Note: Descriptions are shown in the official language in which they were submitted.
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
CHECKPOINT BLOCKADE AND MICROSATELLITE
INSTABILITY
[on This invention was made with government support under CA43460 and
CA62924
awarded by the National Institutes of Health. The government has certain
rights in the
invention.
TECHNICAL FIELD OF THE INVENTION
[02] This invention is related to the area of cancer. In particular, it
relates to cancer
therapy.
BACKGROUND OF THE INVENTION
[03] Microsatellite instability (MSI) is the accumulation of sequencing errors
in
microsatellites. This occurs in tumors with deficiency in DNA mismatch repair.
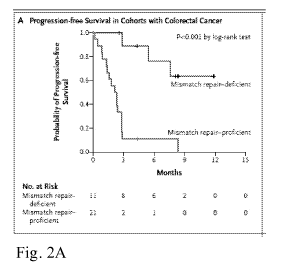
MSI
is present in Lynch Syndrome which is an inherited cancer syndrome that
predisposes
patients to colon, endometrial, gastric cancer, ovarian, small intestine,
liver,
hepatobiliary, upper urinary tract, brain, and prostate cancer. MSI is also
present in
10-20% of sporadic colorectal, gastric, prostate, lung, ampullary, and
endometrial
cancers. Between 0.3% and 13% of pancreatic cancers are reported to be MSI as
well.
[04] The importance of intact immune surveillance in controlling outgrowth of
neoplastic
transformation has been known for decades. Accumulating evidence shows a
correlation between tumor-infiltrating lymphocytes (TILs) in cancer tissue and
favorable prognosis in various malignancies. In particular, the presence of
CD8+ T-
cells and the ratio of CD8+ effector T-cells / FoxP3+ regulatory T-cells seems
to
correlate with improved prognosis and long-term survival in solid malignancies
such
as ovarian, colorectal and pancreatic cancer, hepatocellular carcinoma,
malignant
MEL and RCC. TILs can be expanded ex vivo and re-infused, inducing durable
objective tumor responses in cancers such as melanoma.
[05] The PD-1 receptor-ligand interaction is a major pathway hijacked by
tumors to
suppress immune control. The normal function of PD-1, expressed on the cell
surface
1
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
of activated T-cells under healthy conditions, is to down-modulate unwanted or
excessive immune responses, including autoimmune reactions. The ligands for PD-
1
(PD-Li and PD-L2) are constitutively expressed or can be induced in various
tumors.
Binding of either PD-1 ligand to PD-1 inhibits T-cell activation triggered
through the
T-cell receptor. PD-Li is expressed at low levels on various non-hematopoietic
tissues, most notably on vascular endothelium, whereas PD-L2 protein is only
detectably expressed on antigen-presenting cells found in lymphoid tissue or
chronic
inflammatory environments. PD-L2 is thought to control immune T-cell
activation in
lymphoid organs, whereas PD-Li serves to dampen unwarranted T-cell function in
peripheral tissues. Although healthy organs express little (if any) PD-L1, a
variety of
cancers were demonstrated to express abundant levels of this T-cell inhibitor.
High
expression of PD-Li on tumor cells (and to a lesser extent of PD-L2) has been
found
to correlate with poor prognosis and survival in various cancer types,
including renal
cell carcinoma (RCC), pancreatic carcinoma, hepatocellular carcinoma, ovarian
carcinoma and non-small cell lung cancer (NSCLC). Furthermore, PD-1 has been
suggested to regulate tumor-specific T cell expansion in patients with
malignant MEL.
The observed correlation of clinical prognosis with PD-Li expression in
multiple
cancers suggests that the PD- 1/PD-L1 pathway plays a critical role in tumor
immune
evasion and should be considered as an attractive target for therapeutic
intervention.
[06] Blockade of immune checkpoints such as cytotoxic T-lymphocyte antigen-4
(CTLA-
4) and programmed death-1 (PD-1) is showing promise in patients with cancer.
CTLA-4 and PD-1 are upregulated on activated T cells and provide inhibitory
signals
to T cells undergoing activation. Inhibitory antibodies directed at these
receptors have
been shown to break immune tolerance and promote anti-tumor immunity. MK-3475
is a humanized monoclonal IgG4 antibody against PD-1 and is showing activity
in
multiple tumor types including melanoma and non-small cell lung cancer
(NSCLC).
Previously, activity of a different PD-1 blocking antibody, BMS-936558, a
fully
humanized monoclonal IgG4 antibody, also showed activity in melanoma, NSCLC,
and a complete response in a single patient with colorectal cancer.
[07] MK-3475 (previously known as SCH 900475) is a potent and highly-selective
humanized mAb of the IgG4/kappa isotype designed to directly block the
interaction
2
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
between PD-1 and its ligands, PD-Li and PD-L2. MK-3475 contains the S228P
stabilizing mutation and has no antibody-dependent cell-mediated cytotoxicity
(ADCC) or complement-dependent cytotoxicity (CDC) activity. MK-3475 strongly
enhances T lymphocyte immune responses in cultured blood cells from healthy
human
donors, cancer patients, and primates. In T- cell activation assays using
human donor
blood cells, the EC50 was in the range of 0.1 to 0.3 nM. MK-3475 also
modulates the
level of interleukin-2 (IL-2), tumor necrosis factor alpha (TNFa), interferon
gamma
(IFNy), and other cytokines. The antibody potentiates existing immune
responses only
in the presence of antigen and does not nonspecifically activate T- cells.
[08] The programmed death 1 (PD-1) pathway is a negative feedback system
repressing
Thl cytotoxic immune responses that, if unregulated, could damage the host1-3.
It is
upregulated in many tumors and their surrounding microenvironment. Blockade of
this pathway with antibodies to PD-1 or its ligands has led to remarkable
clinical
responses in some patients with many different cancer types, including
melanomas,
non-small cell lung cancer, renal cell carcinoma, bladder cancer and Hodgkin's
lymphoma4-1 . The expression of ligands to PD-1 (PD-Li or PD-L2) on the
surface of
tumor cells or immune cells is important but not a definitive predictive
biomarker for
response to PD-1 blockade4,6-8,11.
[09] We were intrigued that, in reports of the effects of PD-1 blockade in
human tumors,
only one of 33 colorectal cancer (CRC) patients responded to this treatment,
in
contrast to substantial fractions of patients with melanomas, renal cell
cancers, and
lung tumors.1 '12. What was different about this single patient? We
hypothesized that
this patient had MMR-deficiency, because MMR-deficiency occurs in a small
fraction
of advanced CRCs,13'14 somatic mutations found in tumors can be recognized by
the
patient's own immune system,15 and MMR-deficient cancers have 10- to 100-fold
more somatic mutations than MMR-proficient CRC.16-18 Moreover, MMR-deficient
cancers contain prominent lymphocyte infiltrates, consistent with an immune
response19-22. And two of the tumor types that were most responsive to PD-1
blockade
in a study by Topalian et al.1 had high numbers of somatic mutations as a
result of
exposure to cigarette smoke (lung cancers) or UV radiation (melanomas)23,24.
Our
hypothesis was correct: the tumor of the single CRC patient who responded to
PD-1
3
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
blockade was MMR-deficient25. We therefore hypothesized that MMR-deficient
tumors are more responsive to PD-1 blockade than are MMR-proficient tumors.
[10] To test this hypothesis, we initiated a phase 2 clinical trial to
evaluate immune
checkpoint blockade in patients whose tumors had or did not have MMR-
deficiency.
Since MMR deficiency in tumors arises through two routes26-28, we recruited
patients
with Hereditary Non-Polyposis Colorectal Cancer (HNPCC, also known as Lynch
Syndrome), which results from an inherited germline defect in one of four MMR
genes followed by a second inactivating somatic change in the remaining wild-
type
allele. We also recruited patients with sporadic MMR-deficient tumors, where
both
alleles of a MMR gene are inactivated by somatic mutations or by epigenetic
silencing29. In either case, the neoplasms that arise harbor hundreds or
thousands of
mutations16'18.
[11] There is a continuing need in the art to improve cancer treatments so
that the lives of
patients are not curtailed and so that the quality of life is not diminished.
SUMMARY OF THE INVENTION
[12] According to one embodiment of the invention a method of treating a
cancer patient is
provided. The cancer patient has a high mutational burden, such as found in
microsatellite instable cancer (MSI). An immune checkpoint inhibitory antibody
is
administered to the cancer patient.
[13] According to another embodiment of the invention a method of treating a
cancer
patient is provided. A sample from a cancer patient is tested for one or more
microsatellite markers selected from the group consisting of BAT-25, BAT-26,
MONO-27, NR-21, NR-24, Penta C, and Penta D, and determined to have
microsatellite instability. The cancer is selected from the group consisting
of: colon,
gastric, endometrial, cholangiocarcinoma, pancreatic, and prostate cancers. An
anti-
PD-1 antibody is administered to the cancer patient.
4
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
[14] According to another embodiment of the invention a method is provided for
categorizing a tumor of a human. A sample from the human is tested to evaluate
stability of one or more microsatellite markers.
Microsatellite instability is
determined in the sample. The tumor is identified as a good candidate for
treatment
with an immune checkpoint inhibitory antibody.
[15] According to yet another embodiment of the invention a method is provided
for
categorizing a tumor of a human. A sample from the human is tested to evaluate
stability of one or more microsatellite markers. Microsatellite stability in
the sample
is determined. The tumor is identified as a bad candidate for treatment with
an
immune checkpoint inhibitory antibody.
