Note: Descriptions are shown in the official language in which they were submitted.
CA 02911712 2015-11-10
F..\=
1
TITLE OF .THE INVENTION
.PYROPHOSPHOROLYSIS ACTIVATED POLYMERIZATION (PAP)
=
BACKGROUND OF THE INVENTION
[0001] This invention relates to nucleic acid polymerization and
amplification. In particular, it
relates .to a novel and general method for nucleic acid amplification, in
which
pyrophosphorolySis and polymerization are serially-coupled. The method has
been adapted for
allele-specific amplification and can greatly increase the specificity to
detect an extremely rare
allele in the presence of wild-type alleles. We refer to the method as
pyrophosphorolysis
ió activated polymerization (PAP).
[00021 The publications and other materials used herein to illnminate the
background of the
invention or provide additional details respecting the practice
are for convenience respectively grouped in the appended Bibliography.
[0003] Multiple methods for detecting mutations present in less than 10% of
cells (i.e. rare
is alleles) have been developed, including PCR amplification of
specific. alleles. (PASA), peptide
nucleic acid (['NA) clamping blocker PCR,.. allele-specific competitive
bIocker PCR, mismatch
amplification mutation assay (MAMA), restriction fragment-length-polymoiphism
(RFLP)/PCR
(Parsons and Heflich, 1997) and QE-PCR. (Ronai and Min. amoto, 1997). These
methods: i)
amplifY the rare allele. selectively, ii) destroy the abundant wild-type
allele, or spatially
20 separate the rare allele frOm the wild-type allele. The specificity
achievable under typical
research/clinical conditions is 10-3 (Parsons and Heflich, 1997), although a
few publications
= reported higher specificity: of detection (Pourzand and Cemtd, 1993;
Knoll et al., 1996). These
methods either do not generally achieve the higher specificity or are not
suitable for routine
analysis.
zs [00041 A robust method of detecting one mutant allele in. 104409
wild-type alleles would be
advantageous for many applications including detecting -minimal residual
disease (recurrence
= after remission or rare remaining cancer cells in lymph nodes and other
neighboring tissues) and
= measurement of mutation load (the frequency and pattern of somatic
mutations present in
normal tissues). Individuals with. a high mutation load may be at increased
risk for cancer due to
30 either environmental exposure or endogenous defects in any of
hundreds of genes necessary to
maintain the integrity of the genome. For those individuals found to have a
high. mutation load,
clues to etiology can be obtained, by defining the mutation pattern.
[00051 There are many DNA sequencing methods and their variants, such as the
Sanger
. _sequencing using dideoxy termination and denaturing gel electrophoresis
(Sanger et al., 1977),
CA 02911712 2015-11-10
=
2
Maxam-Gilbert sequencing using chemical cleavage and denatiring gel
electrophoresis (Maxam
and Gilbert, 1977), pyro-sequencing detecting pyrophosphate (PP) released
during the DNA
polym.erase reaction (Ronaghi et al., 1998), and sequencing by hybridi7ation
(SBH) using
oligonucleotides (Lysov et at, 1988; Bains and Smith, 1988; Dmianac et al.,
1989; Khrapko et
s al., 1989; Pevzner et al., 1989; Southern et al., 1992).
[00061 There are multiple gel-based methods for scanning for unknown mutations
including
single stranded conformation polymorphism (SSCP) and the SSCP-hybrid methods
of dideoxy
fingerprinting (ddF), restriction endonuclease -6ngetprinting (REF), and
Detection Of Virtually
All Mutations-SSCP (DOV AM-S), denaturing gradient gel electrophoresis (DGGE),
denaturing'
io
HPLC (dTTPLC) chemical or enzymatic cleavage (Sarkar et al., 1992; Liu and.
Sommer, 1995;
" Liu et at., 1999; Myers et al., 1985; Cotton et al., 1988; Liu et al., 1999;
Buzin et at, 2000;
Spiegelman et al., 2000). DOVAM-S and chemical cleavage reactions have been
shown in
blinded analyses to identify essentially all mutations (Bnzin et at, 2000).
dHPLC, which is
based on. reverse phase chromatography, also may identify essentially all
mutations under
is appropriate conditions (O'Donovan et al., 1998; Oefner and
Underhill, 1998; Spiegelman et al.,
2000). Efforts are under way to develop general scanning methods with higher
throughput.
[00071 Sequencing by hybridization (SBH) is being adapted to scanning or
resequencing for
unknown mutations on microarrays (Southern, 1996). This continues to be a
promising area Of
intense study. However it is not possible as yet to detect most
microinsertions and deletions with
20 this approach and the signal to noise ratio for single base changes
precludes detection of 5-10% -
of single nucleotide changes (Hacia, 1999). Alternative _approaches warrant
exploration.
[00081 It is becoming increasingly apparent that in vivo chromatin structure
is crucial for
mammalian gene regulation and development Stable changes in chromatin
structure often
involve changes in methylaiion and/or changes in histone acetylation.
Somatically heritable
25 changes in chromatin structure are commonly called epigenetic
changes (Russo and Riggs,
1996) and it is now clear that epigenetic "mistakee' or epimutations are
frequently an important
contributing factor to the development of cancer (Jones and Laird, 1999).
[00091 One of the few methods for assaying in vivo chromatin structure, and
the onlr. method
with resolution at the single nucleotide level, is ligation-mediated PCR (LM-
PCR.) (Mueller and
30 Wold, 1989; Pfeifer et at, 1989) and its variant of terminal
transferase-mediated PCR (fl)-PCR)
(Komura and Riggs, 1998). Many aspects of chromatin struCture can be
determined by LM-
PCR, such as the location of methylated cytosine residues, bound transcription
factors, or
positioned nucleosomes. It is readily apparent that LM-PCR works better with
some primer sets
CA 02911712 2015-11-10
3
than with others. Thus, it is desired to develop a more robust method of
measuring chromatin
structure.
=
[0010] Thus, it is an object of the present invention to develop alternative
methods for
amplification of DNA, fot sequencing DNA and for analysis of chromatin
structure. This object
is accomplished by the use of-the novel pyrophosphorolysis activated
polymerization (PAP) as
described herein. PAP has the potential to enhance dramatically the
specificity of the
amplification of specific alleles, for resequencing DNA and for chromatin
structure analysis.
SUMIVIARY OF THE INVENTION
o [0011] The invention is a pyrophosphorolysis activated polymerization
(PAP) method of
synthesizing a desired nucleic acid strand on a nucleic acid template strand.
The method
comprises the following steps carried out serially.
[0012] (a) Annealing to the template strand a complementary activatable
oligonucleotide P*.
This activatable oligonucleotide has a non-extendible 3' terminus that is
activatable by
is pyrophosphorolysis (hereinafter referred to as a non-extendible 3' terminus
or a 3' non-
extendible end or a non-extendible 3' end). The non-extendible 3' terminus (or
end) is a
nucleotide or nucleotide analog which has the capacity to form a Watson-Crick
base bair with a
complementary nucleotide and which lacks a 3' OH capable of being extended by
a nucleic acid
=
polymerase. In one embodiment, the non-extendible 3' terminus May be a non-
extendible '
20 3 'deoxynucleotide, such as a dideol.rynucleotide. In a second
embodiment, the non.-estendible 3'
terminus may be a chemically modified. nucleotide lacking the 3' hydroxyl
group, such as an
acyclonucleotide. Acyclonucleotides- substitute a 2-hydroxyethoxyhaethyl group
for the 2'-
deoxyribofuranosyl sugar normally present in dNMPs. In other embodiments, the
non-
extendible 3' terminus may be other blockers as described herein. In one
embodiment, the
25 activatable oligonucleotide P* has no nucleotides at or near its 3'
terminus that mismatch the
corresponding nucleotide's on the template strand. In. a second embodiment,
the activatable,
oligonucleotide P* has a mismatch at or within 16 nucleotides of its 3'
terminus with respect to a
corresponding nucleotide on- the template strand. The tentiftal 3'-
deoxyntteleotide is hybridiZed
to the template strand when the oligonucleotide P* is a-nnealed.
30 [0013] (b) Pyrophosphorolyzing the annealed activatable oligonucleotide
P* with
pyrophosphate and an enzyme that has pyrophosphorolysis activity. ThiS
activates the
oligonucleotide P* by removal of the hybridized non-extendible 3' terminus. =
CA 02911712 2015-11-10
4
=
[00141 (c) Polymerizing by extending the activated oligonucleotide P* on the
template strand in
presence of four nucleoside triphosphates of their analogs and a nucleic acid
polymerase to
synthesize the desired nucleic acid. strand.
[00151 The PAP method can be applied to amplify a desired nucleic acid strand
by the following
additional steps.
[00161 (d) Separating the desired nucleic acid strand of step (c) from the
template strand, and
[0017] (e). Repeating steps (a)-(d) until a desired level of amplification of
the desired nucleic
acid strand is achieved.
[0018] In a preferred aspect, the PAP method as described above is applied to
allele-specific
amplification (PAP-A). In this application, the nucleic acid template strand
is a sense or
antisense strand of one allele and is present in admixture With the
corresponding (sense or
antisense) nucleic acid strand of the second allele (the allelelic strand).
The activatable
oligonucleotide P* has at least one nucleotide or analog at or near its 3'
termirmS, e.g., within .16
nucleotides of the 3' terminus, that mismatches the corresponding nucleotide
of the allelic strand.
is Because of the mismatch, in step (a) of the PAP. method the non-
extendible 3' tenninus of
oligonucleotide P* is not substantially hybridized to the allelelic strand. In
step (b) the
pyrophosphorolysis does not substantially remove the non-hybridized non-
extendible 3'
terminus from the activatable oligonucleotide P* annealed to the allelic
strand. In step (c) the
oligonucleotide P* is not substantially extended by polymerization on the
allelic strand. As a
1:-) result, the desired nucleic acid strand synthesized on the
template strand is amplified
preferentially over any nucleic acid, strand synthesized on the allelelic
strand.
[00191 In a second preferred aspect, the PAP-A method described above can be.
performed -
bidirectionolly (Bi-PAP-A). Bidirectional-PAP (Bi-PAP) is a novel design that
preferably uses
two opposing pyrophosphorolysis activatable oligonucleotides (P*) with one
nucleotide overlap
25
at their 3' termini. Thus, in Bi-PAP, PAP-A is performed with a pair of
opposing activatable
oligonucleotide P*s. Both the downstream and upstream P*s are specific for the
nucleotide of
interest at the 3' termini (e.g., an A:T base pair). In the initial round of
amplification from
genoraic DNA, segments of undefined size are generated. In subsequent rounds,
a seginent
equal to the combined lengths of the oligonucleotides minus one is amplified
exponentially.
30 Nonspecific amplification occurs at lower frequencies because- this design
elimitatog
misincorporation error from an unblocked upstream. The P's may be 3060
nucleotides for
most efficient amplification.
CA 02911712 2015-11-10
[90201 The PAP method can be used to amplify either RNA or DNA. When used to
amplify
DNA, the activatable oligonucleotide P* may be a 2'-deoxyoligonucleotide, the
non-extendible
= 3' terminus may be, e.g., a 2',3'-dideoxynucleotide or an
acyclonucleotide or other blOckers as
described herein, the four nucleoside triphoSphates are 2'-deoxynticleoside
triphosphates or their
s
analogs, and the nucleic acid polymerase is a DNA polymerase. The DNA
polyinerase used in
= step (c) can also be the enzyme having pyrophosphorolysis activity used
in step (b). Preferred
DNA polymerases having pyrophosphorolysis.activity are thermostable Tfl, Mg,
and genetically
engineered DNA polymerases, such as ArapliTaqFs and ThermoSequenaseTm. These
genetically engineered DNA polymerases have the mutation F667Y or an
equivalent mutation in
their active sites. The use of genetically engineered DNA polymerases, such as
AmpliTaqFs
and ThennoSequenaseTm, greatly improves the efficiency of PAP. These Family I
DNA
polymerases can be used when the activatable oligonucleotide P* is a 3'
dideoxynucleotide or an
acyclonucleotide. When the activatable oligonucleotide P* is an
acyclonucleotide, Family
archaeon DNA polyinerases can also be used. Examples of such polymerases
include, but are
not limited to, Vent (exo-) and Pfu (exo-). These polymerases efficiently =
amplify
3 'acyclonucleotide blocked P*. Two or more polymerases can also be used in
one reaction. If
the template is RNA, the nucleic acid polymerase may be RNA polymerase,
reverse
transcriptase, or their variants. The activatable oligonucleotide P* may be a
ribornicleotide or a
T-d.eoxynucleotide. The non-extendible 3' terminus may be a 3' deoxyribo-
nucleotide or an
acyclonucleotide. The four nucleoside triphosphates may be ribonucleoside
triphosphates., 2'
deoxynucleoside triphosphates or their analogs. For convenience, the
description that follows
uses DNA as the template. However, RNA is also included, such as described for
the Present
aspect.
= [00211 Amplification by the PAP method can be linear or expOnential.
Linear amplification is
obtained when the .activatable oligonucleotide P* is the only complementary
oligonucleotide
. used, Exponential amplification is obtained when a second opposing
oligonucleotide, which may
be a P*, is present that is complementary to the desired nucleic acid strand.
The activatable
oligonucleotide P* and the second oligonnclebtide flank the region that is
targeted for
amplification: In step (a) the second oligonucleotide anneals to the separated
desired nucleic
acid strand product of step (d). In step (c) polymerization extends the second
oligonucleotide on
the desired nucleic acid strand to synthesize a copy of the nucleic acid
template strand. In. step
= (d) the synthesized nucleic acid template strand is separated from the
desired nucleic acid strand.
CA 02911712 2015-11-10
6
Steps (a) through (d) are repeated until the desired level exponential
amplification has been
= achieved.
[0022] In the PAP method, a mismatch between the activatable oligonucleotide
P* and the
template strand results in no substantial amplification, if the mismatch
occurs in the 3' specific
s subsequence of P* at the .3' terminus of P* or within 16 nucleotides
of the 3' terminus of P*.
This lack of amplification for such mismatches in the 3' specific subsequence
of P* provides
four billion different and specific oligonucleotides with one base
substitution resolution.
[00231 In a preferred aspect, the PAP. method is used for exponential
amplification of a rare,
- mutant allele in a mixture containing one or more wild-type alleles, Strands
of the alleles are
io separated to provide single-stranded nucleic acid, and then the
following steps are carried out
serially.
[00241 (a) Annealing to the sense or antisense strands of each allele a
complementary
activatable =T-deoxyoligonucleotide P* that has a non-extendible 3' terminus.
The non-
extendible 3' terminus may be, e.g., a non-extendible 2',3'-dideoxynucleotide
or an
is acyclonucleotide. P* has no 2'-deoxymicleotides at or near its 3'
terminus that mismatch the
corresponding 2'-deoxynncleotides on the mutant strand, but has at least one
2'-deoxynucleotide
at or near its 3' terminus that mismatches the corresponding 2'-
deoxynucleotide on the wild-type
stand. Consequently, the non-extendible 3' terminus is hybridized to the
mutant strand but not to
the wild-type strand when the oligonucleotide P* is annealed. Simultaneously,
a second
20 2'-deoxyoligonucleotide that is complementary to the anti-parallel
strands of each allele is
annealed to the anti-parallel strands. The activatable T-deoxyoligonucleotide
P* and the second
2'-deoxyoligonucleotide fin.* the region of the gene to be amplified.
[00251 (b) Pyrophosphorolyzing the activatable 2'-deoxyoligonucleotide P* that
is annealed to a
mutant stiand with pyrophosphate and an enzyme that has pyrophosphorolysis
activity. This
25 activates the 2'-deoxyoligonucleotide P* that is annealed to the
mutant strand by removal of the
hybrie1i7ed non-extendible 3' terminus. = It does not substantially activate
the
2'.-deoxyoligonucleotide P-* when it is armealed to the mutant strand because
the non-hybridized
non-extendible 3' terminus is not substantially removed by the
pyrophosphorolysis.
[0026] (c) Polymerizing by extending the activated oligonucleotide P* on the
mutant strand in
30 presence of four nucleoside triphosphates or their analogs and a DNA
polymerase and extending
the second 2'-deoxyoligonucleotide on both mutant and wild-type anti-parallel
strands.
[00271 (d) Separating the extension products of step (c);
CA 02911712 2015-11-10
7
[00281 (e) Repeating steps (a)-(d) until the desired level of amplification of
the mutant allele has
been achieved.
[0029] The activatable 2'-deoxyoligonucleotide P* is annealed to the antisense
strands of the
alleles and the second 2`-deoxyoligonucleotide is annealed to the sense
strands, or vice versa.
[00301 Steps (a) to (c) of PAP can be conducted sequentially as two or more
temperature stages
on a thermocycler, or they can be conducted as one temperature stage on a
thermocycler.
[00311 Nucleoside triphosphates and 2'-deoxynucleoside triphosphates or their
chemically
' modified versions may be used as substrates for multiple-nucleotide
extension by PAP, i.e.,
when one nucleotide is incorporated the extending strand can be further
extended,
i.o 2',3'-dideoxynucleoside triphosphates, their chemically modified
versions, acyclonucleotides or
other blocked nucleotides which are terminators for further extension may be
used for single-
nucleotide extension. 2`,3'-dideoxynucleoside triphosphates may be labeled
with radioactivity or
dye for differentiation from the 3' terminal dideoxynucleotide, if present, of
oligonucleotide P*.
Mixtures of nucleoside triphosphates or 2'-deoxynucleotide triphosphates or
their analogs, and
2',3'-dideoxynucleoside triphosphates or their analogs may also be used.
[00321 PAP can be used in a novel method of DNA sequence determination. In
PAP,.
pyrophosph.orolysis and polymerization by DNA polymerase are coupled serially
by using P*,
an oligonucleotide containing a non-extendible 3' terminus. The non-extendible
3' terminus may
be, e.g., a non-extendible 3' -deoxynucleotide or an acyclonucleotide. This
principle is based on
the specificity of PAP and in, turn on the base pairing specificity of the 3'
specific subsequence.
This property of the 3' specific subsequence can be applied to scan or
resequence for unknown
sequence variants, to determine de novo DNA sequence, to compare two DNA
sequences, and to
monitor gene expression profiling in large scale. A P* array is possible in
these methods. That
is, each of the P*s can he immobilized. at an individual dot or a solid
support, thus allowing all
the PAP reactions to be processed in parallel.
[00331 Thus in one aspect, the PAP method is used for scanning or resequencing
unknown
sequence variants within a predetermined sequence by carrying out the
following steps serially.
[00341 (a) Mixing under hybridization conditions a template strand of the
nucleic acid with.
multiple sets of four activatable oligonucleotides P* which are sufficiently
complementary to the
template strand to hybridize therewith. Within each set the oligonucleotides
P* differ, from
each other in having a different non-extendible 3' terminus; so that the non-
extendible 3'
terminus is hybridized to the template. strand if the template strand is
complementary to the non-
extendible 3' terminus. The number of sets corresponds to the number of
nucleotides in the
CA 02911712 2015-11-10
8
sequence. The non-extendible 3' terminus may be, e.g., a non-extendible 3 '-
deoxynucleotide or
an acyclonucleotide.
[00351 (b) Treating the resulting duplexed P*s with pyrophosphate and an
enzyme that has
pyrophosphorolysiS activity to activate by pyrophosphorolysis only those
oligonucleotides P*
which have a non-extendible 3' terminus that is hybridized to the template
strand.
[0036] (c) Polymering by extending the activated oligOrticleotides P* on the
template strand
in presence of four nucleoside hiphosphateS or their analogs and a nucleic
acid polymerase.
[00371 (d) Separating the nucleic acid strands synthesized in step (c) from
the template strand.
[00381 (e) Repeating steps (a)-(d) until a desired level of amplification is
achieved, and
ic [00391 (f) Arranging the nucleic acid sequence in order by analyzing
overlaps of oligonuclotides
P* that produced amplifications.
10040] In a second aspect, the PAP method is used fer determining de novo the
sequence of a
nucleic acid by carrying out the following steps serially.
[00411 (a) Mixing under hybridization conditions a template strand of the
nucleic add with
is multiple activatable oligonucleotides P*. All of the oligonucleotides P*
have the same number n
of nucleotides as the template and constitute collectively all possible
sequence's having n
nucleotides. All of the oligonucleotides P* have a non-extendible 3' terminus.
The non-
extendible 3' terminus may be, e.g., a non-extendible3,-deoxynucleotide or an
acyclonucleotide.
Any oligonucleotides P* that are sufficiently complementary will hybridize to
the template
20 strand. The non-extendible 3' terminus -will hybridize to the template
strand only if the template
strand is complementary at the position corresponding tO the 3' terminus'.
[00421 (b) Treating the resulting duplexed P*s. with pyrophosphate and an
enzyme that has
pyrOphosphorolysis activity to activate only those hybridized oligonucleotides
P* which have a
non-extendible 3' terminus that is hybridized to the template strand, by
pyrophosphorolysis of
25 those hybridized non-extendible 3' termini.
[00431 (c) Polymerizing by extending the activated oligonucleotides P* on the
template strand
in presence of four nucleoside triphosphates or their analogs and a nucleic
acid polymerase.
[00441 (d) Separating the nucleic acid strands synthesized in step (c) from
the template stand.
[00451 (e) Repeating steps (a)-(d) until a desired level of amplification has
been achieved, and
30 [00461 (f) Detennining the sequence of oligonucleotides P* that produced
amplifications, then
arranging the nucleic acid sequence in order by analyzing overlaps of these
oligonucleotides.
[00471 PAP can also be used to study chromatii structure analogously to
ligation-mediated PCR
(LM-PCR) by carrying out the following steps serially. LM-PAP has been used
for the
CA 02911712 2015-11-10
9
determination of primary nucleotide sequence, cytosine methylation patterns,
DNA lesion
formation and repair and in vivo protein-DNA footprints (Dai et al., 2000;
Mueller and Wold,
1989; Pfeifer et al, 1989; Pfeifer et al.; 1999; Becker and Grossman, 1993).
Ligation-mediated
PAP (LM-PAP) involves cleavage, primer extension, linker ligation and PAP that
can be applied
s
for analysis of in vivo chromatin structure, such as, methylated state of
chromesomes, and for
other nucleic acid. analysis as for LM-PCR:
[00481 The nature of LM-PAP is that the template is synthesized before PAP,
such as by
ligation reaction or by extension using terminal transferase. PAP may be any
type of PAP: with
only one P*, with two opposing oligonucleotides where at least one is P*, Bi-
PAP, matched
io
PAP, mismatched PAP, and. so on. Thus, at its simplest, LM-PAP is the
application of PAP to a
=
presynthesized template. LM-PAP may be performed by steps (i), (iv) and
(v), by
steps (i),
(iii) and (Vi); by steps (ii), (iii), (iv) and (v) or by steps (ii), (iii) and
(vi), where the -
steps are as follows.
[0049] (i) The cleavage occurs chemically, enzymatically or naturally to
"breakdown" nucleic
. 15 acid strands: The nucleic acid usually is genomic DNA that may
have lesions or nicks produced
in vivo.
100501 (ii) The oligonucleotide P1 is gene-specific and its extension
includes: 1) annealing to
. the template strand a substantially complementary oligonucleotide; 2)
extending the
oligonucleotide on the template strand in the presence of nucleoside
triphosphates or their
zo analogs and a nucleic acid polymerase, the extension "runs off' at
the cleavage site on the
template strand. Steps 1) and 2) may be repeated:
[00511 The primer extension may be replaced by a P* extension, in which the
above PAP is
performed with only one activatable oligonucleotide P*.
[00521 (iii) The linker ligation step includes " ligation of a linker to the
3' terminus of the
25
synthesized nucleic acid strand. The linker ligation step may be replaced by a
terminal
transferase extension that is non-template dependent polymerization and an
extra nucleic acid
sequence is added to the 3' terminus .of the synthesized nucleic acid strand.
[00531 (iv) PCR. is performed with a second gene-specific oligenucleotide (P2)
together with an.
oligonucleotide specifie for the linker or the added Sequence by terminal
transferase.
3 [00541 (v) A third gene-specific P* (P3) is used to detect the PCR
generated fragments. PAP
method is applied with only one activatable oligonucleotide P*. The extension
of the activated
oligonucleotide P* "runs off" at the end of the template strand generated in
IV. The PAP
method may be applied in an allele-specific manlier. The activatable
oligonucleotide P* may
CA 02911712 2015-11-10
=
contain one or more nucleotides that are not complementary to the template
strand. The
uncomplimentary nucleotide(s) of P* may locate at the 3' terminus of P*.
[00551 (vi) Instead of steps (iv) and (v), PAP method can be applied with two
opposing
oligonucleotides of which at least one is the activatable oligonucleotide P*.
The activatable
5 oligonucleotide P*(P3) is gene-specific. The second oligonucleotide is
specific for the linker or
the added sequence by terminal transferase. The second oligonucleotide may be
another
activatable oligonucleotide P* or a regular oligonucleotide. The PAP method
may be applied in
an allele-specific manner. The activatable oligonucleotide P* (P3) may contain
one or more
nucleotides that are not complementary to the template strand. The
uncomplimentary
o nucleotide(s) of P* may locate at the 3' terminus of P* (P3).
