Note: Descriptions are shown in the official language in which they were submitted.
CA 02963740 2017-04-05
WO 2016/055780 PCT/GB2015/052920
1
SPIRO-INDOLINES FOR THE TREATMENT AND PROPHYLAXIS OF RESPIRATORY SYNCYTIAL
VIRUS INFECTION (RSV)
Field of the Invention
The present invention relates to benzimidazole compounds and to their use in
treating or
preventing a respiratory syncytial virus (RSV) infection
Background to the Invention
RSV is a negative-sense, single-stranded RNA virus of the Paramyxoviridae
family.
RSV is readily transmitted by secretions from an infected person via surfaces
or hand-
to-hand transfer. Unlike influenza, it is not transmitted by small-particle
aerosols.
Following successful inoculation, the incubation period is between four and
six days
during which time the virus spreads from the nasopharynx to the lower
respiratory tract
by fusion of infected with uninfected cells and by sloughing of the necrotic
epithelium.
In infants, coupled with increased mucus secretion and oedema, this can lead
to mucus
plugging causing hyper-inflation and collapse of distal lung tissue indicative
of
bronchiolitis. Hypoxia is common and the ability to feed is often impaired
because of
respiratory distress In RSV pneumonia, inflammatory infiltration of the
airways
consists of mononuclear cells and is more generalised, with involvement of the
bronchioles, bronchi and alveoli. The duration and degree of viral shedding
has been
found to correlate with the clinical signs and severity of disease.
RSV is the leading cause of serious respiratory tract infections in infants
and young
children throughout the world. The highest morbidity and mortality occurs in
those
born prematurely and for those with chronic lung or heart disease, although
many
infants hospitalised for RSV infection are otherwise healthy. Severe RSV
infection in
infancy can lead to several years of recurrent wheezing and is linked to the
later
development of asthma.
RSV is also a major cause of morbidity and mortality in the elderly and in
immunocompromised children and adults as well as those with chronic
obstructive
pulmonary disease (COPD) and congestive heart failure (CHF)
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
2
RSV has a seasonal incidence; it is highly predictable and occurs in the
winters of both
hemispheres, from September to May in Europe and North America, peaking in
December and January, and can occur throughout the year in tropical countries.
It
affects >90% of infants and young children by the age of two years and as
natural
immunity is short-lived; many will be re -infected each year. As with
influenza, in
elderly people, RSV causes around 10% of winter hospitalisations with an
associated
mortality of 10%.
Current anti-RSV treatment involves the use of a monoclonal antibody to RSV,
called
palivizumab. Such use of palivizumab is a prophylactic, rather than
therapeutic,
treatment of RSV. Although this antibody is often effective, its use is
restricted to
preterm infants and infants at high risk. Indeed, its limited utility means
that it is
unavailable for many people in need of anti-RSV treatment. There is therefore
an urgent
need for effective alternatives to existing anti-RSV treatment.
Additionally, several compounds have been proposed as inhibitors of RSV,
including
benzimidazole-based compounds. For example, K D Combrink et al., Bioorganic &
Medicinal Chemistry Letters, 17 (2007), 4784-4790 discloses the compound BMS-
433771 and variants thereof. Further benzimidazole-based compounds are
disclosed in
WO-02/062290, WO-03/053344 and WO-10/103306.
WO 2013/068769 discloses spirocyclic compounds having activity against RSV.
However there exists a need to identify further compounds, and in particular
compounds
having favourable pharmacokinetic profiles
Summary of the Invention
It has now been found that a novel series of benzimidazoles are active as RSV
inhibitors
with favourable pharmacokinetics.
Accordingly, the present invention provides a compound which is a
benzimidazole of
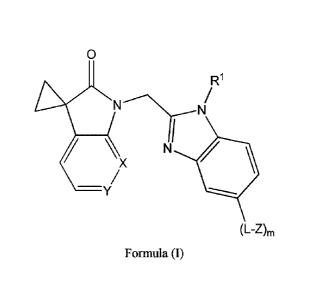
formula (I):
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
3
0
R1
iC\ X
11110
-y
(L-Z),,
Formula (I)
wherein:
one of X and Y is an N atom or a substituted C atom, and the other is
CH,
- L is a single bond, C1.3 alkylene, C2_3 alkenylene or C2_3 alkynylene,
- Rl is C1_6 alkyl, C2_6 alkenyl, C2_6 alkynyl, 3- to 10-membered cycloa141
5- to 10-membered heterocyclyl or 5- to 12- membered heteroaryl, each
of which is unsubstituted or substituted;
- Z is halo, Ci_6 haloalkyl, nitro, -CN, -N(R2)2, -0R2, -SR2, -S(=0)R2, or -
S(=0)2R2;
- each R2 is independently hydrogen, C1_6 alkyl, C2_6 alkenyl or C2-6
alkynyl, wherein said alkyl, alkenyl and alkynyl groups are
unsubstituted or substituted; and
- m is 0 or 1;
or a pharmaceutically acceptable salt theteof.
Detailed Description of the Invention
When any group, ring, substituent or moiety defined herein is substituted, it
is typically
substituted by Q as defined below.
A C1.6 alkyl group or moiety is linear or branched. A C1_6 alkyl group is
typically a C14
alkyl group, or a C44 alkyl group. Examples of C1_6 alkyl groups and moieties
include
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
4
methyl, ethyl, n-propyl, i-propyl, n-butyl, i-butyl, t-butyl, n-pentyl, i-
pentyl (i.e. 3-
methylbut-1-y1), t-pentyl (i.e. 2-methylbut-2-y1), neopentyl (i.e. 2,2-
dimethylpropan-1-
y1), n-hexyl, i-hexyl (i.e. 4-methylpentan-1-y1), t-hexyl (i.e. 3-methylpentan-
3-y1) and
neopentyl (i.e. 3,3-dimethylbutan-1-y1). For the avoidance of doubt, where two
alkyl
moieties are present in a group, the alkyl moieties may be the same or
different. A C1-6
alkyl group is unsubstituted or substituted, typically by one or more groups Q
as defined
below. For example, a Ci_6 alkyl group is unsubstituted or substituted by 1, 2
or 3
groups Q as defined below.
Q is halo, nitro, -CN, 01-1, CI-6 alkoxy, CI _6 hydroxyalkyl, CI.6 alkylthio,
C1_6 haloalkyl,
C1-4 haloalkoxy, -CO2R", -NR'"2, -SR", -S(=0)R'", -S(=0)2R", C3-Cio
cycloalkyl, 5 to
10-membered heterocyclyl, 5- to 12-membered aryl or 5- to 12-membered
heteroaryl,
wherein each R" is independently selected from H, C1-6 alkyl, C3-10
cycloalkyl, 5 to 10-
membered heterocyclyl, 5- to 12-membered aryl and 5- to 12-membered
heteroaryl.
A C1.3 alkylene group or moiety is an unsubstituted or substituted, linear or
branched,
saturated divalent aliphatic hydrocarbon group or moiety containing 1 to 3
carbon
atoms. Examples include methylene, ethylene, n-propylene and i-propylene
groups and
moieties. When the alkylene group is substituted it is typically substituted
by a group Q
as defined above.
A C2.6 alkenyl group is an unsubstituted or substituted, linear or branched
hydrocarbon
radical of two to six carbon atoms with at least one site of unsaturation,
i.e., a carbon-
carbon sp2 double bond. An alkenyl group may have "cis" or "trans"
orientation, or
alternatively "E" or "Z" orientation. Typically it is a C24 alkenyl group or a
C4_6 alkenyl
group. Examples include ethyl enyl or vinyl (-CH=CH2), and allyl (-CH2CH=CH2).
When the alkenyl group is substituted it is typically substituted by a group Q
as defined
above.
A C7-3 alkenylene group or moiety is linear or branched, unsaturated divalent
aliphatic
hydrocarbon group or moiety containing two or three carbon atoms with at least
one
carbon-carbon sp2 double bond. An alkenylene group may have "cis" or "trans"
orientation, or alternatively "E" or "Z" orientation. Examples include -CH=CH-
,
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
-CH=CHCH2- and -CH2CH=CH- groups and moieties.
A C2-6 alkynyl group is an unsubstituted or substituted, linear or branched
hydrocarbon
radical of two to six carbon atoms with at least one site of unsaturation,
i.e., a carbon-
5 .. carbon sp triple bond. Typically it is a C24 alkynyl group or a C4-6
alkynyl group. An
alkynyl group may have "cis" or "trans" orientation, or alternatively "E" or
orientation. Examples include ethynyl (-CCH) or propynyl (propargyl, -CH2CCH).
When an alkynyl group is substituted it is typically substituted by one or
more groups Q
as defined above
A C2.3 alkynylene group is a linear, unsaturated divalent aliphatic
hydrocarbon group or
moiety containing two or three carbon atoms with one carbon-carbon sp triple
bond. An
alkynylene group may have "cis" or "trans" orientation, or alternatively "E"
or "Z"
orientation. Examples include -CCCH2- and -CH2CC- groups and moieties.
A C1.6 alkoxy group is linear or branched. It is typically a C1-4 alkoxy
group, for
example a methoxy, ethoxy, propoxy, i-propoxy, n-propoxy, n-butoxy, sec-butoxy
or
tert-butoxy group. A C1_6 alkoxy group is unsubstituted or substituted,
typically by one
or more groups Q as defined.
A C1.6 alkylthio group is linear or branched. It is typically a Ci4 alkylthio
group, for
example a methylthio, ethylthio, propylthio, i-propylthio, n-propylthio, n-
butylthio,
sec-butylthio or tert-butylthio group. A C1.6 alkyltho group is unsubstituted
or
substituted, typically by one or more groups Q as defined.
A halogen or halo group is F, Cl, Br or I. Preferably it is F, Cl or Br. A
Ci_6 alkyl
group substituted by halogen may be denoted "Cif, haloalkyl", which means a
Ci_6 alkyl
group as defined above in which one or more hydrogens is replaced by halo.
Likewise a
C1-6 alkoxy group substituted by halogen may be denoted "C1-6 haloalkoxy",
which
means a C1_6 alkoxy group as defined above in which one or more hydrogens is
replaced
by halo. Typically, C1-6 haloalkyl or C1_6 haloalkoxy is substituted by 1, 2
or 3 said
halogen atoms. Haloalkyl and haloalkoxy groups include perhaloalkyl and
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
6
perhaloalkoxy groups such as -CX3 and -OCX3 wherein X is a halogen, for
example -
CF3 -CC13 -0CF3 and -OCC13.
A C1.6 hydroxyalkyl group is a C1_6 alkyl group as defined above, substituted
by one or
more OH groups. Typically, it is substituted by one, two or three OH groups.
Preferably, it is substituted by a single OH group.
A 5- to 12-membered aryl group is an aromatic carbocyclic group containing
from 5 to
12 carbon atoms, for instance from 6 to 10 carbon atoms, such as 6 or 10
carbon atoms
It is monocyclic or a fused bicyclic ring system in which an aromatic ring is
fused to
another aromatic carbocyclic ring. Examples of a 5-to 12-membered aryl group
include
phenyl and naphthalenyl. When substituted, an aryl group is typically
substituted by CI.
4 alkyl or a group Q as defined above, for instance by 1, 2 or 3, groups
selected from a
C1-4 alkyl group and a group Q as defined above
An aralkyl group is an aryl group, as defined above, attached to an alkyl
group, as
defined above. Examples include benzyl.
A C3_10 cycloalkyl group is a saturated hydrocarbon ring having from 3 to 10
carbon
atoms. A C3_10 cycloalkyl group may be, for instance, C3-C7 cycloalkyl such as
cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, or cycloheptyl. Typically it
is C3-C6
cycloalkyl, for example cyclopropyl, cyclobutyl or cyclopentyl. In one
embodiment it is
cyclopropyl. A C3-10 cycloalkyl group is unsubstituted or substituted,
typically by one
or more groups Q as defined above.
A 5- to 12- membered heteroaryl group or moiety is a 5- to 12-membered
aromatic
heterocyclic group which contains 1, 2, 3, or 4 heteroatoms selected from 0, N
and S. It
is monocyclic or bicyclic. Typically it contains one N atom and 0, 1, 2 or 3
additional
heteroatoms selected from 0, S and N. It may be, for example, a 5- to 7-
membered
heteroaryl group, for instance a 5- or 6-membered N-containing heteroaryl
group.
Examples include pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, furanyl,
thienyl,
pyrazolidinyl, pyrrolyl, oxadiazolyl, oxazolyl, isoxazolyl, thiazolyl,
thiadiazolyl,
imidazolyl and pyrazolyl groups. Furanyl, thienyl, pyridyl and pyrimidyl
groups are
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
7
preferred. When substituted, a heteroaryl group is typically substituted by
one or more,
e.g. 1, 2 or 3, groups selected from C14 alkyl and a group Q as defined above.
A 5- to 10-membered heterocyclyl moiety is a monocyclic or bicyclic non-
aromatic,
saturated or unsaturated C5-10 carbocyclic ring, in which at least one, for
example 1, 2 or
3, carbon atoms in the ring are replaced with an atom or group selected from
0, S, SO,
SO2, CO and N. Typically, it is a saturated C540 ring in which 1, 2 or 3 of
the carbon
atoms in the ring are replaced with an atom or group selected from 0, S, SO2,
CO and
NH. More typically it is a monocyclic ring, preferably a monocyclic C5-C6
ring.
Examples include piperidyl, piperidin-2,6-dionyl, piperidin-2-onyl,
piperazinyl,
morpholinyl, thiomorpholinyl, S,S-dioxothiomorpholinyl, 1,3-dioxolanyl,
pyrrolidinyl,
imidazol-2-onyl, pyrrolidin-2-onyl, tetrahydrofuranyl and tetrahydropyranyl
moieties.
For the avoidance of doubt, although the above definitions of heteroaryl and
heterocyclyl groups refer to an "N" atom which can be present in the ring, as
will be
evident to a skilled chemist the N atom will be protonated (or will carry a
substituent as
defined above) if it is attached to each of the adjacent ring atoms via a
single bond. Such
protonated forms are embraced within the present definitions of heteroaryl and
heterocyclyl groups.
Typically, when one of X and Y is a substituted C atom, it is a C atom
substituted with
Q as defined above. More typically, when one of X and Y is a substituted C
atom, it is
a C atom substituted with halo, nitro, -CN, OH, C1_6 alkoxy, C1-6
hydroxyalkyl, C1-6
alkylthio, Ci_6 haloalkyl, C14 haloalkoxy, -CO2R", -NR"2, -S(=0)R1", or -
S(=0)2R", wherein each R" is independently selected from H and C1-6 alkyl.
Preferably, when one of X and Y is a substituted C atom, it is a C atom
substituted with
halo, nitro, -CN, OH, C14 alkoxy, C14 hydroxyalkyl, C1_4 alkylthio, C14
haloalkyl, C1-4
haloalkoxy, -CO2R", -NR"'2, -SR", -S(=0)R", or -S(=0)2R", wherein each R" is
independently selected from H and Ci_4 alkyl. More preferably, when one of X
and Y is
a substituted C atom, it is a C atom substituted with halo, nitro, -CN, OH.
Still more
preferably, when one of X and Y is a substituted C atom, it is a C atom
substituted with
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
8
a halo atom. Most preferably, when one of X and Y is a substituted C atom, it
is a C
atom substituted with a fluorine atom.
Thus, in some embodiments, one of X and Y is an N atom or a C atom substituted
with
a halogen atom, and the other is CH. Typically, in such embodiments one of X
and Y is
an N atom or C substituted with a fluorine atom, and the other is CH.
In some embodiments, RI is C1_6 alkyl, C2_6 alkenyl, C2-6 alkynyl, 5- to 10-
membered
heterocyclyl or 5-to 12- membered heteroaryl, each of which is unsubstituted
or
substituted.
Typically, when RI is a heterocyclyl or heteroaryl group, said heterocyclyl or
heteroaryl
group contains one, two or three heteroatoms, more typically one or two
heteroatoms,
and preferably one heteroatom. Typically, said heteroatoms are selected from
N, 0, and
S. More typically heteroatoms are selected from N and 0. Preferably, said
heteroatoms
are 0.
Typically, when Rl is substituted, it is substituted with one or more groups
(e.g. 1, 2, 3
or 4 groups) Q as defined above. More typically when R is substituted, it is
substituted
with one or more groups (e.g. 1, 2, 3 or 4 groups) selected from halo, nitro, -
CN, OH,
C1-6 alkoxy, C1-6 hydroxyalkyl, C1_6 alkylthio, Co haloalkyl, C14 haloalkoxy, -
CO2R", -
NR'"2, -SR", -S(=0)R"', and -S(=0)2R"', wherein each R"' is independently
selected
from H and C1_6 alkyl.
Still more typically, when R1 is substituted, it is substituted with one or
more groups
(e.g. 1, 2, 3 or 4 groups) selected from halo, nitro, -CN, 01-1, Ci-4 alkoxy,
C1-4
hydroxyalkyl, C1_4 alkylthio, C14 haloalkyl, C1-4 haloalkoxy, -CO,R", -NR"2, -
SR"', -
S(=0)R", and -S(=0)2R'", wherein each R" is independently selected from H and
C1-4
alkyl.
Preferably, when RI is substituted, it is substituted with one or more groups
(e.g. 1, 2, 3
or 4 groups) selected from halo, nitro, -CN, OH. More preferably, when Rl is
substituted, it is substituted with one or more (e.g. 1, 2, 3 or 4, typically
3) halo atoms.
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
9
Still more preferably, when It' is substituted, it is substituted with one or
more (e.g. 1,
2, 3 or 4, typically 3) fluorine atoms.
Preferably, in some embodiments, when le is substituted it is substituted with
one or
more groups (e.g. 1, 2, 3 or 4 groups) selected from halo, nitro, -CN, OH.
More
preferably, when le is substituted, it is substituted with one or more (e.g.
1, 2, 3 or 4,
typically 3) halo atoms or one or more (e.g. 1, 2, 3 or 4, typically 1) -OH
groups. Still
more preferably, when le is substituted, it is substituted with one or more
(e.g. 1, 2, 3 or
4, typically 3) fluorine atoms or one or more (e.g. 1, 2, 3 or 4, typically 1)
-OH groups.
Typically, le is C3-6 alkyl, C3_6 alkenyl, 5- or 6-membered heterocyclyl or 5-
or 6-
membered heteroaryl, each of which is unsubstituted or substituted. More
typically R1
is C3.6 alkyl, C3.6 alkenyl, or 5- or 6-membered heterocyclyl each of which is
unsubstituted or substituted (e.g. substituted with 1 or more halogen atoms).
