Note: Descriptions are shown in the official language in which they were submitted.
CA 02938180 2016-08-04
SIRP-ALPHA VARIANT CONSTRUCTS AND USES THEREOF
BACKGROUND OF THE INVENTION
Signal-regulatory protein a (SIRP-a) is a protein widely expressed on the
membrane of myeloid
cells. SIRP-a interacts with CD47, a protein broadly expressed on many cell
types in the body. The
interaction of SIRP-a with CD47 prevents engulfment of "self" cells, which
could otherwise be recognized
by the immune system. SIRP-a was first discovered as a binder of SHP-2 (an SH-
2 domain containing
tyrosine phosphatase). CD47 was early characterized as an overexpressed
antigen on ovarian
carcinoma cells.
In 2000, Oldenborg et al. showed that administration of CD47-deficient red
blood cells (RBCs) in
a mouse model resulted in rapid clearance of the RBCs from the system,
demonstrating CD47 to be a
"protective" signal on some subset of "self" cells. Subsequently, the
potential link between the SIRP-a
and cancer was further explored. It was found that high CD47 expression on
tumor cells acted, in acute
myeloid leukemia (AML) and several solid tumor cancers, as a negative
prognostic factor for survival.
Strategies focused on disrupting the interaction between CD47 and SIRP-a, such
as administration of
agents that mask either CD47 or SIRP-a, have been explored as potential
anticancer therapies.
However, in considering these therapeutic strategies, it is a concerning issue
that SIRP-a could
bind to CD47 on many different cell types in the human body. Thus, there
exists a need to engineer
SIR P-a to preferentially bind to CD47 only on diseased cells or on cells at a
diseased site.
SUMMARY OF THE INVENTION
The invention features signal-regulatory protein a (SIRP-a) variant
constructs. The SIRP-a
variant constructs include SIRP-a variants. In some embodiments, the SIRP-a
variant constructs have
.. preferential activity at a diseased site (e.g., at the site of a tumor than
at a non-diseased site). In certain
embodiments, the SIRP-a variant constructs have higher binding affinity to
C047 on diseased cells (e.g.,
tumor cells). In some embodiments, the SIRP-a variants bind with higher
affinity to CD47 under acidic pH
(e.g., less than around pH 7) and/or under hypoxic condition than under
physiological conditions. In
some embodiments, the SIRP-a variants contain one or more substitutions of
amino acids with histidine
.. residues or with other amino acids that allow preferential binding of SIRP-
a variant constructs at a
diseased site. In some embodiments, the SIRP-a variant constructs are
prevented from binding to CD47
in a non-diseased site by a blocking peptide. In some embodiments, the SIRP-a
variant constructs are
targeted to the diseased site (e.g., the tumor) by a targeting moiety (e.g.,
an antibody directed to a tumor-
associated antigen or an antibody-binding peptide). The invention also
features methods and
pharmaceutical compositions containing SIRP-a variant constructs to treat
various diseases, such as
cancer, preferably solid tumor cancer and hematological cancer.
In one aspect, the invention features a signal-regulatory protein a (SIRP-a)
variant construct,
wherein the SIRP-a variant construct preferentially binds CD47 on diseased
cells or at a diseased site
than on non-diseased cells. In some embodiments, the SIRP-a variant construct
binds to CD47 on
diseased cells or at a diseased site with higher affinity than it binds CD47
on non-diseased cells.
In some embodiments, the SIRP-a variant construct includes a SIRP-a variant
attached to a
blocking peptide. In some embodiments, the blocking peptide binds with higher
affinity to a wild-type
1
CA 02938180 2016-08-04
SIRP-a than to the SIRP-a variant. In some embodiments, the SIRP-a variant
binds with higher affinity to
a wild-type CD47 than to the blocking peptide.
In some embodiments, the blocking peptide is a CD47-based blocking peptide. In
some
embodiments, the CD47-based blocking peptide includes a portion that has at
least 80% amino acid
.. sequence identity to the sequence of the wild-type, IgSF domain of CD47
(SEQ ID NO: 35) or a fragment
thereof. In some embodiments, the CD47-based blocking peptide has the sequence
of SEQ ID NO: 38 or
40.
Provided herein are SIRP-a variant constructs comprising a SIRP-a variant
described herein,
wherein said SIRP-a variant is attached to a blocking peptide described herein
by use of at least one
linker (e.g, a cleavable linker). In some embodiments, the SIRP-a variant may
comprise the same CD47
binding site as a wild type SIRP-a. In some embodiments, the SIRP-a variant
may comprise one or more
mutations, or insertions as compared to a wild type SIRP-a. In some
embodiments, the SIRP-a variant
may be a truncated form of the wild type SIRP-a. In some embodiments, the
blocking peptide may be a
CD47 mimic, variant, or fragment described herein. In some embodiments, the
blocking peptide may
exhibit a higher affinity for a wild-type SIRP-a, as compared to the SIRP-a
variant in the S1RP-a variant
construct. In some embodiments, the blocking peptide may be a CD47 variant
polypeptide that
demonstrates a lower affinity for a SIRP-a variant as compared to the wild-
type CD47. In some
embodiments, the linker between the SIRP-a variant and the blocking peptide
may be at least one linker
that is optionally cleavable by one or more protease& In some embodiments, the
linker optionally also
comprises one or more spacers.
In some embodiments, the SIRP-a variant is attached to a blocking peptide by
way of a cleavable
linker and optionally one or more spacers. In some embodiments, the cleavable
linker is cleaved under
acidic pH and/or hypoxic condition. In some embodiments, the cleavable linker
is cleaved by a tumor-
associated enzyme. In some embodiments, the tumor-associated enzyme is a
protease. In some
embodiments, the protease is selected from the group consisting of matriptase
(MTSP1), urinary-type
plasminogen activator (uPA), legumain, PSA (also called KLK3, kallikrein-
related peptidase-3), matrix
metalloproteinase-2 (MMP-2), MMP9, human neutrophil elastase (FINE), and
proteinase 3 (Pr3). In some
embodiments, the protease is matriptase. In some embodiments, the cleavable
linker has the sequence
of LSGRSDNH (SEQ ID NO: 47) or any one of the sequences listed in Table 7. In
some embodiments,
the cleavable linker includes one or a combination of the following sequences
(see, e.g., Table 7):
PRFKIIGG, PRFRIIGG, SSRHRRALD, RKSSIIIRMRDVVL, SSSFDKGKYKKGDDA,
SSSFDKGKYKRGDDA, IEGR, IDGR, GGSIDGR, PLGLWA, GPLGIAGI, GPEGLRVG, YGAGLGVV,
AGLGVVER, AGLGISST, DVAQFVUT, VAQFVLTE, AQFVLTEG, PVQPIGPQ, US/G-/R--/S-
/D/N/H, -
/s/gs/Rk-/rv/-/-/-, SGR-SA, US/G-/R--/S-/D/N/H, r/-/-/Rk-/v-/-/g/-, RQAR-VV,
r/-/-/Rk/v/-/g,
.. /Kr/RKQ/gAS/RK/A, US/G-/RIS-/D/N/H, -/-/-/NI-/-/-, AAN-L, ATN-L, si/sq/-
/yqrs/s/-/-, S/S/K/UQ, -/p/-/-
g/pa/-/g1/-/g/-, G/P/UG/I/A/G/Q, P/V/G/UI/G, H/P/V/G/UUNR, -/-/-/viat-/-/-/-,
and -/y/y/vta -/-/-/-, where
"¨" means any amino acid (i.e., any naturally occurring amino acid), capital
case indicates an strong
preference for that amino acid, lower case indicates a minor preference for
that amino acid, and "I"
separates amino acid positions in cases where more than one amino acid at a
position adjacent to the
"/"is possible.
In some embodiments, the SIRP-a variant is attached to an antibody-binding
peptide. In some
embodiments, the antibody-binding peptide binds to a constant region of an
antibody reversibly or
2
CA 02938180 2016-08-04
irreversibly. In some embodiments, the antibody-binding peptide binds to the
fragment antigen-binding
(Fab) region of an antibody reversibly or irreversibly. In some embodiments,
the antibody-binding peptide
binds to a variable region of an antibody reversibly or irreversibly. In some
embodiments, the antibody is
Cetuximab. In some embodiments, the antibody-binding peptide has at least 75%
amino acid sequence
identity to the sequence of a disease localization peptide (DLP) (CQFDLSTRRLKC
(SEQ ID NO: 64) or
CQYNLSSRALKC (SEQ ID NO: 65)) or a fragment thereof. In some embodiments, the
antibody-binding
peptide has the sequence of SEQ ID NO: 64.
In some embodiments, the SIRP-a variant is attached to an Fc domain monomer.
In some
embodiments, the SIRP-a variant is attached to a human serum albumin (HSA). In
some embodiments,
the HSA includes amino acid substitution C345 and/or K573P, relative to SEQ ID
NO: 67. In some
embodiments, the HSA has the sequence of
DAHKSEVAHRFKDLGEENFKALVLIAFAQYLQQSPFEDHVKLVNEVTEFAKTCVADESAENCDKSLHTLF
GDKLCIVATLRETYGEMADCCAKQEPERNECFLQHKDDNPNLPRLVRPEVDVMCTAFHDNEETFLKKYL
YEIARRHPYFYAPELLFFAKRYKAAFTECCQAADKAACLLPKLDELRDEGKASSAKQRLKCASLQKFGER
AFKAWAVARLSQRFPKAEFAEVSKLVTDLTKVHTECCHGDLLECADDRADLAKYICENQDSISSKLKECC
EKPLLEKSHCIAEVENDEMPADLPSLAADFVESKDVCKNYAEAKDVFLGMFLYEYARRHPDYSVVLLLRL
AKTYETTLEKCCAAADPHECYAKVFDEFKPLVEEPQNLIKQNCELFEQLGEYKFQNALLVRYTKKVPQVS
TPTLVEVSRNLGKVGSKCCKHPEAKRMPCAEDYLSVVLNQLCVLHEKTPVSDRVTKCCTESLVNRRPCF
SALEVDETYVPKEFNAETFTFHADICTLSEKERQIKKQTALVELVKHKPKATKEQLKAVMDDFAAFVEKCC
KADDKETCFAEEGKKLVAASQAALGL (SEQ ID NO: 68).
In some embodiments, the SIRP-a variant is attached to an albumin-binding
peptide. In some
embodiments, the albumin-binding peptide has the sequence of SEQ ID NO: 2. In
some embodiments,
the SIRP-a variant is attached to a polymer, wherein the polymer is
polyethylene glycol (PEG) chain or
polysialic acid chain.
In some embodiments, the SIRP-a variant is attached to an antibody. In some
embodiments, the
antibody is a tumor-specific antibody. In some embodiments, the antibody
(e.g., a tumor-specific
antibody) is selected from the group consisting of cetuximab, pembrolizumab,
nivolumab, pidilizumab,
MEDI0680, MEDI6469, Ipilimumab, tremelimumab, urelumab, vantictumab,
varlilumab, mogamalizumab,
anti-CD20 antibody, anti-CD19 antibody, anti-CS1 antibody, herceptin,
trastuzumab, and pertuzumab. In
some embodiments, the antibody (e.g., a tumor-specific antibody) may bind to
one or more of the
following: 5T4, AGS-16, ALK1, ANG-2, B7-H3, B7-H4, c-fms, c-Met, CA6, CD123,
CD19, CD20, CD22,
EpCAM, CD30, CD32b, CD33, CD37, CD38, CD40, CD52, CD70, CD74, CD79b, CD98,
CEA,
CEACAM5, CLDN18.2, CLDN6, CS1, CXCR4, DLL-4, EGFR, EGP-1, ENPP3, EphA3, ETBR,
FGER2,
fibronectin, FR-alpha, GCC, GD2, glypican-3, GPNMB, HER-2, HER3, HLA-DR, ICAM-
1, IGF-1R, 1L-3R,
LIV-1, mesothelin, MUC16, MUC1, NaPi2b, Nectin-4, Notch 2, Notch 1, PD-L1, PD-
L2, PDGFR-a, PS,
PSMA, SLTRK6, STEAP1, TEM1, VEGFR, CD25, CD27L, DKK-1, and/or CSF-1R.
In some embodiments, the SIRP-a variant in the SIRP-a variant construct has at
least 80% (e.g.,
at least 85%, 87%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99%)
sequence identity to a
sequence of any one of SEQ ID NOs: 3-12 and 24-34.
In some embodiments, the SIRP-a variant in the SIRP-a variant construct has a
sequence of
EEEX1QX2.1QPDKSVLVAAGETX3TLRCTX.ITSLX5PVGPIQWFRGAGPGRX6LIYNQX7X8GX9FPRVITVSD
X10TX11RNNMDFSIRIGX121TX13ADAGTYYCX14KX15RKGSPDDVEX16KSGAGTELSVRAKPS (SEQ ID
3
CA 02938180 2016-08-04
!
NO: 13), wherein X, is L, I, or V; X2 IS V, L, or, I; X3 is A or V; X4 is A,
I, or L; X5 is I, T, S, or F; X6 is E, V,
or L; X7 is K or R; X8 is E or Q; X9 is H, P, or R; X10 is L, T, or G; X11 is
K or R; X12 is N, A, C, D, E, F,
H, I, K, L, M, P, Q, R, S, T, V, W, or Y; X13 is P, A, C, D, E, F, G, H, I, K,
L, M, N, Q, R, S, T, V, W, or Y;
X14 is V or I; X15 is F, L, or V; and X16 is F or V.
In some embodiments, the SIRP-a variant in the SIRP-a variant construct has a
sequence of
EEGX1QX21QPDKSVSVAAGESX3ILHCTX4TSLX9PVGPIQWFRGAGPGRX6LIYNQX7X9GX9FPRVTTVSD
Xi0TX11RNNMDFSIRIGX121TX13ADAGTYYCX14KX19RKGSPDDVEX16KSGAGTELSVRAKPS (SEQ ID
NO: 16), wherein Xi is L, I, or V; X2 is V, L, or, I; X3 is A or V; X4 is A,
I, or L; X5 is I, T, S, or F; X6 is E, V,
or L; X7 is K or R; X8 is E or Q; Xg is H, P, or R; X10 is L, T, or G; X11 is
K or R; X12 is N, A, C, D, E, F,
H, I, K, L, M, P, 0, R, S, T, V, W, or Y; X13 is P, A, C, D, E, F, G, H, I, K,
L, M, N, Q, R, S, T, V, W, or Y;
X14 iS V or I; X15 is F, L, or V; and X16 is F or V.
In some embodiments, the SIRP-a variant in the SIRP-a variant construct has a
sequence of
EEEX1QX21QPDKFVLVAAGETX3TLRCTX4TSLX9PVGPIQWFRGAGPGRX6LIYNQX7X8GX9FPRVTIVSD
Xi0TX11RNNMDFSIRIGX121TX13ADAGTYYCX14KX19RKGSPDDVEXie,KSGAGTELSVRAKPS (SEQ ID
NO: 17), wherein X1 is L, I, or V; X2 IS V, L, or, I; X3 is A or V; X4 is A,
I, or L; X5 is I, T, 5, or F; X6 is E, V,
or L; X7 is K or R; X8 is E or 0; X9 is H, P, or R; X10 is L, T, or G; X11 is
K or R; X12 is N, A, C, D, E, F, G,
H, I, K, L, M, P, Q, R, S, T, V, W, or Y; X13 is P, A, C, D, E, F, G, H, I, K,
L, M, N, Q, R, S, T, V, W, or Y;
X14 iS V or I, X15 is F, L, or V; and X16 is F or V.
In some embodiments, the SIRP-a variant in the SIRP-a variant construct has a
sequence of
EEEX1QX2IQPDKSVLVAAGETX3TLRCTX4TSLX9PVGPIQWFRGAGPGRX0LIYNQX7X8GX9FPRVTTVSD
X10TX11RNNMDFPIRIGX121TX19ADAGTYYCX14KX19RKGSPDDVEX16KSGAGTELSVRAKPS (SEQ ID
NO: 18), wherein Xi is L, I, or V; X2 is V, L, or, I; X3 is A or V; X4 is A,
I, or L; X5 is I, T, 5, or F; X6 is E, V,
or L; X7 is K or R; X8 is E or Q; X9 is H, P, or R; X10 is L, T, or G; X11 is
K or R; X12 is N, A, C, D, E, F, G,
H, I, K, L, M, P, Q, R, S, T, V, W, or Y; X13 is P, A, C, D, E, F, G, H, I, K,
L, M, N, Q, R, S, 7, V, W, or Y;
X14 iS V or I; X15 is F, L, or V; and X16 is F or V.
In some embodiments, the SIRP-a variant in the SIRP-a variant construct has a
sequence of
EEEX1QX21QPDKSVLVAAGETX3TLRCTX4TSLX9PVGPIQWFRGAGPGRX6LIYNQX7X0GX9FPRVTIVSD
X10TX11RNNMDFSIRISX121TX13ADAGTYYCX14KX19RKGSPDDVEX16KSGAGTELSVRAKPS (SEQ ID
NO: 21), wherein X1 is L, I, or V; X2 is V, L, or, I; X3 is A or V; X.4 is A,
I, or L; X9 is I, T, 5, or F; X6 is E, V,
or L; X7 is K or R; X8 is E or Q; X9 is H, P, or R; Xio is L, T, or G; Xii is
K or R; X12 is N, A, C, D, E, F, G,
H, I, K, L, M, P, Q, R, S, T, V, W, or Y; X13 is P, A, C, D, E, F, G, H, I, K,
L, M, N, Q, R, S, T, V, W, or Y;
X14 iS V or I; X15 is F, L, or V; and X16 is F or V.
In some embodiments, the SIRP-a variant in the SIRP-a variant construct has a
sequence of
EEEX1QX21QPDKSVSVAAGESX3ILHCTX4TSLX9PVGPIQWFRGAGPARX6LIYNQX7X9GX9FPRVTTVSEX
10TX11RENMDFSISISX12ITX13ADAGTYYCX14KX19RKGSPDTEX16KSGAGTELSVRAKPS (SEQ ID NO:
14), wherein Xi is L, I, or V; X2 is V, L, or, I; X3 is A or V; X4 is V, I, or
L; X5 is I, T, S, or F; X6 is E, V, or L;
X7 is K or R; X8 is E or Q; Xg is H, P, or R; X10 is S, T, or G; Xii is K or
R; X12 is N, A, C, D, E, F, G, H, I,
K, L, M, P, Q, R, S, T, V, W, or Y; X13 is P, A, C, D, E, F, G, H, I, K, L, M,
N, Q, R, S, T, V, W, or Y; X14 is
V or I; X15 is F, L, or V; and X16 is F or V;.
In some embodiments, the SIRP-a variant in the SIRP-a variant construct has a
sequence of
EEEX1QX21QPDKSVSVAAGESX3ILLCTX4TSLX9PVGPIQWFRGAGPARX6LIYNQX7X9GX9FPRVTIVSEX
10TX11RENMDFSISISX121TX13ADAGTYYCX14KX19RKGSPDTEX16KSGAGTELSVRAKPS (SEQ ID NO:
4
CA 02938180 2016-08-04
15), wherein X1 is L, I, or V; X2 is V, L, or, I; X3 is A or V; X4 is V, I, or
L; X5 is I, T, S, or F; X6 is E, V, or L;
X7 is K or R; X8 is E or Q; X9 is H, P, or R; X10 is S, T, or G; Xii is K or
R; X12 is N, A, C, D, E, F, G, H, I,
K, L, M, P, Q, R, S, T, V, W, or Y; X13 is P, A, C, D, E, F, G, H, I, K, L, M,
N, Q, R, S, T, V, W, or Y; X14 is
V or I; X15 is F, L, or V; and X16 is F or V.
In some embodiments, the SIRP-a variant in the S1RP-a variant construct has a
sequence of
EEEX1QX21QPDKSVSVAAGESX3ILHCTX4TSLX5PVGPIQWFRGAGPARX6LIYNQX7X8GX0FPRVTIVSEX
10TX11RENMDFSISISX121TX13ADAGTYYCX14KX10RKGSPDTEX16KSGAGTELSVRGKPS (SEQ ID NO:
19), wherein X1 is L, I, or V; X2 is V, L, or, I; X3 is A or V; X4 is V, 1, or
L; X5 is I, T, S, or F; X6 is E, V, or L;
X7 is K or R; X8 is E or Q; X9 is H, P, or R; X10 is S, T, or G; X11 is K or
R; X12 is N, A, C, D, E, F, G, H, I,
K, L, M, P, Q, R, S, T, V, W, or Y; X13 is P, A, C, D, E, F, G, H, I, K, L, M,
N, Q, R, S, T, V, W, or Y; X14 is
V or I; X15 is F, L, or V; and X16 is F or V.
In some embodiments, the SIRP-a variant in the SIRP-a variant construct has a
sequence of
EEEX1QX21QPDKSVSVAAGESX3ILHCTX4TSLX5PVGPIQWERGAGPARX6LIYNQX7X0GX0FPRVITVSEX
10TX11RENMDFSISISX12ITX13ADAGTYYCX1.4KX10RKGSPDTEX16KSGAGTELSVRAKPS (SEQ ID
NO:
22), wherein X1 is L, I, or V; X2 is V, L, or, I; X3 is A or V; X4 is V, I, or
L; X5 is I, T, S, or F; X6 is E, V, or L;
X7 is K or R; X8 is E or Q; X9 is H, P, or R; Xio is S, T, or G; X11 is K or
R; X12 is N, A, C, D, E, F, G, H, I,
K, L, M, P, 0, R, 5, T, V, W, or Y; X13 is P, A, C, D, E, F, G, H, I, K, L, M,
N, 0, R, S, T, V, W, or Y; X14 is
V or 1; X15 is F, L, or V; and X16 is F or V.
In some embodiments, the SIRP-a variant in the SIRP-a variant construct has a
sequence of
EEEX1QX21QPDKSVLVAAGETX3TLRCTX4TSLX5PVGPIQWFRGAGPARX6LIYNQX7X0GX0FPRVTTVSE
Xi0TX11RENMDFSISISX121TX13ADAGTYYCX14KX10RKGSPDTEX16KSGAGTELSVRAKPS (SEQ ID
NO:
20), wherein Xi is L, 1, or V; X2 is V, L, or, I; X3 is A or V; X4 is A, I, or
L; X5 is I, T, S, or F; X6 is E, V, or L;
X7 is K or R; X8 is E or Q; X9 is H, P, or R; X10 is S, T, or G; X11 is K or
R; X12 is N, A, C, D, E, F, G, H, I,
K, L, M, P, Q, R, S, T, V, W, or Y; X13 is P, A, C, D, E, F, G, H, I, K, L, M,
N, Q, R, S, T, V, W, or Y; X14 is
V or I; X15 is F, L, or V; and X16 is F or V.
In some embodiments, the SIRP-a variant in the SIRP-a variant construct has a
sequence of
EEX1X2QX31QPDKXIVX0VAAGEX6X7X0LX0CTX10T5LX11PVGPIQWFRGAGPX12RX13LIYNQX14X13GX16
F
PRVTIVSX17X18TX10RX20NMDFX211X221X23X241TX25ADAGTYYCX26KX27RKGSPDX28X20EX30KSGA
GTEL
SVRX31KPS (SEQ ID NO: 23), wherein X1 is E or G; X2 is L, I, or V; X3 is V, L,
or, I; X4 is S or F; X5 is L or
.. 5; X6 is S or T; X7 is A or V; X8 is I or T; X9 is H or R; Xio is A, V, I,
or L; X11 is 1, T, S, or F; X12 is A or G;
X13 is E, V. or L; X14 is K or R; X15 is E or Q; X16 is H, P, or R; X17 is D
or E; X18 is S, L, T, or G; X19 is K or
R; X20 is E or N; X21 is S or P; X22 is S or R; X23 is S or G; X24 is N, A, C,
D, E, F, G, H, I, K, L, M, P, Q, R,
S, T, V, W, or Y; X25 is P, A, C, D, E, F, G, H, I, K, L, M, N, 0, R, S, T, V,
W, or Y; X26 is V or I; X27 is F, L,
V; X28 is D or absent; X29 is T or V; X30 is F or V; and X31 is A or G.
In some embodiments, the SIRP-a variant in the SIRP-a variant construct has at
least 80% (e.g.,
at least 85%, 87%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99%)
sequence identity to a
sequence of any one of SEQ ID NOs: 13-23.
In some embodiments, a SIRP-a variant in the SIRP-a variant construct does not
include the
sequence of any one of SEQ ID NOs: 3-12 and 24-34.
In some embodiments, the SIRP-a variant in the SIRP-a variant construct
includes one or more
substitutions of amino acid residues with histidine residues. In some
embodiments, the one or more
substitutions of amino acid residues with histidine residues are located at
one or more of the following
5
= CA 02938180 2016-08-04
amino acid positions: 29, 30, 31, 32, 33, 34, 35, 52, 53, 54, 66, 67, 68, 69,
74, 93, 96, 97, 98, 100, 4,6,
27, 36, 39, 47, 48, 49, 50, 57, 60, 72, 74, 76, 92, 94, 103, relative to a
sequence of any one of SEQ ID
NOs: 3-12.
In some embodiments, the SIRP-a variant construct binds with at least two, at
least four, or at
least six fold higher affinity to CD47 on diseased cells or at a diseased site
than on non-diseased cells.
In some embodiments, the SIRP-a variant construct binds with at least two, at
least four, or at
least six fold higher affinity to CD47 under acidic pH than under neutral pH.
In some embodiments, the SIRP-a variant construct binds with at least two, at
least four, or at
least six fold higher affinity to CD47 under hypoxic condition than under
physiological condition.
In some embodiments, the diseased cell is a cancer cell of a cancer disease.
In some embodiments, the acidic pH is a pH between about 4 to about 7.
In another aspect, the invention features a nucleic acid molecule encoding a
SIRP-a variant
construct described herein.
In another aspect, the invention features a vector including the nucleic acid
molecule encoding a
SIRP-a variant construct described herein.
In another aspect, the invention features a host cell that expresses a SIRP-a
variant construct
described herein, wherein the host cell includes a nucleic acid molecule
encoding a S1RP-a variant
construct described herein or a vector including the nucleic acid molecule,
wherein the nucleic acid
molecule or vector is expressed in the host cell.
In another aspect, the invention features a method of preparing a SIRP-a
variant construct
described herein, wherein the method includes: a) providing a host cell
including a nucleic acid molecule
of encoding a SIRP-a variant construct described herein or a vector including
the nucleic acid molecule;
b) expressing the nucleic acid molecule or vector in the host cell under
conditions that allow for the
formation of the SIRP-a variant construct; and c) recovering the SIRP-a
variant construct.