[00561 The third gene-specific oligonucleotide (P3) is then usually used to
label and allow
visualintion of the PCR generated fragments. P3 is labeled at the 5' terminus
with 32P or, more
recently, with near infrared fluorochromes such as [RD 700 or [RD 800 (Li-Cor
Inc.) (Dai et al.,
2000).
is [0057) PAP can be used to detect a target nucleic acid. In one
embodiment this method involves
the following steps:
[00581 (a) -adding to a nucleic acid containing sample an oligonucleotide P*,
wherein the
oligonucleotide P* has a non-extendible 3' terminus, wherein the 3' terminal
residue of
oligonucleotide P* is removable by pyrophosphorolysis, and wherein the
oligonucleotide P*
anneals to a substantially complementary strand of the target nucleic acid
present in the sample;
[0059] (b) removing the 3' non-extendible terminus of the oligonucleotide P*
annealed to the
substantially complementary strand of the target nucleic acid by
pyrophosphorolysis to unblock
the oligonucleotide P* to produce an unblocked oligonucleotide; and
[0060] (c) detecting the presence of the target nucleic acid, wherein the
sequence of the target
nucleic acid is substantially complementary to the sequence of the
oligonucleotide P*.
[00611 The method of the first embodiment may further include before the
detection step the
step: (b1) extending the unblocked oligonucleotide using a nucleic acid
polymerase to produce
an extended oligonucleotide. The method may also include the addition of a
second
oligonucleotide which may or may not have a 3' non-extendible terminus. The
second
oligon.ucleotide may anneal to the substantially complementary strand of the
target nucleic acid
or it may anneal to the complement of the substantially complementarty strand
of the target
nucleci acid.
CA 02911712 2015-11-10
= 11
[0062] In a Second embodiment for detecting a nucleic acid. the method
involves the following
steps:
[0063J (a) adding to a nucleic acid containing sample two oligonucleotide P*s,
wherein each
oligonucleotide P* has a non-extendable 3' terminus, wherein the 3' terminal
residue of each
s oligonucleotide P* is removable by pyrophosphorolysis, wherein one
oligonucleotide -P*
overlaps with the other oligonucleotide P* by at least one nucleotide at their
respective 3' ends,
and wherein one oligonucleotide P* anneals to a substantially complementary
strand of the
rargot acid-present in. -the- sample- and-- the -other-
oligonucieotide--P*- -anneals-ta-. a -
complement of the substantially complementary strand of the target nucleic
acid;
[0064] (b) removing the 3' non-extendable terminus of the oligonucleotide P*s
annealed to the
target nucleic acid by pyrophosphorolysis to unblock the' oligonucleotide P*s
to produce
unblocked oligonucleotides; and
[0065] (c) detecting the presence of the target nucleic acid, wherein the
sequence of the target
nucleic acid is substantially complementary to the sequence of the
oligonucleotide P*s.
= 15 [0066] The method of the second embodiment may thither include before
the detection step the
step: (bl) extending the unblocked oligonucleotide using a nucleic acid
polymerase to produce
an 'extended oligonucleotide. =
- [0067] In one embodiment, the detection of the nucleic acid in step (c)
is performed by detecting
the tinblOcking of oligonucleotide P*. In one aspect, the unblocking is
detected by loss of a
label contained in the 3' terminal residue of oligonucleotide P*. In a second
aspect, the
unblocking is detected by detecting the presence of a 3' OH on the 3' terminal
residue that is
capable of extension or ligation. In this aspect, the detection is determined
by extending the =
unblocked oligonucleotide or by ligating the unblocked oligonucleotide to an
oligonucleotide.
In a second embodiment, the detection of the nucleic acid in step (0) is.
performed by detecting
the extended oligonucleotide. In one aspect, the extended oligonucleotide is
detected by the
presence of a label in the extended oligonteleotide. The label is part of a
nucleotide or
nucleotide analog used in the extension step. In a second aspect, the extended
oligonucleetide is
detected by gel electrophoresis: In a third aspect, the extended
oligonucleotide is detected by the
binding or incorporation of A dye or spectral' material.
[00681 The P* oligonncleotides are selected to be "substantiallyi!
complementary" to the
= different strands of each specific sequence to be smpiiiied, Therefore,
the P* oligonucleotide
sequence need. not reflect the exact sequence of the template. For example, a
non-
complementary nucleotide segment may be attached to the 5'-end of the P*
oligonucleotide, with
CA 02911712 2015-11-10
12
the remainder of the P* oligonucleotide sequence being complementary to the
strand.
Alternatively, non-complementary bases or longer sequences can be interspersed
into the P*
oligonucleotide, provided that the P* oligonucleotide sequence has sufficient
complementarity
with the sequence of the strand to be amplified to hybridize therewith and
form a template for
s synthesis of the extension product of the other P* oligonucleotide.
The ability to detect
nucleic acid sequences which are substantially complementary to
oligonucleotide P* is
particularly useful for the detection of multiple mutations; such as seen in
high viral load, where
the detection of the presence of the virus is important and not necessarily
the exact nucleic acid
sequence of the virus. This method is also capable of detecting nucleic acids
that are completely
o complementary.
[0069] The present invention also includes other modifications of PAP.
[00701. The activatable oligonucleotide P* may contain blocked nucleotides at
other positions
in addition to the 3' terminus.
[00711 = The introduction of internal blocking nucleotides results in. an
interface between
15 amplification and PAP which would permit PAP to amplify in a non-
exponential manner (e.g.,
quadratic or geometric) with higher fidelity, i.e., errors made by the
polymerase would not be
prop agatable.
[0072] = The activatable oligonucleotide P* may contain modified nucleotides
that are
extendible as well as the 3' blocked nucleotide. Thus, anywhere 5' to the 3'
terminus, there may
20 be either blocking or non-blocicing modified nucleotides.
[0073] = A polymerase that pyrophosphorolyzes the mismatched primer rather
than the matched
primer could be used to detect rare mutations in which the II* that mismatched
at the 3' terminus
is activated and extended.
[0074] = The detection of a rare mutation is based on no mismatch anywhere
along the length of
25 the oligonuoleotide because a Mismatch inhibits the activation of P*s.
[00751 = Activation may occur by another mechsnism, such as a 3' exonuelease.
The 3'
exonuclease may have specificity for the matched primer or the mismatched
primer so that it
discriminates as to whether there is a mismatch at the 3' end. The 3'
exon.uclease can be used
either way. If it prefers a mismatch, it can be used as described above, but
its ability to detect
30 uncommon mutations would depend on some specificity for activation,
although that specificity
may come partly from internal mismatches.
CA 02911712 2015-11-10
13
[00761 = The extension reaction can be performed by a DNA polymerase, an RNA
polymerase
or a reverse transcripta.se, the template may be a DNA or an RNA, and the
oligonucleotide P*
may be a DNA, an RNA, or a DNA/RNA heteromer.
[0077] = Pyrophosphorolysis and the extension can be performed by different
polyrnerases. For
s example, the P* may include a penultimate modified oligonucleotide that
could not be extended
by pyrophosphorolyzing polymerase but could be extended by another polymerase.
One
example is a 3' dideoxy that could be pyrophosphorolyzed by a DNA polymerase,
but the
presence of a ribonucleotide in the penultimate position would require
extension by an RNA
polymerase. =
[0078] = PAP can. be generalized as an inactive oligonucleotide that is
activated by a nucleic
acid metabolizing enzyme, such as helicases, top oisomerases, telomerases,
RNase H or
restriction enzymes.
[0079] = Methylases would detect the presence or absence of a methyl group in
genomic DNA.
Methylase,s could be coupled with truncating amplification which forces the
polymerase back to
the template.
[0080] = A P* in which the 3' end is a dideoxy and penultimate few nucleotides
are ribos can be
used as a tool for differentially making a protein product derived from a
specific mutation that
was desired, or for making a protein product whose expression is linked, to
the presence of a
particular sequence. Pyrophosphorolysis would activate the P* if there was a
precise match to
the mutation at the 3' end. The activated oligonucleotide is then a substrate
for the generation of
RNA by an RNA polyinerase. The RNA could then be translated in vitro to
produce the protein
product
[0081] = PAP (PAP, Bi-PAP, matched or mismatched PAP, simplex PAP, multiplex
PAP and.
others) can be used for quantification. The yield of the amplification
products is quantitatively
associated with the amount of input template. The association may be
proportional or otherwise.
[00821 = In PAP, product may accumulate linearly, exponentially or otherwise.
BRIEF DESCRIPTION OF THE FIGURES
[0083] Figure 1 shows a schematic of the detection of a rare mutation by
allele-specific PAP
(PAP-A).
= [00841 Figure 2 shows a schematic of bidirectional PAP-A (Bi-PAP-A).
[00851 Figure 3 shows a schematic of PAP-based resequeucing (PAP-k) performed
on a
microarray with programmable photochemical oligonucleotide.
CA 02911712 2015-11-10
14
[0086] Figure 4 shows a schematic of microarray-based .resequencing to detect
a G to A
mutation.
=
[0087] Figure 5 shows a schematic of ligation-mediated PR (LM-PCR).
[0088] Figures 6A and 613 are a schematic illustrating use of PAP to detect
the G allele at
=
s nucleotide 229 of the D1. dopamine receptor gene. The procedure is
described in detail in
Example 1 below..
[0089] Figure 6C is an autoradiogram of PAP from the GIG, A/A and G/A
genotypes of the
huthan dopamine receptor gene.
[0090] Figures 7A and 7B are diagrams illustrating enhanced specificity of PAP
relative to
o PASA.
[0091] Figures 8A and 813 are autoradiograms showing the results of
electrophoresis of samples
obtained in Example 1 below.
[0092] Figure 9 is an autoradiogram showing the results of electrophoresis of
samples obtained
in Example 1 be/ow.
is [0093] Figure 10 is an autoradiogram showing the results of
electrophoresis of samples obtained
in Example 1 below.
[0094] Figure 11A is a schematic illustrating enhancement of PAP efficiency.
[0095] Figure 1113 is an autoradiogram of PAP from the GIG, .AJA and G/A
genotypes of the
human dopamine receptor gene.
20 [0096] Figures 12A-12E are autoradiogra-ms showing the results of
electrophoresis of samples
obtained in Example 2 below.
[0097] Figure 13 is an. autoracliograrn showing the results of electrophoresis
of samples obtained
in Example 2 below..
[0098] Figure 14 is an autoradiograni showing the results of eleetrophoresis
of-samples obtained =
25 in. Example 2 below.
[0099] Figure 15 is an autoradiograrn showing the results of electrophoresis
of samples obtained
in. Example 3 below.
[0100] Figures 16A-16B show UV footprinting by LM-PAP. Fig. 16A shows allele-
specific
LM-PAP versus allele-specific LM-PCR for the dopamine D1 receptor gene. Fig.
16B shows
30 LM-PAP for the pgic gene.
[01011 Figures 17A-17B show PAP amplification directly from human .(Fig. 17A)
and mouse
(Fig. 17B) genonaic DNA using PAP and Bi-PAP, respectively.
CA 02911712 2015-11-10
=
[01021 Figures 18A-18E show PAP amplification using 3' terminal
acyclonucleotide blocked
P*. Fig 18A: Model: A duplexed DNA template of the lad gene is shown. The
mutated
template contains a G at the nucleotide position 369, while the wild-type
template contains a T
at the nucleotide position 369 of the /ad" gene. P*
pyrophosphorolysis activatable
5
oligonucleotide. The P* has an acycloN1VIT or a ddNIAP at the 3' terminus. The
P* is specific to
the mutated template but mismatches to the wild-type template at the 3'
terminus (Table 6). 0 =
oligodeoxynucleotide. PAP was performed with P*1 and 01, P*2 and 02, or P*1
and P*2,
respectively. Fig. 18B: PAP with 30 mer P*s: The P*s are specific for the
mutated template but
mismatch the wild-type template at their 3' terminus. In lanes 1-8 are 3'
terminal
io
acyclonucleotide blocked P*s. In lanes 9-16 are 3' terminal dideoxyn_ucleotide
blocked P*s for
comparison. In lanes 1-4 and 9-12, the mutated template is used. In lanes 5-8
and 13-16, the
wild-type template is used. The PAP product and P* are indicated with their
sizes. Lane M is
12Ong of 4a174-PUC19/HaeLTI DNA marker. Fig. 18C: PAP with 35-mer P*s: The
experiment
is the same as in Fig. 18B except with 35-mer P*s that are 3' co-terminal with
the 30-mer P*s
is and
five nucleotides longer at their 5' termini. Fig. 18D: PAP with Vent (exo-)
polymerase. The
experiment is the same as in Fig. I8B except that Vent (exo-) was used. Fig.
18E: PAP with Pfu
(exo-) polymerase. The experiment is the same as in Fig. I9B except that Pfu
(exo-) was used.
[0103] Figure 19 shows that PAP has high selectivity to detect rare mutations
in the abundance
of the wild-type template. In the example of nucleotide position 190, the
mutation-specific P*
matches the mutated A template but mismatches the wild-type T template at the
3' terminus.
Specific and efficient amplification is indicated, by thick arrows. When
hybridized to the
mutated A template, the P* cannot extend directly from the 3' terminal
dideoxynucleotide, the 3'
terminal ddTIVf2 must be removed by pyrophosphorolysis and the activated
oligonucleotide is
then extended efficiently. Two types of nonspecific amplification ikom the T
template are
zs
indicated as Types I and 11 The nonspecific amplification -occurs rarely when
mismatch
pyrophosphorolysis occurs to generate a/ wild-type product that will not
support efficient
amplification as template for subsequent cycles (Type I) (the error is
indicated by thin arrow and
estimated frequency of as low as 10-5). When both mismatch pyrophosphorolysis
and
misincorporation occur extremely rarely to generate a mutated product (Type
11) (the errors are
3o
indicated by thin arrowe and estimated. coupled frequency of 3.3x 101). Once
the errors occur,
the mutated product can be amplified exponentially in subsequent cycles and so
it determines
the selectivity.
CA 02911712 2015-11-10
16
[0104j Figures 20A-20B show Bi-PAP amplification. Fig. 20A: Schematic of Bi-
PAP to
detection a rare mutation: The mutation-specific assay with two mutated P* for
nucleotide 190 is
shown. The downstream and upstream P*s contain a dideoxy T and a dideoxy A at
their 3'
termini, respectively. They are specific for the T:A allele at nucleotide 190
(on the right), but are
mismatehed to the A:T wild-type allele at their 3' termini (on the left). The
P*s are 40
nucleotides long and overlap at their 3' termini by one nucleotide. On the
left, no substantial
product is generated from the wild-type template because of the mismatch. On
the right, the
mutated product is generated efficiently frorri the mutated template. Fig.
2013: Bi-PAP
amplification directly from X DNA. Each of the wild-type and mutation-specific
Bi-PAP assays =
o at
nucleotide 190 was used to amplify a 79-bp segment of the lad gene from X
DNAs. For the
wild-type assay in. lanes 1-3, the two wild-type P*s have 3' terminal ddA and
ddT, respectively.
For the mutation-specific assay in lanes 4-6 and lanes 7-9, the two mutated
P*s are with ddT and
ddA at their 3' termini, respectively. In lanes, 1, 4 and 7, 2000 copies of
the wild-type template
were added to each reaction. In lanes 2, 5 and 8, 2000 copies of the mutated
template were
added to each reaction. In lanes 3, 6 and 9; no template was added. In lanes 7-
9, 200 ng of
human genomic DNA was added as carrier. The product and P* are indicated Lane
M is 12Ong
of +X174-PUC19/Haelli DNA marker.
[01051 Figures 21A-21C show titration of template for sensitivity and
selectivity of Bi-PAP.
With the mutated P*s, the wild-type template was amplified to generate tfie
ifintataproduct in
Experiment I. The mutated template was = amplified to generate the mutated
product in
Experiments II, III and W. Fig. 21A: The mutation-specific Bi-PAP assay for
A190T. In
Experiment I, the copies of the wild-type A, DNA are indicated in lanes 1-5.
Lane 6 is a negative
contror 'With no DNA. In Experiment II, the copies of the mutated 2t, DNA are
indicated in lane
7-11: Lane 11(0.2 copy) is a negative control to support the dilution accuracy
of copy number.
Lane 12 is a negative control with no DNA. In Experiment Ili, the copies of
the mutated X DNA
in the presence of 2x109 copies of the wild-type X DNA are indicated in lane
13-17. Lane 18 is a
negative control with only the wild-type X DNA. In Experiment IV, the copies
of the mutated X
DNA. in the presence of 100 rig of human genomic DNA are indicated in lanes 19-
23.-Lane 24 is
negative control only with the human genomic DNA. Lane "C WT" is the wild-type
product
30.
control in which the wild-type P*s were used to amplify 2000 copies of the
.wild-type A, DNA.
Lane "C Muf' is the mutated product control in which the mutated P*s were used
to alnplify
2000 copies of the mutated I DNA. The wild-type and. mutated products with
unique mobilities
=
CA 02911712 2015-11-10
= =
17
are indiCated. Fig. 2113: The mutation-specific Bi-PAP assay for T369G. Fig.
21C: The
mutation-specific Bi-PAP assay for T369C.
[0106] Figure 22 shows a design of P* rnicroarray for Bi-PAP resequencing. Bi-
PAP can be
used. for resequencing to detect unknown mutations in a known region on a
microarray. The P*s
s are
designed according to the wild-type template. The two opposing P*s for each Bi-
PAP are
anchored in a micro array spot. Each pair of arrows represents four Bi-PAPs
for one nucleotide
position. A mutation is indicated On the template, and it spans six overlapped
P*s. On the
micro array, many Bi-PAPs can be processed in a parallel way.
[01071 Figures- 23A-23B show a schematic of 13i-PAP resequencing. Fig. 23A:
Detection of the
. 10 wild-
type sequence:. This is a close look at the microanay. The P*s are designed
according to
the wild-type sequence. On the position of nucleotide A, four Bi-PAPs are
synthesized with four
pairs of P*s. The four downstream P*s have identical sequence, except that at
the 3' terminus
either ddAMP; ddTIVIP, ddGMP or ddCMP, corresponds to the wild-type sequence
and the three
possible single base substitutions. The four corresponding upstream P*s have
identical sequence,
15
except that at the 3' terminus either ddTMP, ddAMP, ddCMP or ddGMP. Each pair
of P*s have
one nucleotide overlap at their. 3' termini. On the next nucleotide C, another
four pairs of P*s are
synthesized (not shown). If the wild-type sample is added, only the. wild-type
Bi-PAPs generates
the- specific product that is labeled by fluorescence. In this way, to scan a
I kb region, you need
89.00 P*s. Fig. 23B: Detection of an A to T mutation. On the mutated
nucleotide T, the
20
mutation-specific Bi-PAP generates the mutated product. On the next nucleotide
G; no produet
. of Bi-PA_P is generated because each pair of P* contains one or two
mismatches (not shown).
= [0108] Figures 24A-24B -show Bi-PAP resequencing naicroarray. Fig. 24A:
Detection of the
wild-type sequence. Four pairs of P*s are designed for each nucleotide
position according to the
= wild-type sequence. Each pair of P*s are downstream and upstream
directed, and have one
25 overlap and complementary nucleotide at their 3' teniaini. The wild-type
P* pair are specifically õ
amplified on each nucleotide position. If all of the wild-type P* pairs
specifically amplified, the
wild-type sequence can be determined. Fig. 2413: Detection of the A to T
mutation. With the
mutated template; the mutation-specific Bi-PAP is P7cip1i19 ed. There is a
window of no Bi-PAP
signals centered by the mutation-specific Bi-PAP and three successive
nucleotides on each side.
30 The
paired specific subsequoace is supposed to be seven nucleotides long. Any
unknown single-
base substitution peal be determined, even if if is a heterozygous mutation.
Also, small deletions
and insertions can be detected and localized.
CA 02911712 2015-11-10
18
[01 09j Figure 25 shows PAP de novo sequencing on microarray. PAP can also be
used for de
novo DNA sequencing of an unknown region. The paired specific subsequence is
supposed to
fifteen nucleotides long. P* pairs of a complete set of the paired specific
subsequence are on a
microarray with known addresses. After the unknown DNA sample is added, Bi-PAP
is
= s performed. All the amplified B-PAP products are collected and. then the
paired specific
subsequences of the amplified P* pairs are assembled by one-nucleotide
overlapping. Thus, the
unknown complementary sequence is reconstructed.
[01101 Figures 26A-26C show the detection of somatic mutations. Fig. 26A:
Eighteen genomic
DNA samples of the laar transgenic mice were chosen. 2 pg genomic DNA of each
sattple was
amplified with the assay B to detect the T369G Mutation two times. Samples 1-
10 are from
livers of 25-month old mice. Samples 11-14 are from hearts (samples 11;13 and
14) and adipose
(sample 12) of 6-month *old mice. Samples 15-18 are from brains of 10-day old
mice. P =
positive control that amplified the mutated X DNA, N = negative control with
no DNA, + =
" amplified product, - = no product Fig. 26B: The assay B was performed. In
lanes 11-12, 13-16
as and 17-20, 2 lig, 0.5 pg and 0.125 pg of the lad- mouse genomic DNA
of sample 12 were used
in each reaction, respectively. Lanes 1-10 are controls; the copy number of
the mutated X DNA
per reaction was reconstructed by two-fold serial dilutions. In lanes 1-10 and
13-20, each
reaction also contained 1 pg of the lad- mouse genomic DNA carrier. ss =
single-stranded, ds =
double-stranded. Fig, 26C: The assay B was performed. In. lanes 11-14, 2 lig
of the Tact
mouse genomic DNA of sample 3 was used in each reaction. In lanes 15-18, 2 pg
of the lad+
mouse genomic DNA of sample 9 was used in each reaction. Lanes 1-10 are
controls; the copy
number of the mutated X. DNA per reaction is indicated. Each control reaction
also contained 1
pg of the lad r mouse genomic DNA carrier.
,DETAMED DESCRIPTION OF TBE INVENTION
[01111 The following temainology is used herein.
[0112] Pyrophosphorolysis: zornoval of the 3' nucleotide from a nucleotide
strand chain by
to generate the nucleotide triphosphate.
DNA polymerase in the presence of pyroplolVilait, (PP)
This is the reverse of the polymerization reaction.
[01131 PAP: Pyrophosphorolysi activated polymerization. PAP c.tet.ise one P*
or can use two
opposing oligonucleotides in which at least one is P.
[01141 P*: an oligonudeotide with a non-extendible 3' terminus (or end) that
is actii-,-,Asible
by-
pyrophosphorolysis.
=
CA 02911712 2015-11-10
19
[0115] PAP-A: PAP-based allele-specific amplification that can be used for
detection of rare
= mutations (Fig. 1).
- [01161)31-PAP-A: PAP-A perfomied with a pair of opposing P*, i.e.,
bidirectional (Fig. 2) with
atleast one nucleotide overlap at their T-termini.
s [01171 PAP-R: PAP-based rese,quencing for detection of unknown mutations
within a known
sequence (Figs. 3 and 4).
= [01181 LM-PAP: ligation-mediated PAP. The nature of LM-PAP is that the
template is
synthesized before PAP, such as by ligation reaction or by extension using
terminal transferase.
[0119] LM-PCR: ligation-mediated PCR (Fig, 5).
[01201 G'w or As' alleles: alleles of the common polymorphism of the dopamine
DI receptor gene
that was used as a model system herein (also referred to herein as G or A
alleles).
[0121] Linear PAP: PAP with only one P* for linear product accumulation.
[0122] Exponential PAP: PAP with two opposing oligonucleotides for exponential
product
accumulation, and at least one is P*.
[0123] Noise rate (%): the relative yield of the mismatched product to the
matched product. A
specific signal for PAP is defined as a noise rate of less than 10%.
[01241 PASA: PCR amplification of specific. alleles (also known as allele-
specific PCR or
AR1V1S).
[0125] Resequeneing: scanning for unknown mutations and determining the
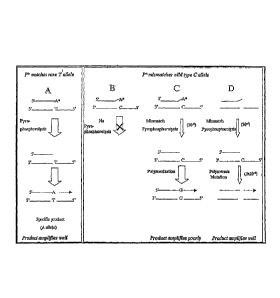
precise sequence
o changes within aknown sequence. Resequencing is distinguished from de
novo sequencing.
[0126] Mutation toad: the frequency and pattern of somatic Mutations within a
tissue.
[0127] Minimal residual disease: e.g., rare remaining cancer cells in lymph
nodes and other
neighboring tissues or early recurrence after remission.
[0128] Non-extendible 3' terminus (or end): a nucleotide or 'nucleotide analog
at the 3'
terminus (or end) of oligonucleotide P* that is non-extendible but that is
actiVatable by
pyrophosphorolysis. Examples of a non-extendible 3' termini (or ends) include,
but are not
limited to, a 2'3'-dideoxynucleotide, an acycionucleotide, 31-deoxyadenosine
(cordycepin), 3'-
azido-3r-deoxythymidine (AZT); 2',3'41ideoxyitioSine = (ddl), 2',3'-dideoxy-3'-
thiacytidine (3TC)
and 2',3'-didehydra-2',3'-dideoxythymidine (d4T).
[0129] Simplex PAP: one PAP (PAP, Bi-PAP, matched or mismatched PAP, and
others) in one
= reaction tube or on. a solid support.
[0130] Multiplex PAP: more than oue PAP (PAP, Bi-PAP, matched or mismatched
PAP, and
others) in one reaction tube or on a solid support; e.g., rnicroarray.
CA 02911712 2015-11-10
' [0131] Matched PAP: PAP having a match between P* and its template.
[0132] Mismatched PAP: PAP having a mismatch between P* and its template.
[0133] Nested PAP: PAP using two or more pairs of P* in which one pair is
located inside a
second pair on a template nucleic acid.
s [0134] Hotstart PAP: PAP in which an essential reaction component is
withheld until
denaturation temperatures are approached (Charo et al, 1992; Kellogg et al.,
1994; Mullis,
1991; D'Aquila et al., 1991). Essential reaction components can be 'withheld
by, e.g., a
neutralizing antibody bound. to the polymerase, sequestering a component such
as the polymers
or MgC12 in wax, chemically modifying the polymerase to prevent activation
until high
10 temperature incubation or separating components by wax.