Typically, in some embodiments, le is C3-6 alkyl, C3-6 alkenyl, 5- or 6-
membered
cycloalkyl, 5- or 6-membered heterocyclyl or 5- or 6- membered heteroaryl,
each of
which is unsubstituted or substituted. More typically le is C3_6 alkyl, C3.6
alkenyl, 5- or
6-membered cycloalkyl or 5- or 6-membered heterocyclyl each of which is
unsubstituted or substituted (e.g. substituted with 1 or more groups each of
which is
independently selected from a halogen atom and an -OH group).
Preferably, le is C3_6 alkyl, or 5- or 6-membered heterocyclyl each of which
is
unsubstituted or substituted (e.g. substituted with 1 or more halogen atoms).
More
preferably is C5_6 alkyl which is unsubstituted or substituted (e.g.
substituted with 1,
2, 3 or 4 halogen atoms), or unsubstituted 6-membered heterocyclyl. Still more
preferably le is isopentyl or n-butyl which is unsubstituted or substituted
with 3
fluorine atoms, or le is unsubstituted tetrahydropyranyl. Most preferably Ill
is
unsubstituted isopentyl, n-butyl which is substituted with 3 fluorine atoms,
or le is
unsubstituted tetrahydropyranyl.
Preferably, in some embodiments, le is C3-6 alkyl, 5- or 6-membered cycloalkyl
or 5- or
6-membered heterocyclyl each of which is unsubstituted or substituted (e.g.
substituted
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
with 1 or more groups each of which is independently selected from a halogen
atom and
an -OH group). More preferably le is C5-6 alkyl which is unsubstituted or
substituted
(e.g. substituted with 1, 2, 3 or 4 halogen atoms), unsubstituted 6-membered
heterocyclyl, or substituted 6-membered cycloalkyl (e.g. substituted with 1,
2, 3 or 4
5 -OH groups). Still more preferably Rl is unsubstituted tetrahydropyranyl,
hydroxycyclohexyl, or isopentyl or n-butyl which isopentyl or n-butyl group is
unsubstituted or substituted with 3 fluorine atoms. Most preferably RI is
unsubstituted
tetrahydropyranyl, hydroxycyclohexyl, unsubstituted isopentyl, or n-butyl
which is
substituted with 3 fluorine atoms.
Typically, L is a single bond or C1_3 alkylene. Preferably, L is a single bond
or Ci
alkylene.
Typically, Z is halo, C1_6 haloalkyl, nitro, -CN, -N(R2)2, -0R2, or -SR2. More
typically
Z is halo, C1_6 haloalkyl (e.g. C1.4 haloalkyl, C1_2 haloalkyl or C1
haloalkyl), -N(R2)2 or -
OR2. Still more typically Z is -N(R2)2, -0R2, a halogen atom or C1-6
haloalkyl.
Preferably Z is halo or -N(R2)2. More preferably Z is fluoro, chloro, NH2,
NHCH3, or
N(CH3)2. Still more preferably Z is chloro or NH2.
Typically, when R2 is a substituted alkyl, alkenyl or alkynyl group, said
alkyl, alkenyl
or alkynyl group is substituted with one or more groups (e.g. 1, 2, 3 or 4
groups)
selected from halo, nitro, -CN, OH, C1_4 alkoxy, C14 hydroxyalkyl, C1_4
alkylthio, C1-4
haloalkyl, C14 haloalkoxy, -CO2R"', -NR"'2, -SR", -S(=0)R", and -S(=0)2R"',
wherein
each R"' is independently selected from H and C1_4 alkyl. More typically, when
R2 is a
substituted alkyl, alkenyl or alkynyl group, said alkyl, alkenyl or alkynyl
group is
substituted with one or more groups (e.g. 1, 2, 3 or 4 groups) selected from
halo, nitro, -
CN, OH.
Typically, each R2 is independently hydrogen, C14 alkyl, C2-4 alkenyl or C24
alkynyl,
wherein said alkyl, alkenyl and alkynyl groups are unsubstituted or
substituted.
Preferably each R2 is independently hydrogen or C14 alkyl, wherein said alkyl
group is
unsubstituted or substituted. More preferably each R2 is independently
hydrogen or
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
11
unsubstituted C1-4 alkyl. Still more preferably each R2 is independently
hydrogen or
unsubstituted methyl. Most preferably each R2 is hydrogen.
Typically, m is 1.
In certain preferred embodiments, L is a direct bond and Z is a halogen atom
(e.g. Z is a
chlorine atom). In other preferred embodiments, L is a Ci alkylene group and Z
is
-N(R2)2 (e.g. Z is -NH2).
In one particularly preferred embodiment of the compounds of the invention, in
formula
(I):
- one of X and Y is an N atom or C substituted with a fluorine atom,
and the other
is CH;
- R1 is C5_6 alkyl, which is unsubstituted or substituted with one,
two, three or four
fluorine atoms, or unsubstituted tetrahydropyan
- m is 1
- L is a single bond or CI alkylene; and
- Z isl\--H2 or a chlorine atom.
In another particularly preferred embodiment of the compounds of the
invention, in
formula (1):
- one of X and Y is an N atom or C substituted with a fluorine atom, and
the other
is CH;
- It' is C6 alkyl, which is unsubstituted or substituted with one, two,
three or four
fluorine atoms, R is cyclohexyl which is unsubstrituted or substituted with
one
or two -OH groups, or RI is unsubstituted tetrahydropyan;
- m is 1
- L is a single bond or CI alkylene; and
- Z is1X-H2 or a chlorine atom.
Specific compounds of the invention include:
- [5-(aminomethyl)-1-(4,4,4-trifluorobuty1)-1H-1,3-benzodiazol-2-
yl]methyl} -
6'-fluoro-1',2'-dihydrospiro[cyclopropane-1,31-indole1-2'-one;
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
12
- 1'-((5-(aminomethyl)-1-(4,4,4-trifluorobuty1)-1H-benzordlimidazol-2-
y1)methyl)spiro[cyclopropane-1,3'-pyrrolo[2,3-b]pyridin]-21(111-1)-one,
- 1'-((5-chloro-1-(tetrahydro-2H-pyran-4-y1)-1H-benzo[d]imidazol-2-
yl)methyl)spiro[cyclopropane-1,3'-pyrrolo[2,3-c]pyridin]-21(1111)-one;
- 114(5-(aminomethyl)-1-isopentyl-1H-benzo[d]imidazol-2-
yl)methyl)spiro[cyclopropane-1,3'-pyrrolo[2,3-b]pyridin]-2'(PH)-one,
- 1'-((5-(aminomethyl)-1-isopenty1-1H-benzo[d]imidazol-2-
yl)methyl)spiro[cyclopropane-1,3'-pyrrolo[2,3-c]pyridin]-21(111-1)-one; and
- 1'-((5-(aminom ethyl )-1-(tetrahydro-2H-pyran-4-y1)-1H-
benzo[d]imidazol-2-
yl)methyl)-6'-fluorospiro[cyclopropane-1,31-indolin]-21-one,
and the pharmaceutically acceptable salts thereof
In certain embodiments, specific compounds of the invention include:
- [5-(aminomethyl)-1-(4,4,4-trifluorobuty1)-1H-1,3 -b enzodiazol-2-
Am ethy1}-
6'-fluoro-1',2'-dihydrospiro[cyclopropane-1,3'-indole]-2'-one;
- 1'-45-(aminomethyl)-1-(4,4,4-trifluorobuty1)-1H-benzo[d]imidazol-2-
y1)methyl)spiro[cyclopropane-1,3'-pyrrolo[2,3-b]pyridin]-2'(1'H)-one,
- 1'-((5-chloro-1-(tetrahydro-2H-pyran-4-y1)-1H-benzo[d]imidazol-2-
yl)methyl)spiro[cyclopropane-1,3'-pyrrolo[2,3-c]pyridin]-21(11H)-one,
- 1'-((5-(aminomethyl)-1-isopenty1-1H-benzo[d]imidazol-2-
y1)methyl)spiro[cyclopropane-1,3'-pyrrolo[2,3-b]pyridin]-2'(i'H)-one,
- 1'-((5-(aminomethyl)-1-isopenty1-1H-benzo[d]imidazol-2-
y1)methyl)spiro[cyclopropane-1,3'-pyrrolo[2,3-c]pyridin]-2'(l 'H)-one; and
- 1'-((5-(aminomethyl)-1-(tetrahydro-2H-pyran-4-y1)-1H-benzo[d]imidazol-2-
yl)m ethyl)-6'-fluorospiro[cyclopropane-1,3'-indoli n]-2'-one;
- 1'-45-(Aminomethyl)-1-((1R,4R)-4-hydroxycyclohexyl)-1H-benzo[d]imidazol-
2-y1)methyl)-6'-fluorospiro[cyclopropane-1,31-indolin]-2'-one
and the pharmaceutically acceptable salts thereof
Preferred compounds of the invention include 1'-115-(aminomethyl)-1-(4,4,4-
tri fluorobuty1)-1H-1,3 -benzodi azol-2-yl] methyl 1-6'fluoro-1',2'-
dihydrospiro[cyclopropane-1,3'-indole]-2'-one.
CA 02963740 2017-04-05
WO 2016/055780 PCT/GB2015/052920
13
In some embodiments, when -L-Z is C1_6alkoxy, halogen, trifluoromethyl or
cyano, R1
is not C1-6 alkyl substituted by Itl ,
wherein RI' is selected from C1_6 alkylsulfonylphenyl, thietan-3-yl,
dioxothietan-3-yl, oxetan-3-yl, aminooxetan-3-yl, hydroxy, C1_6 alkylsufinyl,
0 NH
µ,
(3-CS Ci_6 alkyl
trifluoromethyl-C1-6alkylene-aminocarbonyloxy,
Ci_o alkyl
\1<
0
alkyl
and S09R108;
wherein 1V 8 is Ci_6 alkyl, cycloalkyl, C1-6 alkylcarbonylamino, C1-6
alkylamino
0
N /NH
di-C16alkylamino, amino, morpholinyl, pyrrolidinyl, piperazinyl, 1¨\
NH
1-6
Ci_6alkyl
or C1-6 alkylene-COR1 9, wherein
Itl 9 is C1_6 alkoxy, amino, hydroxy, cycloalkylsulfonylamino,
cycloalkylsulfonylamino(C1-6 alkyl) C1-6 alkylsulfonylamino(C1-6 alkyl), or C2-
6 alkyl-
wherein R11 is hydrogen, Rill is hydrogen, C1_6 alkoxycarbonyl, C1_6
alkylcarbonyl, C1_6 alkylsulfonyl, or hydroxy-C1_6 alkyl, or RH and R",
together with
the nitrogen atom to which they are attached, form_
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
14
0
0
\ S
\õ.0
i_N¨ ¨N S ¨ ¨N NH
0
0
NH
or --N\ ___________ , which is unsubstituted or substituted by hydroxy, C1-6
alkylcarbonyl or C1_6 alkyl sulfonyl.
In some embodiments, when -L-Z is C1-6alkoxy, halogen, trifluoromethyl or
cyano, RI
is C2.6 alkenyl, C2_6 alkynyl, 5- to 10-membered heterocyclyl or 5- to 12-
membered
heteroaryl, each of which is unsubstituted or substituted (e.g. substituted by
one or more
groups Q as defined above), or Rl is C1.6 alkyl which is unsubstituted or
substituted by
one or more groups (e.g. 1, 2, 3 or 4 groups) selected from halo, nitro, -CN,
C1_6 alkoxy,
C1-6 alkylthio, C1-6 haloalkyl, C1-4 haloalkoxy, -0O21r, -NR"2, -SR"', C3-C10
cycloalkyl,
5 to 10-membered heterocyclyl, or 5-to 12-membered heteroaryl, wherein each R"
is
independently selected from H, C1_6 alkyl, C3-10 cycloalkyl, 5 to 10-membered
heterocyclyl, 5- to 12-membered aryl and 5- to 12-membered heteroaryl.
Typically, in
such embodiments, when -L-Z is C1_6 alkoxy, halogen, trifluoromethyl or cyano,
RI is 5-
to 10-membered heterocyclyl or 5-to 12- membered heteroaryl, each of which is
unsubstituted or substituted (e.g substituted by one or more groups Q as
defined
above), or R' is a Ci_6 alkyl, C2_6 alkenyl or C2-6 alkynyl group, which
alkyl, alkenyl or
alkynyl group is unsubstituted or substituted by one or more groups (e.g. 1,
2, 3 or 4
groups) selected from halo, nitro, -CN, C1_6 alkoxy, C1_6 alkylthio, C1-6
haloalkyl, C1-4
haloalkoxy, -CO2R", -NR1"2, -SR", C3-Cw cycloalkyl, 5 to 10-membered
heterocyclyl,
or 5-to 12-membered heteroaryl, wherein each R" is independently selected from
H,
C1_6 alkyl, C3-10 cycloalkyl, 5 to 10-membered heterocyclyl, 5- to 12-membered
aryl and
5- to 12-membered heteroaryl.
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
In some embodiments, -L-Z is not C1-6 alkoxy, halogen, trifluoromethyl or
cyano. For
example, in some embodiments Z is nitro, -N(R2)2, -SR2, -S(=0)R2, or -
S(=0)2R2, and L
is as defined above.
5 In some embodiments, RI is not C1-6 alkyl substituted by R100, wherein
RH)" is as
defined above. For example, in some embodiments, RI is C2-6 alkenyl, C2-6
alkynyl, 5-
to 10-membered heterocyclyl or 5-to 12- membered heteroaryl, each of which is
unsubstituted or substituted (e.g. substituted by one or more groups Q as
defined
above), or R' is C1-6 alkyl which is unsubstituted or substituted by one or
more groups
10 (e.g. 1, 2, 3 or 4 groups) selected from halo, nitro, -CN, Ci_6 alkoxy,
C1_6 alkylthio, C1-6
haloalkyl, C14 haloalkoxy, -CO2R", -NR"2, -SW", C3-Cio cycloalkyl, 5 to 10-
membered
heterocyclyl, or 5- to 12-membered heteroaryl, wherein each R" is
independently
selected from H, Ci alkyl, C3-10 cycloalkyl, 5 to 10-membered heterocyclyl, 5-
to 12-
membered aryl and 5- to 12-membered heteroaryl.
In some embodiments, when ¨L-Z is chloro, RI is not C1-6 alkyl substituted by -
S(=0)2R", wherein R" is H or C1_4 alkyl.
In some embodiments, when -L-Z is chloro, R1 is C26 alkenyl, C2_6 alkynyl, 5-
to 10-
membered heterocyclyl or 5- to 12- membered heteroaryl, each of which is
unsubstituted or substituted (e.g. substituted by one or more groups Q as
defined
above), or R' is Ci-6 alkyl which is unsubstituted or substituted by one or
more groups
(e.g. 1, 2, 3 or 4 groups) selected from halo, nitro, -CN, OH, C1_6 alkoxy, C1-
6
hydroxyalkyl, C1_6 alkylthio, C1_6 haloalkyl, C14 haloalkoxy, -CO2R", -NR"2, -
SW", -
S(=0)R", C3-Cm cycloalkyl, 5 to 10-membered heterocyclyl, 5- to 12-membered
aryl
or 5- to 12-membered heteroaryl, wherein each R" is independently selected
from H,
C1-6 alkyl, C3-10 cycloalkyl, 5 to 10-membered heterocyclyl, 5- to 12-membered
aryl and
5- to 12-membered heteroaryl.
In some embodiments, ¨L-Z is not chloro. For example, in some embodiments, Z
is F,
Br, I, C1_6 haloalkyl, nitro, -CN, -N(R2)2, -0R2, -SR2, -S(=0)R2, or
¨S(=0)2R2, and L is
as defined above. Typically, in such embodiments, Z is C1_6 haloalkyl, nitro, -
CN, -
N(R2)2, -0R2, -5R2, -S(=0)R2, or ¨S(=0)2R2, and L is as defined above.
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
16
In some embodiments, RI is not C1_6 alkyl substituted by -S(=0)2R", wherein
R"' is H
or C14 alkyl. For example, in some embodiments, R.' is C2-6 alkenyl, C2-6
alkynyl, 5- to
10-membered heterocyclyl or 5-to 12- membered heteroaryl, each of which is
unsubstituted or substituted (e.g. substituted by one or more groups Q as
defined
above), or Rl is C1-6 alkyl which is unsubstituted or substituted by one or
more groups
(e.g. 1, 2, 3 or 4 groups) selected from halo, nitro, -CN, OH, C1-6 alkoxy, C1-
6
hydroxyalkyl, C1_6 alkylthio, C1_6 haloalkyl, C14 haloalkoxy, -CO2R", -NR"2, -
SR.", -
S(=0)R", C3-C10 cycloalkyl, 5 to 10-membered heterocyclyl, 5- to 12-membered
aryl
or 5-to 12-membered heteroaryl, wherein each R" is independently selected from
H,
C1_6 alkyl, C3-10 cycloalkyl, 5 to 10-membered heterocyclyl, 5- to 12-membered
aryl and
5- to 12-membered heteroaryl. Typically, in such embodiments, R1 is 5- to 10-
membered heterocyclyl or 5- to 12- membered heteroaryl, each of which is
unsubstituted or substituted (e.g. substituted by one or more groups Q as
defined
above), or Rl is C1_6 alkyl, C2_6 alkenyl, or C2_6 alkynyl which alkyl,
alkenyl or alkynyl
group is unsubstituted or substituted by one or more groups (e.g. 1, 2, 3 or 4
groups)
selected from halo, nitro, -CN, OH, C1_6 alkoxy, C1-6 hydroxyalkyl, C1_6
alkylthio, C1-6
haloalkyl, Ci_4 haloalkoxy, -CO2R", -NR"2, -SR", -S(=0)R", C3-Cio cycloalkyl,
5 to
10-membered heterocyclyl, 5- to 12-membered aryl or 5- to 12-membered
heteroaryl,
wherein each R" is independently selected from H, C1_6 alkyl, C.3-10
cycloalkyl, 5 to 10-
membered heterocyclyl, 5- to 12-membered aryl and 5- to 12-membered
heteroaryl.
