Note: Descriptions are shown in the official language in which they were submitted.
CA 02884685 2015-03-10
HIGH EFFICIENCY GENE TRANSFER AND EXPRESSION IN MAMMALIAN CELLS
BY A MULTIPLE TRANSFECTION PROCEDURE OF MAR SEQUENCES
FIELD OF THE INVENTION
The present invention relates to purified and isolated DNA sequences having
protein
production increasing activity and more specifically to the use of matrix
attachment
regions (MARs) for increasing protein production activity in a eukaryotic
cell. Also
disclosed is a method for the identification of said active regions, in
particular MAR
nucleotide sequences, and the use of these characterized active MAR sequences
in a
new multiple transfection method.
BACKGROUND OF THE INVENTION
Nowadays, the model of loop domain organization of eukaryotic chromosomes is
well
accepted (Boulikas T, "Nature of DNA sequences at the attachment regions of
genes to
the nuclear matrix", J. Cell Biochem., 52:14-22, 1993). According to this
model
chromatin is organized in loops that span 50-100 kb attached to the nuclear
matrix, a
proteinaceous network made up of RNPs and other nonhistone proteins (Bode J,
Stengert-lber M, Kay V, Schalke T and Dietz-Pfeilstetter A, Crit. Rev. Euk.
Gene Exp.,
6:115-138, 1996).
The DNA regions attached to the nuclear matrix are termed SAR or MAR for
respectively scaffold (during metaphase) or matrix (interphase) attachment
regions
(Hart C and Laemmli U (1998), "Facilitation of chromatin dynamics by SARs"
Curr Opin
Genet Dev 8, 519-525.)
As such, these regions may define boundaries of independent chromatin domains,
such that only the encompassing cis-regulatory elements control the expression
of the
genes within the domain.
However, their ability to fully shield a chromosomal locus from nearby
chromatin
elements, and thus confer position-independent gene expression, has not been
seen in
stably transfected cells (Poljak L, Seum C, Mattioni land Laemmli U. (1994)
"SARs
stimulate but do not confer position independent gene expression", Nucleic
Acids Res
CA 02884685 2015-03-10
111111111111VOINV ____________________________________
22, 4386-4394). On the other hand, MAR (or S/MAR) sequences have been shown to
interact with enhancers to increase local chromatin accessibility (Jenuwein T,
Forrester
W, Fernandez-Herrero L, Laible G, Dull M, and Grosschedl R. (1997) "Extension
of
chromatin accessibility by nuclear matrix attachment regions" Nature 385, 269-
272).
Specifically, MAR elements can enhance expression of heterologous genes in
cell
culture lines (Kalos M and Fournier R (1995) "Position-independent transgene
expression mediated by boundary elements from the apolipoprotein B chromatin
domain" Mo/ Cell Biol 15,198-207), transgenic mice (Castilla J, Pintado B,
Sola, I,
Sanchez-Morgado J, and Enjuanes L (1998) "Engineering passive immunity in
transgenic mice secreting virus-neutralizing antibodies in milk" Nat
Biotechnol 16, 349-
354) and plants (Allen G, Hall GJ, Michalowski S, Newman W, Spiker S,
Weissinger A,
and Thompson W (1996), "High-level transgene expression in plant cells:
effects of a
strong scaffold attachment region from tobacco" Plant Cell 8, 899-913). The
utility of
MAR sequences for developing improved vectors for gene therapy is also
recognized
(Agarwal M, Austin T, Morel F, Chen J, Bohnlein E, and Plavec 1(1998),
"Scaffold
attachment region-mediated enhancement of retroviral vector expression in
primary T
cells" J Virol 72, 3720-3728).
Recently, it has been shown thatchromatin-structure modifying sequences
including
MARs, as exemplified by the chicken lysozyme 5' MAR is able to significantly
enhance
reporter expression in pools of stable Chinese Hamster Ovary (CHO) cells (Zahn-
Zabal
M, et al., "Development of stable cell lines for production or regulated
expression using
matrix attachment regions" J Biotechnol, 2001, 87(1): p. 29-42). This property
was used
to increase the proportion of high-producing clones, thus reducing the number
of clones
that need to be screened. These benefits have been observed both for
constructs with
MARs flanking the transgene expression cassette, as well as when constructs
are co-
transfected with the MAR on a separate plasmid. However, expression levels
upon co-
transfection with MARs were not as high as those observed for a construct in
which two
MARs delimit the transgene expression unit. A third and preferable process was
shown
to be the transfection of transgenes with MARs both linked to the transgene
and on a
separate plasmid (Girod et al., submitted for publication). However, one
persisting
limitation of this technique is the quantity of DNA that can be transfected
per cell.
Many multiples transfection protocols have been developed in order to achieve
a high
transfection efficiency to characterize the function of genes of interest. The
protocol
applied by Yamamoto et al, 1999 ("High efficiency gene transfer by multiple
transfection
2
CA 02884685 2015-03-10
protocol", Histochem. J. 31(4), 241-243) leads to a transfection efficiency of
about 80%
after 5 transfections events, whereas the conventional transfection protocol
only
achieved a rate of <40%. While this technique may be useful when one wishes to
increase the proportion of expressing cells, it does not lead to cells with a
higher
intrinsic productivity. Therefore, it cannot be used to generate high producer
monoclonal cell lines. Hence, the previously described technique has two major
drawbacks:
i) this technique does not generate a homogenous population of transfected
cells, since it cannot favour the integration of further gene copy, nor does
it
direct the transgenes to favorable chromosomal loci,
ii) the use of the same selectable marker in multiple transfection events
does
not permit the selection of doubly or triply transfected cells.
In patent application W002/074969, the utility of MARs for the development of
stable
eukaryotic cell lines has also been demonstrated. However, this application
does not
disclose neither any conserved homology for MAR DNA element nor any technique
for
predicting the ability for a DNA sequence to be a MAR sequence.
In fact no clear-cut MAR consensus sequence has been found (Boulikas T,
"Nature of
DNA sequences at the attachment regions of genes to the nuclear matrix", J.
Cell
Biochem., 52:14-22, 1993) but evolutionarily, the structure of these sequences
seem to
be functionally conserved in eukaryotic genomes, since animal MARs can bind to
plant
nuclear scaffolds and vice versa (Mielke C, Kohwi Y, Kohwi-Shigematsu T and
Bode J,
"Hierarchical binding of DNA fragments derived from scaffold-attached regions:
correlation of properties in vitro and function in vivo", Biochemistry,
29:7475-7485,
1990) .
The identification of MARs by biochemical studies is a long and unpredictable
process;
various results can be obtained depending on the assay (Razin SV, "Functional
architecture of chromosomal DNA domains", Crit Rev Eukaryot Gene Expr., 6:247-
269,
1996). Considering the huge number of expected MARs in a eukaryotic genome and
the amount of sequences issued from genome projects, a tool able to filter
potential
MARS in order to perform targeted experiments would be greatly useful.
Currently two different predictive tools for MARs are available via the
Internet.
3
CA 02884685 2015-03-10
The fist one, MAR-Finder (http://futuresoft.org/MarFinder; Singh GB, Kramer JA
and
Krawetz SA, "Mathematical model to predict regions of chromatin attachment to
the
nuclear matrix", Nucleic Acid Research, 25:1419-1425, 1997) is based on set of
patterns identified within several MARs and a statistical analysis of the co-
occurrence of
these patterns. MAR-Finder predictions are dependent of the sequence context,
meaning that predicted MARs depend on the context of the submitted sequence.
The
other predictive software, SMARTest (http://www. genomatix.de; Frisch M, Frech
K,
Klingenhoff A, Cartharius K, Liebich I and Werner T, "In silico prediction of
scaffold/matrix attachment regions in large genomic sequences", Genome
Research,
12:349-354, 2001), use weight-matrices derived from experimentally identified
MARs.
SMARTest is said to be suitable to perform large-scale analyses. But actually
aside its
relative poor specificity, the amount of hypothetical MARs rapidly gets huge
when doing
large scale analyses with it, and in having no way to increase its specificity
to restrain
the number of hypothetical MARs, SMARTest becomes almost useless to screen for
potent MARs form large DNA sequences.
Some other softwares, not available via the Internet, also exists; they are
based as well
on the frequency of MAR motifs (MRS criterion;Van Drunen CM et al., "A
bipartite
sequence element associated with matrix/scaffold attachment regions", Nucleic
Acids
Res, 27:2924-2930, 1999), (ChrClass; Glazko GV et al., "Comparative study and
prediction of DNA fragments associated with various elements of the nuclear
matrix",
Biochim. Biophys. Acta, 1517:351-356, 2001) or based on the identification of
sites of
stress-induced DNA duplex (SIDD; Benham C and al., "Stress-induced duplex DNA
destabilization in scaffold/matrix attachment regions", J. Mol. Biol., 274:181-
196, 1997).
However, their suitability to analyze complete genome sequences remains
unknown,
and whether these tools may allow the identification of protein production-
increasing
sequences has not been reported.
Furthermore, due to the relatively poor specificity of these softvvares
(Frisch M, Frech K,
Klingenhoff A, Cartharius K, Liebich I and Werner T, "In silico prediction of
scaffold/matrix attachment regions in large genomic sequences", Genome
Research,
12:349-354, 2001), the amount of hypothetical MARs identified in genomes
rapidly gets
unmanageable when doing large scale analyses, especially if most of these have
no or
poor activity in practice. Thus, having no way to increase prediction
specificity to
restrain the number of hypothetical MARs, many of the available programs
become
4
CA 02884685 2015-03-10
almost useless to identify potent genetic elements in view of efficiently
increasing
recombinant protein production,
Since all the above available predictive methods have some drawbacks that
prevent
large-scale analyses of genomes to identify reliably novel and potent MARs,
the object
of this invention is to 1) understand the functional features of MARs that
allow improved
recombinant protein expression; 2) get a new Bioinformatic tool compiling MAR
structural features as a prediction of function, in order to 3) perform large
scale
analyses of genomes to identify novel and more potent MARs, and, finally 4) to
demonstrate improved efficiency to increase the production of recombinant
proteins -
from eukaryotic cells or organisms when using the newly identified MAR
sequences.
SUMMARY OF THE INVENTION
This object has been achieved by providing an improved and reliable method for
the
identification of DNA sequences having protein production increasing activity,
in
particular MAR nucleotide sequences, and the use of these characterized active
MAR
sequences in a new multiple transfection method to increase the production of
recombinant proteins in eukaryotic cells.
BRIEF DESCRIPTION OF THE FIGURES
Fig. 1 shows the distribution plots of MARs and non-MARs sequences. Histograms
are
density plots (relative frequency divided by the bin width) relative to the
score of the
observed parameter. The density histogram for human MARs in the SMARt DB
database is shown in black, while the density histogram for the human
chromosome 22
are in grey.
Fig. 2 shows Scatterplots of the four different criteria used by SMAR Scan
and the
AT-content with human MARs from SMARt DB.
Fig. 3 shows the distribution plots of MAR sequences by organism. MAR
sequences
from SMARt DB of other organisms were retrieved and analyzed. The MAR
sequences
5
CA 02884685 2015-03-10
density distributions for the mouse, the chicken, the sorghum bicolor and the
human
are plotted jointly.
Fig. 4 shows SMAR Scan predictions on human chromosome 22 and on shuffled
chromosome 22. Top plot: Average number of hits obtained by SMAR Scan with
five:
rubbled, scrambled, shuffled within nonoverlapping windows of 10 bp, order 1
Markov
chains model and with the native chromosome 22. Bottom plot: Average number of
MARs predicted by SMAR Scan in five: rubbled, scrambled, shuffled within non-
overlapping windows of 10 bp, order 1 Markov chains model and with the native
chromosome 22.
Fig. 5 shows the dissection of the ability of the chicken lysozyme gene 5'-MAR
to
stimulate transgene expression in CHO-DG44 cells. Fragments B, K and F show
the
highest ability to stimulate transgene expression. The indicated relative
strength of the
elements was based on the number of high-expressor cells.
Fig. 6 shows the effect of serial-deletions of the 5'-end (upper part) and the
3'-end
(lower part) of the 5'-MAR on the loss of ability to stimulate transgene
expression. The
transition from increased to decreased activity coincide with B-, K- and F-
fragments.
Fig. 7 shows that portions of the F fragment significantly stimulate transgene
expression. The F fragment regions indicated by the light grey arrow were
multimerized,
inserted in pGEGFP Control and transfected in CHO cells. The element that
displays
the highest activity is located in the central part of the element and
corresponds to
fragment Fill (black bar labelled minimal MAR). In addition, an enhancer
activity is
located in the 3'-flanking part of the Fill fragment (dark grey bar labelled
MAR
enhancer).
Fig. 8 shows a map of locations for various DNA sequence motifs within the
cLysMAR.
Fig. 8 (B) represents a Map of locations for various DNA sequence motifs
within the
cLysMAR. Vertical lines represent the position of the computer-predicted sites
or
sequence motifs along the 3034 base pairs of the cLysMAR and its active
regions, as
presented in Fig. 5. The putative transcription factor sites, (MEF2 05, Oct-1,
USF-02,
GATA, NFAT) for activators and (CDP, SATB1, CTCF, ARBP/MeCP2) for repressors
of
transcription, were identified using Matlnspector (Genomatix), and CpG islands
were
6
CA 02884685 2015-03-10
identifed with CPGPLOT. Motifs previously associated with MAR elements are
labelled
in black and include CpG dinucleotides and CpG islands, unwinding motifs
(AATATATT
and AATATT), poly As and Is, poly Gs and Cs, Drosophila topoisomerase II
binding
sites (GINWAYATTNATTNATNNR) which had identity to the 6 bp core and High
mobility group I (HMG-I/Y) protein binding sites. Other structural motifs
include
nucleosome-binding and nucleosome disfavouring sites and a motif thought to
relieve
the superhelical strand of DNA. Fig. 8(A) represents the comparison of the
ability of
portions of the cLysMAR to activate transcription with MAR prediction score
profiles
with MarFinder. The top diagram shows the MAR fragment activity as in Fig. 5,
while
the middle and bottom curves show MARFinder-predicted potential for MAR
activity and
for bent DNA structures respectively.
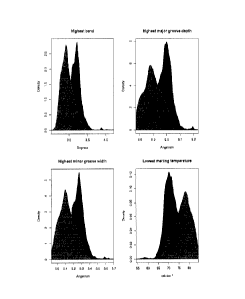
Fig. 9 shows the correlation of DNA physico-chemical properties with MAR
activity. Fig.
9(A), represents the DNA melting temperature, double helix bending, major
groove
depth and minor groove width profiles of the 5'-MAR and were determined using
the
algorithms of Levitsky et al (Levitsky VG, Ponomarenko MP, Ponomarenko JV,
Frolov
AS, Kolchanov NA "Nucleosomal DNA property database", Bioinformatics, 15;
582592,
1999). The most active B, K and F fragments depicted at the top are as shown
as in
Figure 1. Fig. 9(B), represents the enlargement of the data presented in panel
A to
display the F fragment map aligned with the tracings corresponding to the
melting
temperature (top curve) and DNA bending (bottom curve). The position of the
most
active FIB fragment and protein binding site for specific transcription
factors are as
indicated.
Fig. 10 shows the distribution of putative transcription factor binding sites
within the 5'-
cLysMAR. Large arrows indicate the position of the CUE elements as identified
with
SMAR Scan .
Fig. 11 shows the scheme of assembly of various portions of the MAR. The
indicated
portions of the cLysMAR were amplified by PCR, introducing EgIll-BamH1 linker
elements at each extremity, and assembled to generate the depicted composite
elements. For instance, the top construct consists of the assembly of all CUE
and
flanking sequences at their original location except that Bg11-Bam H11 linker
sequences
separate each element.
