Note: Descriptions are shown in the official language in which they were submitted.
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
METHOD AND SYSTEM FOR ELECTROCHEMICAL REDUCTION OF CARBON
DIOXIDE EMPLOYING A GAS DIFFUSION ELECTRODE
TECHNICAL FIELD
Noon The present disclosure generally relates to the field of electrochemical
reactions, and more particularly to a method and system for reducing carbon
dioxide to a reduction product.
BACKGROUND
[0002] The combustion of fossil fuels in activities such as electricity
generation,
transportation, and manufacturing produces billions of tons of carbon dioxide
annually. Research since the 1970s indicates increasing concentrations of
carbon
dioxide in the atmosphere may be responsible for altering the Earth's climate,
changing the pH of the ocean, and other potentially damaging effects.
Countries
around the world, including the United States, may be seeking ways to mitigate
emissions of carbon dioxide.
[0003] One implementation may be to convert carbon dioxide into economically
valuable materials such as fuels and industrial chemicals. If the carbon
dioxide
may be converted using energy from renewable sources, it will be possible to
both mitigate carbon dioxide emissions and to convert renewable energy into a
chemical form that may be stored for later use. Electrochemical and
photochemical pathways may be likely mechanisms for carbon dioxide
conversion.
SUMMARY OF THE PREFERRED EMBODIMENTS
[0004] The present disclosure is a method and system for the reduction of
carbon dioxide. The method may include receiving hydrogen gas at an anolyte
region of an electrochemical cell including an anode, the anode including a
gas
diffusion electrode, receiving an anolyte feed at an anolyte region of the
1
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
electrochemical cell, and receiving a catholyte feed including carbon dioxide
and an alkali metal bicarbonate at a catholyte region of the electrochemical
cell including a cathode. The method may include applying an electrical
potential between the anode and cathode sufficient to reduce the carbon
dioxide to at least one reduction product.
[0005] It is to be understood that both the foregoing general description and
the
following detailed description are exemplary and explanatory only and are not
necessarily restrictive of the present disclosure. The accompanying drawings,
which are incorporated in and constitute a part of the specification,
illustrate
subject matter of the disclosure. Together, the descriptions and the drawings
serve to explain the principles of the disclosure.
BRIEF DESCRIPTION OF THE DRAWINGS
[0006] The numerous advantages of the present disclosure may be better
understood by those skilled in the art by reference to the accompanying
figures
in which:
Fig. 1A shows a system for production of oxalic acid starting with the
electrochemical generation of carbon monoxide from carbon dioxide in
accordance with an embodiment of the present disclosure;
Fig. 1B shows a system for the production of oxalic acid utilizing a hydrogen
halide in the anolyte to co-produce a halogen in accordance with an
embodiment of the present disclosure;
Fig. 2A shows a system for production of oxalic acid starting with the
electrochemical generation of formate using carbon dioxide in accordance with
an embodiment of the present disclosure;
Fig. 2B shows a system for production of oxalic acid via electrochemical
generation of formate using carbon dioxide and utilizing a halogen halide in
the
2
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
anolyte to co-produce a halogen in accordance with an embodiment of the
present disclosure;
Fig. 3 shows a system for production of alkali metal formate using carbon
dioxide in accordance with an embodiment of the present disclosure;
Fig. 4 shows a system for electrochemical acidification of alkali metal
oxalate
in accordance with an embodiment of the present disclosure;
Fig. 5 shows a system for production of alkali metal formate using carbon
dioxide and utilizing a alkali metal chloride brine in the anolyte to co-
produce
chlorine and alkali metal bicarbonate in accordance with an embodiment of the
present disclosure;
Fig. 6 shows a system for production of alkali metal formate using carbon
dioxide and co-generating chlorine, alkali metal hypochlorite (MOCl) and
oxalic
acid in accordance with an embodiment of the present disclosure;
Fig. 7 shows a system for the production of a formic acid solution and oxygen
co-product using carbon dioxide and a three compartment electrochemical cell
configuration in accordance with an embodiment of the present disclosure;
Fig. 8 shows a system for the production of a formic acid solution and a
chlorine co-product using carbon dioxide and HCl and a three compartment
electrochemical cell configuration in accordance with an embodiment of the
present disclosure;
Fig. 9 shows a system for the production of an oxalic acid solution product
and an oxygen co-product using carbon dioxide, a thermal reactor to convert
formate to oxalate, and a three compartment electrochemical cell configuration
in accordance with an embodiment of the present disclosure;
3
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
Fig. 10 shows a system for the production of an oxalic acid solution product
and a chlorine co-product using carbon dioxide, a thermal reactor to convert
formate to oxalate, and a three compartment electrochemical cell configuration
in accordance with an embodiment of the present disclosure;
Fig. 11 shows a system in utilizing a formic acid produced in the
electrochemical system for use in an off peak power energy storage system in
accordance with an embodiment of the present disclosure;
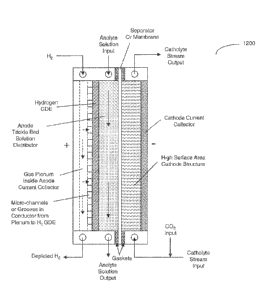
Fig. 12 is a schematic illustrating an electrochemical cell utilizing a
hydrogen
GDE for the anode reaction in producing oxalic acid from the reduction of
carbon dioxide in accordance with an embodiment of the present disclosure;
Fig. 13 is a schematic illustrating an electrochemical cell utilizing a
hydrogen
GDE for the anode reaction and a carbon dioxide GDE for the cathode reaction
in producing alkali metal formate from the reduction of carbon dioxide in
accordance with an embodiment of the present disclosure;
Fig. 14 is a schematic illustrating three different anode GDE constructions
used in an electrochemical cell in producing alkali metal formate from the
reduction of carbon dioxide in accordance with an embodiment of the present
disclosure;
Fig. 15 is a schematic illustrating three different cathode GDE constructions
used in an electrochemical cell in producing alkali metal formate from the
reduction of carbon dioxide in accordance with an embodiment of the present
disclosure;
Fig. 16 is a schematic illustrating an electrochemical cell utilizing a
hydrogen
GDE and a weir solution distribution system for the anode reaction in
producing
alkali metal formate from the reduction of carbon dioxide in accordance with
an
embodiment of the present disclosure;
4
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
Fig. 17 is a schematic illustrating an electrochemical cell utilizing a
hydrogen
GDE for the anode reaction and a carbon dioxide GDE for the cathode reaction
and showing a weir solution distribution system in producing alkali metal
formate from the reduction of carbon dioxide in accordance with an
embodiment of the present disclosure;
Fig. 18 shows potential chemical derivatives starting with oxalic acid as an
initial chemical feedstock in accordance with an embodiment of the present
disclosure;
Fig. 19 shows a system for electrochemically converting carbon dioxide to
oxalic acid which is then converted to glyoxylic acid electrochemically and
the
glyoxylic acid is converted to glycine by reductive annination in accordance
with
an embodiment of the present disclosure in accordance with an embodiment of
the present disclosure;
Fig. 20 shows an electrochemical cell for reducing oxalic acid to produce a
glyoxylic acid product in accordance with an embodiment of the present
disclosure; and
Fig. 21 shows a batch reactor system for the conversion of glyoxylic acid to
glycine in accordance with an embodiment of the present disclosure.
DETAILED DESCRIPTION
[0007] Reference will now be made in detail to the subject matter disclosed,
which is illustrated in the accompanying drawings.
[00os] The present disclosure describes a method and system for the reduction
of carbon dioxide. The method may include receiving hydrogen gas at an
anolyte region of an electrochemical cell including an anode, the anode
including a gas diffusion electrode, receiving an anolyte feed at an anolyte
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
region of the electrochemical cell, and receiving a catholyte feed including
carbon dioxide and an alkali metal bicarbonate at a catholyte region of the
electrochemical cell including a cathode. The method may include applying an
electrical potential between the anode and cathode sufficient to reduce the
carbon dioxide to at least one reduction product.
[0009] Additionally, the present disclosure describes a method and system for
production of carboxylic based chemicals, including carboxylic acids and
salts.
The method may employ an electrochemical cell, cathode reduction reaction to
produce carbon monoxide (CO) or alkali metal formate from a carbon dioxide
feedstock. A thermal reaction with the addition of a small amount of an alkali
metal hydroxide catalyst may be used to combine, for example, two alkali metal
formate molecules, into an alkali metal oxalate product.
[0olo] The alkali metal oxalate may be then converted to oxalic acid by a
membrane based electrochemical acidification process, where protons (H+ ions)
formed at the anode may be used to replace the alkali metal ions, and the
alkali
metal ions (M+) may be captured as alkali metal hydroxide (MOH) at the
cathode, and may be recycled to be used as the alkali metal hydroxide used in
a
thermal intermolecular condensation process unit operation.
[001 1] Alternatively, the alkali metal oxalate may be converted to oxalic
acid
through treatment with mineral acid, such as HCl, HBr, HI, H2SO4, H3PO4, and
the like. For example, sodium oxalate may be treated with aqueous HCl may
produce an oxalic acid solution including NaCl. The oxalic acid may be
extracted
from the solution via extraction with an organic solvent such as alcohol,
ether,
halo-organic, ketone, amide, or ester. Useful solvents include, but are not
limited to, methanol, ethanol, propanol, diethyl ether, methyl ethyl ether,
methyl tert-butyl ether, tetrahydrofuran, dioxane, methylene chloride,
chloroform, carbon tetrachloride, chlorobenzene, di-chlorobenzene, methyl
acetate, ethyl acetate, methyl propionate, ethyl propionate, acetone,
butanone, dinnethylfornnannide, N-methyl pyrrolidone, and the like. The oxalic
6
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
acid may also be recovered from the solution through crystallization from the
aqueous solution. Crystallization may require concentrating the solution by
evaporation and/or by cooling the solution.
[0012] After removal of oxalic acid, the aqueous solution including salt (NaCl
for
example) may be recycled by sending it to an anolyte compartment of an
electrochemical cell. Halide ions (for example chloride) may be oxidized to
form
halogen (for example chlorine). The halogen may be isolated from an anolyte
stream after exiting an anolyte compartment of an electrochemical cell. The
halogen may be reacted with hydrogen, for example hydrogen produced during a
thermal alkali metal formate to alkali metal oxalate calcination reaction.
Hydrogen may also be obtained from another source. The mineral acid (HCl for
example) formed by the reaction of hydrogen with halogen may be used to
acidify alkali metal oxalate, completing the cycle. The energy produced (heat
or electrical energy) by reacting halogen with hydrogen may be captured and
used in other processes (in the thermal calcination reaction for example) or
may
be used elsewhere.
[0013] Before any embodiments of the disclosure are explained in detail, it is
to
be understood that the embodiments may not be limited in application per the
details of the structure or the function as set forth in the following
descriptions
or illustrated in the figures. Different embodiments may be capable of being
practiced or carried out in various ways. Also, it is to be understood that
the
phraseology and terminology used herein is for the purpose of description and
should not be regarded as limiting. The use of terms such as "including,"
"comprising," or "having" and variations thereof herein are generally meant to
encompass the item listed thereafter and equivalents thereof as well as
additional items. Further, unless otherwise noted, technical terms may be used
according to conventional usage. It is further contemplated that like
reference
numbers may describe similar components and the equivalents thereof.
7
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
[0014] Referring to Fig. 1A, a system 100 for production of a dicarboxylic
acid,
such as oxalic acid, starting with the electrochemical generation of formate
from the electrochemical reduction of carbon dioxide in accordance with an
embodiment of the present disclosure is shown. System 100 may include an
electrochemical cell 110. Electrochemical cell 110 (also referred as a
container, electrolyzer, or cell) may be implemented as a divided cell. The
divided cell may be a divided electrochemical cell and/or a divided photo-
electrochemical cell. Electrochemical cell 110 may include an anolyte region
and a catholyte region. Anolyte region and catholyte region may refer to a
compartment, section, or generally enclosed space, and the like without
departing from the scope and intent of the present disclosure.
[0015] Catholyte region may include a cathode. Anolyte region may include an
anode. An energy source (not shown) may generate an electrical potential
between the anode and the cathode of electrochemical cell 110. The electrical
potential may be a DC voltage. Energy source may be configured to supply a
variable voltage or constant current to electrochemical cell 110. A separator
may selectively control a flow of ions between the anolyte region and the
catholyte region. Separator may include an ion conducting polymer based
membrane, an ion conducting inorganic material, a combination
polymer/inorganic based membrane, or a diaphragm material such as expanded
PTFE (polytetrafluoroethylene) and the like.
[0016] Electrochemical cell 110 may operate to perform an electrochemical
reduction of carbon dioxide in an electrochemical cell producing carbon
monoxide (CO) and hydrogen as cathode products and oxygen as an anode
product when using an anolyte including sulfuric acid (H2SO4).
[0017] The CO generated from electrochemical cell 110 may be separated from
the hydrogen and then passed to a thermal reactor 120. Thermal reactor 120
may react the carbon monoxide with an alkali metal hydroxide, such as KOH via
a thermal intermolecular condensation reaction to form alkali metal formate.
8
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
Thermal reactor 120 may operate to perform a thermal decomposition reaction
or a carbonylation reaction, which may be reactions which incorporate CO into
organic and inorganic chemical structures.
[0018] Alkali metal formate formed from thermal reactor 120 may be passed to
another thermal reactor 130. Thermal reactor 130 may perform a second
thermal intermolecular condensation reaction employing an alkali metal
hydroxide (e.g. KOH) that may promote the reaction to produce alkali metal
oxalate. While system 100 of Fig. 1 depicts a thermal reactor 120 and thermal
reactor 130, it is contemplated that a single thermal reactor may be employed
with system 100 without departing from the scope and intent of the present
disclosure.
[0019] Alkali metal oxalate from thermal reactor 130 may be dissolved in water
and may be passed to an electrochemical acidification electrolyzer 140.
Electrochemical acidification electrolyzer 140 may produce a dicarboxylic
acid,
such as oxalic acid, and KOH along with oxygen and hydrogen byproducts.
Electrochemical acidification electrolyzer 140 may be a membrane based unit
including of at least three regions, including an anode region, one or more
central ion exchange regions, and a cathode region. It is contemplated that an
energy source (not shown) may generate an electrical potential between the
anode and the cathode of electrochemical acidification electrolyzer 140
sufficient to produce oxalic acid. Alkali metal oxalate may be passed through
the central ion exchange region where alkali metal ions may be replaced with
protons, and the displaced alkali metal ions pass through the adjoining
membrane into the cathode region to form alkali metal hydroxide (e.g. KOH).
The anode reaction may utilize an acid, such as sulfuric acid, producing
oxygen
and hydrogen ions.
[0020] The hydrogen byproduct resulting from electrochemical acidification
electrolyzer 140, as an alternative embodiment, may be used as a fuel to
9
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
produce steam or used in another chemical process that may utilize hydrogen,
such as a hydrogenation process.
[0021] The dicarboxylic acid, such as an oxalic acid product may be purified
to
produce a final purified product, or may be further processed as a chemical
intermediate to produce another product, such as nnonoethylene glycol, using a
reduction process such as an electrochemical reduction or a catalytic
hydrogenation process.
[0022] Aqueous KOH from electrochemical acidification electrolyzer 140 may be
passed to an evaporator 150. Evaporator 150 may evaporate the water from
aqueous KOH product using steam or another heat source, converting it into a
concentrated aqueous solution and/or solid with 5% or less water content as
needed in electrochemical cell 110 and thermal reactor 120.
[0023] Referring to Fig. 1B, a system 105 for production of dicarboxylic acid,
such as oxalic acid, utilizing a hydrogen halide, such as HBr, in anolyte to
co-
produce bromine in accordance with an embodiment of the present disclosure is
shown. System 105 may operate with a less energy intensive electrochemical
process, using HBr as the anolyte in the anode region of electrochemical cell
110 and electrochemical acidification electrolyzer 140, producing bromine and
hydrogen ions at a significantly lower anode potential. The bromine may then
be used, for example, in reactions to produce bronninated chemical products,
such as bronninated organic compounds, for example bronnoethane, which may
then be converted into alcohols such as ethanol, or converted to nnonoethylene
glycol in a series of thernnochennical reactions. It is contemplated that
system
105 shown with thermal reactor 120 and thermal reactor 130 could be
implemented with a single thermal reactor without departing from the scope
and intent of the present disclosure.
[0024] Referring to Fig. 2A, a system 200 for production of dicarboxylic acid,
such as oxalic acid, starting with the electrochemical generation of formate
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
using carbon dioxide in accordance with an embodiment of the present
disclosure is shown. System 200 may provide an alternative system for
production of oxalic acid as produced by systems 100 of Fig. 1A and 105 Fig.
1B
respectively.
[0025] System 200 may include an electrochemical cell 110. Electrochemical
cell 110 may operate to perform an electrochemical reduction of carbon dioxide
with an alkali metal carbonate and/or alkali metal bicarbonate cathode feed,
which may be formed from the reaction of CO2 with alkali metal hydroxide, to
produce alkali metal formate along with oxygen as an anode product when using
an anolyte including sulfuric acid (H2SO4). The alkali metal formate product
solution concentration from the catholyte compartment of electrochemical cell
110 may range from 1 wt% to 30 wt% or more based on the formate ion, and
preferably range from 5 wt% to 20 wt% as formate. The corresponding % weight
as the alkali metal formate, for example alkali metal formate may be based on
the molecular weight of the alkali metal compound.
[0026] Alkali metal formate may be passed to a thermal reactor 120. Thermal
reactor 120 may perform a thermal intermolecular condensation reaction with
the addition of a small amount of catalyst, such as an alkali metal hydroxide
(e.g. KOH) which may increase the conversion yield to produce alkali metal
oxalate.
[0027] Alkali metal oxalate from thermal reactor 120 may be dissolved in water
and may be passed to an electrochemical acidification electrolyzer 140.
Electrochemical acidification electrolyzer 140 may produce dicarboxylic acid,
such as oxalic acid, and KOH along with oxygen and hydrogen byproducts.
Electrochemical acidification electrolyzer 140 may be a membrane based unit
including of at least three regions, including an anode region, one or more
central ion exchange regions, and a cathode region. Alkali metal oxalate may
be passed through the central ion exchange region where alkali metal ions may
be replaced with protons, and the displaced alkali metal ions pass through the
11
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
adjoining membrane into the cathode region to form an alkali metal hydroxide,
such as KOH. The anode reaction may utilize an acid, such as sulfuric acid,
producing oxygen and hydrogen ions.
[0028] The hydrogen byproduct resulting from electrochemical acidification
electrolyzer 140, as an alternative embodiment, may be used as a fuel to
produce steam or used in a side process that may utilize hydrogen, such as in
a
chemical hydrogenation process.
[0029] The dicarboxylic acid, such as oxalic acid product may be purified to
produce a final purified product, or may be further processed as a chemical
intermediate to produce another product, such as nnonoethylene glycol, using
an
electrochemical reduction or thernnochennical process.
[0030] Aqueous KOH from electrochemical acidification electrolyzer 140 may be
passed to an evaporator 150. Evaporator 150 may evaporate the water from
aqueous KOH product using steam or another heat source, converting it into a
concentrated aqueous solution and/or solid with 5% or less water content as
needed in the electrochemical cell 110 or thermal reactor 120.
[0031] Referring to Fig. 2B, a system 205 for production of dicarboxylic acid,
such as oxalic acid, via electrochemical generation of formate using carbon
dioxide and utilizing a halogen halide in the anolyte to co-produce a halogen,
such as bromine, in accordance with an embodiment of the present disclosure is
shown. System 205 may be similar to system 200, where system 205 may use a
hydrogen halide, such as HBr as the anolyte in the anode regions of
electrochemical cell 110 and electrochemical acidification electrolyzer 140.
Electrochemical cell 110 may produce bromine and hydrogen ions at a
significantly lower anode potential. Bromine may then be used, for example, in
reactions to produce bronninated chemical products, such as bronnoethane,
which may then be converted into alcohols such as ethanol, or converted to
nnonoethylene glycol in a series of thernnochennical reactions.
