Note: Descriptions are shown in the official language in which they were submitted.
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
LIQUID INHALATION FORMULATION COMPRISING RPL554
FIELD OF THE INVENTION
The present invention relates to a liquid pharmaceutical composition
comprising a respiratory
drug.
BACKGROUND OF THE INVENTION
The toxicokinetic properties of liquid pharmaceutical compositions are
unpredictable. It is
important that an active pharmaceutical ingredient (API) is correctly
formulated so that a
safe, effective and controlled dosage is provided when the composition is
delivered to a
patient. This is particularly the case for inhaled compositions. Furthermore,
liquid
pharmaceutical compositions must have reliable long term stability to ensure
that the dosage
profile of the composition will be maintained after storage. This avoids the
administration of
incorrect dosages. Pharmaceutical compositions must also be formulated in such
a way that
administration is not unpleasant for a patient, for instance as regards taste
and acidity.
RPL554 (9, 10-Dimethoxy-2-(2,4,6-trimethylphenylimino)-3-(N-carbamoy1-2-
aminoethyl)-
3,4,6,7-tetrahydro-2H-pyrimido[6,1-a]isoquinolin-4-one) is a dual PDE3/PDE4
inhibitor and
is described in WO 00/58308 (and is also sometimes referred to as RPL554). As
a combined
PDE3/PDE4 inhibitor, RPL554 has both anti-inflammatory and bronchodilatory
activity and
is useful in the treatment of respiratory disorders such as asthma and chronic
obstructive
pulmonary disease (COPD). The structure of RPL554 is shown below.
0 0
I NO
Y:
-NNH2
it
N
,
It is often preferable to administer RPL554 by inhalation because of its
efficacy in the
treatment of respiratory disorders. An effective method of administration is
nebulisation.
Franciosi et al. disclose a solution of RPL554 in a citrate-phosphate buffer
(Efficacy and
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
2
safety of RPL554, a dual PDE3 and PDE4 inhibitor, in healthy volunteers and in
patients
with asthma or chronic obstructive pulmonary disease. findings from four
clinical trials, The
Lancet: Respiratory Medicine 11/2013, 1(9):714-27. DOI 10.1016/S2213-
2600(13)70187-5).
SUMMARY OF THE INVENTION
It is a surprising finding of the present invention that the optimal
composition for inhalation
comprising RPL554 is a liquid pharmaceutical composition comprising a
suspension of
particles of RPL554. Suspensions of RPL554 have been found to have highly
desirable
properties for a clinical setting. A number of benefits have been found to be
associated with
a liquid pharmaceutical composition which comprises a diluent and a suspension
of particles
of RPL554 or a pharmaceutically acceptable salt thereof.
The liquid pharmaceutical composition of the invention has greatly improved
properties over
previous formulations, for instance when compared with a solution formulation
of RPL554
Regarding toxicokinetic properties, the suspension formulation has been found
to have
delayed release characteristics compared to those of a solution. This can
reduce the number
of treatments that must be administered to a patient. The suspension has also
been found to
allow a much greater dosage of RPL554 to be administered, as can be seen from
plasma Cmax
and AUC values. This can also reduce the frequency of administrations
required.
The liquid pharmaceutical composition of the invention also shows excellent
stability, with
compositions showing no degradation after 12 months at ambient conditions (25
C), nor after
6 months at accelerated conditions (40 C). Degradation is observed in a
comparable solution
composition under the same conditions. The compositions may show no
degradation after 12
months at ambient conditions (25 C/60%RH), nor after 6 months at accelerated
conditions
(40 C/75 70RH).
Accordingly, the present invention provides a liquid pharmaceutical
composition comprising
a diluent and a suspension of particles of 9,10-dimethoxy-2-(2,4,6-
trimethylphenylimino)-3-
(N-carbamoy1-2-aminoethyl)-3,4,6,7-tetrahydro-2H-pyrimido[6,1-a]isoquinolin-4-
one
(RPL554) or a pharmaceutically acceptable salt thereof.
The invention also provides a liquid pharmaceutical composition according to
the invention
for use in the treatment of the human or animal body. A liquid pharmaceutical
composition
according to the invention may be used in the treatment or prevention of a
disease or
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
3
condition selected from asthma, allergic asthma, hay fever, allergic rhinitis,
bronchitis,
emphysema, bronchiectasis, chronic obstructive pulmonary disease (COPD), adult
respiratory
distress syndrome (ARDS), steroid resistant asthma, severe asthma, paediatric
asthma, cystic
fibrosis, lung fibrosis, pulmonary fibrosis, interstitial lung disease, skin
disorders, atopic
dermatitis, psoriasis, ocular inflammation, cerebral ischaemia, inflammatory
diseases and
auto-immune diseases.
Typically, said disease or condition is asthma or COPD, more typically COPD.
The invention also provides a method of treating or preventing a disease or
condition as
defined herein in a subject, which method comprises administering to said
subject an
effective amount of a liquid pharmaceutical composition according according to
the
invention.
BRIEF DESCRIPTION OF THE FIGURES
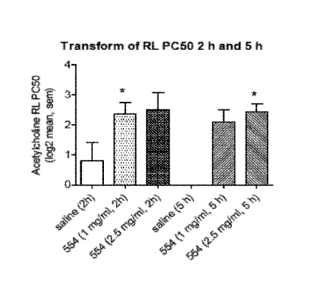
Figure 1 - The provocation concentration (PC) of acetylcholine which
cumulatively produced
a 50% increase in total airway resistance (Acetylcholine RL PC50) in animals
treated with
saline or RPL554 solution formulation: 2 and 5 h time point: Acetylcholine RL
PC50 was
log transformed for the purposes of statistical analysis. Post hoc t-test of
the untransformed
data showed differences between groups vs saline. *Non parametric analysis did
show a
significant difference between 2 h (1 mg/mL RPL554 solution, P = 0.0190
unadjusted) and 5
h (2.5 mg/mL solution, P = 0.0095 unadjusted) vs saline group. N values = 6,
4, 4; 4, 4 left to
right (Note 5 h control not plotted).
Figure 2 - Acetylcholine RL PC50 in animals treated with vehicle or the RPL554
suspension
formulation: 2 and 5 h time point: Acetylcholine RL PC50 was log transformed
for the
purposes of statistical analysis (upper panel). Non parametric analysis did
show a significant
difference between 2 h (10 mg/mL RPL554 suspension, P = 0.0268 unadjusted) vs
vehicle
control group. Re-analysis of the data by combining the vehicle data (2 + 5
h), revealed an
overall significant bronchoprotection (P=0.0016) which was reflected by a
significant degree
of bronchoprotection with 2.5 mg/mL RPL554 suspension (2.52 fold (1-5,72), P <
0.05 and
mg/mL RPL554 suspension (4.67 fold (2-11), *P < 0.05) vs vehicle. At the 5 h
time point,
RPL554 10 mg/mL suspension caused a significant degree of bronchoprotection
(2.77 fold
(1.3-6.0), *P < 0.05). (N=8,4,4,3, 6, left to right) (lower panel).
WO 2016/042313
PCT/GB2015/052668
4
Figure 3 shows mean concentrations of RPL554 in plasma from healthy human
volunteers
following administration of either (i) the suspension formulation or (ii) the
solution
formulation by nebuliser at a nominal dose-level of about 1.5 mg.
DETAILED DESCRIPTION OF THE INVENTION
The particles of RPL554 may be of any suitable size for a use in a liquid
pharmaceutical
composition suitable for inhalation. Typically, the particles of RPL554 are
micronized
particles. For instance, the particles of RPL554 may have a Dv50 (median
particle size by
volume) value of less than or equal to 10 um or from about 0.1 um to about 8
um.
Typically, the particles have a particle size distribution with a Dv50 value
of from about 0.2
[an to about 5 um. More typically, the particles of RPL554 have a particle
size distribution
with a Dv50 value of from about 0.7 um to about 3.0 um. For instance, the
particles of
RPL554 may have a particle size distribution with a Dv50 value of from 0.9 um
to 1.7 um, or
alternatively from 1.7 um to 2.7 um Often, the particles of RPL554 have a
particle size
distribution with a Dv50 value of from about 1.1 um to about 2.6 um.
The Dv50 value is the median particle size for a volume distribution. Thus,
half the volume
of the particles is comprised in particles having diameters of less than the
Dv50 value and
half the volume of the particles is comprised in particles having diameters of
greater than the
Dv50 value. This is a well known manner in which to describe particle size
distributions.
The particles typically have a particle size distribution with a Dv10 value of
from about 0.4
um to about 1.0 um. The particles typically have a particle size distribution
with a Dv90
value of from about 2.0 um to about 4.0 um. The Dv10 value reflects the
particle diameter
where 10% of the volume of the sample is in particles having a particle
diameter less than the
Dvl 0 value The Dv90 value reflects the particle diameter where 90% of the
volume of the
sample is in particles having a particle diameter less than the Dv90 value.
The technique used to measure the Dv50 value is typically laser diffraction.
For instance, the
RPL554 particles typically have a particle size distribution with a Dv50 value
of from about
0.2 um to about 5 um as measured by laser diffraction. The particle size
distribution analysis
can be performed by laser diffraction using the Malvern SprayteTcmin
conjunction with a wet
dispersion cell. Typically, the instrument parameters for the Malvern Spraytec
are as
follows:
Date Recue/Date Received 2022-01-20
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
= particle ¨ standard opaque particle;
= refractive index Particle ¨ 1.50;
= refractive index (imaginary) ¨ 0.50;
= density of particle ¨ 1.00;
= refractive index of dispersant ¨ 1.33;
= controller unit ¨ 1000RPM;
= measurement type ¨ timed;
= initial sampling time ¨ 30s,
= obscuration ¨ 20% - 30%;
= dispersant ¨ % Polysorbate 20 in deionised water.
The particles of RPL554 may be produced by any pharmaceutically acceptable
size reduction
process or particle size controlled production process. For instance, the
particles may be
produced by spray-drying a solution of RPL554, or by controlled
crystallisation, or by size
reduction of a solid form of RPL554, for example by air jet milling,
mechanical
micronisation or media milling.
