Note: Descriptions are shown in the official language in which they were submitted.
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
FACTOR VIII MUTATION REPAIR AND TOLERANCE INDUCTION AND
RELATED cDNAs, COMPOSITIONS, METHODS AND SYSTEMS
CROSS REFERENCE TO RELATED APPLICATIONS
[0001] This application claims priority to U.S. Provisional Application
62/011,019, entitled
"Factor VIII mutation repair and tolerance induction" and filed on June 11,
2014, and is also
a continuation-in-part application of U.S. Non-Provisional Application No.
14/649,910, filed
on June 4, 2015, which, in turn, is a U.S. national stage entry of
International Patent
Application No. PCT/U52013/073751, filed on December 6, 2013, which, in turn,
claims
priority from U.S. Provisional Application No. 61/734,678, filed on December
7, 2012, and
U.S. Provisional Application No. 61/888,424, filed on October 8, 2013. All
such applications
are incorporated herein by reference in their entirety.
STATEMENT OF GOVERNMENT GRANT
[0002] The U.S. government has certain rights in the inventions pursuant to
Grant Nos grant
# 1R41MD008156-01A1 and 1R41MD008808-01 awarded by the National Institutes of
Health (NIH).
FIELD
[0003] The present disclosure relates to gene mutation repairs and related
materials, methods
and systems, and in particular relates to Factor VIII mutation repair and
tolerance induction
and related cDNAs compositions, methods and systems.
BACKGROUND
[0004] Factor VIII (FVIII) is a blood-clotting protein, also known as anti-
hemophilic factor
(AHF), encoded by a Factor VIII gene (F8 gene or F8).
[0005] Certain mutations in the F8 gene (F8) result in production of a
dysfunctional version
of the Factor VIII protein (qualitative deficiency), and/or in production of
Factor VIII in
insufficient amounts (quantitative deficiency) which cause hemophilia in
subjects having the
mutations.
1
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
[0006] Despite developments of various options to manage hemophilia,
prophylaxis and
treatment of hemophilia in subjects remains challenging.
SUMMARY
[0007] Provided herein are methods and systems and related cDNA,
polynucleotides,
vehicles and compositions which allow in several embodiments to selectively
target and
repair one or more mutations in the sequence of Factor VIII gene of a subject,
and in
particular the one or more mutations of the Factor VIII gene resulting in
hemophilia.
[0008] According to a first aspect, a method for repairing one or more
mutations in a Factor
VIII gene (F8 gene) sequence of a subject is described. The method comprises
introducing
into a cell of the subject one or more polynucleotides encoding a DNA scission
enzyme
(DNA-SE) such as a nuclease or nickase and one or more repair vehicles (RVs)
containing at
least a cDNA-repair sequence (RS) comprising a repaired version of the F8 gene
sequence of
the subject comprising the one or more mutations within a cDNA sequence
encoding for a
truncated Factor VIII.
The DNA-SE is selected to be capable of targeting a portion of the F8 gene of
the subject and
to create a first break in one strand of the F8 gene and a second break in the
other strand of
the F8 gene for subsequent repair by the cDNA-RS. The cDNA-RS is comprised in
each of
the one or more repair vehicles (RVs) flanked by an upstream flanking sequence
(uFS) and a
downstream flanking sequence (dFS) to form a DNA donor within the RVs. The
upstream
flanking sequence (uFS) is homologous to a nucleic acid sequence upstream of
the first break
in the one strand of the F8 gene and the downstream flanking sequence (dFS)
homologous to
a nucleic acid sequences downstream of the second break in the other strand of
the F8 gene.
In the method, introducing into a cell of the subject one or more
polynucleotides encoding a
DNA scission enzyme (DNA-SE) and one or more repair vehicles (cDNA-RS) is
performed
to allow insertion of the cDNA-RS through homologous recombination of the
upstream
flanking sequence (uFS) and the downstream flanking sequence (dFS) with the
subject's F8
gene (sF8) to provide a repaired F8 gene (rF8). In the method, the repaired F8
gene (rF8)
upon expression forms functional FVIII that confers improved coagulation
functionality to
the FVIII protein encoded by the sF8 without the repair.
[0009] According to a second aspect, a system for repairing one or more
mutations in a
2
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
Factor VIII gene (F8 gene) sequence of a subject is described. The system
comprises one or
more polynucleotides encoding a DNA scission enzyme (DNA-SE) herein described
and one
or more repair vehicles (RVs) herein described.
In the system, the DNA scission enzyme (DNA-SE), and the and one or more
repair vehicles
(RVs) are selected and configured so that upon insertion of the cDNA-RS
through
homologous recombination of the upstream flanking sequence (uFS) and the
downstream
flanking sequence (dFS) of the DNA donor sequence with the subject's F8 gene
(sF8) a
repaired F8 gene (rF8) is provided. In the system, the repaired F8 gene (rF8)
upon expression
forms functional FVIII that confers improved coagulation functionality to the
FVIII protein
encoded by the sF8 without the repair.
[0010] According to a third aspect, a cDNA is described configured to be used
as a cDNA-
RS in methods and systems of the disclosure for repairing one or more
mutations in a Factor
VIII gene (F8 gene) sequence of a subject. The cDNA encodes a truncated Factor
VIII
polypeptide consisting essentially of the amino acid sequence encoded by each
of exons 1, 2,
3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23,
24, 25, 26 of a F8 gene
or an in frame combination thereof In some embodiments, the each of the exons
has a
sequence of a corresponding exon in the F8 gene of the subject.
[0011] According to a fourth aspect a repair vehicle (RV) is described
configured to be used
in methods and systems of the disclosure for repairing one or more mutations
in a Factor VIII
gene (F8 gene) sequence of a subject. The repair vehicle is a polynucleotide
configured for
use in combination with a DNA scission enzyme (DNA-SE) selected to target a
portion of the
F8 gene of the subject and to create a first break in one strand of the F8
gene and a second
break in the other strand of the F8 gene. The repair vehicle comprises a cDNA-
repair
sequence (RS) comprising a repaired version of the F8 gene sequence of the
subject
comprising the one or more mutations within a cDNA sequence encoding for a
truncated
Factor VIII. In the repair vehicle (RV), the cDNA-RS is flanked by an upstream
flanking
sequence (uFS) and a downstream flanking sequence (dFS) to form a DNA donor
within the
RV. The upstream flanking sequence (uFS) is homologous to a nucleic acid
sequence
upstream of the first break in the one strand of the F8 gene and the
downstream flanking
sequence (dFS) homologous to a nucleic acid sequences downstream of the second
break in
the other strand of the F8 gene.
3
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
[0012] According to a fifth aspect a polynucleotide encoding a DNA scission
enzyme
(DNA-SE) is described configured for use in methods and systems of the
disclosure for
repairing one or more mutations in a Factor VIII gene (F8 gene) sequence of a
subject. The
DNA scission enzyme is selected to be capable of targeting a portion of the F8
gene of the
subject and to create a first break in one strand of the F8 gene and a second
break in the other
strand of the F8 gene for subsequent repair by the cDNA-RS.
[0013] According to a sixth aspect, a cell is described comprising one or more
repair vehicles
(RVs) herein described and one or more polynucleotide encoding a DNA scission
enzyme
(DNA-SE) herein described.
[0014] According to a seventh aspect, a composition for repairing one or more
mutations in a
Factor VIII gene (F8 gene) sequence of a subject is described. The composition
comprises
one or more polynucleotides encoding a DNA scission enzyme (DNA-SE) herein
described
and one or more repair vehicles (RVs) herein described together with a
suitable excipient. In
some embodiments, the composition is a pharmaceutical composition for
treatment of
hemophilia and/or promotion of immune tolerance to a Factor VIII replacement
protein in a
subject and the suitable excipient is a pharmaceutically acceptable excipient.
[0015] Methods and systems and related cDNA, polynucleotides, vehicles and
compositions
are expected in several embodiments to provide a repaired F8 gene and
corresponding
functional Factor VIII in a subject with hemophilia in a form and amount
remedying the
qualitative and/or quantitative deficiencies of the Factor VIII of the
subject, thus allowing
treatment of the hemophilia in the subject.
[0016] Methods and systems and related cDNA, polynucleotides, vehicles and
compositions
are expected in several embodiments to provide a repaired F8 and corresponding
functional
Factor VIII formed by sequences of the subject thus minimizing production of
Factor VIII
inhibitor in the subject.
[0017] Methods and systems and related cDNA, polynucleotides, vehicles and
compositions
are expected in several embodiments to provide a repaired F8 gene expressing a
functional
FVIII which allows inducing immune tolerance to a FVIII replacement product
((r)FVIII) in
a subject having a FVIII deficiency and who will be administered, is being
administered, or
has been administered a (r)FVIII product.
4
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
[0018] The methods and systems and related cDNA, polynucleotides, vehicles and
compositions herein described, can be used in connection with applications
wherein repair of
mutations in Factor VIII gene of a subject is desired, in particular in
connection with
treatment and/or prophylaxis of various forms of hemophilia and in particular
hemophilia A,
in subjects. Exemplary applications comprise medical applications, biological
analysis,
research and diagnostics including but not limited to clinical, therapeutic
and pharmaceutical
applications, and additional applications identifiable by a skilled person.
[0019] The details of one or more embodiments of the disclosure are set forth
in the
accompanying drawings and the description below. Other features and objects
will be
apparent from the description and drawings, and from the appended claims.
BRIEF DESCRIPTION OF THE DRAWINGS
[0020] The accompanying drawings, which are incorporated into and constitute a
part of this
specification, illustrate one or more embodiments of the present disclosure
and, together with
the description of example embodiments, serve to explain the principles and
implementations
of the disclosure.
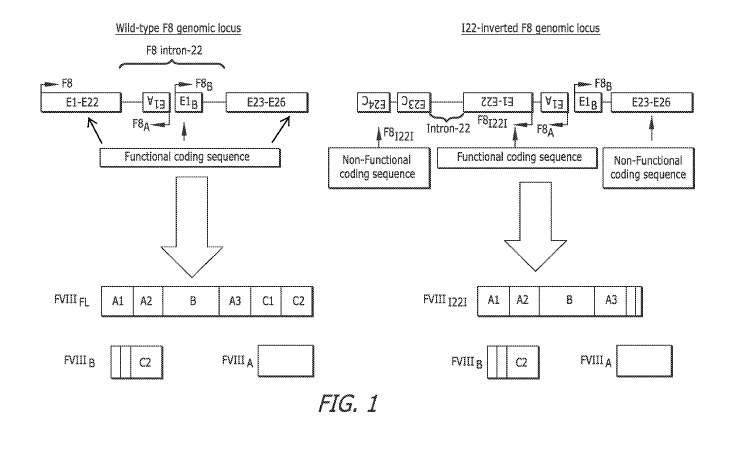
[0021] FIG. 1 is a schematic illustration of the wild-type and intron-22-
inverted FVIII loci
(F8 & F81221) and their expressed protein products (FVIIIFL & FVIIIB for F8
and FVIIII22I
& FVIIIB for F8I 221).
[0022] FIG. 2 is a schematic illustration of a TALEN-mediated genomic editing
that can be
used to repair the human intron-22 (I22)-inverted F8 locus, F81221.
[0023] FIG. 3 shows a functional heterodimeric TALEN, comprised of its left
and right
monomer subunits (TALEN-L and TALEN-R), targeting the human F8 gene.
[0024] FIG. 4 shows a functional heterodimeric TALEN, comprised of its left
and right
monomer subunits (TALEN-L and TALEN-R) targeting the canine F8 gene
[0025] FIG. 5 illustrates the TALEN approach linking Exon 22 of the F8 gene to
a nucleic
acid encoding a truncated FVIII polypeptide encoding exons 23-26.
[0026] FIG. 6 illustrates the TALEN approach linking Intron 22 to a F8 3'
splice acceptor
site operably linked to a nucleic acid encoding a truncated FVIII polypeptide.
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
[0027] FIG. 7 shows a comparison of expected genomic DNA, spliced RNA and
proteins pre
and post repair.
[0028] FIG. 8 shows PCR primer design to confirm correct integration of exons
23-26 to
repair the human intron-22 0221-inverted F8 locus, F81221.
[0029] FIG. 9 illustrates the donor plasmid targeting the F8 Exon22/Intron22
junction using
a TALEN, ZFN, CRISPR/Cas, CRISPR-PN, and CRISPR-RFN approach.
[0030] FIG. 10 illustrates the donor plasmid targeting the F8 Exonl/Intronl
junction using a
TALEN, ZFN, CRISPR/Cas, CRISPR-PN, and CRISPR-RFN approach.
[0031] FIG. 11 illustrates the donor plasmid targeting the F8 Intron 22 region
using a
TALEN, ZFN, CRISPR/Cas, CRISPR-PN, and CRISPR-RFN approach.
[0032] FIG. 12 illustrates the donor plasmid targeting the F8 Intron 1 region
using a TALEN,
ZFN, CRISPR/Cas, CRISPR-PN, and CRISPR-RFN approach.
[0033] FIG. 13 illustrates the CRISPR/Cas9-mediated F8 repair strategy
targeting intron 1.
[0034] FIG. 14 illustrates examples of severe HA-causing F8 mutations that can
be cured
with the exon-2 I targeted CasPN therapeutics of our personalized 3' gene
repair system.
[0035] FIG. 15 is a schematic diagram of exon-21 targeted, CasPN mediated
personalized
repair of the intron-22 inversion mutation (F8I221).
[0036] FIG. 16 is a schematic diagram of the repair vehicle, donor sequence
used in the
repair of FIG. 15.
[0037] FIG. 17 shows a series of graphs displaying results obtained from flow
cytometry
using CRISPR/Cas9 plasmids pH0007, pH0009 as well as a repair plasmid (labeled
as
"Donor").
[0038] FIG. 18 is an image of an agarose gel electrophoresis assay displaying
results from a
T7E1 assay done on cells transfected with CRISPR/Cas9 plasmids pH0007, pH0009,
pH0011
and pH0013.
[0039] FIG. 19 is a bar graph showing estimated NHEJ rates for CRISPR
constructs
6
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
pH0007, pH0009, pH0011 and pH0013.
[0040] FIG. 20 is an image of an agarose gel electrophoresis assay displaying
results from a
RFLP assay done on cells transfected with CRISPR/Cas9 plasmids pH0007, pH0009
as well
as a repair plasmid (labeled as "Donor").
[0041] FIG. 21 is a bar graph showing the percentage of homologous
recombination in cells
following Intron 22-targeted CRISPR treatment.
DETAILED DESCRIPTION
[0042] Provided herein are methods and systems and related cDNA,
polynucleotides,
vehicles and compositions which allow in several embodiments to selectively
target and
repair one or more mutations in the sequence of Factor VIII gene of a subject.
[0043] The term "Factor VIII" or "FVIII" as used herein indicates an essential
cofactor in the
blood coagulation pathway provided by a large plasma glycoprotein that
functions in the
blood coagulation cascade as a cofactor for the factor IXa-dependent
activation of factor X.
Factor VIII is tightly associated in the blood with von Willebrand factor
(VWF), which
serves as a protective carrier protein for factor VIII. In particular Factor
VIII circulates in the
bloodstream in an inactive form, bound to von Willebrand factor (VWF). Upon
injury, FVIII
is activated. The activated protein (FVIIIa) interacts with coagulation factor
IX, leading to
clotting as will be understood by a skilled person.
[0044] FVIII is encoded in a subject by a F8 gene containing 26 exons and
spanning 186 kb
(Gitschier, et al. Nature 314: 738-740, 1985). In human the F8 gene is located
in the X
chromosome. In some subjects (e.g. humans, monkeys, rats) the sequences F8
gene also
contains an F8A gene and an F8B gene within intron 22. The F8A gene is intron-
less, is
contained entirely in intron 22 of the F8 gene in reverse orientation to the
F8 gene, and is
therefore transcribed in the opposite direction to F8. The F8B gene is also
located in intron 22
and is transcribed in opposite direction from F8A gene; its first exon lies
within intron 22 and
is spliced to exons 23-26.
[0045] The term "orientation" with reference to a gene indicates the direction
of the 5' ¨> 3'
DNA strand which provides the sense strand in the double stranded
polynucleotide
comprising the gene. Accordingly, 5'->3' DNA strand is designated, for a given
gene, as
7
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
'sense', 'plus' or 'coding' strand when its sequence is identical to the
sequence of the
premessenger (premRNA), except for uracil (U) in RNA, instead of thymine (T)
in DNA. An
antisense strand is instead the 3'->5' strand complementary to the sense
strand in a double
stranded polynucleotide coding for the gene. The antisense transcribed by the
RNA
polymerase and is also designated as "template" DNA. Accordingly two genes or
sequences
thereof within the F8 genomic locus encoded by a same polynucleotide are in a
same
orientation when their respective sense strands are located on a same strand
of the
polynucleotide and are in in reverse or opposite orientation when respective
sense strands are
located on different strand of the polynucleotide. Accordingly two genes or
coding sequences
within the F8 genomic locus encoded by a same polynucleotide are in a same
orientation
when their respective sense strands are located on a same strand of the
polynucleotide. Two
genes or coding sequences within the F8 genomic locus are in reverse or
opposite orientation
when their respective sense strands are located on the opposing strand of the
polynucleotide.
[0046] FVIII is synthesized primarily in the liver of s subject and the
primary translation
product of 2332 amino acids undergoes extensive post-translational
modification, including
N- and 0-linked glycosylation, sulfation, and proteolytic cleavage. The latter
event divides
the initial multi-domain protein (A 1 -A2-B-A3-C1-C2) into a heavy chain (A 1 -
A2-B) and a
light chain (A3-C1-C2) and the protein is secreted as a two-chain molecule
associated
through a metal ion bridge (Lenting et al., The life cycle of coagulation
FVIII in view of its
structure and function. Blood 1998; 92: 3983-96).
[0047] Mutations in the F8 gene can result in production of a dysfunctional
version of the
Factor VIII protein (qualitative deficiency), and/or in production of Factor
VIII in insufficient
amounts (quantitative deficiency) causing hemophilia in subjects having the
mutations.
[0048] Accordingly, a Factor VIII is indicated as functional when it is
produced in a form
and an amount allowing a coagulation functionality comparable with the
coagulation
functionality of the wild type FVIII protein in a healthy subject. FVIII
function is evaluated
by routine clinical laboratory methods that are well established in the art
and apparent to one
of ordinary skill in the art (Barrowcliffe TW, Raut S, Sands D, Hubbard AR:
Coagulation and
chromogenic assays of factor VIII activity: general aspects, standardization,
and
recommendations. Semin Thromb Hemost 2002 Jun;28(3):247-256).
[0049] A non-functional Factor VIII instead indicates an FVIII protein
functioning aberrantly
8
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
or FVIII proteins present in circulating blood in a reduced or absent amount,
leading to the
reduction of or absence of the ability to clot in response to injury by the
subject. FVIII
function is evaluated by routine clinical laboratory methods that are well
established in the art
and apparent to one of ordinary skill in the art (Banowcliffe TW, Raut S,
Sands D, Hubbard
AR: Coagulation and chromogenic assays of factor VIII activity: general
aspects,
standardization, and recommendations. Semin Thromb Hemost 2002 Jun;28(3):247-
256).
[0050] Over 2100 different hemophilia A (HA)-causing mutations have thus far
been
identified in the F8 loci of unrelated patients which result in the expression
of a non-
functional and/or deficient FVIII protein. In particular, defects within the
F8 affect about one
in 5000 newborn males (Jones et al., Identification and removal of promiscuous
CD4+ T cell
epitope from the Cl domain of factor VIII. J. Throm. Haemost. 2005; 3: 991-
1000).
[0051] Mutations of the F8 gene resulting in a non-functional Factor VIII
include point
mutations, deletions, insertion and inversion as will be understood by a
skilled person. For
example, of the 2100 unique mutations identified in human F8 gene, over 980 of
them being
missense mutations, i.e., a point mutation wherein a single nucleotide is
changed, resulting in
a codon that codes for a different amino acid than its wild-type counterpart
(see HAMSTeRS
Database: at the http :// web page: hadb.org.uk/WebP ages/PublicF
iles/Mutation
Summary.htm). One of the most common mutations resulting in a non-functional
and/or
deficient FVIII protein includes inversion of intron 22, which leads to a
severe type of HA.
[0052] Accordingly, a mutation in an F8 gene of a subject resulting in a non-
functional
Factor VIII results in an F8 gene comprising at least one Factor VIII
functional coding
sequence and at least one Factor VIII non-functional coding sequence.
[0053] The wording "functional coding sequence" of Factor VIII refers to an F8
gene
sequence that is configured to be transcribed and contains one or more exons
of the F8 gene
with an open reading frame resulting in a functional Factor VIII or in a
portion thereof
Exemplary functional coding sequences comprise the sequence of E1-E22 and E23-
E26 of
the wild type F8 genomic locus in FIG. 1, the sequence of E1-E22 of the Intron-
22 inverted
F8 locus of FIG. 1, the sequence of human F8 cDNA of FIG. 2, the sequence of
Exons 1-22
and Ex 23-26 of the normal F8 gene in FIG. 7, the sequence of Ex 1-22 of the
Intron 22
inversion of the F8 gene in FIG. 7, the sequence of Ex 1-22 and Ex 23-26 of
the repaired F8
gene of FIG. 7, the cDNA sequence of Exons 23-26 of the repair vehicle of FIG.
9, the cDNA
9
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
sequence of Exons 2-26 of the repair vehicle of FIG. 10, the cDNA sequence of
Exons 23-26
of the repair vehicle of FIG. 11, the cDNA sequence of Exons 2-26 of the
repair vehicle of
FIG. 12, the cDNA of exons 23-26 of the repair vehicle of Table 51, the cDNA
sequence of
exons 23-26 of the repair vehicle of Table 52, the cDNA sequence of exons 2-26
or 2-13 of
the repair vehicle of Tables 53 and 54, respectively.
[0054] Functional coding sequences can include introns or be formed by exons
only or a
portion thereof Exemplary functional coding sequences comprise the sequence of
E1-E22
and E23-E26 of the wild type F8 genomic locus in FIG. 1, the sequence of E1-
E22 of the
Intron-22 inverted F8 locus of FIG. 1, Exons 1-22 and respective intervening
introns of the
Intron-22 inversion human F8 locus of FIG. 2, the sequence of Exons 1-22 and
Exons 23-26
of the normal F8 gene in FIG. 7, the sequence of Exons 1-22 of the Intron 22
inversion of the
F8 gene in FIG. 7, the sequence of Exons 1-22 and Exons 23-26 of the repaired
F8 gene of
FIG. 7.
[0055] Functional coding sequences can be included in the same orientation as
the wild type
F8 gene or in an opposite orientation as the wild type F8 gene. Exemplary
functional coding
sequences in a same orientation as the wild type F8 gene comprise the sequence
of E1-E22
and E23-E26 of the wild type F8 genomic locus in FIG. 1, the sequence of Exons
1-22 and
Exons 23-26 of the normal F8 gene in FIG. 7, the cDNA sequence of Exons 2-26
of the repair
vehicle of FIG. 10, the cDNA sequence of Exons 2-26 of the repair vehicle of
FIG. 12, the
cDNA of exons 23-26 of the repair vehicle of Table 51, the cDNA sequence of
exons 23-26
of the repair vehicle of Table 52, the cDNA sequence of exons 2-26 or 2-13 of
the repair
vehicle of Tables 53 and 54, respectively. Exemplary functional coding
sequences in an
opposite orientation as compared to wild type F8 gene comprise the sequence of
E1-E22 of
the Intron-22 inverted F8 locus of FIG. 1, the sequence of human F8 cDNA of
FIG. 2, the
sequence of Ex 1-22 of the Intron 22 inversion of the F8 gene in FIG. 7, the
sequence of Ex
1-22 and Ex 23-26 of the repaired F8 gene of FIG. 7, the cDNA sequence of
Exons 23-26 of
the repair vehicle of FIG. 9, the cDNA sequence of Exons 2-26 of the repair
vehicle of FIG.
10, the cDNA sequence of Exons 23-26 of the repair vehicle of FIG. 11, the
cDNA sequence
of Exons 2-26 of the repair vehicle of FIG. 12.
[0056] The wording "non-functional coding sequence" of the F8 gene refers to
an F8 gene
sequence that is not configured to be transcribed and/or contains one or more
exons of the F8
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
gene with an open reading frame resulting in a non-functional Factor VIII or
in a portion
thereof. In particular, coding sequences can be non-functional, and therefore
result in a non-
functional Factor VIII, due to point mutations resulting in a sequence coding
for an amino
acid, in an insertion or deletion of coding sequences resulting in frame shift
or a different
open reading frame, with respect to an open reading frame (such as the open
reading frame of
the wild type F8 gene), which results in a functional Factor VIII.
[0057] Exemplary non-functional coding sequences resulting from F8 gene
mutations
comprise the sequence of E24 in the case of a F8 c.6761 T>A nonsense mutation
that results
in a stop codon at codon 2178 in place of the leucine (Leu)-encoding codon
that is present at
codon 2178 in the non-mutated form of the F8 gene as seen in FIG. 14, the
sequence of E25
in the case of a F8 c.6917 T>G missense mutation that results in a codon
encoding arginine
(Arg) at codon 2230 in place of the leucine (Leu)-encoding codon that is
present at that codon
2230 in the non-mutated form of the F8 gene as seen in FIG. 14, the sequence
of sequence of
E24, E25 and E26 in the case of a F8 IVS-23 +1 G>A splice site mutation that
results in a
non-functional pre-mRNA splice site immediately downstream of exon 23 of the
F8 gene as
seen in FIG. 14, sequence of E26 in the case of a F8 Exon 26 del.[A] small
deletion and
frameshift mutation that results in a frameshift of the gene-encoding sequence
which changes
the downstream sequence by a single base-pair deletion frameshift and
introduction of a
novel terminating stop codon in the gene-encoding sequence as seen in FIG. 14.
[0058] Non-functional coding sequences can be included in the same orientation
as the wild
type F8 gene or in an opposite orientation of the wild type F8 gene. Exemplary
non-
functional coding sequences in a same orientation of the wild type F8 gene
comprise the
sequence of El B and the sequence of E23-E26 of the Intron-22 inverted F8
genomic locus of
FIG. 1, the sequence of exons 23c and 24c of the Intron-22 inverted human
locus of FIG. 2A,
the sequence of Exons 23-26 of the Intron 22 Inversion of the F8 gene in FIG.
7, the sequence
of E24 in the case of a F8 c.6761 T>A nonsense mutation that results in a stop
codon at
codon 2178 in place of the leucine (Leu)-encoding codon that is present at
codon 2178 in the
non-mutated form of the F8 gene as seen in FIG. 14, the sequence of E25 in the
case of a F8
c.6917 T>G missense mutation that results in a codon encoding arginine (Arg)
at codon 2230
in place of the leucine (Leu)-encoding codon that is present at that codon
2230 in the non-
mutated form of the F8 gene as seen in FIG. 14, the sequence of sequence of
E24, E25 and
E26 in the case of a F8 IVS-23 +1 G>A splice site mutation that results in a
non-functional
11
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
pre-mRNA splice site immediately downstream of exon 23 of the F8 gene as seen
in FIG. 14,
sequence of E26 in the case of a F8 Exon 26 del.[A] small deletion and
frameshift mutation
that results in a frameshift of the gene-encoding sequence which changes the
downstream
sequence by a single base-pair deletion frameshift and introduction of a novel
terminating
stop codon in the gene-encoding sequence as seen in FIG. 14. Exemplary non-
functional
coding sequences comprise in opposite orientation of the wild type F8 gene
comprise the
sequence of exons E23C and E24C of the Intron-22 inverted F8 genomic locus of
FIG. 1.
[0059] In embodiments, herein described non-functional coding sequences are
replaced by a
cDNA-repair sequence (RS).
[0060] The term cDNA or complementary DNA indicates double-stranded DNA that
can be
synthesized from a messenger RNA (mRNA) template in a reaction catalysed by
the enzyme
reverse transcriptase. Accordingly cDNA can be synthesized from mature (fully
spliced)
mRNA using the enzyme reverse transcriptase or be synthesized synthetically
based on the
mRNA sequence as will be understood by a skilled person.
[0061] The terms "polynucleotide", "oligonucleotide" and "nucleic acid," are
used
interchangeably and refer to an organic polymer composed of two or more
monomers
including nucleotides, nucleosides or analogs thereof. The term "nucleotide"
refers to any of
several compounds that consist of a ribose or deoxyribose sugar joined to a
purine or
pyrimidine base and to a phosphate group and that is the basic structural unit
of nucleic acids.
The term "nucleoside" refers to a compound (such as guanosine or adenosine)
that consists of
a purine or pyrimidine base combined with deoxyribose or ribose and is found
especially in
nucleic acids. The term "nucleotide analog" or "nucleoside analog" refers
respectively to a
nucleotide or nucleoside in which one or more individual atoms have been
replaced with a
different atom or a with a different functional group. Exemplary functional
groups that can be
comprised in an analog include methyl groups and hydroxyl groups and
additional groups
identifiable by a skilled person. In general, an analogue of a particular
nucleotide has the
same base-pairing specificity; i.e., an analogue of A will base-pair with T.
[0062] Exemplary monomers of a polynucleotide comprise deoxyribonucleotide,
and
ribonucleotides. The term "deoxyribonucleotide" refers to the monomer, or
single unit, of
DNA, or deoxyribonucleic acid. Each deoxyribonucleotide comprises three parts:
a
nitrogenous base, a deoxyribose sugar, and one or more phosphate groups. The
nitrogenous
12
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
base is typically bonded to the l' carbon of the deoxyribose, which is
distinguished from
ribose by the presence of a proton on the 2' carbon rather than an -OH group.
The phosphate
group is typically bound to the 5' carbon of the sugar. The term
"ribonucleotide" refers to the
monomer, or single unit, of RNA, or ribonucleic acid. Ribonucleotides have
one, two, or
three phosphate groups attached to the ribose sugar.
[0063] Accordingly, the term "polynucleotide", "oligonucleotide includes
nucleic acids of
any length, and in particular DNA, RNA, analogs thereof, and fragments thereof
Polynucleotides can typically be provided in single-stranded form or double-
stranded form
(herein also duplex form, or duplex).
[0064] A "single-stranded polynucleotide" refers to an individual string of
monomers linked
together through an alternating sugar phosphate backbone. In particular, the
sugar of one
nucleotide is bond to the phosphate of the next adjacent nucleotide by a
phosphodiester
bond. Depending on the sequence of the nucleotides, a single-stranded
polynucleotide can
have various secondary structures, such as the stem-loop or hairpin structure,
through
intramolecular self-base-paring. A hairpin loop or stem loop structure occurs
when two
regions of the same strand, usually complementary in nucleotide sequence when
read in
opposite directions, base-pairs to form a double helix that ends in an
unpaired loop. The
resulting lollipop-shaped structure is a key building block of many RNA
secondary
structures. The term "small hairpin RNA" or "short hairpin RNA" or "shRNA" as
used herein
indicate a sequence of RNA that makes a tight hairpin turn and can be used to
silence gene
expression via RNAi.
[0065] A "double-stranded polynucleotide", "duplex polynucleotide" refers to
two single-
stranded polynucleotides bound to each other through complementarily binding.
The duplex
typically has a helical structure, such as double-stranded DNA (dsDNA)
molecule or double
stranded RNA, is maintained largely by non-covalent bonding of base pairs
between the
strands, and by base stacking interactions.
[0066] In embodiments, herein described a cDNA-repair sequence (RS) is a
double stranded
polynucleotide comprising a repaired version of the entire F8 gene non-
functional coding
sequence of the subject or of a portion thereof In particular in methods and
compositions
herein described the cDNA-RS comprise at least a repaired version the portion
of the non-
functional sequence of the Factor VIII of the subject comprising the one or
more mutations in
13
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
the Factor VII of the subject. In some embodiments, cDNA-RS described herein
further
comprises introns and/or exons located upstream and/or downstream to the non-
functional
coding sequence. In embodiments described herein, the cDNA-RS is designed so
that once
recombined into the desired region in the F8 genomic locus it remains in-frame
with
functional coding upstream and downstream functional coding sequences.
[0067] Accordingly in methods systems and related cDNA vehicles and
compositions herein
described a cDNA-RS are designed based on the one or more mutations within the
subject's
F8 gene targeted for replacement and repair. For example, when repairing a
point mutation,
the cDNA-RS includes only a small number of replacement nucleotide sequences
compared
with, for example, a cDNA-RS derived for repairing an inversion such as an
intron 22
inversion. Therefore, a cDNA-RS can be of any length, for example between 2
and 10,000
nucleotides in length (or any integer value there between or there above),
e.g. between about
100 and 1,000 nucleotides in length (or any integer there between), between
about 200 and
500 nucleotides in length (or any integer there between). Exemplary cDNA-RS
herein
described comprise the sequence of human F8 cDNA of FIG. 2, the cDNA sequence
of
Exons 23-26 of the repair vehicle of FIG. 9, the cDNA sequence of Exons 2-26
of the repair
vehicle of FIG. 10, the cDNA sequence of Exons 23-26 of the repair vehicle of
FIG. 11, the
cDNA sequence of Exons 2-26 of the repair vehicle of FIG. 12, the cDNA
sequence of exons
23-26 of the repair vehicle of Table 51, the cDNA sequence of exons 23-26 of
the repair
vehicle of Table 52, the cDNA sequence of exons 2-26 or 2-13 of the repair
vehicle of Tables
53 and 54, respectively.
[0068] In an embodiment, the gene mutation targeted for repair is a point
mutation, and the
cDNA-RS includes a nucleic acid sequence that replaces the point mutation with
a functional
sequence for Factor VIII that does not include the point mutation, for
example, the wild-type
F8 sequence. In one embodiment, the gene mutation targeted for repair is a
deletion and the
cDNA-RS includes a nucleic acid sequence that replaces the deletion with a
functional Factor
VIII sequence that does not include the deletion, for example, a corresponding
F8 sequence
of the wild-type F8 sequence.
[0069] In one embodiment, the gene mutation targeted for repair is an
inversion, and the
cDNA-RS includes a nucleic acid sequence that encodes a truncated FVIII
polypeptide that,
upon insertion into the F8 genome, repairs the inversion and provides for the
production of a
14
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
functional FVIII protein. In one embodiment, the gene mutation targeted for
repair is an
inversion of intron 1. In one embodiment, the gene mutation targeted for
repair is an
inversion of intron 22, and the donor sequence includes a nucleic acid that
encodes all of
exons 23-25 and the coding sequence of exon-26 to be inserted in frame with
the inverted
exons 1-22 in opposite orientation with the F8 gene.
[0070] In the methods and compositions described herein, the cDNA-RS can
contain
sequences that are homologous, but not identical (for example, contain nucleic
acid sequence
encoding wild-type amino acids or differing ns-SNP amino acids), to subject's
genomic
sequences in the region of interest, thereby stimulating homologous
recombination to insert a
non-identical sequence in the region of interest.
[0071] The term "homologous" and "homology" when referred to protein or
polynucleotide
sequences is defined in terms of sequence similarities and percent identity
between
sequences. Accordingly homologous sequences indicate sequences having a
percent identify
of at least 80% versus sequences with a percentage identify lower than 80%,
which are
instead indicated as non-homologous. The terms "percent homology" and
"sequence
similarity" are often used interchangeably. Sequence regions that are
homologous are also
called conserved.
[0072] Thus, in certain embodiments, portions of the cDNA-RS that are
homologous to
sequences in the region of interest exhibit between about 80 to about 99%
sequence identity
to the subject's genomic sequence that is replaced. In other embodiments, the
homology
between the cDNA-RS and the subject's genomic sequence is higher than 99%, for
example if
only 1 nucleotide differs as between the cDNA-RS and the subject's genomic
sequences of
over 100 contiguous base pairs. In certain cases, a non-homologous portion of
the cDNA-RS
contains sequences not present in the region of interest, such that new
sequences are
introduced into the region of interest. In these instances, the non-homologous
sequence is
generally flanked by sequences of 50-1,000 base pairs, or any number of base
pairs greater
than 1,000, that are homologous or identical to the subject's sequences in the
region of
interest. In other embodiments, the cDNA-RS containing non-homologous sequence
is
inserted into the subject's genome by homologous recombination mechanisms.
[0073] Accordingly, cDNA-RS herein described can be comprised within a cDNA
sequence
encoding for a truncated Factor VIII. The term "truncated FVIII polypeptide"
refers to a
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
polypeptide that contains less than the full length of FVIII protein. The
truncated FVIII
polypeptide is encoded in a portion of the full length F8 gene such as a
partial F8 cDNA
replacement sequence (cDNA-RS). For example, for FVIII polypeptide that is
truncated from
the corresponding 5' end of the oligonucleotide sequence, a variable amount of
the
oligonucleotide sequence can be missing from the 5' end of the gene. In one
embodiment,
the truncated FVIII polypeptide is encoded by exons 23-26. In one embodiment,
the
truncated FVIII polypeptide is encoded by exons 2-26. In one embodiment, the
truncated
FVIII polypeptide is encoded by exons 15-26.
[0074] In embodiments herein described the cDNA-RS are designed in combination
with the
selection of DNA scission Enzyme (DNA-SE) and the related target site.
