Note: Descriptions are shown in the official language in which they were submitted.
ENERGY STORAGE DEVICE
FIELD OF THE INVENTION
This invention relates generally to an energy storage device, and, more
particularly, to an electro-active electrical component used to store energy
electrostatically in an electric field.
BACKGROUND
There has been a recent trend in the usc of electrochemical capacitors for
enhanced storage of electrical energy. These capacitors derive their enhanced
characteristics from two primary mechanisms: double layer capacitance and
pseudocapacitance. Double layer-type capacitors use an electrical double layer
(explained below) to achieve a very small charge separation (d), which
increases
electric field (E) for a given voltage, increases capacitance (C) and
consequently
increases the energy stored (U) for the given voltage versus a conventional
planar
surface capacitor, as apparent in Eqs. 1 through 3 below.
Eq.] E= ¨dv
where E = electric field, V =potential difference or voltage, and d =
separation
of charged plates.
Eq. 2 C=
where k = relative permittivity or dielectric, C = capacitance,
eo=permittivity
of free space, and A = cross-sectional surface area.
Eq. 3 U = CV2
where U = energy stored, C = capacitance and V = voltage.
Practically, the smaller thickness (d) allows for much more surface area of
the
plates to be packaged (usually rolled or stacked) in a given volume. As
evident from
Eq. 2, this area increase also significantly increases capacitance. Devices of
the above
described nature are commonly referred to as electric double layer capacitors
(EDLCs).
1
Date Recue/Date Received 2020-08-20
In pseudocapacitors, which are a hybrid between double-layer capacitors and
batteries, both the bulk and the surface of the material play key roles. They
thus can
store much more energy than conventional planar surface capacitors, but face
many of
the same reliability and scientific challenges as advanced batteries,
including high
cost due to expensive raw materials and complex processing. Pseudocapacitance
imitates battery technology by storing energy in chemical reactions (oxidation
and
reduction) which take place at or very near the surface of the relevant
electrodes. The
surface nature of the reactions is the distinguishing characteristic from
chemical
battery technology.
Either or both of these effects (i.e., double layer and pseudocapacitance) may
be used in so called "supercapacitors." Advantageously, the invention herein
makes
use of and extends double layer theories in a novel manner, without any formal
"chemical reactions" present.
Also previously explored is the notion of enhancing a double layer capacitor
by the application of an electrically conducting polymer e.g. Hu, U.S. Patent
No.
8,164,881. While the invention described herein certainly makes use of a
polymer
coating, the polymer is sometimes electrically resistive and sometimes
insulating but
is not electrically conducting by design. This significantly differs in
structure, nature
and consequently in function from previous applications.
Current EDLCs can handle only low voltages before breakdown. In order to
attain the higher voltages necessary for many practical applications (such as
electric
vehicles), low voltage EDLCs are connected in series much in the same way
batteries
are series-connected for high voltage use. An energy storage device
constructed
according to principles of the invention can handle higher voltages and be
connected
in series.
The invention is directed towards overcoming one or more of the fundamental
problems with existing designs and solving one or more of the needs as set
forth
above.
2
Date Recue/Date Received 2020-08-20
SUMMARY OF THE INVENTION
To solve one or more of the problems set forth above, in an exemplary
implementation of the invention, an energy storage device comprises a
capacitor
having a first conductive electrode having a first outer side and an opposite
first inner
side; a thin non-porous first nonconductive coating on the first inner side of
the first
conductive electrode; a dielectric material on the first nonconductive
coating, the first
nonconductive coating being disposed between the first conductive electrode
and the
dielectric material; and a second conductive electrode adjacent to the
dielectric
material, the dielectric material being disposed between the second conductive
to electrode and the first nonconductive coating. Optionally, a second
nonconductive
coating may be provided on the second conductive electrode, disposed between
the
second conductive electrode and the dielectric material. The nonconductive
coatings
are thin, having a thickness that is less than 10% of the overall thickness of
the energy
storage device. Illustratively, and without limitation, the nonconductive
coatings may
be comprised of a condensed and polymerized xylylene monomer, a parylene
polymer, PuraleneTM polymer, a metal oxide, or some other insulator that can
be
deposited or otherwise formed in a thin film on the electrode(s).
The nonconductive coatings constitute insulating layers that allow for much
higher voltages to be employed than in traditional EDLCs. This extends the
layers
from just a few (two or three which alternate in charge) to many (possibly
orders of
magnitude more in number) which can reach far into the dielectric medium. The
increase in working voltage, significantly increases the electric field
present in the
capacitor and energy stored in the capacitor.
In one embodiment, the dielectric material is a variable viscosity dielectric
material. In other words, the viscosity may be increased or decreased in a
controlled
manner, such as in response to an applied external stimulus. By way of
example, the
external stimulus may be a force, a pressure, a shear stress, a normal stress,
heat, a
heat sink, a coolant, a magnetic field, or an electric field. The external
stimulus may
comprise a mechanism from the group consisting of a controllable heat source,
a heat
sink, a coolant, a controllable cooling source, a controllable magnetic field
generator,
3
Date Recue/Date Received 2020-08-20
CA 02908178
a controllable electric field generator, a controllable force generator, a
controllable
pressure generator, or a controllable shear stress generator. Viscosity of the
dielectric
can be made to gradually increase from electrode layer to electrode layer
sequentially,
or vice versa. With a viscosity increase, the discharge of the Helmholtz and
Diffuse
Helmholtz layers as thermal energy can be slowed and essentially halted with
complete solidification. Electrical energy can thereby be stored for extended
periods
of time until ready for release. When ready for release, the viscosity may be
reduced
in a controlled manner such as by removing a viscosity-increasing stimulus or
by
applying a viscosity-decreasing stimulus. The reduction of viscosity
facilitates
discharge.
The dielectric material may be comprised of a dielectric substance such as a
conductive polymer, a nonconductive polymer, an inorganic metal oxide, a metal
oxide mixture, a biopolymers or some other dielectric substance with a
changeable
viscosity. Electro-rheological dielectric substances, magneto-rheological
dielectric
substances and Bingham plastic dielectric substances may be used within the
spirit
and scope of the invention.
BRIEF DESCRIPTION OF THE DRAWINGS
The foregoing and other aspects, objects, features and advantages of the
invention will become better understood with reference to the following
description
and accompanying drawings, where:
FIG. 1 is a schematic diagram that conceptually illustrates the Helmholtz
model of an electric double layer; and
FIG. 2 is a schematic diagram that conceptually illustrates the Gouy-Chapman
model of an electric double layer; and
FIG. 3 is a schematic diagram that conceptually illustrates the Stern model of
an electric double layer; and
FIG. 4 is a schematic diagram that conceptually illustrates the Grahame model
of an electric double layer; and
FIG. 5 is a schematic diagram that conceptually illustrates the model of an
electric double layer by Bockris, Devanathan and Muller; and
4
Date Recue/Date Received 2022-08-05
FIG. 6 conceptually illustrates an exemplary energy storage device according
to principles of the invention;
FIG. 7 conceptually illustrates an exemplary energy storage device with
applied forces according to principles of the invention;
FIG. 8 conceptually illustrates a flow diagram for producing a polymer for use
with an exemplary energy storage device according to principles of the
invention;
FIG. 9 is an exemplary flow chart illustrating a method for making a high
permittivity dielectric material for use in an energy storage device according
to
principles of the invention; and
FIG. 10 conceptually illustrates a multi-state electrical circuit diagram in
accordance with one or more embodiments of the present disclosure for the
recovery
of leakage current from an energy storage capacitor;
FIG. 11 conceptually illustrates a multi-state electrical circuit diagram in
accordance with one or more embodiments of the present disclosure for the
recovery
of leakage current from an energy storage capacitor;
FIG. 12 conceptually illustrates a multi-state electrical circuit diagram in
accordance with one or more embodiments of the present disclosure for the
recovery
of leakage current from an energy storage capacitor;
FIG. 13 conceptually illustrates a multi-state electrical circuit diagram in
accordance with one or more embodiments of the present disclosure for the
recovery
of leakage current from an energy storage capacitor;
FIG. 14 illustrates voltages over time for an exemplary energy storage device
according to principles of the invention.