7
CA 02884685 2015-03-10
Fig. 12 represents the plasmid maps.
Fig. 13 shows the effect of re-transfecting primary transfectants on GFP
expression.
Cells (CHO-DG44) were co-transfected with pSV40EGFP (left tube) or pMAR-
SV40EGFP (central tube) and pSVneo as resistance plasmid. Cells transfected
with
pMAR-SV40EGFP were re-transfected 24 hours later with the same plasmid and a
different selection plasmid, pSVpuro (right tube). After two weeks selection,
the
phenotype of the stably transfected cell population was analysed by FACS.
Fig. 14 shows the effect of multiple load of MAR-containing plasmid. The pMAR-
SV40EGFP/ pMAR-SV40EGFP secondary transfectants were used in a third cycle of
transfection at the end of the selection process. The tertiary transfection
was
accomplished with pMAR or pMAR-SV40EGFP to give tertiary transfectants. After
24
hours, cells were transfected again with either plasmid, resulting in the
quaternary
transfectants (see Table 4).
Fig. 15 shows comparative performance of SMAR prediction algorithms
exemplified by
region INP18A10A7. (A) SMAR Scan analysis was performed with default
settings.
(B) SIDD analysis (top curve and left-hand side scale), and the attachment of
several
DNA fragments to the nuclear matrix in vitro (bar-graph, right-hand side
scale) was
taken from Goetze et al ( Goetze S, Gluch A, Benham C, Bode J, "Computational
and
in vitro analysis of destabilized DNA regions in the interferon gene cluster:
potential of
predicting functional gene domains." Biochemistry, 42:154-166, 2003).
Fig. 16 represents the results of a a gene therapy-like protocol using MARs.
The group of mice injected by MAR-network, induced from the beginning of the
experiment, display a better induction of the hematocrit in comparison of mice
injected
by original network without MAR. After 2 months, hematocrits in "MAR-
containing group"
is still at values higher (65%) than normal hematocrit levels (45-55%).
Fig. 17 represents the scatterplot for the 1757 S/MAR sequences of the AT
(top) and
TA (bottom) dinucleotide percentages versus the predicted DNA bending as
computed
by SMAR Scan .
Fig. 18 represents the dinucleotide percentage distribution plots over the
1757 non-
S/MARs sequences.
8
CA 02884685 2015-03-10
Fig.19 shows the effect of various S/MAR elements on the production of
recombinant
green fluorescent protein (GFP). Populations of CHO cells transfected with a
GFP
expression vector containing or a MAR element, as indicated, were analyzed by
a
fluorescence-activated cell sorter (FACS ), and typical profiles are shown.
The profiles
display the cell number counts as a function of the GFP fluorescence levels.
Fig. 20 depicts the effect of the induction of hematocrit in mice injected by
MAR-
network.
DETAILED DESCRIPTION OF THE INVENTION
The present invention relates to a purified and isolated DNA sequence having
protein
production increasing activity characterized in that said DNA sequence
comprises at
least one bent DNA element, and at least one binding site for a DNA binding
protein.
Certain sequences of DNA are known to form a relatively "static curve", where
the DNA
follows a particular 3-dimensional path. Thus, instead of just being in the
normal B-DNA
conformation ("straight"), the piece of DNA can form a flat, planar curve also
defined as
bent DNA (Marini, etal., 1982 "Bent helical structure in kinetoplast DNA",
Proc. Natl.
Acad. Sc!. USA, 79: 7664-7664).
Surprisingly, Applicants have shown that the bent DNA element of a purified
and
isolated DNA sequence having protein production increasing activity of the
present
invention usually contains at least 10% of dinucleotide TA, and/or at least
12% of
dinucleotide AT on a stretch of 100 contiguous base pairs. Preferably, the
bent DNA
element contains at least 33% of dinucleotide TA, and/or at least 33% of
dinucleotide
AT on a stretch of 100 contiguous base pairs. These data have been obtained by
the
method described further.
According to the present invention, the purified and isolated DNA sequence
usually
comprises a MAR nucleotide sequence selected from the group comprising the
sequences SEQ ID Nos 1 to 27 or a cLysMAR element or a fragment thereof.
Preferably, the purified and isolated DNA sequence is a MAR nucleotide
sequence
9
CA 02884685 2015-03-10
selected from the group comprising the sequences SEQ ID Nos 1 to 27, more
preferably the sequences SEQ ID Nos 24 to 27.
Encompassed by the present invention are as well complementary sequences of
the
above-mentioned sequences SEQ ID Nos 1 to 27 and the cLysMAR element or
fragment, which can be produced by using PCR or other means.
An "element" is a conserved nucleotide sequences that bears common functional
properties (i.e. binding sites for transcription factors) or structural (i.e.
bent DNA
sequence) features.
A part of sequences SEQ ID Nos 1 to 27 and the cLysMAR element or fragment
refers
to sequences sharing at least 70% nucleotides in length with the respective
sequence
of the SEQ ID Nos 1 to 27. These sequences can be used as long as they exhibit
the
same properties as the native sequence from which they derive. Preferably
these
sequences share more than 80%, in particular more than 90% nucleotides in
length with
the respective sequence of the SEQ ID Nos 1 to 27.
The present invention also includes variants of the aforementioned sequences
SEQ ID
Nos 1 to 27 and the cLysMAR element or fragment, that is nucleotide sequences
that
vary from the reference sequence by conservative nucleotide substitutions,
whereby
one or more nucleotides are substituted by another with same characteristics.
The sequences SEQ ID Nos 1 to 23 have been identified by scanning human
chromosome 1 and 2 using SMAR Scan , showing that the identification of novel
MAR
sequences is feasible using the tools reported thereafter whereas SEQ ID No 24
to 27
have been identified by scanning the complete human genome using the combined
SMAR Scan method.
In a first step, the complete chromosome 1 and 2 were screened to identify
bent DNA
element as region corresponding to the highest bent, major groove depth, minor
groove
width and lowest melting temperature as shown in figure 3. In a second step,
this
collection of sequence was scanned for binding sites of regulatory proteins
such as
SATB1, GATA, etc. as shown in the figure 8B) yielding sequences SEQ ID 1-23.
CA 02884685 2016-04-27
Furthermore, sequences 21-23 were further shown to be located next to known
gene
. from the Human Genome Data Base.
With regard to SEQ ID No 24 to 27 these sequences have been yielded by
scanning
the human genorne according to the combined method and were selected as
examples among 1757 MAR elements so detected.
Molecular chimera of MAR sequences are also considered in the present
invention. By
molecular chimera is intended a nucleotide sequence that may include a
functional
portion of a MAR element and that will be obtained by molecular biology
methods
known by those skilled in the art.
Particular combinations of MAR elements or fragments or sub-portions thereof
are also
considered in the present invention. These fragments can be prepared by a
variety of
methods known in the art. These methods include, but are not limited to,
digestion with
restriction enzymes and recovery of the fragments, chemical synthesis or
polymerase
chain reactions (PCR).
Therefore, particular combinations of elements or fragments of the sequences
SEQ ID
Nos 1 to 27 and cLysMAR elements or fragments are also envisioned in the
present
Invention, depending on the functional results to be obtained. Elements of the
cLysMAR
are e.g. the B, K and F regions as described in WO 021074969. The preferred
elements of the cLysMAR used In the present invention are the B, K and F
regions.
Only one element might be used or multiple copies of the same or distinct
elements
26 (multimerized elements) might be used (see Fig. BA).
By fragment is intended a portion of the respective nucleotide sequence.
Fragments or
a MAR nucleotide sequence may retain biological activity and hence bind to
purified
nuclear matrices andfor alter the expression patterns of coding sequences
operably
linked to a promoter. Fragments of a MAR nucleotide sequence may range from at
least about 100 to 1000 bp, preferably from about 200 to 700 bp, more
preferably from
about 300 to 500 bp nucleotides. Also envisioned are any combinations of
fragments,
which have the same number of nucleotides present in a synthetic MAR sequence
11
CA 02884685 2015-03-10
consisting of natural MAR element and/or fragments. The fragments are
preferably
assembled by linker sequences. Preferred linkers are BgIII-BamHI linker.
"Protein production increasing activity" refers to an activity of the purified
and isolated
DNA sequence defined as follows: after having been introduced under suitable
conditions into a eukaryotic host cell, the sequence is capable of increasing
protein
production levels in cell culture as compared to a culture of cell transfected
without said
DNA sequence. Usually the increase is 1.5 to 10 fold, preferably 4 to 10 fold.
This
corresponds to a production rate or a specific cellular productivity of at
least 10 pg per
cell per day (see Example 11 and Fig.13).
As used herein, the following definitions are supplied in order to facilitate
the
understanding of this invention.
"Chromatin" is the protein and nucleic acid material constituting the
chromosomes of a
eukaryotic cell, and refers to DNA, RNA and associated proteins.
A "chromatin element" means a nucleic acid sequence on a chromosome having the
property to modify the chromatine structure when integrated into that
chromosome.
"Cis" refers to the placement of two or more elements (such as chromatin
elements) on
the same nucleic acid molecule (such as the same vector, plasmid or
chromosome).
"Trans" refers to the placement of two or more elements (such as chromatin
elements)
on two or more different nucleic acid molecules (such as on two vectors or two
chromosomes).
Chromatin modifying elements that are potentially capable of overcoming
position
effects, and hence are of interest for the development of stable cell lines,
include
boundary elements (BE5), matrix attachment regions (MARs), locus control
regions
(LCRs), and universal chromatin opening elements (UCOEs).
Boundary elements ("BEs"), or insulator elements, define boundaries in
chromatin in
many cases (Bell A and Felsenfeld G. 1999; "Stopped at the border: boundaries
and
insulators, Curr Opin Genet Dev 9, 191-198) and may play a role in defining a
12
CA 02884685 2015-03-10
transcriptional domain in vivo. BEs lack intrinsic promoter/enhancer activity,
but rather
are thought to protect genes from the transcriptional influence of regulatory
elements in
the surrounding chromatin. The enhancer-block assay is commonly used to
identify
insulator elements. In this assay, the chromatin element is placed between an
enhancer
and a promoter, and enhancer-activated transcription is measured. Boundary
elements
have been shown to be able to protect stably transfected reporter genes
against
position effects in Drosophila, yeast and in mammalian cells. They have also
been
shown to increase the proportion of transgenic mice with inducible transgene
expression.
Locus control regions ("LCRs") are cis-regulatory elements required for the
initial
chromatin activation of a locus and subsequent gene transcription in their
native
locations (Grosveld, F. 1999, "Activation by locus control regions?" Curr Opin
Genet
Dev 9, 152-157). The activating function of LCRs also allows the expression of
a
coupled transgene in the appropriate tissue in transgenic mice, irrespective
of the site
of integration in the host genome. While LCRs generally confer tissue-specific
levels of
expression on linked genes, efficient expression in nearly all tissues in
transgenic mice
has been reported for a truncated human T-cell receptor LCR and a rat LAP LCR.
The
most extensively characterized LCR is that of the globin locus. Its use in
vectors for the
gene therapy of sickle cell disease and (3-thalassemias is currently being
evaluated.
"MARs", according to a well-accepted model, may mediate the anchorage of
specific
DNA sequence to the nuclear matrix, generating chromatin loop domains that
extend
outwards from the heterochromatin cores. While MARs do not contain any obvious
consensus or recognizable sequence, their most consistent feature appears to
be an
overall high NT content, and C bases predominating on one strand (Bode J,
Schlake T,
RiosRamirez M, Mielke C, Stengart M, Kay V and KlehrWirth D, "Scaffold/matrix-
attached regions: structural propreties creating transcriptionally active
loci",Structural
and Functional Organization of the Nuclear Matrix: International Review of
Citology,
162A:389453, 1995). These regions have a propensity to form bent secondary
structures that may be prone to strand separation. They are often referred to
as base-
unpairing regions (BURs), and they contain a core-unwinding element (CUE) that
might
represent the nucleation point of strand separation (Benham C and al., Stress
induced
duplex DNA destabilization in scaffold/matrix attachment regions, J. Mol.
Biol., 274:181-
196, 1997). Several simple AT-rich sequence motifs have often been found
within MAR
13
CA 02884685 2015-03-10
sequences, but for the most part, their functional importance and potential
mode of
action remain unclear. These include the A-box (AATAAAYAAA), the T-box
(TTWTWTTWTT), DNA unwinding motifs (AATATATT, AATATT), SATB1 binding sites
(H-box, A/T/C25) and consensus Topoisomerase II sites for vertebrates
(RNYNNCNNGYNGKTNYNY) or Drosophila (GTNWAYATTNATNNR).
Ubiquitous chromatin opening elements ("UCOEs", also known as "ubiquitously-
acting
chromatin opening elements") have been reported in WO 00/05393.
An "enhancer" is a nucleotide sequence that acts to potentiate the
transcription of
genes independent of the identity of the gene, the position of the sequence in
relation
to the gene, or the orientation of the sequence. The vectors of the present
invention
optionally include enhancers.
A "gene" is a deoxyribonucleotide (DNA) sequence coding for a given mature
protein.
As used herein, the term "gene" shall not include untranslated flanking
regions such as
RNA transcription initiation signals, polyadenylation addition sites,
promoters or
enhancers.
A "product gene" is a gene that encodes a protein product having desirable
characteristics such as diagnostic or therapeutic utility. A product gene
includes, e. g.,
structural genes and regulatory genes.
A "structural gene" refers to a gene that encodes a structural protein.
Examples of
structural genes include but are not limited to, cytoskeletal proteins,
extracellular matrix
proteins, enzymes, nuclear pore proteins and nuclear scaffold proteins, ion
channels
and transporters, contractile proteins, and chaperones. Preferred structural
genes
encode for antibodies or antibody fragments.
A "regulatory gene" refers to a gene that encodes a regulatory protein.
Examples of
regulatory proteins include, but are not limited to, transcription factors,
hormones,
growth factors, cytokines, signal transduction molecules, oncogenes, proto-
oncogenes,
transmembrane receptors, and protein kinases.
"Orientation" refers to the order of nucleotides in a given DNA sequence. For
example,
14
CA 02884685 2015-03-10
an inverted orientation of a DNA sequence is one in which the 5' to 3' order
of the
sequence in relation to another sequence is reversed when compared to a point
of
reference in the DNA from which the sequence was obtained. Such reference
points
can include the direction of transcription of other specified DNA sequences in
the
source DNA and/or the origin of replication of replicable vectors containing
the
sequence.
"Eukaryotic cell" refers to any mammalian or non-mammalian cell from a
eukaryotic
organism. By way of non-limiting example, any eukaryotic cell that is capable
of being
maintained under cell culture conditions and subsequently transfected would be
included in this invention. Especially preferable cell types include, e. g.,
stem cells,
embryonic stem cells, Chinese hamster ovary cells (CHO), COS, BHK21, NIH3T3,
HeLa, C2C12, cancer cells, and primary differentiated or undifferentiated
cells. Other
suitable host cells are known to those skilled in the art.
The terms "host cell" and "recombinant host cell" are used interchangeably
herein to
indicate a eukaryotic cell into which one or more vectors of the invention
have been
introduced. It is understood that such terms refer not only to the particular
subject cell
but also to the progeny or potential progeny of such a cell. Because certain
modifications may occur in succeeding generations due to either mutation or
environmental influences, such progeny may not, in fact, be identical to the
parent cell,
but are still included within the scope of the term as used herein.
