Note: Descriptions are shown in the official language in which they were submitted.
CA 02877190 2014-12-18
1
English Translation of PCT/EP2013/063162
Medicament form for release of active ingredients
The present invention relates to a medicament form for the release of active
ingredients, a
method for the production thereof and uses thereof.
This invention is concerned with the provision of medicament forms which
enable an
effective administration of an active ingredient which is usually taken twice
or more daily by
way of a single daily dose.
With the development of new medicament forms it is always important that the
design of the
new medicament form is such that the compliance of the patient can be
improved. This for
example can be achieved by the reduction of single doses of a medicament which
have to be
taken daily. The reason for that is that the compliance of the patient often
considerably
decreases, when several single doses of a medicament have to be taken on one
day, in
particular, when a combination with further medicaments has to be taken. This
is in particular
the case with patients with difficulties in swallowing and also elder patients
who have to take
numerous medicaments due to multimorbidity.
But a sufficient compliance of the patient is constantly necessary for
avoiding side effects and
reduced effectiveness of the medicament. The lack of compliance of numerous
patients is a
significant factor for the increasing health expenditures.
In this respect, the reduction of the frequency of use of a medicament from
doses which have
to be administered a few times daily to a single daily dose has become more
important.
Normally, in such a case the effect should be comparable with an
administration a few times
daily. This can be achieved by the use of mechanisms of retardation such as
embedment of a
drug into a matrix so that the active ingredient is released in a retarded
manner, this means
uniformly over a longer period of time.
But the reduction of the frequency of use requires special medicament
formulations which are
associated with high technological requirements, since they comprise
relatively high amounts
of active ingredient. One technological challenge is the provision of a
medicament form with
sufficient stability and good release behavior, wherein the size of the
medicament form is still
is a tolerable range. The latter is important, so that the medicament form can
be swallowed
easily. Similarly, in the case of high amounts of active ingredient it may be
difficult to
guarantee a tolerable taste.
CA 02877190 2014-12-18
2
English Translation of PCT/EP2013/063162
Till today, predominantly active ingredients having bad solubility have been
the center stage
of such developments. For them numerous medicament forms have been developed,
in
particular matrix-based systems. The resorption of such active ingredients is
determined by
their release from the medicament form so that by the development of suitable
matrix systems
a good resorption of these active ingredients could be achieved. For active
ingredients having
good solubility other concepts are required. Naturally it is much more
difficult to retard the
release of an active ingredient with good solubility than that of an active
ingredient with bad
solubility which already dissolves slowly.
Independently from the solubility of an active ingredient there are in
particular considerable
difficulties, when the processing of the active ingredients in question is
difficult due to their
instability. Such active ingredients are in particular active ingredients with
high
decomposition rates or hygroscopy.
Hygroscopic active ingredients have a tendency of taking up humidity from the
environment,
most often in the form of water vapor from air moisture. This can take place
via different
mechanisms such as absorption and adsorption. This involves the risk of
melting or
agglutinating of the active ingredients or of the formation of undesired
crystal forms. The
latter considerably complicates their processing so that laborious and costly
methods and
intermediate steps are required, for example for excluding humidity. This
disadvantageous
property of some active ingredients also complicates the provision of a
medicament form with
good storability and shelf life. Not only the taking up of humidity from the
environment is a
critical point, because the active ingredient can also extract humidity from
the medicament
form itself. For example, such active ingredients can adsorb humidity from the
capsule
casings, wherein this may result in cracks in the capsule casing.
Due to the hygroscopy of the active ingredient, besides the release of the
active ingredient
from the medicament form also the handling of such medicament forms can be
complicated
considerably. Thus in general it would be required that the medicament forms
are stored in a
manner which guarantees protection from air and smallest amounts of humidity.
But the
handling during the use of the patient cannot be controlled and it can be
assumed that in
particular elder humans store their tablets outside the blister, for example
in pill organizers.
In this respect it is also problematic that many highly effective active
ingredients and active
ingredients for which no alternatives are available are hygroscopic. Such
active ingredients
can for example be found in the classes of active ingredients of
antiepileptics, antibiotics,
CA 02877190 2014-12-18,
3
English Translation of PCT/EP2013/063162
antidiabetics, cardiovascular agents, gastrointestinal therapeutics, lipid-
lowering agents,
antidementives, muscle relaxants, analgesics, broncholytic/antiasthmatic
agents or also
antiallergic agents as well as antiemetic/antivertiginous agents.
As an example, here the antiepileptics valproic acid and carbamazepine are
mentioned. Also
the antibiotics tetracycline, lincomycin, clindamycin or also rifampicin have
hygroscopic
properties. Cardiovascular agents with hygroscopic properties are for example
atenolol and
minoxidil. Betahistine is mentioned as an example for an
antiemetic/antivertiginous agent. An
example of an antiallergic agent is dexamethasone, and an example of an
antidementive is
piracetam. An example of a hygroscopic lipid-lowering agent is gemfibrozil.
In therapy it is not possible to dispense with such drugs. So numerous of such
active
ingredients are also classified by the world health organization as
indispensable medicaments.
Therefore, there exists a high demand for medicament forms which contain such
drugs and
are nevertheless storable and are characterized by a good shelf life and which
also show a
release behavior which enables the single daily intake. In particular, there
is a need for
medicament forms which release such drugs in a manner so that an intake
several times a day
is not necessary and which can be manufactured in a cost-effective manner in a
larger scale.
Considerable difficulties arise in particular with respect to hygroscopic
active ingredients
which at the same time are also highly soluble. The high solubility of such
active ingredients
further complicates the already due to hygroscopy very difficult provision of
such active
ingredients in medicament forms with retarded release. Due to the high
solubility normal
retardation formulations for the retarded release of such active ingredients
are generally
unsuitable. Additional difficulties also result from active ingredients which
besides high
hygroscopy also show high acidity, in particular in the case, when
concurrently the active
ingredient is characterized by high solubility. So in this case there is the
additional risk that
the acidic active ingredient attacks or decomposes the adjuvants. This may
result in
decomposition of the medicament form and in modification of the release
profile. If it is
desired to provide a medicament form showing the respective release, so there
is the risk that
the whole proportion of the active ingredient is quickly released, resulting
in high plasma
levels and thus also strong side effects. Insofar the formulation of such a
drug is further
complicated. In this connection in particular betahistine, for example in the
form of the salts
betahistine dimesylate or betahistine dihydrochloride have to be mentioned.
CA 02877190 2014-12-18
4
English Translation of PCT/EP2013/063162
In many cases medicament forms were provided which however are not suitable
for the
release of hygroscopic drugs, in particular active ingredients with high
solubility and/or
acidity, and insofar do also not address the problems during their processing
as well as the
problems, when medicament forms with sufficient stability and storability are
provided.
A tablet for the release of betahistine in the form of betahistine
dihydrochloride is described in
WO 00/53162 Al. Here, the tablet releases the active ingredient in a retarded
manner over a
period of time of mostly 5 to 8 hours. Thus however, several daily intakes may
be become
necessary. Also generally a combined quick and retarded release in the form of
a two layer
tablet is mentioned, but without any disclosure about its exact design or
production. In
particular there is no disclosure about the cohesion of the layers and how an
acceptable size of
the medicament form can be guaranteed. Also the release behavior of the
medicament form
remains unclear. There is only one general note that an intake two times or
also one time a
day may be possible. Insofar it cannot be deduced, whether in general this
medicament form
would be suitable for mimicking an intake several times a day.
In DE 100 10 509 Al oral medicament forms are described which contain a
sucrose fatty acid
ester in a proportion of 1 to 95 % by weight as the single agent for release
control. The
medicament forms release the contained active ingredients in a quick or
retarded manner. As
possible active ingredients a plurality of substances, including virtually
insoluble active
ingredients are mentioned. For example, also betahistine dimesylate is
mentioned as a
possible active ingredient. A medicament form which is suitable for releasing
one and the
same active ingredient in at least two phases, thus mimicking an intake
several times a day, is
not described in DE 100 10 509 Al. So also the problem of a release in at
least two phases of
especially hygroscopic active ingredients which possibly in addition are
highly soluble and
acidic is not solved in DE 100 10 509 Al.
WO 98/13029 Al on the other hand relates to medicament forms for retarded
release of one
or more active ingredients, including proteins, enzymes or vaccines which are
sensitive to
gastric juice, for guaranteeing the therapeutic action over a sufficient time.
As a possible
active ingredient inter alia betahistine is described. The formulations
contain a water-
insoluble coating. A gastric juice-resistant coating is not described. A
formulation for quick
release or another formulation for retarded release may be combined with this
formulation, for
example in a capsule or as a coating. Exact designs of such a combination of
formulations
containing active ingredient, in particular suitable proportions and ratios of
amounts are not
described. Insofar it cannot be deduced, whether this medicament form would at
all be
CA 02877190 2014-12-18
English Translation of PCT/EP2013/063162
suitable for optimally mimicking an intake several times a day together with
an immediate
release of a proportion of the active ingredient in a first phase.
In DE 101 04 880 Al multi-particulate medicament forms for release in multiple
phases of a
plurality of active ingredients are described, for guaranteeing a uniform
release of the active
ingredients over the intestine region. As a possible active ingredient inter
alia betahistine is
mentioned. The medicament form comprises two different forms of pellets which
can be
combined in a capsule or can be compressed together, wherein a ratio of 1:1 is
described. The
different forms of pellets release the active ingredient or the active
ingredients at different pH
values each. However, this medicament form does not enable an immediate
release of an
active ingredient in a first phase. So with this medicament form it is not
possible to achieve
therapeutic plasma levels in a first phase very quickly. Thus with this the
daily intake of
several medicament forms of active ingredients for which immediately
therapeutic plasma
levels have to be achieved is not dispensable. In addition, the production of
the multi-
particulate medicament forms is laborious and expensive and in particular in
the case of
hygroscopic and possibly also acidic active ingredients less suitable.
Insofar there is a great need for medicament forms for the release of
hygroscopic active
ingredients which at least release one active ingredient after intake in such
a way that an
intake several times a day is dispensable. This in particular relates to
active ingredients with
high solubility and/or high acidity. Here, conventional medicament forms are
stretched to
their limits. In particular, a retardation of active ingredients having the
above mentioned
properties with common retardation means and normally used retardation
principles is only
possible with great effort and is very costly. For such active ingredients an
extension of their
action to more than 10 hours with common retardation principles is hardly
possible. Thus,
normally used retardation mechanisms are not suitable for a cost-effective and
thus in an
industrial scale realizable formulation of such active ingredients in
medicament forms which
make an intake of the active ingredient several times a day dispensable. The
problem
underlying the present invention is solved by the subject matter of the patent
claims.
The present invention provides oral medicament forms for at least biphasic
release of at least
one active ingredient, wherein a part of the active ingredient in a first
phase is immediately
released after the intake into the stomach. Furthermore, the invention relates
to production
methods of such medicament forms as well as suitable uses. Thus, the concept
according to
the present invention does not relate to retarded release in the sense of a
common retardation
formulation, but provides the immediate release of a first proportion of the
active ingredient
CA 02877190 2014-12-18
6
English Translation of PCT/EP2013/063162
immediately after the intake thereof and the release of at least one second
proportion of the
active ingredient a certain time after its intake. Such a pulsed release
optimally simulates the
intake of an active ingredient two or more times a day and makes the intakes
of a medicament
form two or more times a day dispensable.
The medicament form of this invention comprises
= a core
= a gastric juice-resistant intermediate layer which is disposed on the
surface of the core,
and
= a shell which is disposed on the opposite side of the intermediate layer
from the core,
wherein both the core and the shell each contain a proportion of an active
ingredient. In
addition, also the intermediate layer may contain an active ingredient. In
preferable
embodiments the medicament form according to the present invention does not
contain any
additional constituent which comprises an active ingredient, besides the core,
the intermediate
layer and the shell, thus in particular no additional layer containing an
active ingredient is
present. Further preferable, the medicament form according to the present
invention consists
of the core, the intermediate layer and the shell.
The active ingredient is released from the medicament form in an at least
biphasic form.
Further phases of release may subsequently follow. A "phase" according to the
present
invention is a time interval of a release of a proportion of the active
ingredient from the
medicament form.
