Note: Descriptions are shown in the official language in which they were submitted.
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
ANTI-MUCIN ANTIBODIES FOR EARLY DETECTION AND TREATMENT OF
PANCREATIC CANCER
RELATED APPLICATIONS
[001] This application claims the benefit under 35 U.S.C. 119(e) of
provisional U.S. Patent
Application Serial Nos. 61/807,176, filed 4/1/13, 61/818,708, filed 5/2/13 and
61/896,909,
filed 10/29/13. The present application is a continuation-in-part of U.S.
Patent Application
Serial No. 14/036,765, filed 9/25/13. The text of each claimed priority
application is
incorporated herein by reference in its entirety.
SEQUENCE LISTING
[002] The instant application contains a Sequence Listing which has been
submitted in
ASCII format via EFS-Web and is hereby incorporated by reference in its
entirety. Said
ASCII copy, created on March 28, 2014, is named IMM343W0 l_SL.txt and is
55,791 bytes
in size.
BACKGROUND OF THE INVENTION
Field of the Invention
[003] This invention relates to anti-pancreatic cancer antibodies and antigen-
binding
fragments thereof that bind to MUC5ac mucin in pancreatic cancer. More
preferably, the
antibodies or fragments thereof bind to an epitope located within the second
to fourth
cysteine-rich domains of MUC5ac (amino acid residues 1575-2052). The subject
antibodies
or antibody fragments bind with high selectivity to pancreatic cancer cells to
allow detection
and/or diagnosis of pancreatic adenocarcinoma at the earliest stages of the
disease. Most
preferably, antibody-based assays are capable of detecting about 85% or more
of pancreatic
adenocarcinomas, with a false positive rate of about 5% or less for healthy
controls. In
particular embodiments, the methods and compositions can be used to detect
and/or diagnose
pancreatic adenocarcinoma by screening serum samples from subjects and
preferably can
detect 60% or more of stage I pancreatic cancers and 80% or more of stage II
cancers by
serum sample analysis. In other embodiments, immunoassay with an anti-MUC5ac
antibody
may be combined with immunodetection using other pancreatic cancer markers,
such as
CA19.9, to provide improved detection rates for pancreatic cancer without
decreasing
1
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
specificity. In still other embodiments, reactivity with the anti-pancreatic
cancer antibody
can be used to detect occult pancreatic cancer or neoplastic precursor lesions
against a
background of pancreatitis or benign pancreatic hyperplasia.
[004] In preferred embodiments, the anti-pancreatic cancer antibody binds to
the same
epitope as, or competes for binding to MUC5ac with a PAM4 antibody comprising
the light
chain variable region complementarity-determining region (CDR) sequences CDR1
(SASSSVSSSYLY, SEQ ID NO:1); CDR2 (STSNLAS, SEQ ID NO:2); and CDR3
(HQWNRYPYT, SEQ ID NO:3); and the heavy chain CDR sequences CDR1 (SYVLH, SEQ
ID NO:4); CDR2 (YINPYNDGTQYNEKFKG, SEQ ID NO:5)and CDR3 (GFGGSYGFAY,
SEQ ID NO:6). Most preferably, the anti-pancreatic cancer antibody is a
humanized PAM4
(hPAM4) antibody comprising the light chain CDR sequences CDR1 (SASSSVSSSYLY,
SEQ ID NO:1); CDR2 (STSNLAS, SEQ ID NO:2); and CDR3 (HQWNRYPYT, SEQ ID
NO:3); and the heavy chain CDR sequences CDR1 (SYVLH, SEQ ID NO:4); CDR2
(YINPYNDGTQYNEKFKG, SEQ ID NO:5) and CDR3 (GFGGSYGFAY, SEQ ID NO:6),
along with human antibody framework (FR) and constant region sequences.
Related Art
[005] Pancreatic cancer is a malignant growth of the pancreas that mainly
occurs in the cells
of the pancreatic ducts. This disease is the ninth most common form of cancer,
yet it is the
fourth and fifth leading cause of cancer deaths in men and women,
respectively. The number
of patients who succumb to pancreatic cancer each year continues to rise,
unlike other leading
cancers where surveillance and/or screening technologies have led to a
decrease in cancer-
related mortality rates (Jemal et al., 2009, CA Cancer J Clin 59:225-49). For
pancreatic
cancer, the overall survival rate is only 20% after one year and less than 4%
after 5 years. The
major reasons for this poor prognosis include the inability to detect the
disease at an early-
stage, when curative measures may have greater opportunity to provide
successful outcomes,
and the lack of an effective treatment for advanced disease.
[006] In general, patients with early-stage disease have better survival rates
than those with
late-stage disease. Those with surgically resected localized disease have a 5-
year relative
survival of 22% vs. 1-2% for patients with unresectable advanced metastatic
disease (Horner
et al., 2009, SEER Cancer Statistics Review, 1975-2006, NCI, Bethesda, MD).
Although
early detection provides a higher probability for successful therapeutic
intervention, a 22% 5-
year relative survival rate translates to an unacceptably high mortality rate
of 78% for
localized disease (Bilimoria et al., 2007, Ann Surg 246:173-80).
2
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
[007] The most common symptoms of pancreatic cancer include jaundice,
abdominal pain,
and weight loss, which, together with other presenting factors, are
nonspecific in nature.
Thus, diagnosing pancreatic cancer at an early stage of tumor growth is often
difficult and
requires extensive diagnostic work-up, often times including exploratory
surgery.
Endoscopic ultrasonography and computed tomography are the best noninvasive
means
available today for diagnosis of pancreatic cancer. However, reliable
detection of small
tumors, as well as differentiation of pancreatic cancer from focal
pancreatitis, is difficult.
The vast majority of patients with pancreatic cancer are presently diagnosed
at a late stage
when the tumor has already extended outside of the capsule to invade
surrounding organs
and/or has metastasized extensively. (Gold et al., 2001, Crit. Rev.
Oncology/Hematology,
39:147-54).
[008] Current treatment procedures available for pancreatic cancer have not
led to a cure, or
to a substantially improved survival time. Surgical resection has been the
only modality that
offers a chance at survival. However, due to a large tumor burden, only 10% to
25% of
patients are candidates for "curative resection." For those patients
undergoing a surgical
treatment, the five-year survival rate is still poor, averaging only about
10%.
[009] Early detection and diagnosis of pancreatic cancer, as well as
appropriate staging of
the disease, would provide an increased survival advantage. A number of
laboratories have
attempted to develop a diagnostic procedure based upon the release of a tumor-
associated
marker into the bloodstream, as well as detection of the marker substance
within biopsy
specimens. The best previously-characterized tumor associated marker for
pancreatic cancer
has been the immunoassay for CA19.9. Elevated levels of this sialylated Lea
epitope
structure were found in 70% of pancreatic cancer patients but were not found
in any of the
focal pancreatitis specimens examined. However, CA19.9 levels were found to be
elevated
in a number of other malignant and benign conditions, and currently the assay
cannot be used
for diagnosis. The assay is useful for monitoring, with continued increase in
CA19.9 serum
levels after surgery being indicative of a poor prognosis. Many other
monoclonal antibodies
(MAbs) have been reported with immunoassays for diagnosis in varying stages of
development. These include but are not limited to DUPAN2, SPAN1, B72.3, 1a3,
and
various anti-CEA (carcinoembryonic antigen, or CEACAM5) antibodies.
[010] Antibodies, in particular MAbs and engineered antibodies or antibody
fragments,
have been tested widely and shown to be of value in detection and treatment of
various
human disorders, including cancers, autoimmune diseases, infectious diseases,
inflammatory
3
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
diseases, and cardiovascular diseases [Filpula and McGuire, Exp. Opin. Ther.
Patents (1999)
9: 231-245]. The clinical utility of an antibody or an antibody-derived agent
is primarily
dependent on its ability to bind to a specific targeted antigen associated
with a particular
disorder. Selectivity is valuable for delivering a diagnostic or therapeutic
agent, such as
drugs, toxins, cytokines, hormones, hormone antagonists, enzymes, enzyme
inhibitors,
oligonucleotides, growth factors, radionuclides, angiogenesis inhibitors or
metals, to a target
location during the detection and treatment phases of a human disorder,
particularly if the
diagnostic or therapeutic agent is toxic to normal tissue in the body.
Radiolabeled antibodies
have been used with some success in numerous malignancies, including ovarian
cancer, colon
cancer, medullary thyroid cancer, and lymphomas. This technology may also
prove useful
for pancreatic cancer. However, previously reported antibodies against
pancreatic cancer
antigens have not been successfully employed to date for the effective therapy
or early
detection and/or diagnosis of pancreatic cancer.
[011] There remains a need in the art for antibodies that exhibit high
selectivity for
pancreatic cancer and other types of cancers, compared to normal pancreatic
tissues and other
normal tissues. In particular, there remains a need for antibodies that
perform as a useful
diagnostic and/or therapeutic tool for pancreatic cancer, preferably at the
earliest stages of the
disease, and that exhibit enhanced uptake at targeted antigens, decreased
binding to
constituents in the blood of healthy individuals and thereby also optimal
protection of normal
tissues and cells from toxic therapeutic agents when these are conjugated to
such antibodies.
Use of such antibodies to detect pancreatic cancer-associated antigens in body
fluids,
particularly blood, can enable improved earlier diagnosis of this disease, so
long as it
differentiates well from benign diseases, and can also be used for monitoring
response to
therapy and potentially also to enhance prognosis by indicating disease
burden.
SUMMARY
[012] In various embodiments, the present invention concerns antibodies,
antigen-binding
antibody fragments and fusion proteins that bind to the MUC5ac pancreatic
cancer mucin,
preferably to an epitope located within the second to fourth cysteine-rich
domains of
MUC5ac (amino acid residues 1575-2052). More preferably, the subject
antibodies or
fragments thereof bind specifically to pancreatic cancer cells, with little or
no binding to
normal or non-neoplastic pancreatic cells. The antibodies are capable of
binding to the
earliest stages of pancreatic cancer, with detection rates of about 50-60% for
PanIN-1A, 70-
80% for Panl B and 80-90% for PanIN-2. More preferably, the antibodies bind to
80 to 90%
4
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
or more of human invasive pancreatic adenocarcinoma, intraductal papillary
mucinous
neoplasia, PanIN-1A, PanIN-1B and PanIN-2 lesions. Most preferably, the
antibodies can
distinguish between early stage pancreatic cancer and non-malignant conditions
such as
pancreatitis.
[013] Such antibodies are of particular use for early detection of cancer and
differential
diagnosis between early stage pancreatic cancer and benign pancreatic
conditions. In
preferred embodiments, such antibodies are of use for in vivo or ex vivo
analysis of samples
from individuals suspected of having early stage pancreatic or certain other
cancers. More
preferably, the antibodies are of use for detection and diagnosis of early
stage pancreatic
cancer by analysis of serum samples.
[014] In alternative embodiments, the antibodies, antibody fragments or fusion
proteins are
capable of binding to synthetic peptide sequences, for example to phage
display peptides,
such as WTWNITKAYPLP (SEQ ID NO:7) and ACPEWWGTTC (SEQ ID NO:8). Such
synthetic peptides may be linear or cyclic and may or may not compete with
antibody binding
to the endogenous pancreatic cancer antigen. Amino acids in certain positions
of the
synthetic peptide sequences may be less critical for antibody binding than
others. For
example, in SEQ ID NO:7 the residues K, A and L at positions 7, 8 and 11 of
the peptide
sequence may be varied while still retaining antibody binding. Similarly, in
SEQ ID NO:8
the threonine residues at positions 8 and 9 of the sequence may be varied,
although
substitution of the threonine at position 9 may significantly affect antibody
binding to the
peptide.
[015] Binding of the antibodies to a target pancreatic cancer antigen may be
inhibited by
treatment of the target antigen with reagents such as dithiothreitol (DTT)
and/or periodate.
Thus, binding of the antibodies to a pancreatic cancer antigen may be
dependent upon the
presence of disulfide bonds and/or the glycosylation state of the target
antigen. In more
preferred embodiments, the epitope recognized by the subject antibodies is not
cross-reactive
with other reported mucin-specific antibodies, such as the MA5 antibody, the
CLH2-2
antibody and/or the 45M1 antibody (see, e.g., Major et al., J Histochem
Cytochem. 35:139-
48, 1987; Dion et al., Hybridoma 10:595-610, 1991).
[016] The subject antibodies or fragments may be naked antibodies or fragments
or
preferably are conjugated to at least one therapeutic and/or diagnostic agent
for delivery of
the agent to target tissues. In alternative embodiments, the subject
antibodies or fragments
may be part of a bispecific antibody with a first binding site for an epitope
located within the
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
second to fourth cysteine-rich domains of MUC5ac (amino acid residues 1575-
2052) and a
second binding site for a hapten conjugated to a targetable construct. The
targetable
construct may in turn be attached to at least one therapeutic and/or
diagnostic agent, of use in
pretargeting techniques.
[017] In preferred embodiments, the subject antibody, antibody fragment or
fusion protein
is a humanized PAM4 antibody or fragment, comprising the light chain variable
region CDR
sequences CDR1 (SASSSVSSSYLY, SEQ ID NO:1); CDR2 (STSNLAS, SEQ ID NO:2);
and CDR3 (HQWNRYPYT, SEQ ID NO:3); and the heavy chain variable region CDR
sequences CDR1 (SYVLH, SEQ ID NO:4); CDR2 (YINPYNDGTQYNEKFKG, SEQ ID
NO:5) and CDR3 (GFGGSYGFAY, SEQ ID NO:6) and human antibody framework region
(FR) and constant region sequences. More preferably, the FRs of the light and
heavy chain
variable regions of the humanized PAM4 antibody or fragment thereof comprise
at least one
amino acid substituted from amino acid residues 5, 27, 30, 38, 48, 66, 67 and
69 of the
murine PAM4 heavy chain variable region (SEQ ID NO:12) and/or at least one
amino acid
selected from amino acid residues 21, 47, 59, 60, 85, 87 and 100 of the murine
PAM4 light
chain variable region (SEQ ID NO:10). Most preferably, the antibody or
fragment thereof
comprises the hPAM4 VH amino acid sequence of SEQ ID NO:19 and the hPAM4 Vic
amino
acid sequence of SEQ ID NO:16.
[018] In alternative embodiments, the anti-pancreatic cancer antibody may be a
chimeric,
humanized or human antibody that binds to the same antigenic determinant
(epitope) as, or
competes for binding to MUC5ac with, a chimeric PAM4 (cPAM4) antibody. As
discussed
below, the cPAM4 antibody is one that comprises the light chain variable
region CDR
sequences CDR1 (SASSSVSSSYLY, SEQ ID NO:1); CDR2 (STSNLAS, SEQ ID NO:2);
and CDR3 (HQWNRYPYT, SEQ ID NO:3); and the heavy chain variable region CDR
sequences CDR1 (SYVLH, SEQ ID NO:4); CDR2 (YINPYNDGTQYNEKFKG, SEQ ID
NO:5)and CDR3 (GFGGSYGFAY, SEQ ID NO:6). Antibodies that bind to the same
antigenic determinant may be identified by a variety of techniques known in
the art, such as
by competitive binding studies using the cPAM4 antibody as the competing
antibody and
human pancreatic mucin or MUC5ac as the target antigen. Antibodies that block
(compete
for) binding to human pancreatic mucin by a cPAM4 antibody are referred to as
cross-
blocking antibodies. Preferably, such cross-blocking antibodies are ones that
bind to an
epitope located within the second to fourth cysteine-rich domains of MUC5ac,
or that
compete for binding to amino acid residues 1575-2052 with a PAM4 antibody.
6
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
[019] Other embodiments concern cancer cell-targeting therapeutic
immunoconjugates
comprising an antibody or fragment thereof or fusion protein bound to at least
one therapeutic
agent. Preferably, the therapeutic agent is selected from the group consisting
of a
radionuclide, an immunomodulator, a hormone, a hormone antagonist, an enzyme,
an
oligonucleotide such as an anti-sense oligonucleotide or a siRNA, an enzyme
inhibitor, a
photoactive therapeutic agent, a cytotoxic agent such as a drug or toxin, an
angiogenesis
inhibitor and a pro-apoptotic agent. In embodiments where more than one
therapeutic agent
is used, the therapeutic agents may comprise multiple copies of the same
therapeutic agent or
else combinations of different therapeutic agents.
[020] In one embodiment, an oligonucleotide, such as an antisense molecule or
siRNA
inhibiting bc1-2 expression as described in U.S. Pat. No. 5,734,033 (the
Examples section of
which is incorporated herein by reference), may be attached to, or form the
therapeutic agent
portion of an immunoconjugate or antibody fusion protein. Alternatively, the
oligonucleotide
may be administered concurrently or sequentially with a naked or conjugated
anti-pancreatic
cancer antibody or antibody fragment, such as a PAM4 antibody. In a preferred
embodiment,
the oligonucleotide is an antisense oligonucleotide that is directed against
an oncogene or
oncogene product, such as bc1-2, p53, ras or other well-known oncogenes.
[021] Preferably, the therapeutic agent is a cytotoxic agent, such as a drug
or a toxin. Also
preferred, the drug is selected from the group consisting of nitrogen
mustards, ethylenimine
derivatives, alkyl sulfonates, nitrosoureas, gemcitabine, triazenes, folic
acid analogs,
anthracyclines, taxanes, COX-2 inhibitors, pyrimidine analogs, purine analogs,
antibiotics,
enzyme inhibitors, epipodophyllotoxins, platinum coordination complexes, vinca
alkaloids,
substituted ureas, methyl hydrazine derivatives, adrenocortical suppressants,
hormone
antagonists, endostatin, taxols, camptothecins, SN-38, doxorubicins and their
analogs,
antimetabolites, alkylating agents, antimitotics, anti-angiogenic agents,
tyrosine kinase
inhibitors, Bruton tyrosine kinase inhibitors, mTOR inhibitors, heat shock
protein (HSP90)
inhibitors, proteosome inhibitors, HDAC inhibitors, pro-apoptotic agents,
methotrexate, CPT-
11, SN-38, 2-PDOX, pro-2-PDOX, and a combination thereof
[022] In another preferred embodiment, the therapeutic agent is a toxin
selected from the
group consisting of ricin, abrin, alpha toxin, saporin, ribonuclease (RNase),
DNase I,
Staphylococcal enterotoxin-A, pokeweed antiviral protein, gelonin, diphtheria
toxin,
Pseudomonas exotoxin, and Pseudomonas endotoxin and combinations thereof Or an
immunomodulator selected from the group consisting of a cytokine, a stem cell
growth
7
CA 02899811 2015-07-29
WO 2014/165506 PCT/US2014/032513
factor, a lymphotoxin, a hematopoietic factor, a colony stimulating factor
(CSF), an
interferon (IFN), a stem cell growth factor, erythropoietin, thrombopoietin
and a
combinations thereof
[023] Alternatively, the therapeutic agent is an enzyme selected from the
group consisting
of malate dehydrogenase, staphylococcal nuclease, delta-V-steroid isomerase,
yeast alcohol
dehydrogenase, alpha-glycerophosphate dehydrogenase, triose phosphate
isomerase,
horseradish peroxidase, alkaline phosphatase, asparaginase, glucose oxidase,
beta-
galactosidase, ribonuclease, urease, catalase, glucose-6-phosphate
dehydrogenase,
glucoamylase and acetylcholinesterase. Such enzymes may be used, for example,
in
combination with prodrugs that are administered in relatively non-toxic form
and converted
at the target site by the enzyme into a cytotoxic agent. In other
alternatives, a drug may be
converted into less toxic form by endogenous enzymes in the subject but may be
reconverted
into a cytotoxic form by the therapeutic enzyme.
[024] Other therapeutic agents include radionuclides such as 14C, 13N, 150,
32p, 33p, 47se,
51Cr, 57CO, 58CO, 59Fe, 62CU, 67CU, 67Ga, 67Ga, 75 75 75 76 77
77 80m Br, Se, Se, Br, As, Br, Br,
895r, 90Y, 95Ru, 92R11, 99mo, 99mTe, 103mRb, 103Rn, 105Rb, 105Rn, 107Bg,
109pd, 109pt,
111Ag, 1111n , 113m1n, 1195b, 121mTe, 122mTe, 1251, 125mTe, 1261, 1311, 1331,
142pr, 143pr, 149pm,
152Dy, 1535m, 161B0, 161Tb, 165Tm, 166Dy, 166B0, 167Tm, 168Tm, 169Er, 169yb,
177Ln, 186Re,
188Re, 189m05, 189Re, 1921r, 1941r, 197pt 198An, 199An, 199An, 201T1, 203Bg,
211At 211Bi,
211pb, 212Bi, 212pb, 213Bi, 215p0, 217At 219Rn, 221Fr, 223Ra, 224Ac, 225Ac,
255Fm or Th222.
[025] A variety of tyrosine kinase inhibitors are known in the art and any
such known
therapeutic agent may be utilized. Exemplary tyrosine kinase inhibitors
include, but are not
limited to canertinib, dasatinib, erlotinib, gefitinib, imatinib, lapatinib,
leflunomide, nilotinib,
pazopanib, semaxinib, sorafenib, sunitinib, sutent and vatalanib. A specific
class of tyrosine
kinase inhibitor is the Bruton tyrosine kinase inhibitor. Bruton tyrosine
kinase (Btk) has a well-
defined role in B-cell development. Bruton kinase inhibitors include, but are
not limited to, PCI-
32765 (ibrutinib), PCI-45292, GDC-0834, LFM-A13 and RN486.
[026] The subject antibody or fragment may be conjugated to at least one
diagnostic (or
detection) agent. Preferably, the diagnostic agent is selected from the group
consisting of a
radionuclide, a contrast agent, a fluorescent agent, a chemiluminescent agent,
a
bioluminescent agent, a paramagnetic ion, an enzyme and a photoactive
diagnostic agent.
Still more preferred, the diagnostic agent is a radionuclide with an energy
between 20 and
4,000 keV or is a radionuclide selected from the group consisting of 1101n,
111In, 177õ, 18F,
8
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
52Fe, 62 64 67 67
Cu, Cu, Cu, Ga, 68 9 89 94
Ga, 86y, n- -,
-Y Zr, m 94 99
Tc, Tc, m 120 123 124 125
Tc, 1, 1, 1, 1,
1311,
154-158Gd, 32p, 11C, 13N, 150, 186Re, 188Re, 51mn, S2mJ,In 55Co, 72As, 75Br,
76Br, 82mRb, 83Sr, or
other gamma-, beta-, or positron-emitters. In a particularly preferred
embodiment, the
diagnostic radionuclide 18F is used for labeling and PET imaging, as described
in the
Examples below. The 18F may be attached to an antibody, antibody fragment or
peptide by
complexation to a metal, such as aluminum, and binding of the 18F-metal
complex to a
chelating moiety that is conjugated to a targeting protein, peptide or other
molecule.
[027] Also preferred, the diagnostic agent is a paramagnetic ion, such as
chromium (III),
manganese (II), iron (III), iron (II), cobalt (II), nickel (II), copper (II),
neodymium (III),
samarium (III), ytterbium (III), gadolinium (III), vanadium (II), terbium
(III), dysprosium
(III), holmium (III) and erbium (III), or a radiopaque material, such as
barium, diatrizoate,
ethiodized oil, gallium citrate, iocarmic acid, iocetamic acid, iodamide,
iodipamide,
iodoxamic acid, iogulamide, iohexol, iopamidol, iopanoic acid, ioprocemic
acid, iosefamic
acid, ioseric acid, iosulamide meglumine, iosemetic acid, iotasul, iotetric
acid, iothalamic
acid, iotroxic acid, ioxaglic acid, ioxotrizoic acid, ipodate, meglumine,
metrizamide,
metrizoate, propyliodone, and thallous chloride.
[028] In still other embodiments, the diagnostic agent is a fluorescent
labeling compound
selected from the group consisting of fluorescein isothiocyanate, rhodamine,
phycoerytherin,
phycocyanin, allophycocyanin, o-phthaldehyde and fluorescamine, a
chemiluminescent
labeling compound selected from the group consisting of luminol, isoluminol,
an aromatic
acridinium ester, an imidazole, an acridinium salt and an oxalate ester, or a
bioluminescent
compound selected from the group consisting of luciferin, luciferase and
aequorin. In
another embodiment, the diagnostic immunoconjugates are used in
intraoperative,
endoscopic, or intravascular tumor diagnosis.
[029] Also contemplated are multivalent, multispecific antibodies or fragments
thereof
comprising at least one binding site that binds to an epitope located within
the second to
fourth cysteine-rich domains of MUC5ac (amino acid residues 1575-2052) and one
or more
hapten binding sites having affinity towards hapten molecules. Preferably, the
antibody or
fragment thereof is a chimeric, humanized or fully human antibody or fragment
thereof The
hapten molecule may be conjugated to a targetable construct for delivery of
one or more
therapeutic and/or diagnostic agents. In certain preferred embodiments, the
multivalent
antibodies or fragments thereof may be prepared by the DOCK-AND-LOCKTM (DNLTM)
9
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
technique, as described below. An exemplary DNLTM construct incorporating
hPAM4
antibody fragments is designated TF10, as described below.
[030] Also contemplated is a bispecific antibody or fragment thereof
comprising at least one
binding site with an affinity toward an epitope located within the second to
fourth cysteine-
rich domains of MUC5ac (amino acid residues 1575-2052) and at least one
binding site with
an affinity toward a targetable construct which is capable of carrying at
least one diagnostic
and/or therapeutic agent. Targetable constructs suitable for use are
disclosed, for example, in
U.S. Patent Nos. 6,576,746; 6,962,702; 7,052,872; 7,138,103; 7,172,751;
7,405,320;
7,597,876; 7,563,433; 7,993,626; ,147,799; 8,153,100; 8,153,101; 8,202,509;
8,343,460;
8,444,956, 8,496,912; 8,545,809; 8,617,518; and 8,632,752, the Examples
section of each of
which is incorporated herein by reference.
[031] Other embodiments concern fusion proteins comprising at least two anti-
pancreatic
cancer antibodies and fragments thereof as described herein. Alternatively,
the fusion protein
or fragment thereof may comprise at least one first antibody or fragment
thereof that binds to
an epitope located within the second to fourth cysteine-rich domains of MUC5ac
(amino acid
residues 1575-2052), and at least one second MAb or fragment thereof
Preferably, the
second MAb binds to a tumor-associated antigen, for example selected from the
group
consisting of CA19.9, DUPAN2, SPAN1, Nd2, B72.3, CC49, CEA (CEACAM5),
CEACAM6, Lea, the Lewis antigen Le(y), CSAp, insulin-like growth factor (IGF),
epithelial
glycoprotein-1 (EGP-1), epithelial glycoprotein-2 (EGP-2), CD-80, placental
growth factor
(P1GF), carbonic anhydrase IX, tenascin, IL-6, HLA-DR, CD40, CD74 (e.g.,
milatuzumab),
CD138 (syndecan-1), MUC1, MUC2, MUC3, MUC4, MUC5ac, MUC16, MUC17, TAG-72,
EGFR, platelet-derived growth factor (PDGF), angiogenesis factors (e.g., VEGF
and P1GF),
products of oncogenes (e.g., bc1-2, Kras, p53), cMET, HER2/neu, and antigens
associated
with gastric cancer and colorectal cancer. The second MAb may also bind to a
different
epitope of MUC5ac than the second to fourth cysteine-rich domains of MUC5ac
(amino acid
residues 1575-2052). The antibody fusion protein or fragments thereof may
further comprise
at least one diagnostic and/or therapeutic agent.
[032] Also described herein are DNA sequences comprising a nucleic acid
encoding an
anti-pancreatic cancer antibody, fusion protein, multispecific antibody,
bispecific antibody or
fragment thereof as described herein. Other embodiments concern expression
vectors and/or
host cells comprising the antibody-encoding DNA sequences. In certain
preferred
embodiments, the host cell may be an Sp2/0 cell line transformed with a mutant
Bc1-2 gene,
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
for example with a triple mutant Bc1-2 gene (T69E, S70E, S87E), that has been
adapted to
cell transformation and growth in serum free medium. (See, e.g., U.S. Patent
Nos. 7,531,327;
7,537,930; and 7,608,425, the Examples section of each of which is
incorporated herein by
reference.)
[033] Another embodiment concerns methods of delivering a diagnostic or
therapeutic
agent, or a combination thereof, to a target comprising (i) providing a
composition that
comprises an anti-pancreatic cancer antibody or fragment that binds to an
epitope located
within the second to fourth cysteine-rich domains of MUC5ac (amino acid
residues 1575-
2052), conjugated to at least one diagnostic and/or therapeutic agent and (ii)
administering to
a subject in need thereof the diagnostic or therapeutic conjugate of any one
of the antibodies,
antibody fragments or fusion proteins claimed herein.
[034] Also contemplated is a method of delivering a diagnostic agent, a
therapeutic agent,
or a combination thereof to a target, comprising: (a) administering to a
subject any one of the
multivalent, multispecific or bispecific antibodies or fragments thereof that
have an affinity
toward an epitope located within the second to fourth cysteine-rich domains of
MUC5ac
(amino acid residues 1575-2052) and comprise one or more hapten binding sites;
(b) waiting
a sufficient amount of time for antibody that does not bind to MUC5ac to clear
the subject's
blood stream; and (c) administering to said subject a carrier molecule
comprising a diagnostic
agent, a therapeutic agent, or a combination thereof, that binds to a binding
site of the
antibody. Preferably, the carrier molecule binds to more than one binding site
of the
antibody.
[035] Described herein is a method for diagnosing or treating cancer,
comprising: (a)
administering to a subject any one of the multivalent, multispecific
antibodies or fragments
thereof claimed herein that have an affinity toward an epitope located within
the second to
fourth cysteine-rich domains of MUC5ac (amino acid residues 1575-2052) and
comprise one
or more hapten binding sites; (b) waiting a sufficient amount of time for an
amount of the
non-bound antibody to clear the subject's blood stream; and (c) administering
to said subject a
carrier molecule comprising a diagnostic agent, a therapeutic agent, or a
combination thereof,
that binds to a binding site of the antibody. In a preferred embodiment the
cancer is
pancreatic cancer. Also preferred, the method can be used for intraoperative
identification of
diseased tissues, endoscopic identification of diseased tissues, or
intravascular identification
of diseased tissues.
11
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
[036] Another embodiment is a method of treating a malignancy in a subject
comprising
administering to said subject a therapeutically effective amount of an
antibody or fragment
thereof that binds to an epitope located within the second to fourth cysteine-
rich domains of
MUC5ac (amino acid residues 1575-2052), optionally conjugated to at least one
therapeutic
agent. The antibody or fragment thereof may alternatively be a naked antibody
or fragment
thereof In more preferred embodiments, the antibody or fragment is
administered either
before, simultaneously with, or after administration of another therapeutic
agent as described
above.
[037] Contemplated herein is a method of diagnosing a malignancy in a subject,
particularly a pancreatic cancer, comprising (a) administering to said subject
a diagnostic
conjugate comprising an antibody or fragment thereof that binds to an epitope
located within
the second to fourth cysteine-rich domains of MUC5ac (amino acid residues 1575-
2052),
wherein said MAb or fragment thereof is conjugated to at least one diagnostic
agent, and (b)
detecting the presence of labeled antibody bound to pancreatic cancer cells or
other malignant
cells, wherein binding of the antibody is diagnostic for the presence of
pancreatic cancer or
another malignancy. In preferred embodiments, the antibody or fragment binds
to pancreatic
cancer and not to normal pancreatic tissue, pancreatitis or other non-
malignant conditions. In
less preferred embodiments, the antibody or fragment binds at a significantly
higher level to
cancer cells than to non-malignant cells, allowing differential diagnosis of
cancer from non-
malignant conditions. In a most preferred embodiment, the diagnostic agent may
be an F-18
labeled molecule that is detected by PET imaging.
[038] In more preferred embodiments, the use of anti-pancreatic cancer
antibodies that bind
to an epitope located within the second to fourth cysteine-rich domains of
MUC5ac (amino
acid residues 1575-2052) allows the detection and/or diagnosis of pancreatic
cancer with high
specificity and sensitivity at the earliest stages of malignant disease.
Preferably, the
diagnostic antibody or fragment is capable of labeling at least 70%, more
preferably at least
80%, more preferably at least 90%, more preferably at least 95%, most
preferably about
100% of well differentiated, moderately differentiated and poorly
differentiated pancreatic
cancer and 90% or more of invasive pancreatic adenocarcinomas. The anti-
pancreatic cancer
antibody of use is preferably capable of detecting 85% or more of PanIN-1A,
PanIN-1B,
PanIN-2, IPMN and MCN precursor lesions. Most preferably, immunoassays using
the anti-
pancreatic cancer antibody are capable of detecting 89% or more of total
PanIN, 86% or more
of IPMN, and 92% or more of MCN.
12
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
[039] An alternative embodiment is a method of detecting the presence of PAM4-
binding
MUC5ac and/or diagnosing pancreatic cancer in an individual by in vitro
analysis of blood,
plasma or serum samples. Preferably, the sample is subjected to an organic
phase extraction,
using an organic solvent such as butanol, before it is processed for
immunodetection using an
anti-pancreatic cancer antibody, such as a PAM4 antibody. Following organic
phase
extraction, the extracted aqueous phase is analyzed for the presence of the
epitope of
MUC5ac to which PAM4 binds in the sample, using any of a variety of
immunoassay
techniques known in the art, such as ELISA, sandwich immunoassay, solid phase
RIA, and
similar techniques. Surprisingly, the organic phase extraction results in the
removal of an
inhibitor of PAM4 binding to MUC5ac, allowing detection of MUC5ac in fresh
serum
samples. More surprisingly, using the in vitro analysis techniques described
herein, serum
samples may be analyzed to detect and/or diagnose pancreatic cancer in a
subject at the
earliest stages of pancreatic adenocarcinoma. These unexpected results provide
the first
serum-based assay technique that is diagnostic for the presence of early stage
pancreatic
cancer.
[040] Another embodiment is a method of treating a cancer cell in a subject
comprising
administering to said subject a composition comprising a naked antibody or
fragment thereof
or a naked antibody fusion protein or fragment thereof that binds to an
epitope located within
the second to fourth cysteine-rich domains of MUC5ac (amino acid residues 1575-
2052).
Preferably, the method further comprises administering a second naked antibody
or fragment
thereof selected from the group consisting of CA19.9, DUPAN2, SPAN1, Nd2,
B72.3, CC49,
anti-CEA, anti-CEACAM6, anti-EGP-1, anti-EGP-2, anti-Lea, antibodies defined
by the
Lewis antigen Le(y), and antibodies against CSAp, MUC1, MUC2, MUC3, MUC4,
MUC5ac, MUC16, MUC17, TAG-72, EGFR, CD40, HLA-DR, CD74, CD138, angiogenesis
factors (e.g., VEGF and placenta-like growth factor (P1GF), insulin-like
growth factor (IGF),
tenascin, platelet-derived growth factor, IL-6, products of oncogenes, cMET,
and HER2/neu.
[041] Still other embodiments concern a method of diagnosing a malignancy in a
subject
comprising (i) performing an in vitro diagnosis assay on a specimen from said
subject with a
composition comprising an antibody or fragment thereof that binds to an
epitope located
within the second to fourth cysteine-rich domains of MUC5ac (amino acid
residues 1575-
2052); and (ii) detecting the presence of antibody or fragment bound to
malignant cells in the
specimen. Preferably, the malignancy is a cancer. More preferably, the cancer
is pancreatic
cancer.
13
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
BRIEF DESCRIPTION OF THE DRAWINGS
[042] FIG. 1A. Variable region cDNA sequence& (SEQ ID NO:9) and the deduced
amino
acid sequences (SEQ ID NO:10) of the murine PAM4 Vk. Amino acid sequences
encoded
by the corresponding DNA sequences are given as one-letter codes below the
nucleotide
sequence. Numbering of the nucleotide sequence is on the right side. The amino
acid
residues in the CDR regions are shown in bold and underlined. Kabat's Ig
molecule
numbering is used for amino acid residues as shown by the numbering above the
amino acid
residues. The amino acid residues numbered by a letter are the insertion
residues defined by
Kabat numbering scheme. The insertion residues have the same preceding digits
as that of
the previous residue.
[043] FIG. 1B. Variable region cDNA sequence (SEQ ID NO:11) and the deduced
amino
acid sequence (SEQ ID NO:12) of the murine PAM4 VH. Amino acid sequences
encoded by
the corresponding DNA sequences are given as one-letter codes below the
nucleotide
sequence. Numbering of the nucleotide sequence is on the right side. The amino
acid
residues in the CDR regions are shown in bold and underlined. Kabat's Ig
molecule
numbering is used for amino acid residues as shown by the numbering above the
amino acid
residues. The amino acid residues numbered by a letter are the insertion
residues defined by
Kabat numbering scheme. The insertion residues have the same preceding digits
as that of
the previous residue.
[044] FIG. 2A. Amino acid sequence (SEQ ID NO:13) of the chimeric PAM4 (cPAM4)
Vk. The sequences are given as one letter codes. The amino acid residues in
the CDR
regions are shown in bold and underlined. Kabat's Ig molecule number scheme is
used to
number the residues.
[045] FIG. 2B. Amino acid sequence (SEQ ID NO:14) of the cPAM4 VH. The
sequences
are given as one letter codes. The amino acid residues in the CDR regions are
shown in bold
and underlined. Kabat's Ig molecule number scheme is used to number the
residues.
[046] FIG. 3A. Alignment of the Vic amino acid sequences of the human antibody
Walker
(SEQ ID NO:15) with PAM4 (SEQ ID NO:10) and hPAM4 (SEQ ID NO:16). Dots
indicate
the residues of PAM4 that are identical to the corresponding residues of the
human or
humanized antibodies. Boxed regions represent the CDR regions. Both N- and C-
terminal
residues (underlined) of hPAM4 are fixed by the staging vectors used. Kabat's
Ig molecule
number scheme is used to number the residues.
14
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
[047] FIG. 3B. Alignment of the VH amino acid sequences of the human antibody
Wi12
(FR1-3) (SEQ ID NO:17) and NEWM (FR4) (SEQ ID NO:1 8) with PAM4 (SEQ ID NO:12)
and hPAM4 (SEQ ID NO:1 9). Dots indicate the residues of PAM4 that are
identical to the
corresponding residues of the human or humanized antibodies. Boxed regions
represent the
CDR regions. Both N- and C-terminal residues (underlined) of hPAM4 are fixed
by the
staging vectors used. Kabat's Ig molecule number scheme is used to number the
residues.
[048] FIG. 4A. DNA (SEQ ID NO:20) and amino acid (SEQ ID NO:1 6) sequences of
the
humanized PAM4 (hPAM4) Vk. Numbering of the nucleotide sequence is on the
right side.
Amino acid sequences encoded by the corresponding DNA sequences are given as
one-letter
codes. The amino acid residues in the CDR regions are shown in bold and
underlined.
Kabat's Ig molecule numbering scheme is used for amino acid residues.
[049] FIG. 4B. DNA (SEQ ID NO:21) and amino acid (SEQ ID NO:1 9) sequences of
the
hPAM4 VH. Numbering of the nucleotide sequence is on the right side. Amino
acid
sequences encoded by the corresponding DNA sequences are given as one-letter
codes. The
amino acid residues in the CDR regions are shown in bold and underlined.
Kabat's Ig
molecule numbering scheme is used for amino acid residues.
[050] FIG. 5. Binding activity of humanized PAM4 antibody, hPAM4, as compared
to the
chimeric PAM4, cPAM4. hPAM4 is shown by diamonds and cPAM4 is shown by closed
circles. Results indicate comparable binding activity of the hPAM4 antibody
and cPAM4
when competing with 125I-cPAM4 binding to CaPanl antigens.
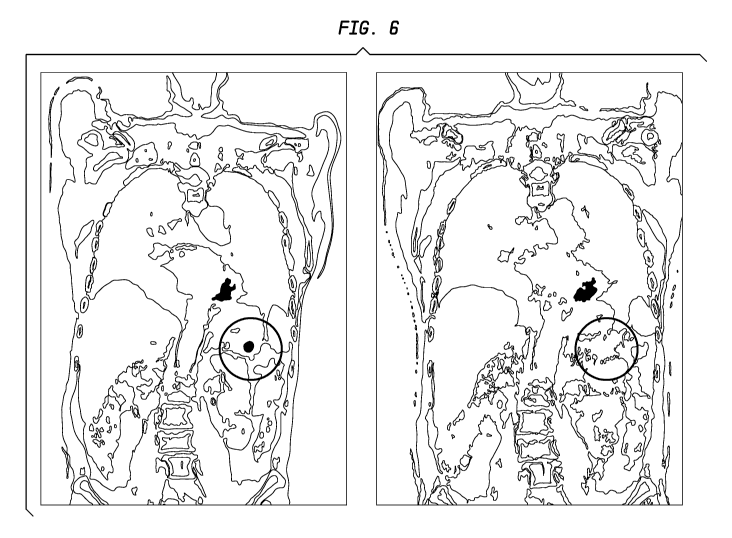
[051] FIG. 6. PET/CT fusion images for a patient with inoperable metastatic
pancreatic
cancer treated with fractionated 9 Y-hPAM4 plus gemcitabine, before therapy
(left side) and
post-therapy (right side). The circle indicates the location of the primary
lesion, which shows
a significant decrease in PET/CT intensity following therapy.
[052] FIG. 7. 3D PET images for a patient with inoperable metastatic
pancreatic cancer
treated with fractionated 90Y-hPAM4 plus gemcitabine, before therapy (left
side) and post-
therapy (right side). Arrows point to the locations of the primary lesion (on
right) and
metastases (on left), each of which shows a significant decrease in PET image
intensity after
therapy with radiolabeled hPAM4 plus gemcitabine.
[053] FIG. 8A. In vivo imaging of tumors using an 1111n-labeled diHSG peptide
(IMP 288)
with or without pretargeting TF10 bispecific anti-pancreatic cancer MUC5ac
antibody. FIG.
8A illustrates mice showing the location of tumors (arrow).
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
[054] FIG. 8B In vivo imaging of tumors using an 1111n-labeled diHSG peptide
(IMP 288)
with or without pretargeting TF10 bispecific anti-pancreatic cancer MUC5ac
antibody. FIG.
8B shows the detected tumors with 1111n-labeled IMP 288 in the presence
(above) or absence
(below) of TF10 bispecific antibody.
[055] FIG. 9. Exemplary binding curves for TF10, PAM4-IgG, PAM4-F(ab')2 and a
monovalent bsPAM4 chemical conjugate (PAM4-Fab' x anti-DTPA-Fab'). Binding to
target
mucin antigen was measured by ELISA assay.
[056] FIG. 10A. Immunoscintigraphy of CaPanl human pancreatic cancer
xenografts (-
0.25 g). An image of mice that were injected with bispecific TF10 (80 g, 5.07
x 10-1 mol)
followed 16 h later by administration of111In-IMP-288 (30 Ci, 5.07 x 10-11
mol). The image
was taken 3 h later. The intensity of the image background was increased to
match the
intensity of the image obtained when 1111n-IMP-288 was administered alone (30
Ci, 5.07 x
10-11 mol).
[057] FIG. 10B. Immunoscintigraphy of CaPanl human pancreatic cancer
xenografts
0.25 g). No targeting was observed in mice given 1111n-IMP-288 alone.
[058] FIG. 10C. Immunoscintigraphy of CaPanl human pancreatic cancer
xenografts (-
0.25 g). An image of mice that were given 1111n-DOTA-PAM4-IgG (20 Ci, 50 lug)
with
imaging done 24 h later. Although tumors are visible, considerable background
activity is
still present at this time point.
[059] FIG. 11A. Extended biodistribution of111In-DOTA-PAM4-IgG (20 Ci, 50 g)
and
TF10-pretargeted1111n-IMP-288 (80 g, 5.07 x 10-10 mol TF10 followed 16 h
later with 30
Ci, 5.07 x 10-11 mol 1111n-IMP-288) in nude mice bearing CaPanl human
pancreatic cancer
xenografts (mean tumor weight +/- SD, 0.28 +/- 0.21 and 0.10 +/- 0.06 g for
the pretargeting
and IgG groups of animals, respectively) . FIG. 11A shows percent of initial
dose per gram
of tissue in tumor with PAM4 IgG (open circles), blood with PAM4 IgG (open
squares),
tumor with pretargeted peptide (closed circles) and blood with pretargeted
peptide (closed
squares).
[060] FIG. 11B. Extended biodistribution of 1111n-DOTA-PAM4-IgG (20 Ci, 50
g) and
TF10-pretargeted1111n-IMP-288 (80 g, 5.07 x 10-10 mol TF10 followed 16 h
later with 30
Ci, 5.07 x 10-11 mol 1111n-IMP-288) in nude mice bearing CaPanl human
pancreatic cancer
xenografts (mean tumor weight +/- SD, 0.28 +/- 0.21 and 0.10 +/- 0.06 g for
the pretargeting
and IgG groups of animals, respectively). FIG. 11B shows percent of initial
dose per per
gram of tissue in liver with PAM4 IgG (open triangles), kidney with PAM4 IgG
(open
16
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
diamonds), liver with pretargeted peptide (closed triangles) and kidney with
pretargeted
peptide (closed diamonds).
[061] FIG. 11C. Extended biodistribution of 1111n-DOTA-PAM4-IgG (20 Ci, 50
g) and
TF10-pretargeted 1111n-IMP-288 (80 g, 5.07 x 1040 mol TF10 followed 16 h
later with 30
Ci, 5.07 x 10-11 mol 1111n-IMP-288) in nude mice bearing CaPanl human
pancreatic cancer
xenografts (mean tumor weight +/- SD, 0.28 +/- 0.21 and 0.10 +/- 0.06 g for
the pretargeting
and IgG groups of animals, respectively). FIG. 11C shows microcuries per gram
of tissue in
tumor with PAM4 IgG (open circles), blood with PAM4 IgG (open squares), tumor
with
pretargeted peptide (closed circles) and blood with pretargeted peptide
(closed squares).
[062] FIG. 11D. Extended biodistribution of 1111n-DOTA-PAM4-IgG (20 Ci, 50
g) and
TF10-pretargeted 1111n-IMP-288 (80 g, 5.07 x 1040 mol TF10 followed 16 h
later with 30
Ci, 5.07 x 10-11 mol 1111n-IMP-288) in nude mice bearing CaPanl human
pancreatic cancer
xenografts (mean tumor weight +/- SD, 0.28 +/- 0.21 and 0.10 +/- 0.06 g for
the pretargeting
and IgG groups of animals, respectively). FIG. 11D shows microcuries per per
gram of
tissue in liver with PAM4 IgG (open triangles), kidney with PAM4 IgG (open
diamonds),
liver with pretargeted peptide (closed triangles) and kidney with pretargeted
peptide (closed
diamonds).
[063] FIG. 12. Therapeutic activity of a single treatment of established (-0.4
cm3) CaPanl
tumors with 0.15 mCi of 90Y-hPAM4 IgG, or 0.25 or 0.50 mCi of TF10-pretargeted
9 Y-IMP-
288.
[064] FIG. 13. Effect of gemcitabine potentiation of PT-RAIT therapy.
[065] FIG. 14. Effect of combination of cetuximab with gemcitabine and PT-
RAIT.
[066] FIG. 15. Differential diagnosis of pancreatic cancer using PAM4-based
immunoassay. The horizontal line shows the cutoff level selected for a
positive result, based
on ROC analysis.
[067] FIG. 16. Frequency distribution of PAM4 antigen in patient sera from
healthy
volunteers and individuals with varying stages of pancreatic cancer.
[068] FIG. 17. ROC curve for PAM4 serum immunoassay, showing sensitivity for
detection of 81.6% and specificity of 84.6%.
[069] FIG. 18. Accuracy of the PAM4-immunoassay was determined to be within
10% of
the nominal concentrations examined at or above the cutoff value of 2.40
units/mL. A linear
trend was calculated with an equation of y = 0.965x + 0.174, and goodness of
fit r2 = 0.999.
17
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
[070] FIG. 19. Frequency distribution of PAM4-reactive antigen in patient sera
by stage of
disease. Cutoff value = 2.4 units/mL (horizontal line). The median values
(units/mL) are
shown for each study group.
[071] FIG. 20. Receiver Operator Characteristics (ROC) curve for the
performance of the
PAM4-based immunoassay; pancreatic adenocarcinoma vs. healthy adults. Values
for the
area under the curves (AUC) and 95% confidence limits are provided.
[072] FIG. 21A. Circulating PAM4 antigen levels correlated with
progression/regression of
tumor volume (CT) following treatment with 90Y-PAM4-IgG plus gemcitabine.