[0135] Truncated Amplification: an amplification method which amplifies non-
exponentially,
e.g., in a quadratic or geometric manner, with over two chimeric
oligonucleotides and produces
truncated terminal products that are no more than three rounds of replication
from the original
template. (Liu et al., 2002).
15 [0136] Reactive '3 011: is a 3' OH that is capable of being extended by
a nucleic acid
polymerase or ligated to an oligonucleotide.
[0137] DNA polymerases, which are critical to nucleic acid amplificadon,
catalyze some or all
of the following reactions: i) polymerization of deoxynucleotide triphosphates
or their analogs;
ii) pyrophosphorolysis of duplexed DNA in the presence of pyrophosphate (PP),
[dNME]I
20 x[PPij + x[dNTP]; 3'-51exonuclease activity (which does not
require PPi), and
iv) 5'-3' exonuclease activity (Duetcher and Komberg, 1969; Komberg and Baker,
1992). For
Tag and Tj/ DNA polymerases, polymerization and 5'-3' exonuclease activity
have been reported
. (Chien et al., 1976; Kaledin et al., 1981; Longley et al., 1990). For T7
Sequenasem and Thermo.
Sequenaseml DNA polymerases, pyrophosphorolysis can lead to the degradation of
specific
dideoxynucleotide- terminated segments in Sanger sequencing reaction (Tabor
and Richardson,
1990; Vander Hom et al., 1997).
[0138] Pyrophosphorolysis is generally of very minor significance because PP i
is degraded by
pyrophosphatase under normal physiological conditions. However, in the
presence of high in
vitro concentrations of PPI, pyrophosphorolysis can be substantial. For
oligonucleotides with a 3'
terminal dideoxy nucleotide, only pyrophosphorolysis is possible. Once the
dideoxy nucleotide
is removed, the activated oligonudeotide can be extended by polymerization.
[0139] Pyropb.osphorolysis activated polymerization (PAP) offers a novel
approach for
retrieving a diversity of information from nucleic acids. The exceptional
specificity of PAP
CA 02911712 2015-11-10
21
derives from the serial coupling of two reactions. PAP involves the activation
by
pyrophosphorolysis of a 3' terminal blocked oligonucleotide (P*) followed by
extension of the
activated oligonucleotide by DNA polymerization. Operationally, PAP involves
the use of an
activatable oligonucleotide (P*) in place of a normal oligonucleotide that can
be directly
s
extended. Examples of P* include an inactive dideoxy terminated
oligonucleotide P* or an
inactive chemically modified nucleotide lacking a 3' hydroxyl group, such as
an
acyclonucleotide, or having a non-extendible nucleotide terminated
oligonucleotide P*.
Acycloclonucleotides (acycloNTPs) in which the sugar ring ig absent are known
to act as chain
terminators in DNA sequencing (Sanger et al., 1977; Trainor, 1996; Gardner and
Jack, 2002).
The activation of P* is inhibited by mismatches throughout the length of the
oligonucleotide.
Mismatches even two nnoleotides from the 5' terminus inhibit PAP
amplification.
[0140] Activation of a P* by pyrophosphorolysis offers extraordinary
specificity throughout the
length of P*. The enhanced specificity can be used to detect rare known
mutations, to elucidate
unknown mutations by resequencing, to determine unknown sequence by de nova
sequencing, to
Is measure gene expressiori levels, to compare two sequences, and to
increase the specificity of in
vivo analysis of chromatin structure. Microarray-based programmable
photochemical
oligonucleotide synthesis and PAP are synergistic technologies. Thus, the
enhanced specificity
. can be used for rapid, microarray-based resequencing, de nova
sequencing, gene expression
profiling and SNP detection.
101411 A number of methods for enzymatic nucleic acid amplification in vitro
have been
developed and can be adapted to detect known sequence variants. These include
polymerase
chain reaction (PCR) (Saild et al., 1985; Saiki et al., 1988), ligase chain
reaction (LCR)
(Landegen, 1998; Barany, 1991) and rolling Girdle amplification (RCA) (Baner
et al., 1998;
Lizardi et al., 1998). Herein, we describe pyrophosphorolysis activated
polymerization (PAP),
an approach that has the potential to enhance dramatically the specificity of
PCR allele-specific
smp1i-Hcation (Sommer et al., 1989). PAP differs from corrections with PCR in
multiple ways: i)
the P* oligonucleotide is= blocked at the 3' terminus and must be activated by
pyrophosphorolysis,
pyrophosphorolysis and polymerization are serially coupled for 'each
apiplification, iii) PAP may be performed with one P* for linear amplification
or with two
oligonucieotides for exponential amplification, iv) PP; is necessary for the
amplification, v)
significant nonspecific amplification would require the serial coupling of
errors of both
mismatch pyrophosphorolysis and misincorporation.
CA 02911712 2015-11-10
22
[0142] The enhanced specificity of PAP relative to PASA is provided by
serially coupling
pyrophosphorolysis and polymerization. Significant nonspecific amplification
requires
mismatch pyrophosphorolysis and misincorporation by DNA polymerase, an
extremely rare
event For example as described herein, DNA polynaerase Was utilized to detect
the G allele at
nucleotide 229 of the DI dopamine receptor gene. P* was synthesized either
With ddA, ddT, ddG
or ddC at the 3' terminus: The 3' terminal dideoxynucleotide inhibits direct
extension by
polymerization, but can be removed by pyrophosphorolysis in the presence of
pyrophosphate
(PP) when the P* is specifically hybridized with the complementary strand of
the G allele, The
activated oligonucleoticte can be extended by polymerization in the 5'-3'
direction. =
a.o
[0143] Evidence is presented that pyrophosphorolysis followed by
polymerization can be used
to increase the specificity of RASA. Significant nonspecific amplification
with PAP requires the
serial coupling of the two types of errors, i.e., mismatched
pyrophosphorolysis and
misincorporation (Fig. 1). The rate of mismatched pyrophosphorolysis is
expressed as the
relative rates of removal of a 3' miSmatch deoxynucleotide relative to the
correct 3'.
as deoxynucleotide. The rate of mismatch pyrophosphorolysis is less
than 104 for T7 DNA
polymerase (Komberg and Baker, 1992; Wong et at, 1991). The misincorporation
rate to create
a substitution mutation by polymerization, expressed- as the incmporation rate
of an incorrect
versus a correct dNMP, was reported to be 10-5 for T7 DNA polymerase and to be
10-4 for E.
coli DNA polymerase I (Komberg and Baker, 1992; Wong et al., 1991; Bebenek et
al., 1990).
20 Similar results were reported for Mq DNA polymerase, 3'-5'
exonuclease-deficient mutants of
Ti DNA polyrnerase and E. coli DNA polymerase I (Komberg and Baker, 1992; Wong
et at,
1991; Bebenek et al., 1990; Eckert and Kunkel, 1990).
[01441 PAP is a method of synthesizing a desired nucleic acid strand on a
nucleOtide acid
template strand. In PAP, pyrophosphorolysis and polymerization are "serially
coupled for nucleic
25 acid- amplification using pyrophosphorolysis activatable oligonncleotides
(P*). P* is an
= oligonucleotide that is composed of N nucleotides or their analogs and
has a non-extendible
nucleotide or its analog at the 3' terminus, such as 3%5' dideoxynacleotide.
When substantially
hybridized on its template strand, P* could not be extended directly from the
3' terminal
nucleotide or its analog by DNA polymerase, the 3' terminal nucleotide or its
analog of the P*
30 can be removed by pyrophosphorolysis and then the activated
oligonucleotide N) can be
extended on. the template.
[0145] The method comprises the following steps canied out serially.
=
CA 02911712 2015-11-10
= =
23
[01461 Annealing to the template strand a substantially complementary
activatable
oligonucleotide P*'. This activatable oligonucleotide P* has a non-extendible
nucleotide or its
analog at the 3' tenninuS.
[01471 (b) - Pyrophosphorolyzing the annealed activatable oligenucleotide P*
with =
pyrophosphate and an. enzyme that has pyrophospliorolysis activity. This
activates
oligonucleotide P* by removal of the 3' terminal non- extendible nucleotide or
its analog.
[01481 (c) PolymeriZina by extending the activated oligonucleotide P* .on the
template strand in
= the presence of nucleoside tiph.osphates or their analogs and a nucleic
acid polymerase to
synthesize the desired nucleic acid strand.
o [01491 The PAP method can be applied to amplify a desired nucleic acid
strand by the following
additional steps.
[01501 (d) Separating the desired nucleic acid strand of step (C) from the
template Strand, and
[01511 (e) Repeating steps (A)-(D) until a desired level of amplification of
the desired nucleic
. acid strand is achieved. =
= is [01521 The above PAP method can be applied to allele-:specific
Amplificatiori. The activatable
oligonucleotide P* has one or more nucleotides that are not complenientary to
the template
strand. The uncomplimentary nucleotide(s) of P* may locate at the 3' terminus
of P*. The above
=- step of (A), (B) or (C) could not occur substantially. As the result,
the desired micleic acid strand
is synthesized substantially less.
' 20 [01531 The above PAP method can be applied with only one
activatable oligonucleotide P. (e)
'Repeating steps (a)-(d); a desired levet of amplificationof the desired
nucleic acid strand may be
achieved li-hearly. The targeted nucleic acid region outside the annealing
region may be of
different sizes or of different sequence contexts, so the synthesized nucleic
acid strands are of
different sizes or of different sequence context.
25 [0154] The above PAP method can be applied with two opposing
oligonucleotides of which at
least one is the activatable oligonucleotide P. The activatable
oligonucleotide P* and the
second oligonucleotide are targeted for amplification of a nucleic acid
region. Steps (a),-(c) occur
to = the activatable oligonucleotide P*. The second oligottucleclide ia
SubStantially
= coMplementary to the other template'strand. If the second oligonucleotide
is another activatable
30 oligonucleotide 154i, steps (a)-(c) occur. If the seCond oligonucleotide
is. a regular extendible
oligonucleotide, steps (a) and (c) occur: (modified a) annealing to its
template strand, followed
by (Modified c) polymerizing by extending the oligonucleotide on its teniplate
Strand in the
preadride of nucleoside triphosphates or their analogs and a nucleic acid
pobijneta.se to
CA 02911712 2015-11-10
24
synthesize the desired nucleic acid strand. (e) Repeating steps (a)-(d), or
steps (a), (c) and (d), a
desired level of amplification of the desired nucleic acid strand may be
achieved, e.gõ
exponentially. The targeted nucleic acid region between the two annealing
regions of the two
opposing oligonucleotides may be of different sizes or of different sequence
contexts, so the
s synthesized nucleic acid strands are of different. sizes or of different
sequence contexts:
[01551 LM-PAP involves cleavage, primer extension, linker ligation and PAP
that can. be
applied for analysis of in vivo chromatin structure, such as, methylated state
of chromosomes.
[015.61 LM-PAP may be performed by steps (1), (ii), (iii), (iv) and (v), by
steps (i), (ii), (iii) and
(vi), by steps (ii), (iv) and (v) or by steps (ii), (iii) and (vi), where
the steps are as follows.
io [01571 The cleavage occurs chemically, enzymatically or naturally to
"breakdown" nucleic acid
strands. The nucleic acid usually is genoraic DNA that may have lesions or
nicks produced in
vivo.
101581 (ii) The primer of P1 is gene-specific and its extension includes: 1)
annealing to the
template strand a substantially complementary primer; 2) extending the primer
on the template
15 strand in the presence of nucleoside triphosphates or their analogs and
a nucleic acid
polymerase, the extension "runs off' at the cleavage site on the template
strand. Steps 1) and 2)
may be repeated.
[01591 The primer extension may be replaced by a P* extension (The above PAP
with only one
activatable oligonucleotide P*).
20 [01601 (iii) The linker ligation step includes ligation of a. linker to
the 3' terminus of the
synthesized nucleic acid strand. The linker ligation step may be replaced by a
terminal
transferase extension that is non-template dependent polymerization and. an
extra nucleic acid
-sequence is added to the 3' terminus of the synthesized nucleic acid strand.
[0161] (iv) PCR is performed with a second gene-specific primer (P2) together
with a primer
25 specific for the linker or the added sequence by. terminal transferase.
[01621 (v) A third gene-specific P* (P3) is used to detect the PCR generated
fragments. PAP
. method is applied with only one activatable oligonucleotide P*. The
extension of the activated
oligonucleotide P* "runs off" at the end of the template strand generated in
step (iv). The PAP
method may be applied in allele-specific manners. The activatable
oligonucleotide P* may
30 contain one or more nucleotides that are not complementary to the
template strand. The
uncomplimentary nucleotide(s) of P* may locate at the 3' terminus of P*.
[01631 (vi) Instead of steps (iv) and (v), PAP method can be applied with two
opposing
oligon.ucleotides a which at least one is the activatable oligonucleotide P*.
The activatable
=
CA 02911712 2015-11-10
oligonucleotide P*(P3) is gene-specific. The second oligonucleotide is
specific for the linker or
the added sequence by terminal transferase. The second oligonucleotide may be
another
activatable oligonucleotide P* or a regular primer. The PAP Method may be
applied in allele-
specific manners. The activatable oligonueleotide P* (P3) may contain one or
more nucleotides
s that are not complementary to the template strand. The
uncomplimentary nucleotide(s) of P*
may locate at the 3' terminus of P* (P3).
[0164] Fig, 1 shows detection of a rare mutation by allele-specific PAP (PAP-
A). PAP-A can
detect a rare allele with extremely high specificity because an allele-
specific oligonucleotide
with a 3' dideoxy terminus (P*) permits the serial coupling of
pyrophosphorolysis and =
o polymerization. For example, if an allele-specific oligonucleotide
has a 3' dideoxy terminus
(P*) that matches a rare "T" allele, activation occurs by pyrophosphorolytic
removal of the
dideoxy nucleotide and is followed by polymerization (Situation A). Activation
by
pyrophosphorolysis does not normally occur with a mismatch at the. Tterminus
as with the wild-
type "C" allele (Situation B). Rarely, pyrophosphorolysis does occur at a
mismatch (estimated
15 frequency 10-5), but the activated oligonucleOtide is extended to
produce wild-type sequence
(Situation C). A product that supports efficient amplification is generated
when mismatch
pyrophosphorolysis occ-urs; a polymerase error that inserts A opposite C in
template DNA
(Situation. 1)). The frequency of mismatch pyrophosphorolysis coupled with the
polymerase
mutation is estimated at 105x3x10-6= 3x10111.
20 [0165] PAP has a potential specificity of 3x10711. Approaching this
potential requires a design
that eliminates confounding sources of error. For example, extension errors
from non-blocked
upstream oligonucleotides can generate a product with the mutation of
interest. If the
misincorporation rate for TaqFS is about 10-5 per nucleotide and. only one of
the three
misincorporations generates the mutation of interest, the error rate is about
3.3x1(16. POIYMeraSeS
25
that contain a proofreading function might have an error rate per specific
mutation of 3x10-7.
Polymerases or polymerase complexes with lower error rates would improve
specificity further.
[0166] One approach utilizes linear PAP. Linear PAP-A may be performed for 40
cycles with
only P* in the presence of a fluorescent or radiolabeled ddNTP. A labeled
terminated product of
defined size will be generated when P* is activated. Linear PAP-A has the
advantage of utilizing
only the original genoraic DNA and eliminating error due to misincorporation
from extension of
an unblocked upstreamprimer. However, the sensitivity of detection is limited
because the level
of amplification is not greater than the number of cycles: For a simple
genorne like lambda
pbage, a detection specificity of 10-6 is possible; The specificity of linear
PAP-A depends
=
CA 02911712 2015-11-10
=
26
critically on the absence of unblocked, extendible oligonucleotides. To
achieve a robust
specificity of 10-6, unblocked extendible oligonucleotides should be present
at le. This may be
achieved by treating gel purified P* (about 99.99% pure with our present
protocol) with a 3' to 5'
exonuclease to degrade unblocked molecules followed by repurification by gel
electrophoresis.
[0167] A secOnd approach is bidirectional PAP-A (Bi-PAP-A; Fig. 2). In Bi-PAP-
A, both the
downstream and upstream oligonucleotides are P*s that are specific for the
nucleotide of
interest. The P*s overlap at their 3' temnai by one nucleotide. This design
eliminates extension
error from a non-blocked upstream oligonucleotide. This design should not he
limited by small
amounts of active contaminating oligonucleotide to which the dideoxy terminus
has not been
io
added (about 0.01% with our Current protocol) because the product generated
will be that of a
control and will not be a substrate for efficient amplification iti subsequent
cycles,
[0168] Bi-PAP-A generates a product that is the size of a primer dimer.
However,, it is not a
primer dimer in the conventional sense, in that template DNA with a mutation
of interest is an
intermediate required to generate a product that is an efficient substrate for
amplification in
Is subsequent cycles. Bidirectional PAP-A eliminates important
bottlenecks to specificity and has
the potential to reach a specificity of
[0169] As shown in Fig. 2, both the downStream and the upstream P*s are
specific for the
nuclecitide of interest at the 3' termini: (an A:T base pair in this example).
In the initial round of
= amplification from genomic DNA, segments of undefined size will be
generated. In subsequent
20 rounds, a segment equal to the combined lengths of the
oligonucleotide mints one will be
amplified exponentially. Nonspecific amplification occurs at lower frequencies
because this
design eliminates misincorporation error fauni an unblocked upstream
oligonucleotide that can
= generate the A:T template from a G:C wild-type template with an error
tate of 3x104. The P*s
may be 30-60 nucleotides for most efficient attplification. Situation A shows -
that a template
25 with a rare A:T allele Will be amplified efficiently. Both the
upstrenm and the downstream P*s
are amplified efficiently. Situation 33 shows that if the DNA template
contains the wild-type
G:C sequence, neither the downstream nor the upstream P* will be activated
substantially.
[0170] Rapid reseqtericing Will facilitate elncidation of genes that
predispose to cancer and
other complex diseases. The specificity of PAP lends itself to resequencing;
154's may be =
30 photochemically synthesized on rnicroarrayS using fiekible digital
micromirror arrays.
[0171] Microarrays of ithrnohili7ed DNA Or oligonncleotides can be fabricated
eitter by in situ
light-directed cOmbinational synthesis or by conventional synthesis (reviewed
by Ramsay, 1998;
Marshall and Hodgson, 1998). Massively parallel analysis can be perfOmaed.
Photochemical
CA 02911712 2015-11-10
27
synthesis of oligonucleotides iS a powerful Means fOr combinatorial parallel
synthesis of
= addressable oligonucleotide microarrays (Singh-Gasson et at, 1999;
LeProust et at, 2000). This
= flexible alternative to a large number of photolithographic Masks for
each chip utilizes a
masIdess array synthesizer with virtual masks generated on a computer. These
virtual masks are
s relayed to a digital micromirror array. An Ultraviolet imageof the
virtual mask is produced on
the active surface of the glass substrate by a 1:1 refiectiVe imaging system.
The glass substrate is
mounted in a flow cell reaction chamber connected to a DNA synthesizer. Cycles
of
programmed chemical coupling occur after light exposure. By repeating the
procedure with
additional virtual masks, it is possible to synthesize oligonucleotide
microarrays with any
o desired sequence. The prototype developed by Singh-Gasson, et al.
synthesized oligonucleotide
microarrays containing more than 76,000 features measuring 16 square microns.
= [01721 By combining programmable photochemical .oligonucleotide synthesis
with digital
minors and oligonucleotide extension of P*, a high throughput and automated
method of
resequencing is possible. PAP-R may detect virtually 100% of single base
substitutions and
..15 other small sequence variants because of its high redundancy; the
mismatch spanned by the
several overlapping P* oligonucleOtides Will prevent activation of :a cluster
of overlapping P*s.
One strategy for resequencing is shown in Figs. 3 and 4. Fig. 3 shows a
schematic of PAP-R
performed on a microan-ay with programmable photochemical oligonucleotide: PAP
can be used
for.resequencing to detect unknown mutations. On this roicroarray, the wild-
type template is
20 indicated. The P.*s are designed according to the wild-type
template. The P*s that overlap -with
the mutation generate little or no signal indicated as "Low" PAP signal.
= [01731 Fig. 4 shows an example of solid support-based, e.g., microarray-
based, resequencing to
detect a G to A mutation. Linear PAP is performed with four different dye-
labeled ddNTPS as
substrates for single-base extensions. P*s have a specific region of 16
nucleotides within the 3'
25 region of the oligonucleotide. Homozygous or hemizygous DNA template
is utilized in the
example. Sets of four P*s, with identical sequence except for the four
ddNIVIE's at the 3'
terminus, are synthesized for each nucleotide position on the sense strand of
the wild-type
sequence. The P* with a ddA at the 3' tennintS generates a PAP signal at the
site of the G-A
mutation. The mutation also creates a 15 base "gap" of no PAP signal for the
subsequent
30 overlapping 15 sets of Psi For heterozygous mutation, the P*s with
ddA and ddG provide PAP
signals. The heterozygous mutation also generates the 15-base "gap" of 50%
signal intensity
(which is flanked by signals of 100% intensity). For added redundancy with
heterozygotes
samples, antisense P*s can be utilized (not shown). An unknown single-base
substitution can be
CA 02911712 2015-11-10
28
determined by combination of the two sets of P*s. Small deletions and
insertions can be detected
and localized.
[0174] With 100,000 oligonucleotides per microarray, about 12 kb can be
resequenced from
downstream. and upstream directions. The detection of virtually all mutations
requires
supplementation of the standard Geniome instrument software. For wild-type
sequence, the
signal intensities may vary. Certain oligonucleatides will generate a weaker
signal due to
secondary structure and other factors. The pattern of signal from wild-type
samples should be '
distinguished reliably from the pattern generated by a given sequence change.
The preliminary
data suggest that almost all mismatches will inhibit aetivation dramatically.
Because of the
redundancy, mutations may be reliably distinguished from the wild-type even if
a significant
minority of single base mismatches does not inhibit activation substantially.
[0175] It is becoming increasingly apparent that in vivo chromatin structure
is crucial for
mammalian gene regulation and development. Stable changes in chromatin
structure often
involve . changes in methylation and/or changes in histone acetylation.
Somatically heritable
changes in chromatin structure are commonly called epigenetic changes (Russo
and Riggs,
1996) and it is now clear that epigenetic "mistakes" or epimutations are
frequently an important
contributing factor to the development of cancer (Tones and Laird, 1999).
[0176] One of the few methods for assaying in vivo chromatin structure, and
the only method
with resolution at the single nucleotide level, is ligation-mediated PCR (LM-
PCR) (Mueller and
Wold, 1989; Pfeifer et al., 1989). LM-PCR has been used to assess chromatin
strueture,
.
inethylation and damaged DNA. Fig. 5 shows a schematic of LM-PCR in which a
DNA lesion
in the starting DNA is indicated by a small diamond. LM-PCR involves cleavage,
primer
= extension, linker ligation and PCR amplification. LM-PAP is similar to LM-
PCR except that
activatable oligonucleotide P*s are used.
[0177] LM-PCR has proven to be an important technique, now having been used in
over 100
published studies (Pfeifer et al., 1999). Many aspects of chromatin stnicture
can be determined
by LM-PCR, such as the location of methylated cytosine residues; bound
transcription factors,
or positioned nucleosomes. Importantly, the- structure is determined in cells
that are intact and
have been minimally perturbed: UV photo-footprinting, for example, is
performed by LTV
irradiating tissue cultuTe cells in a Petri dish, immediately extracting the
DNA, and performing .
LM-PCR to determine the location of thymidine dimers, the formation of which
is affected by
bound transcription. factors.
CA 02911712 2015-11-10
29
-
[0178] Allele-specific LM-PAP can be applied to quantitatively detenrdne the
level of in vivo
methylation. The background of LM-PCR currently limits reliable estimation of
the level of
methylation. It is generally considered that 0 %, 50 % and 100% methylation
can. be determined,
but distinguishing finer gradations is not reliable. With a marked reduction.
in background in
LM-PAP, 0%, 20%, 40%, 60%, 80%, and 100% Methylation standards may be
distinguished
reliably. It will be of particular interest to utilize allele-specific LM-PAP
to examine the level of
methylation in imprinted regions, or in active verses inactive X-chromosomal
genes in females.
It is anticipated that LM-PAP will decrease the skill and. experience needed
to examine
chromatin structure, thereby facilitating analysis of chromatin structure by
more laboratories.
[0179] LM-PAP has a diversity of applications. It will be of particular-
interest to utilize allele-
specific PAP to examine differential methylation and chromatin structure in
imprinted genes or
in active versus inactive X chromosomal genes in females. In addition, the
relationship between
mutagens, DNA damage, and mutagenesis can be examined.
[0180] In PAP, as described above and illustrated herein, pyrophosphorolysis
and
is polymerization by DNA polymerase are coupled serially by using
pyrophosphorolysis
activatable oligonucleotide: In PAP sequencing, the principle is based on the
specificity of PAP
and in turn on the base pairing specificity of the 3' specific subsequence.
This property of the 3'
specific subsequence can be applied to scan for unknown sequence variants, to
determine de
novo DNA sequence, to compare two DNA sequences and to monitor gene expression
profiling.
[0181] PAP is highly sensitive to mismatches along the length of P* in PAP
with one P* and
one opposing unclocked oligonucleotide. The specificity of PAP is also
affected by P* length
and mismatch. If the allele-specific nucleotide of P* is at the 3' terminus,
only the specific allele
is amplified and the specificity is not associated with P* length. If the
allele-specific nucleotide.
is not at the 3' terminus of P*, the specificity is associated with P* length.