In a first phase a first proportion of the amount of active ingredient is
released. In a second
phase a second proportion of the amount of active ingredient is released.
According to the
present invention the first phase directly starts after the entry of the
medicament form into the
stomach. Thus, the first phase relates to the immediate release of the active
ingredient after
the ingestion into the gastric juice. Preferably, this first phase of the
release takes place from
the shell, wherein the shell preferably disintegrates in the stomach.
Preferably, the second
phase of the release takes place from the core. The core preferably does not
disintegrate in the
stomach and is preferably transported into the small intestine by the so-
called "housekeeper
waves", thus by contractions of the stomach, for releasing the proportion of
the active
ingredient of the core there.
CA 02877190 2014-12-18
7
English Translation of PCT/EP2013/063162
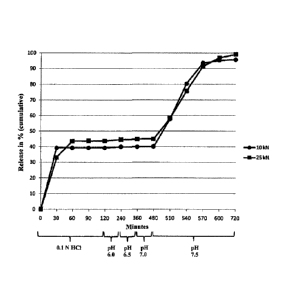
"Immediate release" preferably means in this case that at least 65 % of the
first proportion of
the amount of active ingredient, preferably at least 75 % and further
preferably at least 80 %
of the first proportion of the amount of active ingredient are released into
the gastric juice
after 60 minutes. Particularly preferably more than 60 %, further preferably
more than 65 %
of the first proportion of the amount of active ingredient are already
released into the gastric
juice after 30 minutes. This is determined in vitro using common apparatuses
such as a bogie
basket, stirrer blade or flow apparatus in 0.1 N HC1 or water as test medium
under
physiological conditions (rotation; at about 36 to 38 C). The exact conditions
depend on the
active ingredient each and the medicament form and can be found in common
pharmacopoeias.
Thus, the release of the first proportion of the amount of active ingredient
takes place directly
after the intake of the medicament form. Preferably, here the release of this
proportion of
active ingredient is not retarded by coatings. So preferably, before the
release of the first
proportion of the active ingredient initially no special coating, thus for
example a film, has to
be dissolved, ruptured or swelled.
Preferably, the second phase only starts at the earliest 4 hours, further
preferably only at the
earliest 6 hours and most preferably only at the earliest 7 hours after the
intake of the
medicament form. A second phase which starts too early may result in increased
plasma
concentrations, and this may involve the respective risk of side effects and
further includes the
risk that the release of the active ingredient is not sufficiently maintained
in order that the
intake one time a day is therapeutically sufficient. However, the release of
the second
proportion of the active ingredient preferably takes place at the latest 16
hours after the intake,
further preferably at the latest 14 hours after the intake and most preferably
at the latest 12.5
hours after the intake. A release which is too late would result in a release
of the second
proportion of the active ingredient in sections of the intestine which are too
deep so that due
to the reduced resorption area and the limited amount of liquid the resorption
would
considerably be reduced. Particularly preferably, the release of the second
proportion of the
active ingredient takes place in the colon.
The release in the second phase in this case preferably takes place
immediately after
achieving a certain pH range. The second proportion of the active ingredient
is preferably
released from the core of the medicament form. Thus preferably, after
achieving a certain pH
value within 60 minutes at least 65 % of the second proportion of the amount
of active
ingredient, preferably at least 75 % and further preferably at least 80 % of
the second
CA 02877190 2014-12-18
8
English Translation of PCT/EP2013/063162
proportion of the amount of active ingredient are released, determined in
vitro using common
apparatuses in a buffer medium with the respective pH value as test medium
under
physiological conditions (rotation; at about 36 to 38 C). The exact conditions
depend on the
active ingredient each and the medicament form and can be found in common
pharmacopoeias.
But in an alternative the release of the second proportion of the active
ingredient may also
take place in a retarded manner, wherein retarded manner means prolonged over
a certain
time interval, preferably over at least 4 hours, further preferably over at
least 6 hours. In an
embodiment further phases of release in which proportions of the active
ingredient are
released can be provided.
But besides the first active ingredient the medicament form may also comprise
further active
ingredients which are released in one phase or in more phases. A release in
more phases is
preferable. Particularly preferably, the medicament form according to the
present invention
only contains said one active ingredient. So possible incompatibilities during
the production
and storage thereof can be avoided.
The medicament form of the present invention is an oral medicament form,
wherein oral
medicament form means that it is intended for oral intake. According to the
present invention
in this case for example a tablet, caplet, micro-tablet, granules or pellets
may be envisaged.
The medicament form may also be contained in a multi-particulate medicament
form such as
a tablet or capsule or in granules, a sachet, stick or bag. Preferably, the
medicament form
according to the present invention is a tablet.
Exceptionally preferably, the medicament form according to the present
invention is a shell-
core tablet internally containing the core which is surrounded by the
intermediate layer. Said
intermediate layer is in turn surrounded by the shell. Preferably,
"surrounded" by the
intermediate layer with respect to the core means that at least 95 % of the
total surface of the
core, further preferably at least 98 % and exceptionally preferably at least
99 % of the total
surface of the core are covered with the intermediate layer. Still more
preferable is, when the
total surface of the core is "completely" surrounded by the intermediate
layer, wherein
"completely" preferably means that more than 99.5 %, ideally more than 99.8 %
of the total
surface of the core are covered by the intermediate layer. This does not
exclude that pores are
contained in the intermediate layer. Preferably, "surrounded" by the shell
with respect to the
intermediate layer means that at least 95 % of the total surface of the
intermediate layer on the
CA 02877190 2014-12-18
9
English Translation of PCT/EP2013/063162
side which is the opposite side of the intermediate layer from the core,
further preferably at
least 98 % and exceptionally preferably at least 99 % are covered by the
shell. Still more
preferable is, when the total surface of the intermediate layer on the side
which is the opposite
side of the intermediate layer from the core is completely surrounded by the
shell, so
preferably more than 99.5 % and ideally more than 99.8 % of the intermediate
layer on the
side which is the opposite side of the intermediate layer from the core are
covered by the
shell.
However in an alternative the medicament form can also be a layer tablet in
which the
intermediate layer may be present between the layers of the layer tablet,
wherein in this case
the core would be one of the layers and the shell would be the other layer.
It has been shown that the design of a shell-core tablet is particularly
advantageous. With the
design as a shell-core tablet it was possible to achieve an optimum initiation
and an optimal
maintenance of the release of the active ingredient over a sufficient period
of time. With this
shell-core concept it is possible to mimic in a particularly preferable manner
the daily intake
of at least two tablets, preferably in a time interval of 8 to 12 hours.
The aim of such a design of the medicament form as a shell-core tablet is
different from the
aim usually pursued in the prior art. So shell-core tablets are known as an
option for the
formulation of intolerable active ingredients or extremely bad smelling or
extremely instable
active ingredients.
Preferably, the active ingredient is hygroscopic and shows good solubility in
water.
Hygroscopic active ingredients and active ingredients with good water
solubility in particular
profit from the active ingredient design according to the present invention.
According to the
present invention a hygroscopic active ingredient is an active ingredient if
it or the salt of it
has the tendency of taking up water from the environment, for example by
adsorption or
absorption or other mechanisms. So hygroscopic active ingredients are also
active ingredients
which melt by taking up water (deliquescence).
Preferably, according to the present invention hygroscopy is defined as a
property of a
substance, when this substance at a relative air humidity of 75 %, preferably
at a relative air
humidity of 45 % and most preferably already at a relative air humidity of 30
% takes up
more than 1 % by weight of water from the environment. Particularly
preferably, at least 5 %
by weight, further preferably at least 8 % by weight of water are taken up
from the
environment. The determination of the taking up of water is conducted by
placement of 0.5 g
CA 02877190 2014-12-18
English Translation of PCT/EP2013/063162
of a sample of the substance which before has exclusively been stored
airtight, in an
atmosphere with relative air humidity of 75 %, 45 % or 30 % for 24 hours.
Subsequently the
water content of the substance is determined by the Karl-Fischer method in a
Karl-Fischer
titrator under heating to a temperature of at most 300 C. The water content is
compared with
the water content of 0.5 g of the substance originating from the same sample,
but which
further has been stored airtight.
Preferably, an active ingredient is used which, when a sample thereof is dried
under the above
mentioned conditions at 105 C until constant weight is achieved, in a drying
oven or by
means of an infrared dryer, wherein the infrared dryer is preferred, has a
water content of at
least 0.5 % by weight, based on the total mass of the medicament form, further
preferably of
at least 1 % by weight. This method determines the proportion of water of the
sample which is
not chemically bonded. In this case the sample is preferably a medicament
form, comprising
the active ingredient and adjuvants.
Examples of preferable active ingredients which themselves or in the form of
their
pharmaceutically acceptable salts are hygroscopic are valproic acid,
carbamazepine,
tetracycline, lincomycin, clindamycin, rifampicin, erythromycin, metformin,
atenolol,
ranitidine, minoxidil, acetylsalicylic acid, diclofenac, omeprazole,
methyldopa, betahistine,
dexamethasone, prednisolone, piracetam, pravastatin and gemfibrozil.
Preferably, the active ingredient has good solubility. According to the
present invention, good
or high solubility of active ingredients is given, when 1 g of the active
ingredient can be
dissolved in a maximum of 10 ml of water at 15 to 25 C under normal pressure.
It is particularly preferable, when the active ingredient is an active
ingredient of the BCS class
I according to the Biopharmaceutics Classification System. Members of the BCS
class I are
such active ingredients which have high solubility and a high permeation
capability,
determined for example according to the rules of the FDA ("Waiver of In Vivo
Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral
Dosage Forms
Based on a Biopharmaceutics Classification System", 2000).
The active ingredient may also be acidic. According to the present invention,
preferably an
active ingredient is acidic, when its solution in water with a mass proportion
of the active
ingredient of 10 % by weight at standard conditions (DIN 1343) has a pH value
of at most 5,
further preferably a pH value of at most 4 and exceptionally preferably a pH
value of at most
3.
CA 02877190 2014-12-18
11
English Translation of PCT/EP2013/063162
The medicament form according to the present invention in total comprises
preferably
between 1 mg and 200 mg of active ingredient. Preferably, the medicament form
according to
the present invention comprises the active ingredient in an amount of between
1.5 mg and
150 mg, further preferably in an amount of between 2 mg and 120 mg, even more
preferably
in an amount of between 5 mg and 60 mg.
Preferably, the active ingredient is betahistine or a pharmaceutically
acceptable salt of
betahistine. Pharmaceutically acceptable salts of betahistine are in
particular betahistine
dihydrochloride, betahistine dimesylate, betahistine methane sulfonate,
betahistine fumarate
and betahistine citrate. Further preferable are betahistine dihydrochloride,
betahistine
dimesylate, betahistine fumarate and betahistine citrate. Exceptionally
preferable are
betahistine dihydrochloride and betahistine dimesylate. In a particularly
preferable
embodiment the medicament form comprises betahistine dihydrochloride or
betahistine
dimesylate. In this case preferable between 2 mg and 55 mg, based on the base
of the active
ingredient, are contained in the medicament form. The base of the active
ingredient is
betahistine, thus not the salt.
It is exceptionally preferable, when the medicament form according to the
present invention
contains betahistine dihydrochloride, preferably in an amount of between 8 mg
and 100 mg,
exceptionally preferably between 30 and 60 mg, and most preferably 48 mg are
contained in
the medicament form. When in this application absolute values are mentioned,
then this
always also comprises values in a tolerance range of + 2 % around the
mentioned absolute
value.
Here, the active ingredient may be contained in the core, in the intermediate
layer and in the
shell. And in this case it is preferable that the proportion of the active
ingredient in the core
and in the shell each amounts to 40 % to 60 % of the total amount of the
active ingredient in
the medicament form. The proportion of the active ingredient in the core
preferably amounts
to at least 42 % and at most 58 %, further preferably at least 45 % and at
most 55 % of the
total amount of the active ingredient in the medicament form. When the amounts
of the active
ingredient in the core are too low, then there is the risk that the effect
cannot be maintained
for a period of time which is sufficiently long.
Preferably, the proportions of the active ingredient in the core and in the
shell are the same.
This enables a multiphasic release of equal amounts of active ingredient and
is advantageous,
because this kind of release optimally mimics an intake of a medicament
several times a day.
