Note: Descriptions are shown in the official language in which they were submitted.
CA 02877639 2015-01-09
MAIZE ETHYLENE SIGNALING GENES AND MODULATION OF
SAME FOR IMPROVED STRESS TOLERANCE IN PLANTS
TECHNICAL FIELD
The present invention relates generally to plant molecular biology. More
specifically,
it relates to nucleic acids and methods for modulating their expression in
plants.
BACKGROUND OF THE INVENTION
to
Plant hormones have been intensively studied for decades for their diverse and
complex effects on the plant life. Of the five main hormones-auxins, ethylene,
abscisic acid,
cytokinins and gibberellins-the molecular signaling and mode of action of
ethylene has been
the most fully resolved. This progress was made chiefly in the 1990s by the
cloning of
genes corresponding to mutations in ethylene production and signaling.
Ethylene (C2H4) is a gaseous plant hormone that affects myriad developmental
processes and fitness responses in plants, such as germination, flower and
leaf senescence,
fruit ripening, leaf abscission, root nodulation, programmed cell death and
responsiveness to
stress and pathogen attack. Over the past decade, genetic screens have
identified more
than a dozen genes involved in the ethylene response in plants. Ethylene
governs diverse
processes in plants, and these effects are sometimes affected by the action of
other plant
hormones, other physiological signals, and the environment, both biotic and
abiotic. For
example, it is known that cytokinin can cause ethylene like effects through
the action of
ethylene. In addition, abscisic acid can inhibit ethylene production and
signaling. Auxin and
ethylene are also known to cooperate in various physiological phenomena. From
what is
currently known, in general ethylene does not appear to be strictly required
for the plant's life
cycle, but it does significantly modify development and condition response to
stresses.
What is needed in the art is a means to improve agronomic performance in
plants,
particularly cereal crops such as maize, by modulating ethylene mediated
responses in
plants.
SUMMARY OF THE INVENTION
This invention involves the identification of maize genes involved in the
ethylene
signal transduction pathway and the modulation of the same for improving
stress tolerance
in plants. The invention relates to characterization and modulation of four
different maize
1
'
CA 02877639 2015-01-09
genes involved in the ethylene pathway including, EIN3, ERF3, EBF1, EBF2 and
EIN5 to
create plants with an altered response to stress and other ethylene inducing
conditions.
Polynucleotides, related polypeptides and all conservatively modified variants
of the
present maize sequences involved in the ethylene transduction pathway are
presented
herein. Included are novel and partial maize sequences for the ethylene
signaling
associated genes including EIN3, ERF3, EBF1, EBF2 and EIN5.
The invention also includes methods to alter the genetic composition of crop
plants,
especially maize, so that such crops can be more tolerant to stress conditions
and other
ethylene mediated responses. The utility of this class of invention is then
both yield
enhancement and stress tolerance.
Ethylene-mediated responses include those involving: crowding tolerance, seed
set
and development, growth in compacted soils, flooding tolerance, maturation and
senescence
and disease resistance. This invention provides methods and compositions to
effect various
alterations in the ethylene-mediated response in a plant that would result in
improved
agronomic performance, particularly under stress.
Therefore, in one aspect, the present invention relates to an isolated nucleic
acid
comprising an isolated polynucleotide sequence associated with ethylene
signaling in maize.
One embodiment of the invention is an isolated polynucleotide comprising a
nucleotide
sequence selected from the group consisting of: (a) the nucleotide sequence
comprising
SEQ ID NO: 1 (EIN3), 3 (EBF1), 5 (EBF2), 7 (EIN5) or 9 (ERF3); (b) the
nucleotide
sequence encoding an amino acid sequence comprising SEQ ID NO: 2, 4, 6, 8 or
10; (c) a
polynucleotide having a specified sequence identity to a polynucleotide
encoding a
polypeptide of the present invention; (d) a polynucleotide which is
complementary to the
polynucleotide of (a); and, (e) a polynucleotide comprising a specified number
of contiguous
nucleotides from a polynucleotide of (a) or (b). The isolated nucleic acid can
be DNA.
Compositions of the invention include an isolated polypeptide comprising an
amino
acid sequence selected from the group consisting of: (a) the amino acid
sequence
comprising SEQ ID NO: 2, 4, 6, 8, or 10; and (b) the amino acid sequence
comprising a
specified sequence identity to SEQ ID NO: 2, 4, 6, 8 or 10, wherein said
polypeptide has
ethylene signaling activity.
In another aspect, the present invention relates to a recombinant expression
cassette
comprising a nucleic acid as described. Additionally, the present invention
relates to a vector
containing the recombinant expression cassette.
Further, the vector containing the
recombinant expression cassette can facilitate the transcription and
translation of the nucleic
acid in a host cell. The present invention also relates to the host cells able
to express the
2
CA 02877639 2015-01-09
polynucleotide of the present invention. A number of host cells could be used,
such as but
not limited to, microbial, mammalian, plant or insect.
In yet another embodiment, the present invention is directed to a transgenic
plant or
plant cells, containing the nucleic acids of the present invention. Preferred
plants containing
the polynucleotides of the present invention include but are not limited to
maize, soybean,
sunflower, sorghum, canola, wheat, alfalfa, cotton, rice, barley, tomato and
millet. In another
embodiment, the transgenic plant is a maize plant or plant cells. Another
embodiment is a
transgenic seed from the transgenic plant.
The plants of the invention can have altered responses to ethylene as compared
to a
control plant. In some plants, the altered ethylene response is located to a
vegetative tissue,
a reproductive tissue, or a vegetative tissue and a reproductive tissue.
Plants of the
invention can have at least one of the following phenotypes including but not
limited to:
differences in crowding tolerance, seed set and development, growth in
compacted soils,
flooding tolerance, maturation and senescence and disease resistance compared
to non
transformed plants.
Another embodiment of the invention would be plants that have been genetically
modified at a genomic locus, wherein the genomic locus encodes an ethylene
signaling
polypeptide of the invention.
Methods for increasing the activity of ethylene signaling polypeptides in a
plant are
provided. The method can comprise introducing into the plant an ethylene
signaling
polynucleotide of the invention.
Methods for reducing or eliminating the level of ethylene signaling
polypeptide in the
plant are also provided. The level or activity of the polypeptide could also
be reduced or
eliminated in specific tissues, causing alteration in plant growth rate.
Reducing the level
and/or activity of the ethylene signaling gene will lead to plants with
changed responses to
the ethylene hormone.
In a further aspect, the present invention relates to a polynucleotide
amplified from a
Zea mays nucleic acid library using primers which selectively hybridize, under
stringent
hybridization conditions, to loci within polynucleotides of the present
invention.
Definitions
Units, prefixes, and symbols may be denoted in their SI accepted form. Unless
otherwise indicated, nucleic acids are written left to right in 5' to 3'
orientation; amino acid
sequences are written left to right in amino to carboxy orientation,
respectively. Numeric
ranges recited within the specification are inclusive of the numbers defining
the range and
3
CA 02877639 2015-01-09
include each integer within the defined range. Amino acids may be referred to
herein by
either their commonly known three letter symbols or by the one-letter symbols
recommended
by the IUPAC-IUB Biochemical Nomenclature Commission. Nucleotides, likewise,
may be
referred to by their commonly accepted single-letter codes. Unless otherwise
provided for,
software, electrical and electronics terms as used herein are as defined in
The New IEEE
Standard Dictionary of Electrical and Electronics Terms (5th edition, 1993).
The terms
defined below are more fully defined by reference to the specification as a
whole.
By "amplified" is meant the construction of multiple copies of a nucleic acid
sequence
or multiple copies complementary to the nucleic acid sequence using at least
one of the
nucleic acid sequences as a template. Amplification systems include the
polymerase chain
reaction (PCR) system, ligase chain reaction (LCR) system, nucleic acid
sequence based
amplification (NASBA, Cangene, Mississauga, Ontario), Q-Beta Replicase
systems,
transcription-based amplification system (TAS), and strand displacement
amplification (SDA).
See, e.g., Diagnostic Molecular Microbiology: Principles and Applications,
Persing, et al., Ed.,
American Society for Microbiology, Washington, DC (1993). The product of
amplification is
termed an amplicon.
The term "antibody" includes reference to antigen binding forms of antibodies
. The
term "antibody" frequently refers to a polypeptide substantially encoded by an
immunoglobulin gene or immunoglobulin genes, or fragments thereof which
specifically bind
and recognize an analyte (antigen). However, while various antibody fragments
can be
defined in terms of the digestion of an intact antibody, one of skill will
appreciate that such
fragments may be synthesized de novo either chemically or by utilizing
recombinant DNA
methodology. Thus, the term antibody, as used herein, also includes antibody
fragments
such as single chain FV, chimeric antibodies (i.e., comprising constant and
variable regions
from different species), humanized antibodies (i.e., comprising a
complementarity
determining region (CDR) from a non-human source) and heteroconjugate
antibodies (e.g.,
bispecific antibodies).
The term "antigen" includes reference to a substance to which an antibody can
be
generated and/or to which the antibody is specifically immunoreactive. The
specific
immunoreactive sites within the antigen are known as epitopes or antigenic
determinants.
These epitopes can be a linear array of monomers in a polymeric composition-
such as
amino acids in a protein-or consist of or comprise a more complex secondary or
tertiary
structure. Those of skill will recognize that all immunogens (i.e., substances
capable of
eliciting an immune response) are antigens, however some antigens, such as
haptens, are
not immunogens but may be made immunogenic by coupling to a carrier molecule.
An
4
CA 02877639 2015-01-09
antibody immunologically reactive with a particular antigen can be generated
in vivo or by
recombinant methods such as selection of libraries of recombinant antibodies
in phage or
similar vectors. See, e.g., Huse, et al., (1989) Science 246:1275-1281; and
Ward, et al.,
(1989) Nature 341:544-546; and Vaughan, etal., (1996) Nature Biotech. 14:309-
314.
As used herein, "antisense orientation" includes reference to a duplex
polynucleotide
sequence that is operably linked to a promoter in an orientation where the
antisense strand
is transcribed. The antisense strand is sufficiently complementary to an
endogenous
transcription product such that translation of the endogenous transcription
product is often
inhibited.
The term "conservatively modified variants" applies to both amino acid and
nucleic
acid sequences. With respect to particular nucleic acid sequences,
conservatively modified
variants refers to those nucleic acids which encode identical or
conservatively modified
variants of the amino acid sequences. Because of the degeneracy of the genetic
code, a
large number of functionally identical nucleic acids encode any given protein.
For instance,
the codons GCA, GCC, GCG and GCU all encode the amino acid alanine. Thus, at
every
position where an alanine is specified by a codon, the codon can be altered to
any of the
corresponding codons described without altering the encoded polypeptide. Such
nucleic
acid variations are "silent variations" and represent one species of
conservatively modified
variation. Every nucleic acid sequence herein that encodes a polypeptide also,
by reference
to the genetic code, describes every possible silent variation of the nucleic
acid.
One of ordinary skill will recognize that each codon in a nucleic acid (except
AUG,
which is ordinarily the only codon for methionine; and UGG, which is
ordinarily the only
codon for tryptophan) can be modified to yield a functionally identical
molecule. Accordingly,
each silent variation of a nucleic acid which encodes a polypeptide of the
present invention
is implicit in each described polypeptide sequence and is within the scope of
the present
invention.
As to amino acid sequences, one of skill will recognize that individual
substitutions,
deletions or additions to a nucleic acid, peptide, polypeptide, or protein
sequence which
alters, adds or deletes a single amino acid or a small percentage of amino
acids in the
encoded sequence is a "conservatively modified variant" where the alteration
results in the
substitution of an amino acid with a chemically similar amino acid. Thus, any
number of
amino acid residues selected from the group of integers consisting of from 1
to 15 can be so
altered. Thus, for example, 1, 2, 3, 4, 5, 7 or 10 alterations can be made.
Conservatively modified variants typically provide similar biological activity
as the
unmodified polypeptide sequence from which they are derived. For example,
substrate
5
CA 02877639 2015-01-09
specificity, enzyme activity, or ligand/receptor binding is generally at least
30%, 40%, 50%,
60%, 70%, 80% or 90% of the native protein for its native substrate.
Conservative
substitution tables providing functionally similar amino acids are well known
in the art.
The following six groups each contain amino acids that are conservative
substitutions
for one another: 1) Alanine (A), Serine (S), Threonine (T); 2) Aspartic acid
(D), Glutamic acid
(E); 3) Asparagine(N), Glutamine (Q); 4) Arginine (R), Lysine (K);5)
Isoleucine(I), Leucine (L),
Methionine (M), Valine (V); and 6)Phenylalanine (F), Tyrosine (Y), Tryptophan
(W). See
also, Creighton (1984) Proteins W. H. Freeman and Company.
By "encoding" or "encoded", with respect to a specified nucleic acid, is meant
comprising the information for translation into the specified protein. A
nucleic acid encoding
a protein may comprise intervening sequences (e.g., introns) within translated
regions of the
nucleic acid, or may lack such intervening non-translated sequences (e.g., as
in cDNA). The
information by which a protein is encoded is specified by the use of codons.
Typically, the
amino acid sequence is encoded by the nucleic acid using the "universal"
genetic code.
However, variants of the universal code, such as are present in some plant,
animal and
fungal mitochondria, the bacterium Mycoplasma capricolum, or the ciliate
Macronucleus,
may be used when the nucleic acid is expressed therein. When the nucleic acid
is prepared
or altered synthetically, advantage can be taken of known codon preferences of
the intended
host where the nucleic acid is to be expressed.
For example, although nucleic acid sequences of the present invention may be
expressed in both monocotyledonous and dicotyledonous plant species, sequences
can be
modified to account for the specific codon preferences and GC content
preferences of
monocotyledons or dicotyledons as these preferences have been shown to differ
(Murray, et
al., (1989) Nucl. Acids Res. 17:477-498). Thus, the maize preferred codon for
a particular
amino acid may be derived from known gene sequences from maize. Maize codon
usage
for 28 genes from maize plants is listed in Table 4 of Murray, et al., supra.
As used herein "full-length sequence" in reference to a specified
polynucleotide or its
encoded protein means having the entire amino acid sequence of, a native
(nonsynthetic),
endogenous, biologically active form of the specified protein. Methods to
determine whether
a sequence is full-length are well known in the art including such exemplary
techniques as
northern or western blots, primer extension, S 1 protection, and ribonuclease
protection.
See, e.g., Plant Molecular Biology: A Laboratory Manual, Clark, Ed., Springer-
Verlag, Berlin
(1997). Comparison to known full-length homologous (orthologous and/or
paralogous)
sequences can also be used to identify full-length sequences of the present
invention.
Additionally, consensus sequences typically present at the 5' and 3'
untranslated regions of
6
CA 02877639 2015-01-09
mRNA aid in the identification of a polynucleotide as full-length.
For example, the
consensus sequence ANNNNAUGG, where the underlined codon represents the N-
terminal
methionine, aids in determining whether the polynucleotide has a complete 5'
end.
Consensus sequences at the 3' end, such as polyadenylation sequences, aid in
determining
whether the polynucleotide has a complete 3' end.
As used herein, "heterologous" in reference to a nucleic acid is a nucleic
acid that
originates from a foreign species, or, if from the same species, is
substantially modified from
its native form in composition and/or genomic locus by deliberate human
intervention. For
example, a promoter operably linked to a heterologous structural gene is from
a species
different from that from which the structural gene was derived, or, if from
the same species,
one or both are substantially modified from their original form. A
heterologous protein may
originate from a foreign species or, if from the same species, is
substantially modified from
its original form by deliberate human intervention.
By "host cell" is meant a cell which contains a vector and supports the
replication
and/or expression of the vector. Host cells may be prokaryotic cells such as
E. coil, or
eukaryotic cells such as yeast, insect, amphibian or mammalian cells.
Preferably, host cells
are monocotyledonous or dicotyledonous plant cells.
A particularly preferred
monocotyledonous host cell is a maize host cell.
The term "hybridization complex" includes reference to a duplex nucleic acid
structure formed by two single-stranded nucleic acid sequences selectively
hybridized with
each other.
By "immunologically reactive conditions" or "immunoreactive conditions" is
meant
conditions which allow an antibody, reactive to a particular epitope, to bind
to that epitope to
a detectably greater degree (e.g., at least 2-fold over background) than the
antibody binds to
substantially any other epitopes in a reaction mixture comprising the
particular epitope.
Immunologically reactive conditions are dependent upon the format of the
antibody binding
reaction and typically are those utilized in immunoassay protocols. See,
Harlow and Lane,
Antibodies, A Laboratory Manual, Cold Spring Harbor Publications, New York
(1988), for a
description of immunoassay formats and conditions.
The term "introduced" in the context of inserting a nucleic acid into a cell,
means
"transfection" or "transformation" or "transduction" and includes reference to
the
incorporation of a nucleic acid into a eukaryotic or prokaryotic cell where
the nucleic acid
may be incorporated into the genome of the cell (e.g., chromosome, plasmid,
plastid or
mitochondrial DNA), converted into an autonomous replicon, or transiently
expressed (e.g.,
transfected mRNA).
7
CA 02877639 2015-01-09
The term "isolated" refers to material, such as a nucleic acid or a protein,
which is:
(1) substantially or essentially free from components that normally accompany
or interact
with it as found in its naturally occurring environment. The isolated material
optionally
comprises material not found with the material in its natural environment or
(2) if the material
is in its natural environment, the material has been synthetically (non-
naturally) altered by
deliberate human intervention to a composition and/or placed at a location in
the cell (e.g.,
genome or subcellular organelle) not native to a material found in that
environment. The
alteration to yield the synthetic material can be performed on the material
within or removed
from its natural state. For example, a naturally occurring nucleic acid
becomes an isolated
Hi nucleic acid if it is altered, or if it is transcribed from DNA which
has been altered, by means
of human intervention performed within the cell from which it originates. See,
e.g.,
Compounds and Methods for Site Directed Mutagenesis in Eukaryotic Cells,
Kmiec, US
Patent Number 5,565,350; In Vivo Homologous Sequence Targeting in Eukaryotic
Cells;
Zarling, et al., PCT/US93/03868. Likewise, a naturally occurring nucleic acid
(e.g., a
promoter) becomes isolated if it is introduced by nonnaturally occurring means
to a locus of
the genome not native to that nucleic acid. Nucleic acids which are "isolated"
as defined
herein, are also referred to as "heterologous" nucleic acids.
Unless otherwise stated, the term "EIN3, ERF3, EIN5, EBF1 or EBF2 nucleic
acid" is
a nucleic acid of the present invention and means a nucleic acid comprising a
polynucleotide
of the present invention (a "EIN3, ERF3, EIN5, EBF1 or EBF2 polynucleotide")
encoding a
EIN3, ERF3, EIN5, EBF1 or EBF2 polypeptide. A "EIN3, ERF3, EIN5, EBF1 or EBF2
gene"
is a gene of the present invention and refers to a heterologous genomic form
of a full-length
EIN3, ERF3, EIN5, EBF1 or EBF2 polynucleotide.
As used herein, "localized within the chromosomal region defined by and
including"
with respect to particular markers includes reference to a contiguous length
of a
chromosome delimited by and including the stated markers.
As used herein, "marker" includes reference to a locus on a chromosome that
serves
to identify a unique position on the chromosome. A "polymorphic marker"
includes reference
to a marker which appears in multiple forms (alleles) such that different
forms of the marker,
when they are present in a homologous pair, allow transmission of each of the
chromosomes of that pair to be followed. A genotype may be defined by use of
one or a
plurality of markers.
As used herein, "nucleic acid" includes reference to a deoxyribonucleotide or
ribonucleotide polymer in either single-or double-stranded form, and unless
otherwise limited,
encompasses known analogues having the essential nature of natural nucleotides
in that
8
CA 02877639 2015-01-09
they hybridize to single-stranded nucleic acids in a manner similar to
naturally occurring
nucleotides (e.g., peptide nucleic acids).
By "nucleic acid library" is meant a collection of isolated DNA or RNA
molecules
which comprise and substantially represent the entire transcribed fraction of
a genome of a
specified organism. Construction of exemplary nucleic acid libraries, such as
genomic and
cDNA libraries, is taught in standard molecular biology references such as
Berger and
Kimmel, Guide to Molecular Cloning Techniques, Methods in Enzymology, Vol.
152,
Academic Press, Inc., San Diego, CA (Berger); Sambrook, et al., Molecular
Cloning-A
Laboratory Manual, 2'd ed., Vol. 1-3 (1989); and Current Protocols in
Molecular Biology,
Ausubel, et al., Eds., Current Protocols, a joint venture between Greene
Publishing
Associates, Inc. and John Wiley & Sons, Inc. (1994).
As used herein "operably linked" includes reference to a functional linkage
between a
promoter and a second sequence, wherein the promoter sequence initiates and
mediates
transcription of the DNA sequence corresponding to the second sequence.
Generally,
operably linked means that the nucleic acid sequences being linked are
contiguous and,
where necessary to join two protein coding regions, contiguous and in the same
reading
frame.
As used herein, the term "plant" includes reference to whole plants, plant
organs
(e.g., leaves, stems, roots, etc.), seeds and plant cells and progeny of same.
Plant cell, as
used herein includes, without limitation, seeds, suspension cultures, embryos,
meristematic
regions, callus tissue, leaves, roots, shoots, gametophytes, sporophytes,
pollen and
microspores. The class of plants which can be used in the methods of the
invention is
generally as broad as the class of higher plants amenable to transformation
techniques,
including both monocotyledonous and dicotyledonous plants. A particularly
preferred plant
is Zea mays.
As used herein, "polynucleotide" includes reference to a
deoxyribopolynucleotide,
ribopolynucleotide or analogs thereof that have the essential nature of a
natural
ribonucleotide in that they hybridize, under stringent hybridization
conditions, to substantially
the same nucleotide sequence as naturally occurring nucleotides and/or allow
translation
into the same amino acid(s) as the naturally occurring nucleotide(s). A
polynucleotide can
be full-length or a subsequence of a native or heterologous structural or
regulatory gene.
Unless otherwise indicated, the term includes reference to the specified
sequence as well as
the complementary sequence thereof. Thus, DNAs or RNAs with backbones modified
for
stability or for other reasons are "polynucleotides" as that term is intended
herein. Moreover,
DNAs or RNAs comprising unusual bases, such as inosine, or modified bases,
such as
9
CA 02877639 2015-01-09
tritylated bases, to name just two examples, are polynucleotides as the term
is used herein.
It will be appreciated that a great variety of modifications have been made to
DNA and RNA
that serve many useful purposes known to those of skill in the art.
The term polynucleotide as it is employed herein embraces such chemically,
enzymatically or metabolically modified forms of polynucleotides, as well as
the chemical
forms of DNA and RNA characteristic of viruses and cells, including among
other things,
simple and complex cells.
The terms "polypeptide", "peptide" and "protein" are used interchangeably
herein to
refer to a polymer of amino acid residues. The terms apply to amino acid
polymers in which
one or more amino acid residue is an artificial chemical analogue of a
corresponding
naturally occurring amino acid, as well as to naturally occurring amino acid
polymers. The
essential nature of such analogues of naturally occurring amino acids is that,
when
incorporated into a protein that protein is specifically reactive to
antibodies elicited to the
same protein but consisting entirely of naturally occurring amino acids. The
terms
"polypeptide", "peptide" and "protein" are also inclusive of modifications
including, but not
limited to, glycosylation, lipid attachment, sulfation, gamma-carboxylation of
glutamic acid
residues, hydroxylation and ADP-ribosylation. It will be appreciated, as is
well known and as
noted above, that polypeptides are not always entirely linear. For instance,
polypeptides
may be branched as a result of ubiquitization, and they may be circular, with
or without
branching, generally as a result of posttranslation events, including natural
processing event
and events brought about by human manipulation which do not occur naturally.
Circular,
branched and branched circular polypeptides may be synthesized by non-
translation natural
process and by entirely synthetic methods, as well. Further, this invention
contemplates the
use of both the methionine-containing and the methionine-less amino terminal
variants of the
protein of the invention.
As used herein "promoter" includes reference to a region of DNA upstream from
the
start of transcription and involved in recognition and binding of RNA
polymerase and other
proteins to initiate transcription. A "plant promoter" is a promoter capable
of initiating
transcription in plant cells whether or not its origin is a plant cell.
Exemplary plant promoters
include, but are not limited to, those that are obtained from plants, plant
viruses, and bacteria
which comprise genes expressed in plant cells such Agrobacterium or Rhizobium.
Examples of promoters under developmental control include promoters that
preferentially
initiate transcription in certain tissues, such as leaves, roots, or seeds.
Such promoters are
referred to as "tissue preferred". Promoters which initiate transcription only
in certain tissue
are referred to as "tissue specific". A "cell type" specific promoter
primarily drives
CA 02877639 2015-01-09
expression in certain cell types in one or more organs, for example, vascular
cells in roots or
leaves. An "inducible" or "repressible" promoter is a promoter which is under
environmental
control. Examples of environmental conditions that may effect transcription by
inducible
promoters include anaerobic conditions or the presence of light. Tissue
specific, tissue
preferred, cell type specific and inducible promoters constitute the class of
"non-constitutive"
promoters. A "constitutive" promoter is a promoter which is active under most
environmental
conditions.
The term "EIN3, ERF3, EIN5, EBF1 or EBF2 polypeptide" is a polypeptide of the
present invention and refers to one or more amino acid sequences, in
glycosylated or non-
glycosylated form. The term is also inclusive of fragments, variants,
homologs, alleles or
precursors (e.g., preproproteins or proproteins) thereof. A "EIN3, ERF3, EIN5,
EBF1 or
EBF2 protein" is a protein of the present invention and comprises a EIN3,
ERF3, EIN5,
EBF1 or EBF2 polypeptide.
As used herein "recombinant" includes reference to a cell or vector, that has
been
modified by the introduction of a heterologous nucleic acid or that the cell
is derived from a
cell so modified. Thus, for example, recombinant cells express genes that are
not found in
identical form within the native (non-recombinant) form of the cell or express
native genes
that are otherwise abnormally expressed, under-expressed or not expressed at
all as a
result of deliberate human intervention. The term "recombinant" as used herein
does not
encompass the alteration of the cell or vector by naturally occurring events
(e.g.,
spontaneous mutation, natural transformation/transduction/transposition) such
as those
occurring without deliberate human intervention.
As used herein, a "recombinant expression cassette" is a nucleic acid
construct,
generated recombinantly or synthetically, with a series of specified nucleic
acid elements
which permit transcription of a particular nucleic acid in a host cell. The
recombinant
expression cassette can be incorporated into a plasmid, chromosome,
mitochondrial DNA,
plastid DNA, virus or nucleic acid fragment. Typically, the recombinant
expression cassette
portion of an expression vector includes, among other sequences, a nucleic
acid to be
transcribed, and a promoter.
The term "residue" or "amino acid residue" or "amino acid" are used
interchangeably
herein to refer to an amino acid that is incorporated into a protein,
polypeptide, or peptide
(collectively "protein"). The amino acid may be a naturally occurring amino
acid and, unless
otherwise limited, may encompass non-natural analogs of natural amino acids
that can
function in a similar manner as naturally occurring amino acids.
11
CA 02877639 2015-01-09
The term "selectively hybridizes" includes reference to hybridization, under
stringent
hybridization conditions, of a nucleic acid sequence to a s other biologics.
Thus, under
designated immunoassay conditions, the specified antibodies bind to an analyte
having the
recognized epitope to a substantially greater degree (e.g., at least 2-fold
over background)
than to substantially all analytes lacking the epitope which are present in
the sample.
Specific binding to an antibody under such conditions may require an antibody
that is
selected for its specificity for a particular protein. For example, antibodies
raised to the
polypeptides of the present invention can be selected from to obtain
antibodies specifically
reactive with polypeptides of the present invention. The proteins used as
immunogens can
be in native conformation or denatured so as to provide a linear epitope.
A variety of immunoassay formats may be used to select antibodies specifically
reactive with a particular protein (or other analyte). For example, solid-
phase ELISA
immunoassays are routinely used to select monoclonal antibodies specifically
immunoreactive with a protein. See, Harlow and Lane, Antibodies, A Laboratory
Manual,
Cold Spring Harbor Publications, New York (1988), for a description of
immunoassay
formats and conditions that can be used to determine selective reactivity.
The term "stringent conditions" or "stringent hybridization conditions"
includes
reference to conditions under which a probe will hybridize to its target
sequence, to a
detectably greater degree than to other sequences (e.g., at least 2-fold over
background).
Stringent conditions are sequence-dependent and will be different in different
circumstances.
By controlling the stringency of the hybridization and/or washing conditions,
target
sequences can be identified which are 100% complementary to the probe
(homologous
probing).
Alternatively, stringency conditions can be adjusted to allow some mismatching
in
sequences so that lower degrees of similarity are detected (heterologous
probing).
Generally, a probe is less than about 1000 nucleotides in length, optionally
less than 500
nucleotides in length.
Typically, stringent conditions will be those in which the salt concentration
is less
than about 1.5 M Na ion, typically about 0.01 to 1.0 M Na ion concentration
(or other salts) at
pH 7.0 to 8.3 and the temperature is at least about 30 C for short probes
(e.g., 10 to 50
nucleotides) and at least about 60 C for long probes (e.g., greater than 50
nucleotides).
Stringent conditions may also be achieved with the addition of destabilizing
agents such as
formamide. Exemplary low stringency conditions include hybridization with a
buffer solution
of 30 to 35% formamide,1 M NaCI, 1% SDS (sodium dodecyl sulphate) at 37 C, and
a wash
in 1X to 2X SSC (20X SSC = 3.0 M NaCl/0.3 M trisodium citrate) at 50 to 55 C.
Exemplary
12
CA 02877639 2015-01-09
moderate stringency conditions include hybridization in 40 to 45% formamide, 1
M NaCI, 1%
SDS at 37 C, and a wash in <RTI 0.5X to 1X SSC at 55 to 60 C. Exemplary high
stringency
conditions include hybridization in 50% formamide, 1 M NaCI, 1% SDS at 37 C,
and a wash
in 0.1X SSC at 60 to 65 C. Specificity is typically the function of post-
hybridization washes,
the critical factors being the ionic strength and temperature of the final
wash solution. For
DNA/DNA hybrids, the Tm can be approximated from the equation of Meinkoth and
Wahl,
(1984) Anal. Biochem. 138:267-284: Tm = 81.5 C + 16.6 (log M) + 0.41(% GC) -
0.61 (%
form)-500/L; where M is the molarity of monovalent cations, % GC is the
percentage of
guanosine and cytosine nucleotides in the DNA, % form is the percentage of
formamide in
the hybridization solution, and L is the length of the hybrid in base pairs.
The Tm is the
temperature (under defined ionic strength and pH) at which 50% of a
complementary target
sequence hybridizes to a perfectly matched probe. Tm is reduced by about 1 C
for each 1%
of mismatching; thus, Tm, hybridization and/or wash conditions can be adjusted
to hybridize
to sequences of the desired identity. For example, if sequences with > 90%
identity are
sought, the Tm can be decreased10 C. Generally, stringent conditions are
selected to be
about 5 C lower than the thermal melting point (Tm) for the specific sequence
and its
complement at a defined ionic strength and pH. However, severely stringent
conditions can
utilize a hybridization and/or wash at 1, 2, 3 or 4 C lower than the thermal
melting point
(Tm); moderately stringent conditions can utilize a hybridization and/or wash
at 6, 7, 8, 9 or
10 C lower than the thermal melting point (Tm); low stringency conditions can
utilize a
hybridization and/or wash at 11, 12, 13, 14, 15 or 20 C lower than the thermal
melting point
(Tm). Using the equation, hybridization and wash compositions, and desired Tm,
those of
ordinary skill will understand that variations in the stringency of
hybridization and/or wash
solutions are inherently described. If the desired degree of mismatching
results in a Tm of
less than 45 C (aqueous solution) or 32 C (formamide solution) it is preferred
to increase the
SSC concentration so that a higher temperature can be used. An extensive guide
to the
hybridization of nucleic acids is found in Tijssen, Laboratory Techniques in
Biochemistry and
Molecular Biology--Hybridization with Nucleic Acid Probes, Part I, Chapter 2
"Overview of
principles of hybridization and the strategy of nucleic acid probe assays",
Elsevier, New York
(1993); and Current Protocols in Molecular Biology, Chapter 2, Ausubel, at
al., Eds., Greene
Publishing and Wiley-Interscience, New York (1995).
A "subject plant" or "subject plant cell" is one in which genetic alteration,
such as
transformation, has been effected as to a gene of interest, or is a plant or
plant cell which is
descended from a plant or cell so altered and which comprises the alteration.
A "control" or
13
CA 02877639 2015-01-09
"control plant" or "control plant cell" provides a reference point for
measuring changes in
phenotype of the subject plant or plant cell.
A control plant or plant cell may comprise, for example: (a) a wild-type plant
or cell,
i.e., of the same genotype as the starting material for the genetic alteration
which resulted in
the subject plant or cell; (b) a plant or plant cell of the same genotype as
the starting material
but which has been transformed with a null construct (i.e. with a construct
which has no
known effect on the trait of interest, such as a construct comprising a marker
gene); (c) a
plant or plant cell which is a non-transformed segregant among progeny of a
subject plant or
plant cell; (d) a plant or plant cell genetically identical to the subject
plant or plant cell but
which is not exposed to conditions or stimuli that would induce expression of
the gene of
interest; or (e) the subject plant or plant cell itself, under conditions in
which the gene of
interest is not expressed.