Those skilled in the art will appreciate that the figures are not intended to
be
drawn to any particular scale; nor are the figures intended to illustrate
every
embodiment of the invention. The invention is not limited to the exemplary
embodiments depicted in the figures or the specific components, sequence of
steps,
configurations, shapes, relative sizes, ornamental aspects or proportions as
shown in
the figures.
5
Date Recue/Date Received 2020-08-20
DETAILED DESCRIPTION
In an exemplary embodiment of an energy storage device according to
principles of the invention, an insulating layer directly allows for much
higher
voltages to be employed than in traditional EDLCs. This in turn increases the
number
of layers from just a few (two or three which alternate in charge) to many
(possibly
orders of magnitude more in number) which can reach far into the dielectric
medium.
A treatment as series capacitors (a common way to analyze multilayer
capacitance)
demonstrates that, for a sct amount of charge, adding more layers will
actually
decrease capacitance and increase voltage. This increase in working voltage
(both
directly and indirectly from the use of an insulating layer) along with the
small degree
of charge separation previously observed in EDLCs, significantly increases the
electric field present in the capacitor as can be seen from Eq. 1 (above).
Worth further
notice is the resulting dramatic increase in energy stored in the capacitor
from the
voltage increase as seen in Eq. 3 (above). While parameters can be manipulated
to
retain high capacitance (such as using stacks, rolls, and other "tricks"), the
increase in
voltage clearly outweighs the proportional decrease of capacitance in the
contributions to the amount of stored energy of the overall device.
As background, the first mathematical description of an electrical double
layer
is thought to have been written by Hermann Helmholtz. He depicted two parallel
layers of dissimilar charge along a surface. This model gave a constant
capacitance
based on the separation of the layers and the dielectric properties of the
medium.
Helmholtz proposed that the interface between a metallic electrode and an
electrolyte
solution behaves like a capacitor in that it is capable of storing an electric
charge.
The Helmholtz model is conceptually illustrated in FIG. 1. Helmholtz's
proposed
model is that the electrode possesses a charge density arising from an excess
negative
or deficiency of positive charges at the electrode surface. In the model, the
charge on
the electrode is exactly balanced in solution by an equal but oppositely
charged
amount of ions. This charge originates from the arrangement of electrolyte
ions at the
interface and/or the reorientation of dipoles in solvent molecules. A
potential
difference occurs across the interface, forming an electric field gradient
across a
6
Date Recue/Date Received 2020-08-20
charge separation layer. Ions are electrostatically repelled or attracted
towards the
electrode surface and an excess of either anions or cations is created.
Upon observation that the capacitance was not truly constant with increasing
potential (voltage), the Gouy-Chapman model was introduced. Gouy employed
statistical mechanics to develop his theory and suggested that the thermal
motion of
the medium prevents the formation of an organized layer. The Gouy-Chapman
model
(FIG. 2) employs diffuse layers of charges which are quite unstationary. Gouy
and
Chapman proposed the diffuse double layer model that predicted a dependence of
the
measured capacitance on both potential and electrolyte concentration. They
showed
that the excess charge density in solution is not exclusively situated at the
outer
Helmholtz plane, and thus the double layer may be of variable thickness. In
their
view, a Helmholtz-type rigid double layer would not form because the
attractive and
repulsive electrostatic forces between the field and the charge on the ions
are
counteracted by random thermal motion in the dielectric solution which tends
to
disperse the excess ions from the surface of the electrode. In the Gouy
Chapman
model, the ions are considered as point charges contained within a single
diffuse layer.
This model, like the Helmholtz model, fails under particular conditions.
Failures of the Helmholtz and Gouy -Chapman models prompted the
contributions of Stern and then Grahame. Their work combined the two
previously
mentioned theories into one in which an inner "Stern layer" or "Helmholtz
layer" is
organized on a charged surface with a diffuse layer forming around it. In the
Stern
model, as conceptually illustrated in FIG. 3, the two previous models were
combined,
with some of the ions adhering to the electrode as suggested by Helmholtz and
some
forming a Gouy-Chapman type diffuse layer. Grahame proposed that, although the
closest approach to the electrode is occupied by solvent molecules, it may
also be
possible for some ionic or uncharged species to penetrate into this region.
This model
for the electrode/electrolyte interface (FIG. 4) employs three regions. First,
the inner
Helmholtz plane or layer extends from the electrode to a plane passing through
the
centers of specifically adsorbed ions. Second, the outer Helmholtz plane or
layer
passes through the centers of hydrated ions at their distance of closest
approach to the
7
Date Recue/Date Received 2020-08-20
electrode. Third, the diffuse layer lies beyond the other layers. Potential ip
changes
linearly with distance up to the outer Helmholtz plane and then exponentially
through
the diffuse double layer region.
Still more work has been done to improve upon the theory by Bockris,
Devanathan, and Muller who take into account solvent interactions in the
dielectric.
This model (illustrated in FIG. 5) is yet imperfect and operates on
assumptions such
as the approximation of ions as point charges, the constancy of dielectric
permittivity,
the constancy of viscosity, and the assumption that the significant
interactions are all
Coulombic in nature. Bockris, Devanathan and Muller suggested that
reorientation of
solvent molecules would occur depending on the excess charges at the electrode
and
the presence or absence of specifically adsorbed ions at the surface. The
proposed
variation of the electrostatic potential with distance is qualitatively
similar to that of
the Grahame model. Water molecules cover most of the electrode in an oriented
layer. At certain sites, the water molecules are replaced by a specifically
adsorbed ion
(e.g., an anion) that has shed its hydration shell. The plane going through
the center of
these ions is the inner Helmholtz plane, defining the inner Helmholtz layer.
Ions that
carry a primary hydration shell are found next to and are situated outside of
the first
layer of water molecules adsorbed onto the electrode surface. The plane going
through
the centers of these ions constitutes the outer Helmholtz plane, defining the
outer
Helmholtz layer. None of these models teach or suggest an energy storage
device
with an insulating layer or a variable viscosity dielectric according to
principles of the
invention.
In the prior art it has been assumed that the energy stored in variously named
layers adjacent to the electrodes is non-recoverable. In other words, when an
electric
potential is applied to a flat electrode in contact with a solution that has
ions capable
of movement through the solution, a movement of ions to that surface takes
place.
