Note: Descriptions are shown in the official language in which they were submitted.
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
PROCESS FOR GENERATING HYDROGEN USING PHOTO-CATALYTIC
COMPOSITE MATERIAL
FIELD
[0001] The present disclosure relates to a photo-catalytic composite
material and a process for generating hydrogen gas using the photo-catalytic
composite material. The disclosure also relates to processes for preparing the
photo-catalytic composite material, and an apparatus for using the material to
measure gas evolution or consumption.
INTRODUCTION
[0002] There are current significant global challenges arising from
anthropogenic climate change and environmental degradation associated with
extraction and use of non-renewable fossil fuels for energy production and
transportation. Thus, there exists a global need to develop renewable,
environmentally friendly alternatives fuels such as hydrogen. When produced
from the splitting of a water molecule it can be a clean and renewable source
of
energy. Upon combustion it re-forms into a water molecule (2H2 + 02 4 2H20)
and can act as a fuel for energy generation, transportation and various other
applications. However, before such green technologies can fully realize their
potential, an inexpensive, efficient and renewable means of producing hydrogen
needs to be developed.
[0003] Current technology for the production of hydrogen using
photocatalytic materials and systems are generally dependent upon the UV
component (,-.4%) of the solar spectrum, in contrast to the visible and lower
energy components that comprise the bulk of solar energy reaching the earth's
surface. Often such systems use expensive and rare materials such as platinum
rendering unfeasible the commercial adoption of such technology. Conventional
photocatalysts capable utilizing the visible solar spectrum for the
photocatalytic
decomposition of water are often unstable under the reaction conditions and
can
suffer from photo-corrosion.
- 1 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
SUMMARY
[0004] The present disclosure includes a process for generating
hydrogen
gas from water, comprising contacting the water with a photo-catalytic
composite
material, wherein the photo-catalytic composite material comprises:
(a) at least one semi-conductive material; and
(b) at least one conductive polymer,
and wherein the composite material is contacted with the water under
conditions
sufficient to generate hydrogen gas.
[0005] In one embodiment, the conditions sufficient to generate
hydrogen
gas comprise conditions sufficient to photocatalyze the decomposition of
water,
such as exposure to solar radiation including, but not limited to, UV
radiation,
visible light radiation and IR radiation.
[0006] The disclosure also includes a process for the preparation of
a
photo-catalytic composite material comprising
(I) contacting
(i) an aqueous suspension comprising at least one semi-conductive
material and an electrically conductive dopant;
with
(ii) a solution of a conductive polymer, or corresponding monomers, in an
ionic liquid,
or
(II) contacting
(i) a suspension of at least one semi-conductive material and at least one
conductive polymer in an ionic liquid;
with
(ii) an aqueous solution of an electrically conductive dopant.
[0007] The disclosure also relates to an apparatus for measuring an
amount of gas captured by, or released from, a sample comprising:
i) a sample chamber and a reference sample chamber, each equipped
with a valve for injection and evacuation;
- 2 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
ii) a pressure transducer connected to the chambers;
iii) a temperature control device to control the temperature of the
chambers;
iv) an electromagnetic delivery device to irradiate the chambers,
individually or simultaneously, with electromagnetic radiation; and
v) a digital data recorder to obtain and record data from the pressure
transducer.
[0008] Other features and advantages of the present application will
become
apparent from the following detailed description. It should be understood,
however,
that the detailed description and the specific examples while indicating
preferred
embodiments of the application are given by way of illustration only, since
various
changes and modifications within the spirit and scope of the application will
become
apparent to those skilled in the art from this detailed description.
DRAWINGS
[0009] The disclosure will now be described in greater detail with
reference to
the following drawings in which:
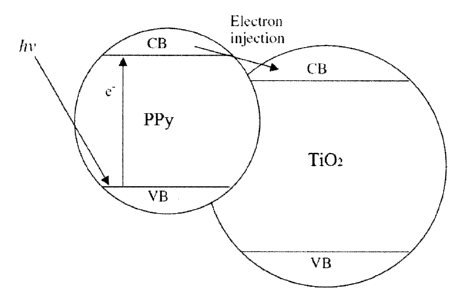
[0010] Fig. 1 shows a schematic energy-level diagram for a
photocatalytic
material of the disclosure and the electron injection process;
[0011] Fig. 2A shows a schematic representation of a nanocomposite
material synthesis under two different non-limiting examples of synthetic
methodologies known as A and B respectively; Fig 2B shows a second
schematic representation of a nanocomposite material synthesis under two
different non-limiting examples of synthetic methodologies known as A and B
respectively;
[0012] Fig. 3 shows a schematic representation of two morphologies of a
PPy/ TiO2 composite: nanoparticles (left) and film (right);
[0013] Fig. 4 shows transmission electron microscopy (TEM)
micrographs
of synthesized TiO2 nanoparticles;
- 3 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
[0014] Fig. 5 shows a histogram of TiO2 nanoparticle size
distribution
calculated from the TEM micrograph in Fig. 4;
[0015] Fig. 6 shows the X-ray diffraction (XRD) spectra of
synthesized
TiO2 nanoparticles in the rutile phase;
[0016] Fig. 7 shows scanning electron microscopy (SEM) micrographs of a
PPy/Ti02 nanocomposite (sample A particles) showing a 3-dimensional network;
[0017] Fig. 8 shows a SEM micrograph of a PPy/Ti02 composite film
(sample A film) showing a 3-dimensional network;
[0018] Fig. 9 shows the energy-dispersive X-ray spectroscopy (EDS)
spectrum of a PPy/TiO2 nanocomposite (sample A particles);
[0019] Fig. 10 shows the energy-dispersive X-ray spectroscopy (EDS)
spectrum of a PPy/Ti02 nanocomposite (sample A films);
[0020] Fig. 11 shows the X-ray diffraction (XRD) spectra of sample A
film,
sample A film bottom, sample C particles, PPy, TiO2 nanoparticles and another
sample A bottom which contains more Ti02;
[0021] Fig. 12 shows the transmission electron microscopy (TEM)
micrographs of a PPy/TiO2 (sample A particle) nanoparticles;
[0022] Fig. 13 shows the histogram of a PPy/T102 nanocomposite size
distribution calculated from the TEM micrograph in Fig. 12;
[0023] Fig. 14 shows the infrared spectrum of a PPy/T102 composite: (a)
sample A particles and (b) sample A film;
[0024] Fig. 15 shows the ultraviolet-visible spectrophotometry (UV-
vis)
absorption spectrum of a PPy/TiO2 composite (sample A particles);
[0025] Fig. 16 shows the differential scanning calorimetry (DSC) plot
of a
PPy/Ti02 composite (sample A particles) showing the curves of three heating
and cooling cycles;
[0026] Fig. 17 shows the differential scanning calorimetry (DSC) plot
of
prepared PPy, showing the second and third heating cycles;
- 4 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
[0027] Fig. 18 shows the amplification of cooling curves of pure PPy
at the
temperature between 95 C. and 115 C;
[0028] Fig. 19 shows the SEM micrograph of a PPy/Ti02 composite film
(sample B film);
[0029] Fig. 20 shows the energy-dispersive X-ray spectroscopy (EDS)
spectrum of a PPy/Ti02 nanocomposite (sample B films);
[0030] Fig. 21 shows the Fourier transform infrared spectroscopy
(FTIR)
spectra of the sample B films: (a) the red curve represents the film in the
interface, and the blue curve represents the film at the bottom layer; (b)
sample B
films at the bottom layer; (c) sample B film at the interface layer;
[0031] Fig. 22A shows the ultraviolet-visible spectrophotometry (UV-
vis)
absorption spectrum of a PPy/TiO2 film (sample B film); Fig. 22B shows a
second
ultraviolet-visible spectrophotometry (UV-vis) absorption spectrum of a
PPy/Ti02
film (sample B film);
[0032] Fig. 23 shows the differential scanning calorimetry (DSC) plot of a
PPy/Ti02 composite from sample B film;
[0033] Fig. 24 is a tabular presentation of the elemental analysis of
pure
PPy and a PPy/Ti02 composite;
[0034] Fig. 25 shows the comparison of mass loss of the samples (a)
sample A particle, b) sample B film, c) sample C particle) under argon flow
and
air flow with increasing temperature recorded by thermogravimetric analysis
(TGA);
[0035] Fig. 26 shows the ultraviolet-visible spectrophotometry (UV-
vis)
absorption spectrum of a PPyiTiO2 composite (sample C particles);
[0036] Fig. 27 shows the thermogravimetric analysis (TGA) curves of a
PPy/Ti02 composite prepared by different methods. The curve (a) represents the
PPy/Ti02 nanoparticles in sample A particles. The curve (b) is the TGA trace
of
the PPy/Ti02 film obtained at the interface in the sample B film. The curve
(c)
represents the PPy/T1O2 composite sample C particles;
- 5 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
[0037] Fig. 28 shows the thermogravimetric analysis (TGA) traces of
(a)
PPy/Ti02 composite (3% Ti02) (red curves), (b) PPy/T102 composite (8% Ti02)
(blue curves), and (c) pure PPy (green curves);
[0038] Fig. 29 shows the amount of hydrogen gas produced over time in
a
sample by a PPy/Ti02 composite (sample A particles);
[0039] Fig. 30 shows the amount of gas produced over time in a sample
by a PPy/Ti02 composite (sample B film);
[0040] Fig. 31 shows the amount of gas produced over time using a
Sample B film versus a Sample A particle;
[0041] Fig. 32 shows the amount of gas produced over time using a PPy-
T102 film composite in a repeated trial;
[0042] Fig. 33 shows the amount of gas produced over time for TiO2
nanoparticles;
[0043] Fig. 34 shows the amount of gas produced over time using a
composite produced from toluene and water;
[0044] Figure 35 shows the circuit diagram of an example data
recorder of
the digital gas capture and release instrument;
[0045] Figure 36 shows the external view of an example data recorder
of
the digital gas capture and release instrument;
[0046] Fig. 37A is a schematic diagram of an apparatus for measuring a
change in an amount of a gas such as measuring H2 gas release rate;
[0047] Fig. 37B is a schematic diagram of the apparatus of FIG. 37A
along
the cross-sectional line A-A;
[0048] Fig. 370 is a close-up schematic diagram of a sample chamber
of
the apparatus of FIG. 37A showing a magnetic stirrer therein;
[0049] Fig. 38 shows a plot of the temperature difference verses
voltage
output based upon linearity testing data presented in table 1, assuming an
ideal
behaviour of gas in the system;
- 6 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
[0050] Fig. 39 shows a plot of the moles injected vs. voltage output
of the
data presented in tabular form in table 2 for the determination of the
voltage/moles relationship of the instrumentation;
[0051] Fig. 40 shows the voltage change vs. time that corresponds to
an
amount of CO2 captured over time in a sample by a solution of NaOH in trial 1;
[0052] Fig. 41 shows a plot of voltage change verses time of the CO2
capture experiment Trial 2; and
[0053] Fig. 42A and 42B are graphs showing CO2 absorption versus
time.