In the present case, for example, changes in the ethylene response, including
changes in amounts or timing of ethylene production, ethylene activity,
ethylene distribution,
ethylene signaling or ethylene recognition or changes in plant or plant cell
phenotype, such
as flowering time, seed set, branching, senescence, stress tolerance or root
mass, could be
measured by comparing a subject plant or plant cell to a control plant or
plant cell.
As used herein, "transgenic plant" includes reference to a plant which
comprises
within its genome a heterologous polynucleotide. Generally, the heterologous
polynucleotide is stably integrated within the genome such that the
polynucleotide is passed
on to successive generations. The heterologous polynucleotide may be
integrated into the
genome alone or as part of a recombinant expression cassette. "Transgenic" is
used herein
to include any cell, cell line, callus, tissue, plant part or plant, the
genotype of which has
been altered by the presence of heterologous nucleic acid including those
transgenics
initially so altered as well as those created by sexual crosses or asexual
propagation from
the initial transgenic. The term "transgenic" as used herein does not
encompass the
alteration of the genome (chromosomal or extra-chromosomal) by conventional
plant
breeding methods or by naturally occurring events such as random cross-
fertilization, non-
recombinant viral infection, non-recombinant bacterial transformation, non-
recombinant
transposition, or spontaneous mutation.
As used herein, "vector" includes reference to a nucleic acid used in
transfection of a
host cell and into which can be inserted a polynucleotide. Vectors are often
replicons.
Expression vectors permit transcription of a nucleic acid inserted therein.
The following terms are used to describe the sequence relationships between a
polynucleotide/polypeptide of the present invention with a reference
14
CA 02877639 2015-01-09
polynucleotide/polypeptide: (a) "reference sequence", (b) "comparison window",
(c)
"sequence identity" and (d) "percentage of sequence identity".
(a) As used herein, "reference sequence" is a defined sequence used as a
basis
for sequence comparison with a polynucleotide/polypeptide of the present
invention. A
reference sequence may be a subset or the entirety of a specified sequence;
for example, as
a segment of a full-length cDNA or gene sequence, or the complete cDNA or gene
sequence.
(b) As used herein, "comparison window" includes reference to a contiguous
and
specified segment of a polynucleotide/polypeptide sequence, wherein the
polynucleotide/polypeptide sequence may be compared to a reference sequence
and
wherein the portion of the polynucleotide/polypeptide sequence in the
comparison window
may comprise additions or deletions (i.e., gaps) compared to the reference
sequence (which
does not comprise additions or deletions) for optimal alignment of the two
sequences.
Generally, the comparison window is at least 20 contiguous nucleotides/amino
acids
residues in length, and optionally can be 30, 40, 50, 100 or longer. Those of
skill in the art
understand that to avoid a high similarity to a reference sequence due to
inclusion of gaps in
the polynucleotide/polypeptide sequence, a gap penalty is typically introduced
and is
subtracted from the number of matches.
Methods of alignment of sequences for comparison are well-known in the art.
Optimal alignment of sequences for comparison may be conducted by the local
homology
algorithm of Smith and Waterman, (1981) Adv. App!. Math. 2:482; by the
homology
alignment algorithm of Needleman and Wunsch, (1970) J. MoL Biol. 48:443; by
the search
for similarity method of Pearson and Lipman, (1988) Proc. Natl. Acad. Sci,
85:2444; by
computerized implementations of these algorithms, including, but not limited
to: CLUSTAL in
the PC/Gene program by Intelligenetics, Mountain View, California; GAP,
BESTFIT, BLAST,
FASTA, and TFASTA in the GCG Wisconsin Package , Version 10 (available from
Accelrys
Inc., 9685 Scranton Road, San Diego, California, USA). The CLUSTAL program is
well
described by Higgins and Sharp, (1988) Gene 73:237-244; Higgins and Sharp,
(1989)
CABIOS 5:151-153; Corpet, et al., (1988) Nucleic Acids Research 16:10881-90;
Huang, et
al., (1992) Computer Applications in the Biosciences 8:155-65, and Pearson, et
al., (1994)
Methods in Molecular Biology 24:307-331.
The BLAST family of programs which can be used for database similarity
searches
includes: BLASTN for nucleotide query sequences against nucleotide database
sequences;
BLASTX for nucleotide query sequences against protein database sequences;
BLASTP for
protein query sequences against protein database sequences; TBLASTN for
protein query
sequences against nucleotide database sequences; and TBLASTX for nucleotide
query
CA 02877639 2015-01-09
sequences against nucleotide database sequences. See, Current Protocols in
Molecular
Biology, Chapter 19, Ausubel, et al., Eds., Greene Publishing and Wiley-
lnterscience, New
York (1995); Altschul, et al., (1990) J. MoL Biol. 215:403-410 and, Altschul,
et al., (1997)
Nucleic Acids Res. 25:3389-3402.
Software for performing BLAST analyses is publicly available, e.g., through
the
National Center for Biotechnology Information, National Library of Medicine,
Building 38A,
Bethesda, Maryland, USA. This algorithm involves first identifying high
scoring sequence
pairs (HSPs) by identifying short words of length W in the query sequence,
which either
match or satisfy some positive-valued threshold score T when aligned with a
word of the
same length in a database sequence. T is referred to as the neighborhood word
score
threshold. These initial neighborhood word hits act as seeds for initiating
searches to find
longer HSPs containing them. The word hits are then extended in both
directions along
each sequence for as far as the cumulative alignment score can be increased.
Cumulative
scores are calculated using, for nucleotide sequences, the parameters M
(reward score for a
pair of matching residues; always >0) and N (penalty score for mismatching
residues;
always <0). For amino acid sequences, a scoring matrix is used to calculate
the cumulative
score.
Extension of the word hits in each direction are halted when: the cumulative
alignment score falls off by the quantity X from its maximum achieved value;
the cumulative
score goes to zero or below, due to the accumulation of one or more negative-
scoring
residue alignments; or the end of either sequence is reached. The BLAST
algorithm
parameters W, T and X determine the sensitivity and speed of the alignment.
The BLASTN
program (for nucleotide sequences) uses as defaults a word length (W) of 11,
an expectation
(E) of 10, a cutoff of 100, M=5, N=-4, and a comparison of both strands. For
amino acid
sequences, the BLASTP program uses as defaults a word length (W) of 3, an
expectation
(E) of 10, and the BLOSUM62 scoring matrix (see, Henikoff and Henikoff, (1989)
Proc. Natl.
Acad. Sci. USA 89:10915).
In addition to calculating percent sequence identity, the BLAST algorithm also
performs a statistical analysis of the similarity between two sequences (see,
e.g., Karlin and
Altschul, (1993) Proc. Nat'l. Acad. ScL USA 90:5873-5877). One measure of
similarity
provided by the BLAST algorithm is the smallest sum probability (P (N)), which
provides an
indication of the probability by which a match between two nucleotide or amino
acid
sequences would occur by chance. BLAST searches assume that proteins can be
modeled
as random sequences. However, many real proteins comprise regions of nonrandom
sequences which may be homopolymeric tracts, short-period repeats or regions
enriched in
16
CA 02877639 2015-01-09
one or more amino acids. Such low-complexity regions may be aligned between
unrelated
proteins even though other regions of the protein are entirely dissimilar. A
number of low-
complexity filter programs can be employed to reduce such low-complexity
alignments. For
example, the SEG (Wooten and Federhen, (1993) Comput Chem. 17:149-163) and XNU
(Claverie and States, (1993) Comput Chem. 17:191-201) low-complexity filters
can be
employed alone or in combination.
Unless otherwise stated, nucleotide and protein identity/similarity values
provided
herein are calculated using GAP (GCG Version 10) under default values. GAP
(Global
Alignment Program) can also be used to compare a polynucleotide or polypeptide
of the
present invention with a reference sequence. GAP uses the algorithm of
Needleman and
Wunsch (J. MoL Biol. 48:443-453 (1970)) to find the alignment of two complete
sequences
that maximizes the number of matches and minimizes the number of gaps. GAP
considers
all possible alignments and gap positions and creates the alignment with the
largest number
of matched bases and the fewest gaps. It allows for the provision of a gap
creation penalty
and a gap extension penalty in units of matched bases. GAP must make a profit
of gap
creation penalty number of matches for each gap it inserts. If a gap extension
penalty
greater than zero is chosen, GAP must, in addition, make a profit for each gap
inserted of
the length of the gap times the gap extension penalty. Default gap creation
penalty values
and gap extension penalty values in Version 10 of the Wisconsin Genetics
Software
Package for protein sequences are 8 and 2, respectively. For nucleotide
sequences the
default gap creation penalty is 50 while the default gap extension penalty is
3. The gap
creation and gap extension penalties can be expressed as an integer selected
from the
group of integers consisting of from 0 to 100. Thus, for example, the gap
creation and gap
extension penalties can each independently be: 3, 4, 5, 6, 7, 8, 9, 10, 15,
20, 30, 40, 50, 60
or greater.
GAP presents one member of the family of best alignments. There may be many
members of this family, but no other member has a better quality. GAP displays
four figures
of merit for alignments: Quality, Ratio, Identity and Similarity. The Quality
is the metric
maximized in order to align the sequences. Ratio is the quality divided by the
number of
bases in the shorter segment. Percent Identity is the percent of the symbols
that actually
match. Percent Similarity is the percent of the symbols that are similar.
Symbols that are
across from gaps are ignored. A similarity is scored when the scoring matrix
value for a pair
of symbols is greater than or equal to 0.50, the similarity threshold. The
scoring matrix used
in Version 10 of the Wisconsin Genetics Software Package is BLOSUM62 (see,
Henikoff
and Henikoff (1989) Proc. Natl. Acad. ScL USA 89:10915).
17
CA 02877639 2015-01-09
Multiple alignment of the sequences can be performed using the CLUSTAL method
of alignment (Higgins and Sharp (1989) CAB/OS 5:151-153) with the default
parameters
(GAP PENALTY=10, GAP LENGTH PENALTY=10). Default parameters for pairwise
alignments using the CLUSTAL method are KTUPLE 1, GAP PENALTY=3, WINDOW=5 and
DIAGONALS SAVED=5.
(c)
As used herein, "sequence identity" or "identity" in the context of two
nucleic
acid or polypeptide sequences includes reference to the residues in the two
sequences
which are the same when aligned for maximum correspondence over a specified
comparison window. When percentage of sequence identity is used in reference
to proteins
it is recognized that residue positions which are not identical often differ
by conservative
amino acid substitutions, where amino acid residues are substituted for other
amino acid
residues with similar chemical properties (e.g., charge or hydrophobicity) and
therefore do
not change the functional properties of the molecule.
Where sequences differ in
conservative substitutions, the percent sequence identity may be adjusted
upwards to
correct for the conservative nature of the substitution. Sequences which
differ by such
conservative substitutions are said to have "sequence similarity" or
"similarity". Means for
making this adjustment are well-known to those of skill in the art. Typically
this involves
scoring a conservative substitution as a partial rather than a full mismatch,
thereby
increasing the percentage sequence identity. Thus, for example, where an
identical amino
acid is given a score of 1 and a non-conservative substitution is given a
score of zero, a
conservative substitution is given a score between zero and 1. The scoring of
conservative
substitutions is calculated, e.g., according to the algorithm of Meyers and
Miller, (1988)
Computer Applic. Biol. ScL 4:11-17, e. <RTI g., as implemented in the program
PC/GENE
(Intelligenetics, Mountain View, California, USA).
(d) As used
herein, "percentage of sequence identity" means the value
determined by comparing two optimally aligned sequences over a comparison
window,
wherein the portion of the polynucleotide sequence in the comparison window
may comprise
additions or deletions (i.e., gaps) as compared to the reference sequence
(which does not
comprise additions or deletions) for optimal alignment of the two sequences.
The
percentage is calculated by determining the number of positions at which the
identical
nucleic acid base or amino acid residue occurs in both sequences to yield the
number of
matched positions, dividing the number of matched positions by the total
number of positions
in the window of comparison and multiplying the result by 100 to yield the
percentage of
sequence identity.
18
CA 02877639 2015-01-09
DESCRIPTION OF THE FIGURES
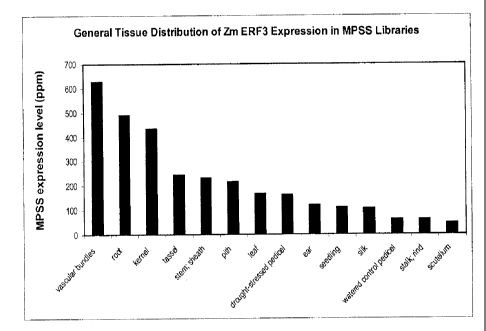
Figure 1 is a graph showing Tissue distribution of Zm ERF3 expression in MPSS
libraries.
Figure 2 is a graph showing the cold-induced time-course of Zm ERF3 expression
in
microarrays.
Figure 3 is a diagram showing the number of genes upregulated or down-
regulated
more than 5-fold both in E3 and E18 of transgenic maize expressing UBI:2mERF3.
DETAILED DESCRIPTION OF THE INVENTION
Overview
The present invention provides, among other things, compositions and methods
for
modulating (i.e., increasing or decreasing) the level of polynucleotides and
polypeptides of
the present invention in plants. In particular, the polynucleotides and
polypeptides of the
present invention can be expressed temporally or spatially, e.g., at
developmental stages, in
tissues, and/or in quantities, which are uncharacteristic of non-recombinantly
engineered
plants. Thus, the present invention provides utility in such exemplary
applications as
provided below.
EIN3 is a nuclear transcription factor that binds to ethylene response
elements
present in the promoters of ethylene response factors, such as ERF1 or ERF3.
Thus down
regulation of EIN3 will reduce ethylene sensitivity.
EBF1 and EBF2 are F-box proteins that bind to EIN3 and target the EIN3 protein
for
degradation through the ubiquitin/proteasome pathway. Thus overexpression of
EBF1 and
EBF2 will decrease ethylene sensitivity.
EIN3 protein levels rapidly increase in response to ethylene and this response
requires several ethylene-signaling pathway components including the ethylene
receptors
(ETR1 and EIN4), CTR1, EIN2, EIN5 and EIN6. In the absence of ethylene, EIN3
is quickly
degraded through a ubiquitin/proteasome pathway mediated by EBF1 and EBF2.
EIN5 has endonuclease activity on EBF1 and EBF2 transcripts and thus
antagonizes
the negative feedback on EIN3 by promoting EBF1 and EBF2 mRNA decay. Thus down
regulation of EIN5 will reduce ethylene sensitivity.
Changes in the ethylene response may include one or more changes in amount or
timing of ethylene production, ethylene activity, ethylene distribution,
ethylene signaling and
ethylene recognition, which may result in phenotypic changes such as, for
example, altered
flowering time, seed set, branching, senescence, stress tolerance, or root
formation,
19
CA 02877639 2015-01-09
In certain embodiments the nucleic acid constructs of the present invention
can be
used in combination ("stacked") with other polynucleotide sequences of
interest in order to
create plants with a desired phenotype. The polynucleotides of the present
invention may
be stacked with any gene or combination of genes, and the combinations
generated can
include multiple copies of any one or more of the polynucleotides of interest.
The desired
combination may affect one or more traits; that is, certain combinations may
be created for
modulation of gene expression affecting ethylene response. Other combinations
may be
designed to produce plants with a variety of desired traits, such as those
described
elsewhere herein.
Crowding Tolerance
The agronomic performance of crop plants is often a function of how well they
tolerate planting density. The stress of overcrowding can be due to simple
limitations of
nutrients, water, and sunlight. Crowding stress may also be due to enhanced
contact
between plants. Plants often respond to physical contact by slowing growth and
thickening
their tissues.
Ethylene has been implicated in plant crowding tolerance. For example,
ethylene
insensitive tobacco plants did not slow growth when contacting neighboring
plants (Knoester,
et al., (1998) PNAS USA 95:1933-1937). There is also evidence that ethylene,
and the
plant's response to it, is involved in water deficit stress, and that ethylene
may be causing
changes in the plant that limit its growth and aggravate the symptoms of
drought stress
beyond the loss of water itself.
The present invention provides for decreasing ethylene sensitivity in a plant,
in
particular cereals such as maize, by modulating the expression or activity of
one or more of
EIN3, ERF3, EBF1, EBF2 or EIN5 genes or gene products to promote tolerance of
close
spacing with reduced stress and minimized yield loss.
Seed Set and Development in Maize
Ethylene plays a number of roles in seed development. For example, in maize
ethylene is linked to programmed cell death of developing endosperm cells
(Young, at al.,
(1997) Plant Physio. 115:737-751). In addition, ethylene is linked to kernel
abortion, such as
occurs at the tips of ears, especially in plants grown under stressful
conditions (Cheng and
Lur, (1996) Physiol. Plant 98:245-252). Reduced kernel seed set is of course a
contributor
to reduced yields. Consequently, the present invention provides plants, in
particular maize
CA 02877639 2015-01-09
plants, that have reduced ethylene action by altering expression of genes
involved in the
ethylene response.
Growth in Compacted Soils
Plant growth is affected by the density and compaction of soils. Denser, more
compacted soils typically result in poorer plant growth. The trend in
agriculture towards
more minimal till planting and cultivation practices, with the goal of soil
and energy
conservation, is increasing the need for crop plants that can perform well
under these
conditions.
Ethylene is well-known to affect plant growth and development, and one effect
of
ethylene is to promote tissue thickening and growth retardation when
encountering
mechanical stress, such as compacted soils. This can affect both the roots and
shoots.
This effect is presumably adaptive in some circumstances in that it results in
stronger, more
compact tissues that can force their way through or around, obstacles such as
compacted
soils. However, in such conditions, the production of ethylene and the
activation of the
ethylene pathway may exceed what is needed for adaptive accommodation to the
mechanical stress of the compacted soils. Any resulting unnecessary growth
inhibition
would be an undesired agronomic result.
The present invention provides for decreasing ethylene sensitivity in a plant,
in
particular cereals such as maize, by modulating the expression or activity of
one or more of
EIN3, ERF3, EBF1, EBF2 or EIN5 genes or gene products. Such modulated plants
grow
and germinate better in compacted soils, resulting in higher stand counts, the
herald of
higher yields.
Flooding Tolerance
Flooding and water-logged soils causes substantial losses in crop yield each
year
around the world. Flooding can be both widespread or local, transitory or
prolonged.
Ethylene has been implicated in flooding mediated damage. In fact, in flooded
conditions
ethylene production can rise. There are two main reasons for this rise: 1)
under such
flooded conditions, which creates hypoxia, plants produce more ethylene, and
2) under
flooded conditions the diffusion of ethylene away from the plant is slowed,
because ethylene
is minimally soluble in water, resulting in a rise of intra-plant ethylene
levels.
In rice, submergence tolerance is known to be imparted through ethylene
signaling
(Perata and Voesenek, (2007) Trends Plant Sci 12(2):43-46).
21
CA 02877639 2015-01-09
Ethylene in flooded maize roots can also inhibit gravitropism, which is
normally
adaptive during germination in that it orients the roots down and the shoots
up. Gravitropism
is a factor in determining root architecture, which in turn plays an important
role in soil
resource acquisition. Manipulation of ethylene levels could be used to impact
root angle for
drought tolerance, flood tolerance, greater standability, and/or improved
nutrient uptake.
For example, a root growing at a more erect angle (steeper) would likely grow
more deeply
in soil and thus obtain water at greater depths, improving drought tolerance.
In the absence
of drought stress a converse argument could be made for more efficient root
uptake of
nutrients and water in the upper layers of the soil profile, by roots which
are more parallel to
the soil surface. In general, roots that have a angle nearer that of vertical
(steep) are also
more susceptible to root lodging than roots with a shallow angle (parallel to
the surface) that
can be more root lodging resistant.
In addition to inhibition of gravitropism, it is likely that ethylene
evolution in flooded
conditions inhibits growth, especially of roots. Such inhibition will likely
contribute to poor
plant growth overall, and consequently is a disadvantageous agronomic trait.
The present invention provides for decreasing ethylene sensitivity in a plant,
in
particular cereals such as maize, by modulating the expression or activity of
one or more of
EIN3, ERF3, EBF1, EBF2 or EIN5 genes or gene products. Such plants should grow
and
germinate better in flooded conditions or water-logged soils, resulting in
higher stand counts.
Plant Maturation and Senescence
Ethylene is known to be involved in controlling senescence, fruit ripening,
and
abscission. The role of ethylene in fruit ripening is well-established and
industrially applied.
It is expected that ethylene underproduction/insensitivity would result in
slower seed
maturation or fruit ripening, and the converse would result in more rapid seed
maturation or
fruit ripening. Abscission is primarily studied for dicot plants and
apparently has little
application to monocots such as cereals. Ethylene mediated senescence also is
mostly
studied in dicots, but control of senescence is agronomically important for
both dicot and
monocot crop species, Ethylene insensitivity can delay, but not arrest,
senescence. The
senescence process mediated by ethylene bears some similarities to the cell
death process
in disease symptoms and in abscission zones. Controlling ethylene sensitivity,
as through
the control of one or more of the ERF3, EIN3, EBF1, EBF2, EIN5 genes could
result in
modulation of maturity rates for crop plants such as maize.
The present invention provides for decreasing ethylene sensitivity in a plant,
in
particular cereals such as maize, by modulating the expression or activity of
one or more of
22
CA 02877639 2015-01-09
EIN3, ERF3, EBF1, EBF2 or EIN5 genes which may contribute to a later maturing
plant,
which is desirable for placing crop varieties in different maturity zones.
Tolerance to Other Abiotic Stresses
Many stresses on plants induce production of ethylene (see, Morgan and Drew,
(1997) Physiol. Plant 100:620-630). These stresses can be, for example, cold,
heat,
wounding, pollution, drought, and hypersalinity. Mechanical impedance (soil
compaction)
and flooding stresses were addressed above. It appears that several of these
stresses
operate through common mechanisms, such as water deficit. Clearly drought
causes water
deficit, crowding stress may also cause water deficit. Additionally, in maize,
chilling can
cause an elevation in ethylene production and activity, and this induction is
apparently due to
chilling causing water deficit in cells (Janowaik and Dorffling, (1995) J.
Plant PhysioL
147:257-262).
Some of the ethylene production following stresses may serve an adaptive
purpose
by regulating ethylene-mediated processes in the plant that result in a plant
reorganized in
such manner to better acclimate to the stress encountered. However, there is
also evidence
that ethylene production during stress can result in an aggravation of
negative symptoms
resulting from the stress, such as yellowing, tissue death, and senescence.
To the extent that ethylene production during stress causes or augments
negative
stress-related symptoms, it would be desirable to create a crop plant that is
less sensitive to
the ethylene. Towards that end, the present invention provides for decreasing
ethylene
sensitivity in a plant, in particular cereals such as maize, by modulating the
expression or
activity of one or more of EIN3, ERF3, EBF1, EBF2 or EIN5 genes or gene
products to
create plants that are less able to produce ethylene mediated effects.
Disease Resistance
Crop plants can be susceptible to a wide variety of pathogens, whether
viruses,
bacteria, fungi or insects. This susceptibility results in large crop yield
losses annually
worldwide. Crop breeders have endeavored to breed more resistant or tolerant
varieties
which can withstand pathogen attack. Additional genetic engineering strategies
seek the
same end. In many plant-pathogen interactions the symptoms of disease, most
often tissue
necrosis and resulting poor plant growth, is known to be the result of an
active plant defense
response to the pathogen. That is, the symptoms are caused directly by the
plant and not
simply by the pathogen. From among the list of all crop plants and their
potential list of
pathogens, resistance is the rule, and susceptibility the exception.
Susceptible interactions
23
CA 02877639 2015-01-09
are often thought to result from an improper or insufficient activation
defense by the plant
that results in increased symptom development and an inability to contain the
pathogen.
Ethylene is known to be associated with plant pathogen defense systems. Many
pathogenesis related genes are induced in expression at the level of mRNA by
ethylene.
The trend in our understanding of the role of ethylene in plant pathogen
defense is towards
ethylene and ethylene mediated effects being viewed as principally part of the
downstream
reactions to pathogen attack, as in symptom development. Ethylene seems to be
involved in
the plant's response to the stress of pathogen attack and in tissue damage
inflicted by the
pathogen. In a susceptible interaction ethylene may actually promote tissue
damage.
Consequently in such situations blocking ethylene production or action may
actually result in
less tissue damage, that is, more apparent resistance, even though the
pathogen is
compatible with the plant. Blocking ethylene action is known to either result
in more
susceptibility (e.g., Knoester, et al., (1988)) or more resistance (e.g.,
Lund, et al., (1998)
Plant Cell 10:371-382), which indicates that the role of ethylene action is
complex, as is to
be expected, for it depends upon the interactions of diverse plants and
pathogens.
The present invention provides for the use of one or more of EIN3, ERF3, EBF1,
EBF2 or EIN5 genes to effect enhanced resistance to plant pathogens, in
particular for
monocots such as maize.
For most applications this will involve the reduction in ethylene signaling by
modulating the expression or activity of EIN3, ERF3, EBF1, EBF2 or EIN5 genes
or gene
products, with the goal of causing plants that responds less to ethylene and
thereby plants
that are less prone to tissue damage following pathogen infection.
It is recognized that for some pathogens, ethylene signaling may be necessary
for
achieving substantial resistance. This can be handled by linking a functional
ethylene
sensitivity gene to a pathogen-inducible promoter, in particular to a promoter
whose
induction is preferentially responsive to the pathogen or pathogens for which
plant ethylene
signaling is desired for achievement of active resistance.
Plant Transformation
The generation of transgenic plants is central to crop plant genetic
engineering
strategies. Transgenesis typically involves the introduction of exogenous DNA
into the
plants cells via a variety of methods, such as particle bombardment or
agrobacterium
infection, which is usually followed by tissue culture and plant regeneration.
Transgenic
plant production remains a costly and rate limiting step in genetic
engineering, especially for
many of the most economically important crop plants, such as the cereals, like
maize.
24
CA 02877639 2015-01-09
Improving the efficiency of this process is therefore of great importance.
It has been accepted for a long time that ethylene action has negative
consequences
for plant transformation. As a result various approaches to bind, trap, or
otherwise block the
accumulation of ethylene are employed in transformation and tissue culture
(see, Songstad,
et al., (1991) Plant Cell Reports 9:694-702). The particle bombardment method
causes
substantial tissue/cell damage, and such damage is known to elicit ethylene
accumulation.
Moreover, in most tissue culture methods, some tissue grows better than
others, as is
designed in chemical selection of transformants. Such dying tissue can emit
ethylene and
cause inhibition of positive transformants. Aggravating these effects is the
confinement of
plant tissues in containers for the purpose of tissue regeneration, that can
result in the
accumulation of ethylene, also causing growth retardation. As ethylene is
known to reduce
tissue growth rates and even advance cell/tissue death, having a means to
block or minimize
ethylene action during transformation is desired.
Consequently, the present invention also provides for the use of an EIN3,
ERF3,
EBF1, EBF2 or EIN5 gene to create transient or stable reductions in ethylene
action by
diminishing the expression and/or activity of one or more of the EIN3, ERF3,
EBF1, EBF2 or
EIN5 genes.
Other Utilities
The present invention also provides isolated nucleic acids comprising
polynucleotides of sufficient length and complementarity to a gene of the
present invention to
use as probes or amplification primers in the detection, quantitation, or
isolation of gene
transcripts. For example, isolated nucleic acids of the present invention can
be used as
probes in detecting deficiencies in the level of mRNA in screenings for
desired transgenic
plants, for detecting mutations in the gene (e.g., substitutions, deletions,
or additions), for
monitoring upregulation of expression or changes in enzyme activity in
screening assays of
compounds, for detection of any number of allelic variants (polymorphisms),
orthologs, or
paralogs of the gene, or for site directed mutagenesis in eukaryotic cells
(see, e.g., US
Patent Number 5,565,350). The isolated nucleic acids of the present invention
can also be
used for recombinant expression of their encoded polypeptides, or for use as
immunogens in
the preparation and/or screening of antibodies. The isolated nucleic acids of
the present
invention can also be employed for use in sense or antisense suppression of
one or more
genes of the present invention in a host cell, tissue, or plant. Attachment of
chemical agents
which bind, intercalate, cleave and/or cross-link to the isolated nucleic
acids of the present
invention can also be used to modulate transcription or translation.
CA 02877639 2015-01-09
The present invention also provides isolated proteins comprising a polypeptide
of the
present invention (e.g., preproenzyme, proenzyme, or enzymes). The present
invention also
provides proteins comprising at least one epitope from a polypeptide of the
present invention.
The proteins of the present invention can be employed in assays for enzyme
agonists or
antagonists of enzyme function, or for use as immunogens or antigens to obtain
antibodies
specifically immunoreactive with a protein of the present invention. Such
antibodies can be
used in assays for expression levels, for identifying and/or isolating nucleic
acids of the
present invention from expression libraries, for identification of homologous
polypeptides
from other species, or for purification of polypeptides of the present
invention.
The isolated nucleic acids and polypeptides of the present invention can be
used
over a broad range of plant types, particularly monocots such as the species
of the family
Gramineae including Hordeum, Secale, Tritium, Sorghum (e.g., S. bicolor) and
Zea (e.g., Z.
mays). The isolated nucleic acid and proteins of the present invention can
also be used in
species from the genera: Cucurbita, Rosa, Vitis, Juglans, Fragaria, Lotus,
Medicago,
Onobrychis, Trifolium, Trigonella, Vigna, Citrus, Linum, Geranium, Manihot,
Daucus,
Arabidopsis, Brassica, Raphanus, Sinapis, Atropa, Capsicum, Datura,
Hyoscyamus,
Lycopersicon, Nicotiana, Solanum, Petunia, Digitalis, Majorana, Ciahorium,
Helianthus,
Lactuca, Bromus, Asparagus, Antirrhinum, Heterocallis, Nemesis, Pelargonium,
Panieum,
Pennisetum, Ranunculus, Senecio, Salpiglossis, Cucumis, Browallia, Glycine,
Pisum,
Phaseolus, Lolium, Oryza and Avena.
Nucleic Acids
The present invention provides, among other things, isolated nucleic acids of
RNA,
DNA, and analogs and/or chimeras thereof, comprising a polynucleotide of the
present
invention.
A polynucleotide of the present invention is inclusive of:
(a) a polynucleotide encoding a polypeptide of SEQ ID NOS: 2, 4, 6, 8 or
10, and
conservatively modified and polymorphic variants thereof, including
exemplary polynucleotides of SEQ ID NOS: 1 (EIN3), 3 (EBF1), 5 (EBF2), 7
(EIN5) or 9 (ERF3);
(b) an isolated polynucleotide which is the product of amplification from a
plant
nucleic acid library using primer pairs which selectively hybridize under
stringent conditions to loci within a polynucleotide of the present invention;
(c) an isolated polynucleotide which selectively hybridizes to a
polynucleotide of
(a) or (b);
26
CA 02877639 2015-01-09
(d) an isolated polynucleotide having a specified sequence identity with
polynucleotides of (a), (b) or (c);
(e) an isolated polynucleotide encoding a protein having a specified number
of
contiguous amino acids from a prototype polypeptide, wherein the protein is
specifically recognized by antisera elicited by presentation of the protein
and
wherein the protein does not detectably immunoreact to antisera which has
been fully immunosorbed with the protein;
(f) complementary sequences of polynucleotides of (a), (b), (c), (d) or
(e); and
(g) an isolated polynucleotide comprising at least a specific number of
contiguous
Ici nucleotides from a polynucleotide of (a), (b), (c), (d), (e) or
(f);
(h) an isolated polynucleotide from a full-length enriched cDNA library
having the
physico-chemical property of selectively hybridizing to a polynucleotide of
(a),
(b), (c),(d), (e), (f) or (g);
(i) an isolated polynucleotide made by the process of: 1) providing a full-
length
enriched nucleic acid library, 2) selectively hybridizing the polynucleotide
to a
polynucleotide of (a), (b), (c), (d),(e), (f), (g) or (h), thereby isolating
the
polynucleotide from the nucleic acid library.
A. Polynucleotides Encoding A Polypeptide of the Present Invention
As indicated in (a), above, the present invention provides isolated nucleic
acids
comprising a polynucleotide of the present invention, wherein the
polynucleotide encodes a
polypeptide of the present invention. Every nucleic acid sequence herein that
encodes a
polypeptide also, by reference to the genetic code, describes every possible
silent variation
of the nucleic acid. One of ordinary skill will recognize that each codon in a
nucleic acid
(except AUG, which is ordinarily the only codon for methionine; and UGG, which
is ordinarily
the only codon for tryptophan) can be modified to yield a functionally
identical molecule.
Thus, each silent variation of a nucleic acid which encodes a polypeptide of
the present
invention is implicit in each described polypeptide sequence and is within the
scope of the
present invention. Accordingly, the present invention includes polynucleotides
of the present
invention and polynucleotides encoding a polypeptide of the present invention.