CA 02877190 2014-12-18
12
English Translation of PCT/EP2013/063162
In an alternative the distribution of the active ingredient in the core and in
the shell may also
be different. According to the present invention, "the same" preferably means
that the mass
ratio of the amount of active ingredient in the core to the amount of active
ingredient in the
shell preferably amounts to at least 1 to 1.1 and at most 1.1 to 1, further
preferably at least 1 to
1.05 and at most 1.05 to 1.
Besides the active ingredient also a second, optionally also a third active
ingredient may be
contained in the core, the shell and/or the intermediate layer. It is
especially preferable that the
intermediate layer does not contain any active ingredient.
The core contains an active ingredient, thus comprises at least the active
ingredient. Besides
the active ingredient, preferably there is contained at least one
pharmaceutically acceptable
adjuvant. Pharmaceutically acceptable adjuvants are substances which with
respect to the
safety of the patient and the processability are generally suitable for the
use in medicament
forms.
It has been shown that it is advantageous, when the core at least comprises a
carrier which is
preferably sorption-active. A sorption-active carrier has the capability of
being adsorbed to a
hygroscopic proportion of an active ingredient, thus "carry" it, and of
removing the
hygroscopy of the active ingredient up to a certain degree. Thereby, the
melting of the
hygroscopic active ingredient is avoided which results in a distinct positive
influence onto the
breaking strength and the shelf life of the achieved medicament form.
Surprisingly it was found that for that in particular carriers are suitable
which themselves are
hygroscopic, thus have a tendency for taking up humidity. Normally it would be
expected that
by the additional uptake of humidity of such carriers the processability would
be
compromised. But in reality it was possible to obtain by the use of such
carriers together with
the active ingredient a hard and stable core which can easily be processed and
compressed.
But it has been shown that for that the carrier or the carriers preferably
have to be present in
an excess with respect to the active ingredient. Therefore preferably, the
mass ratio of the
carrier or the carriers in the core to the active ingredient in the core is at
least 2:1, further
preferably at least 3:1, further preferably at least 3.4:1 and in
exceptionally preferable
embodiments at least 5:1. Obviously the carrier in this case has the effect
that a further up-
take of water from the environment can be considerably reduced. But the mass
ratio should
preferably not be higher than 20:1, preferably not higher than 10:1, further
preferably not
higher than 7.5:1, because otherwise the processability may be complicated. A
mass ratio
CA 02877190 2014-12-18
13
English Translation of PCT/EP2013/063162
which is too high is also connected with a core which is too big. This may
result in an
enlargement of the medicament form according to the present invention which is
too high.
Suitable carriers may be of inorganic or organic kind. Preferable carriers are
selected from
natural and synthetic polymers. Preferably, they are natural or modified
polysaccharides,
preferably consisting of two or more equal or different monosaccharide
entities. Such
substances can be obtained cheaply and can be easily processed. The latter is
in particular
advantageous, when the release of the proportion of the active ingredient from
the core should
take place immediately. It has been shown that polysaccharides containing
glucose units are
particularly advantageous. In this case the natural or modified polysaccharide
is preferably a
disaccharide, particularly preferably this is selected from lactose, sucrose
and mixtures
thereof.
In an alternative this carrier may be a polysaccharide with more than 10
monosaccharide
entities which in contact with water is preferably swellable. Particularly
preferably, here
starch including starch derivatives, cellulose including cellulose derivatives
or mixtures
thereof are used. Powdered cellulose such as microcrystalline cellulose is
exceptionally
preferable. Also starch in native or pre-gelatinized form, in particular corn
starch can be used.
Such carriers are still more capable of taking up water from the proportion of
the active
ingredient.
But it has been shown that the breaking strength of the tablet may decrease
with increasing
storage time, when in the case of the use of certain active ingredients
exclusively only such
polysaccharides are used as carriers. Therefore, in a particularly preferable
embodiment in the
core are contained at least two carriers, wherein in this case exceptionally
preferably at least
one disaccharide and at least one polysaccharide with more than 10
monosaccharide entities
which in contact with water is preferably swellable are used. With this
combination an
optimal breaking strength and an optimal stability were achieved. Also the
proportion of the
active ingredient is optimally fixed, preferably at the mixture of the
carriers. And it is
exceptionally preferable, when the core contains at least three carriers. The
mass ratio of
polysaccharides with more than 10 monosaccharide entities to disaccharides is
preferably at
least 1.1:1, preferably at least 1.25:1 and particularly preferably at least
1.3:1.
There may also be used inorganic carriers, preferably ones having hygroscopic
properties.
Inorganic carriers may also be contained in addition to the above mentioned
organic carriers.
CA 02877190 2014-12-18
14
English Translation of PCT/EP2013/063162
Preferably, the core contains salts of the above mentioned organic or
inorganic substances in a
proportion of at most 5 % by weight, preferably at most 2 % by weight and
exceptionally
preferably in a proportion of at most 1 % by weight. When salts of the above
mentioned
organic substances or salts of inorganic substances are used in the core
according to the
present invention in high amounts, then this may result in reaction of the
active ingredient in
the core with these substances, by what the stability of the active ingredient
in the core can be
negatively influenced. In this case salts in particular comprise alkali and
alkaline earth metal
salts of such substances. Particularly preferably, the core is free of salts
of the above
mentioned organic or inorganic substances, i.e. salts of such substances, in
particular
comprising calcium phosphate, sodium salts of celluloses and/or magnesium
stearate, are only
contained as impurities in a proportion of preferably at most 0.5 % by weight,
preferably at
most 0.1 % by weight of the total mass of the core.
Preferably, the total proportion of the carriers according to the present
invention, based on the
total mass of the core, is at least 50 % by weight, preferably at least 63 %
by weight and
exceptionally preferably at least 65 % by weight as well as further preferably
at least 68 % by
weight. When the amount of the carrier(s) is too low, then the hygroscopy of
the active
ingredient can only be compensated to an insufficient extent. Furthermore, the
carrier(s)
result(s) in a certain retardation effect with respect to the release of the
active ingredient.
Preferably, the core contains at most 90 % by weight of carrier respectively
carriers,
preferably at most 85 % by weight and further preferably at most 80 % by
weight. Mass
proportions of the carrier(s) which are too high complicate the forming
operation of the core.
The carrier may also lessen the acidity of the active ingredient. But for that
a certain amount
of the carrier is required.
In order that the amount of the carrier keeps within reasonable bounds,
according to the
present invention preferably a buffer substance can be used in the core. The
mass of the buffer
substance used is preferably lower than the mass of the carriers. In this case
a mass ratio of
carriers in the core to buffer substances in the core of preferably at least
3:1, further preferably
at least 4:1 and exceptionally preferably at least 4.5:1 is advantageous, and
it was possible to
achieve both, a sufficient compensation of the hygroscopy and a sufficient
lessening of the
acidity, together with good processability of the core. The core preferably
contains at least
1 % by weight of buffer substance, preferably at least 8 % by weight.
Preferably, the core
contains at most 26 % by weight of buffer substance, further preferably at
most 16 % by
CA 02877190 2014-12-18
English Translation of PCT/EP2013/063162
weight. The proportion of buffer substance should not exceed certain values,
because this
component may compromise the stability of the medicament form.
As a buffer substance organic acids are particularly suitable, wherein
preferably they are low-
molecular ones. "Low-molecular" organic acids are organic acids with a molar
mass of lower
than 300 g/mol. The buffer substance is preferably a carboxylic acid, selected
from citric acid,
lactic acid, tartaric acid, fumaric acid, maleic acid and ascorbic acid as
well as mixtures
thereof. Particularly preferable are alpha-hydroxyl carboxylic acids which due
to the alpha-
hydroxyl group show an optimum buffer effect. Preferably they are selected
from lactic acid,
tartaric acid, citric acid and mixtures thereof. Citric acid is exceptionally
preferable, because it
can be processed very easily and it does not significantly modify the release
of the active
ingredient from the core. This is a surprising fact, because citric acid in a
lot of presentation
forms results in an acceleration of the release, which would not be desired
here.
The mass ratio of the active ingredient in the core to the buffer substance in
the core is
preferably at least 0.5:1, preferably at least 0.8:1, further preferably at
least 0.9:1. The mass
ratio is preferably at most 1.5:1, further preferably at most 1.2:1 and
particularly preferably at
most 1.1:1. When the amount of the buffer substance is too low, then it is
possible that the
acidity of the active ingredient cannot be buffered to a sufficient extent.
When the amount of
the buffer substance is too high, then in turn the acidity may increase in an
unfavorable
manner. According to the present invention, the core preferably contains at
least 8 mg of
buffer substance, preferably at least 15 mg and exceptionally preferably at
least 20 mg.
Preferably at most 48 mg and exceptionally preferably at most 30 mg of buffer
substance are
contained in the core.
The core may contain at least one further pharmaceutically acceptable
adjuvant, such as for
example fillers and/or binders. In addition, the core may contain
disintegrating agents, which
are in particular selected from alginic acid, calcium carboxymethyl cellulose,
cross-linked
carboxymethyl cellulose, hydroxypropyl cellulose, sodium carboxymethyl starch,
cross-linked
polyvinylpyrrolidone and mixtures thereof. The addition of such a
disintegrating agent
prevents that the proportion of the active ingredient in the core is only
released in sections of
the intestine which are too deep. Due to the reduced resorption area this
would result in a
considerably worse resorption of the proportion of the active ingredient. It
has been shown
that an addition of cross-linked polyvinylpyrrolidone is particularly
suitable.
CA 02877190 2014-12-18
16
English Translation of PCT/EP2013/063162
Preferably, the disintegrating agent amounts to a weight proportion of the
core of at least 1 %
by weight, further preferably at least 2 % by weight, for effecting a
sufficient stimulation of
the disintegration. The maximum content of disintegrating agent is preferably
8 % by weight,
preferably at most 6.5 % by weight. When the proportions of the disintegrating
agent are too
high, then this results in a release of the active ingredient which is too
fast. In an alternative
the core is preferably free of such disintegrating agents, because with the
special core
composition according to the present invention surprisingly most often already
a sufficiently
quick disintegration is guaranteed.
According to the conditions of the production the core may comprise at least
one granulating
agent. Preferably, the amount of the granulating agent in the core amounts to
at least 0.1 % by
weight, further preferably at least 0.25 % by weight, still further preferably
at least 0.5 % by
weight and still further preferably at least 0.75 % by weight. The core
contains a maximum of
preferably 5 % by weight, further preferably a maximum of 2.5 % by weight of
granulating
agent. Granulating agents which are preferred in the core are mentioned below.
According to the conditions of the production the core preferably contains at
least one
lubricant, preferably in a mass proportion of at most 10 % by weight,
preferably at most 8 %
by weight, based on the total mass of the core. Preferably, at least 0.5 % by
weight, further
preferably at least 1 % by weight of lubricant are contained in the core.
Lubricants which are
preferred in the core are mentioned below.
Thus, in one embodiment the core contains:
a. an active ingredient; and
b. optionally at least one carrier (preferably with a mass proportion of 50 %
by weight to
90 % by weight) which in particular has the purpose "to carry" the active
ingredient,
to lessen the hygroscopy and acidity thereof and also to guarantee a certain
retardation; and
c. optionally at least one buffer substance (preferably with a mass proportion
of 1 % by
weight to 26 % by weight), in particular for lessening the acidity of the
active
ingredient; and
d. optionally a disintegrating agent (preferred mass proportion of 1 % by
weight to 8 %
by weight) for accelerating the disintegration of the core; and
e. optionally according to the conditions of the production at least one
granulating agent
(preferred mass proportion of 0.1 % by weight to 5 % by weight); and
CA 02877190 2014-12-18
17
English Translation of PCT/EP2013/063162
f. optionally according to the conditions of the production at least one
lubricant
(preferred mass proportion of 0.5 % by weight to 10 % by weight); and
g. optionally further adjuvants, such as for example fillers and/or binders.
On the surface of the core is disposed an intermediate layer. It has been
shown that such a
layer is advantageous for providing a time interval between the release of the
active ingredient
from the core and the shell. Preferably, an additional purpose of the
intermediate layer is the
bonding of the core and the shell to one another. Without the intermediate
layer most often no
medicament form with sufficient breaking strength and storability could be
achieved.
