Note: Descriptions are shown in the official language in which they were submitted.
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
ANTI-TROP-2 ANTIBODY-DRUG CONJUGATES AND USES THEREOF
INVENTORS: Serengulam V. Govindan and David M. Goldenberg
ASSIGNEE: IMMUNOMEDICS, INC.
RELATED APPLICATIONS
[001] This application claims priority to U.S. Patent Applications 14/259,469,
filed April
23, 2014; 14/040,024, filed September 27, 2013; and 14/258,228, filed April
22, 2014. The
text of each priority application is incorporated herein by reference in its
entirety.
SEQUENCE LISTING
[002] The instant application contains a Sequence Listing which has been
submitted in
ASCII format via EFS-Web and is hereby incorporated by reference in its
entirety. Said
ASCII copy, created on June 24, 2014, is named IMM184W08_SEtxt and is 44,899
bytes in
size.
BACKGROUND OF THE INVENTION
Field of the Invention
[003] This invention relates to antibody-drug conjugates (ADCs) comprising one
or more
cytotoxic drug moieties conjugated to an antibody or antigen-binding antibody
fragment that
binds to Trop-2 antigen (also known as EGP-1, TACSTD2, M1S1, GP50, or GA733-
1). In
preferred embodiments, the antibody may be a humanized R57 antibody and the
drug may be
SN-38 or pro-2PDox. However, the embodiments are not limiting and any other
known anti-
Trop-2 antibody or cytotoxic drug may be utilized. More preferably, a linker
such as CL2A
may be used to attach the drug to the antibody or antibody fragment. However,
other linkers
and other known methods of conjugating drugs to antibodies may be utilized.
The antibody
or fragment may be attached to 1-12, 1-6, 1-5 or about six copies of drug
moiety or drug-
linker moiety per antibody or fragment. More preferably, the drug to antibody
ratio may vary
between 1.5:1 to 8:1. The anti-Trop-2 ADCs are of use for therapy of Trop-2
expressing
cancers, such as breast, ovarian, cervical, endometrial, lung, prostate,
colorectal, stomach,
esophageal, bladder, renal, pancreatic, thyroid, and head-and-neck cancer. The
ADC may be
of particular use for treatment of cancers that are resistant to one or more
standard anti-cancer
therapies, such as colorectal cancer, pancreatic ductal cancer, triple-
negative breast cancer or
1
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
small-cell lung cancer. The anti-Trop-2 ADCs may be used alone or as a
combination
therapy, along with one or more therapeutic modalities selected from the group
consisting of
surgery, radiation therapy, chemotherapy, immunomodulators, cytokines,
chemotherapeutic
agents, pro-apoptotic agents, anti-angiogenic agents, cytotoxic agents, drugs,
toxins,
radionuclides, RNAi, siRNA, a second antibody or antibody fragment, and an
immunoconjugate. In preferred embodiments, the combination of ADC and other
therapeutic
modalities exhibits a synergistic effect or an additive effect without
increased host toxicities,
and is more effective to induce cancer cell death than either ADC or other
therapeutic
modality alone, or the sum of the effects of ADC and other therapeutic
modality administered
individually. The combination may include one or more therapies directed
against Trop-2,
such as PROXINIUMO (VB4-845, Viventia), IGN-101 (Aphton), adecatumumab (MT201,
Micromet), ING-1 (Xoma) or EMD 273 066 (Lexigen). The combination may also
include
administering an immunotherapy subsequent to tumor reduction with the ADC,
such as
subsequent administration of check point inhibiting agents (including
antibodies) or T-cell (or
NK-cell) redirecting bispecific antibodies. Alternatively, the combination may
be directed to
different target antigens expressed on the same cancer, such as the
combination of anti-Trop-
2 ADC and a radiolabeled anti-MUC5ac antibody, for example 90Y-hPAM4.
[004] In contrast to earlier reports that anti-Trop-2 antibodies did not bind
to normal
epithelial cells and were tumor specific (see, e.g., U.S. Patent No.
5,840,854), we have
observed at least limited Trop-2 expression in numerous types of normal
tissues, including
breast, eye, gastrointestinal tract, kidney, lung, ovary, fallopian tube,
pancreas, parathyroid,
prostate, salivary gland, skin, thymus, tonsil, ureter, urinary bladder and
uterus (see, e.g.,
Example 4 below). It was therefore surprising and unexpected that, as
discussed in the
Examples below, administration of therapeutically effective dosages of anti-
Trop-2 ADCs to
human cancer patients resulted in only limited toxicity, with no formation of
antibodies
against the anti-Trop-2 ADC and no fatal toxicities observed. In preferred
embodiments, the
anti-Trop-2 ADC can be administered to human cancer patients at
therapeutically effective
dosages with only limited toxicity, more preferably < Grade 3 neutropenia,
nausea, diarrhea,
alopecia and vomiting and no more serious side effects. Most preferably, the
anti-Trop-2
ADC can be administered to human cancer patients with tumors that were
previously
resistant to one or more standard anti-cancer therapies, with only limited
toxicity and without
inducing a fatal immune response to the ADC. Surprisingly, the anti-Trop-2 ADC
can also be
effective against tumors that are refractory to topoisomerase-1 or
topoisomerase-2 inhibitors,
such as irinotecan, the parent compound of SN-38. In other preferred
embodiments,
2
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
administration of the anti-Trop-2 ADC to human cancer patients is capable of
inducing
partial response or stable disease of such tumors.
Related Art
[005] Trop-2 (human trophoblast-cell-surface marker) is a cell surface
glycoprotein that was
originally identified in normal and malignant trophoblast cells (Lipinski et
al., 1981, Proc
Natl. Acad Sci USA 78:5147-50). Trop-2 is highly expressed in most human
carcinomas,
particularly in epithelial carcinomas and adenocarcinomas, with reported low
to restricted
expression in normal tissues (see, e.g., Cubas et al., 2010, Molec Cancer
9:253; Stepan et al.,
2011, J Histochem Cytochem 59:701-10; Varughese et al., 2011, Am J Obst Gyn
205:567e-
e7). Expression of Trop-2 is associated with metastasis, increased tumor
aggressiveness and
decreased patient survival (Cubas et al., 2010; Varughese et al., 2011).
Pathogenic effects of
Trop-2 have been reported to be mediated, at least in part, by the ERK 1/2
MAPK pathway
(Cubas et al., 2010).
[006] Overexpression of Trop-2 in many different types of human carcinomas and
adenocarcinomas, squamous cell carcinomas, as well as its transmembrane
location, render it
a potential target for anti-cancer immunotherapy. A need exists for effective
ADCs against
Trop-2 as therapeutic agents for Trop-2 expressing cancers.
SUMMARY
[007] In various embodiments, the present invention concerns treatment of Trop-
2
expressing cancers with anti-Trop-2 antibody-drug conjugates (ADCs). It has
been
discovered that various anti-Trop-2 antibodies can be conjugated to a variety
of drugs, all
having selective efficacy against Trop-2-expressing cancers. The anti-Trop-2
ADC may be
used alone or as a combination therapy with one or more other therapeutic
modalities, such as
surgery, radiation therapy, chemotherapy, immunomodulators, cytokines,
chemotherapeutic
agents, pro-apoptotic agents, anti-angiogenic agents, cytotoxic agents, drugs,
toxins,
radionuclides, RNAi, siRNA, a second antibody or antibody fragment, or an
immunoconjugate. In preferred embodiments, the anti-Trop-2 ADC may be of use
for
treatment of cancers for which standard therapies are not effective or to
which the cancers
have become refractive, such as colorectal cancer, small-cell lung cancer,
pancreatic ductal
and non-ductal (e.g., neuroendocrine), cancers or triple-negative breast
cancer, but including
also non-small cell lung cancers and endocrine- and Her2- responsive breast
cancers. More
preferably, the combination of ADC and other therapeutic modality is more
efficacious than
3
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
either alone, or the sum of the effects of individual treatments, especially
without a
concomitant increase in toxic side effects.
[008] In a specific embodiment, the anti-Trop-2 antibody may be a humanized
RS7
antibody (see, e.g., U.S. Patent No. 7,238,785, the Figures and Examples
section of which are
incorporated herein by reference), comprising the light chain CDR sequences
CDR1
(KASQDVSIAVA, SEQ ID NO:1); CDR2 (SASYRYT, SEQ ID NO:2); and CDR3
(QQHYITPLT, SEQ ID NO:3) and the heavy chain CDR sequences CDR1 (NYGMN, SEQ
ID NO:4); CDR2 (WINTYTGEPTYTDDFKG, SEQ ID NO:5) and CDR3
(GGFGSSYWYFDV, SEQ ID NO:6). However, as discussed below other anti-Trop-2
antibodies are known and may be used in the subject ADCs. A number of
cytotoxic drugs of
use for cancer treatment are well-known in the art and any such known drug may
be
conjugated to the antibody of interest, so long as the conjugation method does
not
compromise the anti-Trop-2 antibody binding property by more than 65%,
preferably not
more than 50%, more preferably not more than 33%. In a more preferred
embodiment, the
drug conjugated to the antibody is a camptothecin or anthracycline, most
preferably SN-38 or
a pro-drug form of 2-pyrrolinodoxorubicin (2-PDox) (see, e.g., U.S. Patent
Application Serial
Nos. 14/175,089 and 14/204,698, the Figures and Examples section of each
incorporated
herein by reference).
[009] The anti-Trop-2 antibody moiety may be a monoclonal antibody, an antigen-
binding
antibody fragment, a bispecific or other multivalent antibody, or other
antibody-based
molecule. The antibody can be of various isotypes, preferably human IgGl,
IgG2, IgG3 or
IgG4, more preferably comprising human IgG1 hinge and constant region
sequences. The
antibody or fragment thereof can be a chimeric, a humanized, or a human
antibody, as well as
variations thereof, such as half-IgG4 antibodies (referred to as "unibodies"),
as described by
van der Neut Kolfschoten et al. (Science 2007; 317:1554-1557). More
preferably, the
antibody or fragment thereof may be designed or selected to comprise human
constant region
sequences that belong to specific allotypes, which may result in reduced
immunogenicity
when the ADC is administered to a human subject. Preferred allotypes for
administration
include a non-Glml allotype (nG1m1), such as G1m3, G1m3,1, G1m3,2 or G1m3,1,2.
More
preferably, the allotype is selected from the group consisting of the nGlml,
G1m3, nG1m1,2
and Km3 allotypes.
[010] The drug to be conjugated to the anti-Trop-2 antibody or antibody
fragment may be
selected from the group consisting of an anthracycline, a camptothecin, a
tubulin inhibitor, a
maytansinoid, a calicheamycin, an auristatin, a nitrogen mustard, an
ethylenimine derivative,
4
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
an alkyl sulfonate, a nitrosourea, a triazene, a folic acid analog, a taxane,
a COX-2 inhibitor, a
pyrimidine analog, a purine analog, an antibiotic, an enzyme inhibitor, an
epipodophyllotoxin, a platinum coordination complex, a vinca alkaloid, a
substituted urea, a
methyl hydrazine derivative, an adrenocortical suppressant, a hormone
antagonist, an
antimetabolite, an alkylating agent, an antimitotic, an anti-angiogenic agent,
a tyrosine kinase
inhibitor, an mTOR inhibitor, a heat shock protein (HSP90) inhibitor, a
proteosome inhibitor,
an HDAC inhibitor, a pro-apoptotic agent, and a combination thereof As used
herein, the
term "drug" does not include protein or peptide toxins, such as ricin, abrin,
ribonuclease
(RNase), DNase I, Staphylococcal enterotoxin-A, pokeweed antiviral protein,
onconase,
gelonin, diphtheria toxin, Pseudomonas exotoxin or Pseudomonas endotoxin.
[011] Specific drugs of use may be selected from the group consisting of 5-
fluorouracil,
afatinib, aplidin, azaribine, anastrozole, anthracyclines, axitinib, AVL-101,
AVL-291,
bendamustine, bleomycin, bortezomib, bosutinib, bryostatin-1, busulfan,
calicheamycin,
camptothecin, carboplatin, 10-hydroxycamptothecin, carmustine, celecoxib,
chlorambucil,
cisplatinum, COX-2 inhibitors, irinotecan (CPT-11), SN-38, carboplatin,
cladribine,
camptothecans, crizotinib, cyclophosphamide, cytarabine, dacarbazine,
dasatinib, dinaciclib,
docetaxel, dactinomycin, daunorubicin, DM1, DM3, DM4, doxorubicin, 2-
pyrrolinodoxorubicine (2-PDox), a pro-drug form of 2-PDox (pro-2-PDox), cyano-
morpholino doxorubicin, doxorubicin glucuronide, endostatin, epirubicin
glucuronide,
erlotinib, estramustine, epidophyllotoxin, erlotinib, entinostat, estrogen
receptor binding
agents, etoposide (VP16), etoposide glucuronide, etoposide phosphate,
exemestane,
fingolimod, floxuridine (FUdR), 3',5'-0-dioleoyl-FudR (FUdR-d0), fludarabine,
flutamide,
farnesyl-protein transferase inhibitors, flavopiridol, fostamatinib,
ganetespib, GDC-0834, GS-
1101, gefitinib, gemcitabine, hydroxyurea, ibrutinib, idarubicin, idelalisib,
ifosfamide,
imatinib, lapatinib, lenolidamide, leucovorin, LFM-A13, lomustine,
mechlorethamine,
melphalan, mercaptopurine, 6-mercaptopurine, methotrexate, mitoxantrone,
mithramycin,
mitomycin, mitotane, monomethylauristatin F (MMAF), monomethylauristatin D
(MMAD),
monomethylauristatin E (MMAE), navelbine, neratinib, nilotinib, nitrosurea,
olaparib,
plicomycin, procarbazine, paclitaxel, PCI-32765, pentostatin, PSI-341,
raloxifene, semustine,
SN-38, sorafenib, streptozocin, SU11248, sunitinib, tamoxifen, temazolomide,
transplatinum,
thalidomide, thioguanine, thiotepa, teniposide, topotecan, uracil mustard,
vatalanib,
vinorelbine, vinblastine, vincristine, vinca alkaloids and ZD1839.
[012] Preferred optimal dosing of the subject ADCs may include a dosage of
between 1
mg/kg and 20 mg/kg, preferably given either weekly, twice weekly, every other
week, or
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
every third week. The optimal dosing schedule may include treatment cycles of
two
consecutive weeks of therapy followed by one, two, three or four weeks of
rest, or alternating
weeks of therapy and rest, or one week of therapy followed by two, three or
four weeks of
rest, or three weeks of therapy followed by one, two, three or four weeks of
rest, or four
weeks of therapy followed by one, two, three or four weeks of rest, or five
weeks of therapy
followed by one, two, three, four or five weeks of rest, or administration
once every two
weeks, once every three weeks, or once a month. Treatment may be extended for
any number
of cycles, preferably at least 2, at least 4, at least 6, at least 8, at least
10, at least 12, at least
14, or at least 16 cycles. This depends on tolerability of the dose as well as
status of the
patient's disease; the less toxic the therapy and the better the control of
the disease, the longer
the therapy can be given in repeated cycles.The dosage may be up to 24 mg/kg.
Exemplary
dosages of use may include 1 mg/kg, 2 mg/kg, 3 mg/kg, 4 mg/kg, 5 mg/kg, 6
mg/kg, 7
mg/kg, 8 mg/kg, 9 mg/kg, 10 mg/kg, 11 mg/kg, 12 mg/kg, 13 mg/kg, 14 mg/kg, 15
mg/kg, 16
mg/kg, 17 mg/kg, 18 mg/kg, 19 mg/kg, 20 mg/kg, 22 mg/kg and 24 mg/kg.
Preferred dosages
are 1, 2, 4, 6, 8, 9, 10, or 12 mg/kg. The person of ordinary skill will
realize that a variety of
factors, such as age, general health, specific organ function or weight, as
well as effects of
prior therapy on specific organ systems (e.g., bone marrow), may be considered
in selecting
an optimal dosage of ADC, and that the dosage and/or frequency of
administration may be
increased or decreased during the course of therapy. The dosage may be
repeated as needed,
with evidence of tumor shrinkage observed after as few as 3 to 8 doses. The
optimized
dosages and schedules of administration disclosed herein show unexpected
superior efficacy
and reduced toxicity in human subjects, which could not have been predicted
from animal
model studies, especially in murine xenograft models where a toxic dose of the
ADC is not
readily established. Surprisingly, the superior efficacy allows treatment of
tumors that were
previously found to be resistant to one or more standard anti-cancer
therapies.
[013] The anti-Trop-2 ADCs are of use for therapy of Trop-2 expressing
cancers, such as
breast, ovarian, cervical, endometrial, lung, prostate, colorectal, stomach,
esophageal, urinary
bladder, renal, pancreatic, thyroid, or head-and-neck cancer. The ADC may be
of particular
use for treatment of cancers that are resistant to one or more standard anti-
cancer therapies,
such as a metastatic colorectal cancer, triple-negative breast cancer, a HER+,
ER+,
progesterone+ breast cancer, metastatic non-small-cell lung cancer (NSCLC),
metastatic
pancreatic cancer, metastatic renal cell carcinoma, metastatic gastric cancer,
metastatic
prostate cancer, or metastatic small-cell lung cancer.
6
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
[014] In contrast to earlier reports that anti-Trop-2 antibodies did not bind
to normal
epithelial cells and were tumor specific (see, e.g., U.S. Patent No.
5,840,854), we have
observed at least limited Trop-2 expression in numerous types of normal
tissues, including
breast, eye, gastrointestinal tract, kidney, lung, ovary, fallopian tube,
pancreas, parathyroid,
prostate, salivary gland, skin, thymus, tonsil, ureter, urinary bladder and
uterus (see, e.g.,
Example 4 below). It was therefore surprising and unexpected that, as
discussed in the
Examples below, administration of therapeutically effective dosages of anti-
Trop-2 ADCs to
human cancer patients resulted in only limited toxicity, with no formation of
antibodies
against the anti-Trop-2 ADC and no fatal toxicities observed. In preferred
embodiments, the
anti-Trop-2 ADC can be administered to human cancer patients at
therapeutically effective
dosages with only limited toxicity, more preferably < Grade 3 neutropenia,
nausea, diarrhea,
alopecia and vomiting and no more serious side effects. Most preferably, the
anti-Trop-2
ADC can be administered to human cancer patients with tumors that were
previously
resistant to one or more standard anti-cancer therapies, with only limited
toxicity and without
inducing a fatal immune response to the ADC. In other preferred embodiments,
administration of the anti-Trop-2 ADC to human cancer patients is capable of
inducing
partial response or stable disease of such tumors.
BRIEF DESCRIPTION OF THE DRAWINGS
[015] FIG. 1. Preclinical in vivo therapy of athymic nude mice, bearing Capan
1 human
pancreatic carcinoma, with SN-38 conjugates of hRS7 (anti-Trop-2), hPAM4 (anti-
MUC5ac),
hMN-14 (anti-CEACAM5) or non-specific control hA20 (anti-CD20).
[016] FIG. 2. Preclinical in vivo therapy of athymic nude mice, bearing BxPC3
human
pancreatic carcinoma, with anti-TROP2-CL2A-SN-38 conjugates compared to
controls.
[017] FIG. 3A. Structure of doxorubicin. "Me" is a methyl group.
[018] FIG. 3B. Structure of 2-pyrrolinodoxorubicin,(2-PDox). "Me" is a methyl
group.
[019] FIG. 3C. Structure of a prodrug form of 2-pyrrolinodoxorubicin,(pro-2-
PDox). "Me"
is a methyl group and "Ac" is an acetyl group.
[020] FIG. 3D. Structure of a maleimide-activated form of pro-2-PDox, for
antibody
coupling. "Me" is a methyl group and "Ac" is an acetyl group.
[021] FIG. 4. Therapy in nude mice bearing s.c. human tumor xenografts using
2.25 mg/kg
protein dose (0.064 mg/kg of drug dose) of MAb-pro-2-PDox conjugates twice
weekly x 2
weeks in nude mice with Capan-1 human pancreatic adenocarcinoma xenografts (n
= 5).
7
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
[022] FIG. 5A. Therapy in nude mice bearing s.c. human tumor xenografts using
2.25
mg/kg protein dose (0.064 mg/kg of drug dose) of MAb-pro-2-PDox conjugates
twice weekly
x 2 weeks in nude mice (n = 7) with NCI-N87 human gastric carcinoma
xenografts.
[023] FIG. 5B. Therapy in nude mice bearing s.c. human tumor xenografts using
2.25
mg/kg protein dose (0.064 mg/kg of drug dose) of MAb-pro-2-PDox conjugates
twice weekly
x 2 weeks in nude mice (n = 7) with MDA-MB-468 human breast carcinoma
xenografts.
[024] FIG. 5C. Therapy in nude mice bearing s.c. human tumor xenografts using
2.25
mg/kg protein dose (0.064 mg/kg of drug dose) of MAb-pro-2-PDox conjugates
twice weekly
x 2 weeks in nude mice (n = 7) with BxPC3 human pancreatic carcinoma
xenografts.
[025] FIG. 6A. In vivo efficacy of pro-2-PDox conjugates in nude mice with NCI-
N87
human gastric cancer xenografts. Mice were administered a saline control.
[026] FIG. 6B. In vivo efficacy of pro-2-PDox conjugates in nude mice with NCI-
N87
human gastric cancer xenografts. Mice were administered 45 tig of hA20-pro-2-
PDox as
indicated by arrows.
[027] FIG. 6C. In vivo efficacy of pro-2-PDox conjugates in nude mice with NCI-
N87
human gastric cancer xenografts. Mice were administered 45 tig of hMN-15-pro-2-
PDox as
indicated by arrows.
[028] FIG. 6D. In vivo efficacy of pro-2-PDox conjugates in nude mice with NCI-
N87
human gastric cancer xenografts. Mice were administered 45 tig of hRS7-pro-2-
PDox as
indicated by arrows.
[029] FIG. 6E. In vivo efficacy of pro-2-PDox conjugates in nude mice with NCI-
N87
human gastric cancer xenografts. Mice were administered 45 tig of hLL1-pro-2-
PDox as
indicated by arrows.
[030] FIG. 6F. In vivo efficacy of pro-2-PDox conjugates in nude mice with NCI-
N87
human gastric cancer xenografts. Mice were administered 45 tig of hMN-14-pro-2-
PDox as
indicated by arrows.
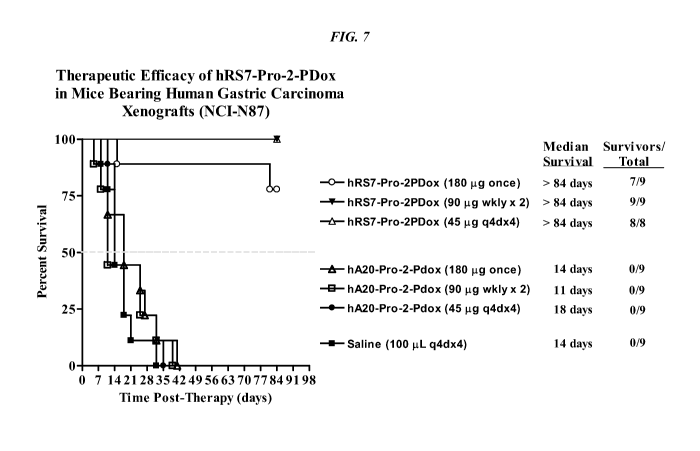
[031] FIG. 7. Effect of different dosing schedules of hRS7-pro-2-PDox on
survival in nude
mice with NCI-N87 human gastric carcinoma xenografts.
[032] FIG. 8A. Dosing schedule study in mice injected with NCI-N87 human
gastric
cancer. Mice were administered a saline control.
[033] FIG. 8B. Dosing schedule study in mice injected with NCI-N87 human
gastric
cancer. Mice were administered 45 tig q4dx4 of hRS7-pro-2-PDox.
[034] FIG. 8C. Dosing schedule study in mice injected with NCI-N87 human
gastric
cancer. Mice were administered 90 tig weekly x 2 of hRS7-pro-2-PDox.
8
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
[035] FIG. 8D. Dosing schedule study in mice injected with NCI-N87 human
gastric
cancer. Mice were administered a single dose of 180 tig hRS7-pro-2-PDox.
[036] FIG. 8E. Dosing schedule study in mice injected with NCI-N87 human
gastric
cancer. Mice were administered 45 tig q4dx4 of hA20-pro-2-PDox.
[037] FIG. 8F. Dosing schedule study in mice injected with NCI-N87 human
gastric
cancer. Mice were administered 90 tig weekly x 2 of hA20-pro-2-PDox.
[038] FIG. 8G. Dosing schedule study in mice injected with NCI-N87 human
gastric
cancer. Mice were administered a single dose of 180 tig hA20-pro-2-PDox.
[039] FIG. 9. Effect of different single doses of hRS7-pro-2-PDox on growth of
human
gastric carcinoma xenografts.
[040] FIG. 10. Effect of different single doses of hRS7-pro-2-PDox on survival
of mice
bearing human gastric carcinoma xenografts.
[041] FIG. 11. ADCC of various hRS7-ADCs vs. hRS7 IgG.
[042] FIG. 12A. Structures of CL2-SN-38 and CL2A-SN-38.
[043] FIG. 12B. Comparative efficacy of anti-Trop-2 ADC linked to CL2 vs. CL2A
linkers
versus hA20 ADC and saline control, using COLO 205 colonic adenocarcinoma.
Animals
were treated twice weekly for 4 weeks as indicated by the arrows. COLO 205
mice (N = 6)
were treated with 0.4 mg/kg ADC and tumors measured twice a week.
[044] FIG. 12C. Comparative efficacy of anti-Trop-2 ADC linked to CL2 vs. CL2A
linkers
versus hA20 ADC and saline control, using Capan-1 pancreatic adenocarcinoma.
Animals
were treated twice weekly for 4 weeks as indicated by the arrows. Capan-1 mice
(N = 10)
were treated with 0.2 mg/kg ADC and tumors measured weekly.
[045] FIG. 13A. Therapeutic efficacy of hRS7-SN-38 ADC in several solid tumor-
xenograft disease models. Efficacy of hRS7-CL2-SN-38 and hRS7-CL2A-SN-38 ADC
treatment was studied in mice bearing human non¨small cell lung, colorectal,
pancreatic, or
squamous cell lung tumor xenografts. All the ADCs and controls were
administered in the
amounts indicated (expressed as amount of SN-38 per dose; long arrows =
conjugate
injections, short arrows = irinotecan injections). Mice bearing Calu-3 tumors
(N = 5-7) were
injected with hRS7-CL2-SN-38 every 4 days for a total of 4 injections (q4dx4).
[046] FIG. 13B. Therapeutic efficacy of hRS7-SN-38 ADC in several solid tumor-
xenograft disease models. Efficacy of hRS7-CL2-SN-38 and hRS7-CL2A-SN-38 ADC
treatment was studied in mice bearing human non¨small cell lung, colorectal,
pancreatic, or
squamous cell lung tumor xenografts. All the ADCs and controls were
administered in the
amounts indicated (expressed as amount of SN-38 per dose; long arrows =
conjugate
9
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
injections, short arrows = irinotecan injections). COLO 205 tumor-bearing mice
(N = 5)
were injected 8 times (q4dx8) with the ADC or every 2 days for a total of 5
injections
(q2dx5) with the MTD of irinotecan.
[047] FIG. 13C. Therapeutic efficacy of hRS7-SN-38 ADC in several solid tumor-
xenograft disease models. Efficacy of hRS7-CL2-SN-38 and hRS7-CL2A-SN-38 ADC
treatment was studied in mice bearing human non¨small cell lung, colorectal,
pancreatic, or
squamous cell lung tumor xenografts. All the ADCs and controls were
administered in the
amounts indicated (expressed as amount of SN-38 per dose; long arrows =
conjugate
injections, short arrows = irinotecan injections). Capan-1 (N= 10) were
treated twice weekly
for 4 weeks with the agents indicated.
[048] FIG. 13D. Therapeutic efficacy of hRS7-SN-38 ADC in several solid tumor-
xenograft disease models. Efficacy of hRS7-CL2-SN-38 and hRS7-CL2A-SN-38 ADC
treatment was studied in mice bearing human non¨small cell lung, colorectal,
pancreatic, or
squamous cell lung tumor xenografts. All the ADCs and controls were
administered in the
amounts indicated (expressed as amount of SN-38 per dose; long arrows =
conjugate
injections, short arrows = irinotecan injections). BxPC-3 tumor-bearing mice
(N= 10) were
treated twice weekly for 4 weeks with the agents indicated.
[049] FIG. 13E. Therapeutic efficacy of hRS7-SN-38 ADC in several solid tumor-
xenograft disease models. Efficacy of hRS7-CL2-SN-38 and hRS7-CL2A-SN-38 ADC
treatment was studied in mice bearing human non¨small cell lung, colorectal,
pancreatic, or
squamous cell lung tumor xenografts. All the ADCs and controls were
administered in the
amounts indicated (expressed as amount of SN-38 per dose; long arrows =
conjugate
injections, short arrows = irinotecan injections). In addition to ADC given
twice weekly for 4
week, SK-MES-1 tumor-bearing (N = 8) mice received the MTD of CPT-11 (q2dx5).
[050] FIG. 14A. Tolerability of hRS7-CL2A-SN-38 in Swiss-Webster mice. Fifty-
six
Swiss-Webster mice were administered 2 i.p. doses of buffer or the hRS7-CL2A-
SN-38 3
days apart (4, 8, or 12 mg/kg of SN-38 per dose; 250, 500, or 750 mg conjugate
protein/kg
per dose). Seven and 15 days after the last injection, 7 mice from each group
were
euthanized, with blood counts and serum chemistries performed. Graphs show the
percent of
animals in each group that had elevated levels of AST.
[051] FIG. 14B. Tolerability of hRS7-CL2A-SN-38 in Swiss-Webster mice. Fifty-
six
Swiss-Webster mice were administered 2 i.p. doses of buffer or the hRS7-CL2A-
SN-38 3
days apart (4, 8, or 12 mg/kg of SN-38 per dose; 250, 500, or 750 mg conjugate
protein/kg
per dose). Seven and 15 days after the last injection, 7 mice from each group
were
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
euthanized, with blood counts and serum chemistries performed. Graphs show the
percent of
animals in each group that had elevated levels of ALT.
[052] FIG. 14C. Tolerability of hRS7-CL2A-SN-38 in Cynomolgus monkeys. Six
monkeys
per group were injected twice 3 days apart with buffer (control) or hRS7-CL2A-
SN-38 at
0.96 mg/kg or 1.92 mg/kg of SN-38 equivalents per dose (60 and 120 mg/kg
conjugate
protein). All animals were bled on day ¨1, 3, and 6. Four monkeys were bled on
day 11 in the
0.96 mg/kg group, 3 in the 1.92 mg/kg group. Changes in neutrophil counts in
Cynomolgus
monkeys.
[053] FIG. 14D. Tolerability of hRS7-CL2A-SN-38 in Cynomolgus monkeys. Six
monkeys
per group were injected twice 3 days apart with buffer (control) or hRS7-CL2A-
SN-38 at
0.96 mg/kg or 1.92 mg/kg of SN-38 equivalents per dose (60 and 120 mg/kg
conjugate
protein). All animals were bled on day ¨1, 3, and 6. Four monkeys were bled on
day 11 in the
0.96 mg/kg group, 3 in the 1.92 mg/kg group. Changes in platelet counts in
Cynomolgus
monkeys.
[054] FIG. 15. In vitro efficacy of anti-Trop-2-paclitaxel ADC against MDA-MB-
468
human breast adenocarcinoma.
[055] FIG. 16. In vitro efficacy of anti-Trop-2-paclitaxel ADC against BxPC-3
human
pancreatic adenocarcinoma.
[056] FIG. 17A. Comparison of in vitro efficacy of anti-Trop-2 ADCs (hRS7-SN-
38 versus
MAB650-SN-38) in Capan-1 human pancreatic adenocarcinoma.
[057] FIG. 17B. Comparison of in vitro efficacy of anti-Trop-2 ADCs (hRS7-SN-
38 versus
MAB650-SN-38) in BxPC-3 human pancreatic adenocarcinoma.
[058] FIG. 17C. Comparison of in vitro efficacy of anti-Trop-2 ADCs (hRS7-SN-
38 versus
MAB650-SN-38) in NCI-N87 human gastric adenocarcinoma.