B. Polynucleotides Amplified from a Plant Nucleic Acid Library
As indicated in (b), above, the present invention provides an isolated nucleic
acid
comprising a polynucleotide of the present invention, wherein the
polynucleotides are
amplified, under nucleic acid amplification conditions, from a plant nucleic
acid library.
27
CA 02877639 2015-01-09
Nucleic acid amplification conditions for each of the variety of amplification
methods
are well known to those of ordinary skill in the art. The plant nucleic acid
library can be
constructed from a monocot such as a cereal crop. Exemplary cereals include
corn,
sorghum, alfalfa, canola, wheat or rice. The plant nucleic acid library can
also be
constructed from a dicot such as soybean. Zea mays lines B73, PHRE1, A632,
BMP2#10,
W23 and Mo17 are known and publicly available. Other publicly known and
available maize
lines can be obtained from the Maize Genetics Cooperation (Urbana, IL).
Wheat lines are available from the Wheat Genetics Resource Center (Manhattan,
KS). The nucleic acid library may be a cDNA library, a genomic library, or a
library generally
constructed from nuclear transcripts at any stage of intron processing. cDNA
libraries can
be normalized to increase the representation of relatively rare cDNAs.
In optional
embodiments, the cDNA library is constructed using an enriched full-length
cDNA synthesis
method. Examples of such methods include Oligo-Capping (Maruyama and Sugano,
(1994)
Gene 138:171-174), Biotinylated CAP Trapper (Carninci, et al., (1996) Genomics
37:327-
336) and CAP Retention Procedure (Edery, et al., (1995) Molecular and Cellular
Biology
15:3363-3371). Rapidly growing tissues or rapidly dividing cells are preferred
for use as an
mRNA source for construction of a cDNA library. Growth stages of corn are
described in
"How a Corn Plant Develops, "Special Report No. 48, Iowa State University of
Science and
Technology Cooperative Extension Service, Ames, Iowa, Reprinted February 1993.
A polynucleotide of this embodiment (or subsequences thereof) can be obtained,
for
example, by using amplification primers which are selectively hybridized and
primer
extended, under nucleic acid amplification conditions, to at least two sites
within a
polynucleotide of the present invention, or to two sites within the nucleic
acid which flank and
comprise a polynucleotide of the present invention, or to a site within a
polynucleotide of the
present invention and a site within the nucleic acid which comprises it.
Methods for
obtaining 5'and/or 3'ends of a vector insert are well known in the art. See,
e.g., RACE
(Rapid Amplification of Complementary Ends) as described in Frohman, in PCR
Protocols: A
Guide to Methods and Applications, Innis, et a/., Eds. (Academic Press, Inc.,
San Diego), pp.
28-38 (1990)); see also, US Patent Number 5,470,722, and Current Protocols in
Molecular
Biology, Unit 15.6, Ausubel, et al., Eds., Greene Publishing and Wiley-
Interscience, New
York (1995); Frohman and Martin, (1989) Techniques 1:165.
Optionally, the primers are complementary to a subsequence of the target
nucleic
acid which they amplify but may have a sequence identity ranging from about
85% to 99%
relative to the polynucleotide sequence which they are designed to anneal to.
As those
skilled in the art will appreciate, the sites to which the primer pairs will
selectively hybridize
28
CA 02877639 2015-01-09
are chosen such that a single contiguous nucleic acid can be formed under the
desired
nucleic acid amplification conditions. The primer length in nucleotides is
selected from the
group of integers consisting of from at least 15 to 50. Thus, the primers can
be at least 15,
18, 20, 25, 30, 40 or 50 nucleotides in length. Those of skill will recognize
that a lengthened
primer sequence can be employed to increase specificity of binding (i.e.,
annealing) to a
target sequence. A non-annealing sequence at the 5' end of a primer (a "tail")
can be
added, for example, to introduce a cloning site at the terminal ends of the
amplicon.
The amplification products can be translated using expression systems well
known to
those of skill in the art. The resulting translation products can be confirmed
as polypeptides
of the present invention by, for example, assaying for the appropriate
catalytic activity (e.g.,
specific activity and/or substrate specificity), or verifying the presence of
one or more
epitopes which are specific to a polypeptide of the present invention. Methods
for protein
synthesis from PCR derived templates are known in the art and available
commercially.
See, e.g., Amersham Life Sciences, Inc, Catalog '97, p. 354.
C. Polvnucleotides Which Selectively Hybridize to a PoIN/nucleotide of
(A) or (B)
As indicated in (c), above, the present invention provides isolated nucleic
acids
comprising polynucleotides of the present invention, wherein the
polynucleotides selectively
hybridize, under selective hybridization conditions, to a polynucleotide of
sections (A) or (B)
as discussed above. Thus, the polynucleotides of this embodiment can be used
for
isolating, detecting, and/or quantifying nucleic acids comprising the
polynucleotides of (A) or
(B). For example, polynucleotides of the present invention can be used to
identify, isolate or
amplify partial or full-length clones in a deposited library.
In some embodiments, the polynucleotides are genomic or cDNA sequences
isolated
or otherwise complementary to a cDNA from a dicot or monocot nucleic acid
library.
Exemplary species of monocots and dicots include, but are not limited to:
maize,
canola, soybean, cotton, wheat, sorghum, sunflower, alfalfa, oats, sugar cane,
millet, barley
and rice. The cDNA library comprises at least 50% to 95% full-length sequences
(for
example, at least 50%, 60%, 70%, 80% 90% or 95% full-length sequences). The
cDNA
libraries can be normalized to increase the representation of rare sequences.
See, e.g., US
Patent Number 5,482,845. Low stringency hybridization conditions are
typically, but not
exclusively, employed with sequences having a reduced sequence identity
relative to
complementary sequences. Moderate and high stringency conditions can
optionally be
employed for sequences of greater identity. Low stringency conditions allow
selective
29
CA 02877639 2015-01-09
hybridization of sequences having about 70% to 80% sequence identity and can
be
employed to identify orthologous or paralogous sequences.
D.
Polynucleotides Having a Specific Sequence Identity with the Polynucleotides
of (A),
(B) or (C)
As indicated in (d), above, the present invention provides isolated nucleic
acids
comprising polynucleotides of the present invention, wherein the
polynucleotides have a
specified identity at the nucleotide level to a polynucleotide as disclosed
above in sections
(A), (B) or (C), above.
Identity can be calculated using, for example, the BLAST,
CLUSTALW or GAP algorithms under default conditions. The percentage of
identity to a
reference sequence is at least 60% and, rounded upwards to the nearest
integer, can be
expressed as an integer selected from the group of integers consisting of from
60 to 99.
Thus, for example, the percentage of identity to a reference sequence can be
at least 70%,
75%, 80%, 85%, 90% or 95%.
Optionally, the polynucleotides of this embodiment will encode a polypeptide
that will
share an epitope with a polypeptide encoded by the polynucleotides of sections
(A), (B) or
(C). Thus, these polynucleotides encode a first polypeptide which elicits
production of
antisera comprising antibodies which are specifically reactive to a second
polypeptide
encoded by a polynucleotide of (A), (B) or (C). However, the first polypeptide
does not bind
to antisera raised against itself when the antisera has been fully
immunosorbed with the first
polypeptide. Hence, the polynucleotides of this embodiment can be used to
generate
antibodies for use in, for example, the screening of expression libraries for
nucleic acids
comprising polynucleotides of (A), (B) or (C), or for purification of, or in
immunoassays for,
polypeptides encoded by the polynucleotides of (A), (B) or (C). The
polynucleotides of this
embodiment comprise nucleic acid sequences which can be employed for selective
hybridization to a polynucleotide encoding a polypeptide of the present
invention.
Screening polypeptides for specific binding to antisera can be conveniently
achieved
using peptide display libraries. This method involves the screening of large
collections of
peptides for individual members having the desired function or structure.
Antibody screening of peptide display libraries is well known in the art. The
displayed
peptide sequences can be from 3 to 5000 or more amino acids in length,
frequently from
5100 amino acids long, and often from about 8 to 15 amino acids long. In
addition to direct
chemical synthetic methods for generating peptide libraries, several
recombinant DNA
methods have been described. One type involves the display of a peptide
sequence on the
surface of a bacteriophage or cell. Each bacteriophage or cell contains the
nucleotide
CA 02877639 2015-01-09
sequence encoding the particular displayed peptide sequence. Such methods are
described
in PCT Patent Publication Numbers 91/17271, 91/18980, 91/19818 and 93/08278.
Other
systems for generating libraries of peptides have aspects of both in vitro
chemical synthesis
and recombinant methods. See, PCT Patent Publication Numbers 92/05258,
92/14843 and
97/20078. See also, US Patent Numbers 5,658,754 and 5,643,768. Peptide display
libraries, vectors, and screening kits are commercially available from such
suppliers as
Invitrogen (Carlsbad, CA).
E.
Polynucleotides Encoding a Protein Having a Subsequence from a Prototype
Polypeptide and Cross-Reactive to the Prototype Polypeptide
As indicated in (e), above, the present invention provides isolated nucleic
acids
comprising polynucleotides of the present invention, wherein the
polynucleotides encode a
protein having a subsequence of contiguous amino acids from a prototype
polypeptide of the
present invention such as are provided in (a), above. The length of contiguous
amino acids
from the prototype polypeptide is selected from the group of integers
consisting of from at
least 10 to the number of amino acids within the prototype sequence. Thus, for
example, the
polynucleotide can encode a polypeptide having a subsequence having at least
10, 15, 20,
25, 30, 35, 40, 45 or 50, contiguous amino acids from the prototype
polypeptide. Further,
the number of such subsequences encoded by a polynucleotide of the instant
embodiment
can be any integer selected from the group consisting of from 1 to 20, such as
2, 3, 4 or 5.
The subsequences can be separated by any integer of nucleotides from 1 to the
number of
nucleotides in the sequence such as at least 5, 10, 15, 25, 50, 100 or 200
nucleotides.
The proteins encoded by polynucleotides of this embodiment, when presented as
an
immunogen, elicit the production of polyclonal antibodies which specifically
bind to a
prototype polypeptide such as but not limited to, a polypeptide encoded by the
polynucleotide of (a) or (b), above.
Generally, however, a protein encoded by a
polynucleotide of this embodiment does not bind to antisera raised against the
prototype
polypeptide when the antisera has been fully immunosorbed with the prototype
polypeptide.
Methods of making and assaying for antibody binding specificity/affinity are
well known in the
art. Exemplary immunoassay formats include ELISA, competitive immunoassays,
radioimmunoassays, Western blots, indirect immunofluorescent assays and the
like.
In a preferred assay method, fully immunosorbed and pooled antisera which is
elicited to the prototype polypeptide can be used in a competitive binding
assay to test the
protein. The concentration of the prototype polypeptide required to inhibit
50% of the binding
of the antisera to the prototype polypeptide is determined. If the amount of
the protein
31
CA 02877639 2015-01-09
required to inhibit binding is less than twice the amount of the prototype
protein, then the
protein is said to specifically bind to the antisera elicited to the
immunogen.
Accordingly, the proteins of the present invention embrace allelic variants,
conservatively modified variants, and minor recombinant modifications to a
prototype
polypeptide.
A polynucleotide of the present invention optionally encodes a protein having
a
molecular weight as the non-glycosylated protein within 20% of the molecular
weight of the
full-length non-glycosylated polypeptides of the present invention. Molecular
weight can be
readily determined by SDS-PAGE under reducing conditions. Optionally, the
molecular
weight is within 15% of a full length polypeptide of the present invention,
more preferably
within 10% or 5%, and most preferably within 3%, 2% or 1% of a full length
polypeptide of
the present invention. Optionally, the polynucleotides of this embodiment will
encode a
protein having a specific enzymatic activity at least 50%, 60%, 80% or 90% of
a cellular
extract comprising the native, endogenous full-length polypeptide of the
present invention.
Further, the proteins encoded by polynucleotides of this embodiment will
optionally
have a substantially similar affinity constant (Km) and/or catalytic activity
(i.e., the
microscopic rate constant, kcat) as the native endogenous, full-length
protein. Those of skill
in the art will recognize that kcat/Km value determines the specificity for
competing
substrates and is often referred to as the specificity constant. Proteins of
this embodiment
can have akcat/Km value at least 10% of a full-length polypeptide of the
present invention as
determined using the endogenous substrate of that polypeptide. Optionally, the
kcat/Km
value will be at least 20%, 30%, 40%, 50% and most preferably at least 60%,
70%, 80%,
90% or 95% the kcat/Km value of the full-length polypeptide of the present
invention.
Determination of kcat, Km, and kcat/Km can be determined by any number of
means
well known to those of skill in the art. For example, the initial rates (i.e.,
the first 5% or less
of the reaction) can be determined using rapid mixing and sampling techniques
(e.g.,
continuous-flow, stopped-flow or rapid quenching techniques), flash photolysis
or relaxation
methods (e.g., temperature jumps) in conjunction with such exemplary methods
of
measuring as spectrophotometry, spectrofluorimetry, nuclear magnetic resonance
or
radioactive procedures. Kinetic values are conveniently obtained using a
Lineweaver Burk
or Eadie-Hofstee plot.
F. Polynucleotides Complementary to the Polynucleotides of (A)-(E)
As indicated in (f), above, the present invention provides isolated nucleic
acids
comprising polynucleotides complementary to the polynucleotides of paragraphs
A-E, above.
32
CA 02877639 2015-01-09
As those of skill in the art will recognize, complementary sequences base-pair
throughout
the entirety of their length with the polynucleotides of sections (A)-(E)
(i.e., have 100%
sequence identity over their entire length). Complementary bases associate
through
hydrogen bonding in double stranded nucleic acids. For example, the following
base pairs
are complementary: guanine and cytosine; adenine and thymine; and adenine and
uracil.
G. Polynucleotides Which are Subsequences of the Polynucleotides of
(A)-(F)
As indicated in (g), above, the present invention provides isolated nucleic
acids
comprising polynucleotides which comprise at least 15 contiguous bases from
the
polynucleotides of sections (A) through (F) as discussed above. The length of
the
polynucleotide is given as an integer selected from the group consisting of
from at least 15 to
the length of the nucleic acid sequence from which the polynucleotide is a
subsequence of.
Thus, for example, polynucleotides of the present invention are inclusive of
polynucleotides
comprising at least 15, 20, 25, 30, 40, 50, 60, 75 or 100 contiguous
nucleotides in length
from the polynucleotides of(A)-(F). Optionally, the number of such
subsequences encoded
by a polynucleotide of the instant embodiment can be any integer selected from
the group
consisting of from 1 to 20, such as 2, 3, 4 or 5. The subsequences can be
separated by any
integer of nucleotides from 1 to the number of nucleotides in the sequence
such as at least
5, 10, 15, 25, 50, 100 or 200 nucleotides.
Subsequences can be made by in vitro synthetic, in vitro biosynthetic or in
vivo
recombinant methods. In optional embodiments, subsequences can be made by
nucleic
acid amplification. For example, nucleic acid primers will be constructed to
selectively
hybridize to a sequence (or its complement) within, or co-extensive with, the
coding region.
The subsequences of the present invention can comprise structura libraries are
known in the art and discussed briefly below. The cDNA library comprises at
least 50% to
95% full-length sequences (for example, at least 50%, 60%, 70%, 80%, 90% or
95% full-
length sequences). The cDNA library can be constructed from a variety of
tissues from a
monocot or dicot at a variety of developmental stages. Exemplary species
include maize,
wheat, rice, canola, soybean, cotton, sorghum, sunflower, alfalfa, oats, sugar
cane, millet,
barley and rice. Methods of selectively hybridizing, under selective
hybridization conditions,
a polynucleotide from a full-length enriched library to a polynucleotide of
the present
invention are known to those of ordinary skill in the art. Any number of
stringency conditions
can be employed to allow for selective hybridization.
In optional embodiments, the
stringency allows for selective hybridization of sequences having at least
70%, 75%, 80%,
85%, 90%, 95% or 98% sequence identity over the length of the hybridized
region. Full-
33
CA 02877639 2015-01-09
length enriched cDNA libraries can be normalized to increase the
representation of rare
sequences.
Polynucleotide Products Made by a cDNA Isolation Process
As indicated in (I), above, the present invention provides an isolated
polynucleotide
made by the process of: 1) providing a full-length enriched nucleic acid
library, 2) selectively
hybridizing the polynucleotide to a polynucleotide of paragraphs (A), (B),
(C), (D), (E), (F),
(G) or (H) as discussed above, and thereby isolating the polynucleotide from
the nucleic acid
library. Full-length enriched nucleic acid libraries are constructed as
discussed in paragraph
(G) and below. Selective hybridization conditions are as discussed in
paragraph (G).
Nucleic acid purification procedures are well known in the art.
Purification can be conveniently accomplished using solid-phase methods; such
methods are well known to those of skill in the art and kits are available
from commercial
suppliers such as Advanced Biotechnologies (Surrey, UK). For example, a
polynucleotide of
paragraphs (A)-(H) can be immobilized to a solid support such as a membrane,
bead or
particle. See, e.g., US Patent Number 5,667,976. The polynucleotide product of
the present
process is selectively hybridized to an immobilized polynucleotide and the
solid support is
subsequently isolated from non-hybridized polynucleotides by methods
including, but not
limited to, centrifugation, magnetic separation, filtration, electrophoresis
and the like.
Construction of Nucleic Acids
The isolated nucleic acids of the present invention can be made using (a)
standard
recombinant methods, (b) synthetic techniques, or combinations thereof.
In some
embodiments, the polynucleotides of the present invention will be cloned,
amplified, or
otherwise constructed from a monocot such as corn, rice or wheat, or a dicot
such as
soybean.
The nucleic acids may conveniently comprise sequences in addition to a
polynucleotide of the present invention. For example, a multi-cloning site
comprising one or
more endonuclease restriction sites may be inserted into the nucleic acid to
aid in isolation of
the polynucleotide. Also, translatable sequences may be inserted to aid in the
isolation of
the translated polynucleotide of the present invention. For example, a
hexahistidine marker
sequence provides a convenient means to purify the proteins of the present
invention. A
polynucleotide of the present invention can be attached to a vector, adapter,
or linker for
cloning and/or expression of a polynucleotide of the present invention.
Additional sequences
may be added to such cloning and/or expression sequences to optimize their
function in
34
CA 02877639 2015-01-09
cloning and/or expression, to aid in isolation of the polynucleotide or to
improve the
introduction of the polynucleotide into a cell. Typically, the length of a
nucleic acid of the
present invention less the length of its polynucleotide of the present
invention is less than 20
kilobase pairs, often less than 15 kb, and frequently less than 10 kb. Use of
cloning vectors,
expression vectors, adapters and linkers is well known and extensively
described in the art.
For a description of various nucleic acids see, for example, Stratagene
Cloning Systems,
Catalogs 1999 (La Jolla, CA); and, Amersham Life Sciences, Inc, Catalog '99
(Arlington
Heights, IL).
to A. Recombinant Methods for Constructing Nucleic Acids
The isolated nucleic acid compositions of this invention, such as RNA, cDNA,
genomic DNA, or a hybrid thereof, can be obtained from plant biological
sources using any
number of cloning methodologies known to those of skill in the art. In some
embodiments,
oligonucleotide probes which selectively hybridize, under stringent
conditions, to the
polynucleotides of the present invention are used to identify the desired
sequence in a cDNA
or genomic DNA library. Isolation of RNA, and construction of cDNA and genomic
libraries
is well known to those of ordinary skill in the art. See, e.g., Plant
Molecular Biology : A
Laboratory Manual, Clark, Ed., Springer-Verlag, Berlin (1997); and Current
Protocols in
Molecular Biology, Ausubel, et al., Eds., Greene Publishing and Wiley-
Interscience, New
York (1995).
Al. Full-length Enriched cDNA Libraries
A number of cDNA synthesis protocols have been described which provide
enriched
full-length cDNA libraries. Enriched full-length cDNA libraries are
constructed to comprise at
least 60%, and more preferably at least 70%, 80%, 90% or 95% full-length
inserts amongst
clones containing inserts. The length of insert in such libraries can be at
least 2, 3, 4, 5, 6, 7,
8, 9, 10 or more kilobase pairs. Vectors to accommodate inserts of these sizes
are known in
the art and available commercially. See, e.g., Stratagene's lambda ZAP Express
(cDNA
cloning vector with 0 to 12 kb cloning capacity). An exemplary method of
constructing a
greater than 95% pure full-length cDNA library is described by Carninci, et
al., (1996)
Genomics 37:327-336. Other methods for producing full-length libraries are
known in the
art. See, e.g., Edery, et a/., (1995) Mo/. Cell Biol. 15(6):3363-3371; and PCT
Application
W096/34981.
CA 02877639 2015-01-09
A2 Normalized or Subtracted cDNA Libraries
A non-normalized cDNA library represents the mRNA population of the tissue it
was
made from. Since unique clones are out-numbered by clones derived from highly
expressed
genes their isolation can be laborious. Normalization of a cDNA library is the
process of
creating a library in which each clone is more equally represented.
Construction of
normalized libraries is described in Ko, (1990) Nucl. Acids. Res. 18(19):5705-
5711;
Patanjali, etal., (1991) Proc. Natl. Acad. USA 88:1943-1947; US Patent Numbers
5,482,685,
5,482,845 and 5,637,685.
In an exemplary method described by Soares, et al.,
normalization resulted in reduction of the abundance of clones from a range of
four orders of
magnitude to a narrow range of only 1 order of magnitude. Proc. Natl. Acad.
Sci. USA,
91:9228-9232 (1994).
Subtracted cDNA libraries are another means to increase the proportion of less
abundant cDNA species. In this procedure, cDNA prepared from one pool of mRNA
is
depleted of sequences present in a second pool of mRNA by hybridization. The
cDNA:
mRNA hybrids are removed and the remaining un-hybridized cDNA pool is enriched
for
sequences unique to that pool. See, Foote, et al., in, Plant Molecular
Biology: A Laboratory
Manual, Clark, Ed., Springer-Verlag, Berlin (1997); Kho and Zarb!, (1991)
Technique
3(2):58-63; Sive and St. John, (1988) Nucl. Acids Res. 16(22):10937; Current
Protocols in
Molecular Biology, Ausubel, et al., Eds., Greene Publishing and Wiley-
lnterscience, New
York (1995); and Swaroop, etal., (1991) Nucl. Acids Res. 19(8):1954. cDNA
subtraction kits
are commercially available. See, e.g., PCR-Select (Clontech, Palo Alto, CA).
To construct genomic libraries, large segments of genomic DNA are generated by
fragmentation, e.g., using restriction endonucleases, and are ligated with
vector DNA to form
concatemers that can be packaged into the appropriate vector.
Methodologies to
accomplish these ends, and sequencing methods to verify the sequence of
nucleic acids are
well known in the art. Examples of appropriate molecular biological techniques
and
instructions sufficient to direct persons of skill through many construction,
cloning, and
screening methodologies are found in Sambrook, et al., Molecular Cloning: A
Laboratory
Manual, 2nd Ed., Cold Spring Harbor Laboratory Vols. 1-3 (1989), Methods in
Enzymology,
Vol. 152: Guide to Molecular Cloning Techniques, Berger and Kimmel, Eds., San
Diego:
Academic Press, Inc. (1987), Current Protocols in Molecular Biology, Ausubel,
et al., Eds.,
Greene Publishing and Wiley-Interscience, New York (1995); Plant Molecular
Biology : A
Laboratory Manual, Clark, Ed., Springer-Verlag, Berlin (1997). Kits for
construction of
genomic libraries are also commercially available.
36
CA 02877639 2015-01-09
The cDNA or genomic library can be screened using a probe based upon the
sequence of a polynucleotide of the present invention such as those disclosed
herein.
Probes may be used to hybridize with genomic DNA or cDNA sequences to isolate
homologous genes in the same or different plant species. Those of skill in the
art will
appreciate that various degrees of stringency of hybridization can be employed
in the assay;
and either the hybridization or the wash medium can be stringent.
The nucleic acids of interest can also be amplified from nucleic acid samples
using
amplification techniques. For instance, polymerase chain reaction (PCR)
technology can be
used to amplify the sequences of polynucleotides of the present invention and
related genes
directly from genomic DNA or cDNA libraries. PCR and other in vitro
amplification methods
may also be useful, for example, to clone nucleic acid sequences that code for
proteins to be
expressed, to make nucleic acids to use as probes for detecting the presence
of the desired
mRNA in samples, for nucleic acid sequencing, or for other purposes. The T4
gene 32
protein (Boehringer Mannheim) can be used to improve yield of long PCR
products.
PCR-based screening methods have been described. Wilfinger, et a/., describe a
PCR-based method in which the longest cDNA is identified in the first step so
that
incomplete clones can be eliminated from study. Bio Techniques 22(3):481-486
(1997).
Such methods are particularly effective in combination with a full-length cDNA
construction
methodology, above.
B. Synthetic Methods for Constructing Nucleic Acids
The isolated nucleic acids of the present invention can also be prepared by
direct
chemical synthesis by methods such as the phosphotriester method of Narang, et
al., (1979)
Meth. Enzymol. 68:90-99; the phosphodiester method of Brown, et al., (1979)
Meth.
EnzymoL 68:109-151; the diethylphosphoramidite method of Beaucage, etal.,
(1981) Tetra.
Lett. 22:1859-1862; the solid phase phosphoramidite triester method described
by Beaucage
and Caruthers, (1981) Tetra. Letts. 22(20):1859-1862, e.g., using an automated
synthesizer,
e.g., as described in Needham-VanDevanter, et al., (1984) Nucleic Acids Res.
12:6159-
6168; and the solid support method of US Patent Number 4,458,066. Chemical
synthesis
generally produces a single stranded oligonucleotide. This may be converted
into double
stranded DNA by hybridization with a complementary sequence, or by
polymerization with a
DNA polymerase using the single strand as a template. One of skill will
recognize that while
chemical synthesis of DNA is best employed for sequences of about 100 bases or
less,
longer sequences may be obtained by the ligation of shorter sequences.
37
CA 02877639 2015-01-09
Recombinant Expression Cassettes
The present invention further provides recombinant expression cassettes
comprising
a nucleic acid of the present invention. A nucleic acid sequence coding for
the desired
polypeptide of the present invention, for example a cDNA or a genomic sequence
encoding
a full length polypeptide of the present invention, can be used to construct a
recombinant
expression cassette which can be introduced into the desired host cell. A
recombinant
expression cassette will typically comprise a polynucleotide of the present
invention operably
linked to transcriptional initiation regulatory sequences which will direct
the transcription of
the polynucleotide in the intended host cell, such as tissues of a transformed
plant.
to
For example, plant expression vectors may include: (1) a cloned plant gene
under
the transcriptional control of 5' and 3' regulatory sequences and (2) a
dominant selectable
marker. Such plant expression vectors may also contain, if desired, a promoter
regulatory
region (e.g., one conferring inducible or constitutive, environmentally-or
developmentally-
regulated, or cell-or tissue-specific/selective expression), a transcription
initiation start site, a
ribosome binding site, an RNA processing signal, a transcription termination
site and/or a
polyadenylation signal.
A plant promoter fragment can be employed which will direct expression of a
polynucleotide of the present invention in all tissues of a regenerated plant.
Such promoters
are referred to herein as "constitutive" promoters and are active under most
environmental
conditions and states of development or cell differentiation. Examples of
constitutive
promoters include the cauliflower mosaic virus (CaMV) 35S transcription
initiation region, the
1'- or 2'-promoter derived from T-DNA of Agrobacterium tumefaciens, the
ubiquitin 1
promoter, the Smas promoter, the cinnamyl alcohol dehydrogenase promoter (US
Patent
Number 5,683,439), the Nos promoter, the pEmu promoter, the rubisco promoter,
theGRP1-
8 promoter and other transcription initiation regions from various plant genes
known to those
of skill.
Alternatively, the plant promoter can direct expression of a polynucleotide of
the
present invention in a specific tissue or may be otherwise under more precise
environmental
or developmental control. Such promoters are referred to here as "inducible"
promoters
Environmental conditions that may effect transcription by inducible promoters
include
pathogen attack, anaerobic conditions, or the presence of light. Examples of
inducible
promoters are the Adhl promoter which is inducible by hypoxia or cold stress,
the Hsp70
promoter which is inducible by heat stress, and the PPDK promoter which is
inducible by
light.
38
CA 02877639 2015-01-09
Examples of promoters under developmental control include promoters that
initiate
transcription only, or preferentially, in certain tissues, such as leaves,
roots, fruit, seeds or
flowers. Exemplary promoters include the anther specific promoter 5126 (US
Patent
Numbers 5,689,049 and 5,689,051), glob-1 promoter, and gamma-zein promoter.
The
operation of a promoter may also vary depending on its location in the genome.
Thus, an
inducible promoter may become fully or partially constitutive in certain
locations.
Both heterologous and non-heterologous (i.e., endogenous) promoters can be
employed to direct expression of the nucleic acids of the present invention.
These
promoters can also be used, for example, in recombinant expression cassettes
to drive
expression of antisense nucleic acids to reduce, increase, or alter
concentration and/or
composition of the proteins of the present invention in a desired tissue.
Thus, in some
embodiments, the nucleic acid construct will comprise a promoter functional in
a plant cell,
such as in Zea mays, operably linked to a polynucleotide of the present
invention.
Promoters useful in these embodiments include the endogenous promoters driving
expression of a polypeptide of the present invention.
A number of promoters can be used in the practice of the invention, including
the
native promoter of a polynucleotide sequence of interest. The promoters can be
selected
based on the desired outcome. The nucleic acids can be combined with
constitutive,
inducible, tissue-preferred or other promoters for expression in plants.
Such constitutive promoters include, for example, the core promoter of the
Rsyn7
promoter and other constitutive promoters disclosed in WO 99/43838 and US
Patent
Number 6,072,050; the core CaMV 35S promoter (Odell, etal., (1985) Nature
313:810-812);
rice actin (McElroy, etal., (1990) Plant Ce// 2:163-171); ubiquitin
(Christensen, etal., (1989)
Plant MoL BioL 12:619-632 and Christensen, et al., (1992) Plant MoL BioL
18:675-689);
pEMU (Last, et al., (1991) Theor. App!. Genet. 81:581-588); MAS (Velten, et
al., (1984)
EMBO J. 3:2723-2730); ALS promoter (US Patent Number 5,659,026), dMMV (double-
enhanced version of the mirabilis mosaic virus promoter; see, Dey and Maiti
(1999) Plant
Molecular Biology 40(5):771-782), LESVBV (enhanced strawberry vein banding
virus
promoter; see, US Patent Application Publication Number 2002/0182593) and the
like.
Other constitutive promoters include, for example, US Patent Numbers
5,608,149;
5,608,144; 5,604,121; 5,569,597; 5,466,785; 5,399,680; 5,268,463; 5,608,142
and
6,177,611.
Tissue-preferred promoters can be utilized to target enhanced expression
within a
particular plant tissue. Tissue-preferred promoters include Yamamoto, et al.,
(1997) Plant J.
12(2):255-265; Kawamata, et al., (1997) Plant Cell Physiol. 38(7):792-803;
Hansen, et al.,
39
CA 02877639 2015-01-09
(1997) MoL Gen Genet. 254(3):337-343; Russell, et al., (1997) Transgenic Res.
6(2):157-
168; Rinehart, et al., (1996) Plant Physiol. 112(3):1331-1341; Van Camp,
etal., (1996) Plant
Physiol. 112(2):525-535; Canevascini, et al., (1996) Plant Physiol. 112(2):513-
524;
Yamamoto, et al., (1994) Plant Cell Physiol. 35(5):773-778; Lam (1994) Results
Probl. Cell
Differ. 20:181-196; Orozco, etal., (1993) Plant Mol Biol. 23(6):1129-1138;
Matsuoka, etal.,
(1993) Proc Natl. Acad. Sci. USA 90(20):9586-9590 and Guevara-Garcia, etal.,
(1993) Plant
J. 4(3):495-505. Such promoters can be modified, if necessary, for weak
expression. See,
also, US Patent Application Number 2003/0074698, herein incorporated by
reference.
Leaf-preferred promoters are known in the art. See, for example, Yamamoto, et
al.,
(1997) Plant J. 12(2):255-265; Kwon, etal., (1994) Plant Physiol. 105:357-67;
Yamamoto, et
al., (1994) Plant Cell PhysioL 35(5):773-778; Gotor, etal., (1993) Plant J.
3:509-18; Orozco,
et al., (1993) Plant Mol. Biol. 23(6):1129-1138; Baszczynski, et al., (1988)
Nucl. Acid Res.
16:4732; Mitra, et al., (1994) Plant Molecular Biology 26:35-93; Kayaya, et
al., (1995)
Molecular and General Genetics 248:668-674; and Matsuoka, etal., (1993) Proc.
Natl. Acad.
Sci. USA 90(20):9586-9590. Senecence regulated promoters are also of use, such
as,
SAM22 (Crowell, et al., (1992) Plant MoL BioL 18:459-466). See, also, US
Patent Number
5,689,042 herein incorporated by reference.