Furthermore it has been shown that fewer adjuvants in the core are required,
when the
intermediate layer exclusively or in addition controls the release of the
active ingredient from
the core. So the size of the core can be reduced and an adequate size of the
whole medicament
form is guaranteed which is necessary for a good compliance of the patient. It
has also been
shown that it is in particularly difficult to exclusively control the release
of the active
ingredient by the core, when the active ingredient is an active ingredient
with high solubility.
So often it was not possible to sufficiently temporally separate the release
of the active
ingredient from the core from the release of the active ingredient from the
shell by the
exclusive use of a retardation matrix. Thus the release could not be
maintained sufficiently
long. Only with the intermediate layer according to the present invention it
was possible to
sufficiently temporally separate the release of the active ingredient from the
core and that
from the shell.
The intermediate layer is preferably designed to be gastric juice-resistant.
"Gastric juice-
resistant" preferably means that the intermediate layer does not dissolve in
the pH range of the
stomach, thus preferably does not dissolve in 0.1 N HC1 at 36 C to 38 C within
120 minutes.
This means preferably that the core on which surface the intermediate layer is
disposed does
not disintegrate in 0.1 N HC1 at 36 to 38 C within 120 minutes. The
measurement of the
disintegration is conducted with common disintegration apparatuses, preferably
with the
stirrer blade apparatus.
Thus the release from the core at the earliest starts with the entry into the
milieu of the small
intestine by dissolving, disintegrating, cracking or other modification of the
intermediate
layer, preferably by the dissolution of the intermediate layer, when a certain
pH value is
reached. So the intermediate layer preferably is dissolved, when a certain pH
value in the
aqueous milieu of the gastrointestinal tract is reached. According to the
present invention the
CA 02877190 2014-12-18
18
English Translation of PCT/EP2013/063162
different possible processes are described by the term "dissolving". The
dissolution of the
intermediate layer preferably starts at a pH value of at least 5, further
preferably at a pH value
of at least 6 and exceptionally preferably at a pH value of at least 6.5. It
is exceptionally
preferable, when the dissolution of the intermediate layer starts at a pH
value of at least 7.0
and still more preferably at a pH value of at least 7.2. A dissolution at pH
values which arc
too low would result in a too early release of the active ingredient in the
second phase even in
the stomach or in upper sections of the duodenum. Depending on the active
ingredient, this
involves the risk of plasma levels which are too high. The intermediate layer
should
preferably be dissolved at the latest at a pH value of 7.65, preferably at the
latest at pH 7.5.
When the intermediate layer is dissolved only at pH values which are too high,
then a
sufficient resorption of the active ingredient can no longer be guaranteed.
The intermediate layer preferably comprises at least one pharmaceutically
acceptable
adjuvant, in particular a film-forming component which preferably guarantees
the gastric
juice-resistance. Optionally, also an active ingredient may be contained in
the intermediate
layer.
It has been shown that suitable film-forming components are natural and
synthetic polymers
having free acid functions. Preferably, these acid functions in salt form are
well soluble.
Preferably, each polymer comprises at least 0.1 free acid functions per
monomer entity,
further preferably at least 0.3 free acid units per monomer entity. Such
polymers are suitable
for being dissolved in the basic pH range of the intestine, but not in the
acidic pH range of the
stomach. Preferably, such free acid functions are carboxyl groups, since these
are most often
sufficiently well soluble in salt form.
Preferred film-forming components are cellulose derivatives, methacrylic acid
polymers,
polyvinyl derivatives and mixtures thereof.
Cellulose derivatives are preferably celluloses which are esterified with
organic acids. The
organic acids may be aliphatic or aromatic. Preferably, these organic acids
comprise at most
carbon atoms, further preferably at most 8 carbon atoms. But preferably they
comprise at
least 2 carbon atoms. The cellulose derivative is particularly preferably
selected from
cellulose acetate, cellulose acetate phthalate, cellulose acetate
trimellitate,
hydroxypropylmethylcellulose phthalate, hydroxypropylmethylcellulose acetate
succinate and
mixtures thereof.
CA 02877190 2014-12-18
19
English Translation of PCT/EP2013/063162
Preferred polyvinyl derivatives are polyvinyl derivatives which are esterified
with organic
acids. The latter may be of aliphatic or aromatic nature and preferably
comprise at least 2
carbon atoms, but preferably at most 10 carbon atoms. Preferably, the
polyvinyl derivative is
esterified with both, aliphatic and also aromatic residues. A particularly
preferable polyvinyl
derivative is polyvinylacetate phthalate.
Under the methacrylic acid polymers those have been shown to be suitable which
are
composed of methacrylic acid monomers with free carboxyl groups and monomers
with
esterified carboxyl group. The monomers with esterified carboxyl group are
preferably
selected from methacrylic acid monomers and acrylic acid monomers. The
monomers with
esterified carboxyl group are preferably esterified aliphatically, preferably
with organic
groups. These organic groups preferably comprise at least 1 carbon atom, but
preferably at
most 5 carbon atoms, further preferably at most 4 carbon atoms. Monomers with
esterified
carboxyl group preferably are selected from methylmethacrylate monomers,
ethylacrylate
monomers and methylacrylate monomers.
Preferred methacrylic acid polymers are polymethacrylic
acid/polymethylmethacrylate
copolymers, polymethacrylic acid/polyethylacrylate copolymers, polymethacrylic
acid/polymethylacrylate/polymethylmethacrylate copolymers and mixtures
thereof. In this
case the molar ratio of monomers with esterified carboxyl groups to
methacrylic acid
monomers with free carboxyl group is preferably at least 0.5:1, particularly
preferably at least
0.9:1. The molar ratio is preferably at most 12:1, further preferably at most
10:1 and
exceptionally preferably at most 2.5:1. Exceptionally preferable methacrylic
acid polymers
are polymethacrylic acid/polymethylmethacrylate copolymers. Particularly
preferably, the
film-forming component comprises at least one methacrylic acid polymer.
Particularly preferred film-forming components are Eudragit L, Eudragit S,
Eudragit L
100-55, Eudragit L30 D-55, Eudragit L 12,5, Eudragit S 12,5, Eudragit FS
30 D and
mixtures thereof. Exceptionally preferred are Eudragit L, Eudragit S and
mixtures thereof,
in particular Eudragit L 100, Eudragit S 100 and mixtures thereof. Eudragit
L 100
conforms with poly(methacrylic acid-co-methylmethacrylate) 1:1 (CAS 25086-15-
1).
Eudragit S 100 conforms with poly(methacrylic acid-co-methylmethacrylate) 1:2
(CAS
25086-15-1). Eudragit L 100-55 conforms with poly(methacrylic acid-co-
ethylacrylate) 1:1
(CAS 25212-88-8). Eudragit L 30 D-55 conforms with poly(methacrylic acid-co-
ethylacrylate) 1:1 (CAS 25212-88-8). Eudragit L 12,5 conforms with
poly(methacrylic acid-
co-methylmethacrylate) 1:1 (CAS 25086-15-1). Eudragit S 12,5 conforms with
CA 02877190 2014-12-18
English Translation of PCT/EP2013/063162
poly(methacrylic acid-co-methylmethacrylate) 1:2 (CAS 25086-15-1). Eudragit
FS 30 D
conforms with poly(methylacrylate-co-methylmethacrylate-co-methacrylic acid)
7:3:1 (CAS
26936-24-3). In further preferable embodiments methacrylic acid polymers are
used which
are polyethylacrylate/polymethylmethacrylate copolymers. Particularly
preferable is
Eudragit NE 30 D. Eudragit NE 30 D conforms with poly(ethylacrylate-co-
methylmethacrylate) 2:1 (CAS 9010-88-2).
The film-forming component is exceptionally preferably a methacrylic acid
polymer,
exceptionally preferably the film-forming component is at least a
polymethacrylic
acid/polymethylmethacrylate copolymer. In a special embodiment exclusively
methacrylic
acid polymers are used as film-forming components. Exceptionally preferably
here a mixture
of different methacrylic acid polymers is used. Through the high solubility of
the polymer in
the milieu of the intestine a targeted release of the active ingredient in the
second phase,
preferably from the core in the small intestine can be guaranteed.
Furthermore, these film-
forming components can be processed cheaply and can be applied onto the
surface of the core
in a quick and easy manner. By mixing different methacrylic acid polymers the
pH value at
which the dissolution of the intermediate layer should take place can be
adjusted in a targeted
manner. Exceptionally preferably is a mixture of Eudragit S 100 and Eudragit
L 100. The
mass ratio of Eudragit S 100 to Eudragit L 100 in this case is preferably at
least 1.5:1,
preferably at least 2.5:1, still more preferably at least 3:1. Preferably, the
mass ratio is at most
10:1, preferably at most 7.5:1 and still more preferably at most 5:1. In
alternative
embodiments the film-forming component is Eudragit S 100 or Eudragit L 100.
In an alternative a natural polymer may be used as a film-forming component,
wherein shellac
is preferred.
The proportion of the film-forming component in the intermediate layer is
preferably at least
20 % by weight, preferably at least 35 % by weight and exceptionally
preferably at least 40 %
by weight and further preferably at least 45 % by weight. A content of the
film-forming
component which is too low may result in dissolution of the intermediate layer
already in the
stomach. But the content of the film-forming component in the intermediate
layer is
preferably at most 75 % by weight, further preferably at most 70 % by weight
and still further
preferably at most 65 % by weight. A content which is too high results in a
very brittle
intermediate layer without sufficient binding effect.
CA 02877190 2014-12-18
21
English Translation of PCT/EP2013/063162
The weight of the intermediate layer is preferably at least 4 mg, further
preferably at least
mg and particularly preferably more than 5 mg. A weight of the intermediate
layer of at
least 8 mg is exceptionally preferable and still more preferably are at least
10 mg. When the
weight of the intermediate layer is too low, then the mechanic stability of
the core may be
reduced. The weight of the intermediate layer is preferably not higher than 55
mg, further
preferably at most 50 mg and still more preferably at most 35 mg.
Exceptionally preferably,
the weight of the intermediate layer is not higher than 28 mg. When the weight
is too high,
thus the amount of the intermediate layer is too high, then there is the risk
that the release of
the active ingredient from the core is compromised. There is also the risk
that the medicament
form in total becomes too large and too heavy and thus worse swallowable. It
has been shown
that a weight of the intermediate layer of 12 to 25 mg is particularly
suitable.
The intermediate layer is suitable for bonding the core and the shell to one
another. The effect
of bonding of the intermediate layer is preferably achieved by the addition of
at least one
plasticizer. According to the present invention, they are liquids at room
temperature and
normal pressure. Particularly suitable are plasticizers which are hygroscopic.
Particularly
preferable are polyalcohols with at least two hydroxyl groups. These
polyalcohols preferably
contain at least 2 carbon atoms, preferably at most 10 carbon atoms. The
hydroxyl groups
may be present in free form. Also all or a part of the hydroxyl groups may be
present in
esterified form, preferably with organic acids. Preferably, these organic
acids are aliphatic
acids, wherein they further preferably comprise at least 2 carbon atoms,
preferably at most 24
carbon atoms, most preferably at most 22 carbon atoms.
Exceptionally preferable plasticizers are selected from glycerol, propylene
glycol, glycerol
triacetate, castor oil, acetylated fatty acid glycerides and mixtures thereof.
According to the
present invention it has also been shown that polyethers of polyalcohols are
suitable, wherein
the polyalcohol entities preferably comprise at least 2 carbon atoms and
preferably at most 10
carbon atoms. Preferable polyethers are polyethylene glycols, preferably
having a mean molar
mass of up to 600 g/mol. By addition of such plasticizers a good bonding
effect of the
intermediate layer could be achieved. Furthermore, these plasticizers are
useful for
guaranteeing the mechanic stability also over longer periods of time.
The proportion of the plasticizer in the intermediate layer is preferably at
least 1 % by weight,
preferably at least 5 % by weight, for achieving a good bonding effect.