Patient 076-
001 was responsive to therapy and serum PAM4 antigen decreased. Serum PAM4
levels
correlated with tumor volume.
[073] FIG. 21B. Circulating PAM4 antigen levels correlated with
progression/regression of
tumor volume (CT) following treatment with 90Y-PAM4-IgG plus gemcitabine.
Patient
1810002 showed an initial response to therapy, followed by recurrence of the
tumor. Serum
PAM4 levels correlated with tumor volume.
[074] FIG. 22. Reactivity of PAM4 with mucin standards in the presence or
absence of
palmitic acid.
[075] FIG. 23A. Sensitivity and specificity for PAM4 detection of PDAC vs.
chronic
pancreatitis (CP).
[076] FIG. 23B. Sensitivity and specificity for PAM4 detection of PDAC vs. all
benign
tissue samples.
[077] FIG. 24. Comparative labeling of PDAC vs. non-neoplastic prostate tissue
with
PAM4 vs. antibodies against MUC1, MUC4, CEACAM6 and CA19-9.
[078] FIG. 25. Reactivity of several anti-mucin MAbs with a high molecular
weight mucin
containing fraction (CPM1) isolated from the Capan-1, human pancreatic
adenocarcinoma.
MAbs are identified by clone name with reactive species of mucin indicated by
horizontal
bars beneath MAb clone names. In addition to PAM4, substantial reactions were
observed for
anti-MUC1, MUC5ac, and CEACAM6 antibodies. All MAbs were employed at a
constant
pg/mL.
[079] FIG. 26. Reaction of several anti-mucin MAbs with PAM4-captured antigen.
Mucin
antigens were captured on hPAM4 coated plates, and then probed with several
murine anti-
mucin MAbs for reaction signal. Both anti-MUC5ac MAbs (2-11M1 and 45M1) bound
to
the hPAM4-captured mucin, whereas the anti-MUC1 MAbs (MA5 and KC4) did not
bind.
The homologous hPAM4/mPAM4, capture/probe immunoassay gave no signal,
suggesting
18
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
the density of PAM4 epitopes within the mucin may be low, possibly only a
single site. A
rabbit polyclonal anti-CPM1, IgG, was used as a positive control for reaction
with hPAM4-
captured antigen.
[080] FIG. 27A. Inhibition of hPAM4/antigen binding reaction by murine anti-
mucin
MAbs. Anti-mucin mMAbs (purified IgG) were added to CPM1-coated plates as
potential
inhibitors prior to addition of hPAM4. mPAM4 provided almost complete
inhibition of the
reaction between hPAM4 and antigen with the 45M1 anti-MUC5ac providing limited
inhibitory affect (ICn,ax = 25.5%). Neither 2-11M1, anti-MUC5ac nor MA5 and
KC4, anti-
MUC1 MAbs were able to inhibit the specific hPAM4/antigen reaction.
[081] FIG. 27B. Inhibition of hPAM4/antigen binding reaction by murine anti-
mucin
MAbs. A similar inhibition study was performed with several anti-MUC5ac MAbs
obtained
as ascites fluids. MAbs 21M1, 62M1, and 463M1, anti-MUC5ac provided
substantial
inhibitory affect similar to that observed with mPAM4, IgG, self-inhibition.
The ascites form
of 45M1 yielded an inhibitory affect similar to that of the purified IgG.
Ascites containing
anti-alpha fetoprotein was employed as a negative control.
[082] FIG. 28. Representation of the domains of the MUC5ac glycoprotein with
reactive
epitopes indicated for several anti-MUC5ac MAbs. Data derived by transfection
with plasmid
vectors containing the cDNA of the 3'-end of MUC5ac, along with derivative
cDNA vectors
obtained by restriction enzyme digestion, have identified the location of
specific epitopes for
anti-MUC5ac MAbs employed in the current studies. Specific blocking studies
suggest the
PAM4-epitope resides within the cysteine-rich C-terminus domain.
DETAILED DESCRIPTION
Definitions
[083] Unless otherwise specified, "a" or "an" means one or more.
[084] As used herein, "about" means plus or minus 10%. For example, "about
100" would
include any number between 90 and 110.
[085] As described herein, the term "PAM4 antibody" includes murine, chimeric,
humanized and human PAM4 antibodies. In preferred embodiments, the PAM4
antibody or
antigen-binding fragment thereof comprises the CDR sequences of SEQ ID NO:1 to
SEQ ID
NO:6.
[086] As used herein, an "anti-pancreatic cancer antibody" is an antibody that
exhibits the
same diagnostic, therapeutic and binding characteristics as the PAM4 antibody.
In preferred
19
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
embodiments, the "anti-pancreatic cancer antibody" binds to an epitope located
within the
second to fourth cysteine-rich domains of MUC5ac (amino acid residues 1575-
2052).
[087] A "non-endocrine pancreatic cancer" generally refers to cancers arising
from the
exocrine pancreatic glands. The term excludes pancreatic insulinomas and
includes
pancreatic carcinoma, pancreatic adenocarcinoma, adenosquamous carcinoma,
squamous cell
carcinoma and giant cell carcinoma and precursor lesions such as pancreatic
intra-epithelial
neoplasia (PanIN), mucinous cyst neoplasms (MCN) and intrapancreatic mucinous
neoplasms (IPMN), which are neoplastic but not yet malignant. The terms
"pancreatic
cancer" and "non-endocrine pancreatic cancer" are used interchangeably herein.
[088] An antibody, as described herein, refers to a full-length (i.e.,
naturally occurring or
formed by normal immunoglobulin gene fragment recombinatorial processes)
immunoglobulin molecule (e.g., an IgG antibody) or an immunologically active
(i.e.,
specifically binding) portion of an immunoglobulin molecule, like an antibody
fragment.
[089] An antibody fragment is a portion of an antibody such as F(ab')2, Fab',
Fab, Fv, sFy
and the like. Regardless of structure, an antibody fragment binds with the
same antigen that
is recognized by the full-length antibody. The term "antibody fragment" also
includes
isolated fragments consisting of the variable regions of antibodies, such as
the "Fv" fragments
consisting of the variable regions of the heavy and light chains and
recombinant single chain
polypeptide molecules in which light and heavy variable regions are connected
by a peptide
linker ("scFy proteins"). Another form of antibody fragment is a single domain
antibody
(nanobody).
[090] A naked antibody is an antibody or fragment thereof that is not
conjugated to a
therapeutic or diagnostic agent. Generally, the Fc portion of the antibody
molecule provides
effector functions, such as complement-mediated cytotoxicity (CDC) and ADCC
(antibody-
dependent cellular cytotoxicity), which set mechanisms into action that may
result in cell
lysis. However, the Fc portion may not be required for therapeutic function,
with other
mechanisms, such as signaling-induced apoptosis, coming into play. Naked
antibodies
include both polyclonal and monoclonal antibodies, as well as fusion proteins
and certain
recombinant antibodies, such as chimeric, humanized or human antibodies.
[091] A chimeric antibody is a recombinant protein that contains the variable
domains
including the complementarity determining regions (CDRs) of an antibody
derived from one
species, preferably a rodent antibody, while the constant domains of the
antibody molecule
are derived from those of a human antibody. For veterinary applications, the
constant
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
domains of the chimeric antibody may be derived from that of other species,
such as a cat or
dog.
[092] A humanized antibody is a recombinant protein in which the CDRs from an
antibody
from one species; e.g., a rodent antibody, are transferred from the heavy and
light variable
chains of the rodent antibody into human heavy and light variable domains
(e.g., framework
region sequences). The constant domains of the antibody molecule are derived
from those of
a human antibody. In certain embodiments, a limited number of framework region
amino
acid residues from the parent (rodent) antibody may be substituted into the
human antibody
framework region sequences.
[093] A human antibody is, e.g., an antibody obtained from transgenic mice
that have been
"engineered" to produce specific human antibodies in response to antigenic
challenge. In this
technique, elements of the human heavy and light chain loci are introduced
into strains of
mice derived from embryonic stem cell lines that contain targeted disruptions
of the
endogenous murine heavy chain and light chain loci. The transgenic mice can
synthesize
human antibodies specific for particular antigens, and the mice can be used to
produce human
antibody-secreting hybridomas. Methods for obtaining human antibodies from
transgenic
mice are described by Green et al., Nature Genet. 7:13 (1994), Lonberg et al.,
Nature 368:856
(1994), and Taylor et al., Int. Immun. 6:579 (1994). A fully human antibody
also can be
constructed by genetic or chromosomal transfection methods, as well as phage
display
technology, all of which are known in the art. See for example, McCafferty et
al., Nature
348:552-553 (1990) for the production of human antibodies and fragments
thereof in vitro,
from immunoglobulin variable domain gene repertoires from unimmunized donors.
In this
technique, antibody variable domain genes are cloned in-frame into either a
major or minor
coat protein gene of a filamentous bacteriophage, and displayed as functional
antibody
fragments on the surface of the phage particle. Because the filamentous
particle contains a
single-stranded DNA copy of the phage genome, selections based on the
functional properties
of the antibody also result in selection of the gene encoding the antibody
exhibiting those
properties. In this way, the phage mimics some of the properties of the B
cell. Phage display
can be performed in a variety of formats, for review, see e.g. Johnson and
Chiswell, Current
Opinion in Structural Biology 3:5564-571 (1993). Human antibodies may also be
generated
by in vitro activated B cells. See U.S. Pat. Nos. 5,567,610 and 5,229,275, the
Examples
sections of which are incorporated herein by reference.
21
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
[094] A therapeutic agent is a compound, molecule or atom which is
administered
separately, concurrently or sequentially with an antibody moiety or conjugated
to an antibody
moiety, i.e., antibody or antibody fragment, or a subfragment, and is useful
in the treatment
of a disease. Examples of therapeutic agents include antibodies, antibody
fragments,
cytotoxic agents, drugs, toxins, nucleases, hormones, immunomodulators, pro-
apoptotic
agents, anti-angiogenic agents, boron compounds, photoactive agents or dyes
and
radioisotopes. Therapeutic agents of use are described in more detail below.
[095] A diagnostic agent is a molecule, atom or other detectable moiety which
may be
administered conjugated to an antibody moiety or targetable construct and is
useful in
detecting or diagnosing a disease by locating cells containing the target
antigen. Useful
diagnostic agents include, but are not limited to, radioisotopes, dyes,
contrast agents,
fluorescent compounds or molecules and enhancing agents (e.g., paramagnetic
ions) for
magnetic resonance imaging (MRI) or positron emission tomography (PET)
scanning.
Preferably, the diagnostic agents are selected from the group consisting of
radioisotopes,
enhancing agents for use in magnetic resonance or PET imaging, and fluorescent
compounds.
In order to load an antibody component with radioactive metals, paramagnetic
ions or other
diagnostic cations, it may be necessary to react it with a reagent having a
long tail to which
are attached a multiplicity of chelating groups for binding the ions. Such a
tail can be a
polymer such as a polylysine, polysaccharide, or other derivatized or
derivatizable chain
having pendant groups to which can be bound chelating groups such as, e.g.,
ethylenediaminetetraacetic acid (EDTA), diethylenetriaminepentaacetic acid
(DTPA),
DOTA, NOTA, NETA, porphyrins, polyamines, crown ethers, bis-
thiosemicarbazones,
polyoximes, and like groups known to be useful for this purpose. Chelates are
coupled to the
antibodies using standard chemistries. The chelate is normally linked to the
antibody by a
group which enables formation of a bond to the molecule with minimal loss of
immunoreactivity and minimal aggregation and/or internal cross-linking. Other,
more
unusual, methods and reagents for conjugating chelates to antibodies are
disclosed in U.S.
Pat. No. 4,824,659, the Examples section of which is incorporated herein by
reference.
Particularly useful metal-chelate combinations include 2-benzyl-DTPA and its
monomethyl
and cyclohexyl analogs, used with diagnostic isotopes in the general energy
range of 60 to
125J, 131J,
123J, 124J,
62 64 18F, 1111n,
67 68 99m 94m 11C,
13N, 4,000 keV, such as I, I, I, I, Cu, Cu, F, In, Ga, Ga, Tc, Tc, C, N,
150, 76Br, for radioimaging. The same chelates, when complexed with non-
radioactive
metals, such as manganese, iron and gadolinium are useful for MRI, when used
along with
22
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
the antibodies of the invention. Macrocyclic chelates such as NOTA (1,4,7-
triazacyclononane-N,N',N"-triacetic acid), DOTA (1,4,7,10-
tetraazacyclododecanetetraacetic
acid), and TETA (p-bromoacetamido-benzyl-tetraethylaminetetraacetic acid) are
of use with
a variety of metals and radiometals, most particularly with radionuclides of
gallium, yttrium
and copper, respectively. Such metal-chelate complexes can be made very stable
by tailoring
the ring size to the metal of interest. Other ring-type chelates such as
macrocyclic polyethers,
which are of interest for stably binding nuclides, such as 223Ra for
radioimmunotherapy
(RAIT) are encompassed by the invention. More recently, techniques of general
utility for
labeling virtually any molecule with an 18F atom of use in PET imaging have
been described
in U.S. Patent Nos. 7,563,433; 7,597,876 and 7,993,626, the Examples section
of each
incorporated herein by reference.
[096] An immunoconjugate is an antibody, antibody fragment or antibody fusion
protein
conjugated to at least one therapeutic and/or diagnostic agent. The diagnostic
agent and/or
therapeutic agent are as defined above.
[097] An expression vector is a DNA molecule comprising a gene that is
expressed in a
host cell. Typically, gene expression is placed under the control of certain
regulatory
elements, including constitutive or inducible promoters, tissue-specific
regulatory elements
and enhancers. Such a gene is said to be "operably linked to" the regulatory
elements.
[098] A recombinant host may be any prokaryotic or eukaryotic cell that
contains either a
cloning vector or expression vector. This term also includes those prokaryotic
or eukaryotic
cells, as well as transgenic animals, that have been genetically engineered to
contain the
cloned gene(s) in the chromosome or genome of the host cell. Suitable
mammalian host cells
include myeloma cells, such as 5P2/0 cells, and NSO cells, as well as Chinese
Hamster Ovary
(CHO) cells, hybridoma cell lines and other mammalian host cell useful for
expressing
antibodies. Also particularly useful to express mAbs and other fusion proteins
are Sp2/0 cells
transfected with an apoptosis inhibitor, such as a Bcl-EEE gene, and adapted
to grow and be
further transfected in serum free conditions, as described in U.S. Patent Nos.
7,531,327;
7,537,930; and 7,608,425, the Examples section of each of which is
incorporated herein by
reference.
PAM4 Antibodies
[099] Various embodiments of the invention concern antibodies that react with
very high
selectivity with pancreatic cancer as opposed to normal or benign pancreatic
tissues. The
anti-pancreatic cancer antibodies and fragments thereof are preferably raised
against a crude
23
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
mucin preparation from a tumor of the human pancreas, although partially
purified or even
purified MUC5ac may be utilized. A non-limiting example of such antibodies is
the PAM4
antibody.
[0100] The murine PAM4 (mPAM4) antibody was developed by employing pancreatic
cancer mucin derived from the xenografted RIP-1 human pancreatic carcinoma as
immunogen. (Gold et al., Int. J. Cancer, 57:204-210, 1994.) As discussed
below, antibody
cross-reactivity and immunohistochemical staining studies indicate that the
PAM4 antibody
recognizes a unique and novel epitope on MUC5ac. Immunohistochemical staining
studies,
such as those described in Example 2, have shown that the PAM4 MAb binds to an
antigen
expressed by breast, pancreas and other cancer cells, with limited binding to
normal human
tissue. However, the highest expression is usually by pancreatic cancer cells.
Thus, the
PAM4 antibodies are relatively specific to pancreatic cancer and
preferentially bind
pancreatic cancer cells. The PAM4 antibody is reactive with a target epitope
which can be
internalized. This epitope is expressed primarily by antigens associated with
pancreatic
cancer and not with focal pancreatitis or normal pancreatic tissue. Binding of
PAM4
antibody to the PAM4 epitope is inhibited by treatment of the antigen with DTT
or periodate.
Localization and therapy studies using a radiolabeled PAM4 MAb in animal
models have
demonstrated tumor targeting and therapeutic efficacy.
[0101] The PAM4 antibodies bind to an epitope of MUC5ac (located within the
second to
fourth cysteine-rich domains), which is expressed by many organs and tumor
types, but is
preferentially expressed in pancreatic cancer cells. Studies with a PAM4 MAb,
such as in
Example 3, indicate that the antibody exhibits several important properties,
which make it a
good candidate for clinical diagnostic and therapeutic applications. The
epitope provides a
useful target for diagnosis and therapy of pancreatic and other cancers. The
PAM4 antibody
apparently recognizes an epitope of MUC5ac that is distinct from the epitopes
recognized by
non-PAM4 anti-pancreatic cancer antibodies (e.g., CA19.9, DUPAN2, SPAN1, Nd2,
CEACAM5, B72.3, anti-Lea, and other anti-Lewis antigens).
[0102] Surprisingly, the Examples below indicate that the MUC5ac epitope to
which PAM4
binds is present in detectable concentrations in serum of patients with very
early stage
pancreatic cancer. Also surprisingly, it appears that an endogenous inhibitor
of PAM4
antibody binding to MUC5ac is present in fresh human serum. The inhibitor is
removed by
long-term frozen storage of serum samples, or by organic phase extraction of
fresh serum.
These unexpected results provide the basis of a relatively non-invasive, early
detection test
24
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
for pancreatic cancer, using blood, serum or plasma samples. In alternative
embodiments, the
PAM4 antibody may be used alone, or else in conjunction with one or more other
antibodies,
such as CA19.9 antibody, to detect pancreatic cancer markers in serum.
[0103] For therapeutic use, antibodies suitable for use in combination or
conjunction with
PAM4 antibodies include, for example, the antibodies CA19.9, DUPAN2, SPAN1,
Nd2,
B72.3, CC49, anti-CEA, anti-CEACAM6, anti-Lea, anti-HLA-DR, anti-CD40, anti-
CD74,
anti-CD138, and antibodies defined by the Lewis antigen Le(y), or antibodies
against colon-
specific antigen-p (CSAp), MUC1, MUC2, MUC3, MUC4, MUC5ac, MUC16, MUC17,
EGP-1, EGP-2, HER2/neu, EGFR, angiogenesis factors (e.g., VEGF and P1GF),
insulin-like
growth factor (IGF), tenascin, platelet-derived growth factor, and IL-6, as
well as products of
oncogenes (bc1-2, Kras, p53), cMET, and antibodies against tumor necrosis
substances, such
as described in patents by Epstein et al. (U.S. Pat. Nos. 6,071,491,
6.017,514, 5,019,368 and
5,882,626). Such antibodies would be useful for complementing PAM4 antibody
immunodetection and immunotherapy methods. These and other therapeutic agents
could act
synergistically with anti-pancreatic cancer antibodies, such as PAM4 antibody,
when
administered before, together with or after administration of PAM4 antibody.
[0104] In therapeutic applications, antibodies that are agonistic or
antagonistic to
immunomodulators involved in effector cell function against tumor cells could
also be useful
in combination with PAM4 antibodies alone or in combination with other tumor-
associated
antibodies, one example being antibodies against CD40. Todryk et al., J.
Immunol Methods,
248:139-147 (2001); Turner et al., J. Immunol, 166:89-94 (2001). Also of use
are antibodies
against markers or products of oncogenes (e.g., bc1-2, Kras, p53, cMET), or
antibodies
against angiogenesis factors, such as VEGFR and placenta-like growth factor
(P1GF).
[0105] The availability of another PAM4-like antibody that binds to a
different epitope of
MUC5ac is important for the development of a double-determinant enzyme-linked
immunosorbent assay (ELISA), of use for MUC5ac in clinical samples. ELISA
experiments
are described in Example 1 and 5.
[0106] The murine, chimeric, humanized and fully human PAM4 antibodies and
fragments
thereof described herein are exemplary of anti-pancreatic cancer antibodies of
use for
diagnostic and/or therapeutic methods. The Examples below disclose a preferred
embodiment of the construction and use of a humanized PAM4 antibody. Because
non-
human monoclonal antibodies can be recognized by the human host as a foreign
protein, and
repeated injections can lead to harmful hypersensitivity reactions,
humanization of a murine
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
antibody sequences can reduce the adverse immune response that patients may
experience.
For murine-based monoclonal antibodies, this is often referred to as a Human
Anti-Mouse
Antibody (HAMA) response. Preferably some human residues in the framework
regions of
the humanized anti-pancreatic cancer antibody or fragments thereof are
replaced by their
murine counterparts. It is also preferred that a combination of framework
sequences from
two different human antibodies is used for VH. The constant domains of the
antibody
molecule are derived from those of a human antibody.
Antibody Preparation
[0107] Monoclonal antibodies for specific antigens may be obtained by methods
known to
those skilled in the art. See, for example, Kohler and Milstein, Nature 256:
495 (1975), and
Coligan et al. (eds.), CURRENT PROTOCOLS IN IMMUNOLOGY, VOL. 1, pages 2.5.1-
2.6.7 (John Wiley & Sons 1991) (hereinafter "Coligan"). Briefly, anti-
pancreatic cancer
MAbs can be obtained by injecting mice with a composition comprising a mixture
of
pancreatic cancer mucins comprising MUC5ac, or a purified MUC5ac, or a peptide
or protein
corresponding to an epitope located within the second to fourth cysteine-rich
domains of
MUC5ac (amino acid residues 1575-2052), verifying the presence of antibody
production by
removing a serum sample, removing the spleen to obtain B-lymphocytes, fusing
the B-
lymphocytes with myeloma cells to produce hybridomas, cloning the hybridomas,
selecting
positive clones which produce antibodies to MUC5ac, culturing the clones that
produce
antibodies to an epitope located within the second to fourth cysteine-rich
domains of
MUC5ac (amino acid residues 1575-2052), and isolating anti-pancreatic cancer
antibodies
from the hybridoma cultures.
[0108] After the initial raising of antibodies to the immunogen, the
antibodies can be
sequenced and subsequently prepared by recombinant techniques to produce
chimeric or
humanized antibodies. Chimerization of murine antibodies and antibody
fragments are well
known to those skilled in the art. The use of antibody components derived from
chimerized
monoclonal antibodies reduces potential problems associated with the
immunogenicity of
murine constant regions.
[0109] General techniques for cloning murine immunoglobulin variable domains
are
described, for example, by the publication of Orlandi et al., Proc Nat'l Acad.
Sci. USA 86:
3833 (1989), incorporated herein by reference. In general, the VK (variable
light chain) and
VH (variable heavy chain) sequences for murine antibodies can be obtained by a
variety of
molecular cloning procedures, such as RT-PCR, 5'-RACE, and cDNA library
screening.
26
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
Specifically, the VH and VK sequences of the murine PAM4 MAb were cloned by
PCR
amplification from the hybridoma cells by RT-PCR, and their sequences
determined by DNA
sequencing. To confirm their authenticity, the cloned VK and VH genes can be
expressed in
cell culture as a chimeric Ab as described by Orlandi et al., (Proc Natl.
Acad. Sci., USA, 86:
3833, 1989).
[0110] In a preferred embodiment, a chimerized PAM4 antibody or antibody
fragment
comprises the complementarity-determining regions (CDRs) and framework regions
(FR) of
a murine PAM4 MAb and the light and heavy chain constant regions of a human
antibody,
wherein the CDRs of the light chain variable region of the chimerized PAM4
comprises
CDR1 (SASSSVSSSYLY, SEQ ID NO:1); CDR2 (STSNLAS, SEQ ID NO:2); and CDR3
(HQWNRYPYT, SEQ ID NO:3); and the CDRs of the heavy chain variable region of
the
chimerized PAM4 MAb comprises CDR1 (SYVLH, SEQ ID NO:4); CDR2
(YINPYNDGTQYNEKFKG, SEQ ID NO:5) and CDR3 (GFGGSYGFAY, SEQ ID NO:6).
The use of antibody components derived from chimerized monoclonal antibodies
reduces
potential problems associated with the immunogenicity of murine constant
regions.
[0111] Humanization of murine antibodies and antibody fragments is also well
known to
those skilled in the art. Techniques for producing humanized MAbs are
disclosed, for
example, by Carter et al., Proc Nat'l Acad. Sci. USA 89: 4285 (1992), Singer
et al., J. Immun.
150: 2844 (1992), Mountain et al. Biotechnol Genet Eng Rev. 10:1 (1992), and
Coligan at
pages 10.19.1-10.19.11, each of which is incorporated herein by reference. For
example,
humanized monoclonal antibodies may be produced by transferring murine
complementary
determining regions from heavy and light variable chains of the mouse
immunoglobulin into
a human variable domain, and then substituting selected human residues in the
framework
regions with their the murine FR counterparts. The use of human framework
region
sequences, in addition to human constant region sequences, further reduces the
chance of
inducing HAMA reactions.
[0112] Humanized antibodies can be designed and constructed as described by
Leung et al.
(Mol Immunol. 32: 1413 (1995)). Example 1 describes the humanization process
utilized for
construction of the hPAM4 MAb.
[0113] The nucleotide sequences of the primers used to prepare the hPAM4
antibodies are
discussed in Example 1, below. In a preferred embodiment, a humanized PAM4
antibody or
antibody fragment comprises the light and heavy chain CDR sequences (SEQ ID
NO:1 to
SEQ ID NO:6) disclosed above. Also preferred, the FRs of the light and heavy
chain variable
27
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
regions of the humanized antibody comprise at least one amino acid substituted
from said
corresponding FRs of the murine PAM4 MAb.
[0114] A fully human antibody, e.g., human PAM4 can be obtained from a
transgenic non-
human animal. See, e.g., Mendez et al., Nature Genetics, 15: 146-156 (1997);
U.S. Pat. No.
5,633,425. For example, a human antibody can be recovered from a transgenic
mouse
possessing human immunoglobulin loci. The mouse humoral immune system is
humanized
by inactivating the endogenous immunoglobulin genes and introducing human
immunoglobulin loci. The human immunoglobulin loci are exceedingly complex and
comprise a large number of discrete segments which together occupy almost 0.2%
of the
human genome. To ensure that transgenic mice are capable of producing adequate
repertoires of antibodies, large portions of human heavy- and light-chain loci
must be
introduced into the mouse genome. This is accomplished in a stepwise process
beginning
with the formation of yeast artificial chromosomes (YACs) containing either
human heavy-
or light-chain immunoglobulin loci in germline configuration. Since each
insert is
approximately 1 Mb in size, YAC construction requires homologous recombination
of
overlapping fragments of the immunoglobulin loci. The two YACs, one containing
the
heavy-chain loci and one containing the light-chain loci, are introduced
separately into mice
via fusion of YAC-containing yeast spheroblasts with mouse embryonic stem
cells.
Embryonic stem cell clones are then microinjected into mouse blastocysts.
Resulting
chimeric males are screened for their ability to transmit the YAC through
their germline and
are bred with mice deficient in murine antibody production. Breeding the two
transgenic
strains, one containing the human heavy-chain loci and the other containing
the human light-
chain loci, creates progeny which produce human antibodies in response to
immunization.
However, these techniques are not limiting and other methods known in the art
for producing
human antibodies, such as the use of phage display, may also be utilized to
produce human
anti-pancreatic cancer antibodies.
[0115] Antibodies can be produced by cell culture techniques using methods
known in the
art. In one example transfectoma cultures are adapted to serum-free medium.
For production
of humanized antibody, cells may be grown as a 500 ml culture in roller
bottles using HSFM.
Cultures are centrifuged and the supernatant filtered through a 0.2 um
membrane. The
filtered medium is passed through a protein-A column (1 x 3 cm) at a flow rate
of 1 ml/min.
The resin is then washed with about 10 column volumes of PBS and protein A-
bound
antibody is eluted from the column with 0.1 M glycine buffer (pH 3.5)
containing 10 mM
28
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
EDTA. Fractions of 1.0 ml are collected in tubes containing 10 IA of 3 M Tris
(pH 8.6), and
protein concentrations determined from the absorbance at 280/260 nm. Peak
fractions are
pooled, dialyzed against PBS, and the antibody concentrated, for example, with
a Centricon
30 filter (Amicon, Beverly, MA). The antibody concentration is determined by
ELISA and
its concentration adjusted to about 1 mg/ml using PBS. Sodium azide, 0.01%
(w/v), is
conveniently added to the sample as preservative.
[0116] Antibodies can be isolated and purified from hybridoma cultures by a
variety of well-
established techniques. Such isolation techniques include affinity
chromatography with
Protein-A Sepharose, size-exclusion chromatography, and ion-exchange
chromatography.
See, for example, Coligan at pages 2.7.1-2.7.12 and pages 2.9.1-2.9.3. Also,
see Baines et
al., "Purification of Immunoglobulin G (IgG)," in METHODS IN MOLECULAR
BIOLOGY, VOL. 10, pages 79-104 (The Humana Press, Inc. 1992).
[0117] Anti-pancreatic cancer MAbs can be characterized by a variety of
techniques that are
well-known to those of skill in the art. For example, the ability of an
antibody to bind to an
epitope located within the second to fourth cysteine-rich domains of MUC5ac
(amino acid
residues 1575-2052) can be verified using an indirect enzyme immunoassay, flow
cytometry
analysis, ELISA or Western blot analysis.
Antibody Fragments
[0118] Antibody fragments are antigen binding portions of an antibody, such as
F(ab') 2, Fab',
F(ab)2, Fab, Fv, sFy, scFy and the like. Antibody fragments which recognize
specific
epitopes can be generated by known techniques. F(ab')2 fragments, for example,
can be
produced by pepsin digestion of the antibody molecule. These and other methods
are
described, for example, by Goldenberg, U.S. Pat. Nos. 4,036,945 and 4,331,647
and
references contained therein. Also, see Nisonoff et al., Arch Biochem.
Biophys. 89: 230
(1960); Porter, Biochem. J. 73: 119 (1959), Edelman et al., in METHODS IN
ENZYMOLOGY VOL. 1, page 422 (Academic Press 1967), and Coligan at pages 2.8.1-
2.8.10 and 2.10.-2.10.4. Alternatively, Fab' expression libraries can be
constructed (Huse et
al., 1989, Science, 246:1274-1281) to allow rapid and easy identification of
monoclonal Fab'
fragments with the desired specificity.
[0119] A single chain Fy molecule (scFv) comprises a VL domain and a VH
domain. The VL
and VH domains associate to form a target binding site. These two domains are
further
covalently linked by a peptide linker (L). A scFy molecule is denoted as
either VL-L-VH if
the VL domain is the N-terminal part of the scFy molecule, or as VH-L-VL if
the VH domain is
29
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
the N-terminal part of the scFy molecule. Methods for making scFy molecules
and designing
suitable peptide linkers are described in U.S. Pat. No. 4,704,692, U.S. Pat.
No. 4,946,778, R.
Raag and M. Whitlow, "Single Chain Fvs." FASEB Vol 9:73-80 (1995) and R. E.
Bird and B.
W. Walker, Single Chain Antibody Variable Regions, TIBTECH, Vol 9: 132-137
(1991).
[0120] Other antibody fragments, for example single domain antibody fragments,
are known
in the art and may be used in the claimed constructs. Single domain antibodies
(VHH) may
be obtained, for example, from camels, alpacas or llamas by standard
immunization
techniques. (See, e.g., Muyldermans et al., TIBS 26:230-235, 2001; Yau et al.,
J Immunol
Methods 281:161-75, 2003; Maass et al., J Immunol Methods 324:13-25, 2007).
The VHH
may have potent antigen-binding capacity and can interact with novel epitopes
that are
inaccessible to conventional VH-VL pairs. (Muyldermans et al., 2001). Alpaca
serum IgG
contains about 50% camelid heavy chain only IgG antibodies (HCAbs) (Maass et
al., 2007).
Alpacas may be immunized with known antigens, such as TNF-a, and VHHs can be
isolated
that bind to and neutralize the target antigen (Maass et al., 2007). PCR
primers that amplify
virtually all alpaca VHH coding sequences have been identified and may be used
to construct
alpaca VHH phage display libraries, which can be used for antibody fragment
isolation by
standard biopanning techniques well known in the art (Maass et al., 2007).
[0121] An antibody fragment can also be prepared by proteolytic hydrolysis of
a full-length
antibody or by expression in E. coli or another host of the DNA coding for the
fragment. An
antibody fragment can be obtained by pepsin or papain digestion of full-length
antibodies by
conventional methods. For example, an antibody fragment can be produced by
enzymatic
cleavage of antibodies with pepsin to provide an approximate 100 Kd fragment
denoted
F(ab')2. This fragment can be further cleaved using a thiol reducing agent,
and optionally a
blocking group for the sulfhydryl groups resulting from cleavage of disulfide
linkages, to
produce an approximate 50 Kd Fab' monovalent fragment. Alternatively, an
enzymatic
cleavage using papain produces two monovalent Fab fragments and an Fc fragment
directly.
[0122] Other methods of cleaving antibodies, such as separation of heavy
chains to form
monovalent light-heavy chain fragments, further cleavage of fragments, or
other enzymatic,
chemical or genetic techniques may also be used, so long as the fragments bind
to the antigen
that is recognized by the intact antibody.
Antibody Fusion Proteins and Multivalent Antibodies
[0123] Fusion proteins comprising the anti-pancreatic cancer antibodies of
interest can be
prepared by a variety of conventional procedures, ranging from glutaraldehyde
linkage to
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
more specific linkages between functional groups. The antibodies and/or
antibody fragments
that comprise the fusion proteins described herein are preferably covalently
bound to one
another, directly or through a linker moiety, through one or more functional
groups on the
antibody or fragment, e.g., amine, carboxyl, phenyl, thiol, or hydroxyl
groups. Various
conventional linkers in addition to glutaraldehyde can be used, e.g.,
diisocyanates,
diiosothiocyanates, bis(hydroxysuccinimide)esters, carbodiimides,
maleimidehydroxy
succinimide esters, and the like.
[0124] A simple method for producing fusion proteins is to mix the antibodies
or fragments
in the presence of glutaraldehyde. The initial Schiff base linkages can be
stabilized, e.g., by
borohydride reduction to secondary amines. A diiosothiocyanate or carbodiimide
can be
used in place of glutaraldehyde as a non-site-specific linker. In one
embodiment, an antibody
fusion protein comprises an anti-pancreatic cancer MAb, or fragment thereof,
wherein the
MAb binds to an epitope located within the second to fourth cysteine-rich
domains of
MUC5ac (amino acid residues 1575-2052). This fusion protein and fragments
thereof
preferentially bind pancreatic cancer cells. This monovalent, monospecific MAb
is useful for
direct targeting of an antigen, where the MAb is attached to a therapeutic
agent, a diagnostic
agent, or a combination thereof, and the protein is administered directly to a
patient.
[0125] The fusion proteins may instead comprise at least two anti-pancreatic
cancer MAbs
that bind to distinct epitopes of MUC5ac. For example, the MAbs can produce
antigen
specific diabodies, triabodies and tetrabodies, which are multivalent but
monospecific to the
MUC5ac. The non-covalent association of two or more scFy molecules can form
functional
diabodies, triabodies and tetrabodies. Monospecific diabodies are homodimers
of the same
scFv, where each scFy comprises the VH domain from the selected antibody
connected by a
short linker to the VL domain of the same antibody. A diabody is a bivalent
dimer formed by
the non-covalent association of two scFvs, yielding two Fy binding sites. A
triabody results
from the formation of a trivalent trimer of three scFvs, yielding three
binding sites, and a
tetrabody is a tetravalent tetramer of four scFvs, resulting in four binding
sites. Several
monospecific diabodies have been made using an expression vector that contains
a
recombinant gene construct comprising VH1-linker-VO. See Holliger et al., Proc
Natl. Acad.
Sci USA 90: 6444-6448 (1993); Atwell et al., Molecular Immunology 33: 1301-
1302 (1996);
Holliger et al., Nature Biotechnology 15: 632-631(1997); Helfrich et al., Int
J Cancer 76:
232-239 (1998); Kipriyanov et al., Int J Cancer 77: 763-772 (1998); Holiger et
al., Cancer
Research 59: 2909-2916(1999)). Methods of constructing scFvs are disclosed in
U.S. Pat.
31
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
No. 4,946,778 (1990) and U.S. Pat. No. 5,132,405 (1992), the Examples section
of each of
which is incorporated herein by reference. Methods of producing multivalent,
monospecific
antibodies based on scFy are disclosed in U.S. Pat. No. 5,837,242 (1998), U.S.
Pat. No.
5,844,094 (1998) and WO-98/44001 (1998), the Examples section of each of which
is
incorporated herein by reference. The multivalent, monospecific antibody
fusion protein
binds to two or more of the same type of epitopes that can be situated on the
same antigen or
on separate antigens. The increased valency allows for additional interaction,
increased
affinity, and longer residence times. These antibody fusion proteins can be
utilized in direct
targeting systems, where the antibody fusion protein is conjugated to a
therapeutic agent, a
diagnostic agent, or a combination thereof, and administered directly to a
patient in need
thereof
[0126] A preferred embodiment is a multivalent, multispecific antibody or
fragment thereof
comprising one or more antigen binding sites having an affinity toward a PAM4
target
epitope and one or more additional binding sites for other epitopes associated
with pancreatic
cancer. This fusion protein is multispecific because it binds at least two
different epitopes,
which can reside on the same or different antigens. For example, the fusion
protein may
comprise more than one antigen binding site, the first with an affinity toward
an epitope
located within the second to fourth cysteine-rich domains of MUC5ac (amino
acid residues
1575-2052) epitope and the second with an affinity toward another target
antigen such as
TAG-72 or CEA. Another example is a bispecific antibody fusion protein which
may
comprise a CA19.9 MAb (or fragment thereof) and a PAM4 MAb (or fragment
thereof).
Such a fusion protein will have an affinity toward CA19.9 as well as MUC5ac.
The antibody
fusion proteins and fragments thereof can be utilized in direct targeting
systems, where the
antibody fusion protein is conjugated to a therapeutic agent, a diagnostic
agent, or a
combination thereof, and administered directly to a patient in need thereof
[0127] Another preferred embodiment is a multivalent, multispecific antibody
comprising at
least one binding site having affinity toward a PAM4 target epitope and at
least one hapten
binding site having affinity towards hapten molecules. For example, a
bispecific fusion
protein may comprise the 679 MAb (or fragment thereof) and the PAM4 MAb (or
fragment
thereof). The monoclonal 679 antibody binds with high affinity to molecules
containing the
tri-peptide moiety histamine succinyl glycyl (HSG). Such a bispecific PAM4
antibody fusion
protein can be prepared, for example, by obtaining a F(ab') 2 fragment from
679, as described
above. The interchain disulfide bridges of the 679 F(ab')2 fragment are gently
reduced with
32
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
DTT, taking care to avoid light-heavy chain linkage, to form Fab'-SH
fragments. The SH
group(s) is (are) activated with an excess of bis-maleimide linker (1,1'-
(methylenedi-4,1-
phenylene)b-is-maleimide). The PAM4 MAb is converted to Fab'-SH and then
reacted with
the activated 679 Fab'-SH fragment to obtain a bispecific antibody fusion
protein. Bispecific
antibody fusion proteins such as this one can be utilized in affinity
enhancing systems, where
the target antigen is pretargeted with the fusion protein and is subsequently
targeted with a
diagnostic or therapeutic agent attached to a carrier moiety (targetable
construct) containing
one or more HSG haptens. In alternative preferred embodiments, a DNLTm-based
hPAM4-
679 construct, such as TF10, may be prepared and used as described in the
Examples below.
[0128] Bispecific antibodies can be made by a variety of conventional methods,
e.g.,
disulfide cleavage and reformation of mixtures of whole IgG or, preferably
F(ab') 2 fragments,
fusions of more than one hybridoma to form polyomas that produce antibodies
having more
than one specificity, and by genetic engineering. Bispecific antibody fusion
proteins have
been prepared by oxidative cleavage of Fab' fragments resulting from reductive
cleavage of
different antibodies. This is advantageously carried out by mixing two
different F(ab')2
fragments produced by pepsin digestion of two different antibodies, reductive
cleavage to
form a mixture of Fab' fragments, followed by oxidative reformation of the
disulfide linkages
to produce a mixture of F(ab') 2 fragments including bispecific antibody
fusion proteins
containing a Fab' portion specific to each of the original epitopes. General
techniques for the
preparation of antibody fusion proteins may be found, for example, in Nisonoff
et al., Arch
Biochem Biophys. 93: 470 (1961), Hammerling et al., J Exp Med. 128: 1461
(1968), and
U.S. Pat. No. 4,331,647.
[0129] More selective linkage can be achieved by using a heterobifunctional
linker such as
maleimidehydroxysuccinimide ester. Reaction of the ester with an antibody or
fragment will
derivatize amine groups on the antibody or fragment, and the derivative can
then be reacted
with, e.g., an antibody Fab fragment having free sulfhydryl groups (or, a
larger fragment or
intact antibody with sulfhydryl groups appended thereto by, e.g., Traut's
Reagent). Such a
linker is less likely to crosslink groups in the same antibody and improves
the selectivity of
the linkage.
[0130] It is advantageous to link the antibodies or fragments at sites remote
from the antigen-
binding sites. This can be accomplished by, e.g., linkage to cleaved
interchain sulfhydryl
groups, as noted above. Another method involves reacting an antibody having an
oxidized
carbohydrate portion with another antibody that has at lease one free amine
function. This
33
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
results in an initial Schiff base linkage, which is preferably stabilized by
reduction to a
secondary amine, e.g., by borohydride reduction, to form the final composite.
Such site-
specific linkages are disclosed, for small molecules, in U.S. Pat. No.
4,671,958, and for larger
addends in U.S. Pat. No. 4,699,784, the Examples section of each of which is
incorporated
herein by reference.
[0131] ScFvs with linkers greater than 12 amino acid residues in length (for
example, 15-or
18-residue linkers) allow interactions between the VH and VL domains on the
same chain and
generally form a mixture of monomers, dimers (termed diabodies) and small
amounts of
higher mass multimers, (Kortt et al., Eur J Biochem. (1994) 221: 151-157).
ScFvs with
linkers of 5 or less amino acid residues, however, prohibit intramolecular
pairing of the VH
and VL domains on the same chain, forcing pairing with VH and VL domains on a
different
chain. Linkers between 3- and 12-residues form predominantly dimers (Atwell et
al., Protein
Engineering (1999) 12: 597-604). With linkers between 0 and 2 residues,
trimeric (termed
triabodies), tetrameric (termed tetrabodies) or higher oligomeric structures
of scFvs are
formed; however, the exact patterns of oligomerization appear to depend on the
composition
as well as the orientation of the V-domains, in addition to the linker length.
[0132] More recently, a novel technique for construction of mixtures of
antibodies, antibody
fragments and/or other effector moieties in virtually any combination has been
described in
U.S. Patent Nos. 7,550,143; 7,521,056; 7,534,866; 7,527,787; and 7,666,400,
the Examples
section of each of which is incorporated herein by reference. The technique,
known generally
as DOCKANDLOCKTM (DNLTM) involves the production of fusion proteins that
comprise
at their N- or C-terminal ends one of two complementary peptide sequences,
called
dimerization and docking domain (DDD) and anchoring domain (AD) sequences. In
preferred embodiments, the DDD sequences are derived from the regulatory
subunits of
cAMP-dependent protein kinase and the AD sequence is derived from the sequence
of A-
kinase anchoring protein (AKAP). The DDD sequences form dimers that bind to
the AD
sequence, which allows formation of trimers, tetramers, hexamers or any of a
variety of other
complexes. By attaching effector moieties, such as antibodies or antibody
fragments, to the
DDD and AD sequences, complexes may be formed of any selected combination of
antibodies or antibody fragments. The DNLTM complexes may be covalently
stabilized by
formation of disulfide bonds or other linkages.
Pretargeting
34
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
[0133] Bispecific or multispecific antibodies may be utilized in pre-targeting
techniques.
Pre-targeting is a multistep process originally developed to resolve the slow
blood clearance
of directly targeting antibodies, which contributes to undesirable toxicity to
normal tissues
such as bone marrow. With pre-targeting, a radionuclide or other therapeutic
agent is
attached to a small delivery molecule (targetable construct or targetable
conjugate) that is
cleared within minutes from the blood. A pre-targeting bispecific or
multispecific antibody,
which has binding sites for the targetable construct as well as a target
antigen, is administered
first, free antibody is allowed to clear from circulation and then the
targetable construct is
administered.
[0134] Pre-targeting methods are well known in the art, for example, as
disclosed in
Goodwin et al., U.S. Pat. No. 4,863,713; Goodwin et al., J. Nucl. Med. 29:226,
1988;
Hnatowich et al., J. Nucl. Med. 28:1294, 1987; Oehr et al., J. Nucl. Med.
29:728, 1988;
Klibanov et al., J. Nucl. Med. 29:1951, 1988; Sinitsyn et al., J. Nucl. Med.
30:66, 1989;
Kalofonos et al., J. Nucl. Med. 31:1791, 1990; Schechter et al., Int. J.
Cancer 48:167, 1991;
Paganelli et al., Cancer Res. 51:5960, 1991; Paganelli et al., Nucl. Med.
Commun. 12:211,
1991; U.S. Pat. No. 5,256,395; Stickney et al., Cancer Res. 51:6650, 1991;
Yuan et al.,
Cancer Res. 51:3119, 1991; U.S. Pat. No. 6,077,499; U.S. Pat. No. 6,090,381;
U.S. Pat. No.
6,472,511; and U.S. Patent No. 6,962,702.
[0135] A pre-targeting method of treating or diagnosing a disease or disorder
in a subject
may be provided by: (1) administering to the subject a bispecific antibody or
antigen binding
antibody fragment; (2) optionally administering to the subject a clearing
composition, and
allowing the composition to clear the antibody from circulation; and (3)
administering to the
subject the targetable construct, containing one or more chelated or
chemically bound
therapeutic or diagnostic agents. The technique may also be utilized for
antibody dependent
enzyme prodrug therapy (ADEPT) by administering an enzyme conjugated to a
targetable
construct, followed by a prodrug that is converted into active form by the
enzyme.
Known Antibodies
[0136] In various embodiments, the claimed methods and compositions may
utilize any of a
variety of antibodies known in the art, for example for combination antibody
therapy.
Antibodies of use may be commercially obtained from a number of known sources.
For
example, a variety of antibody secreting hybridoma lines are available from
the American
Type Culture Collection (ATCC, Manassas, VA). A large number of antibodies
against
various disease targets, including but not limited to tumor-associated
antigens, have been
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
deposited at the ATCC and/or have published variable region sequences and are
available for
use in the claimed methods and compositions. See, e.g., U.S. Patent Nos.
7,312,318;
7,282,567; 7,151,164; 7,074,403; 7,060,802; 7,056,509; 7,049,060; 7,045,132;
7,041,803;
7,041,802; 7,041,293; 7,038,018; 7,037,498; 7,012,133; 7,001,598; 6,998,468;
6,994,976;
6,994,852; 6,989,241; 6,974,863; 6,965,018; 6,964,854; 6,962,981; 6,962,813;
6,956,107;
6,951,924; 6,949,244; 6,946,129; 6,943,020; 6,939,547; 6,921,645; 6,921,645;
6,921,533;
6,919,433; 6,919,078; 6,916,475; 6,905,681; 6,899,879; 6,893,625; 6,887,468;
6,887,466;
6,884,594; 6,881,405; 6,878,812; 6,875,580; 6,872,568; 6,867,006; 6,864,062;
6,861,511;
6,861,227; 6,861,226; 6,838,282; 6,835,549; 6,835,370; 6,824,780; 6,824,778;
6,812,206;
6,793,924; 6,783,758; 6,770,450; 6,767,711; 6,764,688; 6,764,681; 6,764,679;
6,743,898;
6,733,981; 6,730,307; 6,720,155; 6,716,966; 6,709,653; 6,693,176; 6,692,908;
6,689,607;
6,689,362; 6,689,355; 6,682,737; 6,682,736; 6,682,734; 6,673,344; 6,653,104;
6,652,852;
6,635,482; 6,630,144; 6,610,833; 6,610,294; 6,605,441; 6,605,279; 6,596,852;
6,592,868;
6,576,745; 6,572;856; 6,566,076; 6,562,618; 6,545,130; 6,544,749; 6,534,058;
6,528,625;
6,528,269; 6,521,227; 6,518,404; 6,511,665; 6,491,915; 6,488,930; 6,482,598;
6,482,408;
6,479,247; 6,468,531; 6,468,529; 6,465,173; 6,461,823; 6,458,356; 6,455,044;
6,455,040,
6,451,310; 6,444,206; 6,441,143; 6,432,404; 6,432,402; 6,419,928; 6,413,726;
6,406,694;
6,403,770; 6,403,091; 6,395,276; 6,395,274; 6,387,350; 6,383,759; 6,383,484;
6,376,654;
6,372,215; 6,359,126; 6,355,481; 6,355,444; 6,355,245; 6,355,244; 6,346,246;
6,344,198;
6,340,571; 6,340,459; 6,331,175; 6,306,393; 6,254,868; 6,187,287; 6,183,744;
6,129,914;
6,120,767; 6,096,289; 6,077,499; 5,922,302; 5,874,540; 5,814,440; 5,798,229;
5,789,554;
5,776,456; 5,736,119; 5,716,595; 5,677,136; 5,587,459; 5,443,953; 5,525,338,
the Examples
section of each of which is incorporated herein by reference. These are
exemplary only and a
wide variety of other antibodies and their hybridomas are known in the art.