26 mer P* has a 3'
specific subsequence of three-nucleotides = within this region any mismatch
inhibit the
amplification. 18-mer has a 3' specific subsequence of 16 nucleotides.
[0182] Bi-PAP is a form. of PAP. In Bi-PAP with two opposing P*s, each P* has
its own 3'
subsequence, i.e., within this region any mismatch inhibit the amplification
of BiTAP. For
example, when the allele-specific nucleotide of the P* pair is at their 3'
termini, only the specific
allele was amplified, no matter what the lengths of the P*s are 40, 35 or 30
nucleotides. The
length of the paired specific subsequence is addition of the P* pair minus
one.
[0183] The length: of the paired specific subsequence may be affected by the
sequence context
and size of each
the type of the 3' terminal non-extendible nucleotide; the template sequence,
CA 02911712 2015-11-10
=
the DNA polymerase, other components like ions, and cycling conditions. When
the template
contains repeated sequences or homogenous polymer runs longer than the length
of the P* pair,
P* may lose specificity for anchoring.
[0184] Resequencing is the sequencing of a known region to detect unktio-wn
mutations. The
s property of the paired specific subsequence of Bi-PAP can be applied to
scanning for unknown
sequence variants or re-sequencing of predetermined sequences in a parallel
way.
. [0185] A Bi-PAP resequencing is shown in Figs. 22, 23A, 23B, 24A and 23B.
Briefly, the wild-
type sequence can be determined, and any single base substitution can be
determined with the
type and position. An unknown small deletion and insertion can be detected and
localized. = In
io order to identify a specific type of deletion or insertion, it is
possible to add corresponding Bi-
PAPs. For fingerprinting, which can provide infomiation regarding mutation
position, fewer
numbers of Bi-PAPs can be used.
[0186] The concept of Bi-PAP de novo DNA sequencing makes use of the complete
set of
paired specific subsequence of the P* pair to identify the presence of the
paired specific
15 subsequence in the de novo sequence.
[0187] Bi-PAP de nova DNA sequencing on microarray is shown in Fig: 25.
Briefly, the
procedure first collects all the Bi.,PAP amplifications with their P* pairs
and then reconstructs
the unknown DNA sequence from this collection by ordering the paired specific
subsequences.
[0188] For comparison of two DNA sequences to see if they are the same or
different, there is a
20 simple way to reduce the number of P* pairs by using an incomplete set
of the specific
subsequences of the P* pair; By arranging theni in a particular order, it is
possible to identify
the chromosomal locations as well as the sequences.
[0189] To monitor gene expression profiling, where up to 6x104 to 105
transcripts are expressed
and details of the precise sequence are unnecessary, Bi-PAP can be applied. A
set of P* pairs
25 which can specifically amplify unique motifs in genes can be designed
for Bi-PAP.
[01901 This property of the base pairing specificity of Bi-PAP can be applied
to seat for
unknown sequence. variants, to: determine de novo DNA sequence, to compare two
DNA
sequences' and to monitor gene expression profiling, A Bi-PAP array is
possible. Each pair of
two opposing P*s can be immobilized at an individual spot on a solid support,
e.g., mieroarray,
30 thus allowing all the Si-PAP reactions to be processed in parallel.
[0191] For PAP, the activatable oligonucleotide has a non-extendible 3'
terminus that is
activatable by pyrophosphorolysis (hereinafter referred to as a non-extendible
3' tettiainns). Any
3' terminal non-extendible oligonucleotide can be used, if it can hybridite
with the template
CA 02911712 2015-11-10
=
=
31
strand., the 3' terminal. nucleotide can be removed by pyrophosphorolysis, and
the activated
oligonucleotide can be extended. Examples. of non-extendible 3' terminus
include, but are not
limited to, a non-extendible 3' deoxyriucleotide, such. as a
dideoxynucleotide, or a chemically
modified nucleotide lacking the 3' hydroxyl group, such as. art
acycionucleofide.
Acyclonucleotides substitute a 2-hydroxyethoxymethyl group for the
2'41.eoxyribofuratosyl
sugar normally present in. dN1v1Ps:
[0192] Alternative blocking agents may increase the selectivity of
pyrophosphoroloysis for a
complete match, thereby further enhancing the selectivity of PAP for detecting
rare mutations.
Finally, alternative blocking agents may be less .expensive or more readily
autoinatable, thereby
3...o improving the cost-effectiveness of PAP and facilitating PAP
microarray-based resequencing.
[0193] In addition, P*s not blocked with dideoxynucleotides extends. the
selection. of DNA
polymerases which can be used for PAP. As demonstrated herein., Family I
polyinerases maybe
used for PAP when the 3' terminal non-extendible oligonucleotide contains a
dideoxymicleotide
= or an acyclonucleotide. Family II polyineraSes may he used for PAP when
the 3' terminal non-
is extendible oligonucleotide contains an acyclonucleotide.
EXAMPLES =
=
[0194] The invention can be understood from the following Examples, which
illustrate that PAP
can be used, to identify a known mutation in a polymorphic site within the
human Di dOp-triirie
20. receptor gene.
The effects of the dideoxyoligonucleotide sequences, acyclonucleotide
.sequences, DNA polym.erases, PP i concentrations, allele-specific templates,
pH, and dNTP
concentrations- were examined: The experiment reported in the Examples Were
conducted for
proof of principle. The following examples are offered by way of illtistration
and are not
intended to limit, the invention in any manner. Stan-lard techniques well
known in the. art or the
25 techniques specif,cally described therein were utilized. ,
EXAMPLE I
Preparation of template by PCR
[0195] A 640-bp region of the hi-1mm Di dopamine receptor gene was amplified
by PCR with
30 two primers = 5' GAC CTG CAG CAA GGG---AGT CAG AAG 3' (SEQ rD NO:1) wad
U = 51.
TCA TAC CGG- AAA GGG CTG GAG ATA 3' (SEQ ID NO:2)) (Fig. 6A). The TU:UT
duplexed product spans AuCleotides 33 to 672 in GenBank X55760 arid the G+C
content is
55.3% A common A to G polymorphism. is located at nucleotide 229, resulting in
three
=
=
CA 02911712 2015-11-10
32
= genotypes of GIG, A/A and G/A (Liu et al., 1995): The PCR mixture
contains a volume of 50
pd.: 50 mM KC1, lOnalVI Tris/HCI, pH 8.3, 1.5 DIM MgC12, 200 pM each of the
four dNTPs
(Boehringer Mamahefin), 0.1 p.M of each primer, 2% DMSO, 1 U of Taq DNA
polymerase
= (Boehringer Mannheim) and 250 ng of genomic DNA from GIG homozygete, A/A
homozygote
s or G/A heterozygotes. Cycling conditions included: denaturation at 95
C for 15 seconds,
amiealing at 55 C for 30 seconds, and elongation at 72 C for one minute, for a
total of 35 cycles
(Perkin-Elmer GeneAmp PCR system 9600). The PCR product was purified from
primers and
other small molecules by approximately 10,000-fold by three times of retentiOn
on a Centricon
100 microconcentrator (Araicon). The amount of recovered PCR product was
determined by
. o IN absorbance at 260 run.
Synthesis of P* by adding a 3'-dideoxynucleotide.
01961 .The deoxynucleotide oligonucIeotide w.as synthesized by Perseptive
Biosystems 8909
Synthesizer. (Framinsham).and purified by oligopure cartridges (Hamilton) in
the City of Hope
DNA/RNA Chemistry Laboratory. The 3' terminal dideoxynucleotide was added by
terminal
15 transferase. The mixture contained a total volume of 40 p.l: 200 mM
potassium cacodylate, 25
mM Tris/HCI (pH 6.6 at 25 C), 2.5 mM CoC12, 0.25 mg/ml of BSA, 4000 pM of the
oligonucleotide, 2.5mM 2'3'-ddNTP (the molar ratio of the 3'-OH terminus to
ddNTP was 1:25)
Boehringer Mannheim); 125 U of terminal transferase (Boehringer Mannheim). The
reaction
was incubated at 37 C for 1 hour and then stopped by adding EDTA at 5 mM final
20 concentration. After desalting by using butanol, the
dideoxyoligonucIeotide was purified by
preparative 7M urea/20% polyacrylarnide gel electrophoresis in TBE buffer (90
mM Tris/borate,
1mM EDTA, pH 8.3) (Maniatis et al., 1982). The amount of the recovered P* was
determined
by UV absorbance at 260.-nm.
[01971 Since small amounts of =terminated oligonucleotide would result in non-
specificity of
25 pyrophosphorolysis, each dideoxyoligonucleotide was 32P-labeled at
the 5 terminus by T4
polynucleotide kinase and then. was electrophoresed through a 7M urea/20%
polyacrylarnide gel.
Only P* products were visible even when the gel was overexposed (data not
shown). It is
estimated that more than. 99.99% of P* contained a dideokynticleotide at the
3' terminus.
Pyrophosphorolysis activated polymerization
30 [0198] A 469-bp region Within. the TIP.I.TT duplexed template was
amplified by PAP with
oligonncleotides P* and U, or with only one P* (Table I and Fig. 6A). The
PU:UP duplexed
product corresponds -to nucleotides 204 to 672 in GenBank X55760 and the G+C
content is
55.6%. Unless stated, the PAP reaction mixture contained a total voluble of 25
1 for Tfl DNA
CA 02911712 2015-11-10
33
polymerase: 75 mIVI KC1, 20 mM Tiis/HC1 (pH 74), 1.5 mM MgC12, 40 uM each of
the four
DNTPs (dATP, dTTP, dGTP and dCTP), 0.2 tiM P*, 0.05 iM U oligonucleotide, 300
11M
Na4PPi (the 20 KM stock solution was adjusted by Ha to pH 8.0), 1 Ci of [a-
32P1-dCTP
(3000Ciinmole, Arriersham), 1 U of Tfl DNA polymerase (Promega) and 2 ng of
TU:UT. For
s Tag DNA polymerase, the reaction mixture was the same except for 50 rriM
KC1, 10 mM
Tris/HC1 (pH 7.4); 2.0 mM MgC12 and 1 U of Tag DNA polyinerase (Boehringer
Marmheim).
The mixtures of PCR and other controls Were the same except for the primers
added. Cycling
conditions included: 94 C for 15 seconds,µ 55 C for one minute, ramping to 72
C for one
minute and 72 C for two minutes, for a total of 15 cycles. .
TABLE 1
= Oligonucleotides used in PAP
= Tem-
plate 5 L . .AATCTGACTGACCCCTATTCCCTGCTT GGAAC . . 3 ' (SEQ ID NO: 3 )
= A
Name Oligonucleotide Sequence 51-3' (SEQ ID NO:) . Purpose
ACTGACCCCTATTCCCTGCTTb (4) Control
_
ACTGACCCCTATTCCCTGCTTG*b ( 5 ) 3' ddG and G allele
specificity
co-localized
DG' ACTGACCCCTATTCCCTGCTTGG-k ( 6) G allele specificity
5' to ddG
= D3G7 ACTGACCCCTATTCCCTGCTTGGG*
(7) G allele specificity 5' to ddG
= D.4G' ACTGACCCCTATTCCCTGCTTGGGG* (8)= 3' ddG
mismatches template
Da TCTGACTGACCCCTATTCCCTGCTTG* (
9 ) DO, With 51 extended bases "
= D6K TGACTGACCCCTATTCCCTGCT
TA* (10) 3' ddA. and A allele-specificity
co-localized
-;TCATACCGGAAAGGGCTGGAGATA (11). Upstream oligonucleotide
=
Name 3' terminal Allele-specific Size Tõ/ Amplification'
nucleotide' nucleotide (base) ( C)e
Type Match Type From 3' =G allele A allele
terminus (bp)
DI dT Yes - +1 21 64 Yes Yes
. .
ddG Yes G 0 22 68 No No
=
_
ddG Yes G -1 23 72 No No
D3G7 ddG Yes G -2 24 76 Yes No
ddG No G -3 25 80 No No
=
D5A. ddG Yes G 0 26 80 Yes No
= ________________________ -
TK
ddA Yes A 0 24 72 No No
=
CA 02911712 2015-11-10
34
U. dA Yes - 24 72 Yes Yes
a DIG. was produced by adding a G dideOxynucleotide to the 3' tenninus of the
D1,4 a
dideoxynucleotide at the 3' terminus.
b The T means the 3' terminus is T deoxynucIeotide and G* means the 3'
terminus is G
dideoxynucleotide. The bold capital G and A are the G and A bases
corresponding to G and A
alleles, respectively. The first base at the 5' terminus corresponds to
nucleotide 208 in GenBank
X55760.
e The 3' terminal base is a deoxynucleotide or dideoxynucleotide, and creates
a match (Yes) or a
mismatch (No) with the corresponding base on the cornplementary strand of the
template.
d The allele-specific nucleotide is G or A and its distance to the 3' temnnus
is assigned: 0= at the
3' terminus +1 = one base downstream from the 3' terminus, -1 = one base
upstream from the
3' terminus, -2 = two bases upstream from the 3' terminus, and -3 three bases
upstream from the
3' terminus.
e The Tõ for oligonucleotides was estimatecf to be 4 C X (G + C) + 2 C X (T +
A) at 1 M NaC1
(Miyada. and Wallace, 1987).
= The amplification with TT and one P* or with only one P*. =
[0199] The reaction was electrophoresed through a standard 2% agarose gel. The
gel was
stained with ethidium bromide for UV photography by a CCD camera (Bio-Rad Gel
Doc 1000),
dried and subjected to Kodak X-OMATTm AR film for autoradiography.
" 20 Restriction digestion
[0200] Each of the three restriction endonucleases of Acil
(5'CYCGC3'/3'GGCAG5') Rad
(51PyTGGCCPu.31/ 31PuCC.GGAPy5t) and Eco0109I (51PuGYGNCCPy3r/ 3'PyCCNGAGPu5')
has
a restriction site within the PU:UP duplex. The G/G alleles were amplified by
PAP with D5G*
and U; PCR amplification with DI and U Was used as the control. 40 p.1 of the
PAP reaction and
2 ul of the PCR reaction were purified and concentrated with a Cennicon0 100
microconcentrator, and the products digested by the restriction endonucIease:
2.5 U of AciI in =
lx NE buffer 5; or 3 T.; of EaeI in.1X NE buffer 1; or 30 U of Eco0109I in NE
buffer 4 with .
BSA (all of the above enzymes and buffers from New England BioLabs). 10 p1 of
the reaction
was incubated at 37 C for 2 hours. The digestion reaction was electrophoresed
through a
standard 2% agarose gel as described above.
Principle of PAP
[02011 Tfl and Tag DNA polymerases were shown to contain pyrophosphorolysis
activity. 71/
DNA polymerase was utilized to detect the G allele at nucleotide 229 of the DL
dopamine
receptor gene (I-Au et al., 1995) (Fig. 6A). P* was synthesized with either
ddG or ddA. at the =
Tterminus (see Table 1). The nernain.al dideoxynucleotide inhibits direct
extension by
. polymerization, but can be removed by pyrophosphorolysis in the presence
of pyrophosphate
(pp) when the P* is specifically hybridized with the complementary strand of
the G allele. The
degraded oligonucleotide can be extended by polymerization in 5'-3'direction
(Figs. 6B and 6C).
CA 02911712 2015-11-10
[0202] The enhanced specificity of PAP relative to PASA is provided by
serially coupling
pyrophosphorolysis and= polymerization.
Significant nonspecific amplification requires
mismatch pyrophosphorolysis and misincorporation by DNA poly-merase, an
extremely rare
event (Fig. 7).
5 Specific amplification with D5G* and D3G*
[0203] PAP was perforined With two oligonucleotides (13* and U), 1)7 DNA
polymerase and
DNA template of the GIG and A/A alleles. Multiple P* were tested (Table 1).
D5G* (the allele-
specific nucleotide and dideoxynucleotide are co-localized to the 3' terminus
and D3G* (the
allele-specific nucleotide is two bases from the 3' terminus) specifically
amplified the G allele in
o the presence of PP; (Fig. 8A). Without added PP, no specific product
.was observed with D5G*,
indicating that added PP i was an essential component for PAP (Fig. 8B, lanes
6 and 15). Faint
products with D3G* in lane 4 and with D4G* in lane 5 were observed (Fig. 833)
(see below).
Effects of pH, [PP and {dNTP} and enzyme
[0204] Each of the above parameters was examined. PAP was most efficient at pH
between 7.4
is and. 7.7, at {PP] between 200 gM and 400 M, and at [dNTPs] between
25 gM and 50 Al
(Table 2). Taq DNA polymerase can substitute for 7:fl with similar
efficiencies (Table 2).
TABLE 2
Parameters affecting PAP
Parameter PAP efficiencYb
D5G*-U D3G*-U
8.1
pHa= . 7.9' -
7.7 ----
7.5 -H 111
-
7.4 -H-
7.15 +
1000
ppia
800
GEM) 600
400 ++ lIM
200
0
200
All d_NTPs 100
changed'
CA 02911712 2015-11-10
= 36
50 ++ +-H-
25 -H- ++++
100 -H-
dGTP
50 -H-
chanaeec
25 -H-
100
dATP 50
changed'
25 -H-
G allele and PPf
Taq DNA A allele arid PPi
polymerase
G allele and no PPi ___________________________________________________ =
Tfl DNA polymerase was used to amplify the GIG alleles under the conditions in
Materials and Methods, except for the factors indicated
b The PAP efficiency is indicated as: -, no specific product(s); + very weak
specific
product(s);. +, weak specific product(s); +4-, moderate specific product(s);
strong
specific product(s); I ____ , very strong specific product(s).
The indicated concentration was changed but the others were kept at 200 M.
Identity of specific products
[02051 In order to confirm the identity of the specific products, restriction
endon_uclease
digestion was performed (Fig. 9). Each of the three restriction endonucleases
of Acil, Ead and
Eco01.09 has a restriction site with the PU:UP duplex. The expected
restriction fragments were
found. Similar results were observed with D36* and U.
[02061 The specific products of PAP with D5G* and. U revealed two specific
bands on the
= agarose gel, i.e., PU:UP and UP; because U was more efficient than D5G*,
under our
amplification conditions. In order to confinn this, the G/G alleles were
amplified by PAP using
T77 DNA polymerase with D5G* and U as previously. The products were denatured
and
electrophoresed through a denaturing polyacryla-mide gel. Only one specific
band in single-
stranded form was observed, indicating that the specific PAP products contain
the duplexed and
single stranded segments. The same result was observed with D3G* and U.
Linear PAP
[0207] PAP was performed for linear amplification with only one P* from the
GIG and A/A
alleles in the presence of PPi. The specific products of PAP were obtai-ned
With D3G* and with
D5G*, but not with the other P* (Fig. 10, lanes 4 and 6). The efficiency of P*
was affected by
the oligonucleotide size, the 3'-terminal dideoxynucleotide and the position
of the allele-specific
nucleotide.
CA 02911712 2015-11-10
37
[02081 Figs. 6A-60 show schematic of PAP. Pig. 6A. A duplexed DNA template
TU:UT is
amplified with two oligon.ucleotides P* and U, Tfl DNA polymerase, dNTPs,
pyrophosphate and
{06-3211}-dCTP. P* = pyrophosphorolysis activatable oligonucleotide. hi this
example P* is
D5G* and TU:UT is a 640-bp sapient of the dopamine D1 receptor gene. Fig. 6B.
D5G* has a
s G
dideoxynucleotide at the 3' terminus, and it is specific to the complementary
strand of the G
allele, but mismatches the A allele at the 3! terminus (Table 1). Removal of
the dideoxy G by
pyrophosphorolysis is followed by polymerization for each amplification. Fig.
6C.
Autoradiogram of PAP from the GIG, A/A and G/A genotypes. When the G allele is
present,
the radioactively labeled specific products of 469 bases (duplex PU:UP and.
excess antisense
o
strand UP) are produced, since the low rate of pyrophosphorolysis by Tfl
polymerase implies
that Oligonucleotide U has a much higher efficiency than oligonucleotide P.
Electrophoresis
for a longer period separates PU:UP from UP. Other products of UT and UT:TU
are indicated.
Note that TU:UT derives from annealing of excess radioactively labeled UT with
non-
radioactively labeled TU original template. PAP was also perfonned with D3G*
and U from the
is GIG, .AJA and G/A genotypes, and similar results were obtained.
[02091 Figs. 7A.,713 show enhanced specificity of PAP with D5G*. The
specificity of PAP is
compared with that of PASA to exponentially amplify a template pool of G and A
alleles. Fig.
7A. The 'specific amplification of PASA derives from the high efficiency of
primer extension
when the primer matches the G allele. The nonspecific amplification results
from mismatch
20 extension from the A allele. When this occurs, it results in an
efficiency substrate for further
Pmplification. The thickness and position of the arrow represent the
amplification efficiency in
each cycle. Fig. 7B. The specific aniplification of PAP from the G allele
occurs at high
efficiency. Two types of nonspecific amplifications originate from the A
allele: (i) nonspecific
Amplification can occur at low efficiency by mismatch pyrophosphorolysis
resulting in a A:T
25 hoi33o-duplex PU:UP product, which is not an efficient template for
subsequent amplification;
(ii) nonspecific aMplification can occur at extiemely low efficiency by both
mismatch
pyrophosphorolysis and misi-ncorporation to produce a G:T hetero-duplex PU:UP
product, but
once i occurs, it provides an efficiency template for subsequent
amplification. A similar
tendency of nonspecific amplifications is suggested for iii-ear amplification
by PAP with only
30 D5G*. It should be noted that allele-specific nucleotide of P*, such
as D3G*, may be near but
not at the 3' terminus. In that ease nonspecific amplification of PAP requires
both mismatch
pyrophosphorolysis and mismatch extension: While both variations of PAP should
have higher
CA 02911712 2015-11-10
38
specificity than PASA, the highest specificity is. predicted when the 3'
terminal dideoxy
nucleotide is also the allele-specific nucleotide.
[02101 Figs. 8A-8B show specific amplification with D5G* and D3G*. PAP was
performed in
the presence (Fig, 8A) or absence (Fig. 8B) of added PP i with two
oligonucleotides for
s
exponential amplification. The oligonucleatides are listed in Table 1.
Extension controls with
only U identify the positions of TU:UT and UT. Extension. controls with Di
identify the
position of PU. PCR controls of Di and U identify the positions of PU:UP and
PU:UT. Only
20% of the extension reaction with Di and the PCR reaction were loaded
relative to other lanes.
[02111 Fig. 9 shows restriction endonuclease digestion. TO show specificity of
PAP, samples
1.o
from the experiment shown in Fig. 8 were digested with Acil, Eael and.
Eco01091 restriction
endonucleases. Each enzyme has a restriction site within PU:UP. PAP amplified
the GIG
alleles with D5G-* and U, and 5% of PCR reaction with Di and U were taken as
control. Acil
= produces a 236 bp and a 233 bp fragments from PU:UP and a 407 bp and a
233 bp fragments
from TU:UT. EaeI produces a 289 bp and a 180 bp fragments from PU:UP and a 460
bp and a
1.5 180 bp fragments from TU:UT. Eco01.09I produces a 348 bp and a 121
bp fragments from
PU:UP and a 107 bp, a 412 bp and a-121 bp fragments from TU:UT. The arrows
indicate the
digestion products expected from PU:UP.
[02121 Fig. 10 .shows linear PAP. PAP was performed with only one P* in the
presence of
added PP. 20% of the reaction with Di was loaded relative to other lanes
(lanes 1 and 10). No
20 = no oligonucleotide added.
Enhanced specificity of PAP with D5G*
[02131 Example 1 provides evidence that pyrophosphorolysis followed by
polymerization may
be used to increase the specificity of PASA. Significant nonspecific
amplification requires the
serial coupling of the two types of errors (Fig. 7). The mismatch
pyrophosphorolysis rate to
25 remove a mismatch deoxynucleotide at the 3' terminus, expressed as
the removal rate of an
incorrect versus a correct cINMP, was reported at less- than 10-5 for' T7 DNA
polymerase
(Komberg and Baker, 1992; Wong et al.., 1991). The misincorporation rate to
create a
substitution mutation by polymerization, expressed as the incorporation rate
of an incorrect
versus a correct dNMP, Was reported as to be 10-5 for 17 DNA polymerase and to
be 10-4 for
30 E.coli DNA polymerase I (Kornberg and Baker, 1992; Wong et al.,
1991; Bebenek et al., 1990).
Sitailar results were reported for Taq DNA polymerase and for 3'-5'
exotablease-deficient
mutants of 17 DNA polymerase and .E. colt DNA polymerase I (Kornberg and
Baker, 1992;
Wong et al., 1991; Eckert and Kunkel, 1990). The specificity due to the (i)
nonspecific
CA 02911712 2015-11-10
=
39
amplification in. PAP with D5G* is estimated to be 10-5 per cycle, if the
mismatch
pyroph.osphorolysis rate of a ddNMP is the same as dna. The specificity due to
the (ii)
nonspecific amplification is estimated to be 3.3x1041, if the mismatch
pyrophosphordysis and
the misincorporation are serially coupled.
s Essential components of PAP
[0214] Each P* was tested by utilizing 7:11 or Taq DNA polymerases to amplify
the GIG and
A/A alleles. The specific amplification requires the presence of PP; and
allele-specific template.
