Note: Descriptions are shown in the official language in which they were submitted.
1
COMPOSÉS BIFONCTIONNELS CARBAMOYL/ACIDE PHOSPHONIQUE OU PHOSPHONATE
UTILES COMME LIGANDS DE L'URANIUM(VI), LEURS PROCÉDÉS DE SYNTHESE ET LEURS
UTILISATIONS
DESCRIPTION
DOMAINE TECHNIQUE
L'invention se rapporte au domaine de l'extraction de l'uranium de
milieux aqueux contenant de l'acide phosphorique.
Plus spécifiquement, l'invention se rapporte à de nouveaux composés
bifonctionnels qui sont capables d'extraire seuls (c'est-à-dire en l'absence
de toute autre
molécule extractante) l'uranium(VI) d'une solution aqueuse d'acide
phosphorique et ce, à
la fois très efficacement et avec une haute sélectivité vis-à-vis des autres
cations
métalliques susceptibles d'être présents dans cette solution et, en
particulier, vis-à-vis du
fer(III).
Elle se rapporte aussi à des procédés qui permettent de synthétiser ces
composés bifonctionnels.
Elle se rapporte également aux utilisations de ces composés
bifonctionnels comme ligands de l'uranium(VI) et, notamment, pour extraire
l'uranium(VI) d'une solution aqueuse d'acide phosphorique telle qu'une
solution issue de
l'attaque d'un phosphate naturel par de l'acide sulfurique.
Elle se rapporte en outre à un procédé qui permet de récupérer
l'uranium présent dans une solution aqueuse d'acide phosphorique issue de
l'attaque
d'un phosphate naturel par de l'acide sulfurique et qui met en oeuvre lesdits
composés.
L'invention trouve notamment application dans le traitement des
phosphates naturels en vue de valoriser l'uranium présent dans ces phosphates.
ÉTAT DE LA TECHNIQUE ANTÉRIEURE
Les phosphates naturels (ou minerais de phosphate), qui sont utilisés
pour la fabrication de l'acide phosphorique et d'engrais, contiennent de
l'uranium à des
CA 2872084 2020-01-22
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
2
teneurs qui peuvent varier de quelques dizaines de ppm à plusieurs milliers de
ppm ainsi
que des quantités variables d'autres métaux.
L'uranium présent dans les phosphates naturels se retrouve presque
intégralement dans les solutions aqueuses d'acide phosphorique qui sont issues
de
l'attaque sulfurique de ces phosphates.
Le potentiel de récupération de l'uranium contenu dans ces minerais de
phosphate est de 14 000 tonnes/an, soit environ 25% de la production annuelle
actuelle
de l'uranium, ce qui représente une source d'approvisionnement en uranium non
négligeable.
Aussi, de nombreuses équipes de recherche se sont-elles intéressées à
l'extraction de l'uranium à partir d'un milieu phosphorique.
Pour des raisons économiques, la valorisation de l'uranium doit
s'effectuer à partir de solutions aqueuses d'acide phosphorique concentrées et
non
diluées, même si l'extraction de l'uranium serait plus aisée à faible acidité.
L'attaque des phosphates naturels par de l'acide sulfurique transforme
le phosphate tricalcique en acide phosphorique H3PO4 à 30% d'anhydride de
phosphate
P205, ainsi qu'en sulfate de calcium insoluble (gypse). Cette lixiviation
solubilise l'uranium
et, divers autres métaux (fer, vanadium, cadmium, molybdène, etc.).
Dans le procédé, qui a été utilisé par la plupart des unités industrielles
pour récupérer l'uranium présent dans des phosphates naturels et qui est connu
sous le
nom de procédé Oak Ridge (pour avoir été mis au point par le laboratoire
national
d'Oak Ridge (USA), brevet US 3,711,591, référence [1]), le flux aqueux d'acide
phosphorique résultant de l'attaque des phosphates par de l'acide sulfurique
est soumis à
une opération d'oxydation par bullage (l'oxygène de l'air servant alors
d'agent oxydant)
ou par ajout d'un oxydant, notamment une solution de chlorate de sodium NaC103
ou
d'eau oxygénée, afin de convertir la totalité de l'uranium en uranium(V1). La
température
de ce flux est ensuite ramenée à 40-45 C et l'uranium(V1) est extrait, dans un
premier
cycle d'extraction, par un mélange synergique d'acide di-(2-
éthylhexyl)phosphorique (ou
HDEHP) et d'oxyde de trioctylphosphine (ou TOPO).
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
3
L'effet synergique maximal de ce mélange vis-à-vis de l'uranium(VI) (et
de la sélectivité U/Fe) est obtenu dans une proportion relative de 4 molécules
d'HDEHP
pour 1 molécule de TOPO et la composition de la phase organique de référence
est la
suivante : 0,5 mol/L d'HDEHP + 0,125 mol/L de TOPO dans du n-dodécane ou dans
un
diluant aliphatique équivalent.
L'uranium est ensuite désextrait par une solution aqueuse phosphorique
contenant des ions Fe2+ qui réduisent l'uranium(VI) en uranium(IV) et
favorisent ainsi sa
désextraction en phase aqueuse. Cette désextraction permet de concentrer
l'uranium
d'un facteur environ 70.
Dans un second cycle d'extraction, le flux aqueux contenant
l'uranium(IV) ainsi désextrait est soumis à son tour à une opération
d'oxydation pour
convertir à l'état d'oxydation VI la totalité de l'uranium qu'il contient,
puis l'uranium(VI)
est extrait avec le mélange synergique HDEHP/TOPO.
La phase organique résultant de cette extraction est lavée à l'eau pour
éliminer l'acide phosphorique extrait puis soumise à une opération de
désextraction avec
une solution de carbonate d'ammonium (NH4)2CO3 qui conduit in fine à la
précipitation de
tricarbonate d'uranyle et d'ammonium qui, après calcination, donne le
sesquioxyde
d'uranium U308.
Dans le cas de l'utilisation de ce procédé pour purifier l'uranium
.. contenu dans des phosphates naturels très concentrés en uranium, différents
inconvénients sont à noter, à savoir que :
¨ les
coefficients de distribution de l'uranium(VI) se révèlent assez
faibles, ce qui nécessite de travailler avec de fortes concentrations
d'extractants (ou de
forts débits organiques); et
¨ des quantités non
négligeables de fer sont extraites malgré un
facteur de séparation entre l'uranium et le fer élevé (FSue, ¨ 200), pouvant
entraîner la
formation de précipités lors de la désextraction de l'uranium en milieu
carbonate.
De plus, l'utilisation d'un système synergique à deux extractants, avec
un rapport molaire optimum à respecter (4/1), est plus délicate à gérer que
celle d'un
système à un seul extractant.
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
4
Dans les années 1980, il a été proposé de remplacer le mélange
synergique HDEHP/TOPO par un mélange d'acide bis-(di-n-butoxy-1,3-propy1-2)-
phosphorique (ou HBiDiBOPP) et d'oxyde de di-n-hexylméthoxyoctylphosphine (ou
DinHMOPO) (voir les demandes de brevets français 2 396 803 et 2 596 383,
références [2]
et [3]). Ce mélange conduit à des coefficients de distribution de l'uranium
plus élevés que
ceux obtenus avec le mélange HDEHP/TOPO, mais il extrait aussi fortement le
fer et il est
donc moins sélectif de l'uranium.
Une voie d'amélioration de l'extraction de l'uranium d'une solution
aqueuse d'acide phosphorique consisterait donc à remplacer le mélange
synergique
HDEHP/TOPO en réunissant les deux fonctions échangeur cationique et
extractant
solvatant au sein d'un seul et même composé.
Un extractant bifonctionnel présenterait plusieurs avantages, à savoir :
le fait de n'avoir à gérer qu'un seul composé au lieu de deux, et le fait
d'offrir la possibilité
de transposer le système d'extraction liquide-liquide vers un système
d'extraction solide-
liquide, car les propriétés d'un solide sur lequel un seul composé est greffé
(ou adsorbé)
sont plus faciles à maîtriser que celles d'un solide sur lequel deux composés
greffés (ou
adsorbés) agissent en synergie.
Tunick et al. ont proposé, dans le brevet US 4,316,877 (référence [4]),
d'extraire l'uranium d'une solution aqueuse d'acide phosphorique avec un acide
di- ou
triphosphonique porteur d'un groupe alkyle en Cg à C18, avec ou sans ajout
d'un co-
extractant comme le tri-n-butylphosphate (ou TBP) ou le di-n-
butylbutylphosphonate (ou
DBBP).
Dans cette référence, les meilleurs résultats sont obtenus avec une
phase organique qui comprend un acide triphosphonique à groupe nonyle, du TBP
(en
tant que co-extractant) et du kérosène, dans un rapport volumique de 10/40/50,
et qui
conduit à des coefficients de distribution n'excédant pas 5,1 pour
l'uranium(VI) et voisin
de 157 pour l'uranium(IV).
Par ailleurs, la sélectivité de l'extraction de l'uranium vis-à-vis des autres
éléments et, notamment vis-à-vis du fer, ainsi que les conditions nécessaires
pour
désextraire ensuite l'uranium(VI) de la phase organique ne sont pas évoquées.
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
Par ailleurs, Sturtz a proposé, dans les demandes de brevets français
2 460 958 et 2 460 960 (références [5] et 16]), d'extraire l'uranium(IV) d'une
solution
aqueuse d'acide phosphorique en utilisant des diphosphonates (référence [5])
ou des
triphosphonates (référence [6]). Dans les deux cas, l'uranium(VI) présent dans
la solution
5 aqueuse d'acide phosphorique est préalablement réduit en uranium(IV) par
l'action du
fer métallique.
Concernant les diphosphonates, les meilleurs résultats sont obtenus
avec une phase organique qui comprend un diphosphonate dilué à 97% en volume
dans
le kérosène et qui conduit à un coefficient de distribution de 53,6 pour
l'uranium(IV) et à
une sélectivité uranium/fer de 151,6, tandis que, concernant les
triphosphonates, les
meilleurs résultats sont obtenus avec une phase organique qui comprend un
triphosphonate, du kérosène et du chloroforme (en tant que co-solvant), dans
un rapport
volumique 3/94,5/2,5, et qui conduit à un coefficient de distribution de
l'uranium(IV) de
25 et à une sélectivité uranium/fer de 2,5.
Rien n'est dit dans ces deux références sur la capacité des
diphosphonates et des triphosphonates à extraire l'uranium(VI) et o fortiori
sur les
conditions nécessaires pour le désextraire secondairement.
Enfin, Warshawsky et al. ont proposé, dans la demande de brevet
français 2 604 919 (référence [7]), un composé bifonctionnel comprenant une
fonction
oxyde de phosphine et une fonction phosphorique ou thiophosphorique, ces deux
fonctions étant reliées l'une à l'autre par un groupe espaceur éther,
thioéther, polyéther
ou polythioéther.
Ce type de composé présente deux inconvénients. En effet, les tests
ayant été réalisés avec l'un de ces composés ont montré que, si ce composé est
solubilisé
dans du n-dodécane, il se forme une troisième phase lors de l'extraction de
l'uranium,
tandis que, s'il est solubilisé dans du chloroforme, il se forme également une
troisième
phase mais lors de la désextraction de l'uranium. Or l'apparition d'une
troisième phase
est totalement rédhibitoire pour un procédé destiné à être mis en oeuvre à une
échelle
industrielle. Par ailleurs, la présence au sein du groupe espaceur d'une
liaison P-0 ou P-S,
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
6
qui est aisément hydrolysable, rend ces composés extrêmement sensibles à une
hydrolyse.
Compte tenu de ce qui précède, les Inventeurs se sont donc fixé pour
but de fournir de nouveaux composés bifonctionnels qui puissent
avantageusement être
utilisés en lieu et place du système synergique HDEHP/TOPO pour récupérer
l'uranium
présent dans une solution aqueuse d'acide phosphorique obtenue à partir d'un
phosphate naturel, notamment en ce qu'ils présentent une plus grande affinité
pour
l'uranium(VI) doublée, si possible, d'une moins bonne affinité pour le
fer(III) et pour les
éventuels autres cations susceptibles d'être présents dans ce type de
solution.
Ils se sont aussi fixé pour but que ces composés soient exempts des
divers inconvénients présentés par les composés bifonctionnels proposés dans
les
références [4] à [7] précitées et, en particulier, de la nécessité de les
associer à un co-
extractant, de la nécessité de réduire préalablement l'uranium(VI) en
uranium(IV), de la
formation d'une troisième phase et du risque d'hydrolyse.
EXPOSÉ DE L'INVENTION
Ces buts et d'autres encore sont atteints par l'invention qui propose, en
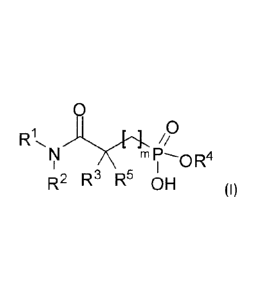
premier lieu, un composé qui répond à la formule générale (I) ci-après :
0
0
R.L
OR4
R2 R3 R5 OH (I)
dans laquelle :
m représente un nombre entier égal à 0, 1 ou 2;
RI- et R2, identiques ou différents, représentent un groupe hydrocarboné,
saturé ou
insaturé, linéaire ou ramifié, comprenant de 6 à 12 atomes de carbone ;
R3 représente :
¨ un atome d'hydrogène ;
¨ un groupe hydrocarboné, saturé ou insaturé, linéaire ou ramifié, comprenant
de 1 à 12 atomes de carbone et éventuellement un ou plusieurs hétéroatomes ;
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
7
¨ un groupe hydrocarboné, saturé ou insaturé, monocyclique, comprenant de 3
à
8 atomes de carbone et éventuellement un ou plusieurs hétéroatomes ; ou
¨ un groupe aryle ou hétéroaryle monocyclique ;
ou bien R2 et R3 forment ensemble un groupe ¨(CH2),¨ dans lequel n est un
nombre
entier allant de 1 à 4;
R4 représente un atome d'hydrogène, un groupe hydrocarboné, saturé ou
insaturé,
linéaire ou ramifié, comprenant de 2 à 8 atomes de carbone, ou un groupe
aromatique
monocyclique ; tandis que
R5 représente un atome d'hydrogène ou un groupe hydrocarboné, saturé ou
insaturé, linéaire ou ramifié, comprenant de là 12 atomes de carbone.
Ainsi, selon la signification de R2 et R3, le composé selon l'invention peut
répondre :
* soit à la formule particulière (l-a) ci-après
0
0
N m P--0
R2 R3 R5 OH (la)
dans laquelle :
m, R1, R4 et R5 sont tels que précédemment définis ;
R2 représente un groupe hydrocarboné, saturé ou insaturé, linéaire ou ramifié,
comprenant de 6 à 12 atomes de carbone; tandis que
R3 représente :
¨ un atome d'hydrogène ;
¨ un groupe hydrocarboné, saturé ou insaturé, linéaire ou ramifié,
comprenant
de 1 à 12 atomes de carbone et éventuellement un ou plusieurs hétéroatomes ;
¨ un groupe hydrocarboné, saturé ou insaturé, monocyclique comprenant de 3
à
8 atomes de carbone et éventuellement un ou plusieurs hétéroatomes ; ou
¨ un groupe aryle ou hétéroaryle monocyclique ;
* soit à la formule particulière (l-b) ci-après :
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
8
0
R1, ,/s,H, /5)
N m POR
--- 4
\ 1
R5 OH
(01-12)n
(I-b)
dans laquelle m, n, RI-, R4 et R5 sont tels que précédemment définis.
Conformément à l'invention, on entend par groupe hydrocarboné,
saturé ou insaturé, linéaire ou ramifié, comprenant de 6 à 12 atomes de
carbone , tout
groupe alkyle, a lcényle ou alcynyle, à chaîne linéaire ou ramifiée, qui
comprend 6, 7, 8, 9,
10, 11 ou 12 atomes de carbone.
De manière analogue, on entend par groupe hydrocarboné, saturé ou
insaturé, linéaire ou ramifié, comprenant de 2 à 8 atomes de carbone , tout
groupe
alkyle, alcényle ou alcynyle, à chaîne linéaire ou ramifiée, qui comprend 2,
3, 4, 5, 6, 7 ou
8 atomes de carbone.
Par ailleurs, on entend par groupe hydrocarboné, saturé ou insaturé,
linéaire ou ramifié, comprenant de 1 à 12 atomes de carbone et éventuellement
un ou
plusieurs hétéroatomes , tout groupe formé d'une chaîne hydrocarbonée,
linéaire ou
ramifiée, qui comprend 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 ou 12 atomes de
carbone, dont la
chaîne peut être saturée ou, au contraire, comporter une ou plusieurs doubles
ou triples
liaisons et dont la chaîne peut être interrompue par un ou plusieurs
hétéroatomes ou
substituée par un ou plusieurs hétéroatomes ou par un ou plusieurs
substituants
comprenant un hétéroatome.
A cet égard, on précise que, par hétéro atome , on entend tout atome
autre que de carbone et d'hydrogène, cet atome étant typiquement un atome
d'azote,
d'oxygène ou de soufre.
Par ailleurs, on entend par groupe hydrocarboné, saturé ou insaturé,
monocyclique, comprenant de 3 à 8 atomes de carbone et éventuellement un ou
plusieurs
hétéroatomes , tout groupe hydrocarboné cyclique qui ne comprend qu'un seul
cycle et
dont le cycle comprend 3, 4, 5, 6, 7 ou 8 atomes de carbone. Ce cycle peut
être saturé ou,
au contraire, comporter une ou plusieurs doubles ou triples liaisons, et peut
comprendre
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
9
un ou plusieurs hétéroatomes ou être substitué par un ou plusieurs
hétéroatomes ou par
un ou plusieurs substituants comprenant un hétéroatome, ce ou ces hétéroatomes
étant
typiquement N, 0 ou S. Ainsi, ce groupe peut notamment être un groupe
cycloalkyle,
cycloalcényle ou cycloalcynyle (par exemple, un groupe cyclopropane,
cyclopentane,
cyclohexane, cyclopropényle, cyclopentényle ou cyclohexényle), un groupe
hétéro-
cyclique saturé (par exemple, un groupe tétrahydrofuryle,
tétrahydrothiophényle,
pyrrolidinyle ou pipéridinyle), un groupe hétérocyclique insaturé mais non
aromatique
(par exemple, pyrrolinyle ou pyridinyle), un groupe aromatique ou encore un
groupe
hétéroaromatique.
A cet égard, on précise que, par groupe aromatique , on entend tout
groupe dont le cycle répond à la règle d'aromaticité de Hückel et présente
donc un
nombre d'électrons TC délocalisés égal à 4n + 2 (par exemple, un groupe
phényle ou
benzyle), tandis qu'on entend par groupe hétéroaromatique , tout groupe
aromatique
tel qu'il vient d'être défini mais dont le cycle comprend un ou plusieurs
hétéroatomes, cet
ou ces hétéroatomes étant typiquement choisis parmi les atomes d'azote,
d'oxygène et
de soufre (par exemple, un groupe furanyle, thiophényle ou pyrrolyle).