[059] FIG. 19. IMMU-132 phase I/II data for best response by RECIST criteria.
[060] FIG. 20. IMMU-132 phase I/II data for time to progression and best
response
(RECIST).
[061] FIG. 21. Therapeutic efficacy of murine anti-Trop-2-SN-38 ADC (162-46.2-
SN-38)
compared to hRS7-SN-38 in mice bearing NCI-N87 human gastric carcinoma
xenografts.
[062] FIG. 22. Therapeutic efficacy of murine anti-Trop-2-pro-2-PDox ADC (162-
46.2-
pro-2-PDox) compared to hRS7-pro-2-PDox in mice bearing NCI-N87 human gastric
carcinoma xenografts.
[063] FIG. 23. Accumulation of SN-38 in tumors of nude mice with Capan-1 human
pancreatic cancer xenografts, when administered as free irinotecan vs. IMMU-
132 ADC.
11
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
[064] FIG. 24. Individual patient demographics and prior treatment for phase
I/II IMMU-
132 anti-Trop-2 ADC in pancreatic cancer patients.
[065] FIG. 25. Response assessment to IMMU-132 anti-Trop-2 ADC in pancreatic
cancer
patients.
[066] FIG. 26. Summary of time to progression (TTP) results in human
pancreatic cancer
patients administered IMMU-132 anti-Trop-2 ADC.
DETAILED DESCRIPTION
Definitions
[067] Unless otherwise specified, "a" or "an" means one or more.
[068] As used herein, "about" means plus or minus 10%. For example, "about
100" would
include any number between 90 and 110.
[069] An antibody, as described herein, refers to a full-length (i.e.,
naturally occurring or
formed by normal immunoglobulin gene fragment recombinatorial processes)
immunoglobulin molecule (e.g., an IgG antibody) or an immunologically active
(i.e.,
specifically binding) portion of an immunoglobulin molecule, like an antibody
fragment.
[070] An antibody fragment is a portion of an antibody such as F(ab')2, Fab',
Fab, Fv, sFv
and the like. Antibody fragments may also include single domain antibodies and
IgG4 half-
molecules, as discussed below. Regardless of structure, an antibody fragment
binds with the
same antigen that is recognized by the full-length antibody. The term
"antibody fragment"
also includes isolated fragments consisting of the variable regions of
antibodies, such as the
"Fv" fragments consisting of the variable regions of the heavy and light
chains and
recombinant single chain polypeptide molecules in which light and heavy
variable regions are
connected by a peptide linker ("scFv proteins").
[071] A chimeric antibody is a recombinant protein that contains the variable
domains
including the complementarity determining regions (CDRs) of an antibody
derived from one
species, preferably a rodent antibody, while the constant domains of the
antibody molecule
are derived from those of a human antibody. For veterinary applications, the
constant
domains of the chimeric antibody may be derived from that of other species,
such as a cat or
dog.
[072] A humanized antibody is a recombinant protein in which the CDRs from an
antibody
from one species; e.g., a rodent antibody, are transferred from the heavy and
light variable
chains of the rodent antibody into human heavy and light variable domains
(e.g., framework
region sequences). The constant domains of the antibody molecule are derived
from those of
12
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
a human antibody. In certain embodiments, a limited number of framework region
amino
acid residues from the parent (rodent) antibody may be substituted into the
human antibody
framework region sequences.
[073] A human antibody is, e.g., an antibody obtained from transgenic mice
that have been
"engineered" to produce specific human antibodies in response to antigenic
challenge. In this
technique, elements of the human heavy and light chain loci are introduced
into strains of
mice derived from embryonic stem cell lines that contain targeted disruptions
of the
endogenous murine heavy chain and light chain loci. The transgenic mice can
synthesize
human antibodies specific for particular antigens, and the mice can be used to
produce human
antibody-secreting hybridomas. Methods for obtaining human antibodies from
transgenic
mice are described by Green et al., Nature Genet. 7:13 (1994), Lonberg et al.,
Nature 368:856
(1994), and Taylor et al., Int. Immun. 6:579 (1994). A fully human antibody
also can be
constructed by genetic or chromosomal transfection methods, as well as phage
display
technology, all of which are known in the art. See for example, McCafferty et
al., Nature
348:552-553 (1990) for the production of human antibodies and fragments
thereof in vitro,
from immunoglobulin variable domain gene repertoires from unimmunized donors.
In this
technique, antibody variable domain genes are cloned in-frame into either a
major or minor
coat protein gene of a filamentous bacteriophage, and displayed as functional
antibody
fragments on the surface of the phage particle. Because the filamentous
particle contains a
single-stranded DNA copy of the phage genome, selections based on the
functional properties
of the antibody also result in selection of the gene encoding the antibody
exhibiting those
properties. In this way, the phage mimics some of the properties of the B
cell. Phage display
can be performed in a variety of formats, for review, see e.g. Johnson and
Chiswell, Current
Opinion in Structural Biology 3:5564-571 (1993). Human antibodies may also be
generated
by in vitro activated B cells. See U.S. Pat. Nos. 5,567,610 and 5,229,275, the
Examples
section of which is incorporated herein by reference.
[074] A therapeutic agent is a compound, molecule or atom which is
administered
separately, concurrently or sequentially with an antibody moiety or conjugated
to an antibody
moiety, i.e., antibody or antibody fragment, or a subfragment, and is useful
in the treatment
of a disease. Examples of therapeutic agents include antibodies, antibody
fragments, drugs,
toxins, nucleases, hormones, immunomodulators, pro-apoptotic agents, anti-
angiogenic
agents, boron compounds, photoactive agents or dyes and radioisotopes.
Therapeutic agents
of use are described in more detail below.
13
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
[075] An immunoconjugate is an antibody, antibody fragment or fusion protein
conjugated
to at least one therapeutic and/or diagnostic agent.
[076] A multispecific antibody is an antibody that can bind simultaneously to
at least two
targets that are of different structure, e.g., two different antigens, two
different epitopes on
the same antigen, or a hapten and/or an antigen or epitope. Multispecific,
multivalent
antibodies are constructs that have more than one binding site, and the
binding sites are of
different specificity.
[077] A bispecific antibody is an antibody that can bind simultaneously to two
different
targets. Bispecific antibodies (bsAb) and bispecific antibody fragments
(bsFab) may have at
least one arm that specifically binds to, for example, a tumor-associated
antigen and at least
one other arm that specifically binds to a targetable conjugate that bears a
therapeutic or
diagnostic agent. A variety of bispecific fusion proteins can be produced
using molecular
engineering.
Anti-Trop-2 Antibodies
[078] The subject ADCs include at least one antibody or fragment thereof that
binds to
Trop-2. In a specific preferred embodiment, the anti-Trop-2 antibody may be a
humanized
RS7 antibody (see, e.g., U.S. Patent No. 7,238,785, incorporated herein by
reference in its
entirety), comprising the light chain CDR sequences CDR1 (KASQDVSIAVA, SEQ ID
NO:1); CDR2 (SASYRYT, SEQ ID NO:2); and CDR3 (QQHYITPLT, SEQ ID NO:3) and
the heavy chain CDR sequences CDR1 (NYGMN, SEQ ID NO:4); CDR2
(WINTYTGEPTYTDDFKG, SEQ ID NO:5) and CDR3 (GGFGSSYWYFDV, SEQ ID
NO:6).
[079] The R57 antibody was a murine IgGi raised against a crude membrane
preparation of
a human primary squamous cell lung carcinoma. (Stein et al., Cancer Res. 50:
1330, 1990)
The R57 antibody recognizes a 46-48 kDa glycoprotein, characterized as cluster
13. (Stein et
al., Int. J. Cancer Supp. 8:98-102, 1994) The antigen was designated as EGP-1
(epithelial
glycoprotein-1), but is also referred to as Trop-2.
[080] Trop-2 is a type-I transmembrane protein and has been cloned from both
human
(Fornaro et al., Int J Cancer 1995; 62:610-8) and mouse cells (Sewedy et al.,
Int J Cancer
1998; 75:324-30). In addition to its role as a tumor-associated calcium signal
transducer
(Ripani et al., Int J Cancer 1998;76:671-6), the expression of human Trop-2
was shown to be
necessary for tumorigenesis and invasiveness of colon cancer cells, which
could be
effectively reduced with a polyclonal antibody against the extracellular
domain of Trop-2
(Wang et al., Mol Cancer Ther 2008;7:280-5).
14
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
[081] The growing interest in Trop-2 as a therapeutic target for solid cancers
(Cubas et al.,
Biochim Biophys Acta 2009;1796:309-14) is attested by further reports that
documented the
clinical significance of overexpressed Trop-2 in breast (Huang et al., Clin
Cancer Res
2005;11:4357-64), colorectal (Ohmachi et al., Clin Cancer Res 2006;12:3057-63;
Fang et al.,
Int J Colorectal Dis 2009;24:875-84), and oral squamous cell (Fong et al.,
Modern Pathol
2008;21:186-91) carcinomas. The latest evidence that prostate basal cells
expressing high
levels of Trop-2 are enriched for in vitro and in vivo stem-like activity is
particularly
noteworthy (Goldstein et al., Proc Natl Acad Sci USA 2008;105:20882-7).
[082] Flow cytometry and immunohistochemical staining studies have shown that
the RS7
MAb detects antigen on a variety of tumor types, with limited binding to
normal human
tissue (Stein et al., 1990). Trop-2 is expressed primarily by carcinomas such
as carcinomas of
the lung, stomach, urinary bladder, breast, ovary, uterus, and prostate.
Localization and
therapy studies using radiolabeled murine R57 MAb in animal models have
demonstrated
tumor targeting and therapeutic efficacy (Stein et al., 1990; Stein et al.,
1991).
[083] Strong R57 staining has been demonstrated in tumors from the lung,
breast, bladder,
ovary, uterus, stomach, and prostate. (Stein et al., Int. J. Cancer 55:938,
1993) The lung
cancer cases comprised both squamous cell carcinomas and adenocarcinomas.
(Stein et al.,
Int. J. Cancer 55:938, 1993) Both cell types stained strongly, indicating that
the R57
antibody does not distinguish between histologic classes of non-small-cell
carcinoma of the
lung.
[084] The R57 MAb is rapidly internalized into target cells (Stein et al.,
1993). The
internalization rate constant for R57 MAb is intermediate between the
internalization rate
constants of two other rapidly internalizing MAbs, which have been
demonstrated to be
useful for immunoconjugate production. (Id.) It is well documented that
internalization of
immunoconjugates is a requirement for anti-tumor activity. (Pastan et al.,
Cell 47:641, 1986)
Internalization of drug immunoconjugates has been described as a major factor
in anti-tumor
efficacy. (Yang et al., Proc. Nat'l Acad. Sci. USA 85: 1189, 1988) Thus, the
R57 antibody
exhibits several important properties for therapeutic applications.
[085] While the hRS7 antibody is preferred, other anti-Trop-2 antibodies are
known and/or
publicly available and in alternative embodiments may be utilized in the
subject ADCs.
While humanized or human antibodies are preferred for reduced immunogenicity,
in
alternative embodiments a chimeric antibody may be of use. As discussed below,
methods of
antibody humanization are well known in the art and may be utilized to convert
an available
murine or chimeric antibody into a humanized form.
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
[086] Anti-Trop-2 antibodies are commercially available from a number of
sources and
include LS-C126418, LS-C178765, LS-C126416, LS-C126417 (LifeSpan BioSciences,
Inc.,
Seattle, WA); 10428-MM01, 10428-MM02, 10428-R001, 10428-R030 (Sino Biological
Inc.,
Beijing, China); MR54 (eBioscience, San Diego, CA); sc-376181, sc-376746,
Santa Cruz
Biotechnology (Santa Cruz, CA); MM0588-49D6, (Novus Biologicals, Littleton,
CO);
ab79976, and ab89928 (ABCAMCD, Cambridge, MA).
[087] Other anti-Trop-2 antibodies have been disclosed in the patent
literature. For example,
U.S. Publ. No. 2013/0089872 discloses anti-Trop-2 antibodies K5-70 (Accession
No. FERM
BP-11251), K5-107 (Accession No. FERM BP-11252), K5-116-2-1 (Accession No.
FERM
BP-11253), T6-16 (Accession No. FERM BP-11346), and T5-86 (Accession No. FERM
BP-
11254), deposited with the International Patent Organism Depositary, Tsukuba,
Japan. U.S.
Patent No. 5,840,854 disclosed the anti-Trop-2 monoclonal antibody BR110 (ATCC
No.
HB11698). U.S. Patent No. 7,420,040 disclosed an anti-Trop-2 antibody produced
by
hybridoma cell line AR47A6.4.2, deposited with the IDAC (International
Depository
Authority of Canada, Winnipeg, Canada) as accession number 141205-05. U.S.
Patent No.
7,420,041 disclosed an anti-Trop-2 antibody produced by hybridoma cell line
AR52A301.5,
deposited with the IDAC as accession number 141205-03. U.S. Publ. No.
2013/0122020
disclosed anti-Trop-2 antibodies 3E9, 6G11, 7E6, 15E2, 18B1. Hybridomas
encoding a
representative antibody were deposited with the American Type Culture
Collection (ATCC),
Accession Nos. PTA-12871 and PTA-12872. U.S. Patent No. 8,715,662 discloses
anti-Trop-2
antibodies produced by hybridomas deposited at the AID-ICLC (Genoa, Italy)
with deposit
numbers PD 08019, PD 08020 and PD 08021. U.S. Patent Application Publ. No.
20120237518 discloses anti-Trop-2 antibodies 77220, KM4097 and KM4590. U.S.
Patent
No. 8,309,094 (Wyeth) discloses antibodies Al and A3, identified by sequence
listing. The
Examples section of each patent or patent application cited above in this
paragraph is
incorporated herein by reference. Non-patent publication Lipinski et al.
(1981, Proc Natl.
Acad Sci USA, 78:5147-50) disclosed anti-Trop-2 antibodies 162-25.3 and 162-
46.2.
[088] Numerous anti-Trop-2 antibodies are known in the art and/or publicly
available. As
discussed below, methods for preparing antibodies against known antigens were
routine in
the art. The sequence of the human Trop-2 protein was also known in the art
(see, e.g.,
GenBank Accession No. CAA54801.1). Methods for producing humanized, human or
chimeric antibodies were also known. The person of ordinary skill, reading the
instant
disclosure in light of general knowledge in the art, would have been able to
make and use the
genus of anti-Trop-2 antibodies in the subject ADCs.
16
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
[089] The drug to be conjugated to the anti-Trop-2 antibody or antibody
fragment may be
selected from the group consisting of an anthracycline, a camptothecin, a
tubulin inhibitor, a
maytansinoid, a calicheamycin, an auristatin, a nitrogen mustard, an
ethylenimine derivative,
an alkyl sulfonate, a nitrosourea, a triazene, a folic acid analog, a taxane,
a COX-2 inhibitor, a
pyrimidine analog, a purine analog, an antibiotic, an enzyme inhibitor, an
epipodophyllotoxin, a platinum coordination complex, a vinca alkaloid, a
substituted urea, a
methyl hydrazine derivative, an adrenocortical suppressant, a hormone
antagonist, an
antimetabolite, an alkylating agent, an antimitotic, an anti-angiogenic agent,
a tyrosine kinase
inhibitor, an mTOR inhibitor, a heat shock protein (HSP90) inhibitor, a
proteosome inhibitor,
an HDAC inhibitor, a pro-apoptotic agent, and a combination thereof
[090] Specific drugs of use may be selected from the group consisting of 5-
fluorouracil,
afatinib, aplidin, azaribine, anastrozole, anthracyclines, axitinib, AVL-101,
AVL-291,
bendamustine, bleomycin, bortezomib, bosutinib, bryostatin-1, busulfan,
calicheamycin,
camptothecin, carboplatin, 10-hydroxycamptothecin, carmustine, celecoxib,
chlorambucil,
cisplatinum, COX-2 inhibitors, irinotecan (CPT-11), SN-38, carboplatin,
cladribine,
camptothecans, crizotinib, cyclophosphamide, cytarabine, dacarbazine,
dasatinib, dinaciclib,
docetaxel, dactinomycin, daunorubicin, DM1, DM3, DM4, doxorubicin, 2-
pyrrolinodoxorubicine (2-PDox), a pro-drug form of 2-PDox (pro-2-PDox), cyano-
morpholino doxorubicin, doxorubicin glucuronide, endostatin, epirubicin
glucuronide,
erlotinib, estramustine, epidophyllotoxin, erlotinib, entinostat, estrogen
receptor binding
agents, etoposide (VP16), etoposide glucuronide, etoposide phosphate,
exemestane,
fingolimod, floxuridine (FUdR), 3',5'-0-dioleoyl-FudR (FUdR-d0), fludarabine,
flutamide,
farnesyl-protein transferase inhibitors, flavopiridol, fostamatinib,
ganetespib, GDC-0834, GS-
1101, gefitinib, gemcitabine, hydroxyurea, ibrutinib, idarubicin, idelalisib,
ifosfamide,
imatinib, lapatinib, lenolidamide, leucovorin, LFM-A13, lomustine,
mechlorethamine,
melphalan, mercaptopurine, 6-mercaptopurine, methotrexate, mitoxantrone,
mithramycin,
mitomycin, mitotane, monomethylauristatin F (MMAF), monomethylauristatin D
(MMAD),
monomethylauristatin E (MMAE), navelbine, neratinib, nilotinib, nitrosurea,
olaparib,
plicomycin, procarbazine, paclitaxel, PCI-32765, pentostatin, PSI-341,
raloxifene, semustine,
SN-38, sorafenib, streptozocin, SU11248, sunitinib, tamoxifen, temazolomide,
transplatinum,
thalidomide, thioguanine, thiotepa, teniposide, topotecan, uracil mustard,
vatalanib,
vinorelbine, vinblastine, vincristine, vinca alkaloids and ZD1839. In
particularly preferred
embodiments, the drug to be conjugated to the anti-Trop-2 antibody may be SN-
38, pro-2-PDox
or paclitaxel.
17
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
[091] Use of antibodies against targets related to Trop-2 has been disclosed
for
immunotherapeutics other than ADCs. The murine anti-Trop-1 IgG2a antibody
edrecolomab
(PANOREXO) has been used for treatment of colorectal cancer, although the
murine
antibody is not well suited for human clinical use (Baeuerle & Gires, 2007,
Br. J Cancer
96:417-423). Low-dose subcutaneous administration of ecrecolomab was reported
to induce
humoral immune responses against the vaccine antigen (Baeuerle & Gires, 2007).
Adecatumumab (MT201), a fully human anti-Trop-1 antibody, has been used in
metastatic
breast cancer and early-stage prostate cancer and is reported to act through
ADCC and CDC
activity (Baeuerle & Gires, 2007). MT110, a single-chain anti-Trop-1/anti-CD3
bispecific
antibody construct has reported efficacy against ovarian cancer (Baeuerle &
Gires, 2007).
Proxinium, an immunotoxin comprising anti-Trop-1 single-chain antibody fused
to
Pseudomonas exotoxin, has been tested in head-and-neck and bladder cancer
(Baeuerle &
Gires, 2007). None of these studies contained any disclosure of the use of
anti-Trop-2
immunoconjugates or of drug-conjugated antibodies.
Antibody Preparation
[092] Techniques for preparing monoclonal antibodies against virtually any
target antigen,
such as Trop-2, are well known in the art. See, for example, Kohler and
Milstein, Nature 256:
495 (1975), and Coligan et al. (eds.), CURRENT PROTOCOLS IN IMMUNOLOGY, VOL.
1, pages 2.5.1-2.6.7 (John Wiley & Sons 1991). Briefly, monoclonal antibodies
can be
obtained by injecting mice with a composition comprising an antigen, removing
the spleen to
obtain B-lymphocytes, fusing the B-lymphocytes with myeloma cells to produce
hybridomas,
cloning the hybridomas, selecting positive clones which produce antibodies to
the antigen,
culturing the clones that produce antibodies to the antigen, and isolating the
antibodies from
the hybridoma cultures.
[093] MAbs can be isolated and purified from hybridoma cultures by a variety
of well-
established techniques. Such isolation techniques include affinity
chromatography with
Protein-A or Protein-G Sepharose, size-exclusion chromatography, and ion-
exchange
chromatography. See, for example, Coligan at pages 2.7.1-2.7.12 and pages
2.9.1-2.9.3. Also,
see Baines et al., "Purification of Immunoglobulin G (IgG)," in METHODS IN
MOLECULAR BIOLOGY, VOL. 10, pages 79-104 (The Humana Press, Inc. 1992).
[094] After the initial raising of antibodies to the immunogen, the antibodies
can be
sequenced and subsequently prepared by recombinant techniques. Humanization
and
18
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
chimerization of murine antibodies and antibody fragments are well known to
those skilled in
the art, as discussed below.
Chimeric Antibodies
[095] A chimeric antibody is a recombinant protein in which the variable
regions of a
human antibody have been replaced by the variable regions of, for example, a
mouse
antibody, including the complementarity-determining regions (CDRs) of the
mouse antibody.
Chimeric antibodies exhibit decreased immunogenicity and increased stability
when
administered to a subject. General techniques for cloning murine
immunoglobulin variable
domains are disclosed, for example, in Orlandi et al., Proc. Nat'l Acad. Sci.
USA 6: 3833
(1989). Techniques for constructing chimeric antibodies are well known to
those of skill in
the art. As an example, Leung et al., Hybridoma /3:469 (1994), produced an LL2
chimera by
combining DNA sequences encoding the V,, and VH domains of murine LL2, an anti-
CD22
monoclonal antibody, with respective human K and IgGi constant region domains.
Humanized Antibodies
[096] Techniques for producing humanized MAbs are well known in the art (see,
e.g., Jones
et al., Nature 321: 522 (1986), Riechmann et al., Nature 332: 323 (1988),
Verhoeyen et al.,
Science 239: 1534 (1988), Carter et al., Proc. Nat'l Acad. Sci. USA 89: 4285
(1992), Sandhu,
Crit. Rev. Biotech. 12: 437 (1992), and Singer et al., 1 Immun. 150: 2844
(1993)). A
chimeric or murine monoclonal antibody may be humanized by transferring the
mouse CDRs
from the heavy and light variable chains of the mouse immunoglobulin into the
corresponding variable domains of a human antibody. The mouse framework
regions (FR) in
the chimeric monoclonal antibody are also replaced with human FR sequences. As
simply
transferring mouse CDRs into human FRs often results in a reduction or even
loss of antibody
affinity, additional modification might be required in order to restore the
original affinity of the
murine antibody. This can be accomplished by the replacement of one or more
human residues
in the FR regions with their murine counterparts to obtain an antibody that
possesses good
binding affinity to its epitope. See, for example, Tempest et al.,
Biotechnology 9:266 (1991) and
Verhoeyen et al., Science 239: 1534 (1988). Preferred residues for
substitution include FR
residues that are located within 1, 2, or 3 Angstroms of a CDR residue side
chain, that are
located adjacent to a CDR sequence, or that are predicted to interact with a
CDR residue.
Human Antibodies
[097] Methods for producing fully human antibodies using either combinatorial
approaches
or transgenic animals transformed with human immunoglobulin loci are known in
the art
19
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
(e.g., Mancini et al., 2004, New Microbiol. 27:315-28; Conrad and Scheller,
2005, Comb.
Chem. High Throughput Screen. 8:117-26; Brekke and Loset, 2003, Curr. Opin.
Pharmacol.
3:544-50). A fully human antibody also can be constructed by genetic or
chromosomal
transfection methods, as well as phage display technology, all of which are
known in the art.
See for example, McCafferty et al., Nature 348:552-553 (1990). Such fully
human
antibodies are expected to exhibit even fewer side effects than chimeric or
humanized
antibodies and to function in vivo as essentially endogenous human antibodies.
[098] In one alternative, the phage display technique may be used to generate
human
antibodies (e.g., Dantas-Barbosa et al., 2005, Genet. MoL Res. 4:126-40).
Human antibodies
may be generated from normal humans or from humans that exhibit a particular
disease state,
such as cancer (Dantas-Barbosa et al., 2005). The advantage to constructing
human
antibodies from a diseased individual is that the circulating antibody
repertoire may be biased
towards antibodies against disease-associated antigens.
[099] In one non-limiting example of this methodology, Dantas-Barbosa et al.
(2005)
constructed a phage display library of human Fab antibody fragments from
osteosarcoma
patients. Generally, total RNA was obtained from circulating blood lymphocytes
(Id.).
Recombinant Fab were cloned from the itt, y and K chain antibody repertoires
and inserted
into a phage display library (Id.). RNAs were converted to cDNAs and used to
make Fab
cDNA libraries using specific primers against the heavy and light chain
immunoglobulin
sequences (Marks et al., 1991,1 Mol. Biol. 222:581-97). Library construction
was performed
according to Andris-Widhopf et al. (2000, In: Phage Display Laboratory Manual,
Barbas et
al. (eds), 1st edition, Cold Spring Harbor Laboratory Press, Cold Spring
Harbor, NY pp. 9.1 to
9.22). The final Fab fragments were digested with restriction endonucleases
and inserted into
the bacteriophage genome to make the phage display library. Such libraries may
be screened
by standard phage display methods, as known in the art. Phage display can be
performed in a
variety of formats, for their review, see e.g. Johnson and Chiswell, Current
Opinion in
Structural Biology 3:5564-571 (1993).
[0100] Human antibodies may also be generated by in vitro activated B-cells.
See U.S. Patent
Nos. 5,567,610 and 5,229,275, incorporated herein by reference in their
entirety. The skilled
artisan will realize that these techniques are exemplary and any known method
for making
and screening human antibodies or antibody fragments may be utilized.
[0101] In another alternative, transgenic animals that have been genetically
engineered to
produce human antibodies may be used to generate antibodies against
essentially any
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
immunogenic target, using standard immunization protocols. Methods for
obtaining human
antibodies from transgenic mice are disclosed by Green et al., Nature Genet.
7:13 (1994),
Lonberg et al., Nature 368:856 (1994), and Taylor et al., Int. Immun. 6:579
(1994). A non-
limiting example of such a system is the XenoMouse0 (e.g., Green et al.,
1999,1 Immunol.
Methods 231:11-23, incorporated herein by reference) from Abgenix (Fremont,
CA). In the
XenoMouse0 and similar animals, the mouse antibody genes have been inactivated
and
replaced by functional human antibody genes, while the remainder of the mouse
immune
system remains intact.
[0102] The XenoMouse0 was transformed with germline-configured YACs (yeast
artificial
chromosomes) that contained portions of the human IgH and Igkappa loci,
including the
majority of the variable region sequences, along with accessory genes and
regulatory
sequences. The human variable region repertoire may be used to generate
antibody producing
B-cells, which may be processed into hybridomas by known techniques. A
XenoMouse0
immunized with a target antigen will produce human antibodies by the normal
immune
response, which may be harvested and/or produced by standard techniques
discussed above.
A variety of strains of XenoMouse0 are available, each of which is capable of
producing a
different class of antibody. Transgenically produced human antibodies have
been shown to
have therapeutic potential, while retaining the pharmacokinetic properties of
normal human
antibodies (Green et al., 1999). The skilled artisan will realize that the
claimed compositions
and methods are not limited to use of the XenoMouse0 system but may utilize
any transgenic
animal that has been genetically engineered to produce human antibodies.
Known Antibodies and Target Antigens
[0103] As discussed above, in preferred embodiments the ADCs are of use for
treatment of
Trop-2-expressing cancer. In certain embodiments, the target cancer may
express one or more
additional tumor-associated antigens (TAAs). Particular antibodies that may be
of use for
therapy of cancer within the scope of the claimed methods and compositions
include, but are
not limited to, LL1 (anti-CD74), LL2 or RFB4 (anti-CD22), veltuzumab (hA20,
anti-CD20),
rituxumab (anti-CD20), obinutuzumab (GA101, anti-CD20), lambrolizumab (anti-PD-
1
receptor), nivolumab (anti-PD-1 receptor), ipilimumab (anti-CTLA-4), RS7 (anti-
epithelial
glycoprotein-1 (EGP-1, also known as Trop-2)), PAM4 or KC4 (both anti-mucin),
MN-14
(anti-carcinoembryonic antigen (CEA, also known as CD66e or CEACAM5), MN-15 or
MN-
3 (anti-CEACAM6), Mu-9 (anti-colon-specific antigen-p), Immu 31 (an anti-alpha-
fetoprotein), R1 (anti-IGF-1R), A19 (anti-CD19), TAG-72 (e.g., CC49), Tn, J591
or HuJ591
(anti-PSMA (prostate-specific membrane antigen)), AB-PG1-XG1-026 (anti-PSMA
dimer),
21
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
D2/B (anti-PSMA), G250 (an anti-carbonic anhydrase IX MAb), L243 (anti-HLA-DR)
alemtuzumab (anti-CD52), bevacizumab (anti-VEGF), cetuximab (anti-EGFR),
gemtuzumab
(anti-CD33), ibritumomab tiuxetan (anti-CD20); panitumumab (anti-EGFR);
tositumomab
(anti-CD20); PAM4 (aka clivatuzumab, anti-mucin) and trastuzumab (anti-ErbB2).
Such
antibodies are known in the art (e.g., U.S. Patent Nos. 5,686,072; 5,874,540;
6,107,090;
6,183,744; 6,306,393; 6,653,104; 6,730.300; 6,899,864; 6,926,893; 6,962,702;
7,074,403;
7,230,084; 7,238,785; 7,238,786; 7,256,004; 7,282,567; 7,300,655; 7,312,318;
7,585,491;
7,612,180; 7,642,239; and U.S. Patent Application Publ. No. 20050271671;
20060193865;
20060210475; 20070087001; the Examples section of each incorporated herein by
reference.)
Specific known antibodies of use include hPAM4 (U.S. Patent No. 7,282,567),
hA20 (U.S.
Patent No. 7,251,164), hAl9 (U.S. Patent No. 7,109,304), hIMMU-31 (U.S. Patent
No.
7,300,655), hLL1 (U.S. Patent No. 7,312,318), hLL2 (U.S. Patent No.
7,074,403), hMu-9
(U.S. Patent No. 7,387,773), hL243 (U.S. Patent No. 7,612,180), hMN-14 (U.S.
Patent No.
6,676,924), hMN-15 (U.S. Patent No. 7,541,440), hR1 (U.S. Patent Application
12/772,645),
hRS7 (U.S. Patent No. 7,238,785), hMN-3 (U.S. Patent No. 7,541,440), AB-PG1-
XG1-026
(U.S. Patent Application 11/983,372, deposited as ATCC PTA-4405 and PTA-4406)
and
D2/B (WO 2009/130575) the text of each recited patent or application is
incorporated herein
by reference with respect to the Figures and Examples sections.
[0104] Other useful tumor-associated antigens that may be targeted include
carbonic
anhydrase IX, B7, CCL19, CCL21, CSAp, HER-2/neu, BrE3, CD1, CD1a, CD2, CD3,
CD4,
CD5, CD8, CD11A, CD14, CD15, CD16, CD18, CD19, CD20 (e.g., C2B8, hA20, 1F5
MAbs), CD21, CD22, CD23, CD25, CD29, CD30, CD32b, CD33, CD37, CD38, CD40,
CD4OL, CD44, CD45, CD46, CD47, CD52, CD54, CD55, CD59, CD64, CD67, CD70,
CD74, CD79a, CD80, CD83, CD95, CD126, CD133, CD138, CD147, CD154, CEACAM5,
CEACAM6, CTLA-4, alpha-fetoprotein (AFP), VEGF (e.g., AVASTINO, fibronectin
splice
variant), ED-B fibronectin (e.g., L19), EGP-1 (Trop-2), EGP-2 (e.g., 17-1A),
EGF receptor
(ErbB1) (e.g., ERBITUXO), ErbB2, ErbB3, Factor H, FHL-1, Flt-3, folate
receptor, Ga
733,GRO-13, HMGB-1, hypoxia inducible factor (HIF), HM1.24, HER-2/neu, histone
H2B,
histone H3, histone H4, insulin-like growth factor (ILGF), IFN-y, IFN-, IFN-
13, IFN-X, IL-
2R, IL-4R, IL-6R, IL-13R, IL-15R, IL-17R, IL-18R, IL-2, IL-6, IL-8, IL-12, IL-
15, IL-17,
IL-18, IL-25, IP-10, IGF-1R, Ia, HM1.24, gangliosides, HCG, the HLA-DR antigen
to which
L243 binds, CD66 antigens, i.e., CD66a-d or a combination thereof, MAGE, mCRP,
MCP-1,
MIP-1A, MIP-1B, macrophage migration-inhibitory factor (MIF), matrix
metalloproteinase-
2, matrix metalloproteinase-9, matrix metalloproteinase-12, MUC1, MUC2, MUC3,
MUC4,
22
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
MUC5ac, placental growth factor (P1GF), PSA (prostate-specific antigen), PSMA,
PAM4
antigen, PD-1 receptor, PD-L1, NCA-95, NCA-90, A3, A33, RNA, DNA, Ep-CAM, KS-
1,
Le(y), mesothelin, S100, tenascin, TAC, Tn antigen, Thomas-Friedenreich
antigens, tumor
necrosis antigens, tumor angiogenesis antigens, TNF-, TRAIL receptor (R1 and
R2), Trop-
2, VEGFR, RANTES, T101, as well as cancer stem cell antigens, complement
factors C3,
C3a, C3b, C5a, C5, and an oncogene product.