The terms "introducing a purified DNA into a eukaryotic host cell" or
"transfection"
denote any process wherein an extracellular DNA, with or without accompanying
material, enters a host cell. The term "cell transfected" or "transfected
cell" means the
cell into which the extracellular DNA has been introduced and thus harbours
the
extracellular DNA. The DNA might be introduced into the cell so that the
nucleic acid is
replicable either as a chromosomal integrant.or as an extra chromosomal
element.
"Promoter" as used herein refers to a nucleic acid sequence that regulates
expression
of a gene.
"Co-transfection" means the process of transfecting a eukaryotic cell with
more than
one exogenous gene, or vector, or plasmid, foreign to the cell, one of which
may confer
CA 02884685 2015-03-10
a selectable phenotype on the cell.
The purified and isolated DNA sequence having protein production increasing
activity
also comprises, besides one or more bent DNA element, at least one binding
site for a
DNA binding protein.
Usually the DNA binding protein is a transcription factor. Examples of
transcription
factors are the group comprising the polyQpolyP domain proteins.
Another example of a transcription factor is a transcription factor selected
from the
group comprising SATB1, NMP4, MEF2, S8, DLX1, FREAC7, BRN2, GATA 1/3, TATA,
Bright, MSX, AP1, C/EBP, CREBP1, FOX, Freac7, HFH1, HNF3alpha, Nkx25,
POU3F2, Pit1, TTF1, XFD1, AR, C/EBPgamma, Cdc5, FOXD3, HFH3, HNF3 beta,
MRF2, Oct1, POU6F1, SRF, V$MTATA_B, XFD2, Bach2, CDP CR3, Cdx2, FOXJ2,
HFL, HP1, Myc, PBX, Pax3, TEF, VBP, XFD3, Brn2, COMP1, Evil, FOXP3, GATA4,
HFN1, Lhx3, NKX3A, POU1F1 , Pax6, TFIIA or a combination of two or more of
these
transcription factors are preferred. Most preferred are SATB1, NMP4, MEF2 and
polyQpolyP domain proteins.
SATB1, NMP4 and MEF2, for example, are known to regulate the development
and/or
tissue-specific gene expression in mammals. These transcription factors have
the
capacity to alter DNA geometry, and reciprocally, binding to DNA as an
allosteric ligand
modifies their structure. Recently, SATB1 was found to form a cage-like
structure
circumscribing heterochromatin (Cai S, Han HJ , and Kohwi-Shigematsu T,
"Tissue-
specific nuclear architecture and gene expression regulated by SATB1" Nat
Genet,
2003. 34(1): p. 42-51).
Yet another object of the present invention is to provide a purified and
isolated
cLysMAR element and/or fragment, a sequence complementary thereof, a part
thereof
sharing at least 70% nucleotides in length, a molecular chimera thereof, a
combination
thereof and variants.
More preferably, the cLysMAR element and/or fragment are consisting of at
least one
nucleotide sequence selected from the B, K and F regions.
16
CA 02884685 2015-03-10
A further object of the present invention is to provide a synthetic MAR
sequence
comprising natural MAR element and/or fragments assembled between linker
sequences.
Preferably, the synthetic MAR sequence comprises a cLysMAR element and/or
fragment a sequence complementary thereof, a part thereof sharing at least 70%
nucleotides in length, a molecular chimera thereof, a combination thereof and
variants.
Also preferably, linker sequences are BgIII-BamHI linker.
An other aspect of the invention is to provide a method for identifying a MAR
sequence
using a Bioinformatic tool comprising the computing of values of one or more
DNA
sequence features corresponding to DNA bending, major groove depth and minor
groove width potentials and melting temperature. Preferably, the
identification of one or
more DNA sequence features further comprises a further DNA sequence feature
corresponding to binding sites for DNA binding proteins, which is also
computed with
this method.
Preferably, profiles or weight-matrices of said bioinformatic tool are based
on
dinucleotide recognition.
The bioinformatic tool used for the present method is preferably, SMAR Scan ,
which
contains algorithms developed by Gene Express
(htto://srs6.bionet.nsc.ru/srs6bin/cgi-
bin/woetz?-e+[FEATURES-Sitela'nn and based on Levitsky etal., 1999. These
algorithms recognise profiles, based on dinucleotides weight-matrices, to
compute the
theoretical values for conformational and physicochemical properties of DNA.
Preferably, SMAR Scan uses the four theoretical criteria also designated as
DNA
sequence features corresponding to DNA bending, major groove depth and minor
groove width potentials, melting temperature in all possible combination,
using scanning
windows of variable size (see Fig. 3). For each function used, a cut-off value
has to be
set. The program returns a hit every time the computed score of a given region
is above
the set cut-off value for all of the chosen criteria. Two data output modes
are available
to handle the hits, the first (called "profile-like") simply returns all hit
positions on the
query sequence and their corresponding values for the different criteria
chosen. The
second mode (called "contiguous hits ") returns only the positions of several
17
CA 02884685 2015-03-10
contiguous hits and their corresponding sequence. For this mode, the minimum
number
of contiguous hits is another cut-off value that can be set, again with a
tunable window
size. This second mode is the default mode of SMAR Scan . Indeed, from a
semantic
point of view, a hit is considered as a core-unwinding element (CUE), and a
cluster of
CUEs accompanied by clusters of binding sites for relevant proteins is
considered as a
MAR. Thus, SMAR Scan considers only several contiguous hits as a potential
MAR.
To tune the default cut-off values for the four theoretical structural
criteria,
experimentally validated MARs from SMARt DB (http://transfac.gbf.de/- SMARt
DB)
were used. All the human MAR sequences from the database were retrieved and
analyzed with SMAR Scan using the "profile-like" mode with the four criteria
and with
no set cut-off value. This allowed the setting of each function for every
position of the
sequences. The distribution for each criterion was then computed according to
these
data (see Fig. 1 and 3).
The default cut-off values of SMAR Scan for the bend, the major groove depth
and
the minor groove width were set at the average of the 75th quantile and the
median.
For the melting temperature, the default cut-off value should be set at the
75th quantile.
The minimum length for the "contiguous-hits" mode should be set to 300 because
it is
assumed to be the minimum length of a MAR (see Fig. 8 and 9). However, one
skilled
in the art would be able to determine the cut-off values for the above-
mentioned criteria
for a given organism with minimal experimentation.
Preferably, DNA bending values are comprised between 3 to 5 (radial degree).
Most
preferably they are situated between 3.8 to 4.4 corresponding to the smallest
peak of
Fig. 1.
Preferably the major groove depth values are comprised between 8.9 to 9.3 A
(Angstrom) and minor groove width values between 5.2 to 5.8 A. Most preferably
the
major groove depth values are comprised between 9.0 to 9.2 A and minor groove
width
values between 5.4 to 5.7 A.
Preferably the melting temperature is comprised between 55 to 75 C (Celsius
degree).
Most preferably, the melting temperature is comprisedbetween 55 to 62 C.
18
CA 02884685 2015-03-10
The DNA binding protein of which values can be computed by the method is
usually a
transcription factor preferably a polyQpolyP domain or a transcription factor
selected
from the group comprising SATB1, NMP4, MEF2, S8, DLX1, FREAC7, BRN2, GATA
1/3, TATA, Bright, MSX, AP1, C/EBP, CREBP1, FOX, Freac7, HFH1, HNF3alpha,
Nkx25, POU3F2, Pit1, TTF1, XFD1, AR, C/EBPgamma, Cdc5, FOXD3, HFH3, HNF3
beta, MRF2, Doti, POU6F1, SRF, V$MTATA_B, XFD2, Bach2, CDP CR3, Cdx2,
FOXJ2, HFL, HP1, Myc, PBX, Pax3, TEF, VBP, XFD3, Brn2, COMP1, Evil, FOXP3,
GATA4, HFN1, Lhx3, NKX3A, POU1F1 , Pax6, THIA or a combination of two or more
of
these transcription factors.
However, one skilled in the art would be able to determine other kinds of
transcription
factors in order to carry out the method according to the present invention.
In case SMAR Scan is envisaged to perform, for example, large scale analysis,
then,
preferably, the above-mentioned method further comprises at least one filter
predicting
DNA binding sites for DNA transcription factors in order to reduce the
computation.
The principle of this method combines SMAR Scan to compute the structural
features
as described above and a filter, such as for example, the pfsearch, (from the
pftools
package as described in Bucher P, Karplus K, Moeri N, and Hofmann K, "A
flexible
search technique based on generalized profiles", Computers and Chemistry,
20:324,
1996) to predict the binding of some transcription factors.
Examples of filters comprise, but are not limited to, pfsearch, Matinspector,
RMatch
Professional and TRANSFAC Professional
This combined method uses the structural features of SMAR Scan and the
predicted
binding of specific transcription factors of the filter that can be applied
sequentially in
any order to select MARs, therefore, depending on the filter is applied at the
beginning
or at the end of the method.
The first level selects sequences out of the primary input sequence and the
second
level, consisting in the filter, may be used to restrain among the selected
sequences
those which satisfy the criteria used by the filter.
19
CA 02884685 2015-03-10
In this combined method the filter detects clusters of DNA binding sites using
profiles or
weightmatrices from, for example, MatInspector (Quandt K, Frech K, Karas H,
Wingender E, Werner T, "MatInd and MatInspector New fast and versatile tools
for
detection of consensus matches in nucleotide sequence data", Nucleic Acids
Research
,23, 48784884, 1995.). The filter can also detect densities of clusters of DNA
binding
sites.
The combined method is actually a "wrapper" written in Pen i for SMAR Scan
and, in
case the pfsearch is used as a filter, from the pftools. The combined method
performs a
twolevel processing using at each level one of these tools (SMAR Scan or
filter) as a
potential "filter", each filter being optional and possible to be used to
compute the
predicted features without doing any filtering.
If SMAR Scan is used in the first level to filter subsequences, it has to be
used with
the "all the contiguous hits" mode in order to return sequences. If the
pfsearch is used
in the first level as first filter, it has to be used with only one profile
and a distance in
nucleotide needs to be provided. This distance is used to group together
pfsearch hits
that are located at a distance inferior to the distance provided in order to
return
sequences; The combined method launches pfsearch, parses its output and
returns
sequences corresponding to pfsearch hits that are grouped together according
to the
distance provided. Then whatever the tool used in the first level, the length
of the sub-
sequences thus selected can be systematically extended at both ends according
to a
parameter called "hits extension".
The second and optional level can be used to filter out sequences (already
filtered
sequences or unfiltered input sequences) or to get the results of SMAR Scan
and/or
pfsearch without doing any filtering on these sequences. If the second level
of
combined method is used to filter, for each criteria considered cutoff values
(hit per
nucleotide)need to be provided to filter out those sequences (see Fig. 20).
Another concern of the present invention is also to provide a method for
identifying a
MAR sequence comprising at least one filter detecting clusters of DNA binding
sites
using profiles or weightmatrices. Preferably, this method comprises two levels
of filters
CA 02884685 2015-03-10
and in this case, SMAR Scan is totally absent from said method. Usually, the
two
levels consist in pfsearch.
Also embraced by the present invention is a purified and isolated MAR DNA
sequence
identifiable according to the method for identifying a MAR sequence using the
described bioinformatic tool, the combined method or the method comprising at
least
one filter.
Analysis by the combined method of the whole human genome yielded a total of
1757
putative MARs representing a total of 1 065 305 base paires. In order to
reduce the
number of results, a dinucleotide analysis was performed on these 1757 MARs,
computing each of the 16 possible dinucleotide percentage for each sequence
considering both strands in the 5' to 3' direction.
Surprisingly, Applicants have shown that all of the "super" MARs detected with
the
combined method contain at least 10% of dinucleotide TA on a stretch of 100
contiguous base pairs. Preferably, these sequences contain at least 33% of
dinucleotide
TA on a stretch of 100 contiguous base pairs.
Applicants have also shown that these same sequences further contain at least
12% of
dinucleotide AT on a stretch of 100 contiguous base pairs. Preferably, they
contain at
least 33% of dinucleotide AT on a stretch of 100 contiguous base pairs.
An other aspect of the invention is to provide a purified and isolated MAR DNA
sequence of any of the preceding described MARs, comprising a sequence
selected
from the sequences SEQ ID Nos 1 to 27, a sequence complementary thereof, a
part
thereof sharing at least 70% nucleotides in length, a molecular chimera
thereof, a
combination thereof and variants.
Preferably, said purified and isolated MAR DNA sequence comprises a sequence
selected from the sequences SEQ ID Nos 24 to 27, a sequence complementary
thereof, a part thereof sharing at least 70% nucleotides in length, a
molecular chimera
thereof, a combination thereof and variants. These sequences 24 to 27
correspond to
those detected by the combined method and show a higher protein production
increasing activity over sequences 1 to 23.
21
CA 02884685 2015-03-10
The present invention also encompasses the use of a purified and isolated DNA
sequence comprising a first isolated matrix attachment region (MAR) nucleotide
sequence which is a MAR nucleotide sequence selected from the group comprising
- a purified and isolated DNA sequence having protein production increasing
activity,
- a purified and isolated MAR DNA sequence identifiable according to
the method
for identifying a MAR sequence using the described bioinformatic tool, the
combined method or the method comprising at least one filter,
- the sequences SEQ ID Nos 1 to 27,
- a purified and isolated cLysMAR element and/or fragment,
- a synthetic MAR sequence comprising natural MAR element and/or
fragments
assembled between linker sequences,
a sequence complementary thereof, a part thereof sharing at least 70%
nucleotides in
length, a molecular chimera thereof, a combination thereof and variants or a
MAR
nucleotide sequence of a cLysMAR element and/or fragment, a sequence
complementary thereof, a part thereof sharing at least 70% nucleotides in
length, a
molecular chimera thereof, a combination thereof and variants for increasing
protein
production activity in a eukaryotic host cell.
Said purified and isolated DNA sequence usually further comprises one or more
regulatory sequences, as known in the art e.g. a promoter and/or an enhancer,
polyadenylation sites and splice junctions usually employed for the expression
of the
protein or may optionally encode a selectable marker. Preferably said purified
and
isolated DNA sequence comprises a promoter which is operably linked to a gene
of
interest.
The DNA sequences of this invention can be isolated according to standard PCR
protocols and methods well known in the art.
Promoters which can be used provided that such promoters are compatible with
the
host cell are, for example, promoters obtained from the genomes of viruses
such as
polyoma virus, adenovirus (such as Adenovirus 2), papilloma virus (such as
bovine
papilloma virus), avian sarcoma virus, cytomegalovirus (such as murine or
human
cytomegalovirus immediate early promoter), a retrovirus, hepatitis-B virus,
and Simian
Virus 40 (such as SV 40 early and late promoters) or promoters obtained from
22
CA 02884685 2015-03-10
heterologous mammalian promoters, such as the actin promoter or an
immunoglobulin
promoter or heat shock promoters. Such regulatory sequences direct
constitutive
expression.
Furthermore, the purified and isolated DNA sequence might further comprise
regulatory
sequences which are capable of directing expression of the nucleic acid
preferentially in
a particular cell type (e. g., tissue-specific regulatory elements are used to
express the
nucleic acid). Tissue-specific regulatory elements are known in the art. Non-
limiting
examples of suitable tissue-specific promoters include the albumin promoter
(liver-
specific; Pinkert,et al., 1987. Genes Dev.1: 268-277), lymphoid-specific
promoters
(Calame and Eaton, 1988. Adv. Immunol. 43: 235-275), in particular promoters
of T cell
receptors (Winoto and Baltimore, 1989. EMBOJ. 8: 729-733) and immunoglobulins
(Banerji, etal., 1983. Cell 33: 729-740; Queen and Baltimore, 1983. Cell
33:741.-748),
neuron-specific promoters (e. g., the neurofilament promoter;Byrne and Ruddle,
1989.