DESCRIPTION OF VARIOUS EMBODIMENTS
(I) DEFINITIONS
[0054] The term "water", as used herein, refers to any form of water
or
aqueous solution which photo-catalytically decomposes to produce hydrogen
gas, and includes substantially pure forms of water, such as distilled water,
well
water, spring water, tap water and the like, and impure water, including, but
not
limited to sea water, lake water, waste water, etc.
[0055] The term "contacting" with respect to generating hydrogen gas
as
used herein refers to the manner in which the water and the photo-catalytic
composite material are intimately combined to effect the photo-catalytic
decomposition of the water. For example, the water and the composite material
are stirred together to ensure intimate contact, resulting in the generation
of
hydrogen gas to the photo-catalytic decomposition of water.
[0056] The term "photo-catalytic composite material" as used herein
refers
to a composite material capable of photo-catalytically decomposing water and
refers to a material containing two or more constituent materials which remain
separate and distinct within the finished material.
[0057] The term "semi-conductive material" as used herein has its
ordinary
technical meaning and include elements or compounds having an electrical
conductivity intermediate between that of conductors, e.g., metals and non-
conductors (insulators).
- 7 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
[0058] The term "conductive polymer" as used herein refers to any
polymer which is capable of electrical conductivity and therefore has a
measurable level of electrical conductivity, and includes, but is not limited
to
inherently electrically conductive polymers, and polymers, which upon addition
of
an electrically conductive dopant, are capable of electrical conductivity.
[0059] The phrase "under conditions sufficient to generate hydrogen
gas"
as used herein refers to any condition in which water decomposes to generate
hydrogen gas (and oxygen) upon contact with the photo-catalytic composite
material including, but not limited to, exposure to solar radiation including
UV
radiation, visible light radiation and/or IR radiation.
[0060] The term "electrically conductive dopant" as used herein
refers to a
substance which is added to a polymer of the present disclosure to alter, or
optionally increase, the electrical conductivity of the polymer.
[0061] The term "ionic liquid" as used herein refers to a liquid salt
consisting solely of ions. In certain embodiments, the ionic liquids are room
temperature ionic liquids, which melt at or close to room temperature, and
typically are salts whose melting point is below 100 C. The term ionic liquid
(IL)
encompasses liquids having organic cations and anions, and may be soluble in
the aqueous suspension resulting in a one-phase reaction, or insoluble in the
aqueous suspension resulting in a two-phase reaction.
[0062] The term "aqueous suspension" as used herein refers to a
suspension in which the liquid medium is primarily aqueous and solid
particles,
for example, a semi-conductive material which is substantially insoluble in
the
aqueous medium and forms the particulate material of the suspension, in which
the solid particles are distributed substantially uniformly throughout the
medium.
The liquid medium is primarily aqueous though it may contain other components
which do not affect the process for preparing the photo-catalytic composite
material.
[0063] The term "suspension" as used herein refers to a suspension in
which the liquid medium is primarily an ionic liquid and solid particles, for
- 8 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
example, a semi-conductive material and a conductive polymer (or monomers for
forming a conductive polymer) which is substantially insoluble in the medium
and
forms the particulate material of the suspension, in which the solid particles
are
distributed substantially uniformly throughout the medium. The liquid medium
is
primarily an ionic liquid though it may contain other components which do not
affect the process for preparing the photo-catalytic composite material.
(II) PHOTO-CATALYTIC COMPOSITE MATERIALS
[0064] The
present disclosure relates to composite materials which photo-
catalyze the decomposition of water to generate hydrogen gas, as well as
oxygen gas. The hydrogen gas can be collected and used as a source of energy
as a result of the combustion of the hydrogen gas in the presence of oxygen to
simply form water. Accordingly, the composites of the present disclosure,
which
do not require heavy metals, are environmentally friendly and are used to
prepare hydrogen gas from water, and therefore, the entire process of
preparing
the composites and using the composites to generate hydrogen gas is
environmentally friendly.
[0065] In
one embodiment of the disclosure, the photo-catalytic composite
material comprises:
(a) at least one semi-conductive material;
(b) at least one conductive polymer; and
(c) at least one ionic liquid.
[0066] In
one embodiment, the composite material consists essentially of,
or consists of:
(a) at least one semi-conductive material;
(b) at least one conductive polymer; and
(c) at least one ionic liquid.
[0067] In
another embodiment, the at least one semi-conductive material
is any shape, for example, a substantially spherical or a spherical particle,
a
cubic particle, a hexagonal particle, an ellipsoidal particle, a rod shaped
particle,
- 9 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
a tubular particle or a wire shaped particle. In one embodiment, the particles
are
generally spherical or non-spherical.
[0068] In other embodiment, the semi-conductive material is in the
form of
a microparticle or a nanoparticle. In one embodiment, the particle size of the
semi-conductive material is between 1 nm and 10 m. In one embodiment, the
particle size is between 1 nm-15 nm, optionally between 5 nm-75 nm, optionally
between 20 nm-300 nm, optionally between 100 nm-1.5 micro-m, or optionally
larger than 1 micro-m. In other embodiments, the particle size is between 1 nm
and 10 10 jim, optionally 1 nm ¨1.5 11M, optionally 5 nm ¨300 nm, optionally
20
nm ¨ 100 nm, or about 20 nm ¨ 75 nm. As used herein, the term "particle size"
refers to the diameter of a particle, such as a substantially spherical
particle, as
determined by microscopy. In the event that a particle of the invention is not
spherical, then size is determined by approximating the shape of the particle
in
the form of a sphere.
[0069] In another embodiment, the at least one semi-conductive material
is any compound or material having an electrical conductivity intermediate
between that of a conductor and a non-conductor. For example, the semi-
conductive material includes transition metal compounds, such as, but not
limited
to, Ti02, W03, SrT103, Ti02¨Si, BaTiO3, LaCr03¨Ti02, LaCr03¨Ru02, 1-102-
1n203, GaAs, GaP, AlGaAs/SiRu02, Pb0, FeTiO3, KTa03, MnTiO3, Sn02, B1203,
Fe203 (including hematite), ZnO, CdS, MoS2, CdTe, CdSe, CdZnTe, ZnTe,
HgTe, HgZnTe, HgSe, ZnTe, ZnS, HgCdTe, HgZnSe, Si,Pt, Pd etc., or
composites of any of the above thereof. The semiconductor material may be
provided in any suitable morphology, phase or arrangement. In some
embodiments the semiconductor material may be composed of more than one
morphology, phase or arrangement. In some embodiments, the semiconductor
material is a transition metal oxide and/or transition metal hydroxide. In
some
embodiments, the semiconductor material is a metal oxide and/or metal
hydroxide. For example, transition metal oxides include, but are not limited
to,
ZnO and Fe203. Examples of transition metal hydroxides, include but are not
limited to, Ti(OH)4 and Fe(OH)2.
-10-
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
[0070] In
certain embodiments, the semi-conductive material is Ti02. In
another embodiment, the semi-conductive material comprises rutile phase,
anastase phase, and/or brookite phase T102, optionally the rutile phase of
Ti02.
[0071] In
embodiments of the disclosure, the at least one conducting
polymer comprises poly(pyrrole) (PPY), polycarbazole, polyindole, polyazepine,
polyanilines (PANI), poly(thiophene) (PT), poly(3,4-ethylenedioxythiophene)
(PEDOT), poly(p-phenylene sulfide) (PPS), Poly(fluorene), polyphenylene,
polypyrene, polyazulene, polynaphthalene, Poly(acetylene) (PAC), Poly(p-
phenylene vinylene) (PPV), or derivatives, co-polymers, or mixtures thereof.
The
conductive polymers include co-polymers, block co-polymers, alternating co-
polymers, random co-polymers or composites thereof of any of the above
polymers, in any suitable morphology, phase or arrangement. The molecular
weights of the conducting polymers of the disclosure is any weight which forms
a
polymer for the composites of the disclosure, and includes, but is not limited
to, a
weight range between about 200 kD to about 20,000,000 kDa, or between about
200 kDa to about 10,000,000 kDa, or between about 1,000 kDa to about
1,000,000 kDa, or between about 10,000 kDa to about 100,000 kDa.
[0072] In
a further embodiment, the at least one conducting polymer
comprises polypyrrole, a polypyrrole co-polymer, or a doped polypryrrole.
[0073] In other embodiments, the at least one conductive polymer further
comprises an electrically conductive dopant or polymerization initiator, to
alter or
increase the electrical conductivity of the conductive polymer. In
some
embodiments, the electrically conductive dopant or polymerization initiator
are
the same compound. In one embodiment, the electrically conductive dopant or
polymerization initiator is an iron(III) or copper(II) complex, such as ferric
chloride
FeCl3, FeC13=6H20, FeBr3, Fe(NO3)=9H20, K3Fe(CN)6, (C8H8)2Fe+FeCI4-, CuCl2,
CuBr2, CuSO4=5H20, Cu(NO3)2=5/2H20 (Myers RE., Journal of Electronic
Materials, March 1986, Volume 15, Issue 2, pp.61-69). Other compounds include
12, H202, Na2S208, lead dioxide and quinone or derivatives thereof. In one
embodiment, non-metallic compounds are used as polymerization initiators to
-11 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
polymerize corresponding monomers of the conducting polymers, and include,
are but are not limited to ammonium persulfate (NH4)S208.