In some embodiments, when ¨L-Z is halo, Rl is not azetidinyl, which is
unsubstituted or
substituted by C1_6 alkylsulfonyl; Ci -6 alkoxycarbonylpyrrolidinyl; C1_6
alkylcarbonylpyrrolidinyl; cycloalkyl, which is unsubstituted or substituted
by Ci_6
alkyl, Ci_6 alkyl sulfonyl, carboxy, halogen or hydroxy; dioxo-
tetrahydrothiophenyl,
which is unsubstituted or substituted by C1.6 alkyl; dioxo-
tetrahydrothiopyranyl; dioxo-
thietanyl; oxo-thietanyl; oxo-pyrrolidinyl, which is unsubstituted or
substituted by Ci_6
alkyl; oxetanyl; oxopiperidinyl; piperidinyl; tetrahydrofuranyl;
tetrahydropyranyl;
CA 02963740 2017-04-05
WO 2016/055780 PCT/GB2015/052920
17
1-6 ___________ OH 0
/11,0
0
NH NH
1-6 or
In some embodiments, when ¨L-Z is halo, le is not alkyl substituted by one or
more
hydroxyl groups, It' is not cycloalkyl, and R.' is not heterocyclyl. For
example, in some
embodiments, when ¨L-Z is halo, RI is C2_6 alkenyl, C2-6 alkynyl, or 5- to 12-
membered heteroaryl, each of which is unsubstituted or substituted (e.g.
substituted by
one or more groups Q as defined above); or Rl is C1-6 alkyl which is
unsubstituted or
substituted by one or more (e.g. 1, 2, 3 or 4) groups selected from halo,
nitro, -CN, C1.6
alkoxy, Ci_6 alkylthio, Ci_6 haloalkyl, C1-4 haloalkoxy, -CO2R", -NR'"2, -
SR''', -
S(=0)R", -S(=0)2R"', C3-Cio cycloalkyl, 5 to 10-membered heterocyclyl, 5-to 12-
membered aryl or 5- to 12-membered heteroaryl, wherein each R"' is
independently
selected from H, C1-6 alkyl, C3-10 cycloalkyl, 5 to 10-membered heterocyclyl,
5-to 12-
membered aryl and 5- to 12-membered heteroaryl. Typically in these
embodiments,
when ¨L-Z is halo, RI is 5- to 12- membered heteroaryl, which is unsubstituted
or
substituted (e.g. substituted by one or more groups Q as defined above); or R'
is C1-6
alkyl C2-6 alkenyl, or C2_6 alkynyl, each of which alkyl, alkenyl or alkynyl
groups is
unsubstituted or substituted by one or more (e.g. 1, 2, 3 or 4) groups
selected from halo,
nitro, -CN, C1-6 alkoxy, C1-6 alkylthio, C1-6 haloalkyl, C1-4 haloalkoxy, -
CO2R'", -NR'"2,
-SR", -S(=0)R", -S(=0)2R"', C3-Cio cycloalkyl, 5 to 10-membered heterocyclyl,
5- to
12-membered aryl or 5- to 12-membered heteroaryl, wherein each R" is
independently
selected from H, C1-6 alkyl, C3-10 cycloalkyl, 5 to 10-membered heterocyclyl,
5-to 12-
membered aryl and 5- to 12-membered heteroaryl.
In some embodiments, -L-Z is not halo. For example, in some embodiments, Z is
C1-6
haloalkyl, nitro, -CN, -N(R2)2, -0R2, -SR2, -S(=0)R2, or -S(=0)2R2, and L is
as defined
above.
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
18
In some embodiments, RI is not alkyl substituted by one or more hydroxyl
groups, R1 is
not cycloalkyl, and R1 is not heterocyclyl. For example, in some embodiments,
R1 is
C2-6 alkenyl, C2o alkynyl, or 5- to 12- membered heteroaryl, each of which is
unsubstituted or substituted (e.g substituted by one or more groups Q as
defined
above); or R1 is C1-6 alkyl which is unsubstituted or substituted by one or
more (e.g. 1,
2, 3 or 4) groups selected from halo, nitro, -CN, C1-6 alkoxy, C1-6 alkylthio,
C1-6
haloalkyl, C1-4 haloalkoxy, -CO2R"', -NR"2, -S(=0)R", -S(=0)2R",
cycloalkyl, 5 to 10-membered heterocyclyl, 5- to 12-membered aryl or 5- to 12-
membered heteroaryl, wherein each R"' is independently selected from H, C1-6
alkyl, C3-
10 cycloalkyl, 5 to 10-membered heterocyclyl, 5- to 12-membered aryl and 5- to
12-
membered heteroaryl. Typically in these embodiments, RI is 5- to 12- membered
heteroaryl which is unsubstituted or substituted (e.g. substituted by one or
more groups
Q as defined above); or le is C1-6 alkyl C2-6 alkenyl, or C/.6 alkynyl, each
of which
alkyl, alkenyl or alkynyl groups is unsubstituted or substituted by one or
more (e.g. 1, 2,
3 or 4) groups selected from halo, nitro, -CN, C1_6 alkoxy, C1_6 alkylthio, C1-
6 haloalkyl,
C1-4 haloalkoxy, -0O21r, -NR'"2, -SR"', -S(=0)R'9, -S(=0)21r, C3-Cio
cycloalkyl, 5 to
10-membered heterocyclyl, 5- to 12-membered aryl or 5- to 12-membered
heteroaryl,
wherein each R" is independently selected from H, C1-6 alkyl, C3-10
cycloalkyl, 5 to 10-
membered heterocyclyl, 5- to 12-membered aryl and 5- to 12-membered
heteroaryl.
In some embodiments, when ¨L-Z is chloro, R1 is not
dioxidotetrahydrothiopyranyl,
tetrahydropyranyl, oxopyrrolidinyl, oxopiperidinvl, tetrahydrofuranyl,
tertbutoxycarbonylpyrrolidinyl, dihydroxypropanyl,
dioxidotetrahydrothiophenyl,
methyloxopyrrolidinyl, ethyloxopyrrolidinyl, piperidinyl, 2-
methylpropanoylpyrrolidinyl, propanoylpyrrolidinyl,
dimethyldioxidotetrahydrothiophenyl, dioxidothiazolidinyl,
oxaazaspiro3.4]octyl,
cyclopentyl, difluorocyclobutyl, hydroxycyclohexyl, hydroxycyclopentyl,
difluorocyclopentyl, cyclohexanecarboxylic acid, (hydroxyl)(methyl)cyclobutyl,
or
hydroxycyclobutyl. In examples of these embodiments, when ¨L-Z is chloro, R"
is not
heterocyclyl, R1 is not cycloalkyl, and R.' is not alkyl substituted by one or
more
hydroxyl groups. Typically, in these embodiments, when ¨L-Z is chloro, R1 is 5-
to 12-
membered heteroaryl which is unsubstituted or substituted (e.g. substituted by
one or
more groups Q as defined above); or RI is Ci_6 alkyl C2_6 alkenyl, or C2-6
alkynyl, each
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
19
of which alkyl, alkenyl or alkynyl groups is unsubstituted or substituted with
one or
more (e.g. 1, 2, 3 or 4) groups selected from halo, nitro, -CN, C1-6 alkoxy,
C1-6 alkylthio,
C1-6 haloalkyl, C1-4 haloalkoxy, -0O21r, -NRI"2, -SR", -S(=0)Rw, -S(=0)2RI",
C3-Cm
cycloalkyl, 5 to 10-membered heterocyclyl, 5- to 12-membered aryl or 5- to 12-
.. membered heteroaryl, wherein each R" is independently selected from H, C1-6
alkyl, C3-
cycloalkyl, 5 to 10-membered heterocyclyl, 5- to 12-membered aryl and 5- to 12-
membered heteroaryl.
In some embodiments R.' is not dioxidotetrahydrothiopyranyl,
tetrahydropyranyl,
10 .. oxopyrrolidinyl, oxopiperidinyl, tetrahydrofuranyl,
tertbutoxycarbonylpyrrolidinyl,
dihydroxypropanyl, dioxidotetrahydrothiophenyl, methyloxopyrrolidinyl,
ethyloxopyrrolidinyl, piperidinyl, 2-methylpropanoylpyrrolidinyl,
propanoylpyrrolidinyl, dimethyldioxidotetrahydrothiophenyl,
dioxidothiazolidinyl,
oxaazaspiro[3.4]octyl, cyclopentyl, difluorocyclobutyl, hydroxycyclohexyl,
hydroxycyclopentyl, difluorocyclopentyl, cyclohexanecarboxylie acid,
(hydroxyl)(methypcyclobutyl, or hydroxycyclobutyl.
In some embodiments, when ¨L-Z is halo, Rl is not pyridine substituted by one
group,
which group is selected from C1-6 alkylsulfonyl, C1-6 alkoxy, -CN and
hydroxyl.
In some embodiments, when ¨L-Z is halo, It' is C1-6 alkyl, C2-6 alkenyl, C2-6
alkynyl, or
3-to 10-membered cycloalkyl or 5- to 10-membered heterocyclyl, each of which
is
unsubstituted or substituted (e.g substituted by one or more groups Q as
defined
above); or 5- to 12- membered heteroaryl which is unsubstituted or substituted
by one or
more groups (e.g 1, 2, 3 or 4 groups) selected from halo, nitro, C1-6
hydroxyalkyl, C1-6
alkylthio, C1-6 haloalkyl, C1_4 haloalkoxy, -CO2R", -NR"2, -SR"', -S(=0)R'",
C3-Cio
cycloalkyl, 5 to 10-membered heterocyclyl, 5- to 12-membered aryl or 5- to 12-
membered heteroaryl, wherein each R"' is independently selected from H, Ci.6
alkyl, C3-
10 cycloalkyl, 5 to 10-membered heterocyclyl, 5- to 12-membered aryl and 5- to
12-
.. membered heteroaryl.
In some embodiments, RI is C1_6 alkyl, C2_6 alkenyl, C2-6 alkynyl, 3- to 10-
membered
cycloalkyl or 5- to 10-membered heterocyclyl, each of which is unsubstituted
or
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
substituted (e.g. substituted by one or more groups Q as defined above); or 5-
to 12-
membered heteroaryl which is unsubstituted or substituted by one or more
groups (e.g.
1, 2, 3 or 4 groups) selected from halo, nitro, Ci_6 hydroxyalkyl, C1_6
alkylthio, C1-6
haloalkyl, CIA haloalkoxy, -0O21r, -NR1"2, -S(=0)R"', C3-
C10 cycloalkyl, 5 to
5 10-membered heterocyclyl, 5- to 12-membered aryl or 5- to 12-membered
heteroaryl,
wherein each R" is independently selected from H, C1-6 alkyl, C3-10
cycloalkyl, 5 to 10-
membered heterocyclyl, 5- to 12-membered aryl and 5- to 12-membered heteroaryl
In some embodiments, when -L-Z is chloro, RI is not pyridine substituted with
a ¨CN,
10 methylsulfonyl, ethylsulfonyl, methoxy, or hydroxy group. For example,
in some
embodiments, when -L-Z is chloro, Rl is C1_6 alkyl, C2_6 alkenyl, C2-6
alkynyl, 3- to 10-
membered cycloalkyl or 5-to 10-membered heterocyclyl, each of which is
unsubstituted
or substituted (e.g. substituted by one or more groups Q as defined above); or
5- to 12-
membered heteroaryl which is unsubstituted or substituted by one or more
groups (e.g.
15 1, 2, 3 or 4 groups) selected from halo, nitro, C1-6 hydroxyalkyl, C1.6
alkylthio, C1-6
haloalkyl, C1.4 haloalkoxy, -0O21r, -NIC2, -SR', -S(=0)R"', C3-Cio cycloalkyl,
5 to
10-membered heterocyclyl, 5- to 12-membered aryl or 5- to 12-membered
heteroaryl,
wherein each R" is independently selected from H, C1_6 alkyl, C3-10
cycloalkyl, 5 to 10-
membered heterocyclyl, 5- to 12-membered aryl and 5- to 12-membered
heteroaryl.
In some embodiments, when -L-Z is C14alkoxy, halogen, trifluoromethyl or
cyano, R1
is a Ci_6 alkyl, C2_6 alkenyl or C2_6 alkynyl group, which alkyl, alkenyl or
alkynyl group
is unsubstituted or substituted by one or more groups (e.g. 1, 2, 3 or 4
groups) selected
from halo, nitro, -CN, C1_6 alkoxy, C1_6 alkylthio, C1_6 haloalkyl, C1-4
haloalkoxy, -
CO211", -NR"2, C3-Cio cycloalkyl, 5 to 10-membered heterocyclyl, or 5- to
12-
membered heteroaryl; or RI is a 5- to 12- membered heteroaryl, which is
unsubstituted
or substituted by one or more groups (e.g. 1, 2, 3 or 4 groups) selected from
halo, nitro,
C1_6 hydroxyalkyl, C1_6 alkylthio, C1-6 haloalkyl, C14 haloalkoxy, -0O21r, -
SR", -S(=0)R", C3-Cio cycloalkyl, 5 to 10-membered heterocyclyl, 5- to 12-
membered aryl or 5- to 12-membered heteroaryl; wherein each R"' is
independently
selected from H, C1-6 alkyl, C3-10 cycloalkyl, 5 to 10-membered heterocyclyl,
5-to 12-
membered aryl and 5- to 12-membered heteroaryl.
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
21
In some embodiments R' is a Ci_6 alkyl, C2_6 alkenyl or C2_6 alkynyl group,
which alkyl,
alkenyl or alkynyl group is unsubstituted or substituted by one or more groups
(e.g. 1, 2,
3 or 4 groups) selected from halo, nitro, -CN, C1_6 alkoxy, C1_6 alkylthio,
C1_6 haloalkyl,
C14 haloalkoxy, -CO2R", -NR"2, -SR", C3-C10 cycloalkyl, 5 to 10-membered
heterocyclyl, or 5- to 12-membered heteroaryl; or Rl is a 5- to 12- membered
heteroaryl,
which is unsubstituted or substituted by one or more groups (e.g. 1, 2, 3 or 4
groups)
selected from halo, nitro, Ci_6 hydroxyalkyl, C1-6 alkylthio, Ci_6 haloalkyl,
C1-4
haloalkoxy, -CO2R", -NR'"2, -SR", -S(=0)R", C3-Clo cycloalkyl, 5 to 10-
membered
heterocyclyl, 5- to 12-membered aryl or 5- to 12-membered heteroaryl; wherein
each
R" is independently selected from H, C14 alkyl, C3-10 cycloalkyl, 5 to 10-
membered
heterocyclyl, 5- to 12-membered aryl and 5- to 12-membered heteroaryl.
In some embodiments, when Z is Cl, R1 is C1-6 alkyl, C24 alkenyl, or C24
alkynyl which
alkyl, alkenyl or alkynyl group is unsubstituted or substituted by one or more
groups
(e.g. 1, 2, 3 or 4 groups) selected from halo, nitro, -CN, C14 alkoxy, C1_6
alkylthio, C1-6
haloalkyl, C14 haloalkoxy, -CO2R', -NR'"2, -SR", -S(=0)R"', C3-Cio cycloalkyl,
5 to
10-membered heterocyclyl, 5- to 12-membered aryl or 5- to 12-membered
heteroaryl;
or Rl is 5- to 12- membered heteroaryl which is unsubstituted or substituted
by one or
more groups (e.g. 1, 2, 3 or 4 groups) selected from halo, nitro, C14
hydroxyalkyl, C1-6
alkylthio, C14 haloalkyl, C14 haloalkoxy, -CO2R", -NR"2, -S(=0)1r, C3-Cio
cycloalkyl, 5 to 10-membered heterocyclyl, 5- to 12-membered aryl or 5- to 12-
membered heteroaryl; wherein each R' is independently selected from H, C1-6
alkyl, C3-
10 cycloalkyl, 5 to 10-membered heterocyclyl, 5- to 12-membered aryl and 5- to
12-
membered heteroaryl.
In some embodiments, RI is C1.6 alkyl, C2.6 alkenyl, or C2_6 alkynyl which
alkyl, alkenyl
or alkynyl group is unsubstituted or substituted by one or more groups (e.g.
1, 2, 3 or 4
groups) selected from halo, nitro, -CN, C1_6 alkoxy, C1_6 alkylthio, C1-6
haloalkyl, C1-4
haloalkoxy, -CO2R', -NR'"2, -SR'", -S(=0)R", C3-Clo cycloalkyl, 5 to 10-
membered
heterocyclyl, 5- to 12-membered aryl or 5- to 12-membered heteroaryl; or R1 is
5- to
12- membered heteroaryl which is unsubstituted or substituted by one or more
groups
(e.g. 1, 2, 3 or 4 groups) selected from halo, nitro, C1_6 hydroxyalkyl, C1_6
alkylthio, C1-6
haloalkyl, C1.4 haloalkoxy, -CO2R', -NR'"2, -SRI", -S(=0)R", C3-C10
cycloalkyl, 5 to
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
22
10-membered heterocyclyl, 5- to 12-membered aryl or 5- to 12-membered
heteroaryl;
wherein each R" is independently selected from H, C1_6 alkyl, C3-10
cycloalkyl, 5 to 10-
membered heterocyclyl, 5- to 12-membered aryl and 5- to 12-membered
heteroaryl.
The compounds of the invention may contain asymmetric or chiral centres, and
therefore exist in different stereoisomeric forms. It is intended that all
stereoisomeric
forms of the compounds of the invention, including but not limited to,
diastereomers,
enantiomers and atropisomers, as well as mixtures thereof such as racemic
mixtures,
form part of the present invention Compounds of Formula (I) containing one or
more
chiral centre may be used in enantiomerically or diastereoisomerically pure
form, or in
the form of a mixture of isomers.
The present invention embraces all geometric and positional isomers of
compounds of
the invention as defined above. For example, if a compound of the invention
incorporates a double bond or a fused ring, the cis- and trans-forms, as well
as mixtures
thereof, are embraced within the scope of the invention. Both the single
positional
isomers and mixture of positional isomers are also within the scope of the
present
invention.
The compounds of the present invention may exist in unsolvated as well as
solvated
forms with pharmaceutically acceptable solvents such as water, ethanol, and
the like,
and it is intended that the invention embrace both solvated and unsolvated
forms.
The compounds of the present invention may exist in different tautomeric
forms, and all
such forms are embraced within the scope of the invention. The term "tautomer"
or
"tautomeric form" refers to structural isomers of different energies which are
interconvertible via a low energy barrier. For example, proton tautomers (also
known as
prototropic tautomers) include interconversions via migration of a proton,
such as keto-
enol tautomerizations. Valence tautomers include interconversions by
reorganization of
some of the bonding electrons.
Compounds of the invention can be prepared according to the reaction schemes
taught
in WO 2013/068769 or by analogy thereto. Compounds of the invention may also
be
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
23
prepared by synthetic methods described in the Examples that follow, or by
analogy
with such methods.
A benzimidazole of formula (I) can be converted into a pharmaceutically
acceptable salt
thereof, and a salt can be converted into the free compound, by conventional
methods.