Enfin, le groupe ¨(CH2)n¨ dans lequel n est un nombre entier allant de 1
à 4 peut être un groupe méthylène, éthylène, propylène ou butylène.
Conformément à l'invention, dans la formule particulière (I-a) ci-avant,
RI- et R2, qui peuvent être identiques ou différents, représentent
avantageusement un
groupe alkyle, linéaire ou ramifié, comprenant de 6 à 12 atomes de carbone.
Plus encore, on préfère que RI- et R2 soient identiques entre eux et qu'ils
représentent tous deux un groupe alkyle ramifié, comprenant de 8 à 10 atomes
de
carbone, le groupe 2-éthylhexyle étant tout particulièrement préféré.
Par ailleurs, dans la formule particulière (I-a) ci-avant :
¨ m est, de préférence, égal à 0;
¨ R3 représente avantageusement un atome d'hydrogène, un groupe
alkyle, linéaire ou ramifié, comprenant de 1 à 12 atomes de carbone, ou un
groupe aryle
monocyclique, de préférence phényle ou ortho-, méta- ou para-tolyle ; tandis
que
¨ Rs représente préférentiellement un atome d'hydrogène.
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
Plus encore, on préfère que R3 représente un atome d'hydrogène, un
groupe méthyle, n-octyle ou phényle.
Enfin, dans la formule particulière (l-a) ci-avant, R4 représente, de
préférence, un groupe alkyle, linéaire ou ramifié, comprenant de 2 à 8 atomes
de carbone
5 et, mieux encore, de 2 à 4 atomes de carbone tel qu'un groupe éthyle, n-
propyle,
isopropyle, n-butyle, sec-butyle, isobutyle ou tert-butyle, les groupes éthyle
et n-butyle
étant tout particulièrement préférés.
Des composés de formule particulière (l-a) ci-avant qui présentent ces
caractéristiques sont notamment :
10 ¨ le 1-(N,N-diéthylhexylcarbamoyl)benzylphosphonate d'éthyle,
qui
répond à la formule particulière (l-a) ci-avant dans laquelle m est égal à 0,
RI- et R2
représentent tous deux un groupe 2-éthyl-hexyle, R3 représente un groupe
phényle, R4
représente un groupe éthyle tandis que R5 représente un atome d'hydrogène ;
¨ le 1-(N,N-diéthylhexylcarbamoyl)éthylphosphonate d'éthyle, qui
répond à la formule particulière (l-a) ci-avant dans laquelle m est égal à 0,
RI- et R2
représentent tous deux un groupe 2-éthyl-hexyle, R3 représente un groupe
méthyle, R4
représente un groupe éthyle tandis que R5 représente un atome d'hydrogène;
¨ le 1-(N,N-diéthylhexylcarbamoyl)nonylphosphonate d'éthyle, qui
répond à la formule particulière (l-a) ci-avant dans laquelle m est égal à 0,
RI- et R2
représentent tous deux un groupe 2-éthyl-hexyle, R3 représente un groupe n-
octyle, R4
représente un groupe éthyle tandis que R5 représente un atome d'hydrogène ;
¨ le 1-(N,N-diéthylhexylcarbamoyl)nonylphosphonate de butyle, qui
répond à la formule particulière (l-a) ci-avant dans laquelle m est égal à 0,
RI- et R2
représentent tous deux un groupe 2-éthyl-hexyle, R3 représente un groupe n-
octyle, R4
représente un groupe n-butyle tandis que R5 représente un atome d'hydrogène ;
et
¨ le 1-(N,N-dioctylcarbamoyl)nonylphosphonate de butyle, qui
répond à la formule particulière (l-a) ci-avant dans laquelle m est égal à 0,
Ri-, R2 et R3
représentent tous trois un groupe n-octyle, R4 représente un groupe n-butyle
tandis que
R5 représente un atome d'hydrogène.
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
11
Parmi ces composés, on préfère tout particulièrement le 1-(N,N-diéthyl-
hexylcarbamoyl)nonylphosphonate d'éthyle et le 1-(N,N-
diéthylhexylcarbamoyl)nonyl-
phosphonate de butyle.
Dans la famille particulière (l-b) ci-avant, R' représente
avantageusement un groupe alkyle, linéaire ou ramifié, comprenant de 6 à 12
atomes de
carbone.
Par ailleurs, dans cette formule particulière :
¨ m est, de préférence, égal à 0;
¨ R4 représente préférentiellement un groupe alkyle, linéaire ou
ramifié, comprenant de 2 à 8 atomes de carbone et, mieux encore, de 2 à 4
atomes de
carbone, tandis que
¨ R5 représente, de préférence, un atome d'hydrogène.
Un composé de formule particulière (l-b) ci-avant qui présente ces
caractéristiques est notamment le (N-dodécylpyrrolidone)-1-phosphonate
d'éthyle qui
répond à la formule particulière (l-b) dans laquelle Ri- représente un groupe
n-dodécyle,
R2 et R3 forment ensemble un groupe éthylène (¨CH2-CH2¨), R4 représente un
groupe
éthyle tandis que R5 représente un atome d'hydrogène.
Les composés selon l'invention peuvent notamment être obtenus par
les procédés qui sont décrits dans les exemples ci-après et dans les figures 1
à 3 jointes en
annexe. Les composés selon l'invention présentent une affinité et une
sélectivité
particulièrement élevées pour l'uranium(VI).
En particulier, les composés selon l'invention sont capables d'extraire
très efficacement l'uranium(VI) d'une solution aqueuse d'acide phosphorique
et,
notamment, d'une solution aqueuse comprenant de 0,01 à 9 mol/L d'acide
phosphorique.
Aussi, l'invention a-t-elle encore pour objet l'utilisation d'un composé
tel que précédemment défini en tant que ligand de l'uranium(VI) et, en
particulier, pour
extraire l'uranium(VI) d'une solution aqueuse d'acide phosphorique, cette
solution
aqueuse comprenant, de préférence, de 0,01 à 9 mol/L d'acide phosphorique.
Une telle solution aqueuse peut notamment être une solution qui
résulte de l'attaque d'un phosphate naturel par de l'acide sulfurique.
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
12
Ainsi, les composés selon l'invention peuvent notamment être utilisés
dans un procédé permettant de récupérer l'uranium présent dans une solution
aqueuse
d'acide phosphorique issue de l'attaque d'un phosphate naturel par de l'acide
sulfurique,
lequel procédé comprend :
a) l'extraction de l'uranium, à l'état d'oxydation VI, de cette solution
aqueuse par mise en contact de cette solution avec une phase organique
comprenant un
composé tel que précédemment défini, puis séparation de ladite solution
aqueuse et de
ladite phase organique ;
b) le lavage de la phase organique obtenue à l'issue de l'étape a) que
l'on réalise, par exemple, avec de l'eau, une solution aqueuse acide, par
exemple une
solution aqueuse d'acide sulfurique ou une solution aqueuse d'oxalate
d'ammonium ;
c) la désextraction de l'uranium(VI) présent dans la phase organique
obtenue à l'issue de l'étape b) par mise en contact de cette phase organique
avec une
solution aqueuse comprenant un carbonate ou un mélange de carbonates, par
exemple
un carbonate d'ammonium ou de sodium, puis séparation de ladite phase
organique et
de ladite solution aqueuse ; et, de manière optionnelle,
d) l'acidification de la phase organique obtenue à l'issue de l'étape c)
par mise en contact de cette phase organique avec une solution aqueuse acide,
par
exemple une solution aqueuse d'acide sulfurique ou une solution aqueuse
d'acide
phosphorique.
Dans ce procédé, le composé est avantageusement utilisé en solution, à
une concentration de 0,01 à 1 mol/L, dans un diluant organique, lequel diluant
est, de
préférence, de type aliphatique tel que le n-dodécane, le tétrapropylène
hydrogéné ou le
kérosène, par exemple celui commercialisé par la société TOTAL sous la
référence
commerciale Isane IP-185.
La solution aqueuse d'acide phosphorique, qui est utilisée à l'étape a),
comprend préférentiellement de 0,01 à 9 mol/L d'acide phosphorique, tandis que
la
solution aqueuse comprenant le carbonate, qui est utilisée à l'étape c),
comprend
préférentiellement de 0,1 à 1,5 mol/L de carbonate(s).
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
13
Par ailleurs, ce procédé permet de concentrer fortement l'uranium(VI),
c'est-à-dire d'obtenir à l'issue de l'étape c) une solution aqueuse dont la
concentration en
uranium(VI) est supérieure à celle que présente la solution aqueuse d'acide
phosphorique
qui est utilisée à l'étape a). A titre d'exemple, cela peut être réalisé par
le choix d'un
rapport volumique entre la phase organique et la solution aqueuse d'acide
phosphorique
inférieur à 1 à l'étape a), et d'un rapport volumique entre la phase organique
et la
solution aqueuse comprenant le(s) carbonate(s) supérieur à 1 à l'étape c).
Cela permet
d'induire une augmentation de la concentration de l'uranium(VI) en phase
organique à
l'étape a) et en phase aqueuse à l'étape c).
D'autres caractéristiques et avantages de l'invention apparaîtront mieux
à la lecture du complément de description qui suit, qui se rapporte à des
exemples de
synthèse de composés selon l'invention et de démonstration de leurs
propriétés.
Bien entendu, ces exemples ne sont donnés qu'a titre d'illustration de
l'objet de l'invention et ne constituent en aucun cas une limitation de cet
objet.
BREVE DESCRIPTION DES FIGURES
La figure 1 illustre les étapes de procédés de synthèse des composés
selon l'invention qui répondent à la formule particulière (l-a).
La figure 2 illustre les étapes de procédés de synthèse des composés
selon l'invention qui répondent à la formule particulière (l-a) dans laquelle
m = 0, R4 est
différent d'un atome d'hydrogène tandis que R5 représente un atome
d'hydrogène.
La figure 3 illustre les étapes de procédés de synthèse des composés
selon l'invention qui répondent à la formule particulière (l-b).
La figure 4 illustre la variation du logarithme du coefficient de
distribution de l'uranium(VI), noté log(Du), tel qu'obtenu lors d'extractions
réalisées avec
un composé selon l'invention, en fonction du logarithme de la concentration
molaire en
ce composé de la phase organique, noté log([DEHCNP13]).
La figure 5 illustre l'isotherme de partage de l'uranium(VI), c'est-à-dire
l'évolution de la concentration de l'uranium en phase organique, notée [U
orgl, en fonction
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
14
de la concentration de cet élément en phase aqueuse, notée [Uaq]), tel
qu'obtenu lors
d'extractions réalisées avec un composé selon l'invention.
La figure 6 illustre l'évolution du logarithme du coefficient de
distribution de l'uranium(VI), noté log(Du), tel qu'obtenu lors d'extractions
réalisées avec
un composé selon l'invention, en fonction du logarithme de la concentration
molaire en
acide de la phase aqueuse à l'équilibre après extraction, noté log([H]éq ). Le
changement
de symbole dans la figure met en évidence le changement de pente.
La figure 7 illustre l'évolution du coefficient de distribution de
l'uranium(VI), noté Du, tel qu'obtenu lors d'extractions réalisées avec un
composé selon
l'invention, en fonction du temps de contact, exprimé en minutes, entre les
phases
aqueuse et organique.
La figure 8 illustre le schéma utilisé pour un essai de mise en uvre en
continu du procédé selon l'invention ayant été réalisé à une échelle pilote,
en utilisant
des batteries de mélangeurs-décanteurs de laboratoire.
EXPOSÉ DÉTAILLÉ DE MODES DE RÉALISATION PARTICULIERS
EXEMPLE I : SYNTHESE DES COMPOSÉS SELON L'INVENTION
1.1¨ Synthèse des composés de formule particulière (I-a) :
Les composés répondant à la formule particulière (l-a) ci-avant dans
laquelle m et R1 à R5 ont la même signification que précédemment peuvent être
synthétisés en suivant le schéma réactionnel illustré sur la figure 1.
Comme visible sur cette figure, cette synthèse consiste à faire réagir
dans une première étape, notée A, une amine, notée 1, avec un halogénure (par
exemple,
un chlorure ou bromure) d'acide, noté 2, qui est fonctionnalisé en a, en 13 ou
en y (selon
la valeur de m dans le composé que l'on souhaite synthétiser) par un groupe
partant X
(par exemple, un atome de chlore ou de brome) pour obtenir le composé 3 dans
lequel R5
est un atome d'hydrogène.
Pour ce faire, on ajoute sous agitation, à une solution de l'amine à
0,7 mol/L dans le dichlorométhane, du carbonate de potassium (2 éq.). On
refroidit la
suspension ainsi obtenue à 0 C et on lui ajoute goutte à goutte l'halogénure
d'acide
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
(1,5 éq.). On laisse le mélange revenir à la température ambiante. Une fois
l'amine
consommée (ce que l'on vérifie par chromatographie sur couche mince (CCM) en
utilisant
de l'acétate d'éthyle comme éluant et de la ninhydrine comme révélateur), on
ajoute
goutte à goutte 4 équivalents d'eau au mélange, ce qui produit une
effervescence.
5 Lorsque cette effervescence est terminée, on ajoute à ce mélange une
quantité d'eau
égale à la moitié du volume de dichlorométhane ayant été utilisé pour
dissoudre l'amine.
Le mélange est maintenu sous agitation pendant 15 minutes. Les phases aqueuse
et
organique sont ensuite séparées et la phase organique est séchée sur Na2SO4,
filtrée et
concentrée. Le composé 3 ainsi obtenu est généralement suffisamment pur pour
pouvoir
10 .. être utilisé tel quel.
Dans une deuxième étape, notée B sur la figure 1, le composé 3 est
soumis à une réaction d'Arbusov pour obtenir le composé 4 dans lequel R4 est
différent
d'un atome d'hydrogène tandis que R5 est un atome d'hydrogène.
Cette réaction d'Arbusov est réalisée en portant un mélange composé
15 du composé 3 et un phosphite P(0R4)3 dans lequel R4 est différent d'un
atome
d'hydrogène (1,2 à 10 éq. selon le cas) à 160 C à reflux pendant 3 à 72 heures
selon le cas.
Une fois le composé 3 consommé (ce que l'on vérifie par CCM en utilisant du
dichlorométhane comme éluant et des UV ou de l'acide phosphomolybdique comme
révélateur), l'excès de phosphite est distillé sous pression réduite. Selon le
cas, le
composé 4 peut être utilisé tel quel dans l'étape suivante ou, au contraire,
nécessite
d'être préalablement purifié, auquel cas cette purification est réalisée par
chromatographie sur colonne avec un gradient d'élution cyclohexane/acétate
d'éthyle :
de 100/0 à 60/40, v/v.
Puis, le composé 4 ainsi obtenu est ensuite soumis :
¨ soit à une étape de C-alkylation, notée C sur la figure 1, par l'action
d'une base forte (par exemple, de l'hydrure de sodium) et d'un halogénure Hal-
R5 dans
lequel R5 est différent d'un atome d'hydrogène, pour alkyler ce composé en a
du groupe
amide et obtenir le composé 5 dans lequel R4 et R5 sont tous deux différents
d'un atome
d'hydrogène ;
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
16
¨ soit à une étape d'hydrolyse, notée D sur la figure 1, par l'action
d'un halogénure (par exemple, un bromure) de triméthylsilane, pour obtenir le
composé
6 dans lequel R4 et R5 sont tous deux des atomes d'hydrogène ;
¨ soit encore à une étape de monosaponification, notée E sur la
figure 1, par l'action d'une base forte (par exemple, de la soude ou de la
potasse), pour
obtenir le composé 7 dans lequel R4 est différent d'un atome d'hydrogène
tandis que R5
est un atome d'hydrogène.
L'étape C (C-alkylation) est réalisée en ajoutant, goutte à goutte et sous
agitation, une solution du composé 4 (préalablement séchée pendant 2,5 heures
à 80 C
sous vide) dans le tétrahydrofurane (THF ¨1 éq. - 1 mol/L) à une suspension
d'hydrure de
sodium (2 éq. ¨ préalablement lavé au pentane) dans le THE anhydre (2 mol/L).
Le
mélange est agité pendant 1 heure à température ambiante puis on refroidit la
solution à
0 C et on ajoute goutte à goutte une solution de l'halogénure Hal-R5 (1.5
éq.). On laisse ce
mélange revenir à la température ambiante qui est alors agité pendant toute
une nuit,
après quoi le brut est acidifié jusqu'à pH 1 à l'aide d'une solution aqueuse
d'acide
chlorhydrique à 1 mol/L et extrait au dichlorométhane. Les phases aqueuse et
organique
sont séparées et la phase organique est séchée sur Na2SO4, filtrée et
concentrée. L'excès
d'halogénure est éliminé par distillation sous vide.
L'étape D (hydrolyse) est réalisée en ajoutant, goutte à goutte et sous
agitation, à une solution du composé 4 à 0,25 mol/L dans le dichlorométhane,
le bromure
de triméthylsilane (6 éq.), puis en maintenant le mélange sous agitation
pendant toute
une nuit. On ajoute ensuite du méthanol au mélange et on le maintient encore
sous
agitation pendant 2 heures. On le concentre. Le brut est ensuite dilué dans du
dichlorométhane, lavé une fois à l'eau et une fois à l'acide chlorhydrique à 1
mol/L. Les
phases aqueuse et organique sont séparées et la phase organique est séchée sur
Na2SO4,
filtrée et concentrée.
L'étape E (monosaponification) est réalisée en ajoutant, à une solution
du composé 4 à 0,4 mol/L dans l'éthanol, une solution de la base forte (6 à 8
éq.). Le
mélange est porté à reflux pendant 2,5 à 12 heures selon le cas. Après
refroidissement, le
mélange est acidifié jusqu'à pH 1 à l'aide d'une solution aqueuse d'acide
chlorhydrique à
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
17
1 mol/L, puis extrait deux fois au dichlorométhane. Les phases aqueuse et
organique sont
séparées et la phase organique est séchée sur Na2SO4, filtrée et concentrée.
Le composé 5 obtenu à l'étape C est ensuite soumis soit à l'étape D
décrite ci-avant pour obtenir le composé qui est noté 8 sur la figure 1 et
dans lequel R4 est
un atome d'hydrogène tandis que R5 est différent d'un atome d'hydrogène, soit
à l'étape
E décrite ci-avant pour obtenir le composé qui est noté 9 sur cette figure et
dans lequel R4
et R5 sont tous deux différents d'un atome d'hydrogène.