[0105] Cancer stem cells, which are ascribed to be more therapy-resistant
precursor
malignant cell populations (Hill and Perris, i _Arad Cancer Inst. 2007;
99:1435-40), have
antigens that can be targeted in certain cancer types, such as CD133 in
prostate cancer
(Maitland et al., Ernst Schering Found. Sympos. Proc. 2006; 5:155-79), non-
small-cell lung
cancer (Donnenberg et al., I Control Release 2007; 122(3):385-91), and
glioblastoma (Beier
et al., Cancer Res. 2007; 67(9):4010-5), and CD44 in colorectal cancer
(Dalerba er al., Proc.
_Arad Acad. Sci. USA 2007; 104(24)10158-63), pancreatic cancer (Li et al.,
Cancer Res. 2007;
67(3):1030-7), and in head and neck squamous cell carcinoma (Prince et al.,
Proc. Natl.
Acad. Sci. USA 2007; 104(3)973-8). Another useful target for breast cancer
therapy is the
LIV-1 antigen described by Taylor et al. (Biochem. J. 2003; 375:51-9). The
CD47 antigen is
a further useful target for cancer stem cells (see, e.g., Naujokat et al.,
2014, Immunotherapy
6:290-308; Goto et al., 2014, Eur J Cancer 50:1836-46; Unanue, 2013, Proc Natl
Acad Sci
USA 110:10886-7).
[0106] Checkpoint inhibitor antibodies have been used in cancer therapy.
Immune
checkpoints refer to inhibitory pathways in the immune system that are
responsible for
maintaining self-tolerance and modulating the degree of immune system response
to
minimize peripheral tissue damage. However, tumor cells can also activate
immune system
checkpoints to decrease the effectiveness of immune response against tumor
tissues.
Exemplary checkpoint inhibitor antibodies against cytotoxic T-lymphocyte
antigen 4
(CTLA4, also known as CD152), programmed cell death protein 1 (PD1, also known
as
CD279) and programmed cell death 1 ligand 1 (PD-L1, also known as CD274), may
be used
in combination with one or more other agents to enhance the effectiveness of
immune
response against disease cells, tissues or pathogens. Exemplary anti-PD1
antibodies include
lambrolizumab (MK-3475, MERCK), nivolumab (BMS-936558, BRISTOL-MYERS
SQUIBB), AMP-224 (MERCK), and pidilizumab (CT-011, CURETECH LTD.). Anti-PD1
antibodies are commercially available, for example from ABCAMO (AB137132),
BIOLEGENDO (EH12.2H7, RMP1-14) and AFFYMETRIX EBIOSCIENCE (J105, J116,
MIH4). Exemplary anti-PD-L1 antibodies include MDX-1105 (MEDAREX), MEDI4736
23
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
(MEDIMMUNE) MPDL3280A (GENENTECH) and BMS-936559 (BRISTOL-MYERS
SQUIBB). Anti-PD-L1 antibodies are also commercially available, for example
from
AFFYMETRIX EBIOSCIENCE (MIH1). Exemplary anti-CTLA4 antibodies include
ipilimumab (Bristol-Myers Squibb) and tremelimumab (PFIZER). Anti-PD1
antibodies are
commercially available, for example from ABCAMO (AB134090), SINO BIOLOGICAL
INC. (11159-H03H, 11159-H08H), and THERMO SCIENTIFIC PIERCE (PAS-29572, PAS-
23967, PAS-26465, MA1-12205, MA1-35914). Ipilimumab has recently received FDA
approval for treatment of metastatic melanoma (Wada et al., 2013, J Transl Med
11:89).
[0107] Macrophage migration inhibitory factor (MIF) is an important regulator
of innate and
adaptive immunity and apoptosis. It has been reported that CD74 is the
endogenous receptor
for MIF (Leng et al., 2003, J Exp Med 197:1467-76). The therapeutic effect of
antagonistic
anti-CD74 antibodies on MIF-mediated intracellular pathways may be of use for
treatment of
a broad range of disease states, such as cancers of the bladder, prostate,
breast, lung, and
colon (e.g., Meyer-Siegler et al., 2004, BMC Cancer 12:34; Shachar & Haran,
2011, Leuk
Lymphoma 52:1446-54). Milatuzumab (hLL1) is an exemplary anti-CD74 antibody of
therapeutic use for treatment of MIF-mediated diseases.
[0108] Various other antibodies of use are known in the art (e.g., U.S. Patent
Nos. 5,686,072;
5,874,540; 6,107,090; 6,183,744; 6,306,393; 6,653,104; 6,730.300; 6,899,864;
6,926,893;
6,962,702; 7,074,403; 7,230,084; 7,238,785; 7,238,786; 7,256,004; 7,282,567;
7,300,655;
7,312,318; 7,585,491; 7,612,180; 7,642,239 and U.S. Patent Application Publ.
No.
20060193865; each incorporated herein by reference.)
[0109] Antibodies of use may be commercially obtained from a wide variety of
known
sources. For example, a variety of antibody secreting hybridoma lines are
available from the
American Type Culture Collection (ATCC, Manassas, VA). A large number of
antibodies
against various disease targets, including tumor-associated antigens, have
been deposited at
the ATCC and/or have published variable region sequences and are available for
use in the
claimed methods and compositions. See, e.g., U.S. Patent Nos. 7,312,318;
7,282,567;
7,151,164; 7,074,403; 7,060,802; 7,056,509; 7,049,060; 7,045,132; 7,041,803;
7,041,802;
7,041,293; 7,038,018; 7,037,498; 7,012,133; 7,001,598; 6,998,468; 6,994,976;
6,994,852;
6,989,241; 6,974,863; 6,965,018; 6,964,854; 6,962,981; 6,962,813; 6,956,107;
6,951,924;
6,949,244; 6,946,129; 6,943,020; 6,939,547; 6,921,645; 6,921,645; 6,921,533;
6,919,433;
6,919,078; 6,916,475; 6,905,681; 6,899,879; 6,893,625; 6,887,468; 6,887,466;
6,884,594;
6,881,405; 6,878,812; 6,875,580; 6,872,568; 6,867,006; 6,864,062; 6,861,511;
6,861,227;
6,861,226; 6,838,282; 6,835,549; 6,835,370; 6,824,780; 6,824,778; 6,812,206;
6,793,924;
24
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
6,783,758; 6,770,450; 6,767,711; 6,764,688; 6,764,681; 6,764,679; 6,743,898;
6,733,981;
6,730,307; 6,720,155; 6,716,966; 6,709,653; 6,693,176; 6,692,908; 6,689,607;
6,689,362;
6,689,355; 6,682,737; 6,682,736; 6,682,734; 6,673,344; 6,653,104; 6,652,852;
6,635,482;
6,630,144; 6,610,833; 6,610,294; 6,605,441; 6,605,279; 6,596,852; 6,592,868;
6,576,745;
6,572;856; 6,566,076; 6,562,618; 6,545,130; 6,544,749; 6,534,058; 6,528,625;
6,528,269;
6,521,227; 6,518,404; 6,511,665; 6,491,915; 6,488,930; 6,482,598; 6,482,408;
6,479,247;
6,468,531; 6,468,529; 6,465,173; 6,461,823; 6,458,356; 6,455,044; 6,455,040,
6,451,310;
6,444,206; 6,441,143; 6,432,404; 6,432,402; 6,419,928; 6,413,726; 6,406,694;
6,403,770;
6,403,091; 6,395,276; 6,395,274; 6,387,350; 6,383,759; 6,383,484; 6,376,654;
6,372,215;
6,359,126; 6,355,481; 6,355,444; 6,355,245; 6,355,244; 6,346,246; 6,344,198;
6,340,571;
6,340,459; 6,331,175; 6,306,393; 6,254,868; 6,187,287; 6,183,744; 6,129,914;
6,120,767;
6,096,289; 6,077,499; 5,922,302; 5,874,540; 5,814,440; 5,798,229; 5,789,554;
5,776,456;
5,736,119; 5,716,595; 5,677,136; 5,587,459; 5,443,953, 5,525,338. These are
exemplary only
and a wide variety of other antibodies and their hybridomas are known in the
art. The skilled
artisan will realize that antibody sequences or antibody-secreting hybridomas
against almost
any disease-associated antigen may be obtained by a simple search of the ATCC,
NCBI
and/or USPTO databases for antibodies against a selected disease-associated
target of
interest. The antigen binding domains of the cloned antibodies may be
amplified, excised,
ligated into an expression vector, transfected into an adapted host cell and
used for protein
production, using standard techniques well known in the art.
Antibody Allotypes
[0110] Immunogenicity of therapeutic antibodies is associated with increased
risk of infusion
reactions and decreased duration of therapeutic response (Baert et al., 2003,
N Engl J Med
348:602-08). The extent to which therapeutic antibodies induce an immune
response in the
host may be determined in part by the allotype of the antibody (Stickler et
al., 2011, Genes
and Immunity 12:213-21). Antibody allotype is related to amino acid sequence
variations at
specific locations in the constant region sequences of the antibody. The
allotypes of IgG
antibodies containing a heavy chain 7-type constant region are designated as
Gm allotypes
(1976, J Immunol 117:1056-59).
[0111] For the common IgG1 human antibodies, the most prevalent allotype is
Glml
(Stickler et al., 2011, Genes and Immunity 12:213-21). However, the G1m3
allotype also
occurs frequently in Caucasians (Stickler et al., 2011). It has been reported
that Glml
antibodies contain allotypic sequences that tend to induce an immune response
when
administered to non-Glml (nG1m1) recipients, such as G1m3 patients (Stickler
et al., 2011).
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
Non-Glml allotype antibodies are not as immunogenic when administered to Glml
patients
(Stickler et al., 2011).
[0112] The human Glml allotype comprises the amino acids aspartic acid at
Kabat position
356 and leucine at Kabat position 358 in the CH3 sequence of the heavy chain
IgGl. The
nGlml allotype comprises the amino acids glutamic acid at Kabat position 356
and
methionine at Kabat position 358. Both Glml and nGlml allotypes comprise a
glutamic acid
residue at Kabat position 357 and the allotypes are sometimes referred to as
DEL and EEM
allotypes. A non-limiting example of the heavy chain constant region sequences
for Glml
and nGlml allotype antibodies is shown below for the exemplary antibodies
rituximab (SEQ
ID NO:7) and veltuzumab (SEQ ID NO:8).
Rituximab heavy chain variable region sequence (SEQ ID NO: 7)
AS TKGP SVFPLAP S SKS T S G GTAALGCLVKDYFPEPVTV SWNS GALT S GVHTFP
AVLQ S SGLYSLS SVVTVPSS SLGTQTYICNVNHKPSNTKVDKKAEPKSCDKTH
TCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWY
VDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALP
APIEKTISKAKGQPREPQVYTLPP SRDELTKNQVSLTCLVKGFYP SDIAVEWES
NGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNH
YTQKSLSLSPGK
Veltuzumab heavy chain variable region (SEQ ID NO:8)
AS TKGP SVFPLAP S SKS T S G GTAALGCLVKDYFPEPVTV SWNS GALT S GVHTFP
AVLQ S S GLY S LS SVVTVPSS SLGTQTYICNVNHKPSNTKVDKRVEPKSCDKTH
TCPP CPAPELLGGP SVFLFPPKPKDTLMI SRTPEVTCVVVDV S HEDPEVKFNWY
VDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALP
APIEKTISKAKGQPREPQVYTLPP SREEMTKNQVSLTCLVKGFYP SDIAVEWES
NGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNH
YTQKSLSLSPGK
[0113] Jefferis and Lefranc (2009, mAbs 1:1-7) reviewed sequence variations
characteristic
of IgG allotypes and their effect on immunogenicity. They reported that the
Glm3 allotype is
characterized by an arginine residue at Kabat position 214, compared to a
lysine residue at
Kabat 214 in the G1m17 allotype. The nG1m1,2 allotype was characterized by
glutamic acid
at Kabat position 356, methionine at Kabat position 358 and alanine at Kabat
position 431.
The Glm1,2 allotype was characterized by aspartic acid at Kabat position 356,
leucine at
Kabat position 358 and glycine at Kabat position 431. In addition to heavy
chain constant
region sequence variants, Jefferis and Lefranc (2009) reported allotypic
variants in the kappa
26
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
light chain constant region, with the Km1 allotype characterized by valine at
Kabat position
153 and leucine at Kabat position 191, the Km1,2 allotype by alanine at Kabat
position 153
and leucine at Kabat position 191, and the Km3 allotype characterized by
alanine at Kabat
position 153 and valine at Kabat position 191.
[0114] With regard to therapeutic antibodies, veltuzumab and rituximab are,
respectively,
humanized and chimeric IgG1 antibodies against CD20, of use for therapy of a
wide variety
of hematological malignancies and/or autoimmune diseases. Table 1 compares the
allotype
sequences of rituximab vs. veltuzumab. As shown in Table 1, rituximab
(G1m17,1) is a DEL
allotype IgGl, with an additional sequence variation at Kabat position 214
(heavy chain
CH1) of lysine in rituximab vs. arginine in veltuzumab. It has been reported
that veltuzumab
is less immunogenic in subjects than rituximab (see, e.g., Morchhauser et al.,
2009, J Clin
Oncol 27:3346-53; Goldenberg et al., 2009, Blood 113:1062-70; Robak & Robak,
2011,
BioDrugs 25:13-25), an effect that has been attributed to the difference
between humanized
and chimeric antibodies. However, the difference in allotypes between the EEM
and DEL
allotypes likely also accounts for the lower immunogenicity of veltuzumab.
Table 1. Allotypes of Rituximab vs. Veltuzumab
Heavy chain position and associated allotypes
Complete allotype 214 (allotype) 356/358 (allotype) 431 (allotype)
Rituximab Glm17,1 K 17 D/L 1 A
Veltuzumab G1m3 R 3 E/M A
[0115] In order to reduce the immunogenicity of therapeutic antibodies in
individuals of
nGlml genotype, it is desirable to select the allotype of the antibody to
correspond to the
G1m3 allotype, characterized by arginine at Kabat 214, and the nG1m1,2 null-
allotype,
characterized by glutamic acid at Kabat position 356, methionine at Kabat
position 358 and
alanine at Kabat position 431. Surprisingly, it was found that repeated
subcutaneous
administration of Glm3 antibodies over a long period of time did not result in
a significant
immune response. In alternative embodiments, the human IgG4 heavy chain in
common with
the G1m3 allotype has arginine at Kabat 214, glutamic acid at Kabat 356,
methionine at
Kabat 359 and alanine at Kabat 431. Since immunogenicity appears to relate at
least in part to
the residues at those locations, use of the human IgG4 heavy chain constant
region sequence
for therapeutic antibodies is also a preferred embodiment. Combinations of
Glm3 IgG1
antibodies with IgG4 antibodies may also be of use for therapeutic
administration.
Nanobodies
27
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
[0116] Nanobodies are single-domain antibodies of about 12-15 kDa in size
(about 110
amino acids in length). Nanobodies can selectively bind to target antigens,
like full-size
antibodies, and have similar affinities for antigens. However, because of
their much smaller
size, they may be capable of better penetration into solid tumors. The smaller
size also
contributes to the stability of the nanobody, which is more resistant to pH
and temperature
extremes than full size antibodies (Van Der Linden et al., 1999, Biochim
Biophys Act
1431:37-46). Single-domain antibodies were originally developed following the
discovery
that camelids (camels, alpacas, llamas) possess fully functional antibodies
without light
chains (e.g., Hamsen et al., 2007, Appl Microbiol Biotechnol 77:13-22). The
heavy-chain
antibodies consist of a single variable domain (VHH) and two constant domains
(CH2 and
CH3). Like antibodies, nanobodies may be developed and used as multivalent
and/or
bispecific constructs. Humanized forms of nanobodies are in commercial
development that
are targeted to a variety of target antigens, such as IL-6R, vWF, TNF, RSV,
RANKL, IL-17A
& F and IgE (e.g., ABLYNXO, Ghent, Belgium), with potential clinical use in
cancer and
other disorders (e.g., Saerens et al., 2008, Curr Opin Pharmacol 8:600-8;
Muyldermans, 2013,
Ann Rev Biochem 82:775-97; Ibanez et al., 2011, J Infect Dis 203:1063-72).
[0117] The plasma half-life of nanobodies is shorter than that of full-size
antibodies, with
elimination primarily by the renal route. Because they lack an Fc region, they
do not exhibit
complement dependent cytotoxicity.
[0118] Nanobodies may be produced by immunization of camels, llamas, alpacas
or sharks
with target antigen, following by isolation of mRNA, cloning into libraries
and screening for
antigen binding. Nanobody sequences may be humanized by standard techniques
(e.g., Jones
et al., 1986, Nature 321: 522, Riechmann et al., 1988, Nature 332: 323,
Verhoeyen et al.,
1988, Science 239: 1534, Carter et al., 1992, Proc. Nat'l Acad. Sci. USA 89:
4285, Sandhu,
1992, Crit. Rev. Biotech. 12: 437, Singer et al., 1993, J. Immun. 150: 2844).
Humanization is
relatively straight-forward because of the high homology between camelid and
human FR
sequences.
[0119] In various embodiments, the subject ADCs may comprise nanobodies for
targeted
delivery of conjugated drug to targeted cancer cells. Nanobodies of use are
disclosed, for
example, in U.S. Patent Nos. 7,807,162; 7,939,277; 8,188,223; 8,217,140;
8,372,398;
8,557,965; 8,623,361 and 8,629,244, the Examples section of each incorporated
herein by
reference.)
28
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
Antibody Fragments
[0120] Antibody fragments are antigen binding portions of an antibody, such as
F(ab') 2, Fab',
F(ab)2, Fab, Fv, sFv, scFv and the like. Antibody fragments which recognize
specific epitopes
can be generated by known techniques. F(ab')2 fragments, for example, can be
produced by
pepsin digestion of the antibody molecule. These and other methods are
described, for
example, by Goldenberg, U.S. Pat. Nos. 4,036,945 and 4,331,647 and references
contained
therein. Also, see Nisonoff et al., Arch Biochem. Biophys. 89: 230 (1960);
Porter, Biochem.
J. 73: 119 (1959), Edelman et al., in METHODS IN ENZYMOLOGY VOL. 1, page 422
(Academic Press 1967), and Coligan at pages 2.8.1-2.8.10 and 2.10.-2.10.4.
Alternatively,
Fab' expression libraries can be constructed (Huse et al., 1989, Science,
246:1274-1281) to
allow rapid and easy identification of monoclonal Fab' fragments with the
desired specificity.
[0121] A single chain Fv molecule (scFv) comprises a VL domain and a VH
domain. The VL
and VH domains associate to form a target binding site. These two domains are
further
covalently linked by a peptide linker (L). A scFv molecule is denoted as
either VL-L-VH if
the VL domain is the N-terminal part of the scFv molecule, or as VH-L-VL if
the VH domain
is the N-terminal part of the scFv molecule. Methods for making scFv molecules
and
designing suitable peptide linkers are described in U.S. Pat. No. 4,704,692,
U.S. Pat. No.
4,946,778, R. Raag and M. Whitlow, "Single Chain Fvs." FASEB Vol 9:73-80
(1995) and R.
E. Bird and B. W. Walker, Single Chain Antibody Variable Regions, TIBTECH, Vol
9: 132-
137 (1991).
[0122] Other antibody fragments, for example single domain antibody fragments,
are known
in the art and may be used in the claimed constructs. Single domain antibodies
(VHH) may
be obtained, for example, from camels, alpacas or llamas by standard
immunization
techniques. (See, e.g., Muyldermans et al., TIBS 26:230-235, 2001; Yau et al.,
J Immunol
Methods 281:161-75, 2003; Maass et al., J Immunol Methods 324:13-25, 2007).
The VHH
may have potent antigen-binding capacity and can interact with novel epitopes
that are
inaccessible to conventional VH-VL pairs. (Muyldermans et al., 2001). Alpaca
serum IgG
contains about 50% camelid heavy chain only IgG antibodies (HCAbs) (Maass et
al., 2007).
Alpacas may be immunized with known antigens, such as TNF-, and VHHs can be
isolated
that bind to and neutralize the target antigen (Maass et al., 2007). PCR
primers that amplify
virtually all alpaca VHH coding sequences have been identified and may be used
to construct
alpaca VHH phage display libraries, which can be used for antibody fragment
isolation by
standard biopanning techniques well known in the art (Maass et al., 2007).
29
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
[0123] An antibody fragment can also be prepared by proteolytic hydrolysis of
a full-length
antibody or by expression in E. coli or another host of the DNA coding for the
fragment. An
antibody fragment can be obtained by pepsin or papain digestion of full-length
antibodies by
conventional methods. For example, an antibody fragment can be produced by
enzymatic
cleavage of antibodies with pepsin to provide an approximate 100 kD fragment
denoted
F(ab')2. This fragment can be further cleaved using a thiol reducing agent,
and optionally a
blocking group for the sulfhydryl groups resulting from cleavage of disulfide
linkages, to
produce an approximate 50 Kd Fab' monovalent fragment. Alternatively, an
enzymatic
cleavage using papain produces two monovalent Fab fragments and an Fc fragment
directly.
[0124] Other methods of cleaving antibodies, such as separation of heavy
chains to form
monovalent light-heavy chain fragments, further cleavage of fragments, or
other enzymatic,
chemical or genetic techniques may also be used, so long as the fragments bind
to the antigen
that is recognized by the intact antibody.
General techniques for antibody cloning and production
[0125] Various techniques, such as production of chimeric or humanized
antibodies, may
involve procedures of antibody cloning and construction. The antigen-binding
Vic (variable
light chain) and VH (variable heavy chain) sequences for an antibody of
interest may be
obtained by a variety of molecular cloning procedures, such as RT-PCR, 5'-
RACE, and
cDNA library screening. The V genes of a MAb from a cell that expresses a
murine MAb can
be cloned by PCR amplification and sequenced. To confirm their authenticity,
the cloned VL
and VH genes can be expressed in cell culture as a chimeric Ab as described by
Orlandi et al.,
(Proc. _Arad Acad. Sci., USA, 86: 3833 (1989)). Based on the V gene sequences,
a humanized
MAb can then be designed and constructed as described by Leung et al. (Mol.
Immunol., 32:
1413 (1995)).
[0126] cDNA can be prepared from any known hybridoma line or transfected cell
line
producing a murine MAb by general molecular cloning techniques (Sambrook et
al.,
Molecular Cloning, A laboratory manual, 2'd Ed (1989)). The Vic sequence for
the MAb may
be amplified using the primers VKlBACK and VK1FOR (Orlandi et al., 1989) or
the
extended primer set described by Leung et al. (BioTechniques, 15: 286 (1993)).
The VH
sequences can be amplified using the primer pair VH1BACKNH1FOR (Orlandi et
al., 1989)
or the primers annealing to the constant region of murine IgG described by
Leung et al.
(Hybridoma, 13:469 (1994)). Humanized V genes can be constructed by a
combination of
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
long oligonucleotide template syntheses and PCR amplification as described by
Leung et al.
(Mol. Immunol., 32: 1413 (1995)).
[0127] PCR products for Vic can be subcloned into a staging vector, such as a
pBR327-based
staging vector, VKpBR, that contains an Ig promoter, a signal peptide sequence
and
convenient restriction sites. PCR products for VH can be subcloned into a
similar staging
vector, such as the pBluescript-based VHpBS. Expression cassettes containing
the Vic and VH
sequences together with the promoter and signal peptide sequences can be
excised from
VKpBR and VHpBS and ligated into appropriate expression vectors, such as pKh
and pG1g,
respectively (Leung et al., Hybridoma, 13:469 (1994)). The expression vectors
can be co-
transfected into an appropriate cell and supernatant fluids monitored for
production of a
chimeric, humanized or human MAb. Alternatively, the Vic and VH expression
cassettes can
be excised and subcloned into a single expression vector, such as pdHL2, as
described by
Gillies et al. (J. Immunol. Methods 125:191 (1989) and also shown in Losman et
al., Cancer,
80:2660 (1997)).
[0128] In an alternative embodiment, expression vectors may be transfected
into host cells
that have been pre-adapted for transfection, growth and expression in serum-
free medium.
Exemplary cell lines that may be used include the Sp/EEE, Sp/ESF and Sp/ESF-X
cell lines
(see, e.g., U.S. Patent Nos. 7,531,327; 7,537,930 and 7,608,425; the Examples
section of each
of which is incorporated herein by reference). These exemplary cell lines are
based on the
Sp2/0 myeloma cell line, transfected with a mutant Bcl-EEE gene, exposed to
methotrexate
to amplify transfected gene sequences and pre-adapted to serum-free cell line
for protein
expression.
Bispecific and Multispecific Antibodies
[0129] In certain embodiments the anti-Trop-2 ADC and one or more other
therapeutic
antibodies may be administered as separate antibodies, either sequentially or
concurrently. In
alternative embodiments, antibodies or antibody fragments may be administered
as a single
bispecific or multispecific antibody. Numerous methods to produce bispecific
or
multispecific antibodies are known, as disclosed, for example, in U.S. Patent
No. 7,405,320,
the Examples section of which is incorporated herein by reference. Bispecific
antibodies can
be produced by the quadroma method, which involves the fusion of two different
hybridomas, each producing a monoclonal antibody recognizing a different
antigenic site
(Milstein and Cuello, Nature, 1983; 305:537-540).
31
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
[0130] Another method for producing bispecific antibodies uses
heterobifunctional cross-
linkers to chemically tether two different monoclonal antibodies (Staerz, et
al. Nature. 1985;
314:628-631; Perez, et al. Nature. 1985; 316:354-356). Bispecific antibodies
can also be
produced by reduction of each of two parental monoclonal antibodies to the
respective half
molecules, which are then mixed and allowed to reoxidize to obtain the hybrid
structure
(Staerz and Bevan. Proc Natl Acad Sci U S A. 1986; 83:1453-1457). Other
methods include
improving the efficiency of generating hybrid hybridomas by gene transfer of
distinct
selectable markers via retrovirus-derived shuttle vectors into respective
parental hybridomas,
which are fused subsequently (DeMonte, et al. Proc Natl Acad Sci U S A. 1990,
87:2941-
2945); or transfection of a hybridoma cell line with expression plasmids
containing the heavy
and light chain genes of a different antibody.
[0131] Cognate VH and VL domains can be joined with a peptide linker of
appropriate
composition and length (usually consisting of more than 12 amino acid
residues) to form a
single-chain Fv (scFv), as discussed above. Reduction of the peptide linker
length to less than
12 amino acid residues prevents pairing of VH and VL domains on the same chain
and forces
pairing of VH and VL domains with complementary domains on other chains,
resulting in the
formation of functional multimers. Polypeptide chains of VH and VL domains
that are joined
with linkers between 3 and 12 amino acid residues form predominantly dimers
(termed
diabodies). With linkers between 0 and 2 amino acid residues, trimers (termed
triabody) and
tetramers (termed tetrabody) are favored, but the exact patterns of
oligomerization appear to
depend on the composition as well as the orientation of V-domains (VH-linker-
VL or VL-
linker-VH), in addition to the linker length.
[0132] These techniques for producing multispecific or bispecific antibodies
exhibit various
difficulties in terms of low yield, necessity for purification, low stability
or the labor-
intensiveness of the technique. More recently, a technique known as "DOCK-AND-
LOCKTM" (DNLTm), discussed in more detail below, has been utilized to produce
combinations of virtually any desired antibodies, antibody fragments and other
effector
molecules. Any of the techniques known in the art for making bispecific or
multispecific
antibodies may be utilized in the practice of the presently claimed methods.
DOCK-AND-LOCKTM (DNLTM)
[0133] Bispecific or multispecific antibodies or other constructs may be
produced using the
DOCK-AND-LOCKTm technology (see, e.g., U.S. Patent Nos. 7,550,143; 7,521,056;
7,534,866; 7,527,787 and 7,666,400, the Examples section of each incorporated
herein by
32
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
reference). Generally, the technique takes advantage of the specific and high-
affinity binding
interactions that occur between a dimerization and docking domain (DDD)
sequence of the
regulatory (R) subunits of cAMP-dependent protein kinase (PKA) and an anchor
domain
(AD) sequence derived from any of a variety of AKAP proteins (Baillie et al.,
FEBS Letters.
2005; 579: 3264. Wong and Scott, Nat. Rev. Mol. Cell Biol. 2004; 5: 959). The
DDD and AD
peptides may be attached to any protein, peptide or other molecule. Because
the DDD
sequences spontaneously dimerize and bind to the AD sequence, the technique
allows the
formation of complexes between any selected molecules that may be attached to
DDD or AD
sequences.
[0134] Although the standard DNLTM complex comprises a trimer with two DDD-
linked
molecules attached to one AD-linked molecule, variations in complex structure
allow the
formation of dimers, trimers, tetramers, pentamers, hexamers and other
multimers. In some
embodiments, the DNLTM complex may comprise two or more antibodies, antibody
fragments or fusion proteins which bind to the same antigenic determinant or
to two or more
different antigens. The DNLTM complex may also comprise one or more other
effectors, such
as proteins, peptides, immunomodulators, cytokines, interleukins, interferons,
binding
proteins, peptide ligands, carrier proteins, toxins, ribonucleases such as
onconase, inhibitory
oligonucleotides such as siRNA, antigens or xenoantigens, polymers such as
PEG, enzymes,
therapeutic agents, hormones, cytotoxic agents, anti-angiogenic agents, pro-
apoptotic agents
or any other molecule or aggregate.
[0135] PKA, which plays a central role in one of the best studied signal
transduction
pathways triggered by the binding of the second messenger cAMP to the R
subunits, was first
isolated from rabbit skeletal muscle in 1968 (Walsh et al., J. Biol. Chem.
1968;243:3763).
The structure of the holoenzyme consists of two catalytic subunits held in an
inactive form by
the R subunits (Taylor, J. Biol. Chem. 1989;264:8443). Isozymes of PKA are
found with two
types of R subunits (RI and RII), and each type has a and isoforms (Scott,
Pharmacol.
Ther. 1991;50:123). Thus, the four isoforms of PKA regulatory subunits are
RIoc, RIP, Ma
and RII13. The R subunits have been isolated only as stable dimers and the
dimerization
domain has been shown to consist of the first 44 amino-terminal residues of
RIIct (Newlon et
al., Nat. Struct. Biol. 1999; 6:222). As discussed below, similar portions of
the amino acid
sequences of other regulatory subunits are involved in dimerization and
docking, each located
near the N-terminal end of the regulatory subunit. Binding of cAMP to the R
subunits leads
to the release of active catalytic subunits for a broad spectrum of
serine/threonine kinase
33
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
activities, which are oriented toward selected substrates through the
compartmentalization of
PKA via its docking with AKAPs (Scott et al., J. Biol. Chem. 1990;265;21561)
[0136] Since the first AKAP, microtubule-associated protein-2, was
characterized in 1984
(Lohmann et al., Proc. Natl. Acad. Sci USA. 1984; 81:6723), more than 50 AKAPs
that
localize to various sub-cellular sites, including plasma membrane, actin
cytoskeleton,
nucleus, mitochondria, and endoplasmic reticulum, have been identified with
diverse
structures in species ranging from yeast to humans (Wong and Scott, Nat. Rev.