12
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
[0032] Referring to Fig. 3, a system 300 for production of a formate, such as
alkali metal formate, using carbon dioxide in accordance with an embodiment of
the present disclosure is shown. Fig. 3 illustrates the electrochemical
reduction
of carbon dioxide in the production of an alkali metal formate as shown in
electrochemical cell 110 of Fig. 2A and Fig. 28. Electrochemical cell 110 may
include an anolyte input feed 310 and a catholyte input feed 312 to produce a
product 314. Product 314 may be a solution of alkali metal formate with excess
alkali metal bicarbonate (KHCO3). Anolyte region 320 may have a titanium
anode 322 having an anode electrode catalyst coating facing cation exchange
membrane 330. Anode mesh screen 332 may be a folded expanded titanium
screen with an anode electrocatalyst coating and provides spacing and contact
pressure between anode 322 and cation exchange membrane 332. Cation
exchange membrane 330 may selectively control a flow of ions between anolyte
region 320 from catholyte region 340.
[0033] Catholyte region 340 may have a mounted cathode 342, which may be a
metal electrode with an active electrocatalyst layer on the front side facing
membrane 330. High surface area cathode structure 344 may be mounted with
direct contact pressure between the face of cathode 342 and cation membrane
330.
[0034] As shown in Fig. 1A and Fig. 2A, feeding anolyte region 320 may be
stream 310 which may include anolyte, the anolyte including an aqueous
sulfuric
acid electrolyte solution. Stream 310 may enter the anolyte region 320 and
flow
by the face of anode 322 through folded anode screen 332. Anode reactions
may include splitting of water into oxygen (02) and hydrogen ions (H+) or
protons. The gases and liquid mixture from anolyte region 320 may leave as
stream 350, which flows by temperature sensor 352 monitoring a solution
temperature in the stream, and into anolyte gas/liquid disengager 354. In
disengager 354, the gas may be vented as stream 356, and excess anolyte
overflow leaves as stream 358. Stream 360 may be a gas-depleted exit stream
13
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
from the anolyte disengager 354, with a deionized water feed stream 362 and a
sulfuric acid make-up feed stream 364 added to the recirculation stream to
maintain anolyte acid strength and volume. Stream 360 with added streams 362
and 364 may then pass through an optional heat exchanger 370 with a cooling
water supply 372, and then becomes stream 310 feeding into the anolyte region
320.
[0035] Electrochemical cell 110 may include a catholyte region 340 which
includes cathode 342 having an electrocatalyst surface facing membrane 330.
High surface area cathode structure 344 may be mounted between membrane
330 and cathode 342, relying on contact pressure with cathode 342 for
conducting electrical current into the structure. The interface between high
surface area structure 344 and membrane 330 may utilize a thin expanded
plastic mesh insulator screen (not shown) to minimize direct contact of the
high
surface area cathode material with the membrane 330.
[0036] Feed stream 312 may feed into catholyte region 340, flowing through the
high surface area structure 344 and across the face of cathode 342 where
cathode reduction reactions between carbon dioxide, electrolyte, and cathode
material at the applied current and voltage potential produce exit stream 314,
the exit stream including a formate.
[0037] Stream 314 may be the exit solution and gas mixture product from the
cathode reaction which flows by pH monitoring sensor 374 and temperature
sensor 352 and then into catholyte gas/liquid disengager 380 where the gas
exits
as stream 382 and formate/electrolyte overflow exits as stream 384, and the
gas-depleted stream leaves the disengager as stream 386. Stream 386 may then
enter an input of catholyte recirculation pump 390, which then passes through
heat exchanger 392 which uses cooling water 372, then passes by temperature
sensor 352. A fresh catholyte electrolyte feed 394 may be metered into stream
386 which may be used to adjust the catholyte flow stream pH into the
catholyte region 340 and control a product overflow rate and sets the formate
14
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
product concentration, with the pH monitored by pH sensor 374. Carbon
dioxide flow stream 396 may be metered into the flow stream which enters the
catholyte region 340 as stream 312.
[0038] In an alternative embodiment, as shown in Figs. 1B and 2B, the anolyte
including sulfuric acid shown in Figs. 1A and 2A may be replaced with an
anolyte
including hydrogen halide (e.g. HBr), producing a halide (e.g. bromine) and
hydrogen ions at a lower voltage potential than required for the generation of
oxygen at the anode. The halide may then be used, for example, in reactions to
produce halide chemical products, such as bronnoethane in the reaction with an
alkane, such as ethane, which may then be converted into alcohols (e.g.
ethanol) or converted to nnonoethylene glycol in a series of thernnochennical
reactions.
[0039] Referring to Fig. 4, system 400 for electrochemical acidification of
alkali
metal oxalate in accordance with an embodiment of the present disclosure is
shown. Electrochemical acidification electrolyzer 140 may include an anolyte
region 402, a central ion exchange region 408 bounded by cation ion exchange
membranes 406a and 406b on each side, and a catholyte region 410 where an
alkali metal hydroxide (e.g. KOH) may be formed. Hydrogen ions (H+) or protons
may be generated in the anolyte region 402, which then may pass through the
adjoining membrane 406a into the central ion exchange region 408 when a
potential and current may be applied to the cell. An alkali metal oxalate
(e.g.
alkali metal oxalate) product solution 405, such as generated in thermal
reactor
120 of Fig. 1A and 130 of Fig. 2B respectively, may pass through the central
ion
exchange region 408, where protons displace the alkali metal ions in the
solution stream, thus acidifying the solution and forming a dicarboxylic acid,
such as oxalic acid. Stream 456, and the displaced alkali metal ions may pass
through the adjoining cation exchange membrane 406b into the catholyte region
410, where they combine with hydroxide ions (OH-) formed from water
reduction reaction at the cathode to form an alkali metal hydroxide (e.g. KOH)
stream 434.
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
[0040] Electrochemical acidification electrolyzer 140 may include input feeds
430 and 432 and may produce a solution of dicarboxylic acid (e.g. oxalic acid)
456, oxygen 420 from the anolyte region 402, and KOH 442 from the catholyte
region 410. Anode region 402 may include a titanium anode 404 with an anode
electrode catalyst coating facing cation exchange membrane 406a. The central
ion exchange region 408 may contain a plastic mesh spacer to maintain the
space in the central ion exchange region between cation exchange membranes
406a and 406b. Optionally, one alternate material used within the central ion
exchange region may be the use of a cation ion exchange material, so that
there
may be increased electrolyte conductivity in the ion exchange region solution
and which may also help capture and exchange cations, such as potassium, to
improve the efficiency of the proton exchange with the cations. Catholyte
region 410 may include a cathode 412.
[0041] Anolyte region 402 may have feed stream input 430 including sulfuric
acid, which may flow through the anolyte region 402 and exit as stream 414
including a gas and liquid, passing by temperature sensor 416 into anolyte
disengager 418, where the gas exits as stream 420 and liquid overflow as
stream
422. Gas-depleted stream 424 may exit the anolyte disengager 418 and
deionized water stream 426 may be metered into the stream 424 as well as
sulfuric acid make-up stream 428 to maintain acid electrolyte strength in the
anolyte region 402. Stream 424 may pass through optional heat exchanger 426
which may have cooling water supply 428 to cool or maintain the stream 424
temperature, and the stream 424 enters the anolyte region 402 as stream 430.
[0042] Catholyte region 410 may include feed stream 432 which may be the
recirculating alkali metal hydroxide (e.g. KOH) in the catholyte loop, which
enters catholyte region 410 and flows by cathode 412, which may generate
hydrogen gas and hydroxide (OH-) ions, and forms a alkali metal hydroxide from
the combination of alkali metal ions crossing the membrane 406b with the
hydroxide ions formed at the cathode 412 from the reduction of water. Exit
16
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
stream 434 from the cathode region 410 may contain alkali metal hydroxide and
hydrogen gas from the cathode reactions, and passes by temperature sensor 436
and then into catholyte disengager 438, where hydrogen gas 440 may be
separated from the catholyte solution, which exits catholyte disengager 438 as
recycle stream 444 and alkali metal hydroxide product overflow stream 442.
Recycle stream 444 may pass through optional recirculation pump 446 and then
through optional heat exchanger 448, which uses cooling water supply 450. The
stream then passes by temperature sensor 452, and then may have a deionized
water addition stream 454 added to the stream to control the alkali metal
hydroxide concentration in the catholyte recirculation loop, and then re-
enters
the catholyte region 410 as stream 432.
[0043] In an alternative embodiment, the anolyte including sulfuric acid may
be
replaced with an anolyte including a hydrogen halide, such as HBr, producing
bromine and hydrogen ions at a much lower voltage potential than required for
the generation of oxygen at the anode.
[0044] Fig. 5 shows schematic drawing of system 500, an alternative
embodiment in operating a system utilizing a sodium-based compound that may
generate, for example, sodium formate from the electrochemical reduction of
carbon dioxide followed by the conversion of the sodium formate to sodium
oxalate, which may then be converted to oxalic acid. The system produces
oxalic acid in addition to two additional co-products, which may be sodium
bicarbonate and sodium hydroxide.
[0045] Electrochemical cell 502 may be similarly configured to the
electrochemical cell as shown and described in Fig. 3 except for modifications
to the feed solutions used in the anolyte and catholyte. Electrochemical cell
502 may include catholyte compartment 506 and anolyte compartment 504, and
ion permeable separator 503, preferably being a cation ion exchange type
membrane. A feed stream 522 of saturated NaCl brine may be introduced into
catholyte compartment 504 of electrochemical cell 502, where the chloride ion
17
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
of the NaCl salt solution may be oxidized to chlorine gas at the anode in
anode
compartment 504. As the chloride ion of the NaCl salt may be oxidized at the
anode, sodium ions migrate in the potential field, and pass through separator
503 into cathode compartment 506.
[0046] The anolyte product stream 508 from catholyte compartment 504
includes a mixture of chlorine gas and NaCl depleted brine solution. The
chlorine gas may then be separated or disengaged as stream 510 from stream
508 as a co-product, and the separated depleted brine solution stream 512 may
then be processed in a series of steps typically used in chlor alkali
processes
including dechlorination of the depleted brine, resaturation of the brine
solution
with NaCl using a bed of solid NaCl salt, followed by a brine purification
step to
remove impurities, such as metals and hardness (such as Ca', Mg', and Ba+),
from the brine solution to impurity levels typically used to achieve long life
operation of separator 503, to produce a purified saturated NaCl brine
solution
stream 522 which is electrolyzed in the anolyte compartment 504 of formate
cell 502.
[0047] Chlorine gas 510 may then be processed in various ways, such as removal
of water from the gas by condensation, and then the chlorine gas may then be
used for producing various useful co-products from the system, for example,
the
generation of sodium hypochlorite by a reaction with NaOH, the generation of
HCl through a reaction with hydrogen, as well as reactions with organics, such
as to produce EDC (ethylene dichloride) by reaction with an external supply of
ethylene. Many other reaction co-products made with the chlorine gas 510 may
be envisioned.
[0048] Brine dechlorination unit 514 may be used to remove residual chlorine
from depleted brine solution 512 using a selected reducing agent, which may
include sodium sulfite, sodium hydrosulfite, activated carbon, and hydrogen
peroxide among others. The dechlorinated brine then may be passed to brine
saturator unit 516, where the depleted brine NaCl concentration may be
18
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
increased from a typical 150 - 240 gnn/L as NaCl to a concentration of 300 -
320
gnn/L as NaCl using a brine saturator, which may include a bed of solid salt
crystals in an apparatus typically called a briner. The saturated brine may
then
be passed through brine purification system 518, which may include chemical
precipitation steps for the removal of most of the hardness in the solution,
typically by the addition of NaOH and sodium carbonate under alkaline
conditions, followed by filtration to remove the precipitated hardness
containing solids, then followed by an ion exchange purification step
utilizing
chelating ion exchange resin beds to reduce the hardness levels in the brine
to
typically 20 - 50 ppb or less. The sulfate component in the brine may be
reduced by the chemical precipitation, or by the use of commercial system that
utilizes nanofiltration to preferentially remove sulfate from brine, for
example
the SRS system - sold by Aker Chennetics. The purification chemicals also may
include HCl and NaOH used for regenerating the chelating ion exchange
columns. Stream 520 may be an effluent stream containing the precipitated
carbonates, sulfates, and metals effluent from the purification of the
saturated
brine solution, which may be processed and recycled back to the process with a
minimum amount of material requiring disposal. Purified brine solution 522 may
then pass into anolyte compartment 504 of electrochemical cell 502. The
recirculation of the anolyte loop is not shown, but the brine flow rate may be
metered so as to maintain the desired brine concentration in the
electrochemical cell anolyte loop and overflow stream 508, with the brine
concentration typically in the range of 150 - 240 gnn/L as NaCl. In an
embodiment, the anolyte brine concentration may be operated lower NaCl
concentrations, to as low as about 100 - 140 gm/L, which may result in a
decrease in chlorine efficiency and the generation of more byproduct oxygen in
the chlorine gas stream, but which may be useful in reducing the brine flow
rate
through the brine purification system with a reduction in brine processing
costs.
[0049] Solution feed stream 548, which may be an aqueous mixture of sodium
formate, sodium bicarbonate, and dissolved carbon dioxide which may include a
gaseous carbon dioxide component which may be in the form of gaseous micro-
19
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
bubbles, may be passed into catholyte compartment of electrochemical cell
502. In catholyte compartment 506, which preferably incorporates a high
surface area cathode structure, carbon dioxide may be electrochemically
reduced to formate, and the formate may combine with the sodium ions (Na)
passing through the adjacent separator 503 to form sodium formate. In
addition, any cathode inefficiency side reactions forming hydrogen (H2) at the
cathode may produce hydroxide ions (OH), and these hydroxide ions may react
with carbon dioxide to form sodium carbonate in the catholyte solution. The
sodium carbonate may then further react with excess carbon dioxide to form
sodium bicarbonate. In addition, it is believed that the other sodium ions may
combine with carbonic acid and the other potential carbon dioxide equilibrium
species at the operating catholyte pH to further form additional sodium
carbonate and sodium bicarbonate.
[0050] The reduction reaction products may exit as stream 524, where they may
be separated or disengaged into gas stream 526 and solution stream 530. Gas
stream 526 may be passed into separator 528, which may separate carbon
dioxide from any byproduct hydrogen so that they may be reused or recycled in
the other system 500 unit operations. Gas separator 528 may be any suitable
membrane-based or molecular sieve pressure swing gas separation unit that may
be capable of the separation of carbon dioxide and hydrogen. The separated
gases may then be further purified and compressed as needed for recycle or
reuse to the process.
[0051] Solution stream 530 including mainly sodium formate and sodium
bicarbonate may then be split into recycle stream 532, which may be recycled
back to electrochemical cell 502 catholyte compartment, and product stream
531 which may go to evaporator-crystallizer 550. Recycle stream 532 may have
several input streams, including the introduction-of-carbon-dioxide stream
534,
optionally a sodium bicarbonate stream 536 from reactor-dissolver unit 560, a
side stream 538 leaving stream 532 which may go into an optional
electrochemical acidification cell 540 and may have an acidified product
stream
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
546 back into stream 532, and may have the addition of water to the stream as
needed to prevent precipitation in stream 532 and catholyte compartment 506,
and having all of the inputs/outputs into stream 532 ending up as solution
stream 548 which may be sent into catholyte compartment 506.
[0052] Electrochemical acidification cell 540 may be used to acidify a small
portion taken from catholyte loop stream 532, and which may then reenter
anolyte recycle stream 532 as stream 546.
[0053] Electrochemical acidification cell 540 may be a similar design as the
acidification cell as shown in Fig. 4. The cell anolyte solution may utilize
sulfuric acid such that the anode reaction produces oxygen and produces
hydrogen ions, which may be used to acidify formate stream 538 as it passes
through the ion exchange compartment in the cell. The cathode reaction in this
cell may be the reduction of water, which produces hydrogen gas and hydroxide
ions (OH). The sodium ions that may be displaced by the hydrogen ions passing
into the ion exchange compartment may pass into the catholyte compartment to
combine with the hydroxide ions to produce a sodium hydroxide co-product. The
hydrogen gas may also be captured for use in the process. Deionized water may
be used in acidification cell 540 as needed to replace electrolyzed water and
in
controlling the concentration of the NaOH in the catholyte compartment.
[0054] Catholyte product stream 531, which may contain high concentrations of
alkali metal formate and alkali metal bicarbonate, may be passed to
evaporator-crystallizer unit 550, which may evaporate sufficient water from
the
solution and continuously precipitate a alkali metal bicarbonate crystal
product
as stream 556, a liquid concentrated alkali metal formate stream 554, and a
water product stream 552 which may be condensed and used elsewhere as
needed in the process, such as in oxalate solution dissolver 572. Evaporator-
crystallizer 550 may utilize steam for providing the energy requirements for
evaporating the water from the stream 531 input stream to the unit.
Evaporator-crystallizer 550 may be a multiple evaporator effect unit,
including
21
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
multiple units to efficiently utilize the energy of the input steam, or any
other
suitable types of units may be utilized. In addition, evaporator-crystallizer
550
may use steam as well as mechanical operation for producing a vacuum to
further reduce the energy requirements for the evaporation of the water from
the solution. Any suitable evaporator-crystallizer unit or system may include
suitable metallurgy for the operating conditions of the system. Alkali metal
formate may have solubility in water that may be about 8 to 10 times more than
that of alkali metal bicarbonate, so the solubility difference may allow the
easy
separation of alkali metal formate from alkali metal bicarbonate using
solution
temperature differences to enhance the separation. Other methods for the
separation of alkali metal formate from alkali metal bicarbonate may be
employed including fractional crystallization, cooling crystallization,
falling film
crystallization, and the like. A continuous process for the separation may be
employed, although batch processing may also be used.
[0055] In an additional embodiment, if the amount or ratio of alkali metal
bicarbonate is small in relation to the alkali metal formate, on the order of
1:10
to 1:20 or lower, the alkali metal bicarbonate may be allowed to be thermally
decomposed to CO2 in the Na-Formate liquid dryer 564 and not recovered for
recycle. This method may reduce the added process costs in conducting the
separating and recycling the alkali metal bicarbonate from the alkali metal
formate.
[0056] The alkali metal bicarbonate crystal stream 556 from unit 550 may be in
the form of an aqueous slurry, which may then be separated, washed, and dried
by any suitable mechanism to produce a dried alkali metal bicarbonate product
558. Equipment such as centrifuges and vacuum belt filters may be used for the
separation of the alkali metal bicarbonate crystals from the 556 stream
slurry,
and the mother liquor from any water rinses may be recycled back to unit 550.
The alkali metal bicarbonate product 558 may also be recrystallized or further
purified by any suitable mechanism to obtain a final product with purity
suitable
for specialty uses, such as food grade quality product. A portion of the
stream
22
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
556 slurry or stream 558 alkali metal bicarbonate product as stream 560 may be
utilized in reactor-dissolver 561, which may be used to convert alkali metal
carbonate to alkali metal bicarbonate using an additional carbon dioxide gas
stream 563. Reactor-dissolver 561 may also have a NaOH input stream 562,
which may then be converted to alkali metal bicarbonate. The NaOH may be
supplied from one or both of the electrochemical acidification units 540 and
576
if required.