The concentration of particles of RPL554 in the liquid pharmaceutical
composition is
typically from about 0.01 mg/mL to about 40 mg/mL. More typically, the
concentration of
particles of RPL554 in the liquid pharmaceutical composition is from about 0.1
mg/mL to
about 20 mg/mL. For instance, the concentration of particles of RPL554 in the
liquid
pharmaceutical composition may be from about 0.01 mg/mL to about 5 mg/mL, or
from 0.1
mg/mL to 5 mg/mL. The concentration of particles of RPL554 in the liquid
pharmaceutical
composition may for instance be from 0.1 mg/mL to 6 mu/mt.
Preferably, the pH of the suspension is from about 6 to about 8, more
preferably from about
6.5 to about 7.
The liquid pharmaceutical composition may further comprise one or more
surfactants. The
surfactants are pharmaceutically acceptable surfactants. The surfactants may
be non-ionic
surfactants, anionic surfactants, cationic surfactants or zwitterionic
surfactants. Preferably,
the one or more surfactants are selected from one or more non-ionic
surfactants
The one or more surfactants are typically selected from polyoxyethylene glycol
alkyl ethers,
polyoxypropylene glycol alkyl ethers, glucoside alkyl ethers, polyoxyethylene
glycol
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
6
octylphenol ethers, polyoxyethylene glycol alkylphenol ethers, glycerol alkyl
esters,
polyoxyethylene glycol sorbitan alkyl esters (polysorbates), sorbitan alkyl
esters, cocamide
MEA, cocamide DEA, dodecyldimethylamine oxide, block copolymers of
polyethylene
glycol and polypropylene glycol (poloxamers) and polyethoxylated tallow amine
(POEA).
Preferably, the one or more surfactants are selected from polyoxyethylene
glycol sorbitan
alkyl esters, for instance polysorbate 20 (polyoxyethylene (20) sorbitan
monolaurate),
polysorbate 40 (polyoxyethylene (20) sorbitan monopalmitate), polysorbate 60
(polyoxyethylene (20) sorbitan monostearate) and polysorbate 80
(polyoxyethylene (20)
sorbitan monooleate), and sorbitan alkyl esters, for instance sorbitan
monolaurate, sorbitan
monopalmitate, sorbitan monostearate, sorbitan tristearate and sorbitan
monooleate.
The total concentration of the one or more surfactants is typically from about
0.01 mg/mL to
about 2 mg/mL. The composition often comprises two or more surfactants, for
instance a
polysorbate and a Span surfactant.
Typically, the liquid pharmaceutical composition further comprises one or more
buffers. The
buffers are pharmaceutically acceptable buffers. The buffers may be any
buffers suitable for
use in a liquid phaunaceutical composition suitable for inhalation. The one or
more buffers
are typically selected from citrate or phosphate buffers. Citrate buffers
include citric acid,
sodium citrate and mixtures thereof Phosphate buffers include phosphoric acid,
monosodium phosphate, dibasic sodium phosphate and mixtures thereof
The total concentration of the one or more buffers is typically from about 5
mg/mL to about
40 mg/mL. The composition often comprises two or more buffer components, for
instance
two phosphate salts, for instance sodium phosphate salts.
A liquid pharmaceutical composition according to the invention may comprise
(a) the
particles of RLP554 at a concentration of from about 0.01 mg/mL to about 40
mg/mL, (b)
one or more surfactants at a concentration of from about 0.01 mg/mL to about 5
mg/mL; and
(c) a buffer at a concentration of from about 5 mg/mL to about 25 mg/mL
The liquid pharmaceutical composition typically further comprises a tonicity
adjuster. The
tonicity adjuster may be any pharmaceutically acceptable tonicity adjuster.
Examples of
tonicity adjusters include simple non toxic salts such as alkali metal
halides, for instance
sodium chloride and potassium iodide. Typically, the tonicity adjuster is
sodium chloride.
WO 2016/042313
PCT/GB2015/052668
7
The concentration of the tonicity adjuster will depend on the amount required
to reach the
desired tonicity, for instance isotonicity with the body or lungs. The
concentration of the
tonicity adjuster is typically from about 2 mg/mL to about 8 mg/mL, and more
typically
from about 3.5 mg/mL to 6 mg/mL.
The liquid pharmaceutical composition may comprise other components.
Alternatively, other
components may be excluded. In some cases, the liquid pharmaceutical
composition does
not comprise al32-adrenergic receptor agonist or a muscarinic receptor
antagonist (or does not
comprise greater than 0.1 wt% total of a f32-adrenergic receptor agonist and a
muscarinic
receptor antagonist).
The diluent may be any pharmaceutically acceptable diluent. The diluent is
suitable for
administration by inhalation. Examples of suitable diluents include water,
ethanol and
glycerol. The diluent is preferably water. The diluent is preferably sterile.
A liquid pharmaceutical composition according to the invention typically
comprises:
(a) the particles of RLP554 at a concentration of 0.01 mg/mL to 40 mgimL;
(b) a polyoxyethylene glycol sorbitan alkyl ester at a concentration of
from
0.1 mg/mL to 2 mg/mL;
(c) a sorbitan alkyl ester at a concentration of from 0.01 mg/mL to 0.1
mg/mL;
(d) a first phosphate buffer component at a concentration of from 5 mg/mL
to 10
mg/mL;
(e) a second phosphate buffer component at a concentration of from 5 mg/mL
to
mg/mL; and
a tonicity adjuster at a concentration of from 2 mg/mL to 8 mg/mL.
For instance, the liquid pharmaceutical composition may comprise:
(a) the particles of RLP554 at a concentration of 0.05 mg/mL to 25 mgimL;
(b) polyoxyethylene (20) sorbitan monolaurate (Polysorbate 20, Tween 205mat
a
concentration of from 0.1 mg/mL to 2 mg/mL;
(c) sorbitan monolaurate (Span 20)t a concentration of from 0.01 mg/mL to
0.1
mg/mL;
(d) monosodium phosphate monohydrate at a concentration of from 5 mg/mL to
10 mg/mL;
Date Recue/Date Received 2022-01-20
WO 2016/042313
PCT/GB2015/052668
8
(e) dibasic
sodium phosphate anhydrous at a concentration of from 5 mg/mL to 10
mg/mL; and
sodium chloride at a concentration of from 2 mg/mL to 8 mg/mL.
Typically, the liquid pharmaceutical composition is suitable for
administration by nebulizer.
The invention also provides a nebulizer comprising a liquid pharmaceutical
composition
according to the invention. The nebulizer is typically loaded with the liquid
pharmaceutical
composition. The nebulizer typically comprises from about 1 mL to about 200
mL, more
typically from 1 mL to 20 mL of the liquid pharmaceutical composition.
Nebulizers use compressed air to aerosolise a liquid pharmaceutical
composition into an
aerosol that is inhaled into a subject's respiratory tract. Examples of
nebulizers include a soft
mist nebulizer, a vibrating mesh nebulizer, a jet nebulizer and an ultrasonic
wave nebulizer.
Suitable nebulizer devices include the Philips InebTM (Philips), the Pad LC
Sprinr(Pari
GmbH), the RTM Pulmonary Delivery System (Aradigm Corp.) and the Pan i LC
PlusTm
Reusable Nebulizer (Pan i GmbH).
The invention also provides a liquid pharmaceutical composition for use in the
treatment of
the human or animal body. The composition is as defined herein. The liquid
pharmaceutical
composition is typically for use in the treatment of the human or animal body,
wherein
treatment comprises administration by inhalation.
The compositions of the invention allow for delayed release of RPL554 into the
bloodstream.
The plasma concentration of RPL554 after a certain time is therefore increased
relative to the
use of known compositions of RPL554. Typically, the blood plasma concentration
of
RPL554 is greater than or equal to 1 ng/mL at a time greater than or equal to
four hours after
a nebulised dose of the composition is administered The nebulised dose is
typically from
0.02 mg/kg to 0.6 mg/kg. For example, the nebulised dose may be from 0.2 mg/kg
to 0.6
mg/kg. For instance, the invention provides a liquid pharmaceutical
composition as defined
herein for use in the treatment of the human or animal body, wherein the blood
plasma
concentration of RPL554 is greater than or equal to 1 ng/mL at a time greater
than or equal to
four hours after a nebulised dose of from 0.2 mg/kg to 0.6 mg/kg is
administered via
inhalation of the nebulised composition of the invention. The blood plasma
concentration of
RPL554 is typically greater than or equal to 1 ng/mL at a time greater than or
equal to three,
four or five hours after a nebulised dose of the composition is administered.
Date Recue/Date Received 2022-01-20
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
9
The invention also provides a method of treating or preventing a disease or
condition as
defined herein in a subject, which method comprises administering to said
subject an
effective amount of a liquid pharmaceutical composition as defined herein.
Typically, the
method is for treating a disease or condition. The disease or condition is
typically selected
from asthma, allergic asthma, hay fever, allergic rhinitis, bronchitis,
emphysema,
bronchiectasis, chronic obstructive pulmonary disease (COPD), adult
respiratory distress
syndrome (ARDS), steroid resistant asthma, severe asthma, paediatric asthma,
cystic fibrosis,
lung fibrosis, pulmonary fibrosis, interstitial lung disease, skin disorders,
atopic dermatitis,
psoriasis, ocular inflammation, cerebral ischaemia and auto-immune diseases.
Preferably the
method comprises treating asthma or chronic obstructive pulmonary disease
(COPD), more
preferably COPD.
An effective amount of RPL554 is typically from about 0.01 mg/kg to 50 mu/kg
for a single
dose. An effective amount of RPL554 is often from about 0.01 mg/kg to 1 mg/kg
for a single
dose. For instance, an effective amount may be a dose of from about 0.1 mg to
about 500
mg, or from about 0.1 mg to 100 mg, preferably from about 0.1 mg to about 6
mg. An
effective amount may be from 0.1 mg to 12 mg. A single dose of RPL554 may be
from 0.5
mg to 3 mg, for instance about 1.5 mg. Doses may be administered daily. For
instance, the
dose of RPL554 may be from 0.01 mg/kg/day to 50 mg/kg/day, typically from 0.01
mg/kg/day to 10 mg/kg/day or from 0.01 mg/kg/day to 1 mg/kg/day. These doses
are
typically the nominal dose emitted from the inhaler. The liquid pharmaceutical
composition
may be administered once, twice or three times a day, or may be administered
twice, three
times, four times or five times a week. The composition may be administered as
often as
required by the patient.