Il est possible d'obtenir des composés de formule particulière (l-a) dans
laquelle R4 représente un groupe différent du groupe R4 qui est présent dans
le composé
4 (et qui est apporté par le phosphite P(0R4)3 à l'étape B) en soumettant soit
le composé
6 soit le composé 8 (selon que R5 doit représenter ou non un atome
d'hydrogène) à une
étape supplémentaire d'O-alkylation, notée F sur la figure 1, par l'action
d'un alcool
R4-0H dans lequel R4 est différent d'un atome d'hydrogène, pour obtenir soit
le composé
10 dans lequel R4 est différent d'un atome d'hydrogène tandis que R5 est un
atome
.. d'hydrogène, soit le composé 11 dans lequel R4 et R5 sont tous deux
différents d'un
atome d'hydrogène.
Sont ainsi synthétisés les composés suivants.
1.1.1 ¨ Acide 1-(N,N-diéthylhexvIcarbamovl)méthylphosphoniclue :
Le composé titre, noté ADEHCMP, qui répond à la formule particulière
(I-a) dans laquelle m = 0, R1 = R2 = 2-éthylhexyle et R3 = R4 = R5 = H, est
synthétisé en
mettant en oeuvre les étapes A, B et D du schéma réactionnel montré sur la
figure 1.
L'étape A est réalisée à partir de 2,2'-diéthylhexylamine et de chlorure
de chloroacétyle et conduit au 2-chloro-N,N-diéthylhexylacétamide (Rdt : 97%)
dont les
caractérisations par RMN 1H et 13C sont données ci-après.
RMN 1H (400 MHz, CDC13) (13Prn) : 0,85 ¨ 0,91 (m, 12H, CH3); 1,23 ¨ 1,33 (m,
16H, CH2);
1,55 ¨ 1,60 (m, 1H, CH-CH2-N) ; 1,67 ¨ 1,73 (m, 1H, CH-CH2-N) ; 3,18 (d, 2H, J
= 7,5 Hz,
CH2-N) ; 3,22¨ 3,32 (m, 2H, CH2-N) ; 4,09 (s, 2H, CH2-CI).
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
18
RMN C (100 MHz, CDCI3) 8 (ppm) : 10,7 ; 11,0 ; 14,1 (CH3) ; 23,1 ; 23,9 ; 24,0
; 28,7 ;
28,9; 30,4 ; 30,6 (CH2) ; 36,8 ; 38,5 (CH) ; 41,6 (CH2-CI) ; 48,8 (CH2-N) ;
51,7 (CH2-N) ;
167,1(C=0).
L'étape B est réalisée en utilisant du triéthylphosphite (1,2 éq. pour
1 éq. de 2-chloro-N,N-diéthylhexylacétamide - reflux de 3 heures) et conduit
au
1-(N,N-diéthylhexylcarbamoyl)méthylphosphonate de diéthyle (Rdt : quantitatif)
dont les
caractérisations par RMN 11-1, 13C et 31P sont données ci-après.
RMN 1H (400 MHz, CDCI3) 8 (ppm) : 0,81 - 0,86 (m, 12H, CH3); 1,21 - 1,32 (m,
22H, CH2,
0-CH2-CH3) ; 1,51 - 1,57 (m, 1H, CH-CH2-N) ; 1,64 - 1,71 (m, 1H, CH-CH2-N) ;
3,02 (d, 2H,
J = 22,0 Hz, CO-CH2-P) ; 3,21- 3,27 (m, 4H, CH2-N) ; 4,08 - 4,16 (m, 4H, 0-CH2-
0-13)=
RMN 13C (100 MHz, CDCI3) 6 (ppm) : 10,6 ; 11,0 ; 14,1 ; 14,2 (CH3) ; 16,3 ;
16,4 (0-CH2-
CH3); 23,1; 23,2 ; 23,5; 23,9; 28,8 ; 28,9; 30,4; 30,6 (CH2) ; 33,1; 34,5 (d,
1 = 134,0 Hz,
CH2-P) ; 37,0; 38,6 (CH) ; 48,9; 52,3 (CH2-N) ; 62,5 (d, J = 6,5 Hz, 0-CH2-
CH3) ; 165,2 (d,
J = 6,0 Hz, C=0).
RMN 31P (160 MHz, CDCI3) ô (ppm) : 21,8.
L'étape D conduit au composé titre (Rdt : quantitatif) dont les
caractérisations par RMN 13C, 311) sont données ci-après.
RMN 1H (400 MHz, CDCI3) 8 (PPrn) 0,86 - 0,93 (m, 12H, CH3) ; 1,22 - 1,37 (m,
16H, CH2) ;
1,59 - 1,65 (m, 1H, CH-CH2-N) ; 1,70 - 1,76 (m, 1H, CH-CH2-N) ; 3,07 (d, 2H; J
= 21,5 Hz,
CO-CH2-P); 3,21- 3,42 (m, 4H, CH2-N) ; 9,56 (Is, 2H, OH).
RMN 13C (100 MHz, CDCI3) ô (ppm) : 10,5; 10,8; 14,0; 14,1 (CI-13) ; 23,0; 23,
5; 23,7;
28,5; 28,7; (CH2) ; 26,8 (CH2-CH2-N) ; 27,1; 27,3 (CH2) ; 28,7 (CH2-CH2-N) ;
29,5; 29,7;
29,1; 32,0 (CH2) ; 32,4; 33,8 (d, J = 131.0 Hz, CH2-P) ; 48,2; 50,1 (CH2-N) ;
168,5 (d,
J = 5,5 Hz, C=0).
RMN 31P (160 MHz, CDCI3) ô (ppm) : 21,0.
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
19
1.1.2 - Acide 1-(N,N-diéthvIhexvIcarbamovnbenzylphosphoniciue :
Le composé titre, noté ADEHMCBP, qui répond à la formule particulière
(l-a) dans laquelle m = 0, RI- = R2= 2-éthylhexyle, R3= phényle et R4= R5= H,
est synthétisé
en mettant en uvre les étapes A, B et D du schéma réactionnel montré sur la
figure 1.
L'étape A est réalisée à partir de 2,2'-diéthylhexylamine et de chlorure
de a-chlorophénylacétyle et conduit au 2-chloro-N,N-bis(2-éthylhexyl)-2-phényl-
acétamide (Rdt : 96%) dont les caractérisations par RMN I-H et 13C sont
données ci-après.
RMN 1H (400 MHz, CDC13) O (PPm) : 0,79 - 0,91 (m, 12H, CH3); 1,09 - 1,37 (m,
16H, CH2);
1,56 - 1,64 (m, 1H, CH-CH2-N) ; 1,65 - 1,73 (m, 1H, CH-CH2-N) ; 3,05 - 3,22
(m, 3H,
CH2-N) ; 3,38 - 3,47 (m, 1H, CH2-N) ; 5,66 (s, 1H, CO-CH(Ph)-CI) ; 7,29 - 7,36
(m, 3H, CHAr) ;
7,47 - 7,49 (m 2H, CHAI).
RMN 13C (100 MHz, CDC13) 6 (PPm) : 10,5 ; 10,7; 10,8; 10,9 (CI-13-CH2-CH) ;
14,1 (CH,);
23,0; 23,7; 23,8 (CH2) ; 28,6; 28,7; 28,8; 28,9 (CH2) ; 30,4; 30,5; 30,6 (CH2)
; 36,9; 37,0;
39,1 ; 39,2 (CH) ; 50,1 ; 50,2 ; 50,3 (CH2-N) ; 51,8; 51,9 (CH2-N) ; 57,6;
57,7 (CO-CH(Ph)-
Cl) ; 128,3; 128,8 (CHAI-); 129,0; 136,6 (CAr) ; 167,7 (C=0).
L'étape B est réalisée en utilisant du triéthylphosphite (10 éq. pour 1 éq.
de 2-chloro-N,N-bis(2-éthylhexyl)-2-phénylacétamide - reflux de 72 heures) et
conduit au
1-(N,N-diéthylhexylcarbamoyl)benzylphosphonate de diéthyle (Rdt : 47%) dont
les
caractérisations par RMN 1-1-1, 13C et 31P sont données ci-après.
RMN 1H (400 MHz, CDC13) O (PPm) : 0,80 - 0,92 (m, 12H, CH3) ; 1,08 - 1,35 (m,
22H, CH2,
0-CH2-CH3) ; 1,54 - 1,75 (m, 2H, CH-CH2-N) ; 2,98 - 3,19 (m, 3H, CH2-N) ; 3,41
- 3,52 (m,
1H, CH2-N) ; 3,97 - 4,07 (m, 2H, 0-CH2-CH3) ; 4,09 - 4,23 (m, 2H, 0-CH2-CH3) ;
4,48 (dd,
1H, J = 23,0 Hz, 3,5 Hz, CO-CH(Ph)-P) ; 7,25 -7,35 (m, 3H, CHAI-) ; 7,48 (m,
2H, CHAr)=
RMN 13C (100 MHz, CDC13) O (ppm) : 10,4; 10,6; 10,7; 10,9; 14,0; 14,1 (CH,);
16,3; 16,4
(0-CH2-CH3) ; 22,9 ; 23,0 ; 23,1 ; 23,2 ; 23,5 ; 23,6 ; 23,7 ; 23,8; 28,6 ;
28,8; 30,3 ; 30,6
(CH2) ; 36,9; 37,0; 37,1; 38,8; 38,9; 39,0 (CH) ; 49,2; 49,4; 49,6; 49,9 (CH2-
N) ; 49,8;
51,3 (d, J = 146 Hz, CH(Ph)-P) ; 51,9; 52,3 (CH2-N) ; 62,7 (d, J = 7,0 Hz, 0-
CH2-CH3) ; 63,0 (d,
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
J = 6,0 Hz, O-CH2-CH3) ; 127,6 (CHAr) ; 128,5 (CHAr) ; 129,5 ; 129,6 (CHAr) ;
132,1 ; 132,2
(CAr) ; 167,7 (dd, 1= 6,0 Hz, 1= 3,5 Hz, C=0).
RMN 31P (160 MHz, CDCI3) 8 (ppm) : 20,6.
5 L'étape D conduit au composé titre (Rdt : 89%) dont les
caractérisations
par RMN 1H, 1-3C et 31P sont données ci-après.
RMN 1H (400 MHz, CDCI3) 8 (13Prn) 0,77 - 0,93 (m, 12H, CH3); 1,05 - 1,44 (m,
16H, CH2) ;
1,53 - 1,61 (m, 1H, CH-CH2-N) ; 1,65 - 1,75 (m, 1H, CH-CH2-N) ; 2,92 - 3,06
(m, 2H,
10 CH2-N) ; 3,15 - 3,21 (m, 1H, C/-12-N) ; 3,56 - 3,65 Cm, 1H, CH2-N) ;
4,42 - 4,49 (dd, 1H,
J = 23,0 Hz, 7,5 Hz, C(0)-CH(Ph)P(0)) ; 7,24 - 7,40 (m, 5H, CHAr) ; 10,55 (ls,
2H, OH).
RMN 13C (100 MHz, CDCI3) 8 (ppm) : 10,2 ; 10,4; 10,5 ; 10,6 ; 10,7 ; 11,0 ;
14,1 ; 14,2
(CH,); 23,0; 23,1; 23,2; 23,5; 23,6; 23,7 ; 23,8 ; 28,7 ; 28,8 ; 28,9 ; 30,3 ;
30,4 ; 30,5 ;
30,6 (CH2) ; 36,8 ; 36,9 ; 37,0 ; 37,1 ; 38,4 ; 38,5 ; 38,7 ; 38,8 (CH) ; 48,7
; 50,0 (d,
15 .. J = 132 Hz; C(0)CH(Ph)P(0)) ; 49,4; 49,7; 50,3; 50,5; 52,0; 52,1; 52,2;
52,4 (CH2-N) ;
127,6; 128,8; 129,0; 129,1 (CHAI-) ; 132,2; 132,3 (CAr) ; 163,8 (CHAr) ; 170,6
(C=0).
RMN P (160 MHz, CDCI3) ô (ppm) : 22,5.
1.1.3 - 1-(N,N-diéthylhexylcarbamoyl)éthylphosphonate d'éthyle :
Le composé titre, noté DEHCEPE, qui répond à la formule particulière
20 (1-a) dans laquelle m = 0, Ri- = R2 = 2-éthylhexyle, R3 = méthyle, R4 =
éthyle et R5 = H, est
synthétisé en mettant en oeuvre les étapes A, B et E du schéma réactionnel
montré sur la
figure 1.
L'étape A est réalisée à partir de 2,2'-diéthylhexylamine et de chlorure
de 2-bromopropionyle et conduit au 2-bromo-N,N-bis(2-éthylhexyl)propanamide
(Rdt :
98%) dont les caractérisations par RMN 1-H et 13C sont données ci-après.
RMN 1H (400 MHz, CDCI3) ô (ppm): 0,83 - 0,89 (m, 12H, CH3); 1,14 - 1,35 (m,
16H, CH2);
1,48 - 1,55 (m, 1H, CH-CH2-N) ; 1,67 - 1,75 (m, 1H, CH-CH2-N) ; 1,78 (d, 3H, J
= 6,5 Hz,
COCH(Br)CH3) ; 2,68 - 2,76 (m, 1H, Cl2-N ) ; 2,68 - 2,76 (dd, 1H, J = 15,0 Hz,
7,0 Hz, CHz-
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
21
N ) ; 3,30 - 2,38 (m, 1H, CH2-N ) ; 3,73 - 3,82 (m, 1H, CH2-N); 4,55 (q, 1H, J
= 6,5 Hz,
COCH(Br)CH3 ).
RMN 13C (100 MHz, CDC13) 8 (ppm): 10,5 ; 10,8; 11,0 (CH3-CH2-CH) ; 14,0 ; 14,1
(CH3) ;
21,6 (COCH(Br)CH3) ; 23,0; 23,5; 23,8; 24,0 (CH2) ; 28,7; 28,8 (CH2) ; 30,2;
30,5 ; 30,6;
30,7 (CH2) ; 37,0; 37,1 (CH) ; 38,9 (COCH(Br)CH3) ; 39,2; 39,3 (CH) ; 49,5 ;
49,6 ; 49,7 ; 49,8
(CH2-N) ; 51,7 ; 51,8 ; 51,9 ; 60,0 (CH2-N) ; 57,6 ; 57,7 (CO-CH (Ph)-Br ) ;
128,3 ; 128,8
(CHAr) ; 129,0; 136,6 (Car); 167,7 (C=0).
L'étape B est réalisée en utilisant du triéthylphosphite (10 éq. pour 1 éq.
de 2-bromo-N,N-bis(2-éthylhexyppropanamide - reflux de 72 heures) et conduit
au
1-(N,N-diéthylhexylcarbamoyl)éthylphosphonate de diéthyle (Rdt : 54%) dont les
caractérisations par RMN 13C et 31P sont données ci-après.
RMN 1H (400 MHz, CDC13) 8 (ppm) : 0,80 - 0,92 (m, 12H, CH3) ; 1,13 - 1,35 (m,
22H, CH2,
O-CH2-CH3) ; 1,42 (dd, 3H, J = 18,5 Hz, J = 7,0 Hz, CH3-CH(C0)-13) ; 1,48 -
1,56 (m, 1H,
CH-CH2-N); 1,68 - 1,76 (m, 1H, CH-CH2-N) ; 2,76 - 2,83 (m, 1H, CH2-N ) ; 2,95 -
3,03 (m,
1H, CH2-N) ; 3,28 (dqd, 1H, J = 21,5 Hz, J = 7,0 Hz, J = 2,0 Hz, CH,-CH(C0)-P)
; 3,41 - 3,52
(m, 1H, CH2-N) ; 3,65 - 3,78 (m, 1H, CH2-N) ; 4,05 -4,17 (m, 4H, O-CH2-CH3).
RMN 13C (100 MHz, CD C13) 8 (ppm) : 10,4; 10,6; 10,9; 11,0 (CH3) ; 12,9 (CH3-
CH(C0)-P) ;
14,0 ; 14,1 (CH,) ; 16,4; 16,5 (0-CH2-CH3) ; 23,0 ; 23,1 ; 23,2 ; 23,6 ; 23,7
; 23,9 ; 28,6 ;
28,7; 28,9; 30,2; 30,3; 30,4; 30,5 ; 30,6 (CH2) ; 35,5; 36,8 (d, J = 134,0 Hz,
CH3-CH(C0)-
P) ; 37,1 ; 37,2 ; 38,8 ; 39,0 (CH-CH2-N) ; 49,3 (d, J = 8,5 Hz, CH2-N) ; 50,1
(d, J = 5,5 Hz,
CH2-N) ; 51,7 (d, J = 7,5 Hz, CH2-N) ; 52,4 (d, J = 9,5 Hz, CH-N) ; 62,2 (d, J
= 6,5 Hz,
O-CH2-CH3) ; 62,6 (d, J = 5,5 Hz, 0-CH2-CH3) ; 169,4 (d, J = 4,5 Hz, C=0).
RMN 31P (160 MHz, CDCI3) 8 (ppm) : 25,3.
L'étape E est réalisée en utilisant de la potasse (4 éq. pour 1 éq. de
1-(N,N-diéthylhexylcarbamoypéthylphosphonate de diéthyle - reflux de 6 heures)
et
conduit au composé titre (Rdt : 97%) dont les caractérisations par RMN 1H, 1-
3C et 31P sont
données ci-après.
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
22
RMN 1H (400 MHz, CDCI3) 3 (PPm) : 0,84 - 0,92 (m, 12H, CH3) ; 1,20 - 1,33 (m,
19H, CI-12,
O-CH2-CH3) ; 1,42 (dd, 3H, J = 18,5 Hz, J = 7,0 Hz, CH3-CH(CC)-P); 1,53 - 1,60
(m, 1H,
CH-CH2-N); 1,68 - 1,78 (m, 1H, CH-CH2-N) ; 2,95 - 3,00 (dd, 1H, J = 13,5 Hz, J
= 6,5 Hz,
CH-N); 3,04 - 3,10 (ddd, 1H, J = 15,0 Hz, J = 6,5 Hz, J = 3,0 Hz, CH2-N ) ;
3,24 - 3,34 (dqd,
1H, 1= 21,5 Hz, J = 7,0 Hz, J = 2,0 Hz, CO-CH(CH3)-P) ; 3,36 - 3,44 (m, 1H,
CH2-N) ; 3,51 -
3,63 (m, 1H, CH2-N) ; 4,09- 4,19 (m, 2H, 0-CH2-CH3), 10,5 (1s, 1H, OH).
RMN 13C (100 MHz, CDCI3) 3 (ppm) : 10,6; 10,7; 11,0; 11,0 (CH,); 12,9 (d, J =
6,0 Hz, CH3-
CH(C0)-P) ; 14,1; 14,2 (CH3) ; 16,5 (d, J = 6,0 Hz, 0-CH2-CH3) ; 23,1 ; 23,2 ;
23,3 ; 23,6 ;
23,8 ; 23,9; 28,8; 29,0; 30,5; 30,6 ; 30,7 (CH2) ; 35,0; 36,4 (d, J = 136,5
Hz, CH3-CH(C0)-
P) ; 37,3 ; 38,9; 39,0; 39,1; 39,2 (CH-CH2-N) ; 49,6; 49,7; 50,3; 50,4; 52,0;
52,1; 52,7;
52,8 (CH2-N) ; 62,2 (d, J = 6,5 Hz, 0-CH2-CH3) ; 170,7 (d, J = 4,5 Hz, C=0).