Once near enough to the electrode, the ions are assumed to be immobilized at
the
surface due to the strong electrostatic forces that bind them in place. The
energy of
collisions with solvent molecules is not sufficient to displace these ions. If
the
electric potential is removed from the surface, these ions are free to move
about in a
8
Date Recue/Date Received 2020-08-20
diffusive manner. It is interesting to note that if the electric potential is
removed from
the electrode surface, the resulting collapse of the electrical bilayer
closest to the
electrode allows the release of energy of the immobilized ions such that the
energy is
not fully released as heat, but instead the electrode can absorb the energy
produced by
the collapsing electrical field and produce an electric potential and current
in that
conductor. This effect is the basis for the energy storage in an electrical
double layer
capacitor (EDLC).
The energy that is stored in the diffuse outer layers of an EDLC is often not
fully recovered. The electrical double layers that are formed close to the
electrode
surface are termed Helmholtz layers, while those that are further away are
termed the
Gouy-Chapman layers. One distinction between these layers is that the ionic
layers
that are not capable of being thermally diffused from the electrical surface
are termed
"Helmholtz" layers. These layers are essentially immobilized at the working
temperature by the application of an electric potential to the surface.
Another
distinction is that the diffuse Helmholtz layers (Gouy-Chapman, but often
referred to
as Diffuse Helmholtz layers), referred to as DH layers herein, are layers
wherein
random thermal movements are able to diffuse the ionic arrangements induced by
the
electric field. Since this is not a sharp boundary, an arbitrary time unit
associated with
a 50% loss of potential energy over a period of 1 second could be used to
define the
boundary layer conditions between the two major macroscopic layers.
Both the Helmholtz and DH layers (which form at constant ambient
temperature) are entropically reduced as compared to the bulk. These
entropically
modified materials display different physical characteristics that have been
noted (e.g.
permittivity). Application of the modified characteristics has been shown in
U.S.
Patent No. 8,633,289, to be issued January 21, 2014, which describes improved
synthesis of the stable intermediate dimer of xylylene ([2,211paracyclophane)
and
derivatives related to that compound and general structure, a method for the
formation
of cyclophanes and related compounds with various substituents, and a method
to
apply the xylylene (or substituted xylylene) monomers to make coatings and
other
polymer products derived from the reactive intermediate. Likewise, US Patent
9
Date Recue/Date Received 2020-08-20
Application No. 13/853,712, published as U.S. Publication No. 2013-0224397 on
August 29, 2013, describes, inter alia, a method for making high permittivity
dielectric material for capacitors using organic polymers to produce low
conductivity
dielectric coatings.
The rationale for enhanced permittivity in entropically reduced materials is
understood by the concept of the charges being "organized" into discrete rows
and
columns. Since each charge layer is energetically optimized to be in the
lowest
energy configuration possible based upon surrounding ionic charges, the
imposition of
an external electric field from the electrode leads to disruption of the
lowest energy
state attainable from its current position. Thus, when the electric field is
applied, the
dipole or ion is moved from its rest position, which in turn leads to a
rearrangement of
the charge distribution in the material. This leads to other rearrangements of
all other
dipoles continuing throughout the dielectric. Thus, energy that is not
converted into
heat is absorbed by the dielectric. When the energy is released, a reverse of
this
process can take place provided the energy stored is not released through
other
mechanisms such as increased thermal motions.
In the case of entropically "normal" materials, the rearrangement of the
dipoles and ions in an electric field is not as certain to cause a
rearrangement of all the
other ions and dipoles in the materials. In other words, there is a
probability that the
rearrangement of the dipole or the ion can take place with little or no net
interaction
with the other dipoles and ions in the material. In these cases, the material
will display
less energy storage capability than in its entropically reduced form.
If the viscosity of the material is such that movement of the molecules is
able
to take place, the energy stored from the formation of the electric field by a
given
dipole or ion is able to dissipate through relaxation mechanisms in which the
energy is
converted into rotation, vibration, translation, and other movements that
manifest
themselves externally as heat. With a low viscosity material, the energy that
has been
stored in the Diffuse Helmholtz layers (DH layers) is thus lost due to random
motions
of the ions and dipoles.
Date Recue/Date Received 2020-08-20
With intermediate to high viscosity materials, the time frame for formation of
the Helmholtz layers (H layers) and the DH layers is substantially increased.
The
thermal motions of molecules (excluding for now vibrations of the lattice as a
macroscopic phenomenon), however, are effectively reduced to near
negligibility. In
these materials, it is possible to store the energy of an electric field in
the H and DH
layers relatively quickly compared to the time required for the energy to be
dissipated
thermally. Thermal dissipation is essentially a first order decaying
exponential in time
similar to radioactive decay or diffusion; if during the charging cycle the
energy is
absorbed over a time period of, for example, 1 second, a high viscosity
material may
require many seconds or even minutes to reach even 90% energy dissipation as
heat.
The thermal decay process is substantially slower than the electrical double
layer energy storage process. Thus, it is possible to utilize the energy
stored by the
formation of both the H and DH layers if the energy is quickly accessed. In
this
situation the release of most of the energy in the formed dipole and ionic
layers is
through the electric field and is subsequently coupled with electric potential
and
current. Since the discharge of the H and DH layers may require the movement
of
molecules and atoms, the discharge process can be relatively slow compared to
charging but still remain fast relative to the relaxation mechanisms that
produce heat.
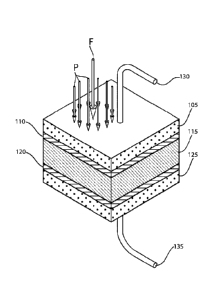
As conceptually illustrated in FIG. 6, in an exemplary embodiment, an energy
storage device according to principles of the invention includes a conductive
electrode
105 having a smooth or rough surface, which, by example, may be comprised of a
smooth metal, a conductive polymer or a rough carbon electrode of high surface
area.
A resistive or insulative coating 110 is applied to one surface of the
electrode 105. By
way of example, the coating 110 may comprise a metal oxide, PuraleneTM, plasma
or
film coating. A method of producing a PuraleneTm coating is describe below.
PuraleneTM is applicant's trademark for the coating substance described below.
A
dielectric material 115, i.e., a high permittivity material or a dipole
containing low
viscosity material, is applied to the outer surface of the coating 110. By way
of
example, the dielectric material 115 may comprise a conductive or
nonconductive
polymer, an inorganic metal oxide, mixed metal oxides, mixed polymer and
organic
11
Date Recue/Date Received 2020-08-20
materials and biopolymers. Nonlimiting examples of other suitable dielectric
compositions are described below. In a preferred embodiment, the low viscosity
of
the dielectric may be increased in a controlled manner by application or
removal of
energy in the form of heat, a force, electric field, magnetic field or other
means of
changing viscosity of the applied dielectric composition. The dielectric 115
may have
its viscosity reduced to aid in the more rapid release of the energy from the
bound
dipole and ionic layers. An opposite conductive electrode 125 (which may be
comprised of a conductor with insulative or resistive coating or without such
a
coating) is applied to the dielectric 115, i.e., the high permittivity
material or dipole
containing low viscosity material. The opposite electrode 125 may be the same
material as the first electrode 105. An insulative or resistive coating 120
between the
opposite conductive electrode 125 and the dielectric 115 is optional. This
coating 120
may be the same as the coating 110 between the first electrode 105 and the
dielectric
115.
The electrodes may be attached to a voltage source, via conductive leads 130,
135 (e.g., conductive wire leads, traces or other pathways), and allowed to
charge.
The viscosity of the dielectric 115 thus charged is optionally increased to
allow for a
longer period of electric charge storage due to the resulting decrease in
random
thermal motions or other viscosity-dependent processes. The dielectric is
discharged
by current flow out of the electrodes 105, 125 by an electrical load.