For instance, a benzimidazole of formula (I) can be contacted with a
pharmaceutically
acceptable acid to form a pharmaceutically acceptable salt. A pharmaceutically
acceptable salt is a salt with a pharmaceutically acceptable acid or base.
Pharmaceutically acceptable acids include both inorganic acids such as
hydrochloric,
sulphuric, phosphoric, diphosphoric, hydrobromic or nitric acid and organic
acids such
as citric, fumaric, maleic, malic, ascorbic, succinic, tartaric, benzoic,
acetic,
methanesulphonic, ethanesulphonic, benzenesulphonic or p-toluenesulphonic
acid.
Pharmaceutically acceptable bases include alkali metal (e.g. sodium or
potassium) and
alkali earth metal (e.g. calcium or magnesium) hydroxides and organic bases
such as
alkyl amines, aralkyl amines and heterocyclic amines.
Compounds of the present invention have been found in biological tests to be
inhibitors
of respiratory syncytial virus (RSV). The compounds are therefore
therapeutically
useful. Accordingly, the present invention further provides a compound which
is a
benzimidazole of formula (1), as defined above, or a pharmaceutically
acceptable salt
thereof, for use in a method of treating the human or animal body by therapy.
The
invention also provides a compound of the invention as defined above for use
in a
method treating or preventing an RSV infection. Still further, the present
invention
provides the use of a compound of the invention as defined above in the
manufacture of
a medicament for use in treating or preventing an RSV infection. A subject
suffering
from or susceptible to an RSV infection may thus be treated by a method
comprising the
administration thereto of a compound of the invention as defined above. The
condition
of the subject may thereby be improved or ameliorated.
The RSV infection is typically a respiratory tract infection. The RSV
infection may be
an infection in a child, for instance a child under ten years of age or an
infant under two
years of age. In one embodiment the invention provides a compound as defined
above
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
24
for use in treating or preventing an RSV infection in paediatric patients.
Alternatively
the infection may be an infection in a mature or elderly adult, for instance
an adult over
60 years of age, an adult over 70 years of age, or an adult over 80 years of
age. The
invention further provides a compound for use in treating or preventing an RSV
infection in geriatric patients.
The RSV infection may be an infection in an immunocompromised individual or an
individual suffering from COPD or CHF. In another embodiment, the RSV
infection is
an infection in a non-compromised individual, for instance an individual who
is
otherwise healthy.
A compound of the present invention can be administered in a variety of dosage
forms,
for example orally such as in the form of tablets, capsules, sugar- or film-
coated tablets,
liquid solutions or suspensions or parenterally, for example intramuscularly,
intravenously or subcutaneously. The compound may therefore be given by
injection,
infusion, or by inhalation or nebulaisation. The compound is preferably given
by oral
administration.
The dosage depends on a variety of factors including the age, weight and
condition of
the patient and the route of administration. Daily dosages can vary within
wide limits
and will be adjusted to the individual requirements in each particular.
Typically,
however, the dosage adopted for each route of administration when a compound
is
administered alone to adult humans is 0.0001 to 650 mg/kg, most commonly in
the
range of 0.001 to 10 mg/kg, body weight, for instance 0.01 to 1 mg/kg. Such a
dosage
may be given, for example, from 1 to 5 times daily. For intravenous injection
a suitable
daily dose is from 0.0001 to 1 mg/kg body weight, preferably from 0.0001 to
0.1 mg/kg
body weight. A daily dosage can be administered as a single dosage or
according to a
divided dose schedule.
A unit dose form such as a tablet or a capsule will usually contain 1-250 mg
of active
ingredient. For example, a compound of formula (I) could be administered to a
human
patient at a dose of between 100-250 mg either once a day, twice or three
times a day.
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
For example, a compound of formula (I) could be administered to a human
patient at a
dose of between 100-250 mg either once a day, twice or three times a day.
The compounds of formula (I) and phaimaceutically acceptable salts thereof may
be
5 used on their own. Alternatively, they may be administered in the form of
a
pharmaceutical composition. The present invention therefore also provides a
phamiaceutical composition comprising a compound of formula (I) or a
phaunaceutically acceptable salt thereof as hereinbefore defined, in
association with a
pharmaceutically acceptable adjuvant, diluent or carrier. Conventional
procedures for
10 the selection and preparation of suitable pharmaceutical formulations
are described in,
for example, "Pharmaceuticals - The Science of Dosage Form Designs", M. E.
Aulton,
Churchill Livingstone, 1988.
Depending on the mode of administration, the pharmaceutical composition will
15 preferably comprise from 0.05 to 99 %w (percent by weight), more
preferably from
0.05 to 80 %w, still more preferably from 0.10 to 70 %w, and even more
preferably
from 0.10 to 50 %w, of active ingredient, all percentages by weight being
based on total
composition.
20 The invention further provides a process for the preparation of a
pharmaceutical
composition of the invention which comprises mixing a compound of formula (I)
or a
pharmaceutically acceptable salt thereof as hereinbefore defined with a
pharmaceutically acceptable adjuvant, diluent or carrier.
25 The compounds of the invention may be administered in a variety of
dosage forms.
Thus, they can be administered orally, for example as tablets, troches,
lozenges,
aqueous or oily suspensions, solutions, dispersible powders or granules. The
compounds
of the invention may also be administered parenterally, whether
subcutaneously,
intravenously, intramuscularly, intrastemally, transdermally, by infusion
techniques or
by inhalation or nebulisation. The compounds may also be administered as
suppositories.
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
26
Solid oral forms of the pharmaceutical composition of the invention may
contain,
together with the active compound, diluents, e.g. lactose, dextrose,
saccharose,
cellulose, corn starch or potato starch; lubricants, e.g. silica, talc,
stearic acid,
magnesium or calcium stearate, and/or polyethylene glycols; binding agents,
e.g.
starches, arabic gums, gelatin, methylcellulose, carboxymethylcellulose or
polyvinyl
pyrrolidone; disaggregating agents, e.g. starch, alginic acid, alginates or
sodium starch
glycolate; effervescing mixtures; dyestuffs; sweeteners; wetting agents, such
as lecithin,
polysorbates, laurylsulfates; and, in general, non toxic and pharmacologically
inactive
substances used in pharmaceutical formulations. Such pharmaceutical
preparations may
.. be manufactured in known manner, for example, by means of mixing,
granulating,
tableting, sugar coating, or film coating processes.
Liquid dispersions for oral administration may be syrups, emulsions and
suspensions.
The syrups may contain as carriers, for example, saccharose or saccharose with
glycerine and/or mannitol and/or sorbitol.
Suspensions and emulsions may contain as carrier, for example a natural gum,
agar,
sodium alginate, pectin, methylcellulose, carboxymethylcellulose, or polyvinyl
alcohol.
The suspension or solutions for intramuscular injections may contain, together
with the
active compound, a pharmaceutically acceptable carrier, e.g. sterile water,
olive oil,
ethyl oleate, glycols, e.g. propylene glycol, and if desired, a suitable
amount of
lidocaine hydrochloride. Further suitable carriers for suspensions include
sterile water,
hydroxypropylmethyl cellulose (HPMC), polysorbate 80, polyvinylpyrrolidone
(PVP),
aerosol AOT (i.e. sodium 1,2-bis(2-ethylhexoxycarbonyl)ethanesulphonate),
pluronic
F127 and/or captisol (i.e. sulfobutylether-beta-cyclodextrin).
The compounds of the invention may, for example, be formulated as aqueous
suspensions in a carrier selected from:
(i) 0.5% w/v hydroxypropylmethyl cellulose (HPMC)/0.1% w/v polysorbate 80;
(ii) 0.67% w/v polyvinylpyrrolidone (PVP)/0.33% w/v aerosol AOT (sodium1,2-
bis(2-
ethylhexoxycarbonyl)ethanesulphonate);
(iii) 1 % w/v pluronic F 127; and
(iv) 0.5% w/v polysorbate 80.
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
27
The carriers may be prepared by standard procedures known to those of skill in
the art.
For example, each of the carriers (i) to (iv) may be prepared by weighing the
required
amount of excipient into a suitable vessel, adding approximately 80% of the
final
volume of water and magnetically stirring until a solution is formed. The
carrier is then
made up to volume with water. The aqueous suspensions of compounds of formula
I
may be prepared by weighing the required amount of a compound of formula I
into a
suitable vessel, adding 100% of the required volume of carrier and
magnetically stirring.
Solutions for injection or infusion may contain as carrier, for example,
sterile water or
preferably they may be in the form of sterile, aqueous, isotonic saline
solutions.
The compounds of the invention may also be administered in conjunction with
other
compounds used for the treatment of viral infections. Thus, the invention
further relates
to combination therapies wherein a compound of the invention, or a
pharmaceutically
acceptable salt thereof, or a pharmaceutical composition or formulation
comprising a
compound of the invention, is administered concurrently or sequentially or as
a
combined preparation with another therapeutic agent or agents, for the
treatment or
prevention of a viral infection, particularly infection by RSV.
Herein, where the term "combination" is used it is to be understood that this
refers to
simultaneous, separate or sequential administration. In one aspect of the
invention
"combination' refers to simultaneous administration. In another aspect of the
invention
"combination'' refers to separate administration. In a further aspect of the
invention
"combination'' refers to sequential administration. Where the administration
is
sequential or separate, the delay in administering the second component should
not be
such as to lose the beneficial effect of the combination.
Suitable therapeutic agents for use in the combination therapies include
(i) RSV nucleocapsid (N)-protein inhibitors;
(ii) other RSV protein inhibitors, such as those that inhibit the
phosphoprotein (P)
protein and large (L) protein;
(iii) anti-RSV monoclonal antibodies, such as the F-protein antibodies;
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
28
(iv) immunomodulating toll-like receptor compounds;
(v) other respiratory virus anti-virals, such as anti-influenza and anti-
rhinovirus
compounds; and/or
(vi) anti-inflammatory compounds.
The RSV nucleocapsid (N)-protein plays a pivotal role in viral transcription
and
replication, mediating the interaction between the genomic RNA and the virally
encoded RNA-dependent RNA polymerase. The RSV P- and L-proteins are components
of RSV's virally encoded RNA-dependent RNA polymerase.
According to a further aspect of the invention, there is provided a compound
of the
formula (I) or a pharmaceutically acceptable salt thereof as hereinbefore
defined in
combination with one or more of the therapeutic agents listed as (i) to (vi)
above for use
in the treatment of RSV.
The following Examples illustrate the invention. They do not however, limit
the
invention in any way.
Examples
Preparatory Example 1
1,3- Diethyl-2-(47fluoro-2-nitrophenyl) propanedioate
To a solution of 1,4-difluoro-2-nitro-benzene (10 06g, 63.23mmo1) and diethyl
propanedioate (13.58mL, 88.99mm01) in 50m1 of dimethylformamide was added
36.4g
of cesium carbonate slowly at room temperature under a nitrogen atmosphere.
The
suspension was stirred for 48h before more dimethylformamide (20m1) and
diethyl
propanedioate (2mL) was added and the mixture stirred for a further 24h at
room
temperature. The reaction mixture was then concentrated under vacuum and
azeotroped
with n-heptane. Water (250m1) was added and extracted with ethyl acetate
(4x75m1).
The organic phase was washed with water (1x300m1), separated, dried over
magnesium
sulphate, filtered and concentrated under vacuum to afford 18.9g (99%) of the
desired
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
29
product as a yellow oil.
LCMS:
M/Z [M+H]+ : 299.96
1H-NMR:
1H NMR (500 MHz, Chloroform-d) 6 7.79 (dd, J = 8.2, 2.7 Hz, 1H), 7.63 ¨7.51
(m,
1H), 7.44 ¨ 7.33 (m, 1H), 5.27 (s, 1H), 4.27 (qd, J = 7.1, 2.2 Hz, 5H), 4.21
(d, J = 7.1
Hz, 1H), 3.36 (d, J= 1.1 Hz, OH), 1.34 ¨1.25 (m, 9H).
13C-NMR:
13C NMR (126 MHz, cdc13) 6 167.00, 166.56, 162.68, 160.67, 133.14, 133.07,
124.24,
124.20, 120.77, 120.60, 112.91, 112.70, 77.26, 77.21, 77.01, 76.75, 62.36,
61.44, 53.74,
41.66, 14.02, 13.94.
Preparatory Example 2
Ethyl 2-(4-Fluoro-2-nitrophenyl)acetate.
A mixture of diethyl 2-(4-fluoro-2-nitrophenyl) propanedioate (18.g,
60.15mmol),
lithium chloride (5.1g, 120.3mmo1) in dimethyl sulfoxide (150mL) and water
(1.08mL,
60.15mmol) was heated with stirring at 100 C for 16 hours. The reaction
mixture was
allowed to cool to room temperature, water (100m1) was added then the mixture
extracted with ethyl acetate (150m1). The aqueous layer was further extracted
with ethyl
acetate (2x75m1) and the combined organic layers were washed with brine
(100m1),
dried over magnesium sulphate, filtered and evaporated under vacuum to leave
the
desired product as a yellow oil (16g, 94%). This crude product was
contaminated with
20% of the starting material and was used directly in the next step.
30
1H NMR (500 MHz, Chloroform-d) 6 7.86 (dd, J = 8.4, 2.6 Hz, 1H), 7.39 - 7.30
(m,
2H), 4.18 (q, J = 7.1 Hz, 2H), 4.00 (s, 3H), 1.27 (t, J = 7.2 Hz, 5H).
Preparatory Example 3
6-Fluoro-2,3-dihydro-1H-indo1-2-one.
Under an atmosphere of nitrogen, iron filings (10.22g, 183.11mmol) were added
in
portions to a solution of ethyl 2-(4-fluoro-2-nitro-phenyl)acetate from
Preparatory
Example 2 (13.g, 45.78mmo1) in acetic acid (200mL). The reaction mixture was
stirred
at 80 C for 48 hours. The reaction mixture was allowed to cool to room
temperature,
filtered through celiteTM, washed with ethyl acetate (100m1) and concentrated
under
vacuum to leave a brown solid. This was dissolved in ethyl acetate (150m1) and
washed
with saturated aqueous sodium bicarbonate (2x75m1). The organic layer was
dried over
magnesium sulphate and concentrated under reduced pressure. This solid was
triturated
with ether and filtered to give solid (4.0g) (58%).
The filtrate was evaporated and the residue was purified by column
chromatography
(silica, 25g, ethyl acetate : petroleum ether 15:85 gradient to 80:20) to
afford a light
yellow solid second crop (1.5g, 22%).
1H-NMR:
1H NMR (500 MHz, DMSO-d6) 6 10.46 (s, 1H), 7.19 (dd, J = 8.1, 5.7 Hz, 1H),
6.71
(ddd, J = 10.3, 8.1, 2.5 Hz, 1H), 6.61 (dd, J = 9.3, 2.4 Hz, 1H), 3.43 (s,
2H).
1H-NMR:
1H NMR (500 MHz, DMSO-d6) 6 10.46 (s, 1H), 7.25 -7.11 (m, 1H), 6.71 (ddd, J =
10.4, 8.2, 2.5 Hz, 1H), 6.61 (dd, J = 9.3, 2.4 Hz, 1H), 3.43 (t, J = 1.5 Hz,
2H).
Preparatory Example 4
6' Fluoro-1 ',2 '-dihydrospiro[cyclopropane-1, 3 '-indole] -2 '-one
Date Recue/Date Received 2022-02-25
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
31
To a stirred solution of 6-fluoro-2,3-dihydro-1H-indo1-2-one from Preparatory
Example
3 (3.g, 19.85mmo1) and diisopropylamine (5.84mL, 41.68mmo1) in tetrahydrofuran
(30mL) under nitrogen at -40 C was added n-butyllithium dropwise over 30
minutes
(2.5M solution in n-hexane, 31.76mL, 79.4mm01). The mixture was warmed to 0 C
in
an ice bath. To this mixture was added dropwise a solution of 1,2-
dibromoethane
(5.13mL, 59.55mmo1) in THF (10m1). The reaction mixture was then left to stir
at room
temperature for 48 hours as a light brown suspension. To the mixture was added
carefully a saturated aqueous solution of ammonium chloride (5m1 then 200m1) .
The
mixture was extracted with acetic acid (4 x75m1). The organics were combined
and
washed with brine (1x150m1), dried using magnesium sulphate, filtered then
concentrated under vacuum to afford 4g of an orange-light brown solid.
This material was combined with the product from a duplicate reaction then
purified
using flash chromatography (100g silica, eluted with Petroleum Ether: ethyl
acetate
100:0 to 40:60 gradient) to afford the desired product 6.31g (89%).
LCMS:
M/Z [M+H]+ : 178.2
1H-NMR:
1H NMR (500 MHz, Chloroform-d) 6 9.18 (s, 1H), 6.88 ¨6.59 (m, 3H), 1.76 (m, J
=
4.4, 4.0 Hz, 2H), 1.53 (m, J = 4.3 Hz, 2H).
Preparatory Example 5
N-1/2-(chloromethyl)-1-(4,4,4-trifluorobu0,1)-1H-1,3-benzodiazol-5-
ylimethylicarbctmate
To a suspension of tert-butyl N-[[2-(hydroxymethyl)-1-(4,4,4-trifluorobuty1)-
1H-1,3-
benzodiazol-5-yl]methyl]carbamate (obtained according to the procedure set out
in WO
2010/103306; 960mg, 2.48mmo1) in tetrahydrofuran (20mL) was added di-
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
32
isopropylethylamine (1.29mL, 7.43mmo1) and this suspension was stirred under
N9 for 5
minutes. This suspension was cooled to 0 C using an ice bath and
methanesulfonyl
chloride (0.25mL, 3.22mmo1) was added drop-wise over 5 minutes. The reaction
was
allowed to warm up to room temperature by removing the ice bath and stirred
under N2
overnight. Water (8m1) was added drop wise to the mixture and the solvent was
removed under vacuum. Further water (60m1) was added and the residue was
extracted
with ethyl acetate (1x75m1) then (3x25m1). The organic phases were combined
and
washed with citric acid solution (1x35m1), saturated aqueous sodium
bicarbonate
(1x60m1), dried over magnesium sulphate, filtered and concentrated under
vacuum to
afford 1.015g of a dark gummy crude material.