[0075] A DNA scission enzyme indicates an enzyme that catalyzes the hydrolytic
cleavage of
phosphodiester linkages in the DNA backbone in a specific target site. DNA
scission refers
to the breaking of the chemical bonds between adjacent nucleotides on a
nucleotide strand or
sequence. DNA scission enzymes comprise nucleases and nickases. "Nucleases" or
"Deoxyribonucleases" are enzymes capable of hydrolyzing phosphodiester bonds
that link
nucleotides. A wide variety of deoxyribonucleases are known, which differ in
their substrate
specificities, chemical mechanisms, and biological functions. DNA-SEs
described herein
break the genomic DNA at a target site on the F8 gene upstream from a region
to be replaced
by a repair vehicle comprising a cDNA-RS. The target site is preferentially
located about 50-
100 base pairs upstream of the desired region to be replaced on the F8 genomic
locus so as to
optimize recombination by the repair vehicle, donor plasmid, or editing
cassette comprising
the cDNA-RS. In studies, it was seen that when a target site is located about
50-100 base
pairs upstream of the desired region to be replaced on the F8 genomic locus,
optimal
recombination was observed by the repair vehicle, donor plasmid, or editing
cassette
comprising the cDNA-RS. Following recombination of the repair vehicle, donor
plasmid, or
editing cassette into the target site, expression of the repaired F8 gene
segment results in
expression of a repaired and functional FVIII protein. DNA-SEs described
herein comprise
nucleases or nickases coupled to nucleotide sequences that specifically guide
the nuclease or
nickase to the target site. DNA-SEs described herein include heterodimeric
nucleases that
bind to specific regions of the F8 gene, nucleases or nickases guided to
specific sites of the
F8 gene by short RNA sequences or combinations thereof. Exemplary nucleases
include
transcription activator¨like effector nuclease (TALEN), a zinc finger nuclease
(ZFN), a
16
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)-associated
(Cas)
nuclease, Paired CRISPR, or CRISPR with ZFN. "Nickases" are enzyme that causes
nicks
(breaks in one strand) of double stranded nucleic acid, allowing it to unwind.
An exemplary
nickase is Cas9n (the DlOA mutant nickase version of Cas9).
[0076] In embodiments described herein, DNA-SEs are designed to comprise
multiple
elements to efficiently target a specific target site within the F8 gene and
function as
heterodimers or heterodimeric nucleases; Such DNA-SEs are referenced in FIG.
2, FIG. 3,
FIG. 4, FIG. 5 and FIG. 6 as TALENL and TALENR. Such heterodimeric nucleases
comprise
two monomers (a left monomer and a right monomer) that each comprise a nuclear
localization signal, a monomer subunit for binding to a specific region of the
F8 gene and a
Fokl nuclease domain. Further, the monomer subunit for binding of the left
monomer binds
upstream (5') of the target site, while the monomer subunit of the right
monomer binds to a
region downstream (3') of the target site, as depicted in FIG. 3 by TALENL and
TALENR. In
such embodiments, a double-stranded break in the DNA of the target region is
mediated by
dimerization of the Fok-1 nucleases. The monomer binding subunits are designed
such that
off-target binding non-specific DNA breaks are minimized and such that the
location of the
target site is optimally placed upstream from a region to be replaced by a
repair vehicle
comprising a cDNA-RS.
[0077] In embodiments described herein, DNA-SEs are designed to efficiently
target a
specific target site within the F8 gene by using a short RNA to guide a
nuclease to the desired
target site; such a DNA-SE is referenced in FIG. 13 as the CRISPR-Associated
Gene Editing
system. Such DNA-SEs comprise at least a complementary single strand RNA
(CRISPR
RNA, labeled as CRISPR g-RNA in FIG. 13, for example) that localizes a Cas9
nuclease to a
target site on F8 gene. The CRISPR RNA binds to a region upstream of a desired
target site,
allowing the Cas9 nuclease to cause a double-strand break. The CRISPR RNA is
designed
such that off-target binding non-specific DNA breaks are minimized and such
that the
location of the target site is optimally placed upstream from a region to be
replaced by a
repair vehicle comprising a cDNA-RS. In embodiments described herein, such a
DNA-SE is
modified to further minimize off-target DNA scission events by modifying the
CRISPR-
Associated Gene editing system DNA-SE described above to carry a mutated Cas9
that
functions as a nickase (Cas9-nickase); such a DNA-SE is referenced in FIG. 14
and in FIG.
15. In such embodiments, CRISPR RNA (labeled as CRISPR gRNAi in FIG. 15) that
is
17
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
longer in length than the CRISPR RNA of the DNA-SE referenced in FIG. 13 is
used to
guide a first Cas9-nickase to a target site. The Cas9-nickase then makes a
single strand break
in the DNA at the target site. A second Cas9-nickase is guided to a second
target on the
complementary DNA strand site by a second CRISPR RNA (labeled as CRISPR g-RNA2
in
FIG. 15) and the second Cas9-nickase makes a single strand break in the
complementary
DNA strand. The two nicking target sites can be separated by 0-30 nucleotides.
[0078] In the methods and compositions set forth herein, the DNA-SEs that
targets a mutation
in F8 for repair are, for example, a transcription activator¨like effector
nuclease (TALEN), a
zinc finger nuclease (ZFN), a CRISPR (Clustered Regularly Interspaced Short
Palindromic
Repeats)-associated (Cas) nuclease, Paired CRISPR, or CRISPR with ZFN, as
described in
detail below.
[0079] In the methods and systems and related compositions set forth herein,
the DNA-SEs is
selected for the DNA-SE ability to target a mutation in the F8 gene for repair
cleaving the F8
gene sequence for subsequent repair by the cDNA-RS. In particular in methods
and systems
and related compositions herein described a DNA-SE is for the capability of
creating a first
break in one strand of the F8 gene and a second break in the other strand of
the F8 gene
defining a target site located in a position of the F8 gene configured to
allow replacement of
the F8 gene non-functional coding sequence by a cDNA-RS.
[0080] In methods and systems herein described, the DNA-SE has a target site
upstream of
the F8 gene nonfunctional coding sequence.
[0081] The wording "upstream" as used herein refers to a position in a
polynucleotide
relative to a 5' end of the reference point in the polynucleotide. Therefore a
sequence or
series of nucleotide residues that is "upstream" relative to a site, region or
sequence indicates
a sequence or series of nucleotides before the 5' end site, region or sequence
of the
polynucleotide in a 5'to 3' direction. Accordingly, making reference to the
exemplary
illustration of FIG. 7, Exons 1-22 are located upstream of Exons 23-26 at the
normal genomic
DNA (gDNA). Additionally, making reference to FIG. 3, TALEN-L binds to a
nucleotide
sequence upstream of the target site.
[0082] The wording "downstream" as used herein refers to a position in a
polynucleotide
relative to a 3' end of the reference point in the polynucleotide. Therefore a
sequence or
18
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
series of nucleotide residues that is "downstream" relative to a site, region
or sequence
indicates a sequence or series of nucleotides after the 3' end site, region or
sequence of the
polynucleotide in a 5' to 3' direction. Accordingly, making reference to the
exemplary
illustration of FIG. 7, Exons 23-26 are located downstream of Exons 1-22 at
the genomic
DNA (gDNA). Additionally, making reference to FIG. 13, the Protospacer
Adjacent Motif
(PAM) is downstream of the target site.
[0083] In methods and systems herein described, the cDNA-RS is designed to
provide a
repaired version of the F8 gene nonfunctional coding sequence or a portion
thereof
encompassing the one or more mutations to be repaired in frame with the F8
gene functional
coding sequence upstream of the DNA-SE target site.
[0084] A sequence or series of nucleotide residues that is "in-frame" or "in
frame" with a F8
gene functional sequence refers to a sequence or series of nucleotide residues
that does not
cause a shift in the open reading frame of the F8 functional sequence. An open
reading frame
(ORF) is the part of a reading frame of a coding sequence that encodes for a
protein or
peptide according to the standard genetic code, in this case a functional
Factor VIII. An ORF
is a continuous stretch of DNA beginning with a start codon, usually
methionine (ATG), and
ending with a stop codon (TAA, TAG or TGA in most genomes) as will be
understood by a
skilled person. Accordingly, sequence or series of nucleotide residues is "out
of frame" or
"out-of-frame" with an F8 functional sequence when to the sequence or series
of nucleotide
residues causes a shift in the open reading frame of the F8 functional
sequence thus resulting
in a sequence coding for a non-functional Factor VIII.
[0085] For example in some embodiments, the cDNA-RS provides a repaired
version of the
F8 nonfunctional sequence in a same orientation with the wild type F8 gene. In
some
embodiments, the cDNA-RS provides a repaired version of the F8 nonfunctional
sequence in
opposite orientation with the wild type F8 gene in frame with the functional
sequence of the
F8 gene following the inversion. In particular in some embodiments the cDNA-RS
for the
inversion of intron 22 provides repaired version of the F8 non-functional
sequence
downstream the inverted exons 1-22 encompassing sequences for exons 23-26 in
opposite
orientation to the F8 gene.
[0086] In embodiments, herein described selection of a suitable DNA-SE is
performed by
selecting a target site among candidate target sites on the F8 gene based on
the one or more
19
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
mutations of the F8 gene to be repaired and based on the features of the cDNA-
RS to be used
on the repair and/or the related donor sequence comprising the cDNA-RS flanked
by flanking
sequence is homologous to nucleic acid sequences of the F8 gene.
[0087] The wording "flanked" as used herein refers to a position relative to
ends of a
reference item. More specifically, in referring to a polynucleotide sequences,
"flanked" refers
to having a sequences upstream and downstream the end of the polynucleotide
sequences. In
particular, a flanked referenced polynucleotide has a first sequence or series
of nucleotide
residues positioned adjacent to the 5' end of the referenced polynucleotide
and a second
sequence or series of nucleotide residues positioned adjacent to the 3' end of
the referenced
polynucleotide. For example, in Figure 2B, the human F8 cDNA is flanked by a
left
homology arm (homologY0 and a right homology arm (homologyr).
[0088] In some embodiments, selection based on the one or more mutations of
the F8 gene to
be repaired can be performed with algorithms or other means directed to
minimize off-target
effects associated with the DNA-SEs. For example, in some embodiments a
program such as
PROGNOS can be used to identify the target site. The PROGNOS algorithm locates
for
example potential TALEN off-target sites by searching through the genome for
sequences
similar to the intended TALEN design. It ranks these similar sequences
according to various
features of TALEN-DNA interactions, including RVD base preferences, polarity
of TALEN
specificity (5' end is more specific), context dependent compensation of
strong RVDs (such
as NN and HD), and a model of dimeric TALEN interactions. The PROGNOS model
has
been shown to accurately predict the majority of all known TALEN off-target
sites as
discussed in Fine et al. Nucleic Acids Research 2013, incorporated herein by
reference. As
another example, an algorithm employed for ranking potential CRISPR off-target
sites
disclosed in Hsu et al. Nature Biotech 2013, incorporate herein by reference,
uses a position-
weight-matrix (PWM) to determine the importance of different types of
mismatches at each
position in the target sequence (both the DNA bases targeted by the guide
strand as well as
the protospacer adjacent motif sequence). This PWM was derived by
experimentally
observing the drop in nuclease activity at a target site of artificial guide
strands (relative to a
perfectly matched guide strand) containing different types of mismatches. This
PWM is then
used to screen potential sites in the genome with homology to the intended
target and assign
them a score indicating their likelihood of off-target activity.
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
[0089] In embodiments herein described a target site is selected based on the
features of a
cDNA-RS used for repair. Factors influencing the location of the target site
include the
desired length and sequence of cDNA-RS, proximity of the target site to
upstream and
downstream functional coding sequences, proximity of the target site to
upstream and
downstream non-functional coding sequences, likelihood of off-target or non-
specific DNA
scission, likelihood of off-target or non-specific homologous recombination of
the cDNA-RS,
homology to off-target genomic sites and nature of the DNA scission enzyme
used.
[0090] In particular in some embodiments the target site is selected to have a
location relative
to the desired region of replacement on the F8 genomic locus that optimizes
the
recombination rate of the cDNA-RS. For instance, in some embodiments, the
target site is
selected to be from 50-100 nucleotides upstream of the desired region of
replacement on the
F8 genomic locus so as to optimize the recombination of the cDNA-RS following
scission of
the genomic DNA. Location of the target site within about 50-100 base pairs
upstream of the
desired region to be replaced on the F8 genomic locus results in optimal
recombination by the
repair vehicle, donor plasmid, or editing cassette comprising the cDNA-RS.
Optimal
recombination is an important aspect as it results in an increase in the
likelihood that the
cDNA-RS will be incorporated at the targeted site within an individual cell
and/or population
of cells following exposure to the cDNA ¨RS. Also, following recombination of
the repair
vehicle, donor plasmid, or editing cassette into the target site, expression
of the repaired F8
gene segment results in expression of a repaired and functional FVIII protein.
Thus,
conditions promoting optimal recombination greatly contribute towards
achieving optimal
expression of a repaired and functional protein for treatment and/or induction
of immune
tolerance.
[0091] In embodiments herein described a target site is also be selected based
on the features
of the donor DNA comprising the cDNA-RS flanked by an upstream flanking
sequence (uFS)
and a downstream flanking sequence (dFS).
[0092] In particular, in embodiments herein described in a donor sequence, the
cDNA-RS is
flanked on each side by regions of nucleic acids which are homologous to the
subject's F8
gene that are called flanking sequences. Each of the flanking sequence can
include about 20,
50, 75, 100, 200, 300, 400, 500, 600, 700, 800, 900, 1000 or more nucleotides
homologous to
regions within the subject's F8 gene. In particular, the upstream flanking
sequence (uFS) is
21
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
homologous to a nucleic acid sequence upstream of the first break in the one
strand of the F8
gene by a selected DNA-SE and the downstream flanking sequence (dFS)
homologous to a
nucleic acid sequences downstream of the second break in the other strand of
the F8 gene by
the selected DNA-SE.
[0093] In some embodiments, each of the homologous regions flanking the donor
sequence is
between about 200 to about 1,200 nucleotides, e.g. between 400 and about 1000,
between
about 600 and about 900, or between about 800 and about 900 nucleotides. Thus,
each
donor sequence includes a cDNA-RS replacing an endogenous mutation in the
subject's F8
gene, and 5' and 3' flanking sequences which are homologous to the F8 gene. In
preferred
embodiments the length of the homologous regions flanking the donor sequence
are between
700 ¨ 800 nucleotides in length. Exemplary homologous regions or arms are the
left and right
homology arms shown in FIG. 9, FIG. 10, FIG. 11 and FIG. 12.
[0094] In some embodiments, the cDNA-RS is comprised within an editing
cassette together
with one or more transcriptional elements and the upstream flanking sequence
(uFS) and
downstream flanking sequence (dFS) are located adjacent at the 5' end and at
3' end of the
editing cassette, respectively.
[0095] The wording "adjacent" as used herein refers to a location and/or
position nearest in
space or position; immediately adjoining without intervening space. More
specifically, when
referring to a sequence or series of nucleotide residues that is "adjacent" to
a site or sequence,
"adjacent" refers to a location and/or position next to or proximate to the
reference site or
position without intervening nucleotide residues. An example is seen in FIG. 9
where the left
homology arm (700 bp) is located adjacent to Exons 23-26 (cDNA sequence).
[0096] In some embodiments, where the cDNA-RS codes for the 3' terminal
sequence of the
F8 gene the cDNA-RS is within an editing cassette also comprising a sequence
for a polyA
site at the 3' end of the cDNA-RS sequence. In some embodiments where the
target site is on
a portion of the F8 gene having downstream intron sequences, the 3' terminal
sequence of the
F8 gene the cDNA-RS is within an editing cassette also comprising a splice
acceptor at the 5'
end of the cDNA-RS sequence. In particular in some embodiment the editing
cassette
comprise (i) a nucleic acid encoding a truncated FVIII polypeptide or (ii) a
native F8 3' splice
acceptor site operably linked to a nucleic acid encoding a truncated FVIII
polypeptide that
contains a non-mutated portion of the FVIII protein.
22
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
[0097] As used throughout, "operably linked" is defined as a functional
linkage between two
or more elements. In particular, the term "operably linked" or "operably
connected" indicates
an operating interconnection between two elements finalized to the expression
and translation
of a sequence. Functional linkages between elements in the sense of the
present disclosure are
identifiable by a skilled person. For example, an operable linkage between a
polynucleotide
of interest and a regulatory sequence (i.e., a promoter) comprise a functional
link that allows
for expression of the polynucleotide of interest. Another example of operable
linkage is
provided by a control sequence ligated to a coding sequence in such a way that
expression of
the coding sequence is achieved under conditions compatible with the control
sequences.
Operably linked elements are contiguous or non-contiguous and comprise
polynucleotides in
a same or different reading frame. In an embodiment, each of the operably
linked
polynucleotide is comprised within the editing cassette. The cassette
additionally contains at
least one additional gene to be co-transformed into the organism (e.g. a
selectable marker
gene). One or more additional genes can also be provided on multiple
expression cassettes
that can further comprise a plurality of restriction sites and/or
recombination sites for
insertion of other polynucleotides.
[0098] In embodiments herein described, editing cassettes refers to a mobile
genetic element
that contains a gene and a sequence used to repair an F8 non-functional coding
sequence.
Editing cassettes carry at least a cDNA-repair sequence (RS) flanked by an
upstream flanking
sequence (uFS) and a downstream flanking sequence (dFS) to form a DNA donor.
The
cDNA-RS is a repaired version of the F8 non-functional F8 gene sequence. The
upstream
flanking sequence (uFS) is homologous to a nucleic acid sequence upstream of a
target site
on the F8 gene and the downstream flanking sequence (dFS) is homologous to a
nucleic acid
sequences downstream of a target site on the F8 gene. In embodiments described
herein, the
cDNA-RS of the editing cassette is designed and oriented such that when
recombined into the
desired region on the F8 gene, it is in-frame with upstream and downstream
functional coding
sequences. Exemplary editing cassettes include the sequence comprising the
left homology
arm, cDNA of Exons 23-26, the human growth hormone polyadenylation signal
sequence and
the right homology arm of the plasmid in FIG. 9, the sequence comprising the
left homology
arm, cDNA of Exons 2-26, the human growth hormone polyadenylation signal
sequence and
the right homology arm of the plasmid in FIG. 10, the sequence comprising the
left homology
arm, cDNA of Exons 23-26, the human growth hormone polyadenylation signal
sequence and
23
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
the right homology arm of the plasmid in FIG. 11, the sequence comprising the
left homology
arm, cDNA of Exons 2-26, the human growth hormone polyadenylation signal
sequence and
the right homology arm of the plasmid in FIG. 12.
[0099] In embodiments herein described, following identification of a target
site a DNA-SE
is configured for binding to the F8 gene at the selected target site. The DNA-
SE is modified
to target a target site that is preferentially located about 50-100 base pairs
upstream of the
desired region to be replaced on the F8 genomic locus so as to optimize
recombination by the
repair vehicle, donor plasmid, editing cassette comprising the cDNA-RS.
Location of the
target site within about 50-100 base pairs upstream of the desired region to
be replaced on the
F8 genomic locus results in optimal recombination by the repair vehicle, donor
plasmid, or
editing cassette comprising the cDNA-RS. Optimal recombination is an important
aspect as it
results in an increase in the likelihood that the cDNA-RS will be incorporated
at the targeted
site within an individual cell and/or population of cells following exposure
to the cDNA ¨RS.
Also, following recombination of the repair vehicle, donor plasmid, or editing
cassette into
the target site, expression of the repaired F8 gene segment results in
expression of a repaired
and functional FVIII protein. Thus, conditions promoting optimal recombination
greatly
contribute towards achieving optimal expression of a repaired and functional
protein for
treatment and/or induction of immune tolerance. DNA-SEs described herein are
modified to
comprise nucleases or nickases coupled to nucleotide sequences that
specifically guide the
nuclease or nickase to the target site. DNA-SEs described herein include
heterodimeric
nucleases that bind to specific regions of the F8 gene, nucleases or nickases
guided to specific
sites of the F8 gene by short RNA sequences or combinations thereof A DNA-SE
can be
designed and assembled using molecular techniques commonly known and available
to one
of ordinary skill in the art and as described in Ran, F. A. et al. Genome
engineering using the
CRISPR-Cas9 system. Nat Protoc 8, 2281-2308 (2013).
[0100] In embodiments described herein, polynucleotides and vectors comprising
the DNA-
SE and the DNA donor are provided for introduction into a cell of a subject
having a mutated
F8 gene. In particular the DNA-SE comprises nucleases or nickases coupled to
nucleotide
sequences that specifically guide the nuclease or nickase to the target site.
DNA-SEs
described herein include heterodimeric nucleases that bind to specific regions
of the F8 gene,
nucleases or nickases guided to specific sites of the F8 gene by short RNA
sequences or
combinations thereof The polynucleotides and vectors comprising the DNA-SE and
DNA
24
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
donor vary in design and function as a function of the type of gene editing
system that is
utilized. For instance, different polynucleotides and vectors are used for
TALENs,
CRISPR/Cas9 nuclease, CRISPR/Cas9n nickase, and CRISPR/Cas9 RFN.
[0101] In embodiments herein described, a "donor plasmid" refers to a mobile
genetic
element in the form of a plasmid, vector, sequence or strand that is be used
as a means to
deliver or donate a polynucleotide sequence to a specific genomic site. The
donor plasmid
contains DNA and/or cDNA. Embodiments of donor plasmids described herein
consist of at
least the following elements: a cDNA-RS for repair of a non-functional F8
coding sequence
flanked by an upstream flanking sequence (uFS) and a downstream flanking
sequence (dFS).
The upstream flanking sequence (uFS) is homologous to a nucleic acid sequence
upstream of
the first break in the one strand of the F8 gene and the downstream flanking
sequence (dFS)
homologous to a nucleic acid sequences downstream of the second break in the
other strand
of the F8 gene. Donor plasmids are designed and configured to optimally
integrate by
homologous recombination at a target site following DNA scission by a DNA-SE.
The
cDNA-RS of donor plasmid designed and oriented such that when recombined into
the
desired region on the F8 gene, it is in-frame with upstream and downstream
functional coding
sequences. Exemplary donor plasmids include the plasmids referenced in FIG. 9,
FIG. 10,
FIG. 11 and FIG. 12.
[0102] In embodiments herein described the DNA donor is comprised within a
repair vehicle
(RV). The RV can be a sequence of DNA in the form of a circular plasmid. The
RV can be a
linear sequence of DNA. The RV provides the template, through which by
homologous
recombination, a targeted DNA sequence can be introduced into the genomic DNA
of the
subject at the site of a targeted double strand break. In addition to a cDNA-
RS, optionally an
editing cassette and flanking sequences of the DNA donor, a RV can also
contain sequences
important for the preparation of the DNA sequence in bacteria, such as an
antibiotic
resistance gene for ampicillin, an antibiotic resistance gene for kanamycin,
and/or other
antibiotic resistance genes. The RV can also contain intervening DNA sequences
important
for the integrity of the plasmid or linear sequence of DNA, such as sequences
that are located
between antibiotic-resistance gene-encoding sequences and cDNA-RS, and which
intervening DNA sequences can contain gene-encoding sequences or alternatively
can
contain sequences that do not encode for a gene.
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
[0103] In methods and systems herein described polynucleotides coding for a
DNA-SE and
one or more repair vehicles are introduced into a cell of a subject having a
mutated F8 for a
time and under condition allowing homologous recombination of the upstream
flanking
sequence (uFS) and the downstream flanking sequence (dFS) of the donor DNA to
corresponding sequences of the F8 gene.
[0104] In particular, in some embodiments herein described, the targeting and
repair of a
mutated F8 gene in a subject, by introducing into a subject's cell one or more
plasmids
encoding a DNA-SE that specifically targets the F8 mutation of the subject.
Each subject's
mutation for targeting and repair can be determined using techniques known in
the art. The
identified mutation in the subject is then directly targeted by DNA-SE for
correction
according e.g. by selecting a DNA-SE target site at the 5' of the mutated non-
functional F8
gene sequence. Alternatively, the subject's F8 gene mutations can be corrected
by targeting a
region of the F8 gene upstream (or 5') from the non-functional coding sequence
(e.g. where
the mutation occurred), and adding back the corresponding downstream coding
regions of the
F8 gene. For example, intron 14 could be targeted by the DNA-SE. This allows
for gene
repair of downstream mutations (i.e. missense mutations in exon 15 to exon 26)
and
inversions (such as the intron 22 inversion), due to the replacement of exons
15 to 26 with the
cDNA-RS discussed above. In other embodiments, the F8 gene can be targeted at
additional
regions upstream, in order to capture an increasing proportion of F8 gene
mutations. Thus,
the DNA-SE can be engineered to specifically target a subject's F8 mutation,
or alternatively,
can target regions upstream of a subject's F8 mutation, in order to correct
the mutation in
combination with a donor sequence which provides cDNA-RS, which is a partial
F8 gene
during homologous recombination that replaces, and thus repairs, the mutated
portion of the
subject's F8 gene and possibly includes functional coding sequences upstream
of the non-
functional coding sequence of the mutated F8 gene.
[0105] In particular in some embodiments of methods and systems herein
described the
repairing is performed introducing into a cell of the subject one or more
nucleic acids
encoding a DNA scission enzyme (DNA-SE) having a DNA-SE target site located
upstream
from a 5' end of at least one Factor VIII non-functional coding sequence to be
repaired, the
DNA-SE target site located about 50 bp to about 100 bp upstream from a 5' end
of the Factor
VIII non-functional coding sequence to be repaired; and introducing into the
cell of the
subject a cDNA repair editing cassette comprising a cDNA repair sequence (cDNA-
RS)
26
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
coding for a repaired version of the Factor VIII non-functional coding
sequence, the cDNA
repair sequence in frame with the Factor VIII functional coding sequence. In
those
embodiments, location of the target site within about 50-100 base pairs
upstream of the
desired region to be replaced on the F8 genomic locus results in optimal
recombination by the
repair vehicle, donor plasmid, or editing cassette comprising the cDNA-RS.
Optimal
recombination is an important aspect as it results in an increase in the
likelihood that the
cDNA-RS will be incorporated at the targeted site within an individual cell
and/or population
of cells following exposure to the cDNA ¨RS. Also, following recombination of
the repair
vehicle, donor plasmid, or editing cassette into the target site, expression
of the repaired F8
gene segment results in expression of a repaired and functional FVIII protein.
Thus,
conditions promoting optimal recombination greatly contribute towards
achieving optimal
expression of a repaired and functional protein for treatment and/or induction
of immune
tolerance.
[0106] Also in those embodiments the cDNA repair editing cassette within a DNA
donor
where the cDNA repair editing cassette is flanked by an upstream flanking
sequence (uFS)
homologous to a genomic nucleic acid sequence of at least 200 bp from the DNA-
SE target
site and a downstream flanking sequence (dFS) homologous to a genomic nucleic
acid
sequences of at least 200 bp downstream of the DNA-SE target site. In those
embodiments
introducing one more nucleic acids encoding a DNA scission enzyme (DNA-SE) and
introducing a cDNA repair editing cassette is performed to allow homologous
recombination
of the upstream flanking sequence (uFS) and the downstream flanking sequence
(dFS) with
corresponding genomic sequences of the Factor VIII gene of the subject.
[0107] In some embodiments, the DNA-SE target site is adjacent to a 3' end of
the Factor
VIII functional coding sequence, and in particular the 3' end of the
functional coding
sequence can be a 3' end of a Factor VIII exon.
[0108] In some embodiments, the upstream flanking sequence (uFS) is homologous
to a
genomic nucleic acid sequence of at least about 400 bp from the DNA-SE target
site and the
downstream flanking sequence (dFS) is homologous to a genomic nucleic acid
sequences of
at least about 400 bp downstream of the DNA-SE target site.
[0109] In some embodiments, the upstream flanking sequence (uFS) is homologous
to a
genomic nucleic acid sequence of at least about 400-800 bp from the DNA-SE
target site and
27
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
the downstream flanking sequence (dFS) is homologous to a genomic nucleic acid
sequences
of at least about 400-800 bp downstream of the DNA-SE target site.
[0110] In some embodiments, the uFS is homologous to a genomic nucleic acid
sequence of
at least about 800-3000 bp from the DNA-SE target site and the dFS is
homologous to a
genomic nucleic acid sequences of at least about 800-3000 bp downstream of the
DNA-SE
target site.
[0111] In some embodiments, the cDNA repair sequence (cDNA-RS) encodes for one
or
more repaired Factor VIII non-functional sequence consisting essentially of
the amino acid
sequence encoded by exons 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15,
26, 17, 18,
19, 20, 21, 22, 23, 24, 25, 26, or an in frame portion or combination thereof
[0112] In some embodiments, the methods and compositions set forth herein, the
DNA-SEs
that targets a mutation in F8 for repair are, for example, a transcription
activator¨like effector
nuclease (TALEN), a zinc finger nuclease (ZFN), a CRISPR (Clustered Regularly
Interspaced
Short Palindromic Repeats)-associated (Cas) nuclease (CasN), a pair of wild-
type CasN each
containing its own CRISPR-single-guide-RNA (CRISPR-sgRNA) targeting a deep
intronic
sequence of a F8 intron flanking the two sides of a large F8 exonic
duplication (to repair a
HA-causing F8 mutation comprised of a large duplication of one or more F8
exons by
introducing a double-stranded DNA (dsDNA) break on each side of large exonic
duplication
such that intervening genomic DNA sequence comprising the duplication can be
deleted,
thereby restoring the transcriptional and post-transcriptional functionality
to the repair F8
sequence), a pair of missense mutant Cas nickases -- each capable of
introducing only a
single-stranded DNA (ssDNA) break -- using paired CRISPR guide RNAs, or CRISPR
with
RFN, as described in detail below.
[0113] To minimize off-target effects associated with the DNA-SEs, a program
such as
PROGNOS is used. The PROGNOS algorithm locates for example potential TALEN off-
target sites by searching through the genome for sequences similar to the
intended TALEN
design. It ranks these similar sequences according to various features of
TALEN-DNA
interactions, including RVD base preferences, polarity of TALEN specificity
(5' end is more
specific), context dependent compensation of strong RVDs (such as NN and HD),
and a
model of dimeric TALEN interactions. The PROGNOS model has been shown to
accurately
predict the majority of all known TALEN off-target sites as discussed in Fine
et al. Nucleic
28
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
Acids Research 2013, incorporated herein by reference in their entirety.
[0114] The algorithm employed for ranking potential CRISPR off-target sites
described in
Hsu et al. Nature Biotech 2013, incorporate herein by reference, uses a
position-weight-
matrix (PWM) to determine the importance of different types of mismatches at
each position
in the target sequence (both the DNA bases targeted by the guide strand as
well as the
protospacer adjacent motif sequence). This PWM was derived by experimentally
observing
the drop in nuclease activity at a target site of artificial guide strands
(relative to a perfectly
matched guide strand) containing different types of mismatches. This PWM is
then used to
screen potential sites in the genome with homology to the intended target and
assign them a
score indicating their likelihood of off-target activity.
[0115] In some embodiments the DNA-SE is Transcription Activator-Like Effector
Nucleases (TALENs) which provides an alternative to zinc finger nucleases
(ZFNs) for
certain types of genome editing. The C-terminus of the TALEN component carries
nuclear
localization signals (NLSs), allowing import of the protein to the nucleus.
Downstream of
the NLSs, an acidic activation domain (AD) is also present, which is probably
involved in the
recruitment of the host transcriptional machinery. The central region harbors
a series of
nearly identical 34/35 amino acids modules repeated in tandem. Residues in
positions 12 and
13 are highly variable and are referred to as repeat-variable di-residues
(RVDs). Studies of
TALENs such as AvrBs3 from X axonopodis pv. vesicatoria and the genomic
regions (e.g.,
promoters) they bind, led two teams to "crack the TALE code" by recognizing
that each RVD
in a repeat of a particular TALE determines the interaction with a single
nucleotide. Most of
the variation between TALEs relies on the number (ranging from 5.5 to 33.5)
and/or the order
of the quasi-identical repeats. Estimates using design criteria derived from
the features of
naturally occurring TALEs suggest that, on average, a suitable TALEN target
site can be
found every 35 base pairs in genomic DNA. Compared with ZFNs, the cloning
process of
TALENs is easier, the specificity of recognized target sequences is higher,
and off-target
effects are lower. In one study, TALENs designed to target chemokine receptor
5 (CCR5)
were shown to have very little activity at the highly homologous chemokine
receptor 2
(CCR2) locus, as compared with CCR5-specific ZFNs that had similar activity at
the two
sites.
[0116] FIG. 2 and FIG. 3 provide exemplary illustrations outlining the use of
a repair vehicle
29
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
encoding a TALEN nuclease that is used to repair the F8 gene in, for example,
a human with
an intron-22 (I22)-inverted F8 locus, F8122I. As illustrated in Figure 2(A),
the major
transcription unit of the F8122I locus consists of 24 exons, which are
designated exons 1-22
(a functional coding sequence) and exons 23C & 24C (a non-functional coding
sequence).
The first 22 are the same as exons 1-22 of the wild-type FVIII structural
locus (F8) but the
last two (exon-23C & exon-24C) are cryptic and non-functional in non-
hemophilic
individuals as well as in patients whose HA is caused by F8 gene abnormalities
other than the
1221-mutation. As illustrated in Figure 2(B) the strategy to repair the 1221-
mutation consists
of introducing in the cell of the subject a repair vehicle encoding a
functional TALEN --
which is a heterodimeric nuclease comprised of a monomer subunit that binds 5'
of the
desired genome editing site (TALEN-L) and one that binds 3' of it (TALEN-R) --
that is
specific for a DNA sequence that is present in only a single copy per haploid
human genome,
which is approximately 1 kb downstream of the 3 '-end of exon-22. Upon
expression, once
both monomers are bound to this specific sequence, their individual Fokl
nuclease domains
dimerize to form the active enzyme that catalyzes a double-stranded (ds) break
in the DNA
between their binding sites. If a ds-DNA break occurs in the presence of a
second nucleic
acid, for example a cDNA-RS (a functional coding sequence) comprising a native
FVIII 3'
splice acceptor site operably linked to a nucleic acid encoding a truncated
FVIII polypeptide
encoding exons 23-26 (i.e., a "donor plasmid (DP)" or donor sequence), which
is flanked by
a stretch of DNA with a left homology (HL) arm and right homology (HL) arm
that have
identical DNA sequences to that in the native chromosomal DNA 5' and 3' of the
region
flanking the break-point, homologous recombination (HR) occurs very
efficiently. Following
HR, the cDNA-RS segment between the left and right homology arms (which as
shown in
Figure 2 contains a partial human F8 cDNA that contains, in-frame, all of
exons 23-25 and
the coding sequence of exon-26, with a functional 3'-splice site at its 5'-
end) becomes
permanently ligated/inserted into the chromosome. Since the cDNA-RS fused at
its 5'-end to
a functional 3'-splice site, this TALEN catalyzes repair and converts F8122I
into wild-type
F8-like locus and restore its ability to drive synthesis of a full-length
fully functional wild-
type FVIII protein. Figure 3 shows the details of a functional heterodimeric
TALEN,
comprised of left and right monomer subunits (TALEN-L and TALEN-R), bound to
its target
"editing" sequence in intron-22 (122) of the human FVIII structural locus
(F8), ¨1 kb
downstream of the 3 '-end of exon-22 (Figure 3).
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
[0117] Likewise, FIG. 4 shows a functional heterodimeric TALEN targeting a F8
mutation in
canine, comprised of its left and right monomer subunits (TALEN-L and TALEN-
R), bound
to its target "editing" sequence in the 122 of the canine F8 structural locus
(cF8), ¨0.25 kb
downstream of the 3'-end of exon-22. Because the target binding sequence of
each monomer
is the same in both a wild-type canine F8 (cF8) and an I22-inverted F8 gene
(cF8-I221), this
TALEN edits each locus equally well. Following binding of this TALEN's
monomeric
subunits to their target I22-sequences in the cF8-122I locus of a dog with
severe HA caused
by the 1221-mutation, their individual Fokl nuclease domains are able to form
a homo-dimer,
i.e. the active form of the enzyme, which catalyzes a double-stranded (ds)
break in the DNA
between the monomer binding sites; this site is labeled as the target site. If
a ds-DNA break
occurs in the presence of a donor sequence or plasmid, which contains a
stretch of DNA with
left and right arms that have identical DNA sequences to that in the native
chromosomal
DNA, in the region flanking the break-point (see Figure 3 for the human F8
locus), homHR
occurs very efficiently. Following HR, the DNA segment between the left and
right
homology arms (which contains a partial cF8 cDNA that contains, in-frame, all
of exons 23-
25 and the coding sequence of exon-26, with a functional 3'-splice site at its
5'-end) becomes
permanently ligated/inserted into the canine X-chromosome. Because the DNA
segment
between the left and right homology arms comprises a partial cF8 cDNA (which,
as shown in
Figure 2 for the human F8-122I, contains, in-frame, all of canine exons 23-25
and the coding
sequence of canine exon-26) fused at its 5'-end to a functional 3'-splice
site, this TALEN
catalyzes repair and converts cF8-122I into a wild-type cF8-like locus that
restores its ability
to drive synthesis of a full-length fully functional wild-type canine FVIII.