[0057] Alkali metal formate stream 554 may be a concentrated alkali metal
formate solution that contains 50 wt% or less water, and preferably 40 wt% or
less water, and more preferably 30 wt% or less water. The formate solution
stream 554 may be viscous and may contain from 0.1 wt% to 30 wt% as alkali
metal bicarbonate depending on the water solubility of alkali metal
bicarbonate
in the alkali metal formate solution. The solution concentrations of the
alkali
metal formate and residual alkali metal carbonate may be varied as needed to
achieve the desired final residual alkali metal bicarbonate concentration in
the
alkali metal formate solution. Alkali metal formate stream 554 may then passed
to alkali metal formate liquid dryer where the residual water may be removed
by any suitable means such as by vacuum evaporation and the like. The alkali
metal formate may be an alkali metal formate melt, including a small
percentage of water, in the range of 0.01 wt% to 5 wt% as water, and may have
between 0.1 wt% to 20 wt% alkali metal bicarbonate. The alkali metal formate
melt stream 566 may then be passed into alkali metal formate thermal reactor
568 for high temperature thermal conversion of the alkali metal formate to
alkali metal oxalate (calcination). A suitable catalyst 567, such as NaOH,
sodium hydride, sodium borohydride, sodium ethoxide, sodium nnethoxide, KOH,
potassium hydride (KH), potassium ethoxide (KOEt), potassium nnethoxide
(KOMe), potassium tert-butoxide (KOtBu) and the like may be added into the
sodium formate before it enters thermal reactor 568. The introduction of
catalyst 567 may help to reduce the calcination temperature and improve the
conversion yield of alkali metal formate to alkali metal oxalate to a range of
50% to 99% or more, and preferably 70% to 99% or more. The reaction may also
23
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
provide suitable yields without the need for the addition of catalyst 567.
Hydrogen may be a major byproduct reaction from thermal reactor 568 and may
be recovered for use in the process. Thermal reactor 568 may be operated in
different configurations, such as under a partial vacuum, under an inert
atmosphere such as nitrogen, or with the use of any suitable gas that may
improve the efficiency of the chemical conversion of the formate to oxalate.
The addition of other chemicals to thermal reactor 568 may also be useful, so
as
to obtain a clean flowing purified product. Thermal reactor 568 may be any
suitable type equipment that can heat the alkali metal formate to suitable
temperatures and control the thermal or calcination atmosphere. Thermal
reactor 568 may include tunnel furnaces, rotary kilns, high temperature spray
dryers, high temperature rotating drunn/flaker units, fluid bed reactors, and
other commercial calcining equipment and designs that may be commercially
available.
[0058] The alkali metal oxalate product stream 570 leaving thermal reactor 568
may be cooled, and passed to oxalate solution dissolver 572, where alkali
metal
oxalate solids are dissolved in water, and may be filtered by various
available
methods to remove any insoluble materials and obtain a clear, filtered product
solution, free of suspended solids. The alkali metal oxalate product may
contain alkali metal carbonate and/or alkali metal bicarbonate as byproduct(s)
of the calcination. The solution may be concentrated sufficiently so that the
alkali metal oxalate - alkali metal bicarbonate solution may not require a
larger
amount of energy or steam for water evaporation in evaporator-crystallizer
576.
[0059] Alkali metal oxalate solution stream 574 may then be passed to
electrochemical acidification cell 576, where the alkali metal oxalate
solution
passes through the ion exchange compartment of the cell and may be converted
to oxalic acid stream 580 and carbon dioxide stream 579 which may produced
from the acidification of any alkali metal carbonate present in alkali metal
oxalate stream 574. Electrochemical acidification cell 576 may utilize the
same
24
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
chemistry and configuration as electrochemical acidification cell 540,
producing
oxygen and hydrogen a co-products, as well as NaOH as stream 578.
[0060] Referring to Fig. 6, in another embodiment, a system 600 for production
of dicarboxylic acid, such as oxalic acid, starting with the electrochemical
generation of formate using carbon dioxide in accordance with an embodiment
of the present disclosure is shown. System 600 may provide an alternative
system for production of oxalic acid as produced by systems 100, 105 of Fig.
1A
and Fig. 1B in addition to the production of alternative co-products.
[0061] System 600 may include an electrochemical cell 610. Electrochemical
cell 610 may operate to perform an electrochemical reduction of carbon dioxide
with a alkali metal carbonate cathode feed, which may be formed from the
reaction of CO2 with NaOH, producing alkali metal formate along with chlorine
gas as an anode product when utilizing hydrochloric acid (HCl) as an anolyte,
which may produced in electrochemical unit 670 which may use a purified NaCl
solution input feed stock.
[0062] Alkali metal formate may be passed to a thermal reactor 620. Alkali
metal formate may be separated from bicarbonate present in the catholyte by
various means as described in Fig. 5 to provide a suitable feed to thermal
reactor 620. Thermal reactor 620 may perform a thermal intermolecular
condensation reaction, or C-C (carbon to carbon bond) coupling reaction, with
an alkali metal hydroxide (e.g. KOH, NaOH) or use other catalysts to produce
alkali metal oxalate.
[0063] Alkali metal oxalate from thermal reactor 620 may then be dissolved in
water and may then be passed to an electrochemical acidification electrolyzer
630. Electrochemical acidification electrolyzer 630 may produce a dicarboxylic
acid, such as oxalic acid, and NaOH along with oxygen and hydrogen byproducts.
Electrochemical acidification electrolyzer 630 may be a membrane based unit
including of at least three regions, an anode region, one or more central ion
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
exchange regions, and a cathode region. Alkali metal oxalate may be passed
through the central ion exchange region, where alkali metal ions may be
replaced with protons, and displaced alkali metal ions pass through the
adjoining membrane into the cathode region to form NaOH. The anode reaction
may produce chlorine gas when utilizing an HCl feed from electrochemical unit
670. Alternative, the anode reaction may utilize a different acid, such as
sulfuric acid, producing oxygen and hydrogen ions. Alternatively,
electrochemical acidification electrolyzer 630 may be an electrochemical
electrodialysis unit, utilizing bipolar membranes, producing oxalic acid as
well
as smaller amounts of hydrogen and NaOH.
[0064] The hydrogen byproduct resulting from electrochemical acidification
electrolyzer 630, as an alternative embodiment, may be used as a fuel to
produce steam or used in a side process that may utilize hydrogen, such as in
a
chemical hydrogenation process. The chemical hydrogenation process may be,
for example, the hydrogenation of an oxalic acid solution or the hydrogenation
of an ester of oxalic acid, such as dinnethyl oxalate (DMO) and diethyl
oxalate
(DEO), that may form high purity nnonoethylene glycol (MEG).
[0065] Aqueous NaOH from electrochemical acidification electrolyzer 630 may
be passed to an evaporator 640. Evaporator 640 may evaporate the water from
aqueous NaOH product using steam or another heat source, converting it into a
concentrated aqueous solution and/or a solid with 5% or less water content.
The
NaOH may be reacted in reactor 680 with CO2 to form an alkali metal
bicarbonate solution with carbon dioxide, which may be passed to the catholyte
compartment in electrochemical cell 610. NaOH may also be converted to a
solid for use as a catalyst in thermal reactor 620.
[0066] Electrochemical unit 670 may be an electrochemical acidification
electrolyzer, a type such as electrochemical acidification electrolyzer 630,
where a purified NaCl brine solution is passed into the ion exchange
compartment and may be acidified, producing an HCl product stream as well as
26
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
co-producing NaOH and hydrogen in the cathode compartment. The anolyte
may utilize sulfuric acid and generate oxygen from the oxidation of water. The
purified brine may be produced by brine purification and recycle unit 660,
utilizing an NaCl solid feed and using various purification chemicals as
needed to
produce the purified brine, suitable for use in electrochemical unit 670.
Electrochemical unit 670 may include other types of electrochemical units,
such
as electrodialysis units which may utilize bipolar membranes, as well as any
other suitable type of electrolyzer that may produce HCl.
[0067] System 600 in another embodiment, may also produce alkali metal
hypochlorite (for example Na0Cl), as a co-product from the system, utilizing
chlorine and NaOH produced from electrochemical unit 670 and electrochemical
acidification electrolyzer 630. Alternatively, chlorine may be reacted with
organics to produce various chlorinated chemical products, such as ethylene
dichloride (EDC). MOH may be a separate product of the process, or may be
converted to alkali metal carbonate or alkali metal bicarbonate, thus
converting
additional carbon dioxide to useful chemicals.
pow In another embodiment, the alkali metal formate, produced in
electrolyzer 610 may be passed directly to electrochemical acidification
electrolyzer 630, bypassing thermal reactor 620, directly producing a formic
acid product. The formic acid be a final product or may be converted to other
suitable chemicals, such as methyl formate, or reacted with various salts to
produce alkali metal formates, such as calcium formate. Methyl formate may
also be converted to produce amides such as fornnannide or
dinnethylfornnannide
via reactions with amines.
[0069] In another embodiment, electrochemical unit 670 may include a two
compartment cell having an anode compartment and a cathode compartment
separated by a separator or membrane. In this embodiment, NaCl may be fed
to the anolyte compartment producing chlorine, and sodium hydroxide and
hydrogen would be produced in the cathode compartment.
27
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
[0070] Referring to Fig. 7, in another embodiment, a system 700 for production
of a carboxylic acid, such as formic acid, starting with the electrochemical
generation of formate using carbon dioxide in accordance with an embodiment
of the present disclosure is shown. The system utilizes a three compartment
electrochemical cell including an anode compartment, a central ion exchange
compartment, and a catholyte compartment.
[0071] Electrochemical cell 701 may include catholyte region or cathode
compartment 704 and anolyte region or anode compartment 712, and two ion
permeable separators 706 and 708, preferably being cation ion exchange type
membranes, forming central ion exchange compartment 714. An anolyte feed
stream 726 including a sulfuric acid electrolyte may be introduced into
anolyte
compartment 712 of electrochemical cell 701, where water may be oxidized to
oxygen and H+ ions at anode 710 in anolyte compartment 712. Anolyte stream
716 exits anolyte compartment 712 and enters anolyte disengager 718, where
oxygen gas 720 may exit as an electrochemical cell anolyte co-product. Gas
separated solution stream 722 may exit disengager 718, and water stream 724
may be added to solution stream 722 to maintain water levels in the anolyte
system loop. Water loss in the anolyte system loop may be due to water
consumed from the anode oxidation reaction and bound water associated with
the H+ ions that may transport into ion exchange compartment 714 through
cation ion exchange membrane 708 by an electrochemical transport process
called electro-osmotic drag. Anode 710 may include any suitable stable
electrode material suitable for water oxidation in a sulfuric acid electrolyte
that
may be stable with a long operating life. The anode may include a metal or non-
metal having an electrocatalyst coating for the efficient oxidation of water.
The anode may also optionally include a gas diffusion electrode (GDE), which
may operate in another electrochemical anode reaction where it is not
producing oxygen, but may be forming water and H+ ions, such as from the
introduction of excess hydrogen to an anode GDE, and which may contain
suitable catalysts for conducting the hydrogen oxidation reaction at the
anode.
28
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
The GDE anode reaction with water operates at a much lower anode potential,
which may be on the order of about 0.100 volts, much lower than the half cell
potential for water oxidation, which is 1.23 volts, and resulting in a much
lower
overall total cell operating potential, corresponding to much lower operating
costs for the electrochemical cell 701. Anolyte circulation loop 726 may
include
a pump (not shown) and a heat exchanger (not shown) for cooling stream 726
before entering anolyte compartment 712 to control the temperature of the
electrolyte in anolyte compartment 712.
[0072] Electrochemical cell 701 may have catholyte region or compartment 704
having a mounted cathode 702, which may be a metal or non-metal electrode
with an active carbon dioxide electrocatalyst layer on the front side facing
membrane 706. Cathode 702 may include a high surface area cathode structure
as shown and described in Fig. 3 such that it may be efficient in the
electrochemical reduction of carbon dioxide to formate. Electrolyte flow
stream
748 containing a pH controlled electrolyte at a suitable flowrate including,
for
example, potassium bicarbonate and dissolved CO2 and optionally CO2 micro-
bubbles may enter catholyte compartment 704, where the electrochemical
reduction reaction at suitable potentials may efficiently produce formate at
cathode 702. H+ ions entering into catholyte compartment 704 through adjoining
cation ion exchange membrane 706 may acidify the bicarbonate electrolyte,
modifying the pH of the catholyte stream in catholyte compartment 704, in
addition to producing CO2 and water as given in reaction 17. Any other
electrode
competing reactions at the cathode, such as the formation of hydrogen from
water reduction reaction 4, may be formed and may be present in the catholyte
electrolyte. The co-transport of K+ ions, as shown in Fig. 7, leaving ion
exchange
compartment 714 through cation ion exchange membrane 706, may supply
potassium cations in the catholyte reactions. The ratio of H+ and K+ ions may
determine the pH of the electrolyte solution flowing through catholyte
compartment 704. Controlling the W/K+ ratio may be implemented by
controlling a rate of flow of the sodium formate solution 714 into ion
exchange
compartment 714.
29
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
[0073] Catholyte compartment stream 730 may then pass by pH monitoring
sensor 731 (and temperature sensor - not shown), which may be used to monitor
and control the catholyte stream pH value, and then enter catholyte disengager
732, where the various gases may be separated from the solution stream as
stream 734, including mainly excess CO2 and hydrogen, in addition to any other
cathode reduction side reaction gas products, such as ethylene, CO, methane,
and the like. These gases may be separated, collected, and recycled to the
process as needed.
[0074] Catholyte disengager stream 738 may then be recycled back to catholyte
compartment 704. Introduced into recycle stream 738 is CO2 as stream 740 in
addition to a metered KHCO3 solution stream 742, with the resulting stream
then passing by pH monitoring sensor 743, which measures and controls the pH
of the stream mixture 744. Stream 744 then enters the input of circulation
pump 746 and exits as solution flow stream 748 entering catholyte compartment
704. The injected CO2 stream flow is such that it provides sufficient and
preferably excess CO2 for the cathode 702 reduction reaction in catholyte
compartment 704. The CO2 may be injected so as to form CO2 micro-bubbles in
the electrolyte solution stream entering catholyte compartment 704 as well as
dissolved CO2 in the electrolyte. KHCO3 metered stream 742 introduced into the
catholyte stream 738 is supplied in a sufficient rate or flow into flow stream
738
to help maintain solution stream 744 in a desired pH range before entering
catholyte compartment 704 as stream 748. The pH range may range from about
2 to 12, and more preferably in a range of about 3 to 11, and even more
preferably in a range of about 4 to 10. The operating pH range may depend on
the electrochemistry of the chosen cathode electrocatalyst materials and the
electrolyte composition used in the catholyte stream. In addition, a heat
exchanger (not shown) may be used after circulation pump 746 to cool the
catholyte solution to a operating range of about -5 C to 80 C, more preferably
a
range of about 0 C to 70 C, and more preferably in a range of about 5 C to
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
60 C. The catholyte temperature and electrolyte composition may affect the
solubility of the CO2 in the catholyte electrolyte solution.
[0075] Catholyte disengager 732 may have a potassium formate product stream
736 including potassium formate (K-Formate) with a potassium bicarbonate
(KHCO3) residual that may be passed to a K-Formate-Bicarbonate Separation
Unit 750, which may be used to separate KHCO3 from the potassium formate.
The separation unit may be an evaporator-crystallizer, falling film
evaporator,
or any suitable commercial equipment using steam or other energy heating
source as well as vacuum that may evaporate the water from the solution to
efficiently conduct the separation using the relative solubilities of the
chemical
components in aqueous solution.
[0076] Separation unit 750 may produce an exit KHCO3 solution product stream
742 that may be metered into catholyte recycle solution steam 738 for pH
control. The solution stream concentration may range from about 5 wt% to 60
wt%, or more preferably in a range of 10 wt% to 55 wt% in order to control
water
input into catholyte electrolyte recycle stream 748. Separation Unit 750 may
have a water vapor output stream 752 from the evaporation and optionally
excess KHCO3 stream 754, if needed, for water or potassium compound balance
in the system that can be added to the system as needed.
[0077] Separation unit 750 may also have K-Formate product stream 756 which
includes a potassium formate solution that may contain levels of KHCO3 in the
range of about 0.1 wt% to 10 wt%, and preferably in a range of about 0.1 wt%
to
wt%, and more preferably in a range of about 0.1 wt% to 1 wt%. The K-
Formate product stream 756 may have a concentration of about 5 wt% to 80 wt%
as potassium formate, more preferably in a range of 10 wt% to 60 wt%, and most
preferably in the range of 10 wt% to 50 wt%.
[0078] K-Formate product stream 756 may be metered into ion exchange
compartment 714 in electrochemical cell 701, where it is acidified to produce
31
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
formic acid solution product stream 728. The flowrate of solution stream 756
and the applied operating current of electrochemical cell 701, which may be
proportional to the H+ ions passing into ion exchange compartment 714 through
cation ion exchange membrane 708, determine the amount of potassium
removal from the potassium formate in the solution in producing formic acid
product 728. Formic acid product 728 may be then further processed and
purified as needed to a final formic acid product, converted to other formate
products as salts, or used as an intermediate in chemical processes.
[0079] In another embodiment, as shown in Fig. 11, formic acid product stream
728 may be used as a hydrogen storage source in an energy storage system for
use in supplying power to an electrical grid during off peak periods.
[0ow] Referring to Fig. 8, a system 800 for production of a carboxylic acid,
such
as formic acid, starting with the electrochemical generation of formate using
carbon dioxide in accordance with an embodiment of the present disclosure is
shown. System 800 may be similar as system 700 but where the anolyte
compartment may produce a halogen, such as chlorine, when utilizing a
hydrogen halide feed, such as HCl, as an anolyte feed. In system 800, anolyte
feed stream 826 contains HCl, which may be fed into anolyte compartment 812,
which includes anode 810 which may be suitable for oxidizing the HCl into
chlorine. Anolyte compartment exit stream 816 may then enter anolyte
disengager 818, where chlorine is separated from the anolyte solution stream.
Overflow stream 819 may be used, if needed, if the anolyte water volume is not
balanced and may later be recycled to the system. Recycle flow stream 822
from anolyte disengager 818 may have an input water stream 824 added to
supply water to the anolyte stream as needed and a metered HCl feed stream
825 to supply the chloride ions to be oxidized to chlorine in the anolyte
compartment at anode 810. The combined feed stream 826 may then enter
anolyte compartment 812.
32
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
[0081] Anolyte feed stream 826 may also contain a portion of the electrolyte
as
sulfuric acid, added as stream 827, which may act as a supporting electrolyte.
The amount of sulfuric acid may range from about 0.5 wt% to 20 wt% in anolyte
solution stream 826. In addition to being a supporting electrolyte, the
sulfuric
acid may allow for variability in the production volume or rate of chlorine
from
anolyte compartment 812, with the production of both chlorine and oxygen
proportional to the HCl metered into anolyte compartment 812. Thus, the mass
or molar ratio of chlorine to formic acid from system 800 may be varied as
needed to meet the requirements of the process employed. The molar ratio of
formic acid product to chlorine co-product may be varied from about 100:1 to
1:1, or more preferably in a range of 90:1 to 1:0.9.
[0082] HCl feed 825 may be aqueous, and may range from about 5 wt% to 36
wt% as HCl, and more preferably in a range of about 10 wt% to 30 wt% as HCl,
and most preferably in a range of about 10 wt% to 20 wt% as HCl. The operating
temperature of the anolyte may be in the range of 5 C to 80 C, and more
preferably in the 10 C to 60 C range. The optimum operating temperature may
be chosen according to the material construction of anode 810, where the use
of some metals as substrates, such as titanium grades containing Pd, such as
ASTM Grades 7, 11, and 17, have a good resistance to HCl, but may have an
upper operating temperature limit.
[0083] In another embodiment, if bromine may be required as a co-product from
electrochemical formate system 800, then HBr would be metered into anolyte
stream 826 entering anolyte compartment 812. The supporting sulfuric acid
electrolyte may be used to ensure that bromine is generated efficiently with
low concentrations of tribronnide in the anolyte solution. The molar ratio of
formic acid product to bromine co-product may be varied from about 100:1 to
1:1, or more preferably in a range of 90:1 to 1:0.9.
[0084] Referring to Fig. 9, in another embodiment, a system 900 for the
production of an oxalic acid solution product and an oxygen co-product using
33
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
carbon dioxide, a thermal reactor to convert formate to oxalate, and a three
compartment electrochemical cell 901 configuration in accordance with an
embodiment of the present disclosure is shown. System 900 may utilize a three
compartment electrochemical cell 901 including an anode compartment, a
central ion exchange compartment, and a catholyte compartment.