Preparatory Example 6
tert-butyl N-[(2-[(6'-fluoro-2'-oxo-1 ',2 '-thhydrospiro [cyclopropane-1, 3'-
indole1-1'-
ylmethy1J-1-(4,4,4-trillitorob0,1)-1H-1,3-benzodiazyl)methylkarbamate
To solution of 6'-fluoro-1,2-spiro[cyclopropane-1,3'-indole]-2'-one from
Preparatory
Example 4 (487.44mg, 2.75mm01) in N,N-dimethylformamide (10mL) at 0 C under
nitrogen was added sodium hydride (0.11mL, 2.75mmo1) in one portion. Once
added,
the cold bath was removed and the cloudy solution was stirred at room
temperature for
1 hour. To this mixture was added drop wise over 5 minutes at room temperature
a
solution of the crude N-[[2-(chloromethyl)-1-(4,4,4-trifluorobuty1)-1H-1,3-
benzodiazol-
5-ylimethyl]carbamate obtained in preparatory Example 5 (1015.mg, 2.5mmo1;
used
without further purification) in DMF (4m1). The mixture was stirred at room
temperature for 16 hours. The reaction was quenched with water (100m1) and
extracted
with ethyl acetate (3x75m1). The combined organics were washed with water
(1x100m1), brine (120m1), then dried over magnesium sulphate and evaporated
under
reduced pressure. The crude oil was purified by flash column chromatography
(25g
silica) eluted with petroleum ether: ethyl acetate (100:0 gradient to 0:100).
Product
containing fractions were evaporated under vacuum and triturated further with
Petroleum ether/ethyl acetate (4:1) (10m1), filtered and dried under vacuum to
afford
1030 mg (75%) of the desired product as a beige solid.
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
33
Preparatory Examples 5 and 6 together gave a yield of 75%.
LCMS:
M/Z [M+H]+ : 547.0
1H-NMR:
NMR (500 MHz, DMSO-d6) 6 7.55 (d, .J= 8.3 Hz, 1H), 7.45 (s, 1H), 7.34 (t, ./=
6.3
Hz, 1H), 7.22- 7.12 (m, 2H), 7.06 (dd, J= 8.2, 5.4 Hz, 1H), 6.81 (ddd, J=
10.3, 8.3,
2.4 Hz, 1H), 5.29 (s, 2H), 4.35 (t, J' 7.7 Hz, 2H), 4.19 (d, J = 5.9 Hz, 2H),
2.33 (ddd, J
= 16.6, 7.8, 4.2 Hz, 2H), 1.84 (dd, J= 10.2, 5.9 Hz, 2H), 1.68 (q, J= 3.9, 3.4
Hz, 2H),
1.58 (q, J= 4.2, 3.8 Hz, 2H), 1.38 (s, 9H).
13C-NMR:
13C NMR (126 MHz, CDC13) 6 177.08, 163.27, 161.32, 148.40, 143.05, 142.96,
142.59, 134.48, 133.84, 127.54, 125.29, 125.27, 123.52, 119.10, 119.03,
118.96,
109.63, 109.22, 109.04, 99.69, 99.46, 77.24,77.19, 76.99, 76.73, 44.85, 42.73,
38.22,
31.23, 30.99, 28.42, 26.68, 22.65, 22.62, 22.60, 19.56.
Preparatory Example 7
Trirnethyl-[2-(pyrrolo[2,3-Npyridin-1-yfrnethavy)ethylisilane
Sodium hydride 60 % dispersion in mineral oil (1.34g, 33.52mm01) was added
portionwise to a solution of 1H-pyrrolo[2,3- b]pyridine (3.3g, 27.93mmo1) in
N,N-
dimethylformamide (25mL) at 0 C and the reaction mixture was stirred at 0 C
for
lhour. After that time, 2-(chloromethoxy)ethyl-trimethylsilane (5.93mL,
33.52mm01)
was added dropwise maintaining the internal temperature of the reaction below
10 C.
The reaction mixture was allowed to slowly warm up to room temperature. LCMS
after
1hr showed the reaction is complete with the expected product present at Rt =
5.18min
(100-500MW, 7min method) m/z 249 [M+H]+. The reaction was quenched with water
(200m1) and extracted into Et0Ac (200m1). The organic layer was washed with
brine
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
34
(3x100m1), dried over MgSO4, filtered and evaporated in vacuo to give the
crude
product as a greenish- yellow oil (8.31g) purified by flash chromatography
(Biotage,
50g) eluting with DCM:Me0H (100:0 to 97:3) to give the desired product in two
batches as a pale oil (4.13g) and as a clear oil (3.35g).
1H NMR (500 MHz, Chloroform-d) 6 8.35 (dd, J = 4.8, 1.6 Hz, 1H), 7.94 (d, J =
7.7
Hz, 1H), 7.36 (d, J = 3.6 Hz, 1H), 7.11 (dd, J = 7.9, 4.7 Hz, 1H), 6.54 (d, J
= 3.6 Hz,
1H), 5.72 (s, 2H), 3.61 - 3.50 (m, 2H), 0.96 - 0.86 (m, 2H), -0.06 (s, 9H).
Preparatory Example 8
1-(2-trimethylsilylethoxymethyl) -3H-pyrrolo12, 3-1) 1pyridin-2 -one
A solution of trimethy142-(pyrrolo[2,3-b]pyridin-l-ylmetboxy)ethyl]silane
(3.36g,
13.51mmol) in 1,4-dioxane (30mL) was added dropwise to a stirring suspension
of
pyridinium bromide perbromide (10.5g, 32.83mmo1) in 1,4-dioxane (30mL) . The
reaction mixture was stirred at room temperature for 3 hours. The reaction
mixture was
quenched with water (100m1) and extracted into Et0Ac (2x100m1). The organic
layer
was washed with brine (2x100m1), dried (MgSO4), filtered and evaporated in
vacuo to
give the expected product 3,3-dibromo-1-(2-
trimethylsilylethoxymethyl)pyrrolo[2,3-
b]pyridin-2- one (4.92g) as a golden oil:
1H NMR (500 MHz, Chloroform-d) 6 8.30 (dd, J = 5.2, 1.6 Hz, 1H), 7.87 (dd, J =
7.4,
1.6 Hz, 1H), 7.15 (dd, J = 7.4, 5.1 Hz, 1H), 5.32 (d, J = 0.9 Hz, 2H), 3.77;
3.68 (m, 2H),
0.98 (dd, J = 9.0, 7.6 Hz, 2H), 0.01 -0.03 (m, 9H).
Zinc dust (5.85g, 89.53mmo1) was added to a solution of 3,3-dibromo-1-(2-
trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyridin-2- one (3.78g, 8.95mmo1) in
tetrahydrofuran (50mL) and ammonium chloride sat solution (15.mL, 8.95mmol)
and
the reaction mixture was stirred at 20 C for 4 hours. LCMS and TLC analysis
(Pet
Ether : Et0Ac, 3:1) showed complete reaction.
The reaction mixture was filtered and concentrated in vacuo, and the residue
partitioned
between Et0Ac (100m1) and water (100m1) which resulted in a foilliation of a
white
precipitate. Both layers were filtered through Celite and separated. The
aqueous layer
was extracted with Et0Ac (2x100m1), the combined organic layer was washed with
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
brine (100m1), dried (MgSO4), filtered and evaporated in vacuo to give the
crude
product, which was purified by flash chromatography (Biotage, 50g) eluting
with Pet
Ether : Et0Ac (75:25 to 50:50) to give the product (1.7g) as a clear oil which
solidified
on standing to a beige solid.
5 1H NMIR (500 MHz, Chloroform-d) 6 8.23 (d, J = 5.4 Hz, 1H), 7.52 (ddd, J
= 7.6, 2.3,
1.1 Hz, 1H), 6.99 (dd, J = 7.3, 5.3 Hz, 1H), 5.27 (s, 2H), 3.70 (dd, J = 9.0,
7.5 Hz, 2H),
3.60 (s, 2H), 1.04 - 0.92 (m, 2H), -0.01 (s, 9H).
LCMS clean product at Rt = 3.99 min (7min method) m/z 264.9 [MH]+
Preparatory Example 9
'-(2-trimetlodsilylethoxymethyl)-spirokyclopropane-1 , 3'-pyrrolo[2 , 3-
NpyridineJ-2
one
To a solution of 1-(2-trimethylsilylethoxymethyl)-3H-pyrrolo[2,3-b]pyridin-2-
one
(1.22g, 4.61mmol) in N,N-dimethylformamide (15mL) at 0 C was added sodium
hydride 60 A) dispersion in mineral oil (461.42mg, 11.54mmo1) and the
reaction was
stirred at 0 C for 30 min. Next 1,2-dibromoethane (0.42mL, 4.84mmo1) was added
and
the reaction mixture was allowed to warm up slowly to room temperature
overnight.
LCMS showed reaction incomplete - SM : product ratio ¨1:2, with the expected
product present at Rt = 4.62 min (7min method) m/z 290.9 [MH]+.
The reaction was quenched with water (50m1), extracted into Et0Ac (100m1), the
organic layer was washed with brine (3x50m1), dried (MgSO4), filtered and
evaporated
in vacuo to give the crude product as an orange oil, which was purified by
flash
chromatography (50g) eluting with pet ether : EtOAC (75:25 to 50:50) to give
the
product (679mg) as a clear oil and the recovered starting material (231mg).
1H NMR (500 MHz, Chloroform-d) 6 8.21 (d, J = 5.3 Hz, 1H), 7.11 (d, J = 7.2
Hz, 11-1),
6.96 (dd, J = 7.3, 5.2 Hz, 1H), 5.34 (s, 2H), 3.72 (dd, J = 9.0, 7.5 Hz, 2H),
1.85 (q, J =
4.2 Hz, 2H), 1.58 (q, J = 4.2 Hz, 2H), 1.06 - 0.94 (m, 2H), -0.01 (s, 9H).
LCMS - product at Rt = 4.72 min m/z 290.9 [M11]+
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
36
Preparatory Example 10
Spiro(1H-pyrrolo[2,3-blpyridine-3,1'-cyclopropane)-2-one
To a solution of 1'-(2-trimethylsilylethoxymethyl)spiro(cyclopropane-1,3'-
pyrrolo[2,3-
b]pyridine)-2'-one (210.mg, 0.72mmo1) in dichloromethane (2mL) was added 2,2,2-
trifluoroacetic acid (1.9mL, 24.81mmol) and the reaction mixture was stirred
at room
temperature for 16 hours. LCMS showed the intermediate is present where SEM
group
had been cleaved to RCH2OH at Rt = 0.67min (7min method) m/z 190.9 [M1-1]+.
The
volatiles were removed in vacuo, the residue was dissolved in dichloromethane
(2mL)
and treated with ethylenediamine (0.19mL, 2.89mm01) and the reaction mixture
was
stirred at room temperature overnight. LCMS showed the reaction complete with
the
expected product present at Rt = 0.88min (100-500MW, 7min method) m/z 161
[MH]+.
The reaction mixture was diluted with sat aqueous NaHCO3 and extracted into
dichloromethane (5x50m1), the organics were dried (MgSO4), filtered and
concentrated
to dryness under reduced pressure to give the crude product as a white solid
which was
purified by flash chromatography (Biotage, 10g) eluting with DCM : Me0H (100:0
to
95:5) to give the final product as a white solid (102mg).
1H NMR (500 MHz, Chloroform-d) 6 8.15 (dd, J = 5.1, 1.6 Hz, 1H), 7.13 (dd, J =
7.1,
1.6 Hz, 1H), 6.96 (dd, J = 7.3, 5.2 Hz, 1H), 1.85 (q, J = 4.3 Hz, 2H), 1.59
(q, J = 4.3 Hz,
2H).
Preparatory Example 11
tert-butyl N-112-1(2'- oxo-spiro[cyclopropane-1,31- pyrrolo[2, 3-171pyridinel-
1 r-
yl)methyl]-1-(4,4,4- trifluorobutyl)benzitnidazol-5- ygmethylicarbamate
To a solution of spiro[1H-pyrrolo[2,3-b]pyridine-3,1'-cyclopropane]-2-one
(Preparatory
example 10, 98.mg, 0.6100mmo1) in N,N-dimethylformamide (2.5mL) at 0 C was
added sodium hydride (60 % dispersion in mineral oil) (36.71mg, 0.92mm01) and
the
.. reaction mixture was stirred for 30 min. Next was added tert-butyl N4[2-
(chloromethyl)-1-(4,4,4-trifluorobutyl)benzimidazol-5- yl]methyl]carbamate
(Preparatory Example 5, 248.3 lmg, 0.6100mmo1) and the reaction was allowed to
warm
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
37
up to room temperature over weekend (for convenience). LCMS showed the
reaction is
essentially complete with the expected intermediate present at Rt = 3.20min
(7min
method) m/z 530 [1VIH]+.
The reaction was quenched with water, diluted with Et0Ac (50m1) and the
organics
were washed with brine (3 x 50m1), dried with MgSO4, filtered and evaporated
in
vacuo. The residue was purified by column chromatography (Biotaae, 10g)
eluting with
DCM : Me0H (100:0 to 95:5) to give the product (187mg) as a yellow oil.
1H NMR (500 MHz, DMSO-d6) 6 8.02 (dd, J = 5.1, 1.6 Hz, 1H), 7.53 (d, J = 8.4
Hz,
1H), 7.47 (dd, J = 7.3, 1.6 Hz, 1H), 7.32 (d, J = 9.9 Hz, 2H), 7.12 (d, J =
8.3 Hz, 1H),
7.01 (dd, J = 7.3, 5.3 Hz, 1H), 5.26 (s, 2H), 4.43 (t, J = 7.4 Hz, 2H), 4.16
(d, J = 6.1 Hz,
2H), 2.42 - 2.35 (m, 3H), 2.01 (q, J = 7.8 Hz, 2H), 1.78 (q, J = 4.0, 3.5 Hz,
2H), 1.66 (q,
J = 3.8 Hz, 2H), 1.36 (s, 9H).
LCMS product at Rt = 3.38 min (7min method) m/z 530 [MH]+
Preparatory Example 12
tert-butyl N-111-isopen0,1-2-1(2' ¨oxo-spirojcyclopropane-1,3'- kyrrolo12,3-
b]pyridine]-1'- yOmethylibenzimidazol-5- yliniethylkarbantate
To spiro[1H-pyrrolo[2,3-b]pyridine-3,11-cyclopropane]-2-one (Preparatory
Example 10,
90.mg, 0.5600mm01) in N,N-dimethylformamide (2mL) to 0 C was added sodium
hydride (60 % dispersion in mineral oil) (29.22me, 0.7300mmo1) and the
reaction
mixture was stirred at that temperature for 1 hour. tert-butyl N4[2-
(chloromethyl)-1-
isopentyl-benzimidazol-5- yl]methyl]carbamate (obtained according to the
procedure
set out in WO 2010/103306, 205.6mg, 0.5600mm01) was added and the reaction
mixture was slowly allowed to warm up to room temperature overnight.
LCMS shows the expected product is present (Rt = 3.24 min m/z 490), plus
unreacted
RHS (Rt = 0.88min m/z 161) and an impurity (Rt = 2.37min m/z 508).
The reaction mixture was quenched with water (1 ml), diluted with Et0Ac
(100m1), and
washed with brine (3 x 50m1), the organic layer was dried (MgSO4), filtered
and
evaporated in vacuo. The crude was purified by column purification (Biotage,
10g)
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
38
eluting with Me0H : DCM (gradient 0:100 to 5:95) to give a light yellow oil,
with was
azeotroped with Petroleum Ether to give the final product as a cream-coloured
foam,
dried to a constant weight under vacuum (115mg).
1H NMR (500 Chloroform-d) 6 8.15 (d, J = 5.4 Hz, 1H), 7.85 (s, 1H),
7.36 (s,
2H). 7.12 (dd, J = 7.4, 1.5 Hz, 1H), 6.95 (dd, J = 7.4, 5.3 Hz, 1H), 5.53 (s,
2H), 4.93 (s,
1H), 4.43 (d, J = 5.8 Hz, 2H), 4.33 (t, J = 8.1 Hz, 2H), 1.90 (q, J = 4.3 Hz,
2H), 1.63
(dq, J = 10.1, 5.9, 4.9 Hz, 5H), 1.46 (s, 9H)
LCMS - clean product at Rt = 3.15 min m/z 490 [MI-1]+.
Preparatory Example 13
Spiro [1 H-pyrrolo[2, 3-c]pyridine-3, 1 '-cyclopropand-2-one
To a red suspension of 1,3-dihydropyrrolo[2,3-c]pyridin-2-one hydrochloride
(767.mg,
4.49 mmol) and diisopropylamine (2.52mL, 17.98 mmol) in tetrahydrofuran (40mL)
under N2 was cooled down at -40 C using a dry ice/acetonitrile bath n-
Butyllithium
solution (9.5 mL, 23.75mmo1) was added drop wise over 60 min, via syringe.
When
addition was complete, the dry ice/acetonitrile bath was changed for an ice
bath and
when the reaction temperature reached 0 C, a solution of 1,2- dibromoethane
(0.77mL,
9.0 mmol) in THF (5m1) was added dropwise over 90min, and a further addition
of THE
(15m1) led to a red suspension. The reaction mixture was allowed to warm
slowly tort
(without removing the ice bath) and it was left stirring at room temperature
from 18:30
overnight. Carefully addition of saturated aqueous NH4C1 solution (60m1) and
phases
were separated. The dark red viscous aqueous phase was extracted with Et0Ac (
5x60m1). Organics were combined, washed with brine (1x50m1) dried using MgSO4,
filtered through a sinter and concentrated under vacuum. The resulting crude
beige solid
material (250mg) was adsorbed onto silica and chromatographed using a lOg pre-
packed Biotave cartridge, gradient elution with a mixture of DCM 90% and DCM
/Me0H/ NH3 (9:1:0.2) 10% with further gradients of this mixture up to 100%.
Fractions
containing product were collected, combined and concentrated under vacuum to
afford
77 mg of the product as a yellow-brown solid.
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
39
LCMS-LCQ: M/Z [M+H]+ : 161.27 RT: 0.45 min
1H-NMR: 1H NMR (500 MHz, DMSO-d6) 6 10.70 (s, 1H), 8.33 - 8.02 (m, 2H), 7.06
(d, J = 4.7 Hz, 1H), 1.70 (q, J = 3.8, 3.4 Hz, 2H), 1.57 (q, J = 3.8 Hz, 2H).