[0074] In another embodiment, the molar ratio of the at least one
conducting polymer to the at least one semi-conductive material is between
0.02:1 to 4000:1. In other embodiments, the molar ratio is between about 1:1
to
about 100:1. In certain embodiments, the molar ratio is between about 8:1 to
about 40:1.
[0075] In another embodiment, the ionic liquid is a protic ionic
liquid or a
phosphonium ionic liquid. In another embodiment, the phosphonium ionic liquid
is trihexyl(tetradecyI)-phosphonium chloride, tri-isobutyl(methyl) phosphonium
salt, or tri(butyl)ethylphosphonium salt.
[0076] Without being bound by theory, Figure 1 schematically depicts
the
general interactions of the electronic structures of the electrically
conductive
polymer and semiconductive materials in the composites of the disclosure,
using
as an example, polypyrrole and titanium dioxide. In this example, the smaller
bandgap of the pyrrole polymer can absorb electromagnetic radiation within,
for
example, the visible spectrum, promoting an electron from its valence band
into
its conduction band upon absorption of electromagnetic radiation. These
electrons are then injected into the conduction band (CB) of the titanium
dioxide
semiconductor material enlarging the separation of electron-hole pairs and
subsequently undergoing photocatalytic reactions upon exposure to, for
example,
visible light. Therefore, these composite materials have improved
photocatalytic
ability and allow for the ability to tune the wavelength(s) or range of
wavelengths
of electromagnetic radiation over which photocatalysis by the composite can
occur by choosing semiconductor and conducting polymer components of band
gaps appropriate to the photocatalytic reaction and electromagnetic radiation
source(s).
[0077] The photogenerated electrons and holes by, for example,
PPy/T102
composites, decomposes water based upon the following reaction under solar
radiation, such as sunlight:
- 12-
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
4h+ +2H20 --3 02 + 4H+ (1)
2e- + 2H20 ¨p H2 + 20H- (2).
[0078] The
photo-catalytic composite materials of the present disclosure
are able to withstand high temperatures, and therefore, are fire-retardant
materials. In one embodiment, the composite materials of the disclosure
withstand temperatures (do not burn) of at least about 500 C, at least about
600 C, at least about 700 C, at least about 800 C, at least about 900 Cor at
least about 1000 C. In one embodiment, as the composite materials are able to
withstand high temperatures without burning or combusting, the composite
materials of the disclosure are fire-retardant materials, and are useful where
materials having fire-retardant properties are used, such as in clothing or
building
materials.
(III) PROCESSES FOR PREPARING PHOTO-CATALYTIC COMPOSITE
MATERIALS
[0079] The
present disclosure also includes environmentally friendly
processes for the preparation of the photocatalytic composite materials of the
present disclosure.
[0080] In
one embodiment, the disclosure includes a process for preparing
a photo-catalytic composite material as described in the disclosure
comprising:
contacting
(i) an aqueous suspension comprising at least one semi-conductive
material and an electrically conductive dopant or polymerization initiator;
with
(ii) a solution of at least one conductive polymer, or its corresponding
monomers, in an ionic liquid,
wherein the at least one semi-conductive material, at least one conductive
polymer, electrically conductive dopant or polymerization initiator and ionic
liquid are all as described above.
-13-
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
[0081] In
another embodiment, the disclosure also includes a process for
preparing a photo-catalytic composite material as described in the disclosure
comprising
contacting
(a) a suspension of at least one semi-conductive material and at least one
conductive polymer, or its corresponding monomers, in an ionic liquid,
with
(b) an aqueous solution of an electrically conductive dopant or radical
initiator,
wherein the at least one semi-conductive material, at least one conductive
polymer, electrically conductive dopant or polymerization initiator and ionic
liquid are all as described above.
[0082] In
one embodiment, the process is conducted using constituent
monomers to form the at least one conductive polymer, such that the
polymerization incurs in situ. For example, when the at least one conducting
polymer is polypyrrole, pyrrole monomers can be used to polymerize the
polypyrrole in situ. Alternatively, pre-polymerized polypyrrole may be used in
the
process to form the composite material.
[0083] In
one embodiment, the ionic liquid is immiscible with the aqueous
suspension or aqueous solution resulting in a two-phase reaction, such that
the
formation of the composite material forms at the interface of the two phases
(the
interface between the ionic liquid and the aqueous suspension or solution). In
one embodiment, the photo-catalytic composite material is produced at the
interface of the aqueous suspension and the solution, or the interface of the
suspension and the aqueous solution. In such embodiments, the ionic liquid is
a
phosphonium ionic liquid such as trihexyl(tetradecyl)phosphonium salt, tri-
isobutyl(methyl) phosphonium salt, or tri(butyl)ethylphosphonium salt.
[0084] In
another embodiment, the ionic liquid is miscible in the aqueous
suspension or aqueous solution, resulting in a one-phase reaction, such that
the
- 14-
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
formation of the composite material forms throughout the one-phase reaction.
In
such embodiment, the ionic liquid is a protic ionic liquid such as imidazolium-
based, ammonium and imidazo, pyridine-based protic ionic liquids.
[0085] In other embodiments, the processes are carried out in the
presence of an electrically conductive dopant as defined above or a
polymerization initiator to initiate polymerization of monomers to form the at
least
one conductive polymer. In some embodiments, the electrically conductive
dopant and the polymerization initiator are the same compound. In one
embodiment, the electrically conductive dopant or polymerization initiator is
an
iron(III) or copper(11) complex, such as ferric chloride FeCI3, FeC13=6H20,
FeBr3,
Fe(NO3).9H20, K3Fe(CN)6, (C5H6)2Fe+FeC14", CuC12, CuBr2, CuSO4.5H20,
Cu(NO3)2=5/2H20 (Myers RE., Journal of Electronic Materials, March 1986,
Volume 15, Issue 2, pp.61-69). Other compounds include 12, H202, Na2S208, lead
dioxide and quinone or derivatives thereof. In one embodiment, non-metallic
compounds are used as polymerization initiators to polymerize corresponding
monomers of the conducting polymers, and include, are but are not limited to
ammonium persulfate (N1-14)S208.
[0086] The processes for preparing the composites of the present
disclosure are carried out at temperatures between about 4 C to about 90 C, or
about 10 C to about 50 C, or about room temperature.
[0087] In another embodiment, the molar ratio of the at least one
conducting polymer to the at least one semi-conductive material is between
0.02:1 to 4000:1. In other embodiments, the molar ratio is between about 1:1
to
about 100:1. In certain embodiments, the molar ratio is between about 8:1 to
about 40:1. It will be understood that if monomers are utilized to form the
conducting polymer in situ, the molar ratio of the monomers will
correspondingly
increase to an amount to provide the desired molar ratio of the polymerized
conducting polymer.
-15-
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
(IV) PROCESSES FOR GENERATING HYDROGEN GAS, AND OTHER
PROCESSES
[0088] The
present disclosure also includes a process for generating
hydrogen gas from water using the photocatalytic composite materials as
described herein. In particular, the process generates hydrogen gas by the
photocatalytic decomposition of water when the composite materials are
contacted with water.
[0089]
Accordingly, in one embodiment, the disclosure includes a process
for generating hydrogen gas from water, comprising contacting the water with a
photo-catalytic composite material, wherein the photo-catalytic composite
material comprises:
(a) at least one semi-conductive material; and
(b) at least one conductive polymer,
wherein the composite material is contacted with the water under conditions
sufficient to generate hydrogen gas.
[0090] In
another embodiment, the composite material consists essentially
of, or consists of:
(a) at least one semi- conductive material; and
(b) at least one conductive polymer.
[0091] In one embodiment, the composite material further comprises an
ionic liquid as described above.
[0092] In
another embodiment, the at least one semi-conductive material
is any shape, for example, a substantially spherical or a spherical particle,
a
cubic particle, a hexagonal particle, an ellipsoidal particle, a rod shaped
particlae,
a tubular particle or a wire shaped particle. In one embodiment, the particles
are
generally spherical or non-spherical.
[0093] In
one embodiment, the particle size of the semi-conductive
material is between 1 nm and 10 tim. In one embodiment, the particle size is
between 1 nm-15 nm, optionally between 5 nm-75 nm, optionally between 20
-16-
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
nm-300 nm, optionally between 100 nm-1.5 micro-m, or optionally larger than 1
micro-m. In other embodiments, the particle size is between 1 nm and 10 10
prn,
optionally 1 nm ¨ 1.5 vtrn, optionally 5 nm ¨300 nm, optionally 20 nm ¨ 100
nm,
or about 20 nm ¨ 75 nm.
[0094] In another embodiment, the at least one semi-conductive material
is any compound or material having an electrical conductivity intermediate
between that of a conductor and a non-conductor. For example, the semi-
conductive material includes transition metal compounds, such as, but not
limited
to, Ti02, W03, SrTiO3, Ti02¨Si, BaTiO3, LaCr03¨Ti02, LaCr03¨Ru02, T102-
In203, GaAs, GaP, AlGaAs/SiRu02, Pb0, FeTiO3, KTa03, MnTiO3, Sn02, 131203,
Fe203 (including hematite), ZnO, CdS, M0S2, CdTe, CdSe, CdZnTe, ZnTe,
HgTe, HgZnTe, HgSe, ZnTe, ZnS, HgCdTe, HgZnSe, Si,Pt, Pd etc., or
composites of any of the above thereof. The semiconductor material may be
provided in any suitable morphology, phase or arrangement. In some
embodiments the semiconductor material may be composed of more than one
morphology, phase or arrangement. In some embodiments, the semiconductor
material is a transition metal oxide and/or transition metal hydroxide. In
some
embodiments, the semiconductor material is a metal oxide and/or metal
hydroxide. For example, transition metal oxides include, but are not limited
to,
ZnO and Fe203. Examples of transition metal hydroxides, include but are not
limited to, Ti(OH)4 and Fe(OH)2.
[0095] In
certain embodiments, the semi-conductive material is Ti02. In
another embodiment, the semi-conductive material comprises rutile phase,
anastase phase, and/or brookite phase Ti02, optionally the rutile phase of
Ti02.