[0118] FIG. 5 illustrates a TALEN-mediated strategies to repair the human
Factor VIII
(FVIII) gene (F8) mutations in >50% of all patients with severe hemophilia-A
(HA),
including the highly recurrent intron-22 (I22)-inversion (1221)-mutation.
Figure 5 highlights
the TALEN approach linking Exon 22 of the F8 gene to a nucleic acid including
exons 23-26
encoding a truncated FVIII polypeptide. Panel A of Figure 5 shows the specific
F8 genomic
DNA sequence (spanning positions 126,625 - 126,693) within which a double-
stranded DNA
break (DSDBs) is introduced (designated "Endonuclease domain" and "target
site" in Panel
B) by this strategy's functional TALEN dimer. The left and right TALEN protein
sequences
for the variable DNA-binding domain are listed as Seq. ID. No. 4 and Seq. ID.
No. 6,
respectively. An example of DNA sequences encoding the left and right TALEN
DNA-
31
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
binding domains are listed as Seq. ID. No 5 and Seq. ID. No. 7, respectively.
Because of the
degeneracy of the genetic code, there are many possible constructs that can be
used to encode
TALEN DNA-binding domains. In some embodiments, the codons are optimized for
expression of the DNA constructs. Panel A in Figure 5 also shows the F8
genomic DNA
sequence containing (i) the recognition sites for the left (TALENL-hF8E22422)
and right
(TALENR-hF8E22n22) TALEN monomers comprising F8-TALEN-5 and (ii) the
intervening
spacer region within which the F8-TALEN-5's endonuclease activity creates the
double-
stranded DNA breaks (DSDBs) required for inducing the physiologic cellular
machinery that
mediates the homology-dependent DNA repair pathway. Panel A in Figure 5 also
shows
important orienting landmarks, including the following: (i) Nucleotide
coordinates of this
region (based on the February, 2009, human genome assembly [UCSC Genome
Browser:
http://genome.ucsc.edu/]) are numbered with respect to the wild-type F8
transcription unit,
where the initial (5'-most) base of the F8 pre-mRNA (5'-base of exon-1 [El])
is designated
+1 or 1 (note that this base corresponds to X-chromosome position 154,250,998)
and includes
the appropriate intronic sequence bases in calculating the genomic base
positioning; (ii)
Relative location of the X-chromosome's centromere (X-Cen) and its long-arm
telomere (Xq-
Tel), as transcription of the wild-type F8 locus and all of its mutant alleles
causing HA
¨with the exception of its two recurrent intronic inversions, the intron-1
(I1)-inversion (In)-
and the 1221-mutations¨ is oriented towards X-Cen. Transcription of the Ii-
and I22-
inverted F8 loci, in contrast, are oriented towards Xq-Tel. This strategy
repairs (i) the highly
recurrent 1221-mutation ¨ also designated F81221 ¨ which causes ¨45% of all
unrelated
patients with severe hemophilia-A (HA) and (ii) mutant F8 loci in ¨20% of all
other patients
with severe HA, who are either known or found to have any one of the >200
distinct
mutations that have been found (according to the HAMSTeRS database of HA-
causing F8
mutations) thus far to reside down-stream (i.e., 3') of exon-22 (E22). The
last codon of exon
22 encodes methionine (Met [M]) as translated residue 2,143 (2,124 in the
mature FVIII
protein secreted into plasma). Most mutations repaired are "previously known"
(literature
and/or HAMSTeRS or other databases), some have never been identified
previously; the F8
abnormalities in this latter category are "private" (found only in this
particular) to the
patient/family.
[0119] Panel B in Figure 5 shows the functional aspects of the TALENs
including the overall
DNA-binding domain (DBD) and the DBD-subunit repeats of the left and right
monomers
32
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
(TALENL-hF8E22422 and TALENR-hF8E22/122). Also shown are the (i) specific DNA
sequences recognized by each TALEN monomer (shown in bold font immediately
below
each DBD-subunit); (ii) the spacer region between the DNA recognition
sequences of the
TALEN monomers contains the sequence within which the dimerized Fokl catalytic
domains, which form a functional endonuclease, introduce a double-stranded DNA
break
(DSDB) ; this site is indicated as the target site. As shown in the lower left
portion of Figure
5, the introduction of a DSDB in the presence of homologous repair vehicle no.
5 (HRV5),
the nucleotide sequence of which is provided below as Seq. ID. No. 12, results
in the in-
frame integration, immediately 3' to exon 22, of the partial human F8 cDNA
comprising
exons 23, 24 and 25 and the protein coding sequence, or CDS, of exon 26
(designated
hF8[E23-E25/E26cDs]). In one embodiment, the TALEN constructs depicted in
Figure 5 can
be used to repair all 1221 inversion mutations (See #1 pathway). In another
embodiment, the
same constructs can be used to repair non-I221 F8 mutations that occur 3'
(i.e. downstream)
of the exon-22/intron-22 junction (See #2 pathway).
[0120] FIG. 6 illustrates a TALEN-mediated strategy to repair the human F8
mutations in
>50% of all patients with severe HA, including the highly recurrent 1221-
mutation. Figure 6
highlights the TALEN approach linking intron-22 of the F8 to a nucleic acid
encoding a
truncated FVIII polypeptide encoding exons 23-26. Panel A shows the specific
F8 genomic
DNA sequence within which a DSDB is introduced (designated "Endonuclease
domain" in
Panel B and "target site") by this strategy's functional TALEN dimer. The left
and right
TALEN protein sequences for the variable DNA-binding domain are listed as Seq.
ID. No. 8
and Seq. ID. No. 10, respectively. Examples of DNA sequences encoding the left
and right
TALEN DNA-binding domains are listed as Seq. ID. No. 9 and Seq. ID. No. 11,
respectively.
Because of the degeneracy of the genetic code, there are many possible
constructs that can be
used to encode TALEN DNA-binding domains. In some embodiments, the codons are
optimized for expression of the DNA constructs. Panel A in Figure 6 also shows
important
orienting landmarks, including the: (i) nucleotide coordinates of this region
(based on the
February, 2009, human genome assembly available at the UCSC Genome Browser:
http://genome.ucsc.edu/) are numbered with respect to the wild-type F8
transcription unit,
where the initial (5'-most) base of the F8 pre-mRNA (5' most base of exon-1
[El]) is
designated +1 or 1 (note that this base corresponds to X-chromosome position
154,250,998)
and includes the appropriate intronic sequence bases in calculating the
genomic base
33
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
positioning; (ii) relative location of the X-chromosome's centromere (X-Cen)
and its long-
arm telomere (Xq-Tel), as transcription of the wild-type F8 locus and all of
its mutant alleles
causing HA ¨with the exception of its two recurrent intronic inversions, Ill-
and the 1221-
mutations ___________________________________________________________ is
oriented towards X-Cen; Transcription of the Ii- and I22-inverted F8 loci, in
contrast, is oriented towards Xq-Tel. This strategy repairs (i) the highly
recurrent 1221-
mutation ¨ also designated F81221 ¨ which causes ¨45% of all unrelated
patients with
severe HA and (ii) mutant F8 loci in ¨20% of all other patients with severe
HA, who are
either known or found to have any one of the >200 distinct mutations that have
been found
(according to the HAMSTeRS database of HA-causing F8 mutations) thus far to
reside
down-stream (i.e., 3') of exon-22 (E22). The last codon of E22 entirely
encodes methionine
(Met [M]) as translated residue 2,143 (2,124 in the mature FVIII secreted into
plasma). Most
mutations repaired are "previously known" (literature and/or HAMSTeRS or other
databases), but some have never been identified previously. The F8
abnormalities in this
latter category are "private" (found only in this particular) to the
patient/family.
[0121] Panel B in Figure 6 shows the functional aspects of the TALENs
including the overall
DBD and the DBD-subunit repeats of the left and right monomers (TALENL-hF822
and
TALENR-hF8I22). Also shown are the (i) specific DNA sequences recognized by
each
TALEN monomer (shown in bold font immediately below each DBD-subunit); (ii)
the spacer
region between the DNA recognition sequences of the TALEN monomers contains
the
sequence within which the dimerized Fokl catalytic domains, which form a
functional
endonuclease, introduce a DSDB; this site is indicated as the target site.. As
shown in the
lower left portion of Figure 6, the introduction of a DSDB in the presence of
a homologous
repair vehicle, the nucleotide sequence of which is listed as Seq. ID. No. 13,
results in the
integration into intron-22 of a native F8 3' splice acceptor site operably
linked to a nucleic
acid encoding F8 exons-23, 24 and 25 and the protein coding sequence, or CDS,
of exon-26
(designated hF8[E23-E25/E26cDs]). In one embodiment, the TALEN constructs
depicted in
Figure 6 can be used to repair all 1221 inversion mutations (See #1 pathway).
In another
embodiment, the same constructs are used to repair non-I221 F8 mutations that
occur 3' (i.e.
downstream) of the exon-22/intron-22 junction (See #2 pathway).
[0122] FIG. 7 shows a comparison of expected genomic DNA, spliced RNA and
proteins pre
and post repair. Several examples of functional and non-functional coding
sequences are
depicted in the gDNA panel of FIG. 7. Example functional coding sequences
include exons
34
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
1-22 and exons 22-23 of the wild-type F8 genomic DNA (Normal), exons 1-22 of
the 1221
mutant F8 genomic DNA (1221), and exons 1-22 of the 1221 mutant F8 genomic DNA
and
exons 23-26 of the wild-type F8 cDNA (Repaired). Example non-functional coding
sequences include exons 23-26 of the 1221 mutant F8 genomic DNA (1221) and
exons 23-26
of the 1221 mutant F8 genomic DNA (right, Repaired).
[0123] In some embodiments, nucleic acids encoding nucleases specifically
target intron-1,
intron-14, or intron-22. In some embodiments, nucleic acids encoding nucleases
specifically
target the exon-1/intron-1 junction; exon-14/intron-14 junction; or the exon-
22/intron-22
junction.
[0124] Figure 9 illustrates an example of a donor plasmid that can be used to
repair the F8 at
the exon-22/intron-22 junction using a TALEN, ZFN, CRISPR/Cas, CRISPR-PN, and
CRISPR-RFN approach. The donor plasmid contains the cDNA sequence for exons 23-
26 of
the F8 (labeled as functional coding sequence) and a polyadenylation signal
sequence flanked
by two regions of homology to the F8. The left homology region contains a DNA
sequence
(approximately 700 base pairs) that is homologous to part of intron-21 and
exon-22 of the F8.
The right homology region contains a DNA sequence (approximately 700 base
pairs) that is
homologous to part of intron-22 of the F8. Upon successful homologous
recombination into
the F8 locus, the integrated construct expresses the resulting mRNA encoding
the wild-type
(corrected) version of the FVIII. The sequence of the plasmid depicted in
Figure 9 is listed as
Seq. ID. No. 12. The annotation of Seq. ID. No. 12 is provided in Table 1
below.
Table 1: Repair vehicle targeted to the Exon 22 ¨ Intron 22 junction of F8
LOCUS RepairVehicle 7753 bp DNA linear
FEATURES Location/Qualifiers
misc_feature 21..327
/note="fl origin (-)"
misc_feature 6765..7625
/note="<= Ampicillin"
misc_feature 471..614
/label=<= lacZ A
misc_feature 626..644
/note="T7 promoter =>"
misc_feature 5564..5583
/note="T3 promoter =>"
misc_feature 6765..7625
/note="<= Orfl"
misc_feature 7667..7695
/note="<= AmpR promoter"
misc_feature 658..740
/note="MCS"
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
Table 1: Repair vehicle targeted to the Exon 22 ¨ Intron 22 junction of F8
misc_feature 1446..2072
/note="Exons 23-26 (cDNA seq)"
misc_feature 1730..1737
/note="Create NotI site"
misc_feature 2082..2707
/note="hGH polyA"
misc_feature 1785..1787
/note="ns-SNP: A6940G (M2238V)"
misc_feature 3408..4160
/note="HSV-TK promoter "
misc_feature 4161..5546
/note="HSV-TK gene and TK pA Terminator "
misc_feature 741..745
/note="Create site for cloning"
misc_feature 5547..5551
/note="Create site for cloning"
misc_feature 746..1445
/note="Left homolgy arm (700 bp)"
misc_feature 1290..1445
/note="Exon 22"
misc_feature 1433..1445
/note="Partial Left TALEN recognition site"
misc_feature 2708..3407
/note="Right homology arm (700 bp)"
misc_feature 2708..2716
/note="Partial Right TALEN recognition site"
misc_feature 2708..3407
/note="Partial Intron 22"
misc_feature 746..1289
/note="Partial Intron 21"
source 1..7753
/dnas_title="RepairVehicle E22-I22 pBluescript"
[0125] FIG. 10 illustrates an example of a donor plasmid that can be used to
repair the F8
using a TALEN, ZFN, CRISPR/Cas, CRISPR-PN, and CRISPR-RFN approach. The donor
plasmid contains the cDNA sequence for exons2-26 of the F8 (labeled as
functional coding
sequence) flanked by two regions of homology to the F8. The left homology
region contains
a DNA sequence that is homologous to part of the F8 promoter and part of exon-
1. The right
homology region contains a DNA sequence that is homologous to part of intron-
1. Upon
successful homologous recombination into the F8, the integrated construct
expresses the
resulting mRNA encoding the wild-type (corrected) version of the FVIII. The
donor sequence
is cloned into plasmid (p)BlueScript-II KS-minus (pBS-II-KS[). The donor
plasmid is used
with a TALEN, ZFN, CRISPR/Cas, CRISPR-PN, and CRISPR-RFN genomic editing
strategy. The sequence of the plasmid depicted in Figure 10 is listed as Seq.
ID. No. 13. The
annotation of Seq. ID. No. 13 is provided in Table 2 below.
Table 2: Repair vehicle targeted to the Exon 1 ¨ Intron 1 junction of F8
LOCUS RepairVehicle 11418 bp DNA linear
36
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
Table 2: Repair vehicle targeted to the Exon 1 ¨ Intron 1 junction of F8
FEATURES Location/Qualifiers
misc_feature 21..327
/note="fl origin (-)"
misc_feature 10430..11290
/note="<= Ampicillin"
misc_feature 471..614
/label=<= lacZ A
misc_feature 626..644
/note="T7 promoter =>"
misc_feature 9229..9248
/note="<= T3 promoter"
misc_feature 10430..11290
/note="<= Orfl"
misc_feature 11332..11360
/note="<= AmpR promoter"
misc_feature 658..740
/note="MCS"
misc_feature 5780..6405
/note="hGH polyA"
misc_feature 7073..7825
/note="HSV-TK promoter "
misc_feature 7826..9211
/note="HSV-TK gene and TK pA Terminator "
misc_feature 740..745
/note="Create site for cloning"
misc_feature 1540..5770
/note="Exons 2-26 BDD (cDNA seq)"
misc_feature 2664..2669
/note="Create ClaI site"
misc_feature 2903..2905
/note="ns-SNP: G1679A (R484H)"
misc_feature 3680..3685
/note="BDD (5er743 - G1n1638)"
misc_feature 5428..5435
/note="Create NotI site"
misc_feature 5768..5768
/dnas_title="Stop"
/vntifkey="21"
/label=Stop
misc_feature 5483..5485
/note="ns-SNP: A6940G (M2238V)"
insertion_seq 3934..5770
/dnas_title="Tg"
/vntifkey="14"
/label=Tg
misc_feature 9212..9217
/note="Create site for cloning"
misc_feature 9212..9212
/note="MCS"
misc_feature 746..1539
/note="Left homolgy arm (794bp)"
misc_feature 746..1237
/note="Partial F8 promoter"
misc_feature 1238..1539
/note="Partial Exon 1"
misc_feature 6406..7072
/note="Right homology arm (667 bp)"
misc_feature 6406..7072
/note="Partial intron 1"
37
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
Table 2: Repair vehicle targeted to the Exon 1 ¨ Intron 1 junction of F8
source 1..11418
/dnas_title="RepairVehicle El-I1 pBluescript"
[0126] Figure 11 illustrates an example of a donor plasmid that is used to
repair the F8 in
intron-22 using a TALEN, ZFN, CRISPR/Cas, CRISPR-PN, and CRISPR-RFN approach.
The donor plasmid contains a 3' splice site, the cDNA sequence for exons 23-26
of the F8
(labeled as functional coding sequence), and a polyadenylation signal sequence
flanked by
two regions of homology to the F8. The left homology region contains a DNA
sequence
(approximately 700 base pairs) that is homologous to part of intron-22 of the
F8. The right
homology region contains a DNA sequence (approximately 700 base pairs) that is
homologous to part of intron-22 of the F8. Upon successful homologous
recombination into
the F8 locus, the integrated construct expresses the resulting mRNA encoding
the wild-type
(corrected) version of the FVIII. The sequence of the plasmid depicted in
Figure 11 is listed
as Seq. ID. No. 14. The annotation of Seq. ID. No. 14 is provided in Table 3
below.
Table 3: Repair vehicle targeted to Intron 22 of F8
LOCUS RepairVehicle 7755 bp DNA linear
FEATURES Location/Qualifiers
misc_feature 21..327
/note="fl origin (-)"
misc_feature 6767..7627
/note="<= Ampicillin"
misc_feature 471..614
/label=<= lacZ A
misc_feature 626..644
/note="T7 promoter =>"
misc_feature 5566..5585
/note="T3 promoter =>"
misc_feature 6767..7627
/note="<= Orfl"
misc_feature 7669..7697
/note="<= AmpR promoter"
misc_feature 658..740
/note="MCS"
misc_feature 1448..2074
/note="Exons 23-26 (cDNA seq)"
misc_feature 1732..1739
/note="Create NotI site"
misc_feature 2084..2709
/note="hGH polyA"
misc_feature 1787..1789
/note="ns-SNP: A6940G (M2238V)"
misc_feature 3410..4162
/note="HSV-TK promoter "
misc_feature 4163..5548
/note="HSV-TK gene and TK pA Terminator "
38
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
Table 3: Repair vehicle targeted to Intron 22 of F8
misc_feature 741..745
/note="Create site for cloning"
misc_feature 5549..5553
/note="Create site for cloning"
misc_feature 746..1445
/note="Left homology arm (700 bp)"
misc_feature 1437..1445
/note="Partial Left TALEN recognition site"
misc_feature 2710..3409
/note="Right homolgy arm (700 bp)"
misc_feature 2710..2719
/note="Partial Right TALEN recognition site"
misc_feature 746..1445
/note="Partial Intron 22"
misc_feature 2710..3409
/note="Partial Intron 22"
misc_feature 1446..1447
/note="3' spice site"
source 1..7755
/dnas title="RepairVehicle 122 pEluescript"
[0127] Figure 12 illustrates an example of a donor plasmid that is used to
repair the F8 in
intron-1 using a TALEN, ZFN, CRISPR/Cas, CRISPR-PN, and CRISPR-RFN approach.
The
donor plasmid contains a 3' splice site, the cDNA sequence of the F8 for exons
2-26 lacking
the B-domain (B-domain deleted (BDD) version of the F8) (labeled as functional
coding
sequence), and a polyadenylation signal sequence flanked by two regions of
homology to the
F8. The left homology region contains a DNA sequence (approximately 700 base
pairs) that
is homologous to part of exon-1 and intron-1 of the F8 gene. The right
homology region
contains a DNA sequence (approximately 700 base pairs) that is homologous to
part of
intron-1 of the F8. Upon successful homologous recombination into the F8
locus, the
integrated construct expresses the resulting mRNA encoding the wild-type
(corrected)
version of the FVIII. The sequence of the plasmid depicted in Figure 12 is
listed as Seq. ID.
No. 15. The annotation of Seq. ID. No. 15 is provided in Table 4 below.
Table 4: Repair vehicle targeted to Intron 1 of F8
LOCUS RepairVehicle 11359 bp DNA linear
FEATURES Location/Qualifiers
misc_feature 21..327
/note="fl origin (-)"
misc_feature 10371..11231
/note="<= Ampicillin"
misc_feature 471..614
/label=<= lacZ A
misc_feature 626..644
/note="T7 promoter =>"
39
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
Table 4: Repair vehicle targeted to Intron 1 of F8
misc_feature 9170..9189
/note="<= T3 promoter"
misc_feature 10371..11231
/note="<= Orfl"
misc_feature 11273..11301
/note="<= AmpR promoter"
misc_feature 658..740
/note="MCS"
misc_feature 874..1187
/note="Exon 1"
misc_feature 1436..1445
/note="Partial Left TALEN recognition site"
misc_feature 5688..6313
/note="hGH polyA"
misc_feature 6314..7013
/note="Right homology arm (700 bp)"
misc_feature 6314..6322
/note="Partial Right TALEN recognition site"
misc_feature 7014..7766
/note="HSV-TK promoter "
misc_feature 7767..9152
/note="HSV-TK gene and TK pA Terminator "
misc_feature 746..1445
/note="Left homolgy arm (700 bp)"
misc_feature 746..873
/note="Partial F8 promoter"
misc_feature 740..745
/note="Create site for cloning"
misc_feature 6314..7013
/note="Partial Intron 1"
misc_feature 1448..5678
/note="Exons 2-26 BDD (cDNA seq)"
misc_feature 1446..1447
/note="3' spice site"
misc_feature 2572..2577
/note="Create ClaI site"
misc_feature 2811..2813
/note="ns-SNP: G1679A (R484H)"
misc_feature 3588..3593
/note="BDD (Ser743 - G1n1638)"
misc_feature 5336..5343
/note="Create NotI site"
misc_feature 5676..5676
/dnas_title="Stop"
/vntifkey="21"
/label=Stop
misc_feature 5391..5393
/note="ns-SNP: A6940G (M2238V)"
insertion_seq 3842..5678
/dnas_title="Tg"
/vntifkey="14"
/label=Tg
misc_feature 9153..9158
/note="Create site for cloning"
misc_feature 9153..9153
/note="MCS"
source 1..11359
/dnas_title="RepairVehicle Ii pBluescript"
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
[0128] In one embodiment, the integration matrix component for each of the
distinct
homologous donor plasmid is either a cDNA that is linked to the immediately
upstream exon
or a cDNA that has a functional 3'-intron-splice-junction so that the cDNA
sequence is linked
through the RNA intermediate following removal of the intron. In one
embodiment, the
donor plasmid is personalized, on an individual basis, so that each patient's
gene that is
repaired expresses the form of the FVIII that they are maximally tolerant of
[0129] In some embodiments the DNA-SE used for F8 targeting is a ZFN. ZFNs are
hybrid
proteins containing the zinc-finger DNA-binding domain present in
transcription factors and
the non-specific cleavage domain of the endonuclease Fokl. (Li et al., In vivo
genome
editing restores hemostasis in a mouse model of hemophilia, Nature 2011 Jun
26;
475(7355):217-21).
[0130] The same sequences targeted by the TALEN approach, discussed above, can
also be
targeted by the ZFN approach for genome editing. ZFNs are a class of
engineered DNA-
binding proteins that facilitate targeted editing of the genome by creating
DSDB at user-
specified locations. Each ZFN consists of two functional domains: 1) a DBD
comprised of a
chain of two-finger modules, each recognizing a unique hexamer (6 bp) sequence
of DNA,
wherein two-finger modules are stitched together to form a ZFN, each with
specificity of?
24 bp, and 2) a DNA-cleaving domain comprised of the nuclease domain of Fok 1.
The
DNA-binding and DNA-cleaving domains are fused together and recognize the
targeted
genomic sequences, allowing the Fokl domains to form a heterodimeric enzyme
that cleaves
the DNA by creating double stranded breaks.
[0131] ZFNs can be readily made by using techniques known in the art (Wright
DA, et al.
Standardized reagents and protocols for engineering zinc finger nucleases by
modular
assembly. Nat Protoc. 2006;1(3):1637-52). Engineered ZFNs can stimulate gene
targeting at
specific genomic loci in animal and human cells. The construction of
artificial zinc finger
arrays using modular assembly has been described. The archive of plasmids
encoding more
than 140 well-characterized zinc finger modules together with complementary
web-based
software for identifying potential zinc finger target sites in a gene of
interest has also been
described. These reagents enable easy mixing-and-matching of modules and
transfer of
assembled arrays to expression vectors without the need for specialized
knowledge of zinc
finger sequences or complicated oligonucleotide design (Wright DA, et al.
Standardized
41
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
reagents and protocols for engineering zinc finger nucleases by modular
assembly. Nat
Protoc. 2006;1(3):1637-52). Any gene in any organism can be targeted with a
properly
designed pair of ZFNs. Zinc-finger recognition depends only on a match to the
target DNA
sequence (Carroll, D. Genome engineering with zinc-finger nucleases. Genetics
Society of
America, 2011, 188(4), pp 773-782).
[0132] In some embodiments the DNA-SE used for F8 gene targeting comprises
Clustered
Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR Associated
(Cas)
Nucleases based on CRISPR technology. (Mali P, Yang L, Esvelt KM, Aach J,
Guell M,
DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via
Cas9.
Science. 2013 Feb 15;339(6121):823-6; Gasiunas G, Barrangou R, Horvath P,
Siksnys V.
Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for
adaptive
immunity in bacteria. Proc Natl Acad Sci U S A. 2012 Sep 25;109(39):E2579-86.
Epub
2012 Sep 4).
[0133] The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)
and
CRISPR Associated (Cas) system was discovered in bacteria and functions as a
defense
against foreign DNA, either viral or plasmid. In bacteria, the endogenous
CRISPR/Cas
system targets foreign DNA with a short, complementary single-stranded RNA
(CRISPR
RNA or crRNA) that localizes the Cas9 nuclease to the target DNA sequence. The
DNA
target sequence can be on a plasmid or integrated into the bacterial genome.
The crRNA can
bind on either strand of DNA and the Cas9 cleaves both strands (double strand
break, DSB).
A recent in vitro reconstitution of the Streptococcus pyogenes type II CRISPR
system
demonstrated that crRNA fused to a normally trans-encoded tracrRNA is
sufficient to direct
Cas9 protein to sequence-specifically cleave target DNA sequences matching the
crRNA.
The fully defined nature of this two-component system allows it to function in
the cells of
eukaryotic organisms such as yeast, plants, and even mammals. By cleaving
genomic
sequences targeted by RNA sequences, such a system greatly enhances the ease
of genome
engineering.
[0134] The crRNA targeting sequences are transcribed from DNA sequences known
as
protospacers. Protospacers are clustered in the bacterial genome in a group
called a CRISPR
array. The protospacers are short sequences (-20bp) of known foreign DNA
separated by a
short palindromic repeat and kept like a record against future encounters. To
create the
42
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
CRISPR targeting RNA (crRNA), the array is transcribed and the RNA is
processed to
separate the individual recognition sequences between the repeats. In the Type
II system, the
processing of the CRISPR array transcript (pre-crRNA) into individual crRNAs
is dependent
on the presence of a trans-activating crRNA (tracrRNA) that has sequence
complementary to
the palindromic repeat. When the tracrRNA hybridizes to the short palindromic
repeat, it
triggers processing by the bacterial double-stranded RNA-specific
ribonuclease, RNase III.
Any crRNA and the tracrRNA can then both bind to the Cas9 nuclease, which then
becomes
activated and specific to the DNA sequence complimentary to the crRNA. (Mali
P, Yang L,
Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided
human
genome engineering via Cas9. Science. 2013 Feb 15;339(6121):823-6; Gasiunas G,
Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex
mediates
specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U
S A. 2012
Sep 25;109(39):E2579-86. Epub 2012 Sep 4).
[0135] The DSDB induced by the TALEN approach overlaps with the 6 distinct
sites of
DSDB induced by Cas9, via targeting by 6 distinct CRISPR-guide RNAs [F8-
CRISPR/Cas9-
1 (F8-Exl/Int1), F8-CRISPR/Cas9-2 (F8-Intl), F8-CRISPR/Cas9-3 (F8-Ex14/Intl
4), F8-
CRISPR/Cas9-4 (F8-Int14), F8-CRISPR/Cas9-5 (F8-Ex22/Int22), F8-CRISPR/Cas9-6
(F8-
Int22)]. This allows use of the same 6 distinct homologous donor sequences
with all three
genome editing approaches, including the TALEN nuclease, ZFN, and the Cas
nuclease.
[0136] Figure 13 illustrates a CRISPR/Cas9-mediated strategy to repair the
human Factor
VIII (FVIII) gene (F8) mutations in ¨95% of all patients with severe
hemophilia-A (HA),
including the highly recurrent intron-1 (I1)-inversion (M)-mutation as well as
the intron-22
(I22)-inversion (122I)-mutation. Figure 13 shows the specific F8 genomic DNA
sequence
(spanning genic base positions 172 ¨ 354 at intron 1) within which a double-
stranded (ds)-
DNA break is introduced (designated "Endonuclease target" or "target site" in
this panel) by
this strategy's wild-type (wt) CRISPR/Cas9 ds-DNase in which both of its
endonuclease
domains are catalytically functional ("hF8-CRISPR/Cas9wt-1"). This panel also
shows
important orienting landmarks, including the following: (i) Nucleotide
coordinates of this
region (based on the February, 2009, human genome assembly [UCSC Genome
Browser:
http://genome.ucsc.edu/]) are numbered with respect to the wild-type F8
transcription unit,
where the initial (5'-most) base of the F8 pre-mRNA (5'-base of exon-1 [El])
is designated
+1 or 1 (note that this base corresponds to X-chromosome position 154,250,998)
and include
43
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
the appropriate intronic sequence bases in calculating the genomic base
positioning; (ii)
Relative location of the X-chromosome's centromere (X-Cen) and its long-arm
telomere (Xq-
Tel), as transcription of the wild-type F8 locus and all of its mutant alleles
causing HA
¨with the exception of its two recurrent intronic inversions, the In- and the
1221-
mutations ___________________________________________________________ is
oriented towards X-Cen. Transcription of the Ii- and I22-inverted F8 loci, in
contrast, are oriented towards Xq-Tel. This strategy repairs (i) the highly
recurrent 1221-
mutation ¨ also designated F81221 ¨ which causes ¨45% of all unrelated
patients with
severe hemophilia-A (HA) and (ii) mutant F8 loci in ¨90-95% of all other
patients with
severe HA, who are either known or found to have any one of the >1,500
distinct mutations
that have been found (according to the HAMSTeRS database of HA-causing F8
mutations)
thus far to reside down-stream (i.e., 3') of exon-1 (El). The last codon of El
partially
encodes the translated residue 48 (29 in the mature FVIII protein secreted
into plasma). Most
mutations repaired are "previously known" (literature and/or HAMSTeRS or other
databases). Some have never been identified previously. These F8 abnormalities
in this latter
category are "private" (found only in this particular) to the patient/family.
Finally, Figure 13
shows the functional aspects of hF8-CRISPR/Cas9wt-1 including the overall DNA-
binding
domain of the CRISPR-associated guide (g)RNA as well as the (i) Protospacer
adjacent motif
(PAM), which is the site at which the DNase function of Cas9 introduces the ds-
DNA break
(DSDB); and (ii) The Transactivating Crispr-RNA (TrCr-RNA), which is
covalently attached
the gRNA as is what brings the Cas9 endonuclease to the genomic DNA target for
digestion.
The introduction of a DSDB in the presence of a homologous repair vehicle,
results in the in-
frame integration, immediately 3' to El, of one of either two partial human F8
cDNAs
comprising either (i) exons 2-25 and the protein coding sequence, or CDS, of
exon 26
(designated hF8[E2-E25/E26cDs]), which effects repair of the F8 gene such that
it now
encodes a full-length wild-type FVIII protein; or (ii) Exons 2-13 entirely
linked next to the
very 5'-most end of exon-14 (E14), which in turn is linked covalently to the
very 3'-most end
of E14 (i.e., a B-domain-deleted [BDD]-F8 cDNA), which is then covalently
linked to Exons
15-25 entirely, and then the protein coding sequence, or CDS, of exon 26
(designated
hF8[E2-E13/E14-BDD/E15-E25/E26cDs]), which effects repair of the F8 gene such
that it
now encodes a BDD-engineered FVIII protein, which is fully functional in
FVIII:C activity.
The homologous repair vehicle is selected to have a F8 cDNA with the
appropriate alleles at
all ns-SNP sites so that the patient can receive a "matched" gene repair or at
least a least
mismatched repair.
44
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
[0137] The left homology arm of the homologous repair vehicle for Homologous
Repair
Vehicle No. 1 (HRV1) for hF8-CRISP/Cas9wt-1 is listed as Seq. ID. No. 17 and
comprises
the first 1114 bases of the human F8 genomic DNA (which is shown here as
single-stranded
and representing the sense strand) and contains 800 bp of the immediately 5'-
promoter region
of the human F8 gene and all 314 bp of the F8 exon-1 (El), including its 171
bp 5'-UTR and
its 143 bp of protein (en)coding sequence (CDS). The actual left homologous
arm (LHA) of
the homologous repair vehicle (HRV1), which is used for this CRISPR/Cas9-
mediated F8
gene repair (that occurs at the El/intron-1 [Ii] junction of a given patient's
endogenous
mutant F8), contains at least 500 bp of this genomic DNA sequence (i.e., from
it's very 3'-
end, which corresponds to the second base of the codon for translated residue
48 of the wild-
type FVIII protein and residue 29 of the mature FVIII protein found in the
circulation) and
could include it all, if, for example, we find that full-length F8 gene repair
can be effected
efficiently in the future. In this instance, the integration matrix would then
follow the LHA
of this HRV1, and be covalently attached to it, and this integration matrix
contains (in-frame
with each other and with the 3'-end of the patient's native exon-1, which is
utilized in situ,
along with his native F8 promoter, to regulate expression of the repaired F8
gene), all of F8
exons 2-25, and the protein CDS of exon-26, followed by the functional mRNA 3'-
end
forming signals of the human growth hormone gene (hGH-pA). The F8 cDNA from
exons 2-
25 and the CDS of exon-26 to be used in the homologous repair vehicle is
listed as Seq. ID.
No. 18 and follows the left homology arm, and in this example represents the
haplotype (H)3
encoding wild-type variant of F8, which can be used to cure, for example,
patients with the
In-mutation and the 1221-mutation, that arose on an H3-background haplotype.
This
following protein encoding cDNA sequence contains 6,909 bp of the entire 7,053
bp of F8
protein encoding sequence (i.e., the first 144 bp of protein CDS from FVIII,
from its initiator
methionine, is not shown, as this is contained in exon-1, which is provided by
the patient's
own endogenous exon-1, providing it is not mutant and thus precluding the
repair event).
The right homology arm of the homologous repair vehicle for the cas nuclease
approach is
listed as Seq. ID. No. 19 and includes 1109 bases of human F8 genomic DNA
(which is
shown here as single-stranded and representing the sense strand) from the F8
gene intron 1.
[0138] In some embodiments, the DNA-SE is a CRISPR Paired Nickase. A single
CRISPR
nuclease targets a total of 22 bp of DNA sequence, which is much less than
what is targeted
by dimeric TALENs (30-40 bp) or ZFNs (30-36 bp); as a result, some CRISPR
nucleases can
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
have substantial off-target activity throughout the rest of the genome. The
Cas9 protein has
two nuclease domains (an HNH domain and a RuvC domain) which each cleave one
of the
strands of the DNA helix in order to cause a double-strand break. By
inactivating one of the
nuclease domains in Cas9 (through the amino acid mutation D1 OA or H840A), the
Cas9
molecule becomes a `nickase' which can only cause a break in one strand of DNA
thereby
creating a nick rather than a double-strand break. However, by targeting to
Cas9-nickase
molecules to nearby regions of DNA, offset nicks can in effect cause a double-
strand break
with DNA overhangs similar to how the two FokI dimers in ZFNs and TALENs come
together to create a double-strand DNA break with overhanging bases.
Guidelines for how to
orient the paired target sites for Cas9-nickases were developed by Ran FA, Hsu
PD et al. Cell
2013, incorporated herein by reference, and it was shown that similar on-
target activity was
able to be achieved by correctly oriented paired Cas9-nickases as by a single
Cas9-
nuclease. Importantly, it was also shown that at sites previously identified
as having off-
target activity when using a certain guide strand with the Cas9 nuclease that
when using the
Cas9-nickase the off-target activity was reduced ¨1400 fold. The hypothesis
for the
reduction in off-target activity is that although at the previously identified
off-target site there
was homology to one of the guide strands (which allowed off-target activity
using the Cas9-
nuclease), in that region of the genome there was not also homology to the
other guide strand
in the pair; binding of a single Cas9-nickase does not induce DNA mutations,
it is only when
both guide strands bind in proper orientation that nicks are made in both DNA
strands to
create a double strand break which can lead to mutations through the NHEJ
pathway. By
creating the requirement that both guide strands bring the two nickases to the
same region of
the genome, the effective targeting length of the paired Cas9-nickase system
is 44 bp,
compared to 22 bp of the Cas9-nuclease system, greatly enhancing specificity
in large
genomes such as the human genome.