Proc.Natl. Acad. Sci. USA 86: 5473-5477), pancreas-specific promoters (Edlund,
et al.,
1985. Science 230: 912-916), and mammary gland-specific promoters (e. g., milk
whey
promoter; U. S. Pat. No. 4,873,316 and European Application No. 264,166).
Developmentally-regulated promoters are also encompassed. Examples of such
promoters include, e.g., the murine hox promoters (Kessel and Gruss, 1990.
Science
249: 374-379) and thea-fetoprotein promoter (Campes and Tilghman, 1989. Genes
Dev. 3: 537-546).
Regulatable gene expression promoters are well known in the art, and include,
by way
of non-limiting example, any promoter that modulates expression of a gene
encoding a
desired protein by binding an exogenous molecule, such as the CRE/LOX system,
the
TET system, the doxycycline system, the NFkappaB/UV light system, the
Leu3p/isopropylmalate system, and theGLVPc/GAL4 system (See e. g., Sauer,
1998, =
Methods 14 (4): 381-92 ; Lewandoski, 2001, Nat. Rev. Genet 2 (10): 743-55;
Legrand-
PoeIs et al., 1998, J. Photochem. Photobiol. B. 45: 18; Guo et al., 1996, FEBS
Lett. 390
(2): 191-5; Wang et al., PNAS USA, 1999,96 (15): 84838).
However, one skilled in the art would be able to determine other kinds of
promoters that
are suitable in carrying out the present invention.
23
CA 02884685 2015-03-10
Enhancers can be optionally included in the purified DNA sequence of the
invention
then belonging to the regulatory sequence, e.g. the promoter.
The "gene of interest" or "transgene" preferably encodes a protein (structural
or
regulatory protein). As used herein "protein" refers generally to peptides and
polypeptides having more than about ten amino acids. The proteins may be
"homologous" to the host (i.e., endogenous to the host cell being utilized),
or
"heterologous," (i.e., foreign to the host cell being utilized), such as a
human protein
produced by yeast. The protein may be produced as an insoluble aggregate or as
a
soluble protein in the periplasmic space or cytoplasm of the cell, or in the
extracellular
medium. Examples of proteins include hormones such as growth hormone or
erythropoietin (EPO), growth factors such as epidermal growth factor,
analgesic
substances like enkephalin, enzymes like chymotrypsin, receptors to hormones
or
growth factors, antibodies and include as well proteins usually used as a
visualizing
marker e.g. green fluorescent protein.
Preferably the purified DNA sequence further comprises at least a second
isolated
matrix attachment region (MAR) nucleotide sequence selected from the group
comprising
- a purified and isolated DNA sequence having protein production increasing
activity,
- a purified and isolated MAR DNA sequence identifiable
according to the method
for identifying a MAR sequence using the described bioinformatic tool, the
combined method or the method comprising at least one filter,
- the sequences SEQ ID Nos 1 to 27,
- a purified and isolated cLysMAR element and/or fragment,
- a synthetic MAR sequence comprising natural MAR element
and/or fragments
assembled between linker sequences,
a sequence complementary thereof, a part thereof sharing at least 70%
nucleotides in
length, a molecular chimera thereof, a combination thereof and variants. The
isolated
matrix attachment region (MAR) nucleotide sequence might be identical or
different.
Alternatively, a first and a second identical MAR nucleotide sequence are
used.
Preferably, the MAR nucleotide sequences are located at both the 5' and the 3'
ends of
the sequence containing the promoter and the gene of interest. But the
invention also
24
CA 02884685 2015-03-10
envisions the fact that said first and or at least second MAR nucleotide
sequences are
located on a sequence distinct from the one containing the promoter and the
gene of
interest.
Embraced by the scope of the present invention is also the purified and
isolated DNA
sequence comprising a first isolated matrix attachment region (MAR) nucleotide
sequence which is a MAR nucleotide sequence selected from the group comprising
- a purified and isolated DNA sequence having protein production
increasing
activity,
- a purified and isolated MAR DNA sequence identifiable according to the
method
for identifying a MAR sequence using the described bioinformatic tool, the
combined method or the method comprising at least one filter,
- the sequences SEQ ID Nos 1 to 27,
- a purified and isolated cLysMAR element and/or fragment,
- a synthetic MAR sequence comprising natural MAR element and/or fragments
assembled between linker sequences,
a sequence complementary thereof, a part thereof sharing at least 70%
nucleotides in
length, a molecular chimera thereof, a combination thereof and variants that
can be
used for increasing protein production activity in a eukaryotic host cell by
introducing
the purified and isolated DNA sequence into a eukaryotic host cell according
to well
known protocols. Usually applied methods for introducing DNA into eukaryotic
host cells
applied are e.g. direct introduction of cloned DNA by microinjection or
microparticle
bombardment; electrotransfer ;use of viral vectors; encapsulation within a
carrier
system; and use of transfecting reagents such as calcium phosphate,
diethylaminoethyl
(DEAE) ¨clextran or commercial transfection systems like the Lipofect-AMINE
2000
(Invitrogen). Preferably, the transfection method used to introduce the
purified DNA
sequence into a eukaryotic host cell is the method for transfecting a
eukaryotic cell as
described below.
The purified and isolated DNA sequence can be used in the form of a circular
vector.
Preferably, the purified and isolated DNA sequence is used in the form of a
linear DNA
sequence as vector.
As used herein, "plasmid" and "vector" are used interchangeably, as the
plasmid is the
most commonly used vector form. However, the invention is intended to include
such
CA 02884685 2015-03-10
other forms of expression vectors, including, but not limited to, viral
vectors (e. g.,
replication defective retroviruses, adenoviruses and adeno-associated
viruses), which
serve equivalent functions.
The present invention further encompasses a method for transfecting a
eukaryotic host
cell, said method comprising
a) introducing into said eukaryotic host cell at least one purified DNA
sequence
comprising at least one DNA sequence of interest and/or at least one purified
and isolated DNA sequence comprising a MAR nucleotide sequence or other
chromatin modifying elements,
b) subjecting within a defined time said transfected eukaryotic host cell to
at least
one additional transfection step with at least one purified DNA sequence
comprising at least one DNA sequence of interest and/or with at least one
purified and isolated DNA sequence comprising a MAR nucleotide sequence or
other chromatin modifying elements
c) selecting said transfected eukaryotic host cell.
Preferably at least two up to four transfecting steps are applied in step b).
In order to select the successful transfected cells, a gene that encodes a
selectable
marker (e. g., resistance to antibiotics) is generally introduced into the
host cells along
with the gene of interest. The gene that encodes a selectable marker might be
located
on the purified DNA sequence comprising at least one DNA sequence of interest
and/or
at least one purified and isolated DNA sequence consisting of a MAR nucleotide
sequence or other chromatin modifying elements or might optionally be co-
introduced in
separate form e.g. on a plasmid. Various selectable markers include those that
confer
resistance to drugs, such as G418, hygromycin and methotrexate. The amount of
the
drug can be adapted as desired in order to increase productivity
Usually, one or more selectable markers are used. Preferably, the selectable
markers
used in each distinct transfection steps are different. This allows selecting
the
transformed cells that are "multi-transformed" by using for example two
different
antibiotic selections.
26
CA 02884685 2015-03-10
Any eukaryotic host cell capable of protein production and lacking a cell wall
can be
used in the methods of the invention. Examples of useful mammalian host cell
lines
include human cells such as human embryonic kidney line (293 or 293 cells
subcloned
for growth in suspension culture, Graham et al., J. Gen Viral 36, 59 (1977)),
human
cervical carcinoma cells (HELA, ATCC CCL 2), human lung cells (W138, ATCC CCL
75), human liver cells (Hep G2, HB 8065); rodent cells such as baby hamster
kidney
cells (BHK, ATCC CCL 10), Chinese hamster ovary cells/-DHFR (CHO, Urlaub and
Chasin, Proc. Natl. Acad. Sci USA, 77, 4216 (1980)), mouse sertoli cells (TM4,
Mather,
Reprod 23, 243-251 (1980)), mouse mammary tumor (MMT 060562, ATCC
CCL51); and cells from other mammals such as monkey kidney CV1 line
transformed
by SV40 (COS-7, ATCC CRL 1651); monkey kidney cells (CV1 ATCC CCL 70); African
green monkey kidney cells (VERO-76, ATCC CRL-1587); canine kidney cells (MDCK,
ATCC CCL 34); buffalo rat liver cells (BRL 3A, ATCC CRL 1442); myeloma (e.g.
NSO)
/hybridoma cells.
Preferably, the selected transfected eukaryotic host cells are high protein
producer cells
with a production rate of at least 10 pg per cell per day.
Most preferred for uses herein are mammalian cells, more preferred are CHO
cells.
The DNA sequence of interest of the purified and isolated DNA sequence is
usually a
gene of interest preferably encoding a protein operably linked to a promoter
as
described above. The purified and isolated DNA sequence comprising at least
one DNA
sequence of interest might comprise additionally to the DNA sequence of
interest MAR
nucleotide sequence or other chromatin modifying elements.
Purified and isolated DNA sequence comprising a MAR nucleotide sequence are
for
example selected from the group comprising the sequences SEQ ID Nos 1 to 27
and/or
particular elements of the cLysMAR e.g. the B, K and F regions as well as
fragment and
elements and combinations thereof as described above. Other chromatin
modifying
elements are for example boundary elements (BEs), locus control regions
(LCRs), and
universal chromatin opening elements (UCOEs) (see Zahn-Zabal et al. already
cited).
An example of multiple transfections of host cells is shown in Example 12
(Table 3).
The first transfecting step (primary transfection) is carried out with the
gene of interest
(SV40EGFP) alone, with a MAR nucleotide sequence (MAR) alone or with the gene
of
interest and a MAR nucleotide sequence (MAR-SV40EGFP). The second transfecting
step (secondary transfection) is carried out with the gene of interest
(SV40EGFP)
27
CA 02884685 2015-03-10
alone, with a MAR nucleotide sequence (MAR) alone or with the gene of interest
and a
MAR nucleotide sequence (MAR-SV40EGFP), in all possible combinations resulting
from the first transfecting step.
Preferably the eukaryotic host cell is transfected by:
a) introducing a purified DNA sequence comprising one DNA sequence of interest
and
additionally a MAR nucleotide sequence,
b) subjecting within a defined time said transfected eukaryotic host cell to
at least one
additional transfection step with the same purified DNA sequence comprising
one DNA
sequence of interest and additionally a MAR nucleotide sequence of step a).
Also preferably, the MAR nucleotide sequence of the of the purified and
isolated DNA
sequence is selected form the group comprising
- a purified and isolated DNA sequence having protein production
increasing
activity,
- a purified and isolated MAR DNA sequence identifiable according to
the method
for identifying a MAR sequence using the described bioinformatic tool, the
combined method or the method comprising at least one filter,
- the sequences SEQ ID Nos 1 to 27,
- a purified and isolated cLysMAR element and/or fragment,
- a synthetic MAR sequence comprising natural MAR element and/or
fragments
assembled between linker sequences,
a sequence complementary thereof, a part thereof sharing at least 70%
nucleotides in
length, a molecular chimera thereof, a combination thereof and variants.
Surprisingly, a synergy between the first and second transfection has been
observed. A
particular synergy has been observed when MAR elements are present at one or
both of
the transfection steps. Multiple transfections of the cells with pMAR alone or
in
combination with various expression plasmids, using the method described above
have
- 30 been carried out. For example, Table 3 shows that transfecting the
cells twice with the
pMAR-SV40EGFP plasmid gave the highest expression of GFP and the highest
degree
of enhancement of all conditions (4.3 fold). In contrast, transfecting twice
the vector
without MAR gave little or no enhancement, 2.8-fold, instead of the expected
two-fold
increase. This proves that the presence of MAR elements at each transfection
step is of
particular interest to achieve the maximal protein synthesis.
28
CA 02884685 2015-03-10
As a particular example of the transfection method, said purified DNA sequence
comprising at least one DNA sequence of interest can be introduced in form of
multiple
unlinked plasmids, comprising a gene of interest operably linked to a
promoter, a
selectable marker gene, and/or protein production increasing elements such as
MAR
sequences.
The ratio of the first and subsequent DNA sequences may be adapted as required
for
the use of specific cell types, and is routine experimentation to one ordinary
skilled in
the art.
The defined time for additional transformations of the primary transformed
cells is
tightly dependent on the cell cycle and on its duration. Usually the defined
time
corresponds to intervals related to the cell division cycle.
Therefore this precise timing may be adapted as required for the use of
specific cell
types, and is routine experimentation to one ordinary skilled in the art.
Preferably the defined time is the moment the host cell just has entered into
the same
phase of a second or a further cell division cycle, preferably the second
cycle.
This time is usually situated between 6h and 48 h, preferably between 20h and
24h
after the previous transfecting event.
Also encompassed by the present invention is a method for transfecting a
eukaryotic
host cell, said method comprising co-transfecting into said eukaryotic host
cell at least
one first purified and isolated DNA sequence comprising at least one DNA
sequence of
interest, and a second purified DNA comprising at least one MAR nucleotide
selected
from the group comprising:
- a purified and isolated DNA sequence having protein production
increasing
activity,
- a purified and isolated MAR DNA sequence identifiable according to
the method
for identifying a MAR sequence using the described bioinformatic tool, the
combined method or the method comprising at least one filter,
- the sequences SEQ ID Nos 1 to 27,
- a purified and isolated cLysMAR element and/or fragment,
- a synthetic MAR sequence comprising natural MAR element and/or
fragments
assembled between linker sequences,
29
CA 02884685 2015-03-10
a sequence complementary thereof, a part thereof sharing at least 70%
nucleotides in
length, a molecular chimera thereof, a combination thereof and variants.
Said first purified and isolated DNA sequence can also comprise at least one
MAR
nucleotide as described above.
Also envisioned is a process for the production of a protein wherein a
eukaryotic host
cell is transfected according to the transfection methods as defined in the
present
invention and is cultured in a culture medium under conditions suitable for
expression of
the protein. Said protein is finally recovered according to any recovering
process known
to the skilled in the art.
Given as an example, the following process for protein production might be
used.
The eukaryotic host cell transfected with the transfection method of the
present
invention is used in a process for the production of a protein by culturing
said cell under
conditions suitable for expression of said protein and recovering said
protein. Suitable
culture conditions are those conventionally used for in vitro cultivation of
eukaryotic cells
as described e.g. in WO 96/39488. The protein can be isolated from the cell
culture by
conventional separation techniques such as e.g. fractionation on
immunoaffinity or ion-
exchange columns; precipitation; reverse phase HPLC; chromatography;
chromatofocusing; SDS-PAGE; gel filtration. One skilled in the art will
appreciate that
purification methods suitable for the polypeptide of interest may require
modification to
account for changes in the character of the polypeptide upon expression in
recombinant
cell culture.
The proteins that are produced according to this invention can be tested for
functionality by a variety of methods. For example, the presence of antigenic
epitopes
and ability of the proteins to bind ligands can be determined by Western blot
assays,
fluorescence cell sorting assays, immunoprecipitation, immunochemical assays
and/or
competitive binding assays, as well as any other assay which measures specific
binding
activity.
The proteins of this invention can be used in a number of practical
applications
including, but not limited to:
1. Immunization with recombinant host protein antigen as a viral/pathogen
antagonist.
2. Production of membrane proteins for diagnostic or screening assays.
3. Production of membrane proteins for biochemical studies.
CA 02884685 2015-03-10
=
4. Production of membrane protein for structural studies.
5. Antigen production for generation of antibodies for immuno-histochemical
mapping,
including mapping of orphan receptors.and ion channels.
