solid by FT-Raman PP415P30.0
PXRD: corresponds to class 2
spectroscopy, PXRD, and TG-FTIR. PXRD: 290a TG-
FTIR: loss of ¨2.1 wt.-% heptane
TG-FTIR: a4347 (and
some Et0Ac) in two steps from
¨50 C to ¨250 C; decomposition at T>
250 C
PP415-P31 suspended 137.6 mg of
PP415-P1 in 2 mL of 9:1 H20/PEG FT-Raman: Raman: corresponds to class 1
400; obtained white suspension; equilibrated suspension at PP415P31.0
PXRD: amorphous, corresponds to class
24 C shaking at 400 rpm; after 5 d recovered solid material PXRD: 320a
1
by vacuum filtration; washed solid three times with small
amount of H20; examined material by FT-Raman
spectroscopy and PXRD.
CID
oc
145
Sample Experimental Description Test Methods
Result! Remarks
PP415-P32 dried material of PP415-P19 under vacuum at 80 C and FT-
Raman: Raman: corresponds to class 2
<1 X 10-3 mbar; after 1 d examined material by TG-FTIR PP415P32.0 PXRD:
corresponds to class 2, less
(a4362); continued drying; after a total of 3 d examined PXRD: 331a
crystalline
material by TG-FTIR (a4365), FT-Raman spectroscopy, and (P32A) TG-FTIR
P32: loss of 2.8 wt.-% heptane
PXRD as P32A. TG-FTIR: (25-250
C), most of it in a step from 170
a4362(P32) C to
200 C; decomposition at T > 250
a4365 (P32A)
C
TG-FTIR P32A: loss of 2.2 wt.-%
heptane (25-250 C), most of it in a step
from 170 C to 200 C; decomposition at
T > 250 C
PP415-P33 dried material of PP415-P19 under vacuum at 80 C and FT-
Raman: Raman: corresponds to class 3
<1 x10-3 mbar; after 3 d examined material by FT-Raman PP415P33.0
PXRD: corresponds to class 3
spectroscopy, PXRD and TG-FTIR. PXRD: 332a TG-FTIR:
loss of ¨4.2 wt.-% 2PrOH (50-
TG-FTIR: a4366 210 C),
most of it in a step from 160 C
to 190 C, another loss of ¨0.5 wt.-%
2PrOH (210 C to 290 C);
decomposition at T > 290 C
PP415-P34 dried material of PP415-P19 under vacuum at 80 C and FT-
Raman: Raman: corresponds to class 2
<1 x10-3 mbar; after 3 d examined material by FT-Raman PP415P34.0 PXRD:
corresponds to class 2, less
spectroscopy, PXRD and TG-FTIR. PXRD: 333a
crystalline
TG-FTIR: a4367 TG-
FTIR: loss of'-2.3 wt.-%
cyclohexane in two steps from 25 C to
k=J
270 C; decomposition at T > 270 C
oc
146
Sample Experimental Description Test Methods
Result! Remarks
PP415-P35 suspended 158.4 mg of PP415-P 1 in 0.2 mL of MeCN/H20 FT-
Raman: Raman: corresponds to class 4, contains
obtained two clear, separated phases; equilibrated solution at
PP415P35.0 solvent signals
24 C shaking at 400 rpm; after 3 d observed thick PXRD: 326a
PXRD: corresponds to class 4
suspension; added 0.1 mL of the solvent mixture; continued TG-FTIR:
a4363 TG-FTIR: loss of 2.9 wt.-% MeCN from
equilibration of suspension at 24 C shaking at 400 rpm; 25 C to
250 C; decomposition at T>
after a total of 5 d recovered solid material by filter
250 C
centrifugation (0.20-ium PTFE membrane); examined
material by FT-Raman spectroscopy; dried material for 10
min under vacuum (10-20 mbar); examined material by
PXRD and TG-FTIR.