[0085] Electrochemical cell 901 may include catholyte region or cathode
compartment 904 and anolyte region or anode compartment 912, and two ion
permeable separators 906 and 908, preferably being cation ion exchange type
membranes, forming central ion exchange compartment 914. An anolyte feed
stream 926 including a sulfuric acid electrolyte may be introduced into
anolyte
compartment 912 of electrochemical cell 901, where water may be oxidized to
oxygen and H+ ions at anode 910 in anode compartment 912. Anolyte product
stream 916 exits anolyte compartment 912 and enters anolyte disengager 918,
where oxygen gas 920 may exit as an electrochemical cell anolyte co-product.
Gas separated solution stream 922 may exit disengager 918, and water stream
924 may be added to solution stream 922 to maintain water levels in the
anolyte
system loop. Water loss in the anolyte system loop may be due to water
consumed from the anode oxidation reaction and the loss of bound water
associated with the H+ ions that may transport into ion exchange compartment
914 through cation ion exchange membrane 908 by an electrochemical transport
process called electro-osmotic drag. Anode 910 may include any suitable stable
electrode material suitable for water oxidation in a sulfuric acid electrolyte
that
may be stable with a long operating life. The anode may include a metal or non-
metal having an electrocatalyst coating for the efficient oxidation of water.
The anode may also optionally include a gas diffusion electrode (GDE), which
may operate in another electrochemical anode reaction where it is not
producing oxygen, but may be forming water and H+ ions, such as from the
introduction of excess hydrogen to an anode GDE, and which may contain
suitable catalysts for conducting the hydrogen oxidation reaction at the
anode.
The GDE anode reaction with water operates at a much lower anode potential,
which may be on the order of about 0.100 volts, much lower than the half cell
34
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
potential for water oxidation, which is 1.23 volts, and resulting in a much
lower
overall total cell operating potential, corresponding to much lower operating
costs for the electrochemical cell 901. Anolyte circulation loop 926 may
include
a pump (not shown) and a heat exchanger (not shown) for cooling stream 926
before entering anolyte compartment 912 to control the temperature of the
electrolyte in anolyte compartment 912.
[0086] Electrochemical cell 901 may have catholyte region or compartment 904
having a mounted cathode 902, which may be a metal or non-metal electrode
with an active carbon dioxide electrocatalyst layer on the front side facing
membrane 706. Cathode 702 may include a high surface area cathode structure
as shown and described in Fig. 3 such that it may be efficient in the
electrochemical reduction of carbon dioxide to formate. Electrolyte flow
stream
948 containing a pH controlled electrolyte at a suitable flowrate including,
for
example, potassium bicarbonate and dissolved CO2 and optionally CO2 micro-
bubbles may enter catholyte compartment 904, where the electrochemical
reduction reaction at suitable potentials may efficiently produce formate at
cathode 902. H+ ions entering into catholyte compartment 904 through adjoining
cation ion exchange membrane 906 may acidify the bicarbonate electrolyte,
modifying the pH of the catholyte stream in catholyte compartment 904, in
addition to producing CO2 and water as given in reaction 17. Any other
electrode
competing reactions at the cathode, such as the formation of hydrogen from
water reduction reaction 4, may be formed and may be present in the catholyte
electrolyte. The co-transport of K+ ions, as shown in Fig. 7, leaving ion
exchange compartment 914 through cation ion exchange membrane 906, may
supply potassium cations in the catholyte reactions. The ratio of H+ and K+
ions
may determine the pH of the electrolyte solution flowing through catholyte
compartment 904. Controlling the W/K+ ratio may be done by controlling the
rate of flow of the sodium oxalate solution 974 into ion exchange compartment
914.
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
[0087] Catholyte compartment stream 930 may then pass by pH monitoring
sensor 931 (and temperature sensor - not shown), which may be used to monitor
and control the catholyte stream pH value, and then enter catholyte disengager
932, where the various gases may be separated from the solution stream as
stream 934, including mainly excess CO2 and hydrogen, in addition to any other
cathode reduction side reaction gas products, such as ethylene, CO, methane,
and the like. These gases may be separated, collected, and recycled to the
process as needed.
[0088] Catholyte disengager stream 938 may then be recycled back to catholyte
compartment 904. Introduced into recycle stream 938 is CO2 as stream 940 in
addition to a metered KHCO3 solution stream 942, with the resulting stream
then passing by pH monitoring sensor 943, which measures and controls the pH
of the stream mixture 944. Stream 944 then enters the input of circulation
pump 946 and exits as solution flow stream 948 entering catholyte compartment
904. The injected CO2 stream flow is such that it provides sufficient and
preferably excess CO2 for the cathode 902 reduction reaction in catholyte
compartment 904. The CO2 may be injected so as to form CO2 micro-bubbles in
the electrolyte solution stream entering catholyte compartment 904 as well as
dissolved CO2 in the electrolyte. KHCO3 metered stream 942 introduced into the
catholyte stream 938 is supplied in a sufficient rate or flow into flow stream
938
to help maintain solution stream 944 in a desired pH range before entering
catholyte compartment 904 as stream 948. The pH range may range from 2 to
12, more preferably in a range of about 3 to 11, and even more preferably in a
range of about 4 to 10.The operating pH range may depend on the
electrochemistry of the chosen cathode electrocatalyst materials and the
electrolyte composition used in the catholyte stream. In addition, a heat
exchanger (not shown) may be used after circulation pump 946 to cool the
catholyte solution to a operating range of about -5 C to 80 C, more preferably
a
range of about 0 C to 70 C, and more preferably in a range of about 5 C to
60 C. Catholyte 904 temperature and composition may affect the solubility of
the CO2 in the catholyte electrolyte solution. The CO2 solubility may be
36
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
increased by reducing electrolyte concentrations, operating at lower
temperatures, and operating electrochemical cell 901 at higher pressures.
Alternatively, formate cell 901 system may employ a cathode 902 using a GDE
based electrode which may operate at higher current densities since the GDE
electrode operates at high partial pressures of CO2 within the electrode
structure and may not be as limited as the solubility of CO2 in the solution
electrolyte, which may be on the order of about 33 nnM as CO2.
[0089] Catholyte disengager 932 may have a potassium formate product stream
936 including potassium formate (K-Formate) with a potassium bicarbonate
(KHCO3) residual that may be passed to a K-Formate-Bicarbonate Separation
Unit 950, which may be used to separate KHCO3 as a solid product from the
potassium formate solution. The separation unit may be an evaporator-
crystallizer, falling film evaporator, or any suitable commercial equipment
using
steam or other energy heating source as well as vacuum that may evaporate the
water from the potassium formate solution to efficiently conduct the KHCO3
separation from the potassium formate using the relative solubilities of the
chemical components in an aqueous solution.
[0090] Separation unit 950 may have an exit KHCO3 solution product stream 980
that may include KHCO3 solid/crystals having some residual potassium formate.
The amount of residual potassium formate in the separated KHCO3 solid may
range from about 0.1% to 10 wt% or less, more preferably about 0.1% to 5 wt%
or
less, and most preferably 0.1 wt% to 2 wt% or less as potassium formate. The
solid/liquid separation methods employed, such as centrifuges, vacuum filter
filtration, and the like with methods for washing/rinsing the potassium
formate
from the KHCO3 crystals may determine the wt% amount of potassium formate
exiting with the KHCO3. Separation Unit 950 may have a water vapor output
stream 952 from the evaporation and optionally an excess KHCO3 stream 954, if
needed, may be used for water or potassium compound balance in the system
that can be added to the system as needed.
37
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
[0091] KHCO3 solid/crystal product stream 980 may then be sent into
carbonation reaction/dilution vessel 976, where KHCO3 solid 980 is dissolved
with the addition of water 941. Additionally, K2CO3 stream 972 from K-Oxalate-
K2CO3 unit 970 is also entering vessel 976. CO2 gas 978 may be injected into
vessel 976 to fully convert any potassium carbonate to potassium bicarbonate
according to reaction (14b).
[0092] Stream 942 may leave vessel 976 and may be metered into catholyte
recycle solution steam 938 for controlling pH in the catholyte loop of
electrochemical cell 901. The solution stream 942 concentration may range from
about 5 wt% to 60 wt% as KHCO3, or more preferably in a range of 10 wt% to 55
wt% as KHCO3 in order to control the total water input into catholyte
electrolyte
recycle stream 948.
[0093] Separation unit 950 may have a K-Formate product stream 956 which
includes a potassium formate solution that may contain levels of KHCO3 in the
range of about 0.1 wt% to 10 wt% or less, and preferably in a range of about
0.1
wt% to 5 wt% or less, and more preferably in a range of about 0.1 wt% to 1
wt%.
The K-Formate product stream may preferably have a concentration of about 90
wt% to 99.9 wt% as potassium formate, and more preferably in a range of 98 wt%
to 99.9 wt% as potassium formate with the remaining balance as water. Most
preferably, potassium formate stream 956 from separation unit 950 may include
as little water as possible, and would basically be a melt of potassium
formate,
which has a melting point of about 167 C.
[0094] Preferably, the K-Formate product stream 956 may be sent as a
potassium formate melt to thermal reactor 958, where it may be converted to
potassium oxalate at a high conversion at specified temperature, atmospheric
gas, and reaction time conditions. Thermal reactor 958 may operate in a range
of about 100 to 550 C, and more preferably at about 200 - 500 C. The
operating temperatures may depend on the decomposition temperatures of the
alkali metal formate salt and the optimum temperature to get the highest
yields
38
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
of the alkali metal oxalate product. A residence time of the reaction at
optimum reaction temperatures may range from 5 seconds to hours, and the
equipment chosen to conduct the reaction may be designed to provide the rate
of heating and cooling to obtain optimal conversion yields. This may include
the
use of cold rotating metal that may rapidly chill the hot thermal product
after
the thermal reaction period may be completed.
[0095] Catalysts that may enhance the conversion efficiency and temperatures
may be used. Catalyst stream 957 may be added to the potassium formate melt
956 in thermal reactor 958. A suitable catalyst 957, for a potassium-based
cation system may include KOH, potassium hydride, potassium borohydride,
potassium ethoxide, potassium nnethoxide, potassium tert-butoxide and the like
may be added into the potassium formate before it enters thermal reactor 958.
The introduction of catalyst 957 may help to reduce the calcination
temperature
and improve the conversion yield of alkali metal formate to alkali metal
oxalate
to a range of 50% to 99% or more, and preferably 70% to 99% or more. The
reaction may also provide suitable yields without the need for the addition of
catalyst 957. Hydrogen 960 may be a major byproduct reaction from thermal
reactor 958 and may be recovered for use in the process. Thermal reactor 958
may be operated in different configurations, such as under a partial vacuum,
under an inert atmosphere such as nitrogen, or with the use of any suitable
gas
that may improve the efficiency of the chemical conversion of the formate to
oxalate. The addition of other chemicals to thermal reactor 958 may also be
useful, so as to obtain a clean flowing purified product. Thermal reactor 958
may be any suitable type equipment that can heat the alkali metal formate to
suitable temperatures and control the thermal or calcination atmosphere.
Thermal reactor 958 may include tunnel furnaces, rotary kilns, high
temperature spray dryers, high temperature rotating drunn Maker units, fluid
bed reactors, and other commercial calcination equipment and designs that may
be commercially available.
39
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
[0096] K-Oxalate product stream 962 leaving thermal reactor 958 may be cooled,
and passed onto K-Oxalate Dissolving Tank 964, where K-Oxalate 962 solids may
be dissolved in water, and may be filtered by various available methods to
remove any insoluble materials and obtain a clear, filtered product solution,
free of suspended solids. Deionized water stream 966 may be added to unit 964
as required. The alkali metal oxalate product may contain potassium carbonate
and/or potassium bicarbonate as byproduct(s) of the calcination. The solution
may be concentrated sufficiently so that the potassium oxalate/alkali metal
bicarbonate solution may not require a larger amount of energy or steam for
water evaporation when it may be passed onto K-Formate-K2CO3 Separation Unit
970 via stream 968.
[0097] K-Formate-K2CO3 Separation Unit 970 may be an evaporator-crystallizer,
removing water and may preferably use some of the physical aqueous solubility
properties of potassium carbonate which may have a much higher water
solubility than potassium oxalate and to precipitate and separate potassium
oxalate crystals, which may be redissolved and exit separation unit 970 as K-
Oxalate Solution stream 974. Stream 972 may be a solution of potassium
carbonate obtained from separation unit 970 that may be passed onto
carbonation reaction/dilution vessel 976, where the potassium carbonate may
be converted with CO2 stream 978 to potassium bicarbonate. The potassium
bicarbonate stream 942 from carbonation reaction/dilution vessel 976 may then
be metered into electrochemical cell 901 catholyte stream 938 for pH control.
[0098] K-Formate-K2CO3 Separation Unit 970 may employ any other suitable
mechanism for separation of potassium oxalate from potassium carbonate,
including nano-filtration, cooling crystallization, and the like.
[0099] K-Oxalate solution 974 obtained from separation unit 970 may then be
passed onto and metered into ion exchange compartment 914 of
electrochemical cell 901, were it may be converted to oxalic acid product
stream 928. K-Oxalate solution stream 974 may have a concentration range of
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
about 5 wt% to 60 wt% as potassium oxalate, and more preferably in a range of
about 10 wt% to 50 wt%. The metered flow rate determines the conversion of
the potassium oxalate to oxalic acid and affects the W:K+ cation ion ratio
transporting from ion exchange compartment 914 through the adjoining cation
ion exchange membrane 906 into catholyte compartment 904, and may have an
effect on the pH of the flowing catholyte electrolyte. The control of pH
within
catholyte compartment 904 may be dependent on balancing the combination of
the metered flow of K-Oxalate solution 974 and metered flow of KHCO3 solution
942. As one option, the flow of the K-Oxalate solution can be put at a set
rate,
and the metered flow of KHCO3 solution 942 can be varied for pH control in
electrochemical cell 901 catholyte flow loop, which may include streams 904,
930, 938, 944 and 948 and using pH sensors 931 and 943 for monitoring and
control.
[0om] Oxalic product stream 928 may then be further purified and
concentrated as needed, including the removal of residual potassium ions to
low
levels using ion exchange, implementation of a smaller electrochemical
acidification system as described in Fig. 4, concentration increase with
removal
of water by evaporation, and any other suitable processing. The oxalic acid
product may then be further converted, in one example, to esters such as
dinnethyl oxalate (DMO), diethyl oxalate (DEO), and dibutyl oxalate (DB0),
which
may then be converted with suitable catalyst in a hydrogenation reactor to
nnonoethylene glycol.
polon Fig. 10 shows a further additional embodiment applied to the system
shown in Fig. 9, where the anolyte compartment may produce a halogen, such
as chlorine, when utilizing a hydrogen halide feed, such as HCl, as an anolyte
feed. In system 1000, anolyte feed stream 1026 contains HCl, which may be fed
into anolyte compartment 1012, which includes anode 1010 which may suitable
for oxidizing the HCl into chlorine. Anolyte compartment exit stream 1016 may
then enter anolyte disengager 1018, where chlorine is separated from the
anolyte solution stream. Overflow stream 1019 may be used, if needed, if the
41
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
anolyte water volume is not balanced and may be later recycled to the system.
Recycle flow stream 1022 from anolyte disengager 1018 may have an input
deionized water stream 1024 added to supply deionized water to the anolyte
stream as needed and a metered HCl feed stream 1025 to supply the chloride
ions to be oxidized to chlorine in the anolyte compartment at anode 1010. The
combined feed stream 1026 may then enter anolyte compartment 1012.
[00102] Anolyte feed stream 1026 may also contain a portion of the electrolyte
as sulfuric acid, added as stream 1027, which may act as a supporting
electrolyte. The amount of sulfuric acid may range from about 0.5 wt% to 20
wt% in anolyte solution stream 1026. In addition to being a supporting
electrolyte, the sulfuric acid may allow for variability in the production
volume
or rate of chlorine from anolyte compartment 1012, with the production of both
chlorine and oxygen proportional to the HCl metered into anolyte compartment
1012. Thus, the mass or molar ratio of chlorine to formic acid from system
1000
may be varied as needed to meet the requirements of the process employed.
The molar ratio of formic acid product to chlorine co-product may be varied
from about 100:1 to 1:1, or more preferably in a range of 90:1 to 1:0.9.
[00103] HCl feed 1025 may be aqueous, and may range from about 5 wt% to 36
wt%, and more preferably in a range of about 5 wt% to 30 wt%, and most
preferably in a range of about 10 wt% to 20 wt%. The operating temperature of
the anolyte may be in the range of 5 C to 80 C, and more preferably in the
C to 60 C range. The optimum operating temperature may be chosen
according to the material construction of anode 1010, where the use of some
metals as substrates, such as titanium grades containing Pd, such as ASTM
Grades 7, 11, and 17, have a good resistance to HCl, but may have an upper
operating temperature limit due to corrosion by HCl.
[00104] In another embodiment, if bromine may be required as a co-product
from electrochemical formate system 1000, then HBr would be metered into
anolyte stream 1026 entering anolyte compartment 1012. The supporting
42
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
sulfuric acid electrolyte may be used to ensure that bromine is generated
efficiently with low concentrations of tribronnide in the anolyte solution.
The
molar ratio of formic acid product to bromine co-product may be varied from
about 100:1 to 1:1, or more preferably in a range of 90:1 to 1:0.9.
[00105] Referring to Fig. 11, a system 1100 utilizing a formic acid produced
in
the electrochemical system for use in an off peak power energy storage system
in accordance with an embodiment of the present disclosure is shown. Carbon
dioxide may be electrochemically reduced to a formic acid product and
employed in an energy storage system to supply electrical power to the
electrical grid in uses such as peak power generation, power load leveling,
and
the like. Energy storage system 1100 may employ electrochemical system 1101,
the electrochemical formic acid system as described in Fig. 7 of this
disclosure
as system 700, preferably using a renewable energy source 1102 as electrical
power, which may be solar, wind, and/or other alternative energy sources, and
CO2 source 1104 in addition to CO2 recycle stream 1126. Formic acid product
1106 from system 1101 may be catalytically decomposed to hydrogen gas in
Decomposition Reactor 1108, which may use a catalyst 1110, which may employ
or one more and combinations of catalysts including, for example, platinum,
silver, ruthenium, rhodium, gold, or palladium based catalysts as their
metals,
oxides, or alloys. In addition, other catalyst compositions that may be
suitable
may include transition metal, transition metal alloys, and transition metal
oxide
compositions. These transition metal catalysts may also include a small amount
of platinum group metals and their oxides in their compositions. The catalysts
may be physically placed on a fixed packed bed in decomposition reactor 1108
or may be suspended in the solution. Decomposition Reactor 1108 may be
heated and circulated using pumps, and may be pressurized. Decomposition
Reactor 1108 may also include a plug flow design with recirculation loop.
[00106] The reacted formic acid in Decomposition Reactor 1108 may be
converted to CO2 as stream 1114, which may be recycled back into CO2 recycle
stream 1126 to again produce formic acid from carbon dioxide. Depleted formic
43
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
acid solution 1112 from Decomposition Reactor 1108 may be recycled to system
1100 to produce formic acid. Preferably, Decomposition Reactor 1108 may be
operated under conditions not requiring extremely high pressures and
temperatures with the chosen catalyst system.
[00107] Hydrogen product 1116 from Decomposition Reactor 1108 may be
pressurized using a compressor (not shown) and may be sent to Fuel Cell System
1118, where the hydrogen may be converted to electrical power 1124 using an
oxidant such as oxygen. Preferably, Fuel Cell System 1118 may utilize a high
temperature solid oxide type, which may employ a natural gas 1120 feed to help
maintain the performance of Fuel Cell System 1118 in maintaining load,
increasing load, and to maintain temperatures and operating conditions in the
system. Fuel Cell System 1118 may have a CO2 effluent stream 1122 which may
be proportional to the amount of natural gas utilized. CO2 stream 1122 is
recycled into CO2 recycle stream 1126. CO2 feed stream 1104 may be used to
balance and provide the additional CO2 needed for Energy Storage System 1100.