Preparatory Example 14
tert-butyl N-1/1-isopen04-2-1-(2'-oxospirokyclopropane-1,3'-pyrrolof2,3-
c7pyr1d1nel-
1'-yl)methylibenzimitlazol-5 -ylimethylkarbamate
To a solution of spiro[1H-pyrrolo[2,3-c]pyridine-3,1'-cyclopropane]-2-one
(Preparatory
Example 13, 77.04mg, 0.4800mmo1) in N,N-dimethylformamide (2mL) cooled to 0 C
using an ice bath, sodium hydride (60 % dispersion in mineral oil) (22.74mg,
0.5700mmo1) was added in one portion and the reaction mixture was stirred at 0
C for 1
hour. A solution of tert-butyl N-R2-(chloromethyl)-1-isopentyl-benzimidazol-5-
yl]methyl]carbamate (160. mg, 0.4400mmo1)tert-butyl N-R2-(chloromethyl)-1-
isopentyl-benzimidazol-5-yl]methyl]carbamate (160.mg, 0.4400mmo1)Llin DMF
(1m1)
was slowly added dropwise for 1 h and the reaction mixture was left stirring
at room
temperature overnight. H20 (3m1) was added and crude material was concentrated
in
vacuo using n-heptane (4x12m1) to remove as much DMF as possible. Addition of
H20
(60m1) and crude was extracted using Et0Ac (5x20m1). Organics were separated,
combined, washed with brine (1x50m1), dried using MgSO4, filtered though a
sinter and
concentrated under vacuum to afford crude material which was triturated with
ether (2x
7m1). The resulting solid adsorbed on silica and purified by flash
chromatography, using
a lOg prepacked Biotage column and gradient elution from DCM 100% to a mixture
with DCM/ Me0H/ NT-I3 (9:1:0.2) from 0% to 60%. The resulting solid on
concentration of fractions was purified using a 5g Grace column, with gradient
elution
from 100% Et0Ac and an increasing gradient of a mixture Et0Ac/ Me0H (95:5)
from
0% up to 100%. This gave the title compound as a white solid (62 mg).
LCMS-LCQ: M/Z [M+H]+: 490.08 RT: 2.57 min
1H NMR (500 MHz, Chloroform-d) 6 8.73 (s, 1H), 8.31 (d, J = 19.9 Hz, 1H), 7.68
(s,
1H). 6.80 (s, 1H), 5.34 (s, 2H), 4.89 (s, 1H), 4.44 (s, 2H), 4.23 (dd, J =
20.0, 12.8 Hz,
2H). 2.24- 1.85 (m, 3H), 1.69 (s, 4H),1.48 (s, 12H), 1.38- 1.13 (m, 12H), 0.97
(d, J=
6.2 Hz, 7H).
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
Preparatory Example 15
2-(Chloromethyl)-1-tetrahydropyran-4-yl-benzimidazole-5-carbonitrile
5 A mixture of 2-chloro-1,1,1-triethoxy-ethane (2.17mL, 12.66mmo1)land 3-
amino-4-
(tetrahydropyran-4-ylamino)benzonitrile (275.mg, 1.27mmo1)Owas heated to 80 C
for
lh. LCMS shows product and an intermediate. Excess reagent was removed under
vacuum and the crude was purified by column chromatography (SiO2 10g, eluent:
20%
Et0Ac in Petroleum Ether to 100% Et0Ac. The second fraction collected was the
10 desired product as a beige solid (100mg)
LCMS-LCQ Rt: 2.08 m/z: 276 [M-4-1]
NMR (500 MHz, DMSO-d6) 6 7.37 - 7.31 (m, 1H), 6.95 (s, 1H), 6.79 (d, J = 8.3
Hz,
1H), 5.04 (d, J = 8.2 Hz, 1H), 4.36 - 4.29 (m, 2H), 4.08 (s, 2H), 3.89 - 3.82
(m, 2H),
15 3.61 (d, J = 11.2 Hz, 1H), 3.42 (t, J = 11.7 Hz, 2H), 1.83 (d, J = 12.0
Hz, 2H), 1.57 -
1.44 (m, 2H), 1.36- 1.27 (m, 3H).
The first fraction collected was the intermediate as a white solid (230mg)
which was
suspended in Et0H (3 ml) and heated for 4h. The solvent was then evaporated to
leave a
brown solid, which was triturated with Et20 and the solid filtered to give a
further
20 quantity of the desired product (87mg)
II-1 NMR. (500 MHz, DMSO-d6) 6 8.22 (d, J = 1.6 Hz, 1H), 7.96 (d, J = 8.5 Hz,
1H),
7.65 (dd, J = 8.5, 1.6 Hz, 1H), 5.19 (s, 3H), 4.79 (tt, J = 12.5, 4.6 Hz, 2H),
4.05 (dd, J =
11.6, 4.5 Hz, 31-1), 3.55 (td, J = 11.7, 2.1 Hz, 3H), 2.46 -2.31 (m, 41-1),
1.94 - 1.82 (m,
3H).
25 LCMS-LCQ Rt: 1.94 m/z: 276 [M+El]
Preparatory Example 16
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
41
2-[(6'7fluoro-2'-oxo-spirokyclopropane-1,31-indoline1-11-yl)methy11-1-
tetrahydropyran-
4-yl-benzimidazole-5-carbonitrile
A mixture of 6'-fluoro[spirocyclopropane-1, 3'-indoline]-21-one (Preparatory
example 3,
128.5mg, 0.7300mmo1) and 2-(chloromethyl)-1-tetrahydropyran-4-yl-benzimidazole-
5-
carbonitrile (Preparatory Example 15, 200.mg, 0.7300mmol) and caesium
carbonate
(354.5mg, 1.09mmo1) in acetonitrile (10mL) was stirred at r.t. overnight.
Volatiles were
removed in vacno, the residue was stirred in water (20m1) and the suspension
filtered to
leave a grey solid, which was then triturated with Et20 and the solid filtered
(228mg)
LCMS-MDAP Rt: 19.0 m/z: 417 [M+H]
11-1 NMR (500 MHz, DMSO-d6) 6 8.18 (s, 1H), 7.90 (d, J = 8.5 Hz, 1H), 7.59 (d,
J = 8.5
Hz, 1H), 7.12 - 7.02 (m, 2H), 6.80 (t, J = 8.7 Hz, 1H), 5.43 (s, 2H), 4.84 (t,
J = 12.6 Hz,
1H), 4.03 (dd, J= 12.3, 4.3 Hz, 2H), 3.51 - 3.44(m, 2H), 243 -2.33 (m, 2H),
1.82 -
1.73 (m, 2H), 1.69 (d, J = 4.3 Hz, 2H), 1.57 (q, J = 4.3, 3.7 Hz, 211).
Preparatory Example 17
N-(4-chloro-2-nitro-phenyl)tetrahydropyran-4-amine
A mixture of 5-Chloro-2-fluoronitrobenzene (3.5g, 19.94mm01), tetrahydropyran-
4-
amine (2.29m1, 21.93mmo1) and potassium carbonate (5.51g, 39.88mmo1) in MeCN
(100mL) was stirred at 25 c over weekend then heated to 50 C until LCMS shows
reaction complete. The reaction mixture was filtered and washed with Et0Ac and
concentrated to dryness to leave an orange solid (5.1g)
1H NMR (500 MHz, Chloroform-d) 6 8.20 (d, J = 2.6 Hz, 1H), 8.06 (d, J = 7.3
Hz, 1H),
7.38 (dd, J = 9.2, 2.6 Hz, 1H), 6.85 (d, J = 9.2 Hz, 1H), 4.03 (dt, J = 12.1,
3.8 Hz, 2H),
3.77 - 3.67 (m, 1H), 3.63 - 3.53 (m, 2H), 2.12 - 2.03 (m, 2H), 1.68 (dtd, J =
14.1, 10.2,
4.1 Hz, 2H).
LCMS Rt: 4.31 m/z: 257 [M+H]
Preparatory Example 18
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
42
4-chloro-NI-tetrahydropyran-4-yl-benzene-1,2-diarnine
An solution of potassium carbonate (16.48g, 119.2 lmmol) and sodium dithionite
(27.67g, 158.95mmo1) in water (30mL) was added dropwise to a solution of N-(4-
chloro-2-nitro-phenyl)tetrahydropyran-4-amine (5.1g, 19.87mmo1) in
acetonitrile
(70m1) and water (30m1) and the reaction mixture was stirred at rt for about
48h.
LCMSRt: 1.97 m/z: 227 [M+H]
Et0Ac (100m1) was added to the reaction, the layers were separated and the
aquous
layer further extracted with Et0Ac (2x5 0m1). The combined organic layer were
washed
with saturated brine solution (lx 60 mL). The organics were dried (MgSO4) and
concentrated to dryness under reduced pressure lo leave a brown solid (4 g)
and the
crude was purified by column chromatography (SiO2 25g, eluent 50% Et0Ac in PE
to
1000/a Et0Ac) to give the title compound (2.0g).
1H NMR (500 MHz, DIVISO-d6) 6 6.54 (d, J = 2.2 Hz, 1H), 6.49 -6.38 (m, 2H),
4.83 (s,
2H). 4.29 (d, J = 7.6 Hz, 1H), 3.86 (dt, J = 11.6, 3.3 Hz, 2H), 3.39 (td, J =
11.3, 2.2 Hz,
3H), 1.92 - 1.83 (m, 2H), 1.37 (qd, J = 11.3, 4.2 Hz, 2H). LCMS-LCQ Rt: 1.79
m/z:
227 [M+H]
Preparatory Example 19
5-chloro-2-(chloromethyl)-1-tetrahydropyran-4- yl-benzimidazole
A mixture of 4-chloro-N1-tetrahydropyran-4-yl-benzene-1,2-diamine (1.g,
4.41mmol)
and 2-chloroacetic acid (0.63g, 6.62mmo1) in 4M HC1(50mL) was heated to 60 C
until
LCMS showed completion (72h). The reaction mixture was allowed to cool to r.t
and
the precipitate that formed was collected by flitration and was washed with
H20
(2x50m1). The white residue was treated with a sat. solution of NaHCO3 and the
product
extracted with Et0Ac (3x100m1) (slightly insoluble), dried (MgSO4) and solvent
evaporated to leave a light brown solid (658mg, N2006-173-1)
1H NMIR (500 MHz, DMSO-d6) 6 7.78 (d, J = 8.8 Hz, 1H), 7.71 (d, J = 2.1 Hz,
1H),
7.29 (dd, J = 8.8, 2.1 Hz, 1H), 5.15 (s, 2H), 4.74 (tt, J = 12.2, 4.4 Hz, 1H),
4.05 (dd, J =
11.5, 4.5 Hz, 2H), 3.54 (td, J= 11.9, 2.0 Hz, 2H), 2.45 -2.34 (m, 2H), 1.88 -
1.80 (m,
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
43
2H).
LCMS-LCQ Rt: 3.24 m/z: 285 [M+]
.. Preparatory Example 20
4-(((I R,4R)-4-Hydroxycyclohexyl)amitio)-3-nitrobenzonitrile
In a flask equipped with a reflux condenser, trans-4-aminocyclohexanol
hydrochloride
.. (2.70g, 17.80mm01es) was suspended in iso-propanol (15m1). To this stirred
suspension,
triethylamine (4.40m1, 31.49mmo1es) was slowly added, followed by 4-chloro-3-
nitrobenzonitrile (2.50g, 13.69mmo1es). The resulting yellow suspension was
heated at
65 C for 36h, allowed to cool down to room temperature before water (10m1) was
added to the reaction mixture. The resulting precipitate was collected by
filtration and
.. successively washed with water and iso-propanol, to yield 4-(((1R,4R)-4-
hydroxycyclohexyl)amino)-3-nitrobenzonitrile as a yellow crystalline solid
(3.28g,
12.55mmo1es, 92%). The resulting solid was further purified by
recrystallization from
hot ethanol.
m/z 262.2 [MH]+
111 NMIR (500 MHz, DMSO-d6) 6 8.49 (s, 1H), 8.18 (d, J = 7.8 Hz, 1H), 7.79 (d,
J = 9.0
Hz, 1H), 7.26 (d, J = 9.1 Hz, 1H), 4.63 (s, 1H), 3.67 (dtd, J = 14.9, 10.4,
4.0 Hz, 1H),
3.57-3.41 (m, 1H), 2.05-1.88 (m, 2H), 1.89-1.75 (m,2H), 1.53-1.19 (m, 4H).
Preparatory Example 21
3-Ainino-4-(((JR,4R)-4-hydroxycyclohexyl)amino)benzoni tri le
.. A flask flushed with nitrogen was successively charged with 4-4(1R,4R)-4-
hydroxycyclohexyl)amino)-3-nitrobenzonitrile from Preparatory Example 20
(1.31g,
5.01mmoles), 10% palladium on carbon (0.131g) and methanol (40m1). The flask
was
then flushed with hydrogen and the reaction mixture left to stir under a
hydrogen
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
44
atmosphere (hydrogen balloon). After 3h, Pd/C 10% was removed by filtration
and the
filtrate concentrated in vacuo. Chromatography on silica
(dichloromethane/ethanol/ammonia, 200/8/1) provided 3-amino-4-(((1R,4R)-4-
hydroxycyclohexyl)amino)benzonitrile as an off-white crystalline solid
(0.253g,
1.09mmo1es, 22%).
m/z 232.2 [lVfE1]+
11-1 NMR (500 MHz, DMSO-d6) 6 6.89 (dd, J = 8.2, 2.0 Hz, 1H), 6.75 (d, J = 2.0
Hz,
1H), 6.49 (d, J = 8.2 Hz, 1H), 5.02 (d, J = 7.4 Hz, 1H), 4.94 (s, 2H), 4.57
(d, J = 4.3 Hz,
1H), 3.44 (d, J = 11.8 Hz, 2H), 3.24 (ddd, J = 10.6, 7.2, 3.7 Hz, 1H), 1.94
(dd, J= 11.8,
4.1 Hz, 3H), 1.89-1.79 (m, 3H), 1.35-1.19 (m, 5H).
Preparatory Example 22
2-(Chloromethyl)-1-WR,4R)-4-hydroxycyclohexyl)-1H-benzo[dflinidazole-5-
carbonitrik
In a flask equipped with a reflux condenser, 2-chloro-1,1,1-triethoxyethane
(544u1,
2.85mmo1es) was added at once to a solution of 3-amino-4-(((1R,4R)-4-
hydroxycyclohexyl)amino)benzonitrile from Preparatory Example 21 (0.220g,
0.95mmo1es) in ethanol (10m1). The resulting solution was heated at 70 C for
24h,
before more 2-chloro-1,1,1-triethoxyethane (544 1, 2.85mm01es) was added and
heating
continued for a further 24h. The resulting solution was allowed to cool down
to room
temperature and the solvent removed in vacua. Chromatography on silica
(dichloromethane/ethanol/ammonia, 200/8/1) provided 2-(chloromethyl)-141R,4R)-
4-
hydroxycyclohexyl)-1H-benzo[d]imidazole-5-carbonitrile as a white crystalline
solid
(0.251g, 0.86mmo1es, 91%).
m/z 289.9 [A/ft+
IHNMR (500 MHz, DMSO-d6) 6 8.20-8.14 (m, 1H), 8.01 (dd, J = 8.6, 2.0 Hz, 1H),
7.65-7.57 (m, 1H), 5.16 (d, J = 1.9 Hz, 2H), 4.74 (s, 1H), 4.47 (tt, J = 12.5,
4.1 Hz, 1H),
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
3.71 (tt, J = 10.4, 4.3 Hz, 1H), 2.26 (ddt, J = 16.3, 12.4, 6.2 Hz, 2H), 2.05-
1.93 (m, 2H),
1.92-1.82 (m, 2H), 1.51-1.38 (m, 2H).
Preparatory Example 23
5
2-((6P-Fluoro-2'-oxospiro kyclopropane- 1 ,3'-indolinkl'-yOmethyl)-1-((JR,4R)-
4-
hydroxycyclohexyl)-1H-benzo[d]imidazole-5-carbonnrile
To a solution of 6'-fluorospiro[cyclopropane-1,3'-indolin]-2'-one from
Preparatory
10 Example 4 (0.084g, 0.475mm01es) ill acetonitrile (5m1) was successively
added 1,8-
diazabicyclo[5.4.0]undec-7-ene (142 1, 0.95mm01es) and 2-(chloromethyl)-1-
((1'R,4R)-4'-hydroxycyclohexyl)-1H-benzo[d]imidazole-5-carbonitrile from
Preparatory Example 22 (0.165g, 0.57mmo1es). The resulting suspension was
heated at
90 C for 2h. The resulting deep purple solution was allowed to cool down to
room
15 temperature before water (20m1) was added. The resulting precipitate was
collected by
filtration, washed with water and purified by chromatography on silica (100%
ethyl
acetate), yielding 2-((6'-fluoro-2'-oxospiro[cyclopropane-1,3'-indolin1-1'-
yl)methyl)-1-
((1R,4R)-4-hydroxycyclohexyl)-1H-benzo[d]imidazole-5-carbonitrile as a white
crystalline solid (0.165g, 0.38mmo1es, 67%).
m/z 431.0 [ME1]+
NMR (500 MHz, DMSO-do) 6 8.17 (d, J = 1.7 Hz, 1H), 7.95 (d, J = 8.6 Hz, 1H),
7.56 (dd, J = 8.6, 1.6 Hz, 1H), 7.09-7.01 (m, 2H), 6.79 (ddd, J = 10.3, 8.3,
2.4 Hz, 1H),
5.38 (s, 2H), 4.72 (d, J = 4.4 Hz, 1H), 4.51 (s, 1H), 3.72-3.60 (m, 1H), 2.27-
2.16 (m,
2H), 1.92 (d, J = 12.2 Hz, 2H), 1.70 (dt, J = 6.9, 4.2 Hz, 4H), 1.58 (q, J =
3.8 Hz, 2H),
1.42-1.29 (m, 2H).
Example 1
1 '-85-(Aminomethyl)-1-(4,4,4-trifluorobuty1)-1H-1,3-benzoa'iazol-2-ylimethy0-
6'-
fluoro-1 ',2 '-dihydrospiro kyclopropane-1, 3 '-indole1-2 '-one
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
46
To a solution of tert-butyl N-[(2-[(6'-fluoro-2'-oxo-1',2'-
dihydro[spirocyclopropane-
1,31-indole]-11-ylmethy1]-1-(4,4,4-trifluorobuty1)-1H-1,3-
benzodiazyl)methyl]carbamate
from Preparatory Example 6 (1030 mg, 1.88mmo1) in dichloromethane (3.5mL)
under
nitrogen was added hydrogen chloride solution (2M in Et20) (12.54mL,
25.08mmo1).
A pink/white solid precipitate formed almost immediately and the heterogenous
mixture
was stirred at room temperature for 6 hours. The reaction mixture was then
concentrated
under vacuum at room temperature and azeotroped further with 3 x 20m1DCM. The
crude product was sonicated and triturated with diethyl ether (2x15m1 then 3 x
10 m1).