2
PP415-P36 dried material of PP415-P35 under vacuum at 80 C and FT-
Raman: Raman: corresponds to class 4
<1x10-3 mbar; after 3 d examined material by FT-Raman PP415P36.0
PXRD: corresponds to class 4
spectroscopy, PXRD and TG-FTTR. PXRD: 339a TG-FTIR:
loss of ¨0.6 wt.-% (probably
TG-FTIR: a4369 H20
and/or MeCN) in two steps from 25
C to 280 C; decomposition at T > 280
C
CID
oc
147
Sample Experimental Description Test Methods
Result! Remarks
PP415-P37 dried material of PP415-P35 under N2 flow at 80 C; after 3 FT-
Raman: Raman: corresponds to class 4
d examined material by FT-Raman spectroscopy, PXRD, PP415P37.0
PXRD: corresponds to class 4
TG-FTIR, DSC, and DVS. PXRD: 340a TG-
FTIR: loss of ¨0.9 wt.-% (probably
TG-FTIR: a4370 H20 and/or MeCN) in two steps from 25
DSC: d_9907 C to
280 C; decomposition at T > 280
DVS: dvs1176
C
post-DVS PXRD: DSC:
sharp endothermic peak at T =
363a
196.1 C (AH = 29.31 J/g); no
decomposition up to 270 C
DVS: slightly hygroscopic; Am = +0.7%
(50%¨>85% r.h.); total mass gain of 2.1
wt.-% from 0% r.h. to 95% r.h.
post-DVS PXRD: corresponds to class 4
PP415-P38 DSC experiment: combined 1.275 mg of PP415-P1 and
DSC: d_9917 DSC
1.344 mg of PP415-P36; equilibrated for 3 min under N2;
heated sample from -50 C to 270 C at 10 K/min.
PP415-P39 DSC experiment: combined 2.17 mg of PP415-P1 and 2.20
DSC: d_9923 DSC
mg of PP415-P36; mixed solids using a spatula; equilibrated
for 3 mm under N2; heated sample from -50 C to 173 C at
10Kimin; held at 173 C for 30 min; heated from 173 C to
270 C at 10 K/min.
PP415-P40 received ¨5 g of 63415, batch #: 2083-69-DC on May 27,
PXRD: 390a PXRD: corresponds to class 2
2011;
c7)
MW ¨ 554.7 g/mol, C33H44F2N203
oc
148
Sample Experimental Description Test Methods
Result! Remarks
PP415-P41 suspended 101.3 mg of PP415-P40 in 0.20 mL of THF/I-120
PXRD: 400a PXRD: corresponds to class 5
(1:1); obtained white suspension; equilibrated suspension at
24 C; after 3 days recovered solid by filter centrifugation
(0.2-pm PTFE membrane); dried solid material under
vacuum for 5 min; examined solid by PXRD.
PP415-P42 dissolved 104.6 mg of PP415-P40 in 0.20 mL of THE;
PXRD: 405a PXRD: amorphous (class 1)
obtained clear solution; evaporated solvent under N2 flow
overnight; obtained white solid; examined solid by PXRD.
PP415-P43 dissolved 101.8 mg of PP415-P40 in 0.20 mL of
PXRD: 429a PXRD: corresponds to class 2
THF/hexane (8:2); obtained clear solution; evaporated
solvent under N2 flow overnight; obtained white solid;
examined solid by PXRD.
PP415-P44 dried material of PP415-P41 under vacuum (-100 mbar) at PXRD:
474a, 482a PXRD: both mainly amorphous (class 1),
80 C; examined solid after 1 day by PXRD (474a); TG-FTIR: a4401 some
broad peaks with low intensity
continued drying overnight; examined solid again by PXRD TG-
FTIR: ¨0.9 wt.-% THE 25-280 C,
(482a) and TG-FTIR.
decomposition at T > 300 C
PP415-P45 suspended 3.03 g of PP415-P40 in 6.0 mL of THF/H20
PXRD: 471a PXRD: corresponds to class 5
(1:1); obtained white suspension; equilibrated suspension at
r.t.; after 1 day recovered small aliquot by filter
centrifugation and examined it by PXRD; recovered solid of
whole sample by vacuum filtration; dried sample for 10 min
under vacuum (-10 mbar).