An effective amount may be a dose of from about 0.1 mg to about 500 mg, or
from about 0.1
mg to 100 mg, preferably from about 0.5 mg to about 6 mg. An effective amount
may be
from 0.1 mg to 12 mg. A single dose of RPL554 may be from 0.5 mg to 24 mg,
which may
be administered by nebuliser. A single dose of RPL554 may be from 0.5 mg to 6
mg, for
instance about 1.5 mg. Doses may be administered once, twice or three times
daily. For
instance, the dose of RPL554 may be from 0.1 mg/day to 50 mg/day, typically
from 1.5
mg/day to 18 mg/day. These doses are typically the nominal dose emitted from
the inhaler.
The liquid pharmaceutical composition may be administered once, twice or three
times a day,
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
or may be administered twice, three times, four times or five times a week.
The composition
may be administered as often as required by the patient.
It is a surprising finding of the present invention that a suspension
formulation of RPL554
allows for high doses of RPL554 to be administered to a patient without a
concomitant
increase in cardiovascular liability. The liquid pharmaceutical composition is
therefore
suitable for use in the treatment of patients suffering from cardiovascular
disorders or at risk
from suffering cardiovascular disorders.
For example, the composition may be used to treat a disease or condition as
defined above in
a patient suffering from cardiovascular vulnerability. Patients suffering from
cardiovascular
vulnerability are typically those at increased risk of adverse events arising
from tachycardia
or hypertension. More typically, patients suffering from cardiovascular
vulnerability are
those suffering from a pre-existing heart condition, or a condition which
would be aggravated
by tachycardia or hypertension. Such patients include those suffering from
coronary artery
disease, cardiomyopathy, hypertension, hypertensive heart disease, heart
failure, arrhythmia,
cardiac dysrhythmias, endocarditis, myocarditis, valvular heart disease and
stroke. Also
included are patients who have suffered a heart attack.
The invention is described in more detail by the following Examples.
WO 2016/042313
PCT/GB2015/052668
11
EXAMPLES
Example 1 ¨ formulation and stability of RPL554 suspension formulation
Liquid pharmaceutical compositions according to the invention were produced
with the
formulations set out Tables 1 and 2 below.
Table I -formulation of variant la (0.4 mg/mL)
Constituent Function Concentration
(mg/mL)
RPL554 (micronized) Active compound 0.4
Polysorbate 20 (Tween 20) Surfactant 0.50
Sorbitan Monolaurate (Span 20) Surfactant 0.05
Monosodium Phosphate Monohydrate Buffer 6.58
Dibasic Sodium Phosphate Buffer 6.80
Anhydrous
Sodium Chloride Tonicity Adjuster 4.80
Water Diluent Q.S.
Table 2 ¨formulation of variant lb (20.0 mg/mL)
Constituent Function Concentration
(mg/mL)
RPL554 (micronized) Active compound 20.0
Polysorbate 20 (Tween 20) Surfactant 0.50
Sorbitan Monolaurate (Span 20) Surfactant 0.05
Monosodium Phosphate Monohydrate Buffer 6.58
Dibasic Sodium Phosphate Buffer 6.80
Anhydrous
Sodium Chloride Tonicity Adjuster 4.80
Water Diluent Q.S.
Assay testing was performed in duplicate. The key method parameters are as
follows:
= mobile phase: acetonitrile:water:TFA (45:55:0.1);
= column: Waters X-Bridgemphenyl, 3.5 pm, 150 x 4.6 mm;
= flow rate: 1.5 ml/min;
= injection volume: 10 uL;
= detection: UV @ 254 nm;
= runtime: 6 minutes; and
= sample and standard concentration: 0.1 mg/mL.
Date Recue/Date Received 2022-01-20
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
12
Variant la was stored under conditions of either (i) a temperature of 25 C and
a relative
humidity (RH) of 60% or (ii) a temperature of 40 C and a RH of 75%. The
appearance, pH,
assay, impurities and particle size distribution (PSD) were measured
initially, at 1 month, at 2
months and at 6 months (6 months only for the 25 C and RH of 60% conditions).
The
amount of impurities were measured by HPLC with the relative retention time
(RRT) of the
impurity indicated.
The results are shown in Tables 3 and 4 below.
Table 3 - Stability of variant la under conditions: 25 C/60% RH
Test Initial 1 month 2 months 6 months
Appearance Light yellow Light yellow Light yellow Light
yellow
suspension free suspension free suspension free
suspension free
from from from from
agglomerates agglomerates agglomerates agglomerates
pH 6.55 6.59 6.68 6.51
Assay (mg/g) 0.372 0.374 0.373 0.372
Impurities ( /0
area)
Total L8 1.8 1.6 1.9
Greatest 0.490 (RRT 0.28) 0.533 (RRT 0.26) 0.429 (RRT 0.26) 0.463 (RRT 0.26)
Second greatest 0.401 (RRT 1.13) 0.408 (RRT 1.12) 0.389 (RRT 1.12) 0.432 (RRT
1.12)
PSD (Dv50, um) 2.282 2.069 2.089 2.133
Table 4 - Stability of variant la under conditions: 40 C/75% RH
Test Initial 1 month 2 months
Appearance Light yellow Light yellow Light yellow
suspension free suspension free suspension free
from from from
agglomerates agglomerates agglomerates
pH 6.55 6.91 6.67
Assay (mg/g) 0.372 0.374 0.377
Impurities (%
area)
Total 1.8 1.7 1.6
Greatest 0.490 (RRT 0.28) 0.445 (RRT 0.26) 0.428 (RRT 0.26)
Second greatest 0.401 (RRT 1.13) 0.409 (RRT 1.12) 0.380 (RRT 1.12)
PSD (Dv50, um) 2.28 2.10 2.14
Variant lb was stored under conditions of either (i) a temperature of 25 C and
a relative
humidity (RH) of 60% or (ii) a temperature of 40 C and a RH of 75%. The
appearance, pH,
impurities and particle size distribution (PSD) were measured initially, at 1
month, and at 2
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
13
months. The amount of impurities were measured by HPLC with the relative
retention time
(RRT) of the impurity indicated.
The results are shown in Tables 5 and 6 below.
Table 5 ¨ Stability of variant lb under conditions: 25 C/60N RH
Test Initial 1 month 2 months
Appearance Yellow Yellow Yellow
suspension free suspension free suspension free
from from from
agglomerates agglomerates agglomerates
pH 6.55 6.67 6.68
Assay (mg/g) 18.72 18.86 18.54
Impurities (%
area)
Total 1.2 1.2 1.2
Greatest 0.399 (RRT 1.13) 0.401 (RRT 1.12) 0.401 (RRT 1.12)
Second greatest 0.306 (RRT 1.11) 0.328 (RRT 1.11) 0.329 (RRT 1.11)
PSD (Dv50, [an) 2.11 2.37 2.05
Table 6 ¨ Stability of variant lb under conditions: 40 C/75% RH
Test Initial 1 month 2 months
Appearance Yellow Yellow Yellow
suspension free suspension free suspension free
from from from
agglomerates agglomerates agglomerates
PH 6.55 6.76 6.3
Assay (mg/g) 18.72 18.99 19.01
Impurities (%
area)
Total 1.2 1.2 1.2
Greatest 0.399 (RRT 1.13) 0.394 (RRT 1.12) 0.388 (RRT 1.13)
Second greatest 0.306 (RRT 1.11) 0.308 (RRT 1.11) 0.322 (RRT 1.11)
PSD (Dv50, atn) 2.11 2.09 2.13
As can be seen from Tables 3 to 6, the liquid pharmaceutical composition
according to the
invention shows excellent long term stability with no significant variation in
pH or the
amount of impurities present. This stability is even observed after 2 months
at 40 C.
Comparative Example 1 ¨ formulation and stability of RPL554 solution
formulation
A similar stability test was performed for the solution formulation of RPL554.
A 1.0 mg/ml
solution of RPL554 in an aqueous citrate/phosphate buffer solution at a pH of
approximately
3.2 was held under conditions of a temperature of 25 C and a relative humidity
(RH) of 600/o
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
14
for six months. The appearance, pH, assay and impurities present were measured
initially
and at 6 months. The results are shown in Table 7 below.
Table 7 ¨ Stability of RPL554 solution under conditions: 25 C/60% RH
Test Initial 6 months
Appearance A clear, slightly coloured A clear, slightly coloured
solution. Free from visible solution. Free from visible
contamination contamination.
pH _ 3.04 3.00
Assay 0.98 mg/mL 0.98 mg/mL
Impurities by HPLC
RRT 0.85 0.19% 0.19%
RRT 0.87 0.15% 0.42%
RRT 1.03 <0.10% <0.10%
RRT 1.09 0.20% 0.16%
RRT 1.12 0.54% 0.57%
RRT 1.14 0.14% <0.10%
RRT 1.15 0.11% <0.10%
While there is no change in the appearance of the solution, there is a
significant increase in
the amount of the impurity with the RRT value of 0.87. This impurity has been
identified as
a hydrolysis product of RPL554. The solution of RPL554 is less stable than the
suspension
formulation according to the invention.
Example 2 ¨ particle size distribution
The particle size distribution of a sample of micronized RPL554 suitable for
use in nebulized
formulations was evaluated using laser diffraction. The particle size
distribution analysis was
performed by laser diffraction using the Malvern Spraytec in conjunction with
a wet
dispersion cell.
The results are as follows: Dv10 = 0.69 wn; Dv50 = 1.35 tun; Dv90 = 2.5 tim.
Example 3 - formulations
RPL554 suspension and solution formulations were prepared as follows. These
formulations
were used in Examples 4 to 6.