Electrochemical Cell Designs Utilizing Gas Diffusion Electrodes
[0olos] Gas diffusion electrodes (GDE's) may be utilized in electrochemical
cells in the reduction of carbon dioxide. In the anode compartment, the
electrochemical cell anode reaction may be the oxidation of hydrogen gas
introduced into the anode GDE to form hydrogen ions (H+) or protons. These
protons may then preferably pass through the adjoining cation ion exchange
compartment as shown in Figs. 3 and 4, or into an adjacent ion exchange
compartment as shown in Figs. 7 and 9.
[00109] Referring to Fig. 12, a schematic illustrating an electrochemical cell
utilizing a hydrogen GDE for the anode reaction in producing oxalic acid from
the reduction of carbon dioxide in accordance with an embodiment of the
present disclosure is shown. Fig. 12 shows one of the hydrogen anode GDE
configurations as an electrochemical cell 1200 in a cross section view, not
44
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
showing some of the internal gas passages within the anode current collector.
Electrochemical cell 1200 may include an anode current collector containing an
internal gas plenum with a multitude of micro-channels or grooves that connect
to the hydrogen GDE, so that hydrogen gas may be evenly distributed and
contact the GDE. The micro-channels may run horizontally or vertically. The
gas plenum may also include a current collector utilizing a metal mesh or
screen
based structure. The hydrogen gas may preferably be introduced into the anode
compartment from an upper entry port in the anode compartment and flow
downwardly into the plenum as shown, and the depleted hydrogen may then
exit at the bottom of the anode compartment. The hydrogen may be humidified
with solvent vapor used in the anolyte compartment as needed. The anode
current collector may be a carbon or graphite material or include metals if
they
are resistant to the anolyte electrolyte reactions and acidity that may be
formed in the anolyte solution. The hydrogen may also be operated in a co-
current or countercurrent flow configuration.
[00110] Adjacent to the right of the anode GDE may be an anode trickle bed
solution distributor or percolator, where an anolyte solution may be
introduced
at a port at the top of the anode compartment such that the solution may be
evenly distributed and the solution is distributed evenly down the length of
the
trickle bed distributor and exits the bottom of the anode compartment. The
solution may be fed a specific flow rate, in the range of 0.001 to 10 liters
per
minute or more depending on the electrochemical cell dimensions, such that the
anode GDE may not be completely flooded with the anolyte solution due to
excessive pressure, and so as to maintain good ionic contact with the anode
GDE
for the transfer of protons into the anolyte solution. The flow and pressure
of
the anolyte flow should be such that minimal amounts of anolyte solution may
pass through the GDE into the hydrogen gas plenum inside the anode current
collector, and the hydrogen gas oxidation within the GDE may be sufficient, so
as to obtain a reasonable anode current density, in the range of 10 nna/cnn2
to
1000 nna/cnn2, or more preferably in a range of about 50 nna/cnn2 to 500
nna/cnn2. It is contemplated that electrochemical cell 1200 may include an
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
energy source (not shown) which may be operably coupled with the anode and
the cathode, the energy source configured to provide power to the anode and
the cathode to reduce carbon dioxide at the cathode. The anode trickle bed
may include a thin construction, between about 0.1 cm to 10 cm in thickness,
and preferably as thin as possible in the range of 0.2 to 5 cm in thickness to
reduce any IR voltage drop to a minimum. The anode trickle bed may be made
from non-conductive corrosion resistant polymer plastics, such as PTFE,
polypropylene, PVDF and the like, in the form of screen-like or convoluted
forms
so to distribute the solution evenly as it passes down the anode GDE
structure.
The entry and exit ports of the anolyte compartment are designed such that the
flow distribution of anolyte liquid is uniform along the cross section of the
trickle bed at the top and bottom. Alternatively, the trickle bed material may
be included of conductive carbon and graphite, and may contain some of the
hydrogen GDE catalyst on its surfaces. The GDE may be partially bonded to the
separator or membrane for improved electrical conduction or contact.
[00111] The separator located between the anode and cathode compartments
may be a membrane type, such as a cation membrane that may ionically
conduct cations, such as H+ ions, through the membrane to the cathode
compartment and may prevent or reduce the amount of back migration of
anions from the catholyte compartment back into the anolyte compartment.
Correspondingly, if the electrochemical cell design may employ an intervening
ion exchange compartment, such as in Fig. 7, the cation membrane may prevent
the same back migration of anions from the adjoining cation membrane of the
ion exchange compartment into the anolyte compartment. The selected cation
membranes may preferably be stable to the solvent and salts in the
electrochemical cell, such as the perfluorinated sulfonic acid membranes under
the trade name of Nafion. The separator may also be, as discussed previously,
a
nnicroporous separator, with fine pores in the 0.001 to 1 micron pore size
range,
such that it limits or controls solution or solvent bulk flow from the
catholyte to
the anolyte. The bulk flow may be controlled by the flow pressure of the
catholyte solution flowing in the catholyte compartment.
46
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
[00112] The catholyte flow stream preferably enters the bottom of the
catholyte
compartment and exits from the top of the catholyte compartment in order to
facilitate the removal of gases in the catholyte compartment. The catholyte
flow rate may range from 0.01 to 10 liters/minute or more depending on the
electrochemical cell dimensions and operating current density. The catholyte
compartment utilizes a high surface area cathode structure for
electrochemically reducing carbon dioxide in the catholyte to formate. The
cathode materials that may be suitable are as described in this application.
The
operating pressure of the catholyte compartment may be in the range of 0.1 to
psig, or in the range of 1 to 30 psig or greater. The operating pressure may
be
a function of the catholyte flow rate and the flow resistance of the high
surface
area cathode structure employed.
[00113] The preferred solvent for the anolyte solution is an aqueous solution
containing an electrolyte such as sulfuric acid or non-oxidizable acid to be a
supporting electrolyte as needed. Organic solvents added to the anolyte in
quantities of 50 wt% or less may be employed, such as methanol or ethanol, but
may not be as preferred if they may interfere with the hydrogen oxidation at
the anode. Salts that may be added to the anolyte electrolyte that may
interfere with the GDE hydrogen oxidation are not preferred.
[00114] As an alternative embodiment, the electrochemical cell configuration
may employ anode and cathode GDE structures that are bonded to or in direct
contact with a cation membrane separator.
[00115] In a further embodiment, ionically conductive materials, such as ion
exchange materials such as resins or iononners that are insoluble to the
solvents
may be used or placed between the anolyte and catholyte GDE's so as to
provide a conductive ionic path for the ions to migrate. Alternatively, gel-
type
membranes that may include ion exchange structures that may hold ionic groups
such as phosphoric acid in its structure and the like may be used. Membranes
of
47
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
these types that may be used are those used or proposed for use in phosphoric
acid fuels and the like.
[00116] Referring to Fig. 13, a schematic illustrating an electrochemical cell
1300 utilizing a hydrogen GDE for the anode reaction and a carbon dioxide GDE
for the cathode reaction in producing alkali metal formate from the reduction
of
carbon dioxide in accordance with an embodiment of the present disclosure is
shown. Electrochemical cell 1300 may employ the same hydrogen GDE anode as
in the electrochemical cell 1200 in Fig. 12, but also employs a carbon dioxide
reduction GDE cathode structure in the catholyte compartment. The anolyte
GDE may operate in the same manner as described in the description of the
electrochemical cell 1200 of Fig. 12.
[00117] The cathode GDE operates in the same manner, except that carbon
dioxide may be reduced at the cathode GDE to form formate. The cathode may
have a carbon dioxide internal gas plenum to distribute the carbon dioxide
evenly into the cathode GDE.
[00118] Adjacent to the right of the cathode GDE is a cathode trickle bed
solution distributor, where the catholyte solution may be introduced at the
top
of the cell catholyte compartment and the solution is distributed evenly down
the cell and exits the bottom of the cathode compartment. Alternatively, the
flow may be reversed, so that the flow is in the vertical direction. The
solution
may be fed a specific rates, in the range of 0.001 to 10 liters per minute or
more depending on the electrochemical cell dimensions, so that the cathode
GDE may not be flooded with the catholyte solution due to excessive pressure,
and so as to maintain good ionic contact with the cathode GDE for the transfer
of electrons into the solution in the reduction of carbon dioxide. The flow
and
pressure of the catholyte flow should be such that minimal amounts of
catholyte
solution pass through the GDE into the carbon dioxide gas plenum inside the
cathode current collector, and that the carbon dioxide gas reduction within
the
GDE is sufficient, so as to obtain a reasonable cathode current density, in
the
48
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
range of 10 nna/cnn2 to 1000 nna/cnn2, or more preferably in a range of about
50
nna/cnn2 to 500 nna/cnn2. It is contemplated that electrochemical cell 1300
may
include an energy source (not shown) which may be operably coupled with the
anode and the cathode, the energy source configured to provide power to the
anode and the cathode to reduce carbon dioxide at the cathode. The cathode
trickle bed may include a thin construction, between about 0.1 cm to 10 cm in
thickness, preferably as thin as possible in the range of 0.2 to 5 cm in
thickness,
made from non-conductive corrosion resistant polymer plastics, such as PTFE,
polypropylene, PVDF and the like, in the form of screen-like or convoluted
forms
so to distribute the solution evenly as it passes down the cathode structure.
The
entry and exit ports of the catholyte compartment are designed such that the
flow distribution of liquid is uniform along the cross section of the trickle
bed at
the top and bottom. Alternatively, the GDE cathode may be able to be operated
in a partially flooded or possibly fully flooded condition, and the flow
conditions
and electrolyte may be adjusted to operate the cathode in this mode.
[00119] Alternatively, the trickle bed material may be included of conductive
carbon and graphite, or potentially of metal, may contain some of the cathode
GDE catalyst on the surfaces. As alternatively, the electrochemical cell
system
1300 in Fig. 13 may be operated with an oxygen generating anode system, and
not a hydrogen GDE, as the anode configuration as shown in Figs. 3, 5, 7, and
9.
[00120] Referring to Fig. 14, a schematic illustrating three different anode
GDE
constructions used in an electrochemical cell in producing alkali metal
formate
from the reduction of carbon dioxide in accordance with an embodiment of the
present disclosure are shown. Fig. 14A shows a carbon cloth support structure
including a thin layer containing a catalyst that may be deposited onto a
carbon
powder base by various methods such as spraying or precipitation and the like
with suitable methods known to the art. The GDE assembly may then be further
processed utilizing a spray application of a PTFE or PVDF emulsion or dipping
the
structure into a PTFE or PVDF emulsion, which may be then be dried to remove
the emulsion solvent. The assembly may then be compressed (under pressure)
49
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
and heated, to a temperature at or near the melting point of the PTFE or PVDF
so that the PTFE or PVDF flows and bonds the various components in the GDE
structure to form a 3-phase structure that is hydrophobic in portions of the
microstructure to liquids and yet allows hydrogen to pass into the pores for
the
oxidation of hydrogen to occur at the catalyst surfaces. Any potential
hydrophobic agents may be used so long as they are not soluble in the solvents
employed in the electrochemical cell anolyte and catholyte. These hydrophobic
compounds or agents may be different for the anolyte GDE versus the catholyte
GDE. Other potential hydrophobic agents, such a polyethylene and
polypropylene waxes, super-hydrophobic agents, and other materials such as
inorganic oxides, silica based materials, nitrides, borides, and others may be
employed that may have hydrophobic properties to the solvents used in the
electrochemical cell anolyte. The hydrophobic material content in the anode
GDE structure may range from 1% to 80%, and preferably from about 5% to 50%
by weight or by volume in the GDE structure to obtain the non-wetting and
anode GDE performance properties required.
[00121] The hydrogen oxidation catalysts used in the anolyte GDE may include
precious metal and precious metal oxides and their mixtures, including
platinum, palladium, gold, ruthenium, iridium, and silver and their alloys and
mixtures. The concentration of the GDE catalyst, such as platinum, is such
that
it is economical, such as 0.5 nng/cnn2 for platinum on the carbon powder
support. Catalyst concentrations may range from about 0.01 to 20 nng/cnn2, and
more preferably in the range of 0.1 to 5 nng/cnn2 for a number of these
precious
metal catalysts. More catalyst may be employed in the composition if lower
cost
catalyst materials are available. The support for the catalyst may be a high
surface area carbon or may be an alternative conductive material, such as
graphene, conductive nitrides, carbon nanotubes, conductive titanium suboxides
such as Ti407 and Ti20 and the like. The catalyst may be applied by various
methods, such as electroplating, chemical reduction, chemical precipitation
and
chemical vapor deposition and the like. A number of GDE's have been described
in the literature and are available commercially, and these may be employed as
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
an anode GDE if they may have suitable hydrophobic properties and catalytic
activity as required for this process.
[00122] Fig. 14B shows the same configuration as Fig. 14A except for an
additional secondary layer containing a carbon powder with the hydrophobic
bonding/filler in its make-up.
[00123] Fig. 14C shows the GDE construction of Fig. 14B except for an
additional
thin metal mesh within the carbon cloth support that may supply mechanical
stability to the structure, as well as provide a better electrical conductive
structure so that the GDE may be operated at high current densities. The metal
may be nickel, silver, or other metal or alloy that is corrosion resistant to
the
anode operating conditions.
[00124] In an alternative embodiment, a separator or membrane may be bonded
to the anode GDE structure on one face or both faces of the anode and/or
cathode GDE, if additional solvent flow rejection is required. In another
embodiment, a Nafion (perfluorinated sulfonic acid) based solution may be also
used in the GDE structures for additional bonding and to provide an ionically
conducting media within the GDE structure. Other ionic monomers similar to
Nafion may be employed, including for example, ion exchange resin materials
and non-halogenated iononners such as sulfonated polystyrene, sulfonated
divinyl benzene and the like. In addition, the anode GDE's may also be post
treated using sprays to add additional hydrophobic material or compounds to
the inside and outside surfaces of the GDE's such as the various super-
hydrophobic materials available commercially.
[00125] Referring to Fig. 15, a schematic illustrating three different cathode
GDE constructions 1500 used in an electrochemical cell in producing alkali
metal
formate from the reduction of carbon dioxide in accordance with an
embodiment of the present disclosure is shown. Figs. 15A, 15B, and 15C have
the same relative corresponding configurations as Figs. 14A, 14B, and 14C
51
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
except that the catalyst for the reduction of carbon dioxide may include, for
example, high hydrogen over potential metals, such as indium, tin, bismuth and
the like, that are suitable electrocatalysts for the reduction of carbon
dioxide to
formate. The preferred metal catalysts may include, for example, indium, tin,
bismuth, lead, silver, gold, zinc, and cadmium including their binary and
ternary
alloys, internnetallics, and combinations as single and multiple composition
coatings deposited on various metals, carbon, or other conductive supports.
Other suitable catalysts may further include other transition metals, such as
copper, cobalt, manganese, vanadium, and nickel that may be combined and
alloyed with the preferred metal catalysts already mentioned such as indium,
tin, and silver. The preferred carbon dioxide reduction product, such as CO or
formate, may determine the catalyst selected for use in the GDE. For example,
a silver based catalyst may be preferred for efficiently converting carbon
dioxide to CO, while an indium-tin alloy may be most suitable for reducing
carbon dioxide to formate. Most importantly, the stability of the catalyst to
the
cathode reaction is the key to providing a long term life cathode for the
electrochemical process.
[00126] The catalyst layer may be a high surface area powder including metal
deposited onto a carbon or conductive ceramic substrate or may include a
composition utilizing a high surface area metal powder that may have a metal
composition that may range from 5 wt% to 90 wt% as the metal(s). The
hydrophobic PTFE or other described hydrophobic and ionically conducting
polymer materials may be used to help provide the bonding in forming the three
dimensional cathode CO2 reduction GDE structure. The carbon cloth may be
substituted with a high surface area metal mesh or cloth structure, or
nnetalized
carbon or polymer material, which may be non-woven or sintered. In addition, a
small percentage of monomers may be added to the GDE composition that may
be polymerized to form a rubber or elastonner type structure that does not
significantly reduce the electrical and ionic conductivity of the GDE
structure.
52
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
[00127] Referring to Fig. 16, a schematic illustrating an electrochemical cell
1600 utilizing a hydrogen GDE and a weir solution distribution system for the
anode reaction in producing alkali metal formate from the reduction of carbon
dioxide in accordance with an embodiment of the present disclosure is shown.
Electrochemical cell 1600 may include a different placement of the anolyte
solution inlet and anolyte solution outlet ports in comparison to
electrochemical
cell 1200. The hydrogen may also be operated in a co-current or countercurrent
flow configuration. It is contemplated that electrochemical cell 1600 may
include an energy source (not shown) which may be operably coupled with the
anode and the cathode, the energy source configured to provide power to the
anode and the cathode to reduce carbon dioxide at the cathode.
[00128] Referring to Fig. 17, a schematic illustrating an electrochemical cell
1700 utilizing a hydrogen GDE for the anode reaction and a carbon dioxide GDE
for the cathode reaction and showing a weir solution distribution system in
producing alkali metal formate from the reduction of carbon dioxide in
accordance with an embodiment of the present disclosure is shown.
Electrochemical cell 1700 includes weir flow anolyte and catholyte solution
distributors that evenly distribute the anolyte and catholyte solutions into
the
anolyte and catholyte trickle bed distributors respectively. Electrochemical
cell
1700 also shows a different placement of the solution inlet and outlet flow
ports
for the anolyte and catholyte in comparison to electrochemical cell 1300. It
is
contemplated that electrochemical cell 1700 may include an energy source (not
shown) which may be operably coupled with the anode and the cathode, the
energy source configured to provide power to the anode and the cathode to
reduce carbon dioxide at the cathode.
[00129] In an alternative embodiment, the cathode carbon dioxide GDE
structure may include a high surface area metallic powder catalyst base that
has
been mixed with a hydrophobic bonding agent that may be compressed and heat
bonded into a thin porous sheet, and then further bonded to the GDE support
structures as shown in Figures 12 - 17, which may be metal or carbon based
53
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
materials or their combination. The percentage of the hydrophobic agent in the
metallic powder catalyst may range from 2% to 95% by weight, and more
preferably 5% to 80% by weight. The metal powder catalyst may include metals
and their alloys suitable for the reduction of carbon dioxide to formate, for
example indium and tin and their alloys. The metal particles may include
multiple electrocatalyst coatings, such as one metal and/or metal oxide plated
or coated onto another metal or metal oxide. Examples of this may be
electrocatalyst materials and structures constructed with a multitude of
layers
that may perform the electrochemical reduction of carbon dioxide that may
have synergistic co-catalytic effects and provide a stable, long term life
performance. Examples of these may be tin particles including an applied
surface coating of indium or an indium alloy with lead or zinc, copper or
nickel
substrate particles with an applied coating of tin with an overlayer of co-
electrocatalyst of indium and the like.
[00130] In addition, the metal powder catalyst may include metal oxides as
well as small amounts of precious metal and precious metal oxides, as mixtures
or coatings on the electrocatalyst particles. The percentages of these added
components, such as the precious metals, may range from 0.001% to 80% or
more in the catalyst composition by weight.
Formate CO2 Reduction Chemistry
[00131] The postulated chemistry of the reduction of CO2 at the cathode may
proceed as follows.
[00132] Hydrogen atoms may be adsorbed at the electrode from the reduction of
water as shown in equation (1).
1-1+ + e- Had (1)
54
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
[00133] Carbon dioxide may be reduced at the cathode surface with an adsorbed
hydrogen atom to form formate, which may be adsorbed on the surface as
shown in equation (2) as follows:
CO2 + Had HCOOad (2)
[00134] The formate adsorbed on the surface then reacts with another adsorbed
hydrogen atom to form formic acid that may be released into the solution as
shown in equation (3):
HCOOad + Had HCOOH (3)
[00135] A competing reaction at the cathode may be the reduction of water
where hydrogen gas may be formed as well as hydroxide ions as shown in
equation (4):
2H20 + 2e- H2 + 20H- (4)
[00136] Operating the electrochemical cell at higher pressures (above
atmospheric), may increase the carbon dioxide to formate current efficiency
and allow operation of the cells at higher current densities.