PP415-P46 dried material of PP415-P45 under vacuum (-100 mbar) at PXRD:
481a, 496a PXRD: both mainly amorphous (class 1),
80 C; examined solid after drying overnight by PXRD TG-FTIR: a4410 some
broad peaks with low intensity
(481a); continued drying; after 4 days examine solid as TG-
FTIR: ¨0.4 wt.-% H20 25-250 C,
PP415-P46a by PXRD (496a) and TG-FTIR.
decomposition at T > 250 C oe
149
Sample Experimental Description Test Methods
Result! Remarks
PP415-P47 dissolved 101.8 mg of PP415-P40 in 0.4 mL of Et0Ac;
PXRD: 492a PXRD: corresponds to class 2
obtained clear solution; evaporated solvent under N2 flow TG-FTIR: a4412
TG-FTIR: ¨6.2 wt.-% Et0Ac 25-170 C,
overnight; obtained white solid; examined solid by PXRD 1.7 wt.-% Et0Ac 170-
240 C,
and TG-FTIR.
decomposition at T > 240 C
PP415-P48 dissolved 101.1 mg of PP415-P40 in 0.4 mL of ethyl
PXRD: 493a PXRD: corresponds to class 2
formate; obtained clear solution; evaporated solvent under TG-FTIR: a4413
TG-FTIR: ¨3.5 wt.-% ethyl formate 25-
N2 flow overnight; obtained white solid; examined solid by .. 200 C,
decomposition at T > 200 C
PXRD and TG-FTIR.
PP415-P49 dissolved 205.3 mg of PP415-P40 in 0.3 mL of acetone;
PXRD: 593a PXRD: amorphous (class 1)
obtained clear solution; added dropwise to 30.0 mL of H20
(pre-cooled to 5 C); obtained thin white suspension; stirred
thin suspension at 5 C overnight; obtained thicker white
suspension; recovered solid by vacuum filtration (pore size
P4); obtained 188.3 mg of white solid; examined solid by
PXRD.
CID
oc
150
Table 31. Parameters of FIG. 51
r.)
Compound NOx Levels (% vs LPS
Controls)
13 mg/kg 25 mg/kg
50 mg/kg
RTA 405 * 44% 26%
18%
63415 30% 18%
16%
Table 32. 63415: Primary In Vivo ADMET ¨ Key Primary ADMET Assays and
Endpoints
Assay Key
Endpoints
Tolerability, body weight, clinical chemistry
14-day mouse toxicity Tissue distribution
Nrf2 target gene mRNA expression & enzyme activation in liver
Tolerability, body weight, clinical chemistry, & limited histopathology
14-day rat toxicity Tissue distribution and plasma
TK
Nrf2 target gene mRNA expression & enzyme activation in liver
Tolerability, body weight, clinical chemistry, & limited histopathology
14-day monkey toxicity Tissue distribution and plasma
TK
Nrf2 target gene mRNA expression and enzyme activation in multiple tissues &
PBMCs
CID
oc
151
Table 33. Parameters of FIG. 54
Vehicle 63415
Dose (mg/kg) 0 10 30
100
4-
ALT (U/L) 100 39 63
91
AST (U/L) 156 98 147
167
ALP (U/L) 120 131 110
98
Tot Bil (mg/dL) <0.2 <0.2 <0.2
<0.2
BUN (mg/dL) 17 15 15
15
Cr (mg/dL) <0.2 <0.2 <0.2
<0.2
2
Glu (mg/dL) 288 307 285
273
C7)
4-
152
0
r.)
Table 34. 63415 is Negative for Genotoxicity in the In Vivo Micronucleus Study
=
0-
w
PCE/Total
,
,-,
Treatment Number of MPCE/1000 PCE
Number of MPCE/PCE =,
w
Erythrocytes Change from Control (%)
w
(n=5/group) (Mean +/- SD)
Scored A
A
(Mean +/- SD)
24-h timepoint
Sesame Oil 0.588 0.04 - 0.2 0.27
2/10000
125 mg/kg 0.543 0.03 -8 0.3 + 0.27
3/10000
250 mg/kg 0.520 0.06 -12 0.3 0.27
3/10000
500 mg,/kg 0.426 0.07 -28 0.0 + 0.00
0/10000 0
1000 mg/kg 0.498 0.05 -15 0.2 0.27
2/10000 2
0
.,
1500 mg/kg 0.499 + 0.06 -15 0.4 + 0.22
4/10000 ..,
0
2000 mg/kg 0.531 0.05 -10 0.2 0.27
2/10000 ,,
.