In another aspect, the invention features a pharmaceutical composition
including a therapeutically
effective amount of a SIRP-a variant construct described herein. In some
embodiments, the
pharmaceutical composition includes one or more pharmaceutically acceptable
carriers or excipients.
In another aspect, the invention features a method of increasing phagocytosis
of a target cell in a
subject including administering to the subject a SIRP-a variant construct
described herein or a
pharmaceutical composition including a therapeutically effective amount of a
SIRP-a variant construct
described herein. In some embodiments, the target cell is a cancer cell.
In another aspect, the invention features a method of eliminating regulatory T-
cells in a subject
including administering to the subject a SIRP-a variant construct described
herein or a pharmaceutical
composition including a therapeutically effective amount of a SIRP-a variant
construct described herein.
In another aspect, the invention features a method for killing a cancer cell,
the method includes
contacting the cancer cell with a SIRP-a variant construct described herein or
the pharmaceutical
composition including a therapeutically effective amount of a SIRP-a variant
construct described herein.
In another aspect, the invention features a method for treating a disease
associated with SIRP-a
and/or CD47 activity in a subject, the method includes administering to the
subject a therapeutically
effective amount of the SIRP-a variant construct described herein or the
pharmaceutical composition
including a therapeutically effective amount of a SIRP-a variant construct
described herein.
6
CA 02938180 2016-08-04
In another aspect, the invention features a method of treating a disease
associated with SIRP-a
and/or CD47 activity in a subject, the method includes: (a) determining the
amino acid sequences of
SIRP-a of the subject; and (b) administering to the subject a therapeutically
effective amount of a SIRP-a
variant construct described herein; wherein the SIRP-a variant in the SIR P-a
variant construct has the
.. same amino acid sequence as that of a SIRP-a of the subject.
In another aspect, the invention features a method of treating a disease
associated with SIRP-a
and/or CD47 activity in a subject, the method includes: (a) determining the
amino acid sequences of
SIRP-a of the subject; and (b) administering to the subject a therapeutically
effective amount of a SIRP-a
variant construct described herein; wherein the SIRP-a variant in the SIRP-a
variant construct has
minimal immunogenicity in the subject.
In another aspect, the invention features a method of treating a disease
associated with SIRP-a
and/or CD47 activity in a subject, the method includes: administering to the
subject a SIRP-a variant
construct described herein, wherein the SIRP-a variant construct
preferentially binds CD47 on diseased
cells or at a diseased site over CD47 on non-diseased cells.
In some embodiments, the disease is cancer. In some embodiments, the cancer is
selected from
solid tumor cancer, hematological cancer, acute myeloid leukemia, chronic
lymphocytic leukemia, chronic
myeloid leukemia, acute lymphoblastic leukemia, non-Hodgkin lymphoma, Hodgkin
lymphoma, multiple
myeloma, bladder cancer, pancreatic cancer, cervical cancer, endometrial
cancer, lung cancer, bronchus
cancer, liver cancer, ovarian cancer, colon and rectal cancer, stomach cancer,
gastric cancer, gallbladder
cancer, gastrointestinal stromal tumor cancer, thyroid cancer, head and neck
cancer, oropharyngeal
cancer, esophageal cancer, melanoma, non-melanoma skin cancer, Merkel cell
carcinoma, virally
induced cancer, neuroblastoma, breast cancer, prostate cancer, renal cancer,
renal cell cancer, renal
pelvis cancer, leukemia, lymphoma, sarcoma, glioma, brain tumor, and
carcinoma. In some
embodiments, the cancer is a solid tumor cancer. In some embodiments, the
cancer is a hematological
cancer.
In some embodiments, the disease is an immunological disease. In some
embodiments, the
immunological disease is an autoimmune disease or an inflammatory disease. In
some embodiments,
the autoimmune or inflammatory disease is multiple sclerosis, rheumatoid
arthritis, a
spondyloarthropathy, systemic lupus erythematosus, an antibody-mediated
inflammatory or autoimmune
disease, graft versus host disease, sepsis, diabetes, psoriasis,
atherosclerosis, Sjogren's syndrome,
progressive systemic sclerosis, scleroderma, acute coronary syndrome, ischemic
reperfusion, Crohn's
Disease, endometriosis, glornerulonephritis, myasthenia gravis, idiopathic
pulmonary fibrosis, asthma,
acute respiratory distress syndrome (ARDS), vasculitis, or inflammatory
autoimmune myositis.
In another aspect, the invention features a method of increasing hematopoietic
stem cell
engraftment in a subject including modulating the interaction between SIRP-a
and CD47 in the subject by
administering to the subject a SIRP-a variant described herein or a
pharmaceutical composition including
a therapeutically effective amount of a SIRP-a variant described herein.
In another aspect, the invention features a method of altering an immune
response in a subject
including administering the subject a SIRP-a variant construct described
herein or a pharmaceutical
composition including a therapeutically effective amount of a SIRP-a variant
construct described herein,
thereby altering the immune response in the subject. In some embodiments, the
immune response
includes suppressing the immune response.
7
CA 02938180 2016-08-04
In some embodiments, the subject is a mammal, preferably, the mammal is a
human.
Definitions
As used herein, the term "diseased cells" and "diseased tissue" refer to, for
example, cancer cells
and tissue. In particular, the cancer may be a solid tumor cancer or a
hematological cancer. For
example, if the cancer is a solid tumor cancer, the diseased cells are the
cells of the solid tumor.
Diseased cells are often living under conditions characteristic of a diseased
site, such as acidic pH and
hypoxia. "Diseased cells" and "diseased tissue" can also be associated with
other diseases including, but
not limited to, cancer. "Diseased cells" and "diseased tissue" can also be
associated with an
immunological disease or disorder, a cardiovascular disease or disorder, a
metabolic disease or disorder,
or a proliferative disease or disorder. An immunological disorder includes an
inflammatory disease or
disorder and an autoimmune disease or disorder.
As used herein, the term "non-diseased cells" refers to normal, healthy cells
of the body. Non-
diseased cells often live under physiological conditions, such as neutral pH
and adequate oxygen
concentration that maintain normal metabolism and regulatory functions of the
cells.
As used herein, the term "diseased site" refers to the location or area
proximal to the location of
the disease in the body. For example, if the disease is solid tumor cancer
located in the liver, then
diseased site is the site of the tumor in the liver and areas close to the
tumor in the liver. Cells at a
diseased site may include diseased cells as well as cells that support the
disease at the diseased site.
For example, if the diseased site is the site of a tumor, cells at the site of
the tumor include both diseased
cells (e.g., tumor cells) and cells supporting tumor growth at the site of the
tumor. Similarly, the term
"cancer site" refers to the location of the cancer in the body.
As used herein, the term "SIRP-a D1 domain" or "D1 domain" refers to the
membrane distal,
extracellular domain of SIRP-a. The SIRP-a D1 domain is located at the N-
terminus of a full-length, wild-
type SIRP-a and mediates binding to CD47. Amino acid sequences of D1 domains
are shown in Table 1.
As used herein, the term "SIRP-a D2 domain" or "D2 domain" refers to the
second extracellular
domain of SIRP-a. The SIRP-a D2 domain includes approximately amino acids 119
to 220 of a full-
length, wild-type S1RP-a.
As used herein, the term "SIRP-a D3 domain" or "D3 domain" refers to the third
extracellular
domain of SIRP-a. The SIRP-a D3 domain includes approximately amino acids 221
to 320 of a full-
length, wild-type SIRP-a.
As used herein, the term "SIRP-a polypeptide" refers to a wild-type SIRP-a as
well as a SIRP-a
variant, as each term is defined and described herein.
As used herein, the term "SIRP-a variant" refers to a polypeptide containing a
SIRP-a D1 domain,
or a CD47-binding portion of a full-length SIRP-a. In some embodiments, the
SIRP-a variant has at least
80% (e.g., at least 85%, 87%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or
99%.) sequence
identity to a sequence of any one of SEQ ID NOs: 3-12 and 24-34. In some
embodiments, a SIRP-a
variant has higher affinity to CD47 than a wild-type S1RP-a. In some
embodiments, a S1RP-a variant
contains a portion of wild-type human SIRP-a (preferably a CD47-binding
portion of the wild-type SIRP-a)
and/or has one or more amino acid substitutions. For example, a S1RP-a variant
may contain
substitutions of one or more (e.g., one, two, three, four, five, six, seven,
eight, nine, ten, etc, with a
maximum of 20) amino acid residues relative to a wild-type SIRP-a. For
example, a SIRP-a variant may
8
CA 02938180 2016-08-04
contain substitutions of one or more (e.g., one, two, three, four, five, six,
seven, eight, nine, ten, etc, with
a maximum of 20) amino acid residues with histidine residues. In some
embodiments, SIRP-a variants
have a portion that has at least 80% (e.g., at least 85%, 87%, 90%, 91%, 92%,
93%, 94%, 95%, 96%,
97%, 98%, or 99%) amino acid sequence identity to a sequence of wild-type
human SIRP-a or to any of
the SIRP-a variants described herein (e.g., to a sequence of a CD47-binding
portion of wild-type human
SIRP-a). A CD47-binding portion of wild-type SIRP-a includes the D1 domain of
the wild-type SIRP-a (a
sequence of any one of SEQ ID NOs: 3-12).
As used herein, the term "SIRP-a variant construct" refers to a polypeptide
containing a SIRP-a
variant attached to, e.g., a blocking peptide, an Fc domain monomer, an HSA,
an albumin-binding
peptide, a polymer, an antibody-binding peptide, an antibody. In some
embodiments, a SIRP-a variant
construct has preferential activity at a diseased site. In some embodiments,
SIRP-a variant constructs
have preferential activity at a diseased site and include a SIRP-a variant
having a portion that has at least
80% (e.g., at least 85%, 87%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or
99%) amino acid
sequence identity to a sequence of wild-type human SIRP-a or to any of the
SIRP-a variants described
herein (e.g., to a sequence of a CD47-binding portion of wild-type human SIRP-
a).
As used herein, the term "percent (/0) identity" refers to the percentage of
amino acid (or nucleic
acid) residues of a candidate sequence, e.g., a SIRP-a variant, that are
identical to the amino acid (or
nucleic acid) residues of a reference sequence, e.g., a wild-type human SIRP-a
or a CD47-binding
portion thereof, after aligning the sequences and introducing gaps, if
necessary, to achieve the maximum
percent identity (i.e., gaps can be introduced in one or both of the candidate
and reference sequences for
optimal alignment and non-homologous sequences can be disregarded for
comparison purposes).
Alignment for purposes of determining percent identity can be achieved in
various ways that are within
the skill in the art, for instance, using publicly available computer software
such as BLAST, ALIGN, or
Megalign (DNASTAR) software. Those skilled in the art can determine
appropriate parameters for
measuring alignment, including any algorithms needed to achieve maximal
alignment over the full length
of the sequences being compared. In some embodiments, the percent amino acid
(or nucleic acid)
sequence identity of a given candidate sequence to, with, or against a given
reference sequence (which
can alternatively be phrased as a given candidate sequence that has or
includes a certain percent amino
acid (or nucleic acid) sequence identity to, with, or against a given
reference sequence) is calculated as
follows:
100 x (fraction of A/B)
where A is the number of amino acid (or nucleic acid) residues scored as
identical in the
alignment of the candidate sequence and the reference sequence, and where B is
the total number of
amino acid (or nucleic acid) residues in the reference sequence. In some
embodiments where the length
of the candidate sequence does not equal to the length of the reference
sequence, the percent amino
acid (or nucleic acid) sequence identity of the candidate sequence to the
reference sequence would not
equal to the percent amino acid (or nucleic acid) sequence identity of the
reference sequence to the
candidate sequence.
In particular embodiments, a reference sequence aligned for comparison with a
candidate
sequence may show that the candidate sequence exhibits from 50% to 100%
identity across the full
length of the candidate sequence or a selected portion of contiguous amino
acid (or nucleic acid)
residues of the candidate sequence. The length of the candidate sequence
aligned for comparison
9
CA 02938180 2016-08-04
purpose is at least 30%, e.g., at least 40%, e.g., at least 50%, 60%, 70%,
80%, 90%, or 100% of the
length of the reference sequence. When a position in the candidate sequence is
occupied by the same
amino acid (or nucleic acid) residue as the corresponding position in the
reference sequence, then the
molecules are identical at that position.
As used herein, the term "tumor-associated protease" or "tumor enzyme" refers
to an enzyme,
e.g., a protease, that is present at an increased level in a cancer, e.g., a
solid tumor cancer. In some
embodiments, the tumor-associated protease may cleave a cleavable linker.
As used herein, the term "blocking peptide" refers to a peptide that can bind
to a SIRP-a variant
and block or "mask" the CD47-binding portion of the SIRP-a variant. In a SIRP-
a variant construct, the
blocking peptide may be attached to a SIRP-a variant by way of a linker that
is optionally cleavable, and
optionally one or more spacers. The blocking peptide may be coupled via non-
covalent bonds to the
SIRP-a variant and cleaved at a diseased site or diseased cell. In some
embodiments, the blocking
peptide may bind to a wild-type SIRP-a at the diseased site or diseased cell.
A blocking peptide can be
used to reduce or minimize binding of the SIRP-a variant with wild-type CD47
under normal physiological
conditions or at a non-diseased site. In some embodiments, the blocking
peptide has higher binding
affinity to a wild-type SIRP-a than a SIRP-a variant. The blocking peptide may
dissociate from the SIRP-
a variant to bind to a wild-type SIRP-a at, for e.g., a diseased site or under
non-physiological conditions.
An example of a blocking peptide is a CD47-based blocking peptide, which is a
peptide derived from
CD47 or a fragment thereof. In some embodiments, a CD47-based blocking peptide
is the extracellular,
SIRP-a binding portion of CD47 (i.e., the IgSF domain of CD47). In some
embodiments, a CD47-based
blocking peptide includes one or more amino acid substitutions, additions,
and/or deletions relative to the
wild-type CD47.
As used herein, the term "cleavable linker" refers to a linker between two
portions of a SIRP-a
variant construct. In some embodiments, a cleavable linker may covalently
attach a blocking peptide to a
SIRP-a variant to block binding of the SIRP-a variant to CD47 under
physiological conditions. In some
embodiments, a cleavable linker may be installed within a blocking peptide,
which may be non-covalently
associated with the SIRP-a variant to block binding of the SIRP-a variant to
CD47 under physiological
conditions. A cleavable linker may be cleaved under certain conditions. If the
cleavable linker is within a
blocking peptide, cleavage of the linker would inactivate the blocking
peptide. The cleavable linker
.. contains a moiety that acts to cleave or induce cleavage of the linker
under conditions characteristic of a
diseased site, such as a cancer site, e.g., inside a solid tumor. The
cleavable linker is stable under
healthy physiological conditions (e.g., neutral pH and adequate oxygen
concentration). The moiety may
be a pH-sensitive chemical functional group (e.g., acetals, ketals,
thiomaleamates, hydrazones, disulfide
bonds) capable of being hydrolyzed under acidic pH. The moiety may also be a
hypoxia-sensitive
.. chemical functional group (e.g., quinones, N-oxides, and heteroaromatic
nitro groups) or amino acid
capable of being reduced under hypoxic condition. The moiety in the cleavable
linker may also be a
protein substrate capable of being recognized and cleaved by a tumor-
associated protease, enzyme, or
peptidase.
As used herein, the term "spacer" refers to a covalent or non-covalent linkage
between two
portions of a SIRP-a variant construct, such as the linker (e.g., cleavable
linker) and the SIRP-a variant,
or the antibody-binding peptide and the SIRP-a variant. Preferably, the spacer
is a covalent linkage. A
spacer can be a simple chemical bond, e.g., an amide bond, or an amino acid
sequence (e.g., a 3-200
CA 02938180 2016-08-04
amino acid sequence). An amino acid spacer is part of the primary sequence of
a polypeptide
joined to the spaced polypeptides or polypeptide domains via the polypeptide
backbone). A spacer
provides space and/or flexibility between the two portions. A spacer is stable
under physiological
conditions (e.g., neutral pH and adequate oxygen concentration) as well as
under conditions
characteristic of a diseased site, e.g., acidic pH and hypoxia. A spacer is
stable at a diseased site, such
as a cancer site, e.g., inside a tumor. Descriptions of spacers are provided
in detail further herein.
As used herein, the term "antibody" refers to intact antibodies, antibody
fragments, provided that
they exhibit the desired biological activity, monoclonal antibodies,
polyclonal antibodies, monospecific
antibodies, and multispecific antibodies (e.g., bispecific antibodies) formed
from at least two intact
antibodies. Preferably, the antibody is specific to a diseased cell, e.g., a
tumor cell. For example, the
antibody may specifically bind to a cell surface protein on a diseased cell,
e.g., a tumor cell.
As used herein, the term "albumin-binding peptide" refers to an amino acid
sequence of 12 to 16
amino acids that has affinity for and functions to bind serum albumin. An
albumin-binding peptide can be
of different origins, e.g., human, mouse, or rat. In some embodiments of the
present invention, a SIRP-a
variant construct may include an albumin-binding peptide that is fused to the
C-terminus of the SIRP-a
variant to increase the serum half-life of the SIRP-a variant. An albumin-
binding peptide can be fused,
either directly or through a spacer, to the SIRP-a variant.
As used herein, the term "human serum albumin (HSA)" refers to the albumin
protein present in
human blood plasma. Human serum albumin is the most abundant protein in the
blood. It constitutes
about half of the blood serum protein. In some embodiments, a human serum
albumin has the sequence
of amino acids 25-609 (SEQ ID NO: 67) of UniProt ID NO: P02768. In some
embodiments, a human
serum albumin further contains C34S relative to the sequence of SEQ ID NO: 67.
As used herein, the term "Fc domain monomer" refers to a polypeptide chain
that includes
second and third antibody constant domains (CH2 and CH3). In some embodiment,
the Fc domain
monomer also includes a hinge domain. The Fc domain monomer can be any
immunoglobulin antibody
isotype, including IgG, IgE, IgM, IgA, or IgD. Additionally, the Fe domain
monomer can be an IgG
subtype (e.g., IgG1, IgG2a, IgG2b, IgG3, or IgG4). An Fc domain monomer does
not include any portion
of an immunoglobulin that is capable of acting as an antigen-recognition
region, e.g., a variable domain or
a complementarity determining region (CDR). Fc domain monomers can include as
many as ten
changes from a wild-type Fc domain monomer sequence (e.g., 1-10, 1-8, 1-6, 1-4
amino acid
substitutions, additions, or deletions) that alter the interaction between an
Fc domain and an Fc receptor.
Examples of suitable changes are known in the art.
As used herein, the term "Fc domain" refers to a dimer of two Fc domain
monomers. In the wild-
type Fc domain, the two Fc domain monomers dimerize by the interaction between
the two CH3 antibody
constant domains, as well as one or more disulfide bonds that form between the
hinge domains of the two
dimerizing Fc domain monomers. In some embodiments, an Fc domain may be
mutated to lack effector
functions, typical of a "dead Fc domain." In certain embodiments, each of the
Fc domain monomers in an
Fc domain includes amino acid substitutions in the CH2 antibody constant
domain to reduce the
interaction or binding between the Fc domain and an Fcy receptor.
As used herein, the term "affinity" or "binding affinity" refers to the
strength of the binding
interaction between two molecules. Generally, binding affinity refers to the
strength of the sum total of
non-covalent interactions between a molecule and its binding partner, such as
a SIRP-a variant and
11
CA 02938180 2016-08-04
CD47. Unless indicated otherwise, binding affinity refers to intrinsic binding
affinity, which reflects a 1:1
interaction between members of a binding pair. The binding affinity between
two molecules is commonly
described by the dissociation constant (KD) or the affinity constant (KA). Two
molecules that have low
binding affinity for each other generally bind slowly, tend to dissociate
easily, and exhibit a large Kr). Two
molecules that have high affinity for each other generally bind readily, tend
to remain bound longer, and
exhibit a small Ko. The KID of two interacting molecules may be determined
using methods and
techniques well known in the art, e.g., surface plasmon resonance. KO is
calculated as the ratio of k0/k0.
As used herein, the term "host cell" refers to a vehicle that includes the
necessary cellular
components, e.g., organelles, needed to express proteins from their
corresponding nucleic acids. The
nucleic acids are typically included in nucleic acid vectors that can be
introduced into the host cell by
conventional techniques known in the art (e.g., transformation, transfection,
electroporation, calcium
phosphate precipitation, direct microinjection, etc.). A host cell may be a
prokaryotic cell, e.g., a bacterial
cell, or a eukaryotic cell, e.g., a mammalian cell (e.g., a CHO cell). As
described herein, a host cell is
used to express one or more SIRP-a variant constructs.
As used herein, the term "pharmaceutical composition" refers to a medicinal or
pharmaceutical
formulation that contains an active ingredient as well as excipients and
diluents to enable the active
ingredient suitable for the method of administration. The pharmaceutical
composition of the present
invention includes pharmaceutically acceptable components that are compatible
with the SIRP-a variant
construct. The pharmaceutical composition may be in tablet or capsule form for
oral administration or in
aqueous form for intravenous or subcutaneous administration.
As used herein, the term "disease associated with SIRP-a and/or CD47 activity"
refers to any
disease or disorder that is caused by and/or related to SIRP-a and/or CD47
activity. For example, any
disease or disorder that is caused by and/or related to the increase and/or
decrease of SIRP-a and/or
CD47 activity. Examples of diseases associated with SIRP-a and/or CD47
activity include, but are not
limited to, cancers and immunological diseases (e.g., autoimmune diseases and
inflammatory diseases).
As used herein, the term "therapeutically effective amount" refers an amount
of a SIRP-a variant
construct of the invention or a pharmaceutical composition containing a SIRP-a
variant construct of the
invention effective in achieving the desired therapeutic effect in treating a
patient having a disease, such
as a cancer, e.g., solid tumor or hematological cancer. In particular, the
therapeutic effective amount of
the SIRP-a variant construct avoids adverse side effects.
As used herein, the term "optimized affinity" or "optimized binding affinity"
refers to an optimized
strength of the binding interaction between a SIRP-a variant and CD47. In some
embodiments, the
SIRP-a variant construct binds primarily or with higher affinity to CD47 on
cells at a diseased site (i.e.,
cancer cells) and does not substantially bind or binds with lower affinity to
CD47 on cells at a non-
diseased site (i.e., non-cancer cells). The binding affinity between the SIRP-
a variant and CD47 is
optimized such that the interaction does not cause clinically relevant
toxicity. In some embodiments, in
order to achieve an optimized binding affinity between the SIRP-a variant and
CD47, the SIRP-a variant
may be developed to have a lower binding affinity to CD47 than which is
maximally achievable.
As used herein, the term "immunogenicity" refers to the property of a protein
(e.g., a therapeutic
protein) which causes an immune response in the host as though it is a foreign
antigen. The
immunogenicity of a protein can be assayed in vitro in a variety of different
ways, in particular through in
12
CA 02938180 2016-08-04
vitro T-cell proliferation assays (see, e.g., Jawa et al., Clinical Immunology
149:534-555, 2013), some of
which are commercially available (see, e.g., immunogenicity assay services
offered by Proimmune).
As used herein, the term "minimal immunogenicity" refers to an immunogenicity
of a protein (e.g.,
a therapeutic protein) that has been modified, i.e., through amino acid
substitutions, to be lower (e.g., at
least 10%, 25%, 50%, or 100% lower) than what could have been before the amino
acid substitutions are
introduced. A protein (e.g., a therapeutic protein) is modified to have
minimal immunogenicity means it
causes no or very little host immune response even though it is a foreign
antigen.
As used herein, the term "optimized pharmacokinetics" refers to that the
parameters that are
generally associated with the pharmacokinetics of a protein are improved and
modified to produce an
optimized protein for in vitro and/or in vivo use. Parameters that are
associated with the
pharmacokinetics of a protein are well-known to a skilled artisan, including,
for examples, KD, valency,
and half-life. In the present invention, the pharmacokinetics of a SIRP-a
variant construct of the invention
are optimized for its interaction with CD47 for use in a therapeutic context.
DESCRIPTION OF THE DRAWINGS
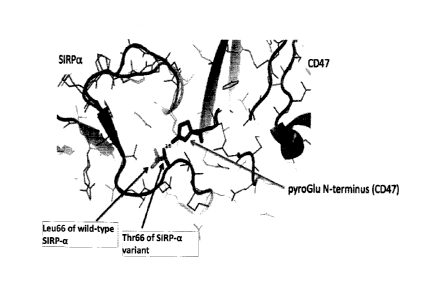
FIG. 1 shows a portion of the co-crystalized structure of CD47:SIRP-a (PDB:
4KJY, 4CMM), the
N-terminus of CD47 exists as a pyro-glutamate and makes hydrogen bonding
interactions with Thr66 of a
SIRP-a variant or Leu66 of a wild-type SIRP-a.
FIG. 2 shows computation models of the interaction site between CD47 having
T102Q and a wild-
type SIRP-a having A27 (left) and the interaction site between CD47 having
T102Q and a SIRP-a variant
having 127.
FIG. 3 shows an SDS-PAGE of SIRP-a variant constructs (SEQ ID NOs: 48-56).
FIG. 4A shows an SDS-PAGE of SIRP-a variant construct (SEQ ID NO: 54) after in
vitro
cleavage with uPA and matriptase.
FIG. 4B shows an SDS-PAGE of SIRP-a variant construct (SEQ ID NO: 54) after in
vitro
cleavage with different amounts of matriptase.
FIG. 4C shows an SDS-PAGE of various SIRP-a variant constructs (SEQ ID NOs: 57-
63) after in
vitro cleavage with matriptase.
FIG. 5A shows a bar graph illustrating the different binding affinities of
various SIRP-a variant
constructs (SEQ ID NOs: 48-55) to CD47 before and after in vitro cleavage with
matriptase.
FIG. 5B shows a bar graph illustrating the different binding affinities of
various SIR P- variant
constructs (SEQ ID NOs: 52-63) and the SIRP-a variant (SEQ ID NO: 31) to CD47
before and after in
vitro cleavage with matriptase.
FIG. 6 shows a sensorgram demonstrating that a SIRP-a variant construct (SEQ
ID NO: 66) can
bind Cetuximab and CD47 simultaneously.