[16] These and other embodiments which will be apparent to those of skill in
the art upon
reading the specification provide the art with methods for treating micro
satellite
instable cancers.
BRIEF DESCRIPTION OF THE DRAWINGS
[17] Figs. 1A-1B. Clinical Responses to pembrolizumab. (Fig. 1A)
Biochemical
Responses. Serum protein biomarker levels were measured with each cycle and
the
values represent percent change from baseline. Patients were included if
baseline
tumor marker values were greater than the upper limit of normal. CA-125 was
used
for a patient with endometrial cancer; CA19-9 was used for one
cholangiocarcinoma
and one ampullary cancer; and CEA was used for all other patients. Green, red,
and
black lines represent patients with MMR-deficient CRCs, MMR-proficient CRCs,
and
MMR-deficient non-CRC, respectively. (Fig. 1B) Radiographic responses. Tumor
responses were measured at regular intervals and values show the best
fractional
change of the sum of longest diameters (SLD) from the baseline measurements of
each measurable tumor.
[18] Figs. 2A-2D. Clinical benefit to pembrolizumab according to MMR status.
Kaplan-
Meier curves are shown for (Fig. 2A) progression-free survival in the
colorectal
cancer cohorts, (Fig. 2B) overall survival in the colorectal cancer cohorts,
(Fig. 2C)
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
progression-free survival of patients with MMR-deficient cancers other than
colorectal (median PFS=5.4 months; 95% CI, 3% to not estimable), and (Fig. 2D)
overall survival of patients with MMR-deficient cancers other than colorectal.
In both
cohorts with MMR-deficient tumors (CRC and non-CRC), median overall survival
was not reached. Patients in the cohort with MMR-proficient cancers had a
median
PFS of 2.2 months (95% CI 1.4 to 2.8%) and a median OS of 5.0 months (95% CI
3.0
to not estimable).
[19] Fig. 3 (Figure S2.) Spider plot of radiographic response. Tumor responses
were
measured at regular intervals and values show percent change of the sum of
longest
diameters (SLD) from the baseline measurements of each measurable tumor.
Patients
were only included if baseline and on study treatment scans were available.
Green
and red represent patients with MMR-deficient and proficient CRCs,
respectively.
Blue represents patients with MMR-deficient cancers other than CRC.
[20] Figs. 4A-4B (Figure S3). MMR-proficient and deficient CRCs have
comparable time
on treatment and duration of metastatic disease prior to study enrollment.
Kaplan-
Meier estimates of ( Fig. 4A) time on therapy immediately prior to study
enrollment
(HR 0.81, 95% CI 0.38 to 1.752, p=.60) and (Fig. 4B) duration of metastatic
disease
prior to enrollment (HR 1.13, 95% CI 0.49 to 2.62, p=.78) on this
pembrolizumab
study were comparable between the MMR-deficient and proficient CRC cohorts.
The
short duration on prior therapy is expected in a treatment refractory CRC
population.
[21] Fig. 5 (Figure S4.) Waterfall plot of biochemical response. Serum protein
biomarker
levels were measured with each cycle and the values represent best percent
change
from baseline. Patients were included if baseline tumor marker values were
greater
than the upper limit of normal. CA-125 was used for a patient with endometrial
cancer; CA19-9 was used for 1 cholangiocarcinoma and 1 ampullary cancer; and
CEA
was used for all other patients. Green and red represent patients with MMR-
deficient
and proficient CRCs, respectively. Blue represents patients with MMR-deficient
cancers other than CRC.
6
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
[22] Figs. 6A-6B (Figure S5.) Somatic mutations in MMR-deficient and
proficient tumors.
Total somatic mutations per tumor identified by exome sequencing of tumor and
matched normal DNA (Fig. 6A) and correlation with objective responses (Fig.
6B)
(non-parametric Wilcoxon test , p= 0.007 and Jonckheere-Terpstra test for
trend,
p=0.02).
[23] Fig. 7 (Figure S6). Immunohistochemistry of CD8 and PD-Li Expression. The
invasive front (yellow dashed line) from a MMR-deficient CRC (subject #16,
top) and
MMR-proficient CRC (subject #3, bottom). The yellow dashed line separates
tumor
(T) and normal (N) tissue. There is marked expression of PD-Li (blue arrows)
and
CD8 (brown dots) in the MMR-deficient tumor (top panels) patient while there
is very
little expression of either marker in the MMR-proficient tumor (bottom
panels).
Representative images of tumor infiltrating lymphocytes (TIL) in another MMR-
deficient CRC (subject #19, top) and MMR-proficient CRC (subject #3, bottom)
immunolabeled with an antibody to CD8 (brown dots). Note the infiltration of
CD8
cells in the MMR-deficient tumor. Invasive front original magnification 10x
and TIL
20x.
[24] Fig. 8 (Figure S7.) CD8 and PD-Li Expression in the MMR-deficient and MMR-
proficient tumor microenvironment. T cell density units are cells/mm2 of
tumor.
Invasive front refers to the immune cells (TILs and macrophages) at the
junction of
the tumor and normal tissue. P-values obtained using an unpaired t-test.
[25] Fig. 9 (Figure S8.) CD8 expression and clinical benefit to pembrolizumab.
Correlation
between the intratumoral CD8 + T cell density (cells/mm2) and objective
response
(Jonckheere-Terpstra test for trend, p=0.02).
[26] Fig. 10 (Table S 1 .) Comparison of immune-related and RECIST response
criteria
(adapted from Wolchok et al. Clin Can Res 2009;15:7412-20.)
7
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
[27] Fig. 11 (Table S2.) Immune-Related response to treatment
[28] Fig. 12 (Table S4.) Correlation of total somatic mutations and mutation
associated
neoantigens (MANA) with clinical outcomes
[29] Fig. 13 (Table S5.) Correlation of immune markers with clinical outcome
DETAILED DESCRIPTION OF THE INVENTION
[30] The inventors have found that immune checkpoint inhibitors work best in
tumors with
high mutation burdens. Furthermore, tumors deficient in mismatch repair are
particularly susceptible to a particular form of immunotherapy because this
phenotype
results in ongoing accumulation of mutations at a high frequency. The
inventors have
developed a treatment for cancer patients that display the microsatellite
instability
phenotype or other high mutational burden. The treatment involves an
inhibitory
antibody for an immune checkpoint. Such checkpoints include PD-1, IDO, CTLA-4,
PD-L1, and LAG-3. Other immune checkpoints can be used as well. Antibodies can
be administered by any means that is convenient, including but not limited to
intravenous infusion, oral administration, subcutaneous administration,
sublingual
administration, ocular administration, nasal administration, etc.
[31] Microsatellite instability (MSI) tumors are deficient in DNA mismatch
repair which
leads to a high rate of spontaneous mutations and the potential for the
expression of
neo-antigens. Furthermore, similar to melanoma, in MSI positive colon cancers,
there
is often prominent lymphocyte infiltration. Any tumors that are MSI or
otherwise high
mutational burden may be treated according to the invention. They may be
tested for
the attribute of MSI according to any method known in the art, including but
not
limited that described in example 1 below. Any of one or more MSI markers can
be
tested to determine an MSI phenotype. Samples may be tested for high
mutational
burden by identifying tumors with at least 100, at least 200, at least 300, at
least 400,
at least 500, at least 600, at least 700, at least 800, at least 900, at least
1000, at least
8
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
1100, at least 1200, at least 1300, at least 1400, at least 1500, or at least
1600
mutations per tumor genome. High mutational burden means a large number of
somatic mutations in the tumor relative to normal tissues of the individual.
An
average number of somatic mutations in a non-MSI tumor is about 70 somatic
mutations.
[32] Any type of tumor that displays the MSI phenotype or a high mutational
burden may
be tested and/or treated according to the invention. These include without
limitation
cancers of the colon, gastric, endometrial, cholangiocarcinoma, pancreatic,
and
prostate cancer. Tumors of the ampulla, biliary, brain, including glioma,
breast, lung,
skin, esophagus, liver, kidney, ovaries, sarcoma, uterus, cervix, bladder,
testes, oral
cavity, tongue, and small and large bowel may also be tested and/or treated.
[33] Testing of MSI can be accomplished by any means known in the art. One or
more of
the following markers may be tested: five nearly monomorphic mononucleotide
repeat markers (BAT-25, BAT-26, MONO-27, NR-21 and NR-24) and two highly
polymorphic pentanucleotide repeat markers (Penta C and Penta D). In one
commercial system which can be used, fluorescently labeled primers (marker
panel)
are used for co-amplification of all seven of the above named markers.
Fragments are
detected after amplification for assignment of genotype/phenotype.
[34] Samples that can be tested for MSI include tumor tissue as well as body
fluids that
contain nucleic acids shed from tumors. Testing for tumor DNA in such tissues
and
body fluids is well known.
[35] Types of antibodies which can be used include any that are developed for
the immune
checkpoint inhibitors. These can be monoclonal or polyclonal. They may be
single
chain fragments or other fragments of full antibodies, including those made by
enzymatic cleavage or recombinant DNA techniques. They may be of any isotype,
including but not limited to IgG, IgM, IgE. The antibodies may be of any
species
source, including human, goat, rabbit, mouse, cow, chimpanzee. The antibodies
may
be humanized or chimeric. The antibodies may be conjugated or engineered to be
9
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
attached to another moiety, whether a therapeutic molecule or a tracer
molecule. The
therapeutic molecule may be a toxin, for example.