Mol. Cell
Biol. 2004;5:959). The AD of AKAPs for PKA is an amphipathic helix of 14-18
residues
(Carr et al., J. Biol. Chem. 1991;266:14188). The amino acid sequences of the
AD are quite
varied among individual AKAPs, with the binding affinities reported for RII
dimers ranging
from 2 to 90 nM (Alto et al., Proc. Natl. Acad. Sci. USA. 2003;100:4445).
AKAPs will only
bind to dimeric R subunits. For human Rlloc, the AD binds to a hydrophobic
surface formed
by the 23 amino-terminal residues (Colledge and Scott, Trends Cell Biol. 1999;
6:216). Thus,
the dimerization domain and AKAP binding domain of human Rlloc are both
located within
the same N-terminal 44 amino acid sequence (Newlon et al., Nat. Struct. Biol.
1999;6:222;
Newlon et al., EMBO J. 2001;20:1651), which is termed the DDD herein.
[0137] We have developed a platform technology to utilize the DDD of human PKA
regulatory subunits and the AD of AKAP as an excellent pair of linker modules
for docking
any two entities, referred to hereafter as A and B, into a noncovalent
complex, which could
be further locked into a DNLTM complex through the introduction of cysteine
residues into
both the DDD and AD at strategic positions to facilitate the formation of
disulfide bonds. The
general methodology of the approach is as follows. Entity A is constructed by
linking a DDD
sequence to a precursor of A, resulting in a first component hereafter
referred to as a.
Because the DDD sequence would effect the spontaneous formation of a dimer, A
would thus
be composed of a2. Entity B is constructed by linking an AD sequence to a
precursor of B,
resulting in a second component hereafter referred to as b. The dimeric motif
of DDD
contained in a2 will create a docking site for binding to the AD sequence
contained in b, thus
facilitating a ready association of a2 and b to form a binary, trimeric
complex composed of
a2b. This binding event is made irreversible with a subsequent reaction to
covalently secure
the two entities via disulfide bridges, which occurs very efficiently based on
the principle of
effective local concentration because the initial binding interactions should
bring the reactive
thiol groups placed onto both the DDD and AD into proximity (Chmura et al.,
Proc. Natl.
Acad. Sci. USA. 2001;98:8480) to ligate site-specifically. Using various
combinations of
34
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
linkers, adaptor modules and precursors, a wide variety of DNLTM constructs of
different
stoichiometry may be produced and used (see, e.g., U.S. Nos. 7,550,143;
7,521,056;
7,534,866; 7,527,787 and 7,666,400.)
[0138] By attaching the DDD and AD away from the functional groups of the two
precursors, such site-specific ligations are also expected to preserve the
original activities of
the two precursors. This approach is modular in nature and potentially can be
applied to link,
site-specifically and covalently, a wide range of substances, including
peptides, proteins,
antibodies, antibody fragments, and other effector moieties with a wide range
of activities.
Utilizing the fusion protein method of constructing AD and DDD conjugated
effectors
described below, virtually any protein or peptide may be incorporated into a
DNLTM
construct. However, the technique is not limiting and other methods of
conjugation may be
utilized.
[0139] A variety of methods are known for making fusion proteins, including
nucleic acid
synthesis, hybridization and/or amplification to produce a synthetic double-
stranded nucleic
acid encoding a fusion protein of interest. Such double-stranded nucleic acids
may be inserted
into expression vectors for fusion protein production by standard molecular
biology
techniques (see, e.g. Sambrook et al., Molecular Cloning, A laboratory manual,
2'd Ed, 1989).
In such preferred embodiments, the AD and/or DDD moiety may be attached to
either the N-
terminal or C-terminal end of an effector protein or peptide. However, the
skilled artisan will
realize that the site of attachment of an AD or DDD moiety to an effector
moiety may vary,
depending on the chemical nature of the effector moiety and the part(s) of the
effector moiety
involved in its physiological activity. Site-specific attachment of a variety
of effector moieties
may be performed using techniques known in the art, such as the use of
bivalent cross-linking
reagents and/or other chemical conjugation techniques.
Structure-Function Relationships in AD and DDD Moieties
[0140] For different types of DNLTm constructs, different AD or DDD sequences
may be
utilized. Exemplary DDD and AD sequences are provided below.
DDD/
SHIQIPPGLTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID
NO:9)
DDD2
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
CGHIQIPPGLTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID
NO:10)
AD/
QIEYLAKQIVDNAIQQA (SEQ ID NO:11)
A 2
CGQIEYLAKQIVDNAIQQAGC (SEQ ID NO:12)
[0141] The skilled artisan will realize that DDD1 and DDD2 are based on the
DDD sequence
of the human Rlloc isoform of protein kinase A. However, in alternative
embodiments, the
DDD and AD moieties may be based on the DDD sequence of the human RIoc form of
protein kinase A and a corresponding AKAP sequence, as exemplified in DDD3,
DDD3C
and AD3 below.
DDD3
SLRECELYVQKHNIQALLKDSIVQLCTARPERPMAFLREYFERLEKEEAK
(SEQ ID NO:13)
3C
MSCGGSLRECELYVQKHNIQALLKDSIVQLCT ARPERPMAFLREYFERLEKEE
AK (SEQ ID NO:14)
A 3
CGFEELAWKIAKMIWSDVFQQGC (SEQ ID NO:15)
[0142] In other alternative embodiments, other sequence variants of AD and/or
DDD
moieties may be utilized in construction of the DNLTM complexes. For example,
there are
only four variants of human PKA DDD sequences, corresponding to the DDD
moieties of
PKA RIct, RIkt, RIP and RIII3. The Mkt DDD sequence is the basis of DDD1 and
DDD2
disclosed above. The four human PKA DDD sequences are shown below. The DDD
sequence represents residues 1-44 of RIkt, 1-44 of RIII3, 12-61 of Rlia and 13-
66 of RII3.
(Note that the sequence of DDD1 is modified slightly from the human PKA RIM,
DDD
moiety.)
PKA RIa
SLRECELYVQKHNIQALLKDVSIVQLCTARPERPMAFLREYFEKLEKEEAK
(SEQ ID NO:16)
36
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
PICA RIfi
SLKGCELYVQLHGIQQVLKDCIVHLCISKPERPMKFLREHFEKLEKEENRQILA
(SEQ ID NO:17)
PICA RIIa
SHIQIPPGLTELLQGYTVEVGQQPPDLVDFAVEYFTRLREARRQ (SEQ ID
NO:18)
PICA RIII6
SIEIPAGLTELLQGFTVEVLRHQPADLLEFALQHFTRLQQENER (SEQ ID
NO:19)
[0143] The structure-function relationships of the AD and DDD domains have
been the
subject of investigation. (See, e.g., Burns-Hamuro et al., 2005, Protein Sci
14:2982-92; Carr
et al., 2001, J Biol Chem 276:17332-38; Alto et al., 2003, Proc Natl Acad Sci
USA 100:4445-
50; Hundsrucker et al., 2006, Biochem J 396:297-306; Stokka et al., 2006,
Biochem J
400:493-99; Gold et al., 2006, Mol Cell 24:383-95; Kinderman et al., 2006, Mol
Cell 24:397-
408, the entire text of each of which is incorporated herein by reference.)
[0144] For example, Kinderman et al. (2006, Mol Cell 24:397-408) examined the
crystal
structure of the AD-DDD binding interaction and concluded that the human DDD
sequence
contained a number of conserved amino acid residues that were important in
either dimer
formation or AKAP binding, underlined in SEQ ID NO:9 below. (See Figure 1 of
Kinderman
et al., 2006, incorporated herein by reference.) The skilled artisan will
realize that in
designing sequence variants of the DDD sequence, one would desirably avoid
changing any
of the underlined residues, while conservative amino acid substitutions might
be made for
residues that are less critical for dimerization and AKAP binding.
SHIQIPPGLTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO:9)
101451 As discussed in more detail below, conservative amino acid
substitutions have been
characterized for each of the twenty common L-amino acids. Thus, based on the
data of
Kinderman (2006) and conservative amino acid substitutions, potential
alternative DDD
sequences based on SEQ ID NO:9 are shown in Table 2. In devising Table 2, only
highly
conservative amino acid substitutions were considered. For example, charged
residues were
only substituted for residues of the same charge, residues with small side
chains were
substituted with residues of similar size, hydroxyl side chains were only
substituted with
other hydroxyls, etc. Because of the unique effect of proline on amino acid
secondary
37
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
structure, no other residues were substituted for proline. A limited number of
such potential
alternative DDD moiety sequences are shown in SEQ ID NO:20 to SEQ ID NO:39
below.
The skilled artisan will realize that an almost unlimited number of
alternative species within
the genus of DDD moieties can be constructed by standard techniques, for
example using a
commercial peptide synthesizer or well known site-directed mutagenesis
techniques. The
effect of the amino acid substitutions on AD moiety binding may also be
readily determined
by standard binding assays, for example as disclosed in Alto et al. (2003,
Proc Natl Acad Sci
USA 100:4445-50).
Table 2. Conservative Amino Acid Substitutions in DDD1 (SEQ ID NO:9).
Consensus
sequence disclosed as SEQ ID NO: 94.
S HI QIPPGL TELL QGY TVE VLR
T K N A SD NA S D K
R
QQP P DL VEF AVE YF T RL RE AR A
NN E D L D S K KDLKL
I I I
V V V
THIQIPPGLTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO :20)
SKIQIPPGLTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO:21)
SRIQIPPGLTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO:22)
SHINIPPGLTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO :23)
SHIQIPPALTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO :24)
SHIQIPPGLSELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO:25)
SHIQIPPGLTDLLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO:26)
SHIQIPPGLTELLNGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO :27)
SHIQIPPGLTELLQAYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO :28)
SHIQIPPGLTELLQGYSVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO:29)
SHIQIPPGLTELLQGYTVDVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO:30)
SHIQIPPGLTELLQGYTVEVLKQQPPDLVEFAVEYFTRLREARA (SEQ ID NO:31)
SHIQIPPGLTELLQGYTVEVLRNQPPDLVEFAVEYFTRLREARA (SEQ ID NO:32)
38
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
SHIQIPPGLTELLQGYTVEVLRQNPPDLVEFAVEYFTRLREARA (SEQ ID NO:33)
SHIQIPPGLTELLQGYTVEVLRQQPPELVEFAVEYFTRLREARA (SEQ ID NO :34)
SHIQIPPGLTELLQGYTVEVLRQQPPDLVDFAVEYFTRLREARA (SEQ ID NO:35)
SHIQIPPGLTELLQGYTVEVLRQQPPDLVEFLVEYFTRLREARA (SEQ ID NO:36)
SHIQIPPGLTELLQGYTVEVLRQQPPDLVEFIVEYFTRLREARA (SEQ ID NO:37)
SHIQIPPGLTELLQGYTVEVLRQQPPDLVEFVVEYFTRLREARA (SEQ ID NO:38)
SHIQIPPGLTELLQGYTVEVLRQQPPDLVEFAVDYFTRLREARA (SEQ ID NO:39)
[0146] Alto et al. (2003, Proc Natl Acad Sci USA 100:4445-50) performed a
bioinformatic
analysis of the AD sequence of various AKAP proteins to design an RII
selective AD
sequence called AKAP-IS (SEQ ID NO:11), with a binding constant for DDD of 0.4
nM. The
AKAP-IS sequence was designed as a peptide antagonist of AKAP binding to PKA.
Residues
in the AKAP-IS sequence where substitutions tended to decrease binding to DDD
are
underlined in SEQ ID NO:11 below. The skilled artisan will realize that in
designing
sequence variants of the AD sequence, one would desirably avoid changing any
of the
underlined residues, while conservative amino acid substitutions might be made
for residues
that are less critical for DDD binding. Table 3 shows potential conservative
amino acid
substitutions in the sequence of AKAP-IS (AD1, SEQ ID NO:19), similar to that
shown for
DDD1 (SEQ ID NO:16) in Table 2 above.
[0147] A limited number of such potential alternative AD moiety sequences are
shown in
SEQ ID NO:40 to SEQ ID NO:57 below. Again, a very large number of species
within the
genus of possible AD moiety sequences could be made, tested and used by the
skilled artisan,
based on the data of Alto et al. (2003). It is noted that Figure 2 of Alto
(2003) shows an even
large number of potential amino acid substitutions that may be made, while
retaining binding
activity to DDD moieties, based on actual binding experiments.
AKAP-IS
QIEYLAKQIVDNAIQQA (SEQ ID NO:11)
Table 3. Conservative Amino Acid Substitutions in AD1 (SEQ ID NO:11).
Consensus
sequence disclosed as SEQ ID NO: 95.
QI E YL AK QI V DN AI QQ A
39
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
NL DF I R N E Q N N L
V T V I
S V
NIEYLAKQIVDNAIQQA (SEQ ID NO:40)
QLEYLAKQIVDNAIQQA (SEQ ID NO:41)
QVEYLAKQIVDNAIQQA (SEQ ID NO:42)
QIDYLAKQIVDNAIQQA (SEQ ID NO:43)
QIEFLAKQIVDNAIQQA (SEQ ID NO:44)
QIETLAKQIVDNAIQQA (SEQ ID NO:45)
QIESLAKQIVDNAIQQA (SEQ ID NO:46)
QIEYIAKQIVDNAIQQA (SEQ ID NO:47)
QIEYVAKQIVDNAIQQA (SEQ ID NO:48)
QIEYLARQIVDNAIQQA (SEQ ID NO:49)
QIEYLAKNIVDNAIQQA (SEQ ID NO:50)
QIEYLAKQIVENAIQQA (SEQ ID NO:51)
QIEYLAKQIVDQAIQQA (SEQ ID NO:52)
QIEYLAKQIVDNAINQA (SEQ ID NO:53)
QIEYLAKQIVDNAIQNA (SEQ ID NO:54)
QIEYLAKQIVDNAIQQL (SEQ ID NO:55)
QIEYLAKQIVDNAIQQI (SEQ ID NO:56)
QIEYLAKQIVDNAIQQV (SEQ ID NO:57)
[0148] Gold et al. (2006, Mol Cell 24:383-95) utilized crystallography and
peptide screening
to develop a SuperAKAP-IS sequence (SEQ ID NO:58), exhibiting a five order of
magnitude
higher selectivity for the RII isoform of PKA compared with the RI isoform.
Underlined
residues indicate the positions of amino acid substitutions, relative to the
AKAP-IS sequence,
which increased binding to the DDD moiety of Mkt. In this sequence, the N-
terminal Q
residue is numbered as residue number 4 and the C-terminal A residue is
residue number 20.
Residues where substitutions could be made to affect the affinity for Ma were
residues 8,
11, 15, 16, 18, 19 and 20 (Gold et al., 2006). It is contemplated that in
certain alternative
embodiments, the SuperAKAP-IS sequence may be substituted for the AKAP-IS AD
moiety
sequence to prepare DNLTM constructs. Other alternative sequences that might
be substituted
for the AKAP-IS AD sequence are shown in SEQ ID NO:59-61. Substitutions
relative to the
AKAP-IS sequence are underlined. It is anticipated that, as with the AD2
sequence shown in
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
SEQ ID NO:12, the AD moiety may also include the additional N-terminal
residues cysteine
and glycine and C-terminal residues glycine and cysteine.
SuperAKAP-IS
QIEYVAKQIVDYAIHQA (SEQ ID NO:58)
Alternative AKAP sequences
QIEYKAKQIVDHAIHQA (SEQ ID NO:59)
QIEYHAKQIVDHAIHQA (SEQ ID NO:60)
QIEYVAKQIVDHAIHQA (SEQ ID NO:61)
[0149] Figure 2 of Gold et al. disclosed additional DDD-binding sequences from
a variety of
AKAP proteins, shown below.
RII-Specific AKAPs
AKAP-KL
PLEYQAGLLVQNAIQQAI (SEQ ID NO:62)
AKAP79
LLIETASSLVKNAIQLSI (SEQ ID NO:63)
AKAP-Lbc
LIEEAASRIVDAVIEQVK (SEQ ID NO:64)
RI-Specific AKAPs
AKAPce
ALYQFADRFSELVISEAL (SEQ ID NO:65)
RIAD
LEQVANQLADQIIKEAT (SEQ ID NO:66)
PV38
FEELAWKIAKMIWSDVF (SEQ ID NO:67)
Dual-Specificity AKAPs
AKAP7
ELVRLSKRLVENAVLKAV (SEQ ID NO:68)
MAP2D
TAEEVSARIVQVVTAEAV (SEQ ID NO:69)
DAKAPI
QIKQAAFQLISQVILEAT (SEQ ID NO:70)
DAKAP2
41
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
LAWKIAKMIVSDVMQQ (SEQ ID NO:71)
[0150] Stokka et al. (2006, Biochem J 400:493-99) also developed peptide
competitors of
AKAP binding to PKA, shown in SEQ ID NO:72-74. The peptide antagonists were
designated as Ht31 (SEQ ID NO:72), RIAD (SEQ ID NO:73) and PV-38 (SEQ ID
NO:74).
The Ht-31 peptide exhibited a greater affinity for the RII isoform of PKA,
while the RIAD
and PV-38 showed higher affinity for RI.
Ht31
DLIEEAASRIVDAVIEQVKAAGAY (SEQ ID NO:72)
RIAD
LEQYANQLADQIIKEATE (SEQ ID NO:73)
PV-38
FEELAWKIAKMIWSDVFQQC (SEQ ID NO:74)
[0151] Hundsrucker et al. (2006, Biochem J 396:297-306) developed still other
peptide
competitors for AKAP binding to PKA, with a binding constant as low as 0.4 nM
to the DDD
of the RII form of PKA. The sequences of various AKAP antagonistic peptides
are provided
in Table 1 of Hundsrucker et al., reproduced in Table 4 below. AKAPIS
represents a
synthetic RII subunit-binding peptide. All other peptides are derived from the
RII-binding
domains of the indicated AKAPs.
Table 4. AKAP Peptide sequences
Peptide Sequence
AKAPIS QIEYLAKQIVDNAIQQA (SEQ ID NO:11)
AKAPIS-P QIEYLAKQIPDNAIQQA (SEQ ID NO:75)
Ht31 KGADLIEEAASRIVDAVIEQVKAAG (SEQ ID NO:76)
Ht31-P KGADLIEEAASRIPDAPIEQVKAAG (SEQ ID NO:77)
AKAP7o-wt-pep PEDAELVRLSKRLVENAVLKAVQQY (SEQ ID NO:78)
AKAP7o-L304T-pep PEDAELVRTSKRLVENAVLKAVQQY (SEQ ID NO:79)
AKAP7o-L308D-pep PEDAELVRLSKRDVENAVLKAVQQY (SEQ ID NO:80)
AKAP7o-P-pep PEDAELVRLSKRLPENAVLKAVQQY (SEQ ID NO:81)
AKAP7o-PP-pep PEDAELVRLSKRLPENAPLKAVQQY (SEQ ID NO:82)
AKAP7o-L314E-pep PEDAELVRLSKRLVENAVEKAVQQY (SEQ ID NO:83)
AKAP1-pep EEGLDRNEEIKRAAFQIISQVISEA (SEQ ID NO:84)
AKAP2-pep LVDDPLEYQAGLLVQNAIQQAIAEQ (SEQ ID NO:85)
AKAP5-pep QYETLLIETASSLVKNAIQLSIEQL (SEQ ID NO:86)
42
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
AKAP9-pep LEKQYQEQLEEEVAKVIVSMSIAFA (SEQ ID NO:87)
AKAP10-pep NTDEAQEELAWKIAKMIVSDIMQQA (SEQ ID NO:88)
AKAP11-pep VNLDKKAVLAEKIVAEAIEKAEREL (SEQ ID NO:89)
AKAP12-pep NGILELETKSSKLVQNIIQTAVDQF (SEQ ID NO:90)
AKAP14-pep TQDKNYEDELTQVALALVEDVINYA (SEQ ID NO:91)
Rab32-pep ETSAKDNINIEEAARFLVEKILVNH (SEQ ID NO:92)
[0152] Residues that were highly conserved among the AD domains of different
AKAP
proteins are indicated below by underlining with reference to the AKAP IS
sequence (SEQ
ID NO:11). The residues are the same as observed by Alto et al. (2003), with
the addition of
the C-terminal alanine residue. (See FIG. 4 of Hundsrucker et al. (2006),
incorporated herein
by reference.) The sequences of peptide antagonists with particularly high
affinities for the
RII DDD sequence were those of AKAP-IS, AKAP76-wt-pep, AKAP76-L304T-pep and
AKAP76-L308D-pep.
AKAP-IS
QIEYLAKQIVDNAIQQA (SEQ ID NO:11)
[0153] Carr et al. (2001, J Biol Chem 276:17332-38) examined the degree of
sequence
homology between different AKAP-binding DDD sequences from human and non-human
proteins and identified residues in the DDD sequences that appeared to be the
most highly
conserved among different DDD moieties. These are indicated below by
underlining with
reference to the human PKA RIM, DDD sequence of SEQ ID NO:9. Residues that
were
particularly conserved are further indicated by italics. The residues overlap
with, but are not
identical to those suggested by Kinderman et al. (2006) to be important for
binding to AKAP
proteins. The skilled artisan will realize that in designing sequence variants
of DDD, it would
be most preferred to avoid changing the most conserved residues (italicized),
and it would be
preferred to also avoid changing the conserved residues (underlined), while
conservative
amino acid substitutions may be considered for residues that are neither
underlined nor
italicized..
SHIQ/PPGLTELLQGYTVEVLRQOPPDLVEFAVEYFTRLREARA (SEQ ID NO:9)
[0154] A modified set of conservative amino acid substitutions for the DDD1
(SEQ ID
NO:9) sequence, based on the data of Carr et al. (2001) is shown in Table 5.
Even with this
reduced set of substituted sequences, there are over 65,000 possible
alternative DDD moiety
sequences that may be produced, tested and used by the skilled artisan without
undue
43
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
experimentation. The skilled artisan could readily derive such alternative DDD
amino acid
sequences as disclosed above for Table 2 and Table 3.
Table 5. Conservative Amino Acid Substitutions in DDD1 (SEQ ID NO:9).
Consensus
sequence disclosed as SEQ ID NO: 96.
S H I QIPPGLTELLQGY TVEVLR
T N S I
L
A
QQPPDL VEFA VE YF T RL RE AR A
N I D S K K L L
L I I
A V V
[0155] The skilled artisan will realize that these and other amino acid
substitutions in the
DDD or AD amino acid sequences may be utilized to produce alternative species
within the
genus of AD or DDD moieties, using techniques that are standard in the field
and only
routine experimentation.
Alternative DNL TM Structures
[0156] In certain alternative embodiments, DNLTM constructs may be formed
using
alternatively constructed antibodies or antibody fragments, in which an AD
moiety may be
attached at the C-terminal end of the kappa light chain (Ck), instead of the C-
terminal end of
the Fc on the heavy chain. The alternatively formed DNLTM constructs may be
prepared as
disclosed in Provisional U.S. Patent Application Serial Nos. 61/654,310, filed
June 1, 2012,
61/662,086, filed June 20, 2012, 61/673,553, filed July 19, 2012, and
61/682,531, filed
August 13, 2012, the entire text of each incorporated herein by reference. The
light chain
conjugated DNLTM constructs exhibit enhanced Fc-effector function activity in
vitro and
improved pharmacokinetics, stability and anti-lymphoma activity in vivo (Rossi
et al., 2013,
Bioconjug Chem 24:63-71).
[0157] Ck-conjugated DNLTM constructs may be prepared as disclosed in
Provisional U.S.
Patent Application Serial Nos. 61/654,310, 61/662,086, 61/673,553, and
61/682,531. Briefly,
Ck-AD2-IgG, was generated by recombinant engineering, whereby the AD2 peptide
was
fused to the C-terminal end of the kappa light chain. Because the natural C-
terminus of CK is
a cysteine residue, which forms a disulfide bridge to CH1, a 16-amino acid
residue "hinge"
44
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
linker was used to space the AD2 from the CK-VH1 disulfide bridge. The
mammalian
expression vectors for Ck-AD2-IgG-veltuzumab and Ck-AD2-IgG-epratuzumab were
constructed using the pdHL2 vector, which was used previously for expression
of the
homologous CH3-AD2-IgG modules. A 2208-bp nucleotide sequence was synthesized
comprising the pdHL2 vector sequence ranging from the Bain HI restriction site
within the
VK/CK intron to the Xho I restriction site 3' of the Ck intron, with the
insertion of the coding
sequence for the hinge linker (EFPKPSTPPGSSGGAP, SEQ ID NO:93) and AD2, in
frame
at the 3'end of the coding sequence for CK. This synthetic sequence was
inserted into the
IgG-pdHL2 expression vectors for veltuzumab and epratuzumab via Bain HI and
Xho I
restriction sites. Generation of production clones with SpESFX-10 were
performed as
described for the CH3-AD2-IgG modules. Ck-AD2-IgG-veltuzumab and Ck-AD2-IgG-
epratuzumab were produced by stably-transfected production clones in batch
roller bottle
culture, and purified from the supernatant fluid in a single step using
MabSelect (GE
Healthcare) Protein A affinity chromatography.
[0158] Following the same DNLTM process described previously for 22-(20)-(20)
(Rossi et
al., 2009, Blood 113:6161-71), Ck-AD2-IgG-epratuzumab was conjugated with CH1-
DDD2-
Fab-veltuzumab, a Fab-based module derived from veltuzumab, to generate the
bsHexAb
22*-(20)-(20), where the 22* indicates the Ck-AD2 module of epratuzumab and
each (20)
symbolizes a stabilized dimer of veltuzumab Fab. The properties of 22*-(20)-
(20) were
compared with those of 22-(20)-(20), the homologous Fc-bsHexAb comprising CH3-
AD2-
IgG-epratuzumab, which has similar composition and molecular size, but a
different
architecture.
[0159] Following the same DNLTM process described previously for 20-2b (Rossi
et al.,
2009, Blood 114:3864-71), Ck-AD2-IgG-veltuzumab, was conjugated with IFNct2b-
DDD2, a
module of IFNct2b with a DDD2 peptide fused at its C-terminal end, to generate
20*-2b,
which comprises veltuzumab with a dimeric IFNct2b fused to each light chain.
The properties
of 20*-2b were compared with those of 20-2b, which is the homologous Fc-IgG-
IFNia.
[0160] Each of the bsHexAbs and IgG-IFNct were isolated from the DNLTM
reaction mixture
by MabSelect affinity chromatography. The two Ck-derived prototypes, an anti-
CD22/CD20
bispecific hexavalent antibody, comprising epratuzumab (anti-CD22) and four
Fabs of
veltuzumab (anti-CD20), and a CD20-targeting immunocytokine, comprising
veltuzumab and
four molecules of interferon-oc2b, displayed enhanced Fc-effector functions in
vitro, as well
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
as improved pharmacokinetics, stability and anti-lymphoma activity in vivo,
compared to
their Fc-derived counterparts.
Amino Acid Substitutions
[0161] In alternative embodiments, the disclosed methods and compositions may
involve
production and use of proteins or peptides with one or more substituted amino
acid residues.
For example, the DDD and/or AD sequences used to make DNLTM constructs may be
modified as discussed above.
[0162] The skilled artisan will be aware that, in general, amino acid
substitutions typically
involve the replacement of an amino acid with another amino acid of relatively
similar
properties (i.e., conservative amino acid substitutions). The properties of
the various amino
acids and effect of amino acid substitution on protein structure and function
have been the
subject of extensive study and knowledge in the art.
[0163] For example, the hydropathic index of amino acids may be considered
(Kyte &
Doolittle, 1982, J. Mol. Biol., 157:105-132). The relative hydropathic
character of the amino
acid contributes to the secondary structure of the resultant protein, which in
turn defines the
interaction of the protein with other molecules. Each amino acid has been
assigned a
hydropathic index on the basis of its hydrophobicity and charge
characteristics (Kyte &
Doolittle, 1982), these are: isoleucine (+4.5); valine (+4.2); leucine (+3.8);
phenylalanine
(+2.8); cysteine/cystine (+2.5); methionine (+1.9); alanine (+1.8); glycine (-
0.4); threonine (-
0.7); serine (-0.8); tryptophan (-0.9); tyrosine (-1.3); proline (-1.6);
histidine (-3.2); glutamate
(-3.5); glutamine (-3.5); aspartate (-3.5); asparagine (-3.5); lysine (-3.9);
and arginine (-4.5).
In making conservative substitutions, the use of amino acids whose hydropathic
indices are
within 2 is preferred, within 1 are more preferred, and within 0.5 are
even more
preferred.
[0164] Amino acid substitution may also take into account the hydrophilicity
of the amino
acid residue (e.g., U.S. Pat. No. 4,554,101). Hydrophilicity values have been
assigned to
amino acid residues: arginine (+3.0); lysine (+3.0); aspartate (+3.0);
glutamate (+3.0); serine
(+0.3); asparagine (+0.2); glutamine (+0.2); glycine (0); threonine (-0.4);
proline (-0.5 ±1);
alanine (-0.5); histidine (-0.5); cysteine (-1.0); methionine (-1.3); valine (-
1.5); leucine (-1.8);
isoleucine (-1.8); tyrosine (-2.3); phenylalanine (-2.5); tryptophan (-3.4).
Replacement of
amino acids with others of similar hydrophilicity is preferred.
[0165] Other considerations include the size of the amino acid side chain. For
example, it
would generally not be preferred to replace an amino acid with a compact side
chain, such as
glycine or serine, with an amino acid with a bulky side chain, e.g.,
tryptophan or tyrosine.
46
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
The effect of various amino acid residues on protein secondary structure is
also a
consideration. Through empirical study, the effect of different amino acid
residues on the
tendency of protein domains to adopt an alpha-helical, beta-sheet or reverse
turn secondary
structure has been determined and is known in the art (see, e.g., Chou &
Fasman, 1974,
Biochemistry, 13:222-245; 1978, Ann. Rev. Biochem., 47: 251-276; 1979,
Biophys. J.,
26:367-384).
[0166] Based on such considerations and extensive empirical study, tables of
conservative
amino acid substitutions have been constructed and are known in the art. For
example:
arginine and lysine; glutamate and aspartate; serine and threonine; glutamine
and asparagine;
and valine, leucine and isoleucine. Alternatively: Ala (A) leu, ile, val; Arg
(R) gln, asn, lys;
Asn (N) his, asp, lys, arg, gln; Asp (D) asn, glu; Cys (C) ala, ser; Gln (Q)
glu, asn; Glu (E)
gln, asp; Gly (G) ala; His (H) asn, gln, lys, arg; Ile (I) val, met, ala, phe,
leu; Leu (L) val, met,
ala, phe, ile; Lys (K) gln, asn, arg; Met (M) phe, ile, leu; Phe (F) leu, val,
ile, ala, tyr; Pro (P)
ala; Ser (S), thr; Thr (T) ser; Trp (W) phe, tyr; Tyr (Y) trp, phe, thr, ser;
Val (V) ile, leu, met,
phe, ala.
[0167] Other considerations for amino acid substitutions include whether or
not the residue is
located in the interior of a protein or is solvent exposed. For interior
residues, conservative
substitutions would include: Asp and Asn; Ser and Thr; Ser and Ala; Thr and
Ala; Ala and
Gly; Ile and Val; Val and Leu; Leu and Ile; Leu and Met; Phe and Tyr; Tyr and
Trp. (See,
e.g., PROWL website at rockefeller.edu) For solvent exposed residues,
conservative
substitutions would include: Asp and Asn; Asp and Glu; Glu and Gln; Glu and
Ala; Gly and
Asn; Ala and Pro; Ala and Gly; Ala and Ser; Ala and Lys; Ser and Thr; Lys and
Arg; Val and
Leu; Leu and Ile; Ile and Val; Phe and Tyr. (Id.) Various matrices have been
constructed to
assist in selection of amino acid substitutions, such as the PAM250 scoring
matrix, Dayhoff
matrix, Grantham matrix, McLachlan matrix, Doolittle matrix, Henikoff matrix,
Miyata
matrix, Fitch matrix, Jones matrix, Rao matrix, Levin matrix and Risler matrix
(Idem.)