In a capacitor formed in this manner, equivalent charges of opposite sign will
flow to each of the electrodes 105, 125. If the dielectric 115 (i.e., high
permittivity
material or a dipole containing low viscosity material applied to the surface
of the
resistive or insulative coating) of low viscosity is used, the charge flow
will be very
substantial for an extended period of time. Very viscous materials require
much
longer charging times at lower rate of charge flow. Once charge has been added
from
a voltage source, removal of the voltage source will then lead to a slow
discharge of
the voltage retained at the electrodes. The leakage current resistively
discharges the
energy stored in the formation of the H and DH layers.
12
Date Recue/Date Received 2020-08-20
The thicker the insulative coating 110, 120 the higher the external applied
voltage needs to be in order to store a given amount of energy at constant
thickness.
Additionally, thicker insulative coatings 110, 120 such as PET (polyethylene
terphthalate) produce an almost order of magnitude reduction in the energy
storage
capabilities. A PuraleneTM coating is preferred due to its characteristics of
reduced
pinholes, i.e., being substantially nonporous, and its ability to be coated
into very thin
layers. This enables the overall thickness of a capacitor to be in the range
of 100
microns and reasonable voltages are thus possible. For example if the
thickness were
1000 microns and the insulative coatings 110, 120 were 1% of that thickness (5
microns each), then to attain a 10V/micron e-field, 10,000V would have to be
applied
on the cap externally. This is too high of a voltage to be used economically
and
safely. Thus, the thinner the nonconductive coating 110, 120, the lower the
voltage
can be while retaining the storage capabilities of the device. Using
PuraleneTM, which
is inexpensive and exhibits very desirable qualities such as reduced pinholes,
flatness,
etc., and the high molecular weight ionic polymers salts described below for
the
dielectric 115, energy densities that are at least 10 times that of typical
EDLCs are
achievable. FIG, 14, which is discussed below, illustrates the differences in
performance of an energy storage device according to principles of the
invention, in
contrast to that of a conventional EDLC and batteries.
Viscosity modifiers, such as solvents, branched polymers, low molecular
weight oligomers, and dendritic polymers may be added to the dielectric
material 115
to reduce viscosity. Ethanol and unreacted starting materials may serve such
purposes.
Due to the viscosity dependence of the charging and discharging
characteristics of the system, it is advantageous to include in the embodiment
of this
technology a method for dynamically varying the viscosity of the dielectric.
There
are a multitude of known methods for efficiently creating a controllable
change in the
viscosity of a fluid, many of which could easily be integrated into the system
implementation by one well-versed in the art of materials sciences,
13
Date Recue/Date Received 2020-08-20
One non-limiting example of a method for controlling the viscosity of the
dielectric is by controlling the temperature. If instead of maintaining the
device
described above at a constant temperature, during or after charging the device
is
cooled from an electrode 105 or 125 inward, then the viscosity of the
dielectric can be
made to gradually increase from electrode layer 105 to electrode layer 125
sequentially, or vice versa. Assuming a viscosity increase with lower
temperature
(although the opposite effect can sometimes be obtained) the discharge of the
H and
DH layers as thermal energy can be slowed and essentially halted with complete
solidification. The electrical energy can thereby be stored for extended
periods of time
until ready for release.
Release of the electrical energy with minimal losses to heating is similar to
the
manner that it was charged. The cooled device can be warmed as necessary with
ambient heat or generated heat to release electrical energy through the
electrodes as
the viscosity of the internal dielectric is reduced. This slow warming has the
added
benefit of preventing rapid discharge of the energy contained in the H and DH
layers.
Coordination of the warming of the electrodes and dielectric can be made to
accommodate the energy demands of the electrical load. Care must be taken in
the
system design in order to prevent a runaway condition in which internal or
external
heating of the dielectric causes the temperature to rise rapidly and in turn
decrease
viscosity at an increasing, uncontrolled rate.
Another well-known method for viscosity control is via exploitation of non-
Newtonian fluid effects. A multitude of materials exhibit, to varying degrees,
a
nonlinear or offset relationship between viscosity and applied stress, shear
rate, time,
or other factors. Applied forces and pressure are conceptually illustrated in
FIG. 7.
Common materials have been noted which exhibit either an increase or decrease
in
apparent viscosity when subjected to mechanical stress. These materials are
often
classed as shear thickening (dilitant) or thinruing (pseudoplastic), depending
on the
sign of the viscosity change. Many materials in this class exhibit
viscoelastic effects,
in that they have a tendency to return to their original shape once stress is
removed.
Additionally, other materials exhibit time dependence on viscosity with stress
14
Date Recue/Date Received 2020-08-20
(thixotropic and rheopectic fluids, again depending on sign), and still others
exhibit an
offset relationship between viscosity and stress. The latter, known as a
Bingham
plastic, is of particular interest in this application due to its specific
characteristics.
A Bingham plastic is a viscoelastic material that behaves as a rigid body at
low
stresses but flows as a viscous fluid at high stress. More specifically, a
Bingham
plastic is known to act as a solid when applied stress is below a given limit,
and
therefore has a measurable yield stress or other factors. By manipulation of
this
feature, a dielectric which acts as a Bingham plastic could be held in a solid
state
under low-stress conditions, preserving H and DH layer formations for an
extended
period of time. When it becomes necessary to release the energy stored within
said
layers, a varying amount of stress would be applied to the dielectric, thereby
controllably lowering it's viscosity.
The makeup of the dielectric could be chosen such that it exhibits a desirable
set of non-Newtonian fluid characteristics. The embodiment of the device could
then
be engineered such that stress could be applied through mechanical or other
means to
appropriately control the viscosity of the dielectric. For a non-limiting
example, the
capacitor stack could be placed between two plates. The bottom plate would be
fixed
in place, while the top plate is attached to a mechanical, electromagnetic,
hydraulic, or
pneumatic actuator. When it becomes desirable to apply stress to the material,
the
actuator could apply force in a linear or rotational direction so as to apply
the optimal
amount, rate, and combination of shear and normal stresses deemed most
suitable to
the fluid application. Alternative methods include using a hydraulic or
pneumatic
bladder to apply stress on the capacitor stack between two fixed plates, as
well as
surrounding the device with a shape memory alloy, electroactive ceramic,
dielectric
elastomer, or other active element.
A combination of these effects may also be used to effect a change in the
dielectric's viscosity. By combining a non-Newtonian shear thickening
dielectric fluid
with low stiffness and compliant electrodes, the capacitor forms what is known
as an
electroactive polymer or specifically a dielectric elastomer actuator. Once a
charge is
applied to this capacitor, the electrostatic force between the electrodes
causes a force
Date Recue/Date Received 2020-08-20
directed normal to both plates. This force effectively "squishes" the
dielectric
together, applying a normal stress to the dielectric. If this dielectric were
also a non-
Newtonian shear thickening fluid, the viscosity would increase as the applied
shear
stress increases.
The viscosity of the dielectric material could also be dynamically controlled
by the modification of its physical characteristics to enhance the
dielectric's
viscoelectric properties. In a viscoelectric or electro-rheological fluid, the
makeup
and structure of thc dielectric fluid causes enhanced reactivity to external
electric
fields. An applied electric field can cause extreme, rapid, and reversible
changes in
viscosity. Electrorheological fluids can behave as a Bingham plastic,
described
previously, such that the yield stress is proportional to the applied electric
field. The
design of the electrode has been shown to increase electro-rheological
effects. Much
in the same way, the dielectric may be designed to exhibit magneto-rheological
effects, which will respond to a magnetic field rather than an electric one.