[36] The data from the small phase 2 trial of pembrolizumab to treat tumors
with and
without deficiency of MMR supports the hypothesis that MMR-deficient tumors
are
more responsive to PD-1 blockade than are MMR-proficient tumors. MMR-
deficiency
occurs in many cancers, including those of the colorectum, uterus, stomach,
biliary
tract, pancreas, ovary, prostate and small intestine18 '34-42 . Patients with
MMR-deficient
tumors of these types also benefit from anti-PD-1 therapy, as may patients
whose
tumors contain other DNA repair deficiencies, such as those with mutations in
POLD,
POLE, or MYH.18'43'"
[37] The hypothesis that MMR-deficient tumors stimulate the immune system is
not a new
idea45, and has been supported by the dense immune infiltration and Thl-
associated
cytokine-rich environment observed in MMR-deficient tumors.19-22,46 A recent
study
refined these classic observations by showing that the MMR-deficient tumor
microenvironment strongly expressed several immune checkpoint ligands
including
PD-1, PD-L1, CTLA-4, LAG-3 and IDO, indicating that their active immune
microenvironment is counterbalanced by immune inhibitory signals that resists
tumor
elimination47. That the immune infiltrate associated with MMR-deficient
carcinomas
was directed at neoantigens was the most likely explanation for both the old
and new
findings. The correlation of higher mutational load and higher response rate
to anti-
CTLA-4 in melanoma41 and anti-PD-1 in lung cancer48 provide further support
for the
idea that MANA recognition is an important component of the endogenous anti-
tumor
immune response.
[38] Based on the results of the current and previous studies, we suggest that
the greatly
(>20-fold) increased number of mutation-associated neoantigens resulting from
MMR
deficiency (Fig.. 12 (Table S4); also available on line at New England Journal
of
Medicine; incorporated by reference herein) is the basis for the enhanced anti-
PD-1
responsiveness of this genetically defined subset of cancers. Though our
estimates for
the number of mutation-associated neoantigens in tumors is based only on in
silky
predictions of binding-affinity, this suggestion is consistent with the
observation that
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
MMR-proficient tumors have far less infiltration of lymphocytes than MMR-
deficient
tumors (Fig. 7 (S6), Fig. 8 (S7) and Fig. 13 (Table S5); available on line at
New
England Journal of Medicine; incorporated by reference herein). Recent
studies49'5
show that only a tiny proportion of predicted neo-epitopes are actually
presented on
the cell surface with MHC and are targets of endogenous T cell responses. It
seems
likely, though that the number of predicted mutation-associated neoantigens is
proportionate to the number of actual mutation-associated neoantigens, and
tumors
with a high number of actual mutation-associated neoantigens are more likely
to
stimulate the immune system to react against the tumor. Alternative mechanisms
underlying the difference in anti-PD-1 responsiveness between MMR-deficient
and
MMR-proficient tumors should also be considered. For example, different
signaling
pathways activated in MMR-deficient and MMR-proficient tumors may result in
differences in secretion of soluble factors that could result in differential
activation of
the PD-1 pathway within the tumor microenvironment26-28. Genetic differences
could
effect epigenetic differences that alter the expression of tumor-associated
self-antigens
that in turn could alter the antigenicity of the tumor. Experimental analyses
of antigen-
specific immune responses as well as changes in immune microenvironments
should
help to define the relative contribution of these factors to the striking
responsiveness
of MMR-deficient tumors to PD-1 antibodies.
[39] Several notable observations were made during the course of this study.
First, changes
in serum protein biomarkers, like CEA, corresponded with clinical benefit
after a
single dose of therapy. Declines in CEA levels preceded objective radiographic
evidence by several months; perhaps other biomarkers such as circulating tumor
DNA
(ctDNA) may also be beneficial as surrogate markers of early response.51'52
Second,
our results suggest that the evaluation of tumor genomes can help guide
immunotherapy. They support the view that the number and type of alterations
may
prove useful for judging the potential utility of immune checkpoint
inhibitors, even in
MMR-proficient cancers41'48'53 Most importantly, our results demonstrate a new
approach for the treatment of a specific class of tumors based solely on
genetic status:
i.e., without regard to underlying tumor type.
ii
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
[40] The above disclosure generally describes the present invention. All
references
disclosed herein are expressly incorporated by reference. A more complete
understanding can be obtained by reference to the following specific examples
which
are provided herein for purposes of illustration only, and are not intended to
limit the
scope of the invention.
EXAMPLE 1
MSI Testing
[41] MSI testing is already standardized and performed in CLIA-certified
laboratories
without need for assay development. Archived tumor samples or newly obtained
biopsies will be used for determining MSI. MSI status will be performed
locally by
CLIA certified immunohistochemistry (IHC) or PCR based tests for eligibility.
Evaluable patients will be confirmed using the MSI Analysis System from
Promega at
Johns Hopkins. This test will determine MSI status through the insertion or
deletion
of repeating units in the five nearly monomorphic mononucleotide repeat
markers
(BAT-25, BAT-26, MONO-27, NR-21 and NR-24). At least 2 MSI loci are required
to
be evaluable in Cohorts A and C. Patients may be assigned to a new cohort
and/or
replaced based on the Promega test results.
EXAMPLE 2
METHODS
[42] Patients
[43] Treatment-refractory progressive metastatic cancer patients for this
phase 2 study were
recruited from three participating centers (Table 1). Three cohorts were
evaluated:
Cohort A was composed of patients with MMR-deficient colorectal
adenocarcinomas;
Cohort B was composed of patients with MMR-proficient colorectal
adenocarcinomas; and Cohort C was composed of patients with MMR-deficient
cancers of types other than colorectal.
[44] Study Oversight
12
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
[45] The protocol, which can be found at NEJM.org, was approved by each site's
institutional review boards, and the study was conducted in accordance with
the
Declaration of Helsinki and the International Conference on Harmonization
Guidelines for Good Clinical Practice. All the patients provided written
informed
consent before study entry. The principal investigator (D.L.) and study
sponsor
(L.A.D.) were responsible for oversight of the study. Merck donated the study
drug,
reviewed the final drafts of the protocol and of this manuscript. The clinical
study was
primarily funded through philanthropic support.
[46] Study Design
[47] This phase 2 trial was conducted using a Green-Dahlberg two-stage design
and
consisted of the three parallel cohorts described above. The study agent,
pembrolizumab (Merck), was administered at 10 mg/kg intravenously every 14
days.
Pembrolizumab is a humanized monoclonal anti-PD-1 antibody of the IgG4/kappa
isotype that blocks the interaction between PD-1 and its ligands, PD-Li and PD-
L2.
[48] Safety assessments were performed before each treatment. Assessments of
total tumor
burden via measurements of serum biomarkers were performed at the start of
each
cycle. Radiologic assessments were made at 12 weeks and every 8 weeks
thereafter.
Further details concerning the clinical protocol are provided in the Example
3.
[49] Analysis of mismatch repair status
[50] Tumors with genetic defects in MMR pathways are known to harbor thousands
of
somatic mutations, especially in regions of repetitive DNA known as
microsatellites.
The accumulation of mutations in these regions of the genome is termed
microsatellite
instability (MSI) 26-28. MMR-status was assessed using the MSI Analysis System
from
Promega in tumors, through the evaluation of selected microsatellite sequences
particularly prone to copying errors when MMR is compromised26-28. See
Supplementary Appendix for additional details.
[51] Genomic & Bioinformatic Analyses
13
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
[52] Primary tumor samples and matched normal peripheral-blood specimens were
obtained from a subset of subjects with MMR-deficient and others with MMR-
proficient carcinomas where sufficient tumor tissue was available for exome
sequencing3 and HLA haplotyping. To assess the potential for mutant peptide
binding, somatic exome data combined with the individual patient's MHC class I
HLA haplotype was applied to the an epitope prediction algorithm31'32. This
algorithm
provided an estimate of the total number of mutation-associated neoantigens in
each
tumor. Additional details are provided in the Supplementary Appendix
(available on
line at New England Journal of Medicine; incorporated by reference herein).
[53] Statistical Analysis
[54] The primary endpoints for Cohorts A and B were immune-related objective
response
rate (irORR) and immune-related progression-free survival (irPFS) rate at 20
weeks
assessed using immune-related response criteria (irRC)33.The primary endpoint
for
Cohort C was irPFS rate at 20 weeks. Immune-related criteria (i.e, criteria
used to
evaluate immune-based therapies) are based on radiographic responses, and
unlike
RECIST criteria, capture extent of disease after disease progression; these
criteria are
defined and compared to RECIST v1.1 in Fig. 10 (Table Si). Response rate and
PFS
rate at 20 weeks were evaluated and reported in this study using RECIST v1.1
and
irRC (Fig. 10 (Table Si)). PFS and overall survival was summarized by Kaplan-
Meier
method. Details of the hypothesis, the decision rules to reject the null
hypotheses and
early-stopping rules for efficacy and futility, and statistical methods are
provided in
the Supplementary Appendix.
EXAMPLE 3
SUPPLEMENTARY METHODS
[55] PATIENTS
[56] To be eligible for participation in this study, patients had to be at
least 18 years of age,
have histologically confirmed evidence of previously-treated, progressive
carcinoma.
All patients underwent MMR status testing prior to enrollment. All patients
had at
14
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
least one measurable lesion as defined by the Response Evaluation Criteria in
Solid
Tumors (RECIST), version 1.1, an Eastern Cooperative Oncology Group (ECOG)
performance-status score of 0 or 1, and adequate hematologic, hepatic, and
renal
function. Eligible patients with CRC must have received at least 2 prior
cancer
therapies and patients with other cancer types must have received at least 1
prior
cancer therapy. Patients with untreated brain metastases, history of HIV,
hepatitis B,
hepatitis C, clinically significant ascites/effusions, or autoimmune disease
were
excluded.