RMN 31P (160 MHz, CDCI3) 3 (ppm) : 27,5.
1.1.4 - 1-(N,N-dihexylcarbamoyl)méthylphosphonate d'éthyle :
Le composé titre, noté DHCMPE, qui répond à la formule particulière
(l-a) dans laquelle m = 0, RI- = R2 = n-hexyle, R3 = R5 = H et R4 = éthyle,
est synthétisé en
mettant en uvre les étapes A, B et E du schéma réactionnel montré sur la
figure 1.
L'étape A est réalisée à partir de dihexylamine et de chlorure de
chloroacétyle et conduit au 2-chloro-N,N-dihexylacétamide (Rdt : 97%) dont les
caractérisations par RMN I-H etI-3C sont données ci-après.
RMN 1H (400 MHz, CDCI3) 3ppm : 0,75 - 0,79 (m, 6H, Hz, CH3); 1,15- 1,22 (m,
12H, CH2);
1,40- 1,51 (m, 4H, CH2-CH2-N) ; 3,13- 3,22 (m, 4H, CH-N) ; 3,94 (s, 2H, CH2-
C1).
RMN 13C (100 MHz, CDCI3) 6 (PPrn) 13,8; 13,9 (CH,); 22,4; 26,3 ; 26,4 (CH2) ;
27,1; 28,9
.. (CH2-CH2-N) ; 31,3; 31,4 (CH2) ; 41,2 (CH2-C1) ; 46,0; 48,1 (CH2-N) ; 165,8
(C=0).
L'étape B est réalisée en utilisant du triéthylphosphite (1,2 éq. pour
1 éq. de 2-chloro-N,N-dihexylacétamide - reflux de 3 heures) et conduit au
1-(N,N-dihexylcarbamoyl)méthylphosphonate de diéthyle (Rdt : quantitatif) dont
les
caractérisations par RMN 1H, I-3C et 31P sont données ci-après.
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
23
RMN 1H (400 MHz, CDCI3) 8 (ppm) : 0,84- 0,90 (m, 6H, Hz, CH3); 1,24- 1,34 (m,
18H, CH2,
O-CH2-CH3) ; 1,48 - 1,78 (m, 4H, CH2-CH2-N) ; 3,00 (d, 2H, J = 22,0 Hz, CO-CH2-
P) ; 3,28 -
3,33 (m, 4H, CH2-N) ; 4,13 -4,20 (m, 4H, 0-CH2-0-13).
RMN C (100 MHz, CDCI3) 3 (ppm) : 14,1; 14,2 (CH3) ; 16,4; 16,5 (CH,-CH2-0) ;
22,7;
26,6; 26,7 (CH2) ; 27,7; 29,1 (CH2-CH2-N) ; 31,7; 31,8 (CH2) ; 32,9; 34,2 (d,
J = 134 Hz,
CH2-P) ; 46,4 (CH2-N) ; 49,0 (CH2-N) ; 62,7 (d, J = 6,0 Hz, O-CH2-CH3) ; 164,4
(d, Jcp = 5,5 Hz,
C=0).
RMN P (160 MHz, CDCI3) 3 (ppm) : 21,7.
L'étape E est réalisée en utilisant de la potasse (4 éq. pour 1 éq. de
1-(N,N-dihexylcarbamoyl)méthylphosphonate de diéthyle - reflux de 2,5 heures)
et
conduit au composé titre (Rdt : 97%) dont les caractérisations par RMN 1-H, 1-
3C, 31P et
HRMS sont données ci-après.
RMN 1H (400 MHz, CDCI3) 8 'ppm) : 0,77 - 0,82 (m, 6H, CH3) ; 1,14 - 1,24 (m,
15H, CH 2, 0-
CH2-CH3) ; 1,40 - 1,51 (m, 4H, CH2-CH2-N) ; 2,95 (d, 2H, J = 22,0 Hz, CO-CH2-
P); 3,20 - 3,26
(m, 4H, CH2-N) ; 4,03 -4,10 (m, 2H, 0-CH2-CH3), 6,94 (Is, 1H, OH).
RMN C (100 MHz, CDCI3) 8 (PPrn) : 13,9; 14,0 (CH3) ; 16,3 (d, J = 6,5 Hz, CH3-
CH2-0) ;
22,6; 26,5; 26,6 (CH2) ; 27,4; 28,9 (CH2-CH2-N) ; 31,5; 31,6 (CH2) ; 32,4;
33,7 (d, J = 134
Hz, CH2-P); 46,6 ; 49,0 (CH2-N) ; 62,6; 62,7 (d, J = 6,5 Hz, O-CH2-CH3) ;
164,8 (d, Jcp = 5,5
Hz, C=0).
RMN 31P (160 MHz, CDCI3) 3 (ppm) : 21,1.
1.1.5 - 1-(N,N-diéthylhexylcarbamoyl)méthylphosphonate d'éthyle :
Le composé titre, noté DEHCMPE, qui répond à la formule particulière
(1-a) dans laquelle m = 0, R1 = R2 = 2-éthylhexyle, R3 = R5 = H et R4 =
éthyle, est synthétisé
en mettant en uvre les étapes A, B et E du schéma réactionnel montré sur la
figure 1.
Les étapes A et B sont identiques aux étapes A et B décrites au point
1.1.1 ci-avant.
CA 02872084 2014-10-30
WO 2013/167516
PCT/EP2013/059352
24
L'étape E est réalisée en utilisant de la potasse (4 éq. pour 1 éq. de
1-(N,N-diéthylhexylcarbamoyl)méthylphosphonate de diéthyle - reflux de 2,5
heures) et
conduit au composé titre (Rdt : 97%) dont les caractérisations par RMN 1H, 1-
3C, 31P sont
données ci-après.
RMN 1H (400 MHz, CDCI3) 6 (ppm) : 0,81 - 0,89 (m, 12H, CH3); 1,17 - 1,33 (m,
19H, CH2,
0-CH2-CH3) ; 1,53 - 1,59 (m, 1H, CH-CH2-N) ; 1,64 - 1,71 (m, 1H, CH-CH2-N) ;
3,02 (d, 2H,
J = 22,0 Hz, CO-CH2-P) ; 3,21 - 3,34 (m, 4H, CH2-N) ; 4,08 - 4,16 (m, 2H, O-
CH2-CH3) ; 10,3
(1s, 1H, OH).
RMN C (100 MHz, CDCI3) 6 (ppm) : 10,7 ; 11,0 ; 14,2 ; 14,3 (CH3) ; 16,5 (d, J
= 6,5 Hz,
0-CH2-CH3) ; 23,1 ; 23,2 ; 23,7 ; 24,0 ; 28,8 ; 28,9 ; 30,5 ; 30,6 (CH2) ;
32,5; 33,8 (d,
J = 134,0 Hz, CH2-P) ; 37,2; 38,7 (CH) ; 48,6 (CH2-N) ; 52,6 (CH2-N) ; 62,1
(d, J = 6,5 Hz, 0-
CH2-CH3) ; 165,1 (d, J = 4,5 Hz, C=0).
RMN 31P (160 MHz, CDCI3) 6 (ppm) : 21,8.
1.1.6 - Acide 1-(N,N-diéthylhexvIcarbamov1)-1-méthyléthylphosphoniaue :
Le composé titre, noté ADEHCEMP, qui répond à la formule particulière
(l-a) dans laquelle m = 0, R1 = R2 = 2-éthylhexyle, R3 = méthyle, R4 = H et R5
= méthyle, est
synthétisé en mettant en oeuvre les étapes A, B, C et D du schéma réactionnel
montré sur
la figure 1.
Les étapes A et B sont identiques aux étapes décrites au point 1.1.3 ci-
avant.
L'étape C est réalisée en utilisant de l'iodométhane et conduit au
1-(N,N-diéthylhexylcarbamoyI)-1-méthyléthylphosphonate de diéthyle (Rdt : 95%)
dont
les caractérisations par RMN 11-1, 13C et 31P sont données ci-après.
RMN 1H (400 MHz, CDC13)4:5 (ppm) : 0,82 - 0,87 (m, 12H, CH3); 1,19 - 1.30 (m,
22H, CH2,
0-C1-12-CH3) ; 1,50 (d, 6H, J -= 17,0 Hz, C(0)-C(-P)-(CH,),); 1,56- 1,63 (m,
2H, CH-CH,-N);
3,17- 3,54 (massif, 4H, CH2-N) ; 4,05 - 4,15 (m, 4H, 0-CH2-0-13).
RMN C (100 MHz, CDCIA 6 (ppm) 10,9 - 11,5 (massif) ; 14,1 (CH,); 16,4; 16,5 (0-
0-12-
CI-13) ; 23,0 ; 23,5 (CH2) ; 23,6 (C(0)-C(-P)-(CH3)2) ; 23,8 ; 28,8 - 29,5
(massif) ; 30,5 (CH2) ;
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
36,2; 37,6 (massif, CH) ; 45,4; 46,7 (d, J = 137,0 Hz, C(0)-C(-P)-(CH3)2) ;
51,0 (massif,
CH2-N) ; 62,4; (0-CH2-CH3) ; 172,4 (C=0).
RMN 31P (160 MHz, CDC13) (ppm) : 29,0.
5 L'étape D conduit au composé titre (Rdt : 95%) dont les
caractérisations
par RMN 1-H, 1-3C et 31P sont données ci-après.
RMN 1H (400 MHz, CD03) (PPm) 0,77 - 0,93 (m, 12H, CH3); 1,05 - 1,44 (m, 16H,
CH2) ;
1,59 (d, 8H, J = 17,0 Hz, CO-C(P)-(CH3)2, et CH-CH2-N) ; 3,23 - 3,35 (massif,
4H, CH2-N) ;
10 10,49 (Is, 2H, OH).
RMN 13C (100 MHz, CDC13) 8 (PPm) : 10,2; 10,4; 14,1; 14,2 (CH3) ; 21,6 (CO-
C(P)-(C1-13)2) ;
22,6; 22,9; 23,0; 23,6; 23,8; 28,3; 28,8 30,4; 30,5 (CH2) ; 36,1; 37,5 (CH) ;
44,4 ; 45,8
(CO-C(P)-(CH3)2) ; 48,6 ; 51,5 (CH2-N) ; 176,4 (C=0).
RMN 31P (160 MHz, CDC13) (ppm) : 32,8.
15 1.1.7 - 1-(N.N-diéthylhexylcarbamoyl)benzylphosphonate d'éthyle :
Le composé titre, noté DEHCBPE, qui répond à la formule particulière
(1-a) dans laquelle m = 0, Ri- = R2 = 2-éthylhexyle, R3 = phényle, R4 = éthyle
et Rs = H, est
synthétisé en mettant en oeuvre les étapes A, B et E du schéma réactionnel
montré sur la
figure 1.
20 Les étapes A et B sont identiques aux étapes A et B décrites au
point
1.1.2 ci-avant.
L'étape E est réalisée en utilisant de la potasse (10 éq. pour 1 éq. de
1-(N,N-diéthylhexylcarbamoyl)benzylphosphonate de diéthyle - reflux de 24
heures) et
conduit au composé titre (Rdt : 91%) dont les caractérisations par RMN 1-H, 1-
3C et 31P sont
25 données ci-après.
RMN 1H (400 MHz, CDC13) (PPm) : 0,76 - 0,89 (m, 12H, CH,) ; 1,06 - 1,32 (m,
19H, CH2,
0-CH2-CH3) ; 1,50 - 1,59 (m, 1H, CH-CH2-N) ; 1,61 - 1,70 (m, 1H, CH-CH,-N);
2,88 - 3,15
(m, 3H, CH2-N) ; 3,24- 3,54 (m, 1H, CH2-N) ; 3,93 -4,04 (m, 2H, 0-CH2-CH,) ;
4,35 (dd, 1H,
J = 23,0 Hz, 5,0 Hz, CO-CH(Ph)-P) ; 7,20 - 7,40 (m, 5H, CHA,-) ; 8,80(s, 1H,
OH).
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
26
RMN C (100 MHz, CDCI3) (ppm) : 10,3 ; 10,6 ; 11,0 ; 14,1 (CH3) ; 16,4 (d, J =
5,5 Hz,
0-CH2-CH3) ; 22,9 ; 23,0 ; 23,5 ; 23,6 ; 23,7 ; 23,8 ; 23,9; 28,6 ; 28,7; 28,8
; 30,3 ; 30,4
(CH2) ; 36,8; 36,9; 37,1; 37,1; 38,5; 38,6; 38,7; 38,9 (CH) ; 48,4; 49,8 (d, 1
= 139,0 Hz;
CH(Ph)-P) ; 49,5 ; 50,2 ; (CH2-N) ; 51,9 ; 52,3 (CH2-N) ; 62,4 (d; J = 7,0 Hz;
0-CH2-0-13) ;
126,6 (CHAr) ; 127,7 (CHAr) ; 128,8; 129,2 (CHAr) ; 131,5; 135,6 (CAr) ; 169,2
(C=0).
RMN 3 (160 MHz, CDCI3) (ppm) : 22,0.
1.1.8- 1-(N.N-diéthylhexylcarbamoyl)nonyl-phosphonate d'éthyle :
Le composé titre, noté DEHCNPE, qui répond à la formule particulière
(l-a) dans laquelle m = 0, R1 = R2 = 2-éthylhexyle, R3 = H, R4 = éthyle et R5
= n-octyle, est
synthétisé en mettant en oeuvre les étapes A, B, Cet E du schéma réactionnel
montré sur
la figure 1.
Les étapes A et B sont les mêmes que les étapes A et B décrites au point
1.1.1 ci-avant.
L'étape C est réalisée en utilisant de l'iodure d'octyle et conduit au 1-
(N,N-diéthylhexylcarbamoyl)nonylphosphonate de diéthyle (Rdt : 99%) dont les
caractérisations par RMN 11-1,13C et 31P sont données ci-après.
RMN 1H (400 MHz, CDCI3) (ppm) : 0,80 - 0,88 (m, 15H, CHA ; 1,13 - 1,40 (m,
34H, CH2,
0-CH2-CH,) ; 1,55 - 1,61 (m, 1H, CH-CH2-N) ; 1,66 - 1,74 (m, 1H, CH-CH2-N) ;
1,76 - 1,84
(m, 1H, C7Hi5-CH2-CH-(CO)P) ; 2,00 - 2,14 (C7H15-CH2-CH-(CO)P) ; 2,82 - 2,84
(m, 1H,
CH2-N) ; 2,96 - 3,04 (m, 1H, CH2-N) ; 3,28 (ddq, 1H, J = 22,0 Hz, J = 10,5 Hz,
J = 3,0 Hz,
CO-CH(Oct)-P) ; 3,39 - 3,51 (m, 1H, CH2-N) ; 3,58 - 3,74 (m, 1H, CH2-N) ; 4,04
- 4,18 (m,
4H, 0-CH2-0-13)=
RMN C (100 MHz, CDCI3) (ppm) : 10,3; 10,6; 10,8; 10,9; 14,0; 14,1 (CH,); 16,4
(C1-13-
CH2-O); 22,6; 23,0; 23,1; 23,2; 23,6; 23,8; 26,9 (CH2) ; 28,1 (C71-115-CH2-CH-
(CO)P) ;
28,2; 28,6; 28,8; 28,9; 29,0; 29,2; 29,4; 29,7; 29,8; 30,3; 30,4; 30,5; 30,7;
31,8 (CH2)
; 37,1; 37,2; 37,3 ; 37,4; 39,1 ; 39,2; 39,3; 39,4 (CH-CH2-N) ; 41,8; 43,1 (d,
J = 132,0 Hz,
CO-CH(Oct)-P) ; 50,0 (d, J = 25,0 Hz, CH2-N) ; 50,8 (d, J = 25,0 Hz, CH2-N) ;
51,9 (d,
J = 11,0 Hz, CH2-N) ; 52,5 (d, J = 11,0 Hz, CH2-N) ; 62,2 (d, J = 7,0 Hz, 0-
CH2- CH2) ; 62,5 (d,
J = 7,0 Hz, 0-CH2- CH2) ; 168,5 (d, J = 5,0 Hz, C=0).
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
27
RMN P (160 MHz, CDCI3) 8 (ppm) : 24,6.
L'étape E est réalisée en utilisant de la soude(8 éq. pour 1 éq. de
1-(N,N-diéthylhexylcarbamoyl)nonylphosphonate de diéthyle - reflux de 12
heures) et
conduit au composé titre (Rdt : 99%) dont les caractérisations par RMN 1H, 13C
et 31P sont
données ci-après.
RMN 1H (400 MHz, CDCI3) 8 (ppm) : 0,81 - 0,88 (m, 15H, CH3) ; 1,20 - 1,34 (m,
31H, CI-12,
O-CH2-CH3) ; 1,55 - 1,74 (m, 2H, CH-CH2-N ; ) ; 1,81 - 1,90 (m, 1H, C71-115-
CH2-CH-(CO)P) ;
1,97 - 2,06 (m, 1H, C71-115-CH2-CH-(CO)P) ; 3,04 - 3,22 (m, 3H, CH2-N, CO-
CH(Oct)-P) ; 3,28
- 3,52 (m, 2H, CH2-N) ; 4,04 - 4,14 (m, 2H, 0-CH2-CH3) ; 9,35 (ls, 1H, OH).
RMN C (100 MHz, CDCI3) 8 (ppm) : 10,4; 10,7; 10,8; 14,0; 14,1 (CH,); 16,4 (d,
J = 6,0 Hz, CH3-CH2-0) ; 22,6; 23,0; 23,1; 23,4; 23,6; 23,8; (CH2) ; 28,3 (d,
J = 3,0 Hz,
C71-115-CH2-CH-(CO)P) ; 28,5; 28,6; 28,7; 28,8; 29,2; 29,4; 29,7; 29,8; 30,4;
30,5; 31,8
(CH2) ; 37,2; 37,3; 38,9; 39,1; 39,3 (CH2-CH2-N) ; 41,0 ; 42,3 (d, J = 133,5
Hz, CO-CH(Oct)-
P) ; 50,0; 50,5; 50,7; 51,2; 52,0; 52,2; 52,6; 52,8 (CH2-N) ; 61,9 (d, J = 7,0
Hz, 0-0-12-
CH2) ; 169,7 (C=0).
RMN P (160 MHz, CDCI3) 8 (ppm) : 26,8.