Formulation 20 mg/ml RPL554 Suspension
constituents Constituent Amount
Concentration (mg/mL)
(Suspension)
RPL554 - Micronised 2.12ga 20 mg/mL
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
1
Wetting Solution 10.0 mL N/A
Buffer Solution To 100mL N/A
a Includes a 6% overage for expected manufacturing losses
Vehicle = as above, omitting RPL554
RPL554 Buffer Solution
Constituent Amount (g) Concentration (mg/mL)
Monosodium phosphate 32.9 6.58
monohydrate
Dibasic sodium phosphate 34.0 6.80
anhydrous
Sodium chloride 24.0 4.80
Water for Injection To 5000m1 N/A
RPL554 Wetting Solution
Constituent Amount (g) Concentration (mg/mL)
Polysorbate 20 (Tween 20) 10.0 5.00
Sorbitan monolaurate 1.0 0.50
(Span 20)
RPL554 Buffer Solution To 2000 mL N/A
Method of RPL554 Buffer Solution: The required amount of vehicle excipients
were
preparation weighed out into a suitable container and made up to the
required volume
(Suspension) with water for injection The excipients were magnetically stirred
for 10
minutes until the buffer salts were fully dissolved. The pH was measured,
recorded and adjusted if necessary to pH 7.0 0.3 using HC1/NaOH and
filtered through a 0.221,.im filter.
RPL554 Wetting Solution: The required amount of polysorbate 20 was
weighed into a suitable container and the required amount of sorbitan
monolaurate was added. The wetting solution was magnetically stirred for
minutes, then transferred to a larger container with buffer solution and
made up to the final volume with the buffer solution. The solution was
magnetically stirred for 10 minutes and filtered through a 0.22 p.m filter.
Vehicle: The required volume of RPL554 Wetting Solution was measured
into a suitable container and made to the final volume with RPL554 Buffer
Solution, mixed with a magnetic stirrer for 10 minutes. The pH was
measured, recorded and adjusted if necessary to pH 7.0 0.3 using
HC1/Na0H.
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
16
20 mg/mL RPL554 Suspension: The required amount of RPL554 was
weighed into a small beaker and the required amount of Wetting Solution
was added. The suspension was mixed using a Silverson mixer fitted with
a 5/8" high sheer screen for approximately 1 minute at 8000 rpm. The
mixture was transferred to a new clean, dry beaker rinsing with Buffer
Solution. The required weight was made up with Buffer Solution and
mixed using a Silverson mixer fitted with a 1" high sheer screen for
approximately 1 minute at 8000 rpm. The pH was measured, recorded and
adjusted if necessary to pH 7.0 0.3 using HC1/Na0H. The solution was
transferred into the final container.
For the lower concentrations of RPL554 suspensions, the required amount
of 20 mg/mL formulation was weighed out and diluted to the required
volume with vehicle using a syringe fitted with a 0.22 lam filter. The
formulation was gently mixed on a magnetic stirrer for 5 minutes.
Formulation 1 mg/mL (pH 3.5) RPL554 Solution
constituents Constituent Amount
(Solution)
RPL554 100 mg
0.1M Citric acid solution 22.5 mL
0.2M Dibasic sodium phosphate 9.7 mL
dodecahydrate solution
0.9% (w/w) Saline solution 67.8 mL
1.5 mg/mL (pH 2.5) RPL554 Solution
Constituent Amount
RPL554 150 mg
0.1M Citric acid solution 45 mL
0.2M Dibasic sodium phosphate 5 mL
dodecahydrate solution
0.9% (w/w) Saline solution 50 mL
Vehicle: As above, omitting RPL554
Method of RPL554 0.1M Citric Acid Solution: The required amount of citric
acid
preparation was weighed into a suitable container, made up to the required
volume
(Solution) with 0.9% saline and magnetically stirred until fully dissolved.
RPL554 0.2M Dibasic Sodium Phosphate Solution: The required
amount of dibasic sodium phosphate anhydrous was weighed into a
suitable container, made up to the required volume with 0.9% saline and
magnetically stirred until fully dissolved.
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
17
Vehicle: The required volume of Citric Acid Solution, Dibasic Sodium
Phosphate Solution and 0.9% saline were measured into a suitable
container and mixed thoroughly. The pH was measured and adjusted if
necessary (pH 3.5 (10.3) or pH 2.5 (--0.2)) by drop wise addition of further
citric acid or sodium phosphate solution as required.
RPL554 Solution: The required amount of test material was weighed into
a suitable container, made up to the final volume with Vehicle and
magnetically stirred until fully dissolved. The pH was measured and
adjusted if necessary.
Example 4 ¨ toxicokinetic profile in rats
Summary
Han Wistar rats (5/sex/group) were dosed by nose-only inhalation once daily at
target doses
of 2.4, 8.4 or 21.6 mg/kg/day of an RPL554 suspension according to the
invention for 7
consecutive days (groups 2, 3 and 4). An additional 5 rats/sex/group received
the vehicle
suspension and acted as controls (group 1). The duration of dose
administration was 240
minutes on each day. At the end of the treatment period, all surviving animals
were
euthanized and necropsied. Satellite animals (3/sex/group) were similarly
dosed and bled on
Day 7 for toxicokinetic analysis. An additional group of satellite animals
(3/sex) were dosed
by nose-only inhalation once daily at a target dose of 2.4 mg/kg/day RPL554
solution for
comparison with the RPL554 suspension and bled on Day 7 for toxicokinetic
analysis (group
5). Mean aerosol concentrations and achieved doses are presented in Table 8
below.
Table 8¨ Mean Aerosol Concentrations and Achieved Doses
Target Achieved
Target Achieved Formulation
Type of exposure exposure
dose dose concentration
Group Formulation level level
(ag/L) (ag/L) (mg/kg/day) (mg/kg/day) (mg/mL)
Vehicle
1 0 0.00 () 0 0
Suspension
2 Suspension 13 18.10 2.4 3.27 1.5
3 Suspension 47 55.18 8.4 9.96 4.5
4 Suspension 121 141.51 21.6 25.49 12
Solution 13 14.47 2.4 2.62 1.6
CA 02959943 2017-03-01
WO 2016/042313 PCT/GB2015/052668
18
Toxicokinetic results
The toxicokinetic profile of the RPL554 suspension was measured at different
doses (groups
2, 3 and 4). This was compared with the toxicokinetic profile of the known
RPL554 solution
(group 5). The dosages administered to group 2 and group 5 are similar. Group
1 is the
control group.
The toxicokinetic parameters of RPL554 in suspension (invention) or solution
(comparative)
form were evaluated on day 7 of 1 week of daily inhalation exposure of the
RPL554
compositions to rats. The results are shown in Tables 10 and 11 below. The
parameters
measured are maximum concentration of RPL554 in the blood plasma of the rats
(C.), the
time taken to reach Cmax (Trna,) and the area under the dose curve after 8
hours (AUC8h).
Table 10 ¨ toxicokinetic results for male rats
Achieved
Group dose C. (ng/mL) T. (hours, median) AUCsh (ng.h/mL)
(mg/kg/day)
1 (vehicle) 0 0 0 0
2 (suspension) 3.27 10.5 4.25 41.9
3 (suspension) 9.96 44.1 4.25 172
4 (suspension) 25.49 94.1 4.083 372
(solution) 2.62 53.4 4.083 139
Table 11 ¨ toxicokinetic results for female rats
Achieved
Group dose C. (ng/mL) T. (hours, median) AUC8h (ng.h/mL)
(mg/kg/day)
1 (vehicle) 0 0 0 0
2 (suspension) 3.27 19.9 4.25 79.1
3 (suspension) 9.96 133 4.25 478
4 (suspension) 25.49 573 4.25 2030
5 (solution) 2.62 52.0 4.083 153
These results demonstrate that it is possible to reach substantially higher C.
values using
the liquid pharmaceutical composition of the invention (e.g groups 3 and 4)
compared with a
solution composition (group 5). The solubility limited dose feasible from a
solution of
RPL554 prevents such Can, values being achieved with a solution composition.
The
suspension compositions are also shown to have a delayed release, with greater
T. values
observed for groups 2 to 4.
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
19
Example 5 ¨ toxicokinetie profile in dogs
The toxicokinetic profile of RPL554 suspensions according to the invention
were evaluated
for beagle dogs. The study design was as set out in Table 12 below.
Table 12 ¨ study design, doses and nominal concentrations
Target dose Target aerosol Nominal
Phase Dose Treatment Formulation of RPL554 concentration*
concentration
(mg/kg/day) l_tg/L (mg/mL)
1 1 RPL554 Suspension 1 19.8 1.5
1 2 RPL554 Suspension 3 59.4 4.5
1 3 RPL554 Suspension 9 178 16
2 RPL554 Suspension 9 178 16
3 RPL554
Suspension 0.5 10 1.5 (suspension)
and solution 1.0 (solution)
* Aerosol concentration calculated assuming an exposure duration of 120
minutes and a body weight
of 12 kg
Animals received the test substance, RPL554, by inhalation administration for
3 days at each
dose in the variable dose phase (Phase 1), with at least 2 days washout period
between doses.
In the constant dose phase (Phase 2), animals received the test substance for
7 consecutive
days. In the crossover phase (Phase 3), animals received the test substance as
a single
exposure to the suspension formulation followed by 2 days off-dose followed by
a single
exposure to the comparative solution formulation.
Toxicokinetic results
Following 7 days of dosing at 982 mg/kg/day with the RPL554 suspension (group
2), the
Cmax values for males and females were 116 ng/mL and 95.9 ng/mL, respectively,
and the
AUC6h values for the males and females were 389 ng.h/mL and 289 ng.h/mL,
respectively.
After a single exposure of RPL554 suspension at 0.395 mg/kg, the Cma, values
for the male
and females were 13.4 ng/mL and 12.7 ng/mL, respectively, and the AUC6h values
for the
male and females were 33.9 ng.h/mL and 31.5 ng.h/mL, respectively.
After a single exposure of RPL554 solution at 0.543 mg/kg, the C. values for
the male and
females were 40.9 ng/mL and 34.2 ng/mL, respectively, and the AUC6h values for
the male
and females were 74.6 ng.h/mL and 62.8 ng.h/mL, respectively.
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
Here it is observed that it is possible to achieve high C. and AUC values
using the RPL554
suspension according to the invention (e.g. 116 ng/mL and 95.9 ng/mL).