The skilled
artisan will realize that antibody sequences or antibody-secreting hybridomas
against almost
any disease-associated antigen may be obtained by a simple search of the ATCC,
NCBI
and/or USPTO databases for antibodies against a selected disease-associated
target of
interest. The antigen binding domains of the cloned antibodies may be
amplified, excised,
ligated into an expression vector, transfected into an adapted host cell and
used for protein
production, using standard techniques well known in the art (see, e.g., U.S.
Patent Nos.
7,531,327; 7,537,930; 7,608,425 and 7,785,880, the Examples section of each of
which is
incorporated herein by reference).
36
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
[0137] Particular antibodies that may be of use for therapy of cancer within
the scope of the
claimed methods and compositions include, but are not limited to, LL1 (anti-
CD74), LL2 or
RFB4 (anti-CD22), veltuzumab (hA20, anti-CD20), rituxumab (anti-CD20),
obinutuzumab
(GA101, anti-CD20), lambrolizumab (anti-PD-1 receptor), nivolumab (anti-PD-1
receptor),
ipilimumab (anti-CTLA-4), RS7 (anti-epithelial glycoprotein-1 (EGP-1, also
known as
TROP-2)), KC4 (anti-mucin), MN-14 (anti-carcinoembryonic antigen (anti-CEA,
also known
as CD66e or CEACAM5), MN-15 or MN-3 (anti-CEACAM6), Mu-9 (anti-colon-specific
antigen-p), Immu 31 (an anti-alpha-fetoprotein), R1 (anti-IGF-1R), A19 (anti-
CD19), TAG-
72 (e.g., CC49), Tn, J591 or HuJ591 (anti-PSMA (prostate-specific membrane
antigen)), AB-
PG1-XG1-026 (anti-PSMA dimer), D2/B (anti-PSMA), G250 (an anti-carbonic
anhydrase IX
MAb), L243 (anti-HLA-DR) alemtuzumab (anti-CD52), bevacizumab (anti-VEGF),
cetuximab (anti-EGFR), gemtuzumab (anti-CD33), ibritumomab tiuxetan (anti-
CD20);
panitumumab (anti-EGFR); tositumomab (anti-CD20); PAM4 (aka clivatuzumab, anti-
MUC5ac) and trastuzumab (anti-ErbB2). Such antibodies are known in the art
(e.g., U.S.
Patent Nos. 5,686,072; 5,874,540; 6,107,090; 6,183,744; 6,306,393; 6,653,104;
6,730.300;
6,899,864; 6,926,893; 6,962,702; 7,074,403; 7,230,084; 7,238,785; 7,238,786;
7,256,004;
7,282,567; 7,300,655; 7,312,318; 7,585,491; 7,612,180; 7,642,239; and U.S.
Patent
Application Publ. No. 20050271671; 20060193865; 20060210475; 20070087001; the
Examples section of each incorporated herein by reference.) Specific known
antibodies of
use include hPAM4 (U.S. Patent No. 7,282,567), hA20 (U.S. Patent No.
7,251,164), hAl9
(U.S. Patent No. 7,109,304), hIMMU-31 (U.S. Patent No. 7,300,655), hLL1 (U.S.
Patent No.
7,312,318, ), hLL2 (U.S. Patent No. 7,074,403), hMu-9 (U.S. Patent No.
7,387,773), hL243
(U.S. Patent No. 7,612,180), hMN-14 (U.S. Patent No. 6,676,924), hMN-15 (U.S.
Patent No.
7,541,440), hR1 (U.S. Patent Application 12/772,645), hRS7 (U.S. Patent No.
7,238,785),
hMN-3 (U.S. Patent No. 7,541,440), AB-PG1-XG1-026 (U.S. Patent Application
11/983,372,
deposited as ATCC PTA-4405 and PTA-4406), D2/B (WO 2009/130575), BWA-3 (anti-
histone H4), LG2-1 (anti-histone H3) and LG2-2 (anti-histone H2B) (U.S. Patent
Application
Serial No. 14/180,646, filed 2/14/14) the text of each recited patent or
application is
incorporated herein by reference with respect to the Figures and Examples
sections.
[0138] Other useful antigens that may be targeted using the described
conjugates include
carbonic anhydrase IX, B7, CCL19, CCL21, CSAp, HER-2/neu, BrE3, CD1, CD1a,
CD2,
CD3, CD4, CD5, CD8, CD11A, CD14, CD15, CD16, CD18, CD19, CD20 (e.g., C2B8,
hA20, 1F5 MAbs), CD21, CD22, CD23, CD25, CD29, CD30, CD32b, CD33, CD37, CD38,
37
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
CD40, CD4OL, CD44, CD45, CD46, CD47, CD52, CD54, CD55, CD59, CD64, CD67,
CD70, CD74, CD79a, CD80, CD83, CD95, CD126, CD133, CD138, CD147, CD154,
CEACAM5, CEACAM6, CTLA-4, CXCR4, alpha-fetoprotein (AFP), VEGF (e.g.,
AVASTINO, fibronectin splice variant), ED-B fibronectin (e.g., L19), EGP-1
(TROP-2),
EGP-2 (e.g., 17-1A), EGF receptor (ErbB1) (e.g., ERBITUXO), ErbB2, ErbB3,
Factor H,
FHL-1, Flt-3, folate receptor, Ga 733,GRO-fl, HMGB-1, hypoxia inducible factor
(HIF),
HM1.24, HER-2/neu, histone H2B, histone H3, histone H4, insulin-like growth
factor
(ILGF), IFN-7, IFN-a, IFN-fl, IFN4,, IL-2R, IL-4R, IL-6R, IL-13R, IL-15R, IL-
17R, IL-18R,
IL-2, IL-6, IL-8, IL-12, IL-15, IL-17, IL-18, IL-25, IP-10, IGF-1R, Ia,
HM1.24, gangliosides,
HCG, the HLA-DR antigen to which L243 binds, CD66 antigens, i.e., CD66a-d or a
combination thereof, MAGE, mCRP, MCP-1, MIP-1A, MIP-1B, macrophage migration-
inhibitory factor (MIF), MUC1, MUC2, MUC3, MUC4, MUC5ac, placental growth
factor
(P1GF), PSA (prostate-specific antigen), PSMA, PD-1, PD-L1, NCA-95, NCA-90,
A3, A33,
Ep-CAM, KS-1, Le(y), mesothelin, S100, tenascin, TAC, Tn antigen, Thomas-
Friedenreich
antigens, tumor necrosis antigens, tumor angiogenesis antigens, TNF-a, TRAIL
receptor (R1
and R2), TROP-2, VEGFR, RANTES, T101, as well as cancer stem cell antigens,
complement factors C3, C3a, C3b, C5a, C5, and an oncogene product.
[0139] A comprehensive analysis of suitable antigen (Cluster Designation, or
CD) targets on
hematopoietic malignant cells, as shown by flow cytometry and which can be a
guide to
selecting suitable antibodies for drug-conjugated immunotherapy, is Craig and
Foon, Blood
prepublished online January 15, 2008; DOL 10.1182/blood-2007-11-120535.
[0140] The CD66 antigens consist of five different glycoproteins with similar
structures,
CD66a-e, encoded by the carcinoembryonic antigen (CEA) gene family members,
BCG,
CGM6, NCA, CGM1 and CEA, respectively. These CD66 antigens (e.g., CEACAM6) are
expressed mainly in granulocytes, normal epithelial cells of the digestive
tract and tumor cells
of various tissues. Also included as suitable targets for cancers are cancer
testis antigens,
such as NY-ESO-1 (Theurillat et al., Int. J. Cancer 2007; 120(11):2411-7), as
well as CD79a
in myeloid leukemia (Kozlov et al., Cancer Genet. Cytogenet. 2005; 163(1):62-
7) and also B-
cell diseases, and CD79b for non-Hodgkin's lymphoma (Poison et al., Blood
110(2):616-
623). A number of the aforementioned antigens are disclosed in U.S.
Provisional Application
Serial No. 60/426,379, entitled "Use of Multi-specific, Non-covalent Complexes
for Targeted
Delivery of Therapeutics," filed November 15, 2002. Cancer stem cells, which
are ascribed
to be more therapy-resistant precursor malignant cell populations (Hill and
Perris, J Natl.
38
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
Cancer Inst. 2007; 99:1435-40), have antigens that can be targeted in certain
cancer types,
such as CD133 in prostate cancer (Maitland et al., Ernst Schering Found.
Sympos. Proc.
2006; 5:155-79), non-small-cell lung cancer (Donnenberg et al., J. Control
Release 2007;
122(3):385-91), and glioblastoma (Beier et al., Cancer Res. 2007; 67(9):4010-
5), and CD44
in colorectal cancer (Dalerba er al., Proc. Natl. Acad. Sci. USA 2007;
104(24)10158-63),
pancreatic cancer (Li et al., Cancer Res. 2007; 67(3):1030-7), and in head and
neck squamous
cell carcinoma (Prince et al., Proc. Natl. Acad. Sci. USA 2007; 104(3)973-8).
Another useful
target for breast cancer therapy is the LIV-1 antigen described by Taylor et
al. (Biochem. J.
2003; 375:51-9).
[0141] For multiple myeloma therapy, suitable targeting antibodies have been
described
against, for example, CD38 and CD138 (Stevenson, Mol Med 2006; 12(11-12):345-
346;
Tassone et al., Blood 2004; 104(12):3688-96), CD74 (Stein et al., ibid.), CS1
(Tai et al.,
Blood 2008; 112(4):1329-37, and CD40 (Tai et al., 2005; Cancer Res.
65(13):5898-5906).
[0142] Macrophage migration inhibitory factor (MIF) is an important regulator
of innate and
adaptive immunity and apoptosis. It has been reported that CD74 is the
endogenous receptor
for MIF (Leng et al., 2003, J Exp Med 197:1467-76). The therapeutic effect of
antagonistic
anti-CD74 antibodies on MIF-mediated intracellular pathways may be of use for
treatment of
a broad range of disease states, such as cancers of the bladder, prostate,
breast, lung, colon
and chronic lymphocytic leukemia (e.g., Meyer-Siegler et al., 2004, BMC Cancer
12:34;
Shachar & Haran, 2011, Leuk Lymphoma 52:1446-54); autoimmune diseases such as
rheumatoid arthritis and systemic lupus erythematosus (Morand & Leech, 2005,
Front Biosci
10:12-22; Shachar & Haran, 2011, Leuk Lymphoma 52:1446-54); kidney diseases
such as
renal allograft rejection (Lan, 2008, Nephron Exp Nephrol. 109:e79-83); and
numerous
inflammatory diseases (Meyer-Siegler et al., 2009, Mediators Inflamm epub
March 22, 2009;
Takahashi et al., 2009, Respir Res 10:33; Milatuzumab (hLL1) is an exemplary
anti-CD74
antibody of therapeutic use for treatment of MIF-mediated diseases.
[0143] Anti-TNF-a antibodies are known in the art and may be of use to treat
immune
diseases, such as autoimmune disease, immune dysfunction (e.g., graft-versus-
host disease,
organ transplant rejection) or diabetes. Known antibodies against TNF-a
include the human
antibody CDP571 (Ofei et al., 2011, Diabetes 45:881-85); murine antibodies
MTNFAI,
M2TNFAI, M3TNFAI, M3TNFABI, M302B and M303 (Thermo Scientific, Rockford, IL);
infliximab (Centocor, Malvern, PA); certolizumab pegol (UCB, Brussels,
Belgium); and
adalimumab (Abbott, Abbott Park, IL). These and many other known anti-TNF-a
antibodies
39
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
may be used in the claimed methods and compositions. Other antibodies of use
for therapy
of immune dysregulatory or autoimmune disease include, but are not limited to,
anti-B-cell
antibodies such as veltuzumab, epratuzumab, milatuzumab or hL243; tocilizumab
(anti-IL-6
receptor); basiliximab (anti-CD25); daclizumab (anti-CD25); efalizumab (anti-
CD11a);
muromonab-CD3 (anti-CD3 receptor); anti-CD4OL (UCB, Brussels, Belgium);
natalizumab
(anti-a4 integrin) and omalizumab (anti-IgE).
[0144] Checkpoint inhibitor antibodies have been used primarily in cancer
therapy. Immune
checkpoints refer to inhibitory pathways in the immune system that are
responsible for
maintaining self-tolerance and modulating the degree of immune system response
to
minimize peripheral tissue damage. However, tumor cells can also activate
immune system
checkpoints to decrease the effectiveness of immune response against tumor
tissues.
Exemplary checkpoint inhibitor antibodies against cytotoxic T-lymphocyte
antigen 4
(CTLA4, also known as CD152), programmed cell death protein 1 (PD1, also known
as
CD279) and programmed cell death 1 ligand 1 (PD-L1, also known as CD274), may
be used
in combination with one or more other agents to enhance the effectiveness of
immune
response against disease cells, tissues or pathogens. Exemplary anti-PD1
antibodies include
lambrolizumab (MK-3475, MERCK), nivolumab (BMS-936558, BRISTOL-MYERS
SQUIBB), AMP-224 (MERCK), and pidilizumab (CT-011, CURETECH LTD.). Anti-PD1
antibodies are commercially available, for example from ABCAMO (AB137132),
BIOLEGENDO (EH12.2H7, RMP1-14) and AFFYMETRIX EBIOSCIENCE (J105, J116,
MIH4). Exemplary anti-PD-L1 antibodies include MDX-1105 (MEDAREX), MEDI4736
(MEDIMMUNE) MPDL3280A (GENENTECH) and BMS-936559 (BRISTOL-MYERS
SQUIBB). Anti-PD-L1 antibodies are also commercially available, for example
from
AFFYMETRIX EBIOSCIENCE (MIH1). Exemplary anti-CTLA4 antibodies include
ipilimumab (Bristol-Myers Squibb) and tremelimumab (PFIZER). Anti-PD1
antibodies are
commercially available, for example from ABCAMO (AB134090), SINO BIOLOGICAL
INC. (11159-H03H, 11159-H08H), and THERMO SCIENTIFIC PIERCE (PAS-29572, PAS-
23967, PAS-26465, MA1-12205, MA1-35914). Ipilimumab has recently received FDA
approval for treatment of metastatic melanoma (Wada et al., 2013, J Transl Med
11:89).
[0145] Other antibodies of use may include anti-histone antibodies and/or
antigen-binding
fragments thereof, such as the BWA-3 (anti-H4), LG2-1 (anti-H3) and LG2-2
(anti-H2B)
antibodies. Exemplary anti-histone antibodies are disclosed, for example, in
U.S. Patent
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
Application Serial No. 14/180,646, filed 2/14/14 (the Examples section of
which is
incorporated herein by reference).
[0146] In another preferred embodiment, antibodies are used that internalize
rapidly and are
then re-expressed, processed and presented on cell surfaces, enabling
continual uptake and
accretion of circulating conjugate by the cell. An example of a most-preferred
antibody/antigen pair is LL1, an anti-CD74 MAb (invariant chain, class II-
specific
chaperone, Ii) (see, e.g., U.S. Patent Nos. 6,653,104; 7,312,318; the Examples
section of each
incorporated herein by reference). The CD74 antigen is highly expressed on B-
cell
lymphomas (including multiple myeloma) and leukemias, certain T-cell
lymphomas,
melanomas, colonic, lung, and renal cancers, glioblastomas, and certain other
cancers (Ong et
al., Immunology 98:296-302 (1999)). A review of the use of CD74 antibodies in
cancer is
contained in Stein et al., Clin Cancer Res. 2007 Sep 15;13(18 Pt 2):5556s-
5563s,
incorporated herein by reference.
[0147] The diseases that are preferably treated with anti-CD74 antibodies
include, but are not
limited to, non-Hodgkin's lymphoma, Hodgkin's disease, melanoma, lung, renal,
colonic
cancers, glioblastome multiforme, histiocytomas, myeloid leukemias, and
multiple myeloma.
Continual expression of the CD74 antigen for short periods of time on the
surface of target
cells, followed by internalization of the antigen, and re-expression of the
antigen, enables the
targeting LL1 antibody to be internalized along with any chemotherapeutic
moiety it carries.
This allows a high, and therapeutic, concentration of LL1-chemotherapeutic
drug conjugate
to be accumulated inside such cells. Internalized LL1-chemotherapeutic drug
conjugates are
cycled through lysosomes and endosomes, and the chemotherapeutic moiety is
released in an
active form within the target cells.
Antibody Use for Treatment and Diagnosis
[0148] Certain embodiments concern methods of diagnosing or treating a
malignancy in a
subject, comprising administering to the subject an anti-pancreatic cancer
MAb, fusion
protein or fragment thereof, wherein the MAb, fusion protein or fragment is
bound to at least
one diagnostic and/or therapeutic agent. Also preferred is a method for
diagnosing or treating
cancer, comprising administering to a subject a multivalent, multispecific
antibody or
fragment thereof comprising one or more antigen binding sites toward an
epitope located
within the second to fourth cysteine-rich domains of MUC5ac (amino acid
residues 1575-
2052) and one or more hapten binding sites, waiting a sufficient amount of
time for non-
bound antibody to clear the subject's blood stream; and then administering to
the subject a
41
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
carrier molecule comprising a diagnostic agent, a therapeutic agent, or a
combination thereof,
that binds to the hapten-binding site of the localized antibody. In a more
preferred
embodiment, the cancer is a non-endocrine pancreatic cancer.
[0149] The use of MAbs for in vitro diagnosis is well-known. See, for example,
Carlsson et
al., Bio/Technology 7 (6): 567 (1989). For example, MAbs can be used to detect
the
presence of a tumor-associated antigen in tissue from biopsy samples. MAbs
also can be
used to measure the amount of tumor-associated antigen in clinical fluid
samples, such as
blood or serum, using techniques such as radioimmunoassay, enzyme-linked
immunosorbent
assay, and fluorescence immunoassay. In vitro and in vivo methods of diagnosis
are
discussed in further detail below.
[0150] Conjugates of tumor-targeted MAbs and toxins can be used to selectively
kill cancer
cells in vivo (Spalding, Bio/Technology 9(8): 701 (1991); Goldenberg,
Scientific American
Science & Medicine 1(1): 64 (1994)). For example, therapeutic studies in
experimental
animal models have demonstrated the anti-tumor activity of antibodies carrying
cytotoxic
radionuclides. (Goldenberg et al., Cancer Res. 41: 4354 (1981), Cheung et al.,
J. Nat'l Cancer
Inst. 77: 739 (1986), and Senekowitsch et al., J. Nucl. Med. 30: 531 (1989)).
In a preferred
embodiment, the conjugate comprises a 9 Y-labeled hPAM4 antibody. The
conjugate may
optionally be administered in conjunction with one or more other therapeutic
agents. In a
preferred embodiment, 9 Y-labeled hPAM4 is administered together with
gemcitabine or 5-
fluorouracil to a patient with pancreatic cancer. In a further preferred
embodiment, 9 Y is
conjugated to a DOTA chelate for attachment to hPAM4. In a more preferred
embodiment,
the 90Y-DOTA-hPAM4 is combined with gemcitabine in fractionated doses
comprising a
treatment cycle, such as with repeated, lower, less-toxic doses of gemcitabine
combined with
lower, fractionated doses of 90Y-DOTA-hPAM4. Alternatively, a radiolabeled or
other
conjugated PAM4 antibody may be administered in combination with another
immunoconjugate, such as an SN-38 conjugated antibody. A particularly
preferred
combination is 90Y-hPAM4 and SN-38-hRS7 (anti-TROP2 antibody) (see, e.g., U.S.
Patent
No. 8,586,050, the Examples section incorporated herein by reference).
[0151] As tolerated, repeated cycles of a fractionated dose schedule are
indicated. By way of
example, 4 weekly doses of 200 mg/m2 of gemcitabine are combined with three
weekly doses
of 8 mg/m2 of 90Y-DOTA-hPAM4, with the latter commencing in the second week of
gemcitabine administration, constitutes a single therapy cycle. Still other
doses, higher or
lower of each component, may constitute a fractionated dose, which is
determined by
42
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
conventional means of assessing hematopoietic toxicity (see, e.g., U.S. Patent
Nos.
6,649,352; 7,112,409; 7,279,289; 7,465,551), since myelosuppressive effects of
both agents
can be cumulative. A skilled physician in such therapy interventions can
adjust these doses
based on the patient's bone marrow status and general health status based on
many factors,
including prior exposure to myelosuppressive therapeutic agents. These
principles can also
apply to combinations of radiolabeled hPAM4 with other therapeutic agents,
including
radiosensitizing drugs such as 5-fluorouracil and cisplatin.
[0152] Chimeric, humanized and human antibodies and fragments thereof have
been used for
in vivo therapeutic and diagnostic methods. Accordingly contemplated is a
method of
delivering a diagnostic or therapeutic agent, or a combination thereof, to a
target comprising
(i) providing a composition that comprises an anti-pancreatic cancer antibody
or fragment
thereof, such as a chimeric, humanized or human PAM4 antibody, conjugated to
at least one
diagnostic and/or therapeutic agent and (ii) administering to a subject the
diagnostic or
therapeutic antibody conjugate. In a preferred embodiment, the anti-pancreatic
cancer
antibodies and fragments thereof are humanized or fully human.
[0153] Another embodiment concerns a method for treating a malignancy
comprising
administering a naked or conjugated anti-pancreatic cancer antibody, antibody
fragment or
fusion protein that binds to an epitope located within the second to fourth
cysteine-rich
domains of MUC5ac (amino acid residues 1575-2052), such as a PAM4 antibody,
either
alone or in conjunction with one or more other therapeutic agents. The other
therapeutic
agent may be added before, simultaneously with or after the antibody. In a
preferred
embodiment, the therapeutic agent is gemcitabine, and in a more preferred
embodiment,
gemcitabine is given with the hPAM4 radioconjugate in a fractionated dose
schedule at lower
doses than the conventional 800-1,000 mg/m2 doses of gemcitabine given weekly
for 6
weeks. For example, when combined with fractionated therapeutic doses of 90Y-
PAM4,
repeated fractionated doses intended to function as a radiosensitizing agent
of 200-380 mg/m2
gemcitabine are infused. The skilled artisan will realize that the antibodies,
fusion proteins
and/or fragments thereof described and claimed herein may be administered with
any known
or described therapeutic agent, including but not limited to heat shock
protein 90 (Hsp90).
[0154] In another form of multimodal therapy, subjects receive
immunoconjugates in
conjunction with standard cancer chemotherapy. For example, "CVB" (1.5 g/m2
cyclophosphamide, 200-400 mg/m2 etoposide, and 150-200 mg/m2 carmustine) is a
regimen
used to treat non-Hodgkin's lymphoma. Patti et al., Eur. J. Haematol. 51: 18
(1993). Other
43
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
suitable combination chemotherapeutic regimens are well-known to those of
skill in the art.
See, for example, Freedman et al., "Non-Hodgkin's Lymphomas," in CANCER
MEDICINE,
VOLUME 2, 3rd Edition, Holland et al. (eds.), pages 2028-2068 (Lea & Febiger
1993). As
an illustration, first generation chemotherapeutic regimens for treatment of
intermediate-
grade non-Hodgkin's lymphoma (NHL) include C-MOPP (cyclophosphamide,
vincristine,
procarbazine and prednisone) and CHOP (cyclophosphamide, doxorubicin,
vincristine, and
prednisone). A useful second generation chemotherapeutic regimen is m-BACOD
(methotrexate, bleomycin, doxorubicin, cyclophosphamide, vincristine,
dexamethasone and
leucovorin), while a suitable third generation regimen is MACOP-B
(methotrexate,
doxorubicin, cyclophosphamide, vincristine, prednisone, bleomycin and
leucovorin).
Additional useful drugs include phenyl butyrate, bendamustine, and bryostatin-
1.
[0155] The present invention contemplates the administration of anti-
pancreatic cancer
antibodies and fragments thereof, including fusion proteins and fragments
thereof, alone, as a
naked antibody or antibody fragment, or administered as a multimodal therapy.
Preferably,
the antibody is a humanized or fully human PAM4 antibody or fragment thereof
Multimodal
therapies further include immunotherapy with a naked anti-pancreatic cancer
antibody
supplemented with administration of other antibodies in the form of naked
antibodies, fusion
proteins, or as immunoconjugates. For example, a humanized or fully human PAM4
antibody may be combined with another naked antibody, or a humanized PAM4 or
other
antibody conjugated to an isotope, one or more chemotherapeutic agents,
cytokines, toxins or
a combination thereof For example, the present invention contemplates
treatment of a naked
or conjugated PAM4 antibody or fragments thereof before, in combination with,
or after
other pancreatic tumor associated antibodies such as CA19.9, DUPAN2, SPAN1,
Nd2,
B72.3, CC49, anti-Lea antibodies, and antibodies to other Lewis antigens
(e.g., Le(y)), as well
as antibodies against carcinoembryonic antigen (CEA or CEACAM5), CEACAM6,
colon-
specific antigen-p (CSAp), MUC1, MUC2, MUC3, MUC4, MUC5ac, MUC16, MUC17,
HLA-DR, CD40, CD74, CD138, HER2/neu, EGFR, EGP-1, EGP-2, angiogenesis factors
(e.g., VEGF, P1GF), insulin-like growth factor (IGF), tenascin, platelet-
derived growth factor,
and IL-6, as well as products of oncogenes (e.g., bc1-2, Kras, p53), cMET, and
antibodies
against tumor necrosis substances.
[0156] These solid tumor antibodies may be naked or conjugated to, inter alia,
drugs, toxins,
isotopes, radionuclides or immunomodulators. Many different antibody
combinations may
be constructed and used in either naked or conjugated form. Alternatively,
different naked
44
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
antibody combinations may be employed for administration in combination with
other
therapeutic agents, such as a cytotoxic drug or with radiation, given
consecutively,
simultaneously, or sequentially.
[0157] Administration of the antibodies and their fragments can be effected by
intravenous,
intraarterial, intraperitoneal, intramuscular, subcutaneous, intrapleural,
intrathecal, perfusion
through a regional catheter, or direct intralesional injection. When
administering the
antibody by injection, the administration may be by continuous infusion or by
single or
multiple boluses.
[0158] The immunoconjugate of the present invention can be formulated for
intravenous
administration via, for example, bolus injection or continuous infusion.
Preferably, the
antibody of the present invention is infused over a period of less than about
4 hours, and more
preferably, over a period of less than about 3 hours. For example, the first
25-50 mg could be
infused within 30 minutes, preferably even 15 min, and the remainder infused
over the next
2-3 hrs. Formulations for injection can be presented in unit dosage form,
e.g., in ampoules or
in multi-dose containers, with an added preservative. The compositions can
take such forms
as suspensions, solutions or emulsions in oily or aqueous vehicles, and can
contain
formulatory agents such as suspending, stabilizing and/or dispersing agents.
Alternatively,
the active ingredient can be in powder form for constitution with a suitable
vehicle, e.g.,
sterile pyrogen-free water, before use.
[0159] Additional pharmaceutical methods may be employed to control the
duration of action
of the therapeutic conjugate. Control release preparations can be prepared
through the use of
polymers to complex or adsorb the immunoconjugate. For example, biocompatible
polymers
include matrices of poly(ethylene-co-vinyl acetate) and matrices of a
polyanhydride
copolymer of a stearic acid dimer and sebacic acid. Sherwood et al.,
Bio/Technology 10:
1446 (1992). The rate of release of an immunoconjugate or antibody from such a
matrix
depends upon the molecular weight of the immunoconjugate or antibody, the
amount of
immunoconjugate or antibody within the matrix, and the size of dispersed
particles.
Saltzman et al., Biophys. J. 55: 163 (1989); Sherwood et al., supra. Other
solid dosage forms
are described in Ansel et al., PHARMACEUTICAL DOSAGE FORMS AND DRUG
DELIVERY SYSTEMS, 5th Edition (Lea & Febiger 1990), and Gennaro (ed.),
REMINGTON'S PHARMACEUTICAL SCIENCES, 18th Edition (Mack Publishing
Company 1990), and revised editions thereof
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
[0160] More generally, the dosage of an administered immunoconjugate for
humans will
vary depending upon such factors as the patient's age, weight, height, sex,
general medical
condition and previous medical history. It may be desirable to provide the
recipient with a
dosage of immunoconjugate, antibody fusion protein that is in the range of
from about 1
mg/kg to 25 mg/kg as a single intravenous infusion, although a lower or higher
dosage also
may be administered as circumstances dictate. A dosage of 1-20 mg/kg for a 70
kg patient,
for example, is 70-1,400 mg, or 41-824 mg/m2 for a 1.7-m patient. The dosage
may be
repeated as needed, for example, once per week for 4-10 weeks, once per week
for 8 weeks,
or once per week for 4 weeks. It may also be given less frequently, such as
every other week
for several months, or monthly or quarterly for many months, as needed in a
maintenance
therapy.
[0161] Alternatively, an antibody may be administered as one dosage every 2 or
3 weeks,
repeated for a total of at least 3 dosages. Or, the antibodies may be
administered twice per
week for 4-6 weeks. If the dosage is lowered to approximately 200-300 mg/m2
(340 mg per
dosage for a 1.7-m patient, or 4.9 mg/kg for a 70 kg patient), it may be
administered once or
even twice weekly for 4 to 10 weeks. Alternatively, the dosage schedule may be
decreased,
namely every 2 or 3 weeks for 2-3 months. It has been determined, however,
that even
higher doses, such as 20 mg/kg once weekly or once every 2-3 weeks can be
administered by
slow i.v. infusion, for repeated dosing cycles. The dosing schedule can
optionally be repeated
at other intervals and dosage may be given through various parenteral routes,
with
appropriate adjustment of the dose and schedule.
Immunoconjugates
[0162] Anti-pancreatic cancer antibodies and fragments thereof may be
conjugated to at least
one therapeutic and/or diagnostic agent for therapy or diagnosis. For
immunotherapy, the
objective is to deliver cytotoxic doses of radioactivity, toxin, antibody
and/or drug to target
cells, while minimizing exposure to non-target tissues. Preferably, anti-
pancreatic cancer
antibodies are used to diagnose and/or treat pancreatic tumors.
[0163] Any of the antibodies, antibody fragments and fusion proteins can be
conjugated with
one or more therapeutic or diagnostic agents, using a variety of techniques
known in the art.
One or more therapeutic or diagnostic agents may be attached to each antibody,
antibody
fragment or fusion protein, for example by conjugating an agent to a
carbohydrate moiety in
the Fc region of the antibody. If the Fc region is absent (for example with
certain antibody
fragments), it is possible to introduce a carbohydrate moiety into the light
chain variable
46
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
region of either an antibody or antibody fragment to which a therapeutic or
diagnostic agent
may be attached. See, for example, Leung et al., J Immunol. 154: 5919 (1995);
Hansen et al.,
U.S. Pat. No. 5,443,953 (1995), Leung et al., U.S. Pat. No. 6,254,868, the
Examples section
of each patent incorporated herein by reference.
[0164] Methods for conjugating peptides to antibody components via an antibody
carbohydrate moiety are well-known to those of skill in the art. See, for
example, Shih et al.,
Int J Cancer 41: 832 (1988); Shih et al., Int J Cancer 46: 1101 (1990); and
Shih et al., U.S.
Pat. No. 5,057,313, the Examples section of which is incorporated herein by
reference. The
general method involves reacting an antibody component having an oxidized
carbohydrate
portion with a carrier polymer that has at least one free amine function and
that is loaded with
a plurality of therapeutic agents, such as peptides or drugs. This reaction
results in an initial
Schiff base (imine) linkage, which can be stabilized by reduction to a
secondary amine to
form the final conjugate.
[0165] Antibody fusion proteins or multispecific antibodies comprise two or
more antibodies
or fragments thereof, each of which may be attached to at least one
therapeutic agent and/or
diagnostic agent. Accordingly, one or more of the antibodies or fragments
thereof of the
antibody fusion protein can have more than one therapeutic and/or diagnostic
agent attached.
Further, the therapeutic agents do not need to be the same but can be
different therapeutic
agents, for example, one can attach a drug and a radioisotope to the same
fusion protein. For
example, an IgG can be radiolabeled with 1311 and attached to a drug. The 1311
can be
incorporated into the tyrosine of the IgG and the drug attached to the epsilon
amino group of
the IgG lysines. Both therapeutic and diagnostic agents also can be attached
to reduced SH
groups and to the carbohydrate side chains of antibodies. Alternatively, a
bispecific antibody
may comprise one antibody or fragment thereof against a disease antigen and
another against
a hapten attached to a targetable construct, for use in pretargeting
techniques as discussed
above.
[0166] A therapeutic or diagnostic agent can be attached at the hinge region
of a reduced
antibody component via disulfide bond formation. As an alternative, such
agents can be
attached to the antibody component using a heterobifunctional cross-linker,
such as N-
succinyl 3-(2-pyridyldithio)proprionate (SPDP). Yu et al., Int. J. Cancer 56:
244 (1994).
General techniques for such conjugation are well-known in the art. See, for
example, Wong,
CHEMISTRY OF PROTEIN CONJUGATION AND CROSS-LINKING (CRC Press 1991);
Upeslacis et al., "Modification of Antibodies by Chemical Methods," in
MONOCLONAL
47
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
ANTIBODIES: PRINCIPLES AND APPLICATIONS, Birch et al. (eds.), pages 187-230
(Wiley-Liss, Inc. 1995); Price, "Production and Characterization of Synthetic
Peptide-
Derived Antibodies," in MONOCLONAL ANTIBODIES: PRODUCTION, ENGINEERING
AND CLINICAL APPLICATION, Ritter et al. (eds.), pages 60-84 (Cambridge
University
Press 1995).
Click Chemistry
[0167] An alternative method for attaching chelating moieties, drugs or other
functional
groups to an antibody, fragment or fusion protein involves use of click
chemistry reactions.
The click chemistry approach was originally conceived as a method to rapidly
generate
complex substances by joining small subunits together in a modular fashion.
(See, e.g., Kolb
et al., 2004, Angew Chem Int Ed 40:3004-31; Evans, 2007, Aust J Chem 60:384-
95.)
Various forms of click chemistry reaction are known in the art, such as the
Huisgen 1,3-
dipolar cycloaddition copper catalyzed reaction (Tornoe et al., 2002, J
Organic Chem
67:3057-64), which is often referred to as the "click reaction." Other
alternatives include
cycloaddition reactions such as the Diels-Alder, nucleophilic substitution
reactions
(especially to small strained rings like epoxy and aziridine compounds),
carbonyl chemistry
formation of urea compounds and reactions involving carbon-carbon double
bonds, such as
alkynes in thiol-yne reactions.
[0168] The azide alkyne Huisgen cycloaddition reaction uses a copper catalyst
in the
presence of a reducing agent to catalyze the reaction of a terminal alkyne
group attached to a
first molecule. In the presence of a second molecule comprising an azide
moiety, the azide
reacts with the activated alkyne to form a 1,4-disubstituted 1,2,3-triazole.
The copper
catalyzed reaction occurs at room temperature and is sufficiently specific
that purification of
the reaction product is often not required. (Rostoystev et al., 2002, Angew
Chem Int Ed
41:2596; Tornoe et al., 2002, J Org Chem 67:3057.) The azide and alkyne
functional groups
are largely inert towards biomolecules in aqueous medium, allowing the
reaction to occur in
complex solutions. The triazole formed is chemically stable and is not subject
to enzymatic
cleavage, making the click chemistry product highly stable in biological
systems. Although
the copper catalyst is toxic to living cells, the copper-based click chemistry
reaction may be
used in vitro for immunoconjugate formation.
[0169] A copper-free click reaction has been proposed for covalent
modification of
biomolecules. (See, e.g., Agard et al., 2004, J Am Chem Soc 126:15046-47.) The
copper-
free reaction uses ring strain in place of the copper catalyst to promote a [3
+ 2] azide-alkyne
48
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
cycloaddition reaction (Id.) For example, cyclooctyne is a 8-carbon ring
structure
comprising an internal alkyne bond. The closed ring structure induces a
substantial bond
angle deformation of the acetylene, which is highly reactive with azide groups
to form a
triazole. Thus, cyclooctyne derivatives may be used for copper-free click
reactions (Id.)
[0170] Another type of copper-free click reaction was reported by Ning et al.
(2010, Angew
Chem Int Ed 49:3065-68), involving strain-promoted alkyne-nitrone
cycloaddition. To
address the slow rate of the original cyclooctyne reaction, electron-
withdrawing groups are
attached adjacent to the triple bond (Id.) Examples of such substituted
cyclooctynes include
difluorinated cyclooctynes, 4-dibenzocyclooctynol and azacyclooctyne (Id.) An
alternative
copper-free reaction involved strain-promoted alkyne-nitrone cycloaddition to
give N-
alkylated isoxazolines (Id.) The reaction was reported to have exceptionally
fast reaction
kinetics and was used in a one-pot three-step protocol for site-specific
modification of
peptides and proteins (Id.) Nitrones were prepared by the condensation of
appropriate
aldehydes with N-methylhydroxylamine and the cycloaddition reaction took place
in a
mixture of acetonitrile and water (Id.) These and other known click chemistry
reactions may
be used to attach chelating moieties to antibodies or other targeting
molecules in vitro.
Therapeutic Agents
[0171] A wide variety of therapeutic reagents can be administered concurrently
or
sequentially, or advantageously conjugated to the antibodies of the invention,
for example,
drugs, toxins, oligonucleotides (e.g., siRNA), immunomodulators, hormones,
hormone
antagonists, enzymes, enzyme inhibitors, radionuclides, angiogenesis
inhibitors, pro-
apoptotic agents, etc. The therapeutic agents recited here are those agents
that are useful for
either conjugated to an antibody, fragment or fusion protein or for
administration separately
with a naked antibody as described above.
[0172] Therapeutic agents include, for example, chemotherapeutic drugs such as
vinca
alkaloids, anthracyclines, gemcitabine, epipodophyllotoxins, taxanes,
antimetabolites,
alkylating agents, antibiotics, SN-38, COX-2 inhibitors, antimitotics,
antiangiogenic and
apoptotic agents, particularly doxorubicin, methotrexate, taxol, CPT-11,
camptothecans,
proteosome inhibitors, mTOR inhibitors, HDAC inhibitors, tyrosine kinase
inhibitors, and
others from these and other classes of anticancer agents.
[0173] Other useful cancer chemotherapeutic drugs include nitrogen mustards,
alkyl
sulfonates, nitrosoureas, triazenes, folic acid analogs, COX-2 inhibitors,
antimetabolites,
pyrimidine analogs, purine analogs, platinum coordination complexes, mTOR
inhibitors,
49
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
tyrosine kinase inhibitors, proteosome inhibitors, HDAC inhibitors,
camptothecins and
hormones. Suitable chemotherapeutic agents are described in REMINGTON'S
PHARMACEUTICAL SCIENCES, 19th Ed. (Mack Publishing Co. 1995), and in
GOODMAN AND GILMAN'S THE PHARMACOLOGICAL BASIS OF
THERAPEUTICS, 7th Ed. (MacMillan Publishing Co. 1985), as well as revised
editions of
these publications. Other suitable chemotherapeutic agents, such as
experimental drugs, are
known to those of skill in the art.
[0174] Specific drugs of use may include 5-fluorouracil, afatinib, aplidin,
azaribine,
anastrozole, anthracyclines, axitinib, AVL-101, AVL-291, bendamustine,
bleomycin,
bortezomib, bosutinib, bryostatin-1, busulfan, calicheamycin, camptothecin,
carboplatin, 10-
hydroxycamptothecin, carmustine, celebrex, chlorambucil, cisplatin (CDDP), Cox-
2
inhibitors, irinotecan (CPT-11), SN-38, carboplatin, cladribine,
camptothecans, crizotinib,
cyclophosphamide, cytarabine, dacarbazine, dasatinib, dinaciclib, docetaxel,
dactinomycin,
daunorubicin, doxorubicin, 2-pyrrolinodoxorubicine (2-PDOX), pro-2PDOX, cyano-
morpholino doxorubicin, doxorubicin glucuronide, epirubicin glucuronide,
erlotinib,
estramustine, epidophyllotoxin, erlotinib, entinostat, estrogen receptor
binding agents,
etoposide (VP16), etoposide glucuronide, etoposide phosphate, exemestane,
fingolimod,
floxuridine (FUdR), 3',5'-0-dioleoyl-FudR (FUdR-d0), fludarabine, flutamide,
farnesyl-
protein transferase inhibitors, flavopiridol, fostamatinib, ganetespib, GDC-
0834, GS-1101,
gefitinib, gemcitabine, hydroxyurea, ibrutinib, idarubicin, idelalisib,
ifosfamide, imatinib, L-
asparaginase, lapatinib, lenolidamide, leucovorin, LFM-A13, lomustine,
mechlorethamine,
melphalan, mercaptopurine, 6-mercaptopurine, methotrexate, mitoxantrone,
mithramycin,
mitomycin, mitotane, navelbine, neratinib, nilotinib, nitrosurea, olaparib,
plicomycin,
procarbazine, paclitaxel, PCI-32765, pentostatin, PSI-341, raloxifene,
semustine, sorafenib,
streptozocin, 5U11248, sunitinib, tamoxifen, temazolomide (an aqueous form of
DTIC),
transplatinum, thalidomide, thioguanine, thiotepa, teniposide, topotecan,
uracil mustard,
vatalanib, vinorelbine, vinblastine, vincristine, vinca alkaloids and ZD1839.
[0175] In a preferred embodiment, conjugates of camptothecins and related
compounds, such
as SN-38, may be conjugated to hPAM4 or other anti-pancreatic cancer
antibodies, for
example as disclosed in U.S. Patent No. 7,591,994; and U.S. Patent Application
Serial No.
11/388,032, filed March 23, 2006, the Examples section of each of which is
incorporated
herein by reference.
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
[0176] In another preferred embodiment, prodrug forms of 2-PDOX, as disclosed
in U.S.
Patent Application Serial No. 14/175,089 (the Examples section of which is
incorporated
herein by reference) may be used as an immunoconjugate with an anti-pancreatic
cancer
antibody that binds to an epitope located within the second to fourth cysteine-
rich domains of
MUC5ac (amino acid residues 1575-2052).
[0177] In another preferred embodiment, an hPAM4 antibody is given with
gemcitabine,
which may be given before, after, or concurrently with a naked or conjugated
chimeric,
humanized or human PAM4 antibody. Preferably, the conjugated hPAM4 antibody or
antibody fragment is conjugated to a radionuclide.
[0178] A toxin can be of animal, plant or microbial origin. A toxin, such as
Pseudomonas
exotoxin, may also be complexed to or form the therapeutic agent portion of an
immunoconjugate of the anti-pancreatic cancer and hPAM4 antibodies. Other
toxins suitably
employed in the preparation of such conjugates or other fusion proteins,
include ricin, abrin,
ribonuclease (RNase), DNase I, Staphylococcal enterotoxin-A, pokeweed
antiviral protein,
gelonin, diphtheria toxin, ranpimase, Pseudomonas exotoxin, and Pseudomonas
endotoxin.
See, for example, Pastan et al., Cell 47:641 (1986), Goldenberg, CA--A Cancer
Journal for
Clinicians 44:43 (1994), Sharkey and Goldenberg, CA--A Cancer Journal for
Clinicians
56:226 (2006). Additional toxins suitable for use are known to those of skill
in the art and are
disclosed in U.S. Pat. No. 6,077,499, the Examples section of which is
incorporated herein by
reference.
[0179] An immunomodulator, such as a cytokine, may also be conjugated to, or
form the
therapeutic agent portion of the immunoconjugate, or may be administered with,
but
unconjugated to, an antibody, antibody fragment or fusion protein. The fusion
protein may
comprise one or more antibodies or fragments thereof binding to different
antigens. For
example, the fusion protein may bind to an epitope located within the second
to fourth
cysteine-rich domains of MUC5ac (amino acid residues 1575-2052) as well as to
immunomodulating cells or factors. Alternatively, subjects can receive a naked
antibody,
antibody fragment or fusion protein and a separately administered cytokine,
which can be
administered before, concurrently or after administration of the naked
antibodies. As used
herein, the term "immunomodulator" includes a cytokine, a lymphokine, a
monokine, a stem
cell growth factor, a lymphotoxin, a hematopoietic factor, a colony
stimulating factor (CSF),
an interferon (IFN), parathyroid hormone, thyroxine, insulin, proinsulin,
relaxin, prorelaxin,
follicle stimulating hormone (FSH), thyroid stimulating hormone (TSH),
luteinizing hormone
51
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
(LH), hepatic growth factor, prostaglandin, fibroblast growth factor,
prolactin, placental
lactogen, OB protein, a transforming growth factor (TGF), TGF-a, TGF-13,
insulin-like
growth factor (IGF), erythropoietin, thrombopoietin, tumor necrosis factor
(TNF), TNF- a,
TNF-13, a mullerian-inhibiting substance, mouse gonadotropin-associated
peptide, inhibin,
activin, vascular endothelial growth factor, integrin, interleukin (IL),
granulocyte-colony
stimulating factor (G-CSF), granulocyte macrophage-colony stimulating factor
(GM-CSF),
interferon- a, interferon-13, interferon-y, S1 factor, IL-1, IL-1 cc, IL-2, IL-
3, IL-4, IL-5, IL-6,
IL-7, IL-8, IL-9, IL-10, IL-11, IL-12, IL-13, IL-14, IL-15, IL-16, IL-17, IL-
18 IL-21 and IL-
25, LIF, kit-ligand, FLT-3, angiostatin, thrombospondin, endostatin and
lymphotoxin.
[0180] The therapeutic agent may comprise one or more radioactive isotopes
useful for
treating diseased tissue. Particularly useful therapeutic radionuclides
include, but are not
limited to 111In, 177õ, 212Bi, 213Bi, 211At, , 62-u
C Cu, 67CU, 90Y, 1251, 1311, 32p, 33p, 47se,
111Ag,
67Ga, 142pr, 1535m, 161Tb, 166Dy, 166H0, 186Re, 188Re, 189Re, 212pb, 223Ra,
225
c
A, 59Fe, 755e,
77As, 895r, 99Mo, 1o5Rh, 109pd, 143pr, 149pm, 169Er, 1941r, 198Au, 199Au,
211pb 227
a Th. The
therapeutic radionuclide preferably has a decay energy in the range of 20 to
6,000 keV,
preferably in the ranges 60 to 200 keV for an Auger emitter, 100-2,500 keV for
a beta
emitter, and 4,000-6,000 keV for an alpha emitter. Maximum decay energies of
useful beta-
particle-emitting nuclides are preferably 20-5,000 keV, more preferably 100-
4,000 keV, and
most preferably 500-2,500 keV. Also preferred are radionuclides that
substantially decay
with Auger-emitting particles. For example, Co-58, Ga-67, Br-80m, Tc-99m, Rh-
103m, Pt-
109, In-111, Sb-119, 1-125, Ho-161, Os-189m and Ir-192. Decay energies of
useful beta-
particle-emitting nuclides are preferably <1,000 keV, more preferably <100
keV, and most
preferably <70 keV. Also preferred are radionuclides that substantially decay
with
generation of alpha-particles. Such radionuclides include, but are not limited
to: Dy-152, At-
211, Bi-212, Ra-223, Rn-219, Po-215, Bi-211, Ac-225, Fr-221, At-217, Bi-213,
Th-227 and
Fm-255. Decay energies of useful alpha-particle-emitting radionuclides are
preferably 2,000-
10,000 keV, more preferably 3,000-8,000 keV, and most preferably 4,000-7,000
keV.