FIG. 7A shows a scheme of the quaternary complex containing EGFR, Cetuximab, a
SIRP-a
variant construct (SEQ ID NO: 66), and CD47.
FIG. 7B shows a sensorgram demonstrating the formation of the quaternary
complex shown in
FIG. 7A.
FIG. 7C is an image of the sensorgram shown in FIG. 7B.
FIG. 8 is a scatter plot showing phagocytosis induced by the SIRP-a variant
construct (SEQ ID
NO: 66) and the SIRP-a variant (SEQ ID NO: 31).
13
= CA 02938180 2016-08-04
DETAILED DESCRIPTION OF THE INVENTION
The invention features signal-regulatory protein a (SIRP-a) variant constructs
having preferential
activity at a diseased site (e.g., at the site of a tumor than at a non-
diseased site). In certain
embodiments, the SIRP-a variant constructs have higher binding affinity to
CD47 on diseased cells (e.g.,
tumor cells), cells. In some embodiments, the SIRP-a variants may contain one
or more amino acid
substitutions. In some embodiments, the amino acids may be substituted with
histidine residues. In
some embodiments, the amino acids may be substituted with other non-histidine
amino acid residues. In
some embodiments, the SIRP-a variant constructs bind with higher affinity to
CD47 on diseased cells or
at a diseased site than on non-diseased cells and under conditions
characteristic of a diseased site, such
as a cancer site, e.g., at the site of or inside a tumor. In some embodiments,
the SIRP-a variant
constructs bind with higher affinity to CD47 under acidic pH (e.g., less than
around pH 7) and/or under
hypoxic condition than under physiological conditions. In some embodiments,
the SIRP-a variant
constructs include a SIRP-a variant and a blocking peptide; the SIRP-a variant
is prevented from binding
to CD47 by the blocking peptide unless under conditions characteristic of a
diseased site. In some
embodiments, the SIRP-a variants are fused to an Fc domain monomer, a human
serum albumin (HSA),
an albumin-binding peptide, or a polymer (e.g., a polyethylene glycol (PEG)
polymer). In some
embodiments, the SIRP-a variant constructs have their immunogenicity,
affinity, and/or pharmacokinetics
optimized for use in a therapeutic context. In some embodiments, the SIRP-a
variant constructs are
preferentially targeted to diseased sites, e.g., a tumor, by way of a
targeting moiety, e.g., a target-specific
antibody. The invention features methods and pharmaceutical compositions
containing SIRP-a variant
constructs to treat various diseases, such as cancer, preferably solid tumor
or hematological cancer, as
well as methods of killing cancer cells and methods of manufacturing SIRP-a
variant constructs and
pharmaceutical compositions containing such SIRP-a variant constructs.
In some embodiments, a SIRP-a variant construct includes a SIRP-a variant
attached to a
blocking peptide. In some embodiments, the preferential binding of the SIRP-a
variant in the SIRP-a
variant construct to CD47 on diseased cells or diseased sites may be obtained
by attaching the block
peptide to the SIRP-a variant by use of a cleavable linker, which is cleaved
at the diseased cells or
diseased sites. In some embodiments, the preferential binding of the SIRP-a
variant in the SIRP-a
variant construct to CD47 on diseased cells or diseased sites may be obtained
by attaching the block
peptide to the SIRP-a variant, wherein the blocking peptide can be detatched
or simply dissociated from
the SIRP-a variant at the diseased cells or diseased sites.
I. SIRP-a variants
There exist at least ten natural variants of wild-type human SIRP-a. The amino
acid sequences
of the D1 domains of the ten wild-type human SIRP-a variants are shown in SEQ
ID NOs: 3-12 (see
Table 1). In some embodiments, the SIRP-a variant has at least 80% (e.g., at
least 85%, 87%, 90%,
91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99%.) sequence identity to a
sequence of any one of
SEQ ID NOs: 3-12. Table 2 lists possible amino acid substitutions in each D1
domain variant (SEQ ID
NOs: 13-23). In some embodiments, the SIRP-a variant binds with an optimized
binding affinity to CD47.
In some embodiments, the SIRP-a variant construct including a SIRP-a variant
binds primarily or with
higher affinity to CD47 on cancer cells and does not substantially bind or
binds with lower affinity to CD47
14
= CA 02938180 2016-08-04
on non-cancer cells. In some embodiments, the binding affinity between the
SIRP-a variant construct and
CD47 is optimized such that the interaction does not cause clinically relevant
toxicity. In some
embodiments, the SIRP-a variant construct has minimal immunogenicity. In some
embodiments, the
SIRP-a variant has the same amino acids as that of the SIRP-a polypeptide in a
biological sample of the
subject, except for the amino acids changes introduced to increase affinity of
the SIRP-a variant.
Techniques and methods for generating SIRP-a variants and determining their
binding affinities to CD47
are described in detail further herein.
Table 2 lists specific amino acid substitutions in a SIRP-a variant, relative
to each D1 domain
variant sequence. A SIRP-a variant may include one or more (e.g., one, two,
three, four, five, six, seven,
eight, nine, ten) of the substitutions listed in Table 2. In some embodiments,
a SIRP-a variant includes at
most ten amino acid substitutions relative to a wild-type D1 domain. In some
embodiments, a SIRP-a
variant includes at most seven amino acid substitutions relative to a wild-
type D1 domain.
In some embodiments, a SIRP-a variant is a chimeric SIRP-a variant that
includes a portion of
two or more wild-type D1 domain variants (e.g., a portion of one wild-type D1
domain variant and a
portion of another wild-type D1 domain variant). In some embodiments, a
chimeric SIRP-a variant
includes at least two portions (e.g., three, four, five, etc.) of wild-type D1
domain variants, wherein each of
the portions is from a different wild-type D1 domain variant. In some
embodiments, a chimeric SIRP-a
variant further includes one or more amino acid substitutions listed in Table
2. In some embodiments, the
SIRP-a variant has at least 80% (e.g., at least 85%, 87%, 90%, 91%, 92%, 93%,
94%, 95%, 96%, 97%,
98%, or 99%.) sequence identity to a sequence of any one of SEQ ID NOs: 24-34
in Table 3.
Table 1. Sequences of wild-type SIRP-a D1 domains
Wild-type D1 domain
EEELQVIQPDKSVLVAAGETATLRCTATSL I PVGPIQW FRGAGPGREL I YNQKEGHFPRV
variant 1
(SEQ ID NO: 3) TTVSDLTKRNNMDFS IRI GNI
TPADAGTYYCVKFRKGSPDDVEFKSGAGTELSVRAKPS
Wild-type D1 domain
EEELQVIQPDKSVSVAAGESAILHCTVTSLIPVGPIQWFRGAGPARELIYNQKEGHFPRV
variant 2 T TV SESTKRENMDFSI S I SN I
TPADAGTYYCVKFRKGSPDTEFKSGAGTELSVRAKPS
(SEQ ID NO: 4)
Wild-type D1 domain
EEELQVI QPDKSVSVAAGE SAI LLCTVT SL I PVGP I QW ERGAGPAREL TYNQKEGHEPRV
variant 3
(SEQ ID NO: 5) TTVSESTKRENMDESI S I SN
TPADAGTYYCVKFRKGSPDTEEKSGAGTELSVRAKPS
Wild-type D1 domain
EEGLQVIQPDKSVSVAAGESAILHCTATSL PVGP IQWERGAGPGREL I YNQKEGHEPRV
variant 4
NO: 6) TTVSDLTKRNNMDFS IRIGN I
TPADAGTYYCVKERKGSPDDVEEKSCAGTELSVRAKPS
(SEQ ID
Wild-type D1 domain
EEELQVI QPDKEVLVAAGETATLRCTAT SL I PVG P I QWERGAGPGREL I YNQKEGHEPRV
variant 5
NO: 7) T TVSDLTKRNNMDFS I RIGNI
TPADAGTYYCVKFRKGSPDDVEFKSGAGTELSVRAKPS
(SEQ ID
Wild-type D1 domain EEELQV I QPDKSVLVAAGETATLRCTATSL IPVGP I QW FRGAGPGRELI
YNQKEGHFPRV
variant 6 TTVSDLTKRNNMDFPIRI
GNITPADAGTYYCVKFRKGSPDDVEFKSGAGTELSVRAKPS
(SEQ ID NO: 8)
Wild-type D1 domain EEELQVIQ PDKSVSVAAGESAILliCTVT SLIPVGP TQWERGAGPAREL I
YNQKEGHEPRV
variant 7
TTVSESTKRENMDFSISISNITPADAGTYYCVKFRKGSPDTEFKSGAGTELSVRGKPS
(SEQ ID NO: 9)
Wild-type D1 domain
EEELQVIQPDKSVLVAAGETATLRCTATSLIPVGPIQWFRGAGPARELIYNQKEGHFPRV
variant 8
(SEQ ID NO: 10) T TVSESTKRENMDFS I S I SNI
TPADAGTYYCVKFRKGSPDTEFKSGAGTELSVRAKPS
= CA 02938180 2016-08-04
Wild-type D1 domain
EEELQVIQPDKSVLVAAGETATERCTATSLIPVGPIQWFRGAGPGRELIYN0KEGHFPRV
variant 9
T TVSDLTKRNNMDFS I RISN I TPADAGTYYCVKFRKGSPDDVEEKSGAGTELSVRAKPS
(SEQ ID NO: 11)
Wild-type D1 domain
EEELQVIQPDKSVSVAAGESAILHCTVTSLIPVGPIQWFRGAGPARELIYNQKEGHFPRV
variant 10
T TVSES TKRENMDFS I S I SN I TPADAGTYYCVKFRKGSPDTEFKSGAGTELSVRAKPS
(SEQ ID NO: 12)
Table 2. Amino acid substitutions in a S1RP-a variant, relative to each D1
domain variant
EEEX1QX2IQPDKSVLVAAGETX3TLRCTX4TSLX5PVGPIQWERGAGPGRX6L I YNQX7
D1 domain v1
XeGX5FPRVTIVS DXio TX iiRNNMDFSIRIGX12ITX3.3ADAGTYYCX14KX15RKGSPDDV
(SEQ ID NO: 13)
EX16KSGAGTELSVRAKPS
X1=L, I, V; X2=V, L, I; X3=A, V; X4=A, I, L; X5=1, T, S, F; X6=E, V, L; X7=K,
R;
Amino acid
X5=E, Q; X9=H, P, R; X10=L, T, G; Xii=K, R; X12=N, A, C, D, E, F, G, H, I, K,
L,
substitutions relative to
M, P, Q, R, S, T, V, W, Y; X13=P, A, C, D, E, F, G, H, I, K, L, M, N, Q, R, S,
T, V,
SEQ ID NO: 13
W, Y; X14=V, I; X15=F, L, V; X16=F, V
EEE)(3.QX2IQPDKSVSVAAGESX3ILHCTX4TSLX5PVGPIQWFRGAGPARX6LIYNOX7
D1 domain v2 XeGX 9FPRVT TVSEX io TX nRENMDFS I SI SX12I
TX13ADAGTYYCX14 KX15RKGS P DTE
(SEQ ID NO: 14) Xi 6KSGAGTELSVRAKPS
Xi =L, I, V; X2=V, L, I; X3=A, V; X.4=V, I, L; X5=1, T, S, F; X6=E, V, L;
X7=K, R;
Amino acid
X5=E, Q; X9=H, P, R; Xi0=5, T, G;
R; X12=N, A, C, D, E, F, G, H, I, K, L,
substitutions relative to
M, P, Q, R, S, T, V, W, Y; X13=P, A, C, D, E, F, G, H, I, K, L, M, N, 0, R, S,
T, V,
SEQ ID NO: 14
W, Y; X14=V, I; X15=F, L, V; X16=F, V
EEEX iQX2 I QPDKSVSVAAGES X3 ILLCTX4 TSLX5PVGPIQWFRGAGPARX6LIYNQX7
D1 domain v3
X8GX5FPRVTTVSEX1oTXuRENMDFSI S I SX12I TX13ADAGTYYCX14KX3.5RKGSPDTE
(SEQ ID NO: 15)
X16KSGAGTELSVRAKPS
Xi =L, I, V; X2=V, L, I; X3=A, V; X4=V; I, L; X5=1, T, S, F; X6=E, V, L; X7=K,
R;
Amino acid
X5=E, Q; X9=H, P, R; X10=S, T, G; X11=K, R; X12=N, A, C, D, E, F, G, H, I, K,
L,
substitutions relative to
M, P, Q, R, 5, T, V, W, Y; X13=P, A, C, D, E, F, G, H, I, K, L, M, N, 0, R, S,
T, V,
SEQ ID NO: 15
W, Y; X14=V, I; X15=F, L, V; X16=F, V
EEGX1QX2 I QP DKSVSVAAGESX3 ILHCTX4TSLXsPVGPI QW FRGAGPGRX6LIYNQX7
Di domain v4
X8GX9FPRVTTVSD)C1oTX1i.RNNMDFSIRIGX3.2ITX1.3ADAGTYYCX14KX35RKGSPDDV
(SEQ ID NO: 16)
EXI6KSGAGTELSVRAKPS
X1L, 1, V; X2=V, L, I; X3=A, V; X4=A, I, L; X5=1, T, S, F; X6=E, V, L; X7=K,
R;
Amino acid
X5=E, Q; X9=H, P, R; X10=L, T, G; X11=K, R; X12=N, A, C, D, E, F, G, H, I, K,
L,
substitutions relative to
M, P, Q, R, S, T, V, W, Y; X13=P, A, C, D, E, F, G, H, I, K, L, M, N, 0, R, S,
T, V,
SEQ ID NO: 16
W, Y; X14=V, I; X15=F, L, V; X16=F, V
EEEX1QX2 I QPDKFVLVAAGETX3TLRCTX4TSLXsPVGPIQWFRGAGPGRX6LIYNQX7
D1 domain v5
X8GX9FPRVTTVS DX iorXiiRNNMDFSIRIGX12ITX13ADAGTYYCX14KX15RKGSPDDV
(SEQ ID NO: 17)
Exi6KSGAGTELSVRAKPS
Xi =1_, I, V; X2=V, L, I; X3=A, V; X4=A, I, L; X5=1, T, S, F; X6=E, V, L;
X7=K, R;
Amino acid
X5=E, Q; X9=H, P, R; X19=L, T, G; X11=K, R; X12=N, A, C, D, E, F, G, H, I, K,
L,
substitutions relative to M, P, Q, R, 5, T, V, W, Y; X13=P, A, C, D, E, F, G,
H, I, K, L, M, N, Q, R, S, T, V,
SEQ ID NO: 17
W, Y; X1.4=V, I; X15=F, L, V; X15=F, V
EEEX1QX2IQPDKSVLVAAGETX3TLRCTX4TSLX5PVGP I QWFRGAGPGRX6LIYNQX7
D1 domain v6
XBGX9FPRVTTVSDXio TXiiRNNMDFPI RI GX12ITX13ADAGTYYCX141IX15RKGS PDDV
(SEQ ID NO: 18)
EXi 61CSGAGTE LSVRAKP S
Xi=L, I, V; X2=V, L, I; X3=A, V; X4=A, I, L; X5=1, T, S, F; X6=E, V, L; X7=K,
R;
Amino acid
X5=E, Q; X9=H, P, R; X10=L, T, G; Xii=K, R; X12=N, A, C, D, E, F, G, H, I, K,
L,
substitutions relative to
M, P, Q, R, S, T, V, W, Y; X13=P, A, C, D, E, F, G, H, I, K, L, M, N, Q, R, 5,
T, V,
SEQ ID NO: 18
W, Y; X14=V, 1; X15=F, L, V; X15=F, V
EEEX1QX2I QPDKSVSVAAGESX3 ILHCTX4 T SLX5PVGP QW FRGAG PARX6L YNQX7
D1 domain v7
X8GX9FPRVT TVSEXioTXIIRENMDFS IS I SX12I TX13ADAGTYYCX14KXibRKGSPDTE
(SEQ ID NO: 19)
X16KSGAGTELSVRGKPS
16
CA 02938180 2016-08-04
X1=L, I, V; X2=V, L, I; X3=A, V; X4--7-V, I, L; X5=1, T, S, F; X8=E, V, L;
X7=K, R;
Amino acid
X8=E, Q; Xg=H, P, R; Xi0=S, T, G; Xii=K, R; X12=N, A, C, D, E, F, G, H, I, K,
L,
substitutions relative to
M, P, Q, R, S, T, V, W, Y; X13=P, A, C, D, E, F, G, H, I, K, L, M, N, Q, R, S,
T, V,
SEQ ID NO: 19
W, Y; X14=V, 1; X15=F, L, V; X18=F, V
EEEX1QX2IQPDKSVLVAAGETX3TLRCTX4TSLX5PVGPIQWFRGAGPARX6LIYNQX7
D1 domain v8
XeGX9FPRVTTVSEX1oTXiiRENMDFSISIS)(3.2ITX13ADAGTYYCX14K){15RKGSPDTE
(SEQ ID NO: 20)
Xi6KSGAGTELEVRAKPS
X1=L, I, V; X2=V, L, I; X3=A, V; X.4=A, 1, L; X5=1, T, S, F; X6=E, V, L; X7=K,
R;
Amino acid
X8=E, Q; Xg=H, P, R; Xio=S, T, G; R;
X12=N, A, C, D, E, F, G, H, I, K, L,
substitutions relative to
M, P, Q, R, S, T, V, W, Y; Xi8=P, A, C, D, E, F, G, H, I, K, L, M, N, Q, R, S,
T, V,
SEQ ID NO: 20
W, Y; Xi4=V, I; X5F, L, V; X18=F, V
EEExiQx2 I QP DKSVLVA.A.GETX3TLRCTX4TSLX5PVGPIQWFRGAGPGRX6LIYNQX7
01 domain v9
X8GX9FPRVTIVSDX10TX1IRNNMDFSIRISX12ITX13ADAGTYYCX14KX3.5RKGSPDDV
(SEQ ID NO: 21)
EX16KSGAGTELSVRAKPS
X1=L, 1, V; X2=V, L, 1; X3=A, V; )(4=A, I, L; X5=1, T, S, F; X6=E, V, L; X7=K,
R;
Amino acid
X8=E, Q; Xg=H, P, R; Xio=L, T, G; X11=K, R; X12=N, A, C, D, E, F, G, H, I, K,
L,
substitutions relative to
M, P, Q, R, S, T, V, W, Y; X13=P, A, C, D, E, F, G, H, I, K, L, M, N, Q, R, 5,
T, V,
SEQ ID NO: 21
W, Y; X14=V, I; X15=F, L, V; X16=F, V
EEEX1QX2IQPDKSVSVAAGESX3ILHCTX4TSLX5PVGPIQWFRGAGPARX6LIYNQX7
D1 domain v10
x8Gx9FPRva7vsEXI0TX33.RENMDFSISISX2.2ITX13ADAGTYYCX3.410C15RKGSPDTE
(SEQ ID NO: 22)
Xi 6KSGAGTELSVRAKPS
X1=L, 1, V; X2=V, L, I; X3=A, V; X4=V, I, L; X5=1, T, S, F; X8=E, V, L; X7=K,
R;
Amino acid
X8=E, Q; Xg=H, P, R; Xio=S, T, G; X.11=K, R; X12=N, A, C, D, E, F, G, H, I, K,
L,
substitutions relative to
M, P, Q, R, S, T, V, W, Y; X13=P, A, C, D, E, F, G, H, I, K, L, M, N, Q, R, S,
T, V,
SEQ ID NO: 22
W, Y; X14=V, I; X15=F, L, V; X18=F, V
EEXIX2QX3IQPDKX4VX5VAAGEX6X7X8LX9CTX3oTSLXHPVGPIQWFRGAGPX1.2RXJ.3
Pan D1 domain
LIYNQX14X15GX16FPRVTTVSX17XIBTX3.9RX20NMDFX21IX22IX23X24ITX25ADAGTYY
(SEQ ID NO: 23)
CX26KX27RKGSPDX28X29EX3oKSGAGTELSVRX31KPS
Xi=E, G; X2=L, I, V; X3=V, L, 1; )(4=S, F; X5=L, S; X6=S, T; X7=A, V; X8=1, T;
Xg=H, R; X10=A, V, I, L; X11=1, T, 5, F; X12=A, G; X13=E, V, L; X14=K, R;
X15=E,
Amino acid
Q; X18=H, P, R; X17=D, E; X18=S, L, T, G; Xig=K, R; X20=E, N; X21=S, P; X22=S,
substitutions relative to
R; X23=S, G; X24=N, A, C, D, E, F, G, H, I, K, L, M, P, 0, R, 5, T, V, W, Y;
SEQ ID NO: 23
X25=P, A, C, D, E, F, G, H, I, K, L, M, N, Q, R, S, T, V, W, Y; X26=V, I;
X27=F, L,
V; X28=D or absent; X29=T, V; X30=F, V; and X31=A, G
Table 3. SEQ ID NOs: 24-34
SEQ ID NO Sequence
24 EEELQVIQPDKSVSVAAGESAILHCTITS LI PVGP I QW
FRGAGPARELIYNQREGHFPRVTTVSET
TRRENMDFS I S I SNI T PADAGT YYCVKFRKGSPDTEVKSGAGTELEVRAKP S
25 EEEVQVIQPDKSVSVAAGESAILFICTLTSLI PVGPIQWFRGAGPARVL I
YlIQRQGHFPRVTTVSEG
TRRENMDFS IS I SN I PADAGTYYCIKFRKGSPDTEFKSGAGTELSVRAKP S
26 EEEVQIIQPDKSVSVAAGESVI LHCT I
TSLIPVGPIQWERGAGPARLLIYNQREGPFPRVTTVSET
TRRENMDFS I S I SN I TPADAGTYYCVKLIRKGSPDTEFKSGAGTELSVRAKPS
27 EEELQI IQP DKSVSVAAGESAI LHCT I
TSLSPVGPIQWFRGAGPARVLIYNQRQGPFPRVTTVSEG
TKRENMDFS IS I SN I TPADAGTYYCIKLRKGSPDTEFKSGAGTELSVRAKPS
28 EEEIQVIQPDKSVSVAAGESVI IHCTVTSLFPVGPIQWFRGAGPARVLIYNQRQGRFPRVTTVSEG
TKRENMDFS IS I SN IT PADAGTYYCVKVRKGSPDTEVKSGAGTELSVRAKPS
29 EEEVQI I QP DKSVSVAAGES I I LFICTVTSLFPVGPIQWFRGAGPARVLI
YNQREGREPRVTIVSEG
TRRENMDFS I S I SNI TPADAGTYYCIKLRKGSPDTEFKSGAGTELSVRAKPS
30 EEEVQLIQPDKSVSVAAGESA I LHCTVTSLFPVGP I QW FRGAGPARVLIYNQREGP
FPRVITVSEG
TKRENMDFS IS I SN I TPADAGTYYCIKFRKGSPDTEVKSGAGTELSVRAKPS
31 EEELQ I IQP DKSVLVAAGETATLRCT I TSLFPVGP I QW FRGAGPGRVLI
YNQRQGPFPRVTTVSDT
TKRNNMDFS IRIGN IT PADAGTYYCIKFRKGSPDDVEFKSGAGTELSVRAKPS
32 EEELQI I QP DKSVSVAAGESAILIICTI TSLFPVGPIQW FRGAGPARLLI
YNQRQGPFPRVTTVSET
TKRENMDFSISISNITPADAGTYYCVKFRKGSPDTEFKSGAGTELSVRAKPS
17
CA 02938180 2016-08-04
SEQ ID NO Sequence
33 EEEVQIIQPDKSVSVAAGESAI LHCTI TSLFPVGPIQW
FRGAGPARVLIYNQKQGPFPRVT T I SET
TRRENMDFS IS I SN IT PADAGTYYCIKFRKGS PDTEFKSGAGTELSVRAKPS
34 EEELQI IQPDKSVSVAAGESAI LFiCT I TSLTPVGPIQWFRGAGPARVL I
YNQRQGP FPRVTTVSEG
TRRENMDFS IS I SN IT PADAGTYYC IKFRKGS PDTEVKSGAGTELSVRAKPS
Desirably, the SIRP-a variant constructs of the invention bind with higher
affinity to CD47 under
conditions characteristic of a diseased site, such as a cancer site, e.g,,
inside a tumor, than under
physiological conditions (e.g., neutral pH and adequate oxygen concentration).
Conditions characteristic
of a diseased site, such as a cancer site, e.g., inside a tumor, are, e.g.,
acidic pH and hypoxia. In some
embodiments, SIRP-a variant constructs of the invention may be engineered to
preferentially bind to
diseased cells over non-diseased cells. In particular, the disease cells may
be cancer cells of a cancer
disease, e.g., solid tumor or hematological cancer. Preferably, the SIRP-a
variant constructs bind with
higher affinity to CD47 under acidic pH (e.g., less than around pH 7) than
under neutral pH, e.g., pH 7.4.
Preferably, the SIRP-a variant constructs bind with higher affinity to CD47
under hypoxic condition than
under a condition with adequate oxygen concentration. In some embodiments, a
SIRP-a variant
construct includes a SIRP-a variant attached to a blocking peptide. In some
embodiments, the
preferential binding of the SIRP-a variant in the SIRP-a variant construct to
C047 on diseased cells or
diseased sites may be obtained by attaching the block peptide to the SIRP-a
variant by use of a
cleavable linker, which is cleaved at the diseased cells or diseased sites. In
some embodiments, the
preferential binding of the SIRP-a variant in the SIRP-a variant construct to
CD47 on diseased cells or
diseased sites may be obtained by attaching the block peptide to the SIRP-a
variant, wherein the
blocking peptide can be detatched or simply dissociated from the SIRP-a
variant at the diseased cells or
diseased sites.
In some embodiments, a SIRP-a variant construct includes a SIRP-a variant and
a blocking
peptide. In some embodiments, SIRP-a variant may be attached to a blocking
peptide through a linker
(e.g., a cleavable linker). The blocking peptide serves to block the CD47
binding site of the SIRP-a
variant to prevent binding of SIRP-a variant to CD47 under physiological
conditions (e.g., neutral pH and
adequate oxygen concentration). The cleavable linker is a linker capable of
being cleaved only under
conditions characteristic of a diseased site (such as a cancer site, e.g.,
inside a tumor), such as acidic pH
and hypoxia. In some embodiments, the cleavable linker is cleaved by a tumor-
associated protease at a
diseased site. In some embodiments, the linker is not cleaved and the blocking
peptide simply
dissociates from the SIRP-a variant at a diseased site such that the SIRP-a
variant is free to bind to
nearby CD47 on diseased cells, e.g., tumor cells. Therefore, only when the
SIRP-a variant is at a
diseased site would it be released from the blocking peptide and be free to
bind to nearby CD47 on
diseased cells, e.g., tumor cells. Blocking peptides and linkers (e.g.,
cleavable linkers) are described in
detail further herein.