The toxicokinetic results also confirm that the suspension formulation gives a
delayed release
compared to the solution formulation. This can be seen from the data from the
phase 3
experiment (separate single doses of the suspension formulation). These data
are presented in
Table 13 below, where the solution formulation (target dose 0.5 mg/kg/day,
actual 0.543
mg/kg/day) is compared with the suspension formulation (target dose 0.5
mg/kg/day, actual
0.395 mg/mg/day). Table 13 shows plasma concentrations (ng/mL) over time
following
treatment with a single dose of the suspension or a single dose of the
solution. The two
treatments were performed two days apart. Data for all three dogs are
presented: one male
dog (61) and two female dogs (62 and 64). BLQ refers to plasma concentrations
of less than
1.00 ng/mL.
Table 13 - RPL554 plasma concentration values (ng/mL) for 3 dogs following a
dose with an
RPL554 suspension and an RPL554 solution
Dog 61(M) 62 (F) 64 (F)
Time (hours) Suspension Solution Suspension Solution Suspension Solution
0 BLQ BLQ BLQ BLQ BLQ BLQ
0.017 11 39.7 9.83 35.2 13.5 33.1
0.25 13.4 40.9 11.6 26.5 13.7 26.5
0.667 10.4 13 7.26 15.7 10.8 13
1.667 4.57 4.84 2.51 3.31 4.99 3.15
3 2.88 1.23 1.87 1.99 2.42 1.21
4 1.81 BLQ 1.4 BLQ 1.74 BLQ
The data in Table 13 demonstrates that the suspension formulation of RPL554
leads to a
more delayed release profile than the solution formulation of RPL554. For
instance, the
plasma concentration of RPL554 is below the detectable limit 4 hours after
inhalation of the
solution formulation for all dogs, whereas at least 1.4 ng/mL of RPL554 is
present after 4
hours in the blood plasma for all dogs following inhalation of the suspension.
Example 6 - lung function in guinea pigs
Introduction
RPL554 causes bronchoprotection against intravenously administered spasmogenic
agents
when administered as dry power or when prepared as a solution in acidifed (pH
2.5) saline.
WO 2016/042313
PCT/GB2015/052668
21
This functional antagonism (1.5 doubling dilution ie 2.8 fold) against
methacholine has also
been verified in human asthmatic subjects (Franciosi et al, 2013). This
Example evaluated
the bronchoprotection afforded by RPL554 when presented as the new suspension
formulation for nebulisation.
Methodology
Drug exposure
Male Dunkin Hartley guinea-pigs (300-400 g) were placed in a custom built
aerosol chamber
and exposed to either vehicle (0.9% saline pH 2.5), RPL554 (2.5 mg/mL
solution) or RPL554
(1 mg/mL solution) for a 15 min period. Aerosols were generated using an
Ultrasonic
TM
Nebuliser (Ultraneb 99) and a flow rate of 1 L/min was used to direct the
nebulized solution
to an aerosol chamber which contained 4 arms that allows animals to be
restrained with their
upper respiratory tract (ie nose) protruding into the aerosol chamber,
After this 15 min exposure period, animals were allowed to recover and then
prior to the 2 h
and 5 h post drug exposure, animals were anaesthetised with urethane (1.5
g/kg) and a
midline incision made to expose the trachea. A tracheostomy was performed, and
a tracheal
cannula tied in place and attached to a pneumotachograph which in turn was
connected to a
Validynempressure transducer (+ 2 cmH20) for the detection of flow. Breath by
breath
changes in airflow were measured using a Lung Function Recording system (LFR,
Version 9,
Mumed UK) and displayed in real time on a PC. The flow signal was integrated
to give a
measure of tidal volume.
A cannula was inserted into the thoracic cavity between the 3rd and 5th
intercostal space and
connected to the negative side of a Validyne pressure transducer (+ 20 cmH20).
The positive
side of the pressure transducer was connected to the side of the
pneumotachograph proximal
to the animal, in order to obtain a measure of transpulmonary pressure (TPP:
difference
between mouth and thoracic pressure). The lung function parameters, total
airway resistance
(RL; cmH20.s/L) and dynamic lung compliance (mL/cmH20) was derived from each
measure of flow, tidal volume and TPP by the method of integration. The
carotid artery was
cannulated for the measurement of blood pressure, respectively. Cumulative
concentration-
effect curves were established and the provocation concentration (PC) of
acetylcholine which
cumulatively produced a 50% increase in RL (PC50).
Date Recue/Date Received 2022-01-20
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
22
Acetylcholine exposure
Bronchoconstriction to acetylcholine (0.25 to 16 mg/mL, 4 sec exposure) was
monitored 2 h
and 5 post administration of a solution of RPL554 (1 and 2.5 mg/mL) or vehicle
(acidified
saline pH 3.5 or pH 2.5). Aerosols of acetylcholine were generated with an
Aeroneb Lab
Nebulizer (Aerogen Inc). The inflow was directed either via the
pneumotachograph for the
measurement of respiratory lung mechanics, or shunted through the nebulizer
for the
purposes of delivering drug directly into the lung (respiratory mechanics not
measured during
the 4 sec exposure period).
In a separate series of experiments, lung function was perfoi ___ filed 2 and
5 h post inhalation of
the new suspension formulation of RPL554 (2.5 mg/mL and 10 mg/mL) and the
vehicle.
Results
Baseline parameters
Baseline total lung resistance (RL), dynamic lung compliance (Cdyn), mean
arterial blood
pressure (BP) and nebulizer output were measured in animals previously exposed
to vehicle
control (saline pH 2.5), and solution formulations of RPL554 (1 mg/mL or 2.5
mg/mL,
Figure 1) and to new suspension formulations of RPL554 (2.5 mg/mL and 10
mg/mL, Figure
2). Baseline respiratory and cardiovascular parameters were made prior to
acetylcholine
exposure and were monitored 2 and 5 h post saline or RPL554 exposure in
anaesthetised
guinea-pigs.
Lung function parameters
In general RL represents changes in airway diameter in the central airways
following the
activation of muscarinic receptors on airway smooth muscles in this region of
the lung.
Acetylcholine was chosen for its short duration of action.
1. RPL554 Solution Formulation
Acetylcholine administered to anaesthetized guinea-pigs by the inhaled route
caused a dose
dependent bronchoconstrictor response as determined by changes in RL measured
2 h and 5 h
post saline and RPL554 (1 mg/mL, 2.5 mg/mL solution) exposure.
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
23
1.1 - Analysis of Acetylcholine RL PC50
Post hoc analysis of the arithmetic mean demonstrated a significant
bronchoprotection at the
2 h point for RPL554 1 mg/mL solution (mean diff = 3.5 (0.11 ¨ 8.83) mg/mL, P
= 0.0445)
and 2.5 mg/mL at the 2 h (4.7 (0.4 ¨ 9) mg/mL, P = 0.0357) and 5 h time point
(3.4 (0.9 ¨6)
mg/mL, P = 0.0133). The degree of bronchoprotection was less evident at 5 h
for 1 mg/mL
RPL554 solution (2.5 (-0.17-5.2) mg/mL, P = 0.0628).
A significant difference between 2 h (1 mg/mL RPL554, P = 0.0190 unadjusted)
and 5 h (2.5
mg/mL, P = 0.0095 unadjusted) RPL554 solution vs saline group was observed
(Figure 1).
2. New Suspension Formulation of RPL554
Acetylcholine administered to anaesthetized guinea-pigs by the inhaled route
caused a dose
dependent bronchoconstriction as determined by changes in RL measured 2 h and
5 h post
saline and RPL554 suspension (2.5 mg/mL and 10 mg/mL) exposure. Dose dependent
reduction in baseline lung compliance (Cdyn) was also simultaneously recorded
following
increasing concentrations of acetylcholine 2 h and 5 h post saline or RPL554
suspension (2.5
mg/mL and 10 mg/mL) exposure.
2.1 - Analysis of Acetylcholine RL PC50
Analysis of variance of the 1og2 transformed data for Acetylcholine RL PC50
revealed an
overall drug effect at the 2 h (P = 0.0238). For the 2 h time group, this was
reflected by a
significant difference between the 10 mg/mL RPL554 suspension and vehicle
group (Mean
fold difference = 2.67 (1.12 ¨ 6.36), P < 0.05). Despite the greater mean
difference, between
RPL554 (10 mg/mL suspension) versus vehicle at the 5 h time point 14 fold
shift (0.5-400),
the wide confidence interval is the reason why statistical difference was not
achieved (Figure
2, upper panel).
The vehicle data for the 2 and 5 h time points were combined and re-analyzed
(Figure 2
lower panel). There was an overall significant treatment effect (P=0.0016)
which was
reflected by a significant degree of bronchoprotection with 2.5 mg/mL RPL554
suspension
(2.52 fold (1-5.72). P < 0.05 and 10 mg/mL RPL554 (4.67 fold (2-11), P <
0.05). At the 5 h
time point, RPL554 10 mg/mL suspension caused a significant degree of
bronchoprotection
(2.77 fold (1.3-6.0) (Figure 2, lower panel).
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
24
Summary
RPL554 inhibited bronchoconstriction induced by aerosolized administered
acetylcholine
when measured 2 and 5 h post drug exposure. Both the RPL554 solution
formulations (1
mg/mL, 2.5 mg/mL) and the new RPL554 suspension formulations (2.5 mg/mL, 10
mg/mL)
produced a statistically significant bronchoprotecti on.
Example 7 ¨ pharmacokinetic study in humans
A phase I, randomised, double-blind, placebo-controlled, study was performed
to assess the
safety, tolerability and pharmacokinetics of single inhaled doses of RPL554
suspension
formulation according to the invention, administered by nebuliser to healthy
male subjects
aged 18 to 50 inclusive.
The nominal dose of suspension formulation was approximately 1.5 mg, being
essentially
equivalent to the 0.018 mg/kg dose previously shown to be well-tolerated in
clinical studies
with the solution formulation.