1.1.9 - 1-(N,N-diéthylhexvIcarbamovl)méthylphosphonate de 2-éthylhexyle :
Le composé titre, noté DEHCMPEH, qui répond à la formule particulière
(l-a) dans laquelle m = 0, R1 = R2 = 2-éthylhexyle, R3 = R5 = H, R4 = 2-
éthylhexyle, est
synthétisé à partir de l'acide 1-(N,N-diéthylhexylcarbamoyl)méthylphosphonique
obtenu
au point 1.1.1 ci-avant, en mettant en oeuvre l'étape F.
Pour ce faire, on ajoute, à une solution d'acide 1-(N,N-diéthylhexyl-
carbamoyl)méthylphosphonique (1,84 g - 5,7 mmol) dans le dichlorométhane (15
mL)
sous atmosphère inerte, 5% molaire de DMF. Le mélange est refroidi à 0 C et on
lui
ajoute, goutte à goutte, du chlorure d'oxalyle (1,1 mL - 12,6 mmol). Puis, le
mélange est
chauffé à reflux pendant une heure. On distille ensuite le dichlorométhane et
l'excès de
chlorure d'oxalyle. Le brut est dissous dans 10 mL de toluène et de
l'imidazole est ajouté
(40 mg - 0,5 mmol). On ajoute à ce mélange, goutte à goutte, une solution de
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
28
N,N-diisopropyléthylamine (2,2 ml - 12,6 mmol) et 2-éthylhexanol (900 1_ -
5,7 mmol)
dans le toluène (10 mL). L'ensemble est laissé pendant 12 heures sous
agitation puis 5 mL
d'acide chlorhydrique à 1 mol/L sont ajoutés. Le mélange est extrait au
dichlorométhane
(2 fois). La phase organique est séchée sur Na2SO4, filtrée et concentrée. Le
produit est
isolé après purification par chromatographique sur colonne avec un gradient
d'élution
dichlorométhane/méthanol : de 100/0 à 90/10, v/v.
On obtient ainsi le composé titre (Rdt : 57%) dont les caractérisations
par RMN 1-3C et 31P sont données ci-après.
RMN 1H (400 MHz, CDCI3) 8 (ppm) : 0,81 - 0,87 (m, 18H, CH3); 1,19 - 1,39 (m,
25H, CH2,
O-CH2-CH); 1,50 - 1,60 (m, 2H, CH-CH2-N; P-O-CH2-CH) ; 1,64 - 1,71 (m, 1H, CH-
CH2-N) ;
3,00 - 3,36 (m, 5H, CO-CH(Oct)-P, CH2-N) ; 3,90 - 3,99 (m, 2H, O-CH2-CH) ;
8,04 (Is, 1H,
OH).
RMN 13C (100 MHz, CDCI3) 8 (ppm) : 10,5 ; 10,9 ; 14,0; 14,1 (CH3) ; 16,5 (d, J
= 6,5 Hz,
O-CH2-CH) ; 23,0; 23,1; 23,2 ; 23,5 ; 23,7 ; 28,6; 28,7; 28,9 ; 29,8; 30,4 ;
30,5 (CH2) ;
31,8; 33,1 (d, J = 133,0 Hz, CH2-P) ; 37,0 ; 38,6 (CH) ; 40,1 (d, 1 = 6,5 Hz,
P-O-CH2-CH ) ;
49,6 (CH2-N) ; 52,4 (CH2-N) ; 67,8 (d, J = 5,5 Hz, O-CH2-CH) ; 167,4 (C=0).
RMN P (160 MHz, CDCI3) 8 (ppm) : 21,6.
1.1.10 - 1-(N,N-diéthylhexylcarbamoyl)méthylphosphonate de butyle:
Le composé titre, noté DEHCMP13, qui répond à la formule particulière
(1-a) dans laquelle m = 0, R1 = R2 = 2-éthylhexyle, R3 = R5 = H, R4 = n-
butyle, est synthétisé
en mettant en oeuvre les étapes A, B et E du schéma réactionnel montré sur la
figure 1.
L'étape A est la même que l'étape A décrite au point 1.1.1 ci-avant.
L'étape B est réalisée en utilisant du tributylphosphite (3 éq. pour 1 éq.
de 2-chloro-N,N-diéthylhexylacétamide - reflux de 4 heures) et conduit au 1-
(N,N-diéthyl-
hexylcarbamoyl)méthylphosphonate de dibutyle (Rdt : quantitatif)
dont les
caractérisations par RMN 11-1, 13C et 31P sont données ci-après.
RMN 1H (400 MHz, CDC13) 8 (ppm): 0,83 - 0,94 (m, 18H, CH3); 1,23 - 1,33 (m,
16H, CH2);
.. 1,34- 1,43 (m, 4H, O-CH2-CH2-CH2-CH3) ; 1,53 - 1,59 (m, 1H, CH-CH2-N) ;
1,61 - 1,72 (m,
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
29
5H, CH-CH2-N, 0-CH2-CH2-CH2-CH3) ; 3,04 (d, 2H, J = 22,0 Hz, CO-CH2-P) ; 3,23 -
3,33 (m,
4H, CH2-N) ; 4,05 -4,12 (m, 4H, 0-CH2-CH2-0-12-0-13).
RMN 13C (100 MHz, CDCI3) (ppm) 10,4; 10,8 (CH3) ; 13,5 (0-CH2-CH2-CH2-CH3) ;
14,1;
14,2 (CH3) ; 18,2 (0-CH2-CH2-CH2-CH3) ; 22,9; 23,0, 23,3; 23,8 ; 28,6 ; 28,7;
30,2 ; 30,4
(CH2) ; 32,4 (d, J = 6,0 Hz, 0-CH2-CH2-CH2-CH3) ; 32,8; 34,1 (d, J = 133,0 Hz,
CH2-P) ; 36,9;
38,5 (CH) ; 48,8 ; 52,2 (CH2-N) ; 66,1 (d, J = 6,5 Hz, 0-CH2-CH2-CH2-CH3) ;
165,0 (d,
J = 6,0 Hz, C=0).
RMN 31P (160 MHz, CDCI3) 8 (ppm) : 21,9.
L'étape E est réalisée en utilisant de la soude 6 éq. pour 1 éq. de
1-(N,N-diéthylhexylcarbamoyl)méthylphosphonate de dibutyle - reflux de 3
heures dans
un mélange dioxane-eau) et conduit au composé titre (Rdt : 99%) dont les
caractérisations par RMN 11-1, 13C et 31P sont données ci-après.
RMN 1H (400 MHz, CDCI3) (PPrn) : 0,83 - 0,94 (m, 15H, CH3); 1,23 - 1,33 (m,
16H, CH2);
1,35 - 1,43 (m, 2H, 0-CH2-CH2-CH2-CH3) ; 1,55 - 1,73 (m, 4H, CH-CH2-N, 0-CH2-
CH2-CH2-
CH3) ; 3,03 (d, 2H, J = 21,5 Hz, CO-CH2-P) ; 3,27 - 3,37 (m, 4H, CH2-N) ; 4,05
- 4,10 (q, 4H,
J = 7,0 Hz, 0-CH2-CH2-CH2-CH3) ; 11,94 (1s, 1H, OH).
RMN 13C (100 MHz, CDCI3) 3 (ppm) 10,4; 10,8 (CH3) ; 13,5 (0-CH2-CH2-CH2-CH3) ;
14,1;
14,2 (CH3) ; 18,2 (0-CH2-CH2-CH2-CH3) ; 22,9; 23,0, 23,3; 23,8 ; 28,6 ; 28,7;
30,2 ; 30,4
(CH2) ; 32,4 (d, J = 6,0 Hz, 0-CH2-CH2-CH2-CH3) ; 32,8; 34,1 (d, J = 133,0 Hz,
CH2-P) ; 36,9;
38,5 (CH) ; 48,8 ; 52,2 (CH2-N) ; 66,1 (d, J = 6,5 Hz, 0-CH2-CH2-CH2-CH3) ;
165,0 (d,
J = 6,0 Hz, C=0).
RMN P (160 MHz, CDCI3) 3 (ppm) : 21,9.
1.1.11- 1-(N,N-diéthylhexylcarbamoyl)nonylphosphonate de butyle:
Le composé titre, noté DEHCNPB, qui répond à la formule particulière
(1-a) dans laquelle m = 0, RI- = R2 = 2-éthylhexyle, R3 = H, R4 = n-butyle et
R5 = n-octyle, est
synthétisé en mettant en oeuvre les étapes A, B, C et E du schéma réactionnel
montré sur
la figure 1.
L'étape A est la même que l'étape A décrite au point 1.1.1 ci-avant.
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
L'étape B est la même que l'étape B décrite au point 1.1.10 ci-ayant.
L'étape C est réalisée en utilisant de l'iodure d'octyle et conduit au
1-(N,N-diéthylhexylcarbamoypnonylphosphonate de dibutyle (Rdt : 99%) dont les
caractérisations par RMN 13C et 31P sont données ci-après.
5
RMN 1H (400 MHz, CDCI3) 8 (PPrn) : 0,81 - 0,93 (m, 21H, CH3) ; 1,17 - 1,42 (m,
32H, CH2,
0-CH2-CH2-CH2-CH,); 1,59 - 1,66 (m, 5H, CH-CH2-N, 0-CH2-CH2-CH2-CH3) ; 1,69 -
1,84 (m,
2H, CH-CH2-N, C71-115-CH2-CH-(CO)P) ; 2,02 - 2,13 (m, 1H, C71-115-CH2-CH-
(CO)P); 2,80 - 2,92
(m, 1H, CH2-N) ; 2,95 - 3,03 (m, 1H, CH2-N) ; 3,14 - 3,22 (m, 1H, CO-CH(Oct)-
P) ; 3,43 -
10 3,55 (m, 1H, CH2-N) ; 3,62- 3,78 (m, 1H, CH2-N) ; 3,96- 4,11 (m, 4H, 0-
CH2-CH2-C1-12-0-13)=
RMN 13C (100 MHz, CDCI3) 8 (PPm) : 10,4; 10,7; 10,9; 11,0 (CH3) ; 13,8 (0-CH2-
CH2-CH2-
CH3) ; 14,2; 14,3 (CH3) ; 18,9 (O-CH2-CH2-CH2-CH,); 22,8; 23,2; 23,3; 23,7;
23,9; 24,0;
(CH2) ; 28,2 (C71-115-CH2-CH-(C0)13) ; 28,8; 28,9; 29,0; 29,1; 29,4; 29,5;
29,8; 29,9; 30,4;
30,5; 30,7; 30,8; 32,0 (CH2) ; 32,7; 32,8 (d, J = 6,5 Hz, 0-CH2-CH2-CH2-CH,);
37,2; 37,3;
15 37,4 ; 39,2 ; 39,3 ; 39,5 ; 39,6 (CH-CH2-N) ; 41,9 ; 43,2 (d, J = 131,0
Hz, CO-CH(Oct)-P) ;
50,0; 50,2; 50,9; 51,1; 51,9; 52,0; 52,6; 52,7 (CH2-N) ; 66,1 (d, J = 6,5 Hz,
0-CH2- CH2) ;
66,3 (d, J = 6,5 Hz, 0-CH2- CH2) ; 168,5 (d, J = 5,0 Hz, C=0).
RMN 31P (160 MHz, CDCI3) 8 (ppm) : 24,6.
20 L'étape E est réalisée en utilisant de la soude (8 éq. pour 1 éq. de
1-(N,N-diéthylhexylcarbamoyl)nonylphosphonate de dibutyle - reflux de 15
heures dans
un mélange dioxane-eau) et conduit au composé titre (Rdt : 99%) dont les
caractérisations par RMN 11-1,13C et 31P sont données ci-après.
25 RMN 1H (400 MHz, CDCI3) (PPm) : 0,83 - 0,94 (m, 18H, CH3) ; 1,20 - 1,41
(m, 30H, CH2,
0-CH2-CH2-CH2-CH,); 1,59 - 1,73 (m, 4H, 0-CH2-CH2-CH2-CH,, C71-115-CH2-CH-
(C0)13) ; 1,85
- 2,06 (m, 2H, CH-CH2-N) ; 3,07 - 3,21 (m, 3H, CH2-N, CO-CH(Oct)-P) ; 3,24 -
3,50 (m, 2H,
CH2-N) ; 3,98 - 4,10 (m, 2H, 0-CH2-CH2-CH2-CH,); 9,34 (s, 1H, OH).
RMN 13C (100 MHz, CDCI3) 8 (ppm) : 10,5; 10,8; 10,9; (CF-1,); 13,7 (O-CH2-CH2-
CH2-CH,);
30 14,1; 14,2 (CH,); 18,8 (O-CH2-CH2-CH2-CH,); 22,8; 23,2; 23,3; 23,7;
23,8; 23,9; (CH2) ;
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
31
28,2 (C7H15-CH2-CH-(CO)P) ; 28,7; 28,9; 29,4; 29,5; 29,8; 29,9; 30,4; 30,5;
30,7; 32,0
(CH2) ; 32,6 (d, _I = 6,5 Hz, 0-CH2-CH2-CH2-CH,); 37,2; 37,3; 39,0; 39,2; 39,4
(CH-CH2-N) ;
41,9 ; 43,2 (d, J = 134,0 Hz, CO-CH(Oct)-P) ; 49,9; 50,5; 50,7; 51,2; 52,0;
52,1 ; 52,7;
52,8 (CH2-N) ; 65,6 (d, J = 6,5 Hz, 0-CH2- CH2) ; 169,3 (d, J = 5,0 Hz, C=0).
RMN 31P (160 MHz, CDCI3)45 (ppm) : 26,8.
1.1.12 - 1-(N.N-dioctylcarbamoyl)nonylphosphonate de butyle:
Le composé titre, noté DOCNPB, qui répond à la formule particulière
(l-a) dans laquelle m = 0, R1 = R2 = n-octyle, R3 = H, R4 = n-butyle et R5 = n-
octyle est
synthétisé en mettant en oeuvre les étapes A, B, C et E du schéma réactionnel
montré sur
la figure 1.
L'étape A est réalisée à partir de dioctylamine et de chlorure de
chloroacétyle et conduit au 2-chloro-N,N-dioctylacétamide (Rdt : 96%) dont les
caractérisations par RMN 1H et13C sont données ci-après.
RMN 1H (400 MHz, CDC13)45 (PPm) 0,81 - 0,86 (m, 6H, CH,); 1,21 - 1,27 (m, 20H,
CH2) ;
1,45 - 1,58 (m, 4H, CH2-CH2N) ; 3,20- 3,29 (m, 4H, CH2-N) ; 4,01 (s, 2H,
CH2CI).
RMN 13C (100 MHz, CDCI3) ô (ppm) : 14,1(CH3) ; 22,6; 26,8; 26,9 (CH2) ; 27,4;
29,1 (CH2-
CH2-N) ; 29,2; 29,3; 29,4 ; 31,7; 31,8 (CH2) ; 41,3 (CH2-CI) ; 46,3; 48,3 (CH2-
N) ; 166,0
(C=0).
L'étape B est réalisée en utilisant du tributylphosphite (3 éq. pour 1 éq.
de 2-chloro-N,N-dioctylacétamide - reflux de 4 heures) et conduit au 1-(N,N-
dioctyl-
carbamoyl)méthylphosphonate de dibutyle (Rdt : quantitatif) dont les
caractérisations par
RMN 11-1, 13C et 31P sont données ci-après.
RMN 1H (400 MHz, CDCI.3) (PPIn) : 0,83 - 0,94 (m, 12H, CH3); 1,23 - 1,33 (m,
20H, CH2);
1,34 - 1,44 (m, 4H, 0-CH2-CH2-CH2-CH,); 1,47 - 1,58 (m, 4H, CH2-CH2-N) ; 1,61 -
1,69 (m,
4H, 0-CH2-CH2-0-12-0-13) ; 3,00 (d, 2H, J = 22,0 Hz, CO-CH2-P) ; 3,27 - 3,33
(m, 4H, CH2-N) ;
4,05 -4,12 (q, 4H, J = 7,0 Hz; 0-CH2-CH2-CH2-CH,).
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
32
RMN 13C (100 MHz, CDC13)45 (13Pm) : 13,4 (0-CH2-CH2-CH2-CH3) ; 13,9 (CH3) ;
18,5 (O-CH2-
CH2-CH2-CH,); 22,4; 26,6, 26,7 (CH2); 27,5; 28,8 (CH2-CH2-N) ; 29,0; 29,1;
29,1; 29,2;
31,5; 31,6 (CH2) ; 32,4 (d, J = 6,0 Hz, 0-CH2-CH2-CH2-CH3) ; 32,5; 33,8 (d, J
= 133,0 Hz,
CH2-P) ; 46,0 ; 48,6 (CH2-N) ; 66,0 (d, J = 6,5 Hz, 0-CH2-CH2-CH2-CH,); 164,0
(d, J = 6,0 Hz,
C=0).
RMN 31P (160 MHz, CDC13) (ppm) : 21,6.
L'étape C est réalisée en utilisant de l'iodure d'octyle et conduit au
1-(N,N-dioctylcarbamoyl)nonylphosphonate de dibutyle (Rdt : 99%) dont les
caractérisations par RMN 13C et 31P sont données ci-après.
RMN 1H (400 MHz, CDC13) (13Pm) : 0,80 - 0,88 (m, 15H, CH3); 1,13 - 1,40 (m,
36H, CH2) ;
1,35 - 1,43 (m, 4H, 0-CH2-CH2-CH2-CH,); 1,49 - 1,56 (m, 3H, CH2-CH2-N) ; 1,59 -
1,69 (m,
5H, CH2-CH2-N ; 0-CH2-CH2-CH2-CH,); 1,75 - 1,84 (m, 1H, C71-115-CH2-CH-(CO)P)
; 2,02 -
2,13 (m, 1H, C7H15-CH2-CH-(CO)P) ; 3,04- 3,21 (m, 3H, CH2-CH2-N ; C71-115-CH2-
CH-(CO)P) ;
3,38- 3,57 (m, 2H, CH2-CH2-N) ; 3,98 - 4,13 (m, 4H, 0-CH2-CH2-CH2-CH,).
RMN 13C (100 MHz, CDCIA (ppm) : 13,5 (cH3-0-12-cH2-0-12-0) ; 14,1 (cH3) ; 18,7
(CH3-
CH2-CH2-CH2-0) ; 22,6; 26,8; 26,9; 27,0; 27,5 ; 27,6; 27,9; 28,4; 29,6; 29,1;
29,2; 29,3
; 29,4 ; 31,31,7 ; 31,8 (CH2) ; 32,6 (d, J = 6,0 Hz, CH,-CH2-CH2-CH2-O); 41,5
; 42,8 (d,
J =132,0 Hz, C7H15CHC(0)P(0)) ; 46,9; 48,4 (CH2-N) ; 66,0 (d, J = 6,5 Hz, 0-
CH2-CH2-CH2-
CH3)) ; 66,2 (d,1 = 6,5 Hz, 0-CH2-CH2-CH2-CH3)) ; 167,6 (d, 1= 4,5 Hz, C=0).
RMN 31P (160 MHz, CDC13) (ppm) : 24,6.