In some embodiments, a SIRP-a variant construct includes a SIRP-a variant and
a targeting
moiety. In some embodiments, a SIRP-a variant may be attached to a targeting
moiety, such as an
antibody, e.g., a tumor-specific antibody, or another protein or peptide,
e.g., an antibody-binding peptide,
that exhibit binding affinity to a diseased cell. After administration, the
tumor-specific antibody or
antibody-binding peptide serves as a targeting moiety to bring the SIRP-a
variant to the diseased site,
such as a cancer site, e.g., inside a solid tumor, where the SIRP-a can
interact specifically with CD47 on
18
diseased cells. In some embodiments, a SIRP-a variant may be fused to a
protein or peptide, e.g., an
antibody-binding peptide, capable of binding to an antibody (e.g., tumor-
specific antibody), i.e., binding to
a constant or variable region of the antibody. SIRP-a variants capable of
binding to one or more
antibodies are described in detail further herein. In other embodiments, other
SIRP-a variants, such as
the ones described in International Publication No. W02013109752, may be
attached to a tumor-specific
antibody or to a protein or peptide, e.g., an antibody-binding peptide,
capable of binding to a tumor-
specific antibody. In some embodiments, the SIRP-a variant may be attached to
the antibody either in
vitro (prior to administration to a human) or in vivo (after administration).
In some embodiments, a SIRP-a variant may further include a D2 and/or D3
domain of a wild-
type human SIRP-a. In some embodiments, a SIRP-a variant may be attached to an
Fc domain
monomer, a human serum albumin (HSA), a serum-binding protein or peptide, or
an organic molecule,
e.g., a polymer (e.g., a polyethylene glycol (PEG)), in order to improve the
pharmacokinetic properties of
the SIRP-a variant, e.g., increase serum half-life. Fc domain monomers, HSA
proteins, serum-binding
proteins or peptides, and organic molecules such as a PEG that serve to
increase the serum half-life of
the SIRP-a variants of the invention are described in detail further herein.
In some embodiments, a S1RP-
a variant does not include the sequence of any one of SEQ ID NOs:3-12 and 24-
34.
IL Amino acid substitutions with histidine residues in SIRP-a variants
In some embodiments, in addition to the amino acid substitutions in a SIRP-a
variant listed in
Table 2, the SIRP-a variant may include one or more amino acid substitutions
with histidine residues.
The SIRP-a variant constructs including a SIRP-a variant bind with higher
affinity to CD47 on diseased
cells or at a diseased site than on non-diseased cells and under conditions
characteristic of a diseased
site (e.g., acidic pH, hypoxia) than under physiological conditions. Amino
acid residues to be substituted
with histidine residues may be identified using histidine scanning
mutagenesis, protein crystal structures,
and computational design and modeling methods. Techniques and methods that may
be used to
generate SIRP-a variants and ways to determine their binding affinities to
CD47 on diseased and non-
diseased cells are described in detail further herein. The histidine residue
substitutions may be located at
the interface of a S1RP-a variant and CD47 or may be at internal regions of a
SIRP-a variant.
Preferentially, histidine residue substitutions are located at the interface
of a SIRP-a variant and CD47.
Table 4 lists specific SIRP-a amino acids that may be substituted with
histidine residues. The amino acid
numbering in Table 4 is relative to the sequence of SEQ ID NO: 3; one or more
amino acids at the
corresponding positions in any one of the sequences of SEQ ID NOs: 4-12 may
also be substituted with
histidine residues. Contact residues are the amino acids located at the
interface of a SIRP-a variant and
CD47. Core residues are the internal amino acids not directly involved in the
binding between a SIR P-a
variant and CD47. The SIRP-a variants may include one or more (e.g., one, two,
three, four, five, six,
seven, eight, nine, ten, etc, or all) of the substitutions listed in Table 4.
The SIRP-a variants may contain
a maximum of 20 histidine substitutions.
Table 4. SIRP-a amino acid substitutions (amino acid numbering is relative to
the sequence of
SEQ ID NO: 3)
S29H, L3OH, 131H, P32H, V33H, G34H, P35H, Q52H, K53H, E54H, L66H, T67H,
Contact residues
K68H, R69H, F74H, K93H, K96H, G97H, S98H, D100H
C L4H, V6H, A27H, 136H, F39H, E47H, L48H, I49H, Y5OH, F57H,
V6OH, M72H,
ore residues
F74H, 176H, V92H, F94H, E103H
19
Date Regue/Date Received 2022-11-28
III. pH-dependent binding
Studies have shown that tumor cell mediated oncogenic metabolism generates a
large amount of
lactic acid and protons, leading to the reduction in the extracellular pH
values to as low as 6 in tumor
tissue (Icard et al., Biochim. Biophys. Acta, 1826:423-433, 2012). In some
embodiments, the SIRP-a
variant constructs including a SIRP-a variant are engineered to bind with high
affinity to CD47 under
acidic pH than under neutral pH (e.g., around pH 7.4). .Thus, the SIRP-a
variant constructs of the
invention are engineered to selectively bind to CD47 on diseased cells (e.g.,
tumor cells) or on cells at a
diseased site (e.g., cells in the tumor rnicro-environment supporting tumor
growth), over CD47 on non-
diseased cells.
In one embodiment, to engineer pH-dependent binding of a SIRP-a variant
construct of the
invention, histidine mutagenesis may be performed on the SIRP-a variant,
especially on the region of
SIRP-a that interacts with CD47. Crystal structures of a SIRP-a and CD47
complex (see, e.g., PDB ID
No. 2JJS) and computer modeling may be used to visualize the three-dimensional
binding site of SIRP-a
and CD47. Computational design and modeling methods useful in designing a
protein with pH-sensitive
binding properties are known in the literature and described in, e.g., Strauch
et al., Ptoc Nat! Acad Sci 111
:675-80, 2014. In some embodiments, computer modeling may be used to identify
key contact residues at
the interface of SIRP-a and CD47. Identified key contact residues may be
substituted with histidine
residues using available protein design software (e.g., RosettaDesign), which
can generate various
protein designs that can be optimized, filtered, and ranked based on computed
binding energy and shape
complementarity. Therefore, energetically favorable histidine substitutions at
certain amino acid positions
may be identified using computational design methods. Computer modeling may be
also be used to
predict the change in the three-dimensional structure of SIRP-a. Histidine
substitutions that generate a
significant change in the three-dimensional structure of SIRP-a may be
avoided.
Once energetically and structurally optimal amino acid substitutions are
identified, the amino
acids may be systematically substituted with histidine residues. In some
embodiments, one or more (e.g.,
one, two, three, four, five, six, seven, eight, nine, ten, etc, with a maximum
of 20) amino acids of SIRP-a
may be substituted with histidine residues. In particular, amino acids located
at the interface of SIRP-a
and CD47, preferably, amino acids directly involved in the binding of SIRP-a
to C047, may be substituted
with histidine residues. The SIRP-a variant may include one or more (e.g.,
one, two, three, four, five, six,
seven, eight, nine, ten, etc, with a maximum of 20) histidine residue
substitutions. In other embodiments,
naturally occurring histidine residues of SIRP-a may be substituted with other
amino acid residues. In yet
other embodiments, one or more amino acids of SIRP-a may be substituted with
non-histidine residues in
order to affect the binding of naturally occurring or substituted histidine
residues with CD47. For example,
substituting amino acids surrounding a naturally occurring histidine residue
with other amino acids may
"bury" the naturally occurring histidine residue. In some embodiments, amino
acids not directly involved in
binding with CD47, i.e., internal amino acids (e.g., amino acids located at
the core of SIRP-a) may also be
substituted with histidine residues. Table 4 lists specific SIRP-a amino acids
that may be substituted with
histidine or non-histidine residues. Contact residues are the amino acids
located at the interface of
SIRP-a and CD47. Core residues are the internal amino acids not directly
involved in the binding
Date Regue/Date Received 2022-11-28
between SIRP-a and CD47. The SIRP-a variants may include one or more (e.g.,
one, two, three, four,
five, six, seven, eight, nine, ten, etc, or all) of the substitutions listed
in Table 4,
SI RP-a variants containing one or more (e.g., one, two, three, four, five,
six, seven, eight, nine,
ten, etc, with a maximum number of 20) histidine residue substitutions may be
tested for their binding to
C047 under different pH conditions (e.g., at pH 5, 5.5, 6, 6.5, 7, 7.4, 8). In
some embodiments, purified
C047 protein may be used to test binding. Various techniques known to those
skilled in the art may be
used to measure the affinity constant (KA) or dissociation constant (Ko) of a
SIRP-a variant/CD47 complex
under different pH conditions (e.g., at pH 5, 5.5, 6, 6.5, 7, 7.4, 8). In a
preferred embodiment, the binding
affinity of a SIRP-a variant to a CD47 may be determined using surface plasmon
resonance (e.g.,
Biacore30001m surface plasmon resonance (SPR) system, Biacore, INC, Piscataway
N.J.). In an
exemplary embodiment, a SIRP-a variant with pH-dependent binding, which
specifically binds a CD47
with higher affinity at pH 6 than at pH 7.4, exhibits a lower Ko at pH 6 than
at pH 7.4.
IV. Hypoxia-dependent binding
Tumor hypoxia is the condition in which tumor cells have been deprived of
oxygen. As a tumor
grows, its blood supply is constantly redirect to the most fast growing parts
of the tumor, leaving portions
of the tumor with oxygen concentration significantly lower than in healthy
tissues.
In some embodiments, a SIRP-a variant may be attached to a hypoxia-activated
prodrug, which
may act to increase the efficacy of a SIRP-alpha variant against the relevant
diseased cells under
specifically hypoxic conditions. Hypoxia-activated prodrugs are known in the
literature, such as those
described by Kling et al. (Nature Biotechnology, 30:381, 2012).
V. Antibody binding
Another strategy to provide selective SIRP-a activity at a diseased site than
at a non-diseased
site is to attach the SIRP-a protein to a protein or peptide that can bind to
a region of an antibody.
Preferably, the antibody is specific to a diseased cell, elg., a tumor cell.
For example, the antibody may
specifically bind to a cell surface protein on a diseased cell, e.g., a tumor
cell. The SIRP-a protein may
bind to the antibody reversibly or irreversibly.
General antibody binding
In some embodiments, to engineer a SIRP-a protein that can bind to different
antibodies
regardless of antibody specificity, the SIRP-a protein may be fused to a
protein or peptide that recognizes
the constant region of an antibody, e.g., the CH2 or CH3 constant domain of
the Fc domain of an antibody.
A SIRP-a protein is capable of binding CD47 and has at least 50% amino acid
sequence identity to a
sequence of a wild-type SIRP-a (e.g., variant 1 (SEQ ID NO: 1, shown below))
or to a sequence of a
C047-binding portion of a wild-type SIRP-a (e.g., a sequence of any one of SEQ
ID NOs: 3-12 listed in
Table 1).
SEQ ID NO: 1
1 MEPAGPAPGR LGPLLCLLLA ASCAWSGVAG EEELQVIQPD KSVLVAAGET ATLRCTATSL
61 IPVGPIQWFR GAGPGRELIY NQKEGHFPRV TTVSDLTKRN NMDFSIRIGN ITPADAGTYY
121 CVKFRKGSPD DVEEKSGAGT ELSVRAKPSA PVVSGPAARA TPQRTVSFTC ESHGFSPRDI
181 TLKWFKNGNE LSDFQTNVDP VGESVSYSIH STAKVVLTRE DVHSQVICEV AHVTLQGDPL
241 RGTANLSETI RVPPTLEVTQ QPVRAENQVN VTCQVRKFYP QRLQLTWLEN GNVSRTETAS
301 TVTENKDGTY NWMSWLLVNV SAHRDDVKLT CQVEHDGQPA VSKSHDLKVS AHPKEQGSNT
21
Date Regue/Date Received 2022-11-28
361 AAENTGSNER NIYIVVGVVC TLLVALLMAA LYLVRIRQKK AQGSTSSTRL HEPEKNAREI
421 TQDTNDITYA DLNLPKGKKP APQAAEPNNH TEYASIQTSP QPASEDTLTY ADLDMVHLNR
481 TPKQPAPKPE PSFSEYASVQ VPRK
A CD47-binding portion of a wild-type SIRP-a includes the D1 domain of a wild-
type SIRP-a (e.g.,
a sequence of any one of SEQ ID NOs: 3-12 listed in Table 1). Proteins and
peptides exhibiting general
binding to the constant region of an antibody are known in the art. For
example, the bacterial antibody-
binding proteins, e.g., Proteins A, G, and L, bind to the constant regions of
an antibody. Proteins A and G
bind to the Fc domains, while Protein L binds to the constant region of the
light chain. In an exemplary
embodiment, Protein A, G, or L may be fused to the N- or C-terminus of a SIRP-
a protein. Preferentially,
in this embodiment, the fusion protein of Protein A, G, or L and the SIRP-a
protein may be attached, i.e.,
through chemical conjugation, to an antibody prior to administration to
prevent the fusion protein from
binding to various other antibodies in serum. Protein A, G, or L may also be
evolved and screened using
conventional techniques in the field (i.e., directed evolution and display
libraries) for higher binding affinity
to the constant regions of an antibody. In some embodiments, a SIRP-a protein
may be directly attached
to an antibody using conventional genetic or chemical conjugation techniques
in the art. In other
embodiments, a SIRP-a protein may also be attached to an antibody by way of a
spacer, which allows for
additional structural and spatial flexibility of the protein. Various spacers
are described in detail further
herein. In some embodiments, the SIRP-a protein may bind, either directly or
through an antibody-
binding protein or peptide, to the antibody reversibly or irreversibly.
Further, screening of modified
antibodies which can be utilized in accordance with the embodiments of the
invention described herein
can be carried out as described in, e.g., US Patent Publication No. US
20100189651.
Other proteins or peptides capable of binding to a constant region of an
antibody and methods of
screening for such proteins or peptides are described in US Patent Publication
No. US20120283408.
Specific antibody binding
In some embodiments, to provide selective targeting of SIRP-a variants at a
diseased site and to
engineer a SIRP-a variant capable of binding to a specific antibody, e.g., a
tumor-specific antibody, the
SIRP-a variant construct may include a SIRP-a variant and an antibody-specific
protein or peptide. The
SIRP-a variant may be fused to an antibody-specific protein or peptide (e.g.,
an antibody-binding =
peptide). Preferably, the protein or peptide specifically binds to a tumor-
specific antibody. In some
embodiments, the fusion protein of the SIRP-a variant and the antibody-binding
protein or peptide may be
co-administered with the tumor-specific antibody in a combination therapy. In
other embodiments, the
fusion protein and the tumor-specific antibody may, be administrated
separately, i.e., within hours of each
other, preferably, the antibody is administered first. In yet other
embodiments, prior to administration, the
fusion protein may be covalently attached to the tumor-specific antibody using
genetic or chemical
methods commonly known in the art.
Examples of antibody-binding peptides include a disease localization peptide
(DLP) (SEQ ID NO:
64 or 65), a small peptide that can bind to the center of the fragment antigen-
binding (Fab) region of
Cetuximab (see, e.g., Donaldson et al., Proc Nall Aced Sc! U S A. 110: 17456-
17461, 2013). Cetuximab
is an anti-epidermal growth factor receptor (EGFR) IgG1 antibody. Antibody-
binding peptides that can be
fused to a SIRP-a variant also include, but are not limited to, peptides
having at least 75% amino acid
22
Date Regue/Date Received 2022-11-28
sequence identity to the sequence of the DLP (SEQ ID NO: 64 or 65) or a
fragment thereof. In some
embodiments, the antibody-binding peptide has the sequence of SEQ ID NO: 64.
In a recent study, SIRP-a has been shown to enhance in vitro phagocytosis of
OLD-1 cells in
combination with the antibody Cetuximab (Weiskopf et al., Science 341: 88-91,
2013). In some
embodiments, a SIRP-a variant may be fused to a specific antibody-binding
peptide, e.g., a DLP having
the sequence of SEQ ID NO: 64. In these embodiments, the SIRP-a variant
construct including a SIRP-a
variant and a DLP may target its activity in Cetuximab-bound, EGFR expressing
tumors. This in turn may
further improve the delivery of the SIRP-a variant construct including a SIRP-
a variant and DLP and
Cetuximab to anti-EGFR responsive patients. An example of a SIRP-a variant
construct including a
SIRP-a variant and DLP is shown in SEQ ID NO: 66, in which single-underlined
portion indicates the DLP
and bold portion indicates the SIRP-a variant. The sequence of the SIRP-a
variant (bold portion) in SEQ
ID NO: 66 may be replaced by a sequence of any SIRP-a variant described
herein. Other antibody-
binding peptides may also be fused to a SIRP-a variant. Such antibody-binding
peptides include, but are
not limited to, peptides that can specifically bind to antibodies such as
cetuximab, pembrolizumab,
nivolumab, pidilizumab, MEDI0680, MEDI6469, Ipilimumab, tremelimumab,
urelumab, vantictumab,
varlilumab, mogamalizumab, anti-CD20 antibody, anti-CD19 antibody, anti-CS1
antibody, herceptin,
trastuzumab, and/or pertuzumab.
SEQ ID NO: 66
CQFDLSTRRLKCGGGGSGGGGSGGGGSGGGGSEEELQ I IQ PDKSVLVAAGE TATLRC T I TSLFPVGP I
QWF
RGAGPGRVLIYNQRQGPFPRVTTVSDTTERNNMDFS I RI GN I TPADAGTYYC I KFRKGS PDDVE
FKSGAGT
ELSVRAKPSGGGGSGGGGSGGGGSGGGGSCUIDLSTRRLKC
In some embodiments, a SIRP-a variant construct including a SIRP-a variant and
a DLP may be
further combined with a CD47-based blocking peptide described herein to block
the binding of the SIRP-a
variant in the construct before the construct reaches the diseased site where
the cleavable linker may be
cleaved. In these embodiments, the therapeutic window can be expanded as the
SIRP-a variant
construct containing a SIRP-a variant, a CD47-based blocking peptide, and a
DLP accumulates at the
diseased site and is only active at the diseased site after linker cleavage
induced by a protease (e.g., a
tumor-specific protease) or other characteristics of the diseased site (e.g.,
acidic pH, hypoxia).
In some embodiments, proteins or peptides capable of binding to tumor-specific
antibodies may
be identified using techniques commonly used in the art, such as directed
evolution and display libraries,
e.g., a phage display library. Methods and techniques directed to identifying
proteins and peptides
capable of binding to tumor-specific antibodies are known in the art, such as
those described by
Donaldson et al. (Proc Nati Acad Sci 110:17456-61,2013). In a phage display
library, a potential
antibody-specific protein or peptide is typically covalently linked to a
bacteriophage coat protein. The
linkage results from translation of a nucleic acid encoding the protein or
peptide fused to the coat protein.
Bacteriophage displaying the peptide can be grown and harvested using standard
phage preparatory
methods, e.g. PEG precipitation from growth media. These displaying phages can
then be screened
against other proteins, e.g., tumor-specific antibodies, in order to detect
interaction between the displayed
protein and the tumor-specific antibodies. Once the tumor-specific protein or
peptide is identified, the
nucleic acid encoding the selected tumor-
23
Date Regue/Date Received 2022-11-28
specific protein or peptide can be isolated from cells infected with the
selected phages or from the phage
themselves, after amplification. Individual colonies or plaques can be picked
and the nucleic acid can be
isolated and sequenced. After identifying and isolating the antibody-specific
protein or peptide, the
protein or peptide may be fused to the N- or C-terminus of a SIRP-a variant.
In some embodiments, a
.. SIRP-a variant may be directly attached to a tumor-specific antibody using
conventional genetic or
chemical conjugation techniques in the art. In other embodiments, a SIR P-a
variant may also be
attached to a tumor-specific antibody by way of a spacer, which allows for
additional structural and spatial
flexibility of the protein. Various spacers are described in detail further
herein. In some embodiments,
the SIRP-a variant may bind, either directly or through an antibody-binding
protein or peptide, to the
antibody reversibly or irreversibly.
In other embodiments, a wild-type SIRP-a or the extracellular D1 domain of the
wild-type SIRP-a
(e.g., a sequence of any one of SEQ ID NOs: 3-12 listed in Table 1) may be
attached to a tumor-specific
antibody. Preferably, the D1 domain of SIRP-a is attached to the tumor-
specific antibody. The tumor-
specific antibody serves as a targeting moiety to bring the wild-type SIRP-a
or the D1 domain to the
diseased site, e.g., a cancer site, e.g., inside a solid tumor, where the wild-
type SIRP-a or the D1 domain
can interact with C047 on diseased cells. In some embodiments, a wild-type
SIRP-a or the extracellular
D1 domain of the wild-type SIRP-a may be directly attached to a tumor-specific
antibody using
conventional genetic or chemical conjugation techniques in the art. In other
embodiments, a wild-type
SIRP-a or the extracellular D1 domain of the wild-type SIRP-a may also be
attached to a tumor-specific
antibody by way of a spacer, which allows for additional structural and
spatial flexibility of the protein.
Various spacers are described in detail further herein. In other embodiments,
a wild-type SIRP-a or the
extracellular D1 domain of the wild-type SIRP-a may be fused to the
aforementioned protein or peptide
capable of binding to a tumor-specific antibody. In yet other embodiments,
other SIRP-a polypeptides,
such as the ones described in International Publication No. W02013109752, may
be attached to a
tumor-specific antibody or to a protein or peptide capable of binding to a
tumor-specific antibody. In some
embodiments, the wild-type SIRP-a or the D1 domain may bind, either directly
or through an
antibody-binding protein or peptide, to the antibody reversibly or
irreversibly,
VI. Blocking peptides
A blocking peptide may be attached a SIRP-a variant by way of a cleavable
linker. In some
embodiments, a blocking peptide may also be non-covalently attached to a SIRP-
a variant. The blocking
peptide acts to block the C047 binding site of the SIRP-a variant such that
the SIRP-a variant cannot
bind to C047 on the cell surface of non-diseased cells under physiological
conditions (e.g., neutral pH
and adequate oxygen concentration). Under abnormal conditions (i.e., an acidic
and/or hypoxic
environment or an environment with increased protease expression) at a
diseased site, such as a cancer
site, e.g., inside a tumor, the cleavable linker may be cleaved to release the
SIRP-a variant from the
blocking peptide. The SIRP-a variant would then be free to bind to CD47 on
nearby tumor cells.
Examples of cleavable linkers are described in detail further herein.
In some embodiments, the blocking peptide has higher affinity towards wild-
type SIR P-a than
.. engineered SIR P-a variant. Once the linker is cleaved, the blocking
peptide dissociates from the SIRP-a
variant and may bind to a wild-type SIRP-a. A blocking peptide with different
binding affinities to wild-type
SIRP-a and SIRP-a variant may be identified using methods and techniques
commonly known in the art,
24
Date Regue/Date Received 2022-11-28
CA 02938180 2016-08-04
e.g., directed evolution and display libraries (e.g., phage or yeast display).
In one exemplary
embodiment, a nucleotide encoding the SIRP-a binding region of CD47 or a
nucleotide encoding the
variable region of an anti-SIRP-a antibody may be mutated and/or recombined at
random to create a
large library of gene variants using techniques such as, e.g., error-prone PCR
and DNA shuffling. Once a
genetic library is created, the mutant peptides encoded by the nucleotides may
be screened for their
ability to bind to wild-type SIRP-a and SIRP-a variant using, e.g., phage or
yeast display. Identified
peptides that can bind to both wild-type SIRP-a and SIRP-a variant may undergo
a second screening
process such that the proteins that bind with higher affinity to wild-type
SIRP-a than to SIRP-a variant
may be isolated. The identified peptides, once bound to wild-type SIRP-a or
SIRP-a variant should
prevent the binding of CD47 to wild-type SIRP-a or SIRP-a variant. Various
techniques known to those
skilled in the art may be used to measure the affinity constant (KA) or
dissociation constant (Ka) of a
SIRP-a variant/blocking peptide complex or a wild-type SIRP-a/blocking peptide
complex. A blocking
peptide may bind with at least three fold higher affinity to a wild-type SIRP-
a than a SIRP-a variant.
CD47-based blocking peptides
A blocking peptide may be a CD47 mimic polypeptide, or a CD47 fragment that
can bind a SIRP-
a variant described herein. Some blocking peptides may bind a SIRP-a variant
at a site that is different
from the CD47 binding site. Some blocking peptides may bind a SIRP-a variant
in a manner that is
different from CD47. In some cases, the blocking peptide may comprise at least
one stabilizing disulfide
bond. A blocking peptide may comprise a polypeptide sequence of CERVIGTGVVVRC,
or a fragment or
variant thereof. A variant blocking peptide may contain one or more
conservative or non-conservative
modification. In some cases a variant blocking peptide may contain
modifications of a cysteine to a
serine and/or one or more modifications of an asparagine to a glutamine. A
blocking peptide may bind to
the SIRP-a variant at the same site as a peptide that comprises a polypeptide
sequence of
CERVIGTGWVRC, or a variant or fragment thereof. A blocking peptide may
comprise a polypeptide
sequence of GNYTCEVTELTREGETIIELK, or a fragment or variant thereof. A
blocking peptide may bind
to the SIRP-a variant at the same site as a peptide that comprises a
polypeptide sequence of
GNYTCEVTELTREGETIIELK, or a variant or fragment thereof. In some cases a
blocking peptide may
comprise a polypeptide sequence of EVTELTREGE, or a fragment or variant
thereof. A blocking peptide
may bind to the SIRP-a variant at the same site as a peptide that comprises a
polypeptide sequence of
EVTELTREGE, or a variant or fragment thereof. In some cases a blocking peptide
may comprise a
polypeptide sequence of CEVTELTREGEC, or a fragment or variant thereof. A
blocking peptide may
bind to the SIRP-a variant at the same site as a peptide that comprises a
polypeptide sequence of
CEVTELTREGEC, or a variant or fragment thereof.