Preferably, the
proportion of the plasticizer in the intermediate layer is at least 8 % by
weight and further
preferably at least 12 % by weight. Preferably, a maximum of 30 % by weight,
further
CA 02877190 2014-12-18
22
English Translation of PCT/EP2013/063162
preferably a maximum of 25 % by weight and still further preferably a maximum
of 20 % by
weight of a plasticizer are contained in the intermediate layer. When the
content of the
plasticizer is too high, then a sufficient strength of the intermediate layer
and also a sufficient
gastric juice-resistance cannot longer be guaranteed.
The intermediate layer may contain at least one further pharmaceutically
acceptable adjuvant
which can simplify the processing and the application of the layer and/or is
suitable for
adjusting the consistency thereof. In particular, normal fillers, pore-formers
and/or further
plasticizers may be contained. Further plasticizers may be esters of organic
acids, wherein
these organic acids preferably comprise at least 2 carbon atoms and preferably
at most 10
carbon atoms. Preferred are citric acid and phthalic acid. Such plasticizers
are for example
tributyl citrate, triethyl citrate, diethyl phthalate, dibutyl phthalate.
For facilitating the application of the intermediate layer, the intermediate
layer preferably
contains fillers which are preferably selected from talcum, titanium dioxide,
magnesium
stearate, colorants, glycerol monostearate, lactose, microcrystalline
cellulose,
polyvinylpyrrolidone and mixtures thereof. Preferably, the filler is talcum.
Particularly
preferably, the filler is suitable for perforating the intermediate layer so
that there is an
additional possibility for controlling the release of the active ingredient in
the core. But the
mass proportion of the filler has to be restricted to preferably at most 40 %
by weight, further
preferably at most 35 % by weight, in particular in the case, when the
intermediate layer
should have a bonding effect. The filler may reduce the bonding effect of the
intermediate
layer. The minimum content of the filler preferably amounts to at least 5 % by
weight, further
preferably at least 15 % by weight.
The mass ratio of core to intermediate layer should preferably be at least
2:1, further
preferably at least 3:1, still further preferably at least 6.5:1 and
exceptionally preferably at
least 7.5:1. Thus, the intermediate layer in relation to the core should not
have a mass
proportion which is too high, in particular in the case, when the intermediate
layer does not
contain any active ingredient. So it is guaranteed that also the total weight
and thus the
dimensions of the medicament form are suitable for oral intake. The mass ratio
should
preferably be at most 60:1, further preferably at most 45:1. In a preferred
embodiment the
mass ratio is at most 40:1 and still further preferably at most 30:1 as well
as especially
preferably at most 20:1. When the ratio becomes too high, then the thickness
of the
intermediate layer is no longer sufficient for the desired function.
CA 02877190 2014-12-18
23
English Translation of PCT/EP2013/063162
The intermediate layer preferably covers at least 40 % of the total surface of
the core. This is
in particularly advantageous, when the core at the non-covered area is covered
with another
layer. Particularly preferably, at least 95 % of the total surface of the core
are covered by the
intermediate layer, further preferably at least 98 % and particularly
preferably at least 99 %.
In particularly preferable embodiments the core is completely covered by the
intermediate
layer. In a preferred embodiment the intermediate layer preferably has no
pores. "Pores"
means holes in the intermediate layer, through which the active ingredient may
be
prematurely released from the core.
Thus, in one embodiment the intermediate layer contains:
a. an active ingredient,
b. optionally a film-forming component (preferred mass proportion of 20 to 75
% by
weight), in particular for guaranteeing a gastric juice-resistance of the
intermediate
layer, and
c. optionally a plasticizer (preferred mass proportion of 1 to 30 % by weight)
which
preferably is hygroscopic, in particular for guaranteeing a good bonding
effect of the
intermediate layer; and
d. optionally a filler, in particular talcum, in a preferred mass proportion
of 5 % by
weight to 40 % by weight; and
e. optionally further pharmaceutically acceptable adjuvants, such as for
example further
fillers, pore-formers and/or further plasticizers.
The shell contains active ingredient, thus contains at least the active
ingredient. Besides the
active ingredient preferably at least one pharmaceutically acceptable adjuvant
is contained.
The mass ratio of shell to core is preferably at least 0.5:1, further
preferably at least 1.1:1,
more preferably at least 1.8:1, further preferably at least 2:1 and
particularly preferably at
least 2.2:1. This enables a sufficient processability of the shell. The mass
ratio is preferably at
most 10:1, preferably at most 5:1, preferably at most 4.5:1 and further
preferably at most 4:1.
When the mass ratios are too high, then a size of the medicament form which is
suitable for
intake cannot longer be guaranteed. Preferably, the shell in comparison to the
core contains a
higher proportion of pharmaceutically acceptable adjuvants, for guaranteeing a
certain volume
of the shell and also for embedding the active ingredient and so sufficiently
shielding it from
influences from outside, for example from humidity and in particular also from
light. The
CA 02877190 2014-12-18
24
English Translation of PCT/EP2013/063162
mass ratio of the adjuvants in the shell to the proportion of the adjuvants in
the core is
preferably at least 1.8:1, further preferably even at least 2.5:1.
In particular, it has also been shown that the use of at least one carrier as
an adjuvant in the
shell is advantageous. Preferably, the carrier in the shell is hygroscopic.
Normally it would be
expected that this would compromise the stability of the medicament form,
since the shell
preferably does not comprise a further coating and insofar the constituents of
the shell are not
shielded from humidity and light. The mass ratio of the carrier in the shell
to the active
ingredient in the shell is preferably at least 7.5:1, preferably at least
10:1, further preferably at
least 14:1, and in exceptionally preferable embodiments it is at least 15:1.
Then, the active
ingredient is preferably distributed and matrix-likely embedded in the
relatively higher
proportion of the carrier in the shell so that a further up-take of water from
the environment
can considerably be reduced. However, the mass ratio is preferably at most
50:1, further
preferably at most 35:1, for guaranteeing a suitable size of the medicament
form. Further
preferably, the mass ratio is at most 30:1 and still further preferably at
most 28:1.
Suitable carriers in the shell may be of inorganic or organic kind. Preferable
carriers in the
shell are selected from natural and synthetic polymers. Preferably, they are
natural or
modified polysaccharides, preferably consisting of two or more equal or
different
monosaccharide entities. Such substances can be obtained cheaply and can be
easily
processed. It has been shown that polysaccharides containing glucose units are
particularly
advantageous. Preferably, the carrier is a disaccharide, particularly
preferably this is selected
from lactose, sucrose and mixtures thereof.
In an alternative this carrier in the shell may be a polysaccharide with more
than 10
monosaccharide entities which in contact with water is preferably swellable.
Particularly
preferably, here starch including starch derivatives, cellulose including
cellulose derivatives
or mixtures thereof are used. Powdered cellulose such as microcrystalline
cellulose is
exceptionally preferable. Also starch in native or pre-gelatinized form, in
particular corn
starch can be used. Such carriers are still more capable of taking up water
from the proportion
of the active ingredient. But it has been shown that the breaking strength of
the tablet with
increasing storage time may decrease, when in the case of the use of certain
active ingredients
exclusively only such polysaccharides are used as carriers in the shell.
Therefore, in a particularly preferable embodiment at least two carriers are
contained in the
shell, wherein exceptionally preferably in this case they are at least one
disaccharide and at
CA 02877190 2014-12-18
English Translation of PCT/EP2013/063162
least one polysaccharide with more than 10 monosaccharide entities which in
contact with
water is preferably swellable. With this combination an optimum breaking
strength and an
optimal stability were achieved. Also the proportion of the active ingredient
is optimally
fixed, preferably on the carrier mixture. Exceptionally preferably, the shell
contains at least
three carriers. The mass ratio of polysaccharides with more than 10
monosaccharide entities
to disaccharides is preferably at least 1.1:1, preferably at least 1.25:1 and
exceptionally
preferably at least 1.3:1.
Also inorganic carriers, preferably having hygroscopic properties can be used
in the shell.
Inorganic carriers may also be contained in the shell in addition to the above
mentioned
carriers.
Preferably, the shell contains salts of the above mentioned organic or
inorganic substances in
a proportion of at most 5 % by weight, preferably at most 2 % by weight and
exceptionally
preferably in a proportion of at most 1 % by weight. When salts of the above
mentioned
organic substances or salts of inorganic substances are used in the shell
according to the
present invention, then this may result in reaction of the active ingredient
in the shell with
these substances, by what the stability of the active ingredient in the shell
can negatively be
influenced. In this case in particular salts comprise alkali and alkaline
earth metal salts of such
substances. It is particularly preferable, when the shell is free of salts of
the above mentioned
organic or inorganic substances, i.e. salts of such substances, in particular
comprising calcium
phosphate, sodium salts of celluloses and magnesium stearate, are only
contained as
impurities in a proportion of preferably at most 0.5 % by weight, preferably
at most 0.1 % by
weight of the total mass of the shell. However, salts of such substances may
be contained in
an optional coating of the shell.
It is particularly preferable, when at least two carriers are contained in the
shell, and
exceptionally preferable are three carriers. Preferably, the total proportion
of the carriers with
respect to the total mass of the shell is at least 70 % by weight, preferably
at least 77.5 % by
weight and exceptionally preferably at least 80 % by weight, for sufficiently
compensating the
hygroscopy of the active ingredient in the shell and for guaranteeing a good
processability of
the shell at the same time. Preferably, the shell contains at most 97 % by
weight of carrier,
further preferably at most 95 % by weight. When the amounts of carrier are too
high, then this
complicates the processability of the shell.
CA 02877190 2014-12-18
26
English Translation of PCT/EP2013/063162
Preferably, the shell contains a buffer substance. As buffer substances
organic acids are
particularly suitable, wherein preferably they are low-molecular ones. "Low-
molecular"
organic acids are organic acids with a molar mass of lower than 300 g/mol. The
buffer
substance in the shell is preferably a carboxylic acid, selected from citric
acid, lactic acid,
tartaric acid, fumaric acid, maleic acid and ascorbic acid as well as mixtures
thereof.
Particularly preferable are alpha-hydroxyl carboxylic acids which due to the
alpha-hydroxyl
group show an optimum buffer effect. Preferably they are selected from lactic
acid, tartaric
acid, citric acid and mixtures thereof. Citric acid is exceptionally
preferable, because it can be
processed very easily and it does not significantly modify the release of the
active ingredient
from the shell. This is a surprising fact, because citric acid in a lot of
presentation forms
results in an acceleration of the release, which would not be desired here.
The mass ratio of the active ingredient in the shell to the buffer substance
in the shell is
preferably at least 0.5:1, preferably at least 0.8:1 and further preferably at
least 0.9:1. The
mass ratio is preferably at most 1.5:1, further preferably at most 1.2:1 and
particularly
preferably at most 1.1:1. When the amount of the buffer substance is too low,
then it is
possible that the acidity of the active ingredient in the shell cannot be
buffered to a sufficient
extent. When the amount of the buffer substance is too high, then in turn the
acidity increases
in an unfavorable manner. Preferably, the shell contains at least 0.5 % by
weight, further
preferably at least 1 % by weight and still further preferably at least 2.5 %
by weight of buffer
substance. Preferably, the shell contains at most 15 % by weight, further
preferably at most
% by weight and still further preferably at most 8 % by weight of buffer
substance.
Surprisingly it has been found that citric acid as a buffer substance is also
particularly suitable
for the shell. So a sufficient lessening of the acidity could be achieved,
without any
recognizable acceleration of the release of the proportion of the active
ingredient from the
shell. Normally it would be expected that the buffer substance accelerates the
release of the
active ingredient. According to the present invention, the shell preferably
contains at least
8 mg of buffer substance, preferably at least 15 mg and exceptionally
preferably at least
mg. Preferably, in the shell are contained at most 48 mg and exceptionally
preferably at
most 30 mg of buffer substance.
The shell may contain at least one further pharmaceutically acceptable
adjuvant, such as for
example fillers and binders. However preferably, no disintegrating agent is
contained in the
shell, because such an agent may result in a release of the active ingredient
from the shell
which is too fast.
CA 02877190 2014-12-18
27
English Translation of PCT/EP2013/063162
According to the conditions of the production the shell may comprise at least
one granulating
agent. Preferably, the amount of the granulating agent in the shell amounts to
at least 0.02 %
by weight, further preferably at least 0.1 % by weight. Preferably, the shell
contains a
maximum of 2.5 % by weight of granulating agent. Granulating agents which are
preferred in
the shell are mentioned below.