Also provided by the present invention is a eukaryotic host cell transfected
according to
any of the preceding transfection methods. Preferably, the eukaryotic host
cell is a
mammalian host cell line.
As already described, example of useful mammalian host cell lines include
human cells
such as human embryonic kidney line (293 or 293 cells subcloned for growth in
suspension culture, Graham et al., J. Gen Virol 36, 59 (1977)), human cervical
carcinoma cells (HELA, ATCC CCL 2), human lung cells (W138, ATCC CCL 75),
human liver cells (Hep G2, HB 8065); rodent cells such as baby hamster kidney
cells
(BHK, ATCC CCL 10), Chinese hamster ovary cells/-DHFR (CHO, Urlaub and Chasin,
Proc. Natl. Acad. Sci. USA, 77, 4216 (1980)), mouse sertoli cells (TM4,
Mather, Biol.
Reprod 23, 243-251 (1980)), mouse mammary tumor (MMT 060562, ATCC CCL51);
and cells from other mammals such as monkey kidney CV1 line transformed by
SV40
(COS-7, ATCC CRL 1651); monkey kidney cells (CV1 ATCC CCL 70); African green
monkey kidney cells (VERO-76, ATCC CRL-1587); canine kidney cells (MDCK, ATCC
CCL 34); buffalo rat liver cells (BRL 3A, ATCC CRL 1442); myeloma (e.g. NSO)
/hybridoma cells.
Most preferred for uses herein are CHO cells.
The present invention also provides for a cell transfection mixture or Kit
comprising at
least one purified and isolated DNA sequence according to the invention.
The invention further comprises a transgenic organism wherein at least some of
its cells
have stably incorporated at least one DNA sequence of
- a purified and isolated DNA sequence having protein production
increasing
activity,
- a purified and isolated MAR DNA sequence identifiable according to the
method
for identifying a MAR sequence using the described bioinformatic tool, the
combined method or the method comprising at least one filter,
- the sequences SEQ ID Nos 1 to 27,
- a purified and isolated cLysMAR element and/or fragment,
31
CA 02884685 2015-03-10
- a synthetic MAR sequence comprising natural MAR element and/or
fragments
assembled between linker sequences,
a sequence complementary thereof, a part thereof sharing at least 70%
nucleotides in
length, a molecular chimera thereof, a combination thereof and variants.
Preferably, some of the cells of the transgenic organisms have been
transfected
according the methods described herein.
Also envisioned in the present invention is a transgenic organism wherein its
genome
has stably incorporated at least one DNA sequence of
- a purified and isolated DNA sequence having protein production increasing
activity,
- a purified and isolated MAR DNA sequence identifiable according to
the method
for identifying a MAR sequence using the described bioinformatic tool, the
combined method or the method comprising at least one filter,
- the sequences SEQ ID Nos 1 to 27,
- a purified and isolated cLysMAR element and/or fragment,
- a synthetic MAR sequence comprising natural MAR element and/or
fragments
assembled between linker sequences,
a sequence complementary thereof, a part thereof sharing at least 70%
nucleotides in
length, a molecular chimera thereof, a combination thereof and variants.
Transgenic eukaryotic organisms which can be useful for the present invention
are for
example selected form the group comprising mammals (mouse, human, monkey etc)
and in particular laboratory animals such as rodents in general, insects
(drosophila,
etc), fishes (zebra fish, etc.), amphibians (frogs, newt, etc..) and other
simpler
organisms such as c. elegans, yeast, etc....
Yet another object of the present invention is to provide a computer readable
medium
comprising computer-executable instructions for performing the method for
identifying a
MAR sequence as described in the present invention.
The foregoing description will be more fully understood with reference to the
following
Examples. Such Examples, are, however, exemplary of methods of practising the
present invention and are not intended to limit the scope of the invention.
32
CA 02884685 2015-03-10
EXAMPLES
Example 1: SMAR Scan and MAR sequences
A first rough evaluation of SMAR Scan was done by analyzing experimentally
defined
human MARs and non-MAR sequences. As MAR sequences, the previous results from
the analysis of human MARS from SMARt Db were used to plot a density histogram
for
each criterion as shown in Fig. 1. Similarly, non-MAR sequences were also
analyzed
and plotted. As non-MAR sequences, all Ref-Seq-contigs from the chromosome 22
were used, considering that this latter was big enough to contain a negligible
part of
MAR sequences regarding the part of non-MAR sequences.
The density distributions shown in Fig. 1 are all skewed with a long tail. For
the highest
bend, the highest major groove depth and the highest minor groove width, the
distributions are right skewed. For the lowest melting temperature, the
distributions are 1
left-skewed which is natural given the inverse correspondence of this
criterion regarding
the three others. For the MAR sequences, biphasic distributions with a second
weak
peak, are actually apparent. And between MAR and non-MAR sequences
distributions,
a clear shift is also visible in each plot.
Among all human MAR sequences used, in average only about 70% of them have a
value greater than the 75th quantile of human MARs distribution, this for the
four
different criteria. Similarly concerning the second weak peak of each human
MARs
distribution, only 15% of the human MAR sequences are responsible of these
outlying
values. Among these 15% of human MAR sequences, most are very well documented
MARs, used to insulate transgene from position effects, such as the interferon
locus
MAR, the beta-globin locus MAR (Ramezani A, Hawley TS, Hawley RG, "Performance-
and safety-enhanced lentiviral vectors containing the human interferon-beta
scaffold
attachment region and the chicken beta-globin insulator", Blood, 101:4717-
4724, 2003), or the
apolipoprotein MAR (Namciu, S, Blochinger KB, Fournier REK, "Human matrix
attachment regions in-sulate transgene expression from chromosomal position
effects
in Drosophila melanogaster", Mot Cell. Biol., 18:2382-2391, 1998).
Always with the same data, human MAR sequences were also used to determine the
association between the four theoretical structural properties computed and
the AT-
33
CA 02884685 2015-03-10
content. Fig. 2 represents the scatterplot and the corresponding correlation
coefficient r
for every pair of criteria.
Example 2: Distribution plots of MAR sequences by organism
MAR sequences from SMARt DB of other organisms were also retrieved and
analyzed
similarly as explained previously. The MAR sequences density distributions for
the
mouse, the chicken, the sorghum bicolor and the human are plotted jointly in
Fig. 3.
= 10 Example 3: MAR prediction of the whole chromosome 22
All RefSeq contigs from the chromosome 22 were analyzed by SMAR Scan using
the
default settings this time. The result is that SMAR Scan predicted a total of
803
MARs, their average length being 446 bp, which means an average of one MAR
predicted per 42 777 bp. The total length of the predicted MARs corresponds to
1% of
the chromosome 22 length. The AT-content of the predicted regions ranged from
65,1% to 93.3%; the average AT-content of all these regions being 73.5%. Thus,
predicted MARs were AT-rich, whereas chromosome 22 is not AT-rich (52.1% AT).
SMARTest was also used to analyze the whole chromosome 22 and obtained 1387
MAR candidates, their average length being 494 bp representing an average of
one
MAR predicted per 24 765 bp. The total length of the predicted MARs
corresponds to
2% of the chromosome 22. Between all MARs predicted by the two softwares, 154
predicted MARs are found by both programs, which represents respectively 19%
and
11% of SMAR Scan and SMARTest predicted MARs. Given predicted MARs mean
length for SMAR Scan and SMARTest, the probability to have by chance an
overlapping between SMAR Scan and SMARTest predictions is 0.0027% per
prediction.
To evaluate the specificity of SMAR Scan predictions, SMAR Scan analyses
were
performed on randomly shuffled sequences of the chromosome 22 (Fig. 4).
Shuffled
sequences were generated using 4 different methods: by a segmentation of the
chromosome 22 into nonoverlapping windows of 10 bp and by separately shuffling
the
nucleotides in each window; by "scrambling" which means a permutation of all
nucleotides of the chromosome; by "rubbling" which means a segmentation of the
chro-
mosome in fragments of 10 bp and a random assembling of these fragments and
finally
by order 1 Markov chains, the different states being the all the different DNA
34
CA 02884685 2015-03-10
dinucleotides and the transition probabilities between these states being
based on the
chromosome 22 scan. For each shuffling method, five shuffled chromosome 22
were
generated and analyzed by SMAR Scan using the default settings. Concerning
the
number hits, an average of 3 519 170 hits (sd: 18 353) was found for the
permutated
chromosome 22 within nonoverlapping windows of 10 bp, 171 936,4 hits (sd: 2
859,04)
for the scrambled sequences and 24708,2 hits (sd: 1191,59) for the rubbled
chromosome 22 and 2 282 hits in average (sd: 334,7) for the chromosomes
generated
according to order 1 Markov chains models of the chromosome 22, which
respectively
represents 185% (sd: 0.5% of the mean), 9% (sd: 1.5%), 1% (sd: 5%) and 0.1%
(sd:
15%) of the number of hits found with the native chromosome 22. For the number
of
MARs predicted, which thus means contiguous hits of length greater than 300, 1
997
MARs were predicted with the shuffled chromosome 22 within windows of 10 bp
(sd:
31.2), only 2.4 MARs candidates were found in scrambled sequences (sd: 0.96)
and
none for the rubbled and for the sequences generated according to Markov
chains
model, which respectively represents 249% and less than 0.3% of the number of
predicted MARs found with the native chromosome 22. These data provide
indications
that SMAR Scan detects specific DNA elements which organization is lost when
the
DNA sequences are shuffled.
Example 4: Analysis of known matrix attachment regions in the Interferon locus
with SMAR Scan
The relevance of MAR prediction by SMAR Scan was investigated by analyzing
the
recently published MAR regions of the human interferon gene cluster on the
short arm
of chromosome 9 (9p22). Goetze et al. (already cited) reported an exhaustive
analysis
of the WP18A10A7 locus to analyze the suspected correlation between BURs
(termed
in this case stress-induced duplex destabilization or SIDD) and in vitro
binding to the
nuclear matrix (Fig. 9, lower part). Three of the SIDD peaks were in agreement
with the
in vitro binding assay, while others did not match matrix attachment sites.
Inspection of
the interferon locus with SMAR Scan (Fig. 9, top part) indicated that three
majors
peaks accompanied by clusters of SATB1, NMP4 and MEF2 regulators binding sites
correlated well with the active MARs. Therefore, we conclude that the
occurrence of
predicted CUEs and binding sites for these transcription factors is not
restricted to the
cLysMAR but may be a general property of all MARs. These results also imply
that the
SMAR Scan program efficiently detects MAR elements from genomic sequences.
CA 02884685 2015-03-10
Example 5: Accuracy of SMAR Scan prediction and comparison with other
predictive tools
The accuracy of SMAR Scan was evaluated using six genomic sequences for which
experimentally determined MARs have been mapped. In order to perform a
comparison
with other predictive tools, the sequences analyzed are the same with the
sequences
previously used to compare MAR-Finder and SMARTest. These genomic sequences
are three plant and three human sequences (Table 1) totalizing 310 151 bp and
37
experimentally defined MARs. The results for SMARTest and MAR-Finder in Table
1
come from a previous comparison (Frisch M, Frech K, Klingenhoff A, Cartharius
K,
Liebich I and Werner T, In silico pre-diction of scaffold/matrix attachment
regions in
large genomic sequences, Genome Research, 12:349-354, 2001.).
MAR-Finder has been used with the default parameters excepted for the
threshold
that has been set to 0.4 and for the analysis of the protamine locus, the AT-
richness
rule has been excluded (to detect the non AT-rich MARs as was done for the
protamine
locus).
36
CA 02884685 2015-03-10
- Sequence, description Length Experiment- SMARle- -- MAR-Finder
SMAR Scan
and reference ally defined prediction
prediction prediction
MARs positions positions positions
positions
I (kb) (kb) (kb) (kb) (kb)
Oryza Sativa putative 30.034 0.0-1.2 _ _ -
ADP-glucose pyro- 5.4-7.4 6.5-7.0 -
_
phosphorylase subunit 15.2-15.7 15.7-15.9
15.6-16
SH2 and putative 16.2-16.6 -
NADPH dependant 17.3-16.5 17.6-18.3 17.5-
18.4 17.6-18.2
reductase A1 genes 20.0-23.1 19.6-20.1 19.8-
20.4 21.6-22
(U70541),[4] 20.7-21.3 21.3-21.5
23.6-23.9 23.9-24.2 23.4-
23.8
25.0-25.4 24.7-25.1 -
27.5-27,9 - -
Sorghum bicolor ADP- 42.446 0.0-1.5 - - -
glucose pyrophopho= 7.1-9.7 = - 7.4-7.7
rylase subunit SH2, 21.3-21.9 - 21.5-
21.6
NADPH-dependant 22.4-24.7 22.9-24.0 23.2-
24.2 22.9-23.2
reducatse Al-b genes - 23.6-
24.0
(AF010283), [41 27.3-27.6 26.9-27.5
27.3-27.6
32.5-33.7 - 33.4-33.9
41.6-42.3 - -
. Sorghum bicolor BAG 78.195 -.Ø9 - - _
= clone 110K5 ,5.8 ..
- _
(AF 124045) , [37] --. 6.3 - - -
-93 - - -
,15.0 15.1-15.6 - -
-.18.5 - - -
-..21.9 21.7-22.0 - 21.4-21.9
-23.3 - - _
-.25.6 - - ..
-.29.1 - - 29.2-29.5
34.6 - - -
- - 39.0-
40.0 .
,44.1 44.1-44.5 -
-:48.5 47.9-49.5 47.9-49.4 48.1-48.6
- - 48.8-
49.3
...57.0 - -
-62.9 63.1-63.7 - -
- - -
.:69.3 - - -
.73.7 74.3-74.7 - 74.3-74,6
Human alpha-l-antitry- 30.461 2.6-6.3 5.5-6.0 3.0-3.2
5.4-5.8
sin and corticosteroicl . 5.1-6.0
binding globulin 22.0-30.4 25.7-26.2 24.9-
25.3 25.8-26.4
intergenic region 27.5-27.8 25.5-25.8
(AF156545), [35] 26.2-26.4 .
- 27.5-28.2 -
Human protamine locus 53.060 8.8-9.7 - 8.0-8.9' -
(U15422). [24] 32.6-33.6 _ 33.9-
34.8 _
37.2-39.4 - 33.9-34.8' -
51.8-53.0 - _
Human beta-globin 75.955 1.5-3.0 - - 2.3-2.6
locus 15.6-19.0 18.0-18.4 15.5-
16.0 15.3-15.6
(U01317). [211 - 18.0-18.4 .
34.4-34.0 - .
44.7-52.7 - 50.6-50.8 -
56.6-57.1 56.5-57.2 _
60.0-70.0 59.8-60.3 58.1-58.5 62.8-63.1
65.6-66.0 63.0-63.6
37
CA 02884685 2015-03-10
' 67.6-67.9 68.7-69.3 66.3-66.7
68.8-69.1
Sum(kb) 310 151 at least 561 145 13.8 9.5
Total numbers : 37 28 25 22
Average kb predicted 11.076 12.406 14.097
MAR
True positives [number 191141 20[12] 17[14]
of experimentally
defined MAR found]
False positives 9 5 5
False negatives 23 25 23
Specificity 19128= 68% 20125= 80% 17122= 77%
Sensitivity 14/37= 38% 12/37= 32% 14137= 38%
Table 1: Evaluation of SMAR Scan accuracy
Six different genomic sequences, three plant and three human sequences, for
which
experimentally defined MARs are known, were analyzed with MAR-Finder, SMARTest
and SMAR Scan . True positive matches are printed in bold, minus (-) indicates
false
negative matches. Some of the longer experimentally defined MARs contained
more
than one in silico prediction, each of them was counted as true positive
match.