Provided herein are SIRP-a variant constructs comprising a SIRP-a variant and
a blocking
peptide, wherein the blocking peptide may comprise a polypeptide sequence of
SEVTELTREGET, or a
fragment or variant thereof. A blocking peptide may bind to the SIRP-a variant
at the same site as a
peptide that comprises a polypeptide sequence of SEVTELTREGET, or a variant or
fragment thereof. In
some cases, the blocking peptide may comprise a polypeptide sequence of
GQYTSEVTELTREGETIIELK, or a fragment or variant thereof. A blocking peptide
may bind to the SIRP-
a variant at the same site as a peptide that comprises a polypeptide sequence
of
GQYTSEVTELTREGETIIELK, or a variant or fragment thereof.
CA 02938180 2016-08-04
In some cases, the blocking peptide may be a CD47 variant polypeptide, that
exhibits a higher
affinity for wild-type SIRP-a, as compared to the SIRP-a variant. As compared
to wild-type CD47, the
blocking polypeptide may comprise at least one of the following mutations:
T102Q, T102H, L101Q,
L101H, and L101Y. As compared to wild-type CD47, the blocking polypeptide may
comprise an
introduction of an additional glycine residue at or near the N-terminus. The
glycine may be introduced
adjacent to a glutamine and/or a leucine at or near the N-terminus of CD47. In
some cases, a blocking
peptide may be a CD47 variant polypeptide that demonstrates a lower affinity
for a SIRP-a variant as
compared to a wild-type CD47. Such CD47 variant polypeptides are easily
identified and tested using
methods described herein.
Provided herein are SIRP-a variant constructs comprising a SIRP-a variant
described herein,
wherein said SIRP-a variant is connected to a blocking peptide described
herein by use of at least one
linker. The SIRP-a variant may comprise the same CD47 binding site as a wild
type SIRP-a. The SIRP-a
variant may comprise one or more mutations, or insertions as compared to a
wild type SIRP-a. The
SIRP-a variant may be a truncated form of the wild type SIRP-a. The blocking
peptide maybe a CD47
mimic, variant, or fragment described herein. The blocking peptide may exhibit
a higher affinity for wild-
type SIRP-a, as compared to the SIRP-a variant in the SIRP-a variant
construct. The blocking peptide
may be a CD47 variant polypeptide that demonstrates a lower affinity for a
SIRP-a variant as compared
to wild-type CD47. The linker may be at least one linker that is optionally
cleavable by one or more
proteases, and optionally also comprises one or more spacers. The cleavable
linker may comprise the
sequence LSGRSDNH. The spacers may comprise one or more units of glycine-
serine spacers, each
unit of which may comprise the sequence GGGGS.
In some embodiments, the blocking peptide that is attached to a SIRP-a variant
by way of a
cleavable linker is a SIRP-a-binding peptide derived from CD47 (i.e., a CD47-
based blocking peptide). In
some embodiments, the CD47-based blocking peptide is derived from the SIRP-a
binding portion of
CD47. The SIRP-a binding portion of CD47 is often referred to as the
immunoglobulin superfamily (IgSF)
domain of CD47, the sequence of which is shown below (SEQ ID NO: 35; Ref. NP
0017681).
SEQ ID NO: 35: wild-type, IgSF domain of human CD47
1-50 QLLFNKTKSV EFTFCNDTVV IPCFVTNMEA QNTTEVYVKW KFKGRDIYTF
51-100 DGALNKSTVP TDFSSAKIEV SQLLKGDASL KMDKSDAVSH TGNYTCEVTE
101-123 LTRFGETTIE LKYRVVSWFS PNE
In some embodiments, the CD47-based blocking peptide contains the full-length,
IgSF domain of
C047 (SEQ ID NO: 35) or a fragment thereof. In some embodiments, the CD47-
based blocking peptide
contains one or more amino acid substitutions, deletions, and/or additions
relative to the wild-type, IgSF
domain of CD47 (SEQ ID NO: 35) or a fragment thereof. In some embodiments, a
CD47-based blocking
peptide has at least 80% (e.g., 83%, 86%, 90%, 93%, 96%, etc) amino acid
sequence identity to the
sequence of the wild-type, IgSF domain of CD47 (SEQ ID NO: 35) or a fragment
thereof.
In some embodiments, the amino acid substitutions, deletions, and/or additions
in the CD47-
based blocking peptide results in the CD47-based blocking peptide having low
binding affinity for a SIRP-
a variant and relatively higher binding affinity for the wild-type SIRP-a. In
some embodiments, the amino
acid substitutions in the CD47-based blocking peptide are located at the
interface of CD47 and SIRP-a.
26
CA 02938180 2016-08-04
For example, amino acid substitution T102Q in the CD47 IgSF domain sterically
clashes with amino acid
substitution A27I in a SIRP-a variant, while a wild-type SIRP-a having A27
would not sterically clash with
the amino acid substitution T102Q (see FIG. 2). Thus, a CD47-based blocking
peptide having T102Q
would bind with higher affinity to a wild-type SIRP-a having A27 than to a
SIRP-a variant having A27I.
Examples of amino acid substitutions in a CD47-based blocking peptide that may
create steric clashes
with specific amino acids in a SIRP-a variant are listed in Table 5. Each of
these amino acid substitutions
in a CD47-based blocking peptide may reduce the binding affinity of the CD47-
based blocking peptide to
a SIRP-a variant, depending on the specific amino acid in the SIRP-a variant
at the SIRP-a-CD47
interaction site.
Table 5: Examples of amino acid substitutions in a CD47-based blocking peptide
that may create
steric clashes with specific amino acids in a SIRP-a variant
Amino acid substitution in a CD47-based blocking Amino acid in a SIRP-a
variant
peptide (amino acid numbering is relative
to any one of
(amino acid numbering is relative to SEQ ID NO: SEQ ID NOs: 13-23)
35)
T102Q 271
T102H ¨271
_L101Q 31F
L101H 31F
L101Y 31F
In addition to creating steric clashes between a CD47-based blocking peptide
and a SIRP-a
variant, amino acid substitutions, additions, and/or deletions can also be
used to break specific non-
covalent interactions between a CD47-based blocking peptide and a SIRP-a
variant, thus, reducing the
binding affinity of the CD47-based blocking peptide to the SIRP-a variant. In
some embodiments,
extending the N-terminus of the CD47-based blocking peptide by one or more
amino acids (e.g., one
amino acid), either by adding the one or more amino acids directly to the N-
terminus and/or by inserting
the one or more amino acids between other amino acids at the N-terminus,
breaks non-covalent
interactions (e.g., hydrogen bonding interactions) between the N-terminus of
the CD47-based blocking
peptide and a SIRP-a variant. For example, an amino acid addition, e.g., a
glycine addition, at the N-
terminus of the CD47-based blocking peptide will prevent cyclization of
glutamine to pyroglutamate at the
N-terminus and also create unwanted contacts and interactions that will likely
will disrupt the hydrogen
bonding interactions between the N-terminal pyroglutamate of the CD47-based
blocking peptide and the
amino acid L66 in a wild-type SIRP-a or amino acid substitution L66T in a SIRP-
a variant (see also
Example 5). In some embodiments, an amino acid residue, e.g., glycine, is
added at the N-terminus of
the CD47-based blocking peptide such that the N-terminus of CD47 is changed
from QLLFNK to
GOLLFNK or QGLLFNK. The choice of the amino acid substitutions, deletions,
and/or additions in a
CD47-based blocking peptide would depend on the specific amino acid
substitutions in a SIRP-a variant.
Furthermore, fusing the N-terminus of the CD47-based blocking peptide to the C-
terminus of a
SIRP-a variant through a cleavable linker and optionally one or more spacers
also affects the binding
interactions between the CD47-based blocking peptide and the SIRP-a variant
and reduces the binding
affinity of the CD47-based blocking peptide to the SIRP-a variant. In some
embodiments, in a SIRP-a
variant construct, the N-terminus of a CD47-based blocking peptide is fused to
the C-terminus of a SIRP-
a variant by way of a cleavable linker and optionally one or more spacers. In
some embodiments, in a
SIRP-a variant construct, the C-terminus of a CD47-based blocking peptide is
fused to the N-terminus of
27
= CA 02938180 2016-08-04
a SIRP-a variant by way of a cleavable linker and optionally one or more
spacers. Examples of cleavable
linkers and spacers are described in detail further herein.
Exemplary CD47-based blocking peptides are shown in Table 6. In some
embodiments, the
CD47-based blocking peptide has or includes the sequence SEVTELTREGET (SEQ ID
NO: 38). In some
embodiments, the CD47-based blocking peptide has or includes the sequence
GQYTSEVTELTREGETIIELK (SEQ ID NO: 40).
Table 6
SEC) ID CD47-based blocking peptides Portion of the
CD47
NO IgSF domain
36 EVTELTREGE Amino acids 97-
106 of
SEQ ID NO: 35
37 CEVTELTREGEC Amino acids 96-
107 of
SEQ ID NO: 35 with
T125C
38 SEVTELTREGET Amino acids 96-
107 of
SEQ ID NO: 35 with
C96S
39 GNYTCEVTELTREGETIIELK Amino adds 92-
112 of
SEQ ID NO: 35
40 GQYTSEVTELTREGETIIELK Amino acids 92-
112 of
SEQ ID NO: 35 with
N93Q and C965
41 QLLFNKTKSV EFTFCNDTVV IPCFVTNMEA QNTTEVYVKW CD47 IgSF
domain
KFKGRDIYTF DGALNKSTVP TDFSSAKIEV SQLLKGDASL with L101Q
KMDKSDAVSH TGNYTCEVTE QTREGETIIE LKYRVVSWFS PNE
42 QLLFNKTKSV EFTFCNDTVV IPCFVTNMEA QNTTEVYVKW CD47 IgSF
domain
KFKGRDIYTF DGALNKSTVP TDFSSAKIEV SQLLKGDASL with L101Y
KMDKSDAVSH TGNYTCEVTE YTREGETIIE LKYRVVSWFS PNE
43 QLLFNKTKSV EFTFCNDTVV IPCFVTNMEA QNTTEVYVKW CD47 IgSF
domain
KFKGRDIYTF DGALNKSTVP TDFSSAKIEV SQLLKGDASL with L101H
KMDKSDAVSH TGNYTCEVTE HTREGETIIE LKYRVVSWFS PNE
44 QLLFNKTKSV EFTFCNDTVV IPCFVTNMEA QNTTEVYVKW CD47 IgSF
domain
KFKGRDIYTF DGALNKSTVP TDFSSAKIEV SQLLKGDASL with T102Q
KMDKSDAVSH TGNYTCEVTE LQREGETIIE LKYRVVSWFS PNE
45 QLLFNKTKSV EFTFCNDTVV IPCFVTNMEA QNTTEVYVKW CD47 IgSF
domain
KFKGRDIYTF DGALNKSTVP TDFSSAKIEV SQLLKGDASL with T102H
KMDKSDAVSH TGNYTCEVTE LHREGETIIE LKYRVVSWFS PNE
46 GQLLFNKTKSV EFTFCNDTVV IPCFVTNMEA QNTTEVYVKW CD47 IgSF
domain
KFKGRDIYTF DGALNKSTVP TDFSSAKIEV SQLLKGDASL with N-
terminal glycine
KMDKSDAVSH TGNYTCEVTE LTREGETIIE LKYRVVSWFS PNE addition
VII. Cleavable linkers
In some embodiments, a SIRP-a variant construct includes a SIRP-a variant
attached to a
blocking peptide. In some embodiments, a SIRP-a variant construct includes a
wild-type SIRP-a attached
to a blocking peptide. A linker used to fuse a SIRP-a variant or a wild-type
SIRP-a and a blocking peptide
can be a cleavable linker or a non-cleavable linker. In some embodiments, the
preferential binding of the
SIRP-a variant in the SIRP-a variant construct to CD47 on diseased cells or
diseased sites may be
obtained by attaching the block peptide to the SIRP-a variant by use of a
cleavable linker, which is
cleaved at the diseased cells or diseased sites.
In some embodiments, a cleavable linker is used between a SIRP-a variant and a
blocking
peptide. In some embodiments, a cleavable linker may be installed within a
blocking peptide, which may
28
be non-covalently associated with the SIRP-a variant to block binding of the
SIRP-a variant to CD47
under physiological conditions. A cleavable linker may be cleaved under
certain conditions. If the
cleavable linker is within a blocking peptide, cleavage of the linker would
inactivate the blocking peptide.
Under conditions characteristic of a diseased site, such as a cancer site,
e.g., inside a tumor, the linker is
cleaved to release the SIRP-a variant from the blocking peptide such that the
SIRP-a variant can bind to
nearby C047 on the cell surface of diseased cells, e.g., tumor cells. In this
manner, in a SIRP-a construct
that includes a SIRP-a variant and a blocking peptide, the SIRP-a variant can
only bind to CD47 on
diseased cells (e.g., tumor cells) or cells at a diseased site (e.g., cells in
the tumor micro-environment
supporting tumor growth), and is unable to bind to CD47 on non-diseased cells
under physiological
.. conditions, since the cleavable linker remains stable under physiological
conditions and the 0D47-binding
site of the SIRP-a variant would be blocked by the blocking peptide. A
cleavable linker may include
amino acids, organic small molecules, or a combination of amino acids and
organic small molecules that
cleave or induce cleavage of the linker under conditions characteristic of a
diseased site, such as acidic
pH, hypoxia, and increased protease expression. Cleavable linkers are stable
at physiological conditions
.. (e.g., neutral pH and adequate oxygen concentration). In some embodiments,
a cleavable linker may not
be cleaved and the blocking peptide may simply dissociate from the SIRP-a
variant at a diseased site
such that the SIRP-a variant is free to bind to nearby CD47 on diseased cells,
e.g., tumor cells. In these
embodiments, the SIRP-a variants may be engineered to have pH-dependent
binding to C047, the
details of which are described previously. The SIRP-a variants may be
engineered to bind with high
affinity to CD47 under acidic pH of a diseased site than under neutral pH
(e.g., around pH 7.4) of a non-
diseased site. Thus, the blocking peptide (e.g.., a CD-47 based blocking
peptide or a CD47 IgSF domain
blocking protein) may dissociate away from the SIRP-a variant under the acidic
pH of a diseased site. In
some embodiments, to engineer pH-dependent binding of a SIRP-a variant to CD47
at a diseased site,
histidine mutagenesis may be performed on the SIRP-a, especially on the region
of SIRP-a that interacts
.. with CD47.
pH-dependent cleavable linkers
One of the characteristics of a cancer site, e.g., inside a tumor, is acidic
pH. In some
embodiments, a linker may be cleaved under acidic pH (e.g., less than around
pH 7). An acid-sensitive õ
linker is stable at physiological pH (e.g., around pH 7.4). The cleavage at
acidic pH may be through acid
hydrolysis or by proteins present and active at acidic pH of a diseased site,
such as a cancer site, e.g.,
inside a tumor. Acid-sensitive linkers may include a moiety, such as a
chemical functional group or
compound, capable of being hydrolyzed under acidic pH. Acid-sensitive chemical
functional groups and
compounds include, but are not limited to, e.g., acetals, ketals,
thiomaleamates, hydrazones, and
disulfide bonds. Acid-sensitive linkers, as well as acid-sensitive chemical
groups and compounds, which
may be used in the construction of acid-sensitive linkers, are well known in
the art and described in US
Patent Nos. 8,748,399, 5,306,809, and 5,505,931, Laurent et al., (Bioconjugate
Chem. 21:5-13, 2010),
Castaneda et al. (Chem. Commun. 49:8187-8189, 2013), and Ducry et al.
(Bioconjug. Chem. 21:5-13,
2010). In one embodiment, a disulfide bond may be installed in a cleavable
linker using a peptide
synthesizer and/or conventional chemical synthesis techniques. In another
embodiment, a thiomaleamic
acid linker (Castaneda et al. Chem. Commun. 49:8187-8189, 2013) may be used as
the cleavable linker.
In this embodiment, to insert a
29
Date Regue/Date Received 2022-11-28
thiomalemic acid linker between a SIRP-a variant and a blocking peptide, one
of the two thiol groups of
the thiomalemic acid linker (see, e.g., Scheme 2, Castaneda et al.) may be
attached to the C-terminus of
a SIRP-a variant, while the ester group of the thiomalemic linker may be
attached to the N-terminus of the
blocking peptide.
Hypoxia-dependent cleavable linkers
In some embodiments, a linker may be cleaved under hypoxic condition, which is
another characteristic of
a cancer site, e.g., inside a tumor. A SIRP-a variant attached to a blocking
peptide by way of a
hypoxia-sensitive linker is prevented from binding to 0047 on non-diseased
cells while the linker remains
stable under physiological conditions (e.g., neutral pH and adequate oxygen
concentration). Once the
fusion protein is at the site of cancer, e.g., inside a tumor, where oxygen
concentration is significantly
lower than in healthy tissues, the linker is cleaved to release the SIRP-a
variant from the blocking peptide,
which can then bind to cell surface CD47 on tumor cells. The hypoxia-sensitive
linker may include a
moiety, e.g., an amino acid or a chemical functional group, capable of being
cleaved under hypoxic
condition. Some examples of chemical moieties that may be cleaved, i.e.,
cleaved through reduction,
under hypoxic condition include, but are not limited to, quinones, N-oxides,
and heteroaromatic nitro
groups. These chemical moieties may be installed in the cleavable linker using
conventional chemical
and peptide synthesis techniques. Examples of hypoxia-sensitive amino acids
are also known in the art,
such as those described by Shigenaga et al. (European Journal of Chemical
Biology 13:968¨ 971, 2012).
In a preferred embodiment, the hypoxia-sensitive amino acid described by
Shigenaga et al.
(European J. Chem, Biol. 13:968-971, 2012) may be inserted between a SIRP-a
variant and a blocking
peptide. For example, the amino group of the hypoxia-sensitive amino acid may
be attached to the C-
terminus of the SIRP-a variant through a peptide bond, and similarly, the
carboxylic acid group of the
hypoxia-sensitive amino acid may be attached to the N-terminus of the blocking
peptide through a peptide
bond. Under hypoxic condition, the reduction of the nitro group induces the
cleavage of the peptide bond
between the hypoxia-sensitive amino acid and the N-terminus of the blocking
peptide, thus, successfully
releasing the SIRP-a variant from the blocking peptide. The SIRP-a variant can
then bind to CD47 on
tumor cells.
In another embodiment, the hypoxia-sensitive 2-nitroimidazole group described
by Duan et al. (J.
Med. Chem. 51:2412-2420, 2008) may be inserted between a SIRP-a variant and a
blocking peptide or
installed in a cleavable linker inserted between a SIRP-a variant and a
blocking peptide. Under hypoxic
condition, the reduction of the nitro group induces further reduction, which
eventually leads to elimination
of the 2-nitroimidazole group from its attachment, e.g., the SIRP-g variant,
the blocking peptide, or the
cleavable linker.
Tumor-associated enzyme-dependent cleavable linkers
In other embodiments, a SIRP-a variant construct may include a SIRP-a variant
attached to a blocking
peptide by way of a linker (e.g., a cleavable linker) and optionally one or
more spacers
(examples of spacers are described in detail further herein). In some
embodiments, the linker (e.g., a
cleavable linker) may be cleaved by a tumor-associated enzyme. In some
embodiments, a linker, which
Date Regue/Date Received 2022-11-28
= CA 02938180 2016-08-04
=
can be cleaved by a tumor-associated enzyme, may be contained within a
blocking peptide, which may
be non-covalently attached to a SIRP-a variant. Once the fusion protein is at
the site of cancer, e.g.,
inside a tumor, the linker is cleaved by a tumor-associated enzyme to release
the SIRP-a variant from the
blocking peptide, which can then bind to cell surface CD47 on tumor cells. A
linker sensitive to a tumor-
associated enzyme may contain a moiety, e.g., a protein substrate, capable of
being specifically cleaved
by an enzyme, e.g., a protease, that is only present at the cancer site, e.g.,
inside a tumor. The moiety
may be selected based on the type of enzyme, e.g., a protease, present at the
cancer site, e.g., inside a
tumor. An exemplary cleavable linker that can be cleaved by a tumor-associated
enzyme is LSGRSDNH
(SEQ ID NO: 47), which can be cleaved by multiple proteases, e.g., matriptase
(MTSP1), urinary-type
plasminogen activator (uPA), legumain, PSA (also called KLK3, kallikrein-
related peptidase-3), matrix
metalloproteinase-2 (MMP-2), MMP9, human neutrophil elastase (NNE), and
proteinase 3 (Pr3).. Other
cleavable linkers that are susceptible to cleavage by enzymes (e.g.,
proteases) are also available. In
addition to the aforementioned proteases, other enzymes (e.g., proteases) that
can cleave a cleavable
linker include, but are not limited to, urokinase, tissue plasminogen
activator, trypsin, plasmin, and
another enzyme having proteolytic activities. According to some embodiments of
the present invention, a
SIRP-a variant or a wild-type SIRP-a is attached to a blocking peptide by way
of a linker (e.g., a cleavable
linker) susceptible to cleavage by enzymes having proteolytic activities, such
as a urokinase, a tissue
plasminogen activator, plasmin, or trypsin.
In some embodiments, sequences of cleavable linkers can be derived and
selected by putting
together several sequences based on different enzyme preferences. Non-limited
examples of several
potential proteases and their corresponding protease sites are shown in Table
7. In Table 7, "¨" means
any amino acid (i.e., any naturally occurring amino acid), capital case
indicates an strong preference for
that amino acid, lower case indicates a minor preference for that amino acid,
and "I" separates amino acid
positions in cases where more than one amino acid at a position adjacent to
the /is possible. Other
cleavable sequences include, but are not limited to, a sequence from a human
liver collagen (a1(III) chain
(e.g., GPLGIAGI (SEQ ID NO: 100))), a sequence from a human PZP (e.g.,
YGAGLGVV (SEQ ID NO:
101), AGLGVVER (SEQ ID NO: 102), or AGLGISST(SEQ ID NO: 103)), and other
sequences that are
autolytic VAQFVLTE(SEQ ID NO: 104), AQFVLTEG (SEQ ID NO: 105), or
PVQPIGPQ (SEQ ID
NO: 106)).
Table 7
Protease Potential protease sites
uPA: US/G-/RLiS-/D/N/H (SEQ ID NO: 69); -/s/gs/Rk-
/rv/-/-/- (SEQ ID NO: 70);
SGR-SA (SEQ ID NO: 71)
Matriptase: L/S/G-/R--/S-/D/N/H (SEQ ID NO: 72); ri-/--
/Rk-Iv-/-/g/- (SEQ ID NO: 73);
RQAR-VV (SEQ ID NO: 74); r/-/-/Rk/v/-/g (SEQ ID NO: 75);
/Kr/RKQ/gAS/RK/A (SEQ ID NO: 76)
Legumain: L/S/G-/R--/S-/D/N/H (SEQ ID NO: 77); ---/--/-
/N/-/-/- (SEQ ID NO: 78);
AAN-L (SEQ ID NO: 79); ATN-L (SEQ ID NO: 80)
PSA si/sq/-/yqr s/s/-/- (SEQ ID NO: 81);
S/S/K/L/Q (SEQ ID NO: 82)
MMP2 -/p/-/- /Ii/-/-/- (SEQ ID NO: 83)
31
Protease Potential protease sites
MMP9 g/pa/-/g1/-/g/- (SEQ ID NO: 84); G/P/UG/I/A/G/Q (SEQ
ID NO: 85);
PN/G/L/1/G (SEQ ID NO: 86); H/PN/G/UUA/R (SEQ ID NO: 87)
HNE -/-/-/viat-/-/-/- (SEQ ID NO: 88)
Pr3 -/y/y/vta -/-/-/- (SEQ ID NO: 89)
Pro-urokinase PRFKIIGG (SEQ ID NO: 90); PRFRI1GG (SEQ ID NO: 91)
TGF13 SSRHRRALD (SEQ ID NO: 92)
Plasminogen RKSSIIIRMRDVVL (SEQ ID NO: 93)
Staphylokinase SSSFDKGKYKKGDDA (SEQ ID NO: 94); SSSFDKGKYKRGDDA (SEQ
ID NO: 95)
Factor Xa IEGR; IDGR (SEQ ID NO: 96); GGSIDGR (SEQ ID NO: 97)
Gelatinase PLGLWA (SEQ ID NO: 98)
Human fibroblast DVAQFVLT (SEQ ID NO: 99)
collagenase
There are reports in the literature of increased levels of enzymes having
known substrates in
various types of cancers, e.g., solid tumors. See, e.g., La Rocca et al.,
Brit. J. Cancer 90:1414-1421 and
Ducry et al., Bioconjeig. Chem 21:5-13, 2010. Tumor-associated enzymes may
also be identified using
conventional techniques known in the art, e.g., immunohistoChemistry of tumor
cells. In one exemplary
embodiment, the enzyme-sensitive moiety in a linker may be a matrix
metalloproteinase (MMP) substrate,
which may be cleaved by an MMP present at the cancer site, e.g., inside a
tumor. In another exemplary
embodiment, the enzyme-sensitive moiety in a linker may be a maleimido-
containing dipeptide linker
(see, e.g., Table 1 in Ducry et al.), which may be cleaved through proteolysis
by proteases (e.g.,
cathepsin or plasmin) present at elevated levels in certain tumors (Koblinski
et al., Chim. Acta
291:113-135, 2000). In this embodiment, the maleimide group of the maleimido-
containing dipeptide
linker may be conjugated to a cysteine residue of the SIRP-a variant and the
carboxylic acid group at the
C-terminus of the maleimido-containing dipeptide linker may be conjugated to
the amino group at the
N-terminus of the blocking peptide. Similarly, the maleimide group of the
maleimido-containing dipeptide
linker may be conjugated to a cysteine residue of the blacking peptide and the
carboxylic acid group at
the C-terminus of the maleimido-containing dipeptide linker may be conjugated
to the amino group at the
N-terminus of the SIRP-a variant. Mass-spectrometry and other available
techniques in the field of
proteomics may be used to confirm the cleavage of the tumor-associated enzyme-
dependent cleavable
linkers. Other enzyme-sensitive moieties are described in US Patent No.
8,399,219. In some
embodiments, the moiety sensitive to a tumor-associated enzyme, e.g., a
protein substrate, may be
inserted between a SIRP-a variant and a blocking peptide using conventional
molecule cell biology and
chemical conjugation techniques well known in the art.