The
magneto-rheological effects may be even more applicable through lack of
interference
with the energy storage mechanism of the device.
In the case of both the dielectric elastomer actuator and viscoelectric
embodiment, care must be taken to avoid a runaway condition. Because the
viscosity
of the fluid is determined by the field magnitude applied to it, specific
conditions such
as short circuit are especially dangerous. If a short circuit condition is
allowed to
exist, the charge on the electrodes would be rapidly removed and therefore the
viscosity of the dielectric fluid would decrease rapidly. This decrease in
viscosity
would greatly increase the mobility of the H and DH layers, causing rapid
discharge
of stored energy into a potentially dangerous (i.e. short circuiting) load.
In one exemplary capacitor according to principles of the invention one 105 or
both electrodes 105, 125 (each of which may be a smooth copper electrode for
example) may be coated using a PuraleneTM coating process. Referring now to
FIG.
8, a high level flowchart that illustrates an exemplary method of producing an
augmented permittivity material, e.g., PuraleneTM, for use as a coating in a
capacitor
according to principles of the invention is shown. Sections, referred to as
chambers,
16
Date Recue/Date Received 2020-08-20
may comprise tanks having an inlet and an outlet or tubular structures with an
inlet
and an outlet. Chamber 210 is a heated tube or other evaporation device
intended to
volatilize starting material feed 200. Starting material feed 200 is
evaporated and
mixed with inert gas 205 in chamber 210. Inert gas 205 may be any of a group,
or a
mixture of, inert or essentially inert gases, such as, but not limited to,
argon or
nitrogen. Substitution of nitrogen for argon and/or other essentially inert
gases is
possible. Pumps and valves may be used to propel and control the flow of
fluids from
one station to another.
By way of example and not limitation, chamber 210 may comprise an
electrically heated Inconel (nickel alloy 600) pyrolysis reaction tube. The
tube is
heated to a temperature of about 450 C to 630 C at atmospheric pressure. A
flowing
stream of argon gas alone, or with a reactive compound such as nitrous oxide,
is
supplied to the pyrolysis reaction tube. The starter material feed 200 may be
xylene
vapor (Aldrich #W4449-411a. If the carrier gas 205 includes a reactive species
or
compound (e.g., N20), the ratio of gases is adjusted to provide approximately
molar
stoichiometric ratios of 1:1 of the reactive species or compounds (xylene to
nitrous
oxide).
The heated starter material 200 in the volatile mixture with inert gas reacts
with monatomic oxygen in reaction chamber 215. Being very reactive and
transient,
monatomic oxygen must be available to react with the volatile mixture in the
reaction
chamber 215. As discussed above, the source of monatomic oxygen may be a
gaseous
compound supplied with the carrier gas 205, or a gaseous compound supplied
separately 240, or another source, such as a plasma generator 227.
Monatomic oxygen plasma may be created by exposing oxygen (02) gas to an
ionizing energy source, such as an RF discharge, which ionizes the gas.
Alternatively,
a compound such as Nitrous Oxide (N20) may supply monatomic oxygen for the
reaction through thermal, catalyzed, and/or other decomposition. Thus, a
monatomic
oxygen plasma generator 227, or a monatomic oxygen chemical compound (e.g.,
N20) feed 240, or another suitable source of monatomic oxygen is provided.
17
Date Recue/Date Received 2020-08-20
A plasma gas can be used with the aforementioned starting materials to form
the intermediate oxidized products that may subsequently react to form
reaction
products that are oxidized forms of the starting materials which may be
monomers,
dimers, trimers, oligomers, or polymers. The plasma generator 227 includes a
gas
feed 230 that supplies gas to a plasma reaction chamber 220. A plasma driver
225
provides energy to ionize the gas.
The ratio of gases is adjusted to provide approximately molar stoichiometric
ratios of 1:1 (xylem to nitrous oxide or xylenc to monatomic oxygen).
Illustratively,
increased amounts of nitrous oxide result in partial and/or complete oxidation
of
xylene with reduced formation of the desired cyclophane or its polymer. Close
control of the stoichiometric ratios of the reactants is desired in this
reaction.
The reaction products are supplied to a reaction chamber 235, which is heated
to approximately 450 C to 800 C to facilitate vaporization of the reaction
products.
The vaporized reaction products 245 are expelled onto a lower temperature
collection
surface 250, where the reaction products condense and form a solid. At higher
temperatures (650 C to 800 C) the output of the reaction chamber 235 is
sufficiently
hot enough to maintain the monomer p-xylylene in monomeric form.
Condensation of the gas into a cooled glass vessel resulted in the deposition
of
a colorless to cream colored solid. This solid is partially soluble in 95%
ethanol. The
solid was compared to a sample of [2,2]paracyclophane (Aldrich #P225-5G-A) by
Gas Chromatography analysis (SRI#310, 15m megabore column, FID detector) and
was shown to give identical retention times.
Rapidly cooling of the monomer onto a lower temperature collection surface
250 (also referred to as a surface) (which, such surface, may comprise a
surface of an
electrode 105, 125) results in a liquid condensation of the monomer and rapid
polymerization of the monomer into a polymer. Comparison of the film thus
produced appears to be identical to parylene film formed by the conventional
vacuum
pyrolysis of dimers produced by the Gorham process. Without augmentation of
the
PuraleneTM polymer, permittivity of both solidified products is about 3,
electric
18
Date Recue/Date Received 2020-08-20
breakdown strengths are about identical at 100 V/micron, and solubility in
both hot
and cold solvents are below detectable levels.
In this reaction it is believed that the reactive p-xylylene reactive
intermediate
is formed and subsequently may be dimerized in the reaction chamber 235 (also
referred to as a reaction tube) or during condensation onto the lower
temperature
collection surface 250 (also referred to as a substrate). This reaction used
to
synthesize the dimer, in comparison with the known "Gorham process", results
in a
vast improvement in the overall synthesis yield of the dimer and also results
in a vast
improvement in the purity of the dimer directly from the reaction. It is
understood
that variation in the stoichiometric amounts of the reactants may be adjusted
to
provide for greater or lesser yield with associated purities varying to
provide a more
economical process or better overall production efficiency without
substantially
deviating from the scope of this invention. Subsequent purifications of the
materials
from this reaction can be performed on this material in a manner that is much
easier to
accomplish than with previously taught processes. The reaction is shown below.
trroiysls, 4.51'10'
Pb 633' C. 1 atrn.4
floiN
NI 4,..74-0116
+1/20 Eacu<Damcq¨
k1/400-cHe
+Nstinsa
As the reaction temperature at reaction chamber 235 (also referred to as a
station) is increased to >650 C, the deposition of the xylylene monomer can
proceed
directly onto a solid substrate target without necessity for isolating the
intermediate
dimer. Deposition of the exit gas at above 650 C reaction temperature upon a
cool
glass plate resulted in formation of an ethanol insoluble substance that
displays
characteristics of a parylene polymer. However, observed solubility
characteristics
clearly show that the material is insoluble in all common solvents (i.e.
hexane, xylene,
ethyl acetate, ethanol, water).