[0096] In embodiments of the disclosure, the at least one conducting
polymer comprises poly(pyrrole) (PPY), polycarbazole, polyindole, polyazepine,
polyanilines (PANI), poly(thiophene) (PT), poly(3,4-ethylenedioxythiophene)
(PEDOT), poly(p-phenylene sulfide) (PPS), Poly(fluorene), polyphenylene,
polypyrene, polyazulene, polynaphthalene, Poly(acetylene) (PAC), Poly(p-
vinylene) (PPV), or derivatives, co-polymers, or mixtures thereof. The
-17-
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
conductive polymers include co-polymers, block co-polymers, alternating co-
polymers, random co-polymers or composites thereof of any of the above
polymers, in any suitable morphology, phase or arrangement.
[0097] In
a further embodiment, the at least one conducting polymer
comprises polypyrrole, a polypyrrole co-polymer, or a doped polypryrrole.
[0098] In
another embodiment, the molar ratio of the at least one
conducting polymer to the at least one semi-conductive material is between
0.02:1 to 4000:1. In other embodiments, the molar ratio is between about 1:1
to
about 100:1. In certain embodiments, the molar ratio is between about 8:1 to
about 40:1.
[0099] In
other embodiments, the at least one conductive polymer further
comprises an electrically conductive dopant or polymerization initiator, to
alter or
increase the electrical conductivity of the conductive polymer. In
some
embodiments, the electrically conductive dopant or polymerization initiator
are
the same compound. In one embodiment, the electrically conductive dopant or
polymerization initiator is an iron(III) or copper(II) complex, such as ferric
chloride
FeCI3, FeC13=6H20, FeBr3, Fe(NO3)=9H20, K3Fe(CN)6, (C6H8)2Fe+FeC14", CuCl2,
CuBr2, CuSO4=5H20, Cu(NO3)2=5/2H20 (Myers RE., Journal of Electronic
Materials, March 1986, Volume 15, Issue 2, pp.61-69). Other compounds include
12, H202, Na2S208, lead dioxide and quinone or derivatives thereof. In one
embodiment, non-metallic compounds are used as polymerization initiators to
polymerize corresponding monomers of the conducting polymers, and include,
are but are not limited to ammonium persulfate (NH4)S208.
[00100] In
another embodiment, the ionic liquid is a protic ionic liquid or a
phosphonium ionic liquid. In another embodiment, the phosphonium ionic liquid
is trihexyl(tetradecyI)-phosphonium chloride, tri-isobutyl(methyl) phosphonium
salt, or tri(butyl)ethylphosphonium salt.
[00101] In
another embodiment, the photocatalytic composite material is
contacted with water under conditions sufficient to generate hydrogen gas as a
- 18-
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
result of the photocatalytic decomposition of water based on the following
reaction:
4h+ +2H20 --4 02 + 4H+ (1)
2e- + 2H20 -- H2 + 20H- (2).
[00102] In one embodiment, the conditions sufficient to generate hydrogen
gas comprise exposing the water and composite material to solar radiation
including UV radiation, visible light radiation and IR radiation. In
one
embodiment, the solar radiation includes visible light electromagnetic
radiation,
for example at a wavelength between 380 nm and 750 nm. In one embodiment,
the solar radiation comprises a wavelength of between 200 nm and 750 nm.
[00103] The
photocatalytic composite material as described in the present
disclosure is also useful for the photocatalysis of other materials, as a
result of
the oxidative or reductive degradation which occurs during photocatalysis. For
example, in one embodiment, the photocatalytic composite material as described
herein is useful for the oxidative or reductive degradation of organic dyes
upon
exposure to electromagnetic radiation (UV, solar, visible light or IR
radiation) by
contacting the organic dye with the composite material.
[00104] In
other embodiments, the photocatalytic composite materials as
described herein have anti-microbial and/or anti-fungal activity through the
photocatalytic production of reactive oxygen species (ROS) production,
especially hydroxyl free radicals and peroxide that disrupt cell walls and
biological membranes, oxidize proteins and damage DNA. Accordingly, in one
embodiment, the photocatalytic composite material is coated or contacted with
any device or surface, such as a medical device, or hospital surface such as a
floor or wall, to provide anti-microbial activity to the surface by exposure
of the
surface to electromagnetic radiation (UV, visible light or IR radiation). In
certain
embodiments, the photocatalytic composite materials having anti-microbial
and/or anti-fungal activity comprise the semi-conductive material at greater
than
about 10%, or greater than about 25%, or greater than about 30%.
-19-
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
[00105] In other embodiments, the photocatalytic composite materials
are
able to impart self-cleaning properties to a surface, such as glass (car
windows,
glass panes, building windows), solar panels, roofing materials, by the
photocatalytic decomposition of hydrocarbons, such as oil and/or grease, upon
exposure of the photocatalytic composite materials to electromagnetic
radiation
(UV, solar, visible light or IR radiation). Accordingly, in one embodiment,
the
photocatalytic composite materials are coated or incorporated on a surface,
such
as glass, to provide self-cleaning properties to the surface by exposure of
the
surface to electromagnetic radiation (UV, visible light or IR radiation). In
other
embodiments, the composite materials can be incorporated into the starting
materials of such surfaces, or into paints, which will impart the surface with
the
self-cleaning properties.
(V) APPARATUS FOR MEASURING CHANGE IN AMOUNT OF GAS
[00106] The present disclosure also includes an apparatus for
measuring a
change in an amount of a gas such as gas that is captured by, or released
from, a
sample. Accordingly, in one embodiment, the amount of hydrogen gas released
from water upon contact with a photocatalytic composite material as described
herein, can be measured using an apparatus made in accordance with an
embodiment of the present disclosure.
[00107] Accordingly, in one embodiment, there is included an apparatus for
measuring an amount of gas captured by, or released from, a sample comprising:
i) a sample chamber and a reference sample chamber, each equipped with a
valve for injection and evacuation;
ii) a pressure transducer connected to the chambers;
iii) a temperature control device to control the temperature of the chambers;
iv) an electromagnetic delivery device to irradiate the chambers, individually
or simultaneously, with electromagnetic radiation; and
-20-
CA 02946327 2016-10-19
WO 2014/169373 PCT/CA2014/000352
V) a digital data recorder to obtain and record data from the pressure
transducer.
[00108] In one embodiment, the gas is hydrogen, carbon dioxide, sulfur
oxide(s) or nitrogen oxide(s).
[00109] In another embodiment, the sample in the sample chamber is water
and the water is contacted with a photo-catalytic composite material as
described
herein in the sample chamber to generate hydrogen gas.
[00110] In one embodiment, the sample and reference chambers are made
from glass, metal, ceramic, polymer or a composite.
[00111] In one embodiment, the temperature of the sample and reference
chambers can be controlled by changing the pressure or molar content of the
gasses according to the ideal gas law.
[00112] In another embodiment, the temperature control device is a
fluid, solid,
or gaseous bath, which surrounds the chambers.
[00113] In another embodiment, the temperature control device is a heating
and cooling sheath.
[00114] In a further embodiment, the chambers comprise mixing means,
such
a magnetic stirrer, or by mechanical mixing means such as agitation.
[00115] In one embodiment, the apparatus measures a change in the
pressure
in the sample chamber to determine how much gas has been captured or evolved.
The pressure transducer measures the change in pressure in the chambers, and
conveys this change as a voltage reading. The voltage reading is calibrated to
a
particular number of moles of gas based on the following equation:
pressure ---> voltage--> (voltage) x (calibration constant (moles of
gas per volt))
[00116] Referring now to FIGS. 37A-37B, there is an apparatus 100 for
measuring a change in an amount of a gas made in accordance with an embodiment
of the present disclosure. The apparatus 100 includes a sample chamber 110 for
- 21 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
containing at least one sample gas, and a reference chamber 112 for containing
a
reference gas. The sample gas and reference gas may be the same gas.
[00117] The sample chamber 110 and the reference chamber 112 have
substantially similar internal volumes. For example, the internal volumes of
the
sample chamber 110 and the reference chamber 112 may be within 5% of each
other, or more particularly, within 1% of each other.
[00118] The sample chamber 110 and the reference chamber 112 may be
made from a suitable material such as glass, metal, ceramic, polymer, a
composite
or combination thereof.
[00119] As shown in FIG. 37B, the apparatus 100 includes a temperature
control device 120 for maintaining the sample chamber 110 and the reference
chamber 112 at substantially similar temperatures. For example, the
temperature
control device 120 may be configured to maintain the temperatures of the
sample
chamber 110 and the reference chamber 112 within 5% of each other, or more
particularly, within 1% of each other.
[00120] As an example, the temperature control device 120 may include
a
coolant surrounding one or both of the sample chamber 110 and the reference
chamber 112. The coolant may directly cool the chambers 110, 112 such as by
immersing the chambers 110, 112 in a solid, liquid or gaseous coolant. For
example,
in the illustrated embodiment, the coolant is air that is circulated within an
enclosure
122 using a fan 124.
[00121] In other embodiments, the coolant could indirectly cool the
chambers
110, 112 such as by circulating the coolant through a coiled pipe or another
type of
heat exchanger using a refrigeration system. The temperature control device
120
could also have other configurations such as a heating or cooling sheath.
[00122] As shown, the apparatus 100 also includes a pressure sensor
130 for
measuring a pressure difference between the sample chamber 110 and the
reference chamber 112. For example, the pressure sensor 130 could be a
differential pressure transducer having a first port fluidly coupled to the
sample
chamber 110 and a second port fluidly coupled to the reference chamber 112.
The
- 22 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
ports may be coupled to the chambers 110, 112 using fluid conduits such as
pipes
132, 134. The differential pressure transducer may generate an output voltage
proportional to the pressure difference.
[00123] The apparatus 100 also includes a processor 140 for
calculating a
change in amount of sample gas within the sample chamber 110 based on the
pressure difference between the sample chamber 110 and the reference chamber
112. For example, the processor 140 may include a voltmeter 141, a digital
data
recorder 142 configured to obtain and record data from the pressure sensor 130
in
real-time. The processor 140 may also include a computer 144 for receiving the
data
from the digital data recorder 142.