[57] STUDY OVERSIGHT
[58] Initial drafts of the manuscript were prepared by a subset of the authors
and all authors
contributed to the final manuscript. All the authors made the decision to
submit the
manuscript for publication. The principal investigator and study sponsor vouch
for
the accuracy and completeness of the data reported as well as adherence to the
protocol.
[59] HLA TYPING
[60] HLA-A, HLA-B and HLA-C Sequence Based Typing can be divided into three
distinct steps, as described below. A generic, A*02 specific, B generic, B
group
specific, C generic and C*07 specific PCR and sequencing mixes were made in
the
JHU core facility. Celera' s AlleleSEQR HLA-B Sequence Based Typing kit was
used
for B generic SBT. The HLA-A typing scheme is composed of two PCR reactions, A
generic and A*02 specific. A generic amplicon encompasses partial exon 1-
partial
exon 5. A*02 amplicon encompasses partial intron 1 - partial exon 5. HLA-B
typing
scheme is composed of two PCR reactions, B generic and B group specific. The B
generic PCR is a multiplexed reaction containing two PCR amplicons
encompassing
exon 2 ¨ exon 3 and exon 4 ¨ exon 7. B group specific amplicon encompasses
partial
intron 1 - partial exon 5. HLA-C typing scheme is composed of two PCR
reactions, C
generic and C*07 specific. C generic and C*07 specific amplicons encompasses
exons 1 - 7.
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
[61] The specificity of the HLA-A and B PCR employed AmpliTaq Gold DNA
polymerase. The GeneAmp High Fidelity enzyme is used for the HLA-C and C*07
PCR mixes. This enzyme is a mix of two polymerases: AmpliTaq DNA polymerase
(non-proofreading polymerase) and a proofreading polymerase. This enzyme mix
is
necessary to produce efficient and robust amplification of the larger full
length HLA-
C amplicon.
[62] PCR product purification was performed using Exonuclease I and Shrimp
Alkaline
Phosphatase The A generic and B generic amplicons were bi-directionally
sequenced
for exons 2,3,4. The C generic amplicon was bi-directionally sequenced for
exons 2,3
and sequenced in a single direction for exons 1,4,5,6,7. A*02 specific, B
group
specific and C*07 specific amplicons were sequenced in a single direction for
exons
2,3. All sequencing reactions were performed with Big Dye Terminator V1.1 from
Applied Biosystems and sequenced with an ABI Prism 3500XL Genetic Analyzer.
Conexio Genomic' s "Assign SBT" allele assignment software was used to process
the
data files.
[63] MISMATCH REPAIR STATUS TESTING1'2
[64] Six slides of tumor and normal (uninvolved lymph node or margin of
resection) were
cut (5 microns each), deparaffinized (xylene), and one stained with
hematoxylin and
eosin (H+E). A tumor area containing at least 20% neoplastic cells, designated
by a
board-certified Anatomic Pathologist was macrodissected using the Pinpoint DNA
isolation system (Zymo Research, Irvine, CA), digested in proteinase K for 8
hours
and DNA was isolated using a QIAamp DNA Mini Kit (Qiagen, Valencia, CA). MSI
was assessed using the MSI Analysis System (Promega, Madison, WI), composed of
5
pseudomonomorphic mononucleotide repeats (BAT-25, BAT-26, NR-21, NR-24 and
MONO-27) to detect MSI and 2-pentanucleotide repeat loci (PentaC and PentaD)
to
confirm identity between normal and tumor samples, per manufacturer's
instructions.
Following amplification of 50-100 ng DNA, the fluorescent PCR products were
sized
on an Applied Biosystems 3130x1 capillary electrophoresis instrument
(Invitrogen,
Calsbad, CA). Pentanucleotide loci confirmed identity in all cases. Controls
included
water as a negative control and a mixture of 80% germline DNA with 20% MSI
16
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
cancer DNA as a positive control. The size in bases was determined for each
microsatellite locus and tumors were designated as MSI if two or more
mononucleotide loci varied in length compared to the germline DNA.
[65] SEQUENCING ANALYSIS
[66] Samples
[67] Samples provided as FFPE blocks or frozen tissue underwent pathological
review to
determine tumor cellularity. Tumors were macrodissected to remove
contaminating
normal tissue, resulting in samples containing >20% neoplastic cells. Matched
normal samples were provided as blood, saliva or normal tissue obtained from
surgery.
[68] Sample Preparation and Next-Generation Sequencing3
[69] Sample preparation, library construction, exome capture, next generation
sequencing,
and bioinformatics analyses of tumor and normal samples were performed at
Personal
Genome Diagnostics, Inc. (Baltimore, Maryland). In brief, DNA was extracted
from
frozen or formalin-fixed paraffin embedded (FFPE) tissue, along with matched
blood
or saliva samples using the Qiagen DNA FFPE tissue kit or Qiagen DNA blood
mini
kit (Qiagen, CA). Genomic DNA from tumor and normal samples were fragmented
and used for IIlumina TruSeq library construction (IIlumina, San Diego, CA)
according to the manufacturer's instructions or as previously described4.
Briefly, 50
nanograms (ng) - 3 micrograms (lug) of genomic DNA in 100 microliters (0) of
TE
was fragmented in a Covaris sonicator (Covaris, Woburn, MA) to a size of 150-
450bp. To remove fragments smaller than 150bp, DNA was purified using
Agencourt
AMPure XP beads (Beckman Coulter, IN) in a ratio of 1.0 to 0.9 of PCR product
to
beads twice and washed using 70% ethanol per the manufacturer's instructions.
Purified, fragmented DNA was mixed with 36 [a of H20, 10 [a of End Repair
Reaction Buffer, 5 [a of End Repair Enzyme Mix (cat# E6050, NEB, Ipswich, MA).
The 100 [a end-repair mixture was incubated at 20 C for 30 min, and purified
using
Agencourt AMPure XP beads (Beckman Coulter, IN) in a ratio of 1.0 to 1.25 of
PCR
17
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
product to beads and washed using 70% ethanol per the manufacturer's
instructions.
To A-tail, 42 [a of end-repaired DNA was mixed with 5 [a of 10X dA Tailing
Reaction Buffer and 3 [a of Klenow (exo-)(cat# E6053, NEB, Ipswich, MA). The
50
[a mixture was incubated at 37 C for 30 min and purified using Agencourt
AMPure
XP beads (Beckman Coulter, IN) in a ratio of 1.0 to 1.0 of PCR product to
beads and
washed using 70% ethanol per the manufacturer's instructions. For adaptor
ligation,
25 [a of A-tailed DNA was mixed with 6.7 [a of H20, 3.3 [a of PE-adaptor
(I1lumina), 10 [a of 5X Ligation buffer and 5 [a of Quick T4 DNA ligase (cat#
E6056,
NEB, Ipswich, MA). The ligation mixture was incubated at 20 C for 15 min and
purified using Agencourt AMPure XP beads (Beckman Coulter, IN) in a ratio of
1.0
to 0.95 and 1.0 of PCR product to beads twice and washed using 70% ethanol per
the
manufacturer's instructions. To obtain an amplified library, twelve PCRs of 25
[a
each were set up, each including 15.5 [a of H20, 5 [a of 5 x Phusion HF
buffer, 0.5 [a
of a dNTP mix containing 10 mM of each dNTP, 1.25 [a of DMSO, 0.25 [a of
Illumina PE primer #1, 0.25 [a of Illumina PE primer #2, 0.25 [a of Hotstart
Phusion
polymerase, and 2 [a of the DNA. The PCR program used was: 98 C for 2 minutes;
12 cycles of 98 C for 15 seconds, 65 C for 30 seconds, 72 C for 30 seconds;
and
72 C for 5 min. DNA was purified using Agencourt AMPure XP beads (Beckman
Coulter, IN) in a ratio of 1.0 to 1.0 of PCR product to beads and washed using
70%
ethanol per the manufacturer's instructions. Exonic or targeted regions were
captured
in solution using the Agilent SureS elect v.4 kit according to the
manufacturer's
instructions (Agilent, Santa Clara, CA). The captured library was then
purified with a
Qiagen MinElute column purification kit and eluted in 17 [a of 70 C EB to
obtain 15
[a of captured DNA library. (5) The captured DNA library was amplified in the
following way: Eight 30uL PCR reactions each containing 19 [a of H20, 6 [a of
5 x
Phusion HF buffer, 0.6 [a of 10 mM dNTP, 1.5 [a of DMSO, 0.30 [a of Illumina
PE
primer #1, 0.30 1 of Illumina PE primer #2, 0.30 [a of Hotstart Phusion
polymerase,
and 2 [a of captured exome library were set up. The PCR program used was: 98 C
for
30 seconds; 14 cycles (exome) or 16 cycles (targeted) of 98 C for 10 seconds,
65 C
for 30 seconds, 72 C for 30 seconds; and 72 C for 5 min. To purify PCR
products, a
NucleoSpin Extract II purification kit (Macherey-Nagel, PA) was used following
the
manufacturer's instructions. Paired-end sequencing, resulting in 100 bases
from each
end of the fragments for exome libraries and 150 bases from each end of the
fragment
18
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
for targeted libraries, was performed using Illumina HiSeq 2000/2500 and
IIlumina
MiSeq instrumentation (I1lumina, San Diego, CA).