Anode Reactions
[00137] The anode reaction may be the oxidation of water into oxygen and
hydrogen ions as shown in equation (5) as follows:
2H20 4H+ + 4e- + 02 (5)
[00138] Below may be the various preferred and alternative embodiments for
the process, arranged in different categories.
Formate Formation From CO
[00139] The thermal intermolecular reaction of alkali metal formate CO with
KOH may be as shown in equation (6) follows:
CO + KOH HCOOK (6)
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
[00140] The KOH may be consumed in the reaction. Under the right conditions,
both formate and oxalate may both be produced, and which may decrease the
number of process steps. The production of both would require the separation
of these carboxylic acids from each other.
[00141] Carbon monoxide may also be selectively absorbed in a alkali metal
carbonate and bicarbonate aqueous solutions to produce formate, where M may
be an alkali metal which may be shown as in equations (7) and (8) as follows:
CO + MHCO3 MOOCH + CO2 (7)
2C0 + M2CO3 + H20 2MCOCH + CO2 (8)
[00142] These reactions may not require MOH, such as NaOH or KOH, in the
reaction for the formation of M-formate as catalysts.
Oxalate From Formate
[00143] The thermal intermolecular reaction of alkali metal formate with KOH
may be as shown in equation (9) as follows:
2HCOOK + KOH K2C204 + H2 (9)
[00144] Optionally, sodium or potassium carbonate may also be used for
converting formate to oxalate, but the conversion yields have been shown to be
significantly lower. Under the right operating conditions and temperatures,
the
yields may be significantly improved.
Anode Oxidation Reactions
[00145] The anode reaction when utilizing sulfuric acid in the anolyte, is the
oxidation of water generating hydrogen ions and oxygen as shown in equation
(10) as follows:
2H20 02 + 4H+ + 4e- (10)
56
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
[00146] If hydrobronnic acid, HBr, is used in the anolyte, the reaction is the
oxidation of the bromide to bromine as follows:
2HBr Br2 + 2H+ + 2e- (11)
[00147] If sodium chloride, NaCl, may be used in the anolyte, the anode
reaction, such as in the formate cell in Fig. 5, is the oxidation of the
chloride
ion as shown in equation (12) as follows:
2 NaCl C12 + 2Na+ + 2e- (12)
[00148] Sodium ions may pass through the ion permeable separator from the
anolyte compartment to the catholyte compartment and combine with any
formate from the reduction of carbon dioxide to form sodium formate and any
by-product hydroxide ions formed from the reduction of water at the cathode
may form NaOH.
[00149] If hydrochloric acid, HCl, may be used in the anolyte, the reaction
may
be the oxidation of the chloride to chlorine with the co-production of
hydrogen
ions as shown in equation (13) as follows:
2HCl ---> C12 + 2H+ + 2e- (13)
Carbonate and Bicarbonate Reactions
[00150] Sodium carbonate, Na2CO3, dissolved in solution may be converted to
sodium bicarbonate, Na2HCO3, with reaction with CO2 as shown in equation
(14a) as follows:
Na2CO3 + CO2 + H20 2Na HCO3 (14a)
Similarly, potassium carbonate, K2CO3, dissolved in solution may be converted
to
potassium bicarbonate with reaction with CO2 as shown in reaction (14b) as
follows:
K2CO3 + CO2 + H20 2KHCO3 (14b)
57
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
[00151] Sodium hydroxide, NaOH, reaction with CO2 in solution may be
converted to sodium carbonate, Na2CO3, as shown in equation (15) as follows:
2NaOH + CO2 + H20 2Na2CO3 + H20 (15)
Chlorine Reaction With NaOH
[00152] Sodium hydroxide, NaOH, may be reacted with chlorine to produce
sodium hypochlorite, Na0Cl, as shown in equation (16) as follows:
2NaOH + C12 Na0Cl + NaCl + H20 (16)
Bicarbonate Reaction With H+ Ions
[00153] Potassium bicarbonate, KHCO3, may react with H+ ions generated in the
electrochemical cell to produce CO2 and water, such as in the cathode cell
compartment using a potassium bicarbonate electrolyte as shown in equation
(17) as follows:
2KHCO3 + 2H+ 2 CO2 + 2H20 + 2 K+ (17)
Electrolyzer Configurations
[00154] The following present various exemplary combinations of cell
configurations, electrode structures, and anolyte/catholyte compositions that
may be used in the electrochemical CO and/or formate, and electrochemical
acidification (EA) electrolyzers in the above described processes.
[00155] The cathode of the electrochemical cell 110 and electrochemical
acidification electrolyzer 140 may be a high surface area electrode. The void
volume for the cathode may be from about 30% to 98%. The surface area of the
cathode may be from 2 cnn2/cnn3 to 500 cnn2/cnn3 or higher. The surface areas
may be further defined as a total area in comparison to the current
distributor/conductor back plate area with a preferred range of from 2 to 1000
times the current distributor/conductor back plate area.
[00156] The cathode of the electrochemical cell 110 may be electrolessly
plated
indium or tin on a copper woven mesh, screen or fiber structure. Indium-copper
58
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
internnetallics may be formed on the copper woven mesh, screen or fiber
structure. The internnetallics may be harder than the soft indium metal, and
allow better mechanical properties in addition to usable catalytic properties.
[00157] In the electrochemical reduction of carbon dioxide, metals including
Pb,
Sn, Ag, Au, Hg, Tl, In, Bi, and Cd among others may produce formic acid (or
formate) as a major C1 product in aqueous solutions. Utilizing alloy
combinations of these metals such as Hg-Cu, Sn-Cd, Pb-Bi, Sn-Zn, Cu-Sn, In-Sn
and the like may have improved Faradaic performance efficiencies in addition
to
improved catalyst life stability. Some of these individual metal non-alloyed
catalysts by themselves, such as Sn and Cu, may have surface changes that
deactivates or shows a decline in the Faradaic conversion activity in
producing
formate or CO. The metal catalyst surface may then may have to be reactivated
by a reverse current or polarity. In the potential cathodic production of C2+
chemicals from CO2 or the electrochemical reduction of oxalic acid, to
products
such as glyoxalic and glycolic acid, metals such as Ti, Nb, Cr, Mo, Ag, Cd,
Hg, Tl,
As, and Pb as well as Cr-Ni-Mo steel alloys among many others may have the
beneficial result in the formation of these higher C2+ products at high
Faradaic
efficiencies.
[00158] In another embodiment, the cathode surfaces may be renewed by the
periodic addition of indium salts or a mix of indium/tin salts in situ during
the
electrochemical cell operation. Electrochemical cell 110 may be operated at
full rate during operation, or temporarily operated at a lower current density
with or without any carbon dioxide addition during the injection of the metal
salts.
[00159] In another exemplary embodiment, in preparing cathode materials for
the production of C2+ chemicals, the addition of metal salts that may reduce
on
the surfaces of the cathode structure may be also used, such as the addition
of
Ag, Au, Mo, Cd, Sn, etc. to provide a catalytic surface that may be difficult
to
59
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
prepare directly during cathode fabrication or for renewal of the catalytic
surfaces.
[00160] In another embodiment, the cathode structure may include a gas
diffusion electrode (GDE), which may be in direct contact with a CO2 gas
stream
in a different cell construction using an additional compartment with a gas
plenum, and allowing for an increase in current density and Faradaic
efficiency
for formate production. In one embodiment for the GDE construction, the GDE
electrode may include a mixture of indium particles or indium coated tin metal
particles that may be processed into a paste matrix with a PTFE binder and a
small percentage of some non-conductive ceramics, utilizing a central metal
screen made from indium plated tin, which is compressed and heated to form a
GDE structure that is gas permeable, but resistant to liquid flooding. The
structure may also include the addition of other commercial chemical agents
used for making materials non-wettable, such as super-hydrophobic materials
from UltraEvershield from Ultratech, International and NeverWet from
NeverWet International as examples. Other binders such as graphite and
graphene may also be used, but may not be as preferred.
[00161] Cathode 412 for the electrochemical acidification electrolyzer 140 may
include stainless steels and nickel electrodes.
Cathode 412 may include
coatings on the cathode to reduce the hydrogen overpotential.
[00162] An alkali metal hydroxide range for the electrochemical acidification
electrolyzer 140 may be 5% to 50% by weight, and more preferably 10% to 45%
by weight. The alkali metal hydroxide examples may be NaOH, KOH, CsOH and
the like.
[00163] Cathode materials for the cathode of electrochemical cell 110 for
carbon monoxide production from CO2 may include precious and noble metals as
well as Cu, Ag, Au, and their oxides, and specifically the oxides of copper.
Ag
and Ag oxides in combination and alloys with the other metals may provide
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
better and longer term electrocatalyst performance. Other d-block metals,
such as Zn and Ni, may be selective for CO reduction in aqueous media.
Regardless of specificity for CO as a CO2 reduction product, a cathode for
electrochemical cell 110 for an aqueous system for CO2 reduction to CO may
have a high hydrogen overpotential to prevent competing H2 formation.
[00164] In another embodiment, the proposed carbon dioxide GDE cathode
structures including metal particle electrocatalysts and multi-coating layer
electrocatalysts may also be used for the conversion of carbon dioxide to
carbon
monoxide, CO. Suitable electrocatalysts for this reaction may be silver and
silver alloys, in combination and alloys with other metals such as copper,
zinc,
gold, and other metals such as transition metals. Other gases may be co-
generated such as useful mixtures of CO and hydrogen in ratios suitable for
use
in Fischer Tropsch reactions producing fuels and organics.
[00165] Anions used for CO production at the cathode may be any species stable
at working potentials such as sulfate, chloride or bicarbonate. CO2 reduction
to
CO may favor high pH due to limited competing H2 formation; however there
may be a practical pH maximum at around 8.5 for a saturated CO2 solution due
to the formation of carbonic acid on dissolution. There may be no strict lower
limit that may have been observed. Depending on the chemistry of the system,
the pH of the catholyte region of electrochemical cell 110 may range from 3 to
12. The pH may be a function of the catalysts used, such that there may be no
corrosion at the electrochemical cell 110 and catholyte operating conditions.
[00166] Electrolytes for the electrochemical cell 110 for forming CO and
formates may include alkali metal bicarbonates, carbonates, sulfates, and
phosphates, borates, ammonium, hydroxides, chlorides, bromides, and other
organic and inorganic salts. The electrolytes may also include non-aqueous
electrolytes, such as propylene carbonate, nnethanesulfonic acid, methanol,
and
other ionic conducting liquids, which may be in an aqueous mixture, or as a
non-
aqueous mixture in the catholyte. The introduction of micro bubbles of carbon
61
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
dioxide into the catholyte stream may improve carbon dioxide transfer to the
cathode surfaces.
[00167] Electrolytes for the anolyte region of the electrochemical cell 110
may
include: alkali metal hydroxides, (e.g. as KOH, NaOH, Li0H) in addition to
ammonium hydroxide; inorganic acids such as sulfuric, phosphoric, and the
like;
organic acids such as nnethanesulfonic acid in both non-aqueous and aqueous
solutions; and alkali halide salts, such as the chlorides, bromides, and
iodine
salts such as NaF, NaCl, NaBr, LiBr, KF, KCl, KBr, KI, and Nal, as well as
their
acid halide forms, such as HCl, and HBr. The alkali halide salts may produce,
for example, fluorine, chlorine, bromine, or iodine as halide gas or dissolved
aqueous products from the anolyte region. Methanol or other hydrocarbon non-
aqueous liquids may also be used, and they would form some oxidized organic
products from the anolyte. Selection of the anolyte would be determined by
the process chemistry product and requirements for lowering the overall
operating cell voltage. For example, using HBr as the anolyte, with the
formation of bromine at the anode, which require a significantly lower anode
voltage potential than chlorine formation. Hydriodic acid, HI, may form iodine
at anode potential voltages even lower than that of bromine.
[00168] Catholyte cross sectional area flow rates may range from 2 to 3,000
gpnn/ft2 or more (0.0076 - 11.36 m3/m2). Flow velocities may range from 0.002
to 20 ft/sec (0.0006 to 6.1 nn/sec).
[00169] Catholyte region of the electrochemical cell 110 may include at least
one catalyst. The catalyst may be a homogenous heterocyclic catalyst which
may be utilized in the catholyte region to improve the Faradaic yield to
formate. Homogenous heterocyclic catalysts may include, for example, one or
more of pyridine, tin 2-picoline, 4-hydroxy pyridine, adenine, a heterocyclic
amine containing sulfur, a heterocyclic amine containing oxygen, an azole, a
benzinnidazole, a bipyridine, a furan, an innidazole, an innidazole related
species
with at least one five-member ring, an indole, a lutidine, nnethylinnidazole,
an
62
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
oxazole, a phenanthroline, a pterin, a pteridine, pyridine, a pyridine related
species with at least one six-member ring, a pyrrole, a quinoline, or a
thiazole,
and mixtures thereof.
[00170] Operating electrochemical cell 110 at a higher operating pressure in
the
catholyte region may allow more dissolved CO2 to dissolve in the aqueous
electrolyte. Typically, electrochemical cells may operate at pressures up to
about 20 to 30 psig in multi-cell stack designs, although with modifications,
they
could operate at up to 100 psig. The electrochemical cell 110 anolyte may also
be operated in the same pressure range to minimize the pressure differential
on
the membrane separating the two electrode regions. Special electrochemical
designs may be required to operate electrochemical units at higher operating
pressures up to about 60 to 100 atmospheres or greater, which may be in the
liquid CO2 and supercritical CO2 operating range.
[00171] In another embodiment, a portion of the catholyte recycle stream may
be separately pressurized using a flow restriction with back pressure or using
a
pump 390 with CO2 injection such that the pressurized stream may be then
injected into the catholyte region of the electrochemical cell 110, and
potentially increasing the amount of dissolved CO2 in the aqueous solution to
improve the conversion yield.
[00172] catholyte region and anolyte region of electrochemical cell 110 may
have operating temperatures that may range from -10 to 95 C, more preferably
- 60 C. The lower temperature may be limited by the electrolytes used and
their freezing points. In general, the lower the temperature, the higher the
solubility of CO2 in the aqueous solution phase of the electrolyte which may
result in obtaining higher conversion and current efficiencies.
However,
operating electrochemical cell voltages may be higher, such that an
optimization may be required to produce the chemicals at the lowest operating
cost. In addition, the operating temperatures of the anolyte and catholyte may
63
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
be different, whereby the anolyte is operated at a higher temperature and the
catholyte is operated at a lower temperature.
[00173] The electrochemical cell 110 and the electrochemical acidification
electrolyzer 140 may be zero gap, flow-through electrolyzers with a
recirculating catholyte electrolyte with various high surface area cathode
materials. For example, flooded co-current packed and trickle bed designs with
various high surface area cathode materials may be employed. The stack cell
design may be bipolar and/or nnonopolar.
[00174] The anode of the electrochemical cell 110 and the electrochemical
acidification electrolyzer 140 may include one or more anode coatings. For
example, for acid anolytes and oxidizing water under acid conditions,
electrocatalytic coatings may include: precious metal and precious metal
oxides such as ruthenium and iridium oxides, as well as platinum and gold and
their combinations as metals and oxides on valve metal conductive substrates
such as titanium, tantalum, or niobium as typically used in the chlor alkali
industry or other electrochemical processes where they may be stable as
anodes. For other anolytes such as alkaline or hydroxide electrolytes, the
electrocatalytic coatings may include carbon, graphite, cobalt oxides, nickel,
stainless steels, and their alloys and combinations which may be stable as
anodes under these alkaline conditions.
[00175] Membrane 330, 406a, 406b may be cation ion exchange type
membranes such as those having a high rejection efficiency to anions. For
example perfluorinated sulfonic acid based ion exchange membranes such as
DuPont Nafion brand unreinforced types N117 and N120 series, more preferred
PTFE fiber reinforced N324 and N424 types, and similar related membranes
manufactured by Japanese companies under the supplier trade names such as
Flennion . Other multi-layer perfluorinated ion exchange membranes used in
the chlor alkali industry and having a bilayer construction of a sulfonic acid
based membrane layer bonded to a carboxylic acid based membrane layer may
64
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
be employed to efficiently operate with an anolyte and catholyte above a pH of
about 2 or higher. These membranes may have a higher anion rejection
efficiency. These may be sold by DuPont under their Nafion trademark as the
N900 series, such as the N90209, N966, N982, and the 2000 series, such as the
N2010, N2020, and N2030 and all of their types and subtypes. Hydrocarbon
based membranes, which may be made from of various cation ion exchange
materials may also be used if the anion rejection may be not as critical, such
as
those sold by Sybron under their trade name lonac , AGC Engineering (Asahi
Glass) under their Selennion trade name, and Tokuyanna Soda among others.
Thermal Conversion of Alkali Metal Formate To Oxalate Experiments
[00176] Experiments were conducted to determine some process conditions in
the thermal conversion of alkali metal formate. Temperature, calcination time,
and the addition of various catalysts that may improve the yields to oxalate
were evaluated. Carbonate was determined by a standard method by titration
using HCl and pH indicators.
Example 1
[00177] Table 1 shows the results of a set of experiments that were conducted
in a thermal furnace using a nitrogen atmosphere. Experiments were conducted
to evaluate the conditions and yields in the thermal conversion of alkali
metal
formate. Temperature was varied as well as calcination time, and the use of
various catalysts were evaluated. These samples were prepared using reagent
grade potassium formate crystal and the addition of reagent grade potassium
hydroxide pellets. The chemical reagents were mixed together, and placed in a
100 nnL nickel crucible. The crucible was calcined at the times and
temperatures as given in Table 1. At 420 C, for time periods of 0.5 to 1.0
hrs,
the percent yield of the potassium formate to potassium oxalate using the
potassium hydroxide catalyst ranged from 73.71% to 78.53%. The oxalate
content was analyzed by both permanganate titration and by Ion
chromatography. At 440 C, the conversion yield to oxalate was about 77%.
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
Table 1
Mass of Percent
Potassium Mass of P Mass Yield
Potassium Hydroxide Percent
Potassium
Temperature Calcination Formate Catalyst of KOH Mass Loss
Oxalate
C Time (hr) (gm) (gm) (%) (grams) (%)
420 0.5 4.0888 0 0.0000 0.1838 7.62
420 0.5 4.1784 0.2244 5.3705 0.2115 76.11
420 0.5 4.0156 0.3348 8.3375 0.1742 73.95
420 0.75 4.0267 0.3246 8.0612 0.2397 73.71
420 1.0 4.1087 0.2268 5.5200 0.2121 78.53
440 0.5 4.2935 0.3323 7.7396 0.2482 77.17
440 1.0 4.0391 0.2008 4.9714 0.2329 77.55
Example 2
[00178] Table 2 shows the results of the same procedure as in Example 1,
except that potassium bicarbonate was added to potassium formate as co-
product or potential catalyst. The calcination temperature was 420 C for 30
minutes in a nitrogen atmosphere in the thermal oven.
Table 2
Catalyst
Sample Wt% % Oxalate
1 10% KHCO3 11.38
1 5% KHCO3 14.44
Example 3
[00179] Table 3 shows the results of the same procedure as in Example 1, KOH
was added to potassium formate as a catalyst. The calcination temperature was
440 C for 30 minutes in a nitrogen atmosphere in the thermal oven. Table 4
shows the results using no KOH catalyst.