[0139] Example of repair at the exon21/intron-21 junction (the 3'-end of exon-
21), using
paired nickase are described below. Repair of the F8 at exon-21/intron-21
junction, i.e. the
3'-end of exon-21 would correct HA in patients with mutations in exons 22, 23,
24, 25, or 26,
as well as the common 1221 mutation. Examples of known patient mutations in
exons 22-26
are detailed in Figure 14, including, but not limited to (i) the F8 c.6761 T>A
nonsense
mutation that results in a stop codon at codon 2178 in place of the leucine
(Leu)-encoding
codon that is present at codon 2178 in the non-mutated form of the F8; (ii)
the F8 c.6917
46
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
T>G missense mutation that results in a codon encoding arginine (Arg) at codon
2230 in
place of the leucine (Leu)-encoding codon that is present at that codon 2230
in the non-
mutated form of the F8; (iii) the F8-122I mutation that is detailed above;
(iv) the F8 IVS-23
+1 G>A splice site mutation that results in a non-functional pre-mRNA splice
site
immediately downstream of exon-23 of the F8; (v) the F8 del exons 24-26 multi-
exonic
deletion mutation that results in deletion of exons 24-26 of the F8; and (vi)
the F8 exon-26
del.[A] small deletion and frameshift mutation that results in a frameshift of
the gene-
encoding sequence which changes the downstream sequence by a single base-pair
deletion
frameshift and introduction of a novel terminating stop codon in the gene-
encoding sequence.
Creating the double-strand break at exon-21/intron-21 junction can be
accomplished by using
DNA-SE including such as TALENs, Cas9-nuclease, paired Cas9-nickases, or RNA-
guided
Fold Nucleases disclosed herein. An example of how to create such a break in
F8 with paired
Cas9-nickases is illustrated in Figure 15. Specifically, Cas9-nickases are
shown binding near
the exon-21/intron-21 junction of F8. The Cas9-nickases create nicks on both
strands of F8
DNA, thereby generating a double-strand break that will trigger homology
directed repair;
the site of the break is indicated as the "target site." An engineered
homologous repair
vehicle (HRV) disclosed herein is then introduced to the cells along with the
DNA-SE in
order to be used as a template in the homology directed repair pathway. An
example of a RV
to be used at the exon-21/intron-21 junction is shown here Figure 16.
Regardless of the
mechanism used to create the DNA-break at the exon-21/intron-21 junction the
same RV can
be used to alter the gene sequence. This RV has a LHA corresponding to the
sequence 5' of
the DNA break labeled as "target break" (exon-21 and a portion of intron-20),
the cDNA
sequence encoding the downstream exons of the F8 (exons 22-26), a
polyadenylation signal
(such as the signal from the hGH gene labeled as "target break," hGH-pA), and
aRHA
corresponding to the sequence 3' of the DNA break (intron-21). After homology
directed
repair takes place, the gDNA sequence now contains a healthy copy of exons 22-
26 fused to
exon-21, allowing expression of the full-length F8. The RV can also contain
SNPs in order
to haplotypically match a certain patient; an example SNP (6940 A>G) is shown
here.
[0140] In some embodiments the DNA-SE comprises CRISPR-RNA-guided Fokl
nucleases
(CRISPR-RFN). Although the paired Cas9-nickases dramatically increased the
specificity of
CRISPR systems, low levels of off-target activity were still observed at some
sites (Ran FA
and Hsu PD et al. Cell 2013), presumably due to the occasional repair of DNA
nicks through
47
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
the error-prone NHEJ pathway rather than the error free base-excision-repair
pathway. In
contrast to a Cas9-nickase, which will cut one strand of DNA even in the
absence of its
corresponding pair, the FokI nuclease requires dimerization in order to cleave
DNA; the
presence of a single Fold monomer will not make any modification to the DNA.
The Cas9
molecule can have all of its DNA cleavage activity removed by mutating both
DNA cleavage
domains (using the amino acid substitutions D 1 OA and H840A) which is known
as "dead"
Cas9 or dCas9. When the FokI domain is fused to dCas9, two properly oriented
guide
strands can bring the two Fold domains in close proximity where they can
dimerize and
create a double-strand break, in a similar manner to ZFNs and TALENs. Tsai SQ
et al
(Nature Biotech 2014), incorporated herein by reference, determined that with
correct
orientation of guide strands and fusing Fold to the N-terminus of dCas9,
double-strand breaks
can be made efficiently by these RNA-guided Fold Nucleases, termed "RFNs".
Tsai et al
further characterized the off-target activity of these RFNs and found that
they had even lower
levels of off-target activity than the paired Cas9-nickases targeted to the
same locations; in
almost all cases the off-target activity of the RFNs was below the detection
limit of the deep-
sequencing-based assay employed. A further method in which RFNs reduce off-
target
activity is that they are more limited in what orientations they can
efficiently cleave DNA
compared to paired Cas9-nickases. This reduces the possibility for off-target
sites, but also
limits the types of sequences which can be targeted by RFNs; several 3' ends
of the exons in
the F8 gene did not contain the required sequence motifs to be able to be
effectively targeted
by RFNs. Overall, RFNs have benefits and drawbacks compared to the paired Cas9-
nickases,
but nonetheless represent another addition to the toolkit of nucleases
available to create
double-strand breaks in order to trigger homology-directed repair.
[0141] In methods and systems and related cDNA, vehicles and composition
herein descried
the gene targeting and repair approaches using the different nucleases of the
disclosure can be
carried out using many different target cells. For example, the transduced
cells can include
endothelial cells, hepatocytes, or stem cells. In one embodiment, the cells
can be targeted in
vivo. In one embodiment, the cells can be targeted using ex vivo approaches
and
reintroduced into the subject.
[0142] In one embodiment, the target cells from the subject are endothelial
cells. In one
embodiment, the endothelial cells are blood outgrowth endothelial cells
(BOECs).
Characteristics that render BOECs attractive for gene repair and delivery
include the: (i)
48
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
ability to be expanded from progenitor cells isolated from blood, (ii) mature
endothelial cell,
stable, phenotype and normal senescence (-65 divisions), (iii) prolific
expansion from a
single blood sample to 1019 BOECs, (iv) resilience, which unlike other
endothelial cells,
permits cryopreservation and hence multiple doses for a single patient
prepared from a single
isolation. Methods of isolation of BOECs are known, where the culture of
peripheral blood
provides a rich supply of autologous, highly proliferative endothelial cells,
also referred to as
blood outgrowth endothelial cells (BOECs). Bodempudi V, et al., Blood
outgrowth
endothelial cell-based systemic delivery of antiangiogenic gene therapy for
solid tumors.
Cancer Gene Ther. 2010 Dec;17(12):855-63.
[0143] Studies in animal models have revealed properties of blood outgrowth
endothelial
cells that indicate that they are suitable for use in ex vivo gene repair
strategies. For example,
a key finding concerning the behavior of canine blood outgrowth endothelial
cells (cBOECs)
is that cBOECs persist and expand within the canine liver after infusion.
Milbauer LC, et al.
Blood outgrowth endothelial cell migration and trapping in vivo: a window into
gene therapy.
2009 Apr;153(4):179-89. Whole blood clotting time (WBCT) in the HA model was
also
improved after administration of engineered cBOECs. WBCT dropped from a
pretreatment
value of under 60 mm to below 40 min and sometimes below 30 min. Milbauer LC,
et al.,
Blood outgrowth endothelial cell migration and trapping in vivo: a window into
gene therapy.
2009 Apr;153(4):179-89.
[0144] In one embodiment, the target cells from the subject are hepatocytes.
In one
embodiment, the cell is a liver sinusoidal endothelial cell (LSECs). Liver
sinusoidal
endothelial cells (LSEC) are specialized endothelial cells that play important
roles in liver
physiology and disease. Hepatocytes and liver sinusoidal endothelial cells
(LSECs) are
thought to contribute a substantial component of FVIII in circulation, with a
variety of extra-
hepatic endothelial cells supplementing the supply of FVIII.
[0145] In one embodiment, the present disclosure targets LSEC cells, as LSEC
cells likely
represent the main cell source of FVIII. Shahani, T, et al., Activation of
human endothelial
cells from specific vascular beds induces the release of a FVIII storage pool.
Blood 2010;
115(23):4902-4909. In addition, LSECs are believed to play a role in induction
of immune
tolerance. Onoe, T, et al., Liver sinusoidal endothelial cells tolerize T
cells across MHC
barriers in mice. J Immunol 2005; 175(1):139-146. Methods of isolation of
LSECs are
49
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
known in the art. Karrar, A, et al., Human liver sinusoidal endothelial cells
induce apoptosis
in activated T cells: a role in tolerance induction. Gut. 2007 February;
56(2): 243-252.
[0146] In one embodiment, the transduced cells from the subject are stem
cells. In one
embodiment, the stem cells are induced pluripotent stem cells (iPSCs). Induced
pluripotent
stem cells (iPSCs) are a type of pluripotent stem cell artificially derived
from a non-
pluripotent cell, typically an adult somatic cell, by inducing expression of
specific genes and
factors important for maintaining the defining properties of embryonic stem
cells. Induced
pluripotent stem cells (iPSCs) have been shown in several examples to be
capable of site
specific gene targeting by nucleases. Ru, R. et al. Targeted genome
engineering in human
induced pluripotent stem cells by penetrating TALENs. Cell Regeneration. 2013,
2:5; Sun
N, Zhao H. Seamless correction of the sickle cell disease mutation of the HBB
gene in
human induced pluripotent stem cells using TALENs. Biotechnol Bioeng. 2013 Aug
8.
Induced pluripotent stem cells (iPSCs) can be isolated using methods known in
the art.
Lorenzo, IM. Generation of Mouse and Human Induced Pluripotent Stem Cells
(iPSC) from
Primary Somatic Cells. Stem Cell Rev. 2013 Aug;9(4):435-50.
[0147] As discussed above, a number of different cells types can be targeted
for repair.
However, in some cases, pure populations of some cell types may not promote
sufficient
homing and implantation upon reintroduction to provide extended and sufficient
expression
of the corrected F8 gene. Therefore, some cell types may be co-cultured with
different cell
types to help promote cell properties (i.e. ability of cells to engraft in the
liver).
[0148] In one embodiment, the transduced cells are from blood outgrowth
endothelial cells
(BOECs) that have been co-cultured with additional cell types. In one
embodiment, the
transduced cells are from blood outgrowth endothelial cells (BOECs) that have
been co-
cultured with hepatocytes or liver sinusoidal endothelial cell (LESCs) or
both. In one
embodiment, the transduced cells are from blood outgrowth endothelial cells
(BOECs) that
have been co-cultured with induced pluripotent stem cells (iPSCs).
[0149] In embodiments of methods and systems herein described and related
vehicles
composition methods and systems, the polynucleotide encoding for the DNA-SE
and repair
vehicles RVs comprising the DNA donor can be delivered to the cells with
methods of
nucleic acid delivery well known in the art. (See, e.g., WO 2012051343). In
the methods
provided herein, the described nuclease encoding nucleic acids can be
introduced into the cell
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
as DNA or RNA, single-stranded or double-stranded and can be introduced into a
cell in
linear or circular form. In one embodiment, the nucleic acids encoding the
nuclease are
introduced into the cell as mRNA. The donor sequence can introduced into the
cell as DNA
single-stranded or double-stranded and can be introduced into a cell in linear
or circular form.
If introduced in linear form, the ends of the nucleic acids can be protected
(e.g., from
exonucleolytic degradation) by methods known to those of skill in the art. For
example, one
or more dideoxynucleotide residues are added to the 3' terminus of a linear
molecule and/or
self-complementary oligonucleotides are ligated to one or both ends. See, for
example,
Chang et al. (1987) Proc. Natl. Acad. Sci. USA 84:4959- 4963; Nehls et al.
(1996) Science
272:886-889.
Additional methods for protecting exogenous polynucleotides from
degradation include, but are not limited to, addition of terminal amino
group(s) and the use of
modified intemucleotide linkages such as, for example, phosphorothioates,
phosphoramidates, and 0-methyl ribose or deoxyribose residues.
[0150] The nucleic acids can be introduced into a cell as part of a vector
molecule having
additional sequences such as, for example, replication origins, promoters and
genes encoding
antibiotic resistance. Moreover, the nucleic acids can be introduced as naked
nucleic acid, as
nucleic acid complexed with an agent such as a liposome or poloxamer, or can
be delivered
by viruses (e.g., adenovirus, AAV, herpesvirus, retrovirus, lentivirus).
[0151] The nucleic acids can be delivered in vivo or ex vivo by any suitable
means. Methods
of delivering nucleic acids are described, for example, in U.S. Patent Nos.
6,453,242;
6,503,717; 6,534,261; 6,599,692; 6,607,882; 6,689,558; 6,824,978; 6,933,113;
6,979,539;
7,013,219; and 7,163,824.
[0152] Any vector systems can be used including, but not limited to, plasmid
vectors,
retroviral vectors, lentiviral vectors, adenovirus vectors, poxvirus vectors;
herpesvirus vectors
and adeno-associated virus vectors, etc. See, also, U.S. Patent Nos.
6,534,261; 6,607,882;
6,824,978; 6,933,113; 6,979,539; 7,013,219; and 7,163,824. Furthermore, any of
these
vectors can comprise one or more of the sequences needed for treatment. Thus,
when one or
more nucleic acids are introduced into the cell, the nucleases and/or donor
sequence nucleic
acids can be carried on the same vector or on different vectors. When multiple
vectors are
used, each vector can comprise a sequence encoding a nuclease, a nickase, or a
donor
sequence nucleic acid. Alternatively, two or more of the nucleic acids can be
contained on a
51
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
single vector.
[0153] Conventional viral and non- viral based gene transfer methods can be
used to
introduce nucleic acids encoding the nucleic acids in cells (e.g., mammalian
cells) and target
tissues. Non- viral vector delivery systems include DNA plasmids, naked
nucleic acid, and
nucleic acid complexed with a delivery vehicle such as a liposome or
poloxamer. Viral
vector delivery systems include DNA and RNA viruses, which have either
episomal or
integrated genomes after delivery to the cell. Methods of non- viral delivery
of nucleic acids
include electroporation, lipofection, microinjection, biolistics, virosomes,
liposomes,
immunoliposomes, polycation or lipid:nucleic acid conjugates, naked DNA,
artificial virions,
and agent- enhanced uptake of DNA. Sonoporation using, e.g., the Sonitron 2000
system
(Rich- Mar) can also be used for delivery of nucleic acids.
[0154] Additional exemplary nucleic acid delivery systems include those
provided by Amaxa
Biosystems (Cologne, Germany), Maxcyte, Inc. (Rockville, Maryland), BTX
Molecular
Delivery Systems (Holliston, MA) and Copernicus Therapeutics Inc, (see for
example
U56008336). Lipofection is described in e.g., U.S. Patent Nos. 5,049,386;
4,946,787; and
4,897,355) and lipofection reagents are sold commercially {e.g., TransfectamTM
and
LipofectinTm). Cationic and neutral lipids that are suitable for efficient
receptor-recognition
lipofection of polynucleotides include those of Feigner, WO 91/17424, WO
91/16024.
[0155] The preparation of lipid:nucleic acid complexes, including targeted
liposomes such as
immunolipid complexes, is well known to one of skill in the art (see, e.g.,
Crystal, Science
270:404-410 (1995); Blaese et al, Cancer Gene Ther. 2:291-297 (1995); Behr et
al,
Bioconjugate Chem. 5:382-389 (1994); Remy et al, Bioconjugate Chem. 5:647-654
(1994);
Gao et al, Gene Therapy 2:710-722 (1995); Ahmad et al, Cancer Res. 52:4817-
4820 (1992);
U.S. Pat. Nos. 4,186,183, 4,217,344, 4,235,871, 4,261,975, 4,485,054,
4,501,728, 4,774,085,
4,837,028, and 4,946,787).
[0156] Additional methods of delivery include the use of packaging the nucleic
acids to be
delivered into EnGeneIC delivery vehicles (EDVs). These EDVs are specifically
delivered to
target tissues using bispecific antibodies where one arm of the antibody has
specificity for the
target tissue and the other has specificity for the EDV. The antibody brings
the EDVs to the
target cell surface and then the EDV is brought into the cell by endocytosis.
Once in the cell,
the contents are released (see MacDiarmid et al (2009) Nature Biotechnology
27(7):643).
52
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
[0157] The use of RNA or DNA viral based systems for the delivery of nucleic
acids take
advantage of highly evolved processes for targeting a virus to specific cells
in the body and
trafficking the viral payload to the nucleus. Viral vectors can be
administered directly to
patients (in vivo) or they can be used to treat cells in vitro and the
modified cells are
administered to patients (ex vivo). Conventional viral based systems for the
delivery of
nucleic acids include, but are not limited to, retroviral, lentivirus,
adenoviral, adeno-
associated, vaccinia and herpes simplex virus vectors for gene transfer.
[0158] The tropism of a retrovirus can be altered by incorporating foreign
envelope proteins,
expanding the potential target population of target cells. Lentiviral vectors
are retroviral
vectors that are able to transduce or infect non-dividing cells and typically
produce high viral
titers. Selection of a retroviral gene transfer system depends on the target
tissue. Retroviral
vectors are comprised of cz's-acting long terminal repeats with packaging
capacity for up to
6-10 kb of foreign sequence. The minimum cz's-acting LTRs are sufficient for
replication
and packaging of the vectors, which are then used to integrate the therapeutic
gene into the
target cell to provide permanent transgene expression. Widely used retroviral
vectors include
those based upon murine leukemia virus (MuLV), gibbon ape leukemia virus
(GaLV), Simian
Immunodeficiency virus (SIV), human immunodeficiency virus (HIV), and
combinations
thereof (see, e.g., Buchscher et al, J. Virol. 66:2731-2739 (1992); Johann et
al, J. Virol.
66:1635-1640 (1992); Sommerfelt et al., Virol. 176:58-59 (1990); Wilson et al,
J. Virol.
63:2374-2378 (1989); Miller et al, J. Virol. 65:2220- 2224 (1991); PCT
U594/05700).
[0159] In applications in which transient expression is preferred, adenoviral
based systems
can be used. Adenoviral based vectors are capable of very high transduction
efficiency in
many cell types and do not require cell division. With such vectors, high
titer and high levels
of expression have been obtained. This vector can be produced in large
quantities in a
relatively simple system. Adeno-associated virus ("AAV") vectors are also used
to transduce
cells with target nucleic acids, e.g., in the in vitro production of nucleic
acids and peptides,
and for in vivo and ex vivo gene therapy procedures (see, e.g., West et al,
Virology 160:38-
47 (1987); U.S. Patent No. 4,797,368; WO 93/24641; Kotin, Human Gene Therapy
5:793-
801 (1994); Muzyczka, J. Clin. Invest. 94:1351(1994). Construction of
recombinant AAV
vectors is described in a number of publications, including U.S. Pat. No.
5,173,414; Tratschin
et al, Mol Cell. Biol. 5:3251-3260 (1985); Tratschin, et al, Mol. Cell. Biol.
4:2072-2081
(1984); Hermonat & Muzyczka, PNAS 81:6466-6470 (1984); and Samulski et al, J.
Virol.
53
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
63:03822-3828 (1989).
[0160] At least six viral vector approaches are currently available for gene
transfer in clinical
trials, which utilize approaches that involve complementation of defective
vectors by genes
inserted into helper cell lines to generate the transducing agent. pLASN and
MFG-S are
examples of retroviral vectors that have been used in clinical trials (Dunbar
et al, Blood
85:3048-305 (1995); Kohn et al, Nat. Med. 1 :1017-102 (1995); Malech et al,
PNAS 94:22
12133-12138 (1997)). PA317/pLASN was the first therapeutic vector used in a
gene therapy
trial. (Blaese et al, Science 270:475-480 (1995)). Transduction efficiencies
of 50% or greater
have been observed for MFG-S packaged vectors. (Ellem et al, Immunol
Immunother.
44(1):10-20 (1997); Dranoff et al, Hum. Gene Ther. 1:111-2 (1997). Recombinant
adeno-
associated virus vectors (rAAV) are an alternative gene delivery systems based
on the
defective and nonpathogenic parvovirus adeno-associated type 2 virus. All
vectors are
derived from a plasmid that retains only the AAV 145 bp inverted terminal
repeats flanking
the transgene expression cassette. Efficient gene transfer and stable
transgene delivery due to
integration into the genomes of the transduced cell are key features for this
vector system.
(Wagner et al, Lancet 351 :9117 1702-3 (1998), Kearns et al, Gene Ther. 9:748-
55 (1996)).
Other AAV serotypes, including AAV1, AAV2, AAV3, AAV4, AAV5, AAV6, AAV7,
AAV8, AAV9 and AAVrh.10 and any novel AAV serotype can also be used in
accordance
with the present disclosure. In a particular embodiment, the vector is based
on a hepatotropic
adeno-associated virus vector, serotype 8 (see, e.g., Nathwani et al., Adeno-
associated viral
vector mediated gene transfer for hemophilia B, Blood 118(21):4-5, 2011).
[0161] Replication-deficient recombinant adenoviral vectors (Ad) can be
produced at high
titer and readily infect a number of different cell types. Most adenovirus
vectors are
engineered such that a transgene replaces the Ad El a, El b, and/or E3 genes;
subsequently the
replication defective vector is propagated in human 293 cells that supply
deleted gene
function in trans. Ad vectors can transduce multiple types of tissues in vivo,
including non-
dividing, differentiated cells such as those found in liver, kidney and
muscle. Conventional
Ad vectors have a large carrying capacity. An example of the use of an Ad
vector in a
clinical trial involved polynucleotide therapy for antitumor immunization with
intramuscular
injection (Sterman et al, Hum. Gene Ther. 7:1083-9 (1998)). Additional
examples of the use
of adenovirus vectors for gene transfer in clinical trials include Rosenecker
et ah, Infection
24:1 5-10 (1996); Sterman et ah, Hum. Gene Ther. 9:7 1083-1089 (1998); Welsh
et ah, Hum.
54
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
Gene Ther. 2:205-18 (1995); Alvarez et al, Hum. Gene Ther. 5:597-613 (1997);
Topf et al,
Gene Ther. 5:507-513 (1998); Sterman et al, Hum. Gene Ther. 7:1083-1089
(1998).
[0162] Packaging cells are used to form virus particles that are capable of
infecting a host
cell. Such cells include 293 cells, which package adenovirus, and tv2 cells or
PA317 cells,
which package retrovirus. Viral vectors used in gene therapy are usually
generated by a
producer cell line that packages a nucleic acid vector into a viral particle.
The vectors
typically contain the minimal viral sequences required for packaging and
subsequent
integration into a host (if applicable), other viral sequences being replaced
by an expression
cassette encoding the protein to be expressed. The missing viral functions are
supplied in
trans by the packaging cell line. For example, AAV vectors used in gene
therapy typically
only possess inverted terminal repeat (ITR) sequences from the AAV genome
which are
required for packaging and integration into the host genome. Viral DNA is
packaged in a cell
line, which contains a helper plasmid encoding the other AAV genes, namely rep
and cap, but
lacking ITR sequences. The cell line is also infected with adenovirus as a
helper. The helper
virus promotes replication of the AAV vector and expression of AAV genes from
the helper
plasmid. The helper plasmid is not packaged in significant amounts due to a
lack of ITR
sequences. Contamination with adenovirus can be reduced by, e.g., heat
treatment to which
adenovirus is more sensitive than AAV.
[0163] In many applications, it is desirable that the g vector be delivered
with a high degree
of specificity to a particular tissue type. Accordingly, a viral vector can be
modified to have
specificity for a given cell type by expressing a ligand as a fusion protein
with a viral coat
protein on the outer surface of the virus. The ligand is chosen to have
affinity for a receptor
known to be present on the cell type of interest. For example, Han et ah,
Proc. Natl. Acad.
Sci. USA 92:9747- 9751 (1995), reported that Moloney murine leukemia virus can
be
modified to express human heregulin fused to gp70, and the recombinant virus
infects certain
human breast cancer cells expressing human epidermal growth factor receptor.
This can be
used with other virus-target cell pairs, in which the target cell expresses a
receptor and the
virus expresses a fusion protein comprising a ligand for the cell- surface
receptor. For
example, filamentous phage can be engineered to display antibody fragments
(e.g., FAB or
Fv) having specific binding affinity for virtually any chosen cellular
receptor. Although the
above description applies primarily to viral vectors, the same principles can
be applied to
non-viral vectors. Such vectors can be engineered to contain specific uptake
sequences
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
which favor uptake by specific target cells.
[0164] Vectors can be delivered in vivo by administration to an individual
patient, typically
by systemic administration (e.g., intravenous, intraperitoneal, intramuscular,
subdermal, or
intracranial infusion) or topical application, as described below.
Alternatively, vectors can be
delivered to cells ex vivo, such as cells explanted from an individual patient
(e.g.,
lymphocytes, bone marrow aspirates, tissue biopsy) or universal donor
hematopoietic stem
cells, followed by re-implantation of the cells into a patient, usually after
selection for cells
which have incorporated the vector.
[0165] Vectors (e.g., retroviruses, adenoviruses, liposomes, etc.) containing
the nucleic acids
described herein can also be administered directly to an organism for
transduction of cells in
vivo. Alternatively, naked DNA can be administered.
[0166] Administration is by any of the routes normally used for introducing a
molecule into
ultimate contact with blood or tissue cells including, but not limited to,
injection, infusion,
topical application and electroporation. Suitable methods of administering
such nucleic acids
are available and well known to those of skill in the art, and, although more
than one route
can be used to administer a particular composition, a particular route can
often provide a
more immediate and more effective reaction than another route.
[0167] Vectors suitable for introduction of the nucleic acids described herein
include non-
integrating lentivirus vectors (IDLV). See, for example, Ory et al. (1996)
Proc. Natl. Acad.
Sci. USA 93:11382-11388; Dull et al. (1998) J. Virol. 72:8463-8471; Zuffery et
al. (1998) J.
Virol. 72:9873-9880; Follenzi et al. (2000) Nature Genetics 25:217-222; U.S.
Patent
Publication No 2009/054985.
[0168] The nucleic acids encoding the monomers of the DNA scission enzymes can
be
expressed either on separate expression constructs or vectors, or can be
linked in one open
reading frame. Expression of the nuclease can be under the control of a
constitutive promoter
or an inducible promoter.
[0169] Administration can be by any means in which the polynucleotides are
delivered to the
desired target cells. For example, both in vivo and ex vivo methods are
contemplated. In one
embodiment, the nucleic acids are introduced into a subject's cells that have
been explanted
from the subject, and reintroduced following F8 gene repair.
56
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
[0170] For in vivo administration, for example, intravenous injection of the
nucleic acids to
the portal vein is a method of administration. Other in vivo administration
modes include, for
example, direct injection into the lobes of the liver or the biliary duct and
intravenous
injection distal to the liver, including through the hepatic artery, direct
injection into the liver
parenchyma, injection via the hepatic artery, and/or retrograde injection
through the biliary
tree. Ex vivo modes of administration include transduction in vitro of
resected hepatocytes or
other cells of the liver, followed by infusion of the transduced, resected
hepatocytes back into
the portal yasculature, liver parenchyma or biliary tree of the human patient,
see e.g.,
Grossman et ah, (1994) Nature Genetics, 6:335-341.
[0171] If ex vivo methods are employed, cells or tissues can be removed and
maintained
outside the body according to standard protocols well known in the art. The
compositions
can be introduced into the cells via any gene transfer mechanism as described
above, such as,
for example, calcium phosphate mediated gene delivery, electroporation,
microinjection,
proteoliposomes, or viral vector delivery. The transduced cells can then be
infused (e.g., in a
pharmaceutically acceptable carrier) or homotopically transplanted back into
the subject per
standard methods for the cell or tissue type. Standard methods are known for
transplantation
or infusion of various cells into a subject.
[0172] In some embodimentsõ the one or more mutations cause hemophilia in the
subject and
the repair results in treatment of the hemophilia in the subject. The term
"treatment" as used
herein indicates any activity that is part of a medical care for, or deals
with, a condition,
medically or surgically.
[0173] The term "subject" as used herein is meant an individual and refers to
a single
biological organism such animals and in particular higher animals and in
particular
vertebrates such as mammals and in particular human beings. . Thus, the
"subject" can
include domesticated animals, such as cats, dogs, etc., livestock (e.g.,
cattle, horses, pigs,
sheep, goats, etc.), laboratory animals (e.g., mouse, rabbit, rat, guinea pig,
etc.) and birds.
Thus, veterinary uses and medical formulations are contemplated herein. In
some
embodiments, the subject is a mammal such as a primate, for example, a human.
[0174] The term "haemophilia" indicates a group of hereditary genetic
disorders that impair
the body's ability to control blood clotting, which is used to stop bleeding
when a blood
vessel is broken.
57
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
[0175] Haemophilia A (HA) (clotting factor VIII deficiency) is the most common
form of the
disorder, present in about 1 in 5,000-10,000 male births and is caused by loss-
of-function
mutations in the X-linked Factor (F) VIII gene. Haemophilia B (HB) (factor IX
deficiency)
occurs in around 1 in about 20,000-34,000 newborn male births.
[0176] The levels of functional FVIII in circulation determine the severity of
the disease,
with plasma levels 5-25% of normal being mild, 1-5% being moderate, and <1%
being severe
(Brettler et al., Clinical aspects of and therapy for hemophilia A. Churchill
Livingstone, New
York, NY 1995; pp. 1648-63). As such, only a small amount of circulating
protein is
necessary to provide protection from spontaneous bleeding episodes.
[0177] The 1221-mutation of the F8 accounts for ¨45% of severe HA and is
caused by an
intra-chromosomal recombination within the gene. FIG. 1 shows a schematic
illustration of
the wild-type and 1221 F8 loci (F8 & F8122I). Indicated in FIG.1 are the exon-
1B (E1B) and
exon-1 to exon-22 (E1-E22) functional coding sequences as well as the exons-
23C (E23C), -
24C (E24C), and exon-23 (E23C), exon-24C (E24C) and exon-23 (E23) to exon-26
(E26)
non-functional coding sequences. Transcription from the F8 promoter of both
the F8 (wild-
type) & F8122I loci, which is normally functioning in both forms, yields
polyadenylated
mRNAs. The F8 (wild-type) mRNA has 26 exons, exon-1 (El) to exon-22 (E22) and
exon-
23 (E23) to exon-26 (E26), all of which encode the amino acids found in the
FVIII.
Conversely, the F8122I mRNA has at least 24 exons, El-E22 (they are the same
in F8 and
thus encode FVIII amino acid sequence), and E23C & E24C (they are cryptic and
encode no
FVIII amino acid sequence). The sequence of intron-22, in both F8 & F8122I,
contains a bi-
directional promoter that transcribes two additional mRNAs from the two genes:
F8A, which
is oriented oppositely to that of F8 & F8122I and contains a single exon (box
designated
ElA), and F8B, which contains five exons that are oriented similarly
transcriptionally to that
of F8 & F8122I and contains a single non-F8 first exon within 122 (box
designated ElB)
followed by four additional exons, which are identical to E23-E26 of F8. The
F8A mRNA
encodes the FVIIIA protein, which is now known as HAP40 (a cytoskeleton-
interacting
protein involved in endocytosis and thus functionally unrelated to the
coagulation system)
and has no FVIII amino acid sequence. The F8B mRNA encodes FVIII B, a protein
with
unknown function that has 8 non-FVIII amino acid residues at its N-terminus
followed by
208 residues that represent FVIII residues 2125-2332.
58
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
[0178] Infusion of replacement plasma-derived (pd) or recombinant (r) FVIII is
the standard
of care to manage this chronic disease. Currently available rFVIII replacement
products
include the commercially available Kogenate0 (Bayer) and Hefixate (ZLB
Behring),
Recombinate0 (Baxter) and Advate0 (Baxter), and the B-domain deleted Refacto0
(Pfizer)
and Xyntha0 (Pfizer). Patients unable to be treated with FVIII experience more
painful, joint
bleeding and over time, a greater loss of mobility than patients whose HA is
able to be
managed with FVIII. Infusion of replacement FVIII, however, is not a cure for
HA.
Spontaneous bleeding remains a serious problem especially for those with
severe HA,
defined as circulating levels of FVIII coagulant activity (FVIII: C) below 1%
of normal.
Furthermore, the formation of anti-FVIII antibodies occurs in about 20% of all
patients and
more often in certain subpopulations of HA patients, such as African Americans
(Viel KR,
Amen i A, Abshire TC, et al. Inhibitors of factor VIII in black patients with
hemophilia. N
Engl J Med. 360: 1618-27, 2009). There is therefore also a critical need to
identify ways to
avoid FVIII inhibitor development and to abate a FVIII inhibitor response.
[0179] In some embodiments herein described, the methods and compositions
described
herein are directed to treating a subject with hemophilia and in particular
hemophilia A
comprising selectively targeting and replacing a portion of the subject's
genomic F8 gene
sequence containing a mutation in the gene with a partial F8 cDNA replacement
sequence
(cDNA-RS). In one embodiment, the resultant repaired F8 gene containing the
cDNA-RS,
upon expression, produces functional FVIII that confers improved coagulation
functionality
to the encoded FVIII protein of the subject. The levels of functional FVIII in
circulation are
believed to obviate or reduce the need for infusions of replacement FVIII in
the subject. In
one embodiment, expression of functional FVIII reduces whole blood clotting
time (WBCT).
In one embodiment, the repaired F8 gene, upon expression, provides for the
immune
tolerance induction (ITI) to an administered replacement FVIII protein
product. In one
embodiment, the subject is a human.
[0180] In one aspect, a method of treating hemophilia A in a subject is
provided comprising
introducing into a cell of the subject one or more repair vehicles (RV)
containing at least a
cDNA-RS and one or more plasmids encoding a DNA scission enzyme (DNA-SE) such
as a
nuclease or nickase. The DNA-SE targets a portion of the F8 gene containing a
mutation that
causes hemophilia A and creates a first break in one strand of the F8 gene and
a second break
in the other strand of the F8 gene for subsequent repair by the cDNA-RS. In
some
59
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
embodiments, the first break and the second break are a double-stranded DNA
break. In
other embodiments, the first break and the second break are off-set paired and
complementary single-stranded DNA nicks. The cDNA-RS comprises (i) a nucleic
acid
encoding a truncated FVIII polypeptide or (ii) a native F8 3' splice acceptor
site operably
linked to a nucleic acid encoding a truncated FVIII polypeptide. The RV
further comprises
flanking sequences comprising an upstream flanking sequence (uFS) that is
homologous to
the nucleic acid sequences upstream of the first break in the DNA of the
subject's F8 gene
and a downstream flanking sequence (dFS) that is homologous to the nucleic
acid sequences
downstream of the second break in the DNA of the subject's F8 gene. The 5' end
of the
cDNA-RS is flanked by the uFS and the 3' end of the cDNA-RS is flanked by dFS
to form a
donor sequence that is a portion of the RV. After insertion of the cDNA-RS
through
homologous recombination into the subject's F8 gene (sF8), a repaired F8 gene
(rF8) is
formed, which upon expression forms functional FVIII that confers improved
coagulation
functionality to the FVIII protein encoded by the sF8 without the repair.
[0181] In one aspect, methods and systems for repairing F8 gene can be used to
induce
immune tolerance to a FVIII replacement product (FVIIIT) such as a recombinant
FVIII
(rFVIII) or a plasma derived FVIII (pdFVIII) in a subject having a FVIII
deficiency and who
will be administered, is being administered, or has been administered a
replacement FVIII
product is disclosed. The method comprises introducing into cells of the
subject one or more
RVs encoding a cDNA-RS and one or more plasmids encoding a DNA-SE. The DNA-SE
targets a portion of the F8 gene containing a mutation that causes hemophilia
A and creates a
first break in one strand of the F8 gene and a second break in the other
strand of the F8 gene
for subsequent repair by the cDNA-RS. In some embodiments, the first break and
the second
break are a double-stranded DNA break. In other embodiments, the first break
and the
second break are off-set paired and complementary single-stranded DNA nicks.
The cDNA-
RS comprises (i) a nucleic acid encoding a truncated FVIII polypeptide or (ii)
a native F8 3'
splice acceptor site operably linked to a nucleic acid encoding a truncated
FVIII polypeptide.
The RV further comprises flanking sequences comprising an upstream flanking
sequence
(uFS) that is homologous to the nucleic acid sequences upstream of the first
break in the
DNA of the subject's F8 gene and a downstream flanking sequence (dFS) that is
homologous
to the nucleic acid sequences downstream of the second break in the DNA of the
subject's F8
gene. The 5' end of the cDNA-RS is flanked by the uFS and the 3' end of the
cNDA-RS is
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
flanked by dFS to form a donor sequence that is a portion of the RV. After
insertion of the
cDNA-RS through homologous recombination into the subject's F8 gene (sF8), a
repaired F8
gene (rF8) is formed, which upon expression forms functional FVIII that
provides immune
tolerance induction (ITI) to an administered replacement FVIII protein
product. In some
cases, the person administered the cells may have no anti-FVIII antibodies or
have anti-FVIII
antibodies as detected by ELISA or Bethesda assays. In one embodiment, the
truncated
FVIII polypeptide amino acid sequence shares homology with a portion of the
FVIIIrp's
amino acid sequence. In one embodiment, the truncated FVIII polypeptide amino
acid
sequence shares homology with a similar portion of the FVIIIrp's amino acid
sequence. In
one embodiment, the truncated FVIII polypeptide amino acid sequence shares
complete
homology with a similar portion of the FVIIIrp's amino acid sequence.