[0181] For example, 67Cu, considered one of the more promising radioisotopes
for
radioimmunotherapy due to its 61.5-hour half-life and abundant supply of beta
particles and
gamma rays, can be conjugated to an antibody using the chelating agent, p-
bromoacetamido-
benzyl-tetraethylaminetetraacetic acid (TETA). Alternatively, 90Y, which emits
an energetic
beta particle, can be coupled to an antibody, antibody fragment or fusion
protein, using
diethylenetriaminepentaacetic acid (DTPA), or more preferably using DOTA.
Methods of
52
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
conjugating 9 Y to antibodies or targetable constructs are known in the art
and any such
known methods may be used. (See, e.g., U.S. Patent No. 7,259,249, the Examples
section of
which is incorporated herein by reference. See also Linden et al., Clin Cancer
Res. 11:5215-
22, 2005; Sharkey et al., J Nucl Med. 46:620-33, 2005; Sharkey et al., J Nucl
Med. 44:2000-
18, 2003.)
[0182] Additional potential therapeutic radioisotopes include 11C, 13N, 150,
75Br, 198AU,
224 126 133 77 113m 95 97 103 105 107 203 121m 122m 125m
Ac, I, I, Br, In, Ru, Ru, Ru, Ru, Hg, Hg, Te, Te, Te,
1651,m, 1671,m, 1681,m, 197pt, 109pd, 105Rb, 142pr, 143pr, 161Tb, 166H0, 199
Au, A, 57Co, 58Co, 51Cr,
59 75 201 225 76 169
Fe, Se, Tl, Ac, Br, Yb, and the like.
[0183] In another embodiment, a radiosensitizer can be used in combination
with a naked or
conjugated antibody or antibody fragment. For example, the radiosensitizer can
be used in
combination with a radiolabeled antibody or antibody fragment. The addition of
the
radiosensitizer can result in enhanced efficacy when compared to treatment
with the
radiolabeled antibody or antibody fragment alone. Radiosensitizers are
described in D. M.
Goldenberg (ed.), CANCER THERAPY WITH RADIOLABELED ANTIBODIES, CRC
Press (1995). Other typical radionsensitizers of interest for use with this
technology include
gemcitabine, 5-fluorouracil, and cisplatin, and have been used in combination
with external
irradiation in the therapy of diverse cancers, including pancreatic cancer.
Therefore, we have
studied the combination of gemcitabine at what is believed to be
radiosensitizing doses (once
weekly 200 mg/m2 over 4 weeks) of gemcitabine combined with fractionated doses
of9 Y-
hPAM4, and have observed objective evidence of pancreatic cancer reduction
after a single
cycle of this combination that proved to be well-tolerated (no grade 3-4
toxicities by NCI-
CTC v. 3 standard).
[0184] Antibodies or fragments thereof that have a boron addend-loaded carrier
for thermal
neutron activation therapy will normally be affected in similar ways. However,
it will be
advantageous to wait until non-targeted immunoconjugate clears before neutron
irradiation is
performed. Clearance can be accelerated using an anti-idiotypic antibody that
binds to the
anti-pancreatic cancer antibody. See U.S. Pat. No. 4,624,846 for a description
of this general
principle. For example, boron addends such as carboranes, can be attached to
antibodies.
Carboranes can be prepared with carboxyl functions on pendant side chains, as
is well-known
in the art. Attachment of carboranes to a carrier, such as aminodextran, can
be achieved by
activation of the carboxyl groups of the carboranes and condensation with
amines on the
carrier. The intermediate conjugate is then conjugated to the antibody. After
administration
53
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
of the antibody conjugate, a boron addend is activated by thermal neutron
irradiation and
converted to radioactive atoms which decay by alpha-emission to produce highly
toxic, short-
range effects.
Interference RNA
[0185] Another type of therapeutic agent is RNAi or siRNA. RNA interference
(RNAi) is
mediated by the RNA-induced silencing complex (RISC) and is initiated by short
double-
stranded RNA molecules that interact with the catalytic RISC component
argonaute (Rand et
al., 2005, Cell 123:621-29). Types of RNAi molecules include microRNA (miRNA)
and
small interfering RNA (siRNA). RNAi species can bind with messenger RNA (mRNA)
through complementary base-pairing and inhibits gene expression by post-
transcriptional
gene silencing. Upon binding to a complementary mRNA species, RNAi induces
cleavage of
the mRNA molecule by the argonaute component of RISC. Among other
characteristics,
miRNA and siRNA differ in the degree of specificity for particular gene
targets, with siRNA
being relatively specific for a particular target gene and miRNA inhibiting
translation of
multiple mRNA species.
[0186] Therapeutic use of RNAi by inhibition of selected gene expression has
been attempted
for a variety of disease states, such as macular degeneration and respiratory
syncytial virus
infection (Sah, 2006, Life Sci 79:1773-80). It has been suggested that siRNA
functions in
host cell defenses against viral infection and siRNA has been widely examined
as an
approach to anti-viral therapy (see, e.g., Zhang et al., 2004, Nature Med
11:56-62; Novina et
al., 2002, Nature Med 8:681-86; Palliser et al., 2006, Nature 439:89-94). The
use of siRNA
for cancer therapy has also been attempted. Fujii et al. (2006, Int J Oncol
29:541-48)
transfected HPV positive cervical cancer cells with siRNA against HPV E6 and
E7 and
suppressed tumor growth. siRNA-mediated knockdown of metadherin expression in
breast
cancer cells was reported to inhibit experimental lung metastasis (Brown and
Ruoslahti,
2004, Cancer Cell 5:365-74).
[0187] Attempts have been made to provide targeted delivery of siRNA to reduce
the
potential for off-target toxicity. Song et al. (2005, Nat Biotechnol 23:709-
17) used
protamine-conjugated Fab fragments against HIV envelope protein to deliver
siRNA to
circulating cells. Schiffelers et al. (2004, Nucl Acids Res 32:e149)
conjugated RGD peptides
to nanoparticles to deliver anti-VEGFR2 siRNA to tumors and inhibited tumor
angiogenesis
and growth rate in nude mice. Dickerson et al. (2010, Cancer 10:10) used
nanogels
functionalized with anti-EphA2 receptor peptides to chemosensitize ovarian
cancer cells with
54
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
siRNA against EGFR. Dendrimer-conjugated magnetic nanoparticles have been
applied to
the targeted delivery of antisense survivin oligodeoxynucleotides (Pan et al.,
2007, Cancer
Res 67:8156-63).
[0188] The skilled artisan will realize that any siRNA or interference RNA
species may be
attached to the subject antibodies. siRNA and RNAi species against a wide
variety of targets
are known in the art, and any such known oligonucleotide species may be
utilized in the
claimed methods and compositions.
[0189] Known siRNA species of potential use include those specific for IKK-
gamma (U.S.
Patent 7,022,828); VEGF, Flt-1 and Flk-1/KDR (U.S. Patent 7,148,342); Bc12 and
EGFR
(U.S. Patent 7,541,453); CDC20 (U.S. Patent 7,550,572); transducin (beta)-like
3 (U.S.
Patent 7,576,196); KRAS (U.S. Patent 7,576,197); carbonic anhydrase II (U.S.
Patent
7,579,457); complement component 3 (U.S. Patent 7,582,746); interleukin-1
receptor-
associated kinase 4 (IRAK4) (U.S. Patent 7,592,443); survivin (U.S. Patent
7,608,7070);
superoxide dismutase 1 (U.S. Patent 7,632,938); MET proto-oncogene (U.S.
Patent
7,632,939); amyloid beta precursor protein (APP) (U.S. Patent 7,635,771); IGF-
1R (U.S.
Patent 7,638,621); ICAM1 (U.S. Patent 7,642,349); complement factor B (U.S.
Patent
7,696,344); p53 (7,781,575), and apolipoprotein B (7,795,421), the Examples
section of each
of which is incorporated herein by reference.
[0190] Additional siRNA species are available from known commercial sources,
such as
Sigma-Aldrich (St Louis, MO), Invitrogen (Carlsbad, CA), Santa Cruz
Biotechnology (Santa
Cruz, CA), Ambion (Austin, TX), Dharmacon (Thermo Scientific, Lafayette, CO),
Promega
(Madison, WI), Mims Bio (Madison, WI) and Qiagen (Valencia, CA), among many
others.
Other publicly available sources of siRNA species include the siRNAdb database
at the
Stockholm Bioinformatics Centre, the MIT/ICBP siRNA Database, the RNAi
Consortium
shRNA Library at the Broad Institute, and the Probe database at NCBI. For
example, there
are 30,852 siRNA species in the NCBI Probe database. The skilled artisan will
realize that
for any gene of interest, either an siRNA species has already been designed,
or one may
readily be designed using publicly available software tools. Any such siRNA
species may be
delivered using the subject DNLTM complexes.
[0191] Exemplary siRNA species that have been reported are listed in Table 1.
Although
siRNA is delivered as a double-stranded molecule, for simplicity only the
sense strand
sequences are shown in Table 1.
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
Table 1. Exemplary siRNA Sequences
Target Sequence SEQ ID NO
VEGF R2 AATGCGGCGGTGGTGACAGTA SEQ ID NO:22
VEGF R2 AAGCTCAGCACACAGAAAGAC SEQ ID NO:23
CXCR4 UAAAAUCUUCCUGCCCACCdTdT SEQ ID NO:24
CXCR4 GGAAGCUGUUGGCUGAAAAdTdT SEQ ID NO:25
PPARC1 AAGACCAGCCUCUUUGCCCAG SEQ ID NO:26
Dynamin 2 GGACCAGGCAGAAAACGAG SEQ ID NO:27
Catenin CUAUCAGGAUGACGCGG SEQ ID NO:28
ElA binding protein UGACACAGGCAGGCUUGACUU SEQ ID NO:29
Plasminogen GGTGAAGAAGGGCGTCCAA SEQ ID NO:30
activator
K-ras GATCCGTTGGAGCTGTTGGCGTAGTT SEQ ID NO:31
CAAGAGACTCGCCAACAGCTCCAACT
TTTGGAAA
Sortilin 1 AGGTGGTGTTAACAGCAGAG SEQ ID NO:32
Apolipoprotein E AAGGTGGAGCAAGCGGTGGAG SEQ ID NO:33
Apolipoprotein E AAGGAGTTGAAGGCCGACAAA SEQ ID NO:34
Bc1-X UAUGGAGCUGCAGAGGAUGdTdT SEQ ID NO:35
Raf-1 TTTGAATATCTGTGCTGAGAACACA SEQ ID NO:36
GTTCTCAGCACAGATATTCTTTTT
Heat shock AATGAGAAAAGCAAAAGGTGCCCTGTCTC SEQ ID NO:37
transcription factor 2
IGFBP3 AAUCAUCAUCAAGAAAGGGCA SEQ ID NO:38
Thioredoxin AUGACUGUCAGGAUGUUGCdTdT SEQ ID NO:39
CD44 GAACGAAUCCUGAAGACAUCU SEQ ID NO:40
MMP14 AAGCCTGGCTACAGCAATATGCCTGTCTC SEQ ID NO:41
MAPKAPK2 UGACCAUCACCGAGUUUAUdTdT SEQ ID NO:42
F GFR1 AAGTCGGACGCAACAGAGAAA SEQ ID NO:43
ERBB2 CUACCUUUCUACGGACGUGdTdT SEQ ID NO:44
BCL2L1 CTGCCTAAGGCGGATTTGAAT SEQ ID NO:45
ABL1 TTAUUCCUUCUUCGGGAAGUC SEQ ID NO:46
CEACAM1 AACCTTCTGGAACCCGCCCAC SEQ ID NO:47
CD9 GAGCATCTTCGAGCAAGAA SEQ ID NO:48
CD151 CATGTGGCACCGTTTGCCT SEQ ID NO:49
Caspase 8 AACTACCAGAAAGGTATACCT SEQ ID NO:50
BRCA1 UCACAGUGUCCUUUAUGUAdTdT SEQ ID NO:51
p53 GCAUGAACCGGAGGCCCAUTT SEQ ID NO:52
CEACAM6 CCGGACAGTTCCATGTATA SEQ ID NO:53
Diagnostic Agents
[0192] In the context of this application, the terms "diagnosis" or
"detection" can be used
interchangeably. Whereas diagnosis usually refers to defining a tissue's
specific histological
status, detection recognizes and locates a tissue, lesion or organism
containing a particular
antigen.
56
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
[0193] The subject antibodies and fragments can be detectably labeled by
linking the
antibody to an enzyme. When the antibody-enzyme conjugate is incubated in the
presence of
the appropriate substrate, the enzyme moiety reacts with the substrate to
produce a chemical
moiety which can be detected, for example, by spectrophotometric, fluorometric
or visual
means. Examples of enzymes that can be used to detectably label antibody
include malate
dehydrogenase, staphylococcal nuclease, delta-V-steroid isomerase, yeast
alcohol
dehydrogenase, alpha-glycerophosphate dehydrogenase, triose phosphate
isomerase,
horseradish peroxidase, alkaline phosphatase, asparaginase, glucose oxidase,
alpha-
galactosidase, ribonuclease, urease, catalase, glucose-6-phosphate
dehydrogenase,
glucoamylase and acetylcholinesterase.
[0194] The immunoconjugate may comprise one or more radioactive isotopes
useful for
detecting diseased tissue. Particularly useful diagnostic radionuclides
include, but are not
limited to, nom, min, 177õ, 18F, 52Fe, 62eu, 64eu, 67cu, 67Ga, 68Ga, 86y, 9n--
'ILl,
89Zr, 94mTC,
94TC, 99mTC, 1201, 1231, 1241, 1251, 1311, 154-158Gd, 321), 11C, 13N, 150,
186Re, 188Re, 51mn, 52mmn,
55CO, 72AS, 75Br, 76Br, 82mRb, 83Sr, or other gamma-, beta-, or positron-
emitters, preferably
with a decay energy in the range of 20 to 4,000 keV, more preferably in the
range of 25 to
4,000 keV, and even more preferably in the range of 25 to 1,000 keV, and still
more
preferably in the range of 70 to 700 keV. Total decay energies of useful
positron-emitting
radionuclides are preferably <2,000 keV, more preferably under 1,000 keV, and
most
preferably <700 keV. Radionuclides useful as diagnostic agents utilizing gamma-
ray
detection include, but are not limited to: 51Cr, 57Co, 58Co, 59Fe, 67Cu, 67Ga,
75Se, 97Ru, 99mTc,
1111n, 1 14min, 1231, 1251, 1311, 169yb, 197,,g
m,
and 201T1. Decay energies of useful gamma-ray
emitting radionuclides are preferably 20-2000 keV, more preferably 60-600 keV,
and most
preferably 100-300 keV.
[0195] Methods of diagnosing cancer in a subject may be accomplished by
administering a
diagnostic immunoconjugate and detecting the diagnostic label attached to an
immunoconjugate that is localized to a cancer or tumor. The antibodies,
antibody fragments
and fusion proteins may be conjugated to the diagnostic agent or may be
administered in a
pretargeting technique using targetable constructs attached to a diagnostic
agent. Radioactive
agents that can be used as diagnostic agents are discussed above. A suitable
non-radioactive
diagnostic agent is a contrast agent suitable for magnetic resonance imaging,
X-rays,
computed tomography or ultrasound. Magnetic imaging agents include, for
example, non-
radioactive metals, such as manganese, iron and gadolinium, complexed with
metal-chelate
57
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
combinations that include 2-benzyl-DTPA and its monomethyl and cyclohexyl
analogs. See
U.S. Ser. No. 09/921,290 (now abandoned) filed on Oct. 10, 2001, the Examples
section of
which is incorporated herein by reference. Other imaging agents such as PET
scanning
nucleotides, preferably 18F, may also be used.
[0196] Contrast agents, such as MRI contrast agents, including, for example,
gadolinium
ions, lanthanum ions, dysprosium ions, iron ions, manganese ions or other
comparable labels,
CT contrast agents, and ultrasound contrast agents may be used as diagnostic
agents.
Paramagnetic ions suitable for use include chromium (III), manganese (II),
iron (III), iron
(II), cobalt (II), nickel (II), copper (II), neodymium (III), samarium (III),
ytterbium (III),
gadolinium (III), vanadium (II), terbium (III), dysprosium (III), holmium
(III) and erbium
(III), with gadolinium being particularly preferred.
[0197] Ions useful in other contexts, such as X-ray imaging, include but are
not limited to
lanthanum (III), gold (III), lead (II) and bismuth (III). Fluorescent labels
include rhodamine,
fluorescein and renographin. Rhodamine and fluorescein are often linked via an
isothiocyanate intermediate.
[0198] Metals are also useful in diagnostic agents, including those for
magnetic resonance
imaging techniques. These metals include, but are not limited to: gadolinium,
manganese,
iron, chromium, copper, cobalt, nickel, dysprosium, rhenium, europium,
terbium, holmium
and neodymium. In order to load an antibody with radioactive metals or
paramagnetic ions,
it may be necessary to react it with a reagent having a long tail to which are
attached a
multiplicity of chelating groups for binding the ions. Such a tail can be a
polymer such as a
polylysine, polysaccharide, or other derivatized or derivatizable chain having
pendant groups
to which can be bound chelating groups such as, e.g.,
ethylenediaminetetraacetic acid
(EDTA), diethylenetriaminepentaacetic acid (DTPA), porphyrins, polyamines,
crown ethers,
bis-thiosemicarbazones, polyoximes, and like groups known to be useful for
this purpose.
[0199] Chelates are coupled to an antibody, fusion protein, or fragments
thereof using
standard chemistries. The chelate is normally linked to the antibody by a
group which
enables formation of a bond to the molecule with minimal loss of
immunoreactivity and
minimal aggregation and/or internal cross-linking. Other, more unusual,
methods and
reagents for conjugating chelates to antibodies are disclosed in U.S. Pat. No.
4,824,659 to
Hawthorne, entitled "Antibody Conjugates", issued Apr. 25, 1989, the Examples
section of
which is incorporated herein by reference. Particularly useful metal-chelate
combinations
include 2-benzyl-DTPA and its monomethyl and cyclohexyl analogs, used with
diagnostic
58
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
isotopes in the general energy range of 20 to 2,000 keV. The same chelates,
when complexed
with non-radioactive metals, such as manganese, iron and gadolinium are useful
for MRI.
Macrocyclic chelates such as NOTA, DOTA, and TETA are of use with a variety of
metals
and radiometals, most particularly with radionuclides of gallium, yttrium and
copper,
respectively. Such metal-chelate complexes can be made very stable by
tailoring the ring
size to the metal of interest. Other ring-type chelates such as macrocyclic
polyethers, which
are of interest for stably binding nuclides, such as 223Ra for RAIT are
encompassed by the
invention.
[0200] Radiopaque and contrast materials are used for enhancing X-rays and
computed
tomography, and include iodine compounds, barium compounds, gallium compounds,
thallium compounds, etc. Specific compounds include barium, diatrizoate,
ethiodized oil,
gallium citrate, iocarmic acid, iocetamic acid, iodamide, iodipamide,
iodoxamic acid,
iogulamide, iohexol, iopamidol, iopanoic acid, ioprocemic acid, iosefamic
acid, ioseric acid,
iosulamide meglumine, iosemetic acid, iotasul, iotetric acid, iothalamic acid,
iotroxic acid,
ioxaglic acid, ioxotrizoic acid, ipodate, meglumine, metrizamide, metrizoate,
propyliodone,
and thallous chloride.
[0201] The antibodies, antibody fragments and fusion proteins also can be
labeled with a
fluorescent compound. The presence of a fluorescent-labeled MAb is determined
by
exposing the antibody to light of the proper wavelength and detecting the
resultant
fluorescence. Fluorescent labeling compounds include Alexa 350, Alexa 430,
AMCA,
aminoacridine, BODIPY 630/650, BODIPY 650/665, BODIPY-FL, BODIPY-R6G,
BODIPY-TMR, BODIPY-TRX, 5-carboxy-4',5'-dichloro-2',7'-dimethoxy fluorescein,
5-
carboxy-2',4',5',7'-tetrachlorofluorescein, 5-carboxyfluorescein, 5-
carboxyrhodamine, 6-
carboxyrhodamine, 6-carboxytetramethyl amino, Cascade Blue, Cy2, Cy3, Cy5,6-
FAM,
dansyl chloride, Fluorescein, fluorescein isothiocyanate, fluorescamine, HEX,
6-JOE, NBD
(7-nitrobenz-2-oxa-1,3-diazole), Oregon Green 488, Oregon Green 500, Oregon
Green 514,
Pacific Blue, phthalic acid, terephthalic acid, isophthalic acid, cresyl fast
violet, cresyl blue
violet, brilliant cresyl blue, para-aminobenzoic acid, erythrosine,
phthalocyanines,
phthaldehyde, azomethines, cyanines, xanthines, succinylfluoresceins, rare
earth metal
cryptates, europium trisbipyridine diamine, a europium cryptate or chelate,
diamine,
dicyanins, La Jolla blue dye, allopycocyanin, allococyanin B, phycocyanin C,
phycocyanin
R, thiamine, phycoerythrocyanin, phycoerythrin R, REG, Rhodamine Green,
rhodamine
isothiocyanate, Rhodamine Red, ROX, TAMRA, TET, TRIT (tetramethyl rhodamine
59
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
isothiol), Tetramethylrhodamine, and Texas Red. Fluorescently-labeled
antibodies are
particularly useful for flow cytometry analysis, but can also be used in
endoscopic and
intravascular detection methods..
[0202] Alternatively, the antibodies, antibody fragments and fusion proteins
can be
detectably labeled by coupling the antibody to a chemiluminescent compound.
The presence
of the chemiluminescent-tagged MAb is determined by detecting the presence of
luminescence that arises during the course of a chemical reaction. Examples of
chemiluminescent labeling compounds include luminol, isoluminol, an aromatic
acridinium
ester, an imidazole, an acridinium salt and an oxalate ester.
[0203] Similarly, a bioluminescent compound can be used to label the
antibodies and
fragments there. Bioluminescence is a type of chemiluminescence found in
biological
systems in which a catalytic protein increases the efficiency of the
chemiluminescent
reaction. The presence of a bioluminescent protein is determined by detecting
the presence of
luminescence. Bioluminescent compounds that are useful for labeling include
luciferin,
luciferase and aequorin.
[0204] Accordingly, a method of diagnosing a malignancy in a subject is
described,
comprising performing an in vitro diagnosis assay on a specimen (fluid, tissue
or cells) from
the subject with a composition comprising an anti-pancreatic cancer MAb,
fusion protein or
fragment thereof Immunohistochemistry can be used to detect the presence of
PAM4
antigen in a cell or tissue by the presence of bound antibody. Preferably, the
malignancy that
is being diagnosed is a cancer. Most preferably, the cancer is pancreatic
cancer.
[0205] Additionally, a chelator such as DTPA, DOTA, TETA, or NOTA or a
suitable
peptide, to which a detectable label, such as a fluorescent molecule, or
cytotoxic agent, such
as a heavy metal or radionuclide, can be conjugated to a subject antibody. For
example, a
therapeutically useful immunoconjugate can be obtained by conjugating a
photoactive agent
or dye to an antibody fusion protein. Fluorescent compositions, such as
fluorochrome, and
other chromogens, or dyes, such as porphyrins sensitive to visible light, have
been used to
detect and to treat lesions by directing the suitable light to the lesion. In
therapy, this has
been termed photoradiation, phototherapy, or photodynamic therapy (Jori et al.
(eds.),
PHOTODYNAMIC THERAPY OF TUMORS AND OTHER DISEASES (Libreria Progetto
1985); van den Bergh, Chem. Britain 22:430 (1986)). Moreover, monoclonal
antibodies have
been coupled with photoactivated dyes for achieving phototherapy. Mew et al.,
J. Immunol.
130:1473 (1983); idem., Cancer Res. 45:4380 (1985); Oseroff et al., Proc Natl.
Acad. Sci.
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
USA 83:8744 (1986); idem., Photochem. Photobiol. 46:83 (1987); Hasan et al.,
Prog. Clin.
Biol. Res. 288:471 (1989); Tatsuta et al., Lasers Surg. Med. 9:422 (1989);
Pelegrin et al.,
Cancer 67:2529 (1991).
[0206] Fluorescent and radioactive agents conjugated to antibodies or used in
bispecific,
pretargeting methods, are particularly useful for endoscopic, intraoperative
or intravascular
detection of the targeted antigens associated with diseased tissues or
clusters of cells, such as
malignant tumors, as disclosed in Goldenberg U.S. Pat. Nos. 5,716,595;
6,096,289 and
6,387,350, the Examples section of each incorporated herein by reference,
particularly with
gamma-, beta- and positron-emitters. Endoscopic applications may be used when
there is
spread to a structure that allows an endoscope, such as the colon.
Radionuclides useful for
positron emission tomography include, but are not limited to: F-18, Mn-51, Mn-
52m, Fe-52,
Co-55, Cu-62, Cu-64, Ga-68, As-72, Br-75, Br-76, Rb-82m, Sr-83, Y-86, Zr-89,
Tc-94m, In-
110, 1-120, and 1-124. Total decay energies of useful positron-emitting
radionuclides are
preferably <2,000 keV, more preferably under 1,000 keV, and most preferably
<700 keV.
Radionuclides useful as diagnostic agents utilizing gamma-ray detection
include, but are not
limited to: Cr-51, Co-57, Co-58, Fe-59, Cu-67, Ga-67, Se-75, Ru-97, Tc-99m, In-
111, In-
114m, 1-123, 1-125, 1-131, Yb-169, Hg-197, and T1-201. Decay energies of
useful gamma-
ray emitting radionuclides are preferably 20-2000 keV, more preferably 60-600
keV, and
most preferably 100-300 keV.
In Vitro Diagnosis
[0207] The present invention contemplates the use of anti-pancreatic cancer
antibodies to
screen biological samples in vitro for the presence of the PAM4 antigen. In
such
immunoassays, the antibody, antibody fragment or fusion protein may be
utilized in liquid
phase or bound to a solid-phase carrier, as described below. For purposes of
in vitro
diagnosis, any type of antibody such as murine, chimeric, humanized or human
may be
utilized, since there is no host immune response to consider.
[0208] One example of a screening method for determining whether a biological
sample
contains MUC5ac is the radioimmunoassay (RIA). For example, in one form of
RIA, the
substance under test is mixed with PAM4 MAb in the presence of radiolabeled
MUC5ac. In
this method, the concentration of the test substance will be inversely
proportional to the
amount of labeled MUC5ac bound to the MAb and directly related to the amount
of free,
labeled MUC5ac. Other suitable screening methods will be readily apparent to
those of skill
in the art.
61
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
[0209] Alternatively, in vitro assays can be performed in which an anti-
pancreatic cancer
antibody, antibody fragment or fusion protein is bound to a solid-phase
carrier. For example,
MAbs can be attached to a polymer, such as aminodextran, in order to link the
MAb to an
insoluble support such as a polymer-coated bead, a plate or a tube.
[0210] Other suitable in vitro assays will be readily apparent to those of
skill in the art. The
specific concentrations of detectably labeled antibody and MUC5ac, the
temperature and time
of incubation, as well as other assay conditions may be varied, depending on
various factors
including the concentration of MUC5ac in the sample, the nature of the sample,
and the like.
The binding activity of a sample of anti-pancreatic cancer antibody may be
determined
according to well-known methods. Those skilled in the art will be able to
determine
operative and optimal assay conditions for each determination by employing
routine
experimentation.
[0211] The presence of the PAM4 antigen in a biological sample can be
determined using an
enzyme-linked immunosorbent assay (ELISA) (e.g., Gold et al. J Clin Oncol.
24:252-58,
2006). In the direct competitive ELISA, a pure or semipure antigen preparation
is bound to a
solid support that is insoluble in the fluid or cellular extract being tested
and a quantity of
detectably labeled soluble antibody is added to permit detection and/or
quantitation of the
binary complex formed between solid-phase antigen and labeled antibody.
[0212] In contrast, a "double-determinant" ELISA, also known as a "two-site
ELISA" or
"sandwich assay," requires small amounts of antigen and the assay does not
require extensive
purification of the antigen. Thus, the double-determinant ELISA is preferred
to the direct
competitive ELISA for the detection of an antigen in a clinical sample. See,
for example, the
use of the double-determinant ELISA for quantitation of the c-myc oncoprotein
in biopsy
specimens. Field et al., Oncogene 4: 1463 (1989); Spandidos et al., AntiCancer
Res. 9: 821
(1989).
[0213] In a double-determinant ELISA, a quantity of unlabeled MAb or antibody
fragment
(the "capture antibody") is bound to a solid support, the test sample is
brought into contact
with the capture antibody, and a quantity of detectably labeled soluble
antibody (or antibody
fragment) is added to permit detection and/or quantitation of the ternary
complex formed
between the capture antibody, antigen, and labeled antibody. In the present
context, an
antibody fragment is a portion of an anti-pancreatic cancer MAb that binds to
an epitope of
MUC5ac. Methods of performing a double-determinant ELISA are well-known. See,
for
example, Field et al., supra, Spandidos et al., supra, and Moore et al., "Twin-
Site ELISAs for
62
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
fos and myc Oncoproteins Using the AMPAK System," in METHODS IN MOLECULAR
BIOLOGY, VOL. 10, pages 273-281 (The Humana Press, Inc. 1992).
[0214] In the double-determinant ELISA, the soluble antibody or antibody
fragment must
bind to a MUC5ac epitope that is distinct from the epitope recognized by the
capture
antibody. The double-determinant ELISA can be performed to ascertain whether
the PAM4
antigen is present in a biopsy sample. Alternatively, the assay can be
performed to quantitate
the amount of MUC5ac that is present in a clinical sample of body fluid. The
quantitative
assay can be performed by including dilutions of purified MUC5ac.
[0215] The anti-pancreatic cancer MAbs, fusion proteins, and fragments thereof
also are
suited for the preparation of an assay kit. Such a kit may comprise a carrier
means that is
compartmentalized to receive in close confinement one or more container means
such as
vials, tubes and the like, each of said container means comprising the
separate elements of the
immunoassay.
[0216] The subject antibodies, antibody fragments and fusion proteins also can
be used to
detect the presence of the PAM4 antigen in tissue sections prepared from a
histological
specimen. Such in situ detection can be used to determine the presence of
MUC5ac and to
determine the distribution of MUC5ac in the examined tissue. In situ detection
can be
accomplished by applying a detectably-labeled antibody to frozen tissue
sections. Studies
indicate that the PAM4 antigen is preserved in paraffin-embedded sections.
General
techniques of in situ detection are well-known to those of ordinary skill.
See, for example,
Ponder, "Cell Marking Techniques and Their Application," in MAMMALIAN
DEVELOPMENT: A PRACTICAL APPROACH 113-38 Monk (ed.) (IRL Press 1987), and
Coligan at pages 5.8.1-5.8.8.
[0217] Antibodies, antibody fragments and fusion proteins can be detectably
labeled with
any appropriate marker moiety, for example, a radioisotope, an enzyme, a
fluorescent label, a
dye, a chromogen, a chemiluminescent label, a bioluminescent labels or a
paramagnetic label.
[0218] The marker moiety can be a radioisotope that is detected by such means
as the use of
a gamma counter or a scintillation counter or by autoradiography. In a
preferred
embodiment, the diagnostic conjugate is a gamma-, beta- or a positron-emitting
isotope. A
marker moiety in the present description refers to a molecule that will
generate a signal under
predetermined conditions. Examples of marker moieties include radioisotopes,
enzymes,
fluorescent labels, chemiluminescent labels, bioluminescent labels and
paramagnetic labels.
63
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
[0219] The binding of marker moieties to anti-pancreatic cancer antibodies can
be
accomplished using standard techniques known to the art. Typical methodology
in this
regard is described by Kennedy et al., Clin Chim Acta 70: 1 (1976), Schurs et
al., Clin. Chim.
Acta 81: 1 (1977), Shih et al., Int J Cancer 46: 1101 (1990).
[0220] The above-described in vitro and in situ detection methods may be used
to assist in
the diagnosis or staging of a pathological condition. For example, such
methods can be used
to detect tumors that express the PAM4 antigen such as pancreatic cancer.
In Vivo Diagnosis/Detection
[0221] Various methods of in vivo diagnostic imaging with radiolabeled MAbs
are well-
known. In the technique of immunoscintigraphy, for example, antibodies are
labeled with a
gamma-emitting radioisotope and introduced into a patient. A gamma camera is
used to
detect the location and distribution of gamma-emitting radioisotopes. See, for
example,
Srivastava (ed.), RADIOLABELED MONOCLONAL ANTIBODIES FOR IMAGING AND
THERAPY (Plenum Press 1988), Chase, "Medical Applications of Radioisotopes,"
in
REMINGTON'S PHARMACEUTICAL SCIENCES, 18th Edition, Gennaro et al. (eds.), pp.
624-652 (Mack Publishing Co., 1990), and Brown, "Clinical Use of Monoclonal
Antibodies,"
in BIOTECHNOLOGY AND PHARMACY 227-49, Pezzuto et al. (eds.) (Chapman & Hall
1993).
[0222] For diagnostic imaging, radioisotopes may be bound to antibody either
directly or
indirectly by using an intermediary functional group. Useful intermediary
functional groups
include chelators such as ethylenediaminetetraacetic acid and
diethylenetriaminepentaacetic
acid. For example, see Shih et al., supra, and U.S. Pat. No. 5,057,313.
[0223] The radiation dose delivered to the patient is maintained at as low a
level as possible
through the choice of isotope for the best combination of minimum half-life,
minimum
retention in the body, and minimum quantity of isotope which will permit
detection and
accurate measurement. Examples of radioisotopes that can be bound to anti-
pancreatic
cancer antibody and are appropriate for diagnostic imaging include 99mTc,
111In and 18F.
[0224] The subject antibodies, antibody fragments and fusion proteins also can
be labeled
with paramagnetic ions and a variety of radiological contrast agents for
purposes of in vivo
diagnosis. Contrast agents that are particularly useful for magnetic resonance
imaging
comprise gadolinium, manganese, dysprosium, lanthanum, or iron ions.
Additional agents
include chromium, copper, cobalt, nickel, rhenium, europium, terbium, holmium,
or
neodymium. Antibodies and fragments thereof can also be conjugated to
ultrasound
64
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
contrast/enhancing agents. For example, one ultrasound contrast agent is a
liposome. Also
preferred, the ultrasound contrast agent is a liposome that is gas filled.
[0225] In a preferred embodiment, a bispecific antibody can be conjugated to a
contrast
agent. For example, the bispecific antibody may comprise more than one image-
enhancing
agent for use in ultrasound imaging. In another preferred embodiment, the
contrast agent is a
liposome. Preferably, the liposome comprises a bivalent DTPA-peptide
covalently attached
to the outside surface of the liposome.
Pharmaceutically Suitable Excipients
[0226] Additional pharmaceutical methods may be employed to control the
duration of
action of an anti-pancreatic cancer antibody in a therapeutic application.
Control release
preparations can be prepared through the use of polymers to complex or adsorb
the antibody,
antibody fragment or fusion protein. For example, biocompatible polymers
include matrices
of poly(ethylene-co-vinyl acetate) and matrices of a polyanhydride copolymer
of a stearic
acid dimer and sebacic acid. Sherwood et al., Bio/Technology 10: 1446 (1992).
The rate of
release of an antibody, antibody fragment or fusion protein from such a matrix
depends upon
the molecular weight of the antibody, antibody fragment or fusion protein, the
amount of
antibody within the matrix, and the size of dispersed particles. Saltzman et
al., Biophys. J.
55: 163 (1989); Sherwood et al., supra. Other solid dosage forms are described
in Ansel et
al., PHARMACEUTICAL DOSAGE FORMS AND DRUG DELIVERY SYSTEMS, 5th
Edition (Lea & Febiger 1990), and Gennaro (ed.), REMINGTON'S PHARMACEUTICAL
SCIENCES, 18th Edition (Mack Publishing Company 1990), and revised editions
thereof
[0227] The antibodies, fragments thereof or fusion proteins to be delivered to
a subject can
comprise one or more pharmaceutically suitable excipients, one or more
additional
ingredients, or some combination of these. The antibody can be formulated
according to
known methods to prepare pharmaceutically useful compositions, whereby the
immunoconjugate or naked antibody is combined in a mixture with a
pharmaceutically
suitable excipient. Sterile phosphate-buffered saline is one example of a
pharmaceutically
suitable excipient. Other suitable excipients are well-known to those in the
art. See, for
example, Ansel et al., PHARMACEUTICAL DOSAGE FORMS AND DRUG DELIVERY
SYSTEMS, 5th Edition (Lea & Febiger 1990), and Gennaro (ed.), REMINGTON'S
PHARMACEUTICAL SCIENCES, 18th Edition (Mack Publishing Company 1990), and
revised editions thereof
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
[0228] The immunoconjugate or naked antibody can be formulated for intravenous
administration via, for example, bolus injection or continuous infusion.
Formulations for
injection can be presented in unit dosage form, e.g., in ampules or in multi-
dose containers,
with an added preservative. The compositions can take such forms as
suspensions, solutions
or emulsions in oily or aqueous vehicles, and can contain formulatory agents
such as
suspending, stabilizing and/or dispersing agents. Alternatively, the active
ingredient can be
in powder form for constitution with a suitable vehicle, e.g., sterile pyrogen-
free water,
before use.
[0229] The immunoconjugate, naked antibody, fragment thereof or fusion protein
may also
be administered to a mammal subcutaneously or by other parenteral routes. In a
preferred
embodiment, the antibody or fragment thereof is administered in a dosage of 20
to 2000
milligrams protein per dose. Moreover, the administration may be by continuous
infusion or
by single or multiple boluses. In general, the dosage of an administered
immunoconjugate,
fusion protein or naked antibody for humans will vary depending upon such
factors as the
patient's age, weight, height, sex, general medical condition and previous
medical history.
Typically, it is desirable to provide the recipient with a dosage of
immunoconjugate, antibody
fusion protein or naked antibody that is in the range of from about 1 mg/kg to
20 mg/kg as a
single intravenous or infusion, although a lower or higher dosage also may be
administered as
circumstances dictate. This dosage may be repeated as needed, for example,
once per week
for four to ten weeks, preferably once per week for eight weeks, and more
preferably, once
per week for four weeks. It may also be given less frequently, such as every
other week for
several months, or more frequently, such as two- or three-time weekly. The
dosage may be
given through various parenteral routes, with appropriate adjustment of the
dose and
schedule.
Kits
[0230] Various embodiments may concern kits containing components suitable for
treating
or diagnosing diseased tissue in a patient. Exemplary kits may contain at
least one antibody,
antigen binding fragment or fusion protein as described herein. If the
composition containing
components for administration is not formulated for delivery via the
alimentary canal, such as
by oral delivery, a device capable of delivering the kit components through
some other route
may be included. One type of device, for applications such as parenteral
delivery, is a
syringe that is used to inject the composition into the body of a subject.
Inhalation devices
may also be used. In certain embodiments, an anti-pancreatic cancer antibody
or antigen
66
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
binding fragment thereof may be provided in the form of a prefilled syringe or
autoinjection
pen containing a sterile, liquid formulation or lyophilized preparation of
antibody (e.g., Kiyitz
et al., Clin. Ther. 2006, 28:1619-29).
[0231] The kit components may be packaged together or separated into two or
more
containers. In some embodiments, the containers may be vials that contain
sterile,
lyophilized formulations of a composition that are suitable for
reconstitution. A kit may also
contain one or more buffers suitable for reconstitution and/or dilution of
other reagents.
Other containers that may be used include, but are not limited to, a pouch,
tray, box, tube, or
the like. Kit components may be packaged and maintained sterilely within the
containers.
Another component that can be included is instructions for use of the kit.
EXAMPLES
[0232] The examples below are illustrative of embodiments of the current
invention and are
not limiting to the scope of the claims. The examples discuss studies
employing an
exemplary anti-pancreatic cancer monoclonal antibody (e.g., PAM4). Clinical
studies with
the PAM4 MAb have shown that a majority of pancreatic cancer lesions were
targeted in
patients and there was no indication of uptake in normal tissues. Dosimetry
indicated that it
was possible to deliver 10 to 20 cGy/mCi to tumors, with a tumor to red marrow
dose ratio of
3:1 to 10:1. The data show that PAM4 is useful for the treatment of pancreatic
cancer.
Example 1. Humanized PAM4 MAb
[0233] In preferred embodiments, the claimed methods and compositions utilize
the antibody
hPAM4 which is a humanized IgG of the murine PAM4 MAb raised against
pancreatic
cancer mucin. Humanization of the murine PAM4 sequences was utilized to reduce
the
human antimouse antibody (HAMA) response. To produce the humanized PAM4,
murine
complementarity determining regions (CDR) were transferred from heavy and
light variable
chains of the mouse immunoglobulin into human framework region (FR) antibody
sequences,
followed by the replacement of some human FR residues with their murine
counterparts.
Humanized monoclonal antibodies are suitable for use in in vitro and in vivo
diagnostic and
therapeutic methods.
[0234] Comparison of the variable region framework sequences of the murine
PAM4 MAb
(FIGS. 1A and 1B) to known human antibodies in the Kabat database showed that
the FRs of
PAM4 Vic and VH exhibited the highest degree of sequence homology to that of
the human
antibodies Walker Vic (FIG. 3A) and Wi12 VH (FIG. 3B), respectively.
Therefore, the
Walker Vic (FIG. 3A) and Wi12 VH (FIG. 3B) FRs were selected as the human
frameworks
67
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
into which the murine CDRs for PAM4 Vic and VH were grafted, respectively. The
FR4
sequence of the human antibody, NEWM, however, was used to replace the Wi12
FR4
sequence for the humanization of the PAM4 heavy chain (FIG. 3B). A few amino
acid
residues in PAM4 FRs that flank the putative CDRs were maintained in hPAM4
based on the
consideration that these residues have more impact on Ag binding than other FR
residues.
These residues were 21M, 47W, 59P, 60A, 85S, 87F, and 100G of Vic (FIG. 3A)
and 27Y,
30P, 38K, 481, 66K, 67A, and 69L of VH (FIG. 3B). The DNA and amino acid
sequences of
hPAM4 Vic (SEQ ID NO:16) and VH (SEQ ID NO:19) are shown in FIGS. 4A and 4B,
respectively.
[0235] A modified strategy as described by Leung et al. (Leung et al., 1994))
was used to
construct the designed Vic (FIG. 4A) and VH (FIG. 4B) genes for hPAM4 using a
combination of long oligonucleotide syntheses and PCR. For the construction of
the hPAM4
VH domain, two long oligonucleotides, hPAM4 VH A (173-mer) and hPAM4 VH B (173-
mer)
were synthesized on an automated DNA synthesizer (Applied Biosystems).
[0236] hPAM4 VH A represents nt 17 to 189 of the hPAM4 VH domain.
5'- AGTCTGGGGC TGAGGTGAAG AAGCCTGGGG CCTCAGTGAA GGTCTCCTGC
GAGGCTTCTG GATACACATT CCCTAGCTAT GTTTTGCACT GGGTGAAGCA
GGCCCCTGGA CAAGGGCTTG AGTGGATTGG ATATATTAAT CCTTACAATG
ATGGTACTCA GTACAATGAG AAG-3' (SEQ ID NO:54)
[0237] hPAM4 VHB represents the minus strand of the hPAM4 VH domain
complementary
to nt 169 to 341.
5'- AGGGTTCCCT GGCCCCAGTA AGCAAATCCG TAGCTACCAC CGAAGCCTCT
TGCACAGTAA TACACGGCCG TGTCGTCAGA TCTCAGCCTG CTCAGCTCCA
TGTAGGCTGT GTTGATGGAC GTGTCCCTGG TCAGTGTGGC CTTGCCTTTG
AACTTCTCAT TGTACTGAGT ACC-3' (SEQ ID NO:55)
[0238] The 3'-terminal sequences (21 nt residues) of hPAM4 VHA and VHB are
complementary to each other. Under defined PCR condition, the 3'-ends of hPAM4
VHA and
VHB anneal to form a short double stranded DNA flanked by the rest of the long
oligonucleotides. Each annealed end serves as a primer for the transcription
of the single
stranded DNA, resulting in a double strand DNA composed of the nt 17 to 341 of
hPAM4
VH. This DNA was further amplified in the presence of two short
oligonucleotides, hPAM4
VHBACK and hPAM4 VHFOR to form the full-length hPAM4 VH. The underlined
portions
are restriction sites for subcloning as shown in FIG. 4B.
68
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
hPAM4 VHBACK 5'-CAG GTG CAG CTG CAG CAG TCT GGG GCT GAG GTG A-3'
(SEQ ID NO:56)
hPAM4 VHFOR 5'-TGA GGA GAC GGT GAC CAG GGT TCC CTG GCC CCA-3' (SEQ
ID NO:57)
[0239] A minimal amount of hPAM4 VHA and VHB (determined empirically) was
amplified
in the presence of 10 1.1,L of 10X PCR Buffer (500 mM KC1, 100 mM Tris HC1
buffer, pH 8.3,
15 mM MgC12), 2 [Imo' of hPAM4 VHBACK and hPAM4 VKFOR, and 2.5 units of Taq
DNA polymerase (Perkin Elmer Cetus, Norwalk, Conn.). This reaction mixture was
subjected to three cycles of polymerase chain reaction (PCR) consisting of
denaturation at
94 C for 1 minute, annealing at 45 C for 1 minute, and polymerization at 72 C
for 1.5
minutes. This procedure was followed by 27 cycles of PCR reaction consisting
of
denaturation at 94 C for 1 minute, annealing at 55 C for 1 minute, and
polymerization at 72 C
for 1 minute. Double-stranded PCR-amplified product for hPAM4 VH was gel-
purified,
restriction-digested with PstI and BstEII restriction sites and cloned into
the complementary
PstI/BstEII restriction sites of the heavy chain staging vector, VHpBS2, in
which the VH
sequence was fully assembled with the DNA sequence encoding the translation
initiation
codon and a secretion signal peptide in-frame ligated at the 5'-end and an
intron sequence at
the 3'-end. VHpBS2 is a modified staging vector of VHpBS (Leung et al.,
Hybridoma,
13:469, 1994), into which a XhoI restriction site was introduced at sixteen
bases upstream of
the translation initiation codon to facilitate the next subcloning step. The
assembled VH gene
was subcloned as a XhoI-BamHI restriction fragment into the expression vector,
pdHL2,
which contains the expression cassettes for both human IgG heavy and light
chains under the
control of IgH enhancer and MT1 promoter, as well as a mouse d/fr gene as a
marker for
selection and amplification. Since the heavy chain region of pdHL2 lacks a
BamHI
restriction site, this ligation requires use of a linker to provide a bridge
between the BamHI
site of the variable chain and the HindIII site present in the pdHL2 vector.
The resulting
expression vectors were designated as hPAM4 VHpdHL2.
[0240] For constructing the full length DNA of the humanized Vic sequence
(FIG. 4A),
hPAM4 VKA (157-mer) and hPAM4 VKB (156-mer) were synthesized as described
above.
hPAM4 VKA and VKB were amplified by two short oligonucleotides hPAM4 VKBACK
and
hPAM4 VKFOR as described above.
[0241] hPAM4 VKA represents nt 16 to 172 of the hPAM4 Vic domain.
5'-CAGTCTCCAT CCTCCCTGTC TGCATCTGTA GGAGACAGAG TCACCATGAC
69
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
CTGCAGTGCC AGCTCAAGTG TAAGTTCCAG CTACTTGTAC TGGTACCAAC
AGAAACCAGG GAAAGCCCCC AAACTCTGGA TTTATAGCAC ATCCAACCTG
GCTTCTG-3' (SEQ ID NO:58)
[0242] hPAM4 VKB represents the minus strand of the hPAM4 Vic domain
complementary
to nt 153 to 308.