48-h timepoint
,.
-
Sesame Oil 0.526 0.05 - 0.3 0.27
3/10000
125 mg/kg 0.453 0.03 -14 0.2 + 0.27
2/10000
250 mg/kg 0.391 0.02 -26 0.2 0.27
2/10000
500 mg/kg 0.339 + 0.05 -36 0.3 0.45
3/10000
1000 mg/kg 0.344 +0.04 -35 0.1 + 0.22
1/10000
od
1500 mg/kg 0.376 0.05 -39 0.4 0.42
4/10000 n
i-i
2000 mg/kg 0.360 0.03 -32 0.1 0.22
1/10000 c7)
0-
w
,
w
Go
A
153
0
Table 35. Parameters of FIG. 35
r.)
o
Tot
ALT
w
ALT AST ALP Tot Bil BUN Cr Albumin
Glucose Chol TG ,
1-
Treatment Day Prot
o
w
(U/L) (U/L) (U/L) (mg/dL) (mg/dL) (mg/dL) (g/dL)
(mg/dL) (mg/dL) (mg/dL) t,4
(g/dL)
A
A
BL 30 29 320 0.15 23 0.63 7.2 4.1 87 124
52
Vehicle Day 37 37 345 0.23 18 0.63 6.9 4.1 63 130 64
14
BL 46 32 351 0.18 35 0.78 7.4 4 74 146
51
mg/kg Day
46 38 382 0.23 27 0.68 7.2 4
39 144 82
14
0
BL 32 32 409 0.18 23 0.7 7.3 4.2 85 125
47 2
0
.,
30 mg/kg Dav
.3-'
47 43 416 0.2 20 0.58 7.2 4
53 122 64 .
,,
.
BL 32 35 381 0.15 24 0.7 6.9 4 96 137
37
i;
100 mg/kg Day
43 37 390 0.18 24 0.55 6 3.2
32 93 61
14
od
n
1-i
C7)
0-
w
,
o
w
Go
o
A
154
Table 36. In Vitro Activity of 63415 and 63355
63415 63355
4-
NO 1050 (nM), RAW264.7 4.0 1 0.63 0.06
WST-1 Ws() (nM), RAW264.7 125 150
NQ01-ARE (fold at 62.5 nM in
5.3 1.0 6.5 0.9
HuH7)
2
Table 37. Parameters of FIG. 52
Compound Plasma Whole Blood Brain Liver
Lung Kidney
RTA 405 (nM) 130 1165 93 1143
1631 2357
63415 (nM) 51 679 1081 985
533 1604
Table 38. Parameters of FIG. 53
Compound Liver Lung
Kidney
RTA 405 1.93 1.48
8.25
63415 10.9 1.75
10.9
c7)
155
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
* * * * * * * * * * * * * * * *
All of the compounds, polymorphs, formulations, and methods disclosed and
claimed herein can be made and executed without undue experimentation in light
of
the present disclosure. While the compounds, polymorphs, formulations, and
methods of this invention have been described in terms of preferred
embodiments, it
will be apparent to those of skill in the art that variations may be applied
to the
compounds, polymorphs, formulations, and methods, as well as in the steps or
in the
sequence of steps of the method described herein without departing from the
concept,
spirit, and scope of the invention. More specifically, it will be apparent
that certain
agents which are both chemically and physiologically related may be
substituted for
the agents described herein while the same or similar results would be
achieved. All
such similar substitutes and modifications apparent to those skilled in the
art are
deemed to be within the spirit, scope and concept of the invention as defined
by the
appended claims.
156
REFERENCES
The following references to the extent that they provide exemplary procedural
or other details supplementary to those set forth herein.
U.S. Patent 6,326,507
U.S. Patent 6,974,801
U.S. Patent 7,915,402
U.S. Patent 7,943,778
U.S. Patent 8,124,799
U.S. Patent 8,129,429
U.S. Patent Publication 2009/0060873
Abraham and Kappas, Free Radical Biol. Med., 39:1-25, 2005.
Aghajan et aL,J Gastroenterol Hepatot , Suppl 2:10-14, 2012.
Ahmad et. al., Cancer Res., 68:2920-2926, 2008.