[0168] In determining amino acid substitutions, one may also consider the
existence of
intermolecular or intramolecular bonds, such as formation of ionic bonds (salt
bridges)
between positively charged residues (e.g., His, Arg, Lys) and negatively
charged residues
(e.g., Asp, Glu) or disulfide bonds between nearby cysteine residues.
[0169] Methods of substituting any amino acid for any other amino acid in an
encoded
protein sequence are well known and a matter of routine experimentation for
the skilled
artisan, for example by the technique of site-directed mutagenesis or by
synthesis and
47
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
assembly of oligonucleotides encoding an amino acid substitution and splicing
into an
expression vector construct.
Pre-Targeting
[0170] Bispecific or multispecific antibodies may be of use in pretargeting
techniques. In this
case, one or more therapeutic agent may be conjugated to a targetable
construct that
comprises one or more haptens. The hapten is recognized by at least one arm of
a bispecific
or multispecific antibody that also binds to a tumor-associated antigen or
other disease-
associated antigen. In this case, the therapeutic agent binds indirectly to
the antibodies, via
the binding of the targetable construct. This process is referred to as
pretargeting.
[0171] Pre-targeting is a multistep process originally developed to resolve
the slow blood
clearance of directly targeting antibodies, which contributes to undesirable
toxicity to normal
tissues such as bone marrow. With pre-targeting, a therapeutic agent is
attached to a small
delivery molecule (targetable construct) that is cleared within minutes from
the blood. A pre-
targeting bispecific or multispecific antibody, which has binding sites for
the targetable
construct as well as a target antigen, is administered first, free antibody is
allowed to clear
from circulation and then the targetable construct is administered.
[0172] Pre-targeting methods are disclosed, for example, in Goodwin et al.,
U.S. Pat. No.
4,863,713; Goodwin et al., J. Nucl. Med. 29:226, 1988; Hnatowich et al., J.
Nucl. Med.
28:1294, 1987; Oehr et al., J. Nucl. Med. 29:728, 1988; Klibanov et al., J.
Nucl. Med.
29:1951, 1988; Sinitsyn et al., J. Nucl. Med. 30:66, 1989; Kalofonos et al.,
J. Nucl. Med.
31:1791, 1990; Schechter et al., Int. J. Cancer 48:167, 1991; Paganelli et
al., Cancer Res.
51:5960, 1991; Paganelli et al., Nucl. Med. Commun. 12:211, 1991; U.S. Pat.
No. 5,256,395;
Stickney et al., Cancer Res. 51:6650, 1991; Yuan et al., Cancer Res. 51:3119,
1991; U.S. Pat.
Nos. 6,077,499; 7,011,812; 7,300,644; 7,074,405; 6,962,702; 7,387,772;
7,052,872;
7,138,103; 6,090,381; 6,472,511; 6,962,702; and 6,962,702, each incorporated
herein by
reference.
[0173] A pre-targeting method of treating or diagnosing a disease or disorder
in a subject
may be provided by: (1) administering to the subject a bispecific antibody or
antibody
fragment; (2) optionally administering to the subject a clearing composition,
and allowing the
composition to clear the antibody from circulation; and (3) administering to
the subject the
targetable construct, containing one or more chelated or chemically bound
therapeutic or
diagnostic agents.
48
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
Targetable Constructs
[0174] In certain embodiments, targetable construct peptides labeled with one
or more
therapeutic or diagnostic agents for use in pre-targeting may be selected to
bind to a
bispecific antibody with one or more binding sites for a targetable construct
peptide and one
or more binding sites for a target antigen associated with a disease or
condition. Bispecific
antibodies may be used in a pretargeting technique wherein the antibody may be
administered
first to a subject. Sufficient time may be allowed for the bispecific antibody
to bind to a target
antigen and for unbound antibody to clear from circulation. Then a targetable
construct, such
as a labeled peptide, may be administered to the subject and allowed to bind
to the bispecific
antibody and localize at the diseased cell or tissue.
[0175] Such targetable constructs can be of diverse structure and are selected
not only for the
availability of an antibody or fragment that binds with high affinity to the
targetable
construct, but also for rapid in vivo clearance when used within the pre-
targeting method and
bispecific antibodies (bsAb) or multispecific antibodies. Hydrophobic agents
are best at
eliciting strong immune responses, whereas hydrophilic agents are preferred
for rapid in vivo
clearance. Thus, a balance between hydrophobic and hydrophilic character is
established.
This may be accomplished, in part, by using hydrophilic chelating agents to
offset the
inherent hydrophobicity of many organic moieties. Also, sub-units of the
targetable construct
may be chosen which have opposite solution properties, for example, peptides,
which contain
amino acids, some of which are hydrophobic and some of which are hydrophilic.
[0176] Peptides having as few as two amino acid residues, preferably two to
ten residues,
may be used and may also be coupled to other moieties, such as chelating
agents. The linker
should be a low molecular weight conjugate, preferably having a molecular
weight of less
than 50,000 daltons, and advantageously less than about 20,000 daltons, 10,000
daltons or
5,000 daltons. More usually, the targetable construct peptide will have four
or more residues
and one or more haptens for binding, e.g., to a bispecific antibody. Exemplary
haptens may
include In-DTPA (indium-diethylene triamine pentaacetic acid) or HSG
(histamine succinyl
glycine). The targetable construct may also comprise one or more chelating
moieties, such as
DOTA (1,4,7,10-tetraazacyclododecane1,4,7,10-tetraacetic acid), NOTA (1,4,7-
triaza-
cyclononane-1,4,7-triacetic acid), TETA (p-bromoacetamido-benzyl-
tetraethylaminetetraacetic acid), NETA ([2-(4,7-
biscarboxymethyl[1,4,7]triazacyclononan-1-
yl-ethyl]-2-carbonylmethyl-amino]acetic acid) or other known chelating
moieties. Chelating
moieties may be used, for example, to bind to a therapeutic and or diagnostic
radionuclide,
paramagnetic ion or contrast agent.
49
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
[0177] The targetable construct may also comprise unnatural amino acids, e.g.,
D-amino
acids, in the backbone structure to increase the stability of the peptide in
vivo. In alternative
embodiments, other backbone structures such as those constructed from non-
natural amino
acids or peptoids may be used.
[0178] The peptides used as targetable constructs are conveniently synthesized
on an
automated peptide synthesizer using a solid-phase support and standard
techniques of
repetitive orthogonal deprotection and coupling. Free amino groups in the
peptide, that are to
be used later for conjugation of chelating moieties or other agents, are
advantageously
blocked with standard protecting groups such as a Boc group, while N-terminal
residues may
be acetylated to increase serum stability. Such protecting groups are well
known to the skilled
artisan. See Greene and Wuts Protective Groups in Organic Synthesis, 1999
(John Wiley and
Sons, N.Y.). When the peptides are prepared for later use within the
bispecific antibody
system, they are advantageously cleaved from the resins to generate the
corresponding C-
terminal amides, in order to inhibit in vivo carboxypeptidase activity.
[0179] Where pretargeting with bispecific antibodies is used, the antibody
will contain a first
binding site for an antigen produced by or associated with a target tissue and
a second
binding site for a hapten on the targetable construct. Exemplary haptens
include, but are not
limited to, HSG and In-DTPA. Antibodies raised to the HSG hapten are known
(e.g. 679
antibody) and can be easily incorporated into the appropriate bispecific
antibody (see, e.g.,
U.S. Patent Nos. 6,962,702; 7,138,103 and 7,300,644, incorporated herein by
reference with
respect to the Examples sections). However, other haptens and antibodies that
bind to them
are known in the art and may be used, such as In-DTPA and the 734 antibody
(e.g., U.S.
Patent No.7,534,431, the Examples section incorporated herein by reference).
Immunoconjugates
[0180] In certain embodiments, a cytotoxic drug or other therapeutic or
diagnostic agent may
be covalently attached to an antibody or antibody fragment to form an
immunoconjugate. In
some embodiments, a drug or other agent may be attached to an antibody or
fragment thereof
via a carrier moiety. Carrier moieties may be attached, for example to reduced
SH groups
and/or to carbohydrate side chains. A carrier moiety can be attached at the
hinge region of a
reduced antibody component via disulfide bond formation. Alternatively, such
agents can be
attached using a heterobifunctional cross-linker, such as N-succinyl 3-(2-
pyridyldithio)propionate (SPDP). Yu et al., Int. 1 Cancer 56: 244 (1994).
General techniques
for such conjugation are well-known in the art. See, for example, Wong,
CHEMISTRY OF
PROTEIN CONJUGATION AND CROSS-LINKING (CRC Press 1991); Upeslacis et al.,
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
"Modification of Antibodies by Chemical Methods," in MONOCLONAL ANTIBODIES:
PRINCIPLES AND APPLICATIONS, Birch et al. (eds.), pages 187-230 (Wiley-Liss,
Inc.
1995); Price, "Production and Characterization of Synthetic Peptide-Derived
Antibodies," in
MONOCLONAL ANTIBODIES: PRODUCTION, ENGINEERING AND CLINICAL
APPLICATION, Ritter et al. (eds.), pages 60-84 (Cambridge University Press
1995).
Alternatively, the carrier moiety can be conjugated via a carbohydrate moiety
in the Fc region
of the antibody.
[0181] Methods for conjugating functional groups to antibodies via an antibody
carbohydrate
moiety are well-known to those of skill in the art. See, for example, Shih et
al., Int.1 Cancer
41: 832 (1988); Shih et al., Int.1 Cancer 46: 1101 (1990); and Shih et al.,
U.S. Patent No.
5,057,313, the Examples section of which is incorporated herein by reference.
The general
method involves reacting an antibody having an oxidized carbohydrate portion
with a carrier
polymer that has at least one free amine function. This reaction results in an
initial Schiff
base (imine) linkage, which can be stabilized by reduction to a secondary
amine to form the
final conjugate.
[0182] The Fc region may be absent if the antibody component of the ADC is an
antibody
fragment. However, it is possible to introduce a carbohydrate moiety into the
light chain
variable region of a full length antibody or antibody fragment. See, for
example, Leung et al.,
Immunol. 154: 5919 (1995); U.S. Patent Nos. 5,443,953 and 6,254,868, the
Examples
section of which is incorporated herein by reference. The engineered
carbohydrate moiety is
used to attach the therapeutic or diagnostic agent.
[0183] An alternative method for attaching carrier moieties to a targeting
molecule involves
use of click chemistry reactions. The click chemistry approach was originally
conceived as a
method to rapidly generate complex substances by joining small subunits
together in a
modular fashion. (See, e.g., Kolb et al., 2004, Angew Chem Int Ed 40:3004-31;
Evans, 2007,
Aust J Chem 60:384-95.) Various forms of click chemistry reaction are known in
the art,
such as the Huisgen 1,3-dipolar cycloaddition copper catalyzed reaction
(Tornoe et al., 2002,
J Organic Chem 67:3057-64), which is often referred to as the "click
reaction." Other
alternatives include cycloaddition reactions such as the Diels-Alder,
nucleophilic substitution
reactions (especially to small strained rings like epoxy and aziridine
compounds), carbonyl
chemistry formation of urea compounds and reactions involving carbon-carbon
double bonds,
such as alkynes in thiol-yne reactions.
[0184] The azide alkyne Huisgen cycloaddition reaction uses a copper catalyst
in the
presence of a reducing agent to catalyze the reaction of a terminal alkyne
group attached to a
51
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
first molecule. In the presence of a second molecule comprising an azide
moiety, the azide
reacts with the activated alkyne to form a 1,4-disubstituted 1,2,3-triazole.
The copper
catalyzed reaction occurs at room temperature and is sufficiently specific
that purification of
the reaction product is often not required. (Rostovstev et al., 2002, Angew
Chem Int Ed
41:2596; Tornoe et al., 2002, J Org Chem 67:3057.) The azide and alkyne
functional groups
are largely inert towards biomolecules in aqueous medium, allowing the
reaction to occur in
complex solutions. The triazole formed is chemically stable and is not subject
to enzymatic
cleavage, making the click chemistry product highly stable in biological
systems. Although
the copper catalyst is toxic to living cells, the copper-based click chemistry
reaction may be
used in vitro for immunoconjugate formation.
[0185] A copper-free click reaction has been proposed for covalent
modification of
biomolecules. (See, e.g., Agard et al., 2004, J Am Chem Soc 126:15046-47.) The
copper-
free reaction uses ring strain in place of the copper catalyst to promote a [3
+ 2] azide-alkyne
cycloaddition reaction (Id.) For example, cyclooctyne is an 8-carbon ring
structure
comprising an internal alkyne bond. The closed ring structure induces a
substantial bond
angle deformation of the acetylene, which is highly reactive with azide groups
to form a
triazole. Thus, cyclooctyne derivatives may be used for copper-free click
reactions (Id.)
[0186] Another type of copper-free click reaction was reported by Ning et al.
(2010, Angew
Chem Int Ed 49:3065-68), involving strain-promoted alkyne-nitrone
cycloaddition. To
address the slow rate of the original cyclooctyne reaction, electron-
withdrawing groups are
attached adjacent to the triple bond (Id.) Examples of such substituted
cyclooctynes include
difluorinated cyclooctynes, 4-dibenzocyclooctynol and azacyclooctyne (Id.) An
alternative
copper-free reaction involved strain-promoted alkyne-nitrone cycloaddition to
give N-
alkylated isoxazolines (Id.) The reaction was reported to have exceptionally
fast reaction
kinetics and was used in a one-pot three-step protocol for site-specific
modification of
peptides and proteins (Id.) Nitrones were prepared by the condensation of
appropriate
aldehydes with N-methylhydroxylamine and the cycloaddition reaction took place
in a
mixture of acetonitrile and water (Id.) These and other known click chemistry
reactions may
be used to attach carrier moieties to antibodies in vitro.
[0187] Agard et al. (2004, J Am Chem Soc 126:15046-47) demonstrated that a
recombinant
glycoprotein expressed in CHO cells in the presence of peracetylated N-
azidoacetylmannosamine resulted in the bioincorporation of the corresponding N-
azidoacetyl
sialic acid in the carbohydrates of the glycoprotein. The azido-derivatized
glycoprotein
reacted specifically with a biotinylated cyclooctyne to form a biotinylated
glycoprotein, while
52
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
control glycoprotein without the azido moiety remained unlabeled (Id.)
Laughlin et al. (2008,
Science 320:664-667) used a similar technique to metabolically label cell-
surface glycans in
zebrafish embryos incubated with peracetylated N-azidoacetylgalactosamine. The
azido-
derivatized glycans reacted with difluorinated cyclooctyne (DIFO) reagents to
allow
visualization of glycans in vivo.
[0188] The Diels-Alder reaction has also been used for in vivo labeling of
molecules. Rossin
et al. (2010, Angew Chem Int Ed 49:3375-78) reported a 52% yield in vivo
between a tumor-
localized anti-TAG72 (CC49) antibody carrying a trans-cyclooctene (TCO)
reactive moiety
and an 1111n-labeled tetrazine DOTA derivative. The TCO-labeled CC49 antibody
was
administered to mice bearing colon cancer xenografts, followed 1 day later by
injection of
1111n-labeled tetrazine probe (Id.) The reaction of radiolabeled probe with
tumor localized
antibody resulted in pronounced radioactivity localization in the tumor, as
demonstrated by
SPECT imaging of live mice three hours after injection of radiolabeled probe,
with a tumor-
to-muscle ratio of 13:1 (Id.) The results confirmed the in vivo chemical
reaction of the TCO
and tetrazine-labeled molecules.
[0189] Antibody labeling techniques using biological incorporation of labeling
moieties are
further disclosed in U.S. Patent No. 6,953,675 (the Examples section of which
is incorporated
herein by reference). Such "landscaped" antibodies were prepared to have
reactive ketone
groups on glycosylated sites. The method involved expressing cells transfected
with an
expression vector encoding an antibody with one or more N-glycosylation sites
in the CH1 or
Vic domain in culture medium comprising a ketone derivative of a saccharide or
saccharide
precursor. Ketone-derivatized saccharides or precursors included N-levulinoyl
mannosamine
and N-levulinoyl fucose. The landscaped antibodies were subsequently reacted
with agents
comprising a ketone-reactive moiety, such as hydrazide, hydrazine,
hydroxylamino or
thiosemicarbazide groups, to form a labeled targeting molecule. Exemplary
agents attached to
the landscaped antibodies included chelating agents like DTPA, large drug
molecules such as
doxorubicin-dextran, and acyl-hydrazide containing peptides. The landscaping
technique is
not limited to producing antibodies comprising ketone moieties, but may be
used instead to
introduce a click chemistry reactive group, such as a nitrone, an azide or a
cyclooctyne, onto
an antibody or other biological molecule.
[0190] Modifications of click chemistry reactions are suitable for use in
vitro or in vivo.
Reactive targeting molecule may be formed either by either chemical
conjugation or by
biological incorporation. The targeting molecule, such as an antibody or
antibody fragment,
may be activated with an azido moiety, a substituted cyclooctyne or alkyne
group, or a
53
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
nitrone moiety. Where the targeting molecule comprises an azido or nitrone
group, the
corresponding targetable construct will comprise a substituted cyclooctyne or
alkyne group,
and vice versa. Such activated molecules may be made by metabolic
incorporation in living
cells, as discussed above.
[0191] Alternatively, methods of chemical conjugation of such moieties to
biomolecules are
well known in the art, and any such known method may be utilized. General
methods of
immunoconjugate formation are disclosed, for example, in U.S. Patent Nos.
4,699,784;
4,824,659; 5,525,338; 5,677,427; 5,697,902; 5,716,595; 6,071,490; 6,187,284;
6,306,393;
6,548,275; 6,653,104; 6,962,702; 7,033,572; 7,147,856; and 7,259,240, the
Examples section
of each incorporated herein by reference.
Other Therapeutic Agents
[0192] A wide variety of therapeutic reagents can be administered concurrently
or
sequentially with the subject ADCs. Alternatively, such agents may be
conjugated to the
antibodies of the invention, for example, drugs, toxins, oligonucleotides,
immunomodulators,
hormones, hormone antagonists, enzymes, enzyme inhibitors, radionuclides,
angiogenesis
inhibitors, etc. The therapeutic agents recited here are those agents that
also are useful for
administration separately with an ADC as described above. Therapeutic agents
include, for
example, cytotoxic drugs such as vinca alkaloids, anthracyclines such as
doxorubicin, 2-
PDox or pro-2-PDox, gemcitabine, epipodophyllotoxins, taxanes,
antimetabolites, alkylating
agents, antibiotics, SN-38, COX-2 inhibitors, antimitotics, anti-angiogenic
and pro-apoptotic
agents, particularly doxorubicin, methotrexate, taxol, CPT-11, camptothecans,
proteosome
inhibitors, mTOR inhibitors, HDAC inhibitors, tyrosine kinase inhibitors, and
others. Other
useful anti-cancer cytotoxic drugs for administering concurrently or
sequentially, or for the
preparation of ADCs include nitrogen mustards, alkyl sulfonates, nitrosoureas,
triazenes, folic
acid analogs, COX-2 inhibitors, antimetabolites, pyrimidine analogs, purine
analogs,
platinum coordination complexes, mTOR inhibitors, tyrosine kinase inhibitors,
proteosome
inhibitors, HDAC inhibitors, camptothecins, hormones, and the like. Suitable
cytotoxic
agents are described in REMINGTON'S PHARMACEUTICAL SCIENCES, 19th Ed. (Mack
Publishing Co. 1995), and in GOODMAN AND GILMAN'S THE PHARMACOLOGICAL
BASIS OF THERAPEUTICS, 7th Ed. (MacMillan Publishing Co. 1985), as well as
revised
editions of these publications. Other suitable cytotoxic agents, such as
experimental drugs,
are known to those of skill in the art. In a preferred embodiment, conjugates
of camptothecins
and related compounds, such as SN-38, may be conjugated to hRS7 or other anti-
Trop-2
54
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
antibodies. In another preferred embodiment, gemcitabine is administered to
the subject in
conjunction with SN-38-hRS7 and/or 90Y-hPAM4.
[0193] A toxin can be of animal, plant or microbial origin. Toxins of use
include ricin, abrin,
ribonuclease (RNase), DNase I, Staphylococcal enterotoxin-A, pokeweed
antiviral protein,
onconase, gelonin, diphtheria toxin, Pseudomonas exotoxin, and Pseudomonas
endotoxin.
See, for example, Pastan et al., Cell 47:641 (1986), Goldenberg, CA--A Cancer
Journal for
Clinicians 44:43 (1994), Sharkey and Goldenberg, CA--A Cancer Journal for
Clinicians
56:226 (2006). Additional toxins suitable for use are known to those of skill
in the art and are
disclosed in U.S. Pat. No. 6,077,499, the Examples section of which is
incorporated herein by
reference.
[0194] As used herein, the term "immunomodulator" includes a cytokine, a
lymphokine, a
monokine, a stem cell growth factor, a lymphotoxin, a hematopoietic factor, a
colony
stimulating factor (CSF), an interferon (IFN), parathyroid hormone, thyroxine,
insulin,
proinsulin, relaxin, prorelaxin, follicle stimulating hormone (FSH), thyroid
stimulating
hormone (TSH), luteinizing hormone (LH), hepatic growth factor, prostaglandin,
fibroblast
growth factor, prolactin, placental lactogen, OB protein, a transforming
growth factor (TGF),
TGF-a, TGF-13, insulin-like growth factor (ILGF), erythropoietin,
thrombopoietin, tumor
necrosis factor (TNF), TNF- a, TNF-13, a mullerian-inhibiting substance, mouse
gonadotropin-associated peptide, inhibin, activin, vascular endothelial growth
factor, integrin,
interleukin (IL), granulocyte-colony stimulating factor (G-CSF), granulocyte
macrophage-
colony stimulating factor (GM-CSF), interferon- a, interferon-13, interferon-
y, interferon-X,
S1 factor, IL-1, IL-lcc, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-
10, IL-11, IL-12, IL-
13, IL-14, IL-15, IL-16, IL-17, IL-18 IL-21 and IL-25, LIF, kit-ligand, FLT-3,
angiostatin,
thrombospondin, endostatin, lymphotoxin, and the like.
[0195] Particularly useful therapeutic radionuclides include, but are not
limited to 111In,
177Lu, 212Bi, 213Bi, 21 ikt, 62cu., 64cu, 67cu, 90y, 1251, 1311, 32p, 33p,
4750, 111Ag, 67Ga, 142pr,
1535m, 161Tb, 166Dy, 166H0, 186Re, 188Re, 189Re, 212pb, 223Ra, 225
0
A, 59Fe, 755e, 77As, 895r, 99Mo,
105Rh, 109pd, 143pr, 149pm, 169Er, 1941r, 198 AA u, 199
Au, 227Th, and 211Pb. The therapeutic
radionuclide preferably has a decay energy in the range of 20 to 6,000 keV,
preferably in the
ranges 60 to 200 keV for an Auger emitter, 100-2,500 keV for a beta emitter,
and 4,000-
6,000 keV for an alpha emitter. Maximum decay energies of useful beta-particle-
emitting
nuclides are preferably 20-5,000 keV, more preferably 100-4,000 keV, and most
preferably
500-2,500 keV. Also preferred are radionuclides that substantially decay with
Auger-emitting
particles. For example, Co-58, Ga-67, Br-80m, Tc-99m, Rh-103m, Pt-109, In-111,
Sb-119, I-
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
125, Ho-161, Os-189m and Ir-192. Decay energies of useful beta-particle-
emitting nuclides
are preferably <1,000 keV, more preferably <100 keV, and most preferably <70
keV. Also
preferred are radionuclides that substantially decay with generation of alpha-
particles. Such
radionuclides include, but are not limited to: Dy-152, At-211, Bi-212, Ra-223,
Rn-219, Po-
215, Bi-211, Ac-225, Fr-221, At-217, Bi-213, Fm-255 and Th-227. Decay energies
of useful
alpha-particle-emitting radionuclides are preferably 2,000-10,000 keV, more
preferably
3,000-8,000 keV, and most preferably 4,000-7,000 keV.
[0196] For example, 90Y, which emits an energetic beta particle, can be
coupled to an
antibody, antibody fragment or fusion protein, using
diethylenetriaminepentaacetic acid
(DTPA), or more preferably using DOTA. Methods of conjugating 90Y to
antibodies or
targetable constructs are known in the art and any such known methods may be
used. (See,
e.g., U.S. Patent No. 7,259,249, the Examples section of which is incorporated
herein by
reference. See also Linden et al., Clin Cancer Res. 11:5215-22, 2005; Sharkey
et al., J Nucl
Med. 46:620-33, 2005; Sharkey et al., J Nucl Med. 44:2000-18, 2003.)
[0197] Additional potential therapeutic radioisotopes include 11C, 13N, 150,
75Br, 198AU,
224 126 133 77 113m 95 97 103 105 107 203 121m 122 125
Ac, I, I, Br, In, Ru, Ru, Ru, Ru, Hg, Hg, Te, Te,m m
Te,
1651,m, 1671,m, 168Tm, 197pt, 109pd, 105Rb, 142pr, 143pr, 161Tb, 166-0,
H 199Au, 57Co, 58Co, 51Cr,
59Fe, 755e, 201T1, 225Ac, 76Br, 169,-+o ,
Y and the like.
[0198] In another embodiment, a radiosensitizer can be used in combination
with a naked or
conjugated antibody or antibody fragment. For example, the radiosensitizer can
be used in
combination with a radiolabeled antibody or antibody fragment. The addition of
the
radiosensitizer can result in enhanced efficacy when compared to treatment
with the
radiolabeled antibody or antibody fragment alone. Radiosensitizers are
described in D. M.
Goldenberg (ed.), CANCER THERAPY WITH RADIOLABELED ANTIBODIES, CRC
Press (1995). Other typical radionsensitizers of interest for use with this
technology include
gemcitabine, 5-fluorouracil, and cisplatin, and have been used in combination
with external
irradiation in the therapy of diverse cancers.
[0199] Antibodies or fragments thereof that have a boron addend-loaded carrier
for thermal
neutron activation therapy will normally be affected in similar ways. However,
it will be
advantageous to wait until non-targeted immunoconjugate clears before neutron
irradiation is
performed. Clearance can be accelerated using an anti-idiotypic antibody that
binds to the
anti-cancer antibody. See U.S. Pat. No. 4,624,846 for a description of this
general principle.
For example, boron addends such as carboranes, can be attached to antibodies.
Carboranes
can be prepared with carboxyl functions on pendant side chains, as is well-
known in the art.
56
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
Attachment of carboranes to a carrier, such as aminodextran, can be achieved
by activation of
the carboxyl groups of the carboranes and condensation with amines on the
carrier. The
intermediate conjugate is then conjugated to the antibody. After
administration of the
antibody conjugate, a boron addend is activated by thermal neutron irradiation
and converted
to radioactive atoms which decay by alpha-emission to produce highly toxic,
short-range
effects.
Formulation and Administration
[0200] Suitable routes of administration of ADCs include, without limitation,
oral, parenteral,
rectal, transmucosal, intestinal administration, intramedullary, intrathecal,
direct
intraventricular, intravenous, intravitreal, intracavitary, intraperitoneal,
or intratumoral
injections. The preferred routes of administration are parenteral, more
preferably intravenous.
Alternatively, one may administer the compound in a local rather than systemic
manner, for
example, via injection of the compound directly into a solid or hematological
tumor.
[0201] ADCs can be formulated according to known methods to prepare
pharmaceutically
useful compositions, whereby the ADC is combined in a mixture with a
pharmaceutically
suitable excipient. Sterile phosphate-buffered saline is one example of a
pharmaceutically
suitable excipient. Other suitable excipients are well-known to those in the
art. See, for
example, Ansel et al., PHARMACEUTICAL DOSAGE FORMS AND DRUG DELIVERY
SYSTEMS, 5th Edition (Lea & Febiger 1990), and Gennaro (ed.), REMINGTON'S
PHARMACEUTICAL SCIENCES, 18th Edition (Mack Publishing Company 1990), and
revised editions thereof
[0202] In a preferred embodiment, the ADC is formulated in Good's biological
buffer (pH 6-
7), using a buffer selected from the group consisting of N-(2-acetamido)-2-
aminoethanesulfonic acid (ACES); N-(2-acetamido)iminodiacetic acid (ADA); N,N-
bis(2-
hydroxyethyl)-2-aminoethanesulfonic acid (BES); 4-(2-hydroxyethyl)piperazine-1-
ethanesulfonic acid (HEPES); 2-(N-morpholino)ethanesulfonic acid (MES); 3-(N-
morpholino)propanesulfonic acid (MOPS); 3-(N-morpholiny1)-2-
hydroxypropanesulfonic
acid (MOPS0); and piperazine-N,N'-bis(2-ethanesulfonic acid) [Pipes]. More
preferred
buffers are MES or MOPS, preferably in the concentration range of 20 to 100
mM, more
preferably about 25 mM. Most preferred is 25 mM MES, pH 6.5. The formulation
may
further comprise 25 mM trehalose and 0.01% v/v polysorbate 80 as excipients,
with the final
buffer concentration modified to 22.25 mM as a result of added excipients. The
preferred
57
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
method of storage is as a lyophilized formulation of the conjugates, stored in
the temperature
range of -20 C to 2 C, with the most preferred storage at 2 C to 8 C.
[0203] The ADC can be formulated for intravenous administration via, for
example, bolus
injection, slow infusion or continuous infusion. Preferably, the antibody of
the present
invention is infused over a period of less than about 4 hours, and more
preferably, over a
period of less than about 3 hours. For example, the first 25-50 mg could be
infused within 30
minutes, preferably even 15 min, and the remainder infused over the next 2-3
hrs.
Formulations for injection can be presented in unit dosage form, e.g., in
ampoules or in multi-
dose containers, with an added preservative. The compositions can take such
forms as
suspensions, solutions or emulsions in oily or aqueous vehicles, and can
contain formulatory
agents such as suspending, stabilizing and/or dispersing agents.
Alternatively, the active
ingredient can be in powder form for constitution with a suitable vehicle,
e.g., sterile
pyrogen-free water, before use.
[0204] Additional pharmaceutical methods may be employed to control the
duration of action
of the therapeutic conjugate. Control release preparations can be prepared
through the use of
polymers to complex or adsorb the ADC. For example, biocompatible polymers
include
matrices of poly(ethylene-co-vinyl acetate) and matrices of a polyanhydride
copolymer of a
stearic acid dimer and sebacic acid. Sherwood et al., Bio/Technology 10: 1446
(1992). The
rate of release of an ADC from such a matrix depends upon the molecular weight
of the
ADC, the amount of ADC within the matrix, and the size of dispersed particles.
Saltzman et
al., Biophys. i 55: 163 (1989); Sherwood et al., supra. Other solid dosage
forms are
described in Ansel et al., PHARMACEUTICAL DOSAGE FORMS AND DRUG
DELIVERY SYSTEMS, 5th Edition (Lea & Febiger 1990), and Gennaro (ed.),
REMINGTON'S PHARMACEUTICAL SCIENCES, 18th Edition (Mack Publishing
Company 1990), and revised editions thereof.
[0205] Generally, the dosage of an administered ADC for humans will vary
depending upon
such factors as the patient's age, weight, height, sex, general medical
condition and previous
medical history. It may be desirable to provide the recipient with a dosage of
ADC that is in
the range of from about 0.3 mg/kg to 5 mg/kg as a single intravenous infusion,
although a
lower or higher dosage also may be administered as circumstances dictate. A
dosage of 0.3-5
mg/kg for a 70 kg patient, for example, is 21-350 mg, or 12-206 mg/m2 for a
1.7-m patient.
The dosage may be repeated as needed, for example, once per week for 2-10
weeks, once per
week for 8 weeks, or once per week for 4 weeks. It may also be given less
frequently, such as
58
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
every other week for several months, or monthly or quarterly for many months,
as needed in
a maintenance therapy. Preferred dosages may include, but are not limited to,
0.3 mg/kg, 0.5
mg/kg, 0.7 mg/kg, 1.0 mg/kg, 1.2 mg/kg, 1.5 mg/kg, 2.0 mg/kg, 2.5 mg/kg, 3.0
mg/kg, 3.5
mg/kg, 4.0 mg/kg, 4.5 mg/kg, and 5.0 mg/kg. More preferred dosages are 0.6
mg/kg for
weekly administration and 1.2 mg/kg for less frequent dosing. Any amount in
the range of 0.3
to 5 mg/kg may be used. The dosage is preferably administered multiple times,
once a week.