[0182] In some embodiments, the repaired version of the Factor VIII non-
functional coding
sequence comprises Factor VIII exons of a replacement FVIII protein product
and the repair
results in inducing immune tolerance to the FVIII replacement product.
[0183] In some embodiments disclosed herein, the cDNA, polynucleotides repair
vehicles
plasmids and vehicles herein described are provided as a part of systems to
repair F8 gene in
a subject. The systems can be provided in the form of a kits of part. In a kit
of parts, the
cDNA, polynucleotides repair vehicles plasmids and vehicles herein described
and other
reagents to repair one or more mutations of the F8 gene can be comprised in
the kit
independently. The cDNA, polynucleotides repair vehicles plasmids and vehicles
herein
described can be included in one or more compositions, and each capture agent
can be in a
composition together with a suitable excipient.
[0184] In some embodiments, additional components of the system include
reagents,
antibodies and enzymes that can be used to verify proper integration and
expression of the
cDNA-RS. Proper integration can be assessed through a variety of means that
would be
apparent to one of ordinary skill in the art, including DNA sequencing by
Sanger technique or
by next-generation sequencing techniques of the desired genomic DNA site of
cDNA-RS
integration to ensure proper integration of the donor sequence. Expression of
a repaired FVIII
can be assessed through a variety of means that would be apparent to one of
ordinary skill in
the art including using ELISA assays to measure repaired FVIII expression both
intracellularly expressed and secreted into the medium and commercially-
available
61
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
coagulation and FVIII assays for measuring coagulation activity.
[0185] In particular, in some embodiments components of the kit are provided,
with suitable
instructions and other necessary reagents, in order to perform the methods
here described.
The kit will normally contain the compositions in separate containers.
Instructions, for
example written or audio instructions, on paper or electronic support such as
tapes or CD-
ROMs, for carrying out the assay, will usually be included in the kit. The kit
can also contain,
depending on the particular method used, other packaged reagents and materials
(e.g.
Chromogenix Coamatic Factor VIII kit, available from Diapharma
(Itttp://www.diaphartna.comlaspiproductdetails.asp?ID=1 00080) can be used for
measuring
FVIII activity).
[0186] In some embodiments, the cDNA, polynucleotides repair vehicles plasmids
and
vehicles herein described herein described can be included in pharmaceutical
compositions
together with an excipient or diluent. In particular, in some embodiments,
disclosed are
pharmaceutical compositions which contain at least one cDNA, polynucleotides
repair
vehicles plasmids and vehicles herein described in combination with one or
more compatible
and pharmaceutically acceptable excipients, and in particular with
pharmaceutically
acceptable diluents or excipients. In those pharmaceutical compositions the
multi-ligand
capture agent can be administered as an active ingredient for treatment or
prevention of a
condition in an individual.
[0187] The term "excipient" as used herein indicates an inactive substance
used as a carrier
for the active ingredients of a medication. Suitable excipients for the
pharmaceutical
compositions herein described include any substance that enhances the ability
of the body of
an individual to absorb the multi-ligand capture agents or combinations
thereof. Suitable
excipients also include any substance that can be used to bulk up formulations
with the
peptides or combinations thereof, to allow for convenient and accurate dosage.
In addition to
their use in the single-dosage quantity, excipients can be used in the
manufacturing process to
aid in the handling of the peptides or combinations thereof concerned.
Depending on the
route of administration, and form of medication, different excipients can be
used. Exemplary
excipients include, but are not limited to, antiadherents, binders, coatings,
disintegrants,
fillers, flavors (such as sweeteners) and colors, glidants, lubricants,
preservatives, sorbents.
[0188] The term "diluent" as used herein indicates a diluting agent which is
issued to dilute
62
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
or carry an active ingredient of a composition. Suitable diluents include any
substance that
can decrease the viscosity of a medicinal preparation.
[0189] Further details concerning the identification of the suitable carrier
agent or auxiliary
agent of the compositions, and generally manufacturing and packaging of the
kit, can be
identified by the person skilled in the art upon reading of the present
disclosure.
EXAMPLES
[0190] The methods and system herein disclosed are further illustrated in the
following
examples, which are provided by way of illustration and are not intended to be
limiting.
[0191] In particular, the following examples illustrate exemplary embodiments
in accordance
with exemplary procedures in accordance to the present disclosure. A person
skilled in the art
will appreciate the applicability of the features described in detail for the
exemplified
embodiments to different methods, different applications and different
reaction conditions
and reagents in accordance with the present disclosure.
Example 1: Ex vivo Gene Repair
[0192] Examples are provided of an ex vivo gene repair strategies that can be
performed
without the use of viral vectors. Genetic materials are delivered to restore
secretion of a wild-
type full-length FVIII to lymphoblastoid cells derived from a human HA patient
with the
F81221, using electroporation and TALENs. A similar strategy can be used as an
example to
repair the naturally-occurring 1221-mutation in cells from an animal model of
HA (dogs of the
HA canine colony located at the University of North Carolina in Chapel Hill).
Canine
(adipose) tissue, which can be induced to acquire many properties of
hepatocytes, can be
used.
[0193] Use of autologous cells is an attractive therapy for several reasons as
levels of blood
clotting proteins needed to maintain hemostasis may be more readily produced
by expansion
of large populations of cells ex vivo and reintroduction into the patient.
Repair of the F8122I
gene residing in a B-lymphoblastoid cell-line derived from a patient with
severe HA caused
by the 1221-mutation is effected by using electroporation to deliver (i) two
distinct mRNAs
encoding a highly specific heterodimeric TALEN that targets a single human
genome site
located in F8 near the 5'-end of 122 and (ii) the corresponding donor plasmid
that carries the
63
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
"editing cassette", which is comprised of a functional 3'-intron splice site
ligated
immediately 5' of a partial F8 cDNA matched in sequence with the wild-type
sequence of
exons 23-26 in the patient's own F8122I locus, flanked by "left" and "right"
homology arms.
[0194] The use of viral-free methods to derive autologous cells of various
phenotypes and to
stably introduce genetic information into the genome is attractive. These
methods can be
effectively used to successfully "repair" the F8122I, which arises through a
highly-recurrent
mutational event essentially restricted to the male germ-line. This same F8
abnormality,
which is widely known as the 1221-mutation, occurs naturally in dogs, and
results in
spontaneous bleeding. Two large colonies of HA dogs have been established, one
at the
University of North Carolina in Chapel Hill. Investigation of F8122I at the
molecular genetic,
biochemical, and cellular levels to characterize its expression products have
been studied in
order to determine the immune response to replacement FVIII. Extensive
sequencing efforts
and analyses of the F8122I and its mRNA transcripts allow for an innovative
gene repair
strategy that exploits nuclease technology, for example, transcription
activator-like effector
TALEN technology to repair the 1221-mutation.
[0195] Lymphoblastoid cells derived from HA patient with the 1221-mutation is
obtained.
The left (TALEN-L) and right (TALEN-R) monomers comprising the heterodimeric
TALEN
is shown in Figure 3, which was specifically designed to cleave within the
human F8 I22-
sequence, ¨1 kb downstream of the 3'-end of exon-22. In alternative
embodiments, the
TALENs target sequences throughout the FVIII gene, with replacement of the
corresponding
FV8 gene sequence on the donor sequence.
[0196] An example of a sequence that can be targeted includes a sequence
within intron 22
(tactatgggatgagttgcagatggcaagtaagacactggggagattaaat (SEQ. ID No.
1)), where the
underlined regions of sequence are recognized by the left TAL Effector DNA-
binding
domain and the right TAL Effector DNA-binding domain). Another example of a
sequence
that can be targeted includes a sequence at the junction of exon 22 with
intron 22
(Iggaaccttaatggtatgtaattagtcatttaaagggaatgcctgaata (SEQ. ID No. 2)), where the
underlined
regions of sequence are recognized by the left TAL Effector DNA-binding domain
and the
right TAL Effector DNA-binding domain). Another example of a sequence that can
be
targeted within intron 22 is depicted in Figure 3
(ttagtattatagtttctcagattatcaccagtgatactatggga
(SEQ. ID No. 3)), where the underlined regions of sequence are recognized by
the left TAL
64
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
Effector DNA-binding domain and the right TAL Effector DNA-binding domain).
The two
TALEN expression plasmids that target these sequences (or the mRNA) are co-
transfected
with the donor plasmid. The donor plasmid contains flanking homology regions
to the intron
22 locus, which allows for recombination of the donor plasmid into the
chromosome. The
cDNA of exons 23 to 26 of the F8 gene is contained between the flanking
homology regions
of the donor plasmid. The donor plasmid can also contain a suicide gene (such
as the
thymidine kinase gene from the herpes simplex virus), which allows counter-
selection to
avoid random and multi-copy integration into the genome.
[0197] Electroporation (AMAXA Nucleofection system) and chemical transfection
(with a
commercial reagent optimized to this cell type) can be used as transfection
methods for the
lymphoblastoid cells. A plasmid containing the green fluorescent protein (GFP)
gene is
introduced into the cells using both methods. The cells are analyzed by
fluorescent
microscopy to obtain an estimate of transfection efficiency, and the cells are
observed by
ordinary light microscopy to determine the health of the transfected cells.
Any transfection
method that gives a desirable balance of high transfection efficiency and
preservation of cell
health in the lymphoblastoid cells can be used. The TALEN mRNAs and the gene
repair
donor plasmid is then introduced into the lymphoblastoid cells using a
transfection method.
The TALENs for the human lymphoblastoid cells and their target site are shown
in Figure 3.
[0198] Repair of the F81221 in the adipose tissue-derived hepatocyte-like
cells from the 1221
HA canine animal model is effected using electroporation to deliver mRNAs
encoding an
analogous TALEN that targets the 5'-end of 122 in canine F8 and an analogous
donor
plasmid carrying a "splice-able" cDNA spanning canine F8 exons 23-26.
[0199] Adipose tissue is collected from these FVIII deficient dogs by standard
liposuction.
Stromal cells from the adipose tissue are reprogrammed into induced
pluripotent stem cells
(iPSC), as described by Sun et al. ("Feeder-free derivation of induced
pluripotent stem cells
from adult human adipose stem cells" Proc Natl Acad Sci USA. 106: 720-5, 2009)
with two
modifications: (i) mRNA of the reprogramming factors are used in place of
lentiviral vectors
and (ii) the reprogramming is performed under conditions of hypoxia, 5% 02,
and in the
presence of small molecules that have been found to increase the reprogramming
efficiency.
Once produced and characterized, pluripotent canine cells are obtained.
[0200] The defective FVIII sequence in iPSC is replaced by the correct
sequence using site-
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
specific TALE nucleases (see Figure 4). The iPSC with repaired Factor VIII are
differentiated into hepatocytes using well established protocols (see, for
example, Hay et al.
"Direct differentiation of human embryonic stem cells to hepatocyte-like cells
exhibiting
functional activities" Cloning Stem Cells. 9: 51-62, 2007; Si-Tayeb et al.
"Highly efficient
generation of human hepatocyte-like cells from induced pluripotent stem cells"
Hepatology.
51: 297-305, 2010; and Cayo et al. "JD induced pluripotent stem cell-derived
hepatocytes
faithfully recapitulate the pathophysiology of familial hypercholesterolemia"
Hepatology.
May 31, 2012). In short, small colonies of iPSC are induced to differentiate
for the first 3
days into definitive endoderm by treatment with 50 ng/mL Wnt3a and 100 ng/mL
Activin A,
and then into the hepatocyte lineage by 20 ng/mL BMP4. Two expression plasmids
necessary to produce mRNAs encoding a functional TALEN are obtained. These are
designed to cleave and yield a double-stranded DNA break at only a single site
within the
canine genome, located within canine F8 122, ¨0.3 kb downstream of the 3'-end
of exon-22.
The left (TALEN-L) and right (TALEN-R) monomers comprising this heterodimeric
TALEN
is shown above in Figure 4.
102011 A donor plasmid containing the sequence of the 3'-end of canine F8
intron-22 and all
of canine F8 exon-22 as the left homologous sequence and the 5'-end of canine
F8 intron-23
as the right homologous sequence to provide an adequate length of genomic DNA
for
efficient homologous recombination at the target site (i.e., the TALEN cut
site) is created.
The TALEN mRNAs and the gene repair donor plasmid are introduced into the
pluripotent
canine cells using a transfection method described herein.
[0202] Likewise, in humans, human iPSCs are electroporated with the human F8
TALENs &
donor plasmid described above, to assess candidate genome-editing tools (which
were
designed to be equally capable of "editing" the I22-sequence in the wild-type
and I22-
inverted F8 loci, F8 and F8122I, respectively) for their efficiency of site-
specific gene repair.
The genomic DNA at the repaired F8 loci, as well as the mRNAs and expression
products
synthesized by, the cells described above are assessed before and after
electroporation.
[0203] The TALEN gene repair method described above inserts F8 exons 23-26
immediately
downstream (telomeric) to F8 exons 1-22 to encode a FVIII protein. Genomic
DNA, spliced
mRNA, and protein sequences differ among normal, repaired, and unrepaired
cells (see
Figure 5). Gene repair is verified in genomic DNA through the use of PCR.
Specific PCR
66
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
primers are designed to amplify across the homologous recombination target
sequence in
unrepaired and repaired cells. A common primer is placed toward the end of
exon-22. An
1221-specific primer is placed in the sequence telomeric to exon-22 in the
1221-inverted cells.
A Repaired-specific primer is placed in the inserted exon 23-26 sequence.
Primer design is
shown in Figure 8. In FIG. 8, Exons 1-22 (top schematic) and Exons 1-22 and 23-
26 (left,
bottom schematic) represent functional coding sequences, while Exons 23-26
(top schematic)
and Exons 23-26 (right, bottom schematic) represent non-functional coding
sequences.
Separate sets of primers are designed for human and canine sequences.
[0204] Characterization of the genomic DNA at the repaired F8 loci, as well as
the mRNAs
and expression products synthesized by, the cells described above, before and
after
electroporation are performed.
[0205] A quantitative RT-PCR test that specifically detects and quantifies the
mRNA
transcripts from normal and 1221 cells is used. The quantitative RT-PCR test
uses three
separate primer sets: one set to detect exons 1-22, one set to detect exons 23-
26, and one set
that spans the exon-22/exon-23 junction. mRNA is purified from cells before
and after
transfection. The existing primer design to probe mRNA from the human cells is
used.
Primers against canine sequences are designed using the same strategy and then
the mRNA
from the canine cells is probed using these new primers. An increased signal
from the exon-
22/exon-23 junction reaction in repaired cells, relative to unrepaired cells
should be observed.
[0206] Monoclonal antibody ESH8, which is specific for the C2-domain of the
FVIII protein,
is be used. NIH3T3 cells were transfected with expression constructs encoding
full-length
and 1221 F8 genes and then assayed by flow cytometry. Signal from the ESH8
antibody was
high in cells transfected with the full-length construct but virtually absent
in cells transfected
with the 1221 construct. The ESH8 antibody is used to test transfected cells.
There should be
an increased signal in repaired cells relative to unrepaired cells. Secreted
FVIII levels, as
measured by ELISA, are dramatically lower in 1221 cells relative to normal
cells. Whole-cell
lysates and supemates from transfected cells are obtained and tested for FVIII
concentration
by ELISA. There should be an increase in FVIII concentration in the supernates
from
repaired cells relative to unrepaired cells.
[0207] In another example, canine blood outgrowth endothelial cells (cBOECs)
and canine
iPSCs derived from canine adipose tissue can be transfected with TALENs that
target the
67
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
F81221 canine gene and a plasmid repair vehicle that carries exons 23-26 of
cF8. TALENs
are expected to make DSBs in the F8122I DNA at the target site to allow
"homologous
recombination and repair" of the canine F8 1221 gene by insertion of exons 23-
26 of the
canine F8. The TALENS are designed to cleave and yield a DSB at only a single
site within
the canine genome, located within canine F8 122, (-0.3 kb) downstream of the
3'-end of
exon-22. The donor plasmid contains the sequence of canine F8 exons 23-26
flanked by the
3 '-end of canine F8 intron-22 and all of canine F8 exon-22 as the left
homologous sequence
and the 5'-end of canine F8 intron-23 as the right homologous sequence to
provide an
adequate length of genomic DNA for efficient homologous recombination at the
target site.
[0208] Feasibility of deriving canine iPSCs is well established. An mRNA
transcript that
enables expression of the so called "Yamanaka" genes coding for transcription
factors OCT4,
SOX2, KLF4 and C-MYC to induce iPSCs from canine adipose derived stem cells
(hADSCs). iPSCs have been transfected using Nucleofector. For transfection,
Qiagen's
Polyfect transfection reagents can be used with TALENs for many cell types,
including
BOECs. Transfection methods can be assessed using commercial reagents and
transfected
cells can be analyzed by fluorescent microscopy to obtain an estimate of
transfection
efficiency, while viability can be determined by Trypan Blue dye exclusion.
The transfection
method that gives the best balance of high transfection efficiency and
preservation of cell
health can be used.
[0209] Prior to commencing transfection with the TALENS and repair plasmid,
the cleavage
activity of the TALENs against the target site can be analyzed. This can be
done by
monitoring TALEN induced mutagenesis (Non-Homologous End Joining Repair) via a
T7
Endonuclease assay. To assess potential risk of unintended genomic
modification induced by
the selected repair method, off-site activity is analyzed following
transfection. In silico
identification based on homologous regions within the genome can be used to
identify the top
20 alternative target sites containing up to two mismatches per target half-
site. PCR primers
can be synthesized for the top 20 alternative sites and Surveyor Nuclease (Cel-
I) assays
(Transgenomics, Inc.) can be performed for each potential off-target site.
[0210] Transfection for expression and secretion of FVIII can be assessed in
the various cell
types before and after transfection. Genomic DNA is isolated from cells before
and after
transfection. Purified genomic DNA is used as template for PCR. Primers are
designed for
68
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
amplification from a FVIII 1221-specific primer only in unrepaired cells, and
amplification
from the repaired-specific primer only in repaired cells. RT-PCR can
specifically detect and
quantify the mRNA hF8 transcripts from normal and 1221 cells. The quantitative
RT-PCR
test uses three separate primer sets: one set to detect exons 1-22, one set to
detect exons 23-
26, and one set that spans the exon-22/exon-23 junction. mRNA is purified from
cells before
and after transfection, with an increased signal from the exon-22/exon-23
junction reaction in
repaired cells, relative to unrepaired cells. Flow-cytometry based assays may
also be used for
FVIII protein in peripheral blood mononuclear cells (PBMCs).
[0211] iPSCs derived from canine adipose tissue engineered can be conditioned
to secrete
FVIII to hepatocyte-like tissue. Canine iPSCs are conditioned toward
hepatocyte like cells
using a three step protocol as described by Chen et al. that incorporates
hepatocyte growth
factor (HGF) in the endodermal induction step (Chen YF, Tseng CY, Wang HW, Kuo
HC,
Yang VW, Lee OK. Rapid generation of mature hepatocyte-like cells from human
induced
pluripotent stem cells by an efficient three-step protocol. Hepatology. 2012
Apr;55(4):1193-
203).
[0212] Subpopulations of cBOECs are segregated and expanded and then
characterized for
the expression of endothelial markers, such as Matrix Metalloproteinases
(MMPs), and cell-
adhesion molecules (JAM-B, JAM-C, Claudin 3, and Claudin 5) using RT-PCR.
Detailed
RT-PCR methods, including primers for detecting expression of mRNA transcripts
of the
cell-adhesion molecules of interest and detailed immunohistochemistry methods
to detect the
proteins of interest, including a list of high affinity antibodies have been
published by Geraud
et al. (Geraud C, et al. Unique cell type-specific junctional complexes in
vascular
endothelium of human and rat liver sinusoids. PLoS One. 2012;7(4):e34206).
Antibodies
that detect JAM-B, JAM-C, Claudin 3, and Claudin 5 may be purchased from
LifeSpan
Biosciences (www.lsbio.com).
[0213] One subpopulation of co-cultured cBOECs can be prepared and segregated
early
(before ¨4 passages of outgrowth). Later segregation of the subpopulation can
occur after
¨10 passages. After 1 week of co-culture, two cBOECs subpopulations can be
compared for
expression and secretion of FVIII, and suitability for engraftment in the
canine liver. Co-
culturing of hepatocytes can be done with several cell types including human
umbilical vein
endothelial cells (HUVECs). cBOECs can be used as surrogates for HUVECS in
this system.
69
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
Once the repaired cBOECs (with the repaired FVIII gene) are obtained, the
cells can be used
to induce immune tolerance in canines with high titer-antibodies to FVIII.
Example 2: Protocol for Factor VIII Gene Repair in Humans
Obtaining a blood sample
[0214] A protocol for gene repair of the F8 gene in blood outgrowth
endothelial cells
(BOECs) is described in the following example. First, a blood sample is
obtained, with 50-
100mL of patient blood samples obtained by venipuncture and collection into
commercially-
available, medical-grade collecting devices that contain anticoagulants
reagents, following
standard medical guidelines for phlebotomy. Anticoagulant reagents that are
used include
heparin, sodium citrate, and/or ethylenediaminetetraacetic acid (EDTA).
Following blood
collection, all steps proceed with standard clinical practices for aseptic
technique.
Isolating appropriate cell populations from blood sample
[0215] Procedures for isolating and growing blood outgrowth endothelial cells
(BOECs) have
been described in detail by Hebbel and colleagues (Lin, Y., Weisdorf, D. J.,
Solovey, A. &
Hebbel, R. P. Origins of circulating endothelial cells and endothelial
outgrowth from blood. J
Clin Invest 105, 71-77 (2000)). Peripheral blood mononuclear cells (PBMCs) are
purified
from whole blood samples by differential centrifugation using density media-
based
separation reagents. Examples of such separation reagents include Histopaque-
1077, Ficoll-
Paque, Ficoll-Hypaque, and Percoll. From these PBMCs multiple cell populations
can be
isolated, including BOECs. PBMCs are resuspended in EGM-2 medium without
further cell
subpopulation enrichment procedures and placed into 1 well of a 6-well plate
coated with
type I collagen. This mixture is incubated at 37 C in a humidified environment
with 5%
CO2. Culture medium is changed daily. After 24 hours, unattached cells and
debris are
removed by washing with medium. This procedure leaves about 20 attached
endothelial cells
plus 100-200 other mononuclear cells. These non-endothelial mononuclear cells
die within
the first 2-3 weeks of culture.
Cell culture for growing target cell population
[0216] BOECs cells are established in culture for 4 weeks with daily medium
changes but
with no passaging. The first passaging occurs at 4 weeks, after approximately
a 100-fold
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
expansion. In the next step, 0.025% trypsin is used for passaging cells and
tissue culture
plates coated with collagen-I as substrate. Following this initial 4-week
establishment of the
cells in culture, the BOECs are passaged again 4 days later (day 32) and 4
days after that (day
36), after which time the cells should number 1 million cells or more.
In vitro gene repair
[0217] In order to affect gene repair in BOECs, cells are transfected with 0.1-
10 micrograms
per million cells of each plasmid encoding left and right TALENs and 0.1-10
micrograms per
million cells of the repair vehicle plasmid. Transfection is done by
electroporation,
liposome-mediated transfection, polycation-mediated transfection, commercially
available
proprietary reagents for transfection, or other transfection methods using
standard protocols.
Following transfection, BOECs are cultured as described above for three days.
Selection of gene-repaired clones
[0218] Using the method of limiting serial dilution, the BOECs are dispensed
into clonal
subcultures, and grown as described above. Cells are examined daily to
determine which
subcultures contain single clones. Upon growth of the subcultures to a density
of >100 cells
per subculture, the cells are trypsinized, re-suspended in medium, and a 1/10
volume of the
cells is used for colony PCR. The remaining 9/10 of the cells are returned to
culture. Using
primers that detect productively repaired F8 genes, each 1/10 volume of
colonies are
screened by PCR for productive gene repair. Colonies that exhibit productive
gene repair are
further cultured to increase cell numbers. Using the top 20 predicted
potential off-site targets
of the TALENs, each of the colonies selected for further culturing is screened
for possible
deleterious off-site mutations. The colonies exhibiting the least number of
off-site mutations
are chosen for further culturing.
Preparation of cells for re-introduction into patients by conditioning and/or
outgrowth
[0219] Prior to re-introducing the cells into patients, the BOECs are grown in
culture to
increase the cell numbers. In addition to continuing cell culture in the
manner described
above, other methods can be used to condition the cells to increase the
likelihood of
successful engraftment of the BOECs in the liver sinusoidal bed of the
recipient patient.
These other methods include: 1) co-culturing the BOECs in direct contact with
hepatocytes,
wherein the hepatocytes are either autologous patient-derived cells, or cells
from another
71
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
donor; 2) co-culturing the BOECs in conditioned medium taken from separate
cultures of
hepatocytes, wherein the hepatocytes that yield this conditioned medium are
either
autologous patient-derived cells, or cells from another donor; or 3) culturing
the BOECs as
spheroids in the absence of other cell types.
[0220] Co-culturing endothelial cells with hepatocytes is described further in
the primary
scientific literature (e.g. Kim, Y. & Rajagopalan, P. 3D hepatic cultures
simultaneously
maintain primary hepatocyte and liver sinusoidal endothelial cell phenotypes.
PLoS ONE 5,
e15456 (2010)). Culturing endothelial cells as spheroids is also described in
the scientific
literature (e.g. Korff, T. & Augustin, H. G. Tensional forces in fibrillar
extracellular matrices
control directional capillary sprouting. J Cell Sci 112 (Pt 19), 3249-3258
(1999)). Upon
growing the colonies of cells to a total cell number of at least 1 billion
cells, the number of
cells needed for injection (>50 million cells) into the patient are separated
from the remainder
of the cells and used in the following step for injection into patients. The
remainder of the
cells are aliqouted and banked using standard cell banking procedures.
Injection of gene-repaired BOECs into patients
[0221] BOECs that have been chosen for injection into patients are resuspended
in sterile
saline at a dose and concentration that is appropriate for the weight and age
of the patient.
Injection of the cell sample is performed in either the portal vein or other
intravenous route of
the patient, using standard clinical practices for intravenous injection.
Example 3: Nuclease sites for repair at different exon-intron junctions
[0222] Because mutations causing Hemophilia A occur throughout the FVIII gene,
different
repair strategies may be employed at different exon-intron junctions in order
to allow the use
of repair vehicles which correct a wider range of patient mutations. All gene
repairs employ
the methodology described herein of using a DNS scission enzyme (DNA-SE) such
as a zinc
finger nuclease, a TALEN, or a CRISPR to induce a double-strand break near the
3' end of
an exon, thereby allowing homologous recombination to incorporate a
therapeutic repair
vehicle encoding the cDNA for the downstream exons of the gene into the genome
in order to
be operably linked to the 3' end of that exon.
[0223] In order to choose CRISPR target sites in exons 1-22, several
considerations were
taken into account. The ¨100 bp of the 3' end of each exon (hg19 human genome
build)
72
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
were searched for CRISPRICas9 binding sites using an online algorithm
described by Hsu et
al. in Nature Biotechnology 2013, incorporated herein by reference. Single
guide RNAs
(sgRNAs) were chosen based on low potential for off-target activity, the
proximity of the
cleavage site to the 3' end of the exon, and guidelines for increasing the
likelihood of high
on-target activity (Wang T et al., Science 2014). Paired nickases were chosen
by adding the
additional consideration that they be orientated to create 5' overhangs and be
spaced apart
within the recommended range for optimal activity (Shen B, et al., Nature
Methods 2014).
[0224] In order to choose TALEN binding sites in exons 1-22, several
considerations were
taken into account. The ¨100 bp of the 3' end of each exon (hg19 human genome
build) were
searched for TALEN binding sites using the SAPTA algorithm as described by Lin
Y, Fine
EJ, et al. in Nucleic Acids 2014, incorporated herein by reference. Potential
binding sites
were then screened using the TALEN v2.0 algorithm of the PROGNOS tool as
described by
Fine EJ et al. in Nucleic Acids Research 2013, incorporated herein by
reference to ensure that
no highly scored potential off-target sites existed in the human genome.
[0225] Sequences listed in Table 5 below contain identified binding sites for
CRISPRs within
exons 1-22 respectively. If a homologous sequence in the canine genome
(canFam3 build)
exists that permits the possibility of CRISPR/Cas9 cleavage using the same
guide strand as
used for the human exon, it is listed with any mismatches in lowercase bold;
if no reasonable
homology exists, it is listed as "N/A".
Table 5
FVIII Gene Genome Editing Genomic Target of SG/PG RNAs
Target of SG/PG RNAs in Dogs
(Region) (Desired Activity) (DNA Sequence) (DNA
Sequence)
single nuclease 5 ' ¨AAGATACTACCTGGGTGCAGtGG 5 ' ¨AAaATACTACCTcGGTGCAGtGG
Exon 1 paired nickase (5') 5' ¨CACTAAAGCAGAATCGCAAAaGG N/A
paired nickase (3') 5' ¨AAGATACTACCTGGGTGCAGtGG N/A
Exon 2 single nuclease 5 ' ¨TTTTCAACATCGCTAAGCCAaGG N/A
paired nickase (5') 5 ' ¨AGTCTTTTTGTACACGACTGaGG N/A
paired nickase (3') 5 ' ¨TTTTCAACATCGCTAAGCCAaGG N/A
Exon 3 single nuclease 5 ' ¨ATGCTGTTGGTGTATCCTACtGG 5 '
¨AcGCTGTTGGTGTATCCTAttGG
paired nickase (5') 5 ' ¨CAGCATGAAGACTGACAGGAtGG N/A
paired nickase (3') 5' ¨ATGCTGTTGGTGTATCCTACtGG N/A
Exon 4 single nuclease 5' ¨GACTTGAATTCAGGCCTCATtGG 5'
¨GACcTGAATTCAGGCCTCATtGG
paired nickase (5') 5 ' ¨TATGAGTAGGTAAGGCACAGtGG N/A
paired nickase (3') 5 ' ¨GACTTGAATTCAGGCCTCATtGG N/A
Exon 5 single nuclease 5 ' ¨AAGTAGTATAAATTTGTGCAaGG N/A
paired nickase (5') 5 ' ¨AAGTAGTATAAATTTGTGCAaGG N/A
73
SUBSTITUTE SHEET (RULE 26)
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
Table 5
FVIII Gene Genome Editing Genomic Target of SG/PG RNAs
Target of SG/PG RNAs in Dogs
(Region) (Desired Activity) (DNA Sequence) (DNA
Sequence)
paired nickase (3') 5 ' -CTTTTTGCTGTATTTGATGAaGG N/A
Exon 6 single nuclease 5 ' -CAGTCAATGGTTATGTAAACaGG 5 ' -
CcaTCAATGGcTATGTAAACaGG
paired nickase (5') 5' -GACTGTGTGCATTTTAGGCCaGG N/A
paired nickase (3') 5' -CAGTCAATGGTTATGTAAACaGG N/A
Exon 7 single nuclease 5' -CAAACACTCTTGATGGACCTtGG N/A
paired nickase (5') 5' -GCGAGATTTCCAAGGACGCCtGG N/A
paired nickase (3') 5' -CAAACACTCTTGATGGACCTtGG N/A
Exon 8 single nuclease 5' -ACATTACATTGCTGCTGAAGaGG N/A
paired nickase (5') 5' -TCTTGGCAACTGAGCGAATTtGG N/A
paired nickase (3') 5' -ACATTACATTGCTGCTGAAGaGG N/A
Exon 9 single nuclease 5' -GAAGCTATTCAGCATGAATCaGG 5' -
GAAGCTATTCAGtATGAATCaGG
paired nickase (5') 5' -AATAGCTTCACGAGTCTTAAaGG N/A
paired nickase (3') 5' -GAAGCTATTCAGCATGAATCaGG N/A
Exon 10 single nuclease 5' -GGACATCAGTGATTCCGTGAgGG N/A
paired nickase (5') 5' -GGACATCAGTGATTCCGTGAgGG N/A
paired nickase (3') 5' -ATGTCCGTCCTTTGTATTCAaGG N/A
Exon 11 single nuclease 5' -GATCTAGCTTCAGGACTCATtGG 5' -
GATCTAGCTTCAGGACTCATtGG
paired nickase (5') 5' -AACGAAACTAGAGTAATAGCgGG N/A
paired nickase (3') 5' -GATCTAGCTTCAGGACTCATtGG N/A
Exon 12 single nuclease 5' -CGCTTTCTCCCCAATCCAGCtGG N/A
paired nickase (5') 5' -AGCGTTGTATATTCTCTGTGaGG N/A
paired nickase (3') 5' -CGCTTTCTCCCCAATCCAGC tGG N/A
Exon 13 single nuclease 5' -AGAAACTGTCTTCATGTCGAtGG 5' -
AGAAACTGTCTTCATGTCaAtGG
paired nickase (5') 5' -ATAGACCATTTTGTGTTTGAaGG 5' -ATAGACCATTTTGTGTTTGAaGG
paired nickase (3') 5' -AGAAACTGTCTTCATGTCGAtGG 5' -AGAAACTGTCTTCATGTCaAtGG
Exon 14 single nuclease 5' -ACACTATTTTATTGCTGCAGtGG 5' -
ACACTATTTcATTGCTGCAGtGG
paired nickase (5') 5' -TTTTCTTTTGAAAGCTGCGGgGG 5' -TTTTCTTTTGAAAGCTGCGGaGG
paired nickase (3') 5' -ACACTATTTTATTGCTGCAGtGG 5' -ACACTATTTcATTGCTGCAGtGG
Exon 15 single nuclease 5' -TCAACTTCTGCTCTTATATAtGG 5' -
TCAACTTCTGCTCTTATATAtGG
paired nickase (5') 5' -ACGGTATAAGGGCTGAGTAAaGG N/A
paired nickase (3') 5' -AAATGAACATTTGGGACTCCtGG N/A
Exon 16 single nuclease 5' -ATGAGTTTGACTGCAAAGCCtGG 5' -
ATGAGTTTGACTGCAAAGCCtGG
paired nickase (5') 5' -CAGTCAAACTCATCTTTAGTgGG 5' -CAGTCAAACTCATCTTTAGTgGG
paired nickase (3') 5' -ATGAGTTTGACTGCAAAGCCtGG 5' -ATGAGTTTGACTGCAAAGCCtGG
Exon 17 single nuclease 5' -GGCTCCCTGCAATATCCAGAtGG 5' -
aGCTCCCTGCAATgTCCAGAaGG
paired nickase (5') 5' -TTCAGTGAAGTACCAGCTTTtGG N/A
paired nickase (3') 5' -GGCTCCCTGCAATATCCAGAtGG N/A
Exon 18 single nuclease 5' -GTTCACTGTACGAAAAAAAGaGG 5' -
GTTCACTGTACGAAAAAAAGaGG
paired nickase (5') 5' -GTCCACTGAAATGAATAGAAtGG N/A
paired nickase (3') 5' -GTTCACTGTACGAAAAAAAGaGG N/A
Exon 19 single nuclease 5' -CAAAGCTGGAATTTGGCGGGtGG N/A
paired nickase (5') 5' -CGCCAAATTCCAGCTTTGGAtGG N/A
paired nickase (3') 5' -ATTGGCGAGCATCTACATGCtGG N/A
Exon 20 single nuclease 5'-TGTCCAGAAGCCATTCCCAGgGG N/A
paired nickase (5') 5'-TGTCCAGAAGCCATTCCCAGgGG N/A
paired nickase (3') 5'-GATTTTCAGATTACAGCTICaGG N/A
Exon 21 single nuclease 5'-AATCAATGCCTGGAGCACCAaGG 5'-
AATCAATGCCTGGAGCACCAaGG
paired nickase (5') 5'-TGATCCGGAATAATGAAGTCtGG 5'-TGATCCGGAATAATGAAGTCtGG
74/1
SUBSTITUTE SHEET (RULE 26)
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
paired nickase (3') 5'-AATCAATGCCTGGAGCACCAaGG 5'-AATCAATGCCTGGAGCACCAaGG
Exon 22 single nuclease 5'-AAGAAGTGGCAGACTTATCGaGG N/A
paired nickase (5') 5'-AGATAAACTGAGAGATGTAGaGG N/A
74/2
SUBSTITUTE SHEET (RULE 26)
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
Table 5
FVIII Gene Genome Editing Genomic Target of SG/PG RNAs
Target of SG/PG RNAs in Dogs
(Region) (Desired Activity) (DNA Sequence) (DNA
Sequence)
paired nickase (3') 5 ' -AAGAAGTGGCAGACTTATCGaGG N/A
[0226] Sequences contain the top 20 potential off-target sites computationally
identified in
the human genome for the previously mentioned CRIPSR binding sites in exons 1-
22 are
listed in tables 6-27, respectively below.
[0227] Top-ranked Potential Off-Target Sites for sgRNAs in Human Genome
[0228] The top twenty potential off-target sites in the human genome (hgl 9
genome build)
for single guide strands were located using an online tool (Hsu et al., Nature
Biotechnology
2013). Mismatches to the intended binding sequence are shown in bold. The
genomic region
is annotated and the gene name given in parentheses.
