Note: Descriptions are shown in the official language in which they were submitted.
CA 02915623 2015-12-18
PAT 103540-1
SEPARATION OF CARBON DIOXIDE FROM FLUE GASES
Technical Field
[0001] The present invention relates to the separation of carbon dioxide
from
flue gases for sequestering the carbon dioxide.
Background
[0002] Sequestration of carbon dioxide as solid carbonates is desirable
to
reduce the levels in or released to the atmosphere. Mineral carbonation, which
is
the process of capturing the CO2 in the atmosphere in the form of solid
carbonates
through the reaction of CO2 with silicates, is a spontaneous,
thermodynamically
favourable process. Solid carbonates are a long-term geological sink of carbon
and
thus their formation is desirable. Unfortunately, the kinetics of natural
mineral
carbonation is very slow and the process is only significant over geological
time
periods, i.e., millions of years.
[0003] Accelerated formation of solid carbonates is observed in
biological
systems, particularly in corals, bivalve molluscs, echinoderms, and
foraminifera.
These organisms have developed mechanisms to induce and accelerate the
precipitation of carbonates, which is required for their skeletons, in natural
saline
waters. Mimicking such biological approaches to achieve the accelerated
formation
of solid carbonates is desirable.
[0004] Precipitation of carbonates in seawater does not occur
spontaneously.
Economically feasible processes to induce and accelerate precipitation are
desirable.
Summary
[0005] According to an aspect of an embodiment, a method of separating
carbon dioxide from a flue gas is provided. The method includes exposing the
flue
gas comprising carbon dioxide to salt water to generate a solution, monitoring
pH
of the solution, when a pH condition is met, adjusting the pH to precipitate a
solid
carbonate material from the solution, separating the solid carbonate material
from
- 1 -
CA 02915623 2015-12-18
PAT 103540-1
the solution, and storing the solid carbonate material to inhibit the carbon
dioxide
from entering the atmosphere.
[0006] According to another aspect of an embodiment, a method of
separating carbon dioxide from a flue gas includes exposing an underground
silicate
deposit to hydrochloric acid to generate a brine, recovering the brine at
surface,
exposing the flue gas comprising carbon dioxide to the brine, monitoring pH of
the
brine, when a pH condition is met, adjusting the pH, precipitating a solid
carbonate
material from the brine, separating the solid carbonate material from the
brine, and
storing the solid carbonate material to inhibit the carbon dioxide from
entering the
atmosphere.
[0007] According to yet another aspect of an embodiment, a method of
separating carbon dioxide from a flue gas includes exposing an underground
silicate
deposit to hydrochloric acid to generate a brine, recovering the brine at
surface,
exposing the flue gas comprising carbon dioxide to the brine, introducing
ammonia
to the brine to generate an ammonium salt and a solid carbonate material,
precipitating the solid carbonate material from the brine, separating the
solid
carbonate material from the brine, and storing the solid carbonate material to
inhibit the carbon dioxide from entering the atmosphere.
[0008] According to still another aspect of an embodiment, a method of
separating carbon dioxide from a flue gas includes exposing the flue gas
comprising
carbon dioxide to salt water to provide a solution, monitoring pH of the
solution,
adjusting the pH to generate a basic solution and precipitate a solid
carbonate
material from the solution, separating the solid carbonate material from the
solution, and storing the solid carbonate material to inhibit carbon dioxide
from
entering the atmosphere.
[0009] According to a further aspect of an embodiment, a method of
sequestering carbon dioxide includes exposing a flue gas comprising carbon
dioxide
to sea water to provide a solution, monitoring the pH of the solution, when a
pH
condition is met, adjusting the pH to precipitate a solid carbonate material
from the
solution, and storing the solid carbonate material in the sea water to inhibit
carbon
dioxide from entering the atmosphere.
- 2 -
CA 02915623 2015-12-18
PAT 103540-1
Brief Description of the Drawings
[0010] Embodiments of the present invention will be described, by way of
example, with reference to the drawings and to the following description, in
which:
[0011] FIG. 1 is a schematic diagram of a system for the capture of CO2
using
sea water and subsequent neutralization of the acidified sea water according
to an
embodiment;
[0012] FIG. 2A is chart showing an equilibrium and operating diagram for
CO2
absorption with sea water at variable pH;
[0013] FIG. 28 is chart showing an equilibrium and operating diagram for
CO2
absorption with sea water at constant pH = 8.2;
[0014] FIG. 3 schematic diagram of an electrochemical cell for the
regeneration of sea water acidified after contact with flue gas according to
an
embodiment;
[0015] FIG. 4 is a chart showing the energy utilized for electrolytic
mineralization of CO2 according to an embodiment;
[0016] FIG. 5 is a schematic diagram of a system for CO2 mineralization
with
silicates according to another embodiment; and
[0017] FIG. 6 is a schematic diagram of a system for CO2 mineralization
with
ammonia and ammonia regeneration according to another embodiment.
Detailed Description
[0018] For simplicity and clarity of illustration, reference numerals may
be
repeated among the figures to indicate corresponding or analogous elements.
Numerous details are set forth to provide an understanding of the examples
described herein. The examples may be practiced without these details. In
other
instances, well-known methods, procedures, and components are not described in
detail to avoid obscuring the examples described. The description is not to be
considered as limited to the scope of the examples described herein.
[0019] The disclosure generally relates to a system and a process for
separating carbon dioxide from a flue gas. The includes exposing a flue gas
that
includes carbon dioxide to salt water to generate a solution, monitoring pH of
the
- 3 -
CA 02915623 2015-12-18
PAT 103540-1
solution, adjusting the pH to precipitate a solid carbonate material from the
solution
when a pH condition is met, separating the solid carbonate material from the
solution; and storing the solid carbonate material to inhibit the carbon
dioxide from
entering the atmosphere.
[0020] Several solvents have been previously proposed and tested for CO2
capture. The use of some solvents such as amines or ionic liquids, however,
increases the overall cost of carbon sequestration because of the cost of the
solvents and the large amount of energy utilized for regeneration of the
solvents.
Water may be utilized for capturing carbon dioxide from flue gas, however the
equilibrium concentration of CO2 in water is relatively low. Nonetheless,
there is a
growing interest in using water, and in particular, particularly sea water, as
a
medium for capturing and sequestering CO2.
[0021] The supply of cations utilized for carbonate precipitations that
exist
within sea water makes sea water a desirable. The dissolution of carbon
dioxide in
water is generally believed to follow the mechanism shown below:
CO2 (g) Ã4 CO2 (aq) AH = ¨19.75 kJ/mol (1)
r
CO2 (am H2O 4-+ H2CO3 AHr = ¨0.56 kJ/mol (2)
CO2+ OH- . HCO3- ATP= ¨46.655 kJ/mol (3)
r
H2CO3 4--* HCO3- + H Aii -= 9.72 kJ/m o I (4)
r
HCO3- 4-4 C032- H Air =14.7 kJ/mol (5)
r
where the enthalpies of reaction at standard conditions were determined using
data
from Dean, IA. Lange's handbook of chemistry, 15th Edition, 1999.
[0022] The overall reaction is:
CO2 (g) H2O 4-- C032- 2H AHr = 4.11 kJ/mol, Aq=103.6kJ/mol (6)
or at higher pH,
CO2 (g) OH-. C032- + H Aiir = ¨51.7 kJ/mol, Aq= 23.77 kJ/mol (7).
- 4 -
CA 02915623 2015-12-18
PAT 103540-1
[0023] The standard Gibbs energy of reaction for the global reactions (6)
and
(7) is positive, indicating that the conversion of CO2 into carbonate is not
spontaneous at 25 C. Reaction (7), however, may be spontaneous at lower
temperatures because this reaction is exothermic.
[0024] If divalent ions are present in solution, e.g., Ca2+, Mg2+, Sr2+,
the
formation of solid carbonates has the potential to occur. For example, calcium
ions
react with carbonate ions in solution to yield calcite or aragonite:
Ca2+ + C032- CaCO3 (calcite or aragonite)
(8).