A minimum dosage schedule of 4 weeks, more preferably 8 weeks, more preferably
16 weeks
or longer may be used, with the dose frequency dependent on toxic side-effects
and recovery
therefrom, mostly related to hematological toxicities. The schedule of
administration may
comprise administration once or twice a week, on a cycle selected from the
group consisting
of: (i) weekly; (ii) every other week; (iii) one week of therapy followed by
two, three or four
weeks off; (iv) two weeks of therapy followed by one, two, three or four weeks
off; (v) three
weeks of therapy followed by one, two, three, four or five week off; (vi) four
weeks of
therapy followed by one, two, three, four or five week off; (vii) five weeks
of therapy
followed by one, two, three, four or five week off; and (viii) monthly. The
cycle may be
repeated 2, 4, 6, 8, 10, or 12 times or more.
[0206] Alternatively, an ADC may be administered as one dosage every 2 or 3
weeks,
repeated for a total of at least 3 dosages. Or, twice per week for 4-6 weeks.
The dosage may
be administered once every other week or even less frequently, so the patient
can recover
from any drug-related toxicities. Alternatively, the dosage schedule may be
decreased,
namely every 2 or 3 weeks for 2-3 months. The dosing schedule can optionally
be repeated at
other intervals and dosage may be given through various parenteral routes,
with appropriate
adjustment of the dose and schedule.
[0207] The methods and compositions described and claimed herein may be used
to treat
malignant or premalignant conditions and to prevent progression to a
neoplastic or malignant
state, including but not limited to those disorders described above. Such uses
are indicated in
conditions known or suspected of preceding progression to neoplasia or cancer,
in particular,
where non-neoplastic cell growth consisting of hyperplasia, metaplasia, or
most particularly,
dysplasia has occurred (for review of such abnormal growth conditions, see
Robbins and
Angell, Basic Pathology, 2d Ed., W. B. Saunders Co., Philadelphia, pp. 68-79
(1976)).
[0208] Dysplasia is frequently a forerunner of cancer, and is found mainly in
the epithelia. It
is the most disorderly form of non-neoplastic cell growth, involving a loss in
individual cell
uniformity and in the architectural orientation of cells. Dysplasia
characteristically occurs
where there exists chronic irritation or inflammation. In preferred
embodiments, the method
59
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
of the invention is used to inhibit growth, progression, and/or metastasis of
cancers, in
particular those listed above.
Kits
[0209] Various embodiments may concern kits containing components suitable for
treating
cancer tissue in a patient. Exemplary kits may contain at least one anti-Trop-
2 ADC as
described herein. If the composition containing components for administration
is not
formulated for delivery via the alimentary canal, such as by oral delivery, a
device capable of
delivering the kit components through some other route may be included. One
type of device,
for applications such as parenteral delivery, is a syringe that is used to
inject the composition
into the body of a subject. Inhalation devices may also be used. In certain
embodiments, an
anti-Trop-2 antibody or antigen binding fragment thereof may be provided in
the form of a
prefilled syringe or autoinjection pen containing a sterile, liquid
formulation or lyophilized
preparation of antibody (e.g., Kivitz et al., Clin. Ther. 2006, 28:1619-29).
[0210] The kit components may be packaged together or separated into two or
more
containers. In some embodiments, the containers may be vials that contain
sterile, lyophilized
formulations of a composition that are suitable for reconstitution. A kit may
also contain one
or more buffers suitable for reconstitution and/or dilution of other reagents.
Other containers
that may be used include, but are not limited to, a pouch, tray, box, tube, or
the like. Kit
components may be packaged and maintained sterilely within the containers.
Another
component that can be included is instructions for use of the kit.
EXAMPLES
[0211] The examples below are illustrative of embodiments of the current
invention and are
not limiting to the scope of the claims.
Example 1. Production and Use of anti-Trop-2-SN-38 Antibody-Drug Conjugate
[0212] The humanized RS7 (hRS7) anti-Trop-2 antibody was produced as described
in U.S.
Patent No. 7,238,785, the Figures and Examples section of which are
incorporated herein by
reference. SN-38 attached to a CL2A linker was produced and conjugated to hRS7
(anti-
Trop-2), hPAM4 (anti-MUC5ac), hA20 (anti-CD20) or hMN-14 (anti-CEACAM5)
antibodies according to U.S. Patent 7,999,083 (Example 10 and 12 of which are
incorporated
herein by reference). The conjugation protocol resulted in a ratio of about 6
SN-38 molecules
attached per antibody molecule.
[0213] Immune-compromised athymic nude mice (female), bearing subcutaneous
human
pancreatic or colon tumor xenografts were treated with either specific CL2A-SN-
38
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
conjugate or control conjugate or were left untreated. The therapeutic
efficacies of the
specific conjugates were observed. FIG. 1 shows a Capan 1 pancreatic tumor
model, wherein
specific CL2A-SN-38 conjugates of hRS7 (anti-Trop-2), hPAM4 (anti-MUC-5ac),
and hMN-
14 (anti-CEACAM5) antibodies showed better efficacies than control hA20-CL2A-
SN-38
conjugate (anti-CD20) and untreated control. Similarly in a BXPC3 model of
human
pancreatic cancer, the specific hRS7-CL2A-SN-38 showed better therapeutic
efficacy than
control treatments (FIG. 2).
Example 2. Efficacy of Anti-Trop-2 Antibody Conjugated to a Prodrug Form of
2-Pyrrolinodoxorubicin (2-PDox)
[0214] A prodrug form of 2-PDox (referred to as pro-2-PDox) was prepared and
conjugated
to antibodies as disclosed in U.S. Patent Application 14/175,089 (Example 1 of
which is
incorporated herein by reference). The structures of doxorubicin, 2-PDox, pro-
2-PDox and a
maleimide activated form of pro-2-PDox that is suitable for conjugation to
sulfhydryl groups
on antibodies or other proteins are shown in FIG. 3. Unless otherwise stated
below, the
number of drug moieties per antibody molecule was in the range of about 6.5 to
about 7.5.
[0215] In vitro cell-binding studies ¨ Retention of antibody binding was
confirmed by cell
binding assays comparing binding of the conjugated to the unconjugated
antibody (Chari,
2008, Acc Chem Res 41:98-107). The potency of the conjugate was tested in a 4-
day MTS
assay using appropriate target cells. The anti-Trop-2 ADC (hRS7-pro-2-PDox)
exhibited IC50
values of 0.35-1.09 nM in gastric (NCI-N87), pancreatic (Capan-1), and breast
(MDA-MB-
468) human cancer cell lines, with free drug exhibiting 0.02-0.07 nM potency
in the same
cell lines. In additional studies, hRS7-pro-2-PDox was observed to be
cytotoxic to MDA-
MB-468, AG S, NCI-N87 and Capan-1 solid tumor cell lines (not shown).
[0216] No significant difference in binding of the antibody moiety to NCI-N87
gastric
carcinoma cells was observed between unconjugated hRS7 and pro-2-PDox-hRS7
conjugated
to 6 molecules of pro-2-PDox per antibody (not shown). It is concluded that
conjugation of
pro-2-PDox to antibodies does not affect antibody-antigen binding activity.
[0217] Serum stability ¨ Serum stability of anti-Trop-2 ADC (hRS7-pro-2-PDox)
was
determined by incubation in human serum at a concentration of 0.2 mg/mL at 37
C. The
incubate was analyzed by HPLC using butyl hydrophobic interaction
chromatography (HIC).
The analysis showed that there was no release of free drug from the conjugate,
suggesting
high serum stability of the conjugate. When the same experiment was repeated
with hRS7-
doxorubicin conjugate, containing the same cleavable linker as hRS7-pro-2-
PDox, and where
61
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
the free drug was independently verified to be released with a half-life of 96
h, clear
formation of a peak corresponding to free doxorubicin was seen on HIC HPLC.
[0218] Surprisingly, it was determined that the pro-2-PDox conjugate was held
tightly to the
antibody because it cross-linked the peptide chains of the antibody together.
The cross-
linking stabilizes the attachment of the drug to the antibody so that the drug
is only released
intracellularly after the antibody is metabolized. The cross-linking assists
in minimizing
toxicity, for example cardiotoxicity, that would result from release of free
drug in circulation.
Previous use of 2-PDox peptide conjugates failed because the drug cross-linked
the peptide to
other proteins or peptides in vivo. With the present anti-Trop-2 ADC, the pro-
2-PDox is
attached to interchain disulfide thiol groups while in the prodrug form. The
prodrug
protection is rapidly removed in vivo soon after injection and the resulting 2-
PDox portion of
the conjugate cross-links the peptide chains of the antibody, forming
intramolecular cross-
linking within the antibody molecule. This both stabilizes the ADC and
prevents cross-
linking to other molecules in circulation.
[0219] In vivo preclinical studies - Tumor size was determined by caliper
measurements of
length (L) and width (W) with tumor volume calculated as (LxW2)/2. Tumors were
measured and mice weighed twice a week. Mice were euthanized if their tumors
reached >1
cm3 in size, lost greater than 15% of their starting body weight, or otherwise
became
moribund. Statistical analysis for the tumor growth data was based on area
under the curve
(AUC) and survival time. Profiles of individual tumor growth were obtained
through linear
curve modeling. Anfitest was employed to determine equality of variance
between groups
prior to statistical analysis of growth curves. A two-tailed t-test was used
to assess statistical
significance between all the various treatment groups and non-specific
controls. For the
saline control analysis a one-tailed t-test was used to assess significance.
Survival studies
were analyzed using Kaplan-Meier plots (log-rank analysis), using the Prism
GraphPad
Software (v4.03) software package (Advanced Graphics Software, Inc.;
Encinitas, CA). All
doses in preclinical experiments are expressed in antibody amounts. In terms
of drug, 100 lug
of antibody (5 mg/kg) in a 20-g mouse, for example, carries 1.4 itig-2.8 lug
(0.14-0.17 mg/kg)
of pro-2-PDox equivalent dose when using an ADC with 3-6 drugs/IgG.
[0220] A single i.v. dose of > 300 lug [¨ 10 lug of pro-2-PDox] of the anti-
Trop-2 ADC was
lethal, but 4 doses of 45 lug given in 2 weeks were tolerated by all animals.
Using this dosing
regimen, we examined the therapeutic effect of anti-Trop-2 hRS7-pro-2-PDox in
2 human
tumor xenograft models, Capan-1 (pancreatic cancer) and NCI-N87 (gastric
cancer). Therapy
62
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
began 7 days after tumor transplantation in nude mice. In the established, 7-
day-old, Capan-1
model, 100% of established tumors quickly regressed, with no evidence of re-
growth (FIG.
4). This result was reproduced in a repeat experiment (not shown). The anti-
Trop-2
conjugate of pro-2-PDox was much more effective than the same drug conjugated
to an
antibody (hMN-14) against CEACAM5, which is also expressed in pancreatic
cancer, or an
antibody against CD20 (hA20), which is not. All treatments were superior to
the saline
control.
[0221] Similar results were observed in the established NCI-N87 model (FIG.
5A), where a
2'd course of therapy, administered after day 70, was safely tolerated and led
to further
regressions of residual tumor (FIG. 5A). The internalizing hRS7-SN-38
conjugate, targeting
Trop-2, provided better therapeutic responses than a conjugate of a poorly
internalizing anti-
CEACAM5 antibody, hMN-14 (FIG. 4, FIG. 5). A non-targeted anti-CD20 ADC, hA20-
pro-
2-PDox, was ineffective, indicating selective therapeutic efficacy (FIG. 4,
FIG. 5). Data
from a breast cancer xenograft (MDA-MB-468) and a second pancreatic cancer
xenograft
(FIG. 5B and FIG. 5C, respectively) showed the same pattern, with the anti-
Trop-2 ADC
significantly more efficacious compared to non-targeting ADC or saline
control. In both
cases, administration of anti-Trop-2 ADC produced a clear inhibition of tumor
growth to the
end of the study.
[0222] PK and toxicity of hRS7-pro-2-PDox with substitutions of 6.8 or 3.7
drug/IgG ¨
Antibody-drug conjugates (ADCs) carrying as much as 8 ultratoxic drugs/MAb are
known to
clear faster than unmodified MAb and to increase off-target toxicity, a
finding that has led to
the current trends to use drug substitutions of < 4 (Hamblett et al., 2004,
Clin Cancer Res
10:7063-70). ADCs were prepared and evaluated with mean drug/MAb substitution
ratios
(MSRs) of ¨6:1 and ¨3:1. Groups of normal mice (n = 5) were administered,
i.v., single doses
of unmodified hRS7 or hRS7-pro-2-PDox with drug substitution of 6.8 or 3.7
(same protein
dose), and serum samples were collected at 30 min, 4 h, 24 h, 72 h, and 168 h
post-injection.
These were analyzed by ELISA for antibody concentration. There were no
significant
differences in serum concentrations at various times, indicating that these
showed similar
clearance from the blood. The PK parameters (Cmax, AUC, etc.) were also
similar. ADCs
with either higher or lower drug substitution had similar tolerability in nude
mice, when the
administered at the same dose of conjugated drug.
[0223] Therapeutic Efficacy at Minimum Effective Dose (MED) ¨ Anti-Trop-2 ADC
(hRS7-
pro-2-PDox), was evaluated in nude mice bearing NCI-N87 human gastric cancer
xenografts
by administering a single bolus protein dose of 9 mg/kg, 6.75 mg/kg, 4.5
mg/kg, 2.25 mg/kg,
63
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
or 1 mg/kg. The therapy was started when the mean tumor volume (mTV) was 0.256
cm3. On
day 21, mTV in the saline control group (non-treatment group) was 0.801
0.181 cm3 which
was significantly larger than that in mice treated with 9, 6.75, 4.5, or 2.25
mg/kg dose with
mTV of 0.211 0.042 cm3, 0.239 0Ø054 cm3, 0.264 0.087 cm3, and 0.567
0.179 cm3,
respectively (P<0.0047, one tailed t-test). From these, the minimum effective
dose was
estimated to be 2.25 mg/kg, while 9 mg/kg represented MTD.
Example 3. Additional Studies With Anti-Trop-2 pro-2-PDox ADC
[0224] Further in vivo efficacy studies were performed in nude mice implanted
with NCI-
N87 human gastric cancer xenografts (FIG. 6A-F). One treatment cycle with 4 x
45 pg of
hRS7-pro-2-PDox rapidly regressed all tumors (FIG. 6D). A second treatment
cycle was
initiated about 2 months after the end of the first cycle, resulting in
complete regression of all
but one of the hRS7-pro-2-PDox treated animals. The hA20 (anti-CD20), hLL1
(anti-CD22)
and hMN-14 (anti-CEACAM5) conjugates had little effect on tumor progression
(FIG. 6B,
6E and 6F) compared to saline control (FIG. 6A). Administration of pro-2-PDox-
hMN-15
(anti-CEACAM6) resulted in a delayed regression of gastric cancer (FIG. 6C),
which was
less effective than the hRS7 conjugate (FIG. 6D).
[0225] The effect of varying dosage schedule of anti-Trop-2 ADC on anti-tumor
efficacy was
examined (FIG. 7, FIG. 8A-G). The experiment began 9 days after tumor
implantation
when mean tumor volume for all groups was 0.383 cm3, and ended on day 93 (84
days after
initiation of therapy). In this study, administration of anti-Trop-2 ADC as a
single dose of
180 tig, two weekly doses of 90 tig, and q4dx4 of 45 tig all resulted in
significantly enhanced
survival (FIG. 7, FIG. 8B-D). For the saline control, 0 of 9 mice survived
(FIG. 8A). For
mice receiving 45 tig q4dx4 of hRS7-pro-2-PDox, 8 of 9 mice were alive at day
94 (FIG.
8B). For mice receiving 90 tig weekly x 2 of hRS7-pro-2-PDox, 9 of 9 mice were
alive at day
94 (FIG. 8C). For mice receiving a single dose of 180 tig of hRS7-pro-2-PDox,
7 of 9 mice
were alive at day 94 (FIG. 8D).
[0226] At the same dosage schedule, the control hA20 (anti-CD20) conjugate had
no effect
on survival (FIG. 7, FIG. 8E-F). A toxicity study showed that the three dosage
schedules of
hRS7-pro-2-PDox resulted in similarly low levels of toxicity (not shown).
[0227] The hRS7-pro-2-PDox conjugate was also effective in Capan-1 pancreatic
cancer (not
shown) and was more effective at inhibiting tumor growth than a hRS7-SN-38
conjugate (not
shown). The hPAM4-pro-2-PDox conjugate was also more effective at inhibiting
growth of
Capan-1 human pancreatic cancer than an hPAM4-SN-38 conjugate (not shown). At
63 days
64
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
after Capan-1 tumor injection (with therapy starting at 1 days post-
innoculation), 0 of 10
mice were alive in the saline control, 10 of 10 mice were alive in mice
treated twice weekly x
2 weeks with 45 ug of hPAM4-pro-2-PDox, 2 of 10 mice were alive in mice
treated twice
weekly x 2 weeks with 45 ug of hA20-pro-2-PDox, 0 of 10 mice were alive in
mice treated
twice weekly x 4 weeks with 250 ug of hPAM4-SN-38, and 0 of 10 mice were alive
in mice
treated twice weekly x 4 weeks with 250 ug of h2O-SN-38.
[0228] hRS7-pro-2-PDox was substantially more effective than hRS7-SN-38 at
inhibiting
growth of PxPC-3 pancreatic cancer (not shown) and was slightly more effective
than hRS7-
SN-38 at inhibiting growth of MDA-MB-468 breast cancer (not shown).
[0229] The effect of different single doses of hRS7-pro-2-PDox on growth of
NCI-N87
gastric carcinoma xenografts is shown in FIG. 9. Using a single dose, the
maximum effect
on tumor growth was observed at 90 ug or higher (FIG. 9).
[0230] Survival curves for mice bearing NCI-N87 human gastric carcinoma
xenografts and
administered a single dose of anti-Trop-2 ADC are shown in FIG. 10. A single
dose of 45 ug
was the minimum required to see a significant survival benefit compared to
saline control
(FIG. 10). Mice administered single doses of 90 ug or higher showed 100%
survival to the
termination of the experiment.
[0231] The ADCC activity of various hRS7-ADC conjugates was determined in
comparison
to hRS7 IgG (FIG. 11). PBMCs were purified from blood purchased from the Blood
Center
of New Jersey. A Trop-2-positive human pancreatic adenocarcinoma cell line
(BxPC-3) was
used as the target cell line with an effector to target ratio of 100:1. ADCC
mediated by hRS7
IgG was compared to hRS7-Pro-2-PDox, hRS7-CL2A-SN-38, and the reduced and
capped
hRS7-NEM. All were used at 33.3 nM.
[0232] Results are shown in FIG. 11. Overall activity was low, but
significant. There was
8.5% specific lysis for the hR57 IgG which was not significantly different
from hRS7-Pro-2-
PDox. Both were significantly better than hLL2 control and hRS7-NEM and hRS7-
SN-38
(P<0.02, two-tailed t-test). There was no difference between hRS7-NEM and hRS7-
SN-38.
Example 4. Efficacy of anti-Trop-2-SN-38 ADC Against Diverse Epithelial
Cancers In Vivo
Abstract
[0233] The purpose of this study was to evaluate the efficacy of an SN-38-anti-
Trop-2
(hRS7) ADC against several human solid tumor types, and to assess its
tolerability in mice
and monkeys, the latter with tissue cross-reactivity to hRS7 similar to
humans. Two SN-38
derivatives, CL2-SN-38 and CL2A-SN-38, were conjugated to the anti-Trop-
2¨humanized
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
antibody, hRS7. The immunoconjugates were characterized in vitro for
stability, binding, and
cytotoxicity. Efficacy was tested in five different human solid tumor-
xenograft models that
expressed Trop-2 antigen. Toxicity was assessed in mice and in Cynomolgus
monkeys.
[0234] The hRS7 conjugates of the two SN-38 derivatives were equivalent in
drug
substitution (-6), cell binding (K , ¨ 1.2 nmol/L), cytotoxicity (IC50¨ 2.2
nmol/L), and
serum stability in vitro (t/, ¨ 20 hours). Exposure of cells to the ADC
demonstrated signaling
pathways leading to PARP cleavage, but differences versus free SN-38 in p53
and p21
upregulation were noted. Significant antitumor effects were produced by hRS7-
SN-38 at
nontoxic doses in mice bearing Calu-3 (P < 0.05), Capan-1 (P < 0.018), BxPC-3
(P < 0.005),
and COLO 205 tumors (P < 0.033) when compared to nontargeting control ADCs.
Mice
tolerated a dose of 2 x 12 mg/kg (SN-38 equivalents) with only short-lived
elevations in ALT
and AST liver enzyme levels. Cynomolgus monkeys infused with 2 x 0.96 mg/kg
exhibited
only transient decreases in blood counts, although, importantly, the values
did not fall below
normal ranges.
[0235] In summary, the anti-Trop-2 hRS7-CL2A-SN-38 ADC provided significant
and
specific antitumor effects against a range of human solid tumor types. It was
well tolerated in
monkeys, with tissue Trop-2 expression similar to humans, at clinically
relevant doses.
Introduction
[0236] Successful irinotecan treatment of patients with solid tumors has been
limited, due in
large part to the low conversion rate of the CPT-11 prodrug into the active SN-
38 metabolite.
Others have examined nontargeted forms of SN-38 as a means to bypass the need
for this
conversion and to deliver SN-38 passively to tumors. We conjugated SN-38
covalently to a
humanized anti-Trop-2 antibody, hRS7. This antibody¨drug conjugate has
specific antitumor
effects in a range of s.c. human cancer xenograft models, including non¨small
cell lung
carcinoma, pancreatic, colorectal, and squamous cell lung carcinomas, all at
nontoxic doses
(e.g., <3.2 mg/kg cumulative SN-38 equivalent dose). Trop-2 is widely
expressed in many
epithelial cancers, but also some normal tissues, and therefore a dose
escalation study in
Cynomolgus monkeys was performed to assess the clinical safety of this
conjugate. Monkeys
tolerated 24 mg SN-38 equivalents/kg with only minor, reversible, toxicities.
Given its
tumor-targeting and safety profile, hRS7-SN-38 provides a significant
improvement in the
management of solid tumors responsive to irinotecan.
Material and Methods
66
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
[0237] Cell lines, antibodies, and chemotherapeutics - All human cancer cell
lines used in
this study were purchased from the American Type Culture Collection. These
include Calu-3
(non¨small cell lung carcinoma), SK-MES-1 (squamous cell lung carcinoma), COLO
205
(colonic adenocarcinoma), Capan-1 and BxPC-3 (pancreatic adenocarcinomas), and
PC-3
(prostatic adenocarcinomas). Humanized RS7 IgG and control humanized anti-CD20
(hA20
IgG, veltuzumab) and anti-CD22 (hLL2 IgG, epratuzumab) antibodies were
prepared at
Immunomedics, Inc. Irinotecan (20 mg/mL) was obtained from Hospira, Inc.
[0238] SN-38 immunoconjugates and in vitro aspects - Synthesis of CL2-SN-38
has been
described previously (Moon et al., 2008, J Med Chem 51:6916-26). Its
conjugation to hRS7
IgG and serum stability were performed as described (Moon et al., 2008, J Med
Chem
51:6916-26; Govindan et al., 2009, Clin Chem Res 15:6052-61). Preparations of
CL2A-SN-
38 (M.W. 1480) and its hRS7 conjugate, and stability, binding, and
cytotoxicity studies, were
conducted as described in the preceding Examples.
[0239] In vivo therapeutic studies - For all animal studies, the doses of SN-
38
immunoconjugates and irinotecan are shown in SN-38 equivalents. Based on a
mean SN-
38/IgG substitution ratio of 6, a dose of 500 tig ADC to a 20-g mouse (25
mg/kg) contains
0.4 mg/kg of SN-38. Irinotecan doses are likewise shown as SN-38 equivalents
(i.e., 40 mg
irinotecan/kg is equivalent to 24 mg/kg of SN-38).
[0240] NCr female athymic nude (nu/nu) mice, 4 to 8 weeks old, and male Swiss-
Webster
mice, 10 weeks old, were purchased from Taconic Farms. Tolerability studies
were
performed in Cynomolgus monkeys (Macaca fascicularis; 2.5-4 kg male and
female) by
SNBL USA, Ltd.
[0241] Animals were implanted subcutaneously with different human cancer cell
lines.
Tumor volume (TV) was determined by measurements in 2 dimensions using
calipers, with
volumes defined as: L x w 2/2, where L is the longest dimension of the tumor
and w is the
shortest. Tumors ranged in size between 0.10 and 0.47 cm3 when therapy began.
Treatment
regimens, dosages, and number of animals in each experiment are described in
the Results.
The lyophilized hRS7-CL2A-SN-38 and control ADC were reconstituted and diluted
as
required in sterile saline. All reagents were administered intraperitoneally
(0.1 mL), except
irinotecan, which was administered intravenously. The dosing regimen was
influenced by our
prior investigations, where the ADC was given every 4 days or twice weekly for
varying
lengths of time (Moon et al., 2008, J Med Chem 51:6916-26; Govindan et al.,
2009, Clin
Chem Res 15:6052-61). This dosing frequency reflected a consideration of the
conjugate's
serum half-life in vitro, to allow a more continuous exposure to the ADC.
67
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
[0242] Statistics - Growth curves are shown as percent change in initial TV
over time.
Statistical analysis of tumor growth was based on area under the curve (AUC).
Profiles of
individual tumor growth were obtained through linear-curve modeling. Anfitest
was
employed to determine equality of variance between groups before statistical
analysis of
growth curves. A 2-tailed t-test was used to assess statistical significance
between the various
treatment groups and controls, except for the saline control, where a 1-tailed
t-test was used
(significance at P < 0.05). Statistical comparisons of AUC were performed only
up to the
time that the first animal within a group was euthanized due to progression.
[0243] Pharmacokinetics and biodistribution - 1111n-radiolabeled hRS7-CL2A-SN-
38 and
hRS7 IgG were injected into nude mice bearing s.c. SK-MES-1 tumors (-0.3 cm3).
One
group was injected intravenously with 20 tiCi (250-lug protein) of 1111n-hRS7-
CL2A-SN-38,
whereas another group received 20 tiCi (250-lug protein) of 1111n-hRS7 IgG. At
various
timepoints mice (5 per timepoint) were anesthetized, bled via intracardiac
puncture, and then
euthanized. Tumors and various tissues were removed, weighed, and counted by y
scintillation to determine the percentage injected dose per gram tissue (%
ID/g). A third
group was injected with 250 lug of unlabeled hRS7-CL2A-SN-38 3 days before the
administration of 1111n-hRS7-CL2A-SN-38 and likewise necropsied. A 2-tailed t-
test was
used to compare hRS7-CL2A-SN-38 and hRS7 IgG uptake after determining equality
of
variance using thefitest. Pharmacokinetic analysis on blood clearance was
performed using
WinNonLin software (Parsight Corp.).
[0244] Tolerability in Swiss-Webster mice and Cynomolgus monkeys - Briefly,
mice were
sorted into 4 groups each to receive 2-mL i.p. injections of either a sodium
acetate buffer
control or 3 different doses of hRS7-CL2A-SN-38 (4, 8, or 12 mg/kg of SN-38)
on days 0
and 3 followed by blood and serum collection, as described in Results.
Cynomolgus monkeys
(3 male and 3 female; 2.5-4.0 kg) were administered 2 different doses of hRS7-
CL2A-SN-
38. Dosages, times, and number of monkeys bled for evaluation of possible
hematologic
toxicities and serum chemistries are described in the Results.
Results
[0245] Stability and potency of hRS7-CL2A-SN-38 - Two different linkages were
used to
conjugate SN-38 to hRS7 IgG (FIG. 12A). The first is termed CL2-SN-38 and has
been
described previously (Moon et al., 2008, J Med Chem 51:6916-26; Govindan et
al., 2009,
Clin Chem Res 15:6052-61). A change in the synthesis of CL2 to remove the
phenylalanine
moiety within the linker was used to produce the CL2A linker. This change
simplified the
synthesis, but did not affect the conjugation outcome (e.g., both CL2-SN-38
and CL2A-SN-
68
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
38 incorporated ¨6 SN-38 per IgG molecule). Side-by-side comparisons found no
significant
differences in serum stability, antigen binding, or in vitro cytotoxicity.
This result was
surprising, since the phenylalanine residue in CL2 is part of a designed
cleavage site for
cathepsin B, a lysosomal protease.
[0246] To confirm that the change in the SN-38 linker from CL2 to CL2A did not
impact in
vivo potency, hRS7-CL2A and hRS7-CL2-SN-38 were compared in mice bearing COLO
205
(FIG. 12B) or Capan-1 tumors (FIG. 12C), using 0.4 mg or 0.2 mg/kg SN-38 twice
weekly x
4 weeks, respectively, and with starting tumors of 0.25 cm3 size in both
studies. Both the
hRS7-CL2A and CL2-SN-38 conjugates significantly inhibited tumor growth
compared to
untreated (AUC,õ,, P< 0.002 vs. saline in COLO 205 model; AUC2,õ, P( 0.001 vs.
saline in
Capan-1 model), and a nontargeting anti-CD20 control ADC, hA20-CL2A-SN-38
(AUC,õ.õ
P< 0.003 in COLO-205 model; AUC,,,: P< 0.002 in Capan-1 model). At the end of
the
study (day 140) in the Capan-1 model, 50% of the mice treated with hRS7-CL2A-
SN-38 and
40% of the hRS7-CL2-SN-38 mice were tumor-free, whereas only 20% of the hA20-
ADC-
treated animals had no visible sign of disease. As demonstrated in FIG. 12,
the CL2A linker
resulted in a somewhat higher efficacy compared to CL2.
[0247] Mechanism of action - In vitro cytotoxicity studies demonstrated that
hRS7-CL2A-
SN-38 had IC50 values in the nmol/L range against several different solid
tumor lines (Table
6). The IC50 with free SN-38 was lower than the conjugate in all cell lines.
Although there was
no apparent correlation between Trop-2 expression and sensitivity to hRS7-CL2A-
SN-38, the
1050 ratio of the ADC versus free SN-38 was lower in the higher Trop-2-
expressing cells,
most likely reflecting the enhanced ability to internalize the drug when more
antigen is
present.
[0248] SN-38 is known to activate several signaling pathways in cells, leading
to apoptosis
(e.g., Cusack et al., 2001, Cancer Res 61:3535-40; Liu et al. 2009, Cancer
Lett 274:47-53;
Lagadec et al., 2008, Br J Cancer 98:335-44). Our initial studies examined the
expression of
2 proteins involved in early signaling events (p21WaniC1P1 and p53) and 1 late
apoptotic event
[cleavage of poly-ADP-ribose polymerase (PARP)] in vitro (not shown). In BxPC-
3, SN-38
led to a 20-fold increase in p21 Wafi/C1P1 expression (not shown), whereas
hRS7-CL2A-SN-38
resulted in only a 10-fold increase (not shown), a finding consistent with the
higher activity
with free SN-38 in this cell line (Table 6). However, hRS7-CL2A-SN-38
increased
p21Wafl/C11l
expression in Calu-3 more than 2-fold over free SN-38 (not shown).
69
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
[0249] A greater disparity between hRS7-CL2A-SN-38- and free SN-38-mediated
signaling
events was observed in p53 expression (not shown). In both BxPC-3 and Calu-3,
upregulation of p53 with free SN-38 was not evident until 48 hours, whereas
hRS7-CL2A-
SN-38 upregulated p53 within 24 hours (not shown). In addition, p53 expression
in cells
exposed to the ADC was higher in both cell lines compared to SN-38 (not
shown).
Interestingly, although hRS7 IgG had no appreciable effect on p21 WafliciP1
expression, it did
induce the upregulation of p53 in both BxPC-3 and Calu-3, but only after a 48-
hour exposure
(not shown). In terms of later apoptotic events, cleavage of PARP was evident
in both cell
lines when incubated with either SN-38 or the conjugate (not shown). The
presence of the
cleaved PARP was higher at 24 hours in BxPC-3 (not shown), which correlates
with high
expression of p21 and its lower IC50. The higher degree of cleavage with free
SN-38 over the
ADC was consistent with the cytotoxicity findings.