According to the conditions of the production the shell preferably contains at
least one
lubricant, preferably in a mass proportion of at most 5 % by weight, further
preferably at most
4.5 % by weight. Preferably, at least 0.25 % by weight, further preferably at
least 0.5 % by
weight of lubricant are contained in the shell. Lubricants which are preferred
in the shell are
mentioned below.
The shell preferably covers at least 40 % of the total surface of the
intermediate layer on the
side which is the opposite side of the intermediate layer from the core. A
part of the surface
may then completely remain uncovered, for example for reducing the size of the
medicament
form, or when the surface should be covered by another layer. It is
particularly preferable,
when at least 95 % of the total surface are covered by the shell, further
preferably at least 98
% and particularly preferably at least 99 %. In a preferable embodiment the
intermediate layer
is completely covered by the shell.
Preferably, the shell itself does not contain a further coating, wherein in
particular there is no
coating on the shell which retards or prolongs the release of the active
ingredient. So
preferably no film is applied on the shell. In an alternative embodiment
however a quickly
disintegrating coating which preferably quickly disintegrates in the aqueous
milieu of the
stomach may be advantageous on the shell. This preferably contains water-
soluble polymers
as film-forming components such as cellulose derivatives,
polyvinylpyrrolidone, polyvidone
acetate. Cellulose derivatives are preferably selected from methylcellulose,
hydroxypropylcellulose, hydroxypropylmethylcellulose, sodium
carboxymethylcellulose and
mixtures thereof. In an alternative the shell may be coated with a sugar-
containing sugar-coat
syrup.
Thus, in one embodiment the shell contains:
a. an active ingredient; and
b. optionally at least one carrier (preferably with a mass proportion of 70 %
by weight to
97 % by weight) which in particular has the purpose "to carry" and to embed
the
CA 02877190 2014-12-18
28
English Translation of PCT/EP2013/063162
active ingredient, to lessen the hygroscopy and acidity thereof and also to
guarantee a
certain retardation; and
c. optionally at least one buffer substance (preferably with a mass proportion
of 0.5 % by
weight to 15 % by weight), in particular for lessening the acidity of the
active
ingredient; and
d. optionally according to the conditions of the production at least one
granulating agent
(preferred range of the amount thereof is 0.02 % by weight to 2.5 % by
weight); and
e. optionally according to the conditions of the production at least one
lubricant
(preferred range of the amount thereof is 0.25 % by weight to 5 % by weight);
and
f. optionally further adjuvants, such as for example fillers and binders.
The invention also relates to a production method for the medicament form
according to the
present invention. This comprises the steps of:
a) producing the core,
b) producing the intermediate layer,
c) producing the shell.
Often directly compressing of the active ingredient, preferably together with
adjuvants, is
difficult due to the typically present hygroscopy of the active ingredient.
Compression of
granulates results in a compressed material with good strength and especially
low abrasion.
Furthermore, the provision of the mixture to be compressed as granules
normally only
guarantees a sufficient adhesion and flowability. Thus, the production of the
core preferably
comprises the production of active ingredient granules from the active
ingredient and
preferably the adjuvants which are preferred for the core. However, the direct
production of
the active ingredient granules was considerably complicated by the hygroscopy
of the active
ingredient. In particular, it was difficult to obtain sufficiently dry and
stable active ingredient
granules with the common granulation methods. Often the obtained active
ingredient granules
were too wet, so that this resulted in insufficient strength, fluctuations of
the dose and capping
of the core, for example during compressing.
Surprisingly it has been found that a combined granulation is advantageous.
According to the
present invention this granulation at first comprises the production of pre-
granules which
according to the present invention are free of active ingredient. The pre-
granules preferably
comprise the carrier of the core and the buffer substance of the core as well
as optionally
CA 02877190 2014-12-18
29
English Translation of PCT/EP2013/063162
further adjuvants. Subsequently, from the pre-granules the active ingredient
granules are
prepared.
It has been shown that this preferred combined granulation in total is more
gentle for the
active ingredient. Surprisingly also hard and stable cores without any
appreciable fluctuations
of the dose could be produced. Here without any appreciable fluctuations of
the dose means
that the content of the active ingredient in the core, measured in 5 cores, is
preferably at least
93 %, preferably at least 95 % and preferably at most 108 %, further
preferably at most 107 %
of the theoretical amount of active ingredient per core. This is a surprising
fact, since
combined granulation is generally connected with the risk of an unequal
distribution of the
active ingredient and an increased tendency to demixing and thus normally is
avoided.
It has been shown that wet granulation is advantageous for the production of
the pre-granules,
wherein the production of adhesive granules is particularly preferable. So the
production of
the pre-granules preferably comprises the step of mixing the carrier and
preferably the buffer
substance with a granulation solution. Surprisingly, with such a method stable
pre-granules
with low residual moisture can be obtained. Since typically the carrier is
hygroscopic,
normally it should be expected that a low residual moisture of the adhesive
granules can only
be achieved with an enormous effort at high drying temperatures, which again
is costly.
Preferably, the granulation solution comprises an aqueous solution of a
granulating agent. A
granulating agent is a substance with adhesive and gelatinizing properties.
Preferably, these
agents are synthetic and/or natural polymers. Preferably, the granulating
agent is selected
from starches, polyvinylpyrrolidone, gelatine, cellulose ethers and mixtures
thereof. It is
exceptionally preferable to use polyvinylpyrrolidone, because with the use of
hygroscopic
active ingredients it was possible to achieve an especially good adhesive
effect. But
preferably, the polyvinylpyrrolidone has to be selected such that the mean
molar mass does
not become higher than 40.000 g/mol. Molecular weights which are too high are
connected
with a high viscosity of the granulation solution so that the granulation
procedure is
considerably complicated. In particular, Povidon K25 is suitable.
=
The production of the pre-granules preferably comprises a drying step. In this
step drying
methods such as spray drying, fluidized bed drying, vacuum drying and/or
lyophilization can
be used. Here it is preferable to use fluidized bed drying, preferably with a
temperature of the
inlet air of at most 75 C, further preferably at most 65 C. The residual
moisture of the pre-
granules is preferably adjusted to a value of lower than 8 % by weight,
further preferably
CA 02877190 2014-12-18
English Translation of PCT/EP2013/063162
lower than 5 % by weight. When the residual moisture of the pre-granules is
too high, then
this results in a core strength which is insufficient. Furthermore,
fluctuations of the dose can
be observed.
The production of the active ingredient granules preferably comprises the
mixing of the pre-
granules with an active ingredient solution. So the active ingredient is
bonded to the pre-
granules. Preferably, the active ingredient solution comprises the active
ingredient and a
solvent. Preferably, the solvent is an alcohol, particularly preferably an
aliphatic alcohol.
Preferably, the aliphatic alcohol is selected from methanol, ethanol, n-
propanol, iso-propanol
and mixtures thereof. Here, methanol shows good solubility at a low boiling
point and is thus
particularly preferable. But the use of methanol is also connected with the
disadvantage that
possible residues have to be removed nearly completely due to the toxicity
thereof.
The method for the production of the active ingredient granules preferably
comprises a drying
step. It has been shown that in particular the fluidized bed drying is
particularly advantageous,
because with fluidized bed drying it was possible to achieve quick drying. So
according to the
present invention it was possible to completely remove the solvent from the
active ingredient
granules. According to the present invention, completely removing the solvent
means a
content of residual solvent of lower than 5,000 ppm (m/m), further preferably
of at most
3,000 ppm (m/m), exceptionally preferably the content of residual solvent is
lower than 3,000
ppm (m/m), based on the total mass of the dried active ingredient granules.
According to the present invention, preferably the production of the core
comprises the
compression of the active ingredient granules. The compression may for example
be
conducted in a rotary pelleting machine, eccentric press or other tableting
facility. But here
the core may adhere and cap at the punch. Preferably, therefore the active
ingredient granules
are mixed with at least one lubricant, for obtaining a compressible core
mixture. It has been
shown that this procedure is advantageous, and hard cores with good
disintegration can be
obtained. This is a surprising fact, because a tendency to demixing would be
expected, when
the highly compact active ingredient granules are mixed with the lubricant.
The addition of
the lubricant already in the step of the preparation of the pre-granules
and/or the active
ingredient granules may result in granules with lower hardness and worse
compressibility.
Therefore preferably the addition of the lubricant is conducted only after the
production of the
active ingredient granules.
CA 02877190 2014-12-18
31
English Translation of PCT/EP2013/063162
It has been shown that in particular fatty acid derivatives are advantageous
lubricants.
According to the present invention, fatty acid derivatives comprise salts of
fatty acids, fatty
acid esters, fatty acids and fats as well as mixtures thereof, wherein the
fatty acids preferably
comprise at least 8 carbon atoms, further preferably at least 12 carbon atoms.
On the other
hand, the use of talcum as lubricant in the method according to the present
invention may
often result in a sub-optimal lubrication effect. Particularly advantageous is
the use of fats
which are liquid at room temperature and thus allow an easy processing. In
this case
particularly preferable is cottonseed oil. When such fats are used, then cores
with optimum
breaking strength without any tendency to capping are obtained.
The lubricant is used in such an amount that a mass proportion of at most 10 %
by weight,
preferably at most 8 % by weight, based on the total mass of the mixture
consisting of active
ingredient granules and lubricant, thus the core mixture, is not exceeded.
When the amounts
of lubricant are too high, then this lowers the wettability of the core, and
thus the
disintegration of the core may be negatively influenced. For achieving a
sufficient lubrication
effect, preferably at least 0.5 % by weight, preferably at least 1 % by weight
of lubricant,
based on the total mass of the core mixture, should be used, which are then
also
correspondingly contained in the core.
The obtained core preferably has a weight of at most 500 mg, preferably at
most 350 mg,
further preferably at most 250 mg. The diameter of the core is preferably at
most 12 mm,
further preferably at most 10 mm and particularly preferably at most 9 mm.
When the cores
are too large, then the further processing is complicated, and most often
medicament forms
are obtained which can be swallowed only with difficulties.
The production of the intermediate layer preferably comprises the dissolution
of the film-
forming component in a solvent. The production of the intermediate layer also
preferably
comprises the addition of optional further adjuvants, in particular of the
plasticizer. The result
is an intermediate layer mixture.
The solvent for the dissolution of the film-forming component is preferably an
aliphatic
alcohol, selected from methanol, ethanol, n-propanol, iso-propanol and
mixtures thereof. Most
preferable is iso-propanol. The intermediate layer mixture is applied onto the
surface of the
core. For that methods such as for example kettle methods in sugar-coating
kettles, drum
boilers, GS coating machines, as well as dip tube methods or fluidized bed
methods can be
used. It is particularly preferable to use GC coaters, because with them a
faster and more
CA 02877190 2014-12-18
32
English Translation of PCT/EP2013/063162
equal coating of the core is possible than in the case of for example a common
sugar-coating
kettle. Here, the application of the intermediate layer mixture is preferably
conducted at a
temperature of the inlet air of at most 75 C, further preferably at most 60 C.
When the
temperatures are too high, then this may result in the decomposition of the
active ingredient in
the core and optionally in the intermediate layer.
The production of the shell preferably comprises the production of active
ingredient granules.
Preferably, these granules are prepared in the same manner as the active
ingredient granules
for the core. Since direct compression of the active ingredient granules is
only possible with
difficulties and for guaranteeing a suitable volume of the shell, preferably
the active
ingredient granules are mixed with at least one pharmaceutically acceptable
adjuvant so that a
shell mixture is obtained. Preferably, the at least one adjuvant is a carrier
in the shell.
Here it has been shown that a mass proportion of the active ingredient
granules in the shell of
preferably at least 10 % by weight, preferably at least 20 % by weight is
advantageous.
Preferably, the mass proportion of the granules is at most 49 % by weight,
preferably at most
42 % by weight.
Normally, with such a procedure actually a strong tendency to demixing would
be expected
between the relatively high proportion of the adjuvant and the compact active
ingredient
granules, which results in bad disintegration. Surprisingly it has been found
that exactly with
this procedure a stable shell with good disintegrating properties can be
obtained.
The production of the medicament form according to the present invention
preferably
comprises the application of the shell mixture onto the intermediate layer.