Root-preferred promoters are known and can be selected from the many available
from the literature or isolated de novo from various compatible species. See,
for example,
Hire, et al., (1992) Plant MoL Biol. 20(2):207-218 (soybean root-specific
glutamine
synthetase gene); Keller and Baumgartner (1991) Plant Cell 3(10):1051-1061
(root-specific
control element in the GRP 1.8 gene of French bean); Sanger, etal., (1990)
Plant MoL BioL
14(3):433-443 (root-specific promoter of the mannopine synthase (MAS) gene of
Agrobacterium tumefaciens); and Miao, etal., (1991) Plant Cell 3(1):11-22
(full-length cDNA
clone encoding cytosolic glutamine synthetase (GS), which is expressed in
roots and root
nodules of soybean). See also, Bogusz, etal., (1990) Plant Cell 2(7):633-641,
where two
root-specific promoters isolated from hemoglobin genes from the nitrogen-
fixing nonlegume
Parasponia andersonii and the related non-nitrogen-fixing nonlegume Trema
tomentosa are
described. The promoters of these genes were linked to a fl-glucuronidase
reporter gene
and introduced into both the nonlegume Nicotiana tabacum and the legume Lotus
comiculatus, and in both instances root-specific promoter activity was
preserved. Leach and
Aoyagi (1991) describe their analysis of the promoters of the highly expressed
roIC and rolD
root-inducing genes of Agrobacterium rhizogenes (see. Plant Science (Limerick)
79(1):69-
76). They concluded that enhancer and tissue-preferred DNA determinants are
dissociated
in those promoters. Teen, et aL, (1989) used gene fusion to lacZ to show that
the
CA 02877639 2015-01-09
Agrobacterium T-DNA gene encoding octopine synthase is especially active in
the epidermis
of the root tip and that the TR2' gene is root specific in the intact plant
and stimulated by
wounding in leaf tissue, an especially desirable combination of
characteristics for use with an
insecticidal or larvicidal gene (see, EMBO J. 8(2):343-350). The TR1' gene,
fused to nptll
(neomycin phosphotransferase II) showed similar characteristics. Additional
root-preferred
promoters include the VfENOD-GRP3 gene promoter (Kuster, et al., (1995) Plant
Mol. Biol.
29(4):759-772); rolB promoter (Capana, etal., (1994) Plant Mol. Biol.
25(4):681-691; and the
CRWAQ81 root-preferred promoter with the ADH first intron (US Patent
Application
Publication NUmber 2005/0097633). See also, US Patent Numbers 5,837,876;
5,750,386;
5,633,363; 5,459,252; 5,401,836; 5,110,732 and 5,023,179.
"Seed-preferred" promoters refers to those promoters active during seed
development and may include expression in seed initials or related maternal
tissue. Such
seed-preferred promoters include, but are not limited to, Cim1 (cytokinin-
induced message);
cZ19B1 (maize 19 kDa zein); milps (myo-inosito1-1-phosphate synthase) (see, WO
00/11177
and US Patent Number 6,225,529; herein incorporated by reference). Gamma-zein
is an
endosperm-specific promoter. Globulin-1 (Glob-1) is a representative embryo-
specific
promoter. For dicots, seed-specific promoters include, but are not limited to,
bean 10-
phaseolin, napin, ig-conglycinin, soybean lectin, cruciferin and the like. For
monocots, seed-
specific promoters include, but are not limited to, maize 15 kDa zein, 22 kDa
zein, 27 kDa
zein, gamma-zein, waxy, shrunken 1 and shrunken 2. See also, WO 00/12733,
where seed-
preferred promoters from endl and end2 genes are disclosed; herein
incorporated by
reference. Additional embryo specific promoters are disclosed in Sato, et al.,
(1996) Proc.
Natl. Acad. Sci. 93:8117-8122; Nakase, et al., (1997) Plant J 12:235-46; and
Postma-
Haarsma, et a/. , (1999) Plant Mol. Biol. 39:257-71. Additional endosperm
specific promoters
are disclosed in Albani, et al., (1984) EMBO 3:1405-15; Albani, et al., (1999)
Theor. App!.
Gen. 98:1253-62; Albani, et al., (1993) Plant J. 4:343-55; Mena, et al.,
(1998) The Plant
Journal 116:53-62, and Wu, etal., (1998) Plant Cell Physiology 39:885-889.
Also of interest are promoters active in meristem regions, such as developing
inflorescence tissues, and promoters which drive expression at or about the
time of anthesis
or early kernel development. This may include, for example, the maize Zag
promoters,
including Zag1 and Zag2 (see, Schmidt, et al., (1993) The Plant Ce// 5:729-37;
GenBank
X80206; Theissen, et al., (1995) Gene 156:155-166; and US Patent Application
Number
10/817,483); maize Zap promoter (also known as ZmMADS; US Patent Application
Number
10/387,937; WO 03/078590); maize ckx1-2 promoter (US Patent Application
Publication
Number 2002/0152500 Al; WO 02/0078438); maize eep1 promoter (US Patent
Application
41
CA 02877639 2015-01-09
Number 10/817,483); maize end2 promoter (US Patent Number 6,528,704 and US
Patent
Application Number 10/310,191); maize led 1 promoter (US Patent Application
Number
09/718,754); maize F3.7 promoter (Baszczynski, et al., (1997) Maydica 42:189-
201); maize
tb1 promoter (Hubbarda, et al., (2002) Genetics 162:1927-1935 and Wang, et
al., (1999)
Nature 398:236-239); maize eep2 promoter (US Patent Application Number
10/817,483);
maize thioredoxinH promoter (US Provisional Patent Application Number
60/514,123); maize
Zm40 promoter (US Patent Number 6,403,862 and WO 01/2178); maize mLIP15
promoter (US
Patent Number 6,479,734); maize ESR promoter (US Patent Application Number
10/786,679);
maize PCNA2 promoter (US Patent Application Number 10/388,359); maize
cytokinin oxidase
to
promoters (US Patent Application Number 11/094,917); promoters disclosed in
Weigel, etal.,
(1992) Cell 69:843-859; Accession Number AJ131822; Accession Number Z71981;
Accession Number AF049870; and shoot-preferred promoters disclosed in McAvoy,
et al.,
(2003) Acta Hort. (ISHS) 625:379-385. Other dividing cell or meristematic
tissue-preferred
promoters that may be of interest have been disclosed in Ito, et al., (1994)
Plant MoL Biol.
24:863-878; Regad, etal., (1995) Mo. Gen. Genet. 248:703-711; Shaul, etal.,
(1996) Proc.
Natl. Acad. ScL 93:4868-4872; Ito, et al., (1997) Plant J. 11:983-992; and
Trehin, et al.,
(1997) Plant Mol. Biol. 35:667-672, all of which are hereby incorporated by
reference herein.
Inflorescence-preferred promoters include the promoter of chalcone synthase
(Van
der Meer, et al., (1990) Plant MoL Biol. 15:95-109), LAT52 (Twell, et al.,
(1989) MoL Gen.
Genet. 217:240-245), pollen specific genes (Albani, et al., (1990) Plant Mol
Biol. 15:605,
Zm13 (Buerrero, et aL, (1993) Mol. Gen. Genet. 224:161-168), maize pollen-
specific gene
(Hamilton, et al., (1992) Plant MoL BioL 18:211-218), sunflower pollen
expressed gene
(Baltz, et al., (1992) The Plant Journal 2:713-721), and B. napus pollen
specific genes
(Arnoldo, etal., (1992) J. Cell. Biochem, Abstract Number Y101204).
Stress-inducible promoters include salt-inducible or water-stress-inducible
promoters
such as P5CS (Zang, at al., (1997) Plant Sciences 129:81-89); cold-inducible
promoters,
such as, cor15a (Hajela, etal., (1990) Plant Physiol. 93:1246-1252), cor15b
(Wlihelm, etal.,
(1993) Plant Mol Biol 23:1073-1077), wsc120 (Ouellet, at al., (1998) FEBS
Lett. 423-324-
328), ci7 (Kirch, et al., (1997) Plant Mol BioL 33:897-909), ci21A (Schneider,
at al., (1997)
Plant Physiol. 113:335-45); drought-inducible promoters, such as, Trg-31
(Chaudhary, et al.,
(1996) Plant MoL BioL 30:1247-57); osmotic inducible promoters, such as, Rab17
(Vilardell,
etal., (1991) Plant MoL Biol. 17:985-93; Busk (1997) Plant J 11(6):1285-1295)
and osmotin
(Raghothama, et al., (1993) Plant Mol Biol 23:1117-28); and, heat inducible
promoters, such
as, heat shock proteins (Barros, etal., (1992) Plant Mol. 19:665-75; Marrs,
etal., (1993) Dev.
Genet. 14:27-41), and smHSP (Waters, et al., (1996) J. Experimental Botany
47:325-338).
42
CA 02877639 2015-01-09
Other stress-inducible promoters include rip2 (US Patent Number 5,332,808 and
US Patent
Application Publication Number 2003/0217393), and rd29a (Yamaguchi-Shinozaki,
et a/.,
(1993) Mol. Gen. Genetics 236:331-340; see also GenBank accession D13044).
Stress-
insensitive promoters can also be used in the methods of the invention.
Nitrogen-responsive promoters can also be used in the methods of the
invention.
Such promoters include, but are not limited to, the 22 kDa Zein promoter
(Spena, et al.,
(1982) EMBO J 1:1589-1594 and Muller, etal., (1995) J. Plant Physiol 145:606-
613); the 19
kDa zein promoter (Pedersen, et al., (1982) Ce// 29:1019-1025); the 14 kDa
zein promoter
(Pedersen, etal., (1986) J. Biol. Chem. 261:6279-6284), the b-32 promoter
(Lohmer, etal.,
(1991) EMBO J 10:617-624); and the nitrite reductase (NiR) promoter (Rastogi,
etal., (1997)
Plant Mol Biol. 34(3):465-76 and Sander, etal., (1995) Plant Mol Biol.
27(1):165-77). For a
review of consensus sequences found in nitrogen-induced promoters, see for
example,
Muller, etal., (1997) The Plant Journal 12:281-291.
Chemically-regulated promoters can be used to modulate the expression of a
gene in
a plant through the application of an exogenous chemical regulator. Depending
upon the
objective, the promoter may be a chemically-inducible promoter, where
application of the
chemical induces gene expression, or a chemical-repressible promoter, where
application of
the chemical represses gene expression. Chemically-inducible promoters are
known in the
art and include, but are not limited to, the maize In2-2 promoter, which is
activated by
benzenesulfonamide herbicide safeners, the maize GST promoter, which is
activated by
hydrophobic electrophilic compounds that are used as pre-emergent herbicides,
and the
tobacco PR-la promoter, which is activated by salicylic acid. Other chemical-
regulated
promoters of interest include steroid-responsive promoters (see, for example,
the
glucocorticoid-inducible promoter in Schena, et al., (1991) Proc. Natl. Acad.
ScL USA
88:10421-10425 and McNellis, et al., (1998) Plant J. 14(2):247-257) and
tetracycline-
inducible and tetracycline-repressible promoters (see, for example, Gatz, et
al., (1991) Mo/.
Gen. Genet. 227:229-237, and US Patent Numbers 5,814,618 and 5,789,156),
herein
incorporated by reference.
Additional inducible promoters include heat shock promoters, such as Gmhsp17.5-
E
(soybean) (Czarnecka, et al., (1989) Mo/ Cell Biol. 9(8):3457-3463); APX1 gene
promoter
(Arabidopsis) (Storozhenko, etal., (1998) Plant PhysioL 118(3):1005-1014): Ha
hsp17.7 G4
(Helianthus annuus) (Almoguera, et al., (2002) Plant Physiol. 129(1):333-341;
and Maize
Hsp70 (Rochester, etal., (1986) EMBO J. 5: 451-8.
In some embodiments, isolated nucleic acids which serve as promoter or
enhancer
elements can be introduced in the appropriate position (generally upstream) of
a non-
43
CA 02877639 2015-01-09
heterologous form of a polynucleotide of the present invention so as to up or
down regulate
expression of a polynucleotide of the present invention. For example,
endogenous
promoters can be altered in vivo by mutation, deletion, and/or substitution
(see, Kmiec, US
Patent Number 5,565,350; Zarling, et al., PCT/US93/03868) or isolated
promoters can be
introduced into a plant cell in the proper orientation and distance from a
gene of the present
invention so as to control the expression of the gene. Gene expression can be
modulated
under conditions suitable for plant growth so as to alter the total
concentration and/or alter
the composition of the polypeptides of the present invention in plant cell.
Thus, the present invention provides compositions, and methods for making,
heterologous promoters and/or enhancers operably linked to a native,
endogenous (i.e.,
nonheterologous) form of a polynucleotide of the present invention.
Methods for identifying promoters with a particular expression pattern, in
terms of,
e.g., tissue type, cell type, stage of development and/or environmental
conditions, are well
known in the art. See, e.g., The Maize Handbook, Chapters 114-115, Freeling
and Walbot,
Eds., Springer, New York (1994); Corn and Corn Improvement, 3rd edition,
Chapter 6,
Sprague and Dudley, Eds., American Society of Agronomy, Madison, Wisconsin
(1988).
A typical step in promoter isolation methods is identification of gene
products that are
expressed with some degree of specificity in the target tissue. Amongst the
range of
methodologies are: differential hybridization to cDNA libraries; subtractive
hybridization;
differential display; differential 2-D protein gel electrophoresis; DNA probe
arrays; and
isolation of proteins known to be expressed with some specificity in the
target tissue. Such
methods are well known to those of skill in the art. Commercially available
products for
identifying promoters are known in the art such as Clontech's (Palo Alto, CA)
Universal
Genome Walker Kit.
For the protein-based methods, it is helpful to obtain the amino acid sequence
for at
least a portion of the identified protein, and then to use the protein
sequence as the basis for
preparing a nucleic acid that can be used as a probe to identify either
genomic DNA directly,
or preferably, to identify a cDNA clone from a library prepared from the
target tissue. Once
such a cDNA clone has been identified, that sequence can be used to identify
the sequence
at the 5' end of the transcript of the indicated gene. For differential
hybridization, subtractive
hybridization and differential display, the nucleic acid sequence identified
as enriched in the
target tissue is used to identify the sequence at the 5' end of the transcript
of the indicated
gene. Once such sequences are identified, starting either from protein
sequences or nucleic
acid sequences, any of these sequences identified as being from the gene
transcript can be
44
CA 02877639 2015-01-09
used to screen a genomic library prepared from the target organism. Methods
for identifying
and confirming the transcriptional start site are well known in the art.
In the process of isolating promoters expressed under particular environmental
conditions or stresses, or in specific tissues, or at particular developmental
stages, a number
of genes are identified that are expressed under the desired circumstances, in
the desired
tissue or at the desired stage. Further analysis will reveal expression of
each particular gene
in one or more other tissues of the plant. One can identify a promoter with
activity in the
desired tissue or condition but that does not have activity in any other
common tissue.
To identify the promoter sequence, the 5' portions of the clones described
here are
analyzed for sequences characteristic of promoter sequences. For instance,
promoter
sequence elements include the TATA box consensus sequence (TATAAT), which is
usually
an AT-rich stretch of 5-10 bp located approximately 20 to 40 base pairs
upstream of the
transcription start site. Identification of the TATA box is well known in the
art. For example,
one way to predict the location of this element is to identify the
transcription start site using
standard RNA-mapping techniques such as primer extension, S 1 analysis, and/or
RNase
protection. To confirm the presence of the AT-rich sequence, a structure-
function analysis
can be performed involving mutagenesis of the putative region and
quantification of the
mutation's effect on expression of a linked downstream reporter gene. See,
e.g., The Maize
Handbook, Chapter 114, Freeling and Walbot, Eds., Springer, New York, (1994).
In plants, further upstream from the TATA box, at positions -80 to -100, there
is
typically a promoter element (i.e., the CAAT box) with a series of adenines
surrounding the
trinucleotide G (or T) N G. J. Messing, et al., in Genetic Engineering in
Plants, Kosage,
Meredith and Hollaender, Eds., pp. 221-227 1983. In maize, there is no well
conserved
CAAT box but there are several short, conserved protein-binding motifs
upstream of the
TATA box. These include motifs for the trans-acting transcription factors
involved in light
regulation, anaerobic induction, hormonal regulation or anthocyanin
biosynthesis, as
appropriate for each gene.
Once promoter and/or gene sequences are known, a region of suitable size is
selected from the genomic DNA that is 5' to the transcriptional start, or the
translational start
site, and such sequences are then linked to a coding sequence. If the
transcriptional start
site is used as the point of fusion, any of a number of possible 5'
untranslated regions can be
used in between the transcriptional start site and the partial coding
sequence. If the
translational start site at the 3' end of the specific promoter is used, then
it is linked directly
to the methionine start codon of a coding sequence.
CA 02877639 2015-01-09
If polypeptide expression is desired, it is generally desirable to include a
polyadenylation region at the 31-end of a polynucleotide coding region. The
polyadenylation
region can be derived from the natural gene, from a variety of other plant
genes, or from T-
DNA. The 3'end sequence to be added can be derived from, for example, the
nopaline
synthase or octopine synthase genes, or alternatively from another plant gene,
or less
preferably from any other eukaryotic gene.
An intron sequence can be added to the 5' untranslated region or the coding
sequence of the partial coding sequence to increase the amount of the mature
message that
accumulates in the cytosol. Inclusion of a spliceable intron in the
transcription unit in both
to plant and animal expression constructs has been shown to increase gene
expression at both
the mRNA and protein levels up to1000-fold. Buchman and Berg, (1988) Mot Cell
Biol. 8:
4395-4405; Callis, et al., (1987) Genes Dev. 1:1183-1200. Such intron
enhancement of
gene expression is typically greatest when placed near the 5' end of the
transcription unit.
Use of maize introns Adhl-S intron 1, 2 and 6, the Bronze-1 intron are known
in the art. See
generally, The Maize Handbook, Chapter 116, Freeling and Walbot, Eds.,
Springer, New
York (1994).
The vector comprising the sequences from a polynucleotide of the present
invention
will typically comprise a marker gene which confers a selectable phenotype on
plant cells.
Usually, the selectable marker gene will encode antibiotic resistance, with
suitable genes
including genes coding for resistance to the antibiotic spectinomycin (e.g.,
the aada gene),
the streptomycin phosphotransferase (SPT) gene coding for streptomycin
resistance, the
neomycin phosphotransferase (NPTII) gene encoding kanamycin or genetic in
resistance,
the hygromycin phosphotransferase (HPT) gene coding for hygromycin resistance,
genes
coding for resistance to herbicides which act to inhibit the action of
acetolactate synthase
(ALS), in particular the sulfonylurea-type herbicides (e.g., the acetolactate
synthase (ALS)
gene containing mutations leading to such resistance in particular the S4
and/or Hra
mutations), genes coding for resistance to herbicides which act to inhibit
action of glutamine
synthase, such as phosphinothricin or basta (e.g., the bar gene), or other
such genes known
in the art. The bar gene encodes resistance to the herbicide basta, the nptll
gene encodes
resistance to the antibiotic kanamycin, and the ALS gene encodes resistance to
the
herbicide chlorsulfuron.
Typical vectors useful for expression of genes in higher plants are well known
in the
art and include vectors derived from the tumor-inducing (Ti) plasmid of
Agrobacterium
tumefaciens described by Rogers, et al., (1987) Meth. in EnzymoL 153:253-277.
These
vectors are plant integrating vectors in that on transformation, the vectors
integrate a portion
46
CA 02877639 2015-01-09
of vector DNA into the genome of the host plant. Exemplary A. tumefaciens
vectors useful
herein are plasmids pKYLX6 and pKYLX7 of Schardl, et al., (1987) Gene 61:1-11
and
Berger, etal., (1989) Proc. Natl. Acad. ScL USA 86:8402-8406. Another useful
vector herein
is plasmid pB1101.2 that is available from Clontech Laboratories, Inc. (Palo
Alto, CA).
A polynucleotide of the present invention can be expressed in either sense or
antisense orientation as desired. It will be appreciated that control of gene
expression in
either sense or anti-sense orientation can have a direct impact on the
observable plant
characteristics. Antisense technology can be conveniently used to inhibit gene
expression in
plants. To accomplish this, a nucleic acid segment from the desired gene is
cloned and
operably linked to a promoter such that the anti-sense strand of RNA will be
transcribed.
The construct is then transformed into plants and the antisense strand of RNA
is produced.
In plant cells, it has been shown that antisense RNA inhibits gene expression
by
preventing the accumulation of mRNA which encodes the enzyme of interest, see,
e.g.,
Sheehy, et al., (1988) Proc. Nat'l. Acad. ScL USA 85:8805-8809; and Hiatt, et
al., US Patent
Number 4,801,340.
Another method of suppression is sense suppression. Introduction of nucleic
acid
configured in the sense orientation has been shown to be an effective means by
which to
block the transcription of target genes. For an example of the use of this
method to
modulate expression of endogenous genes, see, Napoli, et al., (1990) The Plant
Cell 2:279-
289 and US Patent Number 5,034,323.
Catalytic RNA molecules or ribozymes can also be used to inhibit expression of
plant
genes. It is possible to design ribozymes that specifically pair with
virtually any target RNA
and cleave the phosphodiester backbone at a specific location, thereby
functionally
inactivating the target RNA. In carrying out this cleavage, the ribozyme is
not itself altered,
and is thus capable of recycling and cleaving other molecules, making it a
true enzyme. The
inclusion of ribozyme sequences within antisense RNAs confers RNA cleaving
activity upon
them, thereby increasing the activity of the constructs. The design and use of
target RNA-
specific ribozymes is described in Haseloff, et al., (1988) Nature 334:585
591. A variety of
cross-linking agents, alkylating agents and radical generating species as
pendant groups on
polynucleotides of the present invention can be used to bind, label, detect
and/or cleave
nucleic acids. For example, Vlassov, et al., (1986) Nucleic Acids Res 14:4065-
4076,
describe covalent bonding of a single-stranded DNA fragment with alkylating
derivatives of
nucleotides complementary to target sequences. A report of similar work by the
same group
is that by Knorre, etal., (1985) Biochimie 67:785-789. Iverson and Dervan.
47
CA 02877639 2015-01-09
The present invention further provides a protein comprising a polypeptide
having a
specified sequence identity with a polypeptide of the present invention. The
percentage of
sequence identity is an integer selected from the group consisting of from 60
to 99.
Exemplary sequence identity values include 60%, 65%, 70%, 75%, 80%, 85%, 90%
and
95%.
As those of skill will appreciate, the present invention includes
catalytically active
polypeptides of the present invention (i.e., enzymes). Catalytically active
polypeptides have
a specific activity of at least 20%, 30% or 40% and preferably at least 50%,
60% or 70% and
most preferably at least 80%, 90% or 95% that of the native (non-synthetic),
endogenous
polypeptide. Further, the substrate specificity (kcat/Km) is optionally
substantially similar to
the native (non-synthetic), endogenous polypeptide. Typically, the Km will be
at least 30%,
40% or 50%, that of the native (non-synthetic), endogenous polypeptide and
more preferably
at least 60%, 70%, 80% or 90%. Methods of assaying and quantifying measures of
enzymatic activity and substrate specificity (heat/Km) are well known to those
of skill in the
art.
Generally, the proteins of the present invention will, when presented as an
immunogen, elicit production of an antibody specifically reactive to a
polypeptide of the
present invention. Further, the proteins of the present invention will not
bind to antisera
raised against a polypeptide of the present invention which has been fully
immunosorbed
with the same polypeptide. Immunoassays for determining binding are well known
to those
of skill in the art. A preferred immunoassay is a competitive immunoassay as
discussed,
infra. Thus, the proteins of the present invention can be employed as
immunogens for
constructing antibodies immunoreactive to a protein of the present invention
for such
exemplary utilities as immunoassays or protein purification techniques.
Expression of Proteins in Host Cells
Using the nucleic acids of the present invention, one may express a protein of
the
present invention in a recombinantly engineered cell such as bacteria, yeast,
insect,
mammalian or preferably plant cells. The cells produce the protein in a .non-
natural condition
(e.g., in quantity, composition, location and/or time), because they have been
genetically
altered through human intervention to do so.
It is expected that those of skill in the art are knowledgeable in the
numerous
expression systems available for expression of a nucleic acid encoding a
protein of the
present invention. No attempt to describe in detail the various methods known
for the
expression of proteins in prokaryotes or eukaryotes will be made.
48
CA 02877639 2015-01-09
In brief summary, the expression of isolated nucleic acids encoding a protein
of the
present invention will typically be achieved by operably linking, for example,
the DNA or
cDNA to a promoter (which is either constitutive or regulatable), followed by
incorporation
into an expression vector. The vectors can be suitable for replication and
integration in
either prokaryotes or eukaryotes. Typical expression vectors contain
transcription and
translation terminators, initiation sequences and promoters useful for
regulation of the
expression of the DNA encoding a protein of the present invention. To obtain
high level
expression of a cloned gene, it is desirable to construct expression vectors
which contain, at
the minimum, a strong promoter to direct transcription, a ribosome binding
site for
translational initiation and a transcription/translation terminator. One of
skill would recognize
that modifications can be made to a protein of the present invention without
diminishing its
biological activity. Some modifications may be made to facilitate the cloning,
expression or
incorporation of the targeting molecule into a fusion protein.
Such modifications are well known to those of skill in the art and include,
for
example, a methionine added at the amino terminus to provide an initiation
site or additional
amino acids (e.g., poly His) placed on either terminus to create conveniently
located
purification sequences. Restriction sites or termination codons can also be
introduced.
A. Expression in Prokaryotes
Prokaryotic cells may be used as hosts for expression. Prokaryotes most
frequently
are represented by various strains of E. coli; however, other microbial
strains may also be
used. Commonly used prokaryotic control sequences which are defined herein to
include
promoters for transcription initiation, optionally with an operator, along
with ribosome binding
site sequences, include such commonly used promoters as the beta lactamase
(penicillinase) and lactose (lac) promoter systems (Chang, et a/., (1977)
Nature 198:1056),
the tryptophan (trp) promoter system (Goeddel, etal., (1980) Nucleic Acids
Res. 8:4057) and
the lambda derived P L promoter and N-gene ribosome binding site (Shimatake,
et al.,
(1981) Nature 292:128). The inclusion of selection markers in DNA vectors
transfected in E.
coli is also useful. Examples of such markers include genes specifying
resistance to
ampicillin, tetracycline or chloramphenicol.
The vector is selected to allow introduction into the appropriate host cell.
Bacterial
vectors are typically of plasmid or phage origin. Appropriate bacterial cells
are infected with
phage vector particles or transfected with naked phage vector DNA. If a
plasmid vector is
used, the bacterial cells are transfected with the plasmid vector DNA.
Expression systems
for expressing a protein of the present invention are available using Bacillus
sp. and
49
CA 02877639 2015-01-09
Salmonella (PaIva, etal., (1983) Gene 22:229-235; Mosbach, etal., (1983)
Nature 302:543-
545).
B. Expression in Eukaryotes
A variety of eukaryotic expression systems such as yeast, insect cell lines,
plant and
mammalian cells, are known to those of skill in the art. As explained briefly
below, a
polynucleotide of the present invention can be expressed in these eukaryotic
systems. In
some embodiments, transformed/transfected plant cells, as discussed infra, are
employed
as expression systems for production of the proteins of the instant invention.
Synthesis of heterologous proteins in yeast is well known. Sherman, et al.,
Methods
in Yeast Genetics, Cold Spring Harbor Laboratory (1982) is a well recognized
work
describing the various methods available to produce the protein in yeast. Two
widely utilized
yeast for production of eukaryotic proteins are Saccharomyces cerevisiae and
Pichia
pastoris. Vectors, strains and protocols for expression in Saccharomyces and
Pichia are
known in the art and available from commercial suppliers (e.g., Invitrogen).
Suitable vectors usually have expression control sequences, such as promoters,
including 3-phosphoglycerate kinase or alcohol oxidase, and an origin of
replication,
termination sequences and the like as desired.
A protein of the present invention, once expressed, can be isolated from yeast
by
lysing the cells and applying standard protein isolation techniques to the
lysate. The
monitoring of the purification process can be accomplished by using Western
blot techniques
or radioimmunoassay or other standard immunoassay techniques.
The sequences encoding proteins of the present invention can also be ligated
to
various expression vectors for use in transfecting cell cultures of, for
instance, mammalian,
insect or plant origin. Illustrative of cell cultures useful for the
production of the peptides are
mammalian cells. Mammalian cell systems often will be in the form of
monolayers of cells
although mammalian cell suspensions may also be used. A number of suitable
host cell
lines capable of expressing intact proteins have been developed in the art,
and include the
HEK293, BHK21 and CHO cell lines. Expression vectors for these cells can
include
expression control sequences, such as an origin of replication, a promoter
(e.g., the CMV
promoter, a HSVtk promoter or pgk (phosphoglycerate kinase) promoter), an
enhancer
(Queen, et al., (1986) Immunol. Rev. 89:49), and necessary processing
information sites,
such as ribosome binding sites, RNA splice sites, polyadenylation sites (e.g.,
an SV40 large
T Ag poly A addition site), and transcriptional terminator sequences. Other
animal cells
CA 02877639 2015-01-09
useful for production of proteins of the present invention are available, for
instance, from the
American Type Culture Collection.
Appropriate vectors for expressing proteins of the present invention in insect
cells are
usually derived from the SF9 baculovirus. Suitable insect cell lines include
mosquito larvae,
silkworm, army worm, moth and Drosophila cell lines such as a Schneider cell
line (see,
Schneider, (1987)J. Embryo!. Exp. MorphoL 27:353-365).
As with yeast, when higher animal or plant host cells are employed,
polyadenylation
or transcription terminator sequences are typically incorporated into the
vector. An example
of a terminator sequence is the polyadenylation sequence from the bovine
growth hormone
gene. Sequences for accurate splicing of the transcript may also be included.
An example
of a splicing sequence is the VP1 intron from SV40 (Sprague, et al., (1983) J.
ViroL 45:773-
781). Additionally, gene sequences to control replication in the host cell may
be
incorporated into the vector such as those found in bovine papilloma virus
type-vectors.
Saveria-Campo, Bovine Papilloma Virus DNA a Eukaryotic Cloning Vector in DNA
Cloning
Vol. ll a Practical Approach, Glover, Ed., IRL Press, Arlington, Virginia pp.
213-238 (1985).
Increasing the Activity and/or Level of an ethylene signaling associated
Polypeptide
Methods are provided to increase the activity and/or level of the ethylene
signaling
associated polypeptide of the invention. An increase in the level and/or
activity of the
ethylene signaling associated polypeptide of the invention can be achieved by
providing to
the plant an ethylene signaling associated polypeptide. The ethylene signaling
associated
polypeptide can be provided by introducing the amino acid sequence encoding
the ethylene
signaling associated polypeptide into the plant, introducing into the plant a
nucleotide
sequence encoding an ethylene signaling associated polypeptide or
alternatively by
modifying a genomic locus encoding the ethylene signaling associated
polypeptide of the
invention.
As discussed elsewhere herein, many methods are known the art for providing a
polypeptide to a plant including, but not limited to, direct introduction of
the polypeptide into
the plant, introducing into the plant (transiently or stably) a polynucleotide
construct encoding
a polypeptide having enhanced activity, such as . It is also recognized that
the methods of
the invention may employ a polynucleotide that is not capable of directing, in
the transformed
plant, the expression of a protein or an RNA. Thus, the level and/or activity
of an ethylene
signaling associated polypeptide may be increased by altering the gene
encoding the
ethylene signaling associated polypeptide or its promoter. See, e.g., Kmiec,
US Patent
Number 5,565,350; Zarling, et al., PCT/US93/03868. Therefore mutagenized
plants that
51
CA 02877639 2015-01-09
carry mutations in ethylene signaling associated genes, where the mutations
increase
expression of the ethylene signaling associated gene or increase the ethylene
signaling
associated activity of the encoded ethylene signaling associated polypeptide
are provided.
Reducing the Activity and/or Level of an ethylene signaling associated
Polypeptide
Methods are provided to reduce or eliminate the activity of an ethylene
signaling
associated polypeptide of the invention by transforming a plant cell with an
expression
cassette that expresses a polynucleotide that inhibits the expression of the
ethylene
signaling associated polypeptide. The polynucleotide may inhibit the
expression of the
ethylene signaling associated polypeptide directly, by preventing
transcription or translation
of the ethylene signaling associated messenger RNA, or indirectly, by encoding
a
polypeptide that inhibits the transcription or translation of an ethylene
signaling associated
gene encoding ethylene signaling associated polypeptide.
Methods for inhibiting or
eliminating the expression of a gene in a plant are well known in the art, and
any such
method may be used in the present invention to inhibit the expression of
ethylene signaling
associated polypeptide.
In accordance with the present invention, the expression of an ethylene
signaling
associated polypeptide is inhibited if the protein level of the ethylene
signaling associated
polypeptide is less than 70% of the protein level of the same ethylene
signaling associated
polypeptide in a plant that has not been genetically modified or mutagenized
to inhibit the
expression of that ethylene signaling associated polypeptide. In particular
embodiments of
the invention, the protein level of the ethylene signaling associated
polypeptide in a modified
plant according to the invention is less than 60%, less than 50%, less than
40%, less than
30%, less than 20%, less than 10%, less than 5% or less than 2% of the protein
level of the
same ethylene signaling associated polypeptide in a plant that is not a mutant
or that has not
been genetically modified to inhibit the expression of that ethylene signaling
associated
polypeptide. The expression level of the ethylene signaling associated
polype'Ptide may be
measured directly, for example, by assaying for the level of ethylene
signaling associated
polypeptide expressed in the plant cell or plant, or indirectly, for example,
by measuring the
ethylene response in the plant cell or plant, or by measuring the phenotypic
changes in the
plant. Methods for performing such assays are described elsewhere herein.