[0025] By combining reactions (6) or (7) with reaction (8), two global
reactions are possible:
CO2 (g) + H20 + Ca2+ 4-> CaCO3 (calcite) + 2 H+ A11r =14.7kJ/mol, Aq= 56.0
kJ/mol (9)
CO2 (g) OW + Ca2+
CaCO3 (calcite) H+ AHr = -41.1k3/mol, AGr = -24.7 kJ/mol (10)
[0026] From the values of the enthalpy of reaction and Gibbs energy of
reaction, it is clear that reaction (9) is not spontaneous at any temperature,
while
reaction (10) is spontaneous at all temperatures. This means that spontaneous
precipitation of calcite is only spontaneous at high pH. The same pattern
holds for
the other carbonates (i.e. aragonite, magnesite, and dolomite).
[0027] The formation of carbonates may theoretically take place when CO2
is
solubilised in a solution containing divalent cations resulting in the
precipitation of
several different solid carbonates including calcite, aragonite, dolomite, and
magnesite. In practice, however, formation of solid carbonates is rarely
observed.
In fact, sea water is often supersaturated with carbonates. As an example,
Dolomite formation has not been observed in sea water, even though the ionic
product of calcium, magnesium, and carbonate is several orders of magnitude
greater than the solubility product of dolomite. Sedimentary deposits,
however,
are rich in magnesium-calcite and dolomite, indicating that at some time
during the
earth's geological history precipitation of carbonates has occurred on a very
large
scale as described in, for example, Burns, S.J. et. al. (2000) Dolomite
formation
and biogeochemical cycles in the Phanerozoic. Sedimentology 47(1), 49-61
(hereinafter "Burns").
- 5 -
CA 02915623 2015-12-18
PAT 103540-1
[0028] The prior art related to carbonate precipitation in sea water
suggests
that solid carbonate formation is mediated by the presence of living organisms
that
either change the redox potential of the water, as described in, for example,
Burns,
or by the raise the local concentration of ions through the use of biological
membranes, as described in, for example, Al-Horani, F.A. et al (2003) The
mechanism of calcification and its relation to photosynthesis and respiration
in the
scleractinian coral Galaxea fascicularis. Marine Biology 142, 419 - 426
(hereinafter
"Al-Horani").
[0029] The precipitation of calcium and magnesium carbonates in sea water
(pH = 8.2) is catalyzed by several living forms that use those carbonates to
build
their skeletons (i.e. corals, bivalves mollusca, echinoderms, and
foraminifera).
These organisms utilize different strategies to carry out and accelerate
carbonate
precipitation. In the case of corals, calcium transporters are responsible for
pumping calcium ions against their concentration potential causing an increase
in
the oversaturation of calcium (though sea water is already supersaturated in
calcium). The energy to drive the calcium transporters is provided by
photosynthesis, as described in, for example, Al-Horani.
[0030] When a gas with a high concentration of CO2, such as for example
flue
gas, is put in contact with sea water, CO2 will solubilize in the water
increasing the
concentration of soluble carbon species in the sea water, as well as the
concentration of protons (H+). The increased acidity, however, impedes the
precipitation of solid carbonates as shown in the above Equations (9) and
(10).
Typically, the solubility of carbonates increases as pH decreases. Once the
acidified
water re-enters the environment and reaches equilibrium with the predominant
CO2
concentration in the atmosphere, most of the solubilized CO2 will have come
out of
solution and been released into the atmosphere, resulting in a zero net carbon
sequestration. Thus, challenges exist to carbon sequestration through carbon
solubilizing in sea water. To achieve a positive net carbon sequestration
using sea
water, solid carbonates must be formed and removed prior to returning the
water
to the sea.
[0031] To illustrate the effect that CO2 dissolution has on the
solubility of solid
carbonates in sea water, and how to enhance carbonate precipitation, the
- 6 -
CA 02915623 2015-12-18
PAT 103540-1
thermodynamic equilibrium of the CO2-carbonate-sea water system was simulated
using the geochemical software PHREEQC version 3Ø PHREEQC is a computer
program for speciation, batch-reaction, one-dimensional transport, and inverse
geochemical calculation available from the U.S. Geological Survey and is
described
more fully at, for example, Parkhurst, D.L., and Appelo, C.A.J., 2013.
Description of
input and examples for PHREEQC version 3 ¨ A computer program for speciation,
batch-reaction, one-dimensional transport, and inverse geochemical
calculations:
U.S. Geological Survey Techniques and Methods, book 6, chap. A43, 497.
[0032] A gas stream representing flue gas is assumed to contact a liquid
stream representing sea water saturated with CO2 and oxygen at the predominant
atmospheric concentration of these gases. Flue gas is the product of
combusting a
hydrocarbon fuel with air. Normally, air is supplied in excess of the
stoichiometric
requirement in order to ensure complete combustion. The molar composition of
the
flue gas resulting from burning natural gas (CH4), assuming a 15% excess air,
is
shown below:
Component N2 02 CO2 Ar H20
% mol 71.67 2.88 8.21 0.86 16.35
[0033] The composition of the sea water stream used in the simulation is
reported in Table 1 of Appendix 1 to this disclosure. The results from the
PHREEQC
simulation are presented in Appendix 2 to this disclosure. The final pH of the
sea
water solution is about 5.8 after being exposed to flue gas and reaching
equilibrium
conditions, as shown in part B of Appendix 2. This acidified solution is then
neutralized with a 0.5M sodium hydroxide solution to bring the pH close to its
original value (-8.2).
[0034] As set out in Appendix 2, the saturation indexes for different
carbonates are presented for the original sea water solution, for the
acidified sea
water solution, and after neutralization. The saturation index (SI) is defined
as:
SI = log (¨IAP (11)
Ksp
where, TAP is the ion activity product and lc is the equilibrium solubility
constant.
- 7 -
CA 02915623 2015-12-18
PAT 103540-1
[0035] Positive SI values indicate that the IAP is greater than the K51,
and
precipitation should occur. For negative SI values the IAP is smaller than the
K51,
and precipitation is inhibited. Values near zero indicate equilibrium. For
calcite, SI
values around 0.2 log units may suggest equilibrium if pH measurements are
considered to be reliable. The activity of the carbonate component (C032-) is
a
direct function of pH: a decrease in pH of one unit will decrease the activity
of C032
byone log unit, as described at, for example, Ouled Ameur, Z. et. al. (2015)
Stimulation of High Temperature SAGD Producer Wells Using a Novel Chelating
Agent (GLDA) and Subsequent Geochemical Modeling Using PHREEQC. SPE
International Symposium Oilfield Chemistry, The Woodlands, Texas, USA, 12-15
April, 2015.
[0036] As shown in the PHREEQC output file in Appendix 2, sea water is
initially oversaturated with calcite, aragonite, and dolomite. This
oversaturation is
a characteristic of sea water that has been extensively documented, such as
in, for
example, Mitchel, M.J. et. al. (2010) A model of carbon dioxide dissolution
and
mineral carbonation kinetics. Proceedings of the Royal Society A 466, 1265-
1290.
[0037] After equilibrium with flue gas, the sea water is no longer
oversaturated with carbonates. The increased partial pressure of CO2 in the
flue
gas causes a drop in the pH of the sea water, increasing the solubility of
carbonates. After the pH of the acidified sea water has been brought back to
the
original sea water pH by neutralization with NaOH, the saturation indexes for
calcite
(1.26), aragonite (1.11) and domolite (3.40) are greater than one, indicating
that
precipitation of dolomite, calcite, and aragonite is, at least theoretically,
possible.
[0038] As described above, a prerequisite for precipitating carbonates
out of
sea water is to counter the excess of hydronium cations (W). Simple
neutralization
with, for example, a base (hydroxide ion donor) is effective at shifting the
equilibrium to the point at which precipitation of carbonate is feasible.