L'étape E est réalisée en utilisant de la soude (8 éq. pour 1 éq. de
1-(N,N-dioctylcarbamoyl)nonylphosphonate de dibutyle - reflux de 15 heures) et
conduit
au composé titre (Rdt : 99%) dont les caractérisations par RMN 13C
et 31P sont
données ci-après.
RMN 1H (400 MHz, CDC13)15 (PPm) : 0,83 - 0,91 (m, 12H, CH3) ; 1,22 - 1,43 (m,
34H, CH2,
0-CH2-CH2-CH2-CH,) ; 1,49 - 1,56 (m, 3H, CH2-CH2-N) ; 1,59 - 1,69 (m, 3H, (CH2-
CH2-N;
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
33
0-CH2-CH2-CH2-CH3) ; 1,80 - 1,88 (m, 1H, C71-115-CH2-CH-(CO)P) ; 1,99 - 2,10
(m, 1H, C71-1,5-
CH2-CH-(CO)P); 3,07 - 3,23 (m, 3H, CH2-CH2-N ; C7Hi5-CH2-CH-(CO)P) ; 3,36 -
3,48 (m, 2H,
CH2-CH2-N) ; 3,97 - 4,09 (m, 2H, 0-CH2-CH2-CH2-CH3) ; 11,05 (1s, 1H, OH).
RMN 13C (100 MHz, CD03) (PPrn) : 13,8 (CH3-CH2-CH2-CH2-0) ; 14,1 (CH3) ; 18,9
(CH3-
CH2-CH2-CH2-O); 22,8 ; 27,1; 27,2 ; 27,0 ; 27,7 ; 28,0 ; 28,1 ; 28,7 ; 28,8 ;
29,4 ; 29,5 ;
29,6; 29,8 ; 31,9 ; 32,0 (CH2) ; 32,7 (d, J = 6,0 Hz, CH3-CH2-CH2-CH2-O); 41,2
; 42,4 (d,
J =132,0 Hz, C71-115CHC(0)P(0)) ; 47,3; 48,8 (CH2-N) ; 65,8 (d, J = 6,5 Hz, 0-
CH2-CH2-0-12-
CH3)) ; 66,2 (d, _I = 6,5 Hz, 0-CH2-CH2-CH2-CH3)) ; 168,7 (d, 1= 4,5 Hz, C=0).
RMN 31P (160 MHz, CDCIA 5 (ppm) : 26,5.
1.1.13 - Acide 2-(N,N-diéthylhexvIcarbamovnéthylphosphonicuie :
Le composé titre, noté ADEHCEP, qui répond à la formule particulière
(l-a) dans laquelle m = 1, RI- = R2= 2-éthylhexyle, R3= R4= R5= H est
synthétisé en mettant
en oeuvre les étapes A, B et D du schéma réactionnel montré sur la figure 1.
L'étape A est réalisée à partir de 2,2'-diéthylhexylamine et de chlorure
de 3-bromopropanoyle et conduit au 3-bromo-N,N-bis(2-éthylhexyl)propanamide
(Rdt :
97%) dont les caractérisations par RMN I-H et 13C sont données ci-après :
RMN 1H (400 MHz, CDCIA ë'(13Pm): 0,78 - 0,85 (m, 12H, CH3); 1,14 - 1,28 (m,
16H, CH2);
1,49- 1, 56 (m, 1H, CH-CH2-N ) ; 1,58- 1,64 (m, 1H, CH-CH2-N) ; 2,85 (t, 2H, J
= 7,0 Hz, CO-
CH2) ; 3,20 (d, 2H, J = 7,5 Hz, CH2-N); 3,26 - 3,32 (m, 2H, CH2-N) ; 3,60 (t,
2H, J = 7,0 Hz,
CH2-Br).
RMN 13C (100 MHz, CD03) (ppm) : 10,8 (CH3-CH2-CH) ; 14,1 (CH3) ; 23,0 ; 23,1 ;
23,9
(CH2) ; 28,0 (CH2-Br) ; 28,8; 30,5; 30,6 (CH2) ; 36,6 (CO-CH2) ; 37,0 ; 38,5
(CH) ; 49,2 (0-12-
N) ; 51,5 (CH2-N) ; 170,4 (C=0).
L'étape B est réalisée en utilisant du triéthylphosphite (4 éq. pour 1 éq.
de 3-bromo-N,N-bis(2-éthylhexyl)propanamide - reflux de 6 heures) et conduit
au
2-(N,N-diéthylhexylcarbamoyl)éthylphosphonate de diéthyle (Rdt : quantitatif)
dont les
caractérisations par RMN 11-1, 13C et 31P sont données ci-après.
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
34
RMN 1H (400 MHz, CDCI3) O (PM) : 0,81 - 0,89 (m, 12H, CH3) ; 1,18 - 1,35 (m,
22H, CH2,
O-CH2-CH3) ; 1,53 - 1,67 (m, 2H, CH-CH2-N) ; 2,04 - 2,13 (m, 2H, CH2-P) ; 2,55
- 2,61 (m,
2H, CO-CH2) ; 3,12 (d, 2H, J = 7,5 Hz, CH2-N) ; 3,18 - 3,32 (m, 2H, CH2-N) ;
4,02 - 4,12 (m,
4H, 0-CH2-0-13)=
RMN 13C (100 MHz; CDCI3) O (PPm) : 10,6 ; 10,8 ; 14,0 ; 14,1 (CH3) ; 16,4 ;
16,5 (O-CH2-
CH3); 21,2 (d; J = 134,0 Hz; CH2-P) ; 23,0; 23,1 ; 23,8; 23,9 (CH2) ; 26,6 (CO-
CH2) ; 28,7;
28,8; 30,5; 30,6 (CH2) ; 37,0; 38,4 (CH) ; 49,0 (CH2-N) ; 51,2 (CH2-N) ; 61,6
(d, J = 6,5 Hz,
O-CH2-CH3) ; 171,2 (d, J = 18,0 Hz, C=0).
RMN 31P (160 MHz, CDCI3) O (ppm) : 32,1.
L'étape D conduit au composé titre (Rdt : quantitatif) dont les
caractérisations par RMN 1-1-1,13C, 31P sont données ci-après.
RMN 1H (400 MHz, CDCI3) O (PPm): 0,82 - 0,90 (m, 12H, CH3); 1,22 - 1,34 (m,
16H, CH2) ;
1,59 - 1,68 (m, 2H, CH-CH2-N) ; 2,04 (dt, 2H, J = 18,5 HZ, 6,5 Hz, CH2-P) ;
2,76 (dt, 2H,
J = 20,5 HZ, 6,5 Hz, CO-CH2) ; 3,15 (d, 2H, J = 7,5 Hz, CH2-N) ; 3,20 - 3,34
(m, 2H, CH2-N) ;
8,34 (ls, 2H, OH).
RMN 13C (100 MHz; CDCI3) éi (ppm) : 10,6 ; 10,8 ; 14,0 ; 14,1 (CH3) ; 22,0;
23,4 (d,
J = 135,0 Hz, CH2-P) ; 26,8 (d, J = 4,5 Hz, CO-CH2) ; 28,6 ; 28,7 ; 30,4 ;
30,5 (CH2) ; 37,0 ;
38,4 (CH) ; 49,8 (CH2-N) ; 52,0 (CH2-N) ; 174,3 (d, J = 6,0 Hz, C=0).
RMN 31P (160 MHz, CDCI3) O (ppm) : 31,1.
1.1.14 - Acide 3-(diéthylhexvIcarbamovnpropvlphosphonique :
Le composé titre, noté ADEHCPP, qui répond à la formule particulière
(I-a) dans laquelle m = 2, RI- = R2= 2-éthylhexyle, R3= R4= R5= H est
synthétisé en mettant
en oeuvre les étapes A, B et D du schéma réactionnel montré sur la figure 1.
L'étape A est réalisée à partir de 2,2'-diéthylhexylamine et de chlorure
de 4-chlorobutanoyle et conduit au 4-chloro-N,N-bis(2-éthylhexyl)butanamide
(Rdt :
quantitatif) dont les caractérisations par RMN 1-E1 et 1-3C sont données ci-
après :
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
RMN1H (400 MHz, CDCI3) 6 (ppm) : 0,86 - 0,93 (m, 12H, CH3); 1,25 - 1,35 (m,
16H, CH2);
1,58- 1,73 (m, 2H, CH-CH2-N) ; 2,11 - 2,17 (m, 2H, CH2-CH2-CI) ; 2,53 (t, 2H,
J = 7,0 Hz, CO-
CH2) ; 3,18 (d, 2H, J = 7,5 Hz, CH2-N); 3,26 - 3,32 (m, 2H, CH2-N) ; 3,64 (t,
2H, J = 6,0 Hz,
CH2-CI).
5 RMN 13C (100 MHz, CDCI3) 8 (ppm) : 10,7; 10,9 (CH3-CH2-CH) ; 14,1 (CH,);
23,1 ; 23,9
(CH2) ; 28,2 (CH2-CH2-CI) ; 28,7 (CH2) ; 30,0 (CO-CH2) ; 30,6 (CH2) ; 37,0;
38,5 (CH) ; 45,0
(CH2-CI) ; 49,0 (CH2-N) ; 51,4 (CH2-N) ; 172,1 (C=0).
L'étape B est réalisée en utilisant du triéthylphosphite (4 éq. pour 1 éq.
10 de 4-chloro-N,N-bis(2-éthylhexyl)butanamide - reflux de 48 heures) et
conduit au 3-(N,N-
diéthylhexylcarbamoyl)propylphosphonate de diéthyle (Rdt : 68%)
dont les
caractérisations par RMN 13C et 31P sont données ci-après.
RMN 1H (400 MHz, CDCI3) 6 (ppm) : 0,81 - 0,87 (m, 12H, CH3) ; 1,13 - 1,30 (m,
22H, CH2,
15 O-CH2-CH3) ; 1,51 - 1,57 (m, 1H, CH-CH2-N) ; 1,59 - 1,65 (m, 1H, CH-CH2-
N); 1,74 - 1,85
(dd, 2H, J = 18,0 Hz, 8,5 Hz, CO-CH2-CH2-CH2-P) ; 1,87 - 1,97 (m, 2H, CO-CH2-
CH2-CH2-P) ;
2,40 (t, 2H, J = 7,0 Hz, CO-CH2-CH2-CH2-P) ; 3,09 (d, 2H, J = 7,5 Hz, CH2-N) ;
3,16 - 3,30 (m,
2H, CH2-N) ; 4,00 -4,10 (m, 4H, 0-CH2-0-13)=
RMN 13C (100 MHz; CDCI3) 8 (ppm) : 10,7 ; 10,9 ; 14,0 ; 14,1 (CH3) ; 16,4;
16,5 (0-CH2-
20 CH3) ; 18,6 (d, Jc_p = 4,5 Hz, CO-CH2-CH2-CH2-P ), 23,0; 23,1; 23,8;
23,9 (CH2) ; 24,5; 25,9
(d; Jc_p = 140,0 Hz; CO-CH2-CH2-CH2-P) ;28,7; 28,8; 30,5; 30,6 (CH2) ; 33,6
(d,
JC-13 = 14,5 Hz; CO-CH2-CH2-CH2-P) ; 37,0; 38,4 (CH-CH2-N) ; 48,6; 51,3 (CH2-
N) ; 61,5 (d,
J = 6,5 Hz, 0-CH2-CH3) ; 172,3 (C=0).
RMN 31P (160 MHz, CDCI3) 3 (ppm) : 31,8.
L'étape D conduit au composé titre (Rdt : quantitatif) dont les
caractérisations par RMN 11-1, 13C, 31P sont données ci-après.
RMN 1H (400 MHz, CDCI3) 3 (ppm) 0,81 - 0,88 (m, 12H, CH3); 1,15 - 1,31 (m,
16H, CH2) ;
1,52 - 1,58 (m, 1H, CH-CH2-N) ; 1,60- 1,66 (m, 1H, CH-CH2-N) ; 1,77 (dt, 2H, J
= 18,5 Hz,
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
36
7,5 Hz, CO-CH2-CH2-CH2-P) ; 1,90 - 2,01 (m, 2H, CO-CH2-CH2-CH2-P) ; 2,48 (t,
2H, J = 7,5 Hz,
CO-CH2-CH2-CH2-P) ; 3,11 (d, 2H, J = 7,5 Hz, CH2-N); 3,15 - 3,34 (m, 2H, CH2-
N) ; 9,33 (1s,
2H, OH).
RMN 13C (100 MHz, CDC13) O (ppm) : 10,8; 11,1; 14,2; 14,3 (CH3) ; 19,2 (d, J =
4,0 Hz, CO-
S CH2-CH2-CH2-P ); 23,2; 23,3; 24,0 (CH2) ; 25,6; 27,0 (d, 1= 140,0 Hz, CO-
CH2-CH2-CH2-P) ;
28,9; 29,0; 30,7; 30,8 (CH2); 30,5; 30,6 (d, J = 12,0 Hz, CO-CH2-CH2-CH2-P) ;
37,1; 38,6
(CH) ; 49,0 (CH2-N) ; 51,4 (CH2-N) ; 174,0 (C=0).
RMN 31P (160 MHz, CDCI3) 8 (ppm) : 31,5.
1.2 - Synthèse des COMPOSéS de formule particulière (l-a) dans laquelle m = 0,
R4 *
H et R5 = H :
Les composés répondant à la formule particulière (l-a) ci-avant dans
laquelle m = 0, Ri., K-2,
R3 ont la même signification que précédemment, R4 est différent
d'un atome d'hydrogène tandis que R5 représente un atome d'hydrogène peuvent
aussi
être synthétisés en suivant le schéma réactionnel illustré sur la figure 2.
Comme visible sur cette figure, cette synthèse consiste à faire réagir
dans une première étape, notée A, une amine, notée 1, avec un halogénure
d'halogénoacétyle (par exemple, un chlorure de chloroacétyle), noté 2, pour
obtenir le
composé 3.
Cette étape est, par exemple, réalisée en présence de triéthylamine
.. dans un solvant organique du type dichlorométhane.
Dans une deuxième étape, notée B sur la figure 2, le composé 3 est mis
à réagir avec un phosphite HP(0)(0R4)2 dans lequel R4 est différent d'un atome
d'hydrogène, pour obtenir le composé 4 dans lequel R4 est différent d'un atome
d'hydrogène
Cette étape est, par exemple, réalisée en présence de n-butyllithium et
de 2,2'-bipyridine (cette dernière servant d'indicateur coloré), ou d'hydrure
de sodium,
dans un solvant organique du type tétrahydrofurane.
Le composé 4 ainsi obtenu est alors soumis :
- soit à une étape de monosaponification, notée C sur la
figure 2, par
.. l'action d'une base forte, par exemple de la potasse, dans un solvant
composé d'eau et de
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
37
diméthylformamide, pour obtenir le composé 5 dans lequel R3 représente un
atome
d'hydrogène tandis que R4 est différent d'un atome d'hydrogène ;
¨ soit
à une étape de C-alkylation, notée D sur la figure 2, par réaction
du composé 4 avec un halogénure Hal-R3 dans lequel R3 est différent d'un atome
d'hydrogène, pour alkyler ce composé en a du groupe amide et obtenir le
composé 6
dans lequel R3 et R4 sont tous deux différents d'un atome d'hydrogène.
L'étape D est, par exemple, réalisée en présence de n-butyllithium et de
2,2'-pyridine, ou d'hydrure de sodium et de 2,2'-pyridine, dans un solvant
organique du
type tétra hyd rofu ra ne.
Le composé 6 obtenu à l'issue de cette étape est alors soumis à une
étape de monosaponification, notée E sur la figure 2, que l'on peut réaliser
de la même
manière que l'étape C, pour obtenir le composé 7 dans lequel R3 et R4 sont
tous deux
différents d'un atome d'hydrogène.
Sont ainsi synthétisés les composés suivants.
1.2.1 ¨ 1-(MN-diéthylhexylcarbamoynnonylphosphonate de butyle:
Le 1-(N,N-diéthylhexylcarbamoyl)nonylphosphonate de butyle, noté
DEHCNPB, qui répond à la formule particulière (l-a) ci-avant dans laquelle m =
0, = R2=
2-éthylhexyle, R3 = n-octyle, R4 = n-butyle et R5 = H, est synthétisé en
mettant en uvre
les étapes A, B, D et E montrées sur la figure 2.
Étape A : Synthèse du 2-chloro-N,N-diéthylhexylacétamide
On introduit dans un tricol, sous agitation et sous azote, 7,46 mL (2 éq.)
de bis(2-éthylhexyl)-amine, 50 mL de dichlorométhane et 3,5 mL (2 éq.) de
triéthylamine,
que l'on refroidit à -30 C avec un bain de carboglace/acétone. Puis, on ajoute
goutte à
goutte 2 mL (1 éq.) de chlorure de chloroacétyle et le mélange réactionnel est
laissé
pendant 3 heures à température ambiante. Après quoi, il est versé sur 50 mL
d'eau
distillée, décanté et la phase aqueuse est extraite avec 50 mL de
dichlorométhane. Les
phases dichlorométhane sont lavées avec 50 mL d'eau distillée, séchées sur du
sulfate de
magnésium, filtrées et évaporées à sec à l'évaporateur rotatif. Les 7,26 g
d'huile jaune
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
38
ainsi obtenus sont soumis à une chromatographie sur une colonne de gel de
silice (80 g -
63-200 lm de granulométrie) avec un mélange dichlorométhane/méthanol 98/2 v/v
comme éluant.
On obtient ainsi 7,20 g de 2-chloro-N,N-diéthylhexylacétamide sous la
forme d'une huile jaune pâle (Rdt : 92%), dont les caractérisations par RMN 1-
H et 13C ainsi
que par spectrométrie de masse sont données ci-après.
RMN 1H (400 MHz, CDCI3) 8 (ppm) : 4,08 (2H, s, CH2CI) ; 3,23-3,28 (2H, m,
NCH2) ; 3,20
(2H, d, 1= 7,5 Hz, NCH2) ; 1,76-1,65 (1H, m, NCH2CH) ; 1,64-1,52 (1H, m,
NCH2CH) ; 1,40-
1,15 (16H, m, CH3(CH2)3CH(CH2CH3)CH2N); 0,95-0,78 (12H, m, CH3).
RMN 13C (100,13 MHz, CDCI3) 8 (PPrn) : 166,64 (C=0) ; 51,27 (NCH2) ; 48,3
(NCH2) ; 41,17
(CH20I) ; 38,10 (CH) ; 36,40 (CH) ; 30,16 (NCH2CH(Et)CH2(CH2)2CH3) ; 30,00
(NCH2CH(Et)-
CH2(CH2)2CH3) ; 28,40 (NCH2CH(Et)CH2CH2CH2CH3) ; 28,24
(NCH2CH(Et)CH2CH2CH2CH3) ;
23,49 (-CHCH2CH3) ; 23,35 (-CHCH2CH3) ; 22,65 (NCH2CH(Et)-CH2CH2CH2CH3) ;
22,62
(NCH2CH(Et)CH2CH2CH2CH3) ; 13,67 (-CH2CH2CH3) ; 13,63 (-CH2CH2CH3) ; 10,49
(-CHCH2CH3) ; 10,20 (-CHCH2CI-13).