66
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
Table 3
Wt% Potassium Potassium
KOH Oxalate Carbonate
Sample # Catalyst Wt% Wt %
1 2.0 80.4 13.0
2 2.0 72.8 22.6
3 2.0 71.7 20.7
Table 4
Wt% Potassium Potassium
KOH Oxalate Carbonate
Sample # Catalyst Wt% Wt %
1 0 14.3 23.8
2 0 43.9 51.0
Example 4
[001 80] Table 5 shows the results of the same procedure as in Example 1, KOH
was added to potassium formate as a catalyst. The calcination temperature was
480 C for 30 minutes in a nitrogen atmosphere in the thermal oven. Table 6
shows the results using no KOH catalyst.
Table 5
Wt% Potassium Potassium
Sample KOH Oxalate Carbonate
# Catalyst Wt% Wt %
1 2.0 75.9 21.6
2 2.0 75.7 21.7
3 2.0 74.6 21.5
67
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
Table 6
Wt% Potassium Potassium
Sample KOH Oxalate Carbonate
# Catalyst Wt% Wt %
1 2.0 73.3 23.6
2 2.0 72.7 24.1
3 2.0 71.2 24.3
Example 5
[00181] Table 7 shows the results of the same procedure as in Example 1,
magnesium oxide powder was added to potassium formate as a catalyst. The
calcination temperature was 420 C in a nitrogen atmosphere in the thermal
oven.
Table 7
MgO Potassium MgO Calcination Potassium
Sample Catalyst Formate wt% Time in Oxalate
# (gm) (gm) Hrs Wt%
1 0.7567 4.2279 17.9 0.75 19.9
2 0.3603 3.7827 9.5 1.0 54.3
3 0.5644 3.9544 14.3 1.5 48.3
Example 6
[00182] Table 8 shows the results of the same procedure as in Example 1,
sodium borohydride (NaBH4) powder was added to potassium formate as a
catalyst. The calcination temperature was 440 C in a nitrogen atmosphere in
68
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
the thermal oven. Table 9 shows the results using NaBH4 and KOH as co-
catalysts
at the same temperature.
Table 8
NaBH4 Calcination Potassium Potassium
Sample Catalyst Time in Oxalate Carbonate
# wt% min Wt% Wt%
1 2.47 3.5 66.2 11.0
2 2.77 2.66 75.6 11.2
Table 9
Potassium Potassium
Calcination NaBH4 KOH Oxalate Carbonate
Sample # Time wt% wt% wt% wt%
1 5 min 25 sec 2 2 74.2 8.5
2 3 min 2.5 2.5 66.5 10.2
3 2 min 30 sec 2.5 2.5 81.3 10.7
4 3 min 10 sec 2.5 2.5 76.8 9.5
3 min 2.77 0 75.6 11.2
6 2 min 30 sec 2.5 3.0 81.3 11.1
7 2 min 30 sec 2.5 2.5 80.5 11.8
Example 7
[00183] Table 10 shows the results of the same procedure as in Example 1,
except that sodium hydride (NaH) powder was added to sodium formate as a
catalyst. The calcination temperature was 440 C in a nitrogen atmosphere in
the thermal oven.
[00184] Table 11 shows the results using NaH added as a catalyst to potassium
formate at various time and temperatures.
69
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
Table 10
Calcination Calcination Sodium Sodium
Time NaH Temp Oxalate Carbonate
Sample # (min) wt% C wt% wt%
1 3.75 2.86 440 85.49 7.36
2 3.75 2.86 440 84.48 7.14
3 3.75 2.59 440 89.12 5.32
4 4.25 2.96 430 86.99 7.15
3.25 2.19 430 89.46 5.14
Table 11
Calcination Potassium Potassium
Calcination Temp NaH Oxalate Carbonate
Sample # Time C wt% wt% wt%
1 19.66 min 440 2.0 55.18 15.69
2 30 min 400 2.5 47.68 22.96
Conversion of oxalic acid to glyoxylic acid
[00185] Referring to Fig. 18, potential chemical derivatives starting with
oxalic
acid as an initial chemical feedstock in accordance with an embodiment of the
present disclosure are shown. Glycine (anninoacetic acid) is an industrial
chemical having a number of applications. It has extensive use as an additive
for
human and animal food products, as well as an intermediate in the synthesis of
numerous chemicals. At the present time, glycine is manufactured commercially
from the reaction of chloroacetic acid with ammonia. Since carbon dioxide is
one of the least expensive potential chemical feedstocks, economic routes for
producing chemicals from carbon dioxide such as glycine may have an advantage
over current conventional routes. Another important chemical is ethylene
glycol, which may be produced using oxalic acid as a raw material. Ethylene
glycol is used in billions of pounds per year in antifreeze and in making
bottles
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
made from polyethylene terephthalate, which is used in bottled consumer
drinks.
[00186] Referring to Fig. 19, a schematic illustrating a system in a block
diagram
format for the conversion of carbon dioxide to glycine is shown. The system
may include Unit A which includes an electrochemical cell for the conversion
of
a carbon dioxide feed to an oxalate salt which then passes onto Unit B,
converting the oxalate salt to oxalic acid, and then passing the oxalic acid
to
Unit C, where the oxalic acid is electrochemically reduced to glyoxylic acid,
and
passing the glyoxylic acid to a reductive annination unit, with the addition
of
ammonia and hydrogen, which is converted to glycine as a product. The solvent
and intermediates passed between the various units as well any purification
steps in FIG. 19 are not shown.
[00187] Referring to Fig. 20, a schematic illustrating an electrochemical cell
for
reducing oxalic acid to produce a glyoxylic acid product is shown. The
electrochemical cell includes of an anolyte compartment containing an anode, a
catholyte compartment containing a cathode, and a membrane or separator
separating the two compartments. An oxalic acid feed is fed into the catholyte
compartment of the electrochemical cell and is reduced at the cathode to
glyoxylic acid. The glyoxylic product is separated from the recirculating
catholyte stream in the catholyte disengager, which also separates any gaseous
byproduct gases from the reduction reaction. The catholyte solution is
recirculated back to the catholyte compartment using a catholyte recirculation
pump. The catholyte solution may include various selected acids, which may be
inorganic or organic acids in an aqueous or nonaqueous based solution.
[00188] The catholyte compartment includes of a cathode structure including of
a cathode current collector or distributor, and a high surface area cathode
made from metals, alloys, and electrocatalyst coatings on metals that are
suitable for the efficient reduction of glyoxylic acid to glycine. These
include,
71
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
but are not limited to metals such as Cd, Pb, Hg, Bi, Sb, PbSb, and their
alloys
and oxides.
[00189] The anolyte compartment includes of an anode and an optional high
surface area structure. The anode reaction may generate oxygen when using
mineral acids such as sulfuric, phosphoric, and nitric acid as examples, or
may
use organic acids that are stable to the anode reaction in producing oxygen
such
as nnethanesulfonic acid. The anode materials suitable for these acid anolytes
are coatings of precious metal oxides such as ruthenium and iridium oxides, as
well as platinum and gold and their combinations as metals and oxides on valve
metal substrates such as titanium, tantalum, or niobium.
[00190] The anode materials for this electrochemical cell are similar to those
employed in the electrochemical cells producing oxalate as disclosed in this
application, such as the electrochemical cell shown in Figs. 1-3, which
include
carbon materials such as carbon and graphite, which may be in the form of
felts, needled felts, or woven forms. These carbon based materials may have
catalysts impregnated into and onto the surfaces of the high surface area
carbon
structure includes platinum group metals and their oxides, mixtures, and
alloys,
such as gold, platinum, ruthenium dioxide, iridium oxide, and the like that
preferably may be chemically resistant to the anode bromine formation
chemistry and may help to promote or catalyze the oxidation of bromide to
bromine. Other suitable anode materials may be valve metals, such as titanium,
niobium, and tantalum having an electrocatalyst surface coating of the various
precious metal group metals and their oxides, mixtures, and their alloys.
These
anode materials are used when producing halogens in the anolyte compartment
when employing solutions containing acid halides such as HCl and HBr, thus
generating chlorine or bromine respectively as the product from the anode
compartment.
72
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
[00191] The separators and membrane materials used for the glyoxylic acid
reduction cell are also the same types as used in the oxalate generating cells
as
disclosed in this application.
[00192] Referring to Fig. 21, a schematic illustrating a reductive annination
batch reactor system for the conversion of glyoxylic acid to glycine is shown,
including a batch reactor system giving the feed inputs, the recycle streams,
and purification of the final glycine product. The glycine separation from the
solvent may be done by various processes, such as distillation or by
extractive
distillation.
[00193] In one embodiment, the reductive annination of oxalic acid to glycine
is
preferably conducted in a reactor. The proposed chemical reaction for the
reductive annination is as follows:
OHCCOOH + NH3 + H2 NH2CH2COOH + H20 (18)
[00194] The glyoxylic acid product generated utilizing the electrochemical
cell
as shown in Fig. 20 is fed to a reactor containing a water soluble organic
solvent, water, and a selected hydrogenation catalyst. Ammonia and hydrogen
are then metered into the reactor in specific molar ratios, and the reaction
proceeds with hydrogen pressure being kept constant, by the addition of more
hydrogen, during the reaction while it is being consumed.
[00195] The ammonia may be employed either as aqueous ammonia or as liquid
ammonia. The ammonia may be used in a molar excess of that theoretical
required (reaction 18), in the molar ratio range of 1.2 to 20 as NH3 :
glyoxylic
acid, or more preferably in a range of 1.5 to 10, and more preferably in a
range
of 2 to 5.
[00196] A number of water-soluble organic solvents may be used in the reactor
to keep the reactants and products in the reactor in solution. These solvents
73
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
should preferably be nonreactive toward the glyoxylic acid and the glycine
product and easily recoverable from the glycine product in the post separation
and purification steps.
[00197] Suitable solvents include alcohols, including methanol, ethanol,
Isopropanol, and tertiary butanols, as well as other water miscible solvents
such
as 1,4-dioxane, tetrahydrofuran, piperidine, and their mixtures. Methanol and
ethanol may be preferred solvents.
[00198] The ratio of water-soluble organic solvent to water is calculated to
keep
the reactants and reaction products in solution and promote the conversion of
the glyoxylic acid to glycine. When methanol is used as the organic solvent, a
solvent mixture including about 60% methanol and 40% water may be used. The
amount of methanol may range from about 20 to about 70% by weight methanol
in the methanol water solvent mixture.
[00199] The reductive annination reaction reactor also employs a suitable
hydrogenation catalyst. Suitable catalysts are the platinum group metals and
several transition metals, including ruthenium, rhodium, nickel, palladium,
platinum, osmium, and their alloys and mixtures. Rhodium has been found as an
effective catalyst in producing high yields of glycine. The catalyst may be in
the
form of particles of the elemental material deposited on a carrier substrate
such
as carbon, silica, and alumina. The catalyst may be in the form of a slurry
suspension, as may be used in a batch reactor, or in the form of a fixed
catalyst
bed, as used in a continuous reactor, which would be preferred.
[00200] The annination reactor may be a batch or continuous type where the
reactants are placed in a pressure vessel equipped or associated with means
for
vigorous stirring and agitation as well as for the introduction of hydrogen
gas
under the desired pressure within the reactor. The reaction pressures may
range
from 20 psig to 3,000 psig depending on the catalyst and temperature
conditions
employed.
74
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
[00201] The reduction annination reaction temperatures may range from 0 C to
100 C, and preferably in the range of 10 C to 60 C, and more preferably from
about 15 C to 40 C by changing the reaction temperatures, catalyst selection,
or the solvents used in the reaction. The preferred conversion yield is 80% or
greater, and more preferably 90% or greater.
Experimental Results
[00202] Below is a summary of results in determining the products of the
electrochemical reduction of oxalic acid using various cathode materials.
[00203] In this group of tests, the electrochemical reduction of oxalic acid
in
batch reactors with various cathode materials was studied. Best Faradaic
yields
(FY) for the production of glycolic and glyoxylic acids were 60% and 75%,
respectively, for the direct reduction of oxalic acid. A FY of 95% was also
achieved for the reduction of glyoxylic acid to glycolic acid. Mono-ethylene
Glycol (MEG) was observed with Cd cathode in trace amounts. The most
influential factors to the Faradaic yields of glycolic and glyoxylic include
cathode material, potential, and temperature while any electrolyte influence
was found to be negligible. No factors affecting ethylene glycol yields have
been discovered as no systems producing meaningful quantities of MEG are yet
known.
Preparation of the cathodes
[00204] To obtain the reproducible surface, the cathode materials were cleaned
by a standard procedure. In a typical experiment, - 2 cnn2 metal pieces was
cut
from the bulk metal and polished by alumina powder (0.3pnn), rinsed with
deionized water and degreased by acetone. Hence cleaned electrode was
sonicated for 2 minutes in deionized water. Prior to use, the electrode was
dried by pressing in between clean Kinnwipes.
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
Bulk Electrolysis
[00205] Electrolysis was performed using an Arbin MSTAT 167563 Potentiostat
(Arbin Instruments) at constant potentials. Unless otherwise noted, all
electrolysis was carried out in a three chambered glass cell with separated
cathode and anode chambers. For the electrolysis, that involved mercury
cathode, a two chambered electrochemical set up was used, with a glass frit
separated cathode chamber. An insulated copper wire was used to establish the
connection between the liquid mercury on the cathode and the potentiostat.
Various forms of carbon were tested for the anode compatibility.
Analysis of the products
[00206] Glycolic and glyoxylic quantitation was performed by Ion
Chromatography (IC). Other analytes were quantified by NMR. Analytical
samples for NMR were prepared as such: 800uL of sample, 100uL of D20, and
100uL of 1000ppnn acetone in H20. They were run for H1 using solvent
suppression. Quantitation was based on relative peak areas using response
factors calculated from prepared standard mixes. In the tables below, Ox
refers
to oxalic acid.
Table 12 Experimental results with system producing glyoxylate as major
product
Expt. Catho Electroly Reacta Volta Temperat Glyoxyla Glycola
de te nt ge Vs, ure te FY% te FY%
(SCE) ( C)
GL106 Cd 0.5MHBr 10%Ox -0.8 RT 75.36 45.82
GL121 Pb 0.5MHCl 7.5%Ox -1.2 40 41.06 5.75
GL127 Pb 0.5M HBr 7.5%Ox -1.2 60 69.16 17.30
GL134 Cd 0.5M HBr 7.5%Ox -1.2 40 63.26 9.15
GL143 Hg 0.5M HBr 7.5%Ox -1.2 RT 54.68 23.17
76
CA 02950294 2016-11-24
WO 2015/184388
PCT/US2015/033378
Table 13 Experimental results with system producing Glycolate as major
product
Expt. Catho Electroly Reactan Voltag Temperatu Glycol Glyoxyl
de te t e Vs, re ic ic FY
(SCE) ( C) FY(%) (%)
GL116 Bi*a 0.5MHBr 7.5%Gly -0.9 RT 95.63 N/A
Ox
GL120 Bi 0.5MHBr 7.5%Ox -1 40 43.42 41.52
GL122 Cd 0.5MHCl 7.5%Ox -1 40 59.84 20.85
GL123 Cd*b 0.5M 7.5%Ox -1.2 40 48.12 12.71
H2SO4
GL126 Cd*c 0.5M 7.5%Ox -1.4 60 59.01 17.69
H2SO4
GL128 Cd 0.5M HBr 7.5%Ox -1.2 60 38.33 18.35
GL131 Bi 0.5M HCl 7.5%Ox -1.2 60 49.22 23.61
GL132 Cd 0.5M HCl 7.5%Ox -1.2 40 57.11 24.47
GL148 Cd* d 0.25M 2%Ox -1.4 RT 24.25 #VALUE
NaBr !
*Cathodes that made more reduced products
a 0.065% EG, b 0 .17% EG, C 0.19%EG; 4.98% glycolaldehyde, d 0.23% EG
Table 14 Experimental results obtained from the EC reduction of Oxalic or
glycolic acid which lead to highly reduced products
Expt Catho Electroly Reacta Volta Temperat Other Compound
. de te nt ge Vs, ure FY( %)
(SCE) ( C)
GL14 Cd 0.5M 7.5%Ox -1.2 40 0.04 MEG
9 H2SO4
GL15 Cd 0.5M 7.5%Ox -1.4 60 0.04 MEG
77
CA 02950294 2016-11-24
WO 2015/184388
PCT/US2015/033378
0 H2SO4
GL16 Graphi 0.5M HBr 7.5% -1 RT 3.27 Acetic
7 te rod glycolic
GL17 Hg 0.5M HBr 7.5% -1 RT 15.91 ? MEG (
2 glycolic need to
confirm)
GL17 Ru 0.5M HBr 7.5% -1.2 60 12.53 Acetic
6 glycolic
GL17 Co 0.5M HBr 7.5% -1.2 60 15.09 glycoaldeh
7 glycolic yde
GL17 Glassy 0.5M HBr 7.5% -1.0 60 1.02 Acetic
1 Carbon glycolic
Table 15 Experimental results obtained by the electrochemical reduction of
oxalic acid on metal alloys cathodes
Expt. Cathod Electroly Reacta Volta Temperat Glyoxyla Glycolat
e te nt ge Vs, ure te FY%
e FY%
(SCE) ( C)
GL108 AgSn 0.5M HBr 7.5%Ox -0.9 RT 2.1 1.7
GL109 AgSn 0.5M HBr 7.5%Ox -1.0 RT 5.6 0.6
GL146 PbSn 0.5M HBr 7.5%Ox -1.2 RT 25.1 7.8
GL147 PbSn 0.5M HBr 7.5%Ox -1.2 RT --- 6.7
GL118 PbSb 0.5M HBr 7.5%Ox -1.3 RT 56.4 2.0
GL140 Galinst 0.5M HBr 7.5%Ox -1.6 RT
33.5 21.5
an
78
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
Table 16 Electrochemical systems with not any products observed
EXP Cathode Electrolyte Reactant Voltage vs. Temperature
SCE
GL152 Cu 0.5M HBr 7.5% glycolic -1 23
GL154 Pb 0.5M HBr 7.5% glycolic -1.2 23
GL155 Pd 0.5M HBr 7.5% glycolic -1.2 60
GL158 Pd 0.5M HBr 7.5% glycolic -1 23
GL160 Cu 0.5M HBr 7.5% glycolic -1.2 60
GL161 Cd 0.5M HBr 7.5% glycolic -1 60
GL162 Pt 0.5M HBr 7.5% glycolic -1 60
GL163 Sn 0.5M HBr 7.5% glycolic -1 60
GL174 Pb 0.5M HBr 7.5% glycolic -1 60
GL175 Graphite 0.5M HBr 7.5% glycolic -1.2 60
rod
GL179 Bi 0.5M HBr 7.5% glycolic -1.2 80
GL180 Cd 0.5M HBr 7.5% glycolic -1.2 80
GL181 Ru 0.5M HBr 7.5% glycolic -1 23
GL182 Mo 0.5M HBr 7.5% glycolic -1.2 23
GL183 Mo 0.5M HBr 7.5% glycolic -1 60
GL184 Bi 0.5M HBr 7.5% glycolic -1.2 60
GL185 Cd 0.5M HBr 7.5% glycolic -1.2 60
[00207] Alternative anolyte solutions may be employed to generate chemical
products such as bromine at the anode region of electrochemical cell 110,
which
may be used to bronninate organics as intermediates in making ethanol,
ethylene, and other chemicals based on bromine chemistry. The oxidation of
sulfur compounds in the anolyte region, such as sodium sulfide or S02 or the
direct or indirect oxidation of organics, and conducting the partial oxidation
of
organics, such as methanol to formaldehyde, are also contemplated.
79
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
[00208] Various alkali metal hydroxides may be employed at the
electrochemical cell 110 and/or a thermal reactor 120, 130. For example,
hydroxides of lithium, sodium, potassium, and rubidium, and cesium may be
used. Further, alkaline earth metal hydroxides may also be used.