Alternatively
or additionally, a buffer solution may also be used to maintain a constant pH
in the
solution. Alternatively, or additionally, neutralization may be performed by
removing H ions using a membrane. Continued movement of the H ions across
the membrane may be facilitated through a reaction that consumes protons on
one
side of the membrane.
- 8 -
CA 02915623 2015-12-18
PAT 103540-1
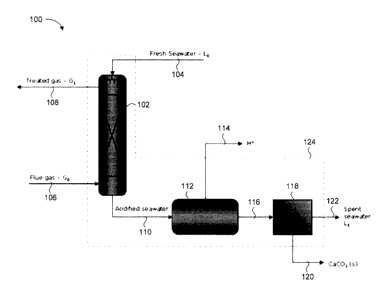
[0039] Fig. 1 shows a schematic diagram of an example system 100 for the
capture of CO2 using sea water and subsequent neutralization of the acidified
sea
water. The system includes a CO2 absorption column 102, into which sea water
104 and flue gas 106 enter. Within the CO2 absorption column 102, CO2 from the
flue gas 106 is absorbed by the sea water 104. Treated gas 108 and acidified
sea
water 110 exit the CO2 absorption column 102. The acidified sea water 110
enters
a neutralizer 112, which removes H+ ions 114 from the acidified sea water 110
to
water's increase the pH. Neutralized sea water 116 enters a separator 118 in
which
CaCO3 precipitate 120 is removed, leaving spent sea water 122.
[0040] The CO2 absorption column 102 may be designed taking into
consideration the equilibrium between CO2 in air and CO2 in sea water, as well
as
the kinetics of CO2 dissolution and hydration. It is possible to carry out the
absorption at constant or variable pH. To maintain a constant pH, it is
necessary to
remove the excess hydronium ions formed by the dissolution of CO2. If the
produced hydronium are not removed or otherwise neutralized, then the pH will
be
lower as the sea water moves along the absorption column 102. The equilibrium
and proposed operating diagram 200 corresponding to a variable pH operation
for
the absorption column is shown in Figure 2A.
[0041] Operation at variable pH requires a very high liquid to gas ratio.
As
shown in FIG. 2A, the minimum molar L'/G' ratio is 1770 mol H20/ mol air,
which is
approximately equivalent to 1050 kg sea water /kg of flue gas. The calculated
minimum liquid to gas ratio at variable pH is much higher than the economical
optimum calculated in previous section. As it is not feasible to operate the
column
at a liquid to gas ratio lower than the minimum (because it will require the
operating line to cross the equilibrium line), then it is concluded that the
absorption
will have to be conducted at a ratio much higher than the economical optimum.
Alternatively, the absorption may be carried out at constant pH, which
effectively
modifies the equilibrium curve 200, as illustrated in chart 202 shown in the
Figure
2B.
[0042] At pH 8.2, the minimum liquid to gas ratio is only 7.8 mol H2O /
mol of
air, or 4.6 kg of sea water per kg of flue-gas. Operating at 1.5 times the
minimum
L/G ratio, or about L'/G' = 12.4, eight ideal separation stages are needed to
- 9 -
CA 02915623 2015-12-18
PAT 103540-1
achieve the proposed absorption. If the operation is performed at the
economical
L/G ratio, near 60 kg of sea water per kg of flue-gas or L'/G' = 100, the
number of
ideal equilibrium stages required may be reduced to two. If only a reduced
number
of separation stages are required, then it might be feasible to perform inter-
stage
neutralization of the excess acid.
[0043] The size (height and diameter) of the absorption column 102
absorbing the CO2 into sea water may be determined by considering the mass
transfer and reaction kinetics limitations. The CO2 hydration reaction is
usually
considered to be the limiting reaction step in the CO2 dissolution and
mineralization
process, while the other reactions are assumed to occur almost instantly. It
must
be noted, however, that the carbonate precipitation and crystallization
reactions are
indeed mass-transfer limited. For the purpose of this preliminary estimation,
it will
be assumed than only the hydration reaction limits the whole process, and all
other
species reach equilibrium instantly.
[0044] From reaction (2), the rate of disappearance of CO2 may be written
in
terms of the forward and reverse reaction constants and the concentration of
the
species intervening in the reaction. Assuming that instantly the equilibrium
between
the CO2 in the gas phase and the CO2 in the liquid phase is reached, as well
as the
equilibrium between carbonic acid and the bicarbonate and hydrogen ions, then
it is
possible to write:
r
rc02 = [CO 2(aq)]+ k2- [14 2CO3 = ¨k;Keqi [CO2( ]-1- 2
PC031141 (Si)
Keq3
[0045] The forward reaction is assumed to have a rate constant k2+ = 0.06
5-1
(see, for example, Bond, G.M. et al (2001) CO2 Capture from coal-fired utility
generation plant exhausts, and sequestration by a biomimet route based on
enzymatic catalysis - Current Status. NETL, First National Conference on
Carbon
Sequestration), while the backward reaction is assumed to have a rate constant
k2-
= 20 s-1 (see, for example, Mitchel, M.J. et al (2010) A model of carbon
dioxide
dissolution and mineral carbonation kinetics. Proceedings of the Royal Society
A
466, 1265-1290).
[0046] If each single stage is treated as a Continuous Stirred Reactor
(CSTR)
(ideal mixing in the stage), then the stage volume may be calculated as:
- 10 -
CA 02915623 2015-12-18
PAT 103540-1
FCO2X
V CSTR
(52)
r rco, 'exit
[0047] The maximum possible conversion is determined by the equilibrium
conditions. At the equilibrium (with X (reaction advancement at anytime) = Xeq
(reaction advancement at equilibrium)), the reaction rate is effectively zero,
which
will require an infinite reactor volume to occur. To calculate the required
volume for
a conversion less than Xeq, the concentration of all relevant species leaving
the
reactor may be determined, as the conditions inside the reactor (or
equilibrium
stage) are assumed to be the same as those of the stream leaving the reactor
(i.e.
ideal mixing). The concentrations of all species at equilibrium may be first
calculated and then the concentrations at the specified conversion point may
be
determined by interpolation. The reaction rate was then calculated using Eq.
(Si)
and the concentrations at the specified conversion point. The equilibrium
conversion
between sea water and a typical power plant flue gas (usually containing 8% of
CO2) at atmospheric pressure was determined to be roughly 60% (see, for
example, Xu, Xiaochun et al. (2003) Separation of CO2 from Power Plant Flue
Gas
Using a Novel CO2 "Molecular Basket" Adsorbent, Fuel Chemistry Division
Preprints
2003, 48(1), 162).
[0048] Table 2 included in Appendix 3 to this disclosure shows the
estimated
reactor volume for different conversions as well as the expected reactor
volume if a
catalyst is used to speed up the reaction. In one case, it is assumed that the
catalyst may provide a 70-fold increase in reaction rate, similar to what is
expected
if the carbonic anhydrase enzyme is used. In the second case, a 14-fold
increase in
the reaction rate is considered, which corresponds to what is expected if
nickel
nanoparticles are used (see, for example, Ouled Ameur, Z. and Husein, M.M.
2013
Electrochemical behavior of Potassium Ferricyanide in aqueous and (w/o)
Microemulsion in the Presence of Dispersed Nickel Nanopartciles. Separation
Science and Technology 48(5): 681-689).
[0049] To determine the amount of H+ ions to be removed or neutralized
from
the acidified sea water to cause precipitation of CaCO3, a total mass balance
and
elemental balances, satisfying all equilibrium conditions, may be performed
around
- 11 -
CA 02915623 2015-12-18
PAT 103540-1
the process envelope, such as the envelop shown as the dashed line 124 in the
example system 100 shown in FIG. 1.