Ahmad et. al., J. Biol. Chem., 281:35764-9, 2006.
Angulo et al., Eur. .1. Immunol., 30:1263-1271, 2000.
Araujo et. al., J. ImmunoL,171(3):1572-1580, 2003.
Arend and Dayer, Arthritis Rheum., 38:151-160, 1995.
Bach, Hum. Immunol, 67(6):430-432, 2006.
Bagasra et aL, Proc. NatL Acad. Sci. USA, 92:12041-12045, 1995.
Blake et. al. Am J Re.spir Cell Mol Biol, 42:524-36, 2010.
Berridge et aL, Biochemica, 4: 14-19, 1996.
Botoman et al., Am. Earn. Physician, 57(1):57-68, 1998.
Brandt et al., Arthritis Rheum., 43:1346-1352, 2000.
Brewerton et al., Lancet., 1:904-907, 1973a.
Brewerton et al., Lancet., 1:956-957, 1973b.
Brontc et al., Trends Immunol., 24:302-306, 2003.
Brown and DuBois .1 Clin. Oncol., 23:2840-2855, 2005.
Brynskov etal., N. Engl. J. Med., 321(13):845-850, 1989.
Cantin et. al., J Gin invest, 79:1665-73, 1987.
Cai etal., Nat. Med., 11(2):183-190, 2005.
157
CA 2869783 2019-09-05
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
Calin and Taurog, In: The Spondylarthritides, CalM et al. (Eds.), Oxford, UK.
Oxford
University Press, 179, 1998.
Car et. al., Am J Respir Crit Care Med, 149:655-9, 1994.
Cemuda-Morollon et. al., J Biol Chem, 276:35530-6, 2001.
Chan and Kan, Proc Natl Acad Sci USA, 96:12731-6, 1999.
Chauhan and Chauhan, Pathophysiology, , 13(3):171-181. 2006.
Chen and Kunsch, Curr Pharm Des, 10:879-91, 2004.
Cho et. al., Am J Respir Cell Mol Biol, 26:175-82, 2002.
Crowell et al., Mol. Cancer Ther., 2:815-823, 2003.
Cuzzocrea et. al., Mol Pharmacol, 61:997-1007, 2002.
Dickerson et. al., Prog Neuropsychopharmacol Biol. Psychiatry, March 6, 2007.
Dinarello, Int. Rev. Immunol., 16:457-499, 1998.
Dinkova-Kostova et. al., Proc. Natl. Acad. ,S'ci. USA, 102(12):4584-4589,
2005.
Dionne et al., Clin. Exp. Imunol., 112(3):435-442, 1998.
Douglas et. al., Am J Respir Crit Care Med, 158:220-5, 1998.
Dudhgaonkar et. al., Eur. J. Pain, 10(7):573-9, 2006.
Eastgate et al., Lancet, 2(8613):706-9, 1988.
Eikelcnboom et al., Glia, 40(2):232-239, 2002.
Ettehadi et al., Clin. Exp. Immunol., 96(1):146-151, 1994.
Fischer et. al.,. Int J Chron Obstruct Pulmon Dis, 6:413-21, 2011.
Forstermann, Biol. Chem., 387:1521, 2006.
Funakoshi et al., Digestion, 59(1):73-78, 1998.
Gebel et. al., Toxicol Sci, 115:238-52, 2010.
Gehrmann etal., Glia, 15(2):141-151, 1995.
Genain and Nauser, Mol. Med., 75:187-197, 1997.
Goodman et al., Kidney Int., 72(8):945-953, 2007.
Graeber et al., Glia, 40(2):252-259, 2002.
Greten et ed., Cell, 118:285-296, 2004.
Grivennikov and Karin, Cvtokine Growth Factor Rev., 21(1):11-19, 2010.
Guilherme et al., Nat. Rev. Mol. Cell Biol., 9(5):367-77, 2008.
Gwee et al., Gut., 44(3):400-406., 1999.
Hallgren et. al., Am Rev Respir Dis, 139:373-7, 1989.
Hahn and Tsao, In: Dubois' Lupus Erythematosus, 4th Ed, Wallace and Hahn
(Eds.),
Lea and Febiger, Philadelphia, 195-201, 1993.