[0250] Efficacy of hRS7-SN-38 - Because Trop-2 is widely expressed in several
human
carcinomas, studies were performed in several different human cancer models,
which started
using the hRS7-CL2-SN-38 linkage, but later, conjugates with the CL2A-linkage
were used.
Calu-3¨bearing nude mice given 0.04 mg SN-38/kg of the hRS7-CL2-SN-38 every 4
days x 4 had a significantly improved response compared to animals
administered the
equivalent amount of non-targeting hLL2-CL2-SN-38 (TV = 0.14 0.22 cm3 vs.
0.80 0.91
cm3, respectively; AUCõ,. P( 0.026; FIG. 13A). A dose¨response was observed
when the
dose was increased to 0.4 mg/kg SN-38 (FIG. 13A). At this higher dose level,
all mice given
the specific hR57 conjugate were "cured" within 28 days, and remained tumor-
free until the
end of the study on day 147, whereas tumors regrew in animals treated with the
irrelevant
ADC (specific vs. irrelevant AUC..: P= 0.05). In mice receiving the mixture of
hRS7 IgG
and SN-38, tumors progressed >4.5-fold by day 56 (TV = 1.10 0.88 cm3; AUG"
P( 0.006
vs. hRS7-CL2-SN-38) (FIG. 13A).
[0251] Efficacy also was examined in human colonic (COLO 205) and pancreatic
(Capan-1)
tumor xenografts. In COLO 205 tumor-bearing animals, (FIG. 13B), hRS7-CL2-SN-
38 (0.4
mg/kg, q4dx8) prevented tumor growth over the 28-day treatment period with
significantly
smaller tumors compared to control anti-CD20 ADC (hA20-CL2-SN-38), or hRS7 IgG
(TV
= 0.16 0.09 cm3, 1.19 0.59 cm3, and 1.77 0.93 cm3, respectively;
AUC28,õ,. P< 0.016).
CA 02920192 2016-02-02
WO 2015/047510 PCT/US2014/045074
Table 6. Expression of Trop-2 in vitro cytotoxicity of SN-38 and hRS7-SN-38 in
various
solid tumor lines
Trop-2 expression via FACS Cytotoxicity
results
Median fluorescence Percent hRS7-SN- ADC/free SN-38
Cell line SN-38 95% CI 95% CI
(background) positive 38 ratio
IC50 IC50 IC50 IC50
(nmol/L) (nmol/L) (nmol/L) (nmol/L)
Calu-3 282.2 (4.7) 99.6% 7.19 5.77-8.95 9.97 8.12-
12.25 1.39
COLO 205 141.5 (4.5) 99.5% 1.02 0.66-1.57 1.95 1.26-
3.01 1.91
Capan-1 100.0 (5.0) 94.2% 3.50 2.17-5.65 6.99
5.02-9.72 2.00
PC-3 46.2 (5.5) 73.6% 1.86 1.16-2.99 4.24
2.99-6.01 2.28
SK-MES-1 44.0 (3.5) 91.2% 8.61 6.30-11.76
23.14 17.98-29.78 2.69
BxPC-3 26.4 (3.1) 98.3% 1.44 1.04-2.00 4.03
3.25-4.98 2.80
[0252] The MTD of irinotecan (24 mg SN-38/kg, q2dx5) was as effective as hRS7-
CL2-SN-
38 in COLO 205 cells, because mouse serum can more efficiently convert
irinotecan to SN-
38 (Morton et al., 2000, Cancer Res 60:4206-10) than human serum, but the SN-
38 dose in
irinotecan (2,400 tig cumulative) was 37.5-fold greater than with the
conjugate (64 tig total).
[0253] Animals bearing Capan-1 (FIG. 13C) showed no significant response to
irinotecan
alone when given at an SN-38-dose equivalent to the hRS7-CL2-SN-38 conjugate
(e.g., on
day 35, average tumor size was 0.04 0.05 cm3 in animals given 0.4 mg SN-
38/kg hRS7-SN-
38 vs. 1.78 0.62 cm3 in irinotecan-treated animals given 0.4 mg/kg SN-38;
AUCdo,
P< 0.001; FIG. 13C). When the irinotecan dose was increased 10-fold to 4 mg/kg
SN-38, the
response improved, but still was not as significant as the conjugate at the
0.4 mg/kg SN-38
dose level (TV = 0.17 0.18 cm3 vs. 1.69 0.47 cm3, AUCõyõP< 0.001) (FIG.
13C). An
equal dose of nontargeting hA20-CL2-SN-38 also had a significant antitumor
effect as
compared to irinotecan-treated animals, but the specific hRS7 conjugate was
significantly
better than the irrelevant ADC (TV = 0.17 0.18 cm3 vs. 0.80 0.68 cm3,
AUCõyõP< 0.018)
(FIG. 13C).
[0254] Studies with the hRS7-CL2A-SN-38 ADC were then extended to 2 other
models of
human epithelial cancers. In mice bearing BxPC-3 human pancreatic tumors (FIG.
13D),
hRS7-CL2A-SN-38 again significantly inhibited tumor growth in comparison to
control mice
treated with saline or an equivalent amount of nontargeting hA20-CL2A-SN-38
(TV = 0.24
0.11 cm3 vs. 1.17 0.45 cm3 and 1.05 0.73 cm3, respectively; AUC,õ,2,P<
0.001), or
irinotecan given at a 10-fold higher SN-38 equivalent dose (TV = 0.27 0.18
cm3vs. 0.90
71
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
0.62 cm3, respectively; AUCw5P< 0.004) (FIG. 13D). Interestingly, in mice
bearing SK-
MES-1 human squamous cell lung tumors treated with 0.4 mg/kg of the ADC (FIG.
13E),
tumor growth inhibition was superior to saline or unconjugated hRS7 IgG (TV =
0.36 0.25
cm3 vs. 1.02 0.70 cm3 and 1.30 1.08 cm3, respectively; AUC28,.õ, P<
0.043), but
nontargeting hA20-CL2A-SN-38 or the MTD of irinotecan provided the same
antitumor
effects as the specific hRS7-SN-38 conjugate (FIG. 13E).
[0255] In all murine studies, the hRS7-SN-38 ADC was well tolerated in terms
of body
weight loss (not shown).
[0256] Biodistribution of hRS7-CL2A-SN-38 - The biodistributions of hRS7-CL2A-
SN-38
or unconjugated hRS7 IgG were compared in mice bearing SK-MES-1 human squamous
cell
lung carcinoma xenografts (not shown), using the respective '"In-labeled
substrates. A
pharmacokinetic analysis was performed to determine the clearance of hRS7-CL2A-
SN-38
relative to unconjugated hRS7 (not shown). The ADC cleared faster than the
equivalent
amount of unconjugated hRS7, with the ADC exhibiting ¨40% shorter half-life
and mean
residence time. Nonetheless, this had a minimal impact on tumor uptake (not
shown).
Although there were significant differences at the 24- and 48-hour timepoints,
by 72 hours
(peak uptake) the amounts of both agents in the tumor were similar. Among the
normal
tissues, hepatic and splenic differences were the most striking (not shown).
At 24 hours
postinjection, there was >2-fold more hRS7-CL2A-SN-38 in the liver than hRS7
IgG (not
shown). Conversely, in the spleen there was 3-fold more parental hRS7 IgG
present at peak
uptake (48-hour timepoint) than hRS7-CL2A-SN-38 (not shown). Uptake and
clearance in
the rest of the tissues generally reflected differences in the blood
concentration (not shown).
[0257] Because twice-weekly doses were given for therapy, tumor uptake in a
group of
animals that first received a predose of 0.2 mg/kg (250 tig protein) of the
hR57 ADC 3 days
before the injection of the "'In-labeled antibody was examined. Tumor uptake
of '"In-hRS7-
CL2A-SN-38 in predosed mice was substantially reduced at every timepoint in
comparison to
animals that did not receive the predose (e.g., at 72 hours, predosed tumor
uptake was 12.5%
3.8% ID/g vs. 25.4% 8.1% ID/g in animals not given the predose; P= 0.0123;
not
shown). Predosing had no appreciable impact on blood clearance or tissue
uptake (not
shown). These studies suggest that in some tumor models, tumor accretion of
the specific
antibody can be reduced by the preceding dose(s), which likely explains why
the specificity
of a therapeutic response could be diminished with increasing ADC doses and
why further
dose escalation is not indicated.
72
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
[0258] Tolerability of hRS7-CL2A-SN-38 in Swiss-Webster mice and Cynomolgus
monkeys
Swiss-Webster mice tolerated 2 doses over 3 days, each of 4, 8, and 12 mg SN-
38/kg of the
hRS7-CL2A-SN-38, with minimal transient weight loss (not shown). No
hematopoietic
toxicity occurred and serum chemistries only revealed elevated aspartate
transaminase (AST,
FIG. 14A) and alanine transaminase (ALT, FIG. 14B). Seven days after
treatment, AST rose
above normal levels (>298 U/L) in all 3 treatment groups (FIG. 14A), with the
largest
proportion of mice being in the 2 x 8 mg/kg group. However, by 15 days
posttreatment, most
animals were within the normal range. ALT levels were also above the normal
range (>77
U/L) within 7 days of treatment (FIG. 14B) and with evidence of normalization
by Day 15.
Livers from all these mice did not show histologic evidence of tissue damage
(not shown). In
terms of renal function, only glucose and chloride levels were somewhat
elevated in the
treated groups. At 2 x 8 mg/kg, 5 of 7 mice had slightly elevated glucose
levels (range of
273-320 mg/dL, upper end of normal 263 mg/dL) that returned to normal by 15
days
postinjection. Similarly, chloride levels were slightly elevated, ranging from
116 to 127
mmoUL (upper end of normal range 115 mmol/L) in the 2 highest dosage groups
(57% in the
2 x 8 mg/kg group and 100% of the mice in the 2 x 12 mg/kg group), and
remained elevated
out to 15 days postinjection. This also could be indicative of
gastrointestinal toxicity, because
most chloride is obtained through absorption by the gut; however, at
termination, there was
no histologic evidence of tissue damage in any organ system examined (not
shown).
[0259] Because mice do not express Trop-2 identified by hRS7, a more suitable
model was
required to determine the potential of the hRS7 conjugate for clinical use.
Immunohistology
studies revealed binding in multiple tissues in both humans and Cynomolgus
monkeys
(breast, eye, gastrointestinal tract, kidney, lung, ovary, fallopian tube,
pancreas, parathyroid,
prostate, salivary gland, skin, thymus, thyroid, tonsil, ureter, urinary
bladder, and uterus; not
shown). Based on this cross-reactivity, a tolerability study was performed in
monkeys.
[0260] The group receiving 2 x 0.96 mg SN-38/kg of hRS7-CL2A-SN-38 had no
significant
clinical events following the infusion and through the termination of the
study. Weight loss
did not exceed 7.3% and returned to acclimation weights by day 15. Transient
decreases were
noted in most of the blood count data (neutrophil and platelet data shown in
FIG 14C and
FIG. 14D), but values did not fall below normal ranges. No abnormal values
were found in
the serum chemistries. Histopathology of the animals necropsied on day 11 (8
days after last
injection) showed microscopic changes in hematopoietic organs (thymus,
mandibular and
mesenteric lymph nodes, spleen, and bone marrow), gastrointestinal organs
(stomach,
duodenum, jejunum, ileum, cecum, colon, and rectum), female reproductive
organs (ovary,
73
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
uterus, and vagina), and at the injection site. These changes ranged from
minimal to moderate
and were fully reversed at the end of the recovery period (day 32) in all
tissues, except in the
thymus and gastrointestinal tract, which were trending towards full recovery
at this later
timepoint (not shown).
[0261] At the 2 x 1.92 mg SN-38/kg dose level of the conjugate, there was 1
death arising
from gastrointestinal complications and bone marrow suppression, and other
animals within
this group showed similar, but more severe adverse events than the 2 x 0.96
mg/kg group (not
shown). These data indicate that dose-limiting toxicities were identical to
that of irinotecan;
namely, intestinal and hematologic. Thus, the MTD for hRS7-CL2A-SN-38 lies
between 2 x
0.96 and 1.92 mg SN-38/kg, which represents a human equivalent dose of 2 x 0.3
to 0.6
mg/kg SN-38.
Discussion
[0262] Trop-2 is a protein expressed on many epithelial tumors, including
lung, breast,
colorectal, pancreas, prostate, and ovarian cancers, making it a potentially
important target
for delivering cytotoxic agents (Ohmachi et al., 2006, Clin Cancer Res 12:3057-
63; Fong et
al., 2008, Br J Cancer 99:1290-95; Cubas et al., 2009, Biochim Biophys Acta
1796:309-14).
The R57 antibody internalizes when bound to Trop-2 (Shih et al., 1995, Cancer
Res
55:5857s-63s), which enables direct intracellular delivery of cytotoxics.
[0263] SN-38 is a potent topoisomerase-I inhibitor, with IC50 values in the
nanomolar range
in several cell lines. It is the active form of the prodrug, irinotecan, that
is used for the
treatment of colorectal cancer, and which also has activity in lung, breast,
and brain cancers.
We reasoned that a directly targeted SN-38, in the form of an ADC, would be a
significantly
improved therapeutic over CPT-11, by overcoming the latter's low and patient-
variable
bioconversion to active SN-38 (Mathijssen et al., 2001, Clin Cancer Res 7:2182-
94).
[0264] The Phe-Lys peptide inserted in the original CL2 derivative allowed for
possible
cleavage via cathepsin B. To simplify the synthetic process, in CL2A the
phenylalanine was
eliminated, and thus the cathepsin B cleavage site was removed. Interestingly,
this product
had a better-defined chromatographic profile compared to the broad profile
obtained with
CL2 (not shown), but more importantly, this change had no impact on the
conjugate's
binding, stability, or potency in side-by-side testing. These data suggest
that SN-38 in CL2
was released from the conjugate primarily by the cleavage at the pH-sensitive
benzyl
carbonate bond to SN-38's lactone ring and not the cathepsin B cleavage site.
74
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
[0265] In vitro cytotoxicity of hRS7 ADC against a range of solid tumor cell
lines
consistently had IC50 values in the nmol/L range. However, cells exposed to
free SN-38
demonstrated a lower IC50 value compared to the ADC. This disparity between
free and
conjugated SN-38 was also reported for ENZ-2208 (Sapra et al., 2008, Clin
Cancer Res
14:1888-96, Zhao et al., 2008, Bioconjug Chem 19:849-59) and NK012 (Koizumi et
al.,
2006, Cancer Res 66:10048-56). ENZ-2208 utilizes a branched PEG to link about
3.5 to 4
molecules of SN-38 per PEG, whereas NK012 is a micelle nanoparticle containing
20% SN-
38 by weight. With our ADC, this disparity (i.e., ratio of potency with free
vs. conjugated
SN-38) decreased as the Trop-2 expression levels increased in the tumor cells,
suggesting an
advantage to targeted delivery of the drug. In terms of in vitro serum
stability, both the CL2-
and CL2A-SN-38 forms of hRS7-SN-38 yielded a il, of ¨20 hours, which is in
contrast to the
short il, of 12.3 minutes reported for ENZ-2208 (Zhao et al., 2008, Bioconjug
Chem 19:849-
59), but similar to the 57% release of SN-38 from NK012 under physiological
conditions
after 24 hours (Koizumi et al., 2006, Cancer Res 66:10048-56).
[0266] Treatment of tumor-bearing mice with hRS7-SN-38 (either with CL2-SN-38
or
CL2A-SN-38) significantly inhibited tumor growth in 5 different tumor models.
In 4 of them,
tumor regressions were observed, and in the case of Calu-3, all mice receiving
the highest
dose of hRS7-SN-38 were tumor-free at the conclusion of study. Unlike in
humans,
irinotecan is very efficiently converted to SN-38 by a plasma esterase in
mice, with a greater
than 50% conversion rate, and yielding higher efficacy in mice than in humans
(Morton et al.,
2000, Cancer Res 60:4206-10; Furman et al., 1999, J Clin Oncol 17:1815-24).
When
irinotecan was administered at 10-fold higher or equivalent SN-38 levels, hRS7-
SN-38 was
significantly better in controlling tumor growth. Only when irinotecan was
administered at its
MTD of 24 mg/kg q2dx5 (37.5-fold more SN-38) did it equal the effectiveness of
hRS7-SN-
38. In patients, we would expect this advantage to favor hRS7-CL2A-SN-38 even
more,
because the bioconversion of irinotecan would be substantially lower.
[0267] We also showed in some antigen-expressing cell lines, such as SK-MES-1,
that using
an antigen-binding ADC does not guarantee better therapeutic responses than a
nonbinding,
irrelevant conjugate. This is not an unusual or unexpected finding. Indeed,
the nonbinding
SN-38 conjugates mentioned earlier enhance therapeutic activity when compared
to
irinotecan, and so an irrelevant IgG-SN-38 conjugate is expected to have some
activity. This
is related to the fact that tumors have immature, leaky vessels that allow the
passage of
macromolecules better than normal tissues (Jain, 1994, Sci Am 271:58-61). With
our
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
conjugate, 50% of the SN-38 will be released in ¨13 hours when the pH is
lowered to a level
mimicking lysosomal levels (e.g., pH 5.3 at 37 C; data not shown), whereas at
the neutral pH
of serum, the release rate is reduced nearly 2-fold. If an irrelevant
conjugate enters an acidic
tumor microenvironment, it is expected to release some SN-38 locally. Other
factors, such as
tumor physiology and innate sensitivities to the drug, will also play a role
in defining this
"baseline" activity. However, a specific conjugate with a longer residence
time should have
enhanced potency over this baseline response as long as there is ample antigen
to capture the
specific antibody. Biodistribution studies in the SK-MES-1 model also showed
that if tumor
antigen becomes saturated as a consequence of successive dosing, tumor uptake
of the
specific conjugate is reduced, which yields therapeutic results similar to
that found with an
irrelevant conjugate.
[0268] Although it is challenging to make direct comparisons between our ADC
and the
published reports of other SN-38 delivery agents, some general observations
can be made. In
our therapy studies, the highest individual dose was 0.4 mg/kg of SN-38. In
the Calu-3
model, only 4 injections were given for a total cumulative dose of 1.6 mg/kg
SN-38 or 32 tig
SN-38 in a 20 g mouse. Multiple studies with ENZ-2208 were done using its MTD
of 10
mg/kg x 5 (Sapra et al., 2008, Clin Cancer Res 14:1888-96; Pastorini et al.,
2010, Clin
Cancer Res 16:4809-21), and preclinical studies with NK012 involved its MTD of
30 mg/kg
x 3 (Koizumi et al., 2006, Cancer Res 66:10048-56). Thus, significant
antitumor effects were
obtained with hRS7-SN-38 at 30-fold and 55-fold less SN-38 equivalents than
the reported
doses in ENZ-2208 and NK012, respectively. Even with 10-fold less hRS7 ADC
(0.04
mg/kg), significant antitumor effects were observed, whereas lower doses of
ENZ-2208 were
not presented, and when the NK012 dose was lowered 4-fold to 7.5 mg/kg,
efficacy was lost
(Koizumi et al., 2006, Cancer Res 66:10048-56). Normal mice showed no acute
toxicity with
a cumulative dose over 1 week of 24 mg/kg SN-38 (1,500 mg/kg of the
conjugate), indicating
that the MTD was higher. Thus, tumor-bearing animals were effectively treated
with 7.5- to
15-fold lower amounts of SN-38 equivalents.
[0269] Biodistribution studies revealed the hRS7-CL2A-SN-38 had similar tumor
uptake as
the parental hRS7 IgG, but cleared substantially faster with 2-fold higher
hepatic uptake,
which may be due to the hydrophobicity of SN-38. With the ADC being cleared
through the
liver, hepatic and gastrointestinal toxicities were expected to be dose
limiting. Although mice
had evidence of increased hepatic transaminases, gastrointestinal toxicity was
mild at best,
with only transient loss in weight and no abnormalities noted upon
histopathologic
examination. Interestingly, no hematological toxicity was noted. However,
monkeys showed
76
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
an identical toxicity profile as expected for irinotecan, with
gastrointestinal and
hematological toxicity being dose-limiting.
[0270] Because Trop-2 recognized by hRS7 is not expressed in mice, it was
important to
perform toxicity studies in monkeys that have a similar tissue expression of
Trop-2 as
humans. Monkeys tolerated 0.96 mg/kg/dose (-12 mg/m2) with mild and reversible
toxicity,
which extrapolates to a human dose of ¨0.3 mg/kg/dose (-11 mg/m2). In a Phase
I clinical
trial of NK012, patients with solid tumors tolerated 28 mg/m2 of SN-38 every 3
weeks with
Grade 4 neutropenia as dose-limiting toxicity (DLT; Hamaguchi et al., 2010,
Clin Cancer Res
16:5058-66). Similarly, Phase I clinical trials with ENZ-2208 revealed dose-
limiting febrile
neutropenia, with a recommendation to administer 10 mg/m2 every 3 weeks or 16
mg/m2 if
patients were administered G-CSF (Kurzrock et al., AACR-NCI-EORTC
International
Conference on Molecular Targets and Cancer Therapeutics; 2009 Nov 15-19;
Boston, MA;
Poster No C216; Patnaik et al., AACR-NCI-EORTC International Conference on
Molecular
Targets and Cancer Therapeutics; 2009 Nov 15-19; Boston, MA; Poster No C221).
Because
monkeys tolerated a cumulative human equivalent dose of 22 mg/m2, it appears
that even
though hRS7 binds to a number of normal tissues, the MTD for a single
treatment of the
hRS7 ADC could be similar to that of the other nontargeting SN-38 agents.
Indeed, the
specificity of the anti¨Trop-2 antibody did not appear to play a role in
defining the DLT,
because the toxicity profile was similar to that of irinotecan. More
importantly, if antitumor
activity can be achieved in humans as in mice that responded with human
equivalent dose of
just at 0.03 mg SN-38 equivalents/kg/dose, then significant antitumor
responses may be
realized clinically.
[0271] In conclusion, toxicology studies in monkeys, combined with in vivo
human cancer
xenograft models in mice, have indicated that this ADC targeting Trop-2 is an
effective
therapeutic in several tumors of different epithelial origin.
Example 5. Anti-Trop-2 ADC Comprising hRS7 and Paclitaxel
[0272] A new antibody-drug conjugate (ADC) was made by conjugating paclitaxel
(TAXOLO) to the hRS7 anti-human Trop-2 antibody (hRS7-paclitaxel). The final
product
had a mean drug to antibody substitution ratio of 2.2. This ADC was tested in
vitro using two
different Trop-2-postive cell lines as targets: BxPC-3 (human pancreatic
adenocarcinoma)
and MDA-MB-468 (human triple negative breast carcinoma). One day prior to
adding the
ADC, cells were harvested from tissue culture and plated into 96-well plates
at 2000 cells per
well. The next day cells were exposed to free paclitaxel (6.1 x 10-11 to 4 x
10-6 M) or the
77
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
drug-equivalent of hRS7-paclitaxel. For comparison, hRS7-SN-38 and free SN-38
were also
tested at a range of 3.84 x 10-12 to 2.5 x 10-7 M. Plates were incubated at 37
C for 96 h.
After this incubation period, an MTS substrate was added to all of the plates
and read for
color development at half-hour intervals until untreated control wells had an
OD492nm reading
of approximately 1Ø Growth inhibition was measured as a percent of growth
relative to
untreated cells using Microsoft Excel and Prism software (non-linear
regression to generate
sigmoidal dose response curves which yield ICso-values).
[0273] The hRS7-paclitaxel ADC exhibited cytotoxic activity in the MDA-MB-468
breast
cell line (FIG. 15), with an ICso-value approximately 4.5-fold higher than
hRS7-SN-38. The
free paclitaxel was much more potent than the free SN-38 (FIG. 15). While the
ICso for free
SN-38 was 1.54x10-9 M, the ICso for free paclitaxel was less than 6.1x10-11 M.
Similar
results were obtained for the BxPC-3 pancreatic cell line (FIG. 16) in which
the hRS7-
paclitaxel ADC had an ICso-value approximately 2.8-fold higher than the hRS7-
SN-38 ADC.
These results show the efficacy of anti-Trop-2 conjugated paclitaxel in vitro,
with ICso-values
in the nanomolar range, similar to the hRS7-SN-38 ADC.
Example 6. Cell Binding Assay of Anti-Trop-2 Antibodies
[0274] Two different murine monoclonal antibodies against human Trop-2 were
obtained for
ADC conjugation. The first, 162-46.2, was purified from a hybridoma (ATCC, HB-
187)
grown up in roller-bottles. A second antibody, MAB650, was purchased from R&D
Systems
(Minneapolis, MN). For a comparison of binding, the Trop-2 positive human
gastric
carcinoma, NCI-N87, was used as the target. Cells (1.5x105/well) were plated
into 96-well
plates the day before the binding assay. The following morning, a
dose/response curve was
generated with 162-46.2, MAB650, and murine R57 (0.03 to 66 nM). These primary
antibodies were incubated with the cells for 1.5 h at 4 C. Wells were washed
and an anti-
mouse-HRP secondary antibody was added to all the wells for 1 h at 4 C. Wells
are washed
again followed by the addition of a luminescence substrate. Plates were read
using Envision
plate reader and values are reported as relative luminescent units.
[0275] All three antibodies had similar KD-values of 0.57 nM for R57, 0.52 nM
for 162-46.2
and 0.49 nM for MAB650. However, when comparing the maximum binding (Bmax) of
162-
46.2 and MAB650 to R57 they were reduced by 25% and 50%, respectively (Bmax
11,250 for
R57, 8,471 for 162-46.2 and 6,018 for MAB650) indicating different binding
properties in
comparison to R57.
Example 7. Cytotoxicity of Anti-Trop-2 ADC (MAB650-SN-38)
78
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
[0276] A novel anti-Trop-2 ADC was made with SN-38 and MAB650, yielding a mean
drug
to antibody substitution ratio of 6.89. Cytotoxicity assays were performed to
compare the
MAB650-SN-38 and hRS7-SN-38 ADCs using two different human pancreatic
adenocarcinoma cell lines (BxPC-3 and Capan-1) and a human triple negative
breast
carcinoma cell line (MDA-MB-468) as targets.
[0277] One day prior to adding the ADCs, cells were harvested from tissue
culture and plated
into 96-well plates. The next day cells were exposed to hRS7-SN-38, MAB650-SN-
38, and
free SN-38 at a drug range of 3.84x10-12 to 2.5x10-7 M. Unconjugated MAB650
was used as
a control at protein equivalent doses as the MAB650-SN-38. Plates were
incubated at 37 C
for 96 h. After this incubation period, an MTS substrate was added to all of
the plates and
read for color development at half-hour intervals until an OD492nm of
approximately 1.0 was
reached for the untreated cells. Growth inhibition was measured as a percent
of growth
relative to untreated cells using Microsoft Excel and Prism software (non-
linear regression to
generate sigmoidal dose response curves which yield 1050-values.
[0278] As shown in FIG. 17, hRS7-SN-38 and MAB650-SN-38 had similar growth-
inhibitory effects with 1050-values in the low nM range which is typical for
SN-38-ADCs in
these cell lines. In the human Capan-1 pancreatic adenocarcinoma cell line
(FIG. 17A), the
hRS7-SN-38 ADC showed an IC50 of 3.5 nM, compared to 4.1 nM for the MAB650-SN-
38
ADC and 1.0 nM for free SN-38. In the human BxPC-3 pancreatic adenocarcinoma
cell line
(FIG. 17B), the hRS7-SN-38 ADC showed an IC50 of 2.6 nM, compared to 3.0 nM
for the
MAB650-SN-38 ADC and 1.0 nM for free SN-38. In the human NCI-N87 gastric
adenocarcinoma cell line (FIG. 17C), the hRS7-SN-38 ADC showed an ICso of 3.6
nM,
compared to 4.1 nM for the MAB650-SN-38 ADC and 4.3 nM for free SN-38.
[0279] In summary, in these in vitro assays, the SN-38 conjugates of two anti-
Trop-2
antibodies, hRS7 and MAB650, showed equal efficacies against several tumor
cell lines,
which was similar to that of free SN-38. Because the targeting function of the
anti-Trop-2
antibodies would be a much more significant factor in vivo than in vitro, the
data support that
anti-Trop-2-SN-38 ADCs as a class would be highly efficacious in vivo, as
demonstrated in
the Examples above for hRS7-SN-38.
Cytotoxicity of Anti-Trop-2 ADC (162-46.2-SN-38)
[0280] A novel anti-Trop-2 ADC was made with SN-38 and 162-46.2, yielding a
drug to
antibody substitution ratio of 6.14. Cytotoxicity assays were performed to
compare the 162-
46.2-SN-38 and hRS7-SN-38 ADCs using two different Trop-2-postive cell lines
as targets,
79
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
the BxPC-3 human pancreatic adenocarcinoma and the MDA-MB-468 human triple
negative
breast carcinoma.
[0281] One day prior to adding the ADC, cells were harvested from tissue
culture and plated
into 96-well plates at 2000 cells per well. The next day cells were exposed to
hRS7-SN-38,
162-46.2-SN-38, or free SN-38 at a drug range of 3.84 x 10-12 to 2.5 x 10-7 M.
Unconjugated
162-46.2 and hRS7 were used as controls at the same protein equivalent doses
as the 162-
46.2-SN-38 and hRS7-SN-38, respectively. Plates were incubated at 37 C for 96
h. After
this incubation period, an MTS substrate was added to all of the plates and
read for color
development at half-hour intervals until untreated control wells had an
OD492nm reading of
approximately 1Ø Growth inhibition was measured as a percent of growth
relative to
untreated cells using Microsoft Excel and Prism software (non-linear
regression to generate
sigmoidal dose response curves which yield ICso-values).
[0282] As shown in FIG. 18A and FIG.18B, the 162-46.2-SN-38 ADC had a similar
1050-
values when compared to hRS7-SN-38. When tested against the BxPC-3 human
pancreatic
adenocarcinoma cell line (FIG. 18A), hRS7-SN-38 had an ICso of 5.8 nM,
compared to 10.6
nM for 162-46.2-SN-38 and 1.6 nM for free SN-38. When tested against the MDA-
MB-468
human breast adenocarcinoma cell line (FIG. 18B), hRS7-SN-38 had an ICso of
3.9 nM,
compared to 6.1 nM for 162-46.2-SN-38 and 0.8 nM for free SN-38. The free
antibodies
alone showed little cytotoxicity to either Trop-2 positive cancer cell line.
[0283] In summary, comparing the efficacies in vitro of three different anti-
Trop-2 antibodies
conjugated to the same cytotoxic drug, all three ADCs exhibited equivalent
cytotoxic effects
against a variety of Trop-2 positive cancer cell lines. These data support
that the class of
anti-Trop-2 antibodies, incorporated into drug-conjugated ADCs, are effective
anti-cancer
therapeutic agents for Trop-2 expressing solid tumors.
Example 9. Clinical Trials With IMMU-132 Anti-Trop-2 ADC Comprising
hRS7 Antibody Conjugated to SN-38
Summary
[0284] The present Example reports results from a phase I clinical trial and
ongoing phase II
extension with IMMU-132, an ADC of the internalizing, humanized, hRS7 anti-
Trop-2
antibody conjugated by a pH-sensitive linker to SN-38 (mean drug-antibody
ratio = 7.6).
Trop-2 is a type I transmembrane, calcium-transducing, protein expressed at
high density (-1
x 105), frequency, and specificity by many human carcinomas, with limited
normal tissue
expression. Preclinical studies in nude mice bearing Capan-1 human pancreatic
tumor
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
xenografts have revealed IMMU-132 is capable of delivering as much as 120-fold
more SN-
38 to tumor than derived from a maximally tolerated irinotecan therapy.
[0285] The present Example reports the initial Phase I trial of 25 patients
who had failed
multiple prior therapies (some including topoisomerase-I/II inhibiting drugs),
and the
ongoing Phase II extension now reporting on 69 patients, including in
colorectal (CRC),
small-cell and non-small cell lung (SCLC, NSCLC, respectively), triple-
negative breast
(TNBC), pancreatic (PDC), esophageal, and other cancers.