The solvent was removed by decantation and triturated further with 10 ml
ether. The
mixture was filtered and dried to give 865mg of the desired product as an off
white solid
as crude HC1 salt. (95% crude yield).
The crude HC1 salt was partitioned between ethyl acetate (80m1) and saturated
aqueous
sodium bicarbonate (80m1). The organics were separated and the aqueous was
extracted
with further ethyl acetate (3x30m1). The organics were combined, dried over
magnesium sulphate, filtered and concentrated under vacuum. The residue was
purified
using flash chromatography, (25g pre-packed Biotage cartridge, silica adsorbed
material
and eluted with Dichloromethane/ethanol/ammonia (100:0:0 gradient to 95:5:1)
to
afford 539 mg (65% yield) of the desired product free base as a white solid.
LCMS:
M/Z [M+H]+ : 447.10
1H-NMR:
1H N1V1R (500 MHz, DMS0d6) 6 7.56 (s, 1H), 7.51 (d, J = 8.3 Hz, 1H), 7.23 (d,
J = 8.3
Hz, 1H), 7.19 - 7.12 (m, 1H), 7.06 (dd, J = 8.3, 5.3 Hz, 1H), 6.85 -6.76 (m,
1H), 5.28
(s, 2H), 4.35 (t, J = 7.5 Hz, 2H), 3.77 (s, 2H), 2.32 (dt, J= 21.6, 8.1 Hz,
2H), 1.83 (p, J
= 8.1 Hz, 2H), 1.67 (q, J= 3.9, 3.4 Hz, 2H), 1.57 (q, J = 4.2, 3.7 Hz, 2H).
Example 2
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
47
1 '-{1.5-(Aininomethyl)-1-(4,4,4-trifluorobutyl)-1H-1,3-benzodiazol-2-
yllmethyll-
6 flitoro-1 ',2 '-dihydrospiro[cyclopropane-1, 3 '-indole]-2 '-one,
hydrochloride
To a solution of 1'-{[5-(aminomethyl)-1-(4,4,4-trifluorobuty1)-1H-1,3-
benzodiazol-2-
yl]methy1}-6'fluoro-1',2'-dihydrospiro[cyclopropane-1,3'-indole]-2'-one from
Example
1 (539.mg, 1.21mmo1) in dichloromethane (10mL) was added hydrogen chloride
solution 2.0 M in diethyl ether (0.6mL, 1 . 21mmol) dropwise and the reaction
mixture
stirred for 30 min. The solvent was then evaporated under vacuum. The residue
was
dissolved in 20m1 Me0H and concentrated under vacuum at room temperature and
dried further at 40 C leading to product as a HC1 salt.
LCMS:
1\4/Z [M+H]+ : 447.05
114-NMR:
1H NMR (500 MHz, DMSO-d6) 6 8.36 (s, 3H), 7.77 (s, 1H), 7.71 (d, J = 8.3 Hz,
1H),
7.42 (d, J = 8.2 Hz, 1H), 7.15 (dd, J = 9.6, 2.4 Hz, 1H), 7.08 (dd, J = 8.3,
5.4 Hz, 1H),
6.82 (ddd, J = 10.5, 8.3, 2.4 Hz, 1H), 5.35 (s, 2H), 4.41 (t, J = 7.7 Hz, 2H),
4.09 (q, J =
5.8 Hz, 2H), 2.43 ¨ 2.26 (m, 2H), 1.87 (p, J = 8.0 Hz, 2H), 1.63 (dq, J =
54.6, 4.2 Hz,
4H),
Example 3
1'-((5-(aininomethyl)- -(4,4,4-trifluorobutyl)-1H-benzoldlimidazol-2-
yl)inethyl)spiro[cyclopropane- ,3'-pyrrolo[2,3-bJpyridinl-2'(1'11)-one
A suspension of tert-butyl N-H2-[(2'-oxo[spirocyclopropane-1,3'-pyrrolo[2,3-
b]pyridine1-1'-yl)methyl]-1-(4,4,4- trifluorobutyl)benzimidazol-5-
yllmethyl]carbamate
(Preparatory Example 11, 184.mg, 0.35mmo1) ill dichloromethane (4mL) was
treated
with trifluoroacetic acid (1.86mL, 24.32mmo1) and the resulting solution was
left
stirring for 1 hr at room temperature. The volatiles were removed under
reduced
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
48
pressure, the residue was purified by SCX-2 cartridge, eluting first with Me0H
and then
with a 2M NH3 solution in Me0H. Fractions containing product were combined,
evaporated in vacuo and further purified by column chromatography (Biotage,
10g)
eluting with DCM : Me0H : NH3 (98:2:0.2 to 90:10:1). 0Fractions containing
product
were combined and solvent evaporated to give a white solid which was further
dried
under vacuum at 40C in vacuum pistol to give the product (113mg).
1H NNIR (500 MHz, DMSO-d6) 6 8.06 -7.98 (m, 1H), 7.51 (d, J = 8.3 Hz, 1H),
7.47
(d, J = 7.2 Hz, 1H), 7.43 (s, 1H), 7.19 (d, J = 8.3 Hz, 1H), 7.00 (dd, J =
7.3, 5.3 Hz, 1H),
5.26 (s, 2H), 4.43 (t, J = 7.4 Hz, 2H), 3.74 (s, 2H), 2.44 - 2.33 (m, 2H),
2.00 (dd, J =
15.1, 7.5 Hz, 2H), 1.78 (q, J = 4.0, 3.5 Hz, 2H), 1.65 (q, J = 4.2, 3.8 Hz,
2H). 0
LCMS Rt: 0.51-0.69min m/z: 430 [M+1-1]+
Example 4
1'45-chloro-1-(tetrahydro-2H-pyran-4-y1)-1H-benzoklfiinidazol-2-
Amethyl)spirokyclopropane-1,3'-pyrrolo[2,3-qpyridinl-2'(1'H)-one
To a solution of spiro[1H-pyrrolo[2,3-c]pyridine-3,1'-cyclopropane]-2-one (30
mg,
0.1900mmol, Preparatory Example 13) in dimethylformamide (3mL) at 0 C was
added
60% sodium hydride (8.99mg, 0.2200mm01) was stirred for 5min at 0 C and for
another 10 at r.t. before adding a solution of 5-chloro-2-(chloromethyl)-1-
tetrahydropyran-4- yl-benzimidazole (53.41mg, 0.1900mmo1, Preparatory Example
19)
in N,N-dimethylformamide (3mL) .
After 30 min LCMS shows no RHS left so the reaction mixture was quenched with
H20
(5m1) and product extracted into Et0Ac (3x30m1). Combined organic layers were
washed with brine, dried (MgSO4) and solvent evaporated to leave an orange
oil.
(82mg) which was purified by column chromatography (SiO2 10g, eluent: 100%
Et0Ac
to 5%Me0H in Et0Ac) and the product re-chromatographed by column
chromatography (6g SiO2; 100 ./oEt0Ac). Fractions containing product were
combined
and solvent evaporated and dried overnight in vacuum pistol at 40 C. A light
yellow
solid was obtained (16mg, N2006-192-2)
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
49
1H NMR (500 MHz, DMSO-d6) 6 8.40 (s, 1H), 8.24 (d, J = 4.8 Hz, 1H), 7.72 (d, J
=
9.9 Hz, 2H), 7.23 (d, J = 8.8 Hz, 1H), 7.15 (d, J = 4.6 Hz, 1H), 5.44 (s, 2H),
4.80 (dq, J
= 12.5, 6.8, 5.0 Hz, 1H), 4.02 (dd, J= 11.1, 4.7 Hz, 3H), 3.45 (t, J = 11.6
Hz, 2H), 2.35
(qd, J = 12.3, 4.4 Hz, 3H), 1.84 (q, J = 4.1, 3.7 Hz, 2H), 1.71 (dq, J = 8.2,
4.2 Hz, 5H).
LCMS-MDAP Rt: 11.84 mz:409 [M+H]
Example 5
1'45-(aniinoinethyl)-1-isopentyl-IH-benzo[dfinfidazol-2-
yl)methyl)Apirocyclopropane-1,3'-pyrrolon,3-Npyridini-2'(1'H)-one
A suspension of tert-butyl N-H1-isopenty1-2-[(21-oxo[spirocyclopropane-1,3'-
pyrrolo[2,3-b]pyridine]-1'-yl)methyl]benzimidazol-5 -yl]methyl]carbamate
(Preparatory
Example 12, 115.mg, 0.2300mmo1) in dichloromethane (3mL) was treated with
trifluoroacetic acid (1.26mL, 16.44mm01) and the resulting solution was left
stirring for
1 hr at room temperature. LCMS showed product present at Rt: 1.24min m/z: 390
[MH]+. The volatiles were removed under reduced pressure, the residue was
purified by
SCX-2 cartridge, eluting first with Me0H and then with a 2M NH3 solution in
Me0H.
Fractions containing product were combined, evaporated in vacuo and further
purified
.. by column chromatography (Biotage, 10g) eluting with DCM : Me0H : NH3
(98:2:0.2
to 90:10:1). Fractions containing product were combined and solvent evaporated
to
give a white solid which was further dried under vacuum at 40C in vacuum
pistol to
give the product (60mg).
.. 1H NMR (500 MHz, DMSO-d6) 6 8.06 (d, J = 5.3 Hz, 1H), 7.53 -7.36 (m, 3H),
7.18
(d, J = 8.1 Hz, 1H), 7.02 (t, J = 6.5 Hz, 1H), 5.25 (s, 2H), 4.31 (t, J = 7.6
Hz, 2H), 3.74
(s, 2H), 1.84 - 1.73 (m, 2H), 1.71 - 1.53 (m, 5H), 0.94 (d, J = 5.8 Hz, 6H).
LCMS Rt: 0.51-0.69min m/z: 390 [M+H]+OLCMS-MDAP: Rt = 10.77min m/z 390
[MH]+
Example 6
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
l'-((5-(aminomethyl)-1-isopenty1-1H-benzokllimidazol-2-
Amethyl)spiro[cyclopropane-1 ,3'-pyrrolo[2, 3-clpyridin]-2'(1'H)-one
A suspension of tert-butyl N-[[1-isopenty1-2-[(2'-oxospiro[cyclopropane-1,3'-
5 pyrrolo[2,3-c]pyridine]-1'-yl)methyl]benzimidazol-5 -yl]methyl]carbamate
(Preparatory
Example 14, 62mg, 0.1300mmol) in dichloromethane (2mL) was treated with
trifluoro
acetic acid (0.68mL, 8.86mmo1) and the resulting solution was left stirring
for 1 hr at
room temperature. The volatiles were removed under reduced pressure, the
residue was
purified by SCX-2 cartridge, eluting first with Me0H and then with a 2M NH3
solution
10 in Me0H. Fractions containing product were combined, evaporated in vacuo
and
further purified by column chromatography (Biotage, 5g) eluting with DCM :
Me0H :
NH3 (98:2:0.2 ) from 0% up to 100% of this mixture. Fractions containing
product were
combined and solvent evaporated to give a white solid. Further ether
trituration (3 x
6m1) was performed to remove additional impurities to give slightly impure
title
15 .. compound (60mg).
LCMS-LCQ: M/Z [114-41]+ : 390.4 RT: 0.47 min0
1H NMR (500 MHz, DMSO-d6) 6 8.42 (s, 1H), 8.24 (d, J = 4.9 Hz, 1H), 7.57 (s,
1H),
7.42 (d, J = 8.4 Hz, 1H), 7.22 (d, J = 8.4 Hz, 1H), 7.16 (d, J = 4.8 Hz, 1H),
5.32 (s, 2H),
4.26 (t, J = 8.2 Hz, 2H), 3.79 (s, 2H), 1.85 (q, J = 4.0 Hz, 2H), 1.71 (q, J =
4.3, 3.9 Hz,
20 .. 2H), 1.68 - 1.58 (m, 1H), 1.46 (q, J = 7.5 Hz, 2H), 0.92 (d, J = 6.5 Hz,
7H).
Example 7
l'-((5-(aminometkv1)-1-(tetrahydro-2H-pyran-4-y1)-1H-benzo[dinnidazol-2-
yinnethyl)-
6'-fluorospiro[cyclopropane-1,3'-indolinl-2'-one
2-[(6'-fluoro-2'-oxo-spiro[cyclopropane-1,31-indoline]-11-yl)methyl]-1-
tetrahydropyran-
4-yl-benzimidazole-5-carbonitrile (Preparatory Example 16, 100mg, 0.2400mmo1)
was
dissolved in a mixture of THF/NH3 (6/0.5m1) and hydrogenated by passing the
solution
through a small cartridge of Ni-Ra in the H-Cube at lml/min at 20bar and 45 C
for
.. about 45min. The solvent was evaporated to leave the title compound as a
white solid
(105mg)
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
51
1H NMR (600 MHz, DMSO-d6) 6 7.59 - 7.54 (m, 2H), 7.16 (dd, J = 8.5, 1.7 Hz,
1H),
7.10 (dd, J = 9.7, 2.4 Hz, 1H), 7.03 (dd, J = 8.2, 5.3 Hz, 1H), 6.78 (ddd, J =
10.4, 8.3,
2.4 Hz, 1H), 5.34(s, 2H), 4.73 (tt, J = 12.2, 4.3 Hz, 1H), 4.00 (dd, J = 11.5,
4.4 Hz, 2H),
3.74 (s, 2H), 3.41 (td, J = 11.9, 1.9 Hz, 2H), 2.36 (qd, J= 12.4, 4.6 Hz, 2H),
1.67(q, J=
3.9 Hz, 2H), 1.62 (dd, J = 13.0, 3.9 Hz, 2H), 1.55 (q, J = 3.8 Hz, 2H).
LCMS-MDAP Rt: 10.68 m/z: 421 [MH-H]
Example 8
/'-((5-(Aminomethyl)-1-0R,410-4-hydroxycyclohexy0-1H-benzoldJimidazol-2-
Ameihyl)-6'-fluorospirokyclopropane-1,3'-indolinl-2'-one
A flask flushed with nitrogen was successively charged with 2-((6'-fluoro-2'-
oxospiro[cyclopropane-1,31-indolin]-1'-yOmethyl)-141R,4R)-4-hydroxycyclohexyl)-
1H-benzokelimidazole-5-carbonitrile (0.139g, 0.32mmo1es), 10% palladium on
carbon
(0.014g), methanol (4m1) and 37% hydrochloric acid (159 1, 1.615mmo1es). The
flask
was flushed with hydrogen and stirred under a hydrogen atmosphere for 3h.
Water
(10m1) was added to the reaction mixture, the catalyst removed by filtration
and the
filtrate concentrated in vacua. Partition of the residue between water (20m1)
and ethyl
acetate (10mL) was followed by extraction of the aqueous phase with ethyl
acetate (2 x
10m1). The aqueous layer was taken to pH= 10 using a 30% aqueous ammonia
solution
and the resulting suspension extracted with ethyl acetate (4 x 20m1). The
combined
organic extracts were washed with brine (50m1), dried over sodium sulfate and
concentrated in vacuo, yielding 1'-((5-(aminomethyl)-1-((1R,4R)-4-
hydroxycyclohexyl)-111-benzo[d]imi dazol-2-yl)m ethyl)-6'-fl uorosp i ro [cycl
op rop ane-
1,3'-indolin]-2'-one as a white solid (0.117g, 0.27mmo1es, 84%). The resulting
solid was
further purified by recrystallization from hot ethanol.
m/z 435.2 [1VIE1]+
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
52
NMR (600 MHz, DMSO-do) 6 7.59 (d, J = 8.3 Hz, 1H), 7.56 (s, 1H), 7.12 (dd, J =
8.5, 1.6 Hz, 1H), 7.07 (dd, J= 9.6, 2.4Hz, 1H), 7.03 (dd, J = 8.2, 5.3 Hz,
1H), 6.77 (ddd,
J = 10.4, 8.2, 2.4 Hz, 1H), 5.29 (s, 2H), 4.65 (d, J = 4.5 Hz, 1H), 4.41 (tt,
J =12.3, 4.0
Hz, 1H), 3.74 (s, 2H), 3.61 (dp, J = 15.6, 5.3, 4.6 Hz, 1H), 2.19 (qd, J =
12.6, 3.3 Hz,
2H), 1.92-1.84 (m, 2H), 1.68 (q, J = 3.8 Hz, 2H), 1.56 (dt, J = 8.4, 3.8 Hz,
4H), 1.36-
1.26 (m, 2H).
Example 9: in vitro Efficacy
Compounds were subjected to RSV fusion assays and plaque reduction assays
according to the following protocols.
RSV fitsion assay
IfEK 293T/17 cells were cultured in 175 culture flasks in Dulbecco's medium
containing 10 % FBS and lx Penicillin-Streptomycin and warmed to 37 C prior
to use.
The cells were passaged by first washing briefly with 3 ml PBS followed by a 4
min
incubation with 3 TrypLE at 37 C. 7 ml media was then added to the flask and
the
cells dispersed via pipetting (x3) against the bottom of the flask. Two
further T75 flasks
were each seeded with 2 x 106 cells in 15 ml fresh media.
Cells were seeded on the T75 plates at the same density as on the 6-well
plates to the
area of a T75 flask and one 1.75 cm radius well from a 6-well plate were
compared.
7.79 x 2 ml of 3 x 105 cells m1-1 was used to seed a single T75 flask.
1-IEK cells were removed from a T75 flask as described above. The cells were
counted
and diluted to 3 x 105 cells/ml in fresh media. Two 175 flasks were each
seeded with
15.58 ml diluted cells.
The plasmid DNA (for pFR-Luc and pcDNA3.1_Ga14/ NFicB ) to be transfected into
the HEK cells was first prepared in serum free media (DMEM + Pen/Strep)
containing
the transfection reagent Fugene 6 (Promega). Transfections were set up as
follows (Luc
= pFR_Luc, Gal4 = pcDNA3.1+_Ga14/NEKB, A2_F = pcDNA3.1+_A2_F)
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
53
Transfections
Luc + A2 F 1
_ _
2 Gal4
Serum free media was placed in a 1.5 ml eppendorf tube then the fugene 6 was
added
into the media. The tube was vortexed for 1 s before being incubated at RT for
5 min.
The plasmid DNA was then added to the tube, vortexed for 1 s, then incubated
at RT for
15 min
The transfection reagents were then added to the appropriate T75 flask by
tipping the
flask on end and adding the reagents directly to the media already in the
flask. The
flask was then tipped on its back so the media could be mixed thoroughly
whilst not
disturbing the cells before placing the flask the right way up and incubating
overnight at
37 C and 5% CO).
Compounds were diluted (in a polypropylene round-bottomed 96 well plate 1:3 in
a
twelve point dilution curve to give top [final] of either 3.3 M, 1 M, 500 nM
200 nM
or 100 nM. The Control compound was always run at a concentration of 100 nM.