158
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
Handbook of Pharmaceutical Salts: Properties, and Use, Stahl and Wermuth
Eds.),
Verlag Helvetica Chimica Acta, 2002.
Hanson et. al., BMC Medical Genetics, 6(7), 2005.
Hansson et al., Annu. Rev. PathoL Mech. Dis., 1:297-329, 2006.
Hayden and Ghosh, Cell, 132:344-62, 2008.
He and Karin, Cell Res., 21(1):159-168, 2011.
Honda et. al. Bioorg. Med. Chem. Lett., 12:1027-1030, 2002.
Honda et. al., Bioorg. Med. Chem. Lett., 16(24):6306-6309, 2006.
Honda et. al., Bioorg. Med. Chem. Lett., 7:1623-1628, 1997.
Honda et. al., Bioorg. Med. Chem. Lett., 8(19):2711-2714, 1998.
Honda et. al., Bioorg. Med. Chem. Lett., 9(24):3429-3434, 1999.
Honda et. al., J. Med. Chem., 43:4233-4246, 2000a.
Honda et. al., J. Med. Chem., 43:1866-1877, 2000b.
Hotamisligil, Nature, 444(7120:860-7, 2006.
Iizuka et. al., Genes Cells, 10:1113-25, 2005.
1kezaki et al., Food Chem Toxicol, 34:327-35, 1996.
Ishii et. al., ,I Immunol, 175:6968-75, 2005.
1shikawa et. al., Circulation, 104(15):1831-1836, 2001.
Tshizawa and Dickson, J. IVeuropathol. Exp. NeuroL, 60(6):647-657, 2001.
Jarvis, Curr. Opin. RheumatoL, 10(5):459-467, 1998.
Jarvis, Pediatr. Ann., 31(7):437-446, 2002.
Jonsson et at., Oral Dis., 8(3):130-140, 2002.
Jonsson et al., Trends ImmunoL, 22(12):653-654, 2001.
Kahle et al., Ann. Rheum. Dis., 51:731-734, 1992.
Kaltschmidt et al., Proc. Natl. Acad. Sci. USA, 94:2642-2647, 1997.
Karin, Nature, 441(7092):431-436, 2006.
Kawakami et. al., Brain Dev., 28(4):243-246, 2006.
Kawamoto et. al., J Biol Chem, 275:11291-9, 2000.
Kendall-Tackett, Trauma Violence Abuse, 8(2):117-126, 2007.
Kensler et. al., Annu Rev Pharmacol Toxicol, 47:89-116, 2007.
Kikuchi et. al., Respir Res,11:31, 2010.
Kim et. aL, Mutat Res, 690:12-23, 2010.
King et. al., Am J Respir Grit Care Med, 184:92-99, 2011.
Kinnula et. al., Am J Respir Crit Care Med, 172:417-22, 2005.
159
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
Klingsberg et. al., Respirology,15:19-31, 2010.
Kortylewski et aL Nat. Med., 11:1314-1321, 2005.
Kotzin and O'Dell, In: Samler's Immunologic Diseases, 5th Ed., Frank et al.
(Eds.),
Little Brown & Co., Boston, 667-697, 1995.
Kotzin, Cell, 85:303-306, 1996.
Kruger et. al., J. Pharmacol. Exp. Tier., 319(3):1144-1152, 2006.
Kuboyama, Kurume Med. J, 45(1):33-37, 1998.
Lee et. al., Mol Cell, 36:131-40, 2009.
Lee et. al., Glia., 55(7):712-22, 2007.
Lencz et. al., Mol. Psychiatry, 12(6):572-80, 2007.
Levonen et. al., Biochem J, 378:373-82, 2004.
Li and Kong, Mo/ Carcinog, 48:91-104, 2009.
Liby et. al., Cancer Res., 65(11):4789-4798, 2005.
Liby et. al., Mol. Cancer Ther., 6(7):2113-9, 2007b.
Liby et. al., Nat. Rev. Cancer, 7(5):357-356, 2007a.
Lipsky, In: Harrison's' principles of internal medicine, Fauci et cd.(Eds.),
14th Ed.,
NY, McGraw-Hill, 1880-1888, 1998.