5'-GTCCCCCCTC CGAACGTGTA CGGGTACCTA TTCCACTGAT GGCAGAAATA
AGAGGCAGAA TCTTCAGGTT GCAGACTGCT GATGGTGAGA GTGAAGTCTG
TCCCAGATCC ACTGCCACTG AAGCGAGCAG GGACTCCAGA AGCCAGGTTG
GATGTG-3' (SEQ ID NO:59)
[0243] The 3'-terminal sequences (20 nt residues) of hPAM4 VKA and VKB are
complementary to each other. Under defined PCR condition, the 3'-ends of hPAM4
VKA and
VKB anneal to form a short double-stranded DNA flanked by the rest of the long
oligonucleotides. Each annealed end served as a primer for the transcription
of the single
stranded DNA, resulting in a double strand DNA composed of nt 16 to 308 of
hPAM4 Vic
This DNA was further amplified in the presence of two short oligonucleotides,
hPAM4
VKBACK and hPAM4 VKFOR to form the full-length hPAM4 Vic The underlined
portions
are restriction sites for subcloning as described below.
hPAM4 VKBACK 5'-GAC ATC CAG CTG ACC CAG TCT CCA TCC TCC CTG-3' (SEQ
ID NO:60)
hPAM4 VKFOR 5'- TTA GAT CTC CAG TCG TGT CCC CCC TCC GAA CGT-3' (SEQ ID
NO:61)
[0244] Gel-purified PCR products for hPAM4 Vic were restriction-digested with
PyuII and
BglII and cloned into the complementary Pvull/Bc1I sites of the light chain
staging vector,
VKpBR2. VKpBR2 is a modified staging vector of VKpBR (Leung et al., Hybridoma,
13:469,
1994), into which a XbaI restriction site was introduced at sixteen bases
upstream of the
translation initiation codon. The assembled Vic genes were subcloned as XbaI-
BamHI
restriction fragments into the expression vector containing the VH sequence,
hPAM4
VHpdHL2. The resulting expression vectors were designated as hPAM4pdHL2.
[0245] Approximately 30 ug of hPAM4pdHL2 was linearized by digestion with Sall
and
transfected into 5p2/0-Ag14 cells by electroporation at 450 V and 25 F. The
transfected
cells were plated into 96-well plates and incubated in a CO2 cell culture
incubator for two
days and then selected for MTX resistance. Colonies surviving selection
emerged in two to
three weeks and were screened for human antibody secretion by ELISA assay.
Briefly,
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
supernatants (-100 ul) from the surviving colonies were added into the wells
of an ELISA
microplate precoated with goat anti-human IgG F(ab')2 fragment-specific Ab.
The plate was
incubated for one hour at room temperature. Unbound proteins were removed by
washing
three times with wash buffer (PBS containing 0.05% Tween-20). Horseradish
peroxidase-
conjugated goat anti-human IgG Fc fragment-specific Ab was added to the wells.
Following
incubation for one hour, a substrate solution (100 p.L/well) containing 4 mM o-
phenylenediamine dihydrochloride (OPD) and 0.04% H202 in PBS was added to the
wells
after washing. Color was allowed to develop in the dark for 30 minutes and the
reaction was
stopped by the addition of 50 1.1,L of 4 N H2504 solution. The bound human IgG
was
measured by reading the absorbance at 490 nm on an ELISA reader. Positive cell
clones
were expanded and hPAM4 was purified from cell culture supernatant by affinity
chromatography on a Protein A column.
[0246] The Ag-binding activity of hPAM4 was confirmed by ELISA assay in a
microtiter
plate coated with pancreas cancer cell extracts. An ELISA competitive binding
assay using
PAM4-antigen coated plates was developed to assess the Ag-binding affinity of
hPAM4 in
comparison with that of a chimeric PAM4 composed of murine V and human C
domains.
Constant amounts of the HRP-conjugated cPAM4 mixed with varying concentrations
of
cPAM4 or hPAM4 were added to the coated wells and incubated at room
temperature for 1-2
h. The amount of HRP-conjugated cPAM4 bound to the CaPanl Ag was revealed by
reading
the absorbance at 490 nm after the addition of a substrate solution containing
4 mM o-
phenylenediamine dihydrochloride and 0.04% H202. As shown by the competition
assays in
FIG. 5, hPAM4 and cPAM4 antibodies exhibited similar binding activities.
Example 2. Immunohistochemistry Staining Studies
[0247] Immunohistochemistry on normal adult tissues showed that the PAM4
reactive
epitope was restricted to the gastrointestinal tract where staining was weak,
yet positive
(Table 2). Normal pancreatic tissue, including ducts, ductules, acini, and
islet cells, were
negative for staining. A PAM4 based enzyme immunoassay with tissue homogenates
as
antigens generally supported the immunohistology data (Table 3). The PAM4
epitope was
absent from normal pancreas and other non-gastrointestinal tissues. In
neoplastic tissues,
PAM4 was reactive with twenty one out of twenty five (85%) pancreatic cancers
(Table 4
and Table 5) and ten out of twenty six colon cancers, but only limited
reactivity with tumors
of the stomach, lung, breast, ovary, prostate, liver or kidney (Table 5). PAM4
reactivity
appeared to correlate with the stage of tumor differentiation, with a greater
percentage of
71
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
staining observed in well differentiated pancreatic cancers than in moderately
differentiated
or poorly differentiated tumors. Generally, poorly differentiated tumors
represent less than
10% of all pancreatic cancers.
[0248] These studies have shown the PAM4 reactivity and tissue distribution
(both normal
and cancer) to be unlike that reported for the CA19.9, DUPAN2, SPAN1, Nd2 and
B72.3
antibodies and antibodies against the Lewis antigens. Together with
crossblocking studies
performed with certain of these MAbs, the data suggests that the PAM4 MAb
recognizes a
unique and novel epitope. When compared to the antigens recognized by the
CA19.9,
DUPAN2, and anti-Lea antibodies, the PAM4 antigen appears to be more
restricted in its
tissue distribution and is reactive with a higher percentage of pancreatic
tumors. Moreover, it
gives a greater overall intensity of reaction at equivalent concentrations and
is reactive with a
higher percentage of cells within the pancreatic tumors. Finally, PAM4 was
found to be only
weakly reactive with three out of twelve chronic pancreatitis specimens,
whereas CA19.9 and
DUPAN2 were strongly reactive with all twelve specimens. Although it is
recognized that
specificity is dependent in part upon the type of assay employed and the range
and number of
tissues examined, the ability of PAM4 to discriminate between normal and
neoplastic
pancreatic tissue, its ability to react with a large percentage of the cancer
specimens, the high
intensity of the reactions, and the ability to distinguish between early stage
pancreatic cancer
and benign conditions such as pancreatitis are important characteristics of
this exemplary
anti-pancreatic cancer antibody.
Table 2. Immunoperoxidase Staining of Normal Adult Tissues with MAb PAM4
Tissue Staining
Reaction
Pancreas (22)a -
Ducts -
Acini -
Islets -
Submaxillary gland (2) -
Esophagus (2) -
Stomach (3) + mucus secreting cells
Duodenum (3) + goblet cells
Jejunum (3) + goblet cells
72
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
Ileum (3) + goblet cells
Colon (5) + goblet cells
Liver (3) -
Gallbladder (2) -
Bronchus (3) -
Lung (3) -
Heart (3) -
Spleen (3) -
Kidney (3) -
Bladder (3) -
Prostate (2) -
Testes (2) -
Uterus (2) -
Ovary (2) -
a
¨ number of individual specimens examined in parentheses
Table 3. Monoclonal Antibody PAM4 Reactivity with Normal Adult Tissue
Homogenates by EIA
Tissue gig tissuea
Pancreas 6.4
Esophagus 8.1
Stomach 61.3
Duodenum 44.7
Jejunum 60.6
Colon 74.5
Liver 0.0
Gallbladder 5.6
Heart 3.7
Spleen 3.4
Kidney 6.6
Bladder 4.9
Thyroid 3.5
Adrenal 1.3
73
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
Ureter 2.6
Testes 3.9
CaPanl Pancreatic Tumor 569
a ¨ values are mean from two autopsy specimens
Table 4. Immunohistochemical Reactivity of Several Monoclonal Antibodies with
Pancreatic Tumors
Differentiation PAM4 CA19.9 Lea DUPAN2
1 W +++- - +++
2 M ++ +++ +++ +
3 M +- + +
4 M +++ +++ +++ +
M ++ +-
-
6 M + ND ND ND
7 M. +++ +++ +++ +++
8 M +- - +++
9 M ++ + ++ -
M* ++ ++ ++ +++
11 M ++ +++ +++ +
12 M ++ + + +++
13 M + +++ +++ +
14 M ++ + + ++
M +++ + + ++
16 M + + ++ -
17 M - + + -
18 M ++ ++ ++ ++
19 M +++ + +++ ++
M +- - -
21 M +++ +++ + ++
22 P + + + +++
23 P - - - -
24 P - - - -
P - - + -
74
CA 02899811 2015-07-29
WO 2014/165506 PCT/US2014/032513
TOTAL 21/25 17/24 18/24 16/24
- : Negative; +: 5-20% of tissue is stained; ++: 21-50% of tissue is stained;
+++ : >50% of tissue is stained; W,M,P : Well, moderate, or poor
differentiation;
* : Metastatic tissue; ND : Not Done
Table 5. Immunoperoxidase Staining of Neoplastic Tissues with MAb PAM4
Cancer Tissue Positive/Total
Pancreas 21/25
Colon 10/26
Stomach 1/5
Lung 1/15
Breast 0/30
Ovarian 0/10
Prostate 0/4
Liver 0/10
Kidney 0/4
Example 3. In Vivo Biodistribution and Tumor Targeting of Radiolabeled PAM4
[0249] Initial biodistribution studies of PAM4 were carried out in a series of
four different
xenografted human pancreatic tumors covering the range of expected
differentiation. Each of
the four tumor lines employed, AsPcl, BxPc3, Hs766T and CaPanl, exhibited
concentrations
of131I-PAM4 within the tumors (range: 21%-48% ID/g on day three) that were
significantly
(P<0.01-0.001) higher than concomitantly administered nonspecific, isotype-
matched Ag8
antibody (range: 3.6%-9.3% ID/g on day three). The biodistribution data were
used to
estimate potential radiation doses to the tumor of 12,230; 10,684; 6,835; and
15,843 cGy/mCi
of injected dose to AsPcl, BxPc3, Hs766T and CaPanl, respectively. With an
actual
maximum tolerated dose (MTD) of 0.7 mCi, PAM4 could provide substantial rad
dose to
each of the xenografted tumor models. In each tumor line the blood levels of
radiolabeled
PAM4 were significantly (P<0.01-0.001) lower than the nonspecific Ag8.
Potential radiation
doses to the blood from PAM4 were 1.4-4.4 fold lower than from Ag8. When
radiation doses
to the tumor from PAM4 were normalized to the blood doses from PAM4, the
tumors
received doses that were 2.2; 3.3; 3.4; and 13.1-fold higher than blood,
respectively.
Importantly, potential radiation doses to non-tumor tissues were minimal.
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
[0250] The biodistribution of PAM4 was compared with an anti-CEA antibody, MN-
14,
using the CaPanl tumor model. The concentration of PAM4 within the tumor was
much
greater than MN-14 at early timepoints, yielding tumonblood ratios at day
three of 12.7 2.3
for PAM4 compared to 2.7 1.9 for MN-14. Although PAM4 uptake within the
tumor was
significantly higher than for MN-14 at early timepoints (day one--P<0.001; day
three--
P<0.01), dosimetry analyses indicated only a 3.2-fold higher dose to the tumor
from PAM4
as compared to MN-14 over the fourteen day study period. This was due to a
rapid clearance
of PAM4 from the tumor, such that at later timepoints similar concentrations
of the two
antibodies were present within the tumors. A rapid clearance of PAM4 from the
tumor was
also noted in the BxPc3 and Hs766T but not AsPc1 tumor models. These
observations were
unlike those reported for other anti-mucin antibodies, as for example G9 and
B72.3 in
colorectal cancer, where each exhibited longer retention times as compared to
the MN-14
antibody. Results from studies on the metabolism of PAM4, indicate that after
initial binding
to the tumor cell, antibody is rapidly released, possibly being catabolized or
being shed as an
antigen:antibody complex. The blood clearance is also very rapid. These data
suggest that
1311 may not be the appropriate choice of isotope for therapeutic
applications. A short-lived
isotope, such as 90Y or 188Re, which can be administered frequently may be a
more effective
reagent.
[0251] PAM4 showed no evidence of targeting to normal tissues, except in the
CaPanl
tumor model, where a small but statistically significant splenic uptake was
observed (range
3.1-7.5% ID/g on day-3). This type of splenic targeting has been observed in
the clinical
application of the anti-mucin antibodies B72.3 and CC49. Importantly, these
studies also
reported that splenic targeting did not affect tumor uptake of antibody nor
did it interfere with
interpretation of the nuclear scans. These studies suggested that splenic
targeting was not due
to crossreactive antigens in the spleen, nor to binding by Fc receptors, but
rather to one or
more of the following possibilities: direct targeting of antigen trapped in
the spleen, or
indirect uptake of antigen:antibody complexes formed either in the blood or
released from the
tumor site. The latter would require the presence of immune complexes in the
blood.
However, these were not observed when specimens as early as five minutes and
as late as
seven days were examined by gel filtration (HPLC, GF-250 column); radiolabeled
antibody
eluted as native material. The former explanation seems more likely in view of
the fact that
the CaPanl tumor produced large quantities of PAM4-reactive antigen, 100- to
1000-fold
higher than for the other tumor cell lines examined. The lack of splenic
targeting by PAM4
76
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
in these other tumor lines suggests that this phenomenon was related to
excessive antigen
production. Splenic targeting can be overcome by increasing the protein dose
to 10 mg from
the original 2 mg dose. A greater amount of the splenic entrapped antigen
presumably was
complexed with unlabeled PAM4 rather than radiolabeled antibody. Increasing
the protein
dose had no adverse effect upon targeting of PAM4 to the tumor or nontumor
tissues. In fact,
an increase of the protein dose to 100 mg more than doubled the concentration
of radiolabeled
PAM4 within the CaPanl tumor.
Example 4. Development of Orthotopic Pancreatic Tumor Model in Athymic
Nude Mice.
[0252] In order to resemble the clinical presentation of pancreatic cancer in
an animal model
more closely, we developed an orthotopic model by injecting tumor cells
directly into the
head of the pancreas. Orthotopic CaPanl tumors grew progressively without
overt symptoms
until the development of ascites and death at ten to fourteen weeks. By three
to four weeks
post-implantation, animals developed a palpable tumor of approximately 0.2 g.
Within eight
weeks of growth, primary tumors of approximately 1.2 g along with metastases
to the liver
and spleen were observed (1-3 metastatic tumors/animal; each tumor <0.1 g). At
ten to
fourteen weeks seeding of the diaphragm with development of ascites were
evident. Ascites
formation and occasional jaundice were usually the first overt indications of
tumor growth.
At this time tumors were quite large, 1 to 2 g, and animals had at most only
three to four
weeks until death occurred.
[0253] Radiolabeled 131I-PAM4, administered to animals bearing four week old
orthotopic
tumors (approximately 0.2 g) showed specific targeting to the primary tumor
with
localization indices of 7.9 3.0 at day one increasing to 22.8 15.3 at day
fourteen. No
evidence of specific targeting to other tissues was noted. In one case where
tumor metastases
to the liver and spleen were observed, both metastases were targeted, and had
high
concentrations of radiolabeled antibody. In addition, approximately half of
the animals
developed a subcutaneous tumor at the incision site. No significant
differences were noted in
the targeting of orthotopic and subcutaneous tumors within the same animal,
and no
significant differences were observed in the targeting of orthotopic tumor
whether or not the
animal had an additional subcutaneous tumor. The estimated radiation doses
from PAM4
were 6,704 and 1,655 cGy/mCi to the primary tumor and blood, respectively.
77
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
Example 5. Radioimmunotherapy of Pancreatic Cancer
[0254] The initial studies on the use of 1311-PAM4 for therapy were carried
out with the
CaPanl tumor, which was grown as a subcutaneous xenograft in athymic mice.
Animals
bearing a 0.25 g tumor were administered 350 uCi, 1311-PAM4 in an experiment
that also
compared the therapeutic effects of a similar dose of nonspecific Ag8. The MTD
for
administration of 1311-PAM4 to animals bearing 1 cm3 tumors is 700 uCi. By
weeks five and
six, the PAM4 treated animals showed a dramatic regression of tumor, and even
at week
twenty seven, five out of eight remained tumor free. The untreated, as well as
Ag8-treated
animals, showed rapid progression of tumor growth although a significant
difference was
noted between these two control groups. At seven weeks, tumors from the
untreated group
had grown 20.0 14.6-fold from the initial timepoint whereas the 1311-Ag8-
treated tumors
had grown only 4.9 1.8-fold. At this time point, the PAM4 tumors had
regressed to 0.1
0.1-fold of their original size, a significant difference from both untreated
(Pí0.001) and
nonspecific Ag8-treated (P< 0.0 1) animals.
[0255] These data show that CaPanl tumors were sensitive to treatment with
1311-PAM4.
The outcome, that is, regression or progression of the tumor, was dependent
upon several
factors including initial tumor size. Thus, groups of animals bearing CaPanl
tumor burdens
of 0.25 g, 0.5 g, 1.0 g, or 2.0 g were treated with a single dose of the 350
uCi 1311-PAM4.
The majority of animals having tumors of initial size 0.25 g and 0.5 g (nine
of ten animals in
each group) showed tumor regression or growth inhibition for at least sixteen
weeks post
treatment. In the 1.0 g tumor group five out of seven showed no tumor growth
for the sixteen
week period and in the 2.0 g tumor group six out of nine showed no tumor
growth for a
period of six weeks before progression occurred. Although a single 350 uCi
dose was not as
effective against larger tumors, a single dose may not be the appropriate
regimen for large
tumors.
[0256] Toxicity studies indicate the ability to give multiple cycles of
radioimmunotherapy,
which may be more effective with a larger tumor burden. Animals bearing CaPanl
tumors
averaging 1.0 g, were given either a single dose of 350 uCi 1311-PAM4, two
doses given at
times zero and four weeks or were left untreated. The untreated group had a
mean survival
time of 3.7 1.0 weeks (survival defined as time for tumor to reach 5 em3).
Animals died as
early as three weeks, with no animal surviving past six weeks. A single dose
of 350 uCi 131I.
PAM4 produced a significant increase in the survival time to 18.8 4.2 weeks
(P<0.0001).
78
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
The range of animal deaths extended from weeks thirteen to twenty five. None
of the
animals were alive at the end of the study period of twenty six weeks.
[0257] A significant increase in survival time was observed for the two dose
group as
compared to the single dose group. Half of the animals were alive at the
twenty six week
timepoint with tumor sizes from 1.0-2.8 cm3, and a mean tumor growth rate of
1.6 0.7 fold
from initial tumor size. For those animals that were non-survivors at twenty
six weeks, the
mean survival time (17.7 5.3 weeks) was similar to the single dose group.
[0258] Therapy studies with PAM4 were also conducted using the orthotopic
tumor model.
Groups of animals bearing four week old orthotopic tumors (estimated tumor
weight of 0.25
g) were either left untreated or treated with a single dose of either 350 laCi
131I-PAM4 or 350
laCi of 131I-nonspecific Ag8. The untreated animals had a 50% death rate by
week ten with
no survivors at week fifteen. Animals administered nonspecific 131I-Ag8 at
four weeks of
tumor growth, showed a 50% death rate at week seven with no survivors at week
fourteen.
Although statistically (logrank analysis) there were no differences between
these two groups,
it is possible that radiation toxicity had occurred in the Ag8 treated
animals. Radiolabeled
PAM4 provided a significant survival advantage (P<0.001) as compared to the
untreated or
Ag8 treated animals, with 70% survival at sixteen weeks, the end of the
experiment. At this
time the surviving animals were sacrificed to determine tumor size. All
animals had tumor
with an average weight of 1.2 g, as well as one or two small (<0.1 g)
metastases evident in
four of the seven animals. At sixteen weeks of growth, these tumors were more
representative of an eight-week-old tumor.
Example 6. Combined Modality GEMZAR Chemotherapy and 1311-PAM4
Experimental Radioimmunotherapy
[0259] Initial studies into the combined use of gemcitabine (GEMZARO) with
131I-PAM4
radioimmunotherapy were performed as a checkerboard array; a single dose of
gemcitabine
(0, 100, 200, 500 mg/kg) versus a single dose of 131I-PAM4 ([MTD=700 [iCi]
100%, 75%,
50%, 0% of the MTD). The combined MTD was found to be 500 mg/kg gemcitabine
with
350 laCi 131I-PAM4 (50% MTD). Toxicity, as measured by loss of body weight,
went to the
maximum considered as nontoxic; that is 20% loss in body weight. Although the
combined
treatment protocol was significantly more effective than gemcitabine alone,
the treatment was
no more effective than radioimmunotherapy alone. The next studies were
performed at a low
dose of gemcitabine and radioimmunotherapy to examine if a true synergistic
therapeutic
effect would be observed. Athymic nude mice bearing tumors of approximately 1
cm3
79
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
(approximately 5% of body weight) were administered gemcitabine, 100 mg/kg on
days zero,
three, six, nine, and twelve, with 100 ,Ci of131I-PAM4 given on day zero. A
therapeutic
effect was observed with statistically significant (P<0.0001) regression (two
of five tumors
less than 0.1 cm3) and/or growth inhibition of the tumors compared to
gemcitabine alone.
Thus, at lower dosages of therapeutic agent, there surprisingly appears to be
a synergistic
effect of the combination of gemcitabine and radioimmunotherapy. Of additional
note, in
terms of body weight, toxicity was not observed. The combination treatment
protocol can, if
necessary, be delivered in multiple cycles, with the second treatment cycle
beginning in
week-four, as was done with the radioimmunotherapy-alone studies described
above.
Example 7. Effects of Reagent Treatment on Immunoreactivity of PAM4
Antigen
[0260] Treatment of pancreatic mucin with DTT (15 min at room temp),
completely
abolished reactivity with PAM4 (DTT-EC50, 0.60 + 0.00 !LIM). The only
cysteines (cystine
bridges) within MUC-1 are present within the transmembrane domain and should
not be
accessible to DTT. The secreted form of MUC-1 does not contain the
transmembrane
domain and therefore has no intramolecular cystine bridges. Data from
periodate oxidation
treatment of pancreatic cancer mucin with 0.05 M sodium periodate for 2 hrs at
room
temperature yielded 40% loss of immunoreactivity with PAM4 antibody (not
shown).
Further periodate studies have shown as high as a 60% loss of immunoreactivity
with PAM4
antibody (not shown). The results of periodate and DTT studies suggest that
the PAM4
epitope is conformationally dependent upon some minimal form of glycosylation,
and may be
affected by intermolecular disulfide bond formation.
Example 8. Distribution and Cross-Reactivity of the PAM4 Antigen
[0261] The expression of the PAM4-epitope within PanINs is atypical for MUC-1.
It is
similar to the expression reported for MUC5ac as detected by the commercially
available
MAb-CLH2-2. However, an attempted sandwich immunoassay with PAM4 capture and
MAb-CLH2-2 as probe gave negative results. Although this possibly suggests the
PAM4 and
CLH2-2 epitopes may overlap and thus block each other, the CLH2-2 was reported
to be
reactive with 42/66 (64%) gastric carcinomas whereas the PAM4 MAb showed
reactivity
with only 6/40 (15%) of gastric carcinomas and, of these, only in focal
reactivity.
[0262] Use of the commercially available 45M1, an anti-MUC5ac MAb, as a probe
reagent
in EIA (with PAM4 as capture) provided positive results, indicating that the
two epitopes
may be present on the same antigenic molecule. Blocking studies (either
direction) indicated
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
that the epitopes bound by 45M1 and PAM4 are in fact two distinct epitopes, as
no blocking
was observed. Labeling of tissue microarrays consisting of cores from invasive
pancreatic
carcinoma has demonstrated significant differences for expression of the 45M1
and PAM4
epitopes in individual patient specimens. 0f28 specimens, concordance was
observed in
only 17 cases (61%). PAM4 was reactive with 24/28 cases (86%) while 45M1 was
reactive
with 13/28 (46%) cases (not shown).
[0263] The results of periodate studies are consistent with glycosylation as a
factor in
MUC5ac immunoreactivity with the PAM4 antibody. Thus, results of studies with
apomucins
may not be definitive for antigen determination.
[0264] Although based on EIA capture, the PAM4 antibody appears to bind to the
same
antigenic protein as the 45M1 anti-MUC5ac MAb, it is noted that MUC5ac is not
specific to
pancreas cancer and it is found in a number of normal tissues (other than the
gastric mucosa
with which PAM4 is reactive). For example, MUC5ac is found in normal lung,
colon and
other tissues. PAM4 antibody does not bind to normal lung tissues, except as
indicated above
in few samples and to a limited or minimal amount.
[0265] With respect to the effects of DTT and periodate, it is probable that
the peptide core
disulfide bridges are identical no matter what tissue produces the protein. A
specific amino
acid sequence should fold in a specific manner, independent of the tissue
source. However,
glycosylation patterns may differ dependent upon tissue source.
Example 9. Phage Display Peptide Binding of PAM4 Antibody
[0266] PAM4 antibody binding was examined with two different phage display
peptide
libraries. The first was a linear peptide library consisting of 12 amino acid
sequences and the
second was a cyclic peptide consisting of 7 amino acid sequences cyclized by a
disulfide
bridge. We panned the individual libraries alternately against hPAM4 and hLL2
(negative
selection with anti-CD22 antibody) for a combined total of 4 rounds, and then
screened the
phage displayed peptide for reactivity with both hPAM4 and mPAM4 with little
to no
reactivity against hLL2. Phage binding in a non-specific manner (i.e., binding
to
epratuzumab [hLL2]) were discarded.
[0267] For the linear phage-displayed peptide, the sequence WTWNITKAYPLP (SEQ
ID
NO:7) was identified 30 times (in 35 sequenced phage), each of which were
shown to have
reactivity with PAM4 antibodies. A mutational analysis was conducted in which
a library
based on this sequence and having 7.5% degeneracy at each position, was
constructed,
panned and screened as before. Variability was noted in the 19 obtained
peptide sequences
81
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
that were positive for PAM4 binding with 7 being identical to the parental
sequence, 5 having
the sequence WTWNITKEYPQP (SEQ ID NO:62) and the rest being uniquely present.
Table 6 shows the results of this mutational analysis. The upper row lists the
sequences
identified and the lower row lists the frequency with which each of the amino
acids was
identified in that position. The parent sequence is most frequent (bold) with
the next highest
variation a substitution of E for A at position 8 and a substitution of Q for
L at position 11. It
does not appear that these substitutions had any great effect upon
immunoreactivity.
Table 6. Phage Display Amino Acid Sequence (SEQ ID NO: 113) Variation with
Linear
Peptide Binding to PAM4 Antibody
WTWN I T K A Y P L P
D R R E T R Q
T T I
N R M
G F C
C
number of 19 19 19 18 19 17 14 10 18 17 11
19
occurrences 1 2 1 5 1 2 5
(out of 19 2 1 1
sequences 1 1 1
analyzed) 1 1 1
1
[0268] Results with the phage displayed cyclic library were significantly
different from the
linear library (Table 7). The sequence ACPEWWGTTC (SEQ ID NO:63) was present
in 33
of 35 peptide sequences examined. Analysis of the cyclic library presented the
following
results (positions with an asterisk were invariant and not subject to
selective pressure in the
library).
Table 7. Phage Display Amino Acid Sequence (SEQ ID NO: 114) Variation with
Linear
Peptide Binding to PAM4 Antibody
82
CA 02899811 2015-07-29
WO 2014/165506 PCT/US2014/032513
A C P E W W G T T C
Y S G M
S S
Q
P
number of * * 33 35 35 35 34 29 28 *
occurrences 2 1 5 4
(out of 19 1 1
sequences 1
analyzed) 1
[0269] The two cysteines (at positions 2 and 10) formed a disulfide bridge.
Substitution of T
at position 9 with any amino acid greatly affected immunoreactivity. The
sequence
GTTGTTC (SEQ ID NO:64) is present within the MUC5ac protein towards the amino
terminus as compared to the cyclic peptide sequence shown above, which shows
homology at
the C-terminal end of the consensus peptide sequence. However, the cyclic
peptide only
showed approximately 10% of the immunoreactivity of the linear sequence with
the PAM4
antibody. Both linear and cyclic consensus sequences are associated with a
cysteine, which
may or may not relate to the effect of DTT on MUC5ac immunoreactivity.
[0270] The results reported herein indicate that the PAM4 epitope is dependent
upon a
specific conformation which may be produced by disulfide bridges, as well as a
specific
glycosylation pattern.
Example 10. Immunohistology of Pancreatic Cancer in a Pancreatitis Specimen
[0231] Several pathologic conditions predispose patients to the development of
pancreatic
carcinoma, such as pancreatitis, diabetes, smoking and others. Within this pre-
selected group
of patients, screening measures are particularly important for the early
detection of pancreatic
neoplasia. We examined 9 specimens of chronic pancreatitis tissue from
patients having
primary diagnosis of this disease. We employed an anti-CD74 MAb, LL1, as an
indicator of
inflammatory infiltrate, and MAb-MA5 as a positive control for pancreatic
ductal and acinar
cells. Whereas the two control MAbs provided immunohistologic evidence
consistent with
pancreatitis, in no instance did PAM4 react with inflamed pancreatic tissue.
However, in one
case, a moderately differentiated pancreatic adenocarcinoma was also present
within the
tissue specimen. PAM4 gave an intense stain of the neoplastic cells within
this tumor. In a
83
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
second case, while the inflamed tissue was negative with PAM4, a small PanIN
precursor
lesion was identified that was labeled with PAM4. Labeling of the PanIN within
this latter
specimen is consistent with early detection of pancreatic neoplasia in a
patient diagnosed
with a non-malignant disease. These results show that detection and/or
diagnosis using the
PAM4 antibody may be performed with high sensitivity and selectivity for
pancreatic
neoplasia against a background of benign pancreatic tissues.
Example 11. Therapy of a Patient With Inoperable and Metastatic Pancreatic
Carcinoma
[0232] Patient 118-001, CWG, is a 63-year-old man with Stage-IV pancreatic
adenocarcinoma
with multiple liver metastases, diagnosed in November of 2007. He agreed to
undertake
combined radioimmunotherapy and gemcitabine chemotherapy as a first treatment
strategy,
and was then given a first therapy cycle of 6.5 mCi/m2 of 9 Y-hPAM4, combined
with 200
mg/m2 gemcitabine, whereby the gemcitabine was given once weekly on weeks 1-4
and 90Y-
hPAM4 was given once-weekly on weeks 2-4 (3 doses). Two months later, the same
therapy
cycle was repeated, because no major toxicities were noted after the first
cycle. Already 4
weeks after the first therapy cycle, CT evidence of a reduction in the
diameters of the primary
tumor and 2 of the 3 liver metastases surprisingly was noted, and this was
consistent with
significant decreases in the SUV values of FDG-PET scans, with 3 of the 4
tumors returning
to normal background SUV levels at this time (FIG. 6 and FIG. 7). The
patient's pre-therapy
CA-19.9 level of 1,297 dropped to a low level of 77, further supportive of the
therapy being
effective. Table 8 shows the effects of combined radioimmunotherapy with 90Y-
hPAM4 and
gemcitabine chemotherapy in this patient. It was surprising and unexpected
that such low
doses of the radionuclide conjugated to the antibody combined with such low,
nontoxic, doses
of gemcitabine showed such antitumor activity even after only a single course
of this therapy.
Table 8. Effects of Combined Radioimmunotherapy with 90Y-hPAM4 and Gemcitabine
Chemotherapy in Metastatic Pancreatic Carcinoma
Tumor Baseline 4 wk post-Tx Baseline PET 4 wk post-Tx
Location Longest Longest (SUV) PET (SUV)
Diameter (cm) Diameter (cm)
Pancreatic tail 4.5 4.3 9.2 4.2
(primary)
L hepatic met 1.9 1.9 4.1 background
84
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
R post hepatic 1.7 1.6 3.7 background
met
R central hepatic 1.9 1.2 3.2 background
met
Example 12. Therapy of a Patient with Inoperable Metastatic Pancreatic
Carcinoma
[0233] A 56-year-old male with extensive, inoperable adenocarcinoma of the
pancreas, with
several liver metastases ranging from 1 to 4 cm in diameter, substantial
weight loss (30 lbs of
weight or more), mild jaundice, lethargy and weakness, as well as abdominal
pains requiring
medication, is given 4 weekly infusions of gemcitabine at doses of 200 mg/m2
each. On the
last three gemcitabine infusions, 90Y-DOTA-hPAM4 radiolabeled humanized
antibody is
administered at a dose of 10 mCi/m2 of 9 Y and 20 mg antibody protein, in a
two-hour i.v.
infusion. Two weeks later, the patient is given a course of gemcitabine
chemotherapy
consisting of 3 weekly doses of 600 mg/m2 by i.v. infusion. The patient is
then evaluated 4
weeks later, and has a mild leukopenia (grade-2), no other major blood or
enzyme changes
over baseline, but shows an improvement in the blood CA19.9 titer from 5,700
to 1,200 and a
decrease in jaundice, with an overall subjective improvement. This follows 3
weeks later
with a repeat of the cycle of lower-dose gemcitabine (weekly x 4), with 3
doses of 9 Y-
DOTA-hPAM4. Four weeks later, the patient is reevaluated, and the CT and PET
scans
confirm an approximately 40% reduction of total tumor mass (primary cancer and
metastases), with a further reduction of the CA19.9 titer to 870. The patient
regains appetite
and activity, and is able to return to more usual daily activities without the
need for pain
medication. He gains 12 lbs after beginning this experimental therapy. A
repeat of the scans
and blood values indicates that this response is maintained 6 weeks later.
Example 13. Preparation of DNLTM Constructs for Pretargeting
DDD and AD Fusion Proteins
[0271] The DNLTM technique can be used to make dimers, trimers, tetramers,
hexamers, etc.
comprising virtually any antibodies or fragments thereof or other effector
moieties. For
certain preferred embodiments, IgG antibodies or Fab antibody fragments may be
produced
as fusion proteins containing either a dimerization and docking domain (DDD)
or anchoring
domain (AD) sequence. Although in preferred embodiments the DDD and AD
moieties are
produced as fusion proteins, the skilled artisan will realize that other
methods of conjugation,
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
such as chemical cross-linking or click chemistry, may be utilized within the
scope of the
claimed methods and compositions.
[0272] Bispecific antibodies may be formed by combining a Fab-DDD fusion
protein of a
first antibody with a Fab-AD fusion protein of a second antibody.
Alternatively, constructs
may be made that combine IgG-AD fusion proteins with Fab-DDD fusion proteins.
The
technique is not limiting and any protein or peptide of use may be produced as
an AD or
DDD fusion protein for incorporation into a DNLTM construct. Where chemical
cross-linking
is utilized, the AD and DDD conjugates are not limited to proteins or peptides
and may
comprise any molecule that may be cross-linked to an AD or DDD sequence using
any cross-
linking technique known in the art. In certain exemplary embodiments, a
polyethylene glycol
(PEG) or other polymeric moiety may be incorporated into a DNLTM construct, as
described
in further detail below.
[0273] For pretargeting applications, an antibody or fragment containing a
binding site for an
antigen associated with a diseased tissue, such as a tumor-associated antigen
(TAA), may be
combined with a second antibody or fragment that binds a hapten on a
targetable construct, to
which a therapeutic and/or diagnostic agent is attached. The DNLTm-based
bispecific
antibody is administered to a subject, circulating antibody is allowed to
clear from the blood
and localize to target tissue, and the conjugated targetable construct is
added and binds to the
localized antibody for diagnosis or therapy.
[0274] Independent transgenic cell lines may be developed for each Fab or IgG
fusion
protein. Once produced, the modules can be purified if desired or maintained
in the cell
culture supernatant fluid. Following production, any DDD-fusion protein module
can be
combined with any AD-fusion protein module to generate a bispecific DNLTM
construct. For
different types of constructs, different AD or DDD sequences may be utilized.
Exemplary
DDD and AD sequences are provided below.
DDD1: SHIQIPPGLTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID
NO:65)
DDD2: CGHIQIPPGLTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID
NO:66)
86
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
AD1: QIEYLAKQIVDNAIQQA (SEQ ID NO:67)
AD2: CGQIEYLAKQIVDNAIQQAGC (SEQ ID NO:68)
[0275] The skilled artisan will realize that DDD1 and DDD2 comprise the DDD
sequence of
the human RIIcc form of protein kinase A. However, in alternative embodiments,
the DDD
and AD moieties may be based on the DDD sequence of the human RIcc form of
protein
kinase A and a corresponding AKAP sequence, as exemplified in DDD3, DDD3C and
AD3
below.
DDD3
SLRECELYVQKHNIQALLKDSIVQLCTARPERPMAFLREYFERLEKEEAK (SEQ ID
NO:69)
DDD3C
MSCGGSLRECELYV QKHNIQALLKDSIV QLCT ARPERPMAFLREYFERLEKEEAK
(SEQ ID NO:70)
AD3
CGFEELAWKIAKMIWSDVFQQGC (SEQ ID NO:71)
Expression Vectors
[0276] The plasmid vector pdHL2 has been used to produce a number of
antibodies and
antibody-based constructs. See Gillies et al., J Immunol Methods (1989),
125:191-202;
Losman et al., Cancer (Phila) (1997), 80:2660-6. The di-cistronic mammalian
expression
vector directs the synthesis of the heavy and light chains of IgG. The vector
sequences are
mostly identical for many different IgG-pdHL2 constructs, with the only
differences existing
in the variable domain (VH and VI) sequences. Using molecular biology tools
known to
those skilled in the art, these IgG expression vectors can be converted into
Fab-DDD or Fab-
AD expression vectors. To generate Fab-DDD expression vectors, the coding
sequences for
the hinge, CH2 and CH3 domains of the heavy chain are replaced with a sequence
encoding
the first 4 residues of the hinge, a 14 residue Gly-Ser linker and the first
44 residues of human
RIIcc (referred to as DDD1). To generate Fab-AD expression vectors, the
sequences for the
hinge, CH2 and CH3 domains of IgG are replaced with a sequence encoding the
first 4
residues of the hinge, a 15 residue Gly-Ser linker and a 17 residue synthetic
AD called
AKAP-IS (referred to as AD1), which was generated using bioinformatics and
peptide array
technology and shown to bind RIIcc dimers with a very high affinity (0.4 nM).
See Alto, et
al. Proc. Natl. Acad. Sci., U.S.A (2003), 100:4445-50.
87
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
[0277] Two shuttle vectors were designed to facilitate the conversion of IgG-
pdHL2 vectors
to either Fab-DDD1 or Fab-AD1 expression vectors, as described below.
Preparation of CH1
[0278] The CH1 domain was amplified by PCR using the pdHL2 plasmid vector as a
template. The left PCR primer consisted of the upstream (5') end of the CH1
domain and a
SacII restriction endonuclease site, which is 5' of the CH1 coding sequence.
The right primer
consisted of the sequence coding for the first 4 residues of the hinge (PKSC
(SEQ ID NO:
115)) followed by four glycines and a serine, with the final two codons (GS)
comprising a
Bam HI restriction site. The 410 bp PCR amplimer was cloned into the PGEMTO
PCR
cloning vector (PROMEGAO, Inc.) and clones were screened for inserts in the T7
(5')
orientation.
[0279] A duplex oligonucleotide, designated (G4S)2DDD1 ('(G45)2' disclosed as
SEQ ID NO:
116), was synthesized by Sigma GENOSYSO (Haverhill, UK) to code for the amino
acid
sequence of DDD1 preceded by 11 residues of the linker peptide, with the first
two codons
comprising a BamHI restriction site. A stop codon and an EagI restriction site
are appended
to the 3'end. The encoded polypeptide sequence is shown below.
GSGGGGSGGGGSHIQIPPGLTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA
(SEQ ID NO:72)
[0280] Two oligonucleotides, designated RIIA1-44 top and RIIA1-44 bottom,
which overlap
by 30 base pairs on their 3' ends, were synthesized and combined to comprise
the central 154
base pairs of the 174 bp DDD1 sequence. The oligonucleotides were annealed and
subjected
to a primer extension reaction with Taq polymerase. Following primer
extension, the duplex
was amplified by PCR. The amplimer was cloned into PGEMTO and screened for
inserts in
the T7 (5') orientation.
[0281] A duplex oligonucleotide was synthesized to code for the amino acid
sequence of
AD1 preceded by 11 residues of the linker peptide with the first two codons
comprising a
BamHI restriction site. A stop codon and an EagI restriction site are appended
to the 3' end.
The encoded polypeptide sequence is shown below.
GSGGGGSGGGGSQIEYLAKQIVDNAIQQA (SEQ ID NO:73)
[0282] Two complimentary overlapping oligonucleotides encoding the above
peptide
sequence, designated AKAP-IS Top and AKAP-IS Bottom, were synthesized and
annealed.
88
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
The duplex was amplified by PCR. The amplimer was cloned into the PGEMTO
vector and
screened for inserts in the T7 (5') orientation.
Ligating DDD1 with CH1
[0283] A 190 bp fragment encoding the DDD1 sequence was excised from PGEMTO
with
BamHI and NotI restriction enzymes and then ligated into the same sites in CH1-
PGEMTO
to generate the shuttle vector CH1-DDD1-PGEMTO.
Ligating AD 1 with CH1
[0284] A 110 bp fragment containing the AD1 sequence was excised from PGEMTO
with
BamHI and NotI and then ligated into the same sites in CH1-PGEMTO to generate
the
shuttle vector CH1-AD1-PGEMTO.
Cloning CH1-DDD1 or CH1-AD1 into pdHL2-based vectors
[0285] With this modular design either CH1-DDD1 or CH1-AD1 can be incorporated
into
any IgG construct in the pdHL2 vector. The entire heavy chain constant domain
is replaced
with one of the above constructs by removing the SacII/EagI restriction
fragment (CH1-CH3)
from pdHL2 and replacing it with the SacII/EagI fragment of CH1-DDD1 or CH1-
AD1,
which is excised from the respective pGemT shuttle vector.
Construction of h679-Fd-AD1-pdHL2
[0286] h679-Fd-AD1-pdHL2 is an expression vector for production of h679 Fab
with AD1
coupled to the carboxyl terminal end of the CH1 domain of the Fd via a
flexible Gly/Ser
peptide spacer composed of 14 amino acid residues. A pdHL2-based vector
containing the
variable domains of h679 was converted to h679-Fd-AD1-pdHL2 by replacement of
the
SacII/EagI fragment with the CH1-AD1 fragment, which was excised from the CH1-
AD1-
SV3 shuttle vector with SacII and EagI.
Construction of C-DDD1-Fd-hMN-14-pdHL2
[0287] C-DDD1-Fd-hMN-14-pdHL2 is an expression vector for production of a
stable dimer
that comprises two copies of a fusion protein C-DDD1-Fab-hMN-14, in which DDD1
is
linked to hMN-14 Fab at the carboxyl terminus of CH1 via a flexible peptide
spacer. The
plasmid vector hMN-14(I)-pdHL2, which has been used to produce hMN-14 IgG, was
converted to C-DDD1-Fd-hMN-14-pdHL2 by digestion with SacII and EagI
restriction
endonucleases to remove the CH1-CH3 domains and insertion of the CH1-DDD1
fragment,
which was excised from the CH1-DDD1-5V3 shuttle vector with SacII and EagI.
89
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
[0288] The same technique has been utilized to produce plasmids for Fab
expression of a
wide variety of known antibodies, such as hLL1, hLL2, hPAM4, hRl, hRS7, hMN-
14, hMN-
15, hA19, hA20 and many others. Generally, the antibody variable region coding
sequences
were present in a pdHL2 expression vector and the expression vector was
converted for
production of an AD- or DDD-fusion protein as described above. The AD- and DDD-
fusion
proteins comprising a Fab fragment of any of such antibodies may be combined,
in an
approximate ratio of two DDD-fusion proteins per one AD-fusion protein, to
generate a
trimeric DNLTM construct comprising two Fab fragments of a first antibody and
one Fab
fragment of a second antibody.
C-DDD2-Fd-hMN-14-pdHL2
[0289] C-DDD2-Fd-hMN-14-pdHL2 is an expression vector for production of C-DDD2-
Fab-
hMN-14, which possesses a dimerization and docking domain sequence of DDD2
appended
to the carboxyl terminus of the Fd of hMN-14 via a 14 amino acid residue
Gly/Ser peptide
linker. The fusion protein secreted is composed of two identical copies of hMN-
14 Fab held
together by non-covalent interaction of the DDD2 domains.
[0290] The expression vector was engineered as follows. Two overlapping,
complimentary
oligonucleotides, which comprise the coding sequence for part of the linker
peptide and
residues 1-13 of DDD2, were made synthetically. The oligonucleotides were
annealed and
phosphorylated with T4 PNK, resulting in overhangs on the 5' and 3' ends that
are compatible
for ligation with DNA digested with the restriction endonucleases BamHI and
PstI,
respectively.
[0291] The duplex DNA was ligated with the shuttle vector CH1-DDD1-PGEMTO,
which
was prepared by digestion with BamHI and PstI, to generate the shuttle vector
CH1-DDD2-
PGEMTO. A 507 bp fragment was excised from CH1-DDD2-PGEMTO with SacII and EagI
and ligated with the IgG expression vector hMN-14(I)-pdHL2, which was prepared
by
digestion with SacII and EagI. The final expression construct was designated C-
DDD2-Fd-
hMN-14-pdHL2. Similar techniques have been utilized to generated DDD2-fusion
proteins
of the Fab fragments of a number of different humanized antibodies.
h679-Fd-AD2-pdHL2
[0292] h679-Fab-AD2, was designed to pair as B to C-DDD2-Fab-hMN-14 as A. h679-
Fd-
AD2-pdHL2 is an expression vector for the production of h679-Fab-AD2, which
possesses
an anchoring domain sequence of AD2 appended to the carboxyl terminal end of
the CH1
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
domain via a 14 amino acid residue Gly/Ser peptide linker. AD2 has one
cysteine residue
preceding and another one following the anchor domain sequence of AD1.
[0293] The expression vector was engineered as follows. Two overlapping,
complimentary
oligonucleotides (AD2 Top and AD2 Bottom), which comprise the coding sequence
for AD2
and part of the linker sequence, were made synthetically. The oligonucleotides
were
annealed and phosphorylated with T4 PNK, resulting in overhangs on the 5' and
3' ends that
are compatible for ligation with DNA digested with the restriction
endonucleases BamHI and
SpeI, respectively.
[0294] The duplex DNA was ligated into the shuttle vector CH1-AD1-PGEMTO,
which was
prepared by digestion with BamHI and SpeI, to generate the shuttle vector CH1-
AD2-
PGEMTO. A 429 base pair fragment containing CH1 and AD2 coding sequences was
excised from the shuttle vector with SacII and EagI restriction enzymes and
ligated into
h679-pdHL2 vector that prepared by digestion with those same enzymes. The
final
expression vector is h679-Fd-AD2-pdHL2.
Example 14. Production of AD- and DDD-linked Fab and IgG Fusion Proteins
From Multiple Antibodies
[0295] Using the techniques described in the preceding Example, the IgG and
Fab fusion
proteins shown in Table 9 were constructed and incorporated into DNLTM
constructs. The
fusion proteins retained the antigen-binding characteristics of the parent
antibodies and the
DNLTM constructs exhibited the antigen-binding activities of the incorporated
antibodies or
antibody fragments.
Table 9. Fusion proteins comprising IgG or Fab
Fusion Protein Binding Specificity
C-AD1-Fab-h679 HSG
C-AD2-Fab-h679 HSG
C-(AD)2-Fab-h679 HSG
C-AD2-Fab-h734 Indium-DTPA
C-AD2-Fab-hA20 CD20
C-AD2-Fab-hA2OL CD20
C-AD2-Fab-hL243 HLA-DR
91
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
C-AD2-Fab-hLL2 CD22
N-AD2-Fab-hLL2 CD22
C-AD2-IgG-hMN-14 CEACAM5
C-AD2-IgG-hR1 IGF-1R
C-AD2-IgG-hRS7 EGP-1
C-AD2-IgG-hPAM4 MUC
C-AD2-IgG-hLL1 CD74
C-DDD1-Fab-hMN-14 CEACAM5
C-DDD2-Fab-hMN-14 CEACAM5
C-DDD2-Fab-h679 HSG
C-DDD2-Fab-hAl9 CD19
C-DDD2-Fab-hA20 CD20
C-DDD2-Fab-hAFP AFP
C-DDD2-Fab-hL243 HLA-DR
C-DDD2-Fab-hLL1 CD74
C-DDD2-Fab-hLL2 CD22
C-DDD2-Fab-hMN-3 CEACAM6
C-DDD2-Fab-hMN-15 CEACAM6
C-DDD2-Fab-hPAM4 MUC
C-DDD2-Fab-hR1 IGF-1R
C-DDD2-Fab-hRS7 EGP-1
N-DDD2-Fab-hMN-14 CEACAM5
Example 15. Sequence variants for DNLTm
[0296] In certain preferred embodiments, the AD and DDD sequences incorporated
into the
DNLTM construct comprise the amino acid sequences of AD1, AD2, AD3, DDD1,
DDD2,
92
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
DDD3 or DDD3C as discussed above. However, in alternative embodiments sequence
variants of AD and/or DDD moieties may be utilized in construction of the
DNLTM
complexes. For example, there are only four variants of human PKA DDD
sequences,
corresponding to the DDD moieties of PKA RIa, Rila, RIP and RII13. The Rila
DDD
sequence is the basis of DDD1 and DDD2 disclosed above. The four human PKA DDD
sequences are shown below. The DDD sequence represents residues 1-44 of Rila,
1-44 of
RII13, 12-61 of RIa and 13-66 of RIP. (Note that the sequence of DDD1 is
modified slightly
from the human PKA RIIa DDD moiety.)