VIII. Serum albumin
Fusion to serum albumins can improve the pharmacokinetics of protein
pharmaceuticals, and in
particular, a SIRP-a variant described here may be joined with a serum
albumin. Serum albumin is a
globular protein that is the most abundant blood protein in mammals. Serum
albumin is produced in the
32
Date Regue/Date Received 2022-11-28
CA 02938180 2016-08-04
liver and constitutes about half of the blood serum proteins. It is monomeric
and soluble in the blood.
Some of the most crucial functions of serum albumin include transporting
hormones, fatty acids, and
other proteins in the body, buffering pH, and maintaining osmotic pressure
needed for proper distribution
of bodily fluids between blood vessels and body tissues. In some embodiments,
a SIRP-a variant may be
.. fused to a serum albumin. In preferred embodiments, serum albumin is human
serum albumin (NSA). In
some embodiments of the present invention, the N-terminus of an HSA is joined
to the C-terminus of the
SiRP-a variant to increase the serum half-life of the SIRP-a variant. An HSA
can be joined, either directly
or through a linker, to the C-terminus of the SIRP-a variant. Joining the N-
terminus of an HSA to the C-
terminus of the SIRP-a variant keeps the N-terminus of the SIRP-a variant free
to interact with CD47 and
the proximal end of the C-terminus of the HSA to interact with FcRn. An HSA
that can be used in the
methods and compositions described here are generally known in the art. In
some embodiments, the
HSA includes amino acids 25-609 (SEQ ID NO: 67) of the sequence of UniProt ID
NO: P02768. In some
embodiments, the HSA includes one or more amino acid substitutions (e.g., C345
and/or K573P), relative
to SEQ ID NO: 67. In some embodiments, the HSA has the sequence of SEQ ID NO:
68.
IX. Albumin-binding peptides
Binding to serum proteins can improve the pharmacokinetics of protein
pharmaceuticals, and in
particular the SIRP-a variants described here may be fused with serum protein-
binding peptides or
proteins. In some embodiments, a SIRP-a variant may be fused to an albumin-
binding peptide that
.. displays binding activity to serum albumin to increase the half-life of the
SIRP-a variant. Albumin-binding
peptides that can be used in the methods and compositions described here are
generally known in the
art. See, e.g., Dennis et al., J. Biol. Chem. 277:35035-35043, 2002 and
Miyakawa et al., J. Pharm.
102:3110-3118, 2013. In one embodiment, the albumin binding peptide includes
the sequence
DICLPRWGCLW (SEQ ID NO: 2). An albumin-binding peptide can be fused
genetically to a SIRP-a
variant or attached to a SIRP-a variant through chemical means, e.g., chemical
conjugation. If desired, a
spacer can be inserted between the SIRP-a variant and the albumin-binding
peptide to allow for
additional structural and spatial flexibility of the fusion protein. Specific
spacers and their amino acid
sequences are described in detail further herein. In some embodiments, an
albumin-binding peptide may
be fused to the N- or C-terminus of a SIRP-a variant. In one example, the C-
terminus of the albumin-
binding peptide may be directly fused to the N-terminus of the SIRP-a variant
through a peptide bond. In
another example, the N-terminus of the albumin-binding peptide may be directly
fused to the C-terminus
of the SIRP-a variant through a peptide bond. In yet another example, the
carboxylic acid at the C-
terminus of the albumin-binding peptide may be fused to an internal amino acid
residue, i.e., the side-
chain amino group of a lysine residue of the SIRP-a variant using conventional
chemical conjugation
techniques. Without being bound to a theory, it is expected that fusion of an
albumin-binding peptide to a
SIRP-a variant may lead to prolonged retention of the therapeutic protein
through its binding to serum
albumin.
X. Fc domains
In some embodiments, a SIRP-a variant construct may include a SIRP-a variant
and an Fc
domain monomer. In some embodiments, a SIRP-a variant may be fused to an Fc
domain monomer of
an immunoglobulin or a fragment of an Fc domain monomer. As conventionally
known in the art, an Fc
33
domain is the protein structure that is found at the C-terminus of an
immunoglobulin. An Fc domain
includes two Fc domain monomers that are dimerized by the interaction between
the CH3 antibody
constant domains. A wild-type Fc domain forms the minimum structure that binds
to an Fc receptor, e.g.,
FcyRI, FcyRIla, FcyRIlb, FcyRIlla, FcyR111b, FcyRIV. In the present invention,
an Fc domain monomer or
a fragment of an Fe domain fused to a SIRP-a variant to increase serum half-
life of the SIRP-a variant
may include a dimer of two Fc domain monomers or an Fc domain monomer,
provided that the Fc
domain monomer can bind to the Fc receptor (e.g., an FcRn receptor).
Furthermore, an Fc domain or a
fragment of the Fc domain fused to a SIRP-a variant to increase serum half-
life of the SIRP-a variant
does not induce any immune system-related response. In some embodiments, an Fc
domain may be
mutated to lack effector functions, typical of a "dead" Fc domain. For
example, an Fc domain may include
specific amino acid substitutions that are known to minimize the interaction
between the Fc domain and
an Fcy receptor. In some embodiments, an Fe domain monomer or a fragment of
the Fc domain may be
fused to the N- or C-terminus of a SIRP-a variant through conventional.
genetic or chemical means, e.g.,
chemical conjugation. If desired, a linker (e.g., a spacer) can be inserted
between the SIRP-a variant and
the Fe domain monomer.
Heterodimerization of Fc domain monomers
In some embodiments, each of the two Fc domain monomers in an Fe domain
includes amino
acid substitutions that promote the heterodimerization of the two monomers.
Heterodimerization of Fc
domain monomers can be promoted by introducing different, but compatible,
substitutions in the two Fc
domain monomers, such as "knob-into-hole" residue pairs and charge residue
pairs. The use of "knob-
into-hole" residue pairs is described by Carter and co-workers (Ridgway et
al., Protein Eng. 9:617-612,
1996; Atwell et al., .1 Mol Biol. 270:26-35, 1997; Merchant et al., Nat
Biotechnol. 16:677-681, 1998). The
knob and hole interaction favors heterodimer formation, whereas the knob-knob
and the hole-hole
interaction hinder homodimer formation due to steric clash and deletion of
favorable interactions. The
"knob-into-hole" technique is also disclosed in U.S. Patent Application
Publication No. 8,216,805,
Merchant et al., Nature Biotechnology 16:677-681, 1998, and Merchant et al.,
PIOC Nat! Acad Sci U S A.
110:E2987¨E2996, 2013. A hole is a void that is created when an original amino
acid in a protein is
replaced with a different amino acid having a smaller side-chain volume. A
knob is a bump that is created
when an original amino acid in a protein is replaced with a different amino
acid having a larger side-chain
volume. Specifically, the amino acid being replaced is in the CH3 antibody
constant domain of an Fc
domain monomer and is involved in the dimerization of two Fc domain monomers.
In some embodiments,
a hole in one CH3 antibody constant domain is created to accommodate a knob in
another CH3 antibody
. constant domain, such that the knob and hole amino acids act to promote
or favor the heterodimerization
of the two Fc domain monomers. In some embodiments, a hole in one CH3 antibody
constant domain is
created to better accommodate an original amino acid in another CH3 antibody
constant domain. In some
embodiments, a knob in one CH3 antibody constant domain is created to form
additional interactions with
original amino acids in another CH3 antibody constant domain.
A hole can be constructed by replacing amino acids having larger side chains
such as tyrosine or
tryptophan with amino acids having smaller side chains such as alanine,
valine, or threonine, such as
Y407V mutation in the CH3 antibody constant domain. Similarly, a knob can be
constructed by replacing
34
Date Regue/Date Received 2022-11-28
amino acids having smaller side chains with amino acids having larger side
chains, such as T366W
mutation in the CH3 antibody constant domain. In a preferred embodiment, one
Fc domain monomer
includes the knob mutation T366W and the other Fc domain monomer includes hole
mutations T366S,
L358A, and Y407V. A SIRP-a D1 variant of the invention may be fused to an Fc
domain monomer
including the knob mutation T366W to limit unwanted knob-knob homodimer
formation. Examples of
knob-into-hole amino acid pairs are included, without limitation, in Table 8.
ladle 8
T3665
Fc domain T394W T394S T366W
Y407T Y407A F405A T394S L358A
monomer 1 Y407T Y407A
T394S
Y407V
Fc domain T366Y
T366W F405W
T366Y T366W T394W F405W T366W
monomer 2 F405A
F405W Y407A
In addition to the knob-into-hole strategy, electrostatic steering strategy
may also be used to
control the dimerization of Fc domain monomers. Electrostatic steering is the
utilization of favorable
electrostatic interactions between oppositely charged amino acids in peptides,
protein domains, and
proteins to control the formation of higher ordered protein molecules. In
particular, to control the
dimerization of Fc domain monomers using electrostatic steering, one or more
amino acid residues that
make up the CH3-CH3 interface are replaced with positively- or negatively-
charged amino acid residues
such that the interaction becomes electrostatically favorable or unfavorable
depending on the specific
charged amino acids introduced. In some embodiments, a positively-charged
amino acid in the interface,
such as lysine, arginine, or histidine, is replaced with a negatively-charged
amino acid such as aspartic
acid or glutamic acid. In some embodiments, a negatively-charged amino acid in
the interface is replaced
with a positively-charged amino acid. The charged amino acids may be
introduced to one of the
' 20 interacting CH3 antibody constant domains, or both. Introducing
charged amino acids to the interacting
CH3 antibody constant domains of the two Fc domain monomers can promote the
selective formation of
heterodimers of Fc domain monomers as controlled by the electrostatic steering
effects resulting from the
interaction between charged amino acids. The electrostatic steering technique
is also disclosed in U.S.
Patent Application Publication No. 20140024111, Gunasekaran et al., J Biol
Chem. 285:19637-46, 2010,
and Martens et al., Clin Cancer Res. 12:6144-52, 2006. Examples of
electrostatic steering amino acid
pairs are included, without limitation, in Table 9.
Table 9
F d
K409 K370E
c omain
K409 K409 K392 K392
K409
monomer K409E K409E K392E K392E
K392
1
D
K439E
D
D399
356
Fc domain
D399 D399 D399 D399 D399 D399 D399 D399
monomer
E357K
2
D356D399
XI. Polyethylene glycol (PEG) polymer
In some embodiments, a SIRP-a variant may also be fused to a polymer, e.g.,
polyethylene glycol
(PEG). The attachment of a polymer to a protein pharmaceutical can "mask" the
protein pharmaceutical
from the host's immune system (Milla et al., Curr Drug Metab. 13:105-119,
2012). In addition, certain
Date Regue/Date Received 2022-11-28
polymers, e.g., hydrophilic polymers, can also provide water solubility to
hydrophobic proteins and drugs
(Gregor6dis et al., Cell Mot Life Sci. 57:1964-1969, 2000; Constantinou et
al., Bioconjug. Chem. 19:643-
650, 2008). Various polymers, such as PEG, polysialic acid chain (Constantinou
et al., Bioconjug. Chem.
19:643-650, 2008), and PAS chain (Schlapschy et al., Protein Eng. Des. SeL
26:489-501, 2013), are
known in the art and can be used in the present invention. In some
embodiments, a polymer, e.g., PEG,
may be covalently attached to a SIRP-a variant, either at the N- or C-terminus
or at an internal location,
using conventional chemical methods, e.g., chemical conjugation. In some
embodiments, a polymer,
e.g., PEG, may be covalently attached to a cysteine substitution or addition
in the SIRP-a variant. The
cysteine substitution in the SIR P-a variant may be I7C, A16C, S20C, T20C,
A45C, G45C, G79C, S79C,
or A84C, relative to the sequence of any one of SEQ ID NOs: 13-23. The
addition of a cysteine residue
in the SIRP-a variant may be introduced using conventional techniques in the
art, e.g., peptide synthesis,
genetic modification, and/or molecular cloning. The polymer, e.g., PEG, may be
attached to the cysteine
residue using cysteine-maleimide conjugation well-known to one of skill in the
art.
In addition to the embodiments described above, other half-life extension
technologies are also
available and may be used in the present invention to increase the serum half-
life of SIRP-a variants. Half-
life extension technologies include, but are not limited to, and EXTEN
(Schellenberger et al., Nat.
BiotechnoL 27:1186-1192, 2009) and Albu tag (Trussel et al., Bioconjug Chem.
20:2286-2292, 2009).
XII. Spacers
In some embodiments, spacers may be used in the SIRP-a variant construct. For
examples, a
SIRP-a variant construct may include a SIRP-a variant attached to a blocking
peptide by way of a linker
(e.g., a cleavable linker). In such SIRP-a constructs, a spacer may be
inserted between the SIRP-a
variant and the linker (e.g., a cleavable linker), and/or between the linker
(e.g., a cleavable linker) and the
blocking peptide. To optimized the spacing between the SIRP-a variant and the
linker, and/or the spacing
between the linker and the blocking peptide, any one or more of the spacers
described below may be
used.
In some embodiments of a SIRP-a variant construct including a SIRP-a variant
attached to a
blocking peptide by way of a linker (e.g., a cleavable linker), the spacer
serves to position the cleavable
linker away from the core of the SIRP-a variant and the blocking peptide such
that the cleavable linker is
more accessible to the enzyme responsible for cleavage. It should be
understood that the attachment of
two elements in a SIRP-a variant construct, for example, a SIRP-a variant and
a linker (e.g., a cleavable
linker) in a SIRP-a variant construct including (e.g., in this order) a SIRP-a
variant, a linker, and a blocking
peptide, need not be of particular mode of attachment or through a particular
reaction. Any reaction
providing a SIRP-a variant construct of suitable stability and biological
compatibility is acceptable.
A spacer refers to a linkage between two elements in a SIRP-a variant
construct, e.g., a SIRP-a
variant and a linker (e.g., a cleavable linker) in a SIRP-a variant construct
including (e.g., in this order) a
SIRP-a variant, a linker, and a blocking peptide, a linker (e.g., a cleavable
linker) and a blocking peptide in
a SIRP-a variant construct including (e.g., in this order) a SIRP-a variant, a
linker, and a blocking peptide,
a SIRP-a variant and a serum protein-binding peptide or protein, e.g., an
albumin-binding peptide. A
spacer may also refer to a linkage that can be inserted between a SIRP-a
variant or a wild-
36
Date Regue/Date Received 2022-11-28
CA 02938180 2016-08-04
type SIRP-a and an antibody, e.g., a tumor-specific antibody, or an antibody-
binding peptide. A spacer
can provide additional structural and/or spatial flexibility of the SIRP-a
variant construct. A spacer can be
a simple chemical bond, e.g., an amide bond, a small, organic molecule (e.g.,
a hydrocarbon chain), an
amino acid sequence (e.g., a 3-200 amino acid sequence), or a combination of a
small, organic molecule
(e.g., a hydrocarbon chain) and an amino acid sequence (e.g., a 3-200 amino
acid sequence). A spacer
is stable under physiological conditions (e.g., neutral pH and adequate oxygen
concentration) as well as
under conditions characteristic of a diseased site, e.g., acidic pH and
hypoxia. A spacer is stable at a
diseased site, such as a cancer site, e.g., inside a tumor.
A spacer may include 3-200 amino acids. Suitable peptide spacers are known in
the art, and
include, for example, peptide linkers containing flexible amino acid residues,
such as glycine and serine.
In certain embodiments, a spacer can contain motifs, e.g., multiple or
repeating motifs, of GS, GGS,
GGGGS, GGSG, or SGGG. In certain embodiments, a spacer can contain 2 to 12
amino acids including
motifs of GS, e.g., GS, GSGS, GSGSGS, GSGSGSGS, GSGSGSGSGS, or GSGSGSGSGSGS.
In
certain other embodiments, a spacer can contain 3 to 12 amino acids including
motifs of GGS, e.g., GGS,
GGSGGS, GGSGGSGGS, and GGSGGSGGSGGS. In yet other embodiments, a spacer can
contain 4
to 12 amino acids including motifs of GGSG, e.g., GGSG, GGSGGGSG, or
GGSGGGSGGGSG. In other
embodiments, a spacer can contain motifs of (GGGGS)õ, wherein n is an integer
from 1 to 10. In other
embodiments, a spacer can also contain amino acids other than glycine and
serine, e.g., GENLYFQSGG,
SACYCELS, RSIAT, RPACKIPNDLKQKVIVINH,
GGSAGGSGSGSSGGSSGASGTGTAGGTGSGSGTGSG, AAANSSIDLISVPVDSR, or
GGSGGGSEGGGSEGGGSEGGGSEGGGSEGGGSGGGS. In some embodiments in the present
invention, one or more 12- or 20-amino acid peptide spacers may be used in a
SIRP-a variant construct.
The 12- and 20-amino acid peptide spacers may contain sequences GGGSGGGSGGGS
and
SGGGSGGGSGGGSGGGSGGG, respectively. In some embodiments, one or more 18-amino
acid
peptide spacers containing sequence GGSGGGSGGGSGGGSGGS may be used in a SIRP-a
variant
construct.
¨
2 n
In some embodiments, a spacer may also have the general structure
wherein W is NH or CH2, Q is an amino acid or a peptide, and n is an integer
from 0 to 20.
XIII. Fusion of a blocking peptide to a SIRP-a variant
A blocking peptide (e.g., a CD47-based blocking peptide having a sequence of
any one of SEQ
ID NOs: 36-46 in Table 6) may be fused to the N- or C-terminus of a SIRP-a
variant by way of a linker,
e.g., a cleavable linker (e.g., LSGRSDNH (SEQ ID NO: 47)), and optionally one
or more spacers (e.g.,
(GGGGS)n, fused genetically to either N- or C-terminus of the linker, wherein
n is an integer from 1 to 10).
Exemplary sequences of SIRP-a variant constructs including a CD47-based
blocking peptide fused to a
SIRP-a variant by way of a cleavable linker and one or more spacers are shown
in sequences of SEQ ID
NOs: 48-63. The length of the spacers may be changed to achieve the most
optimized binding between
the CD47-based blocking peptide and the SIRP-a variant.
37
CA 02938180 2016-08-04
XIV. Methods of producing SIRP-a variant constructs
The SIRP-a variant constructs of the invention can be produced from a host
cell. A host cell
refers to a vehicle that includes the necessary cellular components, e.g.,
organelles, needed to express
the polypeptides and constructs described herein from their corresponding
nucleic acids. The nucleic
acids may be included in nucleic acid vectors that can be introduced into the
host cell by conventional
techniques known in the art (e.g., transformation, transfection,
electroporation, calcium phosphate
precipitation, direct microinjection, infection, etc). The choice of nucleic
acid vectors depends in part on
the host cells to be used. Generally, preferred host cells are of either
prokaryotic (e.g., bacterial) or
eukaryotic (e.g., mammalian) origin.
Nucleic Acid Vector Construction and Host Cells
A polynucleotide sequence encoding the amino acid sequence of a SIRP-a variant
construct may
be prepared by a variety of methods known in the art. These methods include,
but are not limited to,
oligonucleotide-mediated (or site-directed) mutagenesis and PCR mutagenesis. A
polynucleotide
molecule encoding a SIRP-a variant construct of the invention may be obtained
using standard
techniques, e.g., gene synthesis. Alternatively, a polynucleotide molecule
encoding a wild-type SIRP-a
may be mutated to contain specific histidine substitutions using standard
techniques in the art, e.g.,
QuikChangeTM mutagenesis. Polynucleotides can be synthesized using nucleotide
synthesizer or PCR
techniques.
Polynucleotide sequences encoding SIRP-a variant constructs may be inserted
into a vector
capable of replicating and expressing the polynucleotides in prokaryotic or
eukaryotic host cells. Many
vectors are available in the art and can be used for the purpose of the
invention. Each vector may
contain various components that may be adjusted and optimized for
compatibility with the particular host
cell, For example, the vector components may include, but are not limited to,
an origin of replication, a
selection marker gene, a promoter, a ribosome binding site, a signal sequence,
a polynucleotide
sequence encoding a SIRP-a variant construct of the invention, and a
transcription termination sequence.
In some embodiments, a vector can include internal ribosome entry site (IRES)
that allows the expression
of multiple SIRP-a variant constructs. Some examples of bacterial expression
vectors include, but are not
limited to, pGEX series of vectors (e.g., pGEX-2T, pGEX-3X, pGEX-4T, pGEX-5X,
pGEX-6P), pET series
of vectors (e.g., pET-21, pET-21a, pET-21b, pET-23, pET-24), pACYC series of
vectors (e.g., pACYDuet-
1), pDEST series of vectors (e.g., pDEST14, pDEST15, pDEST24, pDEST42), and
p8R322 and its
derivatives (see, e.g., U.S. Patent No. 6,648,237). Some examples of mammalian
expression vectors
include, but are not limited to, pCDNA3, pCDNA4, pNICE, pSELECT, and pFLAG-
CMV. Other types of
nucleic acid vectors include viral vectors for expressing a protein in a cell
(e.g., a cell of a subject). Such
viral vectors include, but are not limited to, retroviral vectors, adenoviral
vectors, poxviral vectors (e.g.,
vaccinia viral vectors, such as Modified Vaccinia Ankara (MVA)), adeno-
associated viral vectors, and
alphaviral vectors.
In some embodiments, E. coli cells are used as host cells for the invention.
Examples of E. coli
strains include, but are not limited to, E. co//294 (ATCC 31,446), E. co/iA
1776 (ATCC 31,537, E. coli
BL21 (DE3) (ATCC BAA-1025), and E. coli RV308 (ATCC 31,608). In other
embodiments, mammalian
cells are used as host cells for the invention. Examples of mammalian cell
types include, but are not
limited to, human embryonic kidney (HEK) cells, Chinese hamster ovary (CHO)
cells, HeLa cells, PC3
38
CA 02938180 2016-08-04
cells, Vero cells, and MC3T3 cells. Different host cells have characteristic
and specific mechanisms for
the posttranslational processing and modification of protein products.
Appropriate cell lines or host
systems may be chosen to ensure the correct modification and processing of the
protein expressed. The
above-described expression vectors may be introduced into appropriate host
cells using conventional
techniques in the art, e.g., transformation, transfection, electroporation,
calcium phosphate precipitation,
and direct microinjection. Once the vectors are introduced into host cells for
protein production, host cells
are cultured in conventional nutrient media modified as appropriate for
inducing promoters, selecting
transformants, or amplifying the genes encoding the desired sequences.
Protein Production, Recoveiy, and Purification
Host cells used to produce the SIRP-a variant constructs of the invention may
be grown in media
known in the art and suitable for culturing of the selected host cells.
Examples of suitable media for
bacterial host cells include Luria broth (LB) plus necessary supplements, such
as a selection agent, e.g.,
ampicillin. Examples of suitable media for mammalian host cells include
Minimal Essential Medium
(MEM), Dulbecco's Modified Eagle's Medium (DMEM), DMEM with supplemented fetal
bovine serum
(FBS), and RPMI-1640.
Host cells are cultured at suitable temperatures, such as from about 20 C to
about 39 C, e.g.,
from 25 C to about 37 'C. The pH of the medium is generally from about 6.8 to
7.4, e.g., 7.0, depending
mainly on the host organism. If an inducible promoter is used in the
expression vector of the invention,
protein expression is induced under conditions suitable for the activation of
the promoter.
Protein recovery typically involves disrupting the host cell, generally by
such means as osmotic
shock, sonication, or lysis. Once the cells are disrupted, cell debris may be
removed by centrifugation or
filtration. The proteins may be further purified, for example, by affinity
resin chromatography. Standard
protein purification methods known in the art can be employed. The following
procedures are exemplary
.. of suitable purification procedures: fractionation on immunoaffinity or ion-
exchange columns, ethanol
precipitation, reverse phase HPLC, chromatography on silica or on a cation-
exchange resin, SOS-PAGE,
and gel filtration.
Alternatively, SIRP-a variant constructs can be produced by the cells of a
subject (e.g., a human),
e.g., in the context of therapy, by administrating a vector (e.g., a
retroviral vector, adenoviral vector,
poxviral vector (e.g., vaccinia viral vector, such as Modified Vaccinia Ankara
(MVA)), adeno-associated
viral vector, and alphaviral vector) containing a nucleic acid molecule
encoding the SIRP-a variant
construct. The vector, once inside a cell of the subject (e.g., by
transformation, transfection,
electroporation, calcium phosphate precipitation, direct microinjection,
infection, etc) will promote
expression of the SIRP-a variant construct, which is then secreted from the
cell.
XV. Pharmaceutical compositions and preparations
In some embodiments, pharmaceutical compositions of the invention may contain
one or more
SIRP-a variant constructs of the invention as the therapeutic proteins. In
addition to a therapeutic amount
of the protein, the pharmaceutical compositions may contain a pharmaceutically
acceptable carrier or
excipient, which can be formulated by methods known to those skilled in the
art. In other embodiments,
pharmaceutical compositions of the invention may contain nucleic acid
molecules encoding one or more
SIRP-a variant constructs of the invention (e.g., in a vector, such as a viral
vector). The nucleic acid
39
molecule encoding a SIRP-a variant construct may be cloned into an appropriate
expression vector,
which may be delivered via well-known methods in gene therapy.
Acceptable carriers and excipients in the pharmaceutical compositions are
nontoxic to recipients
at the dosages and concentrations employed. Acceptable carriers and excipients
may include buffers
.. such as phosphate, citrate, HEPES, and TAE, antioxidants such as ascorbic
acid and methionine,
preservatives such as hexamethonium chloride, octadecyldimethylbenzyl ammonium
chloride, resorcinol,
and benzalkonium chloride, proteins such as human serum albumin, gelatin,
dextran, and
immunoglobulins, hydrophilic polymers such as polyvinylpyrrolidone, amino
acids such as glycine,
glutamine, histidine, and lysine, and carbohydrates such as glucose, mannose,
sucrose, and sorbitol.
Pharmaceutical compositions of the invention can be administered parenterally
in the form of an
injectable formulation. Pharmaceutical compositions for injection can be
formulated using a sterile
solution or any pharmaceutically acceptable liquid as a vehicle.
Pharmaceutically acceptable vehicles
include, but are not limited to, sterile water, physiological saline, and cell
culture media (e.g., Dulbecco's
Modified Eagle Medium (DMEM), a-Modified Eagles Medium (a-MEM), F-12 medium).
The pharmaceutical compositions of the invention may be prepared in
microcapsules, such as
hydroxylmethylcellulose or gelatin-microcapsule and poly-(methylmethacrylate)
microcapsule. The
pharmaceutical compositions of the invention may also be prepared in other
drug delivery systems such
as liposomes, albumin microspheres, microemulsions, nano-particles, and
nanocapsules. Such =
techniques are described in Remington: The Science and Practice of Pharmacy
20th edition (2000). The
pharmaceutical compositions to be used for in vivo administration must be
sterile. This is readily
accomplished by filtration through sterile filtration membranes.