[0050] The total mass balance and elemental balances are given by:
Total mass balance is given by:
Go +Lc,(12a)
= + mcaco,
Carbon balance is given by:
G0[CO2]0 +L1(CO*2]0 +[HCOs]o +[C032]0)= G1[CO2]1 +Li([CO*2], +[HCO]i
+[0:X1)+ncaco,
(12b)
Hydrogen balance is given by:
L00-1C0i]0 +[11-]0+ 2 [H20]3 + [OH-]= (HCOi + [H+ + 2 [H20]1 + [OH-]1 )+ nw_
(12c)
Oxygen balance is given by:
2G0[CO2]0+ Lo picoijo + [H20j0 + [OH- ]0 2 [CO*2 + 3[C03210 =
2G0 [CO2 Jo + (3 [HCOi +[H20]1 + [OH-]1 + 2 [CO*2 + 3 [C032 -]
)+ 3ncaco,
(12d)
where:
Lo is the initial sea water mass flow rate in kg/[t]
Go is the initial flue gas conditions mass flow rate in kg/[t]
L1 is the acidified sea water mass flow rate (after reaching equilibrium) in
kg/[t]
G1 is the flue gas flow rate (at final conditions) in kg/[t]
IT is the mass flow rate of protons ions in kg/[t]
mCaCO3 is the mass flow rate of calcium carbonate in kg/[t]
n
CaCO3 is the molar flow rate of calcium carbonate in mole/[t]
and
nu+ is the molar flow rate of proton ions in mole/[t]
[0051] The equilibrium relations are given by:
- 12 -
CA 02915623 2015-12-18
PAT 103540-1
[CO*2] = IC, fcc,2
(13a)
[HCC30][H'] = K1[CO*2]
(13b)
[C0][H' ]= K2[HCOi]
(13c)
[OH-][H]= K õ,
(13d)
[co;-][ca 2 5 Kfpalcite
(13e)
[ 3il a
c02-c 2 5_ p KsAragonite
(13f)
[ ,CO2-i[mg p
2 < K smagnesite
(13g)
[co[ 2 [ca2 11
mg2 < KsDpolomite
(13h)
[0052] The values for K0 and Kw may be obtained from, for example
Dickson,
A.G. and Goyet, C. 1994, Handbook of methods for the analysis of the various
parameters of the carbon dioxide system in sea water, U.S. Department of
Energy
ORNL/CDIAC-74; the values for K1, K2 may be obtained from Mojica, F.J. and
Miller,
F.J. 2002, The values of pKi + pK2 for the dissociation of carbonic acid in
sea water,
Geochimica et Cosmochimica Acta 66(14), 2529-2540; the solubility product
constants (Ksp) for calcite and aragonite may be obtained from Al-Rawajfeh,
A.E.
(2004) Modelling and simulation of CO2 release in multiple-effect distillers
for sea
water desalination, Martin Luther Universitat Halle Wittenberg, PhD thesis,
2004;
and the values for all other constants may be obtained may be obtained from
Dean.
The fugacity of CO2 may be assumed to be equal to its partial pressure (i.e.,
ideality
may be assumed).
[0053]
Assuming a sea water inlet flowrate of 1 kg/[t] and a flue gas flowrate
of 0.001 kg/[t], the composition and flowrate of the outlet streams may be
calculated as shown in Table 3 included in Appendix 4 to this disclosure. In
the
calculations reported in Table 3, the amount of H+ produced is adjusted to
give the
treated gas a CO2 concentration equal to that of atmospheric air (around
0.039%).
That is, from a greenhouse gas perspective the discharged treated gas may be
considered to be equal to atmospheric air (though depleted in oxygen).
- 13 -
CA 02915623 2015-12-18
PAT 103540-1
[0054] For capturing 1 MM tons of CO2 per year, the following rates are
desired:
CO2 sequestered = 32.15 kg/s = 116 t/h
H+ consumed = 1787 molts
CaCO3 (s) produced = 115 kg/s = 412 t/h
Fresh sea water = 897626 m3/h = 249 m3/s
[0055] In an example in which caustic soda (NaOH) is used to neutralize
the
excess H+, utilizing 2.22 ton of NaOH facilitates capturing one ton of CO2. In
this
example, for a bulk cost for NaOH of $415/ton of NaOH, the cost for CO2
capture
will be $922 per ton. Therefore, cost considerations may make neutralization
utilizing NaOH unfeasible.
[0056] Precipitation of carbonates may be accomplished if a weak base is
added to the acidified sea water, or to the sea water before CO2 absorption.
Ammonia (NH3) may be used in a similar way as it is used in the Solvay process
for
the production of sodium bicarbonate.
[0057] When ammonia is bubbled in water the following reaction takes
place:
NH3 (g) H20 4-> NH4 + OH- AI/;? = -31.5 kYmol, AGr =17.1 kJ/mol (14)
or at lower pH:
NH3 (g) 1-1+ 4--> NH4+ AH = -
87.3 kJ/mol, AG = -62.8 kJ/mol (15)
[0058] If reactions (14) and (15) are coupled to reactions (6) and (7),
the
following global reactions may take place:
2NH3 + Ca2+ + CO2 + H2O-CaCO3(s) + 2 NH4 + AHr = -159.9 k3/mol, AG ¨69.6
kJ/mol
(16)
NH3+ Ca2+ + CO2 + H20 CaCO3(s) + H+ + NH4 + AHr = -72.6 kJ/mol, AG = -6.8
kJ/mol
(17)
- 14 -
CA 02915623 2015-12-18
PAT 103540-1
NH3 + Ca2+ + CO2 + OH- CaCO3(s) + NH4 AH(r)= -128.4 kJ/mol, AG = -86.7
kJ/mol
(18)
[0059] All three reactions (16 to 18) are exothermic and spontaneous. The
pH
of the system will determine which reaction is dominant.
[0060] To determine the amount of ammonia that is desirable to achieve
the
mineralization of CO2, the hydrogen balance formulated in Eq. (12c) may be
modified as follows:
I0(HC0i10 + [H 10 + 2 [H20]0 +[OH-10)+ =
41-1C0i]1 +[H ]1 +2[H20]1 2 [H20], + [OH-], +3[N113]1 + 4[N114],)
(12c*)
and the following equilibrium relation may be utilized to the set of
equilibrium
constraints:
[NH, ][OH ] = Kb [NH3]
(13i)
[0061] Solving the modified system of equations (12* and 13i), the amount
of
ammonia that facilitates the capture 1 ton of CO2 is calculated as 1.2 ton of
NH3. At
a current cost of $600/t of NH3 the cost of carbon capture (due to NH3 only)
may be
$718/ton of CO2. Therefore, cost considerations may make neutralization
utilizing
NH3 economically unfeasible. However, a closed-loop operation with ammonia
regeneration may reduce operating costs and will be described later in this
disclosure.
[0062] As an alternative to neutralization with a base, the excess
hydronium
ions (H+) may be removed from the acidified sea water using a membrane; for
example a polymeric proton exchange like Nafion could be used for this
purpose. An
anionic membrane may also be used to allow negatively charged ions (mainly
chlorides and fluorides) to move out of the acidified water in order to
maintain
electroneutrality. To sustain a continuous flow of protons across the membrane
an
electrolytic cell may be utilized in order to convert the protons into
hydrogen and
the chloride ions into chlorine.
[0063] An example of an electrochemical cell 300 that may be utilized to
sustain a continuous flow of protons is shown in Fig. 3. The electrochemical
cell 300
- 15 -
CA 02915623 2015-12-18
PAT 103540-1
includes a water inlet 302 through which acidified sea water flows into the
cell 300,
and a water outlet 304 through which water exits the cell 300. The cell 300
includes a cathode 306 and an anode that are coupled via power supply 310. A
proton exchange membrane 312 separates the inlet 302 from the cathode 306, and
an anion exchange membrane 314 separates the inlet from the anode 308. The
cell
300 also includes a hydrogen outlet 316 through which hydrogen exits the cell
300,
and a chlorine outlet 318 through which chlorine exits the cell 300.