The term
"application" according to the present invention comprises different methods,
preferably
pressing the shell onto the intermediate layer, preferably using common tablet-
compressing
machines. For avoiding capping, thus the detachment of single pressed layers
and so the
adhesion at the punches, at least one lubricant is preferably added to the
shell mixture.
Preferred lubricants are the same which have already been mentioned for the
use in the
production of the core. But it has been shown that already very low
proportions of lubricants
are sufficient for good compressibility of the shell mixture and that
surprisingly an increase of
the amount quickly has a negative influence onto the wettability and the
disintegration of the
shell. Therefore the amount of the lubricant in the shell should preferably be
restricted to 5 %
by weight, preferably 4.5 % by weight. It has been shown that advantageous
lubricants are in
CA 02877190 2014-12-18
33
English Translation of PCT/EP2013/063162
particular fats which at room temperature and normal pressure are liquid and
thus can be
processed very easily. It is exceptionally preferable to use cottonseed oil.
The pressing of the shell onto the intermediate layer is preferably conducted
such that the
shell mixture is placed in the press. When the core with the intermediate
layer should be
placed at about the center of the medicament form to be produced, then
preferably at least
40 % by weight, further preferably at least 45 % by weight of the shell
mixture is given into
the die. But preferably at most 65 % by weight, further preferably at most 60
% by weight of
the shell mixture are given into the die. Subsequently, the core with the
intermediate layer is
inserted and the form is filled with the remaining proportion of the shell
mixture.
Subsequently pressing power is applied.
When the shell is pressed onto the intermediate layer, then preferably a force
of pressure of at
most 35 kN, further preferably at most 30 kN and particularly preferably at
most 28 kN is
applied. When the used forces of pressure are too high, then often medicament
forms which
are too hard are obtained, which is connected with a worse release of the
active ingredient.
Also pressing with pressing powers which are too high, in particular in the
case of a concave
or biplanar form of the core, may result in deformation of the core. So the
intermediate layer
may crack and the active principle may be destroyed. Pressing powers which are
too high may
also result in fragmentation of the compressed material. But preferably the
force of pressure
should be higher than 7.5 kN, further preferably 8 kN. When the used forces of
pressure are
too low, then it can be observed that the strength of the medicament form is
insufficient.
Preferably, the breaking strength of the medicament form should be at least 60
N, further
preferably at least 100 N and exceptionally preferably at least 120 N. But the
breaking
strength should preferably not exceed 350 N, preferably 300 N. It is
exceptionally preferable,
when the breaking strength of the medicament form is between 140 N and 280 N,
still more
preferably between 170 and 270 N. When the breaking strengths are too high,
then it can be
measured that the disintegration is worse. The breaking strength of a tablet
can be determined
according to common methods under standard conditions with hardness testers
with the
application of a diametrically acting force, normally a sharp or conical
specimen. According
to the present invention the breaking strength was determined with an Ervveka
TBH-30
hardness tester.
The diameter of the core is preferably at least 3 mm, further preferably at
least 4 mm. When
the diameter of the core is too low, then the handling and the processability
may be
CA 02877190 2014-12-18
34
English Translation of PCT/EP2013/063162
complicated. Preferably, the diameter of the core is at most 16 mm, further
preferably at most
15 mm and particularly preferably at most 14 mm. When the diameter of the core
is too high,
then the swallowability of the obtained medicament form may be compromised.
The term
"diameter" means the diameter of the core at its broadest site each.
The layer thickness of the shell is preferably at least 0.7 mm, further
preferably at least
0.8 mm. When the layer thicknesses of the shell are too low, then the handling
of the
medicament form according to the present invention and its production are
complicated.
Preferably, the layer thickness of the shell is at most 5 mm, preferably at
most 3 mm. When
the layer thicknesses of the shell are too high, then the medicament form may
become too
large and the swallowability may be compromised. Layer thickness in this case
is the
thickness of the shell at its broadest site each.
The total diameter of the medicament form depends on the ingredients which are
used
respectively, in particular the active ingredients in the core and the shell.
Also the diameter of
the core is an important feature which influences the total diameter of the
medicament form.
Preferably, the total diameter of the medicament form is at most 20 mm,
further preferably at
most 18 mm, exceptionally preferably at most 16 mm. When the diameters are too
high, then
the swallowability may be compromised. The term "total diameter" relates to
the diameter of
the medicament form at its broadest site each. It has been shown that a
diameter of the
medicament form of between 8 mm and 14 mm, in particular between 11 mm and 13
mm is
advantageous.
Preferably, the medicament form has a mass of at most 1100 mg, preferably at
most 950 mg
and most preferably at most 850 mg, so that it can be taken in very easily.
But the
medicament form according to the present invention preferably has a minimum
weight of
115 mg, further preferably 225 mg, so that the handling of the medicament form
also for elder
persons is no problem. Particularly preferable was a weight of the medicament
form of
between 700 mg and 800 mg.
Also in the case of exposure to air the medicament form according to the
present invention
has preferably a residual moisture, thus an absolute proportion of water of at
most 15 % by
weight, preferably at most 13 % by weight. It is exceptionally preferable,
when the absolute
proportion of water is at most 5 % by weight. Preferably, this proportion is
determined by
drying at 105 C until constant weight is reached, in a drying oven or by means
of an infrared
dryer, the infrared dryer being preferred.
CA 02877190 2014-12-18
English Translation of PCT/EP2013/063162
The medicament forms of the invention have the advantage of particularly good
storage
stability, which means that the criteria for storage stability of the ICH are
at least fulfilled and
are preferably exceeded, i.e. the medicament forms according to the present
invention show
better values than necessary. Storage stability preferably means that during a
storage at certain
storage conditions and for a certain storage time which can be deduced from
the ICH directive
Q3B (R2) (Impurities in New Drug Products) a sufficiently high content of drug
of preferably
more than 90 %, based on the original amount of the active ingredient, is
available, and that
degradation products which may endanger the patients do not exceed a certain
maximum
value. These maximum values can be deduced from the ICH directive Q3B (R2).
The medicament forms according to the present invention have shown a very good
storage
stability in a short-term stress study in common blisters (for example top
foil aluminum foil,
20 [tm strong and bottom foil PVC/PVDC foil, glassy) at 25 C and 60 % relative
air humidity
(corresponds to subtropical to Mediterranean climate), 30 C and 65 % relative
air humidity
(corresponds to hot and wet climate) as well as at 40 C and 75 % relative air
humidity
(corresponds to very hot and particularly wet climate). The medicament forms
according to
the present invention also after the storage at 25 C and 60 % relative air
humidity, 30 C and
65 % relative air humidity or at 40 C and 75 % relative air humidity after 1
month preferably
contain still between 95 % and 105 % of the theoretical amount of the active
ingredient in the
medicament form, thus fulfil the specification. Preferably, the amount of the
active ingredient
also after a storage for 1 month under one of the mentioned conditions (25 C
and 60 %
relative air humidity, 30 C and 65 % relative air humidity or 40 C and 75 %
relative air
humidity) is between 96 % and 103 % and still more preferably between 97 % and
102 %,
based on the amount of active ingredient in the not stored medicament form.
The total mass of
the stored medicament form differs from the mass of the not stored medicament
form
preferably by less than 5 %, still further preferably by less than 4 % and
still more preferably
by less than 2 %, when the storage of the medicament form is conducted for 1
month at 25 C
and 60 % relative air humidity, 30 C and 65 % relative air humidity or 40 C
and 75 %
relative air humidity. This shows that an up-take of water from the
environment can be
avoided with the medicament form according to the present invention, normally
comprising a
hygroscopic active ingredient, also in the case of extreme humidity in the
atmosphere of the
environment. Also, after a storage time of 1 month in one of the given
atmospheres, the
diameter of the medicament form according to the present invention differs
from the diameter
of the not stored medicament form preferably by less than 4 %, further
preferably by less than
3 % and still more preferably by less than 2 %. The medicament forms according
to the
CA 02877190 2014-12-18
36
English Translation of PCT/EP2013/063162
present invention are preferably storage-stable over more than 6 months,
preferably at least 12
months of storage time under standard conditions according to the
international directives of
the ICH.
The medicament forms according to the present invention are characterized by
an excellent
uniformity of the mass and uniformity of the content, which is guaranteed by
the composition
of the medicament forms and the production method. The tests are conducted
according to the
respective methods of the European Pharmacopoeia (Ph. Eur. 7). The medicament
form
preferably shows a uniformity of the mass in such a manner that the mass of 20
such
medicament forms does preferably not differ by more than 5 %, differ further
preferably by
less than 5 %, still further preferably by less than 4 % from the mean value
of the mass of the
medicament form which is deduced from the mass of the 20 medicament forms. The
medicament form according to the present invention preferably shows a
uniformity of the
content in such a manner that the content of the active ingredient in 10 such
medicament
forms each is between 85 % and 115 %, preferably between 87 % and 113 % and
ideally
between 95 % and 105 %, based on the mean value of the content of the active
ingredient in
the 10 medicament forms.
With the medicament form according to the present invention and the production
method
according to the present invention it is thus possible to provide in
particular hygroscopic
active ingredients, thus active ingredients which can only be processed with
difficulties, in
such a manner that an intake several times a day is simulated. Here the first
proportion of the
active ingredient is immediately released in the first phase. Such a release
is in particularly
advantageous in the case of a short-term therapy, when the desired plasma
level should be
reached as quickly as possible and at the same time the effect should be
prolonged by
subsequent phases of release. Such a release system may also be advantageous
in the case of a
long-term therapy, in particular when in the case of long dosing intervals at
the end of the
interval the value falls below the minimum effect concentration. By the fast
initial release of
the active ingredient from the follow-up medicament form the plasma level is
quickly
increased and then in the required range again.
Here, the medicament form has a size which is suitable for oral intake. It is
characterized by
high mechanical stability also in the case of long storage. In particular
hygroscopic active
ingredients which are acidic and/or highly soluble profit from the design of
the medicament
form according to the present invention. So the daily dose of such active
ingredients can in
CA 02877190 2014-12-18
37
English Translation of PCT/EP2013/063162
particular be reduced to a single daily intake which may have a positive
influence onto the
compliance of the patient and directly on the health expenditures.
The medicament form according to the present invention is in particularly
suitable for the
administration of the following active ingredients and/or their
pharmaceutically acceptable
salts: valproic acid, carbamazepine, tetracycline, lincomycin, clindamycin,
erythromycin,
rifampicin, metformin, atenolol, ranitidine, acetylsalicylic acid, diclofenac,
omeprazole,
methyldopa, minoxidil, betahistine, dexamethasone, prednisolone, piracetam,
pravastatin and
gemfibrozil.
Valproic acid and carbamazepine are in particularly used for the treatment of
epilepsy.
Tetracycline, lincomycin, clindamycin, erythromycin and rifampicin are
suitable for the
treatment of bacterial infection diseases. Metformin is used in the case of
diabetes mellitus.
The active ingredient atenolol is used in the case of functional
cardiovascular problems,
arrhythmias, arterial hypertension and angina pectoris. Also methyldopa and
minoxidil are
suitable for the treatment of hypertension. The active ingredients ranitidine
and omeprazole
are in particularly used for the treatment of gastrointestinal ulcers, reflux
esophagitis and
Zollinger-Ellison syndrome. Acetylsalicylic acid and diclofenac are used
against pain.
Acetylsalicylic acid in low doses is also suitable for inhibiting the
aggregation of
thrombocytes in the case of angina pectoris or after acute cardiac infarction.
The active ingredient betahistine is used for the treatment of the Meniere
symptom complex,
the symptoms of which are dizziness, often in combination with nausea and/or
vomiting,
tinnitus and hearing loss. Dexamethasone as a corticosteroid is used in the
case of
autoimmune diseases, cerebral edema and asthma. Prednisolone as a
corticosteroid is in
particularly used in the case of adrenal cortex insufficiency. The active
ingredient piracetam is
in particularly suitable in the case of brain-related performance disorders.
Gemfibrozil and
pravastatin are normally used for the treatment of hypertriglyceridemia and
hypercholesterolemia.