In other embodiments of the invention, the activity of the ethylene signaling
associated polypeptide is reduced or eliminated by transforming a plant cell
with an
expression cassette comprising a polynucleotide encoding a polypeptide that
inhibits the
activity of an ethylene signaling associated polypeptide. The activity of an
ethylene signaling
52
CA 02877639 2015-01-09
associated polypeptide is inhibited according to the present invention if the
activity of the
ethylene signaling associated polypeptide is less than 70% of the activity of
the same
ethylene signaling associated polypeptide in a plant that has not been
modified to inhibit the
ethylene signaling associated activity of that polypeptide. In particular
embodiments of the
invention, the ethylene signaling associated activity of the ethylene
signaling associated
polypeptide in a modified plant according to the invention is less than 60%,
less than 50%,
less than 40%, less than 30%, less than 20%, less than 10% or less than 5% of
the ethylene
signaling associated activity of the same polypeptide in a plant that that has
not been
modified to inhibit the expression of that ethylene signaling associated
polypeptide. The
ethylene signaling associated activity of an ethylene signaling associated
polypeptide is
"eliminated" according to the invention when it is not detectable by the assay
methods
described elsewhere herein. Methods of determining the alteration of activity
of an ethylene
signaling associated polypeptide are described elsewhere herein.
In other embodiments, the activity of an ethylene signaling associated
polypeptide
may be reduced or eliminated by disrupting the gene encoding the ethylene
signaling
associated polypeptide. The invention encompasses mutagenized plants that
carry
mutations in ethylene signaling associated genes, where the mutations reduce
expression of
the ethylene signaling associated gene or inhibit the activity of the encoded
ethylene
signaling associated polypeptide.
Thus, many methods may be used to reduce or eliminate the activity of an
ethylene
signaling associated polypeptide. In addition, more than one method may be
used to reduce
the activity of a single ethylene signaling associated polypeptide.
1. Polynucleotide-Based Methods:
In some embodiments of the present invention, a plant is transformed with an
expression cassette that is capable of expressing a polynucleotide that
inhibits the
expression of an ethylene signaling associated polypeptide of the invention.
The term
"expression" as used herein refers to the biosynthesis of a gene product,
including the
transcription and/or translation of said gene product. For example, for the
purposes of the
present invention, an expression cassette capable of expressing a
polynucleotide that
inhibits the expression of at least one ethylene signaling associated
polypeptide is an
expression cassette capable of producing an RNA molecule that inhibits the
transcription
and/or translation of at least one ethylene signaling associated polypeptide
of the invention.
The "expression" or "production" of a protein or polypeptide from a DNA
molecule refers to
the transcription and translation of the coding sequence to produce the
protein or
53
CA 02877639 2015-01-09
polypeptide, while the "expression" or "production" of a protein or
polypeptide from an RNA
molecule refers to the translation of the RNA coding sequence to produce the
protein or
polypeptide.
Examples of polynucleotides that inhibit the expression of an ethylene
signaling
associated polypeptide are given below.
i. Sense Suppression/Cosuppression
In some embodiments of the invention, inhibition of the expression of an
ethylene
signaling associated polypeptide may be obtained by sense suppression or
cosuppression.
For cosuppression, an expression cassette is designed to express an RNA
molecule
corresponding to all or part of a messenger RNA encoding an ethylene signaling
associated
polypeptide in the "sense" orientation. Over expression of the RNA molecule
can result in
reduced expression of the native gene. Accordingly, multiple plant lines
transformed with
the cosuppression expression cassette are screened to identify those that show
the greatest
inhibition of ethylene signaling associated polypeptide expression.
The polynucleotide used for cosuppression may correspond to all or part of the
sequence encoding the ethylene signaling associated polypeptide, all or part
of the 5' and/or
3' untranslated region of an ethylene signaling associated polypeptide
transcript, or all or
part of both the coding sequence and the untranslated regions of a transcript
encoding an
ethylene signaling associated polypeptide. In some embodiments where the
polynucleotide
comprises all or part of the coding region for the ethylene signaling
associated polypeptide,
the expression cassette is designed to eliminate the start codon of the
polynucleotide so that
no protein product will be translated.
Cosuppression may be used to inhibit the expression of plant genes to produce
plants having undetectable protein levels for the proteins encoded by these
genes. See, for
example, Broin, et al., (2002) Plant Cell 14:1417-1432. Cosuppression may also
be used to
inhibit the expression of multiple proteins in the same plant. See, for
example, US Patent
Number 5,942,657. Methods for using cosuppression to inhibit the
expression of
endogenous genes in plants are described in Flavell, of al., (1994) Proc.
Natl. Acad. Sc!.
USA 91:3490-3496; Jorgensen, et al., (1996) Plant Mol. Biol. 31:957-973;
Johansen and
Carrington, (2001) Plant Physic!. 126:930-938; Broin, etal., (2002) Plant Cell
14:1417-1432;
Stoutjesdijk, of al., (2002) Plant PhysioL 129:1723-1731; Yu, et al., (2003)
Phytochemistry
63:753-763; and US Patent Numbers 5,034,323, 5,283,184 and 5,942,657.
The efficiency of cosuppression may be increased by
including a poly-dT region in the expression cassette at a position 3' to the
sense sequence
54
CA 02877639 2015-01-09
and 5' of the polyadenylation signal. See, US Patent Application Publication
Number
2002/0048814 =
Typically, such a nucleotide sequence has
substantial sequence identity to the sequence of the transcript of the
endogenous gene,
optimally greater than about 65% sequence identity, more optimally greater
than about 85%
sequence identity, most optimally greater than about 95% sequence identity.
See, US
Patent Numbers 5,283,184 and 5,034,323
Ant/sense Suppression
In some embodiments of the invention, inhibition of the expression of the
ethylene
signaling associated polypeptide may be obtained by antisense suppression. For
antisense
suppression, the expression cassette is designed to express an RNA molecule
complementary to all or part of a messenger RNA encoding the ethylene
signaling
associated polypeptide. Over expression of the antisense RNA molecule can
result in
reduced expression of the native gene. Accordingly, multiple plant lines
transformed with
the antisense suppression expression cassette are screened to identify those
that show the
greatest inhibition of ethylene signaling associated polypeptide expression.
The polynucleotide for use in antisense suppression may correspond to all or
part of
the complement of the sequence encoding the ethylene signaling associated
polypeptide, all
or part of the complement of the 5' and/or 3' untranslated region of the
ethylene signaling
associated transcript, or all or part of the complement of both the coding
sequence and the
untranslated regions of a transcript encoding the ethylene signaling
associated polypeptide.
In addition, the antisense polynucleotide may be fully complementary (i.e.,
100% identical to
the complement of the target sequence) or partially complementary (i.e., less
than 100%
identical to the complement of the target sequence) to the target sequence.
Antisense
suppression may be used to inhibit the expression of multiple proteins in the
same plant.
See, for example, US Patent Number 5,942,657. Furthermore, portions of the
antisense
nucleotides may be used to disrupt the expression of the target gene.
Generally, sequences
of at least 50 nucleotides, 100 nucleotides, 200 nucleotides, 300, 400, 450,
500, 550 or
greater may be used. Methods for using antisense suppression to inhibit the
expression of
endogenous genes in plants are described, for example, in Liu, et at., (2002)
Plant PhysioL
129:1732-1743 and US Patent Numbers 5,759,829 and 5,942,657.
Efficiency of antisense suppression may be increased by
including a poly-dT region in the expression cassette at a position 3' to the
antisense
sequence and 5' of the polyadenylation signal. See, US Patent Application
Publication
Number 200210048814.
CA 02877639 2015-01-09
Double-Stranded RNA Interference
In some embodiments of the invention, inhibition of the expression of an
ethylene
signaling associated polypeptide may be obtained by double-stranded RNA
(dsRNA)
interference. For dsRNA interference, a sense RNA molecule like that described
above for
cosuppression and an antisense RNA molecule that is fully or partially
complementary to the
sense RNA molecule are expressed in the same cell, resulting in inhibition of
the expression
of the corresponding endogenous messenger RNA.
Expression of the sense and antisense molecules can be accomplished by
designing
the expression cassette to comprise both a sense sequence and an antisense
sequence.
Alternatively, separate expression cassettes may be used for the sense and
antisense
sequences. Multiple plant lines transformed with the dsRNA interference
expression
cassette or expression cassettes are then screened to identify plant lines
that show the
greatest inhibition of ethylene signaling associated polypeptide expression.
Methods for
using dsRNA interference to inhibit the expression of endogenous plant genes
are described
in Waterhouse, of al., (1998) Proc. Natl. Acad. Sci. USA 95:13959-13964, Liu,
et al., (2002)
Plant Physiol. 129:1732-1743, and WO 99/49029, WO 99/53050, WO 99/61631, and
WO
00/49035 =
iv. Hairpin RNA Interference and lntron-Containing Hairpin RNA
Interference
In some embodiments of the invention, inhibition of the expression of an
ethylene
signaling associated polypeptide may be obtained by hairpin RNA (hpRNA)
interference or
intron-containing hairpin RNA (ihpRNA) interference. These methods are highly
efficient at
inhibiting the expression of endogenous genes. See, Waterhouse and Helliwell,
(2003) Nat.
Rev. Genet. 4:29-38 and the references cited therein.
For hpRNA interference, the expression cassette is designed to express an RNA
molecule that hybridizes with itself to form a hairpin structure that
comprises a single-
stranded loop region and a base-paired stem. The base-paired stem region
comprises a
sense sequence corresponding to all or part of the endogenous messenger RNA
encoding
the gene whose expression is to be inhibited, and an antisense sequence that
is fully or
partially complementary to the sense sequence. Alternatively, the base-paired
stem region
may correspond to a portion of a promoter sequence controlling expression of
the gene to be
inhibited. Thus, the base-paired stem region of the molecule generally
determines the
56
CA 02877639 2015-01-09
specificity of the RNA interference. hpRNA molecules are highly efficient at
inhibiting the
expression of endogenous genes, and the RNA interference they induce is
inherited by
subsequent generations of plants. See, for example, Chuang and Meyerowitz,
(2000) Proc.
Natl. Acad. ScL USA 97:4985-4990; Stoutjesdijk, of al., (2002) Plant Physiol.
129:1723-
1731; and Waterhouse and Helliwell, (2003) Nat. Rev. Genet. 4:29-38. Methods
for using
hpRNA interference to inhibit or silence the expression of genes are
described, for example,
in Chuang arid Meyerowitz, (2000) Proc. Natl. Acad. ScL USA 97:4985-4990;
Stoutjesdijk, et
al., (2002) Plant Physiol. 129:1723-1731; Waterhouse and Helliwell, (2003)
Nat. Rev. Genet.
4:29-38; Pandolfini of al., BMC Biotechnology 3:7 and US Patent Application
Publication
Number 2003/0175965 A transient
assay for the efficiency of hpRNA constructs to silence gene expression in
vivo has been
described by Panstruga, et al., (2003) MoL Biol. Rep. 30:135-140.
For ihpRNA, the interfering molecules have the same general structure as for
hpRNA,
but the RNA molecule additionally comprises an intron that is capable of being
spliced in the
cell in which the ihpRNA is expressed. The use of an intron minimizes the size
of the loop in
the hairpin RNA molecule following splicing, and this increases the efficiency
of interference.
See, for example, Smith, et al., (2000) Nature 407:319-320. In fact, Smith, of
al., show
100% suppression of endogenous gene expression using ihpRNA-mediated
interference.
Methods for using ihpRNA interference to inhibit the expression of endogenous
plant genes
are described, for example, in Smith, et al., (2000) Nature 407:319-320;
Wesley, et al.,
(2001) Plant J. 27:581-590; Wang and Waterhouse, (2001) Curr. Opin. Plant BioL
5:146-
150; Waterhouse and Helliwell, (2003) Nat. Rev. Genet. 4:29-38; Helliwell and
Waterhouse,
(2003) Methods 30:289-295, and US Patent Application Publication Number
2003/0180945.
The expression cassette for hpRNA interference may also be designed such that
the
sense sequence and the antisense sequence do not correspond to an endogenous
RNA. In
this embodiment, the sense and antisense sequence flank a loop sequence that
comprises a
nucleotide sequence corresponding to all or part of the endogenous messenger
RNA of the
target gene. Thus, it is the loop region that determines the specificity of
the RNA
interference. See, for example, WO 02/00904; Mette, et a/., (2000) EMBO J
19:5194-5201;
Matzke, etal., (2001) Curr. Opin. Genet. DeveL 11:221-227; Scheid, etal.,
(2002) Proc. Natl.
Acad. Sc., USA 99:13659-13662; Aufsaftz, etal., (2002) Proc. Natl. Acad. Sci.
99(4):16499-
16506; Sijen, etal., Curr. Biol. (2001) 11:436-440) .
57
CA 02877639 2015-01-09
v. Am plicon-Mediated Interference
Amp!icon expression cassettes comprise a plant virus-derived sequence that
contains all or part of the target gene but generally not all of the genes of
the native virus.
The viral sequences present in the transcription product of the expression
cassette allow the
transcription product to direct its own replication. The transcripts produced
by the amplicon
may be either sense or antisense relative to the target sequence (i.e., the
messenger RNA
for the ethylene signaling associated polypeptide). Methods of using amplicons
to inhibit the
expression of endogenous plant genes are described, for example, in Angell and
Baulcombe,
(1997) EMBO J. 16:3675-3684, Angell and Baulcombe, (1999) Plant J. 20:357-362,
and US
Patent Number 6,646,805 .
vi. Ribozymes
In some embodiments, the polynucleotide expressed by the expression cassette
of
the invention is catalytic RNA or has ribozyme activity specific for the
messenger RNA of the
ethylene signaling associated polypeptide. Thus, the polynucleotide causes the
degradation
of the endogenous messenger RNA, resulting in reduced expression of the
ethylene
signaling associated polypeptide. This method is described, for example, in US
Patent
Number 4,987,071.
vii. Small Interfering RNA or Micro RNA
In some embodiments of the invention, inhibition of the expression of an
ethylene
signaling associated polypeptide may be obtained by RNA interference by
expression of a
gene encoding a micro RNA (miRNA). miRNAs are regulatory agents consisting of
about 22
ribonucleotides. miRNA are highly efficient at inhibiting the expression of
endogenous genes.
See, for example Javier, et al., (2003) Nature 425:257-263.
For miRNA interference, the expression cassette is designed to express an RNA
molecule that is modeled on an endogenous miRNA gene. The miRNA gene encodes
an
RNA that forms a hairpin structure containing a 22-nucleotide sequence that is
complementary to another endogenous gene (target sequence). For suppression of
ethylene signaling associated expression, the 22-nucleotide sequence is
selected from an
ethylene signaling associated transcript sequence and contains 22 nucleotides
of said
ethylene signaling associated sequence in sense orientation and 21 nucleotides
of a
corresponding antisense sequence that is complementary to the sense sequence.
miRNA
molecules are highly efficient at inhibiting the expression of endogenous
genes, and the
RNA interference they induce is inherited by subsequent generations of plants.
58
CA 02877639 2015-01-09
2. Polypeptide-Based Inhibition of Gene Expression
In one embodiment, the polynucleotide encodes a zinc finger protein that binds
to a
gene encoding an ethylene signaling associated polypeptide, resulting in
reduced
expression of the gene. In particular embodiments, the zinc finger protein
binds to a
regulatory region of an ethylene signaling associated gene. In other
embodiments, the zinc
finger protein binds to a messenger RNA encoding an ethylene signaling
associated
polypeptide and prevents its translation. Methods of selecting sites for
targeting by zinc
finger proteins have been described, for example, in US Patent Number
6,453,242, and
methods for using zinc finger proteins to inhibit the expression of genes in
plants are
described, for example, in US Patent Application Publication No. 2003/0037355
3. Polypeptide-Based Inhibition of Protein Activity
In some embodiments of the invention, the polynucleotide encodes an antibody
that
binds to at least one ethylene signaling associated polypeptide, and reduces
the enhanced
activity of the ethylene signaling associated polypeptide. In another
embodiment, the
binding of the antibody results in increased turnover of the antibody-
ethylene signaling
associated complex by cellular quality control mechanisms. The expression of
antibodies in
plant cells and the inhibition of molecular pathways by expression and binding
of antibodies
to proteins in plant cells are well known in the art. See, for example, Conrad
and Sonnewald,
(2003) Nature Biotech. 21:35-36.
4. Gene Disruption
In some embodiments of the present invention, the activity of an ethylene
signaling
associated polypeptide is reduced or eliminated by disrupting the gene
encoding the
ethylene signaling associated polypeptide. The gene encoding the ethylene
signaling
associated polypeptide may be disrupted by any method known in the art. For
example, in
one embodiment, the gene is disrupted by transposon tagging. In another
embodiment, the
gene is disrupted by mutagenizing plants using random or targeted mutagenesis,
and
selecting for plants that have reduced activity.
I. Transposon Tagging
In one embodiment of the invention, transposon tagging is used to reduce or
eliminate the ethylene signaling activity of one or more ethylene signaling
associated
59
CA 02877639 2015-01-09
polypeptide. Transposon tagging comprises inserting a transposon within an
endogenous
ethylene signaling associated gene to reduce or eliminate expression of the
ethylene
signaling associated polypeptide.
In this embodiment, the expression of one or more ethylene signaling
associated
polypeptide is reduced or eliminated by inserting a transposon within a
regulatory region or
coding region of the gene encoding the ethylene signaling associated
polypeptide. A
transposon that is within an exon, intron, 5' or 3' untranslated sequence, a
promoter, or any
other regulatory sequence of an ethylene signaling associated gene may be used
to reduce
or eliminate the expression and/or activity of the encoded ethylene signaling
associated
polypeptide.
Methods for the transposon tagging of specific genes in plants are well known
in the
art. See, for example, Maes, et a/., (1999) Trends Plant Sci. 4:90-96;
Dharmapuri and Sonti,
(1999) FEMS Microbiol. Lett. 179:53-59; Meissner, etal., (2000) Plant J.
22:265-274; Phogat,
etal., (2000) J. BioscL 25:57-63; Walbot, (2000) Curr. Op/n. Plant Biol. 2:103-
107; Gai, of at.,
(2000) Nucleic Acids Res. 28:94-96; Fitzmaurice, etal., (1999) Genetics
153:1919-1928). In
addition, the TUSC process for selecting Mu insertions in selected genes has
been
described in Bensen, et a/., (1995) Plant Cell 7:75-84; Mena, of al., (1996)
Science
274:1537-1540; and US Patent Number 5,962,764.
Mutant Plants with Reduced Activity
Additional methods for decreasing or eliminating the expression of endogenous
genes in plants are also known in the art and can be similarly applied to the
instant invention.
These methods include other forms of mutagenesis, such as ethyl
rnethanesulfonate-
induced mutagenesis, deletion mutagenesis, and fast neutron deletion
mutagenesis used in
a reverse genetics sense (with PCR) to identify plant lines in which the
endogenous gene
has been deleted. For examples of these methods see, Ohshima, et al., (1998)
Virology
243:472-481; Okubara, et al., (1994) Genetics 137:867-874; and Quesada, et
al., (2000)
Genetics 154:421-436 _
In addition, a fast
and automatable method for screening for chemically induced mutations, TILLING
(Targeting Induced Local Lesions In Genomes), using denaturing HPLC or
selective
endonuclease digestion of selected PCR products is also applicable to the
instant invention.
See, McCallum, of al., (2000) Nat. Biotechnol. 18:455-457 =
Mutations that impact gene expression or that interfere with the function
(enhanced
activity) of the encoded protein are well known in the art. Insertional
mutations in gene
CA 02877639 2015-01-09
exons usually result in null-mutants. Mutations in conserved residues are
particularly
effective in inhibiting the activity of the encoded protein. Conserved
residues of plant
ethylene signaling associated polypeptides suitable for mutagenesis with the
goal to
eliminate ethylene signaling associated activity have been described. Such
mutants can be
isolated according to well-known procedures, and mutations in different
ethylene signaling
associated loci can be stacked by genetic crossing. See, for example, Gruis,
et al., (2002)
Plant Ce/I 14:2863-2882.
In another embodiment of this invention, dominant mutants can be used to
trigger
RNA silencing due to gene inversion and recombination of a duplicated gene
locus. See, for
example, Kusaba, et al., (2003) Plant Cell 15:1455-1467.
The invention encompasses additional methods for reducing or eliminating the
activity of one or more ethylene signaling associated polypeptide. Examples of
other
methods for altering or mutating a genomic nucleotide sequence in a plant are
known in the
art and include, but are not limited to, the use of RNA:DNA vectors, RNA:DNA
mutational
IS vectors, RNA:DNA repair vectors, mixed-duplex oligonucleotides, self-
complementary
RNA:DNA oligonucleotides, and recombinogenic oligonucleobases. Such vectors
and
methods of use are known in the art. See, for example, US Patent Numbers
5,565,350;
5,731,181; 5,756,325; 5,760,012; 5,795,972 and 5,871,984
See also, WO 98/49350, WO 99/07865, WO 99/25821 and
Beetham, et al., (1999) Proc. Natl. Acad. Sci. USA 96:8774-8778.
TransfectionfTransformation of Cells
The method of transformation/transfection is not critical to the instant
invention;
various methods of transformation or transfection are currently available. As
newer methods
are available to transform crops or other host cells they may be directly
applied.
Accordingly, a wide variety of methods have been developed to insert a DNA
sequence into the genome of a host cell to obtain the transcription and/or
translation of the
sequence to effect phenotypic changes in the organism. Thus, any method which
provides
for effective transformation/transfection may be employed.
A. Plant Transformation
A DNA sequence coding for the desired polypeptide of the present invention,
for
example a cDNA or a genomic sequence encoding a full length protein, will be
used to
construct a recombinant expression cassette which can be introduced into the
desired plant.
61
CA 02877639 2015-01-09
Isolated nucleic acid acids of the present invention can be introduced into
plants
according to techniques known in the art. Generally, recombinant expression
cassettes as
described above and suitable for transformation of plant cells are prepared.
Techniques for
transforming a wide variety of higher plant species are well known and
described in the
technical, scientific, and patent literature. See, for example, Weising,
etal., (1988) Ann. Rev.
Genet. 22:421-477. For example, the DNA construct may be introduced directly
into the
genomic DNA of the plant cell using techniques such as electroporation,
polyethylene glycol
(PEG), poration, particle bombardment, silicon fiber delivery, or
microinjection of plant cell
protoplasts or embryogenic callus. See, e.g., Tomes, et al., Direct DNA
Transfer into Intact
Plant Cells Via Microprojectile Bombardment. pp. 197213 in Plant Cell, Tissue
and Organ
Culture, Fundamental Methods. eds. Gamborg and Phillips. Springer-Verlag
Berlin
Heidelberg New York, 1995. Alternatively, the DNA constructs may be combined
with
suitable T-DNA flanking regions and introduced into a conventional
Agrobacterium
tumefaciens host vector. The virulence functions of the Agrobacterium
tumefaciens host will
direct the insertion of the construct and adjacent marker into the plant cell
DNA when the cell
is infected by the bacteria. See, US Patent Number 5,591,616.
The introduction of DNA constructs using PEG precipitation is described in
Paszkowski, et al., (1984) Embo J. 3:2717-2722. Electroporation techniques are
described
in Fromm, et al., (1985) Proc. Natl. Acad. ScL (USA) 82:5824. Ballistic
transformation
techniques are described in Klein, etal., (1987) Nature 327:70-73.
Agrobacterium tumefaciens-mediated transformation techniques are well
described
in the scientific literature. See, for example Horsch, etal., (1984) Science
233:496-498, and
Fraley, et al., (1983) Proc. Natl. Acad. ScL (USA) 80:4803. Although
Agrobacterium is
useful primarily in dicots, certain monocots can be transformed by
Agrobacterium. For
instance, Agrobacterium transformation of maize is described in US Patent
Number
5,550,318.
Other methods of transfection or transformation include (1) Agrobacterium
rhizogenes-mediated transformation (see, e. g., Lichtenstein and Fuller In:
Genetic
Engineering, vol. 6, Rigby, Ed., London, Academic Press, 1987; and
Lichtenstein and Draper,
In: DNA Cloning, Vol. II, Glover, Ed., Oxford, IRI Press, 1985), Application
PCT/US87/02512
(WO 88/02405 published Apr. 7,1988) describes the use of A. rhizogenes strain
A4 and its
Ri plasmid along with A. tumefaciens vectors pARC8 orpARC16; (2) liposome-
mediated
DNA uptake (see, e.g., Freeman, et al., (1984) Plant Cell PhysioL 25:1353 );
(3) the
vortexing method (see, e.g., Kindle, (1990) Proc. Natl. Acad. ScL (USA)
87:1228).
62
CA 02877639 2015-01-09
DNA can also be introduced into plants by direct DNA transfer into pollen as
described by Zhou, etal., (1983) Methods in Enzymology 101:433; Hess, (1987)
Intern Rev.
CytoL 107:367; Luo, et al., (1988) Plant MoL Biol. Reporter 6:165. Expression
of polypeptide
coding genes can be obtained by injection of the DNA into reproductive organs
of a plant as
described by Pena, etal., (1987) Nature 325:274.
DNA can also be injected directly into the cells of immature embryos and the
rehydration of desiccated embryos as described by Neuhaus, et al., (1987)
Theor. App!.
Genet. 75:30; and Benbrook, et al., (1986) in Proceedings Bio Expo 1986,
Butterworth,
Stoneham, Mass., pp. 27-54. A variety of plant viruses that can be employed as
vectors are
known in the art and include cauliflower mosaic virus (CaMV), geminivirus,
brome mosaic
virus, and tobacco mosaic virus.
B. Transfection of Prokaryotes, Lower Eukarvotes, and Animal Cells
Animal and lower eukaryotic (e.g., yeast) host cells are competent or rendered
competent for transfection by various means. There are several well-known
methods of
introducing DNA into animal cells. These include: calcium phosphate
precipitation, fusion of
the recipient cells with bacterial protoplasts containing the DNA, treatment
of the recipient
cells with liposomes containing the DNA, DEAE dextran, electroporation,
biolistics, and
micro-injection of the DNA directly into the cells. The transfected cells are
cultured by
means well known in the art. Kuchler, Biochemical Methods in Cell Culture and
Virology,
Dowden, Hutchinson and Ross, Inc. (1977).
Synthesis of Proteins
The proteins of the present invention can be constructed using non-cellular
synthetic
methods. Solid phase synthesis of proteins of less than about 50 amino acids
in length may
be accomplished by attaching the C-terminal amino acid of the sequence to an
insoluble
support followed by sequential addition of the remaining amino acids in the
sequence.
Techniques for solid phase synthesis are described by Barany and Merrifield,
Solid-Phase
Peptide Synthesis, pp. 3-284 in The Peptides: Analysis, Synthesis, Biology
Vol. 2 : Special
Methods in Peptide Synthesis, Part A.; Merrifield, etal., (1963) J. Am. Chem.
Soc. 85:2149-
2156 and Stewart, et al., Solid Phase Peptide Synthesis, 2nd ed., Pierce Chem.
Co.,
Rockford, IL (1984). Proteins of greater length may be synthesized by
condensation of the
amino and carboxy termini of shorter fragments. Methods of forming peptide
bonds by
activation of a carboxy terminal end (e.g., by the use of the coupling reagent
N, N'-
dicycylohexylcarbodiimide) are known to those of skill.
63
CA 02877639 2015-01-09
Purification of Proteins
The proteins of the present invention may be purified by standard techniques
well
known to those of skill in the art. Recombinantly produced proteins of the
present invention
can be directly expressed or expressed as a fusion protein. The recombinant
protein is
purified by a combination of cell lysis (e.g., sonication, French press) and
affinity
chromatography. For fusion products, subsequent digestion of the fusion
protein with an
appropriate proteolytic enzyme releases the desired recombinant protein.
The proteins of this invention, recombinant or synthetic, may be purified to
substantial purity by standard techniques well known in the art, including
detergent
solubilization, selective precipitation with such substances as ammonium
sulfate, column
chromatography, immunopurification methods, and others. See, for instance, R.
Scopes,
Protein Purification: Principles and Practice, Springer-Verlag: New York
(1982); Deutscher,
Guide to Protein Purification, Academic Press (1990). For example, antibodies
may be
raised to the proteins as described herein. Purification from E. coli can be
achieved following
procedures described in US Patent Number 4,511,503. The protein may then be
isolated
from cells expressing the protein and further purified by standard protein
chemistry
techniques as described herein. Detection of the expressed protein is achieved
by methods
known in the art and include, for example, radioimmunoassays, Western blotting
techniques
or immunoprecipitation.
Transgenic Plant Regeneration
Transformed plant cells which are derived by any of the above transformation
techniques can be cultured to regenerate a whole plant which possesses the
transformed
genotype. Such regeneration techniques often rely on manipulation of
certain
phytohormones in a tissue culture growth medium. For transformation and
regeneration of
maize, see, Gordon-Kamm, et al., (1990) The Plant Cell 2:603-618.
Plants cells transformed with a plant expression vector can be regenerated,
e.g.,
from single cells, callus tissue or leaf discs according to standard plant
tissue culture
techniques. It is well known in the art that various cells, tissues, and
organs from almost any
plant can be successfully cultured to regenerate an entire plant. Plant
regeneration from
cultured protoplasts is described in Evans, et al., Protoplasts Isolation and
Culture,
Handbook of Plant Cell Culture, Macmillan Publishing Company, New York, pp.
124-176
(1983); and Binding, Regeneration of Plants, Plant Protoplasts, CRC Press,
Boca Raton, pp,
21-73 (1985).
64
CA 02877639 2015-01-09
The regeneration of plants containing the foreign gene introduced by
Agrobacterium
from leaf explants can be achieved as described by Horsch, etal., (1985)
Science 227:1229-
1231. In this procedure, transformants are grown in the presence of a
selection agent and in
a medium that induces the regeneration of shoots in the plant species being
transformed as
described by Fraley, et a/., (1983) Proc. Natl. Acad. ScL USA 80:4803. This
procedure
typically produces shoots within two to four weeks and these transformant
shoots are then
transferred to an appropriate root-inducing medium containing the selective
agent and an
antibiotic to prevent bacterial growth. Transgenic plants of the present
invention may be
fertile or sterile.
Regeneration can also be obtained from plant callus, explants, organs, or
parts
thereof. Such regeneration techniques are described generally in Kleen, et
al., (1987) Ann.
Rev. of Plant Phys. 38:467-486. The regeneration of plants from either single
plant
protoplasts or various explants is well known in the art. See, for example,
Methods for Plant
Molecular Biology, Weissbach and Weissbach, eds., Academic Press, Inc., San
Diego, Calif.
(1988). This regeneration and growth process includes the steps of
selection of
transformant cells and shoots, rooting the transformant shoots and growth of
the plantlets in
soil. For maize cell culture and regeneration see generally, The Maize
Handbook, Freeling
and Walbot, Eds., Springer, New York (1994); Corn and Corn Improvement, 3rd
edition,
Sprague and Dudley Eds., American Society of Agronomy, Madison, Wisconsin
(1988).
One of skill will recognize that after the recombinant expression cassette is
stably
incorporated in transgenic plants and confirmed to be operable, it can be
introduced into
other plants by sexual crossing. Any of a number of standard breeding
techniques can be
used, depending upon the species to be crossed. In vegetatively propagated
crops, mature
transgenic plants can be propagated by the taking of cuttings or by tissue
culture techniques
to produce multiple identical plants.
Selection of desirable transgenics is made and new varieties are obtained and
propagated vegetatively for commercial use. In seed propagated crops, mature
transgenic
plants can be self crossed to produce a homozygous inbred plant. The inbred
plant
produces seed containing the newly introduced heterologous nucleic acid. These
seeds can
be grown to produce plants that would produce the selected phenotype.
Parts obtained from the regenerated plant, such as flowers, seeds, leaves,
branches,
fruit and the like are included in the invention, provided that these parts
comprise cells
comprising the isolated nucleic acid of the present invention. Progeny and
variants, and
mutants of the regenerated plants are also included within the scope of the
invention,
provided that these parts comprise the introduced nucleic acid sequences.
Transgenic
CA 02877639 2015-01-09
plants expressing the selectable marker can be screened for transmission of
the nucleic acid
of the present invention by, for example, standard immunoblot and DNA
detection
techniques. Transgenic lines are also typically evaluated on levels of
expression of the
heterologous nucleic acid. Expression at the RNA level can be determined
initially to identify
and quantitate expression-positive plants. Standard techniques for RNA
analysis can be
employed and include PCR amplification assays using oligonucleotide primers
designed to
amplify only the heterologous RNA templates and solution hybridization assays
using
heterologous nucleic acid-specific probes. The RNA-positive plants can then
analyzed for
protein expression by Western immunoblot analysis using the specifically
reactive antibodies
of the present invention. In addition, in situ hybridization and
immunocytochemistry
according to standard protocols can be done using heterologous nucleic acid
specific
polynucleotide probes and antibodies, respectively, to localize sites of
expression within
transgenic tissue. Generally, a number of transgenic lines are usually
screened for the
incorporated nucleic acid to identify and select plants with the most
appropriate expression
profiles.