The suspension formulation used was a sterile suspension for nebulisation
containing 1.5 mg
micronised RPL554 drug substance in 5 mL phosphate buffered saline solution at
pH 7, with
surfactants (Tween 20 and Span 20) (as described in Example 1 above, but with
a different
concentration of RPL554). The dosing of the suspension was performed using a
standard Jet
nebuliser (PARI LC Sprint)
Blood samples (4mL at each time point) were collected at appropriate time
intervals after
administration. Samples were collected by venepuncture or via indwelling
cannula in the
forearm into lithium heparin tubes and will be immediately chilled (ice bath).
The blood was
centrifuged within 15 minutes of collection. The plasma was separated in a
refrigerated
centrifuge (about 4 C) at 1100g for 15 minutes and transferred into
polypropylene tubes. 'The
concentration of the active was then measured.
The mean results are presented in Figure 3, which shows the mean concentration
curves for
the subjects after administration of the solution or suspension formulations
The results for
the solution formulation were derived from a previous clinical study (The
Lancet Respiratory
Medicine Volume 1, No.9, p.'714-72'7, November 2013) which used a sterile
solution for
nebulisation containing RPL554 dissolved in ciliate-phosphate buffered saline
at pH 3.2. The
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
quantity administered via nebuliser was such that the nominal RPL554 dose was
0.018 mg/kg
(-1.26 mg for a 70kg person).
Mean pharmacokinetic parameters measured during the study are presented in
Table 14
below.
Table 14
Formulation Cmax (pg/ml) AUCinf (pg.h/m1) Half-life (h)
Solution 870 1833 3
Suspension 393 3795 10
The values in Table 14 demonstrate that the suspension formulation according
to the
invention has a substantially improved pharmacokinetic profile compared with
the
comparative solution formulation. In particular. the area under the curve
(AUC) and the half-
life are both significantly increased. This demonstrates that the suspension
formulation
provides an unexpectedly improved way by which RPL554 may be administered by
inhalation.
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
26
Example 8 ¨ twelve months stability study
Three batches of the suspension formulation of the invention were made with
the
compositions set out in Table 15 below.
Table 15
Constituent Concentration (mg/mL)
RPL554 (heat treated API) 0.5, 2.5 and 10
Polysorbate 20 (Tween 20) 0.5
Sorbitan monolaurate (Span 20) 0.05
Monosodium phosphate monohydrate 6.58
Dibasic sodium phosphate anhydrous 6.80
Sodium chloride 4.80
Water for injection To weight
Analytical testing
Analytical testing was performed after 1, 3, 6 and 12 months storage of the
formulations at
25 C and 60% RH. The 12 months results are described below.
Appearance
Appearance testing was performed visually on a single sample. The placebo
sample was
found to be a clear, colourless solution. The 0.5mg/mL strength samples were a
light yellow
suspension free from visible agglomerates while the 2.5mg/mL and 10mg/mL
strength
samples appeared to be yellow suspensions free from visible agglomerates.
pH
The pH determination was performed on a single sample. The results are shown
in Table 16.
Table 16
Batch Number Strength pH
BN011/14 0.5mg/mL 6.656
BN010/14 2.5mu/mL 6.652
BN009/14 10mg/mL 6.655
BN008/14 Placebo 6.661
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
27
All three active foimulations and placebo were found to have a pH of 6.7 at
the twelve month
time-point. No change was observed from the initial time point.
Assay
Assay testing was performed in duplicate. The key method parameters are
summarised as
follows:
Mobile phase Acetonitrile: water: TFA 45:55:0.1
Waters X-Bridge phenyl, 3.5 um, 150 x 4.6 mm,
Column
(Part Number 186003335)
Column temperature 40 C
Flow rate 1.5 ml/min
Injection volume 10 IA
Detection UV, 254 nm
Runtime 6 minutes
Autosampler temperature Ambient
Standard Concentration 0.1 mg/ml
The results of the assay arc shown in Table 17.
Table 17
Batch Number Strength Prep 1 Prep 2 Mean
BN011/14 0.5mg/mL 0.5180 0.5178 0.52
BN010/14 2.5mg/mL 2.6541 2.6407 2.65
BN009/14 10mg/mL 10.4219 10.4926 10.46
The assay results for the active suspensions of each strength at the twelve
month time point
did not show any change from initial time point.
Related substances
Related substances deteimination was performed in duplicate. The key method
parameters
are summarised as follows.
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
28
A - Purified water/Acetonitrile/TFA (95/5/0.1).
Mobile phase
B - Acetonitrile/Water/TFA (95/5/0.1).
X-Bridge Phenyl 4.6 x 150 mm 3.5 pm particle size (ex -
Column
Waters; 4186003335)
Column temperature 30 C
Autosampler Temperature Ambient
Flow rate 1.0 ml/min
Injection volume 10 1_11
Detection UV, 254 nm
Time (min) %A %B
0 100 0
2 100 0
Gradient 15 0 100
25 0 100
27 100 0
37 100 0
Diluent Acetonitrile : Water (50 : 50)
Sample Concentration 0.2mg/m1
Standard Concentration 2 [tg/m1
The results from the related substances determination of the suspensions are
presented in
Table 18.
Table 18 - Results from the related substances analysis of RPL554 suspensions
(as %LC)
Batch BN011/14 - (0.5mg/mL) - 25 C/60%RH
%LC
RRT Mean
Ti T2
0.78 0.0776 0.0764 0.08
0.86 0.1028 0.1046 0.10
0.92 0.2084 0.2101 0.21
0.93 0.1875 0.1846 0.19
1.09 0.0548 0.0562 0.06
1.11 0.4130 0.4142 0.41
Total 1.05
Batch BN010/14- (2.5mg/mL) - 25 C/60 ,/oRH
%LC
RRT Mean
Ti T2
0.77 0.0779 0.0792 0.08
0.85 0.1041 0.1051 0.10
0.92 0.2184 0.2227 0.22
0.93 0.1768 0.1746 0.18
1.11 0.4226 0.4270 0.42
Total 1.00
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
29
Batch BN009/14 - (10mg/mL) - 25 Ci60%RH
%LC
RRT Mean
Ti T2
0.78 0.0769 0.0789 0.08
0.86 0.1016 0.1031 0.10
0.92 0.2056 0.2194 0.21
0.93 0.1819 0.1684 0.18
1.11 0.4137 0.4250 0.42
Total 0.99
The related substances results for the active suspensions of each strength at
the twelve month
time-point were similar to the initial time point.
Particle size distribution (PSD)
The Particle Size Distribution method was performed using a Spraytec with the
settings
detailed below.
Optical Properties: Particle: RI (real): 1.50 (standard opaque particle); RI
(imaginary): 0.50;
Density: 1.00; Dispersant (water); Dispersant RI: 1.33
Dispersant used for sample analysis: 1% polysorbate 20 in deionised water
Controller unit settings: Stirrer speed 1000rpm
Measurement time. Sampling time: 30s; Background time: lOs
The results are shown in Table 19.
Table 19 - Results from the Particle Size Determination by Laser Diffraction
of RPL554
suspensions (in ,um)
Batch Number Strength Dv10 Dv50 Dv90
BN011/14 0.5 mg/mL 0.7946 1.577 3.168
BN010/14 2.5mg/mL 0.7872 1.589 3.119
BN009/14 10mg/mL 0.8012 1.566 2.976
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
The Particle Size Distribution profiles were similar for all 3 strengths at
the twelve month
time-point and did not show any change compared to the initial time point.
Microscopic evaluation
Microscopic evaluation was performed by visually assessing the formulations
using a G3
microscope. No aggregates were observed in any of the batches tested, although
some large
loose agglomerates were occasionally seen for the 2.5mg/mL and 10mg/mL
suspensions.
The evaluation determined that there was no change compared to the initial
time point.
Delivery rate and total delivered dose
Delivery rate and total delivered dose was performed on 3 vials per batch. The
suspensions
were dispensed and delivered using a PARI LC PLUS Nebuliser / PARI TurboBOY
compressor combination. The results from the delivery rate and total delivered
dose are
presented in Table 20.
Table 20 - Results from the delivery rate and total dose delivered
determination using PARI
LC PLUS nebuliser
Batch BN011/14 0.5mg/mL
Replicate number Ti T2 T3
Initial weight (g) 36.8175 36.7852 36.8030
Final weight (g) 32.6399 32.7369 32.6272
Delivered mass (g) 4.1776 4.0483 4.1758
Total Delivery Time (s) 900 1020 960
Delivery Rate
(mg/min) 0.136 0.130 0.137
(over the first min)
Mean Delivery Rate
0.1
(mg/min)
Assay* (mg) 1.179 1.144 1.198
Mean Assay* (mg) 1.2
% Efficiency 54.50 54.56 55.39
Mean % Efficiency 54.8
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
31
Batch BN010/14 2.5mg/mL
Replicate number Ti T2 T3
Initial weight (g) 37.0977 38.7543 37.2942
Final weight (g) 32.8590 34.9311 32.9187
Delivered mass (g) 4.2387 4.3632 4.3755
Total Delivery Time (s) 1020 1020 1020
Delivery Rate
(mg/min) 0.303 0.229 0.335
(over the first mm)
Mean Delivery Rate 0.3
(mg/min)
Assay* (mg) 6.212 6.684 6.565
Mean Assay* (mg) 6.5
% Efficiency 55.36 57.86 56.68
Mean % Efficiency 56.6
Batch BN009/14 10mg/mL
Replicate number T1 T2 T3
Initial weight (g) 36.9365 38.4396 37.0922
Final weight (g) 32.6608 34.2031 32.7858
Delivered mass (g) 4.2757 4.2365 4.3064
Total Delivery Time (s) 1080 1140 1080
Delivery Rate
(mg/min) 1.326 1.095 1.221
(over the first mm)
Mean Delivery Rate
1.2
(mg/min)
Assay* (mg) 24.279 24.168 24.705
Mean Assay* (mg) 24.4
% Efficiency 54.30 54.55 54.86
Mean % Efficiency 54.6
* assay is total delivered
% Efficiency is total actual dose delivered/theoretical dose delivered
(calculated using
delivered mass and formulation strength from assay)
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
32
The total delivered dose and delivery rate for all strengths were similar to
the initial time
point. The delivery efficiency of all the suspensions was consistent with
previous time
points.