The pharmaceutical compositions of the invention may also be prepared as a
sustained-release
formulation. Suitable examples of sustained-release preparations include
semipermeable matrices of
solid hydrophobic polymers containing the SIRP-a variant constructs of the
invention. Examples of
sustained release matrices include polyesters, hydrogels, polyactides (U.S.
Patent No. 3,773,919),
copolymers of L-glutamic acid and y ethyl-L-glutamate, non-degradable ethylene-
vinyl acetate,
degradable lactic acid-glycolic acid copolymers such as LUPRON DEPOTTm, and
poly-D-(-)-3-
hydroxybutyric acid. Some sustained-release formulations enable release of
molecules over a few
months, e.g., one to six months, while other formulations release
pharmaceutical compositions of the
invention for shorter time periods, e.g., days to weeks.
The pharmaceutical composition may be formed in a unit dose form as needed.
The amount of
an active component, e.g., a SIRP-a variant construct of the invention,
included in the pharmaceutical
preparations is such that a suitable dose within the designated range is
provided (e.g., a dose within the
range of 0.01-100 mg/kg of body weight).
The pharmaceutical composition for gene therapy can be in an acceptable
diluent, or can
comprise a slow release matrix in which the gene delivery vehicle is imbedded.
Vectors that may be used
as in vivo gene delivery vehicle include, but are not limited to, retroviral
vectors, adenoviral vectors,
poxviral vectors (e.g., vaccinia viral vectors, such as Modified Vaccinia
Ankara (MVA)), adeno-associated
viral vectors, and alphaviral vectors. In some embodiments, a vector can
include internal ribosome entry
site (IRES) that allows the expression of multiple SIRP-a variant constructs.
Other vehicles and methods
for gene delivery are described in U.S. Patent Nos. 5,972,707, 5,697,901, and
6,261,554.
Date Regue/Date Received 2022-11-28
Other methods of producing pharmaceutical compositions are described in, e.g.,
U.S. Patent Nos.
5,478,925, 8,603,778, 7,662,367, and 7,892,558.
XVI. Routes, dosage, and timing of administration
Pharmaceutical compositions of the invention that contain one or more SIRP-a
variant constructs
as the therapeutic proteins may be formulated for parenteral administration,
subcutaneous administration,
intravenous administration, intramuscular administration, intra-arterial
administration, intrathecal
administration, or intraperitoneal administration. The pharmaceutical
composition may also be formulated
for, or administered via, nasal, spray, oral, aerosol, rectal, or vaginal
administration. Methods of
administering therapeutic proteins are known in the art. See, for example,
U.S. Patent Nos. 6,174,529,
6,613,332, 8,518,869, 7,402,155, and 6,591,129, and U.S. Patent Application
Publication Nos.
US20140051634, W01993000077, and US20110184145. One or more of these methods
may be used
to administer a pharmaceutical composition of the invention that contains one
or more SIRP-a variant
constructs of the invention. For injectable formulations, various effective
pharmaceutical carriers are
known in the art. See, e.g., Pharmaceutics and Pharmacy Practice, J. B.
Lippincott Company,
Philadelphia, Pa., Banker and Chalmers, eds., pages 238-250 (1982), and ASHP
Handbook on Injectable
Drugs, Toissel, 4th ed., pages 622-630 (1986).
The dosage of the pharmaceutical compositions of the invention depends on
factors including the
route of administration, the disease to be treated, and physical
characteristics, e.g., age, weight, general
health, of the subject. Typically, the amount of a SIRP-a variant construct of
the invention contained
within a single dose may be an amount that effectively treats the disease
without inducing significant
toxicity. A pharmaceutical composition of the invention may include a dosage
of a SIRP-a variant
construct ranging from 0.001 to 500 mg (e.g., 0.05, 0.01,0.1, 0.2, 0.3, 0.5,
0.7, 0.8, 1 mg, 2 mg, 3 mg, 4
mg, 5 mg, 10 mg, 15 mg, 20 mg, 30 mg, 50 mg, 100 mg, 250 mg, or 500 mg) and,
in a more specific
embodiment, about 0.1 to about 100 mg and, in a more specific embodiment,
about 0.2 to about 20 mg.
The dosage may be adapted by the clinician in accordance with conventional
factors such as the extent
of the disease and different parameters of the subject.
A pharmaceutical composition of the invention can be administered in an amount
from about
0.001 mg up to about 500 mg/kg/day (e.g., 0.05, 0.01, 0.1, 0.2, 0.3, 0.5, 0.7,
0.8, 1 mg, 2 mg, 3 mg, 4 mg,
5 mg, 10 mg, 15 mg, 20 mg, 30 mg, 50 mg, 100 mg, 250 mg, or 500 mg/kg/day).
Pharmaceutical
compositions of the invention that contain a SIRP-a variant construct may be
administered to a subject in
need thereof, for example, one or more times (e.g., 1-10 times or more) daily,
weekly, monthly, biannually,
annually, or as medically necessary. Dosages may be provided in either a
single or multiple dosage
regimens. For example, in some embodiments, the effective amount is a dose
that ranges from about 0.1
to about 100 mg/kg/day, from about 0.2 mg to about 20 mg of the SIRP-a variant
construct per day, about
1 mg to about 10 mg of the SIRP-a variant construct per day, from about 0.7 mg
to about 210 mg of the
SIRP-a variant construct per week, 1.4 mg to about 140 mg of the SIRP-a
variant construct per week,
about 0.3 mg to about 300 mg of the SIRP-a variant construct every three days,
about 0.4 mg to about 40
mg of the SIRP-a variant construct every other day, and about 2 mg to about 20
mg of the
41
Date Regue/Date Received 2022-11-28
CA 02938180 2016-08-04
SIR P-a variant construct every other day. The timing between administrations
may decrease as the
medical condition improves or increase as the health of the patient declines.
XVII. Methods of treatment
The invention provides pharmaceutical compositions and methods of treatment
that may be used
to treat patients who are suffering from diseases and disorders associated
with SIRP-a and/or CD47
activity, such as cancers and immunological diseases. In some embodiments, the
SIRP-a variant
constructs described herein may be administered to a subject in a method of
increasing phagocytosis of a
target cell (e.g., a cancer cell) in the subject. In some embodiments, the
SIRP-a variant constructs may
be administered to a subject in a method of eliminating regulatory T-cells in
the subject. In some
embodiments, the SIRP-a variant constructs may be administered to a subject in
a method to kill cancer
cells in the subject. In some embodiments, the SIRP-a variant constructs may
be administered to a
subject in a method of treating a disease associated with SIRP-a and/or CD47
activity in the subject,
wherein the SIRP-a variant construct preferentially binds CD47 on diseased
cells or at a diseased site
over CD47 on non-diseased cells. In some embodiments, the SIRP-a variants may
be administered to a
subject in a method of increasing hematopoietic stem cell engraftment in the
subject, wherein the method
includes modulating the interaction between SIRP-a and CD47 in the subject. In
some embodiments, the
SIRP-a variant constructs may be administered to a subject in a method of
altering an immune response
(i.e., suppressing the immune response) in the subject.
In some embodiments, before treating a disease (e.g., cancer) in a subject,
the amino acid
sequence(s) of SIRP-a in the subject are determined, for example, from each of
the two alleles encoding
the SIRP-a gene. In this method of the invention, the method determines the
amino acid sequences of
SIR P- polypeptide in a biological sample from the subject, and then
administers to the subject a
therapeutically effective amount of a SIRP-a variant construct. In this
method, the SIRP-a variant in the
SIRP-a variant construct has the same amino acid sequence as that of a SIRP-a
polypeptide in the
biological sample of the subject, except for the amino acids changes
introduced to increase affinity of the
SIRP-a variant. The SIRP-a variant construct has minimal immunogenicity in the
subject after it is
administered.
The SIRP-a variant constructs and pharmaceutical compositions of the invention
may be used in
various cancer therapies. The cancers amenable to treatment according to the
invention include, but are
not limited to, solid tumor cancer, hematological cancer, acute myeloid
leukemia, chronic lymphocytic
leukemia, chronic myeloid leukemia, acute lymphoblastic leukemia, non-Hodgkin
lymphoma, Hodgkin
lymphoma, multiple myeloma, bladder cancer, pancreatic cancer, cervical
cancer, enclometrial cancer,
lung cancer, bronchus cancer, liver cancer, ovarian cancer, colon and rectal
cancer, stomach cancer,
gastric cancer, gallbladder cancer, gastrointestinal stromal tumor cancer,
thyroid cancer, head and neck
cancer, oropharyngeal cancer, esophageal cancer, melanoma, non-melanoma skin
cancer, Merkel cell
carcinoma, virally induced cancer, neuroblastoma, breast cancer, prostate
cancer, renal cancer, renal cell
cancer, renal pelvis cancer, leukemia, lymphoma, sarcoma, glioma, brain tumor,
and carcinoma. In some
embodiments, cancerous conditions amenable to treatment according to the
invention include metastatic
cancers. In some embodiments, the cancer amenable to treatment according to
the invention is a solid
tumor or hematological cancer.
42
= CA 02938180 2016-08-04
The SIRP-a variant constructs and pharmaceutical compositions of the invention
may be used in
various therapies to treat immunological diseases. In some embodiments, the
immunological disease is
an autoimmune disease or an inflammatory disease, such as multiple sclerosis,
rheumatoid arthritis, a
spondyloarthropathy, systemic lupus erythematosus, an antibody-mediated
inflammatory or autoimmune
disease, graft versus host disease, sepsis, diabetes, psoriasis,
atherosclerosis, Sjogren's syndrome,
progressive systemic sclerosis, scleroderma, acute coronary syndrome, ischemic
reperfusion, Crohn's
Disease, endometriosis, glomerulonephritis, myasthenia gravis, idiopathic
pulmonary fibrosis, asthma,
acute respiratory distress syndrome (ARDS), vasculitis, or inflammatory
autoimmune myositis.
EXAMPLES
Example 1 ¨ Methods
Production of SIRP-a variant constructs
All gene constructs are generated using gene synthesis and codon optimized for
expression in
mammalian cells (DNA2.0). The genes are cloned into mammalian expression
vectors and expressed
using CMVa-intron promoter. A leader sequence has been engineered at the N-
terminus of the
constructs to ensure appropriate signaling and processing for secretion. The
expression of SIRP-a fusion
proteins is carried out using Expi293FTM cells (Life Technologies). This cell
line is adapted to high
density, serum-free suspension culture in Expi293FTM Expression Medium and is
capable of producing
high levels of recombinant proteins. Transfection procedures have been
performed according to
manufacturer's manual. The supernatant is typically harvested at 5-7 days post
transfection. The protein
constructs are designed to carry a 6xhistidine affinity tag and this allows
purification by affinity
chromatography. The column was first equilibrated with 5 mM imidazole, 100 mM
Tris HCl (pH 8), 500
mM NaCI. The clarified media expressing the various SIRP-a variant constructs
is loaded onto Hi-Trap Ni
Sepharose excel affinity resin on Avant 25 (GE Healthcare). Another
equilibration step is performed.
After that, the column is washed with 40 mM imidazole, 100 mM Tris, 500mM
NaCl, and subsequently
eluted with 250 mM imidazole, 100 mM Tris, 500 mM NaCI. Eluted fractions
containing the SIRP-a
variant constructs are pooled and thereafter buffer exchanged into 1X PBS.
In vitro cleavage of SIRP-a proteins
Recombinant human uPA and matriptase are purchased from R&D systems. 3 pM of
SIRP-a
proteins are added to respective amounts of uPA and matriptase (0.1 to 44 ng)
in 50 mM Tris HCI (pH
8.5), 0.01% Tween as described. The digestion reactions are typically
incubated for 18-24 hours at 37 C.
To stop the reaction, SDS-PAGE loading dye is added to the reaction and heated
at 95 C for 3 minutes.
To assess cleavage, the digested samples are separated on a 4-20% Tris-Glycine
SDS-PAGE.
Example 2 ¨ Design of SIRP-a variant constructs that will be specifically
activated in tumor tissue
The goal is to design SIRP-a variant constructs that will remain inert until
activated locally to bind
to CD47 in tumor tissue. This will limit binding of SIRP-a to CD47 on the cell-
surface of non-diseased
cells and prevent undesirable "on-target" "off tissue" toxicity. To generate
such SIRP-a variant constructs,
the blocking peptides (e.g., a CD47-based blocking peptide) are genetically
fused to the SIRP-a variant
by way of a cleavable linker. The blocking peptides explored are based on CD47
interaction sites to
SIRP-a and the sequences are described below (sections (a)-(c)). Spacers
containing repeated units of
43
= CA 02938180 2016-08-04
GGGGS are designed to flank the cleavable linker, which often encodes a
protease recognition site. In
some embodiments, the protease cleavage site chosen is LSGRSDNH, but many
others are possible.
The protease cleavage site LSGRSDNH is selected for its sensitivity to
numerous proteases that are up-
regulated in a variety of human carcinomas, for example, matriptase (MTSP1),
urinary-type plasminogen
activator (uPA), legumain, PSA (also called KLK3, kallikrein-related peptidase-
3), matrix
metalloproteinase-2 (MMP-2), MMP9, human neutrophil elastase (HNE), and
proteinase 3 (Pr3) (Ulisse et
al., Curr. Cancer Drug Targets 9:32-71, 2009; Uhlancl et al., Cell. Mol. Life
Sci. 63:2968-2978, 2006;
LeBeau et al., Proc. Nett Acad. Sci. USA 110:93-98, 2013; Liu et al., Cancer
Res. 63:2957-2964, 2003).
(a) Blocking S1RP-a by CD47-based blocking peptides
CD47-based blocking peptides are described previously. These peptides bind
SIRP-a with
different affinities and block its function. The N-terminus of CD47 is
important for the interaction with
SIRP-a, therefore, structural analysis predicted fusing SIRP-a to the C-
terminus of CD47. To better
understand the results, both N-terminal and C-terminal fusions are explored
with different lengths of
spacers. Different CD-47 based blocking peptides (e.g., peptides listed in
Table 6) are fused to the N- or
C-terminus of a SIRP-a variant with a cleavable linker and one or more
spacers. Exemplary sequences
of fusion proteins containing a CD47-based blocking peptide fused to a SIRP-a
variant by way of a
cleavable linker and one or more spacers are shown in sequences of SEQ ID NOs:
48-56, in which
single-underlined portion indicates the CD47-based blocking peptide, double-
underlined portion indicates
the cleavable linker, and bold portion indicates the SIRP-a variant. Sequences
of SEQ ID NOs: 48-51
contain CD47-based blocking peptides that include 12 or 21 amino acids and
spacers of 2-3 repeats of
GGGGS. Sequences of SEQ ID NOs: 52-56 contain CD47-based blocking peptides
that include the
CD47 IgSF domain (truncated at VVS) having a C15S substitution and spacers of
2-5 repeats of GGGGS
or 3-6 repeats of GGS. Additionally, in some embodiments, an HSA may be fused
to the C-terminus of
any one of the sequences of SEQ ID NOs: 48-56. Furthermore, in some
embodiments, an Fc domain
monomer or an HSA (SEQ ID NO: 68) may be fused to either the N- or C-terminus
of any one of the
fusion proteins listed in Table 10.
Table 10
SEQ Fusion protein of a CD47-based blocking peptide and a SIRP-a
variant
ID NO
48 SEVTELTREGETGGGGSGGGGSLSGRSDNHGGGGSGGGGSEEELQI IQ PDKSVINAAGE
TATLRC T ITS
LFPVGP I QWFRGAGPGRVL IYNQRQGPFP-RVTTVSDTTKRNNMDFS IRIGNI TPADAGTYYCIKFRKGS
PDDVE FKS GAGTELSVRAKPS
49 SEVTELTREGETGGGGSGGGGSGGGGSLSGRS
DNHGGGGSGGGGSEEELQIIQPDKSVINAAGETATLR
-6-iiTSLFPVGPIQWFRGAGPGRVLIYNQRQ6ii'iRVTTVSDTTKRNNMDFSIRIGNITPADAGTYYCIK
FRKGSPDDVEFKSGAGTELSVRAKPS
50 GQYTSEVTELTREGET I
IELKGGGGSGGGGSLSGRSDNHGGGGSGGGGSEEELQIIQPDKSVLVAAGET
ATLRC T I T SLFPVGP I QWERGAGPGRVL IYNQRQGPFPRVTTVSDTTKRNNMDFS IRI GNI
TPADAGTY
Y C IKFRKGS PDDVE FKS GAGTELSVRAKPS
51 GQYTSEVTELTREGE T I IELKGGGGSGGGGSGGGGSLSGRS
DNHGGGGSGGGGSEEELQIIQPDKSVLV
AAGE TATLRC T I TSLFPVGP I QWFRGAGPGRVL I YNQRQGP FPRVT TVSD TTKRNNMDFS I RI
GNI TPA
DAG TYYC I KFRKGS PDDVE FKSGAGTELSVRAKPS
52 QLLFNKTKSVEFT FSNDTVVI
PCFVTNMEAQNTTEVYVKWKFKGRDIYTFDGALNKSTVPTDFSSAKIE
VSQLLKGDASLKMDKSDAVSHTGNYTCEVTELTREGE T I IELKYRVVSGGGGSGGGGSGGGGSLSGRSD
NHGGGGSGGGGSEEELQ I I QPDKSVLVAAGE TATLRC T I T SLFPVGP I QWFRGAGPGRVL I
YNQRQGP F
PRVTTVSD TTKRNNMDFS I RI GNI TPADAGTYYCIKFRKGSPDDVE FKSGAG TEL SVRAKP S
44
CA 02938180 2016-08-04
SEQ Fusion protein of a CD47-based blocking peptide and a SIRP-a
variant
ID NO
53
QLLFNKTKSVEFTFSNDTVVIPCFVTNMEAONTTEVYVKWKFKGRDIYTFDGALNKSTVPTDFSSAKIE
VSQLLKGDASLKMDKSDAVSHTGNYTCEVTELTREGE T I IELKYRVVSGGGGSGGGGSGGGGSGGGGSL
SGRSDNHGGGGSGGGGSEEELQI I QPDKSVLVAAGE TATLRCT I TS LFPVGPIQWFRGAGPGRVL I YNQ
RQGPFPRVTTVSDTTKRNNMD FS IRI GN I TPADAGTYYC IKFRKGSPDDVEFKSGAGTELSVRAKPS
54
QLL FNKTKSVEFT FSNDTVVI PCFVTNMEAQNTTEVYVKWKFKGRDIYTFDGALNKS TVPTDFSSAKIE
VSQLLKGDASLKMDKSDAVSHTGNYTCEVTELTREGET I IELKYRVVSGGGGSGGGGSGGGGSGGGGSG
GGGSLSGRSDNHGGGGSGGGGSEEELQI IQPDKSVLVAAGE TATLRCT I TSLFPVGPIQWFRGAGPGRV
LIYNQRQGPFPRVTTVSDTTKRNNMDFS IRI GNI T PADAGTYYC IKFRKGSPDDVEFKSGAGTELSVRA
KPS
55
EEELQI IQPDKSVINAAGE TATLRCT I TSLFPVGP I QWFRGAGPGRVL IYNQRQGPFPRVTTVSDTTKR
NNMDFS IRI GNI TPADAGTYYC I KFRKGS PDDVE FKS GAGTELSVRAKPS GG S GG S GG S LS
GRS DNHGG
S GGSGGSGGS QLLFNKTKSVE FTFSN DTVV I PCFVTNMEAQNTTEVYVKWKFKGRDI TI EDGALNKS TV
PTDESSAKIEVSQLLKGDASLKMDKSDAVSHTGNYTCEVTELTREGET I I ELKYRVVS
56
EEELQIIQPDKSVLVAAGETATLRCTITSLFPVGPIQWFRGAGPGRVLIYNQRQGPFPRVTTVSDTTKR
NNMDFS IRI GNI TPADAGTYYC IKFRKGS PDDVE FKS GAGTELSVRAKPS GGSGGSGGS GGS LS GRS
DN
HGGSGGSGGSGGSGGSGGSQLLFNKTKSVEFT FSNDTVVI PCFVTNMEAQNT TEVYVKWKFKGR-D¨I-Y-T
DGALNKSTVPTDFSSAKIEVSQLLKGDASLKMDKS DAVSHTGNYTCEVTELTREGET II ELKYRVVS
(b) Blocking SIRP-a variant by a low affinity CD47 mutant having an extended N-
terminus
Due to the high affinity of the SIRP-a variant for CD47, there is a chance
that after cleaving the
linker, the fusion protein may not dissociate and the SIRP-a variant may
remain blocked. To solve this,
we studied the structure of the S1RP-a-0047 complex and designed CD47 mutants
with reduced binding
affinity for the SIRP-a variant relative to the binding affinity for a wild-
type SIRP-a. Thus, after protease
cleavage of the linker, the CD47 mutant will dissociate away from the SIRP-a
variant. The designed
CD47 mutants are described below. Initial experiments will be performed fusing
the SIRP-a variant to the
wild-type C047. These SIRP-a variant constructs including a SIR P-a variant
and a wild-type CD47 or
CD47 mutant will be cleaved in vitro, analyzed by SDS-page to ensure cleavage,
and measured by
biacore to see their capacity of binding to CD47 (i.e., the binding of the
SIRP-a variant to wild-type CD47
after the CD47 mutant is dissociated from the SIRP-a variant). If the initial
SIRP-a variant constructs
containing a SIRP-a variant fused to the wild-type CD47 are expressed, able to
block CD47 binding
before protease cleavage, and able to bind CD47 after protease cleavage, CD47
mutants may not be
needed. If these initial SIRP-a variant constructs are inactive (i.e., can be
cleaved but do not bind CD47
after protease cleavage due to the lack of dissociation), then other fusion
proteins containing a SIRP-a
variant fused to the low affinity CD47 mutants will be tested.
In the co-crystalized structure of CD47:SIRP-a (PDB: 4KJY, 4CMM), the N-
terminus of CD47
exists as a pyro-glutamate and makes hydrogen bonding interactions with Thr66
of a SIRP-a variant and
Leu66 of a wild-type SIRP-a (FIG. 1). It is hypothesized that extending the N-
terminus of CD47 by adding
an amino acid, e.g., a glycine, will prevent cyclization of glutamine to
pyroglutamate and also create other
unwanted contacts and interactions that will likely will disrupt the hydrogen
bonding interactions with
Thr66 or Leu66, and therefore perturb binding of C047 to SIPR-a. Sequences of
the fusion proteins
containing a low affinity CD47 IgSF domain mutant and a SIRP-a variant are
shown in SEQ ID NOs: 57-
59 in Table 11, in which single-underlined portion indicates the low affinity
CD47 IgSF domain mutant
containing amino acids 1-118 and C15S, relative to SEQ ID NO: 46 in Table 6,
double-underlined portion
indicates the cleavable linker, and bold portion indicates the SIRP-a variant.
SEQ ID NOs: x10-x12 also
include spacers of 3-5 repeats of GGGGS. Sequences similar to SEQ ID NOs: x10-
x12 may be designed
= CA 02938180 2016-08-04
and expressed in which a low affinity CD47 IgSF domain mutant is fused to the
C-terminus of a SIRP-a
variant by way of a cleavable linker and one or more spacers.
Table 11
SEQ ID Fusion protein of a low affinity CD47 IgSF domain mutant having an
extended N-
NO terminal glycine and a SIRP-a variant
57 GQLLFNKTKSVEFTFSNDTVVIPCFVTNMEAQNTTEVYVKWKFKGRDIYT
EDGALNKSTVPTDESSA
KIEVSQLLKGDASLKMDKSDAVSHTGNYTCEVTELTREGET II ELKYRVVSGGGGSGGGGSGGGGSL
SGRSDNHGGGG'SGGGG SEE ELQI IQPDKSVLVAAGE TATLRCT I TSLFPVGP IQWFRGAGPGRVL IY
NQRQGPFPRVTTVSDTTKRNNNEWS IRI GN I T PADAGTYYC IKFRKGS PDDVEFKSGAGTELSVRAK
PS
58 GQLLFNKTKSVEFT FSNDTVV I PCFVTNMEAQNTTEVYVKWKEKGRDI
YTEDGALNKSTVPTDESSA
KIEVSQLLKGDASLKMDKSDAVSHTGNYTCEVTELTREGET I I ELKYRVVSGGGGSGGGGS GGGGS G
GGGSLSGRSDNHGGGGSGGGGSEEELQI IQ PDKSVLVAAGE TATLRCT I TSLFPVGP I QWFRGAGP G
RVLIYNQRQGPFPRVTTVSDT TKRNNMD FS IRI GNI TPADAGTYYC I KFRKGS PDDVE FKS GAGTE L
SVRAKPS
59 GQLLFNKTKSVEFT FSNDTVV I PCFVTNMEAQNT TEVYVKWKFKGRDI YT
FDGALNKSTVPTDFSSA
KIEVSQLLKGDASLKMDKSDAVSHTGNYTCEVTELTREGET I IELKYRVVSGGGGSGGGGSGGGGSG
GGGSGGGGSLSGRSDNHGGGGSGGGGSEEELQIIQPDKSVLVAAGETATLRCTITSLFPVGPIQWFR
GAGPGRVLIYN-Q-RQ6P" F-PR'VT TVSD TTKR.NNMDFS I RI GNI TPADAGTYYCIKFRKGSPDDVE
FKSG
AGTELSVRAKPS
(c) Blocking SIRP-a by a low-affinity CD47 IgSF domain mutant haying amino
acid substitutions
CD47 binds to a deep pocket to SIRP-a (PDB code: 41{,1Y and 4CMM).