[70] Primary Processing of Next-Generation Sequencing Data and Identification
of
Putative Somatic Mutations3
[71] Somatic mutations were identified using VariantDx custom software
(Personal
Genome Diagnostics, Baltimore, Maryland) for identifying mutations in matched
tumor and normal samples. Prior to mutation calling, primary processing of
sequence
data for both tumor and normal samples were performed using Illumina CASAVA
software (v1.8), including masking of adapter sequences. Sequence reads were
aligned against the human reference genome (version hg18) using ELAND with
additional realignment of select regions using the Needleman-Wunsch method 5.
Candidate somatic mutations, consisting of point mutations, insertions, and
deletions
were then identified using VariantDx across the either the whole exome or
regions of
interest. VariantDx examines sequence alignments of tumor samples against a
matched normal while applying filters to exclude alignment and sequencing
artifacts.
In brief, an alignment filter was applied to exclude quality failed reads,
unpaired
reads, and poorly mapped reads in the tumor. A base quality filter was applied
to
limit inclusion of bases with reported phred quality score > 30 for the tumor
and > 20
for the normal. A mutation in the tumor was identified as a candidate somatic
mutation only when (i) distinct paired reads contained the mutation in the
tumor; (ii)
the number of distinct paired reads containing a particular mutation in the
tumor was
at least 10% of read pairs; (iii) the mismatched base was not present in >1%
of the
reads in the matched normal sample as well as not present in a custom database
of
common germline variants derived from dbSNP; and (iv) the position was covered
in
both the tumor and normal at > 150X. Mutations arising from misplaced genome
alignments, including paralogous sequences, were identified and excluded by
searching the reference genome.
[72] Candidate somatic mutations were further filtered based on gene
annotation to identify
those occurring in protein-coding regions. Functional consequences were
predicted
using snpEff and a custom database of CCDS, RefSeq and Ensembl annotations
using
19
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
the latest transcript versions available on hg18 from UCSC
(https://genome.ucsc.edu/). Predictions were ordered to prefer transcripts
with
canonical start and stop codons and CCDS or Refseq transcripts over Ensembl
when
available. Finally mutations were filtered to exclude intronic and silent
changes,
while retaining mutations resulting in missense mutations, nonsense mutations,
frameshifts, or splice site alterations. A manual visual inspection step was
used to
further remove artifactual changes.
[73] MUTANT PEPTIDE MHC BINDING PREDICTION
[74] Somatic frameshift, insertions, deletions, and missense mutations
predicted to result
in an amino acid change were analyzed for potential MHC class I binding based
on
the individual patient's HLA haplotype. Our initial analysis focused on HLA-A
and
HLA-B. Amino acid mutations were linked to their corresponding CCDS accession
number and in instances where this was unavailable, either a Refseq or
ensemble
transcript was used to extract the protein sequence. To identify 8mer, 9mer,
and
10mer epitopes, amino acid fragments surrounding each mutation were
identified.
These 15, 17, and 19 mutant amino acid fragments were analyzed by the epitope
prediction program NetMHC 3.4.6 Epitopes with a predicted affinity of <50nm
were
considered to be strong potential binders and epitopes with a predicted
affinity of
<500nm were considered to be weak potential binders as suggested by the NetMHC
group 6.
[75] To further refine the total neoantigen burden, we repeated that same
process for the
complementary wild-type peptide for each mutant peptide. We then filtered for
mutant peptides that were strong potential binders when the complementary wild-
type
peptide was predicted a weak potential binder. These mutant peptides are
referred to
as mutation-associated neoantigens (MANA). In the event that a patient had a
(e.g.,
cases 1, 17 and 21) single MHC haplotype not supported by NetMHC 3.4, the
individual haplotype was not included in our analysis.
[76] STATISTICAL METHODS
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
[77] Design of the trial'
[78] This trial was conducted using a parallel two-stage design to
simultaneously evaluate
the efficacy of MK-3475 and MSI as a treatment selection marker for anti-PD-1
therapy. It consisted of two-stage phase 2 studies in parallel in the three
cohorts of
patients described in the text. The study agent, MK-3475, was administered at
10
mg/kg intravenously every 14 days.
[79] For each of Cohort A and B, the co-primary endpoints were progression-
free-survival
(irPFS) at 20 weeks and objective response (ir0R) assessed using immune
related
criteria. A step-down gatekeeping procedure was used to preserve the overall
type I
error. A two-stage Green-Dahlberg design was used to evaluate irPFS, with
interim
and final analysis after 15 and 25 patients, respectively. At stage 1, > 1 of
15 free-of-
progression at 20 weeks were required to proceed to the second stage, and > 4
of 25
free-of-progression at 20 weeks were then required to proceed to test for
ir0R, with?
4 of 25 responders (irCR or irPR) indicating promising efficacy in that
cohort. Each
cohort could be terminated for efficacy as soon as > 4 free-of-progression at
20 weeks
and > 4 responses were confirmed, or be terminated for futility as soon as 0
of 15 in
stage 1 were free-of-progression at 20 weeks or > 22 subjects had disease
progression
by 20 weeks. This design achieves 90% power to detect a 20-week irPFS rate of
25%
and 80% power to detect an irOR rate (irORR) of 21%, with an overall type I
error of
0.05 at the null hypothesis of 20-week irPFS rate of 5% and irORR of 5%.
[80] For Cohort C, the primary endpoint was irPFS at 20 weeks. A two-stage
Green-
Dahlberg two-stage design was used, with an interim and final analysis after
14 and
21 patients; at stage 1, > 1 of 14 free-of-progression at 20 weeks were
required to
proceed to the second stage, with > 4 of 21 free-of-progression at 20 weeks at
the end
indicating adequate efficacy in Cohort C. The cohort could be terminated as
soon as?
4 free-of-progression at 20 weeks were confirmed. The design has 81% power to
detect a 20-week irPFS rate of 25% with a 5% type I error at the null
hypothesis of 20-
week irPFS rate of 5%.
[81] Statistical analysis
21
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
[82] Response and progression were evaluated using RECIST v1.1 and the immune-
related
response criteria (irRC) adopted from Wolchok et al.8, which uses the sum of
the
products of bidimensional tumor measurements and incorporates new lesions into
the
sum. Progression-free survival (PFS) rates and irPFS rate at 20-weeks was
estimated
as the proportion of patients who were free-of-disease progression and alive
at 20
weeks after the initiation of pembrolizumab. Patients who had disease
progression
prior to 20 weeks or were enrolled for >20 weeks at the time the study data
were
collated were included in the analysis for estimating 20-week PFS (irPFS)
rate.
Patients who dropped out early due to toxicities or worsening disease and
therefore
did not have 20-week tumor assessment were considered as having progressive
disease. ORR (irORR) was the proportion of patients who achieved best overall
response of CR or PR (irCR or irPR). Patients who were in the study long
enough to
have tumor response evaluations were included in the analysis for estimating
response
rates. Among those who responded (CR or PR), duration of response was the time
of
first RECIST response to the time of disease progression, and was censored at
the last
evaluable tumor assessment for responders who had not progressed.
[83] PFS and irPFS were defined as the time from the date of initial dose to
the date of
disease progression or the date of death due to any cause, whichever occurred
first.
PFS and irPFS were censored on the date of the last evaluable tumor assessment
documenting absence of progressive disease for patients who were alive and
progression-free. Overall survival (OS) was defined as the time from the date
of
initial dose to death due to any cause. For patients who were still alive at
the time of
analysis, the OS time was censored on the last date the patients were known to
be
alive. Survival times were summarized by the Kaplan-Meier method. As a post
hoc
analysis, log-rank tests were used to compare Cohort A and B and hazard ratios
were
estimated based on Cox models.
[84] The association of percent CEA decline after 1 cycle with PFS or OS was
assessed
using landmark analysis based on Cox regression models. For correlative
studies, non-
parametric Wilcoxon test was used to compare mutational load between MMR-
deficient and MMR-proficient patients. The effects of baseline mutational
burden and
22
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
immune markers on response and survival times were examined using logistic
regression and Cox regression, respectively.
[85] IMMUNOHISTOCHEMISTRY & IMAGE ANALYSIS
[86] The fraction of malignant cells exhibiting a membranous pattern of B7-H1
expression
and the percentage at the invasive front were quantified by three pathologists
(R.A.A.,
F.B., and J.M.T.) as previously reported9,10. Image analysis was used to
determine
the number of CD8 diaminobenzidine (DAB)-stained cells. Using the H&E-stained
slide for each case, we identified the following regions: i) tumor, ii)
invasive front (the
boundary between malignant and non-malignant tissue), and iii) normal tissue.
The
CD8-stained slides were scanned at 20x equivalent magnification (0.49
micrometers
per pixel) on an Aperio ScanScope AT. Regions corresponding to tumor, invasive
front and normal tissue (above, from the H&E) were annotated on separate
layers
using Aperio ImageScope v12.1Ø5029.
[87] CD8-positive lymphocyte density was calculated in each of the above
regions using a
custom algorithm implemented in PIP11. Results were converted to Deepzoom
images using the VIPS library12 and visualized using the OpenSeadragon viewer
( http://openseadragon.gi th ub,i o).
[88] References for Example 3 only.
1. Bacher JW, Flanagan LA, Smalley RL, et al. Development of a fluorescent
multiplex
assay for detection of MSI-High tumors. Disease markers 2004;20:237-50.
2. Murphy KM, Zhang S, Geiger T, et al. Comparison of the microsatellite
instability
analysis system and the Bethesda panel for the determination of microsatellite
instability in
colorectal cancers. The Journal of molecular diagnostics : JMD 2006;8:305-11.
3. Jones S, Anagnostou V, Lytle K, et al. Personalized genomic analyses for
cancer
mutation discovery and interpretation. Science translational medicine
2015;7:283ra53.