Aerodynamic Particle Size Distribution (APSD)
Aerodynamic Particle Size Distribution (APSD) determination was performed on 3
vials per
batch per condition using the Next Generation Impactor (NGI). The suspensions
were
dispensed and delivered using a PART LC PLUS Nebuliser / PART TurboBOY S
compressor combination. The results from the APSD determination are presented
in Table
21. The results were input into the CITDAS program (Version 3.10) to calculate
the fine
particle dose using a cut-off value of 51.1m and the results obtained from the
calculations are
also presented Table 21.
Table 21 - Results from the Aerodynamic Particle Size Distribution by111GI (as
mg RPL554)
using PARI LC PLUS nebuliser
Batch BN011/14 - (0.5mg/mL) 25 C/60%RH
Stage NGI 1 NGI 2 NGI 3 Mean
Throat 0.05495 0.09324 0.08583 0.08
Stage 1 0.13433 0.12798 0.12692 0.13
Stage 2 0.26114 0.18827 0.22637 0.23
Stage 3 0.40347 0.29208 0.39028 0.36
Stage 4 0.49538 0.41178 0.46929 0.46
Stage 5 0.23507 0.24668 0.29097 0.26
Stage 6 0.07792 0.06313 0.08504 0.08
Stage 7 0.01105 0.01100 0.01525 0.01
MOC 0 00034 0.00169 0.00151 0.00
Sum 1.67365 1.43585 1.69147 1.60
Delivered Mass (g) 4.2864 4.1465 4.2623 4.23
FPD < 5[1m* 0 735 0.667 0.785 0.7
FPD/delivered mass
0.171 0.161 0.184 0.2
(mg/g)
FPF (FPD as % total
43.9 46.4 46.4 45.6
dose) < 5litm*
GSD* 2.0 2.2 2.0 2.1
MMAD* 5.3 5.0 5.1 5.2
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
33
Batch BN010/14 - (2.5mg/mL) 25 C160%RH
Stage I NGI 1 NGI 2 NGI 3 Mean
Throat 0.35731 0.43867 0.47848 0.42
Stage 1 0.71616 1.04125 0.93292 0.90
Stage 2 1.28121 1.58441 1.43900 1.43
Stage 3 1.98259 2.21051 1.95403 2.05
Stage 4 2.41709 2.37565 2.23993 2.34
Stage 5 1.69766 1.45824 1.39429 1.52
Stage 6 0.54927 0.43922 0.37542 0.45
Stage 7 0.07994 0.07293 0.06443 0.07
MOC 0.00163 0.00151 0.00540 0.00
Sum 9.08286 9.62239 8.88390 9.20
Delivered Mass (g) 4.3881 4.5659 4.3137 4.42
FPD < 5p.m* 4.356 3.945 3.702 4.0
FPD/delivered mass (mg/g) 0.993 0.864 0.858 0.9
FPF (FPD as % total dose)
48.0 41.0 41.7 43.5
GSD* 2.1 2.1 2.1 2.1
MMAD* 5.0 5.7 5.5 5.4
Batch BN009/14 - (10mg/mL) 25"C/60%RH
Stage NGI 1 NGI 2 NGI 3 Mean
Throat 1.82141 1.46478 1.70097 1.66
Stage 1 2.99033 3.28692 3.75069 3.34
Stage 2 5.51909 6.88069 7.01532 6.47
Stage 3 8.46593 10.48679 10.09839 9.68
Stage 4 9.42726 10.16201 9.36013 9.65
Stage 5 6.30967 4.85282 4.67990 5.28
Stage 6 1.84396 0.98008 1.00599 1.28
Stage 7 0.28335 0.13666 0.12275 0.18
MOC 0.00383 0.00710 0.00108 0.00
Sum 36.66483 38.25785 37.73522 , 37.55
Delivered Mass (g) 4.3787 4.6309 4.4044 4.47
FPD < 5p,m* 16.313 14.318 13.490 14.7
FPD/delivered mass (mg-/g) 3.726 3.092 3.063 3.3
FPF (FPD as (?/0 total dose)
44.5 37.4 35.7 39.2
< 5 m*
GSD* 2.0 1.9 1.9 2.0
MMAD* 5.3 5.9 6.1 5.8
* calculated using ClIDAS on unrounded data. Abbreviations: FPD: fine particle
dose;
FPF: fine particle fraction; GSD: geometric size distribution; MMAD: mass
median
aerodynamic diameter; MOC: micro-orifice collector.
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
34
12 months stability
Tables 22 (for 0.5 mg/mL), 23 (for 2.5 mg/mL) and 24 (for 10 mg/mL) below
combine the 12
months data described above with the corresponding data for the 1, 3 and 6
months time
points for the RPL554 suspension formulations of the invention.
(Note: Impurity peaks marked with * in Tables 22, 23 and 24 were observed as a
doublet but
had been observed as a single peak at the initial time point. This is probably
due to a new
column being used which gave better separation and is not believed to be a
sign of
degradation.)
Sumnuiry
RPL554 Suspensions for Nebulisation 0.5 mg/mL (BN011/14), 2.5 mg/mL (BN010/14)
and
mg/mL (BN009/14) and associated placebo (BN008/14) were manufactured and
placed on
stability at 25 C/60%RH and were tested at the 12 month time-point.
The results showed that there was essentially no change in any of the batches
tested at the 12
month time point and confirmed that the RPL554 suspensions for nebulisation
were stable for
12 months at 25 C/60(3/0RH.
Table 22 (0.5 mg/mL) Test Initial 1 month 3 month 6
month 12 month
0
Storage condition N/A , 25 C/60 /oRH , 25
C/60%RH 25 C/60"/012H , 25 C/60"/oRH No
1-,
Pale yellow Pale yellow Pale yellow Pale
O' yellow Pale yellow o
..
Appearance suspensions free from suspensions free from suspensions
free from suspensions free from suspensions free from =P
N?
Co4
visible agglomerates visible agglomerates visible
agglomerates visible agglomerates visible agglomerates
w
pH 6.7 6.7 6.6
6.6 6.7
Mean Assay (n=2) (mg/mL) 0.51 0.51 0.52
0.52 0.52
.
.
Mean Impurities (n=2) (%LC)
Total 0.98 1 0.89 0.95 1.05
Greatest 0.39 (RRT 1.11) 0.23 (RRT 0.92) 0.38 (RRT
1.10) 0.38 (RRT 1.10) 0.41 (RRT 1.11)
0.20 (RRT 1.11)*
P
Second greatest 0.23 (RRT 0.92) 0.18 (RRT 1.10) 0.22 (RRT
0.92) 0.22 (RRT 0.93) 0.21 (RRT 0.92)
*
2
Dv10= 0.7646 Dv10= 0.7721 Dv10= 0.7823 Dv10=
0.8036 Dv10= 0.7946 .
w .
un
,,
PSD (um) Dv50= 1.534 Dv50= 1.542 Dv50= 1.56 Dv50=
1.585 Dv50= 1.577
..,
Dv90= 2.999 Dv90= 2.991 Dv90= 3.084 Dv90=
3.158 Dv90= 3.168 '
Mean FPD (mg) 0.4 0.6 0.6
0.6 0.7 2
Mean FPD/mass delivered
0.1 0.2 0.1
0.1 0.2
(n=3) (mg/g)
Mean MMAD (n=3) 5.4 5.7 5.4
5.6 5.2
Mean GSD (n=3) 2 2 2.1 2
2.1
Mean vial fill weight (n=5) (g) 5.0 (n=10) 5.1
5.1 5.1 5.1
Iv
n
Mean Total delivered dose
1-i
1.1 1.1 1.2 (n=5) 1
1.2
(n=3) (mg)
0
to
r..)
Mean Delivery rate (n=3)
o
0.1 0.1 0.1 (n=5)
0.1 0.1
(mg/min) (over first two min)
rA
vi
Mean %Efficiency (n=3) 53.8 55.5 56.0 (n=5)
51.4 54.8 k4
o
o
ot
Table 23 (2.5 mg/mL) Test Initial 1 month 3 month 6
month 12 month
0
Storage condition N/A 25 C/60 /0RH 25
C/60%RH 25 C/60%RH 25 C/60%RH No
1-,
Yellow suspension Yellow suspension
Yellow suspension Yellow suspension Yellow suspension
cs
O'
Appearance free from visible free from visible free
from visible free from visible free from visible =P
N?
agglomerates agglomerates agglomerates
agglomerates agglomerates w
1-,
w
pH 6.7 6.7 6.7
6.6 6.7
Mean Assay (n=2) (mg/g) 2.6 2.57 2.64
2.63 2.65
Mean Impurities (n=2) (%LC)
Total 1.02 0.92 0.89
0.98 1
Greatest 0.41 (RRT 1.10) 0.23 (RRT 0.92) 0.39
(RRT 1.10) 0.41 (RRT 1.10) 0.42 (RRT 1.11)
0.20 (RRT 1 11)*
Second greatest 0.23 (RRT 0.92) 0.18 (RRT 1.10) 0.22
(RRT 0.93) 0.23 (RRT 0.93) 0.22 (RRT 0.92)
* P
. . .
.
2
PSD (.un) Dv10- 0 7964 Dv10- 0.7981 Dv10-
0.7943 Dv10-0 7851 Dv10-0.7872 .
Dv50= 1.586 Dv50= 1.569 Dv50=
1.561 Dv50= 1.583 Dv50= 1.589
cs
,,
Dv90= 3.075 Dv90= 3 Dv90=2.98
Dv90=3.099 Dv90=3.119 .
...]
,
Mean FPD (mg) 3 3.1 3.4
3.5 4 .
Mean FPD/mass delivered
0.7 0.7 0.8 0.8 0.9
(n=3) (mg/g)
Mean MMAD (n=3) 6.0 6.1 5.8 6
5.4
Mean GSD (n=3) 1.9 1.9 1.9 2
2.1
Mean Vial Content Uniformity Pass EP
2.9.6 and ' .
N/A N/A
N/A N/A
(n=10) 2.9.40 .
.