In. addition, the amplification efficiency is affected by the oligonucleotide
size, the 3' terminal
dideoxynucleotide, the position of the allele-specific nucleotide relative to
the 3' terminus of P*.
o [0215] It is not clear why DIG* and D2G* did not generate the
specilic signals, but it may be
related to a threshold stability of duplex between P* and the template. D6A*,
which contains A
dideoxynucleotide at the 3' terminus, did not generate the specific signal,
which may be
associated with different incorporation efficiencies of ddNTPs by
polymerization. Klenow
fragment of E. coil DNA polymerase I, Taq DNA poiyirterase and Araq DNA
polymerase
as incorPorate ddGTP more efficiently than other ddNTPs (Sanger et at,
1977; Tabor and
Richardson, 1995; Vander Horn et al., 1997). The rate of ddNTP incorporation
also varies
depending on the template sequence and can. be 10-fold higher at some bases
relative to others
(Sanger et at, 1977). Another possibility is that D6A* is shorter in size with
a lower
- [0216] In. PAP without added PP, very faint false signals were generated
with D3G* and with
20 D4G* (Fig. 8B). One possibility is that oligonucleotide dimers can
form and trigger nonspecific
pyrophosphorolYsis of P* in later cycles after "endo-" PP; is released from
the by-polymerization
to generate UT. 3Ttennin.al degraded D3G* and D4G* can be hybridized and
extended as false
signal. Oligonucleotide climers were observed with D3G* and D4G*. Another
possibility with
D3G* is that the specific pyrOphesphorolysis can occur in later cycles after
"endo.," PP; is
25 released. A third possibility is that D30* and D4G* were contami-
nated by minimal D3 and D4
which were not fully added by G dideoxynucleotide at 3' termitit
. Coniparison with other technologies
[02171 A number of methods for enzymatic nucleic acid amplification in vitro
have been
developed and can be adapted"to detect known sequence variants. These include
polymerase
30 chain reaction (PCR) (Saud et al., 1985; Saila et at, 1988), ligase
chain reaction (LCR)
(Landegren et al.; 1988; Barmy, 1991) and rolling circle amplification (RCA)
(Lizardi et al.,
1998; Ban& et al., 1998). PAP is different in many ways: i) pyrophosphOrolysiS
and
polyrnerization are serially cOupled for each amplification, ii) there is at
least one
=
CA 02911712 2015-11-10
dideoxyoligonucleotide for PAP. Other chemically modified nucleotides lacking
the 3"-
hydroxyl group at the 3' terminus, such as acyclonucleotides can serve the
same function (see
Example 12 below), iii) one format is for linear amplification and the other
is for exponential
amplification, iv) PP' is necessary for the amplification; v) significant
nonspecific amplification
5
requires both mismatch pyrophosphorolysis and misincorporation, vi) PAP can
detect known
point mutations and greatly increase the specificity to detect an extremely
rare mutant allele
from the wild-type allele.
[0218] The mechardstic basis is that two or more reactions are serially
coupled for amplification
with increased specificity. The key component of PAP is a pyrophosphorolysis
activatable
10 oligonucleotide. The blocked 3' terminus in these experiments is a
dideoxy nucleotide, but any
non-extendible nucleotide susceptible to pyrophosphorolysis could in principle
be substituted.
Indeed, any enzyme that cleaves an oligonucleotide 5'= to a mismatch could
serve the same
function as pyrophosphorolysis activation. For example, a blocked
oligonucleotide including the
methylated recognition sequence (such as GmATC) is annealed to its target
with. the
15 unmethylated recognition sequence, then restriction endonuclease (such as
Dpnl) can only
cleave the methylated site and so activate the oligonucleotide for extension.
If a mismatch is
located 5' to the cleavage site, significant nonspecific amplification
requires the serial coupling
of mismatch cleavage and a misincorporation, which is a rare event.
Activatable
oligonucleotides may also be combined with "minisequencingu primer extension.
This may
20 provide a more specific assay for detection of single base changes
that might be particularly
amenable to chip technology in which specificity can be a problem (Syva-nen,
1999).
. Demonstration that PAP can occur in the linear format (Fig. 10)
supports the feasibility of this
approach.
[02191 Nucleoside triphosphates and 2'-deoxynucleoside triphosphates or their
chemically
25 modified versions may be used as substrates for multiple-nucleotide
extension by PAP, i.e.,
when one nucleotide is incorporated the extending strand can be further
extended. 2',3'-
dideoxynucleoside triphOsphates or their chemically modified versions that are
terminators for
further extension may be used for single-nucleotide extension. 2',3'-
dideoxynucleoside
triphosphates may be labeled with radioactivity or fluorescence dye for
differentiation from the
30 3' terminal dideoxynucleotide of oligonucleotide P. Mixtures of
nucleoside triphosphates or 2'-
deoxynucleotide triphosphates and 2`,3'-dideoxyaucleoside triphosphates may
also be used,
102201 In PAP, specific nucleic acid sequence is produced by using the nucleic
acid containing
that sequence as a template. If the nucleic acid contains- two strands, it is
necessary to separate
CA 02911712 2015-11-10
41
the strands of the nucleic acid before it can be used as the template, either
as a Separate step or
. .simultaneously. The strand separation can also be accomplished by any Other
suitable method
including physical, chemical or enzymatic means: =
[0221] When. it is desired to produce more than one specific product.dom the
original nucleic
acid Or mixture of nucleic acids, the appropriate number of different
oligonucleotides are
utilized. For example, if two different. specific products are to be produced
exponentially, four
oligonucleotides. are utilized. Two of the oligonucleotides (
are specific for one of the
specific nucleic acid sequel-ides and the other two oligonucleotides ( P*:L1)
are specific for the .
second specific nucleic acid sequence. In this manner, each of the two
different specific
o sequences can be produced exponentially by the present process.
[02221 The DNA or RNA may be single- or double-stranded, may be a relatively
pure species or
a component of a mixture of nucleic acids, and may be linear or cirtular. The
nucleic acid or
acid S may be obtained from. any source, for example, from plasmid,. from
cloned. DNA or RNA,
. , .
or from natural DNA or RNA from any source, including bacteria, yeast,
viruses, and higher
15
organisMs such as plants or animals. DNA Or RNA may be extracted from blood,
tissue material
such as chorionic villi or amniotic cells by a -Variety Of techniques such as
that described by
Maniatis et al. (1982).
[0223] The P* oligonucleotides are selected to be. "substantially"
complementary" to the
different strands of each specific sequence to be amplified. Therefore, the P*
oligOnucleotide
20 seqUence need not reflect the exact sequence of the template. For example,
a
non-complementary nucleotide segment may be attached to the 51-end of the P*
oligdnucleotide,
with the retnailidet of the P* oligon.utleotide sequende= being complementary
to the strand.
AlternatiVeiy, non-compleinentary bases or longer sequences Can be
interspersed into the P*
oligorinclecitide, provided that the P* oligonticleotide sequence has
sufficient coMpleMentarity
25
with the sequence of the strand to be amplified to hybricii76 therewith and
form a template for
'synthesis of the extension product of the other P* Oligonucleotide. As used
in the claims, the
= term. "complementary" should be understood to mean "substantially
complementary," as.
discussed herein.
[02241 Any specifid nucleic acid' sequence can be produced by the present
prOceSs. It is only
30
necessary that a sufficient number of bases at both ends of the sequence be
known in Sufficient =
detail so that.tWO oligonucleotides can hybri1i7e to different strands of the
desired sequence at
relatiVe pOsitIonS along the sequence. The greater the knowledge about the
bases at both ends of
the Seqtiende, the greater can be the specificity of the oligliticleotides for
the target nucleic. acid
=
=
CA 02911712 2015-11-10
= 42 -
sequence, and thus the greater the efficiency of the process. It will be
understood that the word
oligonucleotide as used hereinafter may refer to more than one
oligonucleotide, particularly in
the case where there is some ambiguity in the information regarding the
terminal sequence(s) of
the segment to be amplified.
[0225] The present invention can be performed in a step-wise fashion where
alter each step neW
= reagents are added, or simultaneously, where all reagents are added at
the initial step, or partially
= = step-wise and partially simultaneOus, where fresh reagent is added
after a given nuMber of steps.
The simultaneous method may be utilized when an enzymatic means is used. for
the strand
separation. step: In the simultaneous procedure, the reaction mixture may
contain the
io strand-separating enzyme (e.g., helicase), an appropriate energy
source for the strand-separating
enzyme, such as ATP. Additional materials may be added as necessary.
[0226] The nucleic acid polymerase may be any compound or system that will
function to
accomplish the amplification. Suitable enzymes for this purpose include, for
example, 1)7 DNA
polymerase, Taq DNA polymerase, E. coil DNA polymerase I, Klenow fragment of
E. coil DNA
is polymerase I,. T4 DNA polymerase, T7 DNA polymerase, other available
DNA polymerases,
RNA polymerases or their variants, reverse transcriptase or its variants, and
other genetic
engineered versions. It is predicted on the basis of the relationship between
reverse and forward
reactions that. a DNA polymerase will have high and even pyrophosphorolysis
activity for the P*
activatable oligonucleotide, if it incorporates dciNTPs efficiently (compared
with dNTPs) and
20 evenly (compared among the four ddNTPs). Of all the DNA polymerases,
the genetic
engineered version may be the best in the future, such as ThermoSequenase
(Vander Horn et al.,
= 1997). Generally, the synthesis will be initiated at the 3' end of each
oligonucleotide and proceed
in the 5' direction on the template strand. However, inducing agents which
initiate synthesis at
the 5' end and proceed in the other direction can also be used in the PAP
method as described
2s above.
EXAMPLE 2
Preparation of template by PCR
[02271 A 640-hp region of the human DI dopamine receptor gene was amplified by
PCR with
o two primers (T = 5' GAC CTG CAG CAA GGG AGT CAG AAG 3' (SEQ ID NO:1)
and U= 5'
TCA TAC CGG AAA GGG CTG GAG ATA. 3' (SEQ ID NO:2)). The MITT duplexed product
spans nucleotides 33 to 672 in GenBank X55760 and the G-FC content of the
product is 55%.. A
common A to G polymorphism is located at nucleotide 229, resulting in three
genotypes of G/G,
CA 02911712 2015-11-10
43
= A/A and G/A. The PCR volume is 50 Ill: 50 mM KCI, 10 inM Tris/HC1, pH
8.3, 1.5 mM MgC12,
200 gM each of the four dNTPs, 0.1 1.1M of each primer, 2% DMSO, 1 U of Taq
DNA
polymerase (Boehrhiger Mannheim) and 250 ng of genomic DNA from GIG
homozygote, A/A
.homozygote or G/A heterozygotes. Cycling conditions included: denaturatiOn at
94 C for 15
s
sec., annealing at 55 C for 30 sec., and elongation at 72 C for one min., for
a total of 35 cycles
with a GeneAmpTm PCR: System 9600 (Perkin-Elmer Applied Biosystems). The PCR
product was =
purified from primers and other small molecules by approximately 10,000-fold
by three times of
retention on a Centricons 100 microconcentrator (Amicon). The amount of
recovered PCR
product was determined by UV absorbance at 260 rim:
io Synthesis of P* by adding a 3'. dideoxynucle,otide.
102281 The deoxynucleotide oligonucleotide was synthesized -by Perseptive
BiOsystems 8909
Synthesizer (Framin.sham) and purified by oligopurecartridges (Hamilton) in
the City of Hope
DNA/RNA. Chemistry Laboratory. The 3' terminal dideoxynucleotide was, added by
terminal
transferase. The mixture. contained a total volume of 30 ill: 100 mM potassium
cacodylate (pH
. is 7.2), 2.0 mM CoC12, 0.2 mM DTT, 2500 pM of the oligonucleotide, 2 inM
2`,. 3'-ddNTP (the
. =
molar ratio of the 3'-OH terminus to ddNTP was 1:24)(Boehringer Mannheim), 100
U of
terminal transferase (GIBCO BRL). The reaction was incubated at 37 C for 4 hr
and then
stopped by adding EDTA at 5 ni.M final concentration. After desalting using a
Centri-spinrm
7ooltirrin .(Princeton Separations), P* was purified by preparative 7 M
urea/20% polyacrylamide
20 gel electrophoresis in TBE buffer (90 mM Tris/borate, 1 mM EPTA, pH
8.3) (Mattiatis..et al.,
1982). The amount of the recovered P* was determined by UV abSorbance at 260
tun.
= [02291 Since small amounts of unterminated oligonucleotide would result
in rionspecificity of
pyrophosphorolysis, each P* ra 32P-labeled at the 5' terminus by T4
polyttucleotide kinase and
then was electrophoreSed through a 7 M urea/20% polyacrylamide gel. Only P*
products were
25 visible even when the. gel =was' overexposed. It is estimated that
More than 99.95% of P*
contained a dideoxynucleotide at the 3! terminus: The purity of P* wat
supported by the abseuce
of PCR. product or PAP product at pH 8.3.
Pyrophosphorolysis activated polymerization
. [0230]. Regions from 445 to 469 bp within the TU:UT duplexed template were
amplified by
30 RAF with oligonucleotides P* and U, or with only P*. The PU:UP
duplexed product
corresponds to nucleotides 204-228 to 672 in GenBank X55760 and its (3i-C
content is 56%.
The PAP reaction mixture contained a total volume of 25 RI: SO mM =1, 10 mM
Tris/HCI (pH
7.6), 1.5 mM MgC12, 100 uM each of the four dNTPs (dATP, dTTP, dalP and dCTP),
0.1 ILM
CA 02911712 2015-11-10
44
P*, 6.1 1.1M U oligonucleotide (TCATACCGGAAAGGGCTGGAGATA (SEQlD NO:2)), 300
1.1M Na4PPi, 2% DMSO, 1 CI of {a-32P] dCTP (3000CM:1=01e, Amersham), 1 U of
= AmpliTaqFS DNA polymerase (PE Applied Biosystems) or 0.5 U of each of
ArapliTaqFS and
'Tag DNA polymerases, and 10 ng of TU:UT. ThermoSequenase (Amershani
Pharmacia) was
s
also tested under the same conditions except for 8U ThermoSeqnenase or 4U
ThermoSequenase
plus 0.5U Taq and 2.5mM MgC12. The cycling conditions included: denaturation
at 94 C for 10
sec., annealing at 60 C for 1 min. (at 55 C for TheftuoSequenase), and
elongation at 72 C for 2 .
min., for a total of 15 cycles.
[02311 The product was. electrophoresed through a standard 2% agarose gel. The
gel was stained
lo with ethidiura bromide for UV photography by a CCD camera (Bio-Rad
Gel Doc 1000) and
Multi-Analyst software, dried and subjected to Kodak X-OMATrm AR film for
autoradiography. The PAP. yield was quantitated with a PhosphorImager with
lmageQuantim
software (Molecular Dynamics) as the total number of pixels in the PCR band
mints the
= background, indicated as a random units
15 Enhanced PAP efficiency
[0232] In Example 1, only the P* with ddG at the 3! terminus was amplified
using native V/ or
Taq. DNA polymerase. AmpliTaqFS and ThoimoSequenase DNA polyinerases were
found to
achieve much higher PAP efficieney with much less discrimination against any
kind of
dideoxynncleotide (ddAMP, dd(MP, ddGMP or ddCMP) at the 3' terminus of P*. For
example,
20 P*(212)18G and P*(212)18A ,. which are 18-mers of the dopamine DI
receptor gene but have
ddGMP and ddAIVIP at the 3' termini (Table 3), specifically amplified the G
and A alleles,
respectively. Their yield ratio was 1.4 (compare lanes 9 with 11 in Fig. 11B),
and scis
P*(212)18G is estimated to be 4% more efficient per cycle than: P*(212)18A .
Another =
=
P*(228)26A-24 =5' TAGGAACTTGGGGOGTOTCAGAGCCC*. Y (SEQ ID NO:12), which is
as a 26-tner With. ddCMP at the 3' terminus, was amplified. as
efficiently as a primer without
ddCMP at the 3' terminus, and the yield was estimated to be increased 1,0.00
fold compared with
that by using Tfl or Taq. Moreover, PAP amplified segments directly .from
taiman genoraic
DNA.
. .
=
= .
=
. . ,.
'
= .
-
. TABLE 3
PAP specificity affected by P* length and mismatch
,
_______________________________________________________________________________
________
Name Sequence (SEQ ID NO:) Mismatch T.
Noise
base (
C)d ratio
- Type Distance'
-Template Strand G _
51...AATCTGACTGACCCCTATTCCCTGCTT GG1AC...3' (3)
A
_______________________________________________________________________________
_________ _
-11*(204)26G" 51tCtgac H
tgACCCCTATTCCCTGCTTG*b (13) G 0 - 80 0.0
_
o
--P*(208)22G 51actgACCCCTATTCCCTGCTTG* (14) G
0 68 0.5
0
N.)
P*(210)20GY 5'tgACCCCTATTCCCTGC1'TG* (15) G -
0 62 - 0.1 ko
1-,
P*(212) 1869 . 5 'ACCCCTATTCCCTGCTTG*
(16) G 0 56 0.3 - .4
1-,
_
(A_
-P*(216)26G' 5'ctattcccTGCTTGGGAACTTGAGGG* (17)
G -12 80 107.1 N.)
0
_______________________________________________________________________________
_________ _
P*(220)22G-12- 5'tcccTGCTTGGGAACTTGAGGG* (18) G
-12 70 95.5 (xi
1
1-,
1-,
P*(222)20G41 5'ccTGCTTGGGAACTTGAGGG* (19)
_ G . -12
64 75.8 1
1-,
0
P*(224)18G"" 5 ' TGCTTGGGAACTTGAGGG* (20) G
42 56 7.0
,
P*(206)26A-2- 5'tgactgacCCCTATTCCCTGCTTAGG* (21)
A -2 80 - 30.4 .
P*(210)2.2A-2--- 5'tgacCCCTATTCCCTGCTTAGG* (22) A
-2 68 3.3 .
P*(212)20A-4 5 ' acCCCTAT TCCCTGCTTAGG k ( 2 3
) A -2 62 2.0
P*(214)18A-2 5 ' CCCTATTCCCTGCTTAGG* ( 2 4 )
A -2 56 0.0
'P*(206)26G-9 5'tgactgacCCCTATTCGCTGCTTAGG* (25)
__ C-+G -9 80 95.0 -
P*(210)220-9 = 5 1 tga
cCCCTATTCGCTGCTTAGG* ( 2 6 ) C--->G -9 ^ 68 88.1 '
_
P*(212)20G-9 5'acCCCTATTCGCTGCTTAGG* (27) C---
)-G- -9 62 49.5
-
=
'P*(214)18G-9 5'
CCCTATTCGCTGCTTAGG* (28) C-+G 56
4.7
P*(206)26Y1
51tgactgacCCTTATTCCCTGCTTAGG* (29) C-->T -15 78 89.0
P*(210)22T-15- 51t
gacCCTTATTCCCTGCTTAGG* (30) C--->T -15 66 47.8
13*(212)20T-15 5 '
acCCTTATTCCCTGCTTAGG* (31) C-+T -15 60 - 3.4
P*(214) 18T-15 5 I
CCTTATTCCCTGCT,TAGG* (32) C-->-T -15 54 0.0
- aP*(204)26Gu is a P* with a G dideoxynucleotide at the 31 terminus. "0"
means the allele-specific base is at the 3' terminus:, The -
first base at 5' terminus corresponds to nucleotide 204 in GenBank X55760. Its
length is 26 bases.
b The bold G or A are the G or A allele specific base and the underlined base
is designed mismatch.
The distance from the 3' terminus to the allele-specific base: "0 "----,- at
the 3' terminus, -3 = three bases from the 3' terminus.
d The Tin for oligonuckotide was estimated to be 4 C X (G + C) + 2 C X
(T + A) under condition of 1M NaCI . The length
of 0
each P* is'18 bases.
The noise rate of PAP (%) is defined as the relative yield of non-specific
allele product to specific allele product by the same
13", or as the relative yield of the designated. mutated P* to its native form
by using the same template. A specific signal is denoted
as <10% noise rate.
0
0
=
=
=
=
. = =
CA 02911712 2015-11-10
47
[02331 AmpliTaeS has two mutations compared with native Taq. One mutation in
the 5'
nuclease domain eliminates 5'-3' exonuclease activity and the second mutation
F667Y in the
active site (Innis and Gelfand, 1999). ThernaoSequenase has the same mutation
F667Y in the
active site but a deletion of the 5'-3' exonuclease domain (Tabor and
Richardson, 1995; Van der
s Horn et al, 1997). They do not distinguish between dNTP and ddNTP for
incorporation. The
pyrophosphorolysis of ddNIVIPs, which is the reverse reaction, is supposed to
be much higher
and less discriminated by these enzymes. Although either AmpliTtiqFS or
ThermoSequenase
DNA. polyno.erases used was formulated to contain a thermostable
pyrophosphatase
(manufacturers' instructions) that can hydrolyze PP i in the reaction so as to
decrease PAP
o efficiency, PAP was still amplified under our conditions. AmpliTaeS and
ThermoSequenase
DNA polymerases will work better in their pure form without the contaminated
pyrophosphatase:
The 3' specific subsequence of P*
[02341 Various Ps Were examined with different lengthS and mismatches using
AinplintqFS
is (Table 3). The effect of length and mismatch on PAP efficiency is
expressed as the relative yield
(%) between two P*3 of different lengths from the same template (Fig. 12),
which varied from
0.0% to 201.5% with each two to four less bases in length. The specificity of
PAP is also
affected by P47 length and mismatch (Table 3). 'The noise rate N is defined as
the relative yield
of the mismatch product to the match product, and a specific signal is scored
with <10% noise
20 rate. If the allele-specific base of P* was at the 3' terminus, only the
specific allele was
amplified and the specificity was not associated with P* length (Fig. 12A). If
the allele-specific
base was not at the 3' terminus of P*, the specificity was associated with P*
length. Any
non-3'-terminal mismatch in the 18-mer P*, which was up to 15 bases from the
3' terminus,
caused no amplification (Figs. 12-12E), but even two such mismatches in the 26-
mer
25 caused non-specific amplification.
= [02351 The 18-mers were further examined using "stacked" P*s, which span
the allele-specific
base at different positions (Fig. 13 and. Table 4). The noise rate eio varied
from 9.0% to 7.1%.
. The length of the 3' specific subsequence was 13 bases.
CA 02911712 2015-11-10
48
TABLE 4
PAP specificity with differently positioned. P*s
Nunn Se4ne (SEQ ID NO:)
Template
= 8.1 GACTGACCCCTATTCCCTGCTT-GGAACTTGAGGGGTGTC . . . 3' (33)
A
P*(21.2) 1.8dr¨ 5 'ACCCCTATTCCCTGCTTG* (16)
P*(212)18Ag 5 'ACCCCTATTCCCTGCTTA* (34)
P*(214)18A"` 5 'CCCTATTCCCTGCTTAGG* (24)
P*(218) 18G" 5' TTCCCTGCTTGGGAACT* (35)
P*(221) 18G" 5 CCCTGCTTGGGAACTTGA* (36)
P*(224) 18if" 5' TGCTTGGGAACTTGAGGG* (37)
Name 3' terminal Allele-specific Tm ( C) Noise rate (%)a
dideoxy base =
Type Dist Exponential Linear PAP template
ance PAP
P*(212)1Gu ddG- G 0 56 2.7 0.0
P*212)18.Au ddA A 0 54 3.8 1.1
- =
P*(214)18A ddG A -2 56 4.7 0.0
P*(218)18G-6- ddT G -6 54 0.0 0.0
. = P*(221)18G" ddA. G -9 56 1.7 1.7
P*(224)18G-" ddG G -12 56 7.1 0.6
a The amplificationfrom the G and A templates by PAP with two oligonucleotides
or linear PAP
. with
one P*. The nOise rate of PAP (%) is the relative yield of the non-specific
allele product to
= the specific allele product
[02361 Similar results were obtained by using 1:4s whfch match and mismatch
the G allele at
different positions (Table 5). The noise rate with one mismatch was various
from 0.8% to 5.6%.
The length of the 3' specific subsequence Was 16 bases. The noise rate with
two mismatches
was 0% (compare lane 2 with lanes 10-15 in Fig. 14).
=
.= . ,
,
. .
. 1
TABLE 5
.
. PAP specificity with differently
niinaatched P*s
.
.
_
Name The 3' terminal
iVrisinatcha Noise rate (%)'''
Sequence (SEQ ID NO;) dideoxy
. TB, (.c) *
Exponential Linear
Type Distance = Reg' PAP
__
14(212)18G 51ACCCCTATICCCTOCTrG* (16) = Dda-
- 56 -1 1.0 0.0
_
P*(212)18A,' -51.ACCCCTATTCCCTG4TTG** (38) DdG C-A. -3
54 1.3 0.0 -
._
P*(212)18G' = 51ACCCCTATTCCGTGCTTG* (39) DdG ' C-G -6
56 = 0.8 0.6 o
P*(212)18C'r VACCCCTA,TCCCCTGCTTG* (40) . DdG ' T-C -9
= 58 . 1,8 0.4 0
.
1..)
ko
_
P*(212)18G'" -51ACCCCGATTCCCTGCITG* (41) DdG T-G -12
58 .5.6 :1.7
1-,
-.3
-p- 1-,
P*(212)18Tb YACTCCIATTCCCTG e la G* (42) DdG C-T- -15
54 3.3 12 o "
tv
o
. a match or mismatch with the G allele.
-
b noise rate (%) is the relative yield between a mismatched P''' and
P*(212)18G with the G allele-specific template. 01
1
1-,
1-,
1
1-,
0
. . . .
=
=
CA 02911712 2015-11-10
[0237] Linear PAP was examined using only 18 met P*s and higher specificity
was observed
- with lower noise rate (Tables 4 and 5). Linear PAP takes a different
mechanistic pathway in
Which every non-specific product is generated from the starting template which
requires
mismatched pyrophosphorolysis with the 3' terminal mismatched P*, or both
mismatched
pyrophosphorolysis and mismatched extension with the non-3' terminal
mismatched P.