It is believed that the reaction mechanism proceeds through a route involving
the prior decomposition of nitrous oxide. Nitrous oxide is an energetically
unstable
19
Date Recue/Date Received 2020-08-20
molecule that can be thermally decomposed at elevated temperatures. Products
of the
reaction are diatomic nitrogen and monoatomic oxygen. The monoatomic oxygen is
able to react with itself to form diatomic oxygen, but this reaction is
relatively slow.
Estimates vary determining the temperature that pure thermal decomposition
occurs,
but estimates of 1100 C are often cited. Catalysis of this reaction as shown
below in
equation 1 is known to occur with a variety of metal oxides and mixed metal
oxides.
Some temperatures used for nitrous oxide decomposition with certain catalysts
are as
low as 350 C.
+ -
N=N_O 0(c))
Eq. 4
CH3 CH2
+ 0(co -3.- + H20
Eq. 5 CH3 C H2
The reactive species for the process is very likely the monoatomic oxygen
produced from the decomposition of the nitrous oxide. In this sense, the
nitrous oxide
can be viewed as a convenient carrier for the delivery of the reactive
intermediate,
monoatomic oxygen.
In a similar manner to the nitrous oxide reaction, pure diatomic oxygen can be
utilized as a reactant. However, to produce substantial yields of the desired
products,
activation of the oxygen is necessary. It is believed that activation of the
oxygen is
due to the excitation of the oxygen molecule to produce monoatomic oxygen as
shown in Equation 3.
[plasma]
(0) (0)
0=0 __________________ a 0 -I- 0
Eq. 6
The reaction with monoatomic oxygen produced in this manner thus proceeds
in a manner similar to that of the nitrous oxide decomposition route.
Cooling of the vaporized reaction products 245 (also referred to as elevated
temperature gases) exiting from the reaction chamber 235 (also referred to as
a
Date Recue/Date Received 2020-08-20
reaction tube) is necessary. If the reaction gas is at too high of a
temperature, the
ability of the reactive intermediate to condense and adhere to a surface is
greatly
reduced. To this end, a device to mix cool nonreactive or inert gases into the
hot
reaction stream has been devised. The reaction may proceed at increased or
decreased
pressure (above or below atmospheric pressure). Accordingly, an expansion
valve
may be used at the exit of the reaction chamber 235 (also referred to as a
reaction
tube) to provide Joule-Thomson effect cooling of the hot gas when the gas is
below its
inversion temperature.
The method may be extended to other substituents such as the ones shown
below.
CH3
OCH3 41 CH3
01 CI 'CI H3C 1111111" di CH3
IS CH3
H3C CH3
CH3 H3C
2,5-DICHLORO-PARA-XYLENE - 1,2,4-TRIMETHYLBENZENE
2,5-DIMETHYLANISOLE
2-CHLOR0-1,4-DIMETHYLBENZENE
Substituents such as the ones noted above (chloro, dichloro, methoxy, and
methyl) are not the only aromatic substituents that are capable of being
modified by
this process into reactive intermediates and their subsequent polymers.
Additionally,
paracyclophanes and compounds derived thereof are not exclusive to this
process.
Meta and ortho orientation of the substituents on the aromatic rings are also
viable
reaction starting materials. The reaction can be generalized to include all
compounds
that are capable of reaction with monatomic oxygen produced from a plasma or
from
decomposed oxygen-containing substances or its intermediate reaction products
and
also contain hydrogen atoms stabilized by the presence of an aromatic ring.
Typically
such hydrogen atoms are located in a position alpha to a phenyl ring (benzylic
position). Michael structures removed from the alpha aromatic ring positions
are
known to give similar reactivity to the hydrogen alpha to the aromatic ring
position as
is well known to those versed in organic synthesis. However, the reactivity of
such
21
Date Recue/Date Received 2020-08-20
hydrogen atoms is not limited to alpha and/or Michael positions from an
aromatic ring
or the aromatic ring such as benzene. Other aromatic stabilizations are known
for
many different rings, fused rings, and non-ring systems, as known to those
versed in
the art of organic chemistry. Such starting materials may preferably have the
presence
of two hydrogen atoms that are capable of being removed to form partially
oxidized
starting materials. These preferred materials may optionally have the ability
to
dimerize, trimerize, oligiomerize, or polymerize. The nonlimiting example used
herein is p-xylcnc.
One implementation of the invention augments permittivity of the polymer by
exposing the vaporized reaction products 245 (also referred to as condensing
reaction
products) to a magnetic or electric field. To the output of the reactions
described
above, the gaseous stream of vaporized reaction products 245 (also referred to
as
reaction products) is directed to a lower temperature collection surface250
(also
referred to as a cool solid surface). Illustratively, the lower temperature
collection
surface 250 (also referred to as a surface target) may be immersed in a
magnetic field
255 such as that provided by a Neodymium magnet (S84, K&J Magnetics). Other
magnetic field sources may be utilized and are intended to come within the
scope of
the invention. Condensation of the monomer and subsequent polymerization can
proceed rapidly while in the magnetic field 255. If the target and the magnet
maintain
the same relative orientation during the polymerization process, then a
baseline
increase in the electrical permittivity has been shown to occur. If the
orientation of
the magnetic field 255 relationship to the target is rotated during the
polymerization
or solid phase condensation process, then the resulting permittivity has been
shown to
decrease.
When the reaction is conducted as noted above, using the p-xylylene monomer
as the polymerization molecule, but without the presence of the applied
magnetic field
the relative permittivity of the material deposited is approximately 3. When
the
material is run as described with a magnetic field 255 (also referred to as a
magnetic
flux) density of approximately 200 to 2000 Gauss, the relative permittivity is
approximately 7. Thus, the magnetic field has been shown to substantially
increase
22
Date Recue/Date Received 2020-08-20
the permittivity of the product by over a factor of 2 times. In a similar
manner other
salts, dipoles, and salts of organic acids can be entropically oriented during
solidification or polymerizations to produce enhanced high permittivity
materials.
Improvements in permittivity from 10 to over 1000% may be attained.
In another implementation, the lower temperature collection surface 250 (also
referred to as a surface target) is immersed in a magnetic field 255 (also
referred to as
an electric field) such as that provided by a high voltage power supply (G40,
Emco,
4000V). Condensation of the monomer and subsequent polymerization can proceed
rapidly while in the electric field. If the target and the electric field
maintain the same
relative orientation during the polymerization process, then a baseline
increase in the
electrical permittivity has been shown to occur. If the orientation of the
electric field
relationship to the target is rotated during the polymerization or solid phase
condensation process, then the resulting permittivity has been shown to be
lower.
Condensation of dielectric reaction products in the presence of an electric
and/or magnetic field, has been shown to augment the permittivity of the
condensed
dielectric. This step may be applied to compounds other than parylene
polymers.
When the condensation step is conducted as noted above, using a mixture of
maleic acid salt with guanidine as a high dielectric material, but without the
presence
of the electric field the relative permittivity of the material deposited is
approximately
500. When the material is processed as described with an electric field
density of
10,000 to 30,000 V/m, the relative permittivity is approximately 25000 to
40000.
Thus, the electric field has been shown to substantially increase the
permittivity of the
dielectric field by at least a factor of 25 in that particular case. In a
similar manner
other salts, dipoles, and salts of organic acids can be entropically oriented
during
solidification or polymerizations to produce enhanced high permittivity
materials.
Improvements in permittivity have been shown to range from 5 to over 10000%.