The cells were then counted and diluted to 4 x 105 cells/m1 in fresh media. 50
111 of
transfection population were added to all wells of the assay plates. 100 pl
diluted
compound (2 rows per compound), standard curve (one row) and controls (100 nM
RV
(100 % inhibition, four wells), DMSO (0% inhibition, eight wells)) were added
to the
appropriate wells. 50 la' of the diluted (4 x 105 cells/m1) population 2 cells
when then
added to all wells
The plates were then incubated for 24 hr at 37 C and 5% CO2
Buffers were prepared for the luciferase assay (20 mM tricine, 10 mM MgSO4, 1
mIV1
EDTA, 10 niM DTT ) and lysis and stored at -20oC. (25mM tris-phosphate, 8mM
MgC12,1mM DTT, 1% Triton X-100, 15% glycerol). Luciferin substrate was
prepared
from and stored at -80C ( 100mM Tris-HC1, 15.76 g/L, Coenzyme A, 10.36 g/L,
23.5mM luciferin, 7.48 g/L, 26.6mM ATP, 14.66 g/L
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
54
Luminescence was measured at appropriate time points as follows:
(a) Luciferase
Media was discarded into Virkon and the plates washed with 100u1 PBS per well.
20u1/well of lysis buffer is added to each well and incubated shaking for 5min
at RT.
Luciferin was added to LAAB at a dilution of 1:50 to give a working luciferin
buffer.
Add 100 1.11 working luciferin buffer to each well and luminescence was
measured
immediately.
(b) Resazurin
Media was discarded into Virkon and 100u1 SFM + 20u1 CellTitre-Blue solution
was
added to each well. The plates were incubated 37 "V, 5% CO2 for 2hrs.
Resorufin
fluorescence was measured at 590 nm.
Plaque Reduction Assay:
Vero cells were seeded in 96-well plates in a volume of 100 uL of Optimem
supplemented with 3% FCS at a concentration of' 4 x 104 cells per well. After
an
overnight incubation at 37 C in a humidified 5% CO2 atmosphere, the monolayer
of
cells should be approximately 90% confluent. Antiviral compounds were titrated
in
pre-warmed Serum Free (SF) Optimem in a U-bottom 96 well plate. For compounds
in a DMSO solution, titration in 100% DMSO was performed first and each
concentration added individually to a 2 x final concentration at 4% DMSO in SF
media
before mixing with yin's (2% final DMSO with virus). Media was then removed
from
cells and replaced with PBS (100 1/well). RSV stock was thawed and diluted in
SF
Optimem media to 4000 PFU/mLl. An equal volume of virus was added to
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
compounds on the titration plate. PBS was removed from cells which were then
inoculated with the virus/compound solution (50 uL/well). Cells were incubated
for
2 h in a 37 C + 5% CO2 humidified incubator to allow infection. Inoculum was
removed and media (Optimem + 1% FCS) added to cells (100W/well). Cells were
5 subsequently incubated for 48 h at 37 C + 5% CO2 in a humidified
incubator.
Iminunostaining Procedure:
Media was removed from cells and the monolayer washed with PBS. Cells were
10 fixed with ice cold 80% Acetone in PBS (100 1/well) for 20mins at -20 C.
Fixative was
removed and cells are dried for 30mins with plates inverted. Blocking solution
(5%
skim milk powder in PBS-T) was added to cells (1504/well) and plates were
incubated for 30mins at room temperature. Blocking solution was removed and
plates washed once with PBS-T. Primary antibody in blocking solution was added
to
15 plates (50111/well) and incubated for lh at 37 C. Plates were then
washed 3 times
with PBS-T. Secondary antibody in blocking solution was added to plates
(50uL/well) and incubated for 1 h at 37 C in the dark. Plates were washed as
above and
then dried for 10 mins. Plates were scanned on the Odyssey Imager (Li-Cor
Biosciences) at a resolution of 42 uM, medium quality and level 5 intensity in
the 800
20 nM channel.
Data Analysis:
Images obtained were saved and plaque numbers counted with the aid of computer
25 imaging software ECso values for compounds were derived from dose
response
curves [three variable log(inhibitor) vs response] obtained using Graphpad
Prism
software.
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
56
Results
RSV Fusion
RSV Plaque Reduction assays
Assay
RSV-A5 RSV-A5 RSV-BA
Compound (n-4) (PRNT I ) (6440) (6470) (Brasil)
= (n=4) (n=1) (11=1) (11= I
)
.:=
ICso IC90 IC50 IC90 I ICso IC90 ICso IC90 ICso IC90
(nM) (nM) (nM) (nM) (nM) (nM) (nM) (nM) (nM) (nM)
..........,..
Example I 1 1 9.4 2.1 13.5 5.7 106 10.3 81
2.1 34.5
Example 5 5.3
Example 6 2.3
Example 7 1.6
Example 8 4.8
Table 1
.. Example 10: in vitro Pharmacokinetics
Compounds were subjected to the following assays to investigate liver
microsomal
stability, permeability, plasma protein binding and calculated
partition/distribution
coefficients.
Microsomal incubation: Experimental Procedure
Pooled human liver microsomes (pooled male and female), pooled rat liver
microsomes (male Sprague Dawley rats) and pooled dog liver microsomes (male
Beagle dog) are purchased from a reputable commercial supplier and stored at -
80 C
prior to use.
Microsomes (final protein concentration 0.5 mg/mL), 0.1 M phosphate buffer pH
7.4
and test compound (final substrate concentration 3 [LM; final DMSO
concentration
.. 0.25 %) are pre-incubated at 37 DC prior to the addition of NADPH (final
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
57
concentration 1 mM) to initiate the reaction. The final incubation volume is
50 [IL. A
control incubation is included for each compound tested where 0.1 M phosphate
buffer pH 7.4 is added instead of NADPH (minus NADPH). Two control compounds
are included with each species. All incubations are performed singularly for
each test
compound.
Compounds are incubated for 0, 5, 15, 30 and 45 min. The control (minus NADPH)
is incubated for 45 min only. The reactions are stopped by transferring 25 uL
of
incubate to 500, methanol at the appropriate time points. The termination
plates
are centrifuged at 2,500 rpm for 20 mm at 4 C to precipitate the protein.
Following protein precipitation, the sample supernatants are combined in
cassettes
of up to 4 compounds, internal standard is added and samples analysed by LC-
MS/MS.
From a plot of In peak area ratio (compound peak area/internal standard peak
area) against time, the gradient of the line is determined. Subsequently, half-
life and
intrinsic clearance are calculated
MDRI-1VIDCK Permeability: Experimental Procedure
1VI1DR1-MDCK cells obtained from the NIH (Rockville, MD, USA) are used between
passage numbers 6 - 30. Cells are seeded onto Millipore Multiscreen Transwell
plates
at 3.4 x 105 cells/cm2 The cells are cultured in DMEM and media is changed on
day 3. On day 4 the permeability study is performed. Cell culture and assay
incubations are carried out at 37 C in an atmosphere of 5 % CO2 with a
relative
humidity of 95 %. On the day of the assay, the monolayers are prepared by
rinsing
both basolateral and apical surfaces twice with Hanks Balanced Salt Solution
(HBSS)
at the desired pH warmed to 37 C. Cells are then incubated with HBSS at the
desired
pH in both apical and basolateral compartments for 40 min to stabilise
physiological
parameters.
The dosing solutions are prepared by diluting test compound with assay buffer
to give
a final test compound concentration of 10 1iM (final DMSO concentration of 1 %
v/v).
The fluorescent integrity marker lucifer yellow is also included in the dosing
solution.
Analytical standards are prepared from test compound DMSO dilutions and
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
58
transferred to buffer, maintaining a 1 % v/v DMSO concentration.
For assessment of A-B permeability, FIBSS is removed from the apical
compartment
and replaced with test compound dosing solution The apical compartment insert
is
.. then placed into a companion plate containing fresh buffer (containing 1
'NOT
DMSO). For assessment of B-A permeability, HBSS is removed from the companion
plate and replaced with test compound dosing solution. Fresh buffer
(containing 1 %
v/v DMSO) is added to the apical compartment insert, which is then placed into
the
companion plate.
At 60 min the apical compartment inserts and the companion plates are
separated
and apical and basolateral samples diluted for analysis.
Test compound permeability is assessed in duplicate. Compounds of known
.. permeability characteristics are run as controls on each assay plate.
Test and control compounds are quantified by LC-MS/MS cassette analysis using
an
8-point calibration with appropriate dilution of the samples. The starting
concentration
(CO) is determined from the dosing solution and the experimental recovery
calculated
from CO and both apical and basolateral compartment concentrations.
The integrity of the monolayer throughout the experiment is checked by
monitoring
lucifer yellow permeation using fluorimetric analysis. Lucifer yellow
permeation is high
if monolayers have been damaged.
Protein Binding Determination: Experimental Procedure
Solutions of test compound (51.1M, 0.5 % final DMSO concentration) are
prepared in
buffer (pH 7.4) and 100 % species-specific plasma. The experiment is performed
using
equilibrium dialysis with the two compartments separated by a semi-permeable
membrane. The buffer solution is added to one side of the membrane and the
plasma
solution to the other side. After equilibration, samples are taken from both
sides of the
membrane. Standards are prepared in plasma and buffer and are incubated at 37
C.
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
59
Test compound incubations are performed in duplicate. A control compound is
included in each experiment.
The solutions for each batch of compounds are combined into two groups
(protein-free
and protein-containing), then cassette analysed by LC-MS/MS using two sets of
calibration standards for protein-free (7 points) and protein-containing
solutions
(6 points).
Log!) Determination: Experimental Procedure
0.1 M phosphate buffer pH 7.4 (saturated with octanol) is added to the vial
containing
lmu of solid test compound and the solution mixed and sonicated for
approximately
min. The solution is transferred to tubes, centrifuged and the supernatant is
drawn
off the top, leaving any solid compound in the bottom. This supernatant is
then syringe
15 filtered through 0.2 um filters to produce the initial solution.
Three vials are prepared containing different ratios of octanol and compound
in
phosphate buffer in order to cover a range of logD values. The vials are mixed
to
equilibrium, then centrifuged to ensure the two phases are fully separated
before the
octanol is removed and the buffer samples analysed.
.. The aqueous solutions from the corresponding vials are then combined in
cassettes of
four and analysed using generic LC-MS/MS conditions. The amount of compound in
each vial is quantified against a 6 point standard curve which is produced by
serially
diluting the initial solution. The logD is then calculated from these
concentrations.
LogP Determination
LogP values were calculated with software available from ChemAxon using the
method
described in Viswanadhan et al.; J. Chem. Inf. Comput. Sci. 1989; 29:163-172.
CA 02963740 2017-04-05
WO 2016/055780 PCT/GB2015/052920
Results
Pharmacokinetie Property Value
==============="---::::::".="'"===== ____________________________
xamplc 1: 32
/ 807 532P-
IL: B
Example 5: 219 11 5 I 390
a a
Li v er c rosom al S tab i lit ,
Example 6: 229 125 / 1100
human / rat / dog
:Example 7: 33.4 /246 / 3350 !!!!!!
!!! !!!!! m a
:**:*
Z:xample 8: 3004 14:7 276
Permeability (Human Pgp transfected) MDCK
-6 Example 1: 0.5/73
P (x 10 cm/s) A-B /B-A
app
PPB fraction txamnie cl /0 17/0
= =
Rat / doe. / human
clogP / clogD Example 1:
3.66/1.80
5 Table 2
Example 11: in vivo Pharmacokinetics
The pharmacokinetics of compounds were studied in vivo in male Sprague Dawley
rats
10 at doses of 1 mg/kg (IV) and 10 mg/kg (PO).
Methods
Sprague Dawley rats were treated with experimental compounds via intravenous
and
15 oral administration. Three animals for each route of administration were
used with
serial blood sampling at ten time points post dosing of compound.
An intravenous bolus was administered at a dose of lmg/kg and at a
concentration of
1 mRml in 40:60 dimethyl acetamide/saline (0.9% w/v saline). Animals were
weighed
20 and used if between 200-250g. Serial blood samples were collected at
0.02, 0.08, 0.25,
61
0.50, 1, 2 ,4,6 ,8 and 24 hours post dosing. Animals were observed for any
overt
clinical signs or symptoms. Blood samples were delivered into an anticoagulant
(sodium heparin) and centrifuged at 4 C. Plasma samples were subsequently
stored
frozen at less than -20 C prior to analysis.
Following protein precipitation with acetonitrile, samples were analysed with
tandem
liquid chromatography/mass spectrometry using electrospray ionisation. A full
matrix
curve with internal standards was employed and PK parameters were calculated.
In a similar manner, oral administration was perfoimed by gavage at doses of 5
or 10
mg/kg at a concentration of 5mg/m1 in 1% Methyl cellulose (Sigma M7140), 0.1%
TweenTm 80 in water. Serial samples were taken as described above.
Results
Pharmacokinetic Property Value
Example I: 22
Volume of distribution (L/k)
Example 5: 8.2
Tmax PO (hr) Example 1: 2.7
Bioavailability (%) Example I:
Table 3
Comparative Example 1: in vitro Efficacy
The protocols of Example 9 were repeated for RV039 (1'-((5-(aminomethyl)-1-
isopenty1-1H-benzo Id] imidazol-2-y Omethy Ospiro[cyclopropane-1,3'-indolin1-
2'-one ;
identified in WO 2013/068769 as the compound of Example 2).
Date Recue/Date Received 2022-02-25
CA 02963740 2017-04-05
WO 2016/055780 PCT/GB2015/052920
62
Results
RSV Fusion RSV Plaque
Assay Reduction assay
ICso (nM) ICso (nM)
31
Table 4
Comparative Example 2: in vitro Pharmacokinetics
The protocols of Example 10 were repeated for RV039, which is 1 '-((5-
(aminomethyl)-
1-isopenty1-1H-benzo[d]imidazol-2-y1)methyl)spiro[cyclopropane-1,3'-indolin]-
2'-one
(identified in WO 2013/068769 as the compound of Example 2).
Results
Pharmacokinetic Property Value
!]! tier NI 1 C rosomal StabiIit //
............ õ
("1 -iii: human / rat / dog)
............ ....... .......
...............................
Permeability (Human Pgp transfected) MDCK
-6 042/49
P (x 10 cm/s) A-B / B-A
app
i,õ
õ õ; L
n RnM I- iracuon unuounu."11] gpmm e 0. dipp,m,
Rat / do o- / human
clogP / clouD 3.62/1.75
Table 5
Comparative Example 3: in vivo Pharmacokinetics
The protocols of Example 11 were repeated for RV039 (1'-((5-(aminomethyl)-1-
isopenty1-1H-benzo[alimidazol-2-ylimethylispiro[cyclopropane-1,3'-indolinJ-2'-
one;
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
63
identified in WO 2013/068769 as the compound of Example 2). RV039 was
administered at doses of 1 mg/kg (IV) and 5 mg/kg (PO).
Results
Pharmaeokinetie Property Value
Volume of distribution (L/kg) 47
T,..õ PO (hr) 7.3
Bioavailability (90) 4.500
Table 6
Example 11: Aqueous formulation
The compound of Example 1 is formulated as a solution in 30% w/v captisol
(i.e.
sulfobutylether-beta-cyclodextrin) at pH4 according to the following
procedure.
A carrier of 30% w/v captisol (i.e. sulfobutylether-beta-cyclodextrin) is
prepared by
weighing the required amount of captisol into a suitable vessel, adding
approximately
80% of the final volume of water and magnetically stirring until a solution is
formed.
The carrier is then made up to volume with water.
An aqueous solution of a compound of Example 1 is prepared by weighing 175 mg
of
the compound into a suitable vessel and adding approximately 80% of the
required
volume of the carrier. Using an aqueous solution of hydrochloric acid, the pH
is
adjusted to pH2 and the resulting mixture is magnetically stirred until a
solution is
formed. The formulation is then made up to volume with carrier and the pH is
adjusted
to pH4 using an aqueous solution of sodium hydroxide.
Example 12: Tablet composition
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
64
Tablets, each weighing 0.15 g and containing 25 mg of a compound of the
invention are
manufactured as follows:
Composition for 10,000 tablets
Compound of the invention (250 g)
Lactose (800 g)
Corn starch (415g)
Talc powder (30 g)
Magnesium stearate (5 g)
The compound of the invention, lactose and half of the corn starch are mixed.
The
mixture is then forced through a sieve 0.5 mm mesh size. Corn starch (10 g) is
suspended in warm water (90 mL). The resulting paste is used to granulate the
powder.
The granulate is dried and broken up into small fragments on a sieve of 1.4 mm
mesh
size. The remaining quantity of starch, talc and magnesium is added, carefully
mixed
and processed into tablets.
Example 13: Injectable Formulation
Compound of the invention 200 mg
Hydrochloric Acid Solution 0.1M or
Sodium Hydroxide Solution 0.1M q.s. to pH 4.0 to 7.0
Sterile water q.s. to 10 mL
The compound of the invention is dissolved in most of the water (35 C-40 C)
and the
pH adjusted to between 4.0 and 7.0 with the hydrochloric acid or the sodium
hydroxide
as appropriate. The batch is then made up to volume with water and filtered
through a
sterile micropore filter into a sterile 10 mL amber glass vial (type 1) and
sealed with
sterile closures and overseals.
CA 02963740 2017-04-05
WO 2016/055780
PCT/GB2015/052920
Example 14 Intramuscular Injection
Compound of the invention 200 mg
Benzyl Alcohol 0.10 g
5 Glycofurol 75 1.45g
Water for injection q.s to 3.00 ml
The compound of the invention is dissolved in the glycofurol. The benzyl
alcohol is
then added and dissolved, and water added to 3 mL. The mixture is then
filtered
10 through a sterile micropore filter and sealed in sterile 3 mL glass
vials (type 1).
Example 15 Syrup Formulation
Compound of invention 250 mg
15 Sorbitol Solution 1.50g
Glycerol 2.00g
Sodium benzoate 0.005 g
Flavour 0.0125 mL
Purified Water q.s. to 5.00 mL
The compound of the invention is dissolved in a mixture of the glycerol and
most of the
purified water. An aqueous solution of the sodium benzoate is then added to
the
solution, followed by addition of the sorbital solution and finally the
flavour. The
volume is made up with purified water and mixed well.