Table 6. Targeting Exon 1
Genome Coordinates Sequence Genomic Region
chrX:154250739 AGATACTACCTGGGTGCAGtGG Exon Coding Sequence (F8)
chr5:65751749 AAACACAACCTGGGTGCAGgGG Intergenic
chr9:17600130 AAAAAGTACCTGGGTGCAGaAG Intron (SH3GL2)
chr9:100168533 AGAAACTACATGGGTGCAGaGG Intergenic
chr21:45748293 GGCGACCACCTGGGTGCAGcAG Intergenic
chr2:144598347 ATTTACCAACTGGGTGCAGcAG Intergenic
chr3:89701232 ATTTACCATCTGGGTGCAGgGG Intergenic
chr10:43493946 AGATGCTTCCTGGGTGCAGcAG Intergenic
chr18:37552785 ACAAACTCCCTGGGTGCAGaGG Intergenic
chr7 :63413239 ACACACTGCCTGGGTGCAGcAG Intergenic
chr7:157859920 GGAGACACCCTGGGTGCAGgAG Intron (PTPRN2)
chr22:48920664 AGGAACGCCCTGGGTGCAGaAG Intron (FAM19A5)
chr1:153919242 GGAAGCTACCTGGGTGCAGgGG Promoter (DENND4B)
chr11:71136741 AGATACCCTCTGGGTGCAGaAG Intergenic
chr2:145627680 AGATACCCTCTGGGTGCAGgAG Intron (TEX41)
chr2:145629372 AGATACCCTCTGGGTGCAGgAG Intron (TEX41)
chr4:60481509 AGATACTGCCTGGGTCCAGaGG Intergenic
chr6:35192631 AGATACTCCCTGGGTCCAGcAG Intron (SCUBE3)
chr10:132278858 GGATACTAGATGGGTGCAGaGG Intergenic
chr3:86928921 AGAGACTACAAGGGTGCAGtGG Intergenic
chr5: 61074999 CAACACTACCTGGGTGCAAaAG Intergenic
Table 7. Targeting Exon 2
Genome Coordinates Sequence Genomic Region
chrX:154227766 TTTCAACATCGCTA_AGCCAaGG Exon Coding Sequence (F8)
SUBSTITUTE SHEET (RULE 26)
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
Table 27. Targeting Exon 22:
Genome Coordinates Sequence Genomic Region
chX:154124374 (target) AGAAGTGGCAGACTTATCGaGG Exon
Coding Sequence (F8)
chr21:42038990 AGAAGCAGCAGACTTATCCaGG Intron
(DSCAM)
chr12:69990980 GGAAGTTGCAAACTTATCGaGG Exon
Coding Sequence (CCT2)
chr7:110964978 GGATGTGGCAGACTTATCTtAG Intron
(IMMPL2)
chr8:42174378 CTGAGTGGCAGGCTTATCGgGG Exon
Coding Sequence (IKBKB)
chr3:57930763 AGAACAGGCAGACTTATCTtAG
Intergenic
chr1:52997435 AGAAGAGGCATACTTATCTgAG Intron
(ZCCHC11)
chr15:27460224 GAAACTGGCAGACTTATCTaGG Intron
(GABRG3)
chr2:102965996 AGAAGTGGCAGAGTTATCCtGG Intron
(IL1RL1)
chr20:2306018 AGGAGTGGCTGACTTATCTaAG Intron
(TGM3)
chr8: 92580265 AAAAATGGTAGACTTATCAaAG I
ntergenic
chr13:113875149 AGAAGTCGCAGGCTTATGGgAG Intron
(CUL4A)
chr18:30300891 AGAAGAGGAAGACTTATGGaAG Intron
(KLHL14)
chr2:135308659 AGTGCTGGCAGACTTATTGcAG Intron
(TMEM163)
chr11:133197425 AGGAGGGGCAGATTTATCGaAG Intron
(OPCML)
chr12:102978261 AGAAGTAGAAAACTTATCAtAG I
ntergenic
chr3:30382779 AGCAGTGGCAGACATATTGaAG I
ntergenic
chr6:118027061 AGAAGTGGATGACTTATTGcAG Intron
(NUS1)
chr9 :117888881 GCAAGTGGCAGGCTTATCTgGG Intron
(L0C101928748)
chr2 :51293036 GCAAGTGGCAGACTTTTCCaAG I
ntergenic
chr21:36105270 AAGAGTGGCAGACTTCTCAtGG Non-
coding Exon (LINC00160)
[0229] Sequences listed in Table 28 contain identified binding sites for
TALENs within
exons 1-22 respectively. If a similar sequence existed in the homologous exon
in the canine
genome (canFam3 genome build), that corresponding binding site is shown with
any
mismatches in lowercase red; if insufficient homology to permit a reasonable
possibility of
the TALENs being able to cleave the canine exon, the site is listed as "N/A".
Table 28
FVIII Gene Genome Editing Genomic Target of TALEN Target
of TALEN in Dogs
(Region) Position (DNA Sequence) (DNA Sequence)
5' Half-Site 5 ' -TGGAACTGTCATGGGAC N/A
Exon 1
3' Half-Site 5 ' -TCCACAGGCAGCTCACCGAG N/A
5' Half-Site 5 ' -TCTGTTTGTAGAATTCACGG N/A
Exon 2
3' Half-Site 5 ' -TGGCCTTGGCTTAGCGAT N/A
5' Half-Site 5 ' -TACACTTAAGAACATGGCT N/A
Exon 3
3' Half-Site 5 ' -TACACCAACAGCATGAAGAC N/A
5' Half-Site 5 ' -TGTGCCTTACCTACTCATATCT
N/A
Exon 4
3' Half-Site 5 ' -TGAATTCAAGTCTTTTACCAG N/A
E 5' Half-Site 5 ' -TCTGGCCAAGGAAAAGACACAGAC 5 -
TCTGGCCAAGAAAgGACACAGAC
xon 5
3' Half-Site 5 ' -TTCATCAAATACAGCAAAAAGTAG 5T -
TTCATCAAATACAGCAAAAAGTAG
5' Half-Site 5 ' -TGCTGCATCTGCTCGGG N/A
Exon 6
3' Half-Site 5 ' -TTTACATAACCATTGACTGTGT
N/A
5' Half-Site 5 ' -TCTCGCCAATAACTTTCC N/A
Exon 7
3' Half-Site 5 ' -TGTCCAAGGTCCATCAAGAG N/A
86
SUBSTITUTE SHEET (RULE 26)
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
Table 28
FVIII Gene Genome Editing Genomic Target of TALEN Target
of TALEN in Dogs
(Region) Position (DNA Sequence) (DNA Sequence)
5' Half-Site 5 ' -TCAGTTGCCAAGAAGCATCCTAA 5' -
TCAGTTGCCAAGAAGCATCCTAA
Exon 8
3' Half-Site 5 ' -TCCTCCTCTTCAGCAGCAATGT 5' -TCCTCCTCTCAGCAGCAATT
5' Half-Site 5 ' -TTCAGCATGAATCAGGAA N/A
Exon 9
3' Half-Site 5 ' -TCTCCAACTTCCCCATAA N/A
5' Half-Site 5 ' -TATAACATCTACCCTCACGG N/A
Exon 10
3' Half-Site 5 ' -TCTCCTTGAATACAA_AGGAC N/A
5' Half-Site 5'-TCTAGCTTCAGGACTCAT 5'-TCTAGCTTCAGGACTCAT
Exonll
3' Half-Site 5'-TCTACAGATTCTTTGTAGCAG 5'-TCTACAGATTCTTTGTAGCAG
5' Half-Site 5'-TCACAGAGAATATACAACG N/A
Exon 12
3' Half-Site 5'-TCCTCAAGCTGCACTCCAGCT N/A
E 13 5' Half-Site 5'-TGTCTTCTTCTCTGGAT 5'-
TGTCTTCTTCTCTGGAT
xon
3' Half-Site 5'-TGTGTCTTCATAGACCATTTT 5'-TGTGTCTTCATAGACCATTTT
5' Half-Site 5'-TCAAAAGAAAACACGACACTATTT 5'-TCAAAAGAAAACACGACACTATTT
Exon 14
3' Half-Site 5'-TCATCCCATAATCCCAGAGCCTCT 5'-TCATCCCATAATCCCAGAGaCgCT
5' Half-Site 5'-TCAGCCCTTATACCGTGGAG 5'-TCAGCCCTTATACCGTGGAG
Exon15
3' Half-Site 5'-TATGGCCCCAGGAGTCCCAA 5'-TATGGCCCCAaGAGTCCCAA
5' Half-Site 5T-TATGGCACCCACTAAAGATGAG 5'-TATGGCACCCACTAAAGATGAG
Exon 16
3' Half-Site 5'-TCAGAGAAATAAGCCCAG 5'-TCAGA&AAATAAGCCCAG
5' Half-Site 5'-TCTTTGATGAGACCAAA N/A
Exon 17
3' Half-Site 5'-TCTTTCCATATTTTCAG N/A
5' Half-Site 5'-TCTATTCATTTCAGTGGAC N/A
Exon 18
3' Half-Site 5'-TATACTCCTCTTTTTTTCG N/A
5' Half-Site 5'-TGTTACCATCCAAAGCT N/A
Exon 19
3' Half-Site 5'-TGCTCGCCAATAAGGCATTCC N/A
5' Half-Site 5'-TCCCCTGGGAATGGCTTCTGG N/A
Exon 20
3' Half-Site 5 ' -TGTCCTGAAGCTGTA_ATCTGAA N/A
5' Half-Site 5 ' -TGGGCCCCAAAGCTGGCCAG 5' -TGGGCCCCAAAGCTGGCCAG
Exon 21
3' Half-Site 5' -TGCTCCAGGCATTGATTGAT 5' -TGCTCCAGGCATTGATTGAT
5' Half-Site 5 ' -TCTACATCTCTCAGTTTAT N/A
Exon 22
3' Half-Site 5 ' -TCTGCCACTTCTTCCCATCAAG N/A
[0230] Sequences listed in Tables 29-50 below contain the top 20 potential off-
target sites
computationally identified in the human genome for the previously mentioned
TALEN
binding sites in exons 1-22, respectively. Off-target analysis was performed
using the
PROGNOS algorithm (Fine et al., Nucleic Acids Research 2013) "TALEN v2.0" on
the hg19
build of the human genome. The top 20 potential off-target sites are given for
each TALEN
pair. Homodimers were allowed in the search and spacing between the TALENs of
10-30 bp.
The right half-site is listed as the sequence on the same strand as the left
half-site; the right
half-site is therefore listed in the reverse anti-sense orientation to the
sequence which is
bound by the TALEN. Left and right half-sites are given as the 5' (left) and
3' (right) binding
sites on the positive strand of the chromosome; the "left" and "right"
annotation may
therefore differ from the annotation for TALENs designed to genes on the
negative strand of
87/1
SUBSTITUTE SHEET (RULE 26)
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
chromosomes. Mismatches to the intended binding sequence are depicted in
lowercase
87/2
SUBSTITUTE SHEET (RULE 26)
ak 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
letters.
Table 29. Targeting Exon 1:
Genome Left Half-Site Right Half-Site Genomic Region
Coordinates
chrX:154250691 TCCACAGGCAGCTCACCGAG GTCCCATGACAGTTCCA Exon (F8)
chr14:45095676 TGGAACTcTCATGGaAC GagCaATGACtGTTCCA Intergenic
chr6:26839581 aGGAgCTGTCAgtcaAC GTCtCATGACAGTTaCA Intron (GUSBP2)
chr10:45462110 TGGAACTGTCATGGtgC CTCaGaGAGtTGCCTGgttA Intron (RASSF4)
chr11:101870316 TGaAACTGTCATatGAC tgCCCATGACtccTCCA Exon (KIAA1377)
chr15:20414578 TGaAgCTGTCATGaaAC cTtCCATtAtAGTTttA Intergenic
chr16:33444315 TaaAACTaTaATGGaAg GTttCATGACAGcTtCA Intergenic
chr5:61534127 TGaAgCTGTCATGaaAC cTtCCATtAtAGTTttA Intergenic
chr7:44551672 TGGAcCcagCATGGGgC GTtCCtTGACAtTTCCA Intergenic
chr1:165095506 TGGAACTGTCATGtGAg GTtCCATGgCAGaTaCt Intergenic
chrX:15724565 TaGgACTGTCcTGaGcC GgCtCAgGACAGTcCCA Intergenic
chr7:67809648 TaGAACTaTCATGGGAa GgCttcTGAgAcTTCCA Intergenic
chr6:13204828 TGGcAtTGTCATGGaAC GTCCtAgGtagGTTCCA Intron (PHACTR1)
chr2:37743218 TGaAACccTCATGaGcC GTCCtATGAgAtTTCtA Intergenic
chr10:78301531 TGtAAaTGTCATGGaAC GTCtCATttCAGTgtaA Intron (ClOorf11)
chrX:106781486 TGGAAaTGTCATaGaAC cTCCatTGACAGaTCtt Intergenic
chr12:70809983 TaGgtCTGTCtTGGGtC GctCCATGtCAGTTtCA Intron (KCNMB4)
chr11:46818282 TatAACTGTCAaGaGAC GTCCaATttCAGTcCaA Intron (CKAP5)
chr3:30945924 TGGAgCTGaaAaGcaAC GTCtCcTGACAGcTCCA Intergenic
chr9:13642916 TaGAACTaaCATaaaAC GTgtCATtAtAGTTgCA Intergenic
chr14:27743308 TaGAAaTaTCcTGGGAt aTtgCATGAtAGTTCCA Intergenic
Table 30. Targeting Exon 2:
Genome Left Half-Site Right Half-Site Genomic Region
Coordinates
chrX:154227764 TGGCCTTGGCTTAGCGAT CCGTGAATTCTACAAACAGA Exon (F8)
chr12:51122429 TGGaCTTGGCTTcGCGcT ATgGaaAAGCCAAGGagA Exon (DIP2B)
chr14:83666273 TaGCCTTGGCTTAGaaAa cTgGCTAAGCaAAGataA Intergenic
chr15:99285268 gGaaCTTGaCTTAGCccT cctGCTAAGCCAAGGCtA Intron (IGF1R)
chr15:29750773 TGcCCTgGaCTTgGaGgT AgaGaTAAGCCAAGGtCA Intron (FAM189A1)
chr20:59053322 TGGCCTTGGtTTAGaaAa AgCGaTAAGgaAAGGttA Intergenic
chr1:163956121 TCTaTTTGTAGAATTactaG tTgGtTAAGCCAAttCCA Intergenic
chr2:123622749 TCTtTTTGTAaAAaTgACGa ATtcCgAAGCCAAGGatA Intergenic
chr12:92444873 TGtCCaTGGCcTgGgGgT ATCttgAAGCCAAGGCtA Intron (L0C256021)
chr14:86193436 caGCCTTGGCTTgtgGAT tTtaCTAAGaCAAGGCCA Intergenic
chr8:1184501 TGaCCTctcCTTAaCcAT ATttCTAAaCtAAGGtCA Intergenic
chr4:60350711 TGGCaaTGcCTTAGaaAT ATtGCTAAGtCAAatCaA
Intergenic
chr2:109270631 TttCCTTGGCTTAGtGAT ATtGCTAActCAAtcaCA Promoter (LIMS1)
Promoter (LIMS3-
chr2:110655405 TttCCTTGGCTTAGtGAT ATtGCTAActCAAtcaCA L0C440895)
Promoter (LIMS3-
chr2:111231206 TGtgaTTGagTTAGCaAT ATCaCTAAGCCAAGGaaA L0C440895)
chr7:105518314 ctGCCcTGGCTgAaCcAT ATCGCTAAGCCAgtGttA Intergenic
chrX:12453009 TtGCaTTtaCTcAGCcAT ATCttTtAGCCAAtGCCA
Intron (FRMPD4)
chr9:133831225 TGGCCTgaGCTTtGgGgT ActGCTAAGaCAAGcCCA Intergenic
chr7:27778567 TgTGcTTaTAaAATTCACtG CaGTtAtTTCTACtAcCAGA Promoter (TAX1BP1)
chr8:22054601 TaGggcTGGCTTgGCGAg gTaGCTAAGtCAAGGCtA
Intron (BMP1)
chr6:102761808 TGGCagTaGCTctGCcAT AattCTAAGCtAAGGCCA Intergenic
Table 31. Targeting Exon 3:
Genome Left Half-Site Right Half-Site Genomic Region
Coordinates
88
SUBSTITUTE SHEET (RULE 26)
CA 02951882 2016-12-09
WO 2015/191899 PCT/US2015/035399
Table 31. Targeting Exon 3:
Genome Left Half-Site Right Half-Site Genomic Region
Coordinates
chrX:154225270 TACACCAACAGCATGAAGAC AGCCATGTTCTTAAGTGTA Exon (F8)
chr2:175647194 aACAaTcAgGctCATGGCa AGCCATGTTtTTAAGaGTA Intergenic
chr4:164801896 TAtACTTAAaAACATaGCT AGtgATtTTtTTcAaTGaA Intron (MARCH1)
chr3:1591042 TACAtTTAAaAACATGtCT AGCtATcTTaTTcAtTtTA
Intergenic
chr21:39750804 TACgCTgcAGAgCtgGGCa AGaCATtTTtTTAAGTGTA Intron (ERG)
chrX:46478957 TACACaTAAcAACATGGCT AGCCAgacaCTaAAaTaTA
Intron (SLC9A7)
chrX:99327213 aAtcCTTAAGAACATGaCT AtCCtTGTTCTTAtGTtcA Intergenic
chr8:103196820 cACACTgAAGAcCATGGCT GTCTTCATcaTGTTaGTGTc Intergenic
chr9:76364644 TAgACTTAAtcAtgTaGCT gGCtATGTTCTTAAGTGTc Intergenic
Intron
chr8:19520723 TACACTTgtGAAgATGGaT AGgCtTGTaCTTAAtTGTA (CSGALNACT1)
chr1:7465386 TACACTTAgaAAaAaaGCT GTtTgttTGCTGTTGtTGTt
Intron (CAMTA1)
chrX:151388800 TACACTTAtGtgttTGGCT AtCCATGTTgTTgAGTGTA Intron (GABRA3)
chr8:52110351 aACACTTAAaAACAgGGCT AtCtATtTaCTaAAtTGTt Intergenic
chr11:42440454 aACAaaTAAtAtCATcaCT AtCtATGTTCTTAAGTcTA Intergenic
chr2:74468885 cgCACaaAAaAACATGGaT AGgCATGTTtTTAAGTGgg
Intron (SLC4A5)
chr6:82600824 cACAtTTgAGAACATGGCT GctTTCAgtCTGgTGGTtTA Intergenic
chr2:65094538 TgCACTTAAaAAtATGaCa AGCacaGTgCTTAAGTGcA Intergenic
chrX:87497023 TACACTgAAGAgaATGGag AGCaATGTTtTTAAGTGat Intergenic
chr13:74882688 TtCAtTgAAGAAaAaaGCT aTtTTtATGCTGTTGGaGTA Intergenic
chr21:25077810 TACAtTTAAGcAtATGGCT tGCttTagTCTTAAtTGTA Intergenic
chr10:92935297 TACcCcTgtGAACATGGaa tGCttTGTTCTTAAaTGTA Intron (PCGF5)
Table32.TargetingExon4:
Genome Left Half-Site Right Half-Site Genomic Region
Coordinates
chrX:154221245 TGAATTCAAGTCTTTTACCAG AGATATGAGTAGGTAAGGCACA Exon (F8)
chr5:166223644 TGAATTCAAaTCTTTTtCCtG tTGGaAAAAtcCcTtAATaCA Intergenic
Promoter
chr3:48957213 TGAtTTCtAGTtTTgTgCCAa
tTaGTAAAtGACcTGAATTCA (C3orf71)
chr1:14460511 TGAcaTtAAGaCaTTTAaCAG CTGGgAAAAGAagTGgATTCA Intergenic
chr8:26674607 gaAAggCAAGcCaTaTACtAG
CTGaTAAAtGACTTGtATTCA Intron (ADRA1A)
chr15:41366843 TGcATaCAAtTCcTTTACCAa CTGaTAAAcaAtTTtAATTtA Intron
(IN080)
chr6:134930070 TaAAgTCActTCcTTTACgAc aTGGTtgAtGACTTGAATTCA Intergenic
chr6:121097474 TGAATcCAAaaCTTTTACCtG CTGGgttAAtACaTttATTtA Intergenic
chr11:49119615 gGAATTaAAGTCcTTcACata tTGGTtAcAGACTTGAAgTCA Intergenic
chr1:74307557 gGAATTCAAtTCaaTaACaAG tgGGcAAAAGACcTGAATTgA Intergenic
chr18:38466162 TGtATTCAAGTCcTTaAaaAG tTGGTtAAAattTTGAAcTCA Intergenic
chr20:45113912 atAATTCtAGTCTTaggaCAG CTGGgAAAAGttTgGAATTtA Intergenic
chr5:26641542 TGAATTCcttcCTTgTACCAt tgGaTtAAAGACTTGAATgCA Intergenic
chr3:160034110 TGAAagCAAaTCTTTccCCAG CTGGTcAAtGcCTTGctTgCA Intron
(IFT80)
chr2:241783612 TGAcTTCAAGTCTTTaAaCAa aTcagAAAAtctTTGAATcCA Intergenic
chr6:123852751 gGTcaCTaAtCTACTCtTATCT AGATATGAacAGGTAAGGCACt Intron
(TRDN)
chr2:89343189 TGAATTCAAcTCTTTagaCAG gTaaggAAAGctTTGAATTCA Intergenic
chr2:90195655 TGAATTCAAagCTTTccttAc CTGtctAAAGAgTTGAATTCA Intergenic
chr8:13349868 TGAAaTtgAaTCTgaTtCCAG
tTtGTcAAAGACTTGtATTtA Intron (DLC1)
chrY:4231090 TGAATTCAAtTCTTcagCCAG
tcaGaAAAAtctTTGAATcCA Intergenic
chrX:90035974 TGAATTCAAtTCTTcagCCAG tcaGaAAAAtctTTGAATcCA Intergenic
Table 33. Targeting Exon 5:
Genome Left Half-Site Right Half-Site Genomic Region
Coordinates
chrX:154215513 TTCATCAAATACAGCAAAAAGTAG
GTCTGTGTCTTTTCCTTGGCCAGA Exon (F8)
chr8:65938903 TCTaGCCAAGccAgAGgCACtGAC GgCTcTGTCTTTTCCTctGCCAcA Intergenic
89/1
SUBSTITUTE SHEET (RULE 26)
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
chr1:26774318 TTCAaCAAcaACAaCAAAAAagca
cTCTGTGcCaTgTaCTTGGCCAGA Intron (DHDDS)
chr10:102225665 cTCAcCAAgcAttGCAtAAAGctG
CTACTTTTaGgTGTATTTtATGAA Intron (WNT8B)
89/2
SUBSTITUTE SHEET (RULE 26)
ak 02951882 2016-12-09
WO 2015/191899 PCT/US2015/035399
Table 33. Targeting Exon 5:
Genome Left Half-Site Right Half-Site Genomic Region
Coordinates
chr7:14755743 TTCATCAAcTcCAGgAAAAAcaAc
GTaTaTGTgTTTTCacTGGaCAGA Intron (DGKB)
chr8:124089292 TTCATaAtATcaAGtAAtAcGTga
GTtTGgGTtTTTTtCTTtGaCAGA Intron (WDR67)
chr6:70049288 TCTGGCCAtGacAgAtAaACgctC
aTACTTTTTGCTGTgTTTGATtcA Exon (BAI3)
Promoter
chr17:37764808 TCaaaCCAAGGgAAAGACAgAGAa GTCTGTGcCTcTgCaTgGGCgtGt (NEUROD2)
chr2:92285124 TCTtGCCAcaaAAAAtACACAGAa CTACgTTgTGaTGTgTTTacTcAA Intergenic
chr11:80679047 TTaATaAAgTgaAaCtAAAAGTAa GTCTGTaTgTTTTatTTtGCtAGA Intergenic
chr7:49746821 TCaGaCCAAGccAgAGgtgCAcAC GgCTtTGTCaTTTCCTTGGCCtGt Intergenic
chr2:92283421 TCTGGCCAcaaAAActACACAGAa CTACgTTgTGaTGTgTTTacTcAA Intergenic
chr6:53622618 TCcacCCAAGGAAtAGgCAgAGAg CTAaTcTTTGCTGTATTTtATtgA Intergenic
chr7:64186025 gcCAaCAgcaACAGCAAcAAaaAG GTtTtTGTCTTTTttTTaGaCAGA Intergenic
chr8:76622826 TCatGaaAAatAAAAGAaACAGta GTtTtTtTtTTTTCtTgGGaCAGA Intergenic
chr13:27818295 TCTGtCCAAaaAAAAaAaAaAaAa gTttTgTTTcCTGaATTTGATaAA Intergenic
chr18:68100701 TCaGGCCAAtaAAAAacaACAaAC tgcCTTTTTttTtTtTTTttTGAA Intergenic
chr5:72817667 TCTaGCaAAGaAAAAtAaACAaAa tTaTtTtTCTTTTttTTttCCAGc Intergenic
chr15:43320939 TCaaaCaAAaaAAAAaAaACAaAC
aTaTaTaTaTaTTCCTTGGCCgGA Intron (UBR1)
chr4:12953588 TaCATaAAAcACAaCAAgAAaTAG tTACTTacattTGTATTTGAaGAt Intergenic
chr22:49683417 TCTGGCaAAaGgAtAGcCACAGAt tTgTGTtTCTTTTtCcTGGgCAtg Intergenic
Table 34. Targeting Exon 6:
Genome Left Half-Site Right Half-Site Genomic Region
Coordinates
chrX:154212976 TTTACATAACCATTGACTGTGT CCCGAGCAGATGCAGCA Exon (F8)
chr3:140445224 TGCTGCATtaGCTCaGa CCaGAGCAGAgGCAGCt Intergenic
chr8:56002214 TaCTGCATCTtCTCtGG CtgGAGtAGgcGCtGCA Intergenic
chr12:49424040 gGtgGCATCTGCTCttG CCCGgGCAGAgGCAGCA Exon (MLL2)
chr1:70622888 TtCTaCtTCTGCTttaG tCtGtGtAGATGCAGCA Intron (LRRC40)
chr4:184357162 TtCTGCcTCTGCTCGaG ttttAcaAGATGCAGCA Intergenic
chr5:172342828 TGCaGCcTCTGCTCaGa CCtGAGCtGggGttGCA Intron (ERGIC1)
chr6:115061184 TGtTaCAcCTGCTCtGG gCtGAGCAtATGCAGgA Intergenic
chr12:39726775 TGaTGCATCTGtTtcGa CCtGAGCAGgTGCAtCA Exon (KIF21A)
chr7:88799625 TTTACcTAACCAaTGAaaGTGT CCtttGtAGATGCAGaA
Intron (ZUF804B)
chr20:17949040 TGCTGCAgCaaCTCGGG CtCGAGCAGggGCcGCc Exon (SNX5)
chr1:189751560 TttTcCATCaGCTCaGa CCtGAGCAGcTtCAGCA Intergenic
chr21:42907464 TGCcaCATCaGCTCtGG CCaGAGCAGcaGgAGCA Intergenic
chr5:2548607 TGCTGCcTCTGCcttca CatGAGCAGgTGCAGCA Intergenic
chr8:19923395 TtCTaCATCTGCTCaGa tCCtgGgAagTGCAGCA Intergenic
chr6:15883284 TGCTGtcTCTGCTCaGG CCtGAGCgGAaGCAGag Intergenic
chr17:81092958 TGCaGCcTCTGCTCcaG tCCcAGgAGATGtAGaA Intergenic
chrX:153711226 TGCTGCATCTaCTCctG CCCGgGCAGATctAttg Intergenic
chr1:3370563 TGCaGCcTCTGCcCGGG tCCcAGCAGgcGgAGCA Promoter
(ARHGEF16)
chr17:58495805 TaCTGCATCTtCTCaGa CaaaAGCAGtTtCAaCA Intergenic
chr5:169541385 TGtTGCATCaGCTCGGG CCtGAtCAGcgaCAGCc Intergenic
Table35.TargetingExon7:
Genome Left Half-Site Right Half-Site Genomic Region
Coordinates
chrX:154197644 TGTCCAAGGTCCATCAAGAG GGAAAGTTATTGGCGAGA Exon (F8)
chr2:18105031 TGTCaAAaaTCaATCAAaAa tTaTTGATtGAttTTtGACA
Intron (KCNS3)
chr7:26500117 TGTCCAAaGTCCATtttGAG tTtTTcATGGACacTGGgCA
Intron 1L0C441204)
chr4:27239786 TGTCacAGGTCCtTaAAGAG atAAAGTTATTGGgGtGA Intergenic
chr4:27428400 TCTtaCCAATcACTTTCt GGAAAGgcAgTGGtGAGA Intergenic
chrX:79810036 TGTCCAAaGTCacTtgAGAG GGAAAGTTgTTtGaGAGt Intergenic
chr1:172943650 TaTCCAgacTCCATCcAcAG tTaTgGAaGGAgtTTGGACA Intergenic
SUBSTITUTE SHEET (RULE 26)
ak 02951882 2016-12-09
WO 2015/191899 PCT/US2015/035399
Table 35. Targeting Exon 7:
Genome Left Half-Site Right Half-Site Genomic Region
Coordinates
chr18:40289853 aGTCCAAcaTCCAgCAAGAa CTCTTGATtGAgCTTaGAac Intergenic
chr17:53122291 TCTtttCAATAACTgTCC CTaTTGATGGACaTTaGACt Intron ISTXBP4)
chr1:184048225 TCTgGCCAATAACcgTtC CTCTTaATGatCtTTGGAtA Intergenic
chr19:32600353 TGaCCctGaTCCATCcAGAG GacAAGTTAgTGGCcAGA Intergenic
chr3:29286452 TGcCaAAGagCCATCAAGAa ttAAAGTTATgGGaaAGA Intergenic
chrX:145253799 TGTCCAAGGTCCcaCAgttG CTCTTGATGccCaTTGtAgA Intergenic
chr9:85073714 TcctCAAGGgCaATCtAGAG CTCTTGATtGtCtTgGGtCA Intergenic
chr22:25490404 TGTCCAAGGcCCcTCAgcAG GGgAAGTaAaaGGtGAGA
Intron (KIAA1671)
chr8:61847049 TCcaGagAcTAACTTTgC CcCTTGATtGACCTaGGACA Intergenic
chr4:177996308 TGTCCAgaGTCCAagAAaAa CaCTTGAaGGAtggTGGAaA Intergenic
chr2:63471205 TaTCaAAGGTCtcTCAAaAc CTCTTGAattAttTTGGgCA
Intron (WDPCP)
chr14:101569007 TGTCCAcatTCCcTCcAGAG CcCaTGATGGACCcaGccCA Intergenic
chr2:75005696 ctTCCAAGGcCCAcagAGAG CcCcTGATtGcCtTTGGAtA Intergenic
chr18:36812500 TCTCtCCAATAACTgTga tgCTTcATGtAtCTTGGcCA Intron
(L00647946)
Table36.TargedngExon8:
Genome Left Half-Site Right Half-Site Genomic Region
Coordinates
chrX:154194740 TCCTCCTCTTCAGCAGCAATGT
TTAGGATGCTTCTTGGCAACTGA Exon (F8)
chr5:33245024 cCAGaTtCCAAGAgaCATCaTAA
ACATgGCaGCTGAAGAGGAtGt Intergenic
chr3:159590558 TCCTCCTCaTCAGtAatAATGT
TTAGaATGtTcagTtGCAAtTGt Intron (SCHIP1)
chrY:14031090 TCAtTTtCaAtGgAtCATCCTAA
ACATgGagGagGAgGAGGAGGA Intergenic
chr10:83854828 TCctTTtCCtgGAAGCtTtCTcA
TTtGGATGCTTtTgGGaAcCTGA Intron (NRG3)
chr12:86811646 TCAaaaGCCAAaAAaCAagCaAA
TTAttATGCTcaTTtGCAAaTGA Intron (MGAT4C)
chr6:43379997 TgAGaTaCCAttAcaCATCCTAg
AaAgTGCTGgTGAAGAtGtGGA Intergenic
chr15:60816292 TCtgCCTCcTCccCAcCcATaT
TTAGGcTGCTTCTTGGCAcCTtc Intron (RORA)
chr4:104036767 TtAaaaGCCAgGAAGCATCCTAA
ttATTGaTtaTGAAtgcGAGGA Intron (CENPE)
chr2:220922430 aCAaTTcCacAGAAtCATCCaAA
aatGGATGCTcCTTGGCAtCaGA Intergenic
chr6:151256031 TCAGcTaCCAAGAgaaATtCTAA
TTgGGAcatTTaTTtGCAcCTGg Intron (MTHFD1L)
chr12:14116257 TCtcCCTCaTCAGCAGaAATGa
gCATgaCaGCTGtAGtGGAGGg Intron (GRIN2B)
chr11:41540671 TttTCaTCTTCAtCtGtgATtT
caATTGCTGCTGAAGgtGAGGA Intergenic
chr10:607478 TaCTCCTCTaaAaCcaCAATGg
acAGGATGgTTCTcaGCcACTGA Intron (DIP2C)
chr18:64076819 TCAtTTaCCAAacAGaATtaTAA
gTAaGATGtTTCcTGatttCTGA Intergenic
chr3:159590555 TCaTCCTCcTCAtCAGtAATaa
TTAGaATGtTcagTtGCAAtTGt Intron (SCHIP1)
chr2:25775417 TCCcCaTCaTtAGCAGCAATGc
TcAGGtTtCcTtTTGcaAACaGA Intron (DTNB)
chr5:60672404 aCCTCCaCTTCAGtAatAATGa
TTAGaATGtgTtaTGtCAttTGA Intron (ZSWIM6)
chr2:158235451 TCAaaTGaCAtaAcaCATtCTAA
tCATTatTaCTGAAGtGGAGGt Intergenic
chr11:131914316 TCtGagGCCAAaAAGaAaaaTAA AtgTgtCTGtTcAAGAGGAGGA Intron
(NTM)
chrY:3867095 aCAGTTaCCAAaAAGCAaaaTAA
gCAagatgGCTGAAtAGGAaGA Intergenic
Table 37. Targeting Exon 9:
Genome Left Half-Site Right Half-Site Genomic Region
Coordinates
chrX:154194255 TCTCCAACTTCCCCATAA TTCCTGATTCATGCTGAA Exon (F8)
chr4:150672318 TTCAGCtTaAcaCtGGAt TTCCTGATTCcTGaTGAA Intergenic
chr2:89399484 TgCAGCATagATCAGGgA TcCCTGgTTtcTGCTGAA Intergenic
chr5:19372097 TTCAtCATaAAgCtaaAA TTCtTaATTaATGCTGAA Intergenic
chr4:56376997 TTCAGaATGAAaCAGGAA TTCCTGAgaCAaGaTGgg
Intron (CLOCK)
chr14:98831622 TtTCCtcCTTCCCCATAc gTtCTGATTCATGaTGAA Intergenic
chr20:6216194 TTCAGCATGAAgCAaGAA TTCCTGAaaCATcaacAA Intergenic
chr3:76350178 TTCAGCtTGAATtAGGAA cTtgTGtTTaATGaTGAA Intergenic
chr6:79957598 TTCAGCATaAATaAtaAA TTCtTGtTTaATtCTcAA Intergenic
chr5:129714571 TTCAcCATctATCtGaAA TTtCTGAggCATGtTGAA Intergenic
chr2:183992955 aTCAaCATGtAaCAGaAA TTttTGATTCATGtaGgA Intron (NUP35)
91
SUBSTITUTE SHEET (RULE 26)
CA 02951882 2016-12-09
WO 2015/191899 PCT/US2015/035399
Table 37. Targeting Exon 9:
Genome Left Half-Site Right Half-Site Genomic Region
Coordinates
chr11:100927598 TTCAatATGAtTaAGtAt TTgaTGATTtATGCTGAA Intron (PGR)
chr5:118162509 TgCAGCAgtAAaCAtGAA TTtCTaATTCATGCTaAA Intergenic
chr7:136796091 TgCAGCATaAATtAaGgA aTCCTGggTCATGtTGAA Intron(L0C349160)
chrX:114442244 TTCcaCATaAAaaAGGAc TTCCTGtTgtAgGCTGAA Intron (LRCH2)
chr17:70147587 TTaAaaATGAATCAaaAc TTtCaGATcaATGCTGAA Intergenic
chr22:17414552 TgCAGCATGAATtAGGAg TcCCTGgTTtcTGCTGAt Intergenic
chr1:220485886 TTCAGgAgaAATCgaGAA TTCCTGATatATGtTGAg Intergenic
chr2:89292060 TgCAGCATagATCAGGAg TcCCTGgTTttTGCTGAt Intergenic
chr2:89309611 TgCAGCATagATCAGGAg TcCCTGgTTttTGCTGAt Intergenic