PKA H&c
SLRECELYVQKHNIQALLKDVSIVQLCTARPERPMAFLREYFEKLEKEEAK (SEQ ID
NO:74)
PKA RI /3
SLKGCELYVQLHGIQQVLKDCIVHLCISKPERPMKFLREHFEKLEKEENRQILA (SEQ
ID NO:75)
PKA Rila
SHIQIPPGLTELLQGYTVEVGQQPPDLVDFAVEYFTRLREARRQ (SEQ ID NO:76)
PKA RH fi
SIEIPAGLTELLQGFTVEVLRHQPADLLEFALQHFTRLQQENER (SEQ ID NO:77)
[0297] The structure-function relationships of the AD and DDD domains have
been the
subject of investigation. (See, e.g., Burns-Hamuro et al., 2005, Protein Sci
14:2982-92; Carr
et al., 2001, J Biol Chem 276:17332-38; Alto et al., 2003, Proc Natl Acad Sci
USA 100:4445-
50; Hundsrucker et al., 2006, Biochem J 396:297-306; Stokka et al., 2006,
Biochem J
400:493-99; Gold et al., 2006, Mol Cell 24:383-95; Kinderman et al., 2006, Mol
Cell 24:397-
408, the entire text of each of which is incorporated herein by reference.)
[0298] For example, Kinderman et al. (2006) examined the crystal structure of
the AD-DDD
binding interaction and concluded that the human DDD sequence contained a
number of
conserved amino acid residues that were important in either dimer formation or
AKAP
binding, underlined in SEQ ID NO:65 below. (See Figure 1 of Kinderman et al.,
2006,
incorporated herein by reference.) The skilled artisan will realize that in
designing sequence
variants of the DDD sequence, one would desirably avoid changing any of the
underlined
residues, while conservative amino acid substitutions might be made for
residues that are less
critical for dimerization and AKAP binding.
93
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
SHIQIPPGLTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO: 65)
[0299] Alto et al. (2003) performed a bioinformatic analysis of the AD
sequence of various
AKAP proteins to design an RII selective AD sequence called AKAP-IS (SEQ ID
NO:67),
with a binding constant for DDD of 0.4 nM. The AKAP-IS sequence was designed
as a
peptide antagonist of AKAP binding to PKA. Residues in the AKAP-IS sequence
where
substitutions tended to decrease binding to DDD are underlined in SEQ ID
NO:67. The
skilled artisan will realize that in designing sequence variants of the AD
sequence, one would
desirably avoid changing any of the underlined residues, while conservative
amino acid
substitutions might be made for residues that are less critical for DDD
binding.
AKAP-IS sequence
QIEYLAKQIVDNAIQQA (SEQ ID NO:67)
[0300] Gold (2006) utilized crystallography and peptide screening to develop a
SuperAKAP-
IS sequence (SEQ ID NO:78), exhibiting a five order of magnitude higher
selectivity for the
RII isoform of PKA compared with the RI isoform. Underlined residues indicate
the
positions of amino acid substitutions, relative to the AKAP-IS sequence, which
increased
binding to the DDD moiety of RIIa. In this sequence, the N-terminal Q residue
is numbered
as residue number 4 and the C-terminal A residue is residue number 20.
Residues where
substitutions could be made to affect the affinity for RIIa were residues 8,
11, 15, 16, 18, 19
and 20 (Gold et al., 2006). It is contemplated that in certain alternative
embodiments, the
SuperAKAP-IS sequence may be substituted for the AKAP-IS AD moiety sequence to
prepare DNLTM constructs. Other alternative sequences that might be
substituted for the
AKAP-IS AD sequence are shown in SEQ ID NO:79-81. Substitutions relative to
the
AKAP-IS sequence are underlined. It is anticipated that, as with the AD2
sequence shown in
SEQ ID NO:68, the AD moiety may also include the additional N-terminal
residues cysteine
and glycine and C-terminal residues glycine and cysteine.
SuperAKAP-IS
QIEYVAKQIVDYAIHQA (SEQ ID NO:78)
Alternative AKAP sequences
QIEYKAKQIVDHAIHQA (SEQ ID NO:79)
QIEYHAKQIVDHAIHQA (SEQ ID NO:80)
94
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
QIEYVAKQIVDHAIHQA (SEQ ID NO:81)
[0301] Figure 2 of Gold et al. disclosed additional DDD-binding sequences from
a variety of
AKAP proteins, shown below.
RII-Specific AKAPs
AKAP-KL
PLEYQAGLLVQNAIQQAI (SEQ ID NO:82)
AKAP79
LLIETASSLVKNAIQLSI (SEQ ID NO:83)
AKAP-Lbc
LIEEAASRIVDAVIEQVK (SEQ ID NO:84)
RI-Specific AKAPs
AKAPce
ALYQFADRFSELVISEAL (SEQ ID NO:85)
RIAD
LEQVANQLADQIIKEAT (SEQ ID NO:86)
PV38
FEELAWKIAKMIWSDVF (SEQ ID NO:87)
Dual-Specificity AKAPs
AKAP7
ELVRLSKRLVENAVLKAV (SEQ ID NO:88)
MAP2D
TAEEVSARIVQVVTAEAV (SEQ ID NO:89)
DAKAP1
QIKQAAFQLISQVILEAT (SEQ ID NO:90)
DAKAP2
LAWKIAKMIVSDVMQQ (SEQ ID NO:91)
[0302] Stokka et al. (2006) also developed peptide competitors of AKAP binding
to PKA,
shown in SEQ ID NO:92-94. The peptide antagonists were designated as Ht31 (SEQ
ID
NO:92), RIAD (SEQ ID NO:93) and PV-38 (SEQ ID NO:94). The Ht-31 peptide
exhibited a
greater affinity for the RII isoform of PKA, while the RIAD and PV-38 showed
higher
affinity for RI.
Ht31
DLIEEAASRIVDAVIEQVKAAGAY (SEQ ID NO:92)
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
RIAD
LEQYANQLADQIIKEATE (SEQ ID NO:93)
PV-38
FEELAWKIAKMIWSDVFQQC (SEQ ID NO:94)
[0303] Hundsrucker et al. (2006) developed still other peptide competitors for
AKAP binding
to PKA, with a binding constant as low as 0.4 nM to the DDD of the RII form of
PKA. The
sequences of various AKAP antagonistic peptides are provided in Table 1 of
Hundsrucker et
al., reproduced in Table 10 below. AKAPIS represents a synthetic RII subunit-
binding
peptide. All other peptides are derived from the RII-binding domains of the
indicated
AKAPs.
Table 10. AKAP Peptide sequences
Peptide Sequence
AKAPIS QIEYLAKQIVDNAIQQA (SEQ ID NO:67)
AKAPIS-P QIEYLAKQIPDNAIQQA (SEQ ID NO:95)
Ht31 KGADLIEEAASRIVDAVIEQVKAAG (SEQ ID NO:96)
Ht31-P KGADLIEEAASRIPDAPIEQVKAAG (SEQ ID NO:97)
AKAP76-wt-pep PEDAELVRLSKRLVENAVLKAVQQY (SEQ ID NO:98)
AKAP76-L304T-pep PEDAELVRTSKRLVENAVLKAVQQY (SEQ ID NO:99)
AKAP76-L308D-pep PEDAELVRLSKRDVENAVLKAVQQY (SEQ ID NO:100)
AKAP76-P-pep PEDAELVRLSKRLPENAVLKAVQQY (SEQ ID NO:101)
AKAP76-PP-pep PEDAELVRLSKRLPENAPLKAVQQY (SEQ ID NO:102)
AKAP76-L314E-pep PEDAELVRLSKRLVENAVEKAVQQY (SEQ ID NO:103)
AKAP1-pep EEGLDRNEEIKRAAFQIISQVISEA (SEQ ID NO:104)
AKAP2-pep LVDDPLEYQAGLLVQNAIQQAIAEQ (SEQ ID NO:105)
AKAP5-pep QYETLLIETASSLVKNAIQLSIEQL (SEQ ID NO:106)
AKAP9-pep LEKQYQEQLEEEVAKVIVSMSIAFA (SEQ ID NO:107)
AKAP10-pep NTDEAQEELAWKIAKMIVSDIMQQA (SEQ ID NO:108)
AKAP 11-pep VNLDKKAVLAEKIVAEAIEKAEREL (SEQ ID NO:109)
AKAP12-pep NGILELETKSSKLVQNIIQTAVDQF (SEQ ID NO:110)
AKAP14-pep TQDKNYEDELTQVALALVEDVINYA (SEQ ID NO:111)
Rab32-pep ETSAKDNINIEEAARFLVEKILVNH (SEQ ID NO:112)
96
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
[0304] Residues that were highly conserved among the AD domains of different
AKAP
proteins are indicated below by underlining with reference to the AKAP IS
sequence (SEQ
ID NO:67). The residues are the same as observed by Alto et al. (2003), with
the addition of
the C-terminal alanine residue. (See FIG. 4 of Hundsrucker et al. (2006),
incorporated herein
by reference.) The sequences of peptide antagonists with particularly high
affinities for the
RII DDD sequence were those of AKAP-IS, AKAP76-wt-pep, AKAP76-L304T-pep and
AKAP76-L308D-pep.
AKAP-IS
QIEYLAKQIVDNAIQQA (SEQ ID NO:67)
[0305] Carr et al. (2001) examined the degree of sequence homology between
different
AKAP-binding DDD sequences from human and non-human proteins and identified
residues
in the DDD sequences that appeared to be the most highly conserved among
different DDD
moieties. These are indicated below by underlining with reference to the human
PKA RIIa
DDD sequence of SEQ ID NO:65. Residues that were particularly conserved are
further
indicated by italics. The residues overlap with, but are not identical to
those suggested by
Kinderman et al. (2006) to be important for binding to AKAP proteins. The
skilled artisan
will realize that in designing sequence variants of DDD, it would be most
preferred to avoid
changing the most conserved residues (italicized), and it would be preferred
to also avoid
changing the conserved residues (underlined), while conservative amino acid
substitutions
may be considered for residues that are neither underlined nor italicized..
SHIQ/PPGLTELLQGYTVEVLRQOPPDLVEFAVEYFTRLREARA (SEQ ID NO:65)
[0306] The skilled artisan will realize that these and other amino acid
substitutions in the
antibody moiety or linker portions of the DNLTM constructs may be utilized to
enhance the
therapeutic and/or pharmacokinetic properties of the resulting DNLTM
constructs.
Example 16. Generation of TF2 DNLTM Pretargeting Construct
[0307] A trimeric DNLTM construct designated TF2 was obtained by reacting C-
DDD2-Fab-
hMN-14 with h679-Fab-AD2. A pilot batch of TF2 was generated with >90% yield
as
follows. Protein L-purified C-DDD2-Fab-hMN-14 (200 mg) was mixed with h679-Fab-
AD2
(60 mg) at a 1.4:1 molar ratio. The total protein concentration was 1.5 mg/ml
in PBS
containing 1 mM EDTA. Subsequent steps involved TCEP reduction, HIC
chromatography,
DMSO oxidation, and IMP 291 affinity chromatography. Before the addition of
TCEP, SE-
HPLC did not show any evidence of a2b formation. Addition of 5 mM TCEP rapidly
resulted in the formation of a2b complex consistent with a 157 kDa protein
expected for the
97
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
binary structure. TF2 was purified to near homogeneity by IMP 291 affinity
chromatography
(not shown). IMP 291 is a synthetic peptide containing the HSG hapten to which
the 679 Fab
binds (Rossi et al., 2005, Clin Cancer Res 11:7122s-29s). SE-HPLC analysis of
the IMP 291
unbound fraction demonstrated the removal of a4, az and free kappa chains from
the product
(not shown).
[0308] Non-reducing SDS-PAGE analysis demonstrated that the majority of TF2
exists as a
large, covalent structure with a relative mobility near that of IgG (not
shown). The additional
bands suggest that disulfide formation is incomplete under the experimental
conditions (not
shown). Reducing SDS-PAGE shows that any additional bands apparent in the non-
reducing
gel are product-related (not shown), as only bands representing the
constituent polypeptides
of TF2 were evident (not shown). However, the relative mobilities of each of
the four
polypeptides were too close to be resolved. MALDI-TOF mass spectrometry (not
shown)
revealed a single peak of 156,434 Da, which is within 99.5% of the calculated
mass (157,319
Da) of TF2.
[0309] The functionality of TF2 was determined by BIACOREO assay. TF2, C-DDD1-
hMN-14+h679-AD I (used as a control sample of noncovalent a2b complex), or C-
DDD2-
hMN-14+h679-AD2 (used as a control sample of unreduced az and b components)
were
diluted to 1 ug/m1 (total protein) and passed over a sensorchip immobilized
with HSG. The
response for TF2 was approximately two-fold that of the two control samples,
indicating that
only the h679-Fab-AD component in the control samples would bind to and remain
on the
sensorchip. Subsequent injections of WI2 IgG, an anti-idiotype antibody for
hMN-14,
demonstrated that only TF2 had a DDD-Fab-hMN-14 component that was tightly
associated
with h679-Fab-AD as indicated by an additional signal response. The additional
increase of
response units resulting from the binding of WI2 to TF2 immobilized on the
sensorchip
corresponded to two fully functional binding sites, each contributed by one
subunit of C-
DDD2-Fab-hMN-14. This was confirmed by the ability of TF2 to bind two Fab
fragments of
WI2 (not shown).
Example 17. Production of TF10 Bispecific Antibody for Pretargeting
[0310] A similar protocol was used to generate a trimeric TF10 DNLTM
construct, comprising
two copies of a C-DDD2-Fab-hPAM4 and one copy of C-AD2-Fab-679. The cancer-
targeting antibody component in TF10 was derived from hPAM4, a humanized anti-
MUC5ac
MAb that has been studied in detail as a radiolabeled MAb (e.g., Gold et al.,
Clin. Cancer
Res. 13: 7380-7387, 2007). The hapten-binding component was derived from h679,
a
98
CA 02899811 2015-07-29
WO 2014/165506 PCT/US2014/032513
humanized anti-histaminyl-succinyl-glycine (HSG) MAb. The TF10 bispecific
([hPAM4]2 x 4
h679) antibody was produced using the method disclosed for production of the
(anti CEA)2 x
anti HSG bsAb TF2, as described above. The TF10 construct bears two humanized
PAM4
Fabs and one humanized 679 Fab.
[0311] The two fusion proteins (hPAM4-DDD and h679-AD2) were expressed
4
independently in stably transfected myeloma cells. The tissue culture
supernatant fluids were
combined, resulting in a two-fold molar excess of hPAM4-DDD. The reaction
mixture was
incubated at room temperature for 24 hours under mild reducing conditions
using 1 mM
reduced glutathione. Following reduction, the DNLTM reaction was completed by
mild
oxidation using 2 mM oxidized glutathione. TF10 was isolated by affinity
chromatography
using IMP 291-affigel resin, which binds with high specificity to the h679
Fab.
[0312] A full tissue histology and blood cell binding panel has been examined
for hPAM4
IgG and for an anti-CEA x anti-HSG bsMAb that is entering clinical trials.
hPAM4 binding
was restricted to very weak binding to the urinary bladder and stomach in 1/3
specimens (no
binding was seen in vivo), and no binding to normal tissues was attributed to
the anti-CEA x 4
anti-HSG bsMAb. Furthermore, in vitro studies against cell lines bearing the
H1 and H2
histamine receptors showed no antagonistic or agonistic activity with the IMP
288 di-HSG
peptide, and animal studies in 2 different species showed no pharmacologic
activity of the
peptide related to the histamine component at doses 20,000 times higher than
that used for 4
imaging. Thus, the HSG-histamine derivative does not have pharmacologic
activity.
Example 18. Imaging Studies Using Pretargeting With TF10 Bispecific Antibody
and "In-Labeled Peptides
[0313] The following study demonstrates the feasibility of in vivo imaging
using the
pretargeting technique with bispecific antibodies incorporating hPAM4 and
labeled peptides.
The TF10 bispecific antibody, comprising two copies of a C-DDD2-Fab-hPAM4 and
one
copy of C-AD2-Fab-679, was prepared as described in the preceding Example.
Nude mice
bearing 0.2 to 0.3 g human pancreatic cancer xenografts were imaged, using
pretargeting with
TF10 and 1111n-IMP-288 peptide. The results, shown in FIG. 8A and FIG. 8B,
demonstrate
how clearly delineated tumors can be detected in animal models using a bsMAb
pretargeting
method, with 1111n-labeled di-HSG peptide, IMP-288. The six animals in the top
of FIG. 8A
and FIG. 8B received 2 different doses of TF10 (10:1 and 20:1 mole ratio to
the moles of
peptide given), and the next day they were given an 1111n-labeled diHSG
peptide (IMP 288).
The 3 other animals on the bottom of FIG. 8A and FIG. 8B received only the
1111n-IMP-288
99
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
(no pretargeting). The images shown in FIG. 8B were taken 3 h after the
injection of the
labeled peptide and show clear localization of 0.2 ¨ 0.3 g tumors in the
pretargeted animals,
with no localization in the animals given the 1111n-peptide alone. Tumor
uptake averaged 20-
25% ID/g with tumor/blood ratios exceeding 2000:1, tumor/liver ratios of
170:1, and
tumor/kidney ratios of 18/1.
Example 19. Production of Targeting Peptides for Use in Pretargeting and 18F
Labeling
[0314] In a variety of embodiments, 18F-labeled proteins or peptides are
prepared by a novel
technique and used for diagnostic and/or imaging studies, such as PET imaging.
The novel
technique for 18F labeling involves preparation of an 18F-metal complex,
preferably an 18F-
aluminum complex, which is chelated to a chelating moiety, such as DOTA, NOTA
or NETA
or derivatives thereof Chelating moieties may be attached to proteins,
peptides or any other
molecule using conjugation techniques well known in the art. In certain
preferred
embodiments, the 18F-A1 complex is formed in solution first and then attached
to a chelating
moiety that is already conjugated to a protein or peptide. However, in
alternative
embodiments the aluminum may be first attached to the chelating moiety and the
18F added
later.
Peptide Synthesis
[0315] Peptides were synthesized by solid phase peptide synthesis using the
Fmoc strategy.
Groups were added to the side chains of diamino amino acids by using Fmoc/Aloc
protecting
groups to allow differential deprotection. The Aloc groups were removed by the
method of
Dangles et. al. (J. Org. Chem. 1987, 52:4984-4993) except that piperidine was
added in a 1:1
ratio to the acetic acid used. The unsymmetrical tetra-t-butyl DTPA was made
as described in
McBride et al. (US Patent Application Pub. No. 2005/0002945, the Examples
section of
which is incorporated herein by reference).
[0316] The tri-t-butyl DOTA, symmetrical tetra-t-butyl DTPA, ITC-benzyl DTPA,
p-SCN-
Bn-NOTA and TACN were obtained from MACROCYCLICSO (Dallas, TX). The
DiBocTACN, NODA-GA(tBu)3 and the NO2AtBu were purchased from CheMatech (Dijon,
France). The Aloc/Fmoc Lysine and Dap (diaminopropionic acid derivatives (also
Dpr))
were obtained from CREOSALUSO (Louisville, KY) or BACHEMO (Torrance, CA). The
Sieber Amide resin was obtained from NOVABIOCHEMO (San Diego, CA). The
remaining
Fmoc amino acids were obtained from CREOSALUSO, BACHEMO, PEPTECHO
(Burlington, MA), EMD BIOSCIENCESO (San Diego, CA), CHEM IMPEXO (Wood Dale,
100
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
IL) or NOVABIOCHEMO. The aluminum chloride hexahydrate was purchased from
SIGMA-ALDRICHO (Milwaukee, WI). The remaining solvents and reagents were
purchased
from FISHER SCIENTIFIC (Pittsburgh, PA) or SIGMA-ALDRICHO (Milwaukee, WI).
18F was supplied by IBA MOLECULAR (Somerset, NJ)
18F-Labeling of IMP 272
[0317] The first peptide that was prepared and 18F-labeled was IMP 272:
DTPA-Gln-Ala-Lys(HSG)-D-Tyr-Lys(HSG)-NH2 MH+ 1512
[0318] IMP 272 was synthesized as described (US Patent No. 7,534,431, the
Examples
section of which is incorporated herein by reference).
[0319] Acetate buffer solution - Acetic acid, 1.509 g was diluted in ¨ 160 mL
water and the
pH was adjusted by the addition of 1 M NaOH then diluted to 250 mL to make a
0.1 M
solution at pH 4.03.
[0320] Aluminum acetate buffer solution - A solution of aluminum was prepared
by
dissolving 0.1028 g of AlC13 hexahydrate in 42.6 mL DI water. A 4 mL aliquot
of the
aluminum solution was mixed with 16 mL of a 0.1 M Na0Ac solution at pH 4 to
provide a 2
mM Al stock solution.
[0321] IMP 272 acetate buffer solution - Peptide, 0.0011 g, 7.28 x 10-7 mol
IMP 272 was
dissolved in 364 [IL of the 0.1 M pH 4 acetate buffer solution to obtain a 2
mM stock solution
of the peptide.
[0322] F-18 Labeling of IMP 272 - A 3 [IL aliquot of the aluminum stock
solution was
placed in a REACTI-VIALTm and mixed with 50 [IL 18F (as received) and 3 [IL of
the IMP
272 solution. The solution was heated in a heating block at 110 C for 15 min
and analyzed by
reverse phase HPLC. HPLC analysis (not shown) showed 93% free 18F and 7% bound
to the
peptide. An additional 10 [IL of the IMP 272 solution was added to the
reaction and it was
heated again and analyzed by reverse phase HPLC (not shown). The HPLC trace
showed 8%
18F at the void volume and 92% of the activity attached to the peptide. The
remainder of the
peptide solution was incubated at room temperature with 150 [IL PBS for ¨ lhr
and then
examined by reverse phase HPLC. The HPLC (not shown) showed 58% 18F unbound
and
42% still attached to the peptide. The data indicate that 18F-A1-DTPA complex
may be
unstable when mixed with phosphate.
[0323] The labeled peptide was purified by applying the labeled peptide
solution onto a 1 cc
(30 mg) WATERS HLB column (Part # 186001879) and washing with 300 [IL water
to
remove unbound F-18. The peptide was eluted by washing the column with 2 x 100
[IL 1:1
101
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
Et0H/H20. The purified peptide was incubated in water at 25 C and analyzed by
reverse
phase HPLC (not shown). The HPLC analysis showed that the 18F-labeled IMP 272
was not
stable in water. After 40 min incubation in water about 17% of the 18F was
released from the
peptide, while 83% was retained (not shown).
[0324] The peptide (16 ii,L 2 mM IMP 272, 48 mg) was labeled with 18F and
analyzed for
antibody binding by size exclusion HPLC. The size exclusion HPLC showed that
the peptide
bound hMN-14 x 679 but did not bind to the irrelevant bispecific antibody hMN-
14 x 734
(not shown).
IMP 272 18F Labeling with Other Metals
[0325] A ¨3 ii,L aliquot of the metal stock solution (6 x 10-9 mol) was placed
in a
polypropylene cone vial and mixed with 75 ii,L 18F (as received), incubated at
room
temperature for ¨ 2 min and then mixed with 20 ii,L of a 2 mM (4 x 10-8 mol)
IMP 272
solution in 0.1 M Na0Ac pH 4 buffer. The solution was heated in a heating
block at 100 C
for 15 min and analyzed by reverse phase HPLC. IMP 272 was labeled with indium
(24%),
gallium (36%), zirconium (15%), lutetium (37%) and yttrium (2%) (not shown).
These
results demonstrate that the 18F metal labeling technique is not limited to an
aluminum ligand,
but can also utilize other metals as well. With different metal ligands,
different chelating
moieties may be utilized to optimize binding of an F-18-metal conjugate.
Production and Use of a Serum-Stable 18F-Labeled Peptide IMP 449
[0326] The peptide, IMP 448 D-Ala-D-Lys(HSG)-D-Tyr-D-Lys(HSG)-NH2 MH+ 1009 was
made on Sieber Amide resin by adding the following amino acids to the resin in
the order
shown: Aloc-D-Lys(Fmoc)-0H, Trt-HSG-OH, the Aloc was cleaved, Fmoc-D-Tyr(But)-
0H,
Aloc-D-Lys(Fmoc)-0H, Trt-HSG-OH, the Aloc was cleaved, Fmoc-D-Ala-OH with
final
Fmoc cleavage to make the desired peptide. The peptide was then cleaved from
the resin and
purified by HPLC to produce IMP 448, which was then coupled to ITC-benzyl
NOTA. The
peptide, IMP 448, 0.0757g (7.5 x 10-5 mol) was mixed with 0.0509 g (9.09 x 10-
5 mol) ITC
benzyl NOTA and dissolved in 1 mL water. Potassium carbonate anhydrous (0.2171
g) was
then slowly added to the stirred peptide/NOTA solution. The reaction solution
was pH 10.6
after the addition of all the carbonate. The reaction was allowed to stir at
room temperature
overnight. The reaction was carefully quenched with 1 M HC1 after 14 hr and
purified by
HPLC to obtain 48 mg of IMP 449.
102
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
18F Labeling of IMP 449
[0327] The peptide IMP 449 (0.002 g, 1.37 x 10-6 mol) was dissolved in 686 [IL
(2 mM
peptide solution) 0.1 M Na0Ac pH 4.02. Three microliters of a 2 mM solution of
Al in a pH
4 acetate buffer was mixed with 15 ,L, 1.3 mCi of 18F. The solution was then
mixed with 20
[IL of the 2 mM IMP 449 solution and heated at 105 C for 15 min. Reverse
Phase HPLC
analysis showed 35% (tR ¨ 10 min) of the activity was attached to the peptide
and 65% of the
activity was eluted at the void volume of the column (3.1 min, not shown)
indicating that the
majority of activity was not associated with the peptide. The crude labeled
mixture (5 ,L)
was mixed with pooled human serum and incubated at 37 C. An aliquot was
removed after
15 min and analyzed by HPLC. The HPLC showed 9.8% of the activity was still
attached to
the peptide (down from 35%). Another aliquot was removed after 1 hr and
analyzed by
HPLC. The HPLC showed 7.6% of the activity was still attached to the peptide
(down from
35%), which was essentially the same as the 15 min trace (data not shown).
High Dose 18F Labeling
[0328] Further studies with purified IMP 449 demonstrated that the 18F-labeled
peptide was
highly stable (91%, not shown) in human serum at 37 C for at least one hour
and was
partially stable (76%, not shown) in human serum at 37 C for at least four
hours. Additional
studies were performed in which the IMP 449 was prepared in the presence of
ascorbic acid
as a stabilizing agent. In those studies (not shown), the metal-18F-peptide
complex showed
no detectable decomposition in serum after 4 hr at 37 C. The mouse urine 30
min after
injection of18F-labeled peptide was found to contain 18F bound to the peptide
(not shown).
These results demonstrate that the 18F-labeled peptides disclosed herein
exhibit sufficient
stability under approximated in vivo conditions to be used for 18F imaging
studies.
[0329] For studies in the absence of ascorbic acid, 18F ¨ 21 mCi in ¨ 400 [IL
of water was
mixed with 9 [IL of 2 mM A1C13 in 0.1 M pH 4 Na0Ac. The peptide, IMP 449, 60
[IL (0.01
M, 6 x 10-7 mol in 0.5 NaOH pH 4.13) was added and the solution was heated to
110 C for
15 min. The crude labeled peptide was then purified by placing the reaction
solution in the
barrel of a 1 cc WATERS HLB column and eluting with water to remove unbound
18F
followed by 1:1 Et0H/H20 to elute the 18F-labeled peptide. The crude reaction
solution was
pulled through the column into a waste vial and the column was washed with 3 x
1 mL
fractions of water (18.97 mCi). The HLB column was then placed on a new vial
and eluted
with 2 x 200 [IL 1:1 Et0H/H20 to collect the labeled peptide (1.83 mCi). The
column
103
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
retained 0.1 mCi of activity after all of the elutions were complete. An
aliquot of the purified
18F-labeled peptide (20 ,L) was mixed with 200 [IL of pooled human serum and
heated at 37
C. Aliquots were analyzed by reverse phase HPLC. The results showed the
relative stability
of18F-labeled purified IMP 449 at 37 C at time zero, one hour (91% labeled
peptide), two
hours (77% labeled peptide) and four hours (76% labeled peptide) of incubation
in human
serum (not shown). It was also observed that 18F-labeled IMP 449 was stable in
TFA
solution, which is occasionally used during reverse phase HPLC chromatography.
There
appears to be a general correlation between stability in TFA and stability in
human serum
observed for the exemplary 18F-labeled molecules described herein. These
results
demonstrate that 18F-labeled peptide, produced according to the methods
disclosed herein,
shows sufficient stability in human serum to be successfully used for in vivo
labeling and
imaging studies, for example using PET scanning to detect labeled cells or
tissues. Finally,
since IMP 449 peptide contains a thiourea linkage, which is sensitive to
radiolysis, several
products are observed by RP-HPLC. However, when ascorbic acid is added to the
reaction
mixture, the side products generated were markedly reduced.
Example 20. In Vivo Studies With Pretargeting TF10 DNLTM Construct and
18F-Labeled Peptide
[0330] 18F-labeled IMP 449 was prepared as follows. The 18F, 54.7 mCi in ¨ 0.5
mL was
mixed with 3 [IL 2 mM Al in 0.1 M Na0Ac pH 4 buffer. After 3 min 10 [IL of
0.05 M IMP
449 in 0.5 M pH 4 Na0Ac buffer was added and the reaction was heated in a 96
C heating
block for 15 min. The contents of the reaction were removed with a syringe.
The crude
labeled peptide was then purified by HPLC on a C18 column. The flow rate was 3
mL/min.
Buffer A was 0.1% TFA in water and Buffer B was 90% acetonitrile in water with
0.1%
TFA. The gradient went from 100% A to 75/25 A:B over 15 min. There was about 1
min
difference in retention time (tR) between the labeled peptide, which eluted
first and the
unlabeled peptide. The HPLC eluent was collected in 0.5 min (mL) fractions.
The labeled
peptide had a tR between 6 to 9 min depending on the column used. The HPLC
purified
peptide sample was further processed by diluting the fractions of interest two
fold in water
and placing the solution in the barrel of a 1 cc WATERS HLB column. The
cartridge was
eluted with 3 x 1 mL water to remove acetonitrile and TFA followed by 400 [IL
1:1
Et0H/H20 to elute the 18F-labeled peptide. The purified [A118F] IMP 449 eluted
as a single
peak on an analytical HPLC C18 column (not shown).
104
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
[0331] TACONIC nude mice bearing the four slow-growing sc CaPanl xenografts
were
used for in vivo studies. Three of the mice were injected with TF10 (162 ug)
followed with
[A118F] IMP 449 18 h later. TF10 is a humanized bispecific antibody of use for
tumor
imaging studies, with divalent binding to the PAM-4 defined tumor antigen and
monovalent
binding to HSG (see, e.g., Gold et al., 2007, J. Clin. Oncol. 25(18S):4564).
One mouse was
injected with peptide alone. All of the mice were necropsied at 1 h post
peptide injection.
Tissues were counted immediately. Comparison of mean distributions showed
substantially
higher levels of 18F-labeled peptide localized in the tumor than in any normal
tissues in the
presence of tumor-targeting bispecific antibody (data not shown).
[0332] Tissue uptake was similar in animals given the [A118F] IMP 449 alone or
in a
pretargeting setting (data not shown). Uptake in the human pancreatic cancer
xenograft,
CaPanl, at 1 h was increased 5-fold in the pretargeted animals as compared to
the peptide
alone (4.6 0.9% ID/g vs. 0.89% ID/g). Exceptional tumor/nontumor ratios were
achieved
at this time (e.g., tumor/blood and liver ratios were 23.4 2.0 and 23.5
2.8, respectively).
[0333] The results demonstrate that 18F labeled peptide used in conjunction
with a PAM4
containing antibody construct, such the TF10 DNLTM construct, provide suitable
targeting of
the 18F label to perform in vivo imaging, such as PET imaging analysis.
Example 21. Further Imaging Studies with TF10
Summary
[0334] Preclinical and clinical studies have demonstrated the application of
radiolabeled
mAb-PAM4 for nuclear imaging and radioimmunotherapy of pancreatic carcinoma.
We have
examined herein the ability of the TF10 construct to pretarget a radiolabeled
peptide for
improved imaging and therapy. Biodistribution studies and nuclear imaging of
the
radiolabeled TF10 and/or TF10-pretargeted hapten-peptide (IMP-288) were
conducted in
nude mice bearing CaPanl human pancreatic cancer xenografts.1251-TF10 cleared
rapidly
from the blood, with levels decreasing to <1% injected dose per gram (ID/g) by
16 hours.
Tumor uptake was 3.47 0.66% ID/g at this time point with no accumulation in
any normal
tissue. To show the utility of the pretargeting approach, 1111n-IMP-288 was
administered 16
hours after TF10. At 3 hours postadministration of radiolabeled peptide,
imaging showed
intense uptake within the tumors and no evidence of accretion in any normal
tissue. No
targeting was observed in animals given only the 1111n-peptide. Tumor uptake
of the TF10-
pretargeted111In-IMP-288 was 24.3 1.7% ID/g, whereas for 1111n-IMP-288 alone
it was
only 0.12 0.002% ID/g at 16 hours. Tumor/blood ratios were significantly
greater for the
105
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
pretargeting group (-1,000:1 at 3 hours) compared with 1111n-PAM4-IgG (-5:1 at
24 hours; P
< 0.0003). Radiation dose estimates suggested that TF10/9 Y-peptide
pretargeting would
provide a greater antitumor effect than 90Y-PAM4-IgG. Thus, the results
support that TF10
pretargeting may provide improved imaging for early detection, diagnosis, and
treatment of
pancreatic cancer as compared with directly radiolabeled PAM4-IgG. (Gold et
al., Cancer Res
2008, 68(12):4819-26)
[0335] We have identified a unique biomarker present on mucin expressed by
>85% of
invasive pancreatic adenocarcinomas, including early stage I disease and the
precursor
lesions, pancreatic intraepithelial neoplasia and intraductal papillary
mucinous neoplasia
(Gold et al., Clin Cancer Res 2007, 13:7380-87). The specific epitope, as
detected by mAb-
PAM4 (Gold et al., Int J Cancer 1994, 57:204-10), is absent from normal and
inflamed
pancreatic tissues, as well as most other malignant tissues. Thus, detection
of the epitope
provides a high diagnostic likelihood for the presence of pancreatic
neoplasia. Early clinical
studies using 131I- and 99111Tc-1abe1ed, murine PAM4 IgG or Fab',
respectively, showed
specific targeting in 8 of 10 patients with invasive pancreatic adenocarcinoma
(Mariani et al.,
Cancer Res 1995, 55:5911s-15s; Gold et al., Crit Rev Oncol Hematol 2001,
39:147-54). Of
the two negative patients, one had a poorly differentiated pancreatic
carcinoma that did not
express the PAM4-epitope, whereas the other patient was later found to have
pancreatitis
rather than a malignant lesion.
[0336] Accordingly, the high specificity of PAM4 for pancreatic cancer is of
use for the
detection and diagnosis of early disease. In addition to improved detection,
90Y-PAM4 IgG
was found to be effective in treating large human pancreatic cancer xenografts
in nude mice
(Cardillo et al., Clin Cancer Res 2001, 7:3186-92), and when combined with
gemcitabine,
further improvements in therapeutic response were observed (Gold et al., Clin
Cancer Res
2004, 10:3552-61; Gold et al., Int J Cancer 2004, 109:618-26). A Phase I
therapy trial in
patients who failed gemcitabine treatment was recently completed, finding the
maximum
tolerated dose of 90Y-humanized PAM4 IgG to be 20 mCi/m2 (Gulec et al., Proc
Amer Soc
Clin Onc, 43rd Annual Meeting, J Clin Oncol 2007, 25(18S):636s). Although all
patients
showed disease progression at or after week 8, initial shrinkage of tumor was
observed in
several cases. Clinical studies are now underway to evaluate a fractionated
dosing regimen of
90Y-hPAM4 IgG in combination with a radiosensitizing dose of gemcitabine.
[0337] We report herein the development of a novel recombinant, humanized
bispecific
monoclonal antibody (mAb), TF10, based on the targeting specificity of PAM4 to
pancreatic
106
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
cancer. This construct also binds to the unique synthetic hapten, histamine-
succinyl-glycine
(HSG), which has been incorporated in a number of small peptides that can be
radiolabeled
with a wide range of radionuclides suitable for single-photon emission
computed tomography
(SPECT) and positron emission tomography (PET) imaging, as well as for
therapeutic
purposes (Karacay et al., Clin Cancer Res 2005, 11:7879-85; Sharkey et al.,
Leukemia 2005,
19:1064-9; Rossi et al., Proc Natl Acad Sci U S A 2006, 103:6841-6; McBride et
al., J Nucl
Med 2006, 47:1678-88). These studies illustrate the potential of this new
construct to target
pancreatic adenocarcinoma for imaging or therapeutic applications.
Methods and Materials
[0338] The TF2 and TF10 bispecific DNLTM constructs and the IMP 288 targeting
peptide
were prepared as described above. Sodium iodide (1251) and indium chloride
(111In) were
obtained from PERKIN-ELMER . TF10 was routinely labeled with 1251 by the
iodogen
method, with purification by use of size-exclusion spin columns. Radiolabeling
of DOTA-
peptide and DOTA-PAM4-IgG with 111InC1 was done as previously described (Rossi
et al.,
Proc Natl Acad Sci U S A 2006, 103:6841-6; McBride et al., J Nucl Med 2006,
47:1678-88).
Purity of the radiolabeled products was examined by size-exclusion high-
performance liquid
chromatography with the amount of free, unbound isotope determined by instant
TLC.
[0339] For TF10 distribution studies, female athymic nude mice =20 g (TACONIC
Farms),
bearing s.c. CaPanl human pancreatic cancer xenografts, were injected with
1251-TF10 (10
p.Ci; 40 pg, 2.50 x mol). At
various time points, groups of mice (n = 5) were necropsied,
with tumor and nontumor tissues removed and counted in a gamma counter to
determine the
percentage of injected dose per gram of tissue (%ID/g), with these values used
to calculate
blood clearance rates and tumor/nontumor ratios.
[0340] For pretargeting biodistribution studies, a bispecific mAb/radiolabeled
peptide molar
ratio of 10:1 was used. For example, a group of athymic nude mice bearing s.c.
CaPanl
human pancreatic cancer xenografts was administered TF10 (80 lig, 5.07 x
mol),
whereas a second group was left untreated. At 16 h postinjection of TF10,
111In-IMP-288
hapten-peptide (30 p.Ci, 5.07 x 10-11 mol) was administered. Mice were
necropsied at several
time points, with tumor and nontumor tissues removed and counted in a gamma
counter to
determine the %ID/g. Tumor/nontumor ratios were calculated from these data. In
a separate
study, groups of mice were given 1111n-DOTA-PAM4-IgG (20 !IC, 50 lig, 3.13 x
10-1 mol)
for the purpose of comparing biodistribution, nuclear imaging, and potential
therapeutic
activity. Radiation dose estimates were calculated from the time-activity
curves with the
107
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
assumption of no activity at zero time. Student's t test was used to assess
significant
differences.
[0341] To perform nuclear immunoscintigraphy, at 3 h postinjection of
radiolabeled peptide
or 24 h postinjection of radiolabeled hPAM4-IgG, tumor-bearing mice were
imaged with a
dual-head Solus gamma camera fitted with medium energy collimator for In (ADAC
Laboratories). Mice were imaged for a total of 100,000 cpm or 10 min,
whichever came first.
Results
[0342] In vitro characterization of the bispecific mAb TF10. The binding of
TF10 to the
target mucin antigen was analyzed by ELISA (FIG. 9). The results showed nearly
identical
binding curves for the divalent TF10, PAM4-IgG, and PAM4-F(ab')2 (half-maximal
binding
was calculated as 1.42 0.10, 1.31 0.12, and 1.83 0.16 nmol/L,
respectively; P > 0.05 for
all), whereas the monovalent bsPAM4 chemical conjugate (PAM4-Fab' x anti-DTPA-
Fab')
had a significantly lower avidity (half-maximal binding, 30.61 2.05 nmol/L;
P = 0.0379,
compared with TF10), suggesting that TF10 binds in a divalent manner. The
immunoreactive
fraction of 125I-TF10 bound to MUC5ac was 87%, with 9% found as unbound TF10
and 3%
as free iodide (not shown). Ninety percent of the 1111n-IMP-288 bound to TF10
(not shown).
Of the total 1111n-IMP-288 bound to TF10, 92% eluted at higher molecular
weight when
excess mucin (200 [ig) was added, with only 3% eluting with the non¨mucin-
reactive TF10
fraction. An additional 5% of the radiolabeled peptide eluted in the free
peptide volume. None
of the radiolabeled peptide bound to the mucin antigen in the absence of TF10
(not shown).
[0343] Biodistribution of 125I-TF10 in CaPanl tumor¨bearing nude mice. TF10
showed a
rapid clearance from the blood, starting with 21.03 1.93 %ID/g at 1 hour and
decreasing to
just 0.13 0.02 %ID/g at 16 hours. The biological half-life was calculated to
be 2.19 hours
[95% confidence interval (95% CI), 2.11-2.27 hours]. Tissue uptake revealed
enhanced
activity in the liver, spleen, and kidneys at 1 hour, which cleared just as
quickly by 16 hours
[T112 = 2.09 hours (95% CI, 2.08-2.10), 2.84 hours (95% CI, 2.49-3.29), and
2.44 hours (95%
CI, 2.28-2.63) for liver, spleen, and kidney, respectively]. Activity in the
stomach most likely
reflects the accretion and excretion of radioiodine, suggesting that the
radioiodinated TF10
was actively catabolized, presumably in the liver and spleen, thereby
explaining its rapid
clearance from the blood. Nevertheless, by 16 hours, the concentration of
radioiodine within
the stomach was below 1% ID/g. A group of five non¨tumor-bearing nude mice
given 1251-
TF10 and necropsied at 16 hours showed similar tissue distribution, suggesting
that the tumor
had not affected the bispecific mAb distribution and clearance from normal
tissues (data not
108
CA 02899811 2015-07-29
WO 2014/165506 PCT/US2014/032513
shown). Of course, it is possible that differences occurred before the initial
time point
examined. Tumor uptake of TF10 peaked at 6 hours postinjection (7.16 1.10
%ID/g) and
had decreased to half maximum binding (3.47 0.66 %ID/g) at 16 hours. Tumor
uptake again
decreased nearly 2-fold over the next 32 hours, but then was stable over the
following 24
hours.
[0344] Biodistribution of TF10-pretargeted, 1111n-labeled peptide. Although
maximum tumor
uptake of TF10 occurred at 6 hours, previous experience indicated that the
radiolabeled
peptide would need to be given at a time point when blood levels of TF10 had
cleared to <1%
ID/g (i.e., 16 hours). Higher levels of TF10 in the blood would lead to
unacceptably high
binding of the radiolabeled peptide within the blood (i.e., low tumor/blood
ratios), whereas
administering the peptide at a later time would mean the concentration of TF10
in the tumor
would be decreased with consequently reduced concentration of radiolabeled
peptide within
the tumor. Thus, an initial pretargeting study was done using a 16-hour
interval. With the
amount of the 1111n-IMP-288 held constant (30 laCi, 5.07 x 10-11 mol),
increasing amounts of
TF10 were given so that the administered dose of TF10 and IMP-288 expressed as
mole ratio
varied from 5:1 to 20:1 (Table 11).
Table 11. Biodistribution of 1111n-IMP-288 alone (no TF10) or pretargeted with
varying
amounts of TF10
%ID/g at 3 h (mean SD)
5:1 10:1 20:1 No TF10
Tumor 19.0 3.49 24.3 1.71 28.6 0.73 0.12
0.00
Liver 0.09 0.01 0.21 0.12 0.17 0.01 0.07
0.00
Spleen 0.12 0.04 0.16 0.07 0.26 0.10 0.04
0.01
Kidneys 1.59 0.11 1.72 0.24 1.53 0.14 1.71
0.22
Lungs 0.19 0.06 0.26 0.00 0.29 0.04 0.03
0.00
Blood 0.01 0.00 0.01 0.01 0.01 0.00 0.00
0.00
Stomach 0.03 0.02 0.02 0.02 0.01 0.00 0.02
0.01
Small intestine 0.12 0.08 0.08 0.03 0.04 0.01 0.06
0.02
Large intestine 0.23 0.10 0.39 0.08 0.25 0.08 0.33
0.02
109
CA 02899811 2015-07-29
WO 2014/165506 PCT/US2014/032513
Pancreas 0.02 0.00 0.02 0.01 0.02 0.00 0.02
0.00
Tumor weight (g) 0.12 0.03 0.32 0.09 0.27 0.01 0.35
0.03
[0345] At 3 hours the amount of 1111n-IMP-288 in the blood was barely
detectable (0.01%).
Tumor uptake increased from 19.0 3.49% ID/g to 28.55 0.73% ID/g as the
amount of
bispecific mAb administered was increased 4-fold (statistically significant
differences were
observed for comparison of each TF10/peptide ratio, one group to another; P <
0.03 or better),
but without any appreciable increase in normal tissue uptake. Tumor uptake in
the animals
given TF10 was >100-fold higher than when 1111n-IMP-288 was given alone.
Comparison of
111In activity in the normal tissues of the animals that either received or
did not receive prior
administration of TF10 indicated similar absolute values, which in most
instances were not
significantly different. This suggests that the bispecific mAb had cleared
sufficiently from all
normal tissues by 16 hours to avoid appreciable peptide uptake in these
tissues. Tumor/blood
ratios were >2,000:1, with other tissue ratios exceeding 100:1. Even
tumor/kidney ratios
exceeded 10:1. The highest tumor uptake of radioisotope with minimal targeting
to nontumor
tissues resulted from the 20:1 ratio; however, either of the TF10/peptide
ratios could be used
to achieve exceptional targeting to tumor, both in terms of signal intensity
and contrast ratios.
The 10:1 ratio was chosen for further study because the absolute difference in
tumor uptake of
radiolabeled peptide was not substantially different between the 10:1 (24.3
1.71% ID/g) and
20:1 (28.6 0.73% ID/g) ratios.
[0346] Images of the animals given TF10-pretargeted 1111n-IMP-288 at a
bispecific
mAb/peptide ratio of 10:1, or the 1111n-IMP-288 peptide alone, are shown in
FIG. 10 A, FIG.
10B and FIG. 10C. The majority of these tumors were :10.5 cm in diameter,
weighing -0.25
g. The images show highly intense uptake in the tumor of the TF10-pretargeted
animals (FIG.
10A). The intensity of the image background for the TF10-pretargeted animals
was increased
to match the intensity of the image taken of the animals given the 1111n-IMP-
288 alone (FIG.
10B). However, when the images were optimized for the TF10-pretargeted mice,
the signal
intensity and contrast were so high that no additional activity was observed
in the body. No
tumor localization was seen in the animals given the111In-IMP-288 alone, even
when image
intensity was enhanced (FIG. 10C).
[0347] An additional experiment was done to assess the kinetics of targeting
111 In-hPAM4
whole-IgG compared with that of the TF10-pretargeted111In-IMP-288 peptide.