ESI+ : [M+H] = 318, [M+Na] = 340, [2M+Na] = 657
Étape B: Synthèse du 1-(N,N-diéthylhexylcarbamoyl)méthylphosphonate de
dibutyle
On introduit dans un tricol, sous agitation et sous azote, 1 mL (1 éq.) de
dibutylphosphite, quelques cristaux de 2,2'-bipyridine et 50 mL de
tétrahydrofurane, que
l'on refroidit à -50 C avec un bain de carboglace/acétone. Puis, on ajoute
goutte à goutte
4,6 mL (1,5 éq.) d'une solution de n-butyllithium à 1,6 mol/L dans de
l'hexane. On obtient
un mélange réactionnel rouge limpide. On ajoute ensuite goutte à goutte 1,95 g
(1,25 éq.)
de 2-chloro-N,N-diéthylhexylacétamide. On obtient un mélange réactionnel jaune
qui est
laissé pendant une nuit à température ambiante. Après quoi, il est versé sur
100 mL d'eau
distillée et acidifié par addition d'HCI 1 N en quantité suffisante pour
obtenir un pH acide.
La phase aqueuse est extraite avec 2 fois 50 mL d'éther éthylique. Les phases
éthérées
sont lavées avec 2 fois 50 mL d'eau distillée et 50 mL d'eau saturée en NaCI,
séchées sur
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
39
du sulfate de magnésium, filtrées et évaporées à sec à l'évaporateur rotatif.
Les 2,69 g
d'huile jaune ainsi obtenus sont soumis à une chromatographie sur une colonne
de silice
(125 g - 63-200 pm de granulométrie) en utilisant un mélange
cyclohexane/acétate
d'éthyle 8/2 v/v comme él ua nt.
On obtient ainsi 1,03 g de 1-(N,N-diéthylhexylcarbamoyl)méthyl-
phosphonate de dibutyle sous la forme d'une huile incolore (Rdt : 44%) dont
les
caractérisations par RMN 13C
et 31P ainsi que par spectrométrie de masse sont
données ci-après.
RMN 1H (400 MHz, CDCI3) (PPm) : 4,18-4,04 (4H, m, OCH2); 3,37-3,20 (4H, m,
NCH2) ;
3,07 (2H, d, J = 22 Hz, COCH2P); 1,78-1,51 (6H, m, OCH2CH2, CH) ; 1,47-1,14
(20H, m,
OCH2CH2CH2, C1-13(CH2)3CH(CH2C1-13)CH2N); 0,99-0,82 (18H, m, CH3)=
RMN 13C (100,13 MHz, CDCI3) (5 (ppm) : 165,15 (C=0); 66,15 (d, 2Jpc = 6,6 Hz)
; 52,26
(NCH2); 48,86 (NCH2), 38.58 (CH) ; 36,98 (CH); 33,61 (d, 1Jpc = 133 Hz, CH2P)
; 30,52 et
30,27 (NCH2CH(Et)CH2(CH2)2CH3); 30,17 (OCH2CH2CH2CH3); 28,81 et 28,67
(NCH2CH(Et)CH2CH2CH2CH3), 26,86 (OCH2CH2CH2CH3); 23,83 et 23,39 (-CHCH2CH3);
23,11
et 23,03 (NCH2CH(Et)CH2CH2CH2CH3); 18,72 (OCH2CH2CH2CH3); 14,12 et 14,03
(OCH2CH2CH2CH3) ; 13,61 (CH2CH2CH3) ; 10,91 et 10,51 (-CHCH2CH3).
RMN31P (162 MHz, CDCI3, découplage 1H) (ppm) : 22,03 (s, P=0)
ESI+ : [M+H] =476, [2M+Na] = 973
Étape D : Synthèse du 1-(N,N-diéthylhexylcarbamoyl)nonylphosphonate de
dibutyle
On introduit dans un tricol, sous agitation et sous azote, 1,99 g (1 éq.) de
1-(N,N-diéthylhexylcarbamoyl)méthylphosphonate de dibutyle, 50 mL de
tétrahydro-
furane et quelques cristaux de 2,2'-bipyridine, que l'on refroidit à -50 C
avec un bain de
carboglace/acétone. Puis, on ajoute goutte à goutte 3,92 mL (1,5 éq.) d'une
solution de
n-butyllithium à 1,6 mol/L dans de l'hexane. On obtient un mélange réactionnel
rouge
limpide que l'on laisse pendant 20 minutes à cette température. On ajoute
alors 0,92 mL
(1,25 éq.) de bromooctane. Le bain de carboglace/ acétone est retiré et la
réaction monte
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
doucement à température ambiante. Le mélange réactionnel est ensuite chauffé
pendant
une nuit à 60 C. Après quoi, le mélange réactionnel est versé sur 100 mL d'eau
distillée et
acidifié par addition d'HCI 1 N en quantité suffisante pour obtenir un pH
acide. La phase
aqueuse est extraite avec 2 fois 50 mL d'éther éthylique. Les phases éthérées
sont lavées
5 avec 2
fois 50 mL d'eau distillée et 50 mL d'eau saturée en NaCI, séchées sur du
sulfate de
magnésium, filtrées et évaporées à sec à l'évaporateur rotatif. Les 3,45 g
d'huile jaune
ainsi obtenus sont soumis à une chromatographie sur une colonne de silice (125
g ¨ 63-
200 pim de granulométrie) en utilisant un mélange cyclohexane/acétate d'éthyle
9/1 v/v
comme éluant.
10 On
obtient ainsi 1,1 g de 1-(N,N-diéthylhexylcarbamoyl)nonyl-
phosphonate de dibutyle sous la forme d'une huile incolore (Rdt : 44%), dont
les
caractérisations par RMN 1H, 13C et 31P ont déjà été données au point 1.1.11
ci-avant.
Étape E: Synthèse du 1-(N,N-diéthylhexylcarbamoyl)nonylphosphonate de butyle
15 On
introduit dans un réacteur micro-ondes CEM DiscoverTm 0,4 g (1 éq.)
de 1-(N,N-diéthylhexylcarbamoyl)nonylphosphonate de dibutyle, 5 mL d'eau
distillée,
5 mL de diméthylformamide et 0,23 g (6 éq.) de potasse, que l'on chauffe à 150
C
pendant 15 heures. Après quoi, le mélange réactionnel est versé sur 100 mL
d'eau
distillée et acidifié par addition d'HCI 1 N en quantité suffisante pour
obtenir un pH acide.
20 La phase
aqueuse est extraite avec 2 fois 50 mL d'éther éthylique. Les phases éthérées
sont lavées avec 2 fois 50 mL d'eau distillée, séchées sur du sulfate de
magnésium, filtrées
et évaporées à sec à l'évaporateur rotatif. Les 0,34 g d'huile ainsi obtenus
sont soumis à
une chromatographie sur une colonne de silice (17 g ¨ 63-200 Lm de
granulométrie) en
utilisant un mélange dichlorométhane/méthanol 97/3 v/v comme éluant.
25 On
obtient ainsi 0,14 g du composé titre sous la forme d'une huile
incolore (Rdt : 39%), dont les caractérisations par RMN 1H, 13C et 31P ont
déjà été données
au point 1.1.11 ci-avant.
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
41
1.2.2 ¨ Autres composés :
Les composés suivants :
¨ 1-(N,N-diéthylhexylcarbamoyl)benzylphosphonate d'éthyle, noté
DEHCBPE, qui répond à la formule particulière (l-a) ci-avant dans laquelle m =
0, R2
2-éthylhexyle, R3 = phényle, R4 = éthyle et R5 = H;
¨ 1-
(N,N-diéthylhexylcarba moyl)éthyl phospho nate d'éthyle, noté
DEHCEPE, qui répond à la formule particulière (l-a) ci-avant dans laquelle m =
0, = R2 =
2-éthylhexyle, R3 = méthyle, R4 = éthyle et R5 = H;
¨ 1-(N,N-diéthylhexylcarba moyl)nonylphosphonate d'éthyle, noté
DEHCNPE, qui répond à la formule particulière (l-a) ci-avant dans laquelle m =
0, = R2 =
2-éthylhexyle, R3 = n-octyle, R4 = éthyle et R5 = H ; et
¨ 1-(N,N-dioctylcarbamoyl)nonylphosphonate de butyle, noté
DOCNPB, qui répond à la formule particulière (l-a) ci-avant dans laquelle m =
0, = R2 =
R3 = n-octyle, R4 = n-butyle et R5 = H;
sont synthétisés en mettant en oeuvre les étapes A, B, D et E montrées sur la
figure 2 et
en utilisant, pour chacune de ces étapes, un protocole opératoire similaire à
celui décrit
au point 1.2.1 ci-avant.
Sont également synthétisés :
¨ le 1-(N,N-diéthylhexylcarbamoyl)méthylphosphonate d'éthyle, noté
DEHCMPE, qui répond à la formule particulière (l-a) ci-avant dans laquelle m =
0, R1= R2
2-éthylhexyle, R3 = R5 = H et R4 = éthyle ; et
¨ le 1-(N,N-diéthylhexylcarbamoyl)méthylphosphonate de butyle,
noté DEHCMPB, qui répond à la formule particulière (l-a) ci-avant dans
laquelle m = 0, RI-
= R2 = 2-e'thylhexyle, R3 = R5 = H et R4 = n-butyle ;
en mettant en oeuvre les étapes A, B et C montrées sur la figure 2 et en
utilisant, pour les
étapes A et B, un protocole opératoire similaire à celui décrit au point 1.2.1
ci-avant pour
ces étapes, et, pour l'étape C, un protocole opératoire similaire à celui
décrit au point
1.2.1 ci-avant pour l'étape E.
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
42
1.3 ¨ Synthèse des composés de formule particulière (l-b):
Les composés répondant à la formule particulière (l-b) ci-avant dans
laquelle m, R1, R4 et R5 ont la même signification que précédemment peuvent
être
synthétisés en suivant le schéma réactionnel illustré sur la figure 3.
Comme visible sur cette figure, cette synthèse consiste à faire réagir
dans une première étape, notée A, un lactame, noté 1, avec un halogénure (par
exemple,
un bromure) Hal-RI= pour obtenir le composé 2.
Dans une deuxième étape, notée B sur la figure 3, le composé 2 est mis
à réagir avec un composé Hal-(CH2)m-P(0)(0R4)2 (c'est-à-dire un halogénoalkyl-
phosphonate si m est différent de 0, et un halogénophosphonate si m = 0) dans
lequel R4
est différent d'un atome d'hydrogène pour obtenir le composé 3 dans lequel R4
est
différent d'un atome d'hydrogène.
Puis, le composé 3 ainsi obtenu est ensuite soumis :
¨ soit
à une étape de monosaponification, notée C sur la figure 3, que
l'on réalise de la même manière que l'étape C de la figure 2 décrite ci-avant,
pour obtenir
le composé 4 dans lequel R4 est différent d'un atome d'hydrogène ;
¨ soit
à une étape d'hydrolyse, notée D sur la figure 3, que l'on réalise
de la même manière que l'étape E de la figure 2 décrite ci-avant, pour obtenir
le composé
5 dans lequel R4 est un atome d'hydrogène.
Est ainsi synthétisé le (N-dodécylpyrrolidone)-1-phosphonate d'éthyle,
noté DPPE, qui répond à la formule particulière (l-b) dans laquelle m = 0, RI-
= n-dodécyle,
R2 et R3 forment ensemble un groupe ¨CH2-CH2-, R4= éthyle, R5= H, en mettant
en uvre
les étapes A, B et C du schéma réactionnel montré sur la figure 3.
L'étape A est réalisée en ajoutant, sous agitation, du bromure de
tétrabutylammonium (950 mg ¨ 3 mmol ¨ 0,05 éq.) et de la potasse en poudre
(23,1 g ¨
411 mmol ¨ 7 éq.) à une solution de 2-pyrrolidinone (5,00 g ¨ 58,8 mmol) et de
bromododécane (18,5 mL, 76,4 mmol, 1,3 éq.) dans le toluène (60 mL). Le
mélange est
ensuite chauffé à 50 C pendant une nuit. Après disparition de la 2-
pyrrolidinone (que l'on
vérifie par CCM en utilisant un mélange d'acétate d'éthyle et de cyclohexane
4/1 v/v et
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
43
de l'acide phosphomolybdique comme révélateur) et refroidissement, on ajoute
60 mL
d'eau et le mélange est agité pendant encore 15 minutes. Les phases aqueuse et
organique sont séparées et la phase aqueuse est extraite une fois au
diéthyléther (60 mL).
Les phases organiques sont réunies, séchées sur Na2SO4, filtrée et
concentrées. Après
distillation sous vide (146 C - 0,5 mbars), on obtient ainsi la N-
dodécylpyrrolidin-2-one
(Rdt : 80%) dont les caractérisations par RMN 11-let 13C sont données ci-
après.
RMN 1H (400 MHz, CDCI3) S (ppm) : 0,88 (t, 3H, J = 7,0 Hz, CH3); 1,25 - 1,31
(m, 18H,
CH2) ; 1,47- 1,54 (m, 2H, Alk-CH2-CH2-N) ; 1,97- 2,05 (m, 2H, CO-CH2-CH2-CH2-
N) ; 2,38 (t,
2H, J = 8,0 Hz, CO-CH2-CH2-CH2-N) ; 3,26 (t, 2H, J = 7,5 Hz, Alk-CH2-CH2-N) ;
3,37 (t, 2H,
J =7,0 Hz, CO-CH2-CH2-CH2-N).
RMN 13C (100 MHz, CDCI3) 5 (ppm) : 14,3 (CH3) ; 18,1 (CO-CH2-CH2-CH2-N) ; 22,9
; 27,0
(CH2aik) ; 27,5 (Alk-CH2-CH2-N) ; 29,5 ; 29,7; 29,8; 29,9 (CH24ik) ; 31,3 (CO-
CH2-CH2-C1-12-N) ;
32,1 (CHzaik) ; 42,7 (Alk-CH2-CH2-N) ; 47,2 (CO-CH2-CH2-CH2-N) ; 174,9 (CO).
L'étape B est réalisée en ajoutant, goutte à goutte et sous agitation, une
solution de N-dodécylpyrrolidinone (1,91 g - 7,5 mmol) dans le THF anhydre
(7,5 mL) à
une solution de disopropylamidure de lithium (7,5 mL - 2 M dans le THF) à -80
C sous
argon. Puis, on laisse le mélange revenir à la température ambiante et on le
maintient
sous agitation pendant 1 heure. Il est ensuite refroidi à -80 C et on lui
ajoute le chloro-
phosphate de diéthyle (1,25 mL - 8,6 mmol). Après 15 minutes, on laisse le
mélange
remonter à température ambiante et on le maintient sous agitation pendant une
nuit. Le
mélange est ensuite acidifié jusqu'à pH 1 à l'aide d'une solution d'HCI à 1
mol/L et extrait
au dichlorométhane (2 x 10 mL). Les phases aqueuse et organique sont séparées
et la
phase organique est séchée sur Na2SO4, filtrée et concentrée. Après
purification par
chromatographie éclair (acétate d'éthyle/acétone : de 100/0 à 20/80, v/v), on
obtient le
(N-dodécylpyrrolidinone)-1-phosphonate de diéthyle (Rdt : 37%) dont les
caractérisations
par RMN 11-1, 13C et 31P sont données ci-après.
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
44
RMN 1H (400 MHz, CDCI3) S (ppm): 0,89 (t, 3H, J = 7,0 Hz, CH3) ; 1,26 - 1,38
(m, 24H, CH2,
0-CH2-CH3) ; 1,49 - 1,56 (m, 2H, Alk-CH2-CH2-N) ; 2,25 - 2,47 (m, 2H, COCH(P)-
CH2-0-12-
N) ; 2,95 (ddd, 1H, J = 22,0 Hz, 10,0Hz, 5,5 Hz, COCH(P)-(CH2)2) ; 3,21 - 3,37
(m, 3H,
COCH(P)-CH2-CH2-N, Alk-CH2-N) ; 3,50 - 3,56 (qd, 1H, J =8,0 Hz, J = 1,0 Hz,
COCH(P)-CH2-
CH2-N) ; 4,13 - 4,31(m, 4H, O-CH2-CH3).
RMN 13C (100 MHz, CDCI3) 5 (ppm) : 14,1 (CH,); 16,4 (CH3-CH2-0) ; 20,5 (d, J =
Hz,
COCH(P)-CH2-CH2-N), 22,7; 26,8 (CH2) ; 27,2 (Alk-CH2-CH2-N) ; 29,3; 29,4;
29,5; 31,9
(CF-12); 40,5; 41,9 (d, J = 142 Hz, COCH(P)-(CH2)2N) ; 43,1 (Alk-CH2-N) ; 43,8
(d, J = 4,0 Hz,
COCH(P)-CH2-CH2-N), 62,2 (d, J = 6,5 Hz, 0-CH2-CH3) ; 63,0 (d, J = 6,5 Hz, 0-
CH2-CH,);
169,0 (d, J = 4,0 Hz, C=0).
RMN P (160 MHz, CDCI3) O (ppm) : 24,7.
L'étape C est réalisée en utilisant de la potasse (4 éq. pour 1 éq. de
(N-dodécylpyrrolidinone)-1-phosphonate de diéthyle - reflux de 2,5 heures) et
conduit au
(N-dodécylpyrrolidone)-1-phosphonate d'éthyle (Rdt : 99%) dont les
caractérisations par
RMN 11-1, 13C et 31P sont données ci-après
RMN 1H (400 MHz, CDCI3) O (ppm) : 0,89 (t, 3H, J = 6,5 Hz, CH3); 1,26 - 1,32
(m, 21H, CH2,
0-CH2-CH3) ; 1,44 - 1,51 (m, 2H, Alk-CH2-CH2-N) ; 2,21 - 2,44 (m, 2H, COCH(P)-
CH2-0-12-
N) ; 2,96 (ddd, 1H, J = 22,0 Hz, 10,0Hz, 5,5 Hz, COCH(P)-(CH2)2) ; 3,19 - 3,34
(m, 3H,
COCH(P)-CH2-CH2-N, Alk-CH2-N) ; 3,45 - 3,51 (q, 1H, J = 8,0 Hz, COCH(P)-CH2-
CH2-N) ; 4,11
- 4,19(m, 2H, O-CH2-CH,).