chr2:90260070 aTCAGCAaaAAcCAGGgA cTCCTGATctATGCTGcA Intergenic
Table38.Targeting Exon 10:
Left Half-Site Right Half-Site Genomic
Genome Coordinates Region
chrX:154189360 TCTCCTTGAATACAAAGGAC CCGTGAGGGTAGATGTTATA Exon (F8)
chr6:129821493 TgTCCTTaAAaACAAAGGAC CttTGAGGtTAcATGTTAgA Intron(LAMAZ
chr2:147755789 TtTCCTTGgATACAAAGaAC aaaaTTTaTATgCAAGGAGg Intergenic
chr15:35542434 TATAAgATaTACCCTaAtGG tTCCTgTGTcTTCAAaGAGA Intergenic
chrX:106606342 TCTCCcTGcATACAgAGatC GTtCTTTGTATaagAGGAGg Intergenic
chr11:116391255 TCTCCaaaAATAaAAAaGAa GcCtaTTGTATTCcAGGAaA Intergenic
chr4:174370428 TaTCtTcaAATtCAAAGGAC aTCCTTTGTAgTCAAGGAtg Intergenic
chrX:48388946 TgTCCTTGcATgCAAAatAC cTCtTTTGTtTTtttGGAGA Intergenic
chr1:184030566 TCTtaTTattTACAAAGagC GTCtcTTtTATTgAAGGAGA Intron(TSEN15)
chr8:105838647 aCatCTTaAATACAAAGaAC GgCaTcTGTAaTCAAGtgGA Intergenic
chr14:60101345 TCTCCaTaAATACAAAGGga CaGaGgGGGaAaATtTTAcA Intron(RTN1)
chr6:32447046 gCTCtTTGtgaACAAAGGcC tTCCTTTGTATTtActGAGA Intergenic
chr6_cibl_hap6:3707956 gCTCtTTGtgaACAAAGGcC tTCCTTTGTATTtActGAGA Intergenic
chr6_apd_hap1:3761430 gCTCtTTGtgaACAAAGGcC tTCCTTTGTATTtActGAGA Intergenic
chr6:153043585 TgTAAtATtTtCCCcCAaGc GTatTTTGTATTCAAtGtGA bmn(NWCT1)
chrX:129578399 TCaCCaTcAgTgCAAgaGAC GgCtTTgGTATTaAAtGAGA Intergenic
chr2:237165553 TCTCgTaGAAagCAAAGaAa tTttTcTGTATTtAAaGAGA Mtron(ASB18)
chr14:74504800 TATcttATCTcCCCTaAtaG GTCCTTTGTATTCAttGAaA Mtron(C14off45)
chr14:94651285 TCTCCTgGggaAtgAAGGtC GatacTTGTATTCAAGGAGA Mtron(PPP4R4)
chr14:42051030 TtTCCTaGtATACAAAaGAt aTCtTTTGTATaCtAGGAaA Intergenic
chr11:31557496 caTCCTTGgATACAgAGGgC GattTTgGTATTCAtGGAGt Mtron(ELP4)
Table39.TargetingExon11:
Genome Left Half-Site Right Half-Site Genomic Region
Coordinates
chrX:154185248 TCTACAGATTCTTTGTAGCAG ATGAGTCCTGAAGCTAGA Exon (F8)
chr8:91254790 TCTAGtTTCAGcAgTatT ATGAGTCaTGAAGCTtGA Intron(LINC00534)
chr2:220340352 TCcAtCTTCAGGACTCAc AgGAGcCCTGAAGtTtGg
Intron (SPEG)
chr13:65583211 TtTACAGATgCTTTaTAGCAG CTGgcAatAAacATCTGTAGA Intergenic
chr8:136213502 cCTACAaATcCTTTGTgGCAG ATGgGctCTGgAGCcAGA Intergenic
chr4:79545446 TtcAcCTTCctGACTCAT ATGAGTtCTGggGCTAGA Intergenic
chr6:105454604 TCTcaCTTCAGGACcCAg ATaAGTttTGAAGCagGA
Intron (LIN28B)
chr17:50618031 TCcAaCcTCAGaACTCAT cTGAGTtCTGAgGtTgGg Intergenic
chr21:40482039 TCTAaaaTCAGGACTCcT gTGAtTgtTGAAGCcAGA Intergenic
chr11:132218577 TCTcaCTTaAGGACTtAc tTGAGTCCaGAAGtTtGA Intergenic
chr2:27385297 TCTgtCTTCAGaAgTCcT gTGAGTtCTGAAtCTgGA Intergenic
chr14:22481030 TCTAcCTTCAGcACTCtg tTttGTtCTGAAGCcAGA Intergenic
chr3:31348185 TCTcGCaTCAaGACcCAT tgGAGTtCaGAtGCTAaA Intergenic
chr4:87584049 aCTACAGcTaCTTgGaAGCAG tTGAGcCCaGAAGtTtGA
Intron (PTPN13)
92
SUBSTITUTE SHEET (RULE 26)
ak 02951882 2016-12-09
WO 2015/191899 PCT/US2015/035399
Table39.Targeting Exon 11:
Genome Left Half-Site Right Half-Site Genomic Region
Coordinates
chr4:71281490 TCaAaCTcCtGacCTCAT tTGtTtCAAAtAATtTGTAtA Intergenic
chr2:108857249 TCTctCTcCAGtACTCAT ATGtGTgCTGtgGgTAGA
Intergenic
chrX:47785928 TgTAGCTTCtGtACTacT ATaAGTCtTGAAGtcAGA
Intergenic
chr8:79584265 TCTtGCcTgAGGACTCAT tgGgGaCtTGAAGtTAGA
Intron (ZC2HC1A)
chr1:216023388 TCaAGaTcCAGaACTCAa ATaAGTaCTGAAGCTAtt
Intron (USH2A)
chr17:50619873 TaTAcaTaCAGaACTtAT ATGAGTtCTGAgGtTAGg
Intergenic
chr13:20930589 aCTAGCTTCAttAtTCAT ATtAGTCtTGAAGtatGA
Intergenic
Table40.Targeting Exon 12:
Genome Left Half-Site Right Half-Site Genomic Region
Coordinates
chrX:154182199 TCCTCAAGCTGCACTCCAGCT CGTTGTATATTCTCTGTGA
Exon (F8)
chr7:156430074 TCCaCAAGCTGgACTCCAaCT atTTGaAcAcTtTCTGTGA Intergenic
chr9:43597045 TCACAaAGAATAaACAACt CtaTGTATATaaTCTtTtA
Intergenic
chr10:899227 TCcCAGtGAATATAaAAat tGTTGTATATTtaaTGTGA
Intron(LARP4B)
chr5:44595593 TCAaAGtGgAaATACAACa CtTTGTATATTtTCTtTtA
Intergenic
chr12:13837730 TCcCAGAGAAaATACcAaG CGTTaTcTcTTtTtTGTGA Intron(GRIN2B)
chr10:85585731 TCAtAGAaAATAagaAACt tGTTGTATATTCTgTGTcA Intergenic
chr10:64580474 TCcCAGAGgcTATAaAcCa AaCTGttGTGaAGCTTGAGGA Intergenic
chrX:38783417 TCCTCAAaCTGCtCTCCAaCa CtTccTATtTgtTCTtTGA Intergenic
chr2:193570138 TtACAtAGAATtTACAAta CaTTGTAaATTCTaTGTGA Intergenic
chr7:110741635 TaAtAcAGAATATACAtaG tcTTGTATATTtcCTGTGA Intron(IMMP2L)
chr3:191344909 TCcCAaAGAcTgTtCtAaG gGTgtTATATTCTCTGTGA Intergenic
chr9:39389206 TaAaAGAttATATACAtaG ttTTGTtTATTCTtTGTGA
Intergenic
chr9:39918509 TaAaAGAttATATACAtaG ttTTGTtTATTCTtTGTGA
Intergenic
chr9:40733954 TaAaAGAttATATACAtaG ttTTGTtTATTCTtTGTGA
Intergenic
chr9:41293775 TCACAaAGAATAaACAAaa CtaTGTATATaaTCTtTtA
Intergenic
chr9:65476200 TCACAaAGAATAaACAAaa CtaTGTATATaaTCTtTtA
Intergenic
chrX:50790890 gCACAGActATAggCAgCc CaTgGTATATTCTtTGTGA
Intergenic
chr5:5141262 TCCcCAAcCTttcCTCCttCT CGTTGctTATTCTCaGTGA Intron(ADAMTS16)
Intron(LOC10087306
chrX:22329605 TCAaAtgGAgTAaACAACt CtTTGTAcATTtTCTGTGt 5)
chr7:105616909 TCACAGAGcATATACtcCa ttTaGTATATTCaCaGTcA
Intron (CDHR3)
Table41.Targeting Exon 13:
Genome Left Half-Site Right Half-Site Genomic Region
Coordinates
chrX:154176028 TGTGTCTTCATAGACCATTTT
ATCCAGAGAAGAAGACA Exon (F8)
chr19:31555212 TaTCTTCTTCTCTGGAT cTCCAtgGAAGAAaAaA
Intergenic
chr11:98185196 TaTcTCTTaATAGcCCATTTT ATaCAGAGAAGAAaACA Intergenic
chr9:126179092 TGTGTCTTtATgGAaCAacTa
ATtCAGAGAAtAAGACA Intron (DENND1A)
chr1:197582736 aGTtcTCaTCcCTGtAT cTCCAGAGAAGAAGACA
Intron (DENND1B)
chr9:25886338 TtTtTaCTTCTCaGaAT ATtCAGAGAAGcAGAtA
Intergenic
chr16:65046771 TGcCTTCTTCTCTGaAT cTCtAGAccAaAAGtCA
Intron (CDH11)
chr6:37769405 TGaGTCTTCATAGAaCATTTT AgCtgGAagAGAAGACc Intergenic
chr4:53116406 TGgCTTCTgCTCTGtgT AgCCAGAGAtGAAGtCA
Intergenic
chr10:117955396 acTaaaCTTCTCTGaAT AgCCAGAGAtGAAGACA
Intron (GFRA1)
chr4:157999316 TaTaTTCTTaTaTGGAg AAggTGGTtTATGAAGACACA Intron (GLRB)
chr4:172676113 TGTCaTCTTCTCTGtAT tTtaAGAGAAaAAtACt Intergenic
chr7:70692951 TGcCTTCTTCcCTGGAT cgatAGAGgAGgAGACA
Intron (WBSCR17)
chr1:153460499 TGTCTTCTTCTCTGtcT ATCtAGAGAAtggGAgt Intergenic
chr17:55521352 gGTCaTCaTCTtTGGtT AgCCAGgGAAGAAGACA
Intron (MSI2)
chr15:37159972 TGTtTTCTTCTCTGcAT tAAATaaTCTATGAtGAgAtA Intron(L0C145845)
chr10:81475753 TcTCTTCTTCTCTGtAT AggCAtAGAtGAtGgCA
Intergenic
chr10:88997979 TcTCTTCTTCTCTGtAT AggCAtAGAtGAtGgCA
Intergenic
93
SUBSTITUTE SHEET (RULE 26)
CA 02951882 2016-12-09
WO 2015/191899 PCT/US2015/035399
Table 41. Targeting Exon 13:
Genome Left Half-Site Right Half-Site Genomic Region
Coordinate s
ch r10 :89259535 TGcCaTCaTCTaTGccT ATaCAGAGAAGAAGAgA Intergenic
chr2:12846210 ctTCTTCTTCTCTGaAT ATatAtAGAAGAAtAtA Intergenic
chr13:107009889 TGTCTcCcaCTCTGctg AT a CAGAGAAGAAG gCA Intergenic
Table 42. Targeting Exon 14:
Genome Left Half-Site Right Half-Site Genomic
Coordinates Region
chrX:154156874 TCATCCCATAATCCCAGAGCCTCT AAATAGTGTCGTGTTTTCTTTTGA Exon (F8)
Intron
chr6:17669261 TgAAAAaAAAAaAaaACACTATTa AAATAcctTttTtTTTTtTTTTGA (NUP153)
Intron
chr11:12730893 TaAAAAaAAAAaACcAgAaTAaTT ttATAGTtTtGTtTcTTtTTTTGA ITEAD1)
chill: 68651384 TCAAAAaAAAcCAaaACACTtaTT
AAtTAaTtTtaTtTaTTtTTTTGA Intergenic
Intron
chr5:132729450 TagAAAGgAgACAaGggtCTAgTT AGAaGCTCTGtGAgTtTGGGATGA (FSTL4)
chr5:102197872 TCAAAAaAAAAaAaaAaAaaAaTT AcATAtTGTCtTtTTTTtTTTTaA Intergenic
Intron
chr6:150020193 TCAAAAaAAAAaAaGgCACTATcT AGtaGgTtaGGGtTTcTGaaATGA (LATS1)
Intron
chr8:102067589 TCAgAAaAtAAtAtGACACTtTTg AAATttTGTCaTGTTTgCTTTaGA (FLJ42969)
Intron
chr5:96436598 aaAAAAaAAAAaAaaAgAaTATaT AAtTAGTGTtGTcTTTTCcTgTGA (LIX1)
chr22:31430439 TCAAAAaAAAAaAaGcCcCTgTcc AtATAtTtTttTtTTTTtTTTTGA Intergenic
Intron
chr5:96436600 aaAAAAaAAAAaAaGAataTATaT AAtTAGTGTtGTcTTTTCcTgTGA (LIX1)
chr8:129874245 TtAAAAGAAAcagCGACACTATTT AtAaAaTagCaTtTTcTCTTcTGA Intergenic
chr8:76048195 TaAcAcagAAtCACctCACTATaT tAATAGTtTttTtTTTTtTTTTGA Intergenic
Intron
chr3:167630709 TtAAAAaAAAAaAaaAgcCTATTT AAATtGTGaCaTcTTTTtTTTTaA (L00646168)
chr17:79330592 TCAAAAaAAAAaAaaAaAtTATTT tttTttTGTttTGTTTTgTTTTGt Intergenic
Intron
chr7:56511801 aaAAAAGAAAACtgGtgtCaATTT AAAaAGTGTCGgGTTTTtTTTTtt (L00650226)
Intron
chrX:108947147 TaAAAAaAAAAaAattCACTATgT AAATAtTGTgGgGTTTTtTTgTtg (ACSL4)
chr12:123230886 TCAAtAaAAAtaAaaAtAaaATTT tAATAGTaTttTtTTTTtTTTTGA
Intergenic
chr3:163374286 TaAAccaAAAACtCaACAaTcaTT AAATAtgGTtGgtTTgTtTTTTGA Intergenic
Intron
chr12:9357687 TCAAAAaAAAACAaaACAaagTTT gAAaAGTcTttTcTTTTtTaTTtA (PEP)
chr2:188514899 TCAAAAGtAAAaAgtAaACTATTT tAATAGTGagGTaaTTTCTTTatA Intergenic
Table 43. Targeting Exon 15:
Genome Left Half-Site Right Half-Site Genomic Region
Coordinates
chrX:154134726 TATGGCCCCAGGAGTCCCAA CTCCACGGTATAAGGGCTGA Exon (F8)
chr1:43805061 TATGGCCCCAGagaTCCCAA tcCCACGGTcatAcaGCTGA Exon (MPL)
chr17:48220703 TATaGCCCCcatgGTCaCcA CTtCAgGGcATAgGGGCTGA Intron
(PPP1R9B)
chr6:10659136 TCAatCCTTATgCCaaGGAG TctGGtCTCCTGtGGtCAcA Intergenic
chr4:138564864 TATGaCCCaAaGAaaCCaAA tTCtAtGtTAaAAGtGaTGA Intergenic
chr1:242357075 TgTGaCCCCAGGAGTCatAA CTtCAaGGgcTAtGGGagGA Intron (PLD5)
chr20:53898975 TCAaCCCTaATtCCtTaGAG CTCtAgGGgATAAGGctTcA Intergenic
chr16:10915221 TcTGaCCCtAaGAaTCaCcA TTGGGgtTCCTGGaGtCATg Intergenic
chr10:134224399 TgTGGCCCCAGGgGcCCaAc agGGGACTttTGGGGgCgTA Intron (PWWP2B)
chrX:17609569 TaAGCCCTTATAatGgGtAG tTCCAtGGTATttGGtaTGA Intron (NHS)
chr12:4412126 TggGcCCCaAGGAGTCCCAc TTGGGAaTCtTGGaGCCtaA Exon (CCND2)
c1ir22:48089574 TgTGGgCCCAGGAGTCaCgA CcCCAgGGTATcAGGGtgGc Intergenic
chr17:1538247 TgTGGCCCCAGGAagCCCAg TTGGGgCTCtgGccGaCAgA Exon (SCARF1)
94/1
SUBSTITUTE SHEET (RULE 26)
CA 02951882 2016-12-09
WO 2015/191899 PCT/US2015/035399
chr19:35657806 TAccaCCCCAGcAGTCaCAA tggCAgGGaAcAAGGGCTGA Intron (FXYD5)
chr1:158375793 TcTaGCtCCAtaAGTCCCtA TTGGGtCTCtTGGGatCtgA Intergenic
chr14:99426061 TCAGCaCTTATcCaGTGGAc TTGGGACaCCaGaGaaCAcA Intergenic
chr1:34177797 cATcaCaCCAGGAtTCCCAA TgGGGtCcCCTGGGGtCAgg Intron (CSMD2)
chr13:19522623 cCAcCCCcccTACaGgGGAG TgGGcACTCCTGGGcCCATA Intergenic
chr11:17783271 TcTGGCCCCAtGgaTCCCAA caGaGcCTCCTGGGGCacaA Intron (KCNC1)
chr14:71921590 TCtGCCCTTtTACtGTGGAG acGGGACaCCTGatGtCAcA Intergenic
chr10:132968471 TCAGCCaTTccACCGTGGAa acGGctCTCCgGGGGCCAct Intron (TCERG1L)
94/2
SUBSTITUTE SHEET (RULE 26)
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
Table 44. Targeting Exon 16:
Genome Left Half-Site Right Half-Site Genomic
Coordinates Region
chrX:154133096 TCAGAGAAATAAGCCCAG CTCATCTTTAGTGGGTGCCATA Exon (F8)
chr7:25537263 TCtGtGcAATAAtCtCAG CTGtGCTTATTTaTtTGA Intergenic
chr1:85241221 TaAaAaAAAaAAGCCCAG CTGGGCTTtcTTCTggGA Intergenic
chr17:49365434 TCcaAGAAAcAAaCCCAa CaGGtgTTAcTTCTCTGA Exon (UTP18)
Intron
chr10:15407376 TATGaCAtCaACTAAAGATGcG agGGGCTTAaTTCcCaGA (FAM171A1)
chr6:66455619 cCAGAcAgAgAAcCCCAG CTGGGtTTATTgCaCTGA Intergenic
chr2:168339348 TCAaAaAAgaAAGCCaAG CTGtGCTTATaTCTCTcA Intergenic
Intron
chr8:3275497 TCAGtGAcATAAGCCCAG CTGtGCTTgTTaaaaTGA (CSMD1)
chr1:172577364 TCAtAGtAATAAaCagAG tTGtGtTTATTTCTCTaA Intron (SUCO)
chr9:131943933 gaAGgGgAATAgGCCCAa CTGGcCTTATTTCTCTGt Intergenic
chr14:30487657 TCAtAGAAATAtGCCCAa CTGaGCTcATgggTtTGA Intergenic
chr3:82950355 aCAtAtAAATAAGaaCAt CTtGGCTTATTTtaCTGA Intergenic
Intron
chr22:40341367 TCAGAGAAATgAGCCCct tcGGctTTAaTcCTCTGA (GRAP2)
Intron
chr20:19686090 TtgGAaAAATAAtCCCAG taGGGCTTATTTgctTGA (SLC24A3)
Intron
chr4:20811976 TCAGAGAcAatAtCaaAG gTGGGtTTATTTgTCTGA (KCNIP4)
chrX:97284124 TCAGgGcAATcAGCCCAG CTGGGgTTtcTTgTCTGg Intergenic
chr18:41220996 TCAaAtgAATAAGaCaAt tTGGttTTgTTTCTCTGA Intergenic
chrY:19504648 TCAGgaAAAaAAtCCCAG CTtGttTTATTctcCTGA Intergenic
chr6:11989807 TCAtAtAAATgAGCtCAt CTtGGCTTcTTTCaCTGA Intergenic
Intron
chr11:100111323 TaAaAttAATgAGCCCAG tTtGGCTTATTTCcaTGA (CNTN5)
Intron
chr13:26279732 agAGAGAAAaAgGCCgAG tTGGGtTTATTTtTCTaA (ATP8A2)
Table 45. Targeting Exon 17:
Genome Left Half-Site Right Half-Site Genomic Region
Coordinates
chrX:154132638 TCTTTCCATATTTTCAG TTTGGTCTCATCAAAGA Exon
(F8)
chr11:86435291 aCTTTCCATAgTTTCAG CTGAAAATATtGAAtGA Intergenic
chr17:191390 TCTTaGAgGAcACCAAA TTTGGTgTCATCtAAGA Intron (RPH3AL)
chrX:16807199 TCTaTCCtTtTTTTCAG tTGAAAATATtGAAAGA Intron (TXLNG)
chrX:4909433 TtTTTCCATATTTTCAG TcaGtTtTCtTCAAAGA Intergenic
chr15:98192520 TCTTTCCAcATTTTCAG CTGAAAATATtaAAtaA Intergenic
chr3:65632758 TCTTTGAaaAGACCAAA CTGAcAAcAgGGAAAaA Intron (MAGI1)
chrX:81782933 TCaTTtaATATTTTtgG CTGAAAATgTGGAAAGA Intergenic
chr20:48433923 TCTTTaATGAtACCAAA TTaGGTCTttTCAgAaA
Intron (SLC9A8)
chr8:84366161 TCaTTtCATATTTTCAG CTGAAAtTgTGGAAAGt Intergenic
chr1:93406669 atTTTGATaAGAtCAAA TTTGGTgTCATCtAAGA Intron (FAM69A)
chr3:23702529 TaTTTGATttaAtCAAA TTTGGTtTCATgAAAGA Intergenic
chr4:127360864 TCTTTCCAcATTcTCtG gTTGGTtTCATCcAAGA Intergenic
chr9:10862420 TtTTaGAaGAaAaCAAA TTTGGTgTCAgCAAAGA Intergenic
chr2:30136701 TCTcTCCATATTcTCca CTGAAAATAcaGAAAGA Intron (ALK)
Intron
chr2:8966383 TtTTTaATaAtcCCAAA TTgGGgCTCATtAAAGA (KIDINS220)
chr10:106620765 TCcTgGgTGAGACCcAA TcTGGTtTCATCAAgGA Intron (SORCS3)
chrX:108769761 TaTTTGATGAGACCAAc aTGAgAATATaGcAAGA Intergenic
chr1:111227475 TCaTTtaATATTTTCAG CTGAAAtTATGGAAAGc Intergenic
chr3:114347859 TCTTTGATGAaAaCcAA TTTGtTtTCAcaAAtGA
Intron (ZBTB20)
chr6:24241996 TCTTTCCATATTTTaAt taGAAtATATGaAtAGA Intron (DCDC2)
95/1
SUBSTITUTE SHEET (RULE 26)
CA 02951882 2016-12-09
WO 2015/191899 PCT/US2015/035399
Table 46. Targeting Exon 18:
Genome Left Half-Site Right Half-Site Genomic Region
Coordinates
chrX:154132208 TATACTCCTCTTTTTTTCG GTCCACTGAAATGAATAGA Exon (F8)
chr3:89963270 TCTATTCATTaCtGTttAC GTCCAtTGAAtTGcATAaA Intergenic
95/2
SUBSTITUTE SHEET (RULE 26)
ak 02951882 2016-12-09
WO 2015/191899 PCT/US2015/035399
Table 46. Targeting Exon 18:
Genome Left Half-Site Right Half-Site Genomic Region
Coordinates
chr13:71330234 TtTATTCATTTCAtTGaAa GTCtAtTtAAATaAAgAGA Intergenic
chr7:52504835 TCTATaCATTTCAGaacAC GcaCACTaAAAaGAAcAGA Intergenic
chr7:93233952 aATACTCCTCcTTcTTTtt aTaCACTGAAATGgATAGA Intergenic
chr20:8957392 TATAaaCgTtTaTTTTTCt GTtaACTGAAATGAcTAGA Intergenic
Intron
chr2:55547229 TATACTtCTCTTTTgTTCa tGAAAAAAtGtGtAcTAgA (CCDC88A)
chr6:55916123 cATACTCCTCTTaTTTTCa tgCCACTGAAATGAcTttt Intergenic
chr8:93952422 TCTATcCATgTCAaaGaAC GTCttCTcAAATGtAcAGA Intron (TRIQK)
chr14:61101496 TCTATcCATTTCtGTGtAC tGcAAAtAAaAGtAGTATt Intergenic
chr11:33381162 TATACTtCTaTTTTTTTat aGAAAAAgAGAGtAGTAcA Intergenic
chr6:84078984 TCTATTacTgaCAcTGaAC GTCtACTGAAgTGAActGA Intron (ME1)
chr11:123025415 aATcCcCCTCaTTTTTctG tTCCACTGAAATGAtTAtA Intron (CLMP)
chr1:58698828 TAatCaCCTCTTTTTcTCc GTatAtTGAAATGtAgAGA Intron (DAB1)
chr13:90438048 TCTATTaATaTCAGTaaAC GgCCAaTGAAAcaAATgGc Intergenic
chr3:20841157 TCTtccCATTTCtGTGaAa GTtaAaTGgAATGAATAGA Intergenic
chr5:22000977 TCTATTaAaaTCAaTaGAC GTttACTtAcATtAtTAGA Intron (CDH12)
chr5:69306485 TCTATTaAaaTCAaTaGAC GTttACTtAcATtAtTAGA Intergenic
chr5:70181567 TCTATTaAaaTCAaTaGAC GTttACTtAcATtAtTAGA Intergenic
chr3:62322281 aCTATaCATTTCAaTaGtC tTCCACTGtAATtAgTAtA Intergenic
chr1:239837471 TtaAaTtATTTCcGTGGAa GTCCACaGAtATGAATAtA Intron (CHRM3)
Table 47. Targeting Exon 19:
Genome Left Half-Site Right Half-Site Genomic Region
Coordinates
chrX:154130370 TGCTCGCCAATAAGGCATTCC AGCTTTGGATGGTAACA Exon (F8)
chr4:53352906 TGTgACCATCCAAgGCT AGCaTTGGAgGGgAACA Intergenic
chr21:36529769 TGTTcCCAcCCAAAtCT AGaTTTGGgTGGggACA Intergenic
chr9:76182583 aaTTACaAaCaAAAGCc tGCTTTtGATGGTAAtA Intergenic
chr3:81470457 TGTTACttTgCAAAtgc AatTTTGGATGGTAACA Intergenic
chr1:203239036 TGTTACCAgCCAAAcCT AGggaTGGAgGGTtgCA Intergenic
chr3:65643349 TGTTtCCtTtaAAAtCT AGCTTTGtcTGGTAACA
Intron (MAGI1)
chr2:52456162 TaTTgCCtTCatcAGCT AGCTTTGGAaGGTAtCA Intergenic
chr4:150055809 TtTcACCATCCAAAtCT AttgTTGGgTGGTAAgA Intergenic
Intron
chr11:43851516 TacTACCATaCAAAGCT tGgaTTGGATGtTcACA (HSD17B12)
chr7:114250318 TaTTACtgTCtAtAtCT AGCTTTGaATGGTAAaA
Intron (FOXP2)
chr3:167657104 TGTgAaCATCCAAgGCT AGCTcTtGATGGTcACt Intergenic
chrX:149844333 TGgTgCCtaCCAcAcCT AGCTTTGGATGGTcAgA Intergenic
chr9:29156612 TGaTAaCtTCCAAgaCT gtCTTTGGAaGGTAACA
Intron (LING02)
chr4:70236889 TaTTACCATCaAAAtCa AGCTTTtGtaGGTAAtg Intergenic
chr3:151160745 aaTTcCaAcCCAAAGgT AGCcTTGGATGGTAACc Exon (IGSF10)
chr13:35431619 TtTTACCcTCCAAAcCc AGCTTTGGAaaaTAACA Intergenic
chr4:29377428 TGTTAaaATCCtAAtCc AcCTTTGGATGGTAAtt Intergenic
chr13:62451673 TGTTcCCAcCCAAAtCT AGagTTGGAgGGaAgtA Intergenic
chr12:95616056 TtTTcCCATttAgAtCT AttTTTGtATGGTAACA Intron (VEET)
chr18:28761651 TagaACCATCCAAAaCT AGaTTTGcATGtTtAaA Intergenic
Table 48. Targeting Exon 20:
Genome Left Half-Site Right Half-Site Genomic Region
Coordinates
chrX:154129651 TGTCCTGAAGCTGTAATCTGAA CCAGAAGCCATTCCCAGGGGA Exon (F8)
chr8:31295960 TCCCCTaGGAcTGaCTTCaGa CCAGActCtATTgCCAtGtGg Intergenic
chr2:165151202 aaTCCaGAAGCaGTAAcCaGtA CgtGAAtCCtTTCCCAGGGGA Intergenic
chr15:66216735 TCCCCaGGGAATGGgaTCTGG aCAGggGtCtcTCCCAGtGGt Intron
(MEGF11)
chr14:97246034 TgCCaTGGGAtTtGCTTCTGc CCAGAAGCagTcttCAGGGGA Intergenic
chr1:17425225 TCCaCTGaaAtgacCTTCTGG CCtGtAGtCATgCCCAtGGGA
Intron (PADI2)
96
SUBSTITUTE SHEET (RULE 26)
ak 02951882 2016-12-09
WO 2015/191899 PCT/US2015/035399
Table 48. Targeting Exon 20:
Genome Left Half-Site Right Half-Site Genomic Region
Coordinates
chr19:11752845 TCCCCTGGGAcactCagCTtt CCAGAttCCATTCCttGGGGA Intergenic
chr6:165113924 TCCCtTGGcAATtGCTTCTct CCccAttCCATTCaCAGGGGA Intergenic
chr3:18310932 TtCCCTGattATaGCTTtctG CCAGAAGaCATTtCaAGGaGA Intergenic
chr16:54478454 TCtCCaGaGAgaGGCTTCTaG CCtGAtGtCcTTCCtttGGGA Intergenic
chr2:100885233 TCCtCaGtcAATGGCTTCTGG atgGAAaCCAgTCCaAGGGaA Intergenic
chr6:160576093 TgCtCTtGGgATGtCTTCTGG taAGAAtCCATTCCtAGGatA Intron (SLC22A1)
chr1:888254 TaCCCTGGccATGGCcTCaGG agAGAgGCCcTcCCCtGGGGA Intron (NOC2L)
chr11:24688064 TCCatTGaaAATaGCTcCTGa gCAGgAGCtATTCtCAGacGA Intron (LUZP2)
chr3:188747522 TCCCtTGtGAATGGCTTggtG aCcGtAGtCATTCCCAtGaGA Intergenic
chr10:74502577 TcTCCTGAAGaTGTAATtaGAg CCtGAgGtgATTtCtAGGGGg Intron (MCU)
Intron
chrX:28644076 TCCaCaGaGAATaGtTTaTGc CttGtAcCCATTCCatGGGGA (IL1RAPL1)
chr2:167140954 cGTCCTtAcGCTGTcATCaGAA gCAGAAGCtgTcCattGGGGA Intron (SCN9A)
chr10:3095266 gCaCCTtGaAATGGgcaCTGG CCgGAAGCCATTCCaAatGGA Intergenic
chr5:73250307 TCCCCTGGGAActGCTgaTGG CCAGAAGggATggtaAaGGGA Intergenic
chr1:145822030 TCaCCTGGGAATaGtaTCTaG CaAGAAGaaAacaCtAGaGGA Intron (GPR89A)
Table 49. Targeting Exon 21:
Genome Left Half-Site Right Half-Site Genomic Region
Coordinates
chrX:154128167 TGCTCCAGGCATTGATTGAT
CTGGCCAGCTTTGGGGCCCA Exon (F8)
chr10:123955374 TGGtCCCacAgGCTGGCCAG CTGGaCAGCTcTGGGcCCCA Intron (TACC2)
chr6:73606839 TaCTCCAGGCATaGAagGAg
tTGGaCcaCTTTGGGGCCCA Intron (KCNQ5)
chr15:87990891 aGaGCCCCAtAtCTccCaAG ATCAgTCAtTGtCTGGAGCA Intergenic
chr13:104866433 TGCTtCAGaCAcTGATTGAg aTtGCCAcaTTTGGGGCCCA Intergenic
Intron
chr21:44889451 TGGtCCCCAAAcCTGGCCAa CTGGaCAGaTgccaGGgCCA (LINC00313)
chr15:72922848 gGaGgCCCAAAcgTGGCCtt CTaGCCAGCTcTGGGGCCCA Intergenic
chr8:20252698 TGCTCattGCAcTGgTgGAT CTGGCaAGCTTTGGGGtCtg Intergenic
chr18:32975516 TGtGgCCCAtAGCTGGCCAG CTGGCCAGCTaTGGGttttc Intergenic
chr16:989379 TGcGCCaCAAAGCTGGCCAc
AgCAATaAAaaCCaGGAaCA Intron (LMF1)
chr20:44515651 TGGGCCCCAggcCTGGgCAG
CTGctCAGCTTTctGGCtCA Exon (SPATA25)
chr2:240861687 TaGGCaCCtcAGCTGGCCAa CTGGgCAGCcTgGGaGCCCt Intergenic
chr9:132364724 TGaGCCaCtgAGCTGGCCAG cTtAtTCctTGtCTGGAGaA Intergenic
Intron
chr1:151341446 TGGtCtaCtgAGCTGGCaAG tTGtgCAGCTTTGGGGCCCg (SELENBP1)
Intron
chr12:1996302 TGGaCCCCcAAGaTGGCCAt CaGaaCAGCTTTGGaGCtag (CACNA2D4)
chr16:68354549 TGCTgCAGagATTtgTTtAT
tTGGCCAGaTTTGGGGgCCt Intron (PRMT7)
Intron
chr3:64099060 TGGGgCCCcAgcCTGGCCAc tTGGgtAcCTTgGGGGCCCA (PRICKLE2)
chr12:133199141 TGGtCCCCAcAGCcaGCCAG CTGcCCAGgcTgGGaGtgCA Intergenic
chr12:53741716 TaaGaaCCAAAGCTaatCAG tTcttCAGtTTTGtGGCCCA Intergenic
chr16:3006381 TGGGgCCCAAAtgaaGCCAG CctGCCAGCcTTGGGGtCCt Intergenic
chr5:53389184 aGcaCCCCAAAcCTGGCCtG
tTGGgCAGCaTTtGGcCCCA Intron (ARL15)
Table 50. Targeting Exon 22:
Genome Left Half-Site Right Half-Site Genomic Region
Coordinates
chrX:154124384 TCTGCCACTTCTTCCCATCAAG ATAAACTGAGAGATGTAGA Exon (F8)
chr17:55200444 TCaACATCTgTCAGacgAT ATAAAaTGAGAGtTGTAGc
Intergenic
Intron
chr7:149959793 TCTACATCTaaCAtTTTAT ATAAAtgGAaAacTGgAGA
(ACTR3C)
chr3:182164176 TgcACATCTCTCAcTTTAa AaAAgCTGAGAGAgGTtGA
Intergenic
chr8:85206496 TgTgCtTaTCTaAGTacAT gcAAAtTGAGAGATGTAGA
Intron (RALYL)
97/1
SUBSTITUTE SHEET (RULE 26)
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
Table 50. Targeting Exon 22:
Genome Left Half-Site Right Half-Site Genomic Region
Coordinates
chr1:107949372 TtTACATCTaTCAGTTTAT AaAAACTGAGctAcagAGg Intron (NTNG1)
Promoter
chr3:150421949 TCTtCgTCTCTCAGcTTAT CTTGggtGGAgGAAGTGGCttc (FAM194A)
chr8:22075977 gCTcCATCTCaaAaaTaAT ATAAAaTGAtAGATGcAGA Intergenic
Intron
chr5:56152387 TaTACATtTCTCAtTTTAT tTtAgtcGtGAGATGgAGA (MAP3K1)
chrX:147805582 TtgGCCACTTCTTCCCATCccG tTAAcCTGAaAcATGgAGA
Intron (AFF2)
97/2
SUBSTITUTE SHEET (RULE 26)
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
Table 50. Targeting Exon 22:
Genome Left Half-Site Right Half-Site Genomic Region
Coordinates
chr3:59243225 aCgAtATCaCTatGTTTAc ATAAtCTGAGAGtTGTAtA Intergenic
chr15:88546432 TCTAgATCTaaCtGacaAT ATAAACTGgGAGgcGTAGA Intron (NTRK3)
chr3:101738660 TCTAgATCTCTCAGgTTAa caActCTGtGAGATGaAGA Intergenic
Intron
chr15:64473144 TCTAgtTCTCTCAGTTTAT ATAgACTtAGtGcTGatGt (CSNK1G1)
chr15:96928325 agTACATCTtTtAaTTTAT CcTGATGGGAAGAAtTaGaAGA Intergenic
chr11:85386305 cCatCcTCaCTaAGTTTAa tTAAAgTGAGAGATGTAtA Intergenic
chr5:117743942 TCTcCATCTggCAaTTgAg cTAAACTGgaAGATGTAGA Intergenic
chr1:5052686 TaTACATtTCTCAGTTgAT CTTGtTctGAcGAtGctGCAGA Intergenic
chr6:9920117 caTACATCTCTCAcTTTAT tTAAACTtAGtGAgGaAGg Intergenic
chr1:159052090 TCTcCATgTCTCAGTTTgT ATAgACTaAGtGActTAtA Intergenic
chr20:25560526 TCTACAaaTgTaAaaTTcT AaAAACTGAGAGATtTtGA Intron (NINL)
[0231] In all exons 1-22, favorable sites were able to be located for TALENs,
Cas9-nuclease,
Cas9 paired-nickase, and dCas9 RNA-guided FokI Nucleases (RFNs). These sites
met
guidelines established for predicting high on-target activity (using the SAPTA
algorithm for
TALENs and avoiding stretches of pyrimidines in the PAM-proximal region of the
target).