[0064] The electrochemical reactions of halogens are highly reversible,
as
described in, for example, Srinivasan 5, and Kirby B. Status of fuel cell
technologies. In: Fuel Cells: From Fundamentals to Applications, Editor: S.
Srinivasan. Springer, 2006. For this reason, there has been a great interest
in
developing regenerative fuel cells based on the H2/C12 system for energy
storage.
Cell designs for energy storage applications desirably have a high energy
conversion efficiency; therefore they are a desirable start point for
developing an
electrochemical cell for enhancing carbonate precipitation in sea water.
[0065] Although several designs have been proposed for the H2/C12 system,
as
described in, for example, Thomassen M, Sandnes E, Borresen B, and Tunold R.
(2006) Evaluation of concepts for hydrogen - chlorine fuel cells. Journal of
Applied
Electrochemistry 36, 813-819 (hereinafter "Thomassen"), the H2/Cl2 galvanic
cell is
still considered an immature technology, as discussed more fully in House
K.Z.,
House C. H., Schrag D. P., and Aziz M. J. (2009) Electrochemical acceleration
of
chemical weathering for carbon capture and sequestration. Energy Procedia 1,
4953-4960.
[0066] Thomassen discloses the performance of an ordinary polymer
electrolyte membrane fuel cell based on a Nafion membrane, a fuel cell based
on a
combination of circulating hydrochloric acid and a Nafion membrane, and a
system
based on a phosphoric acid doped Polybenzimidazole (PBI) membrane. Thomassen
concludes that even though the three systems studied achieved open circuit
voltages close to the reversible and exhibited fast electrode kinetics, stable
operation was not possible due to electrocatalyst corrosion.
[0067] More recently, the use of a ruthenium alloy oxide
[(Ru0.09000.90304]
electrocatalyst for the chlorine electrode has been considered, as described
more
- 16 -
CA 02915623 2015-12-18
PAT 103540-1
fully in Huskinson B., Rugolo J., Mondal S.K., and Aziz M.J. (2012) A high
power
density, high efficiency hydrogen-chlorine regenerative fuel cell with a low
precious
metal content catalyst. Energy Environ. Sci. 5, 8690 - 8698.
[0068] In this example system Nafion 112 was used as the proton-exchange
membrane. The ruthenium loading in the chlorine-side electrode was 0.15 mg
Ru/cm2, while the hydrogen-side electrode used a standard ELAT gas diffusion
electrode (GDE) with a Pt loading of 0.5 mg/cm2. The reported power density
was
approximately 0.4 W/cm2 at 90% galvanic efficiency.
[0069] To evaluate the feasibility of using an electrochemical cell for
removing
excess hydronium ions and to promote carbonate precipitation, it may be
desirable
to estimate the energy consumption associated with operating the cell. The
electric
potential, E, that facilitates operating an electrochemical cell may be
determined
using the Nernst equation:
E = E0 - RT In Q
zF
(19)
where E is the standard cell potential, Q is the reaction quotient, z is the
number
of moles of electrons transferred in the cell reaction, R is the universal gas
constant, F is the Faraday constant, and T is the absolute temperature.
[0070] The reaction quotient Q in Eq. (19) may be calculated assuming
that
the activity of the gas species involved in the electrochemical reaction is
equal to
their partial pressure (P,), and the activity of the ionic species is equal to
their
molar concentration in the solution.
[0071] For the example electrochemical cell 300 illustrated in Fig. 3,
the
global reaction may then be written as:
2 H+ + 2 C1 4-4 C12 H2 = -1.36 V
(20)
and cell potential that facilitates the drive reaction (20) forward is
estimated at E=-
1.365 V. In practice, however, an overvoltage is desired to result in
increased
power consumption.
[0072] From the reduction half-reaction utilized in the example
electrochemical cell 300 shown in Fig. 3, it may be seen that for each mole of
H+
reduced, 1 mole of electrons is consumed. Therefore the electron consumption
rate
for sequestering 1MM ton of CO2/year will be 1787 mol
- 17 -
CA 02915623 2015-12-18
PAT 103540-1
[0073] The power utilization may be given by:
P = lie FE = 1787 mol/s x 96484.56 C/mol x1.365 V 106 = 235 MW
[0074] 235 MW relates to the power utilization if no overvoltage is
applied, to
carry out the specified reaction. For an overvoltage of 0.8 V, the power
consumption may be calculated as 373 MW.
[0075] The hydrogen gas produced may be used to generate electricity and
partially off-set the power utilization of the process. Hydrogen may be
converted to
electricity through a combined cycle or in a fuel cell. Alkaline fuel cells
have
demonstrated efficiencies up to 60% (see, for example, Mulder, G. et al.
(2008)
Market-ready stationary 6 kW generator with alkaline fuel cells. ECS
Transactions
12, 743-758), while phosphoric acid fuel cells, proton exchange membrane fuel
cells, and solid oxide fuel cells may be up to 85% efficient when used in co-
generation of electricity and heat (see, for example, Hamelin J., Agbossou K.,
Laperriere A., Laurencelle F, Bose T.K. (2001) Dynamic behavior of a PEM fuel
cell
stack for stationary applications. International Journal of Hydrogen Energy
26(6),
625-629, and Edwards P., Kuznetsov V., David W.I.F., Brandon N.P. (2008)
Hydrogen and fuel cells: Towards a sustainable energy future. Energy Policy
36(12), 4356-4362). The efficiency of combined cycle gas turbines have
increased
in recent years and typical values of newly-build power plants are reported in
around 61% (see, for example, Robb D. (2010) CCGT: Breaking the 60 per cent
efficiency barrier. Power Engineering International 18(3)). Assuming a
conversion
efficiency of 70% the process will potentially produce 179 MW of power.
Consequently, the net energy utilized will be:
0 V overvoltage: Pnet = 56 MW = 1.7 GJ/ton CO2 = $29/ton CO2
0.8 V overvoltage: Pnet = 194 MW = 6.0 GJ/ton CO2 = $101/ton CO2,
where electricity is assumed to cost 6 ct/kWh.
[0076] An alternative cell design aimed at reducing the net energy
consumption, and in turn reducing the cost/ton of CO2 capture, consists of
bubbling
oxygen at the cathode, which suppresses hydrogen gas production. In this case,
the half-cell reaction is:
02 (g) 4 H+ + 4e- *-4 2 H20 E = 1.229 V (21)
- 18 -
CA 02915623 2015-12-18
PAT 103540-1
[0077] And the overall electrochemical reaction is:
4 H+ + 02(9) 4 a- 2 Cl2 + 2 H20 E = -0.131 V (22)
[0078] The energy utilized by this alternative cell design is:
RT PC21
E = E __ in
4F [1-Ff[C1-]2/33 - = -0.136 V (23)
[0079] Consequently, the net energy utilized will be:
0 V overvoltage: Pnet = 23.4 MW = 0.73 G3/ton CO2 = $12/ton CO2
0.8 V overvoltage: Pnet = 161 MW = 5.0 GJ/ton CO2 = $84/ton CO2.
[0080] Depending on the operating temperature of electrolitical cell,
heat
from the produced water or steam could be recovered or integrated to adjoining
processes.
[0081] The calculations in the previous description were performed for an
assumed sea water rate of 1 kg and 0.001 kg of flue gas, for a Lo/Go ratio
equal to
1000. Modifying this ratio affects both the relative amount of sea water
utilized and
the energy utilized for pumping, as well as the final equilibrium points and
the net
energy utilized for electrolysis.