Therefore, the number of true in silico predictions is higher than the number
of
experimentally defined MARs found. Specificity is defined as the ratio of true
positive
predictions, whereas sensitivity is defined as the ratio of experimentally
defined MARs
found. *AT-rich rule excluded using MAR-Finder.
SMARTest predicted 28 regions as MARs, 19 (true positives) of these correlate
with
experimentally defined MARs (specificity: 68%) whereas 9 (32%) are located in
non-
MARs (false positives). As some of the longest experimentally determined
MARs contains more than one in silico prediction, the 19 true positives
correspond
actually to 14 different experimentally defined MARs (sensitivity: 38%).
MARFinder
predicted 25 regions as MARs, 20 (specificity: 80%) of these correlate with
experimentally defined MARs corresponding to 12 different experimentally
defined
MARs (sensitivity: 32%). SMAR Scan predicted 22 regions, 17 being true
positives
(specificity: 77%) matching 14 different experimentally defined MARs
(sensitivity: 38%).
As another example, the same analysis has been applied to human chromosomes 1
and 2 and lead to the determination of 23 MARs sequences (SEQ ID N 1 to 23).
These
sequences are listed in Annex 1 in ST25 format.
38
CA 02884685 2015-03-10
Example 6: Analyses of the whole denome using the combined method (SMAR
Scan -pfsearchl
In order to test the potential correlation between the structural features
computed by
SMAR Scan and the S/MAR functional activity, the whole human genome has been
analyzed with the combined method with very stringent parameters, in order to
get
sequences with the highest values for the theoretical structural features
computed,
which are called "super" S/MARs below. This was done with the hope to obtain
predicted MAR elements with a very potential to increase transgene expression
and
recombinant protein production. The putative S/MARs hence harvested were first
analyzed from the bioinformatics perpective in an attempt to characterize and
classify
them.
6.1 S/MARs predicted from the analysis of the whole human genome
As whole human genome sequence, all human RefSeq (National Center for
Biotechnology Information, The NCBI handbook [Internet]. Bethesda (MD):
National
Library of Medicine (US), Oct. Chapter 17, The Reference Sequence (RefSeq)
Project,
2002 (Available from http://vvww.ncbi.nih.gov/entrez/query.fcgi?db=Books)
contigs
(release 5) were used and analyzed with the combined method, using SMAR Scan
as
filter in the first level processing, employing default settings except for
the highest bend
cutoff value, whereas a stringent threshold of 4.0 degrees (instead of 3.202
degrees)
has been used for the DNA bending criterion.
In the second level processing, predicted transcription factors binding have
been
sought in the sequences selected from the previous step without doing any
filtering on
these sequences.
The analysis by the combined method of the whole human genome came up with a
total of 1757 putative "super" S/MARs representing a total of 1 065 305 bp
(0.35% of
the whole human genome). Table 2 shows for each chromosome: its size, its
number of
genes, its number of S/MARs predicted, its S/MARs density per gene and its kb
per
S/MAR. This table shows that there are very various gene densities per S/MAR
predicted for the different chromosomes (standard deviation represents more
than 50%
of the mean of the density of genes per S/MAR predicted and the fold
difference
39
CA 02884685 2015-03-10
between the higher and the lower density of genes per S/MAR is 6,5). Table 2
also
shows that the kb per S/MAR varies less that the density of genes per S/MAR
(standard
deviation represents 25% of the mean of kb per S/MAR and the fold difference
between
the higher and the lower kb per S/MAR is 3.2).
,
, 5
r¨cirsTmosome ¨ Number of Size of the
I NuMber 4¨ Density olT-16-per ¨
genes per chromosome , S/MARs ,'
genes =' S/MAR
chromosome
= (millions bp)
predicted per S/MAR !
: ............ -----_:
1 2544 230 ----1 85 29.9
, -.1- 2705
i 2 1772 241 143 12.3
1685 '
,
,
3 1406 198 ' 101 13.9
/960
,
i 4 1036 190 ,
118 8,7
1610
1233 180 116 10.6 1551
6 1247 170 94 13.2
1808
7 1383 160 179 7.7
1754 r
8 942 145 77 12.2
1883 :
9 1100 119 48 229
2479 .
1003 133 71 14 1 1873
11 1692 132 , 67 25.2
, 1970
12 1278 131 78 16 3
1679
,
13 506 97 70 7.2
1385 .
14 1168 88 i 36 32.4
2444 =
, .
895 83 ,
:
. 35 25.5 , 2371
,
16 1107 81 .
,
. 41 27 ' 1975 =
17 1421 80 i
!
, 37 38.4 2162
18 396 75
,
,
. 51 7.7 1470 -
,
19 1621 56 ,
,
,
, 36 45.02 1555 .
,
724 60 i , 28 25.8 2142
21355 34 .
, 1 18 19.7
1888
22 707 34 1 28 I 25.2 1214 ,
,
, X 1168 154 170 6,8
1 905
1 V 251 25
_.,..9....................... ,833
3 ...... 833
--- - -
1 SUM 26955 3050 1 757 457
-433 12 .
I Mean 1 123 127 .
73 19
1 804 :
,
I Sd_..510 72.8 ! 45 _....j,
10 462 ,
,
,
,
,
,
,
,
,
,
,
,
,
Table 2: Number of S/MARs predicted per chromosome. The number of genes per
chromosome
1
corresponds to the NCBI human genome statistics (Build 34 Version 3) (National
Center for i
10 Biotechnology Information, The NCBI handbook [Internet]. Bethesda (MD):
National Library
of Medicine (US), Oct. Chapter 17, The Reference Sequence (RefSeq) Project,
2002 (Available
from http://www.nchi.nih.gov/entrez/queryfegi?db=Books) based on GenBank
annotations.
Chromosome sizes are the sum of the corresponding human RefSeq (National
Center for
Biotechnology Information, The NCBI handbook [Internet]. Bethesda (MD):
National Library
15 of Medicine (US), Oct. Chapter 17, The Reference Sequence (RefSeq)
Project, 2002 (Available
from http://www.ncbi.nih.gov/entrez/query.fcgi?db=Books) (release 5) contig
lengths
6.2 Bioinformatics analysis of "super" MARS for transcription factor binding
sites
20 The 1757 predicted "super" S/MARs sequences obtained previously by SMAR
Scan
were then analyzed for potential transcription factors binding sites. This has
been
achieved using RMatch TMProfessional (Kel AE, Gossling E, Reuter 1,
Cheremushkin E,
KelMargoulis OV, Wingender E, MATCH: A tool for searching transcription factor
CA 02884685 2015-03-10
binding sites in DNA sequences, Nucleic Acids Res, 31(13):35769, 2003), a
weight
matrixbased tool based on TRANSFAC (Wingender E, Chen X, Fricke E, Geffers R,
Hehl R, Liebich I, Krull M, Matys V, Michael H, Ohnhauser R, Pruss M.
Schacherer F,
Thiele S, Urbach S, The TRANSFAC system on gene expression regulation, Nucleic
Acids Research , 29(1):2813, 2001). Match TM 2.0 Professional has been used
with most
of the default settings Match Tmanalysis was based on TRANSFAC Professional,
release
8.2 (20040630). The sums of all transcription factors binding prediction on
the 1757
sequences analyzed according to Match 1Mare in Table 3. Based on this table,
only the
transcription factors totalizing at least 20 hits over the 1757 sequences
analyzed were
considered for further analyses.
Hereafter are some of the human transcription factors that are the most often
predicted to bind on the 1757 putative S/MAR sequences and their Match
description: Cdc5 (cell division control protein 5) a transcriptional
regulator/repressor,
Nkx3A a homeodomain protein regulated by androgen, POU1F1 (pituitaryspecific
positive transcription factor 1) which is specific to the pituitary and
stimulates cells
proliferation. Thus, in addition to SATB1, NMP4 and MEF2, other transcription
factors can participate in the activity of MARs.
[API TTAR 2 Bach2 1 Brn2 1
C/EBP 20 C/EBPgamma 5 CDP CR3 1 COMP 1 2
CREBP I 34 Cdc5 858 Cdx2 35 Evil 472
FOX 78 FOXD3 79 FOXJ2 244 FOXP3 29
Freac7 272 GATA1 2 GATA3 142 GATA4 125
HFH1 12 HFH3 I HU' 275 HNF1 337
HNF3alpha 23 HNF3beta 71 1-IP1 2 Lhx3 22
MEF2 114 MRF2 57 Myc 18 NKX3A 849
Nkx25 2 Octl 191 PBX 5 POUIF1 483
POU3F2 11 POU6F1 29 Pax3 3 Pax6 20
Pill 505 SRF 8 TM' 2852 TFI1A 14
1TF1 I VSMTATA _13 4 VBP 53 1 Vmw65 1
XFDI _________ 65 XED2 418 X FD3 ........... 2
Table 3 is a summary of all transcription factors binding prediction
(totalizing 20 hits or
more) on the 1757 sequences analyzed.
6.3 Bioinformatics analysis of predicted "super" MARs for dinucleotide
frequencies
Various computer analysis were performed in order to easily identify "super"
S/MAR
41
CA 02884685 2015-03-10
sequences using an explicit criterion that could be identified without
computing. Among
those, a di-nucleotide analysis was performed on the 1757 superMARs, computing
each of the 16 possible dinucleotide percentage for each sequence considering
both
strands in the 5' > 3' direction.
A summary (min., max., median, mean, 25th percentile and 75th percentile) as
well as
1 the histograms of each dinucleotide percentage over the 1757
S/MAR sequences are
respectively presented in Table 4. A similar analysis was performed on
randomly
I selected sequences from the human genome, representing randomly
selected non-
S/MAR sequences (which might however contain some MARs). Table 5 represents
respectively a summary of the dinucleotide content analysis for these
sequences.
Table 4: Dinucleotide percentages over the 1757 S/MAR sequences
AA % AC % - ----AdT.A,- --.7-----
-A-TTe-------
Minimum 0.000 0,0000 0.0000 __ ...-- ____
18-.50
25th percentile . 4.234 0.9372 0.1408 32.11
,
Median 7.843 2.2408 0.4777 34.68
Mean 7.184 3.2117 1.0865 34.32
75th percentile 10.110 4.7718 1.5096 36.94
Maximum17.290 12.9479 8.1230 50.00
,
_
_....._
CA % CC % CG % CT %
'
Minimum 0.0000 0.00000 0.0000 0.0000
25th percentile 0,9695 0.00000 0.0000 0.1408
Median I .9776 0.00000 000(10 0.4777
Mean 2.6977 0.14123 0.2709 1.0865
1
75th percentile 3.7543 0.09422 0.1256 1.5096
Maximum 10.4061 4.24837 7.4410 8.1230
GA % GC % GG % GT %
-
Minimum 0.00000 0.0000 0.00000 0.0000
25th percentile 0.08696 0.0000 0.00000 0.9372
I
1
Median 0.32616 0.0000 0.00000 2.2408
I
Mean 0.63347 0.2104 0 14123 3.2117
.
75th percentile 0.83333 0.1914 0.09422 4.7718
Maximum 5.77889 9.8795 4.24837 12.9479 ]
,
, TA % Tc % TG % IT %
Minimum 28.63 .
, 0.00000 0.0000
' ----7.666---1
,
25th percentile 33,48 0.08696 0.9693 4.234
Median 35.22 0.32616 1.9776 7.843
Mean 35.29 0.63347 2 6977 7.184
75th percentile 37.14 0.83333 3.7543 10.110
Maximum 50.00 5.77889 10.4061.
17 290
,
Considering the results of the predicted S/MAR elements and of the nonS/MAR se-
quences in the summary tables, noticeable differences can be noticed in the AT
et TA
dinucleotide contents between these two groups of sequences. AT and TA
represent
respectively at least 18,5 % and 28.6 % of the dinucleotide content of the
predicted
S/MAR sequences, whereas the minimum percentages for the same dinucleotides in
42
CA 02884685 2015-03-10
1
1
nonS/MAR sequences are respectively 0.3 % and 0%. Similarly, the maximum CC
and
GG content in S/MAR sequences is 4.2 %, whereas in nonS/MAR sequences the .
percentages for these two dinucleotides can amount up to 20.81%.
The correlation between AT and TA dinucleotide percentages and the DNA highest
I
bend as computed by SMAR Scan is depicted in Fig. 17 for the predicted S/MAR
sequences and in Fig.18 for the nonS/MAR sequences. The different scatterplots
of i
!
i
these figures show that the TA percentage correlates well with the predicted
DNA bend
as predicted by SMAR Scan .
Table 5: Dinucleotide percentages over the 1757 nonS/MAR sequences summary
_________ _ _ _ ________________
AA % AC % AG % AT %
1 __________________________________
I Minimum 0.000 I _735 1.512 - = 0.3257
,
25th percentile 7.096 4.586 6.466 5.1033
Median 9.106 5.016 7.279 6.8695
,
Mean 8.976 5.054 7.184 7.0108
75th percentile 10.939 5.494 7.969 8.7913
'
Maximum 17.922 13.816 12.232 23.1788 .
,
. .
CA % CC % CG % CT %
. .õõ
..
Minimum . 3.571 0.8278 0.0000 1.512
25th percentile 6.765 4.1077 0.4727 6.466
Median ' 7.410 5 5556 0.8439 ' 7.279
Mean 7.411 5.9088 1,2707 7.184
75th percentile 8.010 7.2460 1.5760. 7.969
Maximum 15.714 20.8415 :
, I 2.6074 .
.
. 12.232
. . ___________
' GA % GC % G G % ' GT %
. . .
, . . ___________
Minimum ,
, 1.319 0 4967 0.8278 . 1.735
25th percentile 5,495 3 2615 4.1077 4.586
Median 6.032 4 4092 5,5556 5.016
Mean 6.065 4 7468 5.9088 5.054
75th percentile 6.602 5,8824 7.2460 5.494
,
Maximum 10.423 . . 16.0000 20.8415 13.816
. .
:
TA % TC % TG % Tr%
Minimum 0 000 1.319 3.571 0.000
25th percentile 3,876 5.495 6.765 7.096
Median 5.625 6.032 7.410 9.106
Mean 5.774 6.065 7.411 8.976
75th percentile 7.464 6.602 8.010 10.939
Maximum 24,338 10.423 15.714 17.922
,
_
Four of the novel super MARs were randomly picked and analyzed for AT and TA
dinucleotide content, and compared with the previously known chicken lysMAR,
considering windows of 100 base pairs (Table 6).
Surprinsigly, Applicants have shown that all of the super MARs have AT
dinucleotide
frequencies greater then 12%, and TA dinucleotides greater than 10% of the
total
dinucleotides analysed in a window of 100base pairs of DNA. The most efficient
MARs
43
,
CA 02884685 2015-03-10
display values around 34% of the two dinucleotide pairs.