[0286] As discussed in detail below, Trop-2 was not detected in serum, but was
strongly
expressed (>2 'immunohistochemical staining) in most archived tumors. In a 3+3
trial design,
IMMU-132 was given on days 1 and 8 in repeated 21-day cycles, starting at 8
mg/kg/dose,
then 12 and 18 mg/kg before dose-limiting neutropenia. To optimize cumulative
treatment
with minimal delays, phase II is focusing on 8 and 10 mg/kg (n=30 and 14,
respectively). In
49 patients reporting related AE at this time, neutropenia >Grade 3 occurred
in 28% (4%
Grade 4). Most common non-hematological toxicities initially in these patients
have been
fatigue (55%;>G3 = 9%), nausea (53%;>G3=0%), diarrhea (47%;>G3 = 9%), alopecia
(40%), and vomiting (32%;>G3 = 2%); alopecia also occurred frequently.
Homozygous
UGT1A1 *28/*28 was found in 6 patients, 2 of whom had more severe
hematological and GI
toxicities. In the Phase I and the expansion phases, there are now 48 patients
(excluding
PDC) who are assessable by RECIST/CT for best response. Seven (15%) of the
patients had
a partial response (PR), including patients with CRC (N = 1), TNBC (N = 2),
SCLC (N = 2),
NSCLC (N = 1), and esophageal cancers (N = 1), and another 27 patients (56%)
had stable
disease (SD), for a total of 38 patients (79%) with disease response; 8 of 13
CT-assessable
PDC patients (62%) had SD, with a median time to progression (TTP) of 12.7 wks
compared
to 8.0 weeks in their last prior therapy. The TTP for the remaining 48
patients is 12.6+ wks
(range 6.0 to 51.4 wks). Plasma CEA and CA19-9 correlated with responses who
had
elevated titers of these antigens in their blood. No anti-hRS7 or anti-SN-38
antibodies were
detected despite dosing over months. The conjugate cleared from the serum
within 3 days,
consistent with in vivo animal studies where 50% of the SN-38 was released
daily, with
>95% of the SN-38 in the serum being bound to the IgG in a non-glucoronidated
form, and at
concentrations as much as 100-fold higher than SN-38 reported in patients
given irinotecan.
These results show that the hRS7-SN-38-containing ADC is therapeutically
active in
metastatic solid cancers, with manageable diarrhea and neutropenia.
Pharmacokinetics
81
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
102871 Two ELISA methods were used to measure the clearance of the IgG
(capture with
anti-hRS7 idiotype antibody) and the intact conjugate (capture with anti-SN-38
IgG/probe
with anti-hRS7 idiotype antibody). SN-38 was measured by HPLC. Total IMMU-132
fraction (intact conjugate) cleared more quickly than the IgG (not shown),
reflecting known
gradual release of SN-38 from the conjugate. HPLC determination of SN-38
(Unbound and
TOTAL) showed >95% the SN-38 in the serum was bound to the IgG. Low
concentrations of
SN-38G suggest SN-38 bound to the IgG is protected from glucoronidation.
Comparison of
ELISA for conjugate and SN-38 HPLC revealed both overlap, suggesting the ELISA
is a
surrogate for monitoring SN-38 clearance.
[0288] A summary of the dosing regiment and patient pool is provided in Table
7.
Table 7. Clinical Trial Parameters
Once weekly for 2 weeks administered every 21 days for up to 8
cycles. In the initial enrollment, the planned dose was delayed and
Dosing regimen reduced if > Grade 2 treatment-related toxicity; protocol
was
amended to dose delay and reduction only in the event of > Grade
3 toxicity.
8, 12, 18 mg/kg; later reduced to an intermediate dose level of 10
Dose level cohorts
mg/kg.
Standard Phase I [3+3] design; expansion includes ¨15 patients in
Cohort size
select cancers.
Grade 4 ANC > 7 d; >Grade 3 febrile neutropenia of any duration; G4
DLT Plt > 5 d; G4 Hgb; Grade 4 N/V/D any duration/G3 N/V/D for >
48 h;
G3 infusion-related reactions; related >G3 non-hematological toxicity.
Maximum
Maximum dose where >2/6 patients tolerate 14 21-d cycle w/o delay or
Acceptable Dose
reduction or > G3 toxicity.
(MAD)
Metastatic colorectal, pancreas, gastric, esophageal, lung (NSCLC,
SCLC), triple-negative breast (TNBC), prostate, ovarian, renal, urinary
bladder, head/neck, hepatocellular. Refractory/relapsed after standard
Patients treatment regimens for metastatic cancer. Prior irinotecan-
containing
therapy NOT required for enrollment. No bulky lesion > 5 cm.
Must be 4 weeks beyond any major surgery, and 2 weeks beyond
radiation or chemotherapy regimen. Gilbert's disease or known CNS
82
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
metastatic disease are excluded.
Clinical Trial Status
[0289] A total of 69 patients (including 25 patients in Phase I) with diverse
metastatic
cancers having a median of 3 prior therapies were reported. Eight patients had
clinical
progression and withdrew before CT assessment. Thirteen CT-assessable
pancreatic cancer
patients were separately reported. The median TTP (time to progression) in PDC
patients was
11.9 wks (range 2 to 21.4 wks) compared to median 8 wks TTP for the preceding
last
therapy.
[0290] A total of 48 patients with diverse cancers had at least 1 CT-
assessment from which
Best Response (FIG. 19) and Time to Progression (TTP; FIG. 20) were
determined. To
summarize the Best Response data, of 8 assessable patients with TNBC (triple-
negative
breast cancer), there were 2 PR (partial response), 4 SD (stable disease) and
2 PD
(progressive disease) for a total response [PR + SD] of 6/8 (75%). For SCLC
(small cell lung
cancer), of 4 assessable patients there were 2 PR, 0 SD and 2 PD for a total
response of 2/4
(50%). For CRC (colorectal cancer), of 18 assessable patients there were 1 PR,
11 SD and 6
PD for a total response of 12/18 (67%). For esophageal cancer, of 4 assessable
patients there
were 1 PR, 2 SD and 1 PD for a total response of 3/4 (75%). For NSCLC (non-
small cell lung
cancer), of 5 assessable patients there were 1 PR, 3 SD and 1 PD for a total
response of 4/5
(80%). Over all patients treated, of 48 assessable patients there were 7 PR,
27 SD and 14 PD
for a total response of 34/48 (71%). These results demonstrate that the anti-
TROP-2 ADC
(hRS7-SN-38) showed significant clinical efficacy against a wide range of
solid tumors in
human patients.
[0291] The reported side effects of therapy (adverse events) are summarized in
Table 8. As
apparent from the data of Table 8, the therapeutic efficacy of hRS7-SN-38 was
achieved at
dosages of ADC showing an acceptably low level of adverse side effects.
Table 8.
Related Adverse Events Listing for IMMU-132-01
Criteria: Total > 10% or > Grade 3
83
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
N = 47 patients
TOTAL Grade 3 Grade 4
Fatigue 55% 4 (9%) 0
Nausea 53% 0 0
Diarrhea 47% 4 (9%) 0
Neutropenia 43% 11 (24%) 2 (4%)
Alopecia 40% -- --
Vomiting 32% 1 (2%) 0
Anemia 13% 2 (4%) 0
Dysgeusia 15% 0 0
Pyrexia 13% 0 0
Abdominal pain 11% 0 0
Hypokalemia 11% 1 (2%) 0
WBC Decrease 6% 1 (2%) 0
Febrile Neutropenia 6% 1 (2%) 2 (4%)
Deep vein thrombosis 2% 1 (2%) 0
Grading by CTCAE v 4.0
[0292] Exemplary partial responses to the anti-Trop-2 ADC were confirmed by CT
data (not
shown). As an exemplary PR in CRC, a 62 year-old woman first diagnosed with
CRC
underwent a primary hemicolectomy. Four months later, she had a hepatic
resection for liver
metastases and received 7 mos of treatment with FOLFOX and 1 mo 5FU. She
presented
with multiple lesions primarily in the liver (3+ Trop-2 by immunohistology),
entering the
hRS7-SN-38 trial at a starting dose of 8 mg/kg about 1 year after initial
diagnosis. On her
first CT assessment, a PR was achieved, with a 37% reduction in target lesions
(not shown).
The patient continued treatment, achieving a maximum reduction of 65% decrease
after 10
months of treatment (not shown) with decrease in CEA from 781 ng/mL to 26.5
ng/mL),
before progressing 3 months later.
[0293] As an exemplary PR in NSCLC, a 65 year-old male was diagnosed with
stage IIIB
NSCLC (sq. cell). Initial treatment of caboplatin/etoposide (3 mo) in concert
with 7000 cGy
XRT resulted in a response lasting 10 mo. He was then started on Tarceva
maintenance
therapy, which he continued until he was considered for IMMU-132 trial, in
addition to
84
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
undergoing a lumbar laminectomy. He received first dose of IMMU-132 after 5
months of
Tarceva, presenting at the time with a 5.6 cm lesion in the right lung with
abundant pleural
effusion. He had just completed his 6th dose two months later when the first
CT showed the
primary target lesion reduced to 3.2 cm (not shown).
[0294] As an exemplary PR in SCLC, a 65 year-old woman was diagnosed with
poorly
differentiated SCLC. After receiving carboplatin/etoposide (Topoisomerase-II
inhibitor) that
ended after 2 months with no response, followed with topotecan (Topoisomerase-
I inhibitor)
that ended after 2 months, also with no response, she received local XRT (3000
cGy) that
ended 1 month later. However, by the following month progression had
continued. The
patient started with IMMU-132 the next month (12 mg/kg; reduced to 6.8 mg/kg;
Trop-2
expression 3+), and after two months of IMMU-132, a 38% reduction in target
lesions,
including a substantial reduction in the main lung lesion occurred (not
shown). The patient
progressed 3 months later after receiving 12 doses.
[0295] These results are significant in that they demonstrate that the anti-
Trop-2 ADC was
efficacious, even in patients who had failed or progressed after multiple
previous therapies.
[0296] In conclusion, at the dosages used, the primary toxicity was a
manageable
neutropenia, with few Grade 3 toxicities. IMMU-132 showed evidence of activity
(PR and
durable SD) in relapsed/refractory patients with triple-negative breast
cancer, small cell lung
cancer, non-small cell lung cancer, colorectal cancer and esophageal cancer,
including
patients with a previous history of relapsing on topoisomerase-I inhibitor
therapy. These
results show efficacy of the anti-Trop-2 ADC in a wide range of cancers that
are resistant to
existing therapies.
Example 10. Comparative Efficacy of Different Anti-Trop-2 ADCs
[0297] The therapeutic efficacy of a murine anti-Trop-2 monoclonal antibody
(162-46.2)
conjugated with either SN-38 or Pro-2-PDox was compared to hRS7-SN-38 and hRS7-
Pro-2-
PDox antibody-drug conjugate (ADC) in mice bearing human gastric carcinoma
xenografts
(NCI-N87). NCI-N87 cells were expanded in tissue culture and harvested with
trypsin/EDTA. Female athymic nude mice were injected s.c. with 200 L of NCI-
N87 cell
suspension mixed 1:1 with matrigel such that 1x107 cells was administered to
each mouse.
Once tumors reached approximately 0.25 cm3 in size (6 days later), the animals
were divided
up into seven different treatment groups of nine mice each. For the SN-38
ADCs, mice
received 500 g i.v. injections once a week for two weeks. Control mice
received the non-
tumor targeting hA20-SN-38 ADC at the same dose/schedule. For the Pro-2-PDox-
ADCs,
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
mice were administered 45 g i.v. twice weekly for two weeks. Control mice
received hA20-
Pro-2-PDox ADC at the same dose/schedule. A final group of mice received only
saline and
served as the untreated control. Tumors were measured and mice weighed twice a
week.
Mice were euthanized for disease progression if their tumor volumes exceeded
1.0 cm3 in
size.
[0298] Mean tumor volumes for the SN-38-ADC treated mice are shown in FIG. 21.
As
determined by area under the curve (AUC), both hRS7-SN-38 and 162-46.2-SN-38
significantly inhibited tumor growth when compared to saline and hA20-SN-38
control mice
(P<0.001). Treatment with hRS7-SN-38 achieved stable disease in 7 of 9 mice
with mean
time to tumor progression (TTP) of 18.4 3.3 days. Mice treated with 162-46.2-
SN-38
achieved a positive response in 6 of 9 mice with the remaining 3 achieving
stable disease.
Mean TTP was 24.2 6.0 days, which is significantly longer than hRS7-SN-38
treated
animals (P=0.0382).
[0299] For the Pro-2-PDox ADCs (FIG. 22), mice treated with hRS7-Pro-2-PDox
experienced tumor regressions and an overall anti-tumor response that was
significantly
better than 162-46.2-Pro-2-PDox treatment (P<0.0001; AUC). While the tumors
did not
immediately respond to the 162-46.2-Pro-2-PDox therapy, they did eventually
regress. This
regression did not occur until approximate 11 days after the start of therapy.
Within 39 days
of the start of therapy (day 45 post-tumor cell implantation) there were no
significant
differences in tumor size between hRS7-Pro-2-PDox and 162-46.2-Pro-2-PDox
treated mice
(Mean Tumor Volume = 0.151 0.025 cm3 and 0.190 0.53 cm3, respectively). On
this day,
9 of 9 mice treated with hRS7-Pro-2-PDox had tumors that were smaller than
when therapy
began. Likewise, 7 of 9 mice in the 162-46.2-Pro-2-PDox group had tumors that
were
smaller than when therapy began.
[0300] These results confirm the in vivo efficacy of four different anti-Trop-
2 ADCs for
treatment of human gastric carcinoma.
Example 11. Treatment of Patients with Advanced, Metastatic Pancreatic
Cancer With Anti-Trop-2 ADC
Summary
[0301] IMMU-132 (hRS7-SN-38) is an anti-Trop-2 ADC comprising the cancer cell
internalizing, humanized, anti-Trop-2 hRS7 antibody, conjugated by a pH-
sensitive linker to
SN-38, the active metabolite of irinotecan, at a mean drug-antibody ratio of
7.6. Trop-2 is a
type-I transmembrane, calcium-transducing protein expressed at high density,
frequency, and
86
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
specificity in many epithelial cancers, including pancreatic ductal
adenocarcinoma, with
limited normal tissue expression. All 29 pancreatic tumor microarray specimens
tested were
Trop-2-positive by immunohistochemistry, and human pancreatic cancer cell
lines were
found to express 115k-891k Trop-2 copies on the cell membrane. We reported in
Example 9
above the results from the IMMU-132 Phase I study enrolling patients with 13
different
tumor types using a 3+3 design. The Phase I dose-limiting toxicity was
neutropenia. Over
80% of 24 assessable patients in this study had long-term stable disease, with
partial
responses (RECIST) observed in patients with colorectal (CRC), triple-negative
breast
(TNBC), small-cell and non-small cell lung (SCLC, NSCLC), and esophageal (EAC)
cancers. The present Example reports the results from the IMMU-132 Phase I/II
study cohort
of patients with metastatic PDC. Patients with PDC who failed a median of 2
prior therapies
(range 1-5) were given IMMU-132 on days 1 and 8 in repeated 21-day cycles.
[0302] In the subgroup of PDC patients (N=15), 14 received prior gemcitabine-
containing
regimens. Initial toxicity data from 9 patients found neutropenia [3 of 9 >G3,
33%; and 1 case
of G4 febrile neutropenia), which resulted in dose delays or dose reductions.
Two patients
had Grade 3 diarrhea; no patient had Grade 3-4 nausea or vomiting. Alopecia
(Grades 1-2)
occurred in 5 of 9 patients. Best response was assessable in 13 of 14
patients, with 8 stable
disease for 8 to 21.4 wks (median 12.7 wks; 11.9 wks all 14 patients). One
patient who is
continuing treatment has not yet had their first CT assessment. Five had
progressive disease
by RECIST; 1 withdrew after just 1 dose due to clinical progression and was
not assessable.
Serum CA19-9 titers decreased in 3 of the patients with stable disease by 23
to 72%. Despite
multiple administrations, none of the patients developed an antibody response
to IMMU-132
or SN-38. Peak and trough serum samples showed that IMMU-132 cleared more
quickly than
the IgG, which is expected based on the known local release of SN-38 within
the tumor cell.
Concentrations of SN-38-bound to IgG in peak samples from one patient given 12
mg/kg of
IMMU-132 showed levels of ¨4000 ng/mL, which is 40-times higher than the SN-38
titers
reported in patients given irinotecan therapy.
[0303] We conclude that IMMU-132 is active (long-term stable disease) in 62%
(8/13) of
PDC patients who failed multiple prior therapies, with manageable neutropenia
and little GI
toxicity. Advanced PDC patients can be given repeated treatment cycles (>6) of
8-10 mg/kg
IMMU-132 on days 1 and 8 of a 21-day cycle, with some dose adjustments or
growth factor
support for neutropenia in subsequent treatment cycles. These results agree
with the findings
in patients with advanced CRC, TNBC, SCLC, NSCLC, EAC who have shown partial
responses and long-term stable disease with IMMU-132 administration. In
summary,
87
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
monotherapy IMMU-132 is a novel, efficacious treatment regimen for patients
with PDC,
including those with tumors that were previously resistant to other
therapeutic regimens for
PDC.
Methods and Results
[0304] Trop-2 expression - The expression of Trop-2 on the surface of various
cancer cell
lines was determined by flow cytometry using QUANTBRITEO PE beads. The results
for
number of Trop-2 molecules detected in the different cell lines was: BxPC-3
pancreatic
cancer (891,000); NCI-N87 gastric cancer (383,000); MDA-MB-468 breast cacner
(341,000);
SK-MES-1 squamous cell lung cancer (27,000); Capan-1 pancreatic cancer
(115,000); AGS
gastric cancer (78,000) COLO 205 colon cancer (52,000). Trop-2 expression was
also
observed in 29 of 29 (100%) tissue microarrays of pancreatic adenocarcinoma
(not shown).
[0305] SN-38 accumulation ¨ SN-38 accumulation was determined in nude mice
bearing
Capan-1 human pancreatic cancer xenografts (-0.06-0.27 g). Mice were injected
IV with
irinotecan 40 mg/kg (773 ug; Total SN-38 equivalents = 448 ug). This dose is
MTD in mice.
Human dose equivalent = 3.25 mg/kg or ¨126 mg/m2. Or mice were injected IV
with IMMU-
132 1.0 mg (SN-38:antibody ratio = 7.6; SN-38 equivalents = 20 ug). This dose
is well
below the MTD in mice. Human equivalent dose ¨4 mg/kg IMMU-132 (-80 ug/kg SN-
38
equivalents). Necropsies were performed on 3 animals per interval, in
irinotecan injected
mice at 5 min, 1, 2, 6 and 24 hours or in IMMU-132 injected mice at 1, 6, 24,
48 and 72 h.
Tissues were extracted and analyzed by reversed-phase HPLC analysis for SN-38,
SN-38G,
and irinotecan. Extracts from IMMU-132-treated animals also were acid
hydrolyzed to
release SN-38 from the conjugate (i.e., SN-38 (TOTAL]). The results, shown in
FIG. 23,
demonstrate that the IMMU-132 ADC has the potential to deliver 120 times more
SN-38 to
the tumor compared to irinotecan, even though 22-fold less SN-38 equivalents
were
administered with the ADC.
[0306] IMMU-132 clinical protocol ¨ The protocol used in the phase I/II study
was as
indicated in Table 9 below.
Table 9. Clinical Protocol Using IMMU-132: OVERVIEW
88
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
Once weekly for 2 weeks administered every 21 days for up to 8 cycles.
Patients with objective responses are allowed to continue beyond 8 cycles.
In the initial enrollment, the planned dose was delayed and reduced if >
Grade 2 treatment-related toxicity; protocol was amended later in study to
dose delay and reduction only in the event of > Grade 3 toxicity. The
development of severe toxicities due to treatment requires dose reduction
by 25% of the assigned dose for 1st occurrence, 50% for 2'd occurrence,
and treatment discontinued entirely in the event of a 3rd occurrence.
8, 12, 18 mg/kg; later reduced to an intermediate dose level of 10 mg/kg.
Standard Phase I [3+3] design; expansion includes 15 patients in select
cancers.
Grade 4 ANC > 7 d; >Grade 3 febrile neutropenia of any duration; Grade 4
Platelets > 5 d; Grade 4 Hgb; Grade 4 N/V/D of any duration or any Grade
3 N/V/D for > 48 h; Grade 3 infusion-related reactions; > Grade 3 non-
heme toxicity at least possibly due to study drug.
Maximum dose where >2/6 patients tolerate the full 21-d treatment cycle
without dose delay or reduction or > Grade 3 toxicity.
= Metastatic colorectal, pancreas, gastric, esophageal, lung (NSCLC,
SCLC), triple-negative breast, prostate, ovarian, renal, urinary
bladder, head and neck, hepatocellular.
= Refractory/relapsed after standard treatment regimens for metastatic
cancer.
= Prior irinotecan-containing therapy NOT required for enrollment.
= No bulky lesion > 5 cm.
= Must be 4 weeks beyond any major surgery, and 2 weeks beyond
radiation or chemotherapy regimen.
= Gilbert 's disease or known CNS metastatic disease are excluded.
[0307] Patients were administered IMMU-132 according to the protocol
summarized above.
The response assessment to last prior therapy before IMMU-132 treatment is
summarized in
FIG 24. The response assessment to IMMU-132 administration is shown in FIG.
25. A
89
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
summary of time to progression (TTP) results following administration of IMMU-
132 is
shown in FIG. 26.
[0308] An exemplary case study is as follows. A 34 y/o white male initially
diagnosed with
metastatic pancreatic cancer (liver) had progressed on multiple chemotherapy
regimens,
including gemcitabine/ Erlotinib/FG-3019, FOLFIRINOX and GTX prior to
introduction of
IMMU-132 (8 mg/kg dose given days 1 and 8 of a 21 day cycle). The patient
received the
drug for 4 mo with good symptomatic tolerance, an improvement in pain, a 72%
maximum
decline in CA19-9 (from 15885 U/mL to 4418 U/mL) and stable disease by CT
RECIST
criteria along with evidence of tumor necrosis. Therapy had to be suspended
due to a liver
abscess; the patient expired ¨6 weeks later, 6 mo following therapy
initiation.
Conclusions
[0309] Preclinical studies indicated that IMMU-132 delivers 120-times the
amount of SN-38
to a human pancreatic tumor xenograft than when irinotecan is given. As part
of a larger
study enrolling patients with diverse metastatic solid cancers, the Phase 2
dose of IMMU-132
was determined to be 8 to 10 mg/kg, based on manageable neutropenia and
diarrhea as the
major side effects. No anti-antibody or anti-SN-38 antibodies have been
detected to-date,
even with repeated therapeutic cycles.
[0310] A study of 14 advanced PDC patients who relapsed after a median of 2
prior therapies
showed CT-confirmed antitumor activity consisting of 8/13 (62%) with stable
disease.
Median duration of TTP for 13 CT assessable pts was 12.7 weeks compared to 8.0
weeks
estimated from last prior therapy. This ADC, with a known drug of nanomolar
toxicity,
conjugated to an antibody targeting Trop-2 prevalent on many epithelial
cancers, by a linker
affording cleavage at the tumor site, represents a new efficacious strategy in
pancreatic
cancer therapy with ADCs. In comparison to the present standard of care for
pancreatic
cancer patients, the extension of time to progression in pancreatic cancer
patients, particularly
in those resistant to multiple prior therapies, was surprising and could not
have been
predicted.
Example 12. Treatment of Triple-Negative Breast Cancer With pro-2-PDox-
hRS7 ADC
[0311] pro-2-PDox-hRS7 ADC is prepared as described in the Examples above.
Patients
with triple-negative breast cancer who had failed at least two standard
therapies receive 3
cycles of 70 mg pro-2-PDox-hRS7 injected i.v. every 3 weeks. Objective
responses are
observed at this dose level of pro-2-PDox- hRS7, with an average decrease in
tumor volume
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
of 35%, after two cycles of therapy. All serum samples evaluated for human
anti-hRS7
antibody (HAHA) are negative.
Example 13. Treatment of Metastatic Colon Cancer With pro-2-PDox-hRS7
ADC
[0312] A 52-year old man with metastatic colon cancer (3-5 cm diameters) to
his left and
right liver lobes, as well as a 5 cm metastasis to his right lung, and an
elevated blood CEA
value of 130 ng/mL, is treated with a 100 mg dose of hRS7 anti-Trop-2
conjugated with pro-
2-PDox at 4 drug molecules per IgG, administered by slow intravenous infusion
every other
week for 4 doses. Upon CT evaluation 8 weeks from treatment begin, a 25%
reduction of the
total mean diameters of the 3 target lesions is measured, thus constituting a
good stable
disease response by RECIST1.1 criteria. Repeated courses of therapy continue
as his
neutropenia normalizes.
Example 14. Treatment of Metastatic Pancreatic Cancer With pro-2-PDox-
hRS7 ADC
[0313] A 62-year-old man with metastatic ductal adenocarcinoma of the
pancreas, who has
relapsed after prior therapies with FOLFIRINOX followed by Nab-taxol
(Abraxane0) plus
gemcitabine is given hRS7-pro-2-PDox ADC at a dose of 120 mg every third week
for 4
courses, and after a 3-week delay, another course of 2 injections 2 weeks
apart are given
intravenously. The patient shows some nausea and transient diarrhea with the
therapy, and
also Grade 3 neutropenia after the first course, which recovers before the
second course of
therapy. CT measurements made at 8 weeks following start of therapy show an
18%
shrinkage of the sum of the 3 target lesions in the liver, as compared to the
pretreatment
baseline measurements, constituting stable disease by RECIST 1.1 criteria.
Also, the patient's
CA19-9 blood titer is reduced by 55% from a baseline value of 12,400. His
general
symptoms of weakness, fatigue and abdominal discomfort also improve
considerably,
including regaining his appetite and a weight increase of 2 kg during the
following 6 weeks.
Example 15. Combining Antibody-Targeted Radiation (Radioimmunotherapy)
and Anti-Trop-2-SN-38 ADC Improves Pancreatic Cancer Therapy
[0314] We previously reported effective anti-tumor activity in nude mice
bearing human
pancreatic tumors with 90Y-humanized PAM4 IgG (hPAM4; 90Y-clivatuzumab
tetraxetan)
that was enhanced when combined with gemcitabine (GEM) (Gold et al., Int J.
Cancer
109:618-26, 2004; Clin Cancer Res 9:3929S-37S, 2003). These studies led to
clinical testing
of fractionated 90Y-hPAM4 IgG combined with GEM that is showing encouraging
objective
91
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
responses. While GEM is known for its radiosensitizing ability, alone it is
not a very effective
therapeutic agent for pancreatic cancer and its dose is limited by hematologic
toxicity, which
is also limiting for 90Y-hPAM4 IgG.
[0315] As discussed in the Examples above, an anti-Trop-2 ADC composed of hRS7
IgG
linked to SN-38 shows anti-tumor activity in various solid tumors. This ADC is
very well
tolerated in mice (e.g., > 60 mg), yet just 4.0 mg (0.5 mg, twice-weekly x 4)
is significantly
therapeutic. Trop-2 is also expressed in most pancreatic cancers.
[0316] The present study examined combinations of90Y-hPAM4 IgG with RS7-SN-38
in
nude mice bearing 0.35 cm3 subcutaneous xenografts of the human pancreatic
cancer cell
line, Capan-1. Mice (n=10) were treated with a single dose of90Y-hPAM4 IgG
alone (130
[tCi, i.e., the maximum tolerated dose (MTD) or 75 [tCi), with R57-SN-38 alone
(as above),
or combinations of the 2 agents at the two 90Y-hPAM4 dose levels, with the
first ADC
injection given the same day as the 90Y-hPAM4. All treatments were tolerated,
with < 15%
loss in body weight. Objective responses occurred in most animals, but they
were more
robust in both of the combination groups as compared to each agent given
alone. All animals
in the 0.13-mCi 90Y-hPAM4 IgG + hRS7-SN-38 group achieved a tumor-free state
within 4
weeks, while other animals continued to have evidence of persistent disease.
These studies
provide the first evidence that combined radioimmunotherapy and ADC enhances
efficacy at
safe doses.
[0317] In the ongoing PAM4 clinical trials, a four week clinical treatment
cycle is performed.
In week 1, subjects are administered a dose of 1111n-hPAM4, followed at least
2 days later by
gemcitabine dose. In weeks 2, 3 and 4, subjects are administered a 90Y-hPAM4
dose,
followed at least 2 days later by gemcitabine (200 mg/m2). Escalation started
at 3 x 6.5
mCi/m2. The maximum tolerated dose in front-line pancreatic cancer patients
was 3 x 15
mCi/m2 (hematologic toxicity is dose-limiting). 0f22 CT-assessable patients,
the disease
control rate (CR+PR+SD) was 68%, with 5 (23%) partial responses and 10 (45%)
having
stabilization as best response by RECIST criteria.
Preparation of Antibody-Drug Conjugate (ADC)
[0318] The SN-38 conjugated hRS7 antibody was prepared as described above and
according
to previously described protocols (Moon et al. J Med Chem 2008, 51:6916-6926;
Govindan
et al., Clin Cancer Res 2009. 15:6052-6061). A reactive bifunctional
derivative of SN-38
(CL2A-SN-38) was prepared. The formula of CL2A-SN-38 is (maleimido-[x]-Lys-
PAB000-20-0-SN-38, where PAB is p-aminobenzyl and 'x' contains a short PEG).
92
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
Following reduction of disulfide bonds in the antibody with TCEP, the CL2A-SN-
38 was
reacted with reduced antibody to generate the SN-38 conjugated RS7.
[0319] 90Y-hPAM4 is prepared as previously described (Gold et al., Clin Cancer
Res 2003,
9:3929S-37S; Gold et al., Int J Cancer 2004, 109:618-26).
Combination RAIT + ADC
[0320] The Trop-2 antigen is expressed in most epithelial cancers (lung,
breast, prostate,
ovarian, colorectal, pancreatic) and hRS7-SN-38 conjugates are being examined
in various
human cancer-mouse xenograft models. Initial clinical trials with 90Y-hPAM4
IgG plus
radiosensitizing amounts of GEM are encouraging, with evidence of tumor
shrinkage or
stable disease. However, therapy of pancreatic cancer is very challenging.
Therefore, a
combination therapy was examined to determine whether it would induce a better
response.
Specifically, administration of hRS7-SN-38 at effective, yet non-toxic doses
was combined
with RAIT with 90Y-hPAM4 IgG.
[0321] The results demonstrated that the combination of hRS7-SN-38 with 90Y-
hPAM4 was
more effective than either treatment alone, or the sum of the individual
treatments (not
shown). At a dosage of 75 tiCi 90Y-hPAM4, only 1 of 10 mice was tumor-free
after 20 weeks
of therapy (not shown), the same as observed with hRS7-SN-38 alone (not
shown). However,
the combination of hRS7-SN-38 with 90Y-hPAM4 resulted in 4 of 10 mice that
were tumor-
free after 20 weeks (not shown), and the remaining subjects showed substantial
decrease in
tumor volume compared with either treatment alone (not shown). At 130 tiCi 90Y-
hPAM4 the
difference was even more striking, with 9 of 10 animals tumor-free in the
combined therapy
group compared to 5 of 10 in the RAIT alone group (not shown). These data
demonstrate the
synergistic effect of the combination of hRS7-SN-38 with 90Y-hPAM4. RAIT + ADC
significantly improved time to progression and increased the frequency of
tumor-free
treatment. The combination of ADC with hRS7-SN-38 added to the MTD of RAIT
with 90Y-
hPAM4 had minimal additional toxicity, indicated by the % weight loss of the
animal in
response to treatment (not shown).
[0322] The effect of different sequential treatments on tumor survival
indicated that the
optimal effect is obtained when RAIT is administered first, followed by ADC
(not shown). In
contrast, when ADC is administered first followed by RAIT, there is a decrease
in the
incidence of tumor-free animals (not shown). Neither unconjugated hPAM4 nor
hRS7
antibodies had anti-tumor activity when given alone (not shown).
* * *
93
CA 02920192 2016-02-02
WO 2015/047510
PCT/US2014/045074
[0323] It will be apparent to those skilled in the art that various
modifications and variations
can be made to the products, compositions, methods and processes of this
invention. Thus, it
is intended that the present invention cover such modifications and
variations, provided they
come within the scope of the appended claims and their equivalents.
94