Computational modeling
has been performed to identify amino acid residues in the pocket region of
CD47, which, when mutated
would reduce the binding affinity of CD47 to SIRP-a variant, but maintain the
binding affinity of CD47 to a
wild-type SIPR-a. The CD47 residues identified are L101Q, L.101H, 1_101Y,
T102Q, and T102H. It is
hypothesized that a low-affinity CD47 IgSF domain mutant containing one of
these substitutions will be
able to block the SIRP-a variant efficiently in the tethered mode. However,
upon reaching the tumor site
and cleavage by proteases at the linker locally, the low-affinity CD47 IgSF
domain mutant will dissociate
from the S1RP-a variant to bind to a wild-type SIRP-a, leaving the SIRP-a
variant free to bind CD47 on
the cell-surface of tumor cells. The dissociated, low-affinity CD47 IgSF
domain mutant can now block
activity of wild-type SIRP-a. This will potentially result in enhanced double-
blocking activity from the
released low-affinity CD47 IgSF domain mutant and the SIRP-a variant. To
illustrate how the amino acid
residues are selected and the principle of how this may result in differential
blocking of wild-type SIRP-a
and SIRP-a variant, an example is shown using Ala27 of a wild-type SIRP-a
(FIG. 2). For instance, Ala27
of the wild-type SIRP-a is a smaller residue than 11e27 in the SIRP-a variant.
Therefore, by mutating
Thr102 in the wild-type CD47 to a larger amino acid such as GIn102, Gln102 in
the low-affinity CD47
IgSF domain mutant may result in a steric clash with 11e27 in the SIRP-a
variant at the corresponding
interaction site. However, the interaction between the CD47 mutant having
Thr102GIn substitution and
the wild-type SIRP-a having Ala27 would be preserved. Accordingly, the CD47
mutant would have a low
binding affinity to the SIRP-a variant and a relatively higher binding
affinity to the wild-type S1RP-a.
Sequences of some exemplary low-affinity CD47 IgSF domain mutants are shown in
SEQ ID NOs: 41-45
in Table 6. Sequences of the SIRP-a variant constructs containing a low
affinity CD47 IgSF domain
mutant having amino acid substitutions and a SIRP-a variant are shown in SEQ
ID NOs: 60-63 in Table
12, in which single-underlined portion indicates the low affinity CD47 IgSF
domain mutant containing
amino acid substitution L101Q, L101Y, T102Q, and T102H, respectively, double-
underlined portion
indicates the cleavable linker, and bold portion indicates the SIRP-a variant.
SEQ ID NOs: 60-63 also
46
= CA 02938180 2016-08-04
include spacers of 3-5 repeats of GGGGS. Sequences similar to SEQ ID NOs: 60-
63 may be designed
and expressed in which a low affinity CD47 IgSF domain mutant is fused to the
C-terminus of a SIRP-a
variant by way of a cleavable linker and one or more spacers.
Table 12
SEQ Fusion protein of a low affinity CD47 IgSF domain mutant having
amino acid
ID NO substitutions and a SIRP-a variant
60 QLLFNKTKSVEFT FSN DTVV I PCFVTNMEAQNTTEVYVKWKFKGRDIYTFDGALNKS
TVPTDFS SAKI E
VSQLLKGDASLKMDKSDAVSHTGNYTCEVTEQTREGE T I IELKYRVVSGGGGSGGGGSGGGGSGGGGSL
S GRS DNHGGGGS GGGGSEEELQI IQPDKSVLVAAGE TATLRCT I TS LEPVGPIQWERGAGPGRvLI YNQ
RQGPFPRITTTVSDTTKRNMDFS I RI GN I T PADAGTYYC IKFRKGS PDDVE FKSGAGTELSVRAKPS
61 QLLENKTKSVEFTESNDTVVI
PCFVTNMEAQNTTEVYVKWKFKGRDIYTEDGALNKSTVPTDESSAKIE
VSQLLKGDASLKMDKSDAVSHTGNYTCEVTEYTREGET I IELKYRVVSGGGGSGGGGSGGGGSGGGGSL
SGRSDNHGGGGSGGGGSEEELQI IQPDKSVLVAAGETATLRCTIPSLFPVGPIQWFRGAGPGRVLIYNQ
RQGPFPRVTIVSDTTKRNNMD FS IRI GN I T PADAGTYYC IKFRKGS PDDVE FKSGAGTE LSVRAKPS
62 QLLFNKTKSVEFT FSNDTVVI PCFVTNMEAQNTTEVYVKWKFKGRDIYT FDGALNKS TVPT
DFSSAKIE
VSQLLKGDASLKMDKSDAVSHTGNYTCEVTELQREGET I IELKYRVVSGGGGSGGGGSGGGGSGGGGSL
SGRSDNHGGGGSGGGGSEEELQI I QPDKSVLVAAGE TATLRCTI TSLFPVGP I QWFRGA.GPGRVL IYNQ
RQGPFPRVTTVSDTTKRNNMD FS I RI GN I T PADAGTYYC IKFRKGS PDDVE FKSGAGTE LSVRAKPS
63 QLLFNKTKSVEFT FSNDTVVI PCFVTNMEAQNTTEVYVKWKFKGRDIYTEDGALNKS TVPT
DES SAKIE
VSQLLKGDASLKMDKSDAVSH TGNYTCEVTELHREGET I IELKYRVVSGGGGSGGGGSGGGGSGGGGSL
SQRSDNHGGGGSGGGGSEEELQI IQPDKSVLVAAGE TATLRCT I TSLFPVGP IgWFRGAGPGRVLI YNQ
RQG-Pi=PRVTIVSDTTKRNNMD FS I RI GN I T PADAGTYYC IKFRKGS PDDVEFKSCAGTELSVRAKPS
Example 3 ¨ Expression and production of SIRP-a variant constructs for in
vitro studies
Various SIRP-a variant constructs (SEQ ID NOs: 48-56) including a SIRP-a
variant and a CD47-
based blocking peptide were expressed in Expi293-F mammalian cells. All the
constructs were designed
with a leader sequence that enabled their expression as secreted proteins into
the media. All constructs
were expressed in the soluble form and purified using one-step IMAC isolation
to high degree of purity
(FIGs. 3A and 38). FIG. 3A shows a reduced, SDS-PAGE gel of SIRP-a variant
constructs of SEQ ID
NOs: 48-56 and FIG. 3B shows a non-reduced, SDS-PAGE gel of the SIRP-a variant
constructs. Size
exclusion data indicated that the SIRP-a variant constructs are not aggregated
(data not shown).
Example 4¨ In vitro cleavage of SIRP-a and CD47 fusion proteins
To determine whether the SIRP-a variant constructs (e.g., SEQ ID NOs: 48-63)
would be cleaved
specifically in vivo at tumor tissues, in vitro experiments were performed
using proteases uPA and
matriptase, which are commonly known to be up-regulated in cancers, to cleave
the SIRP-a variant
constructs_ Initial experiments were performed using SIRP-a variant construct
(SEQ ID NO: 54) to
determine protease cleavability and optimize cleavage conditions. FIG. 4A
shows the results of testing
cleavability by uPA and matriptase. 3 pM SIRP-a variant construct (SEQ ID NO:
54) was incubated for 18
hrs at 37 'C using excess uPA or matriptase. Lane 1 of FIG. 4A shows the
control experiment with no
addition of protease and lanes 2 and 3 show incubation of the S1RP-a variant
construct (SEQ ID NO: 54)
with uPA and matriptase, respectively. Data obtained as shown in FIG. 4A
clearly demonstrate that the
SIRP-a variant construct (SEQ ID No: 54) can be cleaved and released in vitro
by digesting the SIRP-a
variant construct with excess uPA and matriptase for 18 hrs at 37 C. The
cleaved SIRP-a variant
migrates as a ¨17 KDa molecular weight band. Cleaved CD47 migrates with smeary
banding, most likely
due to glycosylation, around 36-40 kDa. By comparing the amount of uncut SIRP-
a variant construct in
47
CA 02938180 2016-08-04
lanes 2 and 3 of FIG. 4A, it appears that a more complete cleavage was
achieved using matriptase
compared to using uPA.
Therefore further optimization of cleavage conditions was performed only using
matriptase and
the results are shown in FIG. 4B. Different amounts of matriptase were tested
and the cleavage was
performed for 18 hrs at 37 C. Lane 1 of FIG. 4B shows the control experiment
with no addition of
matriptase and lanes 2-4 each shows cleavage performed with 44 ng, 0.44 ng,
and 0.167 ng of
matriptase, respectively. The data obtained indicate that 0.44 ng enzyme is
sufficient for complete
cleavage under current conditions. Next, we tested cleavage of remaining SIRP-
a variant constructs
using the optimized cleavage conditions. As an example and shown in FIG. 4C,
respective SIRP-a
variant constructs (SEQ ID NOs: 57-63) were cleaved successfully in vitro by
matriptase using the
optimized cleavage conditions. Lanes 1-7 of FIG. 4C correspond to the
uncleaved fusion proteins of SEQ
ID NOs: 57-63, respectively. Lanes 8-14 of FIG. 4C correspond to the fusion
proteins of SEQ ID NOs:
57-63, respective, cleaved with matriptase.
Example 5 ¨ Binding affinities of SIRP-a variant constructs
Binding of human CD47-hFc (R & D Systems, catalog number 4670-CD) to SIRP-a
variant
constructs was analyzed on a Biacore T100 instrument (GE Healthcare) using
phosphate buffered saline
(pH 7.4) supplemented with 0.01% Tween-20 as running buffer.
370 Resonance Unit (RU) of CD47-hFc were immobilized on flow cell 2 of a CM4
sensor chip
(GE Healthcare) by standard amine coupling. Flow cell 1 was activated with
EDC/NHS and blocked (with
ethanolamine) to serve as a reference. All SIRP-a variant constructs were
injected at 50 nM or 100 nM
for two minutes at a flow rate of 30 pL/min and followed by ten minutes of
dissociation time. After each
injection, the surface was regenerated using a 2:1 mixture of Pierce IgG
elution buffer (Life Technologies,
catalog number 21004) and 4 M NaCl. Complete regeneration of the surface was
confirmed by injecting
the SIRP-a variants at the beginning and end of the experiment. All
sensorgrams were double-
referenced using flow cell 1 and a buffer injection.
For all samples, the binding signal at 100 nM after 50 seconds of association
was determined,
normalized by the molecular weight of the SIRP-a variant construct, and
expressed as percent of maximal
binding response. The binding of the SIR P-a variant (SEQ ID NO: 31) at 100 nM
normalized by its MW
was used as maximal binding response. Results in FIG. 5A show that SIRP-a
variant constructs (SEQ ID
NOs: 48-51) do not block SIRP-a variants from binding to CD47 on the chip.
After cleavage of the linker,
the binding activity modestly increases. SIRP-a variant constructs (SEQ ID
NOs: 52-54) efficiently
blocked binding of the SIRP-a variant to CD47 on the chip. However, after
cleavage of the linker, the
binding activity of the SIRP-a variant to CD47 on the chip only modestly
increased, suggesting that the
high affinity of interaction between the SIRP-a variant and the IgSF domain of
CD47 keeps the complex
together and therefore the IgSF domain of CD47 continues block the SIRP-a
variant even after the linker
is cleaved. Surprisingly, when the IgSF domain of CD47 is fused to the C-
terminus of SIRP-a (SEQ ID
NO: 55), the intact SIRP-a variant construct is efficiently blocked from
binding to CD47 on the chip (same
as fusion proteins of SEQ ID NOs: 52-54), but cleavage of the linker restores
100% of the binding of the
SIRP-a variant to CD47 on the chip, suggesting that the IgSF domain of CD47
dissociated from the SIRP-
a variant after linker cleavage, thus, the SIRP-a variant is free to bind to
CD47 on the chip. Another
construct with CD47 fused to the C-terminus of a SIRP-a variant was tested
later (see FIG. 5B). This
48
CA 02938180 2016-08-04
construct (SEQ ID NO: 56), which contains a longer spacer, also recovered
activity after cleaving,
confirming the general approach of linking the N-terminus of a CD47-based
blocking peptide to the C-
terminus of a SIRP-a variant obtain SIRP-a constructs in which the CD47-based
blocking peptide
efficiently blocks the SIRP-a variant and dissociates after cleaving of the
cleavable linker.
To further examine the binding affinity of SIRP-a variant constructs, SIRP-a
variant constructs of
SEQ ID NOs: 52-63 were analyzed on the Biacore instrument following the same
protocol described
previously. SIRP-a variant constructs of SEQ ID NOs: 52-54 contain the C047
IgSF domain having
amino acids 1-117 and C15S, relative to wild-type CD47 (SEQ ID NO: 35) fused
to the N-terminus of the
SIRP-a variant (SEQ ID NO: 31) through the cleavable linker LSGRSDNH and
multiple spacers of
different lengths. SIRP-a variant constructs of SEQ ID NOs: 55 and 56 contain
the CD47 IgSF domain
having amino acids 1-117 and C15S, relative to wild-type CD47 (SEQ ID NO: 35)
fused to the C-terminus
of the S1RP-a variant (SEQ ID NO: 31) through the cleavable linker LSGRSDNH
and multiple spacers of
different lengths. SIRP-a variant constructs of SEQ ID NOs: 57-59 contain the
CD47 IgSF domain having
amino acids 1-118 and C15S, relative to SEQ ID NO: 46 in Table 6 fused to the
N-terminus of the SIRP-a
variant (SEQ ID NO: 31) through the cleavable linker LSGRSDNH and multiple
spacers of different
lengths. S1RP-a variant constructs of SEQ ID NOs: 60-63 contain the CD47 IgSF
domain having amino
acids 1-117 of SEQ ID NOs: 41, 42, 44, and 45 in Table 6, respectively, fused
to the N-terminus of the
SIRP-a variant (SEQ ID NO: 31) through the cleavable linker LSGRSDNH and
multiple spacers of
different lengths.
FIG. 5B shows that SIR P-a variant constructs of SEQ ID NOs: 55 and 56 were
efficiently blocked
from binding to CD47 on the chip before linker cleavage, but cleavage of the
linker restores 100% of the
binding activity. Similar results were observed for SIRP-a variant constructs
of SEQ ID NO: 57-63. We
observed that by extending the N-terminus of the CD47-based blocking peptide
by one glycine residue
generated a SIRP-a variant construct that was efficiently blocked before the
linker was cleaved and
subsequently recovered close to 100% of the CD47 binding activity after
protease treatment (SEQ ID
NOs: 57-59), demonstrating dissociation of the CD47-based blocking peptide
from the SIRP-a variant =
after linker cleavage.
This result suggests SIRP-a variant fused to the N-terminus of CD47-based
blocking peptide
through a cleavable linker and spacers works well. The cleavable linker
stabilizes the fusion complex and
once cleaved, the extended N-terminus of the CD47-based blocking peptide,
which includes a fragment
of the cleavable linker attached to the N-terminus of the CD47-based blocking
peptide, prevents binding
of the C047-based blocking peptide to the SIRP-a variant. The same effect is
obtained by fusing a
CD47-based blocking peptide having one or more amino acid additions, e.g., one
glycine addition (e.g.,
sequences of SEQ ID NO: 46 in Table 6), at the N-terminus of the CD47-blocking
peptide to the C-
terminus of a SIRP-a variant by way of a cleavable linker and one or more
spacers. This same effect is
also observed by fusing a CD47-based blocking peptide having one or more amino
acid substitutions,
e.g., L101Q, L101Y, L101H, T102Q, or T102H (e.g., sequences of SEQ ID NOs: 41-
45 in Table 6), to the
C-terminus of a SIRP-a variant by way of a cleavable linker and one or more
spacers. We have
demonstrated that CD47-based blocking peptides can be fused to the C-terminus
of a SIRP-a variant and
can block SIR P-a variant binding to CD47 before linker cleavage and release
SIRP-a variant after linker
cleavage (see, e.g., SEQ ID NO: 55 in FIG. 5A, and SEQ ID NOs: 55 and 56 in
FIG. 5B).
49
CA 02938180 2016-08-04
Based on this information, we can create fusion proteins of C047-blocked SIRP-
a variants, i.e.,
fusing an Fc domain monomer or HSA to a SIRP-a variant, by choosing the
orientation (e.g., N- or C-
terminal fusion) that gives better results in pharmacokinetics, efficacy,
safety, production, and stability of
the product.
Example 6 - Specific targeting of SIRP-a variants through antibody-binding
peptide
First, we used Cetuximab, which is known to contain a binding site for the DLP
having the
sequence of SEQ ID NO: 64, to check if the SIRP-a variant construct including
a SIRP-a variant and the
DLP is able to concentrate on bound antibody. We immobilized Cetuximab using
EDC/NHS chemistry on
a CM4 biacore chip (2000RU) and flowed the SIRP-a variant construct (SEQ ID
NO: 66) at 100 nM and
50 nM using PBS 0.01% P20 as running and sample buffer at 30 pL/min onto the
chip (biacore T100).
FIG: 6 shows the binding of the SIRP-a variant construct, but not the SIRP-a
variant alone, onto the chip.
We then injected CD47-ECD and saw binding of CD47 in the case where the SIRP-a
variant construct
was used, demonstrating that the SIRP-a variant construct can bind EGFR and
CD47 simultaneously
(FIG. 6). Therefore, the SIRP-a variant construct including a SIRP-a variant
and a DLP injected to a
cancer patient would concentrate at the site where the therapeutic antibody
(e.g., Cetuximab)
accumulates, increasing efficacy and reducing toxicity.
Secondly, we demonstrated that the SIRP-a variant construct including a SIRP-a
variant and a
DLP can first bind Cetuximab that is bound to EGFR, and then bind CD47. A
scheme of the binding
complex is shown in FIG. 7A. We immobilized 3000 RUs of hrEGFR-Fc (R&D
Systems) to a CM4 chip
using EDC/NHS chemistry. Using PBS 0.01% P20 as sample and running buffer at
30 pUmin (biacore
T100), we injected different concentrations (4, 20, and 100 nM) of Cetuximab.
Binding of Cetuximab to
the immobilized hrEGFR-Fc was observed. We then injected the SIRP-a variant
construct (SEQ ID NO:
66) at 100 nM and observed binding. Binding was not observed when the SIRP-a
variant was injected
alone. We then injected CD47-ECD at 100 nM and observed binding. The data is
shown in FIGs. 7B and
7C. Therefore, we demonstrated that the formation of the quaternary complex
EGFR-Cetuximab-SIRP-a
variant construct of SEQ ID NO: 66-CD47 is possible. The SIRP-a variant
construct including a SIRP-a
variant and a DLP is able to bind and inhibit CD47 when the construct pre-
concentrates at the diseased
site by binding specifically to a tumor-specific antibody (e.g., Cetuximab).
Furthermore, following the
same concept, a SIRP-a variant construct including a SIRP-a variant, a DLP,
and a CD47-based blocking
peptide is also able to bind and inhibit CD47 when the construct pre-
concentrates at the diseased site by
binding specifically to a tumor-specific antibody (e.g., Cetuximab).
Example 7 ¨ Phagocytosis assay
SIRP-a variant construct (SEQ ID NO: 66), which includes a SIRP-a variant
attached to DLPs
through spacers, and a SIRP-a variant (SEQ ID NO: 31) were tested in a
phagocytosis assay on DLD1
cells (FIG. 8). Phagocytosis assay was performed as described in, e.g.,
Weiskofp et al, Science 341:88-
91, 2013. An experimental protocol is described below. The SIRP-a variant
construct (SEQ ID NO: 66)
showed higher potency than the SIRP-a variant alone (SEQ ID NO: 31),
presumably due to higher
accumulation on disease cells.
Buffy coats were obtained from the Stanford Blood Center from anonymous
donors, and
peripheral blood mononuclear cells were enriched by density gradient
centrifugation over Ficoll-Paque
Premium (GE Healthcare). Monocytes were purified using Macs Miltenyi Biotec
Monocyte Isolation Kit II
according to the manufacturer's instructions. This is an indirect magnetic
labeling system for the isolation
of monocytes from human PBMCs. The isolated monocytes are differentiated into
macrophages by
culturing in RPM! 1640 media supplemented with 10% heat-inactivated human AB
serum and 1%
GlutaMax and 1% penicillin and streptomycin (GIBCO Life Technologies) for 6-10
days. For
phagocytosis assay, 100,000 GFP+ DLD-1 cells are plated ontovells of Ultra low
attachment U bottom
96 well plate (Corning 7007). 50 pUwell of either 4 pg/ml IgG1k isotype
control or 4 pg/ml Cetuximab
(Absolute Antibody, Ab00279-10.0) are added to OLD-1 tumor cells and pre-
incubated for 30 minutes at
room temp. After that, 50 pL/well SIRP-a variants are added and 50 p1./well
macrophages (1 x 106/m1)
(50,000 macrophages) are also added to each well. Final dilution of antibodies
and SIRP-a construct
samples is 1:4. Cetuximab final concentration is 1pg/ml. The co-culturing of
macrophages, tumor cells,
antibodies and SIRP-a variant constructs are carried out for 2 hours at 37 C.
For analysis, cell samples
were fixed, stained and analyzed by BD FACS Canto. Primary human macrophages
were identified by
flow cytometry using anti-CD14, anti-CD45, or anti-CD206 antibodies
(BioLegend). Dead cells were
excluded from the analysis by staining with DAPI (Sigma). Phagocytosis was
evaluated as the
percentage of GFP+ macrophages and normalized to the maximal response by each
independent donor
against each cell line.
Example 8 ¨ Modeling pH dependent binding of SIRP-a variants to C1347
To engineer pH-dependent binding of a SIRP-a variant of the invention,
histidine mutagenesis
may be performed on the SIRP-a, especially on the region of SIRP-a that
interacts with CD47. Crystal
structures of a SIRP-a and CD47 complex (see, e.g., PDB ID No. 21.IS) and
computer modeling may be
used to visualize the three-dimensional binding site of SIRP-a and CD47.
Computational design and
modeling methods useful in designing a protein with pH-sensitive binding
properties are known in the
literature and described in, e.g., Strauch et al., Proc Nati Acad Sci 111:675-
80, 2014. In some
embodiments, computer modeling may be used to identify key contact residues at
the interface of SIRP-a
and C047. Identified key contact residues may be substituted with histidine
residues using available
protein design software (e.g., RosettaDesign), which can generate various
protein designs that can be
optimized, filtered, and ranked based on computed binding energy and shape
complementarity.
Therefore, energetically favorable histidine substitutions at certain amino
acid positions may be identified
using computational design methods. Computer modeling may be also be used to
predict the change in
the three-dimensional structure of SIRP-a. Histidine substitutions that
generate a significant change in
the three-dimensional structure of SIRP-a may be avoided.
Once energetically and structurally optimal amino acid substitutions are
identified, the amino
acids may be systematically substituted with histidine residues. In some
embodiments, one or more (e.g,,
one, two, three, four, five, six, seven, eight, nine, ten, etc, with a maximum
of 20) amino acids of SIRP-a
may be substituted with histidine residues. In particular, amino acids located
at the interface of SIRP-a
and CD47, preferably, amino acids directly involved in the binding of SIRP-a
to C047, may be substituted
with histidine residues. The SIRP-a variants of the invention may include one
or more (e.g., one, two,
three, four, five, six, seven, eight, nine, ten, etc, with a maximum of 20)
histidine residue substitutions. In
other embodiments, naturally occurring histidine residues of SIRP-a may be
substituted with other amino
51
Date Regue/Date Received 2022-11-28
acid residues. In yet other embodiments, one or more amino acids of SIRP-a may
be substituted with
non-histidine residues in order to affect the binding of naturally occurring
or substituted histidine residues
with CD47. For example, substituting amino acids surrounding a naturally
occurring histidine residue with
other amino acids may "bury" the naturally occurring histidine residue. In
some embodiments, amino
.. acids not directly involved in binding with CD47, i.e., internal amino
acids (e.g., amino acids located at the
core of SIRP-a) may also be substituted with histidine residues. Table 4 lists
specific SIRP-a amino acids
that may be substituted with histidine residues. Contact residues are the
amino acids located at the
interface of SIRP-a and C047. Core residues are the internal amino acids not
directly involved in the
binding between SIRP-a and CD47. The SIRP-a variants of the invention may
include one or more (e.g.,
one, two, three, four, five, six, seven, eight, nine, ten, etc, or all) of the
substitutions listed in Table 4.
Example 9¨ Generating and screening SIRP-a variants with pH-dependent binding
to C047
The SIRP-a variants containing one or more (e.g., one, two, three, four, five,
six, seven, eight,
nine, ten, etc, with a maximum of 20) substitutions of amino acids with
histidine residues may be
generated using conventional molecular cloning and protein expression
techniques. A nucleic acid
molecule encoding a SIR P-a variant of the invention may be cloned into a
vector optimized for expression
in bacteria using well known molecular biology techniques. The vector can then
be transformed into
bacteria cells (e.g., E. coli cells), which may be grown to optimal density
prior to protein expression
induction. After protein expression induction (i.e., using IPTG), bacterial
cells may be allowed to grow for
an additional 24 hours. Cells can be collected and the expressed SIRP-a
variant protein may be purified
from the cell culture supernatant using, e.g., affinity column chromatography.
Purified SIRP-a variant
may be analyzed by SDS-PAGE followed by Coomassie Blue staining to confirm the
presence of protein
bands of expected size.
Purified SIRP-a variants may be screened for pH-dependent binding to CD47
using available
techniques in the art, such as phage display, yeast display, surface plasmon
resonance, scintillation
proximity assays, ELISA, ORIGEN immunoassay (IGEN), fluorescence quenching,
and/or fluorescence
transfer. Binding may also be screened using a suitable bioassay. The desired
SIRP-a variant binds with
higher affinity to CD47 under acidic pH (e.g., less than pH 7 (e.g., pH 6))
than under neutral pH (e.g., pH
7.4). The KD of a SIRP-a/CD47 complex at pH 6 would be lower than KD of a SIRP-
a/CD47 complex at
pH 7.4.
Example 10 ¨ Testing SIRP-a variants with pH-dependent binding to CD47 in mice
Genetically engineered mouse models of various cancers, e.g., solid tumor and
hematological
cancer, may be used to test the pH-dependent binding of SIRP-a variants of the
invention to CD47 at a
diseased site in a mouse model. A SIRP-a variant may be injected directly or
indirectly to the diseased
site in a mouse, which may be dissected at the later time to detect the
presence of the complex of SIRP-a
variant and CD47 at the diseased site. Antibodies specific to SIRP-a variant
or CD47 may be used in the
detection.
Other Embodiments
Various modifications and variations of the described compositions and
52
Date Regue/Date Received 2022-11-28
CA 02938180 2016-08-04
methods of the invention will be apparent to those skilled in the art without
departing from the scope and
spirit of the invention. Although the invention has been described in
connection with specific
embodiments, it should be understood that the invention as claimed should not
be unduly limited to such
specific embodiments. Indeed, various modifications of the described modes for
carrying out the
invention that are obvious to those skilled in the art are intended to be
within the scope of the invention.
Other embodiments are within the following claims.
What is claimed is:
53