[0238] PASA was performed with 17-mer primers without adding a ddNMP at the 3'
terminus
(see Tables 4 and 5). A mismatched 17-mer primer strongly amplified a
nonspecific product
with 30% noise rate when the mismatch was as near as 6 bases to 3' terminus,
showing a much.
shorter 3' specific subsequence. Similar results were reported elsewhere
previously (Sarkar et al.,
10 1990)..
[0239] In "summary, P* (1-length) has two subsequences: a 3' specific
subsequence (n = the
number of bases of the 3' specific subsequence
determines the specificity, i.e.,within this
region any mismatch to its complementary strand of the template results in no
substantial
amplification; and a 5' enhancer subsequence (in = the number of bases of 5'
enhancer
15 subsequence 0) enhances the amplification efficiency. PAP specificity is
co-determined by the
base pairing specificity of the 3' specific subsequence, the
pyrophosphorolysis specificity and
the polymerization specificity. Thus, the base pairing specificity of the 3'
specific subasequence is
a mirrinrum requirement of the PAP specificity.
[0240] The length of the 3' specific subsequence of P* may be affected by the
sequence context
20 and size of the P*, the :type of the 3' terminal dideoxynucleotide, the
template sequence, the
DNA polymerase, other components like ion, and cycling conditions. When. the
template
contains repeated sequences > 1 or homogeneous polymer runs > 1, P* loses
specificity for
anchoring. The length of the 3' specific subsequence of P* may be affected by
the sequence
context and size of the p*, the type of the 3' terminal dideoxynudeotide, the
template sequence,
25 the DNA polymerase, other components like ion, and cycling conditions.
When the template
contains repeated sequences > 1 or homogeneous polymer runs > L P* loses
specificity for
anchoring.
Scanning or Resequen.cing for Unknown. Sequence Variants
[0241] The property of the 3' specific subsequence of P* can be applied to
scanning for
o unknown sequence variants or re-sequencing of predetermined sequences in
a parallel way. Each
nucleotide on. the complementary strand of the predetermined sequence is
queried by four
downstream P*s, such as 18-mers (Fig. 11), which have identical sequence
except that at the 3'
. terminus, either ddAlVfP, ddTMP, ddGIVIP or ddCMP corresponds to the wild-
type sequence and
CA 02911712 2015-11-10
51
the three possible single. base substitutions. The number of P*s scanning the
complementary
strand of X bases is multiplication of 4 and X, which is suitable for either
exponential or linear
PAP. The four downstream P*s can even be hinnobilized on a single dot when.
ddAMP, ddTMP,
ddGMP and ddC1VIP at the 3' termini are labeled differently for
differontiation, such as by four
S fluorescence dyes. The amplification signal can thus be represented by
intensity decrease of
each dye when ddINIMP is removed from P* by pyrophosphorolysis. One advantage
of linear
PAP is that the four ddNTPs can be used as substrates for single base
extensions, with are
labeled with different dyes for differentiation:
[0242] Briefly, if only all the P*s corresponding the wild-type sequence are
specifically
io
amplified, the wild-type sequence can be arranged in order by analyzing
overlaps. A 13* with a
single base substitution at the 3' terminus is amplified at the position of
hemi- or homo-point
mutations. The mutation also creates a "gap" of no PAP signal, which spans a
region of several
successive nucleotides. For single base substitution, the gap size (bases) + 1
= the length of the
3' specific subsequence.
= . is [0243] Furthermore, we can also scan the sense strand by
designing a second set of upstream
P*s. An unknown single .base substitution can be determined by combination of
the two sets of .
P*s, even in lieterozygotes. An unknown. small deletion and insertIon can be
detected and
localized. In order to identify a specific type of deletion or insertion, it
is possible to add
= corresponding P*s. For fingerprinting, which can provide information of
mutation position,
20 there iS a simple stacking way that the stacked region of each two
successive P*s < the 3'
specific subsequence on the array to reduce the number of P*s by up to n.
fold.
Determination of de novo DNA sequence
[0244] The concept of de novo DNA. sequencing by PAP makes use of all the
possible 3'
specific subsequences el)* to identify the presence of the 3' specific
subsequence in de novo
25 sequence. A complete set of the 3' specific subsequences of P* is e.
Each of the 3' specific
subsequence has a complete subset of the 5' enhancer subsequence of 4311. For
example, a
complete set of 16-mer as the 3' specific subsequence and 2-mer as the 5'
enhancer subsequence
can be indicated as (A, T; G, C)(A, T, G, N16 = 418.
[02451 Briefly, the procedure first determines the list of all the specific
PAP amplifications and
30 then reconstructs the unknown. DNA complementary sequence from this
list by ordering the 3'
specific subsequences with the given length by using the Watson-Crick pairing
rules.
[0246] The assembly process ls intenupted wherever a given 3' specific
subsequence of P* is
encountered two or more times. One of the factors influencing the maximum
sequencing length
CA 02911712 2015-11-10
= 52
is the length of the 3' specific subsequence. The length of a random sequence
that can be
reconstructed unambiguously by a complete set of the 3' specific subsequence
with the given
length is approximately the square root of the number of the 3' specific
sequence in the. complete
set with .:5.0% possibility that any given 3' specific subsequence is not
encountered -two or more
times. Octatners of the 3' specific subsequence, of which there are 65;536,
may be useful in the
range up to 200 bases. Decanucleotides, of which there are more than a
million, may analyze up
to a kilObase denovo sequence. 18 mer P*s containing 16 mer as the 3' specific
subsequence,
= which Complete set is 418 of P*s, may sequence maximum 77,332 bases.
= [02471 When there is neighbored known. sequence to deSign an opposite
oligonucleotide for
io PAP With two oligonucleotides. The maximum sequencing length is mainly
limited to the
opposite oligonucleotide, but not to the length of the 3' specific subsequence
of P*, termed
= Conditional de novo DNA sequencing.
Other Applications for PAP
[0248] For fingerprinting which compares two DNA sequences to see if they are
the same or
is different, there is a simple way to reduce the number of P*s by using
an incomplete set of the 3'
specific subsequences, By arranging them in. a particular Order, it is
possible to identify the
Chromosomal locations as well as sequences. Considering the 3 x 1.0? bp DNA in
human
geteme, PAP with two oligonacleotides is preferred over PAP with only one P*
to increase the
specificity.
4
20 [0249.1 To monitor gene expression profiling, where up to 6 x 10 to 105
transcripts are
expressed and details of the precise sequence are nnnecessary, PAP with only
one. P* can be
applied and a set of P* which identify nnique motifs in genes can be designed
with a total length
of up to 22- mer. Between each two Ps, there is at least a sequence difference
at the 3'. terminus
or > 2 sequence differences at the non-3' terminus.
25 Comparison with. Sequence by Hybridization
[0250] In SBH by using oligbitatleotide; the DNA sequence is deteitained- by
the hybridization
and assembly of positively hybridizing probes through overlapping portions. It
has been known
= for a long time that a single oligonucleotide hybridiatioii on. a
immobilized sample can be very
specific in optimal hyblidization ahd washing conditions (Wallace et al.,
1979), -thus it is. =
30 possible to discriminate perfect hybrids- from Cones containing a
single internal mismatch. The
oligonucleotideS- in array ate 11-20 nucleotides in length and have 7-9 bases
specific region in-
the middle, the non-specifie signal is generated by mismatched hybridization.
Under standard
hybridization and washing -coriditiotA the duplex stability between match and
rniSinatch is also
- =
CA 02911712 2015-11-10
=
53
affected by the terminal. mismatch and the flanking sequence (Drmana.c et al.,
1989; Khrapke et
al.; 1989; Ginot, 1997).
[0251] SHB. can be modified with enzymes in several ways (Miyada and Wallace,
1987;
Southern, 1996): Primer extension by DNA polymerase incorporates bases one at
a time only if
s
they match the complement strand. Ligase has similar requirements: twO
oligonacleotides can be
.
joined enzymatically provided they both are complementary to the template at
the position of
joining.
[02521 Figs. 11A-11B show the enhancement of PAP effidericy. Fig. 11A. PAP is
amplified
with two oligonucleoticleS P*. and U from duplex TU:UT template. Each of the
four P*s has a
ddA, dd.T, dd0 and ddC at the 3' terminus. The 3', terminal base is either
specific to the
complementary strand of the G or A alleles, or not matched: Fig. 11B.
Autoradiogram of PAP
from the GIG, .A/A and G/A genotypes of the human dopamine receptor gene. Tho
radioactively
= labeled specific products of 461 bases (dupieX PU:UP and excess antisense
strand UP) are
produced. Other side products UT and UT:TU are indicated. Note that TU:UT
derives from
annealing of excess radioactiVoly labeled UT with non-radioactively labeled.
TU original
template.
[02531 Figs. .12A-12E show the effect of* length and mismatch on PAP
effiCiency. PAP was
amplified with P* and U oligonucleotide (see Table 3): In each of Figs. 12A-
12E, P*s have the .
sample 3' termini but are different in. length Fig. 12A. = In lanes 1-4, the
P*s niatched. and
amplified the G allele. In lanes 5-8, the P*s mismatched at the 3' termini but
amplified the A
allele. Fig. 12B. In lanes 942, the P*s matched and amplified the G allele: In
lanes 13-16, the
P*s, mismatched. at 42 bases to the 3' termini but arciplified the A allele. =
Fig. 12C.. In lanes
1.7-20, the P*s matched and amplified the A allele. In lanes 21-24, the P*s
mismatched at -2,
.
bases to the 3' tennini but amplified the G allele.. Fig: 12D.- In lanes-25-
28, the P*s mismatched
. 25
at -9 bases to the. 3' termith. but amplified the A allele. Fig. 12E. In lanes
29-32, the P*s
= mismatched at -15 bases to the termini but amplified the A allele. The
length effect is
indicated as..the yield ratio in, one lane (4) tO the previous lane (Lõ..1)..
The length effect waS-not
= , she-Wain lanes 5-43 bocanto the. signa1:11re at or close to. the
background,
[02541 Fig. 13 shows PAP specificity with differently positioned P*s. PAP was
amplified. with
a pi= and U oligonucleotide (see Table 4). The P* matched to and amplified the
G allele in lanes
2-7, but mismatched to and amplified the A allele in lanes 9-15. Lanes 1 and 9
were PCR control
with D1(212)17 mer and U. Lanes 8 and 16 were extension control with only U.
=
CA 02911712 2015-11-10
54
[0255] Fig. 14 shows PAP specificity with differently mismatched 134's. PAP
was amplified
with a P* and U oligonudeotide (see Table 5). In lanes 2-7, the P* amplified
the G allele with
match or one mismatch: In lanes 9-15, the P* amplified the A with one or two
Mismatches.
Lanes 1 and 9 were PCR control with D1(212)17 mei: and U. Lanes 8 and 16 \Veit
extension
control with only U.
EXAMPLE 3
PAP Amplification From Genomic DNA
[0256] This example illustrates PAP amplification directly from genoniic DNA.
The
oligonucleatides used in this example are listed below. Lane numbers refer to
lanes in Fig. IS.
[0257] The downstream oligonucleotides in 0.1 AM concentration are:
_ Lane 1: Di(204)25D 5' TCTGACTGACCCCTATTCCCTGCTT 3' (SEQ NO:43) =
Lane 2: P*(206)24A 5' TGACTGACCCCTATTCCCTGCTTA* 3' (A allele
specific; =
SEQ NO:44)
Lane 3: P*(204)26G9 5' TCTGACTGACCCCTATTCCCTGCTTG* 3' (G allele
specific; SEQ NO:45).
Lane 4: P*(206)24G-2 = 5' ACTGACCCCTATTCCCTGCTTGGG* 3' (G allele specific;
= SEQ ID NO:46)
Lane 5: P*(228)26A-24 5' TAGGAACTTGGGGGGTGTCAGAGCCC*. 3' (A allele
specific; SEQ ID NO:47)
= [0258] The opposite upstream oligonucleotide in 0.1 jaM concentration is:
D1(420)24U
5' ACGGCAGCACAGACCAGCGTGTTC 3' (SEQ ID NO:48), which was paired with each
- downstream oligonucleotide. See Footnotes of Table 3 for detail.
= [02591 The other components were the same as in Example 2, except for the
following: 0.5 U of
each of AmpliTaqFS and Tag DNA polymerases, and 100 ng of heterozygous GIA.
allelic
genamic DNA were used per 25 ul reaction by using 30 cycles.
[0260] The PAP product size range from 193bp to 218 bp. One double stranded
and one single
stranded product was observed on the gel, indicating the exhaust of PP i
hydrolyzed by the
contaminated thermostable pyrophosphatase.
CA 02911712 2015-11-10
=
=
EXAMPLE 4
=
Comparison of Specificity of LM-PCR and LM-PAP
[0261] The LM-PCR protocol includes primer extension, linker ligation, PCR
amplification, and
directed labeling in the human dopamine DI receptor gene model system: (Fig.
16). LM-PCR
5 was performed with the addition by terminal deoxynucleotidyl transferas8
(TdT) (this protOcolis =
',Mown as TD-PCR) .on UV-treated genoinic DNA samples essentially as described
(Pfeifer et
al., 1999), except that VentR (exo-) DNA poIymerase was used in the first 10
cycles of Primer
eXtension (P1 primer 5' TTGCCACTCAAGCGGTCCTCTCAT 3 (SEQ ID NO:49)).
Temperature cycles were 1 Min at 95 C, 3 Min. at 63 C, and 3 min at 72 C.
To enhanoe the
.io
signal, terminal transfetase was added to the protocol, and this variation of
LM-PCR is called
TD-PCR. Dynabeads were used to enrich target DNA molecules before terminal
deoxyriucleotidyl transferase (TdT) tailing. PCR was performed using Expand
Long Template
PCR System 3 (Bm-,B) aS described by the manufactiter (P2 primer 5'
GAAGCAATCTGGCT
GTGCAA_AGTC 3' (SEQ ID NO:50)), The PCR products were purified using Q1Aquick
PCR
is Purification Kit (Q1AGEN) before performing the direct labeling. A
portion of the cleaned PCR
product was used for direct labeling with Amplinq DNA Polyinerase (Perkin-
Elmer) with 32P-
labeled plinierst
P3A: (5' TCTGACTGACCCCTATTCCCTGCTTA 3' (SEQ
NO:51; the 3' terminal
deoxynucleotide is A allele specific) and =
20 P3G: (5' TCTGACTGACCCCTA.TTCCCTGCTTG 3' (SEQ ID NO:52; the 3' terminal
deoxynucleotide i G allele specific). =
[02621 LM-PAP was performed as allele-specific PCR except for the direct
labeling step by
PAP (Fig. 16A), The purified PCR product was used for direct labeling with
3211 labeled primers:
P3A: 5' TCTGACTGACCCCTATTCCCTGCTTA* 3' (SEQ ID NO:53; the 3' terminal
25 deoxynucleotide is A allele specific) and
P3G: 5' TCTGACTGACCCCTATTCCCTGCTTG* 3' (SEQ ID NO:54; the 3' terminal
deoxynucleotide is G allele specific)
using PAP reaction conditions in. a 10 ill Volume (50 niM KC1, 10 niM Tris/HC1
(pH 7.6),1.5
niM MgC12, 100 11M of each dNTP, 0.1 tiM P*, 300 i.t.M NA4PPi, 2% DMSO,
0.25U.dath of
30 AinialircieS and AMpliTaq DNA Polynierases (Perkin-Elmer). The
cycling conditions were
940 C, 10 sec.; 60 C, I. mitt. and 72 C, 2 min. Mt a total of 8 or 16
cycles. LM-PAP was
dt aniatically more specific than LM-PCR. The initial data with the &par-line
D1 gene shows a
lower background with LM-PAP than with the identical unblocked oligonucleotide
with LM-
=
CA 02911712 2015-11-10
56
PCR. Also, LM-PAP can be performed with the PGK gene, a gene with a very high
GC rich
region (70%) (Fig. 16B).
10263j Fig, 16A shows a UV footprinting of the dopaniine D1 receptor 'gene
with a eon parison
of allele-specific LM-PA,P and allele-specific LM-PCR. A direct comparison of
LM-PAP with a
s P* and LM-PCR with an. unblocked primer of identical sequence shows
that two alleles can be
distinguished with LM-PAP, but not with LM-PCR. Both methods were performed on
HF46
DNA that was untreated (C), in vitro treated (T) or in vivo treated (V) with
UV. The direct
labeling reaction using PAP conditions (lanes 7-18) with 32P labeled primers
P3A* (lanes 7-9
and 13-15) and P3G* (lanes 10-12 and 16-18) was done with AmpliTaqFS and
AmpliTaq for 8
and 16 cycles. For LM-PCR the direct labeling reaction was done with AmpliTaq
(lanes 1:6)
and 32P-labeled primers P3A (lanes 1-3) and P30 (lanes 4-6) for 8 cycles:
Allelic primers P*s,
P3A* and P3G* for LM-PAP clearly distinguish the two alleles, while unblocked
allelic primers
of identical sequence, P3A and P3G; were unable to distinguish the alleles by
LM-PCR.
[0264] Fig: 16B shows a UV footprinting of the pgK gene. The LM-PAP procedure
for PGK
was essentially the same as for the dopamine Di receptor except that Pfu Turbo
DNA
polYmerase was used in. the primer extension, as well as. 7-deaza-dGTP/dGTP in
a 3:1 ratio.
Temperature cycles were 950 1 min, 60 2 min., and 76 3 min The PCR step was
performed.
using Vent (exo-) DNA Polymerase at 97 1 min., 60 2 nun.,. 76 3 min. also
with deaza dGTP.
The purified PCR products were used for direct labeling with the 32P P3G* and
P3C* primers
lasing PAP reaction conditions in. a 25 pi volume (50 rolVIKCL, 20 mM Hepes,
pH 6.95, 10 mM
(NH4)2SO4, 1.5 mM MgC12, 40 pM dNTP, 150 uM Na4PPi., 4% DMSO, and 1 unit of
AinpliTaq
FS DNA.Polymerase. The conditions for cycling were 94 15 sec., 60 30 sec.,
and 72 1 min:
for 10 cycles.
EXAMPLE 5
Optimization of PAP-A to Detect a Mutation in 1 of 104-105 Templates
[0265] One ug of lambda phage DNA contains 2 x 101 copies of template. The
specificity of
PAP is determined by mixing one part mutant lac templates with 104 to 105
parts control DNA
templates, e.g., wild-type lad. The specificity of PAP-A is a function of the
error rate of the
polymerase, the purity of P* (<2 x le by current purification protocol) and
the potential for
damage of the DNA template in the extraction process. The yield and
specificity of PAP is -
optiroind by testing enzyme type and concentration and the concentrations of
other
=
=
CA 02911712 2015-11-10
57
=
components, such as dNTP, PP, Mgf+ or Mn. Hotstart PAP using antibody-
activated enzyme,
such as DNA polymerase,, at room temperature can be used to eliminate spurious
amplifications.
[0266] Wild-type and mutant lambda phage DNA, which are used. in the
laboratory as a model
system to study Spontaneous mutation in mammals', are prepared from infected
E. colt SCS-8
. 5
cells (Nishino et al., 1996): The lambda phage is graft under high fidelity
cenditions and DNA
is isolated: with. care under conditions with low rateS of DNA damage
(Stratagene manual)
(Nishind et al., 1996; Hill et al., 1999).
[0267] The mutants include one example of each of the two types of
transitions, the four types
of transversions and a= one-base nucleotide deletion. P*s specific for each,
of the mutations is =
synthesized: These DNA. templates. are used for reconstruction experiments in
which mutated
DNA is serially diluted into wild-type DNA. The spiked samples are used to
optimize PAP-A.
The most robust polyrnerases are chosen_ = based on yield, and specificity
using TaqFS,
ThermoSequenase, and SequiThenn. Excel II (Epicentre). Other components of
the. reaction are
optimized systematically; including thermocycling = parameters 5
ofigonucleotide. lengthy and
is
reagent concentrations of PPi, dNTP and Mg-3-4- or Mn. Quantitative detection
of the yield of
PAP product is achieved with antoradiography or fluorescence on a SSCP gel.
Theft data aids in.
= the optimization of PAP-R and LI\CPAP (below). The optimization of these
various parameters
result in a specificity of 1 part in 104-105.
= [0268] The Optimized conditions are also tested for detecting mutations
in the human factor IX
gone by mixing human mutant genomic DNA templates with up= to 104 wild-type
templates. As
= with the lambda experiment, exponential PAP is performed with
appropriately designed
= oligonucleotides (using Oligo5 softWare) for .40 cycles and. strong
signal is achieved by
antoracliography or by fluorescence detection.
=
. EXAMPLE 6
. Optimization of PAP-R
[02691 In a model System, mismatches along the length. of p.* intribitiq
activation, even when
the II *Smatch is two IiiideotiddS. from. the 5' end (Fig,14), An additional
set of 18. meta of P*s, -
whose 5 termini were displaced 2, 6, 9, and 12 nucleotides downstream, also
showed inhibition
of activation (Fig. 13). In addition, 20 and 22 mers also show inhibition with
single nucleotide
mismatches (Fig. 12). To extend these mdings and to lay the foundation for a
robust method of
teg6qtencing,.thd relationship between the location of single base mismatches
and activation of
P4's 15 analyzed further.. . =
CA 02911712 2015-11-10
58
[0270] The factor IX gene is used as a model system. because more than 1,000
DNA samples
from, hemophilia patients and family members have been ascertained from
previous work on the
niolecular epidemiology of gennline mutations in humans (Soinrner, 1995;
Ketterling et al.;
1999). TWO 20-nttclecitide regions of exon B and exon H in the human factor IX
gene are used
as model sygten18. The region of exon B is designed from nucleotides 6460 to
6479 (5'
CGAGAAGTTTTTGAAAACAC 3'.(SEQ JD NO:55;Yoshitake et at, 1985), within which
eight
different single base mutations are available. The region of exon H is from
nucleotides 30845 to
30864 (5' GAACATACAGAGCAAAAGCG 3' (SEQ ID NO:56), within which seven mutations
at different positions are available: P*s identical to wild-type regions B and
H will be
o synthesized. Identical P*s are synthesized, with the exception of a
single nucleotide mismatch:
= [02711 The wild-type factor IX sequence is used in the initial studies. A
few P*s that match the
wild-type sequence or that miSinatch at selected:sites within the 5' third of
the oligonucleotide
sequence are helpful in performing pilot experiments to assess the optimal
length of the
= oligonucleotide. The effects of polymerases and. reaction conditions can
be assessed.
[02721 FrOm preliminary data, it appears that 18 mers or larger may be an
optimal size. It is
= also possible that 25 mers. or even 30 DION may be optimal. For. the
present example, it is
assmiled that 20 mers are an optimal size. Wild type P* and twenty P*S with
one of the possible
single base mismatches at each nucleotide of the position region of exon B are
synthesized.
= Eight of these P* are a perfect match to a Mutation in a patient with
hemophilia B. As positive
=
0011trOlg, it is shown that these P*s activate efficiently when the
appropriate mutated DNA
sample is used. Exponential PAP and linear PAP are performed and the noise
rate is determined.
The noise tate for linear PAP is generally lower and is used.
[0273] To confinn preliminary data in another sequence context; a similar
experiment is
performed in exon H. The seven mutations in that region of exon H are analyzed
in a blinded
*manner to determine if the precise match is detected. The effects of the
position of the mismatch
or the type of mismatch. on P* activation is determined. The effects of
different polymerases,
reaction temperattre, and other reaction conditions can also be determined:
Another set of 20
. P*s provides additional data from misniatcheS 12.20 nucleotides froth.
the 3' terminus:
EXAMPLE 8
Optimization of LM-PAP
[02741 The lairnan dopamine Di receptor gene and the mouse Peci gene are used
as model
= systems to compare the analysis of chromatin structure when LM-PAP or LM-
PCR is utilized.
CA 02911712 2015-11-10
59
The dopamine DI receptor gene has. been described above: X chromosome
inactivation occurs at
an early embryonic stage. Since the two alleles in female cells maintain a
different expression
status, this is an advantageous system for studies of gene regulation. Pgld is
an X-linked
housekeeping gene encoding phosphoglycerate kin ase (PGK). PGK is an
important enzyme in
glycolysis and the gene is expected . to be active all the time except. in the
inactive X
chromosome (Xi) of female somatic cells and in male germ cells.
[02751 The preliminary data shows a dramatic enhancement of specificity.with
LM-PAP relative
to LM-PCR in the dopamine DI receptor gene, a gene not previously analyzed for
chromatin
structure (Fig. 16A). In this example, LM-PAP and LM-PCR are performed. Three
sets of
oligonucleotides that generated LM-PCR profiles and seven sets of primers that
generated LM-
PCR profiles with unacceptable background in the Pgld (and other X-chromosomal
genes) are
used = to compare LM-PAP with LM-PCR. Deoxy-terminating and dideoxy-
tenninating
oligonucleotides of identical sequence are utilized to perfonn LM-PAP and LM-
PCR,
respectively. The level of signal relative to background is also quantitated
by a Phospholmager.
The average signal-to-noise ratio. is determined. Optimization data derived
from analyses with
PAP-A and PAP-R are also useful. in the LM-PAP protocol: LM-PAP is optimized
for the two
regions to determine if the signal-to-noise ratio can be reduced further.
3.