23
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
4. Sausen M, Leary RJ, Jones S, et al. Integrated genomic analyses identify
ARID1A and
ARID1B alterations in the childhood cancer neuroblastoma. Nature genetics
2013;45:12-7.
5. Needleman SB, Wunsch CD. A general method applicable to the search for
similarities in the amino acid sequence of two proteins. Journal of molecular
biology
1970;48:443-53.
6. Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund 0, Nielsen M. NetMHC-
3.0:
accurate web accessible predictions of human, mouse and monkey MHC class I
affinities for
peptides of length 8-11. Nucleic acids research 2008;36:W509-12.
7. Buyse M, Michiels S, Sargent DJ, Grothey A, Matheson A, de Gramont A.
Integrating
biomarkers in clinical trials. Expert review of molecular diagnostics
2011;11:171-82.
8. Wolchok JD, Hoos A, O'Day S, et al. Guidelines for the evaluation of
immune therapy
activity in solid tumors: immune-related response criteria. Clinical cancer
research : an
official journal of the American Association for Cancer Research 2009;15:7412-
20.
9. Llosa NJ, Cruise M, Tam A, et al. The vigorous immune microenvironment
of
microsatellite instable colon cancer is balanced by multiple counter-
inhibitory checkpoints.
Cancer Discov 2015:43-51.
10. Taube JM, Anders RA, Young GD, et al. Colocalization of Inflammatory
Response
with B7-H1 Expression in Human Melanocytic Lesions Supports an Adaptive
Resistance
Mechanism of Immune Escape. Science Translational Medicine 2012;4:127ra37.
11. Cuka N, Hempel H, Sfanos K, De Marzo A, Cornish T. PIP: An Open Source
Framework for Multithreaded Image Analysis of Whole Slide Images. LABORATORY
INVESTIGATION 2014;94:398A-A.
12. Cupitt J, Martinez K. VIPS: an image processing system for large
images. Electronic
Imaging: Science & Technology; 1996: International Society for Optics and
Photonics. p. 19-
28.
24
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
EXAMPLE 4
[89] Patients
[90] 41 consecutive patients were enrolled and treated between September 2013
and
January 2015. (Table 1). Recruitment included patients in pursuit of a
clinical trial
option who were known to have tumors with mismatch repair, or who had tumors
of
unknown status who were then tested. One patient in the MMR-deficient CRC
cohort
was enrolled under an IRB eligibility waiver allowing a grade 3 bilirubin
level. A total
of 32 CRC patients were enrolled into Cohorts A and B. All CRC patients
received
>2 prior chemotherapy regimens (median=4) except for one MMR-proficient
patient
who had received one chemotherapeutic and one (non-PD1-based)
immunotherapeutic
regimen.
[91] Nine subjects diagnosed with MMR-deficient solid tumors other than CRC
were
enrolled onto Cohort C. All Cohort C patients received >1 prior cancer
treatments
(median=2).
EXAMPLE 5
[92] Primary endpoint evaluation
[93] The irORR and irPFS at 20 weeks (Fig. 11 (Table S2)) for Cohort A were
40% (4 of
patients; 95% CI, 12 to 74%) and 78% (7 of 9 patients; 95% CI, 40 to 97%) and
for
Cohort C were 71% (5 of 7 patients; 95% CI, 29 to 96%) and 67% (4 of 6
patients;
95% CI, 22 to 96%). In Cohort B, comprised of patients with MMR-proficient
CRCs,
irORR and 20-week irPFS were 0% (95%CI, 0 to 20%) and 11% (2 of 18 patients;
95% CI, 1 to 35%). Both the MMR-deficient cohorts A and C reached their
predefined early stopping rule for efficacy when four subjects were free-of-
disease
progression at 20 weeks and four objective responses were observed based on
immune-related response criteria (Fig. 11 (Table S2); available on line at New
England Journal of Medicine; incorporated by reference herein; and
supplementary
methods, above).
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
[94] The median time of follow-up for patients was 32 weeks (range, 5-51
weeks) for
patients with MMR-deficient CRC (Cohort A), 12 weeks (range, 2-56 weeks) for
patients with MMR-proficient CRC (Cohort B) and 12 weeks (range, 4-42 weeks)
for
patients with MMR-deficient non-CRC tumors (Cohort C). All patients evaluable
for
20-week irPFS were followed for at least 20 weeks.
EXAMPLE 6
[95] Radiographic evaluation
[96] Of the ten evaluable MMR-deficient CRC patients in Cohort A, four (40%;
95% CI,
12-74%) achieved objective responses by RECIST criteria (Table 2, Fig. 1 and
Fig. 3
(S2)). Patients were considered not evaluable unless they underwent a 12-week
scan.
The disease control rate was defined as the fraction of patients who achieved
an
objective response or whose disease was stable, and was 90% in Cohort A (9 of
10
patients; 95% CI, 55-100%).
[97] Of the seven evaluable patients with MMR-deficient cancer types other
than CRC
enrolled in Cohort C, five (71%; 95% CI, 29-96%) achieved objective responses
(Table 2, Fig. 3 (S2) and Fig. 1) using RECIST criteria and the disease
control rate
was 71% (5 of 7 patients; 95% CI, 29-96%).
[98] Patients in Cohort C responded faster than patients in Cohort A (median
time to
response by RECIST of 12 vs. 28 weeks, p=0.03). Furthermore, all six MMR-
deficient tumors that were not associated with Lynch syndrome (100%) achieved
an
objective response, whereas only three of eleven tumors (27%) associated with
Lynch
Syndrome responded (Table S3; p=0.009; available on-line at New England
Journal of
Medicine; and incorporated by reference herein). No other baseline
characteristics
showed statistically significant association with objective responses.
[99] Of the 18 patients with MMR-proficient CRCs in Cohort B, no objective
responses
were observed (Table 2, Fig. 3 (S2) and Fig. 1) using RECIST criteria and the
disease
control rate was 11% (2 of 18 patients; 95%CI, 1 to 35%).
26
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
[100] All patients who achieved a response by RECIST criteria (Fig. 11 (Table
2)) also
achieved a response by immune-related response criteria (Fig. 11 (Table S2)).
EXAMPLE 7
[101] Survival
[102] In Cohort A, the patients with MMR-deficient CRC, median progression-
free survival
(PFS) and median overall survival (OS) were not reached (Fig. 2). In contrast,
the
patients with MMR-proficient cancers in Cohort B achieved a PFS of only 2.2
months
(95% CI, 1.4-2.8) and a median OS of 5.0 months (95% CI, 3.0 to not
estimable). In
Cohort C (MMR-deficient non-CRC), the median PFS was 5.4 months (95% CI, 3 to
not estimable) and the median OS was not reached.
[103] A post hoc (Fig. 2) comparison of the MMR-deficient and proficient CRC
cohorts
showed hazard ratios (HR) for disease progression (HR=0.10; 95% CI, 0.03-0.37;
p<0.001) and overall survival (HR = 0.22; 95% CI, 0.05-1.00; p=0.05), favoring
patients with MMR-deficient CRC.
[104] To evaluate whether the difference in survival might be due to
prognostic differences,
we measured the duration of time patients had been diagnosed with metastatic
disease
and the clinical performance of patients on their previous regimen prior to
enrollment.
We found that there was no significant difference between MMR-deficient vs.
MMR-
proficient CRC patients with respect to their duration of metastatic disease
(p=0.77;
Log-rank test) or median PFS (p=0.60, Log-rank test) on their prior regimens
(Fig. 4
(S3)). We also performed an additional multivariate analysis of PFS and OS to
examine the difference in outcomes between MMR-deficient CRC and MMR-
proficient tumors adjusting for elapsed time since initial diagnosis. The
magnitude of
the hazard ratios for PFS (HR 0.04, 95% CI 0.01-0.21, P<0.001) and OS (HR
0.18,
95% CI 0.03-1.01, P=0.05), representing the different effect of pembrolizumab
between MMR-deficient and MMR-proficient tumors, was maintained after
adjusting
for this potential difference.
27
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
EXAMPLE 8
[105] Safety Assessment
[106] Adverse events occurring in > 5% of patients are listed in Table 3.
Select adverse
events included rash/pruritus (24%), thyroiditis/hypothyroidism/hypophysitis
(10%),
and asymptomatic pancreatitis (15%). While the numbers were small, thyroid
function
abnormalities were limited to the MMR-deficient cohorts (Table 3).
EXAMPLE 9
[107] Tumor markers
[108] In the two CRC cohorts, baseline CEA levels were evaluable and above the
upper
limit of normal (3 mg/di), in 29 of 32 patients prior to enrollment. Major CEA
declines occurred in seven of the ten patients with MMR-deficient CRC and in
none of
the 19 patients with MMR-proficient CRCin which CEA was evaluable (Fig. 1 and
Fig.
(S4)). In non-CRC MMR-deficient patients, tumor marker levels (CEA, CA19-9 or
CA-125) were elevated above the upper limit of normal in four patients. CA19-9
or
CA-125 declines of > 70% occurred in three of these four patients. Tumor
marker
kinetics of all 3 cohorts are shown in Figure 1. The level of CEA decline
after 1 dose
(between days 14 and 28) of pembrolizumab was predictive of both progression-
free
(p=0.01) and overall survival outcomes (p=0.02). The CEA response occurred
well in
advance of radiographic confirmation of disease control (range, 10 to 35
weeks). In
contrast, patients who progressed showed rapid biomarker elevation within 30
days of
initiating therapy. Thus, changes in CEA levels significantly preceded and
correlated
with ultimate radiographic changes.