Table 6. Summary of %AT and TA dinucleotide frequencies of experimentally
verified
MARs
CLysMAR (average of CUEs) AT`)/0 12.03 ITA /0 : 10.29 SEQ ID No
P1_68 AT% : 33.78 TA% : 33.93 SEQ ID No
P1_6 AT% : 34.67 'TA /0 : 34.38 SEQ ID No
P1_42 AT /0 : 35.65 TA /0 : 35.52 SEQ ID
No
Mean value for all human "superMARs AT% : 34.32 TA% : 35.29
Mean value for all human non-MARs AT% : 7.01 TA% : 5.77
6.4 Analysis of orthologous intergenic regions of human and mouse genomes
In order to get an insight on S/MAR evolution, orthologous intergenic regions
of human
and mouse genomes have been analysed with SMAR Scan . The data set used is
composed of 87 pairs of complete orthologous intergenic regions from the human
and
mouse genomes (Shabalina SA, Ogurtsov AY, Kondrashov VA, Kondrashov AS,
Selective constraint in intergenic regions of human and mouse genomes, Trends
Genet, 17(7):3736, 2001) (average length -12 000 bp) located on 12 human and
on 12
mouse chromosomes, the synteny of these sequences was confirmed by pairwise
sequence alignment and consideration of the annotations of the flanking genes
(experimental or predicted).
Analysis of the 87 human and mouse orthologous intergenic sequences have been
analysed with SMAR Scan using its default settings. Analysis of the human
sequences yielded a total of 12 S/MARs predicted (representing a total length
of 4 750
bp), located on 5 different intergenic sequences.
Among the three human intergenic sequences predicted to contain a "super"
S/MAR
using SMAR Scan stringent settings, one of the corresponding mouse
orthologous
intergenic sequence is also predicted to contain a S/MAR (human EMBL ID:
Z96050,
position 28 010 to 76 951 othologous to mouse EMBL ID: AC015932, positions 59
884
to 89 963). When a local alignement of these two orthologous intergenic
sequences is
performed, the best local alignement of these two big regions correspond to
the regions
44
CA 02884685 2015-03-10
predicted by SMAR Scan to be S/MAR element. A manual search for the mouse
orthologs of the two other human intergenic sequences predicted to contain a
"super"
S/MAR was performed using the Ensembl Genome Browser (http://ensembl.org). The
mouse orthologous intergenic sequences of these two human sequences were
retrieved using Ensembl orthologue predictions (based on gene names),
searching the
orthologous mouse genes for the pairs of human genes flanking these intergenic
regions.
Because SMAR Scan has been tuned for human sequences and consequently yields
little "superMARs with mouse genomic sequences, its default cutoff values were
slightly relaxed for the minimum size of contiguous hits to be considered as
S/MAR
(using 200 bp instead of 300 bp). Analysis by SMAR Scan of these mouse
sequences
predicted several S/MARs having high values for the different computed
structural
features. This finding suggests that the human MAR elements are conserved
across
species.
Example 7: Dissection of the chicken lysozyme gene 5'- MAR
The 3000 base pair 5'-MAR was dissected into smaller fragments that were
monitored
for effect on transgene expression in Chinese hamster ovary (CHO) cells. To do
so,
seven fragments of ¨400 bp were generated by polymerase chain reaction (PCR).
These PCR-amplified fragments were contiguous and cover the entire MAR
sequence
when placed end-to-end. Four copies of each of these fragments were ligated in
a
head-to-tail orientation, to obtain a length corresponding to approximately
half of that of
the natural MAR. The tetramers were inserted upstream of the SV40 promoter in
pGEGFPControl, a modified version of the pGL3Control vector (Promega). The
plasmid
pGEGFPControl was created by exchanging the luciferase gene of pGL3Control for
the
EGFP gene from pEGFP-N1 (Clontech). The 5'-MAR-fragment-containing plasmids
thus created were co-transfected with the resistance plasmid pSVneo in CHO-
D044
cells using LipofectAmine 2000 (lnvitrogen) as transfection reagent, as
performed
previously (Zahn-Zabal, M., et al., "Development of stable cell lines for
production or
regulated expression using matrix attachment regions" J Biotechnol, 2001.
87(1): p.
29-42.). After selection of the antibiotic (G-418) resistant cells, polyclonal
cell
populations were analyzed by FAGS for EGFP fluorescence.
45
CA 02884685 2015-03-10
Transgene expression was expressed at the percentile of high expressor cells,
defined
as the cells which fluorescence levels are at least 4 orders of magnitude
higher than the
average fluorescence of cells transfected with the pGEGFPControl vector
without MAR.
Fig. 5 shows that multimerized fragments B, K and F enhance transgene
expression,
despite their shorter size as compared to the original MAR sequence. In
contrast, other
fragments are poorly active or fully inactive.
Example 8 : Specificity of B, K and F regions in the MAR context
The 5'-MAR was serially deleted from the 5'-end (Fig.6, upper part) or the 3'-
end (Fig.6,
lower part), respectively. The effect of the truncated elements was monitored
in an
assay similar to that described in the previous section. Figure 6 shows that
the loss of
ability to stimulate transgene expression in CHO cells was not evenly
distributed.
In this deletion study, the loss of MAR activity coincided with discrete
regions of
transition which overlap with the 5'-MAR B-, K- and F-fragment, respectively.
In 5'
deletions, activity was mostly lost when fragment K and F were removed. 3'
deletions
that removed the F and b elements had the most pronounced effects. In
contrast,
flanking regions A, D, E and G that have little or no ability to stimulate
transgene
expression on their own (Fig. 5), correspondingly did not contribute to the
MAR activity
in the 5'- and 3'-end deletion studies (Fig. 6).
Example 9:Structure of the F element
The 465 bp F fragment was further dissected into smaller sub-fragments of 234,
243,
213 bp and 122, 125 and 121 bp, respectively. Fragments of the former group
were
octamerized (8 copies) in a head-to-tail orientation, while those of the
latter group were
similarly.hexa-decamerized (16 copies), to maintain a constant length of MAR
sequence. These elements were cloned in pGEGFPControl vector and their effects
were assayed in CHO cells as described previously. Interestingly, fragment
Fill retained
most of the activity of the full-length F fragment whereas fragment FII, which
contains
the right-hand side part of fragment FIII, lost all the ability to stimulate
transgene
expression (Fig. 7). This points to an active region comprised between nt 132
and nt
221 in the FIB fragment. Consistently, multiple copies of fragments Fl and
FIB, which
encompass this region, displayed similar activity. FHA on its own has no
activity.
However, when added to FIB, resulting in Fill, it enhances the activity of the
former.
46
CA 02884685 2015-03-10
Therefore FHA appears to contain an auxiliary sequence that has little
activity on its
own, but that strengthens the activity of the minimal domain located in FIB.
Analysis of the distribution of individual motifs within the lysozyme gene 5'-
MAR is
shown in Fig. 8 A, along with some additional motifs that we added to the
analysis.
Most of these motifs were found to be dispersed throughout the MAR element,
and not
specifically associated with the active portions. For instance, the binding
sites of
transcription factors and other motifs that have been associated with MARs
were not
preferentially localized in the active regions. It has also been proposed that
active MAR
sequences may consist of combination of distinct motifs. Several computer
programs
(MAR Finder, SMARTest, SIDD duplex stability) have been reported to identify
MARs
as regions of DNA that associate with the DNA matrix. They are usually based
on
algorithms that utilizes a predefined series of sequence-specific patterns
that have
previously been suggested as containing MAR activity, as exemplified by MAR
Finder,
now known as MAR Wiz. The output of these programs did not correlate well with
the
transcriptionally active portions of the cLysMAR. For instance, peaks of
activity obtained
with MAR Finder did not clearly match active MAR sub-portion, as for instance
the B
fragment is quite active in vivo but scores negative with MAR Finder (Fig. 8B,
compare
the top and middle panels). Bent DNA structures, as predicted by this program,
did not
correlate well either with activity (Fig. 8B, compare the top and bottom
panels). Similar
results were obtained with the other available programs (data not shown).
The motifs identified by available MAR prediction computer methods are
therefore
unlikely to be the main determinants of the ability of the cLysMAR to increase
gene
expression. Therefore, a number of other computer tools were tested.
Surprisingly,
predicted nucleosome binding sequences and nucleosome disfavouring sequences
were found to be arranged in repetitively interspersed clusters over the MAR,
with the
nucleosome favouring sites overlapping the active B, K and F regions.
Nucleosome
positioning sequences were proposed to consist of DNA stretches that can
easily wrap
around the nucleosomal histones, and they had not been previously associated
with
MAR sequences.
Nucleosome-favouring sequences may be modelled by a collection of DNA features
that include moderately repeated sequences and other physico-chemical
parameters
that may allow the correct phasing and orientation of the DNA over the curved
histone
47
CA 02884685 2015-03-10
surface. Identification of many of these DNA properties may be computerized,
and up
to 38 different such properties have been used to predict potential nucleosome
positions. Therefore, we set up to determine if specific components of
nucleosome
prediction programs might correlate with MAR activity, with the objective to
construct a
tool allowing the identification of novel and possibly more potent MARs from
genomic
sequences.
To determine whether any aspects of DNA primary sequence might distinguish the
active B, K and F regions from the surrounding MAR sequence, we analyzed the
5'-
MAR with MAR Scan . Of the 38 nucleosomal array prediction tools, three were
found
to correlate with the location of the active MAR sub-domains (Fig. 9A).
Location of the
MAR B, K and F regions coincides with maxima for DNA bending, major groove
depth
and minor groove width. A weaker correlation was also noted with minima of the
DNA
melting temperature, as determined by the GC content. Refined mapping over the
MAR
F fragment indicated that the melting temperature valley and DNA bending
summit
indeed correspond the FIB sub-fragment that contains the MAR minimal domain
(Fig.
9B). Thus active MAR portions may correspond to regions predicted as curved
DNA
regions by this program, and we will refer to these regions as CUE-B, CUE-K
and CUE-
F in the text below. Nevertheless, whether these regions correspond to actual
bent DNA
and base-pair unwinding regions is unknown, as they do not correspond to bent
DNA
as predicted by MAR Wiz (Fig.9B).
Example 10: Imprints of other regulatory elements in the F fragment
Nucleosome positioning features may be considered as one of the many specific
chromatin codes contained in genomic DNA. Although this particular code may
contribute to the activity of the F region, it is unlikely to determine MAR
activity alone,
as the 3' part of the F region enhanced activity of the minimal MAR domain
contained in
the FIB portion. Using the Matlnspector program (Genomatix), we searched for
transcription factor binding sites with scores higher than 0.92 and found DNA
binding
sequences for the NMP4 and MEF2 proteins in the 3' part of the F fragment
(Fig. 86).
To determine whether any of these transcription factor-binding sites might
localize close
to the B and K active regions, the entire 5'-MAR sequence was analyzed for
binding by
NMP4 and MEF2 and proteins reported to bind to single-stranded or double-
stranded
form of BURs. Among those, SATB1 (special AT-rich binding protein 1) belongs
to a
class of DNA-binding transcription factor that can either activate or repress
the
48
CA 02884685 2015-03-10
expression of nearby genes. This study indicated that specific proteins such
as SATB1,
NMP4 (nuclear matrix protein 4) and MEF2 (myogenic enhancer factor 2), have a
specific distribution and form a framework around the minimal MAR domains of
cLysMAR (Fig. 10). The occurrence of several of these NMP4 and SATB1 binding
sites
has been confirmed experimentally by the EMSA analysis of purified recombinant
proteins (data not shown).
Example 11: Construction of artificial MARs by combining defined genetic
elements
To further assess the relative roles of the various MAR components, the
cLysMAR was
deleted of all three CUE regions (Fig. 11, middle part), which resulted in the
loss of part
of its activity when compared to the complete MAR sequence similarly assembled
from
all of its components as a control (Fig. 11, top part). Consistently, one copy
of each
CUE alone, or one copy of each of the three CUEs assembled head-to-tail, had
little
activity in the absence of the flanking sequences. These results strengthen
the
conclusion that optimal transcriptional activity requires the combination of
CUEs with of
flanking sequences. Interestingly, the complete MAR sequence generated from
each of
its components, but containing also BgIII-BamHI linker sequences (AGATCC) used
to
assemble each DNA fragment, displayed high transcriptional activity (6 fold
activation)
as compared to the 4.8 fold noted for the original MAR element in this series
of assays
(see Fig. 5).
We next investigated whether the potentially curved DNA regions may also be
active in
an environment different from that found in their natural MAR context.
Therefore, we set
up to swap the CUE-F, CUE-B and CUE-K elements, keeping the flanking sequences
unchanged. The sequences flanking the CUE-F element were amplified by PCR and
assembled to bracket the various CUEs, keeping their original orientation and
distance,
or without a CUE. These engineered -1.8 kb MARs were then assayed for their
ability
to enhance transgene expression as above. All three CUE were active in this
context,
and therefore there action is not restricted to one given set of flanking
sequences.
Interestingly, the CUE-K element was even more active than CUE-F when inserted
between the CUE-F flanking sequences, and the former composite construct
exhibited
an activity as high as that observed for the complete natural MAR (4.8 fold
activation).
What distinguishes the CUE-K element from CUE-F and CUE-B is the presence of
49
CA 02884685 2015-03-10
overlapping binding sites for the MEF-2 and SatB1 proteins, in addition to its
CUE
feature. Therefore, fusing CUE-B with CUE-F-flanking domain results in a
higher
density of all three binding sites, which is likely explanation to the
increased activity.
These results indicate that assemblies of CUEs with sequences containing
binding sites
for proteins such as NMP4, MEF-2, SatB1, and/or polyPpolyQ proteins constitute
potent artificial MAR sequences.
Example 12: Expression vectors
Three expression vectors according to the present invention are represented on
Figure
12.
Plasmid pPAGO1 is a 5640 bp pUC19 derivative. It contains a 2960 bp chicken
DNA
fragment cloned in BamH1 and Xbal restriction sites. The insert comes from the
border
of the 5'-end of the chicken lyzozyme locus and has a high Aft-content.
Plasmid pGEGFP (also named pSV40EGFP) control is a derivative of the pGL3-
control vector (Promega) in which the luciferase gene sequence has been
replaced by
the EGFP gene sequence form the pEGFP-N1 vector (Clontech). The size of pGEGFP
plasmid is 4334bp.
Plasmid pUbCEGFP control is a derivative of the pGL3 wit an Ubiquitin
promoter.
Plasmid pPAGO1GFP (also named pMAR-SV40EGFP) is a derivative of pGEGFP with
the 5'-Lys MAR element cloned in the MCS located just upstream of the SV40
promoter. The size of the pPAGO1EGF plasmid is 7285bp.
Example 13: Effect of the additional transfection of primary transfectant
cells on
transqene expression
One day before transfection, cells were plated in a 24-well plate, in growth
medium at a
density of 1.35 x 105 cells/well for CHO-DG44 cells. 16 hours post-inoculum,
cells were
transfected when they reached 30-40% confluence, using Lipofect-AMINE 2000
(hereinafter LF2000), according to the manufacturer's instructions
(Invitrogen). Twenty-
seven microliters of serum free medium (Opti-MEM; Invitrogen) containing 1.4
pl of
LF2000 were mixed with 27 pl of Opti-MEM containing 830 ng of linear plasmid
DNA.
The antibiotic selection plasmid (pSVneo) amounted to one tenth of the
reporter
plasmid bearing the GFP transgene. The mix was incubated at room temperature
for 20
=
CA 02884685 2015-03-10
min, to allow the DNA-LF2000 complexes to form, The mixture was diluted with
300 pl
of Opti-MEM and poured into previously emptied cell-containing wells.
Following 3
hours incubation of the cells with the DNA mix at 37 C in a CO2 incubator, one
ml of
DMEM-based medium was added to each well. The cells were further incubated for
24
hours in a CO2 incubator at 37 C. The cells were then transfected a second
time
according to the method described above, except that the resistance plasmid
carried
another resistance gene (pSVpuro). Twenty-four hours after the second
transfection,
cells were passaged and expanded into a T-75 flask containing selection medium
supplemented with 500 pg/ml G-418 and 5 pg/ml puromycin. After a two week
selection
period, stably transfected cells were cultured in 6-well plates.