EXAMPLE 8
Optimization of Allele-Specific LM-PAP
[02761 Polymorphic sites of pgIclo .and lb gene in both coding and non-coding
regions have
been reported (Boer et al., 1990)õ These are used to design the allele-
specific P. One allele-
specific oligonucleotide is chosen prospectively from the Pea gene and one is
chosen
prospectively from. the d.opam-hie D1 receptor gene. Blocked and unblocked
oligonucleotides of .
identical sequence are synthesized and allele-specific LM-PAP and LM-PCR are
performed;
respectively. The signal to noise ratio is quantitated and compared,
EXAMPLE 9
PAP-R on. a Microarray
[02771 The initial experiment will focus on the two 20 nucleotide regions of
exons B and H. as
described above. The experimental design of PAP-R is similar to the
experiments described
above, except for digital light-direct synthesis of P* oligonucleotides on a
microarray, e.g., with
the Geniom instrument. A total of 160 oligonucleotides are synthesized
complementary to
CA 02911712 2015-11-10
= 60
=
wild-type and to all the single base mismatches for 20 bp regions of eX011S B
and H of the factor
IX gene. As a positive control; 160 oligouncleotides, each out of registered
by one nucleotide,
are synthesized to match exactly an adjacent 160 bp region Of the faCtor DC
gene. anomie DNA
frorn wild-type and 'mutant .saMples is amplified; annealed to the
oligonncleotides and primer
eXtension will be perfornied with a fluorescent dideoxy terminator. The
protocol is optimized for
the solid support. Adjustment of primer length, enzyme utilized and reaction
conditions is
performed such that most, if not all, of the oligonucleotides that mismatch
the two 20 bp
nucleotide regions of factor IX generate little if any signal, while most of
the 160 control
oligonucleotides generate a strong signal.
io
[0278] One strategy for resequencing is shown. in Figs. 3 and 4. Each
nucleotide in the
complementary strand of the predetermined sequence is queried by four
downstream P*s, such
'as 20 mers, which have identical sequence eXcept for the 3' terminus, which
is either ddA, ddT,
ddG or ddC. For a 1 kb segment, 4,000 P*S are needed in the dow:astream
direction.. In the
second set of experiments, exons B and.H of the factor DC gene are
resequenced. Samples from:
more than 200 patients With different mutations in these regions are available
for. analysis. False
== positives and false negatives are assessed by blinded. analysis.
Heterozygous female samples are
= available for many of the mutations. For the remaining male patient
samples one to one mixing
experiments with, wild-type or a second mutated sample generates the
equivalent of
heterozygotes or compound heterozygotes, respectively. Subsequently, all the
regions ,of likely
functional significance (the putative promoter region, the coding regions, and
the splice
kinction. ) are resequenced (2.2' kb). Since more than 600 independent
mutations are available, it
= is possible to determine whether mere than 99% of all Sequence changes
are identified (the
sequence changes ill these samples have been determhied by direct sequencing
Or the course
of a decade).
[02101 A F*.with a single base substitution at the 3' terminus generates a
signal at the position
of hemizygous or homozygous point mutations: The mutation also createS a "gap"
of no PAP
signal, which spans a region of several successive nucleotides. When a single
base substitution
occurs, the gap, size (nucleotides) + 1 = the length of the 3' specific
subsequence (Figs. 3 and 4).
[0280] To analyze samples with higher G+C content (55%), mutations in the lac/
gene are
utilized : These' Mutations from the Big Blue Transgenie Molise Mutation
Detection System,
have. the pcitential to facilitate the definition of a strategy that detects
more that 99.9% of
nitrtatiotts, since more than 6,000 mutations are available in this system.
The relestantre are.
analyzed with the help of robotic devices. In addition, hundreds of mutations
o 'polyMorphisms
=
=
CA 02911712 2015-11-10
61
are available for aralysis in other genes with (3+C contents of 30-75%. The
dystrophin gene is
particularly amenable to testing perforniance under conditions in which
naegabases of sequence
require scanning. In this gene in which 90 segments are amplified by a robotic
device, virtually
all sequence variants have been defined by DOVAM-S followed by DNA sequencing.
This is
s advantageous because many molecular epidemiological and molecular
diagnostic applications
benefit from resequencing that detects virtually 100% of the mutations.
= EXAMPLE 1.0
PAP Amplification Directly from Haman and Mouse anomie DNAs
io
[02811 PAP was performed with each of two P*s, Pl* (SEQ ID NO:45, G allele
specific)) or
P2* (SEQ ED NO:47, A allele specific) and an upstream unblocked primer (U;
(SEQ ID NO:48)
to amplify 180-bp segments of the D1 dopamine gene. The P* are 26-mers with
ddC and ddG at
the 3' temaini. 100 ng of lun-na3i genomic DNA was amplified for 35 cycles
followed by 2% gel
,
electrophoresis. The PM reaction mixture contained a total volume of 25 ill:
50 mM KC', 20
345
InM HEPES/NaOH (p11 6.9 at 25 C), 10 tn.M (NH4)2804, 1.5 m114 MgC12, 40 uM
each of the
= four dNTPs (dATP, dTTP,,dGTP, dCTP),. 0.1 tM U, 150 uM NaRpi, 2% DMSO,
0.5.,U of
2.0
AmpliTagPS Polymerase (PE Applied Biosystems),.. 0.5 U Tag polymerase and 100
ng of human
4 .2
genoinic, DNA. The cycling conditions were 94 C for 15 sec, 65 C for 30 sec
and 72 C for 1
min.- Fig. 17A shows the results for PAP amplification of the DI 41opainine.
gene. In lanes 2 and
20 5, P1* is specific for the A allele template at 24 nucleotides from
the 3' terminus, so there is
little or no discritnination between the G/G and A/A genotypes.. In lanes 3
and 6, P2* is specific
for the A allele template at 2 nucleotides from the 3' terminus, so there is
specific amplification
of the A/A genotype: Lanes 1 and 5 are PCR controls. Lanes 4 and 8 are
negative controls
without P. Lane M is 120 ng cpx DNA/HAM-Sr marker.
25
[0282] Three Bi-PAP assays were tested directly from. mouse genomic DNA. Bi-
PAP was
performed with two.P*s containing a clideoxynucleotide blocker at the 3'
terminus to amplify an
80-bp segment of the lad gene: The P*S are specific to the Wild-type template
and are 40-42
nucleotides long. In each of the three Bi-PAP assays, two opposite P* with one
nucleotide
.
overlap at their 3' termini were used to amplify 400 copies of the lad gene
using 35 cycles..
30 The sequences of the P*s are as fellows:
5' GAAGCGGCGTCGAAGCCTG-TAAAGCGGCGGTGCACAATCT* 3' (SEQ ID
NO:67) and 5' GCGGATAGTTAATGATCAGCCCACTGACGCGTTGCGCGAGAA.* 3'
(SEQ ID NO:68) in lanes 1. and 2;
=
CA 02911712 2015-11-10
69
=
. .
5' GATGGCGGAGCTGAATTACATTCCC.AACCGCGTGGCACAA* 3' (SEQ ID
NO:69 and 5.' GGCAA.CGCCAATCAGCAACGACTGTTTGCCCGCCAGTTGT* (SEQ ID
NO:70) in lanes a and 4; and
5' TACATTCCCAACCGCGTGGCACAACAACTGGCGGGCAAAC*- 3' (SEQ ID
NO:t71) and 5' GGGCCAG-ACTGGAGGTGGCAACGCCAATCAGCAACGACTG* (SEQ
ED NO:72) in_ lanes 5 and 6:
[02831 The PAP reaction mixture contained a total volume of 25 p.1: 50 niM
KC1, 20 mM
'HEPES/NaOH (pH 6.9 at 25 C), 10 mM (NH4)2SO4, 1.5 rriM MgC12, 40 IrM each of
the four
dNTPs (dA11), dTTP, dGTP, dCTP), 0.1 !IM U, 150 p.M NaiPpi, 4% DMSO, 1.0 U of
lo
AmpliTaqFS polynierase (PE Applied Biosystems) and 400 copies of mouse genomic
DNA,
The cycling conditions were 94 C for 15 sec, '65 C for 30 sec and 72 C for
1. min. The
- unincorporated P*s were separated well fromthe
product on 2% agarose gel. Na diner
was seen. Fig. 17B shows the results for these Bi-PAP assays. In lanes 1, 3
and 5, the wild-typo
templates are amplified. Lanes 2, 4 and 6 are negative controls without mouse
genomic DNA.
15 [0284] Three PAP assays directly amplified .18043p. segment . of the
D1 receptor gene from
human genomic DNA with strong signals of PAP products:, The allele-specificity
of26-mer P*
remains when the mismatch_ is at 2 nucleotides from the terminus, but the
allele-specificity is
lost when the mismatch is at 24 nucleotides from the 3' terminus: Three Bi-PAP
assays directly
amplified as low as four hundred copies of the lac/ gene from mouse genomic
DNA. The P*
20 oligorrticleotides have different deoxynucleotides blocked at the 3'
termini's. and all can be
efficiently activated. Addition of extra human. DNA did not affect the
amplification of the lad =
gene in "mouse genornic DNA. The product of Bi-PAP was easily distinguished
from
"imincorporated P*s. P* does not form dimmers because P* needs long and.
perfectly matched
regions at the 3' terminus for activation.
25
=
EXAMPLE 11
PAP with Acyciomicleotides and Various Polymerases
X Phage DNA Template
[0285] The Wild-type X phage-DNA template that Contains an inserted wild-type
Lc/ gene of E.
30
coil (Kohler et at., 1991) was purchased from Stratagene. The mutant X phage
DNA template
was prepared from 7c phage plaques transforined into SCS-8 E. coil della
according to Maniatis,
et at. (im). It contained a T to G mutation at nucleotide 369 in the lad'
gene. The slnount of X
phage DNA was determined by UV absorbance at 260 urn. =
=
CA 02911712 2015-11-10
63 =
Synthesis of P* by Adding Acyclonucleotide or a Dideoxynucleotide at the 3'
Terminus
[0286] The 3' terminal acyclonucleotide or 3 terminal dideoxynucleotide was
added to a
deoxynucleotide oligonucleotide by terminal transferase. The mixture contained
a total volume
of 25
100 I/1M potassium cacodylate (pH 7.2), 2.0 mM CoCl2, 0.2 mM DTT, 2 n_M of the
oligonucleotide, 2.4 mIVI acycloNTP (the molar ratio of the 3'-OH terminus to
acycloNTP was
1:30) (New England BioLabs), or 2.4 mM 2',31-d.dNTP (the molar ratio of the 3'-
01-1 terminus to
ddNTP was 1:30)(Roche), 100 U of terminal transferase (Invitrogen). The
reaction was
incubated at 37 C for 6 hr and then stopped by adding EDTA to a 5 mM final
concentration.
After desalting using a Centri-spin-2 column (Princeton Separations), P* was
purified by
preparative 7 M urea/18% polyacrylam i de gel electrophoresis with 30 mM
triethanolamine/tricine buffer (pH 7.9 at 25 C) (Maniatis, et al., 1982; Liu,
et al., 1999b). The
amount of recovered P* was determined by UV absorbance at 260 inn.
[02871 Since small amounts of unterminated oligonucleotide would result in
unexpected PCR
,
amplification, the purity of P* was tested by the absence of PCR product at pH
8.3 in which
Is
pyrophosphorolysis is inhibited. It is estimated that more than 99.99% of P*
contained an
acyclonucleotide or a dideoxynucleotide at the 3' terminus.
r:.. PAP Amplification
[02881 PAP was examined with P*1 and 01, with P*2 and. 02, and with P*1 and
P2*
= respectively (Fig. I 8A and Table 6). The P*s were 30 or 35 nucleotide
long and contained an
acyclonucleotide or a dideoxynucleotide at the 3' terminus.
=
=
TABLE 6
List of Oligonucleotides
PAP Amplification
(allele)
Desig. Name' - Sequence (ID NO:)
3' Terminal G: C T:A
P*1 P*(340)30D CGAAGCCTGTAAAGCGGCGGTGCACAATCG* (57) acycloGIVIP .
Yes No
or ddGMP
01 -0(502)25U ACTGTTGATGGGTGTCTGGTCAGAG (58) dGMP
P*2 P *(398)3 OU TGATCAGCCCACTGACGCGTTGCGCGAGAC* (59) acycloCMP
Yes No
or dddMP 0
02 0(190)21D A.CAACTGGCGGGCAAACAGTC (60) dalIP
1.)
a The position of the first nucleotide of the transcript in the lad- gene of
E. coli is assigned the nucleotide position 1
1.)
(Farabaugh, 1978). As an example for P*1, P* = pyrophosphorolysis activatable
bligonucleotide, it may be a 3 terminal 1.)
0
acydonucleotide blocked P* or a 3' terminal dideoxynucleotide blocked P*.
(340)30D = 5' end of the P'' begins at 340,
the length is 30 nucleotides and the direction is downstream (i.e., in the
direction of transcription). The precise sizes and
Locations of the amplified fragment can be obtained from the informative
names. The 30-mer P*s are indicated above.
The 35-mer Pss are 3' co,terminal with the 30-xner P*s and 5 nucleotides
longer at their 5' termini. 0
, =
, .
CA 02911712 2015-11-10
[0289] The PAP reaction mixture with AmpliTaqFS DNA polyinerase contained a
total volume
of 25 pi: 50 inN1 KCI, 20 m_M BEPES/NaOH (PH 6.9 at 25 C), 10 inM (NH4)2SO4,
1.5 niM
= MgC12, 50 jiM each of the four dl\ITPs (dATP, dTTP, dGTP and dCTP), 0.1
plW of each.
oligonucleotide, 150 1.1M Na4PPi, 4% DMSO, 1 U of AmpHMO'S' DNA polyraerase
(PE-
s Applied Biosysterns), 0.1 ng
phage DNA teinplate. The eyeling conditions were 92 C
- =
. fOr 10 see, 65 C for 30 sec, and 72 C for I Min for a total of 30 cycles. A
denaturing step of 92
= C for 1 min was added before the first cyele.
.
[0290] The PAP reaction mixture with Vent (exo..) or Pfu (exo-) contained a
total volmte of
25
10 mM KC1, 20 .n1M EEPES/NaOH (pH 7.19 at 25 C), 10 m.M (NH4)2S03, 1.2 inM
o
MgCl, 50 pM each of the four dNTPS (dATP, dTTP, dGTP and d.CTP), 0.1 pM= of
each
oligonucleotide, 150 1.1A4 Na4PP1, 4% DMSO, 1 U of Vent (exo-) DNA polymerase
(New
England BioLabs) or Pfu (exo-) DNA polyinerase (Stratagene), 0.1 ng = of the.2
phage DNA
template. The cycling conditions were 94 C for 15 sec, 60 C for 30 sec, and
72 C for 1 min
for a total of 30 cycles. A.denaturing step of 94 C for 1 min was added
before the fttst cycle.
15
[02911 The product was electrophoresed through a standard 2% agarose gel. The
gel was stained
with ethidium bromide for UV: photography by a CCD camera (Bio-Rad Gel Doc
1000).
[0292] As shown above, TagFS, a genetically engineered DNA polyinerase (Innis
and Gelfand,
1999), greatly improved the efficiency of PAP. 3' terminal dideoxyMicleotide
blocked P*s can.
be activated by pyrophosphorolysis to remove the 3' terminal dideoXynteleotide
in the presence
20 = of pyrophosphate (IT) and the complementary strand of the allelic
template. Then the actiVated
p* can be extended by DNA polymerization.
[0293] PAP was perfolined= with 3' acyclonucleotide blocked P*s by nail-1g X
phage DNA
containing the lad- gene as model system. P*1 and P*2 are downstream and
npstreant blocked
oligonucleotides, respectively, for the same mutation (Fig. 18A and Table 6):
The P*1 and P*2
25 have an acycloGM2 and acycloCIAll at their 3' termini, respectively.
Amplification products
were absent without pyrophosphate added at pH 8.3 where pyrophosphorolysis is
inhibited,
showing that P*1 and P*2 were not directly extendible.
[0294]. P*1 and P*2 are specific to the mutated template but mismatch to the
wild-type template
at their 3' termini. The mutated template was amplified efficiently by PAP
with one
30 acyclonucleotide blocked P* and one opposing unblocked
oligonucleotide and by PAP with tWO
opposing 3' terminal acyclonucleotide blocked P*s (lanes I. and 2 in Fig.
18B), with two
opposing acyclonucleotide blocked P*s (a special form of PAP where the tWO
Opposing P*s are
.
overlapped at their 3' termini by one nucleotide),(Land 3 in Fig. 1813):
However, no product was
CA 02911712 2015-11-10
=
66 -
generated from the wild-type template because of the misniatch at the 3'
terminus, showing the
= specificity (lanes 5-7 in. Fig. I 813). PAP with the 3' dideoxynucleotide
blocked P* showed
similar results (lanes 9-16 in Fig. 18B). Direct sequencing analysis confirmed
the correct
sequence of the amplified product. The effect of P* length Was also tested.
Similar results were
s
obtained with 35-m.er P*s that are co-terminal with the 30-mer P*8 and five
nucleotides longer at
"
their 5' termini (Fig: 18C), Other P*s specific for the wild-type sequence at
the 3' terminus
(with acycloTMP and ddTMP) were also tested with similar results.
[02951 Family It DNA polymerases Vent (exo-) and PA (exo-) were tested using
the above
model system. With the acycIonucleotide blocker and. perfect match at the 3'
terminus, the
o
mutated template was amplified efficiently by PAP with one P* (lanes 1 and 2
in Figs. 18D and
. 18E)
and one opposing unblocked oligonucleotide and by PAP with two opposing P*s of
P*1
and P*2 (a special form of PAP where the two opposing P*s are overlapped at
their 3' temiini by
one. nucleotide) (lane 3 in Figs. 18D and 18E). However, no product was
generated from the
wild-type template because P*1 and P*2 mismatch the wild-type template at
their 3' termini,
is
showing the specificity (lanes 5-7 in Figs. 1813 and 18E). Vent (exo-) and Pfu
(exo-)
polymerases could not amplify with the 3' dideoxynucleatide blocked P* (lanes
9-16 in Figs.
18D and 18E). Direct sequencing analysis confirmed the con-ect sequence of the
P*1/01 and
P*2/0 2 products. Similar results were obtained With AcycloPol (Perkin-Elmer),
a genetically
engineered Family II archeon DNA polymerase. It is not clear why PAP with Vent
(exo-) and
20. Pfu (exo-) DNA polymerases discriminates against 3'
dideoxyribonucleotide blockers.
=
Other Blockers
[0296] These results demonstrate that two terminators used in Sanger
sequencing can be used as
blockers in PAP. Terminators have also been described as therapies of viral
illnesses, such as
AIDS, and for cancer therapy, such as, 3'-deoxyadenosine (cordycepin), 3'-
azido-3'-
25
deoxythyraidine (AZT), 2',3'-dideoxyinosine (ddl), 2',3'-dideoxy-3'-
thiacytidine (3TC) and 2',3'-
didehydro-2',3'-dideoxythymidirie (d4T). DNA polymerase can incorporate their
triphosphate
form into the synthesizing strand, and the incorporation cause termination of
the extension
(Gardner and Jack, 1999; Cheng et al., 1987; St. Clair et al., 1987; Ueno and
Mitsuya, 1997).
The raonophosphate nucleotides of 3'-azido-3'-deoxythymidine (AZT), T,3'-
dideoxy-3'-
30
thiacytidine (3TC) and 2',3'-didehydro-2',3'-dideoxythymidirie (d4T); when
located at the 3'
termini of oligonucleotides, can be removed by pyrophosphorolysis by MY
reverse transcriptase
or its variants (Anon et al., 1998; Gate et al., 2000; Meyer et al., 2000;
Urban et al., 2001).
=
CA 02911712 2015-11-10
6.7
These results indicate the application of PAP for various types of blocicers
and for RNA.
templates.
[02971 In summary, PAP amplification occurred efficiently and specifically
with 3'
acyclonucleotide and 3' dideOxynucleotide .blockers using TaqFS. DNA
polymerase, and only
s with acyclonucleotide blockers using Vent (exo-) and Pfu (exo-) DNA
polymerases. Other 3'
terminal nonextendible oligonucleotides and other DNA polyinerases can. be
used, if the 3'
terminal nucleotide can be. removed by pyrophosphorolysis, and the activated
oligonucleotide
can be extended.
o EXAMPLE 12
Detection of Extremely Rare Alleles by Bi-PAP
Phage DNA Template
[02981 The wild-type X phage DNA template that contains an inserted wild-type
lad gene of E.
coil (Kohler et- al., 1991) was Purchased from Stratagene, Three mutated X.
phage DNA.
=
IS templatds were prepared from 2t, phage plaques transformed into SCS.-8
E. coil cells according
to Mani atis etal. (1982) . They contain an A to T Mutation at nucleotide
position 190, a T to G
=:
mutation at. nucleotide 369 and a T to C mutation at nucleotide 369 in the lad
gene,
z ¨
respectively. The amount of X phage DNA was determined by UV abSorbance at 260
iitrt.
'Synthesis of P* by Adding a 3' Dideoxyiaucleotide
.20 [02991 The 3' ternainal ditleOxyniicleoticle was added- to an
oligodeoxynucleOtide by terminal
transferase. The mixtUre contained a total volume of 25 1.11: 100 mM potassium
pacodylate(pH
2.0 raM= CoCii, 02 mM DTT, 2 nM of the oligonuclootide, 2:4
3'-dd.NTP (the
molar ratio of the 3'-0H terminus. to .ddl\ITP was 1:30)(Roche), 100 U of
terminal transferase
(Invitrogen). The reaction was incubated at 37 C for 6 hr and then stopped bY
adding EDTA to
25 a 5 mM Thial cdneentration.. After desalting using a Centri-spitia
column i (Princeton
Separations), P* was purified by preparative 7 M urea/16% polyacrylamide gel
electrophoresis
with 30 mM Triethanolarnirie/Tricine buffer (pH 7.9 at 25 C) (Maniatis et al .
, 1982, Lin
et al., I999b). The atiount of recoVeredP* was determined by UV absorbance at
260 um.
[0300] Sirico small amitaintS of untentinafed oligonncleotide would result in
unexpected PCR
30 amplification, P* was =32P-labeled at the 5' terminus by T4
polyn.ucleotide kinase and then was
electrophoresed through a 7 M uted20% polyaciylarnide gel. Only P* prod-acts
Were visible
even when the gel waS=overeXposed. It is estiniated that More than 99.99% of
P.* contained a
=
CA 02911712 2015-11-10
68
=
dideoXynucleotide at the 3' terminus The parity of P* WAS supported by the
absence of PCk
product at pH 8.3 in which pyrophosphorolysis is inhibited. .
PAP Amplification
[0301] Bi-PAP assays for nucleotide 190 and lincleotide 369 of the lad gene
Were examined.
s The P's were 40 nucleotides long except that the upstream P*s = for
'position 369 are 42
nucleotides. Each P* contained the. sequence-specific nucleotide = at the 3
term-in-LIS. The PAP
reaction mixture contained a total volume of 25 ul: 50 inM Kel, 20 in.M
HEPES/Na011 (pH 6.=9
at 25 C), 10 naM (NH4)2504, 1.5 naM MgC12, 40 ILM each of the four dNTPs
(dATP, dTTP,
dGTP and dCTP), 0.1 WA each P*, 150 uM Na4PPi, 4% DMSO, 1 u.Ci of [a-3211-
4:1CTP
(3000Ciinamole, Amersh.ara), 1 U of AmpliTu0S- DNA polymerase (PE Applied
Biosystems),
2,000 copies of the phage DNA template or stated elsewhere. The cycling
conditions were
92 C for 6 sec, 68 C for 20 sec, and 72 C for 20 sec for a total of 15
cycles. A denaturing step
of 92 C for 1 min was, added before the first cycle.
[03.021 The product was electrophoresed through a standard 2.5% agarose gel,
and the gel was
. stained with ethidium bromide for UV photography by a CCD camera (Bia.Rad
Gel Doc 1000):
[0303] In order to differentiate the mutated product from. the wild-type
product of the same size,
non-denaturing SSCP gel electrophoresis was perfonted (Orita et al., 1989):
The reaction was.
=
mixed with two-fold volume of loading buffer (7M urea. and 50% formarnide),
bOiled= and
rapidly cooled on ice. The product in 10 41 of the mixed reaction was
electrophoresed through
an 8% .non-denaturing PAGE-PLUS (A3nresco) gel with 30 In.M
Ethanolarnine/Capsco buffer
(pH 9.6) (Liu et al., I999b) at 4 C. The gel was dried and exposed to Kodak X-
OMATTm AR
film for autoradiography. Three or four bands from each amplified product were
seen on a gel.
=
The upper one or two bands were double strained DNA due to hybridization of de-
natured
single-stranded segments during the electrophoresis as a result of the
substantial amountS of
amplified product present. Increasing the concentration of the amplified
product further increase
the intensity of the upper bands: =
Highly Efficient PAP Amplification
[0304] TaqFS, a genetically engineered DNA polymerase greatly improved, the
efficiency of
PAP. The conditions of PAP were further optimized. for dramatically higher
efficienCieS
allOwing PAP to amplify directly from a few copies of % phage DNA or human
genomic DNA
template. The reaction components and the thennocycling regime were optimized,
including: 1)
decreased concentrations of PPi in that keeping the PPi to dNTP ratio
essentially constant,
CA 02911712 2015-11-10
69
use of low pH HEPES buffer (pH 6.9 at 25 C), iii) addition of (NH4)2803, iv)
increased amount
of Ta0S, and v) higher annealing temperature.