[00124] The underlying theory for using the pressure difference to
calculate the
change in amount of sample gas is based on the ideal gas law, namely:
PV
71 = ¨
RT
[00125] More specifically, the difference in moles of gas, An, can be
calculated
as follows with respect to the sample chamber (subscript 2) and the reference
chamber (subscript 1):
'2"2 P1 V1
An = n2 ¨ ni = ¨ ¨ ¨
R T2 RTi
[00126] As described above, both chambers 110, 112 have substantially
similar volumes (V1=V2) and are maintained at substantially similar
temperatures
(T1=T2). Thus, the equation can be simplified to:
V
An = ¨RT (P2 ¨ P1)
An = UP
where k is a constant. Thus, by measuring the pressure difference, it is
possible to
estimate the release or capture of gas within the sample chamber 110.
[00127]
In some embodiments, the processor 140 may be configured to
calculate a calibration coefficient that correlates the change in the amount
of the
sample gas (e.g. An) based on a change in the pressure difference between the
- 23 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
sample chamber 110 and the reference chamber 112 (e.g. AP). More specifically,
the pressure sensor 130 may output a voltage that is proportion to the
pressure
difference, AP, and thus, the change in the amount of gas in the sample
chamber
110 may be related to the output voltage multiplied by some calibration
constant.
The calibration constant can be calculated directly based on the volume of the
sample chamber 110, the temperature of the sample chamber 110, and the voltage-
to-pressure ratio of the pressure sensor 130. Alternatively, the calibration
constant
can be determined empirically by changing the amount of gas in the sample
chamber 110 by some known amount to determine the relationship between change
in output voltage and the change in the amount of gas. An example of this
empirical
calibration process will be described later below.
[00128] Referring still to FIGS. 37A and 37B, in some embodiments, the
apparatus 100 may include a balancing valve 150 for equalizing initial gas
pressures
between the sample chamber 110 and the reference chamber 112. The balancing
valve 150 may be coupled to the chambers 110, 112 using fluid conduits such as
pipes 152, 154. The balancing valve 150 may be operated manually, or may be
operated automatically by electronics, pneumatics, or otherwise.
[00129] In some embodiments, the apparatus 100 may include a vacuum
suction device 160 for drawing a vacuum within the sample chamber 110 and the
reference chamber 112. As shown, the vacuum suction device 160 may be a
vacuum pump. The vacuum suction device 160 may be coupled to the chambers
110, 112 via fluid conduits such as the pipes 132, 134.
[00130] In some embodiments, the apparatus 100 may include an
electromagnetic delivery device 170 for irradiating the sample chamber with
electromagnetic radiation. For example, the electromagnetic delivery device
170
may include a light source 172 for emitting a beam of light, which may pass
through
an optical lens 174 and through an opening 176 in the enclosure 122 before
irradiating the sample chamber 110. The electromagnetic delivery device 170
may
be useful when the apparatus 100 is being used to measure gas released by a
photo-catalytic composite material as described herein.
- 24 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
[00131] In some embodiments, the apparatus 100 may include a mixer 180
for
mixing the sample gas within the sample chamber 110. For example, as shown in
FIG. 370, the mixer 180 may include a magnetic stirrer. The magnetic stirrer
may
include a magnetic stir bar 182 within the sample chamber 110 and a magnetic
device 184 such as an electro-magnetic for generating a rotating magnetic
field to
cause the magnetic stir bar 182 to spin.
(VI) METHOD FOR MEASURING CHANGE IN AMOUNT OF GAS
[00132] The present disclosure also includes a method for measuring a
change
in an amount of a gas. The method comprising:
(a) equalizing initial gas conditions between a sample chamber and a
reference chamber, the sample chamber and the reference chamber having
substantially similar internal volumes;
(b) isolating the sample chamber from the reference chamber;
(c) initiating a chemical reaction within the sample chamber to cause a
change in an amount of at least one sample gas;
(d) maintaining the sample chamber and the reference chamber at
substantially similar temperatures;
(e) measuring a pressure difference between the sample chamber and the
reference chamber;
(f) calculating a change in the amount of the sample gas within the
sample chamber based on the pressure difference between the sample
chamber and the reference chamber
[00133] The method may be performed using the apparatus 110 described
above, or another suitable apparatus.
[00134] In some embodiments, the sample gas may be at least one of:
hydrogen, carbon dioxide, sulfur oxide and nitrogen oxide.
[00135] In some embodiments, the method may include placing a photo-
catalytic composite material and water into the sample chamber, and the
chemical
- 25 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
reaction may be initiated by irradiating the sample chamber with
electromagnetic
radiation to generate hydrogen gas.
[00136] In some embodiments, the method may include drawing a vacuum
prior to equalizing the initial gas conditions.
[00137] In some embodiments, the change in the amount of the sample gas
may be calculated in real-time using a digital data recorder.
[00138] In some embodiments, the method may include calculating a
calibration coefficient that correlates the change in the amount of the sample
gas
based on a change in the pressure difference between the sample chamber and
the
reference chamber. The calibration coefficient may be used to calculate the
change
in the amount of the sample gas.
[00139] Although the disclosure has been described in conjunction with
specific
embodiments thereof, if is evident that many alternatives, modifications and
variations will be apparent to those skilled in the art. Accordingly, it is
intended to
embrace all such alternatives, modifications and variations that fall within
the spirit
and broad scope of the appended claims. In addition, citation or
identification of any
reference in this application shall not be construed as an admission that such
reference is available as prior art to the present disclosure.
[00140] The operation of the disclosure is illustrated by the
following
representative examples. As is apparent to those skilled in the art, many of
the
details of the examples may be changed while still practicing the disclosure
described herein.
(VII) EXAMPLES
[00141] The operation of the disclosure is illustrated by the
following
representative examples. As is apparent to those skilled in the art, many of
the
details of the examples may be changed while still practicing the disclosure
described herein.
- 26 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
[00142] Materials and Methods: The ionic liquids mentioned in
methodologies A, B the specific ionic liquid used is trihexyl (tetradecyl)
phosphonium dicyanamide (IL 105). However, this ionic liquid hereinafter
referred to as IL 105 can be substituted by any other solvent classified as,
or
possessing the physical and chemical properties that commonly defined as an
ionic liquid by one skilled in the art. This substitution is valid for the
example
methodologies A, B and any other methodology or embodiments to produce the
photocatalytic composite materials covered by this invention that employs
ionic
liquids as a part of the synthetic process.
[00143] For the synthetic methodologies presented, the precursor titanium
dioxide (Ti02) nanoparticles used in all three methodologies A, B and C are
synthesized based upon previously published methodology by Cassaignon,
Koelsch and Jolivet (Cassaignon, S., M. Koelsch, and J.-P. Jolivet, J Mater
Sci,
2007. 42: p. 6689-6695). More specifically the TiO2 nanoparticles were
synthesized by adding 7 ml of nitric acid (70%) to 120 ml deionized water to
adjust the acid concentration at a value of 1 mol L-1. Then 1 ml of TiCla is
slowly
added to this solution at room temperature. The solution is then heated at 95
C
for 24 hours in an oven. The resulting solid TiO2 nanoparticles produced is
washed with deionized water and the suspension centrifuged at 5000 rpm for 10
minutes. These particles are characterized by transmission electron microscopy
(TEM) as shown in Figure 4. A histogram of the calculated particle size
distribution from the TEM micrograph is presented in Figure 5. In the present
example synthesis the measured particle sizes as determined by microscopy and
presented as a histogram in Figure 5 show the TiO2 particles have a diameters
in
the range of 1-9, 11 and 13nm.
[00144] Figure 6 shows the results of X-ray diffraction (XRD) spectrum
of
the above synthesized TiO2 nanoparticles. An analysis of the spectrum
determines the precursor nanoparticles used in the examples A,B and C to be of
rutile phase. In other embodiments, the precursor TiO2 particles can be
composed of other polymorphs of titanium dioxide. Three such known
polymorphs are anatase (tetragonal), brookite (orthorhombic) and rutile
-27-
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
(tetragonal). Multiphase titanium dioxide nanoparticles containing one, two or
all
three of the known phases of titanium dioxide in compositions varying from 0
to
100% of the particle can also be used as a precursor particle for synthetic
methodologies used to produce the example embodiments of the photocatalytic
composite material.
Example 1 ¨ General process for preparing photo-catalytic composite material
[00145] Figure 2 shows two synthetic methodologies (A and B) to
synthesize the photocatalytic composite material. In one embodiment, the
process of Figure 2A produces photocatalytic composite particles or
nanocomposite films, while the process of Figure 2B produces photocatalytic
composite films. The schematic representation of the composite material in
these
two morphological configurations (film and particle) is depicted in figure 3.
[00146] The examples mentioned will hereinafter be referred to as
sample
A (film or particle from Figures 2A or 2B ¨ synthetic methodology A) and
sample
B film (Figure 2A or 2B ¨ synthetic methodology B) or sample C particle based
upon the synthetic methodology used and the film or particle configuration of
the
composite material produced.
[00147] Synthetic methodology A, as depicted by Figures 2A and 2B
produces a polypyrrole, titanium dioxide (PPy/Ti02) photocatalytic composite
material in the form of both a film formed at the interface of the ionic
liquid
denoted as IL and the deionized water and as particles formed in the base of
the
beaker in the deionized water phase of the mixture. In this example, 0.01 g
TiO2
nanoparticles were suspended in 20 ml deionized water and sonicated for 30
minutes. Then, 0.7g FeCI3 is added and dissolved in the aqueous solution. A
second solution comprised of 0.3 ml pyrrole, added into 20 ml of an ionic
liquid is
prepared separately. The two solutions were carefully transferred to a beaker
forming an interface between the ionic liquid and aqueous layers of the
mixture.
After a few minutes the interface started changing color, while the reaction
was
allowed to continue for 8 hours at room temperature. For this methodology a
black film was generated at the interface, and black powder was dispersed in
the
- 28 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
aqueous component of the mixture. The film and powders were isolated and
washed with distilled water and ethanol (or acetone) until the filtrate became
colourless. Following this, the samples were dried in an oven at 70 C
overnight.
[00148] The particles and film were characterized by scanning electron
microscopy (SEM). The SEM micrographs are presented in Figures 7 and 8, for
sample A particles and sample A films respectively. Figure 7 shows the
presence of spherical-like structures of polymer as clusters in the composite.
These structures connect with each other in a 3d-network, comprising a porous
structure as seen in the SEM micrograph. The average dimension of the pores is
around 1 pm, and the average size of particles is about 300 nm. As shown in
Fig.