Alternatively, the cell
population was transfected again using the same method, but pTKhygro
(Clontech) and
pSVdhfr as resistance plasmids. The expression of GFP was analysed with
Fluorescence-activated cell sorter (FAGS) and with a Fluoroscan.
Fig.13 shows that the phenotype of the twice-transfected cells (hereafter
called
secondary transfectants) not only was strongly coloured, such that special
bulb and
filter were not required to visualize the green color from the GFP protein,
but also
contained a majority of producing cells (bottom right-hand side FACS
histogram) as
compared to the parental population (central histogram). This level of
fluorescence
corresponds to specific cellular productivities of at least 10 pg per cell per
day. Indeed,
cells transfected only one time (primary transfectants) that did not express
the marker
protein were almost totally absent from the cell population after re-
transfection. Bars
below 101 unitsof GFP fluorescence amounted 30% in the central histogram and
less
than 5% in the right histogram. This suggested that additional cells had been
transfected and successfully expressed GFP.
Strikingly, the amount of fluorescence exhibited by re-transfected cells
suggested that
the subpopulation of cells having incorporated DNA twice expressed much more
GFP
than the expected two-fold increase. Indeed, the results shown in Table 2
indicate that
the secondary transfectants exhibited, on average, more than the two-fold
increase of
GFP expected if two sets of sequences, one at each successive transfection,
would
have been integrated independently and with similar efficiencies.
Interestingly, this was
not dependent on the promoter sequence driving the reporter gene as bath viral
and
cellular promoter-containing vectors gave a similar GFP enhancement (compare
lane 1
and 2). However, the effect was particularly marked for the MAR-containing
vector as
51
CA 02884685 2015-03-10
compared to plasmids without MAR- (lane 3), where the two consecutive
transfections
resulted in a 5.3 and 4.6 fold increase in expression, in two distinct
experiments.
Type of plasmids ¨ Primary 1 Secondary EGFP
fluorescence
transfection transfection Fold
increase
pUbCEGFP 4'992 14'334 2.8
pSV40EGFP 4'324 _____ 12'237 2.8
_pMAR-SV40EGFP 6996 36748 5.3
Type of plasmids Primary Secondary EGFP
fluorescence
transfection transfection Fold
increase
pUbCEGFP 6452 15794 2.5
pSV40EGFP 4433 11735 2.6
pMAR-SV40EGFP 8116 37475 4.6
Table 7. Effect of re-transfecting primary transfectants at 24 hours interval
on GFP
expression. Two independent experiments are shown. The resistance plasmid
pSVneo was co-transfected with various GFP expression vectors. One day post-
transfection, cells were re-transfected with the same plasmids with the
difference
that the resistance plasmid was changed for pSVpuro. Cells carrying both
resistance
genes were selected on 500 pg/ml G-418 and 5pg/m1 puromycin and the expression
of the reporter gene marker was quantified by Fluoroscan. The fold increases
correspond to the ratio of fluorescence obtained from two consecutive
transfections
as compared to the sum of fluorescence obtained from the corresponding
independent transfections. The fold increases that were judged significantly
higher
are shown in bold, and correspond to fluorescence values that are consistently
over I
2-fold higher than the addition of those obtained from the independent
transfections.
The increase in the level of GFP expression in multiply tranfected cells was
not
expected from current knowledge, and this effect had not been observed
previously.
Taken together, the data presented here support the idea that the plasmid
sequences
that primarily integrated into the host genome would facilitate integration of
other
plasmids by homologous recombination with the second incoming set of plasmid
molecules. Plasmid recombination events occur within a 1-h interval after the
plasmid
DNA has reached the nucleus and the frequency of homologous recombination
between co-injected plasmid molecules in cultured mammalian cells has been
shown to
be extremely high, approaching unity (Folger, K,R., K. Thomas, and M.R.
Capecchi,
Nonreciprocal exchanges of information between DNA duplexes coinjected into
52
CA 02884685 2015-03-10
mammalian cell nuclei. Mol Cell Biol, 1985. 5(1): p. 59-69], explaining the
integration of
multiple plasmid copies. However, homologous recombination between newly
introduced DNA and its chromosomal homolog normally occurs very rarely, at a
frequency of 1 in 103 cells receiving DNA to the most [ Thomas, KR., K.R.
Folger, and
M.R. Capecchi, High frequency targeting of genes to specific sites in the
mammalian
genome. Cell, 1986. 44(3): p. 419-281 Thus, the results might indicate that
the MAR
element surprisingly acts to promote such recombination events. MARs would not
only
modify the organization of genes in vivo, and possibly also allow DNA
replication in
conjunction with viral DNA sequences, but they may also act as DNA
recombination
signals.
Example 14: MARs mediate the unexpectedly high levels of expression in
multiply transfected cells
If MAR-driven recombination events were to occur in the multiple transfections
process,
we expect that the synergy between the primary and secondary plasmid DNA would
be
affected by the presence of MAR elements at one or both of the transfection
steps. We
examined this possibility by multiply transfections of the cells with pMAR
alone or in
combination with various expression plasmids, using the method described
previously..
Table 3 shows that transfecting the cells twice with the pMAR-SV40EGFP plasmid
gave
the highest expression of GFP and the highest degree of enhancement of all
conditions
(4.3 fold). In contrast, transfecting twice the vector without MAR gave little
or no
enhancement, 2.8-fold, instead of the expected two-fold increase. We conclude
that the
presence of MAR elements at each transfection step is necessary to achieve the
maximal protein synthesis.
Table 8
Primary transfection Secondary transfection
Type of plasmid EGFP- Type of plasmid EGFP- Fold
____________________ fluorescence
fluorescence increase
pMAR 0 pMAR 0 0
pSV40EGFP 15437 2.3-
2.5
pMAR-SV40EGFP 30488 2.6-
2.7
pMAR-SV40EGF15 11'278 pMAR-SV40EGFP 47'027 4.3-
5.3
pMAR 12'319 1.0-
1.1
pSV40EGFP 6'114 pSV40EGFP 17200 2.8
pMAR 11169 1.8-
2.3
-------
53
CA 02884685 2015-03-10
Interestingly, when cells were first transfected with pMAR alone, and then re-
transfected with pSV40EGFP or pMAR-SV40EGFP, the GFP levels were more than
doubled as compared to those resulting from the single transfection of the
later
plasmids (2.5 and 2.7 fold respectively, instead of the expected 1-fold). This
indicates
that the prior transfection of the MAR can increase the expression of the
plasmid used
in the second transfection procedure. Because MARs act only locally on
chromatin
structure and gene expresIsion, this implies that the two types of DNA may
have
integrated at a similar chromosomal locus. In contrast, transfecting the GFP
expression
vectors alone, followed by the MAR element in the second step, yielded little
or no
improvement of the GFP levels. This indicates that the order of plasmid
transfection is important, and that the first transfection event should
contain a MAR
element to allow significantly higher levels of transgene expression.
If MAR elements favoured the homologous recombination of the plasmids
remaining in
episomal forms from the first and second transfection procedures, followed by
their co-
integration at one chromosomal locus, one would expect that the order of
plasmid
transfection would not affect GFP levels. However, the above findings indicate
that it is
more favourable to transfect the MAR element in the first rather than in the
second
transfection event. This suggests the following molecular mechanism: during
the first
transfection procedure, the MAR elements may concatemerize and integrate, at
least in
part, in the cellular chromosome. This integrated MAR DNA may in turn favour
the
further integration of more plasmids, during the second transfection
procedure, at the
same or at a nearby chromosomal locus.
Example 15 MARs as long term DNA transfer facilitators
If integrated MARs mediated a persistent recombination-permissive chromosomal
structure, one would expect high levels of expression even if the second
transfection
was performed long after the first one, at a time when most of the transiently
introduced
episomal DNA has been eliminated. To address this possibility, the cells from
Table 3,
selected for antibiotic resistance for three weeks, were transfected again
once or twice
and selected for the incorporation of additional DNA resistance markers. The
tertiary, or
the tertiary and quaternary transfection cycles, were performed with
combinations of
pMAR or pMAR-SV40EGFP, and analyzed for GFP expression as before.
54
CA 02884685 2015-03-10
Table 9
Tertiary transfection Quaternary transfection
Type of plasmid EGFP- Fold Type of plasmid,-.. EFP-
Fold
fluorescence increase
increas
fluorescence
pMAR 18368 2.2 pMAR 43'186
2.4
pMAR- 140000 7.6
SV40EGFP
pMAR-SV40EGF11 16544
2.0 1pMAR- 91000 5.5
ISV40EGFP
______________________________________ pMAR
33'814 J 2.0
Table 9. MARs act as facilitator of DNA integration.
The pMAR-SV40EGFP/ pMAR-SV40EGFP secondary transfectants were used in a
third cycle of transfection at the end of the selection process. The tertiary
transfection
was accomplished with pMAR or pMAR-SV40EGFP, and pTKhygro as selection
plasmid, to give tertiary transfectants. After 24 hours, cells were
transfected again with
either plasmid and pSVdhfr, resulting in the quaternary transfectants which
were
selected in growth medium containing 500 tg/m1 G-418 and 5.ig/m1 puromycin,
300
jig/m1 hygromycin B and 5j.tM methotrexate. The secondary transfectants
initially
exhibited a GFP fluorescence of 8300. The fold increases correspond to the
ratio of
fluorescence obtained from two consecutive transfections as compared to the
sum of
fluorescence obtained from the corresponding independent transfections. The
fold
increases that were judged significantly higher are shown in bold, and
correspond to
fluorescence values that are 2-fold higher than the addition of those obtained
from the
independent transfections.
These results show that loading more copies of pMAR or pMAR-SV40EGFP resulted
in
similar 2-fold enhancements of total cell fluorescence. Loading even more of
the MAR
in the quaternary transfection further enhanced this activity by another 2.4-
fold. This is
consistent with our hypothesis that newly introduced MAR sequences may
integrate at
the chromosomal transgene locus by homologous recombination and thereby
further
increase transgene expression:
CA 02884685 2015-03-10
When the cells were transfected a third and fourth time with the pMAR-SV40EGFP
plasmid, GFP activity further increased, once again to levels not expected
from the
addition of the fluorescence levels obtained from independent transfections.
GFP
expression reached levels that resulted in cells visibly glowing green in day
light
(Fig.14). These results further indicate that the efficiency of the quaternary
transfection
was much higher than that expected from the efficacy of the third DNA
transfer,
indicating that proper timing between transfections is crucial to obtain the
optimal gene
expression increase, one day being preferred over a three weeks period.
We believe that MAR elements favour secondary integration events in increasing
recombination frequency at their site of chromosomal integration by relaxing
closed
chromatin structure, as .they mediate a local increase of histone acetylation
(Yasui, D.,
et al., SATB1 targets chromatin remodelling to regulate genes over long
distances.
Nature, 2002. 419(6907): p. 641-5.1. Alternatively, or concomitantly, MARs
potentially
relocate nearby genes to subnuclear locations thought to be enriched in trans-
acting
factors, including proteins that can participate in recombination events such
as
topoisomerases. This can result in a locus in which the MAR sequences can
bracket
the pSV40EGFP repeats, efficiently shielding the transgenes from chromatin-
mediated
silencing effects.
Example 16: Use of MARs identified with SMAR Scan II to increase the
expression of a recombinant protein.
Four MAR elements were randomly selected from the sequences obtained from the
analysis of the complete human genome sequence with SMAR Scan or the combined
method. These are termed 1_6, 1_42, 1_68, (where the first number represents
the
chromosome from which the sequence originates, and the second number is
specific to
the predicted MAR along this chromosome) and X_S29, a "super" MAR identified
on
chromosome X. These predicted MARs were inserted into the pGEGFPControl vector
upstream of the SV40 promoter and enhancer driving the expression of the green
fluorescent protein and these plasmids were transfected into cultured CHO
cells, as
described previously (Zahn-Zabal, M., et al., Development of stable cell lines
for
production or regulated expression using matrix attachment regions. J
Biotechnol, 2001.
87(1): p. 29-42). Expression of the transgene was then analyzed in the total
population
of stably transfected cells using a fluorescent cell sorter (FAGS) machine. As
can be
seen from Fig. 19, all of these newly identified MARs increased the expression
of the
56
CA 02884685 2015-03-10
transgene significantly above the expression driven by the chicken lysosyme
MAR, the
"super" MAR X_S29 being the most potent of all of the newly identified MARs.
Example 17: Effect on hematocrit of in vivo expression of mEpo by
electrotransfer of Network system with and without Human MAR (1-68).
The therapeutic gene encodes EPO (erythropoietin), an hormone used for the
treatment of anemia. The EPO gene is placed under the control of a doxycycline
inducible promoter, in a gene switch system described previously called below
the
Network system (Imhof, M. 0., Chatellard, P., and Mermod, N. (2000). A
regulatory
network for efficient control of transgene expression. J. Gene. Med. 2, 107-
116.). The
EPO and regulatory genes are then injected in the muscle of mice using an in
vivo
electroporation procedure termed the electrotransfer, so that the genes are
transferred
to the nuclei of the muscle fibers. When the doxycycline antibiotic is added
to the
drinking water of the mice, this compound is expected to induce the expression
of EPO,
which will lead to the elevation of the hematocrit level, due to the increase
in red blood
cell counts mediated by the high levels of circulating EPO. Thus, if the MAR
improved
expression of EPO, higher levels of hematocrit would be expected.
In vivo experiments were carried out on 5 week-old C578L6 female mice (Iffa
Credo-
Charles River, France). 304 of plasmid DNA in normal saline solution was
delivered by
trans-cutaneous injections in the tibialis anterior muscle. All injections
were carried out
under Ketaminol (75 mg/kg) and Narcoxyl (10 mg/kg) anesthesia. Following the
intramuscular injection of DNA, an electrical field was applied to the muscle.
A voltage
of 200 Vicm was applied in 8 ms pulses at 1Hz (Bettan M, Darteil R, Caillaud
JM,
Soubrier F, Delaere P, Branelec D, Mahfoudi A, Duverger N, Scherman D. 2000.
"High-
level protein secretion into blood circulation after electric pulse-mediated
gene transfer
into skeletal muscle". Mol Thor. 2: 204-10).
16 mice were injected by the Network system expressing EPO without the 1_68
MAR
and 16 other mice were injected with the Network system incorporating the MAR
in 5' of
the promoter/enhancer sequences driving the expression of the activator and
EPO
genes. In each group, half of the mice were submitted to doxycycline in
drinking water
from the beginning of the experiment (day 0 ¨ the day of electrotransfer) and
in the
other half, doxycycline was put in drinking water starting at day 21.
57
CA 02884685 2015-03-10
Blood samples were collected using heparinated capillaries by retro-orbital
punction at
different times after the injection of plasmids. Capillaries were
centrifugated 10 minutes
at 5000 rpm at room temperature and the volumetric fraction of blood cells is
assessed
in comparison to the total blood volume and expressed as a percentile,
determining the
hematocrit level.
As can be deduced from Fig. 16 The group of mice injected by MAR-network,
induced
from the beginning of the experiment, display a better induction of the
hematocrit in
comparison of mice injected by original network without MAR. After 2 months,
haematocrits in "MAR-containing group" is still at values higher (65%) than
normal
hematocrit levels (45-55%).
More importantly, late induction (day 21) is possible only in presence of MAR
but not
from mice where the Network wvvas injected without the MAR. Thus the MAR
likely
protects " the transgenes from silencing and allows induction of its
expression even after
prolong period in non-inducing conditions.
Overall, the MAR element is able to increase the expression of the therapeutic
gene as
detected from its increased physiological effect on the hematocrit.
58