[00209] Thermal reactors 120, 130 may perform thermal intermolecular
condensation reactions using alkali metal hydroxides. Such condensation
reactions may include chemical reactions in which two molecules or moieties
(functional groups) combine to form one single molecule, together with the
loss
of a small molecule. When two separate molecules may be reacted, the
condensation may be termed intermolecular. Since the reaction occurs at
elevated temperatures, the reactions may be characterized as a "thermal
intermolecular condensation step". If water may be lost, the reactions may be
characterized as "thermal intermolecular dehydration step". These reactions
may occur in an aqueous solution phase, such as with the reaction of CO with
the alkali metal hydroxide, or as a melt of the alkali metal carboxylic acid
and
the alkali metal hydroxide in the thermal reaction.
[00210] Thermal reactors 120, 130 may operate at about 40 to 500 C, and more
preferably at about 50 - 450 C. The operating temperatures may depend on the
decomposition temperatures of the carboxylic acid and the optimum
temperature to get the highest yields of the carboxylic product. A residence
time of the reaction at optimum reaction temperatures may range from 5
seconds to hours, and the equipment chosen to conduct the reaction may be
designed to provide the rate of heating and cooling required to obtain optimal
conversion yields. This may include the use of cold rotating metal that may
rapidly chill the hot thermal product after the thermal reaction period may be
completed.
[00211] Thermal reactors 120, 130 may operate in air or an enriched oxygen
atmospheres, as well as inert gas atmospheres, such as nitrogen, argon, and
helium. Carbon dioxide and hydrogen atmospheres may also be employed to
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
obtain the highest yield in the reaction, as well as partial CO atmospheres.
Thermal reactors 120, 130 may be operated under a full or partial vacuum.
[00212] The use of CO from other sources, such as from the production of
syngas
from methane or natural gas reforming may be employed. CO may also come
from other sources, such as process waste streams, where may be it separated
from carbon dioxide.
[00213] Alkali metal hydroxide concentration ranges may be 2% to 99%, more
preferably 5 to 98% by weight. The alkali hydroxide may run in molar excess of
the alkali metal carboxylic acid being thermally processed in the initial
reaction
mix or in a continuous process where they may be mixed together. The
anticipated molar ratios of the alkali metal carboxylic acid to alkali metal
hydroxide may range from 0.005 to 100, and more preferably 0.01 to 50. It may
be preferable to use the least amount of alkali metal hydroxide as possible
for
the reaction to reduce the consumption of the hydroxide in the process.
[00214] The process operating equipment that may be employed for thermal
reactors 120, 130 may include various commercially available types. For the CO
reaction with alkali metal hydroxide, the equipment that may be used may be
batch operation equipment, where gas may be injected into a solution mix of
the alkali hydroxide. This may also be done in a continuous manner where there
may be a feed input of fresh alkali metal hydroxide into a continuous stirred
tank reactor (CSTR) with a CO feed into the solution through a gas diffuser
into
the solution. Alternatively, counter-current packed towers may be used where
CO may be injected into the tower counter-current to the flow of alkali metal
hydroxide.
[00215] For an alkali metal oxalate operation, thermal reactors 120, 130 may
include equipment such as rotary kilns, and single pass plug flow reactors
that
may be used if the process required the thermal processing of a mixture of
alkali metal formate and alkali hydroxide as a solid or hot melt mix.
Preferably,
81
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
the equipment would be operated in a continuous fashion, providing the
required residence time for the reaction to go to completion at the selected
temperatures, which may be followed by a cooling section.
[00216] A thermal intermolecular condensation process may also be conducted
to produce higher carbon content carboxylic acids as well as converting the
carboxylic acids into esters, amides, acid chlorides, and alcohols. In
addition,
the carboxylic acid products may be converted to the corresponding halide
compounds using bromine, chlorine, and iodine.
[00217] Catalysts for the thermal conversion of the alkali metal formate may
include various bases, including alkali metal hydroxide as well as other
compounds that are bases. In addition, alkali metal and other hydrides may be
used, since they also act as bases. Any other suitable catalysts that may be
compatible with the formates in the calcination and provide high conversion
yields are suitable for the process.
[00218] It is contemplated that the structure and operation of the
electrochemical cells described in the present disclosure, such as
electrochemical cell 110, may be adjusted to provide desired results.
[00219] For example, the electrochemical cell may operate at higher pressures,
such as pressure above atmospheric pressure which may increase current
efficiency and allow operation of the electrochemical cell at higher current
densities.
[00220] Additionally, the cathode and anode of the electrochemical cell may
include a high surface area electrode structure with a void volume which may
range from 30% to 98%. The electrode void volume percentage may refer to the
percentage of empty space that the electrode is not occupying in the total
volume space of the electrode. The advantage in using a high void volume
electrode is that the structure has a lower pressure drop for liquid flow
through
82
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
the structure. The specific surface area of the electrode base structure may
be
from 2 cnn2/cnn3 to 500 cnn2/cnn3 or higher. The electrode specific surface
area is
a ratio of the base electrode structure surface area divided by the total
physical
volume of the entire electrode. It is contemplated that surface areas also may
be defined as a total area of the electrode base substrate in comparison to
the
projected geometric area of the current distributor/conductor back plate, with
a preferred range of 2x to 1000x or more. The actual total active surface area
of the electrode structure is a function of the properties of the electrode
catalyst deposited on the physical electrode structure which may be 2 to 1000
times higher in surface area than the physical electrode base structure.
[00221] Cathode may be selected from a number of high surface area materials
to include copper, stainless steels, transition metals and their alloys,
carbon,
and silicon, which may be further coated with a layer of material which may be
a conductive metal or semiconductor. The base structure of cathode may be in
the form of fibrous, metal foams, reticulated, or sintered powder materials
made from metals, carbon, or other conductive materials including polymers.
The materials may be a very thin plastic screen incorporated against the
cathode side of the membrane to prevent the membrane from directly touching
the high surface area cathode structure. The high surface area cathode
structure may be mechanically pressed against a cathode current distributor
backplate, which may be composed of material that has the same surface
composition as the high surface area cathode.
[00222] In addition, cathode may be a suitable conductive electrode, such as
Al,
Au, Ag, Bi, C, Cd, Co, Cr, Cu, Cu alloys (e.g., brass and bronze), Ga, Hg, In,
Mo,
Nb, Ni, NiCo204, Ni alloys (e.g., Ni 625, NiHX), Ni-Fe alloys, Pb, Pd alloys
(e.g.,
PdAg), Pt, Pt alloys (e.g., PtRh), Rh, Sn, Sn alloys (e.g., SnAg, SnPb, SnSb),
Ti,
V, W, Zn, stainless steel (SS) (e.g., SS 2205, SS 304, SS 316, SS 321),
austenitic
steel, ferritic steel, duplex steel, nnartensitic steel, Nichronne (e.g., NiCr
60:16
(with Fe)), elgiloy (e.g., Co-Ni-Cr), degenerately doped n-Si, degenerately
doped n-Si:As, degenerately doped n-Si:B, degenerately doped n-Si,
83
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
degenerately doped n-Si:As, and degenerately doped n-Si:B. Other conductive
electrodes may be implemented to meet the criteria of a particular
application.
For photoelectrochennical reductions, cathode 122 may be a p-type
semiconductor electrode, such as p-GaAs, p-GaP, p-InN, p-InP, p-CdTe, p-GaInP2
and p-Si, or an n-type semiconductor, such as n-GaAs, n-GaP, n-InN, n-InP, n-
CdTe, n-GaInP2 and n-Si. Other semiconductor electrodes may be implemented
to meet the criteria of a particular application including, but not limited
to,
CoS, Mo52, TiB, W52, SnS, Ag25, CoP2, Fe3P, Mn3P2, MoP, Ni2Si, MoSi2, W5i2,
CoSi2, Ti407, 5n02, GaAs, GaSb, Ge, and CdSe.
[00223] The catholyte may include a pH range from 1 to 12, preferably from pH
4 to pH 10. The selected operating pH may be a function of any catalysts
utilized in operation of the electrochemical cell. Preferably, catholyte and
catalysts may be selected to prevent corrosion at the electrochemical cell.
The
catholyte may include homogeneous catalysts. Homogeneous catalysts are
defined as aromatic heterocyclic amines and may include, but are not limited
to, unsubstituted and substituted pyridines and innidazoles. Substituted
pyridines and innidazoles may include, but are not limited to mono and
disubstituted pyridines and innidazoles. For example, suitable catalysts may
include straight chain or branched chain lower alkyl (e.g., C1-C10) mono and
disubstituted compounds such as 2-nnethylpyridine, 4-tertbutyl pyridine, 2,6
dinnethylpyridine (2,6-lutidine); bipyridines, such as 4,4'-bipyridine; amino-
substituted pyridines, such as 4- dinnethylannino pyridine; and hydroxyl-
substituted pyridines (e.g., 4-hydroxy-pyridine) and substituted or
unsubstituted
quinoline or isoquinolines. The catalysts may also suitably include
substituted or
unsubstituted dinitrogen heterocyclic amines, such as pyrazine, pyridazine and
pyrinnidine. Other catalysts generally include azoles, innidazoles, indoles,
oxazoles, thiazoles, substituted species and complex multi-ring amines such as
adenine, pterin, pteridine, benzinnidazole, phenonthroline and the like.
[00224] The catholyte may include an electrolyte. Catholyte electrolytes may
include alkali metal bicarbonates, carbonates, sulfates, phosphates, borates,
84
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
and hydroxides. Non-
aqueous electrolytes, such as propylene carbonate,
nnethanesulfonic acid, methanol, and other ionic conducting liquids may be
used
rather than water and using salt addition electrolytes such as alkali metal
salts.
The electrolyte may include one or more of Na2SO4, KCl, NaNO3, NaCl, NaF,
NaCl04, KCl04, K2SiO3, CaCl2, a guanidiniunn cation, a H cation, an alkali
metal
cation, an ammonium cation, an alkylannnnoniunn cation, a tetraalkyl ammonium
cation, a halide anion, an alkyl amine, a borate, a carbonate, a guanidiniunn
derivative, a nitrite, a nitrate, a phosphate, a polyphosphate, a perchlorate,
a
silicate, a sulfate, and a hydroxide.
[00225] The catholyte may further include an aqueous or non-aqueous solvent.
An aqueous solvent may include greater than 5% water. A non-aqueous solvent
may include as much as 5% water. A solvent may contain one or more of water,
a protic solvent, or an aprotic polar solvent. Representative solvents include
methanol, ethanol, acetonitrile, propylene carbonate, ethylene carbonate,
dinnethyl carbonate, diethyl carbonate, dinnethylsulfoxide,
dinnethylfornnannide,
acetonitrile, acetone, tetrahydrofu ran, N,N -
dinnethylacetanninde,
dinnethoxyethane, diethylene glycol dinnethyl ester, butyrolnitrile, 1,2-
difluorobenzene, y-butyrolactone, N-methyl-2-pyrrolidone, sulfolane, 1,4-
dioxane, nitrobenzene, nitronnethane, acetic anhydride, ionic liquids, and
mixtures thereof.
[00226] In one embodiment, a catholyte/anolyte flowrate may include a
catholyte/anolyte cross sectional area flow rate range such as 2 - 3,000
gpnn/ft2
or more ( 0.0076 - 11.36 m3/m2). A flow velocity range may be 0.002 to 20
ft/sec ( 0.0006 to 6.1 nn/sec). Operation of the electrochemical cell
catholyte
at a higher operating pressure allows more dissolved carbon dioxide to
dissolve
in the aqueous solution.
Typically, electrochemical cells can operate at
pressures up to about 20 to 30 psig in multi-cell stack designs, although with
modifications, the electrochemical cells may operate at up to 100 psig. The
electrochemical cell may operate the anolyte and the catholyte at the same
pressure range to minimize the pressure differential on a separator 120 or
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
membrane separating the two regions. Special electrochemical designs may be
employed to operate electrochemical units at higher operating pressures up to
about 60 to 100 atmospheres or greater, which is in the liquid CO2 and
supercritical CO2 operating range.
[00227] In another embodiment, a portion of a catholyte recycle stream may be
separately pressurized using a flow restriction with backpressure or using a
pump, with CO2 injection, such that the pressurized stream is then injected
into
the catholyte region of the electrochemical cell which may increase the amount
of dissolved CO2 in the aqueous solution to improve the conversion yield. In
addition, nnicrobubble generation of carbon dioxide can be conducted by
various
means in the catholyte recycle stream to maximize carbon dioxide solubility in
the solution.
[00228] catholyte may be operated at a temperature range of -10 to 95 C,
more preferably 5 - 60 C. The lower temperature will be limited by the
catholytes used and their freezing points. In
general, the lower the
temperature, the higher the solubility of CO2 in an aqueous solution phase of
the catholyte, which would help in obtaining higher conversion and current
efficiencies. The drawback is that the operating electrochemical cell voltages
may be higher, so there is an optimization that would be done to produce the
chemicals at the lowest operating cost. In addition, the catholyte may require
cooling, so an external heat exchanger may be employed, flowing a portion, or
all, of the catholyte through the heat exchanger and using cooling water to
remove the heat and control the catholyte temperature.
[00229] Anolyte operating temperatures may be in the same ranges as the
ranges for the catholyte, and may be in a range of 0 C to 95 C. In addition,
the
anolyte may require cooling, so an external heat exchanger may be employed,
flowing a portion, or all, of the anolyte through the heat exchanger and using
cooling water to remove the heat and control the anolyte temperature.
86
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
[00230] Electrochemical cells may include various types of designs. These
designs may include zero gap designs with a finite or zero gap between the
electrodes and membrane, flow-by and flow-through designs with a recirculating
catholyte electrolyte utilizing various high surface area cathode materials.
The
electrochemical cell may include flooded co-current and counter-current
packed and trickle bed designs with the various high surface area cathode
materials. Also, bipolar stack cell designs and high pressure cell designs may
also be employed for the electrochemical cells.
[00231] Anode electrodes may be the same as cathode electrodes or different.
For sulfur dioxide and hydrogen sulfide anode oxidation chemistry under acid
conditions, the preferred electrocatalytic coatings may include precious metal
oxides such as ruthenium and iridium oxides, as well as platinum and gold and
their combinations as metals and oxides on valve metal substrates such as
titanium, tantalum, zirconium, or niobium. Carbon and graphite may also be
suitable for use as anodes in addition to boron-doped diamond films on metal
or
other electrically conductive substrates. For other sulfur based reactants in
the
anolyte such as sodium sulfide or hydrogen sulfide being oxidized under
alkaline
conditions, such as in a hydroxide containing electrolyte, selected anode
materials may include carbon, transition metals, transitional metal oxides
carbon steel, stainless steels, and their alloys and combinations which are
stable
as anodes. Anode may include electrocatalytic coatings applied to the surfaces
of the base anode structure. Anolytes may be the same as catholytes or
different. The anolyte electrolytes may be the same as catholyte electrolytes
or different. The anolyte may include solvent. The anolyte solvent may be the
same as catholyte solvent or different. For
example, for acid anolytes
containing S02 as the sulfur-based reactant, the preferred electrocatalytic
coatings may include precious metal oxides such as ruthenium and iridium
oxides, as well as platinum and gold and their combinations as metals and
oxides on valve metal substrates such as titanium, tantalum, zirconium, or
niobium. For
other anolytes, including alkaline or hydroxide electrolytes,
anodes may include carbon, cobalt oxides, stainless steels, transition metals,
87
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
and their alloys, oxides, and combinations. High surface area anode structures
that may be used which would help promote the reactions at the anode. The
high surface area anode base material may be in a reticulated form composed of
fibers, sintered powder, sintered screens, and the like, and may be sintered,
welded, or mechanically connected to a current distributor back plate that is
commonly used in bipolar cell assemblies. In addition, the high surface area
reticulated anode structure may also contain areas where additional applied
catalysts on and near the electrocatalytic active surfaces of the anode
surface
structure to enhance and promote reactions that may occur in the bulk solution
away from the anode surface such as the introduction of SO2 into the anolyte.
The anode structure may be gradated, so that the suitable of the may vary in
the vertical or horizontal direction to allow the easier escape of gases from
the
anode structure. In this gradation, there may be a distribution of particles
of
materials mixed in the anode structure that may contain catalysts, such as
transition metal based oxides, such as those based on the transition metals
such
as Co, Ni, Mn, Zn, Cu and Fe as well as precious metals and their oxides based
on platinum, gold, silver and palladium which may be deposited on inorganic
supports within cathode compartment space or externally, such as in the second
product extractor or a separate reactor.
[00232] A separator of the electrochemical cell, also referred to as a
membrane,
may be placed between an anode region and a cathode region of the
electrochemical cell. Separator may include cation ion exchange type
membranes. Cation ion exchange membranes which have a high rejection
efficiency to anions may be preferred. Examples of such cation ion exchange
membranes may include perfluorinated sulfonic acid based ion exchange
membranes such as DuPont Nafion brand unreinforced types N117 and N120
series, more preferred PTFE fiber reinforced N324 and N424 types, and similar
related membranes manufactured by Japanese companies under the supplier
trade names such as AGC Engineering (Asahi Glass) under their tradenanne
Flennion . Other multi-layer perfluorinated ion exchange membranes used in the
chlor alkali industry may have a bilayer construction of a sulfonic acid based
88
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
membrane layer bonded to a carboxylic acid based membrane layer, which
efficiently operates with an anolyte and catholyte above a pH of about 2 or
higher. These membranes may have a higher anion rejection efficiency. These
are sold by DuPont under their Nafion trademark as the N900 series, such as
the N90209, N966, N982, and the 2000 series, such as the N2010, N2020, and
N2030 and all of their types and subtypes. Hydrocarbon based membranes,
which are made from of various cation ion exchange materials can also be used
if a lower the anion rejection efficiency is not as important, such as those
sold
by Sybron under their trade name lonac , AGC Engineering (Asahi Glass) under
their trade name under their Selennion trade name, and Tokuyanna Soda,
among others on the market. Ceramic based membranes may also be employed,
including those that are called under the general name of NASICON (for sodium
super-ionic conductors) which are chemically stable over a wide pH range for
various chemicals and selectively transports sodium ions, the composition is
Na1-FxZr2SixP3-x012, and well as other ceramic based conductive membranes
based on titanium oxides, zirconium oxides and yttrium oxides, and beta
aluminum oxides. Alternative membranes that may be used are those with
different structural backbones such as polyphosphazene and sulfonated
polyphosphazene membranes in addition to crown ether based membranes.
Preferably, the membrane or separator is chemically resistant to the anolyte
and catholyte.
[00233] A rate of the generation of reactant formed in the anolyte compartment
and the catholyte compartment may be proportional to the applied current to
the electrochemical cell. The operation of an extractor and its selected
separation method, for example fractional distillation or packed tower
scrubbing, the actual products produced, and the selectivity of the wanted
reaction would determine the optimum molar ratio of the reactant to the
generated reactant.
[00234] The electrochemical cell may be easily operated at a current density
of
greater than 3 kA/m2 (300 nnA/cnn2), or in suitable range of 0.5 to 5 kA/m2 or
89
CA 02950294 2016-11-24
WO 2015/184388 PCT/US2015/033378
higher if needed. The anode preferably has a high surface area structure with
a
specific surface area of 50 cnn2/cnn3 or more that fills the gap between the
cathode backplate and the membrane, thus having a zero gap anode
configuration. Metal and/or metal oxide catalysts may be added to the anode in
order to decrease anode potential and/or increase anode current density.
Stainless steels or nickel may also be used as anode materials with for sodium
sulfide oxidation under alkaline conditions.
[00235] It is understood that the specific order or hierarchy of steps in the
methods disclosed are examples of exemplary approaches. Based upon design
preferences, it is understood that the specific order or hierarchy of steps in
the
method can be rearranged while remaining within the disclosed subject matter.
The accompanying method claims present elements of the various steps in a
sample order, and are not necessarily meant to be limited to the specific
order
or hierarchy presented.
[00236] It is believed that the present disclosure and many of its attendant
advantages will be understood by the foregoing description, and it will be
apparent that various changes may be made in the form, construction and
arrangement of the components without departing from the disclosed subject
matter or without sacrificing all of its material advantages. The form
described
is merely explanatory, and it is the intention of the following claims to
encompass and include such changes.