[0082] The electrochemical system described in this disclosure, such as
the
example electrochemical cell 300, may be similar in practice to existing
membrane-
based electrochemical cells for the chlor-alkali process. However, certain
aspects of
the carbonation process are different from the chlor-alkali processes, which
may
utilize a new cell design or special membranes. Among the aspects that a cell
design may take into consideration are:
= Potential formation of precipitates inside the cell
= Ion concentrations lower than in a conventional chlor-alkali system if
untreated sea water is to be used
= Presence of multiple contaminants that may adversely affect membrane
performance
[0083] Cells may be designed to minimize or inhibit the potential
formation of
a solid layer on the membrane surfaces. Equipment may also be designed to
allow
- 19 -
CA 02915623 2015-12-18
PAT 103540-1
recycling of acid effluent into the membrane chambers in case solids are
deposited
on the membrane.
[0084] In Table 4 included in Appendix 5 to this disclosure, the power
utilization and sea water consumption per ton of CO2 captured are shown for
different liquid to gas ratios. As shown in the results shown in Table 4,
decreasing
the L1/G1 ratio improves the overall performance of the operation, with less
energy
per ton of CO2 is utilized to carry out the electrolysis. Moreover, the amount
of sea
water utilized also decreases by decreasing the L1/G1 ratio, which will reduce
the
associated pumping costs (both capital and operational).
[0085] As the L1/G1 ratio decreases, the amount of CO2 mineralized (i.e.
converted into calcium or magnesium carbonates) decreases as a fraction of the
total amount of CO2 captured. This is because sea water is originally
saturated in
CO2 and carbonates, and changing the final concentration of calcium and
magnesium ions (through precipitation) shifts the saturation levels of
carbonated
ions and the solubility of CO2. At higher L1/G1 ratios (e.g., >70), more CO2
is
precipitated as carbonate than what has been effectively removed from the flue
gas, indicating that part of the CO2 originally in the sea water was co-
precipitated.
At lower L1/G1 ratios (e.g., <60) more CO2 is removed from the flue gas than
what
is precipitated as solid carbonates, which indicates that the sea water has
been
enriched in carbonate ions and solubilized CO2. Consequently, it may be
expected
that part of this excess CO2 will came out of solution if the pH is lowered or
if there
are other processes that modify the final equilibrium points. Furthermore, as
the
spent sea water may be returned to the sea, it is desirable to return it at
the same
pH as it was taken up in order to inhibit the disturbance to the natural
processes
occurring in the sea.
[0086] To calculate the energy utilized for pumping sea water is
necessary to
first determine the total pressure drop in the liquid circuit (sea water
pipeline and
process equipment). For the purpose of a preliminary estimation, a 5 bar
maximum
pressure drop was assumed. As a result, the total energy cost for both pumping
and electrolysis may be calculated per ton of CO2. The total cost per ton of
CO2
captured, as well as per ton of CO2 mineralized is shown in the chart 400
shown in
FIG. 4 (For a 0.0 V overvoltage).
- 20 -
CA 02915623 2015-12-18
PAT 103540-1
[0087] The total CO2 captured is a contentious quantity, as it may be
defined
as either the total CO2 removed from the flue gas and stored in the sea water
and
in the solid carbonates, or it may be strictly defined as the CO2 that has
been
permanently sequestered (over geological time) in a mineral form. If the first
definition is followed, then it may be concluded that the total energy
consumption
increases with increasing L/G ratio. However, if the more strict definition is
followed, an optimum L/G ratio may be found (around 50, mass basis) at which
the
energy consumption per ton of CO2 captured is minimized. As pointed out
previously, it is desirable to return the spent sea water to the sea at its
original pH.
Consequently, the preferred L/G ratio will be slightly above the economic
optimum
point (between 50 and 70, mass basis).
[0088] The disclosed electrochemical CO2 capture process has chlorine gas
as
a by-product. Approximately 1.6 ton of Cl2 may be produced per ton of CO2
captured. Current worldwide chlorine production capacity is 60 million metric
tonnes per year (see, for example, World Chlorine Council, 2012). Chlorine is
mainly used for the production of polyvinyl chloride (35%), isocyanate and
propylene oxide (15%), other organic derivatives (20%), inorganic chemicals
(20%), and other uses including sanitation and water treatment (10%). The
global
market for PVC is expected to grow at a rate of 4-5 % per year.
[0089] Industrial production of chlorine is an energy intensive process
that
produces caustic soda (sodium hydroxide) as a by-product. In some areas, the
produced soda does not have a local market and consequently must be
transported
over long distances, adding to the total cost. In these locations, the
proposed
process might be more attractive as only chlorine, but not caustic soda, is
produced
as a by-product of carbon capture.
[0090] The production of large amounts of chlorine associated with the
electrochemical carbon capture system will nonetheless flood the current
chlorine
market. For instance, capturing only 0.12% of the global CO2 emissions (30.8
billion tons in 2009 (see, for example, Friedlingstein P., Houghton R.A.,
Marland G.,
Hackler J., Boden T.A., Conway T. J., Canadell J. G., Raupach M.R., Ciais P.
and Le
Quere C. (2010) Update on CO2 emissions. Nature Geoscience 3, 811-812) would
produce enough chlorine to supply all current worldwide consumption. Current
Free
- 21 -
CA 02915623 2015-12-18
PAT 103540-1
on Board (FOB) prices for chlorine range from $800 to $1600 per ton and this
price
would likely collapse if the market were flooded. A reduction in chlorine
prices,
however, might increase the global demand for PVC and other chlorinated
plastics.
[0091] In Table 5 included at Appendix 6 to this disclosure, a list of
chlorine-
containing polymers is presented together with the relative chlorine content
in the
monomer, and total global consumption. At 29924 kton/year production (Table
5),
PVC is the main chlorinated polymer, with all other chlorinated polymers
accounting
for less than 3% of the chlorinated polymer market share.
[0092] Chlorine may also be used to produce hydrochloric acid than may
later
be used as a leaching agent in mineral extraction or simply be disposed of
after
neutralization with certain minerals. Chlorine gas reacts spontaneously with
water
in a disproportionation reaction:
Cl2 (g) + H2O HCI + HCIO ATP= -2.22 kJ/mol (24)
[0093] The hypochlorous acid is highly reactive and yields HCI in the
presence
of light or transition metal oxides of copper, nickel, or cobalt:
2 HCIO "V > 2 HCI + 02 AFP= 92.5 kJ/mol (25)
[0094] The hydrochloric acid may then be used to dissolve mineral
deposits.
Of particular interest are the deposits of silicates that may be used as end
donors of
calcium and magnesium ions.
[0095] In the above disclosure, the source of calcium and magnesium
cations
was assumed to be from sea water; these cations could also be derived from
land
based rocks. Mineral carbonation is a natural process in which calcium and
magnesium silicates react with atmospheric CO2 or CO2 dissolved in ground
water
to yield calcium or magnesium carbonate and silica (S102). Unfortunately, this
natural process is very slow and only of significance over geological time
frames.
Mining, grinding, and dispersion of silicates have been proposed as ways to
accelerate mineral carbonation. These operations, however, may consume
significant amounts of energy. Leaching of silicates with HCI has the
potential to
reduce energy utilization.
- 22 -
CA 02915623 2015-12-18
PAT 103540-1
[0096] Hydrochloric acid, working in a closed loop manner, may be used to
accelerate the in-situ underground conversion of silicates into silica and the
produced calcium or magnesium brine may then be used for the production of
calcium or magnesium carbonates at the surface. In this arrangement, fresh sea
water consumption may be reduced to only make up the chloride losses to the
underground silicate formation.
[0097] The reaction of silicates with HCI is in all cases exothermic and
spontaneous. As an example, consider the following three silicates:
Mg2SiO4(S) + 4 HCI 2 MgCl2 SiO2() 2 H20 AH
¨236.9 kJ/mot (26)
Ca2SiO4() + 4 HCI -4 2 CaC12 (aq) S102 (s) 2 H20 Ae= -255.3kYmol
(27)
MgSiO3 + 2 HCI MgC12 (am + Si02 (s) + H20 Ae= -109.3 k.1/mol
(28)
[0098] Because the silicate dissolution reaction will occur underground,
it is
unlikely that the heat of reaction could be harvested. On the other hand, the
heat
generated by the chlorine dissolution reaction may potentially be recovered to
at
least partially offset the power consumption during electrolysis.