8, the SEM image of PPy/TiO2 composite film (sample A film) shows a smooth
surface at the upper layer and a porous sphere-like structure bridging with
the
film at the back of the surface, indicating that the solvent with two phases
forms a
smooth PPy film. The porous structure is considered beneficial to the
composite
efficiency in increasing the surface area available for photocatalytic
reactions to
take place.
[00149] The energy-dispersive X-ray spectroscopy (EDS) spectrum of the
sample A particles and sample A film are presented in Figures 9 and 10
respectively. Fig. 9 shows a large amount of TiO2 in the composite sample A
particles which are black in color. The conductive polymer PPy coats the TiO2
nanoparticles forming a composite material. The EDS spectrum presented in Fig.
10 illustrates the presence of a lower concentration of semiconductor TiO2 in
the
sample A film, indicating that the film's upper component is mainly a PPy film
with a majority of the TiO2 nanoparticles, coated with PPy as a nanocomposite,
is present in the bottom layer of the film. This conclusion is similar to that
achieved from X-ray diffraction (XRD) spectrum of the composite depicted in
Figure 11.
[00150] Transmission electron micrographs (TEM) of the composites from
sample A particle are presented in Figure 12. An analysis of the composite
particle size analysis of the TEM micrograph in Figure 12 is presented in
Figure
-29-
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
13. This analysis shows the presence of composite particles of sizes varying
from 62nm to 299nm and further demonstrates the formation of PPy/Ti02
composites in contrast to the input pyrrole monomers, and TiO2 precursor
particle
sizes ranging from mm to 13nm.
[00151] The samples from synthesis methodology A were also
characterized by Fourier Transform Infrared Spectroscopy hereinafter referred
to
as FTIR or IR. The IR spectra of sample A film and sample A particles are
shown
in Fig. 14 as 14a and 14b respectively. Although the morphologies of these two
samples have differences, the essential product is the same ¨Poly Pyrrole
(PPy). The broad peak in the film around 3436 cm-1 is assigned to the N¨H
stretching, but the N-H stretching shifts to 3391 cm-1. This shift may be
ascribed
to the structure change of PPy/TiO2 composites and effect of the ionic liquid
(IL)
solvent. The noisy peaks from 1656 to 1707 cm-1 may be related to a carbonyl
or
hydroxyl group and C=C stretching. The noisy peaks between 1440 and 1560
cm-1 are corresponding to the typical pyrrole rings vibration, N-H vibration,
and C-
N vibration and so on. Particular, the peaks at 1459 and 1543 cm-1 are
attributed
to the C-N and C-C asymmetric and symmetric ring stretching, respectively
(Fig.14b). The peak at 1299 (1290 cm-1 in sample A particles) and 1034 cm-1
(1033 cm-1 in A particles) are related to the in-plane vibrations of =CH.
(Feng, X.,
et al., J. Phys. Chem. C, 2007. 111: p. 8463-8468) The peak at 1167 cm-1 in
sample A particles and broad peak around 1161 cnril in A film may be assigned
for N-C stretching. (Vishnuvardhan, T.K., et al., Bull. Mater. Sci., 2006.
29(1): p.
77-83) The peak at 896 cm-1 is attributed to the C-H out of plane deformation
vibration, and the C-H out of plane ring deformation vibration locates at 784
cm
1.( Xu, P., et al., J. Phys. Chem. B, 2008. 112: p. 10443-10448 and Karim,
M.R.,
C.J. Lee, and M.S. Lee, Polym. Adv. Technol., 2007. 18: p. 916-920)
[00152] Further characterization using ultraviolet-visible
spectrophotometry
(UV-vis) is shown via a UV/Vis absorption spectrum of PPy/Ti02 composite
(sample A particles) in Figure 15. Here, the two bands at 348 nm and 465 nm
are
assigned to pyrrole oligomer and polypyrrole, respectively.
- 30 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
[00153] Fig. 16 shows the differential scanning calorimetry (DSC)
plots of
PPy/TiO2 composite (sample A particles). From this plot, there is no obvious
endothermic peak in the first heating curve. The glass transition phase is not
apparent in this sample, but there may be a wide interval of the endothermic
crest at around 145 C. This suggests that the strong binding between PPy and
TiO2 in the composite structure increases the binding energy of PPy leading to
no detectable glass transition within the temperature range of the experiment.
A
sample of pure PPy prepared according to methodology A, without the presence
of semiconductor TiO2 particles and analysed with DSC for comparison purposes
is depicted in Figure 17at the second and the third heating and cooling
cycles.
The temperature range is from 20 C to 150 C at a rate of 20 C/min. While no
obvious phase transition peaks can be observed, a small step at 105 C during
cooling process is highlighted further in Fig. 18. This small step may be
ascribed
to the glass transition point. Since only 2.15 mg PPy was used for DSC, the
glass
transition may not be obvious.
[00154] Example 2 ¨ Alternative general process for preparing photo-
catalytic composite material
[00155] Another example of the synthetic methodology B, as depicted by
Figure 2A and 2B produces a polypyrrole, titanium dioxide (PPy/T102)
photocatalytic composite material in the form of a film formed at the
interface of
the ionic liquid (IL) and the deionized water and as a film formed at the base
of
the beaker in the deionized water phase of the mixture. In this example, 0.01
g
TiO2 nanoparticles were suspended in 20 ml of IL and sonicated for 30 minutes.
Then, 0.3 ml of pyrrole was added to the IL solution. A separate solution
comprising of 0.7g FeCI3 dissolved in 20m1 of deionized water was prepared
separately. The two solutions were carefully transferred to a beaker to form
an
interface between the ionic liquid and aqueous layers of the mixture. After a
few
minutes the interface started changing color, while the reaction was allowed
to
continue for 8 hours at room temperature. For this methodology, two films
(with
some particles embedded within them) were obtained from the interface and
bottom of the sample. The films were isolated and washed separately with
- 31 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
ethanol (or acetone). The films were then dried at 70 C overnight in an oven.
In
the sample B, the adhesion of white TiO2 particles at the surface of the film
is
observed.
Discussion
[00156] The
film recovered from this synthetic methodology B, was
characterized by scanning electron microscopy (SEM). The SEM micrographs
are presented in Fig. 19 and depict the composite semiconductor-polymer
particles embedded on the surface of a PPy film. The presence of elemental Ti
in
the composite is confirmed by an EDS spectrum presented in Figure 20.
[00157] The
samples from synthesis methodology B were also
characterized by FTIR. The IR spectra of sample B is shown in Figure 21. The
FTIR spectra of the sample B film are similar to that of sample A, with clear
and
more defined peaks. Fig. 21(a) shows the spectrums of the two films obtained
by
methodology B, specifically one at the interface and one at the bottom of the
beaker (the top line is the spectrum of the film from the bottom layer while
the
bottom line is the spectrum of the film from the interface/middle layer). The
transmittance peaks of the two films are almost at the same positions.
Therefore
the spectrum from the bottom layer film is illustrated as an example. C=C and
C¨
N stretching modes for the polypyrrole rings occur at 1543 cm-1 and 1459 cm
1.(Qingzhi Luo, et al., Journal of materials science, 2011. 46(6): p. 1646-
1654)
The bands at 1297 cm-1, 1172 cm-1 and 1043 cm-1 are also attributed to
stretching mode for polypyrrole.(Qingzhi Luo, et al., Journal of materials
science,
2011. 46(6): p. 1646-1654) It is also noticed that the film generated from
interface
shows a peak at 2130 cnn-1, which is due to ILI 05, indicating that the
composite
retains a portion of the ionic liquid IL 105 used in the synthetic methodology
B.
[00158]
Further characterization using ultraviolet-visible spectrophotometry
(UV-vis) is shown via a UVNis absorption spectrum of PPy/TiO2 composite
(sample B film) in Figure 22. In Figure 22A, the two bands at 275 nm and 375
nm
are related to pyrrole oligomers. As the PPy composite film is slightly
dissolved,
the UV-vis peaks of PPy are very weak. Comparing this to the UV-vis of the
- 32 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
sample A particles in Figure 15 where the absorption wavelengths are longer,
it
shows that the PPy synthesized in the sample B film composites have shorter
polymer chains. It is proposed that the phase and/or the dimension of the core
semiconductor particles could affect the polymerization process on the surface
of
said semiconductor particles. Figure 22B shows the Uv-vis absorption spectrum
of the PPy/TiO2 film, in which the two bands at 375 nni and 685 nm are
assigned
to pyrrole oligomer and PPy, respectively.
[00159] A DSC plot of the sample B film is presented in Figure 23.
From
this plot, there is no obvious endothermic peak in the first heating curve.
The
glass transition phase is not apparent in this sample, but there may be a wide
interval of the endothermic crest at around 145 C. The strong binding between
PPy and TiO2 in the composite structure increases the binding energy of PPy
leading to no detectable glass transition within the temperature range of the
experiment.
[00160] The composite films produced by methodology B were in
comparison with sample A particles subjected to an elemental analysis to study
the thermal decomposition and burning processes of the photocatalytic
composites. The element analysis was done for "original" or newly synthesized
samples, and for "burned samples", which were produced by heating the
photocatalytic composites to 1000 C in air. Figure 24 presents in a tabular
form
the element analysis for the ratio of C to N in the samples. The C/N molar
ratio of
prepared PPy is 3.9 0.3, which accords with the PPy structure. The C/N molar
ratio of sample A particles and sample B film also correspond to the structure
of
PPy, implying that the TiO2 particles do not affect the structure of PPy. The
mass
change during heating process was recorded by thermogravimetric analysis
(TGA) and presented in Figure 25. The element analysis was accomplished by
combustion analysis. In this analysis technique, the sample was burnt with
excessive oxygen, and the combustion products, examples of which include CO2,
H20 and NO were collected. The composition of the sample is calculated from
the different combustion products to determine the ratio of elements from the
sample, and infer its structure.
- 33 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
[00161] In order to study the "burning process" of the composites and
the
components of composites, the samples A particles and B film were placed in
the
TGA under air flow. The temperature went to 1000 C at a rate of 15 C/min.