Bi-PAP
[0305] PAP has a potential selectivity of 3.3x1011:1 (Fig. 19). Approaching
this potential
s requires a design that eliminates confounding sources of error. The A190T
mutation of the lac/
gene of A. DNA is used as a model system. In PAP with one downstream P* and
one upstream
unblocked eligonucleotide,' extension errors fitin the non-blocked upstream
oligonucleotide can
produce the rare mutation of interest, thus reducing the selectivity. If the
nfisincorporation rate
of TaqF8 is 104 per incorporated nucleotide and one of the three possible
misincorporations
2.. o generates the A--->T mutation on the newly synthesized upstream
strand, the selectivity decreases
to 3.3x10-5 due to the side effect. In. order to remove this limitation, Bi-
PAP was developed
(Fig. 20A). In Bi-PAP, both the downstream and upstream oligonucleotides are
P*s that are
specific for the nucleotide of interest at "their 3' termini. The P's overlap
at their 3' termini by
= one nucleotide.
-15 [0306] Bi-PAP amplified efficiently and specifically at nucleotide
position 190 using X phage
= DNA containing the lad l gene as template (Fig. 20B). Addition of human
genomic DNA did not
= affect the amplification. The 79-bp product of Bi-PAP was easily
distinguished from
nnincorporated P*s. P* did not form dimers because P* needs a perfectly
matched region at the
3' tenninus" for activation. Similar results were observed at nucleotide
position 369. Direct
20 sequencing analysis confirmed the correct sequence of the amplified
product.
Sensitivity and Selectivity of Bi-PAP
[0307] In order to demonstrate the extremely high selectivity of Bi-PAP, more
than 1010 copies
of DNA template was Used for a Bi-PAP reaction. X DNA containifig the in'fq"
gene of E.coli
was chosen as the model system because I ig of X DNA contains 2x1010 vector
genomes, while
25 lug of human genomic DNA only contains 3.3X.105 genomes. In order to
avoid potential
contamination of the wild-type X DNA in this laboratory, mutation-specific Bi-
PAP assays with
mutated P*8 were chosen to amplify the wild,type X DNA. The relative frequency
of a
spontaneons mutation of the lac/ gene in the wild-type X DNA is estimated to
be less than le
by examining X, phage plaques infecting E. coll.
= 30 [0308] The sensitivity and selectivity of Bi-PAP were examined
using three mutation-specific
131-PAP assays with their corresponding mutated X DNA (see Table 7 footnotes
for definitions).
Four titration experiments were performed for each mutation-specific B1-PAP
assay (Figs. 21A-
.
CA 02911712 2015-11-10
2IC). Experiment I tested how much the mutated pi, can "tolerate" the wild-
type DNA template
(i.e., the maximum copies of the wild-type template without a detectable
Mutated product). The
wild-type 2k. DNA was titrated fi-ana 2x1010 copies to 2x106 copies. The
maximum tolerances
were 2x109 to 2x101 , 2x107 to 2x 108, and 2x107 to 2x 108, respectively, for
the three mutation
= s sPecifie Bi-PAP as8A3is, respectively (Figs. 21A-21C). Experiment
11 tested the sensitivity of Bi-
PAP. The mutated X DNA was titrated from 2x103 to 0 copies: The ratio of the
maximum
tolerance (Experiment I) to the sensitiVity is the selectivity. Experiment II
was repeated in the
presence of large amount of wild-type template (Experiment III) or large
amounts of human
genomic DNA (Experiment IV) without effects (Fig. 21A; data not shown for
T3690 and
10 T369C). A dose response with template copy number was observed.
TABLE 7
Summary of the three mutation-specific Bi-PAP assay?
Assay Position" Type' SensitiVityc
Selectivitityd
A 190 2 109:1 to
101":1
=
369 2 10':1 to
108:1
369 2 101:1 to
108:1
15 'In each of the three mutation-specific Bi-PAP assays, two opposite P*s
with one nucleotide overlap at
their 3' termini were used: The P*s are 40-42 nucleotides long. They are
5' GATGGCGGAGCTGAATTACATTCCCAACCGCGTGGCACAT* (SEQ ID NO:61) and
5' GGCAACGCCAATCAGCAACGACTGrUGCCCGCCAGTIGA* (SEQ NO:62) in Assay A;
5' GAAGCGGCGTCGAAGCCTGTAAAGCGGCGGTGCACAATCW (SEQ
NO:63) and
20 5' GCGGATAGTTAATGATCAGCCCAC TGACGCG1TGCGCGAGAC* (SEQ ID NO:64) in' Assay
B; 5' GAAGCGGCGTCGAA.GCCTGTAAAGCGGCGGTGCACAATCC* (SEQ ID NO:65) and
5' GCGGATAGTTAATGATCAGCCCACTGACGCGTTGCGCGAG AG* (SEQ ID NO:66) in Assay
C.
." The position of the first n.ueleotide of transcript in the /ad gene is
assigned the nucleotide position 1
25 (Farabaugh, 1978), The 3' nucleotide of the P* is located at the
indicated position and is complementary
to the corresponding mutation.
The sensitivity is defined as the minimum copies of the Mutated template from
which a detectable
mutated product is generated when a mutation-specific Bi-PAP assay is used. It
was determined by
Experiment II (Figs. 21A-21C).
30 d The selectivity is the ratio of the Makin:ant copies of the wild-type
template with undetectable product
the minimum copies of the mutated template with detectable product to, when a
mutation-specific Bi-
. PAP assay is used.
[03091 The approximately 100-fold difference in selectivity betWeert the
nucleotide positions
35 190 and 369 my deriVe frOM: i) the preSence of spontaneous mutations at
the position .360 at a
CA 02911712 2015-11-10
71 =
frequency of 10-7 to le in. the wild-type X DNA, ii) iniptirity of P*
oligonucleotides, iii)
specificity of pyrophosphorolysis for a perfect match at. the 3' terminus and
fidelity of DNA
polymerate to incorpota.te a correct nucleotide may be assothated With
sequence context such
that the Type II non-specific amplification occurs at a frequency of 104 to
le. In the latter
= 5 case, a 100-4bld difference in selectiVity could = arise = from a
10-fold , difference in
pyrOphOSphOrolVis specificity aiid a .10-fold difference in DNA polymerase
fidelity with
- sequence context
[05101 The rate of a .spontaneous niutatiOn. of X phage M. E. coil varlet.
from locua to 1od1.18; bii
the average from 10-9 to 104' per incorporated nucleotide. The amplified
signal Seen in.
10. Expotiment I Might be caused by rate spontaneous mutations; =
[0811] There iS a possible side reaction due to the impurity of P*
contamination of unblocked.
= oligonteleotide where the didebxy tenni:ring has not been added, although
no unblocked
= oligonucleotide was detected in the P*. However, this selectiVity may net
be limited severely by
r small 'amounts of unblocked oligonucleotide because, the product
generated Would be much =
= TT- more likely to he the wild-type rather than the specific mutation
(3.3x105:1).
¨.T. [05121 Bi-PAP has = extremely high sensitivity and
selectivity.- Bi-PAP Can
selectively detect two copies of rare mutatedallele with a single base
substitution from Up to.
2X109 copies of the Wild-type allele. Bi-PAP is a simple, rapid, attomatable
method for
detecting any tare allele of interest. =
20 =
EXAMPLE 13 =
- Meaanrement Of Mutation Loadirt Mouse Tisanes by BiTAP
Materials and Methods
[0313] Liver, heart, adipose tisane; cerebrum and cerebellum from 10-day to 25-
Month old mice
25 were snap froten and stated undet liquid nitrogen until used. DNA Was
extracted according. to
the Big Blueprotocol (Stratagene Matructien. Manual). In brief, tisanes "Were
homogenized and
digested with Proteinage K. The genomic DNA Was extratted with
phenol/chloroforin and
precipitated with ethanbl. The DNA Was diStelVecl in TE bti#er (10 MM
Tris/IICI, 1 tnIVI
E15TA"., pH 8.0) aftd stilted at 4 C. The amount of the Iii0i1S6 genoMic DNA
Wag deterrnined by
30 U\i" abSofbatee at 2.60 rim.
[08141 =The nintatiOnApeCific Bi-PAP assay for -T3690 (ASSay 8: the two
opposite P4's are
cadeoxyfiddeotiitte blockedwith one nucleotide OVerlap at their 3' terinir'd.
arci
=
CA 02911712 2015-11-10
72
5cGAAGCGGCGTCGAAGCCTGTAAAGCGGCGGTGCACAATCG*31 (SEQ ID
NO:63) and
5' GCGGATAGTTAATGATCAGCCCACTGACGCGTTGCGCGAGAC*31 (SEQ ID
NO:64)
s of the /Cie/ gene was performed as above except that i) the reaction
contained .2 pg of the mouse
genomic DNA (-20 kb it size), unless otherwise stated; ii) mouse DNA in 20 ul
(1.25x HEP-ES
buffer, 5% DMSO without MgCl2) was heated at 100 C for 2 min and quickly
cooled on ice;
. before the other components added; iii) a denaturing step at 95 C for 1 min
was added before the
first cycle; iv) the denaturing step was 95 C for 10 sec.
o
[0315] 10 p_l of the 25 p_l reaction was mixed with .10 pl of the denaturing
loading buffer, boiled
and rapidly cooled on ice. The product was electrophoresed through a 8% 7M
urea /PAGE gel
with 90 in)\11 T13E buffer at room temperature. The gel was dried and exposed
to Kodak X-
OMATTm AR film for autoradiography.
= - Results and Discussion
15 [0316] Transgenic mouse mutation detection systems permit
determination of the frequency and
pattern of spontaneous or induced mutations in vivo. The Big Blue! system uses
transgenic mice.
harboring chromosornally-integrated A phage. DNA containing the K. coil lad
gene as the
mutational target (Grpssen and Vijg, 1993; Gossen et al.; 1989; Kohler et al.,
1990. The lad
gene is integrated within each mouse diploid genome in 40 tandemly repeated X
DNAs.
20 [0317] The Big Blues mutation detection system assay is performed by
isolating genomic DNA.
fi-om transgenic mouse tissues and mixing it with X packaging extracts. The
packaged X phage
can infect E. coll. In the presence of X-gal substrate, lad mutants give rise
to blue plaques on a
background of colorless wild-type plaques. Observed mutants derive
overwhelmingly from the
mouse (Hill et al., 1999): The mutant frequency is detennined by dividing the
number of circular
25 blue plaques by. the total mrrnber of plaques. Of 5000 sequenced
mutant plaques; 31. T369G
mutants have been found in a total of 149x106 plaques screened from various
ages, genders and
treatments in this laboratory (frequency = 2,1x1e). .
[03181 TO assess the utility of Hi-PAP for measuring ultra-rare mutations in
mammalian cells;
the T369G mutation was analyzed in genomic DNA from the Big Blue mice. Two pg
of mouse
30 genomic DNA was amplified in 25 Pl reaction containing a total of
1.2X107 copies of the lad
gene. The mutation-specific Bi-PAP assay for T369G (Assay B) was performed for
18 samples
in duplicate (Fig. 26A). Three categories of results were denned; each with
similar number of
=
CA 02911712 2015-11-10
73
samples: 1) six samples were positive two times (5, 11-15), 2) seven samples
were positive one
= time (1, 3, 6, 9, 16-18), and 3) five samples were negative two times (2,
4, 7, 8, 10).
[0319] Two samples in each category were studied further (Figs. 261B-26C,
Table 8). In category
1, for the two samples 5 and 12 with the strongest amplified signals (Fig.
26A), a four-fold
s dilution to 0.5 lag and 16-fold dilution to 0.125 pg of mouse genomic
DNA were performed for
further quantitation (Fig. 26B). The T369G mutant frequency for each sample
was estimated
and varies 370-fold among the six samples (Table 8). The average T369G mutant
frequency of
2.9x1(17 was within. 50% of the average T369G mutant frequency of 2.1 x10-7
measured from
4x107 plaques using the Big Blue mutation detection system and confirmed by
direct
sequencing.
TABLE 8
Somatic mutant frequency measured by Bi-PAP
Mouse genomic DNA Frequency of positive amplification" Estimated
Sample' mutant
frequency"
- Tissue Age 2 pg of 0.5 lag of 0.125 pg of
DNA DNA DNA
1 12 Adipose 6 months 8/8 8/8 4/8
(0.69) 9.25x10-7
2 5 Liver 25 months
8/8 7/8 - 5/8
(0.98) 1.31x10-6
3 3 Liver 25 months 8/24 (0.41) 3.38x10-8
4 9 Liver 25 months 13/24 (0.78) 6.50x104
.
5 7 Liver 25 months' 2/24 (0.09) 7.25x10-9
6 10 - Liver 25 months 1/24 (0.04) 3.52x10-9
Average 2.91x10-'
a see Fig. 26A.
"the ratio of the number of positive signals for the T369G mutation relative
to the total number of
reactions.
the average number of T369G mutants per reaction is estimated using a formula
(the frequency of zero
mutants per reaction = e , x is the average number of mutants per reaction)
suppose that the mutant
distributes in the reaction according to a Poisson distribution and that if
one or more mutants are in the
reaction, the amplification is positive, and if zero mutant is in the
reaction, it is negative.
the frequency of the T369G mutant of the lad gene in mouse genome per reaction
is estimated
assuming that the mutant distributes in mouse genomic DNA according to a
Poisson distribution and that
one or more mutants are positive in the detection. For each of samples 12 and
5, a total of ¨6.0x106,
copies of the lad gene are used for the estimate, and for each of samples 3,
9, 7 and 10, ¨2.9x108 copies
are used assuming that 2 lig of the lad- mouse genomic DNA contains -4 .2l
copies of the lad gene.
CA 02911712 2015-11-10
74
=
[0320] The 370-fold variation in mutant frequency was observed in livers of
five mice at 25
months of age. This large variation could be due to difficulties in amplifying
one copy of the
template. To address this issue, each of the analyses was repeated at Mast two
titres with similar
results: For example, in sample 9, seven of 14 reactions with 2 -p.g Of DNA
Were positivehi one
experiment,: three of fbur such reactions were positive in another
eXperithent, and two of four
such reactions were positive hi a third experiment For sample 7, there wag one
positive in eight
and one positive in 14 reactions./ The product was sequenced to donfitni the
T369G mutation
after re-amplification from the positive reaction. hi addition, positive
controls (2 p:g of the lad+
mouse DNA with ¨10 copies of T369G) and negative controls (mouse genorniC DNA
without
io .the lad- target, i.e., the lad mouse DNA) were performed. As
additional positive controls,
reconstruction experiments were performed in that the copy number of the
mutated X DNA per
reaction was serially diluted by two-fold in the presence of the lad" genomic
DNA carrier.
Reproducible amplifications from as low as one copy of template were
demonstrated. (Figs. 26B,
26C).
as [0321] In retrospect, the 370-fold variation in the frequency of
T369G mutant observed among
the six mice may not be surprising because the T369G mutant frequency ainong
mice is over
dispersed, implying a hyper-Poisson distribution (Nishino et al., 1996;
Piegorsch et al., 1994).
Among six mice the inter-animal variation in the overall mutant frequency
assayed by the Big
Blues mutation detection system might be 3 to 4 fold, with significant founder
effects in one or
20 a few of the mice. The variation might be in the range= of 2x1(15 to
8xle which is the sum of
more than 1,000 different mutations_ Here, only the T369G mutation is assayed.
It is anticipated.
that the great majority of the signal derives from duplex mutated templates
(EEll et al., 1999),
but it should be noted that unresolved mismatch intermediates derived
primarily from DNA
replication or DNA repair would also generate a signal. Thus, the physical
limit of sensitivity is
25 actually one half of a duplex DNA molecule per reaction.
[03221 In conclusion, we demonstrate that Bi-PAP can analyze ultra-rare
mutations at
frequencies as low as 10-7 to 109, depending on the assay. It is shown that Bi-
PAP can detect
single copies of the somatic mutation directly front mammalian genomic DNA.
The inter-assay
variation may reflect locus-specific variability hi the assay sensitivity or
in the frequency of the z
30
assayed mutants among the samples. More work is necessary to distinguish
between these
possibilities. In mammalian DNA, the number of copies of template is limited
by the enormous
genome size. Two lig of genbraic DNA contains only 600,000 mouse haploid
genomes, yet the
reaction is viscous. Our analysis of the Big Blue mouse genomic DNA was
facilitated, by the 20
=
CA 02911712 2015-11-10
copies of the /c/c/ gene per haploid gamine. To measure mutation load in
humans, genomic
DNA in one reaction could be increased ,at least three fold by reducing the
viscosity (e.g.,
shearing the DNA into small segments by ultrasonic treatment) and another four
fold by
expanding the reaction_ volume to 100 pl. Mutation load in human genomic DNA
might be
5 facilitated .by analyzing- segments of virtually identical sequence,
e.g.. there are three 9.6 kb
- segments. with 99+% sequence identity on human X chromosome involved in a
common
in-version, mutation in hemophilia A (Lakich et aL, 1993). Less complex
genomes including C-
elegans, Drosophila, and human mitochondria gen.ome or chronic viral
infections (e.g., hepatitis
B) also should be analyzable with this protocol.
[0323] While the invention has been disclosed in this patent application by
reference to the
details of preferred embodiments of the invention, it is to be understood that
the disclosure is
intended in an illustrative rather than in a limiting sense, as it is
contemplated that modifications.
will readily occur to those skilled in the art
BIBLIOGRAPHY =
Anon, D. et al., Biochemistly 37, 1590845917 (1998).
Bains W. and Smith G.C., J27wor Biol 135, 303-307(1988).
Bailer, 3. et al., Nucleic Acids Res 26, 5073-5078 (1998).
Barony, F., ProC Natl Acad Eci USA 88, 189-193 (1991). =
Bebenek, K. et at., IBiol Chem 265, 13878-13887 (1990).
Becker, M.M. and Grossmann, a, in Footprinting of nucleic acid-protein
complexes (ed.
Revzin, A.), pp. 129-159 (Academic Press, New York, 1993).
Boer, P.H. et al., Biochem Genet 28, 299-308 (1990).
Buzin, C.H. et al., BioTechniques 28,746-753 (2000).
cieng, Y.C. et al., J Bid Chem 262, 2187-2189 (1987).
Chien, A. et al., Bacterial 127, 1550-1557 (1976).
Chou, Q. et al.,Nucl Acids Res. 20,1717-1723 (1992).
Cotton, R.G. et al., Proc. Nati Acad Set USA 85, 4397-4401 (1988).
Dal, S.-M. et at., Nature Biotechnology 18:1108-1111 (2000).
R.J. et al., Nucl Acids Ras 19,3749 (1991).
Drmanac, R. et at., Genomics 4, 114-128 (1989).
Duetoher, M.P. and Kornberg, A., J Biol Chem 244, 3019-3028 (1969).
CA 02911712 2015-11-10
76
= =
Eckert, K.A. and Kunkel, T.A., Nucleic Acids Res. 18, 3739-3744 (1990).
Gardner, A.F. and Jack, W.E, Nucleic Acids Res 30:605-613.(2002).
Gardner, A.F. and Jack, W.E., Nucleic Acids Res 27:2545-2553(1999).
Ginot, F., Hum Mutat 10, 1-10 (1997).
Gossen, J. and Vijg, J., Trends Genet 9, 27-31 (1993).
Gossen, J.A. et al., Proc Nad Acad Sci USA 86, 7971-7975 (1989).
Gotte, M. et at, J Viral 74, 3579-3585 (2000).
Hacia, J. et al., Nat Genet 21, 42-47 (1999), ,
Hill, K.A. et al., Mutat Res Mini Reviews 436, 11-19 (1999).
3.o Innis, M.A. and Gelfand, D.Hõ in PCR APPLICATIONS Protocols for
Functional Geizornics
(eds. Innis, MA., Gelfand, D.H. & Sninsky, II), pp. 3-22 ( Academic Press,
1999).
Jones, P.A. and Laird, P.W., Nat Genet 21, 163-167 (1999). =
Kaledin, A.S. et al., Biokl2india 46, 15764584 (1981).
Kellogg, D.E. et at, Biotechniques 16, 1134-1137 (1994).
Ketterling, R.P. et al., Hum Genet 105, 629-640 (1999).
Khrapko, K.R. et al., ,FEBS Letts 256. 118-122 (1989).
Knoll, A. et al., Hum Genet 98, 539-545 (1996).
= Kohler, S.W. et al., Strategies in Mal Bid l 3, 19-21 (1990).
Kohler, S.W. et al., Proc Nati Acad Sci USA 88, 7958-7962 (1991).
20 Komura, I. and Riggs, A.D., Nucleic Acids Res 26, 1807-1811 (1998).
Komberg, A. and Baker, T.A., DNA Replication, (eds., Second Edition), pp. 113-
226 (W.H.
Freeman and Co., New York 1992).
Lakich, D. et at, Nature Genet 5, 236-241 (1993).
Landegren, U. et al., Science 241, 1077-1080 (1988).
=
25 LeProust, E. et al., J Comb Chem 2, 349-354 (2000).
Liu, Q. and Somn-ter, S., BioTechniques 29, 10724083 (2000).
Liu, Q. and Sommer, S., BioTechniques 18,470-477 (1995).
Liu, Q. et al., Am J Med Genet (Neuropsych Genet) 60, 165-171 (1995).
Liu, Q. et al., BioTechniques 26, 932-942 (1999).
3 o Liu, Q. et al., BioTechniques 33, 129-138 (2002).
Liu, Q. et al., Anal Biochem 270, 112-122 (1999b).
Lizardi, P.M. et al., Nature Genetics 19, 225-232 (1998).
Longley, M.J. et al., Nucleic Acids Res 18, 7317-7322 (1990).
Lysov, I. et al., Dokl Akad Nauk SSSR 303, 1508-1511 (1988).
CA 02911712 2015-11-10
77
Mathatis, T. et al., Molecular Cloning.. a Laboratory Manual, Cold Spring
Harbor, New York:
Cold Spring Harbor Laboratory, 1982. .
Marshall, A. and Hodgson, I.., Nat Biotechnol 16; 27-31(1998)
Maxana, A.M. and Gilbert, W., ProcNatl Acad Sci USA 74, 560-564 (1977).
s Meyer, P et al., EMBO J19, 3520-3529 (2000).
.Miyada, C.G. and Wallace, R.B., Methods in. Enzymology 154, 94-107 (1987).
Mueller, P.R. and Wold, B., Science 246, 780-786 (1989). [published erratum,
appears in
Science 248, 802 (1990).]
Mullis, K.B., PCR Methods Appl 1, 11-4 (1991).
io Myers, R.M. et at., Science 230, 1242-1246 1985..
Nishino, H. et al.,.Environ Mal Mutagen 28, 299-312 (1996).
= Nishino, H. et al., Environ Mal MUtagen 28, 414-417 (1996).
= O'Donovan, M.C. et al., Genomics 51, 44-49 (1998).
Oefner, P. and Underhill, P., Current Protocols in Human Genetics Supplement
19:7.10.1-7
15 .10.12 (1998).
. Orita, M. et al., Proc Natl Acad Sci USA 86:2766-2770 (1989).
Parsons, B.L. and Heflich, R.H., Mutat Res 387, 97-121 (1997).
. Pevzuer P.A., JBionzo/ Struct Dyn 7, 63-73 (1989).
Pfeifer, G.F. et al., Science 246, 810-813 (1989).
20 Pfeifer, G.P. et at., Methods Enzymol 304, 548-571 (1999).
Piegorsch, W.W. et al., Environ Mol Mutagen 23, 17-31(1994).
Pourzqnd, C. and Cerutti, P., Mutat Res 288, 113-121 (1993).
Ramsay, G., Nat Biotechnol16, 4044 (1998). .
Ronaghi, M. et al., Science 281, 363, 365 (1998).
2s Ronai, Z. and Minamoto, T., Hum Mutat 10, 322-325 (1997).
= Russo, E. and Riggs, A.D., Epigenetics Mechanics of gene regulation.
Anonymous 1996
Saiki, R.K. et al., Science 230, 1350-1354(1985).
R.K. et al., Science 239, 487-491 (1988).
Sanger, F. et at, Proc Nail Acad Sci USA 74, 5463-5467 (1977).
30 Sarkar, G. et at., Anal Biochem 186, 64-68 (1990).
Sarkar, G. et al., Nucleic Acids Res 20, 871-878 (1992).
Singh-G-asson, S. et al., Nat Biotechnol17, 974-978 (1999).
Sommer, S.S., Trends Genet 11, 141-147 (1995).
= Sommer, S.S. et at, Mayo Clinic Prac. 64, 1361-1372 (1989).
35 Southern, E.M. et al., Genomics 13, 1008-1017 (1992),
/
CA 02911712 2015-11-10
78
Southern, E.M. Trends Genet 12, 110-115 (1996).
Spiegebnan, LI. et al., Biorechniques 29, 10844092(2000).
St. Clair, M.H., Antimicrob Agents Cheinother 31, 1972-1977 (1987).
Syvanen, A.C., Hum Mutat 13, 140 (1999).
s Tabor, S. and Richardson, C.C., J Biol Chem 265, 8322-828 (1990).
Tabor, S. and Richardson, C.C., ProcNatl Acad Sci USA 92, 6339-6343 (1995).
Trainor, GL., US Patent No. 5,558,991 (1996).
Ueno, T. and IVIitsuya, H., Biochemistry 36, 1092-1099 (1997).
Urban, S. et al., Proc Nall Acad Scie USA 98, 4984-4989 (2001).
lo Vander Horn, P.B. et al., BioTechniques 22, 758-762 (1997).
Wallace, R.B.et al., Nucleic Acids Res 6, 3543-3557 (1979).
Wong, I. et al., Biochensishy 30, 526-537 (1991).
Yoshitake, S. et al., Biochemist?), 24, 3736-3750 (1985).
=
=
=
=
=