A preferred embodiment is a transgenic plant that is homozygous for the added
heterologous nucleic acid; i.e., a transgenic plant that contains two added
nucleic acid
sequences, one gene at the same locus on each chromosome of a chromosome pair.
A
homozygous transgenic plant can be obtained by sexually mating (selfing) a
heterozygous
transgenic plant that contains a single added heterologous nucleic acid,
germinating some of
the seed produced and analyzing the resulting plants produced for altered
expression of a
polynucleotide of the present invention relative to a control plant (i.e.,
native, nontransgenic).
Back-crossing to a parental plant and out-crossing with a non-transgenic plant
are also
contemplated.
Modulation of Polvpeptide Levels and/or Composition
The present invention further provides a method for modulating (i.e.,
increasing or
decreasing) the concentration or ratio of the polypeptides of the present
invention in a plant
or part thereof. Modulation can be effected by increasing or decreasing the
concentration
and/or the ratio of the polypeptides of the present invention in a plant.
The method comprises introducing into a plant cell a recombinant expression
cassette comprising a polynucleotide of the present invention as described
above to obtain a
transformed plant cell, culturing the transformed plant cell under plant cell
growing conditions
and inducing or repressing expression of a polynucleotide of the present
invention in the
66
CA 02877639 2015-01-09
plant for a time sufficient to modulate concentration and/or the ratios of the
polypeptides in
the plant or plant part.
In some embodiments, the concentration and/or ratios of polypeptides of the
present
invention in a plant may be modulated by altering, in vivo or in vitro, the
promoter of a gene
to up-or down-regulate gene expression. In some embodiments, the coding
regions of native
genes of the present invention can be altered via substitution, addition,
insertion, or deletion
to decrease activity of the encoded enzyme. See, e.g., Kmiec, US Patent Number
5,565,350; Zarling, et al., PCT/US93/03868. And in some embodiments, an
isolated nucleic
acid (e.g., a vector) comprising a promoter sequence is transfected into a
plant cell.
to,
Subsequently, a plant cell comprising the promoter operably linked to a
polynucleotide of the present invention is selected for by means known to
those of skill in the
art such as, but not limited to, Southern blot, DNA sequencing, or PCR
analysis using
primers specific to the promoter and to the gene and detecting amplicons
produced
therefrom. A plant or plant part altered or modified by the foregoing
embodiments is grown
under plant forming conditions for a time sufficient to modulate the
concentration and/or
ratios of polypeptides of the present invention in the plant. Plant forming
conditions are well
known in the art and discussed briefly, supra.
In general, concentration or the ratios of the polypeptides is increased or
decreased
by at least 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80% or 90% relative to a
native
control plant, plant part or cell lacking the aforementioned recombinant
expression cassette.
Modulation in the present invention may occur during and/or subsequent to
growth of the
plant to the desired stage of development. Modulating nucleic acid expression
temporally
and/or in particular tissues can be controlled by employing the appropriate
promoter
operably linked to a polynucleotide of the present invention in, for example,
sense or
antisense orientation as discussed in greater detail, supra. Induction of
expression of a
polynucleotide of the present invention can also be controlled by exogenous
administration
of an effective amount of inducing compound. Inducible promoters and inducing
compounds
which activate expression from these promoters are well known in the art. In
preferred
embodiments, the polypeptides of the present invention are modulated in
monocots,
particularly maize.
Molecular Markers
The present invention provides a method of genotyping a plant comprising a
polynucleotide of the present invention. Optionally, the plant is a monocot,
such as maize or
sorghum. Genotyping provides a means of distinguishing homologs of a
chromosome pair
67
CA 02877639 2015-01-09
and can be used to differentiate segregants in a plant population. Molecular
marker
methods can be used for phylogenetic studies, characterizing genetic
relationships among
crop varieties, identifying crosses or somatic hybrids, localizing chromosomal
segments
affecting monogenic traits, map based cloning, and the study of quantitative
inheritance.
See, e.g., Clark, Ed., Plant Molecular Biology: A Laboratory Manual. Berlin,
Springer Verlag,
1997. Chapter 7. For molecular marker methods, see generally, "The DNA
Revolution" in:
Paterson, Genome Mapping in Plants (Austin, TX, Academic Press/R. G. Landis
Company,
1996) pp. 7-21.
The particular method of genotyping in the present invention may employ any
number of molecular marker analytic techniques such as, but not limited to,
restriction
fragment length polymorphisms (RFLPs). RFLPs are the product of allelic
differences
between DNA restriction fragments resulting from nucleotide sequence
variability. As is well
known to those of skill in the art, RFLPs are typically detected by extraction
of genomic DNA
and digestion with a restriction enzyme. Generally, the resulting fragments
are separated
according to size and hybridized with a probe; single copy probes are
preferred. Restriction
fragments from homologous chromosomes are revealed.
Differences in fragment size among alleles represent an RFLP. Thus, the
present
invention further provides a means to follow segregation of a gene or nucleic
acid of the
present invention as well as chromosomal sequences genetically linked to these
genes or
nucleic acids using such techniques as RFLP analysis. Linked chromosomal
sequences are
within 50 centiMorgans (cM), often within 40 or 30 cM, preferably within 20 or
10 cM, more
preferably within 5, 3, 2 or 1 cM of a gene of the present invention.
In the present invention, the nucleic acid probes employed for molecular
marker
mapping of plant nuclear genomes selectively hybridize, under selective
hybridization
conditions, to a gene encoding a polynucleotide of the present invention. In
preferred
embodiments, the probes are selected from polynucleotides of the present
invention.
Typically, these probes are cDNA probes or restriction-enzyme treated (e.g.,
Pst I)
genomic clones. The length of the probes is discussed in greater detail,
supra, but are
typically at least 15 bases in length, more preferably at least 20, 25, 30,
35, 40 or 50 bases
in length. Generally, however, the probes are less than about 1 kilobase in
length.
Preferably, the probes are single copy probes that hybridize to a unique locus
in a haploid
chromosome complement. Some exemplary restriction enzymes employed in RFLP
mapping are EcoRI, EcoRv, and Sstl. As used herein the term "restriction
enzyme" includes
reference to a composition that recognizes and, alone or in conjunction with
another
composition, cleaves at a specific nucleotide sequence.
68
CA 02877639 2015-01-09
The method of detecting an RFLP comprises the steps of (a) digesting genomic
DNA
of a plant with a restriction enzyme; (b) hybridizing a nucleic acid probe,
under selective
hybridization conditions, to a sequence of a polynucleotide of the present of
said genomic
DNA; (c) detecting therefrom a RFLP. Other methods of differentiating
polymorphic (allelic)
variants of polynucleotides of the present invention can be had by utilizing
molecular marker
techniques well known to those of skill in the art including such techniques
as: 1) single
stranded conformation analysis (SSCA); 2) denaturing gradient gel
electrophoresis (DGGE);
3) RNase protection assays; 4) allele-specific oligonucleotides (AS0s);5) the
use of proteins
which recognize nucleotide mismatches, such as the E. coli mutS protein and 6)
allele-
specific PCR. Other approaches based on the detection of mismatches between
the two
complementary DNA strands include clamped denaturing gel electrophoresis
(CDGE);
heteroduplex analysis (HA); and chemical mismatch cleavage (CMC). Thus, the
present
invention further provides a method of genotyping comprising the steps of
contacting, under
stringent hybridization conditions, a sample suspected of comprising a
polynucleotide of the
present invention with a nucleic acid probe. Generally, the sample is a plant
sample;
preferably, a sample suspected of comprising a maize polynucleotide of the
present
invention (e.g., gene, mRNA). The nucleic acid probe selectively hybridizes,
under stringent
conditions, to a subsequence of a polynucleotide of the present invention
comprising a
polymorphic marker. Selective hybridization of the nucleic acid probe to the
polymorphic
marker nucleic acid sequence yields a hybridization complex. Detection of the
hybridization
complex indicates the presence of that polymorphic marker in the sample. In
preferred
embodiments, the nucleic acid probe comprises a polynucleotide of the present
invention.
UTRs and Codon Preference
In general, translational efficiency has been found to be regulated by
specific
sequence elements in the 5'non-coding or untranslated region (5' UTR) of the
RNA. Positive
sequence motifs include translational initiation consensus sequences (Kozak,
(1987) Nucleic
Acids Res. 15:8125) and the 7-methylguanosine cap structure (Drummond, et al.,
(1985)
Nucleic Acids Res. 13:7375). Negative elements include stable intramolecular
5'UTR stem-
loop structures (Muesing, at aL, (1987) Cell 48:691) and AUG sequences or
short open
reading frames preceded by an appropriate AUG in the 5' UTR (Kozak, supra,
Rao, et al.,
(1988) Mol. and Cell. Biol. 8:284). Accordingly, the present invention
provides 51and/or
3'untranslated regions for modulation of translation of heterologous coding
sequences.
Further, the polypeptide-encoding segments of the polynucleotides of the
present
invention can be modified to alter codon usage. Altered codon usage can be
employed to
69
CA 02877639 2015-01-09
alter translational efficiency and/or to optimize the coding sequence for
expression in a
desired host such as to optimize the codon usage in a heterologous sequence
for
expression in maize. Codon usage in the coding regions of the polynucleotides
of the
present invention can be analyzed statistically using commercially available
software
packages such as "Codon Preference" available from the University of Wisconsin
Genetics
Computer Group (see, Devereaux, et aL, (1984) Nucleic Acids Res. 12:387-395 )
or
MacVector 4.1 (Eastman Kodak Co., New Haven, Conn.). Thus, the present
invention
provides a codon usage frequency characteristic of the coding region of at
least one of the
polynucleotides of the present invention. The number of polynucleotides that
can be used to
determine a codon usage frequency can be any integer from 1 to the number of
polynucleotides of the present invention as provided herein. Optionally, the
polynucleotides
will be full-length sequences. An exemplary number of sequences for
statistical analysis can
be at least 1,5, 10, 20, 50 or 100.
Sequence Shuffling
The present invention provides methods for sequence shuffling using
polynucleotides
of the present invention, and compositions resulting therefrom. Sequence
shuffling is
described in PCT Publication Number WO 97/20078. See also, Zhang, et al.,
(1997) Proc.
Natl. Acad. Sci. USA 94:4504-4509. Generally, sequence shuffling provides a
means for
generating libraries of polynucleotides having a desired characteristic which
can be selected
or screened for. Libraries of recombinant polynucleotides are generated from a
population
of related sequence polynucleotides which comprise sequence regions which have
substantial sequence identity and can be homologously recombined in vitro or
in vivo. The
population of sequence-recombined polynucleotides comprises a subpopulation of
polynucleotides which possess desired or advantageous characteristics and
which can be
selected by a suitable selection or screening method. The characteristics can
be any
property or attribute capable of being selected for or detected in a screening
system, and
may include properties of: an encoded protein, a transcriptional element, a
sequence
controlling transcription, RNA processing, RNA stability, chromatin
conformation, translation,
or other expression property of a gene or transgene, a replicative element, a
protein-binding
element, or the like, such as any feature which confers a selectable or
detectable property.
In some embodiments, the selected characteristic will be a decreased Km and/or
increased
KCat over the wild-type protein as provided herein. In other embodiments, a
protein or
polynucleotide generated from sequence shuffling will have a ligand binding
affinity greater
CA 02877639 2015-01-09
than the non-shuffled wild-type polynucleotide. The increase in such
properties can be at
least 110%, 120%, 130%, 140% or at least 150% of the wild-type value.
Generic and Consensus Sequences
Polynucleotides and polypeptides of the present invention further include
those
having: (a) a generic sequence of at least two homologous polynucleotides or
polypeptides,
respectively, of the present invention and (b) a consensus sequence of at
least three
homologous polynucleotides or polypeptides, respectively, of the present
invention. The
generic sequence of the present invention comprises each species of
polypeptide or
polynucleotide embraced by the generic polypeptide or polynucleotide sequence,
respectively. The individual species encompassed by a polynucleotide having an
amino acid
or nucleic acid consensus sequence can be used to generate antibodies or
produce nucleic
acid probes or primers to screen for homologs in other species, genera,
families, orders,
classes, phyla or kingdoms. For example, a polynucleotide having a consensus
sequence
from a gene family of Zea mays can be used to generate antibody or nucleic
acid probes or
primers to other Gramineae species such as wheat, rice or sorghum.
Alternatively, a polynucleotide having a consensus sequence generated from
orthologous genes can be used to identify or isolate orthologs of other taxa.
Typically, a
polynucleotide having a consensus sequence will be at least 25, 30 or 40 amino
acids in
length, or 20, 30, 40, 50, 100 or 150 nucleotides in length. As those of skill
in the art are
aware, a conservative amino acid substitution can be used for amino acids
which differ
amongst aligned sequence but are from the same conservative substitution group
as
discussed above. Optionally, no more than 1 or 2 conservative amino acids are
substituted
for each 10 amino acid length of consensus sequence.
Similar sequences used for generation of a consensus or generic sequence
include
any number and combination of allelic variants of the same gene, orthologous,
or paralogous
sequences as provided herein. Optionally, similar sequences used in generating
a
consensus or generic sequence are identified using the BLAST algorithm's
smallest sum
probability (P (N)). Various suppliers of sequence-analysis software are
listed in chapter 7 of
Current Protocols in Molecular Biology, Ausubel, et al., Eds., Current
Protocols, a joint
venture between Greene Publishing Associates, Inc. and John Wiley & Sons, Inc.
(Supplement 30).
A polynucleotide sequence is considered similar to a reference sequence if the
smallest sum probability in a comparison of the test nucleic acid to the
reference nucleic acid
is less than about 0.1, more preferably less than about 0.01 or 0.001 and most
preferably
71
CA 02877639 2015-01-09
less than about 0.0001, or 0.00001. Similar polynucleotides can be aligned and
a
consensus or generic sequence generated using multiple sequence alignment
software
available from a number of commercial suppliers such as the Genetics Computer
Group's
(Madison, WI) PILEUP software, Vector NTI's (North Bethesda, MD) ALIGNX, or
Genecode's (Ann Arbor, MI) SEQUENCHER. Conveniently, default parameters of
such
software can be used to generate consensus or generic sequences.
Machine Applications
The present invention provides machines, articles of manufacture, and
processes for
to identifying, modeling, or analyzing the polynucleotides and polypeptides
of the present
invention. Identification methods permit identification of homologues of the
polynucleotides
or polypeptides of the present invention while modeling and analysis methods
permit
recognition of structural or functional features of interest.
A. Machines: Data Processing Systems
In one embodiment, the present invention provides a machine having: 1) a
memory
comprising data representing at least one genetic sequence, 2) a genetic
identification,
analysis, or modeling program with access to the data, 3) a data processor
which executes
instructions according to the program using the genetic sequence or a
subsequence thereof,
and 4) an output for storing or displaying the results of the data processing.
The machine of the present invention is a data processing system, typically a
digital
computer. The term "computer" includes one or several desktop or portable
computers,
computer workstations, servers (including intranet or internet servers),
mainframes, and any
integrated system comprising any of the above irrespective of whether the
processing,
memory, input, or output of the computer is remote or local, as well as any
networking
interconnecting the modules of the computer. Data processing can thus be
remote or
distributed amongst several processors at one or multiple sites. The data
processing system
comprises a data processor, such as a central processing unit (CPU), which
executes
instructions according to an application program. As used herein, machines,
articles of
manufacture and processes are exclusive of the machines, manufactures and
processes
employed by the United States Patent and Trademark Office or the European
Patent Office
when data representing the sequence of a polypeptide or polynucleotide of the
present
invention is used for patentability searches.
The machine of the present invention includes a memory comprising data
representing at least one genetic sequence. As used herein, "genetic sequence"
refers to
72
CA 02877639 2015-01-09
the primary sequence (i.e., amino acid or nucleotide sequence) of a
polynucleotide or
polypeptide of the present invention. The genetic sequence can represent a
partial
sequence from a full-length protein, genomic DNA, or full-length cDNA/mRNA.
Nucleic acids
or proteins comprising a genetic sequence that is identified, analyzed or
modeled according
to the present invention can be cloned or synthesized.
As those of skill in the art will be aware, the form of memory of a machine of
the
present invention, or the particular embodiment of the computer readable
medium, are not
critical elements of the invention and can take a variety of forms. The memory
of such a
machine includes, but is not limited to, ROM, or RAM or computer readable
media such as,
but not limited to, magnetic media such as computer disks or hard drives, or
media such as
CD-ROMs, DVDs and the like. The memory comprising the data representing the
genetic
sequence includes main memory, a register and a cache. In some embodiments the
data
processing system stores the data representing the genetic sequence in memory
while
processing the data and wherein successive portions of the data are copied
sequentially into
at least one register of the data processor for processing. Thus, the genetic
sequence
stored in memory can be a genetic sequence created during computer runtime or
stored
beforehand. The machine of the present invention includes a genetic
identification, analysis,
or modeling program (discussed below) with access to the data representing the
genetic
sequence. The program can be implemented in software or hardware.
The present invention further contemplates that the machine of the present
invention
will reference, directly or indirectly, a utility or function for the
polynucleotide or polypeptide
of the present invention. For example, the utility/function can be directly
referenced as a
data element in the machine and accessible by the program. Alternatively, the
utility/function
of the genetic can be indirectly referenced to an electronic or written
record. The function or
utility of the genetic sequence can be a function or utility for the genetic
sequence or the
data representing the sequence (i.e., the genetic sequence data).
Exemplary function or utilities for the genetic sequence include: 1) its name
(per
International Union of Biochemistry and Molecular Biology rules of
nomenclature) or the
function of the enzyme or protein represented by the genetic sequence, 2) the
metabolic
pathway that the protein represented by the genetic sequence participates in,
3) the
substrate or product or structural role of the protein represented by the
genetic sequence or
4) the phenotype (e.g., an agronomic or pharmacological trait) affected by
modulating
expression or activity of the protein represented by the genetic sequence.
The machine of the present invention also includes an output for displaying,
printing
or recording the results of the identification, analysis or modeling performed
using a genetic
73
CA 02877639 2015-01-09
sequence of the present invention. Exemplary outputs include monitors,
printers or various
electronic storage mechanisms (e.g., floppy disks, hard drives, main memory)
which can be
used to display the results or employed as a means to input the stored data
into a
subsequent application or device.
In some embodiments, data representing a genetic sequence of the present
invention is a data element within a data structure. The data structure may be
defined by the
computer programs that define the processes of identification, modeling, or
analysis (see
below) or it may be defined by the programming of separate data storage and
retrieval
programs subroutines or systems. Thus, the present invention provides a memory
for
storing a data structure that can be accessed by a computer programmed to
implement a
process for identification, analysis, or modeling of a genetic sequence. The
data structure,
stored within memory, is associated with the data representing the genetic
sequence and
reflects the underlying organization and structure of the genetic sequence to
facilitate
program access to data elements corresponding to logical sub-components of the
genetic
sequence. The data structure enables the genetic sequence to be identified,
analyzed, or
modeled. The underlying order and structure of a genetic sequence is data
representing the
higher order organization of the primary sequence. Such higher order
structures affect
transcription, translation, enzyme kinetics or reflects structural domains or
motifs.
Exemplary logical sub-components which constitute the higher order
organization of
the genetic sequence include but are not limited to: restriction enzyme sites,
endopeptidase
sites, major grooves, minor grooves, beta-sheets, alpha helices, open reading
frames
(ORFs), 5' untranslated regions (UTRs), 3' UTRs, ribosome binding sites,
glycosylation sites,
signal peptide domains, intron-exon junctions, poly-A tails, transcription
initiation sites,
translation start sites, translation termination sites, methylation sites,
zinc finger domains,
modified amino acid sites, preproprotein-proprotein junctions, proprotein-
protein junctions,
transit peptide domains, single nucleotide polymorphisms (SNPs), simple
sequence repeats
(SSRs), restriction fragment length polymorphisms (RFLPs), insertion elements,
transmembrane spanning regions and stem-loop structures.
In another embodiment, the present invention provides a data processing system
comprising at least one data structure in memory where the data structure
supports the
accession of data representing a genetic sequence of the present invention.
The system
also comprises at least one genetic identification, analysis or modeling
program which
directs the execution of instructions by the system using the genetic sequence
data to
identify, analyze or model at least one data element which is a logical sub-
component of the
genetic sequence. An output for the processing results is also provided.
74
CA 02877639 2015-01-09
B. Articles of Manufacture: Computer Readable Media
In one embodiment, the present invention provides a data structure in a
computer
readable medium that contains data representing a genetic sequence of the
present
invention. The data structure is organized to reflect the logical structuring
of the genetic
sequence, so that the sequence can be analyzed by software programs capable of
accessing the data structure. In particular, the data structures of the
present invention
organize the genetic sequences of the present invention in a manner which
allows software
tools to perform an identification, analysis, or modeling using logical
elements of each
genetic sequence.
In a further embodiment, the present invention provides a machine-readable
media
containing a computer program and genetic sequence data. The program provides
instructions sufficient to implement a process for effecting the
identification, analysis, or
modeling of the genetic sequence data. The media also includes a data
structure reflecting
the underlying organization and structure of the data to facilitate program
access to data
elements corresponding to logical sub-components of the genetic sequence, the
data
structure being inherent in the program and in the way in which the program
organizes and
accesses the data.
An example of a data structure resembles a layered hash table, where in one
dimension the base content of the sequence is represented by a string of
elements A, T, C,
G and N. The direction from the 5' end to the 3' end is reflected by the order
from the
position 0 to the position of the length of the string minus one. Such a
string, corresponding
to a nucleotide sequence of interest, has a certain number of substrings, each
of which is
delimited by the string position of its 5' end and the string position of its
3' end within the
parent string. In a second dimension, each substring is associated with or
pointed to one or
multiple attribute fields. Such attribute fields contain annotations to the
region on the
nucleotide sequence represented by the substring.
For example, a sequence under investigation is 520 bases long and represented
by a
string named SeqTarget. There is a minor groove in the 5'upstream non-coding
region from
position 12 to 38, which is identified as a binding site for an enhancer
protein HM-A, which in
turn will increase the transcription of the gene represented by SeqTarget.
Here, the
substring is represented as (12, 38) and has the following attributes:
[upstream uncoded],
[minor groove], [HM-A binding] and [increase transcription upon binding by HM-
A). Similarly,
other types of information can be stored and structured in this manner, such
as information
related to the whole sequence, e.g., whether the sequence is a full length
viral gene, a
CA 02877639 2015-01-09
mammalian house keeping gene or an EST from clone X, information related to
the 3' down
stream non-coding region, e.g., hair pin structure, and information related to
various domains
of the coding region, e.g., Zinc finger.
This data structure is an open structure and is robust enough to accommodate
newly
generated data and acquired knowledge. Such a structure is also a flexible
structure. It can
be trimmed down to al-D string to facilitate data mining and analysis steps,
such as
clustering, repeat-masking, and HMM analysis. Meanwhile, such a data structure
also can
extend the associated attributes into multiple dimensions. Pointers can be
established
among the dimensioned attributes when needed to facilitate data management and
processing in a comprehensive genomics knowledge base. Furthermore, such a
data
structure is object-oriented. Polymorphism can be represented by a family or
class of
sequence objects, each of which has an internal structure as discussed above.
The
common traits are abstracted and assigned to the parent object, whereas each
child object
represents a specific variant of the family or class. Such a data structure
allows data to be
efficiently retrieved, updated and integrated by the software applications
associated with the
sequence database and/or knowledge base.
C. Processes: Identification, Analysis, or Modeling
The present invention also provides a process of identifying, analyzing, or
modeling
data representing a genetic sequence of the present invention. The process
comprises: 1)
providing a machine having a hardware or software implemented genetic sequence
identification, modeling or analysis program with data representing a genetic
sequence, 2)
executing the program while granting it access to the genetic sequence data,
and 3)
displaying or outputting the results of the identification, analysis, or
modeling. Data
structures made by the processes of the present invention and embodied within
a computer
readable medium are also provided herein.
A further process of the present invention comprises providing a memory
embodied
with data representing a genetic sequence and developing within the memory a
data
structure associated with the data and reflecting the underlying organization
and structure of
the data to facilitate program access to data elements corresponding to
logical
subcomponents of the sequence. A computer is programmed with a program
containing
instructions sufficient to implement the process for effecting the
identification, analysis or
modeling of the genetic sequence and the program is executed on the computer
while
granting the program access to the data and to the data structure within the
memory. The
program results are outputted.
76
CA 02877639 2015-01-09
Identification, analysis, and modeling programs are well known in the art and
available commercially. The program typically has at least one application to:
1) identify the
structural role or enzymatic function of the gene which the genetic sequence
encodes or is
translated from, 2) analyzes and identifies higher order structures within the
genetic
sequence or 3) model the physico-chemical properties of a genetic sequence of
the present
invention in a particular environment.
Included amongst the modeling/analysis tools are methods to: 1) recognize
overlapping sequences (e.g., from a sequencing project) with a polynucleotide
of the present
invention and create an alignment called a "contig", 2) identify restriction
enzyme sites of a
polynucleotide of the present invention, 3) identify the products of a TI
ribonuclease digestion
of a polynucleotide of the present invention, 4) identify PCR primers with
minimal self-
complementarity, 5) compute pairwise distances between sequences in an
alignment,
reconstruct phylogentic trees using distance methods and calculate the degree
of
divergence of two protein coding regions, 6) identify patterns such as coding
regions,
terminators, repeats and other consensus patterns in polynucleotides of the
present
invention, 7) identify RNA secondary structure, 8) identify sequence motifs,
isoelectric point,
secondary structure, hydrophobicity and antigenicity in polypeptides of the
present invention,
9) translate polynucleotides of the present invention and backtranslate
polypeptides of the
present invention and 10) compare two protein or nucleic acid sequences and
identifying
points of similarity or dissimilarity between them.
Identification of the function/utility of a genetic sequence is typically
achieved by
comparative analysis to a gene/protein database and establishing the genetic
sequence as a
candidate homologue (i.e., ortholog or paralog) of a gene/protein of known
function/utility.
A candidate homologue has statistically significant probability of having the
same
biological function (e.g., catalyzes the same reaction, binds to homologous
proteins/nucleic
acids, has a similar structural role) as the reference sequence to which it is
compared.
Sequence identity/similarity is frequently employed as a criterion to identify
candidate
homologues. In the same vein, genetic sequences of the present invention have
utility in
identifying homologs in animals or other plant species, particularly those in
the family
Gramineae such as, but not limited to, sorghum, wheat or rice. Function is
frequently
established on the basis of sequence identity/similarity. Exemplary sequence
comparison
systems are provided for in sequence analysis software such as those provided
by the
Genetics Computer Group (Madison, WI) or InforMax</RTI.
The present invention further provides methods for detecting a polynucleotide
of the
present invention in a nucleic acid sample suspected of containing a
polynucleotide of the
77
CA 02877639 2015-01-09
present invention, such as a plant cell lysate, particularly a lysate of
maize. In some
embodiments, a gene of the present invention or portion thereof can be
amplified prior to the
step of contacting the nucleic acid sample with a polynucleotide of the
present invention.
The nucleic acid sample is contacted with the polynucleotide to form a
hybridization complex.
The polynucleotide hybridizes under stringent conditions to a gene encoding a
polypeptide of
the present invention. Formation of the hybridization complex is used to
detect a gene
encoding a polypeptide of the present invention in the nucleic acid sample.
Those of skill will
appreciate that an isolated nucleic acid comprising a polynucleotide of the
present invention
should lack cross-hybridizing sequences in common with non-target genes that
would yield a
false positive result.
Detection of the hybridization complex can be achieved using any number of
well
known methods. For example, the nucleic acid sample, or a portion thereof, may
be
assayed by hybridization formats including but not limited to, solution phase,
solid phase,
mixed phase or in situ hybridization assays. Briefly, in solution (or
liquid) phase
hybridizations, both the target nucleic acid and the probe or primer are free
to interact in the
reaction mixture. In solid phase hybridization assays, probes or primers are
typically linked
to a solid support where they are available for hybridization with target
nucleic in solution. In
mixed phase, nucleic acid intermediates in solution hybridize to target
nucleic acids in
solution as well as to a nucleic acid linked to a solid support. In in situ
hybridization, the
target nucleic acid is liberated from its cellular surroundings in such as to
be available for
hybridization within the cell while preserving the cellular morphology for
subsequent
interpretation and analysis. The following articles provide an overview of the
various
hybridization assay formats: Singer, et al., (1986) Biotechniques 4(3):230-
250; Haase, etal.,
(1984) Methods in Virology 7:189-226; Wilkinson, The theory and practice of in
situ
hybridization in: In situ Hybridization, Wilkinson, Ed., IRL Press, Oxford
University Press,
Oxford; and Nucleic Acid Hybridization: A Practical Approach, Flames, and
Higgins, Eds.,
IRL Press (1987).
Nucleic Acid Labels and Detection Methods
The means by which nucleic acids of the present invention are labeled is not a
critical
aspect of the present invention and can be accomplished by any number of
methods
currently known or later developed. Detectable labels suitable for use in the
present
invention include any composition detectable by spectroscopic, radioisotopic,
photochemical,
biochemical, immunochemical, electrical, optical or chemical means.
78
CA 02877639 2015-01-09
Useful labels in the present invention include biotin for staining with
labeled
streptavidin conjugate, magnetic beads, fluorescent dyes (e.g., fluorescein,
Texas red,
rhodamine, green fluorescent protein and the like), radiolabels (e.g., 3H,
1251, 35S, I4C or
32p), enzymes (e.g., horse radish peroxidase, alkaline phosphatase and others
commonly
used in an ELISA), and colorimetric labels such as colloidal gold or colored
glass or plastic
(e.g., polystyrene, polypropylene, latex, etc.) beads.
Nucleic acids of the present invention can be labeled by any one of several
methods
typically used to detect the presence of hybridized nucleic acids. One common
method of
detection is the use of autoradiography using probes labeled with3H, 1251,
35S, I4C or 32p or
the like. The choice of radioactive isotope depends on research preferences
due to ease of
synthesis, stability and half lives of the selected isotopes. Other labels
include ligands which
bind to antibodies labeled with fluorophores, chemiluminescent agents and
enzymes.
Alternatively, probes can be conjugated directly with labels such as
fluorophores,
chemiluminescent agents or enzymes. The choice of label depends on sensitivity
required,
ease of conjugation with the probe, stability requirements and available
instrumentation. I
Labeling the nucleic acids of the present invention is readily achieved such
as by the use of
labeled PCR primers.
In some embodiments, the label is simultaneously incorporated during the
amplification step in the preparation of the nucleic acids. Thus, for example,
polymerase
chain reaction (PCR) with labeled primers or labeled nucleotides will provide
a labeled
amplification product. In another embodiment, transcription amplification
using a labeled
nucleotide (e.g., fluorescein-labeled UTP and/or CTP) incorporates a label
into the
transcribed nucleic acids.
Non-radioactive probes are often labeled by indirect means. For example, a
ligand I
molecule is covalently bound to the probe. The ligand then binds to an anti-
ligand molecule
which is either inherently detectable or covalently bound to a detectable
signal system, such
as an enzyme, a fluorophore or a chemiluminescent compound. Enzymes of
interest as
labels will primarily be hydrolases, such as phosphatases, esterases and
glycosidases or
oxidoreductases, particularly peroxidases. Fluorescent compounds include
fluorescein and 1
its derivatives, rhodamine and its derivatives, dansyl, umbelliferone, etc.
Chemiluminescers
include luciferin and 2,3-dihydrophthalazinediones, e.g., luminol.
Ligands and anti-ligands may be varied widely. Where a ligand has a natural
anti- I
ligand, namely ligands such as biotin, thyroxine and cortisol, it can be used
in conjunction
with its labeled, naturally occurring anti-ligands. Alternatively, any
haptenic or antigenic
compound can be used in combination with an antibody. Probes can also be
labeled by
79
CA 02877639 2015-01-09
direct conjugation with a label. For example, cloned DNA probes have been
coupled directly
to horseradish peroxidase or alkaline phosphatase.
Means of detecting such labels are well known to those of skill in the art.
Thus, for
example, radiolabels may be detected using photographic film or scintillation
counters,
fluorescent markers may be detected using a photodetector to detect emitted
light.
Enzymatic labels are typically detected by providing the enzyme with a
substrate and
detecting the reaction product produced by the action of the enzyme on the
substrate and
colorimetric labels are detected by simply visualizing the colored label.
Antibodies to Proteins
Antibodies can be raised to a protein of the present invention, including
individual,
allelic, strain or species variants and fragments thereof, both in their
naturally occurring (full-
length) forms and in recombinant forms. Additionally, antibodies are raised to
these proteins
in either their native configurations or in non-native configurations. Many
methods of making
antibodies are known to persons of skill. A variety of analytic methods are
available to
generate a hydrophilicity profile of a protein of the present invention. Such
methods can be
used to guide the artisan in the selection of peptides of the present
invention for use in the
generation or selection of antibodies which are specifically reactive, under
immunogenic
conditions, to a protein of the present invention. See, e.g., Janin, (1979)
Nature 277:491-
492; Wolfenden, etal., (1981) Biochemistry 20:849-855; Kyte and Doolite,
(1982) J. Mol Biol.