After the heating and cooling process, the black composite samples were
transformed into a yellow powdery residue. Since there are no C, N and H
elements in the residue (Figure 24), the PPy should be burned out during the
heating. To estimate the other components of in the yellow residue, the sample
A
particles is used for the following example determination. In the sample A
particles, the weight percentage of element C, N and H are 44.41%, 14.01% and
3.52% respectively, so the total content of these three elements is about 62%.
Since only 0.01g TiO2 nanoparticles was added in to the solvent that contains
about 0.3g pyrrole in the preparation of composite, the weight percentage of
TiO2
should be only 2% in the sample (assuming all the pyrrole was polymerized and
doped with all TiO2 nanoparticles). Therefore, the rest of the weight, which
was
36% in the composite, is expected to be FeCl2 (as Fe2+ and Cl- doping into the
composites). Some unoxidized Fe3+ may also be present in the composite. This
finding may explain the observed color change of black to yellow after heating
to
1000 C. The FeCl2 is oxidized by exposure to air at high temperature,
producing
the red-yellow iron oxide (Fe203). As rutile TiO2 is known to be very stable
at a
thousand degrees Celsius, the produced yellow powder should be the
combination of Fe203and Ti02.
[00162] The abovementioned TGA, measurements of the sample burning
are depicted in Figure 25. It compares the mass loss of the photocatalytic
composites (A particles, B film and C particles) under argon flow and air flow
respectively with increase of temperature. Fig. 25 (a) and (b) show that the
mass
of sample A particles and sample B film under air flow decrease more than the
photocatalytic composites under argon flow. This is probably attributed to two
reasons. One is that all the PPy is "burned" out and forms combustion products
such as 002, NO2, etc. under air flow (based on the element analysis data).
While the composite was under inert gas flow, some of the polymer PPy that is
- 34 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
strongly bound with the semiconductor TiO2 nanoparticles, exhibit a higher
thermal stability, and does not burn.
[00163] An alternative explanation for the observed data can be
ascribed to
the ability of FeCl2 (with the possibility of some FeCI3) to undergo a
different
chemical reaction in air and an inert atmosphere. Under an inert atmosphere, a
part of FeC13 decomposes to FeCl2 and 012. TheC12 can be flushed away in the
gas stream and its weight loss is the main loss for FeC13. While in the air,
FeCl2
reacts with oxygen and produces Fe203 and C12. The weight loss in this
reaction
is larger than the previous reaction, so this may be the other reason that the
decomposition of composites under air atmosphere is more significant than the
decomposition under an inert atmosphere. However, sample C particles, whose
methodology is described later in this patent, exhibit different TGA curves
with
the other two composites. Fig. 25 (c) shows the TGA curves of sample C
particles under air and argon atmosphere. The weight of the composite in argon
atmosphere loses 5% more than the composites in exposure of air. As described
in Figure 27, the comparison between TGA curves verified that the interaction
between PPy and TiO2 nanoparticles in A particles and B film is much stronger
than the C particles (Fig. 27). Because fewer TiO2 nanoparticles are in C
particles, more PPy was decomposed than the other two composites. In this
case, the weight loss of the PPy should be almost the same under two gases. In
addition, it is very possible that there are much fewer Fe2+ ions in this
sample
than other composites because the small amount of TiO2 nanoparticles in the
composite which can absorb iron ions at the surface. Therefore, the 5%
difference could be from the error during mass measurement as only 0.273 mg
sample left after heating under inert gas.
[00164] Comparing the two TGA curves run under air flow and argon flow
of
all the samples in Figure 25 a, b and c respectively, the two decomposition
processes are similar at temperatures below about 250 C. After the
temperature
approaches 250 C, the composite under air flow loses weight rapidly due to
the
oxidation of the PPy. All the PPy is oxidized into gaseous products of
combustion
(e.g. 002, H20) after 700 C.
- 35 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
Example 3 ¨ Second alternative general process for preparing photo-catalytic
composite material
[00165] Another example synthetic methodology C, is capable of
producing
a polypyrrole, titanium dioxide (PPy/Ti02) photocatalytic composite material
in
the form of particles formed by synthesis in an aqueous medium. In this
example
0.01 g prepared rutile TiO2 nanoparticles and 0.3 mL pyrrole were added to 40
mL aqueous FeCI3 (0.7 g) solution. Polymerization was initiated as soon as the
pyrrole was mixed with the FeCI3 (the initiator) solution, and allowed to stir
for 8
hours at room temperature. The product precipitated from the solution as dark
powder. It was then filtered and washed with distilled water, and dried at 70
C
overnight.
Discussion
[00166] A UV-Vis absorption spectrum of the sample C particles is
presented in Figure 26. In this spectrum the bands at 275 nm, 372 nm and 620
nm are assigned to pyrrole oligorners and longer chain polymers of pyrrole as
seen by peaks at both a long and short wavelength on the UV-vis spectrum.
[00167] Thermogravinnetric Analysis (TGA) was then used to compare the
thermal stability of three samples (sample A particle, sample B film, sample C
particle). The TGA curves relating to the thermal stability testing are
presented in
Figure 27. Comparing the TGA curves of sample A particles and sample B film
with the curves sample C which is synthesized in water, sample C loses much
more weight. When the temperature approaches 800 C, 94% of sample C is
decomposed, while 61% of sample B and 51% of sample A are decomposed.
Therefore, the composites synthesized in IL/water solutions have higher
thermal
stability than the photocatalytic composites synthesized in water due to a
possible increase in the polymer-semiconductor interactions present in the
photocatalytic composites synthesized by the A and B example methodologies.
[00168] The thermal stability of the two composites made at different
molar
material ratios in comparison to pure PPy is examined by TGA. The TGA traces
is presented in Figure 28. The curves (a) and (b) represent the two samples
- 36 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
synthesized by the same method, but with different molar material ratios. The
comparison of pure PPy and PPy/Ti02 particles shows that the weight loss of
PPy is much more than the composite. The better stability is ascribed to the
presence of TiO2 nanoparticles within the composite.
[00169] The degradation of PPy shows a three stage decomposition pattern
as three major slope changes are observed. At T < 100 C, the mass loss is
caused by the presence of residual water in the sample. The next stage of the
mass loss is by degradation that lasts until 225 C, and is attributed to the
loss of
dopant ions that are weakly (electrostatically) bound, from the inter-chain
sites of
the polymer. When the temperature goes higher, the more obvious degradation
begins. This weight loss is due to degradation and decomposition of the
polymer
backbone.
[00170] Unlike pure PPy, the degradation processes of PPy/Ti02
composites show two stages. In curve (a) and (b), the mass loss of the
PPyiTi02
composite observed at T < 125 C is also because of the evaporation of
residual
water in the sample. The binding of PPy to TiO2 may affect the water
absorption
of PPy, hence less water absorbed by the composites. Another potential cause
is
the steric effect that could have been due to possible crosslinking of PPy
chains
on the surface of TiO2 nanoparticles. Therefore there is less weight loss in
the
PPy/Ti02 composite at T < 125 C. The second stage from 125 C to 800 C is
due to the degradation and decomposition of the PPy backbone. This weight loss
may also be related to the decomposition of coordination compounds formed as
a result of interaction between the TiO2 and PPy at the interface. [16] There
are
some differences between curves (a) and (b) in the second stage of
degradation.
The major weight loss happens in the curve (a) at 270 C, and after 400 C,
the
mass loses consistently. In the curve (b), although a small amount of weight
loses at 200 C because of the loss of dopant ions, the major loss occurs up
to
400 C which is higher than the temperature in curve (a). This may be ascribed
to
the higher amount of TiO2 in this sample, leading to a stronger backbone of
composites.
- 37 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
[00171] When the temperature approaches 800 C, the weight loss of
PPy/Ti02 is about 50%, while the pure PPy is completely degraded. This is due
to the TiO2 content and the residues bound to TiO2 which are stabilized by
TiO2
nanoparticles. The picture that emerges from comparison of the TGA data (fig.
28) of PPy/Ti02 and PPy is the following: PPy is more like a linear polymer
but
when the polymer is grown on the surface of rutile TiO2 nanoparticles a large
degree of cross linking happens. This causes the shift of decomposition of PPy
to
larger temperatures. At lower temperatures the decomposition is less but at
higher temperatures (after the cross linked network is broken) the
decomposition
is faster, however it is limited to a certain mass that includes TiO2 and
small
oligomers of pyrrole stabilized by T102. The composite material is therefore
useful as a fire-retardant material.
Example 4¨ Generation of Hydrogen Gas from Water
[00172] The ability of the materials of the present disclosure to
photocatalytically decompose water and produce gas under visible light
irradiation was tested using a measurement system described below. The results
of the measurements of hydrogen gas production over time are presented in fig.
29 for the sample A particles and fig. 30 for sample B film when immersed in
water and irradiated with the visible component of sunlight. Sample B film
over a
period of 600s is seen to produce more gas than the sample A particle
configuration.
[00173] FIG. 31 shows the amount of hydrogen gas produced using the
composites sample B film (triangles) and sample A particles (diamonds) at a
light
intensity of 460 IX. FIG. 32 shows the amount of hydrogen gas produced using a
PPy-Ti02 film composite in first(+) and second(o) trials. FIG. 33 shows the
amount of hydrogen gas produced using TiO2 nanoparticles, while FIG. 34 shows
the amount of hydrogen gas produced using sample T (using toluene, water
interface for synthesis at a light intensity of 450 lx. Pure PPy did not
generate
any hydrogen or any other gas under similar experimental conditions.
Example 5¨ Apparatus for Measuring Gas Evolution or Gas Capture
- 38 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
[00174] The measurement of hydrogen gas generation by the
photocatalytic
composite materials of the present disclosure was conducted with an apparatus
for measuring quantitative digital gas capture and digital gas release
measurements. The apparatus comprises a pressure transducer connected to
the sample measurement chambers equipped with evacuating and balancing
valves. The pressure transducer measures the pressure difference between the
two cells, and provides an output that is or can be converted into a digital
signal
by a data recorder linked to a computer system that enables via appropriate
electronic software, a digital recording and analysis of the data
(i) Gas Capture
[00175] In order to test the capability of the apparatus in detecting
CO2
capture, a known agent for absorbing CO2, sodium hydroxide was used to
remove CO2 in the experiment cell, and the apparatus detected and recorded the
pressure changing due to the CO2 being absorbed.