[0099] The global reaction for CO2 sequestration may be summarized as
follows:
Ca2+ + CO2 (g) H2O CaCO3 + 2 H+
(29)
4 Fl+ + 02 (g) 4 a- 2 Cl2 + 2 H2
(30)
2 Cl2 (9) + 2 H20 ¨> 4 HCI + 02
(31)
Ca2+ + CO2 (g) + H20 + 2 a- CaCO3 + 2 HCI Ae= -378.3 k.1/mol
(32)
[00100] As shown above, the global reaction is exothermic; therefore,
there is
the potential (through efficiency improvements and thermal integration) to
carry
out the process without external energy sources. A schematic of a system 500
illustrating for this alternative process is shown in FIG. 5.
[00101] The system includes a CO2 absorption column 502, similar to CO2
absorption column 102 shown in FIG. 1, a electrochemical cell 504, similar to
electrochemical cell 300 shown in FIG. 3, a separator 506, similar to
separator 118
- 23 -
CA 02915623 2015-12-18
PAT 103540-1
shown in FIG. 1, and a tank 508 the chlorine produced in the electrochemical
cell
504 reacts to produce hydrochloric acid. Heat from the reaction occurring
within
the tank 508 may be recovered by a coil 510. Hydrochloric acid is deposited in
a
silicates deposit 512 to accelerate the in-situ underground conversion of
silicates
into silica and the produced calcium or magnesium brine may then be used for
the
production of calcium or magnesium carbonates at the surface, facilitating the
operation of a closed loop to reduce fresh sea water consumption of the
process.
[00102] As shown in reactions (26-28), for each mole of silicate that
reacts,
one mole of silica (Si02) is produced. The molar volume of silica is 22.68
cm3/mol,
which is slightly more than 50% the molar volume of the magnesium silicate
(43.02
cm3/mol) or calcium silicate (40.05 cm3/mol). Therefore, it is expected that
as the
leaching progresses, a cavern will be formed. Final abandonment of the cavern
may
be accomplished by flooding the cavern with carbonate slurry.
[00103] Deposits of magnesium and calcium silicate are relatively common.
In
Canada, biotite and amphibolite -silicate rocks rich in magnesium and calcium-
are
a common occurrence in the bedrock of North Eastern Alberta, South and Central
British Columbia, and Northern Saskatchewan. Higher quality deposits of
wollastonite (CaSiO3) and olivine (Mg2S104) have also been reported in British
Columbia and Quebec. Some of these deposits are currently being commercially
exploited and others are reported as prospects. The Zippa Mountain (56039'10"N-
131018'07"W) in British Columbia has estimated reserves totalling 20 Million
tons of
wollastonite, which could provide enough calcium to permanently sequester up
to
7.58 million tons of CO2.
[00104] As described above, ammonia (NH3) may be used to assist the
precipitation of calcium and magnesium carbonates. The resulting solution will
be
enriched in solubilised ammonium chloride (NH4CI). It is possible to
regenerate the
ammonia from this ammonium chloride solution, by passing the solution through
an
activated carbon (AC) column (see, for example, Huang, H.P. et al (2001) Dual
alkali approaches for the capture and separation of CO2. Energy and Fuels
15(2),
263-268, (hereinafter "Huang")). The absorption of chloride and regeneration
of
ammonia is described as follows:
NH4CI + AC AC=FICI + NH3
AHads,õption 10.6 kJ/mol (33)
- 24 -
CA 02915623 2015-12-18
PAT 103540-1
[00105] The activated carbon may later be regenerated by flowing steam (or
hot water) through the activated carbon (see, for example, Huang):
AC.1-1C1 + H20 AC + HCI + H20 A I I desorption 24.8 kJ/mol (34)
[00106] The system 500 that carries out the carbon mineralization process
previously described with reference to FIG. 5 may be modified to incorporate
the
ammonia-assisted carbonated precipitation and ammonia regeneration, as shown
in
the FIG. 6.
[00107] FIG. 6 shows a system 600 for CO2 mineralization with ammonia and
ammonia regeneration. The system 600 includes a CO2 absorption column 602,
similar to CO2 absorption column 502 shown in FIG. 5, a precipitation column
604
in which carbonate precipitates with ammonia as discussed above, and ammonia
regeneration tanks 606 and 608. Hydrochloric acid that exits the ammonia
regeneration tanks 606 and 608 is deposited in a silicates deposit 610,
similar to
the deposition in the silicates deposit 512 described above with reference to
FIG. 5.
Ammonia regeneration systems are common in several industries, and therefore
the system 600 is not described in further detail herein.
[00108] The regeneration of ammonia described in disclosure is based on
the
results reported by Huang using activated carbon. It is possible, however,
that
other processes for regeneration may be more economical.
[00109] The carbonate precipitation with ammonia is an exothermic process,
as
has been shown in Eqs. (31-33). Based on the material balance equations
presented before, the following energies may calculated:
= 1381 MJ will be released per ton of CO2 that is precipitated;
= 45.4 kmol of NH4 + should be regenerated per ton of CO2;
= 481 MJ per ton of CO2 will be released during ammonia regeneration;
= 1126 MJ per ton of CO2 are utilized for regeneration of the activated
carbon;
= Total energy in: 1126 MJ/ton CO2; and
= Total energy released: 1862 MJ/ton CO2.
[00110] Because more energy may be released during the process than the
amount of energy utilized for regeneration of the activated carbon it is at
least
theoretically possible to design a system that will not require any thermal
energy
- 25 -
CA 02915623 2015-12-18
PAT 103540-1
input. The challenge is to achieve a satisfactory energy integration scheme to
allow
the recovery of most of the energy released in the precipitation column and in
the
ammonia regeneration column.
[00111] A preliminary economic evaluation of the four alternative
processes
described above were carried out in terms of expected revenues and capital and
operating costs. It was found that the four alternative processes described
above
have the potential to be economically feasible for permanently capturing CO2
emissions directly from flue gas.
[00112] The use of a catalyst is a desirable to reduce the size of the CO2
absorption equipment (and associated capital costs) as well as to reduce total
operating costs. Catalysts must be selected based on cost, stability, and
activity
level. The carbonic anhydrase enzyme is the most efficient catalyst known for
the
CO2 hydration reaction in terms of activity but the stability of the enzyme
under
industrial operating conditions and the associated costs of the enzyme are
unknown. As an alternative, nickel nanoparticles have been shown capable of
catalyzing the hydration reaction, but the activity of the nanoparticles was
several
times lower than that of the carbonic anhydrase enzyme (see, for example,
Ouled
Ameur, Z. and Husein, M. (2013) Electrochemical behavior of potassium
ferricyanide in aqueous and (w/o) microemulsion in the presence of dispersed
nickel nanopartciles. Separation Science and Technology 48(5), 681 689).
Therefore, it may be desirable to conduct laboratory and pilot tests, using
sea water
or brines and flue-gas to evaluate the performance of several potential
catalysts to
determine the most cost effective one.
[00113] Permanent sequestration of carbon dioxide as solid carbonates may
be
a feasible solution to the increased levels of CO2 in the atmosphere.
Accelerated
carbonation may be achieved through bio-mimicking approaches in which
catalysts
are used to accelerate the dissolution of CO2 in water and the formation of
bicarbonate ions, while membrane or chemical systems may be used to remove the
excess hydronium ions that are formed and that impede carbonate precipitation.
The economic feasibility of electrochemically assisted precipitation of
carbonates
may be increased if the process is coupled to the production and
commercialization
of chlorine, hydrochloric acid, or polyvinyl chloride.