157:105-132; Rose, et al., (1985) Science 229:834-838. The following
discussion is
presented as a general overview of the techniques available; however, one of
skill will
recognize that many variations upon the following methods are known.
A number of immunogens are used to produce antibodies specifically reactive
with a
protein of the present invention. An isolated recombinant, synthetic, or
native polynucleotide
of the present invention are the preferred antigens for the production of
monoclonal or
polyclonal antibodies. Polypeptides of the present invention are optionally
denatured, and
optionally reduced, prior to formation of antibodies for screening expression
libraries or other
assays in which a putative protein of the present invention is expressed or
denatured in a
non-native secondary, tertiary or quaternary structure.
The protein of the present invention is then injected into an animal capable
of
producing antibodies. Either monoclonal or polyclonal antibodies can be
generated for
subsequent use in immunoassays to measure the presence and quantity of the
protein of the
present invention. Methods of producing polyclonal antibodies are known to
those of skill in
the art. In brief, an antigen, preferably a purified protein, a protein
coupled to an appropriate
CA 02877639 2015-01-09
carrier (e.g., GST, keyhole limpet hemanocyanin, etc.) or a protein
incorporated into an
immunization vector such as a recombinant vaccinia virus (see, US Patent
Number
4,722,848) is mixed with an adjuvant and animals are immunized with the
mixture. The
animal's immune response to the immunogen preparation is monitored by taking
test bleeds
and determining the titer of reactivity to the protein of interest. When
appropriately high titers
of antibody to the immunogen are obtained, blood is collected from the animal
and antisera
are prepared. Further fractionation of the antisera to enrich for antibodies
reactive to the
protein is performed where desired (See, e.g., Coligan, (1991) Current
Protocols in
Immunology, Wiley/Greene, NY; and Harlow and Lane, Antibodies: A Laboratory
Manual,
Cold Spring Harbor Press, NY (1989)).
Antibodies, including binding fragments and single chain recombinant versions
thereof, against predetermined fragments of a protein of the present invention
are raised by
immunizing animals, e. g., with conjugates of the fragments with carrier
proteins as
described above. Typically, the immunogen of interest is a protein of at least
about 5 amino
acids, more typically the protein is 10 amino acids in length, preferably, 15
amino acids in
length and more preferably the protein is 20 amino acids in length or greater.
The peptides
are typically coupled to a carrier protein (e.g., as a fusion protein), or are
recombinantly
expressed in an immunization vector. Antigenic determinants on peptides to
which
antibodies bind are typically 3 to 10 amino acids in length.
Monoclonal antibodies are prepared from hybrid cells secreting the desired
antibody.
Monoclonals antibodies are screened for binding to a protein from which the
antigen was
derived. Specific monoclonal and polyclonal antibodies will usually have an
antibody binding
site with an affinity constant for its cognate monovalent antigen at least
between 106-107,
usually at least 108, preferably at least 109, more preferably at least 110
and most
preferably at least 111 liters/mole.
In some instances, it is desirable to prepare monoclonal antibodies from
various
mammalian hosts, such as mice, rodents, primates, humans, etc. Description of
techniques
for preparing such monoclonal antibodies are found in, e.g., Basic and
Clinical Immunology,
4th ed., Stites, et al., Eds., Lange Medical Publications, Los Altos, CA, and
references cited
therein; Harlow and Lane, supra; Goding, Monoclonal Antibodies: Principles and
Practice,
2nd ed., Academic Press, New York, NY (1986); and Kohler and Milstein, (1975)
Nature
256:495-497. Summarized briefly, this method proceeds by injecting an animal
with an
antigen comprising a protein of the present invention. The animal is then
sacrificed and cells
taken from its spleen, which are fused with myeloma cells. The result is a
hybrid cell or
"hybridoma" that is capable of reproducing in vitro,
81
CA 02877639 2015-01-09
The population of hybridomas is then screened to isolate individual clones,
each of
which secrete a single antibody species to the antigen. In this manner, the
individual
antibody species obtained are the products of immortalized and cloned single B
cells from
the immune animal generated in response to a specific site recognized on the
antigenic
substance.
Other suitable techniques involve selection of libraries of recombinant
antibodies in
phage or similar vectors (see, e.g., Huse, etal., (1989) Science 246:1275-
1281; and Ward,
etal., (1989) Nature 341:544-546 and Vaughan, et al., (1996) Nature
Biotechnology 14:309-
314). Alternatively, high avidity human monoclonal antibodies can be obtained
from
o transgenic mice comprising fragments of the unrearranged human heavy and
light chain Ig
loci (i.e., mini locus transgenic mice). Fishwild, et al., (1996) Nature
Biotech. 14:845-851.
Also, recombinant immunoglobulins may be produced. See, Cabilly, US Patent
Number
4,816,567 and Queen, etal., (1989) Proc. Nat/Acad. ScL 86:10029-10033.
The antibodies of this invention are also used for affinity chromatography in
isolating
proteins of the present invention. Columns are prepared, e.g., with the
antibodies linked to a
solid support, e.g., particles, such as agarose, SEPHADEX, or the like, where
a cell lysate is
passed through the column, washed, and treated with increasing concentrations
of a mild
denaturant, whereby purified protein are released.
The antibodies can be used to screen expression libraries for particular
expression
products such as normal or abnormal protein. Usually the antibodies in such a
procedure
are labeled with a moiety allowing easy detection of presence of antigen by
antibody binding.
Antibodies raised against a protein of the present invention can also be used
to raise anti-
idiotypic antibodies. These are useful for detecting or diagnosing various
pathological
conditions related to the presence of the respective antigens.
Frequently, the proteins and antibodies of the present invention will be
labeled by
joining, either covalently or non-covalently, a substance which provides for a
detectable
signal. A wide variety of labels and conjugation techniques are known and are
reported
extensively in both the scientific and patent literature.
Suitable labels include
radionucleotides, enzymes, substrates, cofactors, inhibitors, fluorescent
moieties,
chemiluminescent moieties, magnetic particles and the like.
Plants exhibiting an altered ethylene-dependent phenotype as compared with
wild-
type plants can be selected among other methods, by visual observation. For
example, an
altered ethylene-dependent phenotype may be detected by utilization of the
"triple
response." The "triple response" consists of three distinct morphological
changes in dark-
grown seedlings upon exposure to ethylene: inhibition of hypocotyl and root
elongation,
82
CA 02877639 2015-01-09
radial swelling of the stem and exaggeration of the apical hook. Thus, a
triple response
displayed in the presence of ethylene inhibitors would indicate one type of
altered ethylene-
dependent phenotype. Ethylene affects a vast array of agriculturally important
plant
processes, including fruit ripening, flower and leaf senescence and leaf
abscission. The
ability to control the sensitivity of plants to ethylene could thus
significantly improve the
quality and longevity of many crops. The invention includes plants produced by
the method
of the invention, as well as plant tissue and seeds.
Although the present invention has been described in some detail by way of
illustration and example for purposes of clarity of understanding, it will be
obvious that
to certain changes and modifications may be practiced within the scope of
the appended
claims.
Example 1
This example describes the construction of the cDNA libraries. Total RNA for
SEQ
ID NO: 1 (EIN3), SEQ ID NO: 3 (EBF1), SEQ ID NO: 5 (EBF2), SEQ ID NO: 7 (EIN5)
or
SEQ ID NO: 9 (ERF3) was obtained from maize genotype Hill (Armstrong and
Phillips,
(1988) Crop Sci. 28:363-369); and for ZmEIN3-2 (SEQ ID NO: 1), from night
harvested leaf
tissue at the V8-V10 stage of maize genotype B75. The total RNA was isolated
from the
maize tissues with TRIzol Reagent (Life Technology Inc. Gaithersburg, MD)
using a
modification of the guanidine isothiocyanate/acid-phenol procedure described
by
Chomczynski and Sacchi (Chomczynski and Sacchi, (1987) Anal. Biochem.
162:156). In
brief, plant tissue samples were pulverized in liquid nitrogen before the
addition of the TRIzol
Reagent, and then were further homogenized with a mortar and pestle. Addition
of
chloroform followed by centrifugation was conducted for separation of an
aqueous phase
and an organic phase. The total RNA was recovered by precipitation with
isopropyl alcohol
from the aqueous phase.
Poly (A) + RNA Isolation
The selection of poly (A) + RNA from total RNA was performed using PolyATact
system (Promega Corporation. Madison, WI). In brief, biotinylated oligo (dT)
primers were
used to hybridize to the 3' poly (A) tails on mRNA. The hybrids were captured
using
streptavidin coupled to paramagnetic particles and a magnetic separation
stand. The mRNA
was washed at high stringency conditions and eluted by RNase-free deionized
water. cDNA
Library Construction cDNA synthesis was performed and unidirectional cDNA
libraries were
constructed using the SuperScript Plasmid System (Life Technology Inc.
Gaithersburg, MD).
83
CA 02877639 2015-01-09
The first strand of cDNA was synthesized by priming an oligo (dT) primer
containing a Not I
site.
The reaction was catalyzed by SuperScript Reverse Transcriptase II at 45 C.
The
second strand of cDNA was labeled withalpha-32P-dCTP and a portion of the
reaction was
analyzed by agarose gel electrophoresis to determine cDNA sizes. cDNA
molecules smaller
than 500 base pairs and unligated adapters were removed bySephacryl-S400
chromatography. The selected cDNA molecules were ligated into pSPORTI vector
in
between of Not I and Sal I sites.
Example 2
This example describes cDNA sequencing and library subtraction. Sequencing
Template Preparation: Individual colonies were picked and DNA was prepared
either by
PCR with M13 forward primers and M13 reverse primers, or by plasmid isolation.
All the
cDNA clones were sequenced using M13 reverse primers.
Q-bot Subtraction Procedure: cDNA libraries subjected to the subtraction
procedure
were plated out on 22 x 22 cm2 agar plate at density of about 3,000 colonies
per plate. The
plates were incubated in a 37 C incubator for 12-24 hours. Colonies were
picked into 384-
well plates by a robot colony picker, 0-bat (GENETIX Limited). These plates
were incubated
overnight at 37 C.
Once sufficient colonies were picked, they were pinned onto 22 x 22 cm2 nylon
membranes using Q-bot. Each membrane contained 9,216 colonies or 36,864
colonies.
These membranes were placed onto agar plate with appropriate antibiotic. The
plates were
incubated at 37 C for overnight.
After colonies were recovered on the second day, these filters were placed on
filter
paper pre-wetted with denaturing solution for four minutes, then were
incubated on top of a
boiling water bath for additional four minutes. The filters were then placed
on filter paper
pre-wetted with neutralizing solution for four minutes. After excess solution
was removed by
placing the filters on dry filter papers for one minute, the colony side of
the filters were place
into Proteinase K solution, incubated at 37 C for 40-50 minutes. The filters
were placed on
dry filter papers to dry overnight. DNA was then cross-linked to nylon
membrane by UV light
treatment.
Colony hybridization was conducted as described by Sambrook, et a/., (in
Molecular
Cloning: A laboratory Manual, 2nd Edition). The following probes were used in
colony
hybridization:
84
CA 02877639 2015-01-09
1. First strand cDNA from the same tissue as the library was made from to
remove the most redundant clones.
2. 48-192 most redundant cDNA clones from the same library based on
previous sequencing data.
3 192 most redundant cDNA clones in the entire maize sequence database.
4. ASal-A20 oligo nucleotide: TCG ACC CAC GCG TCC GAA AAA AAA AAA
AAA AAA AAA, removes clones containing a poly A tail but no cDNA.
5. cDNA clones derived from rRNA.
The image of the autoradiography was scanned into computer and the signal
intensity and cold colony addresses of each colony was analyzed. Re-arraying
of cold
colonies from 384 well plates to 96 well plates was conducted using Q-bot.
Example 3
This example describes identification of the gene from a computer homology
search.
Gene identities were determined by conducting BLAST (Basic Local Alignment
Search Tool;
Altschul, et al., (1993) J. Mol. Biol. 215:403-410; see also, National Center
for Biotechnology
Information, National Library of Medicine, Building 38A, Bethesda, Maryland,
USA) searches
under default parameters for similarity to sequences contained in the BLAST
"nr" database
(comprising all non-redundant GenBank CDS translations, sequences derived from
the 3-
dimensional structure Brookhaven Protein Data Bank, the last major release of
the SWISS-
PROT protein sequence database, EMBL, and DDBJ databases). The cDNA sequences
were analyzed for similarity to all publicly available DNA sequences contained
in the "nr"
database using the BLASTN algorithm.
The DNA sequences were translated in all reading frames and compared for
similarity to all publicly available protein sequences contained in the "nr"
database using the
BLASTX algorithm (Gish and States, (1993) J. Nature Genetics 3:266-272)
provided by the
NCBI. In some cases, the sequencing data from two or more clones containing
overlapping
segments of DNA were used to construct contiguous DNA sequences.
Example 4
Vector Construction and Over Expression of ZM-ERF3 in Maize
PHP21751-UBI:ZM-ERF3
The coding sequence of ZM-ERF3 was amplified by PCR and cloned into pCR2.1
TOPO vector (Invitrogen). ZM-ERF3 was sequence verified and ligated into a
vector
containing the maize UBI promoter and PINII terminator. The gene cassette was
then
CA 02877639 2015-01-09
ligated to generate UBI PRO:ZM-ERF3:PINII TERM + 35S:BAR:PIN11. 35S:BAR is
used as
a herbicide resistance marker. The expression vector was quality checked by
restriction
digestion mapping and transferred into Agrobacterium tumefaciens LB4404JT by
electroporation. This Agrobacterium strain was used to transform GS3 maize
inbred.
Molecular analyses on TO events were performed and single copy transgene
expressing
events were advanced for further experiments.
PHP25534-RAB17:ZM-ERF3
The coding sequence of ZM-ERF3 was amplified by PCR and cloned into pCR2.1
TOPO vector (Invitrogen). ZM-ERF3 was sequence verified and ligated into a
vector
containing the maize RAB17 promoter and GZ-W64A terminator, as well as Gateway
(Invitrogen) AU sites. The entry vector in combination with a destination
vector was used in
a single site Gateway (Invitrogen) reaction to generate RAB17:ZM-EFR3:GZ-W64A
+
UBI:MOPAT:PINII + LTP2:DS-RED:PINII. UBI:MOPAT and LTP2:RFP are used as
herbicide resistance and visible markers, respectively. The expression vector
was quality
checked by restriction digestion mapping and transferred into Agrobacterium
tumefaciens
LB4404JT by electroporation. This Agrobacterium strain was used to transform
GS3 maize
inbred. Molecular analyses on TO events were performed and single copy
transgene
expressing events were advanced for further experiments. Advancement comprises
self-
pollination or pollination with the parent genotype and selection for the
transgenic progeny.
For example, Ti progeny comprises two doses of the parental genotype and may
be
referred to as D2. Advanced lines may be crossed with a tester genotype for
field evaluation.
Hybrid material representing ten events of PH P25534 was planted in replicated
field
trials subjected to drought stress during the grain-fill stage (i.e., within
the R2 to R5 stages
as described in How a Corn Plant Develops, Iowa State University of Science
and
Technology Cooperative Extension Service Special Report No. 48, Reprinted June
1993.)
Four of ten events showed statistically significant improved yield under
drought stress as
compared to controls. One of those four events also demonstrated statistically
significant
improved performance in seedling assays for drought tolerance and for early
vigor under
low-temperature stress.
PHP25536-RYE CBF31 PRO:ZM-ERF3
The coding sequence of ZM-ERF3 was amplified by PCR and cloned into pCR2.1
TOPO vector (Invitrogen). ZM-ERF3 was sequence verified and ligated along with
the RYE
CBF31 promoter (US Patent Application Serial Number 12/256,568 filed 23
October 2008)
86
CA 02877639 2015-01-09
into a vector containing the maize GZ-W64A terminator, as well as Gateway
(Invitrogen)
ATT sites. The entry vector in combination with a destination vector was used
in a single
site Gateway (lnvitrogen) reaction to generate RAB17:ZM-EFR3:GZ-W64A +
UBI:MOPAT:PINII + LTP2:DS-RED:PINII. UBI:MOPAT and LTP2:RFP are used as
herbicide resistance and visible markers, respectively. The expression vector
was quality
checked by restriction digestion mapping and transferred into Agrobacterium
tumefaciens
LB4404JT by electroporation. This Agrobacterium strain was used to transform
GS3 maize
inbred. Molecular analyses on TO events were performed and single copy
transgene
expressing events were advanced for further experiments. Advancement comprises
self-
pollination or pollination with the parent genotype and selection for the
transgenic progeny.
For example, Ti progeny comprises two doses of the parental genotype and may
be
referred to as D2. Advanced lines may be crossed with a tester genotype for
field evaluation.
Hybrid material representing 9 events of PHP25536 was planted in replicated
field
trials subjected to drought stress during the grain-fill stage. Three of nine
events showed
statistically significant improved yield under drought stress as compared to
controls. Two of
those three events, and two additional events, demonstrated statistically
significant improved
performance in seedling assays for early vigor under low-temperature stress.
PHP25537-RYE CBF31 PRO:ZM-ERF3 + RYE CBF31 PRO:ZM-CBF2
The coding sequence of ZM-ERF3 was amplified by PCR and cloned into pCR2.1
TOPO vector (Invitrogen). ZM-ERF3 was sequence verified & ligated along with
the RYE
CBF31 promoter into a vector containing the maize GZ-W64A terminator, as well
as
Gateway (Invitrogen) ATT sites. The entry vector in combination with a RYE
CBF31
PRO:ZM-CBF2 entry vector were used in a multisite Gateway (Invitrogen)
reaction to
generate RYE CBF31 PRO:ZM-EFR3:GZ-W64A + RYE CBF31:ZM-CBF2:PINII +
UBI:MOPAT:PINII. UBI:MOPAT is used as a herbicide resistance marker. The
expression
vector was quality checked by restriction digestion mapping and transferred
into
Agrobacterium tumefaciens LB4404JT by electroporation. This Agrobacterium
strain was
used to transform GS3 maize inbred. Molecular analyses on TO events were
performed and
single copy transgene expressing events were advanced for further experiments.
PHP25538-RYE CBF31 PRO:ZM-ERF3 + RYE CBF31 PRO:RYE CBF31
The coding sequence of ZM-ERF3 was amplified by PCR and cloned into pCR2.1
TOPO vector (Invitrogen). ZM-ERF3 was sequence verified and ligated along with
the RYE
CBF31 promoter into a vector containing the maize GZ-W64A terminator, as well
as
87
CA 02877639 2015-01-09
Gateway (Invitrogen) ATT sites. The entry vector in combination with a RYE
CBF31
PRO:RYE CBF31 entry vector were used in a multisite Gateway (Invitrogen)
reaction to
generate RYE CBF31 PRO:ZM-EFR3:GZ-W64A + RYE CBF31:RYE:CBF31:GZ-W64A +
UBI:MOPAT:PINII. UBI:MOPAT is used as a herbicide resistance marker. The
expression
vector was quality checked by restriction digestion mapping and transferred
into
Agrobacterium tumefaciens LB4404JT by electroporation. This Agrobacterium
strain was
used to transform GS3 maize inbred. Molecular analyses on TO events were
performed and
single copy transgene expressing events were advanced for further experiments.
PHP26620-RD29A PRO:ZM-ERF3 + RD29A:RYE CBF31
The coding sequence of ZM-ERF3 was amplified by PCR and cloned into pCR2.1
TOPO vector (Invitrogen). ZM-ERF3 was sequence verified and ligated along with
the
RD29A promoter into a vector containing the PINII terminator, as well as
Gateway
(Invitrogen) ATT sites. The entry vector in combination with a R029A PRO:RYE
CBF31
entry vector were used in a multisite Gateway (Invitrogen) reaction to
generate RD29A
PRO:ZM-EFR3:PINII + RD29A:RYE:CBF31:GZ-W64A + UBI:MOPAT:PINII. UBI:MOPAT is
used as a herbicide resistance marker. The expression vector was quality
checked by
restriction digestion mapping and transferred into Agrobacterium tumefaciens
LB4404JT by
electroporation. This Agrobacterium strain was used to transform EFO9B maize
inbred.
Molecular analyses on TO events were performed and single copy transgene
expressing
events were advanced for further experiments. Statistically significant yield
improvement
was observed in one of two events tested under separate drought stresses at
anthesis and
during grain-fill.
Example 5
Vector construction and gene silencing of ZM-EIN3 in maize
UBLEIN3:PINII RNAi
Two ¨500 base pair (sense and anti-sense) truncated fragments of the ZM-EIN3
gene were amplified by PCR and cloned into an Invitrogen TOPO vector. The ZM-
EIN3
sense and anti-sense truncated fragments were sequence verified and ligated,
along with an
ADH1 intron loop sequence into a vector containing the maize UBI promoter and
PINII
terminator, as well as Gateway (Invitrogen) ATT sites. The entry vector in
combination with
a destination vector was used in a single site Gateway (Invitrogen) reaction
to generate
UBI:ZM-EIN3:PINII RNAi + UBI:MOPAT:PINII + LTP2:DS-RED:PINII. UBI:MOPAT and
LTP2:RFP are used as herbicide resistance and visible markers, respectively.
The
88
CA 02877639 2015-01-09
expression vector was quality checked by restriction digestion mapping and
transferred into
Agrobacterium tumefaciens LB4404JT by electroporation. This Agrobacterium
strain was
used to transform EFO9B maize inbred. Molecular analyses on TO events were
performed
and single copy transgene expressing events were advanced for further
experiments.
Example 6
Sequence Isolation and Endogenous Expression
Sequence isolation
The ethylene signaling genes EIN3 and EIN5 are being used in down-regulation
constructs using the RNAi strategy. Two RNAi constructs for EIN3 and one for
EIN5 have
been prepared. In the case of EIN3, full-insert sequence from cfp7n.pk010.h4
(PC0642867)
has been used to generate two RNAi constructs with truncated fragments of
approximately
500 bp at the 5' end of the coding sequence. One of the two constructs
included the starting
ATG in the RNAi fragment, while the second avoided the use of the starting ATG
and started
immediately after. The EIN5 RNAi construct was prepared using approximately
500bp at the
5' end of the coding sequence, starting immediately after the first ATG.
Fragments for the
E1N5 RNAI construct were amplified from cfp5n.pk005.c17.f:fis (PC0637491).
Expression information:
The maize ethylene genes ERF3, EIN3, EIN5, EBF1 and EBF2 are expressed in all
tissues in the plant (Table 2). Endogenous expression of ZmERF3 is found to be
highest in
vascular bundles showing an MPSS expression level (Solexa, Hayward, CA;
Brenner, at al.,
(2000) Nature Biotechnology 18:630-634) of 628 ppm, while the gene is
expressed in
practically all maize tissues (Fig 1). The highest expression levels observed
for ZmEIN3,
ZmEIN5, ZmEBF1 and ZmEBF2 are, respectively, 1603 ppm (internodes), 168 ppm
(ear
meristem), 560 ppm (root) and 902 ppm (root).
The genes under consideration here also show differential expression in the
presence of stresses or hormones, as indicated in Table 2. ZmERF3 is observed
to be
induced by drought in most tissues, although one experiment indicated
downregulation of
the gene in leaves and roots under drought. It is also observed to be
downregulated by cold
stress. The expression of this gene appears to be closely related to the time
of exposure to
the stress, as indicated by a microarray experiment conducted to determine the
cold-induced
time-course of gene expression in maize seedling leaves. The expression of the
gene was
found highest at the very early time point of 0.5 hour after exposure to cold
stress, and
thereafter it declined to normal uninduced levels by 24 hours after exposure
to stress (Fig 2).
89
CA 02877639 2015-01-09
ZmEIN3 is induced by drought stress, and to a lesser extent by cold stress, in
aerial
tissues, while it appears to be down-regulated by drought in the root. It also
shows a higher
expression in response to ABA treatment during the early hours (24h) of ABA
exposure. In
contrast to this, ZmEIN5 expression is downregulated in most aerial tissues by
drought and
upregulated in root. It shows enhanced expression upon treatment with both ABA
and
ethylene.
Expression of ZmEBF1 and ZmEBF2 appears to be more or less similarly regulated
in the plant. Both are upregulated by drought in aerial tissues and
downregulated in roots.
In addition, ZmEBF1 is induced by cold stress, while ZmEBF2 is induced by both
ABA and
ethylene treatment.
Downstream gene expression in UBI::Zm ERF3 transoenic maize
Constitutive over-expression of ZmERF3 in maize resulted in a pleiotropic
effect,
where the stems of the plants curved as they grew. The plants also exhibited
"buggy-
whipping", a phenomenon where the newly emerging leaves were tightly curled
and bent,
during the vegetative stage prior to tasseling. However, they recovered from
this phenotype
as they grew towards the reproductive stage. Considering that the highest
endogenous
expression of the gene is in vascular bundles, it is likely that constitutive
overexpression
resulted in adversely affecting vascular formation in the stem and this caused
the curving of
the stalk during growth. We analyzed two events, namely E3 and E18, of
transgenic maize
constitutively expressing ZmERF3 from the UBI promoter, to identify changes in
downstream
gene expression. The event, E18, showed a pronounced pleiotropic effect, while
the event,
E3, did not show such an effect. Transgene expression in event E18 was
confirmed to be
very high by northern blotting, relative to endogenous levels. As ZmERF3 is a
transcription
factor, constitutive over-expression of this gene would result in either
upregulation or
downregulation of genes whose expression is regulated at the transcription
level by
ZmERF3 There was significant overlap between the upregulated and downregulated
genes
in the two events, with more number of genes showing change in E18 than in E3
(Figure 3).
A list of the genes with known functionality that are up- or down-regulated
commonly in the
two events is presented in Table 3. Analysis of downstream gene expression
indicates the
presence of stress and/or ethylene related genes in both the up-regulated and
down-
regulated categories. In attempting to overcome the pleiotropic effect of
UBI:2mERF3,
constructs were designed to express the gene from stress-regulated promoters.
Since
several stress-related genes are down-regulated in UB1:2mERF3 transgenics, one
RNAi
CA 02877639 2015-01-09
construct will also be prepared to assess the effect of this transcription
factor in transgenic
stress tolerance.
91
CA 0287 7639 2015-01-09
Table 2. Endogenous expression of four ethylene signaling genes as
represented in
MPSS libraries.
EXPRESSION DETAILS GENE
General Expression Information ZM ERF3 ZM E1N3 ZM ENS ZM EBF1
ZM EBF1/2
Tissue specificity All Tissues All Tissues All
Tissues All Tissues All Tissues
Vascular
Highest MPSS expression bundles Internode Ear
MeristemRoot (560ppm) Root (902ppm)
(1603ppm) (168ppm)
(628ppm)
MPSS Tissue Libraries MPSS Expression (ppm)
Corn pedicels, drought stressed 162 37 - 173 10
Corn pedicels, watered control 62 8 - - -
Corn leaf, drought stressed 38 763 21 65 239
Corn leaf, watered control 65 339 36- -
Corn root, drought stressed 219 485 53 15 -
Corn root, watered control 410 650 39 58 27
Corn v5 leaves, ABA treated, 24hr 68 814 33 -
-
Corn v5 leaves, ABA treated, 48hr 25 518 79- 45
Corn v5 leaves, Ethephon treated, 24hr 26 297 22-
-
Corn v5 leaves, Ethephon treated, 48hr 95 521 55-
59
Corn v5 leaves, control (no hormone treatment) 57 509 20-
7
Corn seedling, cold stress 28 459- 30 -
Corn seedling, cold-stress recovery 25 380
- - -
Corn seedling, no cold-stress control 109 328- - -
.
Corn immature ear tips, drought stressed 12- 68 50
14
Corn immature ear tips, watered control -- 78 - -
Corn ear leaf, drought stressed 100- . 3 44
Corn ear leaf, watered control 5- 120 27 -
Corn 7-DAP apical kernels, drought stressed 75- 96 105
24
,
Corn 7-DAP apical kernels, watered control 2956 -
13
-
Corn 7-DAP Basal Kernels, drought stressed 82- 128 125
-
Corn 7-DAP Basal Kernels, watered control 58_ 99 25
30
Table 3-a: Top-BLAST hit of genes with known functionality that is commonly
upregulated in both events 3 and 18 of maize transgenics harboring UBLZmERF3.
92
CA 02 87 7 63 9 2 0 15-0 1-0 9
Fold change
Accession Gene Name
in E18
Q6Z2W4 ,AvrRpt2-induced protein 2-like [Oryza sativa (japonica cultivar-
group)] 81.584
Q7XLD7 OSJNBa0070C17.11 protein [Oryza sativa (japonica cultivar-group)]
21.523
Q8SOK1 Selenoprotein-like [Oryza sativa (japonica cultivar-group)]
26.790
Q5VQ37 Leaf senescence protein-like [Oryza sativa (japonica cultivar-
group)] 6.737
008062 Malate dehydrogenase [Zea mays] 9.206
QOJQR6 0s01g0143500 protein [Oryza sativa (japonica cultivar-group)]
9.720
P42390 Indole-3-glycerol phosphate lyase, chloroplast precursor [Zea mays]
54.913
Q69XR7 Putative acyl-CoA oxidase ACX3 [Oryza sativa (japonica cultivar-
group)] 164.089
06K4Y6 Prefoldin-related K 9.165
075196 Putative receptor-like kinase [Oryza saliva (japonica cultivar-
group)] 13.604
Q9XF58 Plasma membrane intrinsic protein [Zea mays] 5.250
043417 Peroxidase precursor [Cenchrus ciliaris] 8.826
000X49 Putative DNA-3-methyladenine glycosylase [Oryza sativa (japonica
cultivar-group)] 10.388
Q6YW60 Zinc finger (C3HC4-type RING finger) protein-like [Oryza sativa
(japonica cultivar-group)] 12.170
Q5NA53 Glycogenin-like protein [Oryza sativa (japonica cultivar-group)]
5.573
048558 60S ribosomal protein L30 [Zea mays] 12.328
Q7XLD7 OSJNBa0070C17.11 protein (Oryza sativa (japonica cultivar-group)]
21.523
A2WNN4 0s01g0287400 protein [Oryza sativa (indica cultivar-group)] 41.096
Q751K0 Putative o-methyltransferase ZRP4 [Oryza sativa (japonica cultivar-
group)] 17.086
93
CA 02 877 63 9 2 015-01-0 9
Table 3-b: Top-BLAST hit of genes with known functionality that is commonly
down-
regulated in both events 3 and 18 of maize transgenics harboring UBLZmERF3.
Fold change
Accession Gene Name
in E18
Q6Z6M4 Isocitrate lyase [Oryza sativa (japonica cultivar-group)] -
16.010
Q75HZ0 Putative late embryogenesis abundant protein [Oryza sativa
(japonica cultivar-group)] -7.250
Q9AVM3 Cytochrome P450 [Triticum aestivum] -
21.360
Q40680 0s0790614500 protein [Oryza sativa] -
11.691
Q1OLJ9 Heavy metal-associated domain containing protein, expressed
[Oryza sativa) -5.053
Q9ZWI4 ZmGR2c protein [Zea mays] -
7.283
Q6J555 MADS16 protein [Dendrocalamus latiflorus] -
9.053
AOS6X4 FT-like protein [Hordeum vulgare subsp. vulgarej -
11.773
Q5VMA5 Putative lipase [Oryza sativa (japonica cultivar-group)) -
6.354
Q9ZSX1 Polyprotein [Zea mays] -
6.338
049010 Herbicide safener binding protein [Zea mays] -
10.906
Q6L5H6 0s05g0537400 protein [Oryza sativa (japonica cultivar-group)] -
6.043
Q10SX1 Sterol desaturase family protein, expressed [Oryza sativa
(japonica cultivar-group)] -5.691
Q2RBL6 Major Facilitator Superfamily protein, expressed [Oryza sativa
(japonica cultivar-group)] -9.974
Q10S44 Basic helix-loop-helix, putative, expressed [Oryza sativa
(japonica cultivar-group)] -9.167
Q8W2K4 Cytochrome b5 reductase isoform II [Zea mays] -
12.870
02R2W1 Adagio-like protein 3 [Oryza sativa] -
6.005
Q69Y12 Putative aminopeptidase C [Oryza sativa (japonica cultivar-
group)] -31.794
Q53JI5 POT family, putative [Oryza sativa (japonica cultivar-group)] -
39.680
Q7EYH1 Putative MDR-like ABC transporter [Oryza sativa (japonica
cultivar-group)] -10.733
Q84ZF7 0s07g0293000 protein [Oryza sativa (japonica cultivar-group)] -
13.433
Q69J29 Pectin methylesterase-like protein [Oryza sativa (japonica
cultivar-group)] -8.375
Q8RZV3 Zinc finger (C3HC4-type RING finger)-like [Oryza sativa (japonica
cultivar-group)] -8.153
Q9LT02 Putative cation-transporting ATPase [Arabidopsis thaliana] -
5.999
Q7XIR1 Carbonyl reductase-like protein [Oryza sativa (japonica cultivar-
group)) -7.132
Example 7
Transformation and Regeneration of Transgenic Plants
Immature maize embryos from greenhouse donor plants are bombarded with a
plasmid containing the Ethylene signaling associated sequence operably linked
to the
drought-inducible promoter RAB17 promoter (Vilardell, et al., (1990) Plant Mol
Biol 14:423-
432) and the selectable marker gene PAT, which confers resistance to the
herbicide
Bialaphos. Alternatively, the selectable marker gene is provided on a separate
plasmid.