Tumor uptake
110
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
of the 1111n-peptide was highest at the initial time point examined, 3 hours
(15.99 4.11%
ID/g), whereas the blood concentration of radiolabeled peptide was only 0.02
0.01% ID/g,
providing a mean tumor/blood ratio of 946.3 383Ø Over time, radiolabeled
peptide cleared
from the tumor with a biological half-life of 76.04 hours. Among nontumor
tissues, uptake
was highest in the kidneys, averaging 1.89 0.42% ID/g at 3 hours with a
steady decrease
over time (biological half-life, 33.6 hours). Liver uptake started at 0.15
0.06% ID/g and
remained essentially unchanged over time. In contrast to the TF10-
pretargeted111In-IMP-288,
the 1111n-hPAM4-IgG had a slower clearance from the blood, albeit there was a
substantial
clearance within the first 24 hours, decreasing from 30.1% ID/g at 3 hours to
just 11.5 1.7%
ID/g at 24 hours. Variable elevated uptake in the spleen suggested that the
antibody was
likely being removed from the blood by targeting of secreted mucin that had
become
entrapped within the spleen. Tumor uptake peaked at 48 hours with 80.4 6.1%
ID/g, and
remained at an elevated level over the duration of the monitoring period. The
high tumor
uptake, paired with a more rapid than expected blood clearance for an IgG,
produced
tumor/blood ratios of 5.2 1.0 within 24 hours. FIG. 10C shows the images of
the animals at
24 hours postadministration of 111In-PAM4-IgG, illustrating that tumors could
be visualized
at this early time, but there was still considerable activity within the
abdomen.
Tumor/nontumor ratios were mostly higher for TF10-pretargeted 1111n-labeled
hapten-peptide
as compared with 1111n-hPAM4-IgG, except for the kidneys, where tumor/kidney
ratios with
the 1111n-IMP-288 and 1111n-hPAM4-IgG were similar at later times. However,
tumor/kidney
ratios for the TF10-pretargeted 1111n-IMP-288 were high enough (e.g., ¨7:1) at
3 hours to
easily discern tumor from normal tissue.
[0348] FIG. 11 A to FIG. 11D illustrates the potential therapeutic capability
of the direct and
pretargeted methods to deliver radionuclide (90Y). Although the concentration
(%ID/g) of
radioisotope within the tumor seems to be much greater when delivered by PAM4-
IgG than
by pretargeted TF10 at their respective maximum tolerated dose (0.15 mCi for 9
Y-hPAM4
and 0.9 mCi for TF10-pretargeted90Y-IMP-288) (FIG. 11A), the radiation dose to
tumor
would be similar (10,080 and 9,229 cGy for 90Y-PAM4-IgG and TF10-pretargeted
90Y-IMP-
288, respectively) (FIG. 11C). The advantage for the pretargeting method would
be the
exceptionally low activity in blood (9 cGy), almost 200-fold less than with
the 90Y-hPAM4
IgG (1,623 cGy) (FIG. 11C). It is also important to note that the radiation
dose to liver, as
well as other nontumor organs, would be much lower with the TF10-pretargeted
90Y-IMP-288
(FIG. 11B, FIG. 11D). The exception would be the kidneys, where the radiation
dose would
111
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
be similar for both protocols at their respective maximum dose (612 and 784
cGy for 9 Y-
PAM4-IgG and TF10-90Y-IMP-288, respectively) (FIG. 11B, FIG. 11D). The data
suggest
that for 90Y-PAM4-IgG, as with most other radiolabeled whole-IgG mAbs, the
dose-limiting
toxicity would be hematologic; however, for the TF10 pretargeting protocol,
the dose-limiting
toxicity would be the kidneys.
Discussion
[0349] Current diagnostic modalities such as ultrasound, computerized
tomography (CT), and
magnetic resonance imaging (MRI) technologies, which provide anatomic images,
along with
PET imaging of the metabolic environment, have routinely been found to provide
high
sensitivity in the detection of pancreatic masses. However, these data are,
for the most part,
based on detection of lesions >2 cm in a population that is already presenting
clinical
symptoms. At this time in the progression of the pancreatic carcinoma, the
prognosis is rather
dismal. To improve patient outcomes, detection of small, early pancreatic
neoplasms in the
asymptomatic patient is necessary.
[0350] Imaging with a mAb-targeted approach, such as is described herein with
mAb-PAM4,
may provide for the diagnosis of these small, early cancers. Of prime
importance is the
specificity of the mAb. We have presented considerable data, including
immunohistochemical studies of tissue specimens (Gold et al., Clin Cancer Res
2007;13:7380-7; Gold et al., Int J Cancer 1994;57:204-10) and immunoassay of
patient sera
(Gold et al., J Clin Oncol 2006;24:252-8), to show that mAb-PAM4 is highly
reactive with a
biomarker, the presence of which provides high diagnostic likelihood of
pancreatic neoplasia.
Furthermore, we determined that PAM4, although not reactive with normal adult
pancreatic
tissues nor active pancreatitis, is reactive with the earliest stages of
neoplastic progression
within the pancreas (pancreatic intraepithelial neoplasia 1 and intraductal
papillary mucinous
neoplasia) and that the biomarker remains at high levels of expression
throughout the
progression to invasive pancreatic adenocarcinoma (Gold et al., Clin Cancer
Res
2007;13:7380-7). Preclinical studies with athymic nude mice bearing human
pancreatic
tumor xenografts have shown specific targeting of radiolabeled murine,
chimeric, and
humanized versions of PAM4.
[0351] In the current studies, we have examined a next-generation,
recombinant, bispecific
PAM4-based construct, TF10, which is divalent for the PAM4 arm and monovalent
for the
anti-HSG hapten arm. There are several important characteristics of this
pretargeting system's
constructs, named DOCK-AND-LOCKTM, including its general applicability and
ease of
112
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
synthesis. However, for the present consideration, the major differences from
the previously
reported chemical construct are the valency, which provides improved binding
to tumor
antigen, and, importantly, its pharmacokinetics. TF10 clearance from nontumor
tissues is
much more rapid than was observed for the chemical conjugate. Time for blood
levels of the
bispecific constructs to reach less than 1% ID/g was 40 hours postinjection
for the chemical
construct versus 16 hours for TF10. A more rapid clearance of the pretargeting
agent has
provided a vast improvement of the tumor/blood ratio, while maintaining high
signal strength
at the tumor site (%ID/g).
[0352] In addition to providing a means for early detection and diagnosis, the
results support
the use of the TF10 pretargeting system for cancer therapy. Consideration of
the effective
radiation dose to tumor and nontumor tissues favors the pretargeting method
over directly
radiolabeled PAM4-IgG. The dose estimates suggest that the two delivery
systems have
different dose-limiting toxicities: myelotoxicity for the directly
radiolabeled PAM4 versus the
kidney for the TF10 pretargeting system. This is of significance for the
future clinical
development of radiolabeled PAM4 as a therapeutic agent.
[0353] Gemcitabine, the frontline drug of choice for pancreatic cancer, can
provide
significant radiosensitization of tumor cells. In previous studies, we showed
that
combinations of gemcitabine and directly radiolabeled PAM4-IgG provided
synergistic
antitumor effects compared with either arm alone (Gold et al., Clin Cancer Res
2004,
10:3552-61; Gold et al., Int J Cancer 2004,109:618-26). The dose-limiting
factor with this
combination was overlapping hematologic toxicity. However, because the dose-
limiting
organ for TF10 pretargeting seems to be the kidney rather than hematologic
tissues,
combinations with gemcitabine should be less toxic, thus allowing increased
administration
of radioisotope with consequently greater antitumor efficacy.
[0354] The superior imaging achieved with TF10 pretargeting in preclinical
models, as
compared with directly radiolabeled DOTA-PAM4-IgG, provides a compelling
rationale to
proceed to clinical trials with this imaging system. The specificity of the
tumor-targeting mAb
for pancreatic neoplasms, coupled with the bispecific antibody platform
technology providing
the ability to conjugate various imaging compounds to the HSG-hapten-peptide
for SPECT
(1111n), PET (68Ga), ultrasound (Au), or other contrast agents, or for that
matter 90Y or other
radionuclides for therapy, provides high potential to improve overall patient
outcomes
(Goldenberg et al., J Nucl Med 2008,49:158-63). In particular, we believe that
a TF10-based
ImmunoPET procedure will have major clinical value to screen individuals at
high-risk for
113
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
development of pancreatic cancer (e.g., genetic predisposition, chronic
pancreatitis, smokers,
etc.), as well as a means for follow-up of patients with suspicious abdominal
images from
conventional technologies and/or with indications due to the presence of
specific
biomarker(s) or abnormal biochemical findings. When used as part of an ongoing
medical
plan for following these patients, early detection of pancreatic cancer may be
achieved.
Finally, in combination with gemcitabine, TF10 pretargeting may provide a
better opportunity
for control of tumor growth than directly radiolabeled PAM4-IgG.
Example 22. Therapy of Pancreatic Cancer Xenografts with Gemcitabine and
90Y-Labeled Peptide Pretargeted Using TF10
Summary
[0355] 90Y-hPAM4 IgG is currently being examined in Phase I/II trials in
combination with
gemcitabine in patients with Stage III/IV pancreatic cancer. We disclose a new
approach for
pretargeting radionuclides that is able to deliver a similar amount of
radioactivity to
pancreatic cancer xenografts, but with less hematological toxicity, which
would be more
amenable for combination with gemcitabine. Nude mice bearing ¨0.4 cm3 sc
CaPanl human
pancreatic cancer were administered a recombinant bsMAb, TF10, followed 1 day
later with
a 90Y-labeled hapten-peptide (IMP-288). Various doses and schedules of
gemcitabine were
added to this treatment, and tumor progression monitored up to 28 weeks. 0.7
mCi PT-RAIT
alone produce only a transient 60% loss in blood counts, and animals given 0.9
mCi of PT-
RAIT alone and 0.7 mCi PT-RAIT + 6 mg gemcitabine (human equivalent ¨1000
mg/m2)
had no histological evidence of renal toxicity after 9 months. A single dose
of 0.25 or 0.5
mCi PT-RAIT alone can completely ablate 20% and 80% of the tumors,
respectively.
Monthly fractionated PT-RAIT (0.25 mCi/dose given at the start of each
gemcitabine cycle)
added to a standard gemcitabine regimen (6 mg wkly x 3; 1 wk off; repeat 3
times)
significantly increased the median time for tumors to reach 3.0 cm3 over PT-
RAIT alone.
Other treatment plans examining non-cytotoxic radiosensitizing dose regimens
of
gemcitabine added to PT-RAIT also showed significant improvements in treatment
response
over PT-RAIT alone. The results show that PT-RAIT is a promising new approach
for
treating pancreatic cancer. Current data indicate combining PT-RAIT with
gemcitabine will
enhance therapeutic responses.
Methods
[0356] TF10 bispecific antibody was prepared as described above. For
pretargeting, TF10
was given to nude mice bearing the human pancreatic adenocarcinoma cell line,
CaPanl.
114
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
After allowing sufficient time for TF10 to clear from the blood (16h), the
radiolabeled
divalent HSG-peptide was administered. The small molecular weight HSG-peptide
(-1.4 kD)
clears within minutes from the blood, entering the extravascular space where
it can bind to
anti-HSG arm of the pretargeted TF10 bsMAb. Within a few hours, >80% of the
radiolabeled
HSG-peptide is excreted in the urine, leaving the tumor localized peptide and
a trace amount
in the normal tissues.
Results
[0357] FIG. 12 illustrates the therapeutic activity derived from a single
treatment of
established (-0.4 cm3) CaPanl tumors with 0.15 mCi of 9 Y-hPAM4 IgG, or 0.25
or 0.50
mCi of TF10-pretargeted 90Y-IMP-288. Similar anti-tumor activity was observed
for the 0.5-
mCi pretargeted dose vs. 0.15-mCi dose of the directly radiolabeled IgG, but
hematological
toxicity was severe at this level of the direct conjugate (not shown), while
the pretargeted
dose was only moderately toxic (not shown). Indeed, the MTD for pretargeting
using 90Y-
IMP-288 is at least 0.9 mCi in nude mice.
[0358] FIG. 13 shows that the combination of gemcitabine and PT-RAIT has a
synergistic
effect on anti-tumor therapy. Human equivalent doses of 1000 mg/m2 (6 mg) of
gemcitabine
(GEM) were given intraperitoneally to mice weekly for 3 weeks, then after
resting for 1
week, this regimen was repeated 2 twice. PT-RAIT (0.25 mCi of TF10-pretargeted
90Y-IMP-
288) was given 1 day after the first GEM dose in each of the 3 cycles of
treatment. Gem
alone had no significant impact on tumor progression (survival based on time
to progress to
3.0 cm3). PT-RAIT alone improved survival compared to untreated animals, but
the
combined GEM with PT-RAIT regimen increased the median survival by nearly 10
weeks.
Because hematological toxicity is NOT dose-limiting for PT-RAIT, but it is one
of the
limitations for gemcitabine therapy, these studies suggest that PT-RAIT could
be added to a
standard GEM therapy with the potential for enhanced responses. The
significant synergistic
effect of gemcitabine plus PT-RAIT was surprising and unexpected.
[0359] A further study examined the effect of the timing of administration on
the potentiation
of anti-tumor effect of gemcitabine plus PT-RAIT. A single 6-mg dose of GEM
was given
one day before or 1 day after 0.25 mCi of TF10-pretargeted 90Y-IMP-288 (not
shown). This
study confirmed what is already well known with GEM, i.e., radiosensitization
is best given
in advance of the radiation. Percent survival of treated mice showed little
difference in
survival time between PT-RAIT alone and PT-RAIT with gemcitabine given 22
hours after
115
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
the radiolabeled peptide. However, administration of gemcitabine 19 hours
prior to PT-RAIT
resulted in a substantial increase in survival (not shown).
[0360] Single PT-RAIT (0.25 mCi) combined with cetuximab (1 mg weekly ip; 7
weeks) or
with cetuximab + GEM (6 mg weekly x 3) in animals bearing CaPanl showed the
GEM +
cetuximab combination with PT-RAIT providing a better initial response (FIG.
14), but the
response associated with just cetuximab alone added to PT-RAIT was encouraging
(FIG. 14),
since it was as good or better than PT-RAIT + GEM. Because the overall
survival in this
study was excellent, with only 2 tumors in each group progressing to >2.0 cm3
after 24
weeks when the study was terminated, these results indicate a potential role
for cetuximab
when added to PT-RAIT.
Example 23. Effect of Fractionated Pretargeted Radioimmunotherapy (PT-
RAIT) for Pancreatic Cancer Therapy
[0361] We evaluated fractionated therapy with 9 Y-DOTA-di-HSG peptide (IMP-
288) and
TF10. Studies using TF10 and radiolabeled IMP-288 were performed in nude mice
bearing
s.c. CaPanl human pancreatic cancer xenografts, 0.32-0.54cm3. For therapy,
TF10-
pretargeted 90Y-IMP-288 was given [A] once (0.6mCi on wk 0) or [B]
fractionated (0.3 mCi
on wks 0 and 1), [C] (0.2 mCi on wks 0, 1 and 2) or [D] (0.2 mCi on wks 0, 1
and 4).
[0362] Tumor regression (>90%) was observed in the majority of mice, 9/10,
10/10, 9/10 and
8/10 in groups [A], [B], [C] and [D], respectively. In group [A], maximum
tumor regression
in 50 % of the mice was reached at 3.7 wks, compared to 6.1, 8.1 and 7.1 wks
in [B], [C] and
[D], respectively. Some tumors showed regrowth. At week 14, the best
therapeutic response
was observed in the fractionated group (2x0.3 mCi), with 6/10 mice having no
tumors (NT)
compared to 3/10 in the 3x0.2 mCi groups and 1/10 in the lx 0.6mCi group. No
major body
weight loss was observed. Fractionated PT-RAIT provides another alternative
for treating
pancreatic cancer with minimum toxicity.
Example 24. 90Y-hPAM4 Radioimmunotherapy (RAIT) Plus Radiosensitizing
Gemcitabine (GEM) Treatment in Advanced Pancreatic Cancer (PC)
[0363] 90Y-hPAM4, a humanized antibody highly specific for PC, showed
transient activity
in patients with advanced disease, and GEM enhanced RAIT in preclinical
studies. This
study evaluated repeated treatment cycles of 90Y-hPAM4 plus GEM in patients
with
untreated, unresectable PC. The 9 Y-dose was escalated by cohort, with
patients repeating 4-
wk cycles (once weekly 200 mg/m2 GEM, 9 Y-hPAM4 once-weekly wks 2-4) until
116
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
progression or unacceptable toxicity. Response assessments used CT, FDG-PET,
and CA19.9
serum levels.
[0364] Of 8 patients (3F/5M, 56-72 y.o.) at the 1st 2 dose levels (6.5 and 9.0
mCi/m290Y-
hPAM4 x 3), hematologic toxicity has been transient Grade 1-2. Two patients
responded to
initial treatment with FDG SUV and CA19.9 decreases, and lesion regression by
CT. Both
patients continue in good performance status now after 9 and 11 mo. and after
a total of 3 and
4 cycles, respectively, without additional toxicity. A 3rd patient with a
stable response by PET
and CT and decreases in CA19.9 levels after initial treatment is now
undergoing a 2nd cycle.
Four other patients had early disease progression and the remaining patient is
still being
evaluated. Dose escalation is continuing after fractionated RAIT with 90Y-
hPAM4 plus low-
dose gemcitabine demonstrated therapeutic activity at the initial 90Y-dose
levels, with
minimal hematologic toxicity, even after 4 therapy cycles.
Example 25. Early Detection of Pancreatic Carcinoma Using Mab-PAM4 and
In Vitro Immunoassay
[0365] Immunohistochemistry studies were performed with PAM4 antibody. Results
obtained with stained tissue sections showed no reaction of PAM4 with normal
pancreatic
ducts, ductules and acinar tissues (not shown). In contrast, use of the MA5
antibody applied
to the same tissue samples showed diffuse positive staining of normal
pancreatic ducts and
acinar tissue (not shown). In tissue sections with well differentiated or
moderately
differentiated pancreatic adenocarcinoma, PAM4 staining was positive, with
mostly
cytoplasmic staining but intensification of at the cell surface. Normal
pancreatic tissue in the
same tissue sections was unstained.
[0366] Table 12 shows the results of immunohistochemical analysis with PAM4
MAb in
pancreatic adenocarcinoma samples of various stages of differentiation.
Overall, there was
an 87% detection rate for all pancreatic cancer samples, with 100% detection
of well
differentiated and almost 90% detection of moderately differentiated
pancreatic cancers.
Table 12 PAM4 Labeling Pattern
Cancer n Focal Diffuse Total
Well Diff. 13 2 11 13 (100%)
Moderately Diff. 24 6 15 21 (88%)
Poorly Diff. 18 5 9 14 (78%)
Total 55 13 35 48 (87%)
117
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
[0367] Table 13 shows that PAM4 immunohistochemical staining also detected a
very high
percentage of precursor lesions of pancreatic cancer, including PanIn-1A to
PanIN-3, IPMN
(intraductal papillary mucinous neoplasms) and MCN (mucinous cystic
neoplasms). Overall,
PAM4 staining detected 89% of all pancreatic precursor lesions. These results
demonstrate
that PAM4 antibody-based immunodetection is capable of detecting almost 90% of
pancreatic cancers and precursor lesions by in vitro analysis. PAM4 expression
was observed
in the earliest phases of PanIN development. Intense staining was observed in
IPMN and
MCN samples (not shown). The PAM4 epitope was present at high concentrations
(intense
diffuse stain) in the great majority of pancreatic adenocarcinomas. PAM4
showed diffuse,
intense reactivity with the earliest stages of pancreatic carcinoma precursor
lesions, including
PanIN-1, IPMN and MCN, yet was non-reactive with normal pancreatic tissue.
Taken
together, these results show that diagnosis and/or detection with the PAM4
antibody is
capable of detecting, with very high specificity, the earliest stages of
pancreatic cancer
development.
Table 13 PAM4 Labeling Pattern
n Focal Diffuse Total
PanIn-1A 27 9 15 24(89%)
PanIn-1B 20 4 16 20(100%)
PanIn-2 11 6 4 1O(91%)
PanIn-3 5 2 0 2 (40%)
Total PanIn 63 21 35 56 (89%)
IPMN 36 6 25 31 (86%)
MCN 27 3 22 25 (92%)
[0368] An enzyme based immunoassay for PAM4 antigen in serum samples was
developed.
FIG. 15 shows the results of differential diagnosis using PAM4 immunoassay for
pancreatic
cancer versus normal tissues and other types of cancer. The results showed a
sensitivity of
detection of pancreatic cancer of 77.4%, with a specificity of detection of
94.3%, comparing
pancreatic carcinoma (n=53) with all other specimens (n=233), including
pancreatitis and
breast, ovarian and colorectal cancer and lymphoma. The data of FIG. 15 are
presented in
tabular form in Table 14.
118
CA 02899811 2015-07-29
WO 2014/165506 PCT/US2014/032513
Table 14. PAM4-Reactive MUC5ac in Patient Sera
n Mean SD Median Range #
Positive
(%)
Normal 43 0.1 0.3 0.0 0-2.0 0 (0)
Pancreatitis 87 3.0 11.5 0.0 0-63.6 4 (5)
Pancreatic CA 53 171 317 31.7 0-1000 41
(77)
Colorectal CA 36 3.3 7.7 0.0 0-37.8 5 (14)
Breast CA 30 3.7 10.1 0.0 0-53.5 2 (7)
Ovarian CA 15 1.8 4.3 0.0 0-16.5 1 (7)
Lymphoma 19 12.3 44.2 0.0 0-194 1 (5)
[0369] An ROC curve (not shown) was constructed with the data from Table 14.
Examining
a total of 283 patients, including 53 with pancreatic carcinoma, and comparing
the presence
of circulating MUC5ac in patients with pancreatic cancer to all other samples,
the ROC curve
provided an AUC of 0.88 0.03 (95% ci, 0.84-0.92) with a P value <0.0001, a
highly
significant difference for discrimination of pancreatic carcinoma from non-
pancreatic
carcinoma samples. Comparing pancreatic CA with other tumors and normal
tissue, the
PAM4 based serum assay showed a sensitivity of 77% and a specificity of 95%.
[0370] A comparison was made of MUC5ac concentration in serum samples from
normal
patients, "early" (stage 1) pancreatic carcinoma and all pancreatic carcinoma
samples. The
specimens included 13 sera from healthy volunteers, 12 sera from stage-1, 13
sera from
stage-2 and 25 sera from stage-3/4 (advanced) pancreatic carcinoma. A cutoff
value of 8.8
units/ml (horizontal line) was used, as determined by ROC curve statistical
analysis. The
frequency distribution of PAM4 antigen concentration is shown in FIG. 16,
which shows that
92% of "early" stage-1 pancreatic carcinomas were above the cutoff line for
diagnosis of
pancreatic cancer. An ROC curve for the PAM4 based assay is shown in FIG. 17,
which
demonstrates a sensitivity of 81.6% and specificity of 84.6% for the PAM4
assay in detection
of pancreatic cancer.
[0371] These results confirm that an enzyme immunoassay based on PAM4 antibody
binding
can detect and quantitate PAM4-reactive antigen in the serum of pancreatic
carcinoma
patients. The immunoassay demonstrates high specificity and sensitivity for
pancreatic
carcinoma. The majority of patients with stage 1 disease were detectable using
the PAM4
immunoassay.
119
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
[0372] In conclusion, an immunohistology procedure employing PAM4 antibody
identified
approximately 90% of invasive pancreatic carcinoma and its precursor lesions,
PanIN, IPMN
and MCN. A PAM4 based enzyme immunoassay to quantitate MUC5ac in human patient
sera showed high sensitivity and specificity for detection of early pancreatic
carcinoma. Due
to the high specificity of PAM4 for pancreatic carcinoma, the mucin biomarker
can also serve
as a target for in vivo targeting of imaging and therapeutic agents. ImmunoPET
imaging for
detection of "early" pancreatic carcinoma is of use for the early diagnosis of
pancreatic
cancer, when it can be more effectively treated. Use of radioimmunotherapy
with a
humanized PAM4 antibody construct, preferably in combination with a
radiosensitizing
agent, is of use for the treatment of pancreatic cancer.
Example 26. Further Studies of In Vitro Detection of PAM4 Antigen in Human
Serum
[0373] In certain embodiments, it is preferred to detect the presence of
MUC5ac and/or to
diagnose the presence of pancreatic cancer in a subject by in vitro analysis
of samples that
can be obtained by non-invasive techniques, such as blood, plasma or serum
samples. Such
ex vivo analysis may be preferred, for example, in screening procedures where
there is no a
priori reason to believe that an individual has a pancreatic tumor in a
specific location. The
objective of the present study was to develop a reliable, accurate, serum-
based assay for
detection of pancreatic cancer at the earliest stages of the disease.
Summary
[0374] A PAM4-based immunoassay was used to quantitate antigen in the serum of
healthy
volunteers (N=19), patients with known diagnosis of pancreatic adenocarcinoma
(N=68), and
patients with a primary diagnosis of chronic pancreatitis (N=29). Sensitivity
for the detection
of pancreatic adenocarcinoma was 82%, with a false-positive rate of 5% for the
healthy
controls. Patients with advanced disease had significantly higher antigen
levels than those
with early-stage disease (P<0.01), with a diagnostic sensitivity of 91%, 86%,
and 62% for
stage 3/4 advanced disease, stage-2, and stage-1, respectively. We also
evaluated chronic
pancreatitis sera, finding 38% positive for antigen. However, this observation
was discordant
with immunohistochemical findings that suggest the PAM4-antigen is not
produced by
inflamed pancreatic tissue. Furthermore, several of the serum-positive
pancreatitis patients,
for whom tissue specimens were available for pathological interpretation, had
evidence of
neoplastic precursor lesions. Immunohistochemistry of additional pancreatitis
specimens
showed 90% to be PAM4-negative with the remainder only weakly positive. This
suggested
120
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
that positive levels of PAM4-antigen within the serum are not derived from
inflamed
pancreatic tissues, but may be an early indicator of pancreatic cancer.
[0375] These results show that the PAM4-serum assay may be used to detect
early-stage
pancreatic adenocarcinoma, and that positive serum levels of PAM4-antigen are
not derived
from inflamed pancreatic tissues, but rather may provide evidence of
subclinical pancreatic
neoplasia.
Materials and Methods
[0376] Human Specimens Sera (N=68) were obtained from patients with a
confirmed
diagnosis of pancreatic adenocarcinoma being treated at the Johns Hopkins
Medical Center,
Baltimore, MD, and stored frozen <5 yrs. Each of these patients underwent
surgical resection
of the pancreas, providing an opportunity for accurate diagnosis and staging.
For stage-1
disease, no neoplastic cells were observed outside of the pancreas. However,
patients with
pancreatic adenocarcinoma are likely to have undetected micrometastatic
disease at
presentation, including those patients reported with stage-1 disease. For this
reason, we
evaluated follow-up survival data. All patients described as having stage-1
disease survived
at least 1 year (time to last recorded follow-up visit), with a median
survival time of 2.70
years (25th percentile = 1.32 years) in comparison to the latest SEER data
(2002-2006), which
reports a 1.42-year median survival for patients having stage-1 disease
treated by surgical
resection.
[0377] A total of 29 sera from patients with a diagnosis of chronic
pancreatitis were obtained
from the Johns Hopkins Medical Center and Zeptometrix Corp. (Franklin, MA).
Healthy
volunteers (N=19) provided blood for control specimens at the Center for
Molecular
Medicine and Immunology. All specimens were de-identified, with the only
clinical data
provided to the investigators being the diagnosis, stage of disease, follow-up
survival time, and
size of the primary tumor.
[0378] Reagents A human pancreatic mucin preparation was isolated from CaPanl,
a human
pancreatic cancer grown as xenografts in athymic nude mice. Briefly, 1 g of
tissue was
homogenized in 10 mL of 0.1 M ammonium bicarbonate containing 0.5 M sodium
chloride.
The sample was then centrifuged to obtain a supernatant that was fractionated
on a
SEPHAROSE0-4B-CL column with the void volume material chromatographed on
hydroxyapatite. The unadsorbed fraction was dialyzed extensively against
deionized water
and then lyophilized. A 1 mg/mL solution was prepared in 0.01 M sodium
phosphate buffer
(pH, 7.2) containing 0.15 M sodium chloride (phosphate-buffered saline [PBS]),
and used as
121
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
the stock solution for the immunoassay standards. A polyclonal, anti-mucin
antiserum was
prepared by immunization of rabbits, as described previously (Gold et al.,
Cancer Res
43:235-38, 1983). An IgG fraction was purified and assessed for purity by
sodium dodecyl
sulfate¨polyacrylamide gel electrophoresis (SDS-PAGE) and molecular-sieve high-
performance liquid chromatography. Murine MA5 antibody reactive with the MUC1
protein
core was obtained from Immunomedics, Inc. (Morris Plains, NJ). A non-binding
isotype-
matched control antibody, Ag8, was purified from the P3X63-Ag8 murine myeloma.
[0379] Sample Preparation All assays were performed in a blinded fashion. To
prepare the
specimens for immunoassay, 300 IAL of serum were placed in a 2.0 mL
microcentrifuge tube
and extracted with an equal volume of 1-butanol. The tubes were vortexed
vigorously for 2
min at which time 300 IAL of chloroform were added and the tubes again
vortexed for 2 min;
this latter step was included in the procedure in order to invert the aqueous
and organic
layers. The tubes were then centrifuged in a microfuge at a setting of 12,000
rpm for 5 min.
The top aqueous layer was removed to a clean tube and the sample diluted 1:2
in 2.0% (w/v)
casein-sodium salt in 0.1 M sodium phosphate buffer, pH 7.2, containing 0.15 M
sodium
chloride (PBS) for immunoassay.
[0380] Enzyme immunoassay The immunoassay was performed in a 96-well polyvinyl
plate
that had been coated with 100 IAL of humanized-PAM4 IgG at 20 1.ig/mL in PBS
with
incubation at 4 C overnight. The wells were then blocked by addition of 200
IAL of a 2.0%
(w/v) solution of casein in PBS and incubated for 1.5 h at 37 C. The blocking
solution was
removed from the wells and the plate washed 5-times with 250 IAL of PBS
containing 0.1%
(v/v) Tween-20. The standards, or unknown specimens, 100 IAL in triplicate,
were added to
the appropriate wells and incubated at 37 C for 1.5 h. The plate was then
washed 5-times
with PBS-Tween-20 as above.
[0381] The polyclonal, rabbit anti-mucin antibody, diluted to 51.ig/mL in 1.0%
(w/v) casein
in PBS containing 501.ig/mL non-specific, human IgG, was added to each well
and incubated
for 1 h at 37 C. The polyclonal antibody was then washed from the wells as
above, and
peroxidase-labeled donkey anti-rabbit IgG (Jackson ImmunoResearch
Laboratories, West
Grove, PA), at a 1:2000 dilution in 1.0% (w/v) casein in PBS, also containing
50 1.ig/mL
human IgG, was added to the wells and incubated at 37 C for 1 h. After washing
the plate as
above, 100 IAL of a 3,3',5,5'-tetramethylbenzidine substrate solution were
added to the wells
and incubated at room temperature for 30 min. The reaction was stopped by the
addition of
122
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
50 IAL 4.0 N sulfuric acid, and the optical density read at a wavelength of
450 nm using a
SPECTRA-MAX 250 spectrophotometer (Molecular Devices, Sunnyvale, CA). Because
of
the considerable microheterogeneity of the PAM4 antigen, we chose to report
our results in
arbitrary units/mL, based on an initial reference standard purified from
xenografted CaPan-1
human pancreatic tumor.
[0382] Immunohistochemistry Paraffin-embedded specimens obtained from the
Cooperative
Human Tissue Network were cut to 4 micron sections on superfrost plus adhesive
slides
(Thermo Scientific, Waltham, MA). Tissue sections were then heated to 95 C for
20 min in a
pH 9.0 Tris buffer, Target Retrieval Solution (Dako, Carpinteria, CA), allowed
to cool to
room temperature, and then quenched with 3% H202 for 15 min at room
temperature.
Primary antibodies were then used at 10 ug/mL with an ABC VECTASTAINO kit
(Vector
Laboratories, Burlingame, CA) for labeling the tissues. The slides were scored
independently
by two pathologists using a paradigm consistent with that reported for earlier
studies on
biomarkers in pancreatic adenocarcinoma (Gold et al., 2007, Clin Cancer Res
13:7380-87): 0-
negative, <1% of the tissue was labeled; 1-a weak, focal labeling of between
1% - 25% of the
tissue; 2-a strong, focal labeling of between 1% - 25% of the tissue; 3-a
weak, diffuse
labeling >25% of the tissue; 4-a strong, diffuse labeling >25% of the tissue.
Only the
appropriate tissue components (e.g., adenocarcinoma cells, normal ducts, etc.)
were
considered for assessment.
[0383] Statistical Analyses Standard curves were generated from the
immunoassay data,
with regression analyses performed to interpolate concentrations of the
unknown samples
(Prism 4.0 software, GraphPad, La Jolla, CA). Receiver operating
characteristic (ROC)
curves were generated by use of the Med-Calc statistical software package
(version 7.5)
(Med-Calc, Mariakerke Belgium). Student's t-test was used to compare variables
in any two
groups. The Cochran-Armitage test was used to detect a trend between detection
rates and
stage of disease.
Results
[0384] Accuracy and precision of the immunoassay A set of control standards
with nominal
concentrations of 15.60, 6.20, 2.50, and 1.00 units/mL was evaluated on
several
nonconsecutive days (N=7) for determination of accuracy and precision. Curve
fitting for the
standards generally gave resultant goodness of fit values for r2 >0.990.
Accuracy was
calculated to be within 8% of the nominal value for the first three
concentrations, but fell to
approximately 22% for the 1.00 units/mL standard. Linear regression of nominal
vs.
123
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
measured units/mL in this series of controls gave a trend-line with a slope of
0.965 and y
intercept of 0.174 (r2= 0.999), where a slope of 1.00 with ay intercept of
0.00 would
constitute 100% accuracy (FIG. 18). An average absolute difference between
nominal and
recovered mass equal to 0.190 0.173 units/mL for the two lowest
concentration standards
suggested a minimum absolute error of approximately 0.2 units/mL for the EIA.
Values for
the coefficient of variation (CV) were 6.40%, 4.85%, 12.0%, and 66.4%,
respectively, for the
4 control standards. Taken together, the data suggest that the PAM4-
immunoassay provides
levels of accuracy and reproducibility that are within the guidelines
suggested for an
immunoassay measurement of an analyte; accuracy and precision were within 15%
for
concentrations above the cutoff value (2.40 units/mL), and within 20% at the
cutoff value. To
further test this, we examined 3 sera, two of which were from healthy
controls, on 3 separate
days. The two healthy controls gave average results of 0.27 0.06 and 0.30
0.27 units/mL,
each of which was close to the minimum absolute error for the EIA with
consequent high CV
of 21.65% and 88.19%, respectively. The other patient serum gave an average of
19.45
2.51 units/mL with a CV of 12.9%.
[0385] Quantitation of antigen in patient sera In a preliminary study reported
in the Example
above, the PAM4 serum-based immunoassay had an apparent sensitivity of 77% and
a
specificity of 94% for pancreatic carcinoma. It should be noted that the
overwhelming
majority of cancer specimens of pancreatic and non-pancreatic origin had been
obtained from
patients enrolled in IRB-approved clinical trials conducted by the Garden
State Cancer Center
and stored frozen at -80 C for more than 10 yrs. However, the specimens of
pancreatitis had
been stored frozen for a considerably shorter time. We evaluated a new group
of 24 sera from
patients diagnosed with pancreatic adenocarcinoma. Only two of the sera had
levels of
PAM4-reactive antigen considered to be positive. Therefore, we considered and
evaluated
reasons why the immunoassay had not performed as expected, including the
quality of the
immunoassay reagents, the possibility that the antigen was being degraded
and/or removed
from the serum, its presence in the form of immune complexes, or being bound
by a blocking
substance. We discovered that there is a substance in fresh human serum and/or
specimens
stored frozen for short periods of time (<5 yrs) that apparently binds to the
PAM4-reactive
epitope and blocks its binding to PAM4 antibody, thus preventing detection by
immunoassay.
Percent recovery of antigen from fresh normal human serum (N=2) spiked with
PAM4-
antigen at concentrations from 5 ¨ 20 units/mL was on the order of 33% or
less.
124
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
[0386] In a series of reports, Slomiany and co-workers disclosed that gastric
mucin had
covalenty bound and/or associated lipids and fatty-acids (Slomiany et al.,
1984, Arch
Biochem Biophys 229:560-67; Slomiany et al., 1986, Biochem Biophys Res Commun
141:387-93; Zalesna et al., 1989, Biochem Int 18:775-84), and that these
lipids and fatty
acids had specific effects upon the physicochemical properties of the mucin.
Furthermore, it
was reported that fatty-acid synthetase levels and activity are significantly
elevated in
pancreatic adenocarcinoma, as is also the case for other forms of cancer and
other pathologic
conditions (Walter et al., 2009, Cancer Epidemiol Biomarkers Prey 19:2380-85).
Because the
blocking substance might be lipid in nature, we performed organic extraction
of sera from the
group of 24 pancreatic adenocarcinoma patients that had been stored frozen for
<5 years. As
was noted above, without prior extraction, only 2 of the 24 specimens (8.3%)
had levels of
PAM4-antigen that were considered positive, whereas after organic extraction,
22 of the 24
specimens (92%) had positive levels of the PAM4-antigen.
[0387] We were also able to re-evaluate, from the study reported in the
Example above, 10
pancreatic adenocarcinoma patient sera that had been stored frozen for >15
years to confirm
the prior results. With or without extraction, all 10 specimens had levels of
antigen that were
considered to be positive . Regression analysis to compare paired results from
extracted and
non-extracted sera gave a trendline with slope of 1.10 (r2 = 0.94),
demonstrating that with or
without extraction of these long-term frozen sera, the results were similar.
It is considered
that long-term storage of the specimens resulted in degradation of the
inhibiting substance or
decreased binding to and unmasking of the epitope. All further testing of sera
was performed
with organic extraction of specimens prior to immunoassay.
[0388] Specimens evaluated for PAM4-reactive antigen included 68 patients with
confirmed
pancreatic adenocarcinoma divided by stage: 21 from stage-1; 14 from stage-2;
and 33 from
stages-3 and -4 (advanced). In addition, 19 sera collected from healthy adult
volunteers and
29 patients diagnosed with chronic pancreatitis were included as control
groups. The
maximum concentration shown in the dot-plot (FIG. 19) was 80 units/mL, because
there
were insufficient volumes of sera to perform additional dilution studies.
Although a cutoff
value of 10.2 units/mL was reported in the Example above, because of the use
of an organic
extraction procedure, as well as differences in the EIA protocol (reagent
concentrations,
inclusion of human IgG in buffers), we chose to treat the current data set
independently of
prior results. A positive cutoff value of 2.4 units/mL was calculated by ROC
curve statistics
(FIG. 20) for the comparison of all pancreatic adenocarcinoma specimens versus
healthy
125
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
adults. The overall sensitivity for detection of pancreatic adenocarcinoma was
82%, with an
area under the curve of 0.92 0.03 (95% CI = 0.84 ¨ 0.97). At this level of
sensitivity, a
false-positive rate of 5% was observed for the healthy control group, the
single positive case
having 3.65 units/mL of circulating antigen, just above the cutoff value.
Insufficient volumes
of sera precluded CA19-9 immunoassays for comparison to the PAM4-immunoassay
results.
[0389] As shown in Table 15, sensitivity for detection of early, stage-1
pancreatic
adenocarcinoma was relatively high, with 13 of 21 (62%) specimens above the
cutoff value.
As expected, this detection rate was lower than that observed for the stage-2
(86%) and
advanced stage-3 and -4 (91%) patient groups. A statistically significant
trend (P <0.01) was
noted for detection rate vs. stage of disease. We considered that this was
most likely due to
tumor size or burden. The average tumor sizes for stage-1, stage-2, and stage-
3/4 groups
were 2.14 1.02 cm3, 3.36 1.18 cm3, and 3.45 1.06 cm3, respectively.
While there was no
statistically significant difference in tumor size between the stage-2 and -
3/4 groups (P
>0.41), a statistically significant difference was observed for each of these
two groups when
compared to stage-1 tumor size (P <0.004 or better). However, it should be
noted that
individual tumor size did not correlate with antigen concentration in the
serum (r2 =0.0065).
[0390] Specimens reported as Stage-1 could be divided into stage-1A (N=13) and
stage-1B
(N=8) subgroups based on tumor size, with detection rates of 54% and 75%,
respectively;
however, caution is emphasized since the number of patients in each subgroup
is small. The
average tumor size for stage-1A was 1.41 0.58 cm3 (range: 0.4 cm3 ¨ 2.0 cm3)
and for
stage-1B was 3.15 0.44 cm3 (range: 2.5 cm3¨ 4 cm3); P <0.001 for comparison
of the two
groups. While, on the whole, tumor sizes were smaller in stage-1A disease than
in stage-1B,
there was no apparent statistical correlation between individual tumor size
and concentration
of the PAM4-antigen in the blood (r2 = 0.03). Furthermore, it is important to
note that of the
13 stage-1A specimens, 4 of the 7 positive cases had PAM4-antigen levels
considerably
higher than the cutoff value, with a range of 17.65 - 32.65 units/mL.
Table 15. PAM4-reactive antigen in the sera of patients
Median T-test
N True- Positive
(units/mL) (P value)a
Total PC 68 9.85 81% <0.001
Stage-1 21 4.53 62% <0.002
126
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
---Stage-1A 13 3.96 54% <0.02
---Stage-1B 8 6.05 75% <0.02
Stage-2 14 10.39 86% <0.005
Stage-3/4 33 13.37 91% <0.001
Chronic Pancreatitis 29 1.28 (38% FP)
Healthy Volunteers 19 1.18 (5% FP)
a - All comparisons are to healthy volunteers
[0391] We also evaluated a set of 29 patient sera with the primary diagnosis
of chronic
pancreatitis. At the 2.4 units/mL cutoff established by ROC evaluation of
normal and
pancreatic adenocarcinoma patients, 11 pancreatitis patients (38%) were
positive. ROC
curve analysis of pancreatitis sera compared directly to the pancreatic
adenocarcinoma
specimens gave an area under the curve of 0.77 0.05 (95% CI = 0.68 ¨ 0.85).
The median
value for the pancreatitis group was 1.28 units/mL, comparable to the healthy
volunteer
group (1.18 units/mL), but considerably lower (3.5-fold) than the stage-1
pancreatic
adenocarcinoma group (4.53 units/mL). It should be noted that our earlier
results for
pancreatitis specimens suggested a considerably lower false-positive rate,
only 5%.
However, those pancreatitis specimens were stored frozen for less than 5
years, and were not
organic phase extracted prior to analysis.
[0392] Biopsy and/or surgical specimens were available from 14 of the chronic
pancreatitis
specimens, 6 of which were from patients who were considered positive for
circulating
MUC5ac. In 3 of these 6 positive cases, precursor lesions were identified
within the tissue
sections. It was then considered whether the positive serum test was due to
pancreatitis or the
presence of neoplastic precursor lesions. We performed immunohistochemistry on
an
additional 30 biopsy specimens from patients diagnosed with pancreatitis. Of
the 30
specimens, one frank invasive pancreatic adenocarcinoma and one large PanIN-2-
3 lesion were
identified (in separate specimens) by use of PAM4 staining, while surrounding
acinar-ductal
metaplasia (ADM) and normal tissues were negative (data not shown). Of the
remaining 28
specimens, 19 had sufficient parenchyma to be evaluated, 16 of which had
evidence of ADM.
PAM4 was negative in all but two of these cases, and in each of these gave
only a very focal,
weak labeling of ADM within the specimens (data not shown).
[0393] Validation studies - We have begun putting together a panel of well-
annotated serum
specimens from patients with known diagnoses. A first set of patient sera (N ¨
450)
127
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
including healthy individuals and patients having invasive pancreatic cancers
(carcinoma,
neuroendocrine, and other forms), benign disease of the pancreas (adenomatous
lesions,
pancreatitis, etc.) and non-pancreatic cancers and benign disease (biliary,
duodenal,
ampullary carcinomas, cholecystitis, gastritis, etc.) is being evaluated
(blind study) to both
confirm and extend the prior results on PAM4 specificity in a much larger
group of patients.
Table 16 presents an interim analysis based upon studies completed to date;
overall, the data
are remarkably similar to our earlier data. Employing a cutoff value
determined by ROC
analysis of PC vs Healthy Adults, the overall sensitivity for detection of
pancreatic carcinoma
was 80% at a specificity of 96%. Only 2 of 16 neuroendocrine tumors were
positive, just over
the cutoff value.
[0394] To date, 14 of 53 (26%) patients with primary diagnosis of pancreatitis
have been
identified as PAM4 positive, lower than that reported in our recent
publication. We are now
attempting to correlate clinical data with results in this pancreatitis group,
as well as provide
for clinical and laboratory follow-up of these patients. Only 2 of 11 patients
with benign
adenomatous lesions (both cystadenoma) were considered positive. One other
cystadenoma
had PAM4-antigen levels greater than 200 units/mL. The pathology report
describes the
biopsy as "very suspicious for cancer".
Table 16. PAM4-reactive antigen in the sera of patients
Positive ROC-AUCb P value'
N Mediana Mean SDa
(%) (95% CI)
Pancreatic
Carcinoma 145 7.84 35.61 64.58 80
(comparisons are to PC)
Neuroendocrine 16 0.00 1.73 4.91 12.5
0.90 0.02
Healthy 27 0.40 0.54 0.53 3.7 <0.0001
(0.85 ¨ 0.94)
0.85 0.28
Pancreatitis 53 0.37 1.56 3.35 26 <0.0001
(0.79 ¨ 0.90)
Pancreatic 0.90 0.03
11 0.00 0.64 0.78 18 <0.0001
Adenoma (0.84 ¨ 0.94)
a ¨ Values for Median and Mean SD are in Units/mL
b ¨ Receiver Operating Characteristic Curves (ROC); Area Under the Curve (AUC)
with
values for 95% Confidence Intervals presented.
Discussion
128
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
[0395] Studies reported in the Example above that employed both
immunohistology of tissue
specimens and EIA of circulating antigen demonstrated that the PAM4-reactive
epitope is a
biomarker for invasive pancreatic adenocarcinoma and is expressed at the
earliest stages of
pancreatic neoplasia (i.e., PanIN-1). It was not detectable within normal
pancreatic tissues
(ducts, acinar and islet cells), nor the majority of non-pancreatic cancers
examined (breast,
lung, gastric, and others). Thus, an elevation of the PAM4-epitope
concentration in the
serum provided a high positive likelihood ratio of 16.8 for pancreatic
adenocarcinoma.
Missing from the prior study was clinical information regarding the stage of
disease.
Consequently, we could not evaluate the value of the immunoassay for detection
of
potentially curable early disease until now.
[0396] We report herein that PAM4-based EIA using serum samples can detect
patients
having early-stage pancreatic adenocarcinoma, and can provide accurate
discrimination from
disease-free individuals. The assay's sensitivity for detection of early
pancreatic
adenocarcinoma was 62% for patients with stage-1 and 86% for patients with
stage-2 disease
and serum levels generally increased with advancing stage of disease. A high
percentage of
patients with stage-1 and -2 disease are clinically asymptomatic. We conclude
that detection
of tumor growth at these early stages using a PAM4 serum assay could provide
improved
prospects for survival.
[0397] The cancer patients in this study all underwent surgical resection,
providing an
opportunity to accurately stage each patient. However, many patients with
pancreatic cancer
are suspected of having micrometastatic disease at presentation, even if they
do not have
histologically-apparent regional lymph node involvement. This highlights a
general problem
in the study of early detection, particularly with a low-incidence disease
such as pancreatic
adenocarcinoma. The accrual of specimens that are well-defined is problematic.
Further
complicating the issue is that many of these pancreatic cancers occur in the
presence of
chronic pancreatitis, cholecystitis, and neoplastic precursor lesions, amongst
other conditions.
[0398] Of 29 sera with a primary diagnosis of chronic pancreatitis, 38% were
identified as
positive for PAM4-antigen. However, several of these serum-positive patients,
for whom
tissue specimens for pathological interpretation were available, had evidence
of neoplastic
precursor lesions. Furthermore, a discrepancy was observed in the comparison
of tissue
reactivity by immunohistology and serum levels of antigen by immunoassay. By
immunohistochemistry, only 10% of the evaluable specimens showed evidence of
PAM4
staining within the ADM, although this was at considerably lower intensity
than observed for
129
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
the overwhelming majority of pancreatic adenocarcinoma specimens (Gold et al.,
2007, Clin
Cancer Res 13:7380-87). Therefore, the results suggest that positive levels of
PAM4-antigen
within the serum may not be derived from inflamed pancreatic tissues, but
rather could
provide evidence of subclinical pancreatic neoplasia, such as PanIN lesions,
and that, at the
very least, positive results provide the rationale for clinical follow-up of
these patients.