Liu et. al., FASEB J., 20(2):207-216, 2006.
Lu et. al., J. Clin. Invest., 121(10):4015-29, 2011.
Lugering et al., Ital. J. Gastroenterol. Hepatol., 30(3):338-344, 1998.
Malhotra et. al., Am J Respir Crit Care Med, 178:592-604, 2008.
March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure,
2007.
Mazur et al., Cell Microbiol., 9(7):1683-94, 2007.
Mazzoni et al., J. Immunol., 168:689-695, 2002.
McAlindon et al., Gut, 42(2):214-219, 1998.
McGeer and McGeer, Brain Res. Brain Res. Rev., 21:195-218, 1995.
McGeer et al., Neurology, 19:331-338, 1996.
McGonagle et al., Arthritis Rheum., 41:694-700, 1998.
McGonagle et al., Curr. Opin. Rheumatol., 11:244-250, 1999.
McIver et. al., Pain, 120(1-2):161-9, 2005.
Mease et al., Lancet, 356:385-390, 2000.
Merrill and Benvenist, Trends Neurosci., 19:331-338, 1996.
Mochizuki et. al., Am J Respir Crit Care Med, 171:1260-6, 2005.
160
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
Morbidity & Mortality: 2009 Chart Book on Cardiovascular, Lung, and Blood
Diseases. National Heart, Lung, and Blood Institute, 2009.
Morris et. al., J. Mol. Med., 80(2):96-104, 2002.
Morse and Choi, Am. I Respir. Crit. Care Med., 172(6):660-670, 2005.
Morse and Choi, Am. I Respir. Crit. Care Med., 27(1):8-16, 2002.
Nath et al., Neurology, 66(1):149-150, 2006.
Neal et al., B311, 314(7083):779-782, 1997.
Nichols, Drug News Perspect., 17(2):99-104, 2004.
Ohnishi et al., Int. Immunol., 6:817-830, 1994.
__ Ogushi et. al., J Med Invest, 44:53-8, 1997.
Pall, Med. Hypoth., 69:821-825, 2007.
Parambil et. al., Chest, 128:3310-5, 2005.
Partsch et al., Br. J. Rheumatol., 24:518-523, 1997.
Pergola et. al., N Engl J Med, 365:327-336, 2011.
Pica et al., Antimicrob Agents Chemother., 44(1):200-4, 2000.
Pimentel et al., Am. I Gastroenterol. , 95(12):3503-3506, 2000.
Place et. al., Clin. Cancer Res., 9(7):2798-806, 2003.
Prochaska and Santamaria, Anal Biochem., 169:328-336, 1988.
Rajakariar et. al., Proc. Natl. Acad. Sci. USA, 104(52):20979-84, 2007.
Rangasamy et. al., J Clin Invest, 114:1248-59, 2004.
Rangasamy et al., J Exp Med, 202:47-59, 2005.
Reimund et al., Eur. I Clin. Invest., 28(2):145-150, 1998.
Renauld, J Clin Pathol, 54:577-89, 2001.
Rogler and Andus, World J. Surg., 22(4):382-389, 1998.
.. Rooney et aL, Rheumato/. Int., 10:217-219, 1990.
Ross et. al., Am. I Clin. PathoL, 120(Suppl):S53-71, 2003.
Ross et. al., Expert Rev. A/IoL Diagn., 3(5):573-585, 2003.
Rossi et. al., Nature, 403:103-8, 2000.
Rostom etal., Ann. Intern. Med., 146, 376-389, 2007.
Ruster et. al., Scand. I Rheumatot, 34(6):460-3, 2005.
Sacerdoti et. al., Curr Neurovase Res., 2(2):103-111, 2005.
Saha et. al., J Biol Chem, 285:40581-92, 2010.
Saiki et al., Scand. 1 Gastroenterol., 33(6):616-622, 1998.
Salomonsson et al., Scand. 1 Immunot, 55(4):336-342, 2002.
161
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
Salvemini et. al., J. Clin. Invest., 93(5):1940-1947, 1994.
Sarchielli et. al., Cephalalgia, 26(9):1071-1079 , 2006.
Satoh et. al., Proc. Natl. Acad. Sci. USA, 103(3):768-773, 2006.