EXAMPLE 10
[109] Genomic Analysis
[110] Analysis of whole-exome sequences showed an average of 1,782 somatic
mutations
per tumor in MMR-deficient patients (n = 9) compared with 73 mutations per
tumor
in MMR-proficient patients (n=6) (non-parametric Wilcoxon test, p=0.007)
(Figs. 6A-
28
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
6B (S5); see also Table S3 which is available on-line at New England Journal
of
Medicine; incorporated by reference herein). Most (63%) of these mutations are
predicted to alter amino acids.
[111] These mutations were then assessed for their immunogenic potential in
the context of
each patient's individual MHC haplotype. We thereby identified an average of
578
and 21 potential mutation-associated neoantigens from the tumors of MMR-
deficient
and MMR-proficient patients, respectively (Table S3; which is available on-
line at
New England Journal of Medicine; incorporated by reference herein). The
fraction of
potential mutation-associated neoantigens among all somatic mutations was
similar in
both cohorts (averaging 32% and 29% in MMR-deficient and -proficient patients,
respectively). High numbers of somatic mutations and potential mutation-
associated
neoantigens were associated with improved progression-free survival and with a
trend
in favor of objective response (Fig. 13 (S5) and Fig. 12 (Table S4); also
available on
line at New England Journal of Medicine; incorporated by reference herein).
EXAMPLE 11
[112] Immunohistochemistry
[113] Expression of CD8 and PD-Li were evaluated by immunohistochemistry
within the
tumor and at the invasive fronts of the tumor in the 30 cases in which tumor
tissue
was available (Fig. 7 (S6); also available on line at New England Journal of
Medicine;
incorporated by reference herein). Tumors from patients in Cohorts A and C
contained a greater density of CD8-positive lymphoid cells than did tumors
from
Cohort B patients (Fig. 8 (S7); p=0.10) and CD8-labeling was associated with a
trend
favoring objective response and stable disease (Fig. 9 (S8) and Fig. 13 (Table
S5);
also available on line at New England Journal of Medicine; incorporated by
reference
herein). This CD8-positive lymphoid infiltrate was especially prominent at the
invasive fronts of the tumors (Fig 8 (S7); p=0.04). Significant membranous PD-
Li
expression only occurred in MMR-deficient patients and was prominent on tumor
infiltrating lymphocytes (TILs) and tumor-associated macrophages located at
the
tumors' invasive fronts (Fig. 8 (S7); p=0.04). Expression of CD8 and PD-Li
were not
statistically associated with PFS or OS (Fig. 13 (Table S5)).
29
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
Table 1. Demographic and Baseline Characteristics of Patients
MMR- MRC-
MMR-
deficient proficient
P values1
deficient
Characteristic
CRC CRC
non-CRC
n=11 n=21
n=9
Age ¨ years
median 46 61 0.02
57
range (24-65) (32-79) (34-92)
Sex ¨ no. (%)
Female 5(45) 8(38) 0.72 4(44)
Male 6(55) 13(62) 5(56)
Race ¨ no. (%)
white 8(73) 17(81) 0.66 8(89)
black 1(9) 3(14) 0(0)
other 2(18) 1(5) 1(11)
ECOG Performance Status ¨ no. (%)2
0 0(0) 6(29) 0.07 2(22)
1 11(100) 15(71) 7(78)
Diagnosis ¨ no. (%)
Colon 9(82) 18(86) >0.99 0(0)
Rectal 2(18) 3(14) 0(0)
Ampullary/Cholangiocarcinoma 0(0) N/A 4(44)
Endometrial 0(0) N/A 2(22)
Small bowel 0(0) N/A 2(22)
Gastric 0(0) N/A 1(11)
Histology ¨ no. (%)
Well/moderately differentiated 7(64) 18(86) 0.20 4(44)
Poorly differentiated 4(36) 3(14) 3(33)
Other 0(0) 0(0) 2(22)
Stage IV ¨ no. (%) (11)100 21(100) >0.99
9(100)
Liver metastases ¨ no. (%) 6(55) 11(52) >0.99
6(67)
Time since first diagnosis - months
median 31 58 0.07
23
range 6-95 27-192 2-105
Prior systemic therapies ¨ no. (%)
0 0(0) 0(0) 0.89 1(11)
2 3(27) 4(19) 5(56)
3 3(27) 5(24) 1(11)
>4 5(45) 12(57) 2(22)
Detected germline mutation or known Lynch ¨ no. (%)
Yes 9(82) 0(0) <0.001 4(44)
No 2(18) 21(100) 4(44)
Unknown 0(0) 0(0) 1(11)
BRAF wild type ¨ no. (%)
Yes 8(73) 11(52) 0.64 4(44)
No 0(0) 1(5) 0(0)
Unknown 3(27) 9(43) 5(56)
KRAS wild type ¨ no. (%)
Yes 6(55) 13(62) 0.72 4(44)
No 5(45) 8(38) 1(11)
Unknown 0(0) 0(0) 4(44)
MMR, mismatch repair; CRC, colorectal cancer
1 MMR-deficient CRC versus MMR-proficient CRC
2 ECOG, Eastern Cooperative Oncology Group
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
Table 2. Objective RECIST responses
MMR-deficient MRC-proficient MMR-deficient
Type of Response ¨ no. (%) CRC CRC
non-CRC
n=10 n=18 n=7
Complete Response 0(0) 0(0) 1(14)1
Partial Response 4(40) 0(0) 4(57)2
Stable Disease (Week 12) 5(50) 2(11) 0(0)
Progressive Disease 1(10) 11(61) 2(29)
Not Evaluable3 0(0) 5(28) 0(0)
Objective Response Rate (`)/0) 40 0 71
95% Cl 12-74 0-19 29-
96
Disease Control Rate (%)4 90 11 71
95% Cl 55-100 1-35 29-
96
Duration of Response ¨ median weeks Not reached N/A5
Not reached
Time to Response, median weeks (range) 28 (13-35) N/A5
11(10-13)
1 Originally PR at 12 weeks that was converted to CR at 20 weeks
2 One PR at 12 weeks
3 Patients were considered not evaluable if they did not undergo a 12 week
scan due to clinical progression.
4 The rate of disease control was defined as the percentage of patients who
had a complete response, partial
response or stable disease for 12 weeks or more.
No responses recorded for MMR-proficient CRC patients
31
CA 02966660 2017-05-02
WO 2016/077553
PCT/US2015/060331
Table 3. Drug-Related Adverse Events
Event ¨ no (%)1 All Grades Grade 3 or 4
N=41 N=41
Any 40(98) 17(41)
Blood and Lymphatic
Anemia 8(20) 7(17)
Lymphopenia 8(20) 8(20)
Cardiac
Sinus tachycardia 4(10) 0
Dermatologic
Dry skin 5(12) 0
Rash/pruritis 10(24) 0
Endocrine Disorders
Thyroiditis/Hypothyroidism/Hypophysitis 4(10) 0
Gastrointestinal
Abdominal Pain 10(24) 0
Anorexia 4(10) 0
Constipation 8(20) 0
Diarrhea 10(24) 2(5)
Dry mouth 5(12) 0
Nausea 5(12) 0
Bowel Obstruction 3(7) 3(7)
Hepatobiliary
ALT, elevated 3(7) 2(5)
Pancreatitis2 6(15) 0
Metabolism and Nutrition
Hypoalbuminemia 4(10) 4(10)
Hyponatremia 3(7) 3(7)
Musculoskeletal
Arthralgia 7(17) 0
Myalgia 6(15) 0
Nervous System
Dizziness 4(10) 0
Headache 7(17) 0
Psychiatric
Insomnia 3(7) 0
Respiratory'
Allergic Rhinitis 12(29) 0
Cough 4(10) 0
Dyspnea 6(15) 0
Upper Respiratory Infection 3(7) 0
Other
Cold intolerance 6(15) 0
Edema 4(10) 0
Fatigue 13(32) 0
Fever 5(12) 0
Pain 14(34) 0
'Adverse Events occurring in greater than 5% of patients
2 All cases of pancreatitis were asymptomatic
3 One incidence of pneumonitis (2%)
32
CA 02966660 2017-05-02
WO 2016/077553
PCT/US2015/060331
References
The disclosure of each reference cited is expressly incorporated herein.
1. Nishimura H, Okazaki T, Tanaka Y, et al. Autoimmune Dilated
Cardiomyopathy in PD-1 Receptor-Deficient Mice. Science 2001;291:319-22.
2. Chen L. Co-inhibitory molecules of the B7-CD28 family in the control of
T-
cell immunity. Nat Rev Immunol 2004;4:336-47.
3. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of Lupus-
like Autoimmune Diseases by Disruption of the PD-1 Gene Encoding an ITIM Motif-
Carrying Immunoreceptor. Immunity 1999;11:141-51.
4. Anse11 SM, Lesokhin AM, Borrello I, et al. PD-1 Blockade with Nivolumab
in
Relapsed or Refractory Hodgkin's Lymphoma. The New England journal of medicine
2015;372:311-9.
5. Hamid 0, Robert C, Daud A, et al. Safety and tumor responses with
lambrolizumab (anti-PD-1) in melanoma. The New England journal of medicine
2013;369:134-44.
6. Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of
response to
the anti-PD-Li antibody MPDL3280A in cancer patients. Nature 2014;515:563-7.
7. Powles T, Eder JP, Fine GD, et al. MPDL3280A (anti-PD-L1) treatment
leads
to clinical activity in metastatic bladder cancer. Nature 2014;515:558-62.
8. Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor
remission, and long-term safety in patients with advanced melanoma receiving
nivolumab. Journal of clinical oncology: official journal of the American
Society of
Clinical Oncology 2014;32:1020-30.
33
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
9. Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-Li
antibody in patients with advanced cancer. The New England journal of medicine
2012;366:2455-65.
10. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune
correlates of anti-PD-1 antibody in cancer. The New England journal of
medicine
2012;366:2443-54.
11. Taube JM, Klein A, Brahmer JR, et al. Association of PD-1, PD-1
ligands, and
other features of the tumor immune microenvironment with response to anti-PD-1
therapy. Clinical cancer research : an official journal of the American
Association for
Cancer Research 2014;20:5064-74.
12. Brahmer JR, Drake CG, Wollner I, et al. Phase I study of single-agent
anti-
programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical
activity,
pharmacodynamics, and immunologic correlates. Journal of clinical oncology:
official journal of the American Society of Clinical Oncology 2010;28:3167-75.
13. Koopman M, Kortman GAM, Mekenkamp L, et al. Deficient mismatch repair
system in patients with sporadic advanced colorectal cancer. Br J Cancer
0000;100:266-73.
14. Goldstein J, Tran B, Ensor J, et al. Multicenter retrospective analysis
of
metastatic colorectal cancer (CRC) with high-level microsatellite instability
(MSI-H).
Annals of oncology: official journal of the European Society for Medical
Oncology /
ESMO 2014;25:1032-8.
15. Segal NH, Parsons DW, Peggs KS, et al. Epitope landscape in breast and
colorectal cancer. Cancer research 2008;68:889-92.
34
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
16. Timmermann B, Kerick M, Roehr C, et al. Somatic mutation profiles of
MSI
and MSS colorectal cancer identified by whole exome next generation sequencing
and
bioinformatics analysis. PloS one 2010;5:e15661.
17. Eshleman JR, Lang EZ, Bowerfind GK, et al. Increased mutation rate at
the
hprt locus accompanies microsatellite instability in colon cancer. Oncogene
1995;10:33-7.
18. Comprehensive molecular characterization of human colon and rectal
cancer.
Nature 2012;487:330-7.
19. Dolcetti R, Viel A, Doglioni C, et al. High prevalence of activated
intraepithelial cytotoxic T lymphocytes and increased neoplastic cell
apoptosis in
colorectal carcinomas with microsatellite instability. The American journal of
pathology 1999;154:1805-13.
20. Alexander J, Watanabe T, Wu TT, Rashid A, Li S, Hamilton SR.
Histopathological identification of colon cancer with microsatellite
instability. The
American journal of pathology 2001;158:527-35.
21. Smyrk TC, Watson P, Kaul K, Lynch HT. Tumor-infiltrating lymphocytes
are
a marker for microsatellite instability in colorectal carcinoma. Cancer
2001;91:2417-
22.
22. Young J, Simms LA, Biden KG, et al. Features of colorectal cancers
with
high-level microsatellite instability occurring in familial and sporadic
settings: parallel
pathways of tumorigenesis. The American journal of pathology 2001;159:2107-16.
23. Berger MF, Hodis E, Heffernan TP, et al. Melanoma genome sequencing
reveals frequent PREX2 mutations. Nature 2012;485:502-6.
24. Lee W, Jiang Z, Liu J, et al. The mutation spectrum revealed by paired
genome
sequences from a lung cancer patient. Nature 2010;465:473-7.
CA 02966660 2017-05-02
WO 2016/077553 PCT/US2015/060331
25. Lipson EJ, Sharfman WH, Drake CG, et al. Durable cancer regression off-
treatment and effective reinduction therapy with an anti-PD-1 antibody.
Clinical
cancer research : an official journal of the American Association for Cancer
Research
2013;19:462-8.
26. Boland CR, Goel A. Microsatellite instability in colorectal cancer.
Gastroenterology 2010;138:2073-87 e3.
27. Lynch HT, de la Chapelle A. Hereditary colorectal cancer. The New
England
journal of medicine 2003;348:919-32.
28. Yamamoto H, Imai K, Perucho M. Gastrointestinal cancer of the
microsatellite
mutator phenotype pathway. Journal of gastroenterology 2002;37:153-63.
29. Herman JG, Umar A, Polyak K, et al. Incidence and functional
consequences
of hMLH1 promoter hypermethylation in colorectal carcinoma. Proceedings of the
National Academy of Sciences of the United States of America 1998;95:6870-5.
30. Jones S, Anagnostou V, Lytle K, et al. Personalized genomic analyses
for
cancer mutation discovery and interpretation. Science translational medicine
2015;7:283ra53.
31. Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund 0, Nielsen M.
NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey
MHC class I affinities for peptides of length 8-11. Nucleic acids research
2008;36:W509-12.
32. Lundegaard C, Lund 0, Nielsen M. Accurate approximation method for
prediction of class I MHC affinities for peptides of length 8, 10 and 11 using
prediction tools trained on 9mers. Bioinformatics 2008;24:1397-8.
33. Wolchok JD, Hoos A, O'Day S, et al. Guidelines for the evaluation of
immune
therapy activity in solid tumors: immune-related response criteria. Clinical
cancer
36
CA 02966660 2017-05-02
WO 2016/077553
PCT/US2015/060331
research : an official journal of the American Association for Cancer Research
2009;15:7412-20.
34. Maple JT, Smyrk TC, Boardman LA, Johnson RA, Thibodeau SN, Chari ST.
Defective DNA mismatch repair in long-term (> or =3 years) survivors with
pancreatic cancer. Pancreatology 2005;5:220-7; discussion 7-8.
35. Meltzer SJ, Yin J, Manin B, et al. Microsatellite instability occurs
frequently
and in both diploid and aneuploid cell populations of Barrett's-associated
esophageal
adenocarcinomas. Cancer research 1994;54:3379-82.
36. Nakata B, Wang YQ, Yashiro M, et al. Prognostic value of microsatellite
instability in resectable pancreatic cancer. Clinical cancer research: an
official journal
of the American Association for Cancer Research 2002;8:2536-40.
37. Comprehensive molecular characterization of gastric adenocarcinoma.
Nature
2014;513:202-9.
38. Agaram NP, Shia J, Tang LH, Klimstra DS. DNA mismatch repair deficiency
in ampullary carcinoma: a morphologic and immunohistochemical study of 54
cases.
American journal of clinical pathology 2010;133:772-80.
39. Kandoth C, Schultz N, Cherniack AD, et al. Integrated genomic
characterization of endometrial carcinoma. Nature 2013;497:67-73.
40. Garg K, Leitao MM, Jr., Kauff ND, et al. Selection of endometrial
carcinomas
for DNA mismatch repair protein immunohistochemistry using patient age and
tumor
morphology enhances detection of mismatch repair abnormalities. The American
journal of surgical pathology 2009;33:925-33.
41. Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical
response
to CTLA-4 blockade in melanoma. The New England journal of medicine
2014;371:2189-99.
37
CA 02966660 2017-05-02
WO 2016/077553
PCT/US2015/060331
42. Williams AS, Huang WY. The analysis of microsatellite instability in
extracolonic gastrointestinal malignancy. Pathology 2013;45:540-52.
43. Jones S, Emmerson P, Maynard J, et al. Biallelic germline mutations in
MYH
predispose to multiple colorectal adenoma and somatic G:C¨>T:A mutations.
Human
Molecular Genetics 2002;11:2961-7.
44. Palles C, Cazier J-B, Howarth KM, et al. Germline mutations affecting
the
proofreading domains of POLE and POLD1 predispose to colorectal adenomas and
carcinomas. Nature genetics 2013;45:136-44.
45. Bodmer W, Bishop T, Karran P. Genetic steps in colorectal cancer.
Nature
genetics 1994;6:217-9.
46. Kim H, Jen J, Vogelstein B, Hamilton SR. Clinical and pathological
characteristics of sporadic colorectal carcinomas with DNA replication errors
in
microsatellite sequences. The American journal of pathology 1994;145:148-56.
47. Llosa NJ, Cruise M, Tam A, et al. The vigorous immune microenvironment
of
microsatellite instable colon cancer is balanced by multiple counter-
inhibitory
checkpoints. Cancer Discov 2015:43-51. [This disclosure was made by some of
the
joint inventors.]
48. Rizvi NA, Hellmann MD, Snyder A, et al. Mutational landscape determines
sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015.
49. Gubin MM, Zhang X, Schuster H, et al. Checkpoint blockade cancer
immunotherapy targets tumour-specific mutant antigens. Nature 2014;515:577-81.
50. Linnemann C, van Buuren MM, Bies L, et al. High-throughput epitope
discovery reveals frequent recognition of neo-antigens by CD4+ T cells in
human
melanoma. Nature medicine 2015;21:81-5.
38
CA 02966660 2017-05-02
WO 2016/077553
PCT/US2015/060331
51. Lipson EJ, Velculescu VE, Pritchard TS, et al. Circulating tumor DNA
analysis as a real-time method for monitoring tumor burden in melanoma
patients
undergoing treatment with immune checkpoint blockade. Journal for
immunotherapy
of cancer 2014;2:42.
52. Diaz LA, Jr., Bardelli A. Liquid biopsies: genotyping circulating tumor
DNA.
Journal of clinical oncology: official journal of the American Society of
Clinical
Oncology 2014;32:579-86.
53. Yadav M, Jhunjhunwala S, Phung QT, et al. Predicting immunogenic tumour
mutations by combining mass spectrometry and exome sequencing. Nature
2014;515:572-6.
39