The use of electrical and/or magnetic fields during the condensation process
modifies the mechanical strength of the product. The material may not be
anisotropic
after condensation in strong fields. Thus, this method could be utilized as a
way of
controlling the mechanical properties of the reaction products made by this
procedure.
23
Date Recue/Date Received 2020-08-20
The thickness of a PuraleneTM coating 110, 120 may range from 5 to 30 nm to
greater than 10 microns. The coated electrode 105, 125 is then used as the
basis for
application of the dielectric material 115.
Dielectrics that may be used to form a capacitor according to principles of
the
invention abound. However, to produce a substantially improved energy storage
device, it requires more than simply making a dielectric and putting it
between two
electrodes. The method whereby the dielectric is selected, transformed, and
applied is
of critical importance and not obvious to those skilled in the art of
capacitor
manufacture.
In an exemplary implementation, a viscosity stratified dielectric for an
energy
storage device according to principles of the invention may be formed from 15
grams
of protein powder (such as Zein, Sigma-Aldrich #Z3625), to which 50 ml of
absolute
ethanol is added. The solution is well stirred under inert atmosphere until
complete
dissolution is obtained. To this solution is added portion-wise 1 Og of maleic
anhydride (Sigma-Aldrich #M188) solid with vigorous stirring for a total
period of 30
mm. The solution is heated to 60 C during this period of time. At the end of
the
period 0.5 g of dicumylperoxide (Sigma-Aldrich #329541) is added portion-wise
over
5 min. The solution is allowed to boil and stir at above 60 C for 1.5h. The
solution
is cooled to room temperature. Then solid guanidine carbonate (Sigma-Aldrich#
G1165-9) is added portion-wise until the solution is neutral to basic. A
resulting
honey colored liquid may be used in the dielectric. Alternatively, other
materials such
as copolymerized maleic acid/acrylic acid (Sigma-Aldrich #416053) may be
neutralized with guanidine to produce similar results. Alternatives to
guanidine may
be used as well. For non-limiting example, Cesium carbonate and Rubidium
carbonate may be used as substitutes. Other organic, polymer, and inorganic
cationic
species may be substituted. Ultrahigh molecular weight acrylic
acid/acrylamides are
also possible dielectrics when they are optionally neutralized to their salts
forms.
FIG. 9 is an exemplary flow chart illustrating a method for making a high
permittivity dielectric material, according to an embodiment of the present
disclosure.
The method begins by dissolving an organic polymer in a solvent to form a
slurry
24
Date Recue/Date Received 2020-08-20
solution (305). The polymer may be shellac, silicone oil, zein, and/or another
organic
polymer. In one embodiment, the undissolved organic polymer is removed from
the
slurry solution (310), for non-limiting example, using a filter or centrifuge.
An
inorganic salt may then be added to the slurry solution (315). The inorganic
salt may
be a transition metal salt, such as a Gd, Sr, Sn, Fe salt, or a mixture
thereof. In one
embodiment, a breakdown voltage adjuvant may be added to the slurry solution
(320).
The breakdown voltage adjuvant may include one or more of Y, Ni, Sm, Sc, Tb,
Yb,
La, Tc, Ti, Zr, Gc, Mg, Pb, Hf, Cu, Ta, Nb, Bi, or a mixture thereof. To
facilitate
screening and drying, a dimethyl formamide and a dimethylsulfoxide may be
added to
the slurry solution (325). The slurry solution may then be heated to a
temperature of
about 150 C to about 300 C to remove or evaporate the solvent (330). This
method
avoids high process temperatures and produces a high dielectric capacitor with
a high
breakdown voltage.
Other suitable dielectric materials include conductive polymers salts, such as
salts of acrylic acid, acrylamides, methacrylates, polypyrole, etc.; inorganic
metal
oxide such as perovskites (i.e. barium titanate, strontium barium titanate,
etc.);
charged ionic liquids such as polymer salts and other electrically charged
liquids or
semi-solids that may have ability to migrate or move to some extent within a
matrix;
or a mixture of these.
The applied dielectric material 115 has a second electrode 125 added that may
be optionally coated with a nonconductive coating 120 such as PuraleneTm,
using a
coating process as described above. Connection of the electrodes 105, 125 to a
voltage source and a load via leads 130, 135 is similar to that of a
traditional
electrostatic capacitor.
In another exemplary embodiment, a high surface area electrode 105, 125 is
used instead of a smooth electrode. This provides for a greater surface
capacitance
and a faster discharge during the first phase of discharge. The high surface
area
electrode may comprise activated carbon or another conductive material which,
when
applied to the surface of the electrode, exhibits high surface area. The
adjacent
electrode may be coated or uncoated.
Date Recue/Date Received 2020-08-20
In another embodiment, an energy storage device according to principles of
the invention may contain a dielectric material that has the property of
changing
viscosity. The methods for introduction of variable viscosity into the
dielectric may
comprise variable temperature, variable electric field, variable magnetic
field, variable
pressure, variable shear and/or normal stress. Variable pressure, shear and
stress are
each a type of application of force. The direction and distribution of the
applied force
determines whether it is a pressure, shear or stress.
An exemplary method of making a magnetorhcological dielectric entails
distributing electrically insulated (or non conducting) magnetic particles
throughout
the dielectric. Once the H and DH layers are formed, a magnetic field would be
applied to increase the viscosity of as well as to prevent particle migration
through the
dielectric and "lock in" the H and DH layers. Altering the magnetic field
strength
would allow controlled dissociation of the layers through charge migration
(current
flow) within the dielectric itself. Also, the applied magnetic field could
potentially
introduce additional layering or entropic changes for energy storage.
An explanation of the mechanism whereby the energy is stored in these
devices is proposed. Although useful for a working theoretical model, no
explanations
offered herein in any way detracts from the inventiveness of the method or the
processes described.
In general the largest mechanism for the initial charging current into the
energy storage devices noted above are through the capacitance-mode of the
device.
During the later energy storage phase of charging, the diffuse Helmholtz layer
formations is the primary mode of energy storage. This DH mode is more easily
accomplished when the dielectric material is less viscous. This general rule
is
tempered by the fact that certain polymers can display more viscous
characteristics
while under an electric field than not. However, the formation of the DH
layers is
more pronounced when the device is under a greater electric field. To prevent
the
dissipation of energy stored in the DH layers, it desirable to have the
viscosity
increase after the electrical energy has been used to form these layers. In
this way
dissipation of the energy is decreased and potentially mitigated.
26
Date Recue/Date Received 2020-08-20
If there is dissipation of the energy thus stored through the electrical field
of
the device, it may be advantageous to use an electronic circuit to recover at
least a
portion of the energy converted by "leakage" and subsequently saved by storage
into
an external energy storage device or consolidated and returned to the device
itself.
Referring now to FIGs. 10-13, a multi-state electrical circuit diagram is
illustrated in various states in accordance with one or more embodiments for
making
an electronic device for the recovery of leakage current from an energy
storage
capacitor. FIGs. 10-13 illustrates four states a novel circuit that has been
developed to
regenerate and recycle the leakage current from a capacitor or capacitor
array, Cl.