Schlosstein et al., NEJ. Medicine, 288:704-706, 1973.
Schulz et. al., Antioxid. Redox. Sig., 10:115, 2008.
Selman et. al., Ann Intern IVIed, 134:136-51, 2001.
Sheppard, J Clin Invest, 107:1501-2, 2001.
Shishodia et. al., Clin Cancer Res, 12:1828-38, 2006.
Simonian and Coyle, Annu. Rev. Pharmacol. Toxicol., 36:83-106, 1996.
Singh et. al., Free Rad Biol Med, 46:376-386, 2009.
Sinha et al., Cancer Res., 67:4507-4513, 2007.
Sporn et. al., JZ\/at Prod, 74:537-45, 2011.
Sriram et. al., Pulm Pharmacol Ther, 22:221-36, 2009.
Stack et al., Lancet, 349(9050:521-524, 1997.
Standiford et. al., Chest, 103:121S, 1993.
Standiford et. al., J Immunol, 151:2852-63 , 1993.
Stewart et al., Neurology, 48:626-632, 1997.
Straus et. al., Proc Nati Acad Sci USA, 97:4844-9, 2000.
Strejan et. al., .1 Neuroimmunol., 7:27, 1984.
Strieter, Am J Respir Crit Care Med, 165:1206-7, 2002.
Suh et. al., Cancer Res., 58:717-723, 1998.
Suh et. al., Cancer Res., 59(2):336-341, 1999.
Sussan et al., Proc Nati Acad Sci USA, 106:250-5, 2009.
Szabo et. al., Nature Rev. Drug Disc., 6:662-680, 2007.
Takahashi et. al., Cancer Res., 57:1233-1237, 1997.
Tamir and Tannebaum, Biochim. Biophys. Acta, 1288:F31-F36, 1996.
Taniguchi et. al., Eur Respir J, 35:821-9, 2010.
Targan et al., N. Engl. J Med., 337(15):1029-1035, 1997.
Thimmulappa et al., Cancer Research, 62: 5196-5203, 2002.
Third Report of the National Cholesterol Education Program Expert Panel on
Detection, Evaluation and Treatment of High Blood Cholesterol in Adults
(Adult Treatment Panel III, or ATP III), National Institutes of Health, 2001,
NIH Publication No. 01-3670.
Touzani et al., J. Neuroimmunol., 100(1-2):203-215, 1999.
162
CA 02869783 2014-10-06
WO 2013/163344
PCT/US2013/038064
Tumlin et al., Am. J Cardiol., 98(6A):14K-20K, 2006.
van den Berg, Semin. Arthritis Rheum., 30(5S-2):7-16, 2001.
van Dullemen etal., Gastroenterol., 109(1):129-135, 1995.
van Hogezand and Verspaget, Drugs, 56(3):299-305, 1998.
Vazquez etal., J Virol., 79(7):4479-91, 2005.
Wakabayashi, et. al., Antioxid Redox Signal, 13:1649-63, 2010.
Wang etal., Cancer Res., 66:10983-10994, 2006.
Wardle, Nephrol. Dial. Transplant., 16(9):1764-8, 2001.
Weyand and Goronzy, Ann. NY Acad. Sci., 987:140-149, 2003.
Williams etal., Clin. Neurosci., 2(3-4):229-245, 1994.
Wordsworth, In: Genes and Arthritis, Brit. Medical Bulletin, 51:249-266, 1995.
Wright, Clin. Orthop. Related Res.,143 :8-14, 1979.
Wu et. al., Toxicol Sci, 123:590-600, 2011.
Xie et. al., J. Biol. Chem., 270(12):6894-6900, 1995.
Yates et al., 1146l. Cancer Ther, 6(1):154-162, 2007.
Yore et. al., Mol Cancer Ther, 5:3232-9, 2006.
Yoh etal., Kidney Int., 60(4):1343-1353, 2001.
Yu et cd., Nat. Rev. lmmunol., 7:41-51, 2007.
Zhou et cll., Am. J Pathol., 166(1):27-37, 2005.
Zhou etal., Cancer Sc., 98:882-889, 2007.
Zingarelli et al., ..I. Immunol., 171(12):6827-6837, 2003.
163