These sites also met guidelines established for being relatively unique
throughout the genome
and having no high-scoring predicted off-target sites. Analysis of TALEN sites
using
PROGNOS yielded no sites generating warnings as scoring substantially similar
to the
designated target site. Analysis of Cas9-nuclease off-target sites found in
almost all cases that
no sites existed with fewer than two mismatches to the target sequence;
furthermore, sites
with few mismatches typically had mismatches in disruptive regions such as the
PAM, or the
12 bp PAM-proximal 'seed region'. Cas9-nickases and RFNs have been shown to
have very
low off-target activity approaching the detection limit of deep-sequencing
assays (Ran & Hsu
et al. Cell 2013, Tsai SQ et al. Nature Biotech 2014).
[0232] Taken together, this example identified the sequences to repair the F8
gene at the 3'
end of any exon 1-22 for TALENs, Cas9-nucleases, Cas9-nickases, or RFNs; by
using the
above-mentioned selected target sites. High on-target activity allows
efficacious clinical
repair of HA and low off-target activity ensures the safety of the proposed
therapy.
Example 4: Homologous Repair Vehicles for repair at different exon-intron
junctions
[0233] Repair at different exon-intron junctions throughout the FVIII gene
employ
methodology similar to example 3 described above, the repair vehicles used
however are
different for each junction. This example describes various repair vehicles.
98/1
SUBSTITUTE SHEET (RULE 26)
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
[0234] All repair vehicles contain the same basic components: a left homology
arm
corresponding to the genomic sequence 5' of the relevant nuclease cut site, a
cDNA sequence
98/2
SUBSTITUTE SHEET (RULE 26)
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
incorporated herein by reference. Final consideration was given to choosing
individual
sgRNAs which each had low potential for off-target activity throughout the
human genome,
as assessed by the online computational tool described by Hsu et al in Nature
Biotechnology
2013, incorporated herein by reference.
[0240] Sequences listed in Table 55 below contain identified binding sites for
paired CRISPR
nickases within exons 1-22 respectively.
Table 55
FVIII Genomic Target of SG/PG RNAs
Genome Editing
Gene
(Desired Activity)
(Region) (DNA Sequence)
paired nickase (5')
Exon 1 5'-CACTAAAGCAGAATCGCAAAaGG
paired nickase (3')
5'-AAGATACTACCTGGGTGCAGtGG
Exon 2 paired nickase (5')
5'-AGTCTTTTTGTACACGACTGaGG
paired nickase (3')
5'-TTTTCAACATCGCTAAGCCAaGG
Exon 3 paired nickase (5')
5'-CAGCATGAAGACTGACAGGAtGG
paired nickase (3')
5'-ATGCTGTTGGTGTATCCTACtGG
Exon 4 paired nickase (5')
5T-TATGAGTAGGTAAGGCACAGtGG
paired nickase (3')
5'-GACTTGAATTCAGGCCTCATtGG
Exon 5 paired nickase (5')
5'-AAGTAGTATAAATTTGTGCAaGG
paired nickase (3')
5'-CTTTTTGCTGTATTTGATGAaGG
Exon6 paired nickase (5')
5'-GACTCTGIGCATITTAGGCCaGG
paired nickase (3')
5'-CAGTCAATGGTTATGTAAACaGG
Exon 7 paired nickase (5')
5'-GCGACATITCCAAGGACGCCaGG
paired nickase (3')
5'-CAAACACTCTTGATGGACCTGG
Exon 8 paired nickase (5')
5'-ICTICGCAACTGAGCGAATTaGG
paired nickase (3')
5'-ACATTACATTGCTGCTGAAGaGG
Exon9 paired nickase (5')
5'-AATACCTTCACGAGTCTTAAaGG
paired nickase (3')
5T-GAASCIATTCAGCATGAATCaGG
Exon 10 paired nickase (5')
5'-GGACATCAGTGATTCCGTGAgGG
paired nickase (3')
s'-ATGICCGICCITIGTATTCAaGG
108/1
SUBSTITUTE SHEET (RULE 26)
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
Exon 11 paired nickase (5')
5'-AACSPAACTAGAGTAATAGCgGG
108/2
SUBSTITUTE SHEET (RULE 26)
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
Table 55
FVIII Genomic Target of SG/PG RNAs
Genome Editing
Gene
(Desired Activity)
(Region) (DNA Sequence)
paired nickase (3')
' -GAICTAGCTICAGGPTI7CATTTGG
Exon 12 paired nickase (5')
5 ' -AGCST IGTATATICICIGTGaGG
paired nickase (3')
5 ' -CGCTT ICICCCCAAT:CAGC-TGG
Exon 13 paired nickase (5')
5 -ATACA CCAT TT TGIGTITGAaGG
paired nickase (3')
5 ' -AGAAPCTGICT TCATGICGATTGG
Exon 14 paired nickase (5')
5 ' -IIIICITIIGAGCICCGGgGG
paired nickase (3')
5 ' -ACATTIAT T TAT TGCTGCAG-TGG
Exon 15 paired nickase (5')
5 ' -ACGSTATAAGGGCTGAGIAAaGG
paired nickase (3')
5 ' -AAATCAACATT TGGGACTCCT_GG
Exon 16 paired nickase (5')
5'-CAGTCAAACTCATCTTTAGTgGG
paired nickase (3')
5 ' -ATGACITTGACTGCAAAGCCcGG
Exon 17 paired nickase (5')
5'-ITCACTGAAGTACCAGCTIT-TGG
paired nickase (3')
5 ' - GGCTCCCIGCAATAT :CAGATTGG
Exon 18 paired nickase (5')
5 ' -GTCCPCTGAAATGAATAGAATTGG
paired nickase (3')
5'-GTTCPCTGTACGAAAAAAAGaGG
Exon 19 paired nickase (5')
5 ' -CGCCPAATICCACCITIGGA-GG
paired nickase (3')
5 ' -AT TSC CGACCATCTACATGCcGG
Exon 20 paired nickase (5')
5 ' -IGIC,CAGAACCCATTT,CCAGgGG
paired nickase (3')
5'-GATTITCAGATTACAGCTICaGG
Exon 21 paired nickase (5')
5 ' -TGATCCGGAATAATGAAGTC-TGG
paired nickase (3')
5'-AAT2PATGCCIGGAGCACCAaGG
Exon 22 paired nickase (5')
5'-A ,A5AL,AC'AC,,,C
paired nickase (3')
5 -AAGAAGTGCCAGACITATCGaGG
10241] The spacing requirements between the sgRNAs differ between paired
CRISPR
nickases and RFNs, but the other considerations regarding on-target and off-
target activity
remain the same and were taken into account when searching for RFN target
sites in exons 1-
22.
109
SUBSTITUTE SHEET (RULE 26)
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
10242] The ¨140 bp of the 3' end of each exon (hg19 human genome build) was
searched for
RFN binding sites matching the spacing distances using the ZiFiT targeter
disclosed in Tsai
SQ et al. Nature Biotech 2014, incorporated herein by reference. For some
exons, there was
no targetable sequence matching the PAM orientation and spacing requirements
of the RFN
system. Sequences in table 56 below contain identified binding sites for RFNs
within exons
1-22 respectively.
Table 56
FVIII Genome
Genomic Target of RFN
Gene Editing
(DNA Sequence)
(Region) Position
5' Half-Site 5'-GCACCCAGGTAGTATCTTCtGG
Exon 1
3' Half-Site 5'-ACTATATGCAAAGTGATCTcGG
5' Half-Site No Compatible Sites
Exon 2
3' Half-Site No Compatible Sites
5' Half-Site No Compatible Sites
Exon 3
3' Half-Site No Compatible Sites
5' Half-Site 5'-ACATGAGAAAGATATGAGTaGG
Exon 4
3' Half-Site 5'-ACTTGAATTCAGGCCTCATtGG
5' Half-Site 5'-AAGGTCTGTGTCTTTTCCTtGG
Exon 5
3' Half-Site 5'-TTTTTGCTGTATTTGATGAaGG
5' Half-Site 5'-TTTTCCCTGATGAGAGAGAaGG
Exon 6
3' Half-Site 5'-ACAAAGAACTCCTTGATGCaGG
5' Half-Site 5'-GTTATTGGCGAGATTTCCAaGG
Exon 7
3' Half-Site 5'-AAACACTCTTGATGGACCTtGG
5' Half-Site No Compatible Sites
Exon 8
3' Half-Site No Compatible Sites
5' Half-Site 5'-ATAGCTTCACGAGTCTTAAaGG
Exon 9
3' Half-Site 5'-TCTTGGGACCTTTACTTTAtGG
5' Half-Site No Compatible Sites
Exon 10
3' Half-Site No Compatible Sites
5' Half-Site 5'-ACGAAACTAGAGTAATAGCgGG
Exon 11
3' Half-Site 5'-ATCTAGCTTCAGGACTCATtGG
5' Half-Site No Compatible Sites
Exon 12
3' Half-Site No Compatible Sites
5' Half-Site No Compatible Sites
Exon 13
3' Half-Site No Compatible Sites
5' Half-Site 5'-TGTTTTCTTTTGAAAGCTGoGG
Exon 14
3' Half-Site 5'-GCTGCAGTGGAGAGGCTCTgGG
5' Half-Site No Compatible Sites
Exon 15
3' Half-Site No Compatible Sites
5' Half-Site 5'-AGTCAAACTCATCTTTAGTgGG
Exon 16
3' Half-Site 5'-TATTTCTCTGATGTTGACCtGG
5' Half-Site 5'-CTTTTGGTCTCATCAAAGAtGG
Exon 17
3' Half-Site 5'-AATATGGAAAGAAACTGCAgGG
5' Half-Site No Compatible Sites
Exon 18
3' Half-Site No Compatible Sites
5' Half-Site 5'-GCCAAATTCCAGCTTTGGAtGG
Exon 19
3' Half-Site 5'-TTGGCGAGCATCTACATGCtGG
5' Half-Site 5'-TGTCCAGAAGCCATTCCCAgGG
Exon 20
3' Half-Site 5'-TTACAGCTTCAGGACAATAtGG
5' Half-Site 5'-GATCCGGAATAATGAAGTCtGG
Exon 21
3' Half-Site 5'-CACCAAGGAGCCCTTTTCTtGG
110
SUBSTITUTE SHEET (RULE 26)
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
Table 56
FVIII Genome
Genomic Target of RFN
Gene Editing
(DNA Sequence)
(Region) Position
5' Half-Site 5'-AGGCTGGAGAACTTCTGACgGG
Exon 22
3' Half-Site 5'-TCATCATGTATAGTCTTGAtGG
Example 6: Additional Methods and Examples for FVIII Gene Repair in Cells
Purifying CRISPR/Cas9 plasmids and repair plasmids (DNA-RS)
[0243] A protocol for preparing CRISPR/Cas9 plasmids (DNA-SE) and repair
plasmids (DNA-RS) using endotoxin-free methods is described in the following
example. For
this protocol, a Qiagen EndoFree Plasmid Maxi Kit is used. The Qiagen EndoFree
Plasmid
Maxi Kit and its contents are stored at room temperature. Once RNAse and
LyseBlue are
added to Buffer P1 from the kit, this buffer is stored at 4 C. The kit also
requires 100%
ethanol and isopropanol (2-propanol).
[0244] According to this protocol, at Day 1, a lmL seed culture of
Escherichia coil
(E. coli) in Luria Broth (LB) and appropriate antibiotic is prepared and
placed on a shaker at
37 C. Whether an antibiotic is appropriate is dependent on the antibiotic
resistance gene that
is present in the plasmid that is being prepared and purified. For example,
such an antibiotic
may be ampicillin, kanamycin, or other antibiotics. Approximately 5 hours from
when the
seed culture is prepared, the seed culture is then used to inoculate a 100 mL
LB culture and
the suspension is left shaking overnight (or for at least about 8 hours) at 37
C.
[0245] At day 2, the 100 mL culture is transferred into 2x50 mL conical tubes
and spun for
min at 4000g; the supernatant is dumped out. The resulting cell pellet can be
stored at -
C for an indefinite period of time. During the spin, Buffer P3 is placed on
ice. Following
the spin and removal of the supernatant, 10 mL of Buffer P1 are added to the
first 50 mL tube
of each prep. This solution is then vortexed to resuspend the pelleted cells.
The resuspended
mixture is poured a second tube and vortexed to resuspend. Next, 10 mL of
Buffer P2 are
added and the suspension is inverted 6X to mix (until mixture is homogenously
blue). This
suspension is incubated for 3 min at room temperature. Next, 10 mL of Buffer
P3 is added to
each tube, and each tube is inverted ¨10X.
[0246] Next, the suspensions are centrifuged for 5 minutes at 4000g. During
the spin, a fresh
111
SUBSTITUTE SHEET (RULE 26)
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
50 mL tube is labeled for each abovementioned prep. A cap is screwed onto a
filter cartridge
and placed in the fresh 50 mL tube. After the spin, a p1000 pipette tip is
used to hold back
debris while pouring the liquid from the spun suspension into the cartridge.
The suspension is
then incubated for 10 minutes at room temperature in the cartridge. Next, the
cartridge is
uncapped and a plunger is used to push the liquid into the 50 mL tube; the
cartridge/plunger
is trashed following this step. Next, 2.5 mL of Buffer ER is added to each
tube, and each tube
is inverted 10X until the liquid becomes cloudy. The suspension is incubated
on ice for 30
minutes. During the incubation, Qiagen-Tip-500 tubes are labeled and placed in
a clamp
draining into a 1000 mL beaker. 10 mL of Buffer QBT is added to Qiagen-Tips to
equilibrate
the system. After the 30 minute incubation, the prep mixture is poured into
the respectively
labeled Qiagen-tips. Buffer QC is used to wash the tips.
[0247] Next, the Qiagen-Tip-Tubes are placed into 50 mL tubes capable of
withstanding
spins @ 15000g. 15 mL of Buffer QN is added to the Qiagen-Tip-Tubes and
centrifuged at
4 C to allow the DNA to elute from the Qiagen-Tip-Tubes as the buffer QN
drains through.
The eluted DNA can be stored at 4 C overnight.
[0248] Next, 10.5 mL of Isopropanol is added and the suspension is inverted
10X to mix. The
samples are then centrifuged at 15000g for 10 min at 4 C; The DNA will be
present as a
pellet. After the supernatant is dumped out, 5 mL of 70% Ethanol (Et0H) is
added to the
pelleted DNA. The samples are centrifuged at 15000g for 10 min at 4 C. Then,
the
supernatant is decanted using a p1000 pipette. The tube is then left to air-
dry for 10 min.
Next, 150 uL of Tris EDTA buffer (TE) is added. Isolated plasmid concentration
is then
determined.
[0249] In the example described, four CRISPR plasmids were prepared using
these methods,
each in triplicate, in addition to the preparation of a pGFP plasmid in
duplicate. These
procedures yielded the results shown in Table 57:
Table 57: Concentration of isolated CRISPR and pGFP plasmid preps
Sample # [DNA] Unit A260 A280 260/280 260/230
pH0007-1 273.7 ng/u1 5.475 2.881 1.9 2.28
112
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
Table 57: Concentration of isolated CRISPR and pGFP plasmid preps
Sample # [DNA] Unit A260 A280 260/280 260/230
pH0007-2 262.8 ng/n1 5.257 2.771 1.9 2.26
pH0007-3 350 ng/n1 7 3.688 1.9 2.27
pH0009-1 328.1 ng/n1 6.561 3.462 1.9 2.26
pH0009-2 345 ng/n1 6.901 3.637 1.9 2.27
pH0009-3 274.9 ng/n1 5.499 2.909 1.89 2.19
pH0011-1 320.4 ng/n1 6.408 3.378 1.9 2.26
pH0011-2 295.2 ng/n1 5.905 3.122 1.89 2.25
pH0011-3 328 ng/n1 6.559 3.469 1.89 2.27
pH0013-1 323.3 ng/n1 6.466 3.388 1.91 2.27
pH0013-2 311 ng/n1 6.22 3.274 1.9 2.22
pH0013-3 306.7 ng/n1 6.135 3.23 1.9 2.28
pGFP-1 273.8 ng/n1 5.477 2.877 1.9 2.28
pGFP-2 341.9 ng/n1 6.838 3.623 1.89 2.2
Nucleofection Conditions and Methods
[0250] A protocol for nucleofection is described in the following example. The
protocol
described uses 20uL Nucleovette Strips (Lonza). The number of cells
recommended for this
technique is 200,000 cells per condition or sample. The maximum mass of DNA
used in this
technique is -1000ng. It is recommended that a significantly greater amount of
repair plasmid
be used compared to the CRISPR/Cas9 plasmid as this minimizes the likelihood
of off-target
effects while maximizing the likelihood of homologous recombination. Typically
a ratio of
4:1 repair plasmid:CRISPR/Cas9 plasmid is used.
[0251] To facilitate all of the analyses involved with these methods, the
following reaction
conditions are recommended. First, for the "experimental" condition, 200ng of
CRISPR/Cas9
plasmid (DNA-SE), 800ng of repair plasmid (DNA-RS), and 4Ong of MaxGFP plasmid
are
used for transfection. Second, for the "no repair plasmid" control condition
(also suitable for
113
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
T7 Endonuclease (T7E1) analysis), 200ng of CRISPR/Cas9 plasmid (DNA-SE), 800ng
of
stiffer plasmid (pUC19), and 40ng of MaxGFP plasmid are used for transfection.
Third, for
the "no CRISPR plasmid" condition, 200ng of stuffer plasmid (pUC19), 800ng of
repair
plasmid (DNA-RS), and 40ng of MaxGFP plasmid are used for transfection.
Fourth, for the
"GFP alone" condition, 1000ng of stuffer plasmid (pUC19) and 40ng of MaxGFP
plasmid
are used for transfection.
[0252] For the method, first, 500u1 of media is added to the required number
of wells in a 24
well plate. This is pre-warmed in an incubator set to 37 C, 5% CO2. Next, lng
of total DNA
in minimum of 2 1 is used. Next, the DNA is setup into a new strip tubes.
[0253] Next, the cells are prepared for nucleofection. 200,000 cells per
nucleofection reaction
are preferred. 1.2X of master mix of cells is prepared to account for cell
loss during media
aspiration and pipetting errors. Next, the cells are pelleted by
centrifugation at 300 xg for 5
minutes. Next, if the Nucleocuvette strip kit is used, a nucleofection
solution provided with
kit is used. All of the supplement is added to Nucleofector solution; 20n1 of
the combined
buffer is required per nucleofection.
[0254] Next, during the spin a plate is labeled. The media is then aspirated
from the cells and
the cells are resuspended in 1.1X Nucleofector buffer (22u1 per nucleofection -
352 uL / 16
nucleofections, 374 uL / 17 reactions). Next, 20u1 of cell suspension
(approx.. 200,000 cells)
is aliquoted to DNA solutions. Next, the Nucleocuvette strip is placed in the
4D Nucleofector
X-module and the corresponding program is selected. Next, the cuvette is
allowed to incubate
for 10 minute following shocking of the cells. Next, 50u1 of media from 24
well plate is
added to the Nucleocuvette. All of the cell/media mix from the cuvette is then
added to the 24
well plate and incubated at 37 C for 72 hours.
Protocol for QuickExtract Method for gDNA Extraction
[0255] A protocol for gDNA extraction is described in the following example.
This method
allows for the extraction of genomic DNA (gDNA) from live cell samples using
QuickExtractTM DNA Extraction Solution (Epicentre). First, about 100,000 cells
are pelleted
by centrifugation. Then 80 lut of the QuickExtract solution is added to the
cells and the
suspension is transferred to a thermocycler tube. The suspension is then
vortexed. The
suspension is then run in a thermocycler for 15 min at 65 C and 8 min at 98 C;
The solution
can then be stored at -20 C and freeze/thawed for at least 40 times. Next, ¨1
lut of this
114
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
solution is used as the genomic DNA template per 50 [EL of PCR reaction.
Protocol for T7E1 Assay
[0256] A protocol for a T7E1 assay is described in the following example.
According to the
protocol, 35 cycles of PCR is used on isolated gDNA to amplify a target locus
at the
exon22/intron22 boundary using T7E1 primers that flank this boundary. The
forward primer
has a sequence of 5'-GGTAATGATGGACACACCTGTAGC-3' and the reverse primer has a
sequence of 5'-GGTTTTGCCCCCTAAACTTGTC-3' and PCR with these primers results in
amplicons of 623 nucleotides in length. The PCR amplicons are then purified
using Wizard
SV Gel and PCR Clean-up System (Promega) according to manufacturer's
instructions.
[0257] Next, 200ng of purified PCR product is placed in lx NEBuffer 2 (New
England
Biolabs, Buffer 2, a component of the T7 Endonuclease 1 kit that is available
from New
England Biolabs) in a total volume of 18uL. Next, the suspension is vortexed
and
centrifuged. Next, the samples are placed in a thermocycler programmed with
the following
protocol: A) 95 C for 5 mm; B) 95-25 C in ¨1 C/s steps; C) hold at 4 C.
[0258] 10 units of T7 Endonuclease 1 is are added to the hybridized PCR
products in a 2uL
volume of lx NEBuffer 2 (for a final reaction volume of 20uL). Note that for
each sample, a
side-by-side negative control (no T7E1 enzyme control) is prepared, wherein
2uL volume of
lx NEBuffer is used in the absence of the enzyme. Next, the suspensions are
vortexed and
centrifuged. The suspensions are then incubated at 37 C for 30 minutes.
Following
incubation, the samples are placed on ice and stop solution is added to them.
The stop
solution is prepared by adding 2.45uL 0.5M EDTA to 4.49 uL 6X loading dye for
each
reaction (6.94uL volume per reaction, resulting in a final concentration of
45mM EDTA and
lx loading dye).
[0259] Next, the samples by agarose gel electrophoresis. The gel image can be
quantified
with ImageJ using the following procedure: 1) the image is inverted; 2) the
background is
subtracted (set to 30 pixels, check light background box); 3) rectangles are
drawn about the
middle of a gel lane, avoid the "smiling" on the end of the gel lanes; 4) in
the analyze gel
lane, "select first lane" option is selected; 5) subsequent lanes are
selected; 6) Quantitative
analysis is performed (fraction cleaved= area cleaved/ area of all); 7)
Calculate % gene
modification with the following equation:
115
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
% gene modification = 100 x (1 ¨ (1 ¨ fraction cleaved)1/2)
Protocol for Restriction Fragment Length Polymorphism (RFLP) Assay
[0260] A protocol for a RFLP assay is described in the following example.
According to the
protocol, 35 cycles of PCR is used on gDNA to amplify a target locus at the
exon22/intron22
boundary using RFLP primers that flank this boundary. The forward primer has a
sequence of
5'- GTTAGGTGACTCAAATGGGTTCAC-3' and the reverse primer has a sequence of 5'-
GAACAAGAAGCAGGGTAGAGAAGC-3' and PCR with these primers results in
amplicons of 1667 nucleotides in length. The PCR amplicons are purified using
Wizard SV
Gel and PCR Clean-up System (Promega) according to manufacturer's
instructions.
[0261] Next, a mixture with 20 [it reaction with 0.5[EL (5U) of restriction
enzyme, 2uL
reaction buffer (provided in the enzyme kit), and then 17.5 [it of the cleaned
PCR reaction is
prepared. This mixture is then incubated at 37 C for 1 hour. Next, the samples
are analyzed
the samples by agarose gel electrophoresis. The gel image is then quantified
with ImageJ
using the following procedure: 1) the image is inverted; 2) the background is
subtracted (set
to 30 pixels, check light background box); 3) rectangles are drawn about the
middle of a gel
lane, avoid the "smiling" on the end of the gel lanes; 4) in the analyze gel
lane, "select first
lane" option is selected; 5) subsequent lanes are selected; 6) Quantitative
analysis is
performed (fraction cleaved= area cleaved/ area of all); 7) Calculation of %
homologous
recombination with the following equation:
%HR = (cut band) / (cut band + uncut band)
Protocol for PCR Amplification at Gene Repair Site
[0262] A protocol for PCR amplification at a gene repair site is described in
the following
example. According to the protocol, as a first qualitative approach, PCR with
RFLP primers
is performed to examine the presence of a band distinct from the main band.
The primers and
procedures in this method are the same as those described above in the section
entitled
"Protocol for Restriction Fragment Length Polymorphism (RFLP) Assay." The main
(uncut)
band is expected to be about 1.7kb in size, wherease the cut band is expected
to be about
1.0kb in size.
[0263] In a second qualitative approach according to this protocol, a reverse
RFLP primer
(with sequence 5'- GAACAAGAAGCAGGGTAGAGAAGC-3') that anneals within exon 22
is paired with a primer that anneals within the gene repair site (with
sequence 5'-
116
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
AAGATGGCCATCAGTGGACTCTC-3') is used. This PCR will only form a product of
about 1.3kb in size if there is successful gene correction.
[0264] Following analysis of the results from the PCR analyses described
above, clonal
colonies are grown out. This is done either through limiting dilution of the
cells or by FACS
sorting of single cells into a 96-well plate. With either method, initially
plate 1 cell into ¨50
uL of media. Then after 1 week add ¨150 uL of new media to the wells. After
about a second
week, or when there are >10,000 cells, use the QuickExtract protocol to
isolate gDNA.
Proceed to perform the same two PCRs described above---the 2nd PCR method will
demonstrate if there is at least monoallelic gene correction, the first PCR
(with the RFLP
primers) will demonstrate if there is biallelic correction (because all of the
PCR product will
be at a different band size) and also serve as a positive control to determine
that the
QuickExtract for that sample is a viable PCR template.
Protocol for gene repair in FVIII
[0265] A protocol for gene repair in FVIII is described in the following
example. According
to the protocol, seed cell cultures were prepared 2 days before transfection,
with a final target
density of 800,000 cells/mL on the day of transfection. Next, CRISPR/Cas9
plasmids (DNA-
SE) and repair plasmids (DNA-RS) were prepared as indicated above in the
protocol for
endotoxin-free plasmid maxiprep. Next, the transfection setup details for
nucleofection, such
as plasmid concentrations and volumes, cell concentrations and volumes were
determined as
discussed above in the protocol for nucleofection conditions and methods.
Next,
nucleofection was performed, followed by culturing the cells for 72 hours as
discussed above
in the protocol for nucleofection conditions and methods.
[0266] Flow cytometry analysis was used to determine % viability and % GFP+
cells in each
sample on one quarter of the cells collected from the nucleofection step.
Results using the
CRISPR/Cas9 plasmids pH0007 and pH0009 as well as a repair plasmid (labeled
"donor")
are shown in Figure 17. In Figure 17 the left-most graph for each sample
displays the
FSC/SSC characteristics of the population and allows for gating on non-debris
in the sample;
the center graph for each sample displays in histogram format the distribution
of live cells in
the sample as evidenced by inclusion of propidium iodide which enters only
dead cells and
yields a red fluorescence; and the right-most graph for each sample displays
in histogram
format the distribution of cells that have been successfully transfected as
evidenced by green
117
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
fluorescence that is due to the presence of GFP. As can be seen from the
results, the
percentages for each parameter are similar across all samples, with a range
for each parameter
of 46.8-51.8% (non-debris), 74.9-85.0% (Live), and 22.6-26.8% (GFP+). Thus the
rates of
successful transfection do not differ substantially as a function of the
plasmid used.
[0267] In this example, gDNA from one quarter of the cells from the
nucleofection event was
isolated following the protocol for gDNA extraction described above. The gDNA
was then
analyzed using the following protocols described above: 1) protocol for T7 El
assay; 2)
protocol for RFLP assay; and 3) protocol for PCR amplification at gene repair
site.
[0268] Results from the analysis following the T7E1 assay are shown in Figure
18 and in
Figure 19. Figure 18 and Figure 19 show results from using_CRISPR/Cas9
plasmids pH0007,
pH0009, pH0011, and pH0013. Figure 18 shows an image from an agarose gel
electrophoresis assay. In Figure 18 the samples names are abbreviated such
that the three
pH0007 are listed as 7-1, 7-2, and 7-3, and this pattern is continued for
pH0009, pH0011, and
pH0013. A negative control (No DNA) and positive control (+ ctrl) in the
analysis. For each
sample there are two lanes: one labeled at the top of the lane with a "+"
which sample
contained the T7E1 enzyme, and a second labeled with a "¨" which sample
contained no
T7E1 enzyme. In the absence of T7E1, no nuclease activity is present and there
is a single
band present in the lane. In the presence of T7E1, some cleavage occurs
resulting in a second
smaller band that appears. This qualitative data demonstrates that pH0007 and
pH0009 yield
the better result than pH0011 and pH0013 as there is a greater relative
abundance of the
smaller band in those samples. This is quantified in Figure 19. Figure 19
shows the calculated
values for percent gene modification by NHEJ (non-homologous end joining),
demonstrating
that pH0007 and pH0009 cause indel formation at the target site at a rate of
66% and 72%
respectively, and that both of these yield statistically significantly
superior rates of indel
formation compared to pH0011 and pH0013. This statistical significance is
evidenced by the
error bars which display the standard error of the mean for each sample.
[0269] Results from the analysis following the RFLP assay are shown in Figure
20 and
Figure 21. Figure 20 and Figure 21 show results from using CRISPR/Cas9
plasmids pH0007,
pH0009, as well as a repair plasmid (labeled "Donor"). Figure 20 shows an
image from an
agarose gel electrophoresis assay. In Figure 20 displays the results of a
simple and standard
RFLP assay demonstrating that only in those samples that contain the donor
plasmid along
118
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
with either pH0007 or pH0009 is there a smaller band which indicates
restriction digestion,
the presence of the restriction site and thus successful recombination in
those samples. In the
other control samples, no such smaller band is seen. Figure 21 shows the
calculated values
for percent gene modification by following Intron 22-targeted CRISPR
treatment. As can be
seen from the data, homologous recombination occurs only in those samples that
were
transfected with the donor plasmid and pH0007 or pH0009 at a rate of 22% and
16%
respectively. The control samples that were transfected with only donor
plasmid, only
pH0007, only pH0009, or none of the three show a rate of homologous
recombination of 0%
for each sample.
[0270] Next, cells were cloned out either by limiting serial dilution or
single-cell FACS.
Clones were cultured until the clonal colonies reach cell numbers of >20,000.
gDNA from
>10,000 cells of each clonal culture using was then extracted. PCR was used to
amplify
across the repair site, using as template each of the extracted gDNA samples
from the clonal
cultures. Next, sanger sequencing methods were used to sequence the repair-
site PCR
amplicons. Next, the DNA sequence immediately upstream (about 25 bases),
immediately
downstream (about 25 bases), and across the repair was analyzed.
[0271] Clones not displaying the desired or expected integration events were
eliminated.
Next, it was determined if any DNA sequence modifications have been made at
sites in the
genome that have been predicted by algorithm to be the top 20 potential off-
target sites in the
genome. Clonal cultures for which DNA sequence modifications have been made at
off-
target sites in the genome we eliminated.
[0272] Remaining clones were cultured out until clonal colonies reach cell
numbers of
>1x106. mRNA was extracted from >100,000 cells of each clonal culture; mRNA
was also
extracted from >100,000 cells of the parent culture (in which no gene repair
has been
performed).
[0273] Quantitative reverse-transcription PCR (qRT-PCR) primers were designed
for the
detection of: a) Transcription of the F8 gene, targeting an exonic site 5' of
the gene repair
site; b) Transcription of the F8 gene, targeting an exonic site 3' of the gene
repair site; c)
Transcription of the F8 gene, targeting a sequence that is unique to the gene
repair site itself,
that furthermore overlaps the junction of (i) the gene repair site and (ii) an
endogenous, non-
repaired exonic site 5' of the gene repair site. This amplified product should
only be detected
119
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
in cells that have been correctly repaired; and d) Transcription of house-
keeping genes that
can be used for normalization of F8 gene transcription, including at least the
genes for beta-
actin (ACTB), gamma-tubulin (TUBG1), and RNA polymerase II (POLR2A).
[0274] Using qRT-PCR methods, transcription of the F8 gene using the mRNA
extracted
from each clonal culture and the parent culture was analyzed; yielded a
quantitative value for
each sample analyzed (ACt value).
[0275] The transcription of the F8 gene across all samples was compared.
Clonal cultures
that exhibit the highest AC t values for transcription of F8 when measured
using qRT-PCR
primers targeting the gene repair site itself were further isolated. These
cells were cultured
until the clonal colonies reach cell numbers of >5x107
[0276] Next, >5x107 cells from each culture were removed and pelleted. Cell
lysate from the
cell pellets was collected. A modified enzyme-linked immunosorbent assay
(mELISA) was
then used to detect the presence of FVIII protein in both the culture medium
and the whole
cell lysates from each culture. This yielded a quantitative value for each
sample analyzed in
units of nanograms of FVIII protein per cell number (ng/5x107 cells). FVIII
protein secretion
across all samples was compared. The culture yielding the highest secretion of
FVIII protein
was chosen to proceed for therapeutic purposes.
[0277] The examples set forth above are provided to give those of ordinary
skill in the art a
complete disclosure and description of how to make and use the embodiments of
the
materials, compositions, systems and methods of the disclosure, and are not
intended to limit
the scope of what the inventors regard as their disclosure.
[0278] All patents and publications mentioned in the specification are
indicative of the levels
of skill of those skilled in the art to which the disclosure pertains.
[0279] The entire disclosure of each document cited (including patents, patent
applications,
journal articles, abstracts, laboratory manuals, books, or other disclosures)
in the Background,
Summary, Detailed Description, and Examples is hereby incorporated herein by
reference.
All references cited in this disclosure are incorporated by reference to the
same extent as if
each reference had been incorporated by reference in its entirety
individually. However, if
any inconsistency arises between a cited reference and the present disclosure,
the present
disclosure takes precedence.
120
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
[0280] The terms and expressions which have been employed herein are used as
terms of
description and not of limitation, and there is no intention in the use of
such terms and
expressions of excluding any equivalents of the features shown and described
or portions
thereof, but it is recognized that various modifications are possible within
the scope of the
disclosure claimed. Thus, it should be understood that although the disclosure
has been
specifically disclosed by embodiments, exemplary embodiments and optional
features,
modification and variation of the concepts herein disclosed can be resorted to
by those skilled
in the art, and that such modifications and variations are considered to be
within the scope of
this disclosure as defined by the appended claims.
[0281] It is also to be understood that the terminology used herein is for the
purpose of
describing particular embodiments only, and is not intended to be limiting. As
used in this
specification and the appended claims, the singular forms "a," "an," and "the"
include plural
referents unless the content clearly dictates otherwise. The term "plurality"
includes two or
more referents unless the content clearly dictates otherwise. Unless defined
otherwise, all
technical and scientific terms used herein have the same meaning as commonly
understood
by one of ordinary skill in the art to which the disclosure pertains.
[0282] When a Markush group or other grouping is used herein, all individual
members of
the group and all combinations and possible subcombinations of the group are
intended to be
individually included in the disclosure. Every combination of components or
materials
described or exemplified herein can be used to practice the disclosure, unless
otherwise
stated. One of ordinary skill in the art will appreciate that methods, device
elements, and
materials other than those specifically exemplified may be employed in the
practice of the
disclosure without resort to undue experimentation. All art-known functional
equivalents, of
any such methods, device elements, and materials are intended to be included
in this
disclosure. Whenever a range is given in the specification, for example, a
temperature range,
a frequency range, a time range, or a composition range, all intermediate
ranges and all
subranges, as well as, all individual values included in the ranges given are
intended to be
included in the disclosure. Any one or more individual members of a range or
group
disclosed herein may be excluded from a claim of this disclosure. The
disclosure illustratively
described herein suitably can be practiced in the absence of any element or
elements,
limitation or limitations which is not specifically disclosed herein.
121
CA 02951882 2016-12-09
WO 2015/191899
PCT/US2015/035399
[0283] A number of embodiments of the disclosure have been described. The
specific
embodiments provided herein are examples of useful embodiments of the
invention and it
will be apparent to one skilled in the art that the disclosure can be carried
out using a large
number of variations of the devices, device components, methods steps set
forth in the
present description. As will be obvious to one of skill in the art, methods
and devices useful
for the present methods can include a large number of optional composition and
processing
elements and steps.
[0284] In particular, it will be understood that various modifications can be
made without
departing from the spirit and scope of the present disclosure. Accordingly,
other
embodiments are within the scope of the following claims.
122