Therefore according to the present invention is also the use of the medicament
form according
to the present invention for the treatment of a patient suffering from a
disease, selected from
epilepsy, bacterial infection disease, diabetes mellitus, functional
cardiovascular problems,
arrhythmias, hypertension, angina pectoris, cardiac insufficiency,
gastrointestinal ulcer, reflux
esophagitis, Zollinger-Ellison syndrome, pains, dizziness, in particular in
connection with the
Meniere symptom complex, autoimmune disease, cerebral edema, asthma, adrenal
cortex
CA 02877190 2014-12-18
38
English Translation of PCT/EP2013/063162
insufficiency, brain-related performance disorders, hypertriglyceridemia and
hypercholesterolemia. Preferably here the medicament form is taken one time a
day.
According to the present invention the use of the medicament form for the
treatment of
dizziness in connection with the Meniere symptom complex is particularly
preferable.
Also according to the present invention is a method for the treatment of a
disease, selected
from epilepsy, bacterial infection disease, diabetes mellitus, functional
cardiovascular
problems, arrhythmias, hypertension, angina pectoris, cardiac insufficiency,
gastrointestinal
ulcer, reflux esophagitis, Zollinger-Ellison syndrome, pains, dizziness in
connection with the
Meniere symptom complex, autoimmune disease, cerebral edema, asthma, brain-
related
performance disorders, hypertriglyceridemia and hypercholesterolemia, wherein
the method
comprises the administration of a medicament form according to the present
invention.
Preferably, this administration takes place one single time a day.
CA 02877190 2014-12-18
39
English Translation of PCT/EP2013/063162
Examples
Example 1: Production of a medicament form according to the present invention
Element Ingredient Amount per Function
medicament
form (mg)
Core betahistine dihydrochloride 24 active ingredient
lactose monohydrate 53 carrier
(Granulac 230)
microcrystalline cellulose 30 carrier
(Vivapur 102)
corn starch 53 carrier
citric acid, anhydrous 24 buffer substance
Povidon K25 (Plasdone K25) 2 granulating agent
hardened cottonseed oil (Lubritab ) 5 lubricant
total mass 191
Intermediate Eudragit S 100 6.67 film-forming
layer component
Eudragit L 100 1.67 film-forming
component
triacetin 2.50 plasticizer
talcum 4.17 filler
total mass 15.01
Shell betahistine dihydrochloride 24 active ingredient
lactose monohydrate 53 carrier
(Granulac 230)
lactose monohydrate 128.5 carrier
(Tablettose 80)
microcrystalline cellulose 230 carrier
(Vivapur 102)
corn starch 53 carrier
citric acid, anhydrous 24 buffer substance
Povidon K25 (Plasdone K25) 2 granulating agent
pre-gelatinized corn starch 30 carrier
(Starch 1500 )
hardened cottonseed oil (Lubritab ) 5.5 lubricant
total mass 550
Medicament mass 756.01
form
CA 02877190 2014-12-18
English Translation of PCT/EP2013/063162
A medicament form according to the present invention in the form of a shell-
core tablet was
prepared. At first the core was prepared. For the production of the core pre-
granules were
prepared. For that the carriers lactose monohydrate, microcrystalline
cellulose and corn starch
and the buffer substance citric acid were intensively mixed in a
mixing/granulating machine
(Diosna P10). Subsequently, the mixture was granulated in the mixing machine
Diosna P10
with a granulation solution. The granulation solution was prepared by
dissolving the Povidon
K25 in purified water so that the solution contained 16 % by weight of Povidon
K25.
Subsequently, the pre-granules were dried in a fluidized bed facility (GPCG-3)
at 60 C to a
residual moisture of < 5 %. Subsequently, the dried pre-granules were sieved
(mesh size 1
mm).
From the pre-granules the active ingredient granules were prepared by the
addition of a
solution of betahistine dihydrochloride in methanol. Here the clear solution
contained 28.5 %
by weight of betahistine dihydrochloride. For that the pre-granules were
placed in the
mixing/granulating machine Diosna 10 and granulated with the active ingredient
solution. The
obtained active ingredient granules were dried in a fluidized bed facility
(GPCG-3) to a
content of residual methanol of < 3000 ppm. Subsequently, the dried active
ingredient
granules were sieved (mesh size 1 mm).
To the active ingredient granules subsequently hardened cottonseed oil was
added and mixed
in a mixing machine. The obtained core mixture was compressed with a rotary
pelleting
machine. Biconvex cores having a diameter of 8 mm were obtained. The obtained
cores had a
weight of 191 mg and a height of 3.85 mm.
Subsequently, the intermediate layer was prepared. For that the film-forming
components
Eudragit S 100 and Eudragit L 100 were dissolved in iso-propanol, wherein
the content of
the film-forming components in the solution was 5.9 % by weight. Into the
solution the
plasticizer triacetin as well as the adjuvant talcum were stirred in. Also
water was added (6.67
m1). Subsequently, the intermediate layer mixture was applied onto the core so
that it
completely covered the core. The application of the intermediate layer mixture
was conducted
in a GS coating machine (GS-10) at a temperature of the inlet air of 50 C.
For the production of the shell the active ingredient granules were prepared
analogously to the
core so that 186 mg of active ingredient granules were available. These active
ingredient
granules were mixed with the carriers lactose monohydrate, microcrystalline
cellulose and
pre-gelatinized corn starch as well as the lubricant hardened cottonseed oil.
This resulted in
CA 02877190 2014-12-18
41
English Translation of PCT/EP2013/063162
the shell mixture. Thus the proportion of the active ingredient granules in
the shell mixture
was 33.8 % by weight.
In the last step the shell mixture was pressed onto the intermediate layer in
a Syl-one press
with tablet feeding system. For that 300 mg of the shell mixture were filled
into a die (round,
biconvex, diameter 12 mm, curvature radius 9.5 mm). Thereafter the core with
the
intermediate layer was centrically positioned. In a second filling step the
residual proportion
of the shell mixture was filled into the die. Subsequently, it was pressed
with a force of
pressure of 10 IN. The shell/core tablet showed a breaking strength of 140 N,
measured with
an Erweka TBH-30 hardness tester. In a further embodiment the same starting
materials,
amount ratios and production methods were used, wherein in the step of
pressing the shell a
force of pressure of 25 kN was applied, so that a shell/core tablet with a
breaking strength of
280 N was obtained.
Example 2: Production of a medicament form according to the present invention
Element Ingredient Amount per Function
medicament form (mg)
Intermediate Eudragit S 100 8.874 film-forming component
layer Eudragit L 100 2.225 film-forming component
triacetin 3.331 plasticizer
talcum 5.556 filler
total mass 19.986
A shell-core tablet with a composition of the core and the shell like in
example 1 was
prepared.
However the composition of the intermediate layer was altered, wherein a
higher weight of
the intermediate layer was chosen. The chosen higher weight of the
intermediate layer further
contributed to an increase of the mechanical strength of the core. The chosen
intermediate
layer is dissolved at a pH value of 7.0 to 7.2. So the intermediate layer
enables the release of
the active ingredient proportion from the core into the middle section of the
intestine.
The production of the core and the shell was conducted as described in example
1. The
production of the intermediate layer comprised the dissolution of the film-
forming
components Eudragit S 100 and Eudragit L 100 in iso-propanol, wherein the
content of the
film-forming components in the solution was 5.9 % by weight. Into the solution
the plasticizer
triacetin as well as the adjuvant talcum were stirred in. Also water was added
(8.86 ml).
CA 02877190 2014-12-18
42
English Translation of PCT/EP2013/063162
Subsequently, the intermediate layer mixture was applied onto the core so that
it completely
covered the core. The application of the intermediate layer mixture was
conducted in a GS
coating machine (GS-10) at a temperature of the inlet air of 50 C.
Example 3: production of a medicament form according to the present invention
Element Ingredient Amount per Function
medicament form (mg)
Intermediate Eudragit S 100 11.099 film-forming component
layer triacetin 3.331 plasticizer
talcum 5.556 filler
total mass 19.986
A shell-core tablet with a composition of the core and the shell like in
example 1 was
prepared. However the composition of the intermediate layer was altered,
wherein a higher
weight of the intermediate layer was chosen, analogously to example 2. The
chosen higher
weight of the intermediate layer further contributed to an increase of the
mechanical strength
of the core. The chosen intermediate layer is dissolved at a pH value of 7.2
to 7.5. So the
intermediate layer enables the release of the active ingredient proportion
from the core into
the lower section of the intestine.
The production of the core and the shell was conducted as described in example
1. The
production of the intermediate layer comprised the dissolution of the film-
forming component
Eudragit S 100 in iso-propanol, wherein the content of the film-forming
component in the
solution was 5.9 % by weight. Into the solution the plasticizer triacetin as
well as the adjuvant
talcum were stirred in. Also water was added (8.86 m1). Subsequently, the
intermediate layer
mixture was applied onto the core so that it completely covered the core. The
application of
the intermediate layer mixture was conducted in a GS coating machine (GS-10)
at a
temperature of the inlet air of 50 C.
Example 4:
In example 4 a test was made for the release of the active ingredient
betahistine
dihydrochloride from a core with intermediate layer having the composition
according to
example 1, however without shell. Thus the core with intermediate layer
comprised 24 mg of
betahistine dihydrochloride. The release was determined with a paddle
apparatus. Figure 2
shows the respective results.
CA 02877190 2014-12-18
43
English Translation of PCT/EP2013/063162
Example 5: Stability of the medicament form according to the present invention
A medicament form according to the present invention in the form of a shell-
core tablet with a
composition of the core and the shell as in example 1 was prepared. Blisters
(top foil
aluminum foil, 20 gm strong, bottom foil PVC/PVDC foil, glassy) comprising the
medicament form were subjected to a stress test for 1 month, thus stored at 25
C/60 %
relative air humidity, 30 C/65 % relative air humidity and 40 C/75 % relative
air humidity.
The medicament form after the storage showed the below listed parameters. It
can be seen
that the medicament form according to the present invention is also stable
under extreme
storage conditions and does not cap and/or swell despite the hygroscopic
active ingredient.
Also after a storage for 1 month at the extreme conditions the impurities
according to the
specification and/or the monograph pursuant to Ph. Eur. 7 were below the
respectively
defined limiting values and/or were not present in measurable amount.
Parameter Not stored Storage for 1 Storage for 1 Storage for 1
medicament month at month at month at
form 25 C/60 A 30 C/65 % 40 C/75 "A
relative air relative air relative air
humidity humidity humidity
Height (mm) 7.1 7.1 7.1 7.2
Diameter (mm) 12.0 12.0 12.0 12.1
Actual weight 755.7 757.6 757.4 767.0
(mg)
Content (% of 100.6 101.1 101.5 100.8
the theoretical
amount of the
active
ingredient)
Description of the figures:
Figure 1 shows the course of the release of two medicament forms according to
the present
invention, prepared according to embodiment example 1, comprising 24 mg of
betahistine
dihydrochloride in the shell and 24 mg of betahistine dihydrochloride in the
core. The shell
was pressed onto the core with the intermediate layer with a force of pressure
of 10 kN or 25
kN. Here a pH course which corresponds to the physiological conditions is
mimicked. A
release of 100 % means a release of 48 mg of betahistine dihydrochloride from
the
medicament form, thus 24 mg from the shell and 24 mg from the core. The
medicament form
according to the present invention releases betahistine dihydrochloride in a
biphasic form,
CA 02877190 2014-12-18
44
English Translation of PCT/EP2013/063162
thus pulsed at first from the shell and starting at a pH value of 7.0 (after
about 8.5 hours) after
the dissolution of the gastric juice-resistant intermediate layer from the
core. So an intake two
times a day is simulated, thus the intake of two commercially available
medicament forms
without any particular release modification in an interval of 8 to 12 hours.
With the special
design of the medicament form an optimum release profile is achieved. It can
be expected that
this will also be confirmed in vivo.
Figure 2 shows the course of the release from the core with intermediate layer
according to
example 4. A release of 50 % means the release of 24 mg of betahistine
dihydrochloride. It
=
can be seen that only starting at a pH value of 7.0 the dissolution of the
intermediate layer
takes place and the proportion of the active ingredient is released from the
core. Thus, the
core with the intermediate layer is stable in the upper sections of the
intestine, wherein the
disintegration only starts at a pH value of 7.0, which corresponds to the
intestine section ileum
to colon and a residence time in the gastrointestinal tract without release of
7 to 12 hours.