In FIGs. 10-13, the following components are described. Cl is a capacitor or
capacitor array that is capable of storing a certain amount of charge. It
displays a
leakage of current when subjected to a given voltage (V+). C2 is a capacitor
(e.g.,
much smaller than Cl) of good storage characteristics that displays a much
lower
leakage current (or could be the same leakage current, but of much smaller
area of
capacitance). D1 is a diode that has the characteristic of being able to
"block" the
voltage from Cl from returning to Vss. When the voltage output from
transformer
Ti's secondary coil exceeds the voltage present on Cl and the forward voltage
drop of
D1, then current will conduct to the Cl capacitor(s). Si is a three position
single pole
switch. Line CL is a control line that controls S 1. Si is switch that is able
to
electrically connect the high voltage side of Cl to the charging voltage, V+.
In one
position it is connected to V+ and in the other position it is an open
connection (NC)
or connected to the load (LOAD). 52 and S3 are electrically controlled
switches that
have the ability to switch between two different outputs. These switches do
not
necessarily need to be high voltage switches able to withstand V+. Ti is a
"flyback"
type of transformer or an equivalent inductor that has the capability of
withstanding a
voltage on the secondary winding that is as great or greater than V+. V+ is a
charging
voltage that is connected to the main energy storage capacitor(s) Cl during
the charge
cycle. Vss is the lower voltage that is present on the opposite electrode of
Cl from
V+that produces the potential difference between the two electrodes.
27
Date Recue/Date Received 2020-08-20
Using the multi-state electrical circuit of FIGs. 10-13, leakage current may
be
recovered and regenerated from a capacitor Cl according to principles of the
invention. Referring to State A of the circuit diagram of FIG. 10, a current
is shown
flowing from the V+ source through Si to the positive plate of Cl. In this
situation S2
is connected to Vss such that the charge can be accumulated on Cl to the
potential
difference between the two. The status of S3 does not matter at this state and
no
current is flowing in the lower part of the circuit.
In State B of the circuit diagram of FIG. 11, V+ has been disconnected from
the positive electrode of Cl and the other electrode of C 1 is connected to
ground
through S2. This illustrates a typical situation where the stored load of the
Cl
capacitor is being used through the Si switch to power an electrical load.
In States C and D of the circuit diagram of FIGs. 12 and 13, two states are
shown where the Cl storage capacitor is not being charged or discharged.
However,
due to the leakage current from one electrode to another, there is a current
flowing
through the non-ideal Cl component to C2 through the S2 switch. This current
will
charge C2 to some voltage at a rate based upon the relative capacitances of Cl
and C2
and the rate of leakage. The switch S2 is disconnected from ground and
connected to
the input of C2. While in State C, the C2 capacitor is charged to some
predetermined
voltage (V1). At that predetermined voltage, the comparator then disconnects
C2 from
Cl Is open "Vss" electrode using S2, and S2 connects to "Vss", and then
subsequently
connects the positive electrode of C2 to the input of Ti transformer using S3,
as
shown in State D of FIG. 13. This discharge current through T1 induces a
voltage on
the secondary of Ti that rises to a voltage value sufficient to return some of
the charge
to Cl through the diode DI. Once the discharge of C2 is complete as determined
by
the comparator's determination of voltage on the positive electrode of C2, the
comparator returns all the switches to State C unless a demand is made to
charge or
discharge Cl.
In the above-described operation, a relatively "leaky" capacitor can return
some of the charge loss through the Cl's leakage when Cl is not in use during
either a
charge or discharge period of time. Due to the efficiency of the circuit
(which can be
28
Date Recue/Date Received 2020-08-20
made to be >90% efficiency), the leakage from the Cl device is effectively
reduced
by a factor of up to 9 times. For production of a large array of capacitors,
this can be a
significant improvement in yield. Often there are unwanted impurities in the
material
that increase the leakage current, and these are often not detected until the
entire
assembly has been completed. In the case of a large array capacitor, this
amounts to a
significant number of good devices being rejected due to a relatively small
number of
failures in the array.
As the graph in FIG. 14 shows, a voltage charges one electrode of the energy
storage device, while a voltage is generated by the other electrode that is
series
connected to ground through a 10K resistor. As the device charges, there is a
rapid
charging of the electrode and low impedance due to the capacitance of the
device. The
capacitance modes of charging are much faster than the Helmholtz layer
formations,
but ultimately much less charge is stored by these mechanisms for energy
storage than
by the DH layer formations.
Referring now to FIG. 14, energy and voltages over time for an exemplary
energy storage device according to principles of the invention are
conceptually
illustrated. The energy storage device is a capacitor that charges through a
resistor
coupled to a 120VDC source. Connection of a PicoScopeTM Model 4262 to each
electrode of the capacitor and utilization of the scope's integrated math
functions
allow calculation and display of the energy flowing into the circuit as shown
by trace
405. The applied voltage to the first electrode is represented by trace 415A-
415B, and
the displacement current is represented by the trace 410. The first charging
voltage of
approximately 120V supplies 8.16J to the capacitor. The second voltage of -
120VDC
applied at approximately the 3 minute mark shows an energy delivery of 8.08J.
In
this particular charge sequence the amount of charge and discharge are
approximately
equal. Integration of the displacement current across the capacitor reveals
that the
energy absorbed and the energy discharged are approximately equal to within
the
error limits of the data acquisition device and integration routine. Longer
charge
cycles could be used, but essentially all of the energy supplied in this
period of time at
this voltage has been absorbed by the capacitor in this time frame. Some droop
in the
29
Date Recue/Date Received 2020-08-20
power supplies are present due to the low reactance of the capacitor during
initial
switching. This voltage drop is accounted for in the calculations of the
scope. In this
example, the energy absorbed is 8.16J. The volume of sample is 0.006333 ml.
The
energy density is 1288 J/m1 or 198 Wh/kg. Integration of the charge reveals
that
essentially a >90% recovery of the charge can be obtained when the discharge
cycle is
at least 10 times longer than the charge cycle.
In another example shown in Table2 below, the charge stored is in the range of
0.41 Wh/kg at very low electric field magnitudes (.34 V per micron).
Ad v E Vol. E/m3 p E/kg E-field
776,000 25,876 50 87 30 1.16E-2 4.35E-9 2.68E+6 1.8 0.413 0.34
Table 2.
q in (nA.$), C in pf, A in mm2,, d in gm, v in volts, E in J, Vol. in m3, p in
g/cm3, E/kg in Wh/kg, E-field in V/tim.
Higher electric field magnitudes than noted in Table 2 above have been used.
Devices charged with larger magnitude electric fields store more charge, and
higher
values for energy density per mass have been obtained.
While an exemplary embodiment of the invention has been described, it
should be apparent that modifications and variations thereto are possible, all
of which
fall within the true spirit and scope of the invention. With respect to the
above
description then, it is to be realized that the optimum relationships for the
components
and steps of the invention, including variations in order, form, content,
function and
manner of operation, are deemed readily apparent and obvious to one skilled in
the
art, and all equivalent relationships to those illustrated in the drawings and
described
in the specification are intended to be encompassed by the present invention.
The
above description and drawings are illustrative of modifications that can be
made
without departing from the present invention, the scope of which is to be
limited only
by the following claims. Therefore, the foregoing is considered as
illustrative only of
the principles of the invention. Further, since numerous modifications and
changes
Date Recue/Date Received 2020-08-20
will readily occur to those skilled in the art, it is not desired to limit the
invention to
the exact construction and operation shown and described, and accordingly, all
suitable modifications and equivalents are intended to fall within the scope
of the
invention as claimed.
31
Date Recue/Date Received 2020-08-20