Mean vial fill weight (n=5) (g) 5.1 (n=10) 5.2
5.2 5.3 5.3 Iv
n
1-3
Mean Total delivered dose
6 6.2 4.5
5.7 6.5 0
(n=3) (mg)
tO
t,..)
Mean Delivers, rate (n-3)
o
1-,
0.3 0.3 0.2 0.3 0.3
r.n
(mg/min) (over the first min)
'1-
vi
Mean %Efficiency (n=3) 56.3 55.5 41.2 55
56.6 k4
cs
cs
ot
Table 24 (10 mg/mL) Test Initial 1 month 3 month 6
month 12 month
0
Storage condition N/A 25 C/60 /0RH 25 C/60%RH 25
C/60%RH 25 C/60%RH No
1-,
Yellow suspension Yellow suspension Yellow
suspension Yellow suspension Yellow suspension cs
O'
Appearance free from visible free from visible
free from visible free from visible free from
visible =P
N?
agglomerates agglomerates agglomerates
agglomerates agglomerates w
1-,
w
pH 6.7 6.7 6.7
6.6 6.7
Mean Assay (n=2) (mg/mL) 10.24 10.16 10.28
10.23 10.46
Mean Impurities (n=2) (%LC)
Total 0.98 0.92 0.89
0.97 0.99
Greatest 0.40 (RRT 1.11) 0.23 (RRT 0.92) 0.39
(RRT 1.10) 0.41 (RRT 1.10) 0.42 (RRT 1.11)
0.21 (RRT 1 11)*
Second greatest 0.23 (RRT 0.92) 0.18 (RRT 1.10)* 0.22
(RRT 0.93) 0.22 (RRT 0.93) 0.21 (RRT 0.92)
P
. . .
.
2
PSD (iii) Dv10- 0 7884 Dv10- 0.7877 Dv10- 0.7954 Dv10-
0.7756 Dv10- 0.8012 .
Dv50= 1.564 Dv50= 1.55 Dv50= 1.545 Dv50=
1.558 Dv50= 1.566
Dv90= 3.02 Dv-90= 2.965 Dv90=2.909
Dv90=3.059 Dv90=2.976 .
...]
,
Mean FPD (mg) 12.2 12.7 14.2
13.3 14.7 .
Mean FPD/mass delivered
2.9 3 3.1 2.2(n2) 3.3
(n=3) (mg/g)
Mean MMAD (n=3) 6.1 5.9 6 5.7
(n=2) 5.8
Mean GSD (n=3) 1.9 1.9 1.9 2.0
(n=2) 2
.
.
Mean Vial Content Uniformity Pass EP 2.9.6 and
N/A N/A
N/A N/A
(n=10) 2.9.40 .
.
Mean vial fill weight (n=5) (g) 5.2 (n=10) 5.1
5.1 5.1 5.1 Iv
n
1-3
Mean Total delivered dose
25.5 23.4 26.4 22.7 24.4
0
(n=3) (mg)
tO
i..)
Mean Delivers, rate (n-3)
o
1-,
1.3 1.4 1.4 1.2 1.2
r.n
(mg/min) (over the first min)
vi
Mean %Efficiency (n=3) 57.3 52.8 56.7
53.5 54.6 k4
cs
cs
ot
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
38
Example 9 ¨ six month analysis at accelerated conditions (40 C/75% RH)
Accelerated stability studies at 40 C/75% RH were also continued to the sixth
month. The
results of these are shown in Table 25 (for 0.5 mg/mL), Table 26 (for 2.5
mg/mL) and Table
27 (for 10 mg/mL).
(As noted in Example 8, impurity peaks marked with * were observed as a
doublet but had
been observed as a single peak at the initial time point. This is probably due
to a new column
being used which gave better separation and is not believed to be a sign of
degradation.)
The results showed that there was essentially no change in any of the batches
tested at the 6
month time point and confirmed that the RPL554 suspensions for nebulisation
have excellent
stability, being stable for 6 months at 40 C/75%RH.
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
39
Table 25
(0.5 mg/mL) Initial 1 month 3 month 6 month
Test
Storage
N/A 40 C/75%RH 40 C/75%RH 40 C/756/0RH
condition
Pale yellow Pale yellow Pale yellow Pale yellow
suspensions free suspensions free suspensions free
suspensions free
Appearance
from visible from visible from visible from visible
agglomerates agglomerates agglomerates agglomerates
pH 6.7 6.7 6.6 6.6
Mean Assay
0.51 0.51 0.52 0.52
(n=2) (mg/mL)
Mean Impurities
(n=2) (%LC)
Total 0.98 0.9 0.9 0.98
Greatest 0.39 (RRT 1.11) 0.23 (RRT 0.92) 0.37 (RRT 1.10)
0.37 (RRT 1.10)
0.20 (RRT 1.11)*
Second greatest 0.23 (RRT 0.92) 0.22 (RRT 0.92) 0.22 (RRT
0.93)
0.18 (RRT 1.10)*
Dv10= 0.7646 Dv10= 0.7902 Dv10= 0.7749 Dv10= 0.8324
PSD (p.m) Dv50= 1.534 Dv50= 1.56 Dv50= 1.586 Dv50= 1.573
Dv90= 2.999 Dv90= 3.026 Dv90= 3.335 Dv90= 2.899
Mean FPD (mg) 0.4 0.6 0.7 0.6
Mean FPD/mass
delivered (n=3) 0.1 0.2 0.2 0.1
(mg/g)
Mean MMAD
5.4 5.8 5.3 5.5
(n=3)
Mean GSD (n=3) 2 1.9 2 2.1
Mean Vial
Content Pass EP 2.9.6 and
N/A N/A N/A
Uniformity 2.9.40
(n=10)
Mean vial fill
5.0(n=10) 5.1 5.1 5.1
weight (n=5) (g)
Mean Total
delivered dose 1.1 1.1 1.2 1.1
(n=3) (mg)
Mean DelWely
rate (n=3)
0.1 0.1 0.1 0.1
(mg/min) (over
first two min)
Mean c',/0
53.8 55.1 54.3 53.4
Efficiency (n=3)
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
Table 26
(2.5 mg/mL) Initial I month 3 month 6 month
Test
Storage
N/A 40 C/75%RII 40 C/75%RII 40 C/75%RH
condition
Yellow suspension Yellow suspension Yellow suspension Yellow suspension
Appearance free from visible free from visible free
from visible free from visible
agglomerates agglomerates agglomerates
agglomerates
pH 6.7 6.7 6.6 6.6
Mean Assay
2.6 2.58 2.64 2.63
(n=2) (mg,/mL)
Mean Impurities
(n=2) (%LC)
Total 1.02 0.92 0.9 0.99
Greatest 0.41 (RRT 1.10) 0.23 (RRT 0.92) 0.38 (RRT 1.10)
0.40 (RRT 1.10)
0.20 (RRT 1.11)*
Second greatest 0.23 (RRT 0.92) 0.18 (RRT 1.10)* 0.22 (RRT
0.93) 0.23 (RRT 0.93)
Dv10= 0.7964 Dv10= 0.7861 Dv10= 0.7994 Dv10=
0.7997
PSD (gm) Dv50= 1.586 Dv50= 1.563 Dv50= 1.577 Dv50= 1.614
Dv90= 3.075 Dv90= 3.023 Dv90=3.058
Dv90=3.183
Mean FPD (mg) 3 3.2 3.6 3.6
Mean FPD/mass
delivered (n=3) 0.7 0.8 0.8 0.8
(mg/g)
Mean MMAD
6 6 5.7 5.9
(n=3)
Mean GSD (n=3) 1.9 1.9 2 2
Mean Vial
Content Pass EP 2.9.6 and
N/A N/A N/A
Uniformity 2.9.40
(n=10)
Mean vial till
5.1 (n=10) 5.3 5.3 5.3
weight (n=5) (g)
Mean Total
delivered dose 6 6.1 5.6 6.4
(n=3) (mg)
Mean Delivery
rate (n=3)
0.3 0.3 0.3 0.4
(mg/min) (over
the first min)
Mean %
56.3 56.1 50.5 56.3
Efficiency (n=3)
CA 02959943 2017-03-01
WO 2016/042313
PCT/GB2015/052668
41
Table 27
(10 mg/mL) Initial 1 month 3 month 6 month
Test
Storage
N/A 40 C/75%RH 40 C/75%RH 40 C/75%1214
condition
Yellow suspension Yellow suspension Yellow suspension Yellow suspension
Appearance free from visible free from visible free
from visible free from visible
agglomerates agglomerates agglomerates
agglomerates
pH 6.7 6.7 6.6 6.6
Mean Assay
10.24 10.1 10.41 10.37
(n=2) (ng/mL)
Mean Impurities
(n=2) (%LC)
Total 0.98 0.97 0.89 0.98
Greatest 0.40 (RRT 1.11) 0.23 (RRT 0.92) 0.38 (RRT 1.10)
0.40 (RRT 1.10)
0.21 (RRT 1.11)*
Second greatest 0.23 (RRT 0.92)
0.18 (RRT 1.10)* 0.23 (RRT 0.93) 0.23 (RRT 0.93)
Dv10= 0.7884 Dv10= 0.7906 Dv10= 0.7882 Dv10=
0.7757
PSD (gm) Dv50= 1.564 Dv50= 1.554 Dv50= 1.565 Dv50= 1.553
Dv90= 3.02 Dv90= 2.971 Dv90= 3.024 Dv90= 3.015
Mean FPD (mg) 12.2 13.1 14.5 13.4
Mean FPD/mass
delivered (n=3) 2.9 3.1 3.4 3.1
(mg/g)
Mean MMAD
6.1 5.8 5.8 5.7
(n=3)
Mean GSD (n=3) 1.9 1.9 2 2
Mean Vial
Content Pass EP 2.9.6 and
N/A N/A N/A
Uniformity 2.9.40
(n=10)
Mean vial fill
5.2 (n=10) 5.2 5.1 5.1
weight (n=5) (g)
Mean Total
delivered dose 25.5 24.1 26.9 22.8
(n=3) (mg)
Mean Delively
rate (n=3)
1.3 1.3 1.2 1.3
(mg/min) (over
the first min)
Mean %
57.3 54.3 56.2 51.6
Efficiency (n=3)