Transformation is performed as follows. Media recipes follow below.
Preparation of Target Tissue:
The ears are husked and surface sterilized in 30% Clorox bleach plus 0.5%
Micro
detergent for 20 minutes, and rinsed two times with sterile water. The
immature embryos
are excised and placed embryo axis side down (scutellum side up), 25 embryos
per plate, on
560Y medium for 4 hours and then aligned within the 2.5-cm target zone in
preparation for
bombardment.
94
CA 02877639 2015-01-09
Preparation of DNA:
A plasmid vector comprising the Ethylene signaling associated sequence
operably
linked to an ubiquitin promoter is made. This plasmid DNA plus plasmid DNA
containing a
PAT selectable marker is precipitated onto 1.1 pm (average diameter) tungsten
pellets using
a CaCl2 precipitation procedure as follows:
100 pl prepared tungsten particles in water
p1(1 pg) DNA in Iris EDTA buffer (1 pg total DNA)
100 p12.5 M CaC12
10 pl 0.1 M spermidine
10
Each reagent is added sequentially to the tungsten particle suspension, while
maintained on the multitube vortexer. The final mixture is sonicated briefly
and allowed to
incubate under constant vortexing for 10 minutes. After the precipitation
period, the tubes
are centrifuged briefly, liquid removed, washed with 500 ml 100% ethanol, and
centrifuged
for 30 seconds. Again the liquid is removed, and 105 p1100% ethanol is added
to the final
tungsten particle pellet. For particle gun bombardment, the tungsten/DNA
particles are
briefly sonicated and 10 pl spotted onto the center of each macrocarrier and
allowed to dry
about 2 minutes before bombardment.
Particle Gun Treatment:
The sample plates are bombarded at level #4 in a particle gun. All samples
receive a
single shot at 650 PSI, with a total of ten aliquots taken from each tube of
prepared
particles/DNA.
Subsequent Treatment:
Following bombardment, the embryos are kept on 560Y medium for 2 days, then
transferred to 560R selection medium containing 3 mg/liter Bialaphos, and
subcultured every
2 weeks. After approximately 10 weeks of selection, selection-resistant callus
clones are
transferred to 288J medium to initiate plant regeneration. Following somatic
embryo
maturation (2-4 weeks), well-developed somatic embryos are transferred to
medium for
germination and transferred to the lighted culture room. Approximately 7-10
days later,
developing plantlets are transferred to 272V hormone-free medium in tubes for
7-10 days
until plantlets are well established. Plants are then transferred to inserts
in flats (equivalent
to 2.5" pot) containing potting soil and grown for 1 week in a growth chamber,
subsequently
grown an additional 1-2 weeks in the greenhouse, then transferred to classic
600 pots (1.6
gallon) and grown to maturity. Plants are monitored and scored for increased
drought
CA 02877639 2015-01-09
tolerance. Assays to measure improved drought tolerance are routine in the art
and include,
for example, increased kernel-earring capacity yields under drought conditions
when
compared to control maize plants under identical environmental conditions.
Alternatively,
the transformed plants can be monitored for a modulation in meristem
development (i.e., a
decrease in spikelet formation on the ear). See, for example, Bruce, et al.,
(2002) Journal of
Experimental Botany 53:1-13.
Bombardment and Culture Media:
Bombardment medium (560Y) comprises 4.0 g/I N6 basal salts (SIGMA C-1416), 1.0
m1/I Eriksson's Vitamin Mix (1000X SIGMA-1511), 0.5 mg/1 thiamine HCI, 120.0
g/I sucrose,
1.0 mg/I 2,4-D, and 2.88 g/I L-proline (brought to volume with D-I H20
following adjustment
to pH 5.8 with KOH); 2.0 g/I Gelrite gelling agent (added after bringing to
volume with D-I
H20); and 8.5 mg/1 silver nitrate (added after sterilizing the medium and
cooling to room
temperature). Selection medium (560R) comprises 4.0 g/I N6 basal salts (SIGMA
C-1416),
1.0 m1/I Eriksson's Vitamin Mix (1000X SIGMA-1511), 0.5 mg/I thiamine HCI,
30.0 g/I
sucrose, and 2.0 mg/I 2,4-D (brought to volume with D-I H20 following
adjustment to pH 5.8
with KOH); 3.0 g/I Gelrite gelling agent (added after bringing to volume with
D-I H20); and
0.85 mg/I silver nitrate and 3.0 mg/I bialaphos (both added after sterilizing
the medium and
cooling to room temperature).
Plant regeneration medium (288J) comprises 4.3 g/I MS salts (GIBCO 11117-074),
5.0 m1/I MS vitamins stock solution (0.100 g nicotinic acid, 0.02 g/I thiamine
HCL, 0.10 g/I
pyridoxine HCL, and 0.40 g/I glycine brought to volume with polished D-I H20)
(Murashige
and Skoog, (1962) Physiol. Plant. 15:473), 100 mg/1 myo-inositol, 0.5 mg/1
zeatin, 60 g/1
sucrose, and 1.0 m1/I of 0.1 mM abscisic acid (brought to volume with polished
D-I H20 after
adjusting to pH 5.6); 3.0 g/I Gelrite gelling agent (added after bringing to
volume with D-I
H20); and 1.0 mg/I indoleacetic acid and 3.0 mg/I bialaphos (added after
sterilizing the
medium and cooling to 60 C). Hormone-free medium (272V) comprises 4.3 g/I MS
salts
(GIBCO 11117-074), 5.0 m1/1 MS vitamins stock solution (0.100 g/I nicotinic
acid, 0.02 g/I
thiamine HCL, 0.10 g/I pyridoxine HCL, and 0.40 g/I glycine brought to volume
with polished
D-I H20), 0.1 g/I myo-inositol, and 40.0 g/I sucrose (brought to volume with
polished D-I H20
after adjusting pH to 5.6); and 6 g/I BactoTm-agar solidifying agent (added
after bringing to
volume with polished D-I H20), sterilized and cooled to 60 C.
96
CA 02877639 2015-01-09
=
Example 8
Agrobacterium-mediated Transformation
For Agrobacterium-mediated transformation of maize with an antisense sequence
of the
Ethylene signaling associated sequence of the present invention, preferably
the method of
Zhao is employed (US Patent Number 5,981,840, and PCT Patent Application
Publication
W098/32326 =
Briefly, immature
embryos are isolated from maize and the embryos contacted with a suspension of
Agrobacterium, where the bacteria are capable of transferring the antisense
Ethylene
signaling associated sequences to at least one cell of at least one of the
immature embryos
(step 1: the infection step). In this step the immature embryos are preferably
immersed in an
Agrobacterium suspension for the initiation of inoculation. The embryos are co-
cultured for a
time with the Agrobacterium (step 2: the co-cultivation step). Preferably the
immature
embryos are cultured on solid medium following the infection step. Following
this co-
cultivation period an optional "resting" step is contemplated.
In this resting step, the
embryos are incubated in the presence of at least one antibiotic known to
inhibit the growth
of Agrobacterium without the addition of a selective agent for plant
transformants (step 3:
resting step). Preferably the immature embryos are cultured on solid medium
with antibiotic,
but without a selecting agent, for elimination of Agrobacterium and for a
resting phase for the
infected cells. Next, inoculated embryos are cultured on medium containing a
selective
agent and growing transformed callus is recovered (step 4: the selection
step). Preferably,
the immature embryos are cultured on solid medium with a selective agent
resulting in the
selective growth of transformed cells. The callus is then regenerated into
plants (step 5: the
regeneration step), and preferably calli grown on selective medium are
cultured on solid
medium to regenerate the plants. Plants are monitored and scored for a
modulation in
meristem development. For instance, alterations of size and appearance of the
shoot and
floral meristems and/or increased yields of leaves, flowers, and/or fruits.
Example 9
Soybean Embryo Transformation
Soybean embryos are bombarded with a plasmid containing an antisense Ethylene
signaling associated sequence operably linked to an ubiquitin promoter as
follows. To
induce somatic embryos, cotyledons, 3-5 mm in length dissected from surface-
sterilized,
immature seeds of the soybean cultivar A2872, are cultured in the light or
dark at 26 C on an
appropriate agar medium for six to ten weeks. Somatic embryos producing
secondary
embryos are then excised and placed into a suitable liquid medium. After
repeated selection
97
CA 02877639 2015-01-09
for clusters of somatic embryos that multiplied as early, globular-staged
embryos, the
suspensions are maintained as described below.
Soybean embryogenic suspension cultures can be maintained in 35 ml liquid
media
on a rotary shaker, 150 rpm, at 26 C with florescent lights on a 16:8 hour
day/night
schedule. Cultures are subcultured every two weeks by inoculating
approximately 35 mg of
tissue into 35 ml of liquid medium.
Soybean embryogenic suspension cultures may then be transformed by the method
of particle gun bombardment (Klein, et at., (1987) Nature (London) 327:70-73,
US Patent
Number 4,945,050). A Du Pont Biolistic PDS1000/HE instrument (helium retrofit)
can be
used for these transformations.
A selectable marker gene that can be used to facilitate soybean transformation
is a
transgene composed of the 35S promoter from Cauliflower Mosaic Virus (Odell,
et al.,
(1985) Nature 313:810-812), the hygromycin phosphotransferase gene from
plasmid
pJR225 (from E. coif; Gritz, et al., (1983) Gene 25:179-188), and the 3'
region of the
nopaline synthase gene from the T-DNA of the Ti plasmid of Agrobacterium
tumefaciens.
The expression cassette comprising an antisense Ethylene signaling associated
sequence
operably linked to the ubiquitin promoter can be isolated as a restriction
fragment. This
fragment can then be inserted into a unique restriction site of the vector
carrying the marker
gene.
To 50 pl of a 60 mg/ml 1 pm gold particle suspension is added (in order): 5 pl
DNA (1
pg/pl), 20 pl spermidine (0.1 M), and 50 pl CaCl2 (2.5 M). The particle
preparation is then
agitated for three minutes, spun in a microfuge for 10 seconds and the
supernatant
removed. The DNA-coated particles are then washed once in 400 pl 70% ethanol
and
resuspended in 40 pl of anhydrous ethanol. The DNA/particle suspension can be
sonicated
three times for one second each. Five microliters of the DNA-coated gold
particles are then
loaded on each macro carrier disk.
Approximately 300-400 mg of a two-week-old suspension culture is placed in an
empty 60x15 mm petri dish and the residual liquid removed from the tissue with
a pipette.
For each transformation experiment, approximately 5-10 plates of tissue are
normally
bombarded. Membrane rupture pressure is set at 1100 psi, and the chamber is
evacuated
to a vacuum of 28 inches mercury. The tissue is placed approximately 3.5
inches away from
the retaining screen and bombarded three times. Following bombardment, the
tissue can be
divided in half and placed back into liquid and cultured as described above.
Five to seven days post bombardment, the liquid media may be exchanged with
fresh media, and eleven to twelve days post-bombardment with fresh media
containing 50
98
CA 02877639 2015-01-09
mg/m1 hygromycin. This selective media can be refreshed weekly. Seven to eight
weeks
post-bombardment, green, transformed tissue may be observed growing from
untransformed, necrotic embryogenic clusters. Isolated green tissue is removed
and
inoculated into individual flasks to generate new, clonally propagated,
transformed
embryogenic suspension cultures. Each new line may be treated as an
independent
transformation event. These suspensions can then be subcultured and maintained
as
clusters of immature embryos or regenerated into whole plants by maturation
and
germination of individual somatic embryos.
Example 10
Sunflower Meristem Tissue Transformation
Sunflower rneristem tissues are transformed with an expression cassette
containing
an antisense Ethylene signaling associated sequences operably linked to a
ubiquitin
promoter as follows (see also, European Patent Number EP 0 486233,
Is and
Malone-Schoneberg, et al., (1994) Plant Science 103:199-207). Mature
sunflower seed (Helianthus annuus L.) are dehulled using a single wheat-head
thresher.
Seeds are surface sterilized for 30 minutes in a 20% Clorox bleach solution
with the
addition of two drops of Tweene 20 per 50 ml of solution. The seeds are rinsed
twice with
sterile distilled water.
Split embryonic axis explants are prepared by a modification of procedures
described
by Schrammeijer, at al. (Schrammeijer, at al., (1990) Plant Cell Rep. 9:55-
60). Seeds are
imbibed in distilled water for 60 minutes following the surface sterilization
procedure. The
cotyledons of each seed are then broken off, producing a clean fracture at the
plane of the
embryonic axis. Following excision of the root tip, the explants are bisected
longitudinally
between the primordial leaves. The two halves are placed, cut surface up, on
GBA medium
consisting of Murashige and Skoog mineral elements (Murashige, et al., (1962)
Physiol.
Plant., 15:473-497), Shepard's vitamin additions (Shepard, (1980) in Emergent
Techniques
for the Genetic Improvement of Crops (University of Minnesota Press, St. Paul,
Minnesota),
40 mg/I adenine sulfate, 30 g/I sucrose, 0.5 mg/I 6-benzyl-aminopurine (BAP),
0.25 mg/I
indole-3-acetic acid (IAA), 0.1 mg/I gibberellic acid (GA3), pH 5.6, and 8 g/I
Phytagar
(Invitrogen, Carlsbad, CA).
The explants are subjected to microprojectile bombardment prior to
Agrobacterium
treatment (Bidney, et al., (1992) Plant Mol. Biol. 18:301-313). Thirty to
forty explants are
placed in a circle at the center of a 60 X 20 mm plate for this treatment.
Approximately 4.7
mg of 1.8 mm tungsten microprojectiles are resuspended in 25 ml of sterile TE
buffer (10
99
CA 02877639 2015-01-09
mM Tris HCI, 1 mM EDTA, pH 8.0) and 1.5 ml aliquots are used per bombardment.
Each
plate is bombarded twice through a 150 mm nytex screen placed 2 cm above the
samples in
a PDS 1000 particle acceleration device.
Disarmed Agrobacterium tumefaciens strain EHA105 is used in all transformation
experiments. A binary plasmid vector comprising the expression cassette that
contains the
ethylene signaling associated gene operably linked to the ubiquitin promoter
is introduced
into Agrobacterium strain EHA105 via freeze-thawing as described by Holsters,
et a/., (1978)
MoL Gen. Genet 163:181-187. This plasmid further comprises a kanamycin
selectable
marker gene (i.e, nptI1). Bacteria for plant transformation experiments are
grown overnight
(28 C and 100 RPM continuous agitation) in liquid YEP medium (10 gm/I yeast
extract, 10
gm/I Bactopeptone, and 5 gm/I NaCI, pH 7.0) with the appropriate antibiotics
required for
bacterial strain and binary plasmid maintenance. The suspension is used when
it reaches
an OD600 of about 0.4 to 0.8. The Agrobacterium cells are pelleted and
resuspended at a
final 0D600 of 0.5 in an inoculation medium comprised of 12.5 mM MES pH 5.7, 1
gm/I NH4CI,
and 0.3 gm/I MgSO4.
Freshly bombarded explants are placed in an Agrobacterium suspension, mixed,
and
left undisturbed for 30 minutes. The explants are then transferred to GBA
medium and co-
cultivated, cut surface down, at 26 C and 18-hour days. After three days of co-
cultivation,
the explants are transferred to 374B (GBA medium lacking growth regulators and
a reduced
sucrose level of 1%) supplemented with 250 mg/I cefotaxime and 50 mg/I
kanamycin sulfate.
The explants are cultured for two to five weeks on selection and then
transferred to fresh
374B medium lacking kanamycin for one to two weeks of continued development.
Explants
with differentiating, antibiotic-resistant areas of growth that have not
produced shoots
suitable for excision are transferred to GBA medium containing 250 mg/I
cefotaxime for a
second 3-day phytohormone treatment. Leaf samples from green, kanamycin-
resistant
shoots are assayed for the presence of NPTII by ELISA and for the presence of
transgene
expression by assaying for a modulation in meristem development (i.e., an
alteration of size
and appearance of shoot and floral meristems).
NPTII-positive shoots are grafted to Pioneer hybrid 6440 in vitro-grown
sunflower
seedling rootstock. Surface sterilized seeds are germinated in 48-0 medium
(half-strength
Murashige and Skoog salts, 0.5% sucrose, 0.3% Gelrite gelling agent, pH 5.6)
and grown
under conditions described for explant culture. The upper portion of the
seedling is removed,
a 1 cm vertical slice is made in the hypocotyl, and the transformed shoot
inserted into the cut.
The entire area is wrapped with Parafilm flexible film to secure the shoot.
Grafted plants
can be transferred to soil following one week of in vitro culture. Grafts in
soil are maintained
100
CA 02877639 2015-01-09
under high humidity conditions followed by a slow acclimatization to the
greenhouse
environment. Transformed sectors of To plants (parental generation) maturing
in the
greenhouse are identified by NPTII ELISA and/or by Ethylene signaling
associated activity
analysis of leaf extracts while transgenic seeds harvested from NPTII-positive
To plants are
identified by Ethylene signaling associated activity analysis of small
portions of dry seed
cotyledon.
An alternative sunflower transformation protocol allows the recovery of
transgenic
progeny without the use of chemical selection pressure. Seeds are dehulled and
surface-
sterilized for 20 minutes in a 20% Clorox bleach solution with the addition
of two to three
drops of Tween 20 per 100 ml of solution, then rinsed three times with
distilled water.
Sterilized seeds are imbibed in the dark at 26 C for 20 hours on filter paper
moistened with
water. The cotyledons and root radical are removed, and the meristem explants
are cultured
on 374E (GBA medium consisting of MS salts, Shepard vitamins, 40 mg/I adenine
sulfate,
3% sucrose, 0.5 mg/I 6-BAP, 0.25 mg/I IAA, 0.1 mg/I GA, and 0.8% Phytagar
(Invitrogen,
Carlsbad, CA) at pH 5.6) for 24 hours under the dark. The primary leaves are
removed to
expose the apical meristem, around 40 explants are placed with the apical dome
facing
upward in a 2 cm circle in the center of 374M (GBA medium with 1.2% Phytagar
(Invitrogen,
Carlsbad, CA)), and then cultured on the medium for 24 hours in the dark.
Approximately 18.8 mg of 1.8 pm tungsten particles are resuspended in 150 pl
absolute ethanol. After sonication, 8 pl of it is dropped on the center of the
surface of
macrocarrier. Each plate is bombarded twice with 650 psi rupture discs in the
first shelf at
26 mm of Hg helium gun vacuum.
The plasmid of interest is introduced into Agrobacterium tumefaciens strain
EHA105
via freeze thawing as described previously. The pellet of overnight-grown
bacteria at 28 C
in a liquid YEP medium (10 g/I yeast extract, 10 g/I Bactopeptone, and 5 g/I
NaCI, pH 7.0) in
the presence of 50 pg/I kanamycin is resuspended in an inoculation medium
(12.5 mM 2-mM
2-(N-morpholino) ethanesulfonic acid, MES, 1 g/I NH4CI and 0.3 g/I MgSO4 at pH
5.7) to
reach a final concentration of 4.0 at 0D600. Particle-bombarded explants are
transferred to
GBA medium (374E), and a droplet of bacteria suspension is placed directly
onto the top of
the meristem. The explants are co-cultivated on the medium for 4 days, after
which the
explants are transferred to 374C medium (GBA with 1% sucrose and no BAP, IAA,
GA3 and
supplemented with 250 pg/ml cefotaxime). The plantlets are cultured on the
medium for
about two weeks under 16-hour day and 26 C incubation conditions.
Explants (around 2 cm long) from two weeks of culture in 374C medium are
screened for a modulation in meristem development (i.e., an alteration of size
and
101
CA 02877639 2015-01-09
appearance of shoot and floral meristems). After positive explants are
identified, those
shoots that fail to exhibit modified Ethylene signaling associated activity
are discarded, and
every positive explant is subdivided into nodal explants. One nodal explant
contains at least
one potential node. The nodal segments are cultured on GBA medium for three to
four days
to promote the formation of auxiliary buds from each node. Then they are
transferred to
374C medium and allowed to develop for an additional four weeks. Developing
buds are
separated and cultured for an additional four weeks on 374C medium. Pooled
leaf samples
from each newly recovered shoot are screened again by the appropriate protein
activity
assay. At this time, the positive shoots recovered from a single node will
generally have
been enriched in the transgenic sector detected in the initial assay prior to
nodal culture.
Recovered shoots positive for modified Ethylene signaling associated
expression are
grafted to Pioneer Hybrid 6440 in vitro-grown sunflower seedling rootstock.
The rootstocks
are prepared in the following manner. Seeds are dehulled and surface-
sterilized for 20
minutes in a 20% Clorox bleach solution with the addition of two to three
drops of Tween
20 per 100 ml of solution, and are rinsed three times with distilled water.
The sterilized
seeds are germinated on the filter moistened with water for three days, then
they are
transferred into 48 medium (half-strength MS salt, 0.5% sucrose, 0.3% Gelrite
gelling
agent pH 5.0) and grown at 26 C under the dark for three days, then incubated
at 16-hour-
day culture conditions. The upper portion of selected seedling is removed, a
vertical slice is
made in each hypocotyl, and a transformed shoot is inserted into a V-cut. The
cut area is
wrapped with Parafilm flexible film. After one week of culture on the medium,
grafted
plants are transferred to soil. In the first two weeks, they are maintained
under high humidity
conditions to acclimatize to a greenhouse environment.
Example 11
Rice Tissue Transformation
One method for transforming DNA into cells of higher plants that is available
to those
skilled in the art is high-velocity ballistic bombardment using metal
particles coated with the
nucleic acid constructs of interest (see, Klein, et al., Nature (1987)
(London) 327:70-73 and
see, US Patent Number 4,945,050). A Biolistic PDS-1000/He (BioRAD
Laboratories,
Hercules, CA) is used for these complementation experiments. The particle
bombardment
technique is used to transform the Ethylene signaling associated mutants and
wild type rice
with DNA fragments
The bacterial hygromycin B phosphotransferase (Hpt II) gene from Streptomyces
hygroscopicus that confers resistance to the antibiotic is used as the
selectable marker for
102
CA 02877639 2015-01-09
rice transformation. In the vector, pML18, the Hpt II gene was engineered with
the 358
promoter from Cauliflower Mosaic Virus and the termination and polyadenylation
signals
from the octopine synthase gene of Agrobacterium tumefaciens. pML18 was
described in
WO 97/47731, which was published on December 18, 1997.
Embryogenic callus cultures derived from the scutellum of germinating rice
seeds
serve as source material for transformation experiments. This material is
generated by
germinating sterile rice seeds on a callus initiation media (MS salts, Nitsch
and Nitsch
vitamins, 1.0 mg/I 2,4-D and 10 jr.M AgNO3) in the dark at 27-28 C.
Embryogenic callus
o proliferating from the scutellurn of the embryos is the transferred to CM
media (NO salts,
Nitsch and Nitsch vitamins, 1 mg/I 2,4-D, Chu, etal., 1985, Sci. Sin/ca 18:
659-668). Callus
cultures are maintained on CM by routine sub-culture at two week intervals and
used for
transformation within 10 weeks of initiation.
Callus is prepared for transformation by subculturing 0.5-1.0 mm pieces
approximately 1 mm apart, arranged in a circular area of about 4 cm in
diameter, in the
center of a circle of Whatman #541 paper placed on CM media. The plates with
callus are
incubated in the dark at 27-28 C for 3-5 days. Prior to bombardment, the
filters with callus
are transferred to CM supplemented with 0.25 M mannitol and 0.25 M sorbitol
for 3 hr in the
dark. The petri dish lids are then left ajar for 20-45 minutes in a sterile
hood to allow
moisture on tissue to dissipate.
Each genomic DNA fragment is co-precipitated with pML18 containing the
selectable
marker for rice transformation onto the surface of gold particles. To
accomplish this, a total
of 10 pg of DNA at a 2:1 ratio of trait:selectable marker DNAs are added to 50
pl aliquot of
gold particles that have been resuspended at a concentration of 60 mg m11.
Calcium
chloride (50 pl of a 2.5 M solution) and spermidine (20 pl of a 0.1 M
solution) are then added
to the gold-DNA suspension as the tube is vortexing for 3 min. The gold
particles are
centrifuged in a microfuge for 1 sec and the supernatant removed. The gold
particles are
then washed twice with 1 ml of absolute ethanol and then resuspended in 50 pl
of absolute
ethanol and sonicated (bath sonicator) for one second to disperse the gold
particles. The
gold suspension is incubated at -70 C for five minutes and sonicated (bath
sonicator) if
needed to disperse the particles. Six pl of the DNA-coated gold particles are
then loaded
onto mylar macrocarrier disks and the ethanol is allowed to evaporate.
At the end of the drying period, a petri dish containing the tissue is placed
in the
chamber of the PDS-1000/He. The air in the chamber is then evacuated to a
vacuum of
28-29 inches Hg. The macrocarrier is accelerated with a helium shock wave
using a rupture
103
CA 02877639 2015-01-09
membrane that bursts when the He pressure in the shock tube reaches 1080-1100
psi. The
tissue is placed approximately 8 cm from the stopping screen and the callus is
bombarded
two times. Two to four plates of tissue are bombarded in this way with the DNA-
coated gold
particles. Following bombardment, the callus tissue is transferred to CM media
without
supplemental sorbitol or mannitol.
Within 3-5 days after bombardment the callus tissue is transferred to SM media
(CM
medium containing 50 mg/I hygromycin). To accomplish this, callus tissue is
transferred
from plates to sterile 50 ml conical tubes and weighed. Molten top-agar at 40
C is added
using 2.5 ml of top agar/100 mg of callus. Callus clumps are broken into
fragments of less
than 2 mm diameter by repeated dispensing through a 10 ml pipet. Three ml
aliquots of the
callus suspension are plated onto fresh SM media and the plates are incubated
in the dark
for 4 weeks at 27-28 C. After 4 weeks, transgenic callus events are
identified, transferred to
fresh SM plates and grown for an additional 2 weeks in the dark at 27-28 C.
Growing callus is transferred to RM1 media (MS salts, Nitsch and Nitsch
vitamins,
2% sucrose, 3% sorbitol, 0.4% Gelrite0 gelling agent +50 ppm hyg B) for 2
weeks in the
dark at 25 C. After 2 weeks the callus is transferred to RM2 media (MS salts,
Nitsch and
Nitsch vitamins, 3% sucrose, 0.4% Gelrite gelling agent + 50 ppm hyg B) and
placed under
cool white light (-40 p.E ril2s -1 ) with a 12 hr photo period at 25 C and 30-
40% humidity. After
2-4 weeks in the light, callus begin to organize, and form shoots. Shoots are
removed from
surrounding callus/media and gently transferred to RM3 media (1/2 x MS salts,
Nitsch and
Nitsch vitamins, 1% sucrose + 50 ppm hygromycin B) in PhytatrayTM disposable
plant cell
culture vessels (Sigma Chemical Co., St. Louis, MO) and incubation is
continued using the
same conditions as described in the previous step.
Plants are transferred from RM3 to 4" pots containing Scotts MetroMix 350
growing
medium after 2-3 weeks, when sufficient root and shoot growth have occurred.
The seed
obtained from the transgenic plants is examined for genetic complementation of
the Ethylene
signaling associated mutation with the wild-type genomic DNA containing the
Ethylene
signaling associated gene.
Example 12
Variants of Ethylene signaling associated Sequences
A. Variant Nucleotide Sequences of Ethylene signaling associated
Proteins That
Do Not Alter the Encoded Amino Acid Sequence
The Ethylene signaling associated nucleotide sequences are used to generate
variant nucleotide sequences having the nucleotide sequence of the open
reading frame
104
CA 02877639 2015-01-09
with about 70%, 75%, 80%, 85%, 90% and 95% nucleotide sequence identity when
compared to the starting unaltered ORF nucleotide sequence of the
corresponding SEQ ID
NO. These functional variants are generated using a standard codon table.
While the
nucleotide sequence of the variants are altered, the amino acid sequence
encoded by the
open reading frames do not change.
B. Variant Amino Acid Sequences of Ethylene signaling associated
Polypeptides
Variant amino acid sequences of the Ethylene signaling associated polypeptides
are
generated. In this example, one amino acid is altered. Specifically, the open
reading frames
are reviewed to determine the appropriate amino acid alteration. The selection
of the amino
acid to change is made by consulting the protein alignment (with the other
orthologs and
other gene family members from various species). An amino acid is selected
that is deemed
not to be under high selection pressure (not highly conserved) and which is
rather easily
substituted by an amino acid with similar chemical characteristics (i.e.,
similar functional
side-chain). Using the protein alignment, an appropriate amino acid can be
changed. Once
the targeted amino acid is identified, the procedure outlined in the following
section C is
followed. Variants having about 70%, 75%, 80%, 85%, 90% and 95% nucleic acid
sequence
identity are generated using this method.
C. Additional Variant Amino Acid Sequences of Ethylene signaling associated
Polypeptides
In this example, artificial protein sequences are created having 80%, 85%,
90%, and
95% identity relative to the reference protein sequence. This latter effort
requires identifying
conserved and variable regions from the alignment and then the judicious
application of an
amino acid substitutions table. These parts will be discussed in more detail
below.
Largely, the determination of which amino acid sequences are altered is made
based
on the conserved regions among Ethylene signaling associated protein or among
the other
Ethylene signaling associated polypeptides. Based on the sequence alignment,
the various
regions of the Ethylene signaling associated polypeptide that can likely be
altered are
represented in lower case letters, while the conserved regions are represented
by capital
letters. It is recognized that conservative substitutions can be made in the
conserved
regions below without altering function. In addition, one of skill will
understand that
functional variants of the Ethylene signaling associated sequence of the
invention can have
minor non-conserved amino acid alterations in the conserved domain.
105
CA 02877639 2015-01-09
Artificial protein sequences are then created that are different from the
original in the
intervals of 80-85%, 85-90%, 90-95% and 95-100% identity. Midpoints of these
intervals are
targeted, with liberal latitude of plus or minus 1%, for example. The amino
acids
substitutions will be effected by a custom Pen l script. The substitution
table is provided
below in Table 4.
106
CA 02877639 2015-01-09
Table 4. Substitution Table
Amino Strongly Similar and Rank of
Acid Optimal Substitution Order to Comment
Change
L,V 1 50:50 substitution
I,V 2 50:50 substitution
V I,L 3 50:50 substitution
A G 4
A 5
6
7
8
9
11
12
13
14
16
First methionine cannot
17 change
Na No good substitutes
Na No good substitutes
Na No good substitutes
First, any conserved amino acids in the protein that should not be changed is
identified and "marked off" for insulation from the substitution. The start
methionine will of
5 course be added to this list automatically. Next, the changes are made.
H, C, and P are not changed in any circumstance. The changes will occur with
isoleucine first, sweeping N-terminal to C-terminal. Then leucine, and so on
down the list
until the desired target it reached. Interim number substitutions can be made
so as not to
cause reversal of changes. The list is ordered 1-17, so start with as many
isoleucine
10 changes as needed before leucine, and so on down to methionine. Clearly
many amino
acids will in this manner not need to be changed. L, I and V will involve a
50:50 substitution
of the two alternate optimal substitutions.
The variant amino acid sequences are written as output. Perl script is used to
calculate the percent identities. Using this procedure, variants of the
Ethylene signaling
15 associated polypeptides are generating having about 80%, 85%, 90% and
95% amino acid
identity to the starting unaltered ORF nucleotide sequence of SEQ ID NOS: 1,
3, 5, 7 or 9.
107
CA 02877639 2015-01-09
Example 13
Transeenic Maize Plants
To transgenic maize plants containing the Ethylene signaling associated
construct
under the control of a promoter were generated. These plants were grown in
greenhouse
conditions, under the FASTCORN system, as detailed in US Patent Application
Publication
Number 2003/0221212, US Patent Application Serial Number 10/367,417.
Each of the plants was analyzed for measurable alteration in one or more of
the
following characteristics in the following manner:
Ti progeny derived from self fertilization of each To plant containing a
single copy of
each Ethylene signaling associated construct that were found to segregate 1:1
for the
transgenic event were analyzed for improved growth rate in low KNO3. Growth
was
monitored up to anthesis when cumulative plant growth, growth rate and ear
weight were
determined for transgene positive, transgene null, and non-transformed control
events. The
distribution of the phenotype of individual plants was compared to the
distribution of a control
set and to the distribution of all the remaining treatments. Variances for
each set were
calculated and compared using an F test, comparing the event variance to a non-
transgenic
control set variance and to the pooled variance of the remaining events in the
experiment.
The greater the response to KNO3, the greater the variance within an event set
and the
greater the F value. Positive results will be compared to the distribution of
the transgene
within the event to make sure the response segregates with the transgene.
All publications and patent applications in this specification are indicative
of the level
of ordinary skill in the art to which this invention pertains.
?5
The scope of the claims should not be limited by the preferred embodiments set
forth
in the examples, but should be given the broadest interpretation consistent
with the
description as a whole.
108