(ii) Gas Evolution
[00176] The apparatus was also used to measure the release of H2 from
water using the photo-catalytic composite materials described herein (Figs 29
and 30).
[00177] A Dycor Validyne DP15 pressure transducer, connected to a pair
of
glass cells, equipped with evacuating and balancing valves. The data recorder
(Figure 35) was built based on an Arduino circuit board and connected to a PC.
The data recorder takes the amplified signal from the transducer indicator,
and
sends them to the PC via the USB port. The Arduino board was connected to PC
via USB, which provides the +5V, the ground, and the data output. The voltage
divider was protected from interference with a magnetic ring. The entire
circuit is
enclosed in an aluminum box, which is grounded and schematically depicted in
Figure 36.
[00178] An example of the software used by the data recorder in this
embodiment of the instrument invention is presented below. The source code of
the program on the data recorder (5th version), in Arduino programming
- 39 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
language (based on Wiring) and the Arduino development environment (based
on Processing):
Wersion 5 of the computeralized data recording for the demodulator.
//By Guy
V/Based on the Arduino example code "AnalogInOutSerial"
//update:
//will show the most recent reading in Gobetwino
//added the standby switch and 2 LEDs
//using only 1 analogue input and the ground for input.
//included the voltage divider and a 2* factor for voltage output.
// These constants won't change. They're used to give names
// to the pins used:
const int analogInPinRed = A2; // Red input from demodulator
const int readingLED = 13;
const int readingSwitch = 7;
float sensorValuePos = 0; // value read from the port
float voltage = 0 //output voltage
double tInterval = 60000; // The time interval between 2 data
(milliseconds) To change the time interval, change this number.
int readingSwitchState = 0;
void setup() {
pinMode(readingLED, OUTPUT); // initialize the led and stitch pins.
pinMode(readingSwitch, INPUT);
// initialize serial communications at 9600 bps:
Serial.begin(9600);
-40 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
void loop() {
read ingSwitchState = digitalRead(readingSwitch);
if (readingSwitchState == HIGH) //When the switch is on "Reading".
digitalVVrite(readingLED, HIGH); //Turn reading LED on
sensorValuePos = analogRead(analogInPinRed);
// read the analog input value in volts:
voltage=(sensorValuePos*5*2)/1023; //Convert to voltage.
// print the results to the serial monitor:
Serial.print("Voltage = " );
Serial.print(voltage);
Serial.println(" V");
Serial.print("#SIVCSVI[");
Serial.print(voltage);
Serial.println("1#");
I/ wait sometime (defined by the tInterval variable) before the next loop
// the interval between 2 reading.
delay(tInterval); }
1 else {digitalVVrite(readingLED, LOW);} //When the switch is on "Standby"
[00179] The PC receives the voltage reading of the recorder, verses
time,
and stores in a .txt or a .csv file. In this example it was done by using the
Gobetwino application developed by MikMo (http://mikmo.dk/gobetwino.html).
Installation of the drivers of the data recorder on PC: based on the
instruction of
the Arduino Duemilanove from http://arduino.cc/en/Guide/Windows#toc4. The
- 41 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
example device and apparatus for measures the release rate of hydrogen gas,
as well as CO2, SO, and NO, capture rates, as depicted in Figure 37.
[00180] Test of linearity: In order to determine the uniformity of the
various
assembled components of the instrument a test of linearity was required before
calibration.
[00181] The whole system was pumped in vacuum for 2 hours, to remove
any possible residual liquid in the connecting tubing. After that both cells
were
filled with air at atmosphere pressure, and then closed to the outside. The
reference cell was kept in room temperature water, while the sample cell
placed
in water with preset temperatures. The temperature difference of 2 cells were
recorded once the voltage reading stabilizes. As the temperature reached
equilibrium, so did the pressure in each cell. These measurements are
presented
in a tabular form below as table 1. A plot of the same data assuming an ideal
behavior of gas in the system is presented in Figure 38. This graph shows a
correlation of temperature of air in each cell to the output voltage is almost
perfectly linear, and there is no significant leaking of gas. The apparatus is
then
ready for calibration.
[00182] Calibration: In order to determine quantitatively the amount
of CO2
being captured, the ratio between the changing of one mole of gas and the
change in output voltage needed to be determined. This will be a factor in the
form x volts per moles of gas, and it is specific to the pair of cells used in
the
calibration.
[00183] A small volume of air at atmospheric pressure and room
temperature was injected to the sample cell, while the reference cell remains
unchanged. The moles of gas injected can be calculated using the ideal gas
law.
Fitting the moles to the voltage reading can give the volts / moles factor.
Each
injection volume was done in triplets and the data presented in tabular format
below as table 2 followed by a sample calculation to determine the moles of
gas
present in the volume of air. A plot of the moles injected vs. voltage with a
linear
-42 -
CA 02946327 2016-10-19
WO 2014/169373 PCT/CA2014/000352
regression analysis applied to the relationship presented in the data (table 2
and
Figure 39) suggests the volts/moles factor is 1 volt per 9.616*10-7 mole of
gas.
[00184] Sample calculation:
1 L
1 atm x 0.025 mix
PV 1000 ml
mole ,---- --= --,-- 0.00000103 mole
RT 0.08206 L atm mot' IC x295.3 K
[00185] CO2 capture test: In trial 1, the results of which are plotted in
Figure
40, 0.0073g NaOH pellet was placed in the sample cell. In order to reduce the
reaction rate to a measureable rate, the balance valve between the 2 cells was
first open after the NaOH pellet being placed, to allow the surface of NaOH
pellet
to react with the CO2, to reduce the CO2 pressure and reduce the reaction
rate.
The balance valve was then closed for 1 second to check the reaction rate for
several time, till a reasonably low rate was observed, and recording started.
In
this trail the voltage went out of the measuring capacity before the reaction
stopped. 9.613*10-6 mole of CO2 was absorbed in 660 seconds, beyond which
the absorption was not measured by the instrument in this trial. This shows
that
the reaction of untreated NaOH surface in high 002 pressure (close to 1 ATM
pure CO2) was too fast at first for the components used in this embodiment of
the
instrument to measure but not with more suitable components.
[00186] In trial 2, 0.0013g NaOH pellet was placed in the sample cell,
but
recording started immediately after that. There was no more balancing after
the
placement of NaOH pellet. The results of trial 2 are shown in Figure 41 as a
voltage change over time, which corresponds to the absorption of CO2 over time
by the system.
[00187] In trials 3 and 4, the CO2 absorbing compounds, imidazolium-
[1,2-
a]-pyridine trifluoroacetate and imidazolium-[1,2-a]-pyridine Mal were
utilized in a
similar fashion as in trials 1 and 2. The results of trials 3 and 4 are shown
in
Figures 42A and 42B, as the amount of 002 absorption (mmol of CO2 absorbed
per 0.1 g of compound) over time. The CO2 absorbing compounds are shown as
follows:
-43-
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
[ImPr][TFA]
_ _
0 H
e/
FL,, e =-=\,.......-N\
0
F
Imidazolium-11,2-4-pyridine trifluoro acetate
[ImPr][Mal]
- o)
H2N H-
eN/ ,,-.,,0
/,------....----
6 N
_
Discussion
[00188] The setup is capable of CO2 capture study, but for CO2
absorption
reaction that is fast, some methods can be used to limit the reaction rate to
a
more measureable rate to improve the quality of the components in the
invention
such that they can detect fast adsorption processes.
[00189] Compared to other methods of tracking and observing CO2
elimination in the literature, the method used in this study has some pros and
cons. Compared to a gas analyzer, used in some studies, the pressure
transducer is low cost, and relatively simpler to setup. As the transducer
measures the CO2 content indirectly from the pressure, it can proceed with a
reference experiment medium (as in this study) or referenced to the
atmosphere,
which allows more flexibility in testing. This is in contrast to other methods
that
rely on the electrical property of CO2 in solvent, pH and electrical
conductivity
(EC) meters, or electrical impedance and capacitance spectroscopy, which can
look into the distribution of CO2 in solvent and may provide more information
than
a transducer. The drawback of these measurement systems however, is that
they are limited to using a solvent medium and cannot be applied to a gaseous
system, or liquid or super critical state 002. An alternative method, where
the
-44 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
reaction product (formic acid) is directly examined requires the analysis
occur at
completion of the chemical reactions, whereas our instrumentation is able to
conduct measurements in real time. Although a production amount verses time
relationship can be constructed by analyzing the reaction products through the
performance of several experiments with varying lengths of time, it would be
very
time consuming as compared to measuring a CO2 elimination rate verses time in
a single experiment.
-45 -
CA 02946327 2016-10-19
WO 2014/169373 PCT/CA2014/000352
Table 1 - Temperature Difference verses Voltage Output
Reference Temp (C) Sample Temp (C) Temp Difference (C) Voltage Output (V)
21.7 25.2 3.5
5.68
21.6 18.8 -2.8 -
4.97
21.6 18.1 -3.5 -
6.02
21.5 17.5 -4.0 -
7.11
21.6 16.9 -4.7 -
8.41
21.7 16.5 -5.2 -
9.00
21.7 16.2 -5.5 -
9.44
21.7 16.0 -5.7 -
10.04
21.7 20.7 -1.0 -
1.88
21.7 21.5 -0.2 -
0.38
21.7 23.0 1.3
2.22
21.7 , 25.0 3.3
5.41
21.7 26.1 4.4
7.18
21.7 27.3 5.6
9.41
21.7 27.7 6.0
10.35
-46 -
CA 02946327 2016-10-19
WO 2014/169373
PCT/CA2014/000352
Table 2 - Gas Injected verses Voltage Output
Room temp = 295.3(K)
Reading Change (V)
Inject Air Volume (ml) Inject Air (Mole) Trial 1 Trial 2 Trial 3
0.025 0.00000103 1.07 1.11 1.09
0.050 0.00000206 2.18 2.20 2.11
0.075 0.00000310 3.21 3.26 3.26
0.100 0.00000413 4.26 4.25 4.30
0.125 0.00000516 5.43 5.39 5.40
0.150 0.00000619 6.33 6.51 6.40
0.175 0.00000722 7.46 7.49 7.55
0.200 0.00000825 8.68 8.50 8.61
0.225 0.00000929 9.76 9.67 9.65
-47 -