- 26 -
CA 02915623 2015-12-18
PAT 103540-1
[00114] Sequestering solid carbonates may involve depositing solid
carbonates
in shallow waters to create artificial islands or expand shore lines (similar
to the
effect of coral reefs). Carbonate brines may be deposited in caverns or other
underground formations or utilized to create new hills or mountains. Another
possible option is to utilize these CO2 capture products of as construction
materials.
[00115] The described embodiments are to be considered in all respects
only
as illustrative and not restrictive. The scope of the claims should not be
limited by
the preferred embodiments set forth in the examples, but should be given the
broadest interpretation consistent with the description as a whole. All
changes that
come with meaning and range of equivalency of the claims are to be embraced
within their scope.
- 27 -
CA 02915623 2015-12-18
PAT 103540-1
Appendix 1
Table 1 ¨ Typical composition of natural sea water, used for simulating the
equilibrium
condition for the CO2 ¨ sea water system
Ion Mol/kg
Cl- 0.54586
S042- 0.02824
Br- 0.00084
F- 0.00007
Na + 0.46906
0.05282
Ca2+ 0.01028
K+ 0.01021
Sr2+ 0.00009
HCO3- 0.00177
C032- 0.00026
B(0H)3 0.00032
B(OH)4- 0.00010
OH- 0.00001
CO2(g) 0.00001
- 28 -
CA 02915623 2015-12-18
PAT 103540-1
Appendix 2
Geochemical Modelling Based - PHREEQC 3.0 output for equilibrium between sea
water and flue gas. A. Carbonate saturation levels for original sea water; B.
Equilibrium
conditions after contact with flue gas; C. Equilibrium conditions after
neutralization with
NaOH.
A. Initial solution 1. Sea water
pH = 8.220
pe = 4.000
Total CO2 (mol/kg) = 2.131e-03
Temperature (deg C) = 25.000
Electrical balance (eq) = 1.308e-13
Phase SI log IAP log KT
Aragonite 0.59 -7.74 -8.34 CaCO3
Calcite 0.74 -7.74 -8.48 CaCO3
CO2(g) -3.39 -4.86 -1.47 CO2
Dolomite 2.36 -14.73 -17.09 CaMg(CO3)2
Strontianite -0.53 -9.81 -9.27 SrCO3
B. Reaction step 1. Using solution 1.
Solution after equilibrium with gas phase 1. Flue-gas
pH = 5.838 Charge balance
pe = 14.948 Adjusted to redox equilibrium
Total CO2 (mol/kg) = 6.045e-03
Temperature (deg C) = 25.000
Electrical balance (eq) = 1.308e-13
Phase SI log IAP log KT
Aragonite -1.69 -10.02 -8.34 CaCO3
Calcite -1.54 -10.02 -8.48 CaCO3
CO2(g) -0.91 -2.37 -1.47 CO2
Dolomite -2.20 -19.29 -17.09 CaMg(CO3)2
Strontianite -2.82 -12.09 -9.27 SrCO3
C. Mixture 1: Neutralization of acidified sea water
9.950e-01 Solution 1 Solution after simulation 2.
5.000e-03 Solution 2 Caustic
pH = 8.302 Charge balance
pe = 12.484 Adjusted to redox equilibrium
Total CO2 (mol/kg) = 6.015e-03
Temperature (deg C) = 25.000
Electrical balance (eq) = -1.887e-03
Phase SI log IAP log KT
Aragonite 1.11 -7.22 -8.34 CaCO3
Calcite 1.26 -7.22 -8.48 CaCO3
CO2(g) -3.03 -4.49 -1.47 CO2
Dolomite 3.40 -13.69 -17.09 CaMg(CO3)2
Strontianite -0.02 -9.29 -9.27 SrCO3
- 29 -
CA 02915623 2015-12-18
PAT 103540-1
Appendix 3
Table 2 - Required reactor volumes for a single stage as a function of CO2
conversion.
Equilibrium conversion = 0.59. Input flow rate = 1 MM ton of CO2 per year.
Conversion CSTR Reactor Volume (m3) PFR Reactor Volume (m3)
Enzyme Nickel Nickel
Base Base Enzyme
% of Xeci . (70- nanop. nanop.
case case (70-fold)
fold) (14-fold) (14-fold)
90% , 24732 353 1767 6538 93 467
80% 11104 159 793 4623 66 330
70% 6544 93 467 3484 50 249
60% 4250 61 304 2671 38 191
50% 2863 41 205 2034 29 145 ¨
40% 1929 28 138 1509 22 108
30% 1253 18 90 1060 15 76
20% 739 11 53 667 10 48
- 30 -
CA 02915623 2015-12-18
PAT 103540-1
Appendix 4
Table 3 - Inlet and outlet concentrations for a sequestration system with pH
regulation and
L/G = 1000
Sea Spent
Variable Units water Flue Sea Treated
gas gas
feed water
Flowrate kg/[t] 1 0.00100
0.999408 0.00087
6.31E-
[H+] mol/kg 09 1.28E-08
1.12E-
[CO2*] mol/kg 05 2.943 1.12E-05 1.3E-05
2.59E-
[HCO3-] mol/kg 03 1.28E-03
4.61E-
[C032] mol/kg 04 1.13E-04
[Ca2+] mol/kg 0.01028 0.00569
[Mg2+] mol/kg 0.05282 5.29E-02
[Na] mol/kg 0.46906 0.46933
[Cr] mol/kg 0.54586 0.53901
[H20] mol/kg 53.5559 53.585
9.61E-
[Ohl-] mol/kg 06 4.75E-06
pH 8.2 7.89
[CO2(g)] %molar 0.0394% 8.210% 0.0394%
CaCO3 (s) = 0.00459 mol/[t]
H+ = 7.165x10-3 mol/[t]
- 31 -
CA 02915623 2015-12-18
PAT 103540-1
Appendix 5
Table 4 - Power utilization for electrolysis and sea water utilization for the
electrolytic
precipitation of carbonates at different UG ratios
0.8 V
0 V Overvolt. Sea
Electro- Overvolt. CO2
water
Final ns Net P
mineraliz
Lo/GoNet P Elec. Elec. flowrat
pH utilized (0.8 ed
(OV) Cost V) Cost
kmol GJ GJ m3
1000 7.89
55.6 0.7 12.15 5.0 83.64 7756 156.6%
500 7.93
50.1 0.7 10.95 4.5 75.38 3878 126.1%
400 7.94
49.1 0.6 10.73 4.4 73.90 3102 120.6%
300 7.95
48.1 0.6 10.52 4.3 72.42 2327 115.1%
200 7.97
47.1 0.62 10.3 4.3 70.91 1551 109.5%
100 8.04 46.1 0.60 10.1 4.2 69.31 776
103.5%
80 8.10 45.8 0.60 10.0 4.1 68.92 620
102.0%
70 8.14 45.6 0.60 10.0 4.1 68.67 543
101.1%
60 8.22 45.4 0.6 9.92 4.1 68.31 465
99.7%
50 8.39 44.9 0.6 9.81 4.1 67.57 388
96.5%
40 8.74 43.5 0.6 9.50 3.9 65.43 310
84.4%
25 9.14 41.6 0.5 9.10 3.8 62.62 194
53.7%
20 9.25 41.4 0.5 9.05 3.7 62.29 155
43.0%
9.50 41.7 0.5 9.11 3.8 62.75 78 21.5%
Note 1: All values (except pH) are given per ton of CO2 captured (in the sea
water and as solid carbonates).
- 32 -
CA 02915623 2015-12-18
PAT 103540-1
Appendix 6
Table 5 - List of chlorine-containing polymers and the relative chlorine
content in the
monomer and total global consumption.
Polymer Chlorine content World Production
(mass %) kton / year Year
PDCA 74.7% -O
PVDC 73.1% 160 2004
CPVC 56 - 74%
PVC 56.7% 29924 2009
Neoprene 40.0% 360 2012
CPE 25 - 40% 260 2003
CSPE 27 - 35%
PCTFE 30.4% 0.4 1998
- 33 -