[0399] Findings from genetically-engineered animal models of pancreatic
adenocarcinoma
suggest that human pancreatic neoplasia may arise before the PanIN-1 lesion
(Leach, 2004,
Cancer Cell 5:7-11). ADM was the earliest change observed in the mutant KRAS
targeted
model described by Zhu et al. (2007, Am J Pathol 171:263-73). On the other
hand, Shi et al.
(2009, Mol Cancer Res 7:230-36) reported that although KRAS gene mutations can
occur
within ADM, they occur predominantly within ADM that are associated with PanIN
lesions.
The authors suggest this may occur by retrograde extension of the PanIN to the
surrounding
ADM. As yet, there is no conclusive evidence that ADM progress to PanIN. The
fact that
PAM4 is reactive with ADM in two patients with pancreatitis is of interest.
[0400] At the present time, screening the general population for pancreatic
cancer is not
considered medically or economically worthwhile, because the disease is simply
too
infrequent. However, there is considerable interest in screening patients
predicted to have an
increased risk of developing pancreatic adenocarcinoma. Several studies have
demonstrated
that screening individuals with strong family histories of pancreatic cancer
can identify
precursor neoplasms of the pancreas that are amenable to surgical resection
(Canto et al.,
2006, Clin Gastroenterol Hepatol 4:766-81; Canto, 2005, Clin Gastroenterol
Hepatol 3:S46-
58; Brentnall et al., 1999, Ann Intern Med 131:247-55). For example, relatives
of pancreatic
cancer patients have a significantly higher risk of developing pancreatic
cancer than the
general population (Shi et al., 2009, Arch Pathol Lab Med 133:365-74). A small
percentage
of patients with familial pancreatic cancer harbor mutations of PALB2 (partner
and localizer
of BRCA2), a susceptibility gene for pancreatic cancer (Tischkowitz et al.,
2009,
Gastroenterology 137:1183-86). Similarly, patients with long-standing chronic
pancreatitis
are at increased risk of developing pancreatic cancer, and the risk is over
30%, among
patients with early-onset (teenage) hereditary pancreatitis (Lowenfels et al.,
1993, New Eng J
Med 328:1433-37; Lowenfels et al., 1997, J Natl Cancer Inst 89:442-46). A 20-
to 34-fold
higher risk has been observed in individuals with familial atypical multiple
mole (FAMMM)
syndrome (Rutter et al., 2004, Cancer 101:2809-16). Also, several studies have
shown a
significantly increased risk of developing pancreatic cancer in diabetic
individuals who meet
130
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
certain criteria (Pannala et al., 2009, Lancet Oncol 10:88-95). Longitudinal
surveillance of
these patients by use of the PAM4-immunoassay may provide for early detection
of
neoplasia. A second potential use of the immunoassay could be as a means to
detect
recurrence of disease post-therapy, and in particular, following surgical
resection for those
patients where the tumor is supposedly confined to the pancreas.
[0401] The relatively high specificity of the PAM4 antibody provides a means
to target both
imaging and therapeutic agents with high tumor uptake and high tumor/nontumor
ratios. We
have demonstrated PAM4's potential as both a directly-radiolabeled or
bispecific,
pretargeting reagent for nuclear imaging and radioimmunotherapy of pancreatic
cancer.
Also, initial results of a clinical phase lb trial to evaluate a fractionated
dosing of 90Y-PAM4
whole IgG (clivatuzumab tetraxetan), in combination with a radiosensitizing
regimen of
gemcitabine, were reported recently (Pennington et al., 2009, J Clin Oncol
27:15s, abstract
4620). Of 22 patients with stage-3/4 disease (mostly stage-4), 68% showed
evidence of
disease control, with 23% of patients having partial responses based on RECIST
criteria.
Thus, positive results by the PAM4-based immunoassay provides a rationale to
pursue
PAM4-targeted imaging and therapy, thus providing a personalized therapy.
[0402] The PAM4-based immunoassay can identify the majority of pancreatic
adenocarcinoma patients of all stages. Although a direct comparison with
CA19.9 was not
possible in the current study, a prior comparison of the two biomarkers in a
limited set of
pancreatic adenocarcinoma sera (N=41) demonstrated a statistically significant
difference
(P<0.01) with PAM4-antigen levels positive in 71% of patient specimens and
CA19.9-
antigen levels positive in 59% of specimens. In general, it is thought that
CA19.9 lacks the
sensitivity and specificity to provide for early detection and/or diagnosis of
pancreatic
adenocarcinoma. However, the assay does have its use for management with
continued
elevation in CA19.9 serum levels post treatment indicative of a poor
prognosis. Similarly, we
recently reported in abstract form (Pennington et al., 2009, J Clin Oncol
27:15s, abstract
4620), the use of circulating PAM4-antigen levels for prediction of anti-tumor
response.
[0403] These results show that the conditions under which specimens are stored
(e.g., the
length of time they are kept frozen) can have significant effects upon
accessibility of the
epitope under study. For the PAM4-based immunoassay, a fatty acid or lipid
substance may
be able to bind the specific epitope and interfere with the immunoassay.
However, it is also
possible this material was a low-molecular weight peptide or other substance
soluble in
organic solvents. The ability to remove this substance by organic extraction
of the serum
131
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
makes the PAM4-immunoassay reproducible. In addition, the question is raised
as to the
biological significance of the circulating inhibitor:MUC5ac interaction.
However, when using
the PAM4 antibody as an in vivo targeting agent (e.g., radioimmunotherapy),
the presence of
circulating PAM4-antigen is not a factor, since targeting of radiolabeled-PAM4
to sites of
tumor growth has been observed in the majority of patients evaluated to date.
Thus, it appears
that the PAM4-antigen within tumor is free of the blocking substance.
Example 27. Phase IB/II Study of "Y-Labeled hPAM4 Antibody and
Gemcitabine in Advanced Pancreatic Cancer
[0404] A phase IB/II study of "Y-labeled hPAM4 antibody (clivatuzumab
tetraxetan) in
advanced pancreatic cancer patients was performed. A total of 100 patients
with previously
untreated Stage III or IV pancreatic cancer were enrolled into this open-label
trial to receive
gemcitabine once-weekly x 4 with 90Y-clivatuzumab tetraxetan on weeks 2, 3 and
4 (therapy
cycle). The therapy cycle could be repeated until disease progression or until
the patient
displayed unacceptable toxicity. Ten patients withdrew early, while 90
patients, of whom 82
had the Stage IV (metastatic) disease, received 1 ¨ 4 therapy cycles. Tumor
responses were
assessed by CT, FDG/PET and serum CA19.9 after each cycle (initially every 4
wks).
[0405] In Part I of this study, 38 patients were treated with 90Y-clivatuzumab
tetraxetan at
6.5, 9, 12 or 15 mCi/m2 x 3, and a low, fixed gemcitabine dose of 200 mg/m2 x
4 for
radiosensitization. Thirteen patients were retreated with the same cycle 1 - 3
times. The
overall disease control rate, which included complete response (CR), partial
response (PR)
and stable disease (SD), by CT-based RECIST criteria, was 58%, including 6
patients (16%)
with PR and 16 patients (42%) with SD as best response.
[0406] The median overall survival (OS) for the 38 treated patients was 7.7
months, which
compares favorably with other regimens for advanced pancreatic cancer. At the
higher
therapy doses (12 and 15 mCi/m2 of "Y-clivatuzumab tetraxetan x 3), a median
OS of 8.0
months was noted. For the 13 patients who received repeated cycles of the
combination
therapy, median OS improved to 11.8 months. Extended survival of up to 14.8
months post
therapy onset has been observed, with 8 patients achieving a survival > 6
months (3 patients
>1 yr). Anecdotal reports indicate performance status and pain level improved
with therapy.
[0407] Fifty-two patients who were treated in Part II of this study received 3
weekly 9 Y
doses of 12 mCi/m2 and gemcitabine doses of 200, 600 or 1000 mg/m2x 4, with 14
patients
receiving repeated therapy cycles at the same gemcitabine dose but 90Y doses
of 6.5, 9 or 12
mCi/m2. Results were available from 47 of the 52 patients. The disease control
rate for the
132
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
200 mg/m2 group was 72%, with 19% PR and 53% SD. For the 600 and 1000 mg/m2
groups,
the disease control rates were 63% (0% PR) and 68% (18% PR), respectively.
Higher
gemcitabine doses appeared to offer no advantage in treatment response over
the lowest dose
of 200 mg/m2. At the time of reporting, survival data were not available for
this group of
patients. Treatments were well tolerated with no infusion reactions to
radiolabeled
clivatuzumab and few non-hematologic side effects. Hematologic suppression was
transient
after cycles 1 and 2.
[0408] These results showed that repeated cycles of fractionated doses of
clivatuzumab
tetraxetan, labeled with yttrium-90 (90Y) and given in combination with
gemcitabine,
demonstrated therapeutic activity in patients with advanced, inoperable,
pancreatic cancer.
Therapy with repeated cycles of clivatuzumab tetraxetan plus low-dose
gemcitabine
improved overall survival over single-cycle therapy in patients with locally
advanced or
metastatic pancreatic cancer.
Example 28. Detection of Early-Stage Pancreatic Ductal Adenocarcinoma
(PDAC): Sensitivity, Specificity, and Discriminatory Properties of Serum-Based
PAM4-Immunoassay
[0409] As disclosed in Example 26, a serum-based enzyme immunoassay employing
the
PAM4 antibody was able to correctly identify 81% of patients with known PDAC
and this
assay had promising sensitivity for detecting early-stage disease. These
findings have been
extended in a much larger patient population that included over 600 sera from
both malignant
and benign diseases of the pancreas and surrounding tissues. In a blinded
analysis, sera from
patients with confirmed PDAC (N=298), other cancers (N=99), benign disease of
the
pancreas (N=126), and healthy adults (N=79) were evaluated by enzyme
immunoassay for
concentration of PAM4-antigen levels.
[0410] Overall sensitivity for detection of PDAC was 76%, with 64% of stage-1
patients
testing positive and a higher sensitivity (85%) for advanced disease. For the
most part, sera
from patients with neuroendocrine tumors of the pancreas or cancers of other
origin
(squamous, GIST, etc.) did not have elevated levels of the PAM4-antigen.
Approximately
half of the patients with ampullary (48%) and extrahepatic biliary (50%)
adenocarcinomas
had positive levels of circulating PAM4-antigen. Of 126 patients diagnosed
with benign
conditions of the pancreas, only 24 (19%) were positive and, in particular, 18
of 80 (23%)
patients with chronic pancreatitis (CP) were positive. ROC curve analysis
demonstrated a
statistically significant difference between the PDAC and CP groups (P <
0.0001), with an
133
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
area under the curve of 0.84 0.02 (95% CI: 0.79 ¨ 0.89). The positive- and
negative-
likelihood ratios for differentiating PDAC from benign conditions of the
pancreas were 4.00
and 0.30, respectively.
[0411] In conclusion, the PAM4-immunoassay detected nearly two-thirds of stage-
1 PDAC
patients, and did so with high discriminatory power with respect to benign
pancreatic disease.
The results provide a rationale for longitudinal surveillance of patients
considered at high-risk
for PDAC (e.g., familial pancreatic cancer, new-onset diabetes, etc.) with the
PAM4 assay.
Example 29. PAM4-Based Assay Differentiates Pancreatic Ductal
Adenocarcinoma (PDAC) From Chronic Pancreatitis and Benign Nonmucinous
Pancreatic Cysts
[0412] We examined the expression of PAM4-reactive MUC5ac in chronic
pancreatitis and
benign non-mucinous cystic lesions of the pancreas. A tissue microarray of
PDAC (N=14), as
well as surgical specimens from chronic pancreatitis (N=32) and benign non-
mucinous cystic
lesions of the pancreas (N=19), were assessed by immunohistochemistry for
expression of the
PAM4-reactive MUC5ac, as well as MUC1 (mAb-MA5), MUC4 (mAb-8G7), and
CEACAM6 (mAb-MN-15).
[0413] PAM4-reactive MUC5ac, MUC1, MUC4 and CEACAM6 were expressed in 79%
(11/14), 100% (14/14), 86% (12/14) and 100% (14/14) of invasive pancreatic
adenocarcinoma. PAM4 only weakly labeled 6% (1/19) of benign non-mucinous
cystic
lesions, 1 of 15 serous cystadenomas (SCAs) and 0 of 4 cysts with squamous
epithelial lining
(2 lymphoepithelial cysts, and 2 retention cysts with squamous metaplasia).
However, the
expression of MUC1, MUC4 and CEACAM6 was detected in 53% (8/15), 0% (0/15) and
13% (2/15) of SCAs, and in 4, 3 and 3 of the 4 cysts with squamous epithelial
lining,
respectively. PAM4 labeled 19% (6/32) of chronic pancreatitis specimens;
however, this
PAM4 reactivity was restricted to the PanIN precursor lesions associated with
chronic
pancreatitis. Inflamed tissue was negative. The expression of MUC1, MUC4 and
CEACAM6
was detected in 90% (27/30), 78% (25/32), and 97% (31/32) of chronic
pancreatitis. In all of
the positively-labeled specimens, the reactivity was present in non-neoplastic
inflamed
pancreatic tissue in addition to PanIN.
[0414] In conclusion, the expression of PAM4 was detected in only 6% of benign
non-
mucinous cystic lesions and in the precursor lesions associated with chronic
pancreatitis.
These results suggest that PAM4, in contrast to MUC1, MUC4, and CEACAM6, may
be
134
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
useful to differentiate benign non-mucinous cystic lesions of the pancreas and
chronic
pancreatitis from PDAC.
Example 30. Combination of the PAM4 and CA19-9 Biomarkers For Improved
Detection of Pancreatic Adenocarcinoma
[0415] Pancreatic ductal adenocarcinoma (PDAC) is almost universally lethal,
due mainly to
the inability to detect early-stage disease. Thus, identification of
biomarkers that can identify
patients with early-stage PDAC may improve overall survival. In a blinded
study, PAM4 and
CA19-9 immunoassays were performed on sera from 480 patients, including those
with
confirmed PDAC (N=234), other cancers (N=84), benign diseases of the pancreas
(N=89),
and healthy adults (N=50).
[0416] Overall sensitivity for PDAC was similar, 74% and 77% for PAM4 and CA19-
9,
respectively. Sensitivity for detection of early, stage-1 disease (N=26),
although somewhat
higher for the PAM4-antigen, was also statistically similar, 65% and 58% for
PAM4 and
CA19-9, respectively (P = 0.5775). However, specificity was significantly
lower for CA19-9,
particularly with respect to chronic pancreatitis (CP): 68% vs.86% for the
PAM4 assay (P =
0.014). Furthermore, CA19-9 results showed considerably higher detection rates
for non-
PDAC neoplasia, including patients with other cancers that metastasized to the
pancreas.
Thus, positive likelihood ratios (+LR) were lower for CA19-9 (+LR = 2.41) than
for the
PAM4 assay (+LR = 5.29).
[0417] PAM4 and CA19-9 antigen levels in PDAC were independent of each other
(r2 =
0.003, P=0.410); however, the positive and negative interpretations were
concordant in 68%
of the cases. Thus, a combined biomarker analysis improved the overall PDAC
detection rate
(84%), without a significant decrease in specificity (83%). Comparison of the
ROC curves
for PDAC vs. CP and PDAC vs. benign disease demonstrated a statistically
significant
improvement for the combined immunoassay, as compared to either assay alone (P
< 0.0001
in both comparisons), to detect and discriminate PDAC from benign disease.
[0418] While the PAM4-immunoassay provided high sensitivity and specificity
for detection
and diagnosis of PDAC, inclusion of the CA19-9 biomarker significantly
enhanced positive
identification of PDAC patients, from 74% to 84%.
Example 31. Use of PAM4-Immunoassay as a Correlate of Tumor Response
[0419] We investigated whether specific trends in PAM4-reactive MUC5ac
concentrations
(within the individual patient) can be used as an indicator of tumor response
after therapy.
Several patients from a 9 Y-hPAM4 phase-lb/II clinical trial now in progress
were evaluated.
135
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
When patients were evaluated 4 weeks after treatment had ended (a treatment
cycle is 4
weeks), a decrease in serum antigen levels of >40% was suggestive of a
response. All of the
patients who had progressive disease had levels of PAM4 antigen that continued
to rise.
Trends are presented for two patients in FIG. 21A and FIG. 21B. In both cases,
trends in the
level of circulating MUC5ac were concordant with the trend in tumor volume as
determined
by CT. These results suggest that serum PAM4 levels are of use to monitor
responsiveness to
anti-cancer treatments for pancreatic cancer.
Example 32. Identification of Target Antigen for PAM4 Antibody
[0420] We performed a set of blocking and capture/probe paired enzyme
immunoassays to
evaluate the relationships between the PAM4 antibody and antibodies reactive
with MUC1
(MA5, KC4, HMFG1, SM3, H23), MUC2 (G9), MUC4 (8G7) and MUC5ac (45M1). A
mucin standard derived from the CaPanl human tumor xenograft was shown to
contain the
reactive mucin species for all of these antibodies except those reactive with
G9 (MUC2). Of
all MAbs examined, only 1 (45M1) reported to be reactive with MUC5ac provided
a positive
reaction in sandwich EIA when PAM4 was used as the capture reagent. The 45M1
antibody
is reactive with a much lower percentage of pancreatic carcinomas than PAM4
(by IHC on
TMA) and so cannot be used as a single probe for the serum-based PAM4-
immunoassay.
[0421] As described above, we performed a peptide-phage-display study by
consecutive
biopanning with the murine and humanized versions of PAM4-IgG. A consensus
sequence
(12mer - WTWNITKAYPLP (SEQ ID NO: 7)) was generated which when input into a
BLAST protein search with query coverage set at 100%, identified MUC5ac and
MUC16
with 7 of 12 and 5 of 12 identical amino acids within the 12mer sequence,
respectively.
[0422] Studies were performed using mass spectrometry to identify PAM4-
immunoprecipitated antigens from credentialed cyst fluids (these fluids were
previously
analyzed by mass spectrometry to identify specific MUCs present in the
mixtures). By PAGE
analyses of the PAM4-immunoprecipitated materials from 3 individual cyst fluid
specimens,
only two identical bands were present in each specimen (not shown). Both of
these bands
contained MUC5ac as the major mucin species.
[0423] We have investigated the nature of the substance within human blood
that binds to the
PAM4-epitope, which necessitates organic extraction prior to immunoassay. As
discussed
above, Slomiany and co-workers have observed that gastric mucin had covalently
bound
and/or associated lipids and fatty-acids. Further, fatty-acid synthetase
levels and activity are
significantly elevated in pancreatic adenocarcinoma, as is also the case for
other forms of
136
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
cancer and other pathologic conditions. Speculating that the blocking
substance might be
lipid in nature, we performed an EIA (FIG. 22) in the presence and absence of
100 M
palmitic acid and observed a statistically signficant 69% reduction in
reactivity at an 0D450
equivalent of 1.0 (P<0.0001). It is noted that the normal adult serum level of
palmitic acid is
in the range of 1,480 to 3,730 M, considerably higher than the concentration
that was used
in this EIA experiment.
Example 33. PAM4 Differentiates Between Pancreatic Ductal Adenocarcinoma
(PDAC) and Chronic Pancreatitis (CP)
[0424] Current practice guidelines suggest that patients who present with
signs and/or
symptoms suspicious of pancreatic cancer undergo a pancreatic protocol CT
imaging study
for detection of tumor mass within the pancreas. Follow-up imaging by
endoscopic
technologies (e.g., EUS, ERCP) can provide high sensitivity for detection of
disease, and
when combined with fine-needle aspiration/biopsy, can provide good diagnostic
accuracy.
However, the majority of these procedures have been performed on patients with
advanced
disease; that is, tumors greater than 2 cm. Detection of early pancreatic
cancer is still
problematic, especially when occurring in a background of pancreatitis. Thus,
the current
reality is that only 7% of all cases detected are early disease. With no
effective treatment for
advanced PC, the prognosis for these patients is dismal.
[0425] Biomarkers that can reliably distinguish between cancer and benign
conditions, and/or
provide means to prioritize patients for follow-up evaluation, would be of
significant clinical
value, especially if the biomarker is capable of detecting early disease. We
have developed
monoclonal antibody PAM4 that demonstrates a high degree of specificity for
pancreatic
ductal adenocarcinoma (PDAC).
[0426] MAbs having defined reactivity with several mucin species, including
MUC1, MUC2,
MUC3, MUC4, MUC5ac, etc., were evaluated for signal response in a heterologous
PAM4-
capture sandwich EIA. The only MAbs able to provide signal response (45M1, 2-
11M1) are
known to react with specific domains of the MUC5ac mucin. Further, three
additional anti-
MUC5ac MAbs (21M1, 62M1, and 463M1) were each able to inhibit the interaction
between
PAM4 and its mucin antigen. These data suggest MUC5ac as an antigen to which
PAM4 is
reactive. PAM4, unlike other anti-MUC5ac MAbs (45M1, 2-11M1, CLH2, and
others),
demonstrates greater specificity for PDAC than cancers originating from other
organs, and
may serve as a useful biomarker for PDAC, as well as a target for antibody-
directed imaging
and therapy.
137
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
Table 17. PAM4-Antigen In the Serum of Patients with Known Disease
Number of Percent of
N Median Positive Positive
_
(units/mL) Cases Cases
Pancreatic Cancer
I I
Ductal Adenocarcinoma 298 10.40 115 76
_
1 1
Neuroendocrine 20 0.08 2 10
Other Morphology 7 0.51 1
_ 14
_
Non-PC, Mets to the
Pancreas 11
_ 0.00 2
_ 18
Ampullary Adenocarcinoma 21
_ 1.52 10 48
Biliary Adenocarcinoma 26 4.41 13 50
Cholangiocarcinoma 7 1.07 2 29
Duodenal Adenocarcinoma 7 2.80 4 57
All Biliary and Periampullary 61 1.78 29 48
Colon Carcinoma 32 0.15 5 16
I
Chronic Pancreatitis (CP) 80 0.41 18 '23
Benign Cystadenoma 15 0.18 1 7
Benign ¨ Other 25 0.20 5 20
All Benign Disease 120 0.26 24 20
_
Healthy Volunteers 79 0.27 3 4
_
All groups are statistically different from the pancreatic adenocarcinoma
group
with P values equal to or better than 0.0001; Mann-Whitney nonparametric test.
Gold DV, Gaedcke J, Ghadimi BM, et al. Cancer. 2013 Feb 1;119(3):522-8.
[0427] The PDAC group consisted of 40% early and 60% advanced stage patients.
Detection
rates were 64% and 85%, respectively. The sensitivity and specificity of the
PAM4 assay
138
CA 02899811 2015-07-29
WO 2014/165506 PCT/US2014/032513
was determined for PDAC vs. CP (FIG. 23A) and for PDAC vs. all benign tissue
samples
(FIG. 23B). The calculated values of AUC were 0.84 and 0.85, respectively.
[0428] Approximately 20% of patients with chronic pancreatitis (CP) are
positive by use of
the serum-based immunoassay. This issue is critical to the interpretation of
the results with
PAM4-positive CP patients being either false positives, or perhaps, the
discovery of occult
neoplasia. Thus, we undertook an extensive immunohistochemical evaluation of
PAM4-
reactivity in CP tissue specimens.
[0429] FIG. 24 shows comparative labeling of PDAC vs. non-neoplastic prostate
tissue by
PAM4 antibody vs. antibodies against MUC1, MUC4, CEACAM6 and CA19-9. Each of
the
antibodies reacted with PDAC. PAM4 showed no reactivity with normal tissue.
The same
antibodies were compared in a sample showing a PanIN-2 lesion arising within a
background
of CP, with partial loss of acinar cells, some fibrosis and PanIN-associated
acinar-ductal
metaplasia (ADM) (not shown). No labeling was observed with PAM4 in any of the
tissues
within CP, including isolated ADM (not shown). Each of the other antibodies
showed some
binding to non-neoplastic tissue (not shown). Table 18 and Table 19 show
comparative
results of labeling with PAM4 vs. antibodies against MUC1, MUC4, CEACAM6 and
CA19-
9.
Table 18. Expression of Biomarkers in Pancreatic Ductal Adenocarcinoma
PAM4 MUC1 MUC4 CEACAM6 CA19-9
Number 43 43 43 42 43
8
Focal Labelinga 1 (2%) 4 (15%) 3 (8%) 2 (5%)
(24%)b
26 42 22
Diffuse Labeling 35 (92%) 37 (95%)
(76%) (98%) (85%)
34 43 26
Total Labeled 38 (90%) 39 (91%)
(79%) (100%) (60%)
Adjacent Normal 14
0 (0%) 6 (43%) 14 (100%)
14 (100%)
(N=14) (100%)
a-Focal labeling, 5% to 25% of the appropriate tissue components labeled with
the indicated
MAb; Diffuse, >25% of the appropriate tissue components labeled with the
indicated MAb;
Total, focal + diffuse. b-value provided in parenthesis is the percentage of
total N PDAC
specimens evaluated
Table 19. Expression of Biomarkers in Chronic Pancreatitis
139
CA 02899811 2015-07-29
WO 2014/165506 PCT/US2014/032513
N ¨PAM4 MUC1 MUC4 CEACAM6 CA19-9
Chronic
Pancreatitis 32
-PanIN1 5 2 2 1 5 5
-PanIN2 5 4 4 3 5 5
-Ducts 32a 0 22 25 31 29
-Acinar cells 32a 0 27 8 30 29
-Isolated ADM 32a 0 24 0 0 26
[0430] We conclude that PAM4 is not reactive with the non-neoplastic tissues
from chronic
pancreatitis (CP) patients, but rather with PDAC and its neoplastic precursor
lesions, such as
PanINs, which are known to develop within the inflamed parenchyma. Together
with results
from a prior study, we have evaluated a total of 51 specimens of CP, finding
that in no
instance was PAM4 reactive with the inflamed parenchyma. On the other hand,
each of the
other biomarkers investigated, MUC1, MUC4, CEACAM6, and CA19-9, were unable to
differentiate PDAC and benign, non-neoplastic tissues. These latter biomarkers
were
expressed to varying extents in CP-associated PanIN lesions, but also in non-
neoplastic ducts
and isolated ADM. A PAM4-based EIA to quantitate antigen in patient sera shows
high
sensitivity and specificity for detection of PDAC. Approximately 2/3 of
patients with stage-1
disease are positive for circulating PAM4-antigen. We speculate that CP
patients (and
perhaps others having disease with high risk for development of PDAC), who are
found to
have positive levels of PAM4-reactive antigen in the circulation, may have
occult PDAC
and/or significant mass of precursor lesions producing the PAM4-biomarker.
Example 34. Mapping the PAM4 Epitope on MUC5ac
Summary
[0431] Indirect and sandwich enzyme immunoassays (ETA) were performed to
compare and
contrast the reactivity of PAM4 with several anti-mucin antibodies having
known reactivity
to specific mucin species (e.g., MUC1, MUC4, MUC5ac, etc.). Studies designed
to block
reactivity of PAM4 with its specific antigen also were performed. We
demonstrated that
MAbs 2-11M1 and 45M1, each reactive with MUC5ac, are able to provide signal in
a
heterologous sandwich immunoassay where PAM4 is the capture antibody. Further,
we
identified MAbs 21M1, 62M1, and 463M1, each reactive with MUC5ac, as
inhibiting the
reaction of PAM4 with its specific epitope. MAbs directed to MUC1, MUC3, MUC4,
140
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
MUC16 and CEACAM6 were not reactive with PAM4-captured antigen, nor are they
able to
block the reaction of PAM4 with its antigen. We concluded that MUC5ac is the
mucin
species to which PAM4 antibody is reactive.
Background
[0432] Mucin glycoproteins are high molecular weight, heavily glycosylated,
proteins that
include at least 19 species categorized on the basis of their unique protein
cores. They can be
found as either transmembrane components of the cell or as secreted products.
Abnormal
expression of mucins is a well-known occurrence in many forms of cancer (see
Hollingsworth & Swanson, 2004, Nat Rev Cancer 4:45-60; Kufe, 2009, Nat Rev
Cancer
9:874-85; Rachagani et al., 2009, Biofactors 35:509-27), including pancreatic
ductal
adenocarcinoma (PDAC) (Ringel & Lohr, 2003, Mo lancer 2:9-13; Andrianifahanana
et al.,
2001, Clin Cancer Res 7:4033-40; Torres et al., 2012, Curr Pharm Des 18:2472-
81). Neo-
expression and/or upregulation/downregulation of specific mucin species, with
and without
the generation of newly transcribed and translated splice variants (Schmid,
2003, Oncol Rep
10:1981-85), have been well-documented in the literature. Alteration of
carbohydrate
moieties through the addition of new terminal sugars (e.g., neuraminic acids),
underglycosylation, and other abnormal biochemical pathways also have been
observed
(Brockhausen, 2006, EMBO Rep 7:599-604; Yue et al., 2009, Mol Cell Proteomics
8:1697-
707; Haab et al., 2010, Ann Surg 251:937-45). These modifications may lead to
changes in
conformational structure and/or appearance or disappearance of specific
epitopes.
Additionally, changes may be observed for the intracellular distribution of
the mucin species
under consideration, such as MUC1, which in normal tissues is a transmembrane
glycoprotein, but with neoplastic transformation is found in the cytoplasm as
well (Jass et al.,
1995, J Pathol 176:143-49; Cao et al., 1997, Virchows Arch 431:159-66). These
events may
prove to be of biological and clinical significance in the process of
neoplastic development
and progression, as well as provide new biomarkers/targets for early detection
and targeted
therapy of cancer.
[0433] Our laboratory initially reported the use of a polyclonal antiserum to
identify a
pancreatic ductal mucin, which at the level of sensitivity provided by
indirect
immunohistochemistry (IHC), was shown to contain an epitope relatively
specific to the
pancreas (Gold et al., 1983, Cancer Res 43:235-38), and ultimately resulted in
the
development of monoclonal antibody (MAb), PAM4 (Gold et al., 1994, Int J
Cancer 57:204-
10), also known as clivatuzumab in its humanized form. PAM4 demonstrates high
specificity
141
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
for PDAC with little to no reactivity towards normal and benign, non-
neoplastic, pancreatic
tissues, although it does show limited reactivity (approximately 10% of all
specimens
examined) with adenocarcinomas originating in certain other organs (e.g.,
stomach, colon,
lung) (Gold et al., 1994, Int J Cancer 57:204-10; Gold et al., 2007, Clin
Cancer Res 13:7380-
87; Gold et al., 2010, Cancer Epidemiol Biomarkers Prey 19:2786-94). PAM4
identifies a
biomarker that, if present, provides a high diagnostic likelihood of the
presence of pancreatic
neoplasia (Gold et al., 2010, Cancer Epidemiol Biomarkers Prey 19:2786-94;
Gold et al.,
2006, J Clin Oncol 24:252-58; Gold et al., 2013, Cancer 119:522-28). Thus,
clinical
applications for detection of early-stage disease (Gold et al., 2010, Cancer
Epidemiol
Biomarkers Prey 19:2786-94; Gold et al., 2013, Cancer 119:522-28), and
antibody-targeted
imaging and therapy, are being pursued (Gulec et al., 2011, Clin Cancer Res
17:4091-4100;
Ocean et al., 2012, Cancer 118:5497-5506). In addition to PDAC, the PAM4-
biomarker is
expressed in the precursor lesions, pancreatic intraepithelial neoplasia
(PanIN, including the
earliest developing lesion, PanIN-1A), and intraductal papillary mucinous
neoplasia (IPMN),
suggesting that there may be oncogenic significance to its expression (Gold et
al., 2007, Clin
Cancer Res 13:7380-87). In the current study, we investigated the identity of
the mucin
species to which this clinically-relevant antibody is reactive, in order to
understand what role
this mucin may play in the development and progression of pancreatic cancers.
Methods
[0434] Antigen and Antibodies - A mucin containing fraction, designated CPM1,
was
isolated, as described previously (Gold et al., 2006, J Clin Oncol 24:252-58),
from the Capan-
1 human PDAC xenograft in athymic nude mice. Briefly, this consisted of
homogenization
of the dissected tumor in 0.1M ammonium bicarbonate containing 0.5M sodium
chloride.
Following high-speed centrifugation (20,000 g x 45 min), the soluble material
was
chromatographed on a SEPHAROSEO 4B-CL column, and then eluted with the
identical
ammonium bicarbonate-sodium chloride solution. The void volume material was
collected,
dialyzed against 0.01M sodium phosphate, pH 7.2, and then passed through
hydroxyapatite to
remove nucleic acids and proteins. The non-binding, mucin-containing fraction
was again
dialyzed extensively to remove salts and used as a source of antigen.
[0435] Antibodies used in the current study are listed in Table 20 with clone
and source
information. For sandwich and blocking studies, PAM4 was available in both
murine
(mPAM4) and humanized (hPAM4; clivatuzumab) versions provided by Immunomedics,
Inc.
(Morris Plains, NJ). All other MAbs were murine IgG. Mouse ascites fluids
containing
142
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
MAbs 21M1, 45M1, 62M1 and 463M1 were kindly provided by Dr. J. Bara, INSERM,
Paris,
France. PAM4 antibodies and ascites fluid containing an anti-alpha-fetoprotein
antibody,
employed as a negative control for the blocking studies (reactive with Hep-G2,
hepatoceullar
carcinoma cells) were provided by Immunomedics, Inc. (Morris Plains, NJ). A
rabbit
polyclonal anti-CPM1 (Gold et al., 1994, Int J Cancer 57:204-210; Gold et al.,
2010, Cancer
Epidemiol Biomarkers Prey 19:2786-94) IgG served as the positive control with
detection by
a horseradish peroxidase (HRP)-labeled donkey anti-rabbit IgG (Jackson
ImmunoResearch,
West Grove, PA).
Table 20. Monoclonal antibodies used
Antigen Clone name Source
MUC1 MA5 Immunomedics
MUC1 KC4 Immunomedics
MUC1 CM1 Gene Tex
MUC2 994/152 Abcam
MUC3 M3.1 Abcam
MUC3 M3A LifeSpan Bio
MUC4 8G7 Santa Cruz Biotech
MUC5ac 2-11M1 Santa Cruz Biotech
MUC5ac 45M1 Santa Cruz Biotech
MUC5ac CLH2 Santa Cruz Biotech
MUC16 X306 Novus Bio
MUC16 X325 Abcam
CEACAM5 MN14 Immunomedics
CEACAM6 MN15 Immunomedics
CA 19-9 CA 19-9 Santa Cruz Biotech
[0436] Immunomedics, Inc. ¨ Morris Plains, NJ; GeneTex ¨ Irvine, CA; Abcam ¨
Cambridge, MA; LifeSpan Biosciences, Inc. - Seattle, WA; Santa Cruz
Biotechnology, Inc. -
Santa Cruz, CA; Novus Biologicals ¨ Littleton, CO.
[0437] Enzyme Immunoassay - Procedures have been described for both indirect
and
sandwich enzyme immunoassays (Gold et al., 1994, Int J Cancer 57:204-210; Gold
et al.,
2010, Cancer Epidemiol Biomarkers Prey 19:2786-94). For indirect immunoassays,
primary
MAbs were used at a concentration of 10 ng/mL to provide high sensitivity for
signal
143
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
detection. For sandwich immunoassays, the capture MAb was coated onto the
wells at a
concentration of 10 p.g/mL, followed by the addition of the CPM1 antigen at
various
concentrations up to 10 p.g/mL. The MAb probe was then added at a high
concentration of 10
p.g/mL for detection of response to captured antigen. Secondary HRP-labeled
anti-species-
specific IgG (Jackson ImmunoResearch, West Grove, PA) was evaluated initially
to
determine optimum concentrations for use in the assay (usually 1:1000 or
1:2000). MAb
inhibition studies were performed by adding the inhibiting MAb to wells coated
with CPM1
antigen, starting at a high concentration of 100 p.g/mL of pure MAb or 1:10
dilution of ascites
fluid, and titrating to lower amounts. After incubating with the inhibiting
antibody at 37 C for
1 h, the plates were washed, and hPAM4 added to the wells at a concentration
of 0.25 p.g/mL.
hPAM4 binding was then detected with a secondary probe, HRP-labeled anti-human
IgG
conjugate.
[0438] SDS-PAGE and Western-blotting - SDS-PAGE was performed under non-
reducing
conditions using 4-20% Tris-Glycine gels at 125V for about 2 h. Resolved
proteins were
transferred onto a nitrocellulose membrane using the Mini TRANS-BLOT cell
system (Bio-
Rad Laboratories, Hercules, CA) at 100 V for 1 h. To examine the identity of
recombinant
proteins, triplicate samples were run in the same gel and membrane with
transferred samples
were cut into three pieces for probing with HRP-anti-Myc, HRP-hPAM4, and 45M1
plus
HRP-GAM, respectively. The signals were developed with SUPERSIGNALTM West Dura
Chemiluminescent Substrate (Thermo Fisher Scientific, Waltham, MA).
Results
[0439] Several MAbs were evaluated by indirect EIA for reactivity with plates
coated with
CPM1 (FIG. 25), a high molecular weight mucin fraction isolated from the Capan-
1 human
pancreatic cancer xenograft. Murine PAM4 and MAbs reactive specifically with
MUC1 and
MUC5ac mucins provided elevated reactivity in this indirect immunoassay, with
minor
reactivity also observed for MAbs directed to MUC3 and CEACAM6. Essentially no
reaction was seen with MAbs to MUC2, MUC4, MUC16, and CEACAM5 glycoproteins,
or
the CA19-9 carbohydrate epitope.
[0440] It should be noted that a negative EIA reaction does not necessarily
indicate absence
of the mucin-antigen, because the specific epitope structure may be present,
but inaccessible
(i.e., cryptic). This is likely the case for MAb-CLH2 anti-MUC5ac generated
against a
peptide derived from the mucin's tandem repeat (Reis et al., 1997, Int J
Cancer 74:112-21),
since the other two anti-MUC5ac MAbs were highly reactive. Similarly, CM1 anti-
MUC1
144
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
was considerably less reactive than MA5 and KC4 anti-MUC1 antibodies. Capan-1
cells
produce well-differentiated tumors with highly glycosylated mucins. Thus, it
is likely that
both CLH2 and CM1, reactive with the tandem repeat domains of their respective
mucins,
would not be reactive with CPM1, since the tandem repeat epitopes are
inaccessible.
[0441] We then evaluated whether the anti-mucin MAbs were reactive with PAM4-
captured
mucin. Humanized PAM4 (hPAM4)-coated plates were used to capture the specific
mucin-
antigen from the CPM1 fraction, which was then probed with various anti-mucin
MAbs.
Murine MAbs (mMAbs) specifically reactive with MUC1, MUC3, MUC4, MUC16 and
CEACAM6 did not provide a signal in these heterologous sandwich immunoassays
(not
shown). On the other hand, both anti-MUC5ac mMAbs tested, 45M1 and 2-11M1,
gave
positive reactions with the hPAM4-captured antigen (FIG. 26), with 45M1
showing
significantly greater reaction than 2-11M1 (Kd = 14.32 1.08 ug/mL and 24.4
7.83 ug/mL,
respectively, for MAbs 45M1 and 2-11M1; P< 0.001). However, neither of these
individual
anti-MUC5ac MAbs provided as strong signal intensity as the rabbit anti-CPM1
polyclonal
IgG fraction. Importantly, mPAM4 did not bind to the hPAM4-captured antigen,
nor did
hPAM4 bind to mPAM4-captured antigen, suggesting that the PAM4 epitope is
present at
low density, possibly only a single site within the mucin-antigen.
[0442] Follow-up studies were designed to inhibit the binding of hPAM4 to CPM1-
coated
plates (FIG. 27A-B). Although 2-11M1 anti-MUC5ac was unable to inhibit hPAM4-
CPM1
binding, 45M1 anti-MUC5ac was able to provide a limited inhibitory effect,
with IC. =
25.5% inhibition (FIG. 27A). mPAM4, included as a positive control, provided
IC.=
92.4% self-inhibition at a concentration 0.1 ug/mL, while the MA5 and KC4 anti-
MUC1
antibodies provided no inhibition, even at the highest concentration evaluated
(10 ug/mL)
(FIG. 27A). hPAM4 was unable to completely block mPAM4 binding to the CPM1
antigen
(ICmax=52.8%) (not shown), a not unexpected finding since the humanized
version of PAM4
is known to have a lower affinity than the murine parent. Ascites fluids
containing mMAbs
with known mapping to MUC5ac were serially diluted as inhibitory reagents,
with results
shown in FIG. 27B. mMAbs 21M1, 62M1, and 463M1 each provided inhibition
similar to
the results shown for mPAM4 self-blocking, with 45M1 ascites providing limited
inhibition,
similar to what was observed with the commercially available 45M1-IgG (FIG.
27B).
Ascites fluid containing a murine anti-alpha-fetoprotein (AFP), included here
as a negative
control, provided no inhibition of the hPAM4 binding to CPM1 (FIG. 27B).
Unfortunately,
145
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
insufficient volumes of ascites precluded determination of MAb concentrations,
so that
relative blocking efficiency could not be calculated.
Discussion
[0443] The current Example suggests that PAM4 is reactive with the MUC5ac
mucin
glycoprotein. FIG. 28 presents a map of the MUC5ac mucin domains with reactive
epitopes
indicated for several of the anti-MUC5ac MAbs employed in our studies (Nollet
et al., 2002,
Int J Cancer 99:336-43; Nollet et al., 2004, Hybrid Hybridomics 23:93-99;
Lidell et al., 2008,
FEBS J 275:481-89). CLH2 is reactive with the peptide core of the tandem
repeat domain
(Reis et al., 1997, Int J Cancer 74:112-21), and is likely a cryptic epitope
within the Capan-1
tumor-derived MUC5ac. 2-11M1 is reactive with the N-terminus of the mucin
(Nollet et al.,
2004, Hybrid Hyridomics 23:93-99), and 45M1 at the furthest N-terminal region
of the
cysteine-rich, C-terminus (Lidell et al., 2008, FEBS J 275:481-89). Both of
these MAbs were
reactive with PAM4-captured mucin, whereas MAbs to MUCs 1, 3, 4, and 16 were
not. We
observed that 45M1 provides a significantly greater signal response than 2-
11M1, suggesting
a greater density of 45M1-epitopes than 2-11M1-epitopes within CPM1. However,
this may
simply be due to a loss of 2-11M1 epitopes through proteolytic digestion of
the relatively
non-glycosylated N-terminus, and/or molecular shear of this very large
glycoprotein during
purification. In any case, the 2-11M1 antibody provided no inhibition of the
hPAM4-CPM1
interaction, suggesting the epitope is located distant to the PAM4-epitope.
[0444] On the other hand, 45M1 did inhibit the hPAM4-CPM1 interaction, albeit
only
partially, suggesting that the PAM4-epitope is within the C-terminal region of
the mucin or
conformationally altered by interaction of this antibody with the mucin
molecule. MAbs
21M1, 62M1, and 463M1 have also been mapped to the C-terminal region of the
MUC5ac
mucin (Nollet et al., 2002 Int J Cancer 99:336-43; et al., 2004, Hybrid
Hybridomics 23:93-
99; Lidell et al., 2008, FEBS J 275:481-89), and each provided significant
inhibition of the
PAM4-mucin reaction. Taken together, our data provide direct evidence that
PAM4 is
reactive with the identical mucin (MUC5ac), and that the PAM4 epitope is
either directly-
blocked, or conformationally modified, by interaction of these MAbs with the
MUC5ac
antigen.
[0445] We had initially reported that PAM4 was reactive with the MUC1 mucin
species
(Gold et al., 2007, Clin Cancer Res 13:7380-87; Gold et al., 2006, J Clin
Oncol 24:252-58).
This was based upon MUC/-gene transfection studies, whereby PAM4 was observed
to react
with the gene-transfected, MUC1+ cell line, but not the MUC1- parental cell
line or vector
146
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
control cell lines. However, other evidence acquired since then has questioned
this
interpretation, suggesting that MUC1 transfection may have upregulated other
mucins as
well. Prior results from our laboratory lend support to the current findings.
The PAM4
epitope was found to be highly sensitive to mild reduction with dithiothreitol
(0.02M, 15 min,
20 C) or heat (100 C, 2 min), suggesting the epitope is peptide in nature, and
highly
dependent upon a specific conformation of the protein core kept intact by
disulfide bridges
(Gold et al., 1994, Int J Cancer 57:204-10). This is unlikely to be MUC1 with
all of the
cysteines located within the transmembrane domain of the mucin, but is
consistent with the
loss of reactivity shown by several anti-MUC5ac MAbs upon reduction of the
mucin antigen.
Further, employing immunohistochemical methods, we reported that frequency of
expression
and morphologic distribution of the PAM4-epitope within PDAC and its precursor
lesions
shared greater similarity to those described for MUC5ac than for MUC1 (Gold et
al., 2007,
Clin Cancer Res 13:7380-87).
[0446] In conclusion, antibodies that bind to the PAM4 epitope of MUC5ac are
of use for
detection and differential diagnosis of pancreatic cancer. Immunoconjugates of
such
antibodies are of use for pancreatic cancer therapy.
Example 35. DOTA Conjugates of PAM4
[0447] The hPAM4 antibody was prepared as described in ExampleThe genes of CDR-
grafted VH and Vic chains of hPAM4 were inserted into the pdHL2 plasmid
vector, a DHFR-
based amplifiable expression system. The plasmid was transfected into the
murine myeloma
cell line, Sp2/0-Ag14 (ATCC, Manassas, VA) to generate the cell clones
producing hPAM4.
The complete mature amino acid sequence is shown below.
hPAM4 Heavy Chain
QVQLQQSGAEVKKPGASVKVSCEASGYTFPSYVLHWVKQAPGQGLEWIGYINPYND
GTQYNEKFKGKATLTRDTSINTAYMELSRLRSDDTAVYYCARGFGGSYGFAYWGQ
GTLVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGV
HTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKRVEPKSCDKTHT
CPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGV
EVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISK
AKGQPREPQVYTLPP SREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTP
PVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK (SEQ
ID NO:117)
hPAM4 light chain
147
CA 02899811 2015-07-29
WO 2014/165506
PCT/US2014/032513
DIQLTQSPSSLSASVGDRVTMTCSASSSVSSSYLYWYQQKPGKAPKLWIYSTSNLASG
VPARFSGSGSGTDFTLTISSLQPEDSASYFCHQWNRYPYTFGGGTRLEIKRTVAAPSV
FIFPP SDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDST
YSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC (SEQ ID NO:118)
[0448] The DNA and amino acid sequences of hPAM4 Vic and VH are shown in FIG.
4A and
FIG. 4B, respectively, with the CDRs identified in bold and underlined.
[0449] The current cell clone name is hPAM4-2E3 and is produced in Sp2/0 host
cells,
DHFR expression system. The antibody is a humanized IgGi,, glycoprotein. A
glycosylation
site on the heavy chain (Asn299) has a composition per mole of hPAM4-DOTA: 0.5
Fuc, 6.3
GlcNAc, 6.3 Man, 0.3 Gal and 0.15 Neu5Gc; glycosylation species: GOF 70%, GlF
23%,
G2F 2%, G1FS1 4%, G2FS1 1%. There are 16 S-S bonds (32 SH), identified and
located
exactly as theoretical prediction based on the above sequence.
[0450] An hPAM4-DOTA product was prepared from purified hPAM4 IgG that was
coupled
with the 12-membered macrocyclic chelating agent 1,4,7,10-
tetraazacyclododecane-N,
NN",N"'-tetraacetic acid (DOTA).
[0451] DOTA was conjugated via one of the carboxyl moieties to reactive sites
on the
hPAM4 antibody to generate a stable conjugate. The coupling is assumed to be
via stable
amide bond to the antibody's lysine side-chain amino group.
[0452] The chemical conjugation was performed by first reacting DOTA with N-
hydroxysulfo-succinimide (sulfo-NHS) in the presence of 1-ethy1-3-(3-
dimethylaminopropyl)carbodiimide (EDC) to generate activated DOTA, then
incubating
activated DOTA with purified hPAM4 antibody. Conditions were optimized to
yield a
substitution ratio of 4-7 DOTA moieties per antibody molecule, as determined
by mass
spectrometry assays.
* * *
[0453] It will be apparent to those skilled in the art that various
modifications and variations
can be made to the products, compositions, methods and processes of this
invention. Thus, it is
intended that the present invention cover such modifications and variations,
provided they
come within the scope of the appended claims and their equivalents.
148