RMN 13C (100 MHz, CDCI3) O (ppm) : 14,1 (CH3) ; 16,4 (d, J = 6,0 Hz, CH 3-CH2-
O) ; 20,5 (d,
J = 3,0 Hz, COCH(P)-CH2-CH2-N), 22,7; 26,8 (CH2) ; 27,2 (Alk-CH2-CH2-N) ;
29,3; 29,4;
29,5; 29,6; 31,9 (CH2); 40,5; 41,9 (d, J = 142 Hz, COCH(P)-(CH2)2N) ; 43,2
(Alk-CH2-N) ;
46,0 (d, J = 4,5 Hz, COCH(P)-CH2-CH2-N), 62,3 (d, J = 6,5 Hz, 0-CH2-CH,);
170,0 (d, J = 4,0
Hz, C=0).
RMN 1P (160 MHz, CDCI3) O (ppm) : 24,9.
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
EXEMPLE II : PROPRIÉTÉS DES COMPOSÉS SELON L'INVENTION
11.1 ¨ Capacité des composés selon l'invention à extraire l'uranium d'une
solution
synthétique d'U-Fe dans H3 PO4 5 M:
La capacité des composés selon l'invention à extraire l'uranium d'une
5 solution aqueuse d'acide phosphorique est appréciée par des tests
d'extraction qui sont
réalisés de la manière suivante.
Chaque composé testé est tout d'abord solubilisé dans du n-dodécane
(sans utilisation d'agent modificateur de phase ni de chauffage) à hauteur de
0,25 mol/L.
Puis, 6 mL de chacune des phases organiques ainsi obtenues sont mis en
10 contact et maintenus sous agitation pendant 1 heure, à température
ambiante (23-24 C),
avec 6 mL d'une phase aqueuse synthétique comprenant 5 mol/L d'acide
phosphorique,
0,25 g/L d'uranium(VI) et 2,5 g/L de fer(III).
Après quoi, ces phases sont séparées par décantation gravitaire en
moins de 3 minutes.
15 Les concentrations en uranium(VI) et en fer(III) sont mesurées :
¨ dans la phase aqueuse initiale avant qu'elle ne soit mise en contact
avec les phases organiques, par fluorescence X et par spectrométrie d'émission
atomique
par torche à plasma (ou ICP-AES),
¨ dans les phases aqueuses obtenues après leur séparation des
20 phases organiques, également par fluorescence X et par ICP-AES, et
¨ dans les phases organiques obtenues après leur séparation des
phases aqueuses, par fluorescence X.
Le tableau I ci-après présente, pour chaque composé testé, le coefficient
de distribution de l'uranium(VI), noté Du, et le facteur de séparation entre
l'uranium(VI)
25 et le fer(110, noté FSuiFe, qui sont obtenus à partir des teneurs en
uranium et en fer ainsi
mesurées.
Sont également précisés dans ce tableau le coefficient de distribution Du
et le facteur de séparation FSuiFe qui sont obtenus dans les mêmes conditions
avec le
mélange synergique HDEHP/TOPO.
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
46
On rappelle que, dans le domaine des extractions liquide-liquide, le
coefficient de distribution Dm d'un élément M correspond au rapport des
concentrations
de cet élément dans les phases organique et aqueuse ayant été mises en contact
l'une
avec l'autre, et que le facteur de séparation FSm1/m2 entre deux éléments
métalliques M1
et M2 correspond à Dm1/Dm2, c'est-à-dire au rapport des coefficients de
distribution des
éléments métalliques M1 et M2 obtenus au cours d'une même extraction.
TABLEAU I
Composés Du FSuiFe
HDEHP/TOPO 3,8 200
DEHCMPE 12,2 10
DEHCMPB 24 10
DEHCBPE 65 1 800
DEHCEPE 120 800
DEHCNPE 120 1 900
DEHCNPB 117 2 800
DOCNPB 116 400
Ce tableau montre que les composés selon l'invention présentent une
capacité à extraire l'uranium(VI) d'une solution aqueuse d'acide phosphorique
bien
supérieure à celle du mélange synergique HDEHP/TOPO et, pour la plupart
d'entre eux,
une sélectivité U/Fe qui est également bien supérieure à celle que présente le
mélange
synergique HDEHP/TOPO.
11.2 ¨ Influence de la concentration à laquelle sont utilisés les composés
selon
l'invention sur leurs propriétés extractantes :
L'influence de la concentration à laquelle sont utilisés les composés
selon l'invention sur leurs propriétés extractantes est appréciée par des
tests d'extraction
que l'on réalise de la même manière qu'au point 11.1 ci-avant, mais en
utilisant ces
composés à différentes concentrations dans les phases organiques.
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
47
Ces tests sont réalisés avec les 5 composés selon l'invention s'étant
révélés les plus performants dans les tests précédents.
Le tableau Il ci-après présente les coefficients de distribution Du et les
facteurs de séparation FSuiFe qui sont obtenus avec chacun des composés testés
et, pour
un même composé, avec chacune des concentrations auxquelles celui-ci a été
utilisé.
Sont également précisés dans ce tableau les coefficients de distribution
Du et les facteurs de séparation FSu/F, qui sont obtenus dans les mêmes
conditions et
pour les mêmes concentrations avec le mélange synergique HDEHP/TOPO, ainsi que
les
rapports entre les Du et les FSue, obtenus avec les composés selon l'invention
et ceux
obtenus avec ce mélange.
TABLEAU Il
(C) Rapport Rapport
Composés Du FS u/Fe
(mol/L) Du FSuee
0,25 3,8 150
TOPO/HDEHP / /
0,1 0,8 230
DEHCBPE 0,25 67 1 450 18 9,7
0,1 16 1 740 20 7,6
0,25 120 800 32 4
DEHCEPE
0,1 71 2 800 89 14
0,25 120 1 900 32 9,5
DEHCNPE 0,1 75 9 400 90 47
0,05 23 26 000 / /
0,25 117 2 800 31 14
DEHCNPB 0,1 70 8 700 88 43,5
0,05 20 30 000 / /
0,25 116 400 30 2
DOCNPB 0,1 62 1 000 77 5
0,05 20 1 300 / /
Ce tableau montre que les rapports entre les coefficients de distribution
Du obtenus avec les composés selon l'invention et ceux obtenus avec le mélange
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
48
HDEHP/TOPO de référence sont d'autant plus élevés que la concentration à
laquelle ces
composés et ce mélange sont utilisés est, elle, plus faible.
A une concentration de 0,1 mol/L, le composé DEHCNPE est 90 fois plus
efficace et 47 fois plus sélectif que le mélange TOPO/HDEHP.
La variation du logarithme du coefficient de distribution de l'U(VI), noté
log(Du), en fonction du logarithme de la concentration molaire en composé
DEHCNPB de
la phase organique, noté log([DEHCNP13]), qui est illustrée sur la figure 4,
donne une
pente d'environ 2, ce qui montre que l'ion U022 serait extrait par le composé
DEHCNPB
sous forme d'un complexe 1 : 2.
La sélectivité uranium/fer atteint ainsi la valeur de 30 000 avec le
composé DEHCNPB lorsque celui-ci est utilisé à une concentration de 0,05
mol/L.
11.3 ¨ Influence de la concentration initiale en uranium(VI) de la solution
aqueuse
d'acide phosphorique sur les propriétés extractantes des COMPOSéS selon
l'invention :
L'influence de la concentration en uranium(VI) que présente
initialement la solution aqueuse d'acide phosphorique sur les propriétés
extractantes des
composés selon l'invention est appréciée par des tests d'extraction que l'on
réalise de la
même manière qu'au point 11.1 ci-avant, mais en faisant varier la
concentration initiale en
uranium(VI) de la phase aqueuse.
Ces tests sont réalisés avec le composé DEHCPNB, à une concentration
de 0,1 mol/L, compte tenu de ses performances.
Avec 0,1 mol/L du composé DEHCNPB dans du n-dodécane, aucune
troisième phase n'a été observée, même pour une concentration en uranium(VI)
de 5 g/L
dans la phase organique, ce qui correspond à 15 g/L d'uranium(VI) dans la
phase aqueuse
initiale.
Les résultats sont illustrés sur la figure 5 qui montre que la
concentration de l'uranium(VI) en phase aqueuse ([Uac]) est inférieure à la
concentration
de l'uranium(VI) en phase organique ([Uord) et ce, dans un domaine de
concentration en
phase organique en uranium(VI) inférieur à 4,5 g/L d'uranium(VI). Dans ce
domaine, le
coefficient de distribution Du observé est supérieur à 1.
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
49
Cet exemple illustre la possibilité de mettre en oeuvre des schémas
concentrants, lors de procédés d'extraction de l'uranium présent dans des
solutions
aqueuses d'acide phosphorique issues de phosphates naturels, en utilisant le
composé
DEHCPNB.
11.4 ¨ Influence de la concentration initiale en acide phosphorique de la
solution
aqueuse d'acide phosphorique sur les propriétés extractantes des composés
selon
l'invention :
L'influence de la concentration en acide phosphorique que présente
initialement la solution aqueuse d'acide phosphorique sur les propriétés
extractantes des
composés selon l'invention est appréciée par des tests d'extraction que l'on
réalise de la
même manière qu'au point 11.1 ci-avant, mais en utilisant une phase aqueuse
contenant
10 fois plus d'uranium que celle utilisée au point 11.1 ci-avant, soit 2,5 g/L
d'uranium, et en
faisant varier la concentration initiale en acide phosphorique de la phase
aqueuse.
Ces tests sont réalisés avec le composé DEHCPNB, à une concentration
de 0,1 mol/L.
La variation du logarithme du coefficient de distribution de
l'uranium(VI), noté log(Du), en fonction du logarithme de la concentration
molaire en
acide à l'équilibre après extraction, noté log([Hlég ), qui est illustrée sur
la figure 6,
montre que l'extraction de l'uranium(VI) diminue lorsque la concentration en
acide
phosphorique augmente de 0,01 à 9 mol/L du fait de la compétition entre les
ions H+ et
U022+. Néanmoins, un coefficient de distribution Du égal à 3,3 est obtenu avec
une
concentration en acide phosphorique de 9 mol/L, ce qui devrait permettre
d'extraire
l'uranium(VI) de solutions aqueuses très concentrées en acide phosphorique.
Cet exemple illustre la possibilité d'opérer dans une large gamme
d'acidités pour extraire l'uranium(VI) d'une solution aqueuse d'acide
phosphorique avec
le composé DEHCPNB.
11.5 ¨ Désextraction de l'uranium(VI) présent dans une phase organique
comprenant un composé selon l'invention :
Des tests de désextraction sont réalisés en utilisant :
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
¨ une pluralité de phases organiques comprenant de 0,01 à 0,1 mol/L
de composé DEHCNPB dans du n-dodécane et qui ont été préalablement chargées en
uranium(VI) et en fer, par contact avec une solution comprenant 5 mol/L
d'acide
phosphorique, 0,25 g/L d'uranium(VI) et 2,5 g/L de fer(III) dans de l'eau ; et
5 ¨ une phase aqueuse comprenant 0,5 mol/L de carbonate
d'ammonium (NH4)2CO3.
Pour ce faire, 6 mL de chacune des phases organiques (qui contiennent
la majorité de l'uranium(VI) qui était initialement présent dans la solution
dont il a été
extrait, ainsi que des traces de fer) sont mis en contact et maintenus sous
agitation
10 pendant 1 heure, à température ambiante (23-24 C), avec 6 mL de phase
aqueuse.
Après quoi, ces phases sont séparées par décantation gravitaire en
moins de 3 minutes.
Comme précédemment, les concentrations en uranium(VI) et en fer(III)
sont mesurées :
15 ¨ dans la phase aqueuse avant qu'elle ne soit mise en contact
avec les
phases organiques, par fluorescence X et par ICP-AES,
¨ dans les phases aqueuses obtenues après séparation des phases
organiques, également par fluorescence X et par ICP-AES, et
¨ dans les phases organiques obtenues après séparation des phases
20 aqueuses, par fluorescence X.
Les résultats montrent que les coefficients de distribution de l'uranium
sont inférieurs à 0,1, ce qui signifie que la totalité de l'uranium présent
dans les phases
organiques se retrouve dans les phases aqueuses après un seul contact entre
phase
aqueuse et phase organique et ce, sans apparition de troisième phase ou de
trouble.
25 Le composé DEHCNPB devrait donc permettre de s'affranchir des
problèmes liés à la présence de fer(III) dans les solutions aqueuses d'acide
phosphorique
qui sont obtenues à partir de phosphates naturels tout en améliorant
l'extraction de
l'uranium(VI) de ces solutions par rapport à celle obtenue avec le mélange
synergique
HDEHP/TOPO.
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
51
11.6 ¨ Cinétique d'extraction :
La cinétique d'extraction de l'uranium(VI) par les composés selon
l'invention est appréciée par des tests d'extraction que l'on réalise de la
même manière
qu'au point 11.1 ci-avant, mais en maîtrisant le fonctionnement aqueux continu
(FAC) et en
utilisant une phase aqueuse contenant 10 fois plus d'uranium que celle
utilisée au point
11.1 ci-avant. Cette phase aqueuse contient donc 5 mol/L d'acide phosphorique,
2,5 g/L
d'uranium et 2,5 g/L de fer.
Les phases organique et aqueuse sont mises en contact dans une cellule
à double enveloppe de 25 mL, thermostatée à 25 C, avec une agitation par pale
et
trébuchet anti-vortex (2000 tr/min).
Ces tests sont réalisés avec le composé DEHCPNB, à une concentration
de 0,1 mol/L.
Les résultats sont illustrés sur la figure 7 qui montre que l'équilibre
d'extraction est rapidement atteint en moins de deux minutes.
La cinétique d'extraction de l'uranium(VI) par le composé DEHCNPB
n'est donc pas un facteur limitant et permet d'envisager une utilisation de ce
composé
comme extractant dans des contacteurs industriels à temps de séjour réduit.
11.7 ¨ Sélectivité de l'extraction de l'uranium (VI) vis-à-vis des autres
cations
métalliques :
La sélectivité de l'extraction de l'uranium(VI) vis-à-vis, non seulement du
fer, mais également des autres cations métalliques qui sont susceptibles
d'être présents
dans des solutions aqueuses d'acide phosphorique obtenues à partir de
phosphates
naturels est appréciée en réalisant un test d'extraction de la même manière
qu'au point
11.1 ci-avant, mais en utilisant comme phase aqueuse, une solution issue de
l'attaque d'un
phosphate naturel par de l'acide sulfurique.
Cette phase aqueuse comprend 5 mol/L d'acide phosphorique. Sa
composition en cations métalliques est présentée dans le tableau III ci-après.
On utilise comme phase organique, une phase comprenant le composé
DEHCPNB, à une concentration de 0,1 mol/L, dans du n-dodécane.
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
52
Dans ce test, la teneur en cations est mesurée par ICP-AES dans la phase
aqueuse initiale, avant qu'elle ne soit mise en contact avec la phase
organique, et dans la
phase aqueuse obtenue après extraction, c'est-à-dire après sa séparation de la
phase
organique.
Les coefficients de distribution Dm obtenus pour l'uranium(VI) et pour
chacun des autres cations métalliques sont présentés dans le tableau III ci-
après.
TABLEAU III
Phase aqueuse initiale Dm après extraction
[C]
Cations
mg/L
U 155 >150
Fe 2470 0,01
As 16 <0,07
Mo 6 0,15
Cr 276 <0,01
Zn 393 <0,01
Cd 19 <0,06
W 14 <0,07
Al 1880 <0,01
V 264 <0,02
Ti 55 <0,02
Zr 44 <0,03
Comme le montre ce tableau, à une concentration de 0,1 mol/L, le
composé DEHCNPB présente une capacité à extraire l'uranium(VI) d'une solution
aqueuse
d'acide phosphorique obtenue à partir d'un phosphate naturel qui est
comparable à celle
observée avec une solution synthétique.
Parmi les deux impuretés majeures, l'aluminium et le fer, seul ce dernier
est extrait de manière quantifiable mais toutefois faiblement.
Parmi les impuretés mineures, seul le molybdène est extrait à des
teneurs mesurables mais faibles également.
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
53
Le composé DEHCNPB présente donc une excellente sélectivité pour
l'uranium(VI) vis-à-vis des autres cations métalliques susceptibles d'être
présents dans
une solution aqueuse d'acide phosphorique obtenue à partir d'un phosphate
naturel.
11.8 ¨ Exemple de mise en uvre en continu du procédé selon l'invention :
Un essai de mise en uvre en continu du procédé selon l'invention a
été réalisé, à une échelle pilote, en utilisant des batteries de mélangeurs-
décanteurs de
laboratoire.
L'objectif de cet essai était de vérifier les performances d'extraction de
l'uranium(VI) et, plus particulièrement, la sélectivité du solvant vis-à-vis
du fer, élément
majoritairement présent dans les minerais. Il s'agissait notamment d'obtenir,
à partir
d'une solution aqueuse d'acide phosphorique, une solution concentrée
d'uranium(VI)
caractérisée par une teneur en fer inférieure à 0,15%.
Le schéma de mise en oeuvre, qui est représenté sur la figure 8,
comportait 3 étapes :
¨ une étape d'extraction de l'uranium présent dans la solution
aqueuse d'acide phosphorique, cette extraction étant réalisée avec le composé
DEHCNPB,
à une concentration de 0,1 mol/L, dans du n-dodécane sur 5 étages ;
¨ une étape de lavage de la phase organique obtenue à l'issue de
cette extraction, ce lavage étant réalisé avec une solution aqueuse d'acide
sulfurique sur
3 étages ; et
¨ une étape de désextraction concentrante de l'uranium(VI) présent
dans la phase organique obtenue à l'issue de ce lavage, cette désextraction
étant réalisée
avec une solution aqueuse de carbonate d'ammonium sur 3 étages.
Les étapes d'extraction et de lavage ont été réalisées à 40 C tandis que
l'étape de désextraction a été réalisée à 45 C.
Le pilotage du procédé a été assuré à partir du suivi des concentrations
en uranium et en fer des solutions aqueuses sortant des trois batteries de
mélangeurs-
décanteurs ainsi que de la phase organique issue de la batterie de mélangeurs-
décanteurs
dévolue à l'extraction.
CA 02872084 2014-10-30
WO 2013/167516 PCT/EP2013/059352
54
L'essai a été réalisé sur une solution d'acide phosphorique industrielle
présentant une concentration en uranium de 119 mg/L, une concentration en fer
de
5,7 g/L et une acidité de 5,2 mol/L.
Les résultats de cet essai sont conformes aux objectifs visés, à savoir
qu'il a été obtenu une solution d'uranium concentrée ([U] 5 g/L dans le
flux de
production uranium issu de l'étape de désextraction) avec une teneur en fer (¨
2,4 mg/L)
très inférieure aux 0,15% visé en masse par rapport à l'uranium (<0,05%).
RÉFÉRENCES CITÉES
[1] US 3,711,591
[2] FR 2 396 803
[3] FR 2 596 383
[4] US 4,316,877
[5] FR 2 460 958
[6] FR 2